JP2022017604A - Morphinan derivative - Google Patents
Morphinan derivative Download PDFInfo
- Publication number
- JP2022017604A JP2022017604A JP2018198525A JP2018198525A JP2022017604A JP 2022017604 A JP2022017604 A JP 2022017604A JP 2018198525 A JP2018198525 A JP 2018198525A JP 2018198525 A JP2018198525 A JP 2018198525A JP 2022017604 A JP2022017604 A JP 2022017604A
- Authority
- JP
- Japan
- Prior art keywords
- group
- substituent
- compound
- nmr
- ppm
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Abstract
Description
本発明は、オピオイドκ受容体アゴニスト作用を有するモルヒナン誘導体に関する。 The present invention relates to a morphinan derivative having an opioid κ receptor agonistic action.
オピオイド受容体にはμ、δ、κの3つのタイプが知られている。μ受容体に対して強い親和性を示すモルヒネは、古くから鎮痛薬として使用されている。しかし、オピオイドμ受容体アゴニストは、μ受容体を介して依存形成、呼吸抑制等の有害事象を引き起こすことが知られている。
一方κ受容体アゴニストも鎮痛作用を示すが、モルヒネで見られる有害事象には関与しないことが知られている。
その一方、κ受容体アゴニストは一般に鎮静作用や薬物嫌悪作用を示すことが知られている。唯一、薬物嫌悪性を分離したκ受容体アゴニストとしてナルフラフィン(特許文献1)があげられるが、ナルフラフィンは鎮痛用量において鎮静作用を示すため、止痒薬としての承認は得られたが、鎮痛薬としての承認は受けていない。つまり、鎮痛薬として承認を受けたオピオイドκ受容体選択的なアゴニストはいまだ存在しない。
従って、鎮静作用や薬物嫌悪作用を示さないκ受容体選択的なアゴニストは、鎮痛薬をはじめとするオピオイドκ受容体に関連する疾患や症状の優れた治療又は改善、予防薬として期待される。
特許文献2には、次式(A)、
Three types of opioid receptors are known: μ, δ, and κ. Morphine, which has a strong affinity for μ receptors, has long been used as an analgesic. However, opioid μ receptor agonists are known to cause adverse events such as dependence formation and respiratory depression via the μ receptor.
On the other hand, κ receptor agonists also have analgesic effects, but are known not to be involved in the adverse events seen in morphine.
On the other hand, κ receptor agonists are generally known to exhibit sedative and drug aversive effects. Nalfurafine (Patent Document 1) is the only κ receptor agonist that separates drug aversion.Since nalfurafine has a sedative effect at an analgesic dose, it has been approved as an antipruritic drug, but as an analgesic drug. Has not been approved. That is, there is still no opioid κ receptor-selective agonist approved as an analgesic.
Therefore, κ receptor-selective agonists that do not show sedative or drug aversive effects are expected to be excellent treatments, ameliorations, or preventive agents for diseases and symptoms related to opioid κ receptors such as analgesics.
In Patent Document 2, the following equation (A),

で表される化合物がオピオイドκ受容体アゴニスト活性を有することが開示されている。しかし、その活性は充分ではなかった。
また、特許文献3には、次式(B)、
It is disclosed that the compound represented by is having opioid κ receptor agonist activity. However, its activity was not sufficient.
Further, in Patent Document 3, the following equation (B),
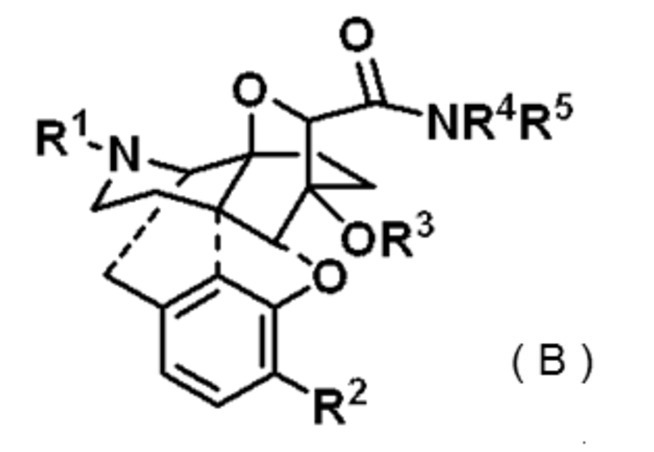
で表わされる化合物が報告されている。この化合物はオピオイドκ受容体選択的な結合及び鎮痛作用を有するとの記載がある。しかし、その鎮痛活性は満足のいくものではなかった。
また、非特許文献1には、次式で表される化合物(C)が、
Compounds represented by are reported. It is stated that this compound has opioid κ receptor selective binding and analgesic activity. However, its analgesic activity was unsatisfactory.
Further, in Non-Patent Document 1, the compound (C) represented by the following formula is described.

極めて高い活性を有することが開示されているが、この化合物は生体内での安定性が十分ではなかった。生体内で化合物が不安定な場合、期待される薬効が発揮されないこと、分解物の生体への影響等の理由により医薬品としての開発は困難となることから、生体内安定性は医薬品の開発において重要な要件となる。
上市されている鎮痛薬のうち高いオピオイドκ受容体アゴニスト活性を有する薬剤はいまだ存在しない。
従って、鎮痛薬を志向する上で、オピオイドκ受容体に対し選択性及び高い活性を示し、かつ生体内での安定性に優れた化合物が望まれている。
Although disclosed to have extremely high activity, this compound was not sufficiently stable in vivo. When a compound is unstable in a living body, it is difficult to develop it as a drug due to reasons such as the expected medicinal effect not being exhibited and the influence of decomposition products on the living body. It is an important requirement.
None of the analgesics on the market yet have high opioid κ receptor agonist activity.
Therefore, a compound that exhibits selectivity and high activity for opioid κ receptors and is excellent in in vivo stability is desired in order to aim for analgesics.
本発明の目的は、鎮静作用や薬物嫌悪作用の抑制された、オピオイドκ受容体に関連する様々な疾患、症状の治療または又は改善、予防に有効な医薬を提供することにある。 An object of the present invention is to provide an agent effective for treating, ameliorating, or preventing various diseases and symptoms related to opioid kappa receptor, which have suppressed sedation and drug aversive effects.
斯かる実情の下、本発明者らは鋭意検討を行った結果、特定のモルヒナン誘導体がオピオイドκ受容体選択性及びオピオイドκ受容体に対する強力なアゴニスト活性を有し、かつ生体内で高い安定性を有することを見出し、本発明を完成するに至った。
[1]即ち、本発明は、次の一般式(I)、
Under such circumstances, as a result of diligent studies by the present inventors, a specific morphinan derivative has opioid κ receptor selectivity and strong agonist activity against opioid κ receptor, and is highly stable in vivo. It was found that the present invention was completed.
[1] That is, the present invention has the following general formula (I),
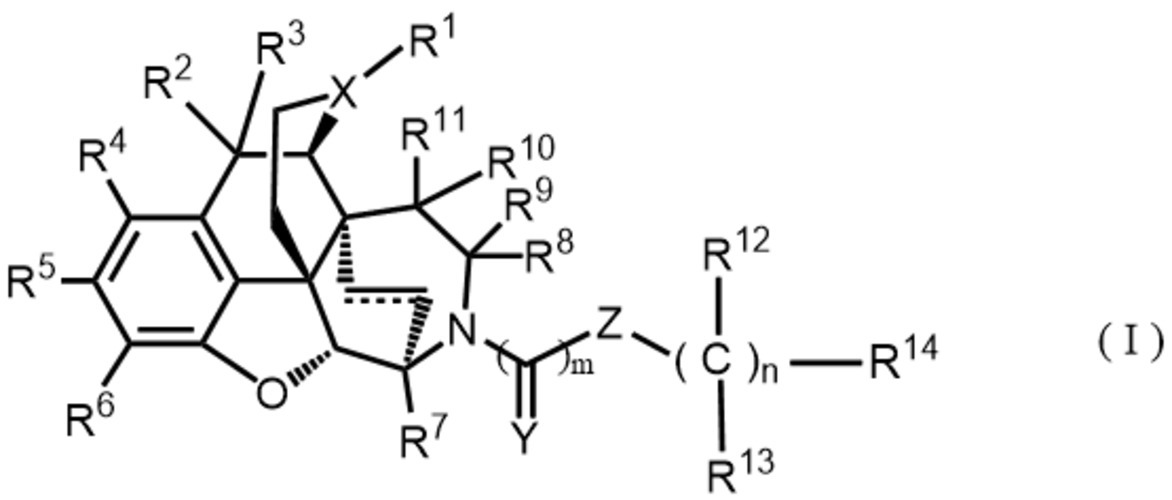
R2及びR3は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基又はR2及びR3が一緒になってカルボニル基を示すかR2及びR3が結合して置換基を有していてもよい環状ケタールを示し、
R4及びR5は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、置換基を有していてもよいアミノ基、ハロゲン原子又はヒドロキシ基を示し、
R6は水素原子、置換基を有していてもよいC1-6アルコキシ基、置換基を有していてもよいアミノ基、ハロゲン原子、ヒドロキシ基、シアノ基、カルボキシ基、カルボン酸エステル基又は置換基を有していてもよいカルバモイル基を示し、
R7は水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基又はヒドロキシ基を示し、
R8及びR9は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基又はR8及びR9が一緒になってカルボニル基、チオカルボニル基を示すかR8及びR9が結合して置換基を有していてもよいC3-6飽和炭化水素環又は置換基を有していてもよい環状ケタールを示し、
R10及びR11は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基又はR10及びR11が一緒になってカルボニル基、チオカルボニル基を示すかR10及びR11が結合して置換基を有していてもよいC3-6飽和炭化水素環又は置換基を有していてもよい環状ケタールを示し、
R12及びR13は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基を示すか、同一炭素上のR12とR13が結合して置換基を有していてもよいC3-6飽和炭化水素環、置換基を有していてもよい飽和複素環を示すか、nが2~3の場合において隣接する一組のR12同士が結合して置換基を有していてもよいC3-6飽和炭化水素環又は置換基を有していてもよい飽和複素環を形成することができ、
R14は水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC3-6シクロアルキル基、置換基を有していてもよいC2-6アルケニル基、置換基を有していてもよいC2-6アルキニル基、置換基を有していてもよいC1-6アルコキシ基、置換基を有していてもよいアミノ基、ハロゲン原子、ヒドロキシ基、置換基を有していてもよいC6-10アリール基、置換基を有していてもよいヘテロアリール基、置換基を有していてもよい環状アミノ基を示し、
Xは窒素原子又はN-オキシドを示し、
Yは酸素原子又は硫黄原子を示し、
ZはNR15、酸素原子、結合手、―CH=CH―又は―C≡C―を示し、
R15は水素原子、置換基を有していてもよいC1-6アルキル基を示すか、R15とR14が結合して置換基を有していてもよい含窒素飽和複素環を示し、
実線と破線からなる二重線は単結合又は二重結合を示し
mは0~1の整数を示し、
nは0~3の整数を示す。)
で表されるモルヒナン誘導体、該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物に関する。
[2]また本発明は、R6がヒドロキシ基又は置換基を有していてもよいC1-6アルコキシ基である前記[1]記載のモルヒナン誘導体、該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物に関する。
[3]また本発明は、R6がヒドロキシ基である前記[1]又は[2]記載のモルヒナン誘導体、該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物に関する。
[4]また本発明は、前記[1]~[3]記載のモルヒナン誘導体、該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物を有効成分として含有する医薬組成物に関する。
[5]また本発明は、オピオイドκ受容体に関連する疾患の治療又は、改善、予防剤である前記[4]記載の医薬に関する。
[6]また本発明は、鎮痛薬である前記[4]又は[5]記載の医薬に関する。
[7]また本発明は、止痒薬である前記[4]又は[5]記載の医薬に関する。。
R 2 and R 3 are the same or different hydrogen atom, C 1-6 alkyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, a halogen atom and a hydroxy group. Alternatively, R 2 and R 3 together indicate a carbonyl group or R 2 and R 3 may be attached to indicate a cyclic ketal which may have a substituent.
R 4 and R 5 have the same or different hydrogen atom, a C 1-6 alkyl group which may have a substituent, a C 1-6 alkoxy group which may have a substituent, and a substituent. Indicates an amino group, a halogen atom or a hydroxy group which may be present.
R 6 is a hydrogen atom, a C 1-6 alkoxy group which may have a substituent, an amino group which may have a substituent, a halogen atom, a hydroxy group, a cyano group, a carboxy group and a carboxylic acid ester group. Alternatively, a carbamoyl group which may have a substituent is shown.
R 7 represents a hydrogen atom, a C 1-6 alkyl group which may have a substituent, a C 1-6 alkoxy group which may have a substituent, or a hydroxy group.
R 8 and R 9 are the same or different hydrogen atoms, C 1-6 alkyl groups which may have substituents, C 1-6 alkoxy groups which may have substituents, halogen atoms, hydroxy groups. Alternatively, R 8 and R 9 may be combined to indicate a carbonyl group, a thiocarbonyl group, or R 8 and R 9 may be bonded to have a substituent C 3-6 saturated hydrocarbon ring or substituent. Indicates a ring-shaped ketal that may have,
R 10 and R 11 are the same or different hydrogen atom, C 1-6 alkyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, a halogen atom and a hydroxy group. Alternatively, R 10 and R 11 together indicate a carbonyl group, a thiocarbonyl group, or R 10 and R 11 may be bonded to have a substituent C 3-6 saturated hydrocarbon ring or substituent. Indicates a ring-shaped ketal that may have,
R 12 and R 13 indicate the same or different C 1-6 alkyl groups that may have the same or different hydrogen atoms, substituents, or R 12 and R 13 on the same carbon have a substituent. It indicates a C 3-6 saturated hydrocarbon ring which may be present, a saturated heterocyclic ring which may have a substituent, or when n is 2 to 3, adjacent sets of R12 are bonded to each other and substituted. It is possible to form a C 3-6 saturated hydrocarbon ring which may have a group or a saturated heterocyclic ring which may have a substituent.
R 14 is a hydrogen atom, a C 1-6 alkyl group which may have a substituent, a C 3-6 cycloalkyl group which may have a substituent, and C 2 which may have a substituent. -6 Alkenyl group, C 2-6 alkynyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, amino group which may have a substituent, halogen. An atom, a hydroxy group, a C 6-10 aryl group which may have a substituent, a heteroaryl group which may have a substituent, and a cyclic amino group which may have a substituent are shown.
X represents a nitrogen atom or N-oxide,
Y represents an oxygen atom or a sulfur atom.
Z indicates NR 15 , oxygen atom, bond, —CH = CH— or —C≡C—,
R 15 indicates a hydrogen atom, a C 1-6 alkyl group which may have a substituent, or indicates a nitrogen-containing saturated heterocycle in which R 15 and R 14 may be bonded and have a substituent. ,
A double line consisting of a solid line and a broken line indicates a single bond or a double bond, and m indicates an integer of 0 to 1.
n represents an integer of 0 to 3. )
It relates to a morphinan derivative represented by, a tautomer of the compound, a stereoisomer, a pharmaceutically acceptable salt thereof, or a solvate thereof.
[2] Further, in the present invention, the morphinan derivative according to the above [1], which is a C 1-6 alkoxy group in which R 6 may have a hydroxy group or a substituent, a tautomer of the compound, and a steric isomer. With respect to the body, or its pharmaceutically acceptable salts or solvates thereof.
[3] In the present invention, the morphinan derivative according to the above [1] or [ 2 ], wherein R6 is a hydroxy group, a tautomer of the compound, a stereoisomer, or a pharmaceutically acceptable salt thereof or the like. Regarding those solvates.
[4] Further, the present invention is effective for the morphinan derivative according to the above [1] to [3], a tautomer of the compound, a stereoisomer, a pharmaceutically acceptable salt thereof, or a solvate thereof. The present invention relates to a pharmaceutical composition contained as an ingredient.
[5] The present invention also relates to the pharmaceutical agent according to the above [4], which is a therapeutic, ameliorating, or preventive agent for a disease related to opioid kappa receptor.
[6] The present invention also relates to the pharmaceutical product according to the above [4] or [5], which is an analgesic.
[7] The present invention also relates to the drug according to the above [4] or [5], which is an antipruritic drug. ..
次に本発明をさらに詳しく説明する。
上記一般式(I)で表されるモルヒナン誘導体、当該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物のうち、好ましくは次のものが挙げられる。
R1~R5、R7~R14で示される置換基を有していてもよいC1-6アルキル基におけるC1-6アルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、ヘキシル基等が挙げられ、好ましくはメチル基、エチル基、プロピル基が挙げられ、より好ましくはメチル基が挙げられる。
Next, the present invention will be described in more detail.
Among the morphinan derivatives represented by the general formula (I), tautomers of the compound, stereoisomers, pharmaceutically acceptable salts thereof, or solvates thereof, the following are preferable. Be done.
The C 1-6 alkyl group in the C 1-6 alkyl group which may have the substituents represented by R 1 to R 5 and R 7 to R 14 includes a methyl group, an ethyl group, a propyl group and an isopropyl group. , Butyl group, isobutyl group, tert-butyl group, pentyl group, hexyl group and the like, preferably a methyl group, an ethyl group, a propyl group and the like, and more preferably a methyl group.
R1及びR14で示される置換基を有していてもよいC3-6シクロアルキル基におけるC3-6シクロアルキル基としては、シクロプロピル基、シクロブチル基、シクロペンチル基、及びシクロヘキシル基が挙げられ、好ましくはシクロプロピル基が挙げられる。 Examples of the C 3-6 cycloalkyl group in the C 3-6 cycloalkyl group which may have the substituents indicated by R 1 and R 14 include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, and a cyclohexyl group. However, a cyclopropyl group is preferable.
R1で示される置換基を有していてもよいC1-6アルキル基及び置換基を有していてもよいC3-6シクロアルキル基における置換基としては、メチル基、エチル基、プロピル基等のC1-6アルキル基、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基等のハロゲン化メチル基、フッ素原子、塩素原子等のハロゲン原子、ヒドロキシ基、C1-6アルキルアミノ基、ジC1-6アルキルアミノ基、アシルアミノ基、保護されたアミノ基等の置換基を有していてもよいアミノ基、ホルミル基、アセチル基、シクロプロピルカルボニル基、ベンゾイル基等のアシル基、アゼチジニル基、ピロリジニル基、ピペラジニル基、モルホリニル基等の環状アミノ基、β-ラクタム、γ-ラクタム、δ-ラクタム等の環状ラクタム基等が挙げられる。 The substituents in the C 1-6 alkyl group which may have a substituent represented by R 1 and the C 3-6 cycloalkyl group which may have a substituent include a methyl group, an ethyl group and a propyl group. C 1-6 alkyl groups such as groups, fluoromethyl groups, difluoromethyl groups, methyl halides such as trifluoromethyl groups, halogen atoms such as fluorine atoms and chlorine atoms, hydroxy groups, C 1-6 alkylamino groups, Di C 1-6 Amino group which may have a substituent such as an alkylamino group, an acylamino group, a protected amino group, a formyl group, an acetyl group, an acyl group such as a cyclopropylcarbonyl group and a benzoyl group, azetidinyl. Examples thereof include a cyclic amino group such as a group, a pyrrolidinyl group, a piperazinyl group and a morpholinyl group, and a cyclic lactam group such as β-lactam, γ-lactam and δ-lactam.
R2~R11及びR14で示される置換基を有していてもよいC1-6アルコキシ基におけるC1-6アルコキシ基としては、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、イソブトキシ基等が挙げられ、好ましくはメトキシ基が挙げられる。
置換基としては、メトキシ基、エトキシ基等のC1-6アルコキシ基、フェノキシ基、フッ素原子、塩素原子等のハロゲン原子が挙げられ、好ましくはフッ素原子であり、具体的にはフルオロメトキシ基、ジフルオロメトキシ基、トリフルオロメトキシ基、2,2,2-トリフルオロエトキシ基等が挙げられる。
The C 1-6 alkoxy group in the C 1-6 alkoxy group which may have the substituents represented by R 2 to R 11 and R 14 includes a methoxy group, an ethoxy group, a propoxy group, an isopropoxy group and a butoxy. Examples thereof include a group, an isobutoxy group and the like, and preferably a methoxy group.
Examples of the substituent include a C 1-6 alkoxy group such as a methoxy group and an ethoxy group, a halogen atom such as a phenoxy group, a fluorine atom and a chlorine atom, preferably a fluorine atom, specifically a fluoromethoxy group and the like. Examples thereof include a difluoromethoxy group, a trifluoromethoxy group and a 2,2,2-trifluoroethoxy group.
R1及びR14で示される置換基を有していてもよいアリール基におけるアリール基としては、フェニル基、ナフチル基が挙げられる。 Examples of the aryl group in the aryl group which may have the substituent represented by R 1 and R 14 include a phenyl group and a naphthyl group.
R1及びR14で示される置換基を有していてもよいヘテロアリール基におけるヘテロアリール基としては、例えばフラニル基、イミダゾリル基、ピラゾリル基、チエニル、オキサゾリル基、イソキサゾリル基、チアゾリル基、イソキサゾリル基等の5員環ヘテロアリール基、ピリジル基、ピリダジニル基、ピラジニル基、ピリミジル基等の6員環ヘテロアリール基、インドリル基、ベンゾイミダゾリル基、ベンゾフラニル基、ベンゾチエニル基、ベンゾオキサゾリル基、ベンゾチアゾリル基等の二環性ヘテロアリール基等の窒素原子、酸素原子及び硫黄原子から選択される1~4個のヘテロ原子を環構成原子として含む単環性又は二環性のヘテロアリール基が挙げられる。
また、これらのヘテロアリール基上の置換基によっては互変異性体が存在しえ、例えばピリジル基上にヒドロキシ基が置換する場合、6-ヒドロキシピリジン-2-イル基と、その互変異性体として6-オキソ-1,6-ジヒドロピリジン-2-イル基、および4-ヒドロキシピリジン-2-イル基と、その互変異性体として4-オキソ-1,4-ジヒドロピリジン-2-イル基が挙げられる。
Examples of the heteroaryl group in the heteroaryl group which may have the substituents indicated by R 1 and R 14 include a furanyl group, an imidazolyl group, a pyrazolyl group, a thienyl, an oxazolyl group, an isoxazolyl group, a thiazolyl group and an isoxazolyl group. 6-membered ring heteroaryl group, indrill group, benzoimidazolyl group, benzofuranyl group, benzothienyl group, benzoxazolyl group, benzothiazolyl group, etc. Examples thereof include a monocyclic or bicyclic heteroaryl group containing 1 to 4 heteroatoms selected from a nitrogen atom, an oxygen atom and a sulfur atom such as a bicyclic heteroaryl group as a ring-constituting atom.
Further, a remutable substance may exist depending on the substituent on these heteroaryl groups. For example, when a hydroxy group is substituted on a pyridyl group, a 6-hydroxypyridine-2-yl group and the remutable body thereof are present. 6-oxo-1,6-dihydropyridine-2-yl group and 4-hydroxypyridine-2-yl group, and 4-oxo-1,4-dihydropyridine-2-yl group as their homomorphs are mentioned. Be done.
R1及びR14で示される置換基を有していてもよいアリール基及び置換基を有していてもよいヘテロアリール基は、環上に1~3の置換基を有していてもよく、その置換基としては、メチル基、エチル基、プロピル基等のC1-6アルキル基、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基等のハロゲン化メチル基、ヒドロキシメチル基、ドロキシエチル基、1-ヒドロキシプロピル基等のヒドロキシアルキル基、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基等のC1-6アルコキシ基、フッ素原子、塩素原子等のハロゲン原子、ヒドロキシ基、ニトロ基、シアノ基、C1-6アルキルアミノ基、ジC1-6アルキルアミノ基、アシルアミノ基、保護されたアミノ基等の置換基を有していてもよいアミノ基、ホルミル基、アセチル基、シクロプロピルカルボニル基、ベンゾイル基等のアシル基、アゼチジニル基、ピロリジニル基、ピペラジニル基、モルホリニル基等の環状アミノ基、β-ラクタム、γ-ラクタム、δ-ラクタム等の環状ラクタム基等が挙げられる。 The aryl group which may have a substituent shown by R 1 and R 14 and the heteroaryl group which may have a substituent may have 1 to 3 substituents on the ring. As the substituents, C 1-6 alkyl groups such as methyl group, ethyl group and propyl group, methyl halide groups such as fluoromethyl group, difluoromethyl group and trifluoromethyl group, hydroxymethyl group and droxyethyl group, Hydroxyalkyl groups such as 1-hydroxypropyl group, methoxy group, ethoxy group, propoxy group, isopropoxy group, C 1-6 alkoxy group such as butoxy group, halogen atom such as fluorine atom and chlorine atom, hydroxy group, nitro group. , Cyan group, C 1-6 alkylamino group, diC 1-6 alkylamino group, acylamino group, amino group which may have a substituent such as a protected amino group, formyl group, acetyl group, cyclo Examples thereof include an acyl group such as a propylcarbonyl group and a benzoyl group, a cyclic amino group such as an azetidinyl group, a pyrrolidinyl group, a piperazinyl group and a morpholinyl group, and a cyclic lactam group such as β-lactam, γ-lactam and δ-lactam.
R1で示される置換基を有していてもよいC3-6シクロアルキルC1-6アルキル基におけるC3-6シクロアルキルとしては、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルが挙げられ、好ましくはシクロプロピル、シクロブチルが挙げられ、より好ましくはシクロプロピルが挙げられる。
また、R1で示されるC3-6シクロアルキルC1-6アルキル基におけるC1-6アルキル基としては、前記と同様のものが挙げられる。
置換基としては、前記置換基を有していてもよいC3-6シクロアルキル基における置換基と同様のものが挙げられる。
Examples of the C 3-6 cycloalkyl in the C 3-6 cycloalkyl C 1-6 alkyl group which may have the substituent represented by R 1 include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, and are preferable. Cyclopropyl and cyclobutyl are mentioned, and more preferably cyclopropyl is mentioned.
Moreover, as the C 1-6 alkyl group in the C 3-6 cycloalkyl C 1-6 alkyl group represented by R 1 , the same thing as above can be mentioned.
Examples of the substituent include the same as the substituent in the C 3-6 cycloalkyl group which may have the above-mentioned substituent.
R1で示される置換基を有していてもよいアラルキル基としては、アリール部分の炭素数がC6~10で、アルキレン部分の炭素数はC1~5を示し、例えばフェニル又はナフチルで置換されたメチル基、エチル基等が挙げられ、好ましくはフェニルで置換されたメチル基(ベンジル基)が挙げられる。 As the aralkyl group which may have the substituent represented by R 1 , the aryl moiety has a carbon number of C 6 to 10 and the alkylene moiety has a carbon number of C 1 to 5 , and is substituted with, for example, phenyl or naphthyl. Examples thereof include a methyl group and an ethyl group, preferably a phenyl-substituted methyl group (benzyl group).
R1で示される置換基を有していてもよいヘテロアリールアルキル基におけるヘテロアリール部分としては、窒素原子、酸素原子及び硫黄原子から選択される1~4個のヘテロ原子を環構成原子として含むヘテロアリールが挙げられ、またアルキル部分としてはメチル基、エチル基、プロピル基等のC1-6アルキル基が挙げられ、例えば、(ピリジン-2-イル)メチル基、(ピリジン-3-イル)メチル基、(ピリジン-4-イル)メチル基、2-(ピリジン-2-イル)エチル基、(フラン-2-イル)メチル基、(フラン-3-イル)メチル基、(イミダゾール-2-イル)メチル基、(イミダゾール-4-イル)メチル基、(イミダゾール-5-イル)メチル基、(チアゾール-2-イル)メチル基、(チアゾール-4-イル)メチル基、(チアゾール-5-イル)メチル基、(チオフェン2-イル)メチル基又は2-(チオフェン-2-イル)エチル基等の単環性ヘテロアリールアルキル基、(キノリン-3-イル)メチル基、(インドール-3-イル)メチル基の二環性ヘテロアリールアルキル基等が挙げられる。 The heteroaryl moiety in the heteroarylalkyl group which may have a substituent represented by R 1 includes 1 to 4 heteroatoms selected from a nitrogen atom, an oxygen atom and a sulfur atom as ring-constituting atoms. Heteroaryl can be mentioned, and examples of the alkyl moiety include C 1-6 alkyl groups such as a methyl group, an ethyl group and a propyl group, for example, (pyridine-2-yl) methyl group and (pyridine-3-yl). Methyl group, (pyridine-4-yl) methyl group, 2- (pyridine-2-yl) ethyl group, (fran-2-yl) methyl group, (fran-3-yl) methyl group, (imidazole-2-) Il) methyl group, (imidazole-4-yl) methyl group, (imidazole-5-yl) methyl group, (thiazol-2-yl) methyl group, (thiazol-4-yl) methyl group, (thiazol-5-yl) Monocyclic heteroarylalkyl groups such as yl) methyl group, (thiophene-2-yl) methyl group or 2- (thiophen-2-yl) ethyl group, (quinolin-3-yl) methyl group, (indol-3-3). Ill) Examples thereof include a bicyclic heteroarylalkyl group of a methyl group.
R1で示される置換基を有していてもよいアラルキル基及び置換基を有していてもよいヘテロアリールアルキル基におけるアリール及びヘテロアリール上に置換基を有してもよく、その置換基としては前記置換基を有していてもよいアリール基及び置換基を有していてもよいヘテロアリール基における置換基と同様のものが挙げられる。 It may have a substituent on the aryl and the heteroaryl in the aralkyl group which may have the substituent represented by R1 and the heteroarylalkyl group which may have the substituent, and may have a substituent as the substituent. Examples thereof include the same as the substituent in the aryl group which may have a substituent and the heteroaryl group which may have a substituent.
R1及びR14で示される置換基を有していてもよいC2-6アルケニル基におけるC2-6アルケニル基としては、C2-6の直鎖又は分岐鎖のアルケニル基が挙げられ、アリル基、ビニル基、1-プロペニル基、2-ブテニル基、3-ブテニル基、2-ペンテニル基、3-ペンテニル基、4-ペンテニル基、2-ヘキセニル基、3-ヘキセニル基、4-ヘキセニル基、5-ヘキセニル基等のアルケニル基が挙げられる。 Examples of the C 2-6 alkenyl group in the C 2-6 alkenyl group which may have the substituents indicated by R 1 and R 14 include a linear or branched alkenyl group of C 2-6 . Allyl group, vinyl group, 1-propenyl group, 2-butenyl group, 3-butenyl group, 2-pentenyl group, 3-pentenyl group, 4-pentenyl group, 2-hexenyl group, 3-hexenyl group, 4-hexenyl group , 5-Hexenyl groups and the like are alkenyl groups.
R1及びR14で示される置換基を有していてもよいC2-6アルキニル基におけるC2-6アルキニル基としては、C2-6の直鎖または分枝鎖状のアルキニル基を示し、たとえば、エチニル基、プロピニル基、ブチニル基などが挙げられる。 The C 2-6 alkynyl group in the C 2-6 alkynyl group which may have the substituents indicated by R 1 and R 14 indicates a linear or branched alkynyl group of C 2-6 . For example, an ethynyl group, a propynyl group, a butynyl group and the like can be mentioned.
斯かるアルケニル基及びアルキニル基に置換し得る基としては、メトキシカルボニル基、エトキシカルボニル基、プロポキシカルボニル基等のアルコキシカルボニル基、ベンジル基、2-フェニルエチル基、3-フェニルプロピル基、4-フェニルブチル基等のアラルキル基、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基等のアルコキシ基、ベンジルオキシ基、2-フェニルエチルオキシ基等のアラルキルオキシ基、C1~6の直鎖または分岐鎖状アルキル基で置換されていてもよいアミノ基、フッ素原子、塩素原子等のハロゲン原子、カルボキシ基及びヒドロキシ基等が挙げられる。 Examples of the group that can be substituted with such an alkenyl group and an alkynyl group include an alkoxycarbonyl group such as a methoxycarbonyl group, an ethoxycarbonyl group and a propoxycarbonyl group, a benzyl group, a 2-phenylethyl group, a 3-phenylpropyl group and a 4-phenyl group. Aralkyl group such as butyl group, methoxy group, ethoxy group, propoxy group, alkoxy group such as butoxy group, aralkyloxy group such as benzyloxy group and 2 - phenylethyloxy group, linear or branched chain of C1-6. Examples thereof include an amino group which may be substituted with an alkyl group, a halogen atom such as a fluorine atom and a chlorine atom, a carboxy group and a hydroxy group.
R1で示される置換基を有していてもよいアシル基としては、ホルミル基、アセチル基、プロピオニル基、ブタノイル基、ペンタノイル基、ヘキサノイル基等のC2-6アルカノイル基、シクロプロピルカルボニル基、シクロブチルカルボニル基、シクロペンチルカルボニル基等のC4―7シクロアルカノイル基、ベンゾイル基、ナフトイル基等のアロイル基又はフロイル基、チオフェンカルボニル基、ニコチニル基、イソニコチノイル基等の5~6員環のヘテロアロイル基等が挙げられる。
また、C2-6アルカノイル基、C4―7シクロアルカノイル基における置換基としては、前記置換基を有していてもよいC1-6アルキル基における置換基と同様のものが挙げられ、アロイル基、ヘテロアロイル基における置換基としては、前記置換基を有していてもよいアリール基及び置換基を有していてもよいヘテロアリール基における置換基と同様のものが挙げられる。
Examples of the acyl group which may have the substituent represented by R 1 include a C 2-6 alkanoyl group such as a formyl group, an acetyl group, a propionyl group, a butanoyl group, a pentanoyl group and a hexanoyl group, and a cyclopropylcarbonyl group. C 4-7 cycloalkanoyl group such as cyclobutylcarbonyl group and cyclopentylcarbonyl group, aloyl group such as benzoyl group and naphthoyl group or floyl group, thiophenecarbonyl group, nicotinyl group, isonicotinoyyl group and other 5- to 6-membered heteroaroyl group. And so on.
Examples of the substituent in the C 2-6 alkanoyl group and the C 4-7 cycloalkanoyl group include the same as the substituent in the C 1-6 alkyl group which may have the above-mentioned substituent, and alloyl. Examples of the substituent in the group and the heteroaroyl group include the same as the substituent in the aryl group which may have the substituent and the heteroaryl group which may have the substituent.
R6で示される置換基を有していてもよいカルバモイル基における置換基及びR4~R6及びR14で示される置換基を有していてもよいアミノ基における置換基としては、メチル基、エチル基、プロピル基、イソプロピル基等の直鎖又は分岐の置換基を有していてもよいC1-6アルキル基(置換基としては、ヒドロキシ基、C1-6アルコキシ基、フッ素原子、塩素原子等のハロゲン原子、アミノ基、シアノ基、ヘテロアリール基等が挙げられる。)が挙げられ、これらの置換基を1又は2有してもよい。
また、R4~R6及びR14示される置換基を有していてもよいアミノ基における置換基としては、上記の他、R1で示されるアミノ保護基と同様のアミノ保護基が挙げられる。
The substituent in the carbamoyl group which may have a substituent represented by R 6 and the substituent in the amino group which may have a substituent represented by R 4 to R 6 and R 14 is a methyl group. , C 1-6 alkyl group which may have a linear or branched substituent such as ethyl group, propyl group, isopropyl group (as the substituent, hydroxy group, C 1-6 alkoxy group, fluorine atom, Examples thereof include a halogen atom such as a chlorine atom, an amino group, a cyano group, a heteroaryl group and the like), and these substituents may have 1 or 2.
In addition to the above, examples of the substituent in the amino group which may have the substituents shown in R 4 to R 6 and R 14 include the same amino protecting group as the amino protecting group shown in R 1 . ..
R1で示されるアミノ保護基としては、メトキシカルボニル基、エトキシカルボニル基、tert-ブトキシカルボニル基、tert-アミロキシカルボニル基、2,2,2-トリクロロエトキシカルボニル基、ベンジルオキシカルボニル基、p-クロロベンジルオキシカルボニル基、p-メトキシベンジルカルボニル基、p-ニトロベンジルオキシカルボニル基、p-フェニルアゾベンジルオキシカルボニル基、p-メトキシフェニルアゾベンジルオキシカルボニル基、3,5-ジメトキシベンジルオキシカルボニル基、3,4,5-トリメトキシベンジルオキシカルボニル基、p-ビフェニルイソプロピルオキシカルボニル基、ジイソプロピルメチロキシカルボニル基、2-(トリメチルシリル)エトキシカルボニル基、9-フルオレニルメチルオキシカルボニル基等のカーバメート系保護基、p-トルエンスルホニル基、2-ニトロベンゼンスルホニル基等スルホンアミド系保護基、フタロイル基等のイミド系保護基、ベンジル基、フェニルエチル基、フェニルプロピル基、トリチル基、ナフチルメチル基等のC7―19のアラルキル基が挙げられる。 Examples of the amino-protecting group represented by R 1 include a methoxycarbonyl group, an ethoxycarbonyl group, a tert-butoxycarbonyl group, a tert-amyloxycarbonyl group, a 2,2,2-trichloroethoxycarbonyl group, a benzyloxycarbonyl group and p-. Chlorobenzyloxycarbonyl group, p-methoxybenzylcarbonyl group, p-nitrobenzyloxycarbonyl group, p-phenylazobenzyloxycarbonyl group, p-methoxyphenylazobenzyloxycarbonyl group, 3,5-dimethoxybenzyloxycarbonyl group, Carbamate protection of 3,4,5-trimethoxybenzyloxycarbonyl group, p-biphenylisopropyloxycarbonyl group, diisopropylmethyloxycarbonyl group, 2- (trimethylsilyl) ethoxycarbonyl group, 9-fluorenylmethyloxycarbonyl group, etc. Group, p-toluenesulfonyl group, 2-nitrobenzenesulfonyl group and other sulfonamide-based protective groups, phthaloyl group and other imide-based protective groups, benzyl group, phenylethyl group, phenylpropyl group, trityl group, naphthylmethyl group and other C7 -19 Aralkyl groups are listed.
R2~R6、R8~R11及びR14で示されるハロゲン原子としては、フッ素原子、塩素原子、臭素原子及びヨウ素原子が挙げられ、好ましくはフッ素原子、塩素原子、より好ましくはフッ素原子が挙げられる。 Examples of the halogen atom represented by R2 to R6 , R8 to R11 and R14 include a fluorine atom, a chlorine atom, a bromine atom and an iodine atom, preferably a fluorine atom, a chlorine atom, and more preferably a fluorine atom. Can be mentioned.
R6で示されるカルボン酸エステル基におけるエステルを形成する基としては、メチル基、エチル基、プロピル基、2-プロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、ヘキシル基等のC1-6アルキル基;ビニル基、アリル基、1-プロペニル基、ブテニル基、ペンテニル基、ヘキセニル基等のC2-6アルケニル基;ベンジル基等のアラルキル基;フェニル基、ナフチル基等のアリール基、アセトキシメチル基、ピバロイルオキシメチル基等のC1-6アルカノイルオキシC1-4低級アルキル基等が挙げられる。 Examples of the group forming the ester in the carboxylic acid ester group represented by R6 include a methyl group, an ethyl group, a propyl group, a 2-propyl group, a butyl group, an isobutyl group, a tert-butyl group, a pentyl group and a hexyl group. C 1-6 alkyl group; C 2-6 alkenyl group such as vinyl group, allyl group, 1-propenyl group, butenyl group, pentenyl group and hexenyl group; aralkyl group such as benzyl group; aryl such as phenyl group and naphthyl group Examples thereof include C 1-6 alkanoyloxy C 1-4 lower alkyl groups such as a group, an acetoxymethyl group and a pivaloyloxymethyl group.
R14で示される置換基を有していてもよい環状アミノ基における環状アミノ基としては、アゼチジニル基、ピロリジニル基、ピペリジニル基、ピペラジニル基、モルホニル基及びチオモルホリニル基等が挙げられる。
置換基としては、メチル基、エチル基等のC1-6アルキル基、メトキシ基、エトキシ基、プロポキシ基等のC1-6アルコキシ基、フッ素原子、塩素原子等のハロゲン原子、ヒドロキシ基等が挙げられる。
Examples of the cyclic amino group in the cyclic amino group which may have the substituent represented by R14 include an azetidinyl group, a pyrrolidinyl group, a piperidinyl group, a piperazinyl group, a morphonyl group and a thiomorpholinyl group.
Examples of the substituent include a C 1-6 alkyl group such as a methyl group and an ethyl group, a C 1-6 alkoxy group such as a methoxy group, an ethoxy group and a propoxy group, a halogen atom such as a fluorine atom and a chlorine atom, and a hydroxy group. Can be mentioned.
R8及びR9が結合して形成される置換基を有していてもよいC3-6飽和炭化水素環、R10及びR11が結合して形成される置換基を有していてもよいC3-6飽和炭化水素環、同一炭素上のR12とR13が結合又はnが2~3の場合において隣接する一組のR12同士が結合して形成される置換基を有していてもよいC3-6飽和炭化水素環におけるC3-6飽和炭化水素環としては、シクロプロパン環、シクロブタン環、シクロペンタン環、シクロヘキサン環が挙げられる。 It may have a substituent formed by bonding R 8 and R 9 , and may have a substituent formed by bonding R 10 and R 11 to a C 3-6 saturated hydrocarbon ring. A good C 3-6 saturated hydrocarbon ring, with substituents formed by the bond of R 12 and R 13 on the same carbon or the bond of adjacent sets of R 12 to each other when n is 2-3. Examples of the C 3-6 saturated hydrocarbon ring in the C 3-6 saturated hydrocarbon ring may include a cyclopropane ring, a cyclobutane ring, a cyclopentane ring, and a cyclohexane ring.
同一炭素上のR12とR13が結合して又はnが2~3の場合において隣接する一組のR12同士が結合して形成される置換基を有していてもよい飽和複素環としては、3~6員環の飽和複素環が挙げられ、例えばアジリジン、アゼチジン、ピロリジン、ピペリジン、ピペラジン、モルホリン、チオモルホリン等の環状アミン、エポキシド、オキセタン、テトラヒドロフラン、テトラヒドロピラン、ジオキサン等の環状エーテル、チエタン、チオラン、チアン等の環状チオエーテル等が挙げられる。
これらの飽和複素環のうち、環の構成原子として窒素原子を有する場合には、C1-6アルキル基、アシル基及びアミノ保護基等の置換基を窒素上に有していてもよい。これらの置換基は前記と同様のものを示す。
As a saturated heterocycle which may have a substituent formed by bonding R 12 and R 13 on the same carbon or by bonding adjacent sets of R 12 to each other when n is 2 to 3. Examples thereof include saturated heterocycles having a 3- to 6-membered ring, such as cyclic amines such as aziridine, azetidine, pyrrolidine, piperidine, piperazine, morpholine and thiomorpholine, and cyclic ethers such as epoxide, oxetane, tetrahydrofuran, tetrahydropyran and dioxane. Cyclic thioethers such as thietan, thiolan, and thian can be mentioned.
When the saturated heterocycle has a nitrogen atom as a constituent atom of the ring, it may have a substituent such as a C 1-6 alkyl group, an acyl group and an amino protecting group on nitrogen. These substituents are the same as described above.
R14とR15が結合して形成される置換基を有していてもよい含窒素飽和複素環としては、3~6員環の含窒素飽和複素環が挙げられ、例えばアジリジン、アゼチジン、ピロリジン、ピペリジン、ピペラジン、モルホリン、チオモルホリン等の環状アミノ基が挙げられる。
これらの含窒素飽和複素環はさらに飽和炭化水素、飽和複素環、不飽和炭化水素又は不飽和複素環と縮環していてもよく、例えばデカヒドロキノリン、デカヒドロイソキノリン、インドリン、イソインドリン、1,2,3,4-テトラヒドロキノリン、1,2,3,4-テトラヒドロイソキノリン、ベンゾモルホリン、ベンゾチオモルホリン等が挙げられる。
これらの含窒素複素環においてR15が結合している窒素原子以外に環の構成原子として窒素原子を有する場合には、C1-6アルキル基、アシル基及びアミノ保護基等の置換基を有していてもよい。これらの置換基は前記と同様のものを示す。
Examples of the nitrogen-containing saturated heterocycle which may have a substituent formed by bonding R 14 and R 15 include a 3- to 6-membered nitrogen-containing saturated heterocycle, for example, aziridine, azetidine, and pyrrolidine. , Piperidine, piperazine, morpholine, thiomorpholine and the like.
These nitrogen-containing saturated heterocycles may be further fused with a saturated hydrocarbon, a saturated heterocycle, an unsaturated hydrocarbon or an unsaturated heterocycle, for example, decahydroquinoline, decahydroisoquinoline, indolin, isoindoline, 1 , 2,3,4-tetrahydroquinoline, 1,2,3,4-tetrahydroisoquinoline, benzomorpholin, benzothiomorpholin and the like.
When the nitrogen-containing heterocycle has a nitrogen atom as a constituent atom of the ring in addition to the nitrogen atom to which R15 is bonded, it has a substituent such as a C 1-6 alkyl group, an acyl group and an amino-protecting group. You may be doing it. These substituents are the same as described above.
同一炭素上のR12とR13が結合して形成される置換基を有していてもよいC3-6飽和炭化水素環、同一炭素上のR12とR13が結合して形成される置換基を有していてもよい飽和複素環、nが2~3の場合において隣接する一組のR12同士が結合して形成される置換基を有していてもよいC3-6飽和炭化水素環又は置換基を有していてもよい飽和複素環及びR14とR15が結合して形成される置換基を有していてもよい含窒素飽和複素環における置換基としては前記環状アミノ基における置換基と同様のものが挙げられる。 A C 3-6 saturated hydrocarbon ring which may have a substituent formed by bonding R 12 and R 13 on the same carbon, and formed by bonding R 12 and R 13 on the same carbon. Saturated heterocycle which may have a substituent, and C 3-6 saturated which may have a substituent formed by bonding adjacent sets of R12 to each other when n is 2 to 3. The cyclic is the substituent in the saturated heterocycle which may have a hydrocarbon ring or a substituent and the nitrogen-containing saturated heterocycle which may have a substituent formed by combining R 14 and R 15 . The same as the substituent in the amino group can be mentioned.
R2とR3、R8とR9及びR10とR11が結合して形成される置換基を有していてもよい環状ケタールとしてはジオキソラン、ジオキサンが挙げられる。置換基としては、メチル基、エチル基、プロピル基等のC1-6アルキル基が挙げられる。 Examples of the cyclic ketal that may have a substituent formed by binding R 2 and R 3 , R 8 and R 9 and R 10 and R 11 include dioxolane and dioxane. Examples of the substituent include C 1-6 alkyl groups such as a methyl group, an ethyl group and a propyl group.
Xは窒素原子又はN-オキシドを示し、窒素原子が好ましい。 X indicates a nitrogen atom or N-oxide, and a nitrogen atom is preferable.
Yは酸素原子又は硫黄原子を示し、好ましくは酸素原子である。 Y represents an oxygen atom or a sulfur atom, and is preferably an oxygen atom.
ZはNR15、酸素原子、結合手、―CH=CH―又は―C≡C―を示し、好ましくはNR15、酸素原子、結合手又はエチレンである。 Z represents NR 15 , oxygen atom, bond, —CH = CH— or —C≡C—, preferably NR 15 , oxygen atom, bond or ethylene.
mは0~1の整数を示し、好ましくは1である。 m represents an integer of 0 to 1, preferably 1.
nは0~3の整数を示し、好ましくは1である。 n represents an integer of 0 to 3, preferably 1.
本発明に用いられる化合物(I)のうち好ましい態様としては、R1は置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC3-6シクロアルキルC1-6アルキル基であり、R2及びR3は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基であり、R4及びR5は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、置換基を有していてもよいアミノ基、ハロゲン原子又はヒドロキシ基であり、R6は置換基を有していてもよいC1-6アルコキシ基又はヒドロキシ基であり、R7は水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基又はヒドロキシ基であり、R8及びR9は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子又はヒドロキシ基であり、R10及びR11は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基であり、R12及びR13は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R14は置換基を有していてもよいC6-10アリール基、置換基を有していてもよいヘテロアリール基又は置換基を有していてもよい環状アミノ基であり、Xは窒素原子又はN-オキシドであり、Yは酸素原子又は硫黄原子であり、ZはNR15、酸素原子、結合手、―CH=CH―又は―C≡C―であり、R15は水素原子、置換基を有していてもよいC1-6アルキル基であり、実線と破線からなる二重線は単結合又は二重結合であり、mは0又は1の整数であり、nは0~3の整数である場合が挙げられる。 In the preferred embodiment of the compound (I) used in the present invention, R 1 may have a substituent C 1-6 alkyl group and may have a substituent C 3-6 cycloalkyl. It is a C 1-6 alkyl group, and R 2 and R 3 may have the same or different hydrogen atom or substituent C 1-6 alkyl group, and may have a substituent C 1- 6 An alkoxy group, a halogen atom, and a hydroxy group, and R4 and R5 may have the same or different hydrogen atom and substituent, and may have a C 1-6 alkyl group and substituent. C 1-6 alkoxy group, an amino group which may have a substituent, a halogen atom or a hydroxy group, and R 6 is a C 1-6 alkoxy group or a hydroxy group which may have a substituent. , R 7 is a hydrogen atom, a C 1-6 alkyl group which may have a substituent, a C 1-6 alkoxy group or a hydroxy group which may have a substituent, and R 8 and R 9 are. The same or different hydrogen atom, C 1-6 alkyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, a halogen atom or a hydroxy group, and R 10 and R 11 is the same or different hydrogen atom, C 1-6 alkyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, a halogen atom, and a hydroxy group. R 12 and R 13 are C 1-6 alkyl groups that may have the same or different hydrogen atoms or substituents, and R 14 is a C 6-10 aryl group that may have substituents, A heteroaryl group which may have a substituent or a cyclic amino group which may have a substituent, X is a nitrogen atom or an N-oxide, Y is an oxygen atom or a sulfur atom, and Z is. Is NR 15 , oxygen atom, bonder, —CH = CH— or —C≡C—, and R15 is a hydrogen atom, a C 1-6 alkyl group that may have a substituent, with a solid line. The double line consisting of a broken line is a single bond or a double bond, m is an integer of 0 or 1, and n is an integer of 0 to 3.
本発明に用いられる化合物(I)のより好ましい態様としては、R1は置換基を有していてもよいC1-6アルキル基又は置換基を有していてもよいC3-6シクロアルキルC1-6アルキル基であり、R2及びR3は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R4及びR5は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R6は置換基を有していてもよいC1-6アルコキシ基又はヒドロキシ基であり、R7は水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基又はヒドロキシ基であり、R8及びR9は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基又は置換基を有していてもよいC1-6アルコキシ基であり、R10及びR11は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R12及びR13は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R14は置換基を有していてもよいC6-10アリール基、置換基を有していてもよいヘテロアリール基又は置換基を有していてもよい環状アミノ基であり、Xは窒素原子又はN-オキシドであり、Yは酸素原子又は硫黄原子であり、ZはNR15、酸素原子、結合手又は―CH=CH―であり、R15は水素原子、置換基を有していてもよいC1-6アルキル基であり、実線と破線からなる二重線は単結合又は二重結合であり、mは0又は1の整数であり、nは0~3の整数である場合が挙げられる。 In a more preferred embodiment of the compound (I) used in the present invention, R 1 may have a C 1-6 alkyl group which may have a substituent or a C 3-6 cycloalkyl which may have a substituent. C 1-6 alkyl groups, R 2 and R 3 are C 1-6 alkyl groups that may have the same or different hydrogen atoms or substituents, and R 4 and R 5 are the same or different. A C 1-6 alkyl group which may have a hydrogen atom or a substituent, R 6 is a C 1-6 alkoxy group or a hydroxy group which may have a substituent, and R 7 is a hydrogen atom. , C 1-6 alkyl group which may have a substituent, C 1-6 alkoxy group or hydroxy group which may have a substituent, and R 8 and R 9 are the same or different hydrogen atoms. , A C 1-6 alkyl group which may have a substituent or a C 1-6 alkoxy group which may have a substituent, and R 10 and R 11 are the same or different hydrogen atoms or substituents. Is a C 1-6 alkyl group that may have, and R 12 and R 13 are C 1-6 alkyl groups that may have the same or different hydrogen atoms or substituents, where R 14 is. A C 6-10 aryl group which may have a substituent, a heteroaryl group which may have a substituent or a cyclic amino group which may have a substituent, and X is a nitrogen atom or N. -Oxide, Y is an oxygen atom or a sulfur atom, Z is an NR 15 , an oxygen atom, a bond or -CH = CH-, and R 15 is a hydrogen atom, which may have a substituent. A 1-6 alkyl group, a double line consisting of a solid line and a broken line is a single bond or a double bond, m is an integer of 0 or 1, and n is an integer of 0 to 3.
さらに本発明に用いられる化合物(I)の好ましい態様としては、R1は置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC3-6シクロアルキルC1-6アルキル基であり、R2及びR3は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R4及びR5は水素原子であり、R6はヒドロキシ基であり、R7は水素原子又はヒドロキシ基であり、R8及びR9は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R10及びR11は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R12及びR13は同一又は異なって水素原子又は置換基を有していてもよいC1-6アルキル基であり、R14は置換基を有していてもよいヘテロアリール基であり、Xは窒素原子であり、Yは酸素原子であり、ZはNR15、酸素原子、結合手又は―CH=CH―であり、R15は水素原子又は置換基を有していてもよいC1-6アルキル基であり、実線と破線からなる二重線は単結合であり、mは0又は1の整数であり、nは0~1の整数である場合が挙げられる。 Further, as a preferred embodiment of the compound (I) used in the present invention, R 1 may have a substituent C 1-6 alkyl group and may have a substituent C 3-6 cycloalkyl. C 1-6 alkyl groups, R 2 and R 3 are C 1-6 alkyl groups that may have the same or different hydrogen atoms or substituents, and R 4 and R 5 are hydrogen atoms. , R 6 is a hydroxy group, R 7 is a hydrogen atom or a hydroxy group, and R 8 and R 9 are C 1-6 alkyl groups which may have the same or different hydrogen atoms or substituents. , R 10 and R 11 are C 1-6 alkyl groups that may have the same or different hydrogen atoms or substituents, and R 12 and R 13 have the same or different hydrogen atoms or substituents. May be a C 1-6 alkyl group, R 14 is a heteroaryl group which may have a substituent, X is a nitrogen atom, Y is an oxygen atom, Z is NR 15 , An oxygen atom, a bond or —CH = CH—, R15 is a C 1-6 alkyl group which may have a hydrogen atom or a substituent, and the double line consisting of a solid line and a broken line is a single bond. Yes, m is an integer of 0 or 1, and n is an integer of 0 to 1.
上記一般式(I)で表されるモルヒナン誘導体、当該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩において、薬学的に許容される塩としては、好ましくは酸付加塩が挙げられ、酸付加塩としては、例えば(イ)塩酸、硫酸、リン酸等の鉱酸との塩、(ロ)ギ酸、酢酸、クエン酸、トリクロロ酢酸、トリフルオロ酢酸、フマール酸、マレイン酸、酒石酸等の有機カルボン酸との塩、(ハ)メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、メシチレンスルホン酸、ナフタレンスルホン酸等のスルホン酸との塩との塩が挙げられる。 In the morphinan derivative represented by the above general formula (I), a homovariant of the compound, a steric isomer, or a pharmaceutically acceptable salt thereof, the pharmaceutically acceptable salt is preferably acid addition. Examples thereof include salts, and examples of the acid addition salt include (a) salts with mineral acids such as hydrochloric acid, sulfuric acid, and phosphoric acid, (b) formic acid, acetic acid, citrate, trichloroacetic acid, trifluoroacetic acid, fumaric acid, and malein. Examples thereof include a salt with an organic carboxylic acid such as acid and tartrate acid, and a salt with a sulfonic acid such as (c) methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, mesitylenesulfonic acid and naphthalenesulfonic acid.
上記一般式(I)で表されるモルヒナン誘導体、当該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物において、立体異性体としてはシス、トランス異性体、ラセミ体や光学活性体等が挙げられる。 In the morphinan derivative represented by the general formula (I), a tautomer of the compound, a stereoisomer, a pharmaceutically acceptable salt thereof, or a mixture thereof, the stereoisomers are cis and trans. Examples include isomers, racemates and optically active substances.
上記一般式(I)で表されるモルヒナン誘導体、当該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物において、水和物又は溶媒和物としても存在することができる。従って、本発明の化合物は、その全ての結晶型及び水和若しくは溶媒和物を含むものである。 As a hydrate or solvate in the morphinan derivative represented by the general formula (I), a tautomer of the compound, a stereoisomer, a pharmaceutically acceptable salt thereof, or a solvate thereof. Can also exist. Therefore, the compounds of the present invention include all their crystalline forms and hydrated or solvated products.
次に、上記一般式(I)で表されるモルヒナン誘導体、当該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物の製造方法を示す。 Next, a method for producing a morphinan derivative represented by the general formula (I), a tautomer of the compound, a stereoisomer, a pharmaceutically acceptable salt thereof, or a solvate thereof will be shown.
(方法A)R7~R11が水素原子、Xが窒素原子、実線と破線からなる二重線が単結合の場合の製造方法
(方法A-1)一般式(I)中、7員環の構成原子である窒素原子がベンジルオキシカルボニル基(Cbz)又は水素原子と結合している場合
以下に記載した方法により、発明化合物(a-10)~(a-13)を得ることができる。
(Method A) A 7-membered ring in the general formula (I) of the manufacturing method (method A-1) in which R 7 to R 11 are hydrogen atoms, X is a nitrogen atom, and a double wire consisting of a solid line and a broken line is a single bond. When the nitrogen atom, which is a constituent atom of the above, is bonded to a benzyloxycarbonyl group (Cbz) or a hydrogen atom, the invention compounds (a-10) to (a-13) can be obtained by the methods described below.

(第一工程)
原料(a-1)及び2-クロロアクリロニトリル1~10当量をベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;;塩化メチレン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類;メタノール、エタノール等のアルコール類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中、炭酸水素ナトリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム等の塩基存在下又は非存在下、80~190℃で3~24時間反応させた後、あるいは封管内で、マイクロウェーブ合成装置によりマイクロウェーブを照射して反応させた後、加水分解により化合物(a-2)を合成することができる。加水分解反応は、一般公知の方法により酸や塩基を用いて行うことができるが、塩基の方が好ましく、例えばテトラヒドロフラン、ジオキサン等のエーテル類;メタノール、エタノール等のアルコール溶媒中に、1~10mol/Lの水酸化リチウム、水酸化ナトリウム、水酸化カリウム水溶液等の無機塩基性水溶液を1~6当量加え、室温~加熱還流下で1~24時間反応させることにより行うことができる。
出発原料(a-1)は一般公知の方法により合成することができる。例えばJ.Chem.Soc.C,1966,617あるいはJ.Chem.Soc.C,1969,2569、J.Chem.Soc.Perkin Trans.I,1994,911記載の方法により合成することができる。
(First step)
Aromatic hydrocarbons such as benzene, toluene and xylene with 1 to 10 equivalents of the raw material (a-1) and 2-chloroacrylonitrile; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and jigglime; Halogenated hydrocarbons such as carbon tetrachloride, 1,2-dichloroethane; alcohols such as methanol and ethanol; aliphatic hydrocarbons such as pentane, hexane, heptane and ligroin; acetonitrile, N, N-dimethylformamide, dimethyl In an aprotonic polar solvent such as sulfoxide, in the presence or absence of a base such as sodium hydrogen carbonate, lithium carbonate, sodium carbonate, potassium carbonate, etc., after reacting at 80 to 190 ° C. for 3 to 24 hours, or in a sealed tube. The compound (a-2) can be synthesized by hydrolysis after irradiating with a hydrocarbon by a hydrocarbon synthesizer to react. The hydrolysis reaction can be carried out using an acid or a base by a generally known method, but a base is preferable, for example, ethers such as tetrahydrofuran and dioxane; 1 to 10 mol in an alcohol solvent such as methanol and ethanol. It can be carried out by adding 1 to 6 equivalents of an inorganic basic aqueous solution such as / L lithium hydroxide, sodium hydroxide, or potassium hydroxide aqueous solution and reacting at room temperature to heating and reflux for 1 to 24 hours.
The starting material (a-1) can be synthesized by a generally known method. For example, J. Chem. Soc. C, 1966, 617 or J.M. Chem. Soc. C, 1969, 2569, J.M. Chem. Soc. Perkin Trans. It can be synthesized by the method described in I, 1994, 911.
(第二工程)
化合物(a-2)を、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;水、酢酸等の溶媒;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中又はこれらの混合溶媒中、トリメチルアミン、トリエチルアミン、トリブチルアミン、ピリジン、N,N-ジメチルアニリン、N,N-ジメチルアミノピリジン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン、ナトリウムメトキシド、ナトリウムエトキシド等の有機塩基;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、水酸化ナトリウム、水酸化カリウム等の無機塩基の存在下、ヒドロキシルアミン(例えば、ヒドロキシルアミン水溶液、ヒドロキシルアミン塩酸塩、ヒドロキシルアミン硫酸塩等)と、室温~加熱還流下で1~24時間反応させることにより、化合物(a-3)を合成することができる。
(Second step)
Aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; alcohols such as methanol, ethanol and 2-propanol; water, Solvents such as acetic acid; trimethylamine, triethylamine, tributylamine, pyridine, N, N-dimethylaniline, N, N in aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide, dimethylsulfoxide, etc. -Organic bases such as dimethylaminopyridine, N-methylpiperidin, N-methylmorpholin, diethylamine, cyclohexylamine, procaine, sodium methoxydo, sodium ethoxydo; lithium carbonate, sodium carbonate, potassium carbonate, sodium hydroxide, potassium hydroxide In the presence of an inorganic base such as, the compound (a-3) is reacted with hydroxylamine (for example, an aqueous hydroxylamine solution, a hydroxylamine hydrochloride, a hydroxylamine sulfate, etc.) at room temperature to heated reflux for 1 to 24 hours. ) Can be synthesized.
(第三工程)
化合物(a-3)を、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中又は無溶媒中、カリウムビス(トリメチルシリル)アミド(KHMDS)、リチウムジイソプロピルアミド(LDA)等の塩基;トリメチルアミン、トリエチルアミン、トリブチルアミン、ピリジン、N,N-ジメチルアニリン、N,N-ジメチルアミノピリジン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン、ナトリウムメトキシド、ナトリウムエトキシド等の有機塩基;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、水酸化ナトリウム、水酸化カリウム等の無機塩基の存在下、L-Cl又はL2Oで表される試薬(Lはメタンスルホニル基、p-トルエンスルホニル基、トリフルオロメタンスルホニル基、2-ニトロベンゼンスルホニル基等を表す)と、-78℃~加熱還流下で1~24時間反応させたのち、加水分解により化合物(a-4)を合成することができる。加水分解反応は、一般公知の方法により酸を用いて行うことができ、例えばテトラヒドロフラン、ジオキサン等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒;酢酸等の溶媒中又は無溶媒中に、1~10mol/Lの塩酸又は硫酸等の無機酸性水溶液を1~6当量又は溶媒量加え、室温~加熱還流下で1~24時間反応させることにより行うことができる。
(Third step)
Aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; halogenated hydrocarbons such as methylene chloride, chloroform and carbon tetrachloride. Classes; aliphatic hydrocarbons such as pentane, hexane, heptane, ligroin; potassium bis (trimethylsilyl) amide (KHMDS) in aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide, dimethylsulfoxide, etc. , Lithium diisopropylamide (LDA) and other bases; trimethylamine, triethylamine, tributylamine, pyridine, N, N-dimethylaniline, N, N-dimethylaminopyridine, N-methylpiperidin, N-methylmorpholin, diethylamine, cyclohexylamine, Organic bases such as prokine, sodium methoxydo and sodium ethoxydo; reagents represented by L - Cl or L2O in the presence of inorganic bases such as lithium carbonate, sodium carbonate, potassium carbonate, sodium hydroxide and potassium hydroxide. (L represents a methanesulfonyl group, a p-toluenesulfonyl group, a trifluoromethanesulfonyl group, a 2-nitrobenzenesulfonyl group, etc.) and a compound after reacting with the compound at −78 ° C. to heated reflux for 1 to 24 hours. (A-4) can be synthesized. The hydrolysis reaction can be carried out using an acid by a generally known method, for example, ethers such as tetrahydrofuran and dioxane; alcohols such as methanol, ethanol and 2-propanol; acetonitrile, N, N-dimethylformamide and dimethyl. Aprotic polar solvent such as sulfoxide; 1 to 6 equivalents or an amount of an inorganic acidic aqueous solution such as sulfuric acid or 1 to 10 mol / L hydrochloric acid or sulfuric acid is added in a solvent such as acetic acid or in the absence of a solvent, and the mixture is heated at room temperature to reflux. This can be done by reacting for 1 to 24 hours.
(第四工程)
水素雰囲気下又はギ酸カリウム(1~5当量)等存在下、化合物(a-4)をジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;水、酢酸等の溶媒中又はこれらの混合溶媒中、金属触媒、例えばラネーニッケル等のニッケル触媒、パラジウム-活性炭素(Pd/C)及びパールマン触媒(Pearlman′s Catalyst:Pd(OH)2)等のパラジウム触媒、またはアダムス触媒(Adams′ Catalyst:PtO2)等の白金触媒存在下、室温~加熱還流下で1~24時間反応させることにより、化合物(a-5)を合成することができる。
(4th step)
In a hydrogen atmosphere or in the presence of potassium formate (1-5 equivalents) or the like, the compound (a-4) is an ether such as diethyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme; alcohols such as methanol, ethanol and 2-propanol; In a solvent such as water or acetic acid or in a mixed solvent thereof, a metal catalyst such as a nickel catalyst such as lane nickel, palladium-activated carbon (Pd / C) and a Pearlman catalyst (Pearlman's Catalyst: Pd (OH) 2 ) and the like. The compound (a-5) can be synthesized by reacting in the presence of a platinum catalyst such as a palladium catalyst or an Adams'catalyst (PtO 2 ) at room temperature to heating and reflux for 1 to 24 hours.
(第五工程)
化合物(a-5)を、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒;酢酸等の溶媒中、水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウム、水素化トリアセトキシホウ素ナトリウム、水素化トリ(sec-ブチル)ホウ素リチウム、水素化トリ(sec-ブチル)ホウ素カリウム等のヒドリド還元剤と、-30℃~加熱還流下で1~24時間反応させることにより、化合物(a-6)を合成することができる。
(Fifth step)
Arochemical hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; alcohols such as methanol, ethanol and 2-propanol; methylene chloride. , Hydrocarbons such as chloroform and carbon tetrachloride; aliphatic hydrocarbons such as pentane, hexane, heptane and ligroin; aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide and dimethylsulfoxide; acetic acid and the like. In the solvent of, hydride reducing agents such as sodium hydride, sodium cyanohydride, sodium hydride triacetoxyboron, hydride tri (sec-butyl) boron lithium, hydride tri (sec-butyl) boron potassium, and the like. The compound (a-6) can be synthesized by reacting at −30 ° C. to heating and reflux for 1 to 24 hours.
(第六工程)
化合物(a-6)を、ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類中、水素化アルミニウムリチウム、ボラン-テトラヒドロフラン錯体、ボラン-ジメチルスルフィド等の還元剤と、0℃~加熱還流下で1~24時間反応させることにより、化合物(a-7)を合成することができる。
(Sixth step)
The compound (a-6) is mixed with a reducing agent such as lithium aluminum hydride, borane-tetrahydrofuran complex, borane-dimethylsulfide in ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and jigglime, and heated at 0 ° C. to reflux. The compound (a-7) can be synthesized by reacting in 1 to 24 hours.
(第七工程)
化合物(a-7)を、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒;水等の溶媒中又はこれらの混合溶媒中、トリメチルアミン、トリエチルアミン、トリブチルアミン、ピリジン、N,N-ジメチルアニリン、N,N-ジメチルアミノピリジン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン、ナトリウムメトキシド、ナトリウムエトキシド等の有機塩基;炭酸水素ナトリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、水酸化ナトリウム、水酸化カリウム等の無機塩基の存在下、クロロギ酸ベンジルと、0℃~室温で1~24時間反応させることにより、化合物(a-8)を合成することができる。
(7th step)
Aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; halogenated hydrocarbons such as methylene chloride, chloroform and carbon tetrachloride. Kind; Aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide, dimethylsulfoxide; In a solvent such as water or a mixed solvent thereof, trimethylamine, triethylamine, tributylamine, pyridine, N, N-dimethylaniline, N. , N-dimethylaminopyridine, N-methylpiperidine, N-methylmorpholine, diethylamine, cyclohexylamine, procaine, sodium methoxydo, sodium ethoxydo and other organic bases; sodium hydrogencarbonate, lithium carbonate, sodium carbonate, potassium carbonate, water The compound (a-8) can be synthesized by reacting with benzyl chloride at 0 ° C. to room temperature for 1 to 24 hours in the presence of an inorganic base such as sodium oxide or potassium hydroxide.
(第八工程)
化合物(a-8)を、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中、カリウムビス(トリメチルシリル)アミド(KHMDS)、リチウムジイソプロピルアミド(LDA)等の塩基;トリメチルアミン、トリエチルアミン、トリブチルアミン、ピリジン、N,N-ジメチルアニリン、N,N-ジメチルアミノピリジン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン、ナトリウムメトキシド、ナトリウムエトキシド、カリウムtert-ブトキシド等の有機塩基;水素化リチウム、水素化ナトリウム、水素化カリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、水酸化ナトリウム、水酸化カリウム等の無機塩基の存在下、L-Cl又はL2Oで表される試薬(Lは前記と同様のものを示す。)と、-78℃~加熱還流下で1~24時間反応させることで、化合物(a-9)を合成することができる。
(8th step)
Aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; halogenated hydrocarbons such as methylene chloride, chloroform and carbon tetrachloride. Classes; aliphatic hydrocarbons such as pentane, hexane, heptane, ligroin; potassium bis (trimethylsilyl) amide (KHMDS), lithium diisopropylamide in aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide, dimethylsulfoxide, etc. Bases such as (LDA); trimethylamine, triethylamine, tributylamine, pyridine, N, N-dimethylaniline, N, N-dimethylaminopyridine, N-methylpiperidine, N-methylmorpholine, diethylamine, cyclohexylamine, procaine, sodium methoxy. Organic bases such as di, sodium ethoxydo, potassium ether-butoxide; in the presence of inorganic bases such as lithium hydride, sodium hydride, potassium hydride, lithium carbonate, sodium carbonate, potassium carbonate, sodium hydroxide, potassium hydroxide, etc. , L — Cl or L2O (L indicates the same as above) by reacting with the compound (a-9) at −78 ° C. to heating and reflux for 1 to 24 hours. Can be synthesized.
(第九工程)
化合物(a-9)を、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中、カリウムビス(トリメチルシリル)アミド(KHMDS)、リチウムジイソプロピルアミド(LDA)等の塩基;トリメチルアミン、トリエチルアミン、トリブチルアミン、ピリジン、N,N-ジメチルアニリン、N,N-ジメチルアミノピリジン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン、ナトリウムメトキシド、ナトリウムエトキシド、カリウムtert-ブトキシド等の有機塩基;水素化リチウム、水素化ナトリウム、水素化カリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、水酸化ナトリウム、水酸化カリウム等の無機塩基と、-78℃~加熱還流下で1~24時間反応させることで、発明化合物(a-10)を合成することができる。
(Ninth step)
Aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; halogenated hydrocarbons such as methylene chloride, chloroform and carbon tetrachloride. Classes; aliphatic hydrocarbons such as pentane, hexane, heptane, ligroine; potassium bis (trimethylsilyl) amide (KHMDS), lithium diisopropylamide in aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide, dimethylsulfoxide, etc. Bases such as (LDA); trimethylamine, triethylamine, tributylamine, pyridine, N, N-dimethylaniline, N, N-dimethylaminopyridine, N-methylpiperidine, N-methylmorpholine, diethylamine, cyclohexylamine, procaine, sodium methoxy. De, sodium ethoxydo, potassium ether-organic bases such as butoxide; inorganic bases such as lithium hydride, sodium hydride, potassium hydride, lithium carbonate, sodium carbonate, potassium carbonate, sodium hydroxide, potassium hydroxide, and- The invention compound (a-10) can be synthesized by reacting at 78 ° C. to heating and reflux for 1 to 24 hours.
(第十工程)
水素雰囲気下、発明化合物(a-10)をジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;水、酢酸等の溶媒中又はこれらの混合溶媒中、金属触媒、例えばラネーニッケル等のニッケル触媒、パラジウム-活性炭素(Pd/C)及びパールマン触媒(Pearlman′s Catalyst:Pd(OH)2)等のパラジウム触媒、またはアダムス触媒(Adams′ Catalyst:PtO2)等の白金触媒存在下、室温~加熱還流下で1~24時間反応させることにより、発明化合物(a-11)を合成することができる。
(10th step)
Under a hydrogen atmosphere, the invention compound (a-10) is mixed with ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; alcohols such as methanol, ethanol and 2-propanol; in a solvent such as water and acetic acid or a mixture thereof. In the solvent, a metal catalyst such as a nickel catalyst such as Raney nickel, a palladium catalyst such as palladium-activated carbon (Pd / C) and a Pearlman catalyst (Pd (OH) 2 ), or an Adams'catalyst: The invention compound (a-11) can be synthesized by reacting in the presence of a platinum catalyst such as PtO 2 ) at room temperature to heating and reflux for 1 to 24 hours.
(第十一工程)
R6がC1-6アルコキシ基の場合、発明化合物(a-11)を、塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;酢酸、酢酸エチル等の有機溶媒;アセトニトリル等の非プロトン性極性溶媒中又は無溶媒中、三臭化ホウ素、トリメチルシリルヨージド、臭化水素、塩酸ピリジニウム等と、-30~180℃で30分~5時間反応させるか、あるいはN,N-ジメチルホルムアミド、ジメチルアセトアミド、N-メチルピロリドン、ジメチルスルホキシド等の非プロトン性極性溶媒中、カリウムtert-ブトキシド、ナトリウムtert-ブトキシド等の有機塩基存在下、1-プロパンチオール、1-ドデカンチオール等を100℃~180℃で30分~24時間反応させることにより、発明化合物(a-12)を得ることができる。
(11th step)
When R 6 is a C 1-6 alkoxy group, the invention compound (a-11) is used as a halogenated hydrocarbon such as methylene chloride, chloroform or carbon tetrachloride; an organic solvent such as acetic acid or ethyl acetate; or a non-acetaken or the like. React with boron tribromide, trimethylsilyl iodide, hydrogen bromide, pyridinium hydrochloride, etc. in a protonic polar solvent or no solvent at -30 to 180 ° C. for 30 minutes to 5 hours, or N, N-dimethylformamide. , 1-Propanethiol, 1-dodecanethiol, etc. in the presence of organic bases such as potassium tert-butoxide, sodium tert-butoxide, etc. in aprotonic polar solvents such as dimethylacetamide, N-methylpyrrolidone, dimethylsulfoxide, etc. from 100 ° C. The invention compound (a-12) can be obtained by reacting at 180 ° C. for 30 minutes to 24 hours.
(第十二工程)
R6がC1-6アルコキシ基の場合、発明化合物(a-10)に対し、上記第十一工程と同様の反応を実施することにより、発明化合物(a-13)を得ることができる。
(Twelfth step)
When R 6 is a C 1-6 alkoxy group, the invention compound (a-13) can be obtained by carrying out the same reaction as in the eleventh step above with respect to the invention compound (a-10).
(第十三工程)
発明化合物(a-13)に対し、上記第十工程と同様の反応を実施することにより、発明化合物(a-12)を得ることができる。
また、化合物(a-5)は以下に記載の方法によっても合成することができる。
(13th step)
The invention compound (a-12) can be obtained by carrying out the same reaction as in the tenth step of the invention compound (a-13).
The compound (a-5) can also be synthesized by the method described below.

(第一工程)
水素雰囲気下又はギ酸カリウム(1~5当量)等存在下、化合物(a-2)をジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;水、酢酸等の溶媒又はこれらの混合溶媒中、金属触媒、例えばラネーニッケル等のニッケル触媒、パラジウム-活性炭素(Pd/C)及びパールマン触媒(Pearlman′s Catalyst:Pd(OH)2)等のパラジウム触媒、またはアダムス触媒(Adams′ catalyst:PtO2)等の白金触媒存在下、室温~加熱還流下で1~24時間反応させることにより、化合物(a-14)を合成することができる。
(First step)
In a hydrogen atmosphere or in the presence of potassium formate (1 to 5 equivalents) or the like, the compound (a-2) is an ether such as diethyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme; alcohols such as methanol, ethanol and 2-propanol; In a solvent such as water or acetic acid or a mixed solvent thereof, a metal catalyst such as a nickel catalyst such as lane nickel, palladium such as palladium-activated carbon (Pd / C) and a Pearlman catalyst (Pearlman's Catalyst: Pd (OH) 2 ) The compound (a-14) can be synthesized by reacting in the presence of a catalyst or a platinum catalyst such as Adams'catalyst (PtO 2 ) at room temperature to heating and reflux for 1 to 24 hours.
(第二工程)
化合物(a-14)を、前記化合物(a-2)から化合物(a-3)の合成における第二工程と同様にして、化合物(a-15)を合成することができる。
(Second step)
The compound (a-14) can be synthesized from the compound (a-2) in the same manner as in the second step in the synthesis of the compound (a-3).
(第三工程)及び(第四工程)
化合物(a-15)を、前記化合物(a-3)から化合物(a-4)の合成における第三工程と同様にして、化合物(a-16)及び化合物(a-5)を合成することができる。
(Third step) and (Fourth step)
The compound (a-15) is synthesized from the compound (a-3) in the same manner as in the third step in the synthesis of the compound (a-4) to synthesize the compound (a-16) and the compound (a-5). Can be done.
(方法A-2)一般式(I)中、Zが結合手、―CH=CH―又は―C≡C―、mが1の場合
以下に記載した方法により、発明化合物(a-19)~(a-22)を得ることができる。
(Method A-2) In the general formula (I), when Z is a bond, -CH = CH- or -C≡C-, and m is 1, the invention compound (a-19) to the method described below. (A-22) can be obtained.

(第一工程)
(方法A-1)で得られた発明化合物(a-11)にベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類:塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類:メタノール、エタノール等のアルコール類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中、(a-18)で表される酸ハロゲン化物をN,N-ジメチルアミノピリジン,トリメチルアミン、トリエチルアミン、トリブチルアミン、N,N-ジイソプロピルエチルアミン、ピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン等の有機塩基:炭酸カリウム、炭酸リチウム等の無機塩基の存在下又は非存在下、0℃~加熱還流下で1~12時間反応させるか、又は(a-17)で表されるカルボン酸を、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロホスファート(HATU)、O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロホスファート(HBTU)、N,N’-ジシクロへキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(WSC)、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルフォリニウムクロライドn水和物(DMT-MM)等の縮合剤存在下又は非存在下、0℃~加熱還流下で1~12時間反応させることにより、発明化合物(a-19)を合成することができる。また、発明化合物(a-19)はベンゼン、トルエン、キシレン等の芳香族炭化水素類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類中、五硫化二リン、ローソン試薬、デービー試薬、ジャパニーズ試薬、ベレオー試薬等の一般公知の硫化剤を用い、0℃~加熱還流下で1~12時間反応させることにより、発明化合物(a-20)に変換することができる。
(First step)
Aromatic hydrocarbons such as benzene, toluene and xylene to the inventive compound (a-11) obtained in (Method A-1); ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme: methylene chloride, chloroform. Hydrocarbons such as carbon tetrachloride: alcohols such as methanol and ethanol; aliphatic hydrocarbons such as pentane, hexane, heptane and ligroin; aprotons such as acetonitrile, N, N-dimethylformamide and dimethylsulfoxide. In the sexual polar solvent, the acid halide represented by (a-18) is N, N-dimethylaminopyridine, trimethylamine, triethylamine, tributylamine, N, N-diisopropylethylamine, pyridine, N, N-dimethylaniline, N. -Organic bases such as methylpiperidin, N-methylmorpholine, diethylamine, cyclocarbonate, prokine: Reaction in the presence or absence of inorganic bases such as potassium carbonate, lithium carbonate, 0 ° C. to heated reflux for 1 to 12 hours. Alternatively, the carboxylic acid represented by (a-17) can be replaced with O- (7-azabenzotriazole-1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate (HATU). ), O- (benzotriazole-1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate (HBTU), N, N'-dicyclohexylcarbodiimide (DCC), 1- Ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC), 4- (4,6-dimethoxy-1,3,5-triazine-2-yl) -4-methylmorpholinium chloride n hydrate The compound of invention (a-19) can be synthesized by reacting the compound (DMT-MM) in the presence or absence of a condensing agent at 0 ° C. to heating and refluxing for 1 to 12 hours. In addition, the invented compound (a-19) is an aromatic hydrocarbon such as benzene, toluene or xylene, ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme; halogenation of methylene chloride, chloroform, carbon tetrachloride and the like. Hydrocarbons; among aliphatic hydrocarbons such as pentane, hexane, heptane, and ligroin, heat from 0 ° C. using a generally known sulfide agent such as diphosphorus pentasulfide, Lawesson's reagent, Davy reagent, Japanese reagent, and bereo reagent. It can be converted to the invention compound (a-20) by reacting under reflux for 1 to 12 hours.
(第二工程)
R6がC1-6アルコキシ基の場合、発明化合物(a-19)又は(a-20)に(方法A-1)の第十一工程と同様の反応を実施することにより、発明化合物(a-21)又は(a-22)をそれぞれ得ることができる。
(Second step)
When R 6 is a C 1-6 alkoxy group, the invention compound (a-19) or (a-20) is subjected to the same reaction as in the eleventh step of (Method A-1). a-21) or (a-22) can be obtained, respectively.
(方法A-3)一般式(I)中、Yが酸素原子、ZがNR15又は酸素原子、mが1の場合
以下に記載した方法により、発明化合物(a-23)、(a-24)及び(a-27)~(a-28)を得ることができる。
(Method A-3) In the general formula (I), when Y is an oxygen atom, Z is NR 15 or an oxygen atom, and m is 1, the invention compounds (a-23) and (a-24) are according to the methods described below. ) And (a-27) to (a-28) can be obtained.

(第一工程)
(方法A-1)で得られた発明化合物(a-11)に、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類:塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類:メタノール、エタノール等のアルコール類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中、N,N-ジメチルアミノピリジン,トリメチルアミン、トリエチルアミン、トリブチルアミン、N,N-ジイソプロピルエチルアミン、ピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン等の有機塩基:炭酸カリウム、炭酸リチウム等の無機塩基の存在下、カルボジイミダゾールを0℃~加熱還流下で1~12時間反応させることにより、発明化合物(a-23)を合成することができる。
(First step)
In the compound of invention (a-11) obtained in (Method A-1), aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme: methylene chloride, Halogenated hydrocarbons such as chloroform and carbon tetrachloride: alcohols such as methanol and ethanol; aliphatic hydrocarbons such as pentane, hexane, heptane and ligroin; non-alkoxy, N, N-dimethylformamide, dimethylsulfoxide and the like. N, N-dimethylaminopyridine, trimethylamine, triethylamine, tributylamine, N, N-diisopropylethylamine, pyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylmorpholine, diethylamine, cyclohexyl in a protonic polar solvent. Organic bases such as amines and prokines: In the presence of inorganic bases such as potassium carbonate and lithium carbonate, the invention compound (a-23) is synthesized by reacting carbodiimidazole at 0 ° C. to heating and reflux for 1 to 12 hours. can do.
(第二工程)
発明化合物(a-23)に、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類:塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類:メタノール、エタノール等のアルコール類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中、N,N-ジメチルアミノピリジン,トリメチルアミン、トリエチルアミン、トリブチルアミン、N,N-ジイソプロピルエチルアミン、ピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン等の有機塩基:炭酸カリウム、炭酸リチウム等の無機塩基の存在下又は非存在下、ヨウ化メチル、硫酸ジメチル、トリフルオロメタンスルホネート、meerwein試薬等のメチル化剤を、0℃~加熱還流下で12~48時間反応させることにより、発明化合物(a-24)を合成することができる。
(Second step)
Aromatic hydrocarbons such as benzene, toluene and xylene in the compound of the invention (a-23); ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme: halogenated hydrocarbons such as methylene chloride, chloroform and carbon tetrachloride. Hydrogens: Alcohols such as methanol and ethanol; aliphatic hydrocarbons such as pentane, hexane, heptane and ligroins; N, N- in aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide and dimethylsulfoxide. Organic bases such as dimethylaminopyridine, trimethylamine, triethylamine, tributylamine, N, N-diisopropylethylamine, pyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylmorpholine, diethylamine, cyclohexylamine, prokine: potassium carbonate , In the presence or absence of an inorganic base such as lithium carbonate, a methylating agent such as methyl iodide, dimethyl sulfate, trifluoromethanesulfonate, meerween reagent, etc. is reacted at 0 ° C. to heated reflux for 12 to 48 hours. , The invention compound (a-24) can be synthesized.
(第三工程)
発明化合物(a-24)にベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類;塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中、N,N-ジメチルアミノピリジン、トリメチルアミン、トリエチルアミン、N,N-ジイソプロピルエチルアミン、トリブチルアミン、ピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン等の有機塩基:炭酸カリウム、炭酸リチウム等の無機塩基の存在下、(a-25)で表されるアミン又は(a-26)で表されるアルコールを、0℃~加熱還流下で1~12時間反応させることにより、発明化合物(a-27)を合成することができる。
(Third step)
Aromatic hydrocarbons such as benzene, toluene and xylene to the compound (a-24) of the invention; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and jigglime; halogenated hydrocarbons such as methylene chloride, chloroform and carbon tetrachloride Classes; aliphatic hydrocarbons such as pentane, hexane, heptane, ligroin; N, N-dimethylaminopyridine, trimethylamine, triethylamine, N in aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide, dimethylsulfoxide, etc. , N-diisopropylethylamine, tributylamine, pyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylmorpholine, diethylamine, cyclohexylamine, prokine and other organic bases: presence of inorganic bases such as potassium carbonate and lithium carbonate The compound (a-27) of the invention is synthesized by reacting the amine represented by (a-25) or the alcohol represented by (a-26) below at 0 ° C. to heated reflux for 1 to 12 hours. can do.
(第四工程)、
R6がC1-6アルコキシ基の場合、発明化合物(a-27)に(方法A-1)の第十一工程と同様にして、発明化合物(a-28)を得ることができる。
(4th step),
When R 6 is a C 1-6 alkoxy group, the invention compound (a-28) can be obtained from the invention compound (a-27) in the same manner as in the eleventh step of (method A-1).
(方法A-4)一般式(I)中mが0、Zが結合手の場合
以下に記載した方法により、発明化合物(a-30)~(a-31)を得ることができる。
(Method A-4) When m is 0 and Z is a bond in the general formula (I) The invention compounds (a-30) to (a-31) can be obtained by the methods described below.
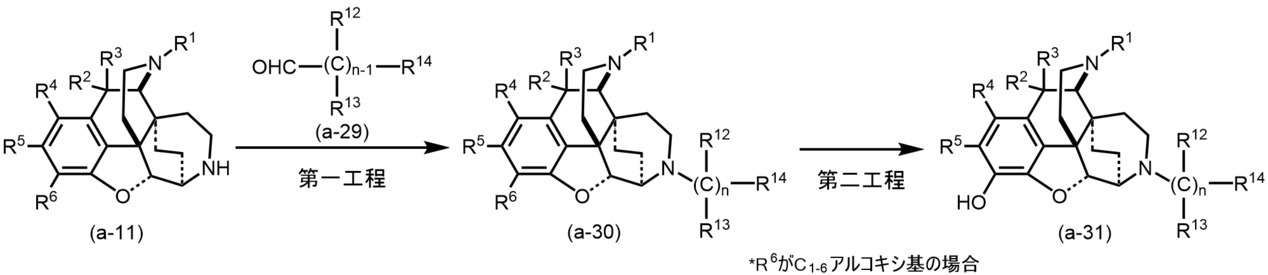
(第一工程)
(方法A-1)で得られた発明化合物(a-11)に、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類:塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類:メタノール、エタノール等のアルコール類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒;酢酸等の溶媒中又はこれらの混合溶媒中、水素化ホウ素ナトリウム、シアノ化水素化ホウ素ナトリウム、水素化トリアセトキシホウ素ナトリウム、水素化トリ(sec-ブチル)ホウ素リチウム、水素化トリ(sec-ブチル)ホウ素カリウム等のヒドリド還元剤の存在下、(a-23)で表されるアルデヒドを0℃~加熱還流下で1~12時間反応させることにより、発明化合物(a-30)を合成することができる。
(First step)
In the compound of invention (a-11) obtained in (Method A-1), aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme: methylene chloride, Halogenated hydrocarbons such as chloroform and carbon tetrachloride: alcohols such as methanol and ethanol; aliphatic hydrocarbons such as pentane, hexane, heptane and ligroin; non-alkoxy, N, N-dimethylformamide, dimethylsulfoxide and the like. Protonic polar solvent; in a solvent such as acetic acid or a mixed solvent thereof, sodium borohydride, sodium borohydride borohydride, sodium triacetoxyborohydride, tri (sec-butyl) boron borohydride, tri borohydride. The invented compound (a-30) is obtained by reacting the aldehyde represented by (a-23) at 0 ° C. to heating and refluxing for 1 to 12 hours in the presence of a hydride reducing agent such as (sec-butyl) potassium borohydride. Can be synthesized.
(第二工程)
R6がC1-6アルコキシ基の場合、発明化合物(a-30)に(方法A-1)の第十一工程と同様にして、発明化合物(a-31)を得ることができる。
(Second step)
When R 6 is a C 1-6 alkoxy group, the invention compound (a-31) can be obtained from the invention compound (a-30) in the same manner as in the eleventh step of (method A-1).
(方法A-5)一般式(I)中、m及びnが0、Zが結合手、R14が置換基を有していてもよいC6-10アリール基又はヘテロアリール基の場合
以下に記載した方法により、発明化合物(a-32)~(a-33)を得ることができる。
(Method A-5) In the general formula (I), when m and n are 0, Z is a bond, and R 14 may have a substituent, C 6-10 aryl group or heteroaryl group may have the following. The invention compounds (a-32) to (a-33) can be obtained by the method described.
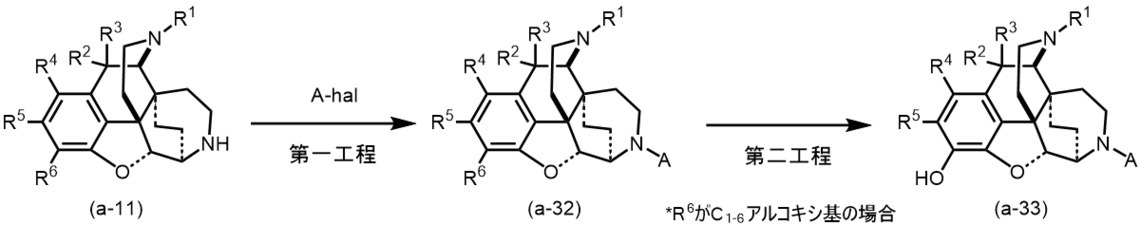
(第一工程)
(方法A-1)で得られた発明化合物(a-11)に、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類:塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類:メタノール、エタノール等のアルコール類;ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒中又はこれらの混合溶媒中、
テトラキストリフェニルホスフィンパラジウム(0)、酢酸パラジウム(II)、ビス(ジベンジリデンアセトン)パラジウム(0)、ビス(トリフェニルホスフィン)パラジウム(0)ジクロリド、パラジウム(II)クロリド等のパラジウム触媒存在下又は非存在下、(±)-2,2′-ビス(ジフェニルホスフィノ)-1,1′-ビナフチル(BINAP)、1,1′-ビス(ジフェニルホスフィノ)フェロセン(dppf)、2-ジシクロヘキシルホスフィノ-2′,4′,6′-トリイソプロピルビフェニル(Xphos)、2-ジシクロヘキシルホスフィノ-2′,6′-ジメトキシビフェニル(Sphos)、4,5′-ビス(ジフェニルホスフィノ)-9,9′-ジメチルキサンテン(xantphos)等のリン配位子存在下又は非存在下、N,N-ジメチルアミノピリジン,トリメチルアミン、トリエチルアミン、トリブチルアミン、N,N-ジイソプロピルエチルアミン、ピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルモルホリン、ジエチルアミン、シクロヘキシルアミン、プロカイン、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウムtert-ブトキシド、カリウムtert-ブトキシド等の有機塩基;炭酸カリウム、炭酸リチウム等の無機塩基等の塩基の存在下又は非存在下、A-halと室温~加熱還流下で1~24時間反応させることにより、発明化合物(a-32)を合成することができる。
(First step)
In the compound of invention (a-11) obtained in (Method A-1), aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme: methylene chloride, Halogenated hydrocarbons such as chloroform and carbon tetrachloride: alcohols such as methanol and ethanol; aliphatic hydrocarbons such as pentane, hexane, heptane and ligroin; non-polymers such as acetonitrile, N, N-dimethylformamide and dimethylsulfoxide. In a protonic polar solvent or in a mixed solvent thereof,
In the presence of palladium catalysts such as tetrakistriphenylphosphine palladium (0), palladium (II) acetate, bis (dibenzilidenacetone) palladium (0), bis (triphenylphosphine) palladium (0) dichloride, palladium (II) chloride or In the absence of (±) -2,2'-bis (diphenylphosphino) -1,1'-binaphthyl (BINAP), 1,1'-bis (diphenylphosphino) ferrocene (dppff), 2-dicyclohexylphos Fino-2', 4', 6'-triisopropylbiphenyl (Xphos), 2-dicyclohexylphosphino-2', 6'-dimethoxybiphenyl (Sphos), 4,5'-bis (diphenylphosphino) -9, N, N-dimethylaminopyridine, trimethylamine, triethylamine, tributylamine, N, N-diisopropylethylamine, pyridine, N, N-dimethyl in the presence or absence of a phosphorus ligand such as 9'-dimethylxanthene (xantphos). Organic bases such as aniline, N-methylpiperidin, N-methylmorpholin, diethylamine, cyclohexylamine, prokine, sodium methoxydo, sodium ethoxydo, sodium tert-butoxide, potassium tert-butoxide; inorganic bases such as potassium carbonate and lithium carbonate. The compound of invention (a-32) can be synthesized by reacting A-hal with A-hal at room temperature to heating and refluxing for 1 to 24 hours in the presence or absence of such a base.
(第二工程)
R6がC1-6アルコキシ基の場合、発明化合物(a-32)に(方法A-1)の第十一工程と同様にして、発明化合物(a-33)を得ることができる。
(Second step)
When R 6 is a C 1-6 alkoxy group, the invention compound (a-33) can be obtained from the invention compound (a-32) in the same manner as in the eleventh step of (method A-1).
(方法B)一般式(I)中、R7がヒドロキシ基、R8とR9が一緒になってカルボニル基、R10及びR11が水素原子、Xが窒素原子、実線と破線からなる二重線が単結合の場合の製造方法
以下に記載した方法により、発明化合物(b-1)~(b-2)を得ることができる。
(Method B) In the general formula (I), R 7 is a hydroxy group, R 8 and R 9 are combined to form a carbonyl group, R 10 and R 11 are hydrogen atoms, X is a nitrogen atom, and a solid line and a broken line are formed. Production method when the heavy wire is a single bond The invention compounds (b-1) to (b-2) can be obtained by the method described below.
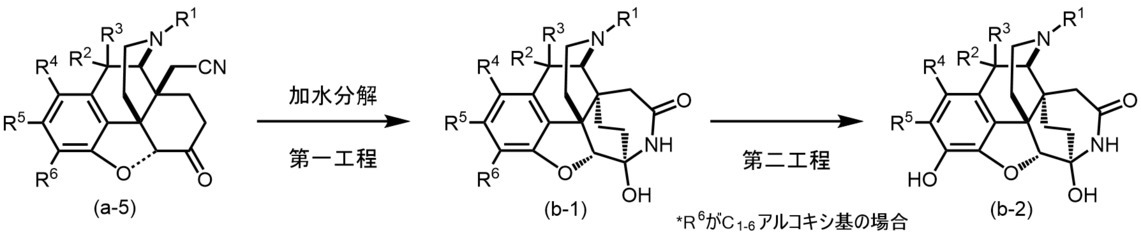
(第一工程)
化合物(a-5)から発明化合物(b-1)への変換は、シアノ基の加水分解反応により実施可能である。加水分解反応は、一般公知の方法により酸又は塩基を用いて行うことができる。酸による加水分解の場合は、例えばテトラヒドロフラン、ジオキサン等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;アセトニトリル、N,N-ジメチルホルムアミド、ジメチルスルホキシド等の非プロトン性極性溶媒;酢酸等の溶媒中、これらの混合溶媒中又は無溶媒中に、1~12mol/Lの塩酸又は硫酸等の無機酸性水溶液を1~6当量又は溶媒量加え、室温~加熱還流下で1~24時間反応させることにより行うことができる。塩基による加水分解の場合は、例えばテトラヒドロフラン、ジオキサン等のエーテル類;メタノール、エタノール、2-プロパノール等のアルコール類;ジメチルスルホキシド等の非プロトン性極性溶媒;水等の溶媒中又はこれらの混合溶媒中に、水酸化ナトリウム、水酸化カリウム等の無機塩基又はこれらの水溶液1~6当量を、室温~加熱還流下で1~24時間反応させることにより行うことができる。
(First step)
The conversion of the compound (a-5) to the invention compound (b-1) can be carried out by a hydrolysis reaction of a cyano group. The hydrolysis reaction can be carried out using an acid or a base by a generally known method. In the case of acid hydrolysis, for example, ethers such as tetrahydrofuran and dioxane; alcohols such as methanol, ethanol and 2-propanol; aprotonic polar solvents such as acetonitrile, N, N-dimethylformamide and dimethylsulfoxide; acetic acid and the like. 1 to 6 equivalents or an amount of an inorganic acidic aqueous solution such as 1 to 12 mol / L hydrochloric acid or sulfuric acid is added to the above solvent, a mixed solvent thereof, or no solvent, and the reaction is carried out at room temperature to heating and reflux for 1 to 24 hours. It can be done by letting it. In the case of hydrolysis with a base, for example, ethers such as tetrahydrofuran and dioxane; alcohols such as methanol, ethanol and 2-propanol; aprotonic polar solvents such as dimethylsulfoxide; in a solvent such as water or a mixed solvent thereof. It can be carried out by reacting 1 to 6 equivalents of an inorganic base such as sodium hydroxide or potassium hydroxide or an aqueous solution thereof for 1 to 24 hours at room temperature to heating and reflux.
(第二工程)
R6がC1-6アルコキシ基の場合、発明化合物(b-1)に(方法A-1)の第十一工程と同様にして、発明化合物(b-2)を得ることができる。
(Second step)
When R 6 is a C 1-6 alkoxy group, the invention compound (b-2) can be obtained from the invention compound (b-1) in the same manner as in the eleventh step of (method A-1).
(方法C)一般式(I)中、R7がヒドロキシ基、R8~R11が水素原子、Xが窒素原子、実線と破線からなる二重線が単結合の場合の製造方法
以下に記載した方法により、発明化合物(c-1)~(c-3)を得ることができる。
(Method C) In the general formula (I), R 7 is a hydroxy group, R 8 to R 11 are hydrogen atoms, X is a nitrogen atom, and a double wire consisting of a solid line and a broken line is a single bond. The invention compounds (c-1) to (c-3) can be obtained by the above-mentioned method.

(第一工程)
(方法B)で得られた発明化合物(b-1)に、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、テトラヒドロフラン、ジオキサン、モノグライム、ジグライム等のエーテル類:塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類:ペンタン、ヘキサン、ヘプタン、リグロイン等の脂肪族炭化水素類中、ボラン-ジメチルスルフィド錯体、ボラン-テトラヒドロフラン錯体テトラヒドロフラン溶液又は水素化アルミニウムリチウム等の還元剤を、室温~加熱還流下で2~24時間反応させることにより、発明化合物(c-1)を合成することができる。
(First step)
In the compound of invention (b-1) obtained in (Method B), aromatic hydrocarbons such as benzene, toluene and xylene; ethers such as diethyl ether, tetrahydrofuran, dioxane, monoglyme and diglyme: methylene chloride, chloroform, etc. Hydrocarbons such as carbon tetrachloride: Among aliphatic hydrocarbons such as pentane, hexane, heptane, and ligroin, a reducing agent such as borane-dimethylsulfide complex, borane-tetratetracomplex tetrahydrofuran solution or lithium aluminum hydride. The invention compound (c-1) can be synthesized by reacting at room temperature to heating and reflux for 2 to 24 hours.
(第二工程)
発明化合物(c-1)に対し、上記(方法A-2)~(方法A-5)に記載の方法と同様の反応を実施することにより、発明化合物(c-2)を合成することができる。
(Second step)
The invention compound (c-2) can be synthesized by carrying out the same reaction with the invention compound (c-1) as described in the above (method A-2) to (method A-5). can.
(第三工程)
R6がC1-6アルコキシ基の場合、発明化合物(c-2)に(方法A-1)と同様にして、発明化合物(c-3)を得ることができる。
(Third step)
When R 6 is a C 1-6 alkoxy group, the invention compound (c-3) can be obtained from the invention compound (c-2) in the same manner as in (method A-1).
(方法D)一般式(I)中、XがNオキシド、実線と破線からなる二重線が単結合の場合の製造方法
以下に記載した方法により、発明化合物(d-2)を得ることができる。
(Method D) In the general formula (I), the invention compound (d-2) can be obtained by the method described below when X is an N oxide and the double bond consisting of a solid line and a broken line is a single bond. can.
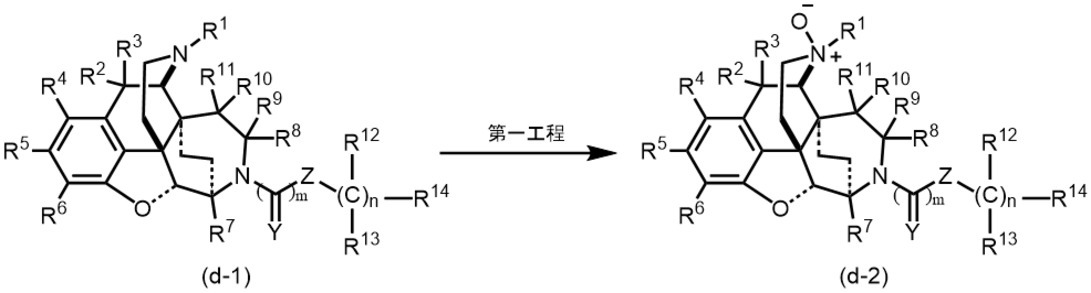
(第一工程)
上記(方法A-1)~(方法A-5)、(方法B)、(方法C)の記載の方法により得られた発明化合物(d-1)を、塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化炭化水素溶媒中、過酸化水素(H2O2)水溶液又はm-クロロ過安息香酸(mCPBA)等の酸化剤1~2当量を、0℃~室温で30分~24時間反応させることにより、発明化合物(d-2)を合成することができる。
(First step)
The invention compound (d-1) obtained by the methods described in (Method A-1) to (Method A-5), (Method B), and (Method C) can be used as methylene chloride, chloroform, carbon tetrachloride, or the like. 1 to 2 equivalents of an oxidizing agent such as an aqueous solution of hydrogen peroxide (H 2 O 2 ) or m-chloroperbenzoic acid (mCPBA) is reacted at 0 ° C. to room temperature for 30 minutes to 24 hours in the halogenated hydrocarbon solvent. Thereby, the invention compound (d-2) can be synthesized.
前記各工程により得られる化合物については、必要に応じ、例えばシリカゲルカラムクロマトグラフィーにより精製することができる。
さらに必要に応じて常法により酸付加塩を形成することができ、例えば発明化合物(I)を、酢酸エチル等の有機溶媒:メタノール、エタノール等のようなアルコール類:あるいは水等の極性溶媒中、本発明化合物(I)を塩酸、硫酸、リン酸等の鉱酸、ギ酸、酢酸、クエン酸、トリクロロ酢酸、トリフルオロ酢酸、フマール酸、マレイン酸等の有機カルボン酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、メシチレンスルホン酸、ナフタレンスルホン酸等の有機スルホン酸等の存在下、室温又は適宜加熱することにより行われる。
The compound obtained by each of the above steps can be purified, for example, by silica gel column chromatography, if necessary.
Further, if necessary, an acid addition salt can be formed by a conventional method. For example, the invention compound (I) can be contained in an organic solvent such as ethyl acetate: alcohols such as methanol and ethanol: or a polar solvent such as water. The compound (I) of the present invention is a mineral acid such as hydrochloric acid, sulfuric acid or phosphoric acid, an organic carboxylic acid such as formic acid, acetic acid, citric acid, trichloroacetic acid, trifluoroacetic acid, fumaric acid or maleic acid, methanesulfonic acid or benzenesulfon. It is carried out by heating at room temperature or appropriately in the presence of an organic sulfonic acid such as an acid, p-toluene sulfonic acid, mesitylane sulfonic acid or naphthalene sulfonic acid.
上記一般式(I)で表されるモルヒナン誘導体、当該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物は、ヒトに対して非経口投与、固形若しくは液体形態での経口投与等のための製薬上許容し得る担体とともに組成物を処方することができる。また、他の鎮痛薬と併用することも可能である。 The morphinan derivative represented by the above general formula (I), tautomer, stereoisomer of the compound, or a pharmaceutically acceptable salt thereof or a solvate thereof is orally administered to humans. The composition can be formulated with a pharmaceutically acceptable carrier for oral administration, etc. in solid or liquid form. It can also be used in combination with other analgesics.
経口投与のための固形製剤としてはカプセル剤、錠剤、丸剤、散剤及び顆粒剤等が挙げられる。この固形製剤の調製にあたっては賦形剤、崩壊剤、結合剤、滑沢剤、色素などを用いることができる。ここで、賦形剤としては、乳糖、D-マンニトール、結晶セルロース、ブドウ糖などが、崩壊剤としては、デンプン、カルボキシメチルセルロースカルシウム(CMC-Ca)などが、滑沢剤としては、ステアリン酸マグネシウム、タルクなどが、結合剤としては、ヒドロキシプロピルセルロース(HPC)、ゼラチン、ポリビニルピロリドン(PVP)などが挙げられる。カプセル剤、錠剤及び丸剤の場合には、更に、緩衝剤を用いてもよい。錠剤及び丸剤には腸溶性被膜を施してもよい。 Examples of the solid preparation for oral administration include capsules, tablets, pills, powders and granules. Excipients, disintegrants, binders, lubricants, dyes and the like can be used in the preparation of this solid preparation. Here, as an excipient, lactose, D-mannitol, crystalline cellulose, glucose and the like are used, starch, carboxymethyl cellulose calcium (CMC-Ca) and the like are used as a disintegrant, and magnesium stearate is used as a lubricant. Examples of the binder include hydroxypropyl cellulose (HPC), gelatin, polyvinylpyrrolidone (PVP) and the like. In the case of capsules, tablets and pills, a buffer may be further used. Tablets and pills may be enteric coated.
注射剤のための本発明組成物の形態としては、製薬上許容し得る無菌水若しくは非水溶液、懸濁液若しくは乳濁液が挙げられる。適当な非水担体、希釈剤、溶媒又はビヒクルの例としては、プロピレングリコール、ポリエチレングリコール、植物油、例えばオリーブ油及び注射可能な有機エステル、例えばオレイン酸エチルが挙げられる。このような組成物は補助剤、例えば防腐剤、湿潤剤、乳化剤、無痛化剤、緩衝剤、保存剤及び分散剤をも含有することができる。
これら組成物は例えば細菌保持フィルターによる濾過により、又は使用直前に減菌剤あるいは若干の他の減菌注射可能な媒質に溶解し得る無菌固形組成物の形態で減菌剤を混入することにより減菌することができる。
Forms of the compositions of the invention for injections include pharmaceutically acceptable sterile water or non-aqueous solutions, suspensions or emulsions. Examples of suitable non-aqueous carriers, diluents, solvents or vehicles include propylene glycol, polyethylene glycol, vegetable oils such as olive oil and injectable organic esters such as ethyl oleate. Such compositions can also contain auxiliaries such as preservatives, wetting agents, emulsifiers, soothing agents, buffers, preservatives and dispersants.
These compositions are reduced, for example, by filtration through a bacterial retention filter or by mixing the sterilizer in the form of a sterile solid composition that can be dissolved in a sterilizer or some other sterilized injectable medium immediately prior to use. Can be fungus.
点眼投与のための製剤は、好ましくは本発明化合物に加えて、溶解補助剤、保存剤、等張化剤及び増粘剤等を加えることができる。 The preparation for eye drop administration is preferably added with a lysis aid, a preservative, an isotonic agent, a thickener and the like in addition to the compound of the present invention.
経口投与のための液体製剤には、当業者間で普通に使用される不活性希釈剤、例えば水を含む製薬上許容し得る乳剤、溶液、懸濁剤、シロップ剤及びエリキシール剤が挙げられる。かかる不活性希釈剤に加えて、組成物には補助剤例えば湿潤剤、乳化、懸濁剤、ならびに甘味、調味及び香味剤も配合することができる。 Liquid formulations for oral administration include inactive diluents commonly used by those of skill in the art, such as pharmaceutically acceptable emulsions, solutions, suspensions, syrups and elixirs containing water. In addition to such Inactive Diluents, the composition may also contain auxiliaries such as wetting agents, emulsifying and suspending agents, as well as sweetening, seasoning and flavoring agents.
経直腸投与のための製剤は、好ましくは本発明化合物に加えて賦形剤例えばカカオ脂若しくは坐剤ワックスを含有していてもよい。 The preparation for transrectal administration may preferably contain an excipient such as cocoa butter or a suppository wax in addition to the compound of the present invention.
投与量は、通常成人においては、有効成分である上記一般式(I)で表されるモルヒナン誘導体、当該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物を、注射剤においては、0.01μg~1g/日、好ましくは0.0001~200mg/日、経口投与においては、0.1μg~10g/日、好ましくは0.001~2000mg/日投与されるが、年齢、症状等により増減することができる。また、所望によりこの一日量を2~4回に分割して投与することもできる。 The dose is usually the active ingredient, a morphinan derivative represented by the above general formula (I), a homovariant of the compound, a stereoisomer, or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable salt thereof. The solvent mixture is 0.01 μg to 1 g / day, preferably 0.0001 to 200 mg / day for injection, and 0.1 μg to 10 g / day, preferably 0.001 to 2000 mg / day for oral administration. It is administered, but it can be increased or decreased depending on age, symptoms, etc. Further, if desired, this daily dose may be administered in 2 to 4 divided doses.
オピオイドκ受容体に関連する疾患や症状としては、例えば心血管系障害、消化器系疾患、血液系疾患、呼吸器系疾患、肝疾患、神経系障害、泌尿器系障害、疼痛、咳嗽、掻痒、虚血性脳疾患、薬物依存などが挙げられる。本発明化合物は、高いオピオイドκ受容体選択性及びオピオイドκ受容体に対する強力なアゴニスト活性を有することから、これらの疾患や症状の治療や改善、予防に有効である。 Diseases and symptoms related to opioid κ receptors include, for example, cardiovascular disorders, digestive disorders, hematological disorders, respiratory disorders, liver disorders, nervous system disorders, urinary disorders, pain, coughing, pruritus, etc. Ischemic brain disease, drug dependence, etc. may be mentioned. Since the compound of the present invention has high opioid κ receptor selectivity and strong agonist activity against opioid κ receptors, it is effective for the treatment, amelioration, and prevention of these diseases and symptoms.
次に、参考例、実施例及び試験例を挙げ、本発明を更に詳細に説明するが、本発明はこれらに限定されるものではない。 Next, the present invention will be described in more detail with reference to reference examples, examples and test examples, but the present invention is not limited thereto.
(参考例1)
(4R,4aR,7S,7aR,12bS)-3-(シクロプロピルメチル)-7,9-ジメトキシ-1,2,3,4,7,7a-ヘキサヒドロ-4a,7-エタノ-4,12-メタノベンゾフロ[3,2-e]イソキノリン-14-オン(1)の合成
(Reference example 1)
(4R, 4aR, 7S, 7aR, 12bS) -3- (Cyclopropylmethyl) -7,9-dimethoxy-1,2,3,4,7,7a-Hexahydro-4a, 7-Etano-4,12- Synthesis of methanobenzoflo [3,2-e] isoquinoline-14-one (1)

(4R,7aR,12bS)-3-(シクロプロピルメチル)-7,9-ジメトキシ-2,3,4,7a-テトラヒドロ-1H-4,12-メタノベンゾフロ[3,2-e]イソキノリン(J.Chem.Soc.C,1966,617、J.Chem.Soc.C,1969,2569およびJ.Chem.Soc.Perkin Trans.I,1994,911に記載の方法で合成)(2.0g,5.63mmol)の1,2-ジクロロエタン(10mL)溶液をマイクロウェーブ反応用のバイアルに入れ、2-クロロアクリロニトリル(4.5mL,56.6mmol)を加えて密封したものを3本用意した。マイクロフェーブ合成装置にて、それぞれマイクロウェーブを照射し、180°C、10barの条件下で30分間反応させた。放冷後、3本のバイアルの内容物を合わせ、減圧下にて濃縮した。残渣をシリカゲルカラムクロマトグラフィー(25-50%酢酸エチル/ヘキサン)にて精製した。得られた濃縮残渣をエタノール(144mL)に溶解し、1M水酸化ナトリウム水溶液(36mL)を加えて3時間加熱還流した。放冷後、水(200mL)を加え、ジエチルエーテルで二回抽出した。有機層を飽和食塩水で2回洗浄し、硫酸マグネシウムで乾燥後、減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(25-50%酢酸エチル/ヘキサン)で精製し、表題化合物(2.5g,44%)を無色アモルファスとして得た。
(方法b)
(4R,7aR,12bS)-3-(シクロプロピルメチル)-7,9-ジメトキシ-2,3,4,7a-テトラヒドロ-1H-4,12-メタノベンゾフロ[3,2-e]イソキノリン(19g,54mmol)のキシレン(180mL)溶液に、炭酸水素ナトリウム(42g,540mmol)及び2-クロロアクリロニトリル(34.4mL,432mmol)を加え、140°Cで6時間撹拌した。放冷後、反応液に水を加え酢酸エチルで二回抽出した。合わせた抽出物を飽和食塩水で洗浄し、硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物に対し、(方法a)と同様の加水分解反応を行うことで、表題化合物(15.2g,71%)を淡黄色固体として得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.08-0.20(m,2H),0.45-0.60(m,2H),0.77-0.91(m,1H),1.87(dd,J=3,13Hz,1H),2.07(ddd,J=5,13,13Hz,1H),2.17(d,J=19Hz,1H),2.32-2.55(m,4H),2.77(dd,J=5,12Hz,1H),3.17(d,J=18Hz,1H),3.32(d,J=19Hz,1H),3.63(s,3H),3.65(d,J=7Hz,1H),3.83(s,3H),4.68(d,J=1Hz,1H),5.70(d,J=9Hz,1H),5.88-5.93(m,1H),6.57(d,J=8Hz,1H),6.66(d,J=8Hz,1H).
(4R, 7aR, 12bS) -3- (Cyclopropylmethyl) -7,9-dimethoxy-2,3,4,7a-tetrahydro-1H-4,12-methanobenzoflo [3,2-e] isoquinoline (J. Synthesized by the method described in Chem. Soc. C, 1966, 617, J. Chem. Soc. C, 1969, 2569 and J. Chem. Soc. Perkin Trans. I, 1994, 911) (2.0 g, 5. A solution of 1,2-dichloroethane (10 mL) (63 mmol) was placed in a vial for a microwave reaction, 2-chloroacrylonitrile (4.5 mL, 56.6 mmol) was added, and three sealed bottles were prepared. Each of them was irradiated with microwaves in a microfave synthesizer and reacted at 180 ° C. and 10 bar for 30 minutes. After allowing to cool, the contents of the three vials were combined and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (25-50% ethyl acetate / hexane). The obtained concentrated residue was dissolved in ethanol (144 mL), a 1 M aqueous sodium hydroxide solution (36 mL) was added, and the mixture was heated under reflux for 3 hours. After allowing to cool, water (200 mL) was added, and the mixture was extracted twice with diethyl ether. The organic layer was washed twice with saturated brine, dried over magnesium sulfate, and concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (25-50% ethyl acetate / hexane) to give the title compound (2.5 g, 44%) as a colorless amorphous substance.
(Method b)
(4R, 7aR, 12bS) -3- (Cyclopropylmethyl) -7,9-dimethoxy-2,3,4,7a-tetrahydro-1H-4,12-methanobenzoflo [3,2-e] isoquinoline (19g, Sodium hydrogen carbonate (42 g, 540 mmol) and 2-chloroacrylonitrile (34.4 mL, 432 mmol) were added to a solution of (54 mmol) xylene (180 mL), and the mixture was stirred at 140 ° C. for 6 hours. After allowing to cool, water was added to the reaction solution, and the mixture was extracted twice with ethyl acetate. The combined extracts were washed with saturated brine, dried over sodium sulfate, and concentrated under reduced pressure. The obtained crude product was subjected to the same hydrolysis reaction as in (Method a) to give the title compound (15.2 g, 71%) as a pale yellow solid.
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.08-0.20 (m, 2H), 0.45-0.60 (m, 2H), 0.77-0.91 (m) , 1H), 1.87 (dd, J = 3,13Hz, 1H), 2.07 (ddd, J = 5,13,13Hz, 1H), 2.17 (d, J = 19Hz, 1H), 2 .32-2.55 (m, 4H), 2.77 (dd, J = 5,12Hz, 1H), 3.17 (d, J = 18Hz, 1H), 3.32 (d, J = 19Hz, 1H), 3.63 (s, 3H), 3.65 (d, J = 7Hz, 1H), 3.83 (s, 3H), 4.68 (d, J = 1Hz, 1H), 5.70 (D, J = 9Hz, 1H), 5.88-5.93 (m, 1H), 6.57 (d, J = 8Hz, 1H), 6.66 (d, J = 8Hz, 1H).
(参考例2)
(4R,4aR,7R,7aR,12bS)-3-(シクロプロピルメチル)-7,9-ジメトキシ-1,2,3,4,7,7a-ヘキサヒドロ-4a,7-エタノ-4,12-メタノベンゾフロ[3,2-e]イソキノリン-14-オン オキシム(2)の合成
(Reference example 2)
(4R, 4aR, 7R, 7aR, 12bS) -3- (Cyclopropylmethyl) -7,9-dimethoxy-1,2,3,4,7,7a-Hexahydro-4a, 7-Etano-4,12- Synthesis of methanobenzoflo [3,2-e] isoquinoline-14-one oxime (2)

1H-NMR(400MHz,CDCl3)δ(ppm):0.11-0.19(m,2H),0.47-0.58(m,2H),0.83-0.93(m,1H),1.80(dd,J=2,13Hz,1H),2.02(ddd,J=6,13,13Hz,1H),2.10(d,J=18Hz,1H),2.34-2.49(m,4H),2.75(dd,J=5,12Hz,1H),3.14(d,J=18Hz,1H),3.64(d,J=6Hz,1H),3.66(s,3H),3.71(d,J=18Hz,1H),3.83(s,3H),4.64(d,J=1Hz,1H),5.62(d,J=9Hz,1H),6.00(dd,J=1,9Hz,1H),6.54(d,J=8Hz,1H),6.63(d,J=8Hz,1H),7.57(br s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.11-0.19 (m, 2H), 0.47-0.58 (m, 2H), 0.83-0.93 (m) , 1H), 1.80 (dd, J = 2,13Hz, 1H), 2.02 (ddd, J = 6,13,13Hz, 1H), 2.10 (d, J = 18Hz, 1H), 2 .34-2.49 (m, 4H), 2.75 (dd, J = 5,12Hz, 1H), 3.14 (d, J = 18Hz, 1H), 3.64 (d, J = 6Hz, 1H), 3.66 (s, 3H), 3.71 (d, J = 18Hz, 1H), 3.83 (s, 3H), 4.64 (d, J = 1Hz, 1H), 5.62 (D, J = 9Hz, 1H), 6.00 (dd, J = 1,9Hz, 1H), 6.54 (d, J = 8Hz, 1H), 6.63 (d, J = 8Hz, 1H) , 7.57 (br s, 1H).
(参考例3)
2-((4R,4aR,7aR,12bS)-3-(シクロプロピルメチル)-9-メトキシ-7-オキソ-1,2,3,4,7,7a-ヘキサヒドロ-4aH-4,12-メタノベンゾフロ[3,2-e]イソキノリン-4a-イル)アセトニトリル(3)の合成
(Reference example 3)
2-((4R, 4aR, 7aR, 12bS) -3- (cyclopropylmethyl) -9-methoxy-7-oxo-1,2,3,4,7,7a-hexahydro-4aH-4,12-methanobenzoflo) [3,2-e] Isoquinoline-4a-yl) Synthesis of acetonitrile (3)

1H-NMR(400MHz,CDCl3)δ(ppm):0.11-0.19(m,2H),0.52-0.61(m,2H),0.80-0.90(m,1H),1.71-1.77(m,1H),2.14-2.29(m,2H),2.34-2.47(m,3H),2.78-2.83(m,1H),3.11(d,J=17Hz,1H),3.14(d,J=16Hz,1H),3.38(d,J=5Hz,1H),3.55(d,J=16Hz,1H),3.84(s,3H),4.93(s,1H),6.24(d,J=11Hz,1H),6.61(d,J=8Hz,1H),6.67(d,J=11Hz,1H),6.69(d,J=8Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.11-0.19 (m, 2H), 0.52-0.61 (m, 2H), 0.80-0.90 (m) , 1H), 1.71-1.77 (m, 1H), 2.14-2.29 (m, 2H), 2.34-2.47 (m, 3H), 2.78-2.83 (M, 1H), 3.11 (d, J = 17Hz, 1H), 3.14 (d, J = 16Hz, 1H), 3.38 (d, J = 5Hz, 1H), 3.55 (d) , J = 16Hz, 1H), 3.84 (s, 3H), 4.93 (s, 1H), 6.24 (d, J = 11Hz, 1H), 6.61 (d, J = 8Hz, 1H) ), 6.67 (d, J = 11Hz, 1H), 6.69 (d, J = 8Hz, 1H).
(参考例4)
2-((4R,4aS,7aR,12bS)-3-(シクロプロピルメチル)-9-メトキシ-7-オキソ-1,2,3,4,5,6,7,7a-オクタヒドロ-4aH-4,12-メタノベンゾフロ[3,2-e]イソキノリン-4a-イル)アセトニトリル(4)の合成
(Reference example 4)
2-((4R, 4aS, 7aR, 12bS) -3- (cyclopropylmethyl) -9-methoxy-7-oxo-1,2,3,4,5,6,7,7a-octahydro-4aH-4 , 12-Methylnobenzoflo [3,2-e] isoquinoline-4a-yl) Synthesis of acetonitrile (4)

1H-NMR(400MHz,CDCl3)δ(ppm):0.10-0.21(m,2H),0.50-0.59(m,2H),0.82-0.92(m,1H),1.52-1.60(m,1H),1.69(ddd,J=4,14,14Hz,1H),1.96-2.18(m,3H),2.31-2.53(m,4H),2.55-2.64(m,1H),2.70-2.78(m,1H),2.97(d,J=16Hz,1H),3.02(d,J=18Hz,1H),3.35(d,J=6Hz,1H),3.89(s,3H),4.09(dd,J=1,16Hz,1H),4.61(s,1H),6.67(d,J=8Hz,1H),6.70(d,J=8Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.10-0.21 (m, 2H), 0.50-0.59 (m, 2H), 0.82-0.92 (m) , 1H), 1.52-1.60 (m, 1H), 1.69 (ddd, J = 4,14,14Hz, 1H), 1.96-2.18 (m, 3H), 2.31 -2.53 (m, 4H), 2.55-2.64 (m, 1H), 2.70-2.78 (m, 1H), 2.97 (d, J = 16Hz, 1H), 3 .02 (d, J = 18Hz, 1H), 3.35 (d, J = 6Hz, 1H), 3.89 (s, 3H), 4.09 (dd, J = 1,16Hz, 1H), 4 .61 (s, 1H), 6.67 (d, J = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H).
(参考例5)
2-((4R,4aS,7S,7aR,12bS)-3-(シクロプロピルメチル)-7-ヒドロキシ-9-メトキシ-1,2,3,4,5,6,7,7a-オクタヒドロ-4aH-4,12-メタノベンゾフロ[3,2-e]イソキノリン-4a-イル)アセトニトリル(5)の合成
(Reference example 5)
2-((4R, 4aS, 7S, 7aR, 12bS) -3- (cyclopropylmethyl) -7-hydroxy-9-methoxy-1,2,3,4,5,6,7,7a-octahydro-4aH -4,12-Methylanobenzoflo [3,2-e] isoquinoline-4a-yl) Synthesis of acetonitrile (5)
1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.17(m,2H),0.49-0.60(m,2H),0.85-0.92(m,1H),1.09-1.18(m,1H),1.38-1.47(m,1H),1.49-1.54(m,1H),1.67-1.80(m,2H),1.99(ddd,J=5,12,12Hz,1H),2.20(ddd,J=4,12,12Hz,1H),2.27-2.40(m,4H),2.56(dd,J=6,19Hz,1H),2.67(dd,J=5,12Hz,1H),2.98(d,J=19Hz,1H),3.38(d,J=6Hz,1H),3.87(s,3H),3.99-4.06(m,2H),4.60(d,J=5Hz,1H),6.65(d,J=8Hz,1H),6.74(d,J=8Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.17 (m, 2H), 0.49-0.60 (m, 2H), 0.85-0.92 (m) , 1H), 1.09-1.18 (m, 1H), 1.38-1.47 (m, 1H), 1.49-1.54 (m, 1H), 1.67-1.80 (M, 2H), 1.99 (ddd, J = 5,12,12Hz, 1H), 2.20 (ddd, J = 4,12,12Hz, 1H), 2.27-2.40 (m, 4H), 2.56 (dd, J = 6,19Hz, 1H), 2.67 (dd, J = 5,12Hz, 1H), 2.98 (d, J = 19Hz, 1H), 3.38 ( d, J = 6Hz, 1H), 3.87 (s, 3H), 3.99-4.06 (m, 2H), 4.60 (d, J = 5Hz, 1H), 6.65 (d, J = 8Hz, 1H), 6.74 (d, J = 8Hz, 1H).
(参考例6)
ベンジル (2-((4R,4aS,7S,7aR,12bS)-3-(シクロプロピルメチル)-7-ヒドロキシ-9-メトキシ-1,2,3,4,5,6,7,7a-オクタヒドロ-4aH-4,12-メタノベンゾフロ[3,2-e]イソキノリン-4a-イル)エチル)カーバメート(6)の合成
(Reference example 6)
Benzyl (2-((4R, 4aS, 7S, 7aR, 12bS) -3- (cyclopropylmethyl) -7-hydroxy-9-methoxy-1,2,3,4,5,6,7,7a-octahydro Synthesis of -4aH-4,12-methanobenzoflo [3,2-e] isoquinoline-4a-yl) ethyl) carbamate (6)

1H-NMR(400MHz,CDCl3)δ(ppm):0.02-0.10(m,2H),0.35-0.51(m,2H),0.79-0.90(m,1H),0.91-1.04(m,1H),1.12-1.22(m,1H),1.37-1.49(m,2H),1.51-1.69(m,2H),2.02-2.15(m,2H),2.26(ddd,J=6,13,13Hz,1H),2.39-2.48(m,3H),2.75-2.85(m,2H),2.92(d,J=19Hz,1H),3.06(d,J=6Hz,1H),3.22-3.42(m,2H),3.86(s,3H),4.04-4.15(m,1H),4.61(d,J=5Hz,1H),5.08(d,J=12Hz,1H),5.12(d,J=12Hz,1H),5.38(br s,1H),6.60(d,J=8Hz,1H),6.71(d,J=8Hz,1H),7.27-7.40(m,5H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.02-0.10 (m, 2H), 0.35-0.51 (m, 2H), 0.79-0.90 (m) , 1H), 0.91-1.04 (m, 1H), 1.12-1.22 (m, 1H), 1.37-1.49 (m, 2H), 1.51-1.69 (M, 2H), 2.02-2.15 (m, 2H), 2.26 (ddd, J = 6,13,13Hz, 1H), 2.39-2.48 (m, 3H), 2 .75-2.85 (m, 2H), 2.92 (d, J = 19Hz, 1H), 3.06 (d, J = 6Hz, 1H), 3.22-3.42 (m, 2H) , 3.86 (s, 3H), 4.04-4.15 (m, 1H), 4.61 (d, J = 5Hz, 1H), 5.08 (d, J = 12Hz, 1H), 5 .12 (d, J = 12Hz, 1H), 5.38 (br s, 1H), 6.60 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 7. 27-7.40 (m, 5H).
(参考例7)
(4R,4aS,7S,7aR,12bS)-4a-(2-(((ベンジロキシ)カルボニル)アミノ)エチル)-3-(シクロプロピルメチル)-9-メトキシ-2,3,4,4a,5,6,7,7a-オクタヒドロ-1H-4,12-メタノベンゾフロ[3,2-e]イソキノリン-7-イル メタンスルホネート(7)の合成
(Reference example 7)
(4R, 4aS, 7S, 7aR, 12bS) -4a- (2-(((Bendiroxy) carbonyl) amino) ethyl) -3- (cyclopropylmethyl) -9-methoxy-2,3,4,4a, 5 , 6,7,7a-Octahydro-1H-4,12-methanobenzoflo [3,2-e] Isoquinoline-7-yl methanesulfonate (7) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.10(m,2H),0.41-0.51(m,2H),0.79-0.85(m,1H),1.25-1.60(m,5H),1.80-1.90(m,1H),2.05-2.24(m,3H),2.36-2.46(m,2H),2.70-2.80(m,1H),2.90-3.00(m,2H),3.09(s,3H),3.10-3.13(m,1H),3.19-3.40(m,2H),3.86(s,3H),4.69(d,J=6Hz,1H),5.00-5.23(m,4H),6.60(d,J=8Hz,1H),6.72(d,J=8Hz,1H),7.29-7.41(m,5H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-0.10 (m, 2H), 0.41-0.51 (m, 2H), 0.79-0.85 (m) , 1H), 1.25-1.60 (m, 5H), 1.80-1.90 (m, 1H), 2.05-2.24 (m, 3H), 2.36-2.46 (M, 2H), 2.70-2.80 (m, 1H), 2.90-3.00 (m, 2H), 3.09 (s, 3H), 3.10-3.13 (m) , 1H), 3.19-3.40 (m, 2H), 3.86 (s, 3H), 4.69 (d, J = 6Hz, 1H), 5.00-5.23 (m, 4H) ), 6.60 (d, J = 8Hz, 1H), 6.72 (d, J = 8Hz, 1H), 7.29-7.41 (m, 5H).
(実施例1)
ベンジル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(8)の合成
(Example 1)
Benzyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxylate (8)

化合物7(173mg,0.29mmol)のテトラヒドロフラン(5mL)溶液に、カリウムtert-ブトキシド(39mg,0.35mmol)を加え、室温で30分間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液加え、酢酸エチルで三回抽出した。合わせた抽出物を硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(40-80%酢酸エチル/へプタン)で精製し、表題化合物(102.3mg,71%)を無色アモルファスとして得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.43-0.52(m,2H),0.73-0.83(m,1H),1.00-1.13(m,1H),1.21-1.52(m,4H),1.61-1.78(m,1H),2.14-2.43(m,5H),2.62(dd,J=5,11Hz,1H),2.76-2.90(m,1H),2.97(d,J=18Hz,1H),3.01(d,J=6Hz,1H),3.34-3.45(m,1H),3.86(s,1.8H),3.87(s,1.2H),4.00-4.15(m,1H),4.23-4.35(m,1H),4.46-4.56(m,1H),5.10-5.23(m,2H),6.55-6.61(m,1H),6.71(d,J=8Hz,1H),7.29-7.40(m,5H).
Potassium tert-butoxide (39 mg, 0.35 mmol) was added to a solution of compound 7 (173 mg, 0.29 mmol) in tetrahydrofuran (5 mL), and the mixture was stirred at room temperature for 30 minutes. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted three times with ethyl acetate. The combined extracts were dried over sodium sulfate and then concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (40-80% ethyl acetate / heptane) to give the title compound (102.3 mg, 71%) as a colorless amorphous substance.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.43-0.52 (m, 2H), 0.73-0.83 (m) , 1H), 1.00-1.13 (m, 1H), 1.21-1.52 (m, 4H), 1.61-1.78 (m, 1H), 2.14-2.43 (M, 5H), 2.62 (dd, J = 5,11Hz, 1H), 2.76-2.90 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.01 (D, J = 6Hz, 1H), 3.34-3.45 (m, 1H), 3.86 (s, 1.8H), 3.87 (s, 1.2H), 4.00-4 .15 (m, 1H), 4.23-4.35 (m, 1H), 4.46-4.56 (m, 1H), 5.10-15.23 (m, 2H), 6.55 -6.61 (m, 1H), 6.71 (d, J = 8Hz, 1H), 7.29-7.40 (m, 5H).
(実施例2)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン(9)の合成
(Example 2)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-methoxy-1,2,3,4,6,7,8,8a-octahydro-5H-4a, 8-ethano- Synthesis of 4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine (9)
化合物8(614.9mg,1.23mmol)のメタノール(12mL)溶液に、20%水酸化パラジウム-活性炭素(50%wet)(120mg)を加え、水素雰囲気下、室温で40分撹拌した。反応混合物に20%水酸化パラジウム-活性炭素(50%wet)(300mg)を追加し、更に2時間撹拌した。反応混合物をセライトろ過し、ろ液を減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(0-10% 10%アンモニア-メタノール/クロロホルム)で精製し、表題化合物(422.2mg,94%)を黒色油状物質として得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.14(m,2H),0.42-0.55(m,2H),0.76-0.83(m,1H),0.99-1.09(m,1H),1.25-1.42(m,3H),1.50-1.58(m,1H),1.61-1.65(m,1H),2.21(dd,J=7,12Hz,1H),2.26-2.41(m,4H),2.58-2.65(m,1H),2.76-2.86(m,1H),2.95(d,J=18Hz,1H),2.96-3.03(m,2H),3.12-3.22(m,2H),3.86(s,3H),4.50-4.52(m,1H),6.57(d,J=8Hz,1H),6.71(d,J=8Hz,1H).
20% Palladium hydroxide-activated carbon (50% wet) (120 mg) was added to a solution of compound 8 (614.9 mg, 1.23 mmol) in methanol (12 mL), and the mixture was stirred at room temperature for 40 minutes under a hydrogen atmosphere. 20% Palladium hydroxide-activated carbon (50% wet) (300 mg) was added to the reaction mixture, and the mixture was further stirred for 2 hours. The reaction mixture was filtered through Celite and the filtrate was concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (0-10% 10% ammonia-methanol / chloroform) to give the title compound (422.2 mg, 94%) as a black oily substance.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.14 (m, 2H), 0.42-0.55 (m, 2H), 0.76-0.83 (m) , 1H), 0.99-1.09 (m, 1H), 1.25-1.42 (m, 3H), 1.50-1.58 (m, 1H), 1.61-1.65 (M, 1H), 2.21 (dd, J = 7, 12Hz, 1H), 2.26-2.41 (m, 4H), 2.58-2.65 (m, 1H), 2.76 -2.86 (m, 1H), 2.95 (d, J = 18Hz, 1H), 2.96-3.03 (m, 2H), 3.12-3.22 (m, 2H), 3 .86 (s, 3H), 4.50-4.52 (m, 1H), 6.57 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H).
(実施例3)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(1H-イミダゾール-1-イル)メタノン(10)の合成
(Example 3)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano -4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (1H-imidazole-1-yl) Synthesis of methanone (10)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.44-0.54(m,2H),0.72-0.82(m,1H),1.11-1.21(m,1H),1.25-1.37(m,1H),1.43-1.55(m,2H),1.62-1.65(m,1H),1.72-1.84(m,1H),2.17-2.45(m,5H),2.66(dd,J=5,12Hz,1H),2.89-2.98(m,1H),3.01(d,J=18Hz,1H),3.06(d,J=6Hz,1H),3.58-3.67(m,1H),3.80-3.87(m,1H),3.88(s,3H),4.46(br s,1H),4.68(s,1H),6.62(d,J=8Hz,1H),6.75(d,J=8Hz,1H),7.10-7.13(m,1H),7.24-7.26(m,1H),7.91(s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.44-0.54 (m, 2H), 0.72-0.82 (m) , 1H), 1.11-1.21 (m, 1H), 1.25-1.37 (m, 1H), 1.43-1.55 (m, 2H), 1.62-1.65 (M, 1H), 1.72-1.84 (m, 1H), 2.17-2.45 (m, 5H), 2.66 (dd, J = 5,12Hz, 1H), 2.89 -2.98 (m, 1H), 3.01 (d, J = 18Hz, 1H), 3.06 (d, J = 6Hz, 1H), 3.58-3.67 (m, 1H), 3 .80-3.87 (m, 1H), 3.88 (s, 3H), 4.46 (br s, 1H), 4.68 (s, 1H), 6.62 (d, J = 8Hz, 1H), 6.75 (d, J = 8Hz, 1H), 7.10-7.13 (m, 1H), 7.24-7.26 (m, 1H), 7.91 (s, 1H) ..
(実施例4)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボニル)-3-メチル-1H-イミダゾール-3-イウム ヨージド(11)の合成
(Example 4)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,6,7,8,8a-octahydro-5H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carbonyl) -3-methyl-1H-imidazole-3-ium iodide (11)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.14(m,2H),0.42-0.54(m,2H),0.70-0.82(m,1H),1.07-1.18(m,1H),1.37-1.71(m,4H),1.96-2.12(m,1H),2.20-2.37(m,4H),2.43(dd,J=6,18Hz,1H),2.65(d,J=8Hz,1H),2.86-2.99(m,1H),2.99(d,J=18Hz,1H),3.10(d,J=6Hz,1H),3.88(s,3H),3.90-4.00(m,1H),4.07-4.24(m,1H),4.27(s,3H),4.36(br s,1H),4.69(br s,1H),6.63(d,J=8Hz,1H),6.75(d,J=8Hz,1H),7.33-7.37(m,1H),7.66-7.70(m,1H),10.64(br s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-0.14 (m, 2H), 0.42-0.54 (m, 2H), 0.70-0.82 (m) , 1H), 1.07-1.18 (m, 1H), 1.37-1.71 (m, 4H), 1.96-2.12 (m, 1H), 2.20-2.37 (M, 4H), 2.43 (dd, J = 6,18Hz, 1H), 2.65 (d, J = 8Hz, 1H), 2.86-2.99 (m, 1H), 2.99 (D, J = 18Hz, 1H), 3.10 (d, J = 6Hz, 1H), 3.88 (s, 3H), 3.90-4.00 (m, 1H), 4.07-4 .24 (m, 1H), 4.27 (s, 3H), 4.36 (br s, 1H), 4.69 (br s, 1H), 6.63 (d, J = 8Hz, 1H), 6.75 (d, J = 8Hz, 1H), 7.33-7.37 (m, 1H), 7.66-7.70 (m, 1H), 10.64 (br s, 1H).
(参考例8)
tert-ブチル (2-((メチルアミノ)メチル)ピリジン-3-イル)カーバメート(12)の合成
(Reference example 8)
Synthesis of tert-butyl (2-((methylamino) methyl) pyridin-3-yl) carbamate (12)

1H-NMR(400MHz,CDCl3)δ(ppm):1.54(s,9H),2.45(s,3H),4.04(s,2H),7.18(dd,J=5,8Hz,1H),8.13(d,J=5Hz,1H),8.34(d,J=8Hz,1H),9.92(br s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 1.54 (s, 9H), 2.45 (s, 3H), 4.04 (s, 2H), 7.18 (dd, J = 5,8Hz, 1H), 8.13 (d, J = 5Hz, 1H), 8.34 (d, J = 8Hz, 1H), 9.92 (br s, 1H).
(実施例5)
tert-ブチル (2-(((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-N―メチル-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド)メチル)ピリジン-3-イル)カーバメート(13)の合成
(Example 5)
tert-Butyl (2-(((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-N-methyl-1,2,3,4,6,7,8, 8a-Octahydro-5H-4a, 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide) methyl) pyridine-3-yl) carbamate (13) Synthesis of

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.45-0.53(m,2H),0.74-0.84(m,1H),1.04-1.10(m,1H),1.25-1.71(m,14H),2.20-2.42(m,5H),2.60-2.64(m,1H),2.85-2.94(m,1H),2.94(s,3H),2.98(d,J=18Hz,1H),3.05(d,J=6Hz,1H),3.41(ddd,J=4,11,14Hz,1H),3.54(ddd,J=4,4,14Hz,1H),3.87(s,3H),4.09-4.12(m,1H),4.52(s,2H),4.59(s,1H),6.58(d,J=8Hz,1H),6.72(d,J=8Hz,1H),7.22(dd,J=4,8Hz,1H),8.20(dd,J=1,4Hz,1H),8.42(d,J=8Hz,1H),10.1(s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.45-0.53 (m, 2H), 0.74-0.84 (m) , 1H), 1.04-1.10 (m, 1H), 1.25-1.71 (m, 14H), 2.20-2.42 (m, 5H), 2.60-2.64 (M, 1H), 2.85-2.94 (m, 1H), 2.94 (s, 3H), 2.98 (d, J = 18Hz, 1H), 3.05 (d, J = 6Hz) , 1H), 3.41 (ddd, J = 4,11,14Hz, 1H), 3.54 (ddd, J = 4,4,14Hz, 1H), 3.87 (s, 3H), 4.09 -4.12 (m, 1H), 4.52 (s, 2H), 4.59 (s, 1H), 6.58 (d, J = 8Hz, 1H), 6.72 (d, J = 8Hz) , 1H), 7.22 (dd, J = 4.8Hz, 1H), 8.20 (dd, J = 1,4Hz, 1H), 8.42 (d, J = 8Hz, 1H), 10.1 (S, 1H).
(実施例6)
(4R,4aS,8R,8aR,13bS)-N-((3-アセタミドピリジン-2-イル)メチル)-3-(シクロプロピルメチル)-10-メトキシ-N―メチル-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(14)の合成
(Example 6)
(4R, 4aS, 8R, 8aR, 13bS) -N-((3-acetamidopyridin-2-yl) methyl) -3- (cyclopropylmethyl) -10-methoxy-N-methyl-1,2, 3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (14) Synthesis of

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.45-0.54(m,2H),0.75-0.84(m,1H),1.05-1.12(m,1H),1.25-1.71(m,5H),2.13-2.43(m,8H),2.61-2.65(m,1H),2.86-2.95(m,4H),2.99(d,J=18Hz,1H),3.07(d,J=6Hz,1H),3.42(ddd,J=4,11,14Hz,1H),3.55(ddd,J=4,4,14Hz,1H),3.87(s,3H),4.09-4.12(m,1H),4.51(d,J=14Hz,1H),4.60(d,J=14Hz,1H),4.61(s,1H),6.60(d,J=8Hz,1H),6.73(d,J=8Hz,1H),7.26(dd,J=5,8Hz,1H),8.25(dd,J=1,5Hz,1H),8.71(dd,J=1,8Hz,1H),11.4(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.45-0.54 (m, 2H), 0.75-0.84 (m) , 1H), 1.05-1.12 (m, 1H), 1.25-1.71 (m, 5H), 2.13-2.43 (m, 8H), 2.61-2.65 (M, 1H), 2.86-2.95 (m, 4H), 2.99 (d, J = 18Hz, 1H), 3.07 (d, J = 6Hz, 1H), 3.42 (ddd) , J = 4,11,14Hz, 1H), 3.55 (ddd, J = 4,4,14Hz, 1H), 3.87 (s, 3H), 4.09-4.12 (m, 1H) , 4.51 (d, J = 14Hz, 1H), 4.60 (d, J = 14Hz, 1H), 4.61 (s, 1H), 6.60 (d, J = 8Hz, 1H), 6 .73 (d, J = 8Hz, 1H), 7.26 (dd, J = 5,8Hz, 1H), 8.25 (dd, J = 1,5Hz, 1H), 8.71 (dd, J = 1.8Hz, 1H), 11.4 (s, 1H).
(実施例7)
(4R,4aS,8R,8aR,13bS)-N-((3-アセタミドピリジン-2-イル)メチル)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(15)の合成
(Example 7)
(4R, 4aS, 8R, 8aR, 13bS) -N-((3-acetamidopyridin-2-yl) methyl) -3- (cyclopropylmethyl) -10-hydroxy-N-methyl-1,2, 3,4,5,6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-Methylnobenzoflo [2,3-c] Pyridine [4,3-d] Azepine-7-Carboxamide (15) Synthesis of

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.45-0.54(m,2H),0.74-0.84(m,1H),1.04-1.12(m,1H),1.25-1.72(m,5H),2.21-2.40(m,8H),2.61-2.65(m,1H),2.85-2.93(m,4H),2.96(d,J=18Hz,1H),3.06(d,J=6Hz,1H),3.43(ddd,J=4,11,14Hz,1H),3.55(ddd,J=4,4,14Hz,1H),4.06-4.09(m,1H),4.53(d,J=14Hz,1H),4.60(s,1H),4.64(d,J=14Hz,1H),6.53(d,J=8Hz,1H),6.70(d,J=8Hz,1H),7.27(dd,J=5,8Hz,1H),8.26(dd,J=1,5Hz,1H),8.74(dd,J=1,8Hz,1H),11.3(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.45-0.54 (m, 2H), 0.74-0.84 (m) , 1H), 1.04-1.12 (m, 1H), 1.25-1.72 (m, 5H), 2.21-2.40 (m, 8H), 2.61-2.65 (M, 1H), 2.85-2.93 (m, 4H), 2.96 (d, J = 18Hz, 1H), 3.06 (d, J = 6Hz, 1H), 3.43 (ddd) , J = 4,11,14Hz, 1H), 3.55 (ddd, J = 4,4,14Hz, 1H), 4.06-4.09 (m, 1H), 4.53 (d, J = 14Hz, 1H), 4.60 (s, 1H), 4.64 (d, J = 14Hz, 1H), 6.53 (d, J = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H), 7.27 (dd, J = 5,8Hz, 1H), 8.26 (dd, J = 1,5Hz, 1H), 8.74 (dd, J = 1,8Hz, 1H), 11. 3 (s, 1H).
(実施例8)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-N-(2-フルオロ-6-ヒドロキシベンジル)-10-ヒドロキシ-N―メチル-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(16)の合成
(Example 8)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -N- (2-Fluoro-6-Hydroxybenzyl) -10-Hydroxy-N-Methyl-1,2,3,4,5 , 6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] synthesis of azepine-7-carboxamide (16)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.41-0.53(m,2H),0.73-0.82(m,1H),1.01-1.10(m,1H),1.21-1.30(m,1H),1.33-1.55(m,3H),1.57-1.70(m,1H),2.12-2.40(m,5H),2.52-2.66(m,1H),2.82(s,3H),2.85-2.98(m,2H),3.02(d,J=6Hz,1H),3.38-3.47(m,1H),3.55-3.62(m,1H),4.05(br s,1H),4.35(d,J=15Hz,1H),4.45(d,J=15Hz,1H),4.64(br s,1H),6.49-6.61(m,2H),6.65-6.78(m,2H),7.09-7.17(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.41-0.53 (m, 2H), 0.73-0.82 (m) , 1H), 1.01-1.10 (m, 1H), 1.21-1.30 (m, 1H), 1.33-1.55 (m, 3H), 1.57-1.70 (M, 1H), 2.12-2.40 (m, 5H), 2.52-2.66 (m, 1H), 2.82 (s, 3H), 2.85-2.98 (m) , 2H), 3.02 (d, J = 6Hz, 1H), 3.38-3.47 (m, 1H), 3.55-3.62 (m, 1H), 4.05 (br s, 1H), 4.35 (d, J = 15Hz, 1H), 4.45 (d, J = 15Hz, 1H), 4.64 (br s, 1H), 6.49-6.61 (m, 2H) ), 6.65-6.78 (m, 2H), 7.09-7.17 (m, 1H).
(参考例9)
6-(トリフルオロメチル)ピコリンアルデヒド(17)の合成
(Reference example 9)
Synthesis of 6- (trifluoromethyl) picoline aldehyde (17)

1H-NMR(400MHz,CDCl3)δ(ppm):7.93(dd,J=1,8Hz,1H),8.10(dd,J=8,8Hz,1H),8.16(dd,J=1,8Hz,1H),10.1(s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 7.93 (dd, J = 1,8 Hz, 1H), 8.10 (dd, J = 8,8 Hz, 1H), 8.16 (dd) , J = 1.8Hz, 1H), 10.1 (s, 1H).
(参考例10)
N-メチル-1-(6-(トリフルオロメチル)ピリジン-2-イル)メタンアミン(18)の合成
(Reference example 10)
Synthesis of N-methyl-1- (6- (trifluoromethyl) pyridin-2-yl) methaneamine (18)

1H-NMR(400MHz,CDCl3)δ(ppm):2.45(s,3H),3.95(s,2H),7.53-7.57(m,2H),7.83(dd,J=8,8Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 2.45 (s, 3H), 3.95 (s, 2H), 7.53-7.57 (m, 2H), 7.83 ( dd, J = 8,8 Hz, 1H).
(実施例9)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-N-((6-(トリフルオロメチル)ピリジン-2-イル)メチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(19)の合成
(Example 9)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-N-methyl-N-((6- (trifluoromethyl) pyridin-2-yl) methyl) -1, 2,3,4,5,6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-Methylnobenzoflo [2,3-c] Pyridine [4,3-d] Azepine-7-Carboxamide ( 19) Synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.42-0.52(m,2H),0.71-0.80(m,1H),0.99-1.07(m,1H),1.19-1.65(m,5H),2.15-2.37(m,5H),2.54-2.62(m,1H),2.78-2.86(m,1H),2.88(s,3H),2.92(d,J=18Hz,1H),3.01(d,J=6Hz,1H),3.35-3.44(m,1H),3.61-3.70(m,1H),4.10-4.13(m,1H),4.53(d,J=17Hz,1H),4.56(d,J=17Hz,1H),4.60(s,1H),6.48(d,J=8Hz,1H),6.73(d,J=8Hz,1H),7.56(dd,J=8,8Hz,2H),7.82(dd,J=8,8Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.42-0.52 (m, 2H), 0.71-0.80 (m) , 1H), 0.99-1.07 (m, 1H), 1.19-1.65 (m, 5H), 2.15-2.37 (m, 5H), 2.54-2.62 (M, 1H), 2.78-2.86 (m, 1H), 2.88 (s, 3H), 2.92 (d, J = 18Hz, 1H), 3.01 (d, J = 6Hz) , 1H), 3.35-3.44 (m, 1H), 3.61-3.70 (m, 1H), 4.10-4.13 (m, 1H), 4.53 (d, J) = 17Hz, 1H), 4.56 (d, J = 17Hz, 1H), 4.60 (s, 1H), 6.48 (d, J = 8Hz, 1H), 6.73 (d, J = 8Hz) , 1H), 7.56 (dd, J = 8,8Hz, 2H), 7.82 (dd, J = 8,8Hz, 1H).
(参考例11)
2-((メチルアミノ)メチル)ピリジン-3-オール(20)の合成
(Reference example 11)
Synthesis of 2-((Methylamino) Methyl) Pyridine-3-ol (20)
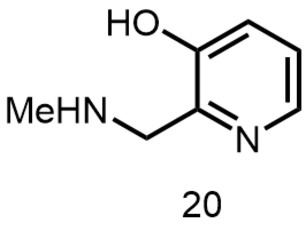
1H-NMR(400MHz,CDCl3)δ(ppm):2.52(s,3H),4.18(s,2H),5.10-6.00(m,2H),7.09-7.10(m,2H),8.00-8.01(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 2.52 (s, 3H), 4.18 (s, 2H), 5.10-16.00 (m, 2H), 7.09- 7.10 (m, 2H), 8.00-8.01 (m, 1H).
(実施例10)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N-((3-ヒドロキシピリジン-2-イル)メチル)-N-メチル-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(21)の合成
(Example 10)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-N-((3-hydroxypyridin-2-yl) methyl) -N-methyl-1,2,3 Synthesis of 4,5,6,8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (21)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.15(m,2H),0.42-0.53(m,2H),0.73-0.83(m,1H),1.02-1.14(m,1H),1.22-1.71(m,5H),2.20-2.41(m,5H),2.60-2.67(m,1H),2.85-3.00(m,2H),2.94(s,3H),3.04(d,J=6Hz,1H),3.39-3.50(m,1H),3.57-3.65(m,1H),4.02-4.06(m,1H),4.39(d,J=14Hz,1H),4.63(d,J=14Hz,1H),4.65(s,1H),6.53(d,J=8Hz,1H),6.71(d,J=8Hz,1H),7.18(dd,J=5,8Hz,1H),7.25-7.28(m,1H),8.06(dd,J=1,5Hz,1H),11.5(br s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.15 (m, 2H), 0.42-0.53 (m, 2H), 0.73-0.83 (m) , 1H), 1.02-1.14 (m, 1H), 1.22-1.71 (m, 5H), 2.20-2.41 (m, 5H), 2.60-2.67 (M, 1H), 2.85-3.00 (m, 2H), 2.94 (s, 3H), 3.04 (d, J = 6Hz, 1H), 3.39-3.50 (m) , 1H), 3.57-3.65 (m, 1H), 4.02-4.06 (m, 1H), 4.39 (d, J = 14Hz, 1H), 4.63 (d, J) = 14Hz, 1H), 4.65 (s, 1H), 6.53 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 7.18 (dd, J = 5) , 8Hz, 1H), 7.25-7.28 (m, 1H), 8.06 (dd, J = 1,5Hz, 1H), 11.5 (br s, 1H).
(参考例12)
tert-ブチルメチル(ピリミジン-2-イルメチル)カーバメート(22)の合成
(Reference example 12)
Synthesis of tert-butylmethyl (pyrimidine-2-ylmethyl) carbamate (22)

1H-NMR(400MHz,CDCl3)δ(ppm):1.33-1.50(m,9H),2.99-3.04(m,3H),4.60-4.70(m,2H),7.17-7.18(m,1H),8.70-8.71(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 1.33-1.50 (m, 9H), 2.99-3.04 (m, 3H), 4.60-4.70 (m) , 2H), 7.17-7.18 (m, 1H), 8.70-8.71 (m, 2H).
(参考例13)
N-メチル-1-(ピリミジン-2-イル)メタンアミン二塩酸塩(23)の合成
(Reference example 13)
Synthesis of N-Methyl-1- (pyrimidine-2-yl) methaneamine dihydrochloride (23)

1H-NMR(400MHz,CD3OD)δ(ppm):2.85(s,3H),4.47(s,2H),7.45-7.49(m,1H),8.81-8.85(m,2H).
1 1 H-NMR (400 MHz, CD 3 OD) δ (ppm): 2.85 (s, 3H), 4.47 (s, 2H), 7.45-7.49 (m, 1H), 8.81 -8.85 (m, 2H).
(実施例11)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N-メチル-N-(ピリミジン-2-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(24)の合成
(Example 11)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Pyrimidine-2-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (24)
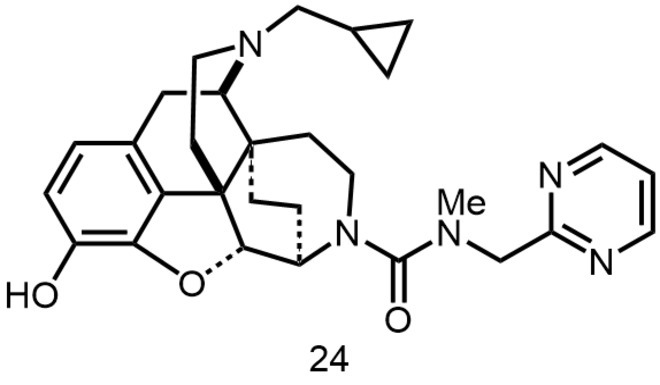
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.14(m,2H),0.42-0.53(m,2H),0.72-0.83(m,1H),0.99-1.09(m,1H),1.19-1.42(m,3H),1.48-1.54(m,1H),1.63-1.76(m,1H),2.18-2.39(m,5H),2.56-2.66(m,1H),2.82-2.93(m,1H),2.94(d,J=17Hz,1H),2.96(s,3H),3.01(d,J=6Hz,1H),3.42(ddd,J=4,11,14Hz,1H),3.73(ddd,J=4,5,14Hz,1H),4.10-4.15(m,1H),4.58(d,J=17Hz,1H),4.65(d,J=17Hz,1H),4.71(s,1H),6.51(d,J=8Hz,1H),6.69(d,J=8Hz,1H),7.20(dd,J=5,5Hz,1H),8.73(d,J=5Hz,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.14 (m, 2H), 0.42-0.53 (m, 2H), 0.72-0.83 (m) , 1H), 0.99-1.09 (m, 1H), 1.19-1.42 (m, 3H), 1.48-1.54 (m, 1H), 1.63-1.76 (M, 1H), 2.18-2.39 (m, 5H), 2.56-2.66 (m, 1H), 2.82-2.93 (m, 1H), 2.94 (d). , J = 17Hz, 1H), 2.96 (s, 3H), 3.01 (d, J = 6Hz, 1H), 3.42 (ddd, J = 4,11,14Hz, 1H), 3.73 (Ddd, J = 4,5,14Hz, 1H), 4.10-4.15 (m, 1H), 4.58 (d, J = 17Hz, 1H), 4.65 (d, J = 17Hz, 1H), 4.71 (s, 1H), 6.51 (d, J = 8Hz, 1H), 6.69 (d, J = 8Hz, 1H), 7.20 (dd, J = 5,5Hz, 1H), 8.73 (d, J = 5Hz, 2H).
(実施例12)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N-メチル-N-(ピリミジン-4-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(25)の合成
(Example 12)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Pyrimidine-4-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (25)
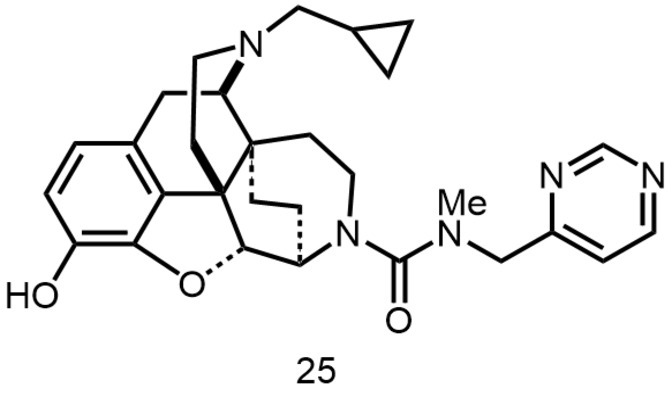
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.17(m,2H),0.43-0.55(m,2H),0.72-0.83(m,1H),1.02-1.12(m,1H),1.23-1.46(m,3H),1.51-1.80(m,2H),2.17-2.41(m,5H),2.59-2.66(m,1H),2.83-3.00(m,2H),2.92(s,3H),3.04(d,J=6Hz,1H),3.37-3.48(m,1H),3.61-3.69(m,1H),4.05-4.11(m,1H),4.46(s,2H),4.64(s,1H),5.42(br s,1H),6.53(d,J=8Hz,1H),6.70(d,J=8Hz,1H),7.39(d,J=6Hz,1H),8.73(d,J=6Hz,1H),9.17(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.17 (m, 2H), 0.43-0.55 (m, 2H), 0.72-0.83 (m) , 1H), 1.02-1.12 (m, 1H), 1.23-1.46 (m, 3H), 1.51-1.80 (m, 2H), 2.17-2.41 (M, 5H), 2.59-2.66 (m, 1H), 2.83-3.00 (m, 2H), 2.92 (s, 3H), 3.04 (d, J = 6Hz) , 1H), 3.37-3.48 (m, 1H), 3.61-3.69 (m, 1H), 4.05-4.11 (m, 1H), 4.46 (s, 2H) ), 4.64 (s, 1H), 5.42 (br s, 1H), 6.53 (d, J = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H), 7.39. (D, J = 6Hz, 1H), 8.73 (d, J = 6Hz, 1H), 9.17 (s, 1H).
(実施例13)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N-メチル-N-(ピリミジン-5-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(26)の合成
(Example 13)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Pyrimidine-5-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (26)
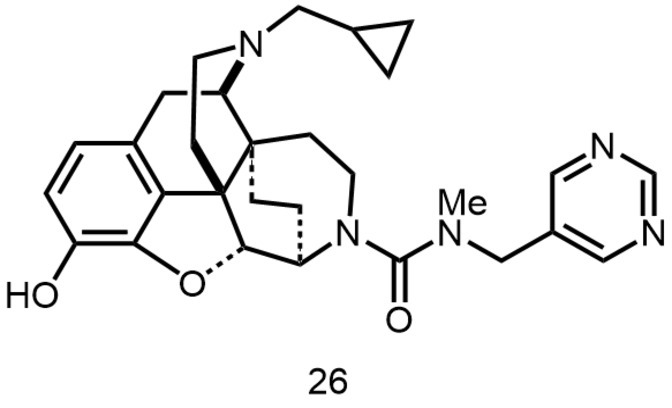
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.43-0.54(m,2H),0.74-0.84(m,1H),1.02-1.13(m,1H),1.22-1.45(m,3H),1.50-1.56(m,1H),1.62-1.73(m,1H),2.16-2.41(m,5H),2.62(dd,J=5,12Hz,1H),2.80-2.95(m,1H),2.82(s,3H),2.95(d,J=18Hz,1H),3.04(d,J=6Hz,1H),3.41(ddd,J=4,12,16Hz,1H),3.66(ddd,J=3,5,14Hz,1H),4.03-4.07(m,1H),4.34(d,J=16Hz,1H),4.38(d,J=16Hz,1H),4.57(s,1H),6.53(d,J=8Hz,1H),6.71(d,J=8Hz,1H),8.76(s,2H),9.16(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.43-0.54 (m, 2H), 0.74-0.84 (m) , 1H), 1.02-1.13 (m, 1H), 1.22-1.45 (m, 3H), 1.50-1.56 (m, 1H), 1.62-1.73 (M, 1H), 2.16-2.41 (m, 5H), 2.62 (dd, J = 5,12Hz, 1H), 2.80-2.95 (m, 1H), 2.82 (S, 3H), 2.95 (d, J = 18Hz, 1H), 3.04 (d, J = 6Hz, 1H), 3.41 (ddd, J = 4,12,16Hz, 1H), 3 .66 (ddd, J = 3,5,14Hz, 1H), 4.03-4.07 (m, 1H), 4.34 (d, J = 16Hz, 1H), 4.38 (d, J = 16Hz, 1H), 4.57 (s, 1H), 6.53 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 8.76 (s, 2H), 9 .16 (s, 1H).
(参考例14)
tert-ブチルメチル(ピラジン-2-イルメチル)カーバメート(27)の合成
(Reference example 14)
Synthesis of tert-butylmethyl (pyrazine-2-ylmethyl) carbamate (27)

1H-NMR(400MHz,CDCl3)δ(ppm):1.44-1.50(m,9H),2.94-3.00(m,3H),4.54-4.60(m,2H),8.49-8.57(m,3H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 1.44-1.50 (m, 9H), 2.94-3.00 (m, 3H), 4.54-4.60 (m) , 2H), 8.49-8.57 (m, 3H).
(参考例15)
N-メチル-1-(ピラジン-2-イル)メタンアミン二塩酸塩(28)の合成
(Reference example 15)
Synthesis of N-Methyl-1- (pyrazine-2-yl) methaneamine dihydrochloride (28)
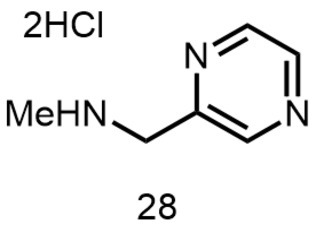
1H-NMR(400MHz,CD3OD)δ(ppm):2.84(s,3H),4.49(s,2H),8.67(d,J=2Hz,1H),8.75-8.78(m,2H).
1 1 H-NMR (400 MHz, CD 3 OD) δ (ppm): 2.84 (s, 3H), 4.49 (s, 2H), 8.67 (d, J = 2Hz, 1H), 8.75 -8.78 (m, 2H).
(実施例14)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N-メチル-N-(ピラジン-2-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(29)の合成
(Example 14)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Pyrazine-2-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (29)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.16(m,2H),0.42-0.54(m,2H),0.72-0.84(m,1H),1.01-1.11(m,1H),1.22-1.44(m,3H),1.50-1.57(m,1H),1.60-1.79(m,1H),2.17-2.41(m,5H),2.59-2.66(m,1H),2.81-2.95(m,1H),2.91(s,3H),2.95(d,J=19Hz,1H),3.03(d,J=6Hz,1H),3.42(ddd,J=4,12,15Hz,1H),3.69(ddd,J=4,4,14Hz,1H),4.04-4.10(m,1H),4.53(s,2H),4.63(s,1H),5.47(br s,1H),6.52(d,J=8Hz,1H),6.70(d,J=8Hz,1H),8.50(d,J=3Hz,1H),8.53-8.55(m,1H),8.68(s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.06-0.16 (m, 2H), 0.42-0.54 (m, 2H), 0.72-0.84 (m) , 1H), 1.01-1.11 (m, 1H), 1.22-1.44 (m, 3H), 1.50-1.57 (m, 1H), 1.60-1.79 (M, 1H), 2.17-2.41 (m, 5H), 2.59-2.66 (m, 1H), 2.81-2.95 (m, 1H), 2.91 (s) , 3H), 2.95 (d, J = 19Hz, 1H), 3.03 (d, J = 6Hz, 1H), 3.42 (ddd, J = 4,12,15Hz, 1H), 3.69 (Ddd, J = 4,4,14Hz, 1H), 4.04-4.10 (m, 1H), 4.53 (s, 2H), 4.63 (s, 1H), 5.47 (br) s, 1H), 6.52 (d, J = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H), 8.50 (d, J = 3Hz, 1H), 8.53-8. 55 (m, 1H), 8.68 (s, 1H).
(参考例16)
N-メチル-1-(ピリダジン-3-イル)メタンアミン(30)の合成
(Reference example 16)
Synthesis of N-Methyl-1- (pyridazine-3-yl) methaneamine (30)

参考例8に記載した方法に従い、ピリダジン-3-カルバルデヒドより表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm):2.51(s,3H),4.09(s,2H),7.45(dd,J=5,8Hz,1H),7.57(dd,J=2,8Hz,1H)9.11(dd,J=2,5Hz,1H).
The title compound was obtained from pyridazine-3-carbaldehyde according to the method described in Reference Example 8.
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 2.51 (s, 3H), 4.09 (s, 2H), 7.45 (dd, J = 5,8 Hz, 1H), 7. 57 (dd, J = 2.8Hz, 1H) 9/11 (dd, J = 2.5Hz, 1H).
(実施例15)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N-メチル-N-(ピリダジン-3-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(31)の合成
(Example 15)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Pyridazine-3-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (31)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.16(m,2H),0.43-0.54(m,2H),0.72-0.83(m,1H),1.01-1.11(m,1H),1.22-1.45(m,3H),1.50-1.57(m,1H),1.60-1.73(m,1H),2.18-2.41(m,5H),2.58-2.65(m,1H),2.82-2.99(m,2H),2.92(s,3H),3.04(d,J=6Hz,1H),3.42(ddd,J=4,11,14Hz,1H),3.63(ddd,J=4,4,14Hz,1H),4.05-4.10(m,1H),4.61(s,1H),4.67(d,J=16Hz,1H),4.72(d,J=16Hz,1H),6.52(d,J=8Hz,1H),6.71(d,J=8Hz,1H),7.49(dd,J=5,9Hz,1H),7.71(dd,J=2,9Hz,1H),9.12(dd,J=2,5Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.16 (m, 2H), 0.43-0.54 (m, 2H), 0.72-0.83 (m) , 1H), 1.01-1.11 (m, 1H), 1.22-1.45 (m, 3H), 1.50-1.57 (m, 1H), 1.60-1.73 (M, 1H), 2.18-2.41 (m, 5H), 2.58-2.65 (m, 1H), 2.82-2.99 (m, 2H), 2.92 (s) , 3H), 3.04 (d, J = 6Hz, 1H), 3.42 (ddd, J = 4,11,14Hz, 1H), 3.63 (ddd, J = 4,4,14Hz, 1H) , 4.05-4.10 (m, 1H), 4.61 (s, 1H), 4.67 (d, J = 16Hz, 1H), 4.72 (d, J = 16Hz, 1H), 6 .52 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 7.49 (dd, J = 5,9Hz, 1H), 7.71 (dd, J = 2, 9Hz, 1H), 9.12 (dd, J = 2.5Hz, 1H).
(実施例16)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N-メチル-N-((6-オキソ-1,6-ジヒドロピリジン-2-イル)メチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(32)の合成
(Example 16)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- ((6-oxo-1,6-dihydropyridine-2-yl) methyl) -1 , 2,3,4,5,6,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide Synthesis of (32)
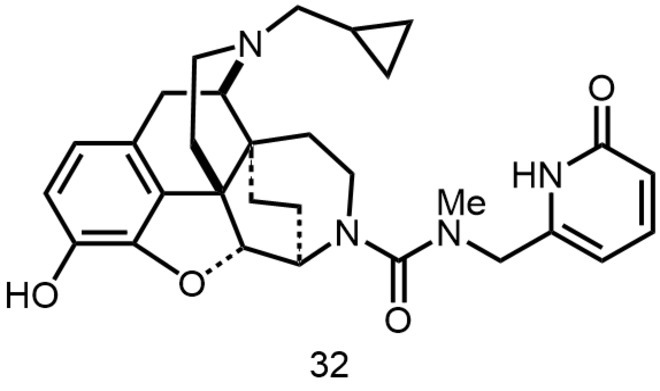
1H-NMR(400MHz,DMSO-d6)δ(ppm):0.04-0.11(m,2H),0.40-0.48(m,2H),0.73-0.80(m,1H),0.88-0.96(m,1H),1.13-1.39(m,4H),1.62-1.69(m,1H),2.10-2.39(m,5H),2.49-2.56(m,1H),2.61-2.78(m,1H),2.72(s,3H),2.86(d,J=19Hz,1H),2.98(d,J=6Hz,1H),3.18-3.49(m,1H),3.52-3.63(m,1H),3.89(s,1H),4.03(d,J=16Hz,1H),4.08(d,J=16Hz,1H),4.49(s,1H),6.13(d,J=7Hz,1H),6.23(d,J=9Hz,1H),6.45(d,J=8Hz,1H),6.57(d,J=8Hz,1H),7.40(dd,J=6,9Hz,1H).
1 1 H-NMR (400MHz, DMSO-d 6 ) δ (ppm): 0.04-0.11 (m, 2H), 0.40-0.48 (m, 2H), 0.73-0.80 (M, 1H), 0.88-0.96 (m, 1H), 1.13-1.39 (m, 4H), 1.62-1.69 (m, 1H), 2.10-2 .39 (m, 5H), 2.49-2.56 (m, 1H), 2.61-2.78 (m, 1H), 2.72 (s, 3H), 2.86 (d, J) = 19Hz, 1H), 2.98 (d, J = 6Hz, 1H), 3.18-3.49 (m, 1H), 3.52-3.63 (m, 1H), 3.89 (s) , 1H), 4.03 (d, J = 16Hz, 1H), 4.08 (d, J = 16Hz, 1H), 4.49 (s, 1H), 6.13 (d, J = 7Hz, 1H) ), 6.23 (d, J = 9Hz, 1H), 6.45 (d, J = 8Hz, 1H), 6.57 (d, J = 8Hz, 1H), 7.40 (dd, J = 6). , 9Hz, 1H).
(参考例17)
N-メチル-1-(1-((2-(トリメチルシリル)エトキシ)メチル)-1H-イミダゾール-2-イル)メタンアミン(33)の合成
(Reference example 17)
Synthesis of N-methyl-1- (1-((2- (trimethylsilyl) ethoxy) methyl) -1H-imidazol-2-yl) methaneamine (33)

参考例8に記載した方法に従い、1-((2-(トリメチルシリル)エトキシ)メチル)-1H-イミダゾール-2-カルバルデヒド(Journal of Organic Chemistry 2006,71,8807に記載の方法により合成)より表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm):-0.01(s,9H),0.90(t,J=8Hz,2H),2.44(s,3H),3.50(t,J=8Hz,2H),3.86(s,2H),5.33(s,2H),6.96(s,2H).
Titled from 1-((2- (trimethylsilyl) ethoxy) methyl) -1H-imidazole-2-carbaldehyde (synthesized by the method described in Journal of Organic Chemistry 2006, 71, 8807) according to the method described in Reference Example 8. The compound was obtained.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): -0.01 (s, 9H), 0.90 (t, J = 8Hz, 2H), 2.44 (s, 3H), 3.50 (T, J = 8Hz, 2H), 3.86 (s, 2H), 5.33 (s, 2H), 6.96 (s, 2H).
(実施例17)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-N―メチル-N-((1-((2-(トリメチルシリル)エトキシ)メチル)-1H-イミダゾール-2-イル)メチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(34)の合成
(Example 17)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Methoxy-N-Methyl-N-((1-((2- (trimethylsilyl) ethoxy) methyl) -1H-imidazole- 2-yl) Methyl) -1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3 -D] Synthesis of azepine-7-carboxamide (34)
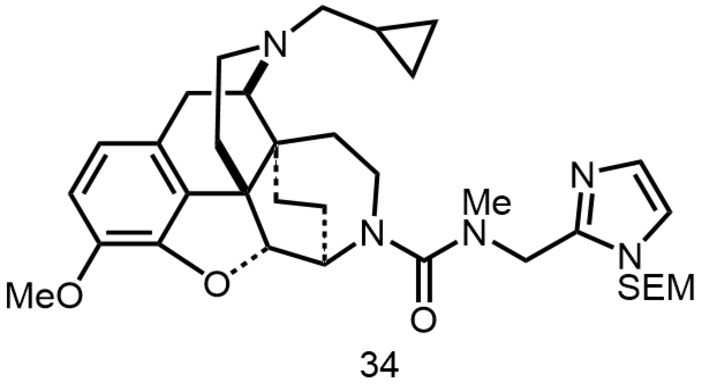
実施例5に記載した方法に従い、化合物11及び化合物33より表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm): -0.01(s,9H),0.00-0.15(m,2H),0.40-0.50(m,2H),0.70-0.85(m,1H),0.85-0.95(m,2H),1.00-1.10(m,1H),1.20-1.80(m,4H),2.20-2.45(m,6H),2.55-2.65(m,1H),2.80-2.90(m,1H),2.89(s,3H),2.96(d,J=18Hz,1H),3.03(d,J=6Hz,1H),3.30-3.40(m,1H),3.40-3.50(m,2H),3.50-3.60(m,1H),3.87(s,3H),4.00-4.10(m,1H),4.47(d,J=15Hz,1H),4.54(d,J=15Hz,1H),4.60-4.70(m,1H),5.34(d,J=10Hz,1H),5.37(d,J=10Hz,1H),6.58(d,J=8Hz,1H),6.71(d,J=8Hz,1H),6.96-7.00(m,2H).
The title compound was obtained from compound 11 and compound 33 according to the method described in Example 5.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): -0.01 (s, 9H), 0.00-0.15 (m, 2H), 0.40-0.50 (m, 2H) , 0.70-0.85 (m, 1H), 0.85-0.95 (m, 2H), 1.00-1.10 (m, 1H), 1.20-1.80 (m, 4H), 2.20-2.45 (m, 6H), 2.55-2.65 (m, 1H), 2.80-2.90 (m, 1H), 2.89 (s, 3H) , 2.96 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 1H), 3.30-3.40 (m, 1H), 3.40-3.50 (m, 2H), 3.50-3.60 (m, 1H), 3.87 (s, 3H), 4.00-4.10 (m, 1H), 4.47 (d, J = 15Hz, 1H) , 4.54 (d, J = 15Hz, 1H), 4.60-4.70 (m, 1H), 5.34 (d, J = 10Hz, 1H), 5.37 (d, J = 10Hz, 1H), 6.58 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 6.96-7.00 (m, 2H).
(実施例18)
(4R,4aS,8R,8aR,13bS)-N-((1H-イミダゾール-2-イル)メチル)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(35)の合成
(Example 18)
(4R, 4aS, 8R, 8aR, 13bS) -N-((1H-imidazol-2-yl) methyl) -3- (cyclopropylmethyl) -10-hydroxy-N-methyl-1,2,3,4 , 5,6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-Methylnobenzoflo [2,3-c] pyrido [4,3-d] Synthesis of azepine-7-carboxamide (35)
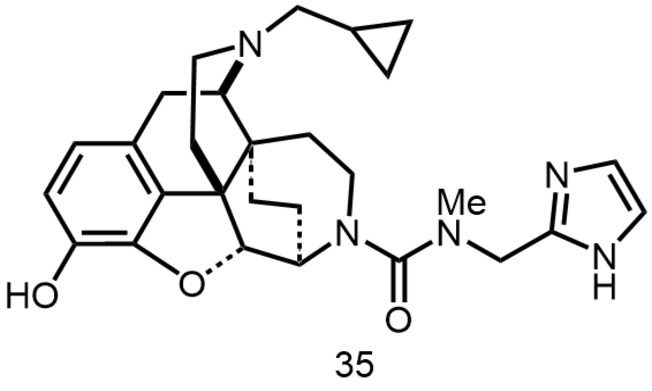
実施例7に記載した方法に従い、化合物34より表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm): 0.00-0.20(m,2H),0.40-0.60(m,2H),0.75-0.90(m,1H),1.00-1.15(m,1H),1.20-2.00(m,8H),2.20-2.50(m,3H),2.60-2.80(m,1H),2.83(s,3H),2.97(d,J=18Hz,1H),3.00-3.20(m,1H),3.43(dt,J=4,12Hz,1H),3.60-3.70(m,1H),4.00-4.10(m,1H),4.19(d,J=15Hz,1H),4.41(d,J=15Hz,1H),4.60-4.70(m,1H),6.54(d,J=8Hz,1H),6.73(d,J=8Hz,1H),6.99(s,2H).
The title compound was obtained from compound 34 according to the method described in Example 7.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.20 (m, 2H), 0.40-0.60 (m, 2H), 0.75-0.90 (m) , 1H), 1.00-1.15 (m, 1H), 1.20-2.00 (m, 8H), 2.20-2.50 (m, 3H), 2.60-2.80 (M, 1H), 2.83 (s, 3H), 2.97 (d, J = 18Hz, 1H), 3.00-3.20 (m, 1H), 3.43 (dt, J = 4) , 12Hz, 1H), 3.60-3.70 (m, 1H), 4.00-4.10 (m, 1H), 4.19 (d, J = 15Hz, 1H), 4.41 (d) , J = 15Hz, 1H), 4.60-4.70 (m, 1H), 6.54 (d, J = 8Hz, 1H), 6.73 (d, J = 8Hz, 1H), 6.99 (S, 2H).
(実施例19)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-N―メチル-N-((1-メチル-1H-イミダゾール-2-イル)メチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(36)の合成
(Example 19)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-N-methyl-N-((1-methyl-1H-imidazol-2-yl) methyl) -1,2 , 3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (36) ) Synthesis

実施例5に記載した方法に従い、化合物11およびN-メチル-1-(1-メチル-1H-イミダゾール-2-イル)メタンアミンより表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm): 0.00-0.15(m,2H),0.40-0.55(m,2H),0.70-0.85(m,1H),1.07(t,J=12Hz,1H),1.20-1.50(m,3H),1.50-1.60(m,1H),2.20-2.45(m,5H),2.60-2.70(m,1H),2.80-2.93(m,1H),2.87(s,3H),2.97(d,J=18Hz,1H),3.05(d,J=5Hz,1H),3.39(dt,J=4,11Hz,1H),3.55-3.65(m,1H),3.66(s,3H),3.70(s,1H),3.87(s,3H),4.00-4.10(m,1H),4.42(d,J=15Hz,1H),4.53(d,J=15Hz,1H),4.58-4.62(m,1H),6.58(d,J=8Hz,1H),6.72(d,J=8Hz,1H),6.85(d,J=1Hz,1H),6.96(d,J=1Hz,1H).
The title compound was obtained from compound 11 and N-methyl-1- (1-methyl-1H-imidazol-2-yl) methaneamine according to the method described in Example 5.
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.00-0.15 (m, 2H), 0.40-0.55 (m, 2H), 0.70-0.85 (m) , 1H), 1.07 (t, J = 12Hz, 1H), 1.20-1.50 (m, 3H), 1.50-1.60 (m, 1H), 2.20-2.45 (M, 5H), 2.60-2.70 (m, 1H), 2.80-2.93 (m, 1H), 2.87 (s, 3H), 2.97 (d, J = 18Hz) , 1H), 3.05 (d, J = 5Hz, 1H), 3.39 (dt, J = 4,11Hz, 1H), 3.55-3.65 (m, 1H), 3.66 (s) , 3H), 3.70 (s, 1H), 3.87 (s, 3H), 4.00-4.10 (m, 1H), 4.42 (d, J = 15Hz, 1H), 4. 53 (d, J = 15Hz, 1H), 4.58-4.62 (m, 1H), 6.58 (d, J = 8Hz, 1H), 6.72 (d, J = 8Hz, 1H), 6.85 (d, J = 1Hz, 1H), 6.96 (d, J = 1Hz, 1H).
(実施例20)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-N-((1-メチル-1H-イミダゾール-2-イル)メチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(37)の合成
(Example 20)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-N-methyl-N-((1-methyl-1H-imidazol-2-yl) methyl) -1,2 , 3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (37) ) Synthesis
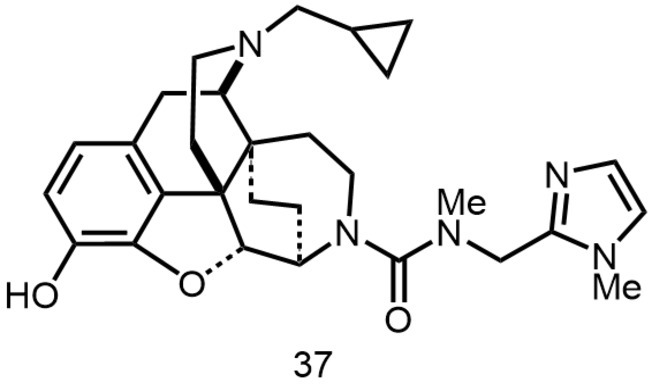
1H-NMR(400MHz,CDCl3)δ(ppm): 0.00-0.15(m,2H),0.40-0.55(m,2H),0.70-0.90(m,2H),1.00-2.00(m,6H),2.15-2.45(m,4H),2.55-2.65(m,1H),2.86(s,3H),2.80-3.10(m,3H),3.49(s,3H),3.30-3.70(m,2H),4.04(s,1H),4.44(d,J=14Hz,1H),4.51(d,J=14Hz,1H),4.61(s,1H),6.52(d,J=8Hz,1H),6.70(d,J=8Hz,1H),6.86(d,J=1Hz,1H),6.97(d,J=1Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.00-0.15 (m, 2H), 0.40-0.55 (m, 2H), 0.70-0.90 (m) , 2H), 1.00-2.00 (m, 6H), 2.15-2.45 (m, 4H), 2.55-2.65 (m, 1H), 2.86 (s, 3H) ), 2.80-3.10 (m, 3H), 3.49 (s, 3H), 3.30-3.70 (m, 2H), 4.04 (s, 1H), 4.44 ( d, J = 14Hz, 1H), 4.51 (d, J = 14Hz, 1H), 4.61 (s, 1H), 6.52 (d, J = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H), 6.86 (d, J = 1Hz, 1H), 6.97 (d, J = 1Hz, 1H).
(実施例21)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-N―メチル-N-((1-メチル-1H-ピラゾール-3-イル)メチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(38)の合成
(Example 21)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Methoxy-N-Methyl-N- ((1-Methyl-1H-Pyrazole-3-yl) Methyl) -1,2 , 3,4,5,6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-Methylnobenzoflo [2,3-c] Pyrazole [4,3-d] Azepine-7-Carboxamide (38) ) Synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.44-0.53(m,2H),0.75-0.84(m,1H),1.02-1.08(m,1H),1.26-1.78(m,5H),2.20-2.41(m,5H),2.58-2.66(m,1H),2.80(s,3H),2.83-2.93(m,1H),2.97(d,J=18Hz,1H),3.03(d,J=6Hz,1H),3.40(ddd,J=4,11,14Hz,1H),3.65(ddd,J=4,4,14Hz,1H),3.87(s,6H),4.09-4.11(m,1H),4.32(s,2H),4.62(s,1H),6.21(d,J=8Hz,1H),6.57(d,J=8Hz,1H),6.71(d,J=2Hz,1H),7.29(d,J=2Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.44-0.53 (m, 2H), 0.75-0.84 (m) , 1H), 1.02-1.08 (m, 1H), 1.26-1.78 (m, 5H), 2.20-2.41 (m, 5H), 2.58-2.66 (M, 1H), 2.80 (s, 3H), 2.83-2.93 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz) , 1H), 3.40 (ddd, J = 4,11,14Hz, 1H), 3.65 (ddd, J = 4,4,14Hz, 1H), 3.87 (s, 6H), 4.09 -4.11 (m, 1H), 4.32 (s, 2H), 4.62 (s, 1H), 6.21 (d, J = 8Hz, 1H), 6.57 (d, J = 8Hz) , 1H), 6.71 (d, J = 2Hz, 1H), 7.29 (d, J = 2Hz, 1H).
(実施例22)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-N-((1-メチル-1H-ピラゾール-3-イル)メチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(39)の合成
(Example 22)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- ((1-Methyl-1H-Pyrazole-3-yl) Methyl) -1,2 , 3,4,5,6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-Methylnobenzoflo [2,3-c] Pyrazole [4,3-d] Azepine-7-Carboxamide (39) ) Synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.13(m,2H),0.43-0.53(m,2H),0.73-0.82(m,1H),1.01-1.08(m,1H),1.24-1.72(m,5H),2.18-2.39(m,5H),2.59-2.64(m,1H),2.79(s,3H),2.83-2.91(m,1H),2.95(d,J=18Hz,1H),3.02(d,J=6Hz,1H),3.41(ddd,J=4,11,14Hz,1H),3.68(ddd,J=4,4,14Hz,1H),3.88(s,3H),4.08-4.11(m,1H),4.29(d,J=15Hz,1H),4.33(d,J=15Hz,1H),4.65(s,1H),6.22(d,J=2Hz,1H),6.52(d,J=8Hz,1H),6.70(d,J=8Hz,1H),7.30(d,J=2Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.13 (m, 2H), 0.43-0.53 (m, 2H), 0.73-0.82 (m) , 1H), 1.01-1.08 (m, 1H), 1.24-1.72 (m, 5H), 2.18-2.39 (m, 5H), 2.59-2.64 (M, 1H), 2.79 (s, 3H), 2.83-2.91 (m, 1H), 2.95 (d, J = 18Hz, 1H), 3.02 (d, J = 6Hz) , 1H), 3.41 (ddd, J = 4,11,14Hz, 1H), 3.68 (ddd, J = 4,4,14Hz, 1H), 3.88 (s, 3H), 4.08 -4.11 (m, 1H), 4.29 (d, J = 15Hz, 1H), 4.33 (d, J = 15Hz, 1H), 4.65 (s, 1H), 6.22 (d) , J = 2Hz, 1H), 6.52 (d, J = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H), 7.30 (d, J = 2Hz, 1H).
(参考例18)
N-メチル-1-(オキサゾール-2-イル)メタンアミン(40)の合成
(Reference Example 18)
Synthesis of N-Methyl-1- (oxazole-2-yl) methaneamine (40)

1H-NMR(400MHz,CDCl3)δ(ppm):2.47(s,3H),3.90(s,2H),7.07(s,1H),7.62(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 2.47 (s, 3H), 3.90 (s, 2H), 7.07 (s, 1H), 7.62 (s, 1H) ..
(実施例23)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-N―メチル-N-(オキサゾール-2-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(41)の合成
(Example 23)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Methoxy-N-Methyl-N- (Oxazole-2-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (41)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.14(m,2H),0.44-0.53(m,2H),0.74-0.84(m,1H),1.04-1.10(m,1H),1.26-1.72(m,5H),2.21-2.42(m,5H),2.58-2.66(m,1H),2.86-2.94(m,4H),2.97(d,J=18Hz,1H),3.04(d,J=6Hz,1H),3.43(ddd,J=4,11,14Hz,1H),3.68(ddd,J=4,4,14Hz,1H),3.87(s,3H),4.09-4.12(m,1H),4.44(d,J=16Hz,1H),4.49(d,J=16Hz,1H),4.64-4.65(m,1H),6.58(d,J=8Hz,1H),6.72(d,J=8Hz,1H),7.08(d,J=1Hz,1H),7.64(d,J=1Hz,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.14 (m, 2H), 0.44-0.53 (m, 2H), 0.74-0.84 (m) , 1H), 1.04-1.10 (m, 1H), 1.26-1.72 (m, 5H), 2.21-2.42 (m, 5H), 2.58-2.66 (M, 1H), 2.86-2.94 (m, 4H), 2.97 (d, J = 18Hz, 1H), 3.04 (d, J = 6Hz, 1H), 3.43 (ddd) , J = 4,11,14Hz, 1H), 3.68 (ddd, J = 4,4,14Hz, 1H), 3.87 (s, 3H), 4.09-4.12 (m, 1H) , 4.44 (d, J = 16Hz, 1H), 4.49 (d, J = 16Hz, 1H), 4.64-4.65 (m, 1H), 6.58 (d, J = 8Hz, 1H), 6.72 (d, J = 8Hz, 1H), 7.08 (d, J = 1Hz, 1H), 7.64 (d, J = 1Hz, 1H).
(実施例24)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-N-(オキサゾール-2-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(42)の合成
(Example 24)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Oxazole-2-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (42)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.14(m,2H),0.44-0.52(m,2H),0.75-0.82(m,1H),1.03-1.09(m,1H),1.24-1.72(m,5H),2.20-2.39(m,5H),2.60-2.64(m,1H),2.84-2.91(m,4H),2.95(d,J=18Hz,1H),3.03(d,J=6Hz,1H),3.40-3.48(m,1H),3.67-3.73(m,1H),4.08-4.11(m,1H),4.46(s,2H),4.66(s,1H),6.52(d,J=8Hz,1H),6.70(d,J=8Hz,1H),7.10(s,1H),7.65(s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.14 (m, 2H), 0.44-0.52 (m, 2H), 0.75-0.82 (m) , 1H), 1.03-1.09 (m, 1H), 1.24-1.72 (m, 5H), 2.20-2.39 (m, 5H), 2.60-2.64 (M, 1H), 2.84-2.91 (m, 4H), 2.95 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 1H), 3.40-3 .48 (m, 1H), 3.67-3.73 (m, 1H), 4.08-4.11 (m, 1H), 4.46 (s, 2H), 4.66 (s, 1H) ), 6.52 (d, J = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H), 7.10 (s, 1H), 7.65 (s, 1H).
(参考例19)
N-メチル-1-(オキサゾール-4-イル)メタンアミン(43)の合成
(Reference example 19)
Synthesis of N-Methyl-1- (oxazole-4-yl) methaneamine (43)

1H-NMR(400MHz,CDCl3)δ(ppm):2.46(s,3H),3.71(s,2H),7.57(d,J=1Hz,1H),7.85(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 2.46 (s, 3H), 3.71 (s, 2H), 7.57 (d, J = 1Hz, 1H), 7.85 ( s, 1H).
(実施例25)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-N-(オキサゾール-4-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(44)の合成
(Example 25)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Oxazole-4-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (44)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.42-0.54(m,2H),0.72-0.83(m,1H),1.00-1.10(m,1H),1.19-1.44(m,3H),1.49-1.56(m,1H),1.58-1.80(m,1H),2.16-2.41(m,5H),2.58-2.65(m,1H),2.81-2.94(m,1H),2.86(s,3H),2.95(d,J=18Hz,1H),3.02(d,J=6Hz,1H),3.41(ddd,J=4,12,15Hz,1H),3.70(ddd,J=4,4,14Hz,1H),4.06-4.11(m,1H),4.22(d,J=16Hz,1H),4.28(d,J=16Hz,1H),4.62(s,1H),5.28(br s,1H),6.52(d,J=8Hz,1H),6.80(d,J=8Hz,1H),7.68(s,1H),7.87(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.42-0.54 (m, 2H), 0.72-0.83 (m) , 1H), 1.00-1.10 (m, 1H), 1.19-1.44 (m, 3H), 1.49-1.56 (m, 1H), 1.58-1.80 (M, 1H), 2.16-2.41 (m, 5H), 2.58-2.65 (m, 1H), 2.81-2.94 (m, 1H), 2.86 (s) , 3H), 2.95 (d, J = 18Hz, 1H), 3.02 (d, J = 6Hz, 1H), 3.41 (ddd, J = 4,12,15Hz, 1H), 3.70 (Ddd, J = 4,4,14Hz, 1H), 4.06-4.11 (m, 1H), 4.22 (d, J = 16Hz, 1H), 4.28 (d, J = 16Hz, 1H), 4.62 (s, 1H), 5.28 (br s, 1H), 6.52 (d, J = 8Hz, 1H), 6.80 (d, J = 8Hz, 1H), 7. 68 (s, 1H), 7.87 (s, 1H).
(実施例26)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-N―メチル-N-(チアゾール-2-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(45)の合成
(Example 26)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Methoxy-N-Methyl-N- (Thiazole-2-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (45)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.44-0.53(m,2H),0.74-0.84(m,1H),1.04-1.11(m,1H),1.25-1.73(m,5H),2.21-2.42(m,5H),2.61-2.66(m,1H),2.87-2.93(m,4H),2.98(d,J=18Hz,1H),3.05(d,J=6Hz,1H),3.44(ddd,J=4,11,14Hz,1H),3.67(ddd,J=4,4,14Hz,1H),3.87(s,3H),4.11-4.13(m,1H),4.59-4.71(m,3H),6.58(d,J=8Hz,1H),6.72(d,J=8Hz,1H),7.31(d,J=3Hz,1H),7.71(d,J=3Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.44-0.53 (m, 2H), 0.74-0.84 (m) , 1H), 1.04-1.11 (m, 1H), 1.25-1.73 (m, 5H), 2.21-2.42 (m, 5H), 2.61-2.66 (M, 1H), 2.87-2.93 (m, 4H), 2.98 (d, J = 18Hz, 1H), 3.05 (d, J = 6Hz, 1H), 3.44 (ddd) , J = 4,11,14Hz, 1H), 3.67 (ddd, J = 4,4,14Hz, 1H), 3.87 (s, 3H), 4.11-4.13 (m, 1H) , 4.59-4.71 (m, 3H), 6.58 (d, J = 8Hz, 1H), 6.72 (d, J = 8Hz, 1H), 7.31 (d, J = 3Hz, 1H), 7.71 (d, J = 3Hz, 1H).
(実施例27)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-N―メチル-N-(チアゾール-2-イルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキサミド(46)の合成
(Example 27)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-N-Methyl-N- (Thiazole-2-ylmethyl) -1,2,3,4,5,6 Synthesis of 8,8a-octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxamide (46)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.16(m,2H),0.44-0.53(m,2H),0.75-0.84(m,1H),1.04-1.10(m,1H),1.25-1.45(m,3H),1.52-1.57(m,1H),1.66-1.72(m,1H),2.21-2.42(m,5H),2.60-2.67(m,1H),2.84-2.92(m,4H),2.96(d,J=18Hz,1H),3.05(d,J=6Hz,1H),3.45(ddd,J=4,11,14Hz,1H),3.67-3.74(m,1H),4.09-4.12(m,1H),4.61-4.70(m,3H),6.53(d,J=8Hz,1H),6.71(d,J=8Hz,1H),7.32(d,J=3Hz,1H),7.73(d,J=3Hz,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.16 (m, 2H), 0.44-0.53 (m, 2H), 0.75-0.84 (m) , 1H), 1.04-1.10 (m, 1H), 1.25-1.45 (m, 3H), 1.52-1.57 (m, 1H), 1.66-1.72 (M, 1H), 2.21-2.42 (m, 5H), 2.60-2.67 (m, 1H), 2.84-2.92 (m, 4H), 2.96 (d). , J = 18Hz, 1H), 3.05 (d, J = 6Hz, 1H), 3.45 (ddd, J = 4,11,14Hz, 1H), 3.67-3.74 (m, 1H) , 4.09-4.12 (m, 1H), 4.61-4.70 (m, 3H), 6.53 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 7.32 (d, J = 3Hz, 1H), 7.73 (d, J = 3Hz, 1H).
(実施例28)
ベンジル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(47)の合成
(Example 28)
Benzyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxylate (47)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.17(m,2H),0.42-0.53(m,2H),0.70-0.93(m,1H),0.99-1.12(m,1H), 1.18-1.76(m,5H), 2.14-2.41(m,5H),2.57-2.67(m,1H), 2.74-3.05(m,3H),3.30-3.44(m,1H),4.02-4.32(m,2H),4.44-4.90(m,2H),5.10-5.25(m,2H),6.48-6.56(m,1H),6.64-6.73(m,1H),7.26-7.45(m,5H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-0.17 (m, 2H), 0.42-0.53 (m, 2H), 0.70-0.93 (m) , 1H), 0.99-1.12 (m, 1H), 1.18-1.76 (m, 5H), 2.14-2.41 (m, 5H), 2.57-2.67 (M, 1H), 2.74-3.05 (m, 3H), 3.30-3.44 (m, 1H), 4.02-4.32 (m, 2H), 4.44-4 .90 (m, 2H), 5.10-15.25 (m, 2H), 6.48-6.56 (m, 1H), 6.64-6.73 (m, 1H), 7.26 -7.45 (m, 5H).
(実施例29)
フェニル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(48)の合成
(Example 29)
Phenyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxylate (48)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.16(m,2H),0.44-0.56(m,2H),0.75-0.86(m,1H),1.07-1.18(m,1H),1.30-1.40(m,1H),1.40-1.51(m,2H),1.52-1.67(m,1H),1.72-1.85(m,1H),2.22-2.41(m,4H),2.42(dd,J=6,18Hz,1H),2.61-2.71(m,1H),2.87-3.01(m,1H)3.00(d,J=18Hz,1H),3.04-3.09(m,1H),3.45(ddd,J=4,11,14Hz,0.7H),3.58(ddd,J=4,12,14Hz,0.3H),3.87(s,3H),4.13-4.25(m,1H),4.33-4.38(m,0.3H),4.42-4.48(m,0.7H),4.63(s,0.3H),4.66(s,0.7H),6.59(d,J=9Hz,0.3H),6.62(d,J=9Hz,0.7H),6.73(d,J=9Hz,0.3H),6.74(d,J=9Hz,0.7H),7.10-7.15(m,2H),7,19(dd,J=7,7Hz,1H),7.32-7.39(m,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.16 (m, 2H), 0.44-0.56 (m, 2H), 0.75-0.86 (m) , 1H), 1.07-1.18 (m, 1H), 1.30-1.40 (m, 1H), 1.40-1.51 (m, 2H), 1.52-1.67 (M, 1H), 1.72-1.85 (m, 1H), 2.22-2.41 (m, 4H), 2.42 (dd, J = 6,18Hz, 1H), 2.61 -2.71 (m, 1H), 2.87-3.01 (m, 1H) 3.00 (d, J = 18Hz, 1H), 3.04-3.09 (m, 1H), 3. 45 (ddd, J = 4,11,14Hz, 0.7H), 3.58 (ddd, J = 4,12,14Hz, 0.3H), 3.87 (s, 3H), 4.13-4 .25 (m, 1H), 4.33-4.38 (m, 0.3H), 4.42-4.48 (m, 0.7H), 4.63 (s, 0.3H), 4 .66 (s, 0.7H), 6.59 (d, J = 9Hz, 0.3H), 6.62 (d, J = 9Hz, 0.7H), 6.73 (d, J = 9Hz, 0.3H), 6.74 (d, J = 9Hz, 0.7H), 7.10-7.15 (m, 2H), 7,19 (dd, J = 7,7Hz, 1H), 7. 32-7.39 (m, 2H).
(実施例30)
フェニル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(49)の合成
(Example 30)
Phenyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxylate (49)
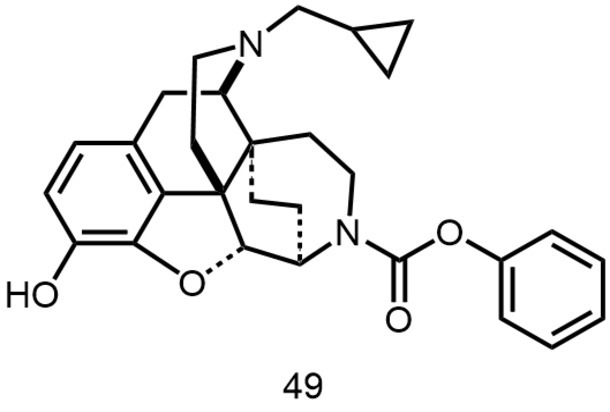
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.17(m,2H),0.44-0.55(m,2H),0.74-0.86(m,1H),1.06-1.17(m,1H),1.24-1.49(m,3H),1.55-1.61(m,1H),1.71-1.85(m,1H),2.22-2.44(m,5H),2.61-2.71(m,1H),2.86-3.01(m,1H),2.98(d,J=18Hz,1H),3.03-3.09(m,1H),3.46(ddd,J=4,11,14Hz,0.6H),3.54(ddd,J=3,11,14Hz,0.4H),4.11-4.32(m,1.4H),4.35-4.41(m,0.6H),4.61(s,0.4H),4.65(s,0.6H),6.54(d,J=8Hz,0.4H),6.55(d,J=8Hz,0.6H),6.70(d,J=8Hz,0.6H),6.71(d,J=8Hz,0.4H),7.11-7.17(m,2H),7.20(t,J=7Hz,1H),7.37(t,J=8Hz,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.17 (m, 2H), 0.44-0.55 (m, 2H), 0.74-0.86 (m) , 1H), 1.06-1.17 (m, 1H), 1.24-1.49 (m, 3H), 1.55-1.61 (m, 1H), 1.71-1.85 (M, 1H), 2.22-2.44 (m, 5H), 2.61-2.71 (m, 1H), 2.86-3.01 (m, 1H), 2.98 (d) , J = 18Hz, 1H), 3.03-3.09 (m, 1H), 3.46 (ddd, J = 4,11,14Hz, 0.6H), 3.54 (ddd, J = 3, 11, 14Hz, 0.4H), 4.11-4.32 (m, 1.4H), 4.35-4.41 (m, 0.6H), 4.61 (s, 0.4H), 4.65 (s, 0.6H), 6.54 (d, J = 8Hz, 0.4H), 6.55 (d, J = 8Hz, 0.6H), 6.70 (d, J = 8Hz) , 0.6H), 6.71 (d, J = 8Hz, 0.4H), 7.11-7.17 (m, 2H), 7.20 (t, J = 7Hz, 1H), 7.37 (T, J = 8Hz, 2H).
(実施例31)
オキサゾール-2-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(50)の合成
(Example 31)
Oxazole-2-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] synthesis of azepine-7-carboxylate (50)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.14(m,2H),0.44-0.52(m,2H),0.74-0.83(m,1H),1.04-1.11(m,1H),1.26-1.90(m,5H),2.18-2.42(m,5H),2.60-2.66(m,1H),2.80-2.91(m,1H),2.97(d,J=18Hz,1H),3.02(d,J=7Hz,1H),3.36-3.44(m,1H),3.85(s,2.1H),3.87(s,0.9H),3.90-4.05(m,0.3H),4.09-4.15(m,0.7H),4.22-4.25(m,0.7H),4.29-4.32(m,0.3H),4.54(s,1H),5.24(s,2H),6.57-6.60(m,1H),6.72(d,J=8Hz,1H),7.12(s,0.7H),7.14(s,0.3H),7.66(s,0.7H),7.68(s,0.3H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.14 (m, 2H), 0.44-0.52 (m, 2H), 0.74-0.83 (m) , 1H), 1.04-1.11 (m, 1H), 1.26-1.90 (m, 5H), 2.18-2.42 (m, 5H), 2.60-2.66 (M, 1H), 2.80-2.91 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.02 (d, J = 7Hz, 1H), 3.36-3 .44 (m, 1H), 3.85 (s, 2.1H), 3.87 (s, 0.9H), 3.90-4.05 (m, 0.3H), 4.09-4 .15 (m, 0.7H), 4.22-4.25 (m, 0.7H), 4.29-4.32 (m, 0.3H), 4.54 (s, 1H), 5 .24 (s, 2H), 6.57-6.60 (m, 1H), 6.72 (d, J = 8Hz, 1H), 7.12 (s, 0.7H), 7.14 (s) , 0.3H), 7.66 (s, 0.7H), 7.68 (s, 0.3H).
(実施例32)
オキサゾール-2-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(51)の合成
(Example 32)
Oxazole-2-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] synthesis of azepine-7-carboxylate (51)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.12(m,2H),0.44-0.52(m,2H),0.73-0.82(m,1H),1.03-1.09(m,1H),1.26-1.72(m,5H),2.18-2.39(m,5H),2.58-2.66(m,1H),2.78-2.90(m,1H),2.95(d,J=18Hz,1H),3.01(d,J=7Hz,1H),3.34-3.44(m,1H),4.02-4.24(m,2H),4.54(s,1H),5.24(s,0.8H),5.27(s,1.2H),6.53(d,J=8Hz,1H),6.69(d,J=8Hz,0.6H),6.71(d,J=8Hz,0.4H),7.14(s,0.6H),7.15(s,0.4H),7.67(s,0.6H),7.68(s,0.4H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.12 (m, 2H), 0.44-0.52 (m, 2H), 0.73-0.82 (m) , 1H), 1.03-1.09 (m, 1H), 1.26-1.72 (m, 5H), 2.18-2.39 (m, 5H), 2.58-2.66 (M, 1H), 2.78-2.90 (m, 1H), 2.95 (d, J = 18Hz, 1H), 3.01 (d, J = 7Hz, 1H), 3.34-3 .44 (m, 1H), 4.02-4.24 (m, 2H), 4.54 (s, 1H), 5.24 (s, 0.8H), 5.27 (s, 1.2H) ), 6.53 (d, J = 8Hz, 1H), 6.69 (d, J = 8Hz, 0.6H), 6.71 (d, J = 8Hz, 0.4H), 7.14 (s). , 0.6H), 7.15 (s, 0.4H), 7.67 (s, 0.6H), 7.68 (s, 0.4H).
(実施例33)
オキサゾール-5-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(52)の合成
(Example 33)
Oxazole-5-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] synthesis of azepine-7-carboxylate (52)
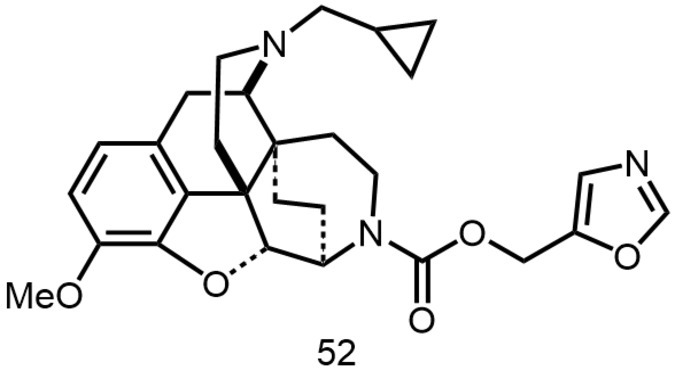
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.13(m,2H),0.43-0.53(m,2H),0.73-0.82(m,1H),1.01-1.11(m,1H),1.24-1.75(m,5H),2.13-2.42(m,5H),2.59-2.65(m,1H),2.76-2.89(m,1H),2.97(d,J=18Hz,1H),3.01(d,J=6Hz,1H),3.37(ddd,J=4,11,14Hz,1H),3.86(s,1.8H),3.88(s,1.2H),3.93-3.99(m,0.4H),4.07-4.14(m,0.6H),4.16-4.19(m,0.6H),4.28-4.32(m,0.4H),4.40-4.43(m,0.6H),4.51-4.53(m,0.4H),5.12-5.23(m,2H),6.57(d,J=8Hz,0.4H),6.59(d,J=8Hz,0.6H),6.72(d,J=8Hz,1H),7.14(s,1H),7.88(s,0.6H),7.89(s,0.4H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.13 (m, 2H), 0.43-0.53 (m, 2H), 0.73-0.82 (m) , 1H), 1.01-1.11 (m, 1H), 1.24-1.75 (m, 5H), 2.13-2.42 (m, 5H), 2.59-2.65 (M, 1H), 2.76-2.89 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.01 (d, J = 6Hz, 1H), 3.37 (ddd) , J = 4,11,14Hz, 1H), 3.86 (s, 1.8H), 3.88 (s, 1.2H), 3.93-3.99 (m, 0.4H), 4 .07-4.14 (m, 0.6H), 4.16-4.19 (m, 0.6H), 4.28-4.32 (m, 0.4H), 4.40-4. 43 (m, 0.6H), 4.51-4.53 (m, 0.4H), 5.12-5.23 (m, 2H), 6.57 (d, J = 8Hz, 0.4H) ), 6.59 (d, J = 8Hz, 0.6H), 6.72 (d, J = 8Hz, 1H), 7.14 (s, 1H), 7.88 (s, 0.6H), 7.89 (s, 0.4H).
(実施例34)
オキサゾール-5-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(53)の合成
(Example 34)
Oxazole-5-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] synthesis of azepine-7-carboxylate (53)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.13(m,2H),0.43-0.53(m,2H),0.73-0.82(m,1H),1.01-1.11(m,1H),1.24-1.72(m,5H),2.13-2.40(m,5H),2.59-2.65(m,1H),2.74-2.89(m,1H),2.95(d,J=18Hz,1H),3.01(d,J=6Hz,1H),3.30-3.43(m,1H),3.95-4.02(m,0.4H),4.07-4.15(m,1.2H),4.20-4.24(m,0.4H),4.39-4.41(m,0.6H),4.49-4.52(m,0.4H),5.13-5.26(m,2H),6.53(d,J=8Hz,1H),6.69(d,J=8Hz,0.6H),6.71(d,J=8Hz,0.4H),7.17(s,1H),7.90(s,0.4H),7.91(s,0.6H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.13 (m, 2H), 0.43-0.53 (m, 2H), 0.73-0.82 (m) , 1H), 1.01-1.11 (m, 1H), 1.24-1.72 (m, 5H), 2.13-2.40 (m, 5H), 2.59-2.65 (M, 1H), 2.74-2.89 (m, 1H), 2.95 (d, J = 18Hz, 1H), 3.01 (d, J = 6Hz, 1H), 3.30-3 .43 (m, 1H), 3.95-4.02 (m, 0.4H), 4.07-4.15 (m, 1.2H), 4.20-4.24 (m, 0. 4H), 4.39-4.41 (m, 0.6H), 4.49-4.52 (m, 0.4H), 5.13-5.26 (m, 2H), 6.53 ( d, J = 8Hz, 1H), 6.69 (d, J = 8Hz, 0.6H), 6.71 (d, J = 8Hz, 0.4H), 7.17 (s, 1H), 7. 90 (s, 0.4H), 7.91 (s, 0.6H).
(実施例35)
チアゾール-2-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(54)の合成
(Example 35)
Thiazole-2-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] synthesis of azepine-7-carboxylate (54)
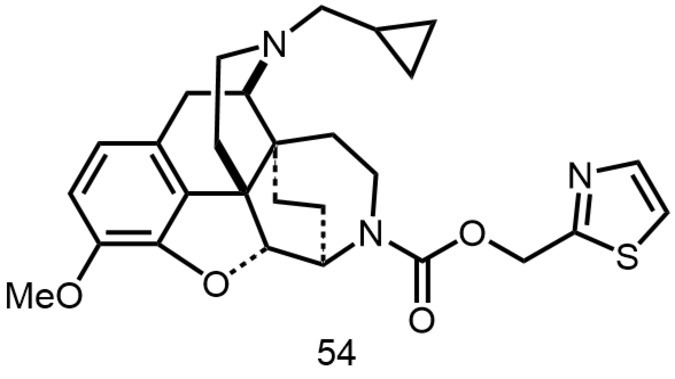
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.44-0.53(m,2H),0.75-0.84(m,1H),1.04-1.13(m,1H),1.26-1.75(m,5H),2.16-2.42(m,5H),2.59-2.67(m,1H),2.80-2.93(m,1H),2.98(d,J=18Hz,1H),3.03(d,J=6Hz,1H),3.38-3.48(m,1H),3.86(s,1.8H),3.88(s,1.2H),4.01-4.18(m,1H),4.25-4.34(m,1H),4.54(s,1H),5.41-5.51(m,2H),6.58(d,J=8Hz,0.4H),6.59(d,J=8Hz,0.6H),6.72(d,J=8Hz,1H),7.34(d,J=3Hz,0.6H),7.36(d,J=3Hz,0.4H),7.77(d,J=3Hz,0.6H),7.79(d,J=3Hz,0.4H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.44-0.53 (m, 2H), 0.75-0.84 (m) , 1H), 1.04-1.13 (m, 1H), 1.26-1.75 (m, 5H), 2.16-2.42 (m, 5H), 2.59-2.67 (M, 1H), 2.80-2.93 (m, 1H), 2.98 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 1H), 3.38-3 .48 (m, 1H), 3.86 (s, 1.8H), 3.88 (s, 1.2H), 4.01-4.18 (m, 1H), 4.25-4.34 (M, 1H), 4.54 (s, 1H), 5.41-5.51 (m, 2H), 6.58 (d, J = 8Hz, 0.4H), 6.59 (d, J) = 8Hz, 0.6H), 6.72 (d, J = 8Hz, 1H), 7.34 (d, J = 3Hz, 0.6H), 7.36 (d, J = 3Hz, 0.4H) , 7.77 (d, J = 3Hz, 0.6H), 7.79 (d, J = 3Hz, 0.4H).
(実施例36)
チアゾール-2-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(55)の合成
(Example 36)
Thiazole-2-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] synthesis of azepine-7-carboxylate (55)
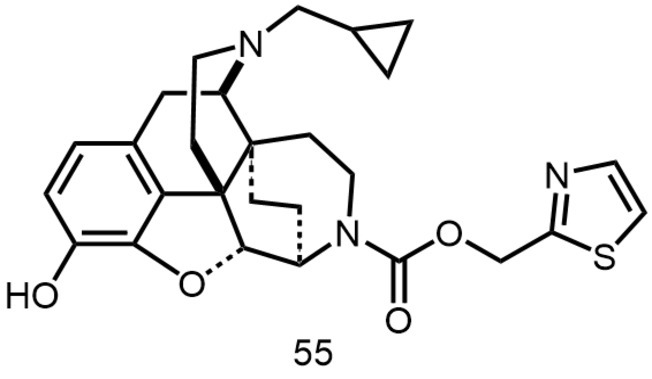
1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.43-0.53(m,2H),0.74-0.83(m,1H),1.03-1.12(m,1H),1.25-1.74(m,5H),2.18-2.41(m,5H),2.59-2.67(m,1H),2.79-2.91(m,1H),2.96(d,J=18Hz,1H),3.03(d,J=6Hz,1H),3.36-3.46(m,1H),4.05-4.32(m,2H),4.53(s,1H),5.41-5.55(m,2H),6.54(d,J=8Hz,1H),6.70(d,J=8Hz,0.6H),6.72(d,J=8Hz,0.4H),7.35-7.39(m,1H),7.78-7.82(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.43-0.53 (m, 2H), 0.74-0.83 (m) , 1H), 1.03-1.12 (m, 1H), 1.25-1.74 (m, 5H), 2.18-2.41 (m, 5H), 2.59-2.67 (M, 1H), 2.79-2.91 (m, 1H), 2.96 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 1H), 3.36-3 .46 (m, 1H), 4.05-4.32 (m, 2H), 4.53 (s, 1H), 5.41-5.55 (m, 2H), 6.54 (d, J) = 8Hz, 1H), 6.70 (d, J = 8Hz, 0.6H), 6.72 (d, J = 8Hz, 0.4H), 7.35-7.39 (m, 1H), 7 .78-7.82 (m, 1H).
(実施例37)
ピリミジン-2-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(56)の合成
(Example 37)
Pyrimidine-2-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-Metanobenzoflo [2,3-c] Pyrimidine [4,3-d] Synthesis of azepine-7-carboxylate (56)
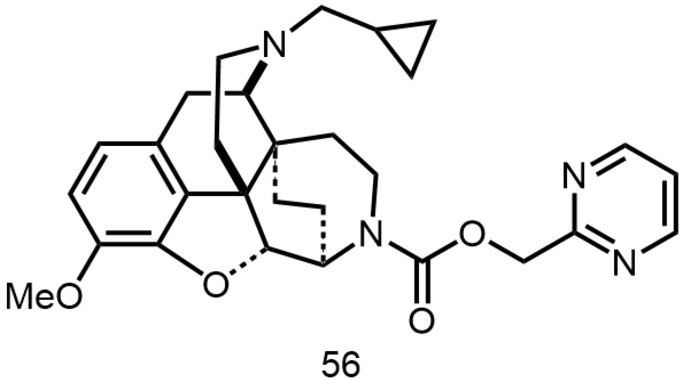
1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.19(m,2H),0.43-0.54(m,2H),0.74-0.92(m,2H),1.01-1.82(m,5H),2.14-2.45(m,5H),2.57-2.69(m,1H),2.81-3.07(m,3H),3.25-3.62(m,1H),3.82-3.89(m,3H),4.10-4.66(m,2H),4.83-5.02(m,1H),5.22-5.53(m,2H),6.55-6.64(m,1H),6.69-6.74(m,1H),7.16-7.26(m,1H),8.67-8.77(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.19 (m, 2H), 0.43-0.54 (m, 2H), 0.74-0.92 (m) , 2H), 1.01-1.82 (m, 5H), 2.14-2.45 (m, 5H), 2.57-2.69 (m, 1H), 2.81-3.07 (M, 3H), 3.25-3.62 (m, 1H), 3.82-3.89 (m, 3H), 4.10-4.66 (m, 2H), 4.83-5 .02 (m, 1H), 5.22-5.53 (m, 2H), 6.55-6.64 (m, 1H), 6.69-6.74 (m, 1H), 7.16 -7.26 (m, 1H), 8.67-8.77 (m, 2H).
(実施例38)
ピリミジン-2-イルメチル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(57)の合成
(Example 38)
Pyrimidine-2-ylmethyl (4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a , 8-Etano-4,13-Metanobenzoflo [2,3-c] Pyrimidine [4,3-d] Synthesis of azepine-7-carboxylate (57)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.18(m,2H),0.41-0.54(m,2H),0.70-0.93(m,1H),1.01-1.83(m,6H),2.14-2.45(m,5H),2.58-2.71(m,1H),2.80-3.09(m,3H),3.26-3.60(m,1H),4.15-4.33(m,2H),4.55-5.06(m,2H),5.22-5.59(m,2H)6.49-6.57(m,1H),6.65-6.74(m,1H),7.15-7.29(m,1H),8.68-8.74(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.18 (m, 2H), 0.41-0.54 (m, 2H), 0.70-0.93 (m) , 1H), 1.01-1.83 (m, 6H), 2.14-2.45 (m, 5H), 2.58-2.71 (m, 1H), 2.80-3.09 (M, 3H), 3.26-3.60 (m, 1H), 4.15-4.33 (m, 2H), 4.55-5.06 (m, 2H), 5.22-5 .59 (m, 2H) 6.49-6.57 (m, 1H), 6.65-6.74 (m, 1H), 7.15-7.29 (m, 1H), 8.68- 8.74 (m, 2H).
(実施例39)
2-(ヒドロキシメチル)ピリジン-3-イル (4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボキシレート(58)の合成
(Example 39)
2- (Hydroxymethyl) Pyridine-3-yl (4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-1,2,3,4,5,6,8,8a -Synthesis of Octahydro-7H-4a, 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carboxylate (58)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.18(m,2H),0.43-0.57(m,2H),0.74-0.94(m,1H),1.07-1.87(m,6H),2.19-2.45(m,5H),2.61-2.72(m,1H),2.86-3.11(m,3H),3.41-3.66(m,1H),4.06-4.37(m,3H),4.54-4.87(m,4H),6.52-6.60(m,1H),6.72(d,J=8Hz,1H),7.24-7.33(m,1H),7.54-7.62(m,1H),8.42-8.47(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.04-0.18 (m, 2H), 0.43-0.57 (m, 2H), 0.74-0.94 (m) , 1H), 1.07-1.87 (m, 6H), 2.19-2.45 (m, 5H), 2.61-2.72 (m, 1H), 2.86-3.11 (M, 3H), 3.41-3.66 (m, 1H), 4.06-4.37 (m, 3H), 4.54-4.87 (m, 4H), 6.52-6 .60 (m, 1H), 6.72 (d, J = 8Hz, 1H), 7.24-7.33 (m, 1H), 7.54-7.62 (m, 1H), 8.42 -8.47 (m, 1H).
(実施例40)
(4R,4aS,8R,8aR,13bS)-7-ベンジル-3-(シクロプロピルメチル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-オール(59)の合成
(Example 40)
(4R, 4aS, 8R, 8aR, 13bS) -7-Benzyl-3- (cyclopropylmethyl) -1,2,3,4,6,7,8,8a-octahydro-5H-4a, 8-ethano- Synthesis of 4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-10-ol (59)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.15(m,2H),0.41-0.54(m,2H),0.73-0.85(m,1H),0.89-0.99(m,1H),1.00-1.11(m,1H),1.16-1.28(m,1H),1.29-1.38(m,1H),1.49-1.70(m,2H),2.22(dd,J=7,12Hz,1H),2.25-2.47(m,4H),2.63(dd,J=5,12Hz,1H),2.74-2.88(m,4H),2.93(d,J=18Hz,1H),2.98(d,J=6Hz,1H),3.66(d,J=14Hz,1H),3.77(d,J=14Hz,1H),4.55(s,1H),6.48(d,J=8Hz,1H),6.64(d,J=8Hz,1H),7.20-7.40(m,5H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.15 (m, 2H), 0.41-0.54 (m, 2H), 0.73-0.85 (m) , 1H), 0.89-0.99 (m, 1H), 1.00-1.11 (m, 1H), 1.16-1.28 (m, 1H), 1.29-1.38 (M, 1H), 1.49-1.70 (m, 2H), 2.22 (dd, J = 7,12Hz, 1H), 2.25-2.47 (m, 4H), 2.63 (Dd, J = 5,12Hz, 1H), 2.74-2.88 (m, 4H), 2.93 (d, J = 18Hz, 1H), 2.98 (d, J = 6Hz, 1H) , 3.66 (d, J = 14Hz, 1H), 3.77 (d, J = 14Hz, 1H), 4.55 (s, 1H), 6.48 (d, J = 8Hz, 1H), 6 .64 (d, J = 8Hz, 1H), 7.20-7.40 (m, 5H).
(実施例41)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-オール(60)の合成
(Example 41)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -1,2,3,4,6,7,8,8a-Octahydro-5H-4a, 8-Etano-4,13- Synthesis of methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-10-ol (60)

1H-NMR(400MHz,CD3OD)δ(ppm):0.12-0.22(m,2H),0.46-0.58(m,2H),0.79-0.92(m,1H),1.11-1.21(m,1H),1.30-1.45(m,2H),1.49-1.61(m,1H),1.69(ddd,J=5,5,16Hz,1H),1.75-1.88(m,1H),2.29-2.45(m,4H),2.50(dd,J=6,19Hz,1H),2.68-2.81(m,1H),3.03(d,J=19Hz,1H),3.03-3.14(m,1H),3.19-3.40(m,3H),3.62(d,J=2Hz,1H),4.73(s,1H),6.57(d,J=8Hz,1H),6.67(d,J=8Hz,1H).
1 1 H-NMR (400 MHz, CD 3 OD) δ (ppm): 0.12-0.22 (m, 2H), 0.46-0.58 (m, 2H), 0.79-0.92 ( m, 1H), 1.11-1.21 (m, 1H), 1.30-1.45 (m, 2H), 1.49-1.61 (m, 1H), 1.69 (ddd, J = 5,5,16Hz, 1H), 1.75-1.88 (m, 1H), 2.29-2.45 (m, 4H), 2.50 (dd, J = 6,19Hz, 1H) ), 2.68-2.81 (m, 1H), 3.03 (d, J = 19Hz, 1H), 3.03-3.14 (m, 1H), 3.19-3.40 (m) , 3H), 3.62 (d, J = 2Hz, 1H), 4.73 (s, 1H), 6.57 (d, J = 8Hz, 1H), 6.67 (d, J = 8Hz, 1H) ).
(実施例42)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(3-フルオロピリジン-2-イル)メタノン(61)の合成
(Example 42)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (3-fluoropyridin-2-yl) methanone (61)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.12(m,2H),0.43-0.52(m,2H),0.71-0.86(m,1H),1.05-1.20(m,1H),1.23-1.35(m,2H),1.50-1.61(m,2H),1.68-1.86(m,1H),2.19-2.44(m,5H),2.61-2.68(m,1H),2.83-2.92(m,0.7H),2.97-3.04(m,0.3H),2.99(d,J=19Hz,1H),3.04(d,J=6Hz,0.7H),3.08(d,J=6Hz,0.3H),3.39-3.62(m,1.7H),3.78-3.82(m,0.3H),3.79(s,0.9H),3.90(s,2.1H),4.51(ddd,J=5,5,14Hz,0.3H),4.68-4.71(m,0.3H),4.73-4.75(m,0.7H),4.80-4.83(m,0.7H),6.60(d,J=8Hz,1H),6.68(d,J=8Hz,0.3H),6.74(d,J=8Hz,0.7H),7.33-7.40(m,1H),7.46-7.53(m,1H),8.44(ddd,J=1,1,5Hz,0.3H),8.47(ddd,J=1,1,5Hz,0.7H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.12 (m, 2H), 0.43-0.52 (m, 2H), 0.71-0.86 (m) , 1H), 1.05-1.20 (m, 1H), 1.23-1.35 (m, 2H), 1.50-1.61 (m, 2H), 1.68-1.86 (M, 1H), 2.19-2.44 (m, 5H), 2.61-2.68 (m, 1H), 2.83-2.92 (m, 0.7H), 2.97 -3.04 (m, 0.3H), 2.99 (d, J = 19Hz, 1H), 3.04 (d, J = 6Hz, 0.7H), 3.08 (d, J = 6Hz, 0.3H), 3.39-3.62 (m, 1.7H), 3.78-3.82 (m, 0.3H), 3.79 (s, 0.9H), 3.90 ( s, 2.1H), 4.51 (ddd, J = 5,5,14Hz, 0.3H), 4.68-4.71 (m, 0.3H), 4.73-4.75 (m) , 0.7H), 4.80-4.83 (m, 0.7H), 6.60 (d, J = 8Hz, 1H), 6.68 (d, J = 8Hz, 0.3H), 6 .74 (d, J = 8Hz, 0.7H), 7.33-7.40 (m, 1H), 7.46-7.53 (m, 1H), 8.44 (ddd, J = 1, 1,5Hz, 0.3H), 8.47 (ddd, J = 1,1,5Hz, 0.7H).
(実施例43)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(3-フルオロピリジン-2-イル)メタノン(62)の合成
(Example 43)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (3-fluoropyridin-2-yl) methanone (62)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.15(m,2H),0.40-0.52(m,2H),0.69-0.78(m,1H),1.14-1.18(m,1H),1.24-1.34(m,2H),1.46-1.88(m,3H),2.17-2.40(m,5H),2.60-2.67(m,1H),2.80-2.91(m,0.8H),2.92-3.00(m,0.2H),2.97(d,J=18Hz,1H),3.03(d,J=6Hz,0.8H),3.07(d,J=6Hz,0.2H),3.39-3.58(m,1.8H),3.69-3.72(m,0.2H),4.51-4.58(m,0.2H),4.68-4.70(m,0.2H),4.71-4.76(m,1.6H),6.52(d,J=8Hz,0.2H),6.55(d,J=8Hz,0.8H),6.64(d,J=8Hz,0.2H),6.73(d,J=8Hz,0.8H),7.39(ddd,J=4,4,8Hz,1H), 7.52(ddd,J=1,8,8Hz,1H),8.14(ddd,J=1,1,4Hz,0.2H),8.49(ddd,J=1,1,4Hz,0.8H),
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.15 (m, 2H), 0.40-0.52 (m, 2H), 0.69-0.78 (m) , 1H), 1.14-1.18 (m, 1H), 1.24-1.34 (m, 2H), 1.46-1.88 (m, 3H), 2.17-2.40 (M, 5H), 2.60-2.67 (m, 1H), 2.80-2.91 (m, 0.8H), 2.92-3.00 (m, 0.2H), 2 .97 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 0.8H), 3.07 (d, J = 6Hz, 0.2H), 3.39-3.58 ( m, 1.8H), 3.69-3.72 (m, 0.2H), 4.51-4.58 (m, 0.2H), 4.68-4.70 (m, 0.2H) ), 4.71-4.76 (m, 1.6H), 6.52 (d, J = 8Hz, 0.2H), 6.55 (d, J = 8Hz, 0.8H), 6.64 (D, J = 8Hz, 0.2H), 6.73 (d, J = 8Hz, 0.8H), 7.39 (ddd, J = 4,4,8Hz, 1H), 7.52 (ddd, J = 1,8,8Hz, 1H), 8.14 (ddd, J = 1,1,4Hz, 0.2H), 8.49 (ddd, J = 1,1,4Hz, 0.8H),
(実施例44)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピリミジン-2-イル)メタノン(63)の合成
(Example 44)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano -4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyrimidine-2-yl) synthesis of methanone (63)

化合物9(14mg,0.038mmol)、ピリミジン-2-カルボン酸(14.2mg,0.11mmol)、N,N-ジメチルアミノピリジン(2.3mg,0.019mmol),1-ヒドロキシベンズトリアゾール一水和物(17.5mg,0.11mmol)、N,N-ジイソプロピルエチルアミン(20μL,0.11mmol)及び1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(22mg,0.11mmol)のN,N-ジメチルホルムアミド(1mL)溶液を、室温で15時間撹拌した。反応混合物に酢酸エチルを加え、飽和炭酸水素ナトリウム水溶液で三回、飽和食塩水で一回洗浄した。有機層を硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物のジクロロメタン(1mL)溶液に、氷冷下、1M三臭化ホウ素-ジクロロメタン溶液(200μL,0.20mmoL)を加え、室温で1時間撹拌した。反応混合物に28%アンモニア水溶液を加え、クロロホルムで三回抽出した。合わせた抽出液を硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(0-10%メタノール/クロロホルム)で精製し、表題化合物(9.2mg,53%)を無色油状物質として得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.15(m,2H),0.40-0.55(m,2H),0.70-0.86(m,1H),1.04-1.33(m,3H),1.45-1.61(m,2H),1.69-1.87(m,1H),2.18-2.41(m,5H),2.60-2.69(m,1H),2.86-2.99(m,1H),2.96(d,J=19Hz,1H),3.04(d,J=6Hz,0.7H),3.08(d,J=6Hz,0.3H),3.38-3.54(m,1.7H),3.64-3.68(m,0.3H),4.54-4.61(m,0.3H),4.69-4.75(m,0.7H),4.75(s,0.7H),4.84(s,0.3H),6.51(d,J=8Hz,0.3H),6.54(d,J=8Hz,0.7H),6.65(d,J=8Hz,0.3H),6.73(d,J=8Hz,0.7H),7.37(t,J=5Hz,1H),8.84(d,J=5Hz,0.6H),8.85(d,J=5Hz,1.4H).
Compound 9 (14 mg, 0.038 mmol), pyrimidin-2-carboxylic acid (14.2 mg, 0.11 mmol), N, N-dimethylaminopyridine (2.3 mg, 0.019 mmol), 1-hydroxybenztriazole monohydrate. Japanese products (17.5 mg, 0.11 mmol), N, N-diisopropylethylamine (20 μL, 0.11 mmol) and 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (22 mg, 0.11 mmol). The N, N-dimethylformamide (1 mL) solution was stirred at room temperature for 15 hours. Ethyl acetate was added to the reaction mixture, and the mixture was washed three times with saturated aqueous sodium hydrogen carbonate solution and once with saturated brine. The organic layer was dried over sodium sulfate and then concentrated under reduced pressure. A 1M boron tribromide-dichloromethane solution (200 μL, 0.20 mmoL) was added to a solution of the obtained crude product in dichloromethane (1 mL) under ice-cooling, and the mixture was stirred at room temperature for 1 hour. A 28% aqueous ammonia solution was added to the reaction mixture, and the mixture was extracted three times with chloroform. The combined extracts were dried over sodium sulfate and then concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (0-10% methanol / chloroform) to give the title compound (9.2 mg, 53%) as a colorless oily substance.
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.04-0.15 (m, 2H), 0.40-0.55 (m, 2H), 0.70-0.86 (m) , 1H), 1.04-1.33 (m, 3H), 1.45-1.61 (m, 2H), 1.69-1.87 (m, 1H), 2.18-2.41 (M, 5H), 2.60-2.69 (m, 1H), 2.86-2.99 (m, 1H), 2.96 (d, J = 19Hz, 1H), 3.04 (d) , J = 6Hz, 0.7H), 3.08 (d, J = 6Hz, 0.3H), 3.38-3.54 (m, 1.7H), 3.64-3.68 (m, 0.3H), 4.54-4.61 (m, 0.3H), 4.69-4.75 (m, 0.7H), 4.75 (s, 0.7H), 4.84 ( s, 0.3H), 6.51 (d, J = 8Hz, 0.3H), 6.54 (d, J = 8Hz, 0.7H), 6.65 (d, J = 8Hz, 0.3H) ), 6.73 (d, J = 8Hz, 0.7H), 7.37 (t, J = 5Hz, 1H), 8.84 (d, J = 5Hz, 0.6H), 8.85 (d). , J = 5Hz, 1.4H).
(実施例45)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピリミジン-4-イル)メタノン(64)の合成
(Example 45)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyrimidine-4-yl) methanone (64)
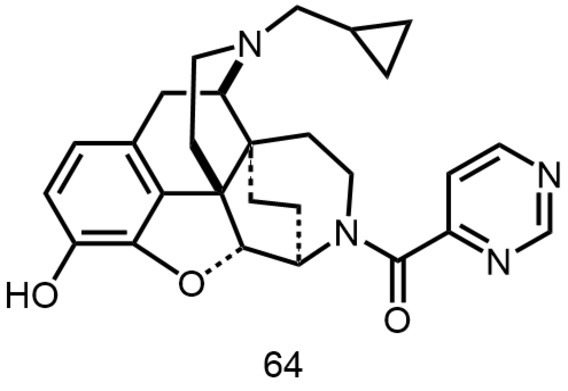
化合物60(5.7mg,0.016mmol)、ピリミジン-4-カルボン酸(6mg,0.049mmol)、N,N-ジメチルアミノピリジン(1.1mg,0.008mmol),1-ヒドロキシベンズトリアゾール一水和物(6.6mg,0.049mmol)、N,N-ジイソプロピルエチルアミン(8.4μL,0.049mmol)及び1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(8.3mg,0.049mmol)のN,N-ジメチルホルムアミド(0.5mL)溶液を、室温で1時間撹拌した。反応混合物に2M水酸化ナトリウム水溶液を加え、1時間撹拌した。反応混合物をクロロホルムで三回抽出後、合わせた抽出物を硫酸ナトリウムで乾燥し、減圧下にて濃縮した。得られた粗生成物を分取薄層クロマトグラフィー(クロロホルム:メタノール=97:3)で精製し、表題化合物(5.9mg,80%)を無色油状物質として得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.43-0.54(m,2H),0.71-0.81(m,1H),1.04-1.18(m,1H),1.25-1.39(m,2H),1.46-1.84(m,3H),2.20-2.43(m,5H),2.61-2.68(m,1H),2.89-2.99(m,1H),2.97(d,J=18Hz,1H),3.06(d,J=6Hz,0.7H),3.08(d,J=6Hz,0.3H),3.43-3.58(m,1H),3.67-3.76(m,0.7H),3.92-3.96(m,0.3H),4.49-4.56(m,0.3H),4.65-4.71(m,1.4H),4.89-4.92(m,0.3H),5.28(br s,1H),6.53(d,J=8Hz,0.3H),6.56(d,J=8Hz,0.7H),6.67(d,J=8Hz,0.3H),6.73(d,J=8Hz,0.7H),7.58(dd,J=1,5Hz,0.7H),7.61(dd,J=1,5Hz,0.3H),8.89(d,J=5Hz,0.7H),8.90(d,J=5Hz,0.3H),9.28(br s,0.7H),9.32(d,J=1Hz,0.3H).
Compound 60 (5.7 mg, 0.016 mmol), pyrimidin-4-carboxylic acid (6 mg, 0.049 mmol), N, N-dimethylaminopyridine (1.1 mg, 0.008 mmol), 1-hydroxybenztriazole monohydrate. Japanese product (6.6 mg, 0.049 mmol), N, N-diisopropylethylamine (8.4 μL, 0.049 mmol) and 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (8.3 mg, 0). A solution of N, N-dimethylformamide (0.5 mL) (0.049 mmol) was stirred at room temperature for 1 hour. A 2M aqueous sodium hydroxide solution was added to the reaction mixture, and the mixture was stirred for 1 hour. The reaction mixture was extracted three times with chloroform, and the combined extracts were dried over sodium sulfate and concentrated under reduced pressure. The obtained crude product was purified by preparative thin layer chromatography (chloroform: methanol = 97: 3) to give the title compound (5.9 mg, 80%) as a colorless oily substance.
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.43-0.54 (m, 2H), 0.71-0.81 (m) , 1H), 1.04-1.18 (m, 1H), 1.25-1.39 (m, 2H), 1.46-1.84 (m, 3H), 2.20-2.43 (M, 5H), 2.61-2.68 (m, 1H), 2.89-2.99 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.06 (d) , J = 6Hz, 0.7H), 3.08 (d, J = 6Hz, 0.3H), 3.43-3.58 (m, 1H), 3.67-3.76 (m, 0. 7H), 3.92-3.96 (m, 0.3H), 4.49-4.56 (m, 0.3H), 4.65-4.71 (m, 1.4H), 4. 89-4.92 (m, 0.3H), 5.28 (br s, 1H), 6.53 (d, J = 8Hz, 0.3H), 6.56 (d, J = 8Hz, 0. 7H), 6.67 (d, J = 8Hz, 0.3H), 6.73 (d, J = 8Hz, 0.7H), 7.58 (dd, J = 1,5Hz, 0.7H), 7.61 (dd, J = 1,5Hz, 0.3H), 8.89 (d, J = 5Hz, 0.7H), 8.90 (d, J = 5Hz, 0.3H), 9.28 (Br s, 0.7H), 9.32 (d, J = 1Hz, 0.3H).
(実施例46)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピリミジン-5-イル)メタノン(65)の合成
(Example 46)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyrimidine-5-yl) methanone (65)
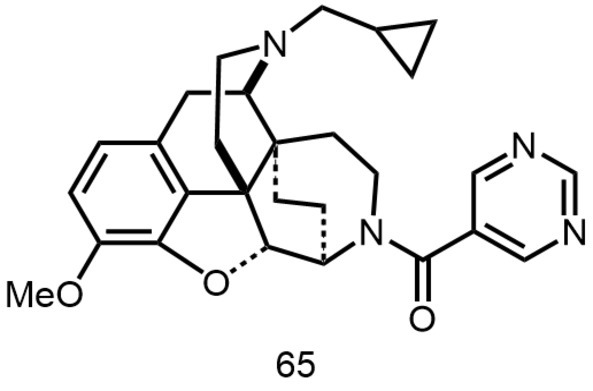
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.16(m,2H),0.43-0.55(m,2H),0.71-0.82(m,1H),1.10-1.43(m,3H),1.50-1.85(m,3H),2.15-2.41(m,4H),2.42(dd,J=6,18Hz,1H),2.66(dd,J=5,12Hz,1H),2.83-2.94(m,1H),3.01(d,J=19Hz,1H),3.05(d,J=6Hz,1H),3.57-3.67(m,2H),3.90(s,3H),4.66(s,1H),4.76-4.82(m,1H),6.62(d,J=8Hz,1H),6.76(d,J=8Hz,1H),8.83(s,2H),9.28(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.16 (m, 2H), 0.43-0.55 (m, 2H), 0.71-0.82 (m) , 1H), 1.10-1.43 (m, 3H), 1.50-1.85 (m, 3H), 2.15-2.41 (m, 4H), 2.42 (dd, J) = 6,18Hz, 1H), 2.66 (dd, J = 5,12Hz, 1H), 2.83-2.94 (m, 1H), 3.01 (d, J = 19Hz, 1H), 3 .05 (d, J = 6Hz, 1H), 3.57-3.67 (m, 2H), 3.90 (s, 3H), 4.66 (s, 1H), 4.76-4.82 (M, 1H), 6.62 (d, J = 8Hz, 1H), 6.76 (d, J = 8Hz, 1H), 8.83 (s, 2H), 9.28 (s, 1H).
(実施例47)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピリミジン-5-イル)メタノン(65)の合成
(Example 47)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyrimidine-5-yl) methanone (65)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.15(m,2H),0.42-0.54(m,2H),0.70-0.81(m,1H),1.11-1.21(m,1H),1.22-1.34(m,1H),1.34-1.42(m,1H),1.48-1.59(m,1H),1.62(dd,J=2,14Hz,1H),1.73-1.85(m,1H),2.13-2.44(m,5H),2.65(dd,J=4,11Hz,1H),2.81-2.92(m,1H),2.98(d,J=18Hz,1H),3.03(d,J=6Hz,1H),3.52-3.70(m,2H),4.64(s,1H),4.68-4.74(m,1H),6.55(d,J=8Hz,1H),6.74(d,J=8Hz,1H),8.84(s,2H),9.29(s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-0.15 (m, 2H), 0.42-0.54 (m, 2H), 0.70-0.81 (m) , 1H), 1.11-1.21 (m, 1H), 1.22-1.34 (m, 1H), 1.34-1.42 (m, 1H), 1.48-1.59 (M, 1H), 1.62 (dd, J = 2,14Hz, 1H), 1.73-1.85 (m, 1H), 2.13-2.44 (m, 5H), 2.65 (Dd, J = 4,11Hz, 1H), 2.81-2.92 (m, 1H), 2.98 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 1H) , 3.52-3.70 (m, 2H), 4.64 (s, 1H), 4.68-4.74 (m, 1H), 6.55 (d, J = 8Hz, 1H), 6 .74 (d, J = 8Hz, 1H), 8.84 (s, 2H), 9.29 (s, 1H).
(実施例48)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピリダジン-3-イル)メタノン(67)の合成
(Example 48)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyridazine-3-yl) methanone (67)

1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.18(m,2H),0.45-0.55(m,2H),0.70-1.00(m,2H),1.10-1.20(m,1H),1.20-1.90(m,4H),2.20-2.45(m,5H),2.60-2.70(m,1H),2.95-3.10(m,3H),3.40-3.70(m,1H),3.85-4.00(m,0.7H),4.13(s,0.3H),4.50-4.65(m,0.3H),4.70-4.80(m,1.4H),5.01(s,0.3H),6.50-6.60(m,1H),6.67(d,J=8Hz,0.3H),6.73(d,J=8Hz,0.7H),7.60-7.65(m,1H),7.80-7.90(m,1H)9.23-9.28(m,1H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.18 (m, 2H), 0.45-0.55 (m, 2H), 0.70-1.00 (m) , 2H), 1.10-1.20 (m, 1H), 1.20-1.90 (m, 4H), 2.20-2.45 (m, 5H), 2.60-2.70 (M, 1H), 2.95-3.10 (m, 3H), 3.40-3.70 (m, 1H), 3.85-4.00 (m, 0.7H), 4.13 (S, 0.3H), 4.50-4.65 (m, 0.3H), 4.70-4.80 (m, 1.4H), 5.01 (s, 0.3H), 6 .50-6.60 (m, 1H), 6.67 (d, J = 8Hz, 0.3H), 6.73 (d, J = 8Hz, 0.7H), 7.60-7.65 ( m, 1H), 7.80-7.90 (m, 1H) 9.23-9.28 (m, 1H).
(実施例49)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピリダジン-4-イル)メタノン(68)の合成
(Example 49)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyridazine-4-yl) methanone (68)

1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.20(m,2H), 0.40-0.60(m,2H),0.60-1.00(m,4H),1.00-2.00(m,3H),2.22-2.50(m,5H),2.60-2.70(m,1H),2.80-2.90(m,1H),2.90-3.20(m,2H),3.40-3.70(m,2H),,4.60-4.80(m,2H),6.56(d,J=8Hz,1H),6.75(d,J=8Hz,1H),7.45-7.60(m,1H),9.20-9.28(m,1H),9.28-9.60(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.20 (m, 2H), 0.40-0.60 (m, 2H), 0.60-1.00 (m) , 4H), 1.00-2.00 (m, 3H), 2.22-2.50 (m, 5H), 2.60-2.70 (m, 1H), 2.80-2.90 (M, 1H), 2.90-3.20 (m, 2H), 3.40-3.70 (m, 2H), 4.60-4.80 (m, 2H), 6.56 ( d, J = 8Hz, 1H), 6.75 (d, J = 8Hz, 1H), 7.45-7.60 (m, 1H), 9.20-9.28 (m, 1H), 9. 28-9.60 (m, 1H).
(実施例50)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピラジン-2-イル)メタノン(69)の合成
(Example 50)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyrazine-2-yl) methanone (69)

実施例42に記載した方法に従い、化合物9及びピラジン-2-カルボン酸より表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm): 0.00-0.15(m,2H),0.40-0.70(m,2H),0.70-1.00(m,1H),1.00-2.00(m,6H),2.10-2.50(m,5H),2.55-2.70(m,1H),2.80-3.20(m,2.7H),3.60-4.00(m,2H),3.79(s,0.9H),3.90(s,2.1H),4.12(s,0.3H),4.40-4.60(m,0.3H),4.72(s,0.7H),4.87(s,1H),6.55-6.65(m,1H),6.69(d,J=8Hz,0.3H),6.75(d,J=8Hz,0.7H),7.55-7.65(m,1H),7.74(dt,J=2,8Hz,0.3H),7.82(dt,J=2,8Hz,0.7H),9.20-9.30(m,1H).
The title compound was obtained from compound 9 and pyrazine-2-carboxylic acid according to the method described in Example 42.
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.15 (m, 2H), 0.40-0.70 (m, 2H), 0.70-1.00 (m) , 1H), 1.00-2.00 (m, 6H), 2.10-2.50 (m, 5H), 2.55-2.70 (m, 1H), 2.80-3.20 (M, 2.7H), 3.60-4.00 (m, 2H), 3.79 (s, 0.9H), 3.90 (s, 2.1H), 4.12 (s, 0) .3H), 4.40-4.60 (m, 0.3H), 4.72 (s, 0.7H), 4.87 (s, 1H), 6.55-6.65 (m, 1H) ), 6.69 (d, J = 8Hz, 0.3H), 6.75 (d, J = 8Hz, 0.7H), 7.55-7.65 (m, 1H), 7.74 (dt). , J = 2.8Hz, 0.3H), 7.82 (dt, J = 2.8Hz, 0.7H), 9.20-9.30 (m, 1H).
(実施例51)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(ピラジン-2-イル)メタノン(70)の合成
(Example 51)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano -4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (pyrazine-2-yl) synthesis of methanone (70)

1H-NMR(400MHz,CDCl3)δ(ppm): 0.00-0.15(m,2H),0.40-0.55(m,2H),0.70-1.00(m,1.3H),1.00-1.90(m,5.4H),2.20-2.45(m,5.3H),2.60-2.70(m,1H),2.88-3.10(m,3H),3.40-3.60(m,1H),3.70-3.80(m,0.7H),4.01(s,0.3H),4.50-4.60(m,0.3H),4.68(s,0.7H),4.72(s,0.7H),4.92(s,0.3H),6.50-6.58(m,1H),6.67(d,J=8Hz,0.3H),6.73(d,J=8Hz,0.7H),8.55-8.65(m,2H),8.89(d,J=1Hz,0.7H),8.94(d,J=1Hz,0.3H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.15 (m, 2H), 0.40-0.55 (m, 2H), 0.70-1.00 (m) , 1.3H), 1.00-1.90 (m, 5.4H), 2.20-2.45 (m, 5.3H), 2.60-2.70 (m, 1H), 2 .88-3.10 (m, 3H), 3.40-3.60 (m, 1H), 3.70-3.80 (m, 0.7H), 4.01 (s, 0.3H) , 4.50-4.60 (m, 0.3H), 4.68 (s, 0.7H), 4.72 (s, 0.7H), 4.92 (s, 0.3H), 6 .50-6.58 (m, 1H), 6.67 (d, J = 8Hz, 0.3H), 6.73 (d, J = 8Hz, 0.7H), 8.55-8.65 ( m, 2H), 8.89 (d, J = 1Hz, 0.7H), 8.94 (d, J = 1Hz, 0.3H).
(実施例52)
6-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボニル)ピリジン-2(1H)-オン(71)の合成
(Example 52)
6-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,6,7,8a-octahydro-5H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carbonyl) pyridin-2 (1H) -one (71)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.12(m,2H),0.43-0.52(m,2H),0.72-0.82(m,1H),1.08-1.18(m,1H),1.22-1.59(m,4H),1.72-1.87(m,1H),2.20-2.45(m,5H),2.59-2.66(m,1H),2.85-2.96(m,1H),2.99(d,J=19Hz,1H),3.06(d,J=6Hz,1H),3.56-3.66(m,1H),3.72-3.84(m,1H),3.88(s,3H),4.62-4.73(m,2H),6.36(d,J=6Hz,1H),6.60(d,J=9Hz,1H),6.61(d,J=8Hz,1H),6.74(d,J=8Hz,1H),7.43(dd,J=6,9Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.12 (m, 2H), 0.43-0.52 (m, 2H), 0.72-0.82 (m) , 1H), 1.08-1.18 (m, 1H), 1.22-1.59 (m, 4H), 1.72-1.87 (m, 1H), 2.20-2.45 (M, 5H), 2.59-2.66 (m, 1H), 2.85-2.96 (m, 1H), 2.99 (d, J = 19Hz, 1H), 3.06 (d). , J = 6Hz, 1H), 3.56-3.66 (m, 1H), 3.72-3.84 (m, 1H), 3.88 (s, 3H), 4.62-4.73 (M, 2H), 6.36 (d, J = 6Hz, 1H), 6.60 (d, J = 9Hz, 1H), 6.61 (d, J = 8Hz, 1H), 6.74 (d). , J = 8Hz, 1H), 7.43 (dd, J = 6,9Hz, 1H).
(実施例53)
6-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボニル)ピリジン-2(1H)-オン(72)の合成
(Example 53)
6-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,6,7,8,8a-octahydro-5H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carbonyl) pyridin-2 (1H) -one (72)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.13(m,2H),0.41-0.51(m,2H),0.69-0.81(m,1H),1.03-1.79(m,6H),2.10-2.40(m,5H),2.53-2.63(m,1H),2.75-2.90(m,1H),2.93(d,J=18Hz,1H),3.02(d,J=5Hz,1H),3.46-3.60(m,2H),3.75-3.90(m,1H),4.48-4.70(m,1H),6.32(br s,1H),6.51(d,J=8Hz,1H),6.63(br s,1H),6.74(br s,1H),7.38(br s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.13 (m, 2H), 0.41-0.51 (m, 2H), 0.69-0.81 (m) , 1H), 1.03-1.79 (m, 6H), 2.10-2.40 (m, 5H), 2.53-2.63 (m, 1H), 2.75-2.90 (M, 1H), 2.93 (d, J = 18Hz, 1H), 3.02 (d, J = 5Hz, 1H), 3.46-3.60 (m, 2H), 3.75-3 .90 (m, 1H), 4.48-4.70 (m, 1H), 6.32 (br s, 1H), 6.51 (d, J = 8Hz, 1H), 6.63 (br s) , 1H), 6.74 (br s, 1H), 7.38 (br s, 1H).
(実施例54)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(6-ヒドロキシピリジン-3-イル)メタノン(73)の合成
(Example 54)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (6-hydroxypyridin-3-yl) methanone (73)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.14(m,2H),0.44-0.54(m,2H),0.74-0.83(m,1H),1.08-1.17(m,1H),1.22-1.36(m,1H),1.37-1.63(m,3H),1.66-1.78(m,1H),2.15-2.45(m,5H),2.65(dd,J=5,11Hz,1H),2.81-2.92(m,1H),2.99(d,J=19Hz,1H),3.06(d,J=6Hz,1H),3.54-3.64(m,1H),3.75-3.88(m,1H),3.88(s,3H),4.50-4.73(m,2H),6.60(d,J=8Hz,1H),6.61(d,J=10Hz,1H),6.73(d,J=8Hz,1H),7.58(d,J=10Hz,0.4H),7.59(d,J=10Hz,0.6H),7.65(s,0.6H),7.66(s,0.4H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.14 (m, 2H), 0.44-0.54 (m, 2H), 0.74-0.83 (m) , 1H), 1.08-1.17 (m, 1H), 1.22-1.36 (m, 1H), 1.37-1.63 (m, 3H), 1.66-1.78 (M, 1H), 2.15-2.45 (m, 5H), 2.65 (dd, J = 5,11Hz, 1H), 2.81-2.92 (m, 1H), 2.99 (D, J = 19Hz, 1H), 3.06 (d, J = 6Hz, 1H), 3.54-3.64 (m, 1H), 3.75-3.88 (m, 1H), 3 .88 (s, 3H), 4.50-4.73 (m, 2H), 6.60 (d, J = 8Hz, 1H), 6.61 (d, J = 10Hz, 1H), 6.73 (D, J = 8Hz, 1H), 7.58 (d, J = 10Hz, 0.4H), 7.59 (d, J = 10Hz, 0.6H), 7.65 (s, 0.6H) , 7.66 (s, 0.4H).
(実施例55)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(6-ヒドロキシピリジン-3-イル)メタノン(74)の合成
(Example 55)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (6-hydroxypyridin-3-yl) methanone (74)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.14(m,2H),0.43-0.53(m,2H),0.71-0.90(m,1H),1.07-1.14(m,1H),1.20-1.33(m,1H),1.35-1.61(m,3H),1.63-1.77(m,1H),2.14-2.42(m,5H),2.59-2.67(m,1H),2.77-2.89(m,1H),2.96(d,J=18Hz,1H),3.04(d,J=6Hz,1H),3.51-3.66(m,1H),3.75-3.93(m,1H),4.26-4.32(m,0.3H),4.41-4.60(m,1.7H),6.54(d,J=8Hz,1H),6.62(d,J=10Hz,1H),6.74(d,J=8Hz,1H),7.59(dd,J=2,10Hz,1H),7.81(br s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-0.14 (m, 2H), 0.43-0.53 (m, 2H), 0.71-0.90 (m) , 1H), 1.07-1.14 (m, 1H), 1.20-1.33 (m, 1H), 1.35-1.61 (m, 3H), 1.63-1.77 (M, 1H), 2.14-2.42 (m, 5H), 2.59-2.67 (m, 1H), 2.77-2.89 (m, 1H), 2.96 (d). , J = 18Hz, 1H), 3.04 (d, J = 6Hz, 1H), 3.51-3.66 (m, 1H), 3.75-3.93 (m, 1H), 4.26 -4.32 (m, 0.3H), 4.41-4.60 (m, 1.7H), 6.54 (d, J = 8Hz, 1H), 6.62 (d, J = 10Hz, 1H), 6.74 (d, J = 8Hz, 1H), 7.59 (dd, J = 2,10Hz, 1H), 7.81 (br s, 1H).
(実施例56)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(2-ヒドロキシピリジン-3-イル)メタノン(75)の合成
(Example 56)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (2-hydroxypyridin-3-yl) methanone (75)
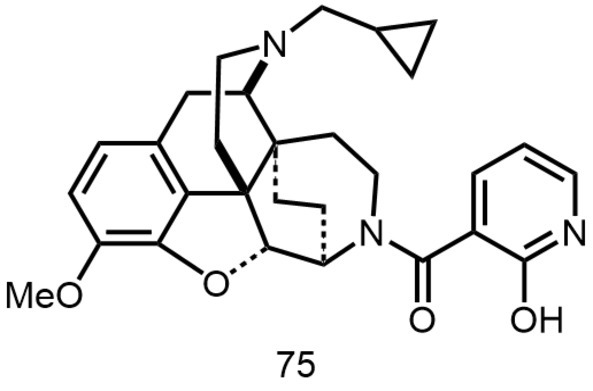
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.16(m,2H),0.41-0.55(m,2H),0.69-0.78(m,1H),1.07-1.16(m,1H),1.23-1.36(m,2H),1.47-1.62(m,2H),1.69-1.80(m,1H),2.15-2.46(m,5H),2.56-2.67(m,1H),2.91-3.14(m,3H),3.43-3.62(m,1.8H),3.79(s,0.6H),3.89(s,2.4H),3.90-3.93(m,0.2H),4.41-4.59(m,0.2H),4.63-4.69(m,1H),4.77(s,0.8H),6.29-6.36(m,1H),6.59(d,J=8Hz,1H),6.69(d,J=8Hz,0.2H),6.73(d,J=8Hz,0.8H),7.51-7.61(m,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.16 (m, 2H), 0.41-0.55 (m, 2H), 0.69-0.78 (m) , 1H), 1.07-1.16 (m, 1H), 1.23-1.36 (m, 2H), 1.47-1.62 (m, 2H), 1.69-1.80 (M, 1H), 2.15-2.46 (m, 5H), 2.56-2.67 (m, 1H), 2.91-3.14 (m, 3H), 3.43-3 .62 (m, 1.8H), 3.79 (s, 0.6H), 3.89 (s, 2.4H), 3.90-3.93 (m, 0.2H), 4.41 -4.59 (m, 0.2H), 4.63-4.69 (m, 1H), 4.77 (s, 0.8H), 6.29-6.36 (m, 1H), 6 .59 (d, J = 8Hz, 1H), 6.69 (d, J = 8Hz, 0.2H), 6.73 (d, J = 8Hz, 0.8H), 7.51-7.61 ( m, 2H).
(実施例57)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(2-ヒドロキシピリジン-3-イル)メタノン(76)の合成
(Example 57)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (2-hydroxypyridin-3-yl) methanone (76)

1H-NMR(400MHz,CD3OD)δ(ppm):0.06-0.16(m,2H),0.44-0.53(m,2H),0.74-0.83(m,1H),1.07-1.15(m,1H),1.25-1.53(m,4H),1.73-1.85(m,1H),2.22-2.43(m,5H),2.61-2.67(m,1H),2.82-2.99(m,1H),2.97(d,J=18Hz,1H),3.06(d,J=6Hz,0.8H),3.13(d,J=6Hz,0.2H),3.48-3.64(m,2H),4.50-4.60(m,2H),6.43-6.49(m,1H),6.51(d,J=8Hz,1H),6.58(d,J=8Hz,0.2H),6.63(d,J=8Hz,0.8H),7.54(d,J=6Hz,1H),7.60-7.73(m,1H).
1 1 H-NMR (400 MHz, CD 3 OD) δ (ppm): 0.06-0.16 (m, 2H), 0.44-0.53 (m, 2H), 0.74-0.83 ( m, 1H), 1.07-1.15 (m, 1H), 1.25-1.53 (m, 4H), 1.73-1.85 (m, 1H), 2.22-2. 43 (m, 5H), 2.61-2.67 (m, 1H), 2.82-2.99 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.06 ( d, J = 6Hz, 0.8H), 3.13 (d, J = 6Hz, 0.2H), 3.48-3.64 (m, 2H), 4.50-4.60 (m, 2H) ), 6.43-6.49 (m, 1H), 6.51 (d, J = 8Hz, 1H), 6.58 (d, J = 8Hz, 0.2H), 6.63 (d, J) = 8Hz, 0.8H), 7.54 (d, J = 6Hz, 1H), 7.60-7.73 (m, 1H).
(実施例58)
6-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボニル)-1-メチルピリジン-2(1H)-オン(77)の合成
(Example 58)
6-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,6,7,8a-octahydro-5H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carbonyl) -1-methylpyridine-2 (1H) -one (77)
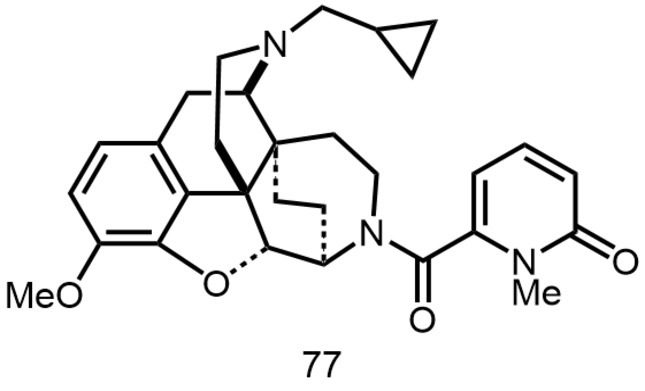
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.17(m,2H),0.43-0.56(m,2H),0.70-0.84(m,1H),1.12-1.80(m,6H),1.98-2.49(m,5H),2.64(dd,J=5,12Hz,1H),2.76-2.88(m,1H),2.97-3.02(m,1H),,3.03(d,J=6Hz,0.4H),3.08(d,J=7Hz,0.6H),3.45-3.60(m,2H),3.47(s,1.8H),3.57(s,1.2H),3.90(s,3H),4.55-4.58(m,0.6H),4.60-4.63(m,0.4H),4.68-4.75(m,1H),6.09(dd,J=1,7Hz,0.4H),6.13(dd,J=1,7Hz,0.6H),6.56-6.65(m,2H),7.26(d,J=8Hz,1H),7.32(dd,J=7,9Hz,0.4H),7.37(dd,J=7,9Hz,0.6H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.17 (m, 2H), 0.43-0.56 (m, 2H), 0.70-0.84 (m) , 1H), 1.12-1.80 (m, 6H), 1.98-2.49 (m, 5H), 2.64 (dd, J = 5,12Hz, 1H), 2.76-2 .88 (m, 1H), 2.97-3.02 (m, 1H), 3.03 (d, J = 6Hz, 0.4H), 3.08 (d, J = 7Hz, 0.6H) ), 3.45-3.60 (m, 2H), 3.47 (s, 1.8H), 3.57 (s, 1.2H), 3.90 (s, 3H), 4.55- 4.58 (m, 0.6H), 4.60-4.63 (m, 0.4H), 4.68-4.75 (m, 1H), 6.09 (dd, J = 1,7Hz) , 0.4H), 6.13 (dd, J = 1,7Hz, 0.6H), 6.56-6.65 (m, 2H), 7.26 (d, J = 8Hz, 1H), 7 .32 (dd, J = 7.9Hz, 0.4H), 7.37 (dd, J = 7.9Hz, 0.6H).
(実施例59)
6-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-カルボニル)-1-メチルピリジン-2(1H)-オン(78)の合成
(Example 59)
6-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,6,7,8,8a-octahydro-5H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-carbonyl) -1-methylpyridine-2 (1H) -one (78)

1H-NMR(400MHz,CDCl3)δ(ppm): 0.05-0.16(m,2H),0.42-0.56(m,2H),0.69-0.81(m,1H),1.11-1.43(m,3H),1.45-1.81(m,3H),2.03-2.44(m,5H),2.60-2.70(m,1H),2.75-2.86(m,1H),2.97(d,J=18Hz,0.4H),2.98(d,J=18Hz,0.6H),3.02(d,J=6Hz,0.4H),3.08(d,J=6Hz,0.6H),3.46-3.61(m,2H),3.48(s,1.8H),3.59(s,1.2H),4.53-4.57(m,0.6H),4.60-4.67(m,1.4H),5.50(br s,1H),6.10(dd,J=1,7Hz,0.4H),6.14(dd,J=1,7Hz,0.6H),6.56(d,J=8Hz,1H),6.63(dd,J=1,9Hz,0.4H),6.63(dd,J=1,9Hz,0.6H),6.75(d,J=8Hz,0.4H),6.75(d,J=8Hz,0.6H),7.34(dd,J=7,9Hz,0.4H),7.38(dd,J=7,9Hz,0.6H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.16 (m, 2H), 0.42-0.56 (m, 2H), 0.69-0.81 (m) , 1H), 1.11-1.43 (m, 3H), 1.45-1.81 (m, 3H), 2.03-2.44 (m, 5H), 2.60-2.70 (M, 1H), 2.75-2.86 (m, 1H), 2.97 (d, J = 18Hz, 0.4H), 2.98 (d, J = 18Hz, 0.6H), 3 .02 (d, J = 6Hz, 0.4H), 3.08 (d, J = 6Hz, 0.6H), 3.46-3.61 (m, 2H), 3.48 (s, 1. 8H), 3.59 (s, 1.2H), 4.53-4.57 (m, 0.6H), 4.60-4.67 (m, 1.4H), 5.50 (br s) , 1H), 6.10 (dd, J = 1,7Hz, 0.4H), 6.14 (dd, J = 1,7Hz, 0.6H), 6.56 (d, J = 8Hz, 1H) , 6.63 (dd, J = 1,9Hz, 0.4H), 6.63 (dd, J = 1,9Hz, 0.6H), 6.75 (d, J = 8Hz, 0.4H), 6.75 (d, J = 8Hz, 0.6H), 7.34 (dd, J = 7, 9Hz, 0.4H), 7.38 (dd, J = 7.9Hz, 0.6H).
(実施例60)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(オキサゾール-2-イル)メタノン(79)の合成
(Example 60)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (oxazole-2-yl) metanone (79)
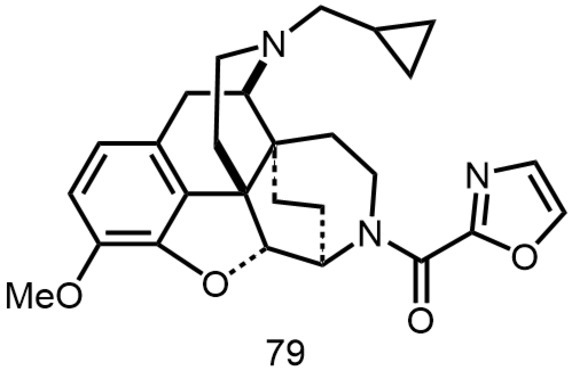
1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.43-0.54(m,2H),0.74-0.85(m,1H),1.09-1.83(m,6H),2.17-2.47(m,5H),2.60-2.67(m,1H),2.89-3.10(m,3H),3.49(ddd,J=4,11,14Hz,0.3H),3.79(ddd,J=4,11,14Hz,0.7H),3.86(s,0.9H),3.89(s,2.1H),4.47-4.55(m,0.3H),4.63-4.67(m,0.7H),4.69-4.77(m,1.3H),4.81-4.84(m,0.7H),6.60(d,J=8Hz,0.7H),6.61(d,J=8Hz,0.3H),6.73(d,J=8Hz,0.3H),6.74(d,J=8Hz,0.7H),7.26-7.30(m,1H),7.75-7.77(m,1H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.43-0.54 (m, 2H), 0.74-0.85 (m) , 1H), 1.09-1.83 (m, 6H), 2.17-2.47 (m, 5H), 2.60-2.67 (m, 1H), 2.89-3.10 (M, 3H), 3.49 (ddd, J = 4,11,14Hz, 0.3H), 3.79 (ddd, J = 4,11,14Hz, 0.7H), 3.86 (s, 0.9H), 3.89 (s, 2.1H), 4.47-4.55 (m, 0.3H), 4.63-4.67 (m, 0.7H), 4.69- 4.77 (m, 1.3H), 4.81-4.84 (m, 0.7H), 6.60 (d, J = 8Hz, 0.7H), 6.61 (d, J = 8Hz) , 0.3H), 6.73 (d, J = 8Hz, 0.3H), 6.74 (d, J = 8Hz, 0.7H), 7.26-7.30 (m, 1H), 7 .75-7.77 (m, 1H).
(実施例61)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(オキサゾール-2-イル)メタノン(80)の合成
(Example 61)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (oxazole-2-yl) metanone (80)
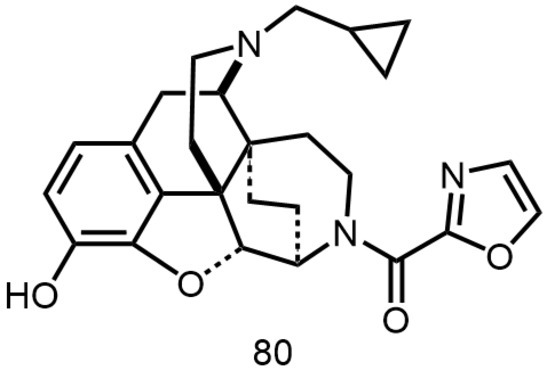
1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.41-0.53(m,2H),0.72-0.85(m,1H),1.07-1.82(m,6H),2.17-2.42(m,5H),2.60-2.68(m,1H),2.90-3.09(m,3H),3.41(ddd,J=4,11,14Hz,0.4H),3.75(ddd,J=4,11,14Hz,0.6H),4.57-4.88(m,3H),6.55(d,J=8Hz,1H),6.71(d,J=8Hz,0.4H),6.73(d,J=8Hz,0.6H),7.27(s,0.6H),7.34(s,0.4H),7.78(s,0.6H),7.80(s,0.4H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.41-0.53 (m, 2H), 0.72-0.85 (m) , 1H), 1.07-1.82 (m, 6H), 2.17-2.42 (m, 5H), 2.60-2.68 (m, 1H), 2.90-3.09 (M, 3H), 3.41 (ddd, J = 4,11,14Hz, 0.4H), 3.75 (ddd, J = 4,11,14Hz, 0.6H), 4.57-4. 88 (m, 3H), 6.55 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 0.4H), 6.73 (d, J = 8Hz, 0.6H), 7.27 (s, 0.6H), 7.34 (s, 0.4H), 7.78 (s, 0.6H), 7.80 (s, 0.4H).
(実施例62)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(オキサゾール-4-イル)メタノン(81)の合成
(Example 62)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (oxazole-4-yl) methanone (81)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.43-0.53(m,2H),0.75-0.85(m,1H),1.07-1.18(m,1H),1.26-1.80(m,5H),2.18-2.44(m,5H),2.60-2.67(m,1H),2.86-3.10(m,3H),3.36-3.46(m,0.4H),3.69-3.78(m,0.6H),3.86(s,1.2H),3.89(s,1.8H),4.46-4.82(m,3H),6.59(d,J=8Hz,0.6H),6.61(d,J=8Hz,0.4H),6.73(d,J=8Hz,0.4H),6.74(d,J=8Hz,0.6H),7.89(s,0.6H),7.91(s,0.4H),8.20(s,1H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.43-0.53 (m, 2H), 0.75-0.85 (m) , 1H), 1.07-1.18 (m, 1H), 1.26-1.80 (m, 5H), 2.18-2.44 (m, 5H), 2.60-2.67 (M, 1H), 2.86-3.10 (m, 3H), 3.36-3.46 (m, 0.4H), 3.69-3.78 (m, 0.6H), 3 .86 (s, 1.2H), 3.89 (s, 1.8H), 4.46-4.82 (m, 3H), 6.59 (d, J = 8Hz, 0.6H), 6 .61 (d, J = 8Hz, 0.4H), 6.73 (d, J = 8Hz, 0.4H), 6.74 (d, J = 8Hz, 0.6H), 7.89 (s, 0.6H), 7.91 (s, 0.4H), 8.20 (s, 1H).
(実施例63)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(オキサゾール-4-イル)メタノン(82)の合成
(Example 63)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (oxazole-4-yl) metanone (82)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.14(m,2H),0.43-0.53(m,2H),0.74-0.84(m,1H),1.06-1.16(m,1H),1.25-1.80(m,5H),2.18-2.41(m,5H),2.60-2.67(m,1H),2.83-3.09(m,3H),3.34(ddd,J=4,11,14Hz,0.4H),3.69(ddd,J=4,11,14Hz,0.6H),4.54-4.83(m,3H),6.54(d,J=8Hz,0.6H),6.55(d,J=8Hz,0.4H),6.71(d,J=8Hz,0.4H),6.72(d,J=8Hz,0.6H),7.90(s,0.6H),7.95(s,0.4H),8.21(s,0.6H),8.27(s,0.4H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.14 (m, 2H), 0.43-0.53 (m, 2H), 0.74-0.84 (m) , 1H), 1.06-1.16 (m, 1H), 1.25-1.80 (m, 5H), 2.18-2.41 (m, 5H), 2.60-2.67 (M, 1H), 2.83-3.09 (m, 3H), 3.34 (ddd, J = 4,11,14Hz, 0.4H), 3.69 (ddd, J = 4,11, 14Hz, 0.6H), 4.54-4.83 (m, 3H), 6.54 (d, J = 8Hz, 0.6H), 6.55 (d, J = 8Hz, 0.4H), 6.71 (d, J = 8Hz, 0.4H), 6.72 (d, J = 8Hz, 0.6H), 7.90 (s, 0.6H), 7.95 (s, 0.4H) ), 8.21 (s, 0.6H), 8.27 (s, 0.4H).
(実施例64)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(イソキサゾール-5-イル)メタノン(83)の合成
(Example 64)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (isoxazole-5-yl) metanone (83)
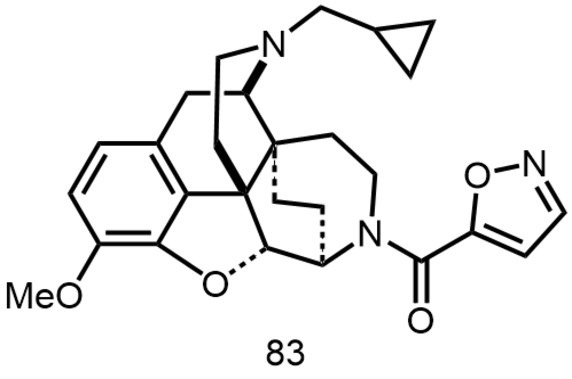
1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.44-0.54(m,2H),0.74-0.85(m,1H),1.10-1.81(m,6H),2.17-2.46(m,5H),2.61-2.68(m,1H),2.93-3.11(m,3H),3.63(ddd,J=4,11,14Hz,1H),3.86-3.96(m,3.7H),4.18-4.21(m,0.3H),4.26-4.34(m,0.3H),4.60-4.63(m,0.7H),4.67-4.69(m,0.3H),4.71-4.74(m,0.7H),6.61(d,J=8Hz,0.7H),6.62(d,J=8Hz,0.3H),6.72-6.77(m,2H),8.31(d,J=2Hz,0.3H),8.32(d,J=2Hz,0.7H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.44-0.54 (m, 2H), 0.74-0.85 (m) , 1H), 1.10-1.81 (m, 6H), 2.17-2.46 (m, 5H), 2.61-2.68 (m, 1H), 2.93-3.11 (M, 3H), 3.63 (ddd, J = 4,11,14Hz, 1H), 3.86-3.96 (m, 3.7H), 4.18-4.21 (m, 0. 3H), 4.26-4.34 (m, 0.3H), 4.60-4.63 (m, 0.7H), 4.67-4.69 (m, 0.3H), 4. 71-4.74 (m, 0.7H), 6.61 (d, J = 8Hz, 0.7H), 6.62 (d, J = 8Hz, 0.3H), 6.72-6.77 (M, 2H), 8.31 (d, J = 2Hz, 0.3H), 8.32 (d, J = 2Hz, 0.7H).
(実施例65)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(イソキサゾール-5-イル)メタノン(84)の合成
(Example 65)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (isoxazole-5-yl) metanone (84)

実施例7に記載した方法に従い、化合物83より表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.44-0.54(m,2H),0.73-0.83(m,1H),1.09-1.81(m,6H),2.18-2.42(m,5H),2.62-2.67(m,1H),2.89-3.10(m,3H),3.44-3.67(m,1H),3.91-3.98(m,0.7H),4.18-4.21(m,0.3H),4.41-4.49(m,0.3H),4.59-4.61(m,0.7H),4.64-4.67(m,0.7H),4.71-4.73(m,0.3H),6.56(d,J=8Hz,1H),6.70-6.76(m,2H),8.33(d,J=2Hz,0.7H),8.35(d,J=2Hz,0.3H).
The title compound was obtained from compound 83 according to the method described in Example 7.
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.44-0.54 (m, 2H), 0.73-0.83 (m) , 1H), 1.09-1.81 (m, 6H), 2.18-2.42 (m, 5H), 2.62-2.67 (m, 1H), 2.89-3.10 (M, 3H), 3.44-3.67 (m, 1H), 3.91-3.98 (m, 0.7H), 4.18-4.21 (m, 0.3H), 4 .41-4.49 (m, 0.3H), 4.59-4.61 (m, 0.7H), 4.64-4.67 (m, 0.7H), 4.71-4. 73 (m, 0.3H), 6.56 (d, J = 8Hz, 1H), 6.70-6.76 (m, 2H), 8.33 (d, J = 2Hz, 0.7H), 8.35 (d, J = 2Hz, 0.3H).
(実施例66)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(チアゾール-2-イル)メタノン(85)の合成
(Example 66)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano -4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (thiazole-2-yl) synthesis of methanone (85)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.42-0.56(m,2H),0.72-0.97(m,1H),1.07-1.90(m,6H),2.14-2.49(m,5H),2.59-2.68(m,1H),2.84-3.12(m,3H),3.33-3.90(m,1H),4.55-5.01(m,3H),5.21-5.36(m,1H),6.55(d,J=8Hz,1H),6.69-6.76(m,1H),7.50-7.60(m,1H),7.91(d,J=3Hz,0.6H),7.99(d,J=3Hz,0.4H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.42-0.56 (m, 2H), 0.72-0.97 (m) , 1H), 1.07-1.90 (m, 6H), 2.14-2.49 (m, 5H), 2.59-2.68 (m, 1H), 2.84-3.12 (M, 3H), 3.33-3.90 (m, 1H), 4.55-5.01 (m, 3H), 5.21-5.36 (m, 1H), 6.55 (d) , J = 8Hz, 1H), 6.69-6.76 (m, 1H), 7.50-7.60 (m, 1H), 7.91 (d, J = 3Hz, 0.6H), 7 .99 (d, J = 3Hz, 0.4H).
(参考例20)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-7-(チアゾール-4-カルボニル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-イル チアゾール-4-カルボキシレート(86)の合成
(Reference example 20)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -7- (Thiazole-4-carbonyl) -1,2,3,4,6,7,8,8a-Octahydro-5H- Synthesis of 4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-10-ylthiazole-4-carboxylate (86)
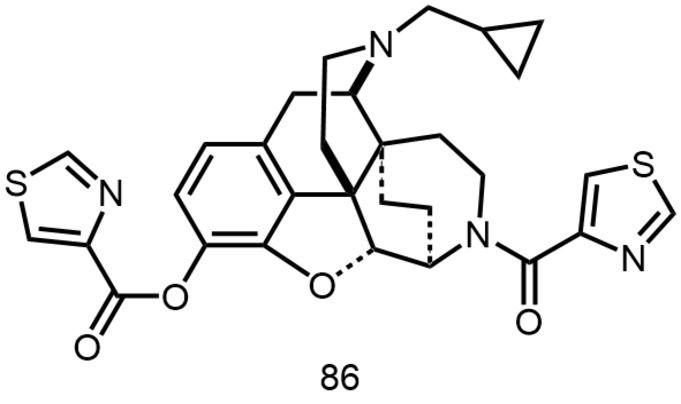
1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.42-0.55(m,2H),0.72-0.93(m,1H),1.05-1.86(m,6H),2.20-2.51(m,5H),2.62-2.71(m,1H),2.86-3.16(m,3H),3.40-3.72(m,1H),4.07-4.98(m,3H),6.68(d,J=8Hz,1H),6.91-7.03(m,1H),7.87-8.00(m,1H),8.41-8.50(m,1H),8.74-8.84(m,1H),8.89-8.95(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.42-0.55 (m, 2H), 0.72-0.93 (m) , 1H), 1.05-1.86 (m, 6H), 2.20-2.51 (m, 5H), 2.62-2.71 (m, 1H), 2.86-3.16 (M, 3H), 3.40-3.72 (m, 1H), 4.07-4.98 (m, 3H), 6.68 (d, J = 8Hz, 1H), 6.91-7 .03 (m, 1H), 7.87-8.00 (m, 1H), 8.41-8.50 (m, 1H), 8.74-8.84 (m, 1H), 8.89 -8.95 (m, 1H).
(実施例67)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(チアゾール-4-イル)メタノン(87)の合成
(Example 67)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (thiazole-4-yl) metanone (87)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.42-0.54(m,2H),0.71-0.90(m,1H),1.03-1.89(m,6H),2.17-2.41(m,5H),2.59-2.68(m,1H),2.86-3.11(m,3H),3.31-3.81(m,1H),4.18-4.29(m,0.7H),4.45-4.96(m,3.3H),6.55(d,J=8Hz,1H),6.66-6.75(m,1H),7.93(d,J=2Hz,0.7H),8.04(d,J=2Hz,0.3H),8.82(d,J=2Hz,0.7H),8.87(d,J=2Hz,0.3H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.42-0.54 (m, 2H), 0.71-0.90 (m) , 1H), 1.03-1.89 (m, 6H), 2.17-2.41 (m, 5H), 2.59-2.68 (m, 1H), 2.86-3.11 (M, 3H), 3.31-3.81 (m, 1H), 4.18-4.29 (m, 0.7H), 4.45-4.96 (m, 3.3H), 6 .55 (d, J = 8Hz, 1H), 6.66-6.75 (m, 1H), 7.93 (d, J = 2Hz, 0.7H), 8.04 (d, J = 2Hz, 0.3H), 8.82 (d, J = 2Hz, 0.7H), 8.87 (d, J = 2Hz, 0.3H).
(実施例68)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(チアゾール-5-イル)メタノン(88)の合成
(Example 68)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (thiazole-5-yl) methanone (88)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.18(m,2H),0.43-0.54(m,2H),0.71-0.92(m,1H),1.08-1.85(m,6H),2.17-2.43(m,5H),2.60-2.69(m,1H),2.82-3.14(m,3H),3.45-3.82(m,1H),3.90-4.16(m,1H),4.55-4.96(m,3H),6.56(d,J=8Hz,1H),6.73(d,J=8Hz,1H),8.06-8.26(m,1H),8.90(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.18 (m, 2H), 0.43-0.54 (m, 2H), 0.71-0.92 (m) , 1H), 1.08-1.85 (m, 6H), 2.17-2.43 (m, 5H), 2.60-2.69 (m, 1H), 2.82-3.14 (M, 3H), 3.45-3.82 (m, 1H), 3.90-4.16 (m, 1H), 4.55-4.96 (m, 3H), 6.56 (d) , J = 8Hz, 1H), 6.73 (d, J = 8Hz, 1H), 8.06-8.26 (m, 1H), 8.90 (s, 1H).
(実施例69)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(イソチアゾール-4-イル)メタノン(89)の合成
(Example 69)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano -4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (isothiazole-4-yl) synthesis of methanone (89)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.42-0.55(m,2H),0.69-0.91(m,1H),1.06-1.86(m,6H),2.15-2.45(m,5H),2.62-2.71(m,1H),2.79-3.12(m,3H),3.33-3.67(m,1H),3.72-4.06(m,1H),4.38-5.00(m,3H),6.56(d,J=8Hz,1H),6.66-6.77(m,1H),8.60-8.73(m,1H),8.80-8.93(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.42-0.55 (m, 2H), 0.69-0.91 (m) , 1H), 1.06-1.86 (m, 6H), 2.15-2.45 (m, 5H), 2.62-2.71 (m, 1H), 2.79-3.12 (M, 3H), 3.33-3.67 (m, 1H), 3.72-4.06 (m, 1H), 4.38-5.00 (m, 3H), 6.56 (d) , J = 8Hz, 1H), 6.66-6.77 (m, 1H), 8.60-8.73 (m, 1H), 8.80-8.93 (m, 1H).
(実施例70)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(イソチアゾール-5-イル)メタノン(90)の合成
(Example 70)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (isothiazole-5-yl) methanone (90)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.15(m,2H),0.41-0.56(m,2H),0.70-0.90(m,1H),
1.07-1.89(m,6H),2.12-2.48(m,5H),2.60-2.70(m,1H),2.79-3.19(m,3H),3.45-3.68(m,1H),3.76-4.85(m,6H),6.57-6.66(m,1H),6.69-6.78(m,1H),7.32-7.51(m,1H),8.44-8.52(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-0.15 (m, 2H), 0.41-0.56 (m, 2H), 0.70-0.90 (m) , 1H),
1.07-1.89 (m, 6H), 2.12-2.48 (m, 5H), 2.60-2.70 (m, 1H), 2.79-3.19 (m, 3H) ), 3.45-3.68 (m, 1H), 3.76-4.85 (m, 6H), 6.57-6.66 (m, 1H), 6.69-6.78 (m). , 1H), 7.32-7.51 (m, 1H), 8.44-8.52 (m, 1H).
(実施例71)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(イソチアゾール-5-イル)メタノン(91)の合成
(Example 71)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (isothiazole-5-yl) methanone (91)
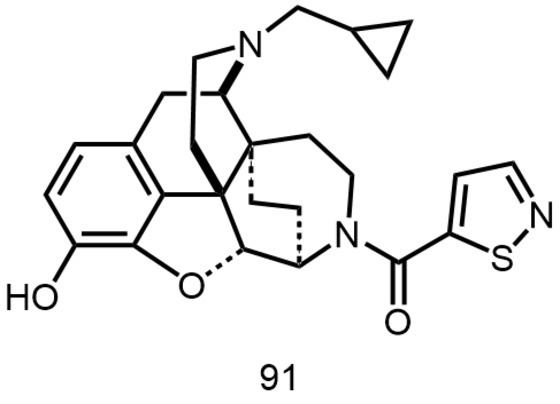
1H-NMR(400MHz,CDCl3)δ(ppm):0.04-1.78(m,2H),0.42-0.55(m,2H),0.70-0.93(m,1H),1.07-1.82(m,6H),2.14-2.44(m,5H),2.60-2.71(m,1H),2.77-3.14(m,3H),3.41-3.93(m,2H),4.40-4.92(m,3H),6.56(d,J=8Hz,1H),6.67-6.77(m,1H),7.35-7.55(m,1H),8.46-8.53(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-1.78 (m, 2H), 0.42-0.55 (m, 2H), 0.70-0.93 (m) , 1H), 1.07-1.82 (m, 6H), 2.14-2.44 (m, 5H), 2.60-2.71 (m, 1H), 2.77-3.14 (M, 3H), 3.41-3.93 (m, 2H), 4.40-4.92 (m, 3H), 6.56 (d, J = 8Hz, 1H), 6.67-6 .77 (m, 1H), 7.35-7.55 (m, 1H), 8.46-8.53 (m, 1H).
(実施例72)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(1H-インドール-2-イル)メタノン(92)の合成
(Example 72)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano -4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (1H-indole-2-yl) Synthesis of methanone (92)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.18(m,2H),0.44-0.57(m,2H),0.75-0.92(m,1H),1.07-1.89(m,6H),2.12-2.49(m,5H),2.58-2.70(m,1H),2.82-3.16(m,3H),3.32-3.52(m,0.4H),3.75-3.99(m,3.6H),4.52-4.87(m,3H),6.55-6.68(m,1H),6.71-6.81(m,1H),6.84-7.33(m,3H),7.42(d,J=7Hz,1H),7.60-7.83(m,1H),9.01-9.42(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.04-0.18 (m, 2H), 0.44-0.57 (m, 2H), 0.75-0.92 (m) , 1H), 1.07-1.89 (m, 6H), 2.12-2.49 (m, 5H), 2.58-2.70 (m, 1H), 2.82-3.16 (M, 3H), 3.32-3.52 (m, 0.4H), 3.75-3.99 (m, 3.6H), 4.52-4.87 (m, 3H), 6 .55-6.68 (m, 1H), 6.71-6.81 (m, 1H), 6.84-7.33 (m, 3H), 7.42 (d, J = 7Hz, 1H) , 7.60-7.83 (m, 1H), 9.01-9.42 (m, 1H).
(実施例73)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(1H-インドール-2-イル)メタノン(93)の合成
(Example 73)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano -4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (1H-indole-2-yl) Synthesis of methanone (93)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.18(m,2H),0.43-0.56(m,2H),0.75-0.90(m,1H),1.09-1.86(m,6H),2.17-2.47(m,5H),2.59-2.71(m,1H),2.83-3.15(m,3H),3.23-3.89(m,1H),4.50-4.94(m,4H),6.54-6.62(m,1H),6.74(d,J=8Hz,1H),6.84-7.35(m,3H),7.40-7.47(m,1H),7.62-7.83(m,1H),8.95-9.50(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.18 (m, 2H), 0.43-0.56 (m, 2H), 0.75-0.90 (m) , 1H), 1.09-1.86 (m, 6H), 2.17-2.47 (m, 5H), 2.59-2.71 (m, 1H), 2.83-3.15 (M, 3H), 3.23-3.89 (m, 1H), 4.50-4.94 (m, 4H), 6.54-6.62 (m, 1H), 6.74 (d). , J = 8Hz, 1H), 6.84-7.35 (m, 3H), 7.40-7.47 (m, 1H), 7.62-7.83 (m, 1H), 8.95 -9.50 (m, 1H).
(実施例74)
(5-クロロピリミジン-2-イル)((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)メタノン(94)の合成
(Example 74)
(5-Chloropyrimidine-2-yl) ((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a -Synthesis of octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) metanone (94)

1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.16(m,2H),0.41-0.56(m,2H),0.70-0.93(m,1H),1.04-1.90(m,5H),2.11-2.45(m,6H),2.59-2.69(m,1H),2.76-3.12(m,3H),3.34-3.56(m,1.6H),3.68-3.75(m,0.4H),3.81(s,1.2H),3.90(s,1.8H),4.49-4.60(m,0.4H),4.68-4.87(m,1.6H),6.57-6.62(m,1H),6.70(d,J=8Hz,0.4H).6.75(d,J=8Hz,0.6H),8.74-8.82(m,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.03-0.16 (m, 2H), 0.41-0.56 (m, 2H), 0.70-0.93 (m) , 1H), 1.04-1.90 (m, 5H), 2.11-2.45 (m, 6H), 2.59-2.69 (m, 1H), 2.76-3.12 (M, 3H), 3.34-3.56 (m, 1.6H), 3.68-3.75 (m, 0.4H), 3.81 (s, 1.2H), 3.90 (S, 1.8H), 4.49-4.60 (m, 0.4H), 4.68-4.87 (m, 1.6H), 6.57-6.62 (m, 1H) , 6.70 (d, J = 8Hz, 0.4H). 6.75 (d, J = 8Hz, 0.6H), 8.74-8.82 (m, 2H).
(実施例75)
(5-クロロピリミジン-2-イル)((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)メタノン(95)の合成
(Example 75)
(5-Chloropyrimidine-2-yl) ((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a -Synthesis of octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) methanone (95)
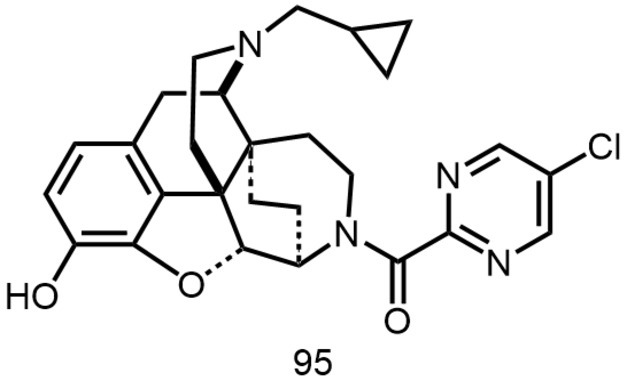
1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.16(m,2H),0.40-0.55(m,2H),0.65-0.96(m,1H),1.03-1.91(m,5H),2.07-2.44(m,6H),2.58-2.69(m,1H),2.84-3.11(m,3H),3.36-3.66(m,2H),4.46-5.00(m,3H),6.50-6.58(m,1H),6.65(d,J=8Hz,0.3H),6.73(d,J=8Hz,0.7H),8.77-8.12(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.03-0.16 (m, 2H), 0.40-0.55 (m, 2H), 0.65-0.96 (m) , 1H), 1.03-1.91 (m, 5H), 2.07-2.44 (m, 6H), 2.58-2.69 (m, 1H), 2.84-3.11 (M, 3H), 3.36-3.66 (m, 2H), 4.46-5.00 (m, 3H), 6.50-6.58 (m, 1H), 6.65 (d) , J = 8Hz, 0.3H), 6.73 (d, J = 8Hz, 0.7H), 8.77-8.12 (m, 1H).
(実施例76)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(5-フルオロピリミジン-2-イル)メタノン(96)の合成
(Example 76)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (5-fluoropyrimidine-2-yl) methanone (96)

1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.18(m,2H),0.41-0.57(m,2H),0.69-0.92(m,1H),1.05-1.92(m,6H),2.13-2.47(m,5H),2.85-3.14(m,3H),2.58-2.69(m,1H),3.35-3.59(m,1.6H),3.67-3.73(m,0.4H),3.80(s,1.2H),3.90(s,1.8H),4.49-4.60(m,0.4H),4.69-4.86(m,1.6H),6.57-6.63(m,1H),6.70(d,J=8Hz,0.4H),6.75(d,J=8Hz,0.6H),8.64-8.71(m,2H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.03-0.18 (m, 2H), 0.41-0.57 (m, 2H), 0.69-0.92 (m) , 1H), 1.05-1.92 (m, 6H), 2.13-2.47 (m, 5H), 2.85-3.14 (m, 3H), 2.58-2.69 (M, 1H), 3.35-3.59 (m, 1.6H), 3.67-3.73 (m, 0.4H), 3.80 (s, 1.2H), 3.90 (S, 1.8H), 4.49-4.60 (m, 0.4H), 4.69-4.86 (m, 1.6H), 6.57-6.63 (m, 1H) , 6.70 (d, J = 8Hz, 0.4H), 6.75 (d, J = 8Hz, 0.6H), 8.64-8.71 (m, 2H).
(実施例77)
((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)(5-フルオロピリミジン-2-イル)メタノン(97)の合成
(Example 77)
((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano Synthesis of -4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) (5-fluoropyrimidine-2-yl) metanone (97)
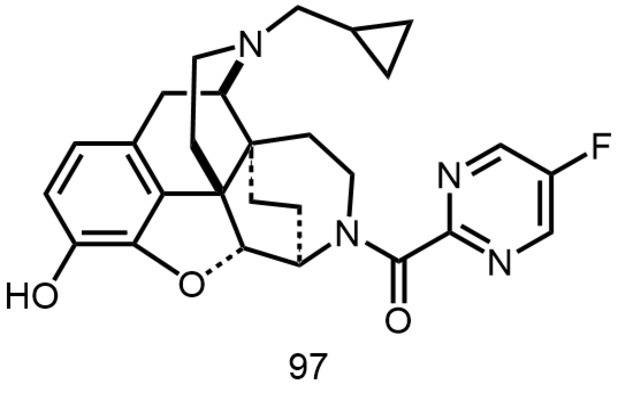
1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.41-0.56(m,2H),0.68-0.94(m,1H),1.03-1.87(m,6H),2.13-2.44(m,5H),2.58-2.71(m,1H),2.84-3.12(m,3H),3.34-3.69(m,2H),4.38-4.92(m,3H),6.50-6.58(m,1H),6.65(d,J=8Hz,0.3H),6.73(d,J=8Hz,0.7H),8.66-8.73(m,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.41-0.56 (m, 2H), 0.68-0.94 (m) , 1H), 1.03-1.87 (m, 6H), 2.13-2.44 (m, 5H), 2.58-2.71 (m, 1H), 2.84-3.12 (M, 3H), 3.34-3.69 (m, 2H), 4.38-4.92 (m, 3H), 6.50-6.58 (m, 1H), 6.65 (d) , J = 8Hz, 0.3H), 6.73 (d, J = 8Hz, 0.7H), 8.66-8.73 (m, 2H).
(実施例78)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(ピリミジン-2-イル)エタン-1-オン(98)の合成
(Example 78)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (pyrimidine-2-yl) ethane-1-one (98)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.13(m,2H),0.43-0.51(m,2H),0.70-0.81(m,1H),1.03-1.12(m,1H),1.17-1.33(m,1H),1.34-1.54(m,3H),1.60-1.75(m,1H),2.11-2.42(m,5H),2.55-2.72(m,1.7H),2.79-2.89(m,0.3H),2.90-3.05(m,2H),3.37-3.55(m,1H),3.74-3.83(m,0.7H),4.17-4.31(m,2H),4.33-4.42(m,0.3H)4.55-4.61(m,2H),6.50(d,J=8Hz,0.7H),6.52(d,J=8Hz,0.3H),6.69(d,J=8Hz,0.3H),6.70(d,J=8Hz,0.7H),7.19-7.23(m,1H),8.72(d,J=8Hz,0.6H),8.74(d,J=8Hz,1.4H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.13 (m, 2H), 0.43-0.51 (m, 2H), 0.70-0.81 (m) , 1H), 1.03-1.12 (m, 1H), 1.17-1.33 (m, 1H), 1.34-1.54 (m, 3H), 1.60-1.75 (M, 1H), 2.11-2.42 (m, 5H), 2.55-2.72 (m, 1.7H), 2.79-2.89 (m, 0.3H), 2 .90-3.05 (m, 2H), 3.37-1.55 (m, 1H), 3.74-3.83 (m, 0.7H), 4.17-4.31 (m, 2H), 4.33-4.42 (m, 0.3H) 4.55-4.61 (m, 2H), 6.50 (d, J = 8Hz, 0.7H), 6.52 (d) , J = 8Hz, 0.3H), 6.69 (d, J = 8Hz, 0.3H), 6.70 (d, J = 8Hz, 0.7H), 7.19-7.23 (m, 1H), 8.72 (d, J = 8Hz, 0.6H), 8.74 (d, J = 8Hz, 1.4H).
(実施例79)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(ピリミジン-5-イル)エタン-1-オン(99)の合成
(Example 79)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (pyrimidine-5-yl) ethane-1-one (99)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.16(m,2H),0.44-0.55(m,2H),0.72-0.85(m,1H),1.06-1.18(m,1H),1.19-1.88(m,5H),1.96-2.13(m,1H),2.18-2.47(m,4H),2.58-2.70(m,1H),2.74-2.90(m,1H),2.94-3.10(m,2H),3.37(ddd,J=5,11,15Hz,0.5H),3.56(ddd,J=4,11,15Hz,0.5H),3.71-3.82(m,0.5H),3.76(s,2H),3.87(s,1.5H),3.89(s,1.5H),4.07-4.12(m,0.5H),4.41(ddd,J=5,5,14Hz,0.5H),4.50(s,1H),4.60-4.65(m,0.5H),6.58(d,J=8Hz,0.5H),6.65(d,J=8Hz,0.5H),6.73(d,J=8Hz,0.5H),6.76(d,J=8Hz,0.5H),8.68(s,2H),9.13(s,0.5H),9.14(s,0.5H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.16 (m, 2H), 0.44-0.55 (m, 2H), 0.72-0.85 (m) , 1H), 1.06-1.18 (m, 1H), 1.19-1.88 (m, 5H), 1.96-2.13 (m, 1H), 2.18-2.47 (M, 4H), 2.58-2.70 (m, 1H), 2.74-2.90 (m, 1H), 2.94-3.10 (m, 2H), 3.37 (ddd) , J = 5,11,15Hz, 0.5H), 3.56 (ddd, J = 4,11,15Hz, 0.5H), 3.71-3.82 (m, 0.5H), 3. 76 (s, 2H), 3.87 (s, 1.5H), 3.89 (s, 1.5H), 4.07-4.12 (m, 0.5H), 4.41 (ddd, J = 5,5,14Hz, 0.5H), 4.50 (s, 1H), 4.60-4.65 (m, 0.5H), 6.58 (d, J = 8Hz, 0.5H) ), 6.65 (d, J = 8Hz, 0.5H), 6.73 (d, J = 8Hz, 0.5H), 6.76 (d, J = 8Hz, 0.5H), 8.68. (S, 2H), 9.13 (s, 0.5H), 9.14 (s, 0.5H).
(実施例80)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(ピリミジン-5-イル)エタン-1-オン(100)の合成
(Example 80)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (pyrimidine-5-yl) ethane-1-one (100)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.16(m,2H),0.43-0.55(m,2H),0.71-0.83(m,1H),1.06-1.19(m,1H),1.20-1.77(m,5H),2.09(ddd,J=6,12,12Hz,1H),2.16-2.45(m,4H),2.58-2.69(m,1H),2.72-2.91(m,1H),2.96(d,J=19Hz,0.7H),2.98(d,J=18Hz,0.3H),3.03(d,J=6Hz,0.7H),3.06(d,J=6Hz,0.3H),3.45(ddd,J=5,11,15Hz,0.3H),3.58(ddd,J=4,12,15Hz,0.7H),3.72-3.86(m,0.7H),3.77(s,1.4H),3.78(s,0.6H),4.04-4.08(m,0.3H),4.32(ddd,J=5,5,14Hz,0.3H),4.47(s,0.7H),4.49(s,0.3H),4.51-4.57(m,0.7H),5.10(br s,0.7H),5.29(br s,0.3H),6.54(d,J=8Hz,0.7H),6.58(d,J=8Hz,0.3H),6.72(d,J=8Hz,0.7H),6.73(d,J=8Hz,0.3H),8.69(s,1.4H),8.70(s,0.6H),9.13(s,0.3H),9.15(s,0.7H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.16 (m, 2H), 0.43-0.55 (m, 2H), 0.71-0.83 (m) , 1H), 1.06-1.19 (m, 1H), 1.20-1.77 (m, 5H), 2.09 (ddd, J = 6,12,12Hz, 1H), 2.16 -2.45 (m, 4H), 2.58-2.69 (m, 1H), 2.72-2.91 (m, 1H), 2.96 (d, J = 19Hz, 0.7H) , 2.98 (d, J = 18Hz, 0.3H), 3.03 (d, J = 6Hz, 0.7H), 3.06 (d, J = 6Hz, 0.3H), 3.45 ( ddd, J = 5,11,15Hz, 0.3H), 3.58 (ddd, J = 4,12,15Hz, 0.7H), 3.72-3.86 (m, 0.7H), 3 .77 (s, 1.4H), 3.78 (s, 0.6H), 4.04-4.08 (m, 0.3H), 4.32 (ddd, J = 5,5,14Hz, 0.3H), 4.47 (s, 0.7H), 4.49 (s, 0.3H), 4.51-4.57 (m, 0.7H), 5.10 (br s, 0) .7H), 5.29 (br s, 0.3H), 6.54 (d, J = 8Hz, 0.7H), 6.58 (d, J = 8Hz, 0.3H), 6.72 ( d, J = 8Hz, 0.7H), 6.73 (d, J = 8Hz, 0.3H), 8.69 (s, 1.4H), 8.70 (s, 0.6H), 9. 13 (s, 0.3H), 9.15 (s, 0.7H).
(実施例81)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(ピラジン-2-イル)エタン-1-オン(101)の合成
(Example 81)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (pyrazine-2-yl) ethane-1-one (101)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.14(m,2H),0.44-0.52(m,2H),0.73-0.80(m,1H),1.03-1.15(m,1H),1.19-1.29(m,1H),1.30-1.56(m,3H),1.60-1.75(m,1H),2.08(ddd,J=6,13,13Hz,1H),2.20-2.43(m,4H),2.57-2.75(m,1.7H),2.81-2.90(m,0.3H),2.94(d,J=18Hz,0.7H),2.97(d,J=18Hz,0.3H),2.99(d,J=6Hz,0.7H),3.05(d,J=6Hz,0.3H),3.43(ddd,J=4,10,14Hz,0.3H),3.55(ddd,J=4,12,15Hz,0.7H),3.86-3.93(m,0.7H),4.01(d,J=15Hz,1H),4.06(d,J=15Hz,1H),4.20-4.23(m,0.3H),4.32(ddd,J=5,5,15Hz,0.3H),4.48-4.52(m,0.7H),4.53-4.57(m,0.7H),4.58-4.61(m,0.3H),6.52(d,J=8Hz,0.7H),6.56(d,J=8Hz,0.3H),6.70(d,J=8Hz,0.7H),6.71(d,J=8Hz,0.3H),8.46-8.52(m,1.3H),8.53-8.55(m,0.7H),8.65-8.67(m,1H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.14 (m, 2H), 0.44-0.52 (m, 2H), 0.73-0.80 (m) , 1H), 1.03-1.15 (m, 1H), 1.19-1.29 (m, 1H), 1.30-1.56 (m, 3H), 1.60-1.75 (M, 1H), 2.08 (ddd, J = 6,13,13Hz, 1H), 2.20-2.43 (m, 4H), 2.57-2.75 (m, 1.7H) , 2.81-2.90 (m, 0.3H), 2.94 (d, J = 18Hz, 0.7H), 2.97 (d, J = 18Hz, 0.3H), 2.99 ( d, J = 6Hz, 0.7H), 3.05 (d, J = 6Hz, 0.3H), 3.43 (ddd, J = 4,10,14Hz, 0.3H), 3.55 (dddd) , J = 4,12,15Hz, 0.7H), 3.86-3.93 (m, 0.7H), 4.01 (d, J = 15Hz, 1H), 4.06 (d, J = 15Hz, 1H), 4.20-4.23 (m, 0.3H), 4.32 (ddd, J = 5,5,15Hz, 0.3H), 4.48-4.52 (m, 0) .7H), 4.53-4.57 (m, 0.7H), 4.58-4.61 (m, 0.3H), 6.52 (d, J = 8Hz, 0.7H), 6 .56 (d, J = 8Hz, 0.3H), 6.70 (d, J = 8Hz, 0.7H), 6.71 (d, J = 8Hz, 0.3H), 8.46-8. 52 (m, 1.3H), 8.53-8.55 (m, 0.7H), 8.65-8.67 (m, 1H).
(実施例82)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(1H-イミダゾール-2-イル)エタン-1-オン(102)の合成
(Example 82)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (1H-imidazol-2-yl) ethane-1-one (102) Synthetic

実施例60に記載した方法に従い、化合物9及び2-(1H-イミダゾール-2-イル)酢酸塩酸塩より表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.15(m,2H),0.45-0.65(m,2H),0.70-0.85(m,1H),1.00-1.15(m,1H),1.20-2.00(m,6H),2.00-2.10(m,0.6H),2.10-2.45(m,4.4H),2.55-2.67(m,1H),2.67-2.88(m,1H),2.90-3.08(m,2H),3.30-3.40(m,0.4H),3.50-3.60(m,0.6H),3.70-4.00(m,5.6H),4.10-4.20(s,0.4H),4.30-4.43(m,0.4H),4.48(s,1H),4.60-4.65(s,0.6H),6.57(d,J=8Hz,0.6H),6.62(d,J=8Hz,0.4H),6.70-6.76(m,1H),6.93(s,0.4H),6.96(s,0.6H),7.58(s,0.4H),7.59(s,0.6H).
The title compound was obtained from compounds 9 and 2- (1H-imidazol-2-yl) acetate according to the method described in Example 60.
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.15 (m, 2H), 0.45-0.65 (m, 2H), 0.70-0.85 (m) , 1H), 1.00-1.15 (m, 1H), 1.20-2.00 (m, 6H), 2.00-2.10 (m, 0.6H), 2.10-2 .45 (m, 4.4H), 2.55-2.67 (m, 1H), 2.67-2.88 (m, 1H), 2.90-3.08 (m, 2H), 3 .30-3.40 (m, 0.4H), 3.50-3.60 (m, 0.6H), 3.70-4.00 (m, 5.6H), 4.10-4. 20 (s, 0.4H), 4.30-4.43 (m, 0.4H), 4.48 (s, 1H), 4.60-4.65 (s, 0.6H), 6. 57 (d, J = 8Hz, 0.6H), 6.62 (d, J = 8Hz, 0.4H), 6.70-6.76 (m, 1H), 6.93 (s, 0.4H) ), 6.96 (s, 0.6H), 7.58 (s, 0.4H), 7.59 (s, 0.6H).
(実施例83)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(1H-イミダゾール-2-イル)エタン-1-オン二塩酸塩(103)の合成
(Example 83)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (1H-imidazol-2-yl) ethane-1-one dihydrochloride ( 103) Synthesis

1H-NMR(400MHz,CD3OD)δ(ppm):0.46-0.56(m,1H),0.58-0.68(m,1H),0.68-0.80(m,1H),0.80-1.00(m,2H),1.06-1.74(m,6H),1.74-1.96(m,1H),2.45-2.65(m,1H),2.75-2.85(m,0.7H),2.95-3.20(m,3H),3.20-3.50(m,3.3H),3.65-3.80(m,0.7H),4.00-4.20(m,3.6H),4.45-4.55(m,1.7H),6.50-6.80(m,2H),7.42(s,1H),8.79(s,1H).
1 H-NMR (400 MHz, CD 3 OD) δ (ppm): 0.46-0.56 (m, 1H), 0.58-0.68 (m, 1H), 0.68-0.80 ( m, 1H), 0.80-1.00 (m, 2H), 1.06-1.74 (m, 6H), 1.74-1.96 (m, 1H), 2.45-2. 65 (m, 1H), 2.75-2.85 (m, 0.7H), 2.95-3.20 (m, 3H), 3.20-3.50 (m, 3.3H), 3.65-3.80 (m, 0.7H), 4.00-4.20 (m, 3.6H), 4.45-4.55 (m, 1.7H), 6.50-6 .80 (m, 2H), 7.42 (s, 1H), 8.79 (s, 1H).
(実施例84)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(イソキサゾール-3-イル)エタン-1-オン(104)の合成
(Example 84)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (isoxazole-3-yl) ethane-1-one (104)
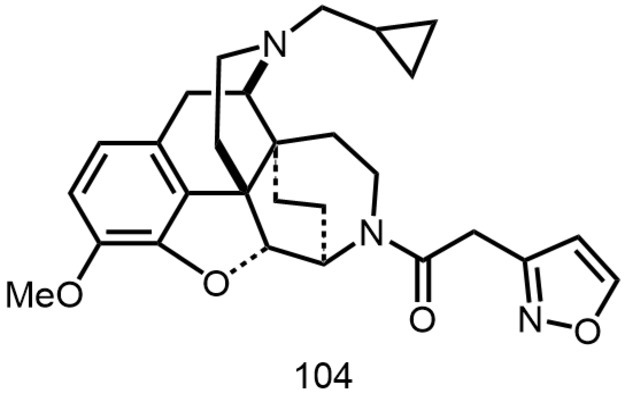
1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.16(m,2H),0.43-0.54(m,2H),0.72-0.85(m,1H),1.03-1.16(m,1H),1.18-1.82(m,5H),1.98-2.13(m,1H),2.17-2.46(m,4H),2.55-2.68(m,1H),2.69-2.79(m,0.6H),2.80-2.90(m,0.4H),2.92-3.03(m,1.6H),3.06(d,J=6Hz,0.4H),3.35-3.45(m,0.4H),3.50-3.60(m,0.6H),3.78-3.97(m,2.6H),3.87(s,3H),4.11-4.16(s,0.4H),4.31-4.39(m,0.4H),4.48(s,0.6H),4.52(s,0.4H),4.59-4.64(m,0.6H),6.46(s,1H),6.57(d,J=9Hz,0.6H),6.63(d,J=9Hz,0.4H),6.72(d,J=9Hz,0.6H),6.75(d,J=9Hz,0.4H),8.36(s,0.4H),8.40(s,0.6H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.16 (m, 2H), 0.43-0.54 (m, 2H), 0.72-0.85 (m) , 1H), 1.03-1.16 (m, 1H), 1.18-1.82 (m, 5H), 1.98-2.13 (m, 1H), 2.17-2.46 (M, 4H), 2.55-2.68 (m, 1H), 2.69-2.79 (m, 0.6H), 2.80-2.90 (m, 0.4H), 2 .92-3.03 (m, 1.6H), 3.06 (d, J = 6Hz, 0.4H), 3.35-3.45 (m, 0.4H), 3.50-3. 60 (m, 0.6H), 3.78-3.97 (m, 2.6H), 3.87 (s, 3H), 4.11-4.16 (s, 0.4H), 4. 31-4.39 (m, 0.4H), 4.48 (s, 0.6H), 4.52 (s, 0.4H), 4.59-4.64 (m, 0.6H), 6.46 (s, 1H), 6.57 (d, J = 9Hz, 0.6H), 6.63 (d, J = 9Hz, 0.4H), 6.72 (d, J = 9Hz, 0) .6H), 6.75 (d, J = 9Hz, 0.4H), 8.36 (s, 0.4H), 8.40 (s, 0.6H).
(実施例85)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(イソキサゾール-3-イル)エタン-1-オン(105)の合成
(Example 85)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (isoxazole-3-yl) ethane-1-one (105)
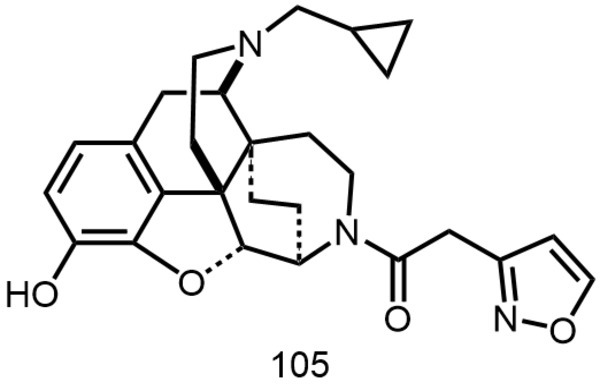
1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.18(m,2H),0.41-0.55(m,2H),0.71-0.84(m,1H),1.01-1.16(m,1H),1.16-1.54(m,4H),1.56-1.73(m,1H),2.01-2.12(m,0.8H),2.13-2.43(m,4.2H),2.51-2.66(m,1H),2.67-2.77(m,0.8H),2.78-2.89(m,0.2H),2.90-3.07(m,2H),3.40(s,J=4,4,10,14Hz,0.2H),3.54(d,J=3,3,11,14Hz,0.8H),3.79-3.99(m,2.8H),4.09-4.15(m,0.2H),4.28-4.38(m,0.2H),4.46(s,0.8H),4.48(s,0.2H),4.51-4.58(m,0.8H),6.43-6.58(m,2H),6.72(d,J=8Hz,1H),8,38(s,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.18 (m, 2H), 0.41-0.55 (m, 2H), 0.71-0.84 (m) , 1H), 1.01-1.16 (m, 1H), 1.16-1.54 (m, 4H), 1.56-1.73 (m, 1H), 2.01-2.12 (M, 0.8H), 2.13-2.43 (m, 4.2H), 2.51-2.66 (m, 1H), 2.67-2.77 (m, 0.8H) , 2.78-2.89 (m, 0.2H), 2.90-3.07 (m, 2H), 3.40 (s, J = 4,4,10,14Hz, 0.2H), 3.54 (d, J = 3,3,11,14Hz, 0.8H), 3.79-3.99 (m, 2.8H), 4.09-4.15 (m, 0.2H) , 4.28-4.38 (m, 0.2H), 4.46 (s, 0.8H), 4.48 (s, 0.2H), 4.51-4.58 (m, 0. 8H), 6.43-6.58 (m, 2H), 6.72 (d, J = 8Hz, 1H), 8,38 (s, 1H).
(実施例86)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(オキサゾール-4-イル)エタン-1-オン(106)の合成
(Example 86)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (oxazole-4-yl) ethane-1-one (106)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.43-0.55(m,2H),0.73-0.90(m,1H),1.05-1.81(m,6H),1.96-2.51(m,5H),2.55-2.69(m,1H),2.75-2.89(m,1H),2.91-3.12(m,2H),3.30-3.96(m,6.6H),4.16-4.22(m,0.4H),4.33-4.67(m,2H),6.54-6.65(m,1H),6.69-6.78(m,1H),7.67-7.75(m,1H),7.79-7.88(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.43-0.55 (m, 2H), 0.73-0.90 (m) , 1H), 1.05-1.81 (m, 6H), 1.96-2.51 (m, 5H), 2.55-2.69 (m, 1H), 2.75-2.89 (M, 1H), 2.91-3.12 (m, 2H), 3.30-3.96 (m, 6.6H), 4.16-4.22 (m, 0.4H), 4 .33-4.67 (m, 2H), 6.54-6.65 (m, 1H), 6.69-6.78 (m, 1H), 7.67-7.75 (m, 1H) , 7.79-7.88 (m, 1H).
(実施例87)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(オキサゾール-4-イル)エタン-1-オン(107)の合成
(Example 87)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (oxazole-4-yl) ethane-1-one (107)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.43-0.57(m,2H),0.71-0.91(m,1H),1.02-1.79(m,6H),2.04-2.45(m,5H),2.56-2.69(m,1H),2.72-3.10(m,3H),3.33-3.63(m,1H),3.67-3.96(m,2.8H),4.13-4.21(m,0.2H),4.30-5.04(m,3H),6.50-6.59(m,1H),6.71(d,J=8Hz,1H),7.73(s,1H),7.82-7.90(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.43-0.57 (m, 2H), 0.71-0.91 (m) , 1H), 1.02-1.79 (m, 6H), 2.04-2.45 (m, 5H), 2.56-2.69 (m, 1H), 2.72-3.10 (M, 3H), 3.33-3.63 (m, 1H), 3.67-3.96 (m, 2.8H), 4.13-4.21 (m, 0.2H), 4 .30-5.04 (m, 3H), 6.50-6.59 (m, 1H), 6.71 (d, J = 8Hz, 1H), 7.73 (s, 1H), 7.82 -7.90 (m, 1H).
(実施例88)
2-(ベンゾ[d]オキサゾール-2-イル)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)エタン-1-オン(108)の合成
(Example 88)
2- (Benzo [d] Oxazole-2-yl) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-methoxy-1,2,3,4,5 , 6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) ethane-1-one (108) ) Synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.19(m,2H),0.40-0.55(m,2H),0.67-0.93(m,1H),1.04-1.82(m,6H),2.07-2.48(m,5H),2.53-3.12(m,4H),3.31-3.92(m,5H),4.04-4.44(m,2.4H),4.55-4.68(m,1.6H),6.57(d,J=8Hz,0.6H),6.64(d,J=8Hz,0.4H),6.69-6.76(m,1H),7.15-7.37(m,2H),7.48-7.57(m,1H),7.66-7.74(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.03-0.19 (m, 2H), 0.40-0.55 (m, 2H), 0.67-0.93 (m) , 1H), 1.04-1.82 (m, 6H), 2.07-2.48 (m, 5H), 2.53-3.12 (m, 4H), 3.31-3.92 (M, 5H), 4.04-4.44 (m, 2.4H), 4.55-4.68 (m, 1.6H), 6.57 (d, J = 8Hz, 0.6H) , 6.64 (d, J = 8Hz, 0.4H), 6.69-6.76 (m, 1H), 7.15-7.37 (m, 2H), 7.48-7.57 ( m, 1H), 7.66-7.74 (m, 1H).
(実施例89)
2-(ベンゾ[d]オキサゾール-2-イル)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)エタン-1-オン(109)の合成
(Example 89)
2- (Benzo [d] Oxazole-2-yl) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Hydroxy-1,2,3,4,5 , 6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) ethane-1-one (109) ) Synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.02-0.17(m,2H),0.40-0.54(m,2H),0.65-0.95(m,1H),1.01-1.82(m,6H),2.06-2.45(m,5H),2.54-3.10(m,4H),3.36-3.64(m,1H),3.77-3.92(m,0.8H),4.07-4.80(m,5.2H),6.52(d,J=8Hz,0.8H),6.57(d,J=8Hz,0.2H),6.67-6.74(m,1H),7.26-7.37(m,2H),7.49-7.57(m,1H),7.67-7.74(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.02-0.17 (m, 2H), 0.40-0.54 (m, 2H), 0.65-0.95 (m) , 1H), 1.01-1.82 (m, 6H), 2.06-2.45 (m, 5H), 2.54-3.10 (m, 4H), 3.36-3.64 (M, 1H), 3.77-3.92 (m, 0.8H), 4.07-4.80 (m, 5.2H), 6.52 (d, J = 8Hz, 0.8H) , 6.57 (d, J = 8Hz, 0.2H), 6.67-6.74 (m, 1H), 7.26-7.37 (m, 2H), 7.49-7.57 ( m, 1H), 7.67-7.74 (m, 1H).
(実施例90)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(1H-インドール-3-イル)エタン-1-オン(110)の合成
(Example 90)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (1H-indole-3-yl) ethane-1-on (110) Synthetic

1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.16(m,2H),0.38-0.54(m,2H),0.67-1.89(m,6H),2.13-2.68(m,7H),2.77-3.08(m,3H),3.22-3.51(m,1H),3.75-4.00(m,5.7H),4.18-4.25(m,0.3H),4.38-4.53(m,1.3H),4.65-4.72(m,0.7H),6.54(d,J=8Hz,0.7H),6.61(d,J=8Hz,0.3H),6.66-6.77(m,1H),7.06-7.39(m,4H),7.58-7.71(m,1H),7.99-8.33(m,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.03-0.16 (m, 2H), 0.38-0.54 (m, 2H), 0.67-1.89 (m) , 6H), 2.13-2.68 (m, 7H), 2.77-3.08 (m, 3H), 3.22-3.51 (m, 1H), 3.75-4.00 (M, 5.7H), 4.18-4.25 (m, 0.3H), 4.38-4.53 (m, 1.3H), 4.65-4.72 (m, 0. 7H), 6.54 (d, J = 8Hz, 0.7H), 6.61 (d, J = 8Hz, 0.3H), 6.66-6.77 (m, 1H), 7.06- 7.39 (m, 4H), 7.58-7.71 (m, 1H), 7.99-8.33 (m, 1H).
(実施例91)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(1H-インドール-3-イル)エタン-1-オン(111)の合成
(Example 91)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (1H-indole-3-yl) ethane-1-on (111) Synthetic

1H-NMR(400MHz,CDCl3)δ(ppm):0.02-0.18(m,2H),0.39-0.54(m,2H),0.65-1.79(m,8H),2.10-2.67(m,6H),2.75-3.10(m,2H),3.26-3.53(m,1H),3.78-4.01(m,3H),4.41-5.18(m,3H),6.45-6.58(m,1H),6.69(d,J=8Hz,1H),7.10-7.43(m,4H),7.64-7.73(m,1H),8.05-8.20(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.02-0.18 (m, 2H), 0.39-0.54 (m, 2H), 0.65-1.79 (m) , 8H), 2.10-2.67 (m, 6H), 2.75-3.10 (m, 2H), 3.26-3.53 (m, 1H), 3.78-4.01 (M, 3H), 4.41-5.18 (m, 3H), 6.45-6.58 (m, 1H), 6.69 (d, J = 8Hz, 1H), 7.10-7 .43 (m, 4H), 7.64-7.73 (m, 1H), 8.05-8.20 (m, 1H).
(実施例92)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(チアゾール-2-イル)エタン-1-オン(112)の合成
(Example 92)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (thiazole-2-yl) ethane-1-one (112)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.14(m,2H),0.44-0.54(m,2H),0.73-0.84(m,1H),1.04-1.16(m,1H),1.19-1.76(m,5H),2.05(ddd,J=5,12,12Hz,0.6H),2.18-2.45(m,4.4H),2.57-2.75(m,1.6H),2.82-3.07(m,2.4H),3.45(ddd,J=4,11,14Hz,0.4H),3.57(ddd,J=4,11,14Hz,0.6H),3.85-3.93(m,3.6H),4.18-4.28(m,2.4H),4.33-4.39(m,0.4H),4.51-4.55(m,1H),4.62-4.65(m,0.6H),6.57(d,J=8Hz,0.6H),6.62(d,J=8Hz,0.4H),6.72(d,J=8Hz,0.6H),6.74(d,J=8Hz,0.4H),7.29(d,J=3Hz,0.4H),7.32(d,J=3Hz,0.6H),7.71(d,J=3Hz,0.4H),7.75(d,J=3Hz,0.6H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.14 (m, 2H), 0.44-0.54 (m, 2H), 0.73-0.84 (m) , 1H), 1.04-1.16 (m, 1H), 1.19-1.76 (m, 5H), 2.05 (ddd, J = 5,12,12Hz, 0.6H), 2 .18-2.45 (m, 4.4H), 2.57-2.75 (m, 1.6H), 2.82-3.07 (m, 2.4H), 3.45 (ddd, J = 4,11,14Hz, 0.4H), 3.57 (ddd, J = 4,11,14Hz, 0.6H), 3.85-3.93 (m, 3.6H), 4.18 -4.28 (m, 2.4H), 4.33-4.39 (m, 0.4H), 4.51-4.55 (m, 1H), 4.62-4.65 (m, 0.6H), 6.57 (d, J = 8Hz, 0.6H), 6.62 (d, J = 8Hz, 0.4H), 6.72 (d, J = 8Hz, 0.6H), 6.74 (d, J = 8Hz, 0.4H), 7.29 (d, J = 3Hz, 0.4H), 7.32 (d, J = 3Hz, 0.6H), 7.71 (d) , J = 3Hz, 0.4H), 7.75 (d, J = 3Hz, 0.6H).
(実施例93)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(チアゾール-2-イル)エタン-1-オン(113)の合成
(Example 93)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (thiazole-2-yl) ethane-1-one (113)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.13(m,2H),0.43-0.53(m,2H),0.72-0.82(m,1H),1.03-1.14(m,1H),1.19-1.72(m,5H),2.05(ddd,J=6,13,13Hz,0.7H),2.13-2.41(m,4.3H),2.55-2.73(m,1.7H),2.80-3.05(m,2.3H),3.45(ddd,J=4,11,14Hz,0.3H),3.56(ddd,J=4,11,14Hz,0.7H),3.88-3.94(m,0.7H),4.18-4.36(m,2.6H),4.48-4.51(m,1H),4.54-4.57(m,0.7H),6.52(d,J=8Hz,0.7H),6.54(d,J=8Hz,0.3H),6.71(d,J=8Hz,1H),7.32(d,J=3Hz,0.3H),7.33(d,J=3Hz,0.7H),7.73(d,J=3Hz,0.3H),7.75(d,J=3Hz,0.7H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.13 (m, 2H), 0.43-0.53 (m, 2H), 0.72-0.82 (m) , 1H), 1.03-1.14 (m, 1H), 1.19-1.72 (m, 5H), 2.05 (ddd, J = 6,13,13Hz, 0.7H), 2 .13-2.41 (m, 4.3H), 2.55-2.73 (m, 1.7H), 2.80-3.05 (m, 2.3H), 3.45 (ddd, J = 4,11,14Hz, 0.3H), 3.56 (ddd, J = 4,11,14Hz, 0.7H), 3.88-3.94 (m, 0.7H), 4.18 -4.36 (m, 2.6H), 4.48-4.51 (m, 1H), 4.54-4.57 (m, 0.7H), 6.52 (d, J = 8Hz, 0.7H), 6.54 (d, J = 8Hz, 0.3H), 6.71 (d, J = 8Hz, 1H), 7.32 (d, J = 3Hz, 0.3H), 7. 33 (d, J = 3Hz, 0.7H), 7.73 (d, J = 3Hz, 0.3H), 7.75 (d, J = 3Hz, 0.7H).
(実施例94)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(チアゾール-5-イル)エタン-1-オン(114)の合成
(Example 94)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (thiazole-5-yl) ethane-1-one (114)

1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.17(m,2H),0.42-0.56(m,2H),0.71-0.92(m,1H),1.03-1.79(m,5H),1.94-2.47(m,6H),2.55-3.09(m,4H),3.28-4.12(m,7H),4.39-4.66(m,2H),6.57(d,J=8Hz,0.5H),6.64(d,J=0.5H),6.68-6.80(m,1H),7.23(s,1H),8.71-8.79(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.03-0.17 (m, 2H), 0.42-0.56 (m, 2H), 0.71-0.92 (m) , 1H), 1.03-1.79 (m, 5H), 1.94-2.47 (m, 6H), 2.55-3.09 (m, 4H), 3.28-4.12 (M, 7H), 4.39-4.66 (m, 2H), 6.57 (d, J = 8Hz, 0.5H), 6.64 (d, J = 0.5H), 6.68 -6.80 (m, 1H), 7.23 (s, 1H), 8.71-8.79 (m, 1H).
(実施例95)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(チアゾール-5-イル)エタン-1-オン(115)の合成
(Example 95)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (thiazole-5-yl) ethane-1-one (115)

1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.17(m,2H),0.43-0.56(m,2H),0.70-0.95(m,1H),1.03-1.78(m,5H),1.97-2.45(m,6H),2.54-3.13(m,4H),3.34-3.63(m,1H),3.72-3.85(m,0.6H),3.96-4.10(m,2H),4.33-5.14(m,3.4H),6.49-6.61(m,1H),6.68-6.76(m,1H),7.70-7.78(m,1H),8.74-8.80(m,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.03-0.17 (m, 2H), 0.43-0.56 (m, 2H), 0.70-0.95 (m) , 1H), 1.03-1.78 (m, 5H), 1.97-2.45 (m, 6H), 2.54-3.13 (m, 4H), 3.34-3.63 (M, 1H), 3.72-3.85 (m, 0.6H), 3.96-4.10 (m, 2H), 4.33-5.14 (m, 3.4H), 6 .49-6.61 (m, 1H), 6.68-6.76 (m, 1H), 7.70-7.78 (m, 1H), 8.74-8.80 (m, 1H) ..
(実施例96)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(チアゾール-4-イル)エタン-1-オン(116)の合成
(Example 96)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (thiazole-4-yl) ethane-1-one (116)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.44-0.52(m,2H),0.73-0.83(m,1H),1.04-1.90(m,6H),2.04(ddd,J=6,13,13Hz,0.6H),2.20-2.44(m,4.4H),2.57-2.69(m,1.6H),2.80-3.07(m,2.4H),3.35-3.43(m,0.4H),3.51-3.58(m,0.6H),3.87(s,3H),3.89-4.09(m,2.6H),4.24-4.28(m,0.4H),4.37-4.43(m,0.4H),4.51-4.57(m,1H),4.62-4.65(m,0.6H),6.56(d,J=8Hz,0.6H),6.62(d,J=8Hz,0.4H),6.72(d,J=8Hz,0.6H),6.74(d,J=8Hz,0.4H),7.23(s,0.4H),7.27(s,0.6H),8.75(d,J=2Hz,0.4H),8.80(d,J=2Hz,0.6H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.44-0.52 (m, 2H), 0.73-0.83 (m) , 1H), 1.04-1.90 (m, 6H), 2.04 (ddd, J = 6,13,13Hz, 0.6H), 2.20-2.44 (m, 4.4H) , 2.57-2.69 (m, 1.6H), 2.80-3.07 (m, 2.4H), 3.35-3.43 (m, 0.4H), 3.51- 3.58 (m, 0.6H), 3.87 (s, 3H), 3.89-4.09 (m, 2.6H), 4.24-4.28 (m, 0.4H), 4.37-4.43 (m, 0.4H), 4.51-4.57 (m, 1H), 4.62-4.65 (m, 0.6H), 6.56 (d, J) = 8Hz, 0.6H), 6.62 (d, J = 8Hz, 0.4H), 6.72 (d, J = 8Hz, 0.6H), 6.74 (d, J = 8Hz, 0. 4H), 7.23 (s, 0.4H), 7.27 (s, 0.6H), 8.75 (d, J = 2Hz, 0.4H), 8.80 (d, J = 2Hz, 0.6H).
(実施例97)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(チアゾール-4-イル)エタン-1-オン(117)の合成
(Example 97)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (thiazole-4-yl) ethane-1-one (117)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.13(m,2H),0.43-0.52(m,2H),0.73-0.81(m,1H),1.02-1.12(m,1H),1.18-1.71(m,5H),2.04(ddd,J=5,13,13Hz,0.7H),2.17-2.41(m,4.3H),2.56-2.67(m,1.7H),2.79-3.05(m,2.3H),3.35-3.42(m,0.3H),3.49-3.56(m,0.7H),3.92-4.15(m,2.7H),4.24-4.27(m,0.3H),4.34-4.41(m,0.3H),4.49-4.58(m,1.7H),6.51(d,J=8Hz,0.7H),6.54(d,J=8Hz,0.3H),6.70(d,J=8Hz,1H),7.26-7.29(m,1H),8.77(d,J=2Hz,0.3H),8.80(d,J=2Hz,0.7H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.13 (m, 2H), 0.43-0.52 (m, 2H), 0.73-0.81 (m) , 1H), 1.02-1.12 (m, 1H), 1.18-1.71 (m, 5H), 2.04 (ddd, J = 5,13,13Hz, 0.7H), 2 .17-2.41 (m, 4.3H), 2.56-2.67 (m, 1.7H), 2.79-3.05 (m, 2.3H), 3.35-3. 42 (m, 0.3H), 3.49-3.56 (m, 0.7H), 3.92-4.15 (m, 2.7H), 4.24-4.27 (m, 0) .3H), 4.34-4.41 (m, 0.3H), 4.49-4.58 (m, 1.7H), 6.51 (d, J = 8Hz, 0.7H), 6 .54 (d, J = 8Hz, 0.3H), 6.70 (d, J = 8Hz, 1H), 7.26-7.29 (m, 1H), 8.77 (d, J = 2Hz, 0.3H), 8.80 (d, J = 2Hz, 0.7H).
(実施例98)
2-(5-クロロピリミジン-2-イル)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)エタン-1-オン(118)の合成
(Example 98)
2- (5-Chloropyrimidine-2-yl) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5, 6,8,8a-Octahydro-7H-4a, 8-Etano-4,13-Metanobenzoflo [2,3-c] Pyrimidine [4,3-d] Azepine-7-yl) Ethane-1-one (118) Synthesis of
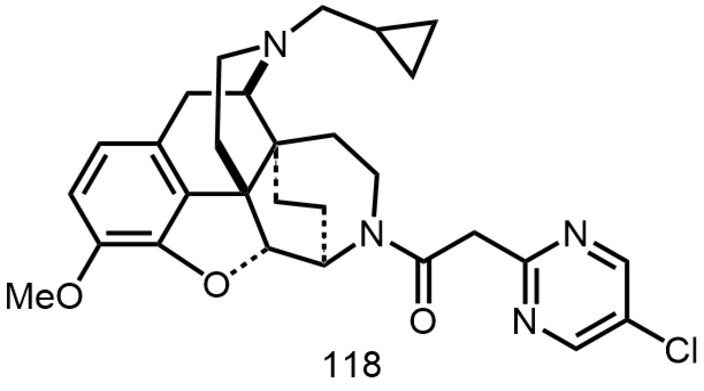
1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.17(m,2H),0.43-0.55(m,2H),0.71-0.92(m,1H),1.03-1.80(m,6H),2.11-2.48(m,5H),2.57-3.10(m,4H),3.33-3.62(m,1H),3.70-3.80(m,0.6H),3.87(s,3H),4.06-4.23(m,2.4H),4.34-4.46(m,0.4H),4.54-4.69(m,1.6H),6.57(d,J=8Hz,0.6H),6.63(d,J=8Hz,0.4H),6.68-6.77(m,1H),8.64-8.71(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.05-0.17 (m, 2H), 0.43-0.55 (m, 2H), 0.71-0.92 (m) , 1H), 1.03-1.80 (m, 6H), 2.11-2.48 (m, 5H), 2.57-3.10 (m, 4H), 3.33-3.62 (M, 1H), 3.70-3.80 (m, 0.6H), 3.87 (s, 3H), 4.06-4.23 (m, 2.4H), 4.34-4 .46 (m, 0.4H), 4.54-4.69 (m, 1.6H), 6.57 (d, J = 8Hz, 0.6H), 6.63 (d, J = 8Hz, 0.4H), 6.68-6.77 (m, 1H), 8.64-8.71 (m, 2H).
(実施例99)
2-(5-クロロピリミジン-2-イル)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)エタン-1-オン(119)の合成
(Example 99)
2- (5-Chloropyrimidine-2-yl) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5, 6,8,8a-Octahydro-7H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) ethane-1-one (119) Synthesis of

1H-NMR(400MHz,CDCl3)δ(ppm):0.03-0.19(m,2H),0.43-0.55(m,2H),0.72-0.94(m,1H),1.01-1.90(m,6H),2.11-2.45(m,5H),2.58-3.11(m,4H),3.35-3.59(m,1H),3.70-3.82(m,0.8H),4.04-4.25(m,2H),4.30-4.42(m,0.2H),4.48-4.85(m,3H),6.50-6.58(m,1H),6.68-6.73(m,1H),8.67(s,0.4H),8.69(s,1.6H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.03-0.19 (m, 2H), 0.43-0.55 (m, 2H), 0.72-0.94 (m) , 1H), 1.01-1.90 (m, 6H), 2.11-2.45 (m, 5H), 2.58-3.11 (m, 4H), 3.35-3.59 (M, 1H), 3.70-3.82 (m, 0.8H), 4.04-4.25 (m, 2H), 4.30-4.42 (m, 0.2H), 4 .48-4.85 (m, 3H), 6.50-6.58 (m, 1H), 6.68-6.73 (m, 1H), 8.67 (s, 0.4H), 8 .69 (s, 1.6H).
(実施例100)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(1H-イミダゾール-1-イル)エタン-1-オン(120)の合成
(Example 100)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (1H-imidazol-1-yl) ethane-1-one (120) Synthetic

1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.15(m,2H),0.42-0.55(m,2H),0.72-0.92(m,2H),1.05-1.80(m,6H),1.95-2.45(m,4H),2.60-2.70(m,1H),2.75-3.10(m,3H),3.50-3.70(m,1.7H),3.90(s,0.3H),4.15-4.35(m,0.3H),4.35-4.50(m,1.7H),4.80-5.00(m,2H),6.53(d,J=8Hz,0.7H),6.57(d,J=8Hz,0.3H),6.70-6.80(m,1H),6.90-7.00(m,1H),7.05-7.15(m,1H),7.58(s,0.7H),7.63(s,0.3H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.15 (m, 2H), 0.42-0.55 (m, 2H), 0.72-0.92 (m) , 2H), 1.05-1.80 (m, 6H), 1.95-2.45 (m, 4H), 2.60-2.70 (m, 1H), 2.75-3.10 (M, 3H), 3.50-3.70 (m, 1.7H), 3.90 (s, 0.3H), 4.15-4.35 (m, 0.3H), 4.35 -4.50 (m, 1.7H), 4.80-5.00 (m, 2H), 6.53 (d, J = 8Hz, 0.7H), 6.57 (d, J = 8Hz, 0.3H), 6.70-6.80 (m, 1H), 6.90-7.00 (m, 1H), 7.05-7.15 (m, 1H), 7.58 (s, 0.7H), 7.63 (s, 0.3H).
(実施例101)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(1H-ピラゾール-1-イル)エタン-1-オン(121)の合成
(Example 101)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (1H-pyrazole-1-yl) ethane-1-one (121) Synthetic

1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.15(m,2H),0.45-0.55(m,2H),0.70-0.85(m,1H),1.00-1.80(m,4.3H),2.00-2.15(m,0.7H),2.15-2.45(m,5H),2.55-2.75(m,2H),2.80-3.10(m,3H),3.40-3.60(m,1H),3.75-3.83(m,0.7H),3.86(s,0.9H),3.88(s,2.1H),4.08(s,0.3H),4.20-4.32(m,0.3H),4.48(s,0.7H),4.55-4.65(m,1H),5.04(d,J=16Hz,1H),5.10(d,J=16Hz,1H),6.30-6.38(m,1H),6.57(d,J=8Hz,0.7H),6.63(d,J=8Hz,0.3H),6.70-6.78(m,1H),7.50-7.60(m,2H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.15 (m, 2H), 0.45-0.55 (m, 2H), 0.70-0.85 (m) , 1H), 1.00-1.80 (m, 4.3H), 2.00-2.15 (m, 0.7H), 2.15-2.45 (m, 5H), 2.55 -2.75 (m, 2H), 2.80-3.10 (m, 3H), 3.40-3.60 (m, 1H), 3.75-3.83 (m, 0.7H) , 3.86 (s, 0.9H), 3.88 (s, 2.1H), 4.08 (s, 0.3H), 4.20-4.32 (m, 0.3H), 4 .48 (s, 0.7H), 4.55-4.65 (m, 1H), 5.04 (d, J = 16Hz, 1H), 5.10 (d, J = 16Hz, 1H), 6 .30-6.38 (m, 1H), 6.57 (d, J = 8Hz, 0.7H), 6.63 (d, J = 8Hz, 0.3H), 6.70-6.78 ( m, 1H), 7.50-7.60 (m, 2H).
(実施例102)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(1H-ピラゾール-1-イル)エタン-1-オン(122)の合成
(Example 102)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (1H-pyrazole-1-yl) ethane-1-one (122) Synthetic

1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.20(m,2H),0.40-0.55(m,2H),0.70-0.85(m,1H),1.00-1.80(m,6H),2.00-2.45(m,5.3H),2.55-2.75(m,1.7H),2.75-3.05(m,2.3H),3.40-3.55(m,0.7H),3.75-3.85(m,0.7H),4.07(s,0.3H),4.20-4.35(m,0.3H),4.40-4.55(m,1.7H),5.04(d,J=16Hz,1H),5.16(d,J=16Hz,1H),6.30-6.38(m,1H),6.51(d,J=8Hz,0.7H),6.55(d,J=8Hz,0.3H),6.66-6.74(m,1H),7.52-7.58(m,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.20 (m, 2H), 0.40-0.55 (m, 2H), 0.70-0.85 (m) , 1H), 1.00-1.80 (m, 6H), 2.00-2.45 (m, 5.3H), 2.55-2.75 (m, 1.7H), 2.75 -3.05 (m, 2.3H), 3.40-3.55 (m, 0.7H), 3.75-3.85 (m, 0.7H), 4.07 (s, 0. 3H), 4.20-4.35 (m, 0.3H), 4.40-4.55 (m, 1.7H), 5.04 (d, J = 16Hz, 1H), 5.16 ( d, J = 16Hz, 1H), 6.30-6.38 (m, 1H), 6.51 (d, J = 8Hz, 0.7H), 6.55 (d, J = 8Hz, 0.3H) ), 6.66-6.74 (m, 1H), 7.52-7.58 (m, 2H).
(実施例103)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(ピリミジン-2-イル)プロプ-2-エン-1-オン(123)の合成
(Example 103)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (pyrimidine-2-yl) prop-2-en-1 -On (123) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.45-0.53(m,2H),0.75-0.85(m,1H),1.08-1.16(m,1H),1.22-1.38(m,2H),1.45-1.55(m,2H),1.68-1.79(m,1H),2.13-2.46(m,5H),2.59-2.68(m,1H),2.87-2.99(m,1.6H),3.00-3.09(m,1.4H),3.50(ddd,J=4,10,14Hz,0.6H),3.64(ddd,J=4,11,14Hz,0.4H),3.89(s,3H),3.98-4.05(m,0.4H),4.30-4.32(m,0.6H),4.40-4.48(m,0.6H),4.58(s,0.6H),4.61(s,0.4H),4.73-4.78(m,0.4H),6.58(d,J=8Hz,0.4H),6.63(d,J=8Hz,0.6H),6.74(d,J=8Hz,0.4H),6.75(d,J=8Hz,0.6H),7.17-7.21(m,1H),7.59(d,J=15Hz,0.4H),7.72(d,J=15Hz,0.6H),7.79(d,J=15Hz,0.4H),7.81(d,J=15Hz,0.6H),8.75(d,J=4Hz,1.2H),8.76(d,J=4Hz,0.8H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.45-0.53 (m, 2H), 0.75-0.85 (m) , 1H), 1.08-1.16 (m, 1H), 1.22-1.38 (m, 2H), 1.45-1.55 (m, 2H), 1.68-1.79 (M, 1H), 2.13-2.46 (m, 5H), 2.59-2.68 (m, 1H), 2.87-2.99 (m, 1.6H), 3.00 -3.09 (m, 1.4H), 3.50 (ddd, J = 4,10,14Hz, 0.6H), 3.64 (ddd, J = 4,11,14Hz, 0.4H), 3.89 (s, 3H), 3.98-4.05 (m, 0.4H), 4.30-4.32 (m, 0.6H), 4.40-4.48 (m, 0) .6H), 4.58 (s, 0.6H), 4.61 (s, 0.4H), 4.73-4.78 (m, 0.4H), 6.58 (d, J = 8Hz) , 0.4H), 6.63 (d, J = 8Hz, 0.6H), 6.74 (d, J = 8Hz, 0.4H), 6.75 (d, J = 8Hz, 0.6H) , 7.17-7.21 (m, 1H), 7.59 (d, J = 15Hz, 0.4H), 7.72 (d, J = 15Hz, 0.6H), 7.79 (d, J = 15Hz, 0.4H), 7.81 (d, J = 15Hz, 0.6H), 8.75 (d, J = 4Hz, 1.2H), 8.76 (d, J = 4Hz, 0) .8H).
(実施例104)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(ピリミジン-2-イル)プロプ-2-エン-1-オン(124)の合成
(Example 104)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (pyrimidine-2-yl) prop-2-en-1 -On (124) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.12(m,2H),0.45-0.53(m,2H),0.73-0.89(m,1H),1.07-1.15(m,2H),1.24-1.33(m,2H),1.39-1.51(m,2H),1.68-1.78(m,1H),2.14-2.43(m,5H),2.60-2.67(m,1H),2.85-2.93(m,1H),2.97(d,J=18Hz,1H),3.03(d,J=6Hz,0.6H),3.06(d,J=6Hz,0.4H),4.01-4.09(m,0.4H),4.25-4.29(m,0.6H)4.39-4.46(m,0.4H),4.57-4.60(m,1H),4.67-4.69(m,0.6H),6.54(d,J=8Hz,0.4H),6.56(d,J=8Hz,0.6H),6.71(d,J=8Hz,0.4H),6.73(d,J=8Hz,0.6H),7.21(t,J=5Hz,1H),7.60(d,J=15Hz,0.6H),7.73(d,J=15Hz,0.4H),7.80(d,J=15Hz,0.6H),7.81(d,J=15Hz,0.4H),8.77(d,J=5Hz,1.2H),8.78(d,J=5Hz,0.8H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.12 (m, 2H), 0.45-0.53 (m, 2H), 0.73-0.89 (m) , 1H), 1.07-1.15 (m, 2H), 1.24-1.33 (m, 2H), 1.39-1.51 (m, 2H), 1.68-1.78 (M, 1H), 2.14-2.43 (m, 5H), 2.60-2.67 (m, 1H), 2.85-2.93 (m, 1H), 2.97 (d). , J = 18Hz, 1H), 3.03 (d, J = 6Hz, 0.6H), 3.06 (d, J = 6Hz, 0.4H), 4.01-4.09 (m, 0. 4H), 4.25-4.29 (m, 0.6H) 4.39-4.46 (m, 0.4H), 4.57-4.60 (m, 1H), 4.67-4 .69 (m, 0.6H), 6.54 (d, J = 8Hz, 0.4H), 6.56 (d, J = 8Hz, 0.6H), 6.71 (d, J = 8Hz, 0.4H), 6.73 (d, J = 8Hz, 0.6H), 7.21 (t, J = 5Hz, 1H), 7.60 (d, J = 15Hz, 0.6H), 7. 73 (d, J = 15Hz, 0.4H), 7.80 (d, J = 15Hz, 0.6H), 7.81 (d, J = 15Hz, 0.4H), 8.77 (d, J) = 5Hz, 1.2H), 8.78 (d, J = 5Hz, 0.8H).
(実施例105)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(フラン-3-イル)プロプ-2-エン-1-オン(125)の合成
(Example 105)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (fran-3-yl) prop-2-en-1 -On (125) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.16(m,2H),0.42-0.55(m,2H),0.73-0.85(m,1H),1.06-1.15(m,1H),1.23-1.39(m,1H),1.40-1.57(m,3H),1.65-1.78(m,1H),2.11-2.43(m,5H),2.57-2.68(m,1H),2.81-2.91(m,1H),2.96(d,J=18Hz,1H),3.03-3.07(m,1H),3.39-3.49(m,0.5H),3.53-3.63(m,0.5H),3.93-4.01(m,0.5H),4.16-4.22(m,0.5H),4.41-4.48(m,0.5H),4.52-4.59(m,1H),4.66(s,0.5H),6.51-6.76(m,4H),7.43(s,1H),7.58(d,J=16Hz,0.5H),7.64(d,J=6Hz,1H),7.68(d,J=15Hz,0.5H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.16 (m, 2H), 0.42-0.55 (m, 2H), 0.73-0.85 (m) , 1H), 1.06-1.15 (m, 1H), 1.23-1.39 (m, 1H), 1.40-1.57 (m, 3H), 1.65-1.78 (M, 1H), 2.11-2.43 (m, 5H), 2.57-2.68 (m, 1H), 2.81-2.91 (m, 1H), 2.96 (d). , J = 18Hz, 1H), 3.03-3.07 (m, 1H), 3.39-3.49 (m, 0.5H), 3.53-3.63 (m, 0.5H) , 3.93-4.01 (m, 0.5H), 4.16-4.22 (m, 0.5H), 4.41-4.48 (m, 0.5H), 4.52- 4.59 (m, 1H), 4.66 (s, 0.5H), 6.51-6.76 (m, 4H), 7.43 (s, 1H), 7.58 (d, J = 16Hz, 0.5H), 7.64 (d, J = 6Hz, 1H), 7.68 (d, J = 15Hz, 0.5H).
(実施例106)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(オキサゾール-2-イル)プロプ-2-エン-1-オン(126)の合成
(Example 106)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (oxazole-2-yl) prop-2-en-1 -Synthesis of on (126)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.45-0.54(m,2H),0.75-0.85(m,1H),1.09-1.77(m,6H),2.12-2.45(m,5H),2.60-2.66(m,1H),2.86-2.94(m,1H),2.97-3.08(m,2H),3.44-3.67(m,0.6H),3.60-3.67(m,0.4H),3.89(s,3H),3.93-3.98(m,0.4H),4.22-4.25(m,0.6H),4.41-4.47(m,0.6H),4.53-4.58(m,1H),4.71-4.74(m,0.4H),6.59(d,J=8Hz,0.4H),6.63(d,J=8Hz,0.6H),6.74(d,J=8Hz,0.4H),6.76(d,J=8Hz,0.6H),7.23-7.69(m,4H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.45-0.54 (m, 2H), 0.75-0.85 (m) , 1H), 1.09-1.77 (m, 6H), 2.12-2.45 (m, 5H), 2.60-2.66 (m, 1H), 2.86-2.94 (M, 1H), 2.97-3.08 (m, 2H), 3.44-3.67 (m, 0.6H), 3.60-3.67 (m, 0.4H), 3 .89 (s, 3H), 3.93-3.98 (m, 0.4H), 4.22-4.25 (m, 0.6H), 4.41-4.47 (m, 0. 6H), 4.53-4.58 (m, 1H), 4.71-4.74 (m, 0.4H), 6.59 (d, J = 8Hz, 0.4H), 6.63 ( d, J = 8Hz, 0.6H), 6.74 (d, J = 8Hz, 0.4H), 6.76 (d, J = 8Hz, 0.6H), 7.23-7.69 (m) , 4H).
(実施例107)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(オキサゾール-2-イル)プロプ-2-エン-1-オン(127)の合成
(Example 107)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (oxazole-2-yl) prop-2-en-1 -On (127) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.14(m,2H),0.45-0.53(m,2H),0.75-0.86(m,1H),1.09-1.15(m,1H),1.25-1.78(m,5H),2.12-2.43(m,5H),2.60-2.66(m,1H),2.84-2.92(m,1H),2.97(d,J=18Hz,1H),3.03(d,J=6Hz,0.6H),3.06(d,J=6Hz,0.4H),3.46-3.53(m,0.4H),3.58-3.66(m,0.6H),3.95-4.01(m,0.6H),4.18-4.21(m,0.4H),4.38-4.44(m,0.4H),4.52-4.56(m,1H),4.63-4.66(m,0.6H),6.54(d,J=8Hz,0.4H),6.56(d,J=8Hz,0.6H),6.72(d,J=8Hz,0.4H),6.73(d,J=8Hz,0.6H),7.26-7.27(m,1H),7.34(d,J=15Hz,0.4H),7.37(d,J=15Hz,0.4H),7.45(d,J=15Hz,0.6H),7.56(d,J=15Hz,0.6H),7.70(s,1H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.14 (m, 2H), 0.45-0.53 (m, 2H), 0.75-0.86 (m) , 1H), 1.09-1.15 (m, 1H), 1.25-1.78 (m, 5H), 2.12-2.43 (m, 5H), 2.60-2.66 (M, 1H), 2.84-2.92 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 0.6H), 3.06 (D, J = 6Hz, 0.4H), 3.46-3.53 (m, 0.4H), 3.58-3.66 (m, 0.6H), 3.95-4.01 ( m, 0.6H), 4.18-4.21 (m, 0.4H), 4.38-4.44 (m, 0.4H), 4.52-4.56 (m, 1H), 4.63-4.66 (m, 0.6H), 6.54 (d, J = 8Hz, 0.4H), 6.56 (d, J = 8Hz, 0.6H), 6.72 (d) , J = 8Hz, 0.4H), 6.73 (d, J = 8Hz, 0.6H), 7.26-7.27 (m, 1H), 7.34 (d, J = 15Hz, 0. 4H), 7.37 (d, J = 15Hz, 0.4H), 7.45 (d, J = 15Hz, 0.6H), 7.56 (d, J = 15Hz, 0.6H), 7. 70 (s, 1H).
(実施例108)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(オキサゾール-4-イル)プロプ-2-エン-1-オン(128)の合成
(Example 108)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (oxazole-4-yl) prop-2-en-1 -On (128) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.16(m,2H),0.43-0.55(m,2H),0.72-0.85(m,1H),1.06-1.57(m,5H),1.66-1.90(m,1H),2.11-2.47(m,5H),2.58-2.68(m,1H),2.84-3.10(m,3H),3.53(ddd,J=4,10,15Hz,0.5H),3.62(ddd,J=4,11,15Hz,0.5H),3.89(s,3H),4.02(ddd,J=4,4,14Hz,0.5H),4.26-4.31(m,0.5H),4.37(ddd,J=5,5,14Hz,0.5H),4.57(s,0.5H),4.58(s,0.5H),4.72-4.77(m,0.5H),6.58(d,J=8Hz,0.5H),6.62(d,J=8Hz,0.5H),6.73(d,J=8Hz,0.5H),6.75(d,J=8Hz,0.5H),7.21(d,J=15Hz,0.5H),7.24(d,J=15Hz,0.5H),7.55(d,J=15Hz,0.5H),7.61(d,J=15Hz,0.5H),7.77(s,0.5H),7.78(s,0.5H),7.87(s,0.5H),7.90(s,0.5H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.16 (m, 2H), 0.43-0.55 (m, 2H), 0.72-0.85 (m) , 1H), 1.06-1.57 (m, 5H), 1.66-1.90 (m, 1H), 2.11-2.47 (m, 5H), 2.58-2.68 (M, 1H), 2.84-3.10 (m, 3H), 3.53 (ddd, J = 4,10,15Hz, 0.5H), 3.62 (ddd, J = 4,11, 15Hz, 0.5H), 3.89 (s, 3H), 4.02 (ddd, J = 4,4,14Hz, 0.5H), 4.26-4.31 (m, 0.5H), 4.37 (ddd, J = 5,5,14Hz, 0.5H), 4.57 (s, 0.5H), 4.58 (s, 0.5H), 4.72-4.77 (m) , 0.5H), 6.58 (d, J = 8Hz, 0.5H), 6.62 (d, J = 8Hz, 0.5H), 6.73 (d, J = 8Hz, 0.5H) , 6.75 (d, J = 8Hz, 0.5H), 7.21 (d, J = 15Hz, 0.5H), 7.24 (d, J = 15Hz, 0.5H), 7.55 ( d, J = 15Hz, 0.5H), 7.61 (d, J = 15Hz, 0.5H), 7.77 (s, 0.5H), 7.78 (s, 0.5H), 7. 87 (s, 0.5H), 7.90 (s, 0.5H).
(実施例109)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(オキサゾール-4-イル)プロプ-2-エン-1-オン(129)の合成
(Example 109)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (oxazole-4-yl) prop-2-en-1 -On (129) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.43-0.54(m,2H),0.73-0.83(m,1H),1.05-1.15(m,1H),1.23-1.78(m,5H),2.11-2.43(m,5H),2.58-2.66(m,1H),2.82-2.93(m,1H),2.96(d,J=18Hz,1H),3.02(d,J=6Hz,0.6H),3.05(d,J=6Hz,0.4H),3.48(ddd,J=5,11,15Hz,0.4H),3.60(ddd,J=4,12,16Hz,0.6H),4.05(ddd,J=3,4,15Hz,0.6H),4.22-4.27(m,0.4H),4.41(ddd,J=5,5,14Hz,0.4H),4.53-4.59(m,1H),4.64-4.68(m,0.6H),5.15(br s,1H),6.53(d,J=8Hz,0.6H),6.55(d,J=8Hz,0.4H),6.72(d,J=8Hz,0.4H),6.72(d,J=8Hz,0.6H),7.23(d,J=15Hz,0.4H),7.26(d,J=15Hz,0.6H),7.55(d,J=15Hz,0.6H),7.62(d,J=15Hz,0.4H),7.79(s,0.6H),7.81(s,0.4H),7.91(s,0.6H),7.92(s,0.4H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.43-0.54 (m, 2H), 0.73-0.83 (m) , 1H), 1.05-1.15 (m, 1H), 1.23-1.78 (m, 5H), 2.11-2.43 (m, 5H), 2.58-2.66 (M, 1H), 2.82-2.93 (m, 1H), 2.96 (d, J = 18Hz, 1H), 3.02 (d, J = 6Hz, 0.6H), 3.05 (D, J = 6Hz, 0.4H), 3.48 (ddd, J = 5,11,15Hz, 0.4H), 3.60 (ddd, J = 4,12,16Hz, 0.6H), 4.05 (ddd, J = 3,4,15Hz, 0.6H), 4.22-4.27 (m, 0.4H), 4.41 (ddd, J = 5,5,14Hz, 0. 4H), 4.53-4.59 (m, 1H), 4.64-4.68 (m, 0.6H), 5.15 (br s, 1H), 6.53 (d, J = 8Hz) , 0.6H), 6.55 (d, J = 8Hz, 0.4H), 6.72 (d, J = 8Hz, 0.4H), 6.72 (d, J = 8Hz, 0.6H) , 7.23 (d, J = 15Hz, 0.4H), 7.26 (d, J = 15Hz, 0.6H), 7.55 (d, J = 15Hz, 0.6H), 7.62 ( d, J = 15Hz, 0.4H), 7.79 (s, 0.6H), 7.81 (s, 0.4H), 7.91 (s, 0.6H), 7.92 (s, 0.4H).
(実施例110)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(チアゾール-2-イル)プロプ-2-エン-1-オン(130)の合成
(Example 110)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,5,6,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (thiazole-2-yl) prop-2-en-1 -On (130) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.15(m,2H),0.45-0.53(m,2H),0.76-0.84(m,1H),1.10-1.76(m,6H),2.12-2.45(m,5H),2.60-2.67(m,1H),2.88-3.08(m,3H),3.52-3.68(m,1H),3.89(s,3H),3.96-4.03(m,0.5H),4.25-4.29(m,0.5H),4.34-4.40(m,0.5H),4.56-4.59(m,1H),4.72-4.76(m,0.5H),6.59(d,J=8Hz,0.5H),6.63(d,J=8Hz,0.5H),6.73(d,J=8Hz,0.5H),6.75(d,J=8Hz,0.5H),7.30-7.41(m,2H),7.63-7.90(m,2H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.15 (m, 2H), 0.45-0.53 (m, 2H), 0.76-0.84 (m) , 1H), 1.10-1.76 (m, 6H), 2.12-2.45 (m, 5H), 2.60-2.67 (m, 1H), 2.88-3.08 (M, 3H), 3.52-3.68 (m, 1H), 3.89 (s, 3H), 3.96-4.03 (m, 0.5H), 4.25-4.29 (M, 0.5H), 4.34-4.40 (m, 0.5H), 4.56-4.59 (m, 1H), 4.72-4.76 (m, 0.5H) , 6.59 (d, J = 8Hz, 0.5H), 6.63 (d, J = 8Hz, 0.5H), 6.73 (d, J = 8Hz, 0.5H), 6.75 ( d, J = 8Hz, 0.5H), 7.30-7.41 (m, 2H), 7.63-7.90 (m, 2H).
(実施例111)
(E)-1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-(チアゾール-2-イル)プロプ-2-エン-1-オン(131)の合成
(Example 111)
(E) -1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H -4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3- (thiazole-2-yl) prop-2-en-1 -On (131) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.14(m,2H),0.45-0.53(m,2H),0.75-0.83(m,1H),1.09-1.15(m,1H),1.25-1.75(m,5H),2.12-2.42(m,5H),2.60-2.66(m,1H),2.84-2.93(m,1H),2.97(d,J=18Hz,1H),3.03(d,J=6Hz,0.6H),3.06(d,J=6Hz,0.4H),3.48-3.66(m,1H),3.99-4.05(m,0.6H),4.22-4.24(m,0.4H),4.36-4.42(m,0.4H),4.54-4.57(m,1H),4.65-4.68(m,0.6H),6.54(d,J=8Hz,0.6H),6.55(d,J=8Hz,0.4H),6.71(d,J=8Hz,0.4H),6.72(d,J=8Hz,0.6H),7.36-7.43(m,2H),7.74(d,J=8Hz,0.6H),7.85(d,J=8Hz,0.4H),7.91-7.93(m,1H).
1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.14 (m, 2H), 0.45-0.53 (m, 2H), 0.75-0.83 (m) , 1H), 1.09-1.15 (m, 1H), 1.25-1.75 (m, 5H), 2.12-2.42 (m, 5H), 2.60-2.66 (M, 1H), 2.84-2.93 (m, 1H), 2.97 (d, J = 18Hz, 1H), 3.03 (d, J = 6Hz, 0.6H), 3.06 (D, J = 6Hz, 0.4H), 3.48-3.66 (m, 1H), 3.99-4.05 (m, 0.6H), 4.22-4.24 (m, 0.4H), 4.36-4.42 (m, 0.4H), 4.54-4.57 (m, 1H), 4.65-4.68 (m, 0.6H), 6. 54 (d, J = 8Hz, 0.6H), 6.55 (d, J = 8Hz, 0.4H), 6.71 (d, J = 8Hz, 0.4H), 6.72 (d, J) = 8Hz, 0.6H), 7.36-743 (m, 2H), 7.74 (d, J = 8Hz, 0.6H), 7.85 (d, J = 8Hz, 0.4H) , 7.91-7.93 (m, 1H).
(実施例112)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-ヒドロキシ-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-3-フェニルプロプ-2-イン-1-オン(130)の合成
(Example 112)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-hydroxy-1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8 -Synthesis of ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -3-phenylprop-2-in-1-one (130)
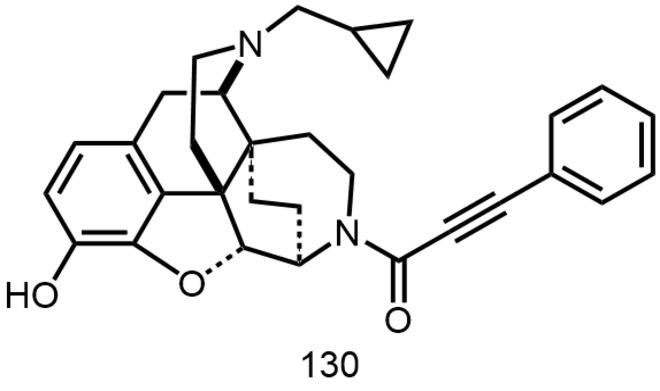
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.43-0.54(m,2H),0.74-0.84(m,1H),1.08-1.17(m,1H),1.25-1.35(m,1H),1.40-1.58(m,2H),1.66-1.84(m,2H),2.16-2.42(m,5H),2.60-2.68(m,1H),2.82-2.98(m,1H),2.97(d,J=18Hz,1H),3.06(d,J=6Hz,1H),3.38(ddd,J=5,11,15Hz,0.4H),3.70(ddd,J=4,11,15Hz,0.6H),4.36-4.45(m,1H),4.54-4.59(m,1.6H),4.64-4.66(m,0.4H),6.54(d,J=8Hz,0.6H),6.56(d,J=8Hz,0.4H),6.73(d,J=8Hz,1H),7.36-7.46(m,3H),7.55-7.63(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.43-0.54 (m, 2H), 0.74-0.84 (m) , 1H), 1.08-1.17 (m, 1H), 1.25-1.35 (m, 1H), 1.40-1.58 (m, 2H), 1.66-1.84 (M, 2H), 2.16-2.42 (m, 5H), 2.60-2.68 (m, 1H), 2.82-2.98 (m, 1H), 2.97 (d). , J = 18Hz, 1H), 3.06 (d, J = 6Hz, 1H), 3.38 (ddd, J = 5,11,15Hz, 0.4H), 3.70 (ddd, J = 4, 11,15Hz, 0.6H), 4.36-4.45 (m, 1H), 4.54-4.59 (m, 1.6H), 4.64-4.66 (m, 0.4H) ), 6.54 (d, J = 8Hz, 0.6H), 6.56 (d, J = 8Hz, 0.4H), 6.73 (d, J = 8Hz, 1H), 7.36-7. .46 (m, 3H), 7.55-7.63 (m, 2H).
(実施例113)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-7-(3-フェニルプロピル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン(133)の合成
(Example 113)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -10-methoxy-7- (3-phenylpropyl) -1,2,3,4,6,7,8,8a-octahydro Synthesis of -5H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine (133)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.42-0.54(m,2H),0.74-0.85(m,1H),0.91-1.01(m,1H),1.16-1.36(m,3H),1.49-1.55(m,1H),1.56-1.69(m,1H),1.76-1.85(m,2H),2.21(dd,J=7,13Hz,1H),2.24-2.43(m,4H),2.53(t,J=7Hz,2H),2.58-2.67(m,1H),2.64(t,J=7Hz,2H),2.68-2.91(m,4H),2.95(d,J=18Hz,1H),2.99(d,J=6Hz,1H),3.85(s,3H),4.55(s,1H),6.55(d,J=8Hz,1H),6.70(d,J=8Hz,1H),7.14-7.22(m,3H),7.24-7.32(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.42-0.54 (m, 2H), 0.74-0.85 (m) , 1H), 0.91-1.01 (m, 1H), 1.16-1.36 (m, 3H), 1.49-1.55 (m, 1H), 1.56-1.69 (M, 1H), 1.76-1.85 (m, 2H), 2.21 (dd, J = 7,13Hz, 1H), 2.24-2.43 (m, 4H), 2.53 (T, J = 7Hz, 2H), 2.58-2.67 (m, 1H), 2.64 (t, J = 7Hz, 2H), 2.68-2.91 (m, 4H), 2 .95 (d, J = 18Hz, 1H), 2.99 (d, J = 6Hz, 1H), 3.85 (s, 3H), 4.55 (s, 1H), 6.55 (d, J) = 8Hz, 1H), 6.70 (d, J = 8Hz, 1H), 7.14-7.22 (m, 3H), 7.24-7.32 (m, 2H).
(実施例114)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-7-(3-フェニルプロピル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-オール(134)の合成
(Example 114)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -7- (3-phenylpropyl) -1,2,3,4,6,7,8a-octahydro-5H-4a , 8-Etano-4,13-Metanobenzoflo [2,3-c] pyrido [4,3-d] Synthesis of azepine-10-ol (134)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.16(m,2H),0.42-0.54(m,2H),0.73-0.85(m,1H),0.90-0.99(m,1H),1.04-1.23(m,2H),1.24-1.36(m,1H),1.49-1.55(m,1H),1.58-1.68(m,1H),1.82(tt,J=7,7Hz,2H),2.20(dd,J=7,13Hz,1H),2.25-2.42(m,4H),2.55(t,J=7Hz,2H),2.57-2.68(m,3H),2.69-2.87(m,4H),2.93(d,J=18Hz,1H),2.99(d,J=6Hz,1H),4.56(s,1H),6.50(d,J=8Hz,1H),6.66(d,J=8Hz,1H),7.15-7.22(m,3H),7.24-7.32(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.06-0.16 (m, 2H), 0.42-0.54 (m, 2H), 0.73-0.85 (m) , 1H), 0.90-0.99 (m, 1H), 1.04-1.23 (m, 2H), 1.24-1.36 (m, 1H), 1.49-1.55 (M, 1H), 1.58-1.68 (m, 1H), 1.82 (tt, J = 7,7Hz, 2H), 2.20 (dd, J = 7,13Hz, 1H), 2 .25-2.42 (m, 4H), 2.55 (t, J = 7Hz, 2H), 2.57-2.68 (m, 3H), 2.69-2.87 (m, 4H) , 2.93 (d, J = 18Hz, 1H), 2.99 (d, J = 6Hz, 1H), 4.56 (s, 1H), 6.50 (d, J = 8Hz, 1H), 6 .66 (d, J = 8Hz, 1H), 7.15-7.22 (m, 3H), 7.24-7.32 (m, 2H).
(実施例115)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-7-(ピリミジン-2-イルメチル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン(135)の合成
(Example 115)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Methoxy-7- (Pyrimidine-2-ylmethyl) -1,2,3,4,6,7,8,8a- Synthesis of Octahydro-5H-4a, 8-Etano-4,13-Methylbenzoflo [2,3-c] Pyrimidine [4,3-d] Azepine (135)

化合物9(14mg,0.038mmol)のジクロロメタン(1mL)溶液に、ピリミジン-2-カルバルデヒド(20.6mg,0.19mmol)及び水素化トリアセトキシホウ素ナトリウム(40.5mg,0.19mmol)を加え、室温で二時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、クロロホルムで三回抽出した。合わせた抽出液を硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(0-5%メタノール/クロロホルム)で精製し、表題化合物(15.3mg,87%)を淡黄色油状物質として得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.14(m,2H),0.43-0.52(m,2H),0.75-0.85(m,1H),0.94-1.14(m,2H),1.20-1.29(m,1H),1.34-1.45(m,1H),1.52(ddd,J=3,13Hz,1H),1.66-1.75(m,1H),2.18-2.40(m,4H),2.47(ddd,J=6,13,13Hz,1H),2.62(dd,J=5,11Hz,1H),2.90-3.02(m,6H),3.81(s,3H),4.00(d,J=15Hz,1H),4.05(d,J=15Hz,1H),4.73(s,1H),6.55(d,J=8Hz,1H),6.67(d,J=8Hz,1H),7.17(t,J=5Hz,1H),8.72(d,J=5Hz,2H).
Pyrimidine-2-carbaldehyde (20.6 mg, 0.19 mmol) and sodium borohydride triacetoxyboron (40.5 mg, 0.19 mmol) are added to a solution of compound 9 (14 mg, 0.038 mmol) in dichloromethane (1 mL). , Stirred for 2 hours at room temperature. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted three times with chloroform. The combined extracts were dried over sodium sulfate and then concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (0-5% methanol / chloroform) to give the title compound (15.3 mg, 87%) as a pale yellow oily substance.
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.06-0.14 (m, 2H), 0.43-0.52 (m, 2H), 0.75-0.85 (m) , 1H), 0.94-1.14 (m, 2H), 1.20-1.29 (m, 1H), 1.34-1.45 (m, 1H), 1.52 (ddd, J) = 3,13Hz, 1H), 1.66-1.75 (m, 1H), 2.18-2.40 (m, 4H), 2.47 (ddd, J = 6,13,13Hz, 1H) , 2.62 (dd, J = 5,11Hz, 1H), 2.90-3.02 (m, 6H), 3.81 (s, 3H), 4.00 (d, J = 15Hz, 1H) , 4.05 (d, J = 15Hz, 1H), 4.73 (s, 1H), 6.55 (d, J = 8Hz, 1H), 6.67 (d, J = 8Hz, 1H), 7 .17 (t, J = 5Hz, 1H), 8.72 (d, J = 5Hz, 2H).
(実施例116)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-7-(ピリミジン-2-イルメチル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-オール(136)の合成
(Example 116)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -7- (Pyrimidine-2-ylmethyl) -1,2,3,4,6,7,8,8a-Octahydro-5H- Synthesis of 4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-10-ol (136)

1H-NMR(400MHz,CDCl3)δ(ppm):0.05-0.14(m,2H),0.40-0.53(m,2H),0.73-0.83(m,1H),0.91-1.07(m,2H),1.16-1.29(m,1H),1.32-1.42(m,1H),1.46-1.54(m,1H),1.61-1.71(m,1H),2.18-2.47(m,5H),2.60(dd,J=5,9Hz,1H),2.82-3.01(m,6H),4.01(d,J=15Hz,1H),4.04(d,J=15Hz,1H),4.68(s,1H),6.48(d,J=8Hz,1H),6.65(d,J=8Hz,1H),7.19(t,J=5Hz,1H),8.73(d,J=5Hz,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.05-0.14 (m, 2H), 0.40-0.53 (m, 2H), 0.73-0.83 (m) , 1H), 0.91-1.07 (m, 2H), 1.16-1.29 (m, 1H), 1.32-1.42 (m, 1H), 1.46-1.54 (M, 1H), 1.61-1.71 (m, 1H), 2.18-2.47 (m, 5H), 2.60 (dd, J = 5,9Hz, 1H), 2.82 -3.01 (m, 6H), 4.01 (d, J = 15Hz, 1H), 4.04 (d, J = 15Hz, 1H), 4.68 (s, 1H), 6.48 (d) , J = 8Hz, 1H), 6.65 (d, J = 8Hz, 1H), 7.19 (t, J = 5Hz, 1H), 8.73 (d, J = 5Hz, 2H).
(実施例117)
(4R,4aS,8R,8aR,13bS)-7-(5-ブロモピリミジン-2-イル)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン(137)の合成
(Example 117)
(4R, 4aS, 8R, 8aR, 13bS) -7- (5-bromopyrimidine-2-yl) -3- (cyclopropylmethyl) -10-methoxy-1,2,3,4,6,7,8 , 8a-Octahydro-5H-4a, 8-Etano-4,13-Metanobenzoflo [2,3-c] Pyrimidine [4,3-d] Synthesis of azepine (137)

化合物9(5.0mg,0.014mmol)のアセトニトリル(0.5mL)溶液に、N,N-ジイソプロピルエチルアミン(10.0μL,0.056mmol)及び5-ブロモ-2-クロロピリミジン(8.0mg,0.042mmol)を加え80°Cで16時間撹拌した。反応混合物に酢酸エチルを加え、飽和炭酸水素ナトリウム水溶液で三回洗浄した。有機層を硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(10-20%酢酸エチル/ヘプタン)で精製し、表題化合物(6.6mg,93%)を淡黄色アモルファスとして得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.20(m,2H),0.40-0.60(m,2H),0.70-0.90(m,1H),1.00-1.20(m,1H),1.20-1.80(m,5H),2.00-2.50(m,5H),2.59(dd,J=6,11Hz,1H),2.70-2.90(m,1H),2.98(d,J=18Hz,1H),3.04(d,J=6Hz,1H),3.40-3.55(m,1H),3.87(s,3H),4.47(s,1H),4.60-4.70(m,2H),6.58(d,J=8Hz,1H),6.72(d,J=8Hz,1H),8.28(s,2H).
N, N-diisopropylethylamine (10.0 μL, 0.056 mmol) and 5-bromo-2-chloropyrimidine (8.0 mg, 8.0 mg,) in an acetonitrile (0.5 mL) solution of compound 9 (5.0 mg, 0.014 mmol). 0.042 mmol) was added, and the mixture was stirred at 80 ° C. for 16 hours. Ethyl acetate was added to the reaction mixture, and the mixture was washed three times with saturated aqueous sodium hydrogen carbonate solution. The organic layer was dried over sodium sulfate and then concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (10-20% ethyl acetate / heptane) to give the title compound (6.6 mg, 93%) as a pale yellow amorphous substance.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.20 (m, 2H), 0.40-0.60 (m, 2H), 0.70-0.90 (m) , 1H), 1.00-1.20 (m, 1H), 1.20-1.80 (m, 5H), 2.00-2.50 (m, 5H), 2.59 (dd, J) = 6,11Hz, 1H), 2.70-2.90 (m, 1H), 2.98 (d, J = 18Hz, 1H), 3.04 (d, J = 6Hz, 1H), 3.40 -3.55 (m, 1H), 3.87 (s, 3H), 4.47 (s, 1H), 4.60-4.70 (m, 2H), 6.58 (d, J = 8Hz) , 1H), 6.72 (d, J = 8Hz, 1H), 8.28 (s, 2H).
(実施例118)
(4R,4aS,8R,8aR,13bS)-7-(5-ブロモピリミジン-2-イル)-3-(シクロプロピルメチル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-オール(138)の合成
(Example 118)
(4R, 4aS, 8R, 8aR, 13bS) -7- (5-bromopyrimidine-2-yl) -3- (cyclopropylmethyl) -1,2,3,4,6,7,8,8a-octahydro Synthesis of -5H-4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-10-ol (138)

1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.20(m,2H),0.40-0.60(m,2H),0.70-0.90(m,1H),1.00-2.40(m,11H),2.61(dd,J=4,11Hz,1H),2.70-2.90(m,1H),2.97(d,J=18Hz,1H),3.05(d,J=6Hz,1H),3.30-3.50(m,1H),4.45(s,1H),4.57(s,1H),4.70-4.90(m,1H),6.55(d,J=8Hz,1H),6.71(d,J=8Hz,1H),8.33(s,2H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.20 (m, 2H), 0.40-0.60 (m, 2H), 0.70-0.90 (m) , 1H), 1.00-2.40 (m, 11H), 2.61 (dd, J = 4,11Hz, 1H), 2.70-2.90 (m, 1H), 2.97 (d) , J = 18Hz, 1H), 3.05 (d, J = 6Hz, 1H), 3.30-3.50 (m, 1H), 4.45 (s, 1H), 4.57 (s, 1H) ), 4.70-4.90 (m, 1H), 6.55 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 8.33 (s, 2H).
(実施例119)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-7-(ピリミジン-2-イル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン(139)の合成
(Example 119)
(4R, 4aS, 8R, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Methoxy-7- (pyrimidine-2-yl) -1,2,3,4,6,7,8,8a- Synthesis of Octahydro-5H-4a, 8-Etano-4,13-Methylbenzoflo [2,3-c] Pyrimidine [4,3-d] Azepine (139)
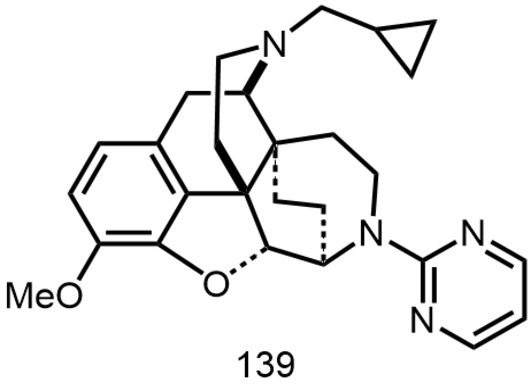
実施例117に記載した方法に従い、化合物9及び2-クロロピリミジンより表題化合物を得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.20(m,2H),0.40-0.60(m,2H),0.70-0.90(m,1H),1.00-1.20(m,1H),1.20-1.90(m,5H),2.10-2.50(m,5H),2.50-2.65(m,1H),2.70-2.90(m,1H),2.98(d,J=18Hz,1H),3.04(d,J=6Hz,1H),3.51(t,J=12Hz,1H),3.88(s,3H),4.53(s,1H),4.75(s,2H),6.45(t,J=5Hz,1H),6.58(d,J=8Hz,1H),6.73(d,J=8Hz,1H),8.30(d,J=5Hz,2H).
The title compound was obtained from compound 9 and 2-chloropyrimidine according to the method described in Example 117.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.00-0.20 (m, 2H), 0.40-0.60 (m, 2H), 0.70-0.90 (m) , 1H), 1.00-1.20 (m, 1H), 1.20-1.90 (m, 5H), 2.10-2.50 (m, 5H), 2.50-2.65 (M, 1H), 2.70-2.90 (m, 1H), 2.98 (d, J = 18Hz, 1H), 3.04 (d, J = 6Hz, 1H), 3.51 (t) , J = 12Hz, 1H), 3.88 (s, 3H), 4.53 (s, 1H), 4.75 (s, 2H), 6.45 (t, J = 5Hz, 1H), 6. 58 (d, J = 8Hz, 1H), 6.73 (d, J = 8Hz, 1H), 8.30 (d, J = 5Hz, 2H).
(実施例120)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-7-(ピリミジン-2-イル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-オール(140)の合成
(Example 120)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -7- (pyrimidine-2-yl) -1,2,3,4,6,7,8a-octahydro-5H- Synthesis of 4a, 8-ethano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-10-ol (140)

1H-NMR(400MHz,CDCl3)δ(ppm):0.00-0.20(m,2H),0.40-0.60(m,2H),0.70-0.90(m,1H),1.12(t,J=12Hz,1H),1.20-1.70(m,4H),1.79(t,J=11Hz,1H),2.10-2.50(m,5H),2.60(dd,J=6,10Hz,1H),2.83(dt,J=5,13Hz,1H),2.96(d,J=18Hz,1H),3.05(d,J=6Hz,1H),3.40-3.50(m,1H),4.50-4.60(m,1H),4.65(q,J=3Hz,1H),4.83(dq,J=3,14Hz,1H),6.48(t,J=5Hz,1H),6.54(d,J=8Hz,1H),6.71(d,J=8Hz,1H),8.35(d,J=5Hz,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.00-0.20 (m, 2H), 0.40-0.60 (m, 2H), 0.70-0.90 (m) , 1H), 1.12 (t, J = 12Hz, 1H), 1.20-1.70 (m, 4H), 1.79 (t, J = 11Hz, 1H), 2.10-2.50 (M, 5H), 2.60 (dd, J = 6,10Hz, 1H), 2.83 (dt, J = 5,13Hz, 1H), 2.96 (d, J = 18Hz, 1H), 3 0.05 (d, J = 6Hz, 1H), 3.40-3.50 (m, 1H), 4.50-4.60 (m, 1H), 4.65 (q, J = 3Hz, 1H) , 4.83 (dq, J = 3,14Hz, 1H), 6.48 (t, J = 5Hz, 1H), 6.54 (d, J = 8Hz, 1H), 6.71 (d, J = 8Hz, 1H), 8.35 (d, J = 5Hz, 2H).
(参考例21)
(4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-7-(2-(ピリミジン-2-イル)アセチル)-1,2,3,4,6,7,8,8a-オクタヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-10-イル トリフルオロメタンスルホネート(141)の合成
(Reference Example 21)
(4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -7- (2- (pyrimidine-2-yl) acetyl) -1,2,3,4,6,7,8,8a -Synthesis of Octahydro-5H-4a, 8-Etano-4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-10-yltrifluoromethanesulfonate (141)

1H-NMR(400MHz,CDCl3)δ(ppm):0.04-0.17(m,2H),0.42-0.58(m,2H),0.61-1.83(m,7H),2.09-3.14(m,9H),3.30-3.91(m,1.7H),4.00-4.49(m,2.3H),4.60-4.79(m,2H),6.64(d,J=8Hz,0.7H),6.69(d,J=8Hz,0.3H),6.98(d,J=8Hz,1H),7.12-7.41(m,1H),8.67-8.79(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.04-0.17 (m, 2H), 0.42-0.58 (m, 2H), 0.61-1.83 (m) , 7H), 2.09-3.14 (m, 9H), 3.30-3.91 (m, 1.7H), 4.00-4.49 (m, 2.3H), 4.60 -4.79 (m, 2H), 6.64 (d, J = 8Hz, 0.7H), 6.69 (d, J = 8Hz, 0.3H), 6.98 (d, J = 8Hz, 1H), 7.12-7.41 (m, 1H), 8.67-8.79 (m, 2H).
(実施例121)
1-((4R,4aS,8R,8aR,13bS)-3-(シクロプロピルメチル)-1,2,3,4,5,6,8,8a-オクタヒドロ-7H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-7-イル)-2-(ピリミジン-2-イル)エタン-1-オン(142)の合成
(Example 121)
1-((4R, 4aS, 8R, 8aR, 13bS) -3- (cyclopropylmethyl) -1,2,3,4,5,6,8,8a-octahydro-7H-4a, 8-ethano-4) , 13-Methylnobenzoflo [2,3-c] pyrido [4,3-d] azepine-7-yl) -2- (pyrimidine-2-yl) ethane-1-one (142) synthesis

1H-NMR(400MHz,CDCl3)δ(ppm):0.02-0.18(m,2H),0.38-0.57(m,2H),0.68-1.88(m,7H),2.06-3.12(m,9H),3.35-3.62(m,1H),3.67-3.87(m,0.7H),4.02-4.44(m,2.3H),4.45-4.69(m,2H),6.49-6.73(m,2H),6.99-7.11(m,1H),7.14-7.25(m,1H),8.65-8.81(m,2H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.02-0.18 (m, 2H), 0.38-0.57 (m, 2H), 0.68-1.88 (m) , 7H), 2.06-3.12 (m, 9H), 3.35-3.62 (m, 1H), 3.67-3.87 (m, 0.7H), 4.02-4 .44 (m, 2.3H), 4.45-4.69 (m, 2H), 6.49-6.73 (m, 2H), 6.99-7.11 (m, 1H), 7 .14-7.25 (m, 1H), 8.65-8.81 (m, 2H).
(参考例22)
(4R,4aS,7S,7aR,12bS)-3-(シクロプロピルメチル)-7,9-ジメトキシ-1,2,3,4,7,7a-ヘキサヒドロ-4a,7-エタノ-4,12-メタノベンゾフロ[3,2-e]イソキノリン-6(5H)-オン(143)の合成
(Reference example 22)
(4R, 4aS, 7S, 7aR, 12bS) -3- (Cyclopropylmethyl) -7,9-dimethoxy-1,2,3,4,7,7a-Hexahydro-4a, 7-Etano-4,12- Synthesis of methanobenzoflo [3,2-e] isoquinoline-6 (5H) -on (143)

1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.13(m,2H),0.45-0.55(m,2H),0.75-0.84(m,1H),0.97-1.06(m,1H),1.26(ddd,J=6,13,13Hz,1H),1.50-1.57(m,1H),1.68(dd,J=3,13Hz,1H),1.71-1.81(m,1H),2.02(ddd,J=6,13,13Hz,1H),2.22(d,J=20Hz,1H),2.30-2.41(m,4H),2.71(dd,J=6,12Hz,1H),3.07(d,J=18Hz,1H),3.13(d,J=7Hz,1H),3.51(dd,J=4,20Hz,1H),3.53(s,3H),3.89(s,3H),4.60(d,J=2Hz,1H),6.63(d,J=8Hz,1H),6.76(d,J=8Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.07-0.13 (m, 2H), 0.45-0.55 (m, 2H), 0.75-0.84 (m) , 1H), 0.97-1.06 (m, 1H), 1.26 (ddd, J = 6,13,13Hz, 1H), 1.50-1.57 (m, 1H), 1.68 (Dd, J = 3,13Hz, 1H), 1.71-1.81 (m, 1H), 2.02 (ddd, J = 6,13,13Hz, 1H), 2.22 (d, J = 20Hz, 1H), 2.30-2.41 (m, 4H), 2.71 (dd, J = 6,12Hz, 1H), 3.07 (d, J = 18Hz, 1H), 3.13 ( d, J = 7Hz, 1H), 3.51 (dd, J = 4,20Hz, 1H), 3.53 (s, 3H), 3.89 (s, 3H), 4.60 (d, J = 2Hz, 1H), 6.63 (d, J = 8Hz, 1H), 6.76 (d, J = 8Hz, 1H).
(参考例23)
(4R,4aS,7R,7aR,12bS)-3-(シクロプロピルメチル)-7,9-ジメトキシ-1,2,3,4,7,7a-ヘキサヒドロ-4a,7-エタノ-4,12-メタノベンゾフロ[3,2-e]イソキノリン-6(5H)-オン オキシム(144)の合成
(Reference example 23)
(4R, 4aS, 7R, 7aR, 12bS) -3- (Cyclopropylmethyl) -7,9-dimethoxy-1,2,3,4,7,7a-Hexahydro-4a, 7-Etano-4,12- Synthesis of methanobenzoflo [3,2-e] isoquinoline-6 (5H) -one oxime (144)

1H-NMR(400MHz,CDCl3)δ(ppm):0.08-0.15(m,2H),0.46-0.56(m,2H),0.80-0.95(m,2H),110-1.20(m,1H),1.55-1.74(m,3H),2.01(ddd,J=6,13,13Hz,1H),2.26-2.38(m,5H),2.69(dd,J=6,12Hz,1H),3.04(d,J=18Hz,1H),3.15(d,J=6Hz,1H),3.55(s,3H),3.70(dd,J=4,21Hz,1H),3.89(s,3H),4.60(d,J=2Hz,1H),6.60(d,J=8Hz,1H),6.73(d,J=8Hz,1H),8.30(br s,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.08-0.15 (m, 2H), 0.46-0.56 (m, 2H), 0.80-0.95 (m) , 2H), 110-1.20 (m, 1H), 1.55-1.74 (m, 3H), 2.01 (ddd, J = 6,13,13Hz, 1H), 2.26-2 .38 (m, 5H), 2.69 (dd, J = 6,12Hz, 1H), 3.04 (d, J = 18Hz, 1H), 3.15 (d, J = 6Hz, 1H), 3 .55 (s, 3H), 3.70 (dd, J = 4,21Hz, 1H), 3.89 (s, 3H), 4.60 (d, J = 2Hz, 1H), 6.60 (d) , J = 8Hz, 1H), 6.73 (d, J = 8Hz, 1H), 8.30 (br s, 1H).
(参考例24)
2-((4R,4aS,7aR,12bS)-3-(シクロプロピルメチル)-7,9-ジメトキシ-1,2,3,4,5,7a-ヘキサヒドロ-4aH-4,12-メタノベンゾフロ[3,2-e]イソキノリン-4a-イル)アセトニトリル(145)の合成
(Reference Example 24)
2-((4R, 4aS, 7aR, 12bS) -3- (cyclopropylmethyl) -7,9-dimethoxy-1,2,3,4,5,7a-hexahydro-4aH-4,12-methanobenzoflo [3] , 2-e] Isoquinoline-4a-yl) Synthesis of acetonitrile (145)

1H-NMR(400MHz,CDCl3)δ(ppm):0.08-0.18(m,2H),0.50-0.61(m,2H),0.86-0.95(m,1H),1.55-1.62(m,1H),1.90-2.01(m,2H),2.11(dd,J=6,16Hz,1H),2.20(ddd,J=4,12,12Hz,1H),2.31(dd,J=7,13Hz,1H),2.40(dd,J=6,13Hz,1H),2.53(dd,J=6,18Hz,1H),2.61(d,J=16Hz,1H),2.67(dd,J=5,12Hz,1H),3.01(d,J=18Hz,1H),3.47(d,J=6Hz,1H),3.52(s,3H),3.71(dd,J=2,16Hz,1H),3.84(s,3H),4.67-4.71(m,1H),4.71(d,J=1Hz,1H),6.63(d,J=8Hz,1H),6.72(d,J=8Hz,1H).
1 1 H-NMR (400 MHz, CDCl 3 ) δ (ppm): 0.08-0.18 (m, 2H), 0.50-0.61 (m, 2H), 0.86-0.95 (m) , 1H), 1.55-1.62 (m, 1H), 1.90-2.01 (m, 2H), 2.11 (dd, J = 6,16Hz, 1H), 2.20 (ddd) , J = 4,12,12Hz, 1H), 2.31 (dd, J = 7,13Hz, 1H), 2.40 (dd, J = 6,13Hz, 1H), 2.53 (dd, J = 6,18Hz, 1H), 2.61 (d, J = 16Hz, 1H), 2.67 (dd, J = 5,12Hz, 1H), 3.01 (d, J = 18Hz, 1H), 3. 47 (d, J = 6Hz, 1H), 3.52 (s, 3H), 3.71 (dd, J = 2,16Hz, 1H), 3.84 (s, 3H), 4.67-4. 71 (m, 1H), 4.71 (d, J = 1Hz, 1H), 6.63 (d, J = 8Hz, 1H), 6.72 (d, J = 8Hz, 1H).
(実施例122)
(4R,4aS,8S,8aR,13bS)-3-(シクロプロピルメチル)-8-ヒドロキシ-10-メトキシ-1,2,3,4,8,8a-ヘキサヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-6(7H)-オン(146)の合成
(Example 122)
(4R, 4aS, 8S, 8aR, 13bS) -3- (Cyclopropylmethyl) -8-hydroxy-10-methoxy-1,2,3,4,8,8a-hexahydro-5H-4a, 8-ethano- Synthesis of 4,13-methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-6 (7H) -on (146)
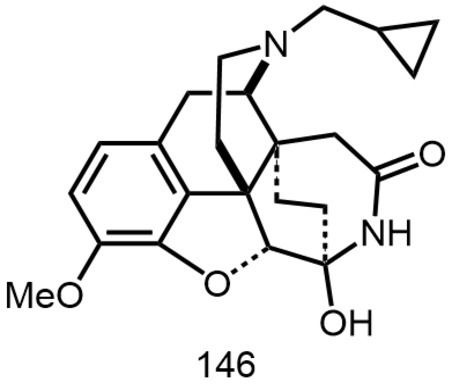
化合物4(219.6mg,0.58mmol)の6M塩酸(5mL)溶液を、70℃で1時間撹拌した。反応混合物を放冷後、飽和炭酸水素ナトリウム水溶液に注ぎ塩基性とし、酢酸エチルで三回抽出した。合わせた抽出物を硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(0-4%メタノール/クロロホルム)で精製し、表題化合物(120.9mg,53%)を無色固体として得た。
(方法b)
化合物145(30mg,0.076mmol)のテトラヒドロフラン(1mL)溶液に、濃塩酸(1mL)を加え、室温で1時間及び50℃で2時間撹拌した。反応混合物を放冷後、飽和炭酸水素ナトリウム水溶液に注ぎ塩基性とし、酢酸エチルで三回抽出した。合わせた抽出物を硫酸ナトリウムで乾燥後、減圧下にて濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(0-6%メタノール/クロロホルム)で精製し、表題化合物(24.8mg,82%)を無色固体として得た。
1H-NMR(400MHz,CDCl3)δ(ppm):0.07-0.13(m,2H),0.47-0.54(m,2H),0.77-0.84(m,1H),1.23-1.50(m,3H),1.56-1.63(m,1H),1.79-1.87(m,1H),2.19-2.26(m,2H),2.27-2.30(m,2H),2.37(d,J=19Hz,1H),2.47(dd,J=6,18Hz,1H),2.63-2.72(m,1H),2.98(d,J=20Hz,1H),3.01(d,J=7Hz,1H),3.36(s,1H),3.88(s,3H),4.03(dd,J=3,20Hz,1H),4.41(d,J=1Hz,1H),6.12(br s,1H),6.66(d,J=8Hz,1H),6.77(d,J=8Hz,1H).
A 6M hydrochloric acid (5 mL) solution of compound 4 (219.6 mg, 0.58 mmol) was stirred at 70 ° C. for 1 hour. The reaction mixture was allowed to cool, poured into saturated aqueous sodium hydrogen carbonate solution to make it basic, and extracted three times with ethyl acetate. The combined extracts were dried over sodium sulfate and then concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (0-4% methanol / chloroform) to give the title compound (120.9 mg, 53%) as a colorless solid.
(Method b)
Concentrated hydrochloric acid (1 mL) was added to a solution of compound 145 (30 mg, 0.076 mmol) in tetrahydrofuran (1 mL), and the mixture was stirred at room temperature for 1 hour and at 50 ° C. for 2 hours. The reaction mixture was allowed to cool, poured into saturated aqueous sodium hydrogen carbonate solution to make it basic, and extracted three times with ethyl acetate. The combined extracts were dried over sodium sulfate and then concentrated under reduced pressure. The obtained crude product was purified by silica gel column chromatography (0-6% methanol / chloroform) to give the title compound (24.8 mg, 82%) as a colorless solid.
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.07-0.13 (m, 2H), 0.47-0.54 (m, 2H), 0.77-0.84 (m) , 1H), 1.23-1.50 (m, 3H), 1.56-1.63 (m, 1H), 1.79-1.87 (m, 1H), 2.19-2.26 (M, 2H), 2.27-2.30 (m, 2H), 2.37 (d, J = 19Hz, 1H), 2.47 (dd, J = 6,18Hz, 1H), 2.63 -2.72 (m, 1H), 2.98 (d, J = 20Hz, 1H), 3.01 (d, J = 7Hz, 1H), 3.36 (s, 1H), 3.88 (s) , 3H), 4.03 (dd, J = 3,20Hz, 1H), 4.41 (d, J = 1Hz, 1H), 6.12 (br s, 1H), 6.66 (d, J = 8Hz, 1H), 6.77 (d, J = 8Hz, 1H).
(実施例123)
(4R,4aS,8S,8aR,13bS)-3-(シクロプロピルメチル)-10-メトキシ-1,2,3,4,6,7-ヘキサヒドロ-5H-4a,8-エタノ-4,13-メタノベンゾフロ[2,3-c]ピリド[4,3-d]アゼピン-8(8aH)-オール(147)の合成
(Example 123)
(4R, 4aS, 8S, 8aR, 13bS) -3- (Cyclopropylmethyl) -10-Methoxy-1,2,3,4,6,7-Hexahydro-5H-4a, 8-Etano-4,13- Synthesis of methanobenzoflo [2,3-c] pyrido [4,3-d] azepine-8 (8aH) -ol (147)

1H-NMR(400MHz,CDCl3)δ(ppm):0.06-0.15(m,2H),0.44-0.53(m,2H),0.73-0.82(m,1H),1.03-1.12(m,1H),1.24-1.37(m,3H),1.53-1.81(m,3H),2.17-2.39(m,5H),2.58-2.65(m,1H),2.69-2.79(m,2H),2.95(d,J=18Hz,1H),2.98-3.05(m,2H),3.16-3.27(m,1H),3.86(s,3H),4.17(d,J=2Hz,1H),6.60(d,J=8Hz,1H),6.73(d,J=8Hz,1H).
1 1 H-NMR (400MHz, CDCl 3 ) δ (ppm): 0.06-0.15 (m, 2H), 0.44-0.53 (m, 2H), 0.73-0.82 (m) , 1H), 1.03-1.12 (m, 1H), 1.24-1.37 (m, 3H), 1.53-1.81 (m, 3H), 2.17-2.39 (M, 5H), 2.58-2.65 (m, 1H), 2.69-2.79 (m, 2H), 2.95 (d, J = 18Hz, 1H), 2.98-3 0.05 (m, 2H), 3.16-3.27 (m, 1H), 3.86 (s, 3H), 4.17 (d, J = 2Hz, 1H), 6.60 (d, J) = 8Hz, 1H), 6.73 (d, J = 8Hz, 1H).
(試験例1)
オピオイド受容体機能試験
本発明化合物のκオピオイド受容体に対する機能活性を調べた。
方法:Lance Ultra cAMP kit(パーキンエルマー社)を用い、所定の方法に従って実施した。アゴニスト活性の評価では、ヒトオピオイドκ受容体発現CHO細胞(Catalog No.CT6606,accession No.NM_000912)と被験化合物を10μM フォルスコリン存在下にて、アッセイバッファー(1×HBSS,5mM HEPES,pH7.4,0.5mM IBMX(Isobutylmethylxanthine),0.1% BSA)中で30分間反応させた。続けて、キット中のcAMP検出試薬を添加し、1時間後にEnVisionプレートリーダー(パーキンエルマー社)を用いて時間分解蛍光測定を行った。被験化合物の評価は、κオピオイド受容体機能試験は10-14~10-7Mの濃度範囲で実施した。
(Test Example 1)
Opioid receptor function test
The functional activity of the compound of the present invention on the kappa opioid receptor was investigated.
Method: A Lance Ultra cAMP kit (PerkinElmer) was used, and the procedure was carried out according to a predetermined method. In the evaluation of agonist activity, human opioid κ receptor-expressing CHO cells (Catalog No. CT6666, accession No. NM_000912) and the test compound were used in the presence of 10 μM forskolin in an assay buffer (1 × HBSS, 5 mM HEPES, pH 7.4). , 0.5 mM IBMX (Isobutylmethylxanthine), 0.1% BSA) for 30 minutes. Subsequently, the cAMP detection reagent in the kit was added, and one hour later, time-resolved fluorescence measurement was performed using an EnVision plate reader (PerkinElmer). The evaluation of the test compound was carried out in the kappa opioid receptor function test in the concentration range of 10-14 to 10-7 M.

(試験例2)
代謝安定性試験
(試験方法)
ヒト肝ミクロソームと被験物質を一定時間 (0~60分) 反応させ、反応試料中の被験物質の未変化体残存量を測定し、残存率を求めた。反応時間0時間における未変化体残存率を100%とし、インキュベーション後の残存率を時間に対してlog-linearプロットし、回帰直線(y=100e-kt、k=直線の傾き:消失速度定数)を求め、以下の式を用いて代謝クリアランスCLint(mL/min/kg)を算出した。
また、比較化合物として段落番号[0007]記載の化合物(C)を用い比較を行った。比較化合物(C)は非特許文献1等に記載の方法により合成した。
CLint*=k(-min)×45(mg MS protein/g liver)×21(g liver/kg)/MS protein(mg MS protein/mL)
*:Yamazaki S.;Skaptason J.; Romero D, Vekich S.;Jones HM.;Tan W.;Wilner KD.;Koudriakova T. Drug Metab.Dispos.2011 Mar;39(3):383-93.
(試験結果)
試験結果を表2に示す。
(Test Example 2)
Metabolic stability test
(Test method)
Human liver microsomes and the test substance were reacted for a certain period of time (0 to 60 minutes), and the residual amount of the test substance in the reaction sample was measured to determine the residual rate. The residual rate of the unchanged form at 0 hours of reaction time is set to 100%, the residual rate after incubation is log-liner plotted with respect to time, and the regression line (y = 100e- kt , k = slope of a line: disappearance rate constant). Was calculated, and the metabolic clearance CL int (mL / min / kg) was calculated using the following formula.
Further, as a comparative compound, the compound (C) described in paragraph number [0007] was used for comparison. The comparative compound (C) was synthesized by the method described in Non-Patent Document 1 and the like.
CL int * = k (-min) x 45 (mg MS protein / g liver) x 21 (g liver / kg) / MS protein (mg MS protein / mL)
*: Yamazaki S. Snaptason J. et al. Romero D, Vekich S. Jones HM. Tan W. Wilner KD. Koudria kova T. Drug Metab. Dispos. 2011 Mar; 39 (3): 383-93.
(Test results)
The test results are shown in Table 2.

Claims (7)

R2及びR3は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基又はR2及びR3が一緒になってカルボニル基を示すかR2及びR3が結合して置換基を有していてもよい環状ケタールを示し、
R4及びR5は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、置換基を有していてもよいアミノ基、ハロゲン原子又はヒドロキシ基を示し、
R6は水素原子、置換基を有していてもよいC1-6アルコキシ基、置換基を有していてもよいアミノ基、ハロゲン原子、ヒドロキシ基、シアノ基、カルボキシ基、カルボン酸エステル基又は置換基を有していてもよいカルバモイル基を示し、
R7は水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基又はヒドロキシ基を示し、
R8及びR9は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基又はR8及びR9が一緒になってカルボニル基、チオカルボニル基を示すかR8及びR9が結合して置換基を有していてもよいC3-6飽和炭化水素環又は置換基を有していてもよい環状ケタールを示し、
R10及びR11は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC1-6アルコキシ基、ハロゲン原子、ヒドロキシ基又はR10及びR11が一緒になってカルボニル基、チオカルボニル基を示すかR10及びR11が結合して置換基を有していてもよいC3-6飽和炭化水素環又は置換基を有していてもよい環状ケタールを示し、
R12及びR13は同一又は異なって水素原子、置換基を有していてもよいC1-6アルキル基を示すか、同一炭素上のR12とR13が結合して置換基を有していてもよいC3-6飽和炭化水素環、置換基を有していてもよい飽和複素環を示すか、nが2~3の場合において隣接する一組のR12同士が結合して置換基を有していてもよいC3-6飽和炭化水素環又は置換基を有していてもよい飽和複素環を形成することができ、
R14は水素原子、置換基を有していてもよいC1-6アルキル基、置換基を有していてもよいC3-6シクロアルキル基、置換基を有していてもよいC2-4アルケニル基、置換基を有していてもよいC2-4アルキニル基、置換基を有していてもよいC1-6アルコキシ基、置換基を有していてもよいアミノ基、ハロゲン原子、ヒドロキシ基、置換基を有していてもよいC6-10アリール基、置換基を有していてもよいヘテロアリール基、置換基を有していてもよい環状アミノ基を示し、
Xは窒素原子又はN-オキシドを示し、
Yは酸素原子又は硫黄原子を示し、
ZはNR15、酸素原子、結合手、―CH=CH―又は―C≡C―を示し、
R15は水素原子、置換基を有していてもよいC1-6アルキル基を示すか、R15とR14が結合して置換基を有していてもよい含窒素飽和複素環を示し、
実線と破線からなる二重線は単結合又は二重結合を示し
mは0~1の整数を示し、
nは0~3の整数を示す。)
で表されるモルヒナン誘導体、該化合物の互変異性体、立体異性体、若しくはその薬学的に許容される塩又はそれらの溶媒和物。 The following general formula (I),
R 2 and R 3 are the same or different hydrogen atom, C 1-6 alkyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, a halogen atom and a hydroxy group. Alternatively, R 2 and R 3 together indicate a carbonyl group or R 2 and R 3 may be attached to indicate a cyclic ketal which may have a substituent.
R 4 and R 5 have the same or different hydrogen atom, a C 1-6 alkyl group which may have a substituent, a C 1-6 alkoxy group which may have a substituent, and a substituent. Indicates an amino group, a halogen atom or a hydroxy group which may be present.
R 6 is a hydrogen atom, a C 1-6 alkoxy group which may have a substituent, an amino group which may have a substituent, a halogen atom, a hydroxy group, a cyano group, a carboxy group and a carboxylic acid ester group. Alternatively, the carbamoyl group which may have a substituent is shown.
R 7 represents a hydrogen atom, a C 1-6 alkyl group which may have a substituent, a C 1-6 alkoxy group which may have a substituent, or a hydroxy group.
R 8 and R 9 are the same or different hydrogen atoms, C 1-6 alkyl groups which may have substituents, C 1-6 alkoxy groups which may have substituents, halogen atoms, hydroxy groups. Alternatively, R 8 and R 9 may be combined to indicate a carbonyl group, a thiocarbonyl group, or R 8 and R 9 may be bonded to have a substituent C 3-6 saturated hydrocarbon ring or substituent. Indicates a ring-shaped ketal that may have,
R 10 and R 11 are the same or different hydrogen atom, C 1-6 alkyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, a halogen atom and a hydroxy group. Alternatively, R 10 and R 11 together indicate a carbonyl group, a thiocarbonyl group, or R 10 and R 11 may be bonded to have a substituent C 3-6 saturated hydrocarbon ring or substituent. Indicates a ring-shaped ketal that may have,
R 12 and R 13 indicate the same or different C 1-6 alkyl groups that may have the same or different hydrogen atoms, substituents, or R 12 and R 13 on the same carbon have a substituent. It indicates a C 3-6 saturated hydrocarbon ring which may be present, a saturated heterocyclic ring which may have a substituent, or when n is 2 to 3, adjacent sets of R12 are bonded to each other and substituted. It is possible to form a C 3-6 saturated hydrocarbon ring which may have a group or a saturated heterocyclic ring which may have a substituent.
R 14 is a hydrogen atom, a C 1-6 alkyl group which may have a substituent, a C 3-6 cycloalkyl group which may have a substituent, and C 2 which may have a substituent. -4 Alkenyl group, C 2-4 alkynyl group which may have a substituent, C 1-6 alkoxy group which may have a substituent, amino group which may have a substituent, halogen. An atom, a hydroxy group, a C 6-10 aryl group which may have a substituent, a heteroaryl group which may have a substituent, and a cyclic amino group which may have a substituent are shown.
X represents a nitrogen atom or N-oxide,
Y represents an oxygen atom or a sulfur atom.
Z indicates NR 15 , oxygen atom, bond, —CH = CH— or —C≡C—,
R 15 indicates a hydrogen atom, a C 1-6 alkyl group which may have a substituent, or indicates a nitrogen-containing saturated heterocycle in which R 15 and R 14 may be bonded and have a substituent. ,
A double line consisting of a solid line and a broken line indicates a single bond or a double bond, and m indicates an integer of 0 to 1.
n represents an integer of 0 to 3. )
A morphinan derivative represented by, a tautomer of the compound, a stereoisomer, a pharmaceutically acceptable salt thereof, or a solvate thereof.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018198525A JP2022017604A (en) | 2018-10-22 | 2018-10-22 | Morphinan derivative |
| PCT/JP2019/041076 WO2020085234A1 (en) | 2018-10-22 | 2019-10-18 | Morphinan derivative |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018198525A JP2022017604A (en) | 2018-10-22 | 2018-10-22 | Morphinan derivative |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2022017604A true JP2022017604A (en) | 2022-01-26 |
Family
ID=70331412
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018198525A Pending JP2022017604A (en) | 2018-10-22 | 2018-10-22 | Morphinan derivative |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2022017604A (en) |
| WO (1) | WO2020085234A1 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20220071223A (en) * | 2019-09-30 | 2022-05-31 | 닛뽕 케미파 가부시키가이샤 | azepane derivatives |
| WO2022127320A1 (en) * | 2020-12-18 | 2022-06-23 | 四川大学 | Intermediate, and preparation method therefor and application thereof |
| WO2022210479A1 (en) * | 2021-03-29 | 2022-10-06 | 日本ケミファ株式会社 | USE OF AZEPANE DERIVATIVE FOR TREATMENT, AMELIORATION OR PREVENTION OF κ-OPIOID RECEPTOR-RELATED DISEASES |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5252391B2 (en) * | 2008-02-21 | 2013-07-31 | 学校法人北里研究所 | Morphinane derivative having oxabicyclo [2.2.2] octane and pharmaceutical use thereof |
| CN104395317A (en) * | 2012-05-02 | 2015-03-04 | 南方研究所 | Heterocycle-fused morphinans, use thereof and preparation thereof |
-
2018
- 2018-10-22 JP JP2018198525A patent/JP2022017604A/en active Pending
-
2019
- 2019-10-18 WO PCT/JP2019/041076 patent/WO2020085234A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020085234A1 (en) | 2020-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11161847B2 (en) | KRAS mutant protein inhibitors | |
| JP6204484B2 (en) | Heteroaryl-substituted pyridyl compounds useful as kinase modulators | |
| AU2022204042A1 (en) | BRM targeting compounds and associated methods of use | |
| JP7307734B2 (en) | Aminopyrrolotriazines as Kinase Inhibitors | |
| JP2019532995A (en) | Tau protein targeted PROTAC and related methods of use | |
| KR20220102156A (en) | A bifunctional molecule containing an E3 ubiquitin ligase binding moiety linked to a BCL6 targeting moiety | |
| KR20200101423A (en) | Sulfonyl urea derivatives as NLRP3 inplasmasome modulators | |
| JP7471348B2 (en) | Bruton's tyrosine kinase inhibitors and methods of use thereof | |
| CN113286794A (en) | KRAS mutein inhibitors | |
| EA039808B1 (en) | Aminotriazolopyridines as kinase inhibitors | |
| CN114423751B (en) | Novel heterocyclic compounds useful as selective AURORA a inhibitors | |
| CN112236423A (en) | Piperidinyl-3- (aryloxy) propanamides and propionates | |
| KR20210106441A (en) | cyclic urea | |
| JP2019523266A (en) | Pharmaceutical compounds | |
| JP2022017604A (en) | Morphinan derivative | |
| US20230088694A1 (en) | Cycloalkylurea derivative | |
| US11352357B2 (en) | Cycloalkylurea derivative | |
| TW202313598A (en) | Phenyl urea derivative | |
| JP2023540548A (en) | Compounds with antitumor activity and their uses | |
| WO2021065898A1 (en) | Azepan derivative | |
| EP3515902A1 (en) | 6-membered cyclic amines or lactames substituted with urea and phenyl | |
| JPWO2019045006A1 (en) | Morhinan derivative | |
| WO2021132524A1 (en) | Epoxy azepan derivative | |
| WO2023015164A1 (en) | Cyanopyridine and cyanopyrimidine bcl6 degraders | |
| JP2023509495A (en) | RORγt INHIBITOR, PRODUCTION METHOD AND USE THEREOF |