JP2021530554A - Methods and ACVR1 Inhibitors for Treatment of Diseases with Abnormal ACVR1 Expression - Google Patents
Methods and ACVR1 Inhibitors for Treatment of Diseases with Abnormal ACVR1 Expression Download PDFInfo
- Publication number
- JP2021530554A JP2021530554A JP2021503828A JP2021503828A JP2021530554A JP 2021530554 A JP2021530554 A JP 2021530554A JP 2021503828 A JP2021503828 A JP 2021503828A JP 2021503828 A JP2021503828 A JP 2021503828A JP 2021530554 A JP2021530554 A JP 2021530554A
- Authority
- JP
- Japan
- Prior art keywords
- dose
- compound
- per week
- pharmaceutical composition
- acvr1
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 413
- 101000799140 Homo sapiens Activin receptor type-1 Proteins 0.000 title claims abstract description 267
- 102100034111 Activin receptor type-1 Human genes 0.000 title claims abstract description 255
- 238000011282 treatment Methods 0.000 title claims abstract description 94
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title claims abstract description 65
- 201000010099 disease Diseases 0.000 title claims abstract description 46
- 230000002159 abnormal effect Effects 0.000 title claims description 6
- 239000003112 inhibitor Substances 0.000 title abstract description 201
- 230000014509 gene expression Effects 0.000 title description 39
- 150000003839 salts Chemical class 0.000 claims abstract description 200
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 161
- 230000035772 mutation Effects 0.000 claims abstract description 147
- 101150045885 Acvr1 gene Proteins 0.000 claims abstract description 38
- 206010018338 Glioma Diseases 0.000 claims abstract description 32
- 208000032612 Glial tumor Diseases 0.000 claims abstract description 31
- 230000000750 progressive effect Effects 0.000 claims abstract description 16
- 239000000203 mixture Substances 0.000 claims description 144
- 150000001875 compounds Chemical class 0.000 claims description 134
- 239000008194 pharmaceutical composition Substances 0.000 claims description 134
- 150000003840 hydrochlorides Chemical group 0.000 claims description 100
- -1 2,2'-bipyridine-3-yl Chemical group 0.000 claims description 70
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 63
- XJOTXKZIRSHZQV-RXHOOSIZSA-N (3S)-3-amino-4-[[(2S,3R)-1-[[(2S)-1-[[(2S)-1-[(2S)-2-[[(2S,3S)-1-[[(1R,6R,12R,17R,20S,23S,26R,31R,34R,39R,42S,45S,48S,51S,59S)-51-(4-aminobutyl)-31-[[(2S)-6-amino-1-[[(1S,2R)-1-carboxy-2-hydroxypropyl]amino]-1-oxohexan-2-yl]carbamoyl]-20-benzyl-23-[(2S)-butan-2-yl]-45-(3-carbamimidamidopropyl)-48-(hydroxymethyl)-42-(1H-imidazol-4-ylmethyl)-59-(2-methylsulfanylethyl)-7,10,19,22,25,33,40,43,46,49,52,54,57,60,63,64-hexadecaoxo-3,4,14,15,28,29,36,37-octathia-8,11,18,21,24,32,41,44,47,50,53,55,58,61,62,65-hexadecazatetracyclo[32.19.8.26,17.212,39]pentahexacontan-26-yl]amino]-3-methyl-1-oxopentan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-4-oxobutanoic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@@H]1CCCN1C(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](Cc1cnc[nH]1)NC(=O)[C@@H](NC(=O)[C@@H](N)CC(O)=O)[C@@H](C)O)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@@H]2CSSC[C@@H]3NC(=O)[C@@H]4CSSC[C@H](NC(=O)[C@H](Cc5ccccc5)NC(=O)[C@@H](NC1=O)[C@@H](C)CC)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc1cnc[nH]1)NC3=O)C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N2)C(=O)NCC(=O)N4)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O XJOTXKZIRSHZQV-RXHOOSIZSA-N 0.000 claims description 62
- 102000018511 hepcidin Human genes 0.000 claims description 62
- 108060003558 hepcidin Proteins 0.000 claims description 62
- 229940066919 hepcidin Drugs 0.000 claims description 62
- 239000003814 drug Substances 0.000 claims description 56
- 239000002253 acid Substances 0.000 claims description 55
- 238000009472 formulation Methods 0.000 claims description 55
- 239000012458 free base Substances 0.000 claims description 55
- 239000007787 solid Substances 0.000 claims description 53
- 239000000872 buffer Substances 0.000 claims description 41
- 230000036961 partial effect Effects 0.000 claims description 40
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical group Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 39
- 102000004338 Transferrin Human genes 0.000 claims description 37
- 108090000901 Transferrin Proteins 0.000 claims description 37
- 239000012581 transferrin Substances 0.000 claims description 37
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 36
- 239000007788 liquid Substances 0.000 claims description 36
- 229940124597 therapeutic agent Drugs 0.000 claims description 36
- 239000002904 solvent Substances 0.000 claims description 34
- 108090000623 proteins and genes Proteins 0.000 claims description 33
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 31
- 230000005764 inhibitory process Effects 0.000 claims description 25
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 claims description 24
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 24
- 239000003755 preservative agent Substances 0.000 claims description 24
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims description 22
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid group Chemical group C(CC(O)(C(=O)O)CC(=O)O)(=O)O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 claims description 21
- 239000000843 powder Substances 0.000 claims description 21
- 238000002360 preparation method Methods 0.000 claims description 21
- 102200104800 rs121912678 Human genes 0.000 claims description 21
- 230000003442 weekly effect Effects 0.000 claims description 20
- 208000035475 disorder Diseases 0.000 claims description 19
- 230000004580 weight loss Effects 0.000 claims description 19
- 239000005711 Benzoic acid Substances 0.000 claims description 18
- 229920000858 Cyclodextrin Polymers 0.000 claims description 18
- 235000010233 benzoic acid Nutrition 0.000 claims description 18
- 230000005855 radiation Effects 0.000 claims description 18
- 230000004083 survival effect Effects 0.000 claims description 18
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 claims description 16
- 239000007903 gelatin capsule Substances 0.000 claims description 16
- 239000001630 malic acid Substances 0.000 claims description 16
- 235000011090 malic acid Nutrition 0.000 claims description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 15
- 239000001116 FEMA 4028 Substances 0.000 claims description 15
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 15
- 239000004376 Sucralose Substances 0.000 claims description 15
- 229960004853 betadex Drugs 0.000 claims description 15
- 239000000796 flavoring agent Substances 0.000 claims description 15
- 235000013355 food flavoring agent Nutrition 0.000 claims description 15
- 210000000056 organ Anatomy 0.000 claims description 15
- 238000006467 substitution reaction Methods 0.000 claims description 15
- 235000019408 sucralose Nutrition 0.000 claims description 15
- BAQAVOSOZGMPRM-QBMZZYIRSA-N sucralose Chemical group O[C@@H]1[C@@H](O)[C@@H](Cl)[C@@H](CO)O[C@@H]1O[C@@]1(CCl)[C@@H](O)[C@H](O)[C@@H](CCl)O1 BAQAVOSOZGMPRM-QBMZZYIRSA-N 0.000 claims description 15
- 230000002335 preservative effect Effects 0.000 claims description 14
- 208000024891 symptom Diseases 0.000 claims description 13
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 12
- 102220198118 rs387906589 Human genes 0.000 claims description 12
- 230000003213 activating effect Effects 0.000 claims description 11
- 208000007502 anemia Diseases 0.000 claims description 11
- 229910052799 carbon Inorganic materials 0.000 claims description 10
- 238000006243 chemical reaction Methods 0.000 claims description 10
- 239000012535 impurity Substances 0.000 claims description 10
- 238000001959 radiotherapy Methods 0.000 claims description 10
- 208000006274 Brain Stem Neoplasms Diseases 0.000 claims description 9
- YAAWASYJIRZXSZ-UHFFFAOYSA-N pyrimidine-2,4-diamine Chemical class NC1=CC=NC(N)=N1 YAAWASYJIRZXSZ-UHFFFAOYSA-N 0.000 claims description 9
- 238000001938 differential scanning calorimetry curve Methods 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 235000015165 citric acid Nutrition 0.000 claims description 7
- 239000013078 crystal Substances 0.000 claims description 7
- 238000002441 X-ray diffraction Methods 0.000 claims description 6
- 125000000539 amino acid group Chemical group 0.000 claims description 6
- 235000011187 glycerol Nutrition 0.000 claims description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 claims description 6
- 208000017667 Chronic Disease Diseases 0.000 claims description 5
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 claims description 5
- 235000011054 acetic acid Nutrition 0.000 claims description 5
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 claims description 5
- 230000000007 visual effect Effects 0.000 claims description 5
- 229920000168 Microcrystalline cellulose Polymers 0.000 claims description 4
- 206010047700 Vomiting Diseases 0.000 claims description 4
- 229960002216 methylparaben Drugs 0.000 claims description 4
- 235000019813 microcrystalline cellulose Nutrition 0.000 claims description 4
- 239000008108 microcrystalline cellulose Substances 0.000 claims description 4
- 229940016286 microcrystalline cellulose Drugs 0.000 claims description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 4
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 claims description 4
- 235000010234 sodium benzoate Nutrition 0.000 claims description 4
- 239000004299 sodium benzoate Substances 0.000 claims description 4
- 230000004614 tumor growth Effects 0.000 claims description 4
- 230000008673 vomiting Effects 0.000 claims description 4
- 229920001450 Alpha-Cyclodextrin Polymers 0.000 claims description 3
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 claims description 3
- 206010019233 Headaches Diseases 0.000 claims description 3
- 206010028813 Nausea Diseases 0.000 claims description 3
- 206010041349 Somnolence Diseases 0.000 claims description 3
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 claims description 3
- 238000009825 accumulation Methods 0.000 claims description 3
- HFHDHCJBZVLPGP-RWMJIURBSA-N alpha-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO HFHDHCJBZVLPGP-RWMJIURBSA-N 0.000 claims description 3
- 229940043377 alpha-cyclodextrin Drugs 0.000 claims description 3
- 230000003542 behavioural effect Effects 0.000 claims description 3
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 claims description 3
- 235000011175 beta-cyclodextrine Nutrition 0.000 claims description 3
- 210000001175 cerebrospinal fluid Anatomy 0.000 claims description 3
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 claims description 3
- 231100000869 headache Toxicity 0.000 claims description 3
- 229960001021 lactose monohydrate Drugs 0.000 claims description 3
- 208000018883 loss of balance Diseases 0.000 claims description 3
- 235000019359 magnesium stearate Nutrition 0.000 claims description 3
- 230000008693 nausea Effects 0.000 claims description 3
- 229940124531 pharmaceutical excipient Drugs 0.000 claims description 3
- 235000013772 propylene glycol Nutrition 0.000 claims description 3
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 claims description 3
- 230000000306 recurrent effect Effects 0.000 claims description 3
- 102220084255 rs863224846 Human genes 0.000 claims description 3
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 claims description 3
- 235000010268 sodium methyl p-hydroxybenzoate Nutrition 0.000 claims description 3
- 239000004290 sodium methyl p-hydroxybenzoate Substances 0.000 claims description 3
- 235000010230 sodium propyl p-hydroxybenzoate Nutrition 0.000 claims description 3
- 239000004404 sodium propyl p-hydroxybenzoate Substances 0.000 claims description 3
- 239000011975 tartaric acid Substances 0.000 claims description 3
- 235000002906 tartaric acid Nutrition 0.000 claims description 3
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 claims description 2
- LQCNTMMEQQVKLK-UHFFFAOYSA-N 5-[3-methoxy-4-(4-methylpiperazin-1-yl)phenyl]pyrimidine-2,4-diamine Chemical compound COC=1C=C(C=CC=1N1CCN(CC1)C)C=1C(=NC(=NC=1)N)N LQCNTMMEQQVKLK-UHFFFAOYSA-N 0.000 claims description 2
- 238000007681 bariatric surgery Methods 0.000 claims description 2
- 125000003275 alpha amino acid group Chemical group 0.000 claims 4
- 201000011510 cancer Diseases 0.000 abstract description 103
- 229940002612 prodrug Drugs 0.000 abstract description 37
- 239000000651 prodrug Substances 0.000 abstract description 37
- 230000008901 benefit Effects 0.000 abstract description 8
- 208000026350 Inborn Genetic disease Diseases 0.000 abstract description 4
- 208000016361 genetic disease Diseases 0.000 abstract description 3
- 235000002639 sodium chloride Nutrition 0.000 description 152
- 229940125782 compound 2 Drugs 0.000 description 139
- 239000000523 sample Substances 0.000 description 57
- 210000004027 cell Anatomy 0.000 description 53
- 238000012360 testing method Methods 0.000 description 43
- 239000000243 solution Substances 0.000 description 40
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 36
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 33
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 33
- 229940112869 bone morphogenetic protein Drugs 0.000 description 33
- 230000000694 effects Effects 0.000 description 32
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical group OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 29
- 238000002411 thermogravimetry Methods 0.000 description 28
- 238000000113 differential scanning calorimetry Methods 0.000 description 27
- 206010068715 Fibrodysplasia ossificans progressiva Diseases 0.000 description 26
- 238000012216 screening Methods 0.000 description 24
- 238000011269 treatment regimen Methods 0.000 description 24
- 238000004255 ion exchange chromatography Methods 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 239000002775 capsule Substances 0.000 description 21
- 208000003174 Brain Neoplasms Diseases 0.000 description 18
- 229940079593 drug Drugs 0.000 description 18
- 125000000623 heterocyclic group Chemical group 0.000 description 18
- 229910052742 iron Inorganic materials 0.000 description 18
- 102000005962 receptors Human genes 0.000 description 18
- 108020003175 receptors Proteins 0.000 description 18
- 208000030760 Anaemia of chronic disease Diseases 0.000 description 16
- 206010027476 Metastases Diseases 0.000 description 16
- 239000003795 chemical substances by application Substances 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 210000004369 blood Anatomy 0.000 description 15
- 239000008280 blood Substances 0.000 description 15
- 238000009826 distribution Methods 0.000 description 15
- 230000004660 morphological change Effects 0.000 description 15
- 230000001225 therapeutic effect Effects 0.000 description 15
- 241000699670 Mus sp. Species 0.000 description 14
- 235000001014 amino acid Nutrition 0.000 description 14
- 150000001413 amino acids Chemical group 0.000 description 14
- 238000010438 heat treatment Methods 0.000 description 14
- 238000004128 high performance liquid chromatography Methods 0.000 description 14
- 210000002381 plasma Anatomy 0.000 description 14
- 230000036470 plasma concentration Effects 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- 210000001519 tissue Anatomy 0.000 description 14
- 206010046766 uterine cancer Diseases 0.000 description 14
- 241000282472 Canis lupus familiaris Species 0.000 description 13
- 241001465754 Metazoa Species 0.000 description 13
- 208000002495 Uterine Neoplasms Diseases 0.000 description 13
- 239000000556 agonist Substances 0.000 description 13
- 208000022400 anemia due to chronic disease Diseases 0.000 description 13
- 239000000463 material Substances 0.000 description 13
- 239000008213 purified water Substances 0.000 description 13
- 230000001105 regulatory effect Effects 0.000 description 13
- 206010006187 Breast cancer Diseases 0.000 description 12
- 208000026310 Breast neoplasm Diseases 0.000 description 12
- 238000012054 celltiter-glo Methods 0.000 description 12
- 238000012512 characterization method Methods 0.000 description 12
- 238000003745 diagnosis Methods 0.000 description 12
- 238000011156 evaluation Methods 0.000 description 12
- 238000001704 evaporation Methods 0.000 description 12
- 230000008020 evaporation Effects 0.000 description 12
- 238000001990 intravenous administration Methods 0.000 description 12
- 229940043355 kinase inhibitor Drugs 0.000 description 12
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 12
- 102000004169 proteins and genes Human genes 0.000 description 12
- 230000011664 signaling Effects 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 11
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 108091000080 Phosphotransferase Proteins 0.000 description 11
- 238000004458 analytical method Methods 0.000 description 11
- 239000002585 base Substances 0.000 description 11
- 230000002068 genetic effect Effects 0.000 description 11
- 125000005843 halogen group Chemical group 0.000 description 11
- 229910052739 hydrogen Inorganic materials 0.000 description 11
- 230000009401 metastasis Effects 0.000 description 11
- 230000035515 penetration Effects 0.000 description 11
- 102000020233 phosphotransferase Human genes 0.000 description 11
- 108090000765 processed proteins & peptides Proteins 0.000 description 11
- 238000002560 therapeutic procedure Methods 0.000 description 11
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 10
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 10
- 238000003556 assay Methods 0.000 description 10
- 210000004556 brain Anatomy 0.000 description 10
- 210000003169 central nervous system Anatomy 0.000 description 10
- 239000003153 chemical reaction reagent Substances 0.000 description 10
- 239000003550 marker Substances 0.000 description 10
- 235000018102 proteins Nutrition 0.000 description 10
- 125000001424 substituent group Chemical group 0.000 description 10
- 125000000217 alkyl group Chemical group 0.000 description 9
- 230000015556 catabolic process Effects 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 239000012669 liquid formulation Substances 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- 102200128192 rs191456345 Human genes 0.000 description 9
- 239000000758 substrate Substances 0.000 description 9
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 8
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical group CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- 238000001994 activation Methods 0.000 description 8
- 239000000443 aerosol Substances 0.000 description 8
- 239000002246 antineoplastic agent Substances 0.000 description 8
- 229940127089 cytotoxic agent Drugs 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 239000003085 diluting agent Substances 0.000 description 8
- 239000002955 immunomodulating agent Substances 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 8
- 229960004768 irinotecan Drugs 0.000 description 8
- 210000002966 serum Anatomy 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 102100024506 Bone morphogenetic protein 2 Human genes 0.000 description 7
- 102100024457 Cyclin-dependent kinase 9 Human genes 0.000 description 7
- 101000980930 Homo sapiens Cyclin-dependent kinase 9 Proteins 0.000 description 7
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 7
- 241000700159 Rattus Species 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 7
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 7
- 208000028919 diffuse intrinsic pontine glioma Diseases 0.000 description 7
- 231100000673 dose–response relationship Toxicity 0.000 description 7
- 239000003446 ligand Substances 0.000 description 7
- 238000004020 luminiscence type Methods 0.000 description 7
- 201000005202 lung cancer Diseases 0.000 description 7
- 208000020816 lung neoplasm Diseases 0.000 description 7
- 238000003860 storage Methods 0.000 description 7
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 7
- 101000621390 Homo sapiens Wee1-like protein kinase Proteins 0.000 description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 6
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 6
- 102100040061 Indoleamine 2,3-dioxygenase 1 Human genes 0.000 description 6
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 6
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 6
- 206010033128 Ovarian cancer Diseases 0.000 description 6
- 206010061535 Ovarian neoplasm Diseases 0.000 description 6
- 229940099471 Phosphodiesterase inhibitor Drugs 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- 102100033912 Retinoic acid receptor gamma Human genes 0.000 description 6
- 102000040856 WT1 Human genes 0.000 description 6
- 108700020467 WT1 Proteins 0.000 description 6
- 102100023037 Wee1-like protein kinase Human genes 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 108010023082 activin A Proteins 0.000 description 6
- 230000008499 blood brain barrier function Effects 0.000 description 6
- 210000001218 blood-brain barrier Anatomy 0.000 description 6
- 210000000988 bone and bone Anatomy 0.000 description 6
- 210000000133 brain stem Anatomy 0.000 description 6
- 210000005013 brain tissue Anatomy 0.000 description 6
- 238000002659 cell therapy Methods 0.000 description 6
- 231100000221 frame shift mutation induction Toxicity 0.000 description 6
- 230000037433 frameshift Effects 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 238000009499 grossing Methods 0.000 description 6
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 6
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 230000004054 inflammatory process Effects 0.000 description 6
- 239000006166 lysate Substances 0.000 description 6
- 229940124302 mTOR inhibitor Drugs 0.000 description 6
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 6
- 201000001441 melanoma Diseases 0.000 description 6
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 6
- 150000007523 nucleic acids Chemical group 0.000 description 6
- 229940023041 peptide vaccine Drugs 0.000 description 6
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 6
- 238000001556 precipitation Methods 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 108091008760 retinoic acid receptors γ Proteins 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- 230000037436 splice-site mutation Effects 0.000 description 6
- 239000003277 telomerase inhibitor Substances 0.000 description 6
- 241000701161 unidentified adenovirus Species 0.000 description 6
- 229960005486 vaccine Drugs 0.000 description 6
- 208000018084 Bone neoplasm Diseases 0.000 description 5
- 229940123189 CD40 agonist Drugs 0.000 description 5
- 206010008342 Cervix carcinoma Diseases 0.000 description 5
- 206010014733 Endometrial cancer Diseases 0.000 description 5
- 206010014759 Endometrial neoplasm Diseases 0.000 description 5
- 206010061218 Inflammation Diseases 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 229940123582 Telomerase inhibitor Drugs 0.000 description 5
- 108010051765 Type I Bone Morphogenetic Protein Receptors Proteins 0.000 description 5
- 102000019044 Type I Bone Morphogenetic Protein Receptors Human genes 0.000 description 5
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 5
- 230000004075 alteration Effects 0.000 description 5
- 238000010171 animal model Methods 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 235000019658 bitter taste Nutrition 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 201000010881 cervical cancer Diseases 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 230000001627 detrimental effect Effects 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 208000005017 glioblastoma Diseases 0.000 description 5
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 5
- 150000004677 hydrates Chemical class 0.000 description 5
- 229940125798 integrin inhibitor Drugs 0.000 description 5
- 230000003902 lesion Effects 0.000 description 5
- 239000002502 liposome Substances 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 5
- 125000004193 piperazinyl group Chemical group 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000001103 potassium chloride Substances 0.000 description 5
- 235000011164 potassium chloride Nutrition 0.000 description 5
- 238000011002 quantification Methods 0.000 description 5
- 230000002285 radioactive effect Effects 0.000 description 5
- 102220104142 rs879253743 Human genes 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 239000004094 surface-active agent Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 4
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Natural products OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 108010049931 Bone Morphogenetic Protein 2 Proteins 0.000 description 4
- 108010040422 Bone Morphogenetic Protein Receptors Proteins 0.000 description 4
- 102000001893 Bone Morphogenetic Protein Receptors Human genes 0.000 description 4
- 206010008805 Chromosomal abnormalities Diseases 0.000 description 4
- 208000031404 Chromosome Aberrations Diseases 0.000 description 4
- 206010009944 Colon cancer Diseases 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- 102100040018 Interferon alpha-2 Human genes 0.000 description 4
- 108010079944 Interferon-alpha2b Proteins 0.000 description 4
- 102000002397 Kinins Human genes 0.000 description 4
- 108010093008 Kinins Proteins 0.000 description 4
- 206010025323 Lymphomas Diseases 0.000 description 4
- 102100026888 Mitogen-activated protein kinase kinase kinase 7 Human genes 0.000 description 4
- 239000012270 PD-1 inhibitor Substances 0.000 description 4
- 239000012668 PD-1-inhibitor Substances 0.000 description 4
- 239000012271 PD-L1 inhibitor Substances 0.000 description 4
- 102000038030 PI3Ks Human genes 0.000 description 4
- 108091007960 PI3Ks Proteins 0.000 description 4
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 4
- 102220473703 Photoreceptor ankyrin repeat protein_F265S_mutation Human genes 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- 206010060862 Prostate cancer Diseases 0.000 description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 102000040945 Transcription factor Human genes 0.000 description 4
- 108091023040 Transcription factor Proteins 0.000 description 4
- 102220534977 Transcriptional repressor protein YY1_H320Y_mutation Human genes 0.000 description 4
- 229940122429 Tubulin inhibitor Drugs 0.000 description 4
- 230000005856 abnormality Effects 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 229940024606 amino acid Drugs 0.000 description 4
- 235000010323 ascorbic acid Nutrition 0.000 description 4
- 239000011668 ascorbic acid Substances 0.000 description 4
- 229960005070 ascorbic acid Drugs 0.000 description 4
- 102220384347 c.298A>G Human genes 0.000 description 4
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 4
- 239000007963 capsule composition Substances 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- 238000012217 deletion Methods 0.000 description 4
- 230000037430 deletion Effects 0.000 description 4
- 229940029030 dendritic cell vaccine Drugs 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 235000005911 diet Nutrition 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 210000003743 erythrocyte Anatomy 0.000 description 4
- 201000010255 female reproductive organ cancer Diseases 0.000 description 4
- 238000000684 flow cytometry Methods 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 238000011835 investigation Methods 0.000 description 4
- TYQCGQRIZGCHNB-JLAZNSOCSA-N l-ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(O)=C(O)C1=O TYQCGQRIZGCHNB-JLAZNSOCSA-N 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 235000011475 lollipops Nutrition 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 230000000877 morphologic effect Effects 0.000 description 4
- 150000007530 organic bases Chemical class 0.000 description 4
- 230000011164 ossification Effects 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 229940121655 pd-1 inhibitor Drugs 0.000 description 4
- 229940121656 pd-l1 inhibitor Drugs 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 102200141947 rs1057516149 Human genes 0.000 description 4
- 102200153431 rs1060501000 Human genes 0.000 description 4
- 102200146616 rs11568361 Human genes 0.000 description 4
- 102200104821 rs121912679 Human genes 0.000 description 4
- 102200060081 rs147241704 Human genes 0.000 description 4
- 102220041797 rs200165598 Human genes 0.000 description 4
- 102200134060 rs28940573 Human genes 0.000 description 4
- 102200153188 rs756928158 Human genes 0.000 description 4
- 102200134068 rs796053439 Human genes 0.000 description 4
- 102220103427 rs878854244 Human genes 0.000 description 4
- 102220127315 rs886044540 Human genes 0.000 description 4
- 230000001953 sensory effect Effects 0.000 description 4
- 239000012453 solvate Substances 0.000 description 4
- 238000013268 sustained release Methods 0.000 description 4
- 239000012730 sustained-release form Substances 0.000 description 4
- 108091008743 testicular receptors 4 Proteins 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 108010082126 Alanine transaminase Proteins 0.000 description 3
- 206010003571 Astrocytoma Diseases 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 206010005949 Bone cancer Diseases 0.000 description 3
- 206010006143 Brain stem glioma Diseases 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 208000021994 Diffuse astrocytoma Diseases 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 102000001554 Hemoglobins Human genes 0.000 description 3
- 108010054147 Hemoglobins Proteins 0.000 description 3
- 101000762366 Homo sapiens Bone morphogenetic protein 2 Proteins 0.000 description 3
- 206010065973 Iron Overload Diseases 0.000 description 3
- 102100025744 Mothers against decapentaplegic homolog 1 Human genes 0.000 description 3
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 101700032040 SMAD1 Proteins 0.000 description 3
- 208000000453 Skin Neoplasms Diseases 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 208000037844 advanced solid tumor Diseases 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 239000012296 anti-solvent Substances 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 235000006708 antioxidants Nutrition 0.000 description 3
- 238000003491 array Methods 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- 208000029742 colonic neoplasm Diseases 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000010949 copper Substances 0.000 description 3
- 230000000593 degrading effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 239000008121 dextrose Substances 0.000 description 3
- 230000037213 diet Effects 0.000 description 3
- 208000026144 diffuse midline glioma, H3 K27M-mutant Diseases 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 201000001169 fibrillary astrocytoma Diseases 0.000 description 3
- 235000003599 food sweetener Nutrition 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 230000003394 haemopoietic effect Effects 0.000 description 3
- 201000005787 hematologic cancer Diseases 0.000 description 3
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229940049920 malate Drugs 0.000 description 3
- 238000007726 management method Methods 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 3
- 235000019796 monopotassium phosphate Nutrition 0.000 description 3
- 102000039446 nucleic acids Human genes 0.000 description 3
- 108020004707 nucleic acids Proteins 0.000 description 3
- 230000002611 ovarian Effects 0.000 description 3
- 239000000813 peptide hormone Substances 0.000 description 3
- 230000009038 pharmacological inhibition Effects 0.000 description 3
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 3
- 230000003863 physical function Effects 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- IWZKICVEHNUQTL-UHFFFAOYSA-M potassium hydrogen phthalate Chemical compound [K+].OC(=O)C1=CC=CC=C1C([O-])=O IWZKICVEHNUQTL-UHFFFAOYSA-M 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 238000004393 prognosis Methods 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 238000003757 reverse transcription PCR Methods 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 102220006250 rs121912680 Human genes 0.000 description 3
- 102220211152 rs35993949 Human genes 0.000 description 3
- 102200104824 rs387906588 Human genes 0.000 description 3
- 102200104796 rs387906589 Human genes 0.000 description 3
- 102220012838 rs397516324 Human genes 0.000 description 3
- 102200057533 rs587781511 Human genes 0.000 description 3
- 102220127254 rs886044521 Human genes 0.000 description 3
- 102220186871 rs886053369 Human genes 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 201000000849 skin cancer Diseases 0.000 description 3
- 238000012453 sprague-dawley rat model Methods 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 230000009747 swallowing Effects 0.000 description 3
- 239000003765 sweetening agent Substances 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- BXTJCSYMGFJEID-XMTADJHZSA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-[6-[3-[(2r)-2-amino-2-carboxyethyl]sulfanyl-2,5-dioxopyrrolidin-1-yl]hexanoyl-methylamino]-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-met Chemical compound C([C@H](NC(=O)[C@H](C)[C@@H](OC)[C@@H]1CCCN1C(=O)C[C@H]([C@H]([C@@H](C)CC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCCCN1C(C(SC[C@H](N)C(O)=O)CC1=O)=O)C(C)C)OC)C(O)=O)C1=CC=CC=C1 BXTJCSYMGFJEID-XMTADJHZSA-N 0.000 description 2
- BKWJAKQVGHWELA-UHFFFAOYSA-N 1-[6-(2-hydroxypropan-2-yl)-2-pyridinyl]-6-[4-(4-methyl-1-piperazinyl)anilino]-2-prop-2-enyl-3-pyrazolo[3,4-d]pyrimidinone Chemical group C1CN(C)CCN1C(C=C1)=CC=C1NC1=NC=C2C(=O)N(CC=C)N(C=3N=C(C=CC=3)C(C)(C)O)C2=N1 BKWJAKQVGHWELA-UHFFFAOYSA-N 0.000 description 2
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 2
- VLULRUCCHYVXOH-UHFFFAOYSA-N 11-benzyl-7-[(2-methylphenyl)methyl]-2,5,7,11-tetrazatricyclo[7.4.0.02,6]trideca-1(9),5-dien-8-one Chemical compound CC1=CC=CC=C1CN1C(=O)C(CN(CC=2C=CC=CC=2)CC2)=C2N2CCN=C21 VLULRUCCHYVXOH-UHFFFAOYSA-N 0.000 description 2
- HNLXNOZHXNSSPN-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(OCCOCCOCCOCCOCCOCCOCCO)C=C1 HNLXNOZHXNSSPN-UHFFFAOYSA-N 0.000 description 2
- QCXJEYYXVJIFCE-UHFFFAOYSA-N 4-acetamidobenzoic acid Chemical compound CC(=O)NC1=CC=C(C(O)=O)C=C1 QCXJEYYXVJIFCE-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- 102220504440 Activin receptor type-1_P115S_mutation Human genes 0.000 description 2
- 102100036475 Alanine aminotransferase 1 Human genes 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 108010003415 Aspartate Aminotransferases Proteins 0.000 description 2
- 102000004625 Aspartate Aminotransferases Human genes 0.000 description 2
- BPYKTIZUTYGOLE-IFADSCNNSA-N Bilirubin Chemical compound N1C(=O)C(C)=C(C=C)\C1=C\C1=C(C)C(CCC(O)=O)=C(CC2=C(C(C)=C(\C=C/3C(=C(C=C)C(=O)N\3)C)N2)CCC(O)=O)N1 BPYKTIZUTYGOLE-IFADSCNNSA-N 0.000 description 2
- 102100025423 Bone morphogenetic protein receptor type-1A Human genes 0.000 description 2
- 108010074051 C-Reactive Protein Proteins 0.000 description 2
- 102100032752 C-reactive protein Human genes 0.000 description 2
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 description 2
- XSQKEVGTZSBVBR-UHFFFAOYSA-N CN(CC1)CCN1c(c(OC)c1)ccc1Nc1nc(Nc2c(-c3ccccn3)nccc2)ccn1 Chemical compound CN(CC1)CCN1c(c(OC)c1)ccc1Nc1nc(Nc2c(-c3ccccn3)nccc2)ccn1 XSQKEVGTZSBVBR-UHFFFAOYSA-N 0.000 description 2
- 101100421200 Caenorhabditis elegans sep-1 gene Proteins 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 206010057248 Cell death Diseases 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 102100026810 Cyclin-dependent kinase 7 Human genes 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 2
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 101710158332 Diuretic hormone Proteins 0.000 description 2
- 101710204261 Diuretic hormone class 2 Proteins 0.000 description 2
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 108050000784 Ferritin Proteins 0.000 description 2
- 102000008857 Ferritin Human genes 0.000 description 2
- 238000008416 Ferritin Methods 0.000 description 2
- 101000934638 Homo sapiens Bone morphogenetic protein receptor type-1A Proteins 0.000 description 2
- 101000911952 Homo sapiens Cyclin-dependent kinase 7 Proteins 0.000 description 2
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 2
- 101000777206 Homo sapiens Ubiquitin carboxyl-terminal hydrolase 40 Proteins 0.000 description 2
- 108091006975 Iron transporters Proteins 0.000 description 2
- 102000008133 Iron-Binding Proteins Human genes 0.000 description 2
- 108010035210 Iron-Binding Proteins Proteins 0.000 description 2
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 2
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 2
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 2
- 239000004166 Lanolin Substances 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 206010027452 Metastases to bone Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 201000002481 Myositis Diseases 0.000 description 2
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 2
- 206010029260 Neuroblastoma Diseases 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 229940126682 ONC201 Drugs 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 102220525391 Protein Niban 1_R325A_mutation Human genes 0.000 description 2
- 208000015634 Rectal Neoplasms Diseases 0.000 description 2
- 241000702247 Reoviridae Species 0.000 description 2
- 108091006976 SLC40A1 Proteins 0.000 description 2
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 2
- 108020004459 Small interfering RNA Proteins 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 102220494275 Somatostatin receptor type 5_L251S_mutation Human genes 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 241000779819 Syncarpia glomulifera Species 0.000 description 2
- 108091005735 TGF-beta receptors Proteins 0.000 description 2
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 102100031284 Ubiquitin carboxyl-terminal hydrolase 40 Human genes 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 229950009557 adavosertib Drugs 0.000 description 2
- 229950006322 adegramotide Drugs 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 2
- 229950010817 alvocidib Drugs 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 230000000845 anti-microbial effect Effects 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 239000008122 artificial sweetener Substances 0.000 description 2
- 235000021311 artificial sweeteners Nutrition 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 229960003852 atezolizumab Drugs 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 229950002916 avelumab Drugs 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 238000001574 biopsy Methods 0.000 description 2
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 2
- 239000004327 boric acid Substances 0.000 description 2
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 2
- 229960001948 caffeine Drugs 0.000 description 2
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- 238000003570 cell viability assay Methods 0.000 description 2
- 210000000038 chest Anatomy 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 2
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 2
- 229960005061 crizotinib Drugs 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 229960002448 dasatinib Drugs 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 229950008925 depatuxizumab mafodotin Drugs 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 208000037846 diffuse midline glioma Diseases 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- OSVXSBDYLRYLIG-UHFFFAOYSA-N dioxidochlorine(.) Chemical compound O=Cl=O OSVXSBDYLRYLIG-UHFFFAOYSA-N 0.000 description 2
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical group C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 2
- 229960002768 dipyridamole Drugs 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 238000007323 disproportionation reaction Methods 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 230000007783 downstream signaling Effects 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 229940082789 erbitux Drugs 0.000 description 2
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- 229960005167 everolimus Drugs 0.000 description 2
- 230000004424 eye movement Effects 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 229960000390 fludarabine Drugs 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- 210000004602 germ cell Anatomy 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- 208000029824 high grade glioma Diseases 0.000 description 2
- 238000009396 hybridization Methods 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- 229960001101 ifosfamide Drugs 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- 125000001841 imino group Chemical group [H]N=* 0.000 description 2
- 231100000405 induce cancer Toxicity 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- XMBWDFGMSWQBCA-RNFDNDRNSA-M iodine-131(1-) Chemical compound [131I-] XMBWDFGMSWQBCA-RNFDNDRNSA-M 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000019388 lanolin Nutrition 0.000 description 2
- 229940039717 lanolin Drugs 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 201000011614 malignant glioma Diseases 0.000 description 2
- BAXLBXFAUKGCDY-UHFFFAOYSA-N mebendazole Chemical group [CH]1C2=NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CC=C1 BAXLBXFAUKGCDY-UHFFFAOYSA-N 0.000 description 2
- 229960003439 mebendazole Drugs 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical group OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 2
- 229960001924 melphalan Drugs 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 208000037819 metastatic cancer Diseases 0.000 description 2
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 2
- 208000010658 metastatic prostate carcinoma Diseases 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 2
- 238000002493 microarray Methods 0.000 description 2
- 210000000214 mouth Anatomy 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 238000007481 next generation sequencing Methods 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 229960003301 nivolumab Drugs 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 239000001301 oxygen Chemical group 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000006174 pH buffer Substances 0.000 description 2
- 229960002621 pembrolizumab Drugs 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 230000037081 physical activity Effects 0.000 description 2
- 239000002504 physiological saline solution Substances 0.000 description 2
- 239000001739 pinus spp. Substances 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 230000004983 pleiotropic effect Effects 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 208000037821 progressive disease Diseases 0.000 description 2
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 230000004853 protein function Effects 0.000 description 2
- GPTFURBXHJWNHR-UHFFFAOYSA-N protopine Chemical compound C1=C2C(=O)CC3=CC=C4OCOC4=C3CN(C)CCC2=CC2=C1OCO2 GPTFURBXHJWNHR-UHFFFAOYSA-N 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 206010038038 rectal cancer Diseases 0.000 description 2
- 201000001275 rectum cancer Diseases 0.000 description 2
- 239000013074 reference sample Substances 0.000 description 2
- 238000002271 resection Methods 0.000 description 2
- 230000029058 respiratory gaseous exchange Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 238000005096 rolling process Methods 0.000 description 2
- 102200077327 rs104894832 Human genes 0.000 description 2
- 102200077719 rs119463992 Human genes 0.000 description 2
- 102200026584 rs121434536 Human genes 0.000 description 2
- 102200104820 rs387906590 Human genes 0.000 description 2
- 102220031616 rs398124537 Human genes 0.000 description 2
- 102200123125 rs758785463 Human genes 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- 239000012679 serum free medium Substances 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- 238000010583 slow cooling Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 235000010413 sodium alginate Nutrition 0.000 description 2
- 239000000661 sodium alginate Substances 0.000 description 2
- 229940005550 sodium alginate Drugs 0.000 description 2
- 229940079827 sodium hydrogen sulfite Drugs 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- 239000008247 solid mixture Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000013112 stability test Methods 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 125000003107 substituted aryl group Chemical group 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000001502 supplementing effect Effects 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 239000008399 tap water Substances 0.000 description 2
- 235000020679 tap water Nutrition 0.000 description 2
- 235000019640 taste Nutrition 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 210000002105 tongue Anatomy 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 229940036248 turpentine Drugs 0.000 description 2
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 2
- 229960000604 valproic acid Drugs 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- PGOHTUIFYSHAQG-LJSDBVFPSA-N (2S)-6-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-4-methylsulfanylbutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-hydroxypropanoyl]amino]-4-methylpentanoyl]amino]-3-sulfanylpropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-oxopentanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-oxopentanoyl]amino]-3-phenylpropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-5-oxopentanoyl]amino]hexanoic acid Chemical compound CSCC[C@H](N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1cnc[nH]1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(O)=O PGOHTUIFYSHAQG-LJSDBVFPSA-N 0.000 description 1
- ZADWXFSZEAPBJS-SNVBAGLBSA-N (2r)-2-amino-3-(1-methylindol-3-yl)propanoic acid Chemical group C1=CC=C2N(C)C=C(C[C@@H](N)C(O)=O)C2=C1 ZADWXFSZEAPBJS-SNVBAGLBSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- 0 *c1cc(Nc2nc(NC3=C(c4ccccn4)N=IC=C3)ccn2)cc(*)c1* Chemical compound *c1cc(Nc2nc(NC3=C(c4ccccn4)N=IC=C3)ccn2)cc(*)c1* 0.000 description 1
- 125000005988 1,1-dioxo-thiomorpholinyl group Chemical group 0.000 description 1
- SJJCQDRGABAVBB-UHFFFAOYSA-N 1-hydroxy-2-naphthoic acid Chemical compound C1=CC=CC2=C(O)C(C(=O)O)=CC=C21 SJJCQDRGABAVBB-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- 125000005987 1-oxo-thiomorpholinyl group Chemical group 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- BUEPKUNNPRRSKV-UHFFFAOYSA-N 2-(dimethylamino)ethyl 2-hydroxy-2,2-diphenylacetate Chemical compound C=1C=CC=CC=1C(O)(C(=O)OCCN(C)C)C1=CC=CC=C1 BUEPKUNNPRRSKV-UHFFFAOYSA-N 0.000 description 1
- PRQQFQIDKDPUEU-UHFFFAOYSA-N 2-N-[3-methoxy-4-(4-methylpiperazin-1-yl)phenyl]-4-(2-pyridin-2-ylpyridin-3-yl)-1H-pyrimidine-2,4-diamine Chemical compound N1=C(C(=CC=C1)C1(NC(=NC=C1)NC1=CC(=C(C=C1)N1CCN(CC1)C)OC)N)C1=NC=CC=C1 PRQQFQIDKDPUEU-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000003229 2-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- 125000004637 2-oxopiperidinyl group Chemical group O=C1N(CCCC1)* 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- RHXHGRAEPCAFML-UHFFFAOYSA-N 7-cyclopentyl-n,n-dimethyl-2-[(5-piperazin-1-ylpyridin-2-yl)amino]pyrrolo[2,3-d]pyrimidine-6-carboxamide Chemical compound N1=C2N(C3CCCC3)C(C(=O)N(C)C)=CC2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 RHXHGRAEPCAFML-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 102220504439 Activin receptor type-1_S41F_mutation Human genes 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 208000036832 Adenocarcinoma of ovary Diseases 0.000 description 1
- 241000473391 Archosargus rhomboidalis Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241001331781 Aspergillus brasiliensis Species 0.000 description 1
- 206010060971 Astrocytoma malignant Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-N Betaine Natural products C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- ROFVEXUMMXZLPA-UHFFFAOYSA-N Bipyridyl Chemical compound N1=CC=CC=C1C1=CC=CC=N1 ROFVEXUMMXZLPA-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 101800000407 Brain natriuretic peptide 32 Proteins 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N Camphoric acid Natural products CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- 206010058019 Cancer Pain Diseases 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 1
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 239000004155 Chlorine dioxide Substances 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 206010062344 Congenital musculoskeletal anomaly Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UHFFFAOYSA-N D-OH-Asp Natural products OC(=O)C(N)CC(O)=O CKLJMWTZIZZHCS-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 208000003164 Diplopia Diseases 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 206010014967 Ependymoma Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 206010049811 Extraskeletal ossification Diseases 0.000 description 1
- 206010068737 Facial asymmetry Diseases 0.000 description 1
- 206010051267 Facial paresis Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 231100001273 GLP toxicology study Toxicity 0.000 description 1
- 102100027988 GTP-binding protein Rhes Human genes 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 201000004066 Ganglioglioma Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000009139 Gilbert Disease Diseases 0.000 description 1
- 208000022412 Gilbert syndrome Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- BCCRXDTUTZHDEU-VKHMYHEASA-N Gly-Ser Chemical compound NCC(=O)N[C@@H](CO)C(O)=O BCCRXDTUTZHDEU-VKHMYHEASA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical class OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 238000010268 HPLC based assay Methods 0.000 description 1
- 102100036284 Hepcidin Human genes 0.000 description 1
- 208000028782 Hereditary disease Diseases 0.000 description 1
- 208000034970 Heterotopic Ossification Diseases 0.000 description 1
- 101000578396 Homo sapiens GTP-binding protein Rhes Proteins 0.000 description 1
- 101001021253 Homo sapiens Hepcidin Proteins 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000035752 Live birth Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 208000002720 Malnutrition Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 235000019596 Masking bitterness Nutrition 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 208000024556 Mendelian disease Diseases 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- 101100437777 Mus musculus Bmpr1a gene Proteins 0.000 description 1
- 206010064470 Muscle swelling Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 102400001263 NT-proBNP Human genes 0.000 description 1
- 208000003788 Neoplasm Micrometastasis Diseases 0.000 description 1
- 208000005890 Neuroma Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 206010061328 Ovarian epithelial cancer Diseases 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 201000007286 Pilocytic astrocytoma Diseases 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 208000009052 Precursor T-Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 241000283080 Proboscidea <mammal> Species 0.000 description 1
- 102000012515 Protein kinase domains Human genes 0.000 description 1
- 108050002122 Protein kinase domains Proteins 0.000 description 1
- 108010094028 Prothrombin Proteins 0.000 description 1
- 102100027378 Prothrombin Human genes 0.000 description 1
- ODHCTXKNWHHXJC-GSVOUGTGSA-N Pyroglutamic acid Natural products OC(=O)[C@H]1CCC(=O)N1 ODHCTXKNWHHXJC-GSVOUGTGSA-N 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 108020005067 RNA Splice Sites Proteins 0.000 description 1
- 238000003559 RNA-seq method Methods 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 102000007374 Smad Proteins Human genes 0.000 description 1
- 108010007945 Smad Proteins Proteins 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 108010073771 Soybean Proteins Proteins 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 208000002847 Surgical Wound Diseases 0.000 description 1
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 229920002807 Thiomer Polymers 0.000 description 1
- 108010000499 Thromboplastin Proteins 0.000 description 1
- 102000002262 Thromboplastin Human genes 0.000 description 1
- 108010033576 Transferrin Receptors Proteins 0.000 description 1
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108050009309 Tuberin Proteins 0.000 description 1
- 102000044633 Tuberous Sclerosis Complex 2 Human genes 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 1
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 1
- 208000008385 Urogenital Neoplasms Diseases 0.000 description 1
- 208000006593 Urologic Neoplasms Diseases 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 229950001573 abemaciclib Drugs 0.000 description 1
- 150000003869 acetamides Chemical class 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 201000011186 acute T cell leukemia Diseases 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 230000010398 acute inflammatory response Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 229960000250 adipic acid Drugs 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940027983 antiseptic and disinfectant quaternary ammonium compound Drugs 0.000 description 1
- 101150010487 are gene Proteins 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- OGBUMNBNEWYMNJ-UHFFFAOYSA-N batilol Chemical class CCCCCCCCCCCCCCCCCCOCC(O)CO OGBUMNBNEWYMNJ-UHFFFAOYSA-N 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 150000003936 benzamides Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- 210000004958 brain cell Anatomy 0.000 description 1
- 238000011095 buffer preparation Methods 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical compound CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 description 1
- 230000004611 cancer cell death Effects 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 230000005907 cancer growth Effects 0.000 description 1
- 229960001631 carbomer Drugs 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000012578 cell culture reagent Substances 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 201000007455 central nervous system cancer Diseases 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 201000007335 cerebellar astrocytoma Diseases 0.000 description 1
- 210000003710 cerebral cortex Anatomy 0.000 description 1
- 229960001927 cetylpyridinium chloride Drugs 0.000 description 1
- NFCRBQADEGXVDL-UHFFFAOYSA-M cetylpyridinium chloride monohydrate Chemical compound O.[Cl-].CCCCCCCCCCCCCCCC[N+]1=CC=CC=C1 NFCRBQADEGXVDL-UHFFFAOYSA-M 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 230000001055 chewing effect Effects 0.000 description 1
- 229940112822 chewing gum Drugs 0.000 description 1
- 235000015218 chewing gum Nutrition 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 235000019398 chlorine dioxide Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000012084 conversion product Substances 0.000 description 1
- 230000010485 coping Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940109239 creatinine Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 208000030381 cutaneous melanoma Diseases 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 125000005507 decahydroisoquinolyl group Chemical group 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 230000010339 dilation Effects 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 229950009791 durvalumab Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000013265 extended release Methods 0.000 description 1
- 238000013213 extrapolation Methods 0.000 description 1
- 230000004438 eyesight Effects 0.000 description 1
- 208000010770 facial weakness Diseases 0.000 description 1
- 206010016256 fatigue Diseases 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 201000010103 fibrous dysplasia Diseases 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000011194 food seasoning agent Nutrition 0.000 description 1
- 150000003948 formamides Chemical class 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 235000011389 fruit/vegetable juice Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000005021 gait Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 150000002315 glycerophosphates Chemical class 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 210000004565 granule cell Anatomy 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000020169 heat generation Effects 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- DKAGJZJALZXOOV-UHFFFAOYSA-N hydrate;hydrochloride Chemical compound O.Cl DKAGJZJALZXOOV-UHFFFAOYSA-N 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 229940079826 hydrogen sulfite Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 230000002267 hypothalamic effect Effects 0.000 description 1
- 210000003016 hypothalamus Anatomy 0.000 description 1
- 208000036260 idiopathic disease Diseases 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 229950009034 indoximod Drugs 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960001375 lactose Drugs 0.000 description 1
- 210000003041 ligament Anatomy 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 238000002595 magnetic resonance imaging Methods 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 230000001071 malnutrition Effects 0.000 description 1
- 235000000824 malnutrition Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 108010082117 matrigel Proteins 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 210000001767 medulla oblongata Anatomy 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 210000001259 mesencephalon Anatomy 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229940102396 methyl bromide Drugs 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 229960001047 methyl salicylate Drugs 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000037230 mobility Effects 0.000 description 1
- 230000003990 molecular pathway Effects 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000036438 mutation frequency Effects 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- UZWDCWONPYILKI-UHFFFAOYSA-N n-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine Chemical compound C1CN(CC)CCN1CC(C=N1)=CC=C1NC1=NC=C(F)C(C=2C=C3N(C(C)C)C(C)=NC3=C(F)C=2)=N1 UZWDCWONPYILKI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 238000013188 needle biopsy Methods 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 238000002610 neuroimaging Methods 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 229950010203 nimotuzumab Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- 230000005937 nuclear translocation Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 208000015380 nutritional deficiency disease Diseases 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 229950004053 octoxinol Drugs 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 238000002966 oligonucleotide array Methods 0.000 description 1
- 230000000174 oncolytic effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 229940100691 oral capsule Drugs 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000000010 osteolytic effect Effects 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 208000013371 ovarian adenocarcinoma Diseases 0.000 description 1
- 201000006588 ovary adenocarcinoma Diseases 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FWZRWHZDXBDTFK-ZHACJKMWSA-N panobinostat Chemical compound CC1=NC2=CC=C[CH]C2=C1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FWZRWHZDXBDTFK-ZHACJKMWSA-N 0.000 description 1
- 229960005184 panobinostat Drugs 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N phenylalanine group Chemical group N[C@@H](CC1=CC=CC=C1)C(=O)O COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229920000151 polyglycol Polymers 0.000 description 1
- 239000010695 polyglycol Substances 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 108010008064 pro-brain natriuretic peptide (1-76) Proteins 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000029983 protein stabilization Effects 0.000 description 1
- 229940039716 prothrombin Drugs 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 229940076788 pyruvate Drugs 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000009103 reabsorption Effects 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 239000013643 reference control Substances 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229950003687 ribociclib Drugs 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 235000002020 sage Nutrition 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 238000007480 sanger sequencing Methods 0.000 description 1
- 108010038379 sargramostim Proteins 0.000 description 1
- 229960002530 sargramostim Drugs 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 201000003708 skin melanoma Diseases 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 229940001941 soy protein Drugs 0.000 description 1
- 238000012289 standard assay Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 239000010902 straw Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L sulfate group Chemical group S(=O)(=O)([O-])[O-] QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229910052717 sulfur Chemical group 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 238000004441 surface measurement Methods 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 150000004685 tetrahydrates Chemical class 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000005985 thienyl[1,3]dithianyl group Chemical group 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229960004906 thiomersal Drugs 0.000 description 1
- DHCDFWKWKRSZHF-UHFFFAOYSA-L thiosulfate(2-) Chemical compound [O-]S([S-])(=O)=O DHCDFWKWKRSZHF-UHFFFAOYSA-L 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000820 toxicity test Toxicity 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 231100000041 toxicology testing Toxicity 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- BSVBQGMMJUBVOD-UHFFFAOYSA-N trisodium borate Chemical compound [Na+].[Na+].[Na+].[O-]B([O-])[O-] BSVBQGMMJUBVOD-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- SZCZSKMCTGEJKI-UHFFFAOYSA-N tuberin Natural products COC1=CC=C(C=CNC=O)C=C1 SZCZSKMCTGEJKI-UHFFFAOYSA-N 0.000 description 1
- 239000003744 tubulin modulator Substances 0.000 description 1
- 208000025421 tumor of uterus Diseases 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 230000004222 uncontrolled growth Effects 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 229940116269 uric acid Drugs 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 208000037964 urogenital cancer Diseases 0.000 description 1
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- BPQMGSKTAYIVFO-UHFFFAOYSA-N vismodegib Chemical group ClC1=CC(S(=O)(=O)C)=CC=C1C(=O)NC1=CC=C(Cl)C(C=2N=CC=CC=2)=C1 BPQMGSKTAYIVFO-UHFFFAOYSA-N 0.000 description 1
- 229960004449 vismodegib Drugs 0.000 description 1
- 230000002747 voluntary effect Effects 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 229920003176 water-insoluble polymer Polymers 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 208000016261 weight loss Diseases 0.000 description 1
- 238000002689 xenotransplantation Methods 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images






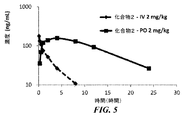








































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/40—Cyclodextrins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0087—Galenical forms not covered by A61K9/02 - A61K9/7023
- A61K9/0095—Drinks; Beverages; Syrups; Compositions for reconstitution thereof, e.g. powders or tablets to be dispersed in a glass of water; Veterinary drenches
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4816—Wall or shell material
- A61K9/4825—Proteins, e.g. gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Abstract
以下の式(I)(その立体異性体、互変異性体、薬学的に許容される塩、およびプロドラッグが含まれ、式中、R1、R2、R3、およびR4は本明細書で定義されたとおりである)を有するACVR1阻害剤を投与することによる、それを必要とする対象の疾患を処置するための方法が開示される(I)。処置から利益を得ることができる対象は、そのACVR1遺伝子中に変異を有する場合がある。がん(例えば、びまん性真性橋膠腫(DIPG))および遺伝的障害(例えば、進行性骨化性線維異形成症(FOP))を含む様々な疾患が、説明される方法を使用して処置され得る。Formula (I), the stereoisomers thereof, tautomers, pharmaceutically acceptable salts, and prodrugs are included, wherein R1, R2, R3, and R4 are defined herein. Disclosed is a method for treating a disease of interest in need thereof by administering an ACVR1 inhibitor having (I). Subjects who can benefit from the treatment may have mutations in their ACVR1 gene. Various diseases, including cancer (eg, diffuse true pontine glioma (DIPG)) and genetic disorders (eg, progressive ossifying fibrodysplasia (FOP)), are described using the methods described. Can be treated.
Description
相互参照
本出願は、参照によりその全体が本明細書に組み込まれる2018年7月26日に出願された米国仮出願第62/703,862号の利益を主張する。
Cross-references This application claims the interests of US Provisional Application No. 62 / 703,862, filed July 26, 2018, which is incorporated herein by reference in its entirety.
発明の分野
本開示は、ACVR1阻害剤としての活性を有する化合物の治療上の用途およびその化合物を投与することを含む処置の方法を、提供する。本開示は、ACVR1阻害剤としての活性を有する化合物を含む、医薬組成物も提供する。さらに本開示は、ACVR1阻害剤としての活性を有する化合物の新規な形態を提供する。
INDUSTRIAL APPLICABILITY The present disclosure provides therapeutic uses of a compound having activity as an ACVR1 inhibitor and methods of treatment including administration of the compound. The present disclosure also provides a pharmaceutical composition comprising a compound having activity as an ACVR1 inhibitor. Furthermore, the present disclosure provides a novel form of a compound having activity as an ACVR1 inhibitor.
発明の背景
骨形成タンパク質(BMP)は、体内の様々な臓器全体にわたって組織機構を協調させるのに必須の役割を果たす、多面発現増殖因子である。BMP配位子は、セリン/トレオニンキナーゼ受容体の形質転換増殖因子ベータ(TGF−b)スーパーファミリーに属する、骨形成タンパク質受容体(BMPR)と、相互に作用する(Ikushima, H. and K. Miyazono, Biology of Transforming Growth Factor-beta Signalin. Curr Pharm Biotechnol, 2011、これは、そのような背景の教示に関して、参照により本明細書に組み込まれる)。配位子はII型受容体と結合し、次いでヘテロマー錯体を形成するI型受容体を補充する。錯体として、II型受容体はI型受容体をリン酸化し、このI型受容体は活性になりかつ下流のシグナル伝達分子をリン酸化する。これらの受容体を活性化する下流の作用は、主にタンパク質のSMADファミリーによって行われる。SMADはリン酸化されたものになり、細胞膜からのシグナルを核に伝達し、そこで遺伝子発現を調節するための転写因子として機能する(Massague, J., J. Seoane, and D. Wotton, Smad transcription factors. Genes Dev, 2005. 19(23): p. 2783-810、これは、そのような背景の教示に関して、参照により本明細書に組み込まれる)。
Background of the Invention Bone morphogenetic protein (BMP) is a pleiotropic growth factor that plays an essential role in coordinating tissue mechanisms throughout various organs in the body. BMP ligands interact with bone morphogenetic protein receptors (BMPRs), which belong to the transforming growth factor beta (TGF-b) superfamily of serine / threonine kinase receptors (Ikushima, H. and K. Miyazono, Biology of Transforming Growth Factor-beta Signalin. Curr Pharm Biotechnol, 2011, which is incorporated herein by reference with respect to such background teachings). The ligand binds to the type II receptor and then replenishes the type I receptor to form a heteromer complex. As a complex, the type II receptor phosphorylates the type I receptor, which becomes active and phosphorylates downstream signaling molecules. The downstream action of activating these receptors is primarily carried out by the SMAD family of proteins. SMAD becomes phosphorylated and transmits signals from the cell membrane to the nucleus, where it functions as a transcription factor for regulating gene expression (Massague, J., J. Seoane, and D. Wotton, Smad transcription). factors. Genes Dev, 2005. 19 (23): p. 2783-810, which is incorporated herein by reference with respect to such background teachings).
がんおよび炎症などの慢性疾患を持つ個体では、BMPシグナル伝達が恒常的に活性化されて貧血症をもたらす。この状態を一般に、慢性疾患の貧血(ACD)と呼び、これはがん患者に関連する衰弱性の症状である(Cullis, J.O., Diagnosis and management of anaemia of chronic disease: current status. Br J Haematol, 2011. 154(3): p. 289-300、これは、そのような背景の教示に関して、参照により本明細書に組み込まれる)。がん患者の慢性貧血症は、極端な脱力感および疲労をもたらし、これらの個体のクオリティーオブライフを不十分なものにする。これらの患者において、2種のBMP I型受容体、ALK2(記述されるように、ACVR1としても公知である)およびALK3を通したBMPシグナル伝達は、ヘプシジンと呼ばれるペプチドホルモンの肝臓発現を誘発させる(Steinbicker, A.U., et al., Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood, 2011. 118(15): p. 4224-30、これは、そのような背景の教示に関し、参照により本明細書に組み込まれる)。 In individuals with chronic diseases such as cancer and inflammation, BMP signaling is constitutively activated leading to anemia. This condition is commonly referred to as anemia of chronic disease (ACD), which is a debilitating symptom associated with cancer patients (Cullis, JO, Diagnosis and management of anaemia of chronic disease: current status. Br J Haematol, 2011. 154 (3): p. 289-300, which is incorporated herein by reference with respect to such background teachings). Chronic anemia in cancer patients results in extreme weakness and fatigue, resulting in an inadequate quality of life for these individuals. In these patients, BMP signaling through two BMP type I receptors, ALK2 (also known as ACVR1 as described) and ALK3, induces liver expression of a peptide hormone called hepcidin. (Steinbicker, AU, et al., Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood, 2011. 118 (15): p. 4224-30, which relates to the teaching of such background. , Incorporated herein by reference).
ヘプシジンは、鉄輸送体、フェロポルチンの分解を促進させることにより、血清中鉄レベルを低減させ、その結果、マクロファージおよびその他の細胞型に蓄えられる鉄が増加し、ヘモグロビンおよび赤血球(RBC)機能に鉄を利用できなくなる。患者の鉄の摂取を補うことで、活性化されたBMP経路および高い血清中ヘプシジンレベルに起因して消化された鉄は蓄えられるだけであるので、ACDは逆転しない。現在、がんにおけるACDは、患者の身体的活動を制限することによって管理され、輸血は、最も重篤な症例で使用される。これらの患者におけるBMPシグナル伝達の阻害は、そのクオリティーオブライフに実際の相違をもたらす可能性があり、最終的には、治療、放射線または外科手術にどのように応答するかに関して良い影響を及ぼす可能性がある(Steinbicker, A.U., et al., Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood, 2011. 117(18): p. 4915-23; Coyne, D.W., Hepcidin: clinical utility as a diagnostic tool and therapeutic target. Kidney Int, 2011. 80(3): p. 240-4; Theurl, I., et al., Pharmacologic inhibition of hepcidin expression reverses anemia of chronic disease in rats. Blood, 2011、これらは、そのような背景の教示に関して、それぞれが参照により本明細書に組み込まれる)。 Hepcidin reduces serum iron levels by promoting the breakdown of the iron transporter, ferroportin, resulting in increased iron storage in macrophages and other cell types, iron for hemoglobin and red blood cell (RBC) function. Will not be available. Supplementing the patient's iron intake does not reverse the ACD, as only the iron digested due to the activated BMP pathway and high serum hepcidin levels is stored. Currently, ACD in cancer is controlled by limiting the physical activity of the patient, and blood transfusions are used in the most severe cases. Inhibition of BMP signaling in these patients can make a real difference in their quality of life and ultimately can have a positive effect on how they respond to treatment, radiation or surgery. Steinbicker, AU, et al., Inhibition of bone morphogenetic protein signaling immunos anemia associated with inflammation. Blood, 2011. 117 (18): p. 4915-23; Coyne, DW, Hepcidin: clinical utility as a diagnostic tool and therapeutic target. Kidney Int, 2011. 80 (3): p. 240-4; Theurl, I., et al., Pharmacologic inhibition of hepcidin expression reverses anemia of chronic disease in rats. Blood, 2011, Each such background teaching is incorporated herein by reference).
ACDにおけるその機能に加え、BMPシグナル伝達は、がん細胞の成長および転移において、特に乳がん、前立腺がん、および骨に頻繁に転移するその他のがんにおいて、極めて重要な役割を果たす(Ye, L., M.D. Mason, and W.G. Jiang, Bone morphogenetic protein and bone metastasis, implication and therapeutic potential. Front Biosci, 2011. 16: p. 865-97)。BMPおよびBMPRは、転移性乳がん細胞において、転移性の低いものよりも高度に発現し、骨硬化性骨転移を発生させる前立腺がん細胞でも発現する(Bobinac, D., et al., Expression of bone morphogenetic proteins in human metastatic prostate and breast cancer. Croat Med J, 2005. 46(3): p. 389-96)。がん細胞の侵襲性および転移性に影響を及ぼすことに加え、BMP経路は、骨微小環境に影響を及ぼすことも示されてきた。研究は、BMPシグナル伝達の阻害が、前立腺がん骨転移の前臨床モデルにおいて、骨腫瘍負荷および溶骨性疾患を著しく低減させることを示していた。これらの結果は、BMP阻害剤が、骨転移の予防における用途を有する可能性があることを示唆する。
さらに、BMP阻害剤は、がん以外の複数の疾患徴候を処置する可能性を有する。ACDは、リウマチ様関節炎、全身性ループス、慢性腎疾患、および多くのその他の炎症性疾患を含むその他の疾患に罹っている個体に、影響を及ぼす壊滅的状態である。さらに、進行性骨化性線維異形成症(FOP)と呼ばれる、希少な小児の遺伝性疾患は、alk2遺伝子の変異を活性化することによって引き起こされることが示されている(Kaplan, F.S., et al., Investigations of activated ACVR1/ALK2, a bone morphogenetic protein type I receptor, that causes fibrodysplasia ossificans progressiva. Methods Enzymol, 2010. 484: p. 357-73)。この疾患におけるALK2の変異は、線維組織(筋肉、腱、靭帯など)が損傷したときに骨化させる。FOPの動物モデルで行われた研究は、ALK2の阻害が、FOPに関連した「再燃」を低下させ、モデルの修復組織の骨化を予防することを示唆する。
In addition to its function in ACD, BMP signaling plays a vital role in the growth and metastasis of cancer cells, especially in breast cancer, prostate cancer, and other cancers that frequently metastasize to bone (Ye, L., MD Mason, and WG Jiang, Bone morphogenetic protein and bone metastasis, implication and therapeutic potential. Front Biosci, 2011. 16: p. 865-97). BMP and BMPR are more highly expressed in metastatic breast cancer cells than those with low metastasis, and are also expressed in prostate cancer cells that cause osteosclerotic bone metastases (Bobinac, D., et al., Expression of). bone morphogenetic proteins in human metastatic prostate and breast cancer. Croat Med J, 2005. 46 (3): p. 389-96). In addition to affecting the invasiveness and metastasis of cancer cells, the BMP pathway has also been shown to affect the bone microenvironment. Studies have shown that inhibition of BMP signaling significantly reduces bone tumor burden and osteolytic disease in a preclinical model of prostate cancer bone metastasis. These results suggest that BMP inhibitors may have uses in the prevention of bone metastases.
In addition, BMP inhibitors have the potential to treat multiple disease signs other than cancer. ACD is a catastrophic condition that affects individuals with other diseases, including rheumatoid arthritis, systemic lupus, chronic kidney disease, and many other inflammatory diseases. In addition, a rare pediatric hereditary disease called progressive ossifying fibrodysplasia (FOP) has been shown to be caused by activating mutations in the alk2 gene (Kaplan, FS, et). al., Investigations of activated ACVR1 / ALK2, a bone morphogenetic protein type I receptor, that causes fibrodysplasia ossificans progressiva. Methods Enzymol, 2010. 484: p. 357-73). Mutations in ALK2 in this disease cause ossification when fibrous tissue (muscles, tendons, ligaments, etc.) is damaged. Studies conducted in animal models of FOP suggest that inhibition of ALK2 reduces FOP-related "relapse" and prevents ossification of the model's repair tissue.
本開示の概要
一実施形態では、本開示は、びまん性真性橋膠腫(DIPG)の処置を必要とする対象のびまん性真性橋膠腫(DIPG)を処置する方法であって、方法は、治療有効量の式(2):

一態様では、結晶塩は、酸付加塩である。一態様では、酸付加塩は、塩酸塩である。一態様では、塩酸塩は、1価である。一態様では、結晶塩形態は、無水物である。一態様では、対象は、小児患者である。一態様では、対象は、約1週齢から約22歳である。一態様では、対象は、約18歳またはそれよりも若い。一態様では、対象は、約10歳またはそれよりも若い。一態様では、対象は、約8歳またはそれよりも若い。一態様では、対象は、約6歳またはそれよりも若い。一態様では、DIPGは、新たに診断されたものまたは再発である。一態様では、DIPGは、浸潤性グリオーマ グレードIIからIVと組織学的診断がなされた脳橋腫瘍として特徴付けられる。一態様では、方法は、放射線療法を投与することをさらに含む。一態様では、放射線の投与は、化合物またはその結晶塩の投与の前に行われる。一態様では、放射線の投与は、化合物またはその結晶塩の投与の後に行われる。一態様では、放射線の投与は、化合物またはその結晶塩の投与の前および後の両方で行われる。一態様では、方法は、1種またはそれより多種の追加の治療剤を投与することをさらに含む。一態様では、放射線は、構成要素剤(component agent)として投与され得る。一態様では、化合物またはその結晶塩の投与は、DIPGに関連する1つまたはそれより多くの徴候または症状を低減させまたは軽減させる。一態様では、1つまたはそれより多くの徴候または症状は、会話または会話パターンの変容、対象の顔または身体の片側を動かす能力の損失、バランスの損失、協調の損失、歩行または運動に関するトラブル、視覚の問題、聴覚の問題、頭痛、吐き気、嘔吐、異常な眠気、エネルギーレベルの変容、行動変化、および学校での成績の変化からなる群から選択される。一態様では、化合物またはその結晶塩の投与は、無憎悪生存期間を実現する。一態様では、無憎悪生存期間は、1か月もしくはそれよりも長い、2か月もしくはそれよりも長い、3か月もしくはそれよりも長い、4か月もしくはそれよりも長い、5か月もしくはそれよりも長い、6か月もしくはそれよりも長い、7か月もしくはそれよりも長い、8か月もしくはそれよりも長い、9か月もしくはそれよりも長い、10か月もしくはそれよりも長い、11か月もしくはそれよりも長い、1年もしくはそれよりも長い、2年もしくはそれよりも長い、3年もしくはそれよりも長い、または5年もしくはそれよりも長い。一態様では、方法は、腫瘍成長の減量術または脳脊髄液転換の1つまたは複数をさらに含む。一態様では、対象は、ACVR1遺伝子中に1つまたはそれより多くの変異を含む所定の遺伝子プロファイルを有する。一態様では、ACVR1遺伝子中の1つまたはそれより多くの変異は、活性化変異である。一態様では、ACVR1遺伝子中の1つまたはそれより多くの変異は、R206H、G328V、R258G、またはこれらの組合せから選択される、1つまたはそれより多くのアミノ酸残基におけるアミノ酸置換を含むACVR1ポリペプチドをコードする。一態様では、ACVR1ポリペプチド中のアミノ酸置換がR206Hを含む。一態様では、化合物またはその結晶塩が経口投与される。一態様では、化合物またはその結晶塩は、週当たり約10mgから約320mgに及ぶ用量で投与される。一態様では、用量は、週当たり約30mgから約240mgに及ぶ。一態様では、用量は、週当たり約60mgから約180mgに及ぶ。一態様では、用量は、週当たり約30mgから約120mgに及ぶ。一態様では、用量は、週当たり約60mgから約120mgに及ぶ。一態様では、用量は、週当たり約60mgである。一態様では、用量は、週当たり約90mgである。一態様では、用量は、週当たり約120mgである。一態様では、用量は、週当たり約180mgである。一態様では、用量は、週当たり約210mgである。一態様では、用量は、週当たり約240mgである。一態様では、化合物またはその結晶塩は、約320mgもしくはそれよりも少ない、約240mgもしくはそれよりも少ない、約210もしくはそれよりも少ない、約180もしくはそれよりも少ない、約120もしくはそれよりも少ない、約90もしくはそれよりも少ない、約60もしくはそれよりも少ない、または約30もしくはそれよりも少ない1週用量で投与される。一態様では、用量は、週当たり約60mgまたはそれよりも少ない。一態様では、用量は、週当たり約90mgまたはそれよりも少ない。一態様では、用量は、週当たり約120mgまたはそれよりも少ない。一態様では、用量は、週当たり約180mgまたはそれよりも少ない。一態様では、用量は、週当たり約210mgまたはそれよりも少ない。一態様では、用量は、週当たり約240mgまたはそれよりも少ない。一態様では、用量は、週に1回投与される。一態様では、用量は、1週間のクールにわたって2回またはそれを超える回数の部分用量、3回またはそれを超える回数の部分用量、4回またはそれを超える回数の部分用量、5回またはそれを超える回数の部分用量、6回またはそれを超える回数の部分用量、または毎日の部分用量で投与される。一態様では、対象が小児患者であり、用量は、用量範囲の約80%から100%の間である。一態様では、用量は、用量範囲の80%、85%、90%、または95%に調節される。一態様では、用量は、週当たり約8mgから約320mgに及ぶ。一態様では、用量は、週当たり約24mgから約240mgに及ぶ。一態様では、用量は、週当たり約24mgから約120mgに及ぶ。一態様では、用量は、週当たり約48mgから約120mgに及ぶ。一態様では、用量は、週当たり約72mgから約120mgに及ぶ。一態様では、用量は、週当たり約96mgから約120mgに及ぶ。一態様では、対象は、少なくとも約0.1ng/mLの所定のヘプシジンレベルを有する。一態様では、所定のヘプシジンレベルは、約10ng/mLから約200ng/mLに及ぶ。一態様では、方法は、化合物またはその結晶塩が投与された後に、対象のヘプシジンレベルを決定することをさらに含む。一態様では、方法は、化合物またはその結晶塩が投与された後に、対象のトランスフェリン飽和レベルを決定することをさらに含む。一態様では、トランスフェリン飽和レベルは、約50%未満である。一態様では、トランスフェリン飽和レベルは、約45%未満である。一態様では、トランスフェリン飽和レベルは、約40%未満である。一態様では、化合物またはその結晶塩は、1回またはそれを超える回数の処置サイクルにわたり投与され、各サイクルが4週を含む。一態様では、方法は、処置サイクル間に1日またはそれを超える日数の休処置日をさらに含む。一態様では、休処置日は、1日、2日、3日、4日、5日、6日、1週、2週、3週、または4週から選択される。 In one aspect, the crystalline salt is an acid addition salt. In one aspect, the acid addition salt is a hydrochloride salt. In one aspect, the hydrochloride salt is monovalent. In one aspect, the crystalline salt form is anhydrous. In one aspect, the subject is a pediatric patient. In one aspect, the subject is about 1 week to about 22 years old. In one aspect, the subject is about 18 years or younger. In one aspect, the subject is about 10 years or younger. In one aspect, the subject is about 8 years or younger. In one aspect, the subject is about 6 years or younger. In one aspect, DIPG is a newly diagnosed or recurrent. In one aspect, DIPG is characterized as a pontine tumor histologically diagnosed as invasive glioma grade II through IV. In one aspect, the method further comprises administering radiation therapy. In one aspect, the administration of radiation is performed prior to the administration of the compound or a crystalline salt thereof. In one aspect, the administration of radiation is performed after administration of the compound or a crystalline salt thereof. In one aspect, the administration of radiation takes place both before and after administration of the compound or its crystalline salt. In one aspect, the method further comprises administering one or more additional therapeutic agents. In one aspect, the radiation can be administered as a component agent. In one aspect, administration of the compound or crystalline salt thereof reduces or alleviates one or more signs or symptoms associated with DIPG. In one aspect, one or more signs or symptoms are changes in conversation or conversation patterns, loss of ability to move one side of the subject's face or body, loss of balance, loss of coordination, gait or motor problems, Select from the group consisting of visual problems, hearing problems, headache, nausea, vomiting, abnormal drowsiness, altered energy levels, behavioral changes, and changes in school performance. In one aspect, administration of the compound or a crystalline salt thereof achieves a hate-free survival. In one aspect, hate-free survival is one month or longer, two months or longer, three months or longer, four months or longer, five months or longer. Longer, 6 months or longer, 7 months or longer, 8 months or longer, 9 months or longer, 10 months or longer, 11 months or longer, 1 year or longer, 2 years or longer, 3 years or longer, or 5 years or longer. In one aspect, the method further comprises one or more of tumor growth weight loss or cerebrospinal fluid conversion. In one aspect, the subject has a given gene profile that contains one or more mutations in the ACVR1 gene. In one aspect, one or more mutations in the ACVR1 gene are activating mutations. In one aspect, one or more mutations in the ACVR1 gene are selected from R206H, G328V, R258G, or a combination thereof and contain an amino acid substitution at one or more amino acid residues. Encode the peptide. In one aspect, the amino acid substitution in the ACVR1 polypeptide comprises R206H. In one aspect, the compound or a crystalline salt thereof is orally administered. In one aspect, the compound or crystalline salt thereof is administered at a dose ranging from about 10 mg to about 320 mg per week. In one aspect, the dose ranges from about 30 mg to about 240 mg per week. In one aspect, the dose ranges from about 60 mg to about 180 mg per week. In one aspect, the dose ranges from about 30 mg to about 120 mg per week. In one aspect, the dose ranges from about 60 mg to about 120 mg per week. In one aspect, the dose is about 60 mg per week. In one aspect, the dose is about 90 mg per week. In one aspect, the dose is about 120 mg per week. In one aspect, the dose is about 180 mg per week. In one aspect, the dose is about 210 mg per week. In one aspect, the dose is about 240 mg per week. In one aspect, the compound or crystalline salt thereof is about 320 mg or less, about 240 mg or less, about 210 or less, about 180 or less, about 120 or less. , About 90 or less, about 60 or less, or about 30 or less weekly doses. In one aspect, the dose is about 60 mg or less per week. In one aspect, the dose is about 90 mg or less per week. In one aspect, the dose is about 120 mg or less per week. In one aspect, the dose is about 180 mg or less per week. In one aspect, the dose is about 210 mg or less per week. In one aspect, the dose is about 240 mg or less per week. In one aspect, the dose is administered once a week. In one aspect, the dose is a partial dose of 2 or more times over a week of cool, a partial dose of 3 or more times, a partial dose of 4 or more times, 5 times or more. It is administered in excess of partial doses, 6 or more partial doses, or daily partial doses. In one aspect, the subject is a pediatric patient and the dose is between about 80% and 100% of the dose range. In one aspect, the dose is adjusted to 80%, 85%, 90%, or 95% of the dose range. In one aspect, the dose ranges from about 8 mg to about 320 mg per week. In one aspect, the dose ranges from about 24 mg to about 240 mg per week. In one aspect, the dose ranges from about 24 mg to about 120 mg per week. In one aspect, the dose ranges from about 48 mg to about 120 mg per week. In one aspect, the dose ranges from about 72 mg to about 120 mg per week. In one aspect, the dose ranges from about 96 mg to about 120 mg per week. In one aspect, the subject has a predetermined hepcidin level of at least about 0.1 ng / mL. In one aspect, predetermined hepcidin levels range from about 10 ng / mL to about 200 ng / mL. In one aspect, the method further comprises determining the hepcidin level of interest after administration of the compound or crystalline salt thereof. In one aspect, the method further comprises determining the transferrin saturation level of the subject after administration of the compound or crystalline salt thereof. In one aspect, the transferrin saturation level is less than about 50%. In one aspect, the transferrin saturation level is less than about 45%. In one aspect, the transferrin saturation level is less than about 40%. In one aspect, the compound or crystalline salt thereof is administered over one or more treatment cycles, each cycle comprising 4 weeks. In one aspect, the method further comprises one or more rest days between treatment cycles. In one aspect, the rest day is selected from 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, or 4 weeks.
一実施形態では、本開示は、ACVR1の阻害の影響を受け易い疾患または障害を処置するための方法であって、方法は、ACVR1の阻害の影響を受け易い疾患または障害の処置を必要とする対象に、治療有効量の式(2):
一実施形態では、本開示は、1種またはそれより多種の薬学的に許容される賦形剤と、式(2):
一態様では、結晶塩は、酸付加塩である。一態様では、酸付加塩は、塩酸塩である。一態様では、塩酸塩は、1価である。一態様では、結晶塩形態は、無水物である。一態様では、医薬組成物は、ゼラチンカプセルである。一態様では、ゼラチンカプセルは、遊離塩基重量に対して(i)5mg、(ii)25mg、または(iii)125mgの力価である。一態様では、ゼラチンカプセルは、遊離塩基重量に対して(i)30mg、(ii)60mg、(iii)90mg、または(iv)120mgである。一態様では、1種またはそれより多種の医薬賦形剤は、微結晶性セルロース、ラクトース一水和物、クロスカルメロースナトリウム、ステアリン酸マグネシウム、またはこれらの組合せから選択される。 In one aspect, the crystalline salt is an acid addition salt. In one aspect, the acid addition salt is a hydrochloride salt. In one aspect, the hydrochloride salt is monovalent. In one aspect, the crystalline salt form is anhydrous. In one aspect, the pharmaceutical composition is a gelatin capsule. In one aspect, gelatin capsules are titered at (i) 5 mg, (ii) 25 mg, or (iii) 125 mg relative to free base weight. In one aspect, gelatin capsules are (i) 30 mg, (ii) 60 mg, (iii) 90 mg, or (iv) 120 mg relative to free base weight. In one aspect, one or more pharmaceutical excipients are selected from microcrystalline cellulose, lactose monohydrate, sodium croscarmellose, magnesium stearate, or a combination thereof.
一実施形態では、本開示は、式(2):
a)1種またはそれより多種の緩衝剤;
b)必要に応じて、1種またはそれより多種の保存剤;
c)必要に応じて、1種またはそれより多種の溶媒;
d)必要に応じて、1種またはそれより多種の矯味剤;および
e)必要に応じて、1種またはそれより多種のさらなるpH調節剤
とを含む、経口液体医薬組成物を提供する。
In one embodiment, the present disclosure comprises formula (2) :.
a) One or more buffers;
b) One or more preservatives as needed;
c) One or more solvents as needed;
d) Provided are oral liquid pharmaceutical compositions comprising one or more flavoring agents as needed; and e) one or more additional pH regulators as needed.
一態様では、結晶塩は、酸付加塩である。一態様では、酸付加塩は、塩酸塩である。一態様では、塩酸塩は、1価である。一態様では、結晶塩形態は、無水物である。一態様では、組成物が約2.0から約5.0の間のpHを有する。一態様では、組成物が約2.0から約3.5の間のpHを有する。一態様では、組成物が約2.0のpHを有する。一態様では、緩衝剤は、クエン酸、酒石酸、リンゴ酸、または酢酸から選択される。一態様では、緩衝剤は、リンゴ酸である。一態様では、リンゴ酸は、DL−リンゴ酸である。一態様では、組成物は、1種またはそれより多種の保存剤を含む。一態様では、保存剤は、安息香酸、安息香酸ナトリウム、パラヒドロキシ安息香酸メチル、パラヒドロキシ安息香酸プロピル、またはプロピレングリコールから選択される。一態様では、保存剤は、安息香酸である。一態様では、安息香酸は、保存剤および緩衝剤である。一態様では、1種またはそれより多種の矯味剤を含む。一態様では、矯味剤は、スクラロース、グリセリン、シクロデキストリン、HP−β−シクロデキストリン、α−シクロデキストリン、β−シクロデキストリン、またはこれらの組合せから選択される。一態様では、矯味剤は、HP−β−シクロデキストリンおよびスクラロースの組合せである。一態様では、組成物は、式(2)の化合物またはその結晶塩を、約10mg/mLの濃度で含む。一態様では、組成物は、リンゴ酸を最高約6.7mg/mLの濃度で含む。一態様では、組成物は、リンゴ酸を約1.3mg/mLの濃度で含む。一態様では、組成物は、HP−β−シクロデキストリンを最高約300mg/mLの濃度で含む。一態様では、組成物は、HP−β−シクロデキストリンを最高約150mg/mLの濃度で含む。一態様では、組成物は、スクラロースを最高約2.0mg/mLの濃度で含む。一態様では、組成物は、スクラロースを約1.0mg/mLの濃度で含む。一態様では、組成物は、安息香酸を最高約3.0mg/mLの濃度で含む。一態様では、組成物は、安息香酸を最高約2.0mg/mLの濃度で含む。一態様では、pHは、塩酸で約2.0に調整される。一態様では、溶媒は水である。 In one aspect, the crystalline salt is an acid addition salt. In one aspect, the acid addition salt is a hydrochloride salt. In one aspect, the hydrochloride salt is monovalent. In one aspect, the crystalline salt form is anhydrous. In one aspect, the composition has a pH between about 2.0 and about 5.0. In one aspect, the composition has a pH between about 2.0 and about 3.5. In one aspect, the composition has a pH of about 2.0. In one aspect, the buffer is selected from citric acid, tartaric acid, malic acid, or acetic acid. In one aspect, the buffer is malic acid. In one aspect, the malic acid is DL-malic acid. In one aspect, the composition comprises one or more preservatives. In one aspect, the preservative is selected from benzoic acid, sodium benzoate, methyl parahydroxybenzoate, propyl parahydroxybenzoate, or propylene glycol. In one aspect, the preservative is benzoic acid. In one aspect, benzoic acid is a preservative and buffer. In one aspect, it comprises one or more flavoring agents. In one aspect, the flavoring agent is selected from sucralose, glycerin, cyclodextrin, HP-β-cyclodextrin, α-cyclodextrin, β-cyclodextrin, or a combination thereof. In one aspect, the flavoring agent is a combination of HP-β-cyclodextrin and sucralose. In one aspect, the composition comprises the compound of formula (2) or a crystalline salt thereof at a concentration of about 10 mg / mL. In one aspect, the composition comprises malic acid at a concentration of up to about 6.7 mg / mL. In one aspect, the composition comprises malic acid at a concentration of about 1.3 mg / mL. In one aspect, the composition comprises HP-β-cyclodextrin at a concentration of up to about 300 mg / mL. In one aspect, the composition comprises HP-β-cyclodextrin at a concentration of up to about 150 mg / mL. In one aspect, the composition comprises sucralose at a concentration of up to about 2.0 mg / mL. In one aspect, the composition comprises sucralose at a concentration of about 1.0 mg / mL. In one aspect, the composition comprises benzoic acid at a concentration of up to about 3.0 mg / mL. In one aspect, the composition comprises benzoic acid at a concentration of up to about 2.0 mg / mL. In one aspect, the pH is adjusted to about 2.0 with hydrochloric acid. In one aspect, the solvent is water.
一実施形態では、本開示は、化合物(2)
一態様では、塩は、薬学的に許容される塩である。一態様では、薬学的に許容される塩は、HCl塩である。一態様では、結晶形態は、形態Aを含む。一態様では、結晶形態は、形態Aから本質的になる。一態様では、形態Aは、不純物を実質的に含まない。 In one aspect, the salt is a pharmaceutically acceptable salt. In one aspect, the pharmaceutically acceptable salt is an HCl salt. In one aspect, the crystalline form comprises form A. In one aspect, the crystalline morphology essentially consists of morphology A. In one aspect, Form A is substantially free of impurities.
一実施形態では、本開示は、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の、化合物形態Aを提供する。 In one embodiment, the present disclosure, N 4 - (2,2'-bipyridine-3-yl) -N 2 - (3- methoxy-4- (4-methylpiperazin-1-yl) phenyl) pyrimidin-2 , 4-Diamine Anhydrous Hydrochloride, Compound Form A.
一実施形態では、本開示は、13.53、16.14、17.67、18.38、24.96、および28.18から選択される1つまたは複数の2θ値を含むx線回折パターン(XRPD)を特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態Aを提供する。 In one embodiment, the present disclosure is an x-ray diffraction pattern comprising one or more 2θ values selected from 13.53, 16.14, 17.67, 18.38, 24.96, and 28.18. (XRPD), characterized in, N 4 - (2,2'-bipyridine-3-yl) -N 2 - (3- methoxy-4- (4-methylpiperazin-1-yl) phenyl) pyrimidin-2, The compound form A of 4-diamine anhydrous hydrochloride is provided.
一態様では、形態は、列挙される2θ値のうちの2つまたはそれよりも多くを特徴とする。一態様では、形態は、列挙される2θ値のうちの3つまたはそれよりも多くを特徴とする。一態様では、形態は、列挙される2θ値のうちの4つまたはそれよりも多くを特徴とする。一態様では、形態は、列挙される2θ値のうちの5つまたはそれよりも多くを特徴とする。一態様では、形態は、列挙される2θ値の6つ全てを特徴とする。一態様では、形態は、6.71、19.25、23.98、および29.60から選択される1つまたは複数の2θ値をさらに特徴とする。一態様では、形態は、列挙される2θ値のうちの2つまたはそれよりも多くを特徴とする。一態様では、形態は、列挙される2θ値のうちの3つまたはそれよりも多くを特徴とする。一態様では、形態は、列挙される2θ値のうちの4つ全てを特徴とする。一態様では、X線粉末回折計は、Cu kαのX線波長、Kα1(Å):1.540598、Kα2(Å):1.544426を使用して、Kα2/Kα1強度比を0.50、そしてX線管の設定を45kV、40mAにして反射モードで使用される。一態様では、2θ値は、±0.2 2θ以内である。 In one aspect, the morphology is characterized by two or more of the listed 2θ values. In one aspect, the morphology is characterized by three or more of the listed 2θ values. In one aspect, the morphology is characterized by four or more of the listed 2θ values. In one aspect, the morphology is characterized by five or more of the listed 2θ values. In one aspect, the morphology is characterized by all six of the listed 2θ values. In one aspect, the form is further characterized by one or more 2θ values selected from 6.71, 19.25, 23.98, and 29.60. In one aspect, the morphology is characterized by two or more of the listed 2θ values. In one aspect, the morphology is characterized by three or more of the listed 2θ values. In one aspect, the morphology is characterized by all four of the listed 2θ values. In one aspect, the X-ray powder diffractometer uses the X-ray wavelength of Cu kα, Kα1 (Å): 1.540598, Kα2 (Å): 1.5444426, with a Kα2 / Kα1 intensity ratio of 0.50. Then, the X-ray tube is set to 45 kV and 40 mA and used in the reflection mode. In one aspect, the 2θ value is within ± 0.2 2θ.
一実施形態では、本開示は、図12と実質的に同じx線回折パターン(XRPD)を特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態Aを提供する。
In one embodiment, the present disclosure features a 12 substantially the same
一実施形態では、本開示は、196.2℃、214.8℃、および274.0℃のうちの1つまたは複数での吸熱を特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態Aを提供する。 In one embodiment, the present disclosure, 196.2 ° C., characterized by endotherm at one or more of 214.8 ° C., and 274.0 ℃, N 4 - (2,2'- bipyridine - 3 is provided in compound form A of 3-yl) -N 2- (3-methoxy-4- (4-methylpiperazin-1-yl) phenyl) pyrimidin-2,4-diamine anhydride.
一実施形態では、本開示は、198.9℃、218.0℃、および275.9℃のうちの1つまたは複数でのピーク吸熱をさらに特徴とする。一態様では、形態は、274.0℃の開始温度をさらに特徴とする。一態様では、形態は、150℃までで1.7%の重量損失をさらに特徴とする。 In one embodiment, the disclosure is further characterized by peak endotherm at one or more of 198.9 ° C., 218.0 ° C., and 275.9 ° C. In one aspect, the morphology is further characterized by a starting temperature of 274.0 ° C. In one aspect, the form is further characterized by a weight loss of 1.7% up to 150 ° C.
一実施形態では、本開示は、図15と実質的に同じTGA−DSCサーモグラムを特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態Aを提供する。 In one embodiment, the disclosure features a substantially identical TGA-DSC thermogram and FIG 15, N 4 - (2,2'-bipyridine-3-yl) -N 2 - (3- methoxy - Provided is compound form A of 4- (4-methylpiperazin-1-yl) phenyl) pyrimidin-2,4-diamine anhydrous hydrochloride.
一実施形態では、本開示は、本開示の結晶質形態を含む医薬組成物を提供する。一態様では、組成物は固体用量製剤である。特定の態様では、固体用量製剤は、化合物が本開示の結晶質形態である、本開示の経口固体医薬組成物である。一態様では、組成物は液体用量製剤である。特定の態様では、液体用量製剤は、化合物が本開示の結晶質形態である、本開示の経口液体医薬組成物である。 In one embodiment, the present disclosure provides a pharmaceutical composition comprising the crystalline form of the present disclosure. In one aspect, the composition is a solid dose formulation. In a particular aspect, the solid-dose formulation is the oral solid pharmaceutical composition of the present disclosure, wherein the compound is in the crystalline form of the present disclosure. In one aspect, the composition is a liquid dose formulation. In a particular aspect, the liquid-dose formulation is the oral liquid pharmaceutical composition of the present disclosure, wherein the compound is in the crystalline form of the present disclosure.
一実施形態では、本開示は、化合物が本開示の結晶質形態である、本開示の任意の方法を提供する。 In one embodiment, the present disclosure provides any method of the present disclosure, wherein the compound is in crystalline form of the present disclosure.
一実施形態では、本開示は、単独でまたは放射線もしくは追加の治療剤のいずれかと組み合わせて、本開示の結晶質形態を含む、医薬組成物を提供する。一態様では、組成物は固体用量製剤である。一態様では、組成物は液体用量製剤である。 In one embodiment, the disclosure provides a pharmaceutical composition comprising the crystalline form of the present disclosure, either alone or in combination with radiation or an additional therapeutic agent. In one aspect, the composition is a solid dose formulation. In one aspect, the composition is a liquid dose formulation.
本開示の一実施形態は、本開示の任意の化合物を投与することを含む、本開示の任意の方法を含む。 One embodiment of the present disclosure includes any method of the present disclosure, comprising administering any compound of the present disclosure.
本開示の一実施形態は、本開示の1つまたはそれより多くの処置での本開示の任意の化合物の使用を含む。 One embodiment of the present disclosure comprises the use of any compound of the present disclosure in one or more of the treatments of the present disclosure.
本開示の一実施形態は、本明細書に開示される1つまたはそれより多くの疾患または障害の処置のための、本開示による医薬の調製を含む。 One embodiment of the present disclosure comprises the preparation of a medicament according to the present disclosure for the treatment of one or more diseases or disorders disclosed herein.
本開示の一実施形態は、治療における本開示の化合物の使用を含む。 One embodiment of the present disclosure includes the use of the compounds of the present disclosure in treatment.
簡単に言うと、本開示の実施形態は、ACVR1遺伝子中に1つまたはそれより多くの変異を含む所定の遺伝子プロファイルを有する対象に、ACVR1阻害剤を含む処置レジメンを投与することを含む、それを必要とする対象の疾患を処置するための方法であって、ACVR1阻害剤が、以下の式(I):
本開示のさらなる実施形態は、ACVR1阻害剤を含む処置レジメンを対象に投与することを含む、それを必要とする対象のびまん性真性橋膠腫(DIPG)を処置するための方法であって、ACVR1阻害剤が、以下の式(I):
本開示の追加の実施形態は、ACVR1阻害剤および治療剤を含む処置レジメンを対象に投与することを含む、それを必要とする対象の進行性骨化性線維異形成症(FOP)を処置するための方法であって、ACVR1阻害剤が、以下の式(I):
本開示の一実施形態は、DIPGおよびFOPなどの異常なACVR1発現(例えば、発現したタンパク質の変異)を伴う疾患を処置するための、式(I)のACVR1阻害剤の使用を含み、同様に提供される。 One embodiment of the present disclosure comprises the use of an ACVR1 inhibitor of formula (I) to treat diseases associated with aberrant ACVR1 expression (eg, mutations in the expressed protein) such as DIPG and FOP, as well. Provided.
1つまたはそれより多くの態様および実施形態は、異なる実施形態に組み込まれてもよいが、特に記述しない。即ち、全ての態様および実施形態は、任意の方法または組合せで組み合わされてもよい。 One or more embodiments and embodiments may be incorporated into different embodiments, but are not specifically described. That is, all embodiments and embodiments may be combined in any way or combination.
本開示のこれらの態様およびその他の態様は、下記の詳細な記述を参照することにより明らかにされる。 These and other aspects of the disclosure will be apparent by reference to the detailed description below.
図中、同一の参照符号は類似の要素を特定する。図中の要素のサイズおよび相対的位置は、必ずしも縮尺に合わせて描かれておらず、これらの要素のいくつかは、図の見易さを改善するよう任意に拡大されかつ位置決めされる。さらに、描かれる要素の特定の形状は、特定の要素の実際の形状に関する任意の情報を伝えることを意図するものではなく、図における認識を容易にするために選択されただけである。 In the figure, the same reference numerals identify similar elements. The size and relative position of the elements in the figure are not necessarily drawn to scale, and some of these elements are arbitrarily magnified and positioned to improve the visibility of the figure. Moreover, the particular shape of the element being drawn is not intended to convey any information about the actual shape of the particular element, but has only been selected to facilitate recognition in the figure.
本開示の詳細な説明
本開示は、ACVR1阻害剤を投与することによって、異常なACVR1発現(例えば、発現タンパク質の変異)を伴う疾患(例えばがん、例えばびまん性真性橋膠腫(DIPG);遺伝的障害(例えば、進行性骨化性線維異形成症(FOP));など)を処置する方法に関する。
Detailed Description of the Disclosure The present disclosure describes diseases (eg, cancer, eg, diffuse true pontine glioma (DIPG)) associated with abnormal ACVR1 expression (eg, mutations in the expressed protein) by administration of an ACVR1 inhibitor; It relates to a method of treating a genetic disorder (eg, progressive ossifying fibrodysplasia (FOP); etc.).
本開示をより詳細に述べる前に、本明細書で使用されるある特定の用語の定義を提供することが、その理解の助けになると考えられる。追加の定義は、本開示の全体を通して述べられる。 Prior to discussing this disclosure in more detail, it is believed that providing definitions of certain terms as used herein will aid in their understanding. Additional definitions are given throughout this disclosure.
「アミノ」は、−NH2基を指す。 "Amino" refers to two -NH groups.
「シアノ」または「ニトリル」は、−CN基を指す。 "Cyanide" or "nitrile" refers to the -CN group.
「ハロ」は、−F、−Cl、−Br、または−I基を指す。 "Halo" refers to the -F, -Cl, -Br, or -I groups.
「ヒドロキシ」または「ヒドロキシル」は、−OH基を指す。 "Hydroxy" or "hydroxyl" refers to the -OH group.
「イミノ」は、=NH置換基を指す。 "Imino" refers to the = NH substituent.
「ニトロ」は、−NO2基を指す。 "Nitro" refers to two -NOs.
「オキソ」は、=O置換基を指す。 "Oxo" refers to the = O substituent.
「チオキソ」は、=S置換基を指す。 "Tioxo" refers to the = S substituent.
「アルキル」は、炭素および水素原子のみからなる直鎖状または分枝鎖状炭化水素鎖基であって、飽和または不飽和であり(即ち、1つまたはそれより多くの二重(アルケニル)結合および/または三重(アルキニル)結合を含有する)、1から12個の炭素原子を有し(C1〜C12アルキル)、好ましくは1から8個の炭素原子(C1〜C8アルキル)または1から6個の炭素原子(C1〜C6アルキル)を有し、単結合によって分子の残りに結合される基を指し、例えばメチル、エチル、n−プロピル、1−メチルエチル(イソプロピル)、n−ブチル、n−ペンチル、1,1−ジメチルエチル(t−ブチル)、3−メチルヘキシル、2−メチルヘキシル、エテニル、プロパ−1−エニル、ブタ−1−エニル、ペンタ−1−エニル、ペンタ−1,4−ジエニル、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、および同様のものがある。1つまたはそれより多くの炭素−炭素二重結合を含むアルキルは、アルケニルである。1つまたはそれより多くの炭素−炭素三重結合を含むアルキルは、アルキニルである。本明細書において他に特に示さない限り、アルキル基は、必要に応じて置換される。 An "alkyl" is a linear or branched hydrocarbon chain group consisting only of carbon and hydrogen atoms and is saturated or unsaturated (ie, one or more double (alkenyl) bonds. And / or containing a triple (alkynyl) bond), having 1 to 12 carbon atoms (C 1 to C 12 alkyl), preferably 1 to 8 carbon atoms (C 1 to C 8 alkyl) or A group that has 1 to 6 carbon atoms (C 1 to C 6 alkyl) and is attached to the rest of the molecule by a single bond, such as methyl, ethyl, n-propyl, 1-methylethyl (isopropyl), n-butyl, n-pentyl, 1,1-dimethylethyl (t-butyl), 3-methylhexyl, 2-methylhexyl, ethenyl, propa-1-enyl, porcine-1-enyl, penta-1-enyl, There are penta-1,4-dienyl, ethynyl, propynyl, butynyl, pentynyl, hexynyl, and the like. Alkyl containing one or more carbon-carbon double bonds are alkenyl. An alkyl containing one or more carbon-carbon triple bonds is an alkynyl. Unless otherwise specified herein, alkyl groups are substituted as needed.
「アルコキシ」は、式−ORaの基を指し、式中、Raは、1から12個の炭素原子を含有する上記定義されたアルキル基である。本明細書で他に特に示さない限り、アルコキシ基は必要に応じて置換される。 "Alkoxy" refers to a group of formula-OR a, where Ra is the above defined alkyl group containing 1 to 12 carbon atoms. Alkoxy groups are optionally substituted, unless otherwise indicated herein.
「縮合」は、本開示のACVR1阻害剤の既存の環構造と縮合している、本明細書に記述される任意の環構造を指す。縮合環がヘテロシクリル環である場合、縮合ヘテロシクリル環または縮合ヘテロアリール間の部分になる、既存の環構造上の任意の炭素原子は、窒素原子で置き換えられてもよい。 "Condensation" refers to any ring structure described herein that is fused to the existing ring structure of the ACVR1 inhibitor of the present disclosure. When the fused ring is a heterocyclyl ring, any carbon atom on the existing ring structure that becomes the portion between the condensed heterocyclyl ring or the condensed heteroaryl may be replaced with a nitrogen atom.
「ヘテロシクリル」または「複素環式環」は、2から12個の炭素原子と、窒素、酸素、および硫黄からなる群から選択される1から6個のヘテロ原子とを含む、安定な3から18員非芳香環基を指す。本明細書で他に特に示さない限り、ヘテロシクリル基は、縮合または架橋環系を含み得る単環式、二環式、三環式、または四環式環系であってもよく;ヘテロシクリル基中の窒素、炭素、または硫黄原子は、必要に応じて酸化されてもよく;窒素原子は、必要に応じて四級化されてもよく;ヘテロシクリル基は、部分的に飽和または完全に飽和であり得る。そのようなヘテロシクリル基の例には、ジオキソラニル、チエニル[1,3]ジチアニル、デカヒドロイソキノリル、イミダゾリニル、イミダゾリジニル、イソチアゾリジニル、イソオキサゾリジニル、モルホリニル、オクタヒドロインドリル、オクタヒドロイソインドリル、2−オキソピペラジニル、2−オキソピペリジニル、2−オキソピロリジニル、オキサゾリジニル、ピペリジニル、ピペラジニル、4−ピペリドニル、ピロリジニル、ピラゾリジニル、キヌクリジニル、チアゾリジニル、テトラヒドロフリル、トリチアニル、テトラヒドロピラニル、チオモルホリニル、チアモルホリニル、1−オキソ−チオモルホリニル、および1,1−ジオキソ−チオモルホリニルが含まれる。本明細書で他に特に示さない限り、ヘテロシクリル基は必要に応じて置換されてもよい。 A "heterocyclyl" or "heterocyclic ring" is a stable 3 to 18 containing 2 to 12 carbon atoms and 1 to 6 heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur. Refers to a member non-aromatic ring group. Unless otherwise specified herein, the heterocyclyl group may be a monocyclic, bicyclic, tricyclic, or tetracyclic ring system which may include a condensed or crosslinked ring system; in the heterocyclyl group. Nitrogen, carbon, or sulfur atoms may be oxidized as needed; nitrogen atoms may be quaternized as needed; heterocyclyl groups are partially saturated or fully saturated. obtain. Examples of such heterocyclyl groups include dioxolanyl, thienyl [1,3] dithianyl, decahydroisoquinolyl, imidazolinyl, imidazolidinyl, isothiazolidinyl, isooxazolidinyl, morpholinyl, octahydroindolyl, octahydro. Isoindrill, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidinyl, oxazolidinyl, piperidinyl, piperazinyl, 4-piperidonyl, pyrrolidinyl, pyrazolydinyl, quinucridinyl, thiazolidinyl, tetrahydrofuryl, trithianil, tetrahydropyrani Includes le, thiomorpholinyl, thiamorpholinyl, 1-oxo-thiomorpholinyl, and 1,1-dioxo-thiomorpholinyl. Unless otherwise specified herein, heterocyclyl groups may be optionally substituted.
本明細書で使用される「置換された」という用語は、少なくとも1個の水素原子が、アミノ、シアノ、ヒドロキシル、イミノ、ニトロ、オキソ、チオキソ、ハロ、アルキル、およびアルコキシなどの非水素ラジカルへの結合に置き換えられている、上記基(例えば、アルキル、アルコキシ、および/またはヘテロシクリル)のいずれかを意味しており、前述のラジカルのそれぞれは、上記置換基の1個または複数個で必要に応じて置換されてもよく、「置換された」は、1個または複数個の水素原子がNRgRh、NRgC(=O)Rh、NRgC(=O)NRgRh、NRgC(=O)ORh、NRgSO2Rh、OC(=O)NRgRh、ORg、SRg、SORg、SO2Rg、OSO2Rg、SO2ORg、NSO2Rg、またはSO2NRgRhで置き換えられている上記基のいずれかも含む。「置換された」は、1個または複数個の水素原子がC(=O)Rg、C(=O)ORg、C(=O)NRgRh、CH2SO2Rg、またはCH2SO2NRgRhで置き換えられている上記基のいずれかも意味する。前述において、RgおよびRhは同じでありまたは異なり、独立して水素であるか、または必要に応じて置換されたアルキルである。 As used herein, the term "substituted" means that at least one hydrogen atom is transferred to non-hydrogen radicals such as amino, cyano, hydroxyl, imino, nitro, oxo, thioxo, halo, alkyl, and alkoxy. Means any of the above groups (eg, alkyl, alkoxy, and / or heterocyclyl) that have been replaced by the bonds of, and each of the above radicals is required by one or more of the above substituents. It may be substituted accordingly, and "substituted" means that one or more hydrogen atoms are NR g R h , NR g C (= O) R h , NR g C (= O) NR g R h. , NR g C (= O) OR h , NR g SO 2 R h , OC (= O) NR g R h , OR g , SR g , SOR g , SO 2 R g , OSO 2 R g , SO 2 OR It also includes any of the above groups replaced by g, NSO 2 R g , or SO 2 NR g R h. “Substituted” means that one or more hydrogen atoms are C (= O) R g , C (= O) OR g , C (= O) NR g R h , CH 2 SO 2 R g , or It also means any of the above groups replaced by CH 2 SO 2 NR g R h. In the above, R g and R h are the same or different, independently hydrogen, or optionally substituted alkyl.
「プロドラッグ」は、生理学的条件下でまたは加溶媒分解によって、本明細書に記述される生物学的に活性な塩に変換され得るACVR1阻害剤を示すことが意図されている。したがって「プロドラッグ」という用語は、薬学的に許容される生物学的に活性なACVR1阻害剤の前駆体を指す。一部の態様では、プロドラッグは、対象に投与されたときに不活性であるが、例えば加水分解によって、in vivoで活性ACVR1阻害剤に変換される。プロドラッグACVR1阻害剤は、対象生物での溶解度、組織適合性、または遅延放出の利点をしばしば提供する(例えば、Bundgard, H., Design of Prodrugs (1985), pp. 7-9, 21-24 (Elsevier, Amsterdam参照)。プロドラッグの考察は、共に本明細書に参照により完全に組み込まれるHiguchi, T., et al., "Pro drugs as Novel Delivery Systems," A.C.S. Symposium Series, Vol. 14、およびBioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987に提示されている。「プロドラッグ」という用語は、そのようなプロドラッグが対象に投与されたときにin vivoで活性ACVR1阻害剤を放出する、任意の共有結合担体を含むことも意図されている。本明細書に記述される活性ACVR1阻害剤のプロドラッグは、親活性ACVR1阻害剤に通常の操作でまたはin vivoでのいずれかで修飾が切断されるような方法で、活性ACVR1阻害剤中に存在する官能基を修飾することにより典型的には調製される。プロドラッグは、ACVR1阻害剤であって、ヒドロキシ、アミノ、またはメルカプト基が、活性ACVR1阻害剤のプロドラッグが対象に投与されたときに切断されて遊離ヒドロキシ、遊離アミノ、または遊離メルカプト基をそれぞれ形成する任意の基に結合している、ACVR1阻害剤を含む。プロドラッグの例には、活性ACVR1阻害剤などにおけるヒドロキシ官能基のアセテート、ホルメート、およびベンゾエート誘導体、またはアミン官能基のアセトアミド、ホルムアミド、およびベンズアミド誘導体が含まれる。 The "prodrug" is intended to indicate an ACVR1 inhibitor that can be converted to the biologically active salts described herein under physiological conditions or by solvolysis. Thus, the term "prodrug" refers to a precursor of a pharmaceutically acceptable biologically active ACVR1 inhibitor. In some embodiments, the prodrug is inactive when administered to the subject, but is converted in vivo to an active ACVR1 inhibitor, for example by hydrolysis. Prodrug ACVR1 inhibitors often provide the benefits of solubility, histocompatibility, or delayed release in the subject organism (eg, Bundgard, H., Design of Prodrugs (1985), pp. 7-9, 21-24). (See Elsevier, Amsterdam). A discussion of prodrugs, both fully incorporated herein by reference, Higuchi, T., et al., "Pro drugs as Novel Delivery Systems," ACS Symposium Series, Vol. 14, And Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987. The term "prodrug" is used when such a prodrug is administered to a subject. It is also intended to include any covalent carrier that releases the active ACVR1 inhibitor on the vivo. The active ACVR1 inhibitor prodrugs described herein are conventional operations on the parentally active ACVR1 inhibitor. Alternatively, it is typically prepared by modifying the functional groups present in the active ACVR1 inhibitor in such a way that the modification is cleaved either in vivo. The prodrug is an ACVR1 inhibitor. The hydroxy, amino, or mercapto group is cleaved when the active ACVR1 inhibitor prodrug is administered to the subject and is attached to any group that forms a free hydroxy, free amino, or free mercapto group, respectively. Examples of prodrugs include acetates, formates, and benzoate derivatives of hydroxy functional groups, such as active ACVR1 inhibitors, or acetamides, formamides, and benzamide derivatives of amine functional groups.
本開示は、1個または複数個の原子が異なる原子質量または質量数を有する原子に置き換えられることによって同位体標識されている、式(I)の、全ての薬学的に許容されるACVR1阻害剤を包含することも意図されている。開示されたACVR1阻害剤に組み込むことができる同位体の例には、水素、炭素、窒素、酸素、リン、フッ素、塩素、およびヨウ素の同位体、例えばそれぞれ2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、31P、32P、35S、18F、36Cl、123I、および125Iが含まれる。これらの放射性標識されたACVR1阻害剤は、例えば動作の部位もしくは形態、または薬理学的に重要な動作部位に対する結合親和性を特徴付けることにより、ACVR1阻害剤の有効性の決定または測定を助けるのに役立てることができる。例えば放射性同位体を組み込んだ、式(I)の、ある特定の同位体標識されたACVR1阻害剤は、薬物および/または基質組織分布研究に有用である。放射性同位体トリチウム、即ち3H、および炭素14、即ち14Cは、その組込みの容易さおよび容易な検出手段の観点から、この目的に特に有用である。 The present disclosure is an isotope-labeled all pharmaceutically acceptable ACVR1 inhibitor of formula (I) in which one or more atoms are replaced by atoms having different atomic masses or mass numbers. Is also intended to include. Examples of isotopes that can be incorporated into the disclosed ACVR1 inhibitors, hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine, chlorine, and iodine, such as each 2 H, 3 H, 11 C , 13 Included are C, 14 C, 13 N, 15 N, 15 O, 17 O, 18 O, 31 P, 32 P, 35 S, 18 F, 36 Cl, 123 I, and 125 I. These radiolabeled ACVR1 inhibitors help determine or measure the efficacy of ACVR1 inhibitors, for example by characterizing the binding affinity for the site or morphology of action, or pharmacologically important sites of action. Can be useful. Certain isotope-labeled ACVR1 inhibitors of formula (I) incorporating, for example, radioisotopes are useful for drug and / or substrate tissue distribution studies. The radioactive isotopes tritium, i.e. 3 H, and carbon-14, i.e. 14 C, from the viewpoint of ease and easy detection means of the incorporation, are particularly useful for this purpose.
重水素、即ち2Hなどのより重い同位体での置換は、より大きな代謝安定性の結果生じるある特定の治療上の利点、例えば長くなったin vivo半減期または削減された投薬要件を提供することができ、したがっていくつかの状況で好ましいと考えられる。 Deuterium, i.e., substitution with heavier isotopes such as 2 H may provide greater metabolic stability resulting certain therapeutic advantages, such as long since in vivo half-life or reduced dosing requirements It can and is therefore considered preferable in some situations.
陽電子放出同位体、例えば11C、18F、15O、および13Nでの置換は、基質受容体占有率を試験するための陽電子放出断層撮影(PET)研究に役立てることができる。式(I)の同位体標識されたACVR1阻害剤は、一般に、当業者に公知の従来の技法によって、またはこれまで用いられた非標識試薬の代わりに適切な同位体標識試薬を使用する、以下に示される調製および実施例で記述されるものに類似のプロセスによって、調製することができる。 Substitution with positron emitting isotopes such as 11 C, 18 F, 15 O, and 13 N can be useful for positron emission tomography (PET) studies to test substrate receptor occupancy. Isotope-labeled ACVR1 inhibitors of formula (I) generally use suitable isotope-labeled reagents by conventional techniques known to those of skill in the art or in place of previously used non-labeled reagents. It can be prepared by a process similar to that described in the preparations and examples set forth in.
本開示は、開示されたACVR1阻害剤のin vivo代謝生成物を包含することも意図されている。そのような生成物は、主に酵素プロセスに起因して、例えば投与されたACVR1阻害剤の酸化、還元、加水分解、アミド化、エステル化などから得ることができる。したがって本開示は、その代謝生成物をもたらすのに十分な期間にわたり、本開示のACVR1阻害剤を対象に投与することを含むプロセスによって生成された、ACVR1阻害剤を含む。そのような生成物は、典型的には、本開示の放射性標識されたACVR1阻害剤を、検出可能な用量で動物に、例えばラット、マウス、モルモット、サル、またはヒトに投与することによって特定され、代謝を生じさせるのに十分な時間が得られ、その変換生成物が、尿、血液、またはその他の生体試料から単離される。 The disclosure is also intended to include in vivo metabolized products of the disclosed ACVR1 inhibitors. Such products can be obtained, for example, from oxidation, reduction, hydrolysis, amidation, esterification, etc. of administered ACVR1 inhibitors, primarily due to enzymatic processes. Accordingly, the present disclosure includes ACVR1 inhibitors produced by a process comprising administering to a subject the ACVR1 inhibitor of the present disclosure for a period sufficient to result in its metabolic products. Such products are typically identified by administering the radiolabeled ACVR1 inhibitor of the present disclosure to animals at detectable doses, such as to rats, mice, guinea pigs, monkeys, or humans. Sufficient time is available to generate metabolism, the conversion product of which is isolated from urine, blood, or other biological sample.
「安定なACVR1阻害剤」および「安定な構造」は、反応混合物から有用な純度に至るまでの単離および効果的な治療剤への形成の後、残存するのに十分堅牢な、ACVR1阻害剤を示すことが意図されている。 "Stable ACVR1 Inhibitors" and "Stable Structures" are ACVR1 inhibitors that are robust enough to survive after isolation from the reaction mixture to useful purity and formation into effective therapeutic agents. Is intended to indicate.
「必要に応じた」または「必要に応じて」という単語の使用は、その後に記述される事象または状況が生じても生じなくてもよいことを意味し、その記述は、事象または状況が生じる場合およびそれが生じない場合を含むことを意味する。例えば、「必要に応じて置換されたアリール」は、アリール基が置換されても置換されなくてもよいことを意味し、その記述は、置換アリール基と、置換されていないアリール基との両方を含むことを意味する。 The use of the words "as needed" or "as needed" means that the event or situation described thereafter may or may not occur, and that description causes the event or situation. Means to include cases and cases where it does not occur. For example, "optionally substituted aryl" means that the aryl group may or may not be substituted, the description of which is both a substituted aryl group and an unsubstituted aryl group. Means to include.
「薬学的に許容される塩」は、酸付加塩および塩基付加塩の両方を含む。 "Pharmaceutically acceptable salts" include both acid and base addition salts.
「薬学的に許容される酸付加塩」は、塩酸、臭化水素酸、硫酸、硝酸、およびリン酸などの無機酸と、酢酸、2,2−ジクロロ酢酸、アジピン酸、アルギン酸、アスコルビン酸、アスパラギン酸、ベンゼンスルホン酸、安息香酸、4−アセトアミド安息香酸、カンファー酸、カンファー−10−スルホン酸、カプリン酸、カプロン酸、カプリル酸、炭酸、ケイ皮酸、クエン酸、シクラミン酸、ドデシル硫酸、エタン−1,2−ジスルホン酸、エタンスルホン酸、2−ヒドロキシエタンスルホン酸、ギ酸、フマル酸、ガラクタル酸、ゲンチジン酸、グルコヘプトン酸、グルコン酸、グルクロン酸、グルタミン酸、グルタル酸、2−オキソ−グルタル酸、グリセロリン酸、グリコール酸、馬尿酸、イソブチル酸、乳酸、ラクトビオン酸、ラウリン酸、マレイン酸、リンゴ酸、マロン酸、マンデル酸、メタンスルホン酸、粘液酸、ナフタレン−1,5−ジスルホン酸、ナフタレン−2−スルホン酸、1−ヒドロキシ−2−ナフトエ酸、ニコチン酸、オレイン酸、オロト酸、シュウ酸、パルミチン酸、パモ酸、プロピオン酸、ピログルタミン酸、ピルビン酸、サリチル酸、4−アミノサリチル酸、セバシン酸、ステアリン酸、コハク酸、酒石酸(例えば、L−(+)−酒石酸)、チオシアン酸、p−トルエンスルホン酸、トリフルオロ酢酸、およびウンデシレン酸などの有機酸とで形成される塩を指す。 "Pharmaceutically acceptable acid addition salts" include inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, and phosphoric acid, acetic acid, 2,2-dichloroacetic acid, adipic acid, alginic acid, ascorbic acid, Asparagic acid, benzenesulfonic acid, benzoic acid, 4-acetamidobenzoic acid, camphoric acid, camphor-10-sulfonic acid, capric acid, caproic acid, capricic acid, carbonic acid, silicate acid, citric acid, cyclamic acid, dodecyl sulfate, Ethan-1,2-disulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid, formic acid, fumaric acid, galactalic acid, gentidic acid, glucoheptonic acid, gluconic acid, glucuronic acid, glutamic acid, glutaric acid, 2-oxo-glutar Acids, glycerophosphates, glycolic acids, horse uric acid, isobutyl acid, lactic acid, lactobionic acid, lauric acid, maleic acid, malic acid, malonic acid, mandelic acid, methanesulfonic acid, mucilage, naphthalene-1,5-disulfonic acid, Naphthalene-2-sulfonic acid, 1-hydroxy-2-naphthoic acid, nicotinic acid, oleic acid, orotoic acid, oxalic acid, palmitic acid, pamoic acid, propionic acid, pyroglutamic acid, pyruvate, salicylic acid, 4-aminosalicylic acid, Refers to salts formed with organic acids such as sebacic acid, stearic acid, succinic acid, tartrate acid (eg L- (+)-tartrate acid), thiosian acid, p-toluenesulfonic acid, trifluoroacetic acid, and undecylene acid. ..
「薬学的に許容される塩基付加塩」は、遊離酸への無機塩基または有機塩基の付加から調製された塩を指す。無機塩基から誘導される塩には、ナトリウム、カリウム、リチウム、アンモニウム、カルシウム、マグネシウム、鉄、亜鉛、銅、マンガン、およびアルミニウム塩などが含まれる。好ましい無機塩は、アンモニウム、ナトリウム、カリウム、カルシウム、およびマグネシウム塩である。有機塩基から誘導された塩には、第一級、第二級、および第三級アミン、天然に存在する置換アミンを含む置換アミン、環状アミン、塩基性イオン交換樹脂、例えばアンモニア、イソプロピルアミン、トリメチルアミン、ジエチルアミン、トリエチルアミン、トリプロピルアミン、ジエタノールアミン、エタノールアミン、デアノール、2−ジメチルアミノエタノール、2−ジエチルアミノエタノール、ジシクロヘキシルアミン、リシン、アルギニン、ヒスチジン、カフェイン、プロカイン、ヒドラバミン、コリン、ベタイン、ベネタミン、ベンザシン、エチレンジアミン、グルコサミン、メチルグルカミン、テオブロミン、トリエタノールアミン、トロメタミン、プリン、ピペラジン、ピペリジン、N−エチルピペリジン、およびポリアミン樹脂などの塩が含まれる。特に好ましい有機塩基は、イソプロピルアミン、ジエチルアミン、エタノールアミン、トリメチルアミン、ジシクロヘキシルアミン、コリン、およびカフェインである。 "Pharmaceutically acceptable base addition salt" refers to a salt prepared from the addition of an inorganic or organic base to a free acid. Salts derived from inorganic bases include sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Preferred inorganic salts are ammonium, sodium, potassium, calcium, and magnesium salts. Salts derived from organic bases include primary, secondary, and tertiary amines, substituted amines containing naturally occurring substituted amines, cyclic amines, basic ion exchange resins such as ammonia, isopropylamine, etc. Trimethylamine, diethylamine, triethylamine, tripropylamine, diethanolamine, ethanolamine, deanol, 2-dimethylaminoethanol, 2-diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, venetamine, Includes salts such as benzacine, ethylenediamine, glucosamine, methylglucamine, theobromine, triethanolamine, tromethamine, purine, piperazine, piperidine, N-ethylpiperidine, and polyamine resins. Particularly preferred organic bases are isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline, and caffeine.
一部の実施形態では、薬学的に許容される塩には、第四級アンモニウム塩、例えば第四級アミンアルキルハロゲン化物塩(例えば、臭化メチル)が含まれる。 In some embodiments, pharmaceutically acceptable salts include quaternary ammonium salts, such as quaternary amine alkyl halide salts (eg, methyl bromide).
しばしば、結晶化は、本開示のACVR1阻害剤の溶媒和物を生成する。本明細書で使用される場合、「溶媒和物」という用語は、溶媒の1個または複数個の分子と共に本開示のACVR1阻害剤の1個または複数個の分子を含む凝集体を指す。溶媒は、水であってもよく、その場合、溶媒和物は水和物であってもよい。あるいは、溶媒は有機溶媒であってもよい。このように、本開示のACVR1阻害剤は、一水和物、二水和物、半水和物、セスキ水和物、三水和物、および四水和物などを含む水和物、ならびに対応する溶媒和形態として存在し得る。本開示のACVR1阻害剤は、真の溶媒和物であってもよく、一方、その他の場合には、本開示のACVR1阻害剤は、単に付随的な水を保持してもよく、または水にいくらかの付随的な溶媒を加えた混合物であってもよい。 Often, crystallization produces a solvate of the ACVR1 inhibitors of the present disclosure. As used herein, the term "solvate" refers to an aggregate comprising one or more molecules of the solvent as well as one or more molecules of the ACVR1 inhibitor of the present disclosure. The solvent may be water, in which case the solvate may be a hydrate. Alternatively, the solvent may be an organic solvent. Thus, the ACVR1 inhibitors of the present disclosure include hydrates, including monohydrates, dihydrates, hemihydrates, sesquihydrates, trihydrates, tetrahydrates, and the like, as well as hydrates. It may exist as the corresponding solvated form. The ACVR1 inhibitor of the present disclosure may be a true solvate, while in other cases the ACVR1 inhibitor of the present disclosure may simply retain ancillary water or in water. It may be a mixture with some incidental solvent added.
本開示のACVR1阻害剤、またはその薬学的に許容される塩もしくは互変異性体は、1つまたはそれより多くの不斉中心を含有してもよく、したがってエナンチオマー、ジアステレオマー、およびその他の立体異性体であって絶対立体化学の観点からアミノ酸に関して(R)−もしくは(S)−または(D)−もしくは(L)−と定義され得るものが生じてもよい。本開示は、そのような可能性のある異性体の全て、ならびにそれらのラセミ体および光学的に純粋な形態を含むことが意図されている。光学的に活性な(+)および(−)、(R)−および(S)−、または(D)−および(L)−異性体は、キラルシントンまたはキラル試薬を使用して調製されてもよく、または従来の技法、例えばクロマトグラフィーおよび分別結晶化を使用して分割されてもよい。個々のエナンチオマーの調製/単離のための従来の技法には、適切な光学的に純粋な前駆体からのキラル合成、または例えばキラル高圧液体クロマトグラフィー(HPLC)を使用したラセミ体(または塩もしくは誘導体のラセミ体)の分割が含まれる。本明細書に記述されるACVR1阻害剤が、オレフィン二重結合またはその他の幾何学的対称性を有する中心を含有する場合、他に指示されない限り、ACVR1阻害剤は、EおよびZ幾何異性体の両方を含むことが意図される。同様に、全ての互変異性体も含まれることが意図される。 The ACVR1 inhibitors of the present disclosure, or pharmaceutically acceptable salts or tautomers thereof, may contain one or more asymmetric centers and thus enantiomers, diastereomers, and other. There may be stereoisomers that can be defined as (R)-or (S)-or (D)-or (L)-with respect to amino acids from the point of view of absolute stereochemistry. The present disclosure is intended to include all such potential isomers, as well as their racemic and optically pure forms. Optically active (+) and (-), (R)-and (S)-, or (D)-and (L) -isomers can also be prepared using chiral synthons or chiral reagents. Well, or may be partitioned using conventional techniques such as chromatography and fractional crystallization. Conventional techniques for the preparation / isolation of individual enantiomers include chiral synthesis from suitable optically pure precursors, or racemates (or salts or salts) using, for example, chiral high performance liquid chromatography (HPLC). Includes division of the racemic form of the derivative. If the ACVR1 inhibitors described herein contain olefin double bonds or other centers with geometric symmetry, unless otherwise indicated, the ACVR1 inhibitors are of E and Z geometric isomers. It is intended to include both. Similarly, all tautomers are intended to be included.
「立体異性体」は、同じ結合によって結合された同じ原子で構成されるが交換可能ではない異なる3次元構造を有する、ACVR1阻害剤を指す。本開示は、様々な立体異性体およびその混合物を企図し、その分子が互いに重ね合わせることができない鏡像である2つの立体異性体を指す「エナンチオマー」を含む。 "Steroisomer" refers to an ACVR1 inhibitor having a different three-dimensional structure composed of the same atoms bonded by the same bond but not exchangeable. The present disclosure includes an "enantiomer" that refers to two stereoisomers that are mirror images of various stereoisomers and mixtures thereof, the molecules of which cannot be superimposed on each other.
「実質的に」という用語は、有意な定性的または定量的範囲を指す。例として、化合物の特定の特徴付けを指す文脈で使用される場合、その用語は、例えばXRPD、DSC、またはTGAなどの参照される特徴付け方法で材料の類似性に基づいて、化学物質を特定する能力を指す。そのような技法の誤差範囲は、当業者に理解されるように、「実質的に」という用語に包含される。さらに、本明細書で使用される場合、「実質的に純粋な」は、形態に関して使用される場合、化合物の重量に対して、化合物2などのACVR1阻害剤の90、91、92、93、94、95、96、97、98、98.5、99、99.1、99.2、99.3、99.4、99.5、99.6、99.7、99.8、99.9重量%よりも大きい値を含み、100重量%に等しい値も含む、90重量%よりも大きい純度を有する化合物を意味する。残りの材料は、その調製から生じるその他の形態(複数可)の化合物、および/または反応不純物、および/または加工不純物を含む。例えば、化合物2の結晶形態は、この時点の公知でありかつ一般に当技術分野で受け入れられる手段によって測定されたとき、90重量%よりも大きい純度を有し、材料の残りの10重量%未満はその他の形態(複数可)の化合物2および/または反応不純物および/または加工不純物を含むので、実質的に純粋と見なし得る。実質的に純粋であることを定義する別の方法は、下記の通りである:本明細書で使用される場合、特定の多形形態に言及する際の「実質的に純粋」という用語は、多形形態が、化合物の任意のその他の物理形態を、10重量%未満、好ましくは5重量%未満、より好ましくは3重量%未満、最も好ましくは1重量%未満含むことを意味する。
The term "substantially" refers to a significant qualitative or quantitative range. As an example, when used in the context of referring to a particular characterization of a compound, the term identifies a chemical based on material similarity with a referenced characterization method such as XRPD, DSC, or TGA. Refers to the ability to do. The margin of error of such techniques is included in the term "substantially" as will be understood by those skilled in the art. Further, as used herein, "substantially pure", when used in terms of form, refers to 90, 91, 92, 93, 90, 91, 92, 93, of an ACVR1 inhibitor such as
「互変異性体」は、分子の1個の原子から同じ分子の別の原子へのプロトンシフトを指し、例えばプロトンシフトを介したケトンからエノールへの変換である。本開示は、任意の前記ACVR1阻害剤の互変異性体を含む。 "Tautomer" refers to a proton shift from one atom of a molecule to another atom of the same molecule, eg, a conversion from a ketone to an enol via a proton shift. The present disclosure includes tautomers of any said ACVR1 inhibitor.
