JP4195065B2 - Novel glucose dehydrogenase and method for producing the dehydrogenase - Google Patents
Novel glucose dehydrogenase and method for producing the dehydrogenase Download PDFInfo
- Publication number
- JP4195065B2 JP4195065B2 JP2007038490A JP2007038490A JP4195065B2 JP 4195065 B2 JP4195065 B2 JP 4195065B2 JP 2007038490 A JP2007038490 A JP 2007038490A JP 2007038490 A JP2007038490 A JP 2007038490A JP 4195065 B2 JP4195065 B2 JP 4195065B2
- Authority
- JP
- Japan
- Prior art keywords
- glucose dehydrogenase
- amino acid
- subunit
- glucose
- enzyme
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images




Description
本発明は、新規なグルコース脱水素酵素及びその製造方法、同酵素をコードするDNA、同酵素をコードするDNAを含有する組換えベクター、同組換えベクターで形質転換された形質転換体、同酵素を生産する新規な微生物、前記酵素、形質転換体又は微生物を含む酵素電極を用いたグルコースセンサ、及びグルコースアッセイキットに関する。 The present invention relates to a novel glucose dehydrogenase and a method for producing the same, a DNA encoding the enzyme, a recombinant vector containing the DNA encoding the enzyme, a transformant transformed with the recombinant vector, and the enzyme A glucose sensor using an enzyme electrode containing the enzyme, a transformant or a microorganism, and a glucose assay kit.
特定の基質に対して特異的に反応する酵素を用いたバイオセンサの開発は、産業の分野を問わず盛んに行われている。その中でも特にバイオセンサの1つであるグルコースセンサは、主に医療分野で測定方法やその方法を利用した装置の開発が盛んに行われている。
グルコースセンサは、1962年にClarkとLyonsによってグルコースオキシダーゼと酸素電極を組み合わせたバイオセンサーの報告(非特許文献1)が最初にされて以来、約40年ほどの歴史を有している。
このように、グルコースセンサに、酵素としてグルコースオキシダーゼを採用してからの歴史は、長い。なぜならグルコースオキシダーゼは、グルコースに対する基質特異性が高く、熱安定性に優れており、更に酵素の量産化が可能であり、生産コストが他の酵素と比べて安価である、からである。
基質特異性が高いということは、酵素がグルコース以外の糖とは反応しないため、測定値に誤差を生じることなく、正確な測定が行なえるという利点に通じる。
また、熱安定性に優れているということは、酵素が熱により変性し酵素活性が失活するという問題を防止することができ、長期間正確な測定が行えるという利点に通じる。
しかし、グルコースオキシダーゼは、基質特異性が高く、熱安定性に優れ、安価に生産できる一方、以下で説明するような溶存酸素の影響を受け、測定結果に影響があるという問題を有する。
一方、グルコースオキシダーゼ以外に、グルコース脱水素酵素(以下、「グルコースデヒドロゲナーゼ」ともいう)を利用したグルコースセンサの開発も行われてきた。そして、酵素も、微生物から発見されている。
例えば、バチルス(Bacillus)属由来のグルコースデヒドロゲナーゼ(EC1.1.1.47)及びクリプトコッカス(Cryptococcus)属由来グルコースデヒドロゲナーゼ(EC1.1.1.119)が知られている。
前者のグルコースデヒドロゲナーゼ(EC1.1.1.47)は、β−D−グルコース+NAD(P)+→D−δ−グルコノラクトン+NAD(P)H+H+の反応を触媒する酵素であり、後者のグルコースデヒドロゲナーゼ(EC1.1.1.119)は、D−グルコース+NADP+→D−δ−グルコノラクトン+NADPH+H+の反応を触媒する酵素であり、前述した微生物由来のグルコースデヒドロゲナーゼは、既に市販もされている。
これらグルコースデヒドロゲナーゼは、測定サンプルの溶存酸素の影響を受けないという利点を有する。このことは、酸素分圧が低い環境下で測定を行ったり、酸素量が多く要求される高濃度サンプルを測定する場合であっても、測定結果に誤差を及ぼさずに正確に測定することができるという利点に通じる。
しかし、グルコースデヒドロゲナーゼは、溶存酸素の影響を受けない一方、熱安定性が悪く、基質特異性がグルコースオキシダーゼよりも劣るという問題点を有する。
そこで、グルコースオキシダーゼやグルコースデヒドロゲナーゼの両欠点を補う酵素の提供が望まれていた。
尚、本発明者は非特許文献2〜4において、温泉近くの土壌より採取した試料を用い、グルコース脱水素酵素についての研究結果を報告している。
しかし、該酵素を産生する能力を有する菌株の同定は該研究段階では行われていなかった。
The glucose sensor has a history of about 40 years since the first report of a biosensor combining glucose oxidase and an oxygen electrode (Non-patent Document 1) was made in 1962 by Clark and Lyons.
Thus, the history since adopting glucose oxidase as an enzyme in a glucose sensor is long. This is because glucose oxidase has a high substrate specificity for glucose, is excellent in thermal stability, can be mass-produced for an enzyme, and is less expensive to produce than other enzymes.
The high substrate specificity leads to the advantage that an accurate measurement can be performed without causing an error in the measurement value because the enzyme does not react with sugars other than glucose.
In addition, the excellent thermal stability can prevent the problem that the enzyme is denatured by heat and the enzyme activity is deactivated, leading to the advantage that accurate measurement can be performed for a long period of time.
However, glucose oxidase has a high substrate specificity, excellent thermal stability, and can be produced at a low cost. However, glucose oxidase is affected by dissolved oxygen as described below and has a problem that measurement results are affected.
On the other hand, in addition to glucose oxidase, a glucose sensor using glucose dehydrogenase (hereinafter also referred to as “glucose dehydrogenase”) has been developed. Enzymes have also been discovered from microorganisms.
For example, glucose dehydrogenase derived from the genus Bacillus (EC1.1.1.17) and glucose dehydrogenase derived from the genus Cryptococcus (EC1.1.1.119) are known.
The former glucose dehydrogenase (EC1.1.1.17) is an enzyme that catalyzes the reaction of β-D-glucose + NAD (P) + → D-δ-gluconolactone + NAD (P) H + H + . Glucose dehydrogenase (EC 1.1.1.119) is an enzyme that catalyzes the reaction of D-glucose + NADP + → D-δ-gluconolactone + NADPH + H + , and the aforementioned microorganism-derived glucose dehydrogenase is already commercially available. ing.
These glucose dehydrogenases have the advantage that they are not affected by the dissolved oxygen of the measurement sample. This means that even when measuring in an environment where the oxygen partial pressure is low or when measuring a high-concentration sample that requires a large amount of oxygen, it is possible to measure accurately without affecting the measurement results. It leads to the advantage of being able to.
However, glucose dehydrogenase is not affected by dissolved oxygen, but has a problem that its thermal stability is poor and its substrate specificity is inferior to that of glucose oxidase.
Therefore, it has been desired to provide an enzyme that compensates for both drawbacks of glucose oxidase and glucose dehydrogenase.
In addition, this inventor has reported the research result about glucose dehydrogenase in the nonpatent literatures 2-4 using the sample extract | collected from the soil near a hot spring.
However, identification of strains having the ability to produce the enzyme has not been performed at the research stage.
本発明は、従来から知られているグルコースオキシダーゼやグルコースデヒドロゲナーゼ両者の欠点をそれぞれ補う性質を持ち、基質特異性が高く、熱安定性に優れ、安価に生産でき、測定サンプルの溶存酸素の影響を受けない酵素を提供することを課題とする。
また本発明は、前記酵素の製造方法、該酵素の特性を活かしたタンパク質および該酵素を生産する新規な微生物を提供することを課題とする。
また本発明は、前記酵素をコードするDNA、同酵素をコードするDNAを含有する組換えベクター、同組換えベクターで形質転換された形質転換体を提供することを課題とする。
また本発明は、前記酵素、形質転換体又は前記微生物を含む酵素電極を用いたグルコースセンサ、及び前記酵素を含むグルコースアッセキットを提供することを課題とする。
本発明者は、温泉近くの土壌より上記の目的に合った酵素を生産するブルクホルデリア・セパシア(Burkhorderia cepacia)を単離することに成功し、本発明に至った。
The present invention has the properties to compensate for the disadvantages of both glucose oxidase and glucose dehydrogenase, which are conventionally known, has high substrate specificity, excellent thermal stability, can be produced at low cost, and has the effect of dissolved oxygen in the measurement sample. It is an object to provide an enzyme that is not affected.
Another object of the present invention is to provide a method for producing the enzyme, a protein utilizing the characteristics of the enzyme, and a novel microorganism that produces the enzyme.
Another object of the present invention is to provide a DNA encoding the enzyme, a recombinant vector containing the DNA encoding the enzyme, and a transformant transformed with the recombinant vector.
Moreover, this invention makes it a subject to provide the glucose sensor using the enzyme electrode containing the said enzyme, a transformant, or the said microorganisms, and the glucose assay kit containing the said enzyme.
The present inventor succeeded in isolating Burkholderia cepacia, which produces an enzyme suitable for the above-mentioned purpose, from soil near a hot spring, and reached the present invention.
すなわち、本発明は以下のとおりである。
(1)ブルクホルデリア属に属し、グルコース脱水素酵素を産生する能力を有する微生物を培地に培養し、同培地又は/及び前記微生物菌体からグルコース脱水素酵素を採取することを特徴とするグルコース脱水素酵素の製造方法。
(2)前記微生物がブルクホルデリア・セパシアである(1)のグルコース脱水素酵素の製造方法。
(3)前記グルコース脱水素酵素が下記性質を有することを特徴とする(1)又は(2)のグルコース脱水素酵素の製造方法。
[1]作用:
グルコースの脱水素反応を触媒する。
[2]還元条件下でのSDS−ポリアクリルアミドゲル電気泳動において、分子量約60kDaと分子量約43kDaを示すサブユニットからなる。
[3]TSK gel G3000SW(東ソー(株)製)を用いたゲル濾過クロマトグラフィーにおいて、分子量約380kDaを示す。
[4]至適反応温度:
45℃付近(Tris−HCl緩衝液、pH8.0)。
(4)前記分子量約43kDaのサブユニットが電子伝達タンパク質であることを特徴とする(3)のグルコース脱水素酵素の製造方法。
(5)前記電子伝達タンパク質がチトクロムCであることを特徴とする(4)のグルコース脱水素酵素の製造方法。
(6)ブルクホルデリア属に属する微生物によって産生され得るグルコース脱水素酵素。
(7)前記微生物がブルクホルデリア・セパシアである(6)のグルコース脱水素酵素。
(8)前記グルコース脱水素酵素が下記性質を有することを特徴とする(6)又は(7)のグルコース脱水素酵素。
[1]作用:
グルコースの脱水素反応を触媒する。
[2]還元条件下でのSDS−ポリアクリルアミドゲル電気泳動において、分子量約60kDaと分子量約43kDaを示すサブユニットからなる。
[3]TSK gel G3000SW(東ソー(株)製)を用いたゲル濾過クロマトグラフィーにおいて、分子量約380kDaを示す。
[4]至適反応温度:
45℃付近 (Tris−HCl緩衝液、pH8.0)。
(9)前記分子量約43kDaのサブユニットが電子伝達タンパク質であることを特徴とする(8)のグルコース脱水素酵素。
(10)前記電子伝達タンパク質がチトクロムCであることを特徴とする(9)のグルコース脱水素酵素。
(11)前記分子量約60kDaのサブユニットが、配列番号3のアミノ酸番号2〜12のアミノ酸配列を含むことを特徴とする(8)〜(10)のいずれかのグルコース脱水素酵素。
(12)前記43kDaのサブユニットのN末端が配列番号5のアミノ酸配列を有する請求項8〜11のいずれか1項に記載のグルコース脱水素酵素。
(13)前記分子量約60kDaのサブユニットが以下の(A)または(B)に示すタンパク質である(11)のグルコース脱水素酵素。
(A)配列番号3のアミノ酸配列を有するタンパク質。
(B)配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿入、又は付加されたアミノ酸配列を有し、かつ、グルコース脱水素酵素活性を有するタンパク質。
(14)45℃付近と75℃付近にそれぞれ活性ピークを有することを特徴とする(6)のグルコース脱水素酵素。
(15)(10)のグルコース脱水素酵素のサブユニットであって、配列番号5のアミノ酸配列を有することを特徴とするチトクロームC。
(16)(15)のチトクロムCの一部をコードし、配列番号8に記載の塩基配列を有するDNA。
(17)(15)のチトクロームCの一部をコードし、配列番号1に記載の塩基配列のうち塩基番号2386〜2467の塩基配列を有するDNA。
(18)(15)のチトクロームCのシグナルペプチドをコードし、配列番号1の塩基配列のうち塩基番号2386〜2451の塩基配列を含むDNA。
(19)チトクロームCのシグナルペプチドであって、配列番号4のアミノ酸配列のうちアミノ酸番号1〜22のアミノ酸配列を有するペプチド。
(20)下記性質を有するタンパク質。
[1]サブユニットとして(6)のグルコース脱水素酵素を構成し得る。
[2]グルコース脱水素酵素活性を有する。
[3]還元条件下でのSDS−ポリアクリルアミドゲル電気泳動において、分子量約60kDaを示す。
[4]至適反応温度:
75℃付近 (Tris−HCl緩衝液、pH8.0)。
(21)配列番号3においてアミノ酸番号2〜12のアミノ酸配列を含むことを特徴とする(20)のタンパク質。
(22)前記タンパク質が以下の(A)または(B)に示すタンパク質である(21)のグルコース脱水素酵素。
(A)配列番号3のアミノ酸配列を有するタンパク質。
(B)配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿入、又は付加されたアミノ酸配列を有し、かつ、グルコース脱水素酵素活性を有するタンパク質。
(23)以下の(A)または(B)に示すタンパク質。
(A)配列番号3のアミノ酸配列を有するタンパク質。
(B)配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿入、又は付加されたアミノ酸配列を有し、かつ、グルコース脱水素酵素活性を有するタンパク質。
(24)以下の(A)または(B)に示すタンパク質をコードするDNA。
(A)配列番号3のアミノ酸配列を有するタンパク質。
(B)配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿入、又は付加されたアミノ酸配列を有し、かつ、グルコース脱水素酵素活性を有するタンパク質。
(25)以下の(a)または(b)に示すDNAである(24)のDNA。
(a)配列番号1の塩基配列のうち、塩基番号764〜2380からなる塩基配列を含むDNA。
(b)配列番号1の塩基配列のうち、塩基番号764〜2380からなる塩基配列又はこの配列から調製され得るプローブとストリンジェントな条件下でハイブリダイズし、かつ、グルコース脱水素酵素活性を有するタンパク質をコードするDNA。
(26)(24)又は(25)のDNAを含有する組換えベクター。
(27)(18)のシグナルペプチド及びβ−サブユニットをコードする塩基配列を含む(26)の組換えベクター。
(28)(24)又は(25)のDNA、又は(26)又は(27)の組換えベクターで形質転換された形質転換体。
(29)(28)の形質転換体を培養して、前記DNAの発現産物としてグルコース脱水素酵素を産生させ、これを採取するグルコース脱水素酵素の製造方法。(30)ブルクホルデリア・セパシアKS1株(FERM BP−7306)。
(31)(6)〜(14)のいずれかのグルコース脱水素酵素、(20)〜(23)のいずれかのタンパク質、(27)の形質転換体、又は(30)の菌株を含む酵素電極を用いたグルコースセンサ。
(32)(6)〜(14)のいずれかのグルコース脱水素酵素、又は(20)〜(23)のいずれかのタンパク質を含むグルコースアッセイキット。
(33)配列番号2のアミノ酸配列を有するタンパク質。
(34)配列番号2のアミノ酸配列を有するタンパク質をコードするDNA。
(35)配列番号1の塩基配列のうち、塩基番号258〜761からなる塩基配列含む(34)のDNA。
(36)(34)又は(35)のDNAと、(24)又は(25)のDNAをこの順に含むDNA。
(37)配列番号1の塩基配列のうち、塩基番号258〜2380からなる塩基配列を含む(36)のDNA。
(38)(36)又は(37)のDNAを含有する組換えベクター。
(39)(18)のシグナルペプチド及びβ−サブユニットをコードする塩基配列を含む(38)の組換えベクター。
(40)(36)又は(37)のDNA又は(38)又は(39)の組換えベクターで形質転換された形質転換体。
(41)(40)の形質転換体を培養して、(36)又は(37)のDNAの発現物質としてグルコース脱水素酵素を産生させ、これを採取するグルコース脱水素酵素の製造方法。
That is, the present invention is as follows.
(1) Glucose belonging to the genus Burkholderia, cultivating a microorganism having an ability to produce glucose dehydrogenase in a medium, and collecting glucose dehydrogenase from the medium or / and the microorganism cells A method for producing a dehydrogenase.
(2) The method for producing glucose dehydrogenase according to (1), wherein the microorganism is Burkholderia cepacia.
(3) The method for producing glucose dehydrogenase according to (1) or (2), wherein the glucose dehydrogenase has the following properties.
[1] Action:
Catalyses the dehydrogenation of glucose.
[2] It consists of subunits having a molecular weight of about 60 kDa and a molecular weight of about 43 kDa in SDS-polyacrylamide gel electrophoresis under reducing conditions.
[3] Gel filtration chromatography using TSK gel G3000SW (manufactured by Tosoh Corporation) shows a molecular weight of about 380 kDa.
[4] Optimum reaction temperature:
Around 45 ° C. (Tris-HCl buffer, pH 8.0).
(4) The method for producing glucose dehydrogenase according to (3), wherein the subunit having a molecular weight of about 43 kDa is an electron transfer protein.
(5) The method for producing glucose dehydrogenase according to (4), wherein the electron transfer protein is cytochrome C.
(6) A glucose dehydrogenase that can be produced by a microorganism belonging to the genus Burkholderia.
(7) The glucose dehydrogenase according to (6), wherein the microorganism is Burkholderia cepacia.
(8) The glucose dehydrogenase according to (6) or (7), wherein the glucose dehydrogenase has the following properties.
[1] Action:
Catalyses the dehydrogenation of glucose.
[2] It consists of subunits having a molecular weight of about 60 kDa and a molecular weight of about 43 kDa in SDS-polyacrylamide gel electrophoresis under reducing conditions.
[3] Gel filtration chromatography using TSK gel G3000SW (manufactured by Tosoh Corporation) shows a molecular weight of about 380 kDa.
[4] Optimum reaction temperature:
Around 45 ° C. (Tris-HCl buffer, pH 8.0).
(9) The glucose dehydrogenase according to (8), wherein the subunit having a molecular weight of about 43 kDa is an electron transfer protein.
(10) The glucose dehydrogenase according to (9), wherein the electron transfer protein is cytochrome C.
(11) The glucose dehydrogenase according to any one of (8) to (10), wherein the subunit having a molecular weight of about 60 kDa comprises the amino acid sequence of
(12) The glucose dehydrogenase according to any one of claims 8 to 11, wherein the N-terminus of the 43 kDa subunit has the amino acid sequence of SEQ ID NO: 5.
(13) The glucose dehydrogenase of (11), wherein the subunit having a molecular weight of about 60 kDa is a protein shown in the following (A) or (B):
(A) A protein having the amino acid sequence of SEQ ID NO: 3.
(B) A protein having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted or added in the amino acid sequence of SEQ ID NO: 3 and having glucose dehydrogenase activity.
(14) The glucose dehydrogenase according to (6), which has activity peaks at around 45 ° C. and around 75 ° C., respectively.
(15) A cytochrome C which is a subunit of glucose dehydrogenase according to (10) and has the amino acid sequence of SEQ ID NO: 5.
(16) A DNA encoding a part of cytochrome C of (15) and having the base sequence set forth in SEQ ID NO: 8.
(17) A DNA encoding a part of cytochrome C of (15) and having a base sequence of base numbers 2386 to 2467 of the base sequence set forth in SEQ ID NO: 1.
(18) A DNA encoding the cytochrome C signal peptide of (15) and comprising the nucleotide sequence of nucleotide numbers 2386 to 2451 of the nucleotide sequence of SEQ ID NO: 1.
(19) A peptide peptide of cytochrome C, which has the amino acid sequence of
(20) A protein having the following properties.
[1] The glucose dehydrogenase of (6) can be constituted as a subunit.
[2] Has glucose dehydrogenase activity.
[3] SDS-polyacrylamide gel electrophoresis under reducing conditions shows a molecular weight of about 60 kDa.
[4] Optimum reaction temperature:
Around 75 ° C. (Tris-HCl buffer, pH 8.0).
(21) The protein according to (20), which comprises the amino acid sequence of
(22) The glucose dehydrogenase according to (21), wherein the protein is a protein shown in the following (A) or (B).
(A) A protein having the amino acid sequence of SEQ ID NO: 3.
(B) A protein having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted or added in the amino acid sequence of SEQ ID NO: 3 and having glucose dehydrogenase activity.
(23) The protein shown in the following (A) or (B).
(A) A protein having the amino acid sequence of SEQ ID NO: 3.
(B) A protein having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted or added in the amino acid sequence of SEQ ID NO: 3 and having glucose dehydrogenase activity.
(24) DNA encoding the protein shown in the following (A) or (B).
(A) A protein having the amino acid sequence of SEQ ID NO: 3.
(B) A protein having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted or added in the amino acid sequence of SEQ ID NO: 3 and having glucose dehydrogenase activity.
(25) The DNA of (24), which is the DNA shown in the following (a) or (b).
(A) DNA containing a base sequence consisting of base numbers 764 to 2380 among the base sequence of SEQ ID NO: 1.
(B) a protein that hybridizes under stringent conditions with a nucleotide sequence consisting of nucleotide numbers 764 to 2380 or a probe that can be prepared from this nucleotide sequence of the nucleotide sequence of SEQ ID NO: 1 and has glucose dehydrogenase activity DNA encoding
(26) A recombinant vector containing the DNA of (24) or (25).
(27) The recombinant vector according to (26) comprising a base sequence encoding the signal peptide of (18) and the β-subunit.
(28) A transformant transformed with the DNA of (24) or (25) or the recombinant vector of (26) or (27).
(29) A method for producing glucose dehydrogenase, comprising culturing the transformant of (28), producing glucose dehydrogenase as an expression product of the DNA, and collecting the glucose dehydrogenase. (30) Burkholderia cepacia KS1 strain (FERM BP-7306).
(31) An enzyme electrode comprising the glucose dehydrogenase of any one of (6) to (14), the protein of any of (20) to (23), the transformant of (27), or the strain of (30) Glucose sensor using
(32) A glucose assay kit comprising the glucose dehydrogenase of any one of (6) to (14) or the protein of any one of (20) to (23).
(33) A protein having the amino acid sequence of SEQ ID NO: 2.
(34) DNA encoding a protein having the amino acid sequence of SEQ ID NO: 2.
(35) The DNA of (34) comprising the nucleotide sequence consisting of nucleotide numbers 258 to 761 among the nucleotide sequence of SEQ ID NO: 1.
(36) A DNA comprising the DNA of (34) or (35) and the DNA of (24) or (25) in this order.
(37) The DNA of (36) comprising a base sequence consisting of base numbers 258 to 2380 among the base sequence of SEQ ID NO: 1.
(38) A recombinant vector containing the DNA of (36) or (37).
(39) The recombinant vector according to (38), comprising a base peptide encoding the signal peptide of (18) and the β-subunit.
(40) A transformant transformed with the DNA of (36) or (37) or the recombinant vector of (38) or (39).
(41) A method for producing glucose dehydrogenase, comprising culturing the transformant of (40), producing glucose dehydrogenase as an expression substance of the DNA of (36) or (37), and collecting this.
以下、本発明を詳細に説明する。
<1>本発明のグルコース脱水素酵素を産生する新規菌株
本発明酵素(以下、「本酵素」または「GDH」ということがある)は、ブルクホルデリア属に属する細菌によって生産され得る。本発明に用いるブルクホルデリア属細菌は、本酵素の生産能を有するブルクホルデリア属細菌であれば特に制限されないが、ブルクホルデリア・セパシア、特にブルクホルデリア・セパシアKS1株が好ましい。この菌株は、後記実施例に示すように、本発明者らが温泉付近の土壌から分離した新規菌株であり、その菌学的性質からブルクホルデリア・セパシアと同定された。従来、ブルクホルデリア属に属する微生物がグルコース脱水素酵素を産生しうることは知られていない。この菌株は、KS1株と命名された。この株は、平成12年9月25日に独立行政法人産業技術総合研究所特許生物寄託センター(〒305−8566 日本国茨城県つくば市東1丁目1番地1 中央第6)に微生物受託番号第FERM BP−7306として寄託されている。
なお、本発明者らはブルクホルデリア・セパシア KS1株以外の株について、財団法人発酵研究所(Institute for Fermentation,Osaka,IFO)又は理化学研究所微生物系統保存施設(Japan Collection of Microorganisms,JCM)に寄託されている同ブルクホルデリア・セパシアのいくつかの菌株を取り寄せてグルコース脱水素酵素活性を測定したところ、いずれの菌株にも活性があることを確認した。
Hereinafter, the present invention will be described in detail.
<1> Novel strain producing the glucose dehydrogenase of the present invention The enzyme of the present invention (hereinafter sometimes referred to as “the present enzyme” or “GDH”) can be produced by bacteria belonging to the genus Burkholderia. The Burkholderia bacterium used in the present invention is not particularly limited as long as it is a bacterium belonging to the genus Burkholderia having the ability to produce this enzyme, but the Burghorderia cepacia, in particular, the Burkholderia cepacia KS1 strain is preferable. This strain is a novel strain isolated from the soil near the hot spring by the present inventors as shown in the examples described later, and was identified as Burkholderia cepacia from its mycological properties. Conventionally, it is not known that microorganisms belonging to the genus Burkholderia can produce glucose dehydrogenase. This strain was named KS1 strain. This strain was established on September 25, 2000, at the National Institute of Advanced Industrial Science and Technology, Patent Biological Depositary Center (1st, 1st, 1st East, 1-chome, Tsukuba, Ibaraki, Japan 305-8586). Deposited as BP-7306.
In addition, the present inventors have established a strain other than the Burkholderia cepacia KS1 strain in the Institute for Fermentation (Institute for Fermentation, Osaka, IFO) or the RIKEN Microbial System Storage Facility (Japan Collection of Microorganisms, JCM). Several strains of the same deposit that was deposited with Burkholderia cepacia were collected and measured for glucose dehydrogenase activity. As a result, it was confirmed that all strains were active.
<2>本発明のグルコース脱水素酵素
本発明のグルコース脱水素酵素は、グルコース脱水素酵素産生能を有するブルクホルデリア属細菌、例えばブルクホルデリア・セパシアKS1株を、通常、微生物の培養に用いられる栄養培地、好ましくは酵素生産能を高めるためにグルコース或はグルコースを含む物質を添加した培地で培養することにより、培養生成物中又は菌体中に生産蓄積されるので、公知の方法で採取することができる。更に本酵素の製造方法を、ブルクホルデリア・セパシアKS1株を例として具体的に説明する。まずブルクホルデリア・セパシアKS1株を適当な栄養培地、例えば適当な炭素源、窒素源、無機塩類、グルコース或はこれらを含む物質などを含む培地で培養して本酵素を培養生成物中か菌体中に生産蓄積させる。
炭素源としては、資化できるものはいずれの物質も利用でき、例えば、D−グルコース、L−アラビソース、D−キシロース、D−マンノース、デンプン、各種ペプトン類などが挙げられる。窒素源としては、酵母エキス、麦芽エキス、各種ペプトン類、各種肉エキス類、コーンスティープリカー、アミノ酸溶液、アンモニウム塩など有機、無機の窒素化合物又はこれらを含有した物質が利用できる。無機塩としては、各種リン酸塩、マグネシウム、カリウム、ナトリウム、カルシウムなどの塩類が使用される。また必要に応じて菌の生育或は酵素生産に必要な各種の無機物や有機物、例えばシリコーン油、ゴマ油、各種界面活性剤などの消泡剤やビタミン類を培地に添加することができる。
培養の形態は、液体培養でも固体培養でもよいが、通常は液体培養が好適である。
こうして得られた培養物の培地中又は/及び菌体中から本発明酵素を得ることが出来る。菌体中にある酵素は、菌体を破砕あるいは溶解することによって、菌体抽出液として得られる。
培養生成物中あるいは菌体抽出液中のグルコース脱水素酵素は、イオン交換体、ゲル濾過担体、ハイドロフォービック(疎水性)担体などを用いたクロマトグラフィーを適宜組み合わせることによって精製することができる。
本酵素の活性は、公知のグルコース脱水素酵素の活性測定と同様の方法で測定することができる。具体的には、例えば後記実施例に示す方法によって測定できる。
<2> Glucose dehydrogenase of the present invention The glucose dehydrogenase of the present invention is a bacterium belonging to the genus Burkholderia that has the ability to produce glucose dehydrogenase, such as the Burkholderia cepacia KS1 strain, which is usually used for culturing microorganisms. It is collected in a known manner since it is produced and accumulated in the culture product or cells by culturing in a nutrient medium, preferably a medium to which glucose or a substance containing glucose is added in order to enhance enzyme production ability. can do. Further, the production method of the present enzyme will be specifically described by taking the Burkholderia cepacia KS1 strain as an example. First, the Burkholderia cepacia KS1 strain is cultured in a suitable nutrient medium, for example, a medium containing a suitable carbon source, nitrogen source, inorganic salts, glucose or a substance containing these, and the enzyme is cultured in the culture product. Accumulate production throughout the body.
As the carbon source, any substance that can be assimilated can be used, and examples thereof include D-glucose, L-arabisose, D-xylose, D-mannose, starch, and various peptones. As a nitrogen source, organic and inorganic nitrogen compounds such as yeast extract, malt extract, various peptones, various meat extracts, corn steep liquor, amino acid solution, ammonium salt, or substances containing these can be used. As the inorganic salt, various salts such as phosphate, magnesium, potassium, sodium, calcium and the like are used. If necessary, various inorganic and organic substances necessary for the growth of bacteria or enzyme production, for example, antifoaming agents such as silicone oil, sesame oil, various surfactants, and vitamins can be added to the medium.
The culture form may be liquid culture or solid culture, but liquid culture is usually preferable.
The enzyme of the present invention can be obtained from the culture medium or / and the cells of the culture thus obtained. The enzyme in the microbial cell is obtained as a microbial cell extract by crushing or dissolving the microbial cell.
Glucose dehydrogenase in the culture product or bacterial cell extract can be purified by appropriately combining chromatography using ion exchangers, gel filtration carriers, hydrophobic (hydrophobic) carriers, and the like.
The activity of this enzyme can be measured by the same method as the activity measurement of a known glucose dehydrogenase. Specifically, for example, it can be measured by the method shown in the examples described later.
次に本発明の新規グルコース脱水素酵素の理化学的性質を示す。
(1)作用:
グルコースの脱水素反応を触媒する。
(2)還元条件下でのSDS−ポリアクリルアミドゲル電気泳動において、分子量約60kDaと分子量約43kDaを示すサブユニットからなる。
(3)TSK gel G3000SW(東ソー(株)製)を用いたゲル濾過クロマトグラフィーにおいて、分子量約380kDaを示す。
(4)至適反応温度:
45℃付近 (Tris−HCl緩衝液、pH8.0)。
尚、上記グルコース脱水素酵素は、上記条件で45℃付近に活性ピークを有するが、75℃付近にも活性ピークを有する(図3(a)参照)。このように、2つの温度領域で活性ピークを示すGDHは知られていない。
尚、分子量及び至適温度は後記実施例に記載の方法で測定できる。
上記、本発明のグルコース脱水素酵素は分子量約60kDaのαサブユニットと分子量約43kDaのβサブユニットの2つの別個のポリペプチドで構成されている(以下、このグルコース脱水素酵素を「多量体酵素」ということがある)が、本発明者らはさらに2つのサブユニットにつき詳細に検討した。
βサブユニットはチトクロムCであることがわかった(後記実施例で示す)。
αサブユニットのみを含むタンパク質は以下の理化学的性質を示す。
(1)サブユニットとして前記多量体酵素を構成し得る。
(2)グルコース脱水素酵素活性を有する。
(3)還元条件下でのSDS−ポリアクリルアミドゲル電気泳動において、分子量60kDaを示す。
(4)至適反応温度:
75℃付近 (Tris−HCl緩衝液、pH8.0)。
尚、至適温度は後記実施例に記載の方法で測定できる。
尚、このタンパク質はそれ自身酵素活性を有しているため、説明の内容に応じ、適宜このタンパク質をペプチド酵素もしくは酵素と言い換えて使用することとする。
Next, the physicochemical properties of the novel glucose dehydrogenase of the present invention will be shown.
(1) Action:
Catalyses the dehydrogenation of glucose.
(2) It consists of subunits having a molecular weight of about 60 kDa and a molecular weight of about 43 kDa in SDS-polyacrylamide gel electrophoresis under reducing conditions.
(3) Gel filtration chromatography using TSK gel G3000SW (manufactured by Tosoh Corporation) shows a molecular weight of about 380 kDa.
(4) Optimal reaction temperature:
Around 45 ° C. (Tris-HCl buffer, pH 8.0).
The glucose dehydrogenase has an activity peak near 45 ° C. under the above conditions, but also has an activity peak near 75 ° C. (see FIG. 3A). Thus, GDH which shows an active peak in two temperature ranges is not known.
The molecular weight and optimum temperature can be measured by the methods described in the examples below.
The glucose dehydrogenase of the present invention is composed of two separate polypeptides, an α subunit having a molecular weight of about 60 kDa and a β subunit having a molecular weight of about 43 kDa (hereinafter, this glucose dehydrogenase is referred to as “multimeric enzyme”). However, the present inventors have further studied in detail about two subunits.
The β subunit was found to be cytochrome C (shown in Examples below).
A protein containing only the α subunit exhibits the following physicochemical properties.
(1) The multimeric enzyme can be constituted as a subunit.
(2) It has glucose dehydrogenase activity.
(3) In SDS-polyacrylamide gel electrophoresis under reducing conditions, the molecular weight is 60 kDa.
(4) Optimal reaction temperature:
Around 75 ° C. (Tris-HCl buffer, pH 8.0).
The optimum temperature can be measured by the method described in Examples below.
In addition, since this protein itself has an enzyme activity, this protein will be used in other words as a peptide enzyme or an enzyme as appropriate according to the content of the explanation.
本発明のペプチド酵素の具体的態様として、配列番号3のアミノ酸配列を有するタンパク質が挙げられる。また同ペプチド酵素は、GDH活性を有する限り、配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿入、又は付加されたアミノ酸配列を有するタンパク質であってもよい。尚、配列番号3には、配列番号1の塩基配列によってコードされ得るアミノ酸配列を示してあるが、N末端のメチオニン残基は、翻訳後に脱落している可能性がある。
また、本発明の多量体酵素の具体的態様として、αサブユニットが配列番号3のアミノ酸配列を有するタンパク質を含む多量体が挙げられる。また同多量体酵素は、GHD活性を有する限り、αサブユニットが配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿入、又は付加されたアミノ酸配列を有するタンパク質を含む多量体であってもよい。
本発明において「1又は複数」とは、1〜10個、好ましくは1〜5個、特に好ましくは1〜3個である。
本発明者らは前記αサブユニット又は前記βサブユニット以外にも、γサブユニットの存在を確認している。
後述する実施例においては、前記γサブユニットは、培養上清あるいは菌体抽出液中から本発明の酵素を精製する段階で除去されて、精製後の酵素ではγサブユニットが確認されなかった。しかし、実施例に示したように、αサブユニットとともにγサブユニットを発現させると、αサブユニットのみを発現させた場合に比べて高い酵素活性が得られた。このことから、γサブユニットは、微生物体内においてαサブユニットが生成する際に何らかの関わりがあるタンパク質であることが示唆された。いずれの場合もαサブユニットの比活性(タンパク質当たりの酵素活性)が同じであるとすれば、酵素活性は酵素量を反映するから、酵素活性が低いことは酵素としてのαサブユニットの量が少ないことを示している。一方、生成したαサブユニットがγサブユニットによって何らかの保護を受けているのか、それともタンパク質としてのαサブユニットの充分発現しているが、γサブユニットが存在しないため酵素活性を示すことができる立体構造を取れずに、結果的に酵素活性が低くなったのかもしれない。いずれにしても、γサブユニットをαサブユニットとともに発現させることにより、高い酵素活性が得られる。
A specific embodiment of the peptide enzyme of the present invention includes a protein having the amino acid sequence of SEQ ID NO: 3. The peptide enzyme may be a protein having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted, or added in the amino acid sequence of SEQ ID NO: 3 as long as it has GDH activity. Although SEQ ID NO: 3 shows an amino acid sequence that can be encoded by the base sequence of SEQ ID NO: 1, the N-terminal methionine residue may be missing after translation.
In addition, a specific embodiment of the multimeric enzyme of the present invention includes a multimer containing a protein in which the α subunit has the amino acid sequence of SEQ ID NO: 3. The multimeric enzyme is a protein having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted, or added in the amino acid sequence of SEQ ID NO: 3 as long as it has GHD activity. It may be a multimer containing.
In the present invention, “one or more” means 1 to 10, preferably 1 to 5, and particularly preferably 1 to 3.
The present inventors have confirmed the presence of a γ subunit in addition to the α subunit or the β subunit.
In the examples described later, the γ subunit was removed from the culture supernatant or bacterial cell extract at the stage of purifying the enzyme of the present invention, and the γ subunit was not confirmed in the purified enzyme. However, as shown in the Examples, when the γ subunit was expressed together with the α subunit, a higher enzyme activity was obtained than when only the α subunit was expressed. From this, it was suggested that the γ subunit is a protein that has some relationship when the α subunit is produced in the microorganism. In either case, if the specific activity of the α subunit (enzyme activity per protein) is the same, the enzyme activity reflects the amount of enzyme, so low enzyme activity means that the amount of α subunit as an enzyme is low. It shows that there are few. On the other hand, the generated α subunit is protected by the γ subunit, or the α subunit as a protein is fully expressed, but since there is no γ subunit, it is a three-dimensional structure that can exhibit enzyme activity. The enzyme activity may have been lowered as a result without taking the structure. In any case, high enzyme activity can be obtained by expressing the γ subunit together with the α subunit.
<3>本発明のDNA
本発明のDNAは、本発明のDNAを含有する微生物、例えばブルクホルデリア・セパシアから取得することができる。本発明のDNAは、本発明を完成する過程においては、ブルクホルデリア・セパシア染色体DNAから単離されたが、本発明によりその塩基配列及び同塩基配列によってコードされるアミノ酸配列が明らかとなったので、これらの配列に基づいて化学合成することによっても取得することができる。また。前記配列に基づいて作製したオリゴヌクレオチドをプローブ又はプライマーとするハイブリダイゼーション又はPCRによって、ブルクホルデリア・セパシア等の染色体DNAから取得することもできる。
本発明のDNAは、配列番号3に示すアミノ酸配列を有するタンパク質をコードするものの他、配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿入、又は付加されたアミノ酸配列を有し、かつ、GDH活性を有するタンパク質をコードするものであってもよい。
本発明のDNAは具体的には、配列番号1の塩基配列のうち、塩基番号764〜2380からなる塩基配列を含むDNAが挙げられる。配列番号1の塩基配列のうち、塩基番号764〜2380からなる塩基配列は、配列番号3のアミノ酸配列を有するGDHのαサブユニットをコードしている。
また本発明のDNAは、配列番号1の塩基配列のうち、塩基番号764〜2380からなる塩基配列又はこの配列から調製され得るプローブとストリンジェントな条件下でハイブリダイズし、かつ、GDH活性を有するタンパク質をコードするDNAであってもよい。
尚、配列番号1の塩基配列のうち、塩基番号258〜761からなる塩基配列は、γサブユニットをコードしていると推定される。そのアミノ酸配列を配列番号2に示す。αサブユニットの上流域にγサブユニットの構造遺伝子が含まれることにより、微生物によってαサブユニットを生産する際に、先ずγサブユニットが発現されてタンパク質として存在することにより微生物体内で効率良くαサブユニットを生産することができると考えられる。したがって、本発明のDNAは、前記DNA以外にも、配列番号2のアミノ酸配列をコードするDNAを含んでいてもよい。
上記のような配列番号3に示すアミノ酸配列を有するタンパク質と実質的に同一のタンパク質をコードするDNAは、例えば部位特異的変異法又は突然変異処理等の方法によって取得することができる。変異が導入されたDNAがコードするタンパク質のGDH活性は、例えば次のようにして測定することができる。
594μMのメチルフェナジンメトサルフェート(mPMS)および5.94μMの2,6−ジクロロフェノールインドフェノール(DCIP)を含む10mMリン酸カリウム緩衝液(pH7.0)に、酵素試料および基質としてグルコースを基質として加え、37℃でインキュベートする。DCIPの600nmの吸光度変化を分光光度計を用いて追跡し、その吸光度の減少速度により、酵素反応速度とする。
さらに、配列番号1の塩基配列のうち、塩基番号2386以降の塩基配列は、β−サブユニットをコードしていると推定される。また、塩基番号2386〜2451の塩基配列は、β−サブユニットのシグナルペプチドをコードしていると推測される。同シグナルペプチドの推定されるアミノ酸配列は、配列番号4のアミノ酸番号1〜22のアミノ酸配列である。シグナルペプチドは、リボソームで合成されたタンパク質が膜を通って分泌される際に必要なペプチドであり、15〜30残基の疎水性アミノ酸残基から成ることが判明されている。よってシグナルペプチドの存在によって、培養上清中に含有するタンパク質量が増加するため、タンパク質の製造方法おいて有効に作用するペプチドである。
<3> DNA of the present invention
The DNA of the present invention can be obtained from a microorganism containing the DNA of the present invention, such as Burkholderia cepacia. In the process of completing the present invention, the DNA of the present invention was isolated from burkholderia cepacia chromosomal DNA, but the present invention revealed the nucleotide sequence and the amino acid sequence encoded by the nucleotide sequence. Therefore, it can also be obtained by chemical synthesis based on these sequences. Also. It can also be obtained from chromosomal DNA such as Burkholderia cepacia by hybridization or PCR using an oligonucleotide prepared based on the sequence as a probe or primer.
The DNA of the present invention encodes a protein having the amino acid sequence shown in SEQ ID NO: 3, as well as amino acids in which one or more amino acid residues are substituted, deleted, inserted or added in the amino acid sequence of SEQ ID NO: 3. It may encode a protein having a sequence and having GDH activity.
Specific examples of the DNA of the present invention include DNA containing a base sequence consisting of base numbers 764 to 2380 in the base sequence of SEQ ID NO: 1. Of the nucleotide sequence of SEQ ID NO: 1, the nucleotide sequence consisting of nucleotide numbers 764 to 2380 encodes the α subunit of GDH having the amino acid sequence of SEQ ID NO: 3.
In addition, the DNA of the present invention hybridizes under stringent conditions with a nucleotide sequence consisting of nucleotide numbers 764 to 2380 or a probe that can be prepared from this nucleotide sequence of the nucleotide sequence of SEQ ID NO: 1, and has GDH activity. It may be DNA encoding a protein.
In addition, it is estimated that the base sequence which consists of base numbers 258-761 among the base sequences of
A DNA encoding a protein substantially identical to the protein having the amino acid sequence shown in SEQ ID NO: 3 as described above can be obtained by a method such as site-specific mutagenesis or mutation treatment. The GDH activity of the protein encoded by the DNA into which the mutation has been introduced can be measured, for example, as follows.
Glucose as a substrate was added to 10 mM potassium phosphate buffer (pH 7.0) containing 594 μM methylphenazine methosulfate (mPMS) and 5.94 μM 2,6-dichlorophenolindophenol (DCIP) as a substrate. Incubate at 37 ° C. The change in absorbance of DCIP at 600 nm is traced using a spectrophotometer, and the enzyme reaction rate is determined by the rate of decrease in the absorbance.
Furthermore, among the base sequence of SEQ ID NO: 1, it is presumed that the base sequence after base number 2386 encodes the β-subunit. Moreover, it is estimated that the base sequence of base number 2386-2451 codes the signal peptide of (beta) -subunit. The deduced amino acid sequence of the signal peptide is the amino acid sequence of
以下に、本発明のDNAを取得する方法を例示する。
ブルクホルデリア・セパシア等の微生物から染色体DNAを分離、精製した後、染色体DNAを超音波処理、制限酵素処理等を用いて切断したものと、リニアーな発現ベクターとをDNAリガーゼなどにより結合閉鎖させて組換えベクターを構築する。得られた組換えベクターを、同ベクターが自律複製可能な宿主微生物に移入した後、形質転換体をベクターのマーカーと酵素活性の発現を指標としてスクリーニングして、GDHをコードする遺伝子を含有する組換えベクターを保持する微生物を得る。得られた微生物が持つ組換えベクターには、少なくともαサブユニットをコードする塩基配列が含まれていると予想される。また、クローン化断片が十分な大きさを有していれば、γサブユニットをコードする塩基配列も含まれている可能性が高い。
次いで、上記組換えベクターを保持する微生物を培養して、該培養微生物の菌体から該組換えベクターを分離、精製し、該発現ベクターからGDHをコードする遺伝子を採取することができる。例えば、遺伝子供与体である染色体DNAは、具体的には以下のようにして採取される。
前記遺伝子供与微生物を、例えば1〜3日間攪拌培養して得られた培養液を遠心分離により集菌し、次いで、これを溶菌させることによりGDH遺伝子の含有溶菌物を調製することができる。溶菌の方法としては、例えばリゾチーム等の溶菌酵素により処理が施され、必要に応じてプロテアーゼや他の酵素やラウリル硫酸ナトリウム(SDS)等の界面活性剤が併用される。さらに、凍結融解やフレンチプレス処理のような物理的破砕方法と組み合わせてもよい。
上記のようにして得られた溶菌物からDNAを分離精製するには、常法に従って、例えばフェノール処理やプロテアーゼ処理による除蛋白処理や、リボヌクレアーゼ処理、アルコール沈殿処理などの方法を適宜組み合わせることにより行うことができる。
微生物から分離、精製されたDNAを切断する方法は、例えば超音波処理、制限酵素処理などにより行うことができる。好ましくは特定のヌクレオチド配列に作用するII型制限酵素が適している。制限酵素は、ベクターの切断末端と適合する末端を生じさせるものを用いてもよく、あるいは任意の制限酵素を用い、切断末端を平滑末端化してベクターと連結してもよい。
クローニングする際のベクターとしては、宿主微生物内で自律的に増殖し得るファージまたはプラスミドから遺伝子組換え用として構築されたものが適している。ファージとしては、例えばエシェリヒア・コリを宿主微生物とする場合には、Lambda gt10、Lambda gt11などが例示される。また、プラスミドとしては、例えば、エシェリヒア・コリを宿主微生物とする場合には、pBR322、pUC18,pUC118,pUC19,pUC119,pTrc99A,pBluescriptあるいはコスミドであるSuperCosIなどが例示される。
クローニングの際、上記のようなベクターを、上述したGDHをコードする遺伝子供与体である微生物DNAの切断に使用した制限酵素で切断してベクター断片を得ることができるが、必ずしも該微生物DNAの切断に使用した制限酵素と同一の制限酵素を用いる必要はない。微生物DNA断片とベクターDNA断片とを結合させる方法は、公知のDNAリガーゼを用いる方法であればよく、例えば微生物DNA断片の付着末端とベクター断片の付着末端とのライゲーションの後、適当なDNAリガーゼの使用により微生物DNA断片とベクターDNA断片との組換えベクターを作製する。必要に応じて、ライゲーションの後、宿主微生物に移入して生体内のDNAリガーゼを利用し組換えベクターを作製することもできる。
クローニングに使用する宿主微生物としては、組換えベクターが安定であり、かつ自律増殖可能で外来性遺伝子の形質発現できるのであれば特に制限されない。一般的には、エシェリヒア・コリDH5α,XL−1BlueMRなどを用いることができる。
宿主微生物に組換えベクターを移入する方法としては、例えば宿主微生物がエシェリヒア・コリの場合には、カルシウム処理によるコンピテントセル法やエレクトロポーレーション法などを用いることができる。
上記の方法により得られたクローン化断片がGDHをコードしていることは、同断片の塩基配列を常法により解読することによって確認することができる。
上記のように得られた形質転換体から、組換えベクターを回収すれば、本発明のDNAが得られる。
本発明のDNA又は同DNAを含む組換えベクターを保持する形質転換体を培養して、同DNAの発現産物としてGDHを産生させ、これを菌体又は培養液から採取することにより、GDHを製造することができる。その際、本発明のDNAは、αサブユニットをコードするDNAであってもよいが、さらにγサブユニットをαサブユニットとともに発現させることによって、発現効率を高めることができる。
GDHを産生させる微生物としては、大腸菌をはじめとする腸内細菌群、シュードモナス属やグルコノバクター属などのグラム陰性細菌、バチルス・サブチリス等のバチルス属細菌をはじめとするグラム陽性細菌、サッカロマイセス・セレビシエ等の酵母、アスペルギルス・ニガー等の糸状菌が挙げられるが、これらに限られず、異種タンパク質生産に適した宿主微生物であれば用いることができる。
一度選択されたGDH遺伝子を保有する組換えベクターより、微生物にて複製できる組換えベクターへの移入は、GDH遺伝子を保持する組換えベクターから制限酵素やPCR法によりGDH遺伝子であるDNAを回収し、他のベクター断片と結合させることにより容易に実施できる。また、これらのベクターによる微生物の形質転換は、例えばエシェリヒア属細菌ではカルシウム処理によるコンピテントセル法、バチルス属細菌ではプロトプラスト法、酵母ではKU法やKUR法、糸状菌ではマイクロマニュピレーション法等の方法によって行うことができる。また、エレクトロポーレーション法も広く用いることができる。
宿主微生物への目的組換えベクターの移入の有無についての選択は、目的とするDNAを保持するベクターの薬剤耐性マーカーとGDH活性を同時に発現する微生物を検索すればよい。例えば、薬剤耐性マーカーに基づく選択培地で生育し、かつGDHを生成する微生物を選択すればよい。
形質転換体の培養形態は、宿主の栄養生理的性質を考慮して培養条件を選択すればよく、多くの場合は液体培養で行う。工業的には通気攪拌培養を行うのが有利である。
培地の栄養源としては、微生物の培養に通常用いられるものが広く使用され得る。炭素源としては資化可能な炭素化合物であればよく、例えば、グルコース、シュークロース、ラクトース、マルトース、ラクトース、糖蜜、ピルビン酸などが使用される。また、窒素源としては利用可能な窒素化合物であればよく、例えば、ペプトン、肉エキス、酵母エキス、カゼイン加水分解物、大豆粕アルカリ抽出物などが使用される。その他、リン酸塩、炭酸塩、硫酸塩、マグネシウム、カルシウム、カリウム、鉄、マンガン、亜鉛などの塩類、特定のアミノ酸、特定のビタミンなどが必要に応じて使用される。
培養温度は菌が生育し、GDHを生産する範囲で適宜変更し得るが、好ましくは20〜42℃程度である。培養時間は条件によって多少異なるが、GDHが最高収量に達する時期を見計らって適当時期に培養を完了すればよく、通常は12〜72時間程度である。培地のpHは菌が発育し、GDHを生産する範囲で適宜変更し得るが、好ましくはpH6.0〜9.0程度の範囲である。
培養物中のGDHを生産する菌体を含む培養液をそのまま採取し、利用することもできるが、一般には、常法に従って、GDHが培養液中に存在する場合はろ過、遠心分離などにより、GDH含有溶液と微生物菌体と分離した後に利用される。GDHが菌体内に存在する場合には、得られた培養物からろ過または遠心分離などの手段により菌体を採取し、次いで、この菌体を機械的方法またはリゾチームなどの酵素的方法で破壊し、また、必要に応じて、EDTA等のキレート剤及び界面活性剤を添加してGDHを可溶化し、水溶液として分離採取する。
上記のようにして得られたGDH含有溶液を、例えば減圧濃縮、膜濃縮、さらに硫酸アンモニウム、硫酸ナトリウムなどの塩析処理、あるいは親水性有機溶媒、例えばメタノール、エタノール、アセトンなどによる分別沈殿法により沈殿せしめればよい。また、加熱処理や等電点処理も有効な精製手段である。その後、吸着剤あるはゲルろ過剤などによるゲルろ過、吸着クロマトグラフィー、イオン交換クロマトグラフィー、アフィニティクロマトグラフィーを適宜組み合わせることによって精製することができる。を行うことにより、精製されたGDHを得ることができる。
カラムクロマイトグラフィーにより分離、精製し、精製酵素標品を得ることができる。該精製酵素標品は、電気泳動(SDS−PAGE)的に単一のバンドを示す程度に純化されていることが好ましいが、γサブユニットが含まれていても良い。
上記のようにして得られた精製酵素を、例えば凍結乾燥、真空乾燥やスプレードライなどにより粉末化して流通させることが可能である。
また、後記実施例に示すαサブユニットのアミノ酸配列と同様にしてβサブユニットのアミノ酸配列を決定し、βサブユニットをコードするDNAを単離することができる。また、得られたDNAを用いて、βサブユニットを製造することができる。さらに、αサブユニットをコードするDNA及びβサブユニットをコードするDNAを用いて、多量体酵素を製造することもできる。
The method for obtaining the DNA of the present invention is exemplified below.
Chromosomal DNA is isolated and purified from microorganisms such as Burkholderia cepacia, and then the chromosomal DNA is cleaved using sonication, restriction enzyme treatment, etc., and linear expression vector is closed by DNA ligase. To construct a recombinant vector. After the obtained recombinant vector is transferred to a host microorganism capable of autonomous replication of the vector, the transformant is screened using the expression of the marker of the vector and the enzyme activity as an index, and a group containing a gene encoding GDH. A microorganism carrying the replacement vector is obtained. The recombinant vector possessed by the obtained microorganism is expected to contain at least a base sequence encoding the α subunit. In addition, if the cloned fragment has a sufficient size, it is highly likely that a nucleotide sequence encoding the γ subunit is also included.
Subsequently, the microorganism holding the recombinant vector can be cultured, the recombinant vector can be isolated and purified from the cells of the cultured microorganism, and the gene encoding GDH can be collected from the expression vector. For example, chromosomal DNA that is a gene donor is specifically collected as follows.
A culture solution obtained by stirring and culturing the gene-donating microorganism for 1 to 3 days, for example, is collected by centrifugation, and then lysed to prepare a GDH gene-containing lysate. As a method for lysis, for example, treatment is performed with a lytic enzyme such as lysozyme, and a protease, other enzyme, or a surfactant such as sodium lauryl sulfate (SDS) is used in combination as necessary. Further, it may be combined with a physical crushing method such as freeze-thawing or French press treatment.
In order to separate and purify DNA from the lysate obtained as described above, a conventional method is used, for example, by appropriately combining methods such as deproteinization treatment by phenol treatment or protease treatment, ribonuclease treatment, and alcohol precipitation treatment. be able to.
A method of cleaving DNA separated and purified from microorganisms can be performed by, for example, ultrasonic treatment, restriction enzyme treatment, or the like. Type II restriction enzymes that act on specific nucleotide sequences are suitable. The restriction enzyme may be a restriction enzyme that generates an end compatible with the cut end of the vector, or an arbitrary restriction enzyme may be used to make the cut end blunt and ligated to the vector.
As a vector for cloning, a vector constructed for gene recombination from a phage or plasmid that can autonomously grow in a host microorganism is suitable. Examples of the phage include Lambda gt10 and Lambda gt11 when Escherichia coli is used as a host microorganism. Examples of plasmids include pBR322, pUC18, pUC118, pUC19, pUC119, pTrc99A, pBluescript, or SuperCosI which is a cosmid when Escherichia coli is used as a host microorganism.
At the time of cloning, a vector fragment can be obtained by cleaving the above-mentioned vector with the restriction enzyme used for cleaving the microbial DNA that is the gene donor encoding GDH described above. It is not necessary to use the same restriction enzyme as that used in the above. The method for binding the microbial DNA fragment and the vector DNA fragment may be a method using a known DNA ligase. For example, after ligation between the sticky end of the microbial DNA fragment and the sticky end of the vector fragment, an appropriate DNA ligase may be used. A recombinant vector of a microbial DNA fragment and a vector DNA fragment is prepared by use. If necessary, after ligation, it can be transferred to a host microorganism and a recombinant vector can be prepared using in vivo DNA ligase.
The host microorganism used for cloning is not particularly limited as long as the recombinant vector is stable, can autonomously grow, and can express a foreign gene. In general, Escherichia coli DH5α, XL-1 Blue MR, or the like can be used.
As a method for transferring the recombinant vector into the host microorganism, for example, when the host microorganism is Escherichia coli, a competent cell method or an electroporation method using calcium treatment can be used.
It can be confirmed that the cloned fragment obtained by the above method encodes GDH by decoding the base sequence of the fragment by a conventional method.
If the recombinant vector is recovered from the transformant obtained as described above, the DNA of the present invention can be obtained.
GDH is produced by culturing a transformant carrying the DNA of the present invention or a recombinant vector containing the DNA, producing GDH as an expression product of the DNA, and collecting this from the cell or culture medium. can do. At this time, the DNA of the present invention may be a DNA encoding an α subunit, but the expression efficiency can be increased by further expressing the γ subunit together with the α subunit.
Examples of microorganisms that produce GDH include enterobacteriaceae such as Escherichia coli, Gram-negative bacteria such as Pseudomonas and Gluconobacter, Gram-positive bacteria including Bacillus bacteria such as Bacillus subtilis, and Saccharomyces cerevisiae. Yeasts such as Aspergillus niger and the like, but are not limited thereto, and any host microorganism suitable for heterologous protein production can be used.
Transfer from a recombinant vector carrying the GDH gene once selected to a recombinant vector that can be replicated in a microorganism is performed by collecting DNA, which is a GDH gene, from the recombinant vector carrying the GDH gene by a restriction enzyme or PCR method. It can be easily carried out by linking with other vector fragments. Moreover, the transformation of microorganisms by these vectors is carried out by, for example, a competent cell method by calcium treatment in Escherichia bacteria, a protoplast method in Bacillus bacteria, a KU method or KUR method in yeast, and a micromanipulation method in filamentous fungi. It can be carried out. Also, electroporation can be widely used.
The selection as to whether or not the target recombinant vector is transferred to the host microorganism may be made by searching for a microorganism that simultaneously expresses the drug resistance marker of the vector holding the target DNA and GDH activity. For example, a microorganism that grows in a selective medium based on a drug resistance marker and generates GDH may be selected.
For the culture form of the transformant, the culture conditions may be selected in consideration of the nutritional physiological properties of the host. Industrially, aeration and agitation culture is advantageous.
As a nutrient source of the medium, those commonly used for culturing microorganisms can be widely used. Any carbon compound that can be assimilated may be used as the carbon source. For example, glucose, sucrose, lactose, maltose, lactose, molasses, pyruvic acid and the like are used. The nitrogen source may be any available nitrogen compound. For example, peptone, meat extract, yeast extract, casein hydrolyzate, soybean cake alkaline extract, and the like are used. In addition, phosphates, carbonates, sulfates, salts such as magnesium, calcium, potassium, iron, manganese, and zinc, specific amino acids, specific vitamins, and the like are used as necessary.
The culture temperature can be appropriately changed within the range in which the bacteria grow and GDH is produced, but is preferably about 20 to 42 ° C. Although the culture time varies slightly depending on the conditions, the culture may be completed at an appropriate time in consideration of the time when GDH reaches the maximum yield, and is usually about 12 to 72 hours. The pH of the medium can be appropriately changed within the range in which the bacteria grow and produce GDH, but is preferably in the range of about pH 6.0 to 9.0.
Although the culture solution containing the cells producing GDH in the culture can be collected and used as it is, generally, when GDH is present in the culture solution according to a conventional method, filtration, centrifugation, etc. It is used after separating the GDH-containing solution and the microbial cells. When GDH is present in the microbial cells, the microbial cells are collected from the obtained culture by means of filtration or centrifugation, and then the microbial cells are destroyed by a mechanical method or an enzymatic method such as lysozyme. Further, if necessary, a chelating agent such as EDTA and a surfactant are added to solubilize GDH and separated and collected as an aqueous solution.
The GDH-containing solution obtained as described above is precipitated by, for example, vacuum concentration, membrane concentration, salting-out treatment with ammonium sulfate, sodium sulfate or the like, or fractional precipitation with a hydrophilic organic solvent such as methanol, ethanol, acetone, etc. You just have to let them know. Heat treatment and isoelectric point treatment are also effective purification means. Then, it can be purified by appropriately combining gel filtration with an adsorbent or gel filter, adsorption chromatography, ion exchange chromatography, and affinity chromatography. By performing the above, purified GDH can be obtained.
A purified enzyme preparation can be obtained by separation and purification by column chromatography. The purified enzyme preparation is preferably purified to the extent that it shows a single band on electrophoresis (SDS-PAGE), but may contain a γ subunit.
The purified enzyme obtained as described above can be pulverized and distributed, for example, by freeze drying, vacuum drying, spray drying, or the like.
In addition, the amino acid sequence of the β subunit can be determined in the same manner as the amino acid sequence of the α subunit shown in Examples below, and DNA encoding the β subunit can be isolated. In addition, β subunit can be produced using the obtained DNA. Furthermore, a multimeric enzyme can be produced using a DNA encoding an α subunit and a DNA encoding a β subunit.
<4>本発明のグルコースセンサ
本発明のグルコースセンサは、本発明の酵素(前記多量体酵素もしくはペプチド酵素、又はγサブユニットが含まれる前記多量体酵素もしくはペプチド酵素)、本発明の形質転換体、又は本発明の微生物(ブルクホルデリア・セパシアKS1株)を、酵素電極として用いることを特徴とする。電極としては、カーボン電極、金電極、白金電極などを用い、この電極上に本発明の酵素を固定化する。固定化方法としては、架橋試薬を用いる方法、高分子マトリックス中に封入する方法、透析膜で被覆する方法、光架橋性ポリマー、導電性ポリマー、酸化還元ポリマーなどがあり、あるいはフェロセンあるいはその誘導体に代表される電子メディエーターとともにポリマー中に固定あるいは電極上に吸着固定してもよく、またこれらを組み合わせて用いてもよい。典型的には、グルタルアルデヒドを用いて本発明のグルコース脱水素酵素をカーボン電極上に固定化した後、アミン基を有する試薬で処理してグルタルアルデヒドをブロッキングする。
グルコースの濃度の測定は、以下のようにして行うことができる。恒温セルに緩衝液を入れ、メディエーターを加えて一定温度に維持する。メディエーターとしては、フェリシアン化カリウム、フェナジンメトサルフェートなどを用いることができる。作用電極として本発明の酵素を固定化した電極を用い、対極(例えば白金電極)および参照電極(例えばAg/AgCl電極)を用いる。カーボン電極に一定の電圧を印加して、電流が定常になった後、グルコースを含む試料を加えて電流の増加を測定する。標準濃度のグルコース溶液により作製したキャリブレーションカーブに従い、試料中のグルコースの濃度を計算することができる。
<4> Glucose sensor of the present invention The glucose sensor of the present invention includes the enzyme of the present invention (the multimeric enzyme or peptide enzyme, or the multimeric enzyme or peptide enzyme containing a γ subunit), or the transformant of the present invention. Alternatively, the microorganism of the present invention (Burkholderia cepacia KS1 strain) is used as an enzyme electrode. As the electrode, a carbon electrode, a gold electrode, a platinum electrode or the like is used, and the enzyme of the present invention is immobilized on this electrode. Immobilization methods include a method using a crosslinking reagent, a method of encapsulating in a polymer matrix, a method of coating with a dialysis membrane, a photocrosslinkable polymer, a conductive polymer, a redox polymer, etc., or ferrocene or a derivative thereof. It may be fixed in a polymer or adsorbed and fixed on an electrode together with a representative electron mediator, or a combination thereof. Typically, after the glucose dehydrogenase of the present invention is immobilized on a carbon electrode using glutaraldehyde, glutaraldehyde is blocked by treatment with a reagent having an amine group.
The concentration of glucose can be measured as follows. Put the buffer in a thermostatic cell and add a mediator to maintain a constant temperature. As the mediator, potassium ferricyanide, phenazine methosulfate, or the like can be used. An electrode on which the enzyme of the present invention is immobilized is used as a working electrode, and a counter electrode (for example, a platinum electrode) and a reference electrode (for example, an Ag / AgCl electrode) are used. After a constant voltage is applied to the carbon electrode and the current becomes steady, a sample containing glucose is added and the increase in current is measured. The concentration of glucose in the sample can be calculated according to a calibration curve prepared with a standard concentration glucose solution.
<5>本発明のグルコースアッセイキット
本発明の糖類アッセイキットは、本発明の酵素(前記多量体酵素もしくはペプチド酵素、又はγサブユニットが含まれる前記多量体酵素もしくはペプチド酵素)を含むことを特徴とする。本発明のグルコースアッセイキットは、本発明の酵素を少なくとも1回のアッセイに十分な量で含む。典型的には、キットは、本発明の酵素に加えて、アッセイに必要な緩衝液、メディエーター、キャリブレーションカーブ作成のためのグルコースなどの標準溶液、ならびに使用の指針を含む。本発明に従う酵素は種々の形態で、例えば、凍結乾燥された試薬として、または適切な保存溶液中の溶液として提供することができる。
<5> Glucose assay kit of the present invention The saccharide assay kit of the present invention comprises the enzyme of the present invention (the multimeric enzyme or peptide enzyme, or the multimeric enzyme or peptide enzyme containing a γ subunit). And The glucose assay kit of the present invention contains the enzyme of the present invention in an amount sufficient for at least one assay. Typically, the kit contains, in addition to the enzyme of the present invention, buffers necessary for the assay, mediators, standard solutions such as glucose for creating a calibration curve, and directions for use. The enzyme according to the invention can be provided in various forms, for example as a lyophilized reagent or as a solution in a suitable storage solution.
以下、実施例により本発明を更に具体的に説明する。
実施例1 グルコース脱水素酵素の生産能を有する菌の取得
〔スクリーニング〕
本発明の微生物は、日本の種々の温泉の近くの土壌を入手し、その土壌中より、グルコースを栄養源とする細菌の中からグルコースデヒドロゲナーゼ活性を示すものを選択して入手した。
この株の形態学的性質、生育特性、生理学特性を調べた結果を次に示す。
菌学的性質;
グラム染色 陰性
細胞の形状 桿菌
極性鞭毛を持つ
運動性 陽性
フラグメントの数 >5
至適増殖温度 45℃
オキシダーゼ 陰性
カタラーゼ 陽性
アセトインの生成 陰性
H2Sの生成 陰性
インドールの生成 陰性
グルコースからの酸 陽性
アルギニンジヒドロラーゼ 陰性
ウレアーゼ 陰性
β−グルコシダーゼ 陰性
プロテアーゼ 陰性
β−ガラクトシダーゼ 陽性
リジンカルボキシラーゼ 陰性
オリニチンカルボキシラーゼ 陰性
硝酸塩の還元 陽性
〔資化性〕
グリセロール 陽性
エリトリトール 陰性
D−アラビノース 陰性
L−アラビノース 陽性
リボース 陽性
D−キシロース 陽性
L−キシロース 陰性
アドニトール 陽性
β−メチル−キシロシド 陰性
ガラクトース 陽性
D−グルコース 陽性
D−フルクトース 陽性
D−マンノース 陽性
L−ソルボース 陰性
ラムノース 陰性
ズルシトール 陽性
イノシトール 陽性
マンニトール 陽性
ソルビトール 陽性
α−メチル−D−マンノシド 陰性
α−メチル−D−グルコシド 陰性
N−アセチル−グルコサミン 陽性
アミグダリン(Amygdaline) 陰性
アルブチン 陰性
エスクリン 陰性
サリシン 陰性
セロビオース 陰性
マルトース 陰性
ラクトース 陰性
メリビオース 陰性
サッカロース 陰性
トレハロース 陽性
イヌリン 陰性
メレチトース 陰性
D−ラフィノース 陰性
アミドン(Amidon) 陰性
グリコーゲン 陰性
キシリトール 陽性
β−ゲンチオビオース 陰性
D−チュラノース 陰性
D−リキソース 陰性
D−タガトース 陰性
D−フコース 陰性
L−フコース 陰性
D−アラビトール 陽性
L−アラビトール 陽性
グルコン酸 陽性
2−ケトグルコン酸 陽性
5−ケトグルコン酸 陰性
カプリン酸 陽性
アジビン酸 陽性
リンゴ酸 陽性
クエン酸 陽性
フェニルアセテート 陽性
〔酸化性〕
グリセロール 陰性
エリトリトール 陰性
D−アラビノース 陰性
L−アラビノース 陽性
リボース 陽性
D−キシロース 陽性
L−キシロース 陰性
アドニトール 陽性
β−メチル−キシロシド 陰性
ガラクトース 陽性
D−グルコース 陽性
D−フルクトース 陽性
D−マンノース 陽性
L−ソルボース 陰性
ラムノース 陰性
ズルシトール 陽性
イノシトール 陽性
マンニトール 陽性
ソルビトール 陽性
α−メチル−D−マンノシド 陰性
α−メチル−D−グルコシド 陰性
N−アセチル−グルコサミン 陰性
アミグダリン(Amygdaline) 陰性
アルブチン 陰性
エスクリン 陽性
サリシン 陰性
セロビオース 陽性
マルトース 陽性
ラクトース 陽性
メリビオース 陰性
サッカロース 陰性
トレハロース 陽性
イヌリン 陰性
メレチトース 陰性
D−ラフィノース 陰性
アミドン(Amidon) 陰性
グリコーゲン 陰性
キシリトール 陰性
β−ゲンチオビオース 陽性
D−チュラノース 陰性
D−リキソース 陰性
D−タガトース 陰性
D−フコース 陽性
L−フコース 陰性
D−アラビトール 陽性
L−アラビトール 陽性
グルコン酸 陰性
2−ケトグルコン酸 陰性
5−ケトグルコン酸 陰性
上記の菌学的性質を有するKS1株の分類学上の位置をバージェイズ・マニュアル・オブ・デターミネイティブ・バクテリオロジー(Bergey’s Manual of Determinative Bacteriology)を参照して検討すると、ブルクホルデリア属に属し、ブルクホルデリア・セパシアである菌株と同定された。
尚、ブルクホルデリア属は、従来シュードモナス属に分類されていたが、現在ではブルクホルデリア属に分かれている(Yabuuchi,E.,Kosako,Y.,Oyaizu,H.,Yano,I.,Hotta,H.,Hashimoto,Y.,Ezaki,T.and Arakawa,M.,Microbiol.Immunol.Vol36(12),1251−1275,1992;International Journal of Systematic Bacteriology,Apr,1993,p398−399)。
また、本発明者らはブルクホルデリア・セパシア KS1株以外の株について、財団法人発酵研究所(Institute for Fermentation,Osaka)又は理化学研究所微生物系統保存施設(Japan Collection of Microorganisms,JCM)に寄託されているブルクホルデリア・セパシアのいくつかの菌株を取り寄せてグルコース脱水素酵素活性を測定したところ、同活性を有することを確認した。グルコース脱水素酵素活性は、後述の実施例2に記載の方法により測定した。KS1株の水溶性画分の酵素活性を100としたときの相対活性を表1に示す。

Example 1 Acquisition of Bacteria Having Ability to Produce Glucose Dehydrogenase [Screening]
The microorganisms of the present invention were obtained by obtaining soils near various hot springs in Japan, and selecting bacteria exhibiting glucose dehydrogenase activity from bacteria using glucose as a nutrient source.
The results of examining the morphological properties, growth characteristics, and physiological characteristics of this strain are shown below.
Mycological properties;
Gram staining Negative cell shape Motility with gonococcal polar flagella Number of positive fragments> 5
Optimal growth temperature 45 ° C
Oxidase Negative catalase Production of positive acetoin Production of negative H 2 S Production of negative indole Acid negative arginine dihydrolase Negative urease Negative glucose Negative β-glucosidase Negative protease Negative β-galactosidase Positive lysine carboxylase Negative ornithine carboxylase Negative nitrate reduction Positive [Utilization]
Glycerol positive erythritol negative D-arabinose negative L-arabinose positive ribose positive D-xylose positive L-xylose negative adonitol positive β-methyl-xyloside negative galactose positive D-glucose positive D-fructose positive D-mannose positive L-sorbose negative rhamnose negative Dulcitol positive inositol positive mannitol positive sorbitol positive α-methyl-D-mannosid negative α-methyl-D-glucoside negative N-acetyl-glucosamine positive amygdalin (Amygdaline) negative arbutin negative esculin negative salobiose negative maltose negative lactose negative lactose negative lactose negative Negative trehalose positive inulin negative melethitose negative D-raffinose negative Don (Amidon) Negative glycogen Negative xylitol Positive β-gentiobiose Negative D-Thuranose Negative D-Lixose Negative D-Tagatose Negative D-Fucose Negative L-Fucose Negative D-Arabitol Positive L-Arabitol Positive Gluconic acid Positive 2-Ketogluconic acid Positive 5-ketogluconic acid negative capric acid positive adivic acid positive malic acid positive citric acid positive phenylacetate positive [oxidative]
Glycerol negative erythritol negative D-arabinose negative L-arabinose positive ribose positive D-xylose positive L-xylose negative adonitol positive β-methyl-xyloside negative galactose positive D-glucose positive D-fructose positive D-mannose positive L-sorbose negative rhamnose negative Dulcitol positive inositol positive mannitol positive sorbitol positive α-methyl-D-mannosid negative α-methyl-D-glucoside negative N-acetyl-glucosamine negative amygdalin (Amygdaline) negative arbutin negative esculin positive salicin negative cellobiose positive maltose positive lactose positive lactose Negative trehalose positive inulin negative melethitose negative D-raffinose negative Don (Amidon) Negative glycogen Negative xylitol Negative β-gentiobiose Positive D-Thuranose Negative D-Lixose Negative D-Tagatose Negative D-Fucose Positive L-Fucose Negative D-arabitol Positive L-arabitol Positive gluconic acid Negative 2-Ketogluconic acid Negative 5-Ketogluconic acid Negative When the taxonomic position of the KS1 strain having the above mycological properties is examined with reference to Bergey's Manual of Detergent Bacteriology , A strain belonging to the genus Burkholderia and being Burkholderia cepacia.
The genus Burkholderia was conventionally classified as the genus Pseudomonas, but is now divided into the genus Burghorderia (Yabuchi, E., Kosako, Y., Oyaizu, H., Yano, I., Hotta). H., Hashimoto, Y., Ezaki, T. and Arakawa, M., Microbiol. Immunol. Vol 36 (12), 1251-1275, 1992; International Journal of Systemic Bacteriology-3, Ap.
In addition, the present inventors have deposited strains other than the Burkholderia cepacia KS1 strain to the Institute for Fermentation (Institute for Fermentation, Osaka) or the Institute of Physical and Chemical Research Microbiology System (Japan Collection of Microorganisms, JCM). We obtained several strains of Burkholderia cepacia and measured glucose dehydrogenase activity, and confirmed that it had the same activity. The glucose dehydrogenase activity was measured by the method described in Example 2 described later. Table 1 shows the relative activities when the enzyme activity of the water-soluble fraction of the KS1 strain is defined as 100.
実施例2 グルコース脱水素酵素の抽出
<1>菌体の培養
本細菌の培養条件は通常通りの好気的培養条件が用いられる。培養液1L中に以下の成分を含む培地7Lで、34℃、8時間、培養した。
ポリペプトン 10g
酵母抽出液 1g
NaCl 5g
KH2PO4 2g
グルコース 5g
Einol(ABLE Co.東京 日本) 0.14g
Total、蒸留水 1L
pH調製 7.2
本培養液7Lを4℃、10分間、9,000×gで遠心分離し、約60gの菌体を得た。
<2>粗精製フラクションの作製
60gの菌体を10mMのリン酸カリウム緩衝液(pH6.0)に分散し、フレンチプレス(大竹製作所 東京 日本)で1,500Kg/cm2の圧力差を加えて、菌体膜を破壊した。細胞抽出液を8,000×gで10分間、遠心分離し、細胞固形物を除いた。さらに、その上清を、4℃で69,800×gで90分間、超遠心し、沈殿物としての膜フラクション、約8gを得た。
<3>酵素の精製
膜フラクションを、最終濃度でTriton−X100が1%になるように、10mMリン酸カリウム緩衝液(pH6.0)で再分散した。そして、4℃、一晩、ゆっくり攪拌した。超遠心後(4℃、69,800g、90分間)、可溶化膜フラクションを、4℃で、15000×gで15分間、再遠心し、上清を得た。
その可溶化膜フラクションに、同量の0.2%Triton−X100を含む10mMリン酸カリウム緩衝液(pH8.0)を加えた。透析後、その溶液を、0.2%Triton−X100を含む10mMリン酸カリウム緩衝液(pH8.0)で等量化されたDEAE−TOYOPEARLカラム(22mmID×20cm 東ソー 東京 日本)に供給した。タンパク質を、10mMリン酸カリウム緩衝液(pH8.0)中のNaClの濃度が0〜0.15Mになるように、直線的グラジエントで溶出した。その流速は5ml/minで行った。GDHは約75mMのNaCl濃度で溶出された。GDH活性をもつフラクションを集め、0.2%Triton−X100を含む10mMリン酸カリウム緩衝液(pH8.0 4℃)で一夜、透析した。
さらに透析調製酵素液を、DEAE−5PWカラム(8.0mmID×7.5cm 東ソー、東京、日本)に通した。そのカラムは予め、0.2%Triton−X100を含む10mMリン酸カリウム緩衝液(pH6.0)で平衡化されている。タンパク質を、10mMリン酸カリウム緩衝液(pH8.0)中のNaClの濃度が0〜100mMになるように、直線的グラジエントで溶出した。その流速は1ml/minで行った。GDH活性のあるフラクションが約20mMのNaCl濃度で溶出した。GDH活性をもつフラクションを集め、0.2%Triton−X100を含む10mMリン酸カリウム緩衝液(pH8.0)で、一夜、脱塩し、本精製酵素を得た。
尚、GDHの活性測定は、本実施例及び以下の実施例を通して、以下の方法に従い行った。
電子受容体として、2,6−ジクロルフェノルインドフェノル(DCIP)及びフェナンジメトサルフェート(PMS)を用いた。反応はポリエチレンチューブ内で所定の温度で実施した。0.75mMPMSと0.75mMDCIP含有25mMトリスHCl緩衝液pH8.0 20μlに酵素溶液5μlを添加した。この混合液を1分間事前定温放置した。2Mグルコース1μl(最終濃度:77mM)の添加により反応を開始させ、2分間定温放置した。次に氷冷蒸留水100μlまたは7.5M尿素120μlを添加して試料を冷却した。超微量計測用セル(100μl)及びこれを用いて計測できる分光光度計(UV160、島津製作所、京都、日本)を用いて、グルコースの脱水素化に基づく、電子受容体の還元反応を追跡した。すなわちDCIP還元にもとづく退色を、DCIPの吸収波長である600nmを時間とともに計測した。DCIPのモル吸光係数(22.23mM×cm−1)を用いた。酵素1単位(U)は標準検定条件下で1分ごとに1μMグルコースを酸化する量と定義した。タンパク濃度はローリー法で測定した。
Example 2 Extraction of Glucose Dehydrogenase <1> Cultivation of Bacteria The usual aerobic culture conditions are used for the culture conditions of this bacterium. The culture was performed at 34 ° C. for 8 hours in 7 L of a medium containing the following components in 1 L of a culture solution.
Polypeptone 10g
Yeast extract 1g
NaCl 5g
KH 2 PO 4 2g
Glucose 5g
Einol (ABLE Co. Tokyo Japan) 0.14g
Total, distilled water 1L
pH adjustment 7.2
7 L of the main culture was centrifuged at 9,000 × g for 10 minutes at 4 ° C. to obtain about 60 g of cells.
<2> Preparation of roughly purified fraction 60 g of cells were dispersed in 10 mM potassium phosphate buffer (pH 6.0), and a pressure difference of 1,500 Kg / cm 2 was applied with a French press (Otake, Tokyo, Japan). The cell membrane was destroyed. The cell extract was centrifuged at 8,000 × g for 10 minutes to remove cell solids. Further, the supernatant was ultracentrifuged at 69,800 × g for 90 minutes at 4 ° C. to obtain about 8 g of a membrane fraction as a precipitate.
<3> The purified membrane fraction of the enzyme was redispersed with 10 mM potassium phosphate buffer (pH 6.0) so that the final concentration of Triton-X100 was 1%. And it stirred slowly at 4 degreeC overnight. After ultracentrifugation (4 ° C., 69,800 g, 90 minutes), the solubilized membrane fraction was recentrifuged at 15000 × g for 15 minutes at 4 ° C. to obtain a supernatant.
A 10 mM potassium phosphate buffer (pH 8.0) containing the same amount of 0.2% Triton-X100 was added to the solubilized membrane fraction. After dialysis, the solution was supplied to a DEAE-TOYOPEARL column (22 mm ID × 20 cm Tosoh Tokyo Japan) which was equalized with 10 mM potassium phosphate buffer (pH 8.0) containing 0.2% Triton-X100. The protein was eluted with a linear gradient such that the concentration of NaCl in 10 mM potassium phosphate buffer (pH 8.0) was 0-0.15M. The flow rate was 5 ml / min. GDH was eluted at a NaCl concentration of about 75 mM. Fractions having GDH activity were collected and dialyzed overnight against 10 mM potassium phosphate buffer (pH 8.0 4 ° C.) containing 0.2% Triton-X100.
Furthermore, the dialyzed enzyme solution was passed through a DEAE-5PW column (8.0 mm ID × 7.5 cm Tosoh, Tokyo, Japan). The column was previously equilibrated with 10 mM potassium phosphate buffer (pH 6.0) containing 0.2% Triton-X100. The protein was eluted with a linear gradient such that the concentration of NaCl in 10 mM potassium phosphate buffer (pH 8.0) was 0-100 mM. The flow rate was 1 ml / min. The fraction with GDH activity eluted at a NaCl concentration of about 20 mM. Fractions having GDH activity were collected and desalted overnight with 10 mM potassium phosphate buffer (pH 8.0) containing 0.2% Triton-X100 to obtain the purified enzyme.
In addition, the activity measurement of GDH was performed according to the following method through the present Example and the following Examples.
As the electron acceptor, 2,6-dichlorophenolindophenol (DCIP) and phenandimethosulphate (PMS) were used. The reaction was carried out at a predetermined temperature in a polyethylene tube. 5 μl of enzyme solution was added to 20 μl of 25 mM Tris HCl buffer pH 8.0 containing 0.75 mM PMS and 0.75 mM DCIP. This mixture was allowed to stand for 1 minute in advance. The reaction was initiated by the addition of 1 μl of 2M glucose (final concentration: 77 mM) and allowed to incubate for 2 minutes. The sample was then cooled by adding 100 μl ice cold distilled water or 120 μl 7.5M urea. The reduction reaction of the electron acceptor based on the dehydrogenation of glucose was traced using a cell for ultra-trace measurement (100 μl) and a spectrophotometer (UV160, Shimadzu Corporation, Kyoto, Japan) that can be measured using this cell. That is, the discoloration due to DCIP reduction was measured over time at 600 nm, which is the absorption wavelength of DCIP. The molar extinction coefficient of DCIP (22.23 mM × cm −1 ) was used. One unit (U) of enzyme was defined as the amount that oxidizes 1 μM glucose per minute under standard assay conditions. Protein concentration was measured by the Raleigh method.
実施例3
本精製酵素について、Native PAGE電気泳動を実施した。本電気泳動の条件は、1%Triton−X100を含むTris−Alanine緩衝液システムを用いた8−25%ポリアクリルアミドグラジエントゲル上で実施した。そのゲルは硝酸銀で染色を行った。タンパク質マーカーとして、チログロブリン(Thyroglobulin):669kDa、フェリチン(Ferritin):440kDa、カタラーゼ(Catalase):232kDa、アルドラーゼ(Aldolase):158kDa、ウシ血清アルブミン(Bovine Serum Albumin):67kDa、オバルブミン(Ovalbumin):43kDa、キモトリプシノーゲン(ChymotrypsinogenA):25kDa、のタンパク質を用いた。
また、そのNative PAGEゲルについて、活性染色を実施した。本ゲルを以下の溶液中に30分間インキュベーションする事により行った。GDHの活性部位はニトロブルーテトラゾリウムが還元され、ホルマザンが生成し、暗紫色に発色した。
200mM グルコース
0.1mM ニトロブルーテトラゾリウム
0.3mM フェナジンメトサルフェート
20mM Tris−HCl緩衝液(pH8.0)
Native PAGEの銀染色の結果より、本酵素は単一酵素であること、その分子量は約400kDaであることが推測された。また、同ゲルを活性染色すると、銀染色と同様の移動度の部位に活性が認められた(図1参照。図中、レーン1は分子量標準マーカータンパク質の銀染色を、レーン2は本酵素の銀染色を、レーン4は、本酵素の活性染色を示す)。本酵素を70℃で30分間、熱処理をすると、以外にも活性は残存し、分子量85kDa付近に活性をもつタンパク質に分離した(図1参照。図中、レーン3は70℃、30min、熱処理された本酵素の銀染色を、レーン5は70℃、30min、熱処理された本酵素の活性染色を示す)。このことは本酵素がサブユニットからなる事を示唆している。
Example 3
The purified enzyme was subjected to Native PAGE electrophoresis. The electrophoresis conditions were performed on an 8-25% polyacrylamide gradient gel using a Tris-Aline buffer system containing 1% Triton-X100. The gel was stained with silver nitrate. As protein markers, thyroglobulin: 669 kDa, ferritin: 440 kDa, catalase: 232 kDa, aldolase: 158 kDa, bovine serum albumin (buvin Serum Albumin): 67 kDa Chymotrypsinogen A: 25 kDa protein was used.
In addition, activity staining was performed on the Native PAGE gel. This gel was incubated for 30 minutes in the following solution. At the active site of GDH, nitroblue tetrazolium was reduced, formazan was produced, and the color developed dark purple.
200 mM glucose 0.1 mM nitroblue tetrazolium 0.3
From the result of silver staining of Native PAGE, it was estimated that this enzyme is a single enzyme and its molecular weight is about 400 kDa. In addition, when the gel was activity-stained, activity was observed at a site having the same mobility as that of silver staining (see Fig. 1. In the figure,
実施例4
本精製酵素液をSDS−PAGEで電気泳動を行った。SDS−PAGEはTris−Tricine緩衝液を用いて8−25%ポリアクリルアミドの勾配ゲル中で実施した。そのゲルのタンパク質は硝酸銀で染色を行った。Phast System(Pharmacia)により、分離と展開を自動的に行った。標準タンパクの相対移動度により分子質量を測定した。SDS−PAGE電気泳動によって、本酵素は約60kDaと43kDaのタンパク質に分離した(図2参照。図2は電気泳動写真である。図中、レーン1は分子量標準マーカータンパク質の硝酸銀染色を、レーン2は本酵素の硝酸銀染色を示す)。従って、本酵素は60kDaのα−サブユニットと43kDaのβ−サブユニットが結合していることが示唆され、かつ、それらが4個結合した8量体を形成していることが予想された。
SDS−PAGEで分離された43kDaのタンパク質であるβ−サブユニットをポリビニリデンフルオリド膜に転写した後、アミノ酸シークエンサー(島津製作所、PPSQ−10)によりβ−サブユニットのN末端アミノ酸配列の決定を行った。その結果、本タンパク質のN末端アミノ酸配列は配列番号5のアミノ酸配列からなる16残基から構成されていることが明かとなった。
また、本酵素を70℃で30分間、熱処理した場合の結果を図2中レーン3で示す。このSDS−PAGEの結果から熱処理後この酵素は分子量60kDaの単一ポリペプチドに変わったことが想定できる。
Example 4
The purified enzyme solution was subjected to electrophoresis by SDS-PAGE. SDS-PAGE was performed in a gradient gel of 8-25% polyacrylamide using Tris-Tricine buffer. The gel protein was stained with silver nitrate. Separation and development were performed automatically by the Phas System (Pharmacia). The molecular mass was measured by the relative mobility of the standard protein. This enzyme was separated into proteins of about 60 kDa and 43 kDa by SDS-PAGE electrophoresis (see FIG. 2. FIG. 2 is an electrophoresis photograph. In the figure,
After transferring the β-subunit, which is a 43 kDa protein separated by SDS-PAGE, onto a polyvinylidene fluoride membrane, the amino acid sequencer (Shimadzu Corporation, PPSQ-10) was used to determine the N-terminal amino acid sequence of the β-subunit. went. As a result, it was revealed that the N-terminal amino acid sequence of this protein is composed of 16 residues consisting of the amino acid sequence of SEQ ID NO: 5.
In addition, the result of heat-treating this enzyme at 70 ° C. for 30 minutes is shown in
実施例5
本酵素のゲルろ過クロマトグラフィーを実施した。ゲルとして、TSK gel G3000SW(東ソー(株)製)を用い、ゲルカラムは(8.0mmID×30cm 東ソー、東京、日本)、10mMリン酸カリウム緩衝液(PH6.0)中の0.3M NaClと0.1%Triton−X100を含む溶液で平衡化されている。フラクション(125μl)を集めた。7つのタンパク質マーカーを本精製酵素の分子量を決定するために用いた。タンパク質マーカーとして、チログロブリン(Thyroglobulin):669kDa、フェリチン(Ferritin):440kDa、カタラーゼ(Catalase):232kDa、アルドラーゼ(Aldolase):158kDa、ウシ血清アルブミン(Bovine Serum Albumin):67kDa、オバルブミン(Ovalbumin):43kDa、キモトリプシノーゲン(ChymotrypsinogenA):25kDa、を用いた。
本酵素の分子量は約380kDaであることが、確認された。
Example 5
Gel filtration chromatography of this enzyme was performed. As the gel, TSK gel G3000SW (manufactured by Tosoh Corporation) was used, and the gel column was (8.0 mm ID × 30 cm Tosoh, Tokyo, Japan) and 0.3 M NaCl in 10 mM potassium phosphate buffer (PH 6.0) and 0 Equilibrated with a solution containing 1% Triton-X100. Fractions (125 μl) were collected. Seven protein markers were used to determine the molecular weight of the purified enzyme. As protein markers, thyroglobulin: 669 kDa, ferritin: 440 kDa, catalase: 232 kDa, aldolase: 158 kDa, bovine serum albumin (buvin Serum Albumin): 67 kDa Chymotrypsinogen A: 25 kDa was used.
It was confirmed that the molecular weight of this enzyme is about 380 kDa.
実施例6
精製した本酵素の至適温度を調べた。
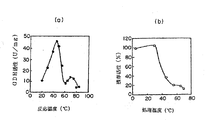
Tris−HCl緩衝液、pH8.0中で、あらかじめ1分間設定温度でインキュベーションした後、反応を開始した。所定の反応温度で活性を測定した。至適温度は45℃付近にみられた(図3(a)参照)。また、45℃付近に比べて活性は低いが、75℃付近にもピークがみられた。
また、本酵素の熱安定性を調べるため、各温度で30分間定温放置後、45℃で残存酵素活性を測定した(図3(b)参照)。
Example 6
The optimum temperature of the purified enzyme was examined.
The reaction was started after incubating in Tris-HCl buffer, pH 8.0 for 1 minute at a preset temperature. Activity was measured at a given reaction temperature. The optimum temperature was found around 45 ° C. (see FIG. 3A). Moreover, although activity was low compared with 45 degreeC vicinity, the peak was seen also at 75 degreeC vicinity.
In addition, in order to examine the thermal stability of the present enzyme, the remaining enzyme activity was measured at 45 ° C. after standing at each temperature for 30 minutes (see FIG. 3B).
実施例7
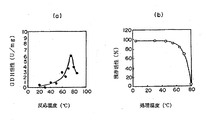
本酵素を70℃で30分間熱処理した場合の分子量60kDaの単一オリゴペプチドを構成する該ペプチド酵素の至適温度及び熱安定性を調べた。
このペプチド酵素は非熱処理酵素よりも高い至適温度を示し、さらに熱安定性も示した。このような温度依存性を示す酵素の報告は未だない。
Tris−HCl緩衝液、pH8.0中で、あらかじめ1分間設定温度でインキュベーションした後、反応を開始した。所定の反応温度で活性を測定した。至適温度は75℃付近にみられた(図4(a)参照)。
また、本酵素の熱安定性を調べるため各温度で30分間定温放置後、70℃で残存酵素活性を測定した(図4(b)参照)。
Example 7
The optimum temperature and thermal stability of the peptide enzyme constituting a single oligopeptide having a molecular weight of 60 kDa when this enzyme was heat-treated at 70 ° C. for 30 minutes were examined.
This peptide enzyme showed a higher optimum temperature than the non-heat-treated enzyme, and also showed thermal stability. There are no reports of enzymes exhibiting such temperature dependence.
The reaction was started after incubating in Tris-HCl buffer, pH 8.0 for 1 minute at a preset temperature. Activity was measured at a given reaction temperature. The optimum temperature was found around 75 ° C. (see FIG. 4A).
Moreover, in order to investigate the thermostability of this enzyme, after standing at each temperature for 30 minutes, the residual enzyme activity was measured at 70 degreeC (refer FIG.4 (b)).
実施例8
それぞれのサブユニットの役割を調査するために、熱処理前後のGDHの分光光度解析を行った。図5(a)(b)は、熱処理前後の(グルコースの存在下で)酸化型及び還元型のGDHの吸収を示している。もともとのGDHである熱処理前の酸化GDHの波長は、409nmに特徴的なピークを示し、また、グルコースの存在下でそれは417nmへと移行し、523nm及び550nmに2つのさらなるピークが見られた(図5a)。対照的に、熱処理後では409nmにおける特徴的なピークが見られなくなり(図5b)、酸化型及び還元型の間に重要な違いが見られなかった。
熱処理前の酸化型GDH、もともとのGDH、の吸収波長はGluconobacter sp.あるいはAcetobacter sp.のデヒドロゲナーゼチトクローム複合体でできているアルコールデヒドロゲナーゼおよびアルデヒドデヒドロゲナーゼの吸収波長と類似していた(以下の文献参照。Adachi,O.,Tayama,K.,Shinagawa,E.,Matsushita,K.and Ameyama,M.(1978)Agr.Biol.Chem.,42,2045−2056.;Adachi,O.,Miyagawa,E.,Matsushita,K.and Ameyama,M.(1978)Agr.Biol.Chem.,42,2331−2340;Ameyama,M.and Adachi,O.,(1982)Methods Enzymol.,89,450−457;Adachi,O.,Tayama,K.,Shinagawa,E.,Matsushita,K.and Ameyama,M.(1980)Agr.Biol.Chem.,44,503−515;Ameyama,M.and Adachi,O.(1982)Methods Enzymol.,89,491−497)。
結果として、本GDHのオリゴマー複合体はチトクロームを含んでいる可能性を示唆していた。従って、観察されたチトクロームC様の波長はβサブユニットに起因するもので、熱処理の間に失われたものと言える。ゆえにβサブユニットはチトクロームCからなっているといえる。
Example 8
In order to investigate the role of each subunit, spectrophotometric analysis of GDH before and after heat treatment was performed. FIGS. 5 (a) and 5 (b) show the absorption of oxidized and reduced GDH before and after heat treatment (in the presence of glucose). The wavelength of oxidized GDH before heat treatment, which is the original GDH, showed a characteristic peak at 409 nm, and in the presence of glucose it moved to 417 nm and two additional peaks were seen at 523 nm and 550 nm ( FIG. 5a). In contrast, no characteristic peak at 409 nm was observed after heat treatment (FIG. 5b), and no significant difference was observed between oxidized and reduced forms.
The absorption wavelength of the oxidized GDH before heat treatment and the original GDH is Gluconobacter sp. Alternatively, Acetobacter sp. The absorption wavelength was similar to that of alcohol dehydrogenase and aldehyde dehydrogenase made of the dehydrogenase cytochrome complex (see the following literature: Adachi, O., Tayama, K., Shinagawa, E., Matsushita, K. and Ameyama, M. (1978) Agr.Biol.Chem., 42, 2045-2056.; Adachi, O., Miyagawa, E., Matsushita, K. and Ameyama, M. (1978) Agr.Biol.Chem., 42, 2331-2340; Ameyama, M. and Adachi, O., (1982) Methods Enzymol., 89, 450-457; Adachi, O., Tayama, K., Shinag Awa, E., Matsushita, K. and Ameyama, M. (1980) Agr. Biol. Chem., 44, 503-515; Ameyama, M. and Adachi, O. (1982) Methods Enzymol., 89, 491-. 497).
As a result, it was suggested that the oligomer complex of the present GDH may contain cytochrome. Therefore, the observed cytochrome C-like wavelength is attributed to the β subunit and can be said to have been lost during the heat treatment. Therefore, it can be said that the β subunit is composed of cytochrome C.
実施例9
実施例4の電気泳動によって得られたβサブユニットを含むバンドを切り取り、ペプチドシークエンサー(島津製作所、PPSQ−10)によりアミノ酸配列を解析したところ、配列番号5に示すN末端の16残基のアミノ酸配列を得ることができた。
前記N末端の16残基のアミノ酸配列から同ペプチド配列をコードする遺伝子領域をPCRにより増幅することを試みた。すなわち、16残基のペプチド鎖のN末端5残基に相当するフォワード側の塩基配列(配列番号6)及び同C末端5残基のアンチセンス鎖に相当するリバース側の塩基配列(配列番号7)を持つ2つのPCRプライマーをデザインした。この1組のPCRプライマーを用い常法に従いKS1株のゲノムに対してPCRを行なったところ、約50bpの遺伝子断片が増幅された。これを常法に従いその塩基配列を決定したところ、上記PCRプライマー1組を含む塩基配列58塩基が解読された。このうち、PCRプライマーを除く18塩基について解析していたところ、前記βサブユニットN末端16残基のN末側から6残基目のProから11残基目のArgに相当する遺伝子配列(配列番号8)が見出され、本増幅遺伝子断片がβサブユニットの遺伝子断片を含むことが明らかとなった。
又、β−サブユニットは、α−サブユニットに続く22アミノ酸残基の後に存在することが分かった。これは、実施例4において決定した、精製されたβ−サブユニットのN末端におけるアミノ酸配列と、配列番号1中の塩基番号2452〜2466の塩基配列によって翻訳される5アミノ酸残基が一致することから、両者が同一であると判明したことに基づいている。
又、配列番号1中の塩基番号2386〜2451の塩基配列は、β−サブユニットのシグナルペプチドであることが推測される。この塩基配列によってコードされるアミノ酸配列は、配列番号4のアミノ酸配列のアミノ酸番号1〜22に相当する。
Example 9
The band containing the β subunit obtained by electrophoresis in Example 4 was cut out, and the amino acid sequence was analyzed by a peptide sequencer (Shimadzu Corporation, PPSQ-10). As a result, the N-terminal 16-residue amino acid shown in SEQ ID NO: 5 was obtained. The sequence could be obtained.
An attempt was made to amplify a gene region encoding the peptide sequence from the N-terminal 16-residue amino acid sequence by PCR. That is, the forward-side base sequence (SEQ ID NO: 6) corresponding to the N-
The β-subunit was found to be present after the 22 amino acid residues following the α-subunit. This is because the amino acid sequence at the N-terminus of the purified β-subunit determined in Example 4 matches the 5 amino acid residues translated by the nucleotide sequence of nucleotide numbers 2452 to 2466 in SEQ ID NO: 1. From this, it is based on the fact that they are found to be the same.
Moreover, it is estimated that the base sequence of base number 2386-2451 in
実施例10
0.1%Triton X−100及び1mMCaCl2を含む50mMリン酸カリウム緩衝液(pH7.5)中に本精製酵素及び市販のNAD補酵素GDH(NAD−GDHと略す)をそれぞれ100U/Lになるように加え混和した。この溶液を60℃の高温漕にいれ、残存活性を測定した。
Example 10
This purified enzyme and commercially available NAD coenzyme GDH (abbreviated as NAD-GDH) are each 100 U / L in 50 mM potassium phosphate buffer (pH 7.5) containing 0.1% Triton X-100 and 1 mM CaCl 2. And mixed. This solution was placed in a high-temperature bath at 60 ° C., and the residual activity was measured.
本酵素は現在市販されているGDH酵素に比べて、驚異的な熱安定性があることが確認できた。本酵素は市販のNAD−GDHとは全く別の新規な酵素であることが判明した。
It was confirmed that this enzyme has an amazing thermostability compared to the currently available GDH enzyme. This enzyme was found to be a completely new enzyme different from commercially available NAD-GDH.
実施例10 GDH αサブユニットをコードする遺伝子の単離
<1>ブルクホルデリア・セパシアKS1株からの染色体DNAの調製
ブルクホルデリア・セパシアKS1株より染色体遺伝子を常法に従って調製した。すなわち、同菌株をTL液体倍地(ポリペプトン 10g、酵母抽出液 1g、NaCl 5g、KH2PO4 2g、グルコース 5g;1L、pH7.2)を用いて、34℃で一晩振盪した。増殖した菌体を遠心分離機により回収した。この菌体を10mM NaCl、20mM Tris−HCl(pH8.0)、1mM EDTA、0.5% SDS、100μg/mlのプロテイナーゼKを含む溶液に懸濁し、50℃で6時間処理した。ここに等量のフェノール−クロロホルムを加えて室温で10分間撹拌した後、遠心分離機により上清を回収した。これに終濃度0.3Mになるように酢酸ナトリウムを加え、2倍量のエタノールを重層して中間層に染色体DNAを析出させた。これをガラス棒を用いてすくいとり、70%エタノールで洗浄した後、適当量のTEバッファーに溶解させ、染色体DNA溶液とした。
<2>GDH αサブユニットのN末端アミノ酸配列の決定
実施例2と同様にして精製したGDHを凍結乾燥によって濃縮後、12.5%ポリアクリルアミドを用いたSDS−電気泳動法を用いて展開し、αサブユニットを分離した。こうして得られたαサブユニットをポリビニリデンフルオリド膜に転写した後、アミノ酸シークエンサー(島津製作所製、PPSQ−10)によりN末端アミノ酸配列の決定を行った。その結果、本酵素には配列番号3のアミノ酸配列においてアミノ酸番号2〜12からなる11残基から構成されるペプチド配列を含むことが明らかとなった。
<3>αサブユニットをコードする遺伝子のクローニング
<1>で調製したDNA1μgを制限酵素Sau3AIで限定分解した。これをCIAP(仔ウシ小腸由来アルカリホスファターゼ)処理した。一方、コスミドであるSuperCosI(ストラジーン社から入手)をBamHI処理し、T4 DNAリガーゼにより、SuperCosIにα−15株由来の染色体DNA断片をSau3AIで限定分解して得られたDNA断片を組み込んだ。得られた組換えDNAでエシェリヒア・コリXL−1 Blue MR(ストラジーン社から入手)を形質転換した。形質転換体はSuperCosI上の抗生物質耐性であるネオマイシン耐性およびアンピシリン耐性にしたがって10μg/mlのネオマイシンおよび25μg/mlのアンピシリンを含むLB寒天培地から選抜した。得られた形質転換体をLB液体培地で培養した。これらの形質転換菌体を集菌後、GDH活性測定試薬に懸濁し、グルコースに対する脱水素酵素活性を指標にクローンを選抜した。その結果、1株のグルコース脱水素酵素活性を示すクローンが得られた。
<4>サブクローニング
<3>で得られたαサブユニットをコードする遺伝子を含むコスミドSuperCosIから、目的遺伝子を含むDNA断片を調製した。同コスミドから挿入遺伝子断片を制限酵素NotIにより切り出した。このDNA断片を制限酵素XbaIで処理し、それらの断片をXbaIで消化したプラスミドpUC18に組み込んだ。各挿入断片を含むプラスミドpUC18でエシェリヒア・コリDH5αMCR株を形質転換し、アンピシリン50μg/mlを含むLB寒天培地で生じるコロニーを採取した。得られた形質転換体を液体のLB培地で培養し、それぞれの細胞のGDH活性を<3>と同様に調べた。その結果、一つの形質転換体にGDH活性を示す株が得られた。この形質転換体からプラスミドを抽出し、その挿入DNA断片を解析したところ、約8.8kbpの挿入断片が確認された。本プラスミドをpKS1と命名した。
<5>塩基配列の決定
pKS1の挿入DNA断片について、制限酵素解析及び常法に従い塩基配列を決定した。その結果、本挿入DNA断片中に、<2>で明かとなったαサブユニットのN末端アミノ酸配列をコードするDNA配列が確認され、この配列を含むオープンリーディングフレームが見つかった。決定した塩基配列および同塩基配列がコードし得るアミノ酸配列は、配列番号1および3に示す通りである。アミノ酸配列から求められるタンパク質の分子量は59,831Daであり、ブルクホルデリア・セパシアKS1株αサブユニットのSDS−PAGEでもとめられた分子量60kDaにほぼ一致した。
αサブユニットの塩基配列が決定されたことにより、前記αサブユニットの構造遺伝子を用いてベクターを作製し、更に前記ベクターにより形質転換体の製造を行った。
先ずベクターに挿入する遺伝子を以下のように調製した。
KS1株由来のゲノム断片をテンプレートとして、所望の制限酵素部位を含むように、PCR反応により増幅した。PCR反応には次の1組のオリゴヌクレオチドプライマーを用いた。
(フォワード)
5’−cccaagcttg ggccgatacc gatacgca−3’(配列番号9)
(リバース)
5’− gagaagcttt ccgcacggtc agacttcc−3’(配列番号10)
PCRにより増幅された遺伝子を制限酵素HindIIIで消化した後、発現ベクターpFLAG−CTS(SIGMA社)のクローニング部位であるHindIII部位に挿入した。得られたプラスミドをpFLAG−CTS/αと命名した。
前記プラスミドpFLAG−CTS/αでエッシェリヒア・コリDH5αMCR株を形質転換し、アンピシリン50μg/mlを含むLB寒天培地で生じるコロニーを採取した。
さらにpKS1挿入断片について、αサブユニットの上流に関してオープンリーディングフレームを検索したところ、新たに配列番号2に記載される168アミノ酸残基から構成されるポリペプチドをコードする507塩基から構成される構造遺伝子(配列番号1中塩基番号258〜761)が見出された。この構造遺伝子は、γサブユニットをコードしていると考えられた。
αサブユニットのコード領域の上流に、γサブユニットをコードする領域の存在が明らかになったことから、γサブユニットとαサブユニットが連続するポリシストロン構造の遺伝子を含む組換えベクターを作製し、同ベクターを導入した形質転換体を構築した。
先ずベクターに挿入する遺伝子を以下のように調製した。
γサブユニットの構造遺伝子およびαサブユニットの構造遺伝子が連続するKS1株由来のゲノム断片をテンプレートとして、所望の制限酵素部位を含むように、PCR反応により増幅した。PCR反応には次の1組のオリゴヌクレオチドプライマーを用いた。
(フォワード)
5’− catgccatgg cacacaacga caacact−3’(配列番号11)
(リバース)
5’− cccaagcttg ggtcagactt ccttcttcag c−3’(配列番号12)
このPCRにより増幅された遺伝子の5’末端をNcoI、3’末端をHindIIIで消化した後、ベクターpTrc99A(Pharmacia社)のクローニング部位である、NcoI/HindIIIに挿入した。得られたプラスミドをpTrc99A/γ+αと命名した。
前記プラスミドpTrc99A/γ+αにより、エシェリヒア・コリDH5αMCR株を形質転換し、アンピシリン50μg/mlを含むLB寒天培地で生じるコロニーを採取した。
Example 10 Isolation of gene encoding GDH α subunit <1> Preparation of chromosomal DNA from Burkholderia cepacia KS1 strain A chromosomal gene was prepared from Burkholderia cepacia KS1 strain according to a conventional method. That is, the strain was shaken overnight at 34 ° C. using TL liquid medium (polypeptone 10 g, yeast extract 1 g, NaCl 5 g, KH 2 PO 4 2 g, glucose 5 g; 1 L, pH 7.2). Proliferated cells were collected by a centrifuge. The cells were suspended in a solution containing 10 mM NaCl, 20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.5% SDS, 100 μg / ml proteinase K, and treated at 50 ° C. for 6 hours. After adding an equal amount of phenol-chloroform and stirring at room temperature for 10 minutes, the supernatant was recovered by a centrifuge. To this, sodium acetate was added to a final concentration of 0.3 M, and twice the amount of ethanol was overlaid to precipitate chromosomal DNA in the intermediate layer. This was scooped using a glass rod, washed with 70% ethanol, and then dissolved in an appropriate amount of TE buffer to obtain a chromosomal DNA solution.
<2> Determination of N-terminal amino acid sequence of GDH α subunit GDH purified in the same manner as in Example 2 was concentrated by freeze-drying and developed using SDS-electrophoresis using 12.5% polyacrylamide. The α subunit was separated. The α subunit thus obtained was transferred to a polyvinylidene fluoride membrane, and then the N-terminal amino acid sequence was determined with an amino acid sequencer (manufactured by Shimadzu Corporation, PPSQ-10). As a result, it was revealed that this enzyme contains a peptide sequence composed of 11 residues consisting of
<3> Cloning of Gene Encoding
<4> Subcloning A DNA fragment containing the target gene was prepared from the cosmid SuperCosI containing the gene encoding the α subunit obtained in <3>. The inserted gene fragment was excised from the cosmid with the restriction enzyme NotI. This DNA fragment was treated with the restriction enzyme XbaI, and these fragments were incorporated into plasmid pUC18 digested with XbaI. Escherichia coli DH5αMCR strain was transformed with plasmid pUC18 containing each inserted fragment, and colonies produced on an LB agar medium containing 50 μg / ml of ampicillin were collected. The obtained transformant was cultured in a liquid LB medium, and the GDH activity of each cell was examined in the same manner as in <3>. As a result, a strain showing GDH activity in one transformant was obtained. When a plasmid was extracted from this transformant and the inserted DNA fragment was analyzed, an inserted fragment of about 8.8 kbp was confirmed. This plasmid was designated as pKS1.
<5> Determination of base sequence The base sequence of the inserted DNA fragment of pKS1 was determined according to restriction enzyme analysis and conventional methods. As a result, a DNA sequence encoding the N-terminal amino acid sequence of the α subunit revealed in <2> was confirmed in this inserted DNA fragment, and an open reading frame containing this sequence was found. The determined nucleotide sequence and the amino acid sequence that can be encoded by the nucleotide sequence are as shown in SEQ ID NOs: 1 and 3. The molecular weight of the protein determined from the amino acid sequence was 59,831 Da, which almost coincided with the molecular weight of 60 kDa determined by SDS-PAGE of the Burkholderia cepacia KS1 strain α subunit.
When the base sequence of the α subunit was determined, a vector was prepared using the structural gene of the α subunit, and a transformant was further produced using the vector.
First, a gene to be inserted into a vector was prepared as follows.
Using a genomic fragment derived from the KS1 strain as a template, amplification was performed by PCR reaction so as to include a desired restriction enzyme site. The following set of oligonucleotide primers was used in the PCR reaction.
(forward)
5'-cccaagcttg ggccgatacc gatacgca-3 '(SEQ ID NO: 9)
(reverse)
5′-gagaagcttt ccgcacggtc agacttcc-3 ′ (SEQ ID NO: 10)
The gene amplified by PCR was digested with the restriction enzyme HindIII, and then inserted into the HindIII site which is the cloning site of the expression vector pFLAG-CTS (SIGMA). The resulting plasmid was named pFLAG-CTS / α.
Escherichia coli DH5α MCR strain was transformed with the plasmid pFLAG-CTS / α, and colonies produced on an LB agar medium containing 50 μg / ml of ampicillin were collected.
Further, when the open reading frame was searched for the upstream of the α subunit for the pKS1 insert fragment, a structural gene composed of 507 bases newly encoding a polypeptide composed of 168 amino acid residues described in SEQ ID NO: 2 (Base numbers 258 to 761 in SEQ ID NO: 1) were found. This structural gene was thought to encode the γ subunit.
Since the existence of a region encoding the γ subunit was found upstream of the coding region of the α subunit, a recombinant vector containing a gene having a polycistron structure in which the γ subunit and the α subunit are continuous was prepared. A transformant into which the vector was introduced was constructed.
First, a gene to be inserted into a vector was prepared as follows.
A genomic fragment derived from the KS1 strain in which the structural gene of the γ subunit and the structural gene of the α subunit are continuous was used as a template to amplify by PCR reaction so as to include a desired restriction enzyme site. The following set of oligonucleotide primers was used in the PCR reaction.
(forward)
5′-catgccatgg cacacaacga caacact-3 ′ (SEQ ID NO: 11)
(reverse)
5'- cccaagcttg ggtcagactt ccttcttcag c-3 '(SEQ ID NO: 12)
The 5 ′ end of the gene amplified by this PCR was digested with NcoI and 3 ′ end with HindIII, and then inserted into NcoI / HindIII, which is the cloning site of vector pTrc99A (Pharmacia). The resulting plasmid was named pTrc99A / γ + α.
Escherichia coli DH5αMCR strain was transformed with the plasmid pTrc99A / γ + α, and colonies produced on an LB agar medium containing 50 μg / ml of ampicillin were collected.
実施例11 組換え大腸菌によるGDH αサブユニットの生産
前記pKS1、pFLAG−CTS/α、pTrc99A/γ+αのそれぞりのプラスミドによって形質転換した大腸菌エシェリヒア・コリDH5αMCR株を用いてαサブユニットの生産を行った。各形質転換体をアンピシリン50μg/mlを含むLB培地3mlに植菌し、37℃で12時間培養を行い、遠心分離機により細胞を集菌した。この細胞をフレンチプレス(1500kgf)で破砕した後、超遠心(4℃、160,400×g、90分)により膜画分(10mMリン酸カリウム緩衝液pH6.0)を分離した。
Example 11 Production of GDH α subunit by recombinant E. coli α subunit using Escherichia coli DH5α MCR strain transformed with the respective plasmids of pKS1, pFLAG-CTS / α and pTrc99A / γ + α Unit production. Each transformant was inoculated into 3 ml of LB medium containing 50 μg / ml of ampicillin, cultured at 37 ° C. for 12 hours, and the cells were collected by a centrifuge. The cells were disrupted with a French press (1500 kgf), and then the membrane fraction (10 mM potassium phosphate buffer pH 6.0) was separated by ultracentrifugation (4 ° C., 160,400 × g, 90 minutes).
実施例12 グルコースのアッセイ
先ず前記各膜分画を用いてGDH活性の確認を行った。具体的には594μMのメチルフェナジンメトサルフェート(mPMS)および5.94μMの2,6−ジクロロフェノールインドフェノール(DCIP)を含む10mMリン酸カリウム緩衝液(pH7.0)により、目視判定を行った。結果は以下のとおりである。+の数は、青色から無色への変化の程度を表す。
pFLAG−CTS/αによる形質転換体培養膜分画 +
pKS1による形質転換体培養膜分画 ++
pTrc99A/γ+αによる形質転換体培養膜分画 +++
αサブユニットのみを組みこんだpFLAG−CTS/αによる形質転換体培養膜分画のGDH活性が最も低く、効率良くベクターを構築したpTrc99A/γ+αによる形質転換体培養膜分画が最も高いGDH活性を示した。
αサブユニットの構造遺伝子のみによるベクターを用いた形質転換体でもαサブユニットは発現されるが、更にγサブユニットの構造遺伝子をαサブユニットの構造遺伝子と合わせたベクターを用いることにより、効率良くαサブユニットを得ることができた。
本発明のグルコース脱水素酵素を用いてグルコースをアッセイした。本発明のグルコース脱水素酵素(αサブユニット)を、各種濃度のグルコースで酵素活性を測定した。GDH活性の測定は594μMのメチルフェナジンメトサルフェート(mPMS)および5.94μMの2,6−ジクロロフェノールインドフェノール(DCIP)を含む10mMリン酸カリウム緩衝液(pH7.0)の中で行った。酵素試料および基質としてグルコースを基質として加え37℃でインキュベートした時のDCIPの600nmの吸光度変化を分光光度計を用いて追跡し、その吸光度の減少速度を酵素反応速度とした。本発明のGDHを用いて、0.01〜1.0mMの範囲でグルコースの定量を行うことができた。
Example 12 Glucose assay First, GDH activity was confirmed using each of the membrane fractions. Specifically, visual determination was performed using a 10 mM potassium phosphate buffer (pH 7.0) containing 594 μM methylphenazine methosulfate (mPMS) and 5.94 μM 2,6-dichlorophenolindophenol (DCIP). The results are as follows. The number of + represents the degree of change from blue to colorless.
Transformant culture membrane fractionation with pFLAG-CTS / α +
Transformant culture membrane fraction with pKS1 ++
Transformant culture membrane fractionation with pTrc99A / γ + α ++
GDH activity of the transformant culture membrane fraction with pFLAG-CTS / α incorporating only the α subunit is the lowest, and the GDH activity of the transformant culture membrane fraction with pTrc99A / γ + α that efficiently constructs the vector is the highest. showed that.
Although the α subunit is expressed even in a transformant using a vector containing only the structural gene of the α subunit, it is more efficient by using a vector in which the structural gene of the γ subunit is combined with the structural gene of the α subunit. The α subunit could be obtained.
Glucose was assayed using the glucose dehydrogenase of the present invention. The enzyme activity of the glucose dehydrogenase (α subunit) of the present invention was measured at various concentrations of glucose. The measurement of GDH activity was performed in 10 mM potassium phosphate buffer (pH 7.0) containing 594 μM methylphenazine methosulfate (mPMS) and 5.94 μM 2,6-dichlorophenolindophenol (DCIP). The absorbance change at 600 nm of DCIP when glucose was added as a substrate and incubated at 37 ° C. as an enzyme sample and substrate was traced using a spectrophotometer, and the rate of decrease in the absorbance was taken as the enzyme reaction rate. Using the GDH of the present invention, glucose could be quantified in the range of 0.01 to 1.0 mM.
実施例13 グルコースセンサーの作製および評価
実施例2で得られた本発明のグルコース脱水素酵素(25単位)にカーボンペースト20mgを加えて凍結乾燥させた。これをよく混合した後、既にカーボンペーストが約40mg充填されたカーボンペースト電極の表面だけに充填し、濾紙上で研磨した。この電極を1%のグルタルアルデヒドを含む10mM MOPS緩衝液(pH7.0)中で室温で30分間処理した後、20mMリジンを含む10mM MOPS緩衝液(pH7.0)中で室温で20分間処理してグルタルアルデヒドをブロッキングした。この電極を10mM MOPS緩衝液(pH7.0)中で室温で1時間以上平衡化させた。電極は4℃で保存した。
前記電極を作用極として、参照極にAg/AgCl、対極にPt電極を用い、グルコース添加による応答電流値を測定した。反応溶液は1mMメトキシPMSを含む10mMリン酸カリウム緩衝液とし、電位100mVを印加し測定を行った。
作製した酵素センサーを用いてグルコースの濃度の測定を行った。本発明のグルコース脱水素酵素を固定化した酵素センサーを用いて、0.05mM〜5.0mMの範囲でグルコースの定量を行うことができた(図6)。
Example 13 Production and evaluation of
Using the electrode as a working electrode, Ag / AgCl as a reference electrode and a Pt electrode as a counter electrode, the response current value due to the addition of glucose was measured. The reaction solution was a 10 mM potassium phosphate buffer solution containing 1 mM methoxy PMS, and measurement was performed by applying a potential of 100 mV.
The concentration of glucose was measured using the prepared enzyme sensor. Using the enzyme sensor to which the glucose dehydrogenase of the present invention was immobilized, glucose could be quantified in the range of 0.05 mM to 5.0 mM (FIG. 6).
実施例14 形質転換体から得られるGDHによるグルコースセンサの作製及び評価
実施例12によって得られた本願発明のαサブユニット(249U/mgタンパク質)10Uをカーボンペースト50mgを加えて凍結乾燥させた。これをよく混合した後、既にカーボンペーストが約40mg充填されたカーボンペースト電極の表面だけに充填し、濾紙上で研磨した。この電極を1%のグルタルアルデヒドを含む10mM MOPS緩衝液(pH7.0)中で室温で30分間処理した後、20mMリジンを含む10mM MOPS緩衝液(pH7.0)中で室温で20分間処理してグルタルアルデヒドをブロッキングした。この電極を10mM MOPS緩衝液(pH7.0)中で室温で1時間以上平衡化させた。電極は4℃で保存した。
前記電極を作用極として、参照極にAg/AgCl、対極にPt電極を用い、グルコース添加による応答電流値を測定した。反応溶液は1mMメトキシPMSを含む10mMリン酸カリウム緩衝液とし、電位100mVを印加しながら各濃度のグルコース水溶液を25℃及び40℃で測定を行った。
作製した酵素センサーを用いてグルコースの濃度の測定を行ったところ各濃度に応じた電流が得られたことを確認した。
Example 14 Production and evaluation of glucose sensor by GDH obtained from transformant 10 U of α subunit (249 U / mg protein) of the present invention obtained in Example 12 was added with 50 mg of carbon paste and lyophilized. I let you. After this was mixed well, it was filled only on the surface of the carbon paste electrode already filled with about 40 mg of carbon paste, and was polished on a filter paper. This electrode was treated in 10 mM MOPS buffer (pH 7.0) containing 1% glutaraldehyde for 30 minutes at room temperature, and then treated in 10 mM MOPS buffer (pH 7.0) containing 20 mM lysine for 20 minutes at room temperature. To block glutaraldehyde. This electrode was equilibrated in a 10 mM MOPS buffer (pH 7.0) at room temperature for 1 hour or longer. The electrode was stored at 4 ° C.
Using the electrode as a working electrode, Ag / AgCl as a reference electrode and a Pt electrode as a counter electrode, the response current value due to the addition of glucose was measured. The reaction solution was a 10 mM potassium phosphate buffer containing 1 mM methoxy PMS, and glucose aqueous solutions of various concentrations were measured at 25 ° C. and 40 ° C. while applying a potential of 100 mV.
When the concentration of glucose was measured using the prepared enzyme sensor, it was confirmed that a current corresponding to each concentration was obtained.
本発明により、基質特異性が高く、安価に生産でき、測定サンプルの溶存酸素の影響を受けない酵素であって、特に熱安定性に優れた新規なグルコース脱水素酵素、及び該酵素の製造方法が提供できた。またこの酵素を生産するブルクホルデリア・セパシアの新規菌株が得られた。本酵素及び菌株を含む酵素電極を用いれば、グルコース測定に有効なグルコースセンサも提供できる。
また、本発明によりグルコース脱水素酵素の遺伝子、及び、同遺伝子を効率良く発現させることのできるペプチド及びそのペプチドをコードするDNAが判明したため、前記遺伝子をもとにGDHを組み換えDNA技術で大量に調製することができる。
INDUSTRIAL APPLICABILITY According to the present invention, a novel glucose dehydrogenase that has high substrate specificity, can be produced at low cost, and is not affected by dissolved oxygen in a measurement sample, and particularly excellent in thermal stability, and a method for producing the enzyme Was able to provide. A new strain of Burkholderia cepacia producing this enzyme was also obtained. If an enzyme electrode containing the present enzyme and strain is used, a glucose sensor effective for glucose measurement can also be provided.
In addition, since the present invention has revealed a glucose dehydrogenase gene, a peptide capable of efficiently expressing the gene, and a DNA encoding the peptide, GDH can be produced in a large amount by recombinant DNA technology based on the gene. Can be prepared.
Claims (13)
に培養し、同培地又は/及び前記微生物菌体からグルコース脱水素酵素を採取することを
特徴とするグルコース脱水素酵素の製造方法であって、該グルコース脱水素酵素が、還元
条件下でのSDS−ポリアクリルアミドゲル電気泳動において、分子量60kDaを示す
αサブユニットと分子量43kDaを示すβサブユニットからなり、該αサブユニットが
、
(A)配列番号3のアミノ酸配列を有するタンパク質、あるいは
(B)配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿
入、又は付加されたアミノ酸配列を有し、かつ、グルコース脱水素酵素活性を有するタン
パク質
であって、該βサブユニットが、
N末端が配列番号5のアミノ酸配列を有する電子伝達タンパク質である、グルコース脱水
素酵素の製造方法。 Glucose dehydrogenase characterized by culturing a microorganism having the ability to produce glucose dehydrogenase in a medium, and collecting glucose dehydrogenase from the same medium or / and the microorganism cells. The glucose dehydrogenase comprises an α subunit having a molecular weight of 60 kDa and a β subunit having a molecular weight of 43 kDa in SDS-polyacrylamide gel electrophoresis under reducing conditions, and the α subunit But,
(A) a protein having the amino acid sequence of SEQ ID NO: 3, or (B) having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted, or added in the amino acid sequence of SEQ ID NO: 3, And a protein having glucose dehydrogenase activity, wherein the β subunit is
A method for producing glucose dehydrogenase, wherein the N-terminus is an electron transfer protein having the amino acid sequence of SEQ ID NO: 5.
製造方法。 The method for producing glucose dehydrogenase according to claim 1, wherein the microorganism is Burkholderia cepacia.
グルコース脱水素酵素の製造方法。
(1)作用:
グルコースの脱水素反応を触媒する。
(2)TSK gel G3000SW(東ソー(株)製)を用いたゲル濾過クロマトグ
ラフィーにおいて、分子量380kDaを示す。
(3)至適反応温度:
45℃(Tris−HCl緩衝液、pH8.0)。 The method for producing glucose dehydrogenase according to claim 1 or 2, wherein the glucose dehydrogenase has the following properties.
(1) Action:
Catalyses the dehydrogenation of glucose.
(2) Gel filtration chromatography using TSK gel G3000SW (manufactured by Tosoh Corporation) shows a molecular weight of 380 kDa.
(3) Optimal reaction temperature:
45 ° C. (Tris-HCl buffer, pH 8.0).
1項に記載のグルコース脱水素酵素の製造方法。 The method for producing glucose dehydrogenase according to any one of claims 1 to 3, wherein the electron transfer protein is cytochrome C.
、該グルコース脱水素酵素が、還元条件下でのSDS−ポリアクリルアミドゲル電気泳動
において、分子量60kDaを示すαサブユニットと分子量43kDaを示すβサブユニ
ットからなり、該αサブユニットが、
(A)配列番号3のアミノ酸配列を有するタンパク質、あるいは
(B)配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿
入、又は付加されたアミノ酸配列を有し、かつ、グルコース脱水素酵素活性を有するタン
パク質
であって、該βサブユニットが、
N末端が配列番号5のアミノ酸配列を有する電子伝達タンパク質である、グルコース脱水
素酵素。 A glucose dehydrogenase that can be produced by a microorganism belonging to the genus Burkholderia, which glucose dehydrogenase has an α subunit having a molecular weight of 60 kDa and a molecular weight of 43 kDa in SDS-polyacrylamide gel electrophoresis under reducing conditions. Β subunits, and the α subunit is
(A) a protein having the amino acid sequence of SEQ ID NO: 3, or (B) having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted, or added in the amino acid sequence of SEQ ID NO: 3, And a protein having glucose dehydrogenase activity, wherein the β subunit is
A glucose dehydrogenase, which is an electron transfer protein having an amino acid sequence of SEQ ID NO: 5 at the N-terminus.
グルコース脱水素酵素。
(1)作用:
グルコースの脱水素反応を触媒する。
(2)TSK gel G3000SW(東ソー(株)製)を用いたゲル濾過クロマトグ
ラフィーにおいて、分子量380kDaを示す。
(3)至適反応温度:
45℃(Tris−HCl緩衝液、pH8.0)。 The glucose dehydrogenase according to claim 5 or 6, wherein the glucose dehydrogenase has the following properties.
(1) Action:
Catalyses the dehydrogenation of glucose.
(2) Gel filtration chromatography using TSK gel G3000SW (manufactured by Tosoh Corporation) shows a molecular weight of 380 kDa.
(3) Optimal reaction temperature:
45 ° C. (Tris-HCl buffer, pH 8.0).
1項に記載のグルコース脱水素酵素。 The glucose dehydrogenase according to any one of claims 5 to 7, wherein the electron transfer protein is cytochrome C.
1項に記載のグルコース脱水素酵素。 The glucose dehydrogenase according to any one of claims 5 to 8, which has activity peaks at 45 ° C and 75 ° C, respectively.
ロムC。 A cytochrome C, which is the β subunit of glucose dehydrogenase according to claim 8.
(1)サブユニットとして請求項5記載のグルコース脱水素酵素を構成し得る。
(2)グルコース脱水素酵素活性を有する。
(3)還元条件下でのSDS−ポリアクリルアミドゲル電気泳動において、分子量60k
Daを示す。
(4)至適反応温度:
75℃(Tris−HCl緩衝液、pH8.0)
を有し、
(A)配列番号3のアミノ酸配列を有するタンパク質、あるいは
(B)配列番号3のアミノ酸配列において、1又は複数のアミノ酸残基が置換、欠失、挿
入、又は付加されたアミノ酸配列を有し、かつ、グルコース脱水素酵素活性を有するタン
パク質
である、タンパク質。 The following properties:
(1) The glucose dehydrogenase according to claim 5 can be constituted as a subunit.
(2) It has glucose dehydrogenase activity.
(3) In SDS-polyacrylamide gel electrophoresis under reducing conditions, the molecular weight is 60 k.
Da is shown.
(4) Optimal reaction temperature:
75 ° C. (Tris-HCl buffer, pH 8.0)
Have
(A) a protein having the amino acid sequence of SEQ ID NO: 3, or (B) having an amino acid sequence in which one or more amino acid residues are substituted, deleted, inserted, or added in the amino acid sequence of SEQ ID NO: 3, And protein which is protein which has glucose dehydrogenase activity.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007038490A JP4195065B2 (en) | 2000-10-31 | 2007-02-19 | Novel glucose dehydrogenase and method for producing the dehydrogenase |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000332085 | 2000-10-31 | ||
| JP2000357102 | 2000-11-24 | ||
| JP2001276832 | 2001-09-12 | ||
| JP2007038490A JP4195065B2 (en) | 2000-10-31 | 2007-02-19 | Novel glucose dehydrogenase and method for producing the dehydrogenase |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002539524A Division JP4107386B2 (en) | 2000-10-31 | 2001-10-31 | Novel glucose dehydrogenase and method for producing the dehydrogenase |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007125047A JP2007125047A (en) | 2007-05-24 |
| JP4195065B2 true JP4195065B2 (en) | 2008-12-10 |
Family
ID=38148228
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007038490A Expired - Fee Related JP4195065B2 (en) | 2000-10-31 | 2007-02-19 | Novel glucose dehydrogenase and method for producing the dehydrogenase |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4195065B2 (en) |
-
2007
- 2007-02-19 JP JP2007038490A patent/JP4195065B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007125047A (en) | 2007-05-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4107386B2 (en) | Novel glucose dehydrogenase and method for producing the dehydrogenase | |
| US7329519B2 (en) | Glucose dehydrogenase β subunit and DNA encoding the same | |
| US5137821A (en) | Gene and process of making glucose-6-phosphate dehydrogenase | |
| JP4195065B2 (en) | Novel glucose dehydrogenase and method for producing the dehydrogenase | |
| JP4036667B2 (en) | Novel glucose dehydrogenase and gene encoding the same | |
| US7713718B1 (en) | Process for producing glucose dehydrogenases | |
| US4960877A (en) | DNA having genetic information of L-α-glycerophosphate oxidase and application thereof | |
| ZA200303208B (en) | Novel glucose dehydrogenase and process for producing the dehydrogenase. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070219 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20070904 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071105 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080311 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080430 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080909 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080924 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4195065 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20111003 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121003 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131003 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |