JP2012246252A - Modified-release type particle pharmaceutical composition for oral administration, method for producing the same, and orally disintegrating tablet including the composition - Google Patents
Modified-release type particle pharmaceutical composition for oral administration, method for producing the same, and orally disintegrating tablet including the composition Download PDFInfo
- Publication number
- JP2012246252A JP2012246252A JP2011119340A JP2011119340A JP2012246252A JP 2012246252 A JP2012246252 A JP 2012246252A JP 2011119340 A JP2011119340 A JP 2011119340A JP 2011119340 A JP2011119340 A JP 2011119340A JP 2012246252 A JP2012246252 A JP 2012246252A
- Authority
- JP
- Japan
- Prior art keywords
- active ingredient
- release
- pharmaceutical composition
- coating
- organic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images









Abstract
Description
本発明は経口投与用の放出制御型粒子状医薬組成物("Modified-release particles":「MRP」と略すことあり。)、該組成物を含有する経口投与用崩壊性粒子状組成物("Orally disintegrating particles"または”Orodispersible particles”:本明細書においては、「ODP」と略すことある。)および口腔内崩壊錠(本明細書においては、経口用崩壊錠ということもあり、または、"Orally disintegrating tablet"もしくは”Orodispersible tablet”:「ODT」と略すことある。)、ならびにそれらの製造方法に関する。さらに詳しくは、服用後、該組成物が口腔内に残存する間は薬剤成分が実質的に放出されず、胃内に移行した後、速やかに放出を開始するよう精密に設計された放出制御型粒子状医薬組成物(MRP)、該組成物を含有する経口投与用崩壊性粒子状組成物(ODP)および口腔内崩壊錠(ODT)、ならびに、それらの製造方法に関する。 The present invention relates to a controlled release particulate pharmaceutical composition for oral administration (“Modified-release particles”: “MRP”), and a collapsible particulate composition for oral administration containing the composition (“ “Orally disintegrating particles” or “Orodispersible particles”: In this specification, “ODP” may be abbreviated as “ODP”) and orally disintegrating tablets (also referred to herein as “orally disintegrating tablets”, or “Orally disintegrating tablet "or" Orodispersible tablet ": abbreviated as" ODT "), and a method for producing them. More specifically, after taking, the drug component is not substantially released while the composition remains in the oral cavity, and is precisely controlled to start releasing immediately after moving into the stomach. The present invention relates to a particulate pharmaceutical composition (MRP), an orally disintegrating particulate composition (ODP) and an orally disintegrating tablet (ODT) containing the composition, and methods for producing them.
合成イミダゾトリアジノン誘導体バルデナフィル(Vardenafil)は、cGMPホスホジエステラーゼ(PDE)5阻害剤として、その使用および活性スペクトルが知られている(例えば、特表2001−522851号公報(特許文献1))。バルデナフィルはレビトラ(Levitra)(登録商標)の名称で購入でき、勃起不全症処置用の即時放出(IR)錠剤として専ら使用されている。この即時放出用であるバルデナフィルの医薬剤形は、それらの迅速な作用開始および比較的短い作用時間を介して、患者の必要に即応できる処置用としての標的プロファイルを満足してきた。この好ましいバルデナフィルの吸収プロファイルは、胃腸管系(Gastrointestinal Tract: ”GIT”と略すことあり。)のうち、胃および十二指腸の部分である上部消化管(Upper Gastrointestinal Tract:"U-GIT"、または「上部GI管」と略すことあり。)で主に吸収されることに由来すると考えられている。バルデナフィルが塩基性薬剤であることから、特に、強酸性状態にある胃がバルデナフィルの主たる溶解部位であると考えられる。 A synthetic imidazotriazinone derivative vardenafil is known as a cGMP phosphodiesterase (PDE) 5 inhibitor and its activity spectrum (for example, JP-T-2001-522851 (Patent Document 1)). Vardenafil can be purchased under the name Levitra® and is exclusively used as an immediate release (IR) tablet for the treatment of erectile dysfunction. This immediate release vardenafil pharmaceutical dosage form has satisfied a targeted profile for treatment that can be adapted to patient needs through their rapid onset of action and relatively short duration of action. The preferred absorption profile of vardenafil is the upper gastrointestinal system (Upper Gastrointestinal Tract: "U-GIT"), which is part of the stomach and duodenum in the gastrointestinal system (Gastrointestinal Tract: "GIT"). It may be abbreviated as “upper GI tube”)). Since vardenafil is a basic drug, the stomach in a strongly acidic state is considered to be the main dissolution site of vardenafil.
一方、バルデナフィルを含有する即時放出錠剤は、速やかな嚥下と有効成分の効率的放出を促すために水と共に口腔適用する必要がある。患者がバルデナフィルを服用する場面を考慮すると、水なしでも服用することができれば患者の利便性を大きく高めることになると思われる。
近年、患者が薬を服用する際の利便性を高めた剤形が注目されるようになり、とりわけ水なしでも服用でき、かつ嚥下が容易な口腔内崩壊錠や口腔内崩壊フィルムが次々と開発されている。しかし、こうした口腔内崩壊製剤の適用が適さない有効成分も数多く存在する。典型は、有効成分が苦味等の不快な味を有する場合で、口腔内で放出された有効成分が患者に強い不快感を与え、服薬コンプライアンスを著しく低下させる。また、有効成分が口腔内吸収される場合、水と共に服用する通常の製剤では認められなかった副作用が発現して、薬効の個体間差拡大等の問題を引き起こす懸念がある。さらに、有効成分が口腔内ないし食道への好ましくない直接作用を有する場合などは、一層適応が困難と考えられる。強い苦味や口腔内吸収が知られているバルデナフィルがこれに該当する。
On the other hand, immediate release tablets containing vardenafil need to be applied orally with water to facilitate rapid swallowing and efficient release of active ingredients. Considering the situation where patients take vardenafil, it seems that the convenience of patients will be greatly enhanced if they can be taken without water.
In recent years, dosage forms that have improved the convenience of patients when taking drugs have attracted attention, and in particular, orally disintegrating tablets and orally disintegrating films that can be taken without water and are easy to swallow have been developed one after another. Has been. However, there are many active ingredients that are not suitable for application of such orally disintegrating preparations. Typically, when the active ingredient has an unpleasant taste such as a bitter taste, the active ingredient released in the oral cavity gives a strong discomfort to the patient and significantly lowers compliance. In addition, when the active ingredient is absorbed into the oral cavity, there are concerns that side effects that have not been observed in normal preparations taken with water will develop and cause problems such as an increase in individual differences in drug efficacy. Furthermore, when the active ingredient has an undesired direct action on the oral cavity or esophagus, it is considered that adaptation is more difficult. This applies to vardenafil, which is known for its strong bitterness and absorption in the oral cavity.
上記の問題を回避しつつ利便性を向上するためには、経口粒子状医薬組成物において、一定時間口腔内で有効成分の放出を抑制することが必要である。一方、有効成分が十分な薬効を発現するためには、経口粒子状医薬組成物から有効成分が放出され、十分量体内に吸収される必要がある。そのためには、一定時間経口粒子状医薬組成物からの有効成分の放出を抑制した後は、経口粒子状医薬組成物が消化管下部へ移行する前に、有効成分が速やかに放出されて消化管上部で吸収されることが望ましい。また、有効成分放出を抑制する時間(ラグタイム)及びその後の溶出速度は、有効成分の特性に応じて適宜コントロールできることが望ましい。 In order to improve convenience while avoiding the above problems, it is necessary to suppress the release of active ingredients in the oral cavity for a certain period of time in an oral particulate pharmaceutical composition. On the other hand, in order for the active ingredient to exhibit a sufficient medicinal effect, it is necessary to release the active ingredient from the oral particulate pharmaceutical composition and absorb it in a sufficient amount. For this purpose, after the release of the active ingredient from the oral particulate pharmaceutical composition for a certain period of time, the active ingredient is rapidly released before the oral particulate pharmaceutical composition moves to the lower part of the digestive tract. Desirably absorbed at the top. Moreover, it is desirable that the time for suppressing the active ingredient release (lag time) and the subsequent dissolution rate can be appropriately controlled according to the characteristics of the active ingredient.
しかしながら、初期溶出の完全抑制とその後の速やかな溶出の両立は、従来技術からはなし得ない。例えば、初期溶出抑制のために徐放性皮膜を用いた場合、その後の速やかな有効成分の放出は難しい。また、胃溶性皮膜で口腔内における初期溶出を完全に抑制した場合、胃低酸症ないし胃無酸症の患者においては有効成分が全く吸収されない。徐放性皮膜と胃溶性皮膜との組み合わせ、または、徐放性皮膜と水溶性皮膜との組み合わせにおいても、初期有効成分溶出の抑制、その後の速やかな有効成分放出、および、ラグタイムのコントロール、を同時に満たすことは、極めて困難である。 However, it is impossible to achieve both the complete suppression of the initial dissolution and the subsequent rapid dissolution from the prior art. For example, when a sustained-release film is used to suppress initial dissolution, it is difficult to quickly release the active ingredient thereafter. In addition, when the initial dissolution in the oral cavity is completely suppressed with a gastric film, the active ingredient is not absorbed at all in patients with gastric hypoacidity or gastric anacidosis. Even in the combination of a sustained-release film and a gastric-soluble film, or a combination of a sustained-release film and a water-soluble film, suppression of initial active ingredient elution, rapid release of the active ingredient thereafter, and control of lag time, It is extremely difficult to satisfy these simultaneously.
従来、初期の有効成分の溶出を抑制した後、速やかに有効成分を放出するために、水溶性有効成分を含有する製剤に対して水不溶性物質と水溶性物質の混合皮膜を適用して被覆する方法が一般的に用いられている(例えば、特表2004−532257号公報(特許文献2))。混合皮膜中の水溶性物質が溶解するまでの間は、製剤内部への水の浸入速度が抑制され、初期の有効成分の物溶出を抑制することができる。その後皮膜中の水溶性物質が溶解すると、皮膜に細孔が形成され、水の浸入速度が加速するため、速やかな有効成分の放出を達成できる。しかしながら、この方法においても結局は、皮膜が薄いとラグタイムが不十分となり、皮膜が厚いと速やかな有効成分放出が抑制され、それらの両立は難しい。 Conventionally, after suppressing the elution of the active ingredient in the initial stage, a mixture containing a water-insoluble substance and a water-soluble substance is applied to the preparation containing the water-soluble active ingredient to quickly release the active ingredient. The method is generally used (for example, Japanese translations of PCT publication No. 2004-532257 (patent document 2)). Until the water-soluble substance in the mixed film is dissolved, the infiltration rate of water into the preparation is suppressed, and the initial elution of active ingredients can be suppressed. Thereafter, when the water-soluble substance in the film is dissolved, pores are formed in the film and the water infiltration speed is accelerated, so that the active ingredient can be released quickly. However, in this method as well, if the film is thin, the lag time is insufficient, and if the film is thick, rapid release of active ingredients is suppressed, making it difficult to achieve both.
一方、特許第4277904号(特許文献3)は、(i)粒子状医薬組成物の中心部に不快な味を有する薬物を含有する核粒子と、(ii)中間層に2種類の水溶性成分である、不溶化促進剤と不溶化物質を含有する層と、(iii)最外層に内部への水浸入速度を制御する水浸入量制御層とを含有してなる、口腔内崩壊錠に用いられる経口投与用時限放出型粒子状医薬組成物を開示している。そして、(ii)の不溶化促進剤と不溶化物質を含有する層の一例として、酸型不溶化促進剤(クエン酸、リンゴ酸、及び酒石酸など)と酸不溶化物質(メタクリル酸コポリマーのL、LD、Sど)を含有する層が開示され、(iii)の水浸入量制御層の具体例としてエチルセルロース、アミノアルキルメタクリレートコポリマーのRS、LD、E、L、LD、Sなどが開示されている。しかしながら、この開示組成物は、環境に有害な有機溶媒を使用した製造工程を用いていること、徐放皮膜強度に難があり、徐放皮膜を厚くすると速やかな薬物放出が難しくなること、口腔内で異物感の少ない小粒子では十分なラグタイム後の速やかな薬物放出が成功していないこと、およびラグタイム後の放出がやや徐放であることから、個別具体的な薬物に関しては未だ課題を残している。即ち、例えばバルデナフィルに関して、この開示組成物を応用しても、所定の制御されたラグタイム後に、患者の必要に即応できる処置用としてのバルデナフィルの標的プロファイルを実現することは難しい。 On the other hand, Japanese Patent No. 4277904 (Patent Document 3) discloses (i) a core particle containing a drug having an unpleasant taste at the center of a particulate pharmaceutical composition, and (ii) two kinds of water-soluble components in an intermediate layer. Orally used for orally disintegrating tablets, comprising a layer containing an insolubilization accelerator and an insolubilizing substance, and (iii) a water infiltration amount control layer for controlling the water infiltration rate in the outermost layer. Disclosed is a time-release particulate pharmaceutical composition for administration. As an example of the layer (ii) containing an insolubilization accelerator and an insolubilizing substance, an acid-type insolubilization accelerator (citric acid, malic acid, tartaric acid, etc.) and an acid insolubilizing substance (L, LD, S of methacrylic acid copolymer) And the like, and specific examples of the water penetration amount control layer of (iii) include ethyl cellulose and aminoalkyl methacrylate copolymer RS, LD, E, L, LD, and S. However, this disclosed composition uses a manufacturing process using an organic solvent harmful to the environment, has difficulty in sustained-release film strength, and increases the thickness of the sustained-release film; In the case of small particles with less foreign body feeling, rapid drug release after sufficient lag time is not successful, and release after lag time is somewhat sustained, so there is still a problem with individual specific drugs. Is leaving. That is, for example with respect to vardenafil, it is difficult to apply the disclosed composition to achieve a target profile of vardenafil for treatment that can quickly meet the needs of the patient after a predetermined controlled lag time.
特許第3350059号(特許文献4)は、消化管において急速に放出し且つ味を遮蔽する3層からなる経口剤形を提示している。即ち、薬剤として具体的には抗性交不能症薬シルデナフィルを選び、(i)該薬剤と微結晶セルロース等とからなる中核粒子と、(ii)水溶性ポリマーのヒドロキシプロピルメチルセルロースと水不溶性ポリマーのメタクリル酸塩コポリマーNEとからなる内側被覆層と、(iii)唾液に不溶性のメタクリル酸アミノアルキルコポリマーEと、要すればタルク等とからなる外側被覆層とを含有する3層粒子剤形を開示している。しかしながら、本発明者らの知見によれば、初期の溶出ラグタイムとその後の急速な放出特性はまだ不十分である。 Japanese Patent No. 3350059 presents a three-layer oral dosage form that releases rapidly in the gastrointestinal tract and masks the taste. Specifically, the drug is selected from the anti-impotence drug sildenafil, (i) core particles composed of the drug and microcrystalline cellulose, and (ii) the water-soluble polymer hydroxypropylmethylcellulose and the water-insoluble polymer methacrylic. Disclosed is a three-layer particle dosage form comprising an inner coating layer comprising an acid salt copolymer NE, and (iii) an aminoalkyl methacrylate copolymer E insoluble in saliva, and an outer coating layer comprising talc, if necessary. ing. However, according to the inventors' knowledge, the initial elution lag time and subsequent rapid release characteristics are still insufficient.
特表2009−524698号公報(特許文献5)は、弱塩基性薬物の、即時放出(IR)ビーズ、徐放性(SR)ビーズ及び/または時限パルス型放出(TPR)ビーズの1つ以上の手段を含む医薬多粒子剤形であって、溶解剤としての有機酸(クエン酸、フマル酸、リンゴ酸など)を薬剤と分離した形で含み、薬剤が放出完了するまで有機酸が枯渇しない医薬多粒子剤形を開示する。ここで用いるIRビーズは、味覚マスクしたIRビーズであって、その層構成の具体的実施態様として、(i)フマル酸で糖質球を積層化した酸コアと、(ii)該酸コアを不溶性のエチルセルロースとトリエチルクエン酸とで被覆したSR層と、(iii)該SR層を血圧降下剤カルベジロールで被覆した薬剤層と、(iv)該薬剤層を腸溶性のエチルセルロース/胃溶性のメタクリル酸アミノアルキルコポリマーEの50/50混合物で被覆した外層とからなる4層構造を有するIRビーズを開示している。しかしながら、本発明者らの知見によれば、例えば、薬剤として抗性交不能症薬バルデナフィルを用いた場合、所期の溶出ラグタイムは得られるものの、その後の急速な放出特性は不十分である。 JP 2009-524698 (Patent Document 5) discloses one or more of weakly basic drugs, immediate release (IR) beads, sustained release (SR) beads and / or timed pulsed release (TPR) beads. A pharmaceutical multiparticulate dosage form comprising means, comprising an organic acid (citric acid, fumaric acid, malic acid, etc.) as a solubilizing agent separated from the drug, and the organic acid is not depleted until the drug is completely released A multiparticulate dosage form is disclosed. The IR beads used here are taste-masked IR beads, and, as a specific embodiment of the layer structure, (i) an acid core in which sugar spheres are laminated with fumaric acid, and (ii) the acid core An SR layer coated with insoluble ethylcellulose and triethylcitric acid; (iii) a drug layer coated with the antihypertensive agent carvedilol; and (iv) the drug layer coated with enteric ethylcellulose / gastric methacrylic acid. An IR bead having a four-layer structure comprising an outer layer coated with a 50/50 mixture of aminoalkyl copolymer E is disclosed. However, according to the knowledge of the present inventors, for example, when the anti-sympathetic drug vardenafil is used as a drug, the desired elution lag time can be obtained, but the subsequent rapid release characteristics are insufficient.
特表2008−531614号公報(特許文献6)は、口中で迅速に溶解し、バイオアベイラビリティーの増加およびプラトー型の血漿濃度曲線を導くバルデナフィルの口腔内崩壊型の新規製剤を開示している。 JP-T-2008-531614 (Patent Document 6) discloses a novel orally disintegrating preparation of vardenafil that dissolves rapidly in the mouth, leading to an increase in bioavailability and a plateau-type plasma concentration curve.
しかしながら、そこで得られるバルデナフィルのバイオアベイラビリティーは、現在レビトラ(登録商標)として専ら適用されている即時放出(IR)錠剤に見られる標的プロファイルからは外れており、かつ、初期のラグタイムは得られない。 However, the bioavailability of vardenafil obtained there deviates from the target profile found in immediate release (IR) tablets currently applied exclusively as Levitra® and the initial lag time is obtained. Absent.
以上述べたように、PDE5阻害剤である抗性交不能症薬バルデナフィルは、現在、勃起不全の急性処置用の即時放出錠剤としての使用しか知られておらず、一方で、今日まで使用されてきたところの専ら即時放出用であるPDE5阻害剤バルデナフィルの医薬剤形は、それらの迅速な作用開始および比較的短い作用時間を介して、一定の要請に即応できる処置用としての標的プロファイルを満足してきた。 As mentioned above, the anti-sympathetic drug vardenafil, a PDE5 inhibitor, is currently only known as an immediate release tablet for acute treatment of erectile dysfunction, while it has been used to date However, pharmaceutical forms of the PDE5 inhibitor vardenafil, which are exclusively for immediate release, have met their targeted profiles for treatments that can respond quickly to certain requirements through their rapid onset of action and relatively short duration of action. .
従って、患者が薬を服用する際の利便性を高めた粒子状医薬組成物および/または該粒子状医薬組成物を含んでなる口腔内崩壊錠は水なしでも服用することができ、かつ嚥下が容易であることから、バルデナフィルの剤形としても、経口投与用放出制御型剤形の開発が待たれる。そのためには、如何にして、該剤形において、バルデナフィルの溶出プロファイルを、市販品の溶出プロファイルに合致させるかが大きな問題点となる。 Therefore, the particulate pharmaceutical composition and / or the orally disintegrating tablet comprising the particulate pharmaceutical composition with improved convenience when the patient takes the medicine can be taken without water and can be swallowed. Since it is easy, development of a controlled-release dosage form for oral administration is awaited as a dosage form of vardenafil. To that end, how to match the dissolution profile of vardenafil with that of a commercial product in the dosage form is a big problem.
本発明の課題は、上記問題点を解決すること、すなわち、薬物の味、口腔内吸収、口腔内ないし食道への直接作用等を回避低減しつつ、同時に経口吸収性を損なわない口腔内即崩壊製剤を提供すること、この目的のために、任意のラグタイムの後、任意の速度で薬物を放出させることにある。即ち、弱酸性ないし塩基性の有効成分を水なしで経口投与すべく設計された粒子状の製剤において、十分な長さのラグタイムを有し、かつラグタイム後に速やかに薬物の放出を開始し、かつ有効成分および製剤の特性および目的に応じて、ラグタイムの長さ及びその後の溶出速度を任意にコントロール可能な放出制御型粒子状医薬組成物(MRP)ならびに該組成物を含有する経口投与用崩壊性粒子状組成物(ODP)および口腔内崩壊錠(ODT)を提供することにある。更に、患者の胃内酸性度に関わらず、水なしで服用した場合にも、所定のラグタイム後に通常錠と同様の速度で有効成分を放出することが可能な放出制御型粒子状医薬組成物(MRP)等を提供することにある。 The object of the present invention is to solve the above-mentioned problems, i.e. avoiding and reducing the taste of the drug, oral absorption, direct action on the oral cavity or esophagus, etc. To provide a formulation, for this purpose, to release the drug at any rate after any lag time. That is, in a granular preparation designed to orally administer a weakly acidic or basic active ingredient without water, it has a sufficiently long lag time and immediately starts to release the drug after the lag time. And a controlled release particulate pharmaceutical composition (MRP) capable of arbitrarily controlling the length of lag time and the subsequent dissolution rate according to the characteristics and purpose of the active ingredient and the preparation, and oral administration containing the composition It is to provide a disintegrating particulate composition (ODP) and an orally disintegrating tablet (ODT). Furthermore, a controlled release particulate pharmaceutical composition capable of releasing an active ingredient at the same rate as a normal tablet after a predetermined lag time even when taken without water regardless of the acidity in the stomach of the patient. (MRP) and the like.
弱酸性ないし塩基性の有効成分として、例えば、塩基性薬物であるバルデナフィルで本発明の課題を具体的に説明すれば、市販品(即時放出型錠剤(IR錠剤)であるレビトラ(登録商標))の溶出プロファイルに合致した溶出プロファイルを示す、水なしで経口投与可能な、バルデナフィルの放出制御型粒子状医薬組成物(MRP)ならびに該組成物を含有する経口投与用崩壊性粒子状組成物(ODP)および口腔内崩壊錠(ODT)の提供にある。バルデナフィルの場合、徐放性皮膜がないと放出が早すぎるので調整が必要となる。 As a weakly acidic or basic active ingredient, for example, the problem of the present invention is specifically described with vardenafil, which is a basic drug, and is a commercially available product (Levitra (registered trademark) which is an immediate release tablet (IR tablet)). Controlled release particulate pharmaceutical composition (MRP) of vardenafil, which can be administered orally without water, showing an elution profile that matches the elution profile of: and an orally disintegrating particulate composition (ODP) containing the composition ) And orally disintegrating tablets (ODT). In the case of vardenafil, if there is no sustained release film, the release is too early and adjustment is necessary.
上記課題を解決するために本発明者らは、役割が明確に異なる複数の層によって被覆する手法が有効ではないかと着想して鋭意検討を進めた結果、驚くべきことに上記課題を解決することを見出し、本発明に到達したのである。
即ち、本発明は、経口投与用の多層構造の粒子状医薬組成物であって、少なくとも、薬学的に許容される有機酸を含有する核粒子、有機酸含有核粒子の外側に弱酸性ないし塩基性の有効成分を含有する有効成分含有層(「薬物含有層」と略すことあり。)、および薬物含有層の外側に胃溶性皮膜層を含んでなる、放出制御型粒子状医薬組成物(Modified-release particles:「MRP」と略すことあり。)を提供する。
更に、本発明は、前記医薬組成物において、医薬組成物からの薬物放出が実質的に口腔では起らず、その後、上部消化管において速やかに起るように構成されていることを特徴とする放出制御型粒子状医薬組成物(MRP)、およびその製造方法、を提供する。
本発明はまた、前記該放出制御型粒子状医薬組成物を含有する経口投与用崩壊性粒子状組成物(ODP)および口腔内崩壊錠剤(ODT)、ならびにそれらの製造方法を提供する。
In order to solve the above-mentioned problems, the present inventors have come up with an idea that a technique of covering with a plurality of layers having clearly different roles is effective, and as a result of diligent research, they have surprisingly solved the above-mentioned problems. The present invention has been reached.
That is, the present invention relates to a multi-layered particulate pharmaceutical composition for oral administration, comprising at least a core particle containing a pharmaceutically acceptable organic acid, a weakly acidic or basic base outside the organic acid-containing core particle. A controlled release particulate pharmaceutical composition (Modified) comprising an active ingredient-containing layer containing an active ingredient having a sexual activity (sometimes abbreviated as “drug-containing layer”) and a gastric soluble coating layer outside the drug-containing layer -release particles: "MRP" may be abbreviated.)
Furthermore, the present invention is characterized in that the pharmaceutical composition is configured such that drug release from the pharmaceutical composition does not substantially occur in the oral cavity and then rapidly occurs in the upper gastrointestinal tract. Provided are a controlled release particulate pharmaceutical composition (MRP) and a method for its production.
The present invention also provides a disintegrating particulate composition for oral administration (ODP) and an orally disintegrating tablet (ODT) containing the controlled release particulate pharmaceutical composition, and a method for producing them.
本発明の多層構造の放出制御型粒子状医薬組成物(MRP)においては、水なしで口腔内投与した場合でも、十分な長さのラグタイムが得られ、かつラグタイム後に速やかに薬物放出を開始させることができる。更に、薬物および製剤の特性および目的に応じて、ラグタイムの長さ及びその後の放出速度を任意にコントロールすることができる。また、胃無酸症患者に水なしで適用した場合にも、同様の効果を得ることができる。
本発明のMRPは、薬学的に許容される有機酸を含有する核粒子、その外側に薬物を含有する薬物含有層、およびその外側には製剤内部への外液(唾液等)の浸入速度を制御する外液(唾液等)浸入量制御層としての胃溶性皮膜層を被覆した粒子状医薬組成物であって、口腔内での十分な長さのラグタイムを有し、ラグタイム後に上部消化管(U−GIT)で速やかに薬物を放出可能で、かつ被覆量や各被覆層の成分を変化させることによって、ラグタイムの長さを約3〜15分の範囲で、好ましくは5〜10分の範囲(例えば、10分)でコントロール可能であるという特徴を有する。ラグタイムの間は弱酸性ないし塩基性の有効成分(例えば、塩基性薬剤であるバルデナフィル)の実質的放出はなく、ラグタイム終了後に直ちに弱酸性または塩基性薬剤(例えば、バルデナフィル)が放出されることが好ましい。
ここで本発明におけるラグタイム(本明細書において、ラグ時間ということもある)の定義について以下に説明する。
ラグタイムの定義に関しては、厚生労働省医薬食品局審査管理課長より平成18年11月24日付各都道府県衛生主管部(局)長宛に発信された薬食審査発第1124004号の「後発医薬品の生物学的同等性試験ガイドライン等の一部改正について」別紙1第19頁に「製剤から薬物が表示含量の5%溶出するまでに要する時間をラグ時間とする」とされている。また、一般的な苦味のマスキングなどでは、例えば20%溶出しても苦味を感じなければ、20%溶出するまでに要する時間と定義するケースも考えられる。しかし、本発明では、実質的に初期漏出がない、より厳しいラグタイム基準が求められる場面、例えば、口腔内吸収の寄与が無視できない場面などを想定している。従って、本発明においては、ラグタイムを「1%溶出するまでに要する時間」と定義する。本発明のラグタイムの計算方法は、厚生労働省医薬食品局審査管理課より平成18年11月24日付各都道府県衛生主管部(局)薬務主管課宛に発信された事務連絡の「「後発医薬品の生物学的同等性試験ガイドラインに関する質疑応答集(Q&A)について」等の改正について」別紙1第24−25頁に記載の「手順1)ラグ時間の計算」に準じたものである。
個々の溶出曲線に対する、溶出率がdA%に到達する時間tAの計算式
tA=t1+(dA−d1)X(t2−t1)/(d2−d1)
ここで、
t1:溶出率がdA%に到達する直前の測定時間
t2:溶出率がdA%を超えた直後の測定時間
d1:時間t1における溶出率
d2:時間t2における溶出率
ただし、dA=1%
In the multilayer controlled-release particulate pharmaceutical composition (MRP) of the present invention, even when administered orally without water, a sufficiently long lag time is obtained, and drug release is promptly performed after the lag time. Can be started. Furthermore, the length of the lag time and the subsequent release rate can be arbitrarily controlled according to the characteristics and purpose of the drug and the preparation. Moreover, the same effect can be acquired also when it applies to a stomach anacidosis patient without water.
The MRP of the present invention has a core particle containing a pharmaceutically acceptable organic acid, a drug-containing layer containing a drug on its outer side, and an outer liquid (saliva etc.) penetration rate inside the preparation on its outer side. A particulate pharmaceutical composition coated with a gastric soluble coating layer as an external fluid (saliva, etc.) infiltration control layer to be controlled, having a sufficiently long lag time in the oral cavity, and upper digestion after the lag time The drug can be rapidly released by a tube (U-GIT), and the length of lag time is changed within a range of about 3 to 15 minutes, preferably 5 to 10 by changing the coating amount and the components of each coating layer. It has a feature that it can be controlled within a range of minutes (for example, 10 minutes). There is no substantial release of a weakly acidic or basic active ingredient (eg, vardenafil, a basic drug) during the lag time, and a weakly acidic or basic drug (eg, vardenafil) is released immediately after the lag time is over It is preferable.
Here, the definition of the lag time in the present invention (sometimes referred to as lag time in the present specification) will be described below.
For the definition of lag time, see “Regarding Generic Drugs” in Drug Medication Review Issue No. 1124004, which was sent to the Director of the Prefectural Sanitation Supervising Department (Department) on November 24, 2006 by the Director of the Examination Management Division, Ministry of Health, Labor and Welfare. “Partial revision of guidelines for bioequivalence testing, etc.” “Appendix 1, page 19” states that “the time required for the drug to elute from the preparation to 5% of the indicated content is the lag time”. Further, in general masking of bitterness, for example, if no bitterness is felt even if 20% is eluted, it may be defined as the time required until 20% is eluted. However, the present invention assumes a scene in which a stricter lag time standard is substantially obtained with substantially no initial leakage, for example, a scene where the contribution of oral absorption cannot be ignored. Therefore, in the present invention, the lag time is defined as “time required for 1% elution”. The calculation method of the lag time of the present invention is as follows: “Ministry of Health, Labor and Welfare, Pharmaceutical and Food Affairs Bureau, Administration and Administration Division,” November 24, 2006. This is in accordance with “Procedure 1) Calculation of lag time” described in Attachment 1 pp. 24-25 “Amendments to“ Q & A ”on Bioequivalence Guidelines for Pharmaceuticals”.
Calculation formula of time tA for elution rate to reach dA% for each elution curve tA = t1 + (dA−d1) X (t2−t1) / (d2−d1)
here,
t1: Measurement time immediately before the elution rate reaches dA% t2: Measurement time immediately after the elution rate exceeds dA% d1: Elution rate at time t1 d2: Elution rate at time t2 where dA = 1%
本発明における有機酸含有核粒子中の有機酸は薬学的に許容される有機酸である。そして、当該有機酸は胃溶性皮膜層を溶解させることにより弱酸性ないし塩基性の有効成分の放出を制御するという機能を有する。即ち、有機酸の溶出開始は弱酸性ないし塩基性の有効成分の溶出開始と完全に同期するが、その後の溶出プロファイルは必ずしも一致しない。有機酸は胃溶性皮膜層を通過して侵入してくる外液(唾液等)によって、所定のラグタイムに相当する期間内に核粒子から外層へと移動し、胃溶性皮膜層の一部ないし全部を溶解させて有機酸自体の溶出は完了する。薬物は胃溶性皮膜の溶解により暴露された表面積に応じた速度で溶出する。 The organic acid in the organic acid-containing core particles in the present invention is a pharmaceutically acceptable organic acid. The organic acid has a function of controlling the release of a weakly acidic or basic active ingredient by dissolving the gastric film layer. That is, the start of elution of the organic acid is completely synchronized with the start of elution of the weakly acidic or basic active ingredient, but the subsequent elution profiles do not necessarily match. The organic acid moves from the core particle to the outer layer within a period corresponding to a predetermined lag time by the external liquid (saliva etc.) entering through the gastric film layer, and a part or part of the gastric film layer is transferred. All are dissolved and the elution of the organic acid itself is complete. The drug elutes at a rate depending on the surface area exposed by dissolution of the gastric film.
本発明により、弱酸性ないし塩基性の有効成分を経口投与すべく設計された製剤において、十分な長さのラグタイムを有し、かつラグタイム後に速やかに薬物を放出でき、かつ薬物および製剤の特性および目的に応じて、ラグタイムの長さを任意にコントロール可能な経口投与用の放出制御型粒子状医薬組成物(MRP)ならびに該組成物を含有する経口投与用崩壊性粒子状組成物(ODP)および口腔内崩壊錠(ODT)を得ることができる。
具体的には、バルデナフィルをMRP、ODPまたはODTとして水なしで服用することができて、かつ嚥下が容易である。更に、バルデナフィルの溶出プロファイルを市販品の溶出プロファイルに合致させることができるため、確立しているバルデナフィルの血中濃度推移、更には有効性及び安全性を損なうことなく適用することができる。
According to the present invention, in a preparation designed for oral administration of a weakly acidic or basic active ingredient, the drug has a sufficiently long lag time, can be released quickly after the lag time, and Depending on the characteristics and purpose, the controlled release particulate pharmaceutical composition (MRP) for oral administration capable of arbitrarily controlling the length of lag time, and the collapsible particulate composition for oral administration containing the composition ( ODP) and orally disintegrating tablets (ODT) can be obtained.
Specifically, vardenafil can be taken without water as MRP, ODP or ODT and is easy to swallow. Furthermore, since the elution profile of vardenafil can be matched with the elution profile of a commercial product, it can be applied without impairing the established blood concentration of vardenafil, and further, the effectiveness and safety.
以下、本発明の実施の形態について、詳細に説明する。
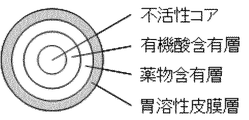
まず、本発明の放出制御型粒子状医薬組成物(MRP)の層構成を説明する。本発明の経口投与用に用いられる放出制御型粒子状医薬組成物(MRP)は、その基本的な層構成として、粒子の中央に有機酸含有核粒子(コア)、その外側に弱酸性ないし塩基性の有効成分の活性薬物成分(API)を含有する薬物含有層(薬物層)、更にその外側に胃溶性皮膜層をこの順に含んでなる多層構造を基本層構成とする粒子である。
放出制御型粒子状医薬組成物(MRP)は、有機酸含有核粒子と薬物含有層との間に徐放性皮膜層(SR層)を有してよい。徐放性皮膜層(SR層)は半透膜としての性質を有するものであれば特に限定するものではないが、エチルセルロースまたはアクリル酸エチル・メタクリル酸メチルコポリマーを含有することが好ましい。
有機酸含有核粒子は、有機酸自体を球形に製するか、不活性コアの外側に有機酸含有層を被覆した粒子であってよい。不活性コアとしては、結晶セルロース粒(結晶セルロース(粒)と同義であり、本明細書においては、結晶セルロース(粒)またはセルロース球ということもある。)、白糖・デンプン球状顆粒、精製白糖球状顆粒、乳糖・結晶セルロース球状顆粒およびマンニトール球状顆粒からなる群から選ばれた少なくとも1つが好ましく、中でも、結晶セルロース(粒)が特に好ましい。
有機酸含有核粒子、徐放性皮膜層、薬物含有層および胃溶性皮膜層の少なくとも1つの表面をシールコート被覆層で被覆してよい。有機酸含有核粒子、徐放性皮膜層および薬物含有層の各層をそれぞれシールコート皮膜層で被覆するのが好ましい。シールコート皮膜層は水溶性被覆層であれば特に限定するものではなく、例えば、ヒプロメロース(ヒドロキシプロピルメチルセルロース)、メチルセルロースなどを含有するシールコート被覆層が挙げられ、必要に応じてそれらにマクロゴール(ポリエチレングリコール)などの可塑剤を加えることができる。また別のシールコート層の例として、エリスリトール等の糖アルコールによる表面改質が挙げられ、通常、ポピドンやヒドロキシプロピルセルロース等の結合剤と混合して使用される。これらの中では、ヒプロメロース又はメチルセルロースを含有するシールコート被覆層が好ましい。
Hereinafter, embodiments of the present invention will be described in detail.
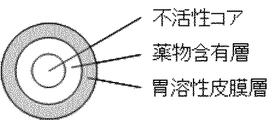
First, the layer structure of the controlled release particulate pharmaceutical composition (MRP) of the present invention will be described. The controlled release particulate pharmaceutical composition (MRP) used for oral administration of the present invention has, as its basic layer structure, an organic acid-containing core particle (core) at the center of the particle, and a weakly acidic or base on the outside. It is a particle having a basic layer constitution of a multilayer structure comprising a drug-containing layer (drug layer) containing an active drug component (API) as an active ingredient and a gastric film layer on the outer side thereof in this order.
The controlled release particulate pharmaceutical composition (MRP) may have a sustained release coating layer (SR layer) between the organic acid-containing core particles and the drug-containing layer. The sustained-release coating layer (SR layer) is not particularly limited as long as it has properties as a semipermeable membrane, but preferably contains ethyl cellulose or ethyl acrylate / methyl methacrylate copolymer.
The organic acid-containing core particles may be particles in which the organic acid itself is made into a spherical shape, or the organic acid-containing layer is coated on the outer side of the inert core. As the inert core, crystalline cellulose grains (synonymous with crystalline cellulose (grains), also referred to as crystalline cellulose (grains) or cellulose spheres in this specification), sucrose / starch spherical granules, purified white sugar spheres At least one selected from the group consisting of granules, lactose / crystalline cellulose spherical granules and mannitol spherical granules is preferred, and crystalline cellulose (grains) is particularly preferred.
At least one surface of the organic acid-containing core particles, sustained-release coating layer, drug-containing layer, and gastric soluble coating layer may be coated with a seal coat coating layer. It is preferable to coat each of the organic acid-containing core particles, the sustained-release coating layer and the drug-containing layer with a seal coat coating layer. The seal coat film layer is not particularly limited as long as it is a water-soluble coating layer, and examples thereof include a seal coat coating layer containing hypromellose (hydroxypropylmethylcellulose), methylcellulose, etc., and macrogol ( A plasticizer such as polyethylene glycol) can be added. Another example of the seal coat layer is surface modification with a sugar alcohol such as erythritol, which is usually used by mixing with a binder such as popidone or hydroxypropylcellulose. In these, the seal-coat coating layer containing a hypromellose or methylcellulose is preferable.
本発明の放出制御型粒子状医薬組成物(MRP)は、医薬組成物からの薬物放出が実質的に口腔内では起らず、その後、上部消化管において速やかに起るように構成されていることを特徴とする。本発明の胃溶性皮膜はpHが弱酸性付近の口腔内では溶解せず、従って、活性薬物成分が口腔内で溶出するのを実質的に阻止し(この期間をラグタイムと称する)、例えば、有効成分の不快な味による患者の不快感を解消する。ラグタイムは3〜15分、好ましくは5〜10分の範囲(例えば、10分)であってよい。粒子外部から胃溶性皮膜を通って外液(唾液等)が侵入する時間によって生じるラグタイム後に、有機酸含有核粒子から有機酸が溶出し、この有機酸が胃溶性皮膜の一部又は全部を所望の速度で溶解させる。
The controlled release particulate pharmaceutical composition (MRP) of the present invention is configured such that drug release from the pharmaceutical composition does not substantially occur in the oral cavity and then rapidly occurs in the upper gastrointestinal tract. It is characterized by that. The gastric film of the present invention does not dissolve in the oral cavity where the pH is near weak acidity, and therefore substantially prevents the active drug component from eluting in the oral cavity (this period is referred to as lag time), for example, Eliminate patient discomfort due to unpleasant taste of active ingredients. The lag time may be in the range of 3-15 minutes, preferably 5-10 minutes (
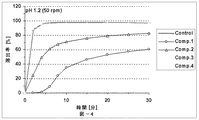
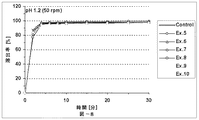
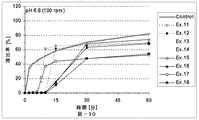
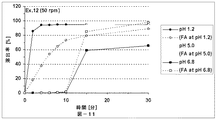
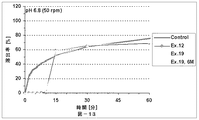
本発明の放出制御型粒子状医薬組成物(MRP)は、前述のとおり、有機酸が胃溶性皮膜層を内側から溶解させることにより弱酸性ないし塩基性の有効成分の放出が始まることを特徴とする。この特徴について以下、説明する。即ち、本発明における有機酸の主要な機能は外層の胃溶性皮膜を溶解させることにあり、低酸症ないし無酸症の患者でもバイオアベイラビリティーを低下させない仕掛けである。具体的に説明すると、図11(実施例4)には、本発明のMRPの、pH6.8(口腔のpHに相当する。)における、活性薬剤成分API(ここでは、バルデナフィル)の溶出率と有機酸(フマル酸)の溶出率とを時間軸に対してプロットした溶出プロファイルも示してある。双方の溶出率が共にほぼゼロである10分間のラグタイムの後に、フマル酸は5分間で約90%溶出するのに対して、バルデナフィルの溶出率は5分間で約60%に止まる。この結果は、フマル酸が外層の胃溶性皮膜層の一部のみを溶解したことを示す。言い換えると、バルデナフィルの溶出率は、フマル酸の配合量並びに徐放性皮膜層の量により適度に調整されている。なお、図11において、バルデナフィルの溶出はpH5.0〜6.8ではフマル酸によって制御されており、pH1.2の強酸性下ではフマル酸とは無関係に速やかに溶出していることが分かる。 As described above, the controlled release particulate pharmaceutical composition (MRP) of the present invention is characterized in that the release of a weakly acidic or basic active ingredient starts when the organic acid dissolves the gastric film layer from the inside. To do. This feature will be described below. That is, the main function of the organic acid in the present invention is to dissolve the gastric film of the outer layer, and is a mechanism that does not reduce bioavailability even in hypoacidic or anacidic patients. Specifically, FIG. 11 (Example 4) shows the dissolution rate of the active drug component API (here, vardenafil) at pH 6.8 (corresponding to the pH of the oral cavity) of the MRP of the present invention. An elution profile in which the elution rate of organic acid (fumaric acid) is plotted against the time axis is also shown. After a lag time of 10 minutes, where both elution rates are both nearly zero, fumaric acid elutes about 90% in 5 minutes, while vardenafil only remains about 60% in 5 minutes. This result indicates that fumaric acid dissolved only a part of the outer gastric film layer. In other words, the dissolution rate of vardenafil is appropriately adjusted by the amount of fumaric acid and the amount of the sustained-release coating layer. In FIG. 11, it can be seen that the elution of vardenafil is controlled by fumaric acid at pH 5.0 to 6.8, and is rapidly eluted under strong acidity at pH 1.2 regardless of fumaric acid.
薬学的に許容される有機酸としては、胃溶性皮膜外層を溶解させるに足るpH(例えば、胃溶性皮膜がアミノアルキルメタアクリレートコポリマーEあるいはポリビニルアセタールジエチルアミノアセテートの場合、pH5以下)を与える酸であって、薬学的に許容されるものであれば特に制限はない。例えば、フマル酸、酒石酸、コハク酸、クエン酸、乳酸、フタル酸、酢酸、蓚酸、マロン酸、アジピン酸、フィチン酸、グルタル酸、マレイン酸、リンゴ酸、マンデル酸、アスコルビン酸、安息香酸およびトルエンスルホン酸等、並びにそれらの水和物から選ばれた少なくとも1つの有機酸が挙げられる。これらの内、フマル酸、酒石酸、コハク酸およびクエン酸、並びにそれらの水和物が好ましく、中でも、フマル酸がその適度な酸性度及び水溶性により制御のしやすさやハンドリングの面から特に好ましい。 The pharmaceutically acceptable organic acid is an acid that provides a pH sufficient to dissolve the outer layer of the gastric film (for example, when the gastric film is aminoalkyl methacrylate copolymer E or polyvinyl acetal diethylaminoacetate, the pH is 5 or less). There is no particular limitation as long as it is pharmaceutically acceptable. For example, fumaric acid, tartaric acid, succinic acid, citric acid, lactic acid, phthalic acid, acetic acid, succinic acid, malonic acid, adipic acid, phytic acid, glutaric acid, maleic acid, malic acid, mandelic acid, ascorbic acid, benzoic acid and toluene Examples thereof include at least one organic acid selected from sulfonic acids and the like, and hydrates thereof. Of these, fumaric acid, tartaric acid, succinic acid and citric acid, and hydrates thereof are preferred, and fumaric acid is particularly preferred from the viewpoint of ease of control and handling due to its moderate acidity and water solubility.
有機酸は、それ自体を球形に造粒するか、賦形剤(例えば結晶セルロース、乳糖、トウモロコシデンプン等)と混合して有機酸含有核粒子を形成するか、核となる不活性コアの外側に被覆して有機酸含有核粒子を形成してもよい。有機酸含有核粒子の大きさは、10〜1000μm、好ましくは、100〜300μmの範囲、例えば、約200μmである。 Organic acids can be granulated themselves or mixed with excipients (eg, crystalline cellulose, lactose, corn starch, etc.) to form organic acid-containing core particles, or outside the core inert core The organic acid-containing core particles may be formed by coating with The size of the organic acid-containing core particles is 10 to 1000 μm, preferably in the range of 100 to 300 μm, for example, about 200 μm.
有機酸含有層を不活性コアの外側に被覆する場合、有機酸単独で被覆層を形成してもよいが、通常は、水溶性の高分子結合剤(例えば、ヒドロキシプロピルセルロース、ピプロメロース、メチルセルロース、ポビドン等)や充填剤(タルク、シリカ、ステアリン酸マグネシウム等)を含んでよい。有機酸および水溶性高分子結合剤を水、アルコール(例えば、エタノール、メタノール)またはそれらの混合溶媒に溶解ないし分散させた溶液に、要すれば、充填剤を分散させ、不活性コアに塗布して被覆層を形成する。塗布方法は、流動層コーティング装置、転動流動層コーティング装置、遠心転動コーティング装置など、粒子状医薬組成物に被覆することが可能ないずれの方法を用いてもよい。例えば、転動流動層コーティング装置中で、不活性コア粒子を流動させながら、スプレーガンにて有機酸等の被覆成分を含有する液を必要量噴霧すればよい。 When the organic acid-containing layer is coated on the outside of the inert core, the organic acid alone may form the coating layer, but usually a water-soluble polymer binder (for example, hydroxypropylcellulose, piperomellose, methylcellulose, Povidone and the like) and fillers (talc, silica, magnesium stearate, etc.). The organic acid and water-soluble polymer binder are dissolved or dispersed in water, alcohol (eg, ethanol, methanol) or a mixed solvent thereof, and if necessary, the filler is dispersed and applied to the inert core. To form a coating layer. As a coating method, any method capable of coating the particulate pharmaceutical composition, such as a fluidized bed coating apparatus, a rolling fluidized bed coating apparatus, and a centrifugal rolling coating apparatus, may be used. For example, a necessary amount of a liquid containing a coating component such as an organic acid may be sprayed with a spray gun while flowing the inert core particles in a rolling fluidized bed coating apparatus.
有機酸の量は、胃溶性皮膜を溶解するのに十分な量、言い換えれば局所的なpHを5以下にするのに十分な量、即ち、化学量論量で云えば、胃溶性皮膜に存する塩基性窒素と等量以上の酸当量であるのが好ましい。所望するラグタイムとその後の溶出速度の長短によっては、胃溶性皮膜に存する塩基性窒素の等量を大きく超える量の有機酸を用いてよい。ラグタイムは3〜15分、好ましくは、5〜10分の範囲(例えば、10分)であってよい。ラグタイムの間は弱酸性ないし塩基性の有効成分、(例えば、塩基性薬物のバルデナフィル)が実質的に放出されず、ラグタイム後、急速に薬物の放出が始まるのが好ましい。バルデナフィルの場合でいえば、3〜15分のラグタイムの後、胃腸管において直ちにバルデナフィルが放出されることにより、市販品(レビトラ(登録商標))の溶出プロファイルに合致させることができるため、確立しているバルデナフィルのバイオアベイラビリティー・プロファイルを損なうことなく適用することができる。
胃溶性皮膜を構成する胃溶性高分子としてアミノアルキルメタクリレートコポリマーE(オイドラギット(登録商標)E100またはEPO)を用い、有機酸としてフマル酸を用いる場合には、例えば、オイドラギット(登録商標)EPOが100重量部に対して、フマル酸が2〜200重量部、好ましくは、5〜100重量部、より好ましくは、15〜50重量部、例えば30重量部であってよい。
The amount of the organic acid is sufficient to dissolve the gastric film, in other words, an amount sufficient to bring the local pH to 5 or less, that is, in stoichiometric amount, it exists in the gastric film. The acid equivalent is preferably equal to or more than that of basic nitrogen. Depending on the desired lag time and the length of the subsequent elution rate, an amount of organic acid greatly exceeding the equivalent amount of basic nitrogen present in the gastric film may be used. The lag time may be in the range of 3-15 minutes, preferably 5-10 minutes (
When aminoalkyl methacrylate copolymer E (Eudragit (registered trademark) E100 or EPO) is used as the gastric polymer constituting the gastric film and fumaric acid is used as the organic acid, for example, Eudragit (registered trademark) EPO is 100. The fumaric acid may be 2 to 200 parts by weight, preferably 5 to 100 parts by weight, more preferably 15 to 50 parts by weight, for example, 30 parts by weight with respect to parts by weight.
有機酸含有核粒子の外側に、弱酸性ないし塩基性の有効成分を含有する有効成分含有層(薬物含有層または薬物層と称することあり。)を被覆する。有効成分は単独で被覆してもよいが、通常は、水溶性の高分子結合剤(例えば、ヒドロキシプロピルセルロース、ヒプロメロース、メチルセルロース、ポビドン等)や滑沢剤(タルク、ステアリン酸マグネシウム等)を含んでよい。薬物および水溶性高分子結合剤を水、アルコール(エタノール、メタノール)またはそれらの混合溶媒に溶解ないし分散させた溶液に、要すれば、充填剤を分散させ、有機酸含有核粒子に塗布して薬物層を形成する。塗布方法は、流動層コーティング装置、転動流動層コーティング装置、遠心転動コーティング装置など、粒子状医薬組成物に被覆することが可能ないずれの方法を用いてもよい。例えば、転動流動層コーティング装置中で、有機酸含有核粒子を流動させながら、スプレーガンにて薬物含有液を必要量噴霧すればよい。 An active ingredient-containing layer containing a weakly acidic or basic active ingredient (sometimes referred to as a drug-containing layer or a drug layer) is coated on the outside of the organic acid-containing core particles. The active ingredient may be coated alone, but usually contains a water-soluble polymer binder (for example, hydroxypropylcellulose, hypromellose, methylcellulose, povidone, etc.) and a lubricant (talc, magnesium stearate, etc.). It's okay. The drug and water-soluble polymer binder are dissolved or dispersed in water, alcohol (ethanol, methanol) or a mixed solvent thereof. If necessary, the filler is dispersed and applied to the organic acid-containing core particles. Form a drug layer. As a coating method, any method capable of coating the particulate pharmaceutical composition, such as a fluidized bed coating apparatus, a rolling fluidized bed coating apparatus, and a centrifugal rolling coating apparatus, may be used. For example, a required amount of the drug-containing liquid may be sprayed with a spray gun while flowing the organic acid-containing core particles in a rolling fluidized bed coating apparatus.
本発明に用いられる薬物としては、唾液(中性の緩衝液)と接触したとき最外層の胃溶性皮膜を直ちに溶解してしまう様な強酸性でなければ何れの薬物でもよく、即ち、弱酸性ないし塩基性の有効成分であって、治療学的に有効な活性成分、あるいは予防学的に有効な活性成分であれば特に限定されない。該弱酸性ないし塩基性の有効成分の飽和水溶液のpHは2.5〜14、望ましくは3.5〜14であり、弱酸性ないし塩基性の有効成分には、フリー体または製薬的に許容され得る塩が含まれる。かかる医薬活性成分としては、cGMP特異的ホスホジエステラーゼ阻害剤、例えば、バルデナフィル、シルデナフィル等の抗勃起不全症薬、塩酸エタンブトール等の抗結核薬、塩酸バルニジピン、塩酸ニカルジピン、塩酸インデノロール、塩酸ヒドララジン等の循環器官用薬、塩酸クロルプロマジン、塩酸アミトリプチリン塩酸モペロン等の抗精神薬、塩酸ラモセトロン、塩酸グラニセトロン、塩酸オンダンセトロン等の制吐剤、塩酸プロパフェノン等の抗不整脈薬、塩酸ジブカイン等の表面麻酔薬、塩酸ラニチジン等の消化管用薬、塩酸チアプリド、塩酸ビフェメラン等の中枢神経系用薬、塩酸アンピシリンフタリジル等の抗生物質、塩酸テラゾシン等のBPH治療剤等が挙げられる。薬物は、強酸性でない限りにおいて、フリー体または製薬的に許容され得る塩のいずれをも用いることができる。また、有効成分は、1種または2種以上組合せて用いることもできる。またこれらの薬物は、本発明に適用でき得る一例であり、限定的に解釈されるべきではない。本発明の粒子状医薬組成物に含有させる有効成分としては、特に時限放出が求められ、かつラグタイム後速やかに溶出することが求められる薬物、特に好ましくは、バルデナフィル、シルデナフィル等の抗勃起不全症薬が挙げられるが、中でもバルデナフィルが好ましく用いられる。 The drug used in the present invention may be any drug as long as it is not strongly acidic so that it immediately dissolves the outermost gastric soluble film when contacted with saliva (neutral buffer), that is, weakly acidic It is not particularly limited as long as it is a basic active ingredient and is a therapeutically effective active ingredient or a prophylactically effective active ingredient. The pH of the saturated aqueous solution of the weakly acidic to basic active ingredient is 2.5 to 14, preferably 3.5 to 14. The weakly acidic to basic active ingredient is free or pharmaceutically acceptable. The resulting salt is included. Such pharmaceutically active ingredients include cGMP-specific phosphodiesterase inhibitors, for example, anti-erectile dysfunction drugs such as vardenafil and sildenafil, antituberculosis drugs such as ethambutol hydrochloride, circulatory organs such as varnidipine hydrochloride, nicardipine hydrochloride, indenolol hydrochloride, hydralazine hydrochloride, etc. Antipsychotics such as chlorpromazine hydrochloride, amitriptyline hydrochloride moperone hydrochloride, antiemetics such as ramosetron hydrochloride, granisetron hydrochloride, ondansetron hydrochloride, antiarrhythmic drugs such as propafenone hydrochloride, surface anesthetics such as dibucaine hydrochloride, ranitidine hydrochloride And other drugs for gastrointestinal tract, central nervous system drugs such as tiapride hydrochloride and bifemelane hydrochloride, antibiotics such as ampicillin phthalidyl hydrochloride, and BPH therapeutic agents such as terazosin hydrochloride. As long as the drug is not strongly acidic, either a free form or a pharmaceutically acceptable salt can be used. Moreover, an active ingredient can also be used 1 type or in combination of 2 or more types. These drugs are examples that can be applied to the present invention, and should not be construed as limiting. As an active ingredient to be contained in the particulate pharmaceutical composition of the present invention, a drug that requires timed release and that is required to elute quickly after a lag time, particularly preferably anti-erectile dysfunction such as vardenafil, sildenafil, etc. Among them, vardenafil is preferably used.
バルデナフィルとしては、塩酸塩および有機酸塩等の酸付加物、ならびに、酸フリー体のいずれも使用できる。中でも、バルデナフィル塩酸塩の水和物(特に、三水和物)が、水溶解性および安定性の点から好ましい。尚、バルデナフィル塩酸塩三水和物の飽和水溶液のpHは4.0であった。 As vardenafil, any of acid adducts such as hydrochloride and organic acid salt, and acid free form can be used. Among these, hydrates (particularly trihydrate) of vardenafil hydrochloride are preferable from the viewpoint of water solubility and stability. The pH of the saturated aqueous solution of vardenafil hydrochloride trihydrate was 4.0.
本発明の放出制御型粒子状医薬組成物(MRP)中の有効成分の配合量は、通常有効成分の種類あるいは医薬用途(適応症)により適宜選択されるが、治療学的に有効な量あるいは予防学的に有効な量であれば特に制限されない。被覆される粒子と等量付近とすると、小型化・高含量化できるため好ましい。好ましくは粒子状医薬組成物全体の0.5〜90重量%であり、より好ましくは1.0〜80重量%、さらに好ましくは5.0〜70重量%である。
具体的には、例えば、抗勃起不全症薬バルデナフィルの場合、バルデナフィルとしてMRP全体の0.5〜90重量%であり、より好ましくは1〜70重量%、さらに好ましくは5〜50重量%、例えば10〜20重量%である。
但し、ここに示した有効成分の配合量は本発明に適用できうる一例であり、限定的に解釈されるべきではない。有効成分含有層(薬物含有層)は、好ましくは、高分子結合剤を含んでよい。高分子結合剤としては、例えば、ヒドロキシプロピルセルロース、ヒプロメロース、メチルセルロース、ポビドン、コーンスターチ、デンプン等の水溶性ポリマーが挙げられるが、中でも、ヒドロキシプロピルセルロースが好ましい。有効成分含有層は、要すれば、タルクやステアリン酸(またはその塩)等の滑沢剤を含んでよい。
本発明において有効成分含有層で被覆するにより形成する有効成分含有層被覆粒子の大きさは、10〜1000μm、好ましくは、100〜300μmの範囲、例えば、約250μmである。
The compounding amount of the active ingredient in the controlled-release particulate pharmaceutical composition (MRP) of the present invention is usually appropriately selected depending on the type of the active ingredient or the pharmaceutical use (indication). The amount is not particularly limited as long as it is a prophylactically effective amount. It is preferable that the amount is about the same as the particles to be coated because the size and content can be increased. Preferably it is 0.5 to 90% by weight of the whole particulate pharmaceutical composition, more preferably 1.0 to 80% by weight, still more preferably 5.0 to 70% by weight.
Specifically, for example, in the case of the anti-erectile dysfunction drug vardenafil, it is 0.5 to 90% by weight of the whole MRP as vardenafil, more preferably 1 to 70% by weight, still more preferably 5 to 50% by weight, for example, 10 to 20% by weight.
However, the compounding amount of the active ingredient shown here is an example that can be applied to the present invention, and should not be interpreted in a limited manner. The active ingredient-containing layer (drug-containing layer) may preferably contain a polymer binder. Examples of the polymer binder include water-soluble polymers such as hydroxypropylcellulose, hypromellose, methylcellulose, povidone, corn starch, and starch, among which hydroxypropylcellulose is preferable. If necessary, the active ingredient-containing layer may contain a lubricant such as talc or stearic acid (or a salt thereof).
In the present invention, the active ingredient-containing layer-coated particles formed by coating with the active ingredient-containing layer have a size of 10 to 1000 μm, preferably 100 to 300 μm, for example, about 250 μm.
本発明の放出制御型粒子状医薬組成物(MRP)は、有効成分含有層の外側を胃溶性皮膜層で被覆する。胃溶性皮膜層は、粒子状医薬組成物が口腔内に存する間は有効成分が溶出されるのを防いで適切なラグタイムを付与する役目を持ち、所定のラグタイム経過後は、外層から粒子内に侵入する外液(唾液等)によって有機酸含有核粒子から溶出される有機酸に接触して一部ないし全部が溶解し、有効成分の溶出を促す役目を有する。即ち、胃溶性皮膜層は、所望の溶出プロファイルを達成するための重要な役目の一つを担っている。この様な機能を満たす胃溶性皮膜材料は、唾液に不溶性のポリマーであり、例えば、メタクリル酸ブチル/(2−ジメチルアミノエチル)メタクリル酸塩/メタクリル酸メチルコポリマーおよびジエチルアミノ酢酸ポリビニルアセタールのようなメタクリル酸アミノアルキルコポリマーが挙げられる。商業的に入手可能な高分子としては、アミノアルキルメタクリレートコポリマーE(オイドラギット(Eudragit)(登録商標)E100およびEPO)、ポリビニルアセタールジエチルアミノアセテート(商品名:AEA「三共」)等が挙げられ、その中でもアミノアルキルメタクリレートコポリマーE又はポリビニルアセタールジエチルアミノアセテートが望ましい。胃溶性皮膜層は、上記胃溶性材料の他に、要すれば、他の助剤を含んでよい。それらの助剤としては、例えば、タルクやステアリン酸金属塩等の滑沢剤、マクロゴール等の可塑剤、ステアリン酸等の乳化剤、およびラウリル硫酸ナトリウム等の界面活性剤等が挙げられる。 The controlled release particulate pharmaceutical composition (MRP) of the present invention covers the outside of the active ingredient-containing layer with a gastric film layer. The gastric film layer serves to prevent the active ingredient from being eluted while the particulate pharmaceutical composition is present in the oral cavity and to provide an appropriate lag time. A part or the whole is dissolved by contact with the organic acid eluted from the organic acid-containing core particles by the external liquid (saliva or the like) entering the inside, thereby promoting the elution of the active ingredient. That is, the gastric film layer plays an important role in achieving a desired dissolution profile. Gastric film materials that fulfill these functions are saliva insoluble polymers such as methacrylic acid such as butyl methacrylate / (2-dimethylaminoethyl) methacrylate / methyl methacrylate copolymer and diethylaminoacetate polyvinyl acetal. Acid aminoalkyl copolymers are mentioned. Examples of commercially available polymers include aminoalkyl methacrylate copolymer E (Eudragit (registered trademark) E100 and EPO), polyvinyl acetal diethylaminoacetate (trade name: AEA “Sankyo”), and the like. Aminoalkyl methacrylate copolymer E or polyvinyl acetal diethylaminoacetate is desirable. The gastric film layer may contain other auxiliaries in addition to the gastric material, if necessary. Examples of such auxiliaries include lubricants such as talc and stearic acid metal salts, plasticizers such as macrogol, emulsifiers such as stearic acid, and surfactants such as sodium lauryl sulfate.
なお、前述の通り、本発明の胃溶性皮膜層は、有機酸核粒子から溶出してくる有機酸により作られる局所的な酸性環境に伴い溶解することによって本発明の効果を発揮するのであって、例えば水不溶性ポリマー等が混在すると、所定のラグタイム後の迅速な有効薬剤の放出という本発明所期の目的が達成されない点に留意する必要がある。具体的には、例えば、米国特許出願公開第2006/0078614号明細書(特表2009−524698号が引用する)で開示されている速溶性(Immediate−release:IR)ビーズの苦みをマスクする工程(味覚マスク工程)は、IRビーズを水不溶性ポリマー(例えば、エチルセルロース)単独で、あるいは胃溶性ポリマー(例えば、オイドラギット E100またはEPO)との組み合わせでコーティングする旨、開示されているが、この方法では、水不溶性ポリマー(エチルセルロース等)が有機酸と接触しても溶解しないことから、有効成分の迅速な溶出は期待し得ない。実際、特表2009−524698号明細書中の段落[0089]〜[0092]には、味覚マスクした即時放出型のIRビーズが開示されているが、外層はEC−10(エチルセルロース)/オイドラギットE100(登録商標)=50/50の混合皮膜となっており、発明者等の知見によれば、有効成分の迅速な溶出は期待できなかった。 As described above, the gastric film layer of the present invention exhibits the effects of the present invention by dissolving with a local acidic environment created by the organic acid eluted from the organic acid core particles. For example, when a water-insoluble polymer or the like is mixed, it should be noted that the intended purpose of the present invention, that is, rapid release of an effective drug after a predetermined lag time is not achieved. Specifically, for example, a process for masking the bitterness of fast-dissolve (Immediate-release: IR) beads disclosed in US Patent Application Publication No. 2006/0078614 (cited in Japanese translations of PCT publication No. 2009-524698) (Taste mask process) discloses that IR beads are coated with a water-insoluble polymer (eg, ethyl cellulose) alone or in combination with a gastric polymer (eg, Eudragit E100 or EPO). Since a water-insoluble polymer (such as ethyl cellulose) does not dissolve even when it comes into contact with an organic acid, rapid elution of the active ingredient cannot be expected. In fact, paragraphs [0089] to [0092] of JP-T-2009-524698 disclose a taste-masked immediate-release IR bead, but the outer layer is EC-10 (ethylcellulose) / eudragit E100. (Registered trademark) = 50/50 mixed film. According to the knowledge of the inventors, rapid elution of the active ingredient could not be expected.
本発明の放出制御型粒子状医薬組成物(MRP)において、胃溶性皮膜層は中性では半透膜として働くが、その量および厚さが増えると、外部からの外液(唾液等)の侵入が遅延し、核粒子からの有機酸の溶出開始が遅延し、胃溶性皮膜層の溶解速度が減少する。その結果、有効成分の溶出開始時間(ラグタイム)が伸びる。胃溶性皮膜層の量および厚さは、目的とする有効成分の溶出プロファイルによって適宜選択することができる。胃溶性皮膜層中の胃溶性ポリマーの量は、通常、MRP全量の5〜80重量%、好ましくは10〜60重量%、例えば、20〜40重量%であってよい。胃溶性皮膜が可塑剤や滑沢剤等の充填剤を含む場合は、胃溶性皮膜層の量は、通常、MRP全量の5〜90重量%、好ましくは20〜80重量%、例えば、30〜60重量%であってよい。
胃溶性皮膜層は、上記組成物を1回で適用して皮膜を形成してもよいが、複数回に分割して適用することができる。例えば、上記組成物を二回に分けて適用してよい。複数回に分割しもラグタイムに大きな影響はない。
本発明において胃溶性皮膜層で被覆することにより形成する胃溶性皮膜層被覆粒子(放出制御型粒子状医薬組成物(MRP))の大きさは、10〜1000μm、好ましくは、100〜500μmの範囲、例えば、約300μmである。
In the controlled release particulate pharmaceutical composition (MRP) of the present invention, the gastric soluble coating layer acts as a semi-permeable membrane in the neutral state, but when the amount and thickness increase, the external liquid (saliva etc.) from the outside is increased. The invasion is delayed, the elution start of the organic acid from the core particles is delayed, and the dissolution rate of the gastric film layer is reduced. As a result, the elution start time (lag time) of the active ingredient is extended. The amount and thickness of the gastric film layer can be appropriately selected depending on the elution profile of the target active ingredient. The amount of gastric polymer in the gastric coating layer may usually be 5-80% by weight of the total MRP, preferably 10-60% by weight, for example 20-40% by weight. When the gastric film contains a filler such as a plasticizer or a lubricant, the amount of the gastric film layer is usually 5 to 90% by weight, preferably 20 to 80% by weight, for example 30 to 30% by weight of the total amount of MRP. It may be 60% by weight.
The gastric film layer may be formed by applying the above composition once to form a film, but can be divided and applied multiple times. For example, the composition may be applied in two portions. Dividing into multiple times does not have a significant effect on lag time.
In the present invention, the size of the gastric soluble coating layer-coated particles (release controlled particulate pharmaceutical composition (MRP)) formed by coating with a gastric coating layer is 10 to 1000 μm, preferably 100 to 500 μm. For example, it is about 300 μm.
本発明は、好ましくは、有機酸含有核粒子と有効成分含有層との間に、徐放性皮膜層(SR層)を有してよい。徐放性皮膜層の存在によって、外部からの外液(唾液等)の侵入速度及び内部からの有機酸の放出速度が抑制され、ラグタイム後の溶出速度の任意の制御が可能となる。
即ち、本発明は、有機酸含有層の外側に徐放性皮膜を被覆してよい。徐放性皮膜層は、有機酸含有核粒子への外部からの侵入外液(唾液等)の到達を抑制し、更には溶解した有機酸の溶出を押さえ、結果として有効成分のラグタイム後の溶出速度を調節する役目を果たす。皮膜量が多い場合には、ラグタイムの長さにも寄与する。有機酸含有層と有効成分含有層の境界をより明確にし、緻密なラグタイム制御を容易にする効果も有する。徐放性皮膜材料としては、水不溶性高分子が好ましい。エチルセルロース、メタクリル酸塩コポリマーならびにアクリル酸エチル/メタクリル酸メチルコポリマーおよびアクリル酸エチル/メタクリル酸メチル/メタクリル酸トリメチルアンモニオエチルコポリマーのようなメタクリル酸アミノアルキルコポリマーが挙げられる。商業的に入手可能な水不溶性高分子としては、エチルセルロースの水分散液であるアクアコート(Aquacoat)(登録商標:FMC社)ECD、オイドラギット(Eudragit)NEおよびオイドラギットRS(エボニック社)の商標名で市販されているものがある。
徐放性皮膜材料に他の添加物を加えても良い。このような添加物としては、例えば、タルクの様な助剤、クエン酸トリエチル(SC−60:登録商標・CBC社)の様な可塑剤等が挙げられる。
The present invention may preferably have a sustained-release coating layer (SR layer) between the organic acid-containing core particles and the active ingredient-containing layer. Due to the presence of the sustained-release film layer, the penetration rate of external liquid (saliva etc.) from the outside and the release rate of organic acid from the inside are suppressed, and the elution rate after the lag time can be arbitrarily controlled.
That is, in the present invention, a sustained-release film may be coated on the outside of the organic acid-containing layer. The sustained-release coating layer suppresses the invasion of external liquids (saliva etc.) from the outside to the organic acid-containing core particles, and further suppresses the dissolution of dissolved organic acids, resulting in a lag time after the active ingredient lag time. Serves to adjust the dissolution rate. When the coating amount is large, it also contributes to the length of lag time. It also has the effect of clarifying the boundary between the organic acid-containing layer and the active ingredient-containing layer and facilitating precise lag time control. As the sustained-release coating material, a water-insoluble polymer is preferable. Mention may be made of ethylcellulose, methacrylate copolymers and aminoalkyl methacrylate copolymers such as ethyl acrylate / methyl methacrylate copolymers and ethyl acrylate / methyl methacrylate / trimethylammonioethyl methacrylate copolymers. Commercially available water-insoluble polymers include Aquacoat (registered trademark: FMC) ECD, Eudragit NE and Eudragit RS (Evonik), which are aqueous dispersions of ethyl cellulose. Some are on the market.
Other additives may be added to the sustained-release coating material. Examples of such additives include auxiliaries such as talc and plasticizers such as triethyl citrate (SC-60: registered trademark, CBC).
徐放性皮膜層の量は、通常、放出制御型粒子状医薬組成物(MRP)全量に対して0.1〜20重量%、好ましくは、0.5〜10重量%、例えば1〜5重量%であってよい。
本発明において徐放性皮膜を被覆することにより形成する徐放性皮膜層被覆粒子の大きさは、10〜1000μm、好ましくは、100〜300μmの範囲、例えば、約200μmである。
The amount of the sustained-release coating layer is usually 0.1 to 20% by weight, preferably 0.5 to 10% by weight, for example 1 to 5% by weight, based on the total amount of the controlled release particulate pharmaceutical composition (MRP). %.
In the present invention, the size of the sustained-release film-layer-coated particles formed by coating the sustained-release film is 10 to 1000 μm, preferably 100 to 300 μm, for example, about 200 μm.
本発明の粒子状医薬組成物は、有機酸含有核粒子、徐放性皮膜層、および有効成分含有層(「薬物含有層」と同義)の少なくとも1つの表面にシールコート皮膜層が被覆してあってよい。有機酸含有核粒子、徐放性皮膜層、および有効成分含有層の各表面をそれぞれシールコート皮膜層が被覆してあるのが好ましい。シールコート皮膜層の存在によって各層の完全分離が果たされ、これにより、相互作用(塩形成等)が回避され、製造再現性が向上し、経時変化(有機酸や有効成分の他層への泳動)が抑制される。即ち、本発明の目的である(1)口腔内での初期有効成分溶出の抑制、(2)その後の上部GI管での速やかな有効成分放出、並びに(3)ラグタイムのコントロール、がより一層精度よく達成することができる。有機酸含有核粒子上のシールコート皮膜層は、有機酸が口腔中の外液(唾液等)によって溶出を開始するまでは、有機酸と有効成分含有層中の有効成分との直接接触を妨げて有機酸が例えば塩基性有効成分と塩を形成して所期の効果(最外層の胃溶性皮膜の溶解)が発揮できなくなることを防ぐ機能を有する。徐放性皮膜層上のシールコートは、特にアクアコートのようにコーティング後の熱処理を必要とする場合、熱処理中の凝集を防止するのに必須である。有効成分含有層上のシールコート皮膜層は、有効成分が最外層の胃溶性皮膜と直接接触して、化学的・物理的変化が生じるのを防ぐ。これら各々の機能が相俟って、本発明の(1)〜(3)の目的の達成が、より一層推進される。 In the particulate pharmaceutical composition of the present invention, at least one surface of an organic acid-containing core particle, a sustained-release film layer, and an active ingredient-containing layer (synonymous with “drug-containing layer”) is coated with a seal coat film layer. It may be. It is preferable that the surfaces of the organic acid-containing core particles, the sustained-release coating layer, and the active ingredient-containing layer are respectively coated with a seal coat coating layer. Each layer is completely separated by the presence of the seal coat layer, thereby avoiding interaction (salt formation, etc.), improving production reproducibility, and changing with time (organic acid and active ingredients to other layers). Migration) is suppressed. That is, the objectives of the present invention are (1) suppression of elution of the initial active ingredient in the oral cavity, (2) subsequent rapid release of the active ingredient in the upper GI tract, and (3) lag time control. It can be achieved with high accuracy. The seal coat film layer on the organic acid-containing core particles prevents direct contact between the organic acid and the active ingredient in the active ingredient-containing layer until the organic acid starts to be eluted by the external liquid (saliva etc.) in the oral cavity. Thus, for example, the organic acid forms a salt with a basic active ingredient and has a function of preventing the intended effect (dissolution of the outermost gastric film) from being exhibited. The seal coat on the sustained-release film layer is essential for preventing aggregation during the heat treatment, particularly when a heat treatment after the coating is required like an aqua coat. The seal coat film layer on the active ingredient-containing layer prevents the active ingredient from coming into direct contact with the outermost gastric soluble film and causing chemical and physical changes. Together with these functions, the achievement of the objects (1) to (3) of the present invention is further promoted.
シールコート皮膜層は溶出を妨げず各層を分離できるものであれば良く、例えば、皮膜形成性の親水性ポリマー水溶液を塗布・乾燥することによって得ることができる。シールコート皮膜材料としては通常の親水性ポリマーを用いることができる。例えば、ヒドロキシプロピルメチルセルロース(HPMC:ヒプロメロース)、メチルセルロース(MC)、ヒドロキシプロピルセルロース(HPC)、ヒドロキシメチルセルロース(HMC)にポリエチレングリコール(PEG)6000(マクロゴール6000)、PEG−4000(マクロゴール4000)等の可塑剤の組み合わせが挙げられる。好ましい例としては、HPMCとPEG−6000の組み合わせ等である。 The seal coat film layer only needs to be capable of separating the respective layers without hindering elution, and can be obtained, for example, by applying and drying a film-forming hydrophilic polymer aqueous solution. A normal hydrophilic polymer can be used as the seal coat film material. For example, hydroxypropylmethylcellulose (HPMC: hypromellose), methylcellulose (MC), hydroxypropylcellulose (HPC), hydroxymethylcellulose (HMC), polyethylene glycol (PEG) 6000 (macrogol 6000), PEG-4000 (macrogol 4000), etc. And a combination of plasticizers. A preferred example is a combination of HPMC and PEG-6000.
本発明はまた、放出制御型粒子状医薬組成物(MRP)の製造方法であって、
(i)不活性コアの外側に有機酸含有層を被覆し有機酸含有核粒子を形成する工程、
(ii)該有機酸含有核粒子の外側に徐放性皮膜を被覆する工程、
(iii)該徐放性皮膜層の外側に薬物を含有する層を被覆する工程、および
(iv)該有効成分含有層の外側を胃溶性皮膜で被覆して経口投与用の放出制御型粒子状医薬組成物を形成する工程
を含むことを特徴とする、放出制御型粒子状医薬組成物の製造方法を提供する。
The present invention is also a method for producing a controlled release particulate pharmaceutical composition (MRP) comprising:
(I) coating the organic acid-containing layer outside the inert core to form organic acid-containing core particles;
(Ii) a step of coating a sustained-release film on the outside of the organic acid-containing core particles;
(Iii) a step of coating a layer containing a drug on the outside of the sustained-release coating layer; and (iv) a controlled-release particulate form for oral administration by coating the outside of the active ingredient-containing layer with a gastric film. A method for producing a controlled release particulate pharmaceutical composition comprising the step of forming a pharmaceutical composition is provided.
前記製造方法において、好ましくは、有機酸含有核粒子および徐放性皮膜層の間、徐放性皮膜層および有効成分含有層の間、ならびに有効成分含有層および胃溶性皮膜層の間の少なくとも1つにシールコート皮膜層を設ける工程を含んでよい。 In the production method, preferably, at least one between the organic acid-containing core particles and the sustained-release coating layer, between the sustained-release coating layer and the active ingredient-containing layer, and between the active ingredient-containing layer and the gastric soluble coating layer. A step of providing a seal coat film layer on one of the layers may be included.
以下、各工程を説明する。
(i)工程では、不活性コア(例えば結晶セルロース(粒)(Celphere(登録商標)CP−102等)を転動流動層造粒機中で流動させながら、有機酸(例えば、フマル酸)、結合剤(例えば、ヒドロキシプロピルセルロース)および滑沢剤(例えば、タルク)等を溶解・分散させた水分散液をノズルからスプレーして不活性コア上に被覆し、有機酸含有核粒子を形成する。有機溶媒の使用は、安全性や環境保全の観点から好ましくない。造粒機としては、例えば、流動層造粒機、転動流動層造粒機または遠心転動造粒機等を用いることができるが、微小粒子を作成するには転動流動層造粒機が好ましい。
不活性コアの粒径は、10〜1000μm、好ましくは、100〜300μmの範囲、例えば、約150μmであってよい。
有機酸含有核粒子の大きさは、10〜1000μm、好ましくは、100〜300μmの範囲、例えば、約200μmである。
有機酸の量は、胃溶性皮膜を溶解するのに十分な量、言い換えれば局所的なpHを5以下にするのに十分な量、即ち、化学量論量で云えば、胃溶性皮膜に存する塩基性窒素と等量以上の酸当量であるのが好ましい。例えば、有機酸としてフマル酸を用いる場合には、オイドラギット(登録商標)EPOが100重量部に対して、フマル酸が2〜200重量部、好ましくは、5〜100重量部、より好ましくは、15〜50重量部、例えば30重量部であってよい。
Hereinafter, each process will be described.
In the step (i), an inert core (for example, crystalline cellulose (grain) (Celphere (registered trademark) CP-102 or the like) is fluidized in a rolling fluidized bed granulator while an organic acid (for example, fumaric acid), An aqueous dispersion in which a binder (for example, hydroxypropylcellulose) and a lubricant (for example, talc) are dissolved and dispersed is sprayed from a nozzle and coated on the inert core to form organic acid-containing core particles. The use of an organic solvent is not preferable from the viewpoint of safety and environmental conservation.For example, a fluidized bed granulator, a rolling fluidized bed granulator, a centrifugal rolling granulator or the like is used as the granulator. However, a rolling fluidized bed granulator is preferable for producing fine particles.
The particle size of the inert core may be in the range of 10 to 1000 μm, preferably 100 to 300 μm, for example about 150 μm.
The size of the organic acid-containing core particles is 10 to 1000 μm, preferably in the range of 100 to 300 μm, for example, about 200 μm.
The amount of the organic acid is sufficient to dissolve the gastric film, in other words, an amount sufficient to bring the local pH to 5 or less, that is, in stoichiometric amount, it exists in the gastric film. The acid equivalent is preferably equal to or more than that of basic nitrogen. For example, when fumaric acid is used as the organic acid, Eudragit (registered trademark) EPO is 100 parts by weight, fumaric acid is 2 to 200 parts by weight, preferably 5 to 100 parts by weight, more preferably 15 parts by weight. It may be ˜50 parts by weight, for example 30 parts by weight.
(ii)工程では、前記で得られた有機酸含有核粒子を転動流動層造粒機中で流動させながら、不溶性ポリマーの水性分散体(例えば、エチルセルロース、可塑剤としてのセチルアルコールおよび界面活性剤としてのラウリル硫酸ナトリウムを含む水性分散液であるアクアコート(Aquacoat:登録商標)ECD−30)およびコーティング助剤(例えば、クエン酸トリエチル(TEC))の水分散液をノズルからスプレーして、徐放性皮膜を被覆する。熱処理により皮膜を熟成させ、徐放性皮膜層被覆粒子を形成する。
不溶性ポリマーの量は、例えば、アクアコート(Aquacoat:登録商標)ECD−30)の固形分の量で示せば、粒子状医薬組成物全体の0.1〜20重量%、好ましくは、0.5〜10重量%、より好ましくは、1.0〜5重量%の範囲であってよい(ここで、アクアコート(登録商標)ECD−30の固形分は約30重量%である)。
本発明において徐放性皮膜を被覆することにより形成する徐放性皮膜層被覆粒子の大きさは、10〜1000μm、好ましくは、100〜300μmの範囲、例えば、約200μmである。
In the step (ii), an aqueous dispersion of an insoluble polymer (for example, ethyl cellulose, cetyl alcohol as a plasticizer, and surface activity) while the organic acid-containing core particles obtained above are fluidized in a rolling fluidized bed granulator. Spray an aqueous dispersion of Aquacoat® ECD-30, which is an aqueous dispersion containing sodium lauryl sulfate as an agent, and a coating aid (eg, triethyl citrate (TEC)) from a nozzle, A sustained release coating is applied. The film is aged by heat treatment to form sustained-release film layer-coated particles.
The amount of the insoluble polymer is, for example, 0.1 to 20% by weight of the total amount of the particulate pharmaceutical composition, preferably 0.5% if expressed by the solid content of Aquacoat (registered trademark) ECD-30. May range from 10 to 10% by weight, more preferably from 1.0 to 5% by weight (where Aquacoat® ECD-30 has a solids content of about 30% by weight).
In the present invention, the size of the sustained-release film-layer-coated particles formed by coating the sustained-release film is 10 to 1000 μm, preferably 100 to 300 μm, for example, about 200 μm.
(iii)工程では、前記で得られた徐放性皮膜層被覆粒子を、例えば、転動流動層造粒機中で流動させながら、弱酸性ないし塩基性の有効成分(例えば、バルデナフィル塩酸塩・3水和物)含有溶液をノズルからスプレーして、有効成分含有層(「薬物含有層」と同義。)を形成する。
有効成分含有溶液は、有効成分を水、アルコール等の溶媒に溶解ないし分散させて用いるが、結合剤としての水溶性ポリマー(例えば、ヒドロキシプロピルセルロース)および滑沢剤(例えば、タルク)を含んでよい。
放出制御型粒子状医薬組成物(MRP)中の有効成分の配合量は、通常有効成分の種類あるいは医薬用途(適応症)により適宜選択されるが、治療学的に有効な量あるいは予防学的に有効な量であれば特に制限されない。具体的には、例えば、抗勃起不全症薬バルデナフィルの場合、バルデナフィルとしてMRP全体の0.5〜90重量%であり、より好ましくは1〜70重量%、さらに好ましくは5〜50重量%、例えば10〜20重量%である。結合剤の量は、有効成分100重量部に対して0.1〜80重量部、好ましくは、1.0〜50重量部、更に好ましくは、5.0〜20重量部の範囲であってよい。滑沢剤の量は、有効成分100重量部に対して0.1〜80重量部、好ましくは、1.0〜50重量部、更に好ましくは、5.0〜20重量部の範囲であってよい。
本発明において有効成分含有層で被覆するにより形成する有効成分含有層被覆粒子の大きさは、10〜1000μm、好ましくは、100〜300μmの範囲、例えば、約250μmである。
In the step (iii), while the sustained-release coating layer-coated particles obtained above are flowed in, for example, a rolling fluidized bed granulator, a weakly acidic or basic active ingredient (for example, vardenafil hydrochloride, A trihydrate) -containing solution is sprayed from a nozzle to form an active ingredient-containing layer (synonymous with “drug-containing layer”).
The active ingredient-containing solution is used by dissolving or dispersing the active ingredient in a solvent such as water or alcohol, and contains a water-soluble polymer (for example, hydroxypropylcellulose) and a lubricant (for example, talc) as a binder. Good.
The compounding amount of the active ingredient in the controlled release particulate pharmaceutical composition (MRP) is usually appropriately selected according to the type of the active ingredient or the pharmaceutical use (indication), but is therapeutically effective or prophylactic. The amount is not particularly limited as long as it is effective. Specifically, for example, in the case of the anti-erectile dysfunction drug vardenafil, it is 0.5 to 90% by weight of the whole MRP as vardenafil, more preferably 1 to 70% by weight, still more preferably 5 to 50% by weight, for example, 10 to 20% by weight. The amount of the binder may be in the range of 0.1 to 80 parts by weight, preferably 1.0 to 50 parts by weight, more preferably 5.0 to 20 parts by weight with respect to 100 parts by weight of the active ingredient. . The amount of lubricant is in the range of 0.1 to 80 parts by weight, preferably 1.0 to 50 parts by weight, more preferably 5.0 to 20 parts by weight, based on 100 parts by weight of the active ingredient. Good.
In the present invention, the active ingredient-containing layer-coated particles formed by coating with the active ingredient-containing layer have a size of 10 to 1000 μm, preferably 100 to 300 μm, for example, about 250 μm.
(iv)工程では、前記で得られた有効成分含有層被覆粒子を転動流動層造粒機中で流動させながら、水、アルコール等の溶媒中に胃溶性皮膜材料(例えば、オイドラギット(登録商標)EPO)を分散させた分散液をノズルからスプレーして、胃溶性皮膜層被覆粒子(放出制御型粒子状医薬組成物(MRP))を形成する。胃溶性皮膜材料含有分散液中には、乳化剤(例えば、ステアリン酸)、界面活性剤(例えば、ラウリル硫酸ナトリウム)および滑沢剤(例えば、タルク)を含有してよい。
胃溶性皮膜層の量および厚さは、目的とする有効成分の溶出プロファイルによって適宜選択することができる。胃溶性皮膜層中の胃溶性ポリマーの量は、通常、MRP全量の5〜80重量%、好ましくは10〜60重量%、例えば、20〜40重量%であってよい。胃溶性皮膜が可塑剤や滑沢剤等の充填剤を含む場合は、胃溶性皮膜層の量は、通常、MRP全量の5〜90重量%、好ましくは20〜80重量%、例えば、30〜60重量%の範囲であってよい。
本発明において胃溶性皮膜層で被覆することにより形成する胃溶性皮膜層被覆粒子(放出制御型粒子状医薬組成物(MRP))の大きさは、10〜1000μm、好ましくは、100〜500μmの範囲、例えば、約300μmである。
In the step (iv), the active ingredient-containing layer-coated particles obtained above are fluidized in a rolling fluidized bed granulator, and a gastric film material (for example, Eudragit (registered trademark) in a solvent such as water or alcohol). ) A dispersion in which EPO) is dispersed is sprayed from a nozzle to form gastric film-coated particles (controlled release particulate pharmaceutical composition (MRP)). The dispersion containing the gastric film material may contain an emulsifier (for example, stearic acid), a surfactant (for example, sodium lauryl sulfate) and a lubricant (for example, talc).
The amount and thickness of the gastric film layer can be appropriately selected depending on the elution profile of the target active ingredient. The amount of gastric polymer in the gastric coating layer may usually be 5-80% by weight of the total MRP, preferably 10-60% by weight, for example 20-40% by weight. When the gastric film contains a filler such as a plasticizer or a lubricant, the amount of the gastric film layer is usually 5 to 90% by weight, preferably 20 to 80% by weight, for example 30 to 30% by weight of the total amount of MRP. It may be in the range of 60% by weight.
In the present invention, the size of the gastric soluble coating layer-coated particles (release controlled particulate pharmaceutical composition (MRP)) formed by coating with a gastric coating layer is 10 to 1000 μm, preferably 100 to 500 μm. For example, it is about 300 μm.
本発明はまた、所定のラグタイム後に急速に有効薬剤成分の放出が始まる上記の放出制御型粒子状医薬組成物(MRP)に、有効成分を含有しない速溶粒子を加えてなる、経口投与用崩壊性粒子状組成物(ODP)を提供する。適当な味を有する速溶粒子を加えることによって唾液の分泌が促され、嚥下を容易にする。投与形態として取り扱いし易くする効果もある。 The present invention also relates to a disintegration for oral administration, wherein fast-dissolving particles containing no active ingredient are added to the above-mentioned controlled release particulate pharmaceutical composition (MRP) in which the release of the active pharmaceutical ingredient starts rapidly after a predetermined lag time. A particulate particulate composition (ODP) is provided. By adding fast-dissolving particles having an appropriate taste, saliva secretion is promoted and swallowing is facilitated. There is also an effect of facilitating handling as a dosage form.
速溶粒子は、適当な味があり、速溶性を有する粒子であれば特に制限はないが、甘味を有する糖アルコール、更には結合剤を含んでなる顆粒であってよい。糖アルコールとしては、例えば、エリスリトール、マンニトール、ソルビトール、マルチトール、キシリトール、および分岐オリゴ糖アルコールなどが挙げられるが、エリスリトールおよびマンニトールが好ましい。結合剤としては特に制限はないが、例えば、乳糖、白糖、ブドウ糖などの糖類、バレイショ、コムギ、トウモロコシなどのデンプン類、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロースなどのセルロース類、並びに、ポビドン(ポリビニルピロリドン)等が挙げられる。これらの内、ヒドロキシプロピルメチルセルロースおよびポビドンが好ましい。 The fast-dissolving particles are not particularly limited as long as they have an appropriate taste and have fast solubility, but may be granules containing a sugar alcohol having a sweet taste and a binder. Examples of the sugar alcohol include erythritol, mannitol, sorbitol, maltitol, xylitol, and branched oligosaccharide alcohol, and erythritol and mannitol are preferable. The binder is not particularly limited, and examples thereof include sugars such as lactose, sucrose and glucose, starches such as potato, wheat and corn, celluloses such as hydroxypropylcellulose and hydroxypropylmethylcellulose, and povidone (polyvinylpyrrolidone). Etc. Of these, hydroxypropylmethylcellulose and povidone are preferred.
本発明はまた、上記の経口投与用崩壊性粒子状組成物(ODP)の製造方法であって、
(i)不活性コアの外側に有機酸含有層を被覆し有機酸含有核粒子を形成する工程、
(ii)第有機酸含有核粒子の外側に徐放性皮膜を被覆する工程、
(iii)該徐放性皮膜層の外側に薬物を含有する層を被覆する工程、
(iv)該有効成分含有層の外側を胃溶性皮膜で被覆して経口投与用の放出制御型粒子状医薬組成物を形成する工程、および
(v)前記放出制御型粒子状医薬組成物および速溶粒子を混合する工程
を含むことを特徴とする、経口投与用粒子状組成物の製造方法を提供する。
The present invention also provides a method for producing the