「医薬組成物」は、1種またはそれより多種の治療剤と、対象、例えばヒトへの生物学的活性剤の送達のために当技術分野で一般に受け入れられる媒体との製剤を指す。そのような媒体には、全ての薬学的に許容される担体、希釈剤、または賦形剤が含まれる。「薬学的に許容される担体、希釈剤、または賦形剤」は、任意のアジュバント、担体、賦形剤、滑剤、甘味剤、希釈剤、保存剤、染料/着色剤、香料増強剤、界面活性剤、湿潤剤、分散剤、懸濁化剤、安定化剤、等張剤、溶媒、または乳化剤であって、ヒトまたは飼い馴らされた動物での使用に許容されることが米国食品医薬品局によって認可されたものが含まれる。 A "pharmaceutical composition" refers to a formulation of one or more therapeutic agents and a medium generally accepted in the art for delivery of a biologically active agent to a subject, eg, a human. Such vehicles include all pharmaceutically acceptable carriers, diluents, or excipients. "Pharmaceutically acceptable carriers, diluents, or excipients" are any adjuvants, carriers, excipients, lubricants, sweeteners, diluents, preservatives, dyes / colorants, fragrance enhancers, surfactants. Activators, wetting agents, dispersants, suspending agents, stabilizers, isotonic agents, solvents, or emulsifiers that may be acceptable for use in humans or domesticated animals. Includes those approved by.
「化学療法剤」または「抗がん剤」は、がん細胞を破壊する、またはがん細胞の成長を止めるかまたは遅くする化学物質である。 A "chemotherapeutic agent" or "antineocancer agent" is a chemical substance that destroys cancer cells or stops or slows the growth of cancer cells.
「腫瘍」を含む「がん」は、細胞の制御されない成長および/または異常に増加した細胞の生存および/またはアポトーシスの阻害であって、身体臓器およびシステムの正常な機能を妨げるものを指す。「がん」(例えば、腫瘍)は、固形および非固形がんを含む。がんまたは腫瘍を有する対象は、対象の体内に存在する、客観的に測定可能な数のがん細胞を有する。「がん」は、良性および悪性がん(例えば、それぞれ良性および悪性腫瘍)、ならびに潜伏腫瘍または微小転移を含む。 "Cancer," including "tumor," refers to the inhibition of uncontrolled growth and / or abnormally increased cell survival and / or apoptosis of cells that interferes with the normal functioning of body organs and systems. "Cancer" (eg, tumor) includes solid and non-solid cancers. A subject with cancer or tumor has an objectively measurable number of cancer cells present in the subject's body. "Cancer" includes benign and malignant cancers (eg, benign and malignant tumors, respectively), as well as latent or micrometastases.
「転移」は、その原発部位から体内のその他の場所へのがんの拡がりを指す。「転移」は、その当初の場所から移動しかつ生命維持に不可欠な臓器に蒔かれるがんであり、最終的には、罹患した臓器の機能の低下により対象は死に至る可能性がある。転移は逐次プロセスであり、がん細胞は原発腫瘍から離脱し、リンパおよび血管に侵入し、血流を経て循環し、体内のその他の場所の正常組織にある遠位巣で成長する可能性がある(転移する)。新しい部位で、細胞は、血液供給を確立し、成長して、生命を脅かす腫瘤を形成する可能性がある。転移は、局所または遠位である可能性がある。腫瘍細胞内の刺激および阻害分子経路は共に、この挙動を規制し、新しい部位での腫瘍細胞と宿主細胞との間の相互作用も著しい。 "Metastasis" refers to the spread of cancer from its primary site to other parts of the body. "Metastasis" is a cancer that moves from its original location and is sown in vital organs, which can eventually lead to death due to functional deterioration of the affected organ. Metastasis is a sequential process that allows cancer cells to leave the primary tumor, invade lymph and blood vessels, circulate through the bloodstream, and grow in distal lesions in normal tissue elsewhere in the body. There is (transfers). At new sites, cells can establish a blood supply and grow to form life-threatening masses. Metastases can be local or distal. Both stimulatory and inhibitory molecular pathways within tumor cells regulate this behavior, and the interaction between tumor cells and host cells at new sites is also significant.
「処置する」または「処置」は、本明細書で使用される場合、目的の疾患または状態、例えばがんを有するヒトなどの対象への、医療または医学的ケアの施用を指し:(i)疾患または状態を阻害すること、即ち、その発症を停止させること;(ii)疾患または状態を緩和すること、即ち、疾患または状態を後退させること;または(iii)根幹をなす疾患または状態に対処することなく疾患または状態の結果生じる症状(例えば、疼痛、体重減少、咳、疲労、衰弱など)を緩和することを含む。本明細書で使用される場合、「疾患」および「状態」という用語は、同義で使用されてもよく、または特定の病弊または状態が公知の病因物質を有していない可能性があり(したがって、病因は、まだ確認されていない)、したがって疾患であるとまだ認識されておらず単に望ましくない状態または症候群として認識されて、多かれ少なかれ特定の症状の組が臨床医により確認されているという点で、異なっていてもよい。 "Treatment" or "treatment," as used herein, refers to the application of medical or medical care to a subject of interest, such as a person with a disease or condition: (i). Inhibiting a disease or condition, i.e. stopping its onset; (ii) alleviating a disease or condition, i.e. retreating a disease or condition; or (iii) coping with an underlying disease or condition. Includes relieving symptoms (eg, pain, weight loss, coughing, fatigue, weakness, etc.) that result from a disease or condition without doing so. As used herein, the terms "disease" and "condition" may be used synonymously, or a particular disease or condition may not have a known pathogen (as used herein). Therefore, the pathogenesis has not yet been identified), and therefore a more or less specific set of symptoms has been identified by the clinician, not yet recognized as a disease but simply as an undesired condition or syndrome. In terms of points, they may differ.
「対象」は、ヒト、飼い馴らされた動物、例えば実験室用動物(例えば、イヌ、サル、ラット、マウスなど)、家庭用ペット(例えば、ネコ、イヌ、ウサギなど)、および家畜(例えば、ブタ、ウシ、ヒツジ、ヤギ、ウマなど)、および飼い馴らされていない動物(例えば、クマ、ゾウ、ヤマアラシなど)を含む。実施形態では、対象は哺乳動物である。実施形態では、対象はヒトである。「患者」という用語は、「対象」という用語と同義で使用され得る。 "Subjects" include humans, domesticated animals such as laboratory animals (eg dogs, monkeys, rats, mice, etc.), domestic pets (eg, cats, dogs, rabbits, etc.), and livestock (eg, cats, dogs, rabbits, etc.). Includes pigs, cows, sheep, goats, horses, etc.), and untamed animals (eg, bears, elephants, yamaarashi, etc.). In embodiments, the subject is a mammal. In embodiments, the subject is a human. The term "patient" may be used synonymously with the term "subject".
FD&C Actは、「小児」を、その診断または処置時に21歳またはそれよりも若い対象と定義する。小児の亜集団は、さらに:(i)新生児−誕生から生後28日まで;(ii)幼児−29日から2歳未満;(iii)子供−2歳から12歳未満;および(iv)青年−12歳から21歳までと特徴付けられる。定義にも関わらず、影響を受け易い患者集団および臨床試験評価に応じて、認可された規制標識は、例えば22歳までの小児患者など、小児集団の範囲を特に修正するという文言を含んでいてもよい。 FD & C Act defines a "child" as a subject 21 years or younger at the time of its diagnosis or treatment. The pediatric subpopulation further includes: (i) newborns-from birth to 28 days; (ii) infants-29 days to less than 2 years; (iii) children-2 years to less than 12 years; and (iv) adolescents- Characterized from 12 to 21 years old. Despite the definition, depending on the vulnerable patient population and clinical trial evaluation, the approved regulatory label contains the wording that it specifically modifies the scope of the pediatric population, for example pediatric patients up to 22 years of age. May be good.
「有効量」または「治療有効量」は、ヒトなどの対象に投与されたときに対象のがんの有効処置に十分な、ACVR1阻害剤または組成物の量を指す。「有効量」を構成するACVR1阻害剤または組成物の量は、ACVR1阻害剤または組成物、処置される状態およびその重症度、投与の方法、処置の持続時間、および/または処置される対象の年齢に応じて変わるが、当業者が自身の知識および本開示に基づいて通常通り決定することができる。実施形態では、「有効量」は、1つまたはそれより多くの徴候、症状、サイン、診断試験、およびバイタルサインなどにおける統計的に有意な変化によって測定された通りに、処置を行う(例えば、処置する、予防する、阻害する、緩和する、促進させる、改善する、増大させる、および低減させるなど)。他の実施形態では、「有効量」は、1つまたはそれより多くの徴候、症状、サイン、診断試験、およびバイタルサインなどにおける統計的に有意な変化の欠如によって測定された通りに状態を抑制し、管理し、または予防する。 An "effective amount" or "therapeutically effective amount" refers to an amount of an ACVR1 inhibitor or composition sufficient for effective treatment of a subject's cancer when administered to a subject such as a human. The amount of the ACVR1 inhibitor or composition constituting the "effective amount" is the amount of the ACVR1 inhibitor or composition, the condition to be treated and its severity, the method of administration, the duration of treatment, and / or the subject to be treated. Depending on age, one of ordinary skill in the art can make decisions as usual based on his or her knowledge and the present disclosure. In embodiments, the "effective amount" is treated as measured by statistically significant changes in one or more signs, symptoms, signs, diagnostic tests, and vital signs (eg,). Treat, prevent, inhibit, alleviate, promote, improve, increase, and reduce, etc.). In other embodiments, the "effective amount" suppresses the condition as measured by the lack of statistically significant changes in one or more signs, symptoms, signs, diagnostic tests, and vital signs, etc. And manage or prevent.
本明細書で使用される場合、「統計的に有意な」は、スチューデントt検定を使用して計算したときの、0.050またはそれよりも小さいp値を指し、測定される特定の事象または結果が偶然生じることがありそうもないことを示す。 As used herein, "statistically significant" refers to a p-value of 0.050 or less as calculated using Student's t-test, a particular event or measured. Indicates that the results are unlikely to occur by chance.
本発明の記述において、任意の濃度範囲、パーセンテージ範囲、比の範囲、または整数範囲は、他に指示しない限り、列挙された範囲内の任意の整数の値、および適切な場合にはその分数(整数の10分の1および100分の1など)を含むことが理解される。また、ポリマーサブユニット、サイズ、または厚さなどの任意の物理的特徴に関して本明細書に列挙される任意の数値範囲も、他に指示しない限り列挙された範囲内の任意の整数を含むと理解される。本明細書で使用される場合、「約」という用語は、他に指示されない限り、指示される範囲、値、または構造の±20%、±10%、±5%、または±1%を意味する。本明細書で使用される場合、「a」および「an」という用語は、列挙される構成要素の「1つまたはそれより多く」を指すことを理解すべきである。代替例(例えば、「または」)の使用は、代替例の1つ、両方、またはその任意の組合せを意味すると理解すべきである。 In the description of the present invention, any concentration range, percentage range, ratio range, or integer range is any integer value within the listed range and, where appropriate, a fraction thereof, unless otherwise indicated. It is understood to include (such as one tenth and one hundredth of an integer). It is also understood that any numerical range listed herein with respect to any physical feature such as polymer subunit, size, or thickness also includes any integer within the listed range unless otherwise indicated. Will be done. As used herein, the term "about" means ± 20%, ± 10%, ± 5%, or ± 1% of the indicated range, value, or structure, unless otherwise indicated. do. As used herein, it should be understood that the terms "a" and "an" refer to "one or more" of the listed components. The use of alternatives (eg, "or") should be understood to mean one, both, or any combination thereof.
文脈が他に必要としない限り、本明細書および特許請求の全体を通して、「含む(comprise)」という用語およびその変形例、例えば「含む(comprises)」および「含む(comprising)」、ならびに「含む(include)」のような同義語、および「有する(have)」およびその変形例は、開放的、包括的な意味で解釈されるものであり;即ち、「〜を含むがそれに限定されない」と解釈され、したがってリストにおける項目の列挙は、この技術の材料、組成物、デバイス、および方法で有用ともなり得るその他同様の項目を排除するものではない。含む、含有する、または有するなどの用語の同義語としての「含む(comprising)」というオープンエンドの用語は、本開示について記述し主張するのに本明細書で使用されるが、本発明の技術またはその実施形態は、あるいは、列挙される成分「からなる」または「から本質的になる」というより限定的な用語を使用して記述されてもよい。 Unless the context requires otherwise, the term "comprise" and examples thereof, such as "comprises" and "comprising", and "include" throughout the specification and claims. Synonyms such as "include" and "have" and its variants are to be interpreted in an open and comprehensive sense; that is, "including but not limited to". The enumeration of items in the list, as interpreted, does not preclude other similar items that may also be useful in the materials, compositions, devices, and methods of the art. The open-ended term "comprising" as a synonym for terms such as include, contain, or have is used herein to describe and assert the disclosure, but the techniques of the invention. Alternatively, embodiments thereof may be described using the more restrictive term "consisting of" or "consisting of essentially" of the listed components.
他に定義されない限り、本明細書の全ての技術的および科学的用語は、本開示が属する分野の当業者によって一般に理解されるものと同じ意味を有する。 Unless otherwise defined, all technical and scientific terms herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure belongs.
「一実施形態」または「実施形態」と本明細書の全体を通して言及する場合、実施形態に関連して記述される特定の特徴、構造、または特性は、本開示の少なくとも1つの実施形態に含まれることを意味する。このように、本明細書の全体を通した様々な場所での「一実施形態では」または「実施形態では」という文言の出現は、全てが同じ実施形態に言及するわけではない。同様に、「できる(can)」および「してもよい(may)」という用語およびそれらの変形例は、非限定的であることが意図され、したがって実施形態がある特定の要素または特徴を含むことができるまたは含んでいてもよいという引用は、それらの要素または特徴を含有しない本発明の技術のその他の実施形態を排除しない。さらに、特定の特徴、構造、または特性は、1つまたはそれより多くの実施形態において任意の適切な手法で組み合わされてもよい。 As used throughout the present specification as "one embodiment" or "embodiment," certain features, structures, or properties described in connection with an embodiment are included in at least one embodiment of the present disclosure. Means to be. Thus, the appearance of the words "in one embodiment" or "in an embodiment" at various locations throughout the specification does not all refer to the same embodiment. Similarly, the terms "can" and "may" and their variants are intended to be non-limiting and thus include certain elements or features of the embodiment. The citation that can or may include does not preclude other embodiments of the art of the invention that do not contain those elements or features. In addition, specific features, structures, or properties may be combined in any suitable manner in one or more embodiments.
以下の記述において、ある特定の詳細は、本開示の様々な実施形態の完全な理解をもたらすために記述される。しかし当業者なら、本開示は、これらの詳細なしで実施され得ることが理解される。
薬理学
In the following description, certain details are provided to provide a complete understanding of the various embodiments of the present disclosure. However, one of ordinary skill in the art will appreciate that this disclosure can be carried out without these details.
Pharmacology
びまん性真性橋膠腫(DIPG)は、脳橋と呼ばれる脳幹の一部に見出される脳腫瘍である。脳橋は、心拍、呼吸、嚥下、眼球運動、視力、および平衡などの本質的な身体機能を制御する。成人で生ずる組織学的に類似した病巣よりもかなり少ない頻度であるが、子供の高度グリオーマには、臨床神経腫瘍学でまだ満たされていない主要なニーズがある。DIPGのそのような背景の教示に関して参照により本明細書に組み込まれる、Jones C et al., Paediatric and adult malignant glioma: close relatives or distant cousins?, Nat Rev Clin Oncol. 2012 May 29; 9(7):400-13を参照されたい。 Diffuse true pons glioma (DIPG) is a brain tumor found in a part of the brain stem called the brain pons. The pons controls essential physical functions such as heartbeat, breathing, swallowing, eye movements, eyesight, and equilibrium. Although much less frequently than histologically similar lesions that occur in adults, advanced glioma in children has a major need that has not yet been met in clinical neuro-oncology. Jones C et al., Paediatric and adult malignant glioma: close relatives or distant cousins ?, Nat Rev Clin Oncol. 2012 May 29; 9 (7), incorporated herein by reference with respect to such background teachings of DIPG. See: 400-13.
DIPG腫瘍は、普遍的に致命的であり、全生存期間の中央値は9〜12か月である。DIPGはびまん的に浸潤し、より低いグレードの組織学の領域を内部に持ち得るが、大脳皮質のWHOグレードIV多形性膠芽細胞腫(GBM)と区別することが非常にできない。したがってこれらの子供での生存率を改善する試みは、これまで失敗している。これらの腫瘍の外科的切開は、その解剖学的部位により可能ではなく、成人GBMの文献からの将来性ある目標に基づく臨床試験は、利益がないことを示した。例えば、Warren, Diffuse intrinsic pontine glioma: poised for progress, Front Oncol. 2012; 2:205、およびTaylor et al., ACVR1 mutations in DIPG: lessons learned from FOP, Cancer Res., 2014 Sep 1: 74(17): 4565-4570を参照されたい(DIPGのそのような背景の教示に関して、それぞれが参照により本明細書に組み込まれる)。 DIPG tumors are universally fatal, with a median overall survival of 9-12 months. DIPGs can be diffusely infiltrated and have lower grade histological areas inside, but are very indistinguishable from WHO grade IV polymorphic glioblastoma (GBM) in the cerebral cortex. Therefore, attempts to improve survival in these children have been unsuccessful. Surgical incisions of these tumors are not possible due to their anatomical site, and clinical trials based on promising goals from the adult GBM literature have shown no benefit. For example, Warren, Diffuse intrinsic pontine glioma: poised for progress, Front Oncol. 2012; 2: 205, and Taylor et al., ACVR1 mutations in DIPG: lessons learned from FOP, Cancer Res., 2014 Sep 1: 74 (17) See: 4565-4570 (each incorporated herein by reference with respect to such background teachings of DIPG).
記述されるように、DIPGは、ほぼ例外なく子供に影響を及ぼす。Michael Mosier Defeat DIPG Foundationによれば、米国内のおよそ200〜400名の子供が、毎年DIPGと診断される。これらの子供は、典型的には4歳から11歳の間である。DIPGは、子供の全ての脳腫瘍のほぼ10〜15%を占める。 As described, DIPG affects children almost without exception. According to the Michael Mosier Defeat DIPG Foundation, approximately 200-400 children in the United States are diagnosed with DIPG each year. These children are typically between the ages of 4 and 11. DIPG accounts for nearly 10-15% of all brain tumors in children.
DIPGは、身体機能を妨げ得る侵襲的腫瘍であり、子供の運動能力、コミュニケーション能力を奪い、さらには飲食する能力さえも奪う。DIPG腫瘍が成長し始めると、脳橋によって調節される本質的な身体機能を制御する神経に圧力を加える可能性がある。DIPGの子供は、複視、眼球運動の低減、顔面脱力または非対称、ならびに腕および脚の脱力を経験する可能性がある。歩行、強調、会話、咀嚼、および嚥下に関する問題も有する可能性がある。腫瘍が進行するにつれ、呼吸および心拍も妨げる可能性があり、最終的には子供の死をもたらす。 DIPG is an invasive tumor that can interfere with physical function and deprives children of their motor skills, communication skills, and even their ability to eat and drink. As DIPG tumors begin to grow, they can put pressure on the nerves that control essential physical functions regulated by the brain pons. Children with DIPG can experience diplopia, reduced eye movements, facial weakness or asymmetry, and weakness in the arms and legs. You may also have problems with walking, emphasis, conversation, chewing, and swallowing. As the tumor progresses, it can also interfere with breathing and heartbeat, ultimately leading to the death of the child.
Buczkowiczらは、臨床的に古典的なDIPGが多様な組織学的スペクトルであることを実証した。DIPGの背景の教示に関連して参照により本明細書に組み込まれる、Buczkowicz et al., Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications, Acta Neuropathol. 2014 Oct;128(4):573-81. doi: 10.1007/s00401-014-1319-6. Epub 2014 Jul 22を参照されたい。したがって文脈において、DIPGは、浸潤性グリオーマ−グレードIIからIVの組織学的診断のある、脳橋腫瘍として特徴付けられてもよい。脳幹の原発性腫瘍は、臨床的治験に基づいておよび磁気共鳴画像法(MRI)を使用した神経画像研究に基づいて診断されてもよい。DIPGの推定診断は、組織学的診断がない状態での、古典的な撮像フィーチャーに基づく可能性がある。ますます、組織学的確認は、研究調査へのエントリーおよび腫瘍の分子の特徴付けの両方を目的に、得られる可能性がある。定位針生検による新しい手法は、生検をより安全にする可能性もある。生検は、診断が撮像の知見に基づいて不確実である場合、脳橋腫瘍に推奨されてもよい。このように、星細胞系腫瘍は、脳幹において優位を占める。毛様細胞性星細胞腫および神経節膠腫などのWHOグレード1腫瘍には好ましい予後があり、中脳蓋を含む脳幹全体にわたって、脳橋内に限局的に、または頚髄延髄接合部で生ずる可能性があり、それらはしばしば外方増殖性である。その他の脳幹部位で脳橋の外側に生ずる低グレードのびまん性星状細胞腫(WHOグレード2)は、より好ましい予後を持つ腫瘍になる傾向がある。DIPGは、診断時で生検されたときにびまん性星状細胞腫(WHOグレード2)から神経膠芽腫(WHOグレード4)に及ぶびまん性星状細胞腫である。死後評価で、DIPGは一般に、形態学的基準により未分化星状細胞腫(WHOグレード3)または神経膠芽腫(WHOグレード4)でもあるが、WHOグレード2領域も明らかにされてもよい。DIPGのおよそ80%は、組織学的グレードとは無関係に、びまん性正中グリオーマとしてWHOによって分類され得る。全てのびまん性正中グリオーマ、H3 K27M−変異体は、組織学的グレードとは無関係にWHOグレード4であり、この診断を持つ子供の不十分な予後を反映している。例えば、Ballester et al., Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol 37 (9): 1357-64, 2013、およびLouis DN et al., The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016を参照されたい(DIPGおよび関連ある疾患の特徴付けおよび診断に関して、それぞれが参照により本明細書に組み込まれる)。
Buczkowiz et al. Demonstrated that clinically classical DIPGs have diverse histological spectra. Buczkowicz et al., Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications, Acta Neuropathol. 2014 Oct; 128 (4): 573- 81. doi: 10.1007 / s00401-014-1319-6. See Epub 2014 Jul 22. Thus, in context, DIPG may be characterized as a pontine tumor with a histological diagnosis of invasive glioma-grade II to IV. Primary tumors of the brainstem may be diagnosed based on clinical trials and on neuroimaging studies using magnetic resonance imaging (MRI). The presumptive diagnosis of DIPG may be based on classical imaging features in the absence of histological diagnosis. Increasingly, histological confirmation may be obtained for both entry into research studies and characterization of tumor molecules. New techniques with stereotactic needle biopsy may also make biopsy safer. A biopsy may be recommended for pontine tumors if the diagnosis is uncertain based on the findings of imaging. Thus, stellate tumors dominate the brainstem.
Taylorらは、ACVR1における体細胞変異を内部に持つためにDIPGの症例の4分の1について明らかにした全ゲノム配列研究を公表した。この遺伝子は、I型骨形成タンパク質(BMP)受容体ALK2をコードし、残りは、生殖細胞系で変異したときに先天性異常症候群である進行性骨化性線維異形成症(FOP)を引き起こすものと同一の影響を受け、その結果、骨への軟組織の変換がもたらされる。この予期せぬリンク点は、腫瘍形成における発生生物学的プロセスの重要性に向かい、小児科神経腫瘍に対してFOP研究者がようやく手に入れた機構的理解および薬物開発での広範な経験をもたらす。例えば、そのような背景の教示に関して参照により本明細書に組み込まれる、Taylor et al., ACVR1 mutations in DIPG: lessons learned from FOP, Cancer Res., 2014 Sep 1: 74(17): 4565-4570を参照されたい。 Taylor et al. Published a whole-genome sequence study that revealed a quarter of DIPG cases due to internal somatic mutations in ACVR1. This gene encodes the type I bone morphogenetic protein (BMP) receptor ALK2, the rest causing progressive ossifying fibrodysplasia (FOP), a congenital anomaly syndrome when mutated in germline. It is affected in the same way as the one, resulting in the conversion of soft tissue into bone. This unexpected link points towards the importance of developmental biological processes in tumorigenesis and provides extensive experience in mechanistic understanding and drug development finally gained by FOP researchers for pediatric neuromas. .. For example, Taylor et al., ACVR1 mutations in DIPG: lessons learned from FOP, Cancer Res., 2014 Sep 1: 74 (17): 4565-4570, which is incorporated herein by reference with respect to such background teachings. Please refer.
骨形成タンパク質(BMP)は、体内の様々な臓器全体にわたって組織機構を協調させる際に本質的な役割を果たす、多面発現増殖因子である。BMP配位子は、セリン/トレオニンキナーゼ受容体の形質転換増殖因子ベータ(TGF−b)スーパーファミリーに属する骨形成タンパク質受容体(BMPR)と相互に作用する(Ikushima, H. and K. Miyazono, Biology of Transforming Growth Factor-beta Signalin. Curr Pharm Biotechnol, 2011、これは、そのような背景の教示に関して参照により本明細書に組み込まれる)。配位子は、II型受容体に結合し、次いでI型受容体が補充されてヘテロマー複合体を形成する。複合体として、II型受容体はI型受容体をリン酸化し、I型受容体は活性になりかつ下流のシグナル伝達分子をリン酸化させる。これらの受容体を活性化する下流の作用は、タンパク質のSMADファミリーによって主に実施される。SMADは、リン酸化されたものになり、細胞膜からのシグナルを核に伝達し、そこで転写因子として機能して遺伝子発現を調節する(Massague, J., J. Seoane, and D. Wotton, Smad transcription factors. Genes Dev, 2005. 19(23): p. 2783-810、これは、そのような背景の教示に関して参照により本明細書に組み込まれる)。 Bone morphogenetic protein (BMP) is a pleiotropic growth factor that plays an essential role in coordinating tissue mechanisms throughout various organs in the body. BMP ligands interact with bone morphogenetic protein receptors (BMPRs) belonging to the transforming growth factor beta (TGF-b) superfamily of serine / threonine kinase receptors (Ikushima, H. and K. Miyazono, Biology of Transforming Growth Factor-beta Signalin. Curr Pharm Biotechnol, 2011, which is incorporated herein by reference with respect to such background teachings). The ligand binds to the type II receptor and then is replenished with the type I receptor to form a heteromer complex. As a complex, type II receptors phosphorylate type I receptors, which become active and phosphorylate downstream signaling molecules. The downstream action of activating these receptors is primarily carried out by the SMAD family of proteins. SMAD becomes phosphorylated and transmits signals from the cell membrane to the nucleus where it functions as a transcription factor to regulate gene expression (Massague, J., J. Seoane, and D. Wotton, Smad transcription). factors. Genes Dev, 2005. 19 (23): p. 2783-810, which is incorporated herein by reference with respect to such background teachings).
がんおよび炎症などの慢性疾患を持つ個体において、BMPシグナル伝達は構成的に活性化されて貧血症をもたらす。この状態は、慢性疾患の貧血症(ACD)と一般に呼ばれ、がん患者に伴う衰弱症状である(Cullis, J.O., Diagnosis and management of anaemia of chronic disease: current status. Br J Haematol, 2011. 154(3): p. 289-300、これは、そのような背景の教示に関して参照により本明細書に組み込まれる)。がん患者の慢性貧血症は、極端な脱力および疲労に繋がり、それがこれらの固体に関する不十分なクオリティーオブライフに繋がる。これらの患者において、2つのBMP I型受容体、ALK2(記述されるように、ACVR1としても公知である)およびALK3を経たBMPシグナル伝達は、ヘプシジンと呼ばれるペプチドホルモンの肝臓発現を誘発させる(Steinbicker, A.U., et al., Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood, 2011. 118(15): p. 4224-30、これは、そのような背景の教示に関して、参照により本明細書に組み込まれる)。 In individuals with chronic diseases such as cancer and inflammation, BMP signaling is constitutively activated leading to anemia. This condition, commonly referred to as chronic disease anemia (ACD), is a debilitating symptom associated with cancer patients (Cullis, JO, Diagnosis and management of anaemia of chronic disease: current status. Br J Haematol, 2011. 154. (3): p. 289-300, which is incorporated herein by reference with respect to such background teachings). Chronic anemia in cancer patients leads to extreme weakness and fatigue, which leads to an inadequate quality of life for these individuals. In these patients, BMP signaling via two BMP type I receptors, ALK2 (also known as ACVR1 as described) and ALK3, induces liver expression of a peptide hormone called hepcidin (Steinbicker). , AU, et al., Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood, 2011. 118 (15): p. 4224-30, which is referred to for such background teaching. Incorporated herein by.
ヘプシジンは、鉄輸送体、フェロポルチンの分解を促進させることによって、血清中鉄レベルを低減させ、その結果、マクロファージおよびその他の細胞型への鉄の貯蔵が増加し、ヘモグロビンおよび赤血球(RBC)機能のために鉄を利用できなくなる。患者の鉄の摂取を補うことは、活性化BMP経路および高血清中ヘプシジンレベルに起因して消化された鉄が貯蔵されるだけであるので、ACDを逆転させない。現在、がんにおけるACDは、患者の身体活動を制限することによって管理され、輸血は、最も深刻な症例において使用される。これらの患者におけるBMPシグナル伝達の阻害には、そのクオリティーオブライフの実際の相違を提供する可能性があり、最終的には、それらが治療、放射線、または外科手術にどのように応答するかに関して良い影響を及ぼすことができる(Steinbicker, A.U., et al., Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood, 2011. 117(18): p. 4915-23; Coyne, D.W., Hepcidin: clinical utility as a diagnostic tool and therapeutic target. Kidney Int, 2011. 80(3): p. 240-4; Theurl, I., et al., Pharmacologic inhibition of hepcidin expression reverses anemia of chronic disease in rats. Blood, 2011、これらは、そのような背景の教示に関し、参照により本明細書に組み込まれる)。 Hepcidin reduces serum iron levels by promoting the degradation of the iron transporter, ferroportin, resulting in increased iron storage in macrophages and other cell types and hemoglobin and red blood cell (RBC) function. Because of this, iron cannot be used. Supplementing a patient's iron intake does not reverse ACD, as only the iron digested due to the activated BMP pathway and high serum hepcidin levels is stored. Currently, ACD in cancer is controlled by limiting the physical activity of the patient, and blood transfusions are used in the most severe cases. Inhibition of BMP signaling in these patients may provide a real difference in their quality of life and ultimately with respect to how they respond to treatment, radiation, or surgery. Steinbicker, AU, et al., Inhibition of bone morphogenetic protein signaling immunos anemia associated with inflammation. Blood, 2011. 117 (18): p. 4915-23; Coyne, DW, Hepcidin: clinical utility as a diagnostic tool and therapeutic target. Kidney Int, 2011. 80 (3): p. 240-4; Theurl, I., et al., Pharmacologic inhibition of hepcidin expression reverses anemia of chronic disease in rats. Blood, 2011 , These are incorporated herein by reference with respect to such background teachings).
上述のように、進行性骨化性線維異形成症(FOP)は、骨格奇形の常染色体優性障害であり、ACVR1における散発的生殖細胞系変異に起因して、1,500,000名の生児出生のうち1名の割合で生じる、異所性骨化を無効にする。FOPのそのような背景の教示に関して参照により本明細書に組み込まれる、Shore EM et al., A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva, Nat Genet. 2006 May; 38(5):525-7を参照されたい。FOPは、筋組織ならびに腱および靭帯などの結合組織が骨によって徐々に置き換えられ(骨化)、骨格の外側に骨が形成され(骨外性または異所骨)、それが運動を拘束する障害である。このプロセスは一般に、幼児において顕著であり、首および肩から始まり身体の下方に進行し、四肢に入る。骨外性骨形成は、関節が罹患するにつれて可動性の損失を進行させる。口を完全に大きく開けることができない状態は、会話および食事を難しくする可能性がある。時間と共に、この障害を持つ人々は、その食事の問題に起因して、栄養失調を経験する可能性がある。肺の拡張を制限する胸郭の周りの骨外形成の結果、呼吸困難にもなる可能性がある。転倒または侵襲的医療手順など、FOPの個体の筋肉に対する任意の外傷は、筋肉の腫脹および炎症(筋炎)のエピソードを引き起こし、その後、より急速な骨化が損傷領域で生じる可能性がある。再発も、インフルエンザなどのウイルス性の病気によって引き起こされる可能性がある。 As mentioned above, progressive ossifying fibrodysplasia (FOP) is an autosomal dominant disorder of skeletal malformations, resulting in sporadic germline mutations in ACVR1 resulting in 1.5 million live births. Disables ectopic ossification, which occurs in 1 out of births. Shore EM et al., A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva, Nat Genet. 2006 May; 38 ( 5): See 525-7. FOP is a disorder in which muscle tissue and connective tissues such as tendons and ligaments are gradually replaced by bone (ossification) and bone is formed outside the skeleton (extraosseous or ectopic bone), which restrains movement. Is. This process is generally prominent in young children, starting at the neck and shoulders, progressing below the body and into the limbs. Extraosseous bone formation develops a loss of mobility as the joint becomes affected. The inability to open the mouth completely can make conversation and eating difficult. Over time, people with this disorder may experience malnutrition due to their dietary problems. Extraosseous formation around the thorax, which limits lung dilation, can also result in dyspnea. Any trauma to the muscles of an individual with FOP, such as a fall or invasive medical procedure, causes episodes of muscle swelling and inflammation (myositis), after which more rapid ossification can occur in the injured area. Recurrence can also be caused by viral illnesses such as influenza.
DIPGで新たに記述される変異の5つの部位も、FOPの症例において見出され、それと共に、コードされたALK2タンパク質のGSおよびキナーゼドメイン全体にわたりさらに5つの部位が見出される。FOPおよびDIPGのそのような背景の教示に関して参照により本明細書に組み込まれる、Kaplan FS et al., Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1, Hum Mutat. 2009 Mar; 30(3):379-90を参照されたい。 Five sites of the mutation newly described in DIPG are also found in cases of FOP, along with an additional five sites across the GS and kinase domains of the encoded ALK2 protein. Kaplan FS et al., Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I, incorporated herein by reference with respect to such background teachings of FOP and DIPG. See receptor ACVR1, Hum Mutat. 2009 Mar; 30 (3): 379-90.
FOPおよびDIPGの両方を管理するのに有効な処置が求められている。放射線は、新たに診断されたDIPGに関する伝統的な治療である。従来の限定領域放射線は、DIPGの子供の90パーセント超で応答をもたらす。これらの応答は短命であるが、平均すると約6から9か月間続く。放射線治療の線量を増加させるいくつかの試験が行われており、いずれも生存率は改善されなかった。外科手術は、両方の状態で除外され、治療抗体は、ALK2/ACVR1に見出される活性化変異が受容体の細胞質部分のみに影響を及ぼすので、不適切であるように見える。 Effective measures are needed to control both FOP and DIPG. Radiation is a traditional treatment for newly diagnosed DIPGs. Conventional limited area radiation provides a response in more than 90 percent of children with DIPG. These responses are short-lived, but on average last for about 6 to 9 months. Several studies have been conducted to increase the dose of radiation therapy, none of which improved survival. Surgery is ruled out in both conditions and therapeutic antibodies appear inappropriate as the activating mutations found in ALK2 / ACVR1 affect only the cytoplasmic portion of the receptor.
ある特定の実施形態では、本開示の方法は、びまん性真性橋膠腫(DIPG)および進行性骨化性線維異形成症(FOP)を含む異常ACVR1発現を処置するのに有用である。 In certain embodiments, the methods of the present disclosure are useful for treating abnormal ACVR1 expression, including diffuse true pontine glioma (DIPG) and progressive ossifying fibrodysplasia (FOP).
本明細書に開示される、ある特定の方法は、それを必要とする対象に関する処置のレジメンを選択する働きをする。即ち、本開示は、処置レジメンを選択するための方法ならびにそれ自体を処置する方法を提供する。他の実施形態は、処置レジメンを選択するためのおよび所定の遺伝子プロファイルを有する対象に基づく対象の疾患を処置するための方法を提供する。様々な実施形態において、本開示の方法はさらに、対象から試料を得ることおよび遺伝子プロファイルを決定することを含む。 Certain methods disclosed herein serve to select a regimen of treatment for the subject in need thereof. That is, the present disclosure provides a method for selecting a treatment regimen as well as a method for treating itself. Other embodiments provide methods for selecting a treatment regimen and for treating a subject's disease based on a subject having a given genetic profile. In various embodiments, the methods of the present disclosure further include obtaining a sample from a subject and determining a genetic profile.
本明細書で提供される実施形態は、対象の遺伝子プロファイルに基づいて対象に関する処置レジメンを選択するための方法を含む。そのような遺伝子プロファイルは、任意の適切な手法(例えば、マイクロアレイ、逆転写ポリメラーゼ連鎖反応(RT−PCR)、RNA/DNA配列決定など)で生成されてもよい。 The embodiments provided herein include methods for selecting treatment regimens for a subject based on the subject's genetic profile. Such gene profiles may be generated by any suitable technique (eg, microarray, reverse transcription polymerase chain reaction (RT-PCR), RNA / DNA sequencing, etc.).
一部の実施形態では、遺伝子プロファイルは、1つまたはそれより多くの変異をACVR1遺伝子中に含む。「遺伝子」という用語は、コード配列だけでなく、プロモーター、エンハンサー、および終端領域などの調節領域を含むこともできる。その用語はさらに、全てのイントロンと、mRNA転写物からスプライスされたその他のDNA配列とを、代替のスプライス部位から得られた変種と共に含むことができる。特定のタンパク質をコードする遺伝子配列は、特定のタンパク質の発現を方向付けるDNAまたはRNAとすることができる。これらの核酸配列は、RNAに転写されるDNA鎖配列または特定のタンパク質に翻訳されたRNA配列であってもよい。核酸配列は、完全長核酸配列ならびに完全長タンパク質から誘導された非完全長配列の両方を含む。 In some embodiments, the gene profile comprises one or more mutations in the ACVR1 gene. The term "gene" can include not only coding sequences, but also regulatory regions such as promoters, enhancers, and termination regions. The term can further include all introns and other DNA sequences spliced from the mRNA transcript, along with variants obtained from alternative splice sites. The gene sequence encoding a particular protein can be a DNA or RNA that directs the expression of the particular protein. These nucleic acid sequences may be DNA strand sequences transcribed into RNA or RNA sequences translated into specific proteins. Nucleic acid sequences include both full-length nucleic acid sequences and non-full-length sequences derived from full-length proteins.
様々な実施形態では、処置を受ける対象は、1つまたはそれより多くの変異をACVR1遺伝子中に有する。実施形態では、対象は、そのような変異(複数可)を含む所定の遺伝子プロファイルを有する。一部の実施形態では、ACVR1遺伝子中の1つまたはそれより多くの変異は、ミスセンス変異、フレームシフト変異、重複(即ち、コピー数多型)、スプライス部位変異、またはこれらの組合せを含む。 In various embodiments, the subject to be treated has one or more mutations in the ACVR1 gene. In embodiments, the subject has a predetermined genetic profile that includes such mutations (s). In some embodiments, one or more mutations in the ACVR1 gene include missense mutations, frameshift mutations, duplications (ie, copy number variation), splice site mutations, or combinations thereof.
実施形態において、1つまたはそれより多くの変異は、(P197F198)L、C509S、D185G、D185N、D433N、E38FS、F265S、G225D、G264S、G328E、G328R、G328V、G328W、G356D、G50C、H320Y、I323V、K31E、K345Q、L196P、L251S、M34I、N100D、N481I、P115S、P455A、Q207E、Q278P、R201I、R206C、R206H、R258G、R258S、R307Q、R325A、R375C、R375P、R401M、R490H、S130F、S226N、S41F、S440G、S469C、S56L、T298S、V234M、V91M、W98R、またはこれらの組合せを含む。実施形態において、ACVR1遺伝子中の1つまたはそれより多くの変異は、R206H、G328V、R258G、またはこれらの組合せを含む。ある特定の実施形態では、ACVR1遺伝子中の1つまたはそれより多くの変異は、R206Hを含む。 In embodiments, one or more mutations are (P197F198) L, C509S, D185G, D185N, D433N, E38FS, F265S, G225D, G264S, G328E, G328R, G328V, G328W, G356D, G50C, H320Y, I. , K31E, K345Q, L196P, L251S, M34I, N100D, N481I, P115S, P455A, Q207E, Q278P, R201I, R206C, R206H, R258G, R258S, R307Q, R325A, R375C, R375P , S440G, S469C, S56L, T298S, V234M, V91M, W98R, or a combination thereof. In embodiments, one or more mutations in the ACVR1 gene include R206H, G328V, R258G, or a combination thereof. In certain embodiments, one or more mutations in the ACVR1 gene include R206H.
一部の実施形態において、ACVR1遺伝子中の1つまたはそれより多くの変異は、ミスセンス変異を含む。一部の実施形態において、ミスセンス変異は、C509S、D185N、D433N、F265S、G225D、H320Y、I323V、K31E、K345Q、M34I、N100D、N481I、P115S、P455A、Q278P、R206C、R401M、S130F、S226N、S41F、S41F、S440G、S469C、S56L、T298S、V234M、V91M、またはW98Rである。一部の実施形態において、ACVR1遺伝子中の1つまたはそれより多くの変異は、フレームシフト変異を含む。一部の実施形態では、フレームシフト変異はE38fsである。一部の実施形態では、ACVR1遺伝子中の1つまたはそれより多くの変異は、スプライス部位変異を含む。一部の実施形態では、スプライス部位変異はG264Sである。 In some embodiments, one or more mutations in the ACVR1 gene include missense mutations. In some embodiments, missense mutations are C509S, D185N, D433N, F265S, G225D, H320Y, I323V, K31E, K345Q, M34I, N100D, N481I, P115S, P455A, Q278P, R206C, R401M, S130F, S226. , S41F, S440G, S469C, S56L, T298S, V234M, V91M, or W98R. In some embodiments, one or more mutations in the ACVR1 gene include frameshift mutations. In some embodiments, the frameshift mutation is E38fs. In some embodiments, one or more mutations in the ACVR1 gene include splice site mutations. In some embodiments, the splice site mutation is G264S.
本明細書で使用される場合、変異に対する簡略表記は、(天然に存在するアミノ酸)(そのアミノ酸の位置)(および変異ペプチド中に存在するアミノ酸)を示す。例えば、P197Lは、197位のプロリンがロイシンで置き換わることを示す。別の実施例では、(P197F198)Lは、197位のプロリンおよび198位のフェニルアラニンが、まとめて単一ロイシンで置き換わることを示す。 As used herein, the abbreviation for mutation indicates (naturally occurring amino acid) (position of that amino acid) (and amino acid present in the mutant peptide). For example, P197L indicates that proline at position 197 is replaced by leucine. In another example, (P197F198) L indicates that proline at position 197 and phenylalanine at position 198 are collectively replaced by a single leucine.
そのような変異は、任意の適切な方法を使用して検出されてもよい。実施形態では、変異は、試料と試薬(例えば、抗体または核酸プライマー)とを接触させ、試薬およびマーカー(複数可)の複合体を発生させ、複合体を検出することによって、検出される。抗体は、受動的結合などの公知の技法に従い、診断アッセイに適した固体支持体とコンジュゲートすることができる。抗体は、マルチカラーフローサイトメトリーを含むフローサイトメトリーなどの公知の技法に従い、診断アッセイのために細胞表面抗原にコンジュゲートすることができる。抗体は、公知の技法に従い、検出可能な標識または基、例えば放射性標識、酵素標識、および蛍光標識にコンジュゲートすることができる。 Such mutations may be detected using any suitable method. In embodiments, mutations are detected by contacting a sample with a reagent (eg, an antibody or nucleic acid primer) to generate a complex of reagents and markers (s) and detecting the complex. Antibodies can be conjugated to solid supports suitable for diagnostic assays according to known techniques such as passive binding. Antibodies can be conjugated to cell surface antigens for diagnostic assays according to known techniques such as flow cytometry, including multicolor flow cytometry. Antibodies can be conjugated to detectable labels or groups, such as radioactive labels, enzyme labels, and fluorescent labels, according to known techniques.
様々な実施形態では、ACVR1阻害剤の有効量が対象に投与される。実施形態では、ACVR1阻害剤は、がんの処置のために投与される。固形腫瘍および白血病を含む広く様々ながんは、本明細書に開示される方法に適している。実施形態では、がんは固形がんである。一部の実施形態では、がんは脳がんである。一部の実施形態では、がんは、子宮、卵巣、または子宮頸がんである。一部の実施形態では、がんは子宮がんである。一部の実施形態では、子宮がんは子宮体がんである。一部の実施形態では、がんは肺がんである。一部の実施形態では、がんは乳がんである。一部の実施形態では、がんは結腸がんである。 In various embodiments, an effective amount of the ACVR1 inhibitor is administered to the subject. In embodiments, the ACVR1 inhibitor is administered for the treatment of cancer. A wide variety of cancers, including solid tumors and leukemias, are suitable for the methods disclosed herein. In embodiments, the cancer is a solid cancer. In some embodiments, the cancer is brain cancer. In some embodiments, the cancer is uterine, ovarian, or cervical cancer. In some embodiments, the cancer is uterine cancer. In some embodiments, uterine cancer is endometrial cancer. In some embodiments, the cancer is lung cancer. In some embodiments, the cancer is breast cancer. In some embodiments, the cancer is colon cancer.
一部の実施形態では、がんは固形腫瘍を含む。一部の実施形態では、がんは、進行固形腫瘍を含む。一部の実施形態では、がんは、進行転移または進行固形腫瘍を含む。実施形態では、がんは、脳腫瘍または子宮腫瘍を含む。実施形態では、がんは、非固形がんである。様々な実施形態では、がんは、前転移がんである。様々な実施形態では、がんは、転移がんである。 In some embodiments, the cancer comprises a solid tumor. In some embodiments, the cancer comprises an advanced solid tumor. In some embodiments, the cancer comprises advanced metastases or advanced solid tumors. In embodiments, the cancer comprises a brain tumor or a uterine tumor. In embodiments, the cancer is a non-solid cancer. In various embodiments, the cancer is a pre-metastatic cancer. In various embodiments, the cancer is a metastatic cancer.
様々な実施形態で処置され得るがんのタイプには:脳がん、子宮がん、卵巣がん、子宮頸がん、肺がん、乳がん、直腸がん、胃腸がん、造血またはリンパ系がん、皮膚がん、および骨がんが含まれる。 Types of cancer that can be treated in various embodiments include: brain cancer, uterine cancer, ovarian cancer, cervical cancer, lung cancer, breast cancer, rectal cancer, gastrointestinal cancer, hematopoietic or lymphoid cancer , Skin cancer, and bone cancer.
一部の実施形態では、がんは、脳がん(例えば、脳幹グリオーマ、小脳星状細胞腫、上衣腫、髄芽腫、原始神経胚葉性腫瘍、視覚路、および視床下部グリオーマ)である。様々な実施形態では、脳がんは脳腫瘍である。様々な実施形態では、脳がんは、グリオーマ(例えば、脳幹、大脳星状細胞腫、視覚路、および視床下部)である。一部の実施形態では、グリオーマは高悪性度グリオーマである。一部の実施形態では、脳がんは、脳幹グリオーマである。一部の実施形態では、脳がんはDIPGである。一部の実施形態では、がんは、中枢神経系がん(例えば、中枢神経系リンパ腫)である。一部の実施形態では、がんは神経芽細胞腫である。 In some embodiments, the cancer is brain cancer (eg, brain stem glioma, cerebellar astrocytoma, ependymoma, medulloblastoma, primordial neuroblastoma, visual tract, and hypothalamic glioma). In various embodiments, the brain cancer is a brain tumor. In various embodiments, the brain cancer is a glioma (eg, brain stem, astrocytoma, visual tract, and hypothalamus). In some embodiments, the glioma is a high-grade glioma. In some embodiments, the brain cancer is a brain stem glioma. In some embodiments, the brain cancer is DIPG. In some embodiments, the cancer is a central nervous system cancer (eg, central nervous system lymphoma). In some embodiments, the cancer is a neuroblastoma.
一部の実施形態では、がんは、子宮、卵巣、または子宮頸がんである。一部の実施形態では、がんは、子宮がんである。一部の実施形態では、子宮がんは、子宮体がんである。一部の実施形態では、子宮体がんは、子宮体子宮内膜がんである。実施形態では、がんは卵巣がん(例えば、卵巣腺癌)である。 In some embodiments, the cancer is uterine, ovarian, or cervical cancer. In some embodiments, the cancer is uterine cancer. In some embodiments, the uterine cancer is endometrial cancer. In some embodiments, the endometrial cancer is endometrial cancer. In embodiments, the cancer is ovarian cancer (eg, ovarian adenocarcinoma).
一部の実施形態では、がんは、肺がん(例えば、非小細胞、小細胞)である。ある特定の実施形態では、肺がんは小細胞肺がんである。一部の実施形態では、がんは乳がんである。 In some embodiments, the cancer is lung cancer (eg, non-small cell, small cell). In certain embodiments, lung cancer is small cell lung cancer. In some embodiments, the cancer is breast cancer.
一部の実施形態では、がんは胃腸がんである。一部の実施形態では、がんは上気道消化管がんである。一部の実施形態では、がんは大腸がんである。一部の実施形態では、がんは直腸がんである。 In some embodiments, the cancer is gastrointestinal cancer. In some embodiments, the cancer is upper airway gastrointestinal cancer. In some embodiments, the cancer is colorectal cancer. In some embodiments, the cancer is rectal cancer.
一部の実施形態では、がんは、尿路がんである。一部の実施形態では、がんは前立腺がんである。 In some embodiments, the cancer is urinary tract cancer. In some embodiments, the cancer is prostate cancer.
一部の実施形態では、がんは、造血またはリンパ系がんである。様々な実施形態では、がんは、白血病(例えば、急性リンパ性白血病、急性骨髄性白血病、慢性リンパ性白血病、慢性骨髄性白血病、毛様細胞、急性T細胞白血病など)またはリンパ腫(例えば、AIDS関連、バーキット、皮膚T細胞ホジキン、非ホジキン、原発性中枢神経系)である。 In some embodiments, the cancer is hematopoietic or lymphoid cancer. In various embodiments, the cancer is leukemia (eg, acute lymphocytic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, hairy cells, acute T-cell leukemia, etc.) or lymphoma (eg, AIDS). Related, Berkit, skin T-cell hodgkin, non-hodgkin, primary central nervous system).
実施形態では、がんは皮膚がんである。様々な実施形態では、がんは、黒色腫(例えば、皮膚黒色腫)である。一部の実施形態では、がんは、骨がんである。一部の実施形態では、がんは骨腫瘍(例えば、骨肉腫、悪性線維性組織球腫)である。 In embodiments, the cancer is skin cancer. In various embodiments, the cancer is melanoma (eg, cutaneous melanoma). In some embodiments, the cancer is bone cancer. In some embodiments, the cancer is a bone tumor (eg, osteosarcoma, malignant fibrous histiocytoma).
特定のタイプのがんに関連付けられる例示的な変異を、表1に示す。
ACVR1阻害剤の有効量は、腫瘍細胞の数を減少させ、転移の数を減少させ、腫瘍体積を減少させ、がん細胞のアポトーシスを誘発させ、がん細胞の死を誘発させ、がん細胞での放射線感受性を誘発させ、がん細胞近くの血管新生を阻害し、がん細胞増殖を阻害し、腫瘍成長を阻害し、転移を予防し、転移の数を低減させ、平均寿命を延ばし、対象の寿命を延ばし、がん関連の疼痛を低減させ、そして/または処置後のがんのぶり返しもしくは再発を低減させることができる。様々な実施形態では、ACVR1阻害剤を投与する影響を、客観的X線評価によって評定することができる。 Effective amounts of ACVR1 inhibitors reduce the number of tumor cells, reduce the number of metastases, reduce tumor volume, induce cancer cell apoptosis, induce cancer cell death, cancer cells Induces radiosensitivity in, inhibits angiogenesis near cancer cells, inhibits cancer cell growth, inhibits tumor growth, prevents metastasis, reduces the number of metastases, prolongs life expectancy, It can extend the lifespan of a subject, reduce cancer-related pain, and / or reduce cancer recurrence or recurrence after treatment. In various embodiments, the effects of administering an ACVR1 inhibitor can be assessed by objective x-ray assessment.
他の実施形態では、疾患は、遺伝子障害である。一部の実施形態では、疾患はFOPである。 In other embodiments, the disease is a genetic disorder. In some embodiments, the disease is FOP.
実施形態において、本明細書に記述される処置の方法は、ACVR1阻害剤の有効量を対象に投与することを含む。様々な実施形態では、ACVR1阻害剤は、以下の式(I):
R1は、HまたはC1〜C6アルコキシであり;
R2は、C1〜C6アルコキシまたはヘテロシクリルであり;
R3は、ハロまたはC1〜C6アルコキシであり;および
R4は、HまたはC1〜C6アルキルである)、
またはその立体異性体、薬学的に許容される塩、互変異性体、もしくはプロドラッグを有する、ACVR1阻害剤である。
In embodiments, the methods of treatment described herein include administering to a subject an effective amount of an ACVR1 inhibitor. In various embodiments, the ACVR1 inhibitor is represented by the following formula (I):
R 1 is H or C 1 to C 6 alkoxy;
R 2 is C 1 -C 6 alkoxy or heterocyclyl;
R 3 is halo or C 1 to C 6 alkoxy; and R 4 is H or C 1 to C 6 alkyl),
Or an ACVR1 inhibitor having a stereoisomer thereof, a pharmaceutically acceptable salt, a tautomer, or a prodrug.
したがって実施形態では、本開示の方法は、ACVR1阻害剤を含む処置レジメンを、ACVR1遺伝子中に1つまたはそれより多くの変異を含む所定の遺伝子プロファイルを有する対象に投与することを含み、ACVR1阻害剤は、以下の式(I):
またはその立体異性体、薬学的に許容される塩、互変異性体、もしくはプロドラッグを有する。
Thus, in embodiments, the methods of the present disclosure include administering a treatment regimen containing an ACVR1 inhibitor to a subject having a predetermined gene profile containing one or more mutations in the ACVR1 gene, comprising administering the ACVR1 inhibitor. The agent has the following formula (I):
Or it has a stereoisomer thereof, a pharmaceutically acceptable salt, a tautomer, or a prodrug.
さらなる実施形態では、本開示の方法は、対象のびまん性真性橋膠腫(DIPG)を処置する方法を含み、この方法は、ACVR1阻害剤を含む処置レジメンを対象に投与することを含み、ACVR1阻害剤は、以下の式(I):
またはその立体異性体、薬学的に許容される塩、互変異性体、もしくはプロドラッグを有する。
In a further embodiment, the methods of the present disclosure include a method of treating diffuse true pontine glioma (DIPG) of a subject, the method comprising administering to the subject a treatment regimen comprising an ACVR1 inhibitor, ACVR1. Inhibitors have the following formula (I):
Or it has a stereoisomer thereof, a pharmaceutically acceptable salt, a tautomer, or a prodrug.
ACVR1阻害剤(I)の、ある特定の実施形態では、R1はHである。 ACVR1 inhibitor (I), in certain embodiments, R 1 is H.
ACVR1阻害剤(I)の一部の実施形態では、R1はC1〜C6アルコキシである。一部の実施形態では、C1〜C6アルコキシはメトキシである。 In some embodiments of ACVR1 inhibitor (I), R 1 is a C 1 -C 6 alkoxy. In some embodiments, C 1 -C 6 alkoxy is methoxy.
ACVR1阻害剤(I)の実施形態では、R2はC1〜C6アルコキシである。特定の実施形態では、C1〜C6アルコキシはメトキシである。 In the embodiment of ACVR1 inhibitor (I), R 2 is a C 1 -C 6 alkoxy. In certain embodiments, C 1 -C 6 alkoxy is methoxy.
ACVR1阻害剤(I)の他の実施形態では、R2はヘテロシクリルである。ある特定の実施形態では、ヘテロシクリルは、必要に応じて置換されたピペラジニルである。特定の実施形態では、必要に応じて置換されたピペラジニルは、C1〜C6アルキルまたはC1〜C6ヒドロキシアルキルである。 In another embodiment of ACVR1 inhibitor (I), R 2 is heterocyclyl. In certain embodiments, the heterocyclyl is a optionally substituted piperazinyl. In certain embodiments, piperazinyl optionally substituted is a C 1 -C 6 alkyl or C 1 -C 6 hydroxyalkyl.
ACVR1阻害剤(I)の一部の実施形態では、R3はハロである。特定の実施形態では、ハロはクロロである。 In some embodiments of ACVR1 inhibitor (I), R 3 is a halo. In certain embodiments, the halo is chloro.
ACVR1阻害剤(I)のさらなる実施形態では、R3はC1〜C6アルコキシである。ある特定の実施形態では、C1〜C6アルコキシはメトキシである。 In a further embodiment of the ACVR1 inhibitor (I), R 3 is a C 1 -C 6 alkoxy. In certain embodiments, the C 1- C 6 alkoxy is methoxy.
ACVR1阻害剤(I)の実施形態では、R4はHである。一部の実施形態では、R4はC1〜C6アルキルである。ある特定の実施形態では、C1〜C6アルキルはメチルである。 In the embodiment of ACVR1 inhibitor (I), R 4 is a H. In some embodiments, R 4 is C 1 -C 6 alkyl. In certain embodiments, C 1 -C 6 alkyl is methyl.
他の、ある特定の実施形態では、ACVR1阻害剤は、表2のACVR1阻害剤から選択される。
特定の実施形態では、ACVR1阻害剤は、
特定の実施形態では、ACVR1阻害剤は、
特定の実施形態では、ACVR1阻害剤は、
特定の実施形態では、ACVR1阻害剤は、
特定の実施形態では、ACVR1阻害剤は、
特定の実施形態では、ACVR1阻害剤は、
様々な実施形態では、薬学的に許容される塩は、酸付加塩である。一部の実施形態では、酸付加塩は塩酸塩である。 In various embodiments, the pharmaceutically acceptable salt is an acid addition salt. In some embodiments, the acid addition salt is a hydrochloride salt.
上述の式(I)のACVR1阻害剤の任意の実施形態、および上述の式(I)のACVR1阻害剤中の、本明細書に記述される特定の置換基(例えば、R1〜R4)のいずれかは、独立して、その他の実施形態および/または式(I)のACVR1阻害剤の置換基と組み合わされて、上記にて特に述べられていない本開示の実施形態を形成することが理解される。さらに、置換基のリストが、特定の実施形態および/または請求項における任意の特定のR基に関して列挙される事象では、個々の置換基それぞれを特定の実施形態および/または請求項から欠失させてもよく、かつ置換基の残りのリストは、本開示の範囲内にあると見なされることが、理解される。本記述において、示される式の置換基および/または変数の組合せは、そのような寄与が、安定なACVR1阻害剤をもたらす場合のみ許容されることが、理解される。 Any embodiments, and wherein in ACVR1 inhibitor (I) above, particular substituents described herein in ACVR1 inhibitors of above formula (I) (e.g., R 1 to R 4) Any of these can be independently combined with other embodiments and / or substituents of the ACVR1 inhibitor of formula (I) to form embodiments of the present disclosure not specifically mentioned above. Understood. In addition, in events where the list of substituents is listed for any particular R group in a particular embodiment and / or claim, each individual substituent is deleted from the particular embodiment and / or claim. It is understood that the remaining list of substituents may be considered to be within the scope of the present disclosure. In this description, it is understood that the substituent and / or variable combinations of the formulas shown are only acceptable if such contributions result in a stable ACVR1 inhibitor.
本開示のACVR1阻害剤は、参照により本明細書に組み込まれるUS2016/0214944に記載される方法を含む、当技術分野で公知の任意の数の方法に従い調製することができる。
The ACVR1 inhibitors of the present disclosure can be prepared according to any number of methods known in the art, including those described in
遊離塩基または酸形態で存在する本開示の全てのACVR1阻害剤は、当業者に公知の方法による、適切な無機または有機塩基での処置によって、それらの薬学的に許容される塩に変換することができる。本開示のACVR1阻害剤の塩は、標準的な技法によって、それらの遊離塩基または酸形態に変換することができる。 All ACVR1 inhibitors presently present in free base or acid form shall be converted to their pharmaceutically acceptable salts by treatment with appropriate inorganic or organic bases by methods known to those of skill in the art. Can be done. The salts of ACVR1 inhibitors of the present disclosure can be converted to their free base or acid form by standard techniques.
本明細書に記述されるACVR1阻害剤(即ち、式(I)のACVR1阻害剤)は、1種またはそれより多種のその他の、追加の治療剤と組み合わせて使用することができる。ACVR1阻害剤の投薬量は、任意の薬物間反応に合わせて調節され得る。 The ACVR1 inhibitors described herein (ie, the ACVR1 inhibitors of formula (I)) can be used in combination with one or more other additional therapeutic agents. The dosage of the ACVR1 inhibitor can be adjusted for any drug-drug response.
したがって実施形態では、本開示の方法は、対象の固形腫瘍、DIPG、またはFOPを処置する方法を含み、この方法は、処置レジメンを対象に投与することを含み、処置レジメンは、ACVR1阻害剤および追加の治療剤を含み、ACVR1阻害剤は、以下の式(I):
またはその立体異性体、薬学的に許容される塩、互変異性体、もしくはプロドラッグを有する。
Thus, in embodiments, the methods of the present disclosure include treating a solid tumor, DIPG, or FOP of interest, the method comprising administering to the subject a treatment regimen, wherein the treatment regimen is an ACVR1 inhibitor and. ACVR1 inhibitors, including additional therapeutic agents, have the following formula (I):
Or it has a stereoisomer thereof, a pharmaceutically acceptable salt, a tautomer, or a prodrug.
様々な実施形態では、追加の治療剤は、レチノイン酸受容体ガンマアゴニスト;mTOR阻害剤;アクチビンA抗体;キナーゼ阻害剤;ACVR1抗体;TAK1阻害剤;ホスホジエステラーゼ阻害剤;HDAC阻害剤;化学療法剤;免疫療法剤;細胞療法;ペプチドまたは腫瘍ライセートワクチン;イリノテカン;GM−CSFを持つTTRNA−DCワクチン、TTRNA−xALT;インテグリン阻害剤;IL−12療法;抗ネオプラストン療法;イミキモド;腫瘍溶解性アデノウイルス;WEE1阻害剤;WT1タンパク質由来ペプチドワクチン;ペグ化インターフェロンアルファ2b;キナーゼ抗体;円滑化阻害剤;チューブリン阻害剤;テロメラーゼ阻害剤;CD40アゴニスト;GM−CSFアゴニスト;IDO阻害剤;および放射性ヨウ素標識モノクローナル抗体8H9.64のうちの1つまたは複数を含む。 In various embodiments, the additional therapeutic agent is a retinoic acid receptor gamma agonist; mTOR inhibitor; activin A antibody; kinase inhibitor; ACVR1 antibody; TAK1 inhibitor; phosphodiesterase inhibitor; HDAC inhibitor; chemotherapeutic agent; Immunotherapeutic agents; Cell therapy; Peptide or tumor lysate vaccines; Irinotecan; TTRNA-DC vaccine with GM-CSF, TTRNA-xALT; Integrin inhibitors; IL-12 therapy; Anti-neoplastone therapy; Imikimod; Tumor-dissolving adenovirus WEE1 inhibitors; WT1 protein-derived peptide vaccines; pegged interferon alpha 2b; kinase antibodies; smoothing inhibitors; tubulin inhibitors; telomerase inhibitors; CD40 agonists; GM-CSF agonists; IDO inhibitors; and radioiodine labeling Includes one or more of the monoclonal antibodies 8H9.64.
実施形態では、追加の治療剤は、レチノイン酸受容体ガンマアゴニスト;mTOR阻害剤;アクチビンA抗体;キナーゼ阻害剤;ACVR1抗体;TAK1阻害剤;およびホスホジエステラーゼ阻害剤のうちの1つまたは複数を含む。一部の実施形態では、追加の治療剤は、HDAC阻害剤;化学療法剤;免疫療法剤;細胞療法;ペプチドまたは腫瘍ライセートワクチン;イリノテカン;GM−CSFを持つTTRNA−DCワクチン、TTRNA−xALT;インテグリン阻害剤;IL−12療法;抗ネオプラストン療法;イミキモド;腫瘍溶解性アデノウイルス;WEE1阻害剤;WT1タンパク質由来ペプチドワクチン;ペグ化インターフェロンアルファ2b;キナーゼ抗体;キナーゼ阻害剤;円滑化阻害剤;チューブリン阻害剤;テロメラーゼ阻害剤;CD40アゴニスト;GM−CSFアゴニスト;IDO阻害剤;および放射性ヨウ素標識モノクローナル抗体8H9のうちの1つまたは複数を含む。 In embodiments, the additional therapeutic agent comprises one or more of a retinoic acid receptor gamma agonist; an mTOR inhibitor; an activin A antibody; a kinase inhibitor; an ACVR1 antibody; a TAK1 inhibitor; and a phosphodiesterase inhibitor. In some embodiments, additional therapeutic agents are HDAC inhibitors; chemotherapeutic agents; immunotherapeutic agents; cell therapy; peptide or tumor lysate vaccines; irinotecan; TTRNA-DC vaccines with GM-CSF, TTRNA-xALT; Integrin inhibitor; IL-12 therapy; anti-neoplastone therapy; imikimod; tumor-soluble adenovirus; WEE1 inhibitor; WT1 protein-derived peptide vaccine; pegged interferon alpha 2b; kinase antibody; kinase inhibitor; smoothing inhibitor; It comprises one or more of a tubulin inhibitor; a telomerase inhibitor; a CD40 agonist; a GM-CSF agonist; an IDO inhibitor; and a radioactive iodine-labeled monoclonal antibody 8H9.
実施形態では、キナーゼ阻害剤は、サイクリン依存性キナーゼ(CDK)を阻害する。一部の実施形態では、CDKは、CDK9またはCDK7である。一部の実施形態では、CDKはCDK9である。様々な実施形態では、CDK9阻害剤は、siRNA、アルボシジブ、またはそのプロドラッグ、ジナシクリブ、またはこれらの組合せである。特定の実施形態では、CDK9阻害剤は、アルボシジム、またはそのプロドラッグである。そのようなプロドラッグの例(例えば、リン酸プロドラッグ)は、参照により本明細書に組み込まれる米国特許第9,758,539号に記載されている。実施形態では、キナーゼ阻害剤はホスホイノシチド3−キナーゼ(PI3K)を阻害する。 In embodiments, the kinase inhibitor inhibits a cyclin-dependent kinase (CDK). In some embodiments, the CDK is CDK9 or CDK7. In some embodiments, the CDK is CDK9. In various embodiments, the CDK9 inhibitor is siRNA, alvocidib, or a prodrug thereof, dynasicrib, or a combination thereof. In certain embodiments, the CDK9 inhibitor is arbocidim, or a prodrug thereof. Examples of such prodrugs (eg, phosphate prodrugs) are described in US Pat. No. 9,758,539, which is incorporated herein by reference. In embodiments, the kinase inhibitor inhibits phosphoinositide 3-kinase (PI3K).
様々な実施形態では、免疫療法剤は、免疫チェックポイント阻害剤である。一部の実施形態では、免疫チェックポイント阻害剤は、PD−1阻害剤である。特定の実施形態では、PD−1阻害剤は、ペムブロリズマブ、ニボルマブ、またはこれらの組合せである。一部の実施形態では、免疫チェックポイント阻害剤は、PD−L1阻害剤である。ある特定の実施形態では、PD−L1阻害剤は、アテゾリズマブ、アベルマブ、デュルバルマブ、またはこれらの組合せである。 In various embodiments, the immunotherapeutic agent is an immune checkpoint inhibitor. In some embodiments, the immune checkpoint inhibitor is a PD-1 inhibitor. In certain embodiments, the PD-1 inhibitor is pembrolizumab, nivolumab, or a combination thereof. In some embodiments, the immune checkpoint inhibitor is a PD-L1 inhibitor. In certain embodiments, the PD-L1 inhibitor is atezolizumab, avelumab, durvalumab, or a combination thereof.
一部の実施形態では、キナーゼ抗体は、薬物コンジュゲートを含む。 In some embodiments, the kinase antibody comprises a drug conjugate.
特定の実施形態では、レチノイン酸受容体ガンマアゴニストはパロバロテンである。特定の実施形態では、mTOR阻害剤は、ラパマイシンまたはエベロリムスである。特定の実施形態では、アクチビンA抗体はREGN2447である。特定の実施形態では、キナーゼ阻害剤は、サラカチニブ、モメロチニブ、ドルソモルフィン、イマチニブ、クリゾチニブ、ダサチニブ、ベバシズマブ、エルロチニブ、バンデタニブ、リボシクリブ、クレノラニブ、アベマシクリブ、ONC201、シレンギチド、アルボシジブ、またはこれらのプロドラッグである。特定の実施形態では、ホスホジエステラーゼ阻害剤は、ジピリダモルである。特定の実施形態では、HDAC阻害剤は、SAHA、ボリノスタット、またはパノビノスタットである。特定の実施形態では、免疫療法剤はMDV9300である。特定の実施形態では、細胞療法は、自家樹状細胞を含む。特定の実施形態では、ペプチドまたは腫瘍ライセートワクチンは、K27Mペプチドまたはリンドペピムトを含む。特定の実施形態では、イリノテカンは、対流強化送達により投与される。特定の実施形態では、インテグリン阻害剤はシレンギチドである。特定の実施形態では、抗ネオプラストン療法は、アテンゲナルまたはアスツゲナルである。特定の実施形態では、腫瘍溶解性アデノウイルスはDNX−2401である。特定の実施形態では、WEE1阻害剤はAZD1775である。特定の実施形態では、WT1タンパク質由来ペプチドワクチンはDSP−7888である。特定の実施形態では、キナーゼ抗体はニモツズマブ、エルビタックス、またはABT−414である。特定の実施形態では、円滑化阻害剤はビスモデギブである。特定の実施形態では、チューブリン阻害剤はメベンダゾールである。特定の実施形態では、テロメラーゼ阻害剤はイメテルスタットである。特定の実施形態では、CD40アゴニストはAPX005Mである。特定の実施形態では、GM−CSFアゴニストは、レオウイルスを持つサルグラモスチンである。特定の実施形態では、IDO阻害剤はインドキシモドである。 In certain embodiments, the retinoic acid receptor gamma agonist is parovarotene. In certain embodiments, the mTOR inhibitor is rapamycin or everolimus. In certain embodiments, the activin A antibody is REGN2447. In certain embodiments, the kinase inhibitors are salacatinib, momerotinib, dolsomorphin, imatinib, crizotinib, dasatinib, bevacizumab, erlotinib, vandetanib, ribociclib, clenoranib, abemaciclib, ONC201, sirengitide, albocitide, syrengitide, albocytide. In certain embodiments, the phosphodiesterase inhibitor is dipyridamol. In certain embodiments, the HDAC inhibitor is SAHA, vorinostat, or panobinostat. In certain embodiments, the immunotherapeutic agent is MDV9300. In certain embodiments, cell therapy comprises autologous dendritic cells. In certain embodiments, the peptide or tumor lysate vaccine comprises a K27M peptide or lindpepimut. In certain embodiments, irinotecan is administered by convection enhanced delivery. In certain embodiments, the integrin inhibitor is sirengitide. In certain embodiments, the anti-neoplaston therapy is attengenal or astugenal. In certain embodiments, the oncolytic adenovirus is DNX-2401. In certain embodiments, the WEE1 inhibitor is AZD1775. In certain embodiments, the WT1 protein-derived peptide vaccine is DSP-7888. In certain embodiments, the kinase antibody is nimotuzumab, erbitux, or ABT-414. In certain embodiments, the smoothing inhibitor is vismodegib. In certain embodiments, the tubulin inhibitor is mebendazole. In certain embodiments, the telomerase inhibitor is imeterstat. In certain embodiments, the CD40 agonist is APX005M. In certain embodiments, the GM-CSF agonist is salgramostin with a reoviridae. In certain embodiments, the IDO inhibitor is indoxymod.
一実施形態では、追加の治療剤は化学療法剤である。特定の実施形態では、化学療法剤は、メルファラン、ゲムシタビン、テモゾロミド、シクロホスファミド、フルダラビン、ドキソルビシン、イリノテカン、レナリドミド、バルプロ酸、クロロキン、カルボプラチン、エトポシド、イフォスファミド、ポマリドミド、またはロムスチンである。 In one embodiment, the additional therapeutic agent is a chemotherapeutic agent. In certain embodiments, the chemotherapeutic agent is melphalan, gemcitabine, temozolomide, cyclophosphamide, fludarabine, doxorubicin, irinotecan, renalidemid, valproic acid, chloroquin, carboplatin, etoposide, ifosfamide, pomalidemid, or lomustine.
上記方法は、放射線療法と組み合わせて実施することもでき、放射線療法と組み合わせたACVR1阻害剤の量は、上記疾患の処置において有効である。 The method can also be performed in combination with radiation therapy, and the amount of ACVR1 inhibitor combined with radiation therapy is effective in treating the disease.
一実施形態では、放射線は、式(I)の化合物での処置前に提供される。一実施形態では、放射線は、式(I)の化合物での処置後に提供される。一実施形態では、放射線は、式(I)の化合物での処置の前および後の両方で提供される。一実施形態では、式(I)の化合物での処置は、長期間が過ぎた後に対象に再照射可能であり、放射線療法に伴う副作用を低減させることが可能である。 In one embodiment, the radiation is provided prior to treatment with the compound of formula (I). In one embodiment, the radiation is provided after treatment with the compound of formula (I). In one embodiment, the radiation is provided both before and after treatment with the compound of formula (I). In one embodiment, treatment with the compound of formula (I) can re-irradiate the subject after a long period of time, reducing the side effects associated with radiation therapy.
放射線療法を投与するための技法は、当技術分野で公知であり、これらの技法は本明細書に記述される併用療法で使用することができる。この併用療法における本開示のACVR1阻害剤の投与は、本明細書に記述されるように決定することができる。 Techniques for administering radiation therapy are known in the art and these techniques can be used in the combination therapies described herein. Administration of the ACVR1 inhibitors of the present disclosure in this combination therapy can be determined as described herein.
一部の実施形態では、本明細書に記述される処置レジメンはさらに、外科的切除を含む。そのような技法は、当技術分野で公知である。 In some embodiments, the treatment regimen described herein further comprises surgical resection. Such techniques are known in the art.
様々な実施形態では、対象は、これらの疾患に関する前処置を受けている。そのような実施形態において、対象は、前処置に対して難治性であるかまたは前処置に不寛容である可能性がある。本明細書で使用される場合、「難治性」は、参照により本明細書に組み込まれるEur J Cancer. 2016 Jul;62:132-7に記載されるように、対象がRECISTによって進行性疾患を表すことを示す。対象は、当業者により理解されるように、標的病変に少なくとも20%の増加がある場合、RECISTに基づいて「進行性疾患」を有すると見なされる。DIPGに関する「完全奏効」は、全ての標的病変の消失を指す。DIPGの「部分奏効」は、標的病変の合計の少なくとも30%の減少を指し、「安定疾患」は、PRに適するよう十分に収縮もしないしPDに適するよう十分に増加もしない。本明細書で使用される場合、「不寛容」は、対象が、有効量の治療剤の有害効果に耐えることができずまたは耐えることを望まないことを意味する。一部の実施形態では、対象は、前処置レジメンに不寛容である。 In various embodiments, the subject is undergoing pretreatment for these diseases. In such embodiments, the subject may be refractory to the pretreatment or intolerant to the pretreatment. As used herein, "refractory" refers to a subject with progressive disease by RECIST, as described in Eur J Cancer. 2016 Jul; 62: 132-7, which is incorporated herein by reference. Indicates to represent. A subject is considered to have "progressive disease" based on RECIST if there is an increase of at least 20% in the target lesion, as will be appreciated by those skilled in the art. "Complete response" for DIPG refers to the disappearance of all target lesions. DIPG "partial response" refers to a reduction of at least 30% of the total target lesions, and "stable disease" does not contract sufficiently to suit PR or increase sufficiently to suit PD. As used herein, "intolerance" means that a subject is unable or unwilling to tolerate the adverse effects of an effective amount of therapeutic agent. In some embodiments, the subject is intolerant of the pretreatment regimen.
特にDIPGに関し、式(I)の化合物による処置に有効性は、週、月、または年に基づいて計算され得る全生存期間(OS)または無進行生存(PFS)に基づいて測定されてもよい。一実施形態では、式(I)の化合物による処置は、6か月もしくはそれよりも長い、9か月もしくはそれよりも長い、1年もしくはそれよりも長い、または5年もしくはそれよりも長いOSを提供する。一実施形態では、式(I)の化合物による処置は、少なくとも6か月、少なくとも9か月、少なくとも1年、または少なくとも5年のPFSを提供する。 Efficacy for treatment with compounds of formula (I), especially with respect to DIPG, may be measured based on overall survival (OS) or progression-free survival (PFS), which can be calculated on a weekly, monthly, or yearly basis. .. In one embodiment, treatment with a compound of formula (I) is 6 months or longer, 9 months or longer, 1 year or longer, or 5 years or longer OS. I will provide a. In one embodiment, treatment with a compound of formula (I) provides at least 6 months, at least 9 months, at least 1 year, or at least 5 years of PFS.
実施形態では、処置がなされる対象は、所定レベルの1つまたはそれより多くのマーカーを有する。一部の実施形態では、1つまたはそれより多くのマーカーは、鉄を調節するためである。一部の実施形態では、1つまたはそれより多くのマーカーは、ヘプシジンレベル、腫瘍負荷、トランスフェリン飽和、c−反応性タンパク質(CRP)、心臓マーカー(B型ナトリウム利尿ペプチド(BNP)、N−末端プロB型ナトリウム利尿ペプチド(NT−proBNP))、血清鉄、総鉄、フェリチン、トランスフェリン、可溶性トランスフェリン受容体(STR)、総鉄結合容量(TIBC)、ホスホ−SMAD、またはこれらの組合せを含む。一部の実施形態では、1つまたはそれより多くのマーカーは、鉄パネル(即ち、血清鉄、フェリチン、トランスフェリン、STR、およびTIBC)を使用して評定される。様々な実施形態では、1つまたはそれより多くのマーカーは、ACVR1の下流のホスホシグナル伝達経路中にある。さらなる実施形態では、1つまたはそれより多くのマーカーは、ACVR1/SMADシグナル伝達の下流の遺伝子発現マーカーである。 In embodiments, the subject being treated has one or more markers at a predetermined level. In some embodiments, one or more markers are for regulating iron. In some embodiments, one or more markers are hepcidin levels, tumor loading, transferrin saturation, c-reactive protein (CRP), cardiac markers (type B sodium diuretic peptide (BNP), N-terminal). Includes proB-type sodium diuretic peptide (NT-proBNP)), serum iron, total iron, ferritin, transferrin, soluble transferrin receptor (STR), total iron binding capacity (TIBC), phospho-SMAD, or a combination thereof. In some embodiments, one or more markers are assessed using iron panels (ie, serum iron, ferritin, transferrin, STR, and TIBC). In various embodiments, one or more markers are in the phosphosignal transduction pathway downstream of ACVR1. In a further embodiment, one or more markers are gene expression markers downstream of ACVR1 / SMAD signaling.
様々な実施形態では、処置がなされる対象は、閾値よりも高い所定のマーカー(例えば、ヘプシジン)のレベルを有する。 In various embodiments, the subject to be treated has a level of a predetermined marker (eg, hepcidin) above the threshold.
ベースラインレベルは、母集団から誘導することができる。「母集団」は、同様の指定された特徴の、対象または試料の任意の群化である。群化は、例えば、臨床パラメーター、臨床的評価、治療レジメン、疾患状態、状態の重症度などに従う。 Baseline levels can be derived from the population. A "population" is any grouping of objects or samples with similar specified characteristics. Grouping follows, for example, clinical parameters, clinical assessments, treatment regimens, disease states, severity of conditions, and the like.
一部の実施形態では、母集団はランダムに選択される。一部の実施形態では、母集団は、約2、約5、約10、約25、約50、約75、または約100の対象を含む群である。一部の実施形態では、母集団は、約200、約300、約500、約1,000、約1,500、約2,000、約3,000、約5,000、または約10,000の対象を含む群である。一部の実施形態では、母集団は、約10,000未満の対象を含む群である。他の実施形態では、母集団は、約10,000の対象よりも多くを含む群である。 In some embodiments, the population is randomly selected. In some embodiments, the population is a group comprising about 2, about 5, about 10, about 25, about 50, about 75, or about 100 subjects. In some embodiments, the population is about 200, about 300, about 500, about 1,000, about 1,500, about 2,000, about 3,000, about 5,000, or about 10,000. It is a group including the target of. In some embodiments, the population is a group containing less than about 10,000 subjects. In other embodiments, the population is a group containing more than about 10,000 subjects.
一部の実施形態では、母集団は、がんを有する個体およびがんを有さない個体を含む。一部の実施形態では、母集団の一部は固形がんを有する。一部の実施形態では、母集団の一部は脳がんを有する。一部の実施形態では、母集団の一部は子宮がんを有する。 In some embodiments, the population includes individuals with and without cancer. In some embodiments, a portion of the population has solid cancer. In some embodiments, a portion of the population has brain cancer. In some embodiments, part of the population has uterine cancer.
一部の実施形態では、母集団は、がんを有さない群である。一部の実施形態では、母集団は、固形がんを有さない。一部の実施形態では、母集団は、脳がんを有さない。一部の実施形態では、母集団は、子宮がんを有さない。 In some embodiments, the population is a cancer-free group. In some embodiments, the population does not have solid cancer. In some embodiments, the population does not have brain cancer. In some embodiments, the population does not have uterine cancer.
一部の実施形態では、母集団は、がんを有する群である。実施形態では、母集団は固形がんを有する。一部の実施形態では、母集団は脳がんを有する。一部の実施形態では、母集団は子宮がんを有する。一部の実施形態では、母集団は、対象と同じタイプのがんを有する。 In some embodiments, the population is a group with cancer. In embodiments, the population has solid cancers. In some embodiments, the population has brain cancer. In some embodiments, the population has uterine cancer. In some embodiments, the population has the same type of cancer as the subject.
実施形態では、マーカー(例えば、ヘプシジン)の発現レベルは、ベースラインの発現レベルよりも少なくとも約10%大きい。一部の実施形態では、発現レベルは、少なくとも約1%大きい、少なくとも約2%大きい、少なくとも約3%大きい、少なくとも約4%大きい、少なくとも約5%大きい、少なくとも約7%大きい、少なくとも約12%大きい、少なくとも約15%大きい、少なくとも約17%大きい、少なくとも約20%大きい、少なくとも約22%大きい、少なくとも約25%大きい、少なくとも約27%大きい、少なくとも約30%大きい、少なくとも約32%大きい、少なくとも約35%大きい、少なくとも約37%大きい、少なくとも約40%大きい、少なくとも約45%大きい、少なくとも約50%大きい、少なくとも約75%大きい、または少なくとも約90%大きい。ある特定の実施形態では、マーカー(例えば、ヘプシジン)は上方調節される。「上方調節」または「上方調節された」は、タンパク質の存在の増加および/またはその遺伝子の発現の増加を指す。 In embodiments, the expression level of the marker (eg, hepcidin) is at least about 10% higher than the baseline expression level. In some embodiments, expression levels are at least about 1% greater, at least about 2% greater, at least about 3% greater, at least about 4% greater, at least about 5% greater, at least about 7% greater, at least about 12 % Large, at least about 15% larger, at least about 17% larger, at least about 20% larger, at least about 22% larger, at least about 25% larger, at least about 27% larger, at least about 30% larger, at least about 32% larger , At least about 35% larger, at least about 37% larger, at least about 40% larger, at least about 45% larger, at least about 50% larger, at least about 75% larger, or at least about 90% larger. In certain embodiments, the marker (eg, hepcidin) is upregulated. "Up-regulated" or "up-regulated" refers to increased presence of a protein and / or increased expression of its gene.
一部の実施形態では、マーカー(例えば、ヘプシジン)の発現レベルは、ベースラインの発現レベル未満である。実施形態において、マーカーの発現レベルは、ベースラインの発現レベルよりも少なくとも10%少ない。一部の実施形態では、発現レベルは、少なくとも約1%少ない、少なくとも約2%少ない、少なくとも約3%少ない、少なくとも約4%少ない、少なくとも約5%少ない、少なくとも約10%少ない、少なくとも約12%少ない、少なくとも約15%少ない、少なくとも約17%少ない、少なくとも約20%少ない、少なくとも約22%少ない、少なくとも約25%少ない、少なくとも約27%少ない、少なくとも約30%少ない、少なくとも約32%少ない、少なくとも約35%少ない、少なくとも約37%少ない、少なくとも約40%少ない、少なくとも約45%少ない、少なくとも約50%少ない、少なくとも約75%少ない、または少なくとも約90%少ない。ある特定の実施形態では、マーカー(例えば、ヘプシジン)は、下方調節される。「下方調節」または「下方調節された」は、タンパク質の存在の減少および/またはその遺伝子の発現の減少を指す。 In some embodiments, the expression level of the marker (eg, hepcidin) is below the baseline expression level. In embodiments, marker expression levels are at least 10% less than baseline expression levels. In some embodiments, expression levels are at least about 1% less, at least about 2% less, at least about 3% less, at least about 4% less, at least about 5% less, at least about 10% less, at least about 12. % Less, at least about 15% less, at least about 17% less, at least about 20% less, at least about 22% less, at least about 25% less, at least about 27% less, at least about 30% less, at least about 32% less , At least about 35% less, at least about 37% less, at least about 40% less, at least about 45% less, at least about 50% less, at least about 75% less, or at least about 90% less. In certain embodiments, the marker (eg, hepcidin) is down-regulated. "Down-regulated" or "down-regulated" refers to a decrease in the presence of a protein and / or a decrease in the expression of its gene.
マーカー(例えば、ヘプシジン)の発現の測定は、当技術分野で公知の任意の方法を使用して、タンパク質または核酸レベルで決定することができる。一部の実施形態では、マーカー発現は、試料と試薬(例えば、抗体または核酸プライマー)とを接触させ、試薬とマーカー(複数可)との複合体を発生させ、複合体を検出することによって、検出される。抗体は、受動的結合などの公知の技法により診断アッセイのために固体支持体とコンジュゲートすることができる。抗体は、マルチカラーフローサイトメトリーを含む、フローサイトメトリーなどの公知の技法に従い、診断アッセイのために細胞表面抗原にコンジュゲートすることができる。抗体は、公知の技法により、検出可能な標識または基、例えば放射性標識、酵素標識、および蛍光標識にコンジュゲートすることができる。 Measurement of the expression of a marker (eg, hepcidin) can be determined at the protein or nucleic acid level using any method known in the art. In some embodiments, marker expression involves contacting the sample with a reagent (eg, an antibody or nucleic acid primer) to generate a complex of the reagent with the marker (s) and detecting the complex. Detected. Antibodies can be conjugated to solid supports for diagnostic assays by known techniques such as passive binding. Antibodies can be conjugated to cell surface antigens for diagnostic assays according to known techniques such as flow cytometry, including multicolor flow cytometry. Antibodies can be conjugated to detectable labels or groups such as radioactive labels, enzyme labels, and fluorescent labels by known techniques.
適切なイムノアッセイの例には、免疫ブロット法、免疫沈降法、免疫蛍光法、化学発光法、電気化学発光法(ECL)、およびELISAが含まれる。マーカーの上方または下方調節は、例えばcDNAアレイ、クローンハイブリダイゼーション、ディファレンシャルディスプレイ、ディファレンシャルスクリーニング、FRET検出、液体マイクロアレイ、PCR、RT−PCR、サンガー配列決定、大規模並列(次世代)配列決定、分子ビーコン、マイクロエレクトリックアレイ、オリゴヌクレオチドアレイ、ポリヌクレオチドアレイ、遺伝子発現の連続解析(SAGE)、および/またはサブトラクティブハイブリダイゼーションを使用して検出することもできる。 Examples of suitable immunoassays include immunoblotting, immunoprecipitation, immunofluorescence, chemiluminescence, electrochemical luminescence (ECL), and ELISA. Up or down regulation of markers includes, for example, cDNA arrays, clone hybridization, differential display, differential screening, FRET detection, liquid microarrays, PCR, RT-PCR, Sanger sequencing, large-scale parallel (next generation) sequencing, molecular beacons. , Microelectric arrays, oligonucleotide arrays, polynucleotide arrays, continuous analysis of gene expression (SAGE), and / or subtractive hybridization can also be used for detection.
発現は、処置前に対象から収集された試料において決定され得る。そのような実施形態で、発現レベルは、特定の処置に対する応答性を予測するのに使用されてもよい。一部の実施形態では、発現レベルは、少なくとも部分的には、対象に投与される処置を決定するのに使用されてもよい。一部の実施形態では、試料は、処置レジメンが投与される約28日前に収集される。一部の実施形態では、試料は、処置レジメンが投与される約14日前に収集される。一部の実施形態では、試料は、処置レジメンが投与される約7日前に収集される。一部の実施形態では、試料は、処置レジメンが投与される約72時間前に収集される。一部の実施形態では、試料は、処置レジメンが投与される最長約6時間前に収集される。 Expression can be determined in a sample collected from the subject prior to treatment. In such embodiments, expression levels may be used to predict responsiveness to a particular treatment. In some embodiments, the expression level may be used, at least in part, to determine the treatment administered to the subject. In some embodiments, the sample is collected approximately 28 days before the treatment regimen is administered. In some embodiments, the sample is collected approximately 14 days before the treatment regimen is administered. In some embodiments, the sample is collected approximately 7 days before the treatment regimen is administered. In some embodiments, the sample is collected approximately 72 hours before the treatment regimen is administered. In some embodiments, the sample is collected up to about 6 hours before the treatment regimen is administered.
特定の実施形態では、試料は、処置の第1のサイクルの1日目、投薬前、投薬の0.5時間後、投薬の2時間後、投薬の4時間後、投薬の6時間後、投薬の8時間後、投薬の12時間後、および/または投薬の24時間後に収集される。特定の実施形態では、試料は、処置の第1のサイクルの1日目、投薬前、投薬の0.5時間後、投薬の2時間後、投薬の4時間後、投薬の6時間後、および/または投薬の24時間後に収集される。特定の実施形態では、試料は、処置の第1のサイクルの1日目、投薬前、投薬の2時間後、投薬の6時間後、および/または投薬の24時間後に収集される。一部の実施形態では、試料は、ある用量の処置が対象に投与された後に収集されてもよい。別の特定の実施形態では、試料は、処置の第1のサイクルの8日目、投薬前にも、収集される。一部の実施形態では、試料は、処置の第1のサイクルの1、7、14、21、および28日目のうちの1つまたは複数において、投薬前に収集される。特定の実施形態では、試料は、処置の第1のサイクルの21日目、投薬前、投薬の0.5時間後、投薬の2時間後、投薬の4時間後、投薬の6時間後、投薬の8時間後、投薬の12時間後、および/または投薬の24時間後に収集される。特定の実施形態では、試料は、処置の第1のサイクルの21日目、投薬前、投薬の0.5時間後、投薬の2時間後、投薬の4時間後、投薬の6時間後、および/または投薬の24時間後に収集される。特定の実施形態では、試料は、処置の第1のサイクルの21日目、投薬前、2時間、および6時間で収集される。特定の実施形態では、試料は、処置の第1のサイクルの21日目、投薬前、投薬の2時間後、投薬の6時間後、および/または投薬の48時間後に収集される。特定の実施形態では、試料は、処置サイクルの8、21、および23日目に収集される。
In certain embodiments, the sample is dosed on
別の特定の実施形態では、試料は、処置の第2のサイクルの1日目、投薬前にも収集される。一部の実施形態では、試料は、第2のサイクルの1日目の最長7日前に、収集される。別の特定の実施形態では、試料は、処置の第2のサイクルの1日目、処置後4時間でも収集される。一部の実施形態では、試料は、処置の第2のサイクルの1、7、14、21、および28日目のうちの1つまたは複数において、投薬前に、収集される。一部の実施形態では、試料は、処置の第2のサイクルの1、8、15、22、および29日目のうちの1つまたは複数において、収集される。一部の実施形態では、試料は、処置の第2のサイクルの21日目、投薬前に、収集される。さらなる特定の実施形態では、試料は、処置の任意の追加のサイクル(例えば、第3、第4、第5など)の1日目、投薬前に、収集される。一部の特定の実施形態では、試料は、処置の任意の追加のサイクル(例えば、第3、第4、第5など)にわたり、投薬前に、本明細書に記述されるスケジュールの1つに従い収集される。別の特定の実施形態では、試料は、処置が終了した後にも収集される。そのような実施形態では、試料は、処置の終了から14日以内に収集されてもよい。
In another particular embodiment, the sample is also collected on the first day of the second cycle of treatment, before dosing. In some embodiments, the sample is collected up to 7 days before the first day of the second cycle. In another particular embodiment, the sample is also collected on the first day of the second cycle of treatment, 4 hours after treatment. In some embodiments, the sample is collected prior to dosing on one or more of
理解されるように、収集される試料のタイプは、試験がなされるマーカーに基づいて選択される。任意の適切な試料(例えば、血漿、血清、末梢血単核細胞(PBMC)など)が使用され得る。 As will be appreciated, the type of sample collected will be selected based on the markers being tested. Any suitable sample (eg, plasma, serum, peripheral blood mononuclear cells (PBMC), etc.) can be used.
上方または下方調節は、値を、関連ある参照レベルと比較することによって評定することができる。例えば、1つまたはそれより多くのマーカーの量は、例えば、行われるアッセイによって試料中のマーカー(複数可)のレベル(複数可)を測定することにより誘導することができる、値として示すことができる。実施形態では、システムおよび方法は、アッセイがなされる試料中にマーカーが存在するか否かについて、定量的検出を提供し、即ち、アッセイがなされる試料中のマーカーの実際の量または相対的存在量の評価または評定がなされる。そのような実施形態では、定量的検出は、絶対的であってもよく、または方法が試料中の2つまたはそれよりも多くの種々のマーカーを検出する方法である場合は、相対的であってもよい。したがって、「定量する」という用語は、試料中のマーカーを定量するという文脈で使用される場合、絶対的なまたは相対的な定量を指すことができる。絶対的な定量は、1つまたはそれより多くの対照マーカーの公知の濃度(複数可)を含めること、およびマーカーの検出されたレベルを公知の対照マーカーと参照することによって、例えば標準化することによって(例えば、標準曲線の作成を通して)、実現することができる。あるいは、相対的定量は、2つまたはそれよりも多くのマーカーのそれぞれの相対的定量が提供されるよう、2つまたはそれよりも多くの種々のマーカーの間で検出されたレベルまたは量を、例えば互いに対して比較することによって、実現することができる。 Up or down adjustments can be assessed by comparing the value with the relevant reference level. For example, the amount of one or more markers can be shown as a value that can be derived, for example, by measuring the level (s) of the markers (s) in the sample by the assay performed. can. In embodiments, the system and method provide quantitative detection of the presence of markers in the sample being assayed, i.e. the actual amount or relative presence of markers in the sample being assayed. A quantity is evaluated or rated. In such embodiments, the quantitative detection may be absolute, or relative if the method is a method of detecting two or more different markers in a sample. You may. Thus, the term "quantitate" can refer to absolute or relative quantification when used in the context of quantifying markers in a sample. Absolute quantification includes, for example, by including known concentrations (s) of one or more control markers and by referring to the detected levels of the markers with known control markers, eg by standardization. It can be achieved (eg, through the creation of standard curves). Alternatively, the relative quantification is the level or amount detected between the two or more different markers so that the relative quantification of each of the two or more markers is provided. This can be achieved, for example, by comparing with each other.
様々な実施形態では、1つまたはそれより多くのマーカーのレベルは、ACVR1阻害剤が投与された後にモニタリングされる。 In various embodiments, the level of one or more markers is monitored after administration of the ACVR1 inhibitor.
実施形態では、ACVR1阻害剤と1つまたはそれより多くのマーカーの投与の間の関係は、スピアマンの順位相関関係統計を使用して、定量されてもよい。 In embodiments, the relationship between administration of the ACVR1 inhibitor and one or more markers may be quantified using Spearman's rank correlation statistics.
一部の実施形態では、1つまたはそれより多くのマーカーは、ヘプシジンを含む。一部の実施形態では、対象は、少なくとも、約0.1ng/mL、約0.2ng/mL、約0.3ng/mL、約0.4ng/mL、約0.5ng/mL、約0.6ng/mL、約0.7ng/mL、約0.8ng/mL、約0.9ng/mL、約1ng/mL、約1.5ng/mL、約2ng/mL、約2.5ng/mL、約3ng/mL、約4ng/mL、約5ng/mL、約6ng/mL、約7ng/mL、約8ng/mL、約9ng/mL、約10ng/mL、約12.5ng/mL、約15ng/mL、約20ng/mL、約25ng/mL、約30ng/mL、約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または約200ng/mLの所定のヘプシジンレベルを有する。 In some embodiments, one or more markers include hepcidin. In some embodiments, the subject is at least about 0.1 ng / mL, about 0.2 ng / mL, about 0.3 ng / mL, about 0.4 ng / mL, about 0.5 ng / mL, about 0. 6 ng / mL, about 0.7 ng / mL, about 0.8 ng / mL, about 0.9 ng / mL, about 1 ng / mL, about 1.5 ng / mL, about 2 ng / mL, about 2.5 ng / mL, about 3 ng / mL, about 4 ng / mL, about 5 ng / mL, about 6 ng / mL, about 7 ng / mL, about 8 ng / mL, about 9 ng / mL, about 10 ng / mL, about 12.5 ng / mL, about 15 ng / mL , About 20 ng / mL, about 25 ng / mL, about 30 ng / mL, about 35 ng / mL, about 40 ng / mL, about 45 ng / mL, about 50 ng / mL, about 55 ng / mL, about 60 ng / mL, about 65 ng / mL , About 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL, about 90 ng / mL, about 95 ng / mL, about 100 ng / mL, about 105 ng / mL about 110 ng / mL, about 115 ng / mL, About 120 ng / mL, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, about 140 ng / mL, about 145 ng / mL, about 150 ng / mL, about 155 ng / mL, about 160 ng / mL, about 165 ng / mL, It has a predetermined hepcidin level of about 170 ng / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, about 190 ng / mL, about 195 ng / mL, or about 200 ng / mL.
種々の実施形態では、対象は、約0.1ng/mL〜約5ng/mL、約10ng/mL、約15ng/mL、約20ng/mL、約25ng/mL、約30ng/mL、約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または200ng/mLの所定のヘプシジンレベルを有する。 In various embodiments, the subject is about 0.1 ng / mL to about 5 ng / mL, about 10 ng / mL, about 15 ng / mL, about 20 ng / mL, about 25 ng / mL, about 30 ng / mL, about 35 ng / mL. , About 40 ng / mL, about 45 ng / mL, about 50 ng / mL, about 55 ng / mL, about 60 ng / mL, about 65 ng / mL, about 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL , About 90 ng / mL, about 95 ng / mL, about 100 ng / mL, about 105 ng / mL, about 110 ng / mL, about 115 ng / mL, about 120 ng / mL, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, About 140 ng / mL, about 145 ng / mL, about 150 ng / mL, about 155 ng / mL, about 160 ng / mL, about 165 ng / mL, about 170 ng / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, It has a predetermined hepcidin level of about 190 ng / mL, about 195 ng / mL, or 200 ng / mL.
種々の実施形態では、対象は、約0.5ng/mL〜約5ng/mL、約10ng/mL、約15ng/mL、約20ng/mL、約25ng/mL、約30ng/mL、約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または約200ng/mLの所定のヘプシジンレベルを有する。 In various embodiments, the subject is about 0.5 ng / mL to about 5 ng / mL, about 10 ng / mL, about 15 ng / mL, about 20 ng / mL, about 25 ng / mL, about 30 ng / mL, about 35 ng / mL. , About 40 ng / mL, about 45 ng / mL, about 50 ng / mL, about 55 ng / mL, about 60 ng / mL, about 65 ng / mL, about 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL , About 90 ng / mL, about 95 ng / mL, about 100 ng / mL, about 105 ng / mL, about 110 ng / mL, about 115 ng / mL, about 120 ng / mL, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, About 140 ng / mL, about 145 ng / mL, about 150 ng / mL, about 155 ng / mL, about 160 ng / mL, about 165 ng / mL, about 170 ng / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, It has a predetermined hepcidin level of about 190 ng / mL, about 195 ng / mL, or about 200 ng / mL.
種々の実施形態では、対象は、約1ng/mL〜約5ng/mL、約10ng/mL、約15ng/mL、約20ng/mL、約25ng/mL、約30ng/mL、約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または約200ng/mLの所定のヘプシジンレベルを有する。 In various embodiments, the subject is about 1 ng / mL to about 5 ng / mL, about 10 ng / mL, about 15 ng / mL, about 20 ng / mL, about 25 ng / mL, about 30 ng / mL, about 35 ng / mL, about. 40 ng / mL, about 45 ng / mL, about 50 ng / mL, about 55 ng / mL, about 60 ng / mL, about 65 ng / mL, about 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL, about 90 ng / mL, about 95 ng / mL, about 100 ng / mL, about 105 ng / mL, about 110 ng / mL, about 115 ng / mL, about 120 ng / mL, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, about 140 ng / ML, about 145 ng / mL, about 150 ng / mL, about 155 ng / mL, about 160 ng / mL, about 165 ng / mL, about 170 ng / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, about 190 ng It has a predetermined hepcidin level of / mL, about 195 ng / mL, or about 200 ng / mL.
種々の実施形態では、対象は、約5ng/mL〜約10ng/mL、約15ng/mL、約20ng/mL、約25ng/mL、約30ng/mL、約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または約200ng/mLの所定のヘプシジンレベルを有する。 In various embodiments, the subject is about 5 ng / mL to about 10 ng / mL, about 15 ng / mL, about 20 ng / mL, about 25 ng / mL, about 30 ng / mL, about 35 ng / mL, about 40 ng / mL, about. 45 ng / mL, about 50 ng / mL, about 55 ng / mL, about 60 ng / mL, about 65 ng / mL, about 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL, about 90 ng / mL, about 95 ng / mL, about 100 ng / mL, about 105 ng / mL about 110 ng / mL, about 115 ng / mL, about 120 ng / mL, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, about 140 ng / mL, about 145 ng / ML, about 150 ng / mL, about 155 ng / mL, about 160 ng / mL, about 165 ng / mL, about 170 ng / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, about 190 ng / mL, about 195 ng It has a predetermined hepcidin level of / mL, or about 200 ng / mL.
種々の実施形態では、対象は、約10ng/mL〜約20ng/mL、約25ng/mL、約30ng/mL、約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または約200ng/mLの所定のヘプシジンレベルを有する。 In various embodiments, the subject is about 10 ng / mL to about 20 ng / mL, about 25 ng / mL, about 30 ng / mL, about 35 ng / mL, about 40 ng / mL, about 45 ng / mL, about 50 ng / mL, about. 55 ng / mL, about 60 ng / mL, about 65 ng / mL, about 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL, about 90 ng / mL, about 95 ng / mL, about 100 ng / mL, about 105 ng / mL about 110 ng / mL, about 115 ng / mL, about 120 ng / mL, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, about 140 ng / mL, about 145 ng / mL, about 150 ng / mL, about 155 ng Prescription of / mL, about 160 ng / mL, about 165 ng / mL, about 170 ng / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, about 190 ng / mL, about 195 ng / mL, or about 200 ng / mL Has hepcidin levels of.
種々の実施形態では、対象は、約12.5ng/mL〜約20ng/mL、約25ng/mL、約30ng/mL、約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または約200ng/mLの所定のヘプシジンレベルを有する。 In various embodiments, the subject is about 12.5 ng / mL to about 20 ng / mL, about 25 ng / mL, about 30 ng / mL, about 35 ng / mL, about 40 ng / mL, about 45 ng / mL, about 50 ng / mL. , About 55 ng / mL, about 60 ng / mL, about 65 ng / mL, about 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL, about 90 ng / mL, about 95 ng / mL, about 100 ng / mL , About 105 ng / mL about 110 ng / mL, about 115 ng / mL, about 120 ng / mL, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, about 140 ng / mL, about 145 ng / mL, about 150 ng / mL, About 155 ng / mL, about 160 ng / mL, about 165 ng / mL, about 170 ng / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, about 190 ng / mL, about 195 ng / mL, or about 200 ng / mL Has a given hepcidin level of.
種々の実施形態では、対象は、約25ng/mL〜約35ng/mL、約40ng/mL、約45ng/mL、約50ng/mL、約55ng/mL、約60ng/mL、約65ng/mL、約70ng/mL、約75ng/mL、約80ng/mL、約85ng/mL、約90ng/mL、約95ng/mL、約100ng/mL、約105ng/mL 約110ng/mL、約115ng/mL、約120ng/mL、約125ng/mL、約130ng/mL、約135ng/mL、約140ng/mL、約145ng/mL、約150ng/mL、約155ng/mL、約160ng/mL、約165ng/mL、約170ng/mL、約175ng/mL、約180ng/mL、約185ng/mL、約190ng/mL、約195ng/mL、または約200ng/mLの所定のヘプシジンレベルを有する。 In various embodiments, the subject is about 25 ng / mL to about 35 ng / mL, about 40 ng / mL, about 45 ng / mL, about 50 ng / mL, about 55 ng / mL, about 60 ng / mL, about 65 ng / mL, about. 70 ng / mL, about 75 ng / mL, about 80 ng / mL, about 85 ng / mL, about 90 ng / mL, about 95 ng / mL, about 100 ng / mL, about 105 ng / mL, about 110 ng / mL, about 115 ng / mL, about 120 ng / ML, about 125 ng / mL, about 130 ng / mL, about 135 ng / mL, about 140 ng / mL, about 145 ng / mL, about 150 ng / mL, about 155 ng / mL, about 160 ng / mL, about 165 ng / mL, about 170 ng It has a given hepsidin level of / mL, about 175 ng / mL, about 180 ng / mL, about 185 ng / mL, about 190 ng / mL, about 195 ng / mL, or about 200 ng / mL.
ヘプシジンレベルは、モル濃度で測定されてもよい。一部の実施形態では、所定のヘプシジンレベルは、少なくとも4nMである。一部のそのような実施形態では、対象はメスである。一部の実施形態では、所定のヘプシジンレベルは少なくとも4.1nMである。一部のそのような実施形態では、対象はメスである。一部の実施形態では、所定のヘプシジンレベルは少なくとも8.5nMである。一部のそのような実施形態では、対象はメスである。一部の実施形態では、所定のヘプシジンレベルは少なくとも7.5nMである。一部のそのような実施形態では、対象はオスである。一部の実施形態では、所定のヘプシジンレベルは少なくとも7.8nMである。一部のそのような実施形態では、対象はオスである。 Hepcidin levels may be measured in molar concentrations. In some embodiments, the predetermined hepcidin level is at least 4 nM. In some such embodiments, the subject is a female. In some embodiments, the predetermined hepcidin level is at least 4.1 nM. In some such embodiments, the subject is a female. In some embodiments, the predetermined hepcidin level is at least 8.5 nM. In some such embodiments, the subject is a female. In some embodiments, the predetermined hepcidin level is at least 7.5 nM. In some such embodiments, the subject is a male. In some embodiments, the predetermined hepcidin level is at least 7.8 nM. In some such embodiments, the subject is a male.
様々な実施形態では、ヘプシジンレベルは、ACVR1阻害剤が投与された後にモニタリングされる。 In various embodiments, hepcidin levels are monitored after administration of the ACVR1 inhibitor.
様々な実施形態では、対象は、閾値よりも低い所定のトランスフェリン飽和を有する。「トランスフェリン飽和」は、血清鉄の値を、総鉄結合容量で除した値を指し、当技術分野で公知の任意の技法を使用して決定され得る。 In various embodiments, the subject has a given transferrin saturation below the threshold. "Transferrin saturation" refers to the value of serum iron divided by the total iron binding volume and can be determined using any technique known in the art.
実施形態では、対象は、約50%未満のトランスフェリン飽和を有する。一部のそのような実施形態では、対象はオスである。実施形態では、対象は、約45%未満のトランスフェリン飽和を有する。一部のそのような実施形態では、対象はメスである。一部の実施形態では、対象は、約40%未満のトランスフェリン飽和を有する。一部の実施形態では、対象は、約35%未満のトランスフェリン飽和を有する。一部の実施形態では、対象は、約30%未満のトランスフェリン飽和を有する。一部の実施形態では、対象は、約25%未満のトランスフェリン飽和を有する。一部の実施形態では、対象は、約20%未満のトランスフェリン飽和を有する。一部の実施形態では、対象は、約15%未満のトランスフェリン飽和を有する。一部のそのような実施形態では、対象はメスである。 In embodiments, the subject has less than about 50% transferrin saturation. In some such embodiments, the subject is a male. In embodiments, the subject has less than about 45% transferrin saturation. In some such embodiments, the subject is a female. In some embodiments, the subject has less than about 40% transferrin saturation. In some embodiments, the subject has transferrin saturation of less than about 35%. In some embodiments, the subject has less than about 30% transferrin saturation. In some embodiments, the subject has transferrin saturation of less than about 25%. In some embodiments, the subject has less than about 20% transferrin saturation. In some embodiments, the subject has transferrin saturation of less than about 15%. In some such embodiments, the subject is a female.
一部の実施形態では、対象は、約12%から約50%に及ぶトランスフェリン飽和を有する。一部の実施形態では、対象は、約15%から約50%に及ぶトランスフェリン飽和を有する。一部のそのような実施形態では、対象はオスである。一部の実施形態では、対象は、約12%から約45%に及ぶトランスフェリン飽和を有する。一部のそのような実施形態では、対象はメスである。一部の実施形態では、対象は、約20%から約40%に及ぶトランスフェリン飽和を有する。一部の実施形態では、対象は、約30%から約45%に及ぶトランスフェリン飽和を有する。一部の実施形態では、対象は、約25%から約35%に及ぶトランスフェリン飽和を有する。一部の実施形態では、対象は、約15%から約25%に及ぶトランスフェリン飽和を有する。一部の実施形態では、対象は、約20%から約30%に及ぶトランスフェリン飽和を有する。 In some embodiments, the subject has transferrin saturation ranging from about 12% to about 50%. In some embodiments, the subject has transferrin saturation ranging from about 15% to about 50%. In some such embodiments, the subject is a male. In some embodiments, the subject has transferrin saturation ranging from about 12% to about 45%. In some such embodiments, the subject is a female. In some embodiments, the subject has transferrin saturation ranging from about 20% to about 40%. In some embodiments, the subject has transferrin saturation ranging from about 30% to about 45%. In some embodiments, the subject has transferrin saturation ranging from about 25% to about 35%. In some embodiments, the subject has transferrin saturation ranging from about 15% to about 25%. In some embodiments, the subject has transferrin saturation ranging from about 20% to about 30%.
様々な実施形態では、トランスフェリン飽和レベルは、ACVR1阻害剤が投与された後にモニタリングされる。 In various embodiments, transferrin saturation levels are monitored after administration of the ACVR1 inhibitor.
様々な実施形態では、対象は、許容される肝機能を有する。そのような実施形態では、対象のビリルビンは≦1.5×正常値上限(ULN)であり(ギルバート症候群を伴わない限り)、対象のアスパラギン酸アミノトランスフェラーゼ(AST/SGOT)、アラニンアミノトランスフェラーゼ(ALT/SGPT)、およびアルカリンホスファターゼは、≦2.5×ULNである(肝転移が存在する場合、≦5×ULNが許される)。 In various embodiments, the subject has acceptable liver function. In such embodiments, the subject's bilirubin is ≤1.5 x upper normal value (ULN) (unless accompanied by Gilbert's syndrome), and the subject's aspartate aminotransferase (AST / SGOT), alanine aminotransferase (ALT). / SGPT), and alanine phosphatase are ≤2.5 x ULN (≤5 x ULN is allowed if liver metastasis is present).
一部の実施形態では、対象は、許容される腎機能を有する。そのような実施形態では、対象の計算されたクレアチニンクリアランスは≧30mL/分である。 In some embodiments, the subject has acceptable renal function. In such an embodiment, the subject's calculated creatinine clearance is ≧ 30 mL / min.
一部の実施形態では、対象は、許容される血液学的状態を有する。そのような実施形態では、対象の顆粒細胞数は≧1500細胞/mm3であり、血小板数は≧100,000(plt/mm3)であり、ヘモグロビンは≧8g/dLである。一部の実施形態では、対象は、ACVR1阻害剤の最初の投与から14日以内に輸血を受けていない。 In some embodiments, the subject has an acceptable hematological condition. In such an embodiment, the number of granule cells of interest is ≧ 1500 cells / mm 3 , the number of platelets is ≧ 100,000 (plt / mm 3 ), and the hemoglobin is ≧ 8 g / dL. In some embodiments, the subject has not received a blood transfusion within 14 days of the initial administration of the ACVR1 inhibitor.
一部の実施形態では、対象は、許容される凝固状態を有する。そのような実施形態では、対象のプロトロンビン時間(PT)は、1.5×正常範囲内であり、活性化部分トロンボプラスチン時間(aPTT)は、1.5×正常範囲内である。 In some embodiments, the subject has an acceptable coagulation state. In such embodiments, the subject's prothrombin time (PT) is in the 1.5 x normal range and the activated partial thromboplastin time (aPTT) is in the 1.5 x normal range.
ある特定の実施形態では、対象は、22歳もしくはそれよりも若い、21歳もしくはそれよりも若い、20歳もしくはそれよりも若い、19歳もしくはそれよりも若い、18歳もしくはそれよりも若い、17歳もしくはそれよりも若い、16歳もしくはそれよりも若い、15歳もしくはそれよりも若い、14歳もしくはそれよりも若い、13歳もしくはそれよりも若い、12歳もしくはそれよりも若い、11歳もしくはそれよりも若い、10歳もしくはそれよりも若い、9歳もしくはそれよりも若い、8歳もしくはそれよりも若い、7歳もしくはそれよりも若い、6歳もしくはそれよりも若い、5歳もしくはそれよりも若い、4歳もしくはそれよりも若い、3歳もしくはそれよりも若い、2歳もしくはそれよりも若い、または1歳もしくはそれよりも若い患者である。一部の実施形態では、患者は、22歳もしくはそれよりも若いまたは18歳もしくはそれよりも若いと定義され得る小児患者である。言い換えれば、一部の実施形態では、患者は、22歳までの小児である。他の実施形態では、患者は18歳までの小児である。一部の実施形態では、患者は、約3から約10歳である。一部の実施形態では、患者は約3から約5歳である。一部の実施形態では、患者は約5から約10歳である。一部の実施形態では、患者は約6から約10歳である。一部の実施形態では、患者は約6から約8歳である。 In certain embodiments, the subject is 22 years or younger, 21 years or younger, 20 years or younger, 19 years or younger, 18 years or younger, 17 years old or younger, 16 years old or younger, 15 years old or younger, 14 years old or younger, 13 years old or younger, 12 years old or younger, 11 years old Or younger, 10 or younger, 9 or younger, 8 or younger, 7 or younger, 6 or younger, 5 or younger Patients younger, 4 years or younger, 3 years or younger, 2 years or younger, or 1 year or younger. In some embodiments, the patient is a pediatric patient who can be defined as 22 years or younger or 18 years or younger. In other words, in some embodiments, the patient is a child up to 22 years of age. In other embodiments, the patient is a child up to 18 years of age. In some embodiments, the patient is about 3 to about 10 years old. In some embodiments, the patient is about 3 to about 5 years old. In some embodiments, the patient is about 5 to about 10 years old. In some embodiments, the patient is about 6 to about 10 years old. In some embodiments, the patient is about 6 to about 8 years old.
様々な実施形態では、ACVR1阻害剤は、式(I)、またはその薬学的に許容される塩もしくはプロドラッグのACVR1阻害剤、および薬学的に許容される担体、希釈剤、または賦形剤を含む、医薬組成物として製剤化される。 In various embodiments, the ACVR1 inhibitor is an ACVR1 inhibitor of formula (I), or a pharmaceutically acceptable salt or prodrug thereof, and a pharmaceutically acceptable carrier, diluent, or excipient. Including, it is formulated as a pharmaceutical composition.
したがって本開示は、本明細書に記述される方法で使用される、ACVR1阻害剤を含む組成物を提供する。実施形態では、ACVR1阻害剤は、経口投与のために製剤化される。様々な実施形態では、ACVR1阻害剤は、錠剤、カプセル(例えば、ゼラチンカプセル)、または溶液として製剤化される。特定の実施形態では、ACVR1阻害剤は、賦形剤として製剤化される。ある特定の実施形態では、ACVR1阻害剤は、ゼラチンカプセルとして製剤化される。一部の実施形態では、ゼラチンカプセルは、5mg、25mg、または125mgの力価で製剤化される。一部の実施形態では、カプセルは、30mg、90mg、または120mgの力価で製剤化される。ある特定の実施形態では、ACVR1阻害剤は、経口溶液として製剤化される。一部の実施形態では、溶液は、5mg/mLの増分、10mg/mLの増分、20mg/mLの増分、または30mg/mLの増分で製剤化される。一部の実施形態では、溶液は、成人用量の約80%から約100%の間の小児用量が提供されるように、製剤化される。一部の実施形態では、溶液は、約24から約30mg、約48から約60mg、約72から約90mg、約96から約120mgの力価で製剤化される。一部の実施形態では、溶液は、30mg、60mg、90mg、または120mgの力価で製剤化される。一部の実施形態では、溶液は、24mg、48mg、72mg、または96mgの力価で製剤化される。 Accordingly, the present disclosure provides compositions containing ACVR1 inhibitors used in the methods described herein. In embodiments, the ACVR1 inhibitor is formulated for oral administration. In various embodiments, the ACVR1 inhibitor is formulated as a tablet, capsule (eg, gelatin capsule), or solution. In certain embodiments, the ACVR1 inhibitor is formulated as an excipient. In certain embodiments, the ACVR1 inhibitor is formulated as a gelatin capsule. In some embodiments, gelatin capsules are formulated with a titer of 5 mg, 25 mg, or 125 mg. In some embodiments, the capsule is formulated with a titer of 30 mg, 90 mg, or 120 mg. In certain embodiments, the ACVR1 inhibitor is formulated as an oral solution. In some embodiments, the solution is formulated in 5 mg / mL increments, 10 mg / mL increments, 20 mg / mL increments, or 30 mg / mL increments. In some embodiments, the solution is formulated to provide a pediatric dose between about 80% and about 100% of the adult dose. In some embodiments, the solution is formulated with a titer of about 24 to about 30 mg, about 48 to about 60 mg, about 72 to about 90 mg, and about 96 to about 120 mg. In some embodiments, the solution is formulated with a titer of 30 mg, 60 mg, 90 mg, or 120 mg. In some embodiments, the solution is formulated with a titer of 24 mg, 48 mg, 72 mg, or 96 mg.
本開示の医薬組成物は、ACVR1阻害剤と適切な薬学的に許容される担体、希釈剤、または賦形剤とを組み合わせることによって調製することができ、固体、半固体、液体、または気体状形態の調製物、例えば錠剤、カプセル、粉末、顆粒、軟膏、溶液、坐薬、注射液、吸入剤、ゲル、微小球、およびエアロゾルに製剤化されてもよい。そのような医薬組成物を投与する典型的な経路には、経口、局所、経皮、吸入、非経口、舌下、頬側、直腸、膣、および鼻内が含まれる。本明細書で使用される場合、非経口という用語は、皮下注射、静脈内、筋肉内、胸骨内注射または注入技法を含む。本開示の医薬組成物は、そこに含有される活性成分が、患者への組成物の投与後に生物学的に利用可能になるように、製剤化される。 The pharmaceutical compositions of the present disclosure can be prepared by combining an ACVR1 inhibitor with a suitable pharmaceutically acceptable carrier, diluent, or excipient and are solid, semi-solid, liquid, or gaseous. It may be formulated into a preparation of the form, for example tablets, capsules, powders, granules, ointments, solutions, suppositories, excipients, inhalants, gels, microspheres, and aerosols. Typical routes for administering such pharmaceutical compositions include oral, topical, transdermal, inhaled, parenteral, sublingual, buccal, rectal, vaginal, and intranasal. As used herein, the term parenteral includes subcutaneous injection, intravenous, intramuscular, intrasternal injection or infusion techniques. The pharmaceutical composition of the present disclosure is formulated so that the active ingredient contained therein is bioavailable after administration of the composition to a patient.
本開示の医薬組成物は、固体または液体の形態をとり得る。一態様では、担体(複数可)は粒状であり、したがって組成物は、例えば錠剤または粉末の形態である。担体(複数可)は、液体であってもよく、組成物は、例えば経口シロップ、注射液、または例えば吸入投与に有用なエアロゾルである。 The pharmaceutical compositions of the present disclosure may take the form of solids or liquids. In one aspect, the carrier (s) are granular and thus the composition is in the form of, for example, tablets or powders. The carrier (s) may be liquid and the composition may be, for example, an oral syrup, an injectable solution, or, for example, an aerosol useful for inhalation administration.
経口投与が意図される場合、医薬組成物は、好ましくは固体または液体のいずれかの形態をとり、半固体、半液体、懸濁液、およびゲル形態は、固体または液体のいずれかとして本明細書で見なされる形態の中に含まれる。 Where oral administration is intended, the pharmaceutical composition preferably takes either solid or liquid form, and the semi-solid, semi-liquid, suspension, and gel forms are herein as either solid or liquid. Included in the form considered in the book.
経口投与のための固体組成物として、医薬組成物は、粉末、顆粒、圧縮錠剤、丸薬、カプセル、チューイングガム、ウエハ、または同様の形態に製剤化されてもよい。そのような固体組成物は、典型的には、1種またはそれより多種の不活性希釈剤または食用単体を含有する。さらに、下記:カルボキシメチルセルロース、エチルセルロース、微結晶性セルロース、トラガカントガム、またはゼラチンなどの結合剤;デンブン、ラクトース、またはデキストリンなどの賦形剤、アルギン酸、アルギン酸ナトリウム、プリモゲル、コーンスターチなどの崩壊剤;ステアリン酸マグネシウムまたはステロテックスなどの滑沢剤;コロイド状二酸化ケイ素などの滑剤;スクロースまたはサッカリンなどの甘味剤;ペパーミント、サリチル酸メチル、またはオレンジフレーバなどの着香剤;および着色剤のうちの1つまたは複数が存在していてもよい。 As a solid composition for oral administration, the pharmaceutical composition may be formulated in powder, granules, compressed tablets, pills, capsules, chewing gum, wafers, or similar forms. Such solid compositions typically contain one or more Inactive Diluents or Elemental Edibles. In addition: Binders such as carboxymethyl cellulose, ethyl cellulose, microcrystalline cellulose, tragacant gum, or gelatin; excipients such as denbun, lactose, or dextrin, disintegrants such as alginic acid, sodium alginate, primogel, corn starch; stearic acid Lubricants such as magnesium or sterotex; lubricants such as colloidal silicon dioxide; sweeteners such as sucrose or saccharin; flavoring agents such as peppermint, methyl salicylate, or orange flavor; and one or more of colorants May be present.
医薬組成物がカプセル、例えばゼラチンカプセルの形態をとる場合、上記タイプの材料に加えて、ポリエチレングリコールまたは油などの液体担体を含有してもよい。 When the pharmaceutical composition is in the form of capsules, such as gelatin capsules, it may contain a liquid carrier such as polyethylene glycol or oil in addition to the above types of materials.
本発明の方法で使用される医薬組成物は、液体、例えばエリキシル、シロップ、溶液、エマルジョン、または懸濁液の形態をとってもよい。液体は、例えば経口投与用であっても注射による送達用であってもよい。経口投与が意図される場合、医薬組成物は、例えば、治療化合物(複数可)に加えて甘味剤、保存剤、色素/着色剤および調味料のうちの1つまたは複数を含有する。注射による投与が意図される組成物では、界面活性剤、保存剤、湿潤剤、分散剤、懸濁化剤、緩衝剤、安定化剤、および等張剤のうちの1つまたは複数が含められてもよい。 The pharmaceutical compositions used in the methods of the invention may be in the form of liquids such as elixirs, syrups, solutions, emulsions or suspensions. The liquid may be, for example, for oral administration or for delivery by injection. When intended for oral administration, the pharmaceutical composition comprises, for example, one or more of a sweetener, a preservative, a pigment / colorant and a seasoning in addition to the therapeutic compound (s). Compositions intended for administration by injection include one or more of surfactants, preservatives, wetting agents, dispersants, suspending agents, buffers, stabilizers, and isotonic agents. You may.
本開示の、ある特定の実施形態で使用される液体医薬組成物は、それらが溶液であろうと懸濁液であろうとまたはその他同様の形であろうと、下記のアジュバント:注射用の水、生理食塩溶液、好ましくは生理学的生理食塩液、リンガー液、等張性塩化ナトリウムなどの滅菌希釈剤、溶媒または懸濁媒体として働くことができる合成モノグリセリドまたは合成ジグリセリド、ポリエチレングリコール、グリセリン、プロピレングリコール、またはその他の溶媒などの固定油;ベンジルアルコールまたはメチルパラベンなどの抗菌剤;アスコルビン酸または亜硫酸水素ナトリウムなどの抗酸化剤;エチレンジアミン四酢酸などのキレート剤;アセテート、シトレート、またはホスフェートなどの緩衝剤、および塩化ナトリウムまたはデキストロースなどの張度を調節するための薬剤のうちの1つまたは複数を含んでいてもよい。非経口調製物は、ガラスまたはプラスチックで作製されたアンプル、使い捨て可能な注射器、または複数回用量バイアルに包封することができる。生理学的生理食塩液は、好ましいアジュバントである。注射可能な医薬組成物は、好ましくは滅菌されている。実施形態では、医薬組成物は、注射用に製剤化される。一部の実施形態では、医薬組成物は、大量注射用に製剤化される。実施形態では、医薬組成物は、輸液用に製剤化される。 The liquid pharmaceutical compositions used in certain embodiments of the present disclosure, whether they are solutions, suspensions or other similar forms, are the following adjuvants: water for injection, physiology. Sterilized diluents such as saline solutions, preferably physiological physiological saline, Ringer's solution, isotonic sodium chloride, synthetic monoglycerides or synthetic diglycerides that can act as solvents or suspension media, polyethylene glycol, glycerin, propylene glycol, or Fixed oils such as other solvents; antibacterial agents such as benzyl alcohol or methylparaben; antioxidants such as ascorbic acid or sodium hydrogen sulfite; chelating agents such as ethylenediaminetetraacetic acid; buffers such as acetate, citrate, or phosphate, and chloride It may contain one or more agents for adjusting tonicity, such as sodium or dextrose. Parenteral preparations can be encapsulated in ampoules made of glass or plastic, disposable syringes, or multi-dose vials. Physiological saline is the preferred adjuvant. The injectable pharmaceutical composition is preferably sterilized. In embodiments, the pharmaceutical composition is formulated for injection. In some embodiments, the pharmaceutical composition is formulated for mass injection. In embodiments, the pharmaceutical composition is formulated for infusion.
非経口または経口投与のいずれかが意図される、本開示の実施形態で使用される液体医薬組成物は、適切な投薬量が得られるような量のACVR1阻害剤を、含有すべきである。 The liquid pharmaceutical composition used in the embodiments of the present disclosure, which is intended for either parenteral or oral administration, should contain an amount of ACVR1 inhibitor that provides an appropriate dosage.
ある特定の実施形態では、本明細書に記述されるACVR1阻害剤は、全身にではなく局所的に投与され、例えば臓器への直接的なACVR1阻害剤の注射によって、しばしばデポー調製物または持続放出製剤として投与される。特定の実施形態では、長時間作用型製剤が、埋込み(例えば、皮下または筋肉内)によって、または筋肉内注射によって投与される。さらに、他の実施形態では、薬物は、標的薬物送達システムで、例えば臓器特異的抗体でコーティングされたリポソームとして送達される。そのような実施形態では、リポソームは、臓器を標的とし、かつ臓器によって選択的に取り込まれる。さらに他の実施形態では、本明細書に記述される化合物は、高速放出製剤の形態で、延長放出製剤の形態で、または中間放出製剤の形態で提供される。 In certain embodiments, the ACVR1 inhibitors described herein are administered topically rather than systemically and are often depot-prepared or sustained release, eg, by direct injection of the ACVR1 inhibitor into an organ. It is administered as a preparation. In certain embodiments, the long-acting formulation is administered by implantation (eg, subcutaneous or intramuscular) or by intramuscular injection. In addition, in other embodiments, the drug is delivered in a targeted drug delivery system, eg, as liposomes coated with an organ-specific antibody. In such embodiments, the liposomes target the organ and are selectively taken up by the organ. In yet another embodiment, the compounds described herein are provided in the form of a fast release formulation, in the form of an extended release formulation, or in the form of an intermediate release formulation.
本開示のある特定の実施形態で使用される医薬組成物は、局所投与が意図されてもよく、その場合、担体は適切に、溶液、エマルジョン、軟膏、またはゲルベースを含み得る。ベースは、例えば、下記:石油、ラノリン、ポリエチレングリコール、蜜蝋、鉱物油、水およびアルコールなどの希釈剤、ならびに乳化剤、および安定化剤のうちの1つまたは複数を含んでいてもよい。増粘剤は、局所投与用の医薬組成物中に存在していてもよい。経皮投与が意図される場合、組成物は、経皮パッチまたはイオン泳動デバイスを含んでいてもよい。 The pharmaceutical compositions used in certain embodiments of the present disclosure may be intended for topical administration, in which case the carrier may appropriately comprise a solution, emulsion, ointment, or gel base. The base may contain, for example, one or more of the following: diluents such as petroleum, lanolin, polyethylene glycol, beeswax, mineral oils, water and alcohols, as well as emulsifiers and stabilizers. The thickener may be present in the pharmaceutical composition for topical administration. If intended for transdermal administration, the composition may include a transdermal patch or iontophoresis device.
本開示のある特定の実施形態で使用される医薬組成物(例えば、固体または液体の形態をとる)は、治療化合物(複数可)に結合し、それによって送達を支援する薬剤を含んでいてもよい。この容量で作用し得る適切な薬剤には、モノクローナル抗体またはポリクローナル抗体、タンパク質、またはリポソームが含まれる。 The pharmaceutical compositions used in certain embodiments of the present disclosure (eg, in the form of solids or liquids) may include agents that bind to the therapeutic compound (s), thereby assisting delivery. good. Suitable agents that can act in this volume include monoclonal or polyclonal antibodies, proteins, or liposomes.
疎水性医薬化合物用の送達システムが用いられてもよい。リポソームおよびエマルジョンは、本明細書で有用な送達ビヒクルまたは担体の例である。ある特定の実施形態では、N−メチルピロリドンなどの有機溶媒も用いられる。追加の実施形態では、本明細書に記述される化合物は、治療剤を含有する固体疎水性ポリマーの半透過性母材などの持続放出システムを使用して送達される。様々な持続放出材料が、本明細書では有用である。一部の実施形態では、持続放出カプセルは、化合物を、数週間から100日超まで放出する。治療用試薬の化学的性質および生物学的安定性に応じて、タンパク質安定化のための追加の戦略が用いられる。 Delivery systems for hydrophobic pharmaceutical compounds may be used. Liposomes and emulsions are examples of delivery vehicles or carriers useful herein. In certain embodiments, organic solvents such as N-methylpyrrolidone are also used. In additional embodiments, the compounds described herein are delivered using a sustained release system, such as a semi-permeable matrix of solid hydrophobic polymers containing therapeutic agents. Various sustained release materials are useful herein. In some embodiments, the sustained release capsule releases the compound from a few weeks to over 100 days. Additional strategies for protein stabilization are used, depending on the chemistry and biological stability of the therapeutic reagents.
実施形態では、ACVR1阻害剤および追加の治療剤は、一緒にリポソーム製剤に製剤化される。 In an embodiment, the ACVR1 inhibitor and additional therapeutic agent are formulated together in a liposome formulation.
本開示のある特定の実施形態で使用される医薬組成物は、医薬品の分野で周知の方法によって調製されてもよい。例えば、注射により投与されることが意図される医薬組成物は、溶液が形成されるように、ACVR1阻害剤と滅菌蒸留水とを合わせることにより、調製することができる。一部の実施形態では、本開示の方法により投与するための医薬組成物(複数可)は、治療剤が溶液中、懸濁液中、またはその両方で存在する液体の形態をとる。一部の実施形態では、治療剤が溶液または懸濁液として投与されるとき、薬剤の第1の部分は溶液中に存在し、薬剤の第2の部分は粒状形態で、液体母材中の懸濁液として存在する。一部の実施形態では、液体組成物は、ゲル製剤を含む。他の実施形態では、液体組成物は水性である。 The pharmaceutical compositions used in certain embodiments of the present disclosure may be prepared by methods well known in the pharmaceutical art. For example, a pharmaceutical composition intended to be administered by injection can be prepared by combining an ACVR1 inhibitor with sterile distilled water so that a solution is formed. In some embodiments, the pharmaceutical composition (s) for administration by the methods of the present disclosure take the form of a liquid in which the therapeutic agent is present in solution, suspension, or both. In some embodiments, when the therapeutic agent is administered as a solution or suspension, the first portion of the agent is present in the solution and the second portion of the agent is in granular form in the liquid base material. Exists as a suspension. In some embodiments, the liquid composition comprises a gel formulation. In other embodiments, the liquid composition is aqueous.
ある特定の実施形態では、有用な水性懸濁液は、1種またはそれより多種のポリマーを懸濁化剤として含有する。有用なポリマーには、セルロースポリマーなどの水溶性ポリマー、例えばヒドロキシプロピルメチルセルロース、および架橋カルボキシル含有ポリマーなどの水不溶性ポリマーが含まれる。本明細書に記述されるある特定の医薬組成物は、例えばカルボキシメチルセルロース、カルボマー(アクリル酸ポリマー)、ポリ(メチルメタクリレート)、ポリアクリルアミド、ポリカルボフィル、アクリル酸/アクリル酸ブチルコポリマー、アルギン酸ナトリウム、およびデキストランから選択される、粘膜付着ポリマーを含む。 In certain embodiments, useful aqueous suspensions contain one or more polymers as suspending agents. Useful polymers include water-soluble polymers such as cellulose polymers, such as hydroxypropyl methylcellulose, and water-insoluble polymers such as crosslinked carboxyl-containing polymers. Certain pharmaceutical compositions described herein include, for example, carboxymethyl cellulose, carbomer (acrylic acid polymer), poly (methyl methacrylate), polyacrylamide, polycarbofyl, acrylic acid / butyl acrylate copolymer, sodium alginate, etc. And a mucoadhesive polymer selected from dextran.
医薬組成物は、必要に応じて、ACVR1阻害剤の溶解性を補助する可溶化剤も含む。「可溶化剤」という用語は、一般に、薬剤のミセル溶液または真溶液の形成をもたらす薬剤を含む。ある特定の許容される非イオン性界面活性剤、例えばポリソルベート80は、眼に許容されるグリコール、ポリグリコール、例えばポリエチレングリコール400、およびグリコールエーテルで可能であるように、可溶化剤として有用である。
The pharmaceutical composition also optionally comprises a solubilizer that assists in the solubility of the ACVR1 inhibitor. The term "solubilizer" generally includes agents that result in the formation of micelle or true solutions of the agent. Certain acceptable nonionic surfactants, such as
さらに、医薬組成物は、必要に応じて、酢酸、ホウ酸、クエン酸、乳酸、リン酸、および塩酸などの酸を含む1種またはそれより多種のpH調節剤または緩衝剤;水酸化ナトリウム、リン酸ナトリウム、ホウ酸ナトリウム、クエン酸ナトリウム、酢酸ナトリウム、乳酸ナトリウム、およびtris−ヒドロキシメチルアミノメタンなどの塩基;およびシトレート/デキストロース、重炭酸ナトリウム、塩化アンモニウムなどの緩衝剤を含む。そのような酸、塩基、および緩衝剤は、許容される範囲で組成物のpHを維持するのに必要とされる量で含まれる。 In addition, the pharmaceutical composition may optionally contain one or more pH adjusters or buffers containing acids such as acetic acid, boric acid, citric acid, lactic acid, phosphoric acid, and hydrochloric acid; sodium hydroxide, Includes bases such as sodium phosphate, sodium borate, sodium citrate, sodium acetate, sodium lactate, and tris-hydroxymethylaminomethane; and buffers such as citrate / dextrose, sodium bicarbonate, ammonium chloride. Such acids, bases, and buffers are included in an acceptable amount to maintain the pH of the composition.
さらに、医薬組成物は、必要に応じて、1種またはそれより多種の塩も、組成物の浸透圧濃度を許容可能な範囲にするのに必要とされる量で含む。そのような塩には、ナトリウム、カリウム、またはアンモニウム陽イオン、および塩化物、クエン酸、アスコルビン酸、ホウ酸、リン酸、重炭酸、硫酸、チオ硫酸、または亜硫酸水素陰イオンを有するものが含まれ;適切な塩には、塩化ナトリウム、塩化カリウム、チオ硫酸ナトリウム、亜硫酸水素ナトリウム、および硫酸アンモニウムが含まれる。 In addition, the pharmaceutical composition will optionally also include one or more salts in the amount required to bring the osmotic concentration of the composition to an acceptable range. Such salts include those having sodium, potassium, or ammonium cations, and chloride, citric acid, ascorbic acid, boric acid, phosphoric acid, bicarbonate, sulfuric acid, thiosulfate, or hydrogen sulfite anion. Suitable salts include sodium chloride, potassium chloride, sodium thiosulfate, sodium hydrogen sulfite, and ammonium sulfate.
その他の医薬組成物は、必要に応じて、微生物活性を阻害する1種またはそれより多種の保存剤を含む。適切な保存剤には、メルフェンおよびチオメルサールなどの水銀含有物質;安定化二酸化塩素;および塩化ベンザルコニウム、臭化セチルトリメチルアンモニウム、および塩化セチルピリジニウムなどの第四級アンモニウム化合物が含まれる。 Other pharmaceutical compositions include, optionally, one or more preservatives that inhibit microbial activity. Suitable preservatives include mercury-containing substances such as melphen and thiomersal; stabilized chlorine dioxide; and quaternary ammonium compounds such as benzalkonium chloride, cetyltrimethylammonium bromide, and cetylpyridinium chloride.
界面活性剤は、均質溶液または懸濁液の形成を容易にするのに添加されてもよい。界面活性剤は、溶解または均質懸濁水性送達システムが容易になるように、治療化合物(複数可)と非共有的に相互に作用する化合物である。実施形態では、医薬組成物は、物理的安定性を高める1種またはそれより多種の界面活性剤を含む。適切な非イオン性界面活性剤は、ポリオキシエチレン脂肪酸グリセリドおよび植物油、例えばポリオキシエチレン(60)水素化ヒマシ油;およびポリオキシエチレンアルキルエーテルおよびアルキルフェニルエーテル、例えばオクトキシノール10、オクトキシノール40などを含む。
Surfactants may be added to facilitate the formation of homogeneous solutions or suspensions. Surfactants are compounds that interact non-covalently with the therapeutic compound (s) to facilitate a soluble or homogeneous suspension aqueous delivery system. In embodiments, the pharmaceutical composition comprises one or more surfactants that enhance physical stability. Suitable nonionic surfactants are polyoxyethylene fatty acid glycerides and vegetable oils such as polyoxyethylene (60) hydrogenated castor oil; and polyoxyethylene alkyl ethers and alkylphenyl ethers such as
さらに他の医薬組成物は、必要な場合に化学的安定性を高める、1種またはそれより多種の抗酸化剤を含む。適切な抗酸化剤には、単なる例としてアスコルビン酸およびメタ重亜硫酸ナトリウムが含まれる。 Yet other pharmaceutical compositions include one or more antioxidants that enhance chemical stability when required. Suitable antioxidants include ascorbic acid and sodium metabisulfite, by way of example only.
ある特定の実施形態では、水性懸濁組成物は、単回用量の再密封できない容器に包装される。あるいは、複数回用量の再密封可能な容器が使用され、その場合、典型的には組成物中に保存剤を含む。 In certain embodiments, the aqueous suspension composition is packaged in a single dose, non-resealable container. Alternatively, multiple dose resealable containers are used, which typically include a preservative in the composition.
本開示の一部の実施形態で使用される医薬組成物は、例えば、直腸内で融解しかつACVR1阻害剤を放出する、坐薬の形態での直腸投与が意図されてもよい。直腸投与用の組成物は、適切な非刺激賦形剤として油性ベースを含有していてもよい。そのようなベースは、ラノリン、ココアバター、およびポリエチレングリコールを含む。 The pharmaceutical compositions used in some embodiments of the present disclosure may be intended for rectal administration, for example, in the form of a suppository that melts in the rectum and releases an ACVR1 inhibitor. Compositions for rectal administration may contain an oily base as a suitable non-irritating excipient. Such bases include lanolin, cocoa butter, and polyethylene glycol.
本開示の実施形態で使用される医薬組成物は、固体または液体の投薬単位の物理的形態を改変する、様々な材料を含んでいてもよい。例えば、組成物は、ACVR1阻害剤の周りにコーティングシェルを形成する材料を含んでいてもよい。コーティングシェルを形成する材料は、典型的には不活性であり、例えば糖、シェラック、およびその他の腸溶性コーティング剤から選択されてもよい。あるいは、活性成分は、ゼラチンカプセルに包封されていてもよい。 The pharmaceutical compositions used in the embodiments of the present disclosure may include a variety of materials that modify the physical form of solid or liquid dosage units. For example, the composition may include a material that forms a coating shell around the ACVR1 inhibitor. The material forming the coating shell is typically inert and may be selected from, for example, sugar, shellac, and other enteric coatings. Alternatively, the active ingredient may be encapsulated in gelatin capsules.
ある特定の実施形態で使用される医薬組成物は、エアロゾルとして投与することができる投薬単位からなるものであってもよい。エアロゾルという用語は、コロイド状の性質のものから加圧パッケージを利用する系に及ぶ、様々な系を示すのに使用される。送達は、液化もしくは圧縮ガスによる、または活性成分を吐出する適切なポンプシステムによるものであってもよい。ACVR1阻害剤のエアロゾルは、活性成分(複数可)を送達するために、単相、2相、または3相系で送達されてもよい。エアロゾルの送達は、一緒になってキットを形成し得る、必要な容器、活性化剤、弁、サブ容器などを含む。当業者なら、過度な実験なしに、好ましいエアロゾルを決定することができる。 The pharmaceutical composition used in certain embodiments may consist of a dosage unit that can be administered as an aerosol. The term aerosol is used to describe a variety of systems, ranging from those of colloidal nature to systems that utilize pressurized packages. Delivery may be by liquefied or compressed gas, or by a suitable pump system that ejects the active ingredient. The aerosol of the ACVR1 inhibitor may be delivered in a single-phase, two-phase, or three-phase system to deliver the active ingredient (s). Delivery of aerosols includes the necessary containers, activators, valves, sub-containers, etc. that can be combined to form a kit. One of ordinary skill in the art can determine the preferred aerosol without undue experimentation.
前述の実施形態のいずれかにおいて、ACVR1阻害剤またはその薬学的に許容される塩は、用いられる特定のACVR1阻害剤の活性;ACVR1阻害剤の代謝安定性および作用の長さ;患者の年齢、体重、全身の健康、性別、および食事;投与する形態および時間;排泄率;薬物の組合せ;特定の障害または状態の重症度;および対象が受ける治療を含む、様々な因子に応じて変わる有効量で、投与される。 In any of the aforementioned embodiments, the ACVR1 inhibitor or pharmaceutically acceptable salt thereof is the activity of the particular ACVR1 inhibitor used; the metabolic stability and length of action of the ACVR1 inhibitor; the age of the patient, Weight, general health, gender, and diet; form and time of administration; excretion rate; combination of drugs; severity of a particular disorder or condition; and effective amount that depends on a variety of factors, including the treatment the subject receives. Is administered.
本明細書に記述される方法の毒性および治療効力は、細胞培養物または実験動物での、標準の薬学的手順によって、例えば投与されたACVR1阻害剤のIC50およびLD50を決定することにより、決定することができる。投与では、有効量(用量とも呼ぶ)は、in vitroアッセイおよび/または動物モデル研究からの結果に基づいて、最初に推定することができる。例えば用量は、特定の標的に対して細胞培養物を決定したときのIC50を含む、循環濃度範囲が実現されるように、動物モデルにおいて製剤化することができる。投薬量は、用いられる剤形および利用される投与経路に応じて変わってもよい。厳密な製剤、投与経路、および投薬量は、患者の状態に鑑み、個々の医師により選択することができる(例えば、Goodman & Gilman's The Pharmacological Basis Of Therapeutics, Ch. 3, 9th ed., Ed. by Hardman, J., and Limbard, L., McGraw-Hill, New York City, 1996, p.46参照)。 The toxicity and therapeutic efficacy of the methods described herein can be determined by standard pharmaceutical procedures in cell cultures or laboratory animals, eg, by determining the administered ACVR1 inhibitors IC 50 and LD 50 . Can be decided. For administration, the effective amount (also referred to as dose) can be initially estimated based on results from in vitro assays and / or animal model studies. For example dose comprises an IC 50 when determining the cell culture for a particular target, as circulating concentration range is achieved, can be formulated in animal models. The dosage may vary depending on the dosage form used and the route of administration used. The exact formulation, route of administration, and dosage, in view of the patient's condition can be chosen by the individual physician (e.g., Goodman &Gilman's The Pharmacological Basis Of Therapeutics, Ch. 3, 9 th ed., Ed. by Hardman, J., and Limbard, L., McGraw-Hill, New York City, 1996, p.46).
対象に投与される組成物は、1つまたはそれより多くの投薬単位の形態をとり、例えば錠剤は、単回投薬単位であってもよく、エアロゾル形態にある本開示の1種またはそれより多種の治療剤の容器は、複数の投薬単位を保持してもよい。そのような剤形を調製する実際の方法は公知であり、または当業者に明らかにされ;例えば、Remington: The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000)を参照されたい。本開示の方法のある特定の実施形態を使用して投与される医薬組成物は、いずれにしても、本開示の実施形態の教示により疾患を処置するために、有効量のACVR1阻害剤またはその薬学的に許容される塩を含有する。 Compositions administered to a subject may take the form of one or more dosing units, eg tablets may be single dosing units, one or more of the present disclosure in aerosol form. The therapeutic agent container may hold multiple dosing units. Practical methods for preparing such dosage forms are known or manifested to those of skill in the art; see, for example, Remington: The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000). sea bream. In any case, a pharmaceutical composition administered using a particular embodiment of the methods of the present disclosure may be an effective amount of an ACVR1 inhibitor or an amount thereof for treating a disease according to the teachings of the embodiments of the present disclosure. Contains pharmaceutically acceptable salts.
本明細書に記述されるACVR1阻害剤は、広い投薬範囲にわたって有効である。例えば、成人の処置では、1日当たり約5mgから約500mg、約10mgから約320mg、約30mgから約240mg、および1日当たり約30mgから約180mgの投薬量が、一部の実施形態で使用される投薬量の例である。一部の実施形態では、用量は、1日当たり約30mgから約120mgである。一部の実施形態では、用量は、1日当たり約30mgから約60mgである。一部の実施形態では、用量は、1日当たり約60mgから約240mgである。一部の実施形態では、用量は、1日当たり約60mgから約180mgである。一部の実施形態では、用量は、1日当たり約60mgから約120mgである。一部の実施形態では、用量は、1日当たり約120mgから約240mgである。一部の実施形態では、用量は、1日当たり約120mgから約180mgである。一部の実施形態では、用量は、1日当たり約180mgから約240mgである。 The ACVR1 inhibitors described herein are effective over a wide range of dosages. For example, in adult treatment, dosages of about 5 mg to about 500 mg, about 10 mg to about 320 mg, about 30 mg to about 240 mg, and about 30 mg to about 180 mg per day are used in some embodiments. This is an example of quantity. In some embodiments, the dose is from about 30 mg to about 120 mg per day. In some embodiments, the dose is from about 30 mg to about 60 mg per day. In some embodiments, the dose is from about 60 mg to about 240 mg per day. In some embodiments, the dose is from about 60 mg to about 180 mg per day. In some embodiments, the dose is from about 60 mg to about 120 mg per day. In some embodiments, the dose is from about 120 mg to about 240 mg per day. In some embodiments, the dose is from about 120 mg to about 180 mg per day. In some embodiments, the dose is from about 180 mg to about 240 mg per day.
特定の実施形態では、用量は、1日当たり約5mgである。特定の実施形態では、用量は、1日当たり約10mgである。特定の実施形態では、用量は、1日当たり約25mgである。他の実施形態では、用量は、1日当たり約30mgである。他の実施形態では、用量は、1日当たり約60mgである。他の実施形態では、用量は、1日当たり約120mgである。特定の実施形態では、用量は、1日当たり約125mgである。他の実施形態では、用量は、1日当たり約180mgである。他の実施形態では、用量は、1日当たり約240mgである。他の実施形態では、用量は、1日当たり約250mgである。他の実施形態では、用量は、1日当たり約320mgである。他の実施形態では、用量は、1日当たり約325mgである。 In certain embodiments, the dose is about 5 mg per day. In certain embodiments, the dose is about 10 mg per day. In certain embodiments, the dose is about 25 mg per day. In other embodiments, the dose is about 30 mg per day. In other embodiments, the dose is about 60 mg per day. In other embodiments, the dose is about 120 mg per day. In certain embodiments, the dose is about 125 mg per day. In other embodiments, the dose is about 180 mg per day. In other embodiments, the dose is about 240 mg per day. In other embodiments, the dose is about 250 mg per day. In other embodiments, the dose is about 320 mg per day. In other embodiments, the dose is about 325 mg per day.
別の例として、成人の処置では、1週当たり約5mgから約500mg、約10mgから約320mg、約30mgから約240mg、および1週当たり約30mgから約180mgの投薬量が、一部の実施形態で使用される投薬量の例である。一部の実施形態では、用量は、1週当たり約30mgから約120mgである。一部の実施形態では、用量は、1週当たり約30mgから約60mgである。一部の実施形態では、用量は、1週当たり約60mgから約240mgである。一部の実施形態では、用量は、1週当たり約60mgから約180mgである。一部の実施形態では、用量は、1週当たり約60mgから約120mgである。一部の実施形態では、用量は、1週当たり約120mgから約240mgである。一部の実施形態では、用量は、1週当たり約120mgから約180mgである。一部の実施形態では、用量は、1週当たり約180mgから約240mgである。 As another example, in adult treatment, dosages of about 5 mg to about 500 mg per week, about 10 mg to about 320 mg, about 30 mg to about 240 mg, and about 30 mg to about 180 mg per week are some embodiments. This is an example of the dosage used in. In some embodiments, the dose is from about 30 mg to about 120 mg per week. In some embodiments, the dose is from about 30 mg to about 60 mg per week. In some embodiments, the dose is from about 60 mg to about 240 mg per week. In some embodiments, the dose is from about 60 mg to about 180 mg per week. In some embodiments, the dose is from about 60 mg to about 120 mg per week. In some embodiments, the dose is from about 120 mg to about 240 mg per week. In some embodiments, the dose is from about 120 mg to about 180 mg per week. In some embodiments, the dose is from about 180 mg to about 240 mg per week.
特定の実施形態では、用量は、1週当たり約5mgである。特定の実施形態では、用量は、1週当たり約10mgである。特定の実施形態では、用量は、1週当たり約25mgである。他の実施形態では、用量は、1週当たり約30mgである。他の実施形態では、用量は、1週当たり約60mgである。他の実施形態では、用量は、1週当たり約120mgである。特定の実施形態では、用量は、1週当たり約125mgである。他の実施形態では、用量は、1週当たり約180mgである。他の実施形態では、用量は、1週当たり約240mgである。他の実施形態では、用量は、1週当たり約250mgである。他の実施形態では、用量は、1週当たり約320mgである。他の実施形態では、用量は、1週当たり約325mgである。 In certain embodiments, the dose is about 5 mg per week. In certain embodiments, the dose is about 10 mg per week. In certain embodiments, the dose is about 25 mg per week. In other embodiments, the dose is about 30 mg per week. In other embodiments, the dose is about 60 mg per week. In other embodiments, the dose is about 120 mg per week. In certain embodiments, the dose is about 125 mg per week. In other embodiments, the dose is about 180 mg per week. In other embodiments, the dose is about 240 mg per week. In other embodiments, the dose is about 250 mg per week. In other embodiments, the dose is about 320 mg per week. In other embodiments, the dose is about 325 mg per week.
本明細書で記述されるように、小児用量は、成人用量の約80%から100%の間であってもよい。 As described herein, the pediatric dose may be between about 80% and 100% of the adult dose.
一部の実施形態では、用量は、上昇させる。一実施形態では、用量は、1週当たり30mgから開始して、30mgずつ増やし、1週当たり120mgまでにする。一実施形態では、用量は、1週当たり30mgで開始して、そのままのレベルである。一実施形態では、用量は、1週当たり30mgで開始して、1週当たり60mgの最終用量まで上昇させる。一実施形態では、用量は、1週当たり30mgで開始し、1週当たり60mgの中間用量に上昇させ、さらに1週当たり90mgの最終用量まで上昇させる。一実施形態では、用量は、1週当たり30mgで開始し、1週当たり60mgの第1の中間用量、1週当たり90mgの第2の中間用量に上昇させ、さらに1週当たり120mgの最終用量に上昇させる。一実施形態では、用量は、1週当たり60mgで開始し、レベルをそのままにする。一実施形態では、用量は、1週当たり60mgで開始し、1週当たり90mgの最終用量に上昇させる。一実施形態では、用量は、1週当たり60mgで開始し、1週当たり90mgの中間用量に上昇させ、さらに1週当たり120mgの最終用量に上昇させる。 In some embodiments, the dose is increased. In one embodiment, the dose starts at 30 mg per week and is increased by 30 mg to 120 mg per week. In one embodiment, the dose starts at 30 mg per week and remains at the same level. In one embodiment, the dose starts at 30 mg per week and is increased to a final dose of 60 mg per week. In one embodiment, the dose is started at 30 mg per week, increased to an intermediate dose of 60 mg per week, and further increased to a final dose of 90 mg per week. In one embodiment, the dose starts at 30 mg per week and is increased to a first intermediate dose of 60 mg per week, a second intermediate dose of 90 mg per week, and a final dose of 120 mg per week. Raise. In one embodiment, the dose is started at 60 mg per week and the levels remain unchanged. In one embodiment, the dose starts at 60 mg per week and is increased to a final dose of 90 mg per week. In one embodiment, the dose is started at 60 mg per week, increased to an intermediate dose of 90 mg per week, and further increased to a final dose of 120 mg per week.
実施形態では、ACVR1阻害剤は、1日当たり約10mg/m2から約500mg/m2に及ぶ用量で投与される。実施形態では、ACVR1阻害剤は、1日当たり約150mg/m2から約350mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1日当たり約200mg/m2から約300mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1日当たり約220mg/m2から約260mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1日当たり約230mg/m2から約250mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1日当たり約235mg/m2から約245mg/m2に及ぶ用量で投与される。特定の実施形態では、ACVR1阻害剤は、1日当たり約240mg/m2の用量で投与される。 In embodiments, ACVR1 inhibitor is administered at a dose ranging from 1 day to about 10 mg / m 2 to about 500 mg / m 2. In embodiments, ACVR1 inhibitor is administered at a dose ranging from 1 day to about 150 mg / m 2 to about 350 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from 1 day to about 200 mg / m 2 to about 300 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from 1 day to about 220 mg / m 2 to about 260 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from 1 day to about 230 mg / m 2 to about 250 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from 1 day to about 235 mg / m 2 to about 245 mg / m 2. In certain embodiments, the ACVR1 inhibitor is administered at a dose of about 240 mg / m 2 per day.
実施形態において、ACVR1阻害剤は、1週当たり約10mg/m2から約500mg/m2に及ぶ用量で投与される。実施形態では、ACVR1阻害剤は、1週当たり約150mg/m2から約350mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1週当たり約200mg/m2から約300mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1週当たり約220mg/m2から約260mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1週当たり約230mg/m2から約250mg/m2に及ぶ用量で投与される。一部の実施形態では、ACVR1阻害剤は、1週当たり約235mg/m2から約245mg/m2に及ぶ用量で投与される。特定の実施形態では、ACVR1阻害剤は、1週当たり約240mg/m2の用量で投与される。 In embodiments, ACVR1 inhibitor is administered at a dose ranging from about 10 mg / m 2 per week to about 500 mg / m 2. In embodiments, ACVR1 inhibitor is administered at a dose ranging from about 150 mg / m 2 per week to about 350 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from about 200 mg / m 2 per week to about 300 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from about 220 mg / m 2 per week to about 260 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from about 230 mg / m 2 per week to about 250 mg / m 2. In some embodiments, ACVR1 inhibitor is administered at a dose ranging from about 235 mg / m 2 per week to about 245 mg / m 2. In certain embodiments, the ACVR1 inhibitor is administered at a dose of about 240 mg / m 2 per week.
厳密な投薬量は、ACVR1阻害剤、投与経路、化合物が投与される形態、処置がなされる対象、標的を含む物理的および生理的因子、体重、状態の重症度、がんのタイプ、事前または同時の治療介入、対象の特発性疾患、ならびに主治医の好みおよび経験に依存する。 The exact dosage is ACVR1 inhibitor, route of administration, form of administration of the compound, subject to be treated, physical and physiological factors including the target, body weight, severity of condition, type of cancer, pre- or It depends on the concurrent intervention, the subject's idiopathic disease, and the preference and experience of the attending physician.
一部の実施形態では、ACVR1阻害剤の有効量が、単回用量で投与される。典型的には、そのような投与は、薬剤を迅速に導入するために、注射、例えば静脈内注射による投与である。しかし、その他の経路が適切に使用される。本開示の化合物の単回用量は、急性状態の処置に使用されてもよい。 In some embodiments, an effective amount of the ACVR1 inhibitor is administered in a single dose. Typically, such administration is by injection, eg, intravenous injection, for rapid introduction of the drug. However, other routes are used appropriately. Single doses of the compounds of the present disclosure may be used in the treatment of acute conditions.
一部の実施形態では、ACVR1阻害剤の有効量が、複数回用量で投与される。一部の実施形態では、投薬は、1日当たり約1回、2回、3回、4回、5回、6回、またはそれを超える回数である。一部の実施形態では、投薬は、1週当たり約1回、2回、3回、4回、5回、6回、またはそれを超える回数である。他の実施形態では、投薬は、1か月に約1回、2週おきに1回、1週に1回、または1日おきに1回である。さらに別の実施形態では、投与は、約6、10、14、28日、2か月、6か月、または1年よりも長く継続される。場合によっては、継続投薬は、必要な限り実現され維持される。一部の実施形態では、ACVR1阻害剤は、1、7、14、21、または28日間連続して投与される。一部の実施形態では、ACVR1阻害剤は、毎週投与される。一部の実施形態では、ACVR1阻害剤は、4週サイクルの1週目、2週目、3週目、および4週目に投与される。一部の実施形態では、4週サイクルは、1または複数の休日を含む。一部の実施形態では、4週サイクルは、休日を含まず、投薬は継続投薬である。
In some embodiments, an effective amount of the ACVR1 inhibitor is administered in multiple doses. In some embodiments, the dosing is about once, twice, three times, four times, five times, six times, or more times per day. In some embodiments, the dosing is about once, twice, three times, four times, five times, six times, or more per week. In other embodiments, the dosing is about once a month, once every two weeks, once a week, or once every other day. In yet another embodiment, administration is continued for more than about 6, 10, 14, 28 days, 2 months, 6 months, or 1 year. In some cases, continuous dosing is realized and maintained as long as necessary. In some embodiments, the ACVR1 inhibitor is administered for 1, 7, 14, 21, or 28 consecutive days. In some embodiments, the ACVR1 inhibitor is administered weekly. In some embodiments, the ACVR1 inhibitor is administered at
様々な実施形態では、ACVR1阻害剤は、毎日投与される。様々な実施形態では、ACVR1阻害剤は、毎週投与される。そのような実施形態のそれぞれにおいて、ACVR1阻害剤は、1日の実質的に同じ時間に摂取される。一部の実施形態では、ACVR1阻害剤は、絶食(例えば、少なくとも6時間)後に投与される。特定の実施形態では、対象は、投与後少なくとも1時間、絶食する。 In various embodiments, the ACVR1 inhibitor is administered daily. In various embodiments, the ACVR1 inhibitor is administered weekly. In each of such embodiments, the ACVR1 inhibitor is ingested at substantially the same time of the day. In some embodiments, the ACVR1 inhibitor is administered after fasting (eg, at least 6 hours). In certain embodiments, the subject fasts for at least 1 hour after administration.
ACVR1阻害剤の投与は、必要な限り継続され得る。一部の実施形態では、ACVR1阻害剤は、1、2、3、4、5、6、7、14、または28日よりも長い間、投与される。一部の実施形態では、ACVR1阻害剤は、28、14、7、6、5、4、3、2、または1日よりも短い間、投与される。一部の実施形態では、ACVR1阻害剤は、1、4、8、12、16、20、24、28、32、36、40、44、48、または52週よりも長い間投与される。一部の実施形態では、ACVR1阻害剤は、52、48、44、40、36、32、28、24、20、16、12、8、4、または1週よりも短い間、投与される。 Administration of the ACVR1 inhibitor can be continued as long as necessary. In some embodiments, the ACVR1 inhibitor is administered for longer than 1, 2, 3, 4, 5, 6, 7, 14, or 28 days. In some embodiments, the ACVR1 inhibitor is administered for less than 28, 14, 7, 6, 5, 4, 3, 2, or 1 day. In some embodiments, the ACVR1 inhibitor is administered for longer than 1, 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, 44, 48, or 52 weeks. In some embodiments, the ACVR1 inhibitor is administered for less than 52, 48, 44, 40, 36, 32, 28, 24, 20, 16, 12, 8, 4, or 1 week.
一部の実施形態では、ACVR1阻害剤は、例えば長期的な影響の処置のために、長期的に継続的に投与される。 In some embodiments, the ACVR1 inhibitor is administered continuously over the long term, for example for the treatment of long-term effects.
一部の実施形態では、追加の治療剤は、長期的に投与されてもよい(例えば、維持療法として)。他のそのような実施形態では、追加の1種またはそれより多種の治療剤が、第2の処置レジメンとして投与される。 In some embodiments, the additional therapeutic agent may be administered in the long term (eg, as maintenance therapy). In other such embodiments, one additional therapeutic agent or more is administered as a second treatment regimen.
一部の実施形態では、ACVR1阻害剤は、投薬量で投与される。化合物の薬物動態の対象間変動に起因して、投薬レジメンの個別化は、ある特定の実施形態において提供される。治療剤に関する投薬は、本開示に照らして通常の実験によって見出すことができ、そして/または当業者により導き出すことができる。 In some embodiments, the ACVR1 inhibitor is administered at a dosage. Due to inter-subject variations in the pharmacokinetics of compounds, individualization of dosing regimens is provided in certain embodiments. Dosings for therapeutic agents can be found by routine experimentation in the light of the present disclosure and / or can be derived by one of ordinary skill in the art.
投薬の量および間隔は、所望の薬理学的作用を維持するのに十分な、活性種の血漿中レベルが提供されるように、個別に調節され得る。これらの血漿中レベルを、最小有効濃度(MEC)と呼ぶ。MECを実現するのに必要な投薬量は、個々の特性および投与経路に依存する。HPLCアッセイまたはバイオアッセイは、血漿中濃度を決定するのに使用することができる。 Dosing amounts and intervals can be individually adjusted to provide plasma levels of the active species sufficient to maintain the desired pharmacological effect. These plasma levels are referred to as the minimum effective concentration (MEC). The dosage required to achieve MEC depends on the individual characteristics and route of administration. HPLC assays or bioassays can be used to determine plasma concentrations.
投薬の間隔は、MEC値を使用して決定されてもよい。一部の実施形態では、処置の方法は、時間の10〜90%にわたりMECよりも高い血漿中レベルを維持することを含む。一部の実施形態では、血漿中レベルは、時間の30〜90%の間でMECよりも高く維持される。一部の実施形態では、血漿中レベルは、時間の50〜90%の間でMECよりも高く維持される。例えば、ある特定の実施形態では、治療剤の有効量は、1日当たりおよそ2.5mg/m2から1500mg/m2に及んでもよい。例えば、ある特定の実施形態では、治療剤の有効量は、1週当たりおよそ2.5mg/m2から1500mg/m2に及んでもよい。追加の例示的な量は、毎日または毎週のいずれかで、0.2〜1000mg、2〜500mg、および20〜250mgに及ぶ。 Dosing intervals may be determined using MEC values. In some embodiments, the method of treatment comprises maintaining plasma levels higher than MEC for 10 to 90% of the time. In some embodiments, plasma levels are maintained above MEC for 30-90% of the time. In some embodiments, plasma levels are maintained above MEC for 50-90% of the time. For example, in certain embodiments, the effective amount of the therapeutic agent may range from 1 day approximately 2.5 mg / m 2 to 1500 mg / m 2. For example, in certain embodiments, the effective amount of the therapeutic agent may range approximately from 2.5 mg / m 2 per week in 1500 mg / m 2. Additional exemplary amounts range from 0.2 to 1000 mg, 2 to 500 mg, and 20 to 250 mg, either daily or weekly.
局所投与または選択的摂取の場合、治療剤の有効局所濃度は、血漿中濃度に関連していなくてもよく、当技術分野で公知のその他の手順は、正しい投薬の量および間隔を決定するのに用いられてもよい。 For topical or selective ingestion, the effective local concentration of therapeutic agent may not be related to plasma concentration, and other procedures known in the art determine the correct dosage and interval of dosing. It may be used for.
一部の実施形態では、医薬組成物中に提供されるACVR1阻害剤の濃度は、100%、90%、80%、70%、60%、50%、40%、30%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.4%、0.3%、0.2%、0.1%、0.09%、0.08%、0.07%、0.06%、0.05%、0.04%、0.03%、0.02%、0.01%、0.009%、0.008%、0.007%、0.006%、0.005%、0.004%、0.003%、0.002%、0.001%、0.0009%、0.0008%、0.0007%、0.0006%、0.0005%、0.0004%、0.0003%、0.0002%、もしくは0.0001%w/w未満、100%、90%、80%、70%、60%、50%、40%、30%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.4%、0.3%、0.2%、0.1%、0.09%、0.08%、0.07%、0.06%、0.05%、0.04%、0.03%、0.02%、0.01%、0.009%、0.008%、0.007%、0.006%、0.005%、0.004%、0.003%、0.002%、0.001%、0.0009%、0.0008%、0.0007%、0.0006%、0.0005%、0.0004%、0.0003%、0.0002%、もしくは0.0001%w/v未満、または100%、90%、80%、70%、60%、50%、40%、30%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.4%、0.3%、0.2%、0.1%、0.09%、0.08%、0.07%、0.06%、0.05%、0.04%、0.03%、0.02%、0.01%、0.009%、0.008%、0.007%、0.006%、0.005%、0.004%、0.003%、0.002%、0.001%、0.0009%、0.0008%、0.0007%、0.0006%、0.0005%、0.0004%、0.0003%、0.0002%、もしくは0.0001%v/v未満である。 In some embodiments, the concentrations of the ACVR1 inhibitor provided in the pharmaceutical composition are 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19 %, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06% , 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005% , 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004% , 0.0003%, 0.0002%, or less than 0.0001% w / w, 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19% , 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2 %, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002%, or less than 0.0001% w / v, or 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19% , 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2 %, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002 %, Or less than 0.0001% v / v.
一部の実施形態では、医薬組成物中に提供されるACVR1阻害剤の濃度は、90%、80%、70%、60%、50%、40%、30%、20%、19.75%、19.50%、19.25% 19%、18.75%、18.50%、18.25% 18%、17.75%、17.50%、17.25% 17%、16.75%、16.50%、16.25% 16%、15.75%、15.50%、15.25% 15%、14.75%、14.50%、14.25% 14%、13.75%、13.50%、13.25% 13%、12.75%、12.50%、12.25% 12%、11.75%、11.50%、11.25% 11%、10.75%、10.50%、10.25% 10%、9.75%、9.50%、9.25% 9%、8.75%、8.50%、8.25% 8%、7.75%、7.50%、7.25% 7%、6.75%、6.50%、6.25% 6%、5.75%、5.50%、5.25% 5%、4.75%、4.50%、4.25%、4%、3.75%、3.50%、3.25%、3%、2.75%、2.50%、2.25%、2%、1.75%、1.50%、125%、1%、0.5%、0.4%、0.3%、0.2%、0.1%、0.09%、0.08%、0.07%、0.06%、0.05%、0.04%、0.03%、0.02%、0.01%、0.009%、0.008%、0.007%、0.006%、0.005%、0.004%、0.003%、0.002%、0.001%、0.0009%、0.0008%、0.0007%、0.0006%、0.0005%、0.0004%、0.0003%、0.0002%、もしくは0.0001%w/wより大きい、90%、80%、70%、60%、50%、40%、30%、20%、19.75%、19.50%、19.25% 19%、18.75%、18.50%、18.25% 18%、17.75%、17.50%、17.25% 17%、16.75%、16.50%、16.25% 16%、15.75%、15.50%、15.25% 15%、14.75%、14.50%、14.25% 14%、13.75%、13.50%、13.25% 13%、12.75%、12.50%、12.25% 12%、11.75%、11.50%、11.25% 11%、10.75%、10.50%、10.25% 10%、9.75%、9.50%、9.25% 9%、8.75%、8.50%、8.25% 8%、7.75%、7.50%、7.25% 7%、6.75%、6.50%、6.25% 6%、5.75%、5.50%、5.25% 5%、4.75%、4.50%、4.25%、4%、3.75%、3.50%、3.25%、3%、2.75%、2.50%、2.25%、2%、1.75%、1.50%、125%、1%、0.5%、0.4%、0.3%、0.2%、0.1%、0.09%、0.08%、0.07%、0.06%、0.05%、0.04%、0.03%、0.02%、0.01%、0.009%、0.008%、0.007%、0.006%、0.005%、0.004%、0.003%、0.002%、0.001%、0.0009%、0.0008%、0.0007%、0.0006%、0.0005%、0.0004%、0.0003%、0.0002%、もしくは0.0001%w/vより大きい、または90%、80%、70%、60%、50%、40%、30%、20%、19.75%、19.50%、19.25% 19%、18.75%、18.50%、18.25% 18%、17.75%、17.50%、17.25% 17%、16.75%、16.50%、16.25% 16%、15.75%、15.50%、15.25% 15%、14.75%、14.50%、14.25% 14%、13.75%、13.50%、13.25% 13%、12.75%、12.50%、12.25% 12%、11.75%、11.50%、11.25% 11%、10.75%、10.50%、10.25% 10%、9.75%、9.50%、9.25% 9%、8.75%、8.50%、8.25% 8%、7.75%、7.50%、7.25% 7%、6.75%、6.50%、6.25% 6%、5.75%、5.50%、5.25% 5%、4.75%、4.50%、4.25%、4%、3.75%、3.50%、3.25%、3%、2.75%、2.50%、2.25%、2%、1.75%、1.50%、125%、1%、0.5%、0.4%、0.3%、0.2%、0.1%、0.09%、0.08%、0.07%、0.06%、0.05%、0.04%、0.03%、0.02%、0.01%、0.009%、0.008%、0.007%、0.006%、0.005%、0.004%、0.003%、0.002%、0.001%、0.0009%、0.0008%、0.0007%、0.0006%、0.0005%、0.0004%、0.0003%、0.0002%、もしくは0.0001%v/vより大きい。 In some embodiments, the concentrations of the ACVR1 inhibitor provided in the pharmaceutical composition are 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19.75%. , 19.50%, 19.25% 19%, 18.75%, 18.50%, 18.25% 18%, 17.75%, 17.50%, 17.25% 17%, 16.75 %, 16.50%, 16.25% 16%, 15.75%, 15.50%, 15.25% 15%, 14.75%, 14.50%, 14.25% 14%, 13. 75%, 13.50%, 13.25% 13%, 12.75%, 12.50%, 12.25% 12%, 11.75%, 11.50%, 11.25% 11%, 10 .75%, 10.50%, 10.25%, 10%, 9.75%, 9.50%, 9.25%, 9%, 8.75%, 8.50%, 8.25%, 8%, 7.75%, 7.50%, 7.25% 7%, 6.75%, 6.50%, 6.25% 6%, 5.75%, 5.50%, 5.25% 5% 4.75%, 4.50%, 4.25%, 4%, 3.75%, 3.50%, 3.25%, 3%, 2.75%, 2.50%, 2.25 %, 2%, 1.75%, 1.50%, 125%, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09% , 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008% , 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007% , 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002%, or greater than 0.0001% w / w, 90%, 80%, 70%, 60%, 50 %, 40%, 30%, 20%, 19.75%, 19.50%, 19.25% 19%, 18.75%, 18.50%, 18.25% 18%, 17.75%, 17.50%, 17.25% 17%, 16.75%, 16.50%, 16.25% 16%, 15.75%, 15.50%, 15.25% 15%, 14.75% , 14.50%, 14.25% 14%, 13.75%, 13.50%, 13.25% 13%, 12.75%, 12.50%, 12.25% 12%, 11.75 % 11.50%, 11.25% 11%, 10.75%, 10.50%, 10.25%, 10%, 9.75%, 9.50%, 9.25%, 9%, 8.75%, 8.50% , 8.25% 8%, 7.75%, 7.50%, 7.25% 7%, 6.75%, 6.50%, 6.25% 6%, 5.75%, 5.50 %, 5.25%, 5%, 4.75%, 4.50%, 4.25%, 4%, 3.75%, 3.50%, 3.25%, 3%, 2.75%, 2.50%, 2.25%, 2%, 1.75%, 1.50%, 125%, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0 .1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0 .009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0 .0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, 0.0002%, or 0.0001% greater than w / v, or 90%, 80 %, 70%, 60%, 50%, 40%, 30%, 20%, 19.75%, 19.50%, 19.25% 19%, 18.75%, 18.50%, 18.25 % 18%, 17.75%, 17.50%, 17.25% 17%, 16.75%, 16.50%, 16.25% 16%, 15.75%, 15.50%, 15. 25% 15%, 14.75%, 14.50%, 14.25% 14%, 13.75%, 13.50%, 13.25% 13%, 12.75%, 12.50%, 12 .25% 12%, 11.75%, 11.50%, 11.25% 11%, 10.75%, 10.50%, 10.25%, 10%, 9.75%, 9.50%, 9.25% 9%, 8.75%, 8.50%, 8.25% 8%, 7.75%, 7.50%, 7.25% 7%, 6.75%, 6.50% , 6.25% 6%, 5.75%, 5.50%, 5.25%, 5%, 4.75%, 4.50%, 4.25%, 4%, 3.75%, 3. 50%, 3.25%, 3%, 2.75%, 2.50%, 2.25%, 2%, 1.75%, 1.50%, 125%, 1%, 0.5%, 0.4%, 0.3%, 0.2%, 0.1%, 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02%, 0.01%, 0.009%, 0.008%, 0.007%, 0.006%, 0.005%, 0.004%, 0.003%, 0.002%, 0.001%, 0.0009%, 0.0008%, 0.0007%, 0.0006%, 0.0005%, 0.0004%, 0.0003%, Greater than 0.0002% or 0.0001% v / v.
一部の実施形態では、医薬組成物中に提供されるACVR1阻害剤の濃度は、約0.0001%から約50%、約0.001%から約40%、約0.01%から約30%、約0.02%から約29%、約0.03%から約28%、約0.04%から約27%、約0.05%から約26%、約0.06%から約25%、約0.07%から約24%、約0.08%から約23%、約0.09%から約22%、約0.1%から約21%、約0.2%から約20%、約0.3%から約19%、約0.4%から約18%、約0.5%から約17%、約0.6%から約16%、約0.7%から約15%、約0.8%から約14%、約0.9%から約12%、約1%から約10%w/wの範囲、約0.0001%から約50%、約0.001%から約40%、約0.01%から約30%、約0.02%から約29%、約0.03%から約28%、約0.04%から約27%、約0.05%から約26%、約0.06%から約25%、約0.07%から約24%、約0.08%から約23%、約0.09%から約22%、約0.1%から約21%、約0.2%から約20%、約0.3%から約19%、約0.4%から約18%、約0.5%から約17%、約0.6%から約16%、約0.7%から約15%、約0.8%から約14%、約0.9%から約12%、約1%から約10%w/vの範囲または約0.0001%から約50%、約0.001%から約40%、約0.01%から約30%、約0.02%から約29%、約0.03%から約28%、約0.04%から約27%、約0.05%から約26%、約0.06%から約25%、約0.07%から約24%、約0.08%から約23%、約0.09%から約22%、約0.1%から約21%、約0.2%から約20%、約0.3%から約19%、約0.4%から約18%、約0.5%から約17%、約0.6%から約16%、約0.7%から約15%、約0.8%から約14%、約0.9%から約12%、約1%から約10%v/vの範囲にある。 In some embodiments, the concentration of ACVR1 inhibitor provided in the pharmaceutical composition is from about 0.0001% to about 50%, from about 0.001% to about 40%, from about 0.01% to about 30. %, About 0.02% to about 29%, about 0.03% to about 28%, about 0.04% to about 27%, about 0.05% to about 26%, about 0.06% to about 25 %, About 0.07% to about 24%, about 0.08% to about 23%, about 0.09% to about 22%, about 0.1% to about 21%, about 0.2% to about 20 %, About 0.3% to about 19%, about 0.4% to about 18%, about 0.5% to about 17%, about 0.6% to about 16%, about 0.7% to about 15 %, About 0.8% to about 14%, about 0.9% to about 12%, about 1% to about 10% w / w, about 0.0001% to about 50%, about 0.001% From about 40%, about 0.01% to about 30%, about 0.02% to about 29%, about 0.03% to about 28%, about 0.04% to about 27%, about 0.05% From about 26%, about 0.06% to about 25%, about 0.07% to about 24%, about 0.08% to about 23%, about 0.09% to about 22%, about 0.1% From about 21%, about 0.2% to about 20%, about 0.3% to about 19%, about 0.4% to about 18%, about 0.5% to about 17%, about 0.6% From about 16%, about 0.7% to about 15%, about 0.8% to about 14%, about 0.9% to about 12%, about 1% to about 10% w / v or about 0 .0001% to about 50%, about 0.001% to about 40%, about 0.01% to about 30%, about 0.02% to about 29%, about 0.03% to about 28%, about 0 .04% to about 27%, about 0.05% to about 26%, about 0.06% to about 25%, about 0.07% to about 24%, about 0.08% to about 23%, about 0 .09% to about 22%, about 0.1% to about 21%, about 0.2% to about 20%, about 0.3% to about 19%, about 0.4% to about 18%, about 0 .5% to about 17%, about 0.6% to about 16%, about 0.7% to about 15%, about 0.8% to about 14%, about 0.9% to about 12%, about 1 It is in the range of% to about 10% v / v.
一部の実施形態では、医薬組成物中に提供されるACVR1阻害剤の濃度は、約0.001%から約10%、約0.01%から約5%、約0.02%から約4.5%、約0.03%から約4%、約0.04%から約3.5%、約0.05%から約3%、約0.06%から約2.5%、約0.07%から約2%、約0.08%から約1.5%、約0.09%から約1%、約0.1%から約0.9%w/wの範囲、約0.001%から約10%、約0.01%から約5%、約0.02%から約4.5%、約0.03%から約4%、約0.04%から約3.5%、約0.05%から約3%、約0.06%から約2.5%、約0.07%から約2%、約0.08%から約1.5%、約0.09%から約1%、約0.1%から約0.9%w/vの範囲、または約0.001%から約10%、約0.01%から約5%、約0.02%から約4.5%、約0.03%から約4%、約0.04%から約3.5%、約0.05%から約3%、約0.06%から約2.5%、約0.07%から約2%、約0.08%から約1.5%、約0.09%から約1%、約0.1%から約0.9%v/vの範囲にある。 In some embodiments, the concentration of ACVR1 inhibitor provided in the pharmaceutical composition is from about 0.001% to about 10%, from about 0.01% to about 5%, from about 0.02% to about 4 5.5%, about 0.03% to about 4%, about 0.04% to about 3.5%, about 0.05% to about 3%, about 0.06% to about 2.5%, about 0 .07% to about 2%, about 0.08% to about 1.5%, about 0.09% to about 1%, about 0.1% to about 0.9% w / w, about 0. 001% to about 10%, about 0.01% to about 5%, about 0.02% to about 4.5%, about 0.03% to about 4%, about 0.04% to about 3.5% , About 0.05% to about 3%, about 0.06% to about 2.5%, about 0.07% to about 2%, about 0.08% to about 1.5%, about 0.09% From about 1%, about 0.1% to about 0.9% w / v, or about 0.001% to about 10%, about 0.01% to about 5%, about 0.02% to about 4.5%, about 0.03% to about 4%, about 0.04% to about 3.5%, about 0.05% to about 3%, about 0.06% to about 2.5%, about It ranges from 0.07% to about 2%, from about 0.08% to about 1.5%, from about 0.09% to about 1%, and from about 0.1% to about 0.9% v / v.
本開示の実施形態で使用されるACVR1阻害剤またはその薬学的に許容される誘導体は、1種またはそれより多種のその他の治療剤の投与と同時に、前に、または後に、投与されてもよい。例えば、第1の治療剤(例えば、ACVR1阻害剤)を投与することができ、十分な期間の後に、第2の治療剤が投与される。そのような実施形態では、第1の治療剤と第2の治療剤の投与の間の期間は、「処置の中断」または「休日」と呼んでもよい。「処置の中断」または「休日」は、処置のサイクル間の期間を指してもよい。一部の実施形態では、そのような処置の中断または休日は、約12時間から約48時間に及ぶ。一部の実施形態では、そのような処置の中断または休日は、約18から約36時間に及ぶ。一部の実施形態では、そのような処置の中断または休日は、約24から約48時間に及ぶ。一部の実施形態では、処置の中断または休日は、約2から約10日間に及ぶ。一部の実施形態では、処置の中断または休日は、約3から約5日間に及ぶ。一部の実施形態では、処置の中断または休日は、約5から約9日間に及ぶ。一部の実施形態では、処置の中断または休日は、約7日である。様々な実施形態では、ACVR1阻害剤は、21日間連続して投与され、その後、7日間の処置の中断または休日が続く。一部の実施形態では、処置の中断または休日は、約30日である。様々な実施形態では、ACVR1阻害剤は、4週連続のサイクルで、サイクルの間、処置の中断または休日なしに、毎週投与される。当業者なら、一般的な技法および知識に基づいて、適切な投薬スケジュールを導き出すことができる。実施形態では、ACVR1阻害剤と、放射線療法および追加の治療剤のうちの1つまたは複数が、順次投与される。 The ACVR1 inhibitor or pharmaceutically acceptable derivative thereof used in the embodiments of the present disclosure may be administered before or after administration of one or more other therapeutic agents. .. For example, a first therapeutic agent (eg, an ACVR1 inhibitor) can be administered, and after a sufficient period of time, the second therapeutic agent is administered. In such embodiments, the period between administration of the first therapeutic agent and the second therapeutic agent may be referred to as "interruption of treatment" or "holiday". “Treatment interruption” or “holiday” may refer to the period between treatment cycles. In some embodiments, interruptions or holidays of such treatment range from about 12 hours to about 48 hours. In some embodiments, interruptions or holidays of such treatment range from about 18 to about 36 hours. In some embodiments, interruptions or holidays of such treatment range from about 24 to about 48 hours. In some embodiments, treatment interruptions or holidays range from about 2 to about 10 days. In some embodiments, treatment interruptions or holidays range from about 3 to about 5 days. In some embodiments, treatment interruptions or holidays range from about 5 to about 9 days. In some embodiments, the discontinuation or holiday of treatment is about 7 days. In various embodiments, the ACVR1 inhibitor is administered for 21 consecutive days, followed by a 7-day discontinuation of treatment or a holiday. In some embodiments, the discontinuation or holiday of treatment is about 30 days. In various embodiments, the ACVR1 inhibitor is administered weekly for a cycle of 4 consecutive weeks, without interruption of treatment or holidays. One of ordinary skill in the art can derive an appropriate dosing schedule based on common techniques and knowledge. In embodiments, the ACVR1 inhibitor and one or more of radiation therapy and additional therapeutic agents are administered sequentially.
(実施例1)
例示的なACVR1阻害剤の試験
野生型および変異ACVR1の阻害
野生型ACVR1、FOP/DIPG変異R206H、およびDIPGに見出される2種の追加の変異キナーゼ(G328VおよびR258G)の、強力な生化学阻害を実証した化合物2を、標準アッセイ手順に従い試験した。
(Example 1)
Experimental ACVR1 Inhibitor Testing Wild-type and Mutant ACVR1 Inhibition Strong biochemical inhibition of wild-type ACVR1, FOP / DIPG mutant R206H, and two additional mutant kinases (G328V and R258G) found in DIPG.
簡単に言うと、カゼインをベースにした基質を調製した。それぞれのACVR1キナーゼを、基質混合物に添加し、温/冷33P−ATP(10μM)を添加して、反応を開始した。次いで活性を、フィルター結合法で測定した。 Simply put, a casein-based substrate was prepared. Each ACVR1 kinase was added to the substrate mixture and hot / cold 33P-ATP (10 μM) was added to initiate the reaction. The activity was then measured by the filter binding method.
キナーゼに対するIC50値を、以下の表3に列挙する。
SMADタンパク質は、TGFβ受容体の下流のシグナル伝達に関わる転写因子である。TGFβ受容体ALK2は、BMP2などの骨形成タンパク質によって活性化されたときにSMADをリン酸化し、それがSMAD核転座、転写開始、およびペプチドホルモンヘプシジンをコードするHAMPのような遺伝子の発現の増加をもたらす。したがって、ALK2を標的とする化合物は、BMP誘発性SMADリン酸化を阻害すべきである。化合物2のin vitro効力を評価するために、HepG2細胞に、様々な濃度の化合物2の存在下または存在しない状態で、ALK2配位子BMP2で刺激を与えた。図1に示される結果は、化合物2によるSMAD1/5リン酸化の明白な濃度依存的阻害(EC50=162nM)を明らかにする。
ヘプシジン発現の阻害
The SMAD protein is a transcription factor involved in signal transduction downstream of the TGFβ receptor. The TGFβ receptor ALK2 phosphorylates SMAD when activated by bone morphogenetic proteins such as BMP2, which expresses genes such as HAMP that encode SMAD nuclear translocation, transcription initiation, and the peptide hormone hepcidin. Bring an increase. Therefore, compounds that target ALK2 should inhibit BMP-induced SMAD phosphorylation. To assess the in vitro potency of
Inhibition of hepcidin expression
化合物2のin vitro効力を評価するために、HepG2細胞を、様々な濃度の化合物2の存在下または存在しない状態で、ALK2配位子BMP−2で刺激し、得られたヘプシジン発現レベルをqPCRにより測定した。図2Aに示される結果は、化合物2によるヘプシジン転写の強力な用量応答性阻害(EC50<1nM)を明らかにする。基礎となるヘプシジン転写に対する影響も、測定し(BMP−2刺激がない状態で)、その結果を図2Bに示す。これらの実験条件下、化合物2は、用量に応答する手法でヘプシジン発現を阻害した(IC50約100nM)。
To assess the in vitro potency of
テレピン油(TO)は、高いヘプシジンレベルを含む、マウスにおける関連のdip検査で急性炎症応答を誘発させることが公知である。本発明者らは、in vivo効力に関して試験化合物を評価するのに、このモデルを使用した(図3)。 Turpentine (TO) is known to elicit an acute inflammatory response on relevant dip tests in mice, including high hepcidin levels. We used this model to evaluate test compounds for in vivo efficacy (Fig. 3).
これらのデータは、化合物2による処置前および/または処置中に評定したときに、ヘプシジンが、価値ある予測的および/または予後的マーカーになり得ることを示唆する。
DIPG細胞系に対する増殖アッセイ
These data suggest that hepcidin can be a valuable predictive and / or prognostic marker when assessed before and / or during treatment with
Proliferation assay for DIPG cell lineage
化合物2を、in vitroでの、ACVR1変異体(G328V変異細胞系)および野性型DIPG細胞系に対する増殖アッセイで試験した。結果は、pSMAD1/5阻害アッセイで観察された値と同様に、変異細胞系に対する選択的活性を実施した。
(実施例2)
ACVR1阻害剤の薬力学的および毒性学試験
(Example 2)
Pharmacodynamic and toxicological studies of ACVR1 inhibitors
DIPGに有効な処置であるために、化合物2は、脳に分布される必要があり、より重要なことは、脳の脳橋領域への非常に深い浸透を実現する必要がある。脳への化合物2の全体的な分布を評定するために、予備的な脳のPK調査を実施した。脳組織濃度は、脳への非常に良好な分布を示す(図4A)。計算されたPKパラメーターを、表4および表5に示す。
さらに、血漿中および脳組織中の化合物2の濃度の比較を行い、その結果は図4Bおよび表6にある。
脳内への化合物2の浸透をさらに探るため、別の調査を実施した。この調査では、マウスに、単回経口用量の化合物2を投与し、脳組織を、投薬の1および2時間後に収集した。化合物2の濃度は、MALDI−TOF質量分析によって脳の組織切片で測定した。
Another study was conducted to further explore the penetration of
マウス脳細胞1グラムを、0.5μgから50μgに及ぶ公知の濃度の化合物2と混合した。4匹のマウスを、100mg/kgで経口投与された化合物2で処置した。試料を、処置の前、ならびに処置の1、2、および4時間後に採取した。次いで化合物2で処置したマウス模倣モデルおよび実際の試料を、一緒に分析した。
1 gram of mouse brain cells was mixed with
追加の薬物動態および組織分布調査を実施して、複数のマウス臓器における化合物2の曝露を包括的に評価した。CD−1オスマウスに、20mg/kg、POを、20%ソルトールおよび80%生理食塩液中に製剤化した化合物2と共に投薬した。血漿および臓器を、9つの時点で収集し、薬物濃度をLC−MS/MSにより決定した。化合物2は、収集された全ての組織で検出可能であり、組織の大部分は、血漿と比較して高い薬物濃度を実証した。結果は、脳および肝臓を含む、臨床上関連ある標的臓器での曝露を確認した(表7)。
化合物2遊離塩基を、ヒトで試験した。化合物2の経口薬物動態を、DIPGの処置に関して1人のヒト患者で決定した。製剤は、遊離塩基化合物2であり、ゼラチンカプセルによって毎日、15日間、その後は20日間の休止期間であった。投薬を、ジュースに溶解することによって再開し、経鼻胃管を介して9日間投与した。公称用量は、240mg/m2(PO)である。血液試料を、4つの時点で収集し、薬物濃度をLC−MS/MSによって決定する。
経口により投薬された化合物2は吸収され、1人のヒトにおいて投薬後24時間で血漿中で検出され、ベースラインへの戻りが長期であった。追加のPKパラメーターを評価した。
Orally administered
化合物2の薬物動態および毒性学を、ラットで試験した。化合物2の静脈内(IV)および経口(PO)薬物動態を、絶食したメスSprague−Dawley(SD)ラットの血漿中で決定した。使用した製剤は、両方の投与経路に関して20%ソルトールおよび80%生理食塩液であった。公称用量は、2mg/kg(IV)および20mg/kg(PO)であった。試料は、8つの時点で採取し、薬物濃度をLC−MS/MSにより決定した。IVおよびPO投与に関する血漿中濃度を図5に示し、このデータからのPK計算を、表8に示す。
非GLP毒性学調査では、SDラットに、400、200、または100mg/kgの、化合物2 HCl塩を20%ソルトールまたはビヒクル対照に溶かしたものを、7日間投与した。
(実施例3)
用量応答の比較試験
In non-GLP toxicology studies, SD rats were administered 400, 200, or 100 mg / kg of
(Example 3)
Dose response controlled trials
7種の細胞系における細胞生存能力に対する化合物2の作用を調査した。50%阻害濃度(IC50)を、種々の試験物濃度でインキュベートした後に、CellTiter−Gloルミネセンス細胞生存能力アッセイを使用して、選択された細胞系で決定した。
The effect of
細胞系を、9つの濃度の化合物2で3回処置し、シスプラチンを参照化合物として使用し、0.5%(v/v)DMSOを、ビヒクル対照として培養培地に添加した。
材料および試薬
一般的な細胞培養試薬およびプラスチック。
96ウェルフラットクリアボトムブラックポリスチレンTC処置マイクロプレート(Cat# 3340、Corning)。
バックシールブラック接着ボトムシール(Cat# 6005189、Perkin Elmer)。
CellTiter−Glo(登録商標)ルミネッセンス細胞生存能力アッセイ(Cat.#.:G7572、Promega.−20℃で保存)
基質は、96ウェルプレートでのアッセイ当たり100μlでの、1,000回のアッセイに十分である。
以下を含む:
・ 1×100mL CellTiter−Glo(登録商標)緩衝液
・ 1×バイアル CellTiter−Glo(登録商標)基質(凍結乾燥)
試薬調製
CellTiter−Glo緩衝液を解凍し、室温に対して平衡にして、その後に使用した。
凍結乾燥したCellTiter−Glo基質を、室温に対して平衡にして、その後に使用した。
適切な体積(100mL)のCellTiter−Glo緩衝液を、CellTiter−Glo基質が入っている琥珀瓶に移して、凍結乾燥された酵素/基質混合物を元に戻した。これにより、CellTiter−Glo試薬が形成された。
内容物を穏やかに撹拌し、渦を形成し、または反転させることにより混合して、均質な溶液を得た。
最大半量阻害濃度IC50の決定
対数的成長期間中に細胞を収集し、カウントスターを使用して細胞数をカウントした。
細胞濃度を、培養培地で3.33×104細胞/mLに調節した。
90μLの細胞懸濁液を、3つの96ウェルプレート(プレートAおよびB)に添加し、最終細胞密度を3×103細胞/ウェルにした(細胞濃度は、データベースまたは密度最適化アッセイに従い調節される)。
T0読取りのプレートに関して:
10μLの培養培地を、T0読取りのためにプレートAの各ウェルに添加した。
プレートおよびその内容物を、室温でおよそ30分間平衡にした。
黒色ステッカーをプレートの底部に配置して、光を遮断した。
100μLのCellTiter−Gloを、各ウェルに添加した。
内容物を、2分間、オービタルシェーカーで混合して、細胞溶解物を誘発させた。
プレートを室温で10分間インキュベートさせて、ルミネッセンスシグナルを安定化させた。
ルミネッセンス(T0)を、EnVisionマルチ標識リーダーを使用して記録した。
試験読取りのプレートに関して:
10×溶液(作用濃度:10μM)の試験物を、培地中に、3.16倍の連続希釈にして、10の用量レベルを実現した。
試験物および参照対照の両方の10μL(10×)薬物溶液を、プレートBの各ウェルに分取した(各薬物濃度ごとに3回)(培養培地中の溶媒最終濃度:0.5%[v/v])。
試験プレートBを72時間、5%CO2を含む37℃の加湿されたインキュベーター内でインキュベートし、次いでCTGアッセイを用いて測定した。
プレートおよびその内容物を、室温でおよそ30分間平衡にした。
黒色ステッカーをプレートの底部に配置して、光を遮断した。
100μLのCellTiter−Gloを、各ウェルに添加した。
内容物を2分間、オービタルシェーカー上で混合して、細胞溶解物を誘発させた。
プレートを、室温で10分間インキュベートして、ルミネッセンスシグナルを安定化させた。
ルミネッセンスを記録した。
データ解析
The cell line was treated 3 times with 9 concentrations of
Materials and Reagents Common cell culture reagents and plastics.
96-well flat clear bottom black polystyrene TC-treated microplate (Cat # 3340, Corning).
Back Seal Black Adhesive Bottom Seal (Cat # 6005189, Perkin Elmer).
CellTiter-Glo® Luminescence Cell Viability Assay (Cat. # .: G7572, Promega. Stored at -20 ° C)
The substrate is sufficient for 1,000 assays in 100 μl per assay in 96-well plates.
Including:
1 × 100 mL CellTiter-Glo® buffer ・ 1 × vial CellTiter-Glo® substrate (lyophilized)
Reagent Preparation CellTiter-Glo buffer was thawed, equilibrated to room temperature and then used.
The lyophilized CellTiter-Glo substrate was equilibrated to room temperature and then used.
The appropriate volume (100 mL) of CellTiter-Glo buffer was transferred to an amber bottle containing the CellTiter-Glo substrate and the lyophilized enzyme / substrate mixture was replaced. As a result, the CellTiter-Glo reagent was formed.
The contents were mixed by gently stirring to form a vortex or inversion to give a homogeneous solution.
Determining Maximum Half Inhibition Concentration IC 50 Cells were harvested during the logarithmic growth period and the number of cells was counted using a count star.
The cell concentration was adjusted to 3.33 × 10 4 cells / mL in culture medium.
90 μL of cell suspension was added to three 96-well plates (plates A and B) to a final cell density of 3 × 10 3 cells / well (cell concentration adjusted according to database or density optimization assay). NS).
Regarding the T0 reading plate:
10 μL of culture medium was added to each well of plate A for T0 reading.
The plate and its contents were equilibrated at room temperature for approximately 30 minutes.
A black sticker was placed on the bottom of the plate to block the light.
100 μL of CellTiter-Glo was added to each well.
The contents were mixed on an orbital shaker for 2 minutes to induce cytolysis.
The plate was incubated at room temperature for 10 minutes to stabilize the luminescence signal.
Luminescence (T0) was recorded using an EnVision multi-label reader.
Regarding the test reading plate:
A 10 × solution (concentration of action: 10 μM) test was serially diluted 3.16 times in medium to achieve 10 dose levels.
10 μL (10 ×) drug solutions of both the test and reference controls were dispensed into each well of plate B (3 times for each drug concentration) (final solvent concentration in culture medium: 0.5% [v). / V]).
Test plate B was incubated for 72 hours in a humidified incubator at 37 ° C. containing 5% CO 2 and then measured using the CTG assay.
The plate and its contents were equilibrated at room temperature for approximately 30 minutes.
A black sticker was placed on the bottom of the plate to block the light.
100 μL of CellTiter-Glo was added to each well.
The contents were mixed on an orbital shaker for 2 minutes to induce cytolysis.
The plate was incubated at room temperature for 10 minutes to stabilize the luminescence signal.
Recorded luminescence.
Data analysis
データを、GraphPad Prism 5.0を使用して、グラフ表示した。 The data were graphed using GraphPad Prism 5.0.
絶対IC50(EC50)を計算するために、用量応答曲線に、非線形回帰モデルを使用してS字状用量応答を当てはめた。生存率を計算するための式を以下に示し、絶対IC50(EC50)を、GraphPad Prism 5.0によって作成された用量応答曲線に従って計算した。 To calculate the absolute IC 50 (EC 50 ), the dose response curve was fitted with an S-shaped dose response using a non-linear regression model. The formula for calculating the survival rate is shown below, and the absolute IC 50 (EC 50 ) was calculated according to the dose response curve created by GraphPad Prism 5.0.
生存率(%)=(Lum試験物−Lum培地対照)/(Lum非処置−Lum培地対照)×100%。 Survival rate (%) = (Lum test-Lum medium control) / (Lum untreated-Lum medium control) x 100%.
図6Aは、IGR−OV1細胞系に関する用量応答曲線を示す。IGR−OV1は、ACVR1変異体(P455A)細胞系である。図6Bは、IGR−OV1(変異体ACVR1)細胞系の、C−33a、SK−OV−3、AN3−CA、CaSki、HeLa、およびHEC−1−A(野生型ACVR1)に対する結果の比較を示す。
(実施例4)
in vivoジャーカット異種移植モデル
FIG. 6A shows the dose response curve for the IGR-OV1 cell line. IGR-OV1 is an ACVR1 mutant (P455A) cell line. FIG. 6B compares the results of the IGR-OV1 (mutant ACVR1) cell line against C-33a, SK-OV-3, AN3-CA, CaSki, HeLa, and HEC-1-A (wild-type ACVR1). show.
(Example 4)
in vivo jar cut xenotransplantation model
異種移植モデルにおける腫瘍体積に対する化合物2の影響を調査した。動物を下記の条件で収容した:
動物を、調整された温度および明/暗サイクルで、換気されたマウスケージシステム(Animal Care Systems、Centennial、CO)に収容した。
温度:22.7〜23.9℃
湿度:10%
寝藁:トウモロコシの穂軸(Animal Care Systems、1/4” 照射)
食餌:全体照射、大豆タンパク質なし、押出し成型済みのげっ歯類用食餌(Envigo、Cat.No:2920X.CS)
水:RO水、随意
The effect of
Animals were housed in a ventilated mouse cage system (Animal Care Systems, Centennial, CO) at regulated temperature and light / dark cycles.
Temperature: 22.7 to 23.9 ° C
Humidity: 10%
Sleeping straw: Corn cob (Animal Care Systems, 1/4 "irradiation)
Diet: Whole irradiation, no soy protein, extruded rodent diet (Envigo, Cat.No: 2920X.CS)
Water: RO water, voluntary
ジャーカット細胞を、10%FBSが補われたRPMI1640培地中で培養し、5%CO2と共に37℃でインキュベートした。細胞を、週当たり2回または必要に応じて継代培養した。 Jarkat cells were cultured in RPMI 1640 medium supplemented with 10% FBS and incubated with 5% CO 2 at 37 ° C. Cells were subcultured twice weekly or as needed.
細胞を、無血清培地で洗浄し、次いで1:1の無血清培地:マトリゲル中に懸濁させた。注射体積は200μLであった。マウスの後側腹に、1×107細胞/マウスを皮下注射した。 Cells were washed with serum-free medium and then suspended in 1: 1 serum-free medium: Matrigel. The injection volume was 200 μL. The flank after the mice, the 1 × 10 7 cells / mouse were injected subcutaneously.
動物腫瘍体積および体重を測定し、毎週2回記録した。腫瘍体積が>100mm3に達したら、処置を開始した。マウスを、腫瘍体積によって層別化して、類似の平均出発腫瘍体積を各動物ケージごとに得、次いで各ケージをランダムに選択して処置群に配置した。 Animal tumor volume and body weight were measured and recorded twice weekly. Treatment was initiated when the tumor volume reached> 100 mm 3. Mice were stratified by tumor volume to obtain a similar average starting tumor volume for each animal cage, and then each cage was randomly selected and placed in the treatment group.
腫瘍体積測定を、長さ(最長直径)および幅(最長直径に直交する)の両方を測定するカリパスを使用して毎週2回行った。腫瘍体積は、L×W×W/2として計算した。 Tumor volume measurements were performed twice weekly using calipers that measure both length (longest diameter) and width (perpendicular to the longest diameter). Tumor volume was calculated as L × W × W / 2.
化合物2のHCl塩50mg/kg(Qd)を、20%ソルトール、80%デキストロース(5%デキストロース溶液)中で製剤化して、マウスに投与した。結果を図7に示す。
(実施例5)
成人がんにおけるACVR1変異の分析
50 mg / kg (Qd) of the HCl salt of
(Example 5)
Analysis of ACVR1 mutations in adult cancer
ACVR1変異の遺伝子構造を分析して、どの変異が、構造(I)の化合物により処置からの利益の前兆となり得るかを評価した。公的に利用可能な配列データベースおよび分析ツール、cBioPortal.orgを使用して、がん患者の組織におけるACVR1変異を分析した。結果を、様々ながんに関連した文献のACVR1の公知の変異または機能獲得活性と比較した。複数の変異がACVR1に見出された。FOPコミュニティーは、ACVR1の異常な活性化を推進させる可能性がある変異を特定し理解するために、広範囲にわたる作業を行った。これらの変異は、L196P、R206H、Q207E、R258S、G328E/R、およびG356Dを含む。成人がんで特定されるACVR1変異は、公知のFOP変異のほとんどを含むが、未知の有意性のより少ない「ホットスポット」および多くの変化(alterations)を持つ、それらのACVR1変異プロファイルのさらなる多様性も実証する。複数のがんタイプにおけるこれらのまれな変異の調査の結果を、本明細書に提示する。 The genetic structure of the ACVR1 mutation was analyzed to assess which mutations could signal benefits from treatment with compounds of structure (I). A publicly available sequence database and analysis tool, cBioPortal. The org was used to analyze ACVR1 mutations in the tissues of cancer patients. Results were compared to known mutations or gain-of-function activity of ACVR1 in various cancer-related literature. Multiple mutations were found in ACVR1. The FOP community has undertaken extensive work to identify and understand mutations that may promote aberrant activation of ACVR1. These mutations include L196P, R206H, Q207E, R258S, G328E / R, and G356D. The ACVR1 mutations identified in adult cancer include most of the known FOP mutations, but have a greater variety of their ACVR1 mutation profiles with less "hot spots" and many alterations of unknown significance. Also demonstrate. The results of investigating these rare mutations in multiple cancer types are presented herein.
生化学的調査は、細胞内ドメインでの変異が、ACVR1/ALK2キナーゼ活性に対する活性化作用を有する可能性があることを示す。公知の変異の多くは、受容体配位子に応答して受容体の活性を高める。図8に示されるように、化合物2は、様々な変異を有するALK2受容体の阻害において有効であった。
Biochemical studies show that mutations in the intracellular domain may have an activating effect on ACVR1 / ALK2 kinase activity. Many of the known mutations increase the activity of the receptor in response to the receptor ligand. As shown in FIG. 8,
cBioPortal.orgブラウザーを使用して、The Cancer Genome Atlas(TCGA)汎がんおよびMemorial Sloan Kettering(MSK)IMPACTデータベースを、ACVR1における変異に関してプローブした。図9Aは、ACVR1変異の分布を示す、ロリポップダイアグラムを示し;ALK2活性の調整で役割を果たすと考えられる、グリシン−セリン調整ドメインおよびタンパク質キナーゼドメインにおけるいくつかの複製変異(ホットスポット)が含まれる。図9Bは、種々の成人腫瘍タイプ全体にわたって見出される、いくつかのタイプの染色体異常(変異、増幅、欠失、および融合)の分布を示す。図9Cは、ACVR1に遺伝子異常を持つ対象が、ACVR1の変化(ACVR1 alterations)がない対象と比較して、6か月短い生存時間を有することを示す、Kaplan−Meier曲線を示す。分析は、ACVR1の変異および融合に焦点を当てた。 cBioPortal. Using an org browser, The Cancer Genome Atlas (TCGA) pan-cancer and Memorial Sloan Kettering (MSK) IMPACT databases were probed for mutations in ACVR1. FIG. 9A shows a lollipop diagram showing the distribution of ACVR1 mutations; it contains several replication mutations (hotspots) in the glycine-serine regulatory domain and the protein kinase domain that are thought to play a role in regulating ALK2 activity. .. FIG. 9B shows the distribution of several types of chromosomal abnormalities (mutations, amplifications, deletions, and fusions) found across various adult tumor types. FIG. 9C shows a Kaplan-Meier curve showing that subjects with genetic abnormalities in ACVR1 have a 6-month shorter survival time compared to subjects without ACVR1 alterations. The analysis focused on mutation and fusion of ACVR1.
表9に示されるように、MSK ImpactおよびTCGAデータベース統計は、21289名の患者の集団から147名の固有対象に、ACVR1の全変異率0.69%が見出されたことを示す。
腫瘍タイプによる変異の分析は、ACVR1変異を有する147名の固有患者のうち、34%が婦人科系がんを有し、18%が胃腸がんを有し、14%が胸部がんを有し、13%が黒色腫を有し、6%が泌尿生殖器がんを有し、5%が神経がんを有し、4%が頭頚部がんを有し、3%がその他のがんを有し、2%が乳がんを有することを示した。 Analysis of mutations by tumor type showed that of the 147 endemic patients with ACVR1 mutations, 34% had gynecologic cancer, 18% had gastrointestinal cancer, and 14% had breast cancer. However, 13% have melanoma, 6% have urogenital cancer, 5% have neurological cancer, 4% have head and neck cancer, and 3% have other cancers. And 2% had breast cancer.
表10は、アミノ酸の位置によって分類される上位の変異頻度を示す。
合計で117の固有の変異が、使用したデータベースの腫瘍タイプ全体にわたって見出された。FOPおよびDIPGに関連したカノニカル変異の多くは、R206H、R375C/H、G356D、G328W/E/V/R、R258G/Wなどの様々な成人がんの中の同じ位置に見出された最も一般的な変異の中にあった。未知の有意性を持つ多くの新規な変異も検出され、がんにおいて役割を果たす可能性がある追加の遺伝的変化を示唆している。Mutation Assessor、SIFT、およびPolyPhen2アルゴリズムを使用したcBioPortal.orgからの影響評価を実施して、タンパク質機能に対する変異の影響を予測した。 A total of 117 unique mutations were found across the tumor types in the database used. Many of the canonical mutations associated with FOP and DIPG are the most common found at the same location in various adult cancers such as R206H, R375C / H, G356D, G328W / E / V / R, R258G / W. Was in a typical mutation. Many novel mutations of unknown significance have also been detected, suggesting additional genetic changes that may play a role in cancer. CBioPortal. Using the Mutation Assessor, SIFT, and PolyPhen2 algorithms. Impact assessment from org was performed to predict the impact of mutations on protein function.
表11は、有害な影響があることがSIFTアルゴリズムにより予測された、データセットで特定されたACVR1変異を示す。SIFTアルゴリズムは、変異の55%が有害であり、変異の35%が許容され、変異の10%が未確認の影響を有すると予測した。
表12は、損傷を与える可能性があることまたは損傷を与えそうであるという影響があることがPolyphen2アルゴリズムにより予測された、データセットで特定されたACVR1変異を示す。Polyphen2アルゴリズムは、変異の48%が損傷を与えそうである(probably damaging)という影響があり、変異の10%が損傷を与える可能性がある(possibly damaging)という影響があり、変異の31%が良性であり、変異の11%が未確認の影響を有すると予測した。
表13は、高い、中程度の、または低い影響を与えることがMutation Assessorアルゴリズムにより予測された、データセットで特定されたACVR1変異を示す。Mutation Assessorアルゴリズムは、変異の8%が高い影響を与え、変異の28%が中程度の影響を与え、変異の35%が低い影響を与え、変異の19%が中立的影響を与え、変異の10%が定義されていない影響を与えることを予測した。
これらのアルゴリズムは、カノニカル変異および固有の変異の両方が、タンパク質機能の影響を及ぼす可能性があることを示す。SIFTアルゴリズムを使用して、本発明者らの調査で見出された全てのカノニカル変異は、有害であると分類され、これはACVR1遺伝子の変異の活性化を予測するのに有用なツールになり得ることを示唆している。ある場合には、特定の部位での種々のアミノ酸置換は、変異の影響に対して種々の作用を有していた。これらの予測的アルゴリズムのさらなる調査は、保証されている。 These algorithms show that both canonical and endemic mutations can affect protein function. Using the SIFT algorithm, all canonical mutations found in our study were classified as detrimental, which would be a useful tool for predicting the activation of mutations in the ACVR1 gene. Suggests to get. In some cases, different amino acid substitutions at specific sites had different effects on the effects of mutations. Further investigation of these predictive algorithms is guaranteed.
次に、特定のがんの亜型を、様々な解剖学的部位に関する最も一般的な変異または公知の活性化変異の存在に関して分析し、SIFT影響スコアを、がん亜型により算出した。 Specific cancer subtypes were then analyzed for the presence of the most common mutations or known activation mutations at various anatomical sites, and SIFT impact scores were calculated by cancer subtype.
婦人科系がん。婦人科系がんは、ほとんどの変異(43)の原因であり、FOPおよびDIPGに見出されるカノニカル変異の多くを含んでいた。SIFTアルゴリズムは、ACVR1変異および婦人科系がんを有する患者のうち(n=50)、患者の45%が有害な影響を持つ変異を有し、40%が許容される影響を持つ変異を有し、15%が定義されていない影響を持つ変異を有することを予測した。婦人科系がんのサブセットで特定された、公知の活性化および/または一般の変異を、表14に示す。
胃腸がん。SIFTアルゴリズムは、ACVR1変異および胃腸がんを有する患者のうち(n=27)、患者の52%が有害な影響を与える変異を有し、40%が許容される影響を与える変異を有し、8%が、まだ定義されていない影響を与える変異を有することを予測した。胃腸がん下位集合で特定された公知の活性化および/または一般的な変異を、表15に示す。
胸部がん。SIFTアルゴリズムは、ACVR1変異および胸部がんを有する患者の中で(n=21)、変異の76%が有害な影響を与え、14%が許容される影響を与え、10%がまだ定義されていない影響を与えることを予測した。胸部がんで特定された公知の活性化および/または一般的な変異を、表16に示す。
黒色腫。SIFTアルゴリズムは、ACVR1変異および黒色腫を有する患者のうち(n=19)、患者の58%が有害な影響を与える変異を有し、32%が許容される影響を与える変異を有し、11%がまだ定義されていない影響を与える変異を有することを予測した。黒色腫の下位集合で特定された公知の活性化および/または一般的な変異を、表17に示す。
神経がん。神経がんは、カノニカル変異の高い出現率(30%)を実証した。SIFTアルゴリズムは、ACVR1変異および神経がんを有する患者のうち(n=7)、患者の100%が有害な影響を与える変異を有することを予測した。神経がんの下位集合で特定された公知の活性化および/または一般的な変異を、表18に示す。
進行固形腫瘍を持つ患者に経口投与される式2の化合物の、ヒト初回調査のための臨床試験調査の設計
Nerve cancer. Neurocancer demonstrated a high incidence (30%) of canonical mutations. The SIFT algorithm predicted that of patients with ACVR1 mutations and neurocancer (n = 7), 100% of the patients would have a detrimental mutation. Known activations and / or common mutations identified in the subgroup of neuronal carcinomas are shown in Table 18.
Design of clinical trial study for initial human study of compound of
試験のIa相部分は、進行または漸進固形腫瘍を持つ患者に関して、3+3修正用量漸増を含む。投薬は、体表面積に基づいて計算され、投薬は、28日の処置サイクルで連続21日間、行われる。最大20名の対象が登録される。アーカイブ組織は、試験のIa相部分に登録された患者の遺伝子検査が要求され、次世代配列決定がアーカイブ組織に関して行われて、ACVR1変異状態を決定する。 The Phase Ia portion of the study comprises a 3 + 3 modified dose escalation for patients with advanced or progressive solid tumors. Dosing is calculated based on body surface area, with dosing taking place for 21 consecutive days in a 28-day treatment cycle. Up to 20 subjects are registered. The archived tissue is required to be genetically tested for patients enrolled in the Phase Ia portion of the study, and next-generation sequencing is performed on the archived tissue to determine the ACVR1 mutant state.
最大耐量の決定の後、追加の対象を試験のIb相部分に登録する。登録された患者は、進行転移または漸進固形腫瘍の、組織的に確認された診断を有する。登録された患者は、それらの状態に臨床上の利益を提供することが公知である確立された治療に対して、効果がなくまたは不寛容にもなる。Ib相部分では、投薬は、体表面積に基づいて計算され、投薬は、28日の処置サイクルで連続21日にわたり行われる。アーカイブ組織には、試験のIb相部分に登録された患者の遺伝子検査が要求され、次世代配列決定が、ACVR1変異状態を決定するのにアーカイブ組織で行われる。 After determining the maximum tolerated dose, additional subjects are enrolled in the Ib phase portion of the test. Enrolled patients have a systematically confirmed diagnosis of advanced metastases or progressive solid tumors. Enrolled patients also become ineffective or intolerant of established treatments that are known to provide clinical benefits to those conditions. In the Ib phase portion, dosing is calculated based on body surface area, with dosing taking place over 21 consecutive days in a 28-day treatment cycle. The archive tissue is required to be genetically tested for patients enrolled in the Ib phase portion of the study, and next-generation sequencing is performed on the archive tissue to determine the ACVR1 mutation status.
代替レジメンは、化合物2の効力および半減期に基づき、週に1回である。他の代替レジメンは、週当たり2用量、週当たり3用量、週当たり4用量、週当たり5用量、または週当たり6用量である。そのようなレジメンのそれぞれは、4週で1サイクルの投薬を含む。したがって1つのレジメンは、4週サイクルにわたって毎週1回であり、休日なしで毎週の連続投薬をもたらし得る。代替のサイクルは、1またはそれより多くの休日を含み、例えば投薬が3週間でありかつ休日が1週間、投薬が2週間でありかつ休日が2週間、投薬が1週間でありかつ休日が3週間、あるいは投薬の週と休日の週が交互にあり、即ち投薬が1週間でありかつ休日が1週間であり、または投薬が隔週である。これらの実施形態の全てに関し、化合物2の用量は、平準であっても上昇してもよい。ある開始用量は、約24から30mgの範囲にあってもよい。ある開始用量、または上昇用量は、約48から60mgの範囲にあってもよい。追加の上昇用量は、約72から90mgおよび約96から120mgの範囲であってもよい。一実施形態では、開始用量は、毎週約60mgであってもよく、この値は維持されるか、または毎週約90mgまで上昇させる。約90mgの1週用量は、維持されてもよく、毎週約120mgまで上昇させてもよい。
(実施例7)
経口固体製剤
The alternative regimen is once a week based on the potency and half-life of
(Example 7)
Oral solid preparation
化合物2の塩酸塩を、3つの経口用量力価(5、25、および125mg用量[遊離塩基に対する])に製剤化した。活性医薬成分の増大した量を、3種の類似したブレンドに製剤化した。表19を参照されたい。生成物を、ブレンド中に一般的な賦形剤を使用して、即時放出用に製剤化した。薬物を、#3、硬質ゼラチンカプセルに入れた。
経口液体製剤
Hydrochloride of
Oral liquid preparation
DIPG療法に向けた小児集団を考慮して、例えば錠剤またはカプセル形態ではなく、経口液体製剤が化合物2用に開発された。化合物2は苦味が際立ち、したがって液体製剤は、かなりの分析および評価を必要とした。
Considering the pediatric population for DIPG therapy, oral liquid formulations have been developed for
まず、表20に記載されるように、様々な賦形剤を、緩衝剤として、保存条件下でスクリーニングした(初期、60℃ 1、2、4W、40℃ 2、4W、5、25℃ 4W)。
賦形剤を、沈殿および分解不純物に関してスクリーニングした。沈殿物に関し、pH3の条件では変化がほとんど観察されなかったが、沈殿は、pH4およびpH5の条件で多くの試料に見出された。不純物に関し、分解はpH依存性であるように見え、pH5およびpH4は、pH3よりも多くの分解を引き起こした。最も少ない分解は、pH3で、10mMリンゴ酸緩衝剤において観察された。リンゴ酸緩衝剤は、他の試験された緩衝剤と比較して、より化学的におよび物理化学的に安定であった。
Excipients were screened for precipitation and degrading impurities. For precipitates, little change was observed under
次に、表21に記載されるように、様々な賦形剤を、保存条件下で保存剤としてスクリーニングし(初期、60℃ 1、2、4W、40℃ 2、4W、25℃ 4W、5℃ 4W)、各試料のpHは、化合物2の溶解後にNaOHまたはHClで、目標pHに調節した。
賦形剤を、沈殿および分解不純物に関してスクリーニングした。沈殿は、クエン酸およびリンゴ酸緩衝剤中に安息香酸ナトリウムを含有する試料で見出された。分解に関し、保存剤は大きな差を実証しなかったが、プロピレングリコールは、最小限の分解をもたらすようであった。安息香酸は、酸性条件下でのその化学的安定性および保存効果に基づいて、保存剤の中で最も適切と考えられた。 Excipients were screened for precipitation and degrading impurities. Precipitation was found in samples containing sodium benzoate in citric acid and malate buffers. In terms of degradation, preservatives did not demonstrate significant differences, but propylene glycol appeared to result in minimal degradation. Benzoic acid was considered to be the most suitable preservative, based on its chemical stability and preservative effect under acidic conditions.
次に、表22に記載されるように、様々な賦形剤を、保存条件下で矯味剤としてスクリーニングし(初期、60℃ 1、2、4W、40℃ 2、4W、25℃ 4W、5℃ 4W)、各試料のpHは、化合物2の溶解後にNaOHまたはHClで目標pHに調節した。
賦形剤を、沈殿および分解不純物に関してスクリーニングした。沈殿に関し、グリセリンは最小限の沈殿をもたらしたが、賦形剤全体は、ほぼ類似しているようであった。分解に関し、グリセリンは最小限の分解をもたらしたが、賦形剤全体は、ほぼ類似しているようであった。スクリーニングの結果は、好ましい薬剤を生成せず、したがってin vitro官能調査は、矯味剤の全ての間で行った。味覚センサーに関する詳細な情報は、Insent(商標)の製造業者、Intelligent Sensor Technology,Inc.から、例えばhttp://www.insent.co.jp/en/index.htmlから見出すことができる。TaharaおよびTokoによって刊行された概説論文、Electronic Tongues - A Review, IEEE Sensors Journal, Volume 13, No. 8, pp 3001-11, 2013年8月を、さらに参照することができる。これらのそれぞれは、味覚センサー技術に関して、参照により本明細書に組み込まれる。
Excipients were screened for precipitation and degrading impurities. With respect to precipitation, glycerin resulted in minimal precipitation, but the overall excipients appeared to be similar. In terms of degradation, glycerin resulted in minimal degradation, but the overall excipients appeared to be similar. Screening results did not produce the preferred drug, so in vitro sensory tests were performed among all of the flavoring agents. For more information on taste sensors, see the manufacturer of Insent ™, Intelligent Sensor Technology, Inc. From, for example, http: // www. insent. co. jp / en / index. It can be found in html. A review article published by Tahara and Toko, Electronic Tongues --A Review, IEEE Sensors Journal,
表23は、in vitro官能調査で調査された試験試料を提供し、「相対値」および「CPA値」により苦味を推定し、次いで試験試料のそれぞれに関してキニン濃度に変換した。
試験の結果を図10に提示する。キニン閾値は約0.03mM(3.00E−02)である。スクラロースなどの人工甘味料による苦味のマスキングは、「官能性マスキング」であると考えられ、したがってセンサー出力が不十分である。しかし、溶液中の人工甘味料濃度が公知である場合、どの程度苦味が抑制されるかに関する予測値およびその時の実際のセンサー出力を、計算することが可能である。センサー応答に基づけば、15%HP−β−CD/0.1%スクラロースは矯味能力を有する。 The results of the test are presented in FIG. The kinin threshold is about 0.03 mM (3.00E-02). Masking bitterness with artificial sweeteners such as sucralose is considered to be "functional masking" and therefore the sensor output is inadequate. However, if the artificial sweetener concentration in the solution is known, it is possible to calculate the predicted value of how much bitterness is suppressed and the actual sensor output at that time. Based on sensor response, 15% HP-β-CD / 0.1% sucralose has a flavoring ability.
表24は、in vitro官能性調査における、第2の組の試験試料調査を提供する。
試験の結果を図11に提示する。センサー応答に基づけば、15%HP−β−CD/0.1%スクラロースは、矯味能力を有する。 The results of the test are presented in FIG. Based on sensor response, 15% HP-β-CD / 0.1% sucralose has a flavoring ability.
保存有効性の調査は、そのようなプロトコールに関して参照により本明細書に組み込まれるUSP40を参照して実施した。全てのプロトタイプ製剤は、カテゴリー3の生成物に必要とされる抗菌剤の有効性の基準に合格した。しかし、Aspergillus brasiliensisの生菌数がモニタリングされ、したがって追加の製剤調査および保存試験を行った。この場合も、USP40を参照する。製剤を、表25に記載する。
全てのプロトタイプ製剤は、カテゴリー3の生成物に必要とされる抗菌剤の有効性の14日および28日の基準に合格した。保存有効性は、pH依存的であることが見出された。0.2%安息香酸およびpH2を有する製剤は、より良好な保存効果をもたらすと考えられる。
All prototype formulations passed the 14-day and 28-day criteria for antimicrobial efficacy required for
経口液体製剤の一実施形態を、表26に記述する。
経口液体製剤に関する代替の実施形態を、表27に記載する。
塩評価および多形スクリーン
Alternative embodiments for oral liquid formulations are shown in Table 27.
Salt rating and polymorphic screen
塩スクリーニングは、塩形態が遊離塩基形態に勝る利益を提供し得るか否かを評定するために、塩基性化合物、化合物2を評価した。特定された任意の適切な塩候補では、予備多形スクリーニングを、その多形リスクを評価するために行うと考えられる。
概要
The salt screening evaluated the basic compound,
Overview
塩スクリーニングは、10種の酸(2モル比のHCl)および3種の溶媒系を使用して、33の条件下で行った。スクリーニング試験の全てから、合計で12の結晶質ヒットを単離し、X線粉末回折(XRPD)、熱重量分析(TGA)、および示差走査熱量測定(DSC)によって特徴付けた。塩ヒットの化学量論比は、プロトン核磁気共鳴(1H NMR)または高性能液体クロマトグラフィー(HPLC)をイオンクロマトグラフィー(IC)と組み合わせたものによって決定された。ヒットの物理的性質に基づいて、無水HCl塩形態Aおよびフマル酸塩形態Aを、評価用の塩の先例として選択した。 Salt screening was performed under 33 conditions using 10 acids (2 mol ratio HCl) and 3 solvent systems. A total of 12 crystalline hits were isolated from all of the screening tests and characterized by X-ray powder diffraction (XRPD), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC). The chemical ratio of salt hits was determined by a combination of proton nuclear magnetic resonance (1 H NMR) or high performance liquid chromatography (HPLC) with ion chromatography (IC). Based on the physical properties of the hits, anhydrous HCl salt form A and fumarate form A were selected as precedents for the salts for evaluation.
HCl塩形態Aおよびフマル酸塩形態Aの塩の先例を、300mgの規模に調製し、吸湿性、pH2、5、および7の緩衝剤中の動力学的溶解度、および40℃/75%RHの下での固体状態溶解度を、1週間評価した。評価結果により示されるように(遊離塩基形態Aを参照として使用して):
a) 遊離塩基形態A、HCl塩形態A、およびフマル酸塩形態Aは全て、DVS試験後に形態変化のない状態でわずかに吸湿性があった;
b) 遊離塩基形態Aと比較すると、HCl塩形態Aは、pH2、5、および7の緩衝剤中で、増大した溶解度を示し、不均化が、pH7の緩衝剤中で観察された。フマル酸塩形態Aは、形態変化が観察された状態で、pH2および5の緩衝剤中で低下した溶解度を示したが、増大した溶解度は、形態変化がない状態で、pH7の緩衝剤中で観察された;そして
c) 遊離塩基形態A、HCl塩形態A、およびフマル酸塩形態Aは全て、40℃/75%RHの下で1週間、良好な物理化学的性質を示した。特徴付けおよび評価の結果を、表28にまとめる。
The precedents for the salts of HCl salt form A and fumarate form A were prepared to a scale of 300 mg, hygroscopic, kinetic solubility in buffers at
a) Free base form A, HCl salt form A, and fumarate form A were all slightly hygroscopic in the absence of morphological changes after the DVS test;
b) Compared to free base form A, HCl salt form A showed increased solubility in buffers of
収集された結果に基づけば、HCl塩形態Aは、好ましい候補形態である。したがって、多形評価調査は、HCl塩(モノ)で行った。HCl塩形態Aで開始して、予備多形スクリーニングを、スラリー変換、蒸発、低速冷却、および逆溶剤添加の種々の方法を使用して、32の条件下で実施した。XRPD比較に基づき、HCl塩形態Aとは別に、5つの追加の結晶質形態(HCl塩形態C〜G)がスクリーニングから得られ、TGAおよびDSCによって特徴付けた。調査結果に基づき、HCl塩形態AおよびCは、それぞれ無水物および水和物であると推測された。蒸発によるHCl塩形態DおよびEの再調製は、それぞれHCl塩形態C+DおよびC+Eを含有する混合物をもたらした。混合物の1H NMRおよび加熱実験の結果、HCl塩形態DおよびEは、水和物であってもよい。限られた量の材料に起因して、潜在的なHCl塩形態FおよびGは特定されなかった。塩および多形スクリーニングの両方から得られたHCl塩形態の詳細な特徴付けデータおよびXRPDオーバーレイを、表29A、図12、ならびに図13Aおよび13Bにまとめる。図12は、HCl塩形態AのXRPDパターンを示す。図13Aは、HCl塩結晶形態A、C、D、およびEのオーバーレイを示す。図13Bは、形態B、F、およびGを示す。各形態を「タイプ」と呼んでもよく、それらの用語は同義で使用される。
*:残された非常に少量の遊離塩基形態Aによって引き起こされ得る
**: 25℃/80%RHでの水摂取に基づく:非常に吸湿性がある−>15%、吸湿性がある−2〜15%、僅かに吸湿性がある−0.2〜2%、非吸湿性−<0.2%
−−:試料の限られた量に起因して測定されていない。
Based on the results collected, HCl salt form A is the preferred candidate form. Therefore, the polymorphism evaluation survey was conducted with the HCl salt (mono). Starting with HCl salt form A, pre-polymorph screening was performed under 32 conditions using various methods of slurry conversion, evaporation, slow cooling, and backsolvent addition. Based on XRPD comparisons, apart from HCl salt form A, five additional crystalline forms (HCl salt forms C to G) were obtained from screening and characterized by TGA and DSC. Based on the findings, the HCl salt forms A and C were presumed to be anhydrides and hydrates, respectively. Repreparation of the HCl salt forms D and E by evaporation resulted in a mixture containing the HCl salt forms C + D and C + E, respectively. As a result of 1 H NMR and heating experiments of the mixture, the HCl salt forms D and E may be hydrates. Due to the limited amount of material, potential HCl salt forms F and G were not identified. Detailed characterization data and XRPD overlays for HCl salt morphology obtained from both salt and polymorphic screening are summarized in Tables 29A, 12 and 13A and 13B. FIG. 12 shows the XRPD pattern of HCl salt form A. FIG. 13A shows overlays of HCl salt crystal forms A, C, D, and E. FIG. 13B shows forms B, F, and G. Each form may be referred to as a "type" and the terms are used synonymously.
−−: Not measured due to the limited amount of sample.
記述されるように、図12は、HCl塩形態AのXRPDパターンを示す。形態Aに関するXRPDの表形式は、表29Bにあるとおりであり、誤差範囲±は、当業者に理解されるように約0.2°2θと記述される:
化合物2の予備塩スクリーニングおよびHCl塩(モノ)の多形スクリーニングの結果、モノHCl塩形態Aは、さらなる開発に好ましい候補である。
詳細:塩スクリーニングおよび塩の先例の再調製
As a result of the preliminary salt screening of
Details: Salt screening and salt precedent re-preparation
推定pKa値7.5および5.1、ならびに遊離塩基(812608−05−A)の室温(室温、25±3℃)での近似溶解度に従い、10種の塩形成剤および3種の溶媒系をスクリーニングに使用した。遊離塩基(約15mg)に、選択された溶媒をガラスバイアル内で分散させ、対応する塩形成剤を、1:1のモル電荷比(HCl/遊離塩基の場合、1:1および2:1の両方の2つの比を使用した)。遊離塩基と酸との混合物を、室温で3.5日間撹拌した。より多くの固体ヒットを得るために、得られた透明溶液(812608−08−B0/B5/B10)を5℃に移し、さらに2.5日間撹拌した。最後に、812608−08−B0/B10の透明溶液に、n−ヘプタンを0.5mL添加し、5℃でさらに2日間撹拌した。 According to the estimated pKa values of 7.5 and 5.1, and the approximate solubility of the free base (812608-05-A) at room temperature (room temperature, 25 ± 3 ° C.), 10 salt forming agents and 3 solvent systems were used. Used for screening. The selected solvent is dispersed in a glass vial on the free base (about 15 mg) and the corresponding salt-forming agent has a 1: 1 molar charge ratio (1: 1 and 2: 1 for HCl / free base). Both two ratios were used). The mixture of free base and acid was stirred at room temperature for 3.5 days. To obtain more solid hits, the resulting clear solution (812608-08-B0 / B5 / B10) was transferred to 5 ° C. and stirred for an additional 2.5 days. Finally, 0.5 mL of n-heptane was added to a clear solution of 812608-08-B0 / B10, and the mixture was stirred at 5 ° C. for another 2 days.
得られた固形分の全てを単離し、50℃で2.5時間乾燥した後にXRPDにより分析した。表30にまとめたように、合計で12の結晶質のヒットが得られ、XRPD、TGA、およびDSCにより特徴付け、その化学量論量を1H NMRまたはHPLC/ICにより決定した。特徴付けデータを、表31にまとめた。
塩の先例の再調製および特徴付け
All of the obtained solids were isolated, dried at 50 ° C. for 2.5 hours and then analyzed by XRPD. As summarized in Table 30, a total of 12 crystalline hits were obtained, characterized by XRPD, TGA, and DSC, and their stoichiometric quantities were determined by 1 H NMR or HPLC / IC. The characterization data are summarized in Table 31.
Precedent repreparation and characterization of salts
特徴付けの結果に基づき、2つの塩の先例(HCl塩形態Aおよびフマル酸塩形態A)は、塩の先例として認められ、数百ミリグラムまで再調製された。選択基準には:1)明らかな非晶質ハロがない、鋭いXRPDピーク、2)TGAにおける、無視できるほどの重量損失、3)DSCにおける鋭い融解ピークを持つ、正味の熱事象が含まれるが、これらに限定するものではない。詳細な調製手順を表32に記述し、特徴付けデータを上記表28にまとめた。
HCl塩形態Aは、図14のXRPD結果により明らかなように、首尾良く再調製された。図15のTGAおよびDSCデータによれば、試料は、150℃まで1.7%の重量損失を示し、3つの吸熱を196.2、214.8、および274.0℃(開始温度)で示した。196.2℃での小さい吸熱は、残された非常に少量の遊離塩基形態Aの融解によって引き起こされ得る。図16に示されるように、HCl塩形態A試料を218℃まで加熱した後、冷却中に202.0℃付近で発熱が観察され、加熱後に得られた試料のDSCは依然として、213.8および273.9℃(開始温度)で吸熱を示した。試料を218℃まで加熱し、元の室温に冷却し、周囲条件に曝露した後に形態変化が観察されなかったという事実と組み合わせて、213.8℃でのシグナルは、形態転移によって引き起こされることが推測された。化学量論比を、HPLC/ICによって0.97(酸/塩基)と決定した。150℃の前の、限られた段階的なTGA重量損失、および190℃の前のDSCにおける非有意な熱事象が観察されたので、試料は、無水HCl塩であることが推測される。
フマル酸塩形態A
HCl salt form A was successfully reprepared, as evidenced by the XRPD results in FIG. According to the TGA and DSC data of FIG. 15, the sample showed a weight loss of 1.7% up to 150 ° C. and three endothermics at 196.2, 214.8, and 274.0 ° C. (starting temperature). rice field. The small endotherm at 196.2 ° C. can be caused by the melting of the very small amount of free base form A left behind. As shown in FIG. 16, after heating the HCl salt form A sample to 218 ° C, heat generation was observed around 202.0 ° C during cooling, and the DSC of the sample obtained after heating was still 213.8 and It showed endothermic at 273.9 ° C. (starting temperature). The signal at 213.8 ° C. can be triggered by morphological transitions, combined with the fact that no morphological changes were observed after heating the sample to 218 ° C., cooling to the original room temperature and exposing to ambient conditions. It was guessed. The stoichiometric ratio was determined by HPLC / IC to be 0.97 (acid / base). A limited gradual TGA weight loss prior to 150 ° C. and a non-significant thermal event at DSC prior to 190 ° C. were observed, suggesting that the sample is an anhydrous HCl salt.
Fumarate form A
フマル酸塩形態Aを、フマル酸塩形態AバッチのXRPDオーバーレイを示す図17の、XRPD結果により明らかなように、首尾良く再調製した。図18のTGAおよびDSCデータによれば、試料は、150℃まで1.0%の重量損失を示し、228.5および233.9℃(ピーク温度)で2つの吸熱を示した。化学量論比は、1H NMR(図19)により0.94(酸/塩基)と決定され、限られたEtOH残留物が検出された(約1.6%)。150℃前の限られたTGA重量損失、200℃前のDSCにおける非有意な熱事象、および限られた溶媒残留物が観察されたので、試料は無水フマル酸塩と推測される。
塩の先例の評価
Fumarate form A was successfully reprepared, as evidenced by the XRPD results in FIG. 17, showing the XRPD overlay of the fumarate form A batch. According to the TGA and DSC data of FIG. 18, the sample showed 1.0% weight loss up to 150 ° C. and two endothermics at 228.5 and 233.9 ° C. (peak temperature). The stoichiometric ratio was determined to be 0.94 (acid / base) by 1 1 H NMR (FIG. 19), and a limited EtOH residue was detected (about 1.6%). A limited TGA weight loss before 150 ° C., a non-significant thermal event at DSC before 200 ° C., and a limited solvent residue were observed, suggesting that the sample is fumarate anhydride.
Evaluation of salt precedent
吸湿性、動力学的溶解度、および固体状態安定性に関するさらなる評価調査を、遊離塩基形態Aを参照として使用して、2つの塩の先例の物理化学的性質をより良く理解するために実施した。
吸湿性
Further assessment studies on hygroscopicity, kinetic solubility, and solid state stability were carried out with reference to free base form A to better understand the physicochemical properties of the precedents of the two salts.
Hygroscopic
DVS等温線プロットを、25℃で収集して、湿度の関数としての固体形態安定性を調査した。無水試料の全てを、0%RHで事前に平衡にして、未結合溶媒または水を除去した後に、開始した。80%RHまで0.60〜1.04%の水の取り込みにより明らかなように、遊離塩基形態Aと、2つの塩形態の両方とは、わずかに吸湿性があり、DVS評価後に固体形態変化は観察されなかった(図20A、20B、20C、20D、20E、および20F)。 DVS isotherm plots were collected at 25 ° C. to investigate solid morphological stability as a function of humidity. All anhydrous samples were pre-equilibriumed at 0% Rhes to remove unbound solvent or water before starting. Both the free base form A and the two salt forms are slightly hygroscopic and solid morphological changes after DVS evaluation, as evidenced by the uptake of 0.60 to 1.04% water up to 80% RH. Was not observed (FIGS. 20A, 20B, 20C, 20D, 20E, and 20F).
水の取り込みの有意な増加(4.56%)は、95%RHでフマル酸塩タイプAに関して観察されたが、HCl塩タイプAでは、わずか1.40%の水の取り込みが95%RHで観察された。
動力学的溶解度
A significant increase in water uptake (4.56%) was observed for fumarate type A at 95% RH, whereas with HCl salt type A only 1.40% water uptake was observed at 95% RH. It was observed.
Kinetic solubility
2種の塩の先例の動力学的溶解度を、pH2、5、および7の緩衝剤中で測定して、遊離塩基形態A(812608−05−A)を対照として使用してそれらの溶解度および不均化リスクを評価した。溶解度試料の全て(初期固体負荷は約10mg/mL)を、ローリングインキュベーター上で25rpmの速度でローリングさせたままにし、それぞれ室温(19±3℃)で、1、2、4、および24時間でサンプリングした。遠心分離後、上澄みをHPLCおよびpH試験用に収集し、湿潤ケーキをXRPD特徴付け用に収集した。透明溶液が得られた場合、正確な濃度を溶液に関して測定した。
The kinetic solubilities of the two salts were measured in buffers of
結果を表33Aにまとめ、動力学的溶解度プロファイルを図21に示す(破線は、透明溶液が得られたことを示す)。遊離塩基形態Aと比較すると、HCl塩形態Aは、pH2、5、および7の緩衝剤中で増大した溶解度を示した。pH2および5の緩衝剤では、固体は得られなかった。pH7の緩衝剤では、遊離塩基形態Aへの形態変化は、HCl塩タイプAが1、2、および4時間にわたり懸濁させた後に観察された(限られた固形分が、1および2時間の時点で得られ、1つの特別なブロードピークが2時間の時点で観察された)。新しい回折ピークは、24時間後に観察された。フマル酸塩形態Aは、pH2および5の緩衝剤中で低下した溶解度を示し、形態変化が観察されたが、増大した溶解度がpH7の緩衝剤中で観察され、形態変化はなかった。遊離塩基形態Aの場合、遊離塩基形態AをpH5で懸濁させた後に、1つの特別なブロードピーク(強調された)が1および2時間の時点で観察されたこと以外、形態変化は観察されなかった。
G:ゲル様物質が得られたので、データを収集しなかった。
N/A:分析用の固体なし。*:1つの余分なブロードピークが2θ≒7.6°で観察された。
固体状態安定性
The results are summarized in Table 33A and the kinetic solubility profile is shown in FIG. 21 (dashed line indicates that a clear solution was obtained). Compared to free base form A, HCl salt form A showed increased solubility in buffers at
G: No data was collected as a gel-like substance was obtained.
N / A: No solid for analysis. *: One extra Broad Peak was observed at 2θ≈7.6 °.
Solid state stability
遊離塩基形態A、HCl塩形態A、およびフマル酸塩形態Aの固体状態安定性を、40℃/75%RHの下で1週間評価した。安定性試料を、任意の固体形態変化をチェックするためにXRPDによって、および純度変化をチェックするためにHPLCによって、特徴付けた。結果の全てを表33Bにまとめ、XRPDデータを図22に示すが、この図は、形態変化または有意なHPLC純度低下が安定性試験後に観察されなかったことを示し、試験条件下で遊離塩基形態Aおよび両方の2つの塩の良好な物理化学的安定性を示している。
収集された結果に基づき、HCl塩形態Aは候補形態である。
HCl塩(モノ)に関する多形評価
Based on the collected results, HCl salt form A is a candidate form.
Polymorphic evaluation of HCl salt (mono)
したがって、多形評価調査をHCl塩(モノ)に関して実施した。予備多形スクリーニングを、スラリー変換、蒸発、低速冷却、および逆溶剤添加の方法を使用して、32の条件下で実施した。XRPD比較に基づき、HCl塩形態Aの他に、5つの新しい結晶質形態(HCl塩形態C〜G)がスクリーニングから得られ、TGAおよびDSCにより特徴付けられた。調査結果に基づき、HCl塩形態Cは、水和物と推測された。蒸発によるHCl塩形態DおよびEの再調製は、それぞれHCl塩形態C+DおよびC+Eを含有する混合物をもたらした。混合物の1H NMRおよび加熱実験の結果、HCl塩形態DおよびEは、水和物である可能性がある。限られた量の材料に起因して、潜在的なHCl塩形態FおよびGは、完全に特定されなかった。
無水HCl塩形態A
Therefore, a polymorphic evaluation study was conducted on the HCl salt (mono). Preliminary polymorph screening was performed under 32 conditions using methods of slurry conversion, evaporation, slow cooling, and backsolvent addition. Based on the XRPD comparison, in addition to HCl salt form A, five new crystalline forms (HCl salt forms C to G) were obtained from screening and characterized by TGA and DSC. Based on the findings, HCl salt form C was presumed to be a hydrate. Repreparation of the HCl salt forms D and E by evaporation resulted in a mixture containing the HCl salt forms C + D and C + E, respectively. As a result of 1 H NMR and heating experiments of the mixture, the HCl salt forms D and E may be hydrates. Due to the limited amount of material, potential HCl salt forms F and G were not completely identified.
Anhydrous HCl salt form A
HCl塩形態A(812608−16−A)を、多形スクリーニング用に再調製し、詳細な調製手順を表34に示した。図23に示されるように、HCl塩タイプAは、首尾良く再調製された。図24のTGAおよびDSCデータによって示されるように、150℃までの2.0%の重量損失、ならびに220.7および274.8℃での2つの吸熱ピーク(開始温度)が観察された。酸/遊離塩基の化学量論量は、HPLC/ICによって1.01と決定された。限られたTGA重量損失および正味のDSC曲線に起因して、HCl塩形態Aは無水物と推定された。
HCl塩形態C(812608−21−A2)を、CHCl3/MeOH(1:1、v/v)溶液の蒸発から得た(図25)。図26のTGAおよびDSCデータによって示されるように、150℃までの3.1%の重量損失、98.8℃でのブロード吸熱ピーク、ならびに267.5℃および275.5℃(ピーク温度)での2つの鋭いピークが観察された。酸/遊離塩基の化学量論量は、HPLC/ICによって1.01と決定された。 HCl salt form C (812608-21-A2) was obtained from evaporation of CHCl 3 / MeOH (1: 1, v / v) solution (FIG. 25). As shown by the TGA and DSC data in FIG. 26, 3.1% weight loss up to 150 ° C., broad endothermic peaks at 98.8 ° C., and 267.5 ° C. and 275.5 ° C. (peak temperatures). Two sharp peaks were observed. The stoichiometric amount of acid / free base was determined by HPLC / IC to be 1.01.
HCl塩形態Cを特定するために、加熱実験を行った。HCl塩タイプC(812608−27−B)をN2下で170℃まで加熱し、30℃に冷却し、周囲条件に曝露した後、形態変化は観察されなかった。しかし、HCl塩形態Cは、蓋を開けた状態で260℃に加熱された後(蓋を締めた状態で、264.9℃での吸熱を越えて)、HCl塩形態Aになった。図27を参照されたい。さらに、HCl塩形態C(812608−27−B)を170℃に加熱し、元の30℃に冷却し、周囲温度に曝露した後、ブロードピークが依然としてDSCで観察され、これは水分の再吸収によって引き起こされ得るものである。XPRD結果と、形態Cを種々の水性系で得ることができるという事実とを組み合わせると、HCl塩形態Cは水和物であると推測された(モノHCl塩一水和物の理論上の含水量は、3.4%であった)。図28は、加熱前および加熱後のHCL形態CのDSCオーバーレイを示す。
完全に特定されていない形態(D〜G)
Heating experiments were performed to identify HCl salt form C. HCl salt type C a (812608-27-B) was heated to 170 ° C. under N 2, it was cooled to 30 ° C., after exposure to ambient conditions, morphological changes were observed. However, the HCl salt form C became the HCl salt form A after being heated to 260 ° C. with the lid open (beyond the endothermic at 264.9 ° C. with the lid closed). See FIG. 27. In addition, after heating HCl salt form C (812608-27-B) to 170 ° C., cooling to the original 30 ° C. and exposing to ambient temperature, Broad Peak is still observed on DSC, which is water reabsorption. Can be caused by. Combining the XPRD results with the fact that form C can be obtained in a variety of aqueous systems, it was speculated that HCl salt form C is a hydrate (theoretical inclusion of monoHCl salt monohydrate). The amount of water was 3.4%). FIG. 28 shows DSC overlays of HCL form C before and after heating.
Unspecified form (DG)
図13Aおよび13BのXRPDオーバーレイを参照されたい。HCl塩形態D(812608−23−A2)が、DCM/MeOH(良溶媒)/DCM(逆溶剤)への逆溶剤添加から得られ、得られた透明溶液を、最初に5℃および−20℃に移し、その後、室温で蒸発させた。TGAおよびDSCデータにより実証されるように、150℃までの2.4%の重量損失および複数回の吸熱が観察された。酸/遊離塩基の化学量論量は、HPLC/ICにより1.02と決定された。 See XRPD overlays in FIGS. 13A and 13B. HCl salt form D (812608-23-A2) was obtained by backsolvent addition to DCM / MeOH (good solvent) / DCM (reverse solvent), and the resulting clear solutions were first subjected to 5 ° C. and -20 ° C. And then evaporated at room temperature. 2.4% weight loss up to 150 ° C. and multiple endotherms were observed, as demonstrated by TGA and DSC data. The stoichiometric amount of acid / free base was determined by HPLC / IC to be 1.02.
HCl塩形態Dを特定するために、DCM/MeOH(良溶媒)/DCM(逆溶剤)への逆溶剤添加とその後の室温での蒸発によって、再調製を試みた。XRPDにより決定されるように、HCl塩タイプC+D(812608−27−C)の混合物を、再調製から得た。2〜3の特別ピークが観察され、これらは新しい形態によるものと考えられるかまたはHCl塩形態Dの参照試料の比較的低い結晶化度に起因すると考えられる。TGAおよびDSCは、150℃まで2ステップの重量損失4.8%を実証し、複数回の吸熱が観察された。HCl塩形態C+Dの混合物を含有する試料が、N2下で170℃に加熱され、30℃に冷却され、周囲条件に曝露された後、形態A+Cの混合物が観察された(図29)。MeOHおよび限られたDCMは、NMRにより検出されなかったので、形態Dは水和物と考えられる。 To identify the HCl salt form D, readjustment was attempted by backsolvent addition to DCM / MeOH (good solvent) / DCM (backsolvent) followed by evaporation at room temperature. A mixture of HCl salt type C + D (812608-27-C) was obtained from the repreparation as determined by XRPD. A few special peaks are observed, which may be due to the new form or due to the relatively low crystallinity of the reference sample of HCl salt form D. TGA and DSC demonstrated weight loss of 4.8% in two steps up to 150 ° C., and multiple endothermic processes were observed. A sample containing a mixture of the HCl salt Form C + D is heated to 170 ° C. under N 2, cooled to 30 ° C., after exposure to ambient conditions, the mixture in the form A + C was observed (Figure 29). MeOH and limited DCM were not detected by NMR, so Form D is considered a hydrate.
再び図13Aおよび13Bを参照すると、HCl塩形態E(812608−23−A5)が、THF/H2O(良溶媒)/EtOH(逆溶剤)への逆溶剤の添加とその後の室温での蒸発から、得られた。TGAおよびDSCにより実証されるように、150℃までの13.5%の重量損失、ならびに105.8℃および273.6℃(ピーク温度)での2つの吸熱ピークが観察された。酸/遊離塩基の化学量論量は、試料の限られた量に起因して決定されなかった。 13A and 13B again, HCl salt form E (812608-23-A5) are evaporated in THF / H 2 O (good solvent) / EtOH (antisolvent) antisolvent addition and subsequent of the room Was obtained from. As demonstrated by TGA and DSC, a weight loss of 13.5% up to 150 ° C. and two endothermic peaks at 105.8 ° C. and 273.6 ° C. (peak temperature) were observed. The stoichiometric amount of acid / free base was not determined due to the limited amount of sample.
HCl塩形態Eを特定するために、THF/H2O(良溶媒)/EtOH(逆溶剤)への逆溶剤添加とその後の室温での蒸発によって、再調製を試みた。XRPDにより決定されるように、HCl塩形態C+E(812608−27−D)の混合物は、再調製から得られた。2〜3の特別なピークが観察され(強調された)、これらは新しい形態によるものと考えられるかまたはHCl塩形態Eの参照試料の比較的低い結晶化度に起因すると考えられる。TGAおよびDSCにより実証されるように、150℃までの2ステップの重量損失7.8%および複数の熱事象が観察された。HCl塩形態C+Eの混合物を含有する試料が、N2下で140℃に加熱され、30℃に冷却され、周囲温度に曝露された後、形態A+Cの混合物が観察された(図30)。THFおよび限られたEtOHはNMRによって検出されなかったので、タイプEは水和物と考えられる。 To identify the HCl salt form E, readjustment was attempted by adding a back solvent to THF / H 2 O (good solvent) / EtOH (back solvent) followed by evaporation at room temperature. As determined by XRPD, a mixture of HCl salt form C + E (812608-27-D) was obtained from repreparation. A few special peaks were observed (emphasized), which may be due to the new form or due to the relatively low crystallinity of the reference sample of HCl salt form E. Two-step weight loss of 7.8% up to 150 ° C. and multiple thermal events were observed, as demonstrated by TGA and DSC. A sample containing a mixture of the HCl salt Form C + E is heated to 140 ° C. under N 2, cooled to 30 ° C., after being exposed to ambient temperature, the mixture in the form A + C was observed (Figure 30). Type E is considered a hydrate as THF and limited EtOH were not detected by NMR.
再び図13Aおよび13Bを参照すると、結晶化度の低い、新しい形態が、H2O/アセトンへの逆溶剤添加およびその後の室温での蒸発から得られ(812608−23−A6)、またはHCl塩形態C(812608−23−A1、MeOH/H2Oからの蒸発によって得られた)が周囲条件(21±1.5℃、60±20%RH)に6日間曝露された後に得られ、これをHCl塩形態Fと称する。TGAおよびDSCにより実証されるように、150℃までの7.3%の重量損失、ならびに120.6および274.7℃(ピーク温度)での2つの吸熱ピークが観察された。 13A and 13B again, low crystallinity, new forms are obtained from antisolvent addition and subsequent evaporation at room temperature to a H 2 O / acetone (812608-23-A6), or HCl salt Form C (obtained by evaporation from 812608-23-A1, MeOH / H 2 O) was obtained after exposure to ambient conditions (21 ± 1.5 ° C., 60 ± 20% RH) for 6 days. Is referred to as HCl salt form F. As demonstrated by TGA and DSC, a 7.3% weight loss up to 150 ° C. and two endothermic peaks at 120.6 and 274.7 ° C. (peak temperatures) were observed.
最後に、図13Aおよび13Bならびに図31も参照すると、HCl塩形態C(812608−21−A3、THF/H2Oからの蒸発によって得られた)が周囲温度(21±1.5℃、60±20%RH)に6日間曝露された後、形態変化が観察され、これをHCl塩形態Gと称する。TGAおよびDSCにより実証されるように、150℃までの8.9%の重量損失および複数の熱事象が、この形態Gに関して観察された。
結果
Finally, referring also to FIG. 13A and 13B and FIGS. 31, HCl salt form C (812608-21-A3, THF /
result
塩のスクリーニングを、遊離塩基化合物2で実施した。合計で12の塩ヒットが、33の塩スクリーニング実験から得られた。無水物HCl塩形態Aおよびフマル酸塩形態Aを、吸湿性、種々のpH緩衝剤中の動力学的溶解度、および固体状態の安定性を含む、さらなる評価のための塩の先例として選択した。その結果、HCl塩形態Aおよびフマル酸塩形態Aの両方が、わずかに吸湿性であり、40℃/75%RH/1週の条件下、良好な物理化学的安定性を示した。遊離塩基形態Aと比較すると、HCl形態Aは、pH2、5、および7の緩衝剤中で増大した溶解度を示したが、不均化および形態変化はpH7の緩衝剤中で観察された。
Salt screening was performed on
特徴付けおよび評価結果に基づけば、無水物HCl塩形態Aは好ましい塩候補であり、多形スクリーニングをHCl塩(モノ)で実施した。XRPD比較に基づけば、無水HCl塩形態Aの他に、5つの新しい結晶質形態(水和HCl塩形態C、およびHCl塩形態D〜G)が、32の条件下で予備多形スクリーニングから得られ、新しい形態が、TGAおよびDSCによって特徴付けられた。 Based on the characterization and evaluation results, anhydrous HCl salt form A was a preferred salt candidate and polymorphic screening was performed on the HCl salt (mono). Based on the XRPD comparison, in addition to anhydrous HCl salt form A, five new crystalline forms (hydrated HCl salt forms C and HCl salt forms D to G) were obtained from preliminary polymorph screening under 32 conditions. And new forms were characterized by TGA and DSC.
化合物2の予備塩スクリーニングおよびHCl塩(モノ)の多形スクリーニングの結果、HCl塩形態Aは、活性医薬成分として開発するための好ましい実施形態である。
塩および多形スクリーニングのための機器および方法
近似溶解度
As a result of the preliminary salt screening of
Equipment and methods for salt and polymorph screening Approximate solubility
遊離塩基形態A(812608−05−A)の近似溶解度を、室温で、12種の溶媒中で測定した。各実験ごとに、試料のおよそ2mgを3mLのガラスバイアルに加えた。次いで表35の溶媒を、固形分が目に見えて溶解するまでまたは全体積が1mLに到達するまで、段階的に(50、50、200、200、500μL)バイアルに添加した。表35にまとめた溶解度の結果を使用して、塩スクリーニングでの溶媒選択を案内した。
使用される溶媒の略称
The approximate solubility of free base form A (812608-05-A) was measured at room temperature in 12 solvents. For each experiment, approximately 2 mg of sample was added to a 3 mL glass vial. The solvents in Table 35 were then added stepwise (50, 50, 200, 200, 500 μL) to the vials until the solids were visibly dissolved or the total volume reached 1 mL. The solubility results summarized in Table 35 were used to guide solvent selection in salt screening.
溶媒の略称を、表36に列挙する。
XRPD
Solvent abbreviations are listed in Table 36.
XRPD分析では、反射モードでのPANalytical X線粉末回折を使用した。XRPDパラメーターを、表37に列挙する。
TGAデータを、TA Instruments製のTA Q500/Q5000 TGAを使用して収集し、DSCを、TA Instruments製のTA Q200/Q2000 DSCを使用して行った。使用した詳細なパラメーターを、表38に列挙する。
Agilent1100/1260HPLCを利用し、純度および溶解度測定に関する詳細なクロマトグラフィー条件を、表39に列挙する。
#:試料812608−08−A1/08−C2/08−A3/08−B4/16−A/21−A2/23−A2に関する化学量論比に使用。
イオンクロマトグラフィー
Detailed chromatographic conditions for purity and solubility measurements utilizing Agent 1100/1260 HPLC are listed in Table 39.
# : Used for stoichiometric ratios for sample 812608-08-A1 / 08-C2 / 08-A3 / 08-B4 / 16-A / 21-A2 / 23-A2.
Ion chromatography
化学量論比を決定する対イオン(陰イオン)含有量測定のための、イオンクロマトグラフィー(IC)法を、表40に列挙した。
動的蒸気収着(DVS)を、SMS(表面測定システム)DVS Intrinsicを介して測定した。25℃での相対湿度を、LiCl、Mg(NO3)2、およびKClの潮解点に対して較正した。DVS試験の実際のパラメーターを、表41に列挙した。
溶液NMRを、DMSO−d6を使用してBruker 400M NMR分光計で収集した。
pH緩衝剤に関する調製手順
ストック溶液の調製
Solution NMR was collected on a Bruker 400M NMR spectrometer using DMSO-d6.
Preparation procedure for pH buffer Preparation of stock solution
0.2M塩酸: 濃塩酸8.25mLを、500mLのメスフラスコに加える。精製水でその体積を希釈し、十分に混合する。 0.2M Hydrochloric Acid: 8.25 mL of concentrated hydrochloric acid is added to a 500 mL volumetric flask. Dilute the volume with purified water and mix well.
0.2M水酸化ナトリウム: 水酸化ナトリウム2.0gを、250mLのメスフラスコに量り取る。これを適切な体積の精製水で溶解させ、体積まで希釈する。十分混合する。 0.2M sodium hydroxide: Weigh 2.0 g of sodium hydroxide into a 250 mL volumetric flask. Dissolve this in an appropriate volume of purified water and dilute to volume. Mix well.
0.2M塩化カリウム: 塩化カリウム2.98gを、200mLのメスフラスコに量り取る。これを適切な体積の精製水で溶解させ、体積まで希釈する。十分混合する。 0.2M Potassium Chloride: 2.98 g of Potassium Chloride is weighed into a 200 mL volumetric flask. Dissolve this in an appropriate volume of purified water and dilute to volume. Mix well.
0.2Mリン酸二水素カリウム: 一塩基性リン酸カリウム(KH2PO4)5.44gを、200mLのメスフラスコに量り取る。適切な体積の精製水を添加し、固形分の全てが完全に溶解するまで超音波処理する。精製水で体積まで希釈し、十分混合する。 0.2M Potassium Dihydrogen Phosphate: Weigh 5.44 g of monobasic potassium dihydrogen phosphate (KH 2 PO 4 ) into a 200 mL volumetric flask. An appropriate volume of purified water is added and sonicated until all of the solids are completely dissolved. Dilute to volume with purified water and mix well.
0.2Mフタル酸水素カリウム溶液: フタル酸水素カリウム[KHC6H4(COO)2]8.17gを、200mLのメスフラスコに量り取る。適切な体積の精製水を添加し、全ての固形分が完全に溶解するまで超音波処理する。精製水で体積まで希釈し、十分混合する。 0.2M Potassium Hydrogen phthalate Solution: Weigh 8.17 g of potassium hydrogen phthalate [KHC 6 H 4 (COO) 2 ] into a 200 mL volumetric flask. Add an appropriate volume of purified water and sonicate until all solids are completely dissolved. Dilute to volume with purified water and mix well.
pH2.0緩衝剤: 0.2M塩化カリウム溶液25mLを、100mLのメスフラスコに移し、0.2M塩酸溶液6.5mLを添加する。十分な精製水を目標体積近くまで添加し、pH2.0に調節する。精製水で体積まで希釈し、十分混合し、pHメーターでpHをチェックする。
pH 2.0 buffer:
pH5.0緩衝剤: 0.2Mフタル酸水素カリウム溶液12.5mL、および0.2M水酸化ナトリウム溶液5.6mLを、50mLのメスフラスコに移す。十分な精製水を、目標体積近くまで添加し、pH5.0に調節する。精製水で体積まで希釈し、十分混合し、pHメーターでpHをチェックする。 pH 5.0 buffer: Transfer 12.5 mL of 0.2 M potassium hydrogen phthalate solution and 5.6 mL of 0.2 M sodium hydroxide solution to a 50 mL volumetric flask. Sufficient purified water is added close to the target volume to adjust the pH to 5.0. Dilute to volume with purified water, mix well and check pH with a pH meter.
pH7.0緩衝剤: 0.2Mリン酸二水素カリウム溶液12.5mL、および0.2M水酸化ナトリウム溶液7.28mLを、50mLのメスフラスコに移す。十分な精製水を目標体積近くまで添加し、pH7.0に調節する。精製水で体積まで希釈し、十分混合し、pHメーターでpHをチェックする。
(実施例10)
脳浸透に関する予測モデリング
pH 7.0 buffer: Transfer 12.5 mL of 0.2 M potassium dihydrogen phosphate solution and 7.28 mL of 0.2 M sodium hydroxide solution to a 50 mL volumetric flask. Sufficient purified water is added close to the target volume to adjust the pH to 7.0. Dilute to volume with purified water, mix well and check pH with a pH meter.
(Example 10)
Predictive modeling of brain penetration
化合物2を、血液脳関門(BBB)を通して化合物の進入を可能にし得る予測特性に関して評価した。複数パラメータースコア(「myMPO」)は、物理化学的性質を考慮に入れて、BBB浸透を予測した。比較群としてCNS薬物を用いて、運動を行った。スコアが高くなるほど(5が理想的である)、CNS浸透の機会がより良好である。種々の結論をその運動から導くことができるが、1つの結論は、化合物2がこのリストの下に向かい、それでも有効になるよう非常に十分に関門を横断する薬物と重なり合う。特に、表42は化合物2に関する分析を提供し、比較群に関しては図32を参照されたい。このように、予測モデリングは、化合物2が脳浸透物であるが比較的弱いことを示唆する。
ビーグル犬における単一経口用量後の、化合物2の2種の懸濁製剤の、薬物動態および相対的バイオアベイラビリティー
Pharmacokinetics and relative bioavailability of the two suspensions of
調査の目的は、ビーグル犬に、強制飼養によって2つの用量レベルで化合物2の2種の懸濁製剤を投薬し、かつ血液試料を収集して、血漿中濃度を決定し、薬物動態パラメーターを誘導し、2種の懸濁製剤の相対的バイオアベイラビリティーを決定することである。化合物2の薬物動態は、カプセルでの経口投与後に評価されると考えられ、経口液体製剤に対するこの剤形の相対的バイオアベイラビリティーが決定された。
The purpose of the study was to administer two suspensions of
この調査では、表43、表44、および表45に概説されるように、7日間のウォッシュアウト期間を間に入れた3つの期間にわたり、経口経路によって、試験項目を投薬する。表45に概説される調査の第3の期間は、必要に応じたものと見なされ、スポンサーの求めによってのみ行われる。投薬日前に、食物は午後4:00にイヌから除去され、投薬から2時間後に再度導入される。
製剤の経口投与では、試験項目を、胃管を介して投与し、その後、水道水10mLを投与した。カプセルの経口投与では、カプセルを舌の裏に置いて、嚥下を誘発させ、その後、水道水10mLを投与した。次いで口腔を検査して、カプセルが飲み込まれたことを確認した。投与された投薬製剤の実際の体積は、動物の最も最近になって一覧に記載された体重に基づいて計算される。 For oral administration of the drug product, the test item was administered via the gastric tube, followed by administration of 10 mL of tap water. For oral administration of capsules, the capsules were placed on the back of the tongue to induce swallowing, followed by administration of 10 mL of tap water. The oral cavity was then examined to confirm that the capsule was swallowed. The actual volume of the administered dosing formulation is calculated based on the most recently listed body weight of the animal.
より詳細には、オスのビーグル犬に、製剤#1(表43)および製剤#2(表44)で溶液として化合物2を2および5mg/kgの用量で経口投与し、これらの用量は十分許容される用量であるが、製剤#2(表44)で5mg/kgの化合物2を投与した唯一のイヌでは軽度の嘔吐があった。
More specifically, male beagle dogs were orally administered with
製剤#1(表43)および製剤#2(表44)として溶液中の化合物2に関する薬物動態パラメーターは、カプセル中の化合物2の粉末(表45)の投薬に関するデータと共に、表46に提示される。
終末期に関する線形相関係数は、0.98〜1.00に及んだ。AUC0−TlastからAUC0−∞への外挿は、1.84〜12.48%に及んだ。
記述的統計を再計算して、データ用に提示されたフォーマットを製剤#1(表43)および#2(表44)と一致させた。
The pharmacokinetic parameters for
The descriptive statistics were recalculated to match the format presented for the data with the formulations # 1 (Table 43) and # 2 (Table 44).
製剤#1(表43)および製剤#2(表44)のそれぞれの溶液としての、ならびにカプセル(表45)中の粉末としての、2および5mg/kgの用量での化合物2の経口投与の後、定量限界(0.5ng/mL)を超えた化合物2の血漿中濃度が全てのイヌで観察され、単一の代表的な種としては、投薬後0.5時間までに観察された。
After oral administration of
両方の用量で、製剤#1(表43)、製剤#2(表44)、およびカプセル中の粉末(表45)に類似したTmax値は、3.0〜4.7時間に及んだ。両方の用量での、製剤#1(表43)、製剤#2(表44)、およびカプセル中の粉末(表45)に関する平均血漿中半減期(t1/2)、全身クリアランス(CL/F)、分布容積(Vz/F)、および平均滞留時間は、それぞれ、2.3〜5.6時間、20〜33L/kg/hr、85〜237L/kg、および5.23〜9.32時間に及ぶ。 At both doses, Tmax values similar to formulation # 1 (Table 43), formulation # 2 (Table 44), and powder in capsules (Table 45) ranged from 3.0 to 4.7 hours. .. Average plasma half-life (t 1/2 ) for formulation # 1 (Table 43), formulation # 2 (Table 44), and powder in capsules (Table 45) at both doses, systemic clearance (CL / F) ), Volume of distribution (Vz / F), and average residence time are 2.3 to 5.6 hours, 20 to 33 L / kg / hr, 85 to 237 L / kg, and 5.23 to 9.32 hours, respectively. To.
各製剤ごとに、平均CmaxおよびAUC0−∞の増大を、2mg/kgから5mg/kg(2.5倍)の用量増加に関して比較した。CmaxおよびAUC0−∞に関する値は、それぞれ、製剤#1(表43)に関して4.1および4.0、製剤#2(表44)に関して1.9および2.1、ならびにカプセル中の粉末(表45)に関して1.7および1.8であった。 For each formulation , increases in mean C max and AUC 0-∞ were compared for dose increases from 2 mg / kg to 5 mg / kg (2.5 fold). Values for C max and AUC 0-∞ are 4.1 and 4.0 for formulation # 1 (Table 43), 1.9 and 2.1 for formulation # 2 (Table 44), and powder in capsule, respectively. It was 1.7 and 1.8 with respect to (Table 45).
製剤#1(表43)、製剤#2(表44)、およびカプセル中粉末(表45)に関する2mg/kgおよび5mg/kgの用量での、化合物2に対する血漿曝露を、化合物2のCmaxおよびAUC0−∞の両方を使用することによって比較した。製剤#1(表43)、製剤#2(表44)、およびカプセル中粉末(表45)を比較したとき、各用量で、平均CmaxおよびAUC0−∞値の間での統計的有意差は観察されなかった。しかし変動する血漿中濃度が、各用量レベルおよび製剤で、3匹のイヌの間で観察された。
Plasma exposure to
製剤#1(表43)から製剤#2(表44)に関するCmaxおよびAUC0−∞に基づいて、化合物2の相対的バイオアベイラビリティーを個々のイヌに関して比較してもよく、それは同じ個々のイヌが製剤#1(表43)および製剤#2(表44)の両方を受けるからである。CmaxおよびAUC0−∞では、平均相対的バイオアベイラビリティーが、それぞれ、2mg/kgの用量で0.70±0.26および0.78±0.24であり、5mg/kgの用量で1.22±0.30および1.34±0.06であった(n=3、±SD)。
The relative bioavailability of
まとめると本開示は、製剤#1(表43)、製剤#2(表44)、およびカプセル中粉末(表45)に調製された、化合物2の経口の強制飼養によるビーグル犬の経口投薬の調査を含む。化合物2は、投薬後0.5時間までに血漿中に現れ、その平均Cmax値は、2および5mg/kgの用量でそれぞれ6.78〜14.90ng/mLおよび22.23〜27.81ng/mLに及び、平均Tmax値は、両方の用量全体を通して3.0〜4.7時間に及ぶ。血漿中半減期、クリアランス、分布容積、および平均滞留時間は、用量および製剤の全体を通して明らかな統計的有意差を示さなかった。しかし追加の検討は、予期せぬ利益を実証し得る。それにも関わらず、データは、2.5倍の用量増加の結果、製剤#1(表43)の化合物2に関して(相乗的)用量比例増加を超える平均Cmaxおよび平均AUC0−∞をもたらし、製剤#2(表44)およびカプセル中粉末(表45)に関して用量比例増加に近い値をもたらすことを実証する。
In summary, the present disclosure is a study of oral dosing of Beagle dogs by oral forced feeding of
2および5mg/kgの用量で、平均Cmax、AUC0−TLast、およびAUC0−∞を比較したとき、製剤#1(表43)、製剤#2(表44)、およびカプセル中粉末(表45)の間に有意差はなかった。CmaxおよびAUC0−∞に基づいて製剤#2(表44)と比較した、製剤#1(表43)中の化合物2の相対的バイオアベイラビリティーの計算は、それぞれ、2mg/kgの用量で0.70および0.78の平均値を、ならびに5mg/kgの用量で1.22および1.34の平均値をもたらした。
When comparing mean C max , AUC0- TLast , and AUC 0-∞ at doses of 2 and 5 mg / kg, formulation # 1 (Table 43), formulation # 2 (Table 44), and powder in capsule (Table 45). There was no significant difference between). Calculations of the relative bioavailability of
このように、イヌ−ヒト薬物動態からの直接相関は入手できないが、経口液体製剤に対する経口カプセル製剤の投与後のイヌ−イヌ薬物動態の直接比較の結果を得ることができる。結果は、カプセル製剤のバイオアベイラビリティーが2種の経口液体製剤に類似することを実証する。図33Aおよび33Bに示されるように、試験された種における化合物2の血漿中濃度は、粉末製剤(表45)と液体製剤(表43)との間で同等である。血漿中半減期、クリアランス、分布容積、および平均滞留時間は、用量および製剤の全体にわたって実質的な相違を示さなかった。
Thus, although no direct correlation from dog-human pharmacokinetics is available, the results of a direct comparison of dog-dog pharmacokinetics after administration of the oral capsule formulation to the oral liquid formulation can be obtained. The results demonstrate that the bioavailability of the capsule formulation is similar to the two oral liquid formulations. As shown in FIGS. 33A and 33B, the plasma concentration of
実施形態および態様:
種々の組み合わせで含まれ得る、本開示の実施形態および態様は、以下を包含する:
実施形態1. びまん性真性橋膠腫(DIPG)の処置を必要とする対象のびまん性真性橋膠腫(DIPG)を処置する方法であって、前記方法は、治療有効量の式(2):
実施形態2. 前記結晶塩が、酸付加塩である、実施形態1に記載の方法。
実施形態3. 前記酸付加塩が、塩酸塩である、実施形態2に記載の方法。
実施形態4. 前記塩酸塩が、1価である、実施形態3に記載の方法。
実施形態5. 結晶塩形態が、無水物である、実施形態1から4のいずれか1つに記載の方法。
実施形態6. 前記対象が、小児患者である、実施形態1から5のいずれか1つに記載の方法。
実施形態7. 前記対象が、約1週齢から約22歳である、実施形態1から5のいずれか1つに記載の方法。
実施形態8. 前記対象が、約18歳またはそれよりも若い、実施形態1から7のいずれか1つに記載の方法。
実施形態9. 前記対象が、約10歳またはそれよりも若い、実施形態1から8のいずれか1つに記載の方法。
実施形態10. 前記対象が、約8歳またはそれよりも若い、実施形態1から9のいずれか1つに記載の方法。
実施形態11. 前記対象が、約6歳またはそれよりも若い、実施形態1から10のいずれか1つに記載の方法。
実施形態12. 前記DIPGが、新たに診断されたものまたは再発である、実施形態1から11のいずれか1つに記載の方法。
実施形態13. 前記DIPGが、浸潤性グリオーマ グレードIIからIVと組織学的診断がなされた脳橋腫瘍として特徴付けられる、実施形態1から12のいずれか1つに記載の方法。
実施形態14. 放射線療法を投与することをさらに含む、実施形態1から13のいずれか1つに記載の方法。
実施形態15. 放射線の投与が、前記化合物またはその結晶塩の投与の前に行われる、実施形態14に記載の方法。
実施形態16. 放射線の投与が、前記化合物またはその結晶塩の投与の後に行われる、実施形態15に記載の方法。
実施形態17. 放射線の投与が、前記化合物またはその結晶塩の投与の前および後の両方で行われる、実施形態16に記載の方法。
実施形態18. 1種またはそれより多種の追加の治療剤を投与することをさらに含む、実施形態1から17のいずれか1つに記載の方法。
実施形態19. 前記化合物またはその結晶塩の投与が、DIPGに関連する1つまたはそれより多くの徴候または症状を低減させまたは軽減させる、実施形態1から18のいずれか1つに記載の方法。
実施形態20. 前記1つまたはそれより多くの徴候または症状が、会話または会話パターンの変容、対象の顔または身体の片側を動かす能力の損失、バランスの損失、協調の損失、歩行または運動に関するトラブル、視覚の問題、聴覚の問題、頭痛、吐き気、嘔吐、異常な眠気、エネルギーレベルの変容、行動変化、および学校での成績の変化からなる群から選択される、実施形態19に記載の方法。
実施形態21. 前記化合物またはその結晶塩の投与が、無憎悪生存期間を実現する、実施形態1から20のいずれか1つに記載の方法。
実施形態22. 前記無憎悪生存期間が、1か月もしくはそれよりも長い、2か月もしくはそれよりも長い、3か月もしくはそれよりも長い、4か月もしくはそれよりも長い、5か月もしくはそれよりも長い、6か月もしくはそれよりも長い、7か月もしくはそれよりも長い、8か月もしくはそれよりも長い、9か月もしくはそれよりも長い、10か月もしくはそれよりも長い、11か月もしくはそれよりも長い、1年もしくはそれよりも長い、2年もしくはそれよりも長い、3年もしくはそれよりも長い、または5年もしくはそれよりも長い、実施形態21に記載の方法。
実施形態23. 腫瘍成長の減量術または脳脊髄液転換の1つまたは複数をさらに含む、実施形態1から22のいずれか1つに記載の方法。
実施形態24. 前記対象が、ACVR1遺伝子中に1つまたはそれより多くの変異を含む所定の遺伝子プロファイルを有する、実施形態1から23のいずれか1つに記載の方法。
実施形態25. ACVR1遺伝子中の前記1つまたはそれより多くの変異が、活性化変異である、実施形態24に記載の方法。
実施形態26. 前記ACVR1遺伝子中の1つまたはそれより多くの変異が、R206H、G328V、R258G、またはこれらの組合せから選択される、1つまたはそれより多くのアミノ酸残基におけるアミノ酸置換を含むACVR1ポリペプチドをコードする、実施形態25に記載の方法。
実施形態27. 前記ACVR1ポリペプチド中のアミノ酸置換がR206Hを含む、実施形態26に記載の方法。
実施形態28. 前記化合物またはその結晶塩が経口投与される、実施形態1から27のいずれか1つに記載の方法。
実施形態29. 前記化合物またはその結晶塩が、週当たり約10mgから約320mgに及ぶ用量で投与される、実施形態1から28のいずれか1つに記載の方法。
実施形態30. 前記用量が、週当たり約30mgから約240mgに及ぶ、実施形態29に記載の方法。
実施形態31. 前記用量が、週当たり約60mgから約180mgに及ぶ、実施形態29に記載の方法。
実施形態32. 前記用量が、週当たり約30mgから約120mgに及ぶ、実施形態29に記載の方法。
実施形態33. 前記用量が、週当たり約60mgから約120mgに及ぶ、実施形態29に記載の方法。
実施形態34. 前記用量が、週当たり約60mgである、実施形態29に記載の方法。
実施形態35. 前記用量が、週当たり約90mgである、実施形態29に記載の方法。
実施形態36. 前記用量が、週当たり約120mgである、実施形態29に記載の方法。
実施形態37. 前記用量が、週当たり約180mgである、実施形態29に記載の方法。
実施形態38. 前記用量が、週当たり約210mgである、実施形態29に記載の方法。
実施形態39. 前記用量が、週当たり約240mgである、実施形態29に記載の方法。
実施形態40. 前記化合物またはその結晶塩が、約320mgもしくはそれよりも少ない、約240mgもしくはそれよりも少ない、約210もしくはそれよりも少ない、約180もしくはそれよりも少ない、約120もしくはそれよりも少ない、約90もしくはそれよりも少ない、約60もしくはそれよりも少ない、または約30もしくはそれよりも少ない1週用量で投与される、実施形態1から28のいずれか1つに記載の方法。
実施形態41. 前記用量が、週当たり約60mgまたはそれよりも少ない、実施形態40に記載の方法。
実施形態42. 前記用量が、週当たり約90mgまたはそれよりも少ない、実施形態40に記載の方法。
実施形態43. 前記用量が、週当たり約120mgまたはそれよりも少ない、実施形態40に記載の方法。
実施形態44. 前記用量が、週当たり約180mgまたはそれよりも少ない、実施形態40に記載の方法。
実施形態45. 前記用量が、週当たり約210mgまたはそれよりも少ない、実施形態40に記載の方法。
実施形態46. 前記用量が、週当たり約240mgまたはそれよりも少ない、実施形態40に記載の方法。
実施形態47. 前記用量が、週に1回投与される、実施形態29から46のいずれか1つに記載の方法。
実施形態48. 前記用量が、1週間のクールにわたって2回またはそれを超える回数の部分用量、3回またはそれを超える回数の部分用量、4回またはそれを超える回数の部分用量、5回またはそれを超える回数の部分用量、6回またはそれを超える回数の部分用量、または毎日の部分用量で投与される、実施形態29から46のいずれか1つに記載の方法。
実施形態49. 前記対象が小児患者であり、前記用量が、前記用量範囲の約80%から100%の間である、実施形態29から48のいずれか1つに記載の方法。
実施形態50. 前記用量が、前記用量範囲の約80%、85%、90%、または95%に調節される、実施形態49に記載の方法。
実施形態51. 前記用量が、週当たり約8mgから約320mgに及ぶ、実施形態49に記載の方法。
実施形態52. 前記用量が、週当たり約24mgから約240mgに及ぶ、実施形態49に記載の方法。
実施形態53. 前記用量が、週当たり約24mgから約120mgに及ぶ、実施形態49に記載の方法。
実施形態54. 前記用量が、週当たり約48mgから約120mgに及ぶ、実施形態49に記載の方法。
実施形態55. 前記用量が、週当たり約72mgから約120mgに及ぶ、実施形態49に記載の方法。
実施形態56. 前記用量が、週当たり約96mgから約120mgに及ぶ、実施形態49に記載の方法。
実施形態57. 前記対象が、少なくとも約0.1ng/mLの所定のヘプシジンレベルを有する、実施形態1から56のいずれか1つに記載の方法。
実施形態58. 前記所定のヘプシジンレベルが、約10ng/mLから約200ng/mLに及ぶ、実施形態57に記載の方法。
実施形態59. 前記化合物またはその結晶塩が投与された後に、前記対象のヘプシジンレベルを決定することをさらに含む、実施形態1から58のいずれか1つに記載の方法。
実施形態60. 前記化合物またはその結晶塩が投与された後に、前記対象のトランスフェリン飽和レベルを決定することをさらに含む、実施形態1から59のいずれか1つに記載の方法。
実施形態61. 前記トランスフェリン飽和レベルが、約50%未満である、実施形態60に記載の方法。
実施形態62. 前記トランスフェリン飽和レベルが、約45%未満である、実施形態60に記載の方法。
実施形態63. 前記トランスフェリン飽和レベルが、約40%未満である、実施形態60に記載の方法。
実施形態64. 前記化合物またはその結晶塩が、1回またはそれを超える回数の処置サイクルにわたり投与され、各サイクルが4週を含む、実施形態1から63のいずれか1つに記載の方法。
実施形態65. 前記方法が、処置サイクル間に1日またはそれを超える日数の休処置日をさらに含む、実施形態64に記載の方法。
実施形態66. 前記休処置日が、1日、2日、3日、4日、5日、6日、1週、2週、3週、または4週から選択される、実施形態65に記載の方法。
実施形態67. ACVR1の阻害の影響を受け易い疾患または障害を処置するための方法であって、前記方法は、ACVR1の阻害の影響を受け易い疾患または障害の処置を必要とする対象に、治療有効量の式(2):
実施形態68. 前記用量が、週当たり約30mgから約240mgに及ぶ、実施形態67に記載の方法。
実施形態69. 前記用量が、週当たり約60mgから約180mgに及ぶ、実施形態67に記載の方法。
実施形態70. 前記用量が、週当たり約30mgから約120mgに及ぶ、実施形態67に記載の方法。
実施形態71. 前記用量が、週当たり約60mgから約120mgに及ぶ、実施形態67に記載の方法。
実施形態72. 前記用量が、週当たり約60mgである、実施形態67に記載の方法。
実施形態73. 前記用量が、週当たり約90mgである、実施形態67に記載の方法。
実施形態74. 前記用量が、週当たり約120mgである、実施形態67に記載の方法。
実施形態75. 前記用量が、週当たり約180mgである、実施形態67に記載の方法。
実施形態76. 前記用量が、週当たり約210mgである、実施形態67に記載の方法。
実施形態77. 前記用量が、週当たり約240mgである、実施形態67に記載の方法。
実施形態78. 前記化合物またはその結晶塩が、約320mgもしくはそれよりも少ない、約240mgもしくはそれよりも少ない、約210もしくはそれよりも少ない、約180もしくはそれよりも少ない、約120もしくはそれよりも少ない、約90もしくはそれよりも少ない、約60もしくはそれよりも少ない、または約30もしくはそれよりも少ない1週用量で投与される、実施形態67から77のいずれか1つに記載の方法。
実施形態79. 前記用量が、週当たり約60mgまたはそれよりも少ない、実施形態78に記載の方法。
実施形態80. 前記用量が、週当たり約90mgまたはそれよりも少ない、実施形態78に記載の方法。
実施形態81. 前記用量が、週当たり約120mgまたはそれよりも少ない、実施形態78に記載の方法。
実施形態82. 前記用量が、週当たり約180mgまたはそれよりも少ない、実施形態78に記載の方法。
実施形態83. 前記用量が、週当たり約210mgまたはそれよりも少ない、実施形態78に記載の方法。
実施形態84. 前記用量が、週当たり約240mgまたはそれよりも少ない、実施形態78に記載の方法。
実施形態85. 前記対象が小児患者であり、前記用量が、前記用量範囲の約80%から100%の間である、実施形態67から84のいずれか1つに記載の方法。
実施形態86. 前記用量が、前記用量範囲の約80%、85%、90%、または95%である、実施形態85に記載の方法。
実施形態87. 前記用量が、週当たり約8mgから約320mgに及ぶ、実施形態85に記載の方法。
実施形態88. 前記用量が、週当たり約24mgから約240mgに及ぶ、実施形態85に記載の方法。
実施形態89. 前記用量が、週当たり約24mgから約120mgに及ぶ、実施形態85に記載の方法。
実施形態90. 前記用量が、週当たり約48mgから約120mgに及ぶ、実施形態85に記載の方法。
実施形態91. 前記用量が、週当たり約72mgから約120mgに及ぶ、実施形態85に記載の方法。
実施形態92. 前記用量が、週当たり約96mgから約120mgに及ぶ、実施形態85に記載の方法。
実施形態93. 前記用量が、1週間に1回投与される、実施形態67から92のいずれか1つに記載の方法。
実施形態94. 前記用量が、1週間のクールにわたって2回の部分用量で投与される、実施形態67から92のいずれか1つに記載の方法。
実施形態95. 前記用量が、1週間のクールにわたって3回の部分用量で投与される、実施形態67から92のいずれか1つに記載の方法。
実施形態96. 前記用量が、1週間のクールにわたって4回の部分用量で投与される、実施形態67から92のいずれか1つに記載の方法。
実施形態97. 前記用量が、1週間のクールにわたって5回の部分用量で投与される、実施形態67から92のいずれか1つに記載の方法。
実施形態98. 前記用量が、1週間のクールにわたって6回の部分用量で投与される、実施形態67から92のいずれか1つに記載の方法。
実施形態99. 前記用量が、毎日の部分用量で投与される、実施形態67から92のいずれか1つに記載の方法。
実施形態100. 前記対象が、ACVR1遺伝子中に1つまたはそれより多くの変異を含む所定の遺伝子プロファイルを有する、実施形態67から99のいずれか1つに記載の方法。
実施形態101. ACVR1遺伝子中の前記1つまたはそれより多くの変異が、活性化変異である、実施形態100に記載の方法。
実施形態102. 前記ACVR1遺伝子中の1つまたはそれより多くの変異が、R206H、G328V、R258G、またはこれらの組合せから選択される、1つまたはそれより多くのアミノ酸残基におけるアミノ酸置換を含むACVR1ポリペプチドをコードする、実施形態101に記載の方法。
実施形態103. 前記ACVR1ポリペプチド中のアミノ酸置換が、R206Hを含む、実施形態102に記載の方法。
実施形態104. 前記化合物またはその結晶塩が、経口投与される、実施形態67から103のいずれか1つに記載の方法。
実施形態105. 前記疾患または障害が、びまん性真性橋膠腫、グレードIIからIVの浸潤性グリオーマと組織学的診断がなされた脳橋腫瘍、固形腫瘍、進行性骨化性線維異形成症、および慢性疾患の貧血症の1つまたは複数から選択される、実施形態67から104のいずれか1つに記載の方法。
実施形態106. 前記化合物またはその結晶塩が、固体用量製剤として投与される、実施形態105に記載の方法。
実施形態107. 前記化合物またはその結晶塩が、液体用量製剤として投与される、実施形態105に記載の方法。
実施形態108. 前記対象が、用量の任意の修正を決定するためにヘプシジンレベルに関してモニタリングされる、実施形態67から107のいずれか1つに記載の方法。
実施形態109. 前記対象が、1つまたはそれより多くの臓器における前記化合物の蓄積に関してモニタリングされる、実施形態67から108のいずれか1つに記載の方法。
実施形態110. 前記結晶塩が、酸付加塩である、実施形態67から109のいずれか1つに記載の方法。
実施形態111. 前記酸付加塩が、塩酸塩である、実施形態110に記載の方法。
実施形態112. 前記塩酸塩が、1価である、実施形態111に記載の方法。
実施形態113. 前記結晶塩形態が、無水物である、実施形態110から112のいずれか1つに記載の方法。
実施形態114. 1種またはそれより多種の薬学的に許容される賦形剤と、式(2):
実施形態115. 前記結晶塩が、酸付加塩である、実施形態114に記載の医薬組成物。
実施形態116. 前記酸付加塩が、塩酸塩である、実施形態115に記載の医薬組成物。
実施形態117. 前記塩酸塩が、1価である、実施形態116に記載の医薬組成物。
実施形態118. 前記結晶塩形態が、無水物である、実施形態115から117のいずれか1つに記載の医薬組成物。
実施形態119. 前記医薬組成物が、ゼラチンカプセルである、実施形態114から118のいずれか1つに記載の医薬組成物。
実施形態120. 前記ゼラチンカプセルが、遊離塩基重量に対して(i)5mg、(ii)25mg、または(iii)125mgの力価である、実施形態119に記載の医薬組成物。
実施形態121. 前記ゼラチンカプセルが、遊離塩基重量に対して(i)30mg、(ii)60mg、(iii)90mg、または(iv)120mgである、実施形態119に記載の医薬組成物。
実施形態122. 前記1種またはそれより多種の医薬賦形剤が、微結晶性セルロース、ラクトース一水和物、クロスカルメロースナトリウム、ステアリン酸マグネシウム、またはこれらの組合せから選択される、実施形態114から121のいずれか1つに記載の医薬組成物。
実施形態123. 式(2):
(i)1種またはそれより多種の緩衝剤;
(ii)必要に応じて、1種またはそれより多種の保存剤;
(iii)必要に応じて、1種またはそれより多種の溶媒;
(iv)必要に応じて、1種またはそれより多種の矯味剤;および
(v)必要に応じて、1種またはそれより多種のさらなるpH調節剤
とを含む、経口液体医薬組成物。
実施形態124. 前記結晶塩が、酸付加塩である、実施形態123に記載の医薬組成物。
実施形態125. 前記酸付加塩が、塩酸塩である、実施形態124に記載の医薬組成物。
実施形態126. 前記塩酸塩が、1価である、実施形態125に記載の医薬組成物。
実施形態127. 前記結晶塩形態が、無水物である、実施形態124から126のいずれか1つに記載の医薬組成物。
実施形態128. 前記組成物が約2.0から約5.0の間のpHを有する、実施形態123から127のいずれか1つに記載の医薬組成物。
実施形態129. 前記組成物が約2.0から約3.5の間のpHを有する、実施形態128に記載の医薬組成物。
実施形態130. 前記組成物が約2.0のpHを有する、実施形態128に記載の医薬組成物。
実施形態131. 前記緩衝剤が、クエン酸、酒石酸、リンゴ酸、または酢酸から選択される、実施形態123から130のいずれか1つに記載の医薬組成物。
実施形態132. 前記緩衝剤が、リンゴ酸である、実施形態131に記載の医薬組成物。
実施形態133. 前記リンゴ酸が、DL−リンゴ酸である、実施形態132に記載の医薬組成物。
実施形態134. 1種またはそれより多種の保存剤を含む、実施形態123から133のいずれか1つに記載の医薬組成物。
実施形態135. 前記保存剤が、安息香酸、安息香酸ナトリウム、パラヒドロキシ安息香酸メチル、パラヒドロキシ安息香酸プロピル、またはプロピレングリコールから選択される、実施形態134に記載の医薬組成物。
実施形態136. 前記保存剤が、安息香酸である、実施形態135に記載の医薬組成物。
実施形態137. 前記安息香酸が、保存剤および緩衝剤である、実施形態136に記載の医薬組成物。
実施形態138. 1種またはそれより多種の矯味剤を含む、実施形態123から137のいずれか1つに記載の医薬組成物。
実施形態139. 前記矯味剤が、スクラロース、グリセリン、シクロデキストリン、HP−β−シクロデキストリン、α−シクロデキストリン、β−シクロデキストリン、またはこれらの組合せから選択される、実施形態138に記載の医薬組成物。
実施形態140. 前記矯味剤が、HP−β−シクロデキストリンおよびスクラロースの組合せである、実施形態138に記載の医薬組成物。
実施形態141. 前記組成物が、式(2)の化合物またはその結晶塩を、約10mg/mLの濃度で含む、実施形態123から140のいずれか1つに記載の医薬組成物。
実施形態142. 前記組成物が、リンゴ酸を約6.7mg/mLまでの濃度で含む、実施形態123から141のいずれか1つに記載の医薬組成物。
実施形態143. 前記組成物が、リンゴ酸を約1.3mg/mLの濃度で含む、実施形態142に記載の医薬組成物。
実施形態144. 前記組成物が、HP−β−シクロデキストリンを約300mg/mLまでの濃度で含む、実施形態123から143のいずれか1つに記載の医薬組成物。
実施形態145. 前記組成物が、HP−β−シクロデキストリンを約150mg/mLまでの濃度で含む、実施形態144に記載の医薬組成物。
実施形態146. 前記組成物が、スクラロースを約2.0mg/mLまでの濃度で含む、実施形態123から145のいずれか1つに記載の医薬組成物。
実施形態147. 前記組成物が、スクラロースを約1.0mg/mLの濃度で含む、実施形態146に記載の医薬組成物。
実施形態148. 前記組成物が、安息香酸を約3.0mg/mLまでの濃度で含む、実施形態123から147のいずれか1つに記載の医薬組成物。
実施形態149. 前記組成物が、安息香酸を約2.0mg/mLまでの濃度で含む、実施形態148に記載の医薬組成物。
実施形態150. 前記pHが、塩酸で約2.0に調節される、実施形態123から149のいずれか1つに記載の医薬組成物。
実施形態151. 前記溶媒が水である、実施形態123から150のいずれか1つに記載の医薬組成物。
実施形態152. 化合物(2)
実施形態153. 前記塩が、薬学的に許容される塩である、実施形態152に記載の結晶形態。
実施形態154. 前記薬学的に許容される塩が、HCl塩である、実施形態153に記載の結晶形態。
実施形態155. 形態Aを含む、実施形態152から154のいずれか1つに記載の結晶形態。
実施形態156. 前記形態Aから本質的になる、実施形態152から154のいずれか1つに記載の結晶形態。
実施形態157. 形態Aが、不純物を実質的に含まない、実施形態155または156に記載の結晶形態。
実施形態158. N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の、化合物形態A。
実施形態159. 13.53、16.14、17.67、18.38、24.96、および28.18から選択される1つまたは複数の2θ値を含むx線回折パターン(XRPD)を特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態A。
実施形態160. 前記形態が、列挙される2θ値のうちの2つまたはそれよりも多くを特徴とする、実施形態159に記載の形態A。
実施形態161. 前記形態が、列挙される2θ値のうちの3つまたはそれよりも多くを特徴とする、実施形態159に記載の形態A。
実施形態162. 前記形態が、列挙される2θ値のうちの4つまたはそれよりも多くを特徴とする、実施形態159に記載の形態A。
実施形態163. 前記形態が、列挙される2θ値のうちの5つまたはそれよりも多くを特徴とする、実施形態159に記載の形態A。
実施形態164. 前記形態が、列挙される2θ値の6つ全てを特徴とする、実施形態159に記載の形態A。
実施形態165. 6.71、19.25、23.98、および29.60から選択される1つまたは複数の2θ値をさらに特徴とする、実施形態157から164のいずれか1つに記載の形態A。
実施形態166. 前記形態が、列挙される2θ値のうちの2つまたはそれよりも多くを特徴とする、実施形態165に記載の形態A。
実施形態167. 前記形態が、列挙される2θ値のうちの3つまたはそれよりも多くを特徴とする、実施形態165に記載の形態A。
実施形態168. 前記形態が、列挙される2θ値のうちの4つ全てを特徴とする、実施形態165に記載の形態A。
実施形態169. 前記X線粉末回折計が、Cu kαのX線波長、Kα1(Å):1.540598、Kα2(Å):1.544426を使用して、Kα2/Kα1強度比を0.50、そしてX線管の設定を45kV、40mAにして反射モードで使用される、実施形態158から168のいずれか1つに記載の形態A。
実施形態170. 前記2θ値が、±0.2 2θ以内である、実施形態158から169のいずれか1つに記載の形態A。
実施形態171. 図12と実質的に同じx線回折パターン(XRPD)を特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態A。
実施形態172. 196.2℃、214.8℃、および274.0℃のうちの1つまたは複数での吸熱を特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態A。
実施形態173. 198.9℃、218.0℃、および275.9℃のうちの1つまたは複数でのピーク吸熱をさらに特徴とする、実施形態172に記載の形態A。
実施形態174. 274.0℃の開始温度をさらに特徴とする、実施形態172または173に記載の形態A。
実施形態175. 150℃までで1.7%の重量損失をさらに特徴とする、実施形態172から174のいずれか1つに記載の形態A。
実施形態176. 図15と実質的に同じTGA−DSCサーモグラムを特徴とする、N4−(2,2’−ビピリジン−3−イル)−N2−(3−メトキシ−4−(4−メチルピペラジン−1−イル)フェニル)ピリミジン−2,4−ジアミン無水塩酸塩の化合物形態A。
実施形態177. 実施形態152から176のいずれか1つに記載の結晶形態を含む医薬組成物。
実施形態178. 固体用量製剤としての、実施形態176に記載の医薬組成物。
実施形態179. 前記化合物が、実施形態152から176のいずれか1つに記載の結晶形態である、実施形態114から122のいずれか1つに記載の経口固体医薬組成物。
実施形態180. 液体用量製剤としての、実施形態176に記載の医薬組成物。
実施形態181. 前記化合物が、実施形態152から176のいずれか1つに記載の結晶形態である、実施形態123から151のいずれか1つに記載の経口液体医薬組成物。
実施形態182. 前記化合物が、実施形態152から176のいずれか1つに記載の結晶形態である、実施形態1から113のいずれか1つに記載の方法。
Embodiments and embodiments:
Embodiments and embodiments of the present disclosure that may be included in various combinations include:
Embodiment 14. The method according to any one of
Embodiment 19. The method of any one of embodiments 1-18, wherein administration of the compound or crystalline salt thereof reduces or alleviates one or more signs or symptoms associated with DIPG.
20. One or more of the above symptoms or symptoms may be changes in conversation or conversation patterns, loss of ability to move one side of the subject's face or body, loss of balance, loss of coordination, walking or motor problems, visual problems. 19. The method of embodiment 19, wherein the method is selected from the group consisting of hearing problems, headache, nausea, vomiting, abnormal drowsiness, altered energy levels, behavioral changes, and altered school performance.
21. The method according to any one of
Embodiment 22. The hateless survival time is one month or longer, two months or longer, three months or longer, four months or longer, five months or longer. Long, 6 months or longer, 7 months or longer, 8 months or longer, 9 months or longer, 10 months or longer, 11
23. The method according to any one of
Embodiment 27. 26. The method of
Embodiment 29. The method of any one of embodiments 1-28, wherein the compound or crystalline salt thereof is administered at a dose ranging from about 10 mg to about 320 mg per week.
Embodiment 31. 29. The method of embodiment 29, wherein the dose ranges from about 60 mg to about 180 mg per week.
Embodiment 32. 29. The method of embodiment 29, wherein the dose ranges from about 30 mg to about 120 mg per week.
Embodiment 33. 29. The method of embodiment 29, wherein the dose ranges from about 60 mg to about 120 mg per week.
Embodiment 34. 29. The method of embodiment 29, wherein the dose is about 60 mg per week.
Embodiment 36. 29. The method of embodiment 29, wherein the dose is about 120 mg per week.
Embodiment 37. 29. The method of embodiment 29, wherein the dose is about 180 mg per week.
Embodiment 38. 29. The method of embodiment 29, wherein the dose is about 210 mg per week.
Embodiment 39. 29. The method of embodiment 29, wherein the dose is about 240 mg per week.
Embodiment 41. 40. The method of
Embodiment 42. 40. The method of
Embodiment 43. 40. The method of
Embodiment 46. 40. The method of
Embodiment 48. The dose is 2 or more partial doses, 3 or more partial doses, 4 or more partial doses, 5 or more partial doses over a week's course. The method of any one of embodiments 29-46, which is administered in a partial dose, 6 or more partial doses, or a daily partial dose.
Embodiment 49. The method of any one of embodiments 29-48, wherein the subject is a pediatric patient and the dose is between about 80% and 100% of the dose range.
Embodiment 51. 49. The method of embodiment 49, wherein the dose ranges from about 8 mg to about 320 mg per week.
Embodiment 52. 49. The method of embodiment 49, wherein the dose ranges from about 24 mg to about 240 mg per week.
Embodiment 53. 49. The method of embodiment 49, wherein the dose ranges from about 24 mg to about 120 mg per week.
Embodiment 54. 49. The method of embodiment 49, wherein the dose ranges from about 48 mg to about 120 mg per week.
Embodiment 55. 49. The method of embodiment 49, wherein the dose ranges from about 72 mg to about 120 mg per week.
Embodiment 56. 49. The method of embodiment 49, wherein the dose ranges from about 96 mg to about 120 mg per week.
Embodiment 57. The method of any one of embodiments 1-56, wherein the subject has a predetermined hepcidin level of at least about 0.1 ng / mL.
Embodiment 58. 58. The method of embodiment 57, wherein the predetermined hepcidin level ranges from about 10 ng / mL to about 200 ng / mL.
Embodiment 59. The method of any one of embodiments 1-58, further comprising determining the hepcidin level of the subject after administration of the compound or crystalline salt thereof.
Embodiment 61. The method of
Embodiment 62. The method of
Embodiment 63. The method of
Embodiment 64. The method of any one of embodiments 1-63, wherein the compound or crystalline salt thereof is administered over one or more treatment cycles, each cycle comprising 4 weeks.
Embodiment 65. 13. The method of embodiment 64, wherein the method further comprises one or more rest days between treatment cycles.
Embodiment 66. The method according to embodiment 65, wherein the rest day is selected from 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, or 4 weeks.
Embodiment 67. A method for treating a disease or disorder that is susceptible to inhibition of ACVR1, said method is a therapeutically effective amount formula for a subject requiring treatment of a disease or disorder that is susceptible to inhibition of ACVR1. (2):
Embodiment 68. 67. The method of embodiment 67, wherein the dose ranges from about 30 mg to about 240 mg per week.
Embodiment 69. 67. The method of embodiment 67, wherein the dose ranges from about 60 mg to about 180 mg per week.
Embodiment 71. 67. The method of embodiment 67, wherein the dose ranges from about 60 mg to about 120 mg per week.
Embodiment 72. 67. The method of embodiment 67, wherein the dose is about 60 mg per week.
Embodiment 73. 67. The method of embodiment 67, wherein the dose is about 90 mg per week.
Embodiment 74. 67. The method of embodiment 67, wherein the dose is about 120 mg per week.
Embodiment 76. 67. The method of embodiment 67, wherein the dose is about 210 mg per week.
Embodiment 77. 67. The method of embodiment 67, wherein the dose is about 240 mg per week.
Embodiment 78. The compound or crystalline salt thereof is about 320 mg or less, about 240 mg or less, about 210 or less, about 180 or less, about 120 or less, about 90. 30. The method of any one of embodiments 67-77, which is administered at a weekly dose of about 60 or less, or about 30 or less.
Embodiment 79. 28. The method of embodiment 78, wherein the dose is about 60 mg or less per week.
80. 28. The method of embodiment 78, wherein the dose is about 90 mg or less per week.
Embodiment 81. 28. The method of embodiment 78, wherein the dose is about 120 mg or less per week.
Embodiment 82. 28. The method of embodiment 78, wherein the dose is about 180 mg or less per week.
Embodiment 83. 28. The method of embodiment 78, wherein the dose is about 210 mg or less per week.
Embodiment 84. 28. The method of embodiment 78, wherein the dose is about 240 mg or less per week.
Embodiment 85. The method of any one of embodiments 67-84, wherein the subject is a pediatric patient and the dose is between about 80% and 100% of the dose range.
Embodiment 86. 85. The method of embodiment 85, wherein the dose is about 80%, 85%, 90%, or 95% of the dose range.
Embodiment 87. The method of embodiment 85, wherein the dose ranges from about 8 mg to about 320 mg per week.
Embodiment 88. The method of embodiment 85, wherein the dose ranges from about 24 mg to about 240 mg per week.
Embodiment 89. The method of embodiment 85, wherein the dose ranges from about 24 mg to about 120 mg per week.
Embodiment 91. The method of embodiment 85, wherein the dose ranges from about 72 mg to about 120 mg per week.
Embodiment 92. The method of embodiment 85, wherein the dose ranges from about 96 mg to about 120 mg per week.
Embodiment 93. The method of any one of embodiments 67-92, wherein the dose is administered once a week.
Embodiment 94. The method of any one of embodiments 67-92, wherein the dose is administered in two partial doses over a one-week course.
Embodiment 96. The method of any one of embodiments 67-92, wherein the dose is administered in four partial doses over a one-week course.
Embodiment 97. The method of any one of embodiments 67-92, wherein the dose is administered in 5 partial doses over a week of cool.
Embodiment 98. The method of any one of embodiments 67-92, wherein the dose is administered in 6 partial doses over a week of cool.
Embodiment 99. The method of any one of embodiments 67-92, wherein the dose is administered in daily partial doses.
Embodiment 101. The method of
Embodiment 102. One or more mutations in the ACVR1 gene encode an ACVR1 polypeptide containing amino acid substitutions at one or more amino acid residues selected from R206H, G328V, R258G, or a combination thereof. The method according to embodiment 101.
Embodiment 103. 10. The method of embodiment 102, wherein the amino acid substitution in the ACVR1 polypeptide comprises R206H.
Embodiment 104. The method according to any one of embodiments 67 to 103, wherein the compound or a crystalline salt thereof is orally administered.
Embodiment 105. The disease or disorder is diffuse true pontine glioma, pontine tumor histologically diagnosed as grade II to IV invasive glioma, solid tumor, progressive ossifying fibrodysplasia, and chronic disease. The method of any one of embodiments 67-104, which is selected from one or more of anemia.
Embodiment 107. The method according to embodiment 105, wherein the compound or a crystalline salt thereof is administered as a liquid dose preparation.
Embodiment 109. The method of any one of embodiments 67-108, wherein the subject is monitored for accumulation of the compound in one or more organs.
Embodiment 110. The method according to any one of embodiments 67 to 109, wherein the crystalline salt is an acid addition salt.
Embodiment 112. 11. The method of
Embodiment 113. The method according to any one of embodiments 110 to 112, wherein the crystalline salt form is anhydrous.
Embodiment 114. One or more pharmaceutically acceptable excipients and formula (2):
Embodiment 115. The pharmaceutical composition according to embodiment 114, wherein the crystalline salt is an acid addition salt.
Embodiment 116. The pharmaceutical composition according to embodiment 115, wherein the acid addition salt is a hydrochloride salt.
Embodiment 117. The pharmaceutical composition according to embodiment 116, wherein the hydrochloride salt is monovalent.
Embodiment 118. The pharmaceutical composition according to any one of embodiments 115 to 117, wherein the crystalline salt form is anhydrous.
Embodiment 119. The pharmaceutical composition according to any one of embodiments 114 to 118, wherein the pharmaceutical composition is a gelatin capsule.
Embodiment 121. 119. The pharmaceutical composition according to embodiment 119, wherein the gelatin capsule is (i) 30 mg, (ii) 60 mg, (iii) 90 mg, or (iv) 120 mg based on the weight of the free base.
Embodiment 122. Any of embodiments 114-121, wherein one or more pharmaceutical excipients are selected from microcrystalline cellulose, lactose monohydrate, sodium croscarmellose, magnesium stearate, or a combination thereof. The pharmaceutical composition according to one.
Embodiment 123. Equation (2):
(I) One or more buffers;
(Ii) One or more preservatives as needed;
(Iii) One or more solvents as needed;
An oral liquid pharmaceutical composition comprising (iv) one or more flavoring agents as needed; and (v) one or more additional pH regulators as needed.
Embodiment 124. The pharmaceutical composition according to embodiment 123, wherein the crystalline salt is an acid addition salt.
Embodiment 126. The pharmaceutical composition according to
Embodiment 127. The pharmaceutical composition according to any one of embodiments 124 to 126, wherein the crystalline salt form is anhydrous.
Embodiment 128. The pharmaceutical composition according to any one of embodiments 123 to 127, wherein the composition has a pH between about 2.0 and about 5.0.
Embodiment 129. The pharmaceutical composition according to embodiment 128, wherein the composition has a pH between about 2.0 and about 3.5.
Embodiment 130. The pharmaceutical composition according to embodiment 128, wherein the composition has a pH of about 2.0.
Embodiment 131. The pharmaceutical composition according to any one of embodiments 123 to 130, wherein the buffer is selected from citric acid, tartaric acid, malic acid, or acetic acid.
Embodiment 132. The pharmaceutical composition according to embodiment 131, wherein the buffer is malic acid.
Embodiment 133. The pharmaceutical composition according to embodiment 132, wherein the malic acid is DL-malic acid.
Embodiment 134. The pharmaceutical composition according to any one of embodiments 123 to 133, comprising one or more preservatives.
Embodiment 135. The pharmaceutical composition according to embodiment 134, wherein the preservative is selected from benzoic acid, sodium benzoate, methyl parahydroxybenzoate, propyl parahydroxybenzoate, or propylene glycol.
Embodiment 136. The pharmaceutical composition according to embodiment 135, wherein the preservative is benzoic acid.
Embodiment 137. The pharmaceutical composition according to embodiment 136, wherein the benzoic acid is a preservative and a buffer.
Embodiment 138. The pharmaceutical composition according to any one of embodiments 123 to 137, comprising one or more flavoring agents.
Embodiment 139. The pharmaceutical composition according to embodiment 138, wherein the flavoring agent is selected from sucralose, glycerin, cyclodextrin, HP-β-cyclodextrin, α-cyclodextrin, β-cyclodextrin, or a combination thereof.
Embodiment 141. The pharmaceutical composition according to any one of embodiments 123 to 140, wherein the composition comprises the compound of formula (2) or a crystalline salt thereof at a concentration of about 10 mg / mL.
Embodiment 142. The pharmaceutical composition according to any one of embodiments 123 to 141, wherein the composition comprises malic acid at a concentration of up to about 6.7 mg / mL.
Embodiment 143. The pharmaceutical composition according to embodiment 142, wherein the composition comprises malic acid at a concentration of about 1.3 mg / mL.
Embodiment 144. The pharmaceutical composition according to any one of embodiments 123 to 143, wherein the composition comprises HP-β-cyclodextrin at a concentration of up to about 300 mg / mL.
Embodiment 145. The pharmaceutical composition according to embodiment 144, wherein the composition comprises HP-β-cyclodextrin at a concentration of up to about 150 mg / mL.
Embodiment 146. The pharmaceutical composition according to any one of embodiments 123 to 145, wherein the composition comprises sucralose at a concentration of up to about 2.0 mg / mL.
Embodiment 147. The pharmaceutical composition according to embodiment 146, wherein the composition comprises sucralose at a concentration of about 1.0 mg / mL.
Embodiment 148. The pharmaceutical composition according to any one of embodiments 123 to 147, wherein the composition comprises benzoic acid at a concentration of up to about 3.0 mg / mL.
Embodiment 149. The pharmaceutical composition according to embodiment 148, wherein the composition comprises benzoic acid at a concentration of up to about 2.0 mg / mL.
Embodiment 151. The pharmaceutical composition according to any one of embodiments 123 to 150, wherein the solvent is water.
Embodiment 152. Compound (2)
Embodiment 153. The crystalline form according to embodiment 152, wherein the salt is a pharmaceutically acceptable salt.
Embodiment 154. The crystalline form according to embodiment 153, wherein the pharmaceutically acceptable salt is an HCl salt.
Embodiment 155. The crystal form according to any one of embodiments 152 to 154, comprising embodiment A.
Embodiment 156. The crystal form according to any one of embodiments 152 to 154, which is essentially the same as the form A.
Embodiment 157. The crystalline form according to embodiment 155 or 156, wherein Form A is substantially free of impurities.
Embodiment 158. Of (3-methoxy-4- (4-methylpiperazin-1-yl) phenyl) pyrimidine-2,4-diamine anhydrous hydrochloride, - N 4 - (2,2'- bipyridine-3-yl) -N 2 Compound form A.
Embodiment 159. N, characterized by an x-ray diffraction pattern (XRPD) containing one or more 2θ values selected from 13.53, 16.14, 17.67, 18.38, 24.96, and 28.18. 4 - (2,2'-bipyridine-3-yl) -N 2 - (3- methoxy-4- (4-methylpiperazin-1-yl) phenyl) compound form of pyrimidine-2,4-diamine in anhydrous hydrochloride A.
Embodiment 161. Embodiment A according to embodiment 159, wherein the embodiment is characterized by three or more of the listed 2θ values.
Embodiment 162. Embodiment A according to embodiment 159, wherein the embodiment is characterized by four or more of the listed 2θ values.
Embodiment 163. Embodiment A according to embodiment 159, wherein the embodiment is characterized by five or more of the listed 2θ values.
Embodiment 164. The embodiment A according to the embodiment 159, wherein the embodiment is characterized by all six of the listed 2θ values.
Embodiment 165. A form according to any one of embodiments 157 to 164, further comprising one or more 2θ values selected from 6.71, 19.25, 23.98, and 29.60.
Embodiment 166. Embodiment A according to embodiment 165, wherein the embodiment is characterized by two or more of the listed 2θ values.
Embodiment 167. Embodiment A according to embodiment 165, wherein the embodiment is characterized by three or more of the listed 2θ values.
Embodiment 168. The embodiment A according to the embodiment 165, wherein the embodiment features all four of the listed 2θ values.
Embodiment 169. The X-ray powder diffractometer uses the X-ray wavelength of Cu kα, Kα1 (Å): 1.540598, Kα2 (Å): 1.5444426, and has a Kα2 / Kα1 intensity ratio of 0.50, and X-rays. The embodiment A according to any one of embodiments 158 to 168, wherein the tube is set to 45 kV, 40 mA and used in the reflection mode.
Embodiment 170. The embodiment A according to any one of embodiments 158 to 169, wherein the 2θ value is within ± 0.2 2θ.
Embodiment 171. And wherein Figure 12 is substantially the same
Embodiment 172. 196.2 ° C., characterized by endotherm at one or more of 214.8 ° C., and 274.0 ℃, N 4 - (2,2'- bipyridine-3-yl) -N 2 - ( Compound form A of 3-methoxy-4- (4-methylpiperazin-1-yl) phenyl) pyrimidin-2,4-diamine anhydride.
Embodiment 173. A.
Embodiment 174. A. Of the embodiment 172 or 173, further characterized by a starting temperature of 274.0 ° C.
Embodiment 175. The A according to any one of embodiments 172 to 174, further characterized by a weight loss of 1.7% up to 150 ° C.
Embodiment 176. Figure 15 is substantially characterized in the same TGA-DSC thermogram and, N 4 - (2,2'-bipyridine-3-yl) -N 2 - (3- methoxy-4- (4-methylpiperazin -1 -Il) Phenyl) Pyrimidine-2,4-Diamine Anhydrous Hydrochloride Compound Form A.
Embodiment 177. A pharmaceutical composition comprising the crystalline form according to any one of embodiments 152 to 176.
Embodiment 178. The pharmaceutical composition according to embodiment 176 as a solid-dose formulation.
Embodiment 179. The oral solid pharmaceutical composition according to any one of embodiments 114 to 122, wherein the compound is in the crystalline form according to any one of embodiments 152 to 176.
Embodiment 181. The oral liquid pharmaceutical composition according to any one of embodiments 123 to 151, wherein the compound is in the crystalline form according to any one of embodiments 152 to 176.
Embodiment 182. The method according to any one of
様々な組合せで組み込まれ得る、本開示のさらなる実施形態および態様は、下記を含む:
実施形態1’. ある疾患の処置を必要とする対象の、ある疾患を処置する方法であって、前記方法は、ACVR1阻害剤を含む処置レジメンを、ACVR1遺伝子中に1つまたはそれより多くの変異を含む所定の遺伝子プロファイルを有する前記対象に投与することを含み、前記ACVR1阻害剤が、下記の構造(I):
R1は、H、またはC1〜C6アルコキシであり;
R2は、C1〜C6アルコキシまたはヘテロシクリルであり;
R3は、ハロ、またはC1〜C6アルコキシであり;
R4は、H、またはC1〜C6アルキルである)
またはその立体異性体、薬学的に許容される塩、互変異性体、もしくはプロドラッグを有する、方法。
実施形態2’. 前記疾患ががんである、実施形態1’に記載の方法。
実施形態3’. 前記がんが固形がんである、実施形態2’に記載の方法。
実施形態4’. 前記がんが、脳がん、子宮がん、卵巣がん、子宮頸がん、肺がん、乳がん、結腸がん、胃腸がん、造血もしくはリンパ系がん、皮膚がん、または骨がんである、実施形態2’または3’に記載の方法。
実施形態5’. 前記がんが脳がんである、実施形態2’〜4’のいずれか1つに記載の方法。
実施形態6’. 前記脳がんが、脳幹グリオーマである、実施形態5’に記載の方法。
実施形態7’. 前記脳がんが、びまん性真性橋膠腫(DIPG)である、実施形態5’または6’に記載の方法。
実施形態8’. 前記がんが、子宮、卵巣、または子宮頸がんである、実施形態2’〜4’のいずれか1つに記載の方法。
実施形態9’. 前記子宮がんが、子宮内膜がんである、実施形態8’に記載の方法。
実施形態10’. 前記がんが、肺がんである、実施形態2’〜4’のいずれか1つに記載の方法。
実施形態11’. 前記肺がんが、非小細胞性肺がんである、実施形態10’に記載の方法。
実施形態12’. 前記がんが、乳がんである、実施形態2’〜4’のいずれか1つに記載の方法。
実施形態13’. 前記がんが、結腸がんである、実施形態2’〜4’のいずれか1つに記載の方法。
実施形態14’. 前記がんが、黒色腫である、実施形態2’〜4’のいずれか1つに記載の方法。
実施形態15’. 前記疾患が、進行性骨化性線維異形成症である、実施形態1’に記載の方法。
実施形態16’. びまん性真性橋膠腫(DIPG)の処置を必要とする対象のびまん性真性橋膠腫(DIPG)を処置する方法であって、前記方法は、ACVR1阻害剤を含む処置レジメンを前記対象に投与することを含み、前記ACVR1阻害剤が、下記の構造(I):
R1は、H、またはC1〜C6アルコキシであり;
R2は、C1〜C6アルコキシまたはヘテロシクリルであり;
R3は、ハロ、またはC1〜C6アルコキシであり;および
R4は、H、またはC1〜C6アルキルである)
またはその立体異性体、薬学的に許容される塩、互変異性体、もしくはプロドラッグを有する、方法。
実施形態17’. 前記処置レジメンが、治療剤を投与することをさらに含む、実施形態1’〜16’のいずれか1つに記載の方法。
実施形態18’. 前記対象が、小児対象である、実施形態1’〜17’のいずれか1つに記載の方法。
実施形態19’. 進行性骨化性線維異形成症(FOP)の処置を必要とする対象の進行性骨化性線維異形成症(FOP)を処置する方法であって、前記方法は、処置レジメンを前記対象に投与することを含み、前記処置レジメンは、ACVR1阻害剤および治療剤を含み、前記ACVR1阻害剤が、下記の構造(I):
R1は、H、またはC1〜C6アルコキシであり;
R2は、C1〜C6アルコキシまたはヘテロシクリルであり;
R3は、ハロ、またはC1〜C6アルコキシであり;および
R4は、H、またはC1〜C6アルキルである)
またはその立体異性体、薬学的に許容される塩、互変異性体、もしくはプロドラッグを有する、方法。
実施形態20’. 前記対象が、ACVR1遺伝子中に1つまたはそれより多くの変異を含む所定の遺伝子プロファイルを有する、実施形態16’〜19’のいずれか1つに記載の方法。
実施形態21’. 前記ACVR1遺伝子中の前記1つまたはそれより多くの変異が、ミスセンス変異、フレームシフト変異、スプライス部位変異、またはこれらの組合せを含む、実施形態1’〜15’または20’のいずれか1つに記載の方法。
実施形態22’. 前記1つまたはそれより多くの変異が、(P197F198)L、C509S、D185G、D185N、D433N、E38FS、F265S、G225D、G264S、G328E、G328R、G328V、G328W、G356D、G50C、H320Y、I323V、K31E、K345Q、L196P、L251S、M34I、N100D、N481I、P115S、P455A、Q207E、Q278P、R201I、R206C、R206H、R258G、R258S、R307Q、R325A、R375C、R375P、R401M、R490H、S130F、S226N、S41F、S440G、S469C、S56L、T298S、V234M、V91M、W98R、またはこれらの組合せを含む、実施形態1’〜15’、20’、または21’のいずれか1つに記載の方法。
実施形態23’. 前記ACVR1遺伝子中の前記1つまたはそれより多くの変異が、ミスセンス変異を含む、実施形態1’〜15’または20’のいずれか1つの方法。
実施形態24’ 前記ミスセンス変異が、C509S、D185N、D433N、F265S、G225D、H320Y、I323V、K31E、K345Q、M34I、N100D、N481I、P115S、P455A、Q278P、R206C、R401M、S130F、S226N、S41F、S41F、S440G、S469C、S56L、T298S、V234M、V91M、またはW98Rである、実施形態21’または23’に記載の方法。
実施形態25’. 前記ACVR1遺伝子中の前記1つまたはそれより多くの変異が、フレームシフト変異を含む、実施形態1’〜16’または20’のいずれか1つに記載の方法。
実施形態26’. 前記フレームシフト変異がE38fsである、実施形態21’または25’に記載の方法。
実施形態27’. 前記ACVR1遺伝子中の前記1つまたはそれより多くの変異が、スプライス部位変異を含む、実施形態1’〜15’または20’のいずれか1つに記載の方法。
実施形態28’. 前記スプライス部位変異が、G264Sである、実施形態21’または27’に記載の方法。
実施形態29’. 前記ACVR1遺伝子中の前記1つまたはそれより多くの変異が、R206H、G328V、R258G、またはこれらの組合せを含む、実施形態1’〜15’または20’〜22’のいずれか1つに記載の方法。
実施形態30’. 前記ACVR1遺伝子中の前記1つまたはそれより多くの変異が、R206Hを含む、実施形態1’〜15’、20’〜22’、または29’のいずれか1つに記載の方法。
実施形態31’. 前記対象が、少なくとも約0.1ng/mLの所定のヘプシジンレベルを有する、実施形態1’〜30’のいずれか1つに記載の方法。
実施形態32’. 前記所定のヘプシジンレベルが、約10ng/mLから約35ng/mLに及ぶ、実施形態31’の方法。
実施形態33’. 前記ACVR1阻害剤が投与された後にヘプシジンレベルを決定することをさらに含む、実施形態1’〜32’のいずれか1つに記載の方法。
実施形態34’. 前記対象が、約50%未満の所定のトランスフェリン飽和レベルを有する、実施形態1’〜33’のいずれか1つに記載の方法。
実施形態35’. 前記対象が、約45%未満の所定のトランスフェリン飽和レベルを有する、実施形態1’〜33’のいずれか1つに記載の方法。
実施形態36’. 前記対象が、約40%未満の所定のトランスフェリン飽和レベルを有する、実施形態1’〜33’のいずれか1つに記載の方法。
実施形態37’. 前記ACVR1阻害剤が、1日当たり約10mgから約320mgに及ぶ用量で投与される、実施形態1’〜36’のいずれか1つに記載の方法。
実施形態38’. 前記用量が、1日当たり約30mgから約240mgに及ぶ、実施形態37’に記載の方法。
実施形態39’. 前記用量が、1日当たり約60mgから約180mgに及ぶ、実施形態37’または38’に記載の方法。
実施形態40’. 前記用量が、1日当たり約25mg;1日当たり約30mg;1日当たり約60mg;1日当たり約120mg;1日当たり約125mg;1日当たり約180mg;1日当たり約240mg;1日当たり約250mg;1日当たり約320mg;または1日当たり約325mgである、実施形態37’〜39’のいずれか1つに記載の方法。
実施形態41’. R1がHである、実施形態1’〜40’のいずれか1つに記載の方法。
実施形態42’. R1がC1〜C6アルコキシである、実施形態1’〜40’のいずれか1つに記載の方法。
実施形態43’. 前記C1〜C6アルコキシがメトキシである、実施形態42’に記載の方法。
実施形態44’. R2がC1〜C6アルコキシである、実施形態1’〜43’のいずれか1つに記載の方法。
実施形態45’. 前記C1〜C6アルコキシがメトキシである、実施形態44’に記載の方法。
実施形態46’. R2がヘテロシクリルである、実施形態1’〜45’のいずれか1つに記載の方法。
実施形態47’. 前記ヘテロシクリルが、必要に応じて置換されたピペラジニルである、実施形態46’に記載の方法。
実施形態48’. 前記必要に応じて置換されたピペラジニルが、C1〜C6アルキルまたはC1〜C6ヒドロキシアルキルで置換される、実施形態47’に記載の方法。
実施形態49’. R3がハロである、実施形態1’〜48’のいずれか1つに記載の方法。
実施形態50’. 前記ハロがクロロである、実施形態49’に記載の方法。
実施形態51’. R3がC1〜C6アルコキシである、実施形態1’〜48’のいずれか1つに記載の方法。
実施形態52’. 前記C1〜C6アルコキシがメトキシである、実施形態51’に記載の方法。
実施形態53’. R4がHである、実施形態1’〜52’のいずれか1つに記載の方法。
実施形態54’. R4がC1〜C6アルキルである、実施形態1’〜52’のいずれか1つに記載の方法。
実施形態55’. 前記C1〜C6アルキルがメチルである、実施形態54’に記載の方法。
実施形態56’. 下記の構造:
実施形態57’. 下記の構造:
実施形態58’. 下記の構造:
実施形態59’. 下記の構造:
実施形態60’. 下記の構造:
実施形態61’. 下記の構造:
実施形態62’. 前記治療剤が:
レチノイン酸受容体ガンマアゴニスト;mTOR阻害剤;アクチビンA抗体;キナーゼ阻害剤;ACVR1抗体;TAK1阻害剤;ホスホジエステラーゼ阻害剤;HDAC阻害剤;化学療法剤;免疫療法剤;細胞療法;ペプチドまたは腫瘍ライセートワクチン;イリノテカン;GM−CSFを含むTTRNA−DCワクチン、TTRNA−xALT;インテグリン阻害剤;IL−12療法;抗ネオプラストン療法;イミキモド;腫瘍溶解性アデノウイルス;WEE1阻害剤;WT1タンパク質由来ペプチドワクチン;ペグ化インターフェロンアルファ2b;キナーゼ抗体;円滑化阻害剤;チューブリン阻害剤;テロメラーゼ阻害剤;CD40アゴニスト;GM−CSFアゴニスト;IDO阻害剤;および
放射性ヨウ素標識モノクローナル抗体8H9.64
から選択される、実施形態17’〜61’のいずれか1つに記載の方法。
実施形態63’. 前記治療剤が:
レチノイン酸受容体ガンマアゴニスト;mTOR阻害剤;アクチビンA抗体;キナーゼ阻害剤;ACVR1抗体;TAK1阻害剤;およびホスホジエステラーゼ阻害剤
から選択される、実施形態17’〜62’のいずれか1つに記載の方法。
実施形態64’. 前記治療剤が:
HDAC阻害剤;化学療法剤;免疫療法剤;細胞療法;ペプチドまたは腫瘍ライセートワクチン;イリノテカン;GM−CSFを含むTTRNA−DCワクチン、TTRNA−xALT;インテグリン阻害剤;IL−12療法;抗ネオプラストン療法;イミキモド;腫瘍溶解性アデノウイルス;WEE1阻害剤;WT1タンパク質由来ペプチドワクチン;
ペグ化インターフェロンアルファ2b;キナーゼ抗体;キナーゼ阻害剤;円滑化阻害剤;チューブリン阻害剤;テロメラーゼ阻害剤;CD40アゴニスト;GM−CSFアゴニスト;
IDO阻害剤;および放射性ヨウ素標識モノクローナル抗体8H9
から選択される、実施形態17’〜62’のいずれか1つに記載の方法。
実施形態65’. 前記キナーゼ阻害剤が、サイクリン依存性キナーゼ(CDK)を阻害する、実施形態17’〜64’のいずれか1つに記載の方法。
実施形態66’. 前記CDKが、CDK9またはCDK7である、実施形態65’に記載の方法。
実施形態67’. 前記CDKがCDK9である、実施形態65’に記載の方法。
実施形態68’. 前記CDK9阻害剤が、siRNA、アルボシジブ、またはそのプロドラッグ、ジナシクリブ、またはこれらの組合せである、実施形態66’または67’に記載の方法。
実施形態69’. 前記キナーゼ阻害剤が、ホスホイノシチド3−キナーゼ(PI3K)を阻害する、実施形態17’〜64’のいずれか1つに記載の方法。
実施形態70’. 前記免疫療法剤が、免疫チェックポイント阻害剤である、実施形態17’〜62’または64’のいずれか1つに記載の方法。
実施形態71’. 前記免疫チェックポイント阻害剤が、PD−1阻害剤である、実施形態70’に記載の方法。
実施形態72’. 前記PD−1阻害剤が、ペムブロリズマブ、ニボルマブ、またはこれらの組合せである、実施形態71’に記載の方法。
実施形態73’. 前記免疫チェックポイント阻害剤が、PD−L1阻害剤である、実施形態70’に記載の方法。
実施形態74’. 前記PD−L1阻害剤が、アテゾリズマブ、アベルマブ、デュラバルマブ、またはこれらの組合せである、実施形態73’に記載の方法。
実施形態75’. 前記キナーゼ抗体が薬物コンジュゲートを含む、実施形態17’〜64’のいずれか1つに記載の方法。
実施形態76’. 前記レチノイン酸受容体ガンマアゴニストがパロバロテンであり;前記mTOR阻害剤がラパマイシンまたはエベロリムスであり;前記アクチビンA抗体がREGN2447であり;前記キナーゼ阻害剤が、サラカチニブ、モメロチニブ、ドルソモルフィン、イマチニブ、クリゾチニブ、ダサチニブ、ベバシズマブ、エルロチニブ、バンデタニブ、リボシクリブ、クレノラニブ、アベマシクリブ、ONC201、シレンギジド、アルボシジブ、またはこれらのプロドラッグであり;前記ホスホジエステラーゼ阻害剤がジピリダモールであり;前記HDAC阻害剤が、SAHA、ボリノスタット、またはパノビノスタットであり;前記化学療法剤が、メルファラン、ゲムシタビン、テモゾロミド、シクロホスファミド、フルダラビン、ドキソルビシン、イリノテカン、レナリドミド、バルプロ酸、クロロキン、カルボプラチン、エトポシド、イフォスファミド、ポマリドミド、またはロムスチンであり;
前記免疫療法剤がMDV9300であり;前記細胞療法が自家樹状細胞を含み;前記ペプチドまたは腫瘍ライセートワクチンが、K27Mペプチドまたはリンドペピムトを含み;前記イリノテカンが、対流強化送達により投与され;前記インテグリン阻害剤がシレンギチドであり;前記抗ネオプラストン療法が、アテンゲナルまたはアスツゲナルであり;前記腫瘍溶解性アデノウイルスがDNX−2401であり;前記WEE1阻害剤がAZD1775であり;前記WT1タンパク質由来ペプチドワクチンがDSP−7888であり;前記キナーゼ抗体が、ニモツズマブ、エルビタックス、またはABT−414であり;前記円滑化阻害剤がビスモデギブであり;前記チューブリン阻害剤がメベンダゾールであり;
前記テロメラーゼ阻害剤がイメテルスタットであり;前記CD40アゴニストがAPX005Mであり;前記GM−CSFアゴニストがレオウイルスを含むサルグラモスチムであり;または前記IDO阻害剤がインドキシモドである、
実施形態17’〜65’のいずれか1つに記載の方法。
実施形態77’. 前記処置レジメンが、外科的切除、放射線療法、またはこれらの組合せをさらに含む、実施形態1’〜76’のいずれか1つに記載の方法。
実施形態78’. 前記薬学的に許容される塩が、酸付加塩である、実施形態1’〜77’のいずれか1つに記載の方法。
実施形態79’. 前記酸付加塩が、塩酸塩である、実施形態78’に記載の方法。
Further embodiments and embodiments of the present disclosure that may be incorporated in various combinations include:
Embodiment 1'. A method of treating a disease in a subject requiring treatment of the disease, wherein the treatment regimen comprising an ACVR1 inhibitor comprises a predetermined mutation containing one or more mutations in the ACVR1 gene. The ACVR1 inhibitor comprises administration to said subject having a genetic profile and has the following structure (I):
R 1 is H, or C 1 to C 6 alkoxy;
R 2 is C 1 -C 6 alkoxy or heterocyclyl;
R 3 is halo or C 1 -C 6 alkoxy;
R 4 is H or C 1 -C 6 alkyl,)
Or a method having a stereoisomer thereof, a pharmaceutically acceptable salt, a tautomer, or a prodrug.
Embodiment 2'. The method according to embodiment 1', wherein the disease is cancer.
Embodiment 3'. The method according to embodiment 2', wherein the cancer is a solid cancer.
Embodiment 4'. The cancer is brain cancer, uterine cancer, ovarian cancer, cervical cancer, lung cancer, breast cancer, colon cancer, gastrointestinal cancer, hematopoietic or lymphoid cancer, skin cancer, or bone cancer. , The method according to embodiment 2'or 3'.
Embodiment 5'. The method according to any one of embodiments 2'to 4', wherein the cancer is brain cancer.
Embodiment 6'. The method according to embodiment 5', wherein the brain cancer is a brain stem glioma.
Embodiment 7'. The method according to embodiment 5'or 6', wherein the brain cancer is diffuse true pontine glioma (DIPG).
Embodiment 8'. The method according to any one of embodiments 2'-4', wherein the cancer is uterine, ovarian, or cervical cancer.
Embodiment 9'. The method according to embodiment 8', wherein the uterine cancer is endometrial cancer.
Embodiment 10'. The method according to any one of embodiments 2'-4', wherein the cancer is lung cancer.
Embodiment 12'. The method according to any one of embodiments 2'-4', wherein the cancer is breast cancer.
Embodiment 13'. The method according to any one of embodiments 2'-4', wherein the cancer is colon cancer.
Embodiment 14'. The method according to any one of embodiments 2'-4', wherein the cancer is melanoma.
Embodiment 15'. The method according to embodiment 1', wherein the disease is progressive ossifying fibrodysplasia.
Embodiment 16'. A method of treating diffuse true pontine glioma (DIPG) in a subject requiring treatment for diffuse true pontine glioma (DIPG), wherein the treatment regimen containing an ACVR1 inhibitor is administered to the subject. The ACVR1 inhibitor comprises the following structure (I):
R 1 is H, or C 1 to C 6 alkoxy;
R 2 is C 1 -C 6 alkoxy or heterocyclyl;
R 3 is halo, or C 1 to C 6 alkoxy; and R 4 is H, or C 1 to C 6 alkyl).
Or a method having a stereoisomer thereof, a pharmaceutically acceptable salt, a tautomer, or a prodrug.
Embodiment 17'. The method of any one of embodiments 1'-16', wherein the treatment regimen further comprises administering a therapeutic agent.
Embodiment 18'. The method according to any one of embodiments 1'to 17', wherein the subject is a pediatric subject.
Embodiment 19'. A method of treating progressive ossifying fibrodysplasia (FOP) in a subject requiring treatment for progressive ossifying fibrodysplasia (FOP), wherein the treatment regimen is applied to the subject. Including administration, the treatment regimen comprises an ACVR1 inhibitor and a therapeutic agent, wherein the ACVR1 inhibitor has the following structure (I):
R 1 is H, or C 1 to C 6 alkoxy;
R 2 is C 1 -C 6 alkoxy or heterocyclyl;
R 3 is halo, or C 1 to C 6 alkoxy; and R 4 is H, or C 1 to C 6 alkyl).
Or a method having a stereoisomer thereof, a pharmaceutically acceptable salt, a tautomer, or a prodrug.
20'. The method of any one of embodiments 16'-19', wherein the subject has a predetermined gene profile that comprises one or more mutations in the ACVR1 gene.
21'. One or more of the mutations in the ACVR1 gene is in any one of embodiments 1'-15' or 20', comprising a missense mutation, a frameshift mutation, a splice site mutation, or a combination thereof. The method described.
Embodiment 22'. One or more of the above mutations are (P197F198) L, C509S, D185G, D185N, D433N, E38FS, F265S, G225D, G264S, G328E, G328R, G328V, G328W, G356D, G50C, H320Y, I323V. K345Q, L196P, L251S, M34I, N100D, N481I, P115S, P455A, Q207E, Q278P, R201I, R206C, R206H, R258G, R258S, R307Q, R325A, R375C, R375P, R401M, R470S The method according to any one of embodiments 1'-15', 20', or 21', comprising S469C, S56L, T298S, V234M, V91M, W98R, or a combination thereof.
23'. The method of any one of embodiments 1'-15' or 20', wherein the one or more mutations in the ACVR1 gene include a missense mutation.
Embodiment 24'The missense mutations are C509S, D185N, D433N, F265S, G225D, H320Y, I323V, K31E, K345Q, M34I, N100D, N481I, P115S, P455A, Q278P, R206C, R401M, S130F, S226. , S440G, S469C, S56L, T298S, V234M, V91M, or W98R, according to embodiment 21'or 23'.
Embodiment 25'. The method of any one of embodiments 1'-16' or 20', wherein the one or more mutations in the ACVR1 gene include a frameshift mutation.
Embodiment 27'. The method according to any one of embodiments 1'-15' or 20', wherein the one or more mutations in the ACVR1 gene include a splice site mutation.
Embodiment 29'. The one or more mutations in the ACVR1 gene according to any one of embodiments 1'-15' or 20'-22', comprising R206H, G328V, R258G, or a combination thereof. Method.
Embodiment 30'. The method according to any one of embodiments 1'-15', 20'-22', or 29', wherein the one or more mutations in the ACVR1 gene comprise R206H.
Embodiment 31'. The method of any one of embodiments 1'-30', wherein the subject has a predetermined hepcidin level of at least about 0.1 ng / mL.
Embodiment 32'. The method of embodiment 31', wherein the predetermined hepcidin level ranges from about 10 ng / mL to about 35 ng / mL.
Embodiment 33'. The method according to any one of embodiments 1'-32', further comprising determining hepcidin levels after administration of the ACVR1 inhibitor.
Embodiment 34'. The method of any one of embodiments 1'-33', wherein the subject has a predetermined transferrin saturation level of less than about 50%.
Embodiment 35'. The method of any one of embodiments 1'-33', wherein the subject has a predetermined transferrin saturation level of less than about 45%.
Embodiment 36'. The method of any one of embodiments 1'-33', wherein the subject has a predetermined transferrin saturation level of less than about 40%.
Embodiment 37'. The method of any one of embodiments 1'-36', wherein the ACVR1 inhibitor is administered at a dose ranging from about 10 mg to about 320 mg per day.
Embodiment 38'. 37'. The method of embodiment 37', wherein the dose ranges from about 30 mg to about 240 mg per day.
Embodiment 39'. 38'. The method of embodiment 37'or 38', wherein the dose ranges from about 60 mg to about 180 mg per day.
Embodiment 40'. The dose is about 25 mg per day; about 30 mg per day; about 60 mg per day; about 120 mg per day; about 125 mg per day; about 180 mg per day; about 240 mg per day; about 250 mg per day; about 320 mg per day; or The method according to any one of embodiments 37'-39', which is about 325 mg per day.
Embodiment 41'. The method according to any one of embodiments 1'-40', wherein R 1 is H.
Embodiment 42'. The method according to any one of embodiments 1'-40', wherein R 1 is C 1 to C 6 alkoxy.
Embodiment 43'. The method according to embodiment 42', wherein the C 1 to C 6 alkoxy is methoxy.
Embodiment 44'. The method according to any one of embodiments 1'to 43', wherein R 2 is C 1 to C 6 alkoxy.
Embodiment 45'. The method according to embodiment 44', wherein the C 1 to C 6 alkoxy is methoxy.
Embodiment 46'. The method according to any one of embodiments 1'to 45', wherein R 2 is heterocyclyl.
Embodiment 48'. Said piperazinyl is optionally substituted, it is substituted by C 1 -C 6 alkyl or C 1 -C 6 hydroxyalkyl, method of embodiment 47 '.
Embodiment 49'. The method according to any one of embodiments 1'to 48', wherein R 3 is halo.
Embodiment 50'. The method according to embodiment 49', wherein the halo is chloro.
Embodiment 51'. The method according to any one of embodiments 1'to 48', wherein R 3 is C 1 to C 6 alkoxy.
Embodiment 52'. The method according to embodiment 51', wherein the C 1 to C 6 alkoxy is methoxy.
Embodiment 53'. The method according to any one of embodiments 1'to 52', wherein R 4 is H.
Embodiment 54'. The method according to any one of embodiments 1'to 52', wherein R 4 is C 1 to C 6 alkyl.
Embodiment 55'. The method according to embodiment 54', wherein the C 1 to C 6 alkyls are methyl.
Embodiment 56'. Structure below:
Embodiment 57'. Structure below:
Embodiment 58'. Structure below:
Embodiment 59'. Structure below:
Embodiment 60'. Structure below:
Embodiment 61'. Structure below:
Embodiment 62'. The therapeutic agent is:
Retinoic acid receptor gamma agonist; mTOR inhibitor; activin A antibody; kinase inhibitor; ACVR1 antibody; TAK1 inhibitor; phosphodiesterase inhibitor; HDAC inhibitor; chemotherapeutic agent; immunotherapeutic agent; cell therapy; peptide or tumor lysate vaccine Irinotecan; TTRNA-DC vaccine containing GM-CSF, TTRNA-xALT; integrin inhibitor; IL-12 therapy; anti-neoplastone therapy; imikimod; tumor-soluble adenovirus; WEE1 inhibitor; WT1 protein-derived peptide vaccine; peg Interferon alpha 2b; kinase antibody; smoothing inhibitor; tuberin inhibitor; telomerase inhibitor; CD40 agonist; GM-CSF agonist; IDO inhibitor; and radioactive iodine-labeled monoclonal antibody 8H9.64
The method according to any one of embodiments 17'to 61', which is selected from.
Embodiment 63'. The therapeutic agent is:
The retinoic acid receptor gamma agonist; mTOR inhibitor; activin A antibody; kinase inhibitor; ACVR1 antibody; TAK1 inhibitor; and phosphodiesterase inhibitor, according to any one of embodiments 17'-62'. Method.
Embodiment 64'. The therapeutic agent is:
HDAC inhibitor; chemotherapeutic agent; immunotherapeutic agent; cell therapy; peptide or tumor lysate vaccine; irinotecan; TTRNA-DC vaccine containing GM-CSF, TTRNA-xALT; integrin inhibitor; IL-12 therapy; anti-neoplaston therapy Imikimod; Tumor-dissolving adenovirus; WEE1 inhibitor; WT1 protein-derived peptide vaccine;
Pegylated interferon alpha 2b; kinase antibody; kinase inhibitor; smoothing inhibitor; tubulin inhibitor; telomerase inhibitor; CD40 agonist; GM-CSF agonist;
IDO inhibitor; and radioiodine-labeled monoclonal antibody 8H9
The method according to any one of embodiments 17'to 62', which is selected from.
Embodiment 65'. The method according to any one of embodiments 17'-64', wherein the kinase inhibitor inhibits a cyclin-dependent kinase (CDK).
Embodiment 66'. 65'. The method of embodiment 65', wherein the CDK is CDK9 or CDK7.
Embodiment 67'. 65'. The method of embodiment 65', wherein the CDK is CDK9.
Embodiment 68'. The method of embodiment 66'or 67', wherein the CDK9 inhibitor is siRNA, alvocidib, or a prodrug, dynasicrib, or a combination thereof.
Embodiment 69'. The method according to any one of embodiments 17'-64', wherein the kinase inhibitor inhibits phosphoinositide 3-kinase (PI3K).
Embodiment 70'. The method according to any one of embodiments 17'-62'or 64', wherein the immunotherapeutic agent is an immune checkpoint inhibitor.
Embodiment 71'. The method according to embodiment 70', wherein the immune checkpoint inhibitor is a PD-1 inhibitor.
Embodiment 72'. The method of embodiment 71', wherein the PD-1 inhibitor is pembrolizumab, nivolumab, or a combination thereof.
Embodiment 73'. The method according to embodiment 70', wherein the immune checkpoint inhibitor is a PD-L1 inhibitor.
Embodiment 74'. The method according to embodiment 73', wherein the PD-L1 inhibitor is atezolizumab, avelumab, duravalumab, or a combination thereof.
Embodiment 75'. The method according to any one of embodiments 17'-64', wherein the kinase antibody comprises a drug conjugate.
Embodiment 76'. The retinoic acid receptor gamma agonist is parovarotene; the mTOR inhibitor is rapamycin or everolimus; the activin A antibody is REGN2447; the kinase inhibitors are salacatinib, momerotinib, dolsomorphin, imatinib, crizotinib, dasatinib. , Bebasizumab, erlotinib, bandetanib, ribocyclib, clenoranib, abemacicrib, ONC201, silengidide, arbosideib, or prodrugs thereof; said phosphodiesterase inhibitor is dipyridamole; said HDAC inhibitor is SAHA, bolinostat, or panobinot The chemotherapeutic agent is melphalan, gemcitabine, temozolomid, cyclophosphamide, fludarabine, doxorubicin, irinotecan, renalidemid, valproic acid, chloroquin, carboplatin, etoposide, ifosfamide, pomalidemid, or lomustine;
The immunotherapeutic agent is MDV9300; the cell therapy comprises autologous dendritic cells; the peptide or tumor lysate vaccine comprises K27M peptide or Lindpepimut; the irinotecan is administered by convection-enhanced delivery; the integrin inhibitor. Is sirengitide; the anti-neoplaston therapy is athenal or astugenal; the tumor-soluble adenovirus is DNX-2401; the WEE1 inhibitor is AZD1775; the WT1 protein-derived peptide vaccine is DSP-7888. The kinase antibody is nimotzumab, erbitux, or ABT-414; the smoothing inhibitor is bismodegib; the tubulin inhibitor is mebendazole;
The telomerase inhibitor is imeterstat; the CD40 agonist is APX005M; the GM-CSF agonist is sargramostim containing a reoviridae; or the IDO inhibitor is indoximod.
The method according to any one of embodiments 17'to 65'.
Embodiment 77'. The method of any one of embodiments 1'-76', wherein the treatment regimen further comprises surgical resection, radiation therapy, or a combination thereof.
Embodiment 78'. The method according to any one of embodiments 1'to 77', wherein the pharmaceutically acceptable salt is an acid addition salt.
Embodiment 79'. The method according to embodiment 78', wherein the acid addition salt is a hydrochloride salt.
上述の様々な実施形態は、さらなる実施形態が提供されるように組み合わされてもよい。本明細書で言及されるおよび/または出願データシートに列挙される、全ての米国特許、米国特許出願公開、米国特許出願、非米国特許、非米国特許出願、および非特許刊行物は、その全体が、参照により本明細書に組み込まれる。実施形態の態様は、さらに他の実施形態を提供するため、様々な特許、出願、および刊行物の概念を用いるように、必要な場合には修正されてもよい。 The various embodiments described above may be combined to provide additional embodiments. All US patents, US patent application publications, US patent applications, non-US patents, non-US patent applications, and non-patent publications referred to herein and / or listed in the application datasheet are in their entirety. Is incorporated herein by reference. The embodiments may be modified, if necessary, to use the concepts of various patents, applications, and publications to provide yet other embodiments.
本明細書に記述される実験用の試験化合物は、注記されるように、遊離形態または塩形態で用いた。 The experimental test compounds described herein were used in free or salt form, as noted.
観察された特定の応答は、選択された特定の活性化合物、または担体が存在するか否か、ならびに製剤のタイプおよび用いられる投与形態に従い、かつこれらに依存して変わってもよく、そのような予測される結果の変動または相違は、本発明の実施により企図される。 The particular response observed may vary depending on and depending on the presence or absence of the particular active compound or carrier selected, as well as the type of formulation and the dosage form used, such. Variations or differences in expected outcomes are contemplated by the practice of the present invention.
これらおよびその他の変更は、上記詳細な記述に照らして実施形態になすことができる。一般に、下記の特許請求の範囲において、使用される用語は、本明細書および特許請求の範囲に開示された特定の実施形態に、特許請求の範囲を限定しないと解釈されるべきであり、そのような特許請求の範囲が与える均等物の全範囲と共に、全ての可能性ある実施形態を含むと解釈すべきである。したがって特許請求の範囲は、本開示により限定されない。
These and other modifications can be made in the embodiment in the light of the above detailed description. In general, the terms used in the claims below should be construed as not limiting the scope of the claims to the particular embodiments disclosed herein and in the claims. It should be construed to include all possible embodiments, as well as the full range of equivalents provided by such claims. Therefore, the scope of claims is not limited by this disclosure.
Claims (182)
(i)1種またはそれより多種の緩衝剤;
(ii)必要に応じて、1種またはそれより多種の保存剤;
(iii)必要に応じて、1種またはそれより多種の溶媒;
(iv)必要に応じて、1種またはそれより多種の矯味剤;および
(v)必要に応じて、1種またはそれより多種のさらなるpH調節剤
とを含む、経口液体医薬組成物。 Equation (2):
(I) One or more buffers;
(Ii) One or more preservatives as needed;
(Iii) One or more solvents as needed;
An oral liquid pharmaceutical composition comprising (iv) one or more flavoring agents as needed; and (v) one or more additional pH regulators as needed.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862703862P | 2018-07-26 | 2018-07-26 | |
| US62/703,862 | 2018-07-26 | ||
| PCT/US2019/043738 WO2020023910A1 (en) | 2018-07-26 | 2019-07-26 | Methods for treating diseases associated with abnormal acvr1 expression and acvr1 inhibitors for use in the same |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021530554A true JP2021530554A (en) | 2021-11-11 |
| JPWO2020023910A5 JPWO2020023910A5 (en) | 2022-08-02 |
Family
ID=69182006
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021503828A Pending JP2021530554A (en) | 2018-07-26 | 2019-07-26 | Methods and ACVR1 Inhibitors for Treatment of Diseases with Abnormal ACVR1 Expression |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US11040038B2 (en) |
| EP (1) | EP3826684A4 (en) |
| JP (1) | JP2021530554A (en) |
| KR (1) | KR20210038906A (en) |
| CN (1) | CN112512597A (en) |
| AU (1) | AU2019310590A1 (en) |
| CA (1) | CA3103995A1 (en) |
| MX (1) | MX2021000977A (en) |
| WO (1) | WO2020023910A1 (en) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2970205B1 (en) | 2013-03-14 | 2019-05-08 | Tolero Pharmaceuticals, Inc. | Jak2 and alk2 inhibitors and methods for their use |
| CN112512597A (en) | 2018-07-26 | 2021-03-16 | 大日本住友制药肿瘤公司 | Methods for treating diseases associated with aberrant ACVR1 expression and ACVR1 inhibitors useful therefor |
| EP3908575A4 (en) * | 2019-01-10 | 2022-09-14 | Sumitomo Pharma Oncology, Inc. | Alk5 inhibitors for treating myelodysplastic syndrome |
| JP2023502264A (en) | 2019-11-22 | 2023-01-23 | スミトモ ファーマ オンコロジー, インコーポレイテッド | Solid dose pharmaceutical composition |
| WO2022232176A1 (en) * | 2021-04-26 | 2022-11-03 | Dnatrix, Inc. | Use of oncolytic adenovirus for the treatment of pediatric brain cancer |
Family Cites Families (186)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2779780A (en) | 1955-03-01 | 1957-01-29 | Du Pont | 1, 4-diamino-2, 3-dicyano-1, 4-bis (substituted mercapto) butadienes and their preparation |
| GB8827305D0 (en) | 1988-11-23 | 1988-12-29 | British Bio Technology | Compounds |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| EP0625200B1 (en) | 1992-02-06 | 2005-05-11 | Chiron Corporation | Biosynthetic binding protein for cancer marker |
| US6177401B1 (en) | 1992-11-13 | 2001-01-23 | Max-Planck-Gesellschaft Zur Forderung Der Wissenschaften | Use of organic compounds for the inhibition of Flk-1 mediated vasculogenesis and angiogenesis |
| US5455258A (en) | 1993-01-06 | 1995-10-03 | Ciba-Geigy Corporation | Arylsulfonamido-substituted hydroxamic acids |
| EP0647450A1 (en) | 1993-09-09 | 1995-04-12 | BEHRINGWERKE Aktiengesellschaft | Improved prodrugs for enzyme mediated activation |
| IL112248A0 (en) | 1994-01-25 | 1995-03-30 | Warner Lambert Co | Tricyclic heteroaromatic compounds and pharmaceutical compositions containing them |
| US5521173A (en) | 1995-01-17 | 1996-05-28 | American Home Products Corporation | Tricyclic benzazepine vasopressin antagonists |
| US5863949A (en) | 1995-03-08 | 1999-01-26 | Pfizer Inc | Arylsulfonylamino hydroxamic acid derivatives |
| CA2218503C (en) | 1995-04-20 | 2001-07-24 | Pfizer Inc. | Arylsulfonyl hydroxamic acid derivatives |
| GB9508538D0 (en) | 1995-04-27 | 1995-06-14 | Zeneca Ltd | Quinazoline derivatives |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US5880141A (en) | 1995-06-07 | 1999-03-09 | Sugen, Inc. | Benzylidene-Z-indoline compounds for the treatment of disease |
| GB9520822D0 (en) | 1995-10-11 | 1995-12-13 | Wellcome Found | Therapeutically active compounds |
| GB9523675D0 (en) | 1995-11-20 | 1996-01-24 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9624482D0 (en) | 1995-12-18 | 1997-01-15 | Zeneca Phaema S A | Chemical compounds |
| DK0780386T3 (en) | 1995-12-20 | 2003-02-03 | Hoffmann La Roche | matrix metalloprotease |
| KR19990082330A (en) | 1996-02-06 | 1999-11-25 | 미즈노 마사루 | Novel compounds and uses thereof |
| PT885198E (en) | 1996-03-05 | 2002-06-28 | Astrazeneca Ab | 4-ANYLINOQUINAZOLINE DERIVATIVES |
| US5795909A (en) | 1996-05-22 | 1998-08-18 | Neuromedica, Inc. | DHA-pharmaceutical agent conjugates of taxanes |
| EP0818442A3 (en) | 1996-07-12 | 1998-12-30 | Pfizer Inc. | Cyclic sulphone derivatives as inhibitors of metalloproteinases and of the production of tumour necrosis factor |
| ID19430A (en) | 1996-07-13 | 1998-07-09 | Glaxo Group Ltd | COMPOUND HETEROSICLIC COMPOUND |
| HRP970371A2 (en) | 1996-07-13 | 1998-08-31 | Kathryn Jane Smith | Heterocyclic compounds |
| AR007857A1 (en) | 1996-07-13 | 1999-11-24 | Glaxo Group Ltd | HETERO-CYCLIC COMPOUNDS FUSED AS PROTEIN INHIBITORS, THYROSINE KINASE, THEIR PREPARATION METHODS, INTERMEDIARY USE IN MEDICINE AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| TR199900066T2 (en) | 1996-07-18 | 1999-04-21 | Pfizer Inc. | Matrix metalloprotateazlar�n phosphinate bazl� inhibit�rleri |
| IL128189A0 (en) | 1996-08-23 | 1999-11-30 | Pfizer | Arylsulfonylamino hydroxamic acid derivatives |
| ID18494A (en) | 1996-10-02 | 1998-04-16 | Novartis Ag | PIRAZOLA DISTRIBUTION IN THE SEQUENCE AND THE PROCESS OF MAKING IT |
| DZ2376A1 (en) | 1996-12-19 | 2002-12-28 | Smithkline Beecham Plc | New sulfonamide derivatives process for their preparation and pharmaceutical compositions containing them. |
| US6077864A (en) | 1997-01-06 | 2000-06-20 | Pfizer Inc. | Cyclic sulfone derivatives |
| PL335027A1 (en) | 1997-02-03 | 2000-03-27 | Pfizer Prod Inc | Derivatives of arylsulphonylamino hydroxamic acid |
| CA2279863A1 (en) | 1997-02-07 | 1998-08-13 | Pfizer Inc. | N-hydroxy-beta-sulfonyl-propionamide derivatives and their use as inhibitors of matrix metalloproteinases |
| BR9807678A (en) | 1997-02-11 | 2000-02-15 | Pfizer | Derivatives of arylsulfonyl hydroxamic acids |
| EP0984930B1 (en) | 1997-05-07 | 2005-04-06 | Sugen, Inc. | 2-indolinone derivatives as modulators of protein kinase activity |
| WO1998054093A1 (en) | 1997-05-30 | 1998-12-03 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| KR100372138B1 (en) | 1997-08-08 | 2003-02-14 | 화이자 프로덕츠 인코포레이티드 | Aryloxyarylsulfonylamino hydroxamic acid derivatives |
| AU8816298A (en) | 1997-08-22 | 1999-03-16 | Zeneca Limited | Oxindolylquinazoline derivatives as angiogenesis inhibitors |
| WO1999016755A1 (en) | 1997-09-26 | 1999-04-08 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| SK6652000A3 (en) | 1997-11-11 | 2002-05-09 | Pfizer Prod Inc | Thienopyrimidine and thienopyridine derivatives useful as anticancer agents |
| GB9725782D0 (en) | 1997-12-05 | 1998-02-04 | Pfizer Ltd | Therapeutic agents |
| GB9800569D0 (en) | 1998-01-12 | 1998-03-11 | Glaxo Group Ltd | Heterocyclic compounds |
| GB9800575D0 (en) | 1998-01-12 | 1998-03-11 | Glaxo Group Ltd | Heterocyclic compounds |
| GB9801690D0 (en) | 1998-01-27 | 1998-03-25 | Pfizer Ltd | Therapeutic agents |
| JP4462654B2 (en) | 1998-03-26 | 2010-05-12 | ソニー株式会社 | Video material selection device and video material selection method |
| PA8469501A1 (en) | 1998-04-10 | 2000-09-29 | Pfizer Prod Inc | HYDROXAMIDES OF THE ACID (4-ARILSULFONILAMINO) -TETRAHIDROPIRAN-4-CARBOXILICO |
| PA8469401A1 (en) | 1998-04-10 | 2000-05-24 | Pfizer Prod Inc | BICYCLE DERIVATIVES OF HYDROXAMIC ACID |
| IL139934A (en) | 1998-05-29 | 2007-10-31 | Sugen Inc | Pyrrole substituted 2-indolinone derivatives and pharmaceutical compositions containing the same |
| UA60365C2 (en) | 1998-06-04 | 2003-10-15 | Пфайзер Продактс Інк. | Isothiazole derivatives, a method for preparing thereof, a pharmaceutical composition and a method for treatment of hyperproliferative disease of mammal |
| PT1004578E (en) | 1998-11-05 | 2004-06-30 | Pfizer Prod Inc | HYDROXAMIDE DERIVATIVES OF 5-OXO-PYRROLIDINE-2-CARBOXYLIC ACID |
| HUP0104693A3 (en) | 1998-12-16 | 2003-12-29 | Warner Lambert Co | Treatment of arthritis with mek inhibitors |
| UA71945C2 (en) | 1999-01-27 | 2005-01-17 | Pfizer Prod Inc | Substituted bicyclic derivatives being used as anticancer agents |
| WO2000047574A1 (en) | 1999-02-11 | 2000-08-17 | Pfizer Products Inc. | Heteroaryl-substituted quinolin-2-one derivatives useful as anticancer agents |
| US6511993B1 (en) | 1999-06-03 | 2003-01-28 | Kevin Neil Dack | Metalloprotease inhibitors |
| EP1081137A1 (en) | 1999-08-12 | 2001-03-07 | Pfizer Products Inc. | Selective inhibitors of aggrecanase in osteoarthritis treatment |
| GB9924862D0 (en) | 1999-10-20 | 1999-12-22 | Celltech Therapeutics Ltd | Chemical compounds |
| MY130363A (en) | 2000-02-15 | 2007-06-29 | Sugen Inc | "pyrrole substituted 2-indolinone protein kinase inhibitors" |
| AU3704101A (en) | 2000-02-17 | 2001-08-27 | Amgen Inc | Kinase inhibitors |
| US6440207B1 (en) | 2000-03-03 | 2002-08-27 | Bayer Corporation | Method for preparing organic pigments |
| EP1275657A4 (en) | 2000-04-14 | 2006-06-28 | Nippon Shinyaku Co Ltd | Peptide derivatives and medicinal compositions |
| CA2411859A1 (en) | 2000-06-05 | 2001-12-13 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| EE05450B1 (en) | 2000-07-19 | 2011-08-15 | Warner-Lambert Company | Oxygenated esters of 4-iodophen laminobenzh droxamic acids, their crystalline forms and pharmaceutical compositions and their use |
| US6995162B2 (en) | 2001-01-12 | 2006-02-07 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| WO2002094766A1 (en) | 2001-05-18 | 2002-11-28 | Nihon Nohyaku Co., Ltd. | Phthalamide derivative, agricultural or horticultural insecticide, and use thereof |
| WO2003003009A1 (en) | 2001-06-29 | 2003-01-09 | 7Tm Pharma A/S | Use of metal-ion chelates in validating biological molecules as drug targets in test animal models |
| US6939874B2 (en) | 2001-08-22 | 2005-09-06 | Amgen Inc. | Substituted pyrimidinyl derivatives and methods of use |
| JP5039268B2 (en) | 2001-10-26 | 2012-10-03 | アベンティス・ファーマスーティカルズ・インコーポレイテツド | Benzimidazoles and analogs and their use as protein kinase inhibitors |
| US6897208B2 (en) | 2001-10-26 | 2005-05-24 | Aventis Pharmaceuticals Inc. | Benzimidazoles |
| WO2003055477A1 (en) | 2001-12-21 | 2003-07-10 | 7Tm Pharma A/S | Method for the treatment of mc receptor related disorders with a chelate and/or a chelator |
| RU2343148C2 (en) | 2002-02-01 | 2009-01-10 | Райджел Фармасьютикалз, Инк | Compounds of 2,4-pyrimidindiamines and their application |
| TWI329105B (en) | 2002-02-01 | 2010-08-21 | Rigel Pharmaceuticals Inc | 2,4-pyrimidinediamine compounds and their uses |
| PL221491B1 (en) | 2002-03-08 | 2016-04-29 | Eisai R&D Man Co | Macrocyclic compounds useful as pharmaceuticals |
| PL401637A1 (en) | 2002-03-13 | 2013-05-27 | Array Biopharma Inc. | N3 alkylated benzimidazole derivatives as MEK inhibitors |
| GB0206215D0 (en) | 2002-03-15 | 2002-05-01 | Novartis Ag | Organic compounds |
| GB0206860D0 (en) | 2002-03-22 | 2002-05-01 | Glaxo Group Ltd | Compounds |
| DE60322857D1 (en) | 2002-03-25 | 2008-09-25 | Nippon Kayaku Kk | NEW ALPHA-AMINO-N- (DIAMINOPHOSPHINYL) LACTAM DERIVATIVE |
| AR039241A1 (en) | 2002-04-04 | 2005-02-16 | Biogen Inc | HETEROARILOS TRISUSTITUIDOS AND METHODS FOR ITS PRODUCTION AND USE OF THE SAME |
| US7005449B2 (en) | 2002-04-23 | 2006-02-28 | Pharmacia & Upjohn Company | Tolterodine salts |
| AU2003231231A1 (en) | 2002-05-06 | 2003-11-11 | Bayer Pharmaceuticals Corporation | Pyridinyl amino pyrimidine derivatives useful for treating hyper-proliferative disorders |
| TWI274055B (en) | 2002-08-09 | 2007-02-21 | Taisho Pharmaceutical Co Ltd | Aryl-5-thio-beta-D-glucopyranoside derivatives and remedies for diabetes containing the same |
| UA80295C2 (en) | 2002-09-06 | 2007-09-10 | Biogen Inc | Pyrazolopyridines and using the same |
| JP4608852B2 (en) | 2002-10-15 | 2011-01-12 | チッソ株式会社 | Liquid crystalline vinyl ketone derivatives and polymers thereof |
| AU2003287880A1 (en) | 2002-12-11 | 2004-06-30 | 7Tm Pharma A/S | Cyclic quinoline compounds for use in mch receptor related disorders |
| WO2004074244A2 (en) | 2003-02-20 | 2004-09-02 | Smithkline Beecham Corporation | Pyrimidine compounds |
| BRPI0300660B8 (en) | 2003-02-28 | 2021-07-27 | Fapesp Fund De Amparo A Pesquisa Do Estado De Sao Paulo | method to quantify mitochondrial release of cytochrome c and kit to detect mitochondrial release of cytochrome c |
| CA2514733A1 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| GB0305929D0 (en) | 2003-03-14 | 2003-04-23 | Novartis Ag | Organic compounds |
| US20040242655A1 (en) | 2003-05-28 | 2004-12-02 | Anziano Paul Q. | Compositions and methods for inhibiting an isoform of human manganese superoxide dismutase |
| US20050070554A1 (en) | 2003-08-27 | 2005-03-31 | Amgen Inc. | Substituted heterocyclic compounds and methods of use |
| US20070105839A1 (en) | 2003-09-18 | 2007-05-10 | Patricia Imbach | 2, 4-Di (phenylamino) pyrimidines useful in the treatment of proliferative disorders |
| WO2005082892A2 (en) | 2004-02-17 | 2005-09-09 | Dr. Reddy's Laboratories Ltd. | Triazole compounds as antibacterial agents and pharmaceutical compositions containing them |
| WO2005092049A2 (en) | 2004-03-22 | 2005-10-06 | Op-Marks, Inc. | Surgical site marking assembly and method of using same |
| EP1737843B1 (en) | 2004-04-09 | 2011-02-23 | Merck Sharp & Dohme Corp. | Inhibitors of akt activity |
| JP2008511631A (en) | 2004-08-31 | 2008-04-17 | バイオジェン・アイデック・エムエイ・インコーポレイテッド | Pyrimidinylimidazoles as TGF-β inhibitors |
| US7786153B2 (en) | 2005-03-02 | 2010-08-31 | Abbott Laboratories Inc. | Compounds that are useful for improving pharmacokinetics |
| CA2604551A1 (en) | 2005-05-03 | 2007-03-08 | Rigel Pharmaceuticals, Inc. | Jak kinase inhibitors and their uses |
| WO2006124874A2 (en) | 2005-05-12 | 2006-11-23 | Kalypsys, Inc. | Inhibitors of b-raf kinase |
| US20070032514A1 (en) | 2005-07-01 | 2007-02-08 | Zahn Stephan K | 2,4-diamino-pyrimidines as aurora inhibitors |
| CA2618218C (en) | 2005-07-21 | 2015-06-30 | Ardea Biosciences, Inc. | N-(arylamino)-sulfonamide inhibitors of mek |
| US8604042B2 (en) | 2005-11-01 | 2013-12-10 | Targegen, Inc. | Bi-aryl meta-pyrimidine inhibitors of kinases |
| US8133900B2 (en) | 2005-11-01 | 2012-03-13 | Targegen, Inc. | Use of bi-aryl meta-pyrimidine inhibitors of kinases |
| NZ592990A (en) | 2005-11-01 | 2013-01-25 | Targegen Inc | Bi-aryl meta-pyrimidine inhibitors of kinases |
| JP2009523724A (en) | 2006-01-13 | 2009-06-25 | ファーマサイクリクス,インコーポレイテッド | Tyrosine kinase inhibitor and method of use thereof |
| JO2660B1 (en) | 2006-01-20 | 2012-06-17 | نوفارتيس ايه جي | PI-3 Kinase inhibitors and methods of their use |
| CA2640398A1 (en) | 2006-01-30 | 2007-08-09 | Exelixis, Inc. | 4-aryl-2-amino-pyrimidines or 4-aryl-2-aminoalkyl-pyrimidines as jak-2 modulators and pharmaceutical compositions containing them |
| WO2007098507A2 (en) | 2006-02-24 | 2007-08-30 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| US20060223820A1 (en) | 2006-03-21 | 2006-10-05 | Chemagis Ltd. | Crystalline aripiprazole salts and processes for preparation and purification thereof |
| CA2652385A1 (en) | 2006-06-08 | 2007-12-21 | Eli Lilly And Company | Novel mch receptor antagonists |
| CN101506175A (en) | 2006-06-15 | 2009-08-12 | 贝林格尔·英格海姆国际有限公司 | 2-anilino-4-(heterocyclic)amino-pyrimidines as inhibitors of protein kinase C-alpha |
| HUE031334T2 (en) | 2006-09-22 | 2017-07-28 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| TWI432427B (en) | 2006-10-23 | 2014-04-01 | Cephalon Inc | Fused bicyclic derivatives of 2,4-diaminopyrimidine as alk and c-met inhibitors |
| US8987233B2 (en) | 2006-11-03 | 2015-03-24 | Pharmacyclics, Inc. | Bruton's tyrosine kinase activity probe and method of using |
| AR065015A1 (en) | 2007-01-26 | 2009-05-13 | Smithkline Beecham Corp | ANTRANILAMIDE DERIVATIVES, PHARMACEUTICAL COMPOSITIONS CONTAINING THEM, AND USES FOR THE TREATMENT OF CANCER |
| ES2702362T3 (en) | 2007-01-31 | 2019-02-28 | Ym Biosciences Australia Pty | Compounds based on thiopyrimidine and uses thereof |
| JP2010520222A (en) | 2007-03-01 | 2010-06-10 | スーパージェン, インコーポレイテッド | Use of pyrimidine-2,4-diamine derivatives and pyrimidine-2,4-diamine derivatives as JAK2 kinase inhibitors |
| CN101657431A (en) | 2007-03-01 | 2010-02-24 | 休普基因公司 | Pyrimidine-2,4-diamine derivatives and their use as JAK2 kinase inhibitors |
| US7834024B2 (en) | 2007-03-26 | 2010-11-16 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US20120101114A1 (en) | 2007-03-28 | 2012-04-26 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| SG10202107066WA (en) | 2007-03-28 | 2021-07-29 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| WO2008124085A2 (en) | 2007-04-03 | 2008-10-16 | Exelixis, Inc. | Methods of using combinations of mek and jak-2 inhibitors |
| EP2139869A2 (en) | 2007-04-13 | 2010-01-06 | SuperGen, Inc. | Axl kinase inhibitors useful for the treatment of cancer or hyperproliferative disorders |
| ATE544761T1 (en) | 2007-05-04 | 2012-02-15 | Irm Llc | PYRIMIDINE DERIVATIVES AND COMPOSITIONS AS C-KIT AND PDGFR KINASE INHIBITORS |
| WO2009010794A1 (en) | 2007-07-19 | 2009-01-22 | Astrazeneca Ab | 2,4-diamino-pyrimidine derivatives |
| KR101584823B1 (en) | 2007-09-12 | 2016-01-22 | 제넨테크, 인크. | Combinations of phosphoinositide 3-kinase inhibitor compounds and chemotherapeutic agents, and methods of use |
| US8354528B2 (en) | 2007-10-25 | 2013-01-15 | Genentech, Inc. | Process for making thienopyrimidine compounds |
| CA2708264C (en) | 2007-12-10 | 2018-07-03 | Sunesis Pharmaceuticals, Inc. | Methods of using (+)-1,4-dihydro-7-[(3s,4s)-3-methoxy-4-(methylamino)-1-pyrrolidinyl]-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acid for treatment of antecedent hematologic disorders |
| US20110028405A1 (en) | 2007-12-20 | 2011-02-03 | Richard John Harrison | Sulfamides as zap-70 inhibitors |
| ES2645689T3 (en) | 2008-05-21 | 2017-12-07 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| RU2536584C2 (en) | 2008-06-27 | 2014-12-27 | Авила Терапьютикс, Инк. | Heteroaryl compounds and using them |
| EP3311818A3 (en) | 2008-07-16 | 2018-07-18 | Pharmacyclics, LLC | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| TW201006839A (en) | 2008-07-18 | 2010-02-16 | Sanofi Aventis | Novel imidazo[1,2-a]pyridine derivatives, process for the preparation thereof, use thereof as medicaments, pharmaceutical compositions and novel use, in particular as MET inhibitors |
| WO2010011349A2 (en) | 2008-07-25 | 2010-01-28 | Supergen, Inc. | Pyrimidine-2,4-diamine jak2 kinase inhibiting anti-inflammation use |
| RU2011108563A (en) | 2008-08-05 | 2012-09-10 | Таргеджен, Инк. (US) | THALASSEMIA TREATMENT METHODS |
| PL2389372T3 (en) | 2009-01-23 | 2016-02-29 | Rigel Pharmaceuticals Inc | Compositions and methods for inhibition of the jak pathway |
| AU2010210986A1 (en) | 2009-02-09 | 2011-08-25 | Supergen, Inc. | Pyrrolopyrimidinyl Axl kinase inhibitors |
| WO2010118986A1 (en) | 2009-04-14 | 2010-10-21 | Cellzome Limited | Fluoro substituted pyrimidine compounds as jak3 inhibitors |
| AU2010292198A1 (en) | 2009-09-09 | 2012-04-05 | Celgene Avilomics Research, Inc. | PI3 kinase inhibitors and uses thereof |
| IN2012DN02534A (en) | 2009-09-16 | 2015-08-28 | Avila Therapeutics Inc | |
| EP2488503A1 (en) | 2009-10-12 | 2012-08-22 | Myrexis, Inc. | Amino - pyrimidine compounds as inhibitors of tbkl and/or ikk epsilon |
| US7718662B1 (en) | 2009-10-12 | 2010-05-18 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of bruton's tyrosine kinase |
| NZ601794A (en) | 2010-01-29 | 2014-04-30 | Otsuka Pharma Co Ltd | Di - substituted pyridine derivatives as anticancers |
| WO2011106168A1 (en) | 2010-02-24 | 2011-09-01 | Dcam Pharma Inc | Purine compounds for treating autoimmune and demyelinating diseases |
| WO2011121223A1 (en) | 2010-03-30 | 2011-10-06 | Sanofi-Aventis | 6-(alkyl- or cycloalkyl-triazolopyridazine-sulfanyl) benzothiazole derivatives: preparation, application as medicaments and use as met inhibitors |
| CN107898791A (en) | 2010-06-03 | 2018-04-13 | 药品循环有限责任公司 | The application of bruton's tyrosine kinase (BTK) inhibitor |
| MX336001B (en) | 2010-06-18 | 2016-01-07 | Genentech Inc | Anti-axl antibodies and methods of use. |
| WO2012135641A2 (en) | 2011-03-30 | 2012-10-04 | H. Lee Moffitt Cancer Center And Research Institute | Aurora kinase inhibitors and methods of making and using thereof |
| EP2694486B1 (en) | 2011-04-01 | 2018-01-10 | University of Utah Research Foundation | Substituted n-(3-(pyrimidin-4-yl)phenyl)acrylamide analogs as tyrosine receptor kinase btk inhibitors |
| PL2693881T3 (en) | 2011-04-01 | 2020-03-31 | University Of Utah Research Foundation | Substituted n-phenylpyrimidin-2-amine analogs as inhibitors of the axl kinase |
| CA2838275C (en) | 2011-06-06 | 2021-08-10 | University Of Iowa Research Foundation | Methods for inhibiting muscle atrophy |
| KR101938368B1 (en) | 2011-07-13 | 2019-01-14 | 주식회사 티움바이오 | 2-pyridyl substituted imidazoles as alk5 and/or alk4 inhibitors |
| KR20140048968A (en) | 2011-07-13 | 2014-04-24 | 파마시클릭스, 인코포레이티드 | Inhibitors of bruton's tyrosine kinase |
| BR112014009276A8 (en) | 2011-10-19 | 2017-06-20 | Pharmacyclics Inc | use of bruton tyrosine kinase inhibitors (btk) |
| CN103087077B (en) | 2011-11-03 | 2016-05-18 | 上海希迈医药科技有限公司 | Thienopyrimidine and furans miazines derivative, its preparation method and in application pharmaceutically |
| US8377946B1 (en) | 2011-12-30 | 2013-02-19 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US8501724B1 (en) | 2012-01-31 | 2013-08-06 | Pharmacyclics, Inc. | Purinone compounds as kinase inhibitors |
| US9133134B2 (en) | 2012-05-16 | 2015-09-15 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| MY194911A (en) | 2012-06-04 | 2022-12-22 | Pharmacyclics Llc | Crystalline forms of a bruton's tyrosine kinase inhibitor |
| MX2015001081A (en) | 2012-07-24 | 2015-10-14 | Pharmacyclics Inc | Mutations associated with resistance to inhibitors of bruton's tyrosine kinase (btk). |
| US8703981B2 (en) | 2012-08-03 | 2014-04-22 | Xerox Corporation | Lignosulfonate compounds for solid ink applications |
| RU2645672C2 (en) | 2012-08-06 | 2018-02-27 | Ацея Байосайенсиз Инк. | New compounds of pyrrolopyrimidine as inhibitors of protein kinases |
| KR101446742B1 (en) | 2012-08-10 | 2014-10-01 | 한국화학연구원 | N2,N4-bis(4-(piperazin-1-yl)phenyl)pyrimidine-2,4-diamine derivatives or pharmaceutically acceptable salt thereof, and pharmaceutical composition for the prevention or treatment of cancer containing the same as an active ingredient |
| US9493454B2 (en) | 2012-09-26 | 2016-11-15 | Tolero Pharmaceuticals, Inc. | Multiple kinase pathway inhibitors |
| CN104854107A (en) | 2012-11-15 | 2015-08-19 | 药品循环公司 | Pyrrolopyrimidine compounds as kinase inhibitors |
| EA201591051A1 (en) | 2013-02-08 | 2016-06-30 | Селджен Авиломикс Рисерч, Инк. | ERK INHIBITORS AND THEIR OPTIONS |
| US10322125B2 (en) | 2013-02-22 | 2019-06-18 | Emory University | TGF-beta enhancing compositions for cartilage repair and methods related thereto |
| US9708326B2 (en) | 2013-02-25 | 2017-07-18 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US20160115167A1 (en) | 2013-03-04 | 2016-04-28 | The Brigham And Women's Hospital, Inc. | Bmp inhibitors and methods of use thereof |
| JP2016512549A (en) | 2013-03-14 | 2016-04-28 | ファーマサイクリックス エルエルシー | Combination of Breton tyrosine kinase inhibitor and CYP3A4 inhibitor |
| EP2970205B1 (en) | 2013-03-14 | 2019-05-08 | Tolero Pharmaceuticals, Inc. | Jak2 and alk2 inhibitors and methods for their use |
| TW201524952A (en) | 2013-03-15 | 2015-07-01 | Araxes Pharma Llc | Covalent inhibitors of KRAS G12C |
| US9745319B2 (en) | 2013-03-15 | 2017-08-29 | Araxes Pharma Llc | Irreversible covalent inhibitors of the GTPase K-Ras G12C |
| AU2014251028A1 (en) | 2013-04-08 | 2015-11-05 | Janssen Pharmaceutica Nv | Ibrutinib combination therapy |
| CA2917167A1 (en) | 2013-07-02 | 2015-01-08 | Pharmacyclics Llc | Purinone compounds as kinase inhibitors |
| TN2016000094A1 (en) | 2013-09-30 | 2017-07-05 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase. |
| JP6559123B2 (en) | 2013-10-10 | 2019-08-14 | アラクセス ファーマ エルエルシー | Inhibitor of KRASG12C |
| TWI582083B (en) | 2014-10-07 | 2017-05-11 | 美國禮來大藥廠 | Aminopyridyloxypyrazole compounds |
| EP3237408B1 (en) | 2014-12-23 | 2018-10-31 | Cephalon, Inc. | Methods for preparing fused bicyclic 2, 4-diaminopyrimidine derivatives |
| WO2016146651A1 (en) | 2015-03-16 | 2016-09-22 | Oncodesign Sa | Macrocyclic activin-like receptor kinase inhibitors |
| CA2982664A1 (en) | 2015-04-16 | 2016-10-20 | Merck Patent Gmbh | 3-(1h-benzimidazol-2-yl)-1h-pyridin-2-one derivatives |
| MX2017014645A (en) | 2015-05-18 | 2018-01-23 | Tolero Pharmaceuticals Inc | Alvocidib prodrugs having increased bioavailability. |
| MX2018012664A (en) | 2016-04-15 | 2019-03-07 | Blueprint Medicines Corp | Inhibitors of activin receptor-like kinase. |
| JP7076741B2 (en) | 2016-12-27 | 2022-05-30 | 国立研究開発法人理化学研究所 | BMP signal inhibitor compound |
| AR112027A1 (en) | 2017-06-15 | 2019-09-11 | Biocryst Pharm Inc | ALK 2 KINASE INHIBITORS CONTAINING IMIDAZOLE |
| BR112020020246A8 (en) | 2018-04-05 | 2022-10-18 | Sumitomo Dainippon Pharma Oncology Inc | AXL KINASE INHIBITORS AND THEIR USE |
| CN112512597A (en) | 2018-07-26 | 2021-03-16 | 大日本住友制药肿瘤公司 | Methods for treating diseases associated with aberrant ACVR1 expression and ACVR1 inhibitors useful therefor |
| EP3908575A4 (en) | 2019-01-10 | 2022-09-14 | Sumitomo Pharma Oncology, Inc. | Alk5 inhibitors for treating myelodysplastic syndrome |
-
2019
- 2019-07-26 CN CN201980049596.5A patent/CN112512597A/en active Pending
- 2019-07-26 EP EP19841400.5A patent/EP3826684A4/en active Pending
- 2019-07-26 MX MX2021000977A patent/MX2021000977A/en unknown
- 2019-07-26 JP JP2021503828A patent/JP2021530554A/en active Pending
- 2019-07-26 KR KR1020217005329A patent/KR20210038906A/en unknown
- 2019-07-26 WO PCT/US2019/043738 patent/WO2020023910A1/en active Application Filing
- 2019-07-26 US US16/523,829 patent/US11040038B2/en active Active
- 2019-07-26 AU AU2019310590A patent/AU2019310590A1/en active Pending
- 2019-07-26 CA CA3103995A patent/CA3103995A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EP3826684A4 (en) | 2022-04-06 |
| KR20210038906A (en) | 2021-04-08 |
| AU2019310590A1 (en) | 2021-01-14 |
| CN112512597A (en) | 2021-03-16 |
| US11040038B2 (en) | 2021-06-22 |
| US20200085823A1 (en) | 2020-03-19 |
| CA3103995A1 (en) | 2020-01-30 |
| WO2020023910A9 (en) | 2020-12-03 |
| MX2021000977A (en) | 2021-04-12 |
| EP3826684A1 (en) | 2021-06-02 |
| WO2020023910A1 (en) | 2020-01-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2021530554A (en) | Methods and ACVR1 Inhibitors for Treatment of Diseases with Abnormal ACVR1 Expression | |
| CN110869357A (en) | Compounds and methods of use thereof for treating cancer | |
| CN101910182B (en) | Antitumor agent | |
| TW202027746A (en) | Combination therapy for the treatment of triple-negative breast cancer | |
| JP6883049B2 (en) | Aminothiazole compounds and their use | |
| CA2993013A1 (en) | Substituted quinazoline compounds and their use as inhibitors of g12c mutant kras, hras and/or nras proteins | |
| JP6513095B2 (en) | Heteroatom-containing deoxyuridine triphosphatase inhibitors | |
| JP2021130682A (en) | Combination therapy for treating malignancies | |
| AU2010276160A1 (en) | Treatment of liver disorders with PI3K inhibitors | |
| CN106928150A (en) | Acrylamide anil and its application pharmaceutically | |
| EA028414B1 (en) | Treatment of cancer with tor kinase inhibitors | |
| CN109640970B (en) | Selective p38α -specific MAPK inhibitors | |
| JP6147246B2 (en) | Combinations of AKT and MEK inhibitor compounds and methods of use | |
| EA018964B1 (en) | PYRIDO[2,3-d]PYRIMIDIN-7-ONE COMPOUNDS AS INHIBITORS OF PI3K-ALPHA FOR THE TREATMENT OF CANCER | |
| CN108026046B (en) | Substituted quinazoline compounds and their use as inhibitors of G12C mutant KRAS, HRAS and/or NRAS proteins | |
| JP2021113819A (en) | METHOD FOR MEASURING INHIBITION OF c-Jun N TERMINAL KINASE IN SKIN | |
| US11224608B2 (en) | Compounds and methods for treating cancer | |
| EA021951B1 (en) | Anticancer combination | |
| EP2387397A2 (en) | Methods for identifying patients who will respond well to cancer treatment | |
| US7045523B2 (en) | Combination comprising N-{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl}-4-(3-pyridyl)-2-pyrimidine-amine and telomerase inhibitor | |
| US11427574B2 (en) | Compounds for treating Rac-GTPase mediated disorder | |
| US10765669B2 (en) | Pharmaceutical composition for preventing or treating DYRK-related diseases, containing pyridine-based compound as active ingredient | |
| US11149300B1 (en) | Methods of treating gastrointestinal malignancies | |
| JP5958753B2 (en) | Wip1 protein phosphatase inhibitor and use thereof | |
| JP2022506289A (en) | Indazole kinase inhibitor and its use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220725 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220725 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20230628 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20230713 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20230713 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20231010 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240115 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240321 |