JP2017001991A - Novel benzoxazolone compound - Google Patents
Novel benzoxazolone compound Download PDFInfo
- Publication number
- JP2017001991A JP2017001991A JP2015118562A JP2015118562A JP2017001991A JP 2017001991 A JP2017001991 A JP 2017001991A JP 2015118562 A JP2015118562 A JP 2015118562A JP 2015118562 A JP2015118562 A JP 2015118562A JP 2017001991 A JP2017001991 A JP 2017001991A
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- substituted
- group
- compound
- independently selected
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CI*(c1ccccc1O1)C1=O Chemical compound CI*(c1ccccc1O1)C1=O 0.000 description 3
- CSKXTTYRNZFYNH-UHFFFAOYSA-N CC(C)(C)c(cc1)cc(N2)c1OC2=O Chemical compound CC(C)(C)c(cc1)cc(N2)c1OC2=O CSKXTTYRNZFYNH-UHFFFAOYSA-N 0.000 description 2
- MPXOEOMIERYCLI-UHFFFAOYSA-N CC(C)(C#N)S(C)(=O)=O Chemical compound CC(C)(C#N)S(C)(=O)=O MPXOEOMIERYCLI-UHFFFAOYSA-N 0.000 description 1
- RKZFMPFEUZIFSK-UHFFFAOYSA-N CC(C)(C)c(cc1)cc(N2CCCN(CCSC)C(OC(C)(C)C)=O)c1OC2=O Chemical compound CC(C)(C)c(cc1)cc(N2CCCN(CCSC)C(OC(C)(C)C)=O)c1OC2=O RKZFMPFEUZIFSK-UHFFFAOYSA-N 0.000 description 1
- RPJUVNYXHUCRMG-UHFFFAOYSA-N CC(C)(C)c(cc1N)ccc1O Chemical compound CC(C)(C)c(cc1N)ccc1O RPJUVNYXHUCRMG-UHFFFAOYSA-N 0.000 description 1
- BFDCRGXAQCTRIJ-UHFFFAOYSA-N CC(C)(C)c(cc1N2CCCNCCSC)ccc1OC2=O Chemical compound CC(C)(C)c(cc1N2CCCNCCSC)ccc1OC2=O BFDCRGXAQCTRIJ-UHFFFAOYSA-N 0.000 description 1
- JKSYLPNQMALDRP-UHFFFAOYSA-N CC(C)(CN)S(C)(=O)=O Chemical compound CC(C)(CN)S(C)(=O)=O JKSYLPNQMALDRP-UHFFFAOYSA-N 0.000 description 1
- TWTYPQMFLIWSLH-UHFFFAOYSA-N CC(C)(CNCCN(c(cc(CCc1ccccc1)cc1)c1O1)C1=O)O Chemical compound CC(C)(CNCCN(c(cc(CCc1ccccc1)cc1)c1O1)C1=O)O TWTYPQMFLIWSLH-UHFFFAOYSA-N 0.000 description 1
- QEDBUZUQGDURSX-UHFFFAOYSA-N CC(C)(CNCCN(c(ccc(Oc(cc1)ccc1F)c1)c1O1)C1=O)O Chemical compound CC(C)(CNCCN(c(ccc(Oc(cc1)ccc1F)c1)c1O1)C1=O)O QEDBUZUQGDURSX-UHFFFAOYSA-N 0.000 description 1
- GWYOVBVNJIYVDK-UHFFFAOYSA-N CS(CCNCCCN(c1cc(-c2ccc(C(F)(F)F)cc2)ccc1O1)C1=O)(=O)=O Chemical compound CS(CCNCCCN(c1cc(-c2ccc(C(F)(F)F)cc2)ccc1O1)C1=O)(=O)=O GWYOVBVNJIYVDK-UHFFFAOYSA-N 0.000 description 1
Abstract
Description
本発明は、ベンズオキサゾロン骨格を有する新規な化合物またはそれらの医薬として許容される塩を有効成分として含有する、Naチャネル、特にSCN9A(Nav1.7)ならびにSCN10A(Nav1.8)が関与する病態に対する治療薬または予防薬に関する。具体的には、神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害または多発性硬化症等の疾患の治療薬または予防薬に関する。 The present invention relates to a pathological condition involving a Na channel, particularly SCN9A (Nav1.7) and SCN10A (Nav1.8), which contain a novel compound having a benzoxazolone skeleton or a pharmaceutically acceptable salt thereof as an active ingredient. It relates to therapeutic or prophylactic drugs. Specifically, therapeutic or prophylactic agents for diseases such as neuropathic pain, nociceptive pain, inflammatory pain, small-fiber neuropathy, limb erythema pain, paroxysmal pain, dysuria or multiple sclerosis About.
電位依存性Naチャネルのポアを形成するαサブユニットは現在9種類存在することが知られている。近年、かかるサブユニットの内、特にSCN9AおよびSCN10Aが急性ならびに慢性疼痛の情報伝達に幅広く関与するエビデンスが得られてきた。
SCN9A(Nav1.7)は、末梢の感覚神経ないしは交感神経に局在するテトロドトキシン(TTX)感受性Naチャネルで、NENAまたはPN1とも呼ばれる。生理的にはNav1.7チャネルは感覚神経終末で疼痛シグナルを増幅する(起動電位を発生する)機能を果たしており、遺伝学的研究から、SCN9A遺伝子に機能喪失変異を有するヒトは先天性無痛症を呈することが明らかとなった。反対に、肢端紅痛症あるいは発作性激痛症といった重篤な希少疾患患者には、SCN9Aの機能獲得変異が認められてきた。更に、小径繊維ニューロパチー患者の実に3割にNav1.7機能が亢進する遺伝子多型が存在するという報告が寄せられている(非特許文献1)。また、慢性疼痛モデル動物のDRGニューロンにおいて発現レベルおよび活性が上昇することや、ノックアウト実験で神経障害性疼痛および炎症性疼痛が減弱することから、Nav1.7チャネル機能が疼痛病態におけるDRGニューロンの過剰興奮に直接関与することが示唆されている(非特許文献2)。
SCN10A(Nav1.8)は、後根神経節の小径ニューロン(C繊維)にドミナントに発現するテトロドトキシン(TTX)抵抗性Naチャネルであり、SNSまたはPN3とも呼ばれる。生理的には、C繊維において活動電位を発生させることで侵害受容情報を伝達し、中でも冷痛覚に対しては重要な役割を果たすことがノックアウトマウスの表現型解析等から示唆されている。慢性疼痛病態ではNav1.7と同様、Nav1.8に関しても発現レベルと活性の両方が上昇し、アンチセンス核酸やsiRNA投与により神経障害性疼痛あるいは炎症性疼痛モデル動物が示す痛覚過敏およびアロディニアが減弱することが報告されている(非特許文献3、4)。更に、臨床においても、小径繊維ニューロパチー患者の一部にNav1.8の機能獲得変異が発見されており、これらの変異がDRGニューロンの興奮性を高めることが確かめられている(非特許文献5)。
従って、Nav1.7あるいはNav1.8の阻害剤、好ましくは両サブタイプを阻害する化合物は、C繊維やAδ繊維等の末梢神経が関与する痛み、しびれ感、灼熱感、鈍痛、刺痛、電撃痛等の自発痛、機械刺激や冷熱刺激に対する痛覚過敏あるいはアロディニアを伴う神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症および発作性激痛症などの疾患に対して鎮痛効果を示す薬剤になり得ると考えられた。さらにNav1.7およびNav1.8が非神経組織や中枢神経系にはほとんど発現しないことから、Nav1.7あるいはNav1.8、好ましくは両サブタイプを選択的に阻害する薬剤は、既存薬で問題となっている心臓や中枢神経由来の副作用を示さない薬剤になり得ると考えられた。
It is known that there are currently nine types of α subunits that form pores of voltage-gated Na channels. In recent years, there has been evidence that such subunits, particularly SCN9A and SCN10A, are widely involved in signaling acute and chronic pain.
SCN9A (Nav1.7) is a tetrodotoxin (TTX) -sensitive Na channel localized in peripheral sensory or sympathetic nerves and is also called NENA or PN1. Physiologically, the Nav1.7 channel functions to amplify pain signals (generate an activation potential) at sensory nerve endings, and from genetic studies, humans with loss-of-function mutations in the SCN9A gene have congenital analgesia It became clear to exhibit. In contrast, SCN9A gain-of-function mutations have been found in patients with severe rare diseases such as limb erythema or paroxysmal pain. Furthermore, it has been reported that 30% of patients with small-diameter fiber neuropathy have a genetic polymorphism that enhances Nav1.7 function (Non-patent Document 1). In addition, since the expression level and activity are increased in DRG neurons of chronic pain model animals and neuropathic pain and inflammatory pain are attenuated in knockout experiments, Nav1.7 channel function is excessive in DRG neurons in pain pathology. It has been suggested to be directly involved in excitement (Non-Patent Document 2).
SCN10A (Nav1.8) is a tetrodotoxin (TTX) -resistant Na channel that is dominantly expressed in small-diameter neurons (C fibers) in the dorsal root ganglion and is also called SNS or PN3. Physiologically, nociceptive information is transmitted by generating action potentials in C fibers, and it is suggested from the phenotypic analysis of knockout mice that it plays an important role for cold pain. Similar to Nav1.7 in chronic pain pathology, both Nav1.8 expression level and activity increased, and administration of antisense nucleic acid or siRNA attenuated hyperalgesia and allodynia exhibited by neuropathic pain or inflammatory pain model animals It has been reported (Non-Patent Documents 3 and 4). Furthermore, also in clinical practice, Nav1.8 gain-of-function mutations have been found in some small fiber neuropathy patients, and these mutations have been confirmed to enhance excitability of DRG neurons (Non-patent Document 5). .
Therefore, an inhibitor of Nav1.7 or Nav1.8, preferably a compound that inhibits both subtypes, is pain associated with peripheral nerves such as C fiber and Aδ fiber, numbness, burning sensation, dull pain, stinging pain, electric shock For diseases such as spontaneous pain such as pain, hyperalgesia to mechanical or cold stimuli, or neuropathic pain with allodynia, nociceptive pain, inflammatory pain, small fiber neuropathy, limb redness and paroxysmal pain It was thought that it could be a drug that shows an analgesic effect. Furthermore, since Nav1.7 and Nav1.8 are hardly expressed in non-neural tissues and central nervous system, drugs that selectively inhibit Nav1.7 or Nav1.8, preferably both subtypes, are a problem with existing drugs. It is thought that it can be a drug that does not show side effects derived from the heart and central nervous system.
また、排尿障害においては、その主症状である頻尿はNav1.8の感作を介したC繊維の過活動に起因すること、すなわち、過活動膀胱や膀胱痛には下部尿路からの求心性知覚神経路の機能異常が関与しており、Naチャネル阻害による膀胱からのC線維知覚神経の抑制が奏効することが示唆されている(非特許文献6)。従って、Nav1.8を阻害する薬剤は、新規な作用点を有する排尿障害の治療薬または予防薬となることが期待される。
一方、通常ではC繊維にしか認められないNav1.8が、多発性硬化症患者の小脳プルキンエ細胞において異所的に発現し、小脳の異常発火パターンの発生に関与することが報告されている(非特許文献7)。従って、Nav1.8阻害薬は、多発性硬化症において、Nav1.8発現に伴う小脳神経の異常発火によって誘発される運動失調などの症状に対する、初めての治療薬または予防薬となることが期待される。
In dysuria, frequent urination, which is the main symptom, is caused by C fiber overactivity through Nav1.8 sensitization, that is, overactive bladder and bladder pain are found from the lower urinary tract. It is suggested that abnormalities of the cardiac sensory nerve tract are involved, and that suppression of C-fiber sensory nerves from the bladder by Na channel inhibition is effective (Non-Patent Document 6). Therefore, a drug that inhibits Nav1.8 is expected to be a therapeutic or preventive drug for dysuria having a novel action point.
On the other hand, it has been reported that Nav1.8, which is usually found only in C fibers, is ectopically expressed in cerebellar Purkinje cells of patients with multiple sclerosis and is involved in the development of abnormal cerebellar firing patterns ( Non-patent document 7). Therefore, Nav1.8 inhibitor is expected to be the first therapeutic or preventive agent for symptoms such as ataxia induced by abnormal cerebellar nerve firing associated with Nav1.8 expression in multiple sclerosis. The
特許文献1には、ベンゾジアゼピンω3受容体リガンドに関する化合物が具体的に開示されている。該特許文献に記載の化合物は、具体的には下記式(A)で表されるように、ベンズオキサゾロン環内窒素原子上の置換基としてアルキルアミド基を有するのが構造的特徴であり、本発明化合物を含まない点において相違する。また、該特許文献1には本発明を示唆する記載は一切なされていない。 Patent Document 1, compounds for benzodiazepine omega 3 receptor ligands are specifically disclosed. Specifically, the compound described in the patent document has an alkylamide group as a substituent on the nitrogen atom in the benzoxazolone ring, as represented by the following formula (A). It differs in that it does not contain the inventive compound. Further, Patent Document 1 does not include any description suggesting the present invention.
本発明の課題は、Nav1.7またはNav1.8が関与する病態、具体的には神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害または多発性硬化症等の疾患に対する治療薬または予防薬を提供することにある。 The subject of the present invention is a pathology involving Nav1.7 or Nav1.8, specifically neuropathic pain, nociceptive pain, inflammatory pain, small fiber neuropathy, limb erythema pain, paroxysmal pain An object of the present invention is to provide a therapeutic or preventive drug for diseases such as dysuria or multiple sclerosis.
本発明者らは上記課題を達成するために、鋭意検討した結果、後述するベンズオキサゾロン環を有する2環性化合物またはその薬学的に許容される塩が、Nav1.7遺伝子発現細胞および/またはNav1.8遺伝子発現細胞において、Naチャネルを介した膜電位変化ないしはNaイオン電流そのものを阻害すること、すなわちNav1.7およびNav1.8の片方又は両方に対して阻害活性を持つことを見出した。更に、該誘導体はまた、神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害または多発性硬化症等の疾患に対する治療薬または予防薬として有用であることを見出し、本発明を完成するに至った。本発明によれば、下記式(1)で表されるベンズオキサゾロン化合物(以下、「本発明の化合物」、「式(1)で表される化合物」または「式(1)の化合物」と称することもある)又はその薬学的に許容される塩が提供される。 As a result of intensive studies to achieve the above-mentioned problems, the present inventors have found that a bicyclic compound having a benzoxazolone ring, which will be described later, or a pharmaceutically acceptable salt thereof is a Nav1.7 gene-expressing cell and / or Nav1. .8 gene-expressing cells were found to inhibit membrane potential change through Na channel or Na ion current itself, that is, to inhibit one or both of Nav1.7 and Nav1.8. In addition, the derivatives may also be used as therapeutic agents for diseases such as neuropathic pain, nociceptive pain, inflammatory pain, small-diameter neuropathy, limb erythema pain, paroxysmal pain, dysuria or multiple sclerosis. It has been found that it is useful as a preventive agent, and the present invention has been completed. According to the present invention, a benzoxazolone compound represented by the following formula (1) (hereinafter referred to as “compound of the present invention”, “compound represented by formula (1)”, or “compound of formula (1)”). Or a pharmaceutically acceptable salt thereof.
[項1]下記式(1); [Item 1] The following formula (1);
R1およびR2は、各々独立して、水素、C1−6アルキル(該アルキルは、ヒドロキシ、ハロゲン、C1−4アルコキシ、C1−4アルキルスルホニル、C1−4アルキルチオ、アミノカルボニル(該アミノカルボニルのアミノ部分は、置換されていてもよいC1−4アルキルによってモノまたはジ置換されていてもよい)、置換されていてもよい4〜7員のヘテロシクロアルキルおよび置換されていてもよい4〜7員のヘテロシクロアルキルカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニルまたは4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニルおよび該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)を表し、
Lは、C1−6アルキレン(該アルキレンは、C1−4アルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)を表し、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜7員の飽和複素環を形成してもよく、
R3は、ハロゲン、C1−6アルキル、C2−6アルケニル、C2−6アルキニル(該アルキル、該アルケニルおよび該アルキニルは、それぞれハロゲン、置換されていてもよいC3−7シクロアルキル、置換されていてもよい4〜7員のヘテロシクロアルキル、C1−4アルコキシ、C1−4ハロアルコキシ、C1−4アルキルスルホニル、C1−4アルキルチオ、置換されていてもよいC6−10アリールおよび置換されていてもよいC6−10アリールオキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニル、4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、置換されていてもよいC1−4アルキル、C1−4ハロアルキル、置換されていてもよいC1−4アルコキシ、C1−4ハロアルコキシ、シアノおよびC1−4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリールオキシ(該アリールオキシのアリール部分は、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ、C1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシおよびC1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)を表し、
R4は、水素、ハロゲンまたはC1−4アルキルを表す]
で表される化合物またはその薬学的に許容される塩;
[項2]
置換されていてもよいC6−10アリール、置換されていてもよいC6−10アリールオキシおよび置換されていてもよいC3−7シクロアルキルにおける置換基が、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ、C1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基であり、
置換されていてもよいC1−4アルキルおよび置換されていてもよいC1−4アルコキシにおける置換基が、C1−4アルコキシ、C1−4ハロアルコキシからなる群から独立して選択される1〜2個の置換基であり、
置換されていてもよい4〜7員のヘテロシクロアルおよび置換されていてもよい4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキルにおける置換基が、C1−4アルキル、ハロゲン、オキソおよびヒドロキシからなる群から独立して選択される1〜3個の置換基である、項1に記載の化合物またはその薬学的に許容される塩;
[項3]
置換されていてもよいC6−10アリール、置換されていてもよいC6−10アリールオキシおよび置換されていてもよいC3−7シクロアルキルにおける置換基が、1〜3個のハロゲンであり、
置換されていてもよいC1−4アルキルおよび置換されていてもよいC1−4アルコキシにおける置換基が、1〜2個のC1−2アルコキシであり、
置換されていてもよい4〜7員のヘテロシクロアルおよび置換されていてもよい4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキルにおける置換基が、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基である、項1に記載の化合物またはその薬学的に許容される塩;
[項4]
R1およびR2が、各々独立して、水素、C1−6アルキル(該アルキルは、ヒドロキシ、ハロゲン、C1−4アルコキシ、C1−4アルキルスルホニル、アミノカルボニル(該アミノカルボニルのアミノ部分は、C1−4アルキルによってモノまたはジ置換されていてもよい)、4〜7員のヘテロシクロアルキルおよび4〜7員のヘテロシクロアルキルカルボニル(該4〜7員のヘテロシクロアルキルおよび該4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキル部分は、それぞれC1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または4〜7員のヘテロシクロアルキル(該ヘテロシクロアルキルは、ハロゲン、およびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)である、項1〜3のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項5]
R1およびR2が、各々独立して、水素、C1−4アルキル(該アルキルは、ヒドロキシ、C1−2アルキルスルホニル、アミノカルボニル(該アミノカルボニルのアミノ部分は、C1−2アルキルによってモノ置換されていてもよい)、4〜5員のヘテロシクロアルキルおよび4〜6員のヘテロシクロアルキルカルボニル(該4〜7員のヘテロシクロアルキルおよび該4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキルは、それぞれC1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)からなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)である、項1〜3のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項6]
R1が、水素またはC1−4アルキルであり、R2が、C1−4アルキル(該アルキルは、ヒドロキシ、C1−2アルキルスルホニルおよびアミノカルボニル(該アミノカルボニルのアミノ部分は、C1−2アルキルによってモノ置換されていてもよい)からなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)である、項1〜3のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項7]
Lが、C1−6アルキレン(該アルキレンは、C1−4アルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜7員の飽和複素環を形成してもよい、項1〜6のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項8]
Lが、C1−4アルキレン(該アルキレンは、C1−2アルキルおよびヒドロキシからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜6員の飽和複素環を形成してもよい、項7に記載の化合物またはその薬学的に許容される塩;
[項9]
Lが、C1−3アルキレン(該アルキレンは、C1−2アルキルおよびヒドロキシからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)である、項1〜6のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項10]
Lが、C1−3アルキレンであり、R1が、C1−6アルキルであり、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共にアゼチジン、ピペリジン、ピロリジンを形成している、項1〜6のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項11]
R3が、ハロゲン、C1−6アルキル、C2−6アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、C3−7シクロアルキル(該シクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)、4〜7員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、C6−10アリールおよびC6−10アリールオキシ(該アリールおよび該アリールオキシは、それぞれ1〜3個のハロゲンによって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニル、4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキルおよびハロゲンからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、C1−4アルキル、C1−4アルコキシ(該アルキルおよび該アルコキシは、それぞれ1〜2個のC1−2アルコキシによって置換されていてもよい)、C1−4ハロアルキル、C1−4ハロアルコキシ、シアノおよびC1−4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシおよびC1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)である、項1〜10のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項12]
R3が、ハロゲン、C1−4アルキル、C2−4アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、C4−6シクロアルキル(該シクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)、置換されていてもよい4〜6員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、C6−10アリールおよびC6−10アリールオキシ(該アリールおよび該アリールオキシは、それぞれ1〜2個のハロゲンによって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−6シクロアルキル、C4−6シクロアルケニル、4〜6員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、C1−2アルキル、C1−2アルコキシ(該アルキルおよび該アルコキシは、それぞれ1〜2個のC1−2アルコキシによって置換されていてもよい)、C1−2ハロアルキル、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、ハロゲンおよびC1−2ハロアルキルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)である、項1〜10のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項13]
R3が、C3−6シクロアルキル、4〜6員のヘテロシクロアルキル(該シクロアルキルおよび該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、フェニル(該フェニルは、ハロゲン、C1−2アルキル、C1−2ハロアルキル、C1−2アルコキシおよびC1−2ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、1〜2個のC1−2ハロアルキルによって置換されていてもよい)である、項1〜10のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項14]
R4が、水素、ハロゲンまたはC1−2アルキルである、項1〜13のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項15]
R4が、水素である、項1〜13のいずれか一項に記載の化合物またはその薬学的に許容される塩;
[項16]
R1およびR2が、各々独立して、水素、C1−6アルキル(該アルキルは、ヒドロキシ、ハロゲン、C1−4アルコキシ、C1−4アルキルスルホニル、アミノカルボニル(該アミノカルボニルのアミノ部分は、C1−4アルキルによってモノまたはジ置換されていてもよい)、4〜7員のヘテロシクロアルキルおよび4〜7員のヘテロシクロアルキルカルボニル(該4〜7員のヘテロシクロアルキルおよび該4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキル部分は、それぞれC1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または4〜7員のヘテロシクロアルキル(該ヘテロシクロアルキルは、ハロゲン、およびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、
Lが、C1−6アルキレン(該アルキレンは、C1−4アルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜7員の飽和複素環を形成してもよく、
R3が、ハロゲン、C1−6アルキル、C2−6アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、C3−7シクロアルキル(該シクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)、4〜7員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、C6−10アリールおよびC6−10アリールオキシ(該アリールおよび該アリールオキシは、それぞれ1〜3個のハロゲンによって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニル、4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキルおよびハロゲンからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、C1−4アルキル、C1−4アルコキシ、シアノおよびC1−4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシおよびC1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、
R4が、水素、ハロゲンまたはC1−2アルキルである、項1に記載の化合物またはその薬学的に許容される塩;
[項17]
R1およびR2が、各々独立して、水素、C1−4アルキル(該アルキルは、ヒドロキシ、C1−2アルキルスルホニル、アミノカルボニル(該アミノカルボニルのアミノ部分は、C1−2アルキルによってモノ置換されていてもよい)、4〜5員のヘテロシクロアルキルおよび4〜6員のヘテロシクロアルキルカルボニル(該4〜7員のヘテロシクロアルキルおよび該4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキルは、それぞれC1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)からなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)であり、
Lが、C1−4アルキレン(該アルキレンは、C1−2アルキルおよびヒドロキシからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜6員の飽和複素環を形成してもよく、
R3が、ハロゲン、C1−4アルキル、C2−4アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、C4−6シクロアルキル(該シクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)、4〜6員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、C6−10アリールおよびC6−10アリールオキシ(該アリールおよび該アリールオキシは、それぞれ1〜2個のハロゲンによって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−6シクロアルキル、C4−6シクロアルケニル、4〜6員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、C1−2アルキル、C1−2アルコキシ(該アルキルおよび該アルコキシは、それぞれ1〜2個のC1−2アルコキシによって置換されていてもよい)、C1−2ハロアルキル、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、ハロゲンおよびC1−2ハロアルキルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
R4が、水素、ハロゲンまたはC1−2アルキルである、項1に記載の化合物またはその薬学的に許容される塩;
[項18]
R1が、水素であり、
R2が、C1−4アルキル(該アルキルは、ヒドロキシおよびC1−2アルキルスルホニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
Lが、C1−3アルキレン(該アルキレンは、C1−2アルキルおよびヒドロキシからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
R3が、C3−6シクロアルキル、4〜6員のヘテロシクロアルキル(該シクロアルキルおよび該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、フェニル(該フェニルは、ハロゲン、C1−2アルキル、C1−2アルコキシ、C1−2ハロアルキル、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、1〜2個のC1−2ハロアルキルによって置換されていてもよい)であり、
R4が、水素である、項1に記載の化合物またはその薬学的に許容される塩;
[項19]
R1が、C1−4アルキルであり、
R2が、C1−4アルキル(該アルキルは、ヒドロキシおよびC1−2アルキルスルホニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
Lが、C1−3アルキレンであり、R1のアルキルとL上の炭素原子が一緒になって、R1が結合する窒素原子と共にアゼチジン、ピペリジンまたはピロリジンを形成しており、
R3が、C3−6シクロアルキル、4〜6員のヘテロシクロアルキル(該シクロアルキルおよび該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、フェニル(該フェニルは、ハロゲン、C1−2アルキル、C1−2アルコキシ、C1−2ハロアルキル、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、1〜2個のC1−2ハロアルキルによって置換されていてもよい)であり、
R4が、水素である、項1に記載の化合物またはその薬学的に許容される塩;
[項20]
以下の化合物群から選択される、項1に記載の化合物またはその薬学的に許容される塩:
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例14);
(S)−2−((2−(2−オキソ−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−3(2H)−イル)エチル)アミノ)プロパンアミド(実施例16);
2−((2−(2−オキソ−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−3(2H)−イル)エチル)アミノ)アセトアミド(実施例19);
(R)−2−((2−(2−オキソ−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−3(2H)−イル)エチル)アミノ)プロパンアミド(実施例20);
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(4−(トリフルオロメトキシ)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例34);
3−(2−(オキセタン−3−イルアミノ)エチル)−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例42);
5−(4−フルオロフェニル)−4−メチル−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例44);
3−(2−メチル−3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例67);
5−(4−フルオロフェニル)−3−(2−ヒドロキシ−3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例70);
5−(4−フルオロフェニル)−3−(3−(オキセタン−3−イルアミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例72);
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−フェニルベンゾ[d]オキサゾール−2(3H)−オン(実施例75);
5−(3−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例76);
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(4−(トリフルオロメトキシ)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例84);
5−(4−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例88);
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(4−(トリフルオロメチル)フェニル) ベンゾ[d]オキサゾール−2(3H)−オン(実施例92);
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例93);
5−(4−フルオロフェニル)−4−メチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例99);
5−(4,4−ジフルオロシクロヘキシル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例100);
6−フルオロ−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(テトラヒドロ−2H−ピラン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例101);
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(テトラヒドロ−2H−ピラン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例102);
5−シクロブチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例111);
5−シクロペンチル−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例118);
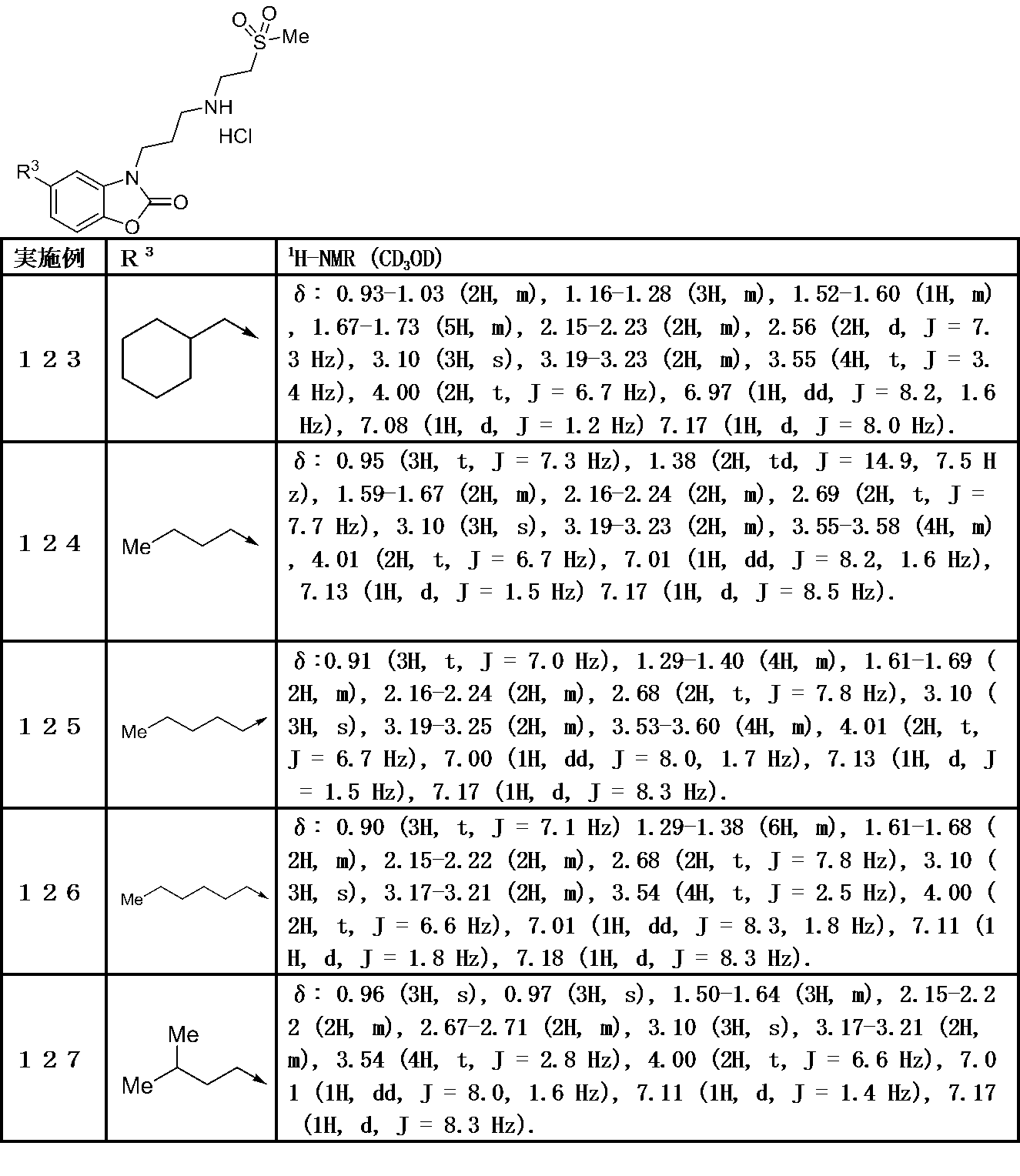
5−ブチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例124);
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−7−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例149);
5−(4−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)ブチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例150);
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例155);
5−(3,3−ジフルオロシクロブチル)−3−(1−(2−(メチルスルホニル)エチル)アゼチジン−3−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例156);
5−(3,3−ジフルオロシクロブチル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例157);
5−(3,3−ジフルオロシクロブチル)−3−(4−((2−(メチルスルホニル)エチル)アミノ)ブチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例158);
5−(3,3−ジフルオロシクロブチル)−3−(1−(2−(メチルスルホニル)エチル)ピペリジン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例159);
2−(3−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)アゼチジン−1−イル)アセトアミド(実施例162);
2−(4−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)ピペリジン−1−イル)アセトアミド(実施例163);または
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−ヒドロキシ−2−メチルプロピル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例165);
[項21]
以下の化合物群から選択される、請求項1に記載の化合物またはその薬学的に許容される塩:
5−(4−フルオロフェニル)−3−(2−ヒドロキシ−3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例70);
5−(4−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例88);
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例93);
5−(4,4−ジフルオロシクロヘキシル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例100);
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(テトラヒドロ−2H−ピラン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例102);
5−シクロブチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例111);
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−7−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例149);
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例155);
5−(3,3−ジフルオロシクロブチル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例157);
5−(3,3−ジフルオロシクロブチル)−3−(1−(2−(メチルスルホニル)エチル)ピペリジン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例159);
2−(4−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)ピペリジン−1−イル)アセトアミド(実施例163);または
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−ヒドロキシ−2−メチルプロピル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン(実施例165);
[項22]
下記式(2);
R 1 And R 2 Each independently represents hydrogen, C 1-6 Alkyl (wherein alkyl is hydroxy, halogen, C 1-4 Alkoxy, C 1-4 Alkylsulfonyl, C 1-4 Alkylthio, aminocarbonyl (the amino moiety of the aminocarbonyl is optionally substituted C 1-4 Independently from the group consisting of optionally substituted 4-7 membered heterocycloalkyl and optionally substituted 4-7 membered heterocycloalkylcarbonyl. Optionally substituted with 1 to 3 selected substituents), C 3-7 Cycloalkyl, C 4-7 Cycloalkenyl or 4- to 7-membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl and the heterocycloalkyl are each C 1-4 Alkyl, C 1-4 Haloalkyl, halogen, hydroxy and C 1-4 Represented by 1 to 3 substituents independently selected from the group consisting of alkoxy),
L is C 1-6 Alkylene (the alkylene is C 1-4 Alkyl, halogen, hydroxy and C 1-4 And optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxy, wherein R 1 But C 1-6 When alkyl, R 1 And carbon atoms on L together, R 1 A 4- to 7-membered saturated heterocyclic ring may be formed together with the nitrogen atom to which
R 3 Is halogen, C 1-6 Alkyl, C 2-6 Alkenyl, C 2-6 Alkynyl (the alkyl, the alkenyl, and the alkynyl are each halogen, optionally substituted C 3-7 Cycloalkyl, optionally substituted 4-7 membered heterocycloalkyl, C 1-4 Alkoxy, C 1-4 Haloalkoxy, C 1-4 Alkylsulfonyl, C 1-4 Alkylthio, optionally substituted C 6-10 Aryl and optionally substituted C 6-10 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of aryloxy), C 3-7 Cycloalkyl, C 4-7 Cycloalkenyl, 4- to 7-membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl, the heterocycloalkyl are each C 1-4 Alkyl, C 1-4 Haloalkyl, halogen, hydroxy and C 1-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxy), C 6-10 Aryl (wherein the aryl is halogen, optionally substituted C 1-4 Alkyl, C 1-4 Haloalkyl, optionally substituted C 1-4 Alkoxy, C 1-4 Haloalkoxy, cyano and C 1-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl), C 6-10 Aryloxy (the aryl moiety of the aryloxy is halogen, C 1-4 Alkyl, C 1-4 Haloalkyl, C 1-4 Alkoxy, C 1-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of haloalkoxy) or 5-10 membered heteroaryl (wherein the heteroaryl is halogen, C 1-4 Alkyl, C 1-4 Haloalkyl, C 1-4 Alkoxy and C 1-4 Which may be substituted with 1 to 3 substituents independently selected from the group consisting of haloalkoxy)
R 4 Is hydrogen, halogen or C 1-4 Represents alkyl]
Or a pharmaceutically acceptable salt thereof;
[Section 2]
C which may be substituted 6-10 Aryl, optionally substituted C 6-10 Aryloxy and optionally substituted C 3-7 The substituent in cycloalkyl is halogen, C 1-4 Alkyl, C 1-4 Haloalkyl, C 1-4 Alkoxy, C 1-4 1 to 3 substituents independently selected from the group consisting of haloalkoxy,
C which may be substituted 1-4 Alkyl and optionally substituted C 1-4 The substituent on alkoxy is C 1-4 Alkoxy, C 1-4 1 to 2 substituents independently selected from the group consisting of haloalkoxy,
Substituents in the optionally substituted 4-7 membered heterocycloalkyl and optionally substituted 4-7 membered heterocycloalkylcarbonyl heterocycloalkyl are C 1-4 Item 2. The compound according to Item 1, or a pharmaceutically acceptable salt thereof, which is 1 to 3 substituents independently selected from the group consisting of alkyl, halogen, oxo and hydroxy;
[Section 3]
C which may be substituted 6-10 Aryl, optionally substituted C 6-10 Aryloxy and optionally substituted C 3-7 The substituent on cycloalkyl is 1-3 halogens;
C which may be substituted 1-4 Alkyl and optionally substituted C 1-4 The substituent in alkoxy is 1-2 C 1-2 Is alkoxy,
Substituents in the optionally substituted 4-7 membered heterocycloalkyl and optionally substituted 4-7 membered heterocycloalkylcarbonyl heterocycloalkyl are C 1-2 Item 2. The compound or a pharmaceutically acceptable salt thereof according to Item 1, wherein the compound is one or two substituents independently selected from the group consisting of alkyl and oxo;
[Claim 4]
R 1 And R 2 Are each independently hydrogen, C 1-6 Alkyl (wherein alkyl is hydroxy, halogen, C 1-4 Alkoxy, C 1-4 Alkylsulfonyl, aminocarbonyl (the amino moiety of the aminocarbonyl is C 1-4 4-7 membered heterocycloalkyl and 4-7 membered heterocycloalkylcarbonyl (the 4-7 membered heterocycloalkyl and the 4-7 membered heterocyclo) The heterocycloalkyl part of alkylcarbonyl is each C 1-2 Substituted with 1 to 3 substituents independently selected from the group consisting of 1 to 2 substituents independently selected from the group consisting of alkyl and oxo) Or 4-7 membered heterocycloalkyl, which is halogen, and C 1-4 The compound according to any one of Items 1 to 3, which may be substituted with 1 to 3 substituents independently selected from the group consisting of alkoxy, or a pharmaceutically acceptable salt thereof salt;
[Section 5]
R 1 And R 2 Are each independently hydrogen, C 1-4 Alkyl (wherein the alkyl is hydroxy, C 1-2 Alkylsulfonyl, aminocarbonyl (the amino moiety of the aminocarbonyl is C 1-2 4-5 membered heterocycloalkyl and 4-6 membered heterocycloalkylcarbonyl (optionally mono-substituted by alkyl) (the 4-7 membered heterocycloalkyl and the 4-7 membered heterocycloalkylcarbonyl) Each heterocycloalkyl is C 1-2 Substituted with 1 to 2 substituents independently selected from the group consisting of 1 to 2 substituents independently selected from the group consisting of alkyl and oxo) Or a 4- to 5-membered heterocycloalkyl, which is optionally substituted by 1 to 2 halogens, or a compound according to any one of items 1 to 3; A pharmaceutically acceptable salt thereof;
[Claim 6]
R 1 Is hydrogen or C 1-4 Alkyl and R 2 But C 1-4 Alkyl (wherein the alkyl is hydroxy, C 1-2 Alkylsulfonyl and aminocarbonyl (the amino moiety of the aminocarbonyl is C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of (optionally monosubstituted by alkyl) or 4 to 5 membered heterocycloalkyl (the heterocycloalkyl is , C 1-2 The compound according to any one of Items 1 to 3, which may be substituted with 1 to 2 substituents independently selected from the group consisting of alkyl and oxo, or a pharmaceutically acceptable salt thereof Salt to be made;
[Claim 7]
L is C 1-6 Alkylene (the alkylene is C 1-4 Alkyl, halogen, hydroxy and C 1-4 And optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxy), wherein R 1 But C 1-6 When alkyl, R 1 And carbon atoms on L together, R 1 The compound or pharmaceutically acceptable salt thereof according to any one of Items 1 to 6, which may form a 4 to 7-membered saturated heterocyclic ring together with the nitrogen atom to which is bonded;
[Section 8]
L is C 1-4 Alkylene (the alkylene is C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of alkyl and hydroxy), wherein R 1 But C 1-6 When alkyl, R 1 And carbon atoms on L together, R 1 Item 8. The compound according to Item 7 or a pharmaceutically acceptable salt thereof, which may form a 4- to 6-membered saturated heterocyclic ring with the nitrogen atom to which is bonded;
[Claim 9]
L is C 1-3 Alkylene (the alkylene is C 1-2 The compound according to any one of Items 1 to 6, which may be substituted with 1 to 2 substituents independently selected from the group consisting of alkyl and hydroxy, or a pharmaceutically acceptable salt thereof Salt to be made;
[Section 10]
L is C 1-3 Alkylene and R 1 But C 1-6 Alkyl and R 1 And carbon atoms on L together, R 1 The compound or a pharmaceutically acceptable salt thereof according to any one of Items 1 to 6, which forms azetidine, piperidine, pyrrolidine together with the nitrogen atom to which is bonded;
[Section 11]
R 3 Is halogen, C 1-6 Alkyl, C 2-6 Alkenyl (the alkyl and the alkenyl are each halogen, C 3-7 Cycloalkyl (the cycloalkyl may be substituted by 1 to 2 halogens), 4-7 membered heterocycloalkyl (the heterocycloalkyl is C 1-2 Optionally substituted by 1 to 2 substituents independently selected from the group consisting of alkyl and oxo), C 6-10 Aryl and C 6-10 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of aryloxy (wherein the aryl and the aryloxy may each be substituted with 1 to 3 halogens) ), C 3-7 Cycloalkyl, C 4-7 Cycloalkenyl, 4- to 7-membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl, the heterocycloalkyl are each C 1-4 Alkyl, C 1-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of haloalkyl and halogen), C 6-10 Aryl (where aryl is halogen, C 1-4 Alkyl, C 1-4 Alkoxy (wherein the alkyl and the alkoxy are each 1-2 C 1-2 Optionally substituted by alkoxy), C 1-4 Haloalkyl, C 1-4 Haloalkoxy, cyano and C 1-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl) or 5-10 membered heteroaryl (wherein the heteroaryl is halogen, C 1-4 Alkyl, C 1-4 Haloalkyl, C 1-4 Alkoxy and C 1-4 The compound according to any one of Items 1 to 10, or a pharmaceutically acceptable salt thereof, which may be substituted with 1 to 3 substituents independently selected from the group consisting of haloalkoxy. Salt;
[Claim 12]
R 3 Is halogen, C 1-4 Alkyl, C 2-4 Alkenyl (the alkyl and the alkenyl are each halogen, C 4-6 Cycloalkyl (the cycloalkyl may be substituted by 1 to 2 halogens), optionally substituted 4-6 membered heterocycloalkyl (the heterocycloalkyl is C 1-2 Optionally substituted by 1 to 2 substituents independently selected from the group consisting of alkyl and oxo), C 6-10 Aryl and C 6-10 Optionally substituted by 1 to 3 substituents independently selected from the group consisting of aryloxy (wherein the aryl and the aryloxy may each be substituted by 1 to 2 halogens) ), C 3-6 Cycloalkyl, C 4-6 Cycloalkenyl, 4-6 membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl, the heterocycloalkyl may be substituted by 1 to 3 halogens), C 6-10 Aryl (where aryl is halogen, C 1-2 Alkyl, C 1-2 Alkoxy (wherein the alkyl and the alkoxy are each 1-2 C 1-2 Optionally substituted by alkoxy), C 1-2 Haloalkyl, C 1-2 Haloalkoxy, cyano and C 1-2 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl) or 6-membered heteroaryl (wherein the heteroaryl is halogen and C 1-2 The compound according to any one of Items 1 to 10, which may be substituted with 1 to 2 substituents independently selected from the group consisting of haloalkyl, or a pharmaceutically acceptable salt thereof salt;
[Claim 13]
R 3 But C 3-6 Cycloalkyl, 4-6 membered heterocycloalkyl (the cycloalkyl and the heterocycloalkyl may be substituted by 1 to 3 halogens), phenyl (the phenyl is halogen, C 1-2 Alkyl, C 1-2 Haloalkyl, C 1-2 Alkoxy and C 1-2 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of haloalkoxy) or 6-membered heteroaryl (wherein the heteroaryl is 1-2 C 1-2 The compound or a pharmaceutically acceptable salt thereof according to any one of Items 1 to 10, which may be substituted with haloalkyl;
[Section 14]
R 4 Is hydrogen, halogen or C 1-2 Item 14. The compound or a pharmaceutically acceptable salt thereof according to any one of Items 1 to 13, which is alkyl;
[Section 15]
R 4 The compound or a pharmaceutically acceptable salt thereof according to any one of Items 1 to 13, wherein is hydrogen.
[Section 16]
R 1 And R 2 Are each independently hydrogen, C 1-6 Alkyl (wherein alkyl is hydroxy, halogen, C 1-4 Alkoxy, C 1-4 Alkylsulfonyl, aminocarbonyl (the amino moiety of the aminocarbonyl is C 1-4 4-7 membered heterocycloalkyl and 4-7 membered heterocycloalkylcarbonyl (the 4-7 membered heterocycloalkyl and the 4-7 membered heterocyclo) The heterocycloalkyl part of alkylcarbonyl is each C 1-2 Substituted with 1 to 3 substituents independently selected from the group consisting of 1 to 2 substituents independently selected from the group consisting of alkyl and oxo) Or 4-7 membered heterocycloalkyl, which is halogen, and C 1-4 May be substituted with 1 to 3 substituents independently selected from the group consisting of alkoxy),
L is C 1-6 Alkylene (the alkylene is C 1-4 Alkyl, halogen, hydroxy and C 1-4 And optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxy), wherein R 1 But C 1-6 When alkyl, R 1 And carbon atoms on L together, R 1 A 4- to 7-membered saturated heterocyclic ring may be formed together with the nitrogen atom to which
R 3 Is halogen, C 1-6 Alkyl, C 2-6 Alkenyl (the alkyl and the alkenyl are each halogen, C 3-7 Cycloalkyl (the cycloalkyl may be substituted by 1 to 2 halogens), 4-7 membered heterocycloalkyl (the heterocycloalkyl is C 1-2 Optionally substituted by 1 to 2 substituents independently selected from the group consisting of alkyl and oxo), C 6-10 Aryl and C 6-10 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of aryloxy (wherein the aryl and the aryloxy may each be substituted with 1 to 3 halogens) ), C 3-7 Cycloalkyl, C 4-7 Cycloalkenyl, 4- to 7-membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl, the heterocycloalkyl are each C 1-4 Alkyl, C 1-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of haloalkyl and halogen), C 6-10 Aryl (where aryl is halogen, C 1-4 Alkyl, C 1-4 Alkoxy, cyano and C 1-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl) or 5-10 membered heteroaryl (wherein the heteroaryl is halogen, C 1-4 Alkyl, C 1-4 Haloalkyl, C 1-4 Alkoxy and C 1-4 May be substituted with 1 to 3 substituents independently selected from the group consisting of haloalkoxy),
R 4 Is hydrogen, halogen or C 1-2 Item 6. The compound according to Item 1, which is alkyl, or a pharmaceutically acceptable salt thereof;
[Section 17]
R 1 And R 2 Are each independently hydrogen, C 1-4 Alkyl (wherein the alkyl is hydroxy, C 1-2 Alkylsulfonyl, aminocarbonyl (the amino moiety of the aminocarbonyl is C 1-2 4-5 membered heterocycloalkyl and 4-6 membered heterocycloalkylcarbonyl (optionally mono-substituted by alkyl) (the 4-7 membered heterocycloalkyl and the 4-7 membered heterocycloalkylcarbonyl) Each heterocycloalkyl is C 1-2 Substituted with 1 to 2 substituents independently selected from the group consisting of 1 to 2 substituents independently selected from the group consisting of alkyl and oxo) Or 4 to 5 membered heterocycloalkyl, which may be substituted by 1 to 2 halogens,
L is C 1-4 Alkylene (the alkylene is C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of alkyl and hydroxy), wherein R 1 But C 1-6 When alkyl, R 1 And carbon atoms on L together, R 1 A 4- to 6-membered saturated heterocyclic ring may be formed together with the nitrogen atom to which
R 3 Is halogen, C 1-4 Alkyl, C 2-4 Alkenyl (the alkyl and the alkenyl are each halogen, C 4-6 Cycloalkyl (the cycloalkyl may be substituted by 1 to 2 halogens), 4 to 6 membered heterocycloalkyl (the heterocycloalkyl is C 1-2 Optionally substituted by 1 to 2 substituents independently selected from the group consisting of alkyl and oxo), C 6-10 Aryl and C 6-10 Optionally substituted by 1 to 3 substituents independently selected from the group consisting of aryloxy (wherein the aryl and the aryloxy may each be substituted by 1 to 2 halogens) ), C 3-6 Cycloalkyl, C 4-6 Cycloalkenyl, 4-6 membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl, the heterocycloalkyl may be substituted by 1 to 3 halogens), C 6-10 Aryl (where aryl is halogen, C 1-2 Alkyl, C 1-2 Alkoxy (wherein the alkyl and the alkoxy are each 1-2 C 1-2 Optionally substituted by alkoxy), C 1-2 Haloalkyl, C 1-2 Haloalkoxy, cyano and C 1-2 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl) or 6-membered heteroaryl (wherein the heteroaryl is halogen and C 1-2 May be substituted with 1 to 2 substituents independently selected from the group consisting of haloalkyl),
R 4 Is hydrogen, halogen or C 1-2 Item 6. The compound according to Item 1, which is alkyl, or a pharmaceutically acceptable salt thereof;
[Section 18]
R 1 Is hydrogen,
R 2 But C 1-4 Alkyl (wherein the alkyl is hydroxy and C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of alkylsulfonyl) or 4 to 5 membered heterocycloalkyl (the heterocycloalkyl is C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of alkyl and oxo),
L is C 1-3 Alkylene (the alkylene is C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of alkyl and hydroxy),
R 3 But C 3-6 Cycloalkyl, 4-6 membered heterocycloalkyl (the cycloalkyl and the heterocycloalkyl may be substituted by 1 to 3 halogens), phenyl (the phenyl is halogen, C 1-2 Alkyl, C 1-2 Alkoxy, C 1-2 Haloalkyl, C 1-2 Haloalkoxy, cyano and C 1-2 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl) or 6-membered heteroaryl (wherein the heteroaryl is 1-2 C 1-2 Optionally substituted by haloalkyl),
R 4 Item 2. The compound according to Item 1 or pharmaceutically acceptable salt thereof, wherein is hydrogen.
[Section 19]
R 1 But C 1-4 Alkyl,
R 2 But C 1-4 Alkyl (wherein the alkyl is hydroxy and C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of alkylsulfonyl) or 4 to 5 membered heterocycloalkyl (the heterocycloalkyl is C 1-2 Optionally substituted with 1 to 2 substituents independently selected from the group consisting of alkyl and oxo),
L is C 1-3 Alkylene and R 1 Together with the alkyl of L and the carbon atom on L 1 Forms azetidine, piperidine or pyrrolidine together with the nitrogen atom to which
R 3 But C 3-6 Cycloalkyl, 4-6 membered heterocycloalkyl (the cycloalkyl and the heterocycloalkyl may be substituted by 1 to 3 halogens), phenyl (the phenyl is halogen, C 1-2 Alkyl, C 1-2 Alkoxy, C 1-2 Haloalkyl, C 1-2 Haloalkoxy, cyano and C 1-2 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl) or 6-membered heteroaryl (wherein the heteroaryl is 1-2 C 1-2 Optionally substituted by haloalkyl),
R 4 Item 2. The compound according to Item 1 or pharmaceutically acceptable salt thereof, wherein is hydrogen.
[Section 20]
Item 1. The compound according to Item 1 or a pharmaceutically acceptable salt thereof selected from the following compound group:
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one (Example 14);
(S) -2-((2- (2-oxo-5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-3 (2H) -yl) ethyl) amino) propanamide (Example 16 );
2-((2- (2-oxo-5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-3 (2H) -yl) ethyl) amino) acetamide (Example 19);
(R) -2-((2- (2-oxo-5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-3 (2H) -yl) ethyl) amino) propanamide (Example 20 );
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (4- (trifluoromethoxy) phenyl) benzo [d] oxazol-2 (3H) -one (Example 34);
3- (2- (oxetane-3-ylamino) ethyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one (Example 42);
5- (4-fluorophenyl) -4-methyl-3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one (Example 44);
3- (2-Methyl-3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazole-2 (3H) -On (Example 67);
5- (4-Fluorophenyl) -3- (2-hydroxy-3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 70);
5- (4-fluorophenyl) -3- (3- (oxetan-3-ylamino) propyl) benzo [d] oxazol-2 (3H) -one (Example 72);
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5-phenylbenzo [d] oxazol-2 (3H) -one (Example 75);
5- (3-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 76);
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (4- (trifluoromethoxy) phenyl) benzo [d] oxazol-2 (3H) -one (Example 84);
5- (4-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 88);
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one (Example 92);
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one Example 93);
5- (4-Fluorophenyl) -4-methyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 99);
5- (4,4-difluorocyclohexyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one (Example 100);
6-Fluoro-3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (tetrahydro-2H-pyran-4-yl) benzo [d] oxazol-2 (3H) -one ( Example 101);
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (tetrahydro-2H-pyran-4-yl) benzo [d] oxazol-2 (3H) -one (Example 102) ;
5-cyclobutyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 111);
5-cyclopentyl-3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one (Example 118);
5-butyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 124);
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -7- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one (implemented) Example 149);
5- (4-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) butyl) benzo [d] oxazol-2 (3H) -one (Example 150);
5- (3,3-difluorocyclobutyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 155);
5- (3,3-difluorocyclobutyl) -3- (1- (2- (methylsulfonyl) ethyl) azetidin-3-yl) benzo [d] oxazol-2 (3H) -one (Example 156);
5- (3,3-difluorocyclobutyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one (Example 157);
5- (3,3-difluorocyclobutyl) -3- (4-((2- (methylsulfonyl) ethyl) amino) butyl) benzo [d] oxazol-2 (3H) -one (Example 158);
5- (3,3-difluorocyclobutyl) -3- (1- (2- (methylsulfonyl) ethyl) piperidin-4-yl) benzo [d] oxazol-2 (3H) -one (Example 159);
2- (3- (5- (3,3-difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) azetidin-1-yl) acetamide (Example 162);
2- (4- (5- (3,3-difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) piperidin-1-yl) acetamide (Example 163); or
5- (3,3-difluorocyclobutyl) -3- (3-((2-hydroxy-2-methylpropyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 165);
[Claim 21]
The compound according to claim 1 or a pharmaceutically acceptable salt thereof selected from the following group of compounds:
5- (4-Fluorophenyl) -3- (2-hydroxy-3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 70);
5- (4-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 88);
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one Example 93);
5- (4,4-difluorocyclohexyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one (Example 100);
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (tetrahydro-2H-pyran-4-yl) benzo [d] oxazol-2 (3H) -one (Example 102) ;
5-cyclobutyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 111);
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -7- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one (implemented) Example 149);
5- (3,3-difluorocyclobutyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 155);
5- (3,3-difluorocyclobutyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one (Example 157);
5- (3,3-difluorocyclobutyl) -3- (1- (2- (methylsulfonyl) ethyl) piperidin-4-yl) benzo [d] oxazol-2 (3H) -one (Example 159);
2- (4- (5- (3,3-difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) piperidin-1-yl) acetamide (Example 163); or
5- (3,3-difluorocyclobutyl) -3- (3-((2-hydroxy-2-methylpropyl) amino) propyl) benzo [d] oxazol-2 (3H) -one (Example 165);
[Item 22]
Following formula (2);
[式(2)中、
R5は、ニトロ、アミノ(該アミノは、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチル、該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)からなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)またはフタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)であり、
R6は、水素またはC1−2アルキル(該アルキルは、C1−2アルコキシ、フェニルおよび4−メトキシフェニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
R7は、水素またはC1−2アルキルであり、
R8は、ヨウ素、臭素、塩素または水素であり、ここにおいて、R8がヨウ素、臭素または塩素であるとき、置換可能な位置に1〜2個置換していてもよく、
A−Aは、C−CまたはC=Cである]
で表される化合物またはその塩;
[項23]
下記式(3);
[In Formula (2),
R 5 represents nitro, amino (the amino is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl moiety in the phenylmethyl, phenylmethyloxycarbonyl is halogen, C 1-2 alkyl And optionally substituted with 1 to 6 substituents independently selected from the group consisting of C 1-2 alkoxy), fluorenylmethylcarbonyl, benzenesulfonyl (wherein the benzenesulfonyl is 1- Optionally substituted by 1 to 2 substituents independently selected from the group consisting of: optionally substituted) or phthalimide (the phenyl portion of the phthalimide is substituted by C 1-2 alkyl) 1 to 3 may be substituted),
R 6 is hydrogen or C 1-2 alkyl, wherein the alkyl is substituted with 1-2 substituents independently selected from the group consisting of C 1-2 alkoxy, phenyl and 4-methoxyphenyl. Is good)
R 7 is hydrogen or C 1-2 alkyl;
R 8 is iodine, bromine, chlorine or hydrogen, and when R 8 is iodine, bromine or chlorine, 1 to 2 may be substituted at substitutable positions;
AA is CC or C = C]
Or a salt thereof;
[Section 23]
Following formula (3);
[式(3)中、
R7は、水素またはC1−2アルキルであり、
R9は、C1−4アルキルオキシカルボニル、フェニルメチルオキシカルボニル(該フェニルメチルオキシカルボニルのフェニルメチル部分は、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニルまたはベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)であり、
R10は、水素またはC1−2アルキルであり、ここにおいて、R9およびR10が一緒になって、R9およびR10が結合する窒素原子と共にフタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)を形成してもよく、
Mは、C1−4アルキレンであり、ここにおいて、R10がC1−2アルキルであるとき、R10とM上の炭素原子が一緒になって、R10が結合する窒素原子と共に4〜6員の飽和複素環を形成してもよく、
Bは、ハロゲンである]
で表される化合物またはその塩;
[項24]
下記式(4);
[In Formula (3),
R 7 is hydrogen or C 1-2 alkyl;
R 9 represents C 1-4 alkyloxycarbonyl, phenylmethyloxycarbonyl (the phenylmethyl moiety of the phenylmethyloxycarbonyl is independently selected from the group consisting of C 1-2 alkyl and C 1-2 alkoxy) Up to 6 substituents), fluorenylmethylcarbonyl or benzenesulfonyl (wherein the benzenesulfonyl may be substituted with 1 to 2 by nitro),
R 10 is hydrogen or C 1-2 alkyl, where R 9 and R 10 are taken together with the nitrogen atom to which R 9 and R 10 are attached together with phthalimide (the phenyl portion of the phthalimide is C 1 1-3 may be substituted by -2 alkyl),
M is C 1-4 alkylene, where when R 10 is C 1-2 alkyl, R 10 and the carbon atom on M are taken together and together with the nitrogen atom to which R 10 is attached, 4- May form a 6-membered saturated heterocycle,
B is halogen]
Or a salt thereof;
[Claim 24]
Following formula (4);
R7は、水素、またはC1−2アルキルであり、
R11およびR12は、それぞれ独立して、水素、C1−4アルキル(該アルキルは、C1−4アルコキシまたはフェニルによって置換されていてもよい)、C1−4ハロアルキル、C1−4ハロアルコキシ、ニトロ、アミノ(該アミノは、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチルおよび該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)からなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、フタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)またはC1−2アルコキシ(該アルコキシにおけるアルキル部分は、C1−2アルコキシ、フェニルおよび4−メトキシフェニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、ここにおいて、R11およびR12が互いにオルト位である場合は、R11とR12がフェニルと一緒になってベンズオキサゾロン環、インドール環またはインダゾール環を形成してもよい]
で表される化合物またはその塩の製造方法であって、下記式(5)
R 7 is hydrogen or C 1-2 alkyl;
R 11 and R 12 are each independently hydrogen, C 1-4 alkyl (wherein the alkyl may be substituted by C 1-4 alkoxy or phenyl), C 1-4 haloalkyl, C 1-4. Haloalkoxy, nitro, amino (wherein the amino is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl moiety in the phenylmethyl and phenylmethyloxycarbonyl is halogen, C 1-2 alkyl and Optionally substituted with 1 to 6 substituents independently selected from the group consisting of C 1-2 alkoxy, fluorenylmethylcarbonyl, benzenesulfonyl (wherein the benzenesulfonyl is 1-2 with nitro) 1 may be independently selected from the group consisting of Number of which may be substituted by a substituent), phthalimide (phenyl moiety of the phthalimide may be 1-3 substituted by C 1-2 alkyl) or C 1-2 alkoxy (alkyl in the alkoxy The moiety is optionally substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkoxy, phenyl and 4-methoxyphenyl, wherein R 11 and R 11 When 12 is ortho to each other, R 11 and R 12 may be combined with phenyl to form a benzoxazolone ring, an indole ring or an indazole ring.
Or a salt thereof, which is represented by the following formula (5):
[式(5)中、
R7、R11およびR12は、上記式(4)と同じであり、
Bは、ハロゲンである]で表される化合物と、下記式(6)
[In Formula (5),
R 7 , R 11 and R 12 are the same as in the above formula (4),
B is a halogen], and the following formula (6)
Xは、ヨウ素、臭素または塩素である]で表される化合物を、パラジウム触媒を用いてカップリングさせる工程を含む製造方法。
[項25]
下記式(7);
X is an iodine, bromine, or chlorine] manufacturing method including a step of coupling a compound represented by a palladium catalyst.
[Claim 25]
Following formula (7);
[式(7)中、
R7は、水素またはC1−2アルキルであり、
R13は、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチルおよび該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)であり、
R14は、水素、C1−2アルキル、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチルおよび該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニルまたはベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)であり、ここにおいて、R13およびR14が一緒になって、R13およびR14が結合する窒素原子と共にフタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)を形成してもよく、
Mは、C1−4アルキレンであり、ここにおいて、R14がC1−2アルキルであるとき、R14とM上の炭素原子が一緒になって、R14が結合する窒素原子と共に4〜6員の飽和複素環を形成してもよい]
で表される化合物またはその塩の製造方法であって、下記式(8)
[In Formula (7),
R 7 is hydrogen or C 1-2 alkyl;
R 13 is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl and the phenylmethyl moiety in the phenylmethyloxycarbonyl are composed of halogen, C 1-2 alkyl and C 1-2 alkoxy. Optionally substituted with 1 to 6 substituents independently selected from the group), fluorenylmethylcarbonyl, benzenesulfonyl (the benzenesulfonyl may be substituted with 1 to 2 by nitro) ) And
R 14 is hydrogen, C 1-2 alkyl, C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl moiety in the phenylmethyl and phenylmethyloxycarbonyl is halogen, C 1-2 alkyl And optionally substituted with 1 to 6 substituents independently selected from the group consisting of C 1-2 alkoxy), fluorenylmethylcarbonyl or benzenesulfonyl (wherein the benzenesulfonyl is 1- In which R 13 and R 14 are taken together with the nitrogen atom to which R 13 and R 14 are attached together with phthalimide (the phenyl moiety of the phthalimide is C 1-2 1 to 3 may be substituted with alkyl) ,
M is C 1-4 alkylene, where when R 14 is C 1-2 alkyl, R 14 and the carbon atom on M are taken together and together with the nitrogen atom to which R 14 is attached, 4 to 4 A 6-membered saturated heterocyclic ring may be formed]
Or a salt thereof, which is represented by the following formula (8):
[式(8)中、
R7、R13、R14およびMは、上記式(7)と同じであり、Bは、ハロゲンである]で表される化合物と、下記式(9)
[In Formula (8),
R 7 , R 13 , R 14 and M are the same as those in the above formula (7), B is a halogen], and the following formula (9)
Xは、ヨウ素、臭素または塩素である]で表される化合物を、パラジウム触媒を用いてカップリングさせる工程を含む製造方法。
[項26]
請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩を含有する医薬組成物;
[項27]
請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩を有効成分として含有する、Nav1.7(SCN9A)、Nav1.8(SCN10A)またはNav1.7およびNav1.8の両方が関与する疾病の治療薬;
[項28]
請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩を有効成分として含有する、Nav1.7(SCN9A)が関与する疾病の治療薬;
[項29]
請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩を有効成分として含有する、Nav1.8(SCN10A)が関与する疾病の治療薬;
[項30]
請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩を有効成分として含有する神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害または多発性硬化症の治療薬;
[項31]
請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩を有効成分として含有する、Nav1.7(SCN9A)、Nav1.8(SCN10A)またはNav1.7およびNav1.8の両方が関与する疾病の治療薬と、抗てんかん薬、抗うつ薬、麻薬性鎮痛薬、抗炎症薬、還元酵素阻害剤、プロスタグランジン誘導体製剤から選択される少なくとも1種以上の薬剤とを組み合わせてなる医薬;
[項32]
神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害または多発性硬化症の治療薬を製造するための、請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩の使用;
[項33]
治療が必要な患者に、治療上の有効量の請求項1〜21のいずれか一項に記載の化合物またはその薬学的に許容される塩を投与することを特徴とする、神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害または多発性硬化症を治療するための方法。
X is an iodine, bromine, or chlorine] manufacturing method including a step of coupling a compound represented by a palladium catalyst.
[Claim 26]
A pharmaceutical composition comprising the compound according to any one of claims 1 to 21 or a pharmaceutically acceptable salt thereof;
[Section 27]
A Nav1.7 (SCN9A), Nav1.8 (SCN10A) or Nav1.7 and Nav1.2 containing the compound according to any one of claims 1 to 21 or a pharmaceutically acceptable salt thereof as an active ingredient. 8 for the treatment of diseases involving both;
[Claim 28]
A therapeutic agent for a disease involving Nav1.7 (SCN9A), comprising as an active ingredient the compound according to any one of claims 1 to 21 or a pharmaceutically acceptable salt thereof;
[Item 29]
A therapeutic agent for a disease involving Nav1.8 (SCN10A) comprising the compound according to any one of claims 1 to 21 or a pharmaceutically acceptable salt thereof as an active ingredient;
[Section 30]
Neuropathic pain, nociceptive pain, inflammatory pain, small-diameter neuropathy, limb redness containing the compound according to any one of claims 1 to 21 or a pharmaceutically acceptable salt thereof as an active ingredient A drug for pain, paroxysmal pain, dysuria or multiple sclerosis;
[Claim 31]
A Nav1.7 (SCN9A), Nav1.8 (SCN10A) or Nav1.7 and Nav1.2 containing the compound according to any one of claims 1 to 21 or a pharmaceutically acceptable salt thereof as an active ingredient. A therapeutic agent for a disease involving both of the above 8 and at least one drug selected from antiepileptic drugs, antidepressants, narcotic analgesics, anti-inflammatory drugs, reductase inhibitors, and prostaglandin derivative preparations. A pharmaceutical comprising a combination of
[Section 32]
Claims 1 to 21 for the manufacture of a therapeutic agent for neuropathic pain, nociceptive pain, inflammatory pain, small fiber neuropathy, limb erythema, paroxysmal pain, dysuria or multiple sclerosis Use of a compound according to any one of the above or a pharmaceutically acceptable salt thereof;
[Section 33]
Neuropathic pain, characterized in that a therapeutically effective amount of a compound according to any one of claims 1 to 21 or a pharmaceutically acceptable salt thereof is administered to a patient in need of treatment, A method for treating nociceptive pain, inflammatory pain, small fiber neuropathy, erythema limb pain, paroxysmal pain, dysuria or multiple sclerosis.
本発明により、新規ベンズオキサゾロンまたはそれらの薬学的に許容される塩を含むNav1.7および/またはNav1.8の阻害剤が提供される。本発明の化合物は、Nav1.7(SCN9A)あるいはNav1.8(SCN10A)、またはその両方が関与する病態全般に対する治療薬または予防薬として有用であり、具体的には、神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害または多発性硬化症等の患者に適用可能である。 The present invention provides inhibitors of Nav1.7 and / or Nav1.8 comprising novel benzoxazolones or pharmaceutically acceptable salts thereof. The compounds of the present invention are useful as therapeutic or prophylactic agents for all pathological conditions involving Nav1.7 (SCN9A) or Nav1.8 (SCN10A), or both, specifically neuropathic pain, nociception Applicable to patients with receptive pain, inflammatory pain, small-fiber neuropathy, erythema acropasis, paroxysmal pain, dysuria or multiple sclerosis.
以下に、本発明をさらに詳細に説明する。本明細書において「置換基」の定義における炭素の数を、例えば、「C1−6」などと表記する場合もある。具体的には、「C1−6アルキル」なる表記は、炭素数1から6のアルキルと同義である。また、本明細書において、「置換されていてもよい」または「置換されている」なる用語を特に明示していない置換基については、「非置換」の置換基を意味する。例えば、「C1−6アルキル」とは、「非置換C1−6アルキル」であることを意味する。 The present invention is described in further detail below. In the present specification, the number of carbons in the definition of “substituent” may be expressed as “C 1-6 ”, for example. Specifically, the notation “C 1-6 alkyl” is synonymous with alkyl having 1 to 6 carbons. In the present specification, a substituent that does not clearly indicate the term “optionally substituted” or “substituted” means an “unsubstituted” substituent. For example, “C 1-6 alkyl” means “unsubstituted C 1-6 alkyl”.
また、本明細書における置換基の説明において、「基」なる用語を省略する場合もある。尚、「置換されていてもよい」で定義される場合において、置換基が存在するときの置換基の数は、置換可能であれば特に制限はなく、1または複数である。すなわち、該当する基における置換可能な炭素原子、又は炭素原子および窒素原子上における置換可能な数の置換基によって置換されていてもよい事を示す。また、特に指示した場合を除き、各々の基の説明はその基が他の基の一部分または置換基である場合にも該当する。 In addition, in the description of substituents in this specification, the term “group” may be omitted. In the case where “optionally substituted” is defined, the number of substituents when a substituent is present is not particularly limited as long as substitution is possible, and is one or more. That is, it may be substituted by a substitutable carbon atom in the corresponding group, or a substitutable number of substituents on the carbon atom and nitrogen atom. In addition, unless otherwise specified, the description of each group also applies when the group is a part of another group or a substituent.
「ハロゲン」としては、例えばフッ素、塩素、臭素またはヨウ素等が挙げられる。好ましくは、フッ素または塩素である。 Examples of “halogen” include fluorine, chlorine, bromine or iodine. Preferred is fluorine or chlorine.
「C1−2アルキル」、「C1−4アルキル」、「C1−6アルキル」とは、それぞれ、炭素数が1〜2個、1〜4個、1〜6個の直鎖状または分枝鎖状の飽和炭化水素基を意味する。例えば、「C1−2アルキル」としては、メチル、エチルが挙げられ、「C1−4アルキル」としては、「C1−2アルキル」に加えn−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチルが挙げられ、「C1−6アルキル」としては、「C1−4アルキル」に加え、ペンチル、イソペンチル、ネオペンチル、n−ヘキシル、ならびにそれらの構造異性体が挙げられる。当該「C1−6アルキル」または「C1−4アルキル」としては、C1−3アルキルが好ましく、メチルおよびエチルがより好ましい。「C1−2アルキル」としては、メチル、エチルが好ましい。「置換されていてもよいC1−4アルキル」における置換基は、下記置換基群[A]から選択され、置換可能な位置にそれぞれ独立して1〜3個置換されていてもよい。 “C 1-2 alkyl”, “C 1-4 alkyl” and “C 1-6 alkyl” are linear or straight chain having 1 to 2, 1 to 4 and 1 to 6 carbon atoms, respectively. It means a branched saturated hydrocarbon group. For example, “C 1-2 alkyl” includes methyl and ethyl, and “C 1-4 alkyl” includes “C 1-2 alkyl” in addition to n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl and tert-butyl are exemplified, and “C 1-6 alkyl” includes “C 1-4 alkyl”, pentyl, isopentyl, neopentyl, n-hexyl, and structural isomers thereof. . As the “C 1-6 alkyl” or “C 1-4 alkyl”, C 1-3 alkyl is preferable, and methyl and ethyl are more preferable. As “C 1-2 alkyl”, methyl and ethyl are preferable. The substituent in “optionally substituted C 1-4 alkyl” is selected from the following substituent group [A], and may be independently substituted at 1 to 3 positions at substitutable positions.
「C3−6シクロアルキル」、「C4−6シクロアルキル」、「C3−7シクロアルキル」とは、それぞれ、炭素数3〜6個、4〜7個、3〜7個を有する、環状の飽和炭化水素基を意味する。「C3−6シクロアルキル」または「C3−7シクロアルキル」として好ましくは、炭素数3〜6個の「C3−6シクロアルキル」である。「C4−6シクロアルキル」として好ましくは、炭素数4〜6個のシクロアルキルである。「C3−6シクロアルキル」の具体例としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル等が挙げられ、「C3−7シクロアルキル」としては、上記に加えシクロヘプチル等が挙げられる。「C4−6シクロアルキル」の具体例としては、例えば、シクロブチル、シクロペンチル、シクロヘキシルが挙げられる。「置換されていてもよいC3−7シクロアルキル」における置換基は、下記置換基群[B]から選択され、置換可能な位置にそれぞれ独立して1〜3個置換されていてもよい。 “C 3-6 cycloalkyl”, “C 4-6 cycloalkyl”, and “C 3-7 cycloalkyl” have 3 to 6 carbon atoms, 4 to 7 carbon atoms, and 3 to 7 carbon atoms, respectively. A cyclic saturated hydrocarbon group is meant. “C 3-6 cycloalkyl” or “C 3-7 cycloalkyl” is preferably “C 3-6 cycloalkyl” having 3 to 6 carbon atoms. “C 4-6 cycloalkyl” is preferably cycloalkyl having 4 to 6 carbon atoms. Specific examples of “C 3-6 cycloalkyl” include, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and the like, and “C 3-7 cycloalkyl” includes cycloheptyl and the like in addition to the above. Specific examples of “C 4-6 cycloalkyl” include, for example, cyclobutyl, cyclopentyl, and cyclohexyl. The substituent in “optionally substituted C 3-7 cycloalkyl” is selected from the following substituent group [B], and may be independently substituted at 1 to 3 positions at substitutable positions.
前記「C3−7シクロアルキル」には、「C3−7シクロアルキル」と、フェニルまたは5員もしくは6員の窒素、硫黄または酸素から選ばれるヘテロ原子を1個または同一または異なって2個以上(例えば2〜4個)含有する環とが縮環した2環式の基も包含される。該基の具体例としては、例えば、下記式で表される基等が挙げられる。 In the “C 3-7 cycloalkyl”, “C 3-7 cycloalkyl” and two or more heteroatoms selected from phenyl or 5-membered or 6-membered nitrogen, sulfur or oxygen are the same or different. A bicyclic group condensed with the ring contained above (for example, 2 to 4) is also included. Specific examples of the group include groups represented by the following formulas.
(式中、結合位置は、シクロアルキル環の炭素原子上であり、化学的に結合可能な任意の位置である。)
(In the formula, the bonding position is on a carbon atom of the cycloalkyl ring and is an arbitrary position capable of being chemically bonded.)
「C6−10アリール」とは、炭素数6〜10個を有する芳香族炭化水素基を意味する。好ましくはフェニルである。「C6−10アリール」の具体例としては、例えば、フェニル、1−ナフチルまたは2−ナフチル等が挙げられる。「置換されていてもよいC6−10アリール」における置換基は、下記置換基群[B]から選択され、置換可能な位置にそれぞれ独立して1〜3個置換されていてもよい。 “C 6-10 aryl” means an aromatic hydrocarbon group having 6 to 10 carbon atoms. Preferred is phenyl. Specific examples of “C 6-10 aryl” include phenyl, 1-naphthyl, 2-naphthyl and the like. The substituent in “optionally substituted C 6-10 aryl” is selected from the following substituent group [B], and may be independently substituted at 1 to 3 positions at substitutable positions.
前記「C6−10アリール」には、「フェニル」と、窒素、硫黄または酸素から選ばれるヘテロ原子を1個または同一または異なって2個以上(例えば2〜4個)含有する5もしくは6員の環、または5〜7員のシクロアルキル環(例えばシクロペンタン、シクロヘキサンまたはシクロヘプタン)とが縮環した基も包含される。該基の具体例としては、例えば、下記式で表される基等が挙げられる。 The “C 6-10 aryl” is a 5- or 6-membered member containing “phenyl” and one or more of the same or different heteroatoms selected from nitrogen, sulfur or oxygen (for example, 2 to 4). Or a group condensed with a 5- to 7-membered cycloalkyl ring (for example, cyclopentane, cyclohexane or cycloheptane). Specific examples of the group include groups represented by the following formulas.
(式中、結合位置は、芳香環の炭素原子上であり、化学的に結合可能な任意の位置である。) (In the formula, the bonding position is on a carbon atom of the aromatic ring and is an arbitrary position capable of chemically bonding.)
「4〜7員のヘテロシクロアルキル」「4〜6員のヘテロシクロアルキル」「4〜5員のヘテロシクロアルキル」とは、環を構成する原子として炭素に加えて、窒素、硫黄または酸素から選ばれるヘテロ原子を1個または同一または異なって2個以上(例えば2〜4個)含有する、それぞれ、4〜7員、4〜6員、4〜5員の飽和の環状基を意味し、一部不飽和であるものも含まれる。「4〜7員のヘテロシクロアルキル」「4〜6員のヘテロシクロアルキル」として好ましくは、ピロリジニル、ピペリジニル、ピペラジニル、テトラヒドロフラニル、テトラヒドロ−2H−ピラニルである。「4〜5員のヘテロシクロアルキル」として好ましくは、ピロリジニル、テトラヒドロフラニルである。「置換されていてもよい4〜7員のヘテロシクロアルキル」における置換基は、下記置換基群[C]から選択され、置換可能な位置にそれぞれ独立して1〜3個置換されていてもよい。 “4-7 membered heterocycloalkyl”, “4-6 membered heterocycloalkyl” and “4-5 membered heterocycloalkyl” are nitrogen, sulfur or oxygen in addition to carbon as atoms constituting the ring. Means a 4-7 membered, 4-6 membered, 4-5 membered saturated cyclic group containing one or the same or different heteroatoms selected from 2 or more (for example, 2-4), Some are partially unsaturated. “4- to 7-membered heterocycloalkyl” and “4- to 6-membered heterocycloalkyl” are preferably pyrrolidinyl, piperidinyl, piperazinyl, tetrahydrofuranyl, and tetrahydro-2H-pyranyl. “4- to 5-membered heterocycloalkyl” is preferably pyrrolidinyl or tetrahydrofuranyl. The substituent in the “optionally substituted 4 to 7-membered heterocycloalkyl” is selected from the following substituent group [C], and may be independently substituted with 1 to 3 at each substitutable position. Good.
「4〜7員のヘテロシクロアルキルカルボニル」とは、前述の「4〜7員のヘテロシクロアルキル」で置換されたカルボニル基を意味し、具体的には、ピロリジニルカルボニル、ピペリジニルカルボニル、ピペラジニルカルボニルが挙げられる。好ましくはピペラジニルカルボニルが挙げられる。「置換されていてもよい4〜7員のヘテロシクロアルキルカルボニル」における置換基は、下記置換基群[C]から選択され、置換可能な位置にそれぞれ独立して1〜3個置換されていてもよい。 “4-7 membered heterocycloalkylcarbonyl” means a carbonyl group substituted with the above “4-7 membered heterocycloalkyl”, specifically, pyrrolidinylcarbonyl, piperidinylcarbonyl. And piperazinylcarbonyl. Preferably, piperazinylcarbonyl is used. The substituent in the “optionally substituted 4 to 7-membered heterocycloalkylcarbonyl” is selected from the following substituent group [C], and 1 to 3 substituents are independently substituted at substitutable positions. Also good.
「C4−6シクロアルケニル」、「C4−7シクロアルケニル」とは、1個以上の2重結合を有する、それぞれ、炭素数4〜6個、4〜7個を有する環状の炭化水素基を意味する。「C4−6シクロアルケニル」として具体的には、シクロペンテニル、シクロヘキセニルが挙げられ、「C4−7シクロアルケニル」としては上記に加え、シクロヘプテニルが挙げられる。好ましくはシクロヘキセニルが挙げられる。 “C 4-6 cycloalkenyl” and “C 4-7 cycloalkenyl” are cyclic hydrocarbon groups having 1 to 6 double bonds and 4 to 6 carbon atoms and 4 to 7 carbon atoms, respectively. Means. Specific examples of “C 4-6 cycloalkenyl” include cyclopentenyl and cyclohexenyl, and “C 4-7 cycloalkenyl” includes cycloheptenyl in addition to the above. Preferably, cyclohexenyl is used.
「5〜10員のヘテロアリール」とは、環を構成する原子として炭素に加えて、窒素、硫黄または酸素から選ばれるヘテロ原子を1個または同一または異なって2個以上(例えば2〜4個)含有する単環式もしくは多環式の芳香族基を意味する。好ましくは5または6員の単環式ヘテロアリールである。 The “5- to 10-membered heteroaryl” is one or the same or different heteroatoms selected from nitrogen, sulfur or oxygen in addition to carbon as a ring-constituting atom (for example, 2 to 4). ) Means a monocyclic or polycyclic aromatic group contained. Preferably it is a 5- or 6-membered monocyclic heteroaryl.
前記5〜10員のヘテロアリールのうち、多環式ヘテロアリールには、単環式へテロアリールと芳香環(例えばベンゼン、ピリジンなど)または非芳香族環(例えばシクロヘキシルなど)とが縮環したものも包含される。「5〜10員のヘテロアリール」の具体例としては、例えば、下記式で表される基が挙げられる。中でも好ましくは、ピラゾリル、イミダゾリル、ピリジル、ピリミジニル、ピラジニル、ピリダジニルである。 Among the 5- to 10-membered heteroaryl, polycyclic heteroaryl is a condensed monocyclic heteroaryl and an aromatic ring (eg, benzene, pyridine) or a non-aromatic ring (eg, cyclohexyl). Are also included. Specific examples of “5- to 10-membered heteroaryl” include, for example, groups represented by the following formulae. Of these, pyrazolyl, imidazolyl, pyridyl, pyrimidinyl, pyrazinyl and pyridazinyl are preferable.
(式中、単環式へテロアリールにおける結合位置は、化学的に結合可能な任意の位置である。また、多環式へテロアリールにおける結合位置は、ヘテロアリール環上であり、化学的に結合可能な任意の位置である。) (In the formula, the bonding position in monocyclic heteroaryl is any position that can be chemically bonded. In addition, the bonding position in polycyclic heteroaryl is on a heteroaryl ring and can be chemically bonded. Any position.)
「4〜6員の飽和複素環」、「4〜7員の飽和複素環」とは、環を構成する原子として炭素に加えて、窒素、酸素および硫黄から選択されるヘテロ原子を1個または同一または異なって2〜3個有する、それぞれ、4〜6員、4〜7員の環状飽和炭化水素基を意味する。「4〜6員の飽和複素環」の具体例としては、ピロリジニル、ピペリジニル、モルホリニル、チオモルホリニル、ジオキソチオモルホリニル、テトラヒドロフラニル、アゼチジニル等が挙げられ、「4〜7員の飽和複素環」としては上記に加え、アゼパニル等が挙げられる。「4〜6員の飽和複素環」または「4〜7員の飽和複素環」として好ましくは、ピペリジニル、ピロリジニル、アゼチジニルである。 The “4- to 6-membered saturated heterocyclic ring” and “4- to 7-membered saturated heterocyclic ring” mean one hetero atom selected from nitrogen, oxygen and sulfur in addition to carbon as an atom constituting the ring, or It means the same or different 2 to 3 cyclic hydrocarbon groups having 4 to 6 members and 4 to 7 members, respectively. Specific examples of “4 to 6-membered saturated heterocycle” include pyrrolidinyl, piperidinyl, morpholinyl, thiomorpholinyl, dioxothiomorpholinyl, tetrahydrofuranyl, azetidinyl and the like, and “4 to 7-membered saturated heterocycle” In addition to the above, azepanyl and the like can be mentioned. As the “4 to 6-membered saturated heterocyclic ring” or “4 to 7-membered saturated heterocyclic ring”, piperidinyl, pyrrolidinyl and azetidinyl are preferable.
「C1−2アルコキシ」、「C1−4アルコキシ」とは、それぞれ、前記「C1−2アルキル」、「C1−4アルキル」で置換されたオキシ基を意味する。「C1−2アルコキシ」の具体例としては、メトキシ、エトキシが挙げられ、「C1−4アルコキシ」としては上記に加え、プロポキシ、イソプロポキシ、ブトキシ、イソブトキシ、sec−ブトキシ、tert−ブトキシ等が挙げられる。「C1−2アルコキシ」として好ましくは、メトキシ、エトキシが挙げられ、「C1−4アルコキシ」として好ましくは、メトキシ、エトキシ、イソプロポキシが挙げられる。「置換されていてもよいC1−4アルコキシ」における置換基は、下記置換基群[A]から選択され、置換可能な位置にそれぞれ独立して1〜3個置換されていてもよい。 “C 1-2 alkoxy” and “C 1-4 alkoxy” mean an oxy group substituted with the above “C 1-2 alkyl” and “C 1-4 alkyl”, respectively. Specific examples of “C 1-2 alkoxy” include methoxy and ethoxy. In addition to the above, “C 1-4 alkoxy” includes propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy and the like. Is mentioned. “C 1-2 alkoxy” preferably includes methoxy and ethoxy, and “C 1-4 alkoxy” preferably includes methoxy, ethoxy and isopropoxy. The substituent in “optionally substituted C 1-4 alkoxy” is selected from the following substituent group [A], and may be independently substituted at 1 to 3 positions at substitutable positions.
「C1−2ハロアルキル」、「C1−4ハロアルキル」とは、それぞれ、ハロゲンで置換されたC1−2アルキル基、C1−4アルキル基を意味する。「C1−2ハロアルキル」、「C1−4ハロアルキル」の具体例としては、トリフルオロメチル、フルオロエチル、トリフルオロエチル、ペンタフルオロエチル等が挙げられる。中でも好ましくは、トリフルオロメチルである。 “C 1-2 haloalkyl” and “C 1-4 haloalkyl” mean a C 1-2 alkyl group and a C 1-4 alkyl group each substituted with a halogen. Specific examples of “C 1-2 haloalkyl” and “C 1-4 haloalkyl” include trifluoromethyl, fluoroethyl, trifluoroethyl, pentafluoroethyl and the like. Of these, trifluoromethyl is preferable.
「C1−2ハロアルコキシ」「C1−4ハロアルコキシ」とは、それぞれ、前記「C1−2ハロアルコキシ」、「C1−4ハロアルキル」で置換されたオキシ基を意味する。「C1−2ハロアルコキシ」、「C1−4ハロアルコキシ」の具体例としては、トリフルオロメチルオキシ、1,1,2,2−テトラフルオロエチルオキシ、ジフルオロメチルオキシ等が挙げられる。中でも好ましくは、トリフルオロメチルである。 “C 1-2 haloalkoxy” and “C 1-4 haloalkoxy” mean an oxy group substituted by the above “C 1-2 haloalkoxy” and “C 1-4 haloalkyl”, respectively. Specific examples of “C 1-2 haloalkoxy” and “C 1-4 haloalkoxy” include trifluoromethyloxy, 1,1,2,2-tetrafluoroethyloxy, difluoromethyloxy and the like. Of these, trifluoromethyl is preferable.
「C6−10アリールオキシ」とは、前記「C6−10アリール」で置換されたオキシ基を意味する。「C6−10アリールオキシ」の具体例としては、例えばフェニルオキシ、ナフチルオキシ等が挙げられ、好ましくはフェニルオキシである。「置換されていてもよいC6−10アリールオキシ」における置換基は、下記置換基群[B]から選択され、置換可能な位置にそれぞれ独立して1〜3個置換されていてもよい。 “C 6-10 aryloxy” means an oxy group substituted with the above “C 6-10 aryl”. Specific examples of “C 6-10 aryloxy” include, for example, phenyloxy, naphthyloxy and the like, preferably phenyloxy. The substituent in “optionally substituted C 6-10 aryloxy” is selected from the following substituent group [B], and may be independently substituted at 1 to 3 positions at substitutable positions.
「C1−3アルキレン」、「C1−4アルキレン」、「C1−6アルキレン」とは、それぞれ、炭素数が1〜3、1〜4、1〜6の、直鎖または分岐鎖の炭素リンカーを意味する。「C1−3アルキレン」の具体例としては、メチレン、エチレン、プロピレンが挙げられ、「C1−4アルキレン」としては、「C1−3アルキレン」に加え、2−メチルプロピレン、ブチレン等が挙げられ、「C1−6アルキレン」としては、「C1−4アルキレン」に加え、2,2−ジメチルプロピレン、ペンチレン、へキシレン等が挙げられる。好ましくエチレン、プロピレンである。 “C 1-3 alkylene”, “C 1-4 alkylene” and “C 1-6 alkylene” are linear or branched, each having 1 to 3, 1 to 4, and 1 to 6 carbon atoms. Means carbon linker. Specific examples of “C 1-3 alkylene” include methylene, ethylene and propylene, and “C 1-4 alkylene” includes “C 1-3 alkylene”, 2-methylpropylene, butylene and the like. Examples of the “C 1-6 alkylene” include 2,2-dimethylpropylene, pentylene, hexylene and the like in addition to “C 1-4 alkylene”. Ethylene and propylene are preferred.
「C2−4アルケニル」、「C2−6アルケニル」とは、それぞれ、1個以上の二重結合を有する炭素数が2〜4個、2〜6個の、直鎖状または分枝鎖状の不飽和の脂肪族炭化水素基を意味する。好ましくは「C2−3アルケニル」である。「C2−4アルケニル」の具体例としては、例えば、エテニル、プロペニル、クロチル、ブテニルが挙げられ、「C2−6アルケニル」としては、上記に加え、ペンテニルおよびヘキセニル等、ならびにそれらの構造異性体や幾何異性体が挙げられる。なお、当該「C2−6アルケニル」における結合手は不飽和炭素原子上にあることが好ましく、例えば、1−エテニルまたは1−プロペニルが挙げられる。 “C 2-4 alkenyl” and “C 2-6 alkenyl” are linear or branched, each having 2 to 4 or 2 to 6 carbon atoms having one or more double bonds. Means an unsaturated aliphatic hydrocarbon group. “C 2-3 alkenyl” is preferable. Specific examples of “C 2-4 alkenyl” include, for example, ethenyl, propenyl, crotyl, butenyl, and “C 2-6 alkenyl” includes, in addition to the above, pentenyl, hexenyl and the like, and structural isomers thereof. Body and geometric isomers. The bond in the “C 2-6 alkenyl” is preferably on an unsaturated carbon atom, such as 1-ethenyl or 1-propenyl.
「C2−6アルキニル」とは、1個以上の三重結合を有する炭素数が2〜6個の直鎖状または分枝鎖状の不飽和の脂肪族炭化水素基を意味する。好ましくは「C2−3アルキニル」である。「C2−6アルキニル」の具体例としては、例えば、エチニル、プロピニル、ブチニル、ペンチニル、およびヘキシニル等、ならびにそれらの構造異性体が挙げられる。なお、当該「C2−6アルキニル」における結合手は不飽和炭素原子上にあることが好ましく、例えば、1−エチニルまたは1−プロピニルが挙げられる。 “C 2-6 alkynyl” means a linear or branched unsaturated aliphatic hydrocarbon group having 2 to 6 carbon atoms and having one or more triple bonds. Preferred is “C 2-3 alkynyl”. Specific examples of “C 2-6 alkynyl” include, for example, ethynyl, propynyl, butynyl, pentynyl, hexynyl and the like, and structural isomers thereof. The bond in the “C 2-6 alkynyl” is preferably on an unsaturated carbon atom, and examples thereof include 1-ethynyl and 1-propynyl.
「C1−2アルキルスルホニル」、「C1−4アルキルスルホニル」とは、それぞれ、前記「C1−2アルキル」、「C1−4アルキル」で置換されたスルホニル基を意味する。「C1−2アルキルスルホニル」の具体例としては、例えば、メチルスルホニル、エチルスルホニルが挙げられ、「C1−4アルキルスルホニル」としては、上記に加え、プロピルスルホニル、イソプロピルスルホニル、ブチルスルホニル等が挙げられる。好ましくはメチルスルホニルである。 "C 1-2 alkylsulfonyl", the "C 1-4 alkylsulfonyl", respectively, the "C 1-2 alkyl" means a substituted sulfonyl group in the "C 1-4 alkyl". Specific examples of “C 1-2 alkylsulfonyl” include, for example, methylsulfonyl and ethylsulfonyl, and “ C1-4 alkylsulfonyl” includes propylsulfonyl, isopropylsulfonyl, butylsulfonyl and the like in addition to the above. Can be mentioned. Preferred is methylsulfonyl.
「C1−4アルキルチオ」とは、前記「C1−4アルキル」で置換されたチオ基を意味する。「C1−4アルキルチオ」の具体例としては、例えば、メチルチオ、エチルチオ等が挙げられ、好ましくはメチルチオである。 “C 1-4 alkylthio” means a thio group substituted with the above “C 1-4 alkyl”. Specific examples of “C 1-4 alkylthio” include, for example, methylthio, ethylthio and the like, preferably methylthio.
上記の置換基群[A]、[B]および[C]を以下に示す。
[A]:C1−4アルコキシ、C1−4ハロアルコキシ
[B]:ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ、C1−4ハロアルコキシ
[C]:C1−4アルキル、ハロゲン、オキソ、ヒドロキシ
The above substituent groups [A], [B] and [C] are shown below.
[A]: C 1-4 alkoxy, C 1-4 haloalkoxy [B]: halogen, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxy, C 1-4 haloalkoxy [C]: C 1-4 alkyl, halogen, oxo, hydroxy
上記の置換基群[A]として好ましくは、C1−2アルコキシまたはC1−2ハロアルコキシであり、[B]として好ましくは、フッ素、C1−2アルキル、C1−2ハロアルキル、C1−2アルコキシまたはC1−4ハロアルコキシであり、[C]として好ましくは、C1−2アルキル、フッ素またはオキソである。 The above substituent group [A] is preferably C 1-2 alkoxy or C 1-2 haloalkoxy, and [B] is preferably fluorine, C 1-2 alkyl, C 1-2 haloalkyl, C 1. -2 alkoxy or C 1-4 haloalkoxy, and [C] is preferably C 1-2 alkyl, fluorine or oxo.
前記式(1)で表される本発明の化合物を更に具体的に開示するため、式(1)において用いられる各種記号につき、その好適な具体例を挙げて更に詳細に説明する。 In order to more specifically disclose the compound of the present invention represented by the formula (1), the various symbols used in the formula (1) will be described in more detail with suitable specific examples.
R1およびR2として好ましくは、それぞれ独立して、水素、C1−6アルキル(該アルキルは、ヒドロキシ、ハロゲン、C1−4アルコキシ、C1−4アルキルスルホニル、アミノカルボニル(該アミノカルボニルのアミノ部分は、置換されていてもよいC1−4アルキルによってモノまたはジ置換されていてもよい)、置換されていてもよい4〜7員のヘテロシクロアルキルおよび置換されていてもよい4〜7員のヘテロシクロアルキルカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または4〜7員のヘテロシクロアルキル(該ヘテロシクロアルキルは、ハロゲン、およびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、より好ましくは、それぞれ独立して、水素、C1−4アルキル(該アルキルは、ヒドロキシ、C1−2アルキルスルホニル、アミノカルボニル(該アミノカルボニルのアミノ部分は、C1−2アルキルによってモノ置換されていてもよい)、置換されていてもよい4〜5員のヘテロシクロアルキルおよび置換されていてもよい4〜6員のヘテロシクロアルキルカルボニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)であり、さらに好ましくは、それぞれ独立して、水素またはC1−4アルキル(該アルキルは、ヒドロキシおよびC1−2アルキルスルホニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)である。
R1およびR2の好ましい具体例としては、水素、メチル、エチル、プロピル、イソプロピル、n−ブチル、イソブチル、tert−ブチル、ジメチルアミノカルボニルメチル、2−ヒドロキシ−2−メチルプロピル、テトラヒドロフラニル、オキセタニル、2−メチルスルホニルエチル、2−エチルスルホニルエチル、2−オキソイミダゾリジニルエチル、2−イソプロピルスルホニルエチル、アミノカルボニルメチル、1−アミノカルボニルエチル、1−アミノカルボニル−1−メチルエチル、4−メチルピペラジニルカルボニルメチル、2−メチルスルホニル−2−メチルプロピル、3−メチルスルホニルプロピルが挙げられ、中でも好ましくは水素、ジメチルアミノカルボニルメチル、2−ヒドロキシ−2−メチルプロピル、テトラヒドロフラニル、オキセタニル、2−メチルスルホニルエチルである。
Preferably, R 1 and R 2 are each independently hydrogen, C 1-6 alkyl (wherein the alkyl is hydroxy, halogen, C 1-4 alkoxy, C 1-4 alkylsulfonyl, aminocarbonyl (of the aminocarbonyl The amino moiety may be mono- or di-substituted by optionally substituted C 1-4 alkyl), optionally substituted 4-7 membered heterocycloalkyl and optionally substituted 4-4 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of 7-membered heterocycloalkylcarbonyl) or 4-7 membered heterocycloalkyl (wherein the heterocycloalkyl is halogen, and C 1-4 optionally substituted with 1-3 substituents independently selected from the group consisting of alkoxy are also be), more favorable Details, each independently, hydrogen, C 1-4 alkyl (said alkyl, hydroxy, amino portion of C 1-2 alkylsulfonyl, aminocarbonyl (the amino carbonyl, optionally mono- substituted by C 1-2 alkyl 1 to 2 independently selected from the group consisting of optionally substituted 4 to 5 membered heterocycloalkyl and optionally substituted 4 to 6 membered heterocycloalkylcarbonyl Optionally substituted with a substituent) or 4 to 5 membered heterocycloalkyl, which may be substituted with 1 to 2 halogens, more preferably each independently. selection Te, hydrogen or C 1-4 alkyl (the alkyl, independently from the group consisting of hydroxy and C 1-2 alkylsulfonyl 1-2 heterocycloalkyl (wherein heterocycloalkyl may be substituted with a substituent), or 4-5 membered to is 1 is independently selected from the group consisting of C 1-2 alkyl and oxo Optionally substituted with 2 substituents).
Preferred examples of R 1 and R 2 include hydrogen, methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, tert-butyl, dimethylaminocarbonylmethyl, 2-hydroxy-2-methylpropyl, tetrahydrofuranyl, oxetanyl. 2-methylsulfonylethyl, 2-ethylsulfonylethyl, 2-oxoimidazolidinylethyl, 2-isopropylsulfonylethyl, aminocarbonylmethyl, 1-aminocarbonylethyl, 1-aminocarbonyl-1-methylethyl, 4-methyl Examples include piperazinylcarbonylmethyl, 2-methylsulfonyl-2-methylpropyl, and 3-methylsulfonylpropyl. Among them, hydrogen, dimethylaminocarbonylmethyl, 2-hydroxy-2-methylpropyl, teto are preferable. Hidorofuraniru, oxetanyl, 2-methylsulfonyl-ethyl.
Lとして好ましくは、C1−6アルキレン(該アルキレンは、C1−4アルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、より好ましくは、C1−4アルキレン(該アルキレンは、C1−2アルキルおよびヒドロキシからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)である。また、R1がC1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に下記式(10); L is preferably C 1-6 alkylene (the alkylene is substituted with 1 to 3 substituents independently selected from the group consisting of C 1-4 alkyl, halogen, hydroxy and C 1-4 alkoxy. More preferably, C 1-4 alkylene (wherein the alkylene is substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and hydroxy). May be). In addition, when R 1 is C 1-6 alkyl, R 1 and the carbon atom on L are combined together with the nitrogen atom to which R 1 is bonded, together with the following formula (10);
(式中、aは、1、2または3であり、bは、1または2であり、R2aは前記〔1〕に記載のR2の定義と同義である。)で表される4〜7員の飽和複素環を形成したものも好ましい。
上記式(10)中、aとして、好ましくは1、2または3であり、より好ましくは1または2である。
bとして、好ましくは、1または2である。
R2aとして、好ましくは、ジメチルアミノカルボニルメチル、アミノカルボニルメチル、2−ヒドロキシ−2−メチルプロピル、テトラヒドロフラニル、オキセタニル、2−メチルスルホニルエチル、2−エチルスルホニルエチルが挙げられ、より好ましくは2−ヒドロキシ−2−メチルプロピル、テトラヒドロフラニル、オキセタニル、2−メチルスルホニルエチル、アミノカルボニルメチルであり、最も好ましくは2−メチルスルホニルエチル、アミノカルボニルメチルである。
上記式(10)で表される4〜7員の飽和複素環の好ましい具体例としては、アゼチジニル、ピロリジニル、ピペリジルが挙げられる。
Lの好ましい具体例としては、エチレン、プロピレン、ブチレン、1−メチルプロピレン、2−メチルプロピレン、3−メチルプロピレン、2−ヒドロキシプロピレン、2,2−ジメチルプロピレン、1−メチルエチレン、2−メチルエチレン、1,1−ジメチルエチレンが挙げられ、中でも好ましくは、エチレン、プロピレン、アゼチジニレン、ピペリジニレンが挙げられる。
Lのうち、上記式(10)を含むリンカーとして好ましい具体例としては、アゼチジニレン、アゼチジニルメチレン、ピペリジニレン、ピロリジニレンが挙げられる。
(Wherein, a is 1, 2 or 3, b is 1 or 2, and R 2a has the same definition as R 2 described in [1] above). What formed the 7-membered saturated heterocyclic ring is also preferable.
In the above formula (10), a is preferably 1, 2 or 3, more preferably 1 or 2.
b is preferably 1 or 2.
R 2a is preferably dimethylaminocarbonylmethyl, aminocarbonylmethyl, 2-hydroxy-2-methylpropyl, tetrahydrofuranyl, oxetanyl, 2-methylsulfonylethyl, 2-ethylsulfonylethyl, more preferably 2- Hydroxy-2-methylpropyl, tetrahydrofuranyl, oxetanyl, 2-methylsulfonylethyl, aminocarbonylmethyl, most preferably 2-methylsulfonylethyl, aminocarbonylmethyl.
Preferable specific examples of the 4- to 7-membered saturated heterocyclic ring represented by the above formula (10) include azetidinyl, pyrrolidinyl, and piperidyl.
Preferable specific examples of L include ethylene, propylene, butylene, 1-methylpropylene, 2-methylpropylene, 3-methylpropylene, 2-hydroxypropylene, 2,2-dimethylpropylene, 1-methylethylene, 2-methylethylene. 1,1-dimethylethylene, among which ethylene, propylene, azetidinylene, and piperidinylene are preferable.
Specific examples of preferable linkers containing the above formula (10) among L include azetidinylene, azetidinylmethylene, piperidinylene and pyrrolidinylene.
R3として好ましくは、ハロゲン、C1−6アルキル、C2−6アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、置換されていてもよいC3−7シクロアルキル、置換されていてもよい4〜7員のヘテロシクロアルキル、置換されていてもよいC6−10アリールおよび置換されていてもよいC6−10アリールオキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニル、4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキルおよびハロゲンからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、置換されていてもよいC1−4アルキル、C1−4ハロアルキル、置換されていてもよいC1−4アルコキシ、C1−4ハロアルコキシ、シアノおよびC1−4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシおよびC1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、より好ましくはハロゲン、C1−4アルキル、C2−4アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、置換されていてもよいC4−6シクロアルキル、置換されていてもよい4〜6員のヘテロシクロアルキル、置換されていてもよいC6−10アリールおよび置換されていてもよいC6−10アリールオキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−6シクロアルキル、C4−6シクロアルケニル、4〜6員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、置換されていてもよいC1−2アルキル、C1−2ハロアルキル、置換されていてもよいC1−2アルコキシ、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲンおよびC1−2ハロアルキルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、更に好ましくはC3−6シクロアルキル、4〜6員のヘテロシクロアルキル(該シクロアルキル、該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、置換されていてもよいC1−2アルキル、C1−2ハロアルキル、置換されていてもよいC1−2アルコキシ、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲンおよびC1−2ハロアルキルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)である。
R3の好ましい具体例としては、水素、フッ素、塩素、臭素、メチル、エチル、イソプロピル、シクロペンチル、シクロヘキシル、フェニル、4−トリフルオロメチルフェニル、4−トリフルオロメトキシフェニル、4−シアノフェニル、4−フルオロ−2−メチルフェニル、2−メチルフェニル、3−メチルフェニル、2−フルオロフェニル、フェノキシ、4−フルオロフェノキシ、4−フルオロフェニル、2−メトキシフェニル、4−トリフルオロメトキシフェニル、3,5−ジフルオロフェニル、3,4−ジフルオロフェニル、2,4−ジフルオロフェニル、4−フルオロフェニル、4−フルオロ−3−メチルフェニル、フェニル、トリフルオロメチル、4−メトキシフェニル、2−フェニルエチル、3−メトキシフェニル、4−ジフルオロメトキシフェニル、3,4,5−トリフルオロフェニル、2−メトキシ−4−トリフルオロメチルフェニル、2−ピリジル、5−フルオロピリジン−2−イル、2−トリフルオロメチルピリジン−5−イル、フェニルエテニル、4−フルオロフェニルメチル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘキセニル、イソブチル、n−ブチル、t−ブチル、n−ペンチル、n−ヘキシル、シクロヘキシルメチル、3,3−ジフルオロシクロブチル、5−メチルピリジン−2−イル、5−トリフルオロメチルピリジン−2−イル、5−メトキシピリジン−2−イル、4−フルオロフェノキシメチル、4−メトキシエトキシ、テトラヒドロピラニル、テトラヒドロピラニルメチル、2−メチルプロピル、3−メチルブチル、4,4−ジフルオロシクロヘキシルが挙げられ、中でも好ましくは、4−トリフルオロメチルフェニル、4−トリフルオロメトキシフェニル、4−フルオロフェニル、シクロブチル、シクロペンチル、シクロヘキシル、n−ブチル、n−ペンチル、n−ヘキシル、3,3−ジフルオロシクロブチル、5−メチルピリジン−2−イル、5−トリフルオロメチルピリジン−2−イル、5−メトキシピリジン−2−イル、テトラヒドロピラニル、4,4−ジフルオロシクロヘキシルであり、最も好ましくは、4−トリフルオロメチルフェニル、4−トリフルオロメトキシフェニル、4−フルオロフェニル、シクロブチル、シクロペンチル、シクロヘキシル、3,3−ジフルオロシクロブチル、5−メチルピリジン−2−イル、5−トリフルオロメチルピリジン−2−イル、5−メトキシピリジン−2−イル、テトラヒドロピラニル、4,4−ジフルオロシクロヘキシルである。
R 3 is preferably halogen, C 1-6 alkyl, C 2-6 alkenyl (the alkyl and the alkenyl are each halogen, optionally substituted C 3-7 cycloalkyl, optionally substituted 4 1 to 3 substituents independently selected from the group consisting of 7-membered heterocycloalkyl, optionally substituted C 6-10 aryl and optionally substituted C 6-10 aryloxy Optionally substituted), C 3-7 cycloalkyl, C 4-7 cycloalkenyl, 4-7 membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl, the heterocycloalkyl are each C 1-4). Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkyl, C 1-4 haloalkyl and halogen), C 6-10 aryl (wherein the aryl is halogen, optionally substituted C 1-4 alkyl, C 1-4 haloalkyl, optionally substituted C 1-4 alkoxy, C 1-4 haloalkoxy, cyano and Optionally substituted with 1 to 3 substituents independently selected from the group consisting of C 1-4 alkoxycarbonyl, or 5-10 membered heteroaryl (wherein the heteroaryl is halogen, C 1- 4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxy and C 1-4 haloalkoxy, optionally substituted with 1 to 3 substituents independently selected from the group consisting of Preferably halogen, C 1-4 alkyl, C 2-4 alkenyl (the alkyl and the alkenyl are each halogen, optionally substituted C 4-6 cycloalkyl Independently selected from the group consisting of kill, optionally substituted 4-6 membered heterocycloalkyl, optionally substituted C 6-10 aryl and optionally substituted C 6-10 aryloxy. 1 to 3 substituents), C 3-6 cycloalkyl, C 4-6 cycloalkenyl, 4- to 6-membered heterocycloalkyl (the cycloalkyl, the cycloalkenyl, the hetero Cycloalkyl may be substituted by 1 to 3 halogens), C 6-10 aryl (wherein the aryl is halogen, optionally substituted C 1-2 alkyl, C 1-2 haloalkyl, substituted) 1 to 2 independently selected from the group consisting of optionally substituted C 1-2 alkoxy, C 1-2 haloalkoxy, cyano and C 1-2 alkoxycarbonyl Optionally substituted with 3 substituents) or 5-10 membered heteroaryl, wherein the heteroaryl is independently selected from the group consisting of halogen and C 1-2 haloalkyl More preferably C 3-6 cycloalkyl, 4-6 membered heterocycloalkyl (wherein the cycloalkyl, the heterocycloalkyl is substituted by 1 to 3 halogens). C 6-10 aryl (the aryl may be halogen, optionally substituted C 1-2 alkyl, C 1-2 haloalkyl, optionally substituted C 1-2 alkoxy, C 1). -2 haloalkoxy, optionally substituted with 1-3 substituents independently selected from the group consisting of cyano and C 1-2 alkoxycarbonyl) or 5-1 Heteroaryl-membered (said heteroaryl, may be substituted with one to two substituents independently selected from the group consisting of halogen and C 1-2 haloalkyl) a.
Preferable specific examples of R 3 include hydrogen, fluorine, chlorine, bromine, methyl, ethyl, isopropyl, cyclopentyl, cyclohexyl, phenyl, 4-trifluoromethylphenyl, 4-trifluoromethoxyphenyl, 4-cyanophenyl, 4- Fluoro-2-methylphenyl, 2-methylphenyl, 3-methylphenyl, 2-fluorophenyl, phenoxy, 4-fluorophenoxy, 4-fluorophenyl, 2-methoxyphenyl, 4-trifluoromethoxyphenyl, 3,5- Difluorophenyl, 3,4-difluorophenyl, 2,4-difluorophenyl, 4-fluorophenyl, 4-fluoro-3-methylphenyl, phenyl, trifluoromethyl, 4-methoxyphenyl, 2-phenylethyl, 3-methoxy Phenyl, 4-diph Fluoromethoxyphenyl, 3,4,5-trifluorophenyl, 2-methoxy-4-trifluoromethylphenyl, 2-pyridyl, 5-fluoropyridin-2-yl, 2-trifluoromethylpyridin-5-yl, Phenylethenyl, 4-fluorophenylmethyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, isobutyl, n-butyl, t-butyl, n-pentyl, n-hexyl, cyclohexylmethyl, 3,3-difluorocyclobutyl 5-methylpyridin-2-yl, 5-trifluoromethylpyridin-2-yl, 5-methoxypyridin-2-yl, 4-fluorophenoxymethyl, 4-methoxyethoxy, tetrahydropyranyl, tetrahydropyranylmethyl, 2-methylpropyl, 3- Examples include butyl butyl and 4,4-difluorocyclohexyl, among which 4-trifluoromethylphenyl, 4-trifluoromethoxyphenyl, 4-fluorophenyl, cyclobutyl, cyclopentyl, cyclohexyl, n-butyl, n-pentyl, n -Hexyl, 3,3-difluorocyclobutyl, 5-methylpyridin-2-yl, 5-trifluoromethylpyridin-2-yl, 5-methoxypyridin-2-yl, tetrahydropyranyl, 4,4-difluorocyclohexyl And most preferably 4-trifluoromethylphenyl, 4-trifluoromethoxyphenyl, 4-fluorophenyl, cyclobutyl, cyclopentyl, cyclohexyl, 3,3-difluorocyclobutyl, 5-methylpyridin-2-yl, 5 -G Fluoromethyl-2-yl, 5-methoxy-2-yl, tetrahydropyranyl, 4,4-difluoro-cyclohexyl.
R4として好ましくは、水素、ハロゲンまたはC1−2アルキルであり、より好ましくは水素である。 R 4 is preferably hydrogen, halogen or C 1-2 alkyl, and more preferably hydrogen.
前記式(2)で表される化合物を更に具体的に開示するため、式(2)において用いられる各種記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the formula (2), the various symbols used in the formula (2) will be described in detail with suitable specific examples.
R5として好ましくは、ニトロ、アミノ(該アミノは、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)からなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)またはフタルイミドであり、中でも好ましくは、ニトロまたはNH2である。
R5の好ましい具体例としては、ニトロ、NH2、C1−4アルキルオキシカルボニルアミノ、ベンジルアミノ、(ジベンジル)アミノ、ベンジルオキシカルボニルアミノ、フルオレニルメチルカルボニルアミノ、2−ニトロベンゼンスルホニルアミノ、4−ニトロベンゼンスルホニルアミノ、2,4−ジニトロベンゼンスルホニルアミノ、フタルイミドが挙げられ、中でも好ましくは、ニトロ、NH2、tert−ブトキシカルボニルアミノ、ベンジルアミノ、ベンジルオキシカルボニルアミノ、2−ニトロベンゼンスルホニルアミノが挙げられる。
R6として、好ましくは、水素、メチル、エチル、メトキシメチル、ベンジル、4−メトキシベンジルであり、中でも好ましくは、メチル、エチル、ベンジルである。
R7として好ましくは、水素又はメチルであり、より好ましくは水素である。
R8として好ましくは、塩素または水素である。
A−Aは、C−CまたはC=Cであり、R8が、塩素である場合、A−Aとして好ましくはC=Cであり、R8が、水素である場合、A−Aとして好ましくはC−Cである。
R 5 is preferably nitro, amino (the amino is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl, fluorenylmethylcarbonyl, benzenesulfonyl (the benzenesulfonyl is one or two by nitro). And optionally substituted with 1 to 2 substituents independently selected from the group consisting of (optionally substituted) or phthalimide, among which nitro or NH 2 is preferred.
Preferred specific examples of R 5 include nitro, NH 2 , C 1-4 alkyloxycarbonylamino, benzylamino, (dibenzyl) amino, benzyloxycarbonylamino, fluorenylmethylcarbonylamino, 2-nitrobenzenesulfonylamino, 4 - nitrobenzenesulfonyl amino, 2,4-nitrobenzenesulfonyl amino, phthalimido, and among them preferably include nitro, NH 2, tert-butoxycarbonylamino, benzylamino, benzyloxycarbonylamino, 2-nitrobenzenesulfonyl amino .
R 6 is preferably hydrogen, methyl, ethyl, methoxymethyl, benzyl or 4-methoxybenzyl, and particularly preferably methyl, ethyl or benzyl.
R 7 is preferably hydrogen or methyl, more preferably hydrogen.
R 8 is preferably chlorine or hydrogen.
AA is C—C or C═C, and when R 8 is chlorine, preferably A = A as C—C, and when R 8 is hydrogen, preferably as AA. Is CC.
前記式(2)で表される化合物としては、下記式(11)に示す化合物が好ましい。 As the compound represented by the formula (2), a compound represented by the following formula (11) is preferable.
前記式(3)で表される化合物を更に具体的に開示するため、式(3)において用いられる各種記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the formula (3), various symbols used in the formula (3) will be described in detail with suitable specific examples.
R7として好ましくは、水素またはメチルであり、より好ましくは水素である。
R9の具体例としては、tert−ブトキシカルボニル、ベンジルオキシカルボニル、フルオレニルメチルカルボニルまたは2−ニトロベンゼンスルホニルが挙げられ、好ましくは、tert−ブトキシカルボニル、ベンジルオキシカルボニルである。
R10の具体例としては、水素、メチルであるか、R9およびR10が一緒になってフタルイミドが挙げられ、好ましくは水素である。
Mの具体例としては、メチレン、エチレン、プロピレン、ブチレン、アゼチジニレン、アゼチジニルメチレン、ピペリジニレン、ピロリジニレンが挙げられる。好ましくはエチレン、プロピレン、アゼチジニレン、ピペリジニレンであり、より好ましくはエチレンまたはプロピレンである。
Bの具体例としては、ヨウ素、臭素、塩素またはフッ素が挙げられ、好ましくはヨウ素、臭素、塩素である。
R 7 is preferably hydrogen or methyl, more preferably hydrogen.
Specific examples of R 9 include tert-butoxycarbonyl, benzyloxycarbonyl, fluorenylmethylcarbonyl and 2-nitrobenzenesulfonyl, preferably tert-butoxycarbonyl and benzyloxycarbonyl.
Specific examples of R 10 are hydrogen and methyl, or R 9 and R 10 taken together include phthalimide, preferably hydrogen.
Specific examples of M include methylene, ethylene, propylene, butylene, azetidinylene, azetidinylmethylene, piperidinylene and pyrrolidinylene. Preferred is ethylene, propylene, azetidinylene, piperidinylene, and more preferred is ethylene or propylene.
Specific examples of B include iodine, bromine, chlorine, and fluorine, preferably iodine, bromine, and chlorine.
前記式(4)で表される化合物を更に具体的に開示するため、式(4)において用いられる各種記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the formula (4), the various symbols used in the formula (4) will be described in detail with suitable specific examples.
R11およびR12おける保護基のついたアミノとは、上記式(2)における定義と同じである。
R11およびR12として好ましくは、それぞれ独立して、水素、C1−4アルキル、ニトロ、アミノ(該アミノは、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)からなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、フタルイミドまたはC1−2アルコキシ(該アルコキシにおけるアルキル部分は、C1−2アルコキシ、フェニルおよび4−メトキシフェニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であるか、互いにオルト位に結合したR11およびR12がフェニルと一緒になって形成したベンズオキサゾロン環、インドール環またはインダゾール環であり、中でも好ましくはニトロ、アミノ(該アミノは、C1−4アルキルオキシカルボニル、フェニルメチルオキシカルボニル、からなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、またはC1−2アルコキシ(該アルコキシにおけるアルキル部分は、C1−2アルコキシおよびフェニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であるか、互いにオルト位に結合したR11およびR12がフェニルと一緒になって形成したベンズオキサゾロン環である。
R11およびR12の好ましい具体例としては、それぞれ独立して、水素、メチル、エチル、メトキシ、メトキシメトキシ、トリフルオロメチル、トリフルオロメトキシ、ニトロ、ベンジルオキシ、4−メトキシベンジルオキシ、tert−ブトキシカルボニルアミノ、ベンジルアミノ、ベンジルオキシカルボニルアミノであるか、互いにオルト位に結合したR11およびR12がフェニルと一緒になったベンズオキサゾロン、インドールまたはインダゾールが挙げられ中でも好ましくは、メトキシ、メトキシメトキシ、ベンジルオキシ、4−メトキシベンジルオキシ、tert−ブトキシカルボニルアミノ、ベンジルオキシカルボニルアミノであるか、互いにオルト位に結合したR11およびR12がフェニルと一緒になって形成したベンズオキサゾロンであり、より好ましくは、メトキシ、ベンジルオキシ、tert−ブトキシカルボニルアミノであるか、R11およびR12が隣接してフェニルに置換して、フェニルと一緒になって形成したベンズオキサゾロンである。
The amino having a protecting group in R 11 and R 12 has the same definition as in the above formula (2).
Preferably, R 11 and R 12 are each independently hydrogen, C 1-4 alkyl, nitro, amino (wherein the amino is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl, fluorenylmethyl). Carbonyl, benzenesulfonyl (which may be substituted with 1 to 2 substituents independently selected from the group consisting of 1 to 2 substituents by nitro), phthalimide Or C 1-2 alkoxy (the alkyl part in the alkoxy may be substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkoxy, phenyl and 4-methoxyphenyl) ) and either, R 11 and R 12 bonded to the ortho-position to each other together with the phenyl The formed benz oxazolone ring, indole ring or indazole ring, among them preferably a nitro, amino (said amino, C 1-4 alkyloxycarbonyl, phenylmethyl oxycarbonyl 1 which are independently selected from the group consisting of Optionally substituted by two substituents), or C 1-2 alkoxy, wherein the alkyl moiety in the alkoxy is independently selected from the group consisting of C 1-2 alkoxy and phenyl R 11 and R 12 bonded to each other in the ortho position may be a benzoxazolone ring formed together with phenyl.
Preferable specific examples of R 11 and R 12 are each independently hydrogen, methyl, ethyl, methoxy, methoxymethoxy, trifluoromethyl, trifluoromethoxy, nitro, benzyloxy, 4-methoxybenzyloxy, tert-butoxy Benzoxazolone, indole or indazole in which R 11 and R 12 bonded to each other in the ortho position with each other, such as carbonylamino, benzylamino, benzyloxycarbonylamino, and phenyl are preferred, among which methoxy, methoxymethoxy, benzyloxy, 4-methoxybenzyloxy, tert- butoxycarbonylamino, or a benzyloxycarbonylamino, Baie which R 11 and R 12 are bonded in the ortho-position to each other to form together with the phenyl A Zuokisazoron, more preferably, methoxy, benzyloxy, or a tert- butoxycarbonylamino, substituted phenyl R 11 and R 12 are adjacent, is benz oxazolone formed together with the phenyl .
前記式(5)で表される化合物を更に具体的に開示するため、式(5)において用いられる各種記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the formula (5), the various symbols used in the formula (5) will be described in detail with suitable specific examples.
R7、R11およびR12は、上記式(4)における定義と同じである。
Bは、上記式(3)における定義と同じである。
R 7 , R 11 and R 12 are the same as defined in the above formula (4).
B is the same as defined in the above formula (3).
前記式(6)で表される化合物を更に具体的に開示するため、式(6)において用いられる記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the formula (6), the symbols used in the formula (6) will be described in detail by giving preferred specific examples.
Xとして好ましくはヨウ素または臭素であり、より好ましくは臭素である。
X is preferably iodine or bromine, more preferably bromine.
上記式(5)の化合物と上記式(6)の化合物との反応において、その好適な反応条件を具体例を挙げて詳細に説明する。
化合物(5)に対して、パラジウム触媒存在下に化合物(6)を作用させることにより化合物(4)を製造することができる。パラジウム触媒としては例えば、ビス(トリス tert−ブチルホスフィン)パラジウム、ビス(トリス o−トリルホスフィン)ジクロロパラジウム、ビス(トリス o−トリルホスフィン)パラジウム、テトラキストリフェニルホスフィンパラジウム、ジクロロパラジウム(アセトニトリル)、ビス(トリス o−トリルホスフィン)ジクロロパラジウム、(1,1’−ビス(ジフェニルホスフィノ)フェロセン)ジクロロパラジウム、PEPPSITM・IPr((1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾリデン) (3−クロロピリジル)パラジウム(II)ジクロライド)、Pd−PEPPSITM−IPent((1,3−ビス(2,6−ジイソペンチルフェニル)イミダゾリデン) (3−クロロピリジル)パラジウム(II)ジクロライド)などの触媒が挙げられる。また、酢酸パラジウムや塩化パラジウムに対して、palladium reagents and catalysts, John Wiley & Sons Inc.(2004年)に記載されている配位子またはその類縁配位子から適切なものを選択して使用することもできる。パラジウム触媒として好ましくは、PEPPSITM・IPr、Pd−PEPPSITM−IPentであり、さらに好ましくは、Pd−PEPPSITM−IPentである。溶媒としては、例えばジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、メチルシクロペンチルエーテル、アニソールまたは1,4−ジオキサン等のエーテル系溶媒、ベンゼン、トルエン、クロロベンゼンまたはキシレン等の芳香族炭化水素系溶媒、酢酸エチルまたは酢酸メチル等のエステル系溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリジノン、1,3−ジメチル−2−イミダゾリジノン、またはジメチルスルホキシド等の非プロトン性極性溶媒、またはそれらの混合物が挙げられる。溶媒として好ましくは、ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、メチルシクロペンチルエーテルである。反応温度は、用いる原料化合物の種類等により異なるが、通常約0℃〜約250℃であり、好ましくは約20℃〜約100℃である。反応時間は、通常1時間から72時間であり、好ましくは1時間から48時間である。
In the reaction of the compound of the above formula (5) and the compound of the above formula (6), suitable reaction conditions will be described in detail with specific examples.
Compound (4) can be produced by reacting compound (5) with compound (6) in the presence of a palladium catalyst. Examples of the palladium catalyst include bis (tris tert-butylphosphine) palladium, bis (tris o-tolylphosphine) dichloropalladium, bis (tris o-tolylphosphine) palladium, tetrakistriphenylphosphine palladium, dichloropalladium (acetonitrile), bis (Tris o-tolylphosphine) dichloropalladium, (1,1′-bis (diphenylphosphino) ferrocene) dichloropalladium, PEPPSI ™ • IPr ((1,3-bis (2,6-diisopropylphenyl) imidazolidene) ( 3-chloropyridyl) palladium (II) dichloride), Pd-PEPPSI ™ -IPent ((1,3-bis (2,6-diisopentylphenyl) imidazolidene) (3-chloropyridyl) para And catalysts such as (dium (II) dichloride). Also, for palladium acetate and palladium chloride, palladium reagents and catalyst, John Wiley & Sons Inc. (2004) or an appropriate ligand selected from the ligands described in (2004) can also be used. Preferable as the palladium catalyst are PEPPSI ™ • IPr and Pd-PEPPSI ™ -IPent, and more preferred is Pd-PEPPSI ™ -IPent. Examples of the solvent include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, methylcyclopentyl ether, anisole or 1,4-dioxane, aromatic hydrocarbon solvents such as benzene, toluene, chlorobenzene or xylene, ethyl acetate or acetic acid. Aprotic polarities such as ester solvents such as methyl, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidinone, 1,3-dimethyl-2-imidazolidinone, or dimethyl sulfoxide A solvent, or a mixture thereof. Preferred solvents are diethyl ether, diisopropyl ether, tetrahydrofuran, and methylcyclopentyl ether. While the reaction temperature varies depending on the kind of raw material compound to be used, etc., it is generally about 0 ° C. to about 250 ° C., preferably about 20 ° C. to about 100 ° C. The reaction time is usually 1 hour to 72 hours, preferably 1 hour to 48 hours.
前記式(7)で表される化合物を更に具体的に開示するため、式(7)において用いられる各種記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the formula (7), the various symbols used in the formula (7) will be described in detail with suitable specific examples.
R7として好ましくは、水素またはメチルであり、より好ましくは水素である。
R13として好ましくは、C1−4アルキルオキシカルボニル、フェニルメチルオキシカルボニル、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)であり、より好ましくはC1−4アルキルオキシカルボニルまたはフェニルメチルオキシカルボニルである。 R14として好ましくは、水素またはメチルである。
Mは、上記式(3)における定義と同じである。
R 7 is preferably hydrogen or methyl, more preferably hydrogen.
R 13 is preferably C 1-4 alkyloxycarbonyl, phenylmethyloxycarbonyl, fluorenylmethylcarbonyl, benzenesulfonyl (the benzenesulfonyl may be substituted by 1 or 2 by nitro), and more Preferably it is C1-4 alkyloxycarbonyl or phenylmethyloxycarbonyl. R 14 is preferably hydrogen or methyl.
M is the same as defined in the above formula (3).
下記式(8)表される化合物を更に具体的に開示するため、式(8)において用いられる各種記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the following formula (8), the various symbols used in the formula (8) will be described in detail with suitable specific examples.
R7、R13およびR14は、上記式(7)における定義と同じである。
R 7 , R 13 and R 14 are the same as defined in the above formula (7).
前記式(9)で表される化合物を更に具体的に開示するため、式(9)において用いられる記号につき、その好適な具体例を挙げて詳細に説明する。 In order to more specifically disclose the compound represented by the formula (9), the symbols used in the formula (9) will be described in detail by giving preferred specific examples.
Xとして好ましくはヨウ素または臭素であり、より好ましくは臭素である。
X is preferably iodine or bromine, more preferably bromine.
上記式(8)と上記式(9)の化合物との反応において、その好適な反応条件を具体例を挙げて詳細に説明する。
化合物(8)に対して、パラジウム触媒存在下に化合物(9)を作用させることにより化合物(7)を製造することができる。好ましい反応条件は、上記化合物(5)と化合物(6)との反応における条件と同じである。
In the reaction of the above formula (8) with the compound of the above formula (9), suitable reaction conditions will be described in detail with specific examples.
Compound (7) can be produced by reacting compound (8) with compound (9) in the presence of a palladium catalyst. Preferable reaction conditions are the same as those in the reaction between the compound (5) and the compound (6).
化合物(1)の製造方法:
次に本発明の化合物の製造方法について以下に説明する。
Production method of compound (1):
Next, the manufacturing method of the compound of this invention is demonstrated below.
式(1)で表される化合物は、公知化合物から、例えば以下に示す製造法1〜12に示す方法、下記の製造法に類似の方法、又は当業者に周知の合成方法を適宜組み合わせて製造することができる。 The compound represented by formula (1) is produced from a known compound by appropriately combining, for example, the methods shown in the following production methods 1 to 12, methods similar to the following production methods, or synthesis methods well known to those skilled in the art. can do.
製造法1:
式(1)で表される化合物のうち、化合物(S−8)またはその薬学的に許容される塩は、例えば下記に示される方法によって製造される。ただし、R3がハロゲンである場合は、X1がハロゲンである化合物を用い、(1−4)の工程を経ないことにより製造される。製造法2〜7においても同様である。
Production method 1:
Among the compounds represented by the formula (1), the compound (S-8) or a pharmaceutically acceptable salt thereof is produced, for example, by the method shown below. However, when R 3 is halogen, it is produced by using a compound in which X 1 is halogen and not passing through the step (1-4). The same applies to the manufacturing methods 2 to 7.
[式中、R1、R2、R3、R4、およびLは、前記項1に定義されるとおりである。X1、X2およびX3は、各々独立し、ハロゲン、トリフルオロメタンスルホニルオキシまたはメタンスルホニルオキシなどの脱離基を表し、Raは、ベンジル、アセチル、ベンゾイル、tert−ブチルジメチルシリル、トリエチルシリル、トリイソプロピルシリルまたはtert−ブチルジフェニルシリル等の保護基を表し、R3aは、C1−6アルキル、C2−6アルケニル、C2−6アルキニル、C3−7シクロアルキル、C4−7シクロアルケニル、4〜7員のヘテロシクロアルキル、C6−10アリール、C6−10アリールオキシまたは5〜10員のヘテロアリールを表す。R3a−Y1は、下記工程(1−4)に示す有機ホウ素化合物、有機亜鉛化合物、アルケニル化合物、アルキニル化合物またはヒドロキシ化合物等を表す。]
[Wherein R 1 , R 2 , R 3 , R 4 , and L are as defined in the above item 1. X 1 , X 2 and X 3 each independently represent a leaving group such as halogen, trifluoromethanesulfonyloxy or methanesulfonyloxy, and R a is benzyl, acetyl, benzoyl, tert-butyldimethylsilyl, triethylsilyl Represents a protecting group such as triisopropylsilyl or tert-butyldiphenylsilyl, and R 3a is C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 3-7 cycloalkyl, C 4-7. Cycloalkenyl represents 4-7 membered heterocycloalkyl, C 6-10 aryl, C 6-10 aryloxy or 5-10 membered heteroaryl. R 3a -Y 1 represents an organic boron compound, an organic zinc compound, an alkenyl compound, an alkynyl compound, a hydroxy compound, or the like shown in the following step (1-4). ]
工程(1−1):
2−ニトロフェノール化合物(s−1)のニトロ基をアミノ基に還元することにより2−アミノフェノール化合物(s−2)を製造することができる。ニトロ基の還元方法としては通常の還元条件が用いられ、例えば接触還元または金属還元等が挙げられる。
Step (1-1):
The 2-aminophenol compound (s-2) can be produced by reducing the nitro group of the 2-nitrophenol compound (s-1) to an amino group. As a method for reducing the nitro group, normal reduction conditions are used, and examples thereof include catalytic reduction and metal reduction.
接触還元を用いる場合、例えばパラジウム炭素、ラネーニッケル、または、酸化白金等の触媒の存在下、ニトロ化合物(s−1)と水素を反応させることによりアミノ化合物(s−2)が得られる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えば、エタノール、メタノールのようなアルコール系溶媒、酢酸エチルまたは酢酸メチル等のエステル系溶媒、テトラヒドロフラン、または1,4−ジオキサン等のエーテル系溶媒、N,N−ジメチルホルムアミド等の非プロトン性極性溶媒、酢酸等のプロトン性極性溶媒、もしくは水、またはそれらの混合物が挙げられる。反応温度は用いる原料化合物の種類等により異なるが、通常約0℃〜約80℃で行われ、好ましくは20℃から50℃であり、常圧または加圧下に行われる。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。 When using catalytic reduction, for example, the amino compound (s-2) is obtained by reacting the nitro compound (s-1) with hydrogen in the presence of a catalyst such as palladium carbon, Raney nickel, or platinum oxide. Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include alcohol solvents such as ethanol and methanol, ester solvents such as ethyl acetate and methyl acetate, ether solvents such as tetrahydrofuran and 1,4-dioxane, and non-solvents such as N, N-dimethylformamide. Examples thereof include a protic polar solvent, a protic polar solvent such as acetic acid, or water, or a mixture thereof. While the reaction temperature varies depending on the type of raw material compound to be used, etc., it is usually carried out at about 0 ° C. to about 80 ° C., preferably 20 ° C. to 50 ° C., and carried out at normal pressure or under pressure. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
金属還元を用いる場合、ニトロ化合物(s−1)を、(1)鉄、亜鉛またはスズ等の金属、または塩化第一スズ等の金属塩と塩酸、酢酸等の酸との組み合わせ、または(2)鉄または塩化第一スズ単独で反応させることにより、アミノ化合物(s−2)が得られる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えば、エタノール、メタノールのようなアルコール系溶媒、テトラヒドロフラン等のエーテル系溶媒、酢酸等のプロトン性極性溶媒、もしくは水、またはそれらの混合物が挙げられる。反応温度は用いる原料化合物の種類等により異なるが、通常約0℃〜約100℃で行われ、好ましくは20℃から80℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。 When metal reduction is used, the nitro compound (s-1) is converted into (1) a metal such as iron, zinc or tin, or a combination of a metal salt such as stannous chloride and an acid such as hydrochloric acid or acetic acid, or (2 ) By reacting with iron or stannous chloride alone, the amino compound (s-2) is obtained. Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include alcohol solvents such as ethanol and methanol, ether solvents such as tetrahydrofuran, protic polar solvents such as acetic acid, water, and mixtures thereof. While the reaction temperature varies depending on the kind of raw material compound to be used, etc., it is generally carried out at about 0 ° C. to about 100 ° C., preferably 20 ° C. to 80 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
工程(1−2):
ベンズオキサゾロン化合物(s−3)は、塩基の存在下あるいは非存在下、不活性溶媒中、2−アミノフェノール化合物(s−2)をカルボニル化反応させることにより製造することができる。カルボニル化試薬としては、例えば、カルボニルジイミダゾール、トリホスゲン、ホスゲン等が用いられる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えば、テトラヒドロフラン、ジエチルエーテル、ジメトキシエタン、1,4−ジオキサン等のエーテル溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド、アセトニトリル等の非プロトン性極性溶媒、ケトン類(アセトン等)、又はこれらの混合溶媒が挙げられる。塩基としては、例えば、N−メチルモルホリン、トリエチルアミン、ジイソプロピルエチルアミン、トリブチルアミン、DBU,DBN、DABCO、ピリジン等の有機塩基が挙げられる。反応温度としては、通常−78〜150℃の範囲から選択することができ、好ましくは0〜100℃の範囲で行われる。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
Step (1-2):
The benzoxazolone compound (s-3) can be produced by subjecting the 2-aminophenol compound (s-2) to a carbonylation reaction in an inert solvent in the presence or absence of a base. As the carbonylation reagent, for example, carbonyldiimidazole, triphosgene, phosgene and the like are used. Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include ether solvents such as tetrahydrofuran, diethyl ether, dimethoxyethane, and 1,4-dioxane, aprotic polar solvents such as N, N-dimethylformamide, N, N-dimethylacetamide, dimethyl sulfoxide, and acetonitrile, Ketones (acetone etc.) or these mixed solvents are mentioned. Examples of the base include organic bases such as N-methylmorpholine, triethylamine, diisopropylethylamine, tributylamine, DBU, DBN, DABCO, and pyridine. As reaction temperature, it can select normally from the range of -78-150 degreeC, Preferably it carries out in the range of 0-100 degreeC. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
工程(1−3):
化合物(s−4)は、塩基の存在下、ベンズオキサゾロン化合物(s−3)とハロゲン化合物を反応させることにより製造することができる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えば、テトラヒドロフラン、ジエチルエーテル、ジメトキシエタン、1,4−ジオキサン等のエーテル溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、アセトニトリル等の非プロトン性極性溶媒が挙げられる。塩基としては、例えば炭酸セシウム、炭酸カリウムのような炭酸アルカリ金属、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属、ナトリウムエトキシド、ナトリウムtert−ブトキシド、カリウムtert−ブトキシド等のアルカリ金属アルコキシド、DBU,DBN、DABCOなどの有機塩基等が挙げられ、好ましくは、炭酸カリウム、水素化ナトリウムが挙げられる。反応温度としては、通常約0〜150℃で行われ、好ましくは0〜100℃の範囲で行われる。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
Step (1-3):
Compound (s-4) can be produced by reacting a benzoxazolone compound (s-3) with a halogen compound in the presence of a base. Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include ether solvents such as tetrahydrofuran, diethyl ether, dimethoxyethane, and 1,4-dioxane, and aprotic polar solvents such as N, N-dimethylformamide, N, N-dimethylacetamide, and acetonitrile. Examples of the base include alkali metal carbonates such as cesium carbonate and potassium carbonate, alkali metal hydrides such as sodium hydride and potassium hydride, alkali metal alkoxides such as sodium ethoxide, sodium tert-butoxide and potassium tert-butoxide, Organic bases such as DBU, DBN, DABCO and the like can be mentioned, and potassium carbonate and sodium hydride are preferable. The reaction temperature is usually about 0 to 150 ° C, preferably 0 to 100 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
工程(1−4):
化合物(s−4)に対して、塩基および金属触媒存在下に有機ホウ素化合物(例えばR3a−B(OH)2等)、有機亜鉛化合物(例えばR3a−ZnBr等)、アルケニル化合物、アルキニル化合物またはヒドロキシ化合物(例えばR3a−OH等)等を作用させることにより種々のカップリング反応を行い、(s−5)の化合物を製造することができる。塩基としては、炭酸カリウム、炭酸セシウム、炭酸ナトリウム、炭酸水素ナトリウム、ナトリウムメトキサイド、ナトリウムtert−ブトキサイド、水酸化ナトリウム、水酸化カリウム、フッ化カリウム等の無機塩基、または、トリエチルアミン、ジイソプロピルエチルアミン、トリブチルアミン、DBN、DABCO、DBU、ピリジン、ジメチルアミノピリジン、ピコリン、またはNMM等の有機塩基が用いられる。カップリングの種類により、塩基を用いない場合もある。金属触媒としては例えば、ビス(トリス tert−ブチルホスフィン)パラジウム、ビス(トリス o−トリルホスフィン)ジクロロパラジウム、ビス(トリス o−トリルホスフィン)パラジウム、テトラキストリフェニルホスフィンパラジウム、ジクロロパラジウム(アセトニトリル)、ビス(トリス o−トリルホスフィン)ジクロロパラジウム、(1,1’−ビス(ジフェニルホスフィノ)フェロセン)ジクロロパラジウム、PEPPSITM・IPr((1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾリデン) (3−クロロピリジル)パラジウム(II)ジクロライド)などの触媒が使用できる。酢酸パラジウムや塩化パラジウムに対して、palladium reagents and catalysts, John Wiley & Sons Inc.(2004年)に記載されている配位子またはその類縁配位子から適切なものを選択して使用することもできる。溶媒としては、例えばジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、メチルシクロペンチルエーテル、アニソールまたは1,4−ジオキサン等のエーテル系溶媒、ベンゼン、トルエン、クロロベンゼンまたはキシレン等の芳香族炭化水素系溶媒、酢酸エチルまたは酢酸メチル等のエステル系溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリジノン、1,3−ジメチル−2−イミダゾリジノン、またはジメチルスルホキシド等の非プロトン性極性溶媒、もしくは水、またはそれらの混合物が挙げられる。反応温度は、用いる原料化合物の種類等により異なるが、通常約0℃〜約250℃であり、好ましくは約20℃〜約200℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
Step (1-4):
With respect to compound (s-4), in the presence of a base and a metal catalyst, an organic boron compound (for example, R 3a -B (OH) 2 etc.), an organic zinc compound (for example, R 3a -ZnBr), an alkenyl compound, an alkynyl compound Alternatively, the compound of (s-5) can be produced by performing various coupling reactions by allowing a hydroxy compound (for example, R 3a —OH or the like) to act. Examples of the base include potassium carbonate, cesium carbonate, sodium carbonate, sodium hydrogen carbonate, sodium methoxide, sodium tert-butoxide, sodium hydroxide, potassium hydroxide, potassium fluoride and other inorganic bases, or triethylamine, diisopropylethylamine, Organic bases such as butylamine, DBN, DABCO, DBU, pyridine, dimethylaminopyridine, picoline, or NMM are used. Depending on the type of coupling, a base may not be used. Examples of the metal catalyst include bis (tris tert-butylphosphine) palladium, bis (tris o-tolylphosphine) dichloropalladium, bis (tris o-tolylphosphine) palladium, tetrakistriphenylphosphine palladium, dichloropalladium (acetonitrile), bis (Tris o-tolylphosphine) dichloropalladium, (1,1′-bis (diphenylphosphino) ferrocene) dichloropalladium, PEPPSI ™ • IPr ((1,3-bis (2,6-diisopropylphenyl) imidazolidene) ( Catalysts such as (3-chloropyridyl) palladium (II) dichloride) can be used. For palladium acetate and palladium chloride, palladium reagents and catalysts, John Wiley & Sons Inc. (2004) or an appropriate ligand selected from the ligands described in (2004) can also be used. Examples of the solvent include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, methylcyclopentyl ether, anisole or 1,4-dioxane, aromatic hydrocarbon solvents such as benzene, toluene, chlorobenzene or xylene, ethyl acetate or acetic acid. Aprotic polarities such as ester solvents such as methyl, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidinone, 1,3-dimethyl-2-imidazolidinone, or dimethyl sulfoxide A solvent or water, or a mixture thereof can be mentioned. While the reaction temperature varies depending on the type of raw material compound used, it is generally about 0 ° C. to about 250 ° C., preferably about 20 ° C. to about 200 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
工程(1−5):
化合物(s−5)の水酸基を脱保護することにより、化合物(s−6)を製造することができる。本工程を実施するには、Protective Groups in Organic Synthesis, John Wiley & Sons Inc.(1981年)に記載されている方法等が挙げられる。
例えば、塩基もしくは酸を用いて化合物(s−6)へ導くことができる。塩基としては、例えばテトラブチルアンモニウムフルオリド、フッ化水素ピリジン錯体等のフッ化物イオン類、水酸化ナトリウム、水酸化カリウム、水酸化リチウム等の各種アルカリまたはアルカリ土類金属アルコキシド等を用いることができ、中でもテトラブチルアンモニウムフルオリドが好ましい。反応温度は、用いる原料化合物の種類等により異なるが、通常約0℃〜約80℃であり、好ましくは約20℃〜約40℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
Step (1-5):
Compound (s-6) can be produced by deprotecting the hydroxyl group of compound (s-5). To carry out this step, Protective Groups in Organic Synthesis, John Wiley & Sons Inc. (1981) and the like.
For example, the compound (s-6) can be derived using a base or an acid. Examples of the base that can be used include fluoride ions such as tetrabutylammonium fluoride and hydrogen fluoride pyridine complex, and various alkali or alkaline earth metal alkoxides such as sodium hydroxide, potassium hydroxide, and lithium hydroxide. Of these, tetrabutylammonium fluoride is preferred. While the reaction temperature varies depending on the type of raw material compound used, it is generally about 0 ° C to about 80 ° C, preferably about 20 ° C to about 40 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
酸としては、例えば酢酸、トリフルオロ酢酸等のカルボン酸類、塩酸、硫酸等の鉱酸類、三臭素化ホウ素、ヨウ化トリメチルシリル等のルイス酸類を用いることができる。反応温度は、0℃から加熱還流の温度範囲であり、好ましくは20℃〜60℃である。反応時間は通常30分間から48時間であり、好ましくは1時間から24時間である。 Examples of the acid include carboxylic acids such as acetic acid and trifluoroacetic acid, mineral acids such as hydrochloric acid and sulfuric acid, and Lewis acids such as boron tribromide and trimethylsilyl iodide. The reaction temperature ranges from 0 ° C. to heating reflux, preferably 20 ° C. to 60 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
工程(1−6):
化合物(s−6)の水酸基をハロゲン化もしくはスルホニルエステル化することにより、化合物(s−7)を製造することができる。ハロゲン化を行う場合は、トリフェニルホスフィンの存在下、(s−6)に四塩化炭素もしくは四臭化炭素を作用させることにより製造することができる。クロロ化を行う場合、塩化チオニル、二塩化オキサリルを用いても製造することができる。溶媒としては、例えばベンゼン、トルエン、クロロベンゼンまたはキシレン等の芳香族炭化水素系溶媒、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン溶媒が挙げられる。反応温度は、用いる原料化合物の種類等により異なるが、通常約0℃〜約150℃であり、好ましくは約20℃〜約50℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
Step (1-6):
Compound (s-7) can be produced by halogenation or sulfonyl esterification of the hydroxyl group of compound (s-6). When performing halogenation, it can manufacture by making carbon tetrachloride or carbon tetrabromide act on (s-6) in presence of triphenylphosphine. When chlorination is performed, it can also be produced using thionyl chloride or oxalyl dichloride. Examples of the solvent include aromatic hydrocarbon solvents such as benzene, toluene, chlorobenzene, and xylene, and halogen solvents such as dichloromethane, chloroform, and carbon tetrachloride. The reaction temperature varies depending on the type of raw material compound used and the like, but is usually about 0 ° C to about 150 ° C, preferably about 20 ° C to about 50 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
スルホニルエステル化を行う場合は、塩基存在下、スルホン酸塩化物と作用させることにより製造することができる。スルホン酸塩化合物としては、例えば、メタンスルホニルクロリド、p−トルエンスルホニルクロリドなどを用いることができる。塩基としては、例えば塩基としては、炭酸カリウム、炭酸セシウム、炭酸ナトリウム、炭酸水素ナトリウム、ナトリウムメトキサイド、ナトリウムtert−ブトキサイド等の無機塩基、または、トリエチルアミン、ジイソプロピルエチルアミン、トリブチルアミン、DBN、DABCO、DBU、ピリジン、ジメチルアミノピリジン、ピコリン、またはNMM等の有機塩基が用いられる。溶媒としては、例えばジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、メチルシクロペンチルエーテル、アニソールまたは1,4−ジオキサン等のエーテル系溶媒、ベンゼン、トルエン、クロロベンゼンまたはキシレン等の芳香族炭化水素系溶媒、酢酸エチルまたは酢酸メチル等のエステル系溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリジノン、1,3−ジメチル−2−イミダゾリジノン、またはジメチルスルホキシド等の非プロトン性極性溶媒、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン溶媒が挙げられる。反応温度は、用いる原料化合物の種類等により異なるが、通常約−10℃〜約100℃であり、好ましくは約0℃〜約40℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。 When sulfonyl esterification is performed, it can be produced by reacting with a sulfonic acid chloride in the presence of a base. As the sulfonate compound, for example, methanesulfonyl chloride, p-toluenesulfonyl chloride and the like can be used. Examples of the base include, for example, potassium carbonate, cesium carbonate, sodium carbonate, sodium bicarbonate, sodium methoxide, sodium tert-butoxide and the like, or triethylamine, diisopropylethylamine, tributylamine, DBN, DABCO, DBU , Organic bases such as pyridine, dimethylaminopyridine, picoline, or NMM are used. Examples of the solvent include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, methylcyclopentyl ether, anisole or 1,4-dioxane, aromatic hydrocarbon solvents such as benzene, toluene, chlorobenzene or xylene, ethyl acetate or acetic acid. Aprotic polarities such as ester solvents such as methyl, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidinone, 1,3-dimethyl-2-imidazolidinone, or dimethyl sulfoxide Examples of the solvent include halogen solvents such as dichloromethane, chloroform, and carbon tetrachloride. While the reaction temperature varies depending on the type of raw material compound used, it is generally about −10 ° C. to about 100 ° C., preferably about 0 ° C. to about 40 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
工程(1−7):
化合物(s−7)に対して、塩基存在下アミン化合物を作用させることにより、化合物(S−8)またはその薬学的に許容される塩を製造することができる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えば、テトラヒドロフラン、ジエチルエーテル、ジメトキシエタン、1,4−ジオキサン等のエーテル溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、アセトニトリル等の非プロトン性極性溶媒が挙げられる。塩基としては、例えば炭酸セシウム、炭酸カリウムのような炭酸アルカリ金属、水素化ナトリウム、水素化カリウム等の水素化アルカリ金属、ナトリウムエトキシド、ナトリウムtert−ブトキシド、カリウムtert−ブトキシド等のアルカリ金属アルコキシド、DBU,DBN、DABCOなどの有機塩基等が挙げられ、好ましくは、炭酸カリウムが挙げられる。反応温度としては、通常約0〜150℃で行われ、好ましくは20〜100℃の範囲で行われる。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
Step (1-7):
Compound (S-8) or a pharmaceutically acceptable salt thereof can be produced by reacting compound (s-7) with an amine compound in the presence of a base. Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include ether solvents such as tetrahydrofuran, diethyl ether, dimethoxyethane, and 1,4-dioxane, and aprotic polar solvents such as N, N-dimethylformamide, N, N-dimethylacetamide, and acetonitrile. Examples of the base include alkali metal carbonates such as cesium carbonate and potassium carbonate, alkali metal hydrides such as sodium hydride and potassium hydride, alkali metal alkoxides such as sodium ethoxide, sodium tert-butoxide and potassium tert-butoxide, Organic bases such as DBU, DBN, DABCO and the like can be mentioned, and potassium carbonate is preferable. The reaction temperature is usually about 0 to 150 ° C, preferably 20 to 100 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
製造法2:
製造法1に記載の化合物(S−8)またはその薬学的に許容される塩のうち、(S−8a)またはその薬学的に許容される塩は、製造法1に記載の化合物(s−3)を用いて、以下の工程(2−1)から(2−4)に従って製造することもできる。
Production method 2:
Of the compound (S-8) or a pharmaceutically acceptable salt thereof described in Production Method 1, (S-8a) or a pharmaceutically acceptable salt thereof is the compound (s- 3) can also be used according to the following steps (2-1) to (2-4).
(式中、R1、R2、R3、R4、およびR3a−Yは、製造法1と同義である。L´は、C1−5アルキレンであり、該アルキレンの置換基は請求項1に記載のLと同義であり、X4は製造法1におけるX2と同義である。RbはC1−4アルキルであり、2個のRbが、一緒になって環を形成してもよい。)
(In the formula, R 1 , R 2 , R 3 , R 4 , and R 3a -Y have the same meaning as in Production Method 1. L ′ is C 1-5 alkylene, and the alkylene substituent is claimed. has the same meaning as L according to claim 1, X 4 has the same meaning as X 2 in the production method 1 .R b is C 1-4 alkyl, the two R b are taken together to form a ring You may do it.)
工程(2−1):
本工程は、化合物(s−3)およびアセタール化合物を用いて、製造法1に記載の工程(1−3)に準じた条件に従い、化合物(s−9)を得る工程である。
Step (2-1):
This step is a step of obtaining compound (s-9) using compound (s-3) and an acetal compound according to the conditions according to step (1-3) described in Production Method 1.
工程(2−2):
本工程は、化合物(s−9)を用いて、製造法1に記載の工程(1−4)に準じた条件に従い、化合物(s−10)を得る工程である。
Step (2-2):
This step is a step of obtaining compound (s-10) using compound (s-9) according to the conditions according to step (1-4) described in Production Method 1.
工程(2−3):
アセタール化合物(s−10)の脱保護反応をおこなうことにより、アルデヒド化合物(s−11)を製造することができる。
本工程を実施するには、Protective Groups in Organic Synthesis, John Wiley & Sons Inc.(1981年)に記載されている方法等が挙げられる。
例えば、以下のような酸加水分解によってアルデヒド化合物(s−11)へと導くことができる。酸としては、塩酸、硫酸等の鉱酸、ギ酸、酢酸、トリフルオロ酢酸、トリフルオロメタンスルホン酸、トルエンスルホン酸等の有機酸などが用いられ、好ましくはトリフルオロ酢酸、トルエンスルホン酸または塩酸が挙げられる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えばジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、ジメトキシエタン、メチルシクロペンチルエーテル、アニソール、1,4−ジオキサン等のエーテル系溶媒、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン溶媒、アセトン、アセトニトリル等の非プロトン性極性溶媒、もしくは水、またはそれらの混合物が挙げられる。反応温度は通常、−20℃から加熱還流の温度範囲で、反応時間は30分間から48時間反応させることにより、式(s−11)の化合物を得ることができる。反応温度として好ましくは0℃〜60℃であり、反応時間として好ましくは1時間から24時間である。
Step (2-3):
An aldehyde compound (s-11) can be produced by performing a deprotection reaction of the acetal compound (s-10).
To carry out this step, Protective Groups in Organic Synthesis, John Wiley & Sons Inc. (1981) and the like.
For example, it can lead to an aldehyde compound (s-11) by the following acid hydrolysis. Examples of the acid include mineral acids such as hydrochloric acid and sulfuric acid, organic acids such as formic acid, acetic acid, trifluoroacetic acid, trifluoromethanesulfonic acid, and toluenesulfonic acid, preferably trifluoroacetic acid, toluenesulfonic acid, and hydrochloric acid. It is done. Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, dimethoxyethane, methylcyclopentyl ether, anisole and 1,4-dioxane, aromatic hydrocarbon solvents such as benzene, toluene and xylene, dichloromethane and chloroform. , Halogen solvents such as carbon tetrachloride, aprotic polar solvents such as acetone and acetonitrile, or water, or a mixture thereof. The reaction temperature is usually in the temperature range of −20 ° C. to heating under reflux, and the reaction time is 30 minutes to 48 hours, whereby the compound of the formula (s-11) can be obtained. The reaction temperature is preferably 0 to 60 ° C., and the reaction time is preferably 1 to 24 hours.
工程(2−4):
化合物(s−11)を用いて、還元剤および酸の存在下、アミン化合物を作用させることにより、化合物(S−8a)またはその薬学的に許容される塩を製造することができる。
還元剤としては、例えばトリアセトキシ水素化ホウ素ナトリウム、水素化ホウ素ナトリウム、水素化シアノホウ素ナトリウム等を用いることができ、中でもトリアセトキシ水素化ホウ素ナトリウムが好ましい。酸としては、酢酸が好ましい。
溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えばジクロロメタン、クロロホルム、1,2−ジクロロエタン等のハロゲン系溶媒、テトラヒドロフラン、ジオキサン、ジエチルエーテル、ジイソプロピルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒、ベンゼン、トルエン、キシレン等の芳香族系溶媒、メタノール、エタノール、n -プロパノール、2-プロパノール等のアルコール系溶媒等を単独でまたはそれらを混合して用いることができ、中でもジクロロメタンが好ましい。
Step (2-4):
Compound (S-8a) or a pharmaceutically acceptable salt thereof can be produced by reacting amine compound with compound (s-11) in the presence of a reducing agent and an acid.
As the reducing agent, for example, sodium triacetoxyborohydride, sodium borohydride, sodium cyanoborohydride and the like can be used, and among these, sodium triacetoxyborohydride is preferable. As the acid, acetic acid is preferred.
Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include halogen solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether and 1,2-dimethoxyethane, and aromatics such as benzene, toluene and xylene. A group solvent, alcohol solvents such as methanol, ethanol, n-propanol, 2-propanol and the like can be used alone or as a mixture thereof. Among them, dichloromethane is preferable.
製造法3:
製造法2に記載の化合物(S−8)またはその薬学的に許容される塩のうちスルホニル化合物(S−15)またはその薬学的に許容される塩は、製造法1に記載の化合物(s−3)を用いて、以下の工程(3−1)から(3−4)に従って製造することもできる。
Production method 3:
Of the compound (S-8) or the pharmaceutically acceptable salt thereof described in Production Method 2, the sulfonyl compound (S-15) or the pharmaceutically acceptable salt thereof is the compound (s 3), it can also be produced according to the following steps (3-1) to (3-4).
(式中、R2、R3、R4、L、およびR3a−Yは、製造法1と同義である。X5は製造法1におけるX2と同義である。R5はC1−4アルキルを表し、Rcはベンジルオキシカルボニル、tert−ブトキシカルボニル等の保護基を表す。)
(In the formula, R 2 , R 3 , R 4 , L, and R 3a -Y have the same meaning as in Production Method 1. X 5 has the same meaning as X 2 in Production Method 1. R 5 is C 1-. 4 represents alkyl, and R c represents a protecting group such as benzyloxycarbonyl, tert-butoxycarbonyl, etc.)
工程(3−1):
化合物(s−3)を用いて、製造法2に記載の工程(2−1)に準じた条件に従い化合物(s−12)を製造することができる。また、化合物(s−12)は化合物(s−3)を用いて、トリフェニルホスフィン存在下、アルキルアルコールおよびジイソプロピルアゾジカルボキシレートまたはジエチルアゾジカルボキシレートを作用させることによっても製造することができる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えばジクロロメタン、クロロホルム、1,2−ジクロロエタン等のハロゲン系溶媒、テトラヒドロフラン、ジオキサン、ジエチルエーテル、ジイソプロピルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒等を単独でまたはそれらを混合して用いることができ、中でもテトラヒドロフランが好ましい。反応温度としては、通常約0℃〜100℃で行われ、好ましくは0〜40℃の範囲で行われる。反応時間は、通常1〜24時間であり、好ましくは2〜12時間である。
Step (3-1):
A compound (s-12) can be manufactured according to the conditions according to the process (2-1) of the manufacturing method 2 using a compound (s-3). Compound (s-12) can also be produced by reacting compound (s-3) with alkyl alcohol and diisopropyl azodicarboxylate or diethyl azodicarboxylate in the presence of triphenylphosphine. . Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include halogen solvents such as dichloromethane, chloroform, and 1,2-dichloroethane, ether solvents such as tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, and 1,2-dimethoxyethane, or a mixture thereof. Of these, tetrahydrofuran is preferred. The reaction temperature is usually about 0 ° C to 100 ° C, preferably 0 to 40 ° C. The reaction time is usually 1 to 24 hours, preferably 2 to 12 hours.
工程(3−2):
本工程は、化合物(s−12)を用いて、製造法1に記載の工程(1−4)に準じた条件に従い、化合物(s−13)を得る工程である。
Step (3-2):
This step is a step of obtaining a compound (s-13) using the compound (s-12) according to the conditions according to the step (1-4) described in the production method 1.
工程(3−3):
化合物(s−14)は、酸又は塩基存在下、化合物(s−13)を脱保護することにより製造することができる。酸としては、例えば塩酸、硫酸等の鉱酸やトリフルオロ酢酸等の有機酸が挙げられ、塩基としては、例えば、水酸化アルカリ金属(水酸化ナトリウム、水酸化カリウム等)が挙げられる。溶媒としては、例えば、メタノール、エタノール、イソプロピルアルコール、1−ブタノール等のアルコール系溶媒、テトラヒドロフラン、ジエチルエーテル、1,2−ジメトキシエタン、1,4−ジオキサン等のエーテル系溶媒、水、又はこれらの混合溶媒が挙げられる。反応温度としては、通常0〜100℃の範囲から選択され、好ましくは0〜50℃の範囲で行われる。反応時間は、通常1〜24時間であり、好ましくは1〜12時間である。
Step (3-3):
Compound (s-14) can be produced by deprotecting compound (s-13) in the presence of an acid or a base. Examples of the acid include mineral acids such as hydrochloric acid and sulfuric acid, and organic acids such as trifluoroacetic acid, and examples of the base include alkali metal hydroxides (sodium hydroxide, potassium hydroxide, etc.). Examples of the solvent include alcohol solvents such as methanol, ethanol, isopropyl alcohol, 1-butanol, ether solvents such as tetrahydrofuran, diethyl ether, 1,2-dimethoxyethane, 1,4-dioxane, water, or these. A mixed solvent is mentioned. The reaction temperature is usually selected from the range of 0 to 100 ° C., preferably 0 to 50 ° C. The reaction time is usually 1 to 24 hours, preferably 1 to 12 hours.
また、化合物(s−13)のRcがベンジルオキシカルボニルの場合、製造法1に記載の工程(1−1)に記載の接触還元法を用いても化合物(s−14)を製造することができる。
工程(3−4):
本工程は、化合物(s−14)を用いて、塩基の存在下あるいは非存在下、アルキルビニルスルホンを作用させることにより、化合物(S−15)またはその薬学的に許容される塩を得る工程である。塩基としては、トリエチルアミン、ジイソプロピルエチルアミン、トリブチルアミン、DBN、DABCO、DBU、ピリジン、ジメチルアミノピリジン、ピコリン、またはNMM等の有機塩基が用いられる。溶媒は、本工程の反応条件で反応しない溶媒であれば使用できる。溶媒としては、例えばジクロロメタン、クロロホルム、1,2−ジクロロエタン等のハロゲン系溶媒、テトラヒドロフラン、ジオキサン、ジエチルエーテル、ジイソプロピルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒、メタノール、エタノール、n -プロパノール、2-プロパノール等のアルコール系溶媒等を単独でまたはそれらを混合して用いることができ、中でもエタノールが好ましい。
In addition, when R c of compound (s-13) is benzyloxycarbonyl, compound (s-14) can also be produced using the catalytic reduction method described in Step (1-1) described in Production Method 1. Can do.
Step (3-4):
This step is a step of obtaining compound (S-15) or a pharmaceutically acceptable salt thereof by reacting alkylvinylsulfone with compound (s-14) in the presence or absence of a base. It is. As the base, organic bases such as triethylamine, diisopropylethylamine, tributylamine, DBN, DABCO, DBU, pyridine, dimethylaminopyridine, picoline, or NMM are used. Any solvent that does not react under the reaction conditions in this step can be used. Examples of the solvent include halogen solvents such as dichloromethane, chloroform, 1,2-dichloroethane, ether solvents such as tetrahydrofuran, dioxane, diethyl ether, diisopropyl ether, 1,2-dimethoxyethane, methanol, ethanol, n-propanol, Alcohol solvents such as 2-propanol can be used alone or as a mixture thereof, and ethanol is particularly preferable.
製造法4:
製造法1に記載の化合物(S−8)またはその薬学的に許容される塩は、製造法1に記載の化合物(s−4)を用いて、以下の工程(4−1)から(4−4)に従って製造することもできる。
Production method 4:
Compound (S-8) described in Production Method 1 or a pharmaceutically acceptable salt thereof is prepared using Compound (s-4) described in Production Method 1 from the following steps (4-1) to (4): -4).
(式中の記号は、製造法1と同義である。)
(The symbols in the formula have the same meaning as in Production Method 1.)
工程(4−1):
本工程は、化合物(s−4)を用いて、製造法1に記載の工程(1−5)に準じた条件に従い、化合物(s−16)を得る工程である。
Step (4-1):
This step is a step of obtaining a compound (s-16) using the compound (s-4) according to the conditions according to the step (1-5) described in the production method 1.
工程(4−2):
本工程は、化合物(s−16)を用いて、製造法1に記載の工程(1−6)に準じた条件に従い、化合物(s−17)を得る工程である。
Step (4-2):
This step is a step of obtaining compound (s-17) using compound (s-16) according to the conditions according to step (1-6) described in Production Method 1.
工程(4−3):
本工程は、化合物(s−17)を用いて、製造法1に記載の工程(1−7)に準じた条件に従い、化合物(s−18)を得る工程である。
Step (4-3):
This step is a step of obtaining compound (s-18) using compound (s-17) according to the conditions according to step (1-7) described in production method 1.
工程(4−4):
本工程は、化合物(s−18)を用いて、製造法1に記載の工程(1−4)に準じた条件に従い、化合物(S−8)またはその薬学的に許容される塩を得る工程である。
Step (4-4):
This step is a step of obtaining compound (S-8) or a pharmaceutically acceptable salt thereof using compound (s-18) according to the conditions according to step (1-4) described in Production Method 1. It is.
製造法5:
製造法2に記載の化合物(S−8a)は、製造法2に記載の化合物(s−9)を用いて、以下の工程(5−1)から(5−3)に従って製造することもできる。
Production method 5:
Compound (S-8a) described in Production Method 2 can also be produced according to the following steps (5-1) to (5-3) using compound (s-9) described in Production Method 2. .
(式中の記号は製造法2と同義である。)
(The symbols in the formula have the same meaning as in Production Method 2.)
工程(5−1):
本工程は、化合物(s−9)を用いて、製造法2に記載の工程(2−3)に準じた条件に従い、化合物(s−19)を得る工程である。
Step (5-1):
This step is a step of obtaining compound (s-19) using compound (s-9) according to the conditions according to step (2-3) described in production method 2.
工程(5−2):
本工程は、化合物(s−19)を用いて、製造法2に記載の工程(2−4)に準じた条件に従い、化合物(s−20)を得る工程である。
Step (5-2):
This step is a step of obtaining compound (s-20) using compound (s-19) according to the conditions according to step (2-4) described in Production Method 2.
工程(5−3):
本工程は、化合物(s−20)を用いて、製造法1に記載の工程(1−4)に準じた条件に従い、化合物(S−8a)またはその薬学的に許容される塩を得る工程である。
Step (5-3):
This step is a step of obtaining compound (S-8a) or a pharmaceutically acceptable salt thereof using compound (s-20) according to the conditions according to step (1-4) described in Production Method 1. It is.
製造法6:
製造法3に記載の化合物(S−15)またはその薬学的に許容される塩は、製造法3に記載の化合物(s−12)を用いて、以下の工程(6−1)から(6−3)に従って製造することもできる。
Production method 6:
Compound (S-15) described in Production Method 3 or a pharmaceutically acceptable salt thereof is prepared by using compound (s-12) described in Production Method 3 in the following steps (6-1) to (6): -3).
(式中の記号は、製造法3と同義である。)
(The symbols in the formula have the same meaning as in Production Method 3.)
工程(6−1):
本工程は、化合物(s−12)を用いて、製造法3に記載の工程(3−3)に準じた条件に従い、化合物(s−21)を得る工程である。
Step (6-1):
This step is a step of obtaining a compound (s-21) using the compound (s-12) according to the conditions according to the step (3-3) described in the production method 3.
工程(6−2):
本工程は、化合物(s−21)を用いて、製造法3に記載の工程(3−4)に準じた条件に従い、化合物(s−22)を得る工程である。
Step (6-2):
This step is a step of obtaining a compound (s-22) using the compound (s-21) according to the conditions according to the step (3-4) described in the production method 3.
工程(6−3):
本工程は、化合物(s−22)を用いて、製造法1に記載の工程(1−4)に準じた条件に従い、化合物(S−15)またはその薬学的に許容される塩を得る工程である。
Step (6-3):
This step is a step of obtaining compound (S-15) or a pharmaceutically acceptable salt thereof using compound (s-22) according to the conditions according to step (1-4) described in Production Method 1. It is.
製造法7:
製造法1に記載の化合物(s−5)は、製造法1に記載の化合物(s−3)を用いて、以下の工程(7−1)から(7−2)に従って製造することもできる。
Production method 7:
Compound (s-5) described in Production Method 1 can also be produced according to the following steps (7-1) to (7-2) using compound (s-3) described in Production Method 1. .
(式中の記号は、製造法1と同義である。)
(The symbols in the formula have the same meaning as in Production Method 1.)
工程(7−1):
本工程は、化合物(s−3)を用いて、製造法1に記載の工程(1−4)に準じた条件に従い、化合物(s−23)を得る工程である。
Step (7-1):
This step is a step of obtaining a compound (s-23) using the compound (s-3) according to the conditions according to the step (1-4) described in the production method 1.
工程(7−2):
本工程は、化合物(s−23)を用いて、製造法1に記載の工程(1−3)に準じた条件に従い、化合物(s−5)を得る工程である。
Step (7-2):
This step is a step of obtaining a compound (s-5) using the compound (s-23) according to the conditions according to the step (1-3) described in the production method 1.
製造法8:
製造法2に記載の化合物(s−10)は、製造法7に記載の化合物(s−23)を用いて、以下の工程(8−1)に従って製造することもできる。
Production method 8:
Compound (s-10) described in Production Method 2 can also be produced according to the following step (8-1) using compound (s-23) described in Production Method 7.
(式中の記号は、製造法2と同義である。)
(The symbols in the formula have the same meaning as in Production Method 2.)
工程(8−1):
本工程は、化合物(s−23)を用いて、製造法2に記載の工程(2−1)に準じた条件に従い、化合物(s−10)を得る工程である。
Step (8-1):
This step is a step of obtaining a compound (s-10) using the compound (s-23) according to the conditions according to the step (2-1) described in the production method 2.
製造法9:
製造法3に記載の化合物(s−13)は、製造法7に記載の化合物(s−23)を用いて、以下の工程(9−1)に従って製造することもできる。
Production method 9:
Compound (s-13) described in Production Method 3 can also be produced according to the following step (9-1) using compound (s-23) described in Production Method 7.
(式中の記号は、製造法3と同義である。)
(The symbols in the formula have the same meaning as in Production Method 3.)
工程(9−1):
本工程は、化合物(s−23)を用いて、製造法3に記載の工程(3−1)に準じた条件に従い、化合物(s−13)を得る工程である。
Step (9-1):
This step is a step of obtaining compound (s-13) using compound (s-23) according to the conditions according to step (3-1) described in production method 3.
製造法10:
製造法1に記載の化合物(s−4)のうち、(s−4a)は、製造法1に記載の化合物(s−2)を用いて、以下の工程(10−1)から(10−2)に従って製造することもできる。
Production method 10:
Among the compounds (s-4) described in the production method 1, (s-4a) is prepared by using the compound (s-2) described in the production method 1 from the following steps (10-1) to (10- It can also be produced according to 2).
(式中、R4、X1、およびRaは製造法1と同義であり、L´は製造法2と同義である。)
(In the formula, R 4 , X 1 and R a have the same meaning as in Production Method 1, and L ′ has the same meaning as in Production Method 2.)
工程(10−1):
本工程は、化合物(s−2)を用いて、製造法2に記載の工程(2−4)に準じた条件に従い、化合物(s−24)を得る工程である。
Step (10-1):
This step is a step of obtaining a compound (s-24) using the compound (s-2) according to the conditions according to the step (2-4) described in the production method 2.
工程(10−2):
本工程は、化合物(s−24)を用いて、製造法1に記載の工程(1−2)に準じた条件に従い、化合物(s−4a)を得る工程である。
Step (10-2):
This step is a step of obtaining compound (s-4a) using compound (s-24) according to the conditions according to step (1-2) described in Production Method 1.
製造法11:
製造法2に記載の化合物(s−9)のうち、(s−9a)は、製造法1に記載の化合物(s−2)を用いて、以下の工程(11−1)から(11−2)に従って製造することもできる。
Production method 11:
Of the compound (s-9) described in Production Method 2, (s-9a) is prepared from the following steps (11-1) to (11-) using the compound (s-2) described in Production Method 1. It can also be produced according to 2).
(式中R4、X1は製造法1と同義であり、Rbは製造法2と同義である。L´´は、C1−4アルキレンであり、該アルキレンの置換基は請求項1に記載のLと同義である。)
(Wherein R 4 and X 1 have the same meaning as in Production Method 1, and R b has the same meaning as in Production Method 2. L ″ is C 1-4 alkylene, and the alkylene substituent is defined as Claim 1. It is synonymous with L of description.)
工程(11−1):
本工程は、化合物(s−2)を用いて、製造法2に記載の工程(2−4)に準じた条件に従い、化合物(s−25)を得る工程である。
Step (11-1):
This step is a step of obtaining a compound (s-25) using the compound (s-2) according to the conditions according to the step (2-4) described in the production method 2.
工程(11−2):
本工程は、化合物(s−25)を用いて、製造法1に記載の工程(1−2)に準じた条件に従い、化合物(s−9a)を得る工程である。
Step (11-2):
This step is a step of obtaining a compound (s-9a) using the compound (s-25) according to the conditions according to the step (1-2) described in the production method 1.
製造法12:
製造法3に記載の化合物(s−12)のうち、(s−12a)は、製造法1に記載の化合物(s−2)を用いて、以下の工程(12−1)から(12−2)に従って製造することもできる。
Production method 12:
Among the compounds (s-12) described in Production Method 3, (s-12a) is prepared using the compound (s-2) described in Production Method 1 from the following steps (12-1) to (12- It can also be produced according to 2).
(式中R4およびX1は製造法1と同義である。Rcは製造法3と同義であり、L´は、製造法2と同義である。)
(Wherein R 4 and X 1 are synonymous with production method 1. R c is synonymous with production method 3 and L ′ is synonymous with production method 2.)
工程(12−1):
本工程は、化合物(s−2)を用いて、製造法2に記載の工程(2−4)に準じた条件に従い、化合物(s−26)を得る工程である。
Step (12-1):
This step is a step of obtaining compound (s-26) using compound (s-2) according to the conditions according to step (2-4) described in production method 2.
工程(12−2):
本工程は、化合物(s−26)を用いて、製造法1に記載の工程(1−2)に準じた条件に従い、化合物(s−12a)を得る工程である。
製造法13:
製造法1に記載の化合物(s−5)は、製造法1に記載の化合物(s−4)を用いて、以下の工程(13−1)から(13−2)に従って製造することもできる。また、製造法2、3、4、5、6、7においても同様に、ベンズオキサゾロン環側の脱離基をホウ素化することによりカップリング反応をすることもできる。
Step (12-2):
This step is a step of obtaining a compound (s-12a) using the compound (s-26) according to the conditions according to the step (1-2) described in the production method 1.
Production method 13:
Compound (s-5) described in Production Method 1 can also be produced according to the following steps (13-1) to (13-2) using compound (s-4) described in Production Method 1. . Similarly, in production methods 2, 3, 4, 5, 6, and 7, a coupling reaction can also be carried out by boring the leaving group on the benzoxazolone ring side.
(式中Ra、R3、R3a、R4、L、およびX1は製造法1と同義である。Rdは水素原子またはC1−4アルキル基を示し、2個のRdがアルキル基である場合は、一緒になって結合する酸素原子およびホウ素原子と共に5〜6員環を形成しても良い。Y2はハロゲン、トリフルオロメタンスルホニルオキシまたはメタンスルホニルオキシなどの脱離基を示す。)
工程(13−1)
化合物(s−4)に対して、塩基および金属触媒存在下に有機ホウ素化合物を作用させることにより(s−27)の化合物を製造することができる。塩基としては、炭酸ナトリウム、炭酸水素ナトリウム、フッ化カリウム、酢酸ナトリウム、酢酸カリウム等の無機塩基が用いられる。金属触媒としては例えば、テトラキストリフェニルホスフィンパラジウム、ジクロロパラジウム(アセトニトリル)、(1,1’−ビス(ジフェニルホスフィノ)フェロセン)ジクロロパラジウム等の触媒が使用できる。有機ホウ素化合物としては、ビスピナコレートジボラン、ビスネオペンチルグリコレートジボラン等が用いられる。溶媒としては、例えばジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、メチルシクロペンチルエーテル、アニソールまたは1,4−ジオキサン等のエーテル系溶媒、ベンゼン、トルエン、クロロベンゼンまたはキシレン等の芳香族炭化水素系溶媒、酢酸エチルまたは酢酸メチル等のエステル系溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリジノン、1,3−ジメチル−2−イミダゾリジノン、またはジメチルスルホキシド等の非プロトン性極性溶媒、またはそれらの混合物が挙げられる。反応温度は、用いる原料化合物の種類等により異なるが、通常約0℃〜約250℃であり、好ましくは約20℃〜約100℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
(In the formula, R a , R 3 , R 3a , R 4 , L, and X 1 have the same meaning as in Production Method 1. R d represents a hydrogen atom or a C 1-4 alkyl group, and two R d are When it is an alkyl group, it may form a 5- to 6-membered ring together with the oxygen atom and boron atom bonded together, and Y 2 represents a leaving group such as halogen, trifluoromethanesulfonyloxy or methanesulfonyloxy. Show.)
Step (13-1)
The compound (s-27) can be produced by reacting the compound (s-4) with an organic boron compound in the presence of a base and a metal catalyst. As the base, inorganic bases such as sodium carbonate, sodium hydrogen carbonate, potassium fluoride, sodium acetate, potassium acetate and the like are used. As the metal catalyst, for example, a catalyst such as tetrakistriphenylphosphine palladium, dichloropalladium (acetonitrile), (1,1′-bis (diphenylphosphino) ferrocene) dichloropalladium can be used. As the organic boron compound, bispinacolate diborane, bisneopentyl glycolate diborane, or the like is used. Examples of the solvent include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, methylcyclopentyl ether, anisole or 1,4-dioxane, aromatic hydrocarbon solvents such as benzene, toluene, chlorobenzene or xylene, ethyl acetate or acetic acid. Aprotic polarities such as ester solvents such as methyl, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidinone, 1,3-dimethyl-2-imidazolidinone, or dimethyl sulfoxide A solvent, or a mixture thereof. While the reaction temperature varies depending on the kind of raw material compound to be used, etc., it is generally about 0 ° C. to about 250 ° C., preferably about 20 ° C. to about 100 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
また、金属触媒を用いずに製造することもできる。その場合、塩基としてn−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウムが用いられる。有機ホウ素化合物としては、トリメトキシボラン、トリエトキシボラン、トリブトキシボラン、トリイソプロポキシボラン、イソプロピルピナコールボレートなどが用いられる。溶媒としては、例えばジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、メチルシクロペンチルエーテル、アニソールまたは1,4−ジオキサン等のエーテル系溶媒、ベンゼン、トルエン、クロロベンゼンまたはキシレン等の芳香族炭化水素系溶媒、またはそれらの混合物が挙げられる。反応温度は、用いる原料化合物の種類等により異なるが、通常約−78℃〜約100℃であり、好ましくは約−78℃〜約30℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。 Moreover, it can also manufacture without using a metal catalyst. In that case, n-butyllithium, sec-butyllithium, or tert-butyllithium is used as the base. As the organoboron compound, trimethoxyborane, triethoxyborane, tributoxyborane, triisopropoxyborane, isopropyl pinacol borate and the like are used. Examples of the solvent include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, methylcyclopentyl ether, anisole and 1,4-dioxane, aromatic hydrocarbon solvents such as benzene, toluene, chlorobenzene and xylene, or mixtures thereof. Is mentioned. While the reaction temperature varies depending on the kind of raw material compound to be used, etc., it is generally about -78 ° C to about 100 ° C, preferably about -78 ° C to about 30 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
得られたアルコキシホウ素化合物は、酸性条件下で後処理することでボロン酸化合物へ導くことができる。酸として、塩酸または硫酸等が用いられる。
工程(13−2)
化合物(s−27)に対して、塩基および金属触媒存在下、R3a−Y2を作用させることにより(s−5)の化合物を製造することができる。塩基としては、炭酸カリウム、炭酸セシウム、炭酸ナトリウム、炭酸水素ナトリウム、ナトリウムメトキサイド、ナトリウムtert−ブトキサイド、水酸化ナトリウム、水酸化カリウム、フッ化カリウム等の無機塩基、または、トリエチルアミン、ジイソプロピルエチルアミン、トリブチルアミン、DBN、DABCO、DBU、ピリジン、ジメチルアミノピリジン、ピコリン、またはNMM等の有機塩基が用いられる。金属触媒としては例えば、ビス(トリス tert−ブチルホスフィン)パラジウム、ビス(トリス o−トリルホスフィン)ジクロロパラジウム、ビス(トリス o−トリルホスフィン)パラジウム、テトラキストリフェニルホスフィンパラジウム、ジクロロパラジウム(アセトニトリル)、ビス(トリス o−トリルホスフィン)ジクロロパラジウム、(1,1’−ビス(ジフェニルホスフィノ)フェロセン)ジクロロパラジウム、PEPPSITM・IPr((1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾリデン) (3−クロロピリジル)パラジウム(II)ジクロライド)などの触媒が使用できる。酢酸パラジウムや塩化パラジウムに対して、palladium reagents and catalysts, John Wiley & Sons Inc.(2004年)に記載されている配位子またはその類縁配位子から適切なものを選択して使用することもできる。溶媒としては、例えばジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、メチルシクロペンチルエーテル、アニソールまたは1,4−ジオキサン等のエーテル系溶媒、ベンゼン、トルエン、クロロベンゼンまたはキシレン等の芳香族炭化水素系溶媒、酢酸エチルまたは酢酸メチル等のエステル系溶媒、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリジノン、1,3−ジメチル−2−イミダゾリジノン、またはジメチルスルホキシド等の非プロトン性極性溶媒、もしくは水、またはそれらの混合物が挙げられる。反応温度は、用いる原料化合物の種類等により異なるが、通常約0℃〜約250℃であり、好ましくは約20℃〜約200℃である。反応時間は、通常30分間から48時間であり、好ましくは1時間から24時間である。
The resulting alkoxyboron compound can be converted into a boronic acid compound by post-treatment under acidic conditions. As the acid, hydrochloric acid or sulfuric acid is used.
Step (13-2)
The compound (s-5) can be produced by reacting the compound (s-27) with R 3a -Y 2 in the presence of a base and a metal catalyst. Examples of the base include potassium carbonate, cesium carbonate, sodium carbonate, sodium hydrogen carbonate, sodium methoxide, sodium tert-butoxide, sodium hydroxide, potassium hydroxide, potassium fluoride and other inorganic bases, or triethylamine, diisopropylethylamine, Organic bases such as butylamine, DBN, DABCO, DBU, pyridine, dimethylaminopyridine, picoline, or NMM are used. Examples of the metal catalyst include bis (tris tert-butylphosphine) palladium, bis (tris o-tolylphosphine) dichloropalladium, bis (tris o-tolylphosphine) palladium, tetrakistriphenylphosphine palladium, dichloropalladium (acetonitrile), bis (Tris o-tolylphosphine) dichloropalladium, (1,1′-bis (diphenylphosphino) ferrocene) dichloropalladium, PEPPSI ™ • IPr ((1,3-bis (2,6-diisopropylphenyl) imidazolidene) ( Catalysts such as (3-chloropyridyl) palladium (II) dichloride) can be used. For palladium acetate and palladium chloride, palladium reagents and catalysts, John Wiley & Sons Inc. (2004) or an appropriate ligand selected from the ligands described in (2004) can also be used. Examples of the solvent include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, methylcyclopentyl ether, anisole or 1,4-dioxane, aromatic hydrocarbon solvents such as benzene, toluene, chlorobenzene or xylene, ethyl acetate or acetic acid. Aprotic polarities such as ester solvents such as methyl, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidinone, 1,3-dimethyl-2-imidazolidinone, or dimethyl sulfoxide A solvent or water, or a mixture thereof can be mentioned. While the reaction temperature varies depending on the type of raw material compound used, it is generally about 0 ° C. to about 250 ° C., preferably about 20 ° C. to about 200 ° C. The reaction time is usually 30 minutes to 48 hours, preferably 1 hour to 24 hours.
上記製造法における出発原料および中間体は、公知化合物であるか、公知化合物から公知の方法により合成することができる。また、上記製造法において、反応点以外の何れかの官能基が、説明した反応条件下で変化するか、または説明した方法を実施するのに不適切な場合は、反応点以外を保護し、反応させた後、脱保護することにより目的化合物を得ることができる。保護基としては、例えば前述のProtective Groups in Organic Synthesis等に記載されているような通常の保護基を用いることができる。具体的には、アミンの保護基としては、例えば、エトキシカルボニル、tert−ブトキシカルボニル、アセチル、ベンジル等を、また水酸基の保護基としては、例えば、トリ低級アルキルシリル、アセチル、ベンジル等を挙げることができる。 The starting materials and intermediates in the above production method are known compounds, or can be synthesized from known compounds by known methods. In the above production method, if any functional group other than the reactive site changes under the described reaction conditions or is inappropriate for carrying out the described method, the other than the reactive site is protected, After the reaction, the target compound can be obtained by deprotection. As the protecting group, for example, a usual protecting group as described in the aforementioned Protective Groups in Organic Synthesis can be used. Specifically, examples of the protecting group for amine include ethoxycarbonyl, tert-butoxycarbonyl, acetyl, benzyl and the like, and examples of the hydroxyl protecting group include tri-lower alkylsilyl, acetyl, benzyl and the like. Can do.
保護基の導入および脱離は、有機合成化学で常用される方法(例えば、上記のプロテクティブ・グループス・イン・オーガニック・シンセシス参照)、あるいはそれらに準じた方法により行うことができる。 Introduction and removal of protecting groups can be carried out by methods commonly used in organic synthetic chemistry (see, for example, the above-mentioned Protective Groups in Organic Synthesis) or methods based thereon.
また、上記製造方法における、中間体、または最終生成物は、その官能基を適宜変換することにより、本発明に含まれる別の化合物へ導く事もできる。官能基の変換は、通常行われる一般的方法(例えば、Comprehensive Organic Transformations, R.C.Larock著(1989年)等参照)により行うことができる。 Moreover, the intermediate body in the said manufacturing method or a final product can also be guide | induced to another compound included in this invention by changing the functional group suitably. The functional group can be converted by a commonly used general method (for example, see Comprehensive Organic Transformations, RC Larock (1989)).
上記各製造法における中間体および目的化合物は、有機合成化学で常用される精製法、例えば中和、濾過、抽出、洗浄、乾燥、濃縮、再結晶、各種クロマトグラフィー等によって単離精製することができる。また、中間体については、特に精製することなく次の反応に用いることも可能である。
また、光学異性体は前記製造法の適切な工程で、光学活性カラムを用いた方法、分別結晶化法などの公知の分離工程を実施することで分離することができる。また、出発原料として光学活性体を使用することもできる。
本発明の化合物は、光学異性体、立体異性体、ケトエノール体のような互変異性体、および/または幾何異性体を有する場合もあるが、本発明の化合物は、これらを含め全ての可能な異性体およびそれらの混合物を包含する。
The intermediates and target compounds in each of the above production methods can be isolated and purified by purification methods commonly used in synthetic organic chemistry, such as neutralization, filtration, extraction, washing, drying, concentration, recrystallization, and various chromatography. it can. Further, the intermediate can be used for the next reaction without any particular purification.
In addition, optical isomers can be separated by performing a known separation step such as a method using an optically active column or a fractional crystallization method in an appropriate step of the production method. An optically active substance can also be used as a starting material.
The compounds of the present invention may have optical isomers, stereoisomers, tautomers such as keto enols, and / or geometric isomers, but the compounds of the present invention may contain all possible It includes isomers and mixtures thereof.
本発明の化合物は、上記の異性体に加え、式(1)で表される化合物もしくはそのプロドラッグ、またはその薬学的に許容される塩を包含する。また、本発明の化合物またはその薬学的に許容される塩は、水あるいは各種溶媒との付加物の形で存在することもあるが、これら付加物も包含する。さらに、本発明の化合物は、あらゆる態様の結晶形のものおよび化合物を構成する原子の一部または全部を同位体に変換した化合物(例えば、水素を重水素化した化合物や、12Cを14Cに変換した化合物)も包含する。 The compound of the present invention includes the compound represented by the formula (1) or a prodrug thereof, or a pharmaceutically acceptable salt thereof, in addition to the above isomer. In addition, the compound of the present invention or a pharmaceutically acceptable salt thereof may exist in the form of an adduct with water or various solvents, and these adducts are also included. Further, the compound of the present invention includes all crystal forms and compounds obtained by converting some or all of the atoms constituting the compound into isotopes (for example, deuterated compounds of 12 C and 14 C of 14 C The compound converted to).
本明細書における「式(1)の化合物のプロドラッグ」なる用語は、生体内における生理条件下で酵素や胃酸等による反応により式(1)の化合物に変換される化合物、すなわち酵素的に酸化、還元、加水分解等を起こして式(1)の化合物に変化する化合物、胃酸等により加水分解を起こして化合物(1)の化合物に変化する化合物を意味する。 In the present specification, the term “prodrug of the compound of formula (1)” is a compound that is converted into a compound of formula (1) by a reaction with an enzyme, gastric acid or the like under physiological conditions in vivo, that is, enzymatically oxidized. It means a compound that undergoes reduction, hydrolysis, etc. to change to a compound of formula (1), or a compound that undergoes hydrolysis by gastric acid or the like to change to a compound of compound (1).
「薬学的に許容される塩」としては、例えば、塩基付加塩または酸付加塩が挙げられる。塩基付加塩としては、例えば、カリウム塩、ナトリウム塩等のアルカリ金属塩、カルシウム塩、マグネシウム塩等のアルカリ土類金属塩、アンモニウム塩、N−メチルグルカミン(メグルミン)等の水溶性アミン付加塩、または有機アミンの低級アルカノールアンモニウム塩が挙げられる。酸付加塩としては、例えば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硝酸塩、硫酸塩、硫酸水素塩、リン酸塩、酢酸塩、乳酸塩、クエン酸塩、酒石酸塩、酒石酸水素塩、コハク酸塩、マレイン酸塩、フマル酸塩、グルコン酸塩、サッカラート、安息香酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、パラトルエンスルホン酸塩、パモエート[1,1’−メチレン−ビス−(2−ヒドロキシ−3−ナフトエート)]等の塩が挙げられる。 Examples of the “pharmaceutically acceptable salt” include base addition salts and acid addition salts. Examples of base addition salts include alkali metal salts such as potassium salts and sodium salts, alkaline earth metal salts such as calcium salts and magnesium salts, ammonium salts, and water-soluble amine addition salts such as N-methylglucamine (meglumine). Or lower alkanol ammonium salts of organic amines. Examples of acid addition salts include hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate, hydrogensulfate, phosphate, acetate, lactate, citrate, tartrate, tartaric acid Hydrogen salt, succinate, maleate, fumarate, gluconate, saccharate, benzoate, methanesulfonate, ethanesulfonate, benzenesulfonate, paratoluenesulfonate, pamoate [1, 1'-methylene-bis- (2-hydroxy-3-naphthoate)] and the like.
本発明の化合物の塩は以下の方法などにより取得することができる。例えば、本発明の化合物が塩の形で得られる場合には、そのまま精製すればよい。また、本発明の化合物が遊離の形で得られる場合には、適当な有機溶媒に溶解または懸濁させ、上記の塩を形成しうる酸または塩基を加えて、通常の方法により塩を形成させればよい。 The salt of the compound of the present invention can be obtained by the following method. For example, when the compound of the present invention is obtained in the form of a salt, it may be purified as it is. When the compound of the present invention is obtained in a free form, it is dissolved or suspended in a suitable organic solvent, and an acid or base capable of forming the above salt is added to form a salt by a usual method. Just do it.
上記で示す製造方法で得られた式(1)の化合物は、抽出、カラムクロマトグラフィー、再結晶、再沈殿のような常法に従って単離・精製される。抽出溶媒としては、例えばジエチルエーテル、酢酸エチル、クロロホルム、ジクロロメタン、トルエン等が挙げられる。カラムクロマトグラフィーによる精製は、例えば、酸性、塩基性もしくは各種化学処理をしたシリカゲルまたはアルミナ等を用いて行われる。溶出溶媒としては、例えば、ヘキサン/酢酸エチル、ヘキサン/クロロホルム、酢酸エチル/メタノール、クロロホルム/メタノール、アセトニトリル/水、メタノール/水等が用いられる。 The compound of formula (1) obtained by the production method shown above is isolated and purified according to conventional methods such as extraction, column chromatography, recrystallization and reprecipitation. Examples of the extraction solvent include diethyl ether, ethyl acetate, chloroform, dichloromethane, toluene and the like. Purification by column chromatography is performed using, for example, silica gel or alumina that has been subjected to acidic, basic, or various chemical treatments. As the elution solvent, for example, hexane / ethyl acetate, hexane / chloroform, ethyl acetate / methanol, chloroform / methanol, acetonitrile / water, methanol / water and the like are used.
本発明のベンズオキサゾロン環を有する新規化合物またはその医薬として許容される塩は、Nav1.7およびNav1.8の一方または両方を阻害する特性を有し、神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症等に対する治療剤または予防剤として使用できる。ここでいう神経障害性疼痛としては、例えば糖尿病性ニューロパチー、帯状疱疹後神経痛、化学療法による神経障害、癌性疼痛、ヒト免疫不全症候群ウイルス感染性感覚神経障害、三叉神経痛、複合性局所疼痛症候群、反射性交感神経性ジストロフィー、腰部術後神経痛、幻肢痛、脊髄損傷後疼痛、遷延性術後疼痛、炎症性の脱髄性多発神経根障害、アルコール性神経障害、絞扼性末梢神経障害、医原性神経障害、突発性感覚神経障害、栄養障害による神経障害、放射線照射後神経障害、神経根障害、有毒性末梢神経障害、外傷性末梢性神経障害、腕神経叢引き抜き損傷、舌因神経痛、自己免疫性神経障害、慢性馬尾障害などが挙げられる。侵害受容性疼痛ないしは炎症性疼痛としては、腰痛、腹痛、慢性関節リウマチ、変形性関節症による疼痛、筋肉痛、急性術後痛、骨折痛、熱傷性疼痛などが挙げられる。また、本発明の化合物またはその医薬として許容される塩は、排尿障害に対する治療剤または予防剤としても使用できる。ここでいう排尿障害としては、頻尿、前立腺肥大による膀胱痛などが挙げられる。さらに、多発性硬化症における小脳の異常神経発火を抑える運動失調に対する治療剤または予防剤としても使用できる。非神経組織や中枢神経系由来の副作用がない薬剤としては、Nav1.7またはNav1.8の選択的阻害活性を有する化合物がより好ましい。 The novel compound having a benzoxazolone ring of the present invention or a pharmaceutically acceptable salt thereof has a property of inhibiting one or both of Nav1.7 and Nav1.8, and is neuropathic pain, nociceptive pain, inflammation It can be used as a therapeutic or prophylactic agent for sexual pain, small-fiber neuropathy, limb erythema, paroxysmal acute pain and the like. Examples of neuropathic pain herein include diabetic neuropathy, postherpetic neuralgia, chemotherapy-induced neuropathy, cancer pain, human immunodeficiency syndrome virus infectious sensory neuropathy, trigeminal neuralgia, complex regional pain syndrome, Reflex sympathetic dystrophy, lumbar postoperative neuralgia, phantom limb pain, post-spinal pain, prolonged postoperative pain, inflammatory demyelinating polyradiculopathy, alcoholic neuropathy, strangulated peripheral neuropathy, medicine Neuropathy, idiopathic sensory neuropathy, neuropathy caused by nutritional disorder, post-irradiation neuropathy, radiculopathy, toxic peripheral neuropathy, traumatic peripheral neuropathy, brachial plexus withdrawal injury, lingual neuropathy, Examples include autoimmune neuropathy and chronic cauda equina disorder. Examples of nociceptive pain or inflammatory pain include low back pain, abdominal pain, rheumatoid arthritis, pain due to osteoarthritis, muscle pain, acute postoperative pain, fracture pain, burn pain, and the like. The compound of the present invention or a pharmaceutically acceptable salt thereof can also be used as a therapeutic or prophylactic agent for dysuria. Examples of dysuria include frequent urination and bladder pain due to enlarged prostate. Furthermore, it can also be used as a therapeutic or prophylactic agent for ataxia that suppresses cerebellar abnormal nerve firing in multiple sclerosis. As a drug having no side effect derived from non-neural tissue or central nervous system, a compound having a selective inhibitory activity of Nav1.7 or Nav1.8 is more preferable.
本発明の化合物の投与経路としては、経口投与、非経口投与又は直腸内投与のいずれでもよく、その一日投与量は、化合物の種類、投与方法、患者の症状・年齢等により異なる。例えば、経口投与の場合は、通常、ヒト又は哺乳動物1kg体重当たり約0.01〜1000mg、更に好ましくは約0.1〜500mgを1〜数回に分けて投与することができる。静注等の非経口投与の場合は、通常、例えば、ヒト又は哺乳動物1kg体重当たり約0.01mg〜300mg、更に好ましくは約1mg〜100mgを投与することができる。 The administration route of the compound of the present invention may be any of oral administration, parenteral administration and rectal administration, and the daily dose varies depending on the type of compound, administration method, patient symptom / age and the like. For example, in the case of oral administration, usually about 0.01 to 1000 mg, more preferably about 0.1 to 500 mg per kg body weight of a human or mammal can be administered in 1 to several divided doses. In the case of parenteral administration such as intravenous injection, usually, for example, about 0.01 mg to 300 mg, more preferably about 1 mg to 100 mg per kg body weight of a human or mammal can be administered.
本発明化合物は、経口投与又は非経口投与により、直接又は適当な剤形を用いて製剤にし、投与することができる。剤形は、例えば、錠剤、カプセル剤、散剤、顆粒剤、液剤、懸濁剤、注射剤、貼付剤、ハップ剤等が挙げられるがこれに限らない。製剤は、薬学的に許容される添加剤を用いて、公知の方法で製造される。添加剤は、目的に応じて、賦形剤、崩壊剤、結合剤、流動化剤、滑沢剤、コーティング剤、溶解剤、溶解補助剤、増粘剤、分散剤、安定化剤、甘味剤、香料等を用いることができる。具体的には、例えば、乳糖、マンニトール、結晶セルロース、低置換度ヒドロキシプロピルセルロース、トウモロコシデンプン、部分α化デンプン、カルメロースカルシウム、クロスカルメロースナトリウム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ステアリン酸マグネシウム、フマル酸ステアリルナトリウム、ポリエチレングリコール、プロピレングリコール、酸化チタン、タルク等が挙げられる。 The compound of the present invention can be formulated and administered by oral administration or parenteral administration, directly or using a suitable dosage form. Examples of the dosage form include, but are not limited to, tablets, capsules, powders, granules, solutions, suspensions, injections, patches, and haptics. The preparation is produced by a known method using a pharmaceutically acceptable additive. Additives are excipients, disintegrants, binders, fluidizers, lubricants, coating agents, solubilizers, solubilizers, thickeners, dispersants, stabilizers, sweeteners depending on the purpose. Perfumes and the like can be used. Specifically, for example, lactose, mannitol, crystalline cellulose, low-substituted hydroxypropyl cellulose, corn starch, partially pregelatinized starch, carmellose calcium, croscarmellose sodium, hydroxypropyl cellulose, hydroxypropyl methylcellulose, polyvinyl alcohol, stearin Examples include magnesium acid, sodium stearyl fumarate, polyethylene glycol, propylene glycol, titanium oxide, and talc.
本発明の化合物およびその薬学的に許容される塩は、その作用の増強を目的として、例えば、セレコキシブ、ボルタレン、イブプロフェン、ロキソプロフェン、アセトアミノフェン、ジクロフェナク、デキサメサゾンなどの非ステロイド系抗炎症薬や、トラマドール、モルヒネ、オキシコドンなどのオピオイド系鎮痛薬とも組み合わせて用いることができる。また、プレガバリン、カルバマゼピンなどの抗てんかん薬、エパルレスタットなどのアルドース還元酵素阻害剤、リマプロスト アルファデクスなどのプロスタグランジン誘導体製剤、アミトリプチリン、デュロキセチンなどの抗うつ薬、抗痙攣薬、抗不安薬、ドーパミン受容体作動薬、パーキンソン病治療薬、ホルモン製剤、偏頭痛治療薬、アドレナリンβ受容体拮抗薬、認知症治療薬、気分障害治療薬などの薬剤とも組み合わせて用いることができる。本発明の化合物およびその薬学的に許容される塩と組み合わせて用いる薬剤として好ましくは、プレガバリン、カルバマゼピンなどの抗てんかん薬、アミトリプチリン、デュロキセチンなどの抗うつ薬、モルヒネ、オキシコドン、トラマドールなどの麻薬性鎮痛薬、アセトアミノフェン、ジクロフェナク、デキサメサゾンなどの抗炎症薬、エパルレスタットなどのアルドース還元酵素阻害剤、リマプロスト アルファデクスなどのプロスタグランジン誘導体が挙げられる。また、その副作用抑制を目的として、制吐剤、睡眠導入剤などの薬剤と組み合わせて用いることができる。本発明の化合物及び併用薬剤の投与時期は限定されず、これらを投与対象に対し、同時に投与してもよいし、適当な間隔をおいて投与してもよい。また、本発明の化合物と併用薬剤の合剤としても良い。併用薬剤の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、本発明化合物と併用薬剤との配合比は、投与対象、投与ルート、対象疾患、症状、組み合わせなどにより適宜選択することができる。例えば投与対象がヒトである場合、本発明の化合物1重量部に対し、併用薬剤を0.01〜1000重量部用いればよい。 The compounds of the present invention and pharmaceutically acceptable salts thereof, for the purpose of enhancing their action, for example, non-steroidal anti-inflammatory drugs such as celecoxib, voltaren, ibuprofen, loxoprofen, acetaminophen, diclofenac, dexamethasone, It can also be used in combination with opioid analgesics such as tramadol, morphine and oxycodone. In addition, antiepileptic drugs such as pregabalin and carbamazepine, aldose reductase inhibitors such as epalrestat, prostaglandin derivative preparations such as limaprost alphadex, antidepressants such as amitriptyline and duloxetine, anticonvulsants, anxiolytics and dopamine receptors It can also be used in combination with drugs such as body agonists, Parkinson's disease drugs, hormone preparations, migraine drugs, adrenergic β receptor antagonists, dementia drugs, and mood disorder drugs. Preferably, the drug used in combination with the compound of the present invention and a pharmaceutically acceptable salt thereof is preferably an antiepileptic drug such as pregabalin or carbamazepine, an antidepressant drug such as amitriptyline or duloxetine, or a narcotic analgesic such as morphine, oxycodone or tramadol. Drugs, anti-inflammatory drugs such as acetaminophen, diclofenac and dexamethasone, aldose reductase inhibitors such as epalrestat, and prostaglandin derivatives such as limaprost alphadex. Moreover, it can be used in combination with drugs such as antiemetics and sleep inducers for the purpose of suppressing the side effects. The administration timing of the compound of the present invention and the concomitant drug is not limited, and these may be administered simultaneously to the administration subject or may be administered at an appropriate interval. Moreover, it is good also as a mixture of the compound of this invention and a concomitant drug. The dose of the concomitant drug can be appropriately selected based on the clinically used dose. The compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination and the like. For example, when the administration subject is a human, the concomitant drug may be used in an amount of 0.01 to 1000 parts by weight per 1 part by weight of the compound of the present invention.
以下に参考例、実施例および試験例を挙げて本発明を更に具体的に説明するが、本願発明の技術的範囲はこれら実施例等に限定されるものではない。参考例および実施例におけるシリカゲルクロマトグラフィーは、山善株式会社製のシリカゲルカラムまたはアミノシリカゲルカラムを用いた。化合物の同定はプロトン核磁気共鳴吸収スペクトル(1H−NMR)、高速液体クロマト質量分析計;LCMS等を用いて行った。1H−NMRは、JNM−LA300(日本電子)またはJNM−AL400(日本電子)を用いて測定した。 The present invention will be described more specifically with reference to the following reference examples, examples and test examples. However, the technical scope of the present invention is not limited to these examples. For silica gel chromatography in Reference Examples and Examples, a silica gel column or amino silica gel column manufactured by Yamazen Co., Ltd. was used. The compound was identified using proton nuclear magnetic resonance absorption spectrum ( 1 H-NMR), high performance liquid chromatography / mass spectrometer; LCMS and the like. 1 H-NMR was measured using JNM-LA300 (JEOL) or JNM-AL400 (JEOL).
高速液体クロマト質量分析計;LCMSの測定条件は、以下の通りであり、観察された質量分析の値[MS(m/z)]をM+Hで示す。
MS detector:ACQITY SQD
HPLC:ACQITY UPLC
カラム:ACQITY BEH C18 1.7 μm 2.1×50 mm
流速:0.75ml/min
測定波長:254nm
移動層:A液;0.05%ギ酸水溶液
B液;アセトニトリル
タイムプログラム:
ステップ 時間(分)
1 0.0−1.3 A液:B液=90:10→1:99
2 1.3−1.5 A液:B液=1:99
3 1.5−2.0 A液:B液=90:10
High-performance liquid chromatograph / mass spectrometer; LCMS measurement conditions are as follows, and the observed mass spectrometry value [MS (m / z)] is indicated by M + H.
MS detector: ACQITY SQD
HPLC: ACQITY UPLC
Column: ACQITY BEH C18 1.7 μm 2.1 × 50 mm
Flow rate: 0.75 ml / min
Measurement wavelength: 254 nm
Moving bed: Liquid A; 0.05% formic acid aqueous solution Liquid B; Acetonitrile time program:
Step time (minutes)
1 0.0-1.3 Liquid A: Liquid B = 90: 10 → 1: 99
2 1.3-1.5 Liquid A: Liquid B = 1: 99
3 1.5-2.0 Liquid A: Liquid B = 90: 10
原料化合物、反応試薬および溶媒は、特に断りのない限り、市販のものを用いるか、または公知の方法に準じて製造したものを使用した。 As the raw material compound, the reaction reagent and the solvent, commercially available ones or those produced according to known methods were used unless otherwise specified.
製造法、参考例および実施例において、本明細書の記載を簡略化するために次に示すような略号を用いることもある。Me:メチル、Et:エチル、Pr:プロピル、Bu:ブチル、Ms:メタンスルホニル、n−:ノルマル、i−:イソ、sec−:セカンダリー、t−またはtert−:ターシャリー、THF:テトラヒドロフラン、DMF:N,N−ジメチルホルムアミド、DBN:1,5−ジアザビシクロ[4.3.0]ノナ−5−エン、DABCO:1,4−ジアザビシクロ[2.2.2]オクタン、DBU:1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン、NMM:N−メチルモルホリン、TBS:tert−ブチルジメチルシリル、J:結合定数、s:一重線、d:二重線、t:三重線、q:四重線、dd:二重の二重線、td:三重の二重線、tt:三重の三重線、m:多重線、br:幅広い。 In manufacturing methods, reference examples, and examples, the following abbreviations may be used to simplify the description of the present specification. Me: methyl, Et: ethyl, Pr: propyl, Bu: butyl, Ms: methanesulfonyl, n-: normal, i-: iso, sec-: secondary, t- or tert-: tertiary, THF: tetrahydrofuran, DMF : N, N-dimethylformamide, DBN: 1,5-diazabicyclo [4.3.0] non-5-ene, DABCO: 1,4-diazabicyclo [2.2.2] octane, DBU: 1,8- Diazabicyclo [5.4.0] undec-7-ene, NMM: N-methylmorpholine, TBS: tert-butyldimethylsilyl, J: coupling constant, s: singlet, d: doublet, t: triplet, q: quadruple line, dd: double double line, td: triple double line, tt: triple triple line, m: multiple line, br: wide.
参考例1:
5−クロロ−6−メチルベンゾ[d]オキサゾール−2(3H)−オン
Reference example 1:
5-Chloro-6-methylbenzo [d] oxazol-2 (3H) -one
工程(i):
化合物1(0.500g)、還元鉄(0.810g)、酢酸(0.481g)、エタノール(10mL)および水(3.0mL)の混合物を80℃で1時間攪拌した。反応混合液をセライトろ過し、ろ液を飽和炭酸水素ナトリウム水溶液および水で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し化合物2(0.270g)を得た。
Step (i):
A mixture of Compound 1 (0.500 g), reduced iron (0.810 g), acetic acid (0.481 g), ethanol (10 mL) and water (3.0 mL) was stirred at 80 ° C. for 1 hour. The reaction mixture was filtered through celite, and the filtrate was washed with saturated aqueous sodium hydrogen carbonate solution and water. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain compound 2 (0.270 g).
工程(ii):
化合物2(0.050g)、1,1’−カルボニルジイミダゾール(0.077g)、ジイソプロピルエチルアミン(0.166mL)およびTHF(1.5mL)の混合物を室温で1時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=3/1)で精製し、表題化合物3(0.041g)を得た。
Step (ii):
A mixture of Compound 2 (0.050 g), 1,1′-carbonyldiimidazole (0.077 g), diisopropylethylamine (0.166 mL) and THF (1.5 mL) was stirred at room temperature for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 3/1) to give the title compound 3 (0.041 g).
1H-NMR (DMSO-d6) δ: 2.30 (3H, s), 7.12 (1H, s), 7.31 (1H, s), 11.68 (1H, s). 1 H-NMR (DMSO-d 6 ) δ: 2.30 (3H, s), 7.12 (1H, s), 7.31 (1H, s), 11.68 (1H, s).
参考例2:
5−ブロモ−4−メチルベンゾ[d]オキサゾール−2(3H)−オン
Reference example 2:
5-Bromo-4-methylbenzo [d] oxazol-2 (3H) -one
工程(i):
化合物4(1.00g)、N−ブロモスクシンイミド(1.74g)、酢酸(0.784g)およびDMF(33mL)の混合物を室温で1時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=6/1)で精製し、化合物5(0.530g)を得た。
Step (i):
A mixture of compound 4 (1.00 g), N-bromosuccinimide (1.74 g), acetic acid (0.784 g) and DMF (33 mL) was stirred at room temperature for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 6/1) to give compound 5 (0.530 g).
工程(ii):
化合物5(0.510g)を用いて、参考例1の工程(i)と同様の方法に従い、化合物6(0.301g)を得た。
Step (ii):
Using compound 5 (0.510 g), compound 6 (0.301 g) was obtained in the same manner as in step (i) of Reference Example 1.
工程(iii):
化合物6(0.050g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物7(0.040g)を得た。
Step (iii):
The title compound 7 (0.040 g) was obtained using the compound 6 (0.050 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (DMSO-d6) δ: 2.29 (3H, s), 7.08-7.10 (1H, m), 7.28-7.31 (1H, m), 11.94 (1H, s). 1 H-NMR (DMSO-d 6 ) δ: 2.29 (3H, s), 7.08-7.10 (1H, m), 7.28-7.31 (1H, m), 11.94 (1H, s).
参考例3:
5−ブロモ−7−メチルベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 3:
5-Bromo-7-methylbenzo [d] oxazol-2 (3H) -one
工程(i):
化合物8(1.00g)および酢酸(0.784g)の混合物に、0℃で、硝酸(0.506g)を加え、同温で1時間攪拌した。反応混合液に氷を加え、析出した固体をろ取し、化合物9(1.01g)を得た。
Step (i):
To a mixture of compound 8 (1.00 g) and acetic acid (0.784 g), nitric acid (0.506 g) was added at 0 ° C. and stirred at the same temperature for 1 hour. Ice was added to the reaction mixture, and the precipitated solid was collected by filtration to obtain Compound 9 (1.01 g).
工程(ii):
化合物9(0.99g)を用いて、参考例1の工程(i)と同様の方法に従い、化合物10(0.700g)を得た。
Step (ii):
Using Compound 9 (0.99 g), Compound 10 (0.700 g) was obtained in the same manner as in Step (i) of Reference Example 1.
工程(iii):
化合物10(0.700g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物11(0.510g)を得た。
Step (iii):
The title compound 11 (0.510 g) was obtained using the compound 10 (0.700 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (DMSO-d6) δ: 2.27 (3H, s), 7.07 (1H, s), 7.13 (1H, s), 11.74 (1H, brs). 1 H-NMR (DMSO-d 6 ) δ: 2.27 (3H, s), 7.07 (1H, s), 7.13 (1H, s), 11.74 (1H, brs).
参考例4:
5−ブロモ−4−フルオロベンゾ[d]オキサゾール−2(3H)−オン
Reference example 4:
5-Bromo-4-fluorobenzo [d] oxazol-2 (3H) -one
工程(i):
化合物12(2.60g)、ベンジルブロミド(8.57g)、炭酸カリウム(6.92g)およびDMF(17mL)の混合物を室温で6時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=9/1)で精製し、化合物13(4.70g)を得た。
Step (i):
A mixture of compound 12 (2.60 g), benzyl bromide (8.57 g), potassium carbonate (6.92 g) and DMF (17 mL) was stirred at room temperature for 6 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 9/1) to obtain Compound 13 (4.70 g).
工程(ii):
化合物13(4.70g)、4mol/L水酸化ナトリウム水溶液(4.20mL)およびメタノール(14mL)の混合物を50℃で14時間攪拌した。反応混合液を減圧濃縮後、水および4mol/L塩酸を加えた後、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1/1)で精製し、化合物14(2.98g)を得た。
Step (ii):
A mixture of Compound 13 (4.70 g), 4 mol / L aqueous sodium hydroxide solution (4.20 mL) and methanol (14 mL) was stirred at 50 ° C. for 14 hours. The reaction mixture was concentrated under reduced pressure, water and 4 mol / L hydrochloric acid were added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 1/1) to obtain Compound 14 (2.98 g).
工程(iii):
化合物14(2.96g)、ジフェニルホスホリルアジド(5.47g)、トリエチルアミン(3.89mL)、tert−ブチルアルコール(10mL)およびトルエン(10mL)の混合物を90℃で15時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=3/1)で精製し、化合物15(2.80g)を得た。
Step (iii):
A mixture of compound 14 (2.96 g), diphenylphosphoryl azide (5.47 g), triethylamine (3.89 mL), tert-butyl alcohol (10 mL) and toluene (10 mL) was stirred at 90 ° C. for 15 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 3/1) to obtain Compound 15 (2.80 g).
工程(iv):
化合物15(1.20g)を用いて、参考例2の工程(i)と同様の方法に従い、化合物16(1.12g)を得た。
Step (iv):
Using compound 15 (1.20 g), compound 16 (1.12 g) was obtained in the same manner as in step (i) of Reference Example 2.
工程(v):
化合物16(1.10g)、12mol/L塩酸(3.00mL)および1,4−ジオキサン(0.5mL)の混合物を80℃で5時間攪拌した。反応混合液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、化合物17(0.510g)を得た。
Step (v):
A mixture of compound 16 (1.10 g), 12 mol / L hydrochloric acid (3.00 mL) and 1,4-dioxane (0.5 mL) was stirred at 80 ° C. for 5 hours. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain Compound 17 (0.510 g).
工程(vi):
化合物17(0.510g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物18(0.420g)を得た。
Step (vi):
The title compound 18 (0.420 g) was obtained using the compound 17 (0.510 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (DMSO-d6) δ: 7.14-7.16 (1H, m), 7.36-7.39 (1H, m), 12.49 (1H, s). 1 H-NMR (DMSO-d 6 ) δ: 7.14-7.16 (1H, m), 7.36-7.39 (1H, m), 12.49 (1H, s).
参考例5:
5−ブロモ−6−フルオロベンゾ[d]オキサゾール−2(3H)−オン
Reference example 5:
5-Bromo-6-fluorobenzo [d] oxazol-2 (3H) -one
工程(i):
化合物19(0.200g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物20(0.168g)を得た。
Step (i):
The title compound 20 (0.168 g) was obtained using the compound 19 (0.200 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (DMSO-d6) δ: 7.36-7.38 (1H, m), 7.54-7.57 (1H, m), 11.88 (1H, brs). 1 H-NMR (DMSO-d 6 ) δ: 7.36-7.38 (1H, m), 7.54-7.57 (1H, m), 11.88 (1H, brs).
参考例6:
5−(トリフルオロメチル)ベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 6:
5- (Trifluoromethyl) benzo [d] oxazol-2 (3H) -one
工程(i):
化合物21(0.500g)、ギ酸アンモニウム(0.761g)、10% Pd/C(0.050g)およびメタノール(12mL)の混合物を50℃で1時間攪拌した。反応混合液をセライトろ過し、減圧濃縮後、残渣に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、化合物22(0.410g)を得た。
Step (i):
A mixture of compound 21 (0.500 g), ammonium formate (0.761 g), 10% Pd / C (0.050 g) and methanol (12 mL) was stirred at 50 ° C. for 1 hour. The reaction mixture was filtered through Celite, concentrated under reduced pressure, saturated aqueous sodium hydrogen carbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain Compound 22 (0.410 g).
工程(ii):
化合物22(0.410g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物23(0.455g)を得た。
Step (ii):
The title compound 23 (0.455 g) was obtained using the compound 22 (0.410 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (DMSO-d6) δ: 7.36 (1H, s), 7.45-7.49 (2H, m), 12.01 (1H, brs). 1 H-NMR (DMSO-d 6 ) δ: 7.36 (1H, s), 7.45-7.49 (2H, m), 12.01 (1H, brs).
参考例7:
3−{[2−(メチルスルホニル)エチル]アミノ}ブタン−1−オール
Reference example 7:
3-{[2- (methylsulfonyl) ethyl] amino} butan-1-ol
工程(i):
化合物24(0.178g)、2−(メタンスルホニル)エチルアミン(0.148g)、トリアセトキシ水素化ホウ素ナトリウム(0.636g)、酢酸(0.090g)およびクロロホルム(5.0mL)の混合物を室温で24時間攪拌した。反応混合液に、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=19/1)で精製し、化合物25(0.250g)を得た。
Step (i):
A mixture of compound 24 (0.178 g), 2- (methanesulfonyl) ethylamine (0.148 g), sodium triacetoxyborohydride (0.636 g), acetic acid (0.090 g) and chloroform (5.0 mL) was stirred at room temperature. For 24 hours. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 19/1) to obtain Compound 25 (0.250 g).
工程(ii):
化合物25(0.100g)を用いて、参考例6の工程(i)と同様の方法に従い、表題化合物26(0.020g)を得た。
Step (ii):
The title compound 26 (0.020 g) was obtained using the compound 25 (0.100 g) according to the same method as in step (i) of Reference Example 6.
1H-NMR (DMSO-d6) δ: 0.96 (3H, d, J = 6.2 Hz), 1.33-1.41 (1H, m), 1.49-1.53 (1H, m), 2.66-2.68 (1H, m), 2.88-2.96 (5H, m), 3.15-3.18 (2H, m), 3.44-3.46 (2H, m), 4.42 (1H, s). 1 H-NMR (DMSO-d 6 ) δ: 0.96 (3H, d, J = 6.2 Hz), 1.33-1.41 (1H, m), 1.49-1.53 (1H, m), 2.66-2.68 (1H, m) , 2.88-2.96 (5H, m), 3.15-3.18 (2H, m), 3.44-3.46 (2H, m), 4.42 (1H, s).
参考例8:
tert−ブチル (3−ヒドロキシ−2−メチルプロピル)カルバメート
Reference Example 8:
tert-Butyl (3-hydroxy-2-methylpropyl) carbamate
工程(i):
化合物27(0.700g)、クロロギ酸イソブチル(0.564g)、トリエチルアミン(0.725mL)およびTHF(17mL)の混合物を0℃で1時間攪拌した。反応混合液をセライトろ過した後、減圧濃縮した。残渣のメタノール(17mL)溶液に、0℃で水素化ホウ素ナトリウムを加え、室温で1時間攪拌した。反応混合液を減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=2/1)で精製し、表題化合物28(0.380g)を得た。
Step (i):
A mixture of compound 27 (0.700 g), isobutyl chloroformate (0.564 g), triethylamine (0.725 mL) and THF (17 mL) was stirred at 0 ° C. for 1 hour. The reaction mixture was filtered through celite and then concentrated under reduced pressure. To a solution of the residue in methanol (17 mL) was added sodium borohydride at 0 ° C., and the mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 2/1) to give the title compound 28 (0.380 g).
1H-NMR (DMSO-d6) δ: 0.77 (3H, d, J = 6.8 Hz), 1.36 (9H, s), 1.58-1.60 (1H, m), 2.72-2.74 (1H, m), 2.88-2.92 (1H, m), 3.19-3.25 (2H, m), 4.37-4.38 (1H, m), 6.73 (1H, br s). 1 H-NMR (DMSO-d 6 ) δ: 0.77 (3H, d, J = 6.8 Hz), 1.36 (9H, s), 1.58-1.60 (1H, m), 2.72-2.74 (1H, m), 2.88 -2.92 (1H, m), 3.19-3.25 (2H, m), 4.37-4.38 (1H, m), 6.73 (1H, br s).
参考例9:
tert−ブチル (3−ヒドロキシ−2,2−ジメチルプロピル)カルバメート
Reference Example 9:
tert-Butyl (3-hydroxy-2,2-dimethylpropyl) carbamate
工程(i):
化合物29(0.345g)、二炭酸tert−ブチル(0.730g)およびTHF(50ml)の混合物を室温で2時間攪拌した。反応液を減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=95/5)で精製し、表題化合物30(0.658g)を得た。
Step (i):
A mixture of compound 29 (0.345 g), tert-butyl dicarbonate (0.730 g) and THF (50 ml) was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 95/5) to give the title compound 30 (0.658 g).
1H-NMR (CDCl3) δ: 0.83 (6H, s), 1.43 (9H, s), 2.94 (2H, d, J = 6.8 Hz), 3.17 (2H, d, J = 7.3 Hz), 3.64 (1H, t, J = 7.3 Hz), 4.81 (1H, br s). 1 H-NMR (CDCl 3 ) δ: 0.83 (6H, s), 1.43 (9H, s), 2.94 (2H, d, J = 6.8 Hz), 3.17 (2H, d, J = 7.3 Hz), 3.64 ( 1H, t, J = 7.3 Hz), 4.81 (1H, br s).
参考例10:
2−メチル−2−(メチルスルホニル)プロパン−1−アミン
Reference Example 10:
2-Methyl-2- (methylsulfonyl) propan-1-amine
工程(i):
化合物31(0.953g)、水素化ナトリウム(0.663g)、ヨウ化メチル(2.04g)およびTHF(40mL)の混合物を0℃で30分攪拌後、室温で1時間攪拌した。反応混合液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=6/1)で精製し、化合物32(0.730g)を得た。
Step (i):
A mixture of compound 31 (0.953 g), sodium hydride (0.663 g), methyl iodide (2.04 g) and THF (40 mL) was stirred at 0 ° C. for 30 minutes and then at room temperature for 1 hour. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 6/1) to obtain Compound 32 (0.730 g).
工程(ii):
化合物32(0.700g)およびTHF(2.0mL)の混合物を45℃で、0.95mol/L ボラン・THF錯体(THF溶液、10mL)に対して滴下し、室温で15時間攪拌した。反応混合液を0℃で、メタノール(20mL)に滴下し、減圧濃縮した。得られた残渣のメタノール(20mL)溶液に、4mol/L 塩化水素−1,4−ジオキサン溶液(40mL)を加え、80℃で1時間攪拌した。混合液に0℃で、クロロホルム(30mL)を加え、固体をろ取した。固体をメタノールに(20mL)溶解させ、ナトリウムメトキシドをpHが9になるまで加えた。反応液にクロロホルムを加え、セライトろ過し、ろ液を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=9/1)で精製し、表題化合物33(0.480g)を得た。
Step (ii):
A mixture of Compound 32 (0.700 g) and THF (2.0 mL) was added dropwise at 45 ° C. to 0.95 mol / L borane / THF complex (THF solution, 10 mL), and the mixture was stirred at room temperature for 15 hours. The reaction mixture was added dropwise to methanol (20 mL) at 0 ° C. and concentrated under reduced pressure. A 4 mol / L hydrogen chloride-1,4-dioxane solution (40 mL) was added to a methanol (20 mL) solution of the obtained residue, and the mixture was stirred at 80 ° C. for 1 hour. Chloroform (30 mL) was added to the mixture at 0 ° C., and the solid was collected by filtration. The solid was dissolved in methanol (20 mL) and sodium methoxide was added until the pH was 9. Chloroform was added to the reaction solution, filtered through celite, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 9/1) to give the title compound 33 (0.480 g).
1H-NMR (DMSO-d6) δ: 1.21 (6H, s), 1.61 (2H, m), 2.78 (2H, m), 2.91 (3H, s). 1 H-NMR (DMSO-d 6 ) δ: 1.21 (6H, s), 1.61 (2H, m), 2.78 (2H, m), 2.91 (3H, s).
参考例11:
4−ヨードベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 11:
4-Iodobenzo [d] oxazol-2 (3H) -one
工程(i):
化合物34(2.00g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物35(1.00g)を得た。
Step (i):
The title compound 35 (1.00 g) was obtained using the compound 34 (2.00 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (CDCl3) δ: 6.91-6.94 (1H, m), 7.16-7.19 (1H, m), 7.45-7.47 (1H, m), 8.70 (1H, s). 1 H-NMR (CDCl 3 ) δ: 6.91-6.94 (1H, m), 7.16-7.19 (1H, m), 7.45-7.47 (1H, m), 8.70 (1H, s).
参考例12:
7−ブロモベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 12:
7-Bromobenzo [d] oxazol-2 (3H) -one
工程(i):
化合物36(1.59g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物37(1.02g)を得た。
Step (i):
The title compound 37 (1.02 g) was obtained using the compound 36 (1.59 g) according to the same method as in step (ii) of Reference Example 1.
LC/MS: m/z = 213, 215 (M+H)+ LC / MS: m / z = 213, 215 (M + H) +
参考例13:
6−(4−フルオロフェノキシ)ベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 13:
6- (4-Fluorophenoxy) benzo [d] oxazol-2 (3H) -one
工程(i):
化合物38(15.9g)を用いて、参考例4の工程(i)と同様の方法に従い、化合物39(24.7g)を得た。
Step (i):
Compound 39 (24.7 g) was obtained using compound 38 (15.9 g) according to the same method as in step (i) of Reference Example 4.
工程(ii):
化合物39(24.7g)、4−フルオロフェノール(13.4g)、炭酸セシウム(49.0g)およびDMF(40mL)の混合物を80℃で4時間攪拌した。反応混合液に水を加え、固体をろ取し、化合物40(33.9g)を得た。
Step (ii):
A mixture of compound 39 (24.7 g), 4-fluorophenol (13.4 g), cesium carbonate (49.0 g) and DMF (40 mL) was stirred at 80 ° C. for 4 hours. Water was added to the reaction mixture, and the solid was collected by filtration to give compound 40 (33.9 g).
工程(iii):
化合物40(5.40g)、10% Pd/C(0.540g)、メタノール(10mL)および酢酸エチル(20mL)の混合物を水素雰囲気下、50℃で12時間攪拌した。反応混合液をセライトろ過度、減圧濃縮し、化合物41(3.49g)を得た。
Step (iii):
A mixture of Compound 40 (5.40 g), 10% Pd / C (0.540 g), methanol (10 mL) and ethyl acetate (20 mL) was stirred at 50 ° C. for 12 hours in a hydrogen atmosphere. The reaction mixture was concentrated through Celite filtration and reduced pressure to obtain Compound 41 (3.49 g).
工程(iv):
化合物41(0.300g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物42(0.300g)を得た。
Step (iv):
The title compound 42 (0.300 g) was obtained using the compound 41 (0.300 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (CDCl3) δ: 6.80-7.07 (7H, m). 1 H-NMR (CDCl 3 ) δ: 6.80-7.07 (7H, m).
参考例14:
5−ブロモ−3−(4,4−ジメトキシブタン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 14:
5-Bromo-3- (4,4-dimethoxybutan-2-yl) benzo [d] oxazol-2 (3H) -one
工程(i):
化合物43(0.661g)、水素化ホウ素ナトリウム(0.378g)およびメタノール(25mL)の混合物を0℃で15分攪拌後、室温にて1時間攪拌した。反応混合液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1/1)で精製し、化合物44(0.620g)を得た。
Step (i):
A mixture of compound 43 (0.661 g), sodium borohydride (0.378 g) and methanol (25 mL) was stirred at 0 ° C. for 15 minutes and then at room temperature for 1 hour. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 1/1) to obtain Compound 44 (0.620 g).
工程(ii):
化合物44(0.620g)、5−ブロモベンズオキサゾロン(0.989g)、ジイソプロピルアゾジカルボキシレート(1.12g)、トリフェニルホスフィン(1.82g)およびTHF(23mL)の混合物を0℃で15分攪拌後、室温にて18時間攪拌した。反応混合液を減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=4/1)で精製し、表題化合物45(0.960g)を得た。
Step (ii):
A mixture of compound 44 (0.620 g), 5-bromobenzoxazolone (0.989 g), diisopropyl azodicarboxylate (1.12 g), triphenylphosphine (1.82 g) and THF (23 mL) at 0 ° C. After stirring for minutes, the mixture was stirred at room temperature for 18 hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 4/1) to give the title compound 45 (0.960 g).
1H-NMR (DMSO-d6) δ: 1.44 (3H, d, J = 7.0 Hz), 1.93-1.96 (1H, m), 2.24-2.28 (1H, m), 3.08 (3H, s), 3.17 (3H, s), 4.29-4.31 (2H, m), 7.29-7.29 (2H, m), 7.63 (1H, s). 1 H-NMR (DMSO-d 6 ) δ: 1.44 (3H, d, J = 7.0 Hz), 1.93-1.96 (1H, m), 2.24-2.28 (1H, m), 3.08 (3H, s), 3.17 (3H, s), 4.29-4.31 (2H, m), 7.29-7.29 (2H, m), 7.63 (1H, s).
参考例15:
3−(メチルスルホニル)プロピル メタンスルホネート
Reference Example 15:
3- (Methylsulfonyl) propyl methanesulfonate
工程(i):
化合物46(2.00g)、メタクロロ過安息香酸(9.40g)および塩化メチレン(60mL)の混合物を室温にて16時間攪拌した。反応混合液をセライトろ過後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=9/1)で精製し、化合物47(0.660g)を得た。
Step (i):
A mixture of compound 46 (2.00 g), metachloroperbenzoic acid (9.40 g) and methylene chloride (60 mL) was stirred at room temperature for 16 hours. The reaction mixture was filtered through celite and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 9/1) to give Compound 47 (0.660 g).
工程(ii):
化合物47(0.100g)、メタンスルホニルクロリド(0.067mL)、トリエチルアミン(0.120mL)および塩化メチレン(2.0mL)の混合物を室温にて1時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を減圧濃縮し、表題化合物48の粗生成物を得た。そのまま全量を次の反応へ用いた。
Step (ii):
A mixture of compound 47 (0.100 g), methanesulfonyl chloride (0.067 mL), triethylamine (0.120 mL) and methylene chloride (2.0 mL) was stirred at room temperature for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was concentrated under reduced pressure to obtain a crude product of the title compound 48. The whole amount was used for the next reaction as it was.
参考例16:
tert−ブチル 3−{[5−ブロモ−2−オキソベンゾ[d]オキサゾール−3(2H)−イル]メチル}アゼチジン−1−カルボキシレート
Reference Example 16:
tert-Butyl 3-{[5-bromo-2-oxobenzo [d] oxazol-3 (2H) -yl] methyl} azetidine-1-carboxylate
工程(i):
化合物49(1.00g)、tert−ブチル 3−ホルミルアゼチジン−1−カルボキシレート(1.00g)、トリアセトキシ水素化ホウ素ナトリウム(2.30g)、テトライソプロポキシチタン(8.00mL)およびTHF(25mL)の混合物を室温で2時間攪拌した。反応混合液に、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1/1)で精製し、化合物50(1.48g)を得た。
Step (i):
Compound 49 (1.00 g), tert-butyl 3-formylazetidine-1-carboxylate (1.00 g), sodium triacetoxyborohydride (2.30 g), tetraisopropoxytitanium (8.00 mL) and THF (25 mL) was stirred at room temperature for 2 hours. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 1/1) to obtain Compound 50 (1.48 g).
工程(ii):
化合物50(0.500g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物51(0.350g)を得た。
Step (ii):
The title compound 51 (0.350 g) was obtained using the compound 50 (0.500 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (CDCl3) δ: 1.42 (9H, s), 2.98-3.12 (1H, m), 3.75-3.78 (2H, m), 4.03-4.06 (4H, m), 7.09-7.13 (2H, m), 7.25-7.28 (1H, m). 1 H-NMR (CDCl 3 ) δ: 1.42 (9H, s), 2.98-3.12 (1H, m), 3.75-3.78 (2H, m), 4.03-4.06 (4H, m), 7.09-7.13 (2H, m), 7.25-7.28 (1H, m).
参考例17:
tert−ブチル {3−[5−ブロモ−2−オキソベンゾ[d]オキサゾール−3(2H)−イル]プロピル}[2−(メチルスルホニル)エチル]カーバメート
Reference Example 17:
tert-Butyl {3- [5-bromo-2-oxobenzo [d] oxazol-3 (2H) -yl] propyl} [2- (methylsulfonyl) ethyl] carbamate
工程(i):
化合物52(2.00g)のメタノール(50mL)溶液にメチルビニルスルホン(2.1mL)を加え、室温で3時間攪拌した。反応液を減圧濃縮して化合物53の粗生成物を得た。このものは精製せずに全量を次工程に用いた。
Step (i):
Methyl vinyl sulfone (2.1 mL) was added to a methanol (50 mL) solution of compound 52 (2.00 g), and the mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure to obtain a crude product of compound 53. This product was used in the next step without purification.
工程(ii):
化合物53の粗生成物、二炭酸tert−ブチル(5.20g)および1,4−ジオキサン(50ml)の混合物を室温で2時間攪拌した。反応液を1mol/L塩酸で酸性にし、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、ろ液を減圧濃縮した。残渣をジクロロメタン(50ml)に溶解し、アミノシリカゲル(40g)を加え、室温で1時間攪拌した。混合液をろ過した後、ろ液を減圧濃縮して表題化合物54(4.80g)を得た。
Step (ii):
A mixture of the crude product of compound 53, tert-butyl dicarbonate (5.20 g) and 1,4-dioxane (50 ml) was stirred at room temperature for 2 hours. The reaction solution was acidified with 1 mol / L hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and the filtrate was concentrated under reduced pressure. The residue was dissolved in dichloromethane (50 ml), amino silica gel (40 g) was added, and the mixture was stirred at room temperature for 1 hour. The mixture was filtered, and the filtrate was concentrated under reduced pressure to give the title compound 54 (4.80 g).
工程(iii):
化合物54(4.22g)を用いて、参考例14の工程(ii)と同様の方法に従い、表題化合物55(7.03g)を得た。
Step (iii):
The title compound 55 (7.03 g) was obtained using the compound 54 (4.22 g) according to the same method as in step (ii) of Reference Example 14.
1H-NMR (CDCl3) δ: 1.42 (9H, s), 2.07-1.99 (2H, m), 2.95 (3H, s), 3.38-3.31 (4H, m), 3.69 (2H, t, J = 7.1 Hz), 3.80 (2H, t, J = 7.3 Hz), 7.08 (1H, d, J = 8.3 Hz), 7.12 (1H, d, J = 2.0 Hz), 7.25 (1H, dd, J = 8.4, 1.8 Hz). 1 H-NMR (CDCl 3 ) δ: 1.42 (9H, s), 2.07-1.99 (2H, m), 2.95 (3H, s), 3.38-3.31 (4H, m), 3.69 (2H, t, J = 7.1 Hz), 3.80 (2H, t, J = 7.3 Hz), 7.08 (1H, d, J = 8.3 Hz), 7.12 (1H, d, J = 2.0 Hz), 7.25 (1H, dd, J = 8.4, 1.8 Hz).
参考例18:
5−(tert−ブチル)ベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 18:
5- (tert-Butyl) benzo [d] oxazol-2 (3H) -one
工程(i):
化合物56(1.00g)を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物57(0.55g)を得た。
Step (i):
The title compound 57 (0.55 g) was obtained using the compound 56 (1.00 g) according to the same method as in step (ii) of Reference Example 1.
1H-NMR (DMSO-D6) δ: 1.27 (9H, s), 7.03 (1H, d, J = 1.8 Hz), 7.08 (1H, dd, J = 1.8, 7.9 Hz), 7.16 (1H, d, J = 7.9 Hz), 11.49 (1H, s). 1 H-NMR (DMSO-D 6 ) δ: 1.27 (9H, s), 7.03 (1H, d, J = 1.8 Hz), 7.08 (1H, dd, J = 1.8, 7.9 Hz), 7.16 (1H, d , J = 7.9 Hz), 11.49 (1H, s).
参考例19:
1−フェニル−5−[(テトラヒドロ−2H−ピラン−4−イル)スルホニル]−1H−テトラゾール
Reference Example 19:
1-phenyl-5-[(tetrahydro-2H-pyran-4-yl) sulfonyl] -1H-tetrazole
工程(i):
化合物58(0.642ml)および1−フェニル−5−メルカプトテトラゾール(1.00g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物59(1.06g)を得た。
Step (i):
Compound 59 (1.06 g) was obtained in the same manner as in step (ii) of Reference Example 14 using compound 58 (0.642 ml) and 1-phenyl-5-mercaptotetrazole (1.00 g).
工程(ii):
化合物59(1.06g)、30%過酸化水素水(4.1mL)、モリブデン酸アンモニウム4水和物(0.999mg)およびエタノール(40mL)の混合物を室温で4時間攪拌した。0℃でチオ硫酸ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=3/1)で精製し、表題化合物60(0.970g)を得た。
Step (ii):
A mixture of Compound 59 (1.06 g), 30% aqueous hydrogen peroxide (4.1 mL), ammonium molybdate tetrahydrate (0.999 mg) and ethanol (40 mL) was stirred at room temperature for 4 hours. An aqueous sodium thiosulfate solution was added at 0 ° C., and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 3/1) to give the title compound 60 (0.970 g).
1H-NMR (CDCl3) δ: 1.95-2.06 (2H, m), 2.12-2.17 (2H, m), 3.49 (2H, m), 4.09-4.17 (3H, m), 7.56-7.67 (5H, m). 1 H-NMR (CDCl 3 ) δ: 1.95-2.06 (2H, m), 2.12-2.17 (2H, m), 3.49 (2H, m), 4.09-4.17 (3H, m), 7.56-7.67 (5H, m).
参考例20:
5−[(テトラヒドロ−2H−ピラン−4−イル)メチル]ベンゾ[d]オキサゾール−2(3H)−オン
Reference Example 20:
5-[(Tetrahydro-2H-pyran-4-yl) methyl] benzo [d] oxazol-2 (3H) -one
工程(i):
化合物61(1.00g)を用いて、参考例4の工程(i)と同様の方法に従い、化合物62(1.15g)を得た。
Step (i):
Compound 62 (1.15 g) was obtained using Compound 61 (1.00 g) according to the same method as in step (i) of Reference Example 4.
工程(ii):
参考例19の化合物60(0.820g)とTHF(30mL)の混合物に対して、−78℃で、1.3mol/L リチウムヘキサメチルジシラジド(THF溶液、2.36mL)を滴下し、10分攪拌した。化合物62(0.862g)のTHF(5.0mL)溶液を、−78℃で滴下し、室温まで自然昇温しながら1.5時間攪拌した。反応混合液に飽和塩化アンモニウム水溶液水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をアミノシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=8/1)で精製し、化合物63(0.740g)を得た。
Step (ii):
1.3 mol / L lithium hexamethyldisilazide (THF solution, 2.36 mL) was added dropwise at −78 ° C. to a mixture of Compound 60 (0.820 g) of Reference Example 19 and THF (30 mL). Stir for 10 minutes. A solution of compound 62 (0.862 g) in THF (5.0 mL) was added dropwise at −78 ° C., and the mixture was stirred for 1.5 hours while naturally warming to room temperature. Saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and then the residue was purified by amino silica gel column chromatography (eluent: hexane / ethyl acetate = 8/1) to obtain Compound 63 (0.740 g).
工程(iii):
化合物63(0.880g)、10% Pd/C(0.094g)およびTHF(3.0mL)の混合物を水素雰囲気下、室温で2時間攪拌した。混合液をセライトろ過し、減圧濃縮を行い、化合物64を粗生成物として得た。全量を次の反応に用いた。
Step (iii):
A mixture of Compound 63 (0.880 g), 10% Pd / C (0.094 g) and THF (3.0 mL) was stirred at room temperature for 2 hours under a hydrogen atmosphere. The mixture was filtered through Celite and concentrated under reduced pressure to obtain Compound 64 as a crude product. The whole amount was used for the next reaction.
工程(iv):
化合物64の粗生成物を用いて、参考例1の工程(ii)と同様の方法に従い、表題化合物65(0.412g)を得た。
Step (iv):
Using the crude product of compound 64, the title compound 65 (0.412 g) was obtained in the same manner as in step (ii) of Reference Example 1.
1H-NMR (DMSO-D6) δ: 1.11-1.22 (2H, m), 1.44 (2H, m), 1.69 (1H, br s), 2.47-2.52 (2H, m), 3.20 (2H, m), 3.79 (2H, m), 6.83-6.88 (2H, m), 7.15 (1H, m), 11.52 (1H, br s). 1 H-NMR (DMSO-D 6 ) δ: 1.11-1.22 (2H, m), 1.44 (2H, m), 1.69 (1H, br s), 2.47-2.52 (2H, m), 3.20 (2H, m ), 3.79 (2H, m), 6.83-6.88 (2H, m), 7.15 (1H, m), 11.52 (1H, br s).
実施例1(A):
5−(4−フルオロフェニル)−3−{2−[(2−ヒドロキシ−2−メチルプロピル)アミノ]エチル}ベンゾ[d]オキサゾール−2(3H)−オン
Example 1 (A):
5- (4-Fluorophenyl) -3- {2-[(2-hydroxy-2-methylpropyl) amino] ethyl} benzo [d] oxazol-2 (3H) -one
工程(i):
化合物I−a(4.20g)、2−(tert-ブチルジメチルシリルオキシ)エチルブロミド(5.10mL)、リチウムブロミド(3.50g)、水素化ナトリウム(0.960g)およびDMF(100mL)の混合物を80℃で2時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=8/1)で精製し、化合物II−a(3.10g)を得た。
Step (i):
Compound Ia (4.20 g), 2- (tert-butyldimethylsilyloxy) ethyl bromide (5.10 mL), lithium bromide (3.50 g), sodium hydride (0.960 g) and DMF (100 mL) The mixture was stirred at 80 ° C. for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 8/1) to obtain Compound II-a (3.10 g).
工程(ii):
化合物II−a(3.10g)、4−フルオロフェニルボロン酸(1.70g)、テトラキストリフェニルホスフィンパラジウム(0.48g)、炭酸カリウム(3.40g)、1,4−ジオキサン(80mL)および水(10mL)の混合物を90℃で6時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=8/1)で精製し、化合物III−a(3.20g)を得た。
Step (ii):
Compound II-a (3.10 g), 4-fluorophenylboronic acid (1.70 g), tetrakistriphenylphosphine palladium (0.48 g), potassium carbonate (3.40 g), 1,4-dioxane (80 mL) and A mixture of water (10 mL) was stirred at 90 ° C. for 6 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and then the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 8/1) to obtain Compound III-a (3.20 g).
工程(iii):
化合物III−a(3.20g)、1.0mol/Lテトラブチルアンモニウムフルオリド(THF溶液、、9.9mL)、酢酸(0.566mL)およびTHF(83mL)の混合物を室温で14時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=4/1)で精製し、化合物IV−a(1.63g)を得た。
Step (iii):
A mixture of Compound III-a (3.20 g), 1.0 mol / L tetrabutylammonium fluoride (THF solution, 9.9 mL), acetic acid (0.566 mL) and THF (83 mL) was stirred at room temperature for 14 hours. . Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 4/1) to obtain Compound IV-a (1.63 g).
工程(iv):
化合物IV−a(1.63g)、四臭化炭素(5.20g)、トリフェニルホスフィン(2.00g)および塩化メチレン(60mL)の混合物を室温で3時間攪拌した。反応混合液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=6/1)で精製し、化合物V−a(1.47g)を得た。
Step (iv):
A mixture of compound IV-a (1.63 g), carbon tetrabromide (5.20 g), triphenylphosphine (2.00 g) and methylene chloride (60 mL) was stirred at room temperature for 3 hours. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 6/1) to obtain Compound Va (1.47 g).
工程(v):
化合物V−a(0.100g)、3−アミノ−2−メチル−2−プロパノール(0.080g)、炭酸カリウム(0.124g)およびアセトニトリル(1.5mL)の混合物を70℃で18時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をアミノシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=6/1)で精製し、表題化合物VI−a(0.081g)を得た。
Step (v):
A mixture of Compound Va (0.100 g), 3-amino-2-methyl-2-propanol (0.080 g), potassium carbonate (0.124 g) and acetonitrile (1.5 mL) was stirred at 70 ° C. for 18 hours. did. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by amino silica gel column chromatography (eluent: hexane / ethyl acetate = 6/1) to give the title compound VI-a (0.081 g). It was.
1H-NMR (CDCl3) δ: 1.13 (6H, s), 2.59 (2H, s), 3.06 (2H, m), 3.97 (2H, m), 7.09-7.26 (5H, m), 7.47-7.52 (2H, m). 1 H-NMR (CDCl 3 ) δ: 1.13 (6H, s), 2.59 (2H, s), 3.06 (2H, m), 3.97 (2H, m), 7.09-7.26 (5H, m), 7.47-7.52 (2H, m).
実施例2〜23:
実施例1(A)と同様にして、表1〜3に示す実施例2〜23の化合物を製造した。
Examples 2 to 23:
In the same manner as in Example 1 (A), the compounds of Examples 2 to 23 shown in Tables 1 to 3 were produced.
実施例24:
6−(4−フルオロフェニル)−3−(2−((2−ヒドロキシ−2−メチルプロピル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン
6−ブロモベンズオキサゾロンを用いて、実施例1(A)と同様にして、表題化合物を製造した。
Example 24:
6- (4-Fluorophenyl) -3- (2-((2-hydroxy-2-methylpropyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one With 6-bromobenzoxazolone, The title compound was produced in the same manner as Example 1 (A).
1H-NMR (CDCl3) δ: 1.16 (6H, s), 2.61 (2H, s), 3.10 (2H, t, J = 6.1Hz), 3.97 (2H, t, J = 6.1Hz), 7.06-7.16 (3H, m), 7.35-7.39 (2H, m), 7.48-7.52 (2H, m).
実施例25:
6−(4−フルオロフェノキシ)−3−(2−((2−ヒドロキシ−2−メチルプロピル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン
参考例13の化合物42を用いて、実施例1(A)と同様にして、表題化合物を製造した。
1 H-NMR (CDCl 3 ) δ: 1.16 (6H, s), 2.61 (2H, s), 3.10 (2H, t, J = 6.1 Hz), 3.97 (2H, t, J = 6.1 Hz), 7.06- 7.16 (3H, m), 7.35-7.39 (2H, m), 7.48-7.52 (2H, m).
Example 25:
6- (4-Fluorophenoxy) -3- (2-((2-hydroxy-2-methylpropyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one Using Compound 42 of Reference Example 13 The title compound was produced in the same manner as in Example 1 (A).
1H-NMR (CDCl3) δ: 1.22 (6H, s), 2.76 (2H, s), 3.19 (2H, t, J = 6.0 Hz), 4.04 (2H, t, J = 6.0 Hz), 6.83-7.03 (7H, m). 1 H-NMR (CDCl 3 ) δ: 1.22 (6H, s), 2.76 (2H, s), 3.19 (2H, t, J = 6.0 Hz), 4.04 (2H, t, J = 6.0 Hz), 6.83- 7.03 (7H, m).
実施例26(B):
5−(4−フルオロフェノキシ)−3−{2−[(2−ヒドロキシ−2−メチルプロピル)アミノ]エチル}ベンゾ[d]オキサゾール−2(3H)−オン
Example 26 (B):
5- (4-Fluorophenoxy) -3- {2-[(2-hydroxy-2-methylpropyl) amino] ethyl} benzo [d] oxazol-2 (3H) -one
工程(i):
化合物II−a(1.10g)、ビス(ピナコレート)ジボロン(1.70g)、ジクロロ[1,1’−ビス(ジフェニルホスフィノ)フェロセン]パラジウム(0.110g)、酢酸カリウム(0.883g)および1,4−ジオキサン(15mL)の混合物を90℃で14時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=8/1)で精製し、化合物I−b(1.26g)を得た。
Step (i):
Compound II-a (1.10 g), bis (pinacolato) diboron (1.70 g), dichloro [1,1′-bis (diphenylphosphino) ferrocene] palladium (0.110 g), potassium acetate (0.883 g) And a mixture of 1,4-dioxane (15 mL) was stirred at 90 ° C. for 14 h. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 8/1) to give Compound Ib (1.26 g).
工程(ii):
化合物I−b(0.420g)、4−フルオロフェノール(0.224g)、酢酸銅(0.272g)、トリエチルアミン(0.697mL)およびアセトニトリル(5mL)の混合物を40℃で16時間攪拌した。反応混合液に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=6/1)で精製し、化合物II−b(0.189g)を得た。
Step (ii):
A mixture of compound Ib (0.420 g), 4-fluorophenol (0.224 g), copper acetate (0.272 g), triethylamine (0.697 mL) and acetonitrile (5 mL) was stirred at 40 ° C. for 16 hours. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and then the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 6/1) to obtain Compound II-b (0.189 g).
工程(iii):
化合物II−b(0.189g)を用いて、実施例Aの工程(iii)と同様の方法に従い、化合物III−b(0.091g)を得た。
Step (iii):
Compound III-b (0.091 g) was obtained using Compound II-b (0.189 g) according to the same method as in step (iii) of Example A.
工程(iv):
化合物III−b(0.091g)を用いて、実施例Aの工程(iv)と同様の方法に従い、化合物IV−b(0.074g)を得た。
Step (iv):
Compound IV-b (0.074 g) was obtained using compound III-b (0.091 g) according to the same method as in step (iv) of Example A.
工程(v):
化合物IV−b(0.036g)を用いて、実施例Aの工程(v)と同様の方法に従い、表題化合物V−b(0.0084g)を得た。
Step (v):
The title compound Vb (0.0084 g) was obtained using the compound IV-b (0.036 g) according to the same method as in step (v) of Example A.
1H-NMR (CDCl3) δ: 1.13 (6H, s), 2.56 (2H, s), 3.03 (2H, t, J = 6.1Hz), 3.87 (2H, t, J = 6.1Hz), 6.68-6.72 (2H, m), 6.93-7.07 (4H, m), 7.13 (1H, m). 1 H-NMR (CDCl 3 ) δ: 1.13 (6H, s), 2.56 (2H, s), 3.03 (2H, t, J = 6.1 Hz), 3.87 (2H, t, J = 6.1 Hz), 6.68- 6.72 (2H, m), 6.93-7.07 (4H, m), 7.13 (1H, m).
実施例27(C):
5−シクロヘキシル−3−{2−[(2−ヒドロキシ−2−メチルプロピル)アミノ]エチル}ベンゾ[d]オキサゾール−2(3H)−オン
Example 27 (C):
5-cyclohexyl-3- {2-[(2-hydroxy-2-methylpropyl) amino] ethyl} benzo [d] oxazol-2 (3H) -one
工程(i):
化合物I-c(実施例7)(0.040g)、10% Pd/C(0.010g)およびメタノール(3.0mL)の混合物を水素雰囲気下、室温で2時間攪拌した。混合液をセライトろ過し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=19/1)で精製し、表題化合物II−c(0.029g)を得た。
Step (i):
A mixture of Compound Ic (Example 7) (0.040 g), 10% Pd / C (0.010 g) and methanol (3.0 mL) was stirred at room temperature for 2 hours under a hydrogen atmosphere. The mixture was filtered through celite and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 19/1) to give the title compound II-c (0.029 g).
1H-NMR (CDCl3) δ: 1.12 (6H, s), 1.36-1.43 (5H, m), 1.77-1.83 (5H, m), 2.53 (1H,m), 2.60 (2H, s), 3.06-3.08 (2H, m), 3.92-3.94 (2H, m), 6.85-6.86 (1H, m), 6.94-6.97 (1H, m), 7.10-7.13 (1H, m). 1 H-NMR (CDCl 3 ) δ: 1.12 (6H, s), 1.36-1.43 (5H, m), 1.77-1.83 (5H, m), 2.53 (1H, m), 2.60 (2H, s), 3.06 -3.08 (2H, m), 3.92-3.94 (2H, m), 6.85-6.86 (1H, m), 6.94-6.97 (1H, m), 7.10-7.13 (1H, m).
実施例28:
3−{2−[(2−ヒドロキシ−2−メチルプロピル)アミノ]エチル}−5−フェネチルベンゾ[d]オキサゾール−2(3H)−オン
実施例11の化合物を用いて、実施例27(C)と同様の方法で、表題化合物を製造した。
Example 28:
3- {2-[(2-hydroxy-2-methylpropyl) amino] ethyl} -5-phenethylbenzo [d] oxazol-2 (3H) -one Using the compound of Example 11, Example 27 (C ) To give the title compound.
1H-NMR (CDCl3) δ: 1.14 (6H, s), 2.59 (2H, s), 2.92-3.06 (6H, m), 3.88 (2H, m), 6.71 (1H, m), 6.89-6.93 (1H, m), 7.09-7.30 (6H, m) 1 H-NMR (CDCl 3 ) δ: 1.14 (6H, s), 2.59 (2H, s), 2.92-3.06 (6H, m), 3.88 (2H, m), 6.71 (1H, m), 6.89-6.93 (1H, m), 7.09-7.30 (6H, m)
実施例29(D):
5−エチル−3−{2−[(2−ヒドロキシ−2−メチルプロピル)アミノ]エチル}ベンゾ[d]オキサゾール−2(3H)−オン
Example 29 (D):
5-ethyl-3- {2-[(2-hydroxy-2-methylpropyl) amino] ethyl} benzo [d] oxazol-2 (3H) -one
工程(i):
化合物I-d(実施例12)(0.033g)、1mol/Lジエチル亜鉛(THF溶液、0.300mL)、ビス(トリ−tert−ブチルホスフィン)パラジウム(0.010g)およびTHF(0.5mL)の混合物を室温で12時間攪拌した。反応混合液をセライトろ過し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=19/1)で精製し、表題化合物II−d(0.014g)を得た。
Step (i):
Compound Id (Example 12) (0.033 g), 1 mol / L diethyl zinc (THF solution, 0.300 mL), bis (tri-tert-butylphosphine) palladium (0.010 g) and THF (0.5 mL) ) Was stirred at room temperature for 12 hours. The reaction mixture was filtered through Celite and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 19/1) to give the title compound II-d (0.014 g).
1H-NMR (DMSO-d6) δ: 1.03 (6H, s), 1.18 (3H, t, J = 7.6 Hz), 2.52 (2H, s), 2.59 (2H, q, J = 7.6 Hz), 2.94 (2H, m), 3.91 (2H, m), 6.92-6.95 (1H, m), 7.18-7.21 (2H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.03 (6H, s), 1.18 (3H, t, J = 7.6 Hz), 2.52 (2H, s), 2.59 (2H, q, J = 7.6 Hz), 2.94 (2H, m), 3.91 (2H, m), 6.92-6.95 (1H, m), 7.18-7.21 (2H, m).
実施例30〜31:
実施例29(D)と同様にして、表4に示す実施例30〜31の化合物を製造した。
Examples 30-31:
In the same manner as in Example 29 (D), the compounds of Examples 30 to 31 shown in Table 4 were produced.
実施例32(E):
5−(2,4−ジフルオロフェニル)−3−(2−{[2−(メチルスルホニル)エチル]アミノ}エチル)ベンゾ[d]オキサゾール−2(3H)−オン
Example 32 (E):
5- (2,4-Difluorophenyl) -3- (2-{[2- (methylsulfonyl) ethyl] amino} ethyl) benzo [d] oxazol-2 (3H) -one
工程(i):
化合物I−a(実施例1)(1.00g)、1mol/Lカリウムヘキサメチルジシラジド(THF溶液、5.00mL)、ブロモアセトアルデヒドジメチルアセタール(0.587mL)およびDMF(5mL)の混合物を90℃で16時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=6/1)で精製し、化合物I−e(1.50g)を得た。
Step (i):
Compound Ia (Example 1) (1.00 g), 1 mol / L potassium hexamethyldisilazide (THF solution, 5.00 mL), bromoacetaldehyde dimethyl acetal (0.587 mL) and DMF (5 mL) were mixed. Stir at 90 ° C. for 16 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 6/1) to give Compound Ie (1.50 g).
工程(ii):
化合物I−e(0.080g)および2,4−ジフルオロフェニルボロン酸(0.084g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合物II−e(0.082g)を得た。
Step (ii):
Using compound Ie (0.080 g) and 2,4-difluorophenylboronic acid (0.084 g) according to the same method as in step (ii) of Example 1 (A), compound II-e (0 .082 g) was obtained.
工程(iii):
化合物II−e(0.082g)、5mol/L塩酸(1.00mL)の混合物を50℃で1時間攪拌した。反応混合液に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた化合物III−eの粗生成物を、全量次の反応へ用いた。
Step (iii):
A mixture of Compound II-e (0.082 g) and 5 mol / L hydrochloric acid (1.00 mL) was stirred at 50 ° C. for 1 hour. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the resulting crude product of compound III-e was used in the next reaction in its entirety.
工程(iv):
化合物III−eの粗生成物、2−(メタンスルホニル)エチルアミン(0.049g)、トリアセトキシ水素化ホウ素ナトリウム(0.168g)、酢酸(0.032g)およびクロロホルム(1.5mL)の混合物を室温で16時間攪拌した。反応混合液に、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=49/1)で精製し、表題化合物IV−e(0.085g)を得た。
Step (iv):
A mixture of the crude product of Compound III-e, 2- (methanesulfonyl) ethylamine (0.049 g), sodium triacetoxyborohydride (0.168 g), acetic acid (0.032 g) and chloroform (1.5 mL). Stir at room temperature for 16 hours. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 49/1) to give the title compound IV-e (0.085 g).
1H-NMR (DMSO-d6) δ: 2.86-2.91 (7H, m), 3.13-3.15 (2H, m), 3.89-3.91 (2H, m), 7.22-7.24 (2H, m), 7.39-7.41 (2H, m), 7.47 (1H, s), 7.59-7.61 (1H, m). 1 H-NMR (DMSO-d 6 ) δ: 2.86-2.91 (7H, m), 3.13-3.15 (2H, m), 3.89-3.91 (2H, m), 7.22-7.24 (2H, m), 7.39- 7.41 (2H, m), 7.47 (1H, s), 7.59-7.61 (1H, m).
実施例33〜42:
実施例32(E)と同様にして、表5に示す実施例33〜42の化合物を製造した。
Examples 33-42:
In the same manner as in Example 32 (E), the compounds of Examples 33 to 42 shown in Table 5 were produced.
実施例43〜47:
参考例1〜5の化合物3、7、11、18および20を用いて、実施例32(E)と同様にして、表6に示す実施例43〜47の化合物を製造した。
Examples 43-47:
Using the compounds 3, 7, 11, 18, and 20 of Reference Examples 1 to 5, the compounds of Examples 43 to 47 shown in Table 6 were produced in the same manner as in Example 32 (E).
実施例48〜49:
参考例14の化合物45を用いて、実施例32(E)と同様にして、表7に示す実施例48〜49の化合物を製造した。
Examples 48-49:
Using the compound 45 of Reference Example 14, the compounds of Examples 48 to 49 shown in Table 7 were produced in the same manner as in Example 32 (E).
実施例50(F):
3−(4−{[2−(メチルスルホニル)エチル]アミノ}ブタン−2−イル)−5−[5−(トリフルオロメチル)ピリジン−2−イル]ベンゾ[d]オキサゾール−2(3H)−オン
Example 50 (F):
3- (4-{[2- (methylsulfonyl) ethyl] amino} butan-2-yl) -5- [5- (trifluoromethyl) pyridin-2-yl] benzo [d] oxazole-2 (3H) -ON
工程(i):
参考例14の化合物45(0.200g)を用いて、実施例26(B)の工程(i)と同様の方法に従い、化合物I−f(0.080g)を得た。
Step (i):
Compound If (0.080 g) was obtained using Compound 45 (0.200 g) of Reference Example 14 according to the same method as in Step (i) of Example 26 (B).
工程(ii):
化合物I−f(0.080g)、2−ブロモ−5−トリフルオロメチルピリジン(0.072g)、ジクロロ[1,1’−ビス(ジフェニルホスフィノ)フェロセン]パラジウム(0.031g)、3mol/L炭酸ナトリウム水溶液(0.212mL)および1,4−ジオキサン(1.0mL)の混合物を80℃で2時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=4/1)で精製し、化合物II−f(0.090g)を得た。
Step (ii):
Compound If (0.080 g), 2-bromo-5-trifluoromethylpyridine (0.072 g), dichloro [1,1′-bis (diphenylphosphino) ferrocene] palladium (0.031 g), 3 mol / A mixture of L aqueous sodium carbonate (0.212 mL) and 1,4-dioxane (1.0 mL) was stirred at 80 ° C. for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and then the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 4/1) to obtain Compound II-f (0.090 g).
工程(iii):
化合物II−f(0.090g)を用いて、実施例32(E)の工程(iii)と同様の方法に従い、化合物III−fを粗生成物として得た。全量をそのまま次の反応に用いた。
Step (iii):
Compound III-f was obtained as a crude product in the same manner as in step (iii) of Example 32 (E) using compound II-f (0.090 g). The whole amount was directly used for the next reaction.
工程(iv):
化合物III−fの粗生成物を用いて、実施例32(E)の工程(iv)と同様の方法に従い、表題化合物IV−f(0.073g)得た。
Step (iv):
The title compound IV-f (0.073 g) was obtained in the same manner as in step (iv) of Example 32 (E) using the crude product of compound III-f.
1H-NMR (DMSO-d6) δ: 1.50 (3H, d, J = 6.8 Hz), 1.91-1.94 (1H, m), 2.13-2.16 (1H, m), 2.81-2.82 (2H, m), 2.93 (3H, s), 3.12-3.14 (2H, m), 4.51-4.54 (1H, m), 7.48-7.51 (1H, m), 7.97-7.99 (1H, m), 8.08 (1H, s), 8.29-8.31 (2H, m), 9.04 (1H, s). 1 H-NMR (DMSO-d 6 ) δ: 1.50 (3H, d, J = 6.8 Hz), 1.91-1.94 (1H, m), 2.13-2.16 (1H, m), 2.81-2.82 (2H, m) , 2.93 (3H, s), 3.12-3.14 (2H, m), 4.51-4.54 (1H, m), 7.48-7.51 (1H, m), 7.97-7.99 (1H, m), 8.08 (1H, s) , 8.29-8.31 (2H, m), 9.04 (1H, s).
実施例51:
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン
実施例32(E)の化合物I−eを用いて、実施例50(F)と同様の方法により表題化合物を製造した。
Example 51:
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one Example The title compound was prepared in the same manner as in Example 50 (F) using 32 (E) compound Ie.
1H-NMR (DMSO-d6) δ: 2.89-2.92 (7H, m), 3.14-3.16 (2H, m), 3.95-3.99 (2H, m), 7.46-7.49 (1H, m), 7.95-7.99 (1H, m), 8.08 (1H, s), 8.26-8.29 (2H, m), 9.03 (1H, s). 1 H-NMR (DMSO-d 6 ) δ: 2.89-2.92 (7H, m), 3.14-3.16 (2H, m), 3.95-3.99 (2H, m), 7.46-7.49 (1H, m), 7.95- 7.99 (1H, m), 8.08 (1H, s), 8.26-8.29 (2H, m), 9.03 (1H, s).
実施例52(G):
5−(4−フルオロフェニル)−3−(2−{[2−(メチルスルホニル)エチル)]アミノ}プロピル)ベンゾ[d]オキサゾール−2(3H)−オン
Example 52 (G):
5- (4-Fluorophenyl) -3- (2-{[2- (methylsulfonyl) ethyl)] amino} propyl) benzo [d] oxazol-2 (3H) -one
工程(i):
化合物I−a(0.214g)およびtert−ブチル (1−ヒドロキシプロパン−2−イル)カルバメート(0.210g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物I−g(0.275g)を得た。
Step (i):
Using compound Ia (0.214 g) and tert-butyl (1-hydroxypropan-2-yl) carbamate (0.210 g), following the same method as in step (ii) of Reference Example 14, compound I- g (0.275 g) was obtained.
工程(ii):
化合物I−g(0.100g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合物II−g(0.095g)を得た。
Step (ii):
Compound II-g (0.095 g) was obtained by using Compound I-g (0.100 g) according to the same method as in Step (ii) of Example 1 (A).
工程(iii):
化合物II−g(0.095g)、トリフルオロ酢酸(2.00mL)およびクロロホルム(0.5mL)の混合物を室温で1時間攪拌した。反応混合液を減圧濃縮後、飽和炭酸水素ナトリウム水溶液を加え、クロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた化合物III−gの粗生成物を、全量次の反応へ用いた。
Step (iii):
A mixture of Compound II-g (0.095 g), trifluoroacetic acid (2.00 mL) and chloroform (0.5 mL) was stirred at room temperature for 1 hour. The reaction mixture was concentrated under reduced pressure, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the resulting crude product of compound III-g was used in the next reaction in its entirety.
工程(iv):
化合物III−gの粗生成物を用いて、参考例17の工程(i)と同様の方法に従い、表題化合物IV−g(0.014g)を得た。
Step (iv):
The title compound IV-g (0.014 g) was obtained in the same manner as in step (i) of Reference Example 17 using the crude product of compound III-g.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.1Hz), 3.13 (3H, s), 3.49-3.54 (4H, m), 3.82-3.86 (1H, m), 4.11-4.23 (2H, m), 7.32-7.41 (4H, m), 7.74-7.77 (3H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.36 (3H, d, J = 6.1 Hz), 3.13 (3H, s), 3.49-3.54 (4H, m), 3.82-3.86 (1H, m), 4.11 -4.23 (2H, m), 7.32-7.41 (4H, m), 7.74-7.77 (3H, m).
実施例53〜59:
実施例52(G)の製造法の工程(i)においては対応するカルバメートを、工程(ii)では対応するボロン酸を用いて、同様の方法により、表8に示す実施例53〜59の化合物を製造した。ただし、実施例58の化合物の製造の際は、原料に6−トリフルオロメチルベンズオキサゾロンを、実施例60の製造の際は参考例18の化合物57をそれぞれ用いた。
Examples 53-59:
The compounds of Examples 53 to 59 shown in Table 8 were prepared in the same manner using the corresponding carbamate in step (i) of the production method of Example 52 (G) and the corresponding boronic acid in step (ii). Manufactured. However, 6-trifluoromethylbenzoxazolone was used as a raw material for the production of the compound of Example 58, and Compound 57 of Reference Example 18 was used for the production of Example 60.
実施例61〜64:
実施例52(G)と同様にして、表9に示す実施例61〜64の化合物を製造した。実施例61においてはエチルビニルスルホンを用いて製造した。また、実施例63の化合物においては、参考例16の化合物51を、実施例64の化合物においては参考例18の化合物57をそれぞれ用いて製造した。
Examples 61-64:
In the same manner as in Example 52 (G), the compounds of Examples 61 to 64 shown in Table 9 were produced. Example 61 was prepared using ethyl vinyl sulfone. The compound of Example 63 was prepared using Compound 51 of Reference Example 16, and the compound of Example 64 was prepared using Compound 57 of Reference Example 18.
実施例65(GG):
5−(tert−ブチル)−3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
Example 65 (GG):
5- (tert-Butyl) -3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) benzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
参考例18の化合物57(0.062g)および参考例17の化合物54(0.110g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物I−gg(0.079g)を得た。
Step (i):
Using Compound 57 (0.062 g) of Reference Example 18 and Compound 54 (0.110 g) of Reference Example 17 and following the same method as in Step (ii) of Reference Example 14, compound I-gg (0.079 g) Got.
工程(ii):
化合物I−ggおよび4mol/L塩化水素−酢酸エチル溶液(2.00mL)の混合物を室温で1時間攪拌した。反応混合液を減圧濃縮後、酢酸エチルを加え、1時間攪拌した。固体をろ取し表題化合物II−gg(0.054g)を得た。
Step (ii):
A mixture of Compound I-gg and 4 mol / L hydrogen chloride-ethyl acetate solution (2.00 mL) was stirred at room temperature for 1 hour. The reaction mixture was concentrated under reduced pressure, ethyl acetate was added, and the mixture was stirred for 1 hr. The solid was collected by filtration to give the title compound II-gg (0.054 g).
1H-NMR (DMSO-D6) δ: 1.31 (9H, s), 2.02-2.10 (2H, m), 3.05 (2H, t, J = 7.6 Hz), 3.10 (3H, s), 3.32 (2H, t, J = 7.9 Hz), 3.55 (2H, t, J = 7.6 Hz), 3.94 (2H, t, J = 6.7 Hz), 7.14 (1H, dd, J = 8.5, 1.8 Hz), 7.23 (1H, d, J = 8.5 Hz), 7.41 (1H, d, J = 1.8 Hz), 9.26 (2H, br s). 1 H-NMR (DMSO-D 6 ) δ: 1.31 (9H, s), 2.02-2.10 (2H, m), 3.05 (2H, t, J = 7.6 Hz), 3.10 (3H, s), 3.32 (2H , t, J = 7.9 Hz), 3.55 (2H, t, J = 7.6 Hz), 3.94 (2H, t, J = 6.7 Hz), 7.14 (1H, dd, J = 8.5, 1.8 Hz), 7.23 (1H , d, J = 8.5 Hz), 7.41 (1H, d, J = 1.8 Hz), 9.26 (2H, br s).
実施例66(H):
5−[(4−フルオロフェノキシ)メチル]−3−(2−{[2−(メチルスルホニル)エチル]アミノ}エチル)ベンゾ[d]オキサゾール−2(3H)−オン
Example 66 (H):
5-[(4-Fluorophenoxy) methyl] -3- (2-{[2- (methylsulfonyl) ethyl] amino} ethyl) benzo [d] oxazol-2 (3H) -one
工程(i):
tert−ブチル (2−ヒドロキシエチル)カルバメート(0.903g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物I−h(0.760g)を得た。
工程(ii):
化合物I−h(1.00g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合物II−hを粗生成物(0.760g)として得た。
Step (i):
Compound Ih (0.760 g) was obtained using tert-butyl (2-hydroxyethyl) carbamate (0.903 g) according to the same method as in step (ii) of Reference Example 14.
Step (ii):
Compound II-h was obtained as a crude product (0.760 g) in the same manner as in step (ii) of Example 1 (A) using compound Ih (1.00 g).
工程(iii):
化合物II−hの粗生成物(0.760g)および塩化メチレン(5.0mL)の混合物に対して、−78℃でオゾンガスを吹き込みながら1時間攪拌した。ジメチルスルフィド(1.00g)を加え、室温にて1時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=2/1)で精製し、化合物III−h(0.120g)を得た。
Step (iii):
The mixture of the crude product of Compound II-h (0.760 g) and methylene chloride (5.0 mL) was stirred for 1 hour while blowing ozone gas at -78 ° C. Dimethyl sulfide (1.00 g) was added and stirred at room temperature for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and then the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 2/1) to obtain Compound III-h (0.120 g).
工程(iv):
化合物III−h(0.120g)を用いて、参考例14の工程(i)と同様の方法に従い、化合物IV−h(0.111g)を得た。
Step (iv):
Compound IV-h (0.111 g) was obtained using Compound III-h (0.120 g) according to the same method as in step (i) of Reference Example 14.
工程(v):
化合物IV−h(0.050g)、4−フルオロフェノール(0.027g)、アゾジカルボニルジピペリジン(0.061g)、トリブチルホスフィン(0.060mL)およびTHF(1.0mL)の混合物を室温にて18時間攪拌した。反応混合液を減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=4/1)で精製し、化合物V−h(0.036g)を得た。
Step (v):
A mixture of compound IV-h (0.050 g), 4-fluorophenol (0.027 g), azodicarbonyldipiperidine (0.061 g), tributylphosphine (0.060 mL) and THF (1.0 mL) was brought to room temperature. And stirred for 18 hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 4/1) to give Compound Vh (0.036 g).
工程(vi):
化合物V−h(0.036g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物VI−hを粗生成物として得た。
Step (vi):
Compound VI-h was obtained as a crude product in the same manner as in step (iii) of Example 52 (G) using compound Vh (0.036 g).
工程(vii):
化合物VI−hの粗生成物を用いて、参考例17の工程(i)と同様の方法に従い、表題化合物VII−h(0.020g)を得た。
Step (vii):
The title compound VII-h (0.020 g) was obtained in the same manner as in step (i) of Reference Example 17 using the crude product of compound VI-h.
1H-NMR (DMSO-d6) δ: 3.11 (3H, s), 3.36-3.46 (4H, m), 3.50-3.57 (2H, m), 4.18 (2H, m), 5.07 (2H, s), 7.04 (2H, m), 7.13 (2H, m), 7.24 (1H, m), 7.38 (1H, m), 7.52 (1H, s), 9.29 (1H, br s). 1 H-NMR (DMSO-d 6 ) δ: 3.11 (3H, s), 3.36-3.46 (4H, m), 3.50-3.57 (2H, m), 4.18 (2H, m), 5.07 (2H, s) , 7.04 (2H, m), 7.13 (2H, m), 7.24 (1H, m), 7.38 (1H, m), 7.52 (1H, s), 9.29 (1H, br s).
実施例67(I):
3−(2−メチル−3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−5−[5−(トリフルオロメチル)ピリジン−2−イル]ベンゾ[d]オキサゾール−2(3H)−オン
Example 67 (I):
3- (2-Methyl-3-{[2- (methylsulfonyl) ethyl] amino} propyl) -5- [5- (trifluoromethyl) pyridin-2-yl] benzo [d] oxazole-2 (3H) -ON
工程(i):
参考例8の化合物28(0.394g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物I−i(0.500g)を得た。
Step (i):
Compound Ii (0.500 g) was obtained using Compound 28 (0.394 g) of Reference Example 8 according to the same method as in Step (ii) of Reference Example 14.
工程(ii):
化合物I−i(0.450g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物II−iを粗生成物として得た。全量を次の反応に用いた。
Step (ii):
Compound II-i was obtained as a crude product in the same manner as in step (iii) of Example 52 (G) using compound Ii (0.450 g). The whole amount was used for the next reaction.
工程(iii):
化合物II−iの粗生成物を用いて、参考例17の工程(i)と同様の方法に従い、化合物III−i(0.320g)を得た。
Step (iii):
Compound III-i (0.320 g) was obtained by using the crude product of Compound II-i and following the same method as in Step (i) of Reference Example 17.
工程(iv):
化合物III−i(0.060g)を用いて、実施例26(B)の工程(i)と同様の方法に従い、化合物IV−iを粗生成物として得た。全量を次の反応に用いた。
Step (iv):
Compound IV-i was obtained as a crude product according to the same method as in step (i) of Example 26 (B) using compound III-i (0.060 g). The whole amount was used for the next reaction.
工程(v):
化合物IV−iの粗生成物を用いて、実施例50(F)の工程(ii)と同様の方法に従い、表題化合物V−i(0.013g)を得た。
Step (v):
The title compound Vi (0.013 g) was obtained using the crude product of compound IV-i according to the same method as in step (ii) of Example 50 (F).
LC/MS: m/z = 458 (M+H)+ LC / MS: m / z = 458 (M + H) +
実施例68〜69:
実施例67(I)と同様にして、表10に示す実施例68〜69の化合物を製造した。
Examples 68-69:
In the same manner as in Example 67 (I), the compounds of Examples 68 to 69 shown in Table 10 were produced.
実施例70(J):
5−(4−フルオロフェニル)−3−(2−ヒドロキシ−3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)ベンゾ[d]オキサゾール−2(3H)−オン
Example 70 (J):
5- (4-Fluorophenyl) -3- (2-hydroxy-3-{[2- (methylsulfonyl) ethyl] amino} propyl) benzo [d] oxazol-2 (3H) -one
工程(i):
グリシドール(0.260g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物I−j(0.080g)を得た。
Step (i):
Compound I-j (0.080 g) was obtained using glycidol (0.260 g) according to the same method as in step (ii) of Reference Example 14.
工程(ii):
化合物I−j(0.076g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合物II−j(0.034g)を得た。
Step (ii):
Compound II-j (0.034 g) was obtained using compound I-j (0.076 g) according to the same method as in step (ii) of Example 1 (A).
工程(iii):
化合物II−j(0.028g)、2−(メタンスルホニル)エチルアミン(0.038g)およびエタノール(1.0mL)の混合物を80℃にて2時間攪拌した。反応混合液を減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=19/1)で精製し、表題化合物III−j(0.013g)を得た。
Step (iii):
A mixture of compound II-j (0.028 g), 2- (methanesulfonyl) ethylamine (0.038 g) and ethanol (1.0 mL) was stirred at 80 ° C. for 2 hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 19/1) to give the title compound III-j (0.013 g).
1H-NMR (DMSO-d6) δ: 2.55-2.68 (2H, m), 2.93 (2H, m), 3.00 (3H, s), 3.22 (2H, m), 3.76-3.99 (3H, m), 5.12 (1H, d, J = 5.1Hz), 7.30 (2H, m), 7.34-7.41 (2H, m), 7.56 (1H, m), 7.69 (2H, m). 1 H-NMR (DMSO-d 6 ) δ: 2.55-2.68 (2H, m), 2.93 (2H, m), 3.00 (3H, s), 3.22 (2H, m), 3.76-3.99 (3H, m) , 5.12 (1H, d, J = 5.1Hz), 7.30 (2H, m), 7.34-7.41 (2H, m), 7.56 (1H, m), 7.69 (2H, m).
実施例71(K):
5−(4−フルオロフェニル)−3−(2−{[3−(メチルスルホニル)プロピル]アミノ}エチル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
Example 71 (K):
5- (4-Fluorophenyl) -3- (2-{[3- (methylsulfonyl) propyl] amino} ethyl) benzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
化合物I−h(実施例66)(0.050g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合物I−k(0.048g)を得た。
Step (i):
Compound Ik (0.048 g) was obtained using Compound Ih (Example 66) (0.050 g) according to the same method as in Step (ii) of Example 1 (A).
工程(ii):
化合物I−k(0.048g)、参考例15の化合物48、炭酸セシウム(0.088g)およびDMF(1.0mL)の混合物を90℃で2時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、得られた化合物II−kの粗生成物を全量次の反応へ用いた。
Step (ii):
A mixture of Compound I-k (0.048 g), Compound 48 of Reference Example 15, cesium carbonate (0.088 g) and DMF (1.0 mL) was stirred at 90 ° C. for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the resulting crude product of compound II-k was used for the next reaction.
工程(iii):
化合物II−kの粗生成物および4mol/L塩化水素−1、4−ジオキサン溶液(1.00mL)の混合物を室温で1時間攪拌した。反応混合液を減圧濃縮後、ジエチルエーテルを加え、1時間攪拌した。固体をろ取し表題化合物III−k(0.014g)を得た。
Step (iii):
A mixture of the crude product of Compound II-k and a 4 mol / L hydrogen chloride-1,4-dioxane solution (1.00 mL) was stirred at room temperature for 1 hour. The reaction mixture was concentrated under reduced pressure, diethyl ether was added, and the mixture was stirred for 1 hr. The solid was collected by filtration to give the title compound III-k (0.014 g).
1H-NMR (DMSO-d6) δ: 1.41-1.62 (2H, m), 2.56-2.68 (2H, m), 3.08 (3H, s), 3.17-3.26 (2H, m), 3.38-3.53 (4H, m), 7.05 (1H, m), 7.21 (1H, m), 7.30-7.45 (3H, m), 7.54 (2H, m), 8.84 (2H, br s). 1 H-NMR (DMSO-d 6 ) δ: 1.41-1.62 (2H, m), 2.56-2.68 (2H, m), 3.08 (3H, s), 3.17-3.26 (2H, m), 3.38-3.53 ( 4H, m), 7.05 (1H, m), 7.21 (1H, m), 7.30-7.45 (3H, m), 7.54 (2H, m), 8.84 (2H, br s).
実施例72(L):
5−(4−フルオロフェニル)−3−[3−(オキセタン−3−イルアミノ)プロピル]ベンゾ[d]オキサゾール−2(3H)−オン
Example 72 (L):
5- (4-Fluorophenyl) -3- [3- (oxetan-3-ylamino) propyl] benzo [d] oxazol-2 (3H) -one
工程(i):
tert−ブチル (3−ヒドロキシプロピル)カルバメート(0.210g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合I−l(0.330g)を得た。
Step (i):
Compound 1-1 (0.330 g) was obtained using tert-butyl (3-hydroxypropyl) carbamate (0.210 g) according to the same method as in step (ii) of Reference Example 14.
工程(ii):
化合物I−l(0.050g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合物II−l(0.040g)を得た。
Step (ii):
Compound II-l (0.040 g) was obtained using Compound I-l (0.050 g) according to the same method as in step (ii) of Example 1 (A).
工程(iii):
化合物II−l(0.040g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物III−lを粗生成物として得た。全量を次の反応に用いた。
Step (iii):
Compound III-l was obtained as a crude product according to the same method as in step (iii) of Example 52 (G) using compound II-l (0.040 g). The whole amount was used for the next reaction.
工程(iv):
化合物III−lの粗生成物、3−オキセタノン(0.015g)、トリアセトキシ水素化ホウ素ナトリウム(0.86g)、酢酸(0.024g)およびクロロホルム(1.0mL)の混合物を室温で16時間攪拌した。反応混合液に、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=49/1)で精製し、表題化合物IV−l(0.019g)を得た。
Step (iv):
A mixture of crude compound III-l, 3-oxetanone (0.015 g), sodium triacetoxyborohydride (0.86 g), acetic acid (0.024 g) and chloroform (1.0 mL) was stirred at room temperature for 16 hours. Stir. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 49/1) to give the title compound IV-1 (0.019 g).
1H-NMR (DMSO-d6) δ: 1.78-1.81 (2H, m), 3.29-3.32 (2H, m), 3.75-3.79 (1H, m), 3.90-3.93 (2H, m), 4.27-4.29 (2H, m), 4.55-4.57 (2H, m), 7.29-7.32 (2H, m), 7.37-7.40 (2H, m), 7.58-7.61 (1H, m), 7.69-7.71 (2H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.78-1.81 (2H, m), 3.29-3.32 (2H, m), 3.75-3.79 (1H, m), 3.90-3.93 (2H, m), 4.27- 4.29 (2H, m), 4.55-4.57 (2H, m), 7.29-7.32 (2H, m), 7.37-7.40 (2H, m), 7.58-7.61 (1H, m), 7.69-7.71 (2H, m ).
実施例73〜74:
実施例52(G)における化合物III−g又はIII−gに相当する化合物を用いて、実施例72(L)の方法を参考に、表11に示す実施例73〜74の化合物を製造した。
Examples 73-74:
Using the compound corresponding to Compound III-g or III-g in Example 52 (G), the compounds of Examples 73 to 74 shown in Table 11 were produced with reference to the method of Example 72 (L).
実施例75(M):
3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−5−フェニルベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
Example 75 (M):
3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) -5-phenylbenzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
化合物I−l(実施例72)(0.500g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物I−mを粗生成物として得た。全量を次の反応に用いた。
Step (i):
Using Compound I-1 (Example 72) (0.500 g) and following the same method as in Step (iii) of Example 52 (G), Compound Im was obtained as a crude product. The whole amount was used for the next reaction.
工程(ii):
化合物I−mの粗生成物を用いて、参考例17の工程(i)と同様の方法に従い、化合物II−m(0.240g)を得た。
Step (ii):
Compound II-m (0.240 g) was obtained using the crude product of Compound Im according to the same method as in Step (i) of Reference Example 17.
工程(iii):
化合物II−m(0.050g)、フェニルボロン酸(0.055g)、ビス(トリ−tert−ブチルホスフィン)パラジウム(0.005g)、炭酸カリウム(0.080g)、アセトニトリル(1.0mL)および水(0.2mL)の混合物を80℃で3時間攪拌した。反応混合液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥し、減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=8/1)で精製した。得られた化合物、酢酸エチル(1.0mL)および4mol/L塩化水素−1,4−ジオキサン溶液(1.00mL)の混合物を室温で1時間攪拌後、混合溶液を減圧濃縮した。析出した結晶を酢酸エチルで懸濁攪拌後、ろ取し表題化合物III−m(0.049g)を得た。
Step (iii):
Compound II-m (0.050 g), phenylboronic acid (0.055 g), bis (tri-tert-butylphosphine) palladium (0.005 g), potassium carbonate (0.080 g), acetonitrile (1.0 mL) and A mixture of water (0.2 mL) was stirred at 80 ° C. for 3 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 8/1). A mixture of the obtained compound, ethyl acetate (1.0 mL) and 4 mol / L hydrogen chloride-1,4-dioxane solution (1.00 mL) was stirred at room temperature for 1 hour, and then the mixed solution was concentrated under reduced pressure. The precipitated crystals were suspended and stirred in ethyl acetate and collected by filtration to give the title compound III-m (0.049 g).
1H-NMR (DMSO-d6) δ: 2.04-2.19 (2H, m), 3.03-3.12 (5H, m), 3.27-3.38 (2H, m), 3.51-3.60 (2H, m), 4.00 (2H, m), 7.33-7.51 (5H, m), 7.67-7.74 (3H, m), 9.32 (2H, br s). 1 H-NMR (DMSO-d 6 ) δ: 2.04-2.19 (2H, m), 3.03-3.12 (5H, m), 3.27-3.38 (2H, m), 3.51-3.60 (2H, m), 4.00 ( 2H, m), 7.33-7.51 (5H, m), 7.67-7.74 (3H, m), 9.32 (2H, br s).
実施例76〜91:
実施例75(M)と同様の方法にて、表12〜13に示す実施例76〜91の化合物を製造した。
Examples 76-91:
The compounds of Examples 76 to 91 shown in Tables 12 to 13 were produced in the same manner as in Example 75 (M).
実施例92:
3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン
実施例75(M)と同様の方法にて、表題化合物を製造した。ただし、工程(iii)の塩酸塩化の操作は行わず、遊離体として得た。
Example 92:
3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one Example 75 (M) In the same manner as described above, the title compound was produced. However, the procedure of hydrochloric acid chlorination in step (iii) was not performed, and the product was obtained as a free form.
1H-NMR (DMSO-d6) δ: 1.83-1.86 (2H, m), 2.56-2.59 (2H, m), 2.88-2.90 (2H, m), 2.98 (3H, s), 3.15-3.18 (2H, m), 3.92-3.95 (2H, m), 7.47-7.48 (2H, m), 7.70 (1H, br s), 7.82-7.92 (4H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.83-1.86 (2H, m), 2.56-2.59 (2H, m), 2.88-2.90 (2H, m), 2.98 (3H, s), 3.15-3.18 ( 2H, m), 3.92-3.95 (2H, m), 7.47-7.48 (2H, m), 7.70 (1H, br s), 7.82-7.92 (4H, m).
実施例93〜97:
実施例67(I)と同様にして、実施例75(M)の化合物II−mを用いて、表14に示す実施例93〜97の化合物を製造した。
Examples 93-97:
In the same manner as in Example 67 (I), compounds of Examples 93 to 97 shown in Table 14 were produced using Compound II-m of Example 75 (M).
実施例98〜99:
実施例67(I)または実施例75(M)と同様にして、参考例2の化合物7を用いて、表15に示す実施例98および99の化合物を製造した。実施例99の化合物の製造の際、実施例75の工程(iii)の塩酸塩化は行わず、遊離体として得た。
Examples 98-99:
In the same manner as in Example 67 (I) or Example 75 (M), the compounds of Examples 98 and 99 shown in Table 15 were produced using the compound 7 of Reference Example 2. In the production of the compound of Example 99, hydrochloric acid chlorination was not performed in step (iii) of Example 75, and the compound was obtained as a free form.
実施例100(MM):
5−(4,4−ジフルオロシクロヘキシル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
Example 100 (MM):
5- (4,4-Difluorocyclohexyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
化合物I−h(実施例66)(0.386g)および2−(4,4−ジフルオロシクロヘキセン−1−イル)−4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン(1.000g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合I−mm(0.720g)を得た。
Step (i):
Compound I-h (Example 66) (0.386 g) and 2- (4,4-difluorocyclohexen-1-yl) -4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1 In the same manner as in step (ii) of Example 1 (A), compound I-mm (0.720 g) was obtained.
工程(ii):
化合物I−mm(0.720g)を用いて、実施例27(C)の工程(i)と同様の方法に従い、化合物II−mm(0.724g)を得た。
Step (ii):
Compound II-mm (0.724 g) was obtained in the same manner as in step (i) of Example 27 (C) using compound I-mm (0.720 g).
工程(iii):
化合物II−mm(0.724mg)、を用いて、実施例65(GG)の工程(ii)と同様の方法に従い、化合物III−mm(0.582g)を得た。
Step (iii):
Using compound II-mm (0.724 mg), compound III-mm (0.582 g) was obtained in the same manner as in step (ii) of Example 65 (GG).
工程(iv):
化合物III−mm(0.619g)、ジイソプロピルアミン(0.721g)およびエタノール(18mL)の混合物に、メチルビニルスルホン(0.237g)を加え、室温にて7時間攪拌した。減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=97/3)で精製し、化合物IV−mmを得た。全量を次の反応に用いた。
Step (iv):
Methyl vinyl sulfone (0.237 g) was added to a mixture of Compound III-mm (0.619 g), diisopropylamine (0.721 g) and ethanol (18 mL), and the mixture was stirred at room temperature for 7 hours. After concentration under reduced pressure, the residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 97/3) to obtain Compound IV-mm. The whole amount was used for the next reaction.
工程(v):
化合物IV−mmを用いて、実施例71(K)の工程(iii)と同様の方法に従い、表題化合物V−mm(0.517g)を得た。
Step (v):
The title compound V-mm (0.517 g) was obtained in the same manner as in step (iii) of Example 71 (K) using compound IV-mm.
1H-NMR (DMSO-D6) δ: 1.67-1.80 (2H, m), 1.84-2.16 (6H, m), 2.74 (1H, t, J = 11.9 Hz), 3.11 (3H, s), 3.35 (2H, m), 3.41-3.46 (2H, m), 3.55 (2H, m), 4.17 (2H, m), 7.03 (1H, d, J = 7.9 Hz), 7.27 (1H, d, J = 7.9 Hz), 7.36 (1H, s), 9.37 (2H, br s). 1 H-NMR (DMSO-D 6 ) δ: 1.67-1.80 (2H, m), 1.84-2.16 (6H, m), 2.74 (1H, t, J = 11.9 Hz), 3.11 (3H, s), 3.35 (2H, m), 3.41-3.46 (2H, m), 3.55 (2H, m), 4.17 (2H, m), 7.03 (1H, d, J = 7.9 Hz), 7.27 (1H, d, J = 7.9 Hz), 7.36 (1H, s), 9.37 (2H, br s).
実施例101〜110:
実施例100(MM)と同様にして、表16〜17に示す実施例101〜110の化合物を製造した。実施例102、103、106,および110に関しては5−ブロモベンズオキサゾロンを、実施例104および105に関しては6−ブロモベンズオキサゾロンを、実施例107および108に関しては参考例20の化合物65を、実施例109に関しては参考例5の化合物を用い、対応するカーバメートを用いて製造した。また、実施例104および106の化合物の製造においては塩酸化は行わず、遊離体として得た。
Examples 101-110:
In the same manner as in Example 100 (MM), the compounds of Examples 101 to 110 shown in Tables 16 to 17 were produced. 5-bromobenzoxazolone for Examples 102, 103, 106, and 110, 6-bromobenzoxazolone for Examples 104 and 105, compound 65 of Reference Example 20 for Examples 107 and 108, 109 was prepared using the compound of Reference Example 5 and the corresponding carbamate. Further, in the production of the compounds of Examples 104 and 106, hydrochloric acid was not used, and it was obtained as a free form.
実施例111(N):
5−シクロブチル−3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
Example 111 (N):
5-cyclobutyl-3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) benzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
化合物II−m(実施例75)(0.050g)および0.5mol/L シクロブチルジンクブロミド(THF溶液、0.260mL)を用いて、実施例29(D)の工程(i)と同様の方法に従い製造した化合物に、酢酸エチル(1.0mL)および4mol/L塩化水素−1,4−ジオキサン溶液(1.0mL)を加え、室温で1時間攪拌後、混合溶液を減圧濃縮した。析出した結晶を酢酸エチルで懸濁攪拌後、ろ取し表題化合物I−n(0.039g)を得た。
Step (i):
Similar to step (i) of Example 29 (D) using Compound II-m (Example 75) (0.050 g) and 0.5 mol / L cyclobutyl zinc bromide (THF solution, 0.260 mL). Ethyl acetate (1.0 mL) and 4 mol / L hydrogen chloride-1,4-dioxane solution (1.0 mL) were added to the compound produced according to the method, and the mixture solution was concentrated under reduced pressure after stirring at room temperature for 1 hour. The precipitated crystals were suspended and stirred in ethyl acetate and collected by filtration to give the title compound In (0.039 g).
1H-NMR (DMSO-d6) δ: 1.75-1.86 (1H, m), 1.90-2.19 (5H, m), 2.25-2.35 (2H, m), 3.01-3.09 (2H, m), 3.10 (3H, s), 3.30-3.36 (2H, m), 3.48-3.60 (3H, m), 3.92 (2H, m), 6.98 (1H, m), 7.24 (1H, m), 7.27 (1H, s), 9.07 (2H, br s). 1 H-NMR (DMSO-d 6 ) δ: 1.75-1.86 (1H, m), 1.90-2.19 (5H, m), 2.25-2.35 (2H, m), 3.01-3.09 (2H, m), 3.10 ( 3H, s), 3.30-3.36 (2H, m), 3.48-3.60 (3H, m), 3.92 (2H, m), 6.98 (1H, m), 7.24 (1H, m), 7.27 (1H, s) , 9.07 (2H, br s).
実施例112〜115:
実施例111(N)と同様にして、表18に示す実施例112〜115の化合物を製造した。
Examples 112-115:
In the same manner as in Example 111 (N), the compounds of Examples 112 to 115 shown in Table 18 were produced.
実施例116(O):
5−(シクロヘキシルメチル)−3−(2−{[2−(メチルスルホニル)エチル]アミノ}エチル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
Example 116 (O):
5- (Cyclohexylmethyl) -3- (2-{[2- (methylsulfonyl) ethyl] amino} ethyl) benzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
化合物I−h(実施例66)(5.71g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物I−oを粗生成物として得た。全量を次の反応に用いた。
Step (i):
Compound Io (Example 66) (5.71 g) was used in the same manner as in Step (iii) of Example 52 (G) to give Compound Io as a crude product. The whole amount was used for the next reaction.
工程(ii):
化合物I−oの粗生成物を用いて、参考例17の工程(i)と同様の方法に従い、化合物II−o(0.936g)を得た。
Step (ii):
Compound II-o (0.936 g) was obtained according to the same method as in step (i) of Reference Example 17 using the crude product of compound Io.
工程(iii):
化合物II−o(1.16g)を用いて、参考例9の工程(i)と同様の方法に従い、化合物III−o(1.25g)を得た。
Step (iii):
Compound III-o (1.25 g) was obtained using Compound II-o (1.16 g) according to the same method as in step (i) of Reference Example 9.
工程(iv):
化合物III−o(0.133g)を用いて、実施例29(D)の工程(i)と同様の方法に従い、化合物IV−o(0.119g)を得た。
Step (iv):
Compound IV-o (0.119 g) was obtained using compound III-o (0.133 g) according to the same method as in step (i) of Example 29 (D).
工程(v):
化合物IV−o(0.119g)を用いて、実施例71(K)の工程(iii)と同様の方法に従い、表題化合物V−o(0.050g)を得た。
Step (v):
The title compound Vo (0.050 g) was obtained using the compound IV-o (0.119 g) according to the same method as in step (iii) of Example 71 (K).
1H-NMR (CD3OD) δ: 0.94-1.04 (2H, m), 1.16-1.22 (3H, m), 1.54-1.61 (1H, m), 1.67-1.73 (5H, m), 2.56 (2H, d, J = 7.1 Hz), 3.11 (3H, s), 3.52-3.59 (4H, m), 3.62-3.67 (2H, m), 4.26 (2H, t, J = 5.6 Hz), 6.99 (1H, dd, J = 8.3, 1.5 Hz), 7.11 (1H, d, J = 1.7 Hz) 7.19 (1H, d, J = 8.3 Hz). 1 H-NMR (CD 3 OD) δ: 0.94-1.04 (2H, m), 1.16-1.22 (3H, m), 1.54-1.61 (1H, m), 1.67-1.73 (5H, m), 2.56 (2H , d, J = 7.1 Hz), 3.11 (3H, s), 3.52-3.59 (4H, m), 3.62-3.67 (2H, m), 4.26 (2H, t, J = 5.6 Hz), 6.99 (1H, dd, J = 8.3, 1.5 Hz), 7.11 (1H, d, J = 1.7 Hz) 7.19 (1H, d, J = 8.3 Hz).
実施例117〜122:
実施例116(O)と同様にして、表19に示す実施例117〜122の化合物を製造した。
Examples 117-122:
In the same manner as in Example 116 (O), the compounds of Examples 117 to 122 shown in Table 19 were produced.
実施例123〜127:
実施例116(O)と同様にして、参考例17の化合物55を用いて表20に示す実施例123〜127の化合物を製造した。
Examples 123-127:
In the same manner as in Example 116 (O), the compounds of Examples 123 to 127 shown in Table 20 were produced using the compound 55 of Reference Example 17.
実施例128および129(P):
3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−5−[5−(トリフルオロメチル)ピリミジン−2−イル]ベンゾ[d]オキサゾール−2(3H)−オン(128)および、3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−5−[5−(トリフルオロメチル)ピリミジン−4−イル]ベンゾ[d]オキサゾール−2(3H)−オン(129)
Examples 128 and 129 (P):
3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) -5- [5- (trifluoromethyl) pyrimidin-2-yl] benzo [d] oxazol-2 (3H) -one (128 ) And 3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) -5- [5- (trifluoromethyl) pyrimidin-4-yl] benzo [d] oxazole-2 (3H)- ON (129)
工程(i):
化合I−l(実施例67)(1.40g)を用いて、実施例26(B)の工程(i)と同様の方法に従い、化合物I−p(1.30g)を得た。
Step (i):
Compound Ip (1.30 g) was obtained according to the same method as in step (i) of Example 26 (B) using Compound I-l (Example 67) (1.40 g).
工程(ii):
化合物I−p(0.125g)および2,4−ジクロロ−5−トリフルオロメチルピリミジン(0.065g)を用いて、実施例50(F)の工程(ii)と同様の方法に従い、化合物II−pおよび化合物III−pを混合物として得た。全量を次の反応に用いた。
Step (ii):
Compound II-p (0.125 g) and 2,4-dichloro-5-trifluoromethylpyrimidine (0.065 g) were used in the same manner as in step (ii) of Example 50 (F) to give compound II. -P and compound III-p were obtained as a mixture. The whole amount was used for the next reaction.
工程(iii):
化合物II−pおよび化合物III−pの混合物を用いて、参考例6の工程(i)と同様の方法に従い、化合物IV−p(0.010g)および化合物V−p(0.028g)をそれぞれ得た。
Step (iii):
Using a mixture of Compound II-p and Compound III-p, Compound IV-p (0.010 g) and Compound Vp (0.028 g) were prepared in the same manner as in Step (i) of Reference Example 6, respectively. Obtained.
工程(iv):
化合物IV−p(0.010g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物VI−pを粗生成物として得た。全量を次の反応に用いた。
Step (iv):
Compound VI-p was obtained as a crude product according to the same method as in step (iii) of Example 52 (G) using compound IV-p (0.010 g). The whole amount was used for the next reaction.
工程(v):
化合物VI−pの粗生成物を用いて、参考例17の工程(i)と同様の方法に従い、表題化合物VII−p(0.0065g)を得た。
Step (v):
The title compound VII-p (0.0065 g) was obtained in the same manner as in step (i) of Reference Example 17 using the crude product of compound VI-p.
1H-NMR (DMSO-d6) δ: 1.82-1.86 (2H, m), 2.55-2.58 (2H, m), 2.86-2.88 (2H, m), 2.98 (3H, s), 3.21-3.23 (2H, m), 3.95-3.97 (2H, m), 7.52-7.55 (1H, m), 8.29-8.32 (2H, m), 9.32-9.35 (2H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.82-1.86 (2H, m), 2.55-2.58 (2H, m), 2.86-2.88 (2H, m), 2.98 (3H, s), 3.21-3.23 ( 2H, m), 3.95-3.97 (2H, m), 7.52-7.55 (1H, m), 8.29-8.32 (2H, m), 9.32-9.35 (2H, m).
工程(vi):
化合物V−p(0.028g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物VIII−pを粗生成物として得た。全量を次の反応に用いた。
Step (vi):
Compound VIII-p was obtained as a crude product according to the same method as in step (iii) of Example 52 (G) using compound Vp (0.028 g). The whole amount was used for the next reaction.
工程(vii):
化合物VIII−pの粗生成物を用いて、参考例17の工程(i)と同様の方法に従い、表題化合物IX−p(0.017g)を得た。
Step (vii):
The title compound IX-p (0.017 g) was obtained in the same manner as in step (i) of Reference Example 17 using the crude product of compound VIII-p.
1H-NMR (DMSO-d6) δ: 1.78-1.82 (2H, m), 2.53-2.56 (2H, m), 2.85-2.87 (2H, m), 2.96 (3H, s), 3.15-3.17 (2H, m), 3.87-3.91 (2H, m), 7.31-7.33 (1H, m), 7.50-7.52 (2H, m), 9.32-9.35 (1H, m), 9.50-9.54 (1H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.78-1.82 (2H, m), 2.53-2.56 (2H, m), 2.85-2.87 (2H, m), 2.96 (3H, s), 3.15-3.17 ( 2H, m), 3.87-3.91 (2H, m), 7.31-7.33 (1H, m), 7.50-7.52 (2H, m), 9.32-9.35 (1H, m), 9.50-9.54 (1H, m).
実施例130〜134:
実施例75(M)と同様にして、参考例11の化合物35を用いて、表21に示す実施例130〜134の化合物を製造した。
Examples 130-134:
In the same manner as in Example 75 (M), using the compound 35 of Reference Example 11, the compounds of Examples 130 to 134 shown in Table 21 were produced.
実施例135:
3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−4−[5−(トリフルオロメチル)ピリジン−2−イル]ベンゾ[d]オキサゾール−2(3H)−オン
実施例75(M)と同様の方法にて、参考例11の化合物35を用いて、表題化合物を製造した。ただし、工程(iii)の塩酸塩化の操作は行わず、遊離体として得た。
Example 135:
3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) -4- [5- (trifluoromethyl) pyridin-2-yl] benzo [d] oxazol-2 (3H) -one Example The title compound was produced using compound 35 of Reference Example 11 in the same manner as in 75 (M). However, the procedure of hydrochloric acid chlorination in step (iii) was not performed, and the product was obtained as a free form.
1H-NMR (DMSO-d6) δ: 1.15-1.18 (2H, m), 2.08-2.10 (2H, m), 2.66-2.74 (2H, m), 2.92 (3H, s), 3.02-3.04 (2H, m), 3.72-3.74 (2H, m), 7.28-7.30 (2H, m), 7.51-7.53 (1H, m), 7.95-7.98 (1H, m), 8.39-8.41 (1H, m), 9.12 (1H, br s). 1 H-NMR (DMSO-d 6 ) δ: 1.15-1.18 (2H, m), 2.08-2.10 (2H, m), 2.66-2.74 (2H, m), 2.92 (3H, s), 3.02-3.04 ( 2H, m), 3.72-3.74 (2H, m), 7.28-7.30 (2H, m), 7.51-7.53 (1H, m), 7.95-7.98 (1H, m), 8.39-8.41 (1H, m), 9.12 (1H, br s).
実施例136〜142:
実施例75(M)と同様にして、市販の6−ブロモベンズオキサゾロンを用いて、表22に示す実施例136〜142の化合物を製造した。
Examples 136-142:
In the same manner as in Example 75 (M), compounds of Examples 136 to 142 shown in Table 22 were produced using commercially available 6-bromobenzoxazolone.
実施例143:
3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−6−[5−(トリフルオロメチル)ピリジン−2−イル]ベンゾ[d]オキサゾール−2(3H)−オン
実施例75(M)と同様の方法にて、市販の6−ブロモベンズオキサゾロンを用いて、表題化合物を製造した。ただし、工程(iii)の塩酸塩化の操作は行わず、遊離体として得た。
Example 143:
3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) -6- [5- (trifluoromethyl) pyridin-2-yl] benzo [d] oxazol-2 (3H) -one Example The title compound was prepared in the same manner as in 75 (M) using commercially available 6-bromobenzoxazolone. However, the procedure of hydrochloric acid chlorination in step (iii) was not performed, and the product was obtained as a free form.
1H-NMR (DMSO-d6) δ: 1.82-1.84 (2H, m), 2.49-2.58 (2H, m), 2.86-2.89 (2H, m), 2.98 (3H, s), 3.18-3.20 (2H, m), 3.90-3.92 (2H, m), 7.46-7.48 (1H, m), 8.11-8.14 (2H, m), 8.23-8.28 (2H, m), 9.01 (1H, br s). 1 H-NMR (DMSO-d 6 ) δ: 1.82-1.84 (2H, m), 2.49-2.58 (2H, m), 2.86-2.89 (2H, m), 2.98 (3H, s), 3.18-3.20 ( 2H, m), 3.90-3.92 (2H, m), 7.46-7.48 (1H, m), 8.11-8.14 (2H, m), 8.23-8.28 (2H, m), 9.01 (1H, br s).
実施例144〜148:
実施例75(M)と同様にして、参考例12の化合物37を用いて、表23に示す実施例144〜148の化合物を製造した。
Examples 144-148:
In the same manner as in Example 75 (M), compounds of Examples 144 to 148 shown in Table 23 were produced using compound 37 of Reference Example 12.
実施例149:
3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)−7−[5−(トリフルオロメチル)ピリジン−2−イル]ベンゾ[d]オキサゾール−2(3H)−オンの製造
実施例75(M)と同様の方法にて、参考例12の化合物37を用いて、表題化合物を製造した。ただし、工程(iii)の塩酸塩化の操作は行わず、遊離体として得た。
Example 149:
Preparation of 3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) -7- [5- (trifluoromethyl) pyridin-2-yl] benzo [d] oxazol-2 (3H) -one The title compound was produced in the same manner as in Example 75 (M) using compound 37 of Reference Example 12. However, the procedure of hydrochloric acid chlorination in step (iii) was not performed, and the product was obtained as a free form.
1H-NMR (DMSO-d6) δ: 1.80-1.85 (2H, m), 2.54-2.56 (2H, m), 2.87-2.89 (2H, m), 2.98 (3H, s), 3.18-3.20 (2H, m), 3.85-3.95 (2H, m), 7.31-7.48 (2H, m), 7.91-7.94 (1H, m), 8.25-8.28 (1H, m), 8.39-8.42 (1H, m), 9.12 (1H, br s).
実施例150(Q):
5−(4−フルオロフェニル)−3−(3−{[2−(メチルスルホニル)エチル]アミノ}ブチル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
1 H-NMR (DMSO-d 6 ) δ: 1.80-1.85 (2H, m), 2.54-2.56 (2H, m), 2.87-2.89 (2H, m), 2.98 (3H, s), 3.18-3.20 ( 2H, m), 3.85-3.95 (2H, m), 7.31-7.48 (2H, m), 7.91-7.94 (1H, m), 8.25-8.28 (1H, m), 8.39-8.42 (1H, m), 9.12 (1H, br s).
Example 150 (Q):
5- (4-Fluorophenyl) -3- (3-{[2- (methylsulfonyl) ethyl] amino} butyl) benzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
参考例7の化合物26(0.280g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物I−q(0.365g)を得た。
Step (i):
Compound Iq (0.365 g) was obtained using Compound 26 (0.280 g) of Reference Example 7 according to the same method as in Step (ii) of Reference Example 14.
工程(ii):
化合物I−q(0.120g)を用いて、実施例1(A)の工程(ii)と同様の方法に従い、化合物II−q(0.077g)を得た。
Step (ii):
Compound II-q (0.077 g) was obtained using compound Iq (0.120 g) according to the same method as in step (ii) of Example 1 (A).
工程(iii):
化合物II−q(0.077g)および4mol/L塩化水素−酢酸エチル溶液(0.40mL)の混合物を室温で1時間攪拌した。固体をろ取し、表題化合物III−q(0.080g)を得た。
Step (iii):
A mixture of Compound II-q (0.077 g) and 4 mol / L hydrogen chloride-ethyl acetate solution (0.40 mL) was stirred at room temperature for 1 hour. The solid was collected by filtration to give the title compound III-q (0.080 g).
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.1Hz), 1.88-1.91 (1H, m), 2.25-2.28 (1H, m), 3.10 (3H, s), 3.32-3.35 (3H, m), 3.47-3.50 (2H, m), 3.99-4.02 (2H, m), 7.30-7.33 (2H, m), 7.42-7.45 (2H, m), 7.67-7.77 (3H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.36 (3H, d, J = 6.1 Hz), 1.88-1.91 (1H, m), 2.25-2.28 (1H, m), 3.10 (3H, s), 3.32 -3.35 (3H, m), 3.47-3.50 (2H, m), 3.99-4.02 (2H, m), 7.30-7.33 (2H, m), 7.42-7.45 (2H, m), 7.67-7.77 (3H, m).
実施例151〜153:
実施例150(Q)と同様にして、表24に示す実施例151〜153の化合物を製造した。
Examples 151-153:
In the same manner as in Example 150 (Q), the compounds of Examples 151 to 153 shown in Table 24 were produced.
実施例154:
3−(3−((2−(メチルスルホニル)エチル)アミノ)ブチル)−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン
実施例150(Q)と同様の方法にて、表題化合物を製造した。ただし、工程(iii)の塩酸塩化の操作は行わず、遊離体として得た。
Example 154:
3- (3-((2- (methylsulfonyl) ethyl) amino) butyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one Example 150 (Q) In the same manner as described above, the title compound was produced. However, the procedure of hydrochloric acid chlorination in step (iii) was not performed, and the product was obtained as a free form.
1H-NMR (DMSO-d6) δ: 1.04 (3H, d, J = 6.2 Hz), 1.73-1.76 (3H, m), 2.48-2.68 (2H, m), 2.98 (3H, s), 3.14-3.16 (2H, m), 3.93-3.96 (2H, m), 7.47-7.48 (2H, m), 7.67 (1H, br s), 7.82-7.92 (4H, m). 1 H-NMR (DMSO-d 6 ) δ: 1.04 (3H, d, J = 6.2 Hz), 1.73-1.76 (3H, m), 2.48-2.68 (2H, m), 2.98 (3H, s), 3.14 -3.16 (2H, m), 3.93-3.96 (2H, m), 7.47-7.48 (2H, m), 7.67 (1H, br s), 7.82-7.92 (4H, m).
実施例155(R):
5−(3,3−ジフルオロシクロブチル)−3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩
Example 155 (R):
5- (3,3-Difluorocyclobutyl) -3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) benzo [d] oxazol-2 (3H) -one hydrochloride
工程(i):
化合物I−r(10.0g)、炭酸セシウム(44.0g)、臭化ベンジル(10.4mL)およびアセトン(100mL)の混合物を2時間加熱還流した。反応液を減圧濃縮し、残渣に水を加え酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、濾過した後、ろ液を減圧濃縮した。得られた固体をヘキサン中で懸濁攪拌後、ろ取し、化合物II−r(15.0g)を得た。
Step (i):
A mixture of Compound Ir (10.0 g), cesium carbonate (44.0 g), benzyl bromide (10.4 mL) and acetone (100 mL) was heated to reflux for 2 hours. The reaction mixture was concentrated under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate and filtered, and then the filtrate was concentrated under reduced pressure. The obtained solid was suspended and stirred in hexane and collected by filtration to obtain Compound II-r (15.0 g).
工程(ii):
化合物II−r(12.0g)、ビニルボロン酸無水物ピリジン錯体(8.70g)、フッ化カリウム(7.00g)、テトラキストリフェニルホスフィンパラジウム(3.50g)、水(10mL)および1,4−ジオキサン(50mL)の混合物を90℃にて2時間攪拌した。反応液に水を加え酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過した後、ろ液を減圧濃縮して得られる残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1/1)で精製し、化合物III−r(8.70g)を得た。
Step (ii):
Compound II-r (12.0 g), vinylboronic acid anhydride pyridine complex (8.70 g), potassium fluoride (7.00 g), tetrakistriphenylphosphine palladium (3.50 g), water (10 mL) and 1,4 A mixture of dioxane (50 mL) was stirred at 90 ° C. for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 1/1) to give compound III-r ( 8.70 g) was obtained.
工程(iii):
化合物III−r(8.70g)、亜鉛−銅合金(25.0g)、ジエチルエーテル(10mL)およびジメトキシエタン(30mL)の混合物にトリクロロアセチルクロライド(14ml)を滴下し、室温で12時間攪拌した。混合液をセライト濾過し、ろ液に水を加え室温で1時間攪拌した後、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過、ろ液を減圧濃縮して化合物IV−rを粗生成物として得た。全量を次工程に用いた。
Step (iii):
Trichloroacetyl chloride (14 ml) was added dropwise to a mixture of compound III-r (8.70 g), zinc-copper alloy (25.0 g), diethyl ether (10 mL) and dimethoxyethane (30 mL), and the mixture was stirred at room temperature for 12 hours. . The mixture was filtered through Celite, water was added to the filtrate, and the mixture was stirred at room temperature for 1 hr, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain compound IV-r as a crude product. The whole amount was used in the next step.
工程(iv):
化合物IV−rの粗生成物、亜鉛粉末(16.5g)、塩化アンモニウム(20.0g)およびメタノール(50mL)の混合物を1時間加熱還流した。反応液をセライト濾過し、ろ液を減圧濃縮した。残渣に水を加え酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過後、ろ液を減圧濃縮し、得られる残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1/1)で精製し、化合物V−r(5.50g)を得た。
Step (iv):
A mixture of the crude product of Compound IV-r, zinc powder (16.5 g), ammonium chloride (20.0 g) and methanol (50 mL) was heated to reflux for 1 hour. The reaction solution was filtered through Celite, and the filtrate was concentrated under reduced pressure. Water was added to the residue and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 1/1) to give compound Vr (5 .50 g) was obtained.
工程(v):
化合物V−r(5.50g)のジクロロメタン(20mL)溶液に−78℃で(ジエチルアミノ)サルファートリフルオライド(9.3mL)を滴下した。反応液を室温で3日間攪拌した。反応液を飽和炭酸水素ナトリウム水溶液に滴下し、ジクロロメタンで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過した後、ろ液を減圧濃縮して得られる残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=2/1)で精製し、化合物VI−r(2.20g)を得た。
Step (v):
(Diethylamino) sulfur trifluoride (9.3 mL) was added dropwise at −78 ° C. to a solution of compound Vr (5.50 g) in dichloromethane (20 mL). The reaction was stirred at room temperature for 3 days. The reaction solution was added dropwise to a saturated aqueous sodium hydrogen carbonate solution and extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate and filtered, and then the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 2/1) to give compound VI-r ( 2.20 g) was obtained.
工程(vi):
化合物VI−r(2.20g)、メタノール(15mL)および2mol/L水酸化ナトリウム水溶液(14mL)の混合物を3時間加熱還流した。反応液を1mol/L塩酸で酸性にし、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過した後、ろ液を減圧濃縮した。得られる残渣をヘキサン中懸濁攪拌後、ろ取、乾燥し化合物VII−r(1.50g)を得た。
Step (vi):
A mixture of compound VI-r (2.20 g), methanol (15 mL) and 2 mol / L aqueous sodium hydroxide solution (14 mL) was heated to reflux for 3 hours. The reaction solution was acidified with 1 mol / L hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered, and then the filtrate was concentrated under reduced pressure. The resulting residue was suspended and stirred in hexane, then collected by filtration and dried to obtain Compound VII-r (1.50 g).
工程(vii):
化合物VII−r(0.500g)、ジフェニルホスホリルアジド(0.5mL)、ジイソプロピルエチルアミン(0.4mL)およびトルエン(10mL)の混合物を室温で30分間攪拌後、1.5時間過熱還流した。反応液にエタノール(3mL)を加えさらに1時間加熱還流した。反応液を1mol/L塩酸で酸性にし、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過した後、ろ液を減圧濃縮し得られる残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=2/1)で精製し、化合物VIII−r(0.320g)を得た。
Step (vii):
A mixture of compound VII-r (0.500 g), diphenylphosphoryl azide (0.5 mL), diisopropylethylamine (0.4 mL) and toluene (10 mL) was stirred at room temperature for 30 minutes and then heated to reflux for 1.5 hours. Ethanol (3 mL) was added to the reaction solution, and the mixture was further heated to reflux for 1 hour. The reaction solution was acidified with 1 mol / L hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 2/1) to give compound VIII-r (0 .320 g) was obtained.
工程(viii):
化合物VIII−r(0.320g)及び10% Pd/C(0.040g)およびエタノール(5mL)の混合物を水素雰囲気下、室温で2時間攪拌した。反応液をセライト濾過し、ろ液を減圧濃縮した。得られた残渣をジエチルエーテル及びヘキサンの混合液より再結晶して化合物IX−r(0.150g)を得た。
Step (viii):
A mixture of compound VIII-r (0.320 g) and 10% Pd / C (0.040 g) and ethanol (5 mL) was stirred at room temperature for 2 hours under a hydrogen atmosphere. The reaction solution was filtered through Celite, and the filtrate was concentrated under reduced pressure. The obtained residue was recrystallized from a mixed solution of diethyl ether and hexane to obtain Compound IX-r (0.150 g).
工程(ix):
化合物IX−r(0.150g)、カリウムtert―ブトキシド(0.120g)およびトルエン(5.0mL)の混合物を1時間加熱還流した。反応液を1mol/L塩酸で酸性にし、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過した後、ろ液を減圧濃縮し得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=3/1)で精製し、化合物X−r(0.110g)を得た
Step (ix):
A mixture of compound IX-r (0.150 g), potassium tert-butoxide (0.120 g) and toluene (5.0 mL) was heated to reflux for 1 hour. The reaction solution was acidified with 1 mol / L hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 3/1) to give compound X-r ( 0.110 g) was obtained.
工程(x):
化合物X−r(0.050g)、参考例17の化合物54(0.080g)、トリフェニルホスフィン(0.075g)、アゾジカルボン酸ジイソプロピル(0.053mL)およびトルエン(2.0mL)の混合物を、室温で3時間攪拌した。反応液に水を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過した後、ろ液を減圧濃縮し得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1/1)で精製し、化合物XI−r(0.046g)を得た
Step (x):
A mixture of Compound Xr (0.050 g), Compound 54 (0.080 g) of Reference Example 17, triphenylphosphine (0.075 g), diisopropyl azodicarboxylate (0.053 mL) and toluene (2.0 mL) was prepared. And stirred at room temperature for 3 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered, and then the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 1/1) to give compound XI-r ( 0.046 g) was obtained.
工程(xi):
化合物XI−r(0.046g)および4mol/L塩化水素−1,4−ジオキサン溶液(3.0mL)の混合物を室温で2時間攪拌した。反応液を減圧濃縮して得られた固体をヘキサン及び酢酸エチルの混合液に懸濁させてろ取、乾燥して表題化合物XII−r(0.031g)を得た。
Step (xi):
A mixture of compound XI-r (0.046 g) and 4 mol / L hydrogen chloride-1,4-dioxane solution (3.0 mL) was stirred at room temperature for 2 hours. The solid obtained by concentrating the reaction solution under reduced pressure was suspended in a mixture of hexane and ethyl acetate, collected by filtration and dried to give the title compound XII-r (0.031 g).
1H-NMR (DMSO-d6) δ: 2.00-2.05 (2H, m), 2.64-2.82 (2H, m), 2.92-3.10 (8H, m), 3.42-3.53 (4H, m), 3.91-3.93 (2H, m), 7.06-7.08 (1H, m), 7.29-7.31 (1H, m), 7.38 (1H, br s), 9.11 (2H, br s). 1 H-NMR (DMSO-d 6 ) δ: 2.00-2.05 (2H, m), 2.64-2.82 (2H, m), 2.92-3.10 (8H, m), 3.42-3.53 (4H, m), 3.91- 3.93 (2H, m), 7.06-7.08 (1H, m), 7.29-7.31 (1H, m), 7.38 (1H, br s), 9.11 (2H, br s).
実施例156(S):
5−(3,3−ジフルオロシクロブチル)−3−{1−[2−(メチルスルホニル)エチル]アゼチジン−3−イル}ベンゾ[d]オキサゾール−2(3H)−オン
Example 156 (S):
5- (3,3-Difluorocyclobutyl) -3- {1- [2- (methylsulfonyl) ethyl] azetidin-3-yl} benzo [d] oxazol-2 (3H) -one
工程(i):
化合物X−r(実施例155)(0.300g)およびtert−ブチル 3−ヒドロキシアゼチジン−1−カルバメート(0.277g)を用いて、参考例14の工程(ii)と同様の方法に従い、化合物I−s(0.178g)を得た。
Step (i):
Using compound Xr (Example 155) (0.300 g) and tert-butyl 3-hydroxyazetidine-1-carbamate (0.277 g), following the same method as in step (ii) of Reference Example 14, Compound Is (0.178 g) was obtained.
工程(ii):
化合物I−s(0.178g)を用いて、実施例52(G)の工程(iii)と同様の方法に従い、化合物II−s(0.100g)を得た。
Step (ii):
Compound II-s (0.100 g) was obtained using Compound Is (0.178 g) according to the same method as in step (iii) of Example 52 (G).
工程(iii):
化合物II−s(0.062g)を用いて、参考例17の工程(i)と同様の方法に従い、表題化合物III−s(0.015g)を得た。
Step (iii):
The title compound III-s (0.015 g) was obtained using the compound II-s (0.062 g) according to the same method as in step (i) of Reference Example 17.
1H-NMR (DMSO-D6) δ: 2.69-2.83 (2H, m), 2.94-3.03 (2H, m), 3.11 (3H, s), 3.35-3.44 (6H, m), 3.57 (2H, t, J = 7.7 Hz), 4.19 (2H, t, J = 5.6 Hz), 7.07 (1H, d, J = 8.3 Hz), 7.30 (1H, d, J = 8.3 Hz), 7.50 (1H, s). 1 H-NMR (DMSO-D 6 ) δ: 2.69-2.83 (2H, m), 2.94-3.03 (2H, m), 3.11 (3H, s), 3.35-3.44 (6H, m), 3.57 (2H, t, J = 7.7 Hz), 4.19 (2H, t, J = 5.6 Hz), 7.07 (1H, d, J = 8.3 Hz), 7.30 (1H, d, J = 8.3 Hz), 7.50 (1H, s) .
実施例157〜161:
実施例156(S)と同様にして、表25に示す実施例157〜161の化合物を製造した。
Examples 157-161:
In the same manner as in Example 156 (S), the compounds of Examples 157 to 161 shown in Table 25 were produced.
実施例162(T):
2−(3−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)アゼチジン−1−イル)アセトアミド
Example 162 (T):
2- (3- (5- (3,3-Difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) azetidin-1-yl) acetamide
工程(i):
化合物II−s(実施例156)(0.022g)、2−ブロモアセトアミド(0.014g)、ジイソプロピルエチルアミン(0.036mL)およびTHF(1.0mL)の混合物を60℃にて3時間攪拌した。反応液に水を加え酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥、濾過した後、ろ液を減圧濃縮して得られる残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=95/5)で精製し、表題化合物I−t(0.016g)を得た。
Step (i):
A mixture of Compound II-s (Example 156) (0.022 g), 2-bromoacetamide (0.014 g), diisopropylethylamine (0.036 mL) and THF (1.0 mL) was stirred at 60 ° C. for 3 hours. . Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 95/5) to give the title compound It ( 0.016 g) was obtained.
1H-NMR (CDCl3) δ: 2.58-2.70 (2H, m), 2.99-3.10 (2H, m), 3.33 (2H, s), 3.35-3.46 (1H, m), 3.87-3.93 (4H, m), 4.84-4.91 (1H, m), 5.75 (1H, br s), 6.68 (1H, br s), 6.98-7.00 (1H, m), 7.14-7.18 (2H, m). 1 H-NMR (CDCl 3 ) δ: 2.58-2.70 (2H, m), 2.99-3.10 (2H, m), 3.33 (2H, s), 3.35-3.46 (1H, m), 3.87-3.93 (4H, m), 4.84-4.91 (1H, m), 5.75 (1H, br s), 6.68 (1H, br s), 6.98-7.00 (1H, m), 7.14-7.18 (2H, m).
実施例163〜164:
実施例156(S)、162(T)および100(MM)を参考にして、表26に示す実施例163〜164の化合物を製造した。
Examples 163 to 164:
The compounds of Examples 163 to 164 shown in Table 26 were produced with reference to Examples 156 (S), 162 (T) and 100 (MM).
実施例165(U):
5−(3,3−ジフルオロシクロブチル)−3−{3−[(2−ヒドロキシ−2−メチルプロピル)アミノ]プロピル}ベンゾ[d]オキサゾール−2(3H)−オン
Example 165 (U):
5- (3,3-Difluorocyclobutyl) -3- {3-[(2-hydroxy-2-methylpropyl) amino] propyl} benzo [d] oxazol-2 (3H) -one
工程(i):
化合物X−r(実施例155)(0.451g)および3−ブロモ−1,1−ジメトキシプロパン(0.549g)を用いて、実施例32(E)の工程(i)と同様の方法に従い化合物I−u(0.655g)を得た。
Step (i):
Using compound Xr (Example 155) (0.451 g) and 3-bromo-1,1-dimethoxypropane (0.549 g) according to the same method as in step (i) of Example 32 (E) Compound Iu (0.655 g) was obtained.
工程(ii):
化合物I−u(0.327g)を用いて、実施例Eの工程(iii)と同様の方法に従い、化合物II−uを粗生成物として得た。全量を次の反応に用いた。
Step (ii):
Compound II-u was obtained as a crude product in the same manner as in step (iii) of Example E using Compound Iu (0.327 g). The whole amount was used for the next reaction.
工程(iii):
化合物II−uの粗生成物および1−アミノ−2−メチルプロパン−2−オール(0.178g)を用いて、参考例17の工程(i)と同様の方法に従い、表題化合物III−u(0.064g)を得た。
Step (iii):
Using the crude product of compound II-u and 1-amino-2-methylpropan-2-ol (0.178 g), in the same manner as in step (i) of Reference Example 17, the title compound III-u ( 0.064 g) was obtained.
1H-NMR (DMSO-D6) δ: 1.17 (6H, s), 2.06-2.16 (2H, m), 2.64-2.84 (4H, m), 2.89-3.11 (4H, m), 3.32 (2H, s), 3.39-3.54 (1H, m), 3.89-3.94 (2H, m), 5.15 (1H, s), 7.06-7.09 (1H, m), 7.29-7.31 (1H, m), 7.39 (1H, m). 1 H-NMR (DMSO-D 6 ) δ: 1.17 (6H, s), 2.06-2.16 (2H, m), 2.64-2.84 (4H, m), 2.89-3.11 (4H, m), 3.32 (2H, s), 3.39-3.54 (1H, m), 3.89-3.94 (2H, m), 5.15 (1H, s), 7.06-7.09 (1H, m), 7.29-7.31 (1H, m), 7.39 (1H, m).
実施例155(R)の化合物X−rは以下方法によっても製造される。
5−(3,3−ジフルオロシクロブチル)ベンゾ[d]オキサゾール−2(3H)−オン(X−r)
Compound Xr of Example 155 (R) is also prepared by the following method.
5- (3,3-Difluorocyclobutyl) benzo [d] oxazol-2 (3H) -one (Xr)
実施例166(V):
4−(4,4−ジクロロ−3,3−ジフルオロシクロ−1−ブテン−1−イル)−1−メトキシ−2−ニトロベンゼン(III−v)
工程(i):
化合物I−v(0.93g)および1,1−ジクロロ−2,2−ジフルオロエテン(9.35g)を耐圧容器に封入し、130℃で7時間撹拌した。室温に冷却後、減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=9/1〜4/1)で精製し、化合物II−v(1.00g)を得た。
Example 166 (V):
4- (4,4-Dichloro-3,3-difluorocyclo-1-buten-1-yl) -1-methoxy-2-nitrobenzene (III-v)
Step (i):
Compound Iv (0.93 g) and 1,1-dichloro-2,2-difluoroethene (9.35 g) were sealed in a pressure vessel and stirred at 130 ° C. for 7 hours. After cooling to room temperature, the residue obtained by concentration under reduced pressure was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 9/1 to 4/1) to obtain Compound II-v (1.00 g). It was.
工程(ii):
化合物II−v(0.75g)、酢酸(26.25mL)および無水酢酸(3.75mL)の混合物に、70%硝酸(410mg)及び濃硫酸(331mg)を加え、室温で3時間攪拌した。反応混合液に氷(100g)を加え析出した固体をろ取し、水で洗浄した。50℃で9時間減圧乾燥した固体をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=8/1〜2/1)で精製し、化合物III−v(0.70g)を得た。
Step (ii):
70% nitric acid (410 mg) and concentrated sulfuric acid (331 mg) were added to a mixture of compound II-v (0.75 g), acetic acid (26.25 mL) and acetic anhydride (3.75 mL), and the mixture was stirred at room temperature for 3 hours. Ice (100 g) was added to the reaction mixture, and the precipitated solid was collected by filtration and washed with water. The solid dried under reduced pressure at 50 ° C. for 9 hours was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 8/1 to 2/1) to obtain Compound III-v (0.70 g).
1H-NMR (CDCl3, 400 MHz) δ4.05 (s, 3H), 6.43 (dd, J = 1.8, 1.8 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 7.98 (dd, J = 8.8, 2.2 Hz, 1H), 8.21 (d, J = 2.2 Hz, 1H) 1 H-NMR (CDCl 3 , 400 MHz) δ4.05 (s, 3H), 6.43 (dd, J = 1.8, 1.8 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 7.98 (dd, J = 8.8, 2.2 Hz, 1H), 8.21 (d, J = 2.2 Hz, 1H)
実施例167(V):
4−(4,4−ジクロロ−3,3−ジフルオロシクロ−1−ブテン−1−イル)−2−ニトロフェノール(IV−v)
工程(iii):
化合物III−v(0.62g)のジクロロメタン(9mL)溶液に氷冷下、1mol/L 三臭化ホウ素(ジクロロメタン溶液、2.4mL)を加え1.5時間撹拌した。反応混合物に水を滴下し、酢酸エチルを加え、室温に昇温した。ヘキサンを加え分液後、得られた有機層を水及び飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧濃縮して得られた固体をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=8/1〜2/1)で精製し、化合物IV−v(0.46g)を得た。
Example 167 (V):
4- (4,4-Dichloro-3,3-difluorocyclo-1-buten-1-yl) -2-nitrophenol (IV-v)
Step (iii):
To a solution of compound III-v (0.62 g) in dichloromethane (9 mL) was added 1 mol / L boron tribromide (dichloromethane solution, 2.4 mL) under ice-cooling, and the mixture was stirred for 1.5 hours. Water was added dropwise to the reaction mixture, ethyl acetate was added, and the temperature was raised to room temperature. Hexane was added for liquid separation, and the obtained organic layer was washed with water and saturated brine. The solid obtained after drying over anhydrous sodium sulfate and concentration under reduced pressure was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 8/1 to 2/1) to give compound IV-v (0.46 g). Got.
1H-NMR (CDCl3, 400 MHz) δ6.46 (dd, J = 1.8, 1.8 Hz, 1H), 7.32 (d, J = 8.8 Hz, 1H), 7.96 (dd, J = 8.8, 2.2 Hz, 1H), 8.55 (d, J = 2.2 Hz, 1H), 10.88 (s, 1H).
1 H-NMR (CDCl 3 , 400 MHz) δ6.46 (dd, J = 1.8, 1.8 Hz, 1H), 7.32 (d, J = 8.8 Hz, 1H), 7.96 (dd, J = 8.8, 2.2 Hz, 1H), 8.55 (d, J = 2.2 Hz, 1H), 10.88 (s, 1H).
実施例168(V):
2−アミノ−4−(3,3−ジフルオロシクロブチル)フェノール(V−v)
工程(iv):
化合物IV−v(0.46g)を用いて、実施例27の工程(i)と同様の方法に従い、化合物V−v(0.33g)を得た。
Example 168 (V):
2-Amino-4- (3,3-difluorocyclobutyl) phenol (Vv)
Step (iv):
Compound Vv (0.33 g) was obtained using Compound IV-v (0.46 g) according to the same method as in step (i) of Example 27.
1H-NMR (CD3OD, 400 MHz) δ2.52-2.69 (m, 2H), 2.91-3.06 (m, 2H), 3.28-3.42 (m, 1H), 6.98 (d, J = 8.4 Hz, 1H), 7.21 (d, J = 2.2 Hz, 1H), 7.23 (dd, J = 8.4, 2.2 Hz, 1H).
1 H-NMR (CD 3 OD, 400 MHz) δ2.52-2.69 (m, 2H), 2.91-3.06 (m, 2H), 3.28-3.42 (m, 1H), 6.98 (d, J = 8.4 Hz, 1H), 7.21 (d, J = 2.2 Hz, 1H), 7.23 (dd, J = 8.4, 2.2 Hz, 1H).
工程(v):
化合物V−v(0.33g)およびテトラヒドロフラン(10mL)の混合物に1,1’−カルボニルジイミダゾール(0.377g)を加え、還流下1.5時間撹拌した。室温に冷却後、酢酸エチルで希釈し、1mol/L塩酸を加え分液した。得られた有機層を水、飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧濃縮して得られた固体をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=4/1〜1/1)で精製し、表題化合物X−r(0.24g)を得た。
Step (v):
1,1′-carbonyldiimidazole (0.377 g) was added to a mixture of compound Vv (0.33 g) and tetrahydrofuran (10 mL), and the mixture was stirred under reflux for 1.5 hours. After cooling to room temperature, the mixture was diluted with ethyl acetate, and 1 mol / L hydrochloric acid was added to separate the layers. The obtained organic layer was washed with water, saturated aqueous sodium hydrogen carbonate solution and saturated brine. The solid obtained after drying over anhydrous sodium sulfate and concentration under reduced pressure was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 4/1 to 1/1) to give the title compound Xr (0.24 g). )
1H-NMR (CDCl3, 400 MHz) δ2.57-2.73 (m, 2H), 2.96-3.10 (m, 2H), 3.42 (ddddd, J = 8.8, 8.8, 8.8, 8.8, 2.4 Hz, 1H), 6.99 (ddd, J = 8.7, 1.8, 0.6 Hz, 1H), 6.99 (dd, J = 1.8, 0.6 Hz, 1H), 7.17 (d, J = 8.7 Hz, 1H). 1H-NMR (CDCl 3 , 400 MHz) δ2.57-2.73 (m, 2H), 2.96-3.10 (m, 2H), 3.42 (ddddd, J = 8.8, 8.8, 8.8, 8.8, 2.4 Hz, 1H), 6.99 (ddd, J = 8.7, 1.8, 0.6 Hz, 1H), 6.99 (dd, J = 1.8, 0.6 Hz, 1H), 7.17 (d, J = 8.7 Hz, 1H).
実施例155(R)の化合物XII−rは以下方法によっても製造される。
5−(3,3−ジフルオロシクロブチル)−3−(3−{[2−(メチルスルホニル)エチル]アミノ}プロピル)ベンゾ[d]オキサゾール−2(3H)−オン 塩酸塩(XII−r)
Compound XII-r of Example 155 (R) is also prepared by the following method.
5- (3,3-difluorocyclobutyl) -3- (3-{[2- (methylsulfonyl) ethyl] amino} propyl) benzo [d] oxazol-2 (3H) -one hydrochloride (XII-r)
実施例169(W):
tert−ブチル (3−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)プロピル)カルバメート(I−w)の製造
工程(i):
1−ブロモ−3,3−ジフルオロシクロブタン(0.130g)、5.0g/dL リーケ亜鉛(THF溶液、1.2mL)およびTHF(0.76mL)の混合物に内部標準液としてドデカン(0.1mL)を加え室温にて5時間撹拌した。その後、化合物I−l(実施例72)(0.225g)、Pd−PEPPSI−IPent(0.0146g)のTHF(1.5mL)溶液を加え室温にて16時間撹拌した。更にPd−PEPPSI−IPent(0.0149g)を追加し室温にて9時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出し、有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=9/1〜1/1)で精製し、化合物I−w(0.0631g)を得た。
Example 169 (W):
Production process (i) of tert-butyl (3- (5- (3,3-difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) propyl) carbamate (Iw):
Dodecane (0.1 mL) as an internal standard solution in a mixture of 1-bromo-3,3-difluorocyclobutane (0.130 g), 5.0 g / dL Likezinc (THF solution, 1.2 mL) and THF (0.76 mL) ) And stirred at room temperature for 5 hours. Thereafter, a solution of compound I-1 (Example 72) (0.225 g) and Pd-PEPPSI-IPent (0.0146 g) in THF (1.5 mL) was added, and the mixture was stirred at room temperature for 16 hours. Further, Pd-PEPPSI-IPent (0.0149 g) was added and stirred at room temperature for 9 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 9/1 to 1/1) to obtain Compound I-w (0.0631 g).
1H-NMR (CDCl3, 400 MHz) δ1.43 (s, 9H), 1.95 (m, 2H), 2.59-2.75 (m, 2H), 2.97-3.10 (m, 2H), 3.18 (q, J = 6.5 Hz, 2H), 3.42 (m, 1H), 3.90 (t, J = 6.5 Hz, 2H), 5.05 (brs, 1H), 6.84 (d, J = 1.0 Hz, 1H), 6.98 (dd, J = 8.0, 1.0 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H). 1 H-NMR (CDCl 3 , 400 MHz) δ1.43 (s, 9H), 1.95 (m, 2H), 2.59-2.75 (m, 2H), 2.97-3.10 (m, 2H), 3.18 (q, J = 6.5 Hz, 2H), 3.42 (m, 1H), 3.90 (t, J = 6.5 Hz, 2H), 5.05 (brs, 1H), 6.84 (d, J = 1.0 Hz, 1H), 6.98 (dd, J = 8.0, 1.0 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H).
工程(ii):
化合物I−w(0.380g)を用いて、実施例71(K)の工程(iii)と同様の方法に従い、化合物II−w(0.240g)を得た。
Step (ii):
Compound II-w (0.240 g) was obtained using Compound Iw (0.380 g) according to the same method as in step (iii) of Example 71 (K).
工程(iii):
化合物II−w(0.050g)、メチルビニルスルホン(0.021ml)およびジイソプロピルエチルアミン(0.080ml)の混合物を、室温で4時間攪拌した。反応混合物を減圧濃縮し得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/メタノール=100/0〜95/5)で精製した。得られた化合物および4mol/L塩化水素−酢酸エチル溶液(1.0mL)の混合物を室温で1時間攪拌した。反応液を減圧濃縮し表題化合物XII−r(0.043g)を得た。
Step (iii):
A mixture of Compound II-w (0.050 g), methyl vinyl sulfone (0.021 ml) and diisopropylethylamine (0.080 ml) was stirred at room temperature for 4 hours. The residue obtained by concentrating the reaction mixture under reduced pressure was purified by silica gel column chromatography (eluent: chloroform / methanol = 100/0 to 95/5). A mixture of the obtained compound and 4 mol / L hydrogen chloride-ethyl acetate solution (1.0 mL) was stirred at room temperature for 1 hour. The reaction solution was concentrated under reduced pressure to obtain the title compound XII-r (0.043 g).
試験例1
電位依存性Naチャネル遺伝子発現細胞を用いた膜電位変化の測定
ヒトSCN9A(テトラサイクリン誘導型)あるいはヒトSCN10A遺伝子を安定発現させたCHO−K1細胞株を用いて、それぞれNav1.7、Nav1.8電流由来の膜電位変化に対する本発明化合物の阻害作用を評価した。
すなわち、膜電位感受性色素を15〜30分間ロード(37℃,5%CO2)したコンフルエントな状態の発現細胞に被験化合物を添加し、そのおよそ5分後に、ベラトリジン(Naチャネル開口薬)を加えることで誘導される蛍光強度変化を測定し、Nav1.7あるいはNav1.8を介した膜電位上昇に対する阻害作用を評価した。測定装置にはFLIPRTETRA(Molecular Device)を使用した。各チャネルの試験条件を表27に示す。
Test example 1
Measurement of membrane potential change using voltage-dependent Na channel gene-expressing cells Using human SCN9A (tetracycline-inducible) or CHO-K1 cell line stably expressing human SCN10A gene, Nav1.7 and Nav1.8 currents, respectively. The inhibitory action of the compound of the present invention on the change in membrane potential derived from the origin was evaluated.
That is, a test compound is added to a confluent expression cell loaded with a membrane potential sensitive dye for 15 to 30 minutes (37 ° C., 5% CO 2 ), and veratridine (Na channel opener) is added approximately 5 minutes later. The change in fluorescence intensity induced by this was measured, and the inhibitory effect on the increase in membrane potential via Nav1.7 or Nav1.8 was evaluated. FLIPR TETRA (Molecular Device) was used as a measuring device. Table 27 shows the test conditions for each channel.
被験化合物のNaチャネル阻害率は以下の計算式によって求めた。
Nav1.7阻害率(%)=100×[(評価化合物非存在下でのベラトリジン刺激のピーク値)−(評価化合物存在下でのベラトリジン刺激のピーク値)]/[(評価化合物非存在下でのベラトリジン刺激のピーク値)−(刺激なしのピーク値)]
Nav1.8阻害率(%)=100×[(評価化合物非存在下でのベラトリジン刺激のAUC)−(評価化合物存在下でのベラトリジン刺激のAUC)]/[(評価化合物非存在下でのベラトリジン刺激のAUC)−(刺激なしのAUC)]
The Na channel inhibition rate of the test compound was determined by the following calculation formula.
Nav1.7 inhibition rate (%) = 100 × [(peak value of veratridine stimulation in the absence of evaluation compound) − (peak value of veratridine stimulation in the presence of evaluation compound)] / [(in the absence of evaluation compound) Peak value of veratridine stimulation)-(peak value without stimulation)]
Nav1.8 inhibition rate (%) = 100 × [(veracridine-stimulated AUC in the absence of evaluation compound) − (veratridine-stimulated AUC in the presence of evaluation compound)] / [(veratridine in the absence of evaluation compound) Stimulus AUC)-(AUC without stimulation)]
試験結果
実施例で得られた化合物について、Naチャネルを介した膜電位上昇に対する阻害作用を評価した結果、本発明化合物がNav1.7およびNav1.8の両方に阻害作用を示すことが観察された。化合物濃度が40μmol/Lのときの各アイソフォーム阻害率(%)を表28〜30に示す。なお、表中に(10μmol/L)と記載している場合は、化合物濃度が10μmol/Lのときの各アイソフォーム阻害率(%)を示し、表中に−と記載している場合は、未測定であることを示す。
Test results As a result of evaluating the inhibitory effect on the increase in membrane potential via the Na channel for the compounds obtained in the Examples, it was observed that the compound of the present invention shows an inhibitory action on both Nav1.7 and Nav1.8. . Each isoform inhibition rate (%) when the compound concentration is 40 μmol / L is shown in Tables 28-30. In addition, when (10 μmol / L) is described in the table, each isoform inhibition rate (%) when the compound concentration is 10 μmol / L is shown, and when − is described in the table, Indicates not measured.
Nav1.7阻害 Nav1.7 inhibition
Nav1.7阻害のつづき Continuation of Nav1.7 inhibition
Nav1.8阻害 Nav1.8 inhibition
試験例2
電位依存性Naチャネル遺伝子発現細胞を用いたNaイオン電流の測定
Nav1.7電流は、ヒトSCN9A安定発現細胞を用いて、オートパッチクランプ法により測定した。
・ヒトSCN9A安定発現細胞
テトラサイクリン誘導型SCN9A安定発現細胞はChanTest社より入手した。細胞は、10%ウシ胎児血清、100units/mL Penicilline−Streptomycin、0.01mg/mL Blasticidin、0.4mg/mL Zeocinを含むHam’s F−12培地で継代維持した。測定前日に、1μg/mL テトラサイクリン、100μmol/L Sodium butyrate、10%ウシ胎児血清、100units/mL Penicilline−Streptomycinを含むHam’s F−12培地に交換し、翌日オートパッチクランプ法にてNaイオン電流を測定した。
・Naイオン電流の電気生理学的測定
Naイオン電流は、オートパッチクランプ法により、以下の細胞外液、細胞内液を用いて測定した。
・細胞外液 (mmol/L):NaCl 130、MgCl2 2、CaCl2 2、CdCl2 0.1、NiCl2 0.1、Tetraethylammonium−Cl 18、4−aminopyridine 1、HEPES 10、(NaOHでpH7.4に調整)
・細胞内液 (mmol/L):CsF 120、EGTA 10、NaCl 15、HEPES 10、(CsOHでpH7.2に調整)
刺激パルスの制御およびデータ取得は、EPC10およびPatch Masterソフトウエア(HEKA)を用いて実施した。データのサンプリングは10kHzで実施し、3kHzのローパスフィルタでノイズ除去した。測定はすべて室温で行った。保持電位はNav1.7チャネルの50%不活性化電位(−60mV付近)とし、20ミリ秒の脱分極パルス(+10mV)を1回与えた。被験化合物の阻害率は、脱分極パルスを与えた時のピーク電流が500pA以上の細胞で、かつ、データ取得の終了までホールセルパラメーターの大きな変化がなかった細胞の結果から算出した。被験化合物のNaイオン電流に対する阻害率は、脱分極パルスによって生じた電流ピーク値をもとに、以下の計算式によって求めた。
Naイオン電流阻害率(%)=100×[(評価化合物非存在下での電流ピーク値)−(評価化合物存在下での電流ピーク値)]/(評価化合物非存在下での電流ピーク値)
Test example 2
Measurement of Na Ion Current Using Voltage-Dependent Na Channel Gene-Expressing Cells Nav1.7 current was measured by auto patch clamp method using human SCN9A stably expressing cells.
-Human SCN9A stably expressing cells Tetracycline-derived SCN9A stably expressing cells were obtained from ChanTest. Cells were maintained in Ham's F-12 medium containing 10% fetal bovine serum, 100 units / mL Penicillin-Streptomycin, 0.01 mg / mL Blasticidin, 0.4 mg / mL Zeocin. The day before the measurement, the medium was replaced with Ham's F-12 medium containing 1 μg / mL tetracycline, 100 μmol / L sodium butyrate, 10% fetal bovine serum, 100 units / mL penicillline-streptomycin, and the following day, the Na ion current was obtained by the autopatch clamp method. Was measured.
-Electrophysiological measurement of Na ion current Na ion current was measured by the auto patch clamp method using the following extracellular fluid and intracellular fluid.
-Extracellular fluid (mmol / L): NaCl 130, MgCl2, CaCl2, 0.1 CdCl2, 0.1, NiCl2 0.1, Tetraethylammonium-Cl 18, 4-aminopyridine 1, HEPES 10, (adjusted to pH 7.4 with NaOH) )
Intracellular fluid (mmol / L): CsF 120, EGTA 10, NaCl 15, HEPES 10, (adjusted to pH 7.2 with CsOH)
Stimulation pulse control and data acquisition were performed using EPC10 and Patch Master software (HEKA). Data sampling was performed at 10 kHz, and noise was removed with a 3 kHz low-pass filter. All measurements were performed at room temperature. The holding potential was 50% inactivation potential (near −60 mV) of the Nav1.7 channel, and a 20-millisecond depolarizing pulse (+10 mV) was applied once. The inhibition rate of the test compound was calculated from the results of the cells having a peak current of 500 pA or more when a depolarizing pulse was given and the cells having no significant change in the whole cell parameters until the end of data acquisition. The inhibition rate of the test compound with respect to the Na ion current was determined by the following formula based on the peak current value generated by the depolarization pulse.
Na ion current inhibition rate (%) = 100 × [(current peak value in the absence of evaluation compound) − (current peak value in the presence of evaluation compound)] / (current peak value in the absence of evaluation compound)
試験結果
実施例で得られた化合物について、Naイオン電流に対する阻害作用を評価した結果、本発明化合物がNav1.7に対して阻害作用を示すことが観察された。化合物濃度が10μmol/Lのときの阻害率(%)を表31に示す。
Test Results As a result of evaluating the inhibitory effect on the Na ion current for the compounds obtained in the Examples, it was observed that the compound of the present invention showed an inhibitory action on Nav1.7. Table 31 shows the inhibition rate (%) when the compound concentration is 10 μmol / L.
Nav1.7阻害 Nav1.7 inhibition
試験例3
電位依存性Naチャネル遺伝子発現細胞を用いたNaイオン電流の測定
Nav1.8由来電流は、ヒトSCN10A一過性発現細胞を用いて、マニュアルパッチクランプ法により測定した。
・ヒトSCN10A一過性発現細胞
宿主細胞であるND7/23細胞は、ECACC(European Collection of Cell Cultures社)より入手した。細胞は、10%ウシ胎児血清、100units/mL Penicilline−Streptomycin、2mmol/L GlutaMAX(Invitrogen)を含むDMEM(Dulbecco’s Modified Eagle’s Medium)培地で継代維持した。スライドグラスに播種したND7/23細胞に、ヒトSCN10A遺伝子を組み込んだNav1.8発現プラスミドとGFP発現プラスミド(Promega)をPlus Reagent(Invitrogen社)とLipofectamine 2000(Invitrogen社)を用いて、2mmol/L GlutaMAX(Invitrogen社)を含むDMEM培地中で導入した。4時間後に継代用培地に交換し、翌日マニュアルパッチクランプ法にてNaイオン電流を測定した。
・Naイオン電流の電気生理学的測定
Naイオン電流は、マニュアルパッチクランプ法により、以下の細胞外液、細胞内液を用いて測定した。
・細胞外液(mmol/L):NaCl 130、MgCl2 2、CaCl2 2、CdCl2 0.1、NiCl2 0.1、Tetraethylammonium−Cl 18、4−aminopyridine 1、HEPES(4−(2−hydroxyethyl)−1−piperazineethanesulfonic acid) 10、TTX 0.0005(NaOHでpH7.4に調整)
・細胞内液(mmol/L):CsF 120、EGTA(ethylene glycol tetraacetic acid) 10、NaCl 15、HEPES 10、(CsOHでpH7.2に調整)
刺激パルスの制御およびデータ取得は、Axopatch 200BおよびClampexソフトウエア(Molecular Devices)、または、EPC10およびPatch Masterソフトウエア(HEKA社製)を用いて実施した。データのサンプリングは20kHzで実施し、2kHzのローパスフィルタでノイズ除去した。測定はすべて室温で行った。保持電位はNav1.8チャネルの20%不活性化電位(−85mV付近)とし、20ミリ秒の脱分極パルス(10mV付近)を10Hzの頻度で30回与えた。被験化合物の阻害率は、上記刺激パルス1回目のピーク電流が500pA以上の細胞で、かつ、データ取得の終了までホールセルパラメーターの大きな変化がなかった細胞の結果から算出した。被験化合物のNaイオン電流に対する阻害率は、30回目の脱分極パルスによって生じた電流ピーク値をもとに、以下の計算式によって求めた。
Naイオン電流阻害率(%)=100×[(評価化合物非存在下での電流ピーク値)−(評価化合物存在下での電流ピーク値)]/(評価化合物非存在下での電流ピーク値)
Test example 3
Measurement of Na ion current using voltage-dependent Na channel gene-expressing cells The Nav1.8-derived current was measured by a manual patch clamp method using human SCN10A transiently expressing cells.
-Human SCN10A transient expression cell ND7 / 23 cell which is a host cell was obtained from ECACC (European Collection of Cell Cultures). Cells were passaged in DMEM (Dulbeco's Modified Eagle's Medium) medium containing 10% fetal bovine serum, 100 units / mL Penicillin-Streptomycin, 2 mmol / L GlutaMAX (Invitrogen). Using ND7 / 23 cells seeded on a slide glass, a Nav1.8 expression plasmid incorporating a human SCN10A gene and a GFP expression plasmid (Promega) were added at 2 mmol / L using Plus Reagent (Invitrogen) and Lipofectamine 2000 (Invitrogen). It introduce | transduced in the DMEM culture medium containing GlutaMAX (Invitrogen). After 4 hours, the medium was replaced with the subculture medium, and the Na ion current was measured by the manual patch clamp method the next day.
Electrophysiological measurement of Na ion current Na ion current was measured by the manual patch clamp method using the following extracellular fluid and intracellular fluid.
Extracellular fluid (mmol / L): NaCl 130, MgCl 2 2, CaCl 2 2, CdCl 2 0.1, NiCl 2 0.1, Tetraethylammonium-Cl 18, 4-aminopyridine 1, HEPES (4- (2- hydroxyethyl) -1-piperazine etheresufonic acid) 10, TTX 0.0005 (adjusted to pH 7.4 with NaOH)
Intracellular fluid (mmol / L): CsF 120, EGTA (ethylene tetraacetic acid) 10, NaCl 15, HEPES 10, (adjusted to pH 7.2 with CsOH)
Stimulation pulse control and data acquisition were performed using Axopatch 200B and Clampex software (Molecular Devices) or EPC10 and Patch Master software (HEKA). Data sampling was performed at 20 kHz, and noise was removed with a 2 kHz low-pass filter. All measurements were performed at room temperature. The holding potential was 20% inactivation potential (near -85 mV) of the Nav1.8 channel, and a 20 millisecond depolarizing pulse (near 10 mV) was applied 30 times at a frequency of 10 Hz. The inhibition rate of the test compound was calculated from the results of cells in which the peak current at the first stimulation pulse was 500 pA or more and the whole cell parameters did not change greatly until the end of data acquisition. The inhibition rate of the test compound with respect to the Na ion current was determined by the following formula based on the current peak value generated by the 30th depolarization pulse.
Na ion current inhibition rate (%) = 100 × [(current peak value in the absence of evaluation compound) − (current peak value in the presence of evaluation compound)] / (current peak value in the absence of evaluation compound)
試験結果
実施例で得られた化合物について、Naイオン電流に対する阻害作用を評価した結果、本発明化合物がNav1.8に対して阻害作用を示すことが観察された。化合物濃度が10μmol/Lのときの阻害率(%)を表32に示す。なお、表中に5umol/Lと記載している場合は、化合物濃度が5umol/Lのときの阻害率(%)を示す。
Test Results As a result of evaluating the inhibitory effect on the Na ion current for the compounds obtained in the Examples, it was observed that the compound of the present invention showed an inhibitory action on Nav1.8. Table 32 shows the inhibition rate (%) when the compound concentration was 10 μmol / L. In addition, when it describes as 5 umol / L in a table | surface, the inhibition rate (%) when a compound concentration is 5 umol / L is shown.
Nav1.8阻害 Nav1.8 inhibition
本発明の化合物は、Nav1.7および/またはNav1.8の関与する病態、具体的には、神経障害性疼痛、侵害受容性疼痛、炎症性疼痛、小径線維ニューロパチー、肢端紅痛症、発作性激痛症、排尿障害、多発性硬化症などの疾患に対して優れた治療剤として使用しうる。従って、本発明の化合物は、非常に有用な医薬となり得る。 The compounds of the present invention can be used in pathologies involving Nav1.7 and / or Nav1.8, specifically neuropathic pain, nociceptive pain, inflammatory pain, small fiber neuropathy, limb erythema pain, seizures It can be used as an excellent therapeutic agent for diseases such as acute pain, dysuria, and multiple sclerosis. Therefore, the compound of the present invention can be a very useful medicament.
Claims (33)
[式(1)中、
R1およびR2は、各々独立して、水素、C1−6アルキル(該アルキルは、ヒドロキシ、ハロゲン、C1−4アルコキシ、C1−4アルキルスルホニル、C1−4アルキルチオ、アミノカルボニル(該アミノカルボニルのアミノ部分は、置換されていてもよいC1−4アルキルによってモノまたはジ置換されていてもよい)、置換されていてもよい4〜7員のヘテロシクロアルキルおよび置換されていてもよい4〜7員のヘテロシクロアルキルカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニルまたは4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニルおよび該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)を表し、
Lは、C1−6アルキレン(該アルキレンは、C1−4アルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)を表し、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜7員の飽和複素環を形成してもよく、
R3は、ハロゲン、C1−6アルキル、C2−6アルケニル、C2−6アルキニル(該アルキル、該アルケニルおよび該アルキニルは、それぞれハロゲン、置換されていてもよいC3−7シクロアルキル、置換されていてもよい4〜7員のヘテロシクロアルキル、C1−4アルコキシ、C1−4ハロアルコキシ、C1−4アルキルスルホニル、C1−4アルキルチオ、置換されていてもよいC6−10アリールおよび置換されていてもよいC6−10アリールオキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニル、4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、置換されていてもよいC1−4アルキル、C1−4ハロアルキル、置換されていてもよいC1−4アルコキシ、C1−4ハロアルコキシ、シアノおよびC1−4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリールオキシ(該アリールオキシのアリール部分は、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ、C1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシおよびC1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)を表し、
R4は、水素、ハロゲンまたはC1−4アルキルを表す]
で表される化合物またはその薬学的に許容される塩。 Following formula (1);
[In Formula (1),
R 1 and R 2 are each independently hydrogen, C 1-6 alkyl (wherein the alkyl is hydroxy, halogen, C 1-4 alkoxy, C 1-4 alkylsulfonyl, C 1-4 alkylthio, aminocarbonyl ( The amino moiety of the aminocarbonyl may be mono- or di-substituted by optionally substituted C 1-4 alkyl), optionally substituted 4-7 membered heterocycloalkyl and substituted Optionally substituted with 1 to 3 substituents independently selected from the group consisting of 4 to 7-membered heterocycloalkylcarbonyl), C 3-7 cycloalkyl, C 4-7 cycloalkenyl or 4-7 membered heterocycloalkyl (wherein cycloalkyl, the cycloalkenyl and the heterocycloalkyl, C 1-4 each represent an alkyl C 1-4 represents haloalkyl, halogen, hydroxy and C 1-4 optionally substituted with 1-3 substituents independently selected from the group consisting of alkoxy),
L is C 1-6 alkylene (the alkylene is substituted with 1 to 3 substituents independently selected from the group consisting of C 1-4 alkyl, halogen, hydroxy and C 1-4 alkoxy; Where R 1 is C 1-6 alkyl, the carbon atom on R 1 and L together, together with the nitrogen atom to which R 1 is attached, 4-7 membered saturation May form a heterocycle,
R 3 is halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl (the alkyl, the alkenyl and the alkynyl are each halogen, optionally substituted C 3-7 cycloalkyl, Optionally substituted 4-7 membered heterocycloalkyl, C 1-4 alkoxy, C 1-4 haloalkoxy, C 1-4 alkylsulfonyl, C 1-4 alkylthio, optionally substituted C 6-6. 10 aryl and optionally substituted with 1 to 3 substituents are also independently selected from the group consisting of optionally C 6-10 aryloxy substituted), C 3-7 cycloalkyl, C 4 -7 cycloalkenyl, 4-7 membered heterocycloalkyl (wherein cycloalkyl, the cycloalkenyl, the heterocycloalkyl, C 1-4 respectively a Kill, C 1-4 haloalkyl, halogen, independently from the group consisting of hydroxy and C 1-4 alkoxy optionally substituted with 1-3 substituents selected), C 6-10 aryl (the Aryl is halogen, optionally substituted C 1-4 alkyl, C 1-4 haloalkyl, optionally substituted C 1-4 alkoxy, C 1-4 haloalkoxy, cyano and C 1-4 alkoxycarbonyl. Optionally substituted with 1 to 3 substituents independently selected from the group consisting of: C 6-10 aryloxy (the aryl moiety of the aryloxy is halogen, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxy, optionally substituted with 1-3 substituents independently selected from the group consisting of C 1-4 haloalkoxy) the 5-10 membered heteroaryl (wherein heteroaryl is 1 are selected from halogen, C 1-4 alkyl, C 1-4 haloalkyl, independently from the group consisting of C 1-4 alkoxy and C 1-4 haloalkoxy ~ Optionally substituted with 3 substituents),
R 4 represents hydrogen, halogen or C 1-4 alkyl]
Or a pharmaceutically acceptable salt thereof.
置換されていてもよいC1−4アルキルおよび置換されていてもよいC1−4アルコキシにおける置換基が、C1−4アルコキシ、C1−4ハロアルコキシからなる群から独立して選択される1〜2個の置換基であり、
置換されていてもよい4〜7員のヘテロシクロアルおよび置換されていてもよい4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキルにおける置換基が、C1−4アルキル、ハロゲン、オキソおよびヒドロキシからなる群から独立して選択される1〜3個の置換基である、請求項1に記載の化合物またはその薬学的に許容される塩。 The substituent in the optionally substituted C 6-10 aryl, the optionally substituted C 6-10 aryloxy and the optionally substituted C 3-7 cycloalkyl is halogen, C 1-4 alkyl, 1 to 3 substituents independently selected from the group consisting of C 1-4 haloalkyl, C 1-4 alkoxy, C 1-4 haloalkoxy,
Substituents in optionally substituted C 1-4 alkyl and optionally substituted C 1-4 alkoxy are independently selected from the group consisting of C 1-4 alkoxy, C 1-4 haloalkoxy. 1 to 2 substituents,
Substituents in heterocycloalkyl of optionally substituted 4-7 membered heterocycloalkyl and optionally substituted 4-7 membered heterocycloalkylcarbonyl are C 1-4 alkyl, halogen, oxo and hydroxy The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein the compound is 1 to 3 substituents independently selected from the group consisting of:
置換されていてもよいC1−4アルキルおよび置換されていてもよいC1−4アルコキシにおける置換基が、1〜2個のC1−2アルコキシであり、
置換されていてもよい4〜7員のヘテロシクロアルおよび置換されていてもよい4〜7員のヘテロシクロアルキルカルボニルのヘテロシクロアルキルにおける置換基が、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基である、請求項1に記載の化合物またはその薬学的に許容される塩。 Optionally substituted C 6-10 aryl, substituent of the C 3-7 cycloalkyl be a good C 6-10 aryloxy and substituted also be substituted, it is a 1-3 halogens ,
The substituent in the optionally substituted C 1-4 alkyl and the optionally substituted C 1-4 alkoxy is 1-2 C 1-2 alkoxy,
The substituent in the optionally substituted 4-7 membered heterocycloalkyl and optionally substituted 4-7 membered heterocycloalkylcarbonyl heterocycloalkyl is selected from the group consisting of C 1-2 alkyl and oxo. The compound according to claim 1 or a pharmaceutically acceptable salt thereof, which is 1 to 2 substituents independently selected.
Lが、C1−6アルキレン(該アルキレンは、C1−4アルキル、ハロゲン、ヒドロキシおよびC1−4アルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜7員の飽和複素環を形成してもよく、
R3が、ハロゲン、C1−6アルキル、C2−6アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、C3−7シクロアルキル(該シクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)、4〜7員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、C6−10アリールおよびC6−10アリールオキシ(該アリールおよび該アリールオキシは、それぞれ1〜3個のハロゲンによって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−7シクロアルキル、C4−7シクロアルケニル、4〜7員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、それぞれC1−4アルキル、C1−4ハロアルキルおよびハロゲンからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、C1−4アルキル、C1−4アルコキシ、シアノおよびC1−4アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または5〜10員のヘテロアリール(該ヘテロアリールは、ハロゲン、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシおよびC1−4ハロアルコキシからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)であり、
R4が、水素、ハロゲンまたはC1−2アルキルである、請求項1に記載の化合物またはその薬学的に許容される塩。 R 1 and R 2 are each independently hydrogen, C 1-6 alkyl (wherein the alkyl is hydroxy, halogen, C 1-4 alkoxy, C 1-4 alkylsulfonyl, aminocarbonyl (the amino moiety of the aminocarbonyl) May be mono or disubstituted by C 1-4 alkyl), 4-7 membered heterocycloalkyl and 4-7 membered heterocycloalkylcarbonyl (the 4-7 membered heterocycloalkyl and the 4 The heterocycloalkyl part of the -7 membered heterocycloalkylcarbonyl may each be substituted by 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and oxo) Optionally substituted with 1 to 3 substituents independently selected from the group) or 4 to 7 membered heterocycloalkyl The heterocycloalkyl, halogen, and a C 1-4 optionally substituted with 1-3 substituents independently selected from the group consisting of alkoxy),
L is C 1-6 alkylene (the alkylene is substituted with 1 to 3 substituents independently selected from the group consisting of C 1-4 alkyl, halogen, hydroxy and C 1-4 alkoxy; Where R 1 is C 1-6 alkyl, the carbon atoms on R 1 and L are taken together to form a 4-7 membered saturation with the nitrogen atom to which R 1 is attached. May form a heterocycle,
R 3 is halogen, C 1-6 alkyl, C 2-6 alkenyl (the alkyl and the alkenyl are each halogen, C 3-7 cycloalkyl (the cycloalkyl is substituted by 1 to 2 halogens) 4-7 membered heterocycloalkyl, which may be substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and oxo. 1), independently selected from the group consisting of C 6-10 aryl and C 6-10 aryloxy, each of which may be substituted by 1 to 3 halogens to three may be substituted with a substituent), C 3-7 cycloalkyl, C 4-7 cycloalkenyl, 4-7 membered heterocycloalkylene (Said cycloalkyl, said cycloalkenyl, said heterocycloalkyl, optionally substituted with 1-3 substituents selected respectively C 1-4 alkyl, from the group consisting of C 1-4 haloalkyl and halogen independently C 6-10 aryl, wherein the aryl is independently selected from the group consisting of halogen, C 1-4 alkyl, C 1-4 alkoxy, cyano and C 1-4 alkoxycarbonyl Optionally substituted with 5 substituents or 5- to 10-membered heteroaryl, which is halogen, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxy and C 1-4 May be substituted with 1 to 3 substituents independently selected from the group consisting of haloalkoxy),
The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein R 4 is hydrogen, halogen or C 1-2 alkyl.
Lが、C1−4アルキレン(該アルキレンは、C1−2アルキルおよびヒドロキシからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、ここにおいて、R1が、C1−6アルキルであるとき、R1とL上の炭素原子が一緒になって、R1が結合する窒素原子と共に4〜6員の飽和複素環を形成してもよく、
R3が、ハロゲン、C1−4アルキル、C2−4アルケニル(該アルキルおよび該アルケニルは、それぞれハロゲン、C4−6シクロアルキル(該シクロアルキルは、1〜2個のハロゲンによって置換されていてもよい)、4〜6員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、C6−10アリールおよびC6−10アリールオキシ(該アリールおよび該アリールオキシは、それぞれ1〜2個のハロゲンによって置換されていてもよい)からなる群から独立して選択される1〜3個の置換基で置換されていてもよい)、C3−6シクロアルキル、C4−6シクロアルケニル、4〜6員のヘテロシクロアルキル(該シクロアルキル、該シクロアルケニル、該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、C6−10アリール(該アリールは、ハロゲン、C1−2アルキル、C1−2アルコキシ(該アルキルおよび該アルコキシは、それぞれ1〜2個のC1−2アルコキシによって置換されていてもよい)、C1−2ハロアルキル、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、ハロゲンおよびC1−2ハロアルキルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
R4が、水素、ハロゲンまたはC1−2アルキルである、請求項1に記載の化合物またはその薬学的に許容される塩。 R 1 and R 2 are each independently hydrogen, C 1-4 alkyl (wherein the alkyl is hydroxy, C 1-2 alkylsulfonyl, aminocarbonyl (the amino moiety of the aminocarbonyl is represented by C 1-2 alkyl) Optionally substituted) 4-5 membered heterocycloalkyl and 4-6 membered heterocycloalkylcarbonyl (the 4-7 membered heterocycloalkyl and the 4-7 membered heterocycloalkylcarbonyl heterozygous) Each cycloalkyl is optionally selected from the group consisting of 1-2 optionally selected from 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and oxo. Or optionally substituted with 4 to 5 membered heterocycloalkyl, which is substituted with 1 to 2 halo It is also may) be substituted by emissions,
L is C 1-4 alkylene, which alkylene may be substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and hydroxy, wherein , When R 1 is C 1-6 alkyl, R 1 and the carbon atom on L may be taken together to form a 4-6 membered saturated heterocycle with the nitrogen atom to which R 1 is attached. ,
R 3 is halogen, C 1-4 alkyl, C 2-4 alkenyl (the alkyl and the alkenyl are each halogen, C 4-6 cycloalkyl (the cycloalkyl is substituted by 1 to 2 halogens) 4-6 membered heterocycloalkyl, which may be substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and oxo. 1) independently selected from the group consisting of C 6-10 aryl and C 6-10 aryloxy, wherein said aryl and said aryloxy may each be substituted by 1 to 2 halogens to three may be substituted with a substituent), C 3-6 cycloalkyl, C 4-6 cycloalkenyl, 4-6 membered heterocycloalkylene (Said cycloalkyl, said cycloalkenyl, said heterocycloalkyl may be substituted with 1 to 3 halogens), C 6-10 aryl (the aryl, halogen, C 1-2 alkyl, C 1 -2 alkoxy (wherein the alkyl and the alkoxy may each be substituted by 1 to 2 C 1-2 alkoxy), C 1-2 haloalkyl, C 1-2 haloalkoxy, cyano and C 1-2 Optionally substituted with 1 to 3 substituents independently selected from the group consisting of alkoxycarbonyl or 6-membered heteroaryl (wherein the heteroaryl is from the group consisting of halogen and C 1-2 haloalkyl) Optionally substituted with 1 to 2 substituents independently selected),
The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein R 4 is hydrogen, halogen or C 1-2 alkyl.
R2が、C1−4アルキル(該アルキルは、ヒドロキシおよびC1−2アルキルスルホニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
Lが、C1−3アルキレン(該アルキレンは、C1−2アルキルおよびヒドロキシからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
R3が、C3−6シクロアルキル、4〜6員のヘテロシクロアルキル(該シクロアルキルおよび該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、フェニル(該フェニルは、ハロゲン、C1−2アルキル、C1−2アルコキシ、C1−2ハロアルキル、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、1〜2個のC1−2ハロアルキルによって置換されていてもよい)であり、
R4が、水素である、請求項1に記載の化合物またはその薬学的に許容される塩。 R 1 is hydrogen;
R 2 is C 1-4 alkyl (which may be substituted with 1 to 2 substituents independently selected from the group consisting of hydroxy and C 1-2 alkylsulfonyl) or 4 to 5-membered heterocycloalkyl, which may be substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and oxo;
L is C 1-3 alkylene (the alkylene may be substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and hydroxy);
R 3 is C 3-6 cycloalkyl, a 4-6 membered heterocycloalkyl (the cycloalkyl and the heterocycloalkyl may be substituted by 1 to 3 halogens), phenyl (the phenyl is 1 to 3 independently selected from the group consisting of, halogen, C 1-2 alkyl, C 1-2 alkoxy, C 1-2 haloalkyl, C 1-2 haloalkoxy, cyano and C 1-2 alkoxycarbonyl Or 6-membered heteroaryl, which may be substituted by 1 to 2 C 1-2 haloalkyls,
The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein R 4 is hydrogen.
R2が、C1−4アルキル(該アルキルは、ヒドロキシおよびC1−2アルキルスルホニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)または4〜5員のヘテロシクロアルキル(該ヘテロシクロアルキルは、C1−2アルキルおよびオキソからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
Lが、C1−3アルキレンであり、R1のアルキルとL上の炭素原子が一緒になって、R1が結合する窒素原子と共にアゼチジン、ピペリジンまたはピロリジンを形成しており、
R3が、C3−6シクロアルキル、4〜6員のヘテロシクロアルキル(該シクロアルキルおよび該ヘテロシクロアルキルは、1〜3個のハロゲンによって置換されていてもよい)、フェニル(該フェニルは、ハロゲン、C1−2アルキル、C1−2アルコキシ、C1−2ハロアルキル、C1−2ハロアルコキシ、シアノおよびC1−2アルコキシカルボニルからなる群から独立して選択される1〜3個の置換基で置換されていてもよい)または6員のヘテロアリール(該ヘテロアリールは、1〜2個のC1−2ハロアルキルによって置換されていてもよい)であり、
R4が、水素である、請求項1に記載の化合物またはその薬学的に許容される塩。 R 1 is C 1-4 alkyl;
R 2 is C 1-4 alkyl (which may be substituted with 1 to 2 substituents independently selected from the group consisting of hydroxy and C 1-2 alkylsulfonyl) or 4 to 5-membered heterocycloalkyl, which may be substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkyl and oxo;
L is C 1-3 alkylene, the alkyl of R 1 and the carbon atom on L are taken together to form an azetidine, piperidine or pyrrolidine with the nitrogen atom to which R 1 is attached;
R 3 is C 3-6 cycloalkyl, a 4-6 membered heterocycloalkyl (the cycloalkyl and the heterocycloalkyl may be substituted by 1 to 3 halogens), phenyl (the phenyl is 1 to 3 independently selected from the group consisting of, halogen, C 1-2 alkyl, C 1-2 alkoxy, C 1-2 haloalkyl, C 1-2 haloalkoxy, cyano and C 1-2 alkoxycarbonyl Or 6-membered heteroaryl, which may be substituted by 1 to 2 C 1-2 haloalkyls,
The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein R 4 is hydrogen.
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン;
(S)−2−((2−(2−オキソ−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−3(2H)−イル)エチル)アミノ)プロパンアミド;
2−((2−(2−オキソ−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−3(2H)−イル)エチル)アミノ)アセトアミド;
(R)−2−((2−(2−オキソ−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−3(2H)−イル)エチル)アミノ)プロパンアミド;
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(4−(トリフルオロメトキシ)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(2−(オキセタン−3−イルアミノ)エチル)−5−(4−(トリフルオロメチル)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4−フルオロフェニル)−4−メチル−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(2−メチル−3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4−フルオロフェニル)−3−(2−ヒドロキシ−3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4−フルオロフェニル)−3−(3−(オキセタン−3−イルアミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−フェニルベンゾ[d]オキサゾール−2(3H)−オン;
5−(3−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(4−(トリフルオロメトキシ)フェニル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(4−(トリフルオロメチル)フェニル) ベンゾ[d]オキサゾール−2(3H)−オン;
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4−フルオロフェニル)−4−メチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4,4−ジフルオロシクロヘキシル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン;
6−フルオロ−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(テトラヒドロ−2H−ピラン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(テトラヒドロ−2H−ピラン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−シクロブチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−シクロペンチル−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−ブチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−7−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)ブチル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(1−(2−(メチルスルホニル)エチル)アゼチジン−3−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(4−((2−(メチルスルホニル)エチル)アミノ)ブチル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(1−(2−(メチルスルホニル)エチル)ピペリジン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
2−(3−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)アゼチジン−1−イル)アセトアミド;
2−(4−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)ピペリジン−1−イル)アセトアミド;または
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−ヒドロキシ−2−メチルプロピル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン。 The compound according to claim 1 or a pharmaceutically acceptable salt thereof selected from the following group of compounds:
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one;
(S) -2-((2- (2-oxo-5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-3 (2H) -yl) ethyl) amino) propanamide;
2-((2- (2-oxo-5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-3 (2H) -yl) ethyl) amino) acetamide;
(R) -2-((2- (2-oxo-5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-3 (2H) -yl) ethyl) amino) propanamide;
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (4- (trifluoromethoxy) phenyl) benzo [d] oxazol-2 (3H) -one;
3- (2- (oxetane-3-ylamino) ethyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one;
5- (4-fluorophenyl) -4-methyl-3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one;
3- (2-Methyl-3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazole-2 (3H) -ON;
5- (4-fluorophenyl) -3- (2-hydroxy-3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
5- (4-fluorophenyl) -3- (3- (oxetan-3-ylamino) propyl) benzo [d] oxazol-2 (3H) -one;
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5-phenylbenzo [d] oxazol-2 (3H) -one;
5- (3-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (4- (trifluoromethoxy) phenyl) benzo [d] oxazol-2 (3H) -one;
5- (4-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (4- (trifluoromethyl) phenyl) benzo [d] oxazol-2 (3H) -one;
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one;
5- (4-fluorophenyl) -4-methyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
5- (4,4-difluorocyclohexyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one;
6-fluoro-3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (tetrahydro-2H-pyran-4-yl) benzo [d] oxazol-2 (3H) -one;
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (tetrahydro-2H-pyran-4-yl) benzo [d] oxazol-2 (3H) -one;
5-cyclobutyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
5-cyclopentyl-3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one;
5-butyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -7- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one;
5- (4-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) butyl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (1- (2- (methylsulfonyl) ethyl) azetidin-3-yl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (4-((2- (methylsulfonyl) ethyl) amino) butyl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (1- (2- (methylsulfonyl) ethyl) piperidin-4-yl) benzo [d] oxazol-2 (3H) -one;
2- (3- (5- (3,3-difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) azetidin-1-yl) acetamide;
2- (4- (5- (3,3-difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) piperidin-1-yl) acetamide; or 5- (3,3-difluoro Cyclobutyl) -3- (3-((2-hydroxy-2-methylpropyl) amino) propyl) benzo [d] oxazol-2 (3H) -one.
5−(4−フルオロフェニル)−3−(2−ヒドロキシ−3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4−フルオロフェニル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−5−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(4,4−ジフルオロシクロヘキシル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)−5−(テトラヒドロ−2H−ピラン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−シクロブチル−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)−7−(5−(トリフルオロメチル)ピリジン−2−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−(メチルスルホニル)エチル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(2−((2−(メチルスルホニル)エチル)アミノ)エチル)ベンゾ[d]オキサゾール−2(3H)−オン;
5−(3,3−ジフルオロシクロブチル)−3−(1−(2−(メチルスルホニル)エチル)ピペリジン−4−イル)ベンゾ[d]オキサゾール−2(3H)−オン;
2−(4−(5−(3,3−ジフルオロシクロブチル)−2−オキソベンゾ[d]オキサゾール−3(2H)−イル)ピペリジン−1−イル)アセトアミド;または
5−(3,3−ジフルオロシクロブチル)−3−(3−((2−ヒドロキシ−2−メチルプロピル)アミノ)プロピル)ベンゾ[d]オキサゾール−2(3H)−オン。 The compound according to claim 1 or a pharmaceutically acceptable salt thereof selected from the following group of compounds:
5- (4-fluorophenyl) -3- (2-hydroxy-3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
5- (4-fluorophenyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -5- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one;
5- (4,4-difluorocyclohexyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one;
3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) -5- (tetrahydro-2H-pyran-4-yl) benzo [d] oxazol-2 (3H) -one;
5-cyclobutyl-3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) -7- (5- (trifluoromethyl) pyridin-2-yl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (3-((2- (methylsulfonyl) ethyl) amino) propyl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (2-((2- (methylsulfonyl) ethyl) amino) ethyl) benzo [d] oxazol-2 (3H) -one;
5- (3,3-difluorocyclobutyl) -3- (1- (2- (methylsulfonyl) ethyl) piperidin-4-yl) benzo [d] oxazol-2 (3H) -one;
2- (4- (5- (3,3-difluorocyclobutyl) -2-oxobenzo [d] oxazol-3 (2H) -yl) piperidin-1-yl) acetamide; or 5- (3,3-difluoro Cyclobutyl) -3- (3-((2-hydroxy-2-methylpropyl) amino) propyl) benzo [d] oxazol-2 (3H) -one.
R5は、ニトロ、アミノ(該アミノは、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチル、該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)からなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)またはフタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)であり、
R6は、水素またはC1−2アルキル(該アルキルは、C1−2アルコキシ、フェニルおよび4−メトキシフェニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、
R7は、水素またはC1−2アルキルであり、
R8は、ヨウ素、臭素、塩素または水素であり、ここにおいて、R8がヨウ素、臭素または塩素であるとき、置換可能な位置に1〜2個置換していてもよく、
A−Aは、C−CまたはC=Cである]
で表される化合物またはその塩。 Following formula (2);
R 5 represents nitro, amino (the amino is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl moiety in the phenylmethyl, phenylmethyloxycarbonyl is halogen, C 1-2 alkyl And optionally substituted with 1 to 6 substituents independently selected from the group consisting of C 1-2 alkoxy), fluorenylmethylcarbonyl, benzenesulfonyl (wherein the benzenesulfonyl is 1- Optionally substituted by 1 to 2 substituents independently selected from the group consisting of: optionally substituted) or phthalimide (the phenyl portion of the phthalimide is substituted by C 1-2 alkyl) 1 to 3 may be substituted),
R 6 is hydrogen or C 1-2 alkyl, wherein the alkyl is substituted with 1-2 substituents independently selected from the group consisting of C 1-2 alkoxy, phenyl and 4-methoxyphenyl. Is good)
R 7 is hydrogen or C 1-2 alkyl;
R 8 is iodine, bromine, chlorine or hydrogen, and when R 8 is iodine, bromine or chlorine, 1 to 2 may be substituted at substitutable positions;
AA is CC or C = C]
Or a salt thereof.
R7は、水素またはC1−2アルキルであり、
R9は、C1−4アルキルオキシカルボニル、フェニルメチルオキシカルボニル(該フェニルメチルオキシカルボニルのフェニルメチル部分は、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニルまたはベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)であり、
R10は、水素またはC1−2アルキルであり、ここにおいて、R9およびR10が一緒になって、R9およびR10が結合する窒素原子と共にフタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)を形成してもよく、
Mは、C1−4アルキレンであり、ここにおいて、R10がC1−2アルキルであるとき、R10とM上の炭素原子が一緒になって、R10が結合する窒素原子と共に4〜6員の飽和複素環を形成してもよく、
Bは、ハロゲンである]
で表される化合物またはその塩。 Following formula (3);
R 7 is hydrogen or C 1-2 alkyl;
R 9 represents C 1-4 alkyloxycarbonyl, phenylmethyloxycarbonyl (the phenylmethyl moiety of the phenylmethyloxycarbonyl is independently selected from the group consisting of C 1-2 alkyl and C 1-2 alkoxy) Up to 6 substituents), fluorenylmethylcarbonyl or benzenesulfonyl (wherein the benzenesulfonyl may be substituted with 1 to 2 by nitro),
R 10 is hydrogen or C 1-2 alkyl, where R 9 and R 10 are taken together with the nitrogen atom to which R 9 and R 10 are attached together with phthalimide (the phenyl portion of the phthalimide is C 1 1-3 may be substituted by -2 alkyl),
M is C 1-4 alkylene, where when R 10 is C 1-2 alkyl, R 10 and the carbon atom on M are taken together and together with the nitrogen atom to which R 10 is attached, 4- May form a 6-membered saturated heterocycle,
B is halogen]
Or a salt thereof.
R7は、水素、またはC1−2アルキルであり、
R11およびR12は、それぞれ独立して、水素、C1−4アルキル(該アルキルは、C1−4アルコキシまたはフェニルによって置換されていてもよい)、C1−4ハロアルキル、C1−4ハロアルコキシ、ニトロ、アミノ(該アミノは、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチルおよび該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)からなる群から独立して選択される1〜2個の置換基によって置換されていてもよい)、フタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)またはC1−2アルコキシ(該アルコキシにおけるアルキル部分は、C1−2アルコキシ、フェニルおよび4−メトキシフェニルからなる群から独立して選択される1〜2個の置換基で置換されていてもよい)であり、ここにおいて、R11およびR12が互いにオルト位である場合は、R11とR12がフェニルと一緒になってベンズオキサゾロン環、インドール環またはインダゾール環を形成してもよい]
で表される化合物またはその塩の製造方法であって、下記式(5)
R7、R11およびR12は、上記式(4)と同じであり、
Bは、ハロゲンである]で表される化合物と、下記式(6)
Xは、ヨウ素、臭素または塩素である]で表される化合物を、パラジウム触媒を用いてカップリングさせる工程を含む製造方法。 Following formula (4);
R 7 is hydrogen or C 1-2 alkyl;
R 11 and R 12 are each independently hydrogen, C 1-4 alkyl (wherein the alkyl may be substituted by C 1-4 alkoxy or phenyl), C 1-4 haloalkyl, C 1-4. Haloalkoxy, nitro, amino (wherein the amino is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl moiety in the phenylmethyl and phenylmethyloxycarbonyl is halogen, C 1-2 alkyl and Optionally substituted with 1 to 6 substituents independently selected from the group consisting of C 1-2 alkoxy, fluorenylmethylcarbonyl, benzenesulfonyl (wherein the benzenesulfonyl is 1-2 with nitro) 1 may be independently selected from the group consisting of Number of which may be substituted by a substituent), phthalimide (phenyl moiety of the phthalimide may be 1-3 substituted by C 1-2 alkyl) or C 1-2 alkoxy (alkyl in the alkoxy The moiety is optionally substituted with 1 to 2 substituents independently selected from the group consisting of C 1-2 alkoxy, phenyl and 4-methoxyphenyl, wherein R 11 and R 11 When 12 is ortho to each other, R 11 and R 12 may be combined with phenyl to form a benzoxazolone ring, an indole ring or an indazole ring.
Or a salt thereof, which is represented by the following formula (5):
R 7 , R 11 and R 12 are the same as in the above formula (4),
B is a halogen], and the following formula (6)
X is an iodine, bromine, or chlorine] manufacturing method including a step of coupling a compound represented by a palladium catalyst.
R7は、水素またはC1−2アルキルであり、
R13は、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチルおよび該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニル、ベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)であり、
R14は、水素、C1−2アルキル、C1−4アルキルオキシカルボニル、フェニルメチル、フェニルメチルオキシカルボニル(該フェニルメチルおよび該フェニルメチルオキシカルボニルにおけるフェニルメチル部分は、ハロゲン、C1−2アルキルおよびC1−2アルコキシからなる群から独立して選択される1〜6個の置換基で置換されていてもよい)、フルオレニルメチルカルボニルまたはベンゼンスルホニル(該ベンゼンスルホニルは、ニトロによって1〜2個置換されていてもよい)であり、ここにおいて、R13およびR14が一緒になって、R13およびR14が結合する窒素原子と共にフタルイミド(該フタルイミドのフェニル部分は、C1−2アルキルによって1〜3個置換されていてもよい)を形成してもよく、
Mは、C1−4アルキレンであり、ここにおいて、R14がC1−2アルキルであるとき、R14とM上の炭素原子が一緒になって、R14が結合する窒素原子と共に4〜6員の飽和複素環を形成してもよい]
で表される化合物またはその塩の製造方法であって、下記式(8)
R7、R13、R14およびMは、上記式(7)と同じであり、Bは、ハロゲンである]で表される化合物と、下記式(9)
Xは、ヨウ素、臭素または塩素である]で表される化合物を、パラジウム触媒を用いてカップリングさせる工程を含む製造方法。 Following formula (7);
R 7 is hydrogen or C 1-2 alkyl;
R 13 is C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl and the phenylmethyl moiety in the phenylmethyloxycarbonyl are composed of halogen, C 1-2 alkyl and C 1-2 alkoxy. Optionally substituted with 1 to 6 substituents independently selected from the group), fluorenylmethylcarbonyl, benzenesulfonyl (the benzenesulfonyl may be substituted with 1 to 2 by nitro) ) And
R 14 is hydrogen, C 1-2 alkyl, C 1-4 alkyloxycarbonyl, phenylmethyl, phenylmethyloxycarbonyl (the phenylmethyl moiety in the phenylmethyl and phenylmethyloxycarbonyl is halogen, C 1-2 alkyl And optionally substituted with 1 to 6 substituents independently selected from the group consisting of C 1-2 alkoxy), fluorenylmethylcarbonyl or benzenesulfonyl (wherein the benzenesulfonyl is 1- In which R 13 and R 14 are taken together with the nitrogen atom to which R 13 and R 14 are attached together with phthalimide (the phenyl moiety of the phthalimide is C 1-2 1 to 3 may be substituted with alkyl) ,
M is C 1-4 alkylene, where when R 14 is C 1-2 alkyl, R 14 and the carbon atom on M are taken together and together with the nitrogen atom to which R 14 is attached, 4 to 4 A 6-membered saturated heterocyclic ring may be formed]
Or a salt thereof, which is represented by the following formula (8):
R 7 , R 13 , R 14 and M are the same as those in the above formula (7), B is a halogen], and the following formula (9)
X is an iodine, bromine, or chlorine] manufacturing method including a step of coupling a compound represented by a palladium catalyst.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015118562A JP2017001991A (en) | 2015-06-11 | 2015-06-11 | Novel benzoxazolone compound |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015118562A JP2017001991A (en) | 2015-06-11 | 2015-06-11 | Novel benzoxazolone compound |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017001991A true JP2017001991A (en) | 2017-01-05 |
Family
ID=57751171
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015118562A Pending JP2017001991A (en) | 2015-06-11 | 2015-06-11 | Novel benzoxazolone compound |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017001991A (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106928212A (en) * | 2017-02-28 | 2017-07-07 | 牡丹江医学院 | A kind of medicine for treating appendicitis and its preparation method and application |
| US11014931B2 (en) | 2018-05-30 | 2021-05-25 | Praxis Precision Medicines, Inc. | 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyrazine as an ion channel modulator |
| US11261188B2 (en) | 2016-11-28 | 2022-03-01 | Praxis Precision Medicines, Inc. | Fused heteroaryl compounds, and methods thereof for treating diseases, disorders, and conditions relating to aberrant function of a sodium channel |
| US11278535B2 (en) | 2017-08-15 | 2022-03-22 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11279700B2 (en) | 2019-05-31 | 2022-03-22 | Praxis Precision Medicines, Inc. | Ion channel modulators |
| US11492345B2 (en) | 2017-02-13 | 2022-11-08 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11505554B2 (en) | 2019-05-31 | 2022-11-22 | Praxis Precision Medicines, Inc. | Substituted pyridines as ion channel modulators |
| WO2023010191A1 (en) | 2021-08-02 | 2023-02-09 | Eurofarma Laboratórios S.A. | N-acylhydrazone compounds capable of inhibiting nav1.7 and/or nav1.8, processes for the preparation thereof, compositions, uses, methods for treatment using same, and kits |
| WO2023010192A1 (en) | 2021-08-02 | 2023-02-09 | Eurofarma Laboratórios S.A. | N-acylhydrazone compounds capable of inhibiting nav1.7 and/or nav1.8, processes for the preparation thereof, compositions, uses, methods for treatment using same, and kits |
| US11629146B2 (en) | 2016-11-28 | 2023-04-18 | Praxis Precision Medicines, Inc. | Substituted [1,2,4]triazolo[4,3-a]pyrazines as modulators of sodium channel activity |
| US11731966B2 (en) | 2017-04-04 | 2023-08-22 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11767325B2 (en) | 2019-11-26 | 2023-09-26 | Praxis Precision Medicines, Inc. | Substituted [1,2,4]triazolo[4,3-a]pyrazines as ion channel modulators |
| US11773099B2 (en) | 2019-05-28 | 2023-10-03 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
-
2015
- 2015-06-11 JP JP2015118562A patent/JP2017001991A/en active Pending
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11261188B2 (en) | 2016-11-28 | 2022-03-01 | Praxis Precision Medicines, Inc. | Fused heteroaryl compounds, and methods thereof for treating diseases, disorders, and conditions relating to aberrant function of a sodium channel |
| US11629146B2 (en) | 2016-11-28 | 2023-04-18 | Praxis Precision Medicines, Inc. | Substituted [1,2,4]triazolo[4,3-a]pyrazines as modulators of sodium channel activity |
| US11492345B2 (en) | 2017-02-13 | 2022-11-08 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| CN106928212B (en) * | 2017-02-28 | 2020-03-17 | 牡丹江医学院 | Medicine for treating appendicitis and preparation method and application thereof |
| CN106928212A (en) * | 2017-02-28 | 2017-07-07 | 牡丹江医学院 | A kind of medicine for treating appendicitis and its preparation method and application |
| US11731966B2 (en) | 2017-04-04 | 2023-08-22 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11278535B2 (en) | 2017-08-15 | 2022-03-22 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11918571B2 (en) | 2017-08-15 | 2024-03-05 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11731976B2 (en) | 2018-05-30 | 2023-08-22 | Praxis Precision Medicines, Inc. | 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyridine as an ion channel modulator |
| US11014931B2 (en) | 2018-05-30 | 2021-05-25 | Praxis Precision Medicines, Inc. | 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyrazine as an ion channel modulator |
| US11731978B2 (en) | 2018-05-30 | 2023-08-22 | Praxis Precision Medicines, Inc. | 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyridine as an ion channel modulator |
| US11866439B2 (en) | 2018-05-30 | 2024-01-09 | Praxis Precision Medicines, Inc. | Ion channel modulators |
| US11773099B2 (en) | 2019-05-28 | 2023-10-03 | Praxis Precision Medicines, Inc. | Compounds and their methods of use |
| US11505554B2 (en) | 2019-05-31 | 2022-11-22 | Praxis Precision Medicines, Inc. | Substituted pyridines as ion channel modulators |
| US11279700B2 (en) | 2019-05-31 | 2022-03-22 | Praxis Precision Medicines, Inc. | Ion channel modulators |
| US11767325B2 (en) | 2019-11-26 | 2023-09-26 | Praxis Precision Medicines, Inc. | Substituted [1,2,4]triazolo[4,3-a]pyrazines as ion channel modulators |
| WO2023010191A1 (en) | 2021-08-02 | 2023-02-09 | Eurofarma Laboratórios S.A. | N-acylhydrazone compounds capable of inhibiting nav1.7 and/or nav1.8, processes for the preparation thereof, compositions, uses, methods for treatment using same, and kits |
| WO2023010192A1 (en) | 2021-08-02 | 2023-02-09 | Eurofarma Laboratórios S.A. | N-acylhydrazone compounds capable of inhibiting nav1.7 and/or nav1.8, processes for the preparation thereof, compositions, uses, methods for treatment using same, and kits |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2017001991A (en) | Novel benzoxazolone compound | |
| US20230103791A1 (en) | 2,3-dihydroquinazolin compounds as nav1.8 inhibitors | |
| CA2799146C (en) | Nitrogen-containing heterocyclic compound having inhibitory effect on production of kynurenine | |
| JP4493503B2 (en) | Heterocyclic compounds and antitumor agents containing the same as active ingredients | |
| JP2016169161A (en) | Novel imidazo pyridine compound | |
| CZ20002349A3 (en) | Inhibition of RAF-kinase by means of aryl- and heteroaryl-substituted heterocyclic ureas | |
| EP3348547B1 (en) | Benzimidazole derivatives as nav 1.7 (sodium channel, voltage-gated, type ix, alpha subunit (scn9a)) inhibitors for treating pain, dysuria and multiple sclerosis | |
| AU2010229142A1 (en) | P2X3, receptor antagonists for treatment of pain | |
| JP2020508327A (en) | 1,4,6-Trisubstituted-2-alkyl-1H-benzo [d] imidazole derivatives as dihydroorotate oxygenase inhibitors | |
| WO2014164756A1 (en) | Novel 5-hydroxytryptamine receptor 7 activity modulators and their method of use | |
| JP2012211085A (en) | Hedgehog signal inhibitor | |
| JP2016124810A (en) | Novel condensed pyrazole derivative and medical uses thereof | |
| EP1908752A1 (en) | Novel 2-quinolone derivative | |
| JP7165501B2 (en) | Pharmaceuticals comprising novel benzimidazole compounds | |
| JP7175878B2 (en) | Novel benzimidazolone compounds and their medicinal uses | |
| CA2926313A1 (en) | Secondary alcohol quinolinyl modulators of roryt | |
| CN110612289B (en) | Deuterated benzimidazole compound and medical application thereof | |
| JP2018154562A (en) | Novel bicyclic heterocyclic compound and pharmaceutical use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20160929 |