JP2017145247A - Infection preventive and/or therapeutic agent comprising c-4" position-substituted macrolide derivative - Google Patents
Infection preventive and/or therapeutic agent comprising c-4" position-substituted macrolide derivative Download PDFInfo
- Publication number
- JP2017145247A JP2017145247A JP2017026792A JP2017026792A JP2017145247A JP 2017145247 A JP2017145247 A JP 2017145247A JP 2017026792 A JP2017026792 A JP 2017026792A JP 2017026792 A JP2017026792 A JP 2017026792A JP 2017145247 A JP2017145247 A JP 2017145247A
- Authority
- JP
- Japan
- Prior art keywords
- formula
- acid
- compound
- therapeutic agent
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000003814 drug Substances 0.000 title claims abstract description 15
- 229940124597 therapeutic agent Drugs 0.000 title claims abstract description 12
- 230000003449 preventive effect Effects 0.000 title claims abstract description 10
- 239000003120 macrolide antibiotic agent Substances 0.000 title abstract description 12
- 208000015181 infectious disease Diseases 0.000 title abstract description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 71
- 150000003839 salts Chemical class 0.000 claims abstract description 18
- 239000012453 solvate Substances 0.000 claims abstract description 11
- 239000004480 active ingredient Substances 0.000 claims abstract description 6
- 238000002360 preparation method Methods 0.000 claims description 15
- 239000007787 solid Substances 0.000 claims description 13
- 239000001913 cellulose Substances 0.000 claims description 10
- 208000035473 Communicable disease Diseases 0.000 claims description 9
- 229920002472 Starch Polymers 0.000 claims description 9
- 239000008107 starch Substances 0.000 claims description 9
- 235000019698 starch Nutrition 0.000 claims description 9
- 239000000843 powder Substances 0.000 claims description 8
- 239000002775 capsule Substances 0.000 claims description 7
- 229920002134 Carboxymethyl cellulose Polymers 0.000 claims description 6
- VJHCJDRQFCCTHL-UHFFFAOYSA-N acetic acid 2,3,4,5,6-pentahydroxyhexanal Chemical compound CC(O)=O.OCC(O)C(O)C(O)C(O)C=O VJHCJDRQFCCTHL-UHFFFAOYSA-N 0.000 claims description 6
- 235000010948 carboxy methyl cellulose Nutrition 0.000 claims description 6
- 229950008138 carmellose Drugs 0.000 claims description 6
- 235000010980 cellulose Nutrition 0.000 claims description 6
- 229920002678 cellulose Polymers 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- 229930006000 Sucrose Natural products 0.000 claims description 5
- 239000008187 granular material Substances 0.000 claims description 5
- 239000005720 sucrose Substances 0.000 claims description 5
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 4
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 claims description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 claims description 4
- 239000008103 glucose Substances 0.000 claims description 4
- 239000001863 hydroxypropyl cellulose Substances 0.000 claims description 4
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 claims description 4
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 4
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 4
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 4
- 239000006187 pill Substances 0.000 claims description 4
- 229920003124 powdered cellulose Polymers 0.000 claims description 4
- 235000019814 powdered cellulose Nutrition 0.000 claims description 4
- 239000003826 tablet Substances 0.000 claims description 4
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 claims description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 claims description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 claims description 3
- 239000005715 Fructose Substances 0.000 claims description 3
- 229930091371 Fructose Natural products 0.000 claims description 3
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 claims description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 claims description 3
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 claims description 3
- 229930195725 Mannitol Natural products 0.000 claims description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 claims description 3
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 claims description 3
- 239000008101 lactose Substances 0.000 claims description 3
- 229940031703 low substituted hydroxypropyl cellulose Drugs 0.000 claims description 3
- 239000000594 mannitol Substances 0.000 claims description 3
- 235000010355 mannitol Nutrition 0.000 claims description 3
- 239000002552 dosage form Substances 0.000 claims description 2
- 229940071676 hydroxypropylcellulose Drugs 0.000 claims description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims 1
- 241000894006 Bacteria Species 0.000 abstract description 33
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 abstract description 25
- 230000000844 anti-bacterial effect Effects 0.000 abstract description 14
- 229960003276 erythromycin Drugs 0.000 abstract description 13
- 241000204031 Mycoplasma Species 0.000 abstract description 9
- 150000004677 hydrates Chemical class 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 96
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 81
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 50
- 210000004072 lung Anatomy 0.000 description 38
- 239000000243 solution Substances 0.000 description 38
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 36
- 238000012360 testing method Methods 0.000 description 29
- 239000007788 liquid Substances 0.000 description 24
- 230000002829 reductive effect Effects 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 18
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 18
- 239000003981 vehicle Substances 0.000 description 18
- 239000000203 mixture Substances 0.000 description 16
- 239000000706 filtrate Substances 0.000 description 14
- 239000012044 organic layer Substances 0.000 description 14
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- 239000002904 solvent Substances 0.000 description 12
- -1 etc.) Substances 0.000 description 11
- 238000011081 inoculation Methods 0.000 description 11
- 239000002609 medium Substances 0.000 description 11
- 238000010898 silica gel chromatography Methods 0.000 description 11
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- 238000000034 method Methods 0.000 description 10
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 239000002054 inoculum Substances 0.000 description 9
- 235000017557 sodium bicarbonate Nutrition 0.000 description 9
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 9
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 239000012153 distilled water Substances 0.000 description 8
- 239000012046 mixed solvent Substances 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 7
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 229940099563 lactobionic acid Drugs 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 241000192125 Firmicutes Species 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- 241000606161 Chlamydia Species 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- 241000606768 Haemophilus influenzae Species 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 229940047650 haemophilus influenzae Drugs 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000035945 sensitivity Effects 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 235000015424 sodium Nutrition 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 3
- YHVUVJYEERGYNU-UHFFFAOYSA-N 4',8-Di-Me ether-5,7,8-Trihydroxy-3-(4-hydroxybenzyl)-4-chromanone Natural products COC1(C)CC(O)OC(C)C1O YHVUVJYEERGYNU-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 229920000881 Modified starch Polymers 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229910000831 Steel Inorganic materials 0.000 description 3
- 241000193998 Streptococcus pneumoniae Species 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 235000015165 citric acid Nutrition 0.000 description 3
- AJSDVNKVGFVAQU-BIIVOSGPSA-N cladinose Chemical group O=CC[C@@](C)(OC)[C@@H](O)[C@H](C)O AJSDVNKVGFVAQU-BIIVOSGPSA-N 0.000 description 3
- 239000007884 disintegrant Substances 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 229940041033 macrolides Drugs 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000010959 steel Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- QCQZFSUBYDWVBG-UHFFFAOYSA-N 2-amino-n-ethylacetamide Chemical compound CCNC(=O)CN QCQZFSUBYDWVBG-UHFFFAOYSA-N 0.000 description 2
- 241000589291 Acinetobacter Species 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 2
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 241000588914 Enterobacter Species 0.000 description 2
- 241000194033 Enterococcus Species 0.000 description 2
- 239000004593 Epoxy Substances 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 241000606790 Haemophilus Species 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 241000588653 Neisseria Species 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 2
- 241000607768 Shigella Species 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 2
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 229960002626 clarithromycin Drugs 0.000 description 2
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 2
- 238000005138 cryopreservation Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000000105 evaporative light scattering detection Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 229940032147 starch Drugs 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 2
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 2
- RXZBMPWDPOLZGW-XMRMVWPWSA-N (E)-roxithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=N/OCOCCOC)/[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 RXZBMPWDPOLZGW-XMRMVWPWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 1
- SPSPIUSUWPLVKD-UHFFFAOYSA-N 2,3-dibutyl-6-methylphenol Chemical compound CCCCC1=CC=C(C)C(O)=C1CCCC SPSPIUSUWPLVKD-UHFFFAOYSA-N 0.000 description 1
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 1
- RVHOBHMAPRVOLO-UHFFFAOYSA-N 2-ethylbutanedioic acid Chemical compound CCC(C(O)=O)CC(O)=O RVHOBHMAPRVOLO-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- QUYFSHOLLKVPGX-UHFFFAOYSA-N 3-methylsulfonylpropan-1-amine Chemical compound CS(=O)(=O)CCCN QUYFSHOLLKVPGX-UHFFFAOYSA-N 0.000 description 1
- 241000588624 Acinetobacter calcoaceticus Species 0.000 description 1
- 241000606125 Bacteroides Species 0.000 description 1
- 241000606124 Bacteroides fragilis Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 241000588807 Bordetella Species 0.000 description 1
- 241000588832 Bordetella pertussis Species 0.000 description 1
- OFISWRJORIMAGZ-UHFFFAOYSA-N CC(C)CN(CCNC)C(C)C Chemical compound CC(C)CN(CCNC)C(C)C OFISWRJORIMAGZ-UHFFFAOYSA-N 0.000 description 1
- DBVADBHSJCWFKI-UHFFFAOYSA-N CC(C)N(CCCl)C(C)C Chemical compound CC(C)N(CCCl)C(C)C DBVADBHSJCWFKI-UHFFFAOYSA-N 0.000 description 1
- UGAMNTGMICNTPS-UHFFFAOYSA-N CCNC(CNC(OCc1ccccc1)=O)=O Chemical compound CCNC(CNC(OCc1ccccc1)=O)=O UGAMNTGMICNTPS-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000589876 Campylobacter Species 0.000 description 1
- 241000589875 Campylobacter jejuni Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 241000588923 Citrobacter Species 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000194032 Enterococcus faecalis Species 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- 229930006677 Erythromycin A Natural products 0.000 description 1
- 241000588722 Escherichia Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 241000606766 Haemophilus parainfluenzae Species 0.000 description 1
- 241000589989 Helicobacter Species 0.000 description 1
- 241000590002 Helicobacter pylori Species 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- 241000588749 Klebsiella oxytoca Species 0.000 description 1
- 241000588747 Klebsiella pneumoniae Species 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 241000589248 Legionella Species 0.000 description 1
- 241000589242 Legionella pneumophila Species 0.000 description 1
- 208000007764 Legionnaires' Disease Diseases 0.000 description 1
- 239000004368 Modified starch Substances 0.000 description 1
- 241001430197 Mollicutes Species 0.000 description 1
- 241000588621 Moraxella Species 0.000 description 1
- 241000588655 Moraxella catarrhalis Species 0.000 description 1
- 241000588771 Morganella <proteobacterium> Species 0.000 description 1
- 241000204022 Mycoplasma gallisepticum Species 0.000 description 1
- 241000204051 Mycoplasma genitalium Species 0.000 description 1
- 241000204048 Mycoplasma hominis Species 0.000 description 1
- 241000204045 Mycoplasma hyopneumoniae Species 0.000 description 1
- 241000202936 Mycoplasma mycoides Species 0.000 description 1
- 241001148556 Mycoplasma ovipneumoniae Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- CJUMAFVKTCBCJK-UHFFFAOYSA-N N-benzyloxycarbonylglycine Chemical compound OC(=O)CNC(=O)OCC1=CC=CC=C1 CJUMAFVKTCBCJK-UHFFFAOYSA-N 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002675 Polyoxyl Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 241000588769 Proteus <enterobacteria> Species 0.000 description 1
- 241000588770 Proteus mirabilis Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000607720 Serratia Species 0.000 description 1
- 241000607715 Serratia marcescens Species 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 241000191940 Staphylococcus Species 0.000 description 1
- 241000191963 Staphylococcus epidermidis Species 0.000 description 1
- 241000194017 Streptococcus Species 0.000 description 1
- 241000193996 Streptococcus pyogenes Species 0.000 description 1
- 241000193990 Streptococcus sp. 'group B' Species 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- 208000037386 Typhoid Diseases 0.000 description 1
- 241000607598 Vibrio Species 0.000 description 1
- 241000607626 Vibrio cholerae Species 0.000 description 1
- 241000607272 Vibrio parahaemolyticus Species 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- PENDGIOBPJLVBT-HMMOOPTJSA-N abt-773 Chemical compound O([C@@H]1[C@@H](C)C(=O)[C@@H](C)C(=O)O[C@@H]([C@]2(OC(=O)N[C@@H]2[C@@H](C)C(=O)[C@H](C)C[C@]1(C)OC\C=C\C=1C=C2C=CC=CC2=NC=1)C)CC)[C@@H]1O[C@H](C)C[C@H](N(C)C)[C@H]1O PENDGIOBPJLVBT-HMMOOPTJSA-N 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 241001148470 aerobic bacillus Species 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- WMGSQTMJHBYJMQ-UHFFFAOYSA-N aluminum;magnesium;silicate Chemical compound [Mg+2].[Al+3].[O-][Si]([O-])([O-])[O-] WMGSQTMJHBYJMQ-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 201000005008 bacterial sepsis Diseases 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000007765 cera alba Substances 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000007330 chocolate agar Substances 0.000 description 1
- 229960004106 citric acid Drugs 0.000 description 1
- 210000003555 cloaca Anatomy 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 238000003113 dilution method Methods 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 229940032049 enterococcus faecalis Drugs 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 210000004211 gastric acid Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 229940037467 helicobacter pylori Drugs 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 229960003943 hypromellose Drugs 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000000752 ionisation method Methods 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 150000002596 lactones Chemical group 0.000 description 1
- 229940115932 legionella pneumophila Drugs 0.000 description 1
- 238000002514 liquid chromatography mass spectrum Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229960003511 macrogol Drugs 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000007721 medicinal effect Effects 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 230000003228 microsomal effect Effects 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 235000019426 modified starch Nutrition 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- MKDYQLJYEBWUIG-UHFFFAOYSA-N n',n'-diethyl-n-methylethane-1,2-diamine Chemical compound CCN(CC)CCNC MKDYQLJYEBWUIG-UHFFFAOYSA-N 0.000 description 1
- IUSXYVRFJVAVOB-UHFFFAOYSA-N n-(2-chloroethyl)-n-propan-2-ylpropan-2-amine;hydron;chloride Chemical compound Cl.CC(C)N(C(C)C)CCCl IUSXYVRFJVAVOB-UHFFFAOYSA-N 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 239000000473 propyl gallate Substances 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- 229940075579 propyl gallate Drugs 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 229960005224 roxithromycin Drugs 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000004328 sodium tetraborate Substances 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229960004793 sucrose Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960003250 telithromycin Drugs 0.000 description 1
- LJVAJPDWBABPEJ-PNUFFHFMSA-N telithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)[C@@H](C)C(=O)O[C@@H]([C@]2(OC(=O)N(CCCCN3C=C(N=C3)C=3C=NC=CC=3)[C@@H]2[C@@H](C)C(=O)[C@H](C)C[C@@]1(C)OC)C)CC)[C@@H]1O[C@H](C)C[C@H](N(C)C)[C@H]1O LJVAJPDWBABPEJ-PNUFFHFMSA-N 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- BPLKQGGAXWRFOE-UHFFFAOYSA-M trimethylsulfoxonium iodide Chemical compound [I-].C[S+](C)(C)=O BPLKQGGAXWRFOE-UHFFFAOYSA-M 0.000 description 1
- 201000008297 typhoid fever Diseases 0.000 description 1
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 1
- 241001148471 unidentified anaerobic bacterium Species 0.000 description 1
- 229940118696 vibrio cholerae Drugs 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
Images


Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Description
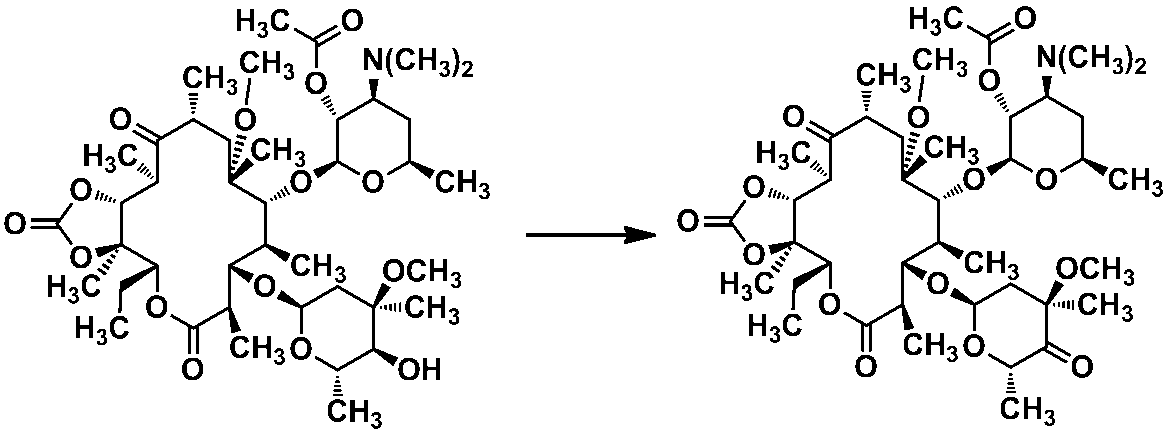
本発明は、エリスロマイシン類似骨格を有する新規抗生物質に関する。より具体的には、本発明は、クラジノースの4”位に窒素原子を有する置換基で置換されたメチル基を有するマクロライド化合物を有効成分として含有する感染症の予防及び/又は治療のために用いる医薬に関するものである。 The present invention relates to a novel antibiotic having an erythromycin-like skeleton. More specifically, the present invention relates to the prevention and / or treatment of infectious diseases containing a macrolide compound having a methyl group substituted with a substituent having a nitrogen atom at the 4 ″ position of cladinose as an active ingredient. It relates to the medicine used.
エリスロマイシンAはグラム陽性菌、マイコプラズマなどに起因する感染症の治療薬として広く使用されている抗生物質である。しかし、エリスロマイシンは胃酸で分解されるため、体内動態が一定しないという欠点があった。そこで酸に対する安定性を増した誘導体が検討され、その結果、クラリスロマイシン、アジスロマイシン(特許文献1及び2)、ロキシスロマイシンなどの体内動態の安定したマクロライド剤が開発されてきた。外来の呼吸器感染症を治療領域とするこれらマクロライド剤は、特に臨床分離頻度の高い肺炎球菌、連鎖球菌並びにインフルエンザ菌に対し強い抗菌活性を有する必要がある。さらに、市中肺炎からマクロライド耐性の肺炎球菌が高頻度に分離されていることから耐性肺炎球菌に有効であることも重要となっている。 Erythromycin A is an antibiotic widely used as a therapeutic agent for infectious diseases caused by Gram-positive bacteria, mycoplasma and the like. However, since erythromycin is decomposed by gastric acid, there is a disadvantage that pharmacokinetics is not constant. Accordingly, derivatives having increased stability to acids have been studied, and as a result, macrolide agents with stable pharmacokinetics such as clarithromycin, azithromycin (Patent Documents 1 and 2), and roxithromycin have been developed. These macrolide agents for treating external respiratory infections need to have strong antibacterial activity against pneumococci, streptococci and Haemophilus influenzae, which are frequently clinically isolated. Furthermore, since macrolide-resistant pneumococci are frequently isolated from community-acquired pneumonia, it is important to be effective against resistant pneumococci.
近年、広範な研究の結果、エリスロマイシン耐性肺炎球菌、エリスロマイシン耐性連鎖球菌のいずれに対しても有効なマクロライドとしてAgouridasらは1995年にHMR3647(テリスロマイシン,特許文献3)を、Orらは1998年にABT−773(セスロマイシン,特許文献4)を相次いで見出した。その後、さらに薬効増強が図られた2−フルオロケトライド(特許文献5)が報告されている。 As a result of extensive research in recent years, Agouridas et al. In 1995 as HMR3647 (Terithromycin, Patent Document 3) and Or et al. 1998 as effective macrolides against both erythromycin-resistant pneumococci and erythromycin-resistant streptococci. In the year, ABT-773 (Cesromycin, Patent Document 4) was found one after another. Thereafter, 2-fluoroketolide (Patent Document 5), which has further enhanced medicinal effects, has been reported.
一方、クラジノースの4”位に窒素原子を有する置換基で置換されたメチル基を有するマクロライド化合物に関しては、ラクトン環内に窒素原子をもつという構造的な特徴を有しているアザライドタイプの化合物がほとんどである(特許文献6)。 On the other hand, the macrolide compound having a methyl group substituted with a substituent having a nitrogen atom at the 4 ″ position of cladinose is an azalide type having a structural feature of having a nitrogen atom in the lactone ring. Most of the compounds are (Patent Document 6).
更に、エリスロマイシン耐性肺炎球菌、エリスロマイシン耐性連鎖球菌のいずれに対しても有効なマクロライドとして、クラジノースの4”位に窒素原子を有する置換基で置換されたメチル基を有するマクロライド化合物については、出願人らも報告している(特許文献7、8及び9)。特にその中でも特許文献7、8に記載された実施例15が好ましい化合物である。
Furthermore, as a macrolide effective against both erythromycin-resistant pneumococci and erythromycin-resistant streptococci, a macrolide compound having a methyl group substituted with a substituent having a nitrogen atom at the 4 ″ position of cladinose is filed. Humans have also reported (
本発明の課題は、従来のエリスロマイシン感受性菌のみならず、エリスロマイシン耐性菌(例えば耐性肺炎球菌、耐性連鎖球菌、及びマイコプラズマ)に対しても有効な化合物を提供することにある。 An object of the present invention is to provide a compound effective not only for conventional erythromycin-sensitive bacteria but also for erythromycin-resistant bacteria (for example, resistant pneumococci, resistant streptococci, and mycoplasma).
そこで、本発明者らは新たなマクロライド化合物の研究を鋭意行った結果、下記に示す化合物が優れた抗菌活性を有することを見出し、本発明を完成した。 Thus, as a result of intensive studies on new macrolide compounds, the present inventors have found that the following compounds have excellent antibacterial activity, and have completed the present invention.
すなわち、本発明により、
(1)式[1]:
That is, according to the present invention,
(1) Formula [1]:
また、上記発明の好ましい態様として、
(2)固形製剤である請求項1に記載の感染症の予防又は治療薬、
(3)剤形が錠剤、丸剤、カプセル剤、顆粒剤、散剤、又は粉剤から選択される上記(2)に記載の感染症の予防又は治療薬、及び
(4)乳糖、ショ糖、ブドウ糖、麦芽糖、果糖、マンニトール、デンプン、粉末セルロース、結晶セルロース、カルメロース、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチルセルロースからなる群から選ばれる1種以上を含む上記(3)に記載の感染症の予防又は治療薬が本発明により提供される。
Moreover, as a preferred embodiment of the above invention,
(2) The preventive or therapeutic agent for infectious diseases according to claim 1, which is a solid preparation,
(3) The preventive or therapeutic agent for infectious diseases according to (2) above, wherein the dosage form is selected from tablets, pills, capsules, granules, powders, or powders, and (4) lactose, sucrose, glucose The infection according to (3) above, comprising at least one selected from the group consisting of maltose, fructose, mannitol, starch, powdered cellulose, crystalline cellulose, carmellose, low-substituted hydroxypropylcellulose, hydroxypropylcellulose and hydroxypropylmethylcellulose A prophylactic or therapeutic agent for the disease is provided by the present invention.
さらに、(5)式[1]で表される化合物若しくはその薬学的に許容される塩、又はそれらの水和物若しくはそれらの溶媒和物の1日あたり投与量が1〜10000mgである、上記(1)乃至(4)に記載の感染症の予防又は治療薬が本発明により提供される。 Further, (5) the daily dose of the compound represented by the formula [1] or a pharmaceutically acceptable salt thereof, or a hydrate or a solvate thereof is 1 to 10,000 mg, The preventive or therapeutic agent for infectious diseases according to (1) to (4) is provided by the present invention.
本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は微生物、好ましくはグラム陽性菌又はグラム陰性菌などの好気性又は嫌気性細菌類、マイコプラズマやクラミジアなどに対して幅広い抗菌活性を有しており、特に従来のマクロライド系抗生物質では十分な抗菌活性が得られなかったエリスロマイシン耐性菌(例えば耐性肺炎球菌、耐性連鎖球菌、及びマイコプラズマ)などに対しても優れた抗菌活性を示すという特徴がある。 The compound of the present invention or a salt thereof, or a hydrate or a solvate thereof has a broad antibacterial activity against microorganisms, preferably aerobic or anaerobic bacteria such as gram-positive bacteria or gram-negative bacteria, mycoplasma and chlamydia In particular, it has excellent antibacterial activity against erythromycin-resistant bacteria (for example, resistant pneumococci, resistant streptococci, and mycoplasma) that were not able to obtain sufficient antibacterial activity with conventional macrolide antibiotics. It has the feature of showing.
本発明において、「その薬学的に許容される塩」とは、酸付加塩又は塩基付加塩のいずれでもよく、酸付加塩としては、例えば酢酸、プロピオン酸、酪酸、ギ酸、トリフルオロ酢酸、マレイン酸、酒石酸、クエン酸、ステアリン酸、コハク酸、エチルコハク酸、ラクトビオン酸、グルコン酸、グルコヘプトン酸、安息香酸、メタンスルホン酸、エタンスルホン酸、2−ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、パラトルエンスルホン酸、ラウリル硫酸、リンゴ酸、アスパラギン酸、グルタミン酸、アジピン酸、システイン、N−アセチルシステイン、塩酸、臭化水素酸、リン酸、硫酸、ヨウ化水素酸、ニコチン酸、シュウ酸、ピクリン酸、チオシアン酸、ウンデカン酸、アクリル酸ポリマー、カルボキシビニルポリマーなどの酸との塩を挙げることができ、塩基付加塩としては、例えばナトリウム塩、カリウム塩、カルシウム塩などの無機塩基との塩、モルホリン、ピペリジンなどの有機アミン、アミノ酸との塩を挙げることができるが、これらに限定されることはない。 In the present invention, the “pharmaceutically acceptable salt” may be either an acid addition salt or a base addition salt. Examples of the acid addition salt include acetic acid, propionic acid, butyric acid, formic acid, trifluoroacetic acid, maleic acid. Acid, tartaric acid, citric acid, stearic acid, succinic acid, ethyl succinic acid, lactobionic acid, gluconic acid, glucoheptonic acid, benzoic acid, methanesulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulfonic acid, paratoluenesulfone Acid, lauryl sulfate, malic acid, aspartic acid, glutamic acid, adipic acid, cysteine, N-acetylcysteine, hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, hydroiodic acid, nicotinic acid, oxalic acid, picric acid, thiocyanate Acid, undecanoic acid, acrylic acid polymer, carboxyvinyl polymer and other acids Examples of base addition salts include salts with inorganic bases such as sodium salts, potassium salts and calcium salts, organic amines such as morpholine and piperidine, and salts with amino acids. It is not limited to.
本発明において、「抗菌剤」とはグラム陽性細菌、グラム陰性細菌やマイコプラズマといった細菌に作用してその生育を抑制又は殺菌する能力を持つ物質を意味する。菌の繁殖を抑えたり、一部の菌を殺してその数を減少させたりするようなものでもよい。グラム陽性細菌としては、例えば、ブドウ球菌属(黄色ブドウ球菌、表皮ブドウ球菌など)、連鎖球菌属(化膿連鎖球菌、B群連鎖球菌、肺炎球菌など)、腸球菌属(エンテロコッカス・フェカーリス、エンテロコッカス・フェシウムなど)が挙げられる。グラム陰性菌としては、例えば、シュードモナス属(緑膿菌など)、大腸菌属(大腸菌など)、クレブシエラ属(肺炎桿菌、クレブシエラ・オキシトカなど)、ヘモフィルス属(インフルエンザ菌、パラインフルエンザ菌など)、ボルデテラ属(百日咳菌、気管支敗血症菌など)、セラチア属(セラチア・マルセッセンスなど)、プロテウス属(プロテウス・ミラビリスなど)エンテロバクター属(エンテロバクター・クロアカなど)、カンピロバクター属(カンピロバクター・ジェジュニなど)、シトロバクター属、ビブリオ属(腸炎ビブリオ、コレラ菌など)、モルガネラ属(モルガネラ・モルガニなど)、サルモネラ属(チフス菌、パラチフス菌など)、シゲラ属(赤痢菌など)、アシネトバクター属(アシネトバクター・バウマニー、アシネトバクター・カルコアセチカスなど)、レジオネラ属(レジオネラ・ニューモフィラなど)、バクテロイデス属(バクテロイデス・フラジリスなど)、ナイセリア属(淋菌、髄膜炎菌など)、モラキセラ属(モラキセラ・カタラーリスなど)、クラミジア属(クラミジア・トラコマティス、クラミジア・シッタシーなど)及びヘリコバクター属(ヘリコバクター・ピロリなど)が挙げられる。マイコプラズマとしては、M. galli septicum、M. genitalium、M. hominis、M. hyopneumoniae、M. laboratorium、M. mycoides、M. ovipneumoniae、M. pneumonia が挙げられる。 In the present invention, the term “antibacterial agent” means a substance having the ability to act on bacteria such as gram positive bacteria, gram negative bacteria, and mycoplasma to suppress or sterilize their growth. It may be something that suppresses the growth of bacteria or kills some bacteria to reduce their number. Gram-positive bacteria include, for example, Staphylococcus (S. aureus, Staphylococcus epidermidis, etc.), Streptococcus (S. pyogenes, Group B Streptococcus, Streptococcus pneumoniae, etc.), Enterococcus (Enterococcus faecalis, Enterococcus Fesium etc.). Gram-negative bacteria include, for example, Pseudomonas genus (such as Pseudomonas aeruginosa), Escherichia genus (such as Escherichia coli), Klebsiella (such as Klebsiella pneumoniae, Klebsiella oxytoca), Haemophilus (such as Haemophilus influenzae and Parainfluenza), Bordetella genus (Such as Bordetella pertussis and Bacterial sepsis), Serratia (such as Serratia marcescens), Proteus (such as Proteus mirabilis), Enterobacter (such as Enterobacter cloaca), Campylobacter (such as Campylobacter jejuni), Citrobacter , Vibrio (Vibrio parahaemolyticus, Vibrio cholerae, etc.), Morganella (Morganella, Morgani, etc.), Salmonella (typhoid, Paratyphi, etc.), Shigella (Shigella, etc.), Acinetobacter (Acinetobacter baumani) Acinetobacter calcoaceticus), Legionella genus (Legionella pneumophila etc.), Bacteroides genus (Bacteroides fragilis etc.), Neisseria genus (gonococcus, meningococcus etc.), Moraxella genus (Moraxella catarrhalis etc.), Chlamydia genus (Chlamydia) -Trachomatis, Chlamydia scitasty, etc.) and Helicobacter genus (Helicobacter pylori, etc.). Examples of mycoplasmas include M. galli septicum, M. genitalium, M. hominis, M. hyopneumoniae, M. laboratorium, M. mycoides, M. ovipneumoniae, and M. pneumonia.
本発明の化合物は、特に従来のマクロライド系抗生物質では十分な抗菌活性が得られなかったエリスロマイシン耐性菌(例えば耐性肺炎球菌、耐性連鎖球菌、及びマイコプラズマ)などに対しても優れた抗菌活性を示すという特徴がある。 The compound of the present invention has excellent antibacterial activity against erythromycin-resistant bacteria (for example, resistant pneumococci, resistant streptococci, and mycoplasma) that have not been able to obtain sufficient antibacterial activity, particularly with conventional macrolide antibiotics. It has the feature of showing.
上記式[1]で表される化合物には光学異性体が存在しうるが、式[1]で表される化合物には、それら光学異性体、及び光学異性体の混合物が含まれる。また、式[1]で表される化合物、若しくはその薬学的に許容される塩、又はそれらの各種水和物、若しくは溶媒和物も本発明の範囲に含まれる。 The compound represented by the formula [1] may have optical isomers, but the compound represented by the formula [1] includes those optical isomers and a mixture of optical isomers. Further, the compound represented by the formula [1], or a pharmaceutically acceptable salt thereof, or various hydrates or solvates thereof are also included in the scope of the present invention.
本発明における「溶媒和物」の「溶媒」とは特に示さない限り、例えば極性溶媒(例えば、メタノール、エタノール、1−プロパノール、2−プロパノール、ブタノール等のアルコール系の溶媒、酢酸エチル等)、不活性溶媒(例えば、クロロホルム若しくは塩化メチレン等のハロゲン化炭化水素系溶媒、ジエチルエーテル、テトラヒドロフラン若しくはジオキサン等のエーテル系溶媒、ジメチルホルムアミド、ジメチルアセトアミド等のアミド系溶媒、ジメチルスルホキシド、アセトニトリル等の非プロトン性溶媒、トルエン等の芳香族炭化水素類、又はシクロヘキサン等の炭化水素類等)、更に2−ブタノン、ヘキサン、イソプロピルエーテル、アセトン、ジクロロメタン等、又はここに例示した溶媒の混合溶媒を意味するが、これらに限定されることはない。 Unless otherwise indicated, the “solvent” of the “solvate” in the present invention is, for example, a polar solvent (for example, alcohol solvents such as methanol, ethanol, 1-propanol, 2-propanol, butanol, ethyl acetate, etc.), Inert solvents (for example, halogenated hydrocarbon solvents such as chloroform or methylene chloride, ether solvents such as diethyl ether, tetrahydrofuran or dioxane, amide solvents such as dimethylformamide and dimethylacetamide, aprotic such as dimethyl sulfoxide and acetonitrile An organic hydrocarbon, aromatic hydrocarbons such as toluene, or hydrocarbons such as cyclohexane), 2-butanone, hexane, isopropyl ether, acetone, dichloromethane, or a mixed solvent of the solvents exemplified here. To these It will not be constant.
上記式[1]で表される本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は、優れた安全性を示す。安全性は、種々の試験によって評価されるが、たとえば、細胞毒性試験、hERG試験、シトクロムP450(CYP)活性阻害試験などで評価することができる。 The compound of the present invention represented by the formula [1] or a salt thereof, or a hydrate or solvate thereof exhibits excellent safety. The safety is evaluated by various tests, and can be evaluated by, for example, a cytotoxicity test, a hERG test, a cytochrome P450 (CYP) activity inhibition test, and the like.
上記式[1]で表される本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は、優れた代謝安定性を示す。代謝安定性は、種々の試験によって評価されるが、たとえば、ヒト肝ミクロソーム代謝安定性試験などで評価することができる。 The compound of the present invention represented by the above formula [1] or a salt thereof, or a hydrate or solvate thereof exhibits excellent metabolic stability. Metabolic stability is evaluated by various tests, and can be evaluated by, for example, a human liver microsomal metabolic stability test.
本発明の化合物は、一つ又は二つ以上の医薬的に許容される担体、賦形剤又は希釈剤と組み合せて医薬的製剤とすることができる。上記式[1]で表される本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は、一般的な医薬製剤として調製される。例えば、製剤上許容しうる担体(賦形剤、結合剤、崩壊剤、矯味剤、乳化剤、希釈剤、溶解補助剤など)と混合、溶解及び/又は分散して医薬組成物とする。この医薬組成物は、錠剤、丸剤、散剤、顆粒剤、カプセル剤、液剤、乳剤、懸濁剤、注射剤、座剤、吸入剤、経皮吸収剤などの製剤として経口または非経口に適した形態で投与される。 経口投与製剤には固形製剤と液状製剤がある。本発明における固形製剤とは、製剤の全体又は集合体を構成する各要素が少なくとも一定の形を有する形態の製剤である。具体的には、例えば錠剤、丸剤、カプセル剤、顆粒剤、散剤又は粉剤が挙げられる。本発明において、内容物が液体のカプセル剤は、全体もしくは複数のカプセルの集合体を構成する一つのカプセルが一定の形を有する場合は、固形製剤に含まれる。また、用時溶解又は懸濁して服用するドライシロップ剤も、保存時に製剤全体又は粉末もしくは顆粒の個々の粒子が一定の形を有する場合、固形製剤に含まれる。それに対して、本発明における液状製剤とは、保存時から投与時まで液体の溶媒又は分散媒に溶解又は分散され、一定の形を有しないため液体として取り扱われる形態の製剤をいう。これら製剤を製造するには賦形剤、希釈剤、結合剤、崩壊剤、滑沢剤、抗酸化剤、安定化剤、保存剤、溶剤、可溶化剤、等張化剤などを添加することができる。医薬的に許容される賦形剤又は希釈剤としては、例えば、乳糖、ショ糖、ブドウ糖、麦芽糖、果糖、マンニトール、キシリトール、ソルビトール、エリスリトール、デンプン、スターチ、カルボキシメチルスターチナトリウム、粉末セルロース、結晶セルロース、カルメロース、結晶セルロース・カルメロースナトリウム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、リン酸水素カルシウム、リン酸水素ナトリウム、リン酸水素カリウム、リン酸二水素カリウム、炭酸カルシウム、軽質無水ケイ酸、酸化チタン、メタケイ酸アルミン酸マグネシウムなどが挙げられる。結合剤としては、例えば、ヒドロキシプロプルセルロース、ヒプロメロース、デンプン、スターチ、アルファー化デンプン、部分アルファー化デンプン、ポリビニルピロリドンなどが挙げられる。崩壊剤としては、例えば、粉末セルロース、結晶セルロース、カルメロース、カルメロースカリウム、カルメロースカルシウム、カルメロースナトリウム、結晶セルロース・カルメロースナトリウム、クロスカルメロースナトリウム、低置換度ヒドロキシプロピルセルロース、デンプン、部分アルファー化デンプン、カルボキシメチルスターチナトリウム、ポビドン、クロスポビドンなどが挙げられる。滑沢剤としては、例えば、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸ポリオキシル、タルク、硬化油、ショ糖脂肪酸エステル、セタノール、ミツロウ、サラシミツロウなどが挙げられる。抗酸化剤としては、例えば、ジブチルヒドロキシトルエン(BHT)、没食子酸プロピル、ブチルヒドロキシアニソール(BHA)、トコフェロール、クエン酸、エデト酸塩などが挙げられる。溶剤としては、例えば水、生理食塩水、エタノールなどが、可溶化剤としては、例えば、ポリオキシエチレン硬化ヒマシ油、ポリソルベート類、ラウリル硫酸ナトリウム、マクロゴール類、ショ糖脂肪酸エステルなど、等張化剤可溶化剤としては、塩化ナトリウム、クエン酸、クエン酸ナトリウム、グリセリン、ソルビトール、ブドウ糖、プロピレングリコール、マクロゴール類、ホウ酸、ホウ砂、リン酸、リン酸水素塩類などが挙げられる。 The compounds of the present invention can be combined with one or more pharmaceutically acceptable carriers, excipients or diluents into a pharmaceutical formulation. The compound of the present invention represented by the above formula [1] or a salt thereof, or a hydrate or solvate thereof is prepared as a general pharmaceutical preparation. For example, a pharmaceutical composition is prepared by mixing, dissolving, and / or dispersing with a pharmaceutically acceptable carrier (excipient, binder, disintegrant, corrigent, emulsifier, diluent, solubilizer, etc.). This pharmaceutical composition is suitable for oral or parenteral preparations such as tablets, pills, powders, granules, capsules, solutions, emulsions, suspensions, injections, suppositories, inhalants, and transdermal absorption agents. Administered in the form. Oral preparations include solid preparations and liquid preparations. The solid preparation in the present invention is a preparation in a form in which each element constituting the whole preparation or aggregate has at least a certain shape. Specific examples include tablets, pills, capsules, granules, powders, and powders. In the present invention, a capsule whose content is a liquid is included in a solid preparation when one capsule constituting the whole or an aggregate of a plurality of capsules has a certain shape. A dry syrup that is dissolved or suspended at the time of use is also included in the solid preparation when the whole preparation or individual particles of powder or granules have a certain shape at the time of storage. On the other hand, the liquid preparation in the present invention refers to a preparation that is dissolved or dispersed in a liquid solvent or dispersion medium from the time of storage to the time of administration and is handled as a liquid because it does not have a certain shape. To produce these preparations, excipients, diluents, binders, disintegrants, lubricants, antioxidants, stabilizers, preservatives, solvents, solubilizers, tonicity agents, etc. should be added. Can do. Examples of the pharmaceutically acceptable excipient or diluent include lactose, sucrose, glucose, maltose, fructose, mannitol, xylitol, sorbitol, erythritol, starch, starch, sodium carboxymethyl starch, powdered cellulose, crystalline cellulose , Carmellose, crystalline cellulose / carmellose sodium, hydroxypropyl cellulose, hydroxypropyl methylcellulose, calcium hydrogen phosphate, sodium hydrogen phosphate, potassium hydrogen phosphate, potassium dihydrogen phosphate, calcium carbonate, light anhydrous silicic acid, titanium oxide, Examples include magnesium aluminate metasilicate. Examples of the binder include hydroxypropylcellulose, hypromellose, starch, starch, pregelatinized starch, partially pregelatinized starch, and polyvinylpyrrolidone. Examples of disintegrants include powdered cellulose, crystalline cellulose, carmellose, carmellose potassium, carmellose calcium, carmellose sodium, crystalline cellulose / carmellose sodium, croscarmellose sodium, low-substituted hydroxypropylcellulose, starch, and partial alpha. Modified starch, sodium carboxymethyl starch, povidone, crospovidone and the like. Examples of the lubricant include stearic acid, magnesium stearate, calcium stearate, polyoxyl stearate, talc, hydrogenated oil, sucrose fatty acid ester, cetanol, beeswax, and white beeswax. Examples of the antioxidant include dibutylhydroxytoluene (BHT), propyl gallate, butylhydroxyanisole (BHA), tocopherol, citric acid, edetate and the like. Solvents include, for example, water, physiological saline, ethanol, etc., and solubilizers include, for example, polyoxyethylene hydrogenated castor oil, polysorbates, sodium lauryl sulfate, macrogol, sucrose fatty acid esters, etc. Examples of the agent solubilizer include sodium chloride, citric acid, sodium citrate, glycerin, sorbitol, glucose, propylene glycol, macrogols, boric acid, borax, phosphoric acid, hydrogen phosphates and the like.
本発明の化合物の投与量は、動物実験の結果に基づき、単回および反復投与したときに、一定量を超えないように定められる。試験例に開示した動物実験のデータに基づけば、成人患者に対して1日の投与量として1〜10000mg、好ましくは5〜1000mgを1日1回又は数回に分けて経口又は非経口で投与することが想定される。さらに、適量と投与回数は、投与方法、年齢、体重、性別、感受性、患者または被処置動物の症状の程度など、種々の要素を勘案し、専門医等によって決定されうる。また、本発明の化合物は、他の薬剤との組み合わせで使用することも可能である。 The dose of the compound of the present invention is determined based on the results of animal experiments so that it does not exceed a certain amount when administered once and repeatedly. Based on the animal experiment data disclosed in the test examples, daily doses of 1 to 10000 mg, preferably 5 to 1000 mg, are administered to adult patients once or several times a day orally or parenterally. It is assumed that Furthermore, the appropriate amount and the number of administrations can be determined by a specialist or the like in consideration of various factors such as the administration method, age, weight, sex, sensitivity, and the degree of symptoms of the patient or treated animal. The compounds of the present invention can also be used in combination with other drugs.
以下に、参考例、実施例及び試験例により本発明をさらに詳細に説明する。本発明の化合物の合成法は以下の方法に限定されず、各工程の順序を入れ替える、官能基の保護・脱保護を経る、等の当業者に周知の方法を用いて合成することもできる。 Hereinafter, the present invention will be described in more detail with reference examples, examples and test examples. The method for synthesizing the compound of the present invention is not limited to the following method, and it can also be synthesized using methods well known to those skilled in the art, such as changing the order of each step, and protecting / deprotecting a functional group.
以下の参考例、実施例記載の各機器データは以下の測定機器で測定した。
NMRスペクトル:日本電子社JNM-ECA600(600MHz)、日本電子社JNM-ECA500(500MHz)
MSスペクトル:島津社LCMS−2010EVあるいはmicromass社 Platform LC
以下の参考例、実施例において、高速液体クロマトグラフィーマススペクトル(LCMS)は以下の条件により測定した。
測定機械:Agilent 2900およびAgilent 6150
カラム:Waters Acquity CSH C18,1.7μm,φ2.1x50mm
溶媒:A液;0.1%ギ酸含有水、B液;0.1%ギ酸含有アセトニトリル
(条件1)
グラジエント:0分(A液/B液=80/20)、1.2−1.4分(A液/B液=1/99)
流速:0.8mL/分、検出法:UV、ELSD
(条件2)
グラジエント:0分(A液/B液=95/5)、1.20分(A液/B液=50/50)、1.0mL/分、1.38分(A液/B液=3/97)
流速:0.8mL/分、検出法:UV、ELSD
イオン化法:ESI
参考例、実施例中の略号を以下に示す。
ESI:エレクトロスプレーイオン化法
MS:マススペクトル
CDCl3:重クロロホルム
NMR:核磁気共鳴
s:シングレット
br:幅広いピーク
d:ダブレット
m:マルチプレット
t:トリプレット
q:カルテット
Each instrument data described in the following reference examples and examples was measured with the following measuring instruments.
NMR spectrum: JEOL JNM-ECA600 (600 MHz), JEOL JNM-ECA500 (500 MHz)
MS spectrum: Shimadzu LCMS-2010EV or micromass Platform LC
In the following Reference Examples and Examples, high performance liquid chromatography mass spectrum (LCMS) was measured under the following conditions.
Measuring machine: Agilent 2900 and Agilent 6150
Column: Waters Acquity CSH C18, 1.7 μm, φ2.1 × 50 mm
Solvent: Solution A; 0.1% formic acid-containing water, Solution B: 0.1% formic acid-containing acetonitrile (Condition 1)
Gradient: 0 minutes (A liquid / B liquid = 80/20), 1.2-1.4 minutes (A liquid / B liquid = 1/99)
Flow rate: 0.8 mL / min, detection method: UV, ELSD
(Condition 2)
Gradient: 0 minutes (A liquid / B liquid = 95/5), 1.20 minutes (A liquid / B liquid = 50/50), 1.0 mL / min, 1.38 minutes (A liquid / B liquid = 3) / 97)
Flow rate: 0.8 mL / min, detection method: UV, ELSD
Ionization method: ESI
Abbreviations in reference examples and examples are shown below.
ESI: electrospray ionization MS: mass spectrum CDCl3: deuterated chloroform NMR: nuclear magnetic resonance s: singlet br: broad peak d: doublet m: multiplet t: triplet q: quartet
参考例1 N,N-ジイソプロピル-N-メチルエタン-1,2-ジアミンの合成
<スキームA>
Reference Example 1 Synthesis of N, N-diisopropyl-N-methylethane-1,2-diamine <Scheme A>
8.9mol/Lメチルアミンのメタノール溶液135mLに氷冷下、ジイソプロピルアミノエチルクロリド塩酸塩24.0gのメタノール72mL溶液を滴下し、室温にて20分間撹拌した。反応液を減圧濃縮して得た残渣をクロロホルムに溶解し、氷冷下2 mol/L水酸化ナトリウム水溶液を加えた。反応液をクロロホルムにて2回抽出し、有機層を減圧下濃縮した後に、得られた残渣をアミノシリカゲルカラムクロマトグラフィー(ヘキサン:クロロホルム=5:1からクロロホルムのみ)にて精製し、標記化合物19.4gを得た。
MS(ESI) m/z= 159 [M+H]+
1H-NMR (400 MHz, CDCl3) δ(ppm) : 0.99 (d, J=1.71 Hz, 6 H) 1.00 (d, J=1.71 Hz, 6 H) 2.43 (s, 3 H) 2.54 -2.57 (m, 4 H) 2.96 -3.03 (m, 2 H)
To 135 mL of a methanol solution of 8.9 mol / L methylamine, 72 mL of methanol in 24.0 g of diisopropylaminoethyl chloride hydrochloride was added dropwise under ice cooling, and the mixture was stirred at room temperature for 20 minutes. The residue obtained by concentrating the reaction solution under reduced pressure was dissolved in chloroform, and 2 mol / L aqueous sodium hydroxide solution was added under ice cooling. The reaction solution was extracted twice with chloroform and the organic layer was concentrated under reduced pressure. The resulting residue was purified by amino silica gel column chromatography (hexane: chloroform = 5: 1 to chloroform only) to give the title compound 19.4. g was obtained.
MS (ESI) m / z = 159 [M + H] +
1H-NMR (400 MHz, CDCl3) δ (ppm): 0.99 (d, J = 1.71 Hz, 6 H) 1.00 (d, J = 1.71 Hz, 6 H) 2.43 (s, 3 H) 2.54 -2.57 (m , 4 H) 2.96 -3.03 (m, 2 H)
参考例2 2-アミノ-N-エチルアセトアミドの合成
<スキームB>
Reference Example 2 Synthesis of 2-amino-N-ethylacetamide <Scheme B>
(1)N-(ベンジルオキシカルボニル)グリシン209gのクロロホルム1.0L溶液に 70%エチルアミン水溶液108mLを加え、氷冷下1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩249gを加えた後、室温にて終夜攪拌した。反応液に飽和重曹水を加え、クロロホルムにて抽出した。有機層を減圧下濃縮した後に、得られた残渣を酢酸エチル400mLに懸濁し、ヘキサン200mLを加え攪拌し、生じた個体をろ取しアミド体150gを得た。 (1) After adding 108 mL of 70% ethylamine aqueous solution to 1.0 L chloroform solution of 209 g N- (benzyloxycarbonyl) glycine and adding 249 g 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride under ice cooling And stirred at room temperature overnight. Saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, and the mixture was extracted with chloroform. After the organic layer was concentrated under reduced pressure, the obtained residue was suspended in 400 mL of ethyl acetate, 200 mL of hexane was added and stirred, and the resulting solid was collected by filtration to obtain 150 g of an amide compound.
<スキームC> <Scheme C>
(2)上記の参考例2−(1)にて得られたアミド体150gのメタノール630mL溶液に 10%パラジウム炭素 15gを加え、水素雰囲気下、室温にて6日間攪拌した。反応液をろ過した後、濾液を減圧下濃縮し、標記化合物64.4gを得た。
MS(ESI) m/z= 103 [M+H]+
1H-NMR (400 MHz, CDCl3) δ(ppm) : 1.17 (t, J=7.2 Hz, 3 H) 1.38 (brs, 2 H) 3.29 - 3.37 (m, 4 H) 7.20 (brs, 1 H)
(2) To a solution of 150 g of the amide obtained in Reference Example 2- (1) above in methanol (630 mL) was added 10% palladium carbon (15 g), and the mixture was stirred at room temperature for 6 days in a hydrogen atmosphere. After the reaction solution was filtered, the filtrate was concentrated under reduced pressure to obtain 64.4 g of the title compound.
MS (ESI) m / z = 103 [M + H] +
1H-NMR (400 MHz, CDCl3) δ (ppm): 1.17 (t, J = 7.2 Hz, 3 H) 1.38 (brs, 2 H) 3.29-3.37 (m, 4 H) 7.20 (brs, 1 H)
参考例3 式[2]で示される化合物の製造
式[2]:
Reference Example 3 Production of Compound represented by Formula [2] Formula [2]:
<スキームD> <Scheme D>
(1)クラリスロマイシン200gをアセトン1.5Lに溶解し、無水酢酸30.3mLを滴下して、室温にて終夜攪拌した。反応液を減圧濃縮して得られた残渣に酢酸エチル、ヘキサン、水酸化ナトリウム水溶液を加えた後、飽和重曹水を加えてpH=9に調整した。析出した固体をグラスフィルターにて濾取、蒸留水で洗浄した後、減圧下乾燥してアセチル体202gを得た。
MS(ESI) m/z= 790.6 [M+H]+
(1) 200 g of clarithromycin was dissolved in 1.5 L of acetone, 30.3 mL of acetic anhydride was added dropwise, and the mixture was stirred overnight at room temperature. The reaction mixture was concentrated under reduced pressure, ethyl acetate, hexane and sodium hydroxide aqueous solution were added to the resulting residue, and then saturated aqueous sodium hydrogen carbonate was added to adjust to pH = 9. The precipitated solid was collected by filtration with a glass filter, washed with distilled water, and then dried under reduced pressure to obtain 202 g of an acetyl compound.
MS (ESI) m / z = 790.6 [M + H] +
<スキームE> <Scheme E>
(2)上記の参考例3−(1)で得られたアセチル体202gをクロロホルム1.8Lに溶解し、ピリジン210mLを加えた後氷冷し、トリホスゲン77.4gのクロロホルム0.8L溶液を40分間かけて滴下した。反応液を室温まで昇温した後、3時間攪拌した。反応液にピリジン158mLを加えて、氷冷下、トリホスゲン57.9gのクロロホルム溶液を滴下して、室温にて15分間攪拌した。反応液に蒸留水、飽和重曹水を加えてクロロホルムにて抽出し、有機層を無水硫酸マグネシウムで乾燥して濾過した。濾液を減圧濃縮して得られた残渣に酢酸エチルとヘキサンの1:1混合溶媒を加えて攪拌し、更にヘキサンを加え室温にて終夜攪拌した。生じた固体を濾取し、酢酸エチルとヘキサンの1:2混合溶媒で洗浄した後、減圧下乾燥してカーボネート体220gを得た。
MS(ESI) m/z= 816.5 [M+H]+
(2) 202 g of the acetyl compound obtained in Reference Example 3- (1) above was dissolved in 1.8 L of chloroform, 210 mL of pyridine was added, and then ice-cooled. A solution of 77.4 g of triphosgene in 0.8 L of chloroform over 40 minutes It was dripped. The reaction mixture was warmed to room temperature and stirred for 3 hours. To the reaction solution, 158 mL of pyridine was added, and a chloroform solution of 57.9 g of triphosgene was added dropwise under ice cooling, followed by stirring at room temperature for 15 minutes. Distilled water and saturated aqueous sodium hydrogen carbonate were added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure, and a 1: 1 mixed solvent of ethyl acetate and hexane was added to the residue and stirred, and further hexane was added and stirred overnight at room temperature. The resulting solid was collected by filtration, washed with a 1: 2 mixed solvent of ethyl acetate and hexane, and then dried under reduced pressure to obtain 220 g of a carbonate body.
MS (ESI) m / z = 816.5 [M + H] +
<スキームF> <Scheme F>
(3)N-クロロコハク酸イミド99.7gをクロロホルム1Lに溶解し、-25℃に冷却した。反応液にジメチルスルフィド210mLのクロロホルム0.2L溶液を20分間かけて滴下して、15分間攪拌した後、上記(2)で得られたカーボネート体のクロロホルム1L溶液を30分間かけて滴下して、15分間攪拌した。反応液にトリエチルアミン136mLのクロロホルム0.2L溶液を加えて、30分間攪拌した。反応液に飽和重曹水を加えて室温まで昇温し、クロロホルムにて分液した。有機層を無水硫酸マグネシウムで乾燥して濾過した後、濾液を減圧濃縮して得られた残渣に酢酸エチルとヘキサンの1:5の混合溶媒を加え室温にて終夜攪拌した。生じた固体を濾取し、酢酸エチルとヘキサンの1:2混合溶媒で洗浄してケトン体109gを得た。濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1からアセトン:ヘキサン:トリエチルアミン = 10:10:0.2)にて精製した後、上記と同様の方法にて結晶化してケトン体59.5gを得た。
MS(ESI) m/z= 814.5 [M+H]+
(3) 99.7 g of N-chlorosuccinimide was dissolved in 1 L of chloroform and cooled to -25 ° C. To the reaction mixture, 210 mL of dimethylsulfide in chloroform (0.2 L) was added dropwise over 20 minutes, stirred for 15 minutes, and then the carbonate body obtained in (2) (1 L) in chloroform (1 L) was added dropwise over 30 minutes. Stir for minutes. To the reaction solution, a 0.2 mL solution of triethylamine 136 mL in chloroform was added and stirred for 30 minutes. Saturated aqueous sodium bicarbonate was added to the reaction mixture, the temperature was raised to room temperature, and the mixture was separated with chloroform. The organic layer was dried over anhydrous magnesium sulfate and filtered, and then the filtrate was concentrated under reduced pressure. To the resulting residue was added a 1: 5 mixed solvent of ethyl acetate and hexane, and the mixture was stirred at room temperature overnight. The resulting solid was collected by filtration and washed with a 1: 2 mixed solvent of ethyl acetate and hexane to obtain 109 g of a ketone body. The residue obtained by concentrating the filtrate under reduced pressure was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 1 to acetone: hexane: triethylamine = 10: 10: 0.2), and then the same method as above. Crystallization gave 59.5 g of ketone body.
MS (ESI) m / z = 814.5 [M + H] +
<スキームG> <Scheme G>
(4)トリメチルスルホキソニウムヨージド210g をジメチルスルホキシドとテトラヒドロフランの5:1混合溶媒1.2Lに溶解し、70%水素化ナトリウム32.6gを少量ずつ加えて、室温にて1.5時間攪拌した。氷冷下、上記(3)で得られたケトン体155gのテトラヒドロフラン0.8L溶液を滴下して、室温にて30分間攪拌した。反応液を氷冷し、蒸留水を加え、酢酸エチルを加えて分液し、得られた有機層を蒸留水で洗浄した。水層を酢酸エチルにて抽出し、有機層を蒸留水で洗浄した。集めた有機層を無水硫酸マグネシウムで乾燥して濾過した。濾液を減圧濃縮してエポキシ体146gを得た。
MS(ESI) m/z= 784.5 [M+H]+
1H-NMR (600 MHz, CDCl3) δ(ppm) : 0.90 (t, J=7.57 Hz, 3 H) 0.97 (d, J=7.34 Hz, 3 H) 1.04 (d, J=6.88 Hz, 3 H) 1.07 (s, 3 H) 1.14 (d, J=6.88 Hz, 3 H) 1.18 (d, J=5.96 Hz, 3 H) 1.21 - 1.36 (m, 7 H) 1.42 (s, 3 H) 1.47 - 1.55 (m, 1 H) 1.67 - 1.73 (m, 1 H) 1.83 - 1.98 (m, 5 H) 2.02 (d, J=1.83 Hz, 6 H) 2.18 - 2.29 (m, 1 H) 2.25 (s, 6 H) 2.58 - 2.69 (m, 1 H) 2.63 (d, J=4.13 Hz, 1 H) 2.80 - 2.89 (m, 1 H) 2.94 (d, J=4.13 Hz, 1 H) 3.12 - 3.26 (m, 1 H) 3.17 (s, 3 H) 3.34 (s, 3 H) 3.43 - 3.51 (m, 1 H) 3.66 (d, J=6.42 Hz, 1 H) 3.94 (brs, 1 H) 4.57 (d, J=7.34 Hz, 1 H) 4.73 (dd, J=10.55, 7.34 Hz, 1 H) 4.80 (q, J=6.42 Hz, 1 H) 4.98 - 5.06 (m, 2 H) 6.50 (s, 1 H)
(4) 210 g of trimethylsulfoxonium iodide was dissolved in 1.2 L of a 5: 1 mixed solvent of dimethylsulfoxide and tetrahydrofuran, 32.6 g of 70% sodium hydride was added little by little, and the mixture was stirred at room temperature for 1.5 hours. Under ice cooling, a solution of 155 g of the ketone obtained in (3) above in 0.8 L in tetrahydrofuran was added dropwise and stirred at room temperature for 30 minutes. The reaction solution was ice-cooled, distilled water was added, ethyl acetate was added for liquid separation, and the obtained organic layer was washed with distilled water. The aqueous layer was extracted with ethyl acetate, and the organic layer was washed with distilled water. The collected organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to obtain 146 g of an epoxy compound.
MS (ESI) m / z = 784.5 [M + H] +
1H-NMR (600 MHz, CDCl3) δ (ppm): 0.90 (t, J = 7.57 Hz, 3 H) 0.97 (d, J = 7.34 Hz, 3 H) 1.04 (d, J = 6.88 Hz, 3 H) 1.07 (s, 3 H) 1.14 (d, J = 6.88 Hz, 3 H) 1.18 (d, J = 5.96 Hz, 3 H) 1.21-1.36 (m, 7 H) 1.42 (s, 3 H) 1.47-1.55 (m, 1 H) 1.67-1.73 (m, 1 H) 1.83-1.98 (m, 5 H) 2.02 (d, J = 1.83 Hz, 6 H) 2.18-2.29 (m, 1 H) 2.25 (s, 6 H) 2.58-2.69 (m, 1 H) 2.63 (d, J = 4.13 Hz, 1 H) 2.80-2.89 (m, 1 H) 2.94 (d, J = 4.13 Hz, 1 H) 3.12-3.26 (m, 1 H) 3.17 (s, 3 H) 3.34 (s, 3 H) 3.43-3.51 (m, 1 H) 3.66 (d, J = 6.42 Hz, 1 H) 3.94 (brs, 1 H) 4.57 (d, J = 7.34 Hz, 1 H) 4.73 (dd, J = 10.55, 7.34 Hz, 1 H) 4.80 (q, J = 6.42 Hz, 1 H) 4.98-5.06 (m, 2 H) 6.50 (s, 1 H)
<スキームH> <Scheme H>
(5)上記参考例3−(4)で得られたエポキシ体138gをテトラヒドロフランとジメチルホルムアミドの1:1混合溶媒1.4Lに溶解し、1,1’-カルボニルジイミダゾール85.6gを加えた。氷冷下、70%水素化ナトリウム18.1gを40分間かけて加えて、室温にて0.5時間攪拌した。反応液を氷冷し、蒸留水を加え、酢酸エチルにて抽出し、有機層を蒸留水で2回洗浄した。水層を酢酸エチルにて抽出し、有機層を蒸留水で2回洗浄した。集めた有機層を無水硫酸マグネシウムで乾燥して濾過した。濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサンからヘキサン:酢酸エチル=1:1からアセトン:ヘキサン:トリエチルアミン=10:10:0.2) にて精製した。得られた精製物に酢酸エチル、ヘキサン(1:1)を加えて、室温にて終夜攪拌した。生じた固体を濾取し、酢酸エチルとヘキサンの1:4混合溶媒にて洗浄し、式[2]で示される化合物87.1gを得た。
MS(ESI) m/z= 878.6 [M+H]+
1H-NMR (600 MHz, CDCl3) δ(ppm) : 0.85 - 1.41 (m, 25 H) 1.64 - 1.78 (m, 3 H) 1.79 (s, 3 H) 1.90 (dd, J=14.67, 5.04 Hz, 4 H) 1.86 (s, 3 H) 2.04 (s, 3 H) 2.19 - 2.28 (m, 1 H) 2.25 (s, 6 H) 2.60 - 2.68 (m, 1 H) 2.65 (d, J=4.13 Hz, 1 H) 2.86 - 2.97 (m, 1 H) 2.95 (d, J=4.13 Hz, 1 H) 3.15 (s, 3 H) 3.22 - 3.29 (m, 1 H) 3.35 (s, 3 H) 3.38 - 3.47 (m, 1 H) 3.66 (d, J=6.42 Hz, 1 H) 3.79 - 3.88 (m, 1 H) 4.56 (d, J=6.88 Hz, 1 H) 4.72 (dd, J=10.32, 7.57 Hz, 1 H) 4.79 (q, J=6.27 Hz, 1 H) 5.01 - 5.09 (m, 1 H) 5.83 (dd, J=10.55, 2.75 Hz, 1 H) 6.66 (s, 1 H) 7.07 (s, 1 H) 7.34 - 7.38 (m, 1 H) 8.08 (s, 1 H)
(5) 138 g of the epoxy compound obtained in Reference Example 3- (4) above was dissolved in 1.4 L of a 1: 1 mixed solvent of tetrahydrofuran and dimethylformamide, and 85.6 g of 1,1′-carbonyldiimidazole was added. Under ice cooling, 18.1 g of 70% sodium hydride was added over 40 minutes, and the mixture was stirred at room temperature for 0.5 hours. The reaction solution was ice-cooled, distilled water was added, extraction was performed with ethyl acetate, and the organic layer was washed twice with distilled water. The aqueous layer was extracted with ethyl acetate, and the organic layer was washed twice with distilled water. The collected organic layer was dried over anhydrous magnesium sulfate and filtered. The residue obtained by concentrating the filtrate under reduced pressure was purified by silica gel column chromatography (hexane to hexane: ethyl acetate = 1: 1 to acetone: hexane: triethylamine = 10: 10: 0.2). Ethyl acetate and hexane (1: 1) were added to the purified product, and the mixture was stirred overnight at room temperature. The resulting solid was collected by filtration and washed with a 1: 4 mixed solvent of ethyl acetate and hexane to obtain 87.1 g of a compound represented by the formula [2].
MS (ESI) m / z = 878.6 [M + H] +
1H-NMR (600 MHz, CDCl3) δ (ppm): 0.85-1.41 (m, 25 H) 1.64-1.78 (m, 3 H) 1.79 (s, 3 H) 1.90 (dd, J = 14.67, 5.04 Hz, 4 H) 1.86 (s, 3 H) 2.04 (s, 3 H) 2.19-2.28 (m, 1 H) 2.25 (s, 6 H) 2.60-2.68 (m, 1 H) 2.65 (d, J = 4.13 Hz , 1 H) 2.86-2.97 (m, 1 H) 2.95 (d, J = 4.13 Hz, 1 H) 3.15 (s, 3 H) 3.22-3.29 (m, 1 H) 3.35 (s, 3 H) 3.38- 3.47 (m, 1 H) 3.66 (d, J = 6.42 Hz, 1 H) 3.79-3.88 (m, 1 H) 4.56 (d, J = 6.88 Hz, 1 H) 4.72 (dd, J = 10.32, 7.57 Hz , 1 H) 4.79 (q, J = 6.27 Hz, 1 H) 5.01-5.09 (m, 1 H) 5.83 (dd, J = 10.55, 2.75 Hz, 1 H) 6.66 (s, 1 H) 7.07 (s, 1 H) 7.34-7.38 (m, 1 H) 8.08 (s, 1 H)
参考例4 式[3]で示される化合物の製造
式[3]:
Reference Example 4 Production of Compound Shown by Formula [3] Formula [3]:
<スキームI> <Scheme I>
(1)参考例3で得られた式[2]で示される化合物360mgをアセトニトリル1.5mLに溶解し、1,8-ジアザビシクロ[5,4,0]-7-ウンデセン280μl、3-メタンスルホニルプロピルアミン塩酸塩273mgを加えて、室温にて1日間攪拌した。反応液に酢酸エチル、飽和塩化アンモニウム水溶液を加えて分液した。有機層を無水硫酸マグネシウムで乾燥して濾過し、濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルムからクロロホルム:メタノール:28%アンモニア水=25:1:0.1から15:1:0.1)にて精製してカーバメート体117mgを得た。 (1) 360 mg of the compound represented by the formula [2] obtained in Reference Example 3 was dissolved in 1.5 mL of acetonitrile, 280 μl of 1,8-diazabicyclo [5,4,0] -7-undecene, 3-methanesulfonylpropyl Amine hydrochloride 273 mg was added and stirred at room temperature for 1 day. Ethyl acetate and a saturated aqueous solution of ammonium chloride were added to the reaction solution for liquid separation. The organic layer was dried over anhydrous magnesium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography (chloroform to chloroform: methanol: 28% aqueous ammonia = 25: 1: 0.1 to 15: 1: 0.1) to obtain 117 mg of carbamate.
<スキームJ> <Scheme J>
(2)上記の参考例4−(1)で得られたカーバメート体115mgをエタノール1mLに溶解し、N,N-ジエチル-N'-メチルエタン-1,2-ジアミン195μlを加えて、封管中100℃にて1日間攪拌した。反応液に酢酸エチル、飽和塩化アンモニウム水溶液を加えて分液した。有機層を無水硫酸マグネシウムで乾燥して濾過し、濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルムからクロロホルム:メタノール:28%アンモニア水=12:1:0.1)、分取用薄層クロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=20:1:0.1)にて精製し、式[3]で示される化合物62.7mgを得た。 (2) 115 mg of the carbamate obtained in the above Reference Example 4- (1) is dissolved in 1 mL of ethanol, and 195 μl of N, N-diethyl-N′-methylethane-1,2-diamine is added to the sealed tube. Stir at 100 ° C. for 1 day. Ethyl acetate and a saturated aqueous solution of ammonium chloride were added to the reaction solution for liquid separation. The organic layer was dried over anhydrous magnesium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography (chloroform to chloroform: methanol: 28% aqueous ammonia = 12: 1: 0.1) for preparative use. Purification by thin layer chromatography (chloroform: methanol: 28% aqueous ammonia = 20: 1: 0.1) gave 62.7 mg of the compound represented by the formula [3].
なお、式[3]で示される化合物は、特許文献7、8に好ましい化合物として記載された実施例15である。
The compound represented by the formula [3] is Example 15 described as a preferable compound in
実施例1 式[1]で示される化合物の製造
式[1]:
Example 1 Production of Compound of Formula [1] Formula [1]:
<スキームK> <Scheme K>
(1)参考例3で得られた式[2]で示される化合物277gをアセトニトリル315mLに溶解 し、参考例2で得られた化合物64.4gおよび1,8-ジアザビシクロ[5,4,0]-7-ウンデセン 191mLを加え、室温にて1.5時間攪拌した。反応液に水500mLを加え、酢酸エチル400mLにて抽出した。有機層を飽和食塩水で洗浄、硫酸マグネシウムにて乾燥濾過後、減圧下濃縮した。残渣を酢酸エチル 300mLとヘキサン 300mLより再結晶し、カーバメート体 83.5gを得た。濾液を減圧下濃縮後、再結晶(酢酸エチル 200mL, ヘキサン 200mL)することでカーバメート体34.4gを得た。さらに濾液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5)で精製した後に、酢酸エチル 100mLとヘキサン 100mLより再結晶し、カーバメート体16.3gを得た。濾液と前記カラムで得られた一部のフラクションを併せて、アミノシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=20:80 から 酢酸エチルのみ)で精製した後に、酢酸エチル 200mLとヘキサン 200mLより再結晶することでカーバメート体55.3gを得た。これらカーバメート体を合わせて189.5g得た。
MS(ESI) m/z= 912.6 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.85 - 0.90 (m, 6 H) 0.95 (d, J=7.55 Hz, 3 H) 0.99 - 1.29 (m, 19 H) 1.33 (s, 3 H) 1.44 (s, 3 H) 1.50 - 1.65 (m, 3 H) 1.68 - 1.73 (m, 1 H) 1.84 - 2.00 (m, 3 H) 2.05 (s, 3 H) 2.21 (dd, J=14.75, 3.09 Hz, 1 H) 2.26 (s, 6 H) 2.53 - 2.60 (m, 1 H) 2.62 (d, J=4.12 Hz, 1 H) 2.63 - 2.70 (m, 1 H) 2.86 - 2.91 (m, 1 H) 2.93 (s, 3 H) 2.94 (d, J=4.12 Hz, 1 H) 3.06 (q, J=6.86 Hz, 1 H) 3.27 - 3.36 (m, 2 H) 3.34 (s, 3 H) 3.41 - 3.50 (m, 1 H) 3.68 (d, J=6.17 Hz, 1 H) 3.73 (d, J=10.29 Hz, 1 H) 3.76 (s, 1 H) 4.20 (d, J=16.81 Hz, 1 H) 4.49 (d, J=16.81 Hz, 1 H) 4.63 (d, J=7.55 Hz, 1 H) 4.68 - 4.77 (m, 2 H) 5.04 (dd, J=4.63, 3.26 Hz, 1 H) 5.25 (dd, J=10.63, 2.40 Hz, 1 H) 6.17 (t, J=5.66 Hz, 1 H)
(1) 277 g of the compound represented by the formula [2] obtained in Reference Example 3 was dissolved in 315 mL of acetonitrile, and 64.4 g of the compound obtained in Reference Example 2 and 1,8-diazabicyclo [5,4,0]- 191 mL of 7-undecene was added and stirred at room temperature for 1.5 hours. 500 mL of water was added to the reaction solution, and the mixture was extracted with 400 mL of ethyl acetate. The organic layer was washed with saturated brine, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was recrystallized from 300 mL of ethyl acetate and 300 mL of hexane to obtain 83.5 g of carbamate. The filtrate was concentrated under reduced pressure and then recrystallized (ethyl acetate 200 mL, hexane 200 mL) to obtain 34.4 g of a carbamate. The filtrate was further concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (chloroform: methanol: 28% aqueous ammonia = 99: 1: 0.1 to 85: 15: 1.5), and then recrystallized from 100 mL of ethyl acetate and 100 mL of hexane. As a result, 16.3 g of a carbamate body was obtained. The filtrate and some fractions obtained from the above column were combined and purified by amino silica gel column chromatography (ethyl acetate: hexane = 20: 80 to ethyl acetate only), then recrystallized from 200 mL of ethyl acetate and 200 mL of hexane As a result, 55.3 g of a carbamate body was obtained. A total of 189.5 g of these carbamate bodies was obtained.
MS (ESI) m / z = 912.6 [M + H] +
1H-NMR (499 MHz, CDCl3) δ (ppm): 0.85-0.90 (m, 6 H) 0.95 (d, J = 7.55 Hz, 3 H) 0.99-1.29 (m, 19 H) 1.33 (s, 3 H ) 1.44 (s, 3 H) 1.50-1.65 (m, 3 H) 1.68-1.73 (m, 1 H) 1.84-2.00 (m, 3 H) 2.05 (s, 3 H) 2.21 (dd, J = 14.75, 3.09 Hz, 1 H) 2.26 (s, 6 H) 2.53-2.60 (m, 1 H) 2.62 (d, J = 4.12 Hz, 1 H) 2.63-2.70 (m, 1 H) 2.86-2.91 (m, 1 H) 2.93 (s, 3 H) 2.94 (d, J = 4.12 Hz, 1 H) 3.06 (q, J = 6.86 Hz, 1 H) 3.27-3.36 (m, 2 H) 3.34 (s, 3 H) 3.41 -3.50 (m, 1 H) 3.68 (d, J = 6.17 Hz, 1 H) 3.73 (d, J = 10.29 Hz, 1 H) 3.76 (s, 1 H) 4.20 (d, J = 16.81 Hz, 1 H ) 4.49 (d, J = 16.81 Hz, 1 H) 4.63 (d, J = 7.55 Hz, 1 H) 4.68-4.77 (m, 2 H) 5.04 (dd, J = 4.63, 3.26 Hz, 1 H) 5.25 ( dd, J = 10.63, 2.40 Hz, 1 H) 6.17 (t, J = 5.66 Hz, 1 H)
<スキームL> <Scheme L>
(2)上記の実施例1−(1)で得られたカーバメート体189gをメタノール 410mLに溶解し、4時間加熱還流した後、室温にて一昼夜攪拌した。反応液を減圧下濃縮した。残渣に酢酸エチル50mLおよびヘキサン300mLを加え、30分間攪拌し、生じた固体を濾取し、脱アセチル体41.2gを得た。濾液をアミノシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=20:80 から 100:0)およびシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5)で3回精製した。得られた粗精製物に酢酸エチル50mLおよびヘキサン 600mLを加え、30分間攪拌し、生じた固体を濾取し、脱アセチル体62.8gを得た。濾液をさらにシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5) で精製し、同様に酢酸エチル 20mLとヘキサン 50mLより再結晶し、脱アセチル体2.99gを得た。
MS(ESI) m/z= 870.6 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.88 (t, J=7.38 Hz, 3 H) 1.00 - 1.08 (m, 9 H) 1.09 - 1.27 (m, 1 H) 1.10 - 1.15 (m, 9 H) 1.18 (d, J=6.17 Hz, 3 H) 1.24 (d, J=7.20 Hz, 3 H) 1.36 (s, 3 H) 1.43 (s, 3 H) 1.58 (ddd, J=14.24, 10.46, 7.20 Hz, 1 H) 1.62 - 1.78 (m, 3 H) 1.88 (dd, J=14.92, 4.97 Hz, 1 H) 1.91 - 2.00 (m, 2 H) 2.23 (dd, J=14.75, 2.74 Hz, 1 H) 2.28 (s, 6 H) 2.42 - 2.50 (m, 1 H) 2.59 (dd, J=7.03, 4.29 Hz, 1 H) 2.62 (d, J=4.12 Hz, 1 H) 2.89 - 2.96 (m, 1 H) 2.93 (d, J=4.12 Hz, 1 H) 2.95 (s, 3 H) 3.08 (q, J=6.86 Hz, 1 H) 3.18 (dd, J=10.29, 7.20 Hz, 1 H) 3.27 - 3.39 (m, 2 H) 3.32 (s, 3 H) 3.42 - 3.50 (m, 1 H) 3.71 (d, J=6.52 Hz, 1 H) 3.76 (d, J=9.95 Hz, 1 H) 3.77 (s, 1 H) 4.21 (d, J=16.81 Hz, 1 H) 4.50 (d, J=10.63 Hz, 1 H) 4.52 (s, 1 H) 4.76 (q, J=6.52 Hz, 1 H) 5.04 (dd, J=4.80, 2.74 Hz, 1 H) 5.21 (dd, J=10.63, 2.40 Hz, 1 H) 6.25 (t, J=5.66 Hz, 1 H)
(2) 189 g of the carbamate obtained in Example 1- (1) above was dissolved in 410 mL of methanol, heated to reflux for 4 hours, and stirred overnight at room temperature. The reaction solution was concentrated under reduced pressure. 50 mL of ethyl acetate and 300 mL of hexane were added to the residue, and the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain 41.2 g of a deacetylated product. The filtrate was analyzed by amino silica gel column chromatography (ethyl acetate: hexane = 20: 80 to 100: 0) and silica gel column chromatography (chloroform: methanol: 28% aqueous ammonia = 99: 1: 0.1 to 85: 15: 1.5). It was purified once. 50 mL of ethyl acetate and 600 mL of hexane were added to the resulting crude product, and the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain 62.8 g of a deacetylated product. The filtrate was further purified by silica gel column chromatography (chloroform: methanol: 28% aqueous ammonia = 99: 1: 0.1 to 85: 15: 1.5) and recrystallized from 20 mL of ethyl acetate and 50 mL of hexane in the same manner to give the deacetylated compound 2.99. g was obtained.
MS (ESI) m / z = 870.6 [M + H] +
1H-NMR (499 MHz, CDCl3) δ (ppm): 0.88 (t, J = 7.38 Hz, 3 H) 1.00-1.08 (m, 9 H) 1.09-1.27 (m, 1 H) 1.10-1.15 (m, 9 H) 1.18 (d, J = 6.17 Hz, 3 H) 1.24 (d, J = 7.20 Hz, 3 H) 1.36 (s, 3 H) 1.43 (s, 3 H) 1.58 (ddd, J = 14.24, 10.46 , 7.20 Hz, 1 H) 1.62-1.78 (m, 3 H) 1.88 (dd, J = 14.92, 4.97 Hz, 1 H) 1.91-2.00 (m, 2 H) 2.23 (dd, J = 14.75, 2.74 Hz, 1 H) 2.28 (s, 6 H) 2.42-2.50 (m, 1 H) 2.59 (dd, J = 7.03, 4.29 Hz, 1 H) 2.62 (d, J = 4.12 Hz, 1 H) 2.89-2.96 (m , 1 H) 2.93 (d, J = 4.12 Hz, 1 H) 2.95 (s, 3 H) 3.08 (q, J = 6.86 Hz, 1 H) 3.18 (dd, J = 10.29, 7.20 Hz, 1 H) 3.27 -3.39 (m, 2 H) 3.32 (s, 3 H) 3.42-3.50 (m, 1 H) 3.71 (d, J = 6.52 Hz, 1 H) 3.76 (d, J = 9.95 Hz, 1 H) 3.77 ( s, 1 H) 4.21 (d, J = 16.81 Hz, 1 H) 4.50 (d, J = 10.63 Hz, 1 H) 4.52 (s, 1 H) 4.76 (q, J = 6.52 Hz, 1 H) 5.04 ( dd, J = 4.80, 2.74 Hz, 1 H) 5.21 (dd, J = 10.63, 2.40 Hz, 1 H) 6.25 (t, J = 5.66 Hz, 1 H)
<スキームM> <Scheme M>
(3)上記の実施例1−(2)で得られた脱アセチル体104gをエタノール 120mLに溶解し、参考例1で得られた化合物56.5gを加え、2時間加熱還流した。反応液を減圧下濃縮した。残渣を酢酸エチルに溶解させ、飽和炭酸水素ナトリウム水溶液にて3回洗浄した後、水を加え分液した。水層を酢酸エチルで再度抽出し、水で洗浄した。あわせた有機層を飽和食塩水で洗浄、硫酸マグネシウムにて乾燥ろ過後、減圧下濃縮した。残渣を酢酸エチル 100mLとヘキサン 600mLより再結晶し、式[1]で示される化合物41.4gを得た。更に、濾液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5)で精製した後に酢酸エチル 100mLと ヘキサン 500mLより再結晶し、式[1]で示される化合物62.1gを得た。このようにして得られた式[1]で示される化合物を合せ、合計で103.5gを得た。
MS(ESI) m/z= 1028.8 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.87 (t, J=7.20 Hz, 3 H) 1.00 (m, J=10.60, 6.50 Hz, 15 H) 1.06 - 1.26 (m, 22 H) 1.38 (s, 3 H) 1.42 (s, 3 H) 1.52 - 1.79 (m, 4 H) 1.84 - 2.07 (m, 5 H) 2.29 (s, 6 H) 2.35 (s, 3 H) 2.39 - 2.55 (m, 5 H) 2.57 - 2.64 (m, 1 H) 2.83 (d, J=14.75 Hz, 1 H) 2.89 (dd, J=9.26, 7.20 Hz, 1 H) 2.94 (s, 3 H) 2.95-3.03 (m, 2 H) 3.08 (q, J=7.09 Hz, 1 H) 3.17 (dd, J=10.12, 7.38 Hz, 1 H) 3.22 - 3.32 (m, 1 H) 3.28 (s, 3 H) 3.34 - 3.48 (m, 3 H) 3.64 (d, J=7.55 Hz, 1 H) 3.73 (d, J=9.61 Hz, 1 H) 3.78 (s, 1 H) 4.08 (q, J=6.40 Hz, 1 H) 4.21 (d, J=17.15 Hz, 1 H) 4.40 (d, J=7.20 Hz, 1 H) 4.57 (d, J=16.81 Hz, 1 H) 4.95 (brs, 1 H) 4.99 (d, J=4.80 Hz, 1 H) 5.11 (dd, J=10.63, 2.06 Hz, 1 H) 6.39 (t, J=5.66 Hz, 1 H)
本発明化合物の作用は以下の薬理試験により確認された。
(3) 104 g of the deacetylated product obtained in Example 1- (2) above was dissolved in 120 mL of ethanol, 56.5 g of the compound obtained in Reference Example 1 was added, and the mixture was heated to reflux for 2 hours. The reaction solution was concentrated under reduced pressure. The residue was dissolved in ethyl acetate and washed three times with a saturated aqueous sodium hydrogen carbonate solution, and water was added to separate the layers. The aqueous layer was extracted again with ethyl acetate and washed with water. The combined organic layers were washed with saturated brine, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was recrystallized from 100 mL of ethyl acetate and 600 mL of hexane to obtain 41.4 g of a compound represented by the formula [1]. The filtrate was further concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (chloroform: methanol: 28% aqueous ammonia = 99: 1: 0.1 to 85: 15: 1.5), and then recrystallized from 100 mL of ethyl acetate and 500 mL of hexane. As a result, 62.1 g of a compound represented by the formula [1] was obtained. The compounds represented by the formula [1] thus obtained were combined to obtain 103.5 g in total.
MS (ESI) m / z = 1028.8 [M + H] +
1H-NMR (499 MHz, CDCl3) δ (ppm): 0.87 (t, J = 7.20 Hz, 3 H) 1.00 (m, J = 10.60, 6.50 Hz, 15 H) 1.06-1.26 (m, 22 H) 1.38 (s, 3 H) 1.42 (s, 3 H) 1.52-1.79 (m, 4 H) 1.84-2.07 (m, 5 H) 2.29 (s, 6 H) 2.35 (s, 3 H) 2.39-2.55 (m , 5 H) 2.57-2.64 (m, 1 H) 2.83 (d, J = 14.75 Hz, 1 H) 2.89 (dd, J = 9.26, 7.20 Hz, 1 H) 2.94 (s, 3 H) 2.95-3.03 ( m, 2 H) 3.08 (q, J = 7.09 Hz, 1 H) 3.17 (dd, J = 10.12, 7.38 Hz, 1 H) 3.22-3.32 (m, 1 H) 3.28 (s, 3 H) 3.34-3.48 (m, 3 H) 3.64 (d, J = 7.55 Hz, 1 H) 3.73 (d, J = 9.61 Hz, 1 H) 3.78 (s, 1 H) 4.08 (q, J = 6.40 Hz, 1 H) 4.21 (d, J = 17.15 Hz, 1 H) 4.40 (d, J = 7.20 Hz, 1 H) 4.57 (d, J = 16.81 Hz, 1 H) 4.95 (brs, 1 H) 4.99 (d, J = 4.80 Hz , 1 H) 5.11 (dd, J = 10.63, 2.06 Hz, 1 H) 6.39 (t, J = 5.66 Hz, 1 H)
The action of the compound of the present invention was confirmed by the following pharmacological test.
試験例1 インビトロ抗菌活性
本発明品、実施例1の式[1]で表される化合物の各種試験菌に対するインビトロ抗菌力は、微量液体希釈法(CLSI法)に準じて測定した。また、参考例4の式[3]で表される化合物も同様に測定した。使用した試験菌を表1に示した。菌体番号A、B、C、D、E、F、G、H、I、J、K及びLの試験菌に対するMIC値(微生物生育最小阻止濃度μg/ml)を表2に示した。
Test Example 1 In Vitro Antibacterial Activity The in vitro antibacterial activity of the product of the present invention, the compound represented by the formula [1] of Example 1 against various test bacteria was measured according to a micro liquid dilution method (CLSI method). The compound represented by the formula [3] in Reference Example 4 was also measured in the same manner. The test bacteria used are shown in Table 1. Table 2 shows the MIC values (microbe growth minimum inhibitory concentration μg / ml) for the test bacteria having the cell numbers A, B, C, D, E, F, G, H, I, J, K, and L.
試験例2 インフルエンザ菌感受性試験
インフルエンザ菌(Haemophilus influenzae)39種類の臨床分離株を用い、試験例1と同様の手法を用いて、薬剤感受性について評価を実施した。表3に結果を示した。
Test Example 2 Haemophilus susceptibility test Using 39 kinds of clinical isolates of Haemophilus influenzae, the drug sensitivity was evaluated using the same method as Test Example 1. Table 3 shows the results.
試験例3 インフルエンザ菌感染動物における治療効果試験
薬理効果の評価は下記に示す方法を用いた。
細菌として、Haemophilus influenzae ATCC43095株(菌体番号A)を用いた。チョコレート寒天培地で1晩培養した菌体を掻き取り、ヘモフィルス感受性試験培地またはフィルズエンリッチメント添加ブレインハートインフュージョン培地に懸濁後、1晩培養した。これをヘモフィルス感受性試験培地またはフィルズエンリッチメント添加ブレインハートインフュージョン培地で希釈し、接種菌液とした。マウス(ICR系、雄性、4週齢)に接種菌液0.05mLを気道内接種して感染させた。接種菌量は2.25x10^6CFU/マウスまたは9.00x10^5CFU/マウスであった。接種翌日から1日1回2日間、実施例1の式[1]で表される化合物(100および200mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。接種3日後の肺内生菌数(1群6例、平均値±標準誤差)を図1に示した。
Test Example 3 Therapeutic effect test in H. influenzae-infected animals The method shown below was used for evaluation of the pharmacological effect.
As a bacterium, Haemophilus influenzae ATCC43095 strain (cell number A) was used. The cells cultured overnight on a chocolate agar medium were scraped off, suspended in a hemophilus sensitivity test medium or a brain heart infusion medium supplemented with Phils enrichment and cultured overnight. This was diluted with a hemophilus sensitivity test culture medium or a brain heart infusion medium supplemented with Phils enrichment to obtain an inoculum. Mice (ICR line, male, 4 weeks old) were infected by inoculating 0.05 ml of the inoculum in the respiratory tract. The amount of inoculum was 2.25 × 10 6 CFU / mouse or 9.00 × 10 6 CFU / mouse. The compound represented by the formula [1] of Example 1 (100 and 200 mg / kg) or vehicle (0.1 mol / L lactobionic acid solution and 0.5 w / v% hydrogen carbonate) once a day for 2 days from the day after the inoculation An equal volume of sodium solution) was orally administered. The number of viable bacteria in the
なお、図1についての備考は以下の通りである。媒体との有意差:Steel検定 *:p<0.05、**:p<0.01 実施例1、式[1]の化合物MIC値は4μg/mL、参考例4、式[3]の化合物MIC値は4μg/mL Remarks about FIG. 1 are as follows. Significant difference from vehicle: Steel test *: p <0.05, **: p <0.01 Example 1, compound MIC value of formula [1] is 4 μg / mL, reference example 4, compound MIC value of formula [3] is 4 μg / mL
以下において、試験結果としての肺内生菌数は、肺内生菌数(CFU/肺)の常用対数(常用対数は以下logと記載する)として表すこととする。
媒体投与群の肺内生菌数は5.88±0.14[log(CFU/肺)]であった。実施例1の式[1]で表される化合物 100および200mg/kg投与群の肺内生菌数は、それぞれ3.54±0.49[log(CFU/肺)]および2.83±0.53[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。同様に、参考例4の式[3]で表される化合物(100および200mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。結果を、肺内生菌数(CFU/肺)の常用対数(常用対数は以下logと記載する)として表すこととすると、接種3日後の肺内生菌数は、媒体投与群で5.67±0.32[log(CFU/肺)]であった。参考例4の式[3]で表される化合物 100および200mg/kg投与群の肺内生菌数は、それぞれ4.37±0.27[log(CFU/肺)]および2.53±0.23[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。以上より、実施例1の式[1]で表される化合物は、当該菌株に対して参考例4の式[3]で表される化合物と同程度の治療効果を示した。
Hereinafter, the pulmonary viable count as a test result is expressed as a common logarithm of the pulmonary viable count (CFU / lung) (the common logarithm is hereinafter referred to as log).
The viable bacterial count in the vehicle administration group was 5.88 ± 0.14 [log (CFU / lung)]. The number of viable bacteria in the lungs of the group of 100 and 200 mg / kg administered with the compound represented by the formula [1] in Example 1 is 3.54 ± 0.49 [log (CFU / lung)] and 2.83 ± 0, respectively. 0.53 [log (CFU / lung)], which was significantly reduced compared to the vehicle administration group. Similarly, the compound represented by the formula [3] of Reference Example 4 (100 and 200 mg / kg) or medium (equal mixture of 0.1 mol / L lactobionic acid solution and 0.5 w / v% sodium bicarbonate solution) Was administered orally. When the result is expressed as a common logarithm of the number of living bacteria in the lung (CFU / lung) (the common logarithm is described as “log” hereinafter), the number of living bacteria in the
試験例4 エリスロマイシン耐性(erm(B)遺伝子保有)肺炎球菌感染動物における治療効果試験
薬理効果の評価は下記に示す方法を用いた。
Test Example 4 Treatment Effect Test in Erythromycin-Resistant (Erm (B) Gene Carrying) Pneumococcal Infected Animal The evaluation of the pharmacological effect was carried out by the following method.
細菌として、 Streptococcus pneumoniae 1101 株(臨床分離株)を用いた。使用菌株の凍結保存液を30vol%非働化ウマ血清添加トッドヒューイット液体培地に添加し、濁度(OD600)が約0.3となるまで培養した。これを30vol%非働化ウマ血清添加トッドヒューイット液体培地で希釈し、接種菌液とした。マウス(CBA/JN系、雄性、5週齢)に接種菌液0.05mLを経鼻接種して感染させた。接種菌量は7.50x10^4CFU/マウスまたは1.65x10^5CFU/マウスであった。接種翌日から1日1回2日間、実施例1の式[1]で表される化合物(30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。接種3日後の肺内生菌数(1群5〜6例、平均値±標準誤差)を図2に示した。
As a bacterium, Streptococcus pneumoniae 1101 strain (clinical isolate) was used. The cryopreservation solution of the strain used was added to Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum and cultured until the turbidity (OD600) was about 0.3. This was diluted with Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum to obtain an inoculum. Mice (CBA / JN system, male, 5 weeks old) were infected with 0.05 mL of the inoculum by nasal inoculation. The amount of inoculum was 7.50 × 10 4 CFU / mouse or 1.65 × 10 5 CFU / mouse. The compound represented by the formula [1] of Example 1 (30 and 100 mg / kg) or vehicle (0.1 mol / L lactobionic acid solution and 0.5 w / v% hydrogen carbonate) once a day for 2 days from the day after inoculation An equal volume of sodium solution) was orally administered. Fig. 2 shows the number of viable bacteria in the
なお、図2についての備考は以下の通りである。媒体との有意差:Steel検定 *:p<0.05、**:p<0.01 実施例1、式[1]の化合物MIC値は0.25μg/mL、参考例4、式[3]の化合物MIC値は0.12μg/mL The remarks about FIG. 2 are as follows. Significant difference from vehicle: Steel test *: p <0.05, **: p <0.01 Example 1, compound MIC value of formula [1] is 0.25 μg / mL, reference example 4, compound MIC of formula [3] The value is 0.12 μg / mL
媒体投与群の肺内生菌数は5.83±0.08[log(CFU/肺)]であった。実施例1の式[1]で表される化合物 30および100mg/kg投与群の肺内生菌数は、それぞれ4.14±0.19[log(CFU/肺)]および2.28±0.24[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。同様に、参考例4の式[3]で表される化合物(10、30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した結果、接種3日後の肺内生菌数は、媒体投与群で5.91±0.18[log(CFU/肺)]であった。参考例4の式[3]で表される化合物 10、30および100mg/kg投与群の肺内生菌数は、それぞれ5.86±0.12[log(CFU/肺)]、5.22±0.16[log(CFU/肺)]および3.65±0.36[log(CFU/肺)]であり、参考例4の式[3]で表される化合物 100mg/kg投与群で媒体投与群と比較して有意に減少した。以上より、実施例1の式[1]で表される化合物は参考例4の式[3]で表される化合物より優れた治療効果を示した。
The viable bacterial count in the vehicle administration group was 5.83 ± 0.08 [log (CFU / lung)]. The numbers of viable bacteria in the lungs of the
試験例5 エリスロマイシン耐性(mef(A)遺伝子保有)肺炎球菌感染動物における治療効果試験
薬理効果の評価は下記に示す方法を用いた。
Test Example 5 Therapeutic effect test in erythromycin resistant (mef (A) gene possessed) pneumococcal infected animals The evaluation of the pharmacological effect was carried out by the following method.
細菌として、 Streptococcus pneumoniae 1028 株(臨床分離株)を用いた。使用菌株の凍結保存液を30vol%非働化ウマ血清添加トッドヒューイット液体培地に添加し、濁度(OD600)が約0.3となるまで培養した。これを30vol%非働化ウマ血清添加トッドヒューイット液体培地で希釈し、接種菌液とした。マウス(CBA/JN系、雄性、5週齢)に接種菌液0.05mLを経鼻接種して感染させた。接種菌量は3.45x10^4CFU/マウスまたは3.90x10^4CFU/マウスであった。接種翌日から1日1回2日間、実施例1の式[1]で表される化合物(3、10、30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。接種3日後の肺内生菌数(1群5〜6例、平均値±標準誤差)を図3に示した。
As a bacterium, Streptococcus pneumoniae 1028 strain (clinical isolate) was used. The cryopreservation solution of the strain used was added to Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum and cultured until the turbidity (OD600) was about 0.3. This was diluted with Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum to obtain an inoculum. Mice (CBA / JN system, male, 5 weeks old) were infected with 0.05 mL of the inoculum by nasal inoculation. The inoculum was 3.45 × 10 4 CFU / mouse or 3.90 × 10 4 CFU / mouse. The compound represented by the formula [1] of Example 1 (3, 10, 30 and 100 mg / kg) or vehicle (0.1 mol / L lactobionic acid solution and 0.5 w / day) once a day for 2 days from the day after the inoculation An equal volume of v% sodium bicarbonate solution) was orally administered. The number of viable bacteria in the
なお、図3についての備考は以下の通りである。媒体との有意差:Steel検定 *:p<0.05、**:p<0.01 実施例1、式[1]の化合物MIC値は0.12μg/mL、参考例4、式[3]の化合物MIC値は0.03μg/mL In addition, the remarks about FIG. 3 are as follows. Significant difference from vehicle: Steel test *: p <0.05, **: p <0.01 Example 1, compound MIC value of formula [1] is 0.12 μg / mL, Reference Example 4, compound MIC of formula [3] The value is 0.03 μg / mL
媒体投与群の肺内生菌数は6.94±0.07[log(CFU/肺)]であった。実施例1の式[1]で表される化合物 3、10、30および100mg/kg投与群の肺内生菌数は、それぞれ6.45±0.18[log(CFU/肺)]、1.30±0.00[log(CFU/肺)]、1.30±0.00[log(CFU/肺)]および1.30±0.00[log(CFU/肺)]であり、実施例1の式[1]で表される化合物 10、30および100mg/kg投与群で肺内生菌数は全例検出限界値以下を示し、媒体投与群と比較して有意に減少した。同様に、参考例4の式[3]で表される化合物(10、30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した結果、接種3日後の肺内生菌数は、媒体投与群で7.03±0.22[log(CFU/肺)]であった。参考例4の式[3]で表される化合物 10、30および100mg/kg投与群の肺内生菌数は、それぞれ5.67±0.38[log(CFU/肺)]、1.30±0.00[log(CFU/肺)]および1.30±0.00[log(CFU/肺)]であり、参考例4の式[3]で表される化合物 10、30および100mg/kg投与群で媒体投与群と比較して有意に減少した。ただし全例で検出限界値以下を示したのは参考例4の式[3]で表される化合物 30および100mg/kg投与群のみであった。以上より、実施例1の式[1]で表される化合物は参考例4の式[3]で表される化合物より優れた治療効果を示した。
The viable bacterial count in the vehicle administration group was 6.94 ± 0.07 [log (CFU / lung)]. The number of viable bacteria in the lungs of the
本発明の化合物又はその薬学的に許容される塩は、グラム陽性細菌、グラム陰性細菌やマイコプラズマに強い抗菌活性を有し、特に従来のマクロライド系抗生物質では十分な抗菌活性が得られなかったエリスロマイシン耐性菌(例えば耐性肺炎球菌、連鎖球菌、及びマイコプラズマ)などに対しても優れた抗菌活性を有し、医薬品として利用することができる。 The compound of the present invention or a pharmaceutically acceptable salt thereof has strong antibacterial activity against gram-positive bacteria, gram-negative bacteria and mycoplasma, and in particular, sufficient antibacterial activity was not obtained with conventional macrolide antibiotics. It has excellent antibacterial activity against erythromycin-resistant bacteria (for example, resistant pneumococci, streptococci, and mycoplasma), and can be used as a pharmaceutical product.
Claims (5)
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016027731 | 2016-02-17 | ||
| JP2016027731 | 2016-02-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017145247A true JP2017145247A (en) | 2017-08-24 |
Family
ID=59680621
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017026792A Pending JP2017145247A (en) | 2016-02-17 | 2017-02-16 | Infection preventive and/or therapeutic agent comprising c-4" position-substituted macrolide derivative |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017145247A (en) |
-
2017
- 2017-02-16 JP JP2017026792A patent/JP2017145247A/en active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20090117924A (en) | Macrocyclic polymorphs, compositions comprising such polymorphs, and methods of use and manufacture thereof | |
| CN114728899A (en) | Novel triphenyl compound salt | |
| EA020733B1 (en) | Actagardine derivatives | |
| EP3037428B1 (en) | Macrolide compound | |
| JP5874871B1 (en) | C-4 "substituted macrolide compounds | |
| WO2015056799A1 (en) | Hydroxamic acid derivative | |
| WO2015056800A1 (en) | Hydroxamic acid derivative | |
| JP2017145247A (en) | Infection preventive and/or therapeutic agent comprising c-4" position-substituted macrolide derivative | |
| KR20130028703A (en) | Certain crystalline hydrates, pharmaceutical compositions thereof and methods for preparation and use thereof | |
| WO2017142002A1 (en) | Crystal forms of free c-4"-substituted macrolide compound and salt thereof, and production methods therefor | |
| EP3719020B1 (en) | Crystal form of beta-lactamase inhibitor and preparation method therefor | |
| JP2019064921A (en) | Crystal forms of c-4"-substituted macrolide compound and production methods therefor | |
| JP2019064922A (en) | Salt of c-4"-substituted macrolide compound, and crystal forms thereof, and production methods therefor | |
| JP2007527434A (en) | Amorphous tacrolimus and its preparation | |
| CN114945577B (en) | Macrolide compounds and their use for the treatment of chronic respiratory diseases | |
| WO2018067663A2 (en) | Ketolides having antibacterial activity | |
| CN115466246B (en) | Pyrrole amide piperidine amine compound and application thereof | |
| WO2015056798A1 (en) | Hydroxamic acid derivative | |
| JP2016199499A (en) | Crystal forms of compounds having (2s)-2-methylamino-n-hydroxy-n',2-dimethylpropanediamide and production method thereof | |
| JP2017105714A (en) | Multiple drug discharge pump inhibitor | |
| JPWO2004078771A1 (en) | 2-Fluoro-6-O-substituted ketolide derivatives | |
| CN110713505A (en) | Gamma-lactam glucoside derivative and synthesis method and application thereof | |
| JP2021178793A (en) | Novel aminoglycoside antimicrobial agents effective against super-multidrug-resistant gram-negative bacteria | |
| CN101328155A (en) | Oxazolidine derivates, preparation and application thereof | |
| JP2016199497A (en) | Medicine containing hydroxamic acid derivative |