JP2014114368A - Thermosetting bismaleimide-based resin composition and prepreg, and method for producing the same - Google Patents
Thermosetting bismaleimide-based resin composition and prepreg, and method for producing the same Download PDFInfo
- Publication number
- JP2014114368A JP2014114368A JP2012268922A JP2012268922A JP2014114368A JP 2014114368 A JP2014114368 A JP 2014114368A JP 2012268922 A JP2012268922 A JP 2012268922A JP 2012268922 A JP2012268922 A JP 2012268922A JP 2014114368 A JP2014114368 A JP 2014114368A
- Authority
- JP
- Japan
- Prior art keywords
- prepreg
- resin composition
- bismaleimide
- component
- reinforced composite
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000011342 resin composition Substances 0.000 title claims abstract description 167
- 229920003192 poly(bis maleimide) Polymers 0.000 title claims abstract description 102
- XQUPVDVFXZDTLT-UHFFFAOYSA-N 1-[4-[[4-(2,5-dioxopyrrol-1-yl)phenyl]methyl]phenyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1C(C=C1)=CC=C1CC1=CC=C(N2C(C=CC2=O)=O)C=C1 XQUPVDVFXZDTLT-UHFFFAOYSA-N 0.000 title claims abstract description 60
- 229920001187 thermosetting polymer Polymers 0.000 title claims abstract description 46
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 34
- -1 bismaleimide compound Chemical class 0.000 claims abstract description 69
- 239000000463 material Substances 0.000 claims abstract description 61
- 239000012783 reinforcing fiber Substances 0.000 claims abstract description 33
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims abstract description 32
- 150000001412 amines Chemical class 0.000 claims abstract description 12
- 229920001577 copolymer Polymers 0.000 claims abstract description 10
- 229920000049 Carbon (fiber) Polymers 0.000 claims description 49
- 239000004917 carbon fiber Substances 0.000 claims description 49
- 229920005992 thermoplastic resin Polymers 0.000 claims description 32
- 239000007790 solid phase Substances 0.000 claims description 14
- 239000000758 substrate Substances 0.000 claims description 9
- 239000000203 mixture Substances 0.000 claims description 8
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 claims 1
- 239000003733 fiber-reinforced composite Substances 0.000 abstract description 45
- 238000000465 moulding Methods 0.000 abstract description 15
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 43
- DSROZUMNVRXZNO-UHFFFAOYSA-K tris[(1-naphthalen-1-yl-3-phenylnaphthalen-2-yl)oxy]alumane Chemical compound C=1C=CC=CC=1C=1C=C2C=CC=CC2=C(C=2C3=CC=CC=C3C=CC=2)C=1O[Al](OC=1C(=C2C=CC=CC2=CC=1C=1C=CC=CC=1)C=1C2=CC=CC=C2C=CC=1)OC(C(=C1C=CC=CC1=C1)C=2C3=CC=CC=C3C=CC=2)=C1C1=CC=CC=C1 DSROZUMNVRXZNO-UHFFFAOYSA-K 0.000 description 39
- 239000011208 reinforced composite material Substances 0.000 description 36
- 229920005989 resin Polymers 0.000 description 35
- 239000011347 resin Substances 0.000 description 35
- 238000003860 storage Methods 0.000 description 30
- 238000000034 method Methods 0.000 description 29
- 229910052757 nitrogen Inorganic materials 0.000 description 26
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 24
- 239000002245 particle Substances 0.000 description 21
- 230000000052 comparative effect Effects 0.000 description 18
- 238000001723 curing Methods 0.000 description 18
- 230000014759 maintenance of location Effects 0.000 description 18
- 239000000835 fiber Substances 0.000 description 16
- 239000011159 matrix material Substances 0.000 description 16
- 229920001971 elastomer Polymers 0.000 description 14
- 239000005060 rubber Substances 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 13
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- 238000010438 heat treatment Methods 0.000 description 11
- 238000012360 testing method Methods 0.000 description 11
- 238000010521 absorption reaction Methods 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 125000003342 alkenyl group Chemical group 0.000 description 8
- 229920001721 polyimide Polymers 0.000 description 8
- 239000009719 polyimide resin Substances 0.000 description 7
- WOCGGVRGNIEDSZ-UHFFFAOYSA-N 4-[2-(4-hydroxy-3-prop-2-enylphenyl)propan-2-yl]-2-prop-2-enylphenol Chemical compound C=1C=C(O)C(CC=C)=CC=1C(C)(C)C1=CC=C(O)C(CC=C)=C1 WOCGGVRGNIEDSZ-UHFFFAOYSA-N 0.000 description 5
- 238000004898 kneading Methods 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 4
- SLJJEYCPTRKHFI-UHFFFAOYSA-N 3-[6-(2,5-dioxopyrrol-3-yl)hexyl]pyrrole-2,5-dione Chemical compound O=C1NC(=O)C(CCCCCCC=2C(NC(=O)C=2)=O)=C1 SLJJEYCPTRKHFI-UHFFFAOYSA-N 0.000 description 4
- 229920013646 Hycar Polymers 0.000 description 4
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 4
- XAZPKEBWNIUCKF-UHFFFAOYSA-N 1-[4-[4-[2-[4-[4-(2,5-dioxopyrrol-1-yl)phenoxy]phenyl]propan-2-yl]phenoxy]phenyl]pyrrole-2,5-dione Chemical compound C=1C=C(OC=2C=CC(=CC=2)N2C(C=CC2=O)=O)C=CC=1C(C)(C)C(C=C1)=CC=C1OC(C=C1)=CC=C1N1C(=O)C=CC1=O XAZPKEBWNIUCKF-UHFFFAOYSA-N 0.000 description 3
- MOSSLXZUUKTULI-UHFFFAOYSA-N 3-[3-(2,5-dioxopyrrol-3-yl)-4-methylphenyl]pyrrole-2,5-dione Chemical compound CC1=CC=C(C=2C(NC(=O)C=2)=O)C=C1C1=CC(=O)NC1=O MOSSLXZUUKTULI-UHFFFAOYSA-N 0.000 description 3
- 239000004695 Polyether sulfone Substances 0.000 description 3
- 239000004697 Polyetherimide Substances 0.000 description 3
- 229920004738 ULTEM® Polymers 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 230000009477 glass transition Effects 0.000 description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 229920002239 polyacrylonitrile Polymers 0.000 description 3
- 229920006393 polyether sulfone Polymers 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- DRWLKECFWWDHJL-UHFFFAOYSA-N C1(C=CC(N1)=O)=O.C1(C=CC(N1)=O)=O.NCCCCCCN Chemical compound C1(C=CC(N1)=O)=O.C1(C=CC(N1)=O)=O.NCCCCCCN DRWLKECFWWDHJL-UHFFFAOYSA-N 0.000 description 2
- 241000579895 Chlorostilbon Species 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 229920000459 Nitrile rubber Polymers 0.000 description 2
- 239000004642 Polyimide Substances 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 239000004760 aramid Substances 0.000 description 2
- 229920006231 aramid fiber Polymers 0.000 description 2
- 239000012965 benzophenone Substances 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229910052876 emerald Inorganic materials 0.000 description 2
- 239000010976 emerald Substances 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 239000003365 glass fiber Substances 0.000 description 2
- 238000013007 heat curing Methods 0.000 description 2
- 239000012943 hotmelt Substances 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000013500 performance material Substances 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229920001601 polyetherimide Polymers 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 239000001294 propane Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000002759 woven fabric Substances 0.000 description 2
- PUKLCKVOVCZYKF-UHFFFAOYSA-N 1-[2-(2,5-dioxopyrrol-1-yl)ethyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1CCN1C(=O)C=CC1=O PUKLCKVOVCZYKF-UHFFFAOYSA-N 0.000 description 1
- IPJGAEWUPXWFPL-UHFFFAOYSA-N 1-[3-(2,5-dioxopyrrol-1-yl)phenyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1C1=CC=CC(N2C(C=CC2=O)=O)=C1 IPJGAEWUPXWFPL-UHFFFAOYSA-N 0.000 description 1
- FXEKNWLZNLIDNR-UHFFFAOYSA-N 1-[3-(2,5-dioxopyrrol-1-yl)propyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1CCCN1C(=O)C=CC1=O FXEKNWLZNLIDNR-UHFFFAOYSA-N 0.000 description 1
- AQGZJQNZNONGKY-UHFFFAOYSA-N 1-[4-(2,5-dioxopyrrol-1-yl)phenyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1C1=CC=C(N2C(C=CC2=O)=O)C=C1 AQGZJQNZNONGKY-UHFFFAOYSA-N 0.000 description 1
- IKFPAKYBSYICFK-UHFFFAOYSA-N 1-[4-(4-propylphenoxy)phenyl]pyrrole-2,5-dione Chemical compound C1=CC(CCC)=CC=C1OC1=CC=C(N2C(C=CC2=O)=O)C=C1 IKFPAKYBSYICFK-UHFFFAOYSA-N 0.000 description 1
- AFTPEBDOGXRMNQ-UHFFFAOYSA-N 2,2,4-Trimethylhexane Chemical compound CCC(C)CC(C)(C)C AFTPEBDOGXRMNQ-UHFFFAOYSA-N 0.000 description 1
- QIRNGVVZBINFMX-UHFFFAOYSA-N 2-allylphenol Chemical compound OC1=CC=CC=C1CC=C QIRNGVVZBINFMX-UHFFFAOYSA-N 0.000 description 1
- PXKLMJQFEQBVLD-UHFFFAOYSA-N Bisphenol F Natural products C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 238000005452 bending Methods 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000011231 conductive filler Substances 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 238000005336 cracking Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000032798 delamination Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 239000003822 epoxy resin Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000007656 fracture toughness test Methods 0.000 description 1
- 229920006015 heat resistant resin Polymers 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000011256 inorganic filler Substances 0.000 description 1
- 229910003475 inorganic filler Inorganic materials 0.000 description 1
- 238000010030 laminating Methods 0.000 description 1
- 238000003475 lamination Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 1
- 239000002557 mineral fiber Substances 0.000 description 1
- 239000006082 mold release agent Substances 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000004745 nonwoven fabric Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000000088 plastic resin Substances 0.000 description 1
- 229920006162 poly(etherimide sulfone) Polymers 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 229920000647 polyepoxide Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- XDOBJOBITOLMFI-UHFFFAOYSA-N pyrrole-2,5-dione;toluene Chemical compound CC1=CC=CC=C1.O=C1NC(=O)C=C1 XDOBJOBITOLMFI-UHFFFAOYSA-N 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 239000011435 rock Substances 0.000 description 1
- 150000003335 secondary amines Chemical group 0.000 description 1
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 1
- 229910010271 silicon carbide Inorganic materials 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000002352 surface water Substances 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 230000000930 thermomechanical effect Effects 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 229920006259 thermoplastic polyimide Polymers 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
Abstract
Description
本発明は、繊維強化複合材料を作製するために用いる熱硬化性ビスマレイミド系樹脂組成物及びその製造方法に関する。また、本発明は、この熱硬化性ビスマレイミド系樹脂組成物を強化繊維基材に含浸させてなるプリプレグ及びその製造方法に関する。 The present invention relates to a thermosetting bismaleimide resin composition used for producing a fiber-reinforced composite material and a method for producing the same. The present invention also relates to a prepreg obtained by impregnating a reinforcing fiber substrate with this thermosetting bismaleimide resin composition and a method for producing the same.
繊維強化複合材料は、その比強度、比弾性が優れているため、航空・宇宙用品等に広く利用されている。繊維強化複合材料の用途が広がるにつれ、200℃以上の高温に耐える耐熱性や優れた破壊靭性を有する繊維強化複合材料の要求が増えている。したがって、耐熱性及び破壊靭性が高い繊維強化複合材料を作製することができるプリプレグが求められている。 Fiber reinforced composite materials are widely used in aerospace products and the like because of their excellent specific strength and specific elasticity. As the use of fiber reinforced composite materials expands, the demand for fiber reinforced composite materials having heat resistance that can withstand high temperatures of 200 ° C. or higher and excellent fracture toughness is increasing. Accordingly, there is a need for a prepreg capable of producing a fiber-reinforced composite material having high heat resistance and high fracture toughness.
プリプレグは、強化繊維基材と該強化繊維基材内に含浸されたマトリックス樹脂とから構成される。従来、耐熱性の高いマトリックス樹脂として、ポリイミド樹脂やビスマレイミド樹脂が用いられている。しかし、ポリイミド樹脂をマトリックス樹脂として用いるプリプレグは、成形加工性が悪いため実用化が遅れている。ビスマレイミド樹脂をマトリックス樹脂として用いるプリプレグは、エポキシ樹脂をマトリックス樹脂として用いる場合と比較して取扱い性や成形加工性が悪い。また、ビスマレイミド樹脂は靭性が乏しいため、ビスマレイミド樹脂をマトリックス樹脂として用いる繊維強化複合材料は、破壊靭性が低い。 The prepreg is composed of a reinforcing fiber base material and a matrix resin impregnated in the reinforcing fiber base material. Conventionally, polyimide resins and bismaleimide resins have been used as matrix resins with high heat resistance. However, prepregs using a polyimide resin as a matrix resin are delayed in practical use because of poor molding processability. A prepreg using a bismaleimide resin as a matrix resin has poor handleability and molding processability compared to a case where an epoxy resin is used as a matrix resin. In addition, since the bismaleimide resin has poor toughness, the fiber-reinforced composite material using the bismaleimide resin as the matrix resin has low fracture toughness.
ビスマレイミド樹脂の靱性を高くする方法として、カルボキシル基末端のゴム化合物をマトリックス樹脂内に分散させる方法や、ビスマレイミド樹脂と相溶性のあるポリイミド等の熱可塑樹脂をマトリックス樹脂に溶解させる方法が検討されている。しかし、従来提案された方法では、靭性が十分に高くならないうえに他の物性が大きく低下している。 As a method to increase the toughness of bismaleimide resin, a method of dispersing a carboxyl group-terminated rubber compound in the matrix resin and a method of dissolving a thermoplastic resin such as polyimide compatible with the bismaleimide resin in the matrix resin are studied. Has been. However, in the conventionally proposed method, the toughness is not sufficiently increased and other physical properties are greatly deteriorated.
特許文献1には、芳香族ビスマレイミド化合物と、アルケニルフェノール又はアルケニルフェノキシ基含有コモノマーと、可溶性の熱可塑性ポリイミド樹脂と、からなるプリプレグが開示されている。このプリプレグを用いて製造される繊維強化複合材料は、破壊靭性は高いが、耐熱酸化特性や、メチルエチルケトン(MEK)によるソルベントクラックなどの耐溶剤特性は十分でない。 Patent Document 1 discloses a prepreg composed of an aromatic bismaleimide compound, an alkenylphenol or alkenylphenoxy group-containing comonomer, and a soluble thermoplastic polyimide resin. The fiber reinforced composite material produced using this prepreg has high fracture toughness, but has insufficient heat resistance and solvent resistance such as solvent cracking due to methyl ethyl ketone (MEK).
特許文献2には、50質量%以上が固形で存在する多官能性マレイミド化合物と、アルケニルフェノール又はアルケニルフェノールエーテル化合物と、熱可塑性樹脂と、からなり、該熱可塑性樹脂がマトリックス樹脂に溶解せずにプリプレグ表面に局在するプリプレグが開示されている。このプリプレグを用いて製造される繊維強化複合材料は、破壊靭性は高いが、耐熱性や耐熱酸化特性は十分でない。 Patent Document 2 includes a polyfunctional maleimide compound in which 50% by mass or more exists in a solid form, an alkenylphenol or an alkenylphenol ether compound, and a thermoplastic resin, and the thermoplastic resin does not dissolve in the matrix resin. Discloses a prepreg localized on the surface of the prepreg. The fiber reinforced composite material produced using this prepreg has high fracture toughness but is not sufficient in heat resistance and heat oxidation resistance.
特許文献3には、ポリフェニルメタンマレイミド、1,6−ビスマレイミド(2,2,4−トリメチル)ヘキサン、ジアリルビスフェノールAを必須成分とするプリプレグが開示されている。このプリプレグは、ジアリルビスフェノールAに溶解しているフェニルメタンマレイミドオリゴマーの結晶が樹脂組成物中に析出し難いため、プリプレグのタック性・ドレープ性といった取扱い性は優れている。しかし、破壊靭性は低い。 Patent Document 3 discloses a prepreg containing polyphenylmethane maleimide, 1,6-bismaleimide (2,2,4-trimethyl) hexane, and diallyl bisphenol A as essential components. This prepreg is excellent in handleability such as tackiness and draping properties of the prepreg because crystals of phenylmethanemaleimide oligomer dissolved in diallyl bisphenol A are difficult to precipitate in the resin composition. However, the fracture toughness is low.
そのため、取扱い性及び成形加工性が優れ、耐熱性や破壊靭性が高い繊維強化複合材料を作製することができるプリプレグが求められている。 Therefore, there is a demand for a prepreg that can produce a fiber-reinforced composite material that is excellent in handleability and moldability, and that has high heat resistance and high fracture toughness.
本発明の課題は、耐熱性や破壊靭性が高い繊維強化複合材料を作製することができ、取扱い性及び成形加工性が高いプリプレグを提供すること、及びそのプリプレグの製造に用いる樹脂組成物を提供することにある。 An object of the present invention is to provide a prepreg that can produce a fiber-reinforced composite material having high heat resistance and high fracture toughness, and that has high handleability and moldability, and a resin composition that is used for manufacturing the prepreg. There is to do.
本発明者らは、ビスマレイミド化合物と、アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物と、アミン末端基を有するアクリロニトリル−ブタジエンコポリマーと、を含んで成る樹脂組成物を強化繊維基材に含浸させて製造したプリプレグは、取扱い性、成形加工性が優れていることを見出した。そして、このプリプレグを用いて作製した繊維強化複合材料は、耐熱性及び破壊靭性が高いことを見出し、本発明を完成するに至った。 The present inventors impregnate a reinforcing fiber substrate with a resin composition comprising a bismaleimide compound, an alkenylphenol and / or alkenylphenol ether compound, and an acrylonitrile-butadiene copolymer having an amine end group. The found prepreg was found to be excellent in handleability and molding processability. And the fiber reinforced composite material produced using this prepreg discovered that heat resistance and fracture toughness were high, and came to complete this invention.
上記課題を解決する本発明は、以下に記載のものである。 The present invention for solving the above problems is as follows.
〔1〕 以下の(a)乃至(c)成分
(a)成分:ビスマレイミド化合物、
(b)成分:アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物、
(c)成分:アミン末端基を有するアクリロニトリル−ブタジエンコポリマー、
を必須成分として含有し、
(c)成分の含有量が3〜30質量%であることを特徴とする熱硬化性ビスマレイミド系樹脂組成物。
[1] The following components (a) to (c) (a) component: bismaleimide compound,
(B) component: alkenylphenol and / or alkenylphenol ether compound,
(C) component: an acrylonitrile-butadiene copolymer having amine end groups,
As an essential component,
(C) Content of component is 3-30 mass%, The thermosetting bismaleimide-type resin composition characterized by the above-mentioned.
〔2〕 前記熱硬化性ビスマレイミド系樹脂組成物中における固相成分の割合が、5〜50質量%である〔1〕に記載の熱硬化性ビスマレイミド系樹脂組成物。 [2] The thermosetting bismaleimide resin composition according to [1], wherein a ratio of a solid phase component in the thermosetting bismaleimide resin composition is 5 to 50% by mass.
〔3〕 (a)成分が、芳香族ビスマレイミド化合物と脂肪族ビスマレイミド化合物とを含んで成る〔1〕又は〔2〕に記載の熱硬化性ビスマレイミド系樹脂組成物。 [3] The thermosetting bismaleimide resin composition according to [1] or [2], wherein the component (a) comprises an aromatic bismaleimide compound and an aliphatic bismaleimide compound.
〔4〕 前記アミン末端基が、第二級アミン末端基である〔1〕〜〔3〕のいずれかに記載の熱硬化性ビスマレイミド系樹脂組成物。 [4] The thermosetting bismaleimide resin composition according to any one of [1] to [3], wherein the amine end group is a secondary amine end group.
〔5〕 熱可塑性樹脂を0.1〜15質量%含有する〔1〕〜〔4〕のいずれかに記載の熱硬化性ビスマレイミド系樹脂組成物。 [5] The thermosetting bismaleimide resin composition according to any one of [1] to [4], which contains 0.1 to 15% by mass of a thermoplastic resin.
〔6〕 以下の(a)乃至(c)成分
(a)成分:ビスマレイミド化合物、
(b)成分:アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物、
(c)成分:アミン末端基を有するアクリロニトリル−ブタジエンコポリマー、
を、温度60〜140℃で混合することを特徴とする〔1〕に記載の熱硬化性ビスマレイミド系樹脂組成物の製造方法。
[6] Components (a) to (c) below (a) Component: Bismaleimide compound,
(B) component: alkenylphenol and / or alkenylphenol ether compound,
(C) component: an acrylonitrile-butadiene copolymer having amine end groups,
Are mixed at a temperature of 60 to 140 ° C., The method for producing a thermosetting bismaleimide resin composition according to [1].
本発明は、上記熱硬化性ビスマレイミド系樹脂組成物を用いて製造される以下の〔7〕〜〔9〕のプリプレグを含む。 This invention contains the prepreg of the following [7]-[9] manufactured using the said thermosetting bismaleimide-type resin composition.
〔7〕 強化繊維基材と、
前記強化繊維基材内に含浸した〔1〕に記載の熱硬化性ビスマレイミド系樹脂組成物と、
から成るプリプレグであって、
前記プリプレグ中における前記熱硬化性ビスマレイミド系樹脂組成物の割合が20〜60質量%であることを特徴とするプリプレグ。
[7] a reinforcing fiber base;
The thermosetting bismaleimide resin composition according to [1] impregnated in the reinforcing fiber base;
A prepreg consisting of
The ratio of the said thermosetting bismaleimide-type resin composition in the said prepreg is 20-60 mass%, The prepreg characterized by the above-mentioned.
〔8〕 前記強化繊維基材が、炭素繊維で構成された強化繊維基材である〔7〕に記載のプリプレグ。 [8] The prepreg according to [7], wherein the reinforcing fiber base is a reinforcing fiber base composed of carbon fibers.
〔9〕 〔1〕に記載の熱硬化性ビスマレイミド系樹脂組成物を温度70〜160℃で強化繊維基材内に含浸させる〔7〕又は〔8〕に記載のプリプレグの製造方法。 [9] The method for producing a prepreg according to [7] or [8], wherein the reinforcing fiber substrate is impregnated with the thermosetting bismaleimide resin composition according to [1] at a temperature of 70 to 160 ° C.
本発明の熱硬化性ビスマレイミド系樹脂組成物を用いて製造されたプリプレグは、タック性、ドレープ性の経時変化が少なく、保存安定性が高い。そのため、長期間保管後のプリプレグであっても取扱い性が良好である。 The prepreg produced using the thermosetting bismaleimide resin composition of the present invention has little change with time in tackiness and drapeability and high storage stability. Therefore, even if it is a prepreg after long-term storage, the handleability is good.
本発明の熱硬化性ビスマレイミド系樹脂組成物を硬化させて得られる樹脂はガラス転移温度が高い。そのため、本発明のプリプレグを用いて作製される繊維強化複合材料は、耐熱性が高い。 The resin obtained by curing the thermosetting bismaleimide resin composition of the present invention has a high glass transition temperature. Therefore, the fiber reinforced composite material produced using the prepreg of the present invention has high heat resistance.
本発明のプリプレグを用いて作製される繊維強化複合材料は、そのマトリックス樹脂内に衝撃を吸収するゴム粒子が分散しているため、破壊靭性が高い。 The fiber-reinforced composite material produced using the prepreg of the present invention has high fracture toughness because rubber particles that absorb impact are dispersed in the matrix resin.
以下、本発明の熱硬化性ビスマレイミド系樹脂組成物と該熱硬化性ビスマレイミド系樹脂組成物を用いて構成される本発明のプリプレグの詳細について説明する。 Hereinafter, the details of the thermosetting bismaleimide resin composition of the present invention and the prepreg of the present invention constituted by using the thermosetting bismaleimide resin composition will be described.
(1)熱硬化性ビスマレイミド系樹脂組成物
本発明の熱硬化性ビスマレイミド系樹脂組成物(以下、「本樹脂組成物」ともいう)は、
(a)成分:ビスマレイミド化合物と、
(b)成分:アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物と、
(c)成分:アミン末端基を有するアクリロニトリル−ブタジエンコポリマーと、
を必須成分として含有する。また、任意成分として熱可塑性樹脂を含むことが好ましい。
(1) Thermosetting Bismaleimide Resin Composition The thermosetting bismaleimide resin composition of the present invention (hereinafter also referred to as “the resin composition”)
(A) component: a bismaleimide compound;
(B) component: an alkenylphenol and / or an alkenylphenol ether compound;
(C) component: an acrylonitrile-butadiene copolymer having amine end groups;
Is contained as an essential component. Moreover, it is preferable that a thermoplastic resin is included as an arbitrary component.
本樹脂組成物は、加熱時に(a)成分と(b)成分とが反応して硬化するとともに、(c)成分のアミン末端基と(a)成分とが反応してマレイミド構造を形成する。したがって、本樹脂組成物中の(c)成分は、硬化反応時に高分子量化して相分離し、マトリックス樹脂中に粒子として析出する。この相分離によって析出した粒子(以下、「ゴム粒子」ともいう)は、ゴムの主鎖を有しており、マトリックス樹脂内に強固に結合している。このゴム粒子は、繊維強化複合材料に対する外部からの衝撃を吸収してクラック伝播を抑制する。その結果、本樹脂組成物を用いて最終的に得られる繊維強化複合材料は、破壊靭性が高くなる。 In the present resin composition, the component (a) and the component (b) react and cure during heating, and the amine end group of the component (c) reacts with the component (a) to form a maleimide structure. Accordingly, the component (c) in the present resin composition has a high molecular weight during the curing reaction, phase-separates, and precipitates as particles in the matrix resin. Particles precipitated by this phase separation (hereinafter also referred to as “rubber particles”) have a main chain of rubber and are firmly bonded in the matrix resin. This rubber particle absorbs the impact from the outside with respect to a fiber reinforced composite material, and suppresses crack propagation. As a result, the fiber reinforced composite material finally obtained using this resin composition has high fracture toughness.
(1−1)ビスマレイミド化合物
本樹脂組成物に配合されるビスマレイミド化合物(以下、BMIともいう)としては、従来公知のビスマレイミド化合物を用いることができる。例えば、下記式(1)で表されるビスマレイミド化合物が挙げられる。
(1-1) Bismaleimide Compound As the bismaleimide compound (hereinafter also referred to as BMI) blended in the resin composition, a conventionally known bismaleimide compound can be used. For example, the bismaleimide compound represented by following formula (1) is mentioned.
[式(1)中、R1〜R4は、それぞれ独立に、−H、−CH3、−C2H5、−C3H7、−F、−Cl、−Br及び−Iからなる群から選ばれる基を表す。Xについては後述する。]
本発明においては、ビスマレイミド化合物は、芳香環構造を含むビスマレイミド化合物と芳香環構造を含まないビスマレイミド化合物とを併用することが好ましい。これらを併用することにより、得られるプリプレグの取扱い性を向上させることができる。
[In Formula (1), R 1 to R 4 each independently include —H, —CH 3 , —C 2 H 5 , —C 3 H 7 , —F, —Cl, —Br, and —I. Represents a group selected from the group; X will be described later. ]
In the present invention, the bismaleimide compound is preferably a combination of a bismaleimide compound containing an aromatic ring structure and a bismaleimide compound not containing an aromatic ring structure. By using these together, the handleability of the obtained prepreg can be improved.
(1−1−1)芳香族ビスマレイミド化合物
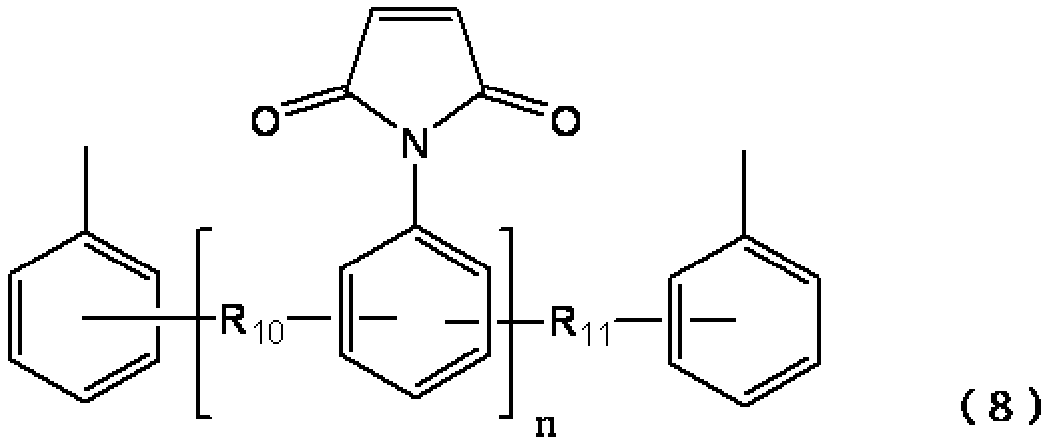
ビスマレイミド化合物が芳香環構造を含む(以下、「芳香族ビスマレイミド化合物」ともいう)場合、式(1)中のXは、以下の式(2)〜(8)に記載する構造であることが好ましい。
(1-1-1) Aromatic Bismaleimide Compound When the bismaleimide compound contains an aromatic ring structure (hereinafter also referred to as “aromatic bismaleimide compound”), X in formula (1) is the following formula (2 ) To (8) are preferred.
[式(5)中、R5は、−CH2−、−C(CH3)2−、−O−、−SO2−を表す。] Wherein (5), R 5 is, -CH 2 -, - C ( CH 3) 2 -, - O -, - SO 2 - represents a. ]
[式(6)中、R5は、−CH2−、−C(CH3)2−、−O−、−SO2−を表す。また、R6〜R9は、それぞれ独立に、−H、−CH3、−C2H5、−C3H7、−F、−Cl、−Br及び−Iからなる群から選ばれる基を表す。] Wherein (6), R 5 is, -CH 2 -, - C ( CH 3) 2 -, - O -, - SO 2 - represents a. R 6 to R 9 are each independently a group selected from the group consisting of —H, —CH 3 , —C 2 H 5 , —C 3 H 7 , —F, —Cl, —Br and —I. Represents. ]
[式(7)中、R5は、−CH2−、−C(CH3)2−、−O−、−SO2−を表す。] Wherein (7), R 5 is, -CH 2 -, - C ( CH 3) 2 -, - O -, - SO 2 - represents a. ]
[式(8)中、R10〜R11は、それぞれ独立に、−CH2−、−C(CH3)2−、−O−、−SO2−を表す。式(8)中、nは0〜0.5である。] Wherein (8), R 10 ~R 11 are each independently, -CH 2 -, - C ( CH 3) 2 -, - O -, - SO 2 - represents a. In formula (8), n is 0-0.5. ]
このような芳香族ビスマレイミド化合物としては、N,N’−4,4’−ジフェニルメタンビスマレイミド、N,N’−4,4’−ジフェニルエーテルビスマレイミド、N,N’−m−フェニレンビスマレイミド、N,N’−p−フェニレンビスマレイミド、N,N’−m−トルイレンビスマレイミド、N,N’−4,4’−ビフェニレンビスマレイミド、N,N’−4,4’−(3,3’−ジメチルビフェニレン)ビスマレイミド、2,2−ビス[4−(4−マレイミドフェノキシ)フェニル]プロパン、3,3’−ジメチル−5,5’−ジエチル-4,4’−ジフェニルメタンビスマレイミド、4−メチル−1,3−フェニレンビスマレイミド、N,N’−4,4’−ジフェニルスルフォンビスマレイミド、N,N’−4,4’−ベンゾフェノンビスマレイミド等を挙げることができる。 Examples of such aromatic bismaleimide compounds include N, N′-4,4′-diphenylmethane bismaleimide, N, N′-4,4′-diphenyl ether bismaleimide, N, N′-m-phenylene bismaleimide, N, N′-p-phenylene bismaleimide, N, N′-m-toluylene bismaleimide, N, N′-4,4′-biphenylene bismaleimide, N, N′-4,4 ′-(3 3′-dimethylbiphenylene) bismaleimide, 2,2-bis [4- (4-maleimidophenoxy) phenyl] propane, 3,3′-dimethyl-5,5′-diethyl-4,4′-diphenylmethane bismaleimide, 4-methyl-1,3-phenylenebismaleimide, N, N′-4,4′-diphenylsulfone bismaleimide, N, N′-4,4′-benzoph Examples include enone bismaleimide.
加熱硬化後の繊維強化複合材料の耐熱性の観点からは、N,N’−4,4’−ジフェニルメタンビスマレイミド、N,N’−4,4’−ジフェニルエーテルビスマレイミド、N,N’−m−トルイレンビスマレイミド、2,2−ビス[4−(4−マレイミドフェノキシ)フェニル]プロパン、4−メチル−1,3−フェニレンビスマレイミド、N,N’−4,4’−ジフェニルスルフォンビスマレイミド、N,N’−4,4’−ベンゾフェノンビスマレイミドが好ましく、N,N’−4,4’−ジフェニルメタンビスマレイミド、N,N’−4,4’−ジフェニルエーテルビスマレイミド、N,N’−m−トルイレンビスマレイミド、2,2−ビス[4−(4−マレイミドフェノキシ)フェニル]プロパン、4−メチル−1,3−フェニレンビスマレイミドが特に好ましい。これらの芳香族ビスマレイミド化合物は、単独で使用しても良く、2種類以上を併用しても良い。 From the viewpoint of heat resistance of the fiber-reinforced composite material after heat curing, N, N′-4,4′-diphenylmethane bismaleimide, N, N′-4,4′-diphenyl ether bismaleimide, N, N′-m -Toluylene bismaleimide, 2,2-bis [4- (4-maleimidophenoxy) phenyl] propane, 4-methyl-1,3-phenylenebismaleimide, N, N'-4,4'-diphenylsulfon bismaleimide N, N′-4,4′-benzophenone bismaleimide, N, N′-4,4′-diphenylmethane bismaleimide, N, N′-4,4′-diphenyl ether bismaleimide, N, N′— m-toluylene bismaleimide, 2,2-bis [4- (4-maleimidophenoxy) phenyl] propane, 4-methyl-1,3-phenylene Sumareimido is particularly preferred. These aromatic bismaleimide compounds may be used alone or in combination of two or more.
本樹脂組成物における芳香族ビスマレイミド化合物の含有量は、本樹脂組成物の全質量に対して20〜80質量%であることが好ましく、35〜65質量%であることがより好ましく、35〜50質量%であることが特に好ましい。芳香族ビスマレイミド化合物の含有量が20質量%未満である場合、本樹脂組成物を用いて得られる繊維強化複合材料の耐熱性が低くなる傾向がある。芳香族ビスマレイミド化合物の含有量が80質量%を超える場合、本樹脂組成物を用いて最終的に得られる繊維強化複合材料の破壊靭性が低下しやすい。 The content of the aromatic bismaleimide compound in the resin composition is preferably 20 to 80% by mass, more preferably 35 to 65% by mass, more preferably 35 to 65% by mass with respect to the total mass of the resin composition. It is especially preferable that it is 50 mass%. When the content of the aromatic bismaleimide compound is less than 20% by mass, the heat resistance of the fiber-reinforced composite material obtained using the resin composition tends to be low. When the content of the aromatic bismaleimide compound exceeds 80% by mass, the fracture toughness of the fiber-reinforced composite material finally obtained using the resin composition tends to decrease.
(1−1−2)脂肪族ビスマレイミド化合物
ビスマレイミド化合物が芳香環構造を含まない(以下、「脂肪族ビスマレイミド化合物」ともいう)場合、式(1)中のXは、以下の式(9)〜(11)に記載する構造であることが好ましい。
(1-1-2) Aliphatic bismaleimide compound When the bismaleimide compound does not contain an aromatic ring structure (hereinafter, also referred to as “aliphatic bismaleimide compound”), X in formula (1) represents the following formula ( The structure described in 9) to (11) is preferable.
[式(9)中、nは10以下の整数であり、1、2、3、4、6が好ましい。] [In Formula (9), n is an integer of 10 or less, and 1, 2, 3, 4, 6 are preferable. ]
このような脂肪族ビスマレイミド化合物としては、1,6’−ビスマレイミド−(2,2,4−トリメチル)ヘキサン、ヘキサメチレンジアミンビスマレイミド、N,N’−1,2−エチレンビスマレイミド、N,N’−1,3−プロピレンビスマレイミド、N,N’−1,4−テトラメチレンビスマレイミドを挙げることができる。1,6’−ビスマレイミド−(2,2,4−トリメチル)ヘキサン、ヘキサメチレンジアミンビスマレイミドは特に好ましい。脂肪族ビスマレイミド化合物は、単独で使用しても良く、2種類以上を併用してもよい。 Examples of such aliphatic bismaleimide compounds include 1,6′-bismaleimide- (2,2,4-trimethyl) hexane, hexamethylenediamine bismaleimide, N, N′-1,2-ethylenebismaleimide, N , N′-1,3-propylene bismaleimide and N, N′-1,4-tetramethylene bismaleimide. 1,6'-bismaleimide- (2,2,4-trimethyl) hexane and hexamethylenediamine bismaleimide are particularly preferred. The aliphatic bismaleimide compound may be used alone or in combination of two or more.
本樹脂組成物における脂肪族ビスマレイミド化合物の含有量は、樹脂組成物の全質量に対して3〜30質量%であることが好ましく、5〜20質量%であることがより好ましく、7〜15質量%であることが特に好ましい。脂肪族ビスマレイミド化合物の配合量が3質量%未満である場合、本樹脂組成物を用いて最終的に得られる繊維強化複合材料の靭性が乏しく、破壊靭性が低下しやすい。脂肪族ビスマレイミド化合物の含有量が30質量%を超える場合、本樹脂組成物を用いて最終的に得られる繊維強化複合材料の耐熱性が低下しやすい傾向がある。 The content of the aliphatic bismaleimide compound in the resin composition is preferably 3 to 30% by mass, more preferably 5 to 20% by mass, and more preferably 7 to 15% with respect to the total mass of the resin composition. It is particularly preferable that the content is% by mass. When the blending amount of the aliphatic bismaleimide compound is less than 3% by mass, the toughness of the fiber-reinforced composite material finally obtained using the resin composition is poor, and the fracture toughness tends to be lowered. When content of an aliphatic bismaleimide compound exceeds 30 mass%, there exists a tendency for the heat resistance of the fiber reinforced composite material finally obtained using this resin composition to fall easily.
(1−2)アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物
本樹脂組成物に配合されるアルケニルフェノールエーテルは、フェノール系化合物とアルケニルハライドとの反応により得られ、アルケニルフェノールエーテルをクライゼン転移することによりアルケニルフェノールが得られる(特開昭52―994号公報)。本発明においては、アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物には、その転移構造体が含まれていてもよい。
(1-2) Alkenylphenol and / or alkenylphenol ether compound The alkenylphenol ether compounded in the present resin composition is obtained by the reaction of a phenolic compound and an alkenyl halide. Phenol is obtained (Japanese Patent Laid-Open No. 52-994). In the present invention, the transition structure may be contained in the alkenylphenol and / or alkenylphenol ether compound.
アルケニルフェノール及び/又はアルケニルフェノールエーテルとしては、アリルフェノール、メタリルフェノール又はそれらのエーテルが好ましい。より好ましくは、アルケニルフェノール又はアルケニルフェノールエーテルは以下の式(12)〜(16)の化合物である。 As the alkenylphenol and / or alkenylphenol ether, allylphenol, methallylphenol or an ether thereof is preferable. More preferably, the alkenylphenol or alkenylphenol ether is a compound of the following formulas (12) to (16).
[式(12)中、R12、R13、R14はそれぞれ独立して水素又は炭素数2〜10のアルケニル基であり、好ましくはアリル基又はプロペニル基である。ただし、R12、R13、R14の少なくとも1個は炭素数2〜10のアルケニル基である。] [In Formula (12), R < 12 >, R < 13 >, R < 14 > is respectively independently hydrogen or a C2-C10 alkenyl group, Preferably they are an allyl group or a propenyl group. However, at least one of R 12 , R 13 , and R 14 is an alkenyl group having 2 to 10 carbon atoms. ]
[式(13)中、R15は、直接結合、−CH2−、−C(CH3)2−、−O−、−S−、−SO−又は−SO2−である。R16、R17、R18、R19はそれぞれ独立して水素又は炭素数2〜10のアルケニル基であり、好ましくはアリル基又はプロペニル基である。ただし、R16、R17、R18、R19の少なくとも1個は炭素数2〜10のアルケニル基である。] [In the formula (13), R 15 represents a direct bond, —CH 2 —, —C (CH 3 ) 2 —, —O—, —S—, —SO— or —SO 2 —. R 16 , R 17 , R 18 and R 19 are each independently hydrogen or an alkenyl group having 2 to 10 carbon atoms, preferably an allyl group or a propenyl group. However, at least one of R 16 , R 17 , R 18 , and R 19 is an alkenyl group having 2 to 10 carbon atoms. ]
式(13)のうち、以下の式(14)の化合物は特に好ましい。 Of the formula (13), compounds of the following formula (14) are particularly preferred.
[式(14)中、R15は、直接結合、−CH2−、−C(CH3)2−、−O−、−S−、−SO−又は−SO2−を表す。] [In the formula (14), R 15 represents a direct bond, —CH 2 —, —C (CH 3 ) 2 —, —O—, —S—, —SO— or —SO 2 —. ]
[式(15)中、R20、R21は、直接結合、−CH2−、−C(CH3)2−、−O−、−S−、−SO−又は−SO2−である。R22、R23、R24、R25、R26、R27は、それぞれ独立して水素、炭素数1〜4のアルキル基、又は炭素数2〜10のアルケニル基であり、好ましくはアリル基又はプロペニル基である。ただし、R22、R23、R24、R25、R26、R27の少なくとも1個は炭素数2〜10のアルケニル基である。Pは0〜10の整数である。] [In the formula (15), R 20 and R 21 are a direct bond, —CH 2 —, —C (CH 3 ) 2 —, —O—, —S—, —SO— or —SO 2 —. R 22 , R 23 , R 24 , R 25 , R 26 , R 27 are each independently hydrogen, an alkyl group having 1 to 4 carbon atoms, or an alkenyl group having 2 to 10 carbon atoms, preferably an allyl group Or it is a propenyl group. However, at least one of R 22 , R 23 , R 24 , R 25 , R 26 and R 27 is an alkenyl group having 2 to 10 carbon atoms. P is an integer of 0-10. ]
[式(16)中、R15は、直接結合、−CH2−、−C(CH3)2−、−O−、−S−、−SO−又は−SO2−を表す。R28、R29は、それぞれ独立して水素、炭素数1〜4のアルキル基、又は炭素数2〜10のアルケニル基であり、好ましくはアリル基又はプロペニル基である。ただし、R28、R29の少なくとも1個は炭素数2〜10のアルケニル基である。] [In the formula (16), R 15 represents a direct bond, —CH 2 —, —C (CH 3 ) 2 —, —O—, —S—, —SO— or —SO 2 —. R 28 and R 29 are each independently hydrogen, an alkyl group having 1 to 4 carbon atoms, or an alkenyl group having 2 to 10 carbon atoms, and preferably an allyl group or a propenyl group. However, at least one of R 28 and R 29 is an alkenyl group having 2 to 10 carbon atoms. ]
このようなアルケニルフェノール又はアルケニルフェノールエーテル化合物としては、O,O’−ジアリルビスフェノールA、4,4’−ジヒドロキシ−3,3’−ジアリルジフェニル、ビス(4−ヒドロキシ−3−アリルフェニル)メタン、2,2’−ビス(4−ヒドロキシ−3,5−ジアリルフェニル)プロパン、2,2’−ジアリルビスフェノールF、4,4’−ジヒドロキシ−3,3’−ジアリルジフェニルエーテル、4,4’−ビス−O−プロペニルフェノキシ−ベンゾフェノン等を挙げることができる。これらの中でも、加熱硬化後の樹脂のガラス転移点が高いため、O,O’−ジアリルビスフェノールA、2,2’−ビス(4−ヒドロキシ−3,5−ジアリルフェニル)プロパン、2,2’−ジアリルビスフェノールF等が好ましい。O,O’−ジアリルビスフェノールAは、本樹脂組成物の粘度を低くするため特に好ましい。本樹脂組成物では、アルケニルフェノール及び/又はアルケニルフェノールエーテルは単独で用いても良く、2種類以上を混合して用いてもよい。 Examples of the alkenylphenol or alkenylphenol ether compound include O, O′-diallylbisphenol A, 4,4′-dihydroxy-3,3′-diallyldiphenyl, bis (4-hydroxy-3-allylphenyl) methane, 2,2′-bis (4-hydroxy-3,5-diallylphenyl) propane, 2,2′-diallylbisphenol F, 4,4′-dihydroxy-3,3′-diallyldiphenyl ether, 4,4′-bis -O-propenylphenoxy-benzophenone and the like can be mentioned. Among these, since the glass transition point of the resin after heat curing is high, O, O′-diallylbisphenol A, 2,2′-bis (4-hydroxy-3,5-diallylphenyl) propane, 2,2 ′ -Diallyl bisphenol F etc. are preferable. O, O'-diallylbisphenol A is particularly preferred because it lowers the viscosity of the resin composition. In the present resin composition, alkenylphenol and / or alkenylphenol ether may be used alone or in combination of two or more.
アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物は、ビスマレイミド化合物の硬化剤として機能する。本樹脂組成物における配合量は、10〜70質量%であることが好ましく、20〜50質量%であることがより好ましく、25〜40質量%であることが特に好ましい。本樹脂組成物は、アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物を上記の所定の範囲内で適宜含有することにより、粘度が調整され、良好な成形加工性を得ることができる。また、強化繊維基材内に本樹脂組成物を十分に含浸させて、強化繊維基材と十分に接着させることができる。 The alkenylphenol and / or alkenylphenol ether compound functions as a curing agent for the bismaleimide compound. The blending amount in the resin composition is preferably 10 to 70% by mass, more preferably 20 to 50% by mass, and particularly preferably 25 to 40% by mass. The resin composition can contain alkenylphenol and / or alkenylphenol ether compound as appropriate within the above predetermined range, thereby adjusting the viscosity and obtaining good moldability. Moreover, the present resin composition can be sufficiently impregnated into the reinforcing fiber base material and can be sufficiently adhered to the reinforcing fiber base material.
本樹脂組成物は、芳香族ビスマレイミド化合物及び/又は脂肪族ビスマレイミド化合物の一部又は全部が、アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に溶解していても良い。本樹脂組成物は、好ましくは液相成分と固相成分を含有して成り、樹脂組成物中の固相成分の割合が5〜50質量%であることが好ましく、15〜45質量%であることがより好ましく、25〜40質量%であることが特に好ましい。なお、本発明において固相成分とは、温度80℃の樹脂組成物中において固形状で存在する成分をいう。固相成分のうちビスマレイミド樹脂は、本樹脂組成物を用いて構成するプリプレグの成形温度以下で樹脂組成物中に溶解する。また、液相成分とは、温度80℃の樹脂組成物中において液状で存在する成分をいう。 In the present resin composition, part or all of the aromatic bismaleimide compound and / or the aliphatic bismaleimide compound may be dissolved in the alkenylphenol and / or alkenylphenol ether compound. The present resin composition preferably contains a liquid phase component and a solid phase component, and the ratio of the solid phase component in the resin composition is preferably 5 to 50% by mass, and preferably 15 to 45% by mass. It is more preferable that the content is 25 to 40% by mass. In addition, in this invention, a solid-phase component means the component which exists in solid form in the resin composition of temperature 80 degreeC. Among the solid phase components, the bismaleimide resin is dissolved in the resin composition at a temperature equal to or lower than the molding temperature of the prepreg formed using the resin composition. The liquid phase component refers to a component that exists in a liquid state in a resin composition at a temperature of 80 ° C.
樹脂組成物中の固相成分の割合が50質量%を超える場合、樹脂組成物の粘度が著しく高くなる場合がある。その結果、樹脂組成物の製造工程やプリプレグの製造工程において取扱い性が著しく悪化する場合がある。樹脂組成物中の固相成分の割合が5質量%未満の場合、ビスマレイミド化合物がアルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に溶解している。そして、この溶解時の加熱によって硬化反応が同時に進行することにより、樹脂組成物が高分子量化して粘度が高くなる場合がある。その結果、樹脂組成物の製造工程やプリプレグの製造工程における取扱い性が著しく悪化する場合がある。 When the ratio of the solid phase component in the resin composition exceeds 50% by mass, the viscosity of the resin composition may be remarkably increased. As a result, the handleability may be significantly deteriorated in the resin composition manufacturing process and the prepreg manufacturing process. When the ratio of the solid phase component in the resin composition is less than 5% by mass, the bismaleimide compound is dissolved in the alkenylphenol and / or alkenylphenol ether compound. And when a hardening reaction advances simultaneously by the heating at the time of melt | dissolution, a resin composition may become high molecular weight and a viscosity may become high. As a result, the handleability in the manufacturing process of the resin composition and the manufacturing process of the prepreg may be significantly deteriorated.
(1−3)アミン末端基を有するアクリロニトリル−ブタジエンコポリマー
本樹脂組成物には、アミン末端基を有するアクリロニトリル−ブタジエンコポリマー(以下、「ATBN」ともいう)が配合される。このATBNは、公知のATBNを用いることができる。その中でも、ニトリル−ブタジエンゴム(NBR)の分子の両末端にアミノ基が導入されたATBNが好ましい。下記式(17)で表されるATBNは特に好ましい。
(1-3) Acrylonitrile-butadiene copolymer having amine end groups An acrylonitrile-butadiene copolymer (hereinafter also referred to as “ATBN”) having amine end groups is blended in the resin composition. As this ATBN, known ATBN can be used. Among them, ATBN in which amino groups are introduced at both ends of a nitrile-butadiene rubber (NBR) molecule is preferable. ATBN represented by the following formula (17) is particularly preferred.
[式(17)中、mは70〜95、nは5〜30である。] [In Formula (17), m is 70-95 and n is 5-30. ]
このようなATBNとしては、「HYPRO ATBN 1300X16(登録商標)」、「HYPRO ATBN 1300X45(登録商標)」(いずれもEmerald Performance Materials社製)のような市販品を好ましく用いることができる。 As such ATBN, commercially available products such as “HYPRO ATBN 1300X16 (registered trademark)” and “HYPRO ATBN 1300X45 (registered trademark)” (both manufactured by Emerald Performance Materials) can be preferably used.
本樹脂組成物におけるATBNの含有量は、3〜30質量%であり、5〜25質量%であることが好ましく、10〜20質量%であることが特に好ましい。ATBNの含有量が3質量%以上である場合、ATBNから形成されるゴム粒子は、繊維強化複合材料が受ける衝撃を効率的に吸収することができる。そのため、繊維強化複合材料の破壊靭性が高くなる。ATBNの含有量が30質量%以下であれば、樹脂組成物の粘度が高くなりすぎず、樹脂組成物の製造工程やプリプレグの製造工程における取扱い性が優れている。上記所定量のATBNから形成されるゴム粒子が、マトリックス樹脂中に均一に分散されている繊維強化複合材料は、衝撃に対するクラック伝播が抑制される。すなわち、破壊靭性が高い。 The content of ATBN in the resin composition is 3 to 30% by mass, preferably 5 to 25% by mass, and particularly preferably 10 to 20% by mass. When the content of ATBN is 3% by mass or more, the rubber particles formed from ATBN can efficiently absorb the impact received by the fiber reinforced composite material. Therefore, the fracture toughness of the fiber reinforced composite material is increased. When the content of ATBN is 30% by mass or less, the viscosity of the resin composition does not become too high, and the handleability in the resin composition manufacturing process and the prepreg manufacturing process is excellent. The fiber reinforced composite material in which the rubber particles formed from the predetermined amount of ATBN are uniformly dispersed in the matrix resin suppresses crack propagation to impact. That is, fracture toughness is high.
本樹脂組成物には、ATBNとビスマレイミド化合物とを予め反応させたマレイミド末端ゴムを配合することもできる。マレイミド末端ゴムは、両末端にアミノ基を有するATBNとビスマレイミド化合物とを、室温〜80℃で30分撹拌することにより得ることができる。又は、両末端にアミノ基を有するATBNとビスマレイミド化合物とを、溶媒中で室温〜80℃で30分撹拌した後、該溶媒を除去することにより得ることができる。用いる溶媒としては、例えば、メチルエチルケトン(MEK)、アセトンが挙げられる。 A maleimide-terminated rubber obtained by reacting ATBN and a bismaleimide compound in advance can also be blended with the resin composition. The maleimide-terminated rubber can be obtained by stirring ATBN having an amino group at both ends and a bismaleimide compound at room temperature to 80 ° C. for 30 minutes. Alternatively, ATBN having an amino group at both ends and a bismaleimide compound can be obtained by stirring in a solvent at room temperature to 80 ° C. for 30 minutes, and then removing the solvent. Examples of the solvent to be used include methyl ethyl ketone (MEK) and acetone.
このようにして得られるマレイミド末端ゴムは、ゴムの主鎖を有し、分子量が大きい。そのため、樹脂組成物の硬化後に得られる樹脂の靱性は高く、力学特性及び耐熱性が優れる。また、分子の両末端の常温における反応性が低い。そのため、室温保存安定性が高い。すなわち、プリプレグの製造用の樹脂組成物として特に好ましい性状が付与される。 The maleimide-terminated rubber thus obtained has a rubber main chain and has a large molecular weight. Therefore, the toughness of the resin obtained after curing of the resin composition is high, and the mechanical properties and heat resistance are excellent. In addition, the reactivity of both ends of the molecule at room temperature is low. Therefore, room temperature storage stability is high. That is, a particularly preferable property is imparted as a resin composition for producing a prepreg.
(1−4)熱可塑性樹脂
本樹脂組成物は、熱可塑性樹脂を含有することが好ましい。熱可塑性樹脂としては、公知の熱可塑性樹脂を使用することができる。アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に可溶な熱可塑性樹脂(以下、「可溶性熱可塑性樹脂」ともいう)、アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に不溶な熱可塑性樹脂(以下、「不溶性熱可塑性樹脂」ともいう)のいずれも用いることができる。
(1-4) Thermoplastic Resin The resin composition preferably contains a thermoplastic resin. A known thermoplastic resin can be used as the thermoplastic resin. Thermoplastic resins soluble in alkenylphenol and / or alkenylphenol ether compounds (hereinafter also referred to as “soluble thermoplastic resins”), thermoplastic resins insoluble in alkenylphenol and / or alkenylphenol ether compounds (hereinafter referred to as “insoluble heat”). Any of the “plastic resin” can be used.
(1−4−1)可溶性熱可塑性樹脂
本発明において、「アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に可溶」とは、繊維強化複合材料を成形する温度条件下で、アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に一部又は全部が溶解することを意味する。繊維強化複合材料の成形過程において、可溶性熱可塑性樹脂は、樹脂組成物が加熱されることによって該樹脂組成物中に溶解し、該樹脂組成物の粘度を増加させる。これにより成形中における樹脂組成物のフローを防止することができる。
(1-4-1) Soluble thermoplastic resin In the present invention, “soluble in alkenylphenol and / or alkenylphenol ether compound” means alkenylphenol and / or alkenyl under the temperature conditions for molding a fiber-reinforced composite material. It means that a part or all of the phenol ether compound is dissolved. In the molding process of the fiber reinforced composite material, the soluble thermoplastic resin dissolves in the resin composition when the resin composition is heated, and increases the viscosity of the resin composition. Thereby, the flow of the resin composition during molding can be prevented.
可溶性熱可塑性樹脂としては、ポリエーテルスルホン、ポリスルホン、ポリエーテルイミド、ポリイミド等を挙げることができる。 Examples of the soluble thermoplastic resin include polyethersulfone, polysulfone, polyetherimide, and polyimide.
可溶性熱可塑性樹脂の含有量は、0.1〜15質量%であることが好ましく、1〜10質量%であることがより好ましい。可溶性熱可塑性樹脂の含有量が0.1質量%未満の場合、樹脂組成物の粘度が十分に上昇せず、樹脂組成物のフローを招くおそれがある。可溶性熱可塑性樹脂の含有量が15質量%を超える場合、樹脂組成物の粘度が高くなり、取扱い性が著しく悪化する場合がある。 The content of the soluble thermoplastic resin is preferably 0.1 to 15% by mass, and more preferably 1 to 10% by mass. When the content of the soluble thermoplastic resin is less than 0.1% by mass, the viscosity of the resin composition is not sufficiently increased, and the resin composition may be flowed. When the content of the soluble thermoplastic resin exceeds 15% by mass, the viscosity of the resin composition increases, and the handleability may be significantly deteriorated.
(1−4−2)不溶性熱可塑性樹脂
不溶性熱可塑性樹脂は、必要に応じて本発明の樹脂組成物に含有される。本発明において「アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に不溶な熱可塑性樹脂」とは、少なくとも繊維強化複合材料を成形する温度条件下で、アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物に溶解しない熱可塑性樹脂をいう。不溶性熱可塑性樹脂としては、ポリイミド樹脂を挙げることができる。
(1-4-2) Insoluble thermoplastic resin An insoluble thermoplastic resin is contained in the resin composition of this invention as needed. In the present invention, the term “thermoplastic resin insoluble in alkenylphenol and / or alkenylphenol ether compound” means a thermoplastic that does not dissolve in alkenylphenol and / or alkenylphenol ether compound at least under the temperature conditions for molding the fiber-reinforced composite material. Refers to resin. An example of the insoluble thermoplastic resin is a polyimide resin.
不溶性熱可塑性樹脂の含有量は、0.1〜15質量%であることが好ましく、1〜10質量%であることがより好ましい。不溶性熱可塑性樹脂の含有量が0.1質量%未満の場合、樹脂組成物の粘度が十分に上昇せず、樹脂組成物のフローを招くおそれがある。不溶性熱可塑性樹脂の含有量が15質量%を超える場合、樹脂組成物の粘度が高くなり取扱い性が著しく悪化する場合がある。不溶性熱可塑性樹脂の粒子径は、特に限定されないが、0.1〜100μmであることが好ましく、1〜50μmであることがより好ましい。 The content of the insoluble thermoplastic resin is preferably 0.1 to 15% by mass, and more preferably 1 to 10% by mass. When the content of the insoluble thermoplastic resin is less than 0.1% by mass, the viscosity of the resin composition is not sufficiently increased, and the flow of the resin composition may be caused. When the content of the insoluble thermoplastic resin exceeds 15% by mass, the viscosity of the resin composition becomes high, and the handleability may be significantly deteriorated. Although the particle diameter of an insoluble thermoplastic resin is not specifically limited, It is preferable that it is 0.1-100 micrometers, and it is more preferable that it is 1-50 micrometers.
(1−5)その他の成分
本樹脂組成物には、耐熱性、成形加工性及び靱性を損なわない限り、他の成分を含有させることができる。他の成分として、重合防止剤、導電性粒子、導電性フィラー、無機フィラー、ゴム状成分、靭性付与剤、安定剤や離型剤、着色剤等が例示される。
(1-5) Other components The present resin composition may contain other components as long as the heat resistance, molding processability and toughness are not impaired. Examples of other components include polymerization inhibitors, conductive particles, conductive fillers, inorganic fillers, rubbery components, toughness imparting agents, stabilizers, mold release agents, and colorants.
(2)熱硬化性ビスマレイミド系樹脂組成物の製造方法
本樹脂組成物を製造するには、必須成分として(a)乃至(c)成分、任意成分として熱可塑性樹脂及びその他の成分を混合して均一に溶解及び/又は分散した組成物とすればよく、その方法に特に制限はない。これらの成分を常温で混合してもよいが、経済的に製造するためには加熱して混合することが好ましい。この場合、加熱温度は通常30〜180℃であり、50〜150℃が好ましく、60〜140℃がより好ましい。180℃を超える温度では重合反応が速くなり、本樹脂組成物が混合中に硬化する可能性がある。
(2) Manufacturing method of thermosetting bismaleimide resin composition To manufacture this resin composition, (a) to (c) components as essential components and thermoplastic resin and other components as optional components are mixed. And a method in which the composition is uniformly dissolved and / or dispersed, and the method is not particularly limited. These components may be mixed at room temperature, but it is preferable to mix by heating in order to produce economically. In this case, heating temperature is 30-180 degreeC normally, 50-150 degreeC is preferable and 60-140 degreeC is more preferable. When the temperature exceeds 180 ° C., the polymerization reaction is accelerated, and the resin composition may be cured during mixing.
本樹脂組成物を混練する場合、(a)成分の一部又は全部を固相成分として配合することが好ましい。即ち、(b)成分に(a)成分の一部又は全部を溶解させないことが好ましい。(a)成分と(b)成分とを完全に加熱溶解させる場合、硬化反応が進む結果、取扱い性や保存安定性が悪化する場合がある。(b)成分に(c)成分や他の成分を加熱溶解した後、温度を下げてから(a)成分を加える等の方法により、(a)成分と(b)成分との硬化反応を抑制できる。 When kneading this resin composition, it is preferable to blend a part or all of the component (a) as a solid phase component. That is, it is preferable not to dissolve part or all of the component (a) in the component (b). When the component (a) and the component (b) are completely dissolved by heating, the handleability and storage stability may deteriorate as a result of the progress of the curing reaction. (B) Component (c) and other components are heated and dissolved in component (b), then the temperature is lowered and then component (a) is added to suppress the curing reaction between component (a) and component (b). it can.
混練は、一段で行ってもよいし、多段で行ってもよい。また、樹脂組成物の各成分の混合順序は限定されないが、固相成分として配合する成分は、樹脂組成物中の他の成分が溶解した後に添加することが好ましい。これにより、固相成分を樹脂組成物中に均一に分散させやすくなる。混練時間は温度により相違するが、10〜180分が好ましい。 Kneading may be performed in one stage or in multiple stages. Moreover, although the mixing order of each component of a resin composition is not limited, It is preferable to add the component mix | blended as a solid-phase component after the other component in a resin composition melt | dissolves. This facilitates uniform dispersion of the solid phase component in the resin composition. The kneading time varies depending on the temperature, but is preferably 10 to 180 minutes.
可溶性熱可塑性樹脂のアルケニルフェノール及び/又はアルケニルフェノールエーテル化合物への溶解は、ビスマレイミド化合物と混練する前段階において、溶解温度80〜130℃で溶解することが好ましい。 The soluble thermoplastic resin is preferably dissolved in the alkenylphenol and / or alkenylphenol ether compound at a dissolution temperature of 80 to 130 ° C. before kneading with the bismaleimide compound.
不溶性熱可塑性樹脂を配合する場合は、粉体を樹脂組成物中に供給し、分散粒子として混合する。 When blending an insoluble thermoplastic resin, the powder is supplied into the resin composition and mixed as dispersed particles.
混練機械装置としては、ロールミル、プラネタリーミキサー、ニーダー、エクストルーダー、バンバリーミキサー等、従来公知の装置を用いることができる。 As the kneading machine apparatus, conventionally known apparatuses such as a roll mill, a planetary mixer, a kneader, an extruder, and a Banbury mixer can be used.
(3)プリプレグ
本発明のプリプレグ(以下、「本プリプレグ」ともいう)は、前述の熱硬化性ビスマレイミド系樹脂組成物が強化繊維基材に含浸しているプリプレグである。
(3) Prepreg The prepreg of the present invention (hereinafter also referred to as “present prepreg”) is a prepreg in which the reinforcing fiber base material is impregnated with the above-mentioned thermosetting bismaleimide resin composition.
本プリプレグの製造において、強化繊維基材を構成する強化繊維としては、炭素繊維、ガラス繊維、アラミド繊維、炭化ケイ素繊維、ポリエステル繊維、セラミック繊維、アルミナ繊維、ボロン繊維、金属繊維、鉱物繊維、岩石繊維及びスラッグ繊維などを挙げることができる。これらの強化繊維の中でも、炭素繊維、ガラス繊維、アラミド繊維が好ましく、比強度、比弾性率が良好で軽量かつ高強度の繊維強化複合材料が得られる炭素繊維がより好ましく、炭素繊維の中でも、引張強度に優れるポリアクリロニトリル(PAN)系炭素繊維が特に好ましい。 In the production of this prepreg, the reinforcing fibers constituting the reinforcing fiber substrate include carbon fibers, glass fibers, aramid fibers, silicon carbide fibers, polyester fibers, ceramic fibers, alumina fibers, boron fibers, metal fibers, mineral fibers, rocks. Examples thereof include fibers and slug fibers. Among these reinforcing fibers, carbon fibers, glass fibers, and aramid fibers are preferable, carbon fibers from which specific strength, specific elastic modulus is good, and a lightweight and high-strength fiber-reinforced composite material is more preferable, among carbon fibers, Polyacrylonitrile (PAN) -based carbon fibers having excellent tensile strength are particularly preferred.
強化繊維にPAN系炭素繊維を用いる場合、引張弾性率は、170〜600GPaであることが好ましく、220〜450GPaであることが特に好ましい。また、引張強度は3920MPa(400kgf/mm2)以上であることが好ましい。このような炭素繊維を用いることにより、繊維強化複合材料の機械的性質を向上できる。 When PAN-based carbon fiber is used as the reinforcing fiber, the tensile elastic modulus is preferably 170 to 600 GPa, and particularly preferably 220 to 450 GPa. The tensile strength is preferably 3920 MPa (400 kgf / mm 2 ) or more. By using such carbon fibers, the mechanical properties of the fiber-reinforced composite material can be improved.
強化繊維基材の形状は限定されないが、シート状物であることが加工性の点から好ましい。強化繊維シートとしては、例えば、多数本の強化繊維を一方向に引き揃えたシート状物や、平織や綾織などの二方向織物、多軸織物、不織布、マット、ニット、組紐、強化繊維を抄紙した紙などを挙げることができる。強化繊維シートの厚さは、0.01〜3mmが好ましく、0.1〜1.5mmがより好ましい。また、強化繊維シートの目付は、70〜400g/m2が好ましく、100〜300g/m2がより好ましい。 The shape of the reinforcing fiber base is not limited, but is preferably a sheet-like material from the viewpoint of workability. As the reinforcing fiber sheet, for example, a sheet-like material in which a large number of reinforcing fibers are aligned in one direction, bi-directional woven fabrics such as plain weave and twill weave, multiaxial woven fabric, non-woven fabric, mat, knit, braid, and reinforcing fiber And the like. The thickness of the reinforcing fiber sheet is preferably 0.01 to 3 mm, and more preferably 0.1 to 1.5 mm. Also, the basis weight of the reinforcing fiber sheet is preferably 70~400g / m 2, 100~300g / m 2 is more preferable.
本プリプレグ中の本樹脂組成物の含有量は、強化繊維基材と本樹脂組成物の合計質量に対して20〜60質量%が好ましく、30〜50質量%がより好ましい。本樹脂組成物の含有量が20質量%未満である場合、このプリプレグを用いて作製される繊維強化複合材料の内部にボイド等が発生する場合がある。本樹脂組成物の含有量が60質量%を超える場合、強化繊維の含有量が不足し、得られる繊維強化複合材料の強度が低下し易い。 20-60 mass% is preferable with respect to the total mass of a reinforced fiber base material and this resin composition, and, as for content of this resin composition in this prepreg, 30-50 mass% is more preferable. When the content of the resin composition is less than 20% by mass, voids or the like may be generated inside the fiber reinforced composite material produced using this prepreg. When content of this resin composition exceeds 60 mass%, content of a reinforced fiber runs short and the intensity | strength of the fiber reinforced composite material obtained falls easily.
本プリプレグの吸水率は、2〜40%が好ましく、4〜25%がより好ましい。本発明において吸水率は、プリプレグ中の空隙率を示す指標であり、吸水率が高いほどプリプレグ中の空隙率が高いことを示す。吸水率が高い場合、プリプレグ中に空隙が多いため、成形時の取扱い性が悪化する。また、製造される繊維強化複合材料に空隙が残りやすいため、その機械特性に悪影響を及す場合がある。吸水率が低い場合、プリプレグ中の空隙が少ないため、ドレープ性が低くなる。そのため、良好な成形加工性(形状追従性)が得られなくなる。なお、吸水率の測定方法は後述する。 The water absorption of the present prepreg is preferably 2 to 40%, more preferably 4 to 25%. In the present invention, the water absorption rate is an index indicating the porosity in the prepreg. The higher the water absorption rate, the higher the porosity in the prepreg. When the water absorption rate is high, there are many voids in the prepreg, and the handleability during molding deteriorates. Moreover, since voids are likely to remain in the manufactured fiber-reinforced composite material, the mechanical properties may be adversely affected. When the water absorption is low, there are few voids in the prepreg, so that the drapability is low. Therefore, good moldability (shape followability) cannot be obtained. In addition, the measuring method of a water absorption rate is mentioned later.
(4)プリプレグの製造方法
本プリプレグは、本樹脂組成物を強化繊維基材内に含浸させることにより製造することができる。本樹脂組成物を強化繊維基材内に含浸させる方法としては、公知の湿式法や乾式法を用いることができる。湿式法は有機溶媒を用いるため、樹脂組成物を含浸した後、有機溶媒を除去する必要がある。したがって、有機溶媒が残存する虞がない乾式法であるホットメルト法を用いることが好ましい。
(4) Manufacturing method of prepreg This prepreg can be manufactured by impregnating this resin composition in a reinforced fiber base material. As a method for impregnating the resin composition into the reinforcing fiber base, a known wet method or dry method can be used. Since the wet method uses an organic solvent, it is necessary to remove the organic solvent after impregnating the resin composition. Therefore, it is preferable to use a hot melt method which is a dry method in which there is no possibility that the organic solvent remains.
ホットメルト法は、本樹脂組成物と積重した強化繊維基材とを加圧下で加熱することにより、本樹脂組成物の粘度を低下させ、本樹脂組成物を強化繊維基材内に含浸させる。強化繊維基材がシート状物の場合、フィルム状に成形した本樹脂組成物を強化繊維基材に積重することが好ましい。 In the hot melt method, the resin composition and the reinforced fiber base material stacked are heated under pressure to reduce the viscosity of the resin composition and impregnate the reinforced fiber base material with the resin composition. . When the reinforcing fiber base is a sheet, it is preferable to stack the resin composition formed into a film on the reinforcing fiber base.
本樹脂組成物は、公知の方法でフィルム状に成形できる。例えば、本樹脂組成物をダイコーター、アプリケーター、リバースロールコーター、コンマコーター、ナイフコーターなどを用いて、離型紙、離型フィルムなどの支持体上に流延させることによりフィルム状に成形することができる。フィルムを製造する温度は、本樹脂組成物の粘度に応じて適宜設定される。通常、温度は60〜130℃が好ましく、80〜110℃がより好ましい。 This resin composition can be formed into a film by a known method. For example, the resin composition can be formed into a film by casting on a support such as release paper or release film using a die coater, applicator, reverse roll coater, comma coater, knife coater, etc. it can. The temperature which manufactures a film is suitably set according to the viscosity of this resin composition. Usually, the temperature is preferably 60 to 130 ° C, more preferably 80 to 110 ° C.
本樹脂組成物のフィルムの厚さは、概ね8〜350μmとすることが好ましく、10〜200μmとすることがより好ましい。 The thickness of the resin composition film is preferably about 8 to 350 μm, more preferably 10 to 200 μm.
強化繊維基材に本樹脂組成物を含浸させる際の加圧条件は、本樹脂組成物の組成や粘度に応じて適宜調整される。通常、線圧0.98〜245N/cmであり、より好ましくは19.6〜147N/cmである。線圧が0.98N/cm未満である場合、樹脂組成物を強化繊維シート内に十分に含浸させるのが困難である。加圧は、1回で行ってもよく、複数回に分けて行ってもよい。 The pressurizing condition for impregnating the reinforcing fiber substrate with the resin composition is appropriately adjusted according to the composition and viscosity of the resin composition. Usually, the linear pressure is 0.98 to 245 N / cm, more preferably 19.6 to 147 N / cm. When the linear pressure is less than 0.98 N / cm, it is difficult to sufficiently impregnate the reinforcing fiber sheet with the resin composition. The pressurization may be performed once or may be performed in a plurality of times.
強化繊維基材に本樹脂組成物を含浸させる際の加熱温度は、本樹脂組成物の粘度に応じて適宜調整される。通常、70〜160℃であり、80〜120℃が好ましい。加熱温度が70℃未満である場合、本樹脂組成物の粘度が低くならず、本樹脂組成物を強化繊維基材内に十分に含浸させることができない。加熱温度が160℃を超える場合、本樹脂組成物中の(a)成分と(b)成分との硬化反応が進行し、プリプレグが硬化をはじめる。そのため、タック性やドレープ性が悪化し易い。 The heating temperature for impregnating the reinforcing fiber substrate with the resin composition is appropriately adjusted according to the viscosity of the resin composition. Usually, it is 70-160 degreeC and 80-120 degreeC is preferable. When heating temperature is less than 70 degreeC, the viscosity of this resin composition does not become low, and this resin composition cannot fully be impregnated in a reinforced fiber base material. When the heating temperature exceeds 160 ° C., the curing reaction of the component (a) and the component (b) in the resin composition proceeds, and the prepreg begins to cure. For this reason, tackiness and drapeability are likely to deteriorate.
プリプレグの工業的生産速度は特に限定されないが、生産性や経済性などを考慮すると、連続生産の場合、0.1m/min以上であることが好ましく、1〜50m/minであることがより好ましく、5〜20m/minであることが特に好ましい。 The industrial production rate of the prepreg is not particularly limited. However, in consideration of productivity and economy, in the case of continuous production, it is preferably 0.1 m / min or more, more preferably 1 to 50 m / min. 5 to 20 m / min is particularly preferable.
(5)本プリプレグの使用方法
本プリプレグは公知の手法により硬化させることにより繊維強化複合材料を作製することができる。本プリプレグを用いて繊維強化複合材料を作製する方法としては、従来公知の方法、例えば、マニュアルレイアップ、自動テープレイアップ(ATL)、自動繊維配置、真空バギング、オートクレーブ硬化、オートクレーブ以外の硬化、流体援用加工、圧力支援プロセス、マッチモールドプロセス、単純プレス硬化、プレスクレーブ硬化、又は連続バンドプレスを使用する方法が挙げられる。
(5) Method of using the present prepreg The present prepreg can be made into a fiber-reinforced composite material by curing by a known method. As a method for producing a fiber-reinforced composite material using the present prepreg, conventionally known methods such as manual layup, automatic tape layup (ATL), automatic fiber placement, vacuum bagging, autoclave curing, curing other than autoclave, Examples include fluid assisted processing, pressure assisted processes, match mold processes, simple press curing, press clave curing, or methods using continuous band presses.
例えば、本プリプレグを積層して、オートクレーブ中で0.2〜1MPaに加圧し、150〜204℃で1〜8時間加熱することによって、成形された繊維強化複合材料を作製することができる。ポストキュアとして180〜280℃の温度範囲で温度を段階的に上昇させながら2〜20時間処理することにより、耐熱性をさらに向上させることができる。 For example, the formed fiber reinforced composite material can be produced by laminating the present prepreg, pressurizing to 0.2 to 1 MPa in an autoclave, and heating at 150 to 204 ° C. for 1 to 8 hours. Heat treatment can be further improved by performing the treatment for 2 to 20 hours while increasing the temperature stepwise in the temperature range of 180 to 280 ° C. as post-cure.
本プリプレグは、高耐熱性の樹脂組成物を用いている。したがって、本プリプレグを用いて作製される繊維強化複合材料は、少なくとも200℃以上の耐熱性を有する。 This prepreg uses a highly heat-resistant resin composition. Therefore, the fiber reinforced composite material produced using this prepreg has a heat resistance of at least 200 ° C. or more.
本樹脂組成物の硬化の過程において、ATBNとビスマレイド化合物とが反応して得られる生成物は、マトリックス樹脂内で相分離してゴム粒子として析出する。このゴム粒子は、マトリックス樹脂内に均一に分散するとともに、マトリックス樹脂と強固に結合している。そのため、このゴム粒子は外部からの衝撃を吸収し、繊維強化複合材料内のクラックの伝播を抑制する。したがって、本プリプレグを用いて作製される繊維強化複合材料は、破壊靭性が高い。 In the curing process of the resin composition, the product obtained by the reaction of ATBN and the bismaleide compound is phase-separated in the matrix resin and precipitated as rubber particles. The rubber particles are uniformly dispersed in the matrix resin and are firmly bonded to the matrix resin. Therefore, the rubber particles absorb external impacts and suppress the propagation of cracks in the fiber reinforced composite material. Therefore, the fiber reinforced composite material produced using this prepreg has high fracture toughness.
本プリプレグを用いて作製される繊維強化複合材料の層間剥離強度(GIc)は、好ましくは175〜800J/m2となり、より好ましくは300〜600J/m2となる。 The delamination strength (GIc) of the fiber reinforced composite material produced using this prepreg is preferably 175 to 800 J / m 2 , more preferably 300 to 600 J / m 2 .
本プリプレグは保存安定性に優れ、本プリプレグの製造後、少なくとも10日間を経過しても、製造直後の成形加工性を維持する。したがって、所定の時間が経過した後も、耐熱性及び破壊靭性が高い繊維強化複合材料を作製することができる。 The present prepreg is excellent in storage stability, and maintains the moldability immediately after production even after at least 10 days have passed since the production of the present prepreg. Therefore, a fiber-reinforced composite material having high heat resistance and high fracture toughness can be produced even after a predetermined time has elapsed.
以下、実施例により本発明をより詳細に説明する。ただし、本発明は以下に記載する実施例に限定されるものではない。 Hereinafter, the present invention will be described in more detail with reference to examples. However, the present invention is not limited to the examples described below.
本樹脂組成物及びプリプレグの原材料として、以下のものを用いた。 The following were used as raw materials for the resin composition and prepreg.
[(a)成分]
[芳香族ビスマレイミド化合物]
・Matrimid 5292 A(商品名)(N,N’−4,4’−ジフェニルメタンビスマレイミド、 ハンツマン・アドバンスト・マテリアルズ社製)
・BMI−1100H(商品名)(N,N’−4,4’−ジフェニルメタンビスマレイミド、 大和化成工業(株)社製)
・BMI−2300(商品名)(フェニルメタンマレイミドオリゴマー、 大和化成工業(株)社製)
・BMI−80(商品名)(2,2’−ビス[4−(4−マレイミドフェノキシ)フェニル]プロパン、 ケイ・アイ化成(株)社製)
・BMI−7000H(商品名)(4−メチル−1,3−フェニレンビスマレイミド、 大和化成工業(株)社製)
[脂肪族ビスマレイミド化合物]
・BMI−TMH(商品名)(1,6’−ビスマレイミド−(2,2,4−トリメチル)ヘキサン、 大和化成工業(株)社製)
・GP−207−R(商品名)(1,6’−ヘキサメチレンジアミンビスマレイミド(HMDA−BMI)、 Cymer社製)
[(A) component]
[Aromatic bismaleimide compounds]
・ Matrimid 5292 A (trade name) (N, N′-4,4′-diphenylmethane bismaleimide, manufactured by Huntsman Advanced Materials)
・ BMI-1100H (trade name) (N, N′-4,4′-diphenylmethane bismaleimide, manufactured by Daiwa Kasei Kogyo Co., Ltd.)
・ BMI-2300 (trade name) (Phenylmethane maleimide oligomer, manufactured by Daiwa Kasei Kogyo Co., Ltd.)
・ BMI-80 (trade name) (2,2′-bis [4- (4-maleimidophenoxy) phenyl] propane, manufactured by KAI Kasei Co., Ltd.)
・ BMI-7000H (trade name) (4-methyl-1,3-phenylenebismaleimide, manufactured by Daiwa Kasei Kogyo Co., Ltd.)
[Aliphatic bismaleimide compounds]
・ BMI-TMH (trade name) (1,6′-bismaleimide- (2,2,4-trimethyl) hexane, manufactured by Daiwa Kasei Kogyo Co., Ltd.)
GP-207-R (trade name) (1,6′-hexamethylenediamine bismaleimide (HMDA-BMI), manufactured by Cymer)
[(b)成分]
・Matrimid 5292 B(商品名)(O,O’−ジアリルビスフェノールA、 ハンツマン・アドバンスト・マテリアルズ社製)
・DABPA(商品名)(2,2’−ジアリルビスフェノールA、 大和化成工業(株)社製)
[Component (b)]
・ Matrimid 5292 B (trade name) (O, O′-diallylbisphenol A, manufactured by Huntsman Advanced Materials)
DABPA (trade name) (2,2'-diallylbisphenol A, manufactured by Daiwa Kasei Kogyo Co., Ltd.)
[(c)成分]
・HYPRO ATBN 1300X16(登録商標)(Emerald Performance Materials社製)
[Component (c)]
・ HYPRO ATBN 1300X16 (registered trademark) (manufactured by Emerald Performance Materials)
[カルボキシル基末端を有するアクリロニトリル−ブタジエンコポリマー]
・HYCAR CTBN 1300X13(登録商標)(B.F.Goodrich社製)
[Acrylonitrile-butadiene copolymer having carboxyl group terminal]
・ HYCAR CTBN 1300X13 (registered trademark) (manufactured by BF Goodrich)
[可溶性熱可塑性樹脂]
・Ultem1000−1000(商品名)粉砕物(ポリエーテルイミド、 SABICイノベーティブプラスチック社製、 平均粒子径15μm)
・スミカエクセル5003P(商品名)粉砕物(ポリエーテルスルホン、 住友化学(株)社製、 平均粒子径15μm)
上記のポリエーテルイミドとポリエーテルスルホンは、(b)成分に可溶である。
[Soluble thermoplastic resin]
-Ultem 1000-1000 (trade name) pulverized product (polyetherimide, manufactured by SABIC Innovative Plastics, average particle size 15 μm)
・ Sumika Excel 5003P (trade name) pulverized product (polyethersulfone, manufactured by Sumitomo Chemical Co., Ltd., average particle size: 15 μm)
The above polyetherimide and polyethersulfone are soluble in the component (b).
[不溶性熱可塑性樹脂]
・AURUM PD450M(商品名)粉砕物(ポリイミド樹脂、 三井化学(株)社製、 平均粒子径14μm)
・P84(商品名)(ポリイミド樹脂、 HP Polymers社製、 平均粒子径15μm)
上記のポリイミド樹脂は、アルケニルフェノール又はアルケニルフェノールエーテル化合物に不溶である。
[Insoluble thermoplastic resin]
・ AURUM PD450M (trade name) pulverized product (polyimide resin, manufactured by Mitsui Chemicals, Inc., average particle size 14 μm)
・ P84 (trade name) (polyimide resin, manufactured by HP Polymers, average particle size: 15 μm)
The polyimide resin is insoluble in alkenylphenol or alkenylphenol ether compound.
[炭素繊維基材]
・IMS 65 E 23 24K 830tex(炭素繊維ストランド、 東邦テナックス(株)社製、 引張強度:5800MPa、 引張弾性率:290GPa)
[Carbon fiber substrate]
IMS 65 E 23 24K 830 tex (carbon fiber strand, manufactured by Toho Tenax Co., Ltd., tensile strength: 5800 MPa, tensile elastic modulus: 290 GPa)
樹脂組成物、プリプレグ及び繊維強化複合材料の物性を以下の方法により測定した。 The physical properties of the resin composition, prepreg and fiber reinforced composite material were measured by the following methods.
[平均粒子径]
各種粒子径は、日機装株式会社製のレーザー回折・散乱式の粒度分析計(マイクロトラック法)MT3300を用いて、粒度分布を測定し、そのD50を平均粒子径とした。
[Average particle size]
Various particle sizes were measured using a laser diffraction / scattering particle size analyzer (Microtrack method) MT3300 manufactured by Nikkiso Co., Ltd., and D50 was defined as the average particle size.
[吸水率]
プリプレグを一辺が100mmの正方形にカットし、質量(W1)を測定した。その後、デシケーター中で、プリプレグを水中に沈めた。デシケーター内を、10kPa以下に減圧した後、常圧に戻すことにより、プリプレグ内部の空気と水とを置換させた。プリプレグを水中から取り出し、表面の水を拭き取り、プリプレグの質量(W2)を測定した。これらの測定値から下記式
吸水率(%)=[(W2−W1)/W1]×100
W1:プリプレグの質量(g)
W2:吸水後のプリプレグの質量(g)
を用いて吸水率を算出した。
[Water absorption rate]
The prepreg was cut into a square having a side of 100 mm, and the mass (W1) was measured. Thereafter, the prepreg was submerged in a desiccator. After reducing the pressure inside the desiccator to 10 kPa or less, the air and water inside the prepreg were replaced by returning to normal pressure. The prepreg was taken out of the water, the surface water was wiped off, and the mass (W2) of the prepreg was measured. From these measured values, the following formula: water absorption (%) = [(W2-W1) / W1] × 100
W1: Mass of prepreg (g)
W2: Mass of prepreg after water absorption (g)
Was used to calculate the water absorption rate.
[室温保存安定性]
プリプレグを、温度26.7℃、湿度65%に10日間保存した後、一辺が100mmの正方形にカットし、後述する金型の折曲げ部を覆って積層した。金型表面形状は、曲率r=12.7mmで金属板をその中央で角度90度に折曲げると共に、折曲げ部を上方に突出した形状であった。プリプレグの金型に対する積層状態を評価した。評価結果は以下の基準(○、△、×)で表した。
○:金型表面形状に十分追従し、取扱い性が製造直後とほとんど変わらない。
△:プリプレグの硬化反応が進行し、タック性・ドレープ性が低下しているが、金型表面形状に追従し、問題なく使用できる。
×:プリプレグの硬化反応が進行し、タック性・ドレープ性が著しく低下しており、金型の表面形状に追従しない。
[Room temperature storage stability]
The prepreg was stored at a temperature of 26.7 ° C. and a humidity of 65% for 10 days, then cut into a square having a side of 100 mm, and laminated so as to cover a bent portion of a mold described later. The mold surface shape was a shape in which the metal plate was bent at an angle of 90 degrees at the center with a curvature r = 12.7 mm, and the bent portion protruded upward. The lamination state of the prepreg mold was evaluated. The evaluation results were represented by the following criteria (◯, Δ, ×).
○: The surface shape of the mold is sufficiently followed, and the handleability is almost the same as that immediately after production.
(Triangle | delta): Although the hardening reaction of a prepreg progresses and tack property and drape property are falling, it follows a metal mold | die surface shape and can be used without a problem.
X: The curing reaction of the prepreg has progressed, the tackiness and draping properties have been remarkably lowered, and do not follow the surface shape of the mold.
[タック性]
プリプレグのタック性は、タッキング試験装置TAC−II(RHESCA CO.,LTD.)を用いて以下のタックプローブ試験により測定した。27℃に保持された試験ステージにプリプレグをセットし、27℃に保持されたφ5のタックプローブで初期荷重100gfの荷重をかけて、10mm/secの試験速度で引き抜いた際の最大の荷重(最大引抜荷重)を求めた。製造直後のプリプレグと、温度26.7℃、湿度65%に10日間保存したプリプレグとについて、それぞれタックプローブ試験を実施した。製造直後のプリプレグの最大引抜荷重に対する10日間保存したプリプレグの最大引抜荷重をタック保持率とした。タック性の評価結果は以下の基準(○、△、×)で表した。
○:製造直後の最大引抜荷重が100gf以上で、10日間保存後のタック保持率が100〜50%
△:製造直後の最大引抜荷重が100gf以上で、10日間保存後のタック保持率が50〜25%
×:製造直後の最大引抜荷重が100gf以上で、10日間保存後のタック保持率が25〜0%
[Tackiness]
The tackiness of the prepreg was measured by the following tack probe test using a tacking test apparatus TAC-II (RHESCA CO., LTD.). Set the prepreg on the test stage held at 27 ° C, apply the initial load of 100 gf with a φ5 tack probe held at 27 ° C, and pull it out at the test speed of 10 mm / sec. Pull-out load) was determined. A tack probe test was performed on each of the prepregs immediately after production and the prepregs stored at a temperature of 26.7 ° C. and a humidity of 65% for 10 days. The maximum pulling load of the prepreg stored for 10 days relative to the maximum pulling load of the prepreg immediately after production was defined as the tack retention. The evaluation results of tackiness were represented by the following criteria (◯, Δ, ×).
○: The maximum pulling load immediately after production is 100 gf or more, and the tack retention after storage for 10 days is 100 to 50%.
Δ: The maximum pull-out load immediately after production is 100 gf or more, and the tack retention after storage for 10 days is 50 to 25%.
X: The maximum pull-out load immediately after production is 100 gf or more, and the tack retention after storage for 10 days is 25 to 0%.
[ドレープ性]
プリプレグのドレープ性は、ASTM D1388に準拠して、以下の試験により評価した。プリプレグを0°繊維方向に対し90°方向にカットし、傾斜角度41.5°の傾斜に対するドレープ性(flexural rigidity, mg*cm)を評価した。この評価は、プリプレグの製造直後と、温度26.7℃、湿度65%で所定の期間保存した後とに、それぞれ実施した。評価結果は以下の基準(○、△、×)で表した。
○:20日間経過後でも、ドレープ性は製造直後と変わらない。
△:10日間経過後でも、ドレープ性は製造直後と変わらない(10日後からは、若干ドレープ性に低下が見られた。)。
×:10日間経過後では、ドレープ性が製造直後よりも低下し、使用するには問題のあるレベルであった。
[Drapability]
The prepreg drapeability was evaluated by the following test in accordance with ASTM D1388. The prepreg was cut in a 90 ° direction with respect to the 0 ° fiber direction, and the drape property (flexural rigidity, mg * cm) with respect to the inclination at an inclination angle of 41.5 ° was evaluated. This evaluation was performed immediately after manufacturing the prepreg and after storage for a predetermined period at a temperature of 26.7 ° C. and a humidity of 65%. The evaluation results were represented by the following criteria (◯, Δ, ×).
○: Even after 20 days, the drapeability is the same as that immediately after production.
(Triangle | delta): Even after 10-day progress, drape property does not change from immediately after manufacture (after 10 days, a slight fall was seen in drape property).
X: After 10 days, the drapeability was lower than that immediately after the production, which was a problem level for use.
[ガラス転移温度]
プリプレグを一辺が100mmの正方形にカットし、これを2枚積層して、積層構成[0]2の積層体を得た。通常の真空オートクレーブ成形法を用い、0.59MPaの圧力下、180℃で6時間成形した。得られた成形物を取り出し、熱風循環乾燥機を用いて、200℃の条件下で12時間、フリースタンドでポストキュアを実施した。
[Glass-transition temperature]
The prepreg was cut into a square with a side of 100 mm, and two of them were laminated to obtain a laminate having a laminate configuration [0] 2 . Using a normal vacuum autoclave molding method, molding was performed at 180 ° C. for 6 hours under a pressure of 0.59 MPa. The obtained molded product was taken out and post-cured on a free stand for 12 hours at 200 ° C. using a hot air circulating dryer.
得られた成形物を幅2.54mm×長さ10.2mmの寸法に切断し、TMA(熱機械分析装置、Bruker社製)を用いて曲げモードにより炭素繊維強化複合材料のTgを測定した。測定は、窒素雰囲気中で、昇温速度10℃/minで400℃まで昇温して行った。 The obtained molded product was cut into dimensions of 2.54 mm wide × 10.2 mm long, and Tg of the carbon fiber reinforced composite material was measured by a bending mode using TMA (thermomechanical analyzer, manufactured by Bruker). The measurement was performed in a nitrogen atmosphere by raising the temperature to 400 ° C. at a rate of temperature increase of 10 ° C./min.
[層間破壊靭性モードI(GIc)]
得られたプリプレグを一辺が360mmの正方形にカットした後、積層し、0°方向に10層積層した積層体を2つ作製した。初期クラックを発生させるために、離型フィルムを2つの積層体の間にはさみ、両者を組み合わせ、積層構成[0]20のプリプレグ積層体を得た。通常の真空オートクレーブ成形法を用い、0.59MPaの圧力下、180℃の条件で6時間成形した。その後、熱風循環オーブンを用いて、フリースタンドで温度200℃の条件下で12時間のポストキュアを行い、成形物を得た。得られた成形物(炭素繊維強化複合材料)を幅12.7mm×長さ304.8mmの寸法に切断し、層間破壊靭性モードI(GIc)の試験片を得た。
[Interlaminar fracture toughness mode I (GIc)]
The obtained prepreg was cut into a square having a side of 360 mm, then laminated, and two laminates were produced in which 10 layers were laminated in the 0 ° direction. In order to generate an initial crack, a release film was sandwiched between two laminates, and both were combined to obtain a prepreg laminate having a laminate configuration [0] 20 . Using a normal vacuum autoclave molding method, molding was performed for 6 hours under the condition of 180 ° C. under a pressure of 0.59 MPa. Thereafter, using a hot air circulating oven, post-cure was performed for 12 hours on a free stand at a temperature of 200 ° C. to obtain a molded product. The obtained molded product (carbon fiber reinforced composite material) was cut into dimensions of 12.7 mm width × 304.8 mm length to obtain a specimen having an interlaminar fracture toughness mode I (GIc).
GIcの試験方法として、双片持ちはり層間破壊靱性試験法(DCB法)を用い、離型フィルムの先端から12.7mmの予亀裂(初期クラック)を発生させた後に、さらに亀裂を進展させる試験を行った。予亀裂の先端から、亀裂進展長さが127mmに到達した時点で試験を終了させた。試験片引張試験機のクロスヘッドスピードは12.7mm/分とし、n=5で測定を行った。亀裂進展長さは顕微鏡を用いて試験片の両端面から測定し、荷重及び亀裂開口変位を計測することにより、GIcを算出した。 A test that uses the double cantilever interlaminar fracture toughness test method (DCB method) as a test method for GIc, and after the pre-crack (initial crack) of 12.7 mm is generated from the tip of the release film, the test further advances the crack. Went. The test was terminated when the crack growth length reached 127 mm from the tip of the pre-crack. The crosshead speed of the test piece tensile tester was 12.7 mm / min, and measurement was performed at n = 5. The crack growth length was measured from both end faces of the test piece using a microscope, and GIc was calculated by measuring the load and crack opening displacement.
[実施例1]
表1に記載する割合で、BMI−2300と、TMH−BMIと、Matrimid 5292Bと、を100℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。その後、80℃に冷却し、Matrimid 5292A、BMI−7000H、HYPRO ATBN 1300X16を添加し、30分間混合撹拌し、熱硬化性ビスマレイミド系樹脂組成物を調製した。
[Example 1]
BMI-2300, TMH-BMI, and Matrimid 5292B were stirred at 100 ° C. for 30 minutes using a stirrer at the ratio shown in Table 1, and completely dissolved. Then, it cooled to 80 degreeC, Matrimid 5292A, BMI-7000H, and HYPRO ATBN 1300X16 were added, and it stirred for 30 minutes, and prepared the thermosetting bismaleimide-type resin composition.
この熱硬化性ビスマレイミド系樹脂組成物を、フィルムコーターを用いて、表1に示す片面樹脂フィルム目付けで離型フィルム上に塗布し、樹脂シートを得た。次に、樹脂シート2枚の間に、炭素繊維ストランド(IMS 65 E 23 24K 830tex)を供給して一方向に均一に配列[目付け(190g/m2)]させることにより、炭素繊維ストランドをシート状にし、ローラーを用いて110℃で加圧及び加熱してプリプレグを得た。プリプレグ全体に対する熱硬化性ビスマレイミド樹脂組成物の含有率は35質量%であった。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表1に示した。 This thermosetting bismaleimide resin composition was applied on a release film with a single-sided resin film basis weight shown in Table 1 using a film coater to obtain a resin sheet. Next, carbon fiber strands (IMS 65 E 23 24K 830 tex) are supplied between two resin sheets and uniformly arranged in one direction [weight per unit area (190 g / m 2 )]. And was pressed and heated at 110 ° C. using a roller to obtain a prepreg. The content rate of the thermosetting bismaleimide resin composition with respect to the whole prepreg was 35 mass%. Table 1 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
[実施例2〜4]
HYPRO ATBN 1300X16の添加量を変更した以外は、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表1に示した。
[Examples 2 to 4]
A prepreg was obtained in the same manner as in Example 1 except that the amount of HYPRO ATBN 1300X16 was changed. Table 1 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
[比較例1]
HYPRO ATBN 1300X16を添加しない以外は、実施例1と同様の方法で樹脂組成物及びプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表1に示した。
[Comparative Example 1]
A resin composition and a prepreg were obtained in the same manner as in Example 1 except that HYPRO ATBN 1300X16 was not added. Table 1 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
樹脂組成物に、HYPRO ATBN 1300X16を添加していない比較例1では、プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であったが、このプリプレグを用いて作製した炭素繊維強化複合材料の層間破壊靭性モードIは、実施例1〜4と比較して低い値となった。 In Comparative Example 1 in which HYPRO ATBN 1300X16 was not added to the resin composition, the prepreg had good room temperature storage stability, tack retention, and draping properties. A carbon fiber reinforced composite material produced using this prepreg The interlaminar fracture toughness mode I was a low value as compared with Examples 1-4.
[比較例2、3]
HYPRO ATBN 1300X16をHYCAR CTBN 1300X13に変更した以外は、実施例1と同様の方法で樹脂組成物及びプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表1に示した。
[Comparative Examples 2 and 3]
A resin composition and a prepreg were obtained in the same manner as in Example 1 except that HYPRO ATBN 1300X16 was changed to HYCAR CTBN 1300X13. Table 1 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であったが、このプリプレグを用いて作製した炭素繊維強化複合材料の層間破壊靭性モードIは、実施例1〜4と比較して低い値となった。 Although the room temperature storage stability, tack retention, and draping property of the prepreg were good, the interlaminar fracture toughness mode I of the carbon fiber reinforced composite material produced using this prepreg was low as compared with Examples 1-4. Value.
[実施例5]
表2に記載する割合で、DABPAと、Ultem1000−1000と、を120℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。得られた樹脂組成物を50℃に冷却し、BMI−2300、BMI−80、GP−207−Rを添加し、120℃で30分間撹拌して完全に溶解させた。その後、得られた樹脂組成物を80℃に冷却し、BMI−1100H、HYPRO ATBN 1300X16、AURUM PD450Mを添加し、30分間混合撹拌し、熱硬化性ビスマレイミド系樹脂組成物を調製した。
[Example 5]
DABPA and Ultem 1000-1000 were stirred at 120 ° C. for 30 minutes using a stirrer at the ratio shown in Table 2 and completely dissolved. The obtained resin composition was cooled to 50 ° C., BMI-2300, BMI-80, and GP-207-R were added, and the mixture was stirred at 120 ° C. for 30 minutes for complete dissolution. Thereafter, the obtained resin composition was cooled to 80 ° C., BMI-1100H, HYPRO ATBN 1300X16, and AURUM PD450M were added, and mixed and stirred for 30 minutes to prepare a thermosetting bismaleimide resin composition.
この熱硬化性ビスマレイミド系樹脂組成物を、フィルムコーターを用いて、表2に示す片面樹脂フィルム目付けで離型フィルム上に塗布し、樹脂シートを得た。次に、樹脂シート2枚の間に、炭素繊維ストランド(IMS 65 E 23 24K 830tex)を供給して一方向に均一に配列[目付け(190g/m2)]させることにより、炭素繊維ストランドをシート状にし、ローラーを用いて120℃で加圧及び加熱してプリプレグを得た。プリプレグ全体に対する樹脂の含有率は35質量%であった。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。 This thermosetting bismaleimide resin composition was applied onto a release film with a single-sided resin film basis weight shown in Table 2 using a film coater to obtain a resin sheet. Next, carbon fiber strands (IMS 65 E 23 24K 830 tex) are supplied between two resin sheets and uniformly arranged in one direction [weight per unit area (190 g / m 2 )]. And pressurizing and heating at 120 ° C. using a roller to obtain a prepreg. The resin content relative to the entire prepreg was 35% by mass. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であり、このプリプレグを用いて作製した炭素繊維強化複合材料は、層間破壊靭性モードIが非常に高い値であった。 The room temperature storage stability, tack retention, and drape of the prepreg were good, and the carbon fiber reinforced composite material produced using this prepreg had a very high value for the interlaminar fracture toughness mode I.
[実施例6〜8]
HYPRO ATBN 1300X16の添加量を変更した以外は、実施例5と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。
[Examples 6 to 8]
A prepreg was obtained in the same manner as in Example 5 except that the addition amount of HYPRO ATBN 1300X16 was changed. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であり、このプリプレグを用いて作製した炭素繊維強化複合材料は、層間破壊靭性モードIが非常に高い値であった。 The room temperature storage stability, tack retention, and drape of the prepreg were good, and the carbon fiber reinforced composite material produced using this prepreg had a very high value for the interlaminar fracture toughness mode I.
[実施例9]
AURUM PD450MをP84に変更した以外は、実施例5と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。
[Example 9]
A prepreg was obtained in the same manner as in Example 5 except that AURUM PD450M was changed to P84. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であり、このプリプレグを用いて作製した炭素繊維強化複合材料は、層間破壊靭性モードIが非常に高い値であった。 The room temperature storage stability, tack retention, and drape of the prepreg were good, and the carbon fiber reinforced composite material produced using this prepreg had a very high value for the interlaminar fracture toughness mode I.
[実施例10]
Ultem1000−1000をスミカエクセル5003Pに変更した以外は、実施例9と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。
[Example 10]
A prepreg was obtained in the same manner as in Example 9 except that Ultem 1000-1000 was changed to Sumika Excel 5003P. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であり、このプリプレグを用いて作製した炭素繊維強化複合材料は、層間破壊靭性モードIが非常に高い値であった。 The room temperature storage stability, tack retention, and drape of the prepreg were good, and the carbon fiber reinforced composite material produced using this prepreg had a very high value for the interlaminar fracture toughness mode I.
[比較例4]
HYPRO ATBN 1300X16を添加しない以外は、実施例5と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。
[Comparative Example 4]
A prepreg was obtained in the same manner as in Example 5 except that HYPRO ATBN 1300X16 was not added. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
樹脂組成物に、HYPRO ATBN 1300X16を添加していない比較例4では、プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であったが、このプリプレグを用いて作製した炭素繊維強化複合材料の層間破壊靭性モードIは、実施例5〜10と比較して低い値となった。 In Comparative Example 4 in which HYPRO ATBN 1300X16 was not added to the resin composition, the prepreg had good room temperature storage stability, tack retention, and draping properties. A carbon fiber reinforced composite material produced using this prepreg The interlaminar fracture toughness mode I was a low value as compared with Examples 5-10.
[比較例5]
HYPRO ATBN 1300X16を添加しない以外は、実施例9と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。
[Comparative Example 5]
A prepreg was obtained in the same manner as in Example 9 except that HYPRO ATBN 1300X16 was not added. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
樹脂組成物に、HYPRO ATBN 1300X16を添加していない比較例5では、プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であったが、このプリプレグを用いて作製した炭素繊維強化複合材料の層間破壊靭性モードIは、実施例5〜10と比較して低い値となった。 In Comparative Example 5 in which HYPRO ATBN 1300X16 was not added to the resin composition, the room temperature storage stability, tack retention and drape of the prepreg were good, but the carbon fiber reinforced composite material produced using this prepreg The interlaminar fracture toughness mode I was a low value as compared with Examples 5-10.
[比較例6]
HYPRO ATBN 1300X16をHYCAR CTBN 1300X13に変更した以外は、実施例5と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。
[Comparative Example 6]
A prepreg was obtained in the same manner as in Example 5 except that HYPRO ATBN 1300X16 was changed to HYCAR CTBN 1300X13. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であったが、このプリプレグを用いて作製した炭素繊維強化複合材料の層間破壊靭性モードIは、実施例5〜10と比較して低い値となった。 Although the prepreg had good room temperature storage stability, tack retention, and draping properties, the interlaminar fracture toughness mode I of the carbon fiber reinforced composite material produced using this prepreg was low compared to Examples 5-10. Value.
[比較例7]
HYPRO ATBN 1300X16をHYCAR CTBN 1300X13に変更した以外は、実施例5と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表2に示した。
[Comparative Example 7]
A prepreg was obtained in the same manner as in Example 5 except that HYPRO ATBN 1300X16 was changed to HYCAR CTBN 1300X13. Table 2 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
プリプレグの室温保存安定性、タック保持性、ドレープ性は良好であったが、このプリプレグを用いて作製した炭素繊維強化複合材料の層間破壊靭性モードIは、実施例5〜10と比較して低い値となった。 Although the prepreg had good room temperature storage stability, tack retention, and draping properties, the interlaminar fracture toughness mode I of the carbon fiber reinforced composite material produced using this prepreg was low compared to Examples 5-10. Value.
[比較例8]
表3に記載する割合で、Matrimid 5292Aと、Matrimid 5292Bと、を120℃で30分間攪拌機を用いて撹拌し、完全に溶解させて樹脂組成物を得た。得られた樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。
[Comparative Example 8]
Matrimid 5292A and Matrimid 5292B were stirred at 120 ° C. for 30 minutes using a stirrer at the ratio shown in Table 3, and completely dissolved to obtain a resin composition. A prepreg was obtained in the same manner as in Example 1 using the obtained resin composition. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
樹脂組成物に、HYPRO ATBN 1300X16を添加していない比較例8では、炭素繊維強化複合材料の層間破壊靭性モードIは、実施例5〜10と比較して低い値となった。また、Matrimid 5292BにMatrimid 5292Aを多量に溶解させることにより、樹脂組成物の硬化反応が進行してしまい、プリプレグを金型に沿わすことができず、室温保存安定性及びドレープ性が悪化した。また、室温保存時に多量に溶解したMatrimid 5292Aが経過時間とともに徐々に析出し、タック保持性が悪化した。 In Comparative Example 8 in which HYPRO ATBN 1300X16 was not added to the resin composition, the interlaminar fracture toughness mode I of the carbon fiber reinforced composite material was a low value as compared with Examples 5-10. Further, when a large amount of Matrimid 5292A was dissolved in Matrimid 5292B, the curing reaction of the resin composition progressed, and the prepreg could not follow the mold, resulting in deterioration in room temperature storage stability and drapeability. Moreover, Matrimid 5292A which melt | dissolved in large quantities at the time of storage at room temperature precipitated gradually with elapsed time, and tack maintenance property deteriorated.
[比較例9]
表3に記載する割合で、Matrimid 5292Aと、Matrimid 5292Bと、を80℃で30分間攪拌機を用いて撹拌し、樹脂組成物を得た。得られた樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。
[Comparative Example 9]
Matrimid 5292A and Matrimid 5292B were stirred with a stirrer at 80 ° C. for 30 minutes at the ratio shown in Table 3 to obtain a resin composition. A prepreg was obtained in the same manner as in Example 1 using the obtained resin composition. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
樹脂組成物に、HYPRO ATBN 1300X16を添加していない比較例9では、実施例5〜10と比較して炭素繊維強化複合材料の層間破壊靭性モードIは低い値となった。また、Matrimid 5292BにMatrimid 5292Aを溶解させていないため、プリプレグ製造時に樹脂組成物の固相成分がプリプレグ表面に多く存在した。そのため、室温保存安定性、タック性が悪化した。 In Comparative Example 9 in which HYPRO ATBN 1300X16 was not added to the resin composition, the interlaminar fracture toughness mode I of the carbon fiber reinforced composite material was lower than in Examples 5 to 10. In addition, since Matrimid 5292A was not dissolved in Matrimid 5292B, many solid phase components of the resin composition were present on the prepreg surface during prepreg production. Therefore, room temperature storage stability and tackiness deteriorated.
[実施例11]
表3に記載する割合で、Matrimid 5292Aと、TMH−BMIと、Matrimid 5292Bと、を120℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。得られた樹脂組成物を80℃に冷却し、HYPRO ATBN 1300X16を添加し、30分間混合撹拌して熱硬化性ビスマレイミド系樹脂組成物を調製した。この熱硬化性ビスマレイミド系樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。
[Example 11]
Matrimid 5292A, TMH-BMI, and Matrimid 5292B were stirred at 120 ° C. for 30 minutes using a stirrer at the ratio shown in Table 3, and completely dissolved. The obtained resin composition was cooled to 80 ° C., HYPRO ATBN 1300X16 was added, and the mixture was stirred for 30 minutes to prepare a thermosetting bismaleimide resin composition. Using this thermosetting bismaleimide resin composition, a prepreg was obtained in the same manner as in Example 1. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
実施例11では、Matrimid 5292BにMatrimid 5292Aを多量に溶解させることにより、樹脂組成物の硬化反応が進行してしまい、プリプレグを金型に沿わすことがやや困難となった。また、室温保存時に多量に溶解したMatrimid 5292Aが経過時間とともに徐々に析出してしまい、タック保持性がやや悪化した。しかし、このプリプレグを用いて作製した繊維強化複合材料の層間破壊靭性モードIは良好な値であった。 In Example 11, when a large amount of Matrimid 5292A was dissolved in Matrimid 5292B, the curing reaction of the resin composition proceeded, making it somewhat difficult to fit the prepreg along the mold. Moreover, Matrimid 5292A which melt | dissolved in large quantities at the time of storage at room temperature gradually precipitated with elapsed time, and tack maintenance property deteriorated a little. However, the interlaminar fracture toughness mode I of the fiber reinforced composite material produced using this prepreg was a good value.
[実施例12]
表3に記載する割合で、BMI−1100Hと、GP−207−Rと、Matrimid 5292Bと、を120℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。この樹脂組成物を80℃に冷却し、Matrimid 5292A、HYPRO ATBN 1300X16を添加し、30分間混合撹拌して熱硬化性ビスマレイミド系樹脂組成物を調製した。この熱硬化性ビスマレイミド系樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。実施例12では、Matrimid 5292BにBMI−1100Hを多量に溶解させることにより、樹脂組成物の硬化反応が進行してしまい、プリプレグを金型に沿わすことがやや困難となった。また、室温保存時に多量に溶解したBMI−1100Hが経過時間とともに徐々に析出してしまい、タック保持性がやや悪化した。しかし、このプリプレグを用いて作製した繊維強化複合材料の層間破壊靭性モードIは良好な値であった。
[Example 12]
BMI-1100H, GP-207-R, and Matrimid 5292B were stirred at 120 ° C. for 30 minutes using a stirrer at the ratio shown in Table 3, and completely dissolved. The resin composition was cooled to 80 ° C., Matrimid 5292A and HYPRO ATBN 1300X16 were added, and mixed and stirred for 30 minutes to prepare a thermosetting bismaleimide resin composition. Using this thermosetting bismaleimide resin composition, a prepreg was obtained in the same manner as in Example 1. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg. In Example 12, when a large amount of BMI-1100H was dissolved in Matrimid 5292B, the curing reaction of the resin composition proceeded, making it somewhat difficult to fit the prepreg along the mold. Moreover, BMI-1100H which melt | dissolved in large quantities at the time of room temperature storage gradually precipitated with elapsed time, and tack maintenance property deteriorated a little. However, the interlaminar fracture toughness mode I of the fiber reinforced composite material produced using this prepreg was a good value.
[実施例13]
表3に記載する割合で、Matrimid 5292Bと、TMH−BMIと、BMI−80と、を120℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。この樹脂組成物を80℃に冷却し、Matrimid 5292A、HYPRO ATBN 1300X16を添加し、30分間混合撹拌して熱硬化性ビスマレイミド樹脂組成物を調製した。この熱硬化性樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。
[Example 13]
Matrimid 5292B, TMH-BMI, and BMI-80 were stirred at 120 ° C. for 30 minutes using a stirrer at the ratio shown in Table 3, and completely dissolved. The resin composition was cooled to 80 ° C., Matrimid 5292A and HYPRO ATBN 1300X16 were added, and mixed and stirred for 30 minutes to prepare a thermosetting bismaleimide resin composition. Using this thermosetting resin composition, a prepreg was obtained in the same manner as in Example 1. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
実施例13で得られたプリプレグは、室温保存安定性、タック保持性、ドレープ性が良好であった。また、このプリプレグを用いて作製した繊維強化複合材料の層間破壊靭性モードIは良好な値であった。 The prepreg obtained in Example 13 had good room temperature storage stability, tack retention, and drape. Moreover, the interlaminar fracture toughness mode I of the fiber reinforced composite material produced using this prepreg was a good value.
[実施例14]
表3に記載する割合で、TMH−BMIと、BMI−1100Hと、Matrimid 5292Bと、を120℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。この樹脂組成物を80℃に冷却し、Matrimid 5292A、HYPRO ATBN 1300X16を添加し、30分間混合撹拌して熱硬化性ビスマレイミド系樹脂組成物を調製した。この熱硬化性ビスマレイミド系樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。
[Example 14]
TMH-BMI, BMI-1100H, and Matrimid 5292B were stirred at 120 ° C. for 30 minutes using a stirrer at the ratio shown in Table 3, and completely dissolved. The resin composition was cooled to 80 ° C., Matrimid 5292A and HYPRO ATBN 1300X16 were added, and mixed and stirred for 30 minutes to prepare a thermosetting bismaleimide resin composition. Using this thermosetting bismaleimide resin composition, a prepreg was obtained in the same manner as in Example 1. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
実施例14では、樹脂組成物中に多量に固相成分を分散させることにより、プリプレグ表面の固相成分が多くなり、樹脂の含浸性が悪くなる傾向が見られ、室温保存安定性、タック保持性がやや悪化した。しかし、このプリプレグを用いて作製した繊維強化複合材料の層間破壊靭性モードIは良好な値であった。 In Example 14, by dispersing a solid phase component in a large amount in the resin composition, the solid phase component on the surface of the prepreg increases, and the resin impregnation tendency tends to deteriorate, and room temperature storage stability and tack retention are observed. Sex was a little worse. However, the interlaminar fracture toughness mode I of the fiber reinforced composite material produced using this prepreg was a good value.
[比較例10]
表3に記載する割合で、BMI−1100Hと、GP−207−Rと、Matrimid 5292Bと、を120℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。この樹脂組成物を80℃に冷却し、Matrimid 5292Aを添加し、30分間混合撹拌して樹脂組成物を調製した。この樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。
[Comparative Example 10]
BMI-1100H, GP-207-R, and Matrimid 5292B were stirred at 120 ° C. for 30 minutes using a stirrer at the ratio shown in Table 3, and completely dissolved. The resin composition was cooled to 80 ° C., Matrimid 5292A was added, and the mixture was stirred for 30 minutes to prepare a resin composition. Using this resin composition, a prepreg was obtained in the same manner as in Example 1. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
樹脂組成物に、HYPRO ATBN 1300X16を添加していない比較例10では、実施例11〜14と比較して、炭素繊維強化複合材料の層間破壊靭性モードIが低い値となった。また、Matrimid 5292BにBMI1100Hを溶解させることにより、樹脂組成物の硬化反応が進行してしまい、プリプレグを金型に沿わすことが困難となった。また、室温保存時に溶解したBMI1100Hが経過時間とともに徐々に析出し、室温保存安定性及びタック保持性が悪化した。 In Comparative Example 10 in which HYPRO ATBN 1300X16 was not added to the resin composition, the interlaminar fracture toughness mode I of the carbon fiber reinforced composite material was a low value as compared with Examples 11-14. Further, by dissolving BMI1100H in Matrimid 5292B, the curing reaction of the resin composition proceeds, making it difficult to fit the prepreg along the mold. In addition, BMI1100H dissolved during storage at room temperature gradually precipitated over time, and room temperature storage stability and tack retention deteriorated.
[比較例11]
表3に記載する割合で、Matrimid 5292Bと、TMH−BMIと、BMI−80と、を、120℃で30分間攪拌機を用いて撹拌し、完全に溶解させた。この樹脂組成物を80℃に冷却し、Matrimid 5292Aを添加し、30分間混合撹拌して樹脂組成物を調製した。この樹脂組成物を用いて、実施例1と同様の方法でプリプレグを得た。得られたプリプレグの各種性能と、このプリプレグを用いて作製した炭素繊維強化複合材料の各種性能を表3に示した。
[Comparative Example 11]
Matrimid 5292B, TMH-BMI, and BMI-80 were stirred with a stirrer at 120 ° C. for 30 minutes and dissolved completely at the ratios shown in Table 3. The resin composition was cooled to 80 ° C., Matrimid 5292A was added, and the mixture was stirred for 30 minutes to prepare a resin composition. Using this resin composition, a prepreg was obtained in the same manner as in Example 1. Table 3 shows various performances of the obtained prepreg and various performances of the carbon fiber reinforced composite material produced using the prepreg.
比較例11で得られたプリプレグは、室温保存安定性、タック保持性、ドレープ性が良好であった。しかし、樹脂組成物に、HYPRO ATBN 1300X16を添加していない比較例11では、実施例11〜14と比較して炭素繊維強化複合材料の層間破壊靭性モードIは低い値となった。 The prepreg obtained in Comparative Example 11 had good room temperature storage stability, tack retention, and drape. However, in Comparative Example 11 in which HYPRO ATBN 1300X16 was not added to the resin composition, the interlaminar fracture toughness mode I of the carbon fiber reinforced composite material was a low value as compared with Examples 11-14.
Claims (9)
(a)成分:ビスマレイミド化合物、
(b)成分:アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物、
(c)成分:アミン末端基を有するアクリロニトリル−ブタジエンコポリマー、
を必須成分として含有し、
(c)成分の含有量が3〜30質量%であることを特徴とする熱硬化性ビスマレイミド系樹脂組成物。 The following (a) to (c) components (a) component: bismaleimide compound,
(B) component: alkenylphenol and / or alkenylphenol ether compound,
(C) component: an acrylonitrile-butadiene copolymer having amine end groups,
As an essential component,
(C) Content of component is 3-30 mass%, The thermosetting bismaleimide-type resin composition characterized by the above-mentioned.
(a)成分:ビスマレイミド化合物、
(b)成分:アルケニルフェノール及び/又はアルケニルフェノールエーテル化合物、
(c)成分:アミン末端基を有するアクリロニトリル−ブタジエンコポリマー、
を、温度60〜140℃で混合することを特徴とする請求項1に記載の熱硬化性ビスマレイミド系樹脂組成物の製造方法。 The following (a) to (c) components (a) component: bismaleimide compound,
(B) component: alkenylphenol and / or alkenylphenol ether compound,
(C) component: an acrylonitrile-butadiene copolymer having amine end groups,
The method for producing a thermosetting bismaleimide resin composition according to claim 1, wherein the composition is mixed at a temperature of 60 to 140 ° C.
前記強化繊維基材内に含浸した請求項1に記載の熱硬化性ビスマレイミド系樹脂組成物と、
から成るプリプレグであって、
前記プリプレグ中における前記熱硬化性ビスマレイミド系樹脂組成物の割合が20〜60質量%であることを特徴とするプリプレグ。 A reinforcing fiber substrate;
The thermosetting bismaleimide resin composition according to claim 1 impregnated in the reinforcing fiber substrate;
A prepreg consisting of
The ratio of the said thermosetting bismaleimide-type resin composition in the said prepreg is 20-60 mass%, The prepreg characterized by the above-mentioned.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012268922A JP2014114368A (en) | 2012-12-10 | 2012-12-10 | Thermosetting bismaleimide-based resin composition and prepreg, and method for producing the same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012268922A JP2014114368A (en) | 2012-12-10 | 2012-12-10 | Thermosetting bismaleimide-based resin composition and prepreg, and method for producing the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2014114368A true JP2014114368A (en) | 2014-06-26 |
Family
ID=51170712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012268922A Pending JP2014114368A (en) | 2012-12-10 | 2012-12-10 | Thermosetting bismaleimide-based resin composition and prepreg, and method for producing the same |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2014114368A (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017210545A (en) * | 2016-05-25 | 2017-11-30 | 日立化成株式会社 | Thermosetting resin composition, and prepreg, copper-clad laminate and printed wiring board using the same |
| EP3170869A4 (en) * | 2014-07-16 | 2018-07-25 | JSR Corporation | Sizing agent, composition, and molded object |
| WO2020095882A1 (en) * | 2018-11-05 | 2020-05-14 | 地方独立行政法人大阪産業技術研究所 | Resin composition for generating allyl phenol-maleimide copolymer for electronic component protective film, and electronic component protective film comprising this copolymer |
| WO2020152906A1 (en) * | 2019-01-24 | 2020-07-30 | 昭和電工株式会社 | Thermosetting resin composition |
-
2012
- 2012-12-10 JP JP2012268922A patent/JP2014114368A/en active Pending
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3170869A4 (en) * | 2014-07-16 | 2018-07-25 | JSR Corporation | Sizing agent, composition, and molded object |
| JP2017210545A (en) * | 2016-05-25 | 2017-11-30 | 日立化成株式会社 | Thermosetting resin composition, and prepreg, copper-clad laminate and printed wiring board using the same |
| WO2020095882A1 (en) * | 2018-11-05 | 2020-05-14 | 地方独立行政法人大阪産業技術研究所 | Resin composition for generating allyl phenol-maleimide copolymer for electronic component protective film, and electronic component protective film comprising this copolymer |
| JP2020075961A (en) * | 2018-11-05 | 2020-05-21 | 地方独立行政法人大阪産業技術研究所 | Resin composition for forming allylphenol-maleimide copolymer for electronic component protection film, and electronic component protection film comprising copolymer |
| CN113195587A (en) * | 2018-11-05 | 2021-07-30 | 地方独立行政法人大阪产业技术研究所 | Resin composition for forming allylphenol-maleimide copolymer for electronic part protective film and electronic part protective film comprising the same |
| JP7181525B2 (en) | 2018-11-05 | 2022-12-01 | 地方独立行政法人大阪産業技術研究所 | Resin composition for producing allylphenol-maleimide copolymer for electronic component protective film, and electronic component protective film comprising the copolymer |
| CN113195587B (en) * | 2018-11-05 | 2024-03-15 | 地方独立行政法人大阪产业技术研究所 | Resin composition for producing allylphenol-maleimide copolymer for electronic part protective film and electronic part protective film comprising the same |
| WO2020152906A1 (en) * | 2019-01-24 | 2020-07-30 | 昭和電工株式会社 | Thermosetting resin composition |
| JPWO2020152906A1 (en) * | 2019-01-24 | 2021-12-02 | 昭和電工株式会社 | Thermosetting resin composition |
| JP7363821B2 (en) | 2019-01-24 | 2023-10-18 | 株式会社レゾナック | thermosetting resin composition |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2935421B1 (en) | Fast cure epoxy resin systems | |
| AU2012356792A1 (en) | Improvements in or relating to fibre reinforced composites | |
| JP2019157097A (en) | Prepreg and fiber-reinforced composite material, and method for producing same | |
| JP2014114368A (en) | Thermosetting bismaleimide-based resin composition and prepreg, and method for producing the same | |
| JP2014114369A (en) | Thermosetting bismaleimide-based resin composition, prepreg using the resin composition, and method for producing the same | |
| JP6949141B2 (en) | Thermosetting resin composition | |
| KR20170099984A (en) | Rapid curing epoxy resin and prepreg obtained therefrom | |
| JP2018062540A (en) | Thermosetting resin composition, prepreg, fiber-reinforced composite material, and method for producing the same | |
| JP7209470B2 (en) | Prepregs and carbon fiber reinforced composites | |
| JP7190258B2 (en) | Epoxy resin composition, prepreg, and fiber reinforced composite | |
| JP7086218B2 (en) | Thermosetting resin compositions, film adhesives, prepregs and methods for producing these | |
| KR102556138B1 (en) | Novel compositions with improved properties | |
| JP2019156981A (en) | Prepreg, fiber reinforced composite material, and manufacturing method therefor | |
| JP7315304B2 (en) | Epoxy resin composition, prepreg, and fiber reinforced composite | |
| JP2014105317A (en) | Resin composition, prepreg using resin composition, and method of manufacturing resin composition | |
| JP2013209474A (en) | Resin composition, prepreg using the same and method for producing resin composition | |
| JP2023084198A (en) | Epoxy resin composition, prepreg, fiber-reinforced composite material and production method | |
| JP2021178902A (en) | Prepreg | |
| WO2024071090A1 (en) | Prepreg, and method for producing fiber-reinforced composite material using said prepreg | |
| JP2022137823A (en) | Epoxy resin composition, resin cured material and fiber-reinforced composite material | |
| JP2022180746A (en) | Composite material and pre-preg | |
| JP2021195473A (en) | Prepreg |