JP4893903B2 - Method for producing bromoisophthalic acid compound - Google Patents
Method for producing bromoisophthalic acid compound Download PDFInfo
- Publication number
- JP4893903B2 JP4893903B2 JP2001167799A JP2001167799A JP4893903B2 JP 4893903 B2 JP4893903 B2 JP 4893903B2 JP 2001167799 A JP2001167799 A JP 2001167799A JP 2001167799 A JP2001167799 A JP 2001167799A JP 4893903 B2 JP4893903 B2 JP 4893903B2
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- acid compound
- acid
- producing
- bromoisophthalic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *OC(c1cc(C(O*)=O)ccc1)=O Chemical compound *OC(c1cc(C(O*)=O)ccc1)=O 0.000 description 1
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
【0001】
【発明の属する技術分野】
本発明は、一般式(1)
【0002】
【化2】
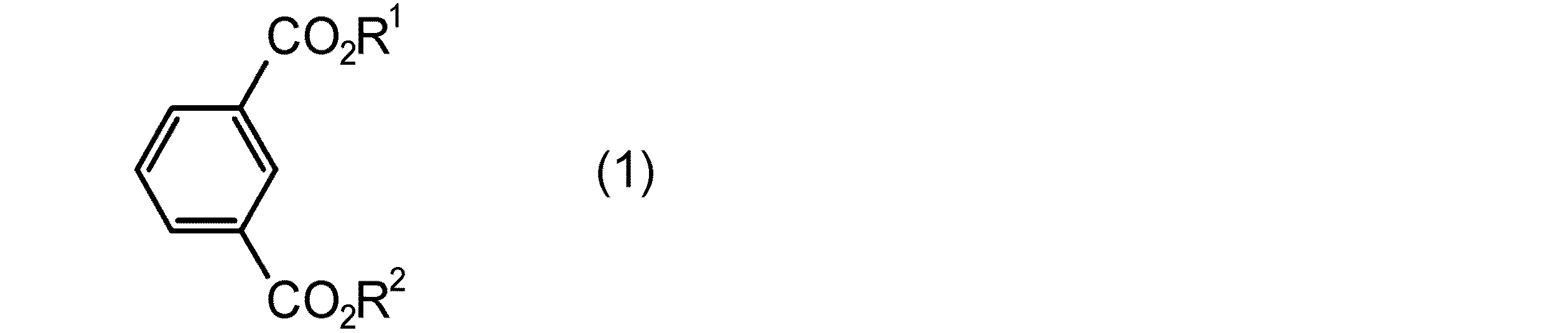
(式中、R1、R2はそれぞれ独立に水素原子もしくは、炭素数1〜6のアルキル基を表す。)で表されるイソフタル酸化合物をブロモ化する、ブロモイソフタル酸化合物の製造方法に関する。ブロモイソフタル酸化合物は、ポリエステル及びポリアミド等に代表される各種ポリマーの改質剤や種々の機能性化学品、及び医農薬品の重要な中間体である。
【0004】
【従来の技術】
従来、電子吸引基を持つ芳香族化合物の選択的ブロモ化は、非常に困難であることが知られている。この様な反応の関連技術としては以下の技術などが知られている。即ち、5−ブロモイソフタル酸ジアルキルを製造する方法としては、BrF3存在下、臭素によりイソフタル酸ジメチルをブロモ化して5−ブロモイソフタル酸ジメチルを収率55%で得ている例[J.Org.Chem.,58,239(1993)]が知られている。しかしこの方法は高価で取り扱いの難しいBrF3を使用し、かつ目的生成物の収率が低く実用的製造ではない。
【0005】
【発明が解決しようとする課題】
本発明の目的は、工業的に安価な臭素を用いて高選択的に5−ブロモイソフタル酸化合物を製造する方法を提供することにある。
【0006】
【課題を解決するための手段】
本発明者らは5−ブロモイソフタル酸化合物の製造方法について鋭意検討を重ねた結果、高選択的に製造できる方法を見出し、本発明のブロモイソフタル酸化合物の製造方法を完成するに至った。
【0007】
即ち、本発明は、一般式(1)
【0008】
【化3】
(式中、R1、R2はそれぞれ独立に水素原子もしくは、C1〜C6アルキル基を表す。)で表されるイソフタル酸化合物を、三酸化硫黄を含有する溶媒中、臭素のみを用いてブロモ化することによる、5−ブロモイソフタル酸化合物の製造方法に関するものである。
【0010】
従来、イソフタル酸のように芳香環上に電子吸引基を持つ化合物のモノブロモ化は非常に困難であり、反応が進行したとしても低い収率でしか対応する化合物を与えることができなかった。本発明者らは、選択的にモノブロモ体を造ることができる三酸化硫黄量、反応温度及び反応時間等の反応条件を見出した。
【0011】
【発明の実施の形態】
以下、本発明を詳細に説明する。原料のイソフタル酸化合物は一般式(1)
【0012】
【化4】
(式中、R1、R2はそれぞれ独立に水素原子もしくは、C1〜C6アルキル基を表す。)
で表され、R1及びR2は、水素原子、メチル基、エチル基、n−プロピル基、i−プロピル基、n−ブチル基、i−ブチル基、s−ブチル基、t−ブチル基、1−ペンチル基、2−ペンチル基、3−ペンチル基、3−メチルブチル基、ネオペンチル基、t−ペンチル基、1−ヘキシル基、2−ヘキシル基、3−ヘキシル基、1−メチル−1−エチルプロピル基、1,1,2−トリメチルプロピル基、1,2,2−トリメチルプロピル基、3,3−ジメチルブチル基等を表す。
具体的には、イソフタル酸或いはイソフタル酸ジメチル、イソフタル酸ジエチル、イソフタル酸メチルエチル、イソフタル酸ジn−プロピル、イソフタル酸ジi−プロピル、イソフタル酸ジn−ブチル、イソフタル酸ジ1−ペンチル、イソフタル酸ジ1−ヘキシル、イソフタル酸ジ1−ペンチル及びイソフタル酸ジ1−ヘキシル等のイソフタル酸ジアルキル化合物が挙げられる。これらの中で工業的な原料の入手し易さからイソフタル酸及びイソフタル酸ジメチル類が好ましい。中でも、イソフタル酸は経済的に製造できる点及び市販品をそのまま使用することができる点から本反応の原料としてふさわしい。
【0014】
本発明の特徴は三酸化硫黄を含有する溶媒中で、臭素によってブロモ化するところにある。従来、イソフタル酸のようにフェニル基に複数の電子吸引基が置換された化合物は臭素のみでは殆ど反応しないことが知られていた。本発明者らは、三酸化硫黄と臭素の錯体を形成させることによって、臭素のブロモ化能を向上させて反応を試みたところモノブロモ体を高選択的に得られることを見出した。
【0015】
三酸化硫黄を含有する溶媒としては、有機溶媒も使用することもできるが、通常は硫酸溶媒中即ち発煙硫酸として使用するのが便利である。又、三酸化硫黄の他の化合物との錯体であるクロロスルホン酸、三酸化硫黄・ジオキサン錯体、三酸化硫黄・ピリジン錯体等を使用することもできる。これらの中で、発煙硫酸が経済的で高い反応性を与えた。
【0016】
三酸化硫黄の使用量は、モノブロモ体製造の場合は臭素のモル数以下で充分であり高い選択性が得られる。
【0017】
一方、ジブロモ体の製造の場合は臭素のモル数以上に存在させることによって有利になる。
【0018】
発煙硫酸の形態としては、三酸化硫黄濃度は広く選択することができるが、通常市販の10〜60重量%品が使用できる。また、10重量%以下の発煙硫酸は、硫酸を混合することで調整することができる。
【0019】
この三酸化硫黄濃度は他の反応条件にもよるが低濃度(1〜30重量%)の方がモノブロモ体製造に適し、高濃度(10〜60重量%)の方がジブロモ体製造に適している。
【0020】
溶媒としては、上記の硫酸の他にハロゲン化炭化水素やスルホランを用いることもできる。
【0021】
ハロゲン化炭化水素の具体例としては、四塩化炭素、1,2−ジクロロエタン(EDC)、1,1,1−トリクロロエタン、1,1,2−トリクロロエタン及び1,1,2,2−テトラクロロエタン等が挙げられる。これらの使用量は、基質に対し1〜20重量倍が好ましく、特には2〜10重量倍が適切である。
【0022】
本発明のブロモ化反剤は工業的に最も経済的な臭素を使用するところに特徴がある。その品質は市販品をそのまま使用することができる。臭素の使用量はモノブロモ体を製造する場合は基質に対し、0.5〜1.5モル倍が好ましい。
【0023】
本反応は、加熱して行なう必要があり通常は加圧下で実施される。反応温度は通常50〜200℃間で行うことができ、特には、100〜160℃の範囲が好ましい。反応時間は、基質の種類、発煙硫酸量、臭素量や反応温度等によって変わるが、通常1〜100時間で実施可能であり通常3〜24時間で終了する。又、ガスクロマトグラフィーや液体クロマトグラフィーで反応の終点を確認することができる。本反応は、回分式でも連続式でも実施することができる。
【0024】
反応終了後、冷却した大過剰の水の中に反応溶液を加え、析出した結晶を濾取、水洗及び乾燥することにより目的のブロモ体の粗結晶が得られる。又、必要により再結晶化させて精製することができる。
【0025】
本発明者らは種々の再結晶溶媒を検討した結果は5−ブロモイソフタル酸は通常の再結晶溶媒では難溶解性のため再結晶が不可能であった。それらに対し、炭素数1〜5の低級アルコール系溶媒が高再結晶収率で高純度品を与えることを見出した。これらの中で特にメタノールが優れた再結晶能力を示し、経済的にも有利であるところから本溶媒として好ましい。
【0026】
生成ブロモ体のもう一つの精製方法として、本発明者らは蒸留法が適用できることを見出した。即ち、反応後単離された5−ブロモイソフタル酸の粗結晶を酸触媒存在下、炭素数1〜5の低級アルコールでジエステル化した後、蒸留することにより目的のブロモ体は未反応原料のエステル体やジブロモ体等から分離することができる。酸触媒としては、硫酸やp−トルエンスルホン酸が使用でき、使用量はイソフタル酸化合物に対し1〜20モル%が好ましい。一方、炭素数1〜5の低級アルコールの中では、ジエステル体の沸点上、メタノール又はエタノールが好ましい。その使用量は、通常5〜100モル倍が用いられる。
【0027】
反応温度は、通常アルコールの沸点で行なうことが出来るが、加圧下でアルコールの沸点以上に上昇して行なうことにより反応が短時間で完結する。メタノールの場合は100〜180℃が好ましい。
【0028】
得られた各エステル体の沸点は高いため減圧下で行うのが好ましい。通常、10〜1000Paで行うのが好ましい。反応生成物の組成によっては単蒸留でも可能であるが、通常精留によって目的のモノブロモ体の高純度品が得られる。
ジブロモ体は、蒸留の後留として留出させることも可能ではあるが、モノブロモ体を留出させ後の釜残から再結晶法で単離することもできる。尚、モノブロモ体を選択的に製造する場合には、本反応は逐次反応であることから、ジブロモ体の生成が懸念される。そこで、反応を途中で止め、反応系内にイソフタル酸化合物及びモノブロモイソフタル酸化合物のみが存在するようにして、原料をリサイクルすることが望ましい。そして前記の様に、メタノールによりエステル化し引き続き蒸留を行なうことで可能となる。
【0029】
【化5】
(Rは炭素数1〜5の低級アルキルを表す。)
以下に実施例により更に具体的に本発明を説明するが、本発明はこれらによって限定されるものではない。
【0031】
【実施例】
実施例1
50ml耐圧硝子製封管チューブにイソフタル酸1.66g(10mmol)、10重量%発煙硫酸6.00g及び臭素1.6g(10mmol)を仕込み、130℃で22時間攪拌した。反応後室温まで冷却し、内容物を氷水の入ったビーカーに取り出すことで固体を得た。生成した固体をろ過、冷却洗浄更に減圧乾燥して目的物の粗結晶2.41g(純度83.5%)を得た(収率81.9%)。その後、10gのメタノールに60℃で溶解させ、室温まで冷却した後濾過することで白色結晶1.61g(純度100%)を得た(再結晶収率80.1%)。この結晶は、MASS、1H−NMR及び融点から5−ブロモイソフタル酸であることを確認した。
【0032】
実施例2〜6
実施例1に於て反応温度及び発煙硫酸濃度を変えた他は同様に行ないモノブロモ体が得られた結果を表1に示す。
【0033】
【表1】
実施例7
50ml耐圧硝子製封管チューブにイソフタル酸1.66g(10mmol)、20重量%発煙硫酸6.00g及び臭素1.6g(10mmol)を仕込み、130℃で22時間攪拌した。反応後室温まで冷却し、内容物を氷水の入ったビーカーに取り出すことで固体を得た。次に、生成した固体をろ過、冷却洗浄更にシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=6/1,v/v)に付すことにより単離を行なった結果、5−ブロモイソフタル酸を収率49.2%、4,5−ジブロモイソフタル酸を収率15.3%、2,5−ジブロモイソフタル酸を痕跡にて得た。これらの結晶は、MASS、1H−NMR及び融点からそれぞれ確認した。
【0035】
実施例8〜11
実施例7に於て反応温度、発煙硫酸濃度及び臭素量を変えた他は同様に行ない、モノブロモ体及びジブロモ体が得られた結果を表2に示す。
【0036】
【表2】
実施例12
50ml耐圧硝子製封管チューブにイソフタル酸1.66g(10mmol)、10重量%発煙硫酸6.00g及び臭素1.6g(10mmol)を仕込み、150℃で7時間攪拌した。反応後室温まで冷却し、内容物を氷水の入ったビーカーに取り出すことで固体を得た。生成した固体をろ過、冷却洗浄更に減圧乾燥して目的物の粗結晶を得た。その後、オートクレーブにメタノール13.1g(408mmol)、硫酸0.35g(30mol%)を加え120℃で加熱撹拌することでジメチルエステルヘと誘導した。引き続き、これを精留に付すことで目的とする5−ブロモイソフタル酸ジメチルを1.78g(収率65.1%、減圧沸点159℃/4.8mmHg)と原料に相当するイソフタル酸ジメチル0.52g(収率26.8%、減圧沸点133℃/4.8mmHg)を得た。得られた結晶は、MASS、1H−NMR及び融点から5−ブロモイソフタル酸ジメチル及び5−ブロモイソフタル酸であることを確認した。
【0040】
【発明の効果】
本発明の方法によれば、選択的かつ安価に5−ブロモイソフタル酸化合物を製造することができる。[0001]
BACKGROUND OF THE INVENTION
The present invention relates to a general formula (1)
[0002]
[Chemical 2]
(In the formula, R 1 and R 2 each independently represents a hydrogen atom or an alkyl group having 1 to 6 carbon atoms.) The present invention relates to a method for producing a bromoisophthalic acid compound, which comprises brominating an isophthalic acid compound. Bromoisophthalic acid compounds are important intermediates for various polymer modifiers such as polyester and polyamide, various functional chemicals, and medical and agrochemical products.
[0004]
[Prior art]
Conventionally, it is known that selective bromination of aromatic compounds having electron-withdrawing groups is very difficult. The following techniques are known as related techniques for such reactions. That is, as a method for producing dialkyl 5-bromoisophthalate, dimethyl isophthalate was brominated with bromine in the presence of BrF 3 to obtain dimethyl 5-bromoisophthalate in a yield of 55% [J. Org. Chem., 58, 239 (1993)] is known. However, this method uses BrF 3 which is expensive and difficult to handle, and the yield of the target product is low, which is not practical production.
[0005]
[Problems to be solved by the invention]
An object of the present invention is to provide a method for producing a high selective 5-bromo-isophthalic acid compound using industrially inexpensive bromine.
[0006]
[Means for Solving the Problems]
As a result of intensive studies on a method for producing a 5-bromoisophthalic acid compound, the present inventors have found a method that can be produced with high selectivity, and have completed the method for producing a bromoisophthalic acid compound of the present invention.
[0007]
That is, the present invention relates to the general formula (1)
[0008]
[Chemical 3]
(Wherein, R 1, R 2 each independently represent a hydrogen atom or, C 1 -C 6 alkyl represents a group.) Isophthalic acid compound represented by the solvent containing sulfur trioxide, with bromine only The present invention relates to a process for producing a 5 -bromoisophthalic acid compound by bromination.
[0010]
Conventionally, monobromination of a compound having an electron withdrawing group on an aromatic ring such as isophthalic acid is very difficult, and even if the reaction proceeds, the corresponding compound can be obtained only in a low yield. The inventors have found sulfur trioxide weight can be produced selectively monobromo, the reaction conditions such as reaction temperature and reaction time.
[0011]
DETAILED DESCRIPTION OF THE INVENTION
Hereinafter, the present invention will be described in detail. The raw material isophthalic acid compound is represented by the general formula (1)
[0012]
[Formula 4]
(Wherein, R 1, R 2 each independently represent a hydrogen atom or represents C 1 -C 6 alkyl group.)
R 1 and R 2 are each a hydrogen atom, methyl group, ethyl group, n-propyl group, i-propyl group, n-butyl group, i-butyl group, s-butyl group, t-butyl group, 1-pentyl group, 2-pentyl group, 3-pentyl group, 3-methylbutyl group, neopentyl group, t-pentyl group, 1-hexyl group, 2-hexyl group, 3-hexyl group, 1-methyl-1-ethyl A propyl group, a 1,1,2-trimethylpropyl group, a 1,2,2-trimethylpropyl group, a 3,3-dimethylbutyl group, etc. are represented.
Specifically, isophthalic acid or dimethyl isophthalate, diethyl isophthalate, methyl ethyl isophthalate, di-n-propyl isophthalate, di-propyl isophthalate, di-n-butyl isophthalate, di-1-pentyl isophthalate, isophthalate Examples thereof include dialkyl phthalate compounds such as di-1-hexyl acid, di-1-pentyl isophthalate and di-1-hexyl isophthalate. Among these, isophthalic acid and dimethyl isophthalate are preferable because of easy availability of industrial raw materials. Among these, isophthalic acid is suitable as a raw material for this reaction because it can be produced economically and a commercially available product can be used as it is.
[0014]
A feature of the present invention is that it is brominated with bromine in a solvent containing sulfur trioxide. Conventionally, it has been known that a compound in which a plurality of electron-withdrawing groups are substituted on a phenyl group such as isophthalic acid hardly reacts only with bromine. The present inventors have found that a monobromo compound can be obtained with high selectivity when a reaction is attempted by improving the bromination ability of bromine by forming a complex of sulfur trioxide and bromine.
[0015]
As the solvent containing sulfur trioxide, an organic solvent can also be used, but it is usually convenient to use it in a sulfuric acid solvent, that is, as fuming sulfuric acid. Further, chlorosulfonic acid, sulfur trioxide / dioxane complex, sulfur trioxide / pyridine complex, etc., which are complexes with other compounds of sulfur trioxide, can also be used. Of these, fuming sulfuric acid gave economic and high reactivity.
[0016]
In the case of monobromo compound production, the amount of sulfur trioxide used is less than the number of moles of bromine, and high selectivity is obtained.
[0017]
On the other hand, in the case of production of a dibromo compound, it is advantageous to make it exist in the number of moles or more of bromine.
[0018]
As the form of fuming sulfuric acid, the sulfur trioxide concentration can be widely selected, but a commercially available 10 to 60% by weight product can be usually used. Moreover, 10 weight% or less fuming sulfuric acid can be adjusted by mixing a sulfuric acid.
[0019]
Although this sulfur trioxide concentration depends on other reaction conditions, a low concentration (1 to 30% by weight) is more suitable for monobromo compound production, and a high concentration (10 to 60% by weight) is suitable for dibromo compound production. Yes.
[0020]
As the solvent, a halogenated hydrocarbon or sulfolane can be used in addition to the above sulfuric acid.
[0021]
Specific examples of the halogenated hydrocarbon include carbon tetrachloride, 1,2-dichloroethane (EDC), 1,1,1-trichloroethane, 1,1,2-trichloroethane, 1,1,2,2-tetrachloroethane, and the like. Is mentioned. The amount of these used is preferably 1 to 20 times by weight, particularly 2 to 10 times by weight based on the substrate.
[0022]
The bromination reaction agent of the present invention is characterized by using industrially most economical bromine. The quality can use a commercial item as it is. The amount of bromine used is preferably 0.5 to 1.5 mol times the substrate when producing a monobromo compound .
[0023]
This reaction must be carried out with heating, and is usually carried out under pressure. Reaction temperature can be normally performed between 50-200 degreeC, and the range of 100-160 degreeC is especially preferable. The reaction time varies depending on the type of substrate, the amount of fuming sulfuric acid, the amount of bromine, the reaction temperature, and the like, but it can usually be carried out in 1 to 100 hours and usually ends in 3 to 24 hours. In addition, the end point of the reaction can be confirmed by gas chromatography or liquid chromatography. This reaction can be carried out either batchwise or continuously.
[0024]
After completion of the reaction, the reaction solution is added to a large amount of cooled water, and the precipitated crystals are collected by filtration, washed with water and dried to obtain the desired crude bromo compound crystals. If necessary, it can be purified by recrystallization.
[0025]
As a result of studying various recrystallization solvents by the present inventors, it was impossible to recrystallize 5-bromoisophthalic acid because it was hardly soluble in a normal recrystallization solvent. In contrast, it has been found that a lower alcohol solvent having 1 to 5 carbon atoms gives a high purity product with a high recrystallization yield. Of these, methanol is particularly preferred as the solvent because it exhibits excellent recrystallization ability and is economically advantageous.
[0026]
As another method for purifying the produced bromo compound, the present inventors have found that a distillation method can be applied. That is, the target bromo compound is an unreacted raw material ester by diesterifying a crude crystal of 5-bromoisophthalic acid isolated after the reaction with a lower alcohol having 1 to 5 carbon atoms in the presence of an acid catalyst, followed by distillation. Can be separated from the body and dibromo form. As the acid catalyst, sulfuric acid or p-toluenesulfonic acid can be used, and the amount used is preferably 1 to 20 mol% with respect to the isophthalic acid compound. On the other hand, among lower alcohols having 1 to 5 carbon atoms, methanol or ethanol is preferable because of the boiling point of the diester. The amount used is usually 5 to 100 moles.
[0027]
The reaction temperature can usually be carried out at the boiling point of the alcohol. However, the reaction is completed in a short time by raising the reaction temperature above the boiling point of the alcohol under pressure. In the case of methanol, 100-180 degreeC is preferable.
[0028]
Since the boiling point of each obtained ester body is high, it is preferable to carry out under reduced pressure. Usually, it is preferable to carry out at 10-1000 Pa. Depending on the composition of the reaction product, simple distillation is possible, but a high purity product of the desired monobromo compound is usually obtained by rectification.
The dibromo compound can be distilled as a post-distillation distillation, but can also be isolated from the kettle residue after the monobromo distillate by recrystallization. In addition, when producing a monobromo body selectively, since this reaction is a sequential reaction, we are anxious about the production | generation of a dibromo body. Therefore, it is desirable to stop the reaction halfway and recycle the raw materials so that only the isophthalic acid compound and the monobromoisophthalic acid compound exist in the reaction system. As described above, it is possible to esterify with methanol and continue distillation.
[0029]
[Chemical formula 5]
(R represents lower alkyl having 1 to 5 carbon atoms.)
The present invention will be described more specifically with reference to the following examples. However, the present invention is not limited to these examples.
[0031]
【Example】
Example 1
A 50 ml pressure-resistant glass sealed tube tube was charged with 1.66 g (10 mmol) of isophthalic acid, 6.00 g of 10 wt% fuming sulfuric acid and 1.6 g (10 mmol) of bromine and stirred at 130 ° C. for 22 hours. After the reaction, the mixture was cooled to room temperature, and the contents were taken out into a beaker containing ice water to obtain a solid. The produced solid was filtered, washed with cooling, and dried under reduced pressure to obtain 2.41 g (purity 83.5%) of the target crude crystal (yield 81.9%). Then, it was dissolved in 10 g of methanol at 60 ° C., cooled to room temperature, and then filtered to obtain 1.61 g (purity: 100%) of white crystals (recrystallization yield: 80.1%). This crystal was confirmed to be 5-bromoisophthalic acid from MASS, 1 H-NMR and melting point.
[0032]
Example 2-6
Table 1 shows the results obtained in the same manner as in Example 1 except that the reaction temperature and the fuming sulfuric acid concentration were changed, and a monobromo compound was obtained.
[0033]
[Table 1]
Example 7
A 50 ml pressure-resistant glass sealed tube tube was charged with 1.66 g (10 mmol) of isophthalic acid, 6.00 g of 20 wt% fuming sulfuric acid and 1.6 g (10 mmol) of bromine and stirred at 130 ° C. for 22 hours. After the reaction, the mixture was cooled to room temperature, and the contents were taken out into a beaker containing ice water to obtain a solid. Next, the resulting solid was filtered, washed with cooling, and further subjected to silica gel column chromatography (chloroform / methanol = 6/1, v / v). As a result, 5-bromoisophthalic acid was obtained in a yield of 49. 2% , 4,5-dibromoisophthalic acid was obtained in a yield of 15.3% , and 2,5-dibromoisophthalic acid was obtained in traces . These crystals were confirmed from MASS, 1 H-NMR and melting point, respectively.
[0035]
Examples 8-11
The same procedure was carried out except that the reaction temperature, fuming sulfuric acid concentration and bromine amount were changed in Example 7 , and the results obtained for the monobromo and dibromo compounds are shown in Table 2.
[0036]
[Table 2]
Example 12
A 50 ml pressure-resistant glass sealed tube tube was charged with 1.66 g (10 mmol) of isophthalic acid, 6.00 g of 10 wt% fuming sulfuric acid and 1.6 g (10 mmol) of bromine and stirred at 150 ° C. for 7 hours. After the reaction, the mixture was cooled to room temperature, and the contents were taken out into a beaker containing ice water to obtain a solid. The produced solid was filtered, washed with cooling, and dried under reduced pressure to obtain crude crystals of the desired product. Thereafter, 13.1 g (408 mmol) of methanol and 0.35 g (30 mol%) of sulfuric acid were added to the autoclave, and the mixture was heated and stirred at 120 ° C. to induce dimethyl ester. Subsequently, by subjecting this to rectification, 1.78 g (yield 65.1%, reduced pressure boiling point 159 ° C./4.8 mmHg) of the target dimethyl 5-bromoisophthalate, and dimethyl isophthalate corresponding to the raw material, 0. 52 g (yield 26.8%, boiling point under reduced pressure 133 ° C./4.8 mmHg) was obtained. The obtained crystals were confirmed to be dimethyl 5-bromoisophthalate and 5-bromoisophthalic acid from MASS, 1 H-NMR and melting point.
[0040]
【Effect of the invention】
According to the method of the present invention, a 5-bromoisophthalic acid compound can be produced selectively and inexpensively.
Claims (4)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001167799A JP4893903B2 (en) | 2000-06-05 | 2001-06-04 | Method for producing bromoisophthalic acid compound |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000167511 | 2000-06-05 | ||
| JP2000167511 | 2000-06-05 | ||
| JP2000-167511 | 2000-06-05 | ||
| JP2001167799A JP4893903B2 (en) | 2000-06-05 | 2001-06-04 | Method for producing bromoisophthalic acid compound |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2002060370A JP2002060370A (en) | 2002-02-26 |
| JP4893903B2 true JP4893903B2 (en) | 2012-03-07 |
Family
ID=26593316
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001167799A Expired - Fee Related JP4893903B2 (en) | 2000-06-05 | 2001-06-04 | Method for producing bromoisophthalic acid compound |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4893903B2 (en) |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3911732B2 (en) * | 1996-10-15 | 2007-05-09 | 日産化学工業株式会社 | Process for producing dialkyl 5-bromo-isophthalate |
-
2001
- 2001-06-04 JP JP2001167799A patent/JP4893903B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002060370A (en) | 2002-02-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| HU193589B (en) | Process for preparing fluorinated phtaloyl- and terephtaloyl compounds | |
| JP5359052B2 (en) | Method for producing fluorine-containing monomer | |
| JP4896040B2 (en) | Method for producing polymerizable hydroxydiamantyl ester compound | |
| JP4893903B2 (en) | Method for producing bromoisophthalic acid compound | |
| KR100787277B1 (en) | Process for preparing 5-bromoisophthalic acid compound | |
| EP1041080A1 (en) | Process for the preparation of pentaacetyl-beta-D-glucopyranose | |
| JP4038657B2 (en) | Method for producing adamantanone | |
| JP2007231002A (en) | Manufacturing method of polymerizable diamantyl ester compound | |
| JPH0678265B2 (en) | Process for producing 1,4-bis- (4-hydroxybenzoyl) -benzene | |
| JP5000031B2 (en) | Method for producing aromatic-o-dialdehyde compound | |
| JPH05286902A (en) | Production of alpha-chloro-beta-ketoester derivative | |
| JP3259893B2 (en) | Method for producing 3,3-dichloro-1,1,1-trifluoropropan-2-one | |
| WO2008075534A1 (en) | Process for producing fluoroalkane ester | |
| US6111130A (en) | Process for the preparation of trifluoromethyl containing derivatives | |
| JP4022413B2 (en) | Perfluoroalkylacrylic acid multimers and multimer compositions and methods for their production | |
| JP4120715B2 (en) | Process for producing 2-bromo and / or dialkyl 2,5-dibromoterephthalate | |
| JPH0967297A (en) | Production of bistrifluoromethylbenzoic acids | |
| JP3312414B2 (en) | Process for producing dienoic halides | |
| JP3997799B2 (en) | Method for producing sulfonic anhydride | |
| JPH11171839A (en) | Production of 1,4-dihydroxy-2-naphthoic aryl ester | |
| JP2005132728A (en) | Method for producing methyltrifluoromethylbenzoic acid derivative | |
| JPH09241184A (en) | Production of friedel-crafts alkylation reaction product | |
| JPH11246542A (en) | New dichlorophthalide and its production, and production of 2-formylbenzoic acid nucleus chlorination products using the same | |
| JP2002363112A (en) | Method for producing chlorinated hydrocarbon | |
| JPH11199543A (en) | Production of benzene nucleus-chlorinated 2-formylbenzoic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080221 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110407 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110606 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20110727 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111024 |
|
| A911 | Transfer of reconsideration by examiner before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20111101 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20111124 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20111207 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150106 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |