JP4504268B2 - Method for coating semiconductor surface and method for producing semiconductor particle - Google Patents
Method for coating semiconductor surface and method for producing semiconductor particle Download PDFInfo
- Publication number
- JP4504268B2 JP4504268B2 JP2005188576A JP2005188576A JP4504268B2 JP 4504268 B2 JP4504268 B2 JP 4504268B2 JP 2005188576 A JP2005188576 A JP 2005188576A JP 2005188576 A JP2005188576 A JP 2005188576A JP 4504268 B2 JP4504268 B2 JP 4504268B2
- Authority
- JP
- Japan
- Prior art keywords
- silicon
- group
- semiconductor
- nanoparticles
- electron
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images






Landscapes
- Silicon Compounds (AREA)
- Liquid Deposition Of Substances Of Which Semiconductor Devices Are Composed (AREA)
- Led Devices (AREA)
Description
本発明はシリコン、ゲルマニウム、炭素、錫、鉛などの4族半導体元素を主たる成分とする4族系半導体の表面を、4族元素または遷移金属元素により被覆する方法、その方法を利用したナノ粒子の製造方法、および、それを用いた発光素子や、電気光学効果に基づく光変調素子などの光素子に関するものである。
The present invention relates to a method for coating the surface of a
光情報通信分野への応用を目指した、光材料としてのシリコンの研究は、歴史的にも旧く、1980年代半ばから現在にかけて長期に亘って行われている。しかしながら、III−V族化合物であるインジウム燐、ガリウムヒ素や、絶縁体のニオブ酸リチウムなど、より新しい光材料の研究と比較すると、シリコンの研究にはあまり進展が見られない。 The research of silicon as an optical material aiming at application in the field of optical information communication is historically old and has been conducted for a long time from the mid-1980s to the present. However, compared to the research of newer optical materials such as III-V group compounds such as indium phosphide, gallium arsenide, and the insulator lithium niobate, there has been little progress in silicon research.
これには2つの大きな理由がある。第一の理由は、シリコンには光を放出する機構が備わっていない、すなわち、発光を示さないことである。第二の理由は、シリコンはポッケルス効果やカー効果などの電気光学効果(EO効果)を示さないことである。これらの理由により、技術的な応用範囲が限られるために、光材料としてシリコンは着目されなかったのであると考えられる。なお、EO効果とは、透明な材料に外部電場を加えたときに屈折率が変化する現象の総称であり、屈折率変化が電場の強さに比例するときをポッケルス効果と呼び、それが電場の二乗に比例するときをカー効果と呼ぶ。 There are two main reasons for this. The first reason is that silicon has no mechanism for emitting light, that is, it does not emit light. The second reason is that silicon does not show an electro-optic effect (EO effect) such as Pockels effect or Kerr effect. For these reasons, since the technical application range is limited, it is considered that silicon was not paid attention as an optical material. The EO effect is a general term for a phenomenon in which the refractive index changes when an external electric field is applied to a transparent material. When the refractive index change is proportional to the strength of the electric field, it is called the Pockels effect. When it is proportional to the square of, it is called the Kerr effect.
ところで、シリコンが発光しない理由は、間接バンドギャップを有する半導体材料であり、半導体レーザーのような効率的な発光素子を作れない結晶構造を持つためである。これとは対照的に、III−V族化合物のインジウム燐やガリウムヒ素は直接バンドギャップを有する半導体材料であることから、半導体レーザーの材料としてよく用いられている。 By the way, the reason why silicon does not emit light is that it is a semiconductor material having an indirect band gap, and has a crystal structure that cannot make an efficient light emitting element such as a semiconductor laser. In contrast, indium phosphide and gallium arsenide, which are III-V compounds, are semiconductor materials having direct band gaps and are often used as semiconductor laser materials.
また、シリコンがEO効果を示さない理由は、結晶構造が高い対称性を持つためである。EO効果を示す材料を用いると、レーザー光の高速変調が可能になる。もう少し具体的に言うと、電気信号を光信号に変換する光変調器を作ることが可能になる。ポッケルス効果を示すニオブ酸リチウムを用いて実際に光変調器が作られており、光ファイバ通信の分野で広く用いられている。 The reason why silicon does not exhibit the EO effect is that the crystal structure has high symmetry. When a material exhibiting the EO effect is used, high-speed modulation of laser light becomes possible. More specifically, it becomes possible to make an optical modulator that converts an electrical signal into an optical signal. An optical modulator is actually made using lithium niobate exhibiting the Pockels effect, and is widely used in the field of optical fiber communication.
表1は、従来検討された、シリコンに発光機能を与える材料技術をまとめたもので、6つの代表的な技術である、液相合成シリコンナノ粒子、希土類イオンドーピング、気相合成シリコンクラスター、ポーラスシリコン、シリサイド半導体、歪み超格子について、課題などをそれぞれ示した。

結論から言うと、いずれの材料技術によっても、シリコン材料を効率的に光らせるには至っていない。1つ目の、液相合成シリコンナノ粒子については、粒径が小さいものしか作れない「小粒径問題」という課題がある。液相合成したシリコンナノ粒子は粒径が小さすぎるため、主に青紫領域で発光を示し、可視発光は得られ難い。 In conclusion, none of the material technologies has led to efficient light emission of silicon materials. As for the first liquid phase synthetic silicon nanoparticles, there is a problem of “small particle size problem” in which only particles having a small particle size can be produced. Since the silicon nanoparticles synthesized in the liquid phase have a particle size that is too small, they emit light mainly in the blue-violet region and it is difficult to obtain visible light emission.
2つ目の希土類イオンドーピング(Erイオンドーピング)については、シリコン結晶中に発光性のErイオンが殆ど溶けないために、ドーピングが困難であり、結果として発光が得づらい課題がある。 The second rare earth ion doping (Er ion doping) has a problem that light emitting Er ions are hardly dissolved in the silicon crystal, so that doping is difficult and as a result, it is difficult to obtain light emission.
3つ目の気相合成シリコンクラスターについては、液相合成シリコンナノ粒子とは対照的に、粒径が大きいものしか作れない「大粒径問題」という課題がある。クラスターは主に赤外領域でしか発光を示さず、可視領域では光らない。 In contrast to the liquid phase synthetic silicon nanoparticles, the third gas phase synthetic silicon cluster has a problem of “large particle size problem” in which only particles having a large particle size can be produced. The cluster mainly emits light only in the infrared region and does not emit light in the visible region.
4つ目のポーラスシリコンについては、気相合成シリコンクラスターと同様、「大粒径問題」という課題がある。陽極酸化では粒径がまだ大きいため、赤領域での発光を示すが、青や緑の発光は得られ難い。 The fourth porous silicon has a problem of “large particle size problem” as in the case of the vapor-phase synthetic silicon cluster. Since the particle size is still large in anodic oxidation, light emission in the red region is exhibited, but blue or green light emission is difficult to obtain.
5つ目のシリサイド半導体(おもにβ−FeSi2を指す)については、材料中に歪みを導入しないと光らないという課題がある。なお、計算と実験の両方から、β−FeSi2は間接バンドギャップの半導体であることが示されており、本質的に光らない物質であることが判明してきた。 The fifth silicide semiconductor (mainly β-FeSi 2 ) has a problem that it does not shine unless strain is introduced into the material. Note that both calculations and experiments have shown that β-FeSi 2 is an indirect bandgap semiconductor, and is essentially a non-luminous substance.
6つ目の歪み超格子(Si/SiGe)については、そのような超格子を有する粒子は殆ど光らないという課題がある。SiGeがそもそも間接バンドギャップの半導体であり、そのために発光し難いことが判明しつつある。 The sixth strained superlattice (Si / SiGe) has a problem that particles having such a superlattice hardly emit light. SiGe is an indirect bandgap semiconductor in the first place, and therefore it is becoming difficult to emit light.
以上の通り、シリコン材料に発光機能を与えることを目的とした従来の材料技術では、実際にシリコン材料を効率的に光らせることができなかった。 As described above, the conventional material technology aiming to give a light emitting function to the silicon material cannot actually cause the silicon material to emit light efficiently.
シリコンに発光機能を与えることができると、以下に述べるような理由で、エレクトロニクスを大きく進化させることが可能になり、豊かな未来社会の実現に大きく貢献すると期待される。 If silicon can be given a light emitting function, it will be possible to make significant progress in electronics for the reasons described below, and it is expected to contribute greatly to the realization of a prosperous future society.
来るべき未来社会では、情報を自在に活用したいという社会的欲求が益々高まると考えられ、LSIを始めとするエレクトロニクスには今以上の超高速性が求められると考えられている。情報処理や情報伝達の担い手は、その主役の座が、現在の電子から、最速の情報速度を持つ光へと代わるものと予測されている。 In the future society to come, the social desire to use information freely is expected to increase, and electronics such as LSIs are expected to have higher speed than ever. Information processing and information transmission players are expected to replace their current role of electrons with the light with the fastest information speed.
光情報通信の分野において、光ファイバ通信をハードウエア面で支える個別の光デバイス(半導体レーザー、光変調器他)が各々シリコンで置き換えられるようになると、LSIと光ファイバがシリコンウエハ上で一体化し、やがて同化する時代が到来するであろう。これは、エレクトロニクスと通信が一体化し、情報処理・伝達の速度が光速の極限にまで速められることを意味する。従って、光デバイスのシリコン化は、広帯域を必要とする、パソコン、検索(インターネット)、画像認識システム、通信、予報・予測(演算や計算)など、様々なアプリケーションや社会インフラの超高速化に大きく貢献するという訳である。 In the field of optical information communication, when individual optical devices (semiconductor lasers, optical modulators, etc.) that support optical fiber communication in terms of hardware are replaced with silicon, LSI and optical fiber are integrated on a silicon wafer. The time to assimilate will come soon. This means that electronics and communication are integrated, and the speed of information processing and transmission can be increased to the limit of the speed of light. Therefore, siliconization of optical devices will greatly increase the speed of various applications and social infrastructures such as personal computers, search (Internet), image recognition systems, communications, forecasting / prediction (calculations and calculations), etc., which require a wide bandwidth. It is a translation.
ところが、従来の材料技術では、シリコンを効率的に光らせることが出来ないという本質的な課題を抱えていることは前記したとおりである。 However, as described above, the conventional material technology has an essential problem that silicon cannot be efficiently illuminated.
本発明は、かかる課題の認識に基づいてなされたものであり、その目的とするところは、4族系半導体材料に発光機能を与える方法として、従来法とは全く異なる、新規な半導体表面の被覆方法と、同様な手法に基づく新規な液相合成ナノ粒子の製造方法とを提供し、かつ、この発光機能を持たせた4族半導体材料をベースとする光素子を提供するものである。
The present invention has been made on the basis of recognition of such a problem, and the object of the present invention is to provide a novel semiconductor surface coating that is completely different from the conventional method as a method for imparting a light emitting function to a
本発明による半導体表面の被覆方法は、
シリコン、ゲルマニウム、炭素、および錫からなる群から選ばれる4族元素を含んでなる半導体材料の表面に、不活性有機溶媒に可溶でありかつ電子供与性の還元剤を前記不活性有機溶媒中で反応させて半導体材料の表面を前記還元剤に由来する金属で被覆し、その後、4族元素および遷移金属元素からなる群から選ばれる元素を含む電子吸引基含有化合物を反応させ、半導体材料の表面を前記電子吸引基含有化合物に由来する4族元素または遷移金属元素で被覆する
ことを特徴とするものである。
The method for coating a semiconductor surface according to the present invention comprises:
On the surface of the semiconductor material containing a
また、本発明による半導体粒子の製造方法は、半導体材料に種結晶を用いて、前記の方法により結晶成長させることを特徴とするものである。 The method for producing semiconductor particles according to the present invention is characterized in that a seed crystal is used as a semiconductor material and crystal growth is performed by the above-described method.
また、本発明による半導体粒子は、前記の半導体粒子の製造方法により製造されたことを特徴とするものである。 The semiconductor particles according to the present invention are manufactured by the above-described method for manufacturing semiconductor particles.
また、本発明による発光素子は、電気的または光学的なエネルギー励起により光を発生する発光部を具備してなる発光素子であって、前記発光部が前記の半導体粒子を有することを特徴とするものである。 The light-emitting device according to the present invention is a light-emitting device including a light-emitting portion that generates light by electrical or optical energy excitation, and the light-emitting portion includes the semiconductor particles. Is.
また、本発明による光変調素子は、電気的または光学的な光変調により光信号を発生する光変調部を具備してなる光変調素子であって、前記光変調部が前記の半導体粒子を有することを特徴とするものである。 The light modulation element according to the present invention is a light modulation element including a light modulation unit that generates an optical signal by electrical or optical light modulation, and the light modulation unit includes the semiconductor particles. It is characterized by this.
本発明の方法によれば、半導体表面に原子レベルで精密に厚さおよび組成が制御された被覆を形成させることができる。さらにその方法を種粒子に適用することによって、粒径制御され、かつ組成制御された4族系半導体ナノ粒子の合成が、はじめて可能になる。他の製造方法で作られた4族系半導体ナノ粒子と比較して、発光波長のテーラーメード化、発光効率の向上など、真に発光特性に優れた4族系半導体ナノ粒子がはじめて実現可能になる。
According to the method of the present invention, it is possible to form a coating whose thickness and composition are precisely controlled at the atomic level on the semiconductor surface. Furthermore, by applying the method to seed particles, synthesis of
そして、本発明の製造方法による4族系半導体ナノ粒子を用いると、光デバイスがシリコン化でき、LSIの光速化が進むため、広帯域を必要とする、パソコン、検索(インターネット)、画像認識システム、通信、予報・予測(演算や計算)など、様々なアプリケーションや社会インフラの超高速化が実現可能になり、その産業上のメリットは多大である。
And when the
以下、図面を参照しつつ、本発明の実施の形態(実施形態)を詳細に説明する。
まず初めに、第1節では、本発明の長所、差別化点を明確にするために、4族系半導体材料に発光機能を与える従来法の1つである、グリニャール法と呼ばれるナノ粒子の液相合成法について、シリコンナノ粒子の合成を例に、その原理を説明する。次に、第2節では、グリニャール法の欠点である「小粒径問題」を考察し、可視発光が可能な粒径にまで成長させることが困難なことを説明する。次に、第3−a節、第3−b節では、本発明のナノ粒子製造方法の原理を説明し、原子レベルでの精密な粒径制御が可能なこと、グリニャール法と異なり大粒径化が可能なことを示し、本発明の目的の1つである4族半導体ナノ粒子での可視発光化が実現可能なことを述べる。そして、第3−c節では、本発明の半導体表面被覆方法の原理を説明し、目的である4族半導体材料への発光機能の付与が実現可能なことを述べる。
Hereinafter, embodiments (embodiments) of the present invention will be described in detail with reference to the drawings.
First of all, in
1)グリニャール法
この方法は、溶液中において、シリコン化合物分子間での縮合反応を利用し、3次元的にSi−Si結合を成長させてナノ粒子を得る手法である。
1) Grignard method This method is a technique for obtaining nanoparticles by three-dimensionally growing Si-Si bonds using a condensation reaction between silicon compound molecules in a solution.
図1は、Si−Si結合の形成と成長の様子を模式的に示すものである。図に示すように、グリニャール法で用いられる出発材料は、塩化シリコン(液体)と金属マグネシウム(粉体)である。不活性有機溶媒としては脱水したエチレングリコールジメチルエーテル(以下、glymeという)が一般に用いられる。glyme中での反応ステップは、以下の通りである。
1.金属マグネシウム粉を入れたglyme中に塩化シリコンを添加する。
2.攪拌によって、塩化シリコンと金属マグネシウムを衝突させる。
3.この衝突によって、反応中間体である、塩化マグネシウムシリコン分子をその場生成させる。
4.下記反応式で表される塩化シリコンと塩化マグネシウムシリコンの分子縮合反応によって、Si−Si結合を形成させる
Cl3Si−MgCl + Cl4Si → Cl3Si−SiCl3 + MgCl2
5.攪拌を続けながら、3次元的にSi−Si結合を成長させる。
FIG. 1 schematically shows the formation and growth of Si—Si bonds. As shown in the figure, the starting materials used in the Grignard method are silicon chloride (liquid) and magnesium metal (powder). As the inert organic solvent, dehydrated ethylene glycol dimethyl ether (hereinafter referred to as “glyme”) is generally used. The reaction steps in glyme are as follows.
1. Silicon chloride is added into glyme containing metal magnesium powder.
2. By stirring, silicon chloride and metal magnesium collide.
3. By this collision, magnesium chloride molecules, which are reaction intermediates, are generated in situ.
4). Cl 3 Si—MgCl + Cl 4 Si → Cl 3 Si—SiCl 3 + MgCl 2 that forms a Si—Si bond by molecular condensation reaction of silicon chloride and magnesium chloride represented by the following reaction formula
5). While continuing stirring, Si—Si bonds are grown three-dimensionally.
反応ステップ4の反応式からわかるように、Si−Si結合は、glyme中でその場生成させた塩化マグネシウムシリコン分子の−MgCl基が反応点となって、塩化シリコン分子の被反応点である−Cl基と縮合反応することで形成される。この化学反応は、−MgCl基は電子供与性の官能基であり、−Cl基は電子吸引性の官能基であることから、両者が電気的に引き合って化学反応が生じるものと考えられる。
As can be seen from the reaction formula of the
なお、塩化マグネシウムシリコン分子は、マグネシウム原子の付加数に応じてタイプ1からタイプ4に分類される。タイプ別の生成確率は、塩化シリコン分子と金属マグネシウムの濃度に強く依存するが、通常の条件(塩化シリコン分子:金属マグネシウムのモル比が1:1乃至1:4の条件)の下では、タイプ1が他のタイプ2,3,4よりも高い割合で生成される。これは以下のように説明出来る。すなわち、塩化マグネシウムシリコン分子は、溶液に溶けた塩化シリコン分子が、溶液に不溶な金属マグネシウム粉に衝突することで生じる。生じた塩化マグネシウムシリコン分子が、再度金属マグネシウムに衝突する確率は極めて低く、その前に溶液中に溶けたほかの塩化シリコン分子や、成長途中のナノ粒子と反応する。このために、タイプ1の塩化マグネシウムシリコン分子が、他のタイプよりも高い割合で生成する。
Note that magnesium silicon chloride molecules are classified into
タイプ1の塩化マグネシウムシリコン分子は、1分子あたり、反応点(≡Si−MgCl)は1点、被反応点(≡Si−Cl)は3点を持ち、反応点と非反応点の数が一致していない。次に述べるが、この不一致が小粒径問題の原因となる。
Each
2)グリニャール法の小粒径問題
図2aは、結晶シリコンの原子構造を模式的に示すものである。結晶シリコンはダイヤモンド構造を持ち、図に示すように、6員環を有する。
図2bは、成長初期のシリコンナノ粒子を模式的に示すものである。この図は成長反応の核となる第1世代(1個目)のシリコン原子から第3世代シリコン原子までSi−Si結合が成長している様子を示している。
2) The small particle size problem of the Grignard method FIG. 2a schematically shows the atomic structure of crystalline silicon. Crystalline silicon has a diamond structure and has a six-membered ring as shown in the figure.
FIG. 2b schematically shows silicon nanoparticles in the early stage of growth. This figure shows a state in which Si—Si bonds are grown from the first generation (first) silicon atom, which is the nucleus of the growth reaction, to the third generation silicon atom.
結晶シリコンと同様、シリコンナノ粒子がダイヤモンド構造をとりながら成長するには、第4世代シリコン原子となるべき塩化マグネシウムシリコン分子が2個の第3世代シリコン原子と反応することで、6員環を形成する必要がある。このため、第3世代のシリコンナノ粒子に反応する塩化マグネシウムシリコン分子は、2箇所の第3世代シリコンの被反応点(≡Si−Cl)と反応する必要がある。 Similar to crystalline silicon, silicon nanoparticles grow in a diamond structure, so that the magnesium chloride silicon molecules that should become fourth-generation silicon atoms react with two third-generation silicon atoms to form a six-membered ring. Need to form. For this reason, the magnesium chloride silicon molecule that reacts with the third generation silicon nanoparticles needs to react with the reaction sites (≡Si—Cl) of two third generation silicon.
しかしながら、通常生成されるタイプ1の塩化マグネシウムシリコン分子は1個のマグネシウム原子しか持たず、反応点(≡Si−MgCl)は1箇所しかない。従って、塩化マグネシウムシリコン分子は、どちらか一方の第3世代シリコン原子とは確実に反応することができない、特異的な状況が生じることが判る。
However, the
いま仮に、片方の第3世代シリコンと反応して、第4世代シリコンが成長したと考える。しかしながら、もう片方の未反応の第3世代シリコン原子に付加する3個の−Cl基と、第4世代シリコン原子に付加する3個の−Cl基との間の立体障害は非常に大きいため、第3・第4世代間のSi−Si結合はエネルギー的に不安定で、この結合は壊れやすい傾向にある。このため、通常の合成条件(塩化シリコン分子:金属マグネシウムのモル比が1:1乃至1:4の条件)の下では、第4世代シリコンは極めて成長しづらい。 Now, suppose that the fourth generation silicon has grown by reacting with one of the third generation silicon. However, the steric hindrance between the three —Cl groups added to the other unreacted third generation silicon atom and the three —Cl groups added to the fourth generation silicon atom is very large. The Si-Si bond between the third and fourth generations is energetically unstable, and this bond tends to be fragile. For this reason, under normal synthesis conditions (conditions where the molar ratio of silicon chloride molecule: metallic magnesium is 1: 1 to 1: 4), the fourth generation silicon is extremely difficult to grow.
第3世代まででナノ粒子の成長が止まるとすると、ナノ粒子はたった17個の原子から構成されることになる。このときのナノ粒子の粒径を見積もると、概ね0.5nm乃至1nm程度となる。フォトルミネッセンスのピーク波長を調べると350〜400nm程度であり、紫外−青紫色領域で発光する粒子の大きさに対応する。 If nanoparticle growth stops until the third generation, the nanoparticle will consist of only 17 atoms. When the particle size of the nanoparticles at this time is estimated, it is approximately 0.5 nm to 1 nm. When the peak wavelength of photoluminescence is examined, it is about 350 to 400 nm, which corresponds to the size of particles emitting in the ultraviolet-blue-violet region.
文献より、シリコンをナノ粒子化することで青色、緑色、赤色発光を得るには、粒径2〜2.5nm、粒径2.5〜3nm、粒径3〜5nmにそれぞれ調整する必要があると見積もられている。発光波長が長くなる、すなわち発光色が紫外→青→緑→赤の順に変化するほど、粒径が大きくなるのは、量子閉じ込め効果によるものである。
According to the literature, in order to obtain blue, green, and red light emission by making silicon into nanoparticles, it is necessary to adjust the particle size to 2 to 2.5 nm, the particle size 2.5 to 3 nm, and the
以上の点から、グリニャール法で得られる粒径は、可視発光を得るには小さすぎることがわかる。これが、グリニャール法に特有の小粒径問題である。同様なナノ粒子の成長反応の抑制は、他の4族元素であるゲルマニウム、炭素、錫でも観察され、グリニャール法を用いた4族系半導体ナノ粒子の合成に共通する現象である。
From the above points, it can be seen that the particle size obtained by the Grignard method is too small to obtain visible light emission. This is a small particle size problem peculiar to the Grignard method. Similar suppression of nanoparticle growth reaction is also observed in
3)本発明による半導体ナノ粒子の製造方法と、半導体表面の被覆方法
4族系半導体ナノ粒子とは異なり、Au、Ag、Pt、Pt、Cu、Fe、Co、Niなどの金属ナノ粒子、CdS、CdSe、CdTe、ZnS、ZnSe、ZnTeなどのII−VI族化合物半導体ナノ粒子、さらにGaAs、InAs、InPなどのIII−V族化合物半導体ナノ粒子では、液相合成法が確立している。これは、構成原子である、Au、Ag、Pt、Pt、Cu、Fe、Co、Ni、Cd、Zn、S、Se、Te、Ga、In、As、Pなどの各原子が、溶液中で、原子イオン状態で比較的安定に存在でき、金属ナノ粒子や化合物半導体ナノ粒子への結晶成長が、時間、試薬濃度、温度などの基本的実験パラメータで比較的容易に制御できるためである。
3) The method for producing semiconductor nanoparticles according to the present invention and the method for coating the semiconductor surface Unlike
これに対して、Si、Ge、C、Snなど4族系原子は、CVDなどで作ったプラズマ状態を除き、原子イオン状態が極めて不安定であり、シリコンを例に取れば、通常塩化シリコンなどの化合物分子の形でしか存在しない。このため、液相合成における化学反応は、他材料系のナノ粒子と異なり、分子縮合反応をベースに組み立てる必要がある。
On the other hand,
先のグリニャール法におけるSi−Si結合形成の本質は、或る分子における1個の電子供与性の反応点(≡Si−MgCl)と、別の分子における1個の電子吸引性の被反応点(≡Si−Cl)との間での分子縮合反応にあった。課題である小粒径問題は、Si−Si結合形成に関わる、塩化マグネシウムシリコン分子と塩化シリコン分子において、その電子供与性基と電子吸引性基の数が不一致であることに起因した。 The essence of Si-Si bond formation in the previous Grignard method is that one electron-donating reactive site (≡Si-MgCl) in one molecule and one electron-withdrawing reactive site in another molecule ( It was in a molecular condensation reaction with (≡Si—Cl). The problem of small particle size, which is a problem, is due to the fact that the number of electron-donating groups and electron-withdrawing groups are inconsistent in magnesium chloride silicon molecules and silicon chloride molecules involved in Si-Si bond formation.
4族半導体ナノ粒子を自在に合成する液相合成法、言い換えると、所望の化学組成で所望の粒径に結晶成長でき、かつ所望の波長で発光させられる、新しい液相合成法を実現するには、縮合反応させる分子の電子供与性基と電子吸引性基の数をつねに一致させるよう、化学反応全体を根本からデザインし直す必要がある。
In order to realize a liquid phase synthesis method for freely synthesizing
そこで、本発明による半導体表面の被覆方法は、シリコン、ゲルマニウム、炭素、および錫からなる群から選ばれる4族元素を含んでなる半導体材料の表面に、不活性有機溶媒に可溶でありかつ電子供与性の還元剤を前記不活性有機溶媒中で反応させて半導体材料の表面を前記還元剤に由来する金属で被覆し、その後、4族元素および遷移金属元素からなる群から選ばれる元素を含む電子吸引基含有化合物を反応させ、半導体材料の表面を前記電子吸引基含有化合物に由来する4族元素または遷移金属元素で被覆することを特徴とするものである。
Therefore, the method for coating a semiconductor surface according to the present invention comprises a surface of a semiconductor material containing a
図3は、本発明に関わる半導体表面の被覆方法を4族半導体粒子に適用した、最も基本的な4族系半導体ナノ粒子の液相における製造方法の模式説明図である。横軸は試薬を投入する手順(合成手順)を表し、縦軸は本手順に対応するナノ粒子の粒径を表す。
FIG. 3 is a schematic explanatory view of the most basic method for producing a
本発明による方法では、電子吸引基含有化合物である4族原子を含む化合物分子(塩化シリコンなど)と、不活性有機溶媒に可溶であり、かつ電子供与性の還元剤(例えばナトリウム・ナフタレンなどの電荷移動錯体、または二ヨウ化サマリウムなどの希土類ヨウ化物)とを用い、溶液中に、この両者を、交互に、必要に応じて繰り返し添加することで半導体表面を被覆する。この方法を半導体からなる種粒子に適用すれば4族系半導体ナノ粒子をつくることができる。本発明による方法により、原子レベルで精密に粒径制御され、かつ組成制御された4族系半導体ナノ粒子をつくることがはじめて可能になる。このようにして作られた4族系ナノ粒子は、従来のナノ粒子と比較して、所望の発光波長で、高い発光効率を持って発光させることが可能になる。
In the method according to the present invention, a compound molecule containing a
以下では、3−a)単一元素からなる1元系の4族系半導体ナノ粒子製造方法と、3−b)2元系の4族系化合物半導体ナノ粒子の製造方法の原理をそれぞれ説明する。例として、前者ではシリコンナノ粒子、後者ではゲルマニウム・カーボン化合物ナノ粒子を取り上げる。
In the following, the principles of 3-a) a
3−a)1元系からなる4族系半導体ナノ粒子の製造方法
本発明によるひとつの半導体粒子の製造方法は、シリコン、ゲルマニウム、炭素、錫から選ばれた4族系半導体材料において、1個の4族原子Aと4個と電子吸引基Xからなる分子(電子吸引基含有化合物)をAX4とし、1個のアルカリ原子Mと1個の芳香族分子Yからなる電荷移動錯体をMYとしたとき、不活性有機溶媒中で、前記分子と前記電荷移動錯体とを化学反応させる際、各々を、AX4とMYの序列、またはMYとAX4の序列で複数回繰り返して加えることで、A原子からなる3次元ナノ構造を原子単位でサイズ制御し、粒子、特に半導体ナノ粒子を成長させることができる。
3-a) Method for
例として、シリコンナノ粒子を形成させる場合について説明する。図3に示した合成手順に従い、glyme中に、シリコン原子の供給源である塩化シリコンと、塩化シリコンを還元する還元剤であるナトリウム・ナフタレン電荷移動錯体(正確には(ナトリウム)+(ナフタレン)―電荷移動錯体。以下、NN錯体という)のglyme溶液とを交互に加えることで、ダイヤモンド構造のシリコンナノ粒子を1原子世代づつ結晶成長させることができる。 As an example, a case where silicon nanoparticles are formed will be described. According to the synthesis procedure shown in FIG. 3, in glyme, silicon chloride as a silicon atom source and sodium naphthalene charge transfer complex as a reducing agent for reducing silicon chloride (exactly (sodium) + (naphthalene) -. a charge transfer complex or less, by adding alternately and glyme solution of NN as complex) can be 1 atomic generation by one crystal growth of silicon nanoparticles having a diamond structure.
さらに具体的に説明すると以下の通りである。
予め塩化シリコンを溶かしたglyme中にNN錯体を徐々に添加し、最初に、種結晶となる、と或る世代のシリコンナノ粒子を生じさせる。この最初に出来上がる種結晶となるナノ粒子は、表面が塩素終端されたナノ粒子である。引き続きNN錯体を添加し続けると、NN錯体はナノ粒子表面にさらに作用し、表面塩素原子はNaClの形に還元されて表面脱離し、また還元剤のナフタレン分子は負イオンから中性イオンに酸化されて溶液中に溶け出し、最終的にナノ粒子表面は全てNN錯体に由来するナトリウム原子で覆われる。この表面がナトリウム終端されたナノ粒子に、改めて塩化シリコン分子を作用させると、表面ナトリウム原子はNaClの形に酸化されて表面脱離し、一方塩化シリコンは塩素原子を失い還元され、塩化シリコンに由来するシリコン原子がナノ粒子表面に化学結合し、丁度1原子世代分だけシリコン原子層が成長した、表面が塩素終端されたシリコンナノ粒子が得られる。言い換えれば、粒子の表面にシリコン原子による単原子膜が形成される。これ以降、NN錯体と塩化シリコンを、規則正しい順番でN回加えれば、N原子世代だけシリコンナノ粒子を結晶成長させることが出来る、すなわち、粒径をN原子世代分だけ選択的に大きく成長させることができる。
More specifically, it is as follows.
An NN complex is gradually added into glyme in which silicon chloride is dissolved in advance, and a silicon nanoparticle of a generation is generated when it first becomes a seed crystal. The first nanoparticle that becomes the seed crystal is a nanoparticle whose surface is terminated with chlorine. When the NN complex is continuously added, the NN complex further acts on the surface of the nanoparticle, the surface chlorine atoms are reduced to the form of NaCl and eliminated, and the naphthalene molecule of the reducing agent is oxidized from a negative ion to a neutral ion. As a result, it dissolves into the solution, and finally the surface of the nanoparticles is entirely covered with sodium atoms derived from the NN complex. When the surface of the sodium-terminated nanoparticle is made to react with silicon chloride molecules, the surface sodium atoms are oxidized to form NaCl and desorbed, while silicon chloride loses chlorine atoms and is reduced, resulting from silicon chloride. The silicon atoms are chemically bonded to the surface of the nanoparticle, and a silicon atom layer in which the silicon atom layer is grown for exactly one atom generation is obtained. In other words, a monoatomic film made of silicon atoms is formed on the surface of the particle. Thereafter, if NN complex and silicon chloride are added N times in a regular order, silicon nanoparticles can be grown by N atom generations, that is, the grain size can be selectively increased by N atom generations. Can do.
なお、溶液中には、ナノ粒子の他に、NaClと中性化したナフタレン分子が混在するが、両者とも安定な状態に化学変化しているため、もはやナノ粒子とは反応しない。 In addition to the nanoparticles, NaCl and neutralized naphthalene molecules are mixed in the solution, but both of them are chemically changed to a stable state, and thus no longer react with the nanoparticles.
図4は、本発明による製造方法で作成した、ダイヤモンド構造シリコンナノ粒子の世代Gに対する粒径Dの計算結果を示す。粒径D(nm)はナノ粒子を球形に見立てたときの換算直径であり、次式で見積もることができる。
D=a×[3M/4π]1/3 (1)
aはバルク結晶シリコンの格子定数(0.543nm)、Mはナノ粒子を構成する全シリコン原子数である。なお、表2に、ダイヤモンド構造のナノ粒子におけるM値を示す。
D = a × [3M / 4π] 1/3 (1)
a is the lattice constant (0.543 nm) of bulk crystalline silicon, and M is the total number of silicon atoms constituting the nanoparticle. Table 2 shows the M values of diamond-structured nanoparticles.
図からわかるように、本方法によってナノ粒子を1世代分成長させると、粒径を約0.3nm増加させることができる。シリコンナノ粒子の可視発光は、一般に10nm以下、特に粒径2〜5nmの間で生じると考えられている。従って、7〜16世代にナノ粒子を成長させればよいことがわかる。なお、最初の種結晶を何世代目にするかは、合成条件(温度、濃度、攪拌速度など)によって調整することが可能である。2〜6世代の間の種結晶が得られやすく、合成条件を高温、低濃度、高速攪拌にすると、小さい世代の、大きさの揃った種結晶に調整することが可能である。本発明の方法により製造した、より大きなナノ粒子、例えば7〜16世代のナノ粒子を種結晶とすることも可能である。 As can be seen from the figure, when one nanoparticle is grown by this method, the particle size can be increased by about 0.3 nm. Visible emission of silicon nanoparticles is generally considered to occur at 10 nm or less, particularly between 2 and 5 nm in particle size. Therefore, it is understood that the nanoparticles should be grown in the 7th to 16th generations. The generation number of the first seed crystal can be adjusted according to the synthesis conditions (temperature, concentration, stirring speed, etc.). It is easy to obtain seed crystals of 2 to 6 generations, and if the synthesis conditions are high temperature, low concentration and high speed stirring, it is possible to adjust to seed crystals of a small generation and uniform size. It is also possible to use larger nanoparticles produced by the method of the present invention, for example, 7 to 16th generation nanoparticles as seed crystals.
還元剤として、不活性有機溶媒に可溶であり、かつ電子供与性の還元剤、例えばナトリウムナフタレンや二ヨウ化サマリウムなどを用いるのは、本発明による製造方法を実現する上で重要な因子の1つである。上記の説明では電荷移動錯体であるNN錯体を還元剤として用いて説明したが、その他の不活性有機溶媒に可溶であり、かつ電子供与性の還元剤、例えば希土類ヨウ化物も用いることができる。表3には、各種還元剤の比較表を示す。
還元剤には、相手に電子を与える電子供与性還元剤と、プロトンを与えるプロトン供与性還元の2種類がある。プロトン供与性の還元剤では、ナノ粒子表面を覆う塩素原子を水素原子に置き換えることは可能であるが、塩化シリコンと水素終端シリコンナノ粒子とは温度の低い溶液中では反応せず、ナノ粒子の成長を制御することができないことがあるので注意が必要である。 There are two types of reducing agents: an electron donating reducing agent that gives electrons to the partner and a proton donating reduction that gives protons. With proton-donating reducing agents, it is possible to replace the chlorine atoms covering the nanoparticle surface with hydrogen atoms, but silicon chloride and hydrogen-terminated silicon nanoparticles do not react in a low temperature solution, Care must be taken because growth may not be controlled.
また、表3より、還元剤には溶媒に不溶な種類と可溶な種類があることが示されている。溶媒に不溶な還元剤では、ナノ粒子と還元剤とが衝突する確率は殆どゼロであり、ナノ粒子表面は塩素終端のままである。塩素終端シリコンナノ粒子は、塩化シリコンと反応しにくいので、やはりナノ粒子の成長を制御しにくい。 Table 3 also shows that there are types of reducing agents that are insoluble and soluble in the solvent. With a reducing agent that is insoluble in the solvent, the probability that the nanoparticles and the reducing agent collide is almost zero, and the nanoparticle surface remains chlorine-terminated. Chlorine-terminated silicon nanoparticles are difficult to react with silicon chloride, so it is still difficult to control nanoparticle growth.
この問題を解決するために、本発明においては還元剤として不活性有機溶媒に可溶であり、かつ電子供与性であるものを用いる。このような還元剤としては、電荷移動錯体(電子供与性の金属原子と、電子受容性の有機分子の組み合わせ)または希土類ヨウ化物が好ましい。より具体的には、電荷移動錯体としてはナトリウム・ナフタレン錯体、リチウム・ナフタレン錯体、リチウム・ジ−t−ブチルビフェニルなど、希土類ヨウ化物としては、二ヨウ化サマリウム、二ヨウ化イットリウム、二ヨウ化ユーロピウムなどが挙げられる。希土類ヨウ化物は特に二価の希土類ヨウ化物が好ましい。ここで、「不活性有機溶媒に可溶である」とは、本発明の方法を実施するのに十分な量が不活性有機溶媒に溶解することを意味する。したがって、本発明の方法に用いる不活性有機溶媒の種類や、還元剤の種類によって必要な溶解度は変化するため、具体的な溶解度は限定されない。しかしながら、本発明に用いられる還元剤の溶解度の一例を挙げれば、glymeに対する溶解度が0.0005モル/リットル以上であることが好ましく、0.001モル/リットル以上であることがより好ましい。 In order to solve this problem, in the present invention, a reducing agent that is soluble in an inert organic solvent and is electron-donating is used. As such a reducing agent, a charge transfer complex (a combination of an electron-donating metal atom and an electron-accepting organic molecule) or a rare earth iodide is preferable. More specifically, sodium-naphthalene complex, lithium-naphthalene complex, lithium-di-t-butylbiphenyl, etc. as charge transfer complexes, samarium diiodide, yttrium diiodide, diiodide as rare earth iodides Examples include europium. The rare earth iodide is particularly preferably a divalent rare earth iodide. Here, “soluble in an inert organic solvent” means that an amount sufficient to carry out the method of the present invention is dissolved in the inert organic solvent. Therefore, since the required solubility varies depending on the type of the inert organic solvent used in the method of the present invention and the type of the reducing agent, the specific solubility is not limited. However, if an example of the solubility of the reducing agent used for this invention is given, it is preferable that the solubility with respect to glyme is 0.0005 mol / liter or more, and it is more preferable that it is 0.001 mol / liter or more.
NN錯体において、ナトリウム自体は、マグネシウム同様、通常は不活性有機溶媒に不溶な金属粉体であるが、溶媒に可溶なナフタレン分子と錯体を組ませることで、還元能力は維持したまま溶媒に可溶化させることができる。 In the NN complex, like magnesium, sodium itself is usually a metal powder that is insoluble in an inert organic solvent. However, by combining the complex with a naphthalene molecule that is soluble in the solvent, the sodium remains in the solvent while maintaining its reducing ability. It can be solubilized.
NN錯体以外の他の電荷移動錯体も、電子供与性で、且つ溶媒可溶であれば、本発明の方法に還元剤として用いることも可能である。電子供与性の金属原子としては、ナトリウムの他に、リチウム、カリウム、ルビジウム、セシウムなどのアルカリ金属原子から選ぶことが可能で、組み合わせる電子受容性の有機分子としては、ナフタレンの他に、ピレン、アントラセン、テトラセン、ペリレン、フェナントレン、フルオレン、ビフェニルをはじめとする各種芳香族有機分子をその分子骨格に含む有機分子の中から選ぶことが可能である。リチウム・ナフタレン錯体は有望な還元剤の1つである。 Other charge transfer complexes other than the NN complex can be used as a reducing agent in the method of the present invention as long as they are electron-donating and soluble in a solvent. In addition to sodium, the electron-donating metal atom can be selected from alkali metal atoms such as lithium, potassium, rubidium and cesium. As the electron-accepting organic molecule to be combined, in addition to naphthalene, pyrene, Various aromatic organic molecules such as anthracene, tetracene, perylene, phenanthrene, fluorene, biphenyl and the like can be selected from organic molecules containing in their molecular skeleton. Lithium naphthalene complexes are one promising reducing agent.
また、本発明の方法に用いることができる還元剤として、希土類ヨウ化物が挙げられる。ここで希土類ヨウ化物は、希土類金属イオンの価数変化を利用した還元剤である。このような還元剤を用いた場合の反応式は、ハロゲン化シランとして臭化シラン(1当量)に対して二ヨウ化サマリウム(3.2当量)を用いた場合、以下のよう示される。
SiBr4 + 3.2*SmI2
→ Siナノ粒子(ハロゲン終端)+3.2*SmI3−xBrx (2)
Moreover, rare earth iodide is mentioned as a reducing agent which can be used for the method of this invention. Here, the rare earth iodide is a reducing agent that utilizes the valence change of the rare earth metal ions. The reaction formula when such a reducing agent is used is as follows when samarium diiodide (3.2 equivalents) is used as the halogenated silane with respect to silane bromide (1 equivalent).
SiBr 4 + 3.2 * SmI 2
→ Si nanoparticles (halogen terminated) + 3.2 * SmI 3-x Br x (2)
希土類ヨウ化物は価数変化を利用した還元剤であるため、NN錯体などの電荷移動錯体とは異なり、副生成物として芳香族分子を生じないため、後工程の抽出・精製が容易になる利点がある。 Since rare earth iodide is a reducing agent that utilizes valence change, unlike charge-transfer complexes such as NN complexes, it does not generate aromatic molecules as a by-product, so it is easy to extract and purify after processes. There is.
シリコン原子の供給源としては、塩化シリコンの他に、臭化シリコン、沃化シリコン、テトラメトシキシシラン、テトラエトキシシランなどから選ぶことが可能である。 As a supply source of silicon atoms, in addition to silicon chloride, silicon bromide, silicon iodide, tetramethoxysilane, tetraethoxysilane, or the like can be selected.
不活性有機溶媒としては、上述した還元剤や塩化シリコンとは反応しない不活性有機溶媒が必須である。このような溶媒として、glyme、テトラヒドロフラン、ジエチルエーテルなど、エーテル結合を有する溶媒を用いることができる。 As the inert organic solvent, an inert organic solvent that does not react with the above-described reducing agent or silicon chloride is essential. As such a solvent, a solvent having an ether bond such as glyme, tetrahydrofuran, or diethyl ether can be used.
なお、ここで説明したシリコンナノ粒子だけでなく、ゲルマニウムナノ粒子、ダイヤモンドナノ粒子、錫ナノ粒子なども基本的には同様な手順で作成可能である。ゲルマニウムナノ粒子は塩化ゲルマニウムとNN錯体から合成可能であり、ダイヤモンドナノ粒子は4臭化炭素とNN錯体から合成可能であり、さらに錫ナノ粒子は塩化錫とNN錯体から合成可能である。いずれも1原子世代ずつ選択的に結晶成長させることが可能である。 In addition to the silicon nanoparticles described here, germanium nanoparticles, diamond nanoparticles, tin nanoparticles, and the like can be basically produced by the same procedure. Germanium nanoparticles can be synthesized from germanium chloride and NN complexes, diamond nanoparticles can be synthesized from carbon tetrabromide and NN complexes, and tin nanoparticles can be synthesized from tin chloride and NN complexes. In any case, it is possible to selectively grow crystals one generation at a time.
3−b)2元系からなる4族系化合物半導体ナノ粒子の製造方法
単一元素からなる半導体粒子の製造方法と異なる点は、4族系化合物を構成する各々の4族系原子の供給源となる、2種類の異なる分子を用いる点である。
3-b) Method for
すなわち、本発明によるもうひとつの半導体粒子の製造方法は、シリコン、ゲルマニウム、炭素、錫、鉛から選ばれた4族系半導体材料において、1個の4族原子Aと4個と電子吸引基Xからなる分子をAX4とし、前記Aと異なる種類の4族原子Bと前記電子吸引基Xからなる分子をBX4とし、1個のアルカリ原子Mと1個の芳香族分子Yからなる電荷移動錯体をMYとしたとき、不活性有機溶媒中で、前記分子と前記電荷移動錯体とを化学反応させる際、各々を、AX4、MY、BX4、MYの序列、またはMY、BX4、MY、AX4の序列、またはBX4、MY、AX4、MYの序列、またはMY、AX4、MY、BX4の序列で複数回繰り返して加えることで、AB2種類の原子からなる3次元ナノ構造を原子単位でサイズ制御し、粒子を成長させることができる。
That is, another method for producing semiconductor particles according to the present invention provides a
以下、例として、ゲルマニウム・カーボン化合物ナノ粒子で説明する。
図5は、この2元系の合成方法を説明する模式図である。図3と同様、横軸が合成手順、縦軸が原子の世代を表す。図から分るように、4塩化ゲルマニウム→NN錯体→4臭化炭素→NN錯体を1サイクルとして、これらの試薬を規則正しく繰り返し添加することで、ゲルマニウム原子世代と炭素原子世代が交互に配列する、ダイヤモンド構造(立方硫化亜鉛構造)のゲルマニウム・カーボン化合物ナノ粒子を結晶成長させることが可能になる。
Hereinafter, germanium / carbon compound nanoparticles will be described as an example.
FIG. 5 is a schematic diagram illustrating this binary system synthesis method. Similar to FIG. 3, the horizontal axis represents the synthesis procedure and the vertical axis represents the generation of atoms. As can be seen from the figure, germanium tetrachloride → NN complex → carbon tetrabromide → NN complex is one cycle, and these reagents are added regularly and repeatedly, so that germanium atom generation and carbon atom generation are alternately arranged. It becomes possible to grow germanium-carbon compound nanoparticles having a diamond structure (cubic zinc sulfide structure).
さらに具体的に説明すると以下の通りである。図5では、4塩化ゲルマニウムから合成を開始しているため、種結晶はゲルマニウムナノ粒子になる。先の1次元の場合と同様、種結晶表面ははじめ塩素終端であるが、NN錯体を添加し続けることで、ナトリウム終端へと変化する。次に4臭化炭素を加えると、表面ナトリウム原子はNaBrの形で表面脱離し、一方4臭化炭素は臭素原子を失い還元されてナノ粒子表面に化学結合し、丁度1原子世代分だけ炭素原子がゲルマニウム種結晶を覆った、最表面が塩素終端されたナノ粒子が得られる。これ以降、NN錯体と4塩化ゲルマニウムと4臭化炭素を規則正しい順番で加え続けると、ゲルマニウム原子と炭素原子が交互に成長したゲルマニウム・カーボンナノ粒子が得られるという訳である。 More specifically, it is as follows. In FIG. 5, since the synthesis is started from germanium tetrachloride, the seed crystal becomes germanium nanoparticles. As in the previous one-dimensional case, the seed crystal surface is initially chlorine-terminated, but changes to sodium-terminated by continuing to add the NN complex. Next, when carbon tetrabromide is added, surface sodium atoms are desorbed in the form of NaBr, while carbon tetrabromide loses bromine atoms and is reduced and chemically bonded to the surface of the nanoparticle, just one atomic generation worth of carbon. Nanoparticles with the outermost surface terminated with chlorine, with atoms covering germanium seed crystals, are obtained. Thereafter, if NN complex, germanium tetrachloride, and carbon tetrabromide are continuously added in a regular order, germanium-carbon nanoparticles in which germanium atoms and carbon atoms are grown alternately are obtained.
なお、最初の種結晶であるゲルマニウムナノ粒子は、合成条件(温度、濃度、攪拌速度など)を調整することで、数原子世代以内の、小さい、大きさの揃った種結晶に調整可能である。 The first seed crystal germanium nanoparticles can be adjusted to a small, uniform seed crystal within several generations by adjusting the synthesis conditions (temperature, concentration, stirring speed, etc.). .
本発明による方法を用いると、種結晶の部分を除けば、2種類以上の4族元素からなる、規則配列した4族系化合物のナノ粒子をつくることが可能になる。一般には、このような規則配列した化合物の合成は極めて難しい。これは、たとえばゲルマニウム・カーボン化合物では、ゲルマニウムも炭素も同じく4配位原子であり、互いによく混ざり合うことから、規則配列を取りづらく、不規則配列となりやすいことに由来する。
By using the method according to the present invention, it is possible to produce nanoparticles of regularly arranged
ゲルマニウム・カーボン化合物ナノ粒子の他にも、シリコン・ゲルマニウム化合物、シリコン・カーボン化合物、シリコン・錫化合物、シリコン・鉛化合物、ゲルマニウム・錫化合物、ゲルマニウム・鉛化合物、錫・鉛化合物などの、2元系の4族系化合物のナノ粒子が作成可能である。
In addition to germanium / carbon compound nanoparticles, binary materials such as silicon / germanium compounds, silicon / carbon compounds, silicon / tin compounds, silicon / lead compounds, germanium / tin compounds, germanium / lead compounds, tin / lead compounds, etc. It is possible to create nanoparticles of
さらには1種類以上の4族元素に加えて、4族系元素以外の遷移金属元素、好ましくは4配位元素となりうるチタン、バナジウム、ジルコニウム、ハフニウム、マンガンなどを選ぶことも可能である。そのような方法によれば、シリコン・チタン化合物、シリコン・バナジウム化合物、シリコン・ジルコニウム化合物、シリコン・ハフニウム化合物、シリコン・マンガン化合物、ゲルマニウム・チタン化合物、ゲルマニウム・バナジウム化合物、ゲルマニウム・ジルコニウム化合物、ゲルマニウム・ハフニウム化合物、ゲルマニウム・マンガン化合物、カーボン・チタン化合物、カーボン・バナジウム化合物、カーボン・ジルコニウム化合物、カーボン・ハフニウム化合物、カーボン・マンガン化合物、錫・チタン化合物、錫・バナジウム化合物、錫・ジルコニウム化合物、錫・ハフニウム化合物、錫・マンガン化合物、鉛・チタン化合物、鉛・バナジウム化合物、鉛・ジルコニウム化合物、鉛・ハフニウム化合物、鉛・マンガン化合物などのナノ粒子が作成可能である。
Furthermore, in addition to one or more kinds of
用途に応じて、規則配列と異なる配列の2元系ナノ粒子も作成可能である。2元系の片方をA原子、もう片方をB原子とすると、規則配列・・ABAB・・以外にも、世代ごとでの任意の原子配列、たとえば・・AABBAABB・・、あるいは・・AABAAB・・、あるいは全くの不規則配列・・AABABBBBABAABBB・・も作成可能である。 Depending on the application, binary nanoparticles having an arrangement different from the regular arrangement can be prepared. Assuming that one atom of the binary system is an A atom and the other is a B atom, in addition to the regular arrangement... ABAB..., An arbitrary atomic arrangement for each generation, for example, AABBAABB, or ABABAAB , Or a completely irregular array... AABABBBBBABAABBBB.
3元系以上の多元系では、規則配列若しくは不規則配列した4族系ナノ粒子を作成することも可能である。
In a ternary system or a multi-element system, it is also possible to create a
すなわち本発明によるもうひとつの半導体粒子の製造方法は、シリコン、ゲルマニウム、炭素、錫、鉛から選ばれた4族系半導体材料において、1個の4族原子Aと4個と電子吸引基Xからなる分子をAX4とし、前記Aと異なる種類の4族原子Bと電子吸引基Xからなる分子をBX4とし、前記ABと異なる種類の4族原子Cと電子吸引基Xからなる分子をCX4とし、前記ABCと異なる種類の4族原子Dと電子吸引基Xからなる分子をDX4とし、前記ABCDと異なる種類の4族原子Eと電子吸引基Xからなる分子をEX4とし、1個のアルカリ原子Mと1個の芳香族分子Yからなる電荷移動錯体をMYとしたとき、不活性有機溶媒中で、前記分子と前記電荷移動錯体とを化学反応させる際、前記分子AX4、BX4、CX4、DX4、EX4から2種類以上の分子を選び、各々を、前記分子、前記電荷移動錯体の序列、または前記電荷移動錯体、前記分子の序列で複数回繰り返して加えることで、2種類以上の任意の4族原子からなる3次元ナノ構造を原子単位でサイズ制御し、粒子成長させることができる。
That is, another method for producing semiconductor particles according to the present invention is based on a
また、本発明による他の半導体粒子の製造方法は、シリコン、ゲルマニウム、炭素、錫、鉛から選ばれた4族系半導体材料において、1個の4族原子Aと4個と電子吸引基Xからなる分子をAX4とし、1個の遷移金属原子Zと4個の前記電子吸引基Xからなる分子をZX4とし、1個のアルカリ原子Mと1個の芳香族分子Yからなる電荷移動錯体をMYとしたとき、不活性有機溶媒中で、前記分子と前記電荷移動錯体とを化学反応させる際、各々を、AX4、MY、ZX4、MYの序列、またはMY、ZX4、MY、AX4の序列、またはZX4、MY、AX4、MYの序列、またはMY、AX4、MY、ZX4の序列で複数回繰り返して加えることで、AZ2種類の原子からなる3次元ナノ構造を原子単位でサイズ制御し、粒子成長させることができる。
In addition, another method for producing semiconductor particles according to the present invention includes a
このような方法により、シリコン・ゲルマニウム・カーボンナノ粒子などが作成可能である。 By such a method, silicon, germanium, carbon nanoparticles and the like can be prepared.
さらに、1元系、2元系、多元系ナノ粒子の一部を発光性の不純物原子で置き換えることも可能である。たとえば、ランタノイド系の希土類イオンや、鉄族の遷移金属イオンなどなどである。希土類イオンは通常3配位であるため、隣接する4個のシリコン原子のうち、1個の原子とは結合出来ない。そこで、5配位の原子、たとえばリン原子を加えることで、これを補償することも可能である。 Furthermore, it is also possible to replace part of the one-component, two-component, and multi-component nanoparticles with luminescent impurity atoms. For example, lanthanoid rare earth ions, iron group transition metal ions, and the like. Since rare earth ions are usually tricoordinate, they cannot bond to one of the four adjacent silicon atoms. Therefore, it is possible to compensate for this by adding a pentacoordinate atom such as a phosphorus atom.
以上より、本発明の方法によって、原子レベルで精密に、好ましくは発光が効率的に得られる球換算で2〜5nmに粒径制御され、かつ組成制御された4族系半導体ナノ粒子の合成が、はじめて可能になる。従来技術による4族系ナノ粒子と比較して、発光波長のテーラーメード化など、発光特性に優れた4族系ナノ粒子が、はじめて実現可能になる。
From the above, by the method of the present invention, the synthesis of
3−c)シリコンウエハ上への成長法
本発明の方法によれば、溶液中で4族系半導体ナノ粒子が合成できるだけでなく、シリコンウエハ上に種々の4族系半導体を膜状あるいは粒子状に結晶成長させることが可能になる。手順を説明すると以下の通りである。
3-c) Growth Method on Silicon Wafer According to the method of the present invention, not only can Group 4 semiconductor nanoparticles be synthesized in solution, but also
まず、シリコンウエハを合成チャンバーに導入し、超高真空状態で表面を清浄化処理し、シリコン原子を析出させる。 First, a silicon wafer is introduced into a synthesis chamber, and the surface is cleaned in an ultrahigh vacuum state to deposit silicon atoms.
次に、真空を破り、大気圧に戻し、合成室にglymeを注入する。この際、表面汚染を避けるため、充分に露点の低い充分に乾燥した不活性ガスを導入する。 Next, break the vacuum, return to atmospheric pressure, and inject glyme into the synthesis chamber. At this time, in order to avoid surface contamination, a sufficiently dry inert gas having a sufficiently low dew point is introduced.
次に、ウエハ表面を臭素(Br2)などのハロゲン、若しくは臭酸(HBr)または塩酸(HCl)などの酸に接触させて、表面のシリコン原子をハロゲン終端させる。一般に未処理のウェハは表面がハロゲン終端されていないため、このような処理によりウェハ表面の4族元素にハロゲンを結合させることにより、ナトリウム原子が吸着しやすくなる。
Next, the wafer surface is brought into contact with a halogen such as bromine (Br 2 ) or an acid such as odorous acid (HBr) or hydrochloric acid (HCl) to terminate the silicon atoms on the surface with halogen. In general, since the surface of an untreated wafer is not terminated with halogen, sodium atoms are easily adsorbed by bonding halogen to the
なお、ウエハ表面をハロゲン化する手法については、上記とは異なる手法によっても実現可能である。例を挙げると、塩素終端化については、フッ酸(HF)、若しくはフッ化アンモニウム(NH4F)で処理し、ウエハ表面を一旦水素終端した後、ラジカル開始剤として過酸化ベンゾイル等を用い、5塩化リン(PCl5)若しくは塩素(Cl2)を適量加えたクロロベンゼン中で処理することによって、塩素終端表面が得られる。また、臭素終端化については、やはり一旦水素終端した後、ラジカル開始剤として過酸化ベンゾイル等を用い、ブロモトリクロロメタン(CBrCl3)で処理することによって、臭素終端表面が得られる。 The method for halogenating the wafer surface can also be realized by a method different from the above. For example, for chlorine termination, after treatment with hydrofluoric acid (HF) or ammonium fluoride (NH 4 F) and once hydrogen termination of the wafer surface, benzoyl peroxide or the like is used as a radical initiator, Treatment in chlorobenzene with the appropriate amount of phosphorus pentachloride (PCl 5 ) or chlorine (Cl 2 ) results in a chlorine terminated surface. As for bromine termination, hydrogen termination is performed once, and then a bromotrichloromethane (CBrCl 3 ) treatment is performed using benzoyl peroxide or the like as a radical initiator to obtain a bromine terminated surface.
次に、NN錯体を添加し、表面シリコン原子にナトリウム原子を吸着させる。要するに、シリコンウェハ表面をナトリウム終端させる。 Next, an NN complex is added to adsorb sodium atoms on the surface silicon atoms. In short, the silicon wafer surface is terminated with sodium.
次に、ウエハ上に1元系の4族半導体、たとえばシリコンを成長させる場合、前節までの要領に従い、塩化シリコンとNN錯体を交互に繰り返し添加すれば良い。また規則配列した2元系4族半導体、たとえばゲルマニウム・カーボン化合物などを成長させる場合、やはり前節までの要領に従い、4塩化ゲルマニウム→NN錯体→4臭化炭素→NN錯体を1サイクルとして、これらの試薬を規則正しく繰り返し添加すれば良い。
Next, in the case of growing a single-
なお、不規則配列した2元系化合物、3元系以上の多元系化合物、さらには1元系・2元系・多元系合成物の一部を発光性の不純物原子で置き換えることも可能である。 In addition, it is also possible to replace a part of an irregularly arranged binary compound, a ternary or more multi-component compound, and a uni-component / bin-ary / multi-component compound with luminescent impurity atoms. .
最終的に得られる合成物の形状は、最初に吸着させるナトリウム原子の面密度に強く依存する。すなわち、ナトリウム原子がシリコン表面上に密に吸着すれば膜成長が生じ、疎であれば粒子成長が起きる。そのため、ナトリウム原子の吸着量の制御が重要である。これは、赤外反射分光法などで、ナトリウム原子の吸着をリアルタイムでモニターすることにより制御可能である。 The shape of the finally obtained composite strongly depends on the areal density of sodium atoms to be adsorbed first. That is, film growth occurs when sodium atoms are densely adsorbed on the silicon surface, and particle growth occurs when they are sparse. Therefore, it is important to control the amount of adsorption of sodium atoms. This can be controlled by monitoring the adsorption of sodium atoms in real time by infrared reflection spectroscopy or the like.
シリコンナノ粒子の合成
図6は、本発明に用いる合成装置を模式的に表したものである。反応容器の四口フラスコ1は、開口部2および3を有し、内部5がアルゴンガス(露点−80度以下)でパージされ、十分に乾燥されている。開口部2または3には図示されていない滴下ロートまたは冷却管が取り付けられている。本実施例を含め、本発明の製造方法では、水や酸素などの大気成分はナノ粒子の汚染に直結することから、このような雰囲気が制御されたフラスコ内で合成を行う必要がある。
Synthesis of Silicon Nanoparticles FIG. 6 schematically shows a synthesis apparatus used in the present invention. The four-
シリコンナノ粒子を合成するために、フラスコ内に塩化シリコンを5ミリモル溶かした無水glyme溶液100mlを用意する。この溶液を室温に保ち、攪拌子6で毎分600回転の速度で攪拌しながら、滴下ロートを用いて開口部2から、NN錯体の0.2モル無水glyme溶液をゆっくり滴下する。
In order to synthesize silicon nanoparticles, 100 ml of an anhydrous glyme solution in which 5 mmol of silicon chloride is dissolved is prepared in a flask. While maintaining this solution at room temperature and stirring with a stirrer 6 at a speed of 600 revolutions per minute, a 0.2 mol anhydrous glyme solution of NN complex is slowly dropped from the
NN錯体の無水glyme溶液は濃緑色の液体であるが、無色透明の塩化シリコンglyme溶液中にこのNN錯体溶液を滴下すると、フラスコ内の溶液4は最初クリーム色に変化する。さらにNN錯体を入れ続け、NN錯体の滴下量が概ね20ミリモルに達したところで、溶液が突然濃赤色に変化する。
Although the anhydrous glyme solution of the NN complex is a dark green liquid, when the NN complex solution is dropped into the colorless and transparent silicon chloride glyme solution, the
フラスコ内の溶液が濃赤色に変化しはじめ、色変化が定常状態に達したら、今度は塩化シリコンをゆっくり滴下する。滴下量がおよそ1ミリモルに達すると、溶液は再度クリーム色に変わる。 When the solution in the flask begins to turn deep red and the color change reaches a steady state, silicon chloride is then slowly added dropwise. When the drop volume reaches approximately 1 mmol, the solution turns cream again.
以後、フラスコ内の溶液色を指標に、クリーム色に変化したら塩化シリコンの滴下を止めてNN錯体溶液を加え、次に濃赤色に変化したらNN錯体溶液の滴下を止めて再び塩化シリコンを加える作業を繰り返す。フラスコに予め仕込んだ塩化シリコンを除き、NN錯体溶液滴下、塩化シリコン滴下の順番で、この操作を4回繰り返すことで塩素終端されたシリコンナノ粒子の合成が可能になる。 Thereafter, using the solution color in the flask as an indicator, when the color changes to cream, the addition of silicon chloride is stopped and the NN complex solution is added. When the color changes to dark red, the addition of NN complex solution is stopped and silicon chloride is added again. repeat. By removing the silicon chloride previously charged in the flask and repeating this operation four times in the order of dropping the NN complex solution and dropping the silicon chloride, it is possible to synthesize chlorine-terminated silicon nanoparticles.
クリーム色の溶液中には、塩素終端されたシリコンナノ粒子、NaCl、中性化したナフタレン分子が混在する。塩素終端されたナノ粒子は、シリコン原子から塩素原子に電子が引き寄せられ、シリコン原子は部分的に正に帯電していると推測される。 In the cream-colored solution, chlorine-terminated silicon nanoparticles, NaCl, and neutralized naphthalene molecules are mixed. In the chlorine-terminated nanoparticle, electrons are attracted from the silicon atom to the chlorine atom, and it is assumed that the silicon atom is partially positively charged.
これに対し、濃赤色の溶液中では、ナトリウム終端シリコンナノ粒子が生成しているものと思われる。負に帯電したシリコンアニオンは、赤い体色を示すことが知られている。このことから、ナトリウム終端シリコンナノ粒子では、ナトリウム原子からシリコン原子に電子が供給され、シリコン原子は部分的に負に帯電し、赤系統の体色を示すものと推測される。NN錯体を入れることで溶液が濃赤色に変化するのは、ナノ粒子表面がナトリウム終端されたことを示す間接的な証拠である。 On the other hand, it is considered that sodium-terminated silicon nanoparticles are generated in the deep red solution. Negatively charged silicon anions are known to exhibit a red body color. From this, in sodium-terminated silicon nanoparticles, it is presumed that electrons are supplied from sodium atoms to silicon atoms, and the silicon atoms are partially negatively charged and exhibit a red body color. The addition of the NN complex turns the solution dark red, which is indirect evidence that the nanoparticle surface is sodium terminated.
フラスコ内に生成した塩素終端シリコンナノ粒子を大気中に取り出すと、容易に加水分解が起こり、シリコンナノ粒子は酸化する。このため、引き続き、フラスコ内で、表面安定化処理を行う。 When the chlorine-terminated silicon nanoparticles generated in the flask are taken out into the atmosphere, hydrolysis easily occurs and the silicon nanoparticles are oxidized. For this reason, the surface stabilization treatment is subsequently performed in the flask.
フラスコ内に滴下ロートを用いて、ブチルアルコールを200ミリモルゆっくり滴下し、室温で24時間撹拌する。こうすることで、フラスコ内には、目的物である、ブトキシ基で覆われ表面安定化された、シリコンナノ粒子が生成可能になる。なお副生成物としては、NaClとナフタレンが生じる。 Using a dropping funnel, 200 mmol of butyl alcohol is slowly dropped into the flask and stirred at room temperature for 24 hours. By doing so, silicon nanoparticles covered with butoxy groups and surface-stabilized can be produced in the flask. As by-products, NaCl and naphthalene are generated.
フラスコにヘキサン100mlを注入し、十分に撹拌して、シリコンナノ粒子とナフタレンを選択的に抽出することができる。抽出したヘキサン溶液は、分液ロートを用いて純水で3回水洗し、残存するNaClを除去・精製することができる。精製したヘキサン溶液は硫酸マグネシウムに潜らせて脱水することができる。 100 ml of hexane is poured into the flask and stirred sufficiently to selectively extract silicon nanoparticles and naphthalene. The extracted hexane solution can be washed three times with pure water using a separatory funnel to remove and purify the remaining NaCl. The purified hexane solution can be dehydrated by immersing it in magnesium sulfate.
その後、真空蒸留でナフタレンとヘキサンをそれぞれ除去・精製し、目的物であるシリコンナノ粒子を得ることができる。 Thereafter, naphthalene and hexane are respectively removed and purified by vacuum distillation to obtain the target silicon nanoparticles.
電子顕微鏡観察によって、このようにして合成したシリコンナノ粒子の、シリコン部分の平均粒径はおよそ2.1nmであることが確認可能である。構成元素の組成分析によれば、塩素と酸素は重量比(wt%)でともに装置の検出限界程度の低い値を示すことから、純度の高いナノ粒子が得られる作成法であると言う事ができる。グリニャール法で作ったナノ粒子と比較すると、各々の不純物原子量(Clは1wt%、Oは10wt%)を大幅に低減させることが可能である。 By observation with an electron microscope, it is possible to confirm that the average particle size of the silicon portion of the silicon nanoparticles synthesized in this way is approximately 2.1 nm. According to the compositional analysis of the constituent elements, both chlorine and oxygen show a low value of about the detection limit of the device in weight ratio (wt%), so it can be said that this is a preparation method for obtaining highly pure nanoparticles. it can. Compared with nanoparticles made by the Grignard method, the amount of each impurity atom (Cl is 1 wt%, O is 10 wt%) can be greatly reduced.
得られたシリコンナノ粒子をヘキサンに溶かし、その蛍光特性を調べることができる。紫外光励起で得られた発光スペクトルからは、ピーク発光波長が525nm、スペクトル半値幅は約60nmの値が得られている。発光量子効率はローダミン6G色素を基準にして求めることができる。その測定によれば、効率が30%から60%程度の値を示す、非常に優れた発光特性を持つシリコンナノ粒子が実現可能である。 The obtained silicon nanoparticles can be dissolved in hexane, and the fluorescence characteristics can be examined. From the emission spectrum obtained by ultraviolet light excitation, a peak emission wavelength of 525 nm and a spectrum half width of about 60 nm are obtained. Luminescent quantum efficiency can be determined based on rhodamine 6G dye. According to the measurement, it is possible to realize silicon nanoparticles having very excellent light emission characteristics, in which efficiency is a value of about 30% to 60%.
粒径制御
第2の実施例では、シリコンナノ粒子の粒径を制御させるために、NN錯体溶液と塩化シリコン各々の滴下回数を4回から12回に増やす点を除き、第1の実施例と同様な手順で、シリコンナノ粒子を合成する。
Particle size control In the second example, in order to control the particle size of the silicon nanoparticles, the number of drops of each of the NN complex solution and silicon chloride is increased from 4 times to 12 times. In the same procedure, silicon nanoparticles are synthesized.
電子顕微鏡観察によって、このようにして合成したシリコンナノ粒子の、シリコン部分の平均粒径はおよそ3nmであることが確認可能である。光励起で得られた蛍光スペクトルから、ピーク発光波長が630nm、スペクトル半値幅は約80nmのスペクトルが測定されている。発光量子効率は10%から40%程度の値を示す、非常に優れた発光特性を持つ、粒径の大きいシリコンナノ粒子が実現可能である。本発明により、滴下回数によって粒径制御が可能であり、延いては発光ピーク波長の制御が可能になる。 By observation with an electron microscope, it can be confirmed that the silicon nanoparticles synthesized in this way have an average particle size of the silicon portion of about 3 nm. From the fluorescence spectrum obtained by photoexcitation, a spectrum having a peak emission wavelength of 630 nm and a spectrum half width of about 80 nm is measured. It is possible to realize silicon nanoparticles having a large particle diameter and having very excellent light emission characteristics, in which the light emission quantum efficiency is about 10% to 40%. According to the present invention, the particle size can be controlled by the number of times of dropping, and the emission peak wavelength can be controlled.
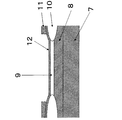
EL発光素子
図7は、本発明の実施例の発光素子を概略的に表す断面図である。この発光素子は、シリコンウエハ上に形成可能であり、p電極としてp+Si層7上に設けたp−Si層8と、対抗するn電極としてアモルファスSi層12と、活性層9としてシリコンナノ粒子の充填層からそれぞれ構成することが可能である。図7の発光素子はさらにSiO2層10とパッド電極11を具備してなる。発光素子は、図示しない電極取出し配線に電気的に接続されている。
EL Light-Emitting Element FIG. 7 is a cross-sectional view schematically showing a light-emitting element of an example of the present invention. This light-emitting element can be formed on a silicon wafer. The p − Si layer 8 provided on the p + Si layer 7 as the p electrode, the
活性層に用いるシリコンナノ粒子の粒径は、発光波長に応じて選択することができる。ここでは、平均粒径2.1nmを有する青色発光ナノ粒子が例として引用されている。
20mA、4Vでこの発光素子を駆動することができ、EL発光効率として5cd/Aと大変良好な値を得ることも可能である。
The particle size of the silicon nanoparticles used for the active layer can be selected according to the emission wavelength. Here, blue light-emitting nanoparticles having an average particle size of 2.1 nm are cited as an example.
This light-emitting element can be driven at 20 mA and 4 V, and it is possible to obtain a very good value of 5 cd / A as the EL luminous efficiency.
ウエハ被覆
本実施例では、シリコンウエハ上にゲルマニウム・カーボン化合物を被膜させる。本発明に用いる合成装置は原理的には図6と同様の構成であり、異なる点は超高真空対応とするために反応容器は全て金属製である点と、ウエハ温度を調整するためにヒーターが追加されている点である。
Wafer coating In this embodiment, a germanium-carbon compound is coated on a silicon wafer. The synthesizer used in the present invention has the same configuration as that shown in FIG. 6 in principle. The difference is that the reaction vessels are all made of metal in order to support ultra-high vacuum, and a heater is used to adjust the wafer temperature. Is added.
合成に先立ち、1cm角のウエハが置かれた反応容器のフラスコを超高真空に引き、この雰囲気でウエハ温度を上げて表面清浄化処理を行い、シリコン原子以外の不純物原子を取り除く。なお、清浄化処理は通常のLSIプロセスに準拠している。 次に、フラスコ内の真空を破り、大気圧に戻し、無水glyme100mlを注入する。この際、表面汚染を避けるため、充分に乾燥したアルゴンガス(露点マイナス80度以下)でパージする。 Prior to the synthesis, the flask in the reaction vessel on which the 1 cm square wafer is placed is evacuated to an ultrahigh vacuum, and the wafer temperature is raised in this atmosphere to perform a surface cleaning process to remove impurity atoms other than silicon atoms. The cleaning process conforms to a normal LSI process. Next, the vacuum in the flask is broken to return to atmospheric pressure, and 100 ml of anhydrous glyme is injected. At this time, purging with sufficiently dried argon gas (dew point minus 80 degrees or less) to avoid surface contamination.
シリコン表面をナトリウム終端するために、NN錯体の0.1モル無水glyme溶液をゆっくり滴下し、溶液を毎分600回転で攪拌させて、NN錯体をウエハ表面に衝突、反応させる。1cm角のシリコンウエハ表面に、シリコン原子は凡そ1015個(約2ナノモル)存在するので、密にナトリウム終端させるために、NN錯体は過剰量の1ミリモル滴下する。 In order to terminate the silicon surface with sodium, a 0.1 molar anhydrous glyme solution of NN complex is slowly dropped, and the solution is stirred at 600 revolutions per minute to cause the NN complex to collide with the wafer surface and react. Since there are approximately 10 15 silicon atoms (about 2 nanomoles) on the surface of a 1 cm square silicon wafer, an excessive amount of 1 millimole of NN complex is dropped in order to terminate sodium densely.
このナトリウム終端されたウエハ表面をゲルマニウム・カーボン膜で被覆するために、4塩化ゲルマニウム→NN錯体→4臭化炭素→NN錯体を1サイクルとして、これらの試薬を規則正しく繰り返し添加する。なお、表面反応が充分進むよう、いずれの試薬も過剰量滴下する。4臭化炭素は固体試薬であるため、予めglymeに溶かし、0.1モル溶液を用意しておく。 In order to coat this sodium-terminated wafer surface with a germanium-carbon film, these reagents are regularly and repeatedly added in one cycle of germanium tetrachloride → NN complex → carbon tetrabromide → NN complex. In addition, an excessive amount of any reagent is dropped so that the surface reaction proceeds sufficiently. Since carbon tetrabromide is a solid reagent, it is dissolved in glyme in advance and a 0.1 molar solution is prepared.
この滴下サイクルを100回繰り返した後、合成したゲルマニウム・カーボン膜表面を安定化させるために、ブチルアルコールを1モルゆっくり滴下し、室温で24時間攪拌する。こうすることで、ウエハ表面には、目的物である、ブトキシ基で覆われ安定化された、ゲルマニウム・カーボン膜が生成可能になる。 After repeating this dropping cycle 100 times, in order to stabilize the surface of the synthesized germanium / carbon film, 1 mol of butyl alcohol is slowly dropped and stirred at room temperature for 24 hours. In this way, a germanium carbon film covered with butoxy groups and stabilized, which is the target, can be formed on the wafer surface.
フラスコからウエハを取り出し、純水とヘキサンを用いて洗浄することで、ウエハに付着するナフタレン、NaCl、未反応試薬、その他副生成物を取り除くことができる。このようにして、目的物である、ゲルマニウム・カーボン膜で被膜したウエハを得ることができる。 By removing the wafer from the flask and washing with pure water and hexane, naphthalene, NaCl, unreacted reagents and other by-products adhering to the wafer can be removed. In this way, a wafer coated with a germanium-carbon film, which is the object, can be obtained.
断面の電子顕微鏡観察によって、合成した被膜層の膜厚はおよそ20nmから30nmの間にあることが確認できる。構成元素の組成分析によれば、被膜層からゲルマニウムとカーボンが各々検出され、その比率はGe:C=1:1であることが示されている。また、塩素、酸素、ナトリウムは装置の検出限界程度の低い値をしめすことから、純度の高い膜が作れる製法と言うことができる。 By observing the cross section with an electron microscope, it can be confirmed that the film thickness of the synthesized coating layer is between about 20 nm and 30 nm. According to the compositional analysis of the constituent elements, germanium and carbon are respectively detected from the coating layer, and the ratio is shown to be Ge: C = 1: 1. In addition, chlorine, oxygen, and sodium have values as low as the detection limit of the apparatus, and thus can be said to be a production method that can produce a highly pure film.
因みに、得られた被膜を紫外線で励起したところ、近赤外領域にピークを持つ発光スペクトルが得られている。発光量子効率はおよそ50%であり、優れた発光特性を持つ4族化合物半導体が実現可能である。
Incidentally, when the obtained film was excited with ultraviolet rays, an emission spectrum having a peak in the near infrared region was obtained. The emission quantum efficiency is about 50%, and a
第5の実施例では、NN錯体とは異なる還元剤、リチウム・ジ−t−ブチルビフェニル電荷移動錯体(LDBB錯体)を用いて合成する点を除き、第1の実施例と同様な手順で、シリコンナノ粒子を合成する。 In the fifth embodiment, a procedure similar to that of the first embodiment is performed except that the synthesis is performed using a reducing agent different from the NN complex, lithium di-t-butylbiphenyl charge transfer complex (LDBB complex). Synthesize silicon nanoparticles.
電子顕微鏡観察によって、このようにして合成したシリコンナノ粒子の、シリコン部分の平均粒径は2.2nmであることが確認可能である。光励起で得られた蛍光スペクトルから、ピーク発光波長が530nm、スペクトル半値幅は約60nmの値が得られている。発光量子効率は30%から50%程度の値を示し、非常に優れた発光特性を持つ、粒径の大きいシリコンナノ粒子が実現可能である。本実施例により、還元剤には、NN錯体以外にも、アルカリ金属と芳香族有機分子からなる電荷移動錯体を用いることが可能であることがわかる。 By observation with an electron microscope, it is possible to confirm that the silicon nanoparticles synthesized in this way have an average particle size of the silicon portion of 2.2 nm. From the fluorescence spectrum obtained by photoexcitation, a peak emission wavelength of 530 nm and a spectrum half width of about 60 nm are obtained. Luminescence quantum efficiency shows a value of about 30% to 50%, and it is possible to realize silicon nanoparticles having a large particle diameter and having very excellent emission characteristics. According to this example, it can be seen that, besides the NN complex, a charge transfer complex composed of an alkali metal and an aromatic organic molecule can be used as the reducing agent.
第6の実施例では、第1の実施例と同様な手順で、還元剤とシラン化合物の種類を変えてシリコンナノ粒子を合成する。還元剤としては二ヨウ化サマリウムを用い、シラン化合物として臭化シリコンを用いてシリコンなの粒子を合成する。 In the sixth example, silicon nanoparticles are synthesized by changing the types of the reducing agent and the silane compound in the same procedure as in the first example. Silicon particles are synthesized using samarium diiodide as the reducing agent and silicon bromide as the silane compound.
第1の実施例と同様な手順でシリコンナノ粒子を表面安定化した後、抽出、精製されたナノ粒子を電子顕微鏡観察すると、第1の実施例同様、平均粒径は2.0nmであることが確認できる。蛍光特性も第1の実施例と同等である。 After stabilizing the surface of the silicon nanoparticles in the same procedure as in the first example, the extracted and purified nanoparticles are observed with an electron microscope. As in the first example, the average particle size is 2.0 nm. Can be confirmed. The fluorescence characteristics are also the same as in the first embodiment.
本実施例により、還元剤として電荷移動錯体の代わりに希土類ヨウ化物を用いることが可能であることがわかる。 This example shows that it is possible to use rare earth iodide as a reducing agent instead of the charge transfer complex.
1 四口フラスコ
2、3 開口部
4 反応溶液
5 フラスコ内部(乾燥アルゴン)
6 攪拌子
7 p+Si層
8 p−Si層(p電極)
9 活性層(シリコンナノ粒子充填層)
10 SiO2層
11 パッド電極
12 アモルファスSi(n電極)
1 Four-
6 Stirrer 7 p + Si layer 8 p - Si layer (p electrode)
9 Active layer (silicon nanoparticle packed layer)
10 SiO 2 layer 11
Claims (8)
ことを特徴とする、半導体表面の被覆方法。 The surface of the semiconductor material comprising silicon, the electron donating reductant dissolved in an inert organic solvent beat drops, the intermediate having a surface coated with metal derived from the reducing agent of a semiconductor material An electron withdrawing group-containing compound containing an element selected from the group consisting of silicon, germanium and carbon is dropped onto the intermediate, and the surface of the semiconductor material is a group 4 element or transition derived from the electron withdrawing group-containing compound. A method for coating a semiconductor surface, comprising coating with a monoatomic film of a metal element.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005188576A JP4504268B2 (en) | 2004-08-30 | 2005-06-28 | Method for coating semiconductor surface and method for producing semiconductor particle |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004250008 | 2004-08-30 | ||
| JP2005188576A JP4504268B2 (en) | 2004-08-30 | 2005-06-28 | Method for coating semiconductor surface and method for producing semiconductor particle |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006100785A JP2006100785A (en) | 2006-04-13 |
| JP4504268B2 true JP4504268B2 (en) | 2010-07-14 |
Family
ID=36240253
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005188576A Expired - Fee Related JP4504268B2 (en) | 2004-08-30 | 2005-06-28 | Method for coating semiconductor surface and method for producing semiconductor particle |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4504268B2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150068055A (en) * | 2013-12-11 | 2015-06-19 | 주식회사 엘지화학 | Encapsulating composition comprising naphthalene derivatives and coordination metal and method for producing capsule reversibly using the same |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009283446A (en) * | 2008-05-23 | 2009-12-03 | Sharp Corp | Manufacturing method of electroluminescent element equipped with silicon nanocrystal embedded silicon oxide film having mid-bandgap transition layer, and operation method of electroluminescent element |
| JP5598809B2 (en) * | 2009-03-06 | 2014-10-01 | 独立行政法人物質・材料研究機構 | Light emitting element |
| JP5822543B2 (en) * | 2011-06-06 | 2015-11-24 | キヤノン株式会社 | Light emitting element |
| WO2013027717A1 (en) | 2011-08-24 | 2013-02-28 | 株式会社 村田製作所 | Solar cell and method for manufacturing same |
| JP2015201621A (en) * | 2014-04-03 | 2015-11-12 | 国立研究開発法人物質・材料研究機構 | Luminescent silicon nanoparticle and current injection light-emitting element |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005503984A (en) * | 2001-09-19 | 2005-02-10 | エバーグリーン ソーラー, インコーポレイテッド | High-yield method for preparing silicon nanocrystals with chemically accessible surfaces |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001014250A2 (en) * | 1999-08-23 | 2001-03-01 | University Of Hawaii | Synthesis of silicon nanoparticles and metal-centered silicon nanoparticles and applications thereof |
-
2005
- 2005-06-28 JP JP2005188576A patent/JP4504268B2/en not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005503984A (en) * | 2001-09-19 | 2005-02-10 | エバーグリーン ソーラー, インコーポレイテッド | High-yield method for preparing silicon nanocrystals with chemically accessible surfaces |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150068055A (en) * | 2013-12-11 | 2015-06-19 | 주식회사 엘지화학 | Encapsulating composition comprising naphthalene derivatives and coordination metal and method for producing capsule reversibly using the same |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2006100785A (en) | 2006-04-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Ling et al. | Composite perovskites of cesium lead bromide for optimized photoluminescence | |
| JP4504268B2 (en) | Method for coating semiconductor surface and method for producing semiconductor particle | |
| Cumberland et al. | Inorganic clusters as single-source precursors for preparation of CdSe, ZnSe, and CdSe/ZnS nanomaterials | |
| Yang et al. | Interfacial synthesis of monodisperse CsPbBr3 nanorods with tunable aspect ratio and clean surface for efficient light-emitting diode applications | |
| Hua et al. | Organically capped silicon nanoparticles with blue photoluminescence prepared by hydrosilylation followed by oxidation | |
| Singh et al. | Magic-sized CdSe nanoclusters: a review on synthesis, properties and white light potential | |
| JP5907544B2 (en) | Method for producing nanoparticles | |
| Ghosh et al. | Colloidal silicon quantum dots: synthesis and luminescence tuning from the near-UV to the near-IR range | |
| Wu et al. | Fabrication and photoluminescence characteristics of single crystalline In2O3 nanowires | |
| JP5169222B2 (en) | Three-layer semiconductor nanoparticles and three-layer semiconductor nanorods | |
| US7553775B2 (en) | Method for coating semiconductor surface, process for production of semiconductor particles using said method, and optical element using said semiconductor particles | |
| Huang et al. | In situ growth of all-inorganic perovskite nanocrystals on black phosphorus nanosheets | |
| Leeb et al. | Colloidal synthesis and electroluminescence properties of nanoporous MnIIZnS films | |
| JP2005325016A (en) | Manufacturing method of metal nanocrystalline sulfide using thiol compound as sulfur precursor | |
| US20100139455A1 (en) | Methods of Forming Nanoparticles | |
| JP2018115315A (en) | Cd-FREE COLLOIDAL QUANTUM DOT EMITTING VISIBLE FLORESCENCE AND METHOD FOR PRODUCING THE SAME | |
| JP4504267B2 (en) | Method for producing semiconductor nanoparticles and method for coating the surface of a semiconductor material | |
| Ji et al. | Colloidal synthesis of size-confined CsAgCl 2 nanocrystals: implications for electroluminescence applications | |
| Xu et al. | Self-assembled catalyst growth and optical properties of single-crystalline ZnGa2O4 nanowires | |
| JP2022527219A (en) | Nanocrystals | |
| Jo et al. | Destabilizing Magic-Sized Clusters for Synthesis of Monodisperse and Highly Luminescent In (Zn) P/ZnSe/ZnS Quantum Dots | |
| CN114945772A (en) | Blue light emitting nanocrystals composed of earth-rich/non-toxic elements | |
| Pahwa et al. | Cadmium Selenide (CdSe) nano-particles for use in light harvesting applications | |
| KR101334694B1 (en) | Method of core-shell structure quantum dots with thick shell and light conversion diode thereof | |
| Lyu | Lanthanide near-infrared luminescence in layered semiconductor nanosheet hosts |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20061110 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090814 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091013 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100105 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100308 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100330 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100422 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130430 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |