JP5115972B2 - Process for producing aromatic amides - Google Patents
Process for producing aromatic amides Download PDFInfo
- Publication number
- JP5115972B2 JP5115972B2 JP2008064793A JP2008064793A JP5115972B2 JP 5115972 B2 JP5115972 B2 JP 5115972B2 JP 2008064793 A JP2008064793 A JP 2008064793A JP 2008064793 A JP2008064793 A JP 2008064793A JP 5115972 B2 JP5115972 B2 JP 5115972B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- aromatic
- dichloro
- less
- represented
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *c(c(Cl)c(c(N)c1)O)c1Cl Chemical compound *c(c(Cl)c(c(N)c1)O)c1Cl 0.000 description 3
Description
本発明は、カラー写真薬シアンカプラーとして用いられる芳香族アミド類の製造法に関する。 The present invention relates to a method for producing an aromatic amide used as a color photographic drug cyan coupler.
カラー写真薬シアンカプラーとして使用される芳香族アミド類の製造法としては、o−アミノフェノール類を酢酸中で、酢酸ソーダの存在下、酸クロリド類と縮合反応させる方法(特許文献1) 、o−アミノフェノール類の塩酸塩をアセトン溶媒中、キノリンの存在下に酸クロリド類と反応させる方法(特許文献2)、Ni系触媒で水添して得られるo−アミノフェノール類を酸クロリド類と縮合反応させる方法(特許文献3)、o−アミノフェノール類の塩酸塩を酸クロリド類と縮合反応させる方法(特許文献4)等が知られている。 As a method for producing an aromatic amide used as a color photographic drug cyan coupler, a method in which o-aminophenols are subjected to a condensation reaction with acid chlorides in the presence of sodium acetate in acetic acid (Patent Document 1), o -Method of reacting hydrochloride of aminophenol with acid chloride in acetone solvent in presence of quinoline (Patent Document 2), o-aminophenol obtained by hydrogenation with Ni-based catalyst and acid chloride A method of performing a condensation reaction (Patent Document 3), a method of performing a condensation reaction of hydrochlorides of o-aminophenols with acid chlorides (Patent Document 4), and the like are known.
しかしながらこれらの方法では、o−アミノフェノール類を塩酸塩や硫酸塩として一旦分離する工程、あるいは、水などの貧溶媒で稀釈晶析して芳香族アミド類を分離する工程が必要であることから、得られた芳香族アミド類の品質がカラー写真薬シアンカプラーとしての使用においては十分なものではないと言う問題点や、溶媒の回収、再使用が容易でない等の問題点があった。 However, these methods require a step of once separating o-aminophenol as a hydrochloride or sulfate, or a step of separating an aromatic amide by diluting crystallization with a poor solvent such as water. However, there are problems that the quality of the obtained aromatic amides is not sufficient for use as a color photographic cyan coupler, and that it is not easy to recover and reuse the solvent.
本発明の目的は、工業薬品、特にカラー写真感光材料に用いられるシアンカプラーとしての使用において十分な高品質の芳香族アミド類を工業的に容易に製造する方法を提供することである。 An object of the present invention is to provide a method for industrially easily producing high-quality aromatic amides sufficient for use as industrial chemicals, particularly cyan couplers used in color photographic light-sensitive materials.
本発明者は、高品質の写真薬カプラーを工業的に有利に得るための方法について検討した結果、本発明者らは、o−アミノフェノール類を塩酸塩または硫酸塩などとして単離することなく、窒素などの不活性ガス雰囲気下でアミド化縮合反応し、芳香族系不活性溶媒を減圧下で濃縮して、芳香族アミド類を晶析することにより、簡便で収率よく、且つ、工業的に有利な生産性に優れた方法で高品質の芳香族アミド化合物が得られることを見いだし本発明を完成した。
すなわち本発明は、式(1)
で示されるo−ニトロフェノール類を炭素数10以下の芳香族系不活性溶媒中、白金系触媒の存在下に接触還元し、式(2)
で示されるo−アミノフェノール類を得、これを単離することなく式(3)
で示される硫黄分含量が0.5%以下(酸クロリド重量基準)の酸クロリド類と、
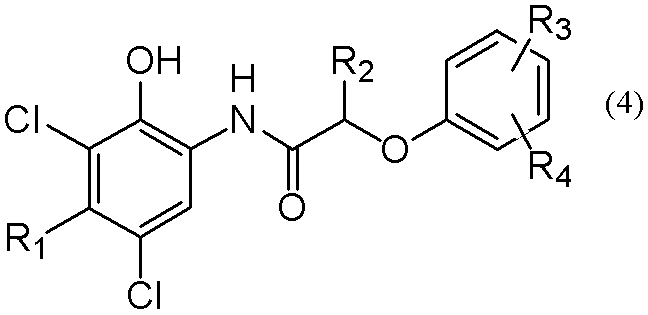
酸素濃度が1%以下の不活性ガス雰囲気下で縮合反応させて、式(4)
で示される芳香族アミド類を得る製造法であって、アミド化縮合反応後、芳香族系不活性溶媒を減圧下で濃縮し、式(4)で示される芳香族アミド類を晶析する工程を含むことを特徴とする式(4)で示される芳香族アミド類の製造法に関する。
As a result of studying a method for industrially obtaining a high-quality photographic drug coupler, the present inventors have found that the present inventors have not isolated o-aminophenols as hydrochlorides or sulfates. By amidating and condensing in an inert gas atmosphere such as nitrogen, concentrating the aromatic inert solvent under reduced pressure, and crystallizing the aromatic amide, the yield is simple, and the industrial The present invention was completed by finding that a high-quality aromatic amide compound can be obtained by a particularly advantageous method with excellent productivity.
That is, the present invention provides the formula (1)
The o-nitrophenol represented by the formula (2) is catalytically reduced in an aromatic inert solvent having 10 or less carbon atoms in the presence of a platinum catalyst.
The o-aminophenol represented by the formula (3) is obtained without isolation.
Acid chlorides having a sulfur content of 0.5% or less (based on the weight of acid chloride),
A condensation reaction is performed in an inert gas atmosphere with an oxygen concentration of 1% or less to obtain a compound of formula (4)
A process for obtaining an aromatic amide represented by the formula (4) by concentrating an aromatic inert solvent under reduced pressure after an amidation condensation reaction: It is related with the manufacturing method of aromatic amides shown by Formula (4) characterized by including this.
本発明の製造方法により、工業薬品、特にカラー写真感光材料に用いられるシアンカプラーとしての使用において十分な高品質の芳香族アミド類を工業的に有利に製造することができる。 According to the production method of the present invention, a sufficiently high-quality aromatic amide can be industrially advantageously produced for use as an industrial chemical, particularly as a cyan coupler used in a color photographic light-sensitive material.
本発明において用いられる式(1)で表されるo−ニトロフェノール類について説明する。
式(1)のo−ニトロフェノール類の置換基であるR1 としては例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチルなどの低級アルキル基が挙げられる。 具体化合物としては、例えば2−ニトロ−4,6−ジクロロ−5−メチルフェノ−ル、2−ニトロ−4,6−ジクロロ−5−エチルフェノ−ル、2−ニトロ−4,6−ジクロロー5−n−プロピルフェノ−ル、2−ニトロ−4,6−ジクロロ−5−イソプロピルフェノ−ル、2−ニトロ−4,6−ジクロロ−5−n−ブチルフェノ−ル、2−ニトロ−4,6−ジクロロ−5−sec −ブチルフェノ−ルが例示される。
The o-nitrophenol represented by the formula (1) used in the present invention will be described.
Examples of R 1 which is a substituent of the o-nitrophenol of the formula (1) include lower alkyl groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl and the like. Specific examples of the compound include 2-nitro-4,6-dichloro-5-methylphenol, 2-nitro-4,6-dichloro-5-ethylphenol, 2-nitro-4,6-dichloro-5-n. -Propylphenol, 2-nitro-4,6-dichloro-5-isopropylphenol, 2-nitro-4,6-dichloro-5-n-butylphenol, 2-nitro-4,6-dichloro An example is -5-sec-butylphenol.
本発明において用いられる式(2)のo−アミノフェノール類について説明する。
式(2)のo−アミノフェノール類の置換基であるR1としては例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチルなどの低級アルキル基が挙げられる。
具体化合物としては、例えば2−アミノ−4,6−ジクロロ−5−メチルフェノール、2−アミノ−4,6−ジクロロ−5−エチルフェノール、2−アミノ−4,6−ジクロロ−5−n−プロピルフェノール、2−アミノ−4,6−ジクロロ−5−イソプロピルフェノール、2−アミノ−4,6−ジクロロ−5−n−ブチルフェノール、2−アミノ−4,6−ジクロロ−5−sec−ブチルフェノールが例示される。
これらの化合物は式(1)で示されるo−ニトロフェノール類を炭素数10以下の芳香族系不活性溶媒中、白金系触媒の存在下に接触還元して得られる。
The o-aminophenol of the formula (2) used in the present invention will be described.
As R < 1 > which is a substituent of o-aminophenol of Formula (2), lower alkyl groups, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, are mentioned, for example.
Specific examples of the compound include 2-amino-4,6-dichloro-5-methylphenol, 2-amino-4,6-dichloro-5-ethylphenol, 2-amino-4,6-dichloro-5-n- Propylphenol, 2-amino-4,6-dichloro-5-isopropylphenol, 2-amino-4,6-dichloro-5-n-butylphenol, 2-amino-4,6-dichloro-5-sec-butylphenol Illustrated.
These compounds are obtained by catalytic reduction of o-nitrophenols represented by the formula (1) in an aromatic inert solvent having 10 or less carbon atoms in the presence of a platinum catalyst.
次に、硫黄分含量が0.5%以下の式(3)の酸クロリド類について説明する。
式(3)の酸クロリド類の置換基R2 、R3 、R4 としては、メチル基、エチル基、n−プロピル基、i−プロピル基、n−ブチル基、i−ブチル基、s−ブチル基、t−ブチル基、n−ペンチル基、i−ペンチル基、s−ペンチル基、t−ペンチル基、neo−ペンチル基、n−ヘキシル基、2−ヘキシル基、3−ヘキシル基、t−ヘキシル基等の低級アルキル基または水素原子があげられる。本発明のアミド化縮合反応では、硫黄分(酸クロリド重量基準でみた量)が多くなると写真用カップラーとして性能が低下する。硫黄分が0.5%以下、より好ましくは0.3%以下の式(3)の酸クロリド類がアミド化縮合反応に用いられる。
Next, the acid chlorides of the formula (3) having a sulfur content of 0.5% or less will be described.
As the substituents R 2 , R 3 and R 4 of the acid chlorides of the formula (3), methyl group, ethyl group, n-propyl group, i-propyl group, n-butyl group, i-butyl group, s- Butyl group, t-butyl group, n-pentyl group, i-pentyl group, s-pentyl group, t-pentyl group, neo-pentyl group, n-hexyl group, 2-hexyl group, 3-hexyl group, t- Examples thereof include a lower alkyl group such as a hexyl group or a hydrogen atom. In the amidation condensation reaction of the present invention, when the sulfur content (amount based on the weight of acid chloride) increases, the performance as a photographic coupler decreases. Acid chlorides of the formula (3) having a sulfur content of 0.5% or less, more preferably 0.3% or less are used in the amidation condensation reaction.
具体的な酸クロリド類としては、2−エチルフェノキシ酢酸クロリド、4−エチルフェノキシ酢酸クロリド、α−(2−イソプロピルフェノキシ)酪酸クロリド、α−(3,5−ジイソプロピルフェノキシ)酪酸クロリド、α−(2−t−アミル−4−メチルフェノキシ)酪酸クロリド、α−(2,4−ジ−t−アミルフェノキシ)酪酸クロリド、α−(2,4−ジ−t−アミルフェノキシ)吉草酸クロリド等のα−置換脂肪族カルボン酸クロリド類が例示される。 Specific acid chlorides include 2-ethylphenoxyacetic acid chloride, 4-ethylphenoxyacetic acid chloride, α- (2-isopropylphenoxy) butyric acid chloride, α- (3,5-diisopropylphenoxy) butyric acid chloride, α- ( 2-t-amyl-4-methylphenoxy) butyric acid chloride, α- (2,4-di-t-amylphenoxy) butyric acid chloride, α- (2,4-di-t-amylphenoxy) valeric acid chloride, etc. α-substituted aliphatic carboxylic acid chlorides are exemplified.
かかる式(3)の酸クロリド類は、例えば硫黄分を含まないホスゲンや塩化オキザリル等をカルボン酸に反応せしめる方法や、オキシ塩化燐をカルボン酸に反応せしめる方法(特開2003−26630号公報)でも得られる。
また、カルボン酸に塩化チオニルを反応させて得られた酸クロリド類の硫黄分を上記の水準まで減らすため蒸留精製して硫黄分含量が0.5%以下、より好ましくは0.3%以下とした物を用いてもよい。
Such acid chlorides of the formula (3) are, for example, a method of reacting phosgene or oxalyl chloride not containing sulfur with a carboxylic acid, or a method of reacting phosphorus oxychloride with a carboxylic acid (Japanese Patent Laid-Open No. 2003-26630). But you can get it.
Further, in order to reduce the sulfur content of the acid chlorides obtained by reacting carboxylic acid with thionyl chloride to the above level, the sulfur content is 0.5% or less, more preferably 0.3% or less. You may use what you did.
次に、式(3)の酸クロリド類を式(2)のo−アミノフェノール類とアミド化縮合反応させる工程について以下説明する。
酸クロリド類の使用量は、式(2)のo−アミノフェノールに対して通常、0.95〜1.05モル倍であり、より好ましくは0.99〜1.00モル倍である。
Next, the step of subjecting the acid chloride of formula (3) to the amidation condensation reaction with the o-aminophenol of formula (2) will be described below.
The usage-amount of acid chloride is 0.95-1.05 mol times normally with respect to o-aminophenol of Formula (2), More preferably, it is 0.99-1.00 mol times.
アミド化縮合反応は式(1)の接触水添で用いた炭素数10以下の芳香族系不活性溶媒中で実施される。不活性溶媒としてはトルエン、キシレン、ベンゼン、エチルベンゼン、クロロベンゼンなどが挙げられ、好ましくはトルエン、キシレンが単独もしくは混合して用いられる。その使用量はo−アミノフェノール類に対して通常5〜20重量倍、好ましくは6〜10重量倍である。 The amidation condensation reaction is carried out in an aromatic inert solvent having 10 or less carbon atoms used in the catalytic hydrogenation of formula (1). Examples of the inert solvent include toluene, xylene, benzene, ethylbenzene, chlorobenzene, and the like. Preferably, toluene and xylene are used alone or in combination. The amount used is usually 5 to 20 times by weight, preferably 6 to 10 times by weight with respect to o-aminophenols.
反応温度は通常30〜40℃である。
反応は通常、o−アミノフェノール類と芳香族系不活性溶媒の中に酸クロリドを滴下するか、10℃以下ですべての試剤を仕込んでから加熱昇温して実施される。
アミド化縮合は、雰囲気中の酸素がo−アミノフェノール類を酸化し、反応に影響を及ぼすので、高純度の芳香族アミド類をより収率よく得るために、酸素濃度1%以下の窒素等の不活性ガス雰囲気下で実施される。
The reaction temperature is usually 30-40 ° C.
The reaction is usually carried out by dropping acid chloride into o-aminophenols and an aromatic inert solvent, or charging all reagents at 10 ° C. or lower and heating to raise the temperature.
In amidation condensation, oxygen in the atmosphere oxidizes o-aminophenol and affects the reaction. Therefore, in order to obtain high-purity aromatic amides with higher yield, nitrogen or the like with an oxygen concentration of 1% or less In an inert gas atmosphere.
この反応においては塩化水素が発生することから、脱酸剤を用いることが好ましい。脱酸剤を用いることにより反応をより容易に行うことができる。
使用できる脱酸剤としては、炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩、炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属重炭酸塩などの無機塩基類があげられる。
その使用量は、二酸塩基を用いたときはo−アミノフェノール類に対して通常0.75モル倍程度までであり、一酸塩基を用いたときは、1.5モル倍程度までである。
Since hydrogen chloride is generated in this reaction, it is preferable to use a deoxidizing agent. The reaction can be performed more easily by using a deoxidizer.
Examples of the deoxidizer that can be used include inorganic bases such as alkali metal carbonates such as sodium carbonate and potassium carbonate, and alkali metal bicarbonates such as sodium hydrogen carbonate and potassium hydrogen carbonate.
The amount used is usually up to about 0.75 mol times with respect to o-aminophenol when a diacid base is used, and is up to about 1.5 mol times with a monoacid base. .
本発明においては、アミド化縮合反応後の反応液から不活性溶媒を減圧下で濃縮し、式(4)で示される芳香族アミド類を晶析する工程を含む。本工程はどの段階で実施しても良い。通常、反応後の第一の工程において実施されるが、反応液を水洗浄し分液により水分離した反応液において実施することが水溶解性の不純物を除去する点からより好ましい。本発明の工程を実施後、ろ過工程に供し目的物である高品質の芳香族アミド類を収率よく取り出すことができる。ろ過工程に供し取り出した芳香族アミド類のウエットケーキ中には水分を実質的に含まないことから乾燥工程で不活性溶媒を煩雑な操作を行うことなく回収することができ、不活性溶媒の再使用が容易に行える。 In this invention, the process of concentrating an inert solvent from the reaction liquid after amidation condensation reaction under reduced pressure, and crystallizing the aromatic amides shown by Formula (4) is included. This step may be performed at any stage. Usually, it is carried out in the first step after the reaction, but it is more preferred to carry out the reaction in a reaction solution that has been washed with water and separated by liquid separation from the viewpoint of removing water-soluble impurities. After carrying out the process of the present invention, it can be subjected to a filtration process to take out a high-quality aromatic amide as a target product with a high yield. Since the wet cake of aromatic amides taken out in the filtration step is substantially free of moisture, the inert solvent can be recovered in the drying step without complicated operations. Easy to use.
本発明において、不活性溶媒を減圧下で濃縮する温度は30〜60℃が好ましい。濃縮温度が30℃未満の場合は濃縮に長時間要し、好ましくない。60℃を超えた場合は得られる芳香族アミド類が着色し易く、好ましくない傾向がある。 In the present invention, the temperature at which the inert solvent is concentrated under reduced pressure is preferably 30 to 60 ° C. When the concentration temperature is less than 30 ° C., it takes a long time for concentration, which is not preferable. When the temperature exceeds 60 ° C., the resulting aromatic amide tends to be colored and tends to be unfavorable.
晶析温度は0〜60℃が好ましく、結晶ろ過温度は0〜25℃が好ましい。ろ過温度が0℃未満の場合は不純物の除去が十分でなく、高品質の芳香族アミド類が得られない傾向がある。ろ過温度が25℃を超える場合には、芳香族アミド類がろ液に溶出するため収率が低下する傾向がある。ろ過した結晶は、必要に応じて洗浄し、乾燥することによって残存溶媒や水分の除かれた芳香族アミド化合物として得られる。 The crystallization temperature is preferably 0 to 60 ° C, and the crystal filtration temperature is preferably 0 to 25 ° C. When the filtration temperature is less than 0 ° C., impurities are not sufficiently removed, and high-quality aromatic amides tend not to be obtained. When the filtration temperature exceeds 25 ° C., aromatic amides are eluted in the filtrate, so the yield tends to decrease. The filtered crystal is obtained as an aromatic amide compound from which residual solvent and moisture have been removed by washing and drying as necessary.
濃縮後の不活性溶媒量は芳香族アミド類に対して、好ましくは1.0〜2.5倍量で実施される。溶媒量が1.0倍量未満の場合は晶析液の流動性が低下し、ろ過工程での不純物除去が不充分で、着色成分が残り易く、高純度の芳香族アミド類が得られない。
溶媒量が2.5倍量を超えると、ろ液に溶出する芳香族アミド類が多くなり収率が低下する傾向がある。
The amount of the inert solvent after concentration is preferably 1.0 to 2.5 times that of the aromatic amide. When the amount of the solvent is less than 1.0 times, the fluidity of the crystallization solution is lowered, the removal of impurities in the filtration step is insufficient, the coloring component tends to remain, and high-purity aromatic amides cannot be obtained. .
When the amount of the solvent exceeds 2.5 times, the amount of aromatic amide eluted in the filtrate tends to increase and the yield tends to decrease.
かかる方法で得られる式(4)の芳香族アミド類としては、例えば、2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミド、2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−メチル−2−ヒドロキシフェニル)ブタンアミド、2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)酢酸アミド、2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−メチル−2−ヒドロキシフェニル)酢酸アミド、2−(2−t−ペンチル4−メチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミド、2−(2−t−ペンチル4−メチルフェノキシ)−N−(3,5−ジクロロ−4−メチル−2−ヒドロキシフェニル)ブタンアミド、2−(2−t−ペンチル4−メチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)酢酸アミド、2−(2−t−ペンチル4−メチルフェノキシ)−N−(3,5−ジクロロ−4−メチル−2−ヒドロキシフェニル)酢酸アミド、2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)吉草酸アミド、2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−メチル−2−ヒドロキシフェニル)吉草酸アミド等が例示される。 Examples of the aromatic amides of the formula (4) obtained by this method include 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxy Phenyl) butanamide, 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-methyl-2-hydroxyphenyl) butanamide, 2- (2,4-di-t- Pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) acetamide, 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4 -Methyl-2-hydroxyphenyl) acetamide, 2- (2-t-pentyl 4-methylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide, 2- (2- t-pe Til 4-methylphenoxy) -N- (3,5-dichloro-4-methyl-2-hydroxyphenyl) butanamide, 2- (2-t-pentyl 4-methylphenoxy) -N- (3,5-dichloro- 4-ethyl-2-hydroxyphenyl) acetamide, 2- (2-t-pentyl 4-methylphenoxy) -N- (3,5-dichloro-4-methyl-2-hydroxyphenyl) acetamide, 2- ( 2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) valeric amide, 2- (2,4-di-t-pentylphenoxy) -N -(3,5-dichloro-4-methyl-2-hydroxyphenyl) valeric acid amide and the like are exemplified.
以上、本発明の方法によりカルボン酸クロリド類とアミノフェノールから式(4)の芳香族アミド類が収率よく簡便な方法で得られ、その品質は色相が良好で、HPLC純度99.5%以上と高く、該芳香族アミド類は優れた画像を形成するシアンカップラーとなる。 As described above, the aromatic amides of the formula (4) can be obtained from the carboxylic acid chlorides and aminophenol by the method of the present invention by a simple method with high yield, the quality is good in hue, and the HPLC purity is 99.5% or more. The aromatic amides become cyan couplers that form excellent images.
(実施例)
次に、実施例を挙げて本発明を更に詳細に説明するが、本発明はこれに限定されるものではない。
本発明において式(1)で示される化合物の含有量は、試料を高速液体クロマトグラフィー(HPLC)により以下の条件で分離し、230nmの検出波長で測定した場合の(1)で示される化合物の面積百分率によって表される。
カラム:Capcellpack C18(5μm、4.0mmφ×250mm)
移動相:A液:アセトニトリル(90%)、B液:水(10%)
流量:1.0ml/min、カラム温度:40℃、検出波長:UV 230nm
(Example)
EXAMPLES Next, although an Example is given and this invention is demonstrated further in detail, this invention is not limited to this.
In the present invention, the content of the compound represented by the formula (1) is determined based on the compound represented by (1) when the sample is separated by high performance liquid chromatography (HPLC) under the following conditions and measured at a detection wavelength of 230 nm. Expressed by area percentage.
Column: Capcellpack C18 (5 μm, 4.0 mmφ × 250 mm)
Mobile phase: Liquid A: acetonitrile (90%), liquid B: water (10%)
Flow rate: 1.0 ml / min, column temperature: 40 ° C., detection wavelength: UV 230 nm
(参考例1)
2−(2,4−ジ−t−ペンチルフェノキシ)酪酸320.5g(1.0モル)とキシレン100gを1Lフラスコに仕込み、N,N−ジメチルホルムアルデヒド24.1g(0.33モル)を仕込む。窒素雰囲気下、35℃に保温してオキシ塩化燐107.3g(0.70モル)を2時間で滴下し、更に50℃で12時間保温撹拌することにより反応を完結せしめた。同温度で0.5時間静置する事によりキシレン層と副生成物層は容易に分液された。次いで、下層に分離する副生成物を分液除去し、2−(2,4−ジ−tert−ペンチルフェノキシ)酪酸クロリドのキシレン溶液430gを得た。含有量を分析した結果、2−(2,4−ジ−tert−ペンチルフェノキシ)酪酸クロリドの濃度は78.0%で、硫黄分は1ppm以下(検出限界以下)であった。
(Reference Example 1)
Charge 320.5 g (1.0 mol) of 2- (2,4-di-t-pentylphenoxy) butyric acid and 100 g of xylene into a 1 L flask, and charge 24.1 g (0.33 mol) of N, N-dimethylformaldehyde. . Under a nitrogen atmosphere, the temperature was kept at 35 ° C., 107.3 g (0.70 mol) of phosphorus oxychloride was added dropwise over 2 hours, and the reaction was further completed by stirring at 50 ° C. for 12 hours. The xylene layer and the by-product layer were easily separated by allowing to stand at the same temperature for 0.5 hour. Subsequently, the by-product separated into the lower layer was separated and removed to obtain 430 g of a xylene solution of 2- (2,4-di-tert-pentylphenoxy) butyric acid chloride. As a result of analyzing the content, the concentration of 2- (2,4-di-tert-pentylphenoxy) butyric chloride was 78.0%, and the sulfur content was 1 ppm or less (below the detection limit).
(参考例2)
2−(2,4−ジ−t−ペンチルフェノキシ)酪酸320.5g(1.0モル)とトルエン100gを1Lフラスコに仕込み、N,N−ジメチルホルムアルデヒド24.1g(0.33モル)を仕込む。窒素雰囲気下、35℃に保温してオキシ塩化燐107.3g(0.70モル)を2時間で滴下し、更に50℃で12時間保温撹拌することにより反応を完結せしめた。同温度で0.5時間静置する事によりトルエン層と副生成物層は容易に分液された。次いで、下層に分離する副生成物を分液除去し、2−(2,4−ジ−tert−ペンチルフェノキシ)酪酸クロリドのトルエン溶液430gを得た。含有量を分析した結果、2−(2,4−ジ−tert−ペンチルフェノキシ)酪酸クロリドの濃度は78.0%で、硫黄分は1ppm以下(検出限界以下)であった。
(Reference Example 2)
Charge 320.5 g (1.0 mol) of 2- (2,4-di-t-pentylphenoxy) butyric acid and 100 g of toluene to a 1 L flask, and charge 24.1 g (0.33 mol) of N, N-dimethylformaldehyde. . Under a nitrogen atmosphere, the temperature was kept at 35 ° C., 107.3 g (0.70 mol) of phosphorus oxychloride was added dropwise over 2 hours, and the reaction was further completed by stirring at 50 ° C. for 12 hours. The toluene layer and the by-product layer were easily separated by allowing to stand at the same temperature for 0.5 hour. Subsequently, the by-product separated into the lower layer was separated and removed to obtain 430 g of a toluene solution of 2- (2,4-di-tert-pentylphenoxy) butyric acid chloride. As a result of analyzing the content, the concentration of 2- (2,4-di-tert-pentylphenoxy) butyric chloride was 78.0%, and the sulfur content was 1 ppm or less (below the detection limit).
2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドの製造
2−ニトロ−4,6−ジクロロ−5−エチルフェノール23.6g(0.1モル)とキシレン100gを500mlオートクレーブに仕込み、3%白金カーボン触媒0.15gを仕込む。窒素置換後、40℃以下、水素ガスで接触水添する。水素吸収が無くなり、還元が終了したら触媒を濾過して、2−アミノ−4,6−ジクロロ−5−エチルフェノールのキシレン溶液144gを得た。2−アミノ−4,6−ジクロロ−5−エチルフェノールの収率は100%であった。
得られた2−アミノ−4,6−ジクロロ−5−エチルフェノールのキシレン溶液124gに、参考例1で得られた2−(2,4−ジ−tert−ペンチルフェノキシ)酪酸クロリドのキシレン溶液43.4g(0.1モル)と7%重曹水174gを、温度30〜50℃で滴下、撹拌し、4時間反応した。反応後、45〜50℃で0.5時間静置することによりキシレン層と水層に容易に分液された。次いで、下層の水層を除去し、同温度で水100gを仕込んで、水洗、分液した。
2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドを含むキシレン層161gを2.0kPaの減圧下、温度30〜60℃で75gまで濃縮した後、5℃まで冷却晶析した。晶析後、5℃で濾過し、40gのキシレンで結晶を洗浄して白色結晶を得た。乾燥後の2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドのアミド化工程収率は94.1%であった。
品質は、mp145−146℃、純度99.8%、10w/v%酢酸エチル溶液の456nm吸光度(1cmセル)0.01と良好であった。
Preparation of 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide 2-nitro-4,6-dichloro-5-ethylphenol 23.6 g (0.1 mol) and 100 g of xylene are charged into a 500 ml autoclave, and 0.15 g of 3% platinum carbon catalyst is charged. After nitrogen substitution, catalytic hydrogenation is performed at 40 ° C. or less with hydrogen gas. When the hydrogen absorption disappeared and the reduction was completed, the catalyst was filtered to obtain 144 g of a xylene solution of 2-amino-4,6-dichloro-5-ethylphenol. The yield of 2-amino-4,6-dichloro-5-ethylphenol was 100%.
To 124 g of the xylene solution of 2-amino-4,6-dichloro-5-ethylphenol obtained, xylene solution 43 of 2- (2,4-di-tert-pentylphenoxy) butyric acid chloride obtained in Reference Example 1 was added. 0.4 g (0.1 mol) and 7% sodium bicarbonate aqueous solution 174 g were dropped and stirred at a temperature of 30 to 50 ° C. and reacted for 4 hours. After the reaction, the mixture was allowed to stand at 45 to 50 ° C. for 0.5 hour, so that it was easily separated into a xylene layer and an aqueous layer. Next, the lower aqueous layer was removed, 100 g of water was charged at the same temperature, washed with water and separated.
161 g of a xylene layer containing 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide was heated at a temperature of 30 to 30 under a reduced pressure of 2.0 kPa. After concentrating to 75 g at 60 ° C, it was cooled and crystallized to 5 ° C. After crystallization, the mixture was filtered at 5 ° C., and the crystals were washed with 40 g of xylene to obtain white crystals. The yield of the amidation step of 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide after drying was 94.1%. It was.
The quality was mp145-146 ° C., purity 99.8%, 10 w / v% ethyl acetate solution 456 nm absorbance (1 cm cell) 0.01 and good.
2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドの製造
2-ニトロ−4,6−ジクロロ−5−エチルフェノール23.6g(0.1モル)とトルエン100gを500mlオートクレーブに仕込み、3%白金カーボン触媒0.15gを仕込む。窒素置換後、40℃以下、水素ガスで接触水添する。水素吸収が無くなり、還元が終了したら触媒を濾過して、2−アミノ−4,6−ジクロロ−5−エチルフェノールのトルエン溶液144gを得た。2−アミノ−4,6−ジクロロ−5−エチルフェノールの収率は100%であった。
得られた2−アミノ−4,6−ジクロロ−5−エチルフェノールのトルエン溶液124gに、参考例2で得られた2−(2,4−ジ−tert−ペンチルフェノキシ)酪酸クロリドのトルエン溶液43.4g(0.1モル)と7%重曹水174gを、温度30〜50℃
で滴下、撹拌し、4時間反応した。反応後、45〜50℃で0.5時間静置することによりトルエン層と水層に容易に分液された。次いで、下層の水層を除去し、同温度で
水100gを仕込んで、水洗、分液した。
2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドを含むトルエン層161gを6.0kPaの減圧下、温度30〜60℃で65gまで濃縮した後、20℃まで冷却晶析した。晶析後、20℃で濾過し、40gのトルエンで結晶を洗浄して白色結晶を得た。乾燥後の2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドのアミド化工程収率は93.9%であった。
品質は mp145−146℃、純度99.9%、10w/v%酢酸エチル溶液の456nm吸光度(1cmセル)0.01と良好であった。
Preparation of 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide 2-nitro-4,6-dichloro-5-ethylphenol 23.6 g (0.1 mol) and 100 g of toluene are charged into a 500 ml autoclave, and 0.15 g of 3% platinum carbon catalyst is charged. After nitrogen substitution, catalytic hydrogenation is performed at 40 ° C. or less with hydrogen gas. When the hydrogen absorption disappeared and the reduction was completed, the catalyst was filtered to obtain 144 g of a toluene solution of 2-amino-4,6-dichloro-5-ethylphenol. The yield of 2-amino-4,6-dichloro-5-ethylphenol was 100%.
The toluene solution 43 of 2- (2,4-di-tert-pentylphenoxy) butyric acid chloride obtained in Reference Example 2 was added to 124 g of the toluene solution of 2-amino-4,6-dichloro-5-ethylphenol obtained. .4 g (0.1 mol) and 174 g of 7% sodium bicarbonate water at a temperature of 30 to 50 ° C.
And stirred for 4 hours. After the reaction, the mixture was allowed to stand at 45 to 50 ° C. for 0.5 hour, so that it was easily separated into a toluene layer and an aqueous layer. Next, the lower aqueous layer was removed, 100 g of water was charged at the same temperature, washed with water and separated.
161 g of toluene layer containing 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide was heated under a reduced pressure of 6.0 kPa at a temperature of 30 to After concentrating to 65 g at 60 ° C., it was cooled and crystallized to 20 ° C. After crystallization, the mixture was filtered at 20 ° C., and the crystals were washed with 40 g of toluene to obtain white crystals. The amidation step yield of 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide after drying was 93.9%. It was.
The quality was mp145-146 ° C., purity 99.9%, 10 w / v% ethyl acetate solution 456 nm absorbance (1 cm cell) 0.01 and good.
(比較例1)
2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドの製造
2-ニトロ−4,6−ジクロロ−5−エチルフェノール23.6g(0.1モル)とトルエン100gを500mlオートクレーブに仕込み、3%白金カーボン触媒0.15gを仕込む。窒素置換後、40℃以下、水素ガスで接触水添する。水素吸収が無くなり、還元が終了したら触媒を濾過して、2−アミノ−4,6−ジクロロ−5−エチルフェノールのトルエン溶液144gを得た。2−アミノ−4,6−ジクロロ−5−エチルフェノールの収率は100%であった。
得られた2−アミノ−4,6−ジクロロ−5−エチルフェノールのトルエン溶液124gに、参考例2で得られた2−(2,4−ジ−tert−ペンチルフェノキシ)酪酸クロリドのトルエン溶液43.4g(0.1モル)と7%重曹水174gを、温度30〜50℃
で滴下、撹拌し、4時間反応した。反応後、45〜50℃で0.5時間静置することによりトルエン層と水層に容易に分液された。次いで、下層の水層を除去し、同温度で
水100gを仕込んで、水洗、分液した。
2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドを含むトルエン層161gを23.0kPaの減圧下、温度65〜80℃で75gまで濃縮した後、5℃まで冷却晶析した。晶析後、5℃で濾過し、40gのトルエンで結晶を洗浄して白色結晶を得た。乾燥後の2−(2,4−ジ−t−ペンチルフェノキシ)−N−(3,5−ジクロロ−4−エチル−2−ヒドロキシフェニル)ブタンアミドのアミド化工程収率は94.0%であった。
品質は mp145−146℃、純度99.7%、10w/v%酢酸エチル溶液の456nm吸光度(1cmセル)0.10と不良であった。
(Comparative Example 1)
Preparation of 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide 2-nitro-4,6-dichloro-5-ethylphenol 23.6 g (0.1 mol) and 100 g of toluene are charged into a 500 ml autoclave, and 0.15 g of 3% platinum carbon catalyst is charged. After nitrogen substitution, catalytic hydrogenation is performed at 40 ° C. or less with hydrogen gas. When the hydrogen absorption disappeared and the reduction was completed, the catalyst was filtered to obtain 144 g of a toluene solution of 2-amino-4,6-dichloro-5-ethylphenol. The yield of 2-amino-4,6-dichloro-5-ethylphenol was 100%.
The toluene solution 43 of 2- (2,4-di-tert-pentylphenoxy) butyric acid chloride obtained in Reference Example 2 was added to 124 g of the toluene solution of 2-amino-4,6-dichloro-5-ethylphenol obtained. .4 g (0.1 mol) and 174 g of 7% sodium bicarbonate water at a temperature of 30 to 50 ° C.
And stirred for 4 hours. After the reaction, the mixture was allowed to stand at 45 to 50 ° C. for 0.5 hour, so that it was easily separated into a toluene layer and an aqueous layer. Next, the lower aqueous layer was removed, 100 g of water was charged at the same temperature, washed with water and separated.
A toluene layer (161 g) containing 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide was heated under a reduced pressure of 23.0 kPa at a temperature of 65-65. After concentrating to 75 g at 80 ° C., it was cooled and crystallized to 5 ° C. After crystallization, the mixture was filtered at 5 ° C., and the crystals were washed with 40 g of toluene to obtain white crystals. The amidation step yield of 2- (2,4-di-t-pentylphenoxy) -N- (3,5-dichloro-4-ethyl-2-hydroxyphenyl) butanamide after drying was 94.0%. It was.
The quality was poor, mp 145-146 ° C., purity 99.7%, 10 w / v% ethyl acetate solution 456 nm absorbance (1 cm cell) 0.10.
Claims (3)
(式中、R1は低級アルキル基を表す。)
で示されるo−ニトロフェノール類を炭素数10以下の芳香族系不活性溶媒中、白金系触媒の存在下に接触還元し、式(2)
で示されるo−アミノフェノール類を得、これを単離することなく式(3)
で示される硫黄分含量が0.5%以下(酸クロリド重量基準)の酸クロリド類と、
酸素濃度が1%以下の不活性ガス雰囲気下で縮合反応させて、式(4)
で示される芳香族アミド類を得る製造法であって、アミド化縮合反応後、芳香族系不活性溶媒を減圧下、60℃以下で濃縮し、式(4)で示される芳香族アミド類を晶析する工程を含むことを特徴とする式(4)で示される芳香族アミド類の製造法。 Formula (1)
(In the formula, R 1 represents a lower alkyl group.)
The o-nitrophenol represented by the formula (2) is catalytically reduced in an aromatic inert solvent having 10 or less carbon atoms in the presence of a platinum catalyst.
The o-aminophenol represented by the formula (3) is obtained without isolation.
Acid chlorides having a sulfur content of 0.5% or less (based on the weight of acid chloride),
A condensation reaction is performed in an inert gas atmosphere with an oxygen concentration of 1% or less to obtain a compound of formula (4)
The aromatic amides represented by formula (4) are obtained by concentrating the aromatic inert solvent under reduced pressure at 60 ° C. or lower after the amidation condensation reaction, A process for producing an aromatic amide represented by the formula (4), which comprises a step of crystallizing.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008064793A JP5115972B2 (en) | 2008-03-13 | 2008-03-13 | Process for producing aromatic amides |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008064793A JP5115972B2 (en) | 2008-03-13 | 2008-03-13 | Process for producing aromatic amides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009221115A JP2009221115A (en) | 2009-10-01 |
| JP5115972B2 true JP5115972B2 (en) | 2013-01-09 |
Family
ID=41238293
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008064793A Expired - Fee Related JP5115972B2 (en) | 2008-03-13 | 2008-03-13 | Process for producing aromatic amides |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5115972B2 (en) |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3014180B2 (en) * | 1991-08-02 | 2000-02-28 | 三菱レイヨン株式会社 | Method for producing crystalline N-methylolacrylamide |
| JP3460264B2 (en) * | 1993-01-29 | 2003-10-27 | 住友化学工業株式会社 | Production method of aromatic amides |
| JP2005289835A (en) * | 2004-03-31 | 2005-10-20 | Konica Minolta Chemical Co Ltd | Method for producing 5-substituted-2-amidophenol |
-
2008
- 2008-03-13 JP JP2008064793A patent/JP5115972B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009221115A (en) | 2009-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3911008B2 (en) | Improved production of rebamipide | |
| JP5465672B2 (en) | Production method of sulfonic acid diamide | |
| US20080171891A1 (en) | Process for preparing carbamic ester derivatives | |
| JP7399850B2 (en) | Method for producing aromatic nitrile compounds | |
| CN109988077A (en) | A kind of synthetic method and intermediate of A Palu amine | |
| JP5115972B2 (en) | Process for producing aromatic amides | |
| US5442114A (en) | Process for producing aromatic amide compounds | |
| JP6915189B1 (en) | High-purity 2-naphthylacetonitrile and its production method | |
| EP0659735B1 (en) | Process for producing aniline derivative | |
| JP3765837B2 (en) | Process for producing aromatic amides | |
| JP3460264B2 (en) | Production method of aromatic amides | |
| CN110734430A (en) | Process for the synthesis of substituted gamma lactams | |
| CN108358866A (en) | A kind of preparation method of Febustat intermediate and its application in preparing Febustat | |
| JP2008169204A (en) | Method for preparing (1r, 2r)-2-amino-1-cyclopentanol | |
| JP2005289835A (en) | Method for producing 5-substituted-2-amidophenol | |
| JP5612977B2 (en) | Process for producing 6-bromo-N-methyl-2-naphthamide | |
| JP4799085B2 (en) | Process for producing optically active N-substituted aminoacyl cyclic urea derivative | |
| JP4114911B2 (en) | Method for producing 4-hydroxybenzenesulfonanilide | |
| CN117285468A (en) | Preparation method of Ensitrelivir intermediate | |
| WO2014037307A1 (en) | Process for preparing 6-iodo-2-oxindole | |
| TW202000640A (en) | Method for producing diaminobenzoic acid ester | |
| JP4028912B2 (en) | Novel cyclopropylanilines and process for producing the same | |
| KR100730766B1 (en) | New method for preparing biphenylacetic acid | |
| JP2018517733A (en) | New production method of teriflunomide | |
| WO2006079857A1 (en) | Diaryl acetic acid derivatives and the preparation thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100901 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120621 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120627 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120725 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121010 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121010 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151026 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |