JP2013236997A - 貴金属担持酸化チタンメソ結晶 - Google Patents
貴金属担持酸化チタンメソ結晶 Download PDFInfo
- Publication number
- JP2013236997A JP2013236997A JP2012110679A JP2012110679A JP2013236997A JP 2013236997 A JP2013236997 A JP 2013236997A JP 2012110679 A JP2012110679 A JP 2012110679A JP 2012110679 A JP2012110679 A JP 2012110679A JP 2013236997 A JP2013236997 A JP 2013236997A
- Authority
- JP
- Japan
- Prior art keywords
- titanium oxide
- noble metal
- mesocrystal
- supported
- oxide mesocrystal
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 title claims abstract description 311
- 239000004408 titanium dioxide Substances 0.000 title abstract 6
- 230000001699 photocatalysis Effects 0.000 claims abstract description 33
- 238000000034 method Methods 0.000 claims abstract description 22
- 150000002736 metal compounds Chemical class 0.000 claims abstract description 19
- 239000002516 radical scavenger Substances 0.000 claims abstract description 5
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 claims description 281
- 238000004519 manufacturing process Methods 0.000 claims description 124
- 229910000510 noble metal Inorganic materials 0.000 claims description 119
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 78
- 239000002082 metal nanoparticle Substances 0.000 claims description 61
- 239000010931 gold Substances 0.000 claims description 40
- 229910052697 platinum Inorganic materials 0.000 claims description 40
- 229910052737 gold Inorganic materials 0.000 claims description 39
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 claims description 35
- 239000002245 particle Substances 0.000 claims description 30
- 239000011941 photocatalyst Substances 0.000 claims description 21
- 150000002500 ions Chemical class 0.000 claims description 19
- 238000005245 sintering Methods 0.000 claims description 9
- 229910052709 silver Inorganic materials 0.000 claims description 6
- 239000004332 silver Substances 0.000 claims description 6
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 claims description 5
- 230000001678 irradiating effect Effects 0.000 claims description 3
- 239000002105 nanoparticle Substances 0.000 abstract description 89
- 239000010970 precious metal Substances 0.000 abstract description 7
- 230000027756 respiratory electron transport chain Effects 0.000 abstract description 5
- 229910021645 metal ion Inorganic materials 0.000 abstract description 2
- 239000013078 crystal Substances 0.000 description 70
- 230000000052 comparative effect Effects 0.000 description 48
- 239000002159 nanocrystal Substances 0.000 description 35
- 239000012298 atmosphere Substances 0.000 description 34
- 238000000354 decomposition reaction Methods 0.000 description 31
- 238000010304 firing Methods 0.000 description 31
- 229910010413 TiO 2 Inorganic materials 0.000 description 27
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 24
- 239000001301 oxygen Substances 0.000 description 24
- 229910052760 oxygen Inorganic materials 0.000 description 24
- 239000007864 aqueous solution Substances 0.000 description 23
- 239000002243 precursor Substances 0.000 description 23
- 229910017855 NH 4 F Inorganic materials 0.000 description 20
- 238000012360 testing method Methods 0.000 description 19
- 239000000758 substrate Substances 0.000 description 17
- 238000004220 aggregation Methods 0.000 description 14
- 230000002776 aggregation Effects 0.000 description 14
- 239000011148 porous material Substances 0.000 description 14
- 239000000243 solution Substances 0.000 description 14
- WXNZTHHGJRFXKQ-UHFFFAOYSA-N 4-chlorophenol Chemical compound OC1=CC=C(Cl)C=C1 WXNZTHHGJRFXKQ-UHFFFAOYSA-N 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- -1 gold ions Chemical class 0.000 description 10
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 9
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 9
- 229940043267 rhodamine b Drugs 0.000 description 9
- 239000010703 silicon Substances 0.000 description 9
- 229910052710 silicon Inorganic materials 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 239000010936 titanium Substances 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 238000002441 X-ray diffraction Methods 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 6
- 230000015556 catabolic process Effects 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- 238000006731 degradation reaction Methods 0.000 description 6
- 238000010586 diagram Methods 0.000 description 6
- 238000002149 energy-dispersive X-ray emission spectroscopy Methods 0.000 description 6
- 239000012535 impurity Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 238000000634 powder X-ray diffraction Methods 0.000 description 6
- 229910052719 titanium Inorganic materials 0.000 description 6
- 238000000921 elemental analysis Methods 0.000 description 5
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 238000011068 loading method Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 4
- 229910052753 mercury Inorganic materials 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000001228 spectrum Methods 0.000 description 4
- 238000004438 BET method Methods 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 230000005540 biological transmission Effects 0.000 description 3
- 238000001354 calcination Methods 0.000 description 3
- 238000000151 deposition Methods 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 230000006798 recombination Effects 0.000 description 3
- 238000005215 recombination Methods 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 239000013081 microcrystal Substances 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- NQTSTBMCCAVWOS-UHFFFAOYSA-N 1-dimethoxyphosphoryl-3-phenoxypropan-2-one Chemical compound COP(=O)(OC)CC(=O)COC1=CC=CC=C1 NQTSTBMCCAVWOS-UHFFFAOYSA-N 0.000 description 1
- ISPYQTSUDJAMAB-UHFFFAOYSA-N 2-chlorophenol Chemical compound OC1=CC=CC=C1Cl ISPYQTSUDJAMAB-UHFFFAOYSA-N 0.000 description 1
- 101710134784 Agnoprotein Proteins 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 229910003771 Gold(I) chloride Inorganic materials 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 230000010718 Oxidation Activity Effects 0.000 description 1
- 239000004280 Sodium formate Substances 0.000 description 1
- 238000003917 TEM image Methods 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960005215 dichloroacetic acid Drugs 0.000 description 1
- 229910001882 dioxygen Inorganic materials 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- 238000002003 electron diffraction Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- FDWREHZXQUYJFJ-UHFFFAOYSA-M gold monochloride Chemical compound [Cl-].[Au+] FDWREHZXQUYJFJ-UHFFFAOYSA-M 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 229910003437 indium oxide Inorganic materials 0.000 description 1
- PJXISJQVUVHSOJ-UHFFFAOYSA-N indium(iii) oxide Chemical compound [O-2].[O-2].[O-2].[In+3].[In+3] PJXISJQVUVHSOJ-UHFFFAOYSA-N 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 229910001867 inorganic solvent Inorganic materials 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 239000002086 nanomaterial Substances 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229940038504 oxygen 100 % Drugs 0.000 description 1
- SOQBVABWOPYFQZ-UHFFFAOYSA-N oxygen(2-);titanium(4+) Chemical class [O-2].[O-2].[Ti+4] SOQBVABWOPYFQZ-UHFFFAOYSA-N 0.000 description 1
- 238000013032 photocatalytic reaction Methods 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000011164 primary particle Substances 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 238000000985 reflectance spectrum Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- HLBBKKJFGFRGMU-UHFFFAOYSA-M sodium formate Chemical compound [Na+].[O-]C=O HLBBKKJFGFRGMU-UHFFFAOYSA-M 0.000 description 1
- 235000019254 sodium formate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000004528 spin coating Methods 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 1
- 229910001887 tin oxide Inorganic materials 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000000101 transmission high energy electron diffraction Methods 0.000 description 1
Images






















Landscapes
- Catalysts (AREA)
- Exhaust Gas Treatment By Means Of Catalyst (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Silicates, Zeolites, And Molecular Sieves (AREA)
Abstract
【課題】高効率な粒子間電子移動を可能とし、コスト面から貴金属ナノ粒子の担持量を低減した場合にも、担持しない場合と比較して劇的に光触媒活性を向上させた貴金属担持酸化チタンメソ結晶を提供することを目的とする。
【解決手段】平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶に、貴金属ナノ粒子が担持されている、貴金属担持酸化チタンメソ結晶。当該貴金属担持酸化チタンメソ結晶は、例えば、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液に、紫外光を照射し、その後焼結する工程を備える方法等により得られる。
【選択図】なし
【解決手段】平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶に、貴金属ナノ粒子が担持されている、貴金属担持酸化チタンメソ結晶。当該貴金属担持酸化チタンメソ結晶は、例えば、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液に、紫外光を照射し、その後焼結する工程を備える方法等により得られる。
【選択図】なし
Description
本発明は、貴金属担持酸化チタンメソ結晶に関する。
紫外光照射によって生じる二酸化チタン(TiO2)ナノ粒子の酸化還元力を、環境浄化型光触媒、機能性コーティング等として応用する試みが注目されている。
近年では、TiO2の光触媒能を向上させるため、貴金属ナノ粒子として金(Au)、銀(Ag)等を表面に担持させたTiO2ナノ粒子の開発が行われている(非特許文献1〜4参照)。また、白金(Pt)ナノ粒子を担持させたTiO2ナノ粒子は光水素発生用の光触媒として機能することが一般によく知られている。
貴金属ナノ粒子担持TiO2ナノ粒子(以下、「M/TiO2」と表記することもある。Mは貴金属ナノ粒子を示す。)では、光照射によってTiO2中に生じた電子が貴金属ナノ粒子に捕集されることで電子と正孔の再結合が抑制され、光触媒等の反応収率が向上すると考えられている。したがって、電子をいかに効率よく貴金属ナノ粒子まで運ぶかが重要な課題となる。
しかしながら、一般に、水溶液中のTiO2ナノ粒子はナノメートルからマイクロメートルサイズの無秩序な凝集体を形成しており、そのような凝集体では界面の不整合によって高効率な粒子間電子移動は期待できず、反応表面積の低下も懸念されている(非特許文献5、図1参照)。さらに、電子を捕集するためには個々のTiO2ナノ粒子に対し最低でも1個の貴金属ナノ粒子を担持する必要があるが、上記の貴金属は非常に高価であり、実用化を考えた場合、コスト面が問題となる。また、コスト面を考慮して貴金属ナノ粒子の担持量を低く抑えた場合には、上記のとおり界面の不整合によって高効率な粒子間電子移動ができないため、光触媒等の十分な反応収率が得られないという問題もある(図2参照)。
近年、粒子間にネットワーク構造を有する多孔質TiO2ナノ材料にPtナノ粒子を担持させた研究例も報告されているが、TiO2ナノ粒子の配列が十分に最適化されているとはいえない(非特許文献6)。
H. Tada, T. Ishida, A. Takao, S. Ito, S. Mukhopadhyay, T. Akita, K. Tanaka, H. Kobayashi, ChemPhysChem 6, 1537-1543 (2005).
T. Kiyonaga, T. Mitsui, M. Torikoshi, M. Takekawa, T. Soejima, H. Tada, J. Phys. Chem. B 110, 10771-10778 (2006).
Y.Z. Yang, C.-H. Chang, H. Idriss, Appl. Catal. B 67, 217-222 (2006).
M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca, H. Idriss, Nat. Chem.3, 489-492 (2011).
N. Lakshminarasimhan, W. Kim, W. Choi, J. Phys. Chem. C 112, 20451-20457 (2008).
A. A. Ismail, D. W. Bahnemann, J. Phys. Chem. C 115, 5784-5791 (2011).
L. Zhou et al., Small 2008, 4, 1566-1574
G. Liu et al., Chem. Commun. 2011, 47, 6763-6783
J. Feng et al., CrystEngComm, 2010, 12, 3425-3429
L. Zhou et al., J. Am. Chem. Soc. 2008, 130, 1309-1320
J. Zhu et al., CrystEngComm, 2010, 12, 2219-2224
T. Tachikawa et al., J. Am. Chem. Soc. 2011, 133, 7197-7204
本発明は、高効率な粒子間電子移動を可能とし、コスト面から貴金属ナノ粒子の担持量を低減した場合にも、担持しない場合と比較して劇的に光触媒活性を向上させた貴金属担持酸化チタンメソ結晶を提供することを目的とする。
上記目的を鑑み、鋭意検討した結果、TiF4、NH4NO3、NH4F及び水を所定量含む前駆体水溶液を、空気雰囲気下で焼成した後に、酸素雰囲気下でより高温で焼成することで得られるサイズ及び比表面積の大きい酸化チタンメソ結晶に、貴金属ナノ粒子を担持することで、上記課題を解決できることを見出した。本発明は、さらに研究を重ね、完成させたものである。すなわち、本発明は以下の構成を包含する。
項1.平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶に、貴金属ナノ粒子が担持されている、貴金属担持酸化チタンメソ結晶。
項2.前記貴金属が、金、白金及び銀よりなる群から選ばれる少なくとも1種である、項1に記載の貴金属担持酸化チタンメソ結晶。
項3.前記貴金属ナノ粒子の担持量が、0.03〜1.5重量%である、項1又は2に記載の貴金属担持酸化チタンメソ結晶。
項4.前記貴金属ナノ粒子の平均粒子径が1〜10nmである、項1〜3のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項5.比表面積が10m2/g以上である、項1〜4のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項6.前記酸化チタンメソ結晶の平均幅が2〜8μmである、項1〜5のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項7.前記酸化チタンメソ結晶の平均厚みが50〜300nmである、項1〜6のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項8.光触媒用である、項1〜7のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項9.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶からなる光触媒。
項10.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶の製造方法であって、
平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液に、紫外光を照射し、その後焼結する工程を備える、製造方法。
項11.前記紫外光の照射時間が10〜100分である、項10に記載の製造方法。
項12.焼結温度が100〜600℃である、項10又は11に記載の製造方法。
項13.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶、又は項9に記載の光触媒を使用する、少量の貴金属量で光触媒活性を向上させる方法。
項14.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶、又は項9に記載の光触媒を使用する、光触媒中の貴金属量を低減する方法。
項1.平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶に、貴金属ナノ粒子が担持されている、貴金属担持酸化チタンメソ結晶。
項2.前記貴金属が、金、白金及び銀よりなる群から選ばれる少なくとも1種である、項1に記載の貴金属担持酸化チタンメソ結晶。
項3.前記貴金属ナノ粒子の担持量が、0.03〜1.5重量%である、項1又は2に記載の貴金属担持酸化チタンメソ結晶。
項4.前記貴金属ナノ粒子の平均粒子径が1〜10nmである、項1〜3のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項5.比表面積が10m2/g以上である、項1〜4のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項6.前記酸化チタンメソ結晶の平均幅が2〜8μmである、項1〜5のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項7.前記酸化チタンメソ結晶の平均厚みが50〜300nmである、項1〜6のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項8.光触媒用である、項1〜7のいずれかに記載の貴金属担持酸化チタンメソ結晶。
項9.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶からなる光触媒。
項10.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶の製造方法であって、
平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液に、紫外光を照射し、その後焼結する工程を備える、製造方法。
項11.前記紫外光の照射時間が10〜100分である、項10に記載の製造方法。
項12.焼結温度が100〜600℃である、項10又は11に記載の製造方法。
項13.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶、又は項9に記載の光触媒を使用する、少量の貴金属量で光触媒活性を向上させる方法。
項14.項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶、又は項9に記載の光触媒を使用する、光触媒中の貴金属量を低減する方法。
本発明によれば、高効率な粒子間電子移動を可能とし、コスト面から貴金属ナノ粒子の担持量を低減した場合にも、貴金属ナノ粒子を担持しない場合と比較して劇的に光触媒活性を向上させた貴金属担持酸化チタンメソ結晶を提供することが可能である。特に光触媒活性については、合成した酸化チタンナノ粒子や市販の酸化チタンナノ粒子に貴金属ナノ粒子を担持した場合と比較しても、2倍以上の活性を有する貴金属担持酸化チタンメソ結晶を提供できる。
1.貴金属担持酸化チタンメソ結晶
本発明の貴金属担持酸化チタンメソ結晶は、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶に、貴金属ナノ粒子が担持されている。
本発明の貴金属担持酸化チタンメソ結晶は、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶に、貴金属ナノ粒子が担持されている。
(1)酸化チタンメソ結晶
本発明で使用する酸化チタンメソ結晶は、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である。
本発明で使用する酸化チタンメソ結晶は、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である。
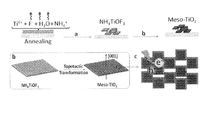
本発明において、「酸化チタンメソ結晶」とは、酸化チタンナノ結晶が規則的に配列したメソサイズ(具体的には1〜10μm程度)の結晶性超構造体を意味する。なお、超構造体とは、ナノ粒子又はナノ結晶が無秩序に凝集しているのではなく、規則的に配列した構造を意味する(図3、非特許文献7のFigure 1参照)。図3において、N-I、N-II及びN-IIIは、酸化チタンナノ粒子が規則的に配列した様々な酸化チタンナノ結晶を表している。そして、M-I、M-II及びM-IIIが、これらが規則的に配列した酸化チタンメソ結晶を表している。この際、酸化チタンメソ結晶において、酸化チタンナノ粒子1つ1つも、規則的に配列している。このように、本発明の酸化チタンメソ結晶は、酸化チタンナノ結晶が無秩序に凝集せず、規則的に配列した大きなサイズの結晶であるため、無秩序な凝集を抑制することができる。
また、本発明で使用する酸化チタンメソ結晶においては、上記のように、酸化チタンナノ結晶が規則的に配列していることから、全体として単結晶とすることも可能である。
なお、図3において、左下に示されているAmorphous Solid及びPolycrystalは、酸化チタンナノ粒子又は酸化チタンナノ結晶が無秩序に凝集した状態を表している。
また、図1にも示されるように、従来の無秩序な凝集体では、酸化チタンナノ結晶同士の接点が少なく(図1では点でのみ接しており)、光照射により発生した電子が伝導しにくかったが、本発明で使用する酸化チタンメソ結晶においては、図4に示されるように、酸化チタンナノ結晶が規則的に配列しており、酸化チタンナノ結晶同士の接点が多く(図4では線で接しており)、電子が伝導しやすくなり、高い電気伝導度が得られる。このため、貴金属ナノ粒子が少量である場合であっても、貴金属ナノ粒子への電子の移動を容易にし、十分な光触媒活性が得られる。また、本発明で使用する酸化チタンメソ結晶では凝集を抑制することができることから、同程度の比表面積を有する酸化チタンナノ粒子を使用した場合と比較しても、高い比表面積を維持することができ、光触媒活性を飛躍的に向上させることもできる。なお、本発明においては、酸化チタンメソ結晶に貴金属ナノ粒子を担持しても、比表面積を維持することが可能である。
本発明で使用する酸化チタンメソ結晶においては、厚みと比較して幅が充分大きいことが特徴の1つである。具体的には、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100、好ましくは40〜50程度である。平均幅の平均厚みに対する比を大きくすることで、酸化チタンメソ結晶のサイズを大きくすることができ、無秩序な凝集を抑制することができるが、大きすぎると壊れやすくなる観点から、上記の範囲が好ましい。
また、本発明の酸化チタンメソ結晶は、上記のとおり、平均幅の平均厚みに対する比が大きいことから、表面エネルギーの高い{001}面を主な結晶面として有することができる。{001}面は、高活性な結晶面として注目されているため(非特許文献8)、本発明で使用する酸化チタンメソ結晶は、{001}面を主な結晶面として有することにより、光触媒活性等を向上させることができる。
このように、平均幅の平均厚みに対する比を大きくすることが好ましいため、平均幅は大きいほうが好ましい。また、平均厚みは小さいほうが好ましい。具体的には、平均幅は、2〜8μm程度が好ましく、4〜5μm程度がより好ましい。また、平均厚みは、50〜300nm程度が好ましく、70〜110nm程度がより好ましい。
なお、本発明で使用する酸化チタンメソ結晶では、平均幅とは、表面が正方形又は長方形である板状結晶と見立てた場合において、見立てた正方形又は長方形の辺の平均値を意味する。また、本発明で使用する酸化チタンメソ結晶の平均厚みとは、板状結晶の場合にはその厚みの平均値、板状でない場合には板状結晶と見立てた場合の厚みの平均値である。これらの幅及び厚みは、例えば、電子顕微鏡観察(SEM等)等により測定することができる。
また、本発明で使用する酸化チタンメソ結晶の比表面積は、10m2/g以上である。酸化チタンメソ結晶の比表面積が小さすぎると、光触媒活性及び光電流寿命に劣る。本発明では、合成した酸化チタンナノ粒子や市販の酸化チタンナノ粒子に貴金属ナノ粒子を担持した場合と比較しても、2倍以上の光触媒活性を有する貴金属担持酸化チタンメソ結晶を得ることもできる。なお、酸化チタンメソ結晶の比表面積は、10〜80m2/g程度が好ましく、50〜70m2/g程度がより好ましい。また、本発明で使用する酸化チタンメソ結晶の比表面積は、例えば、BET法等により測定することができる。
本発明で使用する酸化チタンメソ結晶を構成する酸化チタンナノ結晶としては、アナターゼ型であってもルチル型であってもよい。なかでも、光触媒活性が高いことから、本発明で使用する酸化チタンメソ結晶は、アナターゼ型酸化チタンナノ結晶の集合体とすることが好ましい。なお、酸化チタンナノ結晶の結晶構造は、例えば、粉末X線回折等により測定することができる。
また、本発明で使用する酸化チタンメソ結晶を構成する酸化チタンナノ結晶の平均粒子径は、光触媒活性により優れる観点から、30〜70nm程度が好ましく、35〜40nm程度がより好ましい。酸化チタンナノ粒子の平均粒子径は、例えば、粉末X線回折(シェラーの式を用いて)等により測定することができる。
本発明で使用する酸化チタンメソ結晶における細孔径及び細孔容積は、より比表面積を向上させる観点から大きいほうが好ましい。具体的には、平均細孔径は、3〜8nm程度が好ましい。また、平均細孔容積は、0.1〜0.2cm3/g程度が好ましい。これらは、BJHモデルによる吸着等温線等から測定することができる。
本発明で使用する酸化チタンメソ結晶においては、酸化チタンの純度を向上させ、不純物である窒素、フッ素等を実質的に含まない結晶とすることができる。このことは、本発明で使用する酸化チタンメソ結晶のバンドギャップが、酸化チタンと同程度であることから確認することができる。
本発明で使用する酸化チタンメソ結晶の形状は、板状であっても、他の形状であってもよい。なかでも、表面エネルギーの高い{001}面を主な結晶面として有するためには平均幅の平均厚みに対する比が大きいことが好ましいことから、板状が好ましい。
(2)酸化チタンメソ結晶の製造方法
本発明で使用する酸化チタンメソ結晶は、例えば、TiF4、NH4NO3、NH4F及び水を含み、且つ、TiF4とNH4NO3との含有比率が1:4〜15(モル比)であり、TiF4とNH4Fとの含有比率が1:1〜9(モル比)である前駆体水溶液を、空気雰囲気又は酸素雰囲気下250〜700℃の条件下に焼成した後に、酸素雰囲気下400〜700℃の条件下に焼成する工程を備える方法により、製造することができる。
本発明で使用する酸化チタンメソ結晶は、例えば、TiF4、NH4NO3、NH4F及び水を含み、且つ、TiF4とNH4NO3との含有比率が1:4〜15(モル比)であり、TiF4とNH4Fとの含有比率が1:1〜9(モル比)である前駆体水溶液を、空気雰囲気又は酸素雰囲気下250〜700℃の条件下に焼成した後に、酸素雰囲気下400〜700℃の条件下に焼成する工程を備える方法により、製造することができる。
本発明で使用する酸化チタンメソ結晶の製造方法では、まず、前駆体水溶液を、空気雰囲気又は酸素雰囲気下250〜700℃の条件下に焼成する。
具体的には、前駆体水溶液からなる液層を基板上に形成し、空気雰囲気又は酸素雰囲気下250〜700℃の条件下に焼成すればよい。
基板としては、特に制限はなく、常温において平滑な面を有するものであり、その面は平面あるいは曲面であってもよく、また応力によって変形するものであってもよい。使用できる基板の具体例としては、例えば、シリコン、各種ガラス、透明樹脂等が挙げられる。ただし、後述のように、400℃以上で焼成する必要があることから、シリコン、ガラス等を用いるのが好ましい。
液層の膜厚は、特に制限されないが、通常2mm以下程度が好ましい。
液層の形成法は特に限定されるものではなく、用いる基板の種類により公知の方法を適宜採用することができる。例えば、基板上にディープコート、スピンコート等を施したり、基板材料と前駆体水溶液の混合溶液をシリコン、ガラス等に滴下したりすればよい。
なお、基板としては、上記の他、機能性材料を用いて、本発明で使用する酸化チタンメソ結晶との複合材料を容易に製造することができる。機能性材料としては、スズドープ酸化インジウム(ITO)、フッ素ドープ酸化スズ(FTO)等が挙げられる。
前駆体水溶液に含まれるチタン前駆体としては、TiF4を使用する。TiF4中のF−イオンが酸化チタンナノ結晶の{001}面に強く吸着し、{001}面を主な結晶面として有する、つまり、サイズが大きい酸化チタンメソ結晶が得られる。
チタン前駆体としては、TiF4よりも非常に安価なTiCl4を使用することも知られている(非特許文献9等)が、実際には、酸化チタンナノ結晶が規則的に配列することはなく、つまり酸化チタンメソ結晶が得られることはなく、凝集体しか得られない。その他、チタン前駆体として、Ti(OC4H9)4、Ti(SO4)2等を使用した場合も同様に、凝集体しか得られない。
本発明においては、酸化チタンメソ結晶の結晶構造を制御するためにNH4NO3を使用することが好ましい。この際、NH4NO3の使用量は、TiF4とNH4NO3との含有比率が1:4〜15(モル比)、好ましくは1:6〜9(モル比)が好ましい。NH4NO3の使用量が少なすぎると、結晶構造を有しない酸化チタンナノ粒子の凝集物ができ、酸化チタンメソ結晶を得にくい傾向がある。一方、NH4NO3の使用量が多すぎると、酸化チタンメソ結晶と酸化チタンナノ粒子との混合物となり、酸化チタンナノ粒子の凝集を防ぎにくい傾向がある。NH4NO3の使用量を上記範囲内とすることで、より結晶性の高い酸化チタンメソ結晶が得られる。
本発明においては、酸化チタンメソ結晶のサイズ(幅)と厚みを制御するためにNH4Fを使用することが好ましい。この際、NH4Fの使用量は、TiF4とNH4Fとの含有比率が1:1〜9(モル比)、特に1:3〜5(モル比)が好ましい。NH4Fの使用量が少なすぎると、平均幅が小さく厚みが大きくなり、凝集を防ぎにくい傾向がある。一方、NH4Fの使用量が多すぎると、酸化チタンメソ結晶の幅が大きくなりすぎ、且つ、厚みが薄くなりすぎて形状が崩壊するため凝集を防ぎにくい傾向がある。
溶媒としては水を使用することが好ましい。原料が有機材料の場合には有機溶媒を使用できるが、本発明では無機系材料を使用するため、水性溶媒、特に水が好ましい。水の使用量は、他の成分に対して過剰量とすればよいが、製膜のためには多すぎないほうがよい。具体的には、TiF4と水との含有比率を1:100〜1000(モル比)程度とすることが好ましい。
なお、本発明では、従来の方法(非特許文献10等)で使用されていた界面活性剤を使用せずとも、酸化チタンナノ結晶を規則的に配列して酸化チタンメソ結晶を製造することが可能である。
本発明においては、上記説明した前駆体水溶液を、まず空気雰囲気下250〜700℃、好ましくは250〜300℃、より好ましくは250〜290℃の条件下に焼成することが好ましい(第1焼成)。これにより、NH4TiOF3結晶を製造することができる。この際、酸素雰囲気下でもNH4TiOF3結晶を製造することができる。また、焼成温度が低すぎる場合には、NH4NO3との混合物となる。さらに、焼成温度が高すぎる場合には、比表面積が10m2/g以下の酸化チタン結晶となる傾向がある。第1焼成において、酸素雰囲気下で400〜700℃で焼成すれば、NH4TiOF3結晶を経由することなく、1ポットで本発明の酸化チタンメソ結晶を得ることも可能である。
酸化チタンメソ結晶は、後述のように、上記第1焼成で得られたNH4TiOF3結晶を用いて、トポタクティック反応により得ることができる。このトポタクティック反応においては、結晶の形状はほぼ維持される(非特許文献10等参照)ので、NH4TiOF3結晶の形状は酸化チタンメソ結晶と同様である。具体的には、以下のとおりである。
NH4TiOF3結晶においては、厚みと比較して幅が充分大きいことが特徴である。具体的には、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100、好ましくは40〜50程度である。平均幅の平均厚みに対する比を多くすることで、得られる酸化チタンメソ結晶のサイズを大きくすることができ、酸化チタンメソ結晶の無秩序な凝集を抑制することができるが、大きすぎると壊れやすくなる観点から、上記の範囲が好ましい。
また、NH4TiOF3結晶は、上記のとおり、平均幅の平均厚みに対する比が大きいことから、得られる酸化チタンメソ結晶を、表面エネルギーの高い{001}面を主な結晶面として有することができる。{001}面は、高活性な結晶面として注目されているため(非特許文献8)、酸化チタンメソ結晶は、{001}面を主な結晶面として有することにより、光触媒活性等を向上させることができる。
このように、平均幅の平均厚みに対する比を大きくすることが好ましいため、平均幅は大きいほうが好ましい。また、平均厚みは小さいほうが好ましい。具体的には、平均幅は、2〜8μm程度が好ましく、4〜5μm程度がより好ましい。また、平均厚みは、50〜300nm程度が好ましく、70〜110nm程度がより好ましい。
なお、平均幅及び平均厚みは、上記説明したものである。
NH4TiOF3結晶の形状は、板状であっても、他の形状であってもよい。なかでも、得られる酸化チタンメソ結晶において、表面エネルギーの高い{001}面を主な結晶面として有するためには平均幅の平均厚みに対する比が大きいことが好ましいことから、板状が好ましい。
第1焼成の後、製造したNH4TiOF3結晶を、酸素雰囲気下400〜700℃の条件下に焼成する(第2焼成)ことが好ましい。この第2焼成により、トポタクティック反応(非特許文献10等参照)を起こさせ、酸化チタンメソ結晶が得られる。この際、上記の空気雰囲気下での焼成と同じ炉で行ってもよいし、異なる炉で行ってもよい。なお、上述したように、第1焼成において、酸素雰囲気下で400〜700℃の条件下で焼成した場合には、第2焼成を行わずに酸化チタンメソ結晶を得ることも可能である。
第2焼成(第1焼成しか行わない場合は第1焼成)の雰囲気を酸素雰囲気とすることで、酸化チタンメソ結晶内への窒素、フッ素等の不純物の混入を抑制することができる。なお、本発明において、酸素雰囲気とは、100%の酸素ガスもしくは酸素濃度が90%以上の酸素と空気の混合ガス雰囲気が好ましい。
また、第2焼成の焼成温度は400〜700℃、好ましくは400〜600℃、より好ましくは450〜550℃の条件化に焼成することが好ましい。焼成温度が低すぎると、NH4TiOF3を十分にTiO2に変換することができず、酸化チタンメソ結晶が得られない傾向がある。また、比表面積を大きくすることができない傾向もある。一方、焼成温度が高すぎると、酸化チタンメソ結晶は得られるが、比表面積は低下し、光触媒活性等が悪化する傾向がある。なお、通常は高温で焼成すると酸化チタンは活性の高いアナターゼ型を維持できず、ルチル型に構造変化するが、本発明においては、高温(上記温度範囲の上限近く)で焼成しても、アナターゼ型を維持することができる。
なお、上記説明した酸化チタンメソ結晶の製造方法の概念の一例を図5に示す。
(3)貴金属ナノ粒子
酸化チタンメソ結晶に担持させる貴金属ナノ粒子としては、特に制限されるわけではないが、金、白金、銀、パラジウム等の1種以上が挙げられる。なかでも、金、白金等が好ましい。
酸化チタンメソ結晶に担持させる貴金属ナノ粒子としては、特に制限されるわけではないが、金、白金、銀、パラジウム等の1種以上が挙げられる。なかでも、金、白金等が好ましい。
貴金属ナノ粒子の平均粒子径は、特に制限されるわけではないが、1〜10nm程度が好ましく、2〜5nm程度がより好ましい。貴金属ナノ粒子の平均粒子径をこの範囲とすることで、TiO2中に生成した電子を捕捉することができる。特に、貴金属ナノ粒子の平均粒子径を低く抑えることで、貴金属ナノ粒子の数を多くすることができ、TiO2中に生成した電子をより効率よく捕捉することができる。また、貴金属ナノ粒子の平均粒子径は、例えば、電子顕微鏡観察(TEM等)等により測定することができる。
本発明において、酸化チタンメソ結晶に対する貴金属ナノ粒子の担持量は、担持させる貴金属の種類によっても異なるが、十分に光触媒能を向上させるとともに、貴金属の使用量をできるだけ抑えるという点から、通常0.03〜1.5重量%、特に0.04〜1.0重量%が好ましい。なお、貴金属の種類によってナノ粒子の形成しやすさ(成長速度)が異なるために好ましい担持量の範囲も異なる。具体的には、貴金属ナノ粒子として金ナノ粒子を使用する場合には0.03〜0.05重量%、白金ナノ粒子を使用する場合には1.0〜1.5重量%、が好ましい。
(4)貴金属担持酸化チタンメソ結晶
本発明の貴金属担持酸化チタンメソ結晶においては、図6にも示されるように、酸化チタンナノ結晶が規則的に配列しており、酸化チタンナノ結晶同士の接点が多く(図6では線で接しており)、電子が伝導しやすくなっている。このため、光照射により発生した電子をより確実に、貴金属ナノ粒子に移動させることが可能であるため、光触媒活性を効率的に向上させることができる。具体的には、従来の酸化チタンナノ粒子系に貴金属ナノ粒子を担持させた場合と比較し、2倍以上の光触媒活性を得ることも可能である。これは、本発明では、酸化チタンナノ粒子同士の凝集を抑制することができることから、同程度の比表面積を有する酸化チタンナノ粒子を使用した場合と比較しても、高い比表面積を維持することができ、光触媒活性を飛躍的に向上させることもできることにも起因する。さらに、上記のとおり、酸化チタンナノ結晶界面においても電子の伝導が容易であるため、全ての酸化チタンナノ粒子又は酸化チタンナノ結晶に1つ以上の貴金属ナノ粒子を担持させる必要がなく、担持量を低減できるために、コスト面でも有利である。
本発明の貴金属担持酸化チタンメソ結晶においては、図6にも示されるように、酸化チタンナノ結晶が規則的に配列しており、酸化チタンナノ結晶同士の接点が多く(図6では線で接しており)、電子が伝導しやすくなっている。このため、光照射により発生した電子をより確実に、貴金属ナノ粒子に移動させることが可能であるため、光触媒活性を効率的に向上させることができる。具体的には、従来の酸化チタンナノ粒子系に貴金属ナノ粒子を担持させた場合と比較し、2倍以上の光触媒活性を得ることも可能である。これは、本発明では、酸化チタンナノ粒子同士の凝集を抑制することができることから、同程度の比表面積を有する酸化チタンナノ粒子を使用した場合と比較しても、高い比表面積を維持することができ、光触媒活性を飛躍的に向上させることもできることにも起因する。さらに、上記のとおり、酸化チタンナノ結晶界面においても電子の伝導が容易であるため、全ての酸化チタンナノ粒子又は酸化チタンナノ結晶に1つ以上の貴金属ナノ粒子を担持させる必要がなく、担持量を低減できるために、コスト面でも有利である。
なお、本発明の貴金属担持酸化チタンメソ結晶においては、サイズや比表面積等については酸化チタンメソ結晶と同様である。具体的には、以下のとおりである。
本発明の貴金属担持酸化チタンメソ結晶は、厚みと比較して幅が充分大きいことが特徴の1つである。具体的には、平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100、好ましくは40〜50程度である。平均幅の平均厚みに対する比を大きくすることで、本発明の貴金属担持酸化チタンメソ結晶のサイズを大きくすることができ、無秩序な凝集を抑制することができるが、大きすぎると壊れやすくなる観点から、上記の範囲が好ましい。
このように、平均幅の平均厚みに対する比を大きくすることが好ましいため、平均幅は大きいほうが好ましい。また、平均厚みは小さいほうが好ましい。具体的には、平均幅は、2〜8μm程度が好ましく、4〜5μm程度がより好ましい。また、平均厚みは、50〜300nm程度が好ましく、70〜110nm程度がより好ましい。
なお、本発明の貴金属担持酸化チタンメソ結晶では、平均幅及び平均厚みは、上記説明したものである。
また、本発明の貴金属担持酸化チタンメソ結晶の比表面積は、10m2/g以上であることが好ましい。酸化チタンメソ結晶の比表面積が小さすぎると、光触媒活性及び光電流寿命に劣る傾向がある。本発明の貴金属担持酸化チタンメソ結晶では、合成した酸化チタンナノ粒子や市販の酸化チタンナノ粒子に貴金属ナノ粒子を担持した場合と比較しても、2倍以上の光触媒活性を有する。なお、貴金属担持酸化チタンメソ結晶の比表面積は、10〜80m2/g程度が好ましく、50〜70m2/g程度がより好ましい。また、本発明の貴金属担持酸化チタンメソ結晶の比表面積は、例えば、BET法等により測定することができる。
本発明の貴金属担持酸化チタンメソ結晶における細孔径及び細孔容積は、より比表面積を向上させる観点から大きいほうが好ましい。具体的には、平均細孔径は、3〜8nm程度が好ましい。また、平均細孔容積は、0.1〜0.2cm3/g程度が好ましい。これらは、BJHモデルによる吸着等温線等から測定することができる。
本発明の貴金属担持酸化チタンメソ結晶の形状は、板状であっても、他の形状であってもよい。なかでも、表面エネルギーの高い{001}面を主な結晶面として有するためには平均幅の平均厚みに対する比が大きいことが好ましいことから、板状が好ましい。
2.貴金属担持酸化チタンメソ結晶の製造方法
本発明の貴金属担持酸化チタンメソ結晶の製造方法は、
平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液に、紫外光を照射し、その後焼結する工程を備える。
本発明の貴金属担持酸化チタンメソ結晶の製造方法は、
平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液に、紫外光を照射し、その後焼結する工程を備える。
酸化チタンメソ結晶は、上記説明したものである。
本発明で用いる貴金属イオン又は貴金属化合物は、電子を受容して0価の貴金属に還元されるものである。貴金属イオン又は貴金属化合物を構成する金属としては、具体的には、金、白金、銀、パラジウム等が挙げられる。つまり、貴金属イオンとしては、金イオン、白金イオン、銀イオン、パラジウムイオン等が挙げられる。なお、貴金属イオンは、例えば、後述の貴金属化合物を溶液中に溶解させることで、溶液中に導入することができる。また、貴金属化合物としては、HAuCl4、AuCl3、H2PtCl6、AgNO2、Pd(C5H7O2)2等が挙げられる。これらの貴金属イオン及び貴金属化合物は、単独で用いてもよいし、2種以上を組合せて用いてもよい。また、これにより、2種以上の異なる金属からなる複合金属ナノ粒子を担持させることも可能である。
また、正孔捕捉剤としては、光照射により発生した電子と正孔が再結合するのを抑制するため、正孔を捕捉する機能を有するものであれば特に制限されないが、具体的には、メタノール、エタノール、1−プロパノール等のアルコール;光触媒活エチレンジアミン四酢酸(EDTA);ギ酸ナトリウム等が挙げられ、アルコールが好ましい。なお、アルコールを正孔捕捉剤として使用する場合は、アルコールを溶媒として使用することもできる。
なお、溶媒としては、アルコールを使用することが好ましいが、アルコールと他の溶媒との混合溶媒としてもよい。他の溶媒としては、具体的には、水等が挙げられる。このように、アルコールと他の溶媒との混合溶媒とする場合には、アルコールの含有量を10〜30体積%とすることが好ましい。
本発明では、溶液中の、酸化チタンメソ結晶の濃度は、凝集による光照射の均一性や反応効率の低下を防ぐという点から、0.5〜2.0g/L程度が好ましく、0.5〜1.0g/L程度がより好ましい。また、貴金属イオン又は貴金属化合物の濃度は、最適な貴金属ナノ粒子のサイズを得るという点から、貴金属イオンに換算して、(5〜80)×10−6mol/L程度が好ましく、(6〜60)×10−6mol/L程度がより好ましい。
本発明では、まず、上記の溶液に、紫外光を照射することが好ましい。
紫外光の波長や露光強度は、還元しようとする貴金属イオン又は貴金属化合物の種類、担持量等によって適宜設定すればよいが、具体的には、300〜390nm程度、特に 300〜350nm程度が好ましい。また、露光強度は、10〜500mW/cm2が、さらには50〜200mW/cm2が好ましい。
紫外光の光源としては、Nd:YAGレーザー、エキシマーレーザー等のレーザー光、水銀灯、Xeランプ等のランプ光等を用いることができる。レーザー光にはパルス光と連続発振光(continuous-wave light)があるが、いずれも用いることができる。また、ランプ光は通常連続発振光のみであるが、機械的手段によってパルス化して用いることも可能である。レーザー照射では、ランプによる光照射と比較してより高効率に貴金属ナノ粒子を担持させることができるだけでなく、レーザーの持つ空間分解能を得ることができるため好ましい。
照射時間は、還元しようとする貴金属イオン又は貴金属化合物の種類、担持量等によって適宜設定すればよいが、十分な貴金属ナノ粒子を担持させるためには、10〜100分が好ましく、30〜60分がより好ましい。なお、必要に応じて、紫外光の照射時に加熱することにより、貴金属ナノ粒子の生成を加速させることも可能である。
本発明では、このようにして貴金属ナノ粒子を酸化チタンメソ結晶に担持させた後、焼結をする。これにより、本発明の貴金属担持酸化チタンメソ結晶の強度を向上させるとともに、不純物濃度を低くすることができる。
なお、焼結温度は、析出させようとする貴金属の種類や担持量等によっても異なるが、100〜600℃程度が好ましく、250〜550℃程度がより好ましい。これにより、酸化チタンメソ結晶の配列を乱すことなく、強度を向上させるとともに、不純物濃度を低くすることができる。なお、金ナノ粒子を析出させる場合は400〜500℃程度が好ましく、白金ナノ粒子を析出させる場合は250〜350℃程度が好ましい。
焼結雰囲気は、特に制限されない。例えば、空気中で行ってもよいし、不活性ガス雰囲気下で行ってもよい。また、焼結時間も特に制限されず、10〜60分程度が好ましく、20〜30分程度がより好ましい。
上記説明においては、酸化チタンメソ結晶に紫外光を照射した場合の光触媒的還元析出により貴金属ナノ粒子を担持させる方法について説明したが、担持方法はこの方法に限定されることはない。つまり、結晶上にナノ粒子を担持又は析出させることができる公知の方法をいずれも採用することができる。
例えば、上記説明した酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液中で酸化チタンメソ結晶をよく分散させる。次に、ロータリーエバポレーター等を用いて溶媒を蒸発させ、その後、200〜600℃程度で焼結することによっても、酸化チタンメソ結晶に貴金属ナノ粒子を担持させることができる。
また、酸化チタンメソ結晶を作製するために使用する、TiF4、NH4NO3、NH4F及び水を含む前駆体水溶液中に、適量の貴金属イオン、貴金属化合物、貴金属ナノ粒子等の1種以上を含ませておくことによっても、貴金属ナノ粒子が担持した酸化チタンメソ結晶を得ることができる。この際、貴金属の形態としては、貴金属ナノ粒子の凝集を抑制する観点から、貴金属イオン又は貴金属化合物が好ましい。なお、貴金属イオン、貴金属化合物、貴金属ナノ粒子等については上述したものが挙げられる。また、この際の貴金属ナノ粒子又は貴金属化合物の量は、上述した紫外光照射による手法の場合と同程度が好ましい。つまり、貴金属イオンに換算して、(5〜80)×10−6mol/L程度が好ましく、(6〜60)×10−6mol/L程度がより好ましい。
さらに、上記説明したようにNH4TiOF3結晶を得た後に、当該NH4TiOF3結晶を、貴金属イオン、貴金属化合物、貴金属ナノ粒子等の1種以上と、正孔捕捉剤とを含む溶液に浸漬し、溶媒を蒸発させた後に、酸素雰囲気下400〜700℃の条件下に焼成することによっても得ることができる。なお、貴金属イオン、貴金属化合物、貴金属ナノ粒子、正孔捕捉剤等については上述したものが挙げられる。また、焼成の際の条件は、上記説明した酸化チタンメソ結晶を作製する方法と同様である。また、溶媒を蒸発させる方法は、公知の方法をいずれも採用できる。
これらのなかでも、担持させる貴金属ナノ粒子の粒径を適度に調節することでTiO2中に生成した電子をより効率よく捕捉し、貴金属ナノ粒子の担持量を十分に大きくして光触媒活性をより向上させるとともに、簡便な手法であるという観点から、光触媒的還元析出により貴金属ナノ粒子を担持させるのが簡便である。
3.用途
本発明の貴金属担持酸化チタンメソ結晶は、上記のように、比表面積が大きいとともに、酸化チタンナノ結晶を規則的に配列し、且つ、サイズも大きく凝集を抑制できる酸化チタンメソ結晶を使用していることから、光触媒活性が高く、導電性も高い。また、本発明によれば、酸化チタンナノ粒子1つ1つに貴金属ナノ粒子を担持させずとも、光照射により発生した電子が貴金属ナノ粒子まで移動するための導電性が確保されているため、従来よりも光触媒(環境浄化光触媒、水素発生光触媒等)活性を顕著に向上させることができる。特に、本発明の貴金属担持酸化チタンメソ結晶は、可視光を照射する場合においても、光触媒として機能させることが可能である。さらに、本発明によれば、非常に簡便な方法で貴金属担持酸化チタンメソ結晶を製造することができるため、大量生産性にも優れる。
本発明の貴金属担持酸化チタンメソ結晶は、上記のように、比表面積が大きいとともに、酸化チタンナノ結晶を規則的に配列し、且つ、サイズも大きく凝集を抑制できる酸化チタンメソ結晶を使用していることから、光触媒活性が高く、導電性も高い。また、本発明によれば、酸化チタンナノ粒子1つ1つに貴金属ナノ粒子を担持させずとも、光照射により発生した電子が貴金属ナノ粒子まで移動するための導電性が確保されているため、従来よりも光触媒(環境浄化光触媒、水素発生光触媒等)活性を顕著に向上させることができる。特に、本発明の貴金属担持酸化チタンメソ結晶は、可視光を照射する場合においても、光触媒として機能させることが可能である。さらに、本発明によれば、非常に簡便な方法で貴金属担持酸化チタンメソ結晶を製造することができるため、大量生産性にも優れる。
実施例に基づいて、本発明を具体的に説明するが、本発明は、これらのみに限定されるものではない。
<酸化チタンメソ結晶等>
製造例1:400℃焼成(Meso-TiO 2 -400)
まず、TiF4、NH4NO3、NH4を含む前駆体水溶液(組成はモル比で、TiF4:NH4NO3:NH4:H2O=1:6.6:4:117)からなる液層を、シリコン基板に形成した。具体的には、前駆体水溶液を基板上に滴下した。液層の厚みは、1mm以下となるようにした。シリコン基板上に形成した液層を、空気雰囲気下250℃で2時間焼結することで、シリコン基板上にNH4TiOF3結晶を形成した。その後、酸素雰囲気(酸素100%)下400℃で8時間焼成することで、シリコン基板上に製造例1の酸化チタンメソ結晶を形成した。なお、前駆体水溶液に使用した原料はいずれも市販品を使用した。
製造例1:400℃焼成(Meso-TiO 2 -400)
まず、TiF4、NH4NO3、NH4を含む前駆体水溶液(組成はモル比で、TiF4:NH4NO3:NH4:H2O=1:6.6:4:117)からなる液層を、シリコン基板に形成した。具体的には、前駆体水溶液を基板上に滴下した。液層の厚みは、1mm以下となるようにした。シリコン基板上に形成した液層を、空気雰囲気下250℃で2時間焼結することで、シリコン基板上にNH4TiOF3結晶を形成した。その後、酸素雰囲気(酸素100%)下400℃で8時間焼成することで、シリコン基板上に製造例1の酸化チタンメソ結晶を形成した。なお、前駆体水溶液に使用した原料はいずれも市販品を使用した。
製造例2:500℃焼成(Meso-TiO 2 -500)
酸素雰囲気下での焼成温度を500℃とすること以外は製造例1と同様に、製造例2のNH4TiOF3結晶及び酸化チタンメソ結晶を作製した。
酸素雰囲気下での焼成温度を500℃とすること以外は製造例1と同様に、製造例2のNH4TiOF3結晶及び酸化チタンメソ結晶を作製した。
製造例3:600℃焼成(Meso-TiO 2 -600)
酸素雰囲気下での焼成温度を600℃とすること以外は製造例1と同様に、製造例3のNH4TiOF3結晶及び酸化チタンメソ結晶を作製した。
酸素雰囲気下での焼成温度を600℃とすること以外は製造例1と同様に、製造例3のNH4TiOF3結晶及び酸化チタンメソ結晶を作製した。
製造例4:700℃焼成(Meso-TiO 2 -700)
酸素雰囲気下での焼成温度を700℃とすること以外は製造例1と同様に、製造例4のNH4TiOF3結晶及び酸化チタンメソ結晶を作製した。
酸素雰囲気下での焼成温度を700℃とすること以外は製造例1と同様に、製造例4のNH4TiOF3結晶及び酸化チタンメソ結晶を作製した。
製造例5:500℃焼成、TiF 4 :NH 4 NO 3 =1:9
前駆体水溶液において、TiF4:NH4NO3=1:9(モル比)とすること以外は製造例2と同様に、製造例5の酸化チタンメソ結晶を作製した。
前駆体水溶液において、TiF4:NH4NO3=1:9(モル比)とすること以外は製造例2と同様に、製造例5の酸化チタンメソ結晶を作製した。
製造例6:500℃焼成、TiF 4 :NH 4 F=1:2
前駆体水溶液において、TiF4:NH4F=1:2(モル比)とすること以外は製造例2と同様に、製造例6の酸化チタンメソ結晶を作製した。
前駆体水溶液において、TiF4:NH4F=1:2(モル比)とすること以外は製造例2と同様に、製造例6の酸化チタンメソ結晶を作製した。
製造例7:500℃焼成、TiF 4 :NH 4 F=1:8
前駆体水溶液において、TiF4:NH4F=1:8(モル比)とすること以外は製造例2と同様に、製造例7の酸化チタンメソ結晶を作製した。
前駆体水溶液において、TiF4:NH4F=1:8(モル比)とすること以外は製造例2と同様に、製造例7の酸化チタンメソ結晶を作製した。
比較製造例1:酸化チタンナノ結晶(Nano-TiO 2 )
非特許文献11に従い、比較製造例1の酸化チタンナノ結晶を作製した。この試料は、酸素雰囲気下、600℃で8時間焼成した。
非特許文献11に従い、比較製造例1の酸化チタンナノ結晶を作製した。この試料は、酸素雰囲気下、600℃で8時間焼成した。
比較製造例2:酸化チタン多結晶(Poly-TiO 2 )
まず、TiF4を含む前駆体水溶液(組成はモル比で、TiF4:H2O=1:117)からなる液層を、シリコン基板に形成した。その後、シリコン基板上に形成した液層を、空気雰囲気下500℃で2時間焼結した。さらに、酸素雰囲気下500℃で8時間焼成し、比較製造例2の酸化チタン多結晶を得た。
まず、TiF4を含む前駆体水溶液(組成はモル比で、TiF4:H2O=1:117)からなる液層を、シリコン基板に形成した。その後、シリコン基板上に形成した液層を、空気雰囲気下500℃で2時間焼結した。さらに、酸素雰囲気下500℃で8時間焼成し、比較製造例2の酸化チタン多結晶を得た。
比較製造例3:酸化チタンマイクロ結晶(Micro-TiO 2 )
非特許文献12に従い、比較製造例3の酸化チタンマイクロ結晶を作製した。この試料は、酸素雰囲気下、600℃で8時間焼成した。
非特許文献12に従い、比較製造例3の酸化チタンマイクロ結晶を作製した。この試料は、酸素雰囲気下、600℃で8時間焼成した。
比較製造例4:300℃焼成(TiO 2 -300)
酸素雰囲気下での焼成温度を300℃とすること以外は製造例1と同様に、比較製造例4の酸化チタン結晶を作製した。
酸素雰囲気下での焼成温度を300℃とすること以外は製造例1と同様に、比較製造例4の酸化チタン結晶を作製した。
比較製造例5:800℃焼成(Meso-TiO 2 -800)
酸素雰囲気下での焼成温度を800℃とすること以外は製造例1と同様に、比較製造例5の酸化チタン結晶を作製した。
酸素雰囲気下での焼成温度を800℃とすること以外は製造例1と同様に、比較製造例5の酸化チタン結晶を作製した。
比較製造例6:500℃焼成、TiF 4 :NH 4 NO 3 =1:0
前駆体水溶液において、NH4NO3を使用しない(TiF4:NH4NO3=1:0(モル比))こと以外は製造例2と同様に、比較製造例6の結晶を作製した。
前駆体水溶液において、NH4NO3を使用しない(TiF4:NH4NO3=1:0(モル比))こと以外は製造例2と同様に、比較製造例6の結晶を作製した。
比較製造例7:500℃焼成、TiF 4 :NH 4 NO 3 =1:3
前駆体水溶液において、TiF4:NH4NO3=1:3(モル比)とすること以外は製造例2と同様に、比較製造例7の結晶を作製した。
前駆体水溶液において、TiF4:NH4NO3=1:3(モル比)とすること以外は製造例2と同様に、比較製造例7の結晶を作製した。
比較製造例8:500℃焼成、TiF 4 :NH 4 NO 3 =1:20
前駆体水溶液において、TiF4:NH4NO3=1:20(モル比)とすること以外は製造例2と同様に、比較製造例8の結晶を作製した。
前駆体水溶液において、TiF4:NH4NO3=1:20(モル比)とすること以外は製造例2と同様に、比較製造例8の結晶を作製した。
比較製造例9:500℃焼成、TiF 4 :NH 4 F=1:0
前駆体水溶液において、NH4Fを使用しない(TiF4:NH4F=1:0(モル比))こと以外は製造例2と同様に、比較製造例9の結晶を作製した。
前駆体水溶液において、NH4Fを使用しない(TiF4:NH4F=1:0(モル比))こと以外は製造例2と同様に、比較製造例9の結晶を作製した。
比較製造例10:500℃焼成、TiF 4 :NH 4 F=1:10
前駆体水溶液において、TiF4:NH4F=1:10(モル比)とすること以外は製造例2と同様に、比較製造例10の結晶を作製した。
前駆体水溶液において、TiF4:NH4F=1:10(モル比)とすること以外は製造例2と同様に、比較製造例10の結晶を作製した。
比較製造例11:250℃焼成(TiO 2 -250)
酸素雰囲気下での焼成温度を250℃とすること以外は製造例1と同様に、比較製造例11の酸化チタン結晶を作製した。
酸素雰囲気下での焼成温度を250℃とすること以外は製造例1と同様に、比較製造例11の酸化チタン結晶を作製した。
<酸化チタンメソ結晶の評価>
試験例1:比表面積
製造例1〜4及び比較製造例1〜5の結晶の比表面積をBET法により測定した。結果を表1に示す。
試験例1:比表面積
製造例1〜4及び比較製造例1〜5の結晶の比表面積をBET法により測定した。結果を表1に示す。
試験例2:細孔径及び細孔容積
製造例1〜4及び比較製造例1〜5の結晶の細孔径及び細孔容積をBJH法により測定した。結果を表1に示す。
製造例1〜4及び比較製造例1〜5の結晶の細孔径及び細孔容積をBJH法により測定した。結果を表1に示す。
試験例3:X線回折
製造例1〜3及び比較製造例4、5及び11の結晶の特性を粉末X線回折(XRD)により測定した。また、製造例1〜4及び比較製造例1〜5の結晶について、X線回折ピークから、シェラーの式を用いて、各結晶を構成する酸化チタンナノ結晶の粒子径を評価した。結果を表1及び図7に示す。
製造例1〜3及び比較製造例4、5及び11の結晶の特性を粉末X線回折(XRD)により測定した。また、製造例1〜4及び比較製造例1〜5の結晶について、X線回折ピークから、シェラーの式を用いて、各結晶を構成する酸化チタンナノ結晶の粒子径を評価した。結果を表1及び図7に示す。
参考までに、比較製造例1〜3の結晶のX線回折の結果も図8〜9に示す。いずれも、アナターゼ型の酸化チタンが検出されている。

試験例4:電子顕微鏡観察
製造例2の結晶の特性を走査型電子顕微鏡(SEM)及び透過型電子顕微鏡(TEM)により観察した。結果を図10〜11に示す。
製造例2の結晶の特性を走査型電子顕微鏡(SEM)及び透過型電子顕微鏡(TEM)により観察した。結果を図10〜11に示す。
図10aに示されるように、酸化チタンメソ結晶(特に製造例2)は、表面が略正方形状の板状結晶であった。
また、図10bのように、酸化チタンナノ結晶が規則的に並んでいた。また、数nm程度の細孔が生じていた。細孔構造はTEMでも確認し(図10d)、結晶上の制限視野電子線回折(SAED)パターンから、{001}面に沿った単結晶のアナターゼ型結晶が確認された。酸化チタン粒子の接点の高分解能透過電子顕微鏡(HRTEM)像より、単結晶格子が、0.189nm程度の格子間隔を有するアナターゼ(200)又は(020)結晶面の原子面を示した(図10e)。これらの結果は、本発明において、酸化チタンメソ結晶は、{001}面が表面に露出していることを強く示唆している。さらに、均一格子縞が明確に得られ、完璧に配向した酸化チタンナノ結晶が確認された。ナノ結晶が規則的に並ぶことにより、表面には多数の欠陥や孔が形成されていた(図10b及び11e)。結晶端の孔を有する箇所のTEM像から、ナノ結晶は、正規構造に並んでいることが示された(図10f)。
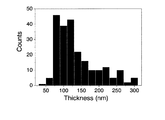
酸化チタンメソ結晶の平均厚みは、約80nmであり(図10c)、50〜300nmの範囲で分布していた(図11)。
他の製造例においても、酸化チタンメソ結晶の形状及び結晶構造は同様の結果が得られた(図7及び12等参照)。
また、製造例5及び比較製造例6〜8の走査型電子顕微鏡(SEM)像を図13に示す。NH4NO3を使用しない場合や、量が少ない場合には、酸化チタン粒子の再結合による一次粒子の凝集体が見られた(図13a〜b)。NH4NO3の量を増やすと、板状の酸化チタンメソ結晶が形成された(図13c)。さらにNH4NO3の量を増やすと、板状結晶と粒子との混合物となった(図13d)。このため、製造例5及び比較製造例6〜8のうち、製造例5のみが、酸化チタンナノ粒子の凝集を抑制できることが示唆されている。
さらに、製造例6〜7及び比較製造例9〜10の走査型電子顕微鏡(SEM)像を図14に示す。NH4Fを使用しない場合は、酸化チタン結晶の厚みは300〜500nm程度で、幅は約1μm程度であった(図14a)。NH4Fの量を増やすと、厚みは小さく、幅は大きくすることができた(図14b〜c)。具体的には、厚みは70〜110nm程度、幅は4〜5μm程度とすることが可能であった。なお、酸化チタンメソ結晶の幅及び厚みは、焼成温度に依存せず、同程度の形状の結晶が得られる。さらにNH4Fの量を増やすと、厚みが小さすぎ、幅が大きすぎて酸化チタン結晶の形状が崩壊し、フラグメント化した小さな塊との混合物となった(図14d)。このため、製造例6〜7が、酸化チタンナノ粒子の凝集を抑制しつつ幅と厚みの比を大きくできることが示唆されている。
参考までに、比較製造例1〜3の結晶の電子顕微鏡(SEM及びTEM)観察の結果も図9(比較製造例3)、15(比較製造例1)及び16(比較製造例2)に示す。比較製造例1〜2は凝集しており、比較製造例3は幅と厚みの比率が小さいものである。
試験例5:元素分析
エネルギー分散型X線分析(EDX)により、製造例2の酸化チタンメソ結晶の元素分析を行った。その結果、窒素、フッ素等のような酸化チタンの構成元素以外の元素は検出されなかった。
エネルギー分散型X線分析(EDX)により、製造例2の酸化チタンメソ結晶の元素分析を行った。その結果、窒素、フッ素等のような酸化チタンの構成元素以外の元素は検出されなかった。
また、定常状態拡散反射スペクトルから、製造例2の酸化チタンメソ結晶のバンドギャップは3.2eVと算出された。酸化チタンのバンドギャップと同程度であるので、このことからも、製造例2の酸化チタンメソ結晶には不純物が混入していないことが示唆されている。
他の製造例においても、酸化チタンメソ結晶の元素分析の結果は同様の結果が得られた。
<貴金属担持酸化チタンメソ結晶>
実施例1:金ナノ粒子担持
HAuCl4を用いて導入した金イオンを含む水溶液とメタノールを混合することにより、金イオンを6×10−6mol/L含む10体積%メタノール水溶液を250mL得た。
実施例1:金ナノ粒子担持
HAuCl4を用いて導入した金イオンを含む水溶液とメタノールを混合することにより、金イオンを6×10−6mol/L含む10体積%メタノール水溶液を250mL得た。
次に、製造例2の酸化チタンメソ結晶0.25gと、上記作製した金イオンを含むメタノール水溶液250mLとを混合した(酸化チタンメソ結晶の濃度:1g/L)。その後、水銀ランプを用いて、波長365nmの紫外光を、140mW/cm2の露光強度で30分間照射し、遠心分離機によって粉末のみを回収した。
さらに、回収した粉末を空気中で、500℃で30分間焼結し、実施例1の金ナノ粒子担持酸化チタンメソ結晶を得た。
実施例2:白金ナノ粒子担持
H2PtCl6を用いて導入した白金イオンを含む水溶液とメタノールを混合することにより、白金イオンを6×10−5mol/L含む10体積%メタノール水溶液を250mL得た。
H2PtCl6を用いて導入した白金イオンを含む水溶液とメタノールを混合することにより、白金イオンを6×10−5mol/L含む10体積%メタノール水溶液を250mL得た。
次に、製造例2の酸化チタンメソ結晶0.25gと、上記作製した白金イオンを含むメタノール水溶液250mLとを混合した(酸化チタンメソ結晶の濃度:1g/L)。その後、水銀ランプを用いて、波長365nmの紫外光を、140mW/cm2の露光強度で30分間照射し、遠心分離機によって粉末のみを回収した。
さらに、回収した粉末を空気中で、300℃で30分間焼結し、実施例2の白金ナノ粒子担持酸化チタンメソ結晶を得た。
なお、比表面積、細孔径及び細孔容積については、実施例1及び2ともに、製造例2の酸化チタンメソ結晶の値をほぼ維持していた。
試験例6:担持量
実施例1の金ナノ粒子担持酸化チタンメソ結晶について、金ナノ粒子の担持量を反応溶液のICP発光分光分析により測定したところ、0.037重量%であった。また、同様に、実施例2の白金ナノ粒子担持酸化チタンメソ結晶について、白金ナノ粒子の担持量を測定したところ、1.2重量%であった。
実施例1の金ナノ粒子担持酸化チタンメソ結晶について、金ナノ粒子の担持量を反応溶液のICP発光分光分析により測定したところ、0.037重量%であった。また、同様に、実施例2の白金ナノ粒子担持酸化チタンメソ結晶について、白金ナノ粒子の担持量を測定したところ、1.2重量%であった。
試験例7:電子顕微鏡観察
実施例1〜2の貴金属担持酸化チタンメソ結晶の特性を、日本電子(株)製のJEM−3000Fを用いて、加速電圧300kVにて、透過型電子顕微鏡(TEM)により観察した。結果を図17に示す。なお、図17A及びBが実施例1の金ナノ粒子担持酸化チタンメソ結晶、図17C及びDが実施例2の白金ナノ粒子担持酸化チタンメソ結晶であり、図17Bは図17Aの拡大図、図17Dは図17Cの拡大図である。
実施例1〜2の貴金属担持酸化チタンメソ結晶の特性を、日本電子(株)製のJEM−3000Fを用いて、加速電圧300kVにて、透過型電子顕微鏡(TEM)により観察した。結果を図17に示す。なお、図17A及びBが実施例1の金ナノ粒子担持酸化チタンメソ結晶、図17C及びDが実施例2の白金ナノ粒子担持酸化チタンメソ結晶であり、図17Bは図17Aの拡大図、図17Dは図17Cの拡大図である。
実施例1の金ナノ粒子担持酸化チタンメソ結晶(図17A及びB)、及び実施例2の白金ナノ粒子担持酸化チタンメソ結晶(図17C及びD)ともに、板状の酸化チタンメソ結晶の全面にわたって、貴金属ナノ粒子の存在が確認された。なお、板状の酸化チタンメソ結晶については、担持前後でサイズはほぼ維持されていた。
この電子顕微鏡観察の結果から、実施例1において、金ナノ粒子の平均粒子径は3.8nmであり、粒子径2〜7nm程度のナノ粒子が分布していた(図18A)。また、実施例2において、白金ナノ粒子の平均粒子径は3.5nmであり、粒子径1〜7nm程度のナノ粒子が分布していた(図18B)。
他の製造例の酸化チタンメソ結晶を用いた場合においても、上記と同様の結果が得られた。
試験例8:元素分析
エネルギー分散型X線分析(EDX)により、実施例1及び2の貴金属担持酸化チタンメソ結晶の元素分析を行った。結果を図19に示す。
エネルギー分散型X線分析(EDX)により、実施例1及び2の貴金属担持酸化チタンメソ結晶の元素分析を行った。結果を図19に示す。
その結果、酸化チタンメソ結晶と同様に、窒素、フッ素等のような不純物元素は検出されなかったとともに、実施例1については担持している金が(図19A)、実施例2については担持している白金が(図19B)、それぞれ検出された。
他の製造例の酸化チタンメソ結晶を用いた場合においても、上記と同様の結果が得られた。
試験例9:拡散反射スペクトル
実施例1及び2の貴金属担持酸化チタンメソ結晶について、日本分光(株)製のV−570を用いて、拡散反射スペクトルを測定した。また、比較用の資料として、製造例2で製造した酸化チタンメソ結晶(担持なし)についても拡散反射スペクトルを測定した。結果を図20に示す。図20において、挿入図は拡大図である。
実施例1及び2の貴金属担持酸化チタンメソ結晶について、日本分光(株)製のV−570を用いて、拡散反射スペクトルを測定した。また、比較用の資料として、製造例2で製造した酸化チタンメソ結晶(担持なし)についても拡散反射スペクトルを測定した。結果を図20に示す。図20において、挿入図は拡大図である。
図20から、製造例2の酸化チタンメソ結晶については、波長が約400nm以上の可視光領域においては、吸収帯はほとんど観測されなかった。それに対して、実施例1の金ナノ粒子担持酸化チタンメソ結晶においては、波長550nm付近に極大を有する金ナノ粒子の表面プラズモンに由来する吸収帯が観測された。また、実施例2の白金ナノ粒子担持酸化チタンメソ結晶においては、可視光領域においても、金ナノ粒子の場合よりも大きくブロードな吸収帯が確認された。
これらの点から、酸化チタンメソ結晶に貴金属ナノ粒子を担持することにより、可視光を照射する場合においても、光触媒として機能する可能性が示唆された。
試験例10:光触媒活性(p−クロロフェノール)その1
実施例1及び2の貴金属担持酸化チタンメソ結晶、並びに製造例1の酸化チタンメソ結晶(担持なし)について、p−クロロフェノールの光触媒酸化を測定した。
実施例1及び2の貴金属担持酸化チタンメソ結晶、並びに製造例1の酸化チタンメソ結晶(担持なし)について、p−クロロフェノールの光触媒酸化を測定した。
p−クロロフェノールを1.0×10−4M含む実施例1〜2又は製造例2の結晶の分散液2mL(0.5g/L)を用いて、20分間超音波分解し、石英キュベット中に移した。光触媒反応を、室温でフィルター(365nm)を用いて水銀光源(REX−120)により紫外光を照射して開始させた。紫外光の強度は、140mW/cm2とし、反応時間は30分間とした。紫外光照射を止めた後、試料を10000rpmで遠心分離し(HITACHI himac CF16RX)、パーティクルを除去した。未反応分子の濃度を、固有波長において紫外分光光度計((株)島津製作所のUV-3100)で分析し、分解率を算出した。
その結果、貴金属ナノ粒子を担持していない場合(製造例2)と比較し、金ナノ粒子を担持した場合(実施例1)は、分解率が186%増加した。また、白金ナノ粒子を担持した場合(実施例2)は、分解率が426%も増加した。なお、分解率の増加率は、
(M/TiO2での基質の分解率−TiO2での基質の分解率)/TiO2での基質の分解率で算出した。以下同様である。
(M/TiO2での基質の分解率−TiO2での基質の分解率)/TiO2での基質の分解率で算出した。以下同様である。
酸化チタンメソ結晶の代わりに、比較製造例1の酸化チタンナノ結晶、市販品(石原産業(株)製のST−01(アナターゼ100%、粒径約7nm、比表面積約300m2/g)、石原産業(株)製のST−21(アナターゼ100%、粒径約20nm、比表面積約50m2/g)、デグッサ社製のP−25(アナターゼ80%+ルチル20%、粒径約20nm、比表面積約50m2/g))を使用した場合には、金ナノ粒子を担持しても分解率は33%以下しか増加せず、白金ナノ粒子を担持しても4〜68%しか増加しなかった。特に、ST−21に金ナノ粒子を担持した場合には、逆に分解率が悪化した。
それに対して、酸化チタンメソ結晶は、元々の分解率がP−25には及ばないにもかかわらず、金ナノ粒子を担持した場合には金ナノ粒子を担持したP−25と同程度の分解率、白金ナノ粒子を担持した場合には白金ナノ粒子を担持したP−25の2倍以上の分解率が得られた。
なお、酸化チタンメソ結晶以外の結晶を用いた場合の、貴金属ナノ粒子担持による分解率増加の程度は、非特許文献6において、ジクロロ酢酸の分解率ではあるが、金ナノ粒子を担持した場合には46%の増加、白金ナノ粒子を担持した場合には87%の増加とされているのと整合性が取れている。
これらの結果を、表2及び図21に示す。
試験例11:光触媒活性(p−クロロフェノール)その2
メタノール水溶液中の貴金属イオンの仕込み量(濃度)を変化させることで金担持酸化チタンメソ結晶及び白金担持酸化チタンメソ結晶の貴金属担持量を変化させ、上記と同様に分解率を測定した。その結果、金ナノ粒子を担持する場合は担持量は0.04重量%程度が、白金ナノ粒子を担持する場合は担持量は1.0重量%程度が好ましいことが分かる。結果を表3及び図22に示す。なお、図22aは金ナノ粒子を担持した場合、図22bは白金ナノ粒子を担持した場合の結果である。
メタノール水溶液中の貴金属イオンの仕込み量(濃度)を変化させることで金担持酸化チタンメソ結晶及び白金担持酸化チタンメソ結晶の貴金属担持量を変化させ、上記と同様に分解率を測定した。その結果、金ナノ粒子を担持する場合は担持量は0.04重量%程度が、白金ナノ粒子を担持する場合は担持量は1.0重量%程度が好ましいことが分かる。結果を表3及び図22に示す。なお、図22aは金ナノ粒子を担持した場合、図22bは白金ナノ粒子を担持した場合の結果である。
酸化チタンメソ結晶の代わりに、比較製造例1の酸化チタンナノ結晶、市販品(石原産業(株)製のST−01(アナターゼ100%、粒径約7nm、比表面積約300m2/g)、石原産業(株)製のST−21(アナターゼ100%、粒径約20nm、比表面積約50m2/g)、デグッサ社製のP−25(アナターゼ80%+ルチル20%、粒径約20nm、比表面積約50m2/g))を使用した場合には、金ナノ粒子の担持量0.04重量%、白金ナノ粒子の担持量1.0重量%では担持量が不十分であり、十分な光触媒活性を得るためには、コスト面が問題となることが示唆された。
また、この試験例の結果から測定した、p−クロロフェノールの分解率が50%となる貴金属ナノ粒子の担持量を表4に示す。なお、表4において、分解率が最大でも50%に達しなかった試料については、最大の分解率を示した担持量とその時の分解率を示した。
金ナノ粒子を担持した場合には、酸化チタンナノ結晶、ST−01、ST−21では分解率が50%に達しなかったのに対し、酸化チタンメソ結晶では約一桁少ない担持量で50%に達した。なお、P−25でも少ない担持量で分解率50%に達しているが、P−25はもともとの分解率が48%であり、酸化チタンメソ結晶の2倍以上であることを考慮すれば、酸化チタンメソ結晶のほうが、少ない担持量で分解率の向上効果が得られることが示唆されている。
また、白金ナノ粒子を担持した場合には、最も少ない担持量(他の試料と比較して約一桁少ない担持量)で50%に達した。酸化チタンメソ結晶においては、もともとの分解率が48%と酸化チタンメソ結晶の2倍以上であるP−25と比較しても、より少ない担持量で50%に到達しており、最も少ない担持量で分解率の向上効果が得られることが示唆されている。
試験例12:光触媒活性(RhB)
実施例1及び2の貴金属担持酸化チタンメソ結晶、並びに製造例1の酸化チタンメソ結晶(担持なし)について、ローダミンB(RhB)の光触媒酸化を測定した。
実施例1及び2の貴金属担持酸化チタンメソ結晶、並びに製造例1の酸化チタンメソ結晶(担持なし)について、ローダミンB(RhB)の光触媒酸化を測定した。
p−クロロフェノールを1.0×10−4M含む分散液ではなく、ローダミンB(RhB)を1.0×10−5M含む分散液を用い、反応時間を100秒としたこと以外はp−クロロフェノールの場合と同様にして、RhBの分解率を算出した。
その結果、貴金属ナノ粒子を担持していない場合(製造例2)と比較し、金ナノ粒子を担持した場合(実施例1)は、分解率が56%増加した。また、白金ナノ粒子を担持した場合(実施例2)は、分解率が133%も増加した。
酸化チタンメソ結晶の代わりに、比較製造例1の酸化チタンナノ結晶、市販品(デグッサ社製のP−25(アナターゼ80%+ルチル20%、粒径約20nm、比表面積約50m2/g))を使用した場合には、金ナノ粒子を担持しても分解率は11%しか増加せず、白金ナノ粒子を担持しても13%しか増加しなかった。
試験例13:光触媒活性(繰り返し使用)
本発明の貴金属担持酸化チタンメソ結晶を、光触媒として繰り返し使用できるか否かを評価するため、繰り返し試験を行った。
本発明の貴金属担持酸化チタンメソ結晶を、光触媒として繰り返し使用できるか否かを評価するため、繰り返し試験を行った。
具体的には、実施例2の白金ナノ粒子酸化チタンメソ結晶について、試験例10と同様に、紫外光照射直後、紫外光照射10分経過後、紫外光照射20分経過後、紫外光照射後30分経過後のp−クロロフェノールの分解率を測定した。また、紫外光照射30分経過した時点で、再度試験例10と同様に紫外光を照射し、再度紫外光照射直後、紫外光照射10分経過後、紫外光照射20分経過後、紫外光照射後30分経過後のp−クロロフェノールの分解率を測定した。以下、このサイクルを何度も繰り返した。結果を図23に示す。
また、実施例2の白金ナノ粒子酸化チタンメソ結晶について、試験例12と同様に、紫外光照射直後、紫外光照射10秒経過後、紫外光照射20秒経過後、紫外光照射後40秒経過後、紫外光照射後60秒経過後、紫外光照射後80秒経過後、紫外光照射後100秒経過後のRhBの分解率を測定した。また、紫外光照射100秒経過した時点で、再度試験例12と同様に紫外光を照射し、紫外光照射直後、紫外光照射10秒経過後、紫外光照射20秒経過後、紫外光照射後40秒経過後、紫外光照射後60秒経過後、紫外光照射後80秒経過後、紫外光照射後100秒経過後のRhBの分解率を測定した。以下、このサイクルを何度も繰り返した。結果を図24に示す。
上記の結果から、本発明の貴金属担持酸化チタンメソ結晶は、光触媒として、何度も繰り返し使用できることが示唆された。
Claims (14)
- 平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶に、貴金属ナノ粒子が担持されている、貴金属担持酸化チタンメソ結晶。
- 前記貴金属が、金、白金及び銀よりなる群から選ばれる少なくとも1種である、請求項1に記載の貴金属担持酸化チタンメソ結晶。
- 前記貴金属ナノ粒子の担持量が、0.03〜1.5重量%である、請求項1又は2に記載の貴金属担持酸化チタンメソ結晶。
- 前記貴金属ナノ粒子の平均粒子径が1〜10nmである、請求項1〜3のいずれかに記載の貴金属担持酸化チタンメソ結晶。
- 比表面積が10m2/g以上である、請求項1〜4のいずれかに記載の貴金属担持酸化チタンメソ結晶。
- 前記酸化チタンメソ結晶の平均幅が2〜8μmである、請求項1〜5のいずれかに記載の貴金属担持酸化チタンメソ結晶。
- 前記酸化チタンメソ結晶の平均厚みが50〜300nmである、請求項1〜6のいずれかに記載の貴金属担持酸化チタンメソ結晶。
- 光触媒用である、請求項1〜7のいずれかに記載の貴金属担持酸化チタンメソ結晶。
- 請求項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶からなる光触媒。
- 請求項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶の製造方法であって、
平均幅の平均厚みに対する比(平均幅/平均厚み)が10〜100であり、比表面積が10m2/g以上である酸化チタンメソ結晶、貴金属イオン若しくは貴金属化合物、並びに正孔捕捉剤を含む溶液に、紫外光を照射し、その後焼結する工程
を備える、製造方法。 - 前記紫外光の照射時間が10〜100分である、請求項10に記載の製造方法。
- 焼結温度が100〜600℃である、請求項10又は11に記載の製造方法。
- 請求項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶、又は請求項9に記載の光触媒を使用する、少量の貴金属量で光触媒活性を向上させる方法。
- 請求項1〜8のいずれかに記載の貴金属担持酸化チタンメソ結晶、又は請求項9に記載の光触媒を使用する、光触媒中の貴金属量を低減する方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012110679A JP2013236997A (ja) | 2012-05-14 | 2012-05-14 | 貴金属担持酸化チタンメソ結晶 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012110679A JP2013236997A (ja) | 2012-05-14 | 2012-05-14 | 貴金属担持酸化チタンメソ結晶 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2013236997A true JP2013236997A (ja) | 2013-11-28 |
Family
ID=49762535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012110679A Pending JP2013236997A (ja) | 2012-05-14 | 2012-05-14 | 貴金属担持酸化チタンメソ結晶 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2013236997A (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016175034A (ja) * | 2015-03-20 | 2016-10-06 | 公立大学法人高知工科大学 | 酸化チタン触媒およびその製造方法 |
| CN111450878A (zh) * | 2020-05-08 | 2020-07-28 | 河南城建学院 | 一种基于TiO2介晶的单原子Ir脱硝催化剂及制备方法 |
| JP2021154233A (ja) * | 2020-03-27 | 2021-10-07 | 大阪瓦斯株式会社 | 金属ナノ粒子担持チタニアナノ粒子及びそれを用いた光触媒 |
| CN114351239A (zh) * | 2021-12-15 | 2022-04-15 | 中国科学院金属研究所 | 一种多孔金属化合物阵列薄膜的制备方法 |
-
2012
- 2012-05-14 JP JP2012110679A patent/JP2013236997A/ja active Pending
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016175034A (ja) * | 2015-03-20 | 2016-10-06 | 公立大学法人高知工科大学 | 酸化チタン触媒およびその製造方法 |
| US10058847B2 (en) | 2015-03-20 | 2018-08-28 | Nippon Pillar Packing Co., Ltd. | Titanium-oxide catalyst and method of producing the same |
| JP2021154233A (ja) * | 2020-03-27 | 2021-10-07 | 大阪瓦斯株式会社 | 金属ナノ粒子担持チタニアナノ粒子及びそれを用いた光触媒 |
| JP7539780B2 (ja) | 2020-03-27 | 2024-08-26 | 大阪瓦斯株式会社 | 金属ナノ粒子担持チタニアナノ粒子及びそれを用いた光触媒 |
| CN111450878A (zh) * | 2020-05-08 | 2020-07-28 | 河南城建学院 | 一种基于TiO2介晶的单原子Ir脱硝催化剂及制备方法 |
| CN111450878B (zh) * | 2020-05-08 | 2023-03-28 | 河南城建学院 | 一种基于TiO2介晶的单原子Ir脱硝催化剂及制备方法 |
| CN114351239A (zh) * | 2021-12-15 | 2022-04-15 | 中国科学院金属研究所 | 一种多孔金属化合物阵列薄膜的制备方法 |
| CN114351239B (zh) * | 2021-12-15 | 2024-05-17 | 中国科学院金属研究所 | 一种多孔金属化合物阵列薄膜的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Sun et al. | Amorphous TiO 2 nanostructures: Synthesis, fundamental properties and photocatalytic applications | |
| Andronic et al. | Black TiO2 synthesis by chemical reduction methods for photocatalysis applications | |
| Zhang et al. | Recent Advances in Surfactant‐Free, Surface‐Charged, and Defect‐Rich Catalysts Developed by Laser Ablation and Processing in Liquids | |
| Mittal et al. | Non-metal modified TiO 2: A step towards visible light photocatalysis | |
| Shi et al. | Fabrication of PAN@ TiO2/Ag nanofibrous membrane with high visible light response and satisfactory recyclability for dye photocatalytic degradation | |
| Fu et al. | Dual Z-scheme charge transfer in TiO2–Ag–Cu2O composite for enhanced photocatalytic hydrogen generation | |
| Huang et al. | Influences of doping on photocatalytic properties of TiO2 photocatalyst | |
| JP6061872B2 (ja) | 酸化チタンメソ結晶 | |
| Zhao et al. | Structure, synthesis, and applications of TiO2 nanobelts | |
| Gao et al. | Structural design of TiO 2-based photocatalyst for H 2 production and degradation applications | |
| CN107075696B (zh) | 通过具有p-n结和等离子体材料的催化剂由水的光催化氢制备 | |
| Liu et al. | Crystal facet engineering of semiconductor photocatalysts: motivations, advances and unique properties | |
| WO2021120359A1 (zh) | 单原子贵金属锚定缺陷型WO3/TiO2纳米管、其制备和应用 | |
| Wang et al. | Improving photocatalytic performance of ZnO via synergistic effects of Ag nanoparticles and graphene quantum dots | |
| Zou et al. | Facile anion exchange to construct uniform AgX (X= Cl, Br, I)/Ag2CrO4 NR hybrids for efficient visible light driven photocatalytic activity | |
| US20140147377A1 (en) | Photocatalyst for water splitting | |
| Zhao et al. | Facile preparation of Z-scheme CdSAgTiO2 composite for the improved photocatalytic hydrogen generation activity | |
| Ravi et al. | CuOCr2O3 core-shell structured co-catalysts on TiO2 for efficient photocatalytic water splitting using direct solar light | |
| Shao et al. | Modification of Ag nanoparticles on the surface of SrTiO3 particles and resultant influence on photoreduction of CO2 | |
| Li et al. | Effect of Pt–Pd hybrid nano-particle on CdS's activity for water splitting under visible light | |
| US20180243727A1 (en) | Hydrogen production using hybrid photonic-electronic materials | |
| Yan et al. | Improving the photocatalytic performance of silver phosphate by thermal annealing: Influence of acetate species | |
| Mohamed et al. | Photodeposition of Ag nanoparticles on mesoporous LaNaTaO3 nanocomposites for promotion H2 evolution | |
| Gu et al. | UV-light aided photoelectrochemical synthesis of Au/TiO 2 NTs for photoelectrocatalytic degradation of HPAM | |
| Hao et al. | Cr-doped TiO2/CuO photocatalytic nanofilms prepared by magnetron sputtering for wastewater treatment |