JP2017105748A - Method for producing 1,5-anhydro-1-substituted phenyl-d-glucitol compounds - Google Patents
Method for producing 1,5-anhydro-1-substituted phenyl-d-glucitol compounds Download PDFInfo
- Publication number
- JP2017105748A JP2017105748A JP2016119447A JP2016119447A JP2017105748A JP 2017105748 A JP2017105748 A JP 2017105748A JP 2016119447 A JP2016119447 A JP 2016119447A JP 2016119447 A JP2016119447 A JP 2016119447A JP 2017105748 A JP2017105748 A JP 2017105748A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- range
- formula
- compound represented
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- -1 1,5-anhydro-1-substituted phenyl-d-glucitol compounds Chemical class 0.000 title claims abstract description 97
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 28
- 150000001875 compounds Chemical class 0.000 claims abstract description 223
- 238000000034 method Methods 0.000 claims description 30
- 230000002401 inhibitory effect Effects 0.000 abstract description 4
- 239000003814 drug Substances 0.000 abstract description 3
- 102100037202 Sodium/myo-inositol cotransporter 2 Human genes 0.000 abstract description 2
- 101710090560 Sodium/myo-inositol cotransporter 2 Proteins 0.000 abstract description 2
- 229940079593 drug Drugs 0.000 abstract 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 111
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 93
- 239000000243 solution Substances 0.000 description 79
- 239000002904 solvent Substances 0.000 description 74
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 57
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 53
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 52
- 238000006243 chemical reaction Methods 0.000 description 52
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 51
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 51
- 239000012044 organic layer Substances 0.000 description 49
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 45
- 239000000203 mixture Substances 0.000 description 40
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 36
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 36
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 36
- 239000012442 inert solvent Substances 0.000 description 34
- 239000007858 starting material Substances 0.000 description 33
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 30
- 239000010410 layer Substances 0.000 description 29
- 239000002994 raw material Substances 0.000 description 28
- 239000002585 base Substances 0.000 description 27
- 239000000047 product Substances 0.000 description 27
- 238000005481 NMR spectroscopy Methods 0.000 description 26
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 26
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 26
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 24
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 24
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 24
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 22
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- 239000002253 acid Substances 0.000 description 21
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 20
- 239000012043 crude product Substances 0.000 description 20
- 239000012046 mixed solvent Substances 0.000 description 20
- BMVXCPBXGZKUPN-UHFFFAOYSA-N 1-hexanamine Chemical compound CCCCCCN BMVXCPBXGZKUPN-UHFFFAOYSA-N 0.000 description 19
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- 238000009835 boiling Methods 0.000 description 19
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 18
- 239000007864 aqueous solution Substances 0.000 description 18
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 17
- 239000004215 Carbon black (E152) Substances 0.000 description 17
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 17
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 17
- 229930195733 hydrocarbon Natural products 0.000 description 17
- 150000002430 hydrocarbons Chemical class 0.000 description 17
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 17
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 16
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 16
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 16
- 239000011734 sodium Substances 0.000 description 16
- 239000008096 xylene Substances 0.000 description 16
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 15
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 239000004210 ether based solvent Substances 0.000 description 15
- KLKFAASOGCDTDT-UHFFFAOYSA-N ethoxymethoxyethane Chemical compound CCOCOCC KLKFAASOGCDTDT-UHFFFAOYSA-N 0.000 description 14
- 150000004795 grignard reagents Chemical class 0.000 description 14
- 238000004949 mass spectrometry Methods 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 13
- ITADIVACIVJFJF-UHFFFAOYSA-N 1-bromo-4-methoxy-2-propan-2-ylbenzene Chemical compound COC1=CC=C(Br)C(C(C)C)=C1 ITADIVACIVJFJF-UHFFFAOYSA-N 0.000 description 13
- 230000015572 biosynthetic process Effects 0.000 description 13
- 238000004587 chromatography analysis Methods 0.000 description 13
- 230000009977 dual effect Effects 0.000 description 13
- 239000011780 sodium chloride Substances 0.000 description 13
- 238000003786 synthesis reaction Methods 0.000 description 13
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 12
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- 229910052736 halogen Inorganic materials 0.000 description 12
- 150000002367 halogens Chemical class 0.000 description 12
- 239000012264 purified product Substances 0.000 description 12
- 239000003153 chemical reaction reagent Substances 0.000 description 11
- 229960002920 sorbitol Drugs 0.000 description 11
- YLTRDVRJBLUXMH-UHFFFAOYSA-N 2-(2-bromo-5-methoxyphenyl)propan-2-ol Chemical compound COC1=CC=C(Br)C(C(C)(C)O)=C1 YLTRDVRJBLUXMH-UHFFFAOYSA-N 0.000 description 10
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 10
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 9
- 238000004566 IR spectroscopy Methods 0.000 description 9
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- 239000012300 argon atmosphere Substances 0.000 description 9
- 239000003638 chemical reducing agent Substances 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 9
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 9
- 229940011051 isopropyl acetate Drugs 0.000 description 9
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 9
- VNGTZLYNGGLPIZ-WCXIOVBPSA-N (3r,4s,5r,6r)-3,4,5-tris(trimethylsilyloxy)-6-(trimethylsilyloxymethyl)oxan-2-one Chemical compound C[Si](C)(C)OC[C@H]1OC(=O)[C@H](O[Si](C)(C)C)[C@@H](O[Si](C)(C)C)[C@@H]1O[Si](C)(C)C VNGTZLYNGGLPIZ-WCXIOVBPSA-N 0.000 description 8
- LCVDMJHIUWFVFH-UHFFFAOYSA-N 2-amino-n-[2-(dimethylamino)ethyl]-2-methylpropanamide;dihydrochloride Chemical compound Cl.Cl.CN(C)CCNC(=O)C(C)(C)N LCVDMJHIUWFVFH-UHFFFAOYSA-N 0.000 description 8
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 8
- 239000005457 ice water Substances 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 238000001953 recrystallisation Methods 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 7
- ZRYZBQLXDKPBDU-UHFFFAOYSA-N 4-bromobenzaldehyde Chemical compound BrC1=CC=C(C=O)C=C1 ZRYZBQLXDKPBDU-UHFFFAOYSA-N 0.000 description 7
- 108091006277 SLC5A1 Proteins 0.000 description 7
- 102100020885 Sodium/glucose cotransporter 1 Human genes 0.000 description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 6
- QACBKCQRNIVLIM-WUWJBHAQSA-N C(C)(=O)O[C@H]1[C@@H](O[C@@H]([C@H]([C@@H]1OC(C)=O)OC(C)=O)COC(C)=O)C1=C(C=C(C(=C1)CC1=CC=C(C=C1)Br)C(C)C)OC Chemical compound C(C)(=O)O[C@H]1[C@@H](O[C@@H]([C@H]([C@@H]1OC(C)=O)OC(C)=O)COC(C)=O)C1=C(C=C(C(=C1)CC1=CC=C(C=C1)Br)C(C)C)OC QACBKCQRNIVLIM-WUWJBHAQSA-N 0.000 description 6
- 239000007818 Grignard reagent Substances 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 6
- 239000012345 acetylating agent Substances 0.000 description 6
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 6
- 235000019270 ammonium chloride Nutrition 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 239000002198 insoluble material Substances 0.000 description 6
- ICAKDTKJOYSXGC-UHFFFAOYSA-K lanthanum(iii) chloride Chemical compound Cl[La](Cl)Cl ICAKDTKJOYSXGC-UHFFFAOYSA-K 0.000 description 6
- 229940098779 methanesulfonic acid Drugs 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 229910000029 sodium carbonate Inorganic materials 0.000 description 6
- 239000012453 solvate Substances 0.000 description 6
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 6
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 6
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 6
- 239000011701 zinc Substances 0.000 description 6
- 229910052725 zinc Inorganic materials 0.000 description 6
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 5
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 5
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 5
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 5
- 239000011777 magnesium Substances 0.000 description 5
- 229910052749 magnesium Inorganic materials 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 5
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 4
- SCFWAOWWAANBPY-UHFFFAOYSA-N 2,2-dimethyl-3-butenoic acid Chemical compound C=CC(C)(C)C(O)=O SCFWAOWWAANBPY-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 230000018044 dehydration Effects 0.000 description 4
- 238000006297 dehydration reaction Methods 0.000 description 4
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 4
- DBTNVRCCIDISMV-UHFFFAOYSA-L lithium;magnesium;propane;dichloride Chemical compound [Li+].[Mg+2].[Cl-].[Cl-].C[CH-]C DBTNVRCCIDISMV-UHFFFAOYSA-L 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- VNDYJBBGRKZCSX-UHFFFAOYSA-L zinc bromide Chemical compound Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 description 4
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 4
- KWEKXPWNFQBJAY-UHFFFAOYSA-N (dimethyl-$l^{3}-silanyl)oxy-dimethylsilicon Chemical compound C[Si](C)O[Si](C)C KWEKXPWNFQBJAY-UHFFFAOYSA-N 0.000 description 3
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 3
- 239000007848 Bronsted acid Substances 0.000 description 3
- 239000002841 Lewis acid Substances 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- IGSUJBNDAWQLST-UHFFFAOYSA-N chloro-di(propan-2-yl)silicon Chemical compound CC(C)[Si](Cl)C(C)C IGSUJBNDAWQLST-UHFFFAOYSA-N 0.000 description 3
- YGHUUVGIRWMJGE-UHFFFAOYSA-N chlorodimethylsilane Chemical compound C[SiH](C)Cl YGHUUVGIRWMJGE-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000006482 condensation reaction Methods 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- OIKHZBFJHONJJB-UHFFFAOYSA-N dimethyl(phenyl)silicon Chemical compound C[Si](C)C1=CC=CC=C1 OIKHZBFJHONJJB-UHFFFAOYSA-N 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- 150000007517 lewis acids Chemical class 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 239000012022 methylating agents Substances 0.000 description 3
- 239000011259 mixed solution Substances 0.000 description 3
- 235000006408 oxalic acid Nutrition 0.000 description 3
- CHNLPLHJUPMEOI-UHFFFAOYSA-N oxolane;trifluoroborane Chemical compound FB(F)F.C1CCOC1 CHNLPLHJUPMEOI-UHFFFAOYSA-N 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- ZGYICYBLPGRURT-UHFFFAOYSA-N tri(propan-2-yl)silicon Chemical compound CC(C)[Si](C(C)C)C(C)C ZGYICYBLPGRURT-UHFFFAOYSA-N 0.000 description 3
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 3
- AKQNYQDSIDKVJZ-UHFFFAOYSA-N triphenylsilane Chemical compound C1=CC=CC=C1[SiH](C=1C=CC=CC=1)C1=CC=CC=C1 AKQNYQDSIDKVJZ-UHFFFAOYSA-N 0.000 description 3
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-bis(diphenylphosphino)propane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 2
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 2
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 2
- FXXACINHVKSMDR-UHFFFAOYSA-N acetyl bromide Chemical compound CC(Br)=O FXXACINHVKSMDR-UHFFFAOYSA-N 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- VYLVYHXQOHJDJL-UHFFFAOYSA-K cerium trichloride Chemical compound Cl[Ce](Cl)Cl VYLVYHXQOHJDJL-UHFFFAOYSA-K 0.000 description 2
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- DEQYTNZJHKPYEZ-UHFFFAOYSA-N ethyl acetate;heptane Chemical compound CCOC(C)=O.CCCCCCC DEQYTNZJHKPYEZ-UHFFFAOYSA-N 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 229910000043 hydrogen iodide Inorganic materials 0.000 description 2
- 239000012336 iodinating agent Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 238000000752 ionisation method Methods 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 2
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 2
- 125000001979 organolithium group Chemical group 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 2
- BWHDROKFUHTORW-UHFFFAOYSA-N tritert-butylphosphane Chemical compound CC(C)(C)P(C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-N 0.000 description 2
- 229940102001 zinc bromide Drugs 0.000 description 2
- 239000011592 zinc chloride Substances 0.000 description 2
- 235000005074 zinc chloride Nutrition 0.000 description 2
- UAYWVJHJZHQCIE-UHFFFAOYSA-L zinc iodide Chemical compound I[Zn]I UAYWVJHJZHQCIE-UHFFFAOYSA-L 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- UKSZBOKPHAQOMP-SVLSSHOZSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 UKSZBOKPHAQOMP-SVLSSHOZSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- XQKQROJYWLWDMP-UHFFFAOYSA-N 2-iodo-1,1-dioxo-1,2-benzothiazol-3-one Chemical compound C1=CC=C2S(=O)(=O)N(I)C(=O)C2=C1 XQKQROJYWLWDMP-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- SYWWBJKPSYWUBN-UHFFFAOYSA-N 6443-90-9 Chemical compound ICl.C1=CC=NC=C1 SYWWBJKPSYWUBN-UHFFFAOYSA-N 0.000 description 1
- JRLTTZUODKEYDH-UHFFFAOYSA-N 8-methylquinoline Chemical group C1=CN=C2C(C)=CC=CC2=C1 JRLTTZUODKEYDH-UHFFFAOYSA-N 0.000 description 1
- 238000006596 Alder-ene reaction Methods 0.000 description 1
- BGEQKRXVLNGNHH-XRIGFGBMSA-N C/[O]=C(/[C@@H]([N]#C)/[O]=[Mn]/C)\C=N Chemical compound C/[O]=C(/[C@@H]([N]#C)/[O]=[Mn]/C)\C=N BGEQKRXVLNGNHH-XRIGFGBMSA-N 0.000 description 1
- PSXLCTPHDAEPLK-UHFFFAOYSA-N CC(C)[Mg] Chemical compound CC(C)[Mg] PSXLCTPHDAEPLK-UHFFFAOYSA-N 0.000 description 1
- 0 CNC(*=C)c1cc(ONC)ccc1Cc1ccc(*)cc1 Chemical compound CNC(*=C)c1cc(ONC)ccc1Cc1ccc(*)cc1 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 229910004664 Cerium(III) chloride Inorganic materials 0.000 description 1
- 206010010774 Constipation Diseases 0.000 description 1
- 108020003264 Cotransporters Proteins 0.000 description 1
- 102000034534 Cotransporters Human genes 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 238000007341 Heck reaction Methods 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- QZRGKCOWNLSUDK-UHFFFAOYSA-N Iodochlorine Chemical compound ICl QZRGKCOWNLSUDK-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 235000010724 Wisteria floribunda Nutrition 0.000 description 1
- OTFQZXXGENGJOF-UHFFFAOYSA-N [I].OO Chemical compound [I].OO OTFQZXXGENGJOF-UHFFFAOYSA-N 0.000 description 1
- ZVAKTWSQIIRIIH-UHFFFAOYSA-N [K].[I] Chemical compound [K].[I] ZVAKTWSQIIRIIH-UHFFFAOYSA-N 0.000 description 1
- MGVXAPSYPXFGNE-UHFFFAOYSA-O [NH4+].[I+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O Chemical compound [NH4+].[I+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O MGVXAPSYPXFGNE-UHFFFAOYSA-O 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000000065 atmospheric pressure chemical ionisation Methods 0.000 description 1
- WXNOJTUTEXAZLD-UHFFFAOYSA-L benzonitrile;dichloropalladium Chemical compound Cl[Pd]Cl.N#CC1=CC=CC=C1.N#CC1=CC=CC=C1 WXNOJTUTEXAZLD-UHFFFAOYSA-L 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000003381 deacetylation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical compound Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- WCHFOOKTKZYYAE-UHFFFAOYSA-N ethoxyperoxyethane Chemical compound CCOOOCC WCHFOOKTKZYYAE-UHFFFAOYSA-N 0.000 description 1
- OAMZXMDZZWGPMH-UHFFFAOYSA-N ethyl acetate;toluene Chemical compound CCOC(C)=O.CC1=CC=CC=C1 OAMZXMDZZWGPMH-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- QFWPJPIVLCBXFJ-UHFFFAOYSA-N glymidine Chemical compound N1=CC(OCCOC)=CN=C1NS(=O)(=O)C1=CC=CC=C1 QFWPJPIVLCBXFJ-UHFFFAOYSA-N 0.000 description 1
- QKGYJVXSKCDGOK-UHFFFAOYSA-N hexane;propan-2-ol Chemical compound CC(C)O.CCCCCC QKGYJVXSKCDGOK-UHFFFAOYSA-N 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- JILPJDVXYVTZDQ-UHFFFAOYSA-N lithium methoxide Chemical compound [Li+].[O-]C JILPJDVXYVTZDQ-UHFFFAOYSA-N 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 1
- CETVQRFGPOGIQJ-UHFFFAOYSA-N lithium;hexane Chemical compound [Li+].CCCCC[CH2-] CETVQRFGPOGIQJ-UHFFFAOYSA-N 0.000 description 1
- YMUBDNJWWYNAOA-UHFFFAOYSA-L lithium;magnesium;butane;dichloride Chemical compound [Li+].[Mg+2].[Cl-].[Cl-].CC[CH-]C YMUBDNJWWYNAOA-UHFFFAOYSA-L 0.000 description 1
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 1
- VXWPONVCMVLXBW-UHFFFAOYSA-M magnesium;carbanide;iodide Chemical compound [CH3-].[Mg+2].[I-] VXWPONVCMVLXBW-UHFFFAOYSA-M 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- VRTQLDFCPNVQNT-UHFFFAOYSA-N methyl 2-bromo-5-methoxybenzoate Chemical compound COC(=O)C1=CC(OC)=CC=C1Br VRTQLDFCPNVQNT-UHFFFAOYSA-N 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000002734 organomagnesium group Chemical group 0.000 description 1
- 238000011170 pharmaceutical development Methods 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- ZBDZVTOURNGNIQ-UHFFFAOYSA-M potassium;hydrogen peroxide;iodide Chemical compound [K+].[I-].OO ZBDZVTOURNGNIQ-UHFFFAOYSA-M 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 210000000512 proximal kidney tubule Anatomy 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 238000010583 slow cooling Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- BPAZVIWQBHZKSY-UHFFFAOYSA-M sodium;1-chloropyrrolidine-2,5-dione;iodide Chemical compound [Na+].[I-].ClN1C(=O)CCC1=O BPAZVIWQBHZKSY-UHFFFAOYSA-M 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000000967 suction filtration Methods 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 210000003437 trachea Anatomy 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 150000008501 α-D-glucopyranosides Chemical class 0.000 description 1
Abstract
Description
本発明は、ナトリウム依存性グルコース共輸送体1(sodium-dependent glucose
cotransporter 1、以下、SGLT1と記載する)阻害活性を有する医薬として有用な1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物、及びその製造中間体の製造方法に関する。さらに詳しくは、(1S)−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール、及びその製造中間体である(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトールの製造方法に関する。
The present invention relates to sodium-dependent glucose cotransporter 1 (sodium-dependent glucose
The present invention relates to a 1,5-anhydro-1-substituted phenyl-D-glucitol compound useful as a pharmaceutical having inhibitory activity (hereinafter referred to as cotransporter 1, hereinafter referred to as SGLT1), and a method for producing an intermediate thereof. More specifically, (1S) -1,5-anhydro-1- {5-[(4-{(1E) -4-[(1-{[2- (dimethylamino) ethyl] amino} -2-methyl) -1-oxopropan-2-yl) amino] -3,3-dimethyl-4-oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl } -D-glucitol and its production intermediate (1S) -2,3,4,6-tetra-O-acetyl-1,5-anhydro-1- {5-[(4-bromophenyl) methyl ] 2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol.
SGLT1は、小腸粘膜上皮、腎臓近位尿細管、骨格筋、心筋、気管及び脳に発現しており、糖の細胞内への吸収・取り込みに関与する能動輸送性の糖輸送担体である。SGLT1は小腸において特に多く存在しており、食事にて摂取した炭水化物の消化により生じたグルコース及びガラクトースの細胞内への吸収に関与している。小腸に発現しているSGLT1の機能を阻害する化合物は、糖の吸収を抑制することから、糖尿病治療薬として有用であるとして医薬品開発が行われている(特許文献1〜3)。また、SGLT1を阻害する化合物は便秘症の予防または治療に有用であることが、特許文献4に開示されている。 SGLT1 is expressed in small intestinal mucosal epithelium, renal proximal tubule, skeletal muscle, myocardium, trachea and brain, and is an active transporting sugar transporter involved in absorption and uptake of sugar into cells. SGLT1 is particularly abundant in the small intestine and is involved in the absorption of glucose and galactose produced by digestion of carbohydrates taken in the diet into cells. Since the compound that inhibits the function of SGLT1 expressed in the small intestine suppresses the absorption of sugar, pharmaceutical development has been carried out as being useful as a therapeutic agent for diabetes (Patent Documents 1 to 3). Patent Document 4 discloses that a compound that inhibits SGLT1 is useful for the prevention or treatment of constipation.
特許文献1〜4に記載された(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(以下、化合物(I)と記載する)の製造方法は、超低温(−80〜−70℃)反応工程及びシリカゲルカラムクロマトグラフィー精製工程を含み、工業的大量生産に適した製造方法とは言えなかった。
例えば、特許文献4に開示されている化合物(I)の製造方法をスキーム1に示す。
スキーム1
(1S) -2,3,4,6-tetra-O-acetyl-1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2- described in Patent Documents 1 to 4 The method for producing methoxy-4- (propan-2-yl) phenyl} -D-glucitol (hereinafter referred to as compound (I)) comprises an ultra-low temperature (−80 to −70 ° C.) reaction step and silica gel column chromatography purification. It was not a manufacturing method suitable for industrial mass production, including processes.
For example, scheme 1 shows a method for producing compound (I) disclosed in Patent Document 4.
Scheme 1
また、特許文献1〜4に記載された(1S)−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(以下、化合物(XI)と記載する)の製造方法も、シリカゲルカラムクロマトグラフィー精製工程を含み、工業的大量生産に適した製造方法とは言えなかった。
例えば、特許文献2及び特許文献4に開示されている、化合物(XI)の製造方法をスキーム2に示す。化合物(XI)は非晶質の化合物であるが、化合物(XI)のエタノール溶媒和物は結晶として得ることができる。化合物(XI)のエタノール溶媒和物は、他の結晶形(A形、B形、C形、二水和物;特許文献2)に容易に変動することが可能である。
スキーム2
In addition, (1S) -1,5-anhydro-1- {5-[(4-{(1E) -4-[(1-{[2- (dimethylamino) ethyl) described in Patent Documents 1 to 4 is used. Amino} -2-methyl-1-oxopropan-2-yl) amino] -3,3-dimethyl-4-oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy-4- ( The method for producing propan-2-yl) phenyl} -D-glucitol (hereinafter referred to as compound (XI)) also includes a silica gel column chromatography purification step and cannot be said to be a production method suitable for industrial mass production. It was.
For example, scheme 2 shows a method for producing compound (XI) disclosed in Patent Document 2 and Patent Document 4. Although compound (XI) is an amorphous compound, an ethanol solvate of compound (XI) can be obtained as crystals. The ethanol solvate of compound (XI) can be easily changed to other crystal forms (A form, B form, C form, dihydrate; Patent Document 2).
Scheme 2
本発明の目的は、医薬として有用な1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の、工業的大量生産に適した製造方法を提供することである。 An object of the present invention is to provide a production method suitable for industrial mass production of a 1,5-anhydro-1-substituted phenyl-D-glucitol compound useful as a medicine.
本発明者らは、上記課題を解決すべく鋭意検討した結果、化合物(I)を製造中間体として経由し、超低温反応工程及びシリカゲルカラムクロマトグラフィー精製工程を回避した化合物(XI)の製造方法を見出し、本発明を完成した。 As a result of intensive studies to solve the above-mentioned problems, the present inventors have devised a method for producing compound (XI) that avoids the ultra-low temperature reaction step and the silica gel column chromatography purification step via compound (I) as a production intermediate. The headline and the present invention were completed.
すなわち、本発明は以下の通りである。
(1)式(XI)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、
That is, the present invention is as follows.
(1) A method for producing a 1,5-anhydro-1-substituted phenyl-D-glucitol compound represented by the formula (XI),
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(I) converting the compound represented by formula (II) into the compound represented by formula (III);
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(Ii) converting the compound represented by the formula (III) into a compound represented by the formula (IV);
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(Iii) converting the compound represented by the formula (IV) into a compound represented by the formula (V);
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
(Iv) converting the compound represented by the formula (V) into a compound represented by the formula (VI);
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(V) converting the compound represented by the formula (VI) into a compound represented by the formula (VII);
(vi)前記式(VII)で表される化合物を式(XII)で表される化合物に変換する工程と、
(Vi) converting the compound represented by the formula (VII) into a compound represented by the formula (XII);
(vii)前記式(XII)で表される化合物を式(XIII)で表される化合物に変換する工程と、
(Vii) converting the compound represented by the formula (XII) into a compound represented by the formula (XIII);
(viii)前記式(XIII)で表される化合物を式(I)で表される化合物に変換する工程と、
(Viii) converting the compound represented by the formula (XIII) into a compound represented by the formula (I);
(ix)前記式(I)で表される化合物を式(XIV)で表される化合物に変換する工程と、
(Ix) converting the compound represented by the formula (I) into a compound represented by the formula (XIV);
(x)前記式(XIV)で表される化合物を式(XV)で表される化合物に変換する工程と、
(X) converting the compound represented by the formula (XIV) into a compound represented by the formula (XV);
(xi)前記式(XV)で表される化合物を前記式(XI)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。
(2)式(I)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、
(Xi) 1,5-anhydro-1-substituted phenyl-D-glucitol compound comprising the step of converting the compound represented by the formula (XV) into the compound represented by the formula (XI) Manufacturing method.
(2) A method for producing a 1,5-anhydro-1-substituted phenyl-D-glucitol compound represented by formula (I),
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(I) converting the compound represented by formula (II) into the compound represented by formula (III);
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(Ii) converting the compound represented by the formula (III) into a compound represented by the formula (IV);
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(Iii) converting the compound represented by the formula (IV) into a compound represented by the formula (V);
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
(Iv) converting the compound represented by the formula (V) into a compound represented by the formula (VI);
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(V) converting the compound represented by the formula (VI) into a compound represented by the formula (VII);
(vi)前記式(VII)で表される化合物を式(VIII)で表される化合物に変換する工程と、
(Vi) converting the compound represented by the formula (VII) into a compound represented by the formula (VIII);
(vii)前記式(VIII)で表される化合物を式(IX)で表される化合物に変換する工程と、
(Vii) converting the compound represented by the formula (VIII) into a compound represented by the formula (IX);
(viii)前記式(IX)で表される化合物を式(X)で表される化合物に変換する工程と、
(Viii) converting the compound represented by the formula (IX) into a compound represented by the formula (X);
(ix)前記式(X)で表される化合物を前記式(I)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。
(Ix) 1,5-anhydro-1-substituted phenyl-D-glucitol compound comprising the step of converting the compound represented by the formula (X) into the compound represented by the formula (I) Manufacturing method.
本発明により、SGLT1阻害活性を有する医薬として有用な化合物(XI)の工業的大量生産が可能になった。 INDUSTRIAL APPLICABILITY According to the present invention, industrial mass production of compound (XI) useful as a pharmaceutical having SGLT1 inhibitory activity has become possible.
以下に、本発明の具体的な態様、置換基の定義等を説明する。 Hereinafter, specific embodiments of the present invention, definitions of substituents and the like will be described.
本発明において、「n」はノルマル(normal)を、「i」はイソ(iso)を、「s」及び「sec」はセカンダリー(secondary)を、「t」及び「tert」はターシャリー(tertiary)を、「c」はシクロ(cyclo)を、「o」はオルト(ortho)を、「m」はメタ(meta)を、「p」はパラ(para)を示す。「Ac」はアセチル(acetyl)を、「TMS」はトリメチルシリル(trimethylsilyl)を、「Piv」はピバロイル(pivaloyl)を示す。 In the present invention, “n” is normal, “i” is iso, “s” and “sec” are secondary, and “t” and “tert” are tertiary. ), “C” represents cyclo, “o” represents ortho, “m” represents meta, and “p” represents para. “Ac” represents acetyl, “TMS” represents trimethylsilyl, and “Piv” represents pivaloyl.
以下、本発明を実施するための形態を具体的に説明する。 Hereinafter, the form for implementing this invention is demonstrated concretely.
本発明は、例えば、以下のスキーム3〜5に示す方法によって実施することができるが、この方法により限定的に解釈されるものではない。 The present invention can be carried out, for example, by the methods shown in the following schemes 3 to 5, but is not construed as being limited by this method.
製造中間体である式(I)で表される化合物は、以下のスキーム3に示す方法によって得ることができる。
スキーム3
The compound represented by the formula (I) which is a production intermediate can be obtained by the method shown in the following scheme 3.
Scheme 3
工程(i):
化合物(1)を不活性溶媒中、添加剤の存在下または非存在下、メチル化剤と反応させることにより、化合物(2)を得ることができる。出発原料となる化合物(1)は、市販品として入手可能である。
メチル化剤としては、例えば、塩化メチルマグネシウム、臭化メチルマグネシウム、よう化メチルマグネシウム、メチルリチウム等を使用することができる。
不活性溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
添加剤としては、例えば、塩化セリウム(III)、塩化亜鉛、塩化ランタン(III)ビス(塩化リチウム)錯体等が挙げられる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜100℃の範囲であり、より好ましくは20〜40℃の範囲である。
メチル化剤の使用量は、原料の化合物(1)に対して2〜6モル当量の範囲で使用することができ、好ましくは2.5〜4モル当量の範囲であり、より好ましくは2.5〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(1)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(2)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
Step (i):
Compound (2) can be obtained by reacting compound (1) with a methylating agent in an inert solvent in the presence or absence of an additive. Compound (1) as a starting material is available as a commercial product.
As the methylating agent, for example, methyl magnesium chloride, methyl magnesium bromide, methyl magnesium iodide, methyl lithium and the like can be used.
Examples of the inert solvent include ether solvents such as diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane, tetrahydrofuran, 2-methyltetrahydrofuran, and tetrahydropyran. A hydrocarbon solvent such as toluene, xylene, benzene, heptane, hexane, or a mixed solvent thereof can be used.
Examples of the additive include cerium (III) chloride, zinc chloride, lanthanum chloride (III) bis (lithium chloride) complex, and the like.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of −40 to 100 ° C., more preferably in the range of 20 to 40 ° C.
The amount of the methylating agent used can be in the range of 2 to 6 molar equivalents relative to the starting compound (1), preferably in the range of 2.5 to 4 molar equivalents, more preferably 2. It is in the range of 5 to 3 molar equivalents.
The solvent can be used in an amount of 1 to 100 times by mass, preferably 1 to 30 times by mass, more preferably 1 to 10 times by mass of the raw material compound (1). It is a range.
The compound (2) can be obtained as a refined product by recrystallization, a refined product by a method such as reslurry or chromatography, or as an unrefined product.
工程(ii):
化合物(2)を不活性溶媒中、または溶媒非存在下、還元剤及び酸と反応させることにより、化合物(3)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜60℃の範囲である。
還元剤の使用量は、原料の化合物(2)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1.5〜3モル当量の範囲である。
酸の使用量は、原料の化合物(2)に対して1〜20モル当量の範囲で使用することができ、好ましくは1〜10モル当量の範囲であり、より好ましくは1.5〜5モル当量の範囲である。
溶媒の使用量は、原料の化合物(2)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(3)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(3)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (ii):
Compound (3) can be obtained by reacting compound (2) with a reducing agent and an acid in an inert solvent or in the absence of a solvent.
Examples of the reducing agent include triethylsilane, triisopropylsilane, triphenylsilane, tert-butyldimethylsilane, chlorodimethylsilane, chlorodiisopropylsilane, dimethylphenylsilane, 1,1,3,3-tetramethyldisiloxane, and the like. Can be used.
Examples of the acid include Lewis acids such as boron trifluoride diethyl ether complex, boron trifluoride tetrahydrofuran complex, aluminum chloride, trifluoromethanesulfonic acid trimethylsilyl ester, or trifluoromethanesulfonic acid, methanesulfonic acid, sulfuric acid, trifluoroacetic acid. Bronsted acids such as can be used.
Examples of the inert solvent include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, heptane, hexane, cyclohexane, tetrahydrofuran, 2-methyltetrahydrofuran, Tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, ether solvents such as 1,2-dimethoxyethane, diethoxymethane, ethyl acetate, acetonitrile, N, N-dimethylformamide, N-methyl Pyrrolidone, dimethyl sulfoxide, acetic acid, water, or a mixed solvent thereof can be used.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 0 to 60 ° C.
The reducing agent can be used in an amount of 1 to 10 molar equivalents relative to the raw material compound (2), preferably 1 to 5 molar equivalents, more preferably 1.5 to 3 A range of molar equivalents.
The amount of the acid used can be in the range of 1 to 20 molar equivalents relative to the raw material compound (2), preferably in the range of 1 to 10 molar equivalents, more preferably 1.5 to 5 moles. Equivalent range.
The amount of the solvent used can be used in the range of 1 to 100 times by mass relative to the raw material compound (2), preferably in the range of 1 to 30 times by mass, more preferably 1 to 10 times by mass It is a range.
Compound (3) can be obtained as a purified product by a method such as distillation or chromatography, or an unpurified product. Compound (3) can be used as a raw material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
工程(iii):
化合物(3)を不活性溶媒中、活性化剤存在下、マグネシウムと反応させてグリニャール試薬(4)を調製後、4−ブロモベンズアルデヒド(5)と反応させることにより、化合物(6)を得ることができる。
活性化剤としては、例えば、よう素、1,2−ジブロモエタン、水素化ジイソブチルアルミニウム等を使用することができる。
不活性溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
グリニャール試薬(4)調製時の反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは20〜100℃の範囲であり、より好ましくは40〜80℃の範囲である。
グリニャール試薬(4)と化合物(5)の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−20〜100℃の範囲であり、より好ましくは0〜30℃の範囲である。
マグネシウムの使用量は、原料の化合物(3)に対して1〜5モル当量の範囲で使用することができ、好ましくは1〜3モル当量の範囲であり、より好ましくは1〜1.2モル当量の範囲である。
活性化剤の使用量は、原料の化合物(3)に対して0.0001〜1モル当量の範囲で使用することができ、好ましくは0.001〜0.1モル当量の範囲であり、より好ましくは0.001〜0.01モル当量の範囲である。
化合物(5)の使用量は、原料の化合物(3)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(3)及び化合物(5)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(6)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(6)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (iii):
Compound (3) is reacted with magnesium in an inert solvent in the presence of an activator to prepare Grignard reagent (4), and then reacted with 4-bromobenzaldehyde (5) to obtain compound (6). Can do.
As the activator, for example, iodine, 1,2-dibromoethane, diisobutylaluminum hydride and the like can be used.
Examples of the inert solvent include ether solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane, A hydrocarbon solvent such as toluene, xylene, benzene, heptane, hexane, cyclohexane, or a mixed solvent thereof can be used.
The reaction temperature during the preparation of the Grignard reagent (4) is usually from 0 ° C. to the boiling point of the solvent used, but is preferably in the range of 20 to 100 ° C., more preferably in the range of 40 to 80 ° C. .
The reaction temperature of the Grignard reagent (4) and the compound (5) is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of −20 to 100 ° C., more preferably 0 to 30. It is in the range of ° C.
The usage-amount of magnesium can be used in the range of 1-5 molar equivalent with respect to the compound (3) of a raw material, Preferably it is the range of 1-3 molar equivalent, More preferably, it is 1-1.2 mol Equivalent range.
The use amount of the activator can be used in the range of 0.0001 to 1 molar equivalent, preferably in the range of 0.001 to 0.1 molar equivalent, relative to the starting compound (3). Preferably it is the range of 0.001-0.01 molar equivalent.
The amount of the compound (5) used can be in the range of 1 to 3 molar equivalents relative to the starting compound (3), preferably in the range of 1 to 1.5 molar equivalents, more preferably 1 It is the range of -1.1 molar equivalent.
The amount of the solvent used can be in the range of 1 to 100 times by mass, preferably in the range of 1 to 30 times by mass, more preferably 1 to the compound (3) and compound (5) of the raw material. It is the range of 10 mass times.
Compound (6) can be obtained as a purified product by a method such as distillation or chromatography, or an unpurified product. Compound (6) can be used as a raw material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
工程(iv):
化合物(6)を不活性溶媒中、または溶媒非存在下、還元剤及び酸と反応させることにより、化合物(7)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜60℃の範囲である。
還元剤の使用量は、原料の化合物(6)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜2モル当量の範囲である。
酸の使用量は、原料の化合物(6)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(6)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(7)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(7)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (iv):
Compound (7) can be obtained by reacting compound (6) with a reducing agent and an acid in an inert solvent or in the absence of a solvent.
Examples of the reducing agent include triethylsilane, triisopropylsilane, triphenylsilane, tert-butyldimethylsilane, chlorodimethylsilane, chlorodiisopropylsilane, dimethylphenylsilane, 1,1,3,3-tetramethyldisiloxane, and the like. Can be used.
Examples of the acid include Lewis acids such as boron trifluoride diethyl ether complex, boron trifluoride tetrahydrofuran complex, aluminum chloride, trifluoromethanesulfonic acid trimethylsilyl ester, or trifluoromethanesulfonic acid, methanesulfonic acid, sulfuric acid, trifluoroacetic acid. Bronsted acids such as can be used.
Examples of the inert solvent include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, heptane, hexane, cyclohexane, tetrahydrofuran, 2-methyltetrahydrofuran, Tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, ether solvents such as 1,2-dimethoxyethane, diethoxymethane, ethyl acetate, acetonitrile, N, N-dimethylformamide, N-methyl Pyrrolidone, dimethyl sulfoxide, acetic acid, water, or a mixed solvent thereof can be used.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 0 to 60 ° C.
The reducing agent can be used in an amount of 1 to 10 molar equivalents, preferably 1 to 5 molar equivalents, more preferably 1 to 2 molar equivalents, relative to the starting compound (6). Range.
The amount of the acid used can be used in the range of 1 to 10 molar equivalents relative to the starting compound (6), preferably in the range of 1 to 5 molar equivalents, more preferably in the range of 1 to 3 molar equivalents. It is a range.
The amount of the solvent used can be used in the range of 1 to 100 times by mass with respect to the raw material compound (6), preferably in the range of 1 to 30 times by mass, more preferably 1 to 10 times by mass. It is a range.
Compound (7) can be obtained as a purified product by a method such as distillation or chromatography, or an unpurified product. Compound (7) can be used as a raw material for the next step without being purified as a concentrated residue after the reaction post-treatment or without being concentrated as a post-treatment solution.
工程(v):
化合物(7)を不活性溶媒中、よう素化剤と反応させることにより、化合物(8)を得ることができる。
よう素化剤としては、例えば、一塩化よう素、ピリジン一塩化よう素、よう素−過酸化水素、よう素−よう素酸カリウム−酸、よう素―硝酸アンモニウムセリウム(IV)、よう化カリウム−過酸化水素、よう化ナトリウム−N−クロロコハク酸イミド、N−ヨードコハク酸イミド、1,3−ジヨード−5,5−ジメチルヒダントイン−酸、N−ヨードサッカリン、ジクロロよう素酸ベンジルトリメチルアンモニウム−塩化亜鉛等を使用することができる。
酸としては、例えば、塩酸、硫酸、酢酸等を使用することができる。
不活性溶媒としては、例えば、メタノール、エタノール、1−プロパノール、2−プロパノール、tert−ブチルアルコール等のアルコール系溶媒、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜80℃の範囲である。
よう素化剤の使用量は、原料の化合物(7)に対して0.1〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
酸の使用量は、原料の化合物(7)に対して0.1〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.1〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(7)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(8)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
Step (v):
Compound (8) can be obtained by reacting compound (7) with an iodinating agent in an inert solvent.
Examples of the iodinating agent include iodine monochloride, pyridine iodine monochloride, iodine-hydrogen peroxide, iodine-potassium iodate-acid, iodine-ammonium cerium (IV) nitrate, potassium iodide- Hydrogen peroxide, sodium iodide-N-chlorosuccinimide, N-iodosuccinimide, 1,3-diiodo-5,5-dimethylhydantoin-acid, N-iodosaccharin, benzyltrimethylammonium dichloroiodate-zinc chloride Etc. can be used.
As the acid, for example, hydrochloric acid, sulfuric acid, acetic acid and the like can be used.
Examples of the inert solvent include alcohol solvents such as methanol, ethanol, 1-propanol, 2-propanol, and tert-butyl alcohol, halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, and chlorobenzene, toluene, and xylene. , Hydrocarbon solvents such as benzene, heptane, hexane, cyclohexane, tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxy Ether solvents such as methane, ethyl acetate, isopropyl acetate, acetone, 2-butanone, acetonitrile, N, N-dimethylformamide, N-methylpyrrolidone, Methyl sulfoxide, acetic acid, water or a mixed solvent thereof and the like can be used.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 0 to 80 ° C.
The amount of the iodizing agent can be used in the range of 0.1 to 10 molar equivalents relative to the raw material compound (7), preferably in the range of 0.1 to 5 molar equivalents, and more preferably Is in the range of 0.2 to 2 molar equivalents.
The amount of the acid used can be 0.1 to 10 molar equivalents relative to the starting compound (7), preferably 0.1 to 5 molar equivalents, more preferably 0.00. It is in the range of 1 to 3 molar equivalents.
The amount of the solvent used can be used in the range of 1 to 100 times by mass relative to the raw material compound (7), preferably in the range of 1 to 30 times by mass, more preferably 1 to 10 times by mass. It is a range.
Compound (8) can be obtained not only as a purified product by recrystallization, but also as a purified product by a method such as reslurry or chromatography, and can also be obtained as an unrefined product.
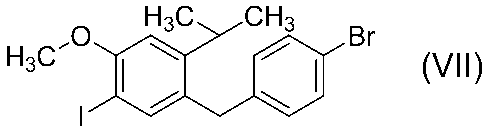
工程(vi):
化合物(8)を不活性溶媒中、有機マグネシウム試薬と反応させてグリニャール試薬(9)を調製後、化合物(10)と反応させることにより、化合物(11)を得ることができる。なお、化合物(10)は、特許文献US2002/0137903A1記載の方法にて合成することができる。
有機マグネシウム試薬としては、例えば、塩化イソプロピルマグネシウム、塩化イソプロピルマグネシウム−塩化リチウム錯体、塩化2−ブチルマグネシウム−塩化リチウム錯体、塩化イソプロピルマグネシウム−ビス[2−(N,N−ジメチルアミノ)エチル]エーテル錯体等を使用することができる。
不活性溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
グリニャール試薬(9)調製時の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜20℃の範囲であり、より好ましくは−20〜0℃の範囲である。グリニャール試薬(9)と化合物(10)の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜20℃の範囲であり、より好ましくは−20〜0℃の範囲である。
有機マグネシウム試薬の使用量は、原料の化合物(8)に対して0.9〜2モル当量の範囲で使用することができ、好ましくは0.95〜1.1モル当量の範囲であり、より好ましくは0.95〜1.05モル当量の範囲である。
化合物(10)の使用量は、原料の化合物(8)に対して0.9〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(8)及び化合物(10)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(11)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、または後処理することなく反応液のまま、次の工程の原料として使用することができる。
Step (vi):
Compound (11) can be obtained by reacting compound (8) with an organic magnesium reagent in an inert solvent to prepare Grignard reagent (9) and then reacting with compound (10). In addition, a compound (10) is compoundable by the method of patent document US2002 / 0137903A1.
Examples of the organic magnesium reagent include isopropylmagnesium chloride, isopropylmagnesium chloride-lithium chloride complex, 2-butylmagnesium chloride-lithium chloride complex, isopropylmagnesium chloride-bis [2- (N, N-dimethylamino) ethyl] ether complex. Etc. can be used.
Examples of the inert solvent include ether solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane, A hydrocarbon solvent such as toluene, xylene, benzene, heptane, hexane, cyclohexane, or a mixed solvent thereof can be used.
The reaction temperature during the preparation of the Grignard reagent (9) is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of −40 to 20 ° C., more preferably −20 to 0 ° C. It is a range. The reaction temperature of the Grignard reagent (9) and the compound (10) is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of −40 to 20 ° C., more preferably −20 to 20 ° C. It is in the range of 0 ° C.
The amount of the organomagnesium reagent used can be used in the range of 0.9 to 2 molar equivalents relative to the starting compound (8), preferably in the range of 0.95 to 1.1 molar equivalents, and more Preferably it is the range of 0.95-1.05 molar equivalent.
The compound (10) can be used in an amount of 0.9 to 3 molar equivalents, preferably 1 to 1.5 molar equivalents, more preferably relative to the starting compound (8). Is in the range of 1 to 1.1 molar equivalents.
The usage-amount of a solvent can be used in the range of 1-100 mass times with respect to the raw material compound (8) and compound (10), Preferably it is the range of 1-30 mass times, More preferably, it is 1 It is the range of 10 mass times.
Compound (11) should be used as a raw material for the next step without being purified as a concentrated residue after post-treatment of the reaction, without being concentrated as a post-treatment solution, or without post-treatment. Can do.
工程(vii):
化合物(11)を酸存在下、メタノール溶媒中にて反応させることにより、化合物(12)を得ることができる。
酸としては、例えば、塩化水素、臭化水素、よう化水素、硫酸、メタンスルホン酸、パラトルエンスルホン酸、ベンゼンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜50℃の範囲である。
酸の使用量は、原料の化合物(11)に対して0.01〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
メタノールの使用量は、原料の化合物(11)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(12)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(12)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (vii):
Compound (12) can be obtained by reacting compound (11) in the presence of an acid in a methanol solvent.
Examples of the acid that can be used include hydrogen chloride, hydrogen bromide, hydrogen iodide, sulfuric acid, methanesulfonic acid, paratoluenesulfonic acid, benzenesulfonic acid, trifluoromethanesulfonic acid, trifluoroacetic acid, and the like.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 0 to 50 ° C.
The acid can be used in an amount of 0.01 to 10 molar equivalents relative to the starting compound (11), preferably 0.1 to 5 molar equivalents, more preferably 0.00. It is in the range of 2 to 2 molar equivalents.
The amount of methanol used can be used in the range of 1 to 100 times by mass with respect to the raw material compound (11), preferably in the range of 1 to 30 times by mass, more preferably 1 to 10 times by mass. It is a range.
Compound (12) can be obtained as a refined product by recrystallization, a refined product by a method such as reslurry or chromatography, or as an unrefined product. The compound (12) can be used as a raw material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
工程(viii):
化合物(12)を不活性溶媒中、塩基存在下、アセチル化剤と反応させることにより、化合物(13)を得ることができる。
アセチル化剤としては、例えば、塩化アセチル、臭化アセチル、無水酢酸等を使用することができる。
塩基としては、例えば、ピリジン、コリジン、2,6−ルチジン、トリエチルアミン、ジイソプロピルエチルアミン等を使用することができる。ピリジンは溶媒としても使用することができる。触媒として4−ジメチルアミノピリジンを使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜40℃の範囲である。
アセチル化剤の使用量は、原料の化合物(12)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
塩基の使用量は、原料の化合物(12)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
溶媒の使用量は、原料の化合物(12)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(13)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(13)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (viii):
Compound (13) can be obtained by reacting compound (12) with an acetylating agent in the presence of a base in an inert solvent.
As the acetylating agent, for example, acetyl chloride, acetyl bromide, acetic anhydride and the like can be used.
As the base, for example, pyridine, collidine, 2,6-lutidine, triethylamine, diisopropylethylamine and the like can be used. Pyridine can also be used as a solvent. 4-Dimethylaminopyridine can be used as a catalyst.
Examples of the inert solvent include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, heptane, hexane, cyclohexane, tetrahydrofuran, 2-methyltetrahydrofuran, Tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane and other ether solvents, ethyl acetate, isopropyl acetate, acetone, 2-butanone, acetonitrile, N , N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, or a mixed solvent thereof can be used.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 0 to 40 ° C.
The amount of the acetylating agent can be used in the range of 4 to 30 molar equivalents relative to the starting compound (12), preferably in the range of 6 to 20 molar equivalents, more preferably 8 to 15 moles. Equivalent range.
The amount of the base used can be 4 to 30 molar equivalents relative to the starting compound (12), preferably 6 to 20 molar equivalents, more preferably 8 to 15 molar equivalents. It is a range.
The solvent can be used in an amount of 1 to 100 times by mass, preferably 1 to 30 times by mass, more preferably 1 to 10 times by mass, relative to the raw material compound (12). It is a range.
Compound (13) can be obtained as a purified product by a method such as chromatography or an unpurified product. Compound (13) can be used as a starting material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
工程(ix):
化合物(13)を不活性溶媒中、水存在下または非存在下、還元剤及び酸と反応させることにより、化合物(I)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜50℃の範囲である。
還元剤の使用量は、原料の化合物(13)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜2モル当量の範囲である。
酸の使用量は、原料の化合物(13)に対して1〜20モル当量の範囲で使用することができ、好ましくは1〜10モル当量の範囲であり、より好ましくは1〜5モル当量の範囲である。
水の使用量は、原料の化合物(13)に対して0.5〜5モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1モル当量である。
溶媒の使用量は、原料の化合物(13)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(I)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(I)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Process (ix):
Compound (I) can be obtained by reacting compound (13) with a reducing agent and an acid in an inert solvent in the presence or absence of water.
Examples of the reducing agent include triethylsilane, triisopropylsilane, triphenylsilane, tert-butyldimethylsilane, chlorodimethylsilane, chlorodiisopropylsilane, dimethylphenylsilane, 1,1,3,3-tetramethyldisiloxane, and the like. Can be used.
Examples of the acid include Lewis acids such as boron trifluoride diethyl ether complex, boron trifluoride tetrahydrofuran complex, aluminum chloride, trifluoromethanesulfonic acid trimethylsilyl ester, or trifluoromethanesulfonic acid, methanesulfonic acid, sulfuric acid, trifluoroacetic acid. Bronsted acids such as can be used.
Examples of the inert solvent include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, heptane, hexane, cyclohexane, tetrahydrofuran, 2-methyltetrahydrofuran, Tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, ether solvents such as 1,2-dimethoxyethane, diethoxymethane, ethyl acetate, acetonitrile, N, N-dimethylformamide, N-methyl Pyrrolidone, dimethyl sulfoxide, acetic acid, water, or a mixed solvent thereof can be used.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 0 to 50 ° C.
The reducing agent can be used in an amount of 1 to 10 molar equivalents relative to the starting compound (13), preferably 1 to 5 molar equivalents, more preferably 1 to 2 molar equivalents. Range.
The amount of the acid used can be in the range of 1 to 20 molar equivalents relative to the starting compound (13), preferably in the range of 1 to 10 molar equivalents, more preferably 1 to 5 molar equivalents. It is a range.
The amount of water used can be in the range of 0.5 to 5 molar equivalents relative to the starting compound (13), preferably in the range of 1 to 2 molar equivalents, more preferably 1 molar equivalent. is there.
The amount of the solvent used can be used in the range of 1 to 100 times by mass, preferably 1 to 30 times by mass, more preferably 1 to 10 times by mass with respect to the raw material compound (13). It is a range.
Compound (I) can be obtained not only as a purified product by recrystallization, but also as a purified product by a method such as reslurry or chromatography, and can also be obtained as an unrefined product. Compound (I) can be used as a raw material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
また、式(I)で表される化合物は、以下のスキーム4に示す方法によっても得ることができる。
スキーム4
The compound represented by the formula (I) can also be obtained by the method shown in the following scheme 4.
Scheme 4
工程(i):
ハロゲン化亜鉛を不活性溶媒中、有機リチウム試薬と反応させた後、化合物(8)と反応させることにより、ジアリール亜鉛試薬(14)を得ることができる。引き続き、化合物(14)にエーテル系溶媒を添加後、化合物(15)と反応させることにより、化合物(16)を得ることができる。
または、化合物(8)を不活性溶媒中、有機リチウム試薬と反応させた後、ハロゲン化亜鉛と臭化リチウムのジブチルエーテル溶液を作用させることにより、ジアリール亜鉛試薬(14)を得、引き続き、化合物(15)と反応させることによっても、化合物(16)を得ることができる。
ハロゲン化亜鉛としては、例えば、塩化亜鉛、臭化亜鉛、ヨウ化亜鉛を使用することができる。
不活性溶媒としては、例えば、トルエン、キシレン、ベンゼン等の炭化水素系溶媒等、及びこれら炭化水素系溶媒とテトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル等のエーテル系溶媒との混合溶媒を使用することができる。
有機リチウム試薬としては、例えば、メチルリチウム、n−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、n−ヘキシルリチウム等を使用することができる。
エーテル系溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル等を使用することができる。
化合物(15)は、特許文献5及び非特許文献1に記載の方法にて合成することができる。
ジアリール亜鉛試薬(14)調製時の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜30℃の範囲であり、より好ましくは−20〜30℃の範囲である。
ジアリール亜鉛試薬(14)と化合物(15)の反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは50〜110℃の範囲であり、より好ましくは90〜100℃の範囲である。
ハロゲン化亜鉛の使用量は、原料の化合物(8)に対して0.3〜2モル当量の範囲で使用することができ、好ましくは0.5〜1モル当量の範囲であり、より好ましくは0.5〜0.6モル当量の範囲である。
有機リチウム試薬の使用量は、原料の化合物(8)に対して0.9〜2モル当量の範囲で使用することができ、好ましくは0.9〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
臭化リチウムの使用量は、原料の化合物(8)に対して0.3〜2モル当量の範囲で使用することができ、好ましくは0.5〜1モル当量の範囲であり、より好ましくは0.5〜0.6モル当量の範囲である。
化合物(15)の使用量は、原料の化合物(8)に対して0.9〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(8)及び化合物(15)に対して0.5〜100質量倍の範囲で使用することができ、好ましくは0.5〜30質量倍の範囲であり、より好ましくは0.5〜10質量倍の範囲である。
化合物(16)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(16)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (i):
A diaryl zinc reagent (14) can be obtained by reacting a zinc halide with an organolithium reagent in an inert solvent and then reacting with the compound (8). Subsequently, the compound (16) can be obtained by adding an ether solvent to the compound (14) and reacting with the compound (15).
Alternatively, the compound (8) is reacted with an organolithium reagent in an inert solvent and then reacted with a dibutyl ether solution of zinc halide and lithium bromide to obtain a diarylzinc reagent (14). Compound (16) can also be obtained by reacting with (15).
As the zinc halide, for example, zinc chloride, zinc bromide, and zinc iodide can be used.
Examples of the inert solvent include hydrocarbon solvents such as toluene, xylene, and benzene, and these hydrocarbon solvents and tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, dibutyl ether, tert-butyl. A mixed solvent with an ether solvent such as methyl ether or cyclopentyl methyl ether can be used.
As the organic lithium reagent, for example, methyl lithium, n-butyl lithium, sec-butyl lithium, tert-butyl lithium, n-hexyl lithium and the like can be used.
As the ether solvent, for example, tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, dibutyl ether, tert-butyl methyl ether, cyclopentyl methyl ether and the like can be used.
Compound (15) can be synthesized by the methods described in Patent Document 5 and Non-Patent Document 1.
The reaction temperature during the preparation of the diarylzinc reagent (14) can usually be from -80 ° C to the boiling point of the solvent used, but is preferably in the range of -40 to 30 ° C, more preferably -20 to 30 ° C. Range.
The reaction temperature of the diaryl zinc reagent (14) and the compound (15) is usually from 0 ° C. to the boiling point of the solvent used, but is preferably in the range of 50 to 110 ° C., more preferably 90 to 100 ° C. Range.
The amount of zinc halide used can be used in the range of 0.3 to 2 molar equivalents relative to the starting compound (8), preferably in the range of 0.5 to 1 molar equivalents, and more preferably The range is 0.5 to 0.6 molar equivalent.
The amount of the organic lithium reagent used can be 0.9 to 2 molar equivalents relative to the starting compound (8), preferably 0.9 to 1.5 molar equivalents, and more Preferably it is the range of 1-1.1 molar equivalent.
The amount of lithium bromide used can be in the range of 0.3 to 2 molar equivalents relative to the starting compound (8), preferably in the range of 0.5 to 1 molar equivalents, more preferably The range is 0.5 to 0.6 molar equivalent.
The amount of the compound (15) used can be used in the range of 0.9 to 3 molar equivalents relative to the starting compound (8), preferably in the range of 1 to 1.5 molar equivalents, and more preferably Is in the range of 1 to 1.1 molar equivalents.
The amount of the solvent used can be used in the range of 0.5 to 100 times by mass, preferably in the range of 0.5 to 30 times by mass with respect to the raw material compound (8) and compound (15), More preferably, it is the range of 0.5-10 mass times.
Compound (16) can be obtained as a purified product by a method such as chromatography or an unpurified product. Compound (16) can be used as a raw material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
工程(ii):
化合物(16)を塩基または酸存在下、メタノール溶媒またはメタノール−水混合溶媒中にて反応させることにより、化合物(17)を得ることができる。
塩基としては、例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、リチウムメトキシド、ナトリウムメトキシド、カリウムメトキシド等を使用することができる。
酸としては、例えば、塩化水素、臭化水素、よう化水素、硫酸、メタンスルホン酸、パラトルエンスルホン酸、ベンゼンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは50〜80℃の範囲である。
塩基または酸の使用量は、原料の化合物(16)に対して0.01〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
溶媒の使用量は、原料の化合物(16)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(17)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(17)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (ii):
Compound (17) can be obtained by reacting compound (16) in the presence of a base or acid in a methanol solvent or a methanol-water mixed solvent.
As the base, for example, lithium hydroxide, sodium hydroxide, potassium hydroxide, lithium methoxide, sodium methoxide, potassium methoxide and the like can be used.
Examples of the acid that can be used include hydrogen chloride, hydrogen bromide, hydrogen iodide, sulfuric acid, methanesulfonic acid, paratoluenesulfonic acid, benzenesulfonic acid, trifluoromethanesulfonic acid, trifluoroacetic acid, and the like.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 50 to 80 ° C.
The amount of the base or acid used can be in the range of 0.01 to 10 molar equivalents relative to the raw material compound (16), preferably in the range of 0.1 to 5 molar equivalents, more preferably The range is 0.2 to 2 molar equivalents.
The amount of the solvent used can be 1 to 100 times by mass, preferably 1 to 30 times by mass, more preferably 1 to 10 times by mass with respect to the raw material compound (16). It is a range.
Compound (17) can be obtained not only as a purified product by recrystallization, but also as a purified product by a method such as reslurry or chromatography, and can also be obtained as an unpurified product. Compound (17) can be used as a raw material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
工程(iii):
化合物(17)を不活性溶媒中、塩基存在下、アセチル化剤と反応させることにより、化合物(I)を得ることができる。
アセチル化剤としては、例えば、塩化アセチル、臭化アセチル、無水酢酸等を使用することができる。
塩基としては、例えば、ピリジン、コリジン、2,6−ルチジン、トリエチルアミン、ジイソプロピルエチルアミン等を使用することができる。ピリジンは溶媒としても使用することができる。触媒として4−ジメチルアミノピリジンを使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜40℃の範囲である。
アセチル化剤の使用量は、原料の化合物(17)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
塩基の使用量は、原料の化合物(17)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
溶媒の使用量は、原料の化合物(17)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(I)は、上述の通り、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(I)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
Step (iii):
Compound (I) can be obtained by reacting compound (17) with an acetylating agent in the presence of a base in an inert solvent.
As the acetylating agent, for example, acetyl chloride, acetyl bromide, acetic anhydride and the like can be used.
As the base, for example, pyridine, collidine, 2,6-lutidine, triethylamine, diisopropylethylamine and the like can be used. Pyridine can also be used as a solvent. 4-Dimethylaminopyridine can be used as a catalyst.
Examples of the inert solvent include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, heptane, hexane, cyclohexane, tetrahydrofuran, 2-methyltetrahydrofuran, Tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane and other ether solvents, ethyl acetate, isopropyl acetate, acetone, 2-butanone, methyl isobutyl ketone , Acetonitrile, N, N-dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide, or a mixed solvent thereof can be used.
The reaction temperature is usually from −80 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 0 to 40 ° C.
The amount of the acetylating agent can be used in the range of 4 to 30 molar equivalents relative to the starting compound (17), preferably in the range of 6 to 20 molar equivalents, more preferably 8 to 15 moles. Equivalent range.
The amount of the base used can be 4 to 30 molar equivalents relative to the starting compound (17), preferably 6 to 20 molar equivalents, more preferably 8 to 15 molar equivalents. It is a range.
The amount of the solvent used can be used in the range of 1 to 100 times by mass, preferably in the range of 1 to 30 times by mass, more preferably 1 to 10 times by mass with respect to the starting compound (17). It is a range.
As described above, compound (I) can be obtained as a refined product by recrystallization, a refined product by a method such as reslurry or chromatography, and can also be obtained as an unrefined product. Compound (I) can be used as a raw material for the next step without being purified as a concentrated residue after the post-treatment of the reaction or without being concentrated as a post-treatment solution.
式(XI)で表される化合物は、以下のスキーム5に示す方法によって得ることができる。
スキーム5
The compound represented by the formula (XI) can be obtained by the method shown in the following scheme 5.
Scheme 5
工程(i):
化合物(I)を不活性溶媒中、パラジウム触媒、ホスフィンリガンド及び塩基存在下、化合物(18)と反応(Heck反応)させた後、不活性溶媒中、n−ヘキシルアミンと塩を形成させて化合物(19)を得ることができる。
化合物(18)は、文献(Tetrahedron、1998、vol.54、4357−4366)記載の方法にて合成することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシド、水またはこれらの混合溶媒等を使用することができる。
パラジウム触媒としては、例えば、酢酸パラジウム、塩化パラジウム、パラジウム炭素、テトラキス(トリフェニルホスフィン)パラジウム、ビス(ジベンジリデンアセトン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム、ビス(トリフェニルホスフィン)パラジウムジクロリド、ビス(ベンゾニトリル)パラジウムジクロリド、ビス(アセトニトリル)パラジウムジクロリド等を使用することができる。
ホスフィンリガンドとしては、例えば、トリフェニルホスフィン、トリ−o−トリルホスフィン、トリ−tert−ブチルホスフィン、1,3−ビス(ジフェニルホスフィノ)プロパン等を使用することができる。
塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、酢酸ナトリウム、カリウム tert−ブトキシド等を使用することができる。
反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは60〜100℃の範囲である。
化合物(18)の使用量は、原料の化合物(I)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.5モル当量の範囲である。
パラジウム触媒の使用量は、原料の化合物(I)に対して0.01〜1モル当量の範囲で使用することができ、好ましくは0.05〜0.5モル当量の範囲であり、より好ましくは0.1〜0.2モル当量の範囲である。
ホスフィンリガンドの使用量は、原料の化合物(I)に対して0.01〜1モル当量の範囲で使用することができ、好ましくは0.05〜0.5モル当量の範囲であり、より好ましくは0.1〜0.3モル当量の範囲である。
塩基の使用量は、原料の化合物(I)に対して1〜10モル当量の範囲で使用することができ、好ましくは2〜6モル当量の範囲であり、より好ましくは3〜5モル当量の範囲である。
反応溶媒の使用量は、原料の化合物(I)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
n−ヘキシルアミンの使用量は、原料の化合物(I)に対して1〜2モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
n−ヘキシルアミン塩(19)を合成する時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシド、水またはこれらの混合溶媒等を使用することができる。
化合物(19)は、結晶化による精製品のほか、リスラリー、再結晶等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
Step (i):
Compound (I) is reacted with Compound (18) in the presence of a palladium catalyst, a phosphine ligand and a base in an inert solvent (Heck reaction), and then a salt is formed with n-hexylamine in an inert solvent. (19) can be obtained.
Compound (18) can be synthesized by the method described in the literature (Tetrahedron, 1998, vol. 54, 4357-4366).
Examples of the inert solvent include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, heptane, hexane, cyclohexane, tetrahydrofuran, 2-methyltetrahydrofuran, Tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane and other ether solvents, ethyl acetate, isopropyl acetate, acetone, 2-butanone, methyl isobutyl ketone , Acetonitrile, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, dimethyl sulfoxide, water or a mixed solvent thereof is used. Rukoto can.
Examples of the palladium catalyst include palladium acetate, palladium chloride, palladium carbon, tetrakis (triphenylphosphine) palladium, bis (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) dipalladium, bis (triphenylphosphine) palladium dichloride, Bis (benzonitrile) palladium dichloride, bis (acetonitrile) palladium dichloride, and the like can be used.
As the phosphine ligand, for example, triphenylphosphine, tri-o-tolylphosphine, tri-tert-butylphosphine, 1,3-bis (diphenylphosphino) propane and the like can be used.
As the base, for example, triethylamine, tri-n-butylamine, N, N-diisopropylethylamine, N-methylmorpholine, potassium carbonate, sodium carbonate, cesium carbonate, sodium acetate, potassium tert-butoxide and the like can be used.
The reaction temperature is usually from 0 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 60 to 100 ° C.
The amount of compound (18) used can be in the range of 1 to 3 molar equivalents, preferably in the range of 1 to 2 molar equivalents, more preferably 1 to 1, relative to the starting compound (I). .5 molar equivalent range.
The amount of the palladium catalyst used can be used in the range of 0.01 to 1 molar equivalent, preferably 0.05 to 0.5 molar equivalent, more preferably relative to the starting compound (I). Is in the range of 0.1 to 0.2 molar equivalents.
The amount of the phosphine ligand used can be used in the range of 0.01 to 1 molar equivalent, preferably 0.05 to 0.5 molar equivalent, more preferably relative to the starting compound (I). Is in the range of 0.1 to 0.3 molar equivalents.
The amount of the base used can be 1 to 10 molar equivalents relative to the starting compound (I), preferably 2 to 6 molar equivalents, more preferably 3 to 5 molar equivalents. It is a range.
The reaction solvent can be used in an amount of 1 to 100 times by mass, preferably 1 to 30 times by mass, more preferably 1 to 10 times by mass of the starting compound (I). Range.
The amount of n-hexylamine used can be in the range of 1 to 2 molar equivalents relative to the starting compound (I), preferably in the range of 1 to 1.5 molar equivalents, more preferably 1 It is the range of -1.1 molar equivalent.
Examples of the inert solvent when synthesizing the n-hexylamine salt (19) include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, and benzene, Ether solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane, ethyl acetate, isopropyl acetate, acetonitrile, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, dimethyl sulfoxide, water, a mixed solvent thereof or the like can be used.
The compound (19) can be obtained as a refined product by a method such as reslurry or recrystallization in addition to a refined product by crystallization, or can be obtained as an unpurified product.
工程(ii):
化合物(19)を不活性溶媒中、酸の水溶液にて処理して化合物(19)のカルボン酸遊離体を得ることができる。化合物(20)を不活性溶媒中、塩基の水溶液にて処理して化合物(20)のアミン遊離体を得ることができる。化合物(19)のカルボン酸遊離体と化合物(20)のアミン遊離体を不活性溶媒中、脱水縮合剤及び塩基の存在下反応させた後、不活性溶媒中、シュウ酸と塩を形成させて化合物(21)を得ることができる。
化合物(20)は、特許文献4に記載の方法で合成することができる。
化合物(19)のカルボン酸遊離体及び化合物(20)のアミン遊離体を得る時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトンまたはこれらの混合溶媒等を使用することができる。
酸の水溶液としては、例えば、塩酸、硫酸水溶液、クエン酸水溶液等を使用することができる。
塩基の水溶液としては、例えば、水酸化ナトリウム水溶液、水酸化カリウム水溶液、水酸化リチウム水溶液、炭酸ナトリウム水溶液、炭酸カリウム水溶液等を使用することができる。
脱水縮合反応時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシドまたはこれらの混合溶媒等を使用することができる。
脱水縮合剤としては、例えば、N,N’−ジシクロヘキシルカルボジイミド、N,N’−ジイソプロピルカルボジイミド、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩及び1−ヒドロキシベンゾトリアゾール一水和物、1,1’−カルボニルジイミダゾール、1−プロパンホスホン酸無水物等を使用することができる。
脱水縮合反応時の塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン等を使用することができる。
シュウ酸塩(21)を合成する時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリルまたはこれらの混合溶媒等を使用することができる。
化合物(19)のカルボン酸遊離体及び化合物(20)のアミン遊離体を得る時の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜50℃の範囲であり、より好ましくは15〜35℃の範囲である。
脱水縮合反応の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは10〜40℃の範囲である。
化合物(20)の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.6モル当量の範囲である。
脱水縮合剤の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.5モル当量の範囲である。
塩基の使用量は、原料の化合物(19)に対して1〜5モル当量の範囲で使用することができ、好ましくは1〜3モル当量の範囲であり、より好ましくは2〜2.5モル当量の範囲である。
シュウ酸の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(19)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(21)は、結晶化による精製品のほか、リスラリー、再結晶等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
Step (ii):
Compound (19) can be treated with an acid aqueous solution in an inert solvent to obtain a carboxylic acid free form of compound (19). Compound (20) can be treated with an aqueous base solution in an inert solvent to obtain the amine free form of compound (20). The carboxylic acid free form of compound (19) and the amine free form of compound (20) are reacted in the presence of a dehydration condensing agent and a base in an inert solvent, and then a salt is formed with oxalic acid in the inert solvent. Compound (21) can be obtained.
Compound (20) can be synthesized by the method described in Patent Document 4.
As the inert solvent for obtaining the carboxylic acid free form of compound (19) and the amine free form of compound (20), for example, halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, toluene, xylene , Hydrocarbon solvents such as benzene, ether solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane , Ethyl acetate, isopropyl acetate, methyl isobutyl ketone, or a mixed solvent thereof can be used.
As the acid aqueous solution, for example, hydrochloric acid, sulfuric acid aqueous solution, citric acid aqueous solution and the like can be used.
Examples of the aqueous base solution include sodium hydroxide aqueous solution, potassium hydroxide aqueous solution, lithium hydroxide aqueous solution, sodium carbonate aqueous solution, potassium carbonate aqueous solution and the like.
Examples of the inert solvent in the dehydration condensation reaction include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, and chlorobenzene, hydrocarbon solvents such as toluene, xylene, and benzene, tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydro Ether solvents such as pyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane, ethyl acetate, isopropyl acetate, methyl isobutyl ketone, acetonitrile, N, N- Dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, dimethyl sulfoxide, or a mixed solvent thereof can be used.
Examples of the dehydrating condensing agent include N, N′-dicyclohexylcarbodiimide, N, N′-diisopropylcarbodiimide, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride, and 1- (3-dimethylaminopropyl). -3-Ethylcarbodiimide hydrochloride and 1-hydroxybenzotriazole monohydrate, 1,1′-carbonyldiimidazole, 1-propanephosphonic anhydride and the like can be used.
As the base in the dehydration condensation reaction, for example, triethylamine, tri-n-butylamine, N, N-diisopropylethylamine, N-methylmorpholine and the like can be used.
As an inert solvent when synthesizing the oxalate (21), for example, halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, tetrahydrofuran, Ether solvents such as 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane, ethyl acetate, isopropyl acetate, acetone, 2- Butanone, acetonitrile, or a mixed solvent thereof can be used.
The temperature at which the carboxylic acid educt of compound (19) and the amine educt of compound (20) are obtained can usually be from 0 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 50 ° C. More preferably, it is the range of 15-35 degreeC.
The temperature of the dehydration condensation reaction is usually from 0 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 100 ° C., more preferably in the range of 10 to 40 ° C.
The amount of the compound (20) used can be in the range of 1 to 3 molar equivalents, preferably in the range of 1 to 2 molar equivalents, more preferably 1 to 1 with respect to the starting compound (19). .6 molar equivalent range.
The amount of the dehydrating condensing agent can be used in the range of 1 to 3 molar equivalents relative to the starting compound (19), preferably in the range of 1 to 2 molar equivalents, more preferably 1-1. The range is 5 molar equivalents.
The amount of the base used can be in the range of 1 to 5 molar equivalents relative to the raw material compound (19), preferably in the range of 1 to 3 molar equivalents, more preferably 2 to 2.5 mols. Equivalent range.
The amount of oxalic acid used can be in the range of 1 to 3 molar equivalents relative to the raw material compound (19), preferably in the range of 1 to 1.5 molar equivalents, more preferably 1 to 1. .1 molar equivalent range.
The solvent can be used in an amount of 1 to 100 times by mass, preferably 1 to 30 times by mass, more preferably 1 to 10 times by mass, relative to the starting compound (19). It is a range.
The compound (21) can be obtained as a refined product by a method such as reslurry or recrystallization in addition to a refined product by crystallization, or it can be obtained as an unrefined product.
工程(iii):
化合物(21)を不活性溶媒中、塩基の水溶液にて処理して化合物(21)のアミン遊離体を得ることができる。化合物(21)のアミン遊離体を溶媒中、塩基の存在下反応させることにより、化合物(XI)を得ることができる。
化合物(21)のアミン遊離体を得る時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトンまたはこれらの混合溶媒等を使用することができる。
塩基の水溶液としては、例えば、水酸化ナトリウム水溶液、水酸化カリウム水溶液、水酸化リチウム水溶液、炭酸ナトリウム水溶液、炭酸カリウム水溶液等を使用することができる。
化合物(XI)を得る時の溶媒としては、例えば、メタノール、エタノール等のアルコール系溶媒またはこれらアルコール系溶媒と水の混合溶媒等を使用することができる。
塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、ナトリウムメトキシド等を使用することができる。
脱アセチル化反応の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜80℃の範囲であり、より好ましくは10〜40℃の範囲である。
塩基の使用量は、原料の化合物(21)に対して0.1〜4モル当量の範囲で使用することができ、好ましくは0.5〜2モル当量の範囲であり、より好ましくは1〜1.6モル当量の範囲である。
溶媒の使用量は、原料の化合物(21)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(XI)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。
特許文献2に記載のように、化合物(XI)は非晶質の化合物であるが、化合物(XI)をエタノールまたはエタノールと混和する性質を有する有機溶媒とエタノールの混合液に溶解させた溶液からの結晶化により、化合物(XI)のエタノール溶媒和物を結晶として得ることができる。
Step (iii):
Compound (21) can be treated with an aqueous base solution in an inert solvent to obtain the amine free form of compound (21). Compound (XI) can be obtained by reacting the amine free form of compound (21) in a solvent in the presence of a base.
Examples of the inert solvent for obtaining the amine free form of the compound (21) include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, hydrocarbon solvents such as toluene, xylene, benzene, tetrahydrofuran, and the like. , 2-methyltetrahydrofuran, tetrahydropyran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, cyclopentyl methyl ether, 1,2-dimethoxyethane, diethoxymethane, and other ether solvents, ethyl acetate, isopropyl acetate, methyl isobutyl ketone Alternatively, a mixed solvent thereof or the like can be used.
Examples of the aqueous base solution include sodium hydroxide aqueous solution, potassium hydroxide aqueous solution, lithium hydroxide aqueous solution, sodium carbonate aqueous solution, potassium carbonate aqueous solution and the like.
As a solvent for obtaining the compound (XI), for example, an alcohol solvent such as methanol or ethanol or a mixed solvent of these alcohol solvent and water can be used.
Examples of the base include triethylamine, tri-n-butylamine, N, N-diisopropylethylamine, N-methylmorpholine, potassium carbonate, sodium carbonate, cesium carbonate, sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium methoxide. Etc. can be used.
The temperature of the deacetylation reaction can usually be from 0 ° C. to the boiling point of the solvent used, but is preferably in the range of 0 to 80 ° C., more preferably in the range of 10 to 40 ° C.
The amount of the base used can be 0.1 to 4 molar equivalents relative to the starting compound (21), preferably 0.5 to 2 molar equivalents, more preferably 1 to 4 molar equivalents. The range is 1.6 molar equivalents.
The amount of the solvent used can be used in the range of 1 to 100 times by mass relative to the raw material compound (21), preferably in the range of 1 to 30 times by mass, more preferably 1 to 10 times by mass. It is a range.
Compound (XI) can be obtained as a purified product by a method such as chromatography or an unpurified product.
As described in Patent Document 2, compound (XI) is an amorphous compound, but from a solution in which compound (XI) is dissolved in ethanol or an organic solvent having a property to be mixed with ethanol and ethanol. By crystallizing, an ethanol solvate of compound (XI) can be obtained as crystals.
以下に実施例を示し、本発明をさらに詳しく具体的に説明するが、本発明はこれらの記載により限定的に解釈されるものではない。また、本発明の範囲を逸脱しない範囲で変化させてもよい。
下記実施例における収率は、反応条件及び後処理・単離条件により影響を受けているものがあり、最適化された反応条件及び後処理・単離条件を選択することによってさらに高い収率にすることが可能である。
The present invention will be described in more detail below with reference to examples, but the present invention should not be construed as being limited by these descriptions. Moreover, you may change in the range which does not deviate from the range of this invention.
The yields in the following examples are affected by the reaction conditions and post-treatment / isolation conditions. By selecting optimized reaction conditions and post-treatment / isolation conditions, higher yields can be obtained. Is possible.
本実施例に記載した機器分析データは、以下の測定機器にて測定した。
融点:融点測定器 B-545(BUCHI)
核磁気共鳴分光分析(NMR):
JNM-ECA600(JEOL RESONANCE);1H 600 MHz、13C 150 MHz
JNM-ECA500(JEOL RESONANCE);1H 500 MHz、13C 125 MHz
質量分析(MS):
GCT(micromass);イオン化法 EI
LCMS-IT-TOF(島津製作所)、LCMS-2010EV(島津製作所);イオン化法 ESI/APCI
CHN元素分析:vario MICRO cube(elementar)
イオンクロマト分析:XS-100(三菱化学)
赤外分光分析(IR):Spectrum One(Perkin Elmer)
The instrumental analysis data described in this example was measured with the following measuring instrument.
Melting point: Melting point measuring instrument B-545 (BUCHI)
Nuclear magnetic resonance spectroscopy (NMR):
JNM-ECA600 (JEOL RESONANCE); 1 H 600 MHz, 13 C 150 MHz
JNM-ECA500 (JEOL RESONANCE); 1 H 500 MHz, 13 C 125 MHz
Mass spectrometry (MS):
GCT (micromass); ionization method EI
LCMS-IT-TOF (Shimadzu Corporation), LCMS-2010EV (Shimadzu Corporation); ionization method ESI / APCI
CHN elemental analysis: vario MICRO cube (elementar)
Ion chromatographic analysis: XS-100 (Mitsubishi Chemical)
Infrared spectroscopy (IR): Spectrum One (Perkin Elmer)
本明細書中で用いられている各略語を次に示す。
NMR:核磁気共鳴(nuclear magentic resonance)
DMSO−d6:ジメチルスルホキシド−d6(dimethylsulfoxide−d6)
s : シングレット(singlet)
d : ダブレット(doublet)
t : トリプレット(triplet)
q : クァルテット(quartet)
m : マルチプレット(multiplet)
dd: ダブレット オブ ダブレッツ(doublet of doublets)
td: トリプレット オブ ダブレッツ(triplet of doublets)
ddd: ダブレット オブ ダブレット オブ ダブレッツ(doublet of doublet of doublets)
J : スピン結合定数(coupling constant)
Hz : ヘルツ(Hertz)
MS:質量分析(mass spectrometry)
HRMS:高分解能質量分析(high resolution mass spectrometry)
EI:電子イオン化法(electron ionization)
ESI :エレクトロスプレーイオン化法(electrospray ionization)
APCI:大気圧化学イオン化法(atomospheric pressure chemical ionization)
mp:融点(melting point)
v/v :体積/体積
wt% :重量パーセント濃度
The abbreviations used in this specification are as follows.
NMR: nuclear magnetic resonance
DMSO-d 6: dimethylsulfoxide -d 6 (dimethylsulfoxide-d 6)
s: singlet
d: doublet
t: triplet
q: quartet
m: multiplet
dd: doublet of doublets
td: triplet of doublets
ddd: doublet of doublets of doublets (doublet of doublets of doublets)
J: Spinning constant (coupling constant)
Hz: Hertz
MS: Mass Spectrometry
HRMS: high resolution mass spectrometry
EI: electron ionization (electron ionization)
ESI: Electrospray ionization method
APCI: Atmospheric pressure chemical ionization
mp: melting point
v / v: volume / volume wt%: weight percent concentration
化合物の命名には、ACD/Name 2015 (Advanced Chemistry Development Inc.)等のソフトを使用している場合がある。 In naming compounds, software such as ACD / Name 2015 (Advanced Chemistry Development Inc.) may be used.
本明細書では、室温とは、特に断りが無い限り、20〜30℃を指す。 In this specification, room temperature refers to 20 to 30 ° C. unless otherwise specified.
式(I)で表される化合物は、下記実施例1〜9に示す方法によって得ることができた。
実施例1
2−(2−ブロモ−5−メトキシフェニル)プロパン−2−オール(2)の合成
The compound represented by the formula (I) could be obtained by the methods shown in Examples 1 to 9 below.
Example 1
Synthesis of 2- (2-bromo-5-methoxyphenyl) propan-2-ol (2)
mp : 99℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.74 ( 6H, s ), 2.69 ( 1H, s, exchangeable with D2O ), 3.80 ( 3H, s ), 6.65 ( 1H, dd, J = 8.7 and 3.2 Hz ), 7.26 ( 1H, d, J = 3.2 Hz ), 7.46 (1H, d, J = 8.7 Hz ).
MS (EI) : m/z 246 [(M+2)+], 244 (M+), 226 (base).
実施例2
1−ブロモ−4−メトキシ−2−(プロパン−2−イル)ベンゼン(3)の合成
mp: 99 ° C.
1 H NMR (600 MHz, CDCl 3 ): δ 1.74 (6H, s), 2.69 (1H, s, exchangeable with D 2 O), 3.80 (3H, s), 6.65 (1H, dd, J = 8.7 and 3.2 Hz), 7.26 (1H, d, J = 3.2 Hz), 7.46 (1H, d, J = 8.7 Hz).
MS (EI): m / z 246 [(M + 2) + ], 244 (M + ), 226 (base).
Example 2
Synthesis of 1-bromo-4-methoxy-2- (propan-2-yl) benzene (3)
1H NMR ( 600 MHz, CDCl3 ) : δ 1.23 ( 6H, d, J = 6.9 Hz ), 3.31 ( 1H, septet, J = 6.9 Hz ), 3.79 ( 3H, s ), 6.61 ( 1H, dd, J = 8.7 and 3.2 Hz ), 6.83 ( 1H, d, J = 3.2 Hz ), 7.41 (1H, d, J = 8.7 Hz ).
MS (EI) : m/z 230 [(M+2)+], 228 (M+), 213 (base).
実施例3
(4−ブロモフェニル)[4−メトキシ−2−(プロパン−2−イル)フェニル]メタノール(6)の合成
1 H NMR (600 MHz, CDCl 3 ): δ 1.23 (6H, d, J = 6.9 Hz), 3.31 (1H, septet, J = 6.9 Hz), 3.79 (3H, s), 6.61 (1H, dd, J = 8.7 and 3.2 Hz), 6.83 (1H, d, J = 3.2 Hz), 7.41 (1H, d, J = 8.7 Hz).
MS (EI): m / z 230 [(M + 2) + ], 228 (M + ), 213 (base).
Example 3
Synthesis of (4-bromophenyl) [4-methoxy-2- (propan-2-yl) phenyl] methanol (6)
窒素雰囲気下、マグネシウム0.099 g及びよう素0.011 gの混合物に1-ブロモ-4-メトキシ-2-(プロパン-2-イル)ベンゼン(3)の粗生成物1.25 gのテトラヒドロフラン(8 mL)溶液を加え、2.5時間加熱還流した。氷水浴にて冷却した反応液に4-ブロモベンズアルデヒド(5)0.756 gのテトラヒドロフラン(2 mL)溶液を3〜18℃にて2分間かけて滴下した。2℃にて1時間撹拌後、氷水浴をはずして室温まで昇温(〜22℃)しながら1.5時間撹拌した。氷水浴にて冷却した反応液に20 wt%塩化アンモニウム水溶液60 mL、引き続きトルエン20 mLを加えて有機層と水層に分液した。有機層を20 wt%塩化アンモニウム水溶液20 mL、引き続き10 wt%塩化ナトリウム水溶液20 mLにて洗浄後、有機層と水層に分液した。有機層を無水硫酸マグネシウムにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して黄色油状物の(4-ブロモフェニル)[4-メトキシ-2-(プロパン-2-イル)フェニル]メタノール(6)の粗生成物1.36 gを得た。
1H NMR ( 600 MHz, CDCl3 ) : δ 1.11 ( 3H, d, J = 6.8 Hz ), 1.18 ( 3H, d, J = 6.8 Hz ), 2.04 ( 1H, d, J = 3.7 Hz, exchangeable with D2O ), 3.21 ( 1H, septet, J = 6.8 Hz ), 3.81 ( 3H, s ), 6.05 ( 1H, d, J = 3.7 Hz ), 6.72 ( 1H, dd, J = 8.7 and 2.9 Hz ), 6.85 ( 1H, d, J = 2.9 Hz ), 7.18-7.23 ( 3H, m ), 7.43-7.46 ( 2H, m ).
MS (EI) : m/z 336 [(M+2)+] , 334 (M+), 222 (base).
実施例4
1−[(4−ブロモフェニル)メチル]−4−メトキシ−2−(プロパン−2−イル)ベンゼン(7)の合成
Under a nitrogen atmosphere, add a solution of crude 1.25 g of 1-bromo-4-methoxy-2- (propan-2-yl) benzene (3) in tetrahydrofuran (8 mL) to a mixture of 0.099 g of magnesium and 0.011 g of iodine. In addition, it was heated to reflux for 2.5 hours. To a reaction solution cooled in an ice-water bath, a solution of 4-bromobenzaldehyde (5) 0.756 g in tetrahydrofuran (2 mL) was added dropwise at 3-18 ° C. over 2 minutes. After stirring at 2 ° C for 1 hour, the ice-water bath was removed and the mixture was stirred for 1.5 hours while warming to room temperature (~ 22 ° C). To the reaction solution cooled in an ice water bath, 60 mL of a 20 wt% ammonium chloride aqueous solution and then 20 mL of toluene were added, and the mixture was separated into an organic layer and an aqueous layer. The organic layer was washed with 20 mL of 20 wt% aqueous ammonium chloride solution and then with 20 mL of 10 wt% aqueous sodium chloride solution, and then separated into an organic layer and an aqueous layer. The organic layer was dried over anhydrous magnesium sulfate, insolubles were filtered off, and the filtrate was concentrated under reduced pressure to give (4-bromophenyl) [4-methoxy-2- (propan-2-yl) phenyl as a yellow oil. ] 1.36 g of a crude product of methanol (6) was obtained.
1 H NMR (600 MHz, CDCl 3 ): δ 1.11 (3H, d, J = 6.8 Hz), 1.18 (3H, d, J = 6.8 Hz), 2.04 (1H, d, J = 3.7 Hz, exchangeable with D 2 O), 3.21 (1H, septet, J = 6.8 Hz), 3.81 (3H, s), 6.05 (1H, d, J = 3.7 Hz), 6.72 (1H, dd, J = 8.7 and 2.9 Hz), 6.85 (1H, d, J = 2.9 Hz), 7.18-7.23 (3H, m), 7.43-7.46 (2H, m).
MS (EI): m / z 336 [(M + 2) + ], 334 (M + ), 222 (base).
Example 4
Synthesis of 1-[(4-bromophenyl) methyl] -4-methoxy-2- (propan-2-yl) benzene (7)
窒素雰囲気下、(4-ブロモフェニル)[4-メトキシ-2-(プロパン-2-イル)フェニル]メタノール(6)の粗生成物1.36 g、トルエン20 mL及びtert-ブチルジメチルシラン0.953 gの混合物にトリフルオロメタンスルホン酸トリメチルシリルエステル1.2 mLを1〜7℃にて3分間かけて滴下した後、1℃にて1時間撹拌した。反応液に15 wt%炭酸ナトリウム水溶液20 mLを1〜9℃にて8分間かけて滴下後、22℃まで昇温しながら70分間撹拌した。有機層と水層に分液し、有機層を10 wt%塩化ナトリウム水溶液20 mLにて洗浄した。分液した有機層を減圧濃縮し、黄色油状物の1-[(4-ブロモフェニル)メチル]-4-メトキシ-2-(プロパン-2-イル)ベンゼン(7)の粗生成物1.26 gを得た。
1H NMR ( 600 MHz, CDCl3 ) : δ 1.11 ( 6H, d, J = 6.8 Hz ), 3.01 ( 1H, septet, J = 6.8 Hz ), 3.80 ( 3H, s ), 3.93 ( 2H, s ), 6.69 ( 1H, dd, J = 8.3 and 2.5 Hz ), 6.85 ( 1H, d, J = 2.5 Hz ), 6.94-6.98 ( 2H, m ), 7.00 (1H, d, J = 8.3 Hz ), 7.34-7.38 ( 2H, m ).
MS (EI) : m/z 320 [(M+2)+], 318 (M+), 275 (base).
実施例5
1−[(4−ブロモフェニル)メチル]−5−ヨード−4−メトキシ−2−(プロパン−2−イル)ベンゼン(8)の合成
A mixture of 1.36 g of crude product of (4-bromophenyl) [4-methoxy-2- (propan-2-yl) phenyl] methanol (6), 20 mL of toluene and 0.953 g of tert-butyldimethylsilane under nitrogen atmosphere To the mixture, 1.2 mL of trimethylsilyl trifluoromethanesulfonate was added dropwise at 1-7 ° C. over 3 minutes, followed by stirring at 1 ° C. for 1 hour. To the reaction solution, 20 mL of a 15 wt% aqueous sodium carbonate solution was added dropwise at 1-9 ° C. over 8 minutes, followed by stirring for 70 minutes while raising the temperature to 22 ° C. The organic layer and the aqueous layer were separated, and the organic layer was washed with 20 mL of a 10 wt% sodium chloride aqueous solution. The separated organic layer was concentrated under reduced pressure, and 1.26 g of a crude product of 1-[(4-bromophenyl) methyl] -4-methoxy-2- (propan-2-yl) benzene (7) as a yellow oil was obtained. Obtained.
1 H NMR (600 MHz, CDCl 3 ): δ 1.11 (6H, d, J = 6.8 Hz), 3.01 (1H, septet, J = 6.8 Hz), 3.80 (3H, s), 3.93 (2H, s), 6.69 (1H, dd, J = 8.3 and 2.5 Hz), 6.85 (1H, d, J = 2.5 Hz), 6.94-6.98 (2H, m), 7.00 (1H, d, J = 8.3 Hz), 7.34-7.38 (2H, m).
MS (EI): m / z 320 [(M + 2) + ], 318 (M + ), 275 (base).
Example 5
Synthesis of 1-[(4-bromophenyl) methyl] -5-iodo-4-methoxy-2- (propan-2-yl) benzene (8)
窒素雰囲気下、1-[(4-ブロモフェニル)メチル]-4-メトキシ-2-(プロパン-2-イル)ベンゼン(7)の粗生成物1.26 gの酢酸(10 mL)溶液によう素酸カリウム水溶液(KIO3 0.281 g / H2O 5 mL)及びよう素0.821 gを加えて49〜51℃にて17時間撹拌した。反応液に酢酸10 mLを加えて52℃にて6.5時間撹拌した。反応液を炭酸カリウム水溶液(K2CO3 30.1 g / H2O 60 mL)に13〜31℃にて24分間かけて滴下後、トルエン40 mLを加えて1時間撹拌した。水10 mLを加えた後、有機層と水層に分液した。有機層をチオ硫酸ナトリウム水溶液(Na2S2O3 8.01 g / H2O 40 mL)、引き続き水40 mLにて順次洗浄した。有機層を減圧濃縮して得られた黄色油状物1.51 gにメタノール6 mLを加え、室温にて61.5時間撹拌した後、氷水浴にて冷却しながら3.5時間撹拌した。析出した固体をろ過、冷メタノール3 mLにて洗浄後、減圧乾燥して白色固体の1-[(4-ブロモフェニル)メチル]-5-ヨード-4-メトキシ-2-(プロパン-2-イル)ベンゼン(8)0.417 g[化合物(2)からの通算換算収率23.0%]を得た。
mp : 93℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.11 ( 6H, d, J = 6.8 Hz ), 3.00 ( 1H, septet, J = 6.8 Hz ), 3.88 ( 5H, s ), 6.74 ( 1H, s ), 6.93-6.97 ( 2H, m ), 7.36-7.40 ( 2H, m ), 7.49 ( 1H, s ).
MS (EI) : m/z 446 [(M+2)+], 444 (M+, base).
実施例6
1−C−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−2,3,4,6−テトラキス−O−(トリメチルシリル)−D−グルコピラノース(11)
Iodic acid was added to a solution of 1.26 g of the crude product of 1-[(4-bromophenyl) methyl] -4-methoxy-2- (propan-2-yl) benzene (7) in acetic acid (10 mL) under a nitrogen atmosphere. A potassium aqueous solution (KIO 3 0.281 g / H 2 O 5 mL) and iodine 0.821 g were added, and the mixture was stirred at 49 to 51 ° C. for 17 hours. 10 mL of acetic acid was added to the reaction solution, and the mixture was stirred at 52 ° C. for 6.5 hours. The reaction solution was added dropwise to an aqueous potassium carbonate solution (K 2 CO 3 30.1 g / H 2 O 60 mL) at 13 to 31 ° C. over 24 minutes, and then 40 mL of toluene was added and stirred for 1 hour. After adding 10 mL of water, it was separated into an organic layer and an aqueous layer. The organic layer was washed successively with an aqueous sodium thiosulfate solution (Na 2 S 2 O 3 8.01 g / H 2 O 40 mL) and subsequently with 40 mL of water. Methanol 6 mL was added to 1.51 g of the yellow oil obtained by concentrating the organic layer under reduced pressure, and the mixture was stirred at room temperature for 61.5 hours and then stirred for 3.5 hours while cooling in an ice-water bath. The precipitated solid was filtered, washed with 3 mL of cold methanol, and dried under reduced pressure to give 1-[(4-bromophenyl) methyl] -5-iodo-4-methoxy-2- (propan-2-yl) as a white solid. ) 0.417 g of benzene (8) [23.0% of total conversion yield from compound (2)] was obtained.
mp: 93 ° C.
1 H NMR (600 MHz, CDCl 3 ): δ 1.11 (6H, d, J = 6.8 Hz), 3.00 (1H, septet, J = 6.8 Hz), 3.88 (5H, s), 6.74 (1H, s), 6.93-6.97 (2H, m), 7.36-7.40 (2H, m), 7.49 (1H, s).
MS (EI): m / z 446 [(M + 2) + ], 444 (M + , base).
Example 6
1-C- {5-[(4-Bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -2,3,4,6-tetrakis-O- (trimethylsilyl) -D -Glucopyranose (11)
アルゴン雰囲気下、1-[(4-ブロモフェニル)メチル]-5-ヨード-4-メトキシ-2-(プロパン-2-イル)ベンゼン(8)2.00 gにテトラヒドロフラン8.0 mLを加えて溶解し、塩化イソプロピルマグネシウム−塩化リチウム錯体のテトラヒドロフラン溶液(1.3 mol / L)3.30 mLを−21〜−18℃にて10分間かけて滴下し、−21℃にて1時間15分間撹拌した。反応液に塩化イソプロピルマグネシウム−塩化リチウム錯体のテトラヒドロフラン溶液(1.3 mol / L)0.15 mLを−21〜−20℃にて1分間かけて滴下し、−21℃にて42分間撹拌した。反応液に塩化イソプロピルマグネシウム−塩化リチウム錯体のテトラヒドロフラン溶液(1.3 mol / L)0.20 mLを−21℃にて2分間かけて滴下し、−21℃にて50分間撹拌した。反応液に2,3,4,6-テトラキス-O-(トリメチルシリル)-D-グルコノ-1,5-ラクトン(10)(97.9 wt%)2.36 gのテトラヒドロフラン(8.0 mL)溶液を−21〜−18℃にて10分間かけて滴下し、−10℃に昇温して1時間撹拌後、0℃に昇温して4時間撹拌した。反応液を飽和塩化アンモニウム水溶液30.0 mL及び水30.0 mLの混合液に加えた後、酢酸エチル100 mLを加えて有機層と水層に分液した。有機層を飽和塩化ナトリウム水溶液30.0 mLにて洗浄後、無水硫酸マグネシウムにて乾燥した。不溶物をろ別し、ろ液を減圧濃縮して淡黄色油状物の1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(トリメチルシリル)-D-グルコピラノース(11)の粗生成物3.80 gを得た。
MS (ESI/APCI dual source) : m/z 809 {[(M+2)+Na]+}, 807 [(M+Na)+].
実施例7
メチル 1−C−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−α−D−グルコピラノシド(12)の合成
Under an argon atmosphere, 1-[(4-bromophenyl) methyl] -5-iodo-4-methoxy-2- (propan-2-yl) benzene (8) (2.00 g) was dissolved in 8.0 mL of tetrahydrofuran, and the solution was chlorinated. 3.30 mL of a tetrahydrofuran solution (1.3 mol / L) of isopropylmagnesium-lithium chloride complex was added dropwise at −21 to −18 ° C. over 10 minutes, and the mixture was stirred at −21 ° C. for 1 hour and 15 minutes. To the reaction solution, 0.15 mL of a isopropylmagnesium chloride-lithium chloride complex tetrahydrofuran solution (1.3 mol / L) was added dropwise at -21 to -20 ° C over 1 minute, and the mixture was stirred at -21 ° C for 42 minutes. To the reaction solution, 0.20 mL of a tetrahydrofuran solution (1.3 mol / L) of isopropylmagnesium chloride-lithium chloride complex was added dropwise at −21 ° C. over 2 minutes and stirred at −21 ° C. for 50 minutes. To the reaction solution, a solution of 2.36 g of 2,3,4,6-tetrakis-O- (trimethylsilyl) -D-glucono-1,5-lactone (10) (97.9 wt%) in tetrahydrofuran (8.0 mL) was added at −21 to − The mixture was added dropwise at 18 ° C. over 10 minutes, heated to −10 ° C. and stirred for 1 hour, then heated to 0 ° C. and stirred for 4 hours. The reaction solution was added to a mixed solution of 30.0 mL of saturated aqueous ammonium chloride and 30.0 mL of water, and then 100 mL of ethyl acetate was added to separate the organic layer and the aqueous layer. The organic layer was washed with 30.0 mL of saturated aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The insoluble material was filtered off, and the filtrate was concentrated under reduced pressure to give 1-C- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl as a pale yellow oil. } -2,3,4,6-tetrakis-O- (trimethylsilyl) -D-glucopyranose (11) crude product 3.80 g was obtained.
MS (ESI / APCI dual source): m / z 809 {[(M + 2) + Na] + }, 807 [(M + Na) + ].
Example 7
Synthesis of methyl 1-C- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -α-D-glucopyranoside (12)
1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(トリメチルシリル)-D-グルコピラノース(11)の粗生成物3.80 gにメタンスルホン酸0.117 gのメタノール(10.0 mL)溶液を加え、アルゴン雰囲気下、24〜25℃にて3時間撹拌した。氷水浴にて冷却した反応液に飽和炭酸水素ナトリウム水溶液10.0 mLを4〜16℃にて2分間かけて滴下後、水15.0 mL及び酢酸エチル70.0 mLを加え、有機層と水層に分液した。水層を酢酸エチル30.0 mLにて抽出した有機層を先の有機層と併せて飽和塩化ナトリウム水溶液30.0 mLにて洗浄した。有機層を無水硫酸マグネシウムにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して淡褐色固体のメチル 1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(12)の粗生成物1.95 gを得た。化合物(12)の粗生成物1.95 gに酢酸エチル4.00 mL及びトルエン12.0 mLを加えて溶解させた溶液に25℃にて化合物(12)の種晶0.5 mgを添加し、25℃にて30分間撹拌した。氷水浴にて冷却し、0〜16℃にて19時間、引き続き0℃にて4時間撹拌した。析出した固体をろ過、トルエン−酢酸エチル混合溶媒(3/1, v/v)6.0 mLにて洗浄後、減圧乾燥して白色固体のメチル 1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(12)0.697 g[化合物(8)からの通算収率30.3%]を得た。
mp : 160℃.
1H NMR ( 600 MHz, DMSO-d6 ) : δ 1.01 ( 3H, d, J = 6.7 Hz ), 1.09 ( 3H, d, J = 6.7 Hz ), 2.99 ( 1H, septet, J = 6.7 Hz ), 3.08 ( 3H, s ), 3.17 ( 1H, td, J = 9.7 and 5.4 Hz ), 3.28-3.33 ( 1H, m ), 3.37 ( 1H, ddd, J = 9.7, 6.0 and 1.9 Hz ), 3.46-3.54 ( 2H, m ), 3.69-3.74 ( 1H, m ), 3.76 ( 3H, s ), 3.89, 3.99 ( 2H, AB quartet, J = 15.9 Hz ), 4.21 ( 1H, d, J = 7.4 Hz, exchangeable with D2O ), 4.27 ( 1H, t, J = 6.0 Hz, exchangeable with D2O ), 4.65 ( 1H, d, J = 5.0 Hz, exchangeable with D2O ), 4.91 ( 1H, d, J = 5.4 Hz, exchangeable with D2O ), 6.90 ( 1H, s ), 7.02-7.07 ( 2H, m ), 7.16 ( 1H, s ), 7.42-7.47 ( 2H, m ).
13C NMR ( 125 MHz, DMSO-d6 ) : δ 23.5, 28.7, 36.9, 48.6, 56.3, 61.3, 70.1, 74.3, 74.4, 75.7, 79.2, 101.5, 110.6, 118.7, 124.4, 127.9, 130.4, 131.0, 131.7, 141.2, 147.7, 156.5.
MS (ESI/APCI dual source) : ポジティブモード;m/z 535 {[(M+2)+Na]+}, 533 [(M+Na)+], ネガティブモード;m/z 511 {[(M+2)-H]−}, 509 [(M-H)−].
実施例8
メチル 2,3,4,6−テトラ−O−アセチル−1−C−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−α−D−グルコピラノシド(13)の合成
1-C- {5-[(4-Bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -2,3,4,6-tetrakis-O- (trimethylsilyl) -D -A solution of 0.117 g of methanesulfonic acid in methanol (10.0 mL) was added to 3.80 g of the crude product of glucopyranose (11), and the mixture was stirred at 24 to 25 ° C for 3 hours under an argon atmosphere. To the reaction solution cooled in an ice-water bath, 10.0 mL of a saturated aqueous sodium bicarbonate solution was added dropwise at 4-16 ° C. over 2 minutes, and then 15.0 mL of water and 70.0 mL of ethyl acetate were added to separate the organic layer and the aqueous layer. . The aqueous layer was extracted with 30.0 mL of ethyl acetate, and the organic layer was combined with the previous organic layer and washed with 30.0 mL of a saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous magnesium sulfate, insolubles were filtered off, and the filtrate was concentrated under reduced pressure to give methyl 1-C- {5-[(4-bromophenyl) methyl] -2-methoxy as a light brown solid. 1.95 g of a crude product of -4- (propan-2-yl) phenyl} -α-D-glucopyranoside (12) was obtained. To a solution obtained by dissolving 4.00 mL of ethyl acetate and 12.0 mL of toluene in 1.95 g of the crude product of compound (12), 0.5 mg of the seed crystal of compound (12) was added at 25 ° C., and 30 minutes at 25 ° C. Stir. The mixture was cooled in an ice-water bath and stirred at 0 to 16 ° C. for 19 hours and subsequently at 0 ° C. for 4 hours. The precipitated solid was filtered, washed with 6.0 mL of a toluene-ethyl acetate mixed solvent (3/1, v / v), and then dried under reduced pressure to obtain methyl 1-C- {5-[(4-bromophenyl) as a white solid. 0.697 g of [methyl] -2-methoxy-4- (propan-2-yl) phenyl} -α-D-glucopyranoside (12) [total yield 30.3% from compound (8)] was obtained.
mp: 160 ° C.
1 H NMR (600 MHz, DMSO-d 6 ): δ 1.01 (3H, d, J = 6.7 Hz), 1.09 (3H, d, J = 6.7 Hz), 2.99 (1H, septet, J = 6.7 Hz), 3.08 (3H, s), 3.17 (1H, td, J = 9.7 and 5.4 Hz), 3.28-3.33 (1H, m), 3.37 (1H, ddd, J = 9.7, 6.0 and 1.9 Hz), 3.46-3.54 ( 2H, m), 3.69-3.74 (1H, m), 3.76 (3H, s), 3.89, 3.99 (2H, AB quartet, J = 15.9 Hz), 4.21 (1H, d, J = 7.4 Hz, exchangeable with D 2 O), 4.27 (1H, t, J = 6.0 Hz, exchangeable with D 2 O), 4.65 (1H, d, J = 5.0 Hz, exchangeable with D 2 O), 4.91 (1H, d, J = 5.4 Hz , exchangeable with D 2 O), 6.90 (1H, s), 7.02-7.07 (2H, m), 7.16 (1H, s), 7.42-7.47 (2H, m).
13 C NMR (125 MHz, DMSO-d 6 ): δ 23.5, 28.7, 36.9, 48.6, 56.3, 61.3, 70.1, 74.3, 74.4, 75.7, 79.2, 101.5, 110.6, 118.7, 124.4, 127.9, 130.4, 131.0, 131.7, 141.2, 147.7, 156.5.
MS (ESI / APCI dual source): positive mode; m / z 535 {[(M + 2) + Na] + }, 533 [(M + Na) + ], negative mode; m / z 511 {[(M +2) -H] − }, 509 [(MH) − ].
Example 8
Methyl 2,3,4,6-tetra-O-acetyl-1-C- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -α- Synthesis of D-glucopyranoside (13)
アルゴン雰囲気下、メチル 1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(12)1.00 gをピリジン5.00 mLに溶解した溶液に4-ジメチルアミノピリジン(DMAP)0.024 g及び無水酢酸1.21 gを加え、21〜38℃にて4.5時間撹拌した。冷却した反応液に水5.00 mLを1〜9℃にて8分間かけて滴下し、1℃にて20分間撹拌した。酢酸エチル60.0 mL及び10 wt%塩酸30.0 mLを加え、有機層と水層に分液した。有機層を飽和炭酸水素ナトリウム水溶液30.0 mL及び飽和塩化ナトリウム水溶液20.0 mLにて順次洗浄し、無水硫酸マグネシウムにて乾燥した。不溶物をろ別し、ろ液を減圧濃縮後、減圧乾燥して白色アモルファス固体のメチル 2,3,4,6-テトラ-O-アセチル-1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(13)の粗生成物1.30 gを得た。
1H NMR ( 500 MHz, CDCl3 ) : δ 1.08 ( 3H, d, J = 6.8 Hz ), 1.14 ( 3H, d, J = 6.8 Hz ), 1.77 ( 3H, s ), 1.97 ( 3H, s ), 2.06 ( 3H, s ), 2.08 ( 3H, s ), 2.98 ( 1H, septet, J = 6.8 Hz ), 3.30 ( 3H, s ), 3.88 ( 3H, s ), 3.92 ( 2H, s ), 4.02 ( 1H, ddd, J = 10.3, 4.7 and 2.4 Hz ), 4.20 ( 1H, dd, J = 12.3 and 2.4 Hz ), 4.30 ( 1H, dd, J = 12.3 and 4.7 Hz ), 5.25 ( 1H, dd, J = 10.3 and 9.8 Hz ), 5.40 ( 1H, d, J = 9.8 Hz ), 5.57 ( 1H, t, J = 9.8 Hz ), 6.82 ( 1H, s ), 6.86-6.91 ( 2H, m ), 7.09 ( 1H, s ), 7.33-7.37 ( 2H, m ).
13C NMR ( 125 MHz, CDCl3 ) : δ 20.3, 20.6, 20.67, 20.74, 23.4, 23.8, 29.4, 37.4, 49.9, 56.0, 62.4, 68.9, 69.0, 71.5, 72.6, 101.1, 110.0, 119.6, 120.6, 128.0, 129.9, 131.30, 131.33, 140.6, 149.7, 156.9, 169.0, 169.7, 170.0, 170.6.
MS (ESI/APCI dual source) : ポジティブモード;m/z 703 {[(M+2)+Na]+}, 701 [(M+Na)+].
実施例9
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(I)の合成
Under an argon atmosphere, 1.00 g of methyl 1-C- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -α-D-glucopyranoside (12) is pyridine To the solution dissolved in 5.00 mL, 0.024 g of 4-dimethylaminopyridine (DMAP) and 1.21 g of acetic anhydride were added, and the mixture was stirred at 21 to 38 ° C. for 4.5 hours. To the cooled reaction solution, 5.00 mL of water was added dropwise at 1-9 ° C. over 8 minutes, and the mixture was stirred at 1 ° C. for 20 minutes. Ethyl acetate 60.0 mL and 10 wt% hydrochloric acid 30.0 mL were added, and the mixture was separated into an organic layer and an aqueous layer. The organic layer was washed successively with 30.0 mL of saturated aqueous sodium hydrogen carbonate solution and 20.0 mL of saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. Insoluble matter was filtered off, and the filtrate was concentrated under reduced pressure and dried under reduced pressure to obtain methyl 2,3,4,6-tetra-O-acetyl-1-C- {5-[(4-bromophenyl) as a white amorphous solid. ) Methyl] -2-methoxy-4- (propan-2-yl) phenyl} -α-D-glucopyranoside (13) 1.30 g of crude product was obtained.
1 H NMR (500 MHz, CDCl 3 ): δ 1.08 (3H, d, J = 6.8 Hz), 1.14 (3H, d, J = 6.8 Hz), 1.77 (3H, s), 1.97 (3H, s), 2.06 (3H, s), 2.08 (3H, s), 2.98 (1H, septet, J = 6.8 Hz), 3.30 (3H, s), 3.88 (3H, s), 3.92 (2H, s), 4.02 (1H , ddd, J = 10.3, 4.7 and 2.4 Hz), 4.20 (1H, dd, J = 12.3 and 2.4 Hz), 4.30 (1H, dd, J = 12.3 and 4.7 Hz), 5.25 (1H, dd, J = 10.3 and 9.8 Hz), 5.40 (1H, d, J = 9.8 Hz), 5.57 (1H, t, J = 9.8 Hz), 6.82 (1H, s), 6.86-6.91 (2H, m), 7.09 (1H, s ), 7.33-7.37 (2H, m).
13 C NMR (125 MHz, CDCl 3 ): δ 20.3, 20.6, 20.67, 20.74, 23.4, 23.8, 29.4, 37.4, 49.9, 56.0, 62.4, 68.9, 69.0, 71.5, 72.6, 101.1, 110.0, 119.6, 120.6, 128.0, 129.9, 131.30, 131.33, 140.6, 149.7, 156.9, 169.0, 169.7, 170.0, 170.6.
MS (ESI / APCI dual source): Positive mode; m / z 703 {[(M + 2) + Na] + }, 701 [(M + Na) + ].
Example 9
(1S) -2,3,4,6-Tetra-O-acetyl-1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propane-2- Yl) phenyl} -D-glucitol (I) synthesis
mp : 127℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.04 ( 3H, d, J = 6.8 Hz ), 1.09 ( 3H, d, J = 6.8 Hz ), 1.76 ( 3H, s ), 2.00 ( 3H, s ), 2.05 ( 3H, s ), 2.06 ( 3H, s ), 2.99 ( 1H, septet, J = 6.8 Hz ), 3.80-3.86 ( 1H, m ), 3.84 ( 3H, s ), 3.89, 3.93 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.4 and 2.5 Hz ), 4.25 ( 1H, dd, J = 12.4 and 4.5 Hz ), 4.88 ( 1H, d, J = 9.1 Hz ), 5.21 ( 1H, t, J = 9.7 Hz ), 5.31-5.40 ( 2H, m ), 6.77 ( 1H, s ), 6.90-6.95 ( 2H, m ), 7.11 ( 1H, s ), 7.33-7.38 ( 2H, m ).
MS (ESI/APCI dual source) : ポジティブモード;m/z 673 {[(M+2)+Na]+}, 671 [(M+Na)+].
Anal.Calcd for C31H37BrO10 : C, 57.32 ; H, 5.74 ; Br, 12.30. Found : C, 57.32 ; H, 5.72 ; Br, 12.26. IR (KBr) cm-1 : 2966, 1747, 1252, 1224, 1042.
mp: 127 ° C.
1 H NMR (600 MHz, CDCl 3 ): δ 1.04 (3H, d, J = 6.8 Hz), 1.09 (3H, d, J = 6.8 Hz), 1.76 (3H, s), 2.00 (3H, s), 2.05 (3H, s), 2.06 (3H, s), 2.99 (1H, septet, J = 6.8 Hz), 3.80-3.86 (1H, m), 3.84 (3H, s), 3.89, 3.93 (2H, AB quartet , J = 16.1 Hz), 4.13 (1H, dd, J = 12.4 and 2.5 Hz), 4.25 (1H, dd, J = 12.4 and 4.5 Hz), 4.88 (1H, d, J = 9.1 Hz), 5.21 (1H , t, J = 9.7 Hz), 5.31-5.40 (2H, m), 6.77 (1H, s), 6.90-6.95 (2H, m), 7.11 (1H, s), 7.33-7.38 (2H, m).
MS (ESI / APCI dual source): Positive mode; m / z 673 {[(M + 2) + Na] + }, 671 [(M + Na) + ].
Anal.Calcd for C 31 H 37 BrO 10 : C, 57.32; H, 5.74; Br, 12.30. Found: C, 57.32; H, 5.72; Br, 12.26. IR (KBr) cm -1 : 2966, 1747, 1252 , 1224, 1042.
また、式(I)で表される化合物は、下記実施例10〜12に示す方法によっても得ることができた。
実施例10
(1S)−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−2,3,4,6−テトラキス−O−(2,2−ジメチルプロパノイル)−D−グルシトール(16)の合成
Moreover, the compound represented by Formula (I) was able to be obtained also by the method shown in the following Examples 10-12.
Example 10
(1S) -1,5-Anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -2,3,4,6-tetrakis Synthesis of —O- (2,2-dimethylpropanoyl) -D-glucitol (16)
アルゴン雰囲気下、臭化亜鉛1.42 gにトルエン10.0 mLを加えた懸濁液にn-ブチルリチウム(2.65 M ヘキサン溶液)4.50 mLを23〜24℃にて2分間かけて滴下し、24〜25℃にて2時間撹拌した。この懸濁液に1-[(4-ブロモフェニル)メチル]-5-ヨード-4-メトキシ-2-(プロパン-2-イル)ベンゼン(8)5.00 gのトルエン20.0 mL溶液を1〜4℃にて20分間かけて滴下した。0℃にて1時間撹拌後、ジブチルエーテル4.00 mLを0〜4℃にて添加して26℃まで昇温しながら、1時間45分間撹拌した。この懸濁液に2,3,4,6-テトラキス-O-(2,2-ジメチルプロパノイル)-α-D-グルコピラノシルブロミド(15)6.54 gのトルエン15.0 mL溶液を25〜26℃にて3分間かけて滴下後、95〜98℃にて4時間撹拌した。反応混合物を26℃まで冷却後、20 wt%塩化アンモニウム水溶液50.0 gを加えて24℃にて15分間撹拌した。トルエン10.0 mLを加えて有機層と水層に分液した。有機層を水25.3 gにて洗浄後、減圧濃縮して褐色油状物の(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(2,2-ジメチルプロパノイル)-D-グルシトール(16)の粗生成物11.0 gを得た。
1H NMR ( 500 MHz, 50℃, acetone-d6 ):δ 0.86 ( 9H, s ), 0.96 ( 3H, d, J = 6.9 Hz ), 1.12 ( 9H, s ), 1.14 ( 3H, d, J = 6.9 Hz ), 1.18 ( 9H, s ), 1.19 ( 9H, s ), 3.07 ( 1H, septet, J = 6.9 Hz ), 3.86 ( 3H, s ), 3.92, 4.01 ( 2H, AB quartet, J = 15.9 Hz ), 4.04 ( 1H, ddd, J = 9.5, 3.8 and 1.9 Hz ), 4.12 ( 1H, dd, J = 12.4 and 3.8 Hz ), 4.23 ( 1H, dd, J = 12.4 and 1.9 Hz ), 5.07 ( 1H, d, J = 9.5 Hz ), 5.32 ( 1H, t, J = 9.5 Hz ), 5.40 ( 1H, t, J = 9.5 Hz ), 5.43 ( 1H, t, J = 9.5 Hz ), 6.90 ( 1H, s ), 7.06-7.11 ( 2H, m ), 7.26 ( 1H, s ), 7.37-7.42 ( 2H, m ).
13C NMR ( 125 MHz, 50℃, acetone-d6 ):δ 23.99, 24.02, 27.3, 27.59, 27.62, 27.8, 30.3, 38.6, 39.3, 39.45, 39.54, 39.6, 56.3, 62.9, 69.5, 73.1, 74.3, 75.2, 77.6, 109.5, 120.1, 123.4, 129.9, 131.7, 132.2, 132.4, 142.3, 150.3, 158.1, 176.9, 177.1, 177.7, 178.1.
MS (ESI/APCI dual source):ポジティブモード;m/z 841 {[(M+2)+Na]+}, 839 [(M+Na)+].
HRMS (ESI/APCI dual source) : ポジティブモード;m/z calcd for C43H61BrNaO10 [(M+Na)+] 839.3340, found :839.3321.
IR (KBr) cm-1 : 2972, 2874, 1744, 1482, 1282, 1174, 1141.
実施例11
(1S)−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(17)の合成
Under argon atmosphere, 4.50 mL of n-butyllithium (2.65 M hexane solution) was added dropwise to a suspension of 1.42 g of zinc bromide and 10.0 mL of toluene at 23-24 ° C over 2 minutes. For 2 hours. To this suspension was added a solution of 10.0 [1-((4-bromophenyl) methyl] -5-iodo-4-methoxy-2- (propan-2-yl) benzene (8) in 20.0 mL of toluene at 1 to 4 ° C. At 20 minutes. After stirring at 0 ° C. for 1 hour, 4.00 mL of dibutyl ether was added at 0 to 4 ° C., and the mixture was stirred for 1 hour and 45 minutes while raising the temperature to 26 ° C. To this suspension, a solution of 6.54 g of 2,4,6-tetrakis-O- (2,2-dimethylpropanoyl) -α-D-glucopyranosyl bromide (15) in 15.0 mL of toluene After dropwise addition at 3 ° C. over 3 minutes, the mixture was stirred at 95 to 98 ° C. for 4 hours. After cooling the reaction mixture to 26 ° C, 50.0 g of a 20 wt% aqueous ammonium chloride solution was added and stirred at 24 ° C for 15 minutes. Toluene 10.0 mL was added and liquid-separated into the organic layer and the water layer. The organic layer was washed with 25.3 g of water and then concentrated under reduced pressure to give (1S) -1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- 11.0 g of a crude product of (propan-2-yl) phenyl} -2,3,4,6-tetrakis-O- (2,2-dimethylpropanoyl) -D-glucitol (16) was obtained.
1 H NMR (500 MHz, 50 ° C, acetone-d 6 ): δ 0.86 (9H, s), 0.96 (3H, d, J = 6.9 Hz), 1.12 (9H, s), 1.14 (3H, d, J = 6.9 Hz), 1.18 (9H, s), 1.19 (9H, s), 3.07 (1H, septet, J = 6.9 Hz), 3.86 (3H, s), 3.92, 4.01 (2H, AB quartet, J = 15.9 Hz), 4.04 (1H, ddd, J = 9.5, 3.8 and 1.9 Hz), 4.12 (1H, dd, J = 12.4 and 3.8 Hz), 4.23 (1H, dd, J = 12.4 and 1.9 Hz), 5.07 (1H , d, J = 9.5 Hz), 5.32 (1H, t, J = 9.5 Hz), 5.40 (1H, t, J = 9.5 Hz), 5.43 (1H, t, J = 9.5 Hz), 6.90 (1H, s ), 7.06-7.11 (2H, m), 7.26 (1H, s), 7.37-7.42 (2H, m).
13 C NMR (125 MHz, 50 ° C., acetone-d 6 ): δ 23.99, 24.02, 27.3, 27.59, 27.62, 27.8, 30.3, 38.6, 39.3, 39.45, 39.54, 39.6, 56.3, 62.9, 69.5, 73.1, 74.3 75.2, 77.6, 109.5, 120.1, 123.4, 129.9, 131.7, 132.2, 132.4, 142.3, 150.3, 158.1, 176.9, 177.1, 177.7, 178.1.
MS (ESI / APCI dual source): positive mode; m / z 841 {[(M + 2) + Na] + }, 839 [(M + Na) + ].
HRMS (ESI / APCI dual source): Positive mode; m / z calcd for C 43 H 61 BrNaO 10 [(M + Na) + ] 839.3340, found: 839.3321.
IR (KBr) cm -1 : 2972, 2874, 1744, 1482, 1282, 1174, 1141.
Example 11
Synthesis of (1S) -1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol (17)
アルゴン雰囲気下、(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(2,2-ジメチルプロパノイル)-D-グルシトール(16)の粗生成物11.0 g、メタノール25.0 mL及び28 wt%ナトリウムメトキシド/メタノール溶液2.17 gの混合物を2時間加熱還流した。室温まで冷却した反応液にメタノール4.00 mL及びヘプタン25.0 mLを加えて撹拌し、上層と下層に分液した。下層にメタノール2.00 mL及びヘプタン25.0 mLを加えて撹拌し、上層と下層に分液した。得られた下層にメタノール1.00 mL、硫酸(97%)1.14 g及び水1.14 gを加えて1.5時間加熱還流後、硫酸(97%)1.15 gを加えて1.5時間加熱還流した。反応液に28 wt%ナトリウムメトキシド/メタノール溶液6.50 gを2〜10℃にて加えて室温まで昇温した。反応混合物にセルロースパウダー3.08 gを添加後、ろ過して固形物をメタノール50.0 mLにて洗浄した。ろ液を減圧濃縮後、酢酸エチル25.0 mLを添加して減圧濃縮する操作を3回実施し、黒色油状物の(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(17)の粗生成物11.5 gを得た。
mp : 157℃.
1H NMR ( 500 MHz, DMSO-d6 ) : δ 1.02 ( 3H, d, J = 6.8 Hz ), 1.06 ( 3H, d, J = 6.8 Hz ), 2.98 ( 1H, septet, J = 6.8 Hz ), 3.09-3.20 ( 2H, m ), 3.27 ( 1H, td, J = 8.5 and 4.7 Hz ), 3.35-3.44 ( 2H, m ), 3.66 ( 1H, ddd, J = 11.8, 5.7 and 1.8 Hz ), 3.76 ( 3H, s ), 3.91, 3.94 ( 2H, AB quartet, J = 15.9 Hz ), 4.37 ( 1H, t, J = 5.7 Hz, exchangeable with D2O ), 4.44 ( 1H, d, J = 9.9 Hz ), 4.61 ( 1H, d, J = 5.7 Hz, exchangeable with D2O ), 4.87 ( 1H, d, J = 5.0 Hz, exchangeable with D2O ), 4.89 ( 1H, d, J = 4.7 Hz, exchangeable with D2O ), 6.84 ( 1H, s ), 7.03-7.09 ( 2H, m ), 7.13 ( 1H, s ), 7.41-7.46 ( 2H, m ).
13C NMR ( 125 MHz, DMSO-d6 ) :δ 23.5, 23.6, 28.8, 37.0, 55.7, 61.5, 70.6, 73.6, 74.2, 78.7, 81.5, 108.5, 118.6, 125.7, 128.1, 130.4, 130.5, 131.0, 141.2, 147.2, 156.7.
MS (ESI/APCI dual source):ポジティブモード;m/z 500 {[(M+2)+NH4]+}, 498 [(M+NH4)+].
MS (ESI/APCI dual source):ネガティブモード;m/z 527 {[(M+2)+HCO2]-}, 525 [(M+HCO2)-].
Anal.Calcd for C23H29BrO6 : C, 57.39 ; H, 6.07 ; Br, 16.60. Found : C, 57.14 ; H, 6.00 ; Br, 16.47.
IR (KBr) cm-1 : 3402, 2961, 2929, 1616, 1508, 1488, 1089, 1072, 1048, 1012.
実施例12
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(I)の合成
Under an argon atmosphere, (1S) -1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -2,3,4 , 6-Tetrakis-O- (2,2-dimethylpropanoyl) -D-glucitol (16) crude product 11.0 g, 25.0 mL of methanol and 2.17 g of 28 wt% sodium methoxide / methanol solution for 2 hours Heated to reflux. To the reaction solution cooled to room temperature, 4.00 mL of methanol and 25.0 mL of heptane were added and stirred, and the mixture was separated into an upper layer and a lower layer. To the lower layer, 2.00 mL of methanol and 25.0 mL of heptane were added and stirred, and the mixture was separated into an upper layer and a lower layer. To the obtained lower layer, 1.00 mL of methanol, 1.14 g of sulfuric acid (97%) and 1.14 g of water were added and heated under reflux for 1.5 hours, then 1.15 g of sulfuric acid (97%) was added and heated under reflux for 1.5 hours. To the reaction solution, 6.50 g of 28 wt% sodium methoxide / methanol solution was added at 2 to 10 ° C, and the temperature was raised to room temperature. To the reaction mixture, 3.08 g of cellulose powder was added, followed by filtration to wash the solid with 50.0 mL of methanol. The filtrate was concentrated under reduced pressure, 25.0 mL of ethyl acetate was added, and the mixture was concentrated under reduced pressure three times to obtain (1S) -1,5-anhydro-1- {5-[(4-bromophenyl) as a black oil. ) 11.5 g of a crude product of methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol (17).
mp: 157 ° C.
1 H NMR (500 MHz, DMSO-d 6 ): δ 1.02 (3H, d, J = 6.8 Hz), 1.06 (3H, d, J = 6.8 Hz), 2.98 (1H, septet, J = 6.8 Hz), 3.09-3.20 (2H, m), 3.27 (1H, td, J = 8.5 and 4.7 Hz), 3.35-3.44 (2H, m), 3.66 (1H, ddd, J = 11.8, 5.7 and 1.8 Hz), 3.76 ( 3H, s), 3.91, 3.94 (2H, AB quartet, J = 15.9 Hz), 4.37 (1H, t, J = 5.7 Hz, exchangeable with D 2 O), 4.44 (1H, d, J = 9.9 Hz), 4.61 (1H, d, J = 5.7 Hz, exchangeable with D 2 O), 4.87 (1H, d, J = 5.0 Hz, exchangeable with D 2 O), 4.89 (1H, d, J = 4.7 Hz, exchangeable with D 2 O), 6.84 (1H, s), 7.03-7.09 (2H, m), 7.13 (1H, s), 7.41-7.46 (2H, m).
13 C NMR (125 MHz, DMSO-d 6 ): δ 23.5, 23.6, 28.8, 37.0, 55.7, 61.5, 70.6, 73.6, 74.2, 78.7, 81.5, 108.5, 118.6, 125.7, 128.1, 130.4, 130.5, 131.0, 141.2, 147.2, 156.7.
MS (ESI / APCI dual source): positive mode; m / z 500 {[(M + 2) + NH 4 ] + }, 498 [(M + NH 4 ) + ].
MS (ESI / APCI dual source): negative mode; m / z 527 {[(M + 2) + HCO 2 ] − }, 525 [(M + HCO 2 ) − ].
Anal.Calcd for C 23 H 29 BrO 6 : C, 57.39; H, 6.07; Br, 16.60. Found: C, 57.14; H, 6.00; Br, 16.47.
IR (KBr) cm -1 : 3402, 2961, 2929, 1616, 1508, 1488, 1089, 1072, 1048, 1012.
Example 12
(1S) -2,3,4,6-Tetra-O-acetyl-1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propane-2- Yl) phenyl} -D-glucitol (I) synthesis
アルゴン雰囲気下、(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(17)の粗生成物5.23 gとピリジン10.0 mLの混合物に無水酢酸5.79 gを加えて23〜26℃にて71時間45分間撹拌した。反応液に水1.00 mLを加えて22〜25℃にて30分間撹拌後、トルエン25.0 mL及び10 wt%塩酸20.0 gを加えて有機層と水層に分液した。有機層に10 wt%塩酸15.1 gを加えて撹拌後、不溶物をろ過してトルエン5.00 mLにて洗浄した。ろ液を有機層と水層に分液し、有機層を2 wt%炭酸水素ナトリウム水溶液15.1 g、引き続き、水10.0 mLにて洗浄した。有機層にトルエン3.00 mL及びNH-シリカゲル(Chromatorex NH、富士シリシア化学株式会社)2.50 gを加えて22〜23℃にて16時間撹拌した。不溶物をろ別後、トルエン10.0 mLにて洗浄し、ろ液を減圧濃縮した。得られた残渣にエタノール5.00 mLを加えて減圧濃縮する操作を2回実施した。濃縮残渣にエタノール3.50 mLを加えて60℃にて加熱溶解後、ヘプタン10.5 mLを57〜59℃にて6分間かけて滴下し、徐冷しながら撹拌した。44℃にて化合物(I)の種晶0.6 mgを添加後、室温(〜21℃)にて19時間撹拌した。析出した固体をろ過、洗浄(ヘプタン:エタノール=3:1(v/v)、6.50 mL)後、50℃にて1時間減圧乾燥して白色粉末の(1S) -2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(I)2.16 g得た[化合物(8)からの通算換算収率64.9%]。
mp : 126℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.04 ( 3H, d, J = 6.9 Hz ), 1.09 ( 3H, d, J = 6.9 Hz ), 1.76 ( 3H, s ), 2.00 ( 3H, s ), 2.05 ( 3H, s ), 2.06 ( 3H, s ), 2.99 ( 1H, septet, J = 6.9 Hz ), 3.80-3.86 ( 1H, m ), 3.84 ( 3H, s ), 3.89, 3.93 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.4 and 2.1 Hz ), 4.25 ( 1H, dd, J = 12.4 and 4.5 Hz ), 4.88 ( 1H, d, J = 9.5 Hz ), 5.22 ( 1H, t, J = 9.7 Hz ), 5.30-5.40 ( 2H, m ), 6.77 ( 1H, s ), 6.90-6.95 ( 2H, m ), 7.11 ( 1H, s ), 7.33-7.38 ( 2H, m ).
13C NMR ( 150 MHz, CDCl3 ) :δ 20.4, 20.7, 20.8, 23.4, 23.8, 29.4, 37.7, 55.7, 62.4, 68.8, 71.8, 73.9, 74.7, 76.1, 108.4, 119.6, 121.5, 128.9, 130.1, 130.6, 131.3, 140.5, 149.3, 156.7, 167.0, 169.6, 170.4, 170.8.
MS (ESI/APCI dual source) : ポジティブモード;m/z 673 {[(M+2)+Na]+}, 671 [(M+Na)+].
Anal.Calcd for C31H37BrO10 : C, 57.32 ; H, 5.74 ; Br, 12.30. Found : C, 57.37 ; H, 5.72 ; Br, 12.28.
IR (KBr) cm-1 : 2966, 1746, 1251, 1224, 1042.
Under an argon atmosphere, (1S) -1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol (17 ) Was added to a mixture of 5.23 g of the crude product and 10.0 mL of pyridine, and the mixture was stirred at 23 to 26 ° C. for 71 hours and 45 minutes. After adding 1.00 mL of water to the reaction solution and stirring at 22 to 25 ° C. for 30 minutes, 25.0 mL of toluene and 20.0 g of 10 wt% hydrochloric acid were added to separate the organic layer and the aqueous layer. To the organic layer, 15.1 g of 10 wt% hydrochloric acid was added and stirred, and then the insoluble material was filtered and washed with 5.00 mL of toluene. The filtrate was separated into an organic layer and an aqueous layer, and the organic layer was washed with 15.1 g of a 2 wt% aqueous sodium hydrogen carbonate solution and subsequently with 10.0 mL of water. To the organic layer, 3.00 mL of toluene and 2.50 g of NH-silica gel (Chromatorex NH, Fuji Silysia Chemical Co., Ltd.) were added and stirred at 22-23 ° C. for 16 hours. The insoluble material was filtered off, washed with 10.0 mL of toluene, and the filtrate was concentrated under reduced pressure. The operation of adding 5.00 mL of ethanol to the obtained residue and concentrating under reduced pressure was performed twice. Ethanol 3.50 mL was added to the concentrated residue and dissolved by heating at 60 ° C., and then 10.5 mL of heptane was added dropwise at 57 to 59 ° C. over 6 minutes, followed by stirring with slow cooling. After adding 0.6 mg of seed crystals of compound (I) at 44 ° C., the mixture was stirred at room temperature (˜21 ° C.) for 19 hours. The precipitated solid was filtered and washed (heptane: ethanol = 3: 1 (v / v), 6.50 mL), and then dried under reduced pressure at 50 ° C. for 1 hour to obtain a white powder (1S) -2,3,4,6 -Tetra-O-acetyl-1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol (I) 2.16 g was obtained [64.9% yield in terms of total conversion from compound (8)].
mp: 126 ° C.
1 H NMR (600 MHz, CDCl 3 ): δ 1.04 (3H, d, J = 6.9 Hz), 1.09 (3H, d, J = 6.9 Hz), 1.76 (3H, s), 2.00 (3H, s), 2.05 (3H, s), 2.06 (3H, s), 2.99 (1H, septet, J = 6.9 Hz), 3.80-3.86 (1H, m), 3.84 (3H, s), 3.89, 3.93 (2H, AB quartet , J = 16.1 Hz), 4.13 (1H, dd, J = 12.4 and 2.1 Hz), 4.25 (1H, dd, J = 12.4 and 4.5 Hz), 4.88 (1H, d, J = 9.5 Hz), 5.22 (1H , t, J = 9.7 Hz), 5.30-5.40 (2H, m), 6.77 (1H, s), 6.90-6.95 (2H, m), 7.11 (1H, s), 7.33-7.38 (2H, m).
13 C NMR (150 MHz, CDCl 3 ): δ 20.4, 20.7, 20.8, 23.4, 23.8, 29.4, 37.7, 55.7, 62.4, 68.8, 71.8, 73.9, 74.7, 76.1, 108.4, 119.6, 121.5, 128.9, 130.1, 130.6, 131.3, 140.5, 149.3, 156.7, 167.0, 169.6, 170.4, 170.8.
MS (ESI / APCI dual source): Positive mode; m / z 673 {[(M + 2) + Na] + }, 671 [(M + Na) + ].
Anal.Calcd for C 31 H 37 BrO 10 : C, 57.32; H, 5.74; Br, 12.30. Found: C, 57.37; H, 5.72; Br, 12.28.
IR (KBr) cm -1 : 2966, 1746, 1251, 1224, 1042.
式(XI)で表される化合物は、下記実施例13〜15に示す方法によって得ることができた。
実施例13
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−[5−({4−[(1E)−3−カルボキシ−3−メチルブタ−1−エン−1−イル]フェニル}メチル)−2−メトキシ−4−(プロパン−2−イル)フェニル]−D−グルシトール=ヘキサン−1−アミン塩(19)の合成
The compound represented by the formula (XI) could be obtained by the methods shown in Examples 13 to 15 below.
Example 13
(1S) -2,3,4,6-Tetra-O-acetyl-1,5-anhydro-1- [5-({4-[(1E) -3-carboxy-3-methylbut-1-ene- Synthesis of 1-yl] phenyl} methyl) -2-methoxy-4- (propan-2-yl) phenyl] -D-glucitol = hexane-1-amine salt (19)
アルゴン雰囲気下、(1S) -2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(I)3.00 g、2,2-ジメチルブタ-3-エン酸(18)0.804 g、トリ−o−トリルホスフィン0.285 g、トリエチルアミン1.88 g、アセトニトリル9.00 mL及び酢酸パラジウム0.112 gの混合物を8.5時間加熱還流した。室温まで冷却後、一晩静置した。反応混合物にアセトニトリル3.00 mLを加えて38℃にて30分間加熱撹拌後、セルロースパウダーパッドにてろ過し、アセトニトリル9.00 mLにて洗浄した。ろ液を減圧濃縮して得た残渣に酢酸エチル20.0mL及び10 wt%塩酸10.0 gを加えて有機層と水層に分液した。有機層を10 wt%塩化ナトリウム水溶液10.0 g、引き続き、水10.0 mLにて順次洗浄した。有機層を減圧濃縮後、酢酸エチル6.00 mLを加えて減圧濃縮する操作を2回実施し、残渣4.15 gを得た。この残渣に酢酸エチル12.0 g及びヘキシルアミン0.495 gを加えて加熱溶解し、ヘプタン31.5 mLを50〜55℃にて8分間かけて滴下した。51℃にて化合物(19)の種晶0.8 mgを添加後、室温(〜23℃)にて21時間45分間撹拌した。析出した固体をろ過、洗浄(ヘプタン:酢酸エチル=3:1(v/v)、12.0 mL)後、室温にて2時間減圧乾燥して白色粉末の(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-[5-({4-[(1E)-3-カルボキシ-3-メチルブタ-1-エン-1-イル]フェニル}メチル)-2-メトキシ-4-(プロパン-2-イル)フェニル]-D-グルシトール=ヘキサン-1-アミン塩(19)3.01 g(収率83.2%)を得た。
mp : 146〜147℃.
1H NMR ( 500 MHz, CDCl3 ) : δ 0.82 ( 3H, t, J = 7.3 Hz ), 1.02 ( 3H, d, J = 6.9 Hz ), 1.07 ( 3H, d, J = 6.9 Hz ), 1.09-1.16 ( 4H, m ), 1.16-1.24 ( 2H, m ), 1.27 ( 6H, s ), 1.43-1.54 ( 2H, m ), 1.76 ( 3H, s ), 2.00 ( 3H, s ), 2.047 ( 3H, s ), 2.052 ( 3H, s ), 2.59-2.66 ( 2H, m ), 3.01 ( 1H, septet, J = 6.9 Hz ), 3.80-3.85 ( 1H, m ), 3.84 ( 3H, s ), 3.91, 3.93 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.3 and 2.1 Hz ), 4.25 ( 1H, dd, J = 12.3 and 4.6 Hz ), 4.85 ( 1H, d, J = 9.5 Hz ), 5.22 ( 1H, t, J = 9.5 Hz ), 5.33 ( 1H, t, J = 9.5 Hz ), 5.41 ( 1H, t, J = 9.5 Hz ), 6.29 ( 1H, d, JAB = 16.3 Hz ), 6.44 ( 1H, d, JAB = 16.3 Hz ), 6.50 ( 3H, br s, exchangeable with D2O ), 6.76 ( 1H, s ), 6.92-6.97 ( 2H, m ), 7.12 ( 1H, s ), 7.20-7.25 ( 2H, m ).
13C NMR ( 150 MHz, CDCl3 ) :δ 14.0, 20.4, 20.7, 20.8, 22.4, 23.5, 23.8, 25.9, 26.2, 28.8, 29.3, 31.3, 38.1, 39.9, 45.5, 55.7, 62.4, 68.8, 71.7, 74.8, 76.1, 108.4, 121.3, 125.8, 126.0, 128.4, 129.5, 130.7, 135.3, 136.7, 140.2, 149.4, 156.6, 169.0, 169.6, 170.4, 170.8, 183.0.
MS (ESI/APCI dual source) : ポジティブモード;m/z 784 [(M+H)+], 705 {[(M−C6H15N)+Na]+}. MS (ESI/APCI dual source) : ネガティブモード;m/z 681 {[(M−C6H15N)−H]−}.
Anal.Calcd for C37H46O12・C6H15N : C, 65.88 ; H, 7.84 ; N, 1.79. Found : C, 65.83 ; H, 7.81 ; N, 1.80.
IR (KBr) cm-1 : 2960, 2933, 2870, 1747, 1254, 1224, 1042.
実施例14
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール一シュウ酸塩(21)の合成
Under an argon atmosphere, (1S) -2,3,4,6-tetra-O-acetyl-1,5-anhydro-1- {5-[(4-bromophenyl) methyl] -2-methoxy-4- ( Propan-2-yl) phenyl} -D-glucitol (I) 3.00 g, 2,2-dimethylbut-3-enoic acid (18) 0.804 g, tri-o-tolylphosphine 0.285 g, triethylamine 1.88 g, acetonitrile 9.00 A mixture of mL and 0.112 g of palladium acetate was heated to reflux for 8.5 hours. After cooling to room temperature, it was allowed to stand overnight. To the reaction mixture was added 3.00 mL of acetonitrile, and the mixture was heated and stirred at 38 ° C. for 30 minutes, filtered through a cellulose powder pad, and washed with 9.00 mL of acetonitrile. The filtrate was concentrated under reduced pressure, 20.0 mL of ethyl acetate and 10.0 g of 10 wt% hydrochloric acid were added to the residue, and the mixture was separated into an organic layer and an aqueous layer. The organic layer was washed sequentially with 10.0 g of a 10 wt% aqueous sodium chloride solution and subsequently with 10.0 mL of water. The organic layer was concentrated under reduced pressure, and then an operation of adding 6.00 mL of ethyl acetate and concentrating under reduced pressure was performed twice to obtain 4.15 g of a residue. To this residue, 12.0 g of ethyl acetate and 0.495 g of hexylamine were added and dissolved by heating, and 31.5 mL of heptane was added dropwise at 50 to 55 ° C. over 8 minutes. After adding 0.8 mg of seed crystals of compound (19) at 51 ° C., the mixture was stirred at room temperature (˜23 ° C.) for 21 hours and 45 minutes. The precipitated solid was filtered and washed (heptane: ethyl acetate = 3: 1 (v / v), 12.0 mL), and then dried under reduced pressure at room temperature for 2 hours to obtain a white powder of (1S) -2,3,4,6 -Tetra-O-acetyl-1,5-anhydro-1- [5-({4-[(1E) -3-carboxy-3-methylbut-1-en-1-yl] phenyl} methyl) -2- Methoxy-4- (propan-2-yl) phenyl] -D-glucitol = hexane-1-amine salt (19) (3.01 g, yield 83.2%) was obtained.
mp: 146-147 ° C.
1 H NMR (500 MHz, CDCl 3 ): δ 0.82 (3H, t, J = 7.3 Hz), 1.02 (3H, d, J = 6.9 Hz), 1.07 (3H, d, J = 6.9 Hz), 1.09- 1.16 (4H, m), 1.16-1.24 (2H, m), 1.27 (6H, s), 1.43-1.54 (2H, m), 1.76 (3H, s), 2.00 (3H, s), 2.047 (3H, s), 2.052 (3H, s), 2.59-2.66 (2H, m), 3.01 (1H, septet, J = 6.9 Hz), 3.80-3.85 (1H, m), 3.84 (3H, s), 3.91, 3.93 (2H, AB quartet, J = 16.1 Hz), 4.13 (1H, dd, J = 12.3 and 2.1 Hz), 4.25 (1H, dd, J = 12.3 and 4.6 Hz), 4.85 (1H, d, J = 9.5 Hz ), 5.22 (1H, t, J = 9.5 Hz), 5.33 (1H, t, J = 9.5 Hz), 5.41 (1H, t, J = 9.5 Hz), 6.29 (1H, d, J AB = 16.3 Hz) , 6.44 (1H, d, J AB = 16.3 Hz), 6.50 (3H, br s, exchangeable with D 2 O), 6.76 (1H, s), 6.92-6.97 (2H, m), 7.12 (1H, s) , 7.20-7.25 (2H, m).
13 C NMR (150 MHz, CDCl 3 ): δ 14.0, 20.4, 20.7, 20.8, 22.4, 23.5, 23.8, 25.9, 26.2, 28.8, 29.3, 31.3, 38.1, 39.9, 45.5, 55.7, 62.4, 68.8, 71.7, 74.8, 76.1, 108.4, 121.3, 125.8, 126.0, 128.4, 129.5, 130.7, 135.3, 136.7, 140.2, 149.4, 156.6, 169.0, 169.6, 170.4, 170.8, 183.0.
MS (ESI / APCI dual source): Positive mode; m / z 784 [(M + H) + ], 705 {[(M−C 6 H 15 N) + Na] + }. MS (ESI / APCI dual source ): Negative mode; m / z 681 {[(M−C 6 H 15 N) −H] − }.
Anal.Calcd for C 37 H 46 O 12・ C 6 H 15 N: C, 65.88; H, 7.84; N, 1.79. Found: C, 65.83; H, 7.81; N, 1.80.
IR (KBr) cm -1 : 2960, 2933, 2870, 1747, 1254, 1224, 1042.
Example 14
(1S) -2,3,4,6-Tetra-O-acetyl-1,5-anhydro-1- {5-[(4-{(1E) -4-[(1-{[2- (dimethyl Amino) ethyl] amino} -2-methyl-1-oxopropan-2-yl) amino] -3,3-dimethyl-4-oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy- Synthesis of 4- (propan-2-yl) phenyl} -D-glucitol monooxalate (21)
(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-[5-({4-[(1E)-3-カルボキシ-3-メチルブタ-1-エン-1-イル]フェニル}メチル)-2-メトキシ-4-(プロパン-2-イル)フェニル]-D-グルシトール=ヘキサン-1-アミン塩(19)25.0 g及び酢酸エチル117 mLの混合物に氷冷下、1M塩酸133 mLを加えた。室温まで昇温後、有機層と水層に分液した。有機層を1M塩酸65.0 mL、引き続き、10 wt%塩化ナトリウム水溶液62.5 gにて順次洗浄した。有機層を無水硫酸マグネシウム6.25 gにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して黄色油状物の化合物(19)のカルボン酸遊離体の粗生成物29.4 gを得た。
N-[2-(ジメチルアミノ)エチル]-2-メチルアラニンアミド二塩酸塩(20)12.1 gに25 wt%水酸化ナトリウム水溶液27.6 g、水5.00 g、酢酸エチル170 g及び塩化ナトリウム5.00 gを順次加えた。有機層と水層に分液し、水層を酢酸エチル162 gにて抽出した。分液した水層を再度酢酸エチル162 gにて抽出し、全ての有機層を合わせた。有機層を無水硫酸マグネシウム6.25 gにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して無色油状物の化合物(20)のアミン遊離体の粗生成物10.6 gを得た。
化合物(19)のカルボン酸遊離体の粗生成物29.4 gのN,N-ジメチルホルムアミド50.0mL溶液、化合物(20)のアミン遊離体の粗生成物10.6 gのN,N-ジメチルホルムアミド50.0mL溶液、N,N-ジメチルホルムアミド127 mL、1-ヒドロキシベンゾトリアゾール一水和物(HOBt・H2O)6.45 g及びトリエチルアミン10.7 mLの混合物に1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩(EDC・HCl)7.70 gを加え、室温にて12.5時間撹拌した。反応液を氷冷した水100 mLに加えた後、トルエン300 mLを加えた。有機層と水層に分液し、有機層を5 wt%塩化ナトリウム水溶液112 mLで2回、5 wt%塩化アンモニウム水溶液56.3 mLで1回、5 wt%塩化ナトリウム水溶液112 mLで1回順次洗浄した。有機層を無水硫酸マグネシウム7.50 gにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して淡黄色液41.15 gを得た。得られた濃縮液にトルエン58.85 gを加えて(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトールの粗生成物トルエン溶液100 gを得た。
得られたトルエン溶液40.0 gを減圧濃縮し、黄色液17.2 gを得た。酢酸エチル90.0 mLを加え、37℃に加熱した。化合物(21)の種晶0.05 g、0.5 Mシュウ酸/酢酸エチル溶液25.4 mL及び酢酸エチル30.0 mLを順次加え、1.5時間同温度にて撹拌した。室温まで放冷撹拌した後、析出した固体をろ過、洗浄(酢酸エチル50.0 mL)後、40℃にて1時間減圧乾燥して白色粉末の(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール一シュウ酸塩(21)10.7 g[化合物(19)からの換算収率90.5%]を得た。
mp : 184℃.
1H NMR ( 500 MHz, CDCl3 ):δ 1.05 ( 3H, d, J = 6.8 Hz ), 1.11 ( 3H, d, J = 6.8 Hz ), 1.35 ( 6H, s ), 1.43 ( 6H, s ), 1.79 ( 3H, s ), 2.00 ( 3H, s ), 2.05 ( 3H, s ), 2.06 ( 3H, s ), 2.87 ( 6H, s ), 3.04 ( 1H, septet, J = 6.8 Hz ), 3.22 ( 2H, t, J = 5.3 Hz ), 3.55-3.65 ( 2H, m ), 3.79-3.86 ( 1H, m ), 3.85 ( 3H, s ), 3.93, 3.99 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.4 and 2.4 Hz ), 4.24 ( 1H, dd, J = 12.4 and 4.5 Hz ), 4.89 ( 1H, d, J = 9.0 Hz ), 5.21 ( 1H, t, J = 9.0 Hz ), 5.33 ( 1H, t, J = 9.0 Hz ), 5.37 ( 1H, t, J = 9.0 Hz ), 6.40 ( 1H, s, exchangeable with D2O ), 6.42 ( 1H, d, J = 16.1 Hz ), 6.49 ( 1H, d, J = 16.1 Hz ), 6.77 ( 1H, s ), 6.98-7.03 ( 2H, m ), 7.13 ( 1H, s ), 7.30-7.35 ( 2H, m ), 8.31 ( 1H, t, J = 5.8 Hz, exchangeable with D2O ).
13C NMR ( 125 MHz, CDCl3 ):δ 20.4, 20.65, 20.68, 20.8, 23.4, 23.9, 24.86, 24.90, 25.06, 25.09, 29.4, 35.0, 37.9, 44.4, 45.0, 55.8, 56.8, 58.8, 62.5, 68.8, 71.9, 73.9, 74.7, 76.1, 108.3, 121.5, 126.5, 128.6, 129.3, 129.6, 130.5, 133.0, 134.2, 141.2, 149.4, 156.6, 163.1, 169.4, 169.6, 170.4, 170.8, 175.5, 177.2.
MS (ESI/APCI dual source) : ポジティブモード;m/z 838 {[(M−C2H2O4)+H]+}.
Anal.Calcd for C45H63N3O12・C2H2O4 : C, 60.83 ; H, 7.06 ; N, 4.53. Found : C, 60.60 ; H, 7.02 ; N, 4.50.
IR (KBr) cm-1 : 3358, 2962, 1756, 1745, 1657, 1505, 1253, 1233.
実施例15
(1S)−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(XI)のエタノール溶媒和物の合成
(1S) -2,3,4,6-Tetra-O-acetyl-1,5-anhydro-1- [5-({4-[(1E) -3-carboxy-3-methylbut-1-ene- 1-yl] phenyl} methyl) -2-methoxy-4- (propan-2-yl) phenyl] -D-glucitol = hexane-1-amine salt (19) 25.0 g and ethyl acetate 117 mL Below, 133 mL of 1M hydrochloric acid was added. After raising the temperature to room temperature, it was separated into an organic layer and an aqueous layer. The organic layer was washed sequentially with 65.0 mL of 1M hydrochloric acid and subsequently with 62.5 g of 10 wt% aqueous sodium chloride solution. The organic layer was dried over 6.25 g of anhydrous magnesium sulfate, the insoluble material was filtered off, and the filtrate was concentrated under reduced pressure to obtain 29.4 g of a crude carboxylic acid free product of compound (19) as a yellow oil.
12.1 g of N- [2- (dimethylamino) ethyl] -2-methylalaninamide dihydrochloride (20) 27.6 g of 25 wt% aqueous sodium hydroxide, 5.00 g of water, 170 g of ethyl acetate and 5.00 g of sodium chloride Added sequentially. The organic layer and the aqueous layer were separated, and the aqueous layer was extracted with 162 g of ethyl acetate. The separated aqueous layer was extracted again with 162 g of ethyl acetate, and all organic layers were combined. The organic layer was dried over 6.25 g of anhydrous magnesium sulfate, the insoluble material was filtered off, and the filtrate was concentrated under reduced pressure to obtain 10.6 g of a crude product of the amine free form of the colorless oily compound (20).
Compound (19) carboxylic acid free product crude product 29.4 g of N, N-dimethylformamide 50.0 mL solution, Compound (20) crude product of amine free product 10.6 g of N, N-dimethylformamide 50.0 mL solution , N, N-dimethylformamide 127 mL, 1-hydroxybenzotriazole monohydrate (HOBt · H 2 O) 6.45 g and triethylamine 10.7 mL in a mixture of 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride 7.70 g of salt (EDC · HCl) was added, and the mixture was stirred at room temperature for 12.5 hours. The reaction solution was added to 100 mL of ice-cooled water, and then 300 mL of toluene was added. Separate the organic layer and aqueous layer, and wash the organic layer twice with 112 mL of 5 wt% aqueous sodium chloride, once with 56.3 mL of 5 wt% aqueous ammonium chloride, and once with 112 mL of 5 wt% aqueous sodium chloride. did. The organic layer was dried over anhydrous magnesium sulfate (7.50 g), insoluble matters were filtered off, and the filtrate was concentrated under reduced pressure to obtain a pale yellow liquid (41.15 g). To the obtained concentrated solution was added 58.85 g of toluene, and (1S) -2,3,4,6-tetra-O-acetyl-1,5-anhydro-1- {5-[(4-{(1E)- 4-[(1-{[2- (dimethylamino) ethyl] amino} -2-methyl-1-oxopropan-2-yl) amino] -3,3-dimethyl-4-oxobut-1-ene-1 100 g of a crude product toluene solution of -yl} phenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol was obtained.
The obtained toluene solution 40.0 g was concentrated under reduced pressure to obtain 17.2 g of a yellow liquid. Ethyl acetate 90.0 mL was added and heated to 37 ° C. 0.05 g of seed crystals of compound (21), 25.4 mL of 0.5 M oxalic acid / ethyl acetate solution and 30.0 mL of ethyl acetate were sequentially added, and the mixture was stirred at the same temperature for 1.5 hours. After standing to cool to room temperature, the precipitated solid was filtered, washed (ethyl acetate 50.0 mL), and dried under reduced pressure at 40 ° C. for 1 hour to obtain a white powder of (1S) -2,3,4,6-tetra- O-acetyl-1,5-anhydro-1- {5-[(4-{(1E) -4-[(1-{[2- (dimethylamino) ethyl] amino} -2-methyl-1-oxo Propan-2-yl) amino] -3,3-dimethyl-4-oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D- 10.7 g of glucitol monooxalate (21) [converted yield from compound (19) 90.5%] was obtained.
mp: 184 ° C.
1 H NMR (500 MHz, CDCl 3 ): δ 1.05 (3H, d, J = 6.8 Hz), 1.11 (3H, d, J = 6.8 Hz), 1.35 (6H, s), 1.43 (6H, s), 1.79 (3H, s), 2.00 (3H, s), 2.05 (3H, s), 2.06 (3H, s), 2.87 (6H, s), 3.04 (1H, septet, J = 6.8 Hz), 3.22 (2H , t, J = 5.3 Hz), 3.55-3.65 (2H, m), 3.79-3.86 (1H, m), 3.85 (3H, s), 3.93, 3.99 (2H, AB quartet, J = 16.1 Hz), 4.13 (1H, dd, J = 12.4 and 2.4 Hz), 4.24 (1H, dd, J = 12.4 and 4.5 Hz), 4.89 (1H, d, J = 9.0 Hz), 5.21 (1H, t, J = 9.0 Hz) , 5.33 (1H, t, J = 9.0 Hz), 5.37 (1H, t, J = 9.0 Hz), 6.40 (1H, s, exchangeable with D 2 O), 6.42 (1H, d, J = 16.1 Hz), 6.49 (1H, d, J = 16.1 Hz), 6.77 (1H, s), 6.98-7.03 (2H, m), 7.13 (1H, s), 7.30-7.35 (2H, m), 8.31 (1H, t, J = 5.8 Hz, exchangeable with D 2 O).
13 C NMR (125 MHz, CDCl 3 ): δ 20.4, 20.65, 20.68, 20.8, 23.4, 23.9, 24.86, 24.90, 25.06, 25.09, 29.4, 35.0, 37.9, 44.4, 45.0, 55.8, 56.8, 58.8, 62.5, 68.8, 71.9, 73.9, 74.7, 76.1, 108.3, 121.5, 126.5, 128.6, 129.3, 129.6, 130.5, 133.0, 134.2, 141.2, 149.4, 156.6, 163.1, 169.4, 169.6, 170.4, 170.8, 175.5, 177.2.
MS (ESI / APCI dual source): positive mode; m / z 838 {[(M−C 2 H 2 O 4 ) + H] + }.
Anal.Calcd for C 45 H 63 N 3 O 12・ C 2 H 2 O 4 : C, 60.83; H, 7.06; N, 4.53. Found: C, 60.60; H, 7.02; N, 4.50.
IR (KBr) cm -1 : 3358, 2962, 1756, 1745, 1657, 1505, 1253, 1233.
Example 15
(1S) -1,5-Anhydro-1- {5-[(4-{(1E) -4-[(1-{[2- (dimethylamino) ethyl] amino} -2-methyl-1-oxo Propan-2-yl) amino] -3,3-dimethyl-4-oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D- Synthesis of ethanol solvate of glucitol (XI)
(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール一シュウ酸塩(21)10.0 g及びトルエン100 mLの混合物に5 wt%炭酸水素ナトリウム水溶液250 g、水50.0 g及びトルエン30.0 mLを加えた。有機層と水層に分液し、有機層を5 wt%塩化ナトリウム水溶液95.0 mLにて洗浄後、減圧濃縮して黄色油状物12.4 gを得た。得られた濃縮物にメタノール100 mL、水2.00 m及びトリエチルアミン2.00 gを加え、室温にて50時間撹拌後、88.5時間静置した。反応液を減圧濃縮し、得られた濃縮物にエタノール50.0 mLを加えて減圧濃縮する操作を3回繰り返し、淡黄色油状物の(1S)-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(XI)13.8 gを得た。エタノール16.2 gを加えて56℃に加熱し、ヘプタン10.0 gを加えた。化合物(XI)のエタノール溶媒和物の種晶0.0025 gを加えた後、6時間かけて25℃にし、同温度にて15時間撹拌した。さらに2時間かけて2℃にし、同温度にて2時間撹拌した。析出した固体をろ過、洗浄(ヘプタン:エタノール=63:37(v/v)、60.0 mL)後、40℃にて2時間減圧乾燥して白色粉末の(1S)-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(XI)のエタノール溶媒和物6.08 g(収率79.4%)を得た。
mp : 105〜106℃.
1H NMR ( 600 MHz, CD3OD ):δ 1.075 ( 3H, d, J = 6.9 Hz ), 1.084 ( 3H, d, J = 6.9 Hz), 1.17 ( 2.4H, t, J = 7.2 Hz ), 1.35 ( 6H, s ), 1.45 ( 6H, s ), 2.22 ( 6H, s ), 2.39 ( 2H, t, J = 6.8 Hz ), 3.09 ( 1H, septet, J = 6.9 Hz ), 3.27 ( 2H, t, J = 6.8 Hz ), 3.36-3.41 ( 2H, m ), 3.45-3.51 ( 1H, m ), 3.57 ( 1H, t, J = 9.2 Hz ), 3.60 ( 1.6H, q, J = 7.2 Hz ), 3.63-3.68 ( 1H, m ), 3.83 ( 3H, s ), 3.85 ( 1H, dd, J = 11.8 and 1.0 Hz ), 3.98 ( 2H, s ), 4.65 ( 1H, d, J = 9.2 Hz ), 6.39 ( 1H, d, JAB = 16.1 Hz ), 6.52 ( 1H, d, JAB = 16.1 Hz ), 6.88 ( 1H, s ), 7.06-7.09 ( 2H, m ), 7.23 ( 1H, s ), 7.30-7.33 ( 2H, m ).
13C NMR ( 150 MHz, CD3OD ):δ 18.5, 24.1, 24.2, 25.2, 25.7, 30.7, 38.4, 39.2, 45.6, 46.0, 56.5, 58.1, 58.5, 59.1, 63.4, 72.3, 75.9, 76.8, 80.3, 82.5, 109.8, 126.3, 127.5, 129.9, 130.4, 130.8, 132.1, 134.8, 136.2, 142.8, 149.7, 158.7, 177.2, 178.3.
MS (ESI/APCI dual source) : ポジティブモード;m/z 670 [(M+H)+].
Anal.Calcd for C37H55N3O8・0.8C2H6O・0.2H2O : C, 65.27 ; H, 8.54 ; N, 5.92. Found : C, 65.13 ; H, 8.46 ; N, 5.85.
IR (KBr) cm-1 : 3357, 2964, 1673, 1634, 1505, 1041.
(1S) -2,3,4,6-Tetra-O-acetyl-1,5-anhydro-1- {5-[(4-{(1E) -4-[(1-{[2- (dimethyl Amino) ethyl] amino} -2-methyl-1-oxopropan-2-yl) amino] -3,3-dimethyl-4-oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy- To a mixture of 10.0 g of 4- (propan-2-yl) phenyl} -D-glucitol monooxalate (21) and 100 mL of toluene, add 250 g of 5 wt% aqueous sodium bicarbonate, 50.0 g of water and 30.0 mL of toluene. It was. The organic layer was separated into an aqueous layer and the organic layer was washed with 95.0 mL of a 5 wt% aqueous sodium chloride solution and then concentrated under reduced pressure to obtain 12.4 g of a yellow oil. To the obtained concentrate, 100 mL of methanol, 2.00 m of water and 2.00 g of triethylamine were added, stirred at room temperature for 50 hours, and allowed to stand for 88.5 hours. The reaction solution was concentrated under reduced pressure, and the operation of adding 50.0 mL of ethanol to the obtained concentrate and concentrating under reduced pressure was repeated three times to obtain (1S) -1,5-anhydro-1- {5-[( 4-{(1E) -4-[(1-{[2- (dimethylamino) ethyl] amino} -2-methyl-1-oxopropan-2-yl) amino] -3,3-dimethyl-4- 13.8 g of oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol (XI) was obtained. 16.2 g of ethanol was added and heated to 56 ° C., and 10.0 g of heptane was added. After adding 0.0025 g of a seed crystal of an ethanol solvate of compound (XI), the mixture was brought to 25 ° C. over 6 hours and stirred at the same temperature for 15 hours. The mixture was further brought to 2 ° C. over 2 hours and stirred at the same temperature for 2 hours. The precipitated solid was filtered and washed (heptane: ethanol = 63: 37 (v / v), 60.0 mL), and then dried under reduced pressure at 40 ° C. for 2 hours to give a white powder of (1S) -1,5-anhydro-1 -{5-[(4-{(1E) -4-[(1-{[2- (dimethylamino) ethyl] amino} -2-methyl-1-oxopropan-2-yl) amino] -3, Ethanol solvate of 3-dimethyl-4-oxobut-1-en-1-yl} phenyl) methyl] -2-methoxy-4- (propan-2-yl) phenyl} -D-glucitol (XI) 6.08 g (Yield 79.4%) was obtained.
mp: 105-106 ° C.
1 H NMR (600 MHz, CD 3 OD): δ 1.075 (3H, d, J = 6.9 Hz), 1.084 (3H, d, J = 6.9 Hz), 1.17 (2.4H, t, J = 7.2 Hz), 1.35 (6H, s), 1.45 (6H, s), 2.22 (6H, s), 2.39 (2H, t, J = 6.8 Hz), 3.09 (1H, septet, J = 6.9 Hz), 3.27 (2H, t , J = 6.8 Hz), 3.36-3.41 (2H, m), 3.45-3.51 (1H, m), 3.57 (1H, t, J = 9.2 Hz), 3.60 (1.6H, q, J = 7.2 Hz), 3.63-3.68 (1H, m), 3.83 (3H, s), 3.85 (1H, dd, J = 11.8 and 1.0 Hz), 3.98 (2H, s), 4.65 (1H, d, J = 9.2 Hz), 6.39 (1H, d, J AB = 16.1 Hz), 6.52 (1H, d, J AB = 16.1 Hz), 6.88 (1H, s), 7.06-7.09 (2H, m), 7.23 (1H, s), 7.30- 7.33 (2H, m).
13 C NMR (150 MHz, CD 3 OD): δ 18.5, 24.1, 24.2, 25.2, 25.7, 30.7, 38.4, 39.2, 45.6, 46.0, 56.5, 58.1, 58.5, 59.1, 63.4, 72.3, 75.9, 76.8, 80.3 , 82.5, 109.8, 126.3, 127.5, 129.9, 130.4, 130.8, 132.1, 134.8, 136.2, 142.8, 149.7, 158.7, 177.2, 178.3.
MS (ESI / APCI dual source): Positive mode; m / z 670 [(M + H) + ].
Anal.Calcd for C 37 H 55 N 3 O 8・ 0.8C 2 H 6 O ・ 0.2H 2 O: C, 65.27; H, 8.54; N, 5.92. Found: C, 65.13; H, 8.46; N, 5.85 .
IR (KBr) cm -1 : 3357, 2964, 1673, 1634, 1505, 1041.
本発明により、SGLT1阻害活性を有する医薬として有用な1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の大量生産に適した製造法を提供することが可能となった。 According to the present invention, it is possible to provide a production method suitable for mass production of a 1,5-anhydro-1-substituted phenyl-D-glucitol compound useful as a pharmaceutical having SGLT1 inhibitory activity.
Claims (2)
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(vi)前記式(VII)で表される化合物を式(XII)で表される化合物に変換する工程と、
(vii)前記式(XII)で表される化合物を式(XIII)で表される化合物に変換する工程と、
(viii)前記式(XIII)で表される化合物を式(I)で表される化合物に変換する工程と、
(ix)前記式(I)で表される化合物を式(XIV)で表される化合物に変換する工程と、
(x)前記式(XIV)で表される化合物を式(XV)で表される化合物に変換する工程と、
(xi)前記式(XV)で表される化合物を前記式(XI)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。 A method for producing a 1,5-anhydro-1-substituted phenyl-D-glucitol compound represented by the formula (XI),
(I) converting the compound represented by formula (II) into the compound represented by formula (III);
(Ii) converting the compound represented by the formula (III) into a compound represented by the formula (IV);
(Iii) converting the compound represented by the formula (IV) into a compound represented by the formula (V);
(Iv) converting the compound represented by the formula (V) into a compound represented by the formula (VI);
(V) converting the compound represented by the formula (VI) into a compound represented by the formula (VII);
(Vi) converting the compound represented by the formula (VII) into a compound represented by the formula (XII);
(Vii) converting the compound represented by the formula (XII) into a compound represented by the formula (XIII);
(Viii) converting the compound represented by the formula (XIII) into a compound represented by the formula (I);
(Ix) converting the compound represented by the formula (I) into a compound represented by the formula (XIV);
(X) converting the compound represented by the formula (XIV) into a compound represented by the formula (XV);
(Xi) 1,5-anhydro-1-substituted phenyl-D-glucitol compound comprising the step of converting the compound represented by the formula (XV) into the compound represented by the formula (XI) Manufacturing method.
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(vi)前記式(VII)で表される化合物を式(VIII)で表される化合物に変換する工程と、
(vii)前記式(VIII)で表される化合物を式(IX)で表される化合物に変換する工程と、
(viii)前記式(IX)で表される化合物を式(X)で表される化合物に変換する工程と、
(ix)前記式(X)で表される化合物を前記式(I)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。 A process for producing a 1,5-anhydro-1-substituted phenyl-D-glucitol compound represented by formula (I), comprising:
(I) converting the compound represented by formula (II) into the compound represented by formula (III);
(Ii) converting the compound represented by the formula (III) into a compound represented by the formula (IV);
(Iii) converting the compound represented by the formula (IV) into a compound represented by the formula (V);
(Iv) converting the compound represented by the formula (V) into a compound represented by the formula (VI);
(V) converting the compound represented by the formula (VI) into a compound represented by the formula (VII);
(Vi) converting the compound represented by the formula (VII) into a compound represented by the formula (VIII);
(Vii) converting the compound represented by the formula (VIII) into a compound represented by the formula (IX);
(Viii) converting the compound represented by the formula (IX) into a compound represented by the formula (X);
(Ix) 1,5-anhydro-1-substituted phenyl-D-glucitol compound comprising the step of converting the compound represented by the formula (X) into the compound represented by the formula (I) Manufacturing method.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015239306 | 2015-12-08 | ||
| JP2015239306 | 2015-12-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017105748A true JP2017105748A (en) | 2017-06-15 |
Family
ID=59059057
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016119447A Pending JP2017105748A (en) | 2015-12-08 | 2016-06-16 | Method for producing 1,5-anhydro-1-substituted phenyl-d-glucitol compounds |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017105748A (en) |
-
2016
- 2016-06-16 JP JP2016119447A patent/JP2017105748A/en active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JPWO2006006496A1 (en) | Method for producing azulene derivatives and synthetic intermediates thereof | |
| JP2021530505A (en) | Chemical process to prepare phenylpiperidinyl indole derivatives | |
| KR20170131508A (en) | METHOD FOR PREPARING LEDIPHASBIR AND ITS DERIVATIVES AND INTERMEDIATE COMPOUND FOR THE PREPARATION OF REDIPASVIR | |
| TW201912656A (en) | Method for preparing sugammadex sodium and crystalline form thereof | |
| KR101308258B1 (en) | A novel method of making Endoxifen | |
| JP5646706B2 (en) | Method for producing C-glycoside derivative | |
| JP2018505184A5 (en) | ||
| CN111471070B (en) | Synthetic method of Reidesciclovir | |
| JP7146067B2 (en) | Preparation of Intermediates Useful for Synthesis of SGLT Inhibitors | |
| KR20120016053A (en) | A method of preparing an alkylamine derivative | |
| KR101112731B1 (en) | Method for preparing 3-iodothyronamine | |
| JP2017105748A (en) | Method for producing 1,5-anhydro-1-substituted phenyl-d-glucitol compounds | |
| US20050159611A1 (en) | Process for producing shogaols and intermediates for the synthesis thereof | |
| Hosono et al. | Enantiospecific synthesis of [(2′ S, 3′ S)-bis (hydroxymethyl) azetidin-1-yl] pyrimidine nucleosides as potential antiviral agents | |
| CN111217709A (en) | Preparation method of (1-fluorocyclopropyl) methylamine hydrochloride | |
| RU2315747C2 (en) | Method for production of acetylene compound | |
| CN108997232B (en) | Method for synthesizing histone deacetylase inhibitor | |
| JP5305697B2 (en) | Method for producing α-D-mannopyranoside derivative | |
| CN108069977B (en) | Synthetic method of fluoroalkyl-substituted pyrrole [1,2-a ] indole | |
| JP4561635B2 (en) | Process for producing 4-alkoxycarbonyltetrahydropyran or tetrahydropyranyl-4-carboxylic acid | |
| CN110845348A (en) | Preparation method of ARV-110 intermediate | |
| JP2021095367A (en) | Method for Producing C-aryl Glycoside Derivative | |
| WO2023168827A1 (en) | Synthesis method for paxlovid intermediate | |
| KR100639705B1 (en) | The preparation method of 1-methoxy-2-deoxy-L-ribose | |
| JP4039026B2 (en) | Method for producing 3-amino-2-thiophenecarboxylic acid ester |