JP2012508014A - CRHR2 peptide agonist and use thereof - Google Patents
CRHR2 peptide agonist and use thereof Download PDFInfo
- Publication number
- JP2012508014A JP2012508014A JP2011535643A JP2011535643A JP2012508014A JP 2012508014 A JP2012508014 A JP 2012508014A JP 2011535643 A JP2011535643 A JP 2011535643A JP 2011535643 A JP2011535643 A JP 2011535643A JP 2012508014 A JP2012508014 A JP 2012508014A
- Authority
- JP
- Japan
- Prior art keywords
- peptide
- instead
- amino acid
- seq
- complex
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 C*(C(CC1*)=O)C1=O Chemical compound C*(C(CC1*)=O)C1=O 0.000 description 1
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/57509—Corticotropin releasing factor [CRF] (Urotensin)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Abstract
本発明は、心不全を含むがこれに限定されない心臓血管病状の治療、改善又は阻害のための、選択的副腎皮質刺激ホルモン放出ホルモン受容体2型(CRHR2)作動薬、及びその組成物である、新しいペプチドに関する。この新しいペプチド作動薬は好ましくは、PEG化ペプチドを含む修飾を含む。更に、本発明はCRHR2活性に関係する疾病又は疾患の治療及び予防のための方法にも関する。 The present invention is a selective corticotropin releasing hormone receptor type 2 (CRHR2) agonist and composition thereof for the treatment, amelioration or inhibition of cardiovascular conditions including but not limited to heart failure, It relates to new peptides. This new peptide agonist preferably includes modifications including PEGylated peptides. The invention further relates to methods for the treatment and prevention of diseases or disorders associated with CRHR2 activity.
Description
(関連出願の相互参照)
本出願は、米国特許仮出願第61/111,233号(2008年11月4日出願)及び同第61/178,890号(2009年5月15日出願)の利益を主張する。
(Cross-reference of related applications)
This application claims the benefit of US Provisional Patent Application Nos. 61 / 111,233 (filed Nov. 4, 2008) and 61 / 178,890 (filed May 15, 2009).
(発明の分野)
本発明は全般に、副腎皮質刺激ホルモン放出ホルモン受容体2活性によって媒介される医学的適応の治療のために、ストレスコピン模倣物質として有用なペプチドと、ペプチド送達のための作成物、それらを含む医薬組成物、及び、副腎皮質刺激ホルモン放出ホルモン受容体2によって媒介される疾病、疾患、又は医学的状態であると診断された被験者を治療する方法に関する。
(Field of Invention)
The present invention generally includes peptides useful as stress copine mimetics and constructs for peptide delivery, for the treatment of medical indications mediated by corticotropin releasing
心不全は一般的な心臓血管疾患であり、米国及び欧州において疾患の大きな割合に達している(Remmeら、Eur.Heart J.,2001,vol.22,pp.1527〜1560)。急性心不全の入院患者数は米国内だけで毎年100万件近くになる。現在、退院後60日以内の再入院率及び死亡率は、30%〜40%に達している(Cuffeeら、JAMA,2002,vol.287(12),pp.1541〜7)。急性心不全では、血行動態機能の悪化、特に非常に高い左心室拡張終期圧が一般的である。 Heart failure is a common cardiovascular disease, reaching a large proportion of the disease in the United States and Europe (Remme et al., Eur. Heart J., 2001, vol. 22, pp. 1527-1560). There are nearly 1 million hospitalized patients with acute heart failure each year in the United States alone. Currently, the readmission rate and mortality within 60 days after discharge have reached 30% to 40% (Cuffee et al., JAMA, 2002, vol. 287 (12), pp. 1541-7). In acute heart failure, impaired hemodynamic function, particularly very high left ventricular end-diastolic pressure, is common.
急性心不全の現在の治療は多くの要素からなり、しばしば患者によって異なる。利尿薬、血管拡張薬、及び強心剤は急性心不全患者の治療において依然として中心であるが、これらの治療は死亡率及び高い再入院率に関連している。 Current treatment of acute heart failure consists of many factors and often varies from patient to patient. Diuretics, vasodilators, and cardiotonics remain central in the treatment of patients with acute heart failure, but these treatments are associated with mortality and high readmission rates.
更に、既存の強心剤治療(例えばドブタミン)は心拍出量の改善をもたらすが、心拍数の増加及び心筋の酸素消費量増加を伴う。これらの強心剤はまた、心不全患者において不整脈誘発の可能性をもたらす。この心臓の傾向は、これら薬剤の直接的強心作用に関連するエネルギー消費とカルシウム駆動に関連するものと考えられる。 Furthermore, existing cardiotonic therapies (eg, dobutamine) provide improved cardiac output, but with increased heart rate and increased myocardial oxygen consumption. These cardiotonics also provide the potential for arrhythmia induction in heart failure patients. This cardiac trend is thought to be related to the energy expenditure and calcium drive associated with the direct cardiotonic effects of these drugs.
拡大するこの未充足の医学的ニーズに対応するため、数多くの新しいアプローチが研究されているが、この症候群の患者において安全に血行動態状態と転帰を改善することについては限定的な成功しか収めていない。そのような薬剤の1つであるペプチドヒトウロコルチン2(h−UCN2)が、健康な被験者及び心不全患者において研究されている。このペプチドは、ヒツジの心不全モデルにおいて左心室駆出率(LVEF)及び心拍出量(CO)を増加させることが示されている(Rademakerら、Circulation,2005,vol.112,pp.3624〜3624)。8人の健康な被験者(Davisら、J.Am.Coll.Cardiol.,2007,vol.49,pp.461〜471)及び8人の心不全を有する被験者(Davisら、Eur.Heart J.,2007,vol.28,pp.2589〜2597)における静脈内注射の後続研究にて、LVEFとCOの増加は、2つの研究それぞれにおいて調べられた投与量の両方で、心拍数の増加と血圧の低下を伴っていた。健康な被験者及び患者に対するh−UCN2の1時間静脈内注射は、十分な忍容性を示したと見られる。 A number of new approaches have been studied to address this growing unmet medical need, but with limited success in safely improving hemodynamic status and outcome in patients with this syndrome. Absent. One such drug, the peptide human urocortin 2 (h-UCN2), has been studied in healthy subjects and patients with heart failure. This peptide has been shown to increase left ventricular ejection fraction (LVEF) and cardiac output (CO) in a sheep heart failure model (Rademaker et al., Circulation, 2005, vol. 112, pp. 3624- 3624). 8 healthy subjects (Davis et al., J. Am. Coll. Cardiol., 2007, vol. 49, pp. 461-471) and 8 subjects with heart failure (Davis et al., Eur. Heart J., 2007) , Vol. 28, pp. 2589-2597) In subsequent studies of intravenous injection in LVEF and CO increased heart rate and decreased blood pressure at both doses examined in each of the two studies. Was accompanied. One hour intravenous injection of h-UCN2 for healthy subjects and patients appears to have been well tolerated.
アミノ酸40個のペプチドであるヒトストレスコピン(h−SCP)は、h−UCN2に関連し、これら両方とも、副腎皮質刺激ホルモン放出ホルモン(CRH)ペプチド群に属する。CRHペプチド群の生物学的作用は、2つの7回膜貫通型Gタンパク質結合受容体、CRH受容体1型(CRHR1)及びCRH受容体2型(CRHR2)によって顕現される。これらの受容体は高い配列相同性を有するが、CRHペプチド群の中の異なる要素は、相対的な結合親和力、受容体活性化の度合、及びこれら2つの受容体の選択性において顕著な違いを示す。 Human stress copine (h-SCP), a 40 amino acid peptide, is associated with h-UCN2, both of which belong to the corticotropin releasing hormone (CRH) peptide family. The biological effects of the CRH peptides are manifested by two 7-transmembrane G protein coupled receptors, CRH receptor type 1 (CRHR1) and CRH receptor type 2 (CRHR2). Although these receptors have a high sequence homology, different elements within the CRH peptide family can make significant differences in the relative binding affinity, the degree of receptor activation, and the selectivity of these two receptors. Show.
CRH群要素の多くのものとは異なり、h−SCPはCRHR2について、より大きな選択性を発現し、生理学的ストレスの開始及び維持を弱めるプロセスを助けるメディエーターとして作用する(Baleら、Nat.Genet.,2000,vol.24,pp.410〜414;Kishimotoら、Nat.Genet.,2000,vol.24,pp.415〜419)。生理学的ストレスにおける明らかな役割に加え、h−SCPは他の数多くの生理学的作用を顕現させることが報告されている。これは内分泌系(Liら、Endocrinology,2003,vol.144,pp.3216〜3224)、中枢神経系、心臓血管系(Baleら、Proc.Natl.Acad.Sci.,2004,vol.101,pp.3697〜3702;Tangら、Eur.Heart J.,2007,vol.28,pp.2561〜2562)、肺系、胃腸系、腎臓系、骨格筋系、及び炎症系(Moffattら、FASEB J.,2006,vol.20,pp.1877〜1879)に対して影響を及ぼす。 Unlike many of the CRH group elements, h-SCP expresses greater selectivity for CRHR2 and acts as a mediator that assists in the process of weakening the initiation and maintenance of physiological stress (Bale et al., Nat. Genet. 2000, vol.24, pp.410-414; Kishimoto et al., Nat.Genet., 2000, vol.24, pp.415-419). In addition to its obvious role in physiological stress, h-SCP has been reported to manifest numerous other physiological effects. This is the endocrine system (Li et al., Endocrinology, 2003, vol. 144, pp. 3216-3224), central nervous system, cardiovascular system (Bale et al., Proc. Natl. Acad. Sci., 2004, vol. 101, pp. 3697-3702; Tang et al., Eur.Heart J., 2007, vol.28, pp. 2561-2562), pulmonary system, gastrointestinal system, renal system, skeletal muscle system, and inflammatory system (Moffatt et al., FASEB J. et al. , 2006, vol. 20, pp. 1877-1879).
加えて、CRHR2活性は、例えば筋肉減少症などの骨格筋消費疾患(Hinkleら、Endocrinology,2003,vol.144(11),pp.4939〜4946)、運動活動及び食物摂取(Ohataら、Peptides,2004,vol.25,pp.1703〜1709)に影響を与え、心臓保護の役割に寄与し(Brarら、Endocrinology,2004,vol.145(1),pp.24〜35)、並びに気管支弛緩及び抗炎症活性を発現する(Moffattら、FASEB J.2006,vol.20,pp.E1181〜E1187)。 In addition, CRHR2 activity is associated with skeletal muscle consumption disorders such as sarcopenia (Hinkle et al., Endocrinology, 2003, vol. 144 (11), pp. 4939-4946), exercise activity and food intake (Ohata et al., Peptides, 2004, vol. 25, pp. 1703-1709) and contribute to the role of cardioprotection (Brar et al., Endocrinology, 2004, vol. 145 (1), pp. 24-35), and bronchial relaxation and It exhibits anti-inflammatory activity (Moffatt et al., FASEB J. 2006, vol. 20, pp. E1181 to E1187).
PEG化は、1つ以上のポリエチレングリコール(PEG)ポリマーを分子に結合するプロセスである。PEG化プロセスはしばしば、抗体、ペプチド、及びタンパク質に適用され、その生物薬理学的特性を改善し、タンパク質分解酵素に対する化合物の感受性、短い循環半減期、短い有効期間、低い溶解度、急速な腎臓クリアランス、及び投与薬剤に対する抗体生成の可能性を克服する(Harris et al.,Nature,2003,vol.2,pp.214〜221;Hamidi et al.,Drug Delivery,2006,3,pp.399〜409;Bailon et al.,PSTT,1998,vol.1(8),pp.352356)。最近FDAは、食物、化粧品、及び薬剤の賦形剤又は基材としての使用についてPEGポリマーを承認した。全体に、PEGポリマーは比較的、非免疫原性であり、毒性はほとんどなく、腎臓により又は糞便中でそのまま排出される。これらの特性は、親分子の親和性、有効性、及び薬理学的プロファイルを保全又は改善するようこのプロセスを開発した場合に、化合物について数多くの臨床的利益をもたらすことができる。 PEGylation is the process of attaching one or more polyethylene glycol (PEG) polymers to a molecule. PEGylation processes are often applied to antibodies, peptides and proteins to improve their biopharmacological properties, sensitivity of compounds to proteolytic enzymes, short circulation half-life, short shelf life, low solubility, rapid renal clearance , And overcoming the potential for antibody generation against the administered drug (Harris et al., Nature, 2003, vol. 2, pp. 214-221; Hamidi et al., Drug Delivery, 2006, 3, pp. 399-409). Bailon et al., PSTT, 1998, vol.1 (8), pp.352356). Recently, the FDA has approved PEG polymers for use as excipients or substrates for food, cosmetics, and drugs. Overall, PEG polymers are relatively non-immunogenic, have little toxicity, and are excreted intact by the kidneys or in feces. These properties can provide numerous clinical benefits for a compound when this process is developed to preserve or improve the affinity, efficacy, and pharmacological profile of the parent molecule.
本発明は、本明細書に添付の独立及び従属請求項によってそれぞれ定義される、一般的かつ好ましい実施形態を対象とし、参照することにより本明細書に組み込む。本発明の好ましい機能及び代表的な機能は、図面の参照と共に下記の「発明を実施するための形態」から明らかとなろう。 The present invention is directed to general and preferred embodiments, each defined by the independent and dependent claims appended hereto, and is incorporated herein by reference. Preferred and representative features of the present invention will become apparent from the following Detailed Description, with reference to the drawings.
特定の実施形態において、本発明は、次のアミノ酸配列を含む副腎皮質刺激ホルモン放出ホルモン受容体2型(CRHR2)に対する作動薬活性を有するペプチドであって:
LSLDV PTNIM NLLFN IAKAK NLRAQ AAANA HLMAQ I、
式中、このペプチドの少なくとも1つのアミノ酸がXで置換され、ただしこの置換はアミノ酸配列のポジション3、29、及び33では行われず、かつここにおいてXはシステイン、チロシン、若しくはグルタミン酸であるペプチド;又はこれらの製薬上許容される塩又はアミドに関する。
In certain embodiments, the present invention is a peptide having agonist activity against adrenocorticotropic hormone releasing hormone receptor type 2 (CRHR2) comprising the following amino acid sequence:
LSLDV PTNIM NLLFN IAKAK NLRAQ AAANA HLMAQ I,
Wherein at least one amino acid of the peptide is replaced with X, provided that this substitution is not made at
本発明で具現されるCRHR2ペプチド作動薬は、ヒトストレスコピン及びヒトウロコルチンIIIの配列に比較的類似のアミノ酸配列を含む。 The CRHR2 peptide agonist embodied in the present invention comprises an amino acid sequence that is relatively similar to the sequences of human stress copine and human urocortin III.
一実施形態において、上記参照のアミノ酸配列に対するアミノ酸置換は、:ポジション1でLの代わりにX;ポジション2でSの代わりにX;ポジション4でDの代わりにX;ポジション5でVの代わりにX;ポジション6でPの代わりにX;ポジション7でTの代わりにX;ポジション8でNの代わりにX;ポジション9でIの代わりにX;ポジション10でMの代わりにX;ポジション11でNの代わりにX;ポジション12でLの代わりにX;ポジション13でLの代わりにX;ポジション14でFの代わりにX;ポジション15でNの代わりにX;ポジション16でIの代わりにX;ポジション17でAの代わりにX;ポジション18でKの代わりにX;ポジション19でAの代わりにX;ポジション20でKの代わりにX;ポジション21でNの代わりにX;ポジション22でLの代わりにX;ポジション23でRの代わりにX;ポジション24でAの代わりにX;ポジション25でQの代わりにX;ポジション26でAの代わりにX;ポジション27でAの代わりにX;ポジション28でAの代わりにX;ポジション30でAの代わりにX;ポジション31でHの代わりにX;ポジション32でLの代わりにX;ポジション34でAの代わりにX;ポジション35でQの代わりにX;及びポジション36でIの代わりにX、からなる群から選択される。
In one embodiment, the amino acid substitutions to the above referenced amino acid sequence are: X at
本発明はまた、このCRHR2ペプチド作動薬の製薬上許容される塩又はアミドに関し、更に、1つ以上の製薬上許容される賦形剤と組み合わせたこのペプチドの医薬組成物を目的とする。 The present invention also relates to pharmaceutically acceptable salts or amides of this CRHR2 peptide agonist, and is further directed to pharmaceutical compositions of this peptide in combination with one or more pharmaceutically acceptable excipients.
更に別の一実施形態において、このCRHR2ペプチド作動薬のアミノ酸配列は、
XLSLD VPTNI MNLLF NIAKA KNLRA QAAAN AHLMA QI−NH2;
XTLSL DVPTN IMNLL FNIAK AKNLR AQAAA NAHLM AQI−NH2;
XFTLS LDVPT NIMNL LFNIA KAKNL RAQAA ANAHL MAQI−NH2;及び
XKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI−NH2からなる群から選択される。
In yet another embodiment, the amino acid sequence of the CRHR2 peptide agonist is
XLSLD VPTNI MNLLF NIAKA KNLRA QAAAN AHLMA QI-NH 2 ;
XTLSL DVPTN IMNLL FNIAK AKNLR AQAAA NAHLM AQI-NH 2 ;
XFTLS LDVPT NIMNL LFNIA KAKNL RAQAA ANAHL MAQI-NH 2 ; and XKFTL SLDVP TNIMN LLFNI AKAKN LRAQA ANAH LMAQ-NH 2 .
別の一実施形態において、複合体は、このアミノ酸配列と、このCRHR2ペプチド作動薬のXに結合されたリンカーとを含む。好ましくはこのXはシステインである。好ましくは、このリンカーはアセトアミド又はN−エチルスクシンイミドである。 In another embodiment, the complex comprises the amino acid sequence and a linker attached to X of the CRHR2 peptide agonist. Preferably this X is cysteine. Preferably, this linker is acetamide or N-ethylsuccinimide.
更に別の一実施形態において、この作動薬ペプチドの複合体は、分子量が80kDa以下のリンカーに結合されたポリエチレングリコール(PEG)を含む。好ましくはこのPEGは、分子量が約2kDa、約5kDa、約12kDa、約20kDa、約30kDa、又は約40kDaのいずれかである。 In yet another embodiment, the agonist peptide conjugate comprises polyethylene glycol (PEG) attached to a linker having a molecular weight of 80 kDa or less. Preferably, the PEG has a molecular weight of either about 2 kDa, about 5 kDa, about 12 kDa, about 20 kDa, about 30 kDa, or about 40 kDa.
リンカーは、ペプチドに対するアミノ酸配列のポジションに関してPEGをより容易かつ選択的に結合するためのものであり、同時に、ペプチドのPEG化は、PEG化したペプチドの半減期を延ばし、これにより患者に対する治療的利益の持続時間が延長される。よって、CRHR2ペプチド作動薬のアミノ酸配列に対する置換は、好ましくは、配列中に少なくとも1つのタイプXのアミノ酸があるようにする。これにより、ペプチドのPEG化が、配列中の少なくとも1つの位置に確実に導かれる。 The linker is for easier and selective attachment of PEG with respect to the position of the amino acid sequence relative to the peptide, while at the same time PEGylation of the peptide extends the half-life of the PEGylated peptide, thereby providing therapeutic therapeutics to the patient. Profit duration is extended. Thus, substitutions to the amino acid sequence of a CRHR2 peptide agonist preferably results in at least one type X amino acid in the sequence. This ensures that the PEGylation of the peptide is directed to at least one position in the sequence.
特定の実施形態において、CRHR2ペプチド作動薬は、配列番号2、3、4、5、6、7、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、35、36、37、39、40、及び41に含まれるアミノ酸配列のうち1つを含む。 In certain embodiments, the CRHR2 peptide agonist is SEQ ID NO: 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 35, 36, 37, 39, 40, and 41.
本発明の別の態様は、代謝性疾患又は心不全である、副腎皮質刺激ホルモン放出ホルモン受容体2型活性によって媒介される疾病、疾患、又は医学的状態で苦しむ被験者、又はこれらであると診断された被験者の治療方法を目的とする。この方法には、そのような治療を必要としている被験者に、本発明で具現されるCRHR2ペプチド作動薬の有効量を投与することが含まれる。
Another aspect of the present invention is a subject suffering from, or diagnosed with, a disease, disorder, or medical condition mediated by corticotropin releasing
本発明の追加の実施形態及び利点は、下記の議論、スキーム、実施例及び請求項から明らかとなろう。 Additional embodiments and advantages of the invention will be apparent from the discussion, schemes, examples, and claims below.
本発明は、心不全並びに代謝性疾患を含みこれらに限定されない心臓血管病状の治療、改善又は阻害のためのCHRH2ペプチド作動薬及びその組成物である新しいペプチド類に関する。 The present invention relates to new peptides which are CHRH2 peptide agonists and compositions thereof for the treatment, amelioration or inhibition of cardiovascular conditions including but not limited to heart failure and metabolic diseases.
本発明の一実施形態において、心不全の治療又は改善を必要としている被験者における心不全の治療又は改善の方法には、少なくとも1つのCRHR2ペプチド作動薬の治療的有効量をその被験者に投与することが含まれる。 In one embodiment of the invention, a method of treating or ameliorating heart failure in a subject in need of treatment or amelioration of heart failure includes administering to the subject a therapeutically effective amount of at least one CRHR2 peptide agonist. It is.
特定の実施形態において、このCRHR2ペプチドは哺乳類ペプチドであり、特に、マウス、ラット、モルモット、ウサギ、イヌ、ネコ、ウマ、ウシ、豚、若しくは霊長類のペプチドであるか、又はこれらの修飾物である。好ましくはこのペプチドは修飾ヒトペプチドである。本発明のCRHR2ペプチド作動薬の例は、下記のセクションで詳細に記述される。 In certain embodiments, the CRHR2 peptide is a mammalian peptide, particularly a mouse, rat, guinea pig, rabbit, dog, cat, horse, cow, pig, or primate peptide, or a modification thereof. is there. Preferably the peptide is a modified human peptide. Examples of CRHR2 peptide agonists of the present invention are described in detail in the following sections.
本発明の別の実施形態には、ペプチド作動薬に共有結合により結合された反応基が含まれる。この反応基は、被験者内のペプチドの循環半減期を延長するようなポリマー又は他の化学的部分と、安定な共有結合を形成することができるよう選択される。一実施形態において、そのようなポリマーには、排出までの被験者の循環中のペプチド持続時間を延長するポリエチレングリコール(PEG)ポリマーが含まれる。この形態において、この反応基は、一方の手をペプチドの1つ以上のアミノ酸と反応させ、もう一方をポリマーと反応させることにより、ペプチド間のリンカーとして作用する。別の一実施形態において、この反応基は最初にPEGと結合してから、ペプチドと化学結合を形成する。修飾ペプチドの好ましい実施形態において、このリンカー基はスクシンイミドであり、より具体的にはN−エチルスクシンイミド、又はアセトアミドである。更に、このリンカーはビニルスルホン又はオルトピリジルジスルフィドであり得る。好ましくは化学修飾は、例えば反応効率を高めるために、単離されたペプチド上で行われる。 Another embodiment of the invention includes a reactive group covalently linked to a peptide agonist. This reactive group is selected so that it can form a stable covalent bond with a polymer or other chemical moiety that extends the circulating half-life of the peptide within the subject. In one embodiment, such polymers include polyethylene glycol (PEG) polymers that extend the duration of the peptide in the subject's circulation to elimination. In this form, the reactive group acts as a linker between the peptides by reacting one hand with one or more amino acids of the peptide and reacting the other with the polymer. In another embodiment, the reactive group is first conjugated with PEG and then forms a chemical bond with the peptide. In a preferred embodiment of the modified peptide, the linker group is succinimide, more specifically N-ethylsuccinimide, or acetamide. Further, the linker can be vinyl sulfone or orthopyridyl disulfide. Preferably, chemical modification is performed on the isolated peptide, for example to increase reaction efficiency.
ペプチド及びPEG部分に結合するのに有用なリンカーは、最小限の免疫原生及び毒性を宿主にもたらし得る。そのようなリンカーの例は、Bailonら、PSTT,1998,vol.1(8),pp.352〜356、又はRobertsら、2002,Adv.Drug Del.Rev.,vol.54,pp.459〜476に見出すことができる。好適な化学部分の例、特にPEG及び同等のポリマーについては、Greenwaldら、2003,Adv.Drug Del.Rev.,vol.55,pp.217〜250に記述されている。例えば、他のポリマー系では、スチレン−無水マレイン酸ネオカルチノスタチンコポリマー、ヒドロキシルプロピルメタクリルアミドコポリマー、デキストラン、ポリグルタミン酸、ヒドロキシルエチルデンプン、及びポリアスパラギン酸は、PEGシステムに類似の送達及び薬物動態学特性を達成するのに使用することができる。 Linkers useful for coupling to the peptide and PEG moieties can provide minimal immunogenicity and toxicity to the host. Examples of such linkers are described in Bailon et al., PSTT, 1998, vol. 1 (8), pp. 352-356, or Roberts et al., 2002, Adv. Drug Del. Rev. , Vol. 54, pp. 459-476. For examples of suitable chemical moieties, particularly PEG and equivalent polymers, see Greenwald et al., 2003, Adv. Drug Del. Rev. , Vol. 55, pp. 217-250. For example, in other polymer systems, styrene-maleic anhydride neocalcinostatin copolymer, hydroxylpropylmethacrylamide copolymer, dextran, polyglutamic acid, hydroxylethyl starch, and polyaspartic acid are similar in delivery and pharmacokinetics to the PEG system. Can be used to achieve properties.
本発明の特定の実施形態において、CRHR2ペプチド作動薬には、アミド化C末端が含まれる。その修飾手順は、単離された精製ペプチド上で実施することができ、又は、固相合成の場合は、合成手順中に実施することができる。その手順は、Ray et al.,Nature Biotech.,1993,vol.11,pp.64〜70;Cottingham et al.,Nature Biotech.,2001,vol.19,pp.974〜977;Walsh et al.,Nature Biotech.,vol.24,pp.1241〜1252;及び米国特許公開第2008/0167231号で検討されている。 In certain embodiments of the invention, the CRHR2 peptide agonist includes an amidated C-terminus. The modification procedure can be performed on the isolated purified peptide or, in the case of solid phase synthesis, during the synthesis procedure. The procedure is described in Ray et al. , Nature Biotech. 1993, vol. 11, pp. 64-70; Cottingham et al. , Nature Biotech. 2001, vol. 19, pp. 974-977; Walsh et al. , Nature Biotech. , Vol. 24, pp. 1241-1252; and U.S. Patent Publication No. 2008/0167231.
本発明の特定の一実施形態において、この化合物は、カルボキシ末端にCONH2を含む配列番号82又は配列番号102に示すアミノ酸配列のペプチドを含み、N−エチルスクシンイミド又はアセトアミドであるリンカーと共に、アミノ酸配列のポジション28でシステイン残基に結合しているリンカー、並びに、約20kDaのPEGポリマーに結合しているリンカーを含む。 In one particular embodiment of the invention, the compound comprises a peptide of the amino acid sequence shown in SEQ ID NO: 82 or SEQ ID NO: 102 containing CONH2 at the carboxy terminus, together with a linker that is N-ethylsuccinimide or acetamide, Includes a linker attached to a cysteine residue at position 28, as well as a linker attached to a PEG polymer of about 20 kDa.
更に、本発明の一実施形態は、CHRH2活性によって媒介される疾病、疾患又は病状を患う、又はこれらであると診断された被験者の、少なくとも1つのCRHR2ペプチド作動薬の治療的有効量を被験者に投与することを含む、治療方法を特徴とする。 Furthermore, one embodiment of the present invention provides a subject with a therapeutically effective amount of at least one CRHR2 peptide agonist of a subject suffering from or diagnosed with a disease, disorder or condition mediated by CHRH2 activity. It features a method of treatment comprising administering.
本発明の別の一実施形態は更に、1つ以上のCHRH2媒介の症状、疾病、又は疾患の進行を治療又は阻害する方法を特徴とし、この方法には、少なくとも1つのCRHR2ペプチド作動薬の薬剤有効量を、治療の必要な患者に投与することが含まれる。 Another embodiment of the invention further features a method of treating or inhibiting one or more CHRH2-mediated symptoms, illnesses, or progression of a disease, the method comprising at least one CRHR2 peptide agonist agent It includes administering an effective amount to a patient in need of treatment.
本発明の更なる一実施形態では、任意のCRHR2ペプチド作動薬と製薬上許容される担体とを混合することを含む、医薬組成物の製造プロセスが提供される。 In a further embodiment of the invention, there is provided a process for producing a pharmaceutical composition comprising mixing any CRHR2 peptide agonist and a pharmaceutically acceptable carrier.
用語及び定義
本発明は、下記の定義、図、及び本明細書に提供されている代表的な開示を参照することにより、最もよく理解される。
Terms and Definitions The invention is best understood by referring to the following definitions, figures, and representative disclosure provided herein.
本明細書では時々、次の略語が使用される:pA50又はpEC50=最大効果の半分を生じさせるのに必要な作動薬濃度の、10を底とした負の対数;SEM=標準誤差;Log DR=作動薬用量比の10を底とした対数;MW=分子量;cAMP=アデノシン3’,5’−環状一リン酸;cDNA=相補的DNA;kb=キロベース(1000個の塩基対);kDa=キロダルトン;ATP=アデノシン5’−三リン酸;nt=ヌクレオチド;bp=塩基対;PAGE=ポリアクリルアミドゲル電気泳動;PCR=ポリメラーゼ連鎖反応;nm=ナノモル。
The following abbreviations are sometimes used herein: pA 50 or pEC 50 = the negative logarithm,
用語「備える」、「含有する」、及び「含む」は本明細書においては、それらの開放された非限定的な意味で用いられる。 The terms “comprising”, “containing”, and “including” are used herein in their open, non-limiting sense.
「投与」又は「投与する」は、薬理学上有用な方法で患者に薬剤を提供することを意味する。 “Administration” or “administering” means providing a drug to a patient in a pharmacologically useful manner.
「組成物」は、本発明の化合物を含む製品(例えば、特定の量で特定の成分を含む製品、及び特定の量での特定の成分のこのような組み合わせから直接的又は間接的に得られる任意の製品など)を意味する。 A “composition” is obtained directly or indirectly from a product comprising a compound of the invention (eg, a product comprising a particular ingredient in a particular amount, and such a combination of a particular ingredient in a particular amount. Means any product).
「化合物」又は「薬剤(drug)」は、CRHR2ペプチド作動薬又はその製薬上許容される形態を意味する。「複合体(Conjugate)」は、2つ以上の化合物の結合により形成された化学的化合物を意味する。 “Compound” or “drug” means a CRHR2 peptide agonist or a pharmaceutically acceptable form thereof. “Conjugate” means a chemical compound formed by the joining of two or more compounds.
「剤形」は、患者への投与に好適な媒体、担体、賦形剤、又は装置内にある1つ以上の化合物を意味する。「経口剤形」は、経口投与に好適な剤形を意味する。 “Dosage form” means one or more compounds in a vehicle, carrier, excipient, or device suitable for administration to a patient. “Oral dosage form” means a dosage form suitable for oral administration.
「用量」は薬剤の単位を意味する。通常、一用量は一剤形として供給される。用量は、さまざまな用量レジメンにしたがって患者に投与され得る。一般的な用量レジメンには、1日1回経口(qd)、1日2回経口(bid)、及び1日3回経口(tid)が挙げられる。 “Dose” means a unit of drug. Usually, a dose is supplied as a single dosage form. The dose can be administered to the patient according to various dosage regimens. Common dosage regimes include once daily oral (qd), twice daily oral (bid), and three times daily oral (tid).
「形態」は、1つ以上のCRHR2ペプチド作動薬のさまざまな異性体及び混合物を意味する。用語「異性体」は、組成及び分子量は同じであるが物理的及び/又は化学的特性が異なる化合物を指す。そのような物質が有する原子の数及び種類は同じであるが、構造は異なる。その構造の差は構成の差(幾何異性体)又は偏光面回転性の差(立体異性体)であり得る。用語「立体異性体」は、構成は同じであるが原子の空間配置が異なる異性体を指す。鏡像異性体及びジアステレオマーは、不斉置換されている炭素原子がキラル中心として働く立体異性体である。用語「キラル」は、分子が鏡像に重なり合わないことを指し、これは、対称軸及び面又は中心が存在しないことを意味する。 “Form” means various isomers and mixtures of one or more CRHR2 peptide agonists. The term “isomer” refers to compounds that have the same composition and molecular weight but differ in physical and / or chemical properties. Such materials have the same number and kind of atoms but different structures. The structural difference can be a structural difference (geometric isomer) or a polarization plane rotation difference (stereoisomer). The term “stereoisomer” refers to isomers that have the same configuration but differ in the arrangement of their atoms in space. Enantiomers and diastereomers are stereoisomers in which the asymmetrically substituted carbon atom acts as a chiral center. The term “chiral” refers to a molecule that does not overlap the mirror image, which means that there is no axis of symmetry and no plane or center.
「薬剤(Medicament)」は、例えば薬物依存、薬物濫用又は薬物誘発性疾患などの薬物関連疾患の予防、治療又は改善に、それらを必要としている被験者において使用するための製品を意味する。 “Medicament” means a product for use in a subject in need thereof for the prevention, treatment or amelioration of drug-related diseases such as drug addiction, drug abuse or drug-induced diseases.
「患者」又は「被験者」は、動物、好ましくは哺乳類、より好ましくはヒトで、治療的介入を必要としている者を意味する。 “Patient” or “subject” means an animal, preferably a mammal, more preferably a human, in need of therapeutic intervention.
「製薬上許容される」は、本発明の組成物又は薬剤の製剤において使用するための、十分な純度と品質を有する、分子実体又は組成物を意味する。ヒトでの使用(臨床及び市販)及び獣医学的使用の両方が本発明の範囲内に等しく含まれることから、製剤には、ヒトでの又は獣医学的用途でのいずれの組成物又は薬剤も含まれるであろう。 “Pharmaceutically acceptable” means a molecular entity or composition of sufficient purity and quality for use in the composition or pharmaceutical formulation of the present invention. Since both human use (clinical and commercial) and veterinary use are equally included within the scope of the present invention, the formulation includes any composition or drug for human or veterinary use. Will be included.
「製薬上許容される賦形剤」は、薬理学的組成物に添加されるか、あるいは薬剤の投与を容易にする賦形剤、担体又は希釈剤として用いられかつ該薬剤と相溶する、例えば不活性な物質のような、毒性を有さないか、生物学的に許容されるか、あるいは患者に投与するうえで生物学的に適した物質を指す。賦形剤の例には炭酸カルシウム、リン酸カルシウム、いろいろな糖及びいろいろな種類の澱粉、セルロース誘導体、ゼラチン、植物油及びポリエチレングリコールが挙げられる。 “Pharmaceutically acceptable excipient” is used as an excipient, carrier or diluent that is added to a pharmacological composition or that facilitates administration of a drug and is compatible with the drug, A substance that is not toxic, biologically acceptable, or biologically suitable for administration to a patient, such as an inert substance. Examples of excipients include calcium carbonate, calcium phosphate, various sugars and various types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols.
「製薬上許容される塩」は、本発明の組成物又は薬剤の製剤に使用するための十分な純度及び品質を有し、かつ医薬品に使用するのに忍容性であり、十分に非毒性である、本発明の化合物の酸塩又は塩基塩を意味する。好適な製薬上許容される塩には、酸付加塩が含まれ、これは例えば薬剤化合物を、塩酸、硫酸、フマル酸、マレイン酸、コハク酸、酢酸、安息香酸、クエン酸、酒石酸、カルボン酸又はリン酸のような好適な製薬上許容される酸と反応させることにより形成することができる。 “Pharmaceutically acceptable salts” have sufficient purity and quality for use in the compositions or pharmaceutical formulations of the present invention, are well tolerated for use in pharmaceuticals, and are sufficiently non-toxic. Is an acid salt or base salt of a compound of the invention. Suitable pharmaceutically acceptable salts include acid addition salts, which include, for example, drug compounds such as hydrochloric acid, sulfuric acid, fumaric acid, maleic acid, succinic acid, acetic acid, benzoic acid, citric acid, tartaric acid, carboxylic acid. Alternatively, it can be formed by reaction with a suitable pharmaceutically acceptable acid such as phosphoric acid.
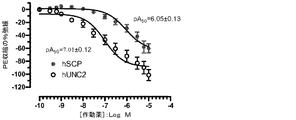
「CRHR2ペプチド作動薬」は、副腎皮質刺激ホルモン放出ホルモン受容体2型(CRHR2)に対する作動活性を呈するペプチド又はその誘導体を意味する。好ましくはCRHR2ペプチド作動薬は、副腎皮質刺激ホルモン放出ホルモン受容体1型(CRHR1)に対する活性も呈し得る、ストレスコピン誘導体又は変異型である。CRHR2ペプチド作動薬は一般に、CRHR1に対して比較的低い活性を備えた選択性CRHR2作動薬である。受容体に対する選択性とは、本明細書において、他の受容体(ペプチドがこれに対しても活性を誘導し得るが有効性はより低い)に対するのと比較して、ペプチドが選択性である受容体に対して活性反応を誘導するような、ペプチドの有効性を指す。CRHR2ペプチド作動薬の定義は作動薬に限定されず、部分的作動薬も含み得る。CRHR2ペプチド作動薬のCRHR1及びCRHR2活性は、例えば、アデノシン3’,5’−環状一リン酸(cAMP)アッセイで評価することができる。
ストレスコピン(h−SCP)のCRHR2活性に極めて似ている、
“CRHR2 peptide agonist” means a peptide or derivative thereof that exhibits agonistic activity against corticotropin releasing hormone receptor type 2 (CRHR2). Preferably, the CRHR2 peptide agonist is a stress copine derivative or variant that may also exhibit activity against adrenocorticotropic hormone releasing hormone receptor type 1 (CRHR1). A CRHR2 peptide agonist is generally a selective CRHR2 agonist with relatively low activity against CRHR1. Selectivity for a receptor, as used herein, is that a peptide is selective compared to other receptors (the peptide can also induce activity against it but is less effective). It refers to the effectiveness of the peptide to induce an active response to the receptor. The definition of CRHR2 peptide agonist is not limited to agonists, and may include partial agonists. The CRHR1 and CRHR2 activities of CRHR2 peptide agonists can be assessed, for example, with an
Very similar to the CRHR2 activity of stress copine (h-SCP),
「治療的有効量」は、研究者、獣医、医師又は他の臨床医により求められている、治療又は予防されている疾病又は疾患の症状の治療的緩和を含む、組織系、動物又はヒト内で生物学的反応又は医薬反応を顕現させる化合物の量を意味する。 A “therapeutically effective amount” is within a tissue system, animal or human, including therapeutic alleviation of the symptoms of the disease or disorder being treated or prevented as sought by a researcher, veterinarian, physician or other clinician. Means the amount of a compound that manifests a biological or pharmaceutical response.
本明細書で使用されるとき、用語「治療する」は、他に記述のない限り、その用語が適用される疾患若しくは状態、又はそのような疾患若しくは状態の1つ以上の症状を、後退させ、緩和し、進行を阻害し、又は予防することを意味する。本明細書で使用されるとき、用語「治療」は、他に記述のない限り、治療する行為を指す。 As used herein, the term “treating”, unless stated otherwise, reverses the disease or condition to which the term applies, or one or more symptoms of such a disease or condition. Means to alleviate, inhibit or prevent progression. As used herein, the term “treatment” refers to the act of treating, unless otherwise stated.
化合物
本発明は、下記のペプチド及びその修飾物に関する。本発明の化合物には更に、新規かつ選択的なCRHR2作動薬ペプチド及びその修飾物が含まれる。
Compound The present invention relates to the following peptides and modified products thereof. The compounds of the present invention further include novel and selective CRHR2 agonist peptides and modifications thereof.
更に、本発明の化合物は、例えばh−SCP又は擬似h−SCPペプチドなどの、CRHR2に結合する、又はCRHR2と錯体を形成する、ペプチド部分を指す。好ましい化合物は、例えば、cAMPアッセイにおいて約7.5以上の範囲内のpA50で測定される、又は、放射性リガンド結合アッセイにおいて約7.5以上の範囲内のpKI(KIの負の対数)で測定される、CRHR2に対する作動薬活性を有するペプチドである。CRHR2ペプチド作動薬は、結合親和性を示すことに加え、ある程度の受容体活性化を示すべきである。よって、h−SCPに相同のペプチドが、類似の物理的特性及び化学的特性を自然に有しているため、好ましい。 Furthermore, the compounds of the invention refer to peptide moieties that bind to CRHR2 or complex with CRHR2, such as h-SCP or pseudo-h-SCP peptides. Preferred compounds are, for example, measured at a pA 50 in the range of about 7.5 or higher in the cAMP assay, or pK I (negative logarithm of K I in the range of about 7.5 or higher in the radioligand binding assay. ), And is a peptide having agonist activity against CRHR2. In addition to showing binding affinity, a CRHR2 peptide agonist should show some degree of receptor activation. Thus, peptides homologous to h-SCP are preferred because they naturally have similar physical and chemical properties.
副腎皮質刺激ホルモン放出因子の群に属するものは、中程度に短い半減期を呈する。CRHR2ペプチド作動薬は、特有の治療特性をもたらす見込みがある。心臓血管疾患及び代謝性疾患を含むがこれらに限定されない、CRHR2によって媒介される疾患の治療について、本発明の一実施形態は、ペプチド作動薬の長時間作用形態を目的とする。長時間作用CRHR2ペプチド作動薬は、継続的な治療曝露が必要な慢性疾患の治療、及び、処方薬治療の患者コンプライアンスに問題がある場合に、特に利益をもたらす。 Those belonging to the group of adrenocorticotropic hormone-releasing factors exhibit a moderately short half-life. CRHR2 peptide agonists are expected to provide unique therapeutic properties. For the treatment of diseases mediated by CRHR2, including but not limited to cardiovascular and metabolic diseases, one embodiment of the present invention is directed to long acting forms of peptide agonists. Long-acting CRHR2 peptide agonists are particularly beneficial when treating chronic diseases that require continuous therapeutic exposure and when there is a problem with patient compliance with prescription drug treatment.
したがって、本発明の一実施形態は、全般に、望ましい治療的特性及び/又はCRHR2に対する構造−活性関係が保持されるような、h−SCPの配列変化、部位特異的配列変化、及び空間的又は立体的な干渉考慮を目的とする。 Accordingly, one embodiment of the present invention generally provides h-SCP sequence changes, site-specific sequence changes, and spatial or spatial, such that desirable therapeutic properties and / or structure-activity relationships for CRHR2 are retained. The purpose is to consider three-dimensional interference.
CRHR2ペプチド作動薬の例は、所望によりC末端でアミド化されており、これらは表1〜5に示されている。反応基又はリンカーは好ましくはスクシンイミド又はアセトアミドである。修飾されたペプチドは所望によりPEG基を含む。このPEGは長さ及び重量が異なり、好ましくは約20kDaである。所望により、反応基の数は1つより多くてもよく、1つの反応基が好ましい。 Examples of CRHR2 peptide agonists are optionally amidated at the C-terminus, and these are shown in Tables 1-5. The reactive group or linker is preferably succinimide or acetamide. The modified peptide optionally contains a PEG group. The PEG varies in length and weight, and is preferably about 20 kDa. If desired, the number of reactive groups may be greater than one, with one reactive group being preferred.

本発明の薬剤化合物はまた、立体異性体の混合物又はそれぞれの純粋な異性体若しくは実質的に純粋な異性体を含む。例えば、本化合物は所望により、任意の1つの置換基を含有する炭素原子に、1つ以上の不斉中心を有してもよい。よって、この化合物は、鏡像異性体若しくはジアステレオマー又はその混合物の形で存在してもよい。本化合物が二重結合を含有するとき、本化合物は、幾何異性(シス−化合物、トランス−化合物)の形で存在してもよく、本化合物がカルボニルのような不飽和結合を含有するとき、本化合物は、互変異性体の形で存在してもよく、本化合物はまた、これらの異性体又はその混合物を含む。ラセミ混合物、鏡像異性体又はジアステレオマーの形の出発化合物を、本化合物を調製する方法で用いてもよい。本化合物がジアステレオマー又は鏡像異性体の形で得られるとき、それらはクロマトグラフィー又は分別晶出のような従来の方法により分離できる。加えて、本化合物は、その分子内塩、水和物、溶媒和物又は同質異像を含む。 The pharmaceutical compounds of the present invention also include a mixture of stereoisomers or the respective pure isomers or substantially pure isomers. For example, the compound may optionally have one or more asymmetric centers on the carbon atom containing any one substituent. Thus, this compound may exist in the form of an enantiomer or diastereomer or a mixture thereof. When the compound contains a double bond, the compound may exist in the form of geometric isomerism (cis-compound, trans-compound), and when the compound contains an unsaturated bond such as carbonyl, The compounds may exist in tautomeric forms and the compounds also include these isomers or mixtures thereof. Starting compounds in the form of racemic mixtures, enantiomers or diastereomers may be used in the processes for preparing the compounds. When the present compounds are obtained in diastereomeric or enantiomeric forms, they can be separated by conventional methods such as chromatography or fractional crystallization. In addition, the present compounds include their inner salts, hydrates, solvates or isogenic.
更に、好適な薬剤化合物は、粘膜に浸透した後、又は経口投与の場合には唾液と共に胃腸管へ運ばれた後に、局所的な生理学的効果、又は全身的効果を及ぼすものである。本発明による処方から調製された剤形は、長期間にわたって活性をもたらす薬剤化合物(とりわけ、少なくとも数時間の半減期を有する薬剤)に特に好適である。 Furthermore, suitable pharmaceutical compounds are those that exert local physiological or systemic effects after penetrating the mucosa or, in the case of oral administration, after being transported with saliva to the gastrointestinal tract. The dosage forms prepared from the formulations according to the invention are particularly suitable for drug compounds that provide activity over a long period of time, especially drugs that have a half-life of at least several hours.
合成経路及び精製
「単離された」ペプチドは、実質的に細胞性物質を含まないか、又は細胞性物質から分離されているか、又は、そのペプチドが産生及び単離された細胞若しくは組織源由来の他の混入タンパク質から分離されているか、あるいは、ペプチドが化学的に合成された場合は化学的前駆体若しくは他の化学物質を実質的に含まない。例えば、細胞性物質を実質的に含まないタンパク質は、混入タンパク質を乾燥重量で約30%未満、又は好ましくは20%未満、又はより好ましくは10%未満、又は更に好ましくは5%未満、又はいっそう好ましくは1%未満有する、タンパク質調製物を含み得る。
Synthetic Routes and Purification An “isolated” peptide is substantially free of cellular material or separated from cellular material, or derived from the cell or tissue source from which the peptide was produced and isolated Is separated from other contaminating proteins or is substantially free of chemical precursors or other chemicals when the peptide is chemically synthesized. For example, a protein that is substantially free of cellular material is less than about 30%, or preferably less than 20%, or more preferably less than 10%, or even more preferably less than 5%, or even more contaminated protein by dry weight. Protein preparations may be included, preferably having less than 1%.
好ましい実施形態において、単離されたペプチドは実質的に純粋である。よって、ペプチドが組換えにより産生された場合、これは培地を実質的に含まず、例えばタンパク質調製物体積の約20%未満、又はより好ましくは10%未満、又は更に好ましくは5%未満、又はいっそう好ましくは1%未満の培地しか含まない。タンパク質が化学合成によって産生された場合、これは化学的前駆体又は他の化学物質を実質的に含まず、すなわち、タンパク質の合成に関与した化学的前駆体又は他の化学物質からは分離されている。したがって、ペプチドのこのような調製は、目的のペプチド以外の化学的前駆体又は化合物を乾燥重量で約30%未満、又は好ましくは20%未満、又はより好ましくは10%未満、又はより好ましくは5%未満、又はいっそう好ましくは1%未満しか含まない。 In preferred embodiments, the isolated peptide is substantially pure. Thus, if the peptide is produced recombinantly, it is substantially free of media, such as less than about 20%, or more preferably less than 10%, or even more preferably less than 5% of the protein preparation volume, or More preferably, it contains less than 1% medium. If the protein is produced by chemical synthesis, it is substantially free of chemical precursors or other chemicals, i.e. separated from the chemical precursors or other chemicals involved in the synthesis of the protein. Yes. Thus, such a preparation of a peptide comprises less than about 30%, or preferably less than 20%, or more preferably less than 10%, or more preferably 5% of a chemical precursor or compound other than the peptide of interest by dry weight. %, Or more preferably less than 1%.
組換えによる産生
細胞環境におけるペプチド発現は、単離されたポリヌクレオチドの利用により達成することができる。「単離された」ポリヌクレオチドは、異なる核酸配列を備えた核酸分子から実質的に分離されているか、又は異なる核酸配列を備えた核酸分子を含まないものである。単離されたポリヌクレオチド分子の実施形態には、cDNA、ゲノムDNA、RNA、及びアンチセンスRNAが挙げられる。好ましいポリヌクレオチドは、例えば組織試料からなど、ヒト由来の生体サンプルから入手される。
Recombinant production Peptide expression in the cellular environment can be achieved by the use of isolated polynucleotides. An “isolated” polynucleotide is one that is substantially separated from nucleic acid molecules with different nucleic acid sequences or does not include nucleic acid molecules with different nucleic acid sequences. Isolated polynucleotide molecule embodiments include cDNA, genomic DNA, RNA, and antisense RNA. Preferred polynucleotides are obtained from biological samples derived from humans, such as from tissue samples.
配列番号1のペプチドをコードするポリヌクレオチドを供給及び伝搬するのに、ベクターを使用することができる。宿主細胞へそのようなベクターを導入することで、模倣ストレスコピンのコードされたmRNA又はタンパク質の産生をもたらすことができる。別の方法としては、発現ベクターを、転写因子、RNAポリメラーゼ、リボソーム、及びアミノ酸などを含むがこれらに限定されない精製要素と組み合わせることにより、無細胞条件で効果的な転写/翻訳反応を生成することができる。結果として生じた反応から発現した模倣ストレスコピンペプチドは、更なる精製、修飾、及び/又は処方のために単離することができる。 Vectors can be used to supply and propagate polynucleotides encoding the peptide of SEQ ID NO: 1. Introduction of such a vector into a host cell can result in production of mimetic stress copine encoded mRNA or protein. Another method is to combine the expression vector with purification elements including, but not limited to, transcription factors, RNA polymerase, ribosomes, and amino acids to produce an effective transcription / translation reaction in cell-free conditions. Can do. The mimetic stress copine peptide expressed from the resulting reaction can be isolated for further purification, modification, and / or formulation.
用語「ベクター」は、連結されたもう1つの核酸を輸送する能力をもつ核酸分子を意味する。ベクターの代表的な一例であるプラスミドは、環状2本鎖DNAループを意味し、追加のDNAセグメントがこれに挿入され得る。ベクターの別の例はウイルスベクターであり、追加のDNAセグメントがこれに挿入され得る。所定のベクターは、それらが導入される宿主細胞(例えば、複製の細菌性起源を有する細菌ベクター及びエピソーム性哺乳類ベクター)において自己複製できる。他のベクター(例えば、非エピソーム性哺乳類ベクター)は、宿主細胞への導入時に、宿主細胞のゲノムに組み込まれ、それによって、宿主ゲノムに沿って複製される。更に、特定のベクター(発現ベクター)は、それらが操作可能に連結される遺伝子の発現を指向することができる。組換えDNA技法における有用なベクターは、プラスミドの形態であり得る。あるいは、例えばウイルスベクター(例えば複製欠陥のレトロウイルス、アデノウイルス、及びアデノ随伴ウイルス)などの、同等の機能を提供する他の形態のベクターを、目的用途に好適なように当業者が選択することができる。 The term “vector” refers to a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked. A plasmid, which is a typical example of a vector, refers to a circular double stranded DNA loop into which additional DNA segments can be inserted. Another example of a vector is a viral vector into which additional DNA segments can be inserted. Certain vectors are capable of self-replication in host cells into which they are introduced (eg, bacterial vectors having a bacterial origin of replication and episomal mammalian vectors). Other vectors (eg, non-episomal mammalian vectors) are integrated into the genome of the host cell upon introduction into the host cell, and are thereby replicated along the host genome. Furthermore, specific vectors (expression vectors) can direct the expression of genes to which they are operably linked. Useful vectors in recombinant DNA techniques can be in the form of plasmids. Alternatively, other forms of vectors that provide equivalent functions, such as viral vectors (eg, replication defective retroviruses, adenoviruses, and adeno-associated viruses) will be selected by those skilled in the art to suit the intended use. Can do.
宿主細胞は、ベクター上にあるか又は細胞染色体内に組み込まれたDNA分子を含む細胞を指す。宿主細胞は、DNA分子を内発的に含むネイティブ宿主細胞か、又は組換え型宿主細胞のいずれかであり得る。宿主細胞の一例は組換え型宿主細胞であり、これは外来のDNA配列により形質転換又は形質移入された細胞である。そのような外来DNAが細胞膜の内側に導入された場合、細胞はその外来DNAによって形質転換される。外来DNAは、細胞のゲノムを構成する染色体DNAに組み込まれて(共有結合して)も、組み込まれなくて(共有結合しなくて)もよい。例えば原核生物及び酵母において、外来DNAは、例えばプラスミドなどのエピソーム要素上に保持され得る。真核細胞については、安定に形質転換又は形質移入された細胞は、外来DNAが染色体に組み込まれ、これが染色体の複製を通して娘細胞に遺伝するようになっているものである。この安定性は、真核細胞の、外因性DNAを含有する娘細胞の集団からなる細胞株又はクローンを確立する能力により示される。クローンは、1つの細胞又は共通祖先から有糸分裂により誘導された細胞群を指す。細胞株は、生体外で数多くの世代にわたって安定に成長できる、初代細胞のクローンを指す。組換え宿主細胞は、大腸菌のような細菌、酵母のような真菌細胞、ヒト、ウシ、ブタ、サル及びげっ歯類起源の細胞株のような哺乳類細胞、ショウジョウバエ及びカイコ由来の細胞株のような昆虫細胞を含む、原核生物又は真核生物であってよい。組換え宿主細胞は、特定の対象細胞だけでなく、かかる細胞の後代又は潜在的後代も指す。特に、変異又は環境の影響のどちらかにより、特定の修飾は後の世代に起こる可能性があるため、かかる後代は親細胞と同一ではない可能性があるが、用語の範囲内に依然として含まれるよう意図されている。 A host cell refers to a cell that contains a DNA molecule that is on a vector or integrated into a cell chromosome. The host cell can be either a native host cell that contains the DNA molecule endogenously or a recombinant host cell. An example of a host cell is a recombinant host cell, which is a cell transformed or transfected with a foreign DNA sequence. When such foreign DNA is introduced inside the cell membrane, the cells are transformed with the foreign DNA. The foreign DNA may or may not be incorporated (covalently linked) into the chromosomal DNA constituting the cell's genome. For example, in prokaryotes and yeast, foreign DNA can be retained on episomal elements such as plasmids. For eukaryotic cells, a stably transformed or transfected cell is one in which foreign DNA is integrated into the chromosome, which is inherited by daughter cells through chromosome replication. This stability is demonstrated by the ability of eukaryotic cells to establish cell lines or clones consisting of a population of daughter cells containing exogenous DNA. A clone refers to a group of cells derived from one cell or common ancestor by mitosis. A cell line refers to a clone of a primary cell that can be stably grown in vitro for many generations. Recombinant host cells include bacteria such as E. coli, fungal cells such as yeast, mammalian cells such as human, bovine, porcine, monkey and rodent cell lines, cell lines derived from Drosophila and silkworms. It can be prokaryotic or eukaryotic, including insect cells. Recombinant host cell refers not only to a particular subject cell, but also to the progeny or potential progeny of such a cell. In particular, such progeny may not be identical to the parental cell, but are still included within the terminology, as certain modifications may occur in later generations, either due to mutations or environmental effects Is intended.
本発明の例示のベクターには、例えば細菌−酵母又は細菌−動物細胞、又は細菌−真菌細胞、又は細菌−無脊椎動物細胞など、宿主間でDNAをやりとりすることができるよう特に設計された発現系も含まれる。当業者には数多くのクローンベクターが知られており、適切なクローンベクターの選択は、当業者の裁量の範囲内である。原核細胞及び真核細胞の両方に好適な他の発現系については、例えば、Maniatisら、(1990),「Molecular Cloning:A Laboratory Manual,」vol.2:16.3〜16.81の第16章及び第17章を参照のこと。 Exemplary vectors of the invention include expression specifically designed to allow DNA to be exchanged between hosts, such as bacteria-yeast or bacteria-animal cells, or bacteria-fungal cells, or bacteria-invertebrate cells. Systems are also included. Many clone vectors are known to those skilled in the art, and the selection of an appropriate clone vector is within the discretion of those skilled in the art. For other expression systems suitable for both prokaryotic and eukaryotic cells, see, for example, Maniatis et al. (1990), “Molecular Cloning: A Laboratory Manual,” vol. 2: See chapters 16 and 17 of 16.3 to 16.81.
クローン遺伝子又は核酸(例えば模倣ストレスコピンペプチドをコードしたcDNA)の高レベルの発現を達成するために、その模倣ストレスコピンペプチド配列に対応するヌクレオチド配列は、好ましくは、転写を誘導する強力なプロモーター、転写/翻訳ターミネーター、及び(タンパク質をコードしている核酸の場合には)翻訳開始のためのリボソーム結合部位を含有する発現ベクターにサブクローンされる。好適な細菌プロモーターは当該技術分野において既知であり、例えばSambrookら、(1989),MOLECULAR CLONING:A LABORATORY MANUAL,Second Edition,Cold Spring Harbor Laboratory,Cold Spring Harbor,New York、及びMacrides,1996,Microbiol.Rev.60(3):512〜38に記述されている。本発明で開示されている模倣ストレスコピンタンパク質を発現するための細菌発現系は、例えば大腸菌、バチルス属菌、及びサルモネラ菌において利用できる(Palvaら、1983,Gene,22:229〜235;Mosbachら、1983,Nature,302:543〜545)。そのような発現系のキットは市販されている。哺乳類細胞、酵母、及び昆虫細胞用の真核生物発現系は当該技術分野において既知であり、これも市販されている。代表的な実施形態において、真核生物発現ベクターは、バキュロウイルスベクター、アデノウイルスベクター、アデノ随伴ベクター、又はレトロウイルスベクターである。 In order to achieve high levels of expression of a clonal gene or nucleic acid (eg, cDNA encoding a mimetic stress copine peptide), the nucleotide sequence corresponding to the mimetic stress copine peptide sequence is preferably a strong promoter that induces transcription, It is subcloned into an expression vector containing a transcription / translation terminator and (in the case of a nucleic acid encoding a protein) a ribosome binding site for translation initiation. Suitable bacterial promoters are known in the art, for example, Sambrook et al., (1989), MOLECULAR CLONING: A LABORATORY MANUAL, Second Edition, Cold Spring Harbor Laboratory, Cold Spring Harbold, Cold Spring. Rev. 60 (3): 512-38. Bacterial expression systems for expressing mimetic stress copine proteins disclosed in the present invention can be used in, for example, E. coli, Bacillus, and Salmonella (Palva et al., 1983, Gene, 22: 229-235; Mosbach et al., 1983, Nature, 302: 543-545). Kits for such expression systems are commercially available. Eukaryotic expression systems for mammalian cells, yeast, and insect cells are known in the art and are also commercially available. In exemplary embodiments, the eukaryotic expression vector is a baculovirus vector, an adenovirus vector, an adeno-associated vector, or a retrovirus vector.
プロモーターは、RNAポリメラーゼに結合して遺伝子の転写を開始することに関与する、DNAの制御配列を指す。プロモーターはしばしば、遺伝子の転写開始部位の上流側(すなわち5’)である。遺伝子は、ペプチド、ペプチド、又はタンパク質の産生に関与するDNAセグメントを参照し、これにはコード領域、コード領域の前(5’UTR)及び後(3’UTR)の非コード領域、並びに、個々のコードセグメント(エクソン)間に介在する非コード配列(イントロン)が含まれる。コーディングは、DNA又はmRNAの3つの塩基トリプレット(コドン)における特定のアミノ酸の使用又は終結シグナルを指す。 A promoter refers to a DNA regulatory sequence involved in binding RNA polymerase and initiating transcription of a gene. The promoter is often upstream (ie 5 ') of the transcription start site of the gene. A gene refers to a DNA segment involved in the production of a peptide, peptide, or protein, including a coding region, a non-coding region before (5′UTR) and after (3′UTR) the coding region, and individual Non-coding sequences (introns) that intervene between the coding segments (exons). Coding refers to the use or termination signal of a particular amino acid in a three base triplet (codon) of DNA or mRNA.
ポリヌクレオチドの発現を誘導するのに用いられるプロモーターは、特定の用途に適合するよう慣例手順で選択することができる。プロモーターは所望により、異種由来の転写開始部位からの距離が、自然の条件での転写開始部位からの距離とほぼ同じ場所に配置される。ただし、当業者には明らかであるように、この距離のある程度の差異は、プロモーター機能を失うことなく許容することができる。 The promoter used to direct expression of the polynucleotide can be selected by routine procedures to suit a particular application. If desired, the promoter is arranged at a position where the distance from the transcription start site derived from a different species is approximately the same as the distance from the transcription start site under natural conditions. However, as will be apparent to those skilled in the art, some difference in this distance can be tolerated without loss of promoter function.
プロモーターに加え、発現ベクターは、宿主細胞内の模倣ストレスコピンをコードしているポリヌクレオチドの発現に必要な、追加の要素すべてを含んだ、転写ユニット又は発現カセットを含有し得る。代表的な発現カセットには、模倣ストレスコピンペプチドをコードしているポリヌクレオチド配列に操作可能に連結されたプロモーター、並びに、転写、リボソーム結合部位、及び翻訳終結の効率的なポリアデニル化のために必要なシグナルが含有される。イヌの模倣ストレスコピンペプチドをコードするポリヌクレオチド配列は、形質移入された細胞によってコードされたタンパク質の分泌を促進する、開裂可能なシグナルペプチド配列に連結され得る。代表的なシグナルペプチドには、組織プラスミノーゲン活性化因子、インスリン、及びニューロン成長因子からのシグナルペプチド、並びにHeliothis virescensの幼若ホルモンエステラーゼが挙げられる。このカセットの追加要素としては、エンハンサー、及び、ゲノムDNAが構造遺伝子として使用されている場合は、機能スプライス供与部位及び受容部位を備えたイントロンが挙げられ得る。 In addition to the promoter, the expression vector may contain a transcription unit or expression cassette that contains all the additional elements required for the expression of the polynucleotide encoding the mimetic stress copine in the host cell. A typical expression cassette contains a promoter operably linked to a polynucleotide sequence encoding a mimetic stress copine peptide, and is required for efficient polyadenylation of transcription, ribosome binding sites, and translation termination Signal is contained. The polynucleotide sequence encoding the canine mimetic stress copine peptide can be linked to a cleavable signal peptide sequence that facilitates secretion of the encoded protein by the transfected cell. Representative signal peptides include signal peptides from tissue plasminogen activator, insulin, and neuron growth factor, and heliothis viruses juvenile hormone esterase. Additional elements of this cassette may include enhancers and introns with functional splice donor and acceptor sites if genomic DNA is used as the structural gene.
プロモーター配列に加え、発現カセットには更に、効率的な終結のために提供された、構造遺伝子の下流にある転写終結部位が含まれ得る。終結部位は、プロモーター配列と同じ遺伝子、ヒトストレスコピン遺伝子、又は別の遺伝子から取得することができる。 In addition to the promoter sequence, the expression cassette may further include a transcription termination site downstream of the structural gene provided for efficient termination. The termination site can be obtained from the same gene as the promoter sequence, the human stress copine gene, or another gene.
代表的な実施形態において、当該技術分野において既知の真核細胞又は原核細胞における発現に好適な任意のベクターが使用され得る。代表的な細菌発現ベクターには、例えばpBR322系プラスミド、pSKF、pET23Dなどのプラスミド、並びに例えばGST及びLacZなどの融合発現系が挙げられる。哺乳動物発現ベクターの例としては、例えばpCDM8(Seed,1987,Nature,329:840)及びpMT2PC(Kaufmanら、1987,EMBO J.,6:187〜195)が挙げられる。本発明のペプチドの組換え発現に好適であり得る、市販されている哺乳類発現ベクターには、例えば、pMAMneo(Clontech(Mountain View,CA))、pcDNA4(Invitrogen(Carlsbad,CA))、pCiNeo(Promega(Madison,WI))、pMC1neo(Stratagene(La Jolla,CA))、pXT1(Stratagene(La Jolla,CA))、pSG5(Stratagene(La Jolla,CA))、EBO−pSV2−neo(ATCC 37593)、pBPV−1(8−2)(ATCC 37110)、pdBPV−MMTneo(342−12)(ATCC 37224)、pRSVgpt(ATCC 37199)、pRSVneo(ATCC 37198)、pSV2−dhfr(ATCC 37146)、pUCTag(ATCC 37460)、及びlZD35(ATCC 37565)が挙げられる。 In an exemplary embodiment, any vector suitable for expression in eukaryotic or prokaryotic cells known in the art can be used. Representative bacterial expression vectors include plasmids such as pBR322 series plasmids, pSKF, pET23D, and fusion expression systems such as GST and LacZ. Examples of mammalian expression vectors include pCDM8 (Seed, 1987, Nature, 329: 840) and pMT2PC (Kaufman et al., 1987, EMBO J., 6: 187-195). Commercially available mammalian expression vectors that may be suitable for recombinant expression of the peptides of the invention include, for example, pMAMneo (Clontech (Mountain View, CA)), pcDNA4 (Invitrogen (Carlsbad, Calif.)), PCiNeo (Promega). (Madison, WI)), pMC1neo (Stratagene (La Jolla, CA)), pXT1 (Stratagene (La Jolla, CA)), pSG5 (Stratagene (La Jolla, CA)), EBO-pSV2-neo (ATCC 37593) pBPV-1 (8-2) (ATCC 37110), pdBPV-MMTneo (342-12) (ATCC 37224), pRSVgpt (ATCC 37199) PRSVneo (ATCC 37198), pSV2-dhfr (ATCC 37146), pUCTag (ATCC 37460), and lZD35 (ATCC 37565).
単離の便利な方法を提供するため、更にエピトープタグも組換えタンパク質に加えることができ、これには例えばc−myc、ヘモグルチニン(HA)タグ、6−Hisタグ、マルトース結合タンパク質、VSV−Gタグ、又は抗FLAGタグ、及び当該技術分野において利用可能なその他のものがある。 In order to provide a convenient method of isolation, an epitope tag can also be added to the recombinant protein, for example c-myc, hemoglutinin (HA) tag, 6-His tag, maltose binding protein, VSV-G. There are tags, or anti-FLAG tags, and others available in the art.
真核細胞性ウイルスからの制御要素を含む発現ベクターは、真核細胞性発現ベクター(例えばSV40ベクター、パピローマウイルスベクター、及びエプスタインバールウイルスから誘導されたベクターなど)において使用され得る。他の代表的な真核細胞性ベクターには、pMSG、pAV009/A+、pMTO10/A+、pMAMneo 5、バキュロウイルスpDSVE、及びその他の、CMVプロモーター、SV40早期プロモーター、SV40後期プロモーター、メタロチオネインプロモーター、ハツカネズミ乳房腫瘍ウイルスプロモーター、ラウス肉腫ウイルスプロモーター、ポリヘドリンプロモーター、その他真核細胞における発現に効果を呈する他のプロモーターの誘導のもとにタンパク質の発現を可能にする他のベクターが挙げられる。
Expression vectors containing control elements from eukaryotic viruses can be used in eukaryotic expression vectors such as vectors derived from SV40 vectors, papillomavirus vectors, and Epstein-Barr viruses. Other representative eukaryotic vectors include pMSG, pAV009 / A +, pMTO10 / A +,
いくつかの発現系は、例えばネオマイシン、チミジンキナーゼ、ハイグロマイシンBホスホトランスフェラーゼ、ジヒドロ葉酸還元酵素など、遺伝子増幅をもたらすマーカーを有する。別の方法としては、遺伝子増幅を用いない高収率発現系も好適であり、例えば、ポリヘドリンプロモーター又はその他の強力なバキュロウイルスプロモーターの誘導のもとに模倣ストレスコピンペプチドをコードする配列を備えた、昆虫細胞におけるバキュロウイルスベクターを使用することが挙げられる。 Some expression systems have markers that provide gene amplification, such as neomycin, thymidine kinase, hygromycin B phosphotransferase, dihydrofolate reductase. As another method, a high-yield expression system that does not use gene amplification is also suitable. For example, a sequence encoding a mimic stress copine peptide under the induction of a polyhedrin promoter or other strong baculovirus promoter is used. Use of the baculovirus vector in insect cells provided.
発現ベクターに含まれ得る要素には更に、大腸菌において機能するレプリコン、組換えプラスミドを宿す細菌の選択を可能にするための抗生物質耐性をコードする遺伝子、及び、真核細胞配列の制御された挿入を可能にするプラスミドの非主要領域における固有の制限部位も挙げられる。特定の抗生物質耐性遺伝子は、当該技術分野において既知の数多くの耐性遺伝子から選択することができる。必要に応じて、又は所望により、真核細胞のDNA複製に干渉しないよう、原核細胞配列を選択することができる。 The elements that can be included in the expression vector further include a replicon that functions in E. coli, a gene encoding antibiotic resistance to allow selection of bacteria harboring the recombinant plasmid, and controlled insertion of eukaryotic sequences. Also included are unique restriction sites in the non-major region of the plasmid that allow A particular antibiotic resistance gene can be selected from a number of resistance genes known in the art. Prokaryotic sequences can be selected so that they do not interfere with eukaryotic DNA replication as needed or desired.
既知の形質移入方法を使用して、大量のSCP模倣物質を発現する細菌、哺乳類、酵母又は昆虫細胞株を産生することができる。このSCP模倣物質は次に、例えば硫酸アンモニウムなどの物質による選択的沈殿、カラムクロマトグラフィー、及び免疫精製法といった標準的技法を用いて、精製される。 Known transfection methods can be used to produce bacterial, mammalian, yeast or insect cell lines that express large amounts of SCP mimetics. This SCP mimetic is then purified using standard techniques such as selective precipitation with substances such as ammonium sulfate, column chromatography, and immunopurification.
真核細胞及び原核細胞の形質転換は、標準技法により実施することができる(例えばMorrison,1977,J Bact.,132:349〜351;Clark−Curtiss & Curtiss,Methods in Enzymology,101:347〜362を参照)。 Transformation of eukaryotic cells and prokaryotic cells can be performed by standard techniques (eg Morrison, 1977, J Bact., 132: 349-351; Clark-Curtis & Curtis, Methods in Enzymology, 101: 347-362). See).
異種のヌクレオチド配列を宿主細胞に導入するのに好適な、任意の既知の手法を使用して、発現ベクターを導入することができる。これには、Superfect(Qiagen)のような試薬の使用、リポソーム、リン酸カルシウムトランスフェクション、ポリブレン、原形質融合、電気穿孔方、顕微注入法、プラスミドベクター、ウイルスベクター、遺伝子銃粒子加速(遺伝子銃)、又はその他の、クローンゲノムDNA、cDNA、合成DNA又はその他の異種遺伝子材料を宿主細胞に導入するための既知の任意の方法が挙げられる(例えばSambrookら(上述)を参照)。選択された、特定の遺伝子操作手法は、模倣ストレスコピンRNA、mRNA、cDNA、又は遺伝子を発現することが可能な宿主細胞に、少なくとも1つの遺伝子をうまく導入できるものであるべきである。 Any known technique suitable for introducing a heterologous nucleotide sequence into a host cell can be used to introduce the expression vector. This includes the use of reagents such as Superfect (Qiagen), liposomes, calcium phosphate transfection, polybrene, protoplast fusion, electroporation, microinjection, plasmid vectors, virus vectors, gene gun particle acceleration (gene gun), Or any other known method for introducing cloned genomic DNA, cDNA, synthetic DNA or other heterologous genetic material into a host cell (see, eg, Sambrook et al., Supra). The particular genetic engineering procedure chosen should be capable of successfully introducing at least one gene into a host cell capable of expressing mimic stress copine RNA, mRNA, cDNA, or gene.
当業者には明らかであり得るように、哺乳類細胞の安定な形質移入のためには、使用される発現ベクター及び形質移入技法に応じて、わずかな部分の細胞だけが、そのゲノム内に異種DNAを統合し得る。これらの構成要素を識別及び選択するために、選択マーカー(例えば抗生物質に対する耐性)をコードする遺伝子を、目的の遺伝子と共に宿主細胞に導入することができる。代表的な選択マーカーには、例えばG−418、プロマイシン、ジェネティシン、ハイグロマイシン及びメトトレキサートなどの薬剤に対する耐性を付与するものが挙げられる。導入された核酸で安定的に形質移入された細胞は、薬剤選択によって選択され、識別され得る(例えば、選択マーカー遺伝子を組み入れた細胞は生き残り、他の細胞は死滅する)。 As will be apparent to those skilled in the art, for stable transfection of mammalian cells, depending on the expression vector and transfection technique used, only a small number of cells will have heterologous DNA in their genome. Can be integrated. In order to identify and select these components, a gene that encodes a selectable marker (eg, resistance to antibiotics) can be introduced into the host cells along with the gene of interest. Representative selectable markers include those that confer resistance to drugs such as G-418, puromycin, geneticin, hygromycin and methotrexate. Cells stably transfected with the introduced nucleic acid can be selected and identified by drug selection (eg, cells incorporating the selectable marker gene will survive and other cells will die).
異種由来の制御要素を安定な細胞株又はクローン微生物に挿入することにより、例えば標的相同組換え(例えば米国特許第5,272,071号及びPCT国際公開特許WO 91/06667号に記述されている)などの技法を用い、内因性遺伝子に操作可能に連結し、この内因性遺伝子発現を活性化することができる。発現ベクターを細胞内に導入した後、この形質移入された細胞は好ましくは、模倣ストレスコピンペプチドの発現に最適な好ましい条件下で培養され、後述の標準技法を用いて培養物から模倣ストレスコピンペプチドが回収される。原核細胞又は真核細胞の培養方法は当該技術分野において既知であり、例えばSambrookら(上述);Freshney,1993,CULTURE OF ANIMAL CELLS,3rd ed.を参照のこと。 By inserting a heterologous regulatory element into a stable cell line or clonal microorganism, for example, target homologous recombination (for example described in US Pat. No. 5,272,071 and PCT International Publication No. WO 91/06667). ) And the like can be operably linked to the endogenous gene to activate this endogenous gene expression. After introducing the expression vector into the cell, the transfected cell is preferably cultured under favorable conditions optimal for expression of the mimetic stress copine peptide, and mimic stress copin peptide from the culture using standard techniques described below. Is recovered. Methods for culturing prokaryotic or eukaryotic cells are known in the art and are described, for example, in Sambrook et al. (Supra); Freshney, 1993, CULTURE OF ANIMAL CELL, 3rd ed. checking ...
ペプチド産生のために細胞系を使用する方法とは別の方法として、無細胞系で、原核細胞系(Zubay G.,Annu Rev Genet.,1973,7:267〜287)及び真核細胞系(Pelhamら、Eur J Biochem.,1976,67:247〜256;Andersonら、Meth Enzymol.,1983,101:635〜644)の遺伝子発現及び合成が可能であることが示されている。これらの系は、ペプチド合成反応のために、mRNA又はDNAヌクレオチドのいずれかに利用することができる。無細胞ペプチド産生のための好ましい技法では、網状赤血球可溶化液、RNAポリメラーゼ、ヌクレオチド、塩、及びリボヌクレアーゼ阻害剤が、迅速に連結された転写/翻訳反応(TNT(登録商標)、Promega(Madison,WI,U.S.A.))において使用される。 As an alternative to using cell lines for peptide production, cell-free systems, prokaryotic cell systems (Zubay G., Annu Rev Genet., 1973, 7: 267-287) and eukaryotic cell systems ( Pelham et al., Eur J Biochem., 1976, 67: 247-256; Anderson et al., Meth Enzymol., 1983, 101: 635-644) have been shown to be possible. These systems can be utilized for either mRNA or DNA nucleotides for peptide synthesis reactions. In a preferred technique for cell-free peptide production, reticulocyte lysate, RNA polymerase, nucleotides, salts, and ribonuclease inhibitors are rapidly linked transcription / translation reactions (TNT®, Promega (Madison, Madison, WI, U.S.A.)).
合成産生
本発明において具現されたペプチドは、Merrifield,J.Am.Chem.Soc.,15:2149〜2154(1963)で最初に記述された固相合成技法を使用して調製することができる。他のペプチド合成技法は例えば、M.Bodanszkyら、(1976)Peptide Synthesis,John Wiley & Sons,2d Ed.;Kent and Clark−Lewis,Synthetic Peptides in Biology and Medicine,p.295〜358,eds.Alitalo,K.ら、Science Publishers,(Amsterdam,1985);並びに、当業者に既知の他の参照研究において見出すことができる。ペプチド合成技法の要約は、J.Stuart and J.D.Young,Solid Phase Peptide Synthelia,Pierce Chemical Company,Rockford,III.(1984)に見出すことができ、これは参考として本明細書に組み込まれる。溶液法によるペプチド合成も使用することができ、これはThe Proteins,Vol.II,3d Ed.,p.105〜237,Neurath,H.ら、編、Academic Press,New York,N.Y.(1976)に記述されている。そのような合成で使用するための適切な保護基は、上記文献、並びにJ.F.W.McOmie,Protective Groups in Organic Chemistry,Plenum Press,New York,N.Y.(1973)に見出され、これは参考として本明細書に組み込まれる。一般に、これらの合成法には、1つ以上のアミノ酸残基又は好適な保護アミノ酸残基を、成長中のペプチド鎖に連続的に加えることが含まれる。通常、第1アミノ酸残基のアミノ基又はカルボキシル基のいずれかが、好適な、選択的に除去可能な保護基によって保護されている。例えばリジンなどの反応側基を含むアミノ酸のために、異なる、選択的に除去可能な保護基が利用される。
Synthetic Production Peptides embodied in the present invention are described in Merrifield, J. et al. Am. Chem. Soc. 15: 2149-2154 (1963), and can be prepared using solid phase synthesis techniques. Other peptide synthesis techniques are described in, for example, M.M. Bodanzky et al. (1976) Peptide Synthesis, John Wiley & Sons, 2d Ed. Kent and Clark-Lewis, Synthetic Peptides in Biology and Medicine, p. 295-358, eds. Alitaro, K.M. Science Publishers, (Amsterdam, 1985); as well as other reference studies known to those skilled in the art. A summary of peptide synthesis techniques can be found in J. Stuart and J.M. D. Young, Solid Phase Peptide Synthelia, Pierce Chemical Company, Rockford, III. (1984), which is incorporated herein by reference. Solution synthesis of peptides can also be used, which is described in The Proteins, Vol. II, 3d Ed. , P. 105-237, Neuroth, H .; Et al., Academic Press, New York, N .; Y. (1976). Suitable protecting groups for use in such syntheses are described in the above references, as well as in J. Am. F. W. McOmie, Protective Groups in Organic Chemistry, Plenum Press, New York, N .; Y. (1973), which is incorporated herein by reference. In general, these synthetic methods involve the sequential addition of one or more amino acid residues or suitable protected amino acid residues to a growing peptide chain. Usually, either the amino group or the carboxyl group of the first amino acid residue is protected by a suitable, selectively removable protecting group. Different, selectively removable protecting groups are utilized for amino acids containing reactive side groups such as lysine.
ペプチド合成の固相法及び溶液法の両方に、ブロック合成法を適用することもできる。単一のアミノ酸残基を連続的に付加するのではなく、順に並んだ2つ以上のアミノ酸残基を含んだあらかじめ形成されたブロックを、開始サブユニットとして使用するか、又は、順に追加されるユニット(単一のアミノ酸残基ではなく)として使用する。 The block synthesis method can also be applied to both the solid phase method and the solution method of peptide synthesis. Rather than sequentially adding a single amino acid residue, a preformed block containing two or more amino acid residues in sequence is used as the starting subunit or added sequentially Used as a unit (not a single amino acid residue).
固相合成を一例として使用し、保護された又は誘導体化されたアミノ酸を、保護されていないカルボキシル基又はアミノ基を介して、不活性の固体支持体に結合する。次に、アミノ基又はカルボキシル基の保護基を選択的に除去し、好適に保護された相補的(アミノ又はカルボキシル)基を有する、配列中で次のアミノ酸を、既に固体支持体に結合されている残基と混合して反応させる。次に、アミノ基又はカルボキシル基の保護基を、この新たに追加されたアミノ酸残基から除去し、更にその次のアミノ酸(好適に保護されている)を付加し、同様に行う。望ましいアミノ酸がすべて適切な配列で連結された後、残った末端及び側基保護基(及び固体支持体)は順次又は同時に除去され、最終生成物のペプチドが得られる。本発明のペプチドは、好ましくはベンジル化又はメチルベンジル化アミノ基を含まない。そのような保護基部分は、合成の過程で使用され得るが、ペプチドを使用する前に除去される。他に記述されているように、コンフォメーションを拘束するために分子内連結を形成する、追加の反応が必要になることがある。 Solid phase synthesis is used as an example to attach a protected or derivatized amino acid to an inert solid support via an unprotected carboxyl or amino group. Next, the amino or carboxyl protecting group is selectively removed and the next amino acid in the sequence having a suitably protected complementary (amino or carboxyl) group is already attached to the solid support. React with the residue. Next, the amino or carboxyl protecting group is removed from the newly added amino acid residue, and the next amino acid (suitably protected) is added, followed by the same procedure. After all of the desired amino acids are linked in the appropriate sequence, the remaining terminal and side group protecting groups (and solid support) are removed sequentially or simultaneously to yield the final product peptide. The peptides of the present invention preferably do not contain benzylated or methylbenzylated amino groups. Such protecting group moieties can be used in the course of synthesis, but are removed before using the peptide. As described elsewhere, additional reactions that form intramolecular linkages to constrain conformation may be required.
固体支持体合成は、自動タンパク質合成装置(Protemist(登録商標)、CellFree Sciences(〒790−8577日本国愛媛県松山市);Symphony SMPS−110,Rainin(Woburn,MA,U.S.A.);ABI 433Aペプチド合成装置、Applied Biosystems(Foster City,CA,U.S.A.))で達成し得る。これらの装置は、合成のより大きな制御及び最適化を可能にする自動化タンパク質反応を実施する能力を有する。 Solid support synthesis was performed using an automated protein synthesizer (Protemist (registered trademark), CellFree Sciences (Matsuyama, Ehime Prefecture, Japan 790-8777); Symphony SMPS-110, Rainin (Woburn, MA, USA). An ABI 433A peptide synthesizer, Applied Biosystems (Foster City, CA, USA)). These devices have the ability to perform automated protein reactions that allow greater control and optimization of synthesis.
精製
本発明のペプチドを単離又は精製するために、数多くの手順が採用され得る。例えば、ペプチドの物理的特性(すなわち疎水性)に基づいて、カラムクロマトグラフィーを使用してペプチドを精製することができる。別の方法としては、確立された分子接着特性を有するタンパク質を、本発明のペプチドに可逆的に融合させることができる。融合タンパク質の適切な配位子により、模倣ストレスコピンペプチドは、精製カラムに選択的に吸着され、次にカラムから実質的に純粋な形態で遊離される。融合タンパク質は次に、酵素活性により除去され得る。別のカラム精製戦略では、模倣ストレスコピンペプチドに対して高められた抗体を採用することができる。これらの抗体は、カラムマトリックスに結合され、この免疫親和性カラムを通してペプチドを精製することができる。
Purification A number of procedures can be employed to isolate or purify the peptides of the present invention. For example, the peptide can be purified using column chromatography based on the physical properties (ie, hydrophobicity) of the peptide. Alternatively, proteins having established molecular adhesion properties can be reversibly fused to the peptides of the present invention. With the appropriate ligand of the fusion protein, the mimetic stress copine peptide is selectively adsorbed onto the purification column and then released from the column in a substantially pure form. The fusion protein can then be removed by enzymatic activity. Another column purification strategy can employ antibodies raised against mimetic stress copine peptides. These antibodies are bound to a column matrix and the peptide can be purified through this immunoaffinity column.
組換え型タンパク質は、例えば塩分留などの好適な分離技法によって、宿主反応から分離することができる。この方法は、目的の組換え型タンパク質から、望ましくない宿主細胞タンパク質(又は細胞培地由来のタンパク質)を分離するのに使用することができる。代表的な塩の例は硫酸アンモニウムであり、これはタンパク質混合物中で水の量を効果的に減少させることにより(タンパク質がその溶解度に基づいて沈殿し)、タンパク質を沈殿させる。タンパク質が疎水性であるほど、低い硫酸アンモニウム濃度で沈殿しやすくなる。代表的な単離プロトコルには、飽和硫酸アンモニウムをタンパク質溶液に加え、結果的に硫酸アンモニウム濃度を20〜30%の間にし、大半の疎水性タンパク質を沈殿させることが含まれる。この沈殿を廃棄し(目的のタンパク質が疎水性でない場合)、この上澄みに、目的のタンパク質が沈殿することが知られている濃度まで硫酸アンモニウムを加える。この沈殿を次に緩衝液に溶かし、過剰な塩を除去して、例えば透析又は膜分離精製法により望ましい純度を達成する。タンパク質の溶解度に依存した他の既知の方法(例えば冷エタノール沈殿法)を使用して、複雑なタンパク質混合物を分別することができる。 The recombinant protein can be separated from the host reaction by a suitable separation technique such as salt fractionation. This method can be used to separate unwanted host cell proteins (or proteins from cell culture media) from the recombinant protein of interest. An example of a typical salt is ammonium sulfate, which precipitates the protein by effectively reducing the amount of water in the protein mixture (the protein precipitates based on its solubility). The more hydrophobic the protein, the more likely it will precipitate at lower ammonium sulfate concentrations. A typical isolation protocol involves adding saturated ammonium sulfate to the protein solution, resulting in an ammonium sulfate concentration between 20-30% and precipitating most hydrophobic proteins. Discard the precipitate (if the protein of interest is not hydrophobic) and add ammonium sulfate to the supernatant to a concentration known to precipitate the protein of interest. This precipitate is then dissolved in a buffer to remove excess salt to achieve the desired purity, for example by dialysis or membrane separation purification methods. Other known methods that depend on the solubility of the protein (eg, cold ethanol precipitation) can be used to fractionate complex protein mixtures.
単離又は精製技法の他の例において、本発明のペプチドの分子量を使用して、種々の孔径の膜(例えばAmicon又はMillipore膜)を通す限外濾過を用い、目的のタンパク質より大きい又は小さい寸法のタンパク質から分離するのに使用することができる。第1段階として、タンパク質混合物を、目的のタンパク質の分子量より小さい分子量カットオフを有する孔径の膜を通して限外濾過を行う。この限外濾過で残ったものを、次に、目的のタンパク質の分子量より大きい分子カットオフを有する膜で、限外濾過を行う。組換え型タンパク質は、膜を通過して濾液内に移り、この濾液をクロマトグラフィーに通すことができる。 In other examples of isolation or purification techniques, the molecular weight of the peptides of the present invention is used to use ultrafiltration through membranes of various pore sizes (eg, Amicon or Millipore membranes) with dimensions larger or smaller than the protein of interest. It can be used to separate from other proteins. As a first step, the protein mixture is ultrafiltered through a membrane with a pore size having a molecular weight cut-off smaller than the molecular weight of the protein of interest. What remains from this ultrafiltration is then subjected to ultrafiltration with a membrane having a molecular cutoff greater than the molecular weight of the protein of interest. The recombinant protein passes through the membrane and into the filtrate, which can be passed through chromatography.
化学修飾
本発明のペプチドは、例えばマレイミドキャッピング、ポリエチレングリコール(PEG)結合、マレイド化(maleidification)、アシル化、アルキル化、エステル化、及びアミド化などの化学修飾により、ペプチドの構造的類似体を産生する対象となり得る。さまざまな化学修飾技法及び部分が存在することが当業者には理解されよう。例えば、米国特許第5,554,728号、同第6,869,932号、同第6,828,401号、同第6,673,580号、同第6,552,170号、同第6,420,339号、米国特許公開第2006/0210526号、及びPCT国際公開特許WO 2006/136586号を参照のこと。好ましくは化学修飾は、例えば反応効率を高めるために、単離されたペプチド上で行われる。
Chemical Modification Peptides of the present invention can be used to modify structural analogs of the peptide by chemical modifications such as maleimide capping, polyethylene glycol (PEG) linkage, maleidification, acylation, alkylation, esterification, and amidation. Can be a target for production. Those skilled in the art will appreciate that there are a variety of chemical modification techniques and moieties. For example, U.S. Pat. Nos. 5,554,728, 6,869,932, 6,828,401, 6,673,580, 6,552,170, See 6,420,339, U.S. Patent Publication No. 2006/0210526, and PCT International Publication No. WO 2006/136586. Preferably, chemical modification is performed on the isolated peptide, for example to increase reaction efficiency.
本発明の特定の実施形態において、本発明のペプチドには、アミド化C末端が含まれる。そのペプチド修飾手順は、単離された精製ペプチド上で実施することができ、又は、固相合成の場合は、合成手順中に実施することができる。そのような手順は、Rayら、Nature Biotechnology,1993,vol.11,pp.64〜70;Cottinghamら、Nature Biotechnology,2001,vol.19,pp.974〜977;Walshら、Nature Biotechnology,2006,vol.24,pp.1241〜1252;米国特許出願公開第2008/0167231号において検討されている。 In certain embodiments of the invention, the peptides of the invention include an amidated C-terminus. The peptide modification procedure can be performed on the isolated purified peptide or, in the case of solid phase synthesis, can be performed during the synthesis procedure. Such a procedure is described in Ray et al., Nature Biotechnology, 1993, vol. 11, pp. 64-70; Cottingham et al., Nature Biotechnology, 2001, vol. 19, pp. 974-977; Walsh et al., Nature Biotechnology, 2006, vol. 24, pp. 1241-1252; U.S. Patent Application Publication No. 2008/0167231.
本発明のペプチドは、ペプチドとPEG部分とを結合させるのに有用な特定の中間リンカーを含有し得る。そのようなリンカーは、宿主に対し、最小限の免疫原性及び毒性をもたらし得る。そのようなリンカーの例は、Bailonら、PSTT,1998,vol.1(8),pp.352〜356に見出し得る。 The peptides of the present invention may contain certain intermediate linkers useful for conjugating the peptide to the PEG moiety. Such linkers can provide minimal immunogenicity and toxicity to the host. Examples of such linkers are described in Bailon et al., PSTT, 1998, vol. 1 (8), pp. 352-356.
特定の実施形態において、本発明は、カルボキシ末端にCONH2を含む配列番号29で示す配列から本質的になる単離されたペプチドと、配列番号29のアミノ酸配列のポジション28でシステイン残基に結合した中間リンカーとを含む、結合を目的とする。特定の実施形態において、この中間リンカーはN−エチルスクシンイミドである。更なる実施形態において、この中間リンカーはビニルスルホンである。更なる実施形態において、この中間リンカーはアセトアミドである。更なる実施形態において、この中間リンカーはオルトピリジルジスルフィドである。 In certain embodiments, the present invention binds to a cysteine residue at position 28 of the amino acid sequence of SEQ ID NO: 29 with an isolated peptide consisting essentially of the sequence shown in SEQ ID NO: 29 containing CONH 2 at the carboxy terminus. Intended for conjugation, including an intermediate linker. In certain embodiments, the intermediate linker is N-ethylsuccinimide. In a further embodiment, the intermediate linker is vinyl sulfone. In a further embodiment, the intermediate linker is acetamide. In a further embodiment, the intermediate linker is orthopyridyl disulfide.
更なる実施形態において、本発明は、カルボキシ末端にCONH2を備えた配列番号29で示すアミノ酸配列を有するペプチドと、配列番号29のポジション28でシステイン残基に結合したN−エチルスクシンイミドリンカーとを含む、結合を目的とし、ここにおいてN−エチルスクシンイミドリンカーは、PEG部分にも結合している。特定の実施形態において、PEG部分の分子量は約2kDa〜約80kDaの範囲であり得る。特定の実施形態において、PEGの質量は約20kDaである。好ましい実施形態において、CRHR2ペプチド作動薬は、配列番号82又は配列番号102のペプチドを含む。特定の実施形態において、PEG質量は約5kDaである。特定の他の実施形態において、PEG質量は約12kDaである。特定の実施形態において、PEG質量は約20kDaである。特定の実施形態において、PEGは質量約30kDaである。特定の実施形態において、PEG質量は約40kDaである。特定の実施形態において、PEG質量は約80kDaである。特定の実施形態において、PEG部分は直鎖状である。他の実施形態において、PEG部分は分枝状である。PEG部分は、当業者に既知の方法にしたがって合成することができる。別の方法としては、PEG部分は市販されており、例えばSUNBRIGHT(登録商標)ME−020MA、SUNBRIGHT(登録商標)ME−050MA、及びSUNBRIGHT(登録商標)ME−200MA(NOF corp.(日本);Sigma Aldrich(St.Louis,MO,U.S.A.))がある。 In a further embodiment, the present invention comprises a peptide having the amino acid sequence shown in SEQ ID NO: 29 with CONH 2 at the carboxy terminus, and an N-ethylsuccinimide linker linked to a cysteine residue at position 28 of SEQ ID NO: 29. For purposes of attachment, including where the N-ethylsuccinimide linker is also attached to the PEG moiety. In certain embodiments, the molecular weight of the PEG moiety can range from about 2 kDa to about 80 kDa. In certain embodiments, the mass of PEG is about 20 kDa. In a preferred embodiment, the CRHR2 peptide agonist comprises the peptide of SEQ ID NO: 82 or SEQ ID NO: 102. In certain embodiments, the PEG mass is about 5 kDa. In certain other embodiments, the PEG mass is about 12 kDa. In certain embodiments, the PEG mass is about 20 kDa. In certain embodiments, PEG has a mass of about 30 kDa. In certain embodiments, the PEG mass is about 40 kDa. In certain embodiments, the PEG mass is about 80 kDa. In certain embodiments, the PEG moiety is linear. In other embodiments, the PEG moiety is branched. The PEG moiety can be synthesized according to methods known to those skilled in the art. Alternatively, the PEG moiety is commercially available, eg, SUNBRIGHT® ME-020MA, SUNBRIGHT® ME-050MA, and SUNBRIGHT® ME-200MA (NOF corp. (Japan); Sigma Aldrich (St. Louis, MO, USA).
本発明は更に、本発明のペプチドの製薬上許容される塩、及びその塩を使用する方法に関する。製薬上許容される塩は、無毒で生物学的に許容されるか又は他の様式で被験者に投与するために生物学的に適した、ペプチドの遊離酸若しくは塩基の塩を指す。一般的には、S.M.Bergeら、「Pharmaceutical Salts」、J.Pharm.Sci.,1977,66:1〜19、及びHandbook of Pharmaceutical Salts,Properties,Selection,and Use、Stahl及びWermuth編、Wiley−VCH and VHCA,Zurich,2002を参照のこと。製薬上許容される好適な塩は、患者の組織に過度の毒性も刺激もアレルギー反応ももたらすことなく接触させるに適した製薬上許容される塩である。製薬上許容される塩の例には、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプロン酸塩、ヘプタン酸塩、プロピオール酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−二酸塩、ヘキシン−1,6−二酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸、フタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、γ−ヒドロキシ酪酸塩、グリコール酸塩、酒石酸塩、メタン−スルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スルホン酸塩及びマンデル酸塩が挙げられる。 The invention further relates to pharmaceutically acceptable salts of the peptides of the invention and methods of using the salts. A pharmaceutically acceptable salt refers to a free acid or base salt of a peptide that is non-toxic, biologically acceptable, or biologically suitable for administration to a subject in another manner. In general, S.M. M.M. Berge et al., “Pharmaceutical Salts”, J. Am. Pharm. Sci. 1977, 66: 1-19, and Handbook of Pharmaceutical Salts, Properties, Selection, and Use, Ed. Stahl and Wermuth, Wiley-VCH and VHCA, Zurich, 2002. Suitable pharmaceutically acceptable salts are pharmaceutically acceptable salts suitable for contacting a patient's tissue without causing excessive toxicity, irritation or allergic reaction. Examples of pharmaceutically acceptable salts include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogen phosphate, dihydrogen phosphate, metaphosphate, pyrroline Acid salt, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate, isobutyrate, caproate, heptanoate, propiolate, sulphate Acid salt, malonate, succinate, suberate, sebacate, fumarate, maleate, butyne-1,4-diacid, hexyne-1,6-diacid, benzoate , Chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoic acid, phthalate, sulfonate, xylenesulfonate, phenylacetate, phenylpropionate, phenylbutyrate ,citric acid , Lactate, γ-hydroxybutyrate, glycolate, tartrate, methane-sulfonate, propanesulfonate, naphthalene-1-sulfonate, naphthalene-2-sulfonate and mandelate .
本発明のペプチドが塩基性窒素を含有する場合、当該技術分野で利用可能な適切な方法のいずれか、例えば遊離塩基を無機酸、例えば塩酸、臭化水素酸、ヨウ化水素酸、過塩素酸、硫酸、スルファミン酸、硝酸、ホウ酸、燐酸などでか、又は有機酸、例えば酢酸、トリフルオロ酢酸、フェニル酢酸、プロピオン酸、ステアリン酸、乳酸、アスコルビン酸、マレイン酸、ヒドロキシマレイン酸、リンゴ酸、パモ酸、イセチオン酸、コハク酸、吉草酸、フマル酸、サッカリン酸、マロン酸、ピルビン酸、シュウ酸、グリコール酸、サリチル酸、オレイン酸、パルミチン酸、ラウリン酸、ピラノシジル酸、例えばグルクロン酸又はガラクツロン酸など、α−ヒドロキシ酸、例えばマンデル酸、クエン酸又は酒石酸など、アミノ酸、例えばアスパラギン酸又はグルタミン酸など、芳香族酸、例えば安息香酸、2−アセトキシ安息香酸、ナフトエ酸又は桂皮酸など、スルホン酸、例えばラウリルスルホン酸、ベンゼンスルホン酸、2−ナフタレンスルホン酸、p−トルエンスルホン酸、メタンスルホン酸、エタンスルホン酸、ヒドロキシエタンスルホン酸、シクロヘキサンスルホン酸、本明細書に例として示した酸などの如き酸の適合し得る任意混合物、及び当該技術分野の技術の通常のレベルに照らして相当物又は許容される代替物であると見なされる他の酸及びこれらの混合物のいずれかで処理する方法などで所望の製薬上許容される塩を調製することができる。 When the peptide of the present invention contains basic nitrogen, any suitable method available in the art, for example free base is converted to an inorganic acid such as hydrochloric acid, hydrobromic acid, hydroiodic acid, perchloric acid. , Sulfuric acid, sulfamic acid, nitric acid, boric acid, phosphoric acid, or organic acids such as acetic acid, trifluoroacetic acid, phenylacetic acid, propionic acid, stearic acid, lactic acid, ascorbic acid, maleic acid, hydroxymaleic acid, malic acid , Pamoic acid, isethionic acid, succinic acid, valeric acid, fumaric acid, saccharic acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, oleic acid, palmitic acid, lauric acid, pyranosidic acid such as glucuronic acid or galacturon Acids, α-hydroxy acids such as mandelic acid, citric acid or tartaric acid, amino acids such as asparagine Aromatic acids such as acid or glutamic acid, such as benzoic acid, 2-acetoxybenzoic acid, naphthoic acid or cinnamic acid, sulfonic acids such as laurylsulfonic acid, benzenesulfonic acid, 2-naphthalenesulfonic acid, p-toluenesulfonic acid, In light of any compatible mixture of acids such as methane sulfonic acid, ethane sulfonic acid, hydroxyethane sulfonic acid, cyclohexane sulfonic acid, acids exemplified herein and the ordinary level of skill in the art Desired pharmaceutically acceptable salts can be prepared, such as by treatment with any of the other acids considered to be equivalent or acceptable substitutes and mixtures thereof.
本発明のペプチドが、例えばカルボン酸又はスルホン酸などの酸基を含む場合、適切な方法のいずれか、例えば遊離酸を無機又は有機塩基、例えばアミン(第一級、第二級又は第三級)、アルカリ金属の水酸化物、アルカリ土類金属の水酸化物、本明細書に例として示した塩基などのような塩基の適合し得る任意混合物、及び当該技術分野の技術の通常のレベルに照らして相当物又は許容できる代替物であると見なされる他の塩基及びこれらの混合物のいずれかで処理する方法などで所望の製薬上許容される塩を調製することができる。適切な塩の具体例には、アミノ酸、例えばグリシン及びアルギニンなど、アンモニア、炭酸塩、重炭酸塩、第一級、第二級及び第三級アミン及び環式アミン、例えばベンジルアミン、ピロリジン、ピペリジン、モルホリン及びピペラジンなどから生じさせた有機塩、並びにナトリウム、カルシウム、カリウム、マグネシウム、マンガン、鉄、銅、亜鉛、アルミニウム及びリチウムから生じさせた無機塩が含まれる。代表的な有機塩基又は無機塩基には更に、ベンザチン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、メグルミン、及びプロカインが挙げられる。 Where the peptides of the invention contain acid groups such as carboxylic acids or sulfonic acids, any suitable method such as free acids can be converted to inorganic or organic bases such as amines (primary, secondary or tertiary). ), Any compatible mixture of bases, such as alkali metal hydroxides, alkaline earth metal hydroxides, bases exemplified herein, and the like, to the ordinary level of skill in the art Desired pharmaceutically acceptable salts can be prepared, such as by treatment with any of the other bases and mixtures thereof that are considered equivalents or acceptable substitutes in the light of. Specific examples of suitable salts include amino acids such as glycine and arginine, such as ammonia, carbonate, bicarbonate, primary, secondary and tertiary amines and cyclic amines such as benzylamine, pyrrolidine, piperidine. Organic salts generated from morpholine, piperazine, and the like, and inorganic salts generated from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum, and lithium. Representative organic or inorganic bases further include benzathine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine, and procaine.
本発明はまた、この化合物の製薬上許容されるプロドラッグ及びかかる製薬上許容されるプロドラッグを用いた治療方法にも関する。用語「プロドラッグ」は、被験者に投与した後に化学的又は生理学的方法、例えば生体内で起こる加溶媒分解又は酵素による開裂などで、又は生理学的条件下で当該化合物になる指定化合物の前駆体を意味する。「製薬上許容されるプロドラッグ」は、無毒であるか、生物学的に許容されるか又は他の様式で当該被験者に投与するに生物学的に適したプロドラッグである。適切なプロドラッグ誘導体の選択及び製造の説明的な操作手順は、例えば、「Design of Prodrugs」、H.Bundgaard,Elsevier,1985に説明される。 The invention also relates to pharmaceutically acceptable prodrugs of this compound and methods of treatment using such pharmaceutically acceptable prodrugs. The term “prodrug” refers to a precursor of a specified compound that becomes a compound in a chemical or physiological manner after administration to a subject, such as solvolysis or enzymatic cleavage that occurs in vivo, or under physiological conditions. means. A “pharmaceutically acceptable prodrug” is a prodrug that is non-toxic, biologically acceptable, or otherwise biologically suitable for administration to a subject. An illustrative operating procedure for the selection and preparation of suitable prodrug derivatives is described, for example, in “Design of Prodrugs”, H.M. Bundgaard, Elsevier, 1985.
代表的なプロドラッグとしては、化合物の遊離アミノ、ヒドロキシ、又はカルボン酸基にアミド又はエステル結合を介して共有結合的に連結したアミノ酸残基、又は2つ以上の(例えば、2〜4の)アミノ酸残基のポリペプチド鎖を有する化合物が挙げられる。アミノ酸残基の例には、天然に存在する20種類のアミノ酸(これらは一般に3文字記号で表わされる)ばかりでなく4−ヒドロキシプロリン、ヒドロキシリシン、デモシン、イソデモシン、3−メチルヒスチジン、ノルバリン、ベータ−アラニン、ガンマ−アミノ酪酸、シトルリン、ホモシステイン、ホモセリン、オルニチン及びメチオニンスルホンが含まれる。 Exemplary prodrugs include amino acid residues covalently linked to the free amino, hydroxy, or carboxylic acid group of the compound via an amide or ester bond, or two or more (eg, 2-4). Examples thereof include compounds having a polypeptide chain of amino acid residues. Examples of amino acid residues include 20 naturally occurring amino acids (which are generally represented by three letter symbols) as well as 4-hydroxyproline, hydroxylysine, demosin, isodesomosin, 3-methylhistidine, norvaline, beta -Alanine, gamma-aminobutyric acid, citrulline, homocysteine, homoserine, ornithine and methionine sulfone are included.
追加的な種類のプロドラッグは、例えば、化合物の構造中の遊離カルボキシル基をアミド又はアルキルエステルに誘導体化することにより製造できる。アミドの例には、アンモニア、第一級C1〜6アルキルアミン及び第二級ジ(C1〜6アルキル)アミンから生じさせたアミドが挙げられる。第二級アミンには、5員若しくは6員のヘテロシクロアルキル又はヘテロアリール環部分が挙げられる。アミドの例には、アンモニア、C1〜3アルキル第一級アミン及びジ(C1〜2アルキル)アミンから生じさせたアミドが挙げられる。本発明のエステルの例には、C1〜7アルキル、C5〜7シクロアルキル、フェニル及びフェニル(C1〜6アルキル)エステルが挙げられる。好適なエステルにはメチルエステルが挙げられる。プロドラッグはまた、Fleisherら、Adv.Drug Delivery Rev.1996,19,115〜130に概説されるもの等の手順の後、ヘミスクシネート、リン酸エステル、ジメチルアミノアセテート、及びホスホリルオキシメチルオキシカルボニルを含む基を使用して、遊離ヒドロキシ基を誘導体化しても調製できる。ヒドロキシ及びアミノ基のカルバメート誘導体も、プロドラッグを産出することができる。また、ヒドロキシ基のカーボネート誘導体、スルホン酸エステル及び硫酸エステルもプロドラッグをもたらし得る。また、ヒドロキシ基に誘導体化を(アシロキシ)−メチル及び(アシロキシ)−エチルエーテル(このアシル基はアルキルエステルであってもよく、場合により1個以上のエーテル、アミン若しくはカルボン酸官能で置換されていてもよいか、又はアシル基は上述したようなアミノ酸エステルである)として受けさせることもプロドラッグを生じさせるに有効である。この種のプロドラッグは、Greenwaldら、J Med Chem.1996,39,10,1938〜40に記述されているように調製することができる。遊離アミンもまた、アミド、スルホンアミド又はホスホンアミドとして誘導体化することができる。そのようなプロドラッグ部分の全部にエーテル、アミン及びカルボン酸官能を包含する基が組み込まれている可能性がある。 Additional types of prodrugs can be made, for example, by derivatizing free carboxyl groups in the structure of the compound to amides or alkyl esters. Examples of amides include amides generated from ammonia, primary C 1-6 alkyl amines and secondary di (C 1-6 alkyl) amines. Secondary amines include 5- or 6-membered heterocycloalkyl or heteroaryl ring moieties. Examples of amides include amides generated from ammonia, C1-3 alkyl primary amines and di ( C1-2 alkyl) amines. Examples of esters of the present invention include C 1-7 alkyl, C 5-7 cycloalkyl, phenyl and phenyl (C 1-6 alkyl) esters. Suitable esters include methyl esters. Prodrugs are also described in Fleisher et al., Adv. Drug Delivery Rev. Following procedures such as those outlined in 1996, 19, 115-130, groups containing hemisuccinate, phosphate ester, dimethylaminoacetate, and phosphoryloxymethyloxycarbonyl may be used to derivatize the free hydroxy group. Can be prepared. Carbamate derivatives of hydroxy and amino groups can also produce prodrugs. Hydroxy carbonate derivatives, sulfonate esters and sulfate esters can also provide prodrugs. Alternatively, the hydroxy group may be derivatized with (acyloxy) -methyl and (acyloxy) -ethyl ether (this acyl group may be an alkyl ester, optionally substituted with one or more ether, amine or carboxylic acid functions. Or the acyl group is an amino acid ester as described above) is also effective to produce a prodrug. This type of prodrug is described in Greenwald et al., J Med Chem. 1996, 39, 10, 1938-40. Free amines can also be derivatized as amides, sulfonamides or phosphonamides. All such prodrug moieties may incorporate groups including ether, amine and carboxylic acid functions.
本発明は更に、本発明の方法に使用され得る化合物の薬学的に活性な代謝物に関する。「薬学的に活性な代謝物」とは、化合物又はその塩の体内の代謝による薬学的に活性な生成物を意味する。化合物のプロドラッグ及び活性な代謝産物の測定は当該技術分野で既知又は利用可能な常規技術を用いて実施可能である。例えば、Bertoliniら、J Med Chem.1997,40,2011〜2016;Shanら、J Pharm Sci.1997,86(7),765〜767;Bagshawe、Drug Dev Res.1995,34,220〜230;Bodor、Adv Drug Res.1984,13,224〜331;Bundgaard、Design of Prodrugs(Elsevier Press,1985);及びLarsen、Design and Application of Prodrugs,Drug Design and Development(Krogsgaard−Larsenら、編、Harwood Academic Publishers,1991)を参照のこと。 The invention further relates to pharmaceutically active metabolites of compounds that can be used in the methods of the invention. “Pharmaceutically active metabolite” means a pharmaceutically active product of metabolism of a compound or salt thereof in the body. Determination of compound prodrugs and active metabolites can be performed using routine techniques known or available in the art. See, for example, Bertolini et al., J Med Chem. 1997, 40, 2011-2016; Shan et al., J Pharm Sci. 1997, 86 (7), 765-767; Bagshawe, Drug Dev Res. 1995, 34, 220-230; Bodor, Adv Drug Res. 1984, 13, 224-331; Bundgaard, Design of Prodrugs (Elsevier Press, 1985); and Larsen, Design and Application of Prodrug, Drug Design. thing.
医薬組成物
本発明の特定の実施形態において、CRHR2ペプチド作動薬は、単独で、又は1つ以上の追加成分と組み合わせて、医薬組成物を処方するのに使用される。医薬組成物は本発明による少なくとも1種の活性薬剤を有効量含有する。
Pharmaceutical Compositions In certain embodiments of the invention, the CRHR2 peptide agonist is used to formulate a pharmaceutical composition alone or in combination with one or more additional ingredients. The pharmaceutical composition contains an effective amount of at least one active agent according to the present invention.
いくつかの実施形態において、この医薬組成物は、配列番号29に示されるアミノ酸配列を有するペプチドを含み、ここにおいてこのペプチドはカルボキシ末端にCONH2を含み、更に、ポジション28でシステイン残基に結合しているN−エチルスクシンイミドリンカー又はアセトアミドリンカーを含み、ここにおいてこのリンカーはPEG部分にも連結している。PEG部分は、その分子量及び物理的特性(例えば直鎖か分枝鎖か)によって分類され、PEGをペプチド基質に結合させるのに使用する1つ以上のリンカー部分を含む。特定の好ましい実施形態において、このペプチドは1つ又は2つのリンカーを含む。 In some embodiments, the pharmaceutical composition comprises a peptide having the amino acid sequence set forth in SEQ ID NO: 29, wherein the peptide comprises CONH 2 at the carboxy terminus and further bound to a cysteine residue at position 28. N-ethylsuccinimide linker or acetamide linker, wherein the linker is also linked to the PEG moiety. A PEG moiety is classified by its molecular weight and physical properties (eg, linear or branched) and includes one or more linker moieties used to attach PEG to the peptide substrate. In certain preferred embodiments, the peptide comprises one or two linkers.
特定の実施形態において、このPEG部分を含む医薬組成物は、約2kDa〜約80kDaの範囲の重量であり得るPEG部分を含み得る。特定の実施形態において、PEG部分の質量は約2kDaである。更なる実施形態において、PEG質量は約5kDaである。特定の実施形態において、PEG質量は約12kDaである。特定の実施形態において、PEG質量は約20kDaである。特定の実施形態において、PEG質量は約30kDaである。特定の実施形態において、PEG質量は約40kDaである。特定の実施形態において、PEG質量は約80kDaである。こうした組成物は更に製薬上許容される賦形剤を含んでもよい。 In certain embodiments, a pharmaceutical composition comprising the PEG moiety can include a PEG moiety that can weigh in a range of about 2 kDa to about 80 kDa. In certain embodiments, the mass of the PEG moiety is about 2 kDa. In a further embodiment, the PEG mass is about 5 kDa. In certain embodiments, the PEG mass is about 12 kDa. In certain embodiments, the PEG mass is about 20 kDa. In certain embodiments, the PEG mass is about 30 kDa. In certain embodiments, the PEG mass is about 40 kDa. In certain embodiments, the PEG mass is about 80 kDa. Such a composition may further comprise a pharmaceutically acceptable excipient.
本開示は更に、本明細書に記述されている化合物又は誘導体と、1つ以上の製薬上許容される担体、賦形剤、及び希釈剤とを含む、組成物(医薬組成物を含む)も提供する。本発明の特定の実施形態において、組成物は更に、少量の湿潤剤若しくは乳化剤、又はpH緩衝剤を含み得る。特定の一実施形態において、この医薬組成物は、ヒトに投与するのに製薬上許容される。特定の実施形態において、この医薬組成物は、本明細書に記述される化合物又は誘導体の治療的有効量又は予防的有効量を含む。治療的に有効又は予防的に有効となる、本発明の化合物又は誘導体の量は、標準の臨床的技法によって決定することができる。代表的な有効量は、下記のセクションで詳しく記述される。本発明の特定の実施形態において、組成物は、安定剤も含み得る。安定剤は、組成物の修飾ペプチドの化学的劣化速度を遅くする化合物である。好適な安定剤には、酸化防止剤(アスコルビン酸など)、pH緩衝剤、又は塩緩衝剤が挙げられるがこれらに限定されない。 The present disclosure further includes compositions (including pharmaceutical compositions) comprising a compound or derivative described herein and one or more pharmaceutically acceptable carriers, excipients, and diluents. provide. In certain embodiments of the invention, the composition may further comprise a small amount of a wetting or emulsifying agent, or a pH buffering agent. In one particular embodiment, the pharmaceutical composition is pharmaceutically acceptable for administration to humans. In certain embodiments, the pharmaceutical composition comprises a therapeutically or prophylactically effective amount of a compound or derivative described herein. The amount of a compound or derivative of the invention that is therapeutically or prophylactically effective can be determined by standard clinical techniques. Representative effective amounts are described in detail in the following sections. In certain embodiments of the invention, the composition may also include a stabilizer. A stabilizer is a compound that slows the rate of chemical degradation of the modified peptide of the composition. Suitable stabilizers include, but are not limited to, antioxidants (such as ascorbic acid), pH buffers, or salt buffers.
医薬組成物は、被験者、特にヒト被験者に投与するのに好適な任意の形態であり得る。特定の実施形態において、この組成物は溶液、懸濁液、乳濁液、錠剤、丸薬、カプセル、粉末、及び持続放出性製剤の形態である。この組成物は更に、特定の単位剤形であり得る。単位剤形の例としては、錠剤;キャプレット;カプセル(例えばソフト弾性ゼラチンカプセル);カシェ剤;トローチ剤;口内錠;分散剤;座薬;軟膏;パップ剤(湿布);ペースト;粉末;包帯;クリーム;ギプス;溶液;パッチ;エアロゾル(例えば鼻スプレー又は吸入器);ゲル;患者に経口又は粘膜投与するのに好適な液体投与形態(懸濁液(例えば水性又は非水性の液体懸濁液、水中油型エマルション、又は油中水型液体エマルション)、溶液、及びエリキシル剤を含む);被験者に非経口投与するのに好適な液体投与形態;被験者に非経口投与するのに好適な液体投与形態に還元できる滅菌固体(例えば結晶又はアモルファス固体)が挙げられるがこれらに限定されない。 The pharmaceutical composition can be in any form suitable for administration to a subject, particularly a human subject. In certain embodiments, the composition is in the form of a solution, suspension, emulsion, tablet, pill, capsule, powder, and sustained release formulation. The composition may further be a specific unit dosage form. Examples of unit dosage forms include tablets; caplets; capsules (eg, soft elastic gelatin capsules); cachets; lozenges; lozenges; dispersions; suppositories; ointments; Creams; casts; solutions; patches; aerosols (eg nasal sprays or inhalers); gels; liquid dosage forms suitable for oral or mucosal administration to patients (suspensions (eg aqueous or non-aqueous liquid suspensions) Oil-in-water emulsion or water-in-oil liquid emulsion), solutions, and elixirs); liquid dosage forms suitable for parenteral administration to subjects; liquid dosage forms suitable for parenteral administration to subjects Examples include, but are not limited to, sterile solids (eg, crystalline or amorphous solids) that can be reduced.
特定の一実施形態において、被験者は、ウシ、ウマ、ヒツジ、ブタ、家禽、ネコ、イヌ、マウス、ラット、ウサギ、又はモルモットなどの哺乳類である。好ましい一実施形態では、被験者はヒトである。好ましくは、この医薬組成物は獣医学的投与及び/又はヒトへの投与に好適である。この実施形態に従い、用語「製薬上許容される」とは、動物における使用について、及びより具体的にはヒトにおける利用について、連邦政府又は州政府の規制機関によって認可されていること、又は、米国薬局方若しくは他の一般に承認されている薬局方に記載されていることを意味する。 In one particular embodiment, the subject is a mammal such as a cow, horse, sheep, pig, poultry, cat, dog, mouse, rat, rabbit, or guinea pig. In a preferred embodiment, the subject is a human. Preferably, the pharmaceutical composition is suitable for veterinary administration and / or administration to humans. In accordance with this embodiment, the term “pharmaceutically acceptable” means approved by a federal or state regulatory agency for use in animals, and more specifically for use in humans, or the United States Means that it is listed in the pharmacopoeia or other generally approved pharmacopoeia.
この組成物における使用に好適な製薬担体は、水及び油(石油、動物、植物又は合成由来のものを含む)などの滅菌液体である。特定の一実施形態において、この油はピーナッツオイル、大豆油、鉱物油、又はゴマ油である。医薬組成物が静注投与される場合には、水が好ましい担体である。生理食塩水並びに水性デキストロース及びグリセロール溶液も、特に注射溶液用の液体担体として採用され得る。好適な製薬担体の更なる例は、当該技術分野において既知であり、例えばE.W.MartinによるRemington’s Pharmaceutical Sciences(1990)18th ed.(Mack Publishing,Easton Pa.)に記述されている。 Pharmaceutical carriers suitable for use in the composition are sterile liquids such as water and oils, including those of petroleum, animal, vegetable or synthetic origin. In one particular embodiment, the oil is peanut oil, soybean oil, mineral oil, or sesame oil. Water is a preferred carrier when the pharmaceutical composition is administered intravenously. Saline solutions and aqueous dextrose and glycerol solutions can also be employed as liquid carriers, particularly for injectable solutions. Further examples of suitable pharmaceutical carriers are known in the art, e.g. W. Martin by Remington's Pharmaceutical Sciences (1990) 18th ed. (Mack Publishing, Easton Pa.).
この組成物に使用するのに好適な賦形剤としては、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、コメ、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、モノステアリン酸グリセロール、タルク、塩化ナトリウム、乾燥スキムミルク、グリセロール、プロピレン、グリコール、水、及びエタノールが挙げられる。特定の賦形剤が医薬組成物への組み込みに好適かどうかは、当該技術分野において周知のさまざまな要素に依存し、例えばその組成物の投与経路及び特定の活性成分が挙げられるがこれらに限定されない。 Suitable excipients for use in this composition include starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, Examples include dry skim milk, glycerol, propylene, glycol, water, and ethanol. Whether a particular excipient is suitable for incorporation into a pharmaceutical composition depends on various factors well known in the art, including but not limited to the route of administration of the composition and the particular active ingredient. Not.
本発明の特定の実施形態において、組成物は無水組成物である。無水組成物は、無水物又は低水分含有の成分と、低水分又は低湿度条件とを用いて調製することができる。第一級アミン又は第二級アミンを有する修飾ペプチドを含む組成物は、製造中、パッケージング中、及び/又は保管中に水分及び/又は湿度に実質的に接触することが予測される場合には、好ましくは無水物である。無水組成物は、無水性が維持されるように調製及び保管されるべきである。したがって、無水組成物は好ましくは、好適な製剤キット内に含まれ得るよう、水への曝露を防ぐことが知られている材料を使用してパッケージされる。好適なパッケージングの例には、例えば密封されたホイル、プラスチック、単位用量容器(例えばバイアル)、ブリスターパック、及びストリップパックが挙げられるがこれらに限定されない。 In certain embodiments of the invention, the composition is an anhydrous composition. An anhydrous composition can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions. A composition comprising a modified peptide having a primary amine or secondary amine is expected to be substantially in contact with moisture and / or humidity during manufacture, packaging, and / or storage. Is preferably an anhydride. An anhydrous composition should be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are preferably packaged using materials known to prevent exposure to water so that they can be included in suitable formulation kits. Examples of suitable packaging include, but are not limited to, sealed foils, plastics, unit dose containers (eg, vials), blister packs, and strip packs.
本明細書に記述されている化合物若しくは誘導体、又はその製薬上許容される塩及び溶媒和物を含む医薬組成物は、意図された投与経路に適合するよう製剤化される。この製剤は、好ましくは皮下投与用であるが、例えば吸入又は吹き込み(口又は鼻を介して)、皮内、経口、頬、非経口、経膣、又は直腸などの他の手段による投与用であり得る。好ましくは、この組成物は更に、保管及び輸送中に化合物の化学的安定性の改善をもたらすよう製剤化される。この製剤は、凍結乾燥することができ、又は液体製剤であってよい。 A pharmaceutical composition comprising a compound or derivative described herein, or a pharmaceutically acceptable salt and solvate thereof, is formulated to be compatible with its intended route of administration. This formulation is preferably for subcutaneous administration, but for administration by other means such as inhalation or insufflation (via mouth or nose), intradermal, oral, buccal, parenteral, vaginal, or rectal. possible. Preferably, the composition is further formulated to provide improved chemical stability of the compound during storage and transportation. The formulation can be lyophilized or can be a liquid formulation.
一実施形態において、この化合物又は誘導体は静脈内投与用に製剤される。静脈内製剤には、生理食塩水溶液などの標準担体が含まれ得る。別の一実施形態において、この化合物又は誘導体は注射用に製剤される。好ましい一実施形態において、この化合物又は誘導体は滅菌凍結乾燥された製剤であり、汚染性の細胞性物質、化学物質、ウイルス、又は毒素を実質的に含まない。特定の一実施形態において、この化合物又は誘導体は、液体形態で製剤される。別の特定の一実施形態において、注射用の製剤は、滅菌された一回用量容器に供給される。特定の一実施形態において、注射用の製剤は、滅菌された一回用量容器に供給される。この製剤は、追加の防腐剤を含んでも含まなくともよい。液体製剤は、油性又は水性溶媒中の懸濁液、溶液又は乳濁液としての形態をとることができ、懸濁剤、安定剤及び/又は分散剤などの処方剤を含み得る。 In one embodiment, the compound or derivative is formulated for intravenous administration. Intravenous formulations can include standard carriers such as saline solution. In another embodiment, the compound or derivative is formulated for injection. In a preferred embodiment, the compound or derivative is a sterile lyophilized formulation and is substantially free of contaminating cellular material, chemicals, viruses, or toxins. In one particular embodiment, the compound or derivative is formulated in a liquid form. In another particular embodiment, the injectable formulation is supplied in a sterile single dose container. In one particular embodiment, the injectable formulation is supplied in a sterile single dose container. This formulation may or may not contain additional preservatives. Liquid formulations can take the form of suspensions, solutions or emulsions in oily or aqueous solvents and may contain formulatory agents such as suspending, stabilizing and / or dispersing agents.
投与方法
本明細書に記述されている化合物若しくは誘導体、又はそれらの製薬上許容される塩は、好ましくは、所望により製薬上許容される溶媒を含む組成物の構成要素として投与される。この化合物又は誘導体は、好ましくは皮下に投与される。別の好ましい投与方法は、化合物又は誘導体の静脈内注射又は連続的静脈内点滴を介した方法である。好ましくは、この投与は、化合物又は誘導体のゆっくりした全身吸収及びクリアランスにより血漿中濃度が安定状態に達し、又は一定時間の所定範囲においてある血漿中濃度レベルを維持する。
Methods of Administration The compounds or derivatives described herein, or pharmaceutically acceptable salts thereof, are preferably administered as a component of a composition that optionally includes a pharmaceutically acceptable solvent. This compound or derivative is preferably administered subcutaneously. Another preferred method of administration is via intravenous injection or continuous intravenous infusion of the compound or derivative. Preferably, this administration achieves a stable plasma concentration due to slow systemic absorption and clearance of the compound or derivative, or maintains a plasma concentration level within a predetermined range of time.
特定の実施形態において、この化合物又は誘導体は、任意の他の簡易経路、例えば注入又はボーラス注射、又は上皮若しくは皮膚粘膜被覆(例えば口内粘膜、直腸、及び腸粘膜)を介した吸収によって投与される。投与方法には、非経口、皮内、筋肉内、腹膜内、静脈内、皮下、鼻孔内、硬膜外、経口、舌下、鼻孔内、大脳内、膣内、経皮、直腸、吸入により、又は局所的に、特に耳、鼻、目若しくは皮膚に対しての投与が含まれるがこれらに限定されない。多くの場合において、投与は、化合物又は誘導体の血中内への放出を生じる。 In certain embodiments, the compound or derivative is administered by any other convenient route, such as infusion or bolus injection, or absorption through epithelial or dermal mucosal coatings (eg, oral mucosa, rectum, and intestinal mucosa). . The methods of administration are parenteral, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, epidural, oral, sublingual, intranasal, intracerebral, intravaginal, transdermal, rectal, by inhalation. Or topically, particularly but not limited to ear, nose, eye or skin administration. In many cases, administration results in the release of the compound or derivative into the blood.
そのような製剤の形態は錠剤、カプセル、小袋、糖衣錠、粉末、顆粒、ロゼンジ、再構成用粉末、液状製剤又は座薬であってもよい。好ましくは、組成物は静脈内注入若しくはボーラス注射、皮下注入若しくはボーラス注射、又は筋肉内注射用に製剤される。 Such dosage forms may be tablets, capsules, sachets, dragees, powders, granules, lozenges, powders for reconstitution, liquid preparations or suppositories. Preferably, the composition is formulated for intravenous or bolus injection, subcutaneous injection or bolus injection, or intramuscular injection.
化合物は好ましくは非経口経路で投与される。例えば、組成物を直腸投与の目的で座薬として構築してもよい。静脈内、筋肉内、腹腔内又は皮下経路を包含する非経口用途の場合、本発明の薬剤を適切なpH及び等張性になるように緩衝剤を入れておいた無菌の水溶液若しくは懸濁液又は非経口的に許容される油として提供してもよい。好ましい水性溶媒には、リンゲル溶液、デキストロース溶液、及び等張性塩化ナトリウムが挙げられる。そのような形態物を単位用量形態物、例えばアンプル又は使い捨て可能注射デバイス、複数単位用量形態、例えば適切な投与量を取り出すことが可能なバイアル瓶など、又は注射可能製剤を製造する目的で使用可能な固体形態物若しくは予濃縮液の形態で提供してもよい。例示した注入用量は、数分から数日間の範囲の期間にわたって投与してもよい。更に別の実施形態において、本発明のペプチドの有効量を、皮下送達に好適なナノ粒子上にコーティングすることができ、又は「貯留物(depot)」内に提供することができる(Hawkinsら、Adv Drug Deliv Rev.,2008,vol.60,pp.876〜885;Montalvoら、Nanotechnology,2008,vol.19,pp.1〜7)。 The compound is preferably administered by a parenteral route. For example, the composition may be constructed as a suppository for rectal administration. For parenteral use, including intravenous, intramuscular, intraperitoneal or subcutaneous routes, a sterile aqueous solution or suspension containing the agent of the present invention in a buffer to ensure proper pH and isotonicity Alternatively, it may be provided as a parenterally acceptable oil. Preferred aqueous solvents include Ringer's solution, dextrose solution, and isotonic sodium chloride. Such forms can be used for the purpose of producing unit dose forms such as ampoules or disposable injection devices, multiple unit dose forms such as vials from which appropriate doses can be removed, or injectable formulations May be provided in the form of a solid form or a preconcentrated liquid. Exemplary infusion doses may be administered over a period ranging from minutes to days. In yet another embodiment, an effective amount of a peptide of the invention can be coated onto nanoparticles suitable for subcutaneous delivery, or provided within a “depot” (Hawkins et al., Adv Drug Deliv Rev., 2008, vol. 60, pp. 876-885; Montalvo et al., Nanotechnology, 2008, vol. 19, pp. 1-7).
活性薬剤は、吸入手法を通じて投与することができる。そのような方法では、乾燥粉末(Johnson K.A.、Adv Drug Del Rev.,1997,vol.26(1),pp.3〜15)及び/又はエアロゾル(Sangwanら、J Aerosol Med.,2001,vol.14(2),pp.185〜195;PCT国際公開特許WO2002/094342号)の製剤技法を使用することができる。 The active agent can be administered through an inhalation procedure. Such methods include dry powder (Johnson KA, Adv Drug Del Rev., 1997, vol. 26 (1), pp. 3-15) and / or aerosol (Sangwan et al., J Aerosol Med., 2001). , Vol.14 (2), pp.185-195; PCT International Patent Publication No. WO2002 / 094342).
本発明による治療方法の実施形態において、本発明による少なくとも1つの活性薬剤の治療的有効量は、例えば心不全、糖尿病、骨格筋消費、及び筋肉減少症などの疾病、疾患又は医学的状態に苦しむ被験者、又はそれらを有すると診断された被験者に投与される。追加の状態としては、不適切な運動活動、食事摂取、又は心臓保護、気管支弛緩、及び/若しくは抗炎症活性の必要が挙げられる。本発明の活性薬剤の治療的有効量若しくは用量を常規方法、例えばモデリング、用量漸増試験又は臨床試験など、及び常規要因、例えば投与様式若しくは経路又は薬剤送達など、薬剤の薬物動態、疾病、疾患、及び状態のひどさ及び過程、被験体が以前又は現在受けている治療、被験体の健康状態及び薬剤に対する反応及び治療を施す医者の判断などを考慮に入れることで確定することができる。代表的な静脈内投与速度は、被験者の体重1kg当たり毎分、ストレスコピン関連活性薬剤を約0.2ng〜約52ngの範囲であり、好ましくは約0.2ng/kg/分〜約22ng/kg/分、又は同等に、1日当たり約0.3μg/kg〜約32μg/kgである。ボーラス注入又は皮下注射の場合、合計投与量は、単回用量単位、又は分割した用量単位で投与することができる(例えばBID、TID、QID、週2回、2週間に1回、又は月1回)。体重70kgのヒトでは、好適な用量の代表的な範囲は、約1μg/日〜約1mg/日である。毎日投与の代わりに、毎週用量レジメンを使用することができる。別の好ましい一実施形態において、約20kDaのPEGがペプチド配列のポジション28でシステイン残基に結合しているアセトアミドリンカーを含む配列番号102のCRHR2ペプチド作動薬が、ボーラス皮下注射によって、10μg/kgの用量で、これを必要としている患者に投与される。この用量の頻度は、被験者の治療的必要性とその他の臨床的考慮事項に応じて、1日1回〜それより少ない頻度の範囲となるべきである。 In an embodiment of the method of treatment according to the invention, the therapeutically effective amount of at least one active agent according to the invention is a subject suffering from a disease, disorder or medical condition such as heart failure, diabetes, skeletal muscle consumption, and muscle loss. Or administered to a subject diagnosed with them. Additional conditions may include inappropriate athletic activity, dietary intake, or the need for cardioprotection, bronchial relaxation, and / or anti-inflammatory activity. Therapeutically effective amounts or doses of the active agents of the invention can be determined by routine methods such as modeling, dose escalation or clinical trials, and routine factors such as mode of administration or route or drug delivery, drug pharmacokinetics, disease, disease, And the severity and course of the condition, the treatment that the subject has received before or at present, the health status of the subject and the response to the medication and the judgment of the treating physician, etc. Typical intravenous administration rates range from about 0.2 ng to about 52 ng of stress copine-related active agents per minute per kg body weight of the subject, preferably from about 0.2 ng / kg / min to about 22 ng / kg. / Min, or equivalently, from about 0.3 μg / kg to about 32 μg / kg per day. In the case of bolus injection or subcutaneous injection, the total dose can be administered in a single dose unit or in divided dose units (eg BID, TID, QID, twice weekly, once every two weeks, or monthly Times). For a human weighing 70 kg, a typical range of suitable doses is from about 1 μg / day to about 1 mg / day. Instead of daily administration, a weekly dosage regimen can be used. In another preferred embodiment, the CRHR2 peptide agonist of SEQ ID NO: 102 comprising an acetamide linker in which about 20 kDa PEG is attached to a cysteine residue at position 28 of the peptide sequence is administered at 10 μg / kg by bolus subcutaneous injection. Dosage is administered to patients in need thereof. The frequency of this dose should range from once a day to less frequently, depending on the subject's therapeutic needs and other clinical considerations.
患者の疾病、疾患又は状態の改善が生じたならば投与量を予防的治療又は維持治療に適した量に調整してもよい。例えば、投与の量及び頻度又は両方を症状の関数として所望の治療若しくは予防効果が維持される度合にまで少なくしてもよい。症状が適切なレベルにまで改善したならば治療を停止してもよい。しかしながら、症状がいくらか再発する時には患者に長期を基準にした断続的治療を受けさせる必要がある。 Once improvement of the patient's disease, disorder or condition has occurred, the dosage may be adjusted to an amount suitable for prophylactic or maintenance treatment. For example, the dose and / or frequency of administration may be reduced to the extent that the desired therapeutic or prophylactic effect is maintained as a function of symptoms. Treatment may be stopped once symptoms have improved to an appropriate level. However, patients need intermittent treatment on a long-term basis when some symptoms recur.
特定の実施形態において、この化合物は、治療レジメンの一部として1つ以上の他の生物学的活性薬剤と組み合わせて投与される。特定の実施形態において、この化合物は、1つ以上の他の生物学的活性薬剤の投与の前に、同時に、又は後に投与される。一実施形態において、1つ以上の他の生物学的活性薬剤は、本明細書に記述される化合物を含む同じ医薬組成物内で投与される。別の一実施形態において、1つ以上の他の生物学的活性薬剤は、本明細書に記述される化合物とは別の医薬組成物内で投与される。この実施形態にしたがって、1つ以上の他の生物学的活性薬剤は、この化合物の投与に使用される投与経路と同じ経路又は異なる経路によって被験者に投与され得る。 In certain embodiments, the compound is administered in combination with one or more other biologically active agents as part of a treatment regimen. In certain embodiments, the compound is administered before, simultaneously with, or after administration of one or more other biologically active agents. In one embodiment, one or more other biologically active agents are administered within the same pharmaceutical composition comprising a compound described herein. In another embodiment, one or more other biologically active agents are administered in a pharmaceutical composition that is separate from the compounds described herein. In accordance with this embodiment, one or more other biologically active agents can be administered to the subject by the same route as the route of administration used to administer the compound or by a different route.
別の一実施形態において、心臓血管疾患のリスク低減又は治療のために、この化合物は1つ以上の他の化合物又は組成物と共に投与される。心臓血管疾患のリスクを低減又は治療する化合物又は組成物には、抗炎症剤、抗血栓剤、抗血小板剤、繊維素溶解剤、血栓溶解剤、脂質低減剤、トロンビン直接阻害剤、抗Xa阻害剤、抗IIa阻害剤、糖タンパク質IIb/IIIa受容体阻害剤及びトロンビン直接阻害剤が挙げられるがこれらに限定されない。本明細書に記述されている化合物と組み合わせて投与され得る薬剤の例には、ビバリルジン、ヒルジン、ヒルゲン、Angiomax、アガトロバン、PPACK、トロンビンアプタマー、アスピリン、GPIlb/IIIa阻害剤(例えばIntegrelin)、P2Y12阻害剤、チエノピリジン、チクロピジン、及びクロピドグレルが挙げられる。 In another embodiment, the compound is administered with one or more other compounds or compositions for risk reduction or treatment of cardiovascular disease. Compounds or compositions that reduce or treat cardiovascular disease risk include anti-inflammatory agents, antithrombotic agents, antiplatelet agents, fibrinolytic agents, thrombolytic agents, lipid reducing agents, thrombin direct inhibitors, anti-Xa inhibitors Agents, anti-IIa inhibitors, glycoprotein IIb / IIIa receptor inhibitors and thrombin direct inhibitors, but are not limited to these. Examples of agents that can be administered in combination with the compounds described herein include bivalirudin, hirudin, hirugen, Angiomax, agatroban, PPACK, thrombin aptamer, aspirin, GPIlb / IIIa inhibitor (eg, Integrelin), P2Y12 inhibition Agents, thienopyridine, ticlopidine, and clopidogrel.
実施形態において、この化合物は、必要としている患者に投与するのに好適な剤形に製剤される。薬剤及び担体粒子の調製のためのプロセス及び機器については、Pharmaceutical Sciences,Remington,17th Ed.,pp.1585〜1594(1985);Chemical Engineers Handbook,Perry,6th Ed.,pp.21−13〜21−19(1984);Journal of Pharmaceutical Sciences,Parrot,Vol.61,No.6,pp.813〜829(1974);及びChemical Engineer,Hixon,pp.94〜103(1990)に開示されている。 In embodiments, the compound is formulated into a dosage form suitable for administration to a patient in need. For processes and equipment for the preparation of drug and carrier particles, see Pharmaceutical Sciences, Remington, 17th Ed. , Pp. 1585-1594 (1985); Chemical Engineers Handbook, Perry, 6th Ed. , Pp. 21-13-21-19 (1984); Journal of Pharmaceutical Sciences, Parrot, Vol. 61, no. 6, pp. 813-829 (1974); and Chemical Engineer, Hixon, pp. 94-103 (1990).
本発明の剤形に組み込まれる化合物の量は、一般に治療適応及び望ましい投与期間(例えば12時間おき、24時間おきなど)に応じて、組成物の重量を基準として約10重量%〜約90重量%で変化し得る。投与したい化合物の用量に応じて、1つ以上の剤形で投与することができる。製剤によっては、この化合物は好ましくは、HCl塩形態、又は遊離塩基形態となる。 The amount of compound incorporated into the dosage forms of the invention will generally range from about 10% to about 90% by weight based on the weight of the composition, depending on the therapeutic indication and the desired period of administration (eg, every 12 hours, every 24 hours, etc.). % Can vary. Depending on the dose of the compound desired to be administered, it can be administered in one or more dosage forms. Depending on the formulation, the compound is preferably in the HCl salt form or the free base form.
更に、本発明はまた、ヒト又はヒト以外の動物体の治療又は診断方法での使用について前述した、医薬組成物又は医薬剤形にも関する。 Furthermore, the present invention also relates to a pharmaceutical composition or pharmaceutical dosage form as described above for use in a method of treatment or diagnosis of a human or non-human animal body.
本発明は更に、治療を必要としている哺乳類に経口投与するための医薬剤形の製造における使用のための医薬組成物に関し、その剤形が、その哺乳類による食事摂取とは独立に、1日のうちいつでも投与できることを特徴とする。 The present invention further relates to a pharmaceutical composition for use in the manufacture of a pharmaceutical dosage form for oral administration to a mammal in need of treatment, wherein the dosage form is independent of daily consumption by the mammal. It can be administered at any time.
本発明は、ヒト又はヒト以外の動物体の治療又は診断方法に関し、これはその動物体に、本明細書に記述されている医薬組成物の治療的又は診断的有効量を投与することを含む。 The present invention relates to a method of treatment or diagnosis of a human or non-human animal body, which comprises administering to the animal body a therapeutically or diagnostically effective amount of a pharmaceutical composition described herein. .
本発明は更に、本明細書で記述される容器、剤形を含む、商業的販売に好適な医薬品パッケージに関し、そのパッケージに、その剤形が食事と共に又は食事なしで投与できるかどうかについてなど、非限定的な書面を伴う。 The invention further relates to a pharmaceutical package suitable for commercial sale comprising the container, dosage form described herein, whether the dosage form can be administered with or without a meal, etc. With non-limiting documentation.
下記の製剤実施例は説明目的に限定され、本発明の範囲をいかなる意味でも制限するものではない。 The following formulation examples are limited for illustrative purposes and are not intended to limit the scope of the invention in any way.
実施例1:ペプチドの合成と精製
配列番号29のペプチドが、Rainin Symphonyマルチプルペプチド合成装置(モデルSMPS−110)上でソフトウェアバージョン3.3.0を使用して、固相ペプチド合成反応によって調製された。ペプチドアミドの合成に使用された樹脂(NovaSyn TGR(登録商標)、440mg、約0.1mmole、0.23mmol/g置換、ロットNo.A33379)は、酸不安定性の修飾されたRinkアミドリンカーで機能付加されたポリエチレングリコールとポリスチレンの複合物であった。
Example 1: Peptide Synthesis and Purification The peptide of SEQ ID NO: 29 was prepared by solid phase peptide synthesis reaction using software version 3.3.0 on a Rainin Symphony multiple peptide synthesizer (model SMPS-110). It was. Resin used for peptide amide synthesis (NovaSyn TGR®, 440 mg, about 0.1 mmole, 0.23 mmol / g substitution, lot No. A33379) functions with acid labile modified Rink amide linker It was a composite of added polyethylene glycol and polystyrene.
合成に使用されたアミノ酸は、C末端にNα−9−フルオレニルメトキシカルボニル(Fmoc)保護基、及び次の側鎖保護基を含んでいた:Arg(2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル、pbf)、Asp(第三級ブトキシ、OtBu)、Asn(トリチル、Trt)、Gln(Trt)、Cys(Trt)、His(Trt)、Lys(t−ブトキシカルボニル、Boc)、Ser(第三級ブチル、tBu)及びThr(tBu)。 The amino acids used in the synthesis contained a Nα-9-fluorenylmethoxycarbonyl (Fmoc) protecting group at the C-terminus and the following side chain protecting group: Arg (2,2,4,6,7- Pentamethyldihydrobenzofuran-5-sulfonyl, pbf), Asp (tertiary butoxy, OtBu), Asn (trityl, Trt), Gln (Trt), Cys (Trt), His (Trt), Lys (t-butoxycarbonyl) , Boc), Ser (tertiary butyl, tBu) and Thr (tBu).
結合反応は次のように実施された:N−メチルピロリジノン(NMP)膨潤済み樹脂(0.1mmole)、DMF(2.5mL)中5倍モル過剰のFmocアミノ酸及び5倍モル過剰のヘキサフルオロリン酸(HBTU)を混合し、DMF(2.5mL)中10倍モル過剰のN−メチルモルホリン(NMM)を加えてから、45分間にわたって結合させた。Fmoc除去のため、反応物を20%ピペリジン/DMF溶液と共に2分間インキュベートした。この溶液を排水し、新たに20%ピペリジン/DMFを加え、18分間インキュベートした。次に反応物をNMPで洗い、続いて、結合工程を繰り返すことによりアミノ酸付加を実施した。C末端を、N末端からの番号でIle40、Gln39、Asn19、Asn12、及びVal9に結合させるため、結合工程を2回実施した。 The conjugation reaction was performed as follows: N-methylpyrrolidinone (NMP) swollen resin (0.1 mmole), 5-fold molar excess of Fmoc amino acid and 5-fold molar excess of hexafluorophosphorus in DMF (2.5 mL). Acid (HBTU) was mixed and a 10-fold molar excess of N-methylmorpholine (NMM) in DMF (2.5 mL) was added prior to binding for 45 minutes. The reaction was incubated with 20% piperidine / DMF solution for 2 minutes for Fmoc removal. The solution was drained and fresh 20% piperidine / DMF was added and incubated for 18 minutes. The reaction was then washed with NMP, followed by amino acid addition by repeating the coupling step. To join the C-terminus to Ile40, Gln39, Asn19, Asn12, and Val9 with numbers from the N-terminus, the coupling step was performed twice.
樹脂からのペプチド開裂は、トリフルオロ酢酸(TFA)(100mL)、1,2−エタンジチオール(EDT)(20.0mL)、フェノール(7.5g)、チオアニソール(5mL)、トリイソプロピルシラン(TIS)(5mL)及び水(5mL)を含む開裂混合物9mLと共に、2時間の開裂プログラム及びインキュベーションを使用して実施された。開裂したペプチドの溶液を50mL BDポリプロピレン遠心管に移し、冷エチルエーテル(40mL)でペプチドを沈殿させた。この混合物を遠心分離にかけ、エチルエーテルをペプチドからデカントした。エチルエーテル(40mL)を加え、混合物を渦流で混合し、遠心分離にかけ、エチルエーテルをデカントした。これらの工程(新たなエチルエーテルの追加、渦流混合、遠心分離、及びデカント)をあと2回繰り返した。このペプチドを減圧下で乾燥させ、408mg(収率92%)の粗生成物を得た。 Peptide cleavage from the resin was performed using trifluoroacetic acid (TFA) (100 mL), 1,2-ethanedithiol (EDT) (20.0 mL), phenol (7.5 g), thioanisole (5 mL), triisopropylsilane (TIS). ) (5 mL) and water (5 mL) and 9 mL of the cleavage mixture was performed using a 2 hour cleavage program and incubation. The cleaved peptide solution was transferred to a 50 mL BD polypropylene centrifuge tube and the peptide was precipitated with cold ethyl ether (40 mL). The mixture was centrifuged and ethyl ether was decanted from the peptide. Ethyl ether (40 mL) was added and the mixture was vortexed, centrifuged and decanted of ethyl ether. These steps (addition of fresh ethyl ether, vortex mixing, centrifugation, and decanting) were repeated two more times. The peptide was dried under reduced pressure to give 408 mg (92% yield) of crude product.
ペプチド精製は、Waters分取HPLCシステム(Waters(MA,U.S.A.))で実施された。粗ペプチド(約100mg)を、0.1% TFAを含む、酢酸/アセトニトリル/水が20/30/50の中に溶かした。この物質を2本のVydac C−18カラム(10mm、2.5×25cm)に注入した。注入後、0〜45%溶媒Bの勾配(溶媒B=0.1% TFAを含む80%アセトニトリル)で5分間、45〜70%溶媒Bで60分間、流量6mL/分を用いて、ペプチドを精製した。分画を集め、分析RP−HPLC、MALDI−TOF MS、及びCEで分析した。最も純粋な分画を蓄積し、凍結乾燥して、23mgの生成物を得た。MALDI−TOF MSでは、4400.5に等しい生成物分子量が測定され、これは、C195H326N56O53S3について計算された分子量4399.2よりも水素原子1つ分大きかった。この液体をアセトン乾燥氷浴中約30分間で急速冷凍することにより、凍結乾燥を実施した。凍結後、開放フラスコ内にある生成物を、濾紙で覆い、高真空下に置いた。高真空下で24時間経過後、乾燥したサンプルを真空から取り出し、後で使用するために保管容器を密封した。 Peptide purification was performed on a Waters preparative HPLC system (Waters (MA, USA)). The crude peptide (approximately 100 mg) was dissolved in 20/30/50 acetic acid / acetonitrile / water containing 0.1% TFA. This material was injected into two Vydac C-18 columns (10 mm, 2.5 × 25 cm). After injection, the peptide was prepared using a gradient of 0-45% solvent B (solvent B = 80% acetonitrile with 0.1% TFA) for 5 minutes, 45-70% solvent B for 60 minutes using a flow rate of 6 mL / min. Purified. Fractions were collected and analyzed by analytical RP-HPLC, MALDI-TOF MS, and CE. The purest fractions were pooled and lyophilized to give 23 mg of product. MALDI-TOF MS measured a product molecular weight equal to 4400.5, which was one hydrogen atom greater than the molecular weight 4399.2 calculated for C 195 H 326 N 56 O 53 S 3 . This liquid was freeze-dried by rapid freezing in an acetone dry ice bath for about 30 minutes. After freezing, the product in the open flask was covered with filter paper and placed under high vacuum. After 24 hours under high vacuum, the dried sample was removed from the vacuum and the storage container was sealed for later use.
実施例2:ペプチドとN−エチルマレイミドとの結合
スキーム1に示すように、部位誘導されたシステイン残基上にキャップするN−エチルマレイミドは、次の条件下で達成された。
Example 2: Conjugation of peptide and N-ethylmaleimide As shown in
2.5mLポリプロピレンバイアル中で、2.0mgの本発明ペプチドを、1.0mLの水に溶かした。次に0.1MのN−エチルマレイミド水溶液20μLをすぐに加えた。反応物を静かに、室温で2時間撹拌した。この反応混合物を、表6に示すプロトコルを用い、Vydac C18 10×250mm(300オングストローム;Grace Davison(IL,U.S.A.))カラムを装着したSummit APS(Dionex(CA,U.S.A.))HPLCで精製した。末端画分を回収し、HPLCで分析し、純粋な画分を蓄積して凍結乾燥した。
In a 2.5 mL polypropylene vial, 2.0 mg of the peptide of the present invention was dissolved in 1.0 mL of water. Next, 20 μL of 0.1 M N-ethylmaleimide aqueous solution was immediately added. The reaction was gently stirred at room temperature for 2 hours. This reaction mixture was subjected to a Summit APS (Dionex (CA, USA) equipped with a
実施例3:ペプチドとヨードアセトアミド−PEGとの結合
ヨードアセトアミド−PEG(ヨードアセトアミド末端を備えた直鎖20kDaポリエチレングリコール鎖であり、弱アルカリ性pHで限定的な量存在し、配列番号29のペプチドを備える)が、スキーム2に示す排他的反応として、システイン修飾を生じた。システインチオールは、ヨードアセトアミド−PEGの結合の選択性ポイントとして作用した。結果として得られた誘導体αスルファヒドリルアセトアミド連結は、アキラル性であった。
Example 3 Conjugation of Peptide with Iodoacetamide-PEG Iodoacetamide-PEG (a linear 20 kDa polyethylene glycol chain with iodoacetamide ends, present in a limited amount at weakly alkaline pH, ) Resulted in cysteine modification as an exclusive reaction shown in
15mLの三角フラスコに、25mg(5.68mmol、1.0当量)の配列番号1のペプチドを加えた。同じフラスコに140mg(6.82mmol、1.2当量、95%活性)のPEG−20ヨードアセトアミド(ロット番号M77592)(Nippon,Oil and Fat(NOF)Corp.製造)を加えた。10mLの水を加え、この溶液を、固体がすべて溶けるまで渦流攪拌した。この濁った溶液に、溶液pH約8.91で、50mLのピリジンを加えた。2時間後、20mLのサンプルのアリコートを除去し、Phenomenex C6−フェニルカラムを用い、溶離剤として0.1% TFA/アセトニトリルを用いた逆相HPLCによって分析した。サンプルは、2時間後に反応がほぼ完了したことを示した(図1A)。この反応混合物を、Phenomenex C6フェニル10×150mmカラムを使用してHPLCで直接精製した。精製のための溶離剤は、0.1% TFA水、及び0.1% TFA水中80%アセトニトリルであった。精製は、2〜3mLのサンプルバッチで行った(図1B)。精製された分画を合わせ、50mL三角フラスコ内で凍結乾燥した。この凍結乾燥した固体を、10mLの水で希釈し、再び凍結乾燥した。約1mgの最終生成物を、1mg/mLに希釈し、マススペクトル分析にかけた(図1C)。配列番号102のPEG化化合物の平均重量は、部分的にはPEGポリマーの長さの不均質性のため、25,449ダルトンであり、化合物は白色非晶質の固体として生じた。 To a 15 mL Erlenmeyer flask, 25 mg (5.68 mmol, 1.0 equiv) of the peptide of SEQ ID NO: 1 was added. To the same flask was added 140 mg (6.82 mmol, 1.2 eq, 95% active) PEG-20 iodoacetamide (Lot No. M77592) (manufactured by Nippon, Oil and Fat (NOF) Corp.). 10 mL of water was added and the solution was vortexed until all solids were dissolved. To this cloudy solution was added 50 mL of pyridine at a solution pH of about 8.91. After 2 hours, an aliquot of a 20 mL sample was removed and analyzed by reverse phase HPLC using a Phenomenex C6-phenyl column and 0.1% TFA / acetonitrile as the eluent. The sample showed that the reaction was almost complete after 2 hours (FIG. 1A). The reaction mixture was directly purified by HPLC using a Phenomenex C6 phenyl 10 × 150 mm column. The eluents for purification were 0.1% TFA water and 80% acetonitrile in 0.1% TFA water. Purification was performed in 2-3 mL sample batches (FIG. 1B). The purified fractions were combined and lyophilized in a 50 mL Erlenmeyer flask. The lyophilized solid was diluted with 10 mL water and lyophilized again. Approximately 1 mg of final product was diluted to 1 mg / mL and subjected to mass spectral analysis (FIG. 1C). The average weight of the PEGylated compound of SEQ ID NO: 102 was 25,449 daltons, partially due to the heterogeneity of the length of the PEG polymer, and the compound resulted as a white amorphous solid.
実施例4:ペプチドのN−エチルマレイミドリンカーとのPEG化
2.5mLのポリプロピレンバイアル中で、2.0mg(約0.44nmol)のペプチドを、2.5mLの水に溶かし、表7に示す量を使用して、次にすぐに、さまざまな分子量の活性化したN−エチルマレイミド誘導ポリエチレングリコールを加えた。
Example 4: PEGylation of peptide with N-ethylmaleimide linker In a 2.5 mL polypropylene vial, 2.0 mg (about 0.44 nmol) of peptide was dissolved in 2.5 mL of water and the amounts shown in Table 7 Then, immediately, activated N-ethylmaleimide-derived polyethylene glycols of various molecular weights were added.
反応混合物を静かに、室温で2時間撹拌した。 The reaction mixture was gently stirred at room temperature for 2 hours.
この反応混合物を、表8に示すプロトコルを用い、Gemini 5u C6−フェニル10×100mm(110オングストローム;Phenomenex(CA,U.S.A.))カラムを装着したSummit APS(Dionex(CA,U.S.A.))HPLCで精製した。
This reaction mixture was subjected to a Summit APS (Dionex (CA, U.S.)) equipped with a Gemini 5u C6-
実施例5:CRHR2及びCRHR1作動薬活性−cAMPアッセイ
CRH群のCRHR2及びCRHR1作動薬活性は、ヒトCRHR2又はヒトCRHR1のいずれかで形質移入されたSK−N−MC(ヒト神経芽細胞腫)の2つの細胞株において、アデノシン3’,5’−環状一リン酸(cAMP)アッセイによって特性付けられた。h−SCP(配列番号1)は、このアッセイにおいてh−UCN2(配列番号115)と等しい効力を有し、CRH群の中で最も選択性のCRHR2作動薬であることが示された(図2)。最大効果の50%に必要な濃度(A50)は0.4nMであった。
Example 5: CRHR2 and CRHR1 agonist activity-cAMP assay The CRHR2 and CRHR1 agonist activity of the CRH group was determined for SK-N-MC (human neuroblastoma) transfected with either human CRHR2 or human CRHR1. Two cell lines were characterized by
ヒトCRHR1(アクセッション番号X72304)又はCRHR2(アクセッション番号U34587)が、pcDNA3.1/Zeo発現ベクターにクローン化され、電気穿孔法により安定にSK−N−MC細胞に形質移入された。細胞は、10% FBS、50I.U.のペニシリン、50μg/mLのストレプトマイシン、2mMのL−グルタミン、1mMのピルビン酸ナトリウム及び0.1mMの非必須アミノ酸、600μg/mLのG418を含んだアール塩(Earl's Salt)入りMEM中で保持された。細胞は5% CO2中、37℃で成長させた。 Human CRHR1 (accession number X72304) or CRHR2 (accession number U34587) was cloned into the pcDNA3.1 / Zeo expression vector and stably transfected into SK-N-MC cells by electroporation. Cells were 10% FBS, 50I. U. Retained in MEM with Earl's Salt containing 50 μg / mL streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate and 0.1 mM non-essential amino acids, 600 μg / mL G418 . Cells were grown at 37 ° C. in 5% CO 2 .
細胞を96ウェル組織培養皿(Biocoat、BD Biosciences販売)に細胞50,000個/ウェルで入れ一晩培養した。細胞をPBSで洗ってから、フェノールレッドを含まず、10μMイソブチルメチルキサンチン(IBMX)を含むDMEM F−12中に再懸濁させた。細胞を、1pM〜10μMの濃度範囲のペプチドと共に、37℃で60分間インキュベートした。続いて、作動薬反応を生じなかったペプチドの任意の拮抗作用活性を評価するため、ペプチドを10μMで20分間プレインキュベートしてから、h−SCPを加えて60分間インキュベートした。フォルスコリン(10μM)(アデニル酸シクラーゼの直接的刺激物質)を陽性対照として使用した。0.5MのHClを加えてアッセイを止め、4℃で2時間、軌道回転で混合した。 The cells were placed in a 96-well tissue culture dish (Biocoat, BD Biosciences) at 50,000 cells / well and cultured overnight. Cells were washed with PBS and then resuspended in DMEM F-12 without phenol red and with 10 μM isobutylmethylxanthine (IBMX). Cells were incubated for 60 minutes at 37 ° C. with peptides in a concentration range of 1 pM to 10 μM. Subsequently, to assess any antagonizing activity of the peptides that did not produce an agonist response, the peptides were preincubated for 20 minutes at 10 μM, then h-SCP was added and incubated for 60 minutes. Forskolin (10 μM) (a direct stimulator of adenylate cyclase) was used as a positive control. The assay was stopped by adding 0.5 M HCl and mixed by orbital rotation at 4 ° C. for 2 hours.
CRHR2での本発明ペプチドの活性を評価するため、Flashプレート放射性アッセイ(カタログ番号Cus56088;Perkin Elmer(MA,U.S.A.))を使用した細胞内cAMP測定試験が採用された。 In order to evaluate the activity of the peptides of the present invention on CRHR2, an intracellular cAMP assay using a Flash plate radioassay (Cat. No. Cus56088; Perkin Elmer (MA, USA)) was employed.
形質移入されたSK−N−MC細胞を、96ウェルBiocoat組織培養皿(BD Biosciences(San Jose,CA,U.S.A))に細胞50,000個/ウェルで入れ一晩培養した。細胞をまずPBSで洗い、次にフェノールレッドを含まず、10μMイソブチルメチルキサンチン(IBMX)を含むDMEM/F−12中に懸濁させた。懸濁した細胞を、シンチラント液でコーティングした96ウェルフラッシュプレートに移した。細胞を、1pM〜1μMの範囲のペプチドと共に、37℃で60分間インキュベートした。10μMのフォルスコリンを陽性対照として使用した。リガンド刺激の後、0.5M HClを加えて細胞を溶解させ、4℃で2時間軌道回転により混合して、細胞内cAMPを培地内に放出させた。 Transfected SK-N-MC cells were placed in a 96-well Biocoat tissue culture dish (BD Biosciences (San Jose, CA, USA)) at 50,000 cells / well and cultured overnight. Cells were first washed with PBS and then suspended in DMEM / F-12 without phenol red and with 10 μM isobutylmethylxanthine (IBMX). The suspended cells were transferred to a 96 well flash plate coated with scintillant fluid. Cells were incubated for 60 minutes at 37 ° C. with peptides ranging from 1 pM to 1 μM. 10 μM forskolin was used as a positive control. After ligand stimulation, 0.5M HCl was added to lyse the cells and mixed by orbital rotation at 4 ° C. for 2 hours to release intracellular cAMP into the medium.
放出された細胞内cAMPを含む培地を、抗cAMP抗体を含むシンチラント液でコーティングした96ウェルフラッシュプレートに移した。このアッセイでは、細胞内cAMPは、抗体に結合しようとする125I−ラベルされたcAMPと競合する。標準曲線を生成するため、2.5〜250pmol/mLの範囲のcAMPを、この実験に含めた。[125I]−cAMPは、TopCountシンチレーションカウンター(Perkin Elmer(MA,U.S.A))で測定された。 The medium containing the released intracellular cAMP was transferred to a 96-well flash plate coated with a scintillant solution containing an anti-cAMP antibody. In this assay, intracellular cAMP competes with 125 I-labeled cAMP for binding to the antibody. In order to generate a standard curve, cAMP in the range of 2.5-250 pmol / mL was included in this experiment. [ 125 I] -cAMP was measured with a TopCount scintillation counter (Perkin Elmer (MA, USA)).
個々の作動薬濃度−反応曲線データは、GraphPad Prism(Graphpad Software(La Jolla,CA,U.S.A.))を使用して下記のHillの式にフィットされ、最大反応の半分を生成するのに必要な作動薬濃度(A50)、極大漸近線(α)及びHill傾き(nH)の各パラメータの見積りを提供した。この式において、[A]は作動薬濃度、Eは測定された効果である: Individual agonist concentration-response curve data is fitted to the Hill equation below using GraphPad Prism (Graphpad Software (La Jolla, CA, USA)) to produce half the maximum response. Provided an estimate of each parameter of the agonist concentration (A 50 ), the maximum asymptote (α) and the Hill slope (n H ) required for. In this equation, [A] is the agonist concentration and E is the measured effect:
表示目的のため、平均のフィットパラメータ見積りは、平均実験データに重ね合わせて示されている単一のE/[A]曲線を生成するのに使用された。作動薬の有効性見積りpA50は、各曲線の中点の負の対数として表され、測定値の標準誤差(SEM)と共にリストされている。作動薬投与量比の底10の対数(Log DR)値は、試験化合物pA50値を、同じアッセイバッチ内の対応するh−SCP(配列番号1)対照pA50値から差し引くことによって計算された。Log DR値のSEM値は、h−SCP(配列番号1)対照及び試験化合物pA50値の2乗SEM値の合計の平方根によって得られる。 For display purposes, the average fit parameter estimate was used to generate a single E / [A] curve shown superimposed on the average experimental data. The agonist efficacy estimate pA 50 is expressed as the negative logarithm of the midpoint of each curve and is listed with the standard error of measurement (SEM). The base 10 logarithm (Log DR) value of the agonist dose ratio was calculated by subtracting the test compound pA 50 value from the corresponding h-SCP (SEQ ID NO: 1) control pA 50 value within the same assay batch. . The SEM value of the Log DR value is obtained by the square root of the sum of the squared SEM values of the h-SCP (SEQ ID NO: 1) control and test compound pA 50 values.
![]()
![]()
h−SCP(配列番号1)に対するCRHR2媒介のcAMP反応は、克服可能な競合拮抗性と一致した濃度依存性状態で、選択的CRHR2拮抗薬、抗ソーバジン−30(SV30、表9に示す配列番号118)によってブロックされた(図3)。抗ソーバジン−30の存在により、配列番号1の化合物についてpA2値は7.82を得た。 The CRHR2-mediated cAMP response to h-SCP (SEQ ID NO: 1) is a concentration-dependent condition consistent with an overcoming competitive antagonistic activity, with a selective CRHR2 antagonist, anti-sorbazine-30 (SV30, SEQ ID NO: shown in Table 9). 118) (FIG. 3). The presence of anti-sorbazine-30 gave a pA 2 value of 7.82 for the compound of SEQ ID NO: 1.
このcAMPフラッシュプレートアッセイにおいて、h−CRHR1又はh−CRHR2形質移入されたSK−N−MC細胞の刺激に、ヒト及びラットペプチド(表10参照)が使用された。ペプチドは37℃で1時間インキュベートされた。曲線は、GraphPad Prismにおいて非線形回帰S字形濃度反応分析計算を使用して算出された。このようにして得られたpA50値を、文献値と共に表11に示す。 In this cAMP flashplate assay, human and rat peptides (see Table 10) were used to stimulate h-CRHR1 or h-CRHR2 transfected SK-N-MC cells. The peptide was incubated at 37 ° C. for 1 hour. Curves were calculated using a non-linear regression sigmoidal concentration response analysis calculation in GraphPad Prism. The pA 50 values thus obtained are shown in Table 11 together with literature values.
1h−CRHR1又は2 m−CRHR2b形質移入CHO−K1細胞(Lewis,K.ら、2001,PNAS,vol.98,pp.7570〜5);
3h−CRHR1又は4 h−CRHR2b形質移入HEK−293細胞、濃度反応曲線から近似された値(Hsu,S.Y.ら、2001,Nat.Med.,vol.7,pp.605〜11);
5m−CRHR2b形質移入されたHEK−293細胞(Brauns,O.ら、2002,Peptides,vol.23,pp.881〜888)。
1 h-CRHR1 or 2 m-CRHR2b transfected CHO-K1 cells (Lewis, K. et al., 2001, PNAS, vol. 98, pp. 7570-5);
3 h-CRHR1 or 4 h-CRHR2b transfected HEK-293 cells, values approximated from concentration response curves (Hsu, SY et al., 2001, Nat. Med., Vol. 7, pp. 605-11) ;
5 m-CRHR2b transfected HEK-293 cells (Brauns, O. et al., 2002, Peptides, vol. 23, pp. 881-888).
組換え型の非アミド化ペプチドライブラリーは、CRHR2形質移入されたSK−N−MC細胞中でアッセイを行うのは難しいため、作動薬活性におけるh−SCPのC末端領域のアミド化の効果が、有効性及び/又は固有の活性の点から調べられた。 Recombinant non-amidated peptide libraries are difficult to assay in CRHR2-transfected SK-N-MC cells, so the effect of amidation of the C-terminal region of h-SCP on agonist activity In terms of efficacy and / or intrinsic activity.
種々のアミノ酸のペプチド作動薬活性寄与を調べるため、いくつかの修飾ペプチドが、N末端配列内で1〜7個の欠失から始めて、合成された。各ペプチドは、ストック濃度1mMで水に溶かし、アリコートに分け−40℃でエッペンドルフチューブ(カタログ番号022364111)に保管した。ペプチドは実験当日に1回だけ解凍し、更にcAMPアッセイ緩衝液で希釈した。 In order to investigate the peptide agonist activity contribution of various amino acids, several modified peptides were synthesized starting with 1-7 deletions within the N-terminal sequence. Each peptide was dissolved in water at a stock concentration of 1 mM, divided into aliquots, and stored in Eppendorf tubes (catalog number 023364111) at -40 ° C. The peptide was thawed only once on the day of the experiment and further diluted with cAMP assay buffer.
各実験複製において、h−CRHR2形質移入されたSK−N−MC細胞中でcAMPを産生したすべてのペプチドが、同様の最大反応を達成した。しかしながら、h−SCP(配列番号1)への極大反応は、毎日の複製間で異なったため、データは、各複製内で得られたh−SCPに対する最大反応に対して正規化された。データは次に、作動薬濃度−効果の曲線パラメータの最終計算のために、3〜5つの複製実験のものを合わせた(図4)。得られたpA50値を、表12にまとめる。 In each experimental replicate, all peptides that produced cAMP in h-CRHR2-transfected SK-N-MC cells achieved a similar maximum response. However, since the maximal response to h-SCP (SEQ ID NO: 1) varied between daily replicates, the data was normalized to the maximum response to h-SCP obtained within each replicate. The data was then combined from three to five replicate experiments for final calculation of agonist concentration-effect curve parameters (FIG. 4). The pA 50 values obtained are summarized in Table 12.
最大反応は判別不能であったが、非アミド化h−SCP(配列番号113)は、アミド化された親ペプチドに比べ、有効性が約200分の1であった。1つのバッチにおいて、親40アミノ酸h−SCPペプチド(配列番号1)はpA50値が9.41±0.03であった。末端アミド化は有効性のために重要であるが必須ではなく、完全に画定された濃度−効果曲線が、非アミド化ペプチドで、アミド化された親ペプチドと同じ最大反応を伴って得られた。
Although the maximum response was indistinguishable, non-amidated h-SCP (SEQ ID NO: 113) was about 200 times more effective than the amidated parent peptide. In one batch, the
1つのアミノ酸欠失(配列番号107)は有効性に対して有意な影響を与えなかったが(pA50 9.24±0.05)、3つ(配列番号108)及び4つ(配列番号109)のアミノ酸欠失は、それぞれ漸進的にpA50値の低下をもたらした(8.49±0.08及び7.33±0.9)。これを表12に示す。5つ以上のアミノ酸欠失(配列番号110、配列番号111及び配列番号112)は、作動薬活性を完全に失わせた(図4)。したがって、後者の3つのペプチドは、濃度10μMで、h−SCPの拮抗薬として試験された(図5)。どのペプチドも、h−SCP濃度−効果曲線に対して有意な影響は与えなかった。これは、これらペプチドが検出可能な固有の有効性を有していないだけでなく、有意な受容体占有性(すなわち10μM未満の親和性)を持たないことを示している。 One amino acid deletion (SEQ ID NO: 107) had no significant effect on efficacy (pA 50 9.24 ± 0.05), three (SEQ ID NO: 108) and four (SEQ ID NO: 109) amino acid deletions) resulted in a decrease in each progressively pA 50 values (8.49 ± 0.08 and 7.33 ± 0.9). This is shown in Table 12. Five or more amino acid deletions (SEQ ID NO: 110, SEQ ID NO: 111 and SEQ ID NO: 112) completely abolished agonist activity (FIG. 4). Therefore, the latter three peptides were tested as antagonists of h-SCP at a concentration of 10 μM (FIG. 5). None of the peptides had a significant effect on the h-SCP concentration-effect curve. This indicates that these peptides not only have no detectable intrinsic efficacy, but also have no significant receptor occupancy (ie, an affinity of less than 10 μM).
h−SCP配列のN末端領域での4つ以上のアミノ酸欠失は、ペプチド有効性に影響する。N末端領域の1〜4つのアミノ酸欠失のペプチドは、有効性が漸進的に低下し、一方で5つ以上のアミノ酸欠失のペプチドは作動薬活性及び受容体親和性を完全に失う結果がもたらされた(KA>10μM)。後者は、h−UCN2について行われた同様の分析の既報に基づいて予測されていたものである(Isfort,R.J.et al.,2006,Peptides,vol.27,pp.1806〜1813)。これは、この欠失が、保持されているポジション6のアミノ酸セリン及びポジション8のアスパラギン酸に近いためである。
Deletion of 4 or more amino acids in the N-terminal region of the h-SCP sequence affects peptide efficacy. Peptides with 1-4 amino acid deletions in the N-terminal region are progressively reduced in effectiveness, whereas peptides with 5 or more amino acid deletions result in complete loss of agonist activity and receptor affinity. (K A > 10 μM). The latter was predicted based on previous reports of similar analyzes performed on h-UCN2 (Isfort, RJ et al., 2006, Peptides, vol. 27, pp. 1806-1813). . This is because the deletion is close to the retained
更に、システイン突然変異、N−エチルマレイミドキャッピング、及びPEG化の、ペプチド作動薬活性に対する影響が調査された。h−SCP(配列番号1)の対照pA50は、種々のアッセイバッチで9.47〜9.74、SEMが0.03〜0.11と変動した。ここでも、いくつかの修飾ペプチドが前述スキームにしたがって合成された。これらペプチドのアッセイ結果を表13に示す。 In addition, the effects of cysteine mutation, N-ethylmaleimide capping, and PEGylation on peptide agonist activity were investigated. control pA 50 of h-SCP (SEQ ID NO: 1) is 9.47 to 9.74 in various assay batches, SEM varies with from 0.03 to 0.11. Again, some modified peptides were synthesized according to the above scheme. The assay results for these peptides are shown in Table 13.
本発明のペプチドのさまざまな修飾の活性プロファイルを例示する結果が、表14に示されている。これにはストレスコピン(h−SCP)ペプチド、ウロコルチン2(h−UCN2)、及びh−SCP−IA−PEGペプチド(配列番号102)が含まれ、ここでh−SCP−IA−PEGは、配列番号29で表されるポジション28にシステイン置換を伴うSCP配列を有し、かつ、ポジション28のシステインにアセトアミド(IA)リンカーを介してPEGポリマーがリンクされているペプチドである。このデータは、1〜3つの複製の平均±SEMであり、各複製実験内のh−SCPに対して得られた最大反応の%で表わされている。
Results illustrating the activity profiles of various modifications of the peptides of the present invention are shown in Table 14. This includes the stress copine (h-SCP) peptide, urocortin 2 (h-UCN2), and h-SCP-IA-PEG peptide (SEQ ID NO: 102), where h-SCP-IA-PEG has the sequence It is a peptide having an SCP sequence with cysteine substitution at position 28 represented by
h−SCP−IA−PEGペプチドはまた、100nMの抗ソーバジン−30(h−CRHR2受容体の選択性競合拮抗薬)の存在下でインキュベートされ、h−SCP−IA−PEGペプチド濃度−反応曲線で右方向へのシフトをもたらし、極大反応が100%に拘束されたときの対応するpA50の近似値は6.89であった。 h-SCP-IA-PEG peptide was also incubated in the presence of 100 nM anti-sorbazine-30 (a selective competitive antagonist of h-CRHR2 receptor), and in the h-SCP-IA-PEG peptide concentration-response curve The corresponding approximate pA 50 value was 6.89 when it resulted in a shift to the right and the maximal response was constrained to 100%.
実施例6:CRHR1及びCRHR2の放射性リガンド結合活性
h−SCP(配列番号1)のCRHR2での結合プロファイルが、[125I]−抗ソーバジン−30を放射性標識として使用しヒトCRHR2で安定に形質移入されたSK−N−MC細胞の膜調製における放射性リガンド結合研究において決定された。この細胞は、細胞擦過によって採取され、得られたペレットをすぐに−80℃で凍結した(細胞約50×106個/ペレット)。
Example 6: Radioligand binding activity of CRHR1 and CRHR2 The binding profile of h-SCP (SEQ ID NO: 1) with CRHR2 is stably transfected with human CRHR2 using [ 125 I] -antisorbazine-30 as a radiolabel. Determined in radioligand binding studies in membrane preparation of cultured SK-N-MC cells. The cells were harvested by cell scraping and the resulting pellet was immediately frozen at −80 ° C. (approximately 50 × 10 6 cells / pellet).
凍結した細胞ペレットを、15mLのアッセイ緩衝液中、氷浴上で解凍した。この緩衝液は10mM HEPES、130mM NaCl、4.7mM KCl、5mM MgCl2、及び0.089mMバシトラシンから構成され、pH 7.2、及び21±3℃であった。この溶液をPolytron組織グラインダーを用い、設定10及び7×3sで均質化した(Brinkmann Instruments(Westbury,NY))。均質化した液を4℃、800×gで5分間遠心分離を行い、ペレットを廃棄した。この上澄みを再び、4℃、26,892×gで25分間遠心分離を行い、最終的なペレットをアッセイ緩衝液中に再懸濁させた。結合アッセイはすべて、0.3% PEI入りのアッセイ緩衝液中にあらかじめ1時間浸しておいた96ウェルMultiscreen GF/Bフィルタープレート(Millipore(Billericay,MA,U.S.A.))内で実施された。研究の完了のため、体積45μLの細胞膜を、CRHR2アッセイ用に体積50μL中の60pM[125I]−抗ソーバジン−30と共に、又は、CRHR1アッセイ用に[125I]−(Tyr0)−ソーバジンと共に、15μLの競合リガンドの存在下で、合計体積150μLとして、120分間インキュベートした。1μMのr−UCN1(配列番号114)を含めることによって、非特異的な結合が決定された。この結合した放射能を、Multiscreen Resistマニフォールド(Millipore Corp.(Billerica,MA,U.S.A))を用いて濾過により分離した。フィルターを、pH 7.5の氷冷PBSで3回洗い、フィルター上に残った放射能を、TopCountカウンター(Packard BioScience(Boston,MA,U.S.A))によって測定した液体シンチレーションによって定量した。すべての実験を3組で実施した。
The frozen cell pellet was thawed on an ice bath in 15 mL assay buffer. This buffer consisted of 10 mM HEPES, 130 mM NaCl, 4.7 mM KCl, 5 mM MgCl 2 , and 0.089 mM bacitracin, pH 7.2, and 21 ± 3 ° C. This solution was homogenized using a Polytron tissue grinder with
個々の競合曲線からのデータは、各実験内で、特異的[125I]−抗ソーバジン−30又は[125I]−(Tyr0)−ソーバジン結合(B)のパーセンテージとして表わされた。これらのデータは次に、GraphPad Prismを使用した4パラメータロジスティックを使用し、それぞれ100%に加重した上漸近線(αmax)及び0%に加重した下漸近線(αmin)で、それぞれ競合物質の最低濃度及び最高濃度から2対数単位上及び下のこれらの値を含めることによって、分析された: Data from individual competition curves were expressed as a percentage of specific [ 125 I] -anti-sorvazine-30 or [ 125 I]-(Tyr 0 ) -sorbazine binding (B) within each experiment. These data are then compared with competitors using a 4-parameter logistic using GraphPad Prism, each with an upper asymptote (α max ) weighted to 100% and a lower asymptote (α min ) weighted to 0%, respectively. Were analyzed by including these values two log units above and below the lowest and highest concentrations of:
![]()
![]()
h−SCP(配列番号1)で得られた競合曲線は二相性であった。これは、50%阻害での濃度の負の対数が高いこと(pIC50)及びpIC50が6.6と低いことによって特徴付けられる、高い親和性及び低い親和性の受容体結合状態を示していた。高親和性部位結合は、100μMグアノシン5’−O−[γ−チオ]三リン酸(GTPγS)によって阻害されることが示された。これに対して、h−UCN2(配列番号115)は高い親和性結合のみを呈した。これは、アッセイ中で、h−UCN2がh−SCP(配列番号1)よりも高い固有有効性を有する作動薬として働いていることを示している。このデータ分析から得られたpKI値を表15に示す。
The competition curve obtained with h-SCP (SEQ ID NO: 1) was biphasic. This indicates a high affinity and low affinity receptor binding state characterized by a high negative logarithm of concentration at 50% inhibition (pIC 50 ) and a low pIC 50 of 6.6. It was. High affinity site binding has been shown to be inhibited by 100
実施例7:血管平滑筋弛緩−ラット大動脈環
フェニレフリン(PE)であらかじめ収縮させた単離ラット大動脈環において、h−SCP(配列番号1)の血管平滑筋を弛緩させる能力が調べられた(図6参照)。このペプチド(配列番号1)は、pA50が6.05±0.12で、濃度依存性の弛緩を生じたが、pA50が7.01±0.13を有するh−UCN2(配列番号115)に比べ有効性が10分の1であった。h−SCP(配列番号1)によって引き起こされた反応は、抗ソーバジン−30(配列番号118)によって阻害された。
Example 7: Relaxation of vascular smooth muscle-rat aortic ring The ability of h-SCP (SEQ ID NO: 1) to relax vascular smooth muscle in isolated rat aortic rings pre-contracted with phenylephrine (PE) was examined (Fig. 6). This peptide (SEQ ID NO: 1) produced a concentration-dependent relaxation with a pA 50 of 6.05 ± 0.12, but h-UCN2 (SEQ ID NO: 115) with a pA 50 of 7.01 ± 0.13. ) Was 1/10 the effectiveness. The response elicited by h-SCP (SEQ ID NO: 1) was inhibited by anti-sorbazine-30 (SEQ ID NO: 118).
実施例8:単離ウサギ心臓における心臓血管特徴付け
ウサギ心臓の逆行性潅流ランゲンドルフアッセイにおいて、心拍数(HR)、左心室(LV)収縮、及び血管緊張に対するh−SCP(配列番号1)の影響が評価された。プラシーボ様対照溶媒又はh−SCP(配列番号1)のボーラスを、潅流ブロック内に直接投与した。h−SCP(配列番号1)は、心拍数に濃度依存性の増加を生じ、左心室が圧力を増し(dP/dtmax)、それぞれ52nM、9.9nM、及び46nMに等しい50%反応の濃度での冠状血管潅流圧力(CPP)が対応して低下したが(図7)、対照溶媒の場合では反応は観察されなかった。
Example 8: Cardiovascular Characterization in Isolated Rabbit Hearts Effect of h-SCP (SEQ ID NO: 1) on heart rate (HR), left ventricular (LV) contraction, and vascular tone in retrograde perfusion Langendorff assay of rabbit heart Was evaluated. A placebo-like control vehicle or h-SCP (SEQ ID NO: 1) bolus was administered directly into the perfusion block. h-SCP (SEQ ID NO: 1) produces a concentration-dependent increase in heart rate, with the left ventricle increasing pressure (dP / dt max ), 50% response concentration equal to 52 nM, 9.9 nM, and 46 nM, respectively. Coronary vascular perfusion pressure (CPP) decreased at the same time (FIG. 7), but no response was observed with the control solvent.
前述の明細書は、例示を目的として提供される実施例と共に、本発明の原理を教示するが、本発明の実践は、以下の特許請求の範囲及びそれらの等価物の範囲内に含まれるすべての通常の変形、改作及び/又は修正を包含することが理解されるであろう。 While the foregoing specification teaches the principles of the invention, along with examples provided for purposes of illustration, the practice of the invention is all included within the scope of the following claims and their equivalents. It will be understood to encompass the usual variations, adaptations and / or modifications of
Claims (32)
LSLDV PTNIM NLLFN IAKAK NLRAQ AAANA HLMAQ I
を含み、式中、該ペプチドの少なくとも1つのアミノ酸がXで置換され、ただし該置換は該アミノ酸配列のポジション3、29、及び33では行われず、かつここにおいてXはシステイン、チロシン、若しくはグルタミン酸であるペプチド、
又はこれらの製薬上許容される塩若しくはアミド。 A peptide having agonist activity against corticotropin releasing hormone receptor type 2, wherein the peptide has the amino acid sequence:
LSLDV PTNIM NLLFN IAKAK NLRAQ AAANA HLMAQ I
Wherein at least one amino acid of the peptide is substituted with X, provided that the substitution is not made at positions 3, 29, and 33 of the amino acid sequence, and where X is cysteine, tyrosine, or glutamic acid A peptide,
Or a pharmaceutically acceptable salt or amide thereof.
ポジション1でLの代わりにX;
ポジション2でSの代わりにX;
ポジション4でDの代わりにX;
ポジション5でVの代わりにX;
ポジション6でPの代わりにX;
ポジション7でTの代わりにX;
ポジション8でNの代わりにX;
ポジション9でIの代わりにX;
ポジション10でMの代わりにX;
ポジション11でNの代わりにX;
ポジション12でLの代わりにX;
ポジション13でLの代わりにX;
ポジション14でFの代わりにX;
ポジション15でNの代わりにX;
ポジション16でIの代わりにX;
ポジション17でAの代わりにX;
ポジション18でKの代わりにX;
ポジション19でAの代わりにX;
ポジション20でKの代わりにX;
ポジション21でNの代わりにX;
ポジション22でLの代わりにX;
ポジション23でRの代わりにX;
ポジション24でAの代わりにX;
ポジション25でQの代わりにX;
ポジション26でAの代わりにX;
ポジション27でAの代わりにX;
ポジション28でAの代わりにX;
ポジション30でAの代わりにX;
ポジション31でHの代わりにX;
ポジション32でLの代わりにX;
ポジション34でAの代わりにX;
ポジション35でQの代わりにX;及び
ポジション36でIの代わりにXからなる群から選択される、請求項1に記載のペプチド。 Said substitution is
X instead of L at position 1;
X instead of S at position 2;
X instead of D at position 4;
X instead of V at position 5;
X instead of P at position 6;
X instead of T at position 7;
X instead of N at position 8;
X instead of I at position 9;
X instead of M at position 10;
X instead of N at position 11;
X instead of L at position 12;
X instead of L at position 13;
X instead of F at position 14;
X instead of N at position 15;
X instead of I at position 16;
X instead of A at position 17;
X instead of K at position 18;
X instead of A at position 19;
X instead of K at position 20;
X instead of N at position 21;
X instead of L at position 22;
X instead of R at position 23;
X instead of A at position 24;
X instead of Q at position 25;
X instead of A at position 26;
X instead of A at position 27;
X instead of A at position 28;
X instead of A at position 30;
X instead of H at position 31;
X instead of L at position 32;
X instead of A at position 34;
2. The peptide of claim 1 selected from the group consisting of X instead of Q at position 35; and X instead of I at position 36.
XLSLD VPTNI MNLLF NIAKA KNLRA QAAAN AHLMA QI−NH2、
XTLSL DVPTN IMNLL FNIAK AKNLR AQAAA NAHLM AQI−NH2、
XFTLS LDVPT NIMNL LFNIA KAKNL RAQAA ANAHL MAQI−NH2、及び
XKFTL SLDVP TNIMN LLFNI AKAKN LRAQA AANAH LMAQI−NH2からなる群から選択される、請求項1に記載のペプチド。 The peptide is
XLSLD VPTNI MNLLF NIAKA KNLRA QAAAN AHLMA QI-NH 2 ,
XTLSL DVPTN IMNLL FNIAK AKNLR AQAAA NAHLM AQI-NH 2 ,
XFTLS LDVPT NIMNL LFNIA KAKNL RAQAA ANAHL MAQI-NH 2 and XKFTL SLDVP TNIMN LLFNI AKAKN LRAQA ANAH LMAQ1-NH 2
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11123308P | 2008-11-04 | 2008-11-04 | |
| US61/111,233 | 2008-11-04 | ||
| US17889009P | 2009-05-15 | 2009-05-15 | |
| US61/178,890 | 2009-05-15 | ||
| PCT/US2009/063276 WO2010053990A2 (en) | 2008-11-04 | 2009-11-04 | Crhr2 peptide agonists and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012508014A true JP2012508014A (en) | 2012-04-05 |
Family
ID=41509080
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011535643A Pending JP2012508014A (en) | 2008-11-04 | 2009-11-04 | CRHR2 peptide agonist and use thereof |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US10040838B2 (en) |
| EP (1) | EP2362881B1 (en) |
| JP (1) | JP2012508014A (en) |
| KR (1) | KR20110091720A (en) |
| CN (1) | CN102272151B (en) |
| AU (1) | AU2009313619A1 (en) |
| BR (1) | BRPI0921640A2 (en) |
| CA (1) | CA2742710A1 (en) |
| CO (1) | CO6341479A2 (en) |
| CR (1) | CR20110305A (en) |
| EA (1) | EA201170647A1 (en) |
| EC (1) | ECSP11011099A (en) |
| ES (1) | ES2723098T3 (en) |
| IL (1) | IL212685A0 (en) |
| MX (1) | MX2011004718A (en) |
| NI (1) | NI201100087A (en) |
| NZ (1) | NZ592670A (en) |
| SG (1) | SG171957A1 (en) |
| WO (1) | WO2010053990A2 (en) |
| ZA (1) | ZA201104160B (en) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG171957A1 (en) | 2008-11-04 | 2011-07-28 | Janssen Pharmaceutica Nv | Crhr2 peptide agonists and uses thereof |
| CA2780163A1 (en) * | 2009-11-04 | 2011-05-12 | Janssen Pharmaceutica Nv | Method for treating heart failure with stresscopin-like peptides |
| HUE036640T2 (en) * | 2012-02-14 | 2018-07-30 | Univ California | Systemic delivery and regulated expression of paracrine genes for cardiovascular diseases and other conditions |
| JOP20170153A1 (en) * | 2016-07-15 | 2019-01-30 | Lilly Co Eli | Novel fatty acid modified urocortin-2 analogs for the treatment of diabetes and chronic kidney disease |
| JOP20190097A1 (en) | 2016-10-27 | 2019-04-28 | Janssen Pharmaceutica Nv | Immunoglobulins and uses thereof |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06506218A (en) * | 1991-03-15 | 1994-07-14 | アムゲン ボールダー,インコーポレイテッド | PEGylation of polypeptides |
| WO2002034934A2 (en) * | 2000-10-26 | 2002-05-02 | The Board Of Trustees Of The Leland Stanford Junior University | Stresscopins and their uses |
| WO2003062268A2 (en) * | 2002-01-16 | 2003-07-31 | The Procter & Gamble Company | Corticotropin releasing factor receptor 2 agonists |
| JP2005505244A (en) * | 2001-03-15 | 2005-02-24 | リサーチ ディベロップメント ファンデーション | Urocortin III and its use |
| WO2007090087A2 (en) * | 2006-01-27 | 2007-08-09 | Research Development Foundation | Use of corticotropin releasing factor receptor-2 inhibitors for treating insulin-related diseases |
| WO2008047241A2 (en) * | 2006-10-16 | 2008-04-24 | Conjuchem Biotechnologies Inc. | Modified corticotropin releasing factor peptides and uses thereof |
| JP2008528050A (en) * | 2005-02-04 | 2008-07-31 | ファイザー・プロダクツ・インク | PYY agonists and uses thereof |
| JP2013510167A (en) * | 2009-11-04 | 2013-03-21 | ヤンセン ファーマシューティカ エヌ.ベー. | Treatment of heart failure with stress-copin-like peptides |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE174058T1 (en) | 1989-11-06 | 1998-12-15 | Cell Genesys Inc | PRODUCTION OF PROTEINS USING HOMOLOGOUS RECOMBINATION |
| US5272071A (en) * | 1989-12-22 | 1993-12-21 | Applied Research Systems Ars Holding N.V. | Method for the modification of the expression characteristics of an endogenous gene of a given cell line |
| US6552170B1 (en) * | 1990-04-06 | 2003-04-22 | Amgen Inc. | PEGylation reagents and compounds formed therewith |
| US5766897A (en) * | 1990-06-21 | 1998-06-16 | Incyte Pharmaceuticals, Inc. | Cysteine-pegylated proteins |
| US5554728A (en) * | 1991-07-23 | 1996-09-10 | Nexstar Pharmaceuticals, Inc. | Lipid conjugates of therapeutic peptides and protease inhibitors |
| EP0922446A1 (en) * | 1997-12-03 | 1999-06-16 | Applied Research Systems Ars Holding N.V. | Solution-phase site-specific preparation of GRF-PEG conjugates |
| US6420339B1 (en) * | 1998-10-14 | 2002-07-16 | Amgen Inc. | Site-directed dual pegylation of proteins for improved bioactivity and biocompatibility |
| US6673580B2 (en) * | 2000-10-27 | 2004-01-06 | Genentech, Inc. | Identification and modification of immunodominant epitopes in polypeptides |
| ES2268044T3 (en) | 2001-05-21 | 2007-03-16 | Injet Digital Aerosols Limited | COMPOSITIONS FOR THE ADMINISTRATION OF PROTEINS BY THE PULMONARY ROUTE. |
| US7144985B2 (en) * | 2001-12-03 | 2006-12-05 | Metabolex, Inc. | Methods and reagents for diagnosis and treatment of diabetes |
| CN100480267C (en) * | 2002-01-16 | 2009-04-22 | 宝洁公司 | Corticotropin releasing factor 2 receptor agonists |
| KR100512483B1 (en) * | 2003-05-07 | 2005-09-05 | 선바이오(주) | Novel Preparation method of PEG-maleimide PEG derivatives |
| GB0316294D0 (en) * | 2003-07-11 | 2003-08-13 | Polytherics Ltd | Conjugated biological molecules and their preparation |
| US7230068B2 (en) * | 2003-10-09 | 2007-06-12 | Ambrx, Inc. | Polymer derivatives |
| WO2005123109A2 (en) * | 2004-06-12 | 2005-12-29 | Bayer Pharmaceuticals Corporation | Pegylation of vasoactive intestinal peptide (vip)/pituitary adenylate cyclase activating peptide (pacap) receptor 2 (vpac2) agonists and methods of use |
| GB2427360A (en) | 2005-06-22 | 2006-12-27 | Complex Biosystems Gmbh | Aliphatic prodrug linker |
| JP2010527989A (en) | 2007-05-25 | 2010-08-19 | ネウトロン リミテッド | CRF complex with extended half-life |
| JP2010539047A (en) | 2007-09-11 | 2010-12-16 | モンドバイオテック ラボラトリーズ アクチエンゲゼルシャフト | Use of peptides as therapeutic agents |
| EP2537940A3 (en) | 2007-09-14 | 2013-04-10 | BASF Plant Science GmbH | Plants having increased yield-related traits and a method for making the same |
| SG171957A1 (en) | 2008-11-04 | 2011-07-28 | Janssen Pharmaceutica Nv | Crhr2 peptide agonists and uses thereof |
| JP2013506697A (en) | 2009-10-06 | 2013-02-28 | アンジオケム インコーポレーテッド | Compositions and methods for delivering therapeutic agents |
| AU2012284122B2 (en) | 2011-07-18 | 2017-06-01 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methods and compositions for inhibiting polyomavirus-associated pathology |
-
2009
- 2009-11-04 SG SG2011040755A patent/SG171957A1/en unknown
- 2009-11-04 WO PCT/US2009/063276 patent/WO2010053990A2/en active Application Filing
- 2009-11-04 BR BRPI0921640A patent/BRPI0921640A2/en not_active IP Right Cessation
- 2009-11-04 MX MX2011004718A patent/MX2011004718A/en not_active Application Discontinuation
- 2009-11-04 ES ES09748922T patent/ES2723098T3/en active Active
- 2009-11-04 JP JP2011535643A patent/JP2012508014A/en active Pending
- 2009-11-04 CA CA2742710A patent/CA2742710A1/en not_active Abandoned
- 2009-11-04 US US12/612,548 patent/US10040838B2/en active Active
- 2009-11-04 NZ NZ592670A patent/NZ592670A/en not_active IP Right Cessation
- 2009-11-04 AU AU2009313619A patent/AU2009313619A1/en not_active Abandoned
- 2009-11-04 EA EA201170647A patent/EA201170647A1/en unknown
- 2009-11-04 EP EP09748922.3A patent/EP2362881B1/en active Active
- 2009-11-04 CN CN200980154228.3A patent/CN102272151B/en not_active Expired - Fee Related
- 2009-11-04 KR KR1020117012435A patent/KR20110091720A/en not_active Application Discontinuation
-
2011
- 2011-05-04 IL IL212685A patent/IL212685A0/en unknown
- 2011-05-04 NI NI201100087A patent/NI201100087A/en unknown
- 2011-05-18 CO CO11061124A patent/CO6341479A2/en active IP Right Grant
- 2011-06-02 EC EC2011011099A patent/ECSP11011099A/en unknown
- 2011-06-03 ZA ZA2011/04160A patent/ZA201104160B/en unknown
- 2011-06-06 CR CR20110305A patent/CR20110305A/en not_active Application Discontinuation
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06506218A (en) * | 1991-03-15 | 1994-07-14 | アムゲン ボールダー,インコーポレイテッド | PEGylation of polypeptides |
| WO2002034934A2 (en) * | 2000-10-26 | 2002-05-02 | The Board Of Trustees Of The Leland Stanford Junior University | Stresscopins and their uses |
| JP2005505244A (en) * | 2001-03-15 | 2005-02-24 | リサーチ ディベロップメント ファンデーション | Urocortin III and its use |
| WO2003062268A2 (en) * | 2002-01-16 | 2003-07-31 | The Procter & Gamble Company | Corticotropin releasing factor receptor 2 agonists |
| EP1724283A2 (en) * | 2002-01-16 | 2006-11-22 | The Procter & Gamble Company | Corticotropin releasing factor receptor 2 agonists |
| JP2008528050A (en) * | 2005-02-04 | 2008-07-31 | ファイザー・プロダクツ・インク | PYY agonists and uses thereof |
| WO2007090087A2 (en) * | 2006-01-27 | 2007-08-09 | Research Development Foundation | Use of corticotropin releasing factor receptor-2 inhibitors for treating insulin-related diseases |
| WO2008047241A2 (en) * | 2006-10-16 | 2008-04-24 | Conjuchem Biotechnologies Inc. | Modified corticotropin releasing factor peptides and uses thereof |
| JP2013510167A (en) * | 2009-11-04 | 2013-03-21 | ヤンセン ファーマシューティカ エヌ.ベー. | Treatment of heart failure with stress-copin-like peptides |
Non-Patent Citations (1)
| Title |
|---|
| JPN6014015899; Journal of the American Chemical Society Vol.129, 2007, p.16102-16114 * |
Also Published As
| Publication number | Publication date |
|---|---|
| ECSP11011099A (en) | 2011-07-29 |
| WO2010053990A2 (en) | 2010-05-14 |
| EP2362881A2 (en) | 2011-09-07 |
| CN102272151A (en) | 2011-12-07 |
| CR20110305A (en) | 2011-11-10 |
| ES2723098T3 (en) | 2019-08-21 |
| NZ592670A (en) | 2013-01-25 |
| MX2011004718A (en) | 2011-07-28 |
| IL212685A0 (en) | 2011-07-31 |
| CN102272151B (en) | 2014-08-20 |
| CA2742710A1 (en) | 2010-05-14 |
| CO6341479A2 (en) | 2011-11-21 |
| WO2010053990A3 (en) | 2010-07-01 |
| US10040838B2 (en) | 2018-08-07 |
| BRPI0921640A2 (en) | 2019-09-24 |
| US20100130424A1 (en) | 2010-05-27 |
| EA201170647A1 (en) | 2011-12-30 |
| SG171957A1 (en) | 2011-07-28 |
| ZA201104160B (en) | 2012-11-28 |
| EP2362881B1 (en) | 2019-03-13 |
| KR20110091720A (en) | 2011-08-12 |
| NI201100087A (en) | 2011-10-25 |
| AU2009313619A1 (en) | 2010-05-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102285377B1 (en) | Trigonal glucagon/GLP-1/GIP receptor agonist | |
| CN103857408B (en) | Glucagon/glp-1 receptor co-agonists | |
| JP2013510167A (en) | Treatment of heart failure with stress-copin-like peptides | |
| KR102378943B1 (en) | Pharmaceutical compositions | |
| JP6622690B2 (en) | Peptide composition | |
| US20170114115A1 (en) | Glucagon receptor agonists | |
| CN102834108A (en) | Glucagon antagonist - gip agonist conjugates and compositions for the treatment of metabolic disorders and obesity | |
| CN102459325A (en) | Gip receptor-active glucagon compounds | |
| MX2012014961A (en) | Glucagon analogues. | |
| TW201307380A (en) | Novel oxyntomodulin derivatives and pharmaceutical composition for treating obesity comprising the same | |
| CN103649115A (en) | Polypeptides | |
| CN110072883A (en) | The relevant peptide of therapeutic MOTS-C | |
| JP2012508014A (en) | CRHR2 peptide agonist and use thereof | |
| ES2287484T3 (en) | CORTICOTROPINE LIBERATING FACTOR 2 RECEIVER AGONISTS. | |
| US9155795B2 (en) | CXCR4 receptor compounds | |
| KR20160075826A (en) | Novel compound for treatment of severe hypoglycemia | |
| KR20160075825A (en) | Novel compound for treatment of severe hypoglycemia | |
| KR20160077215A (en) | Novel compound for treatment of severe hypoglycemia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20121102 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140416 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20141001 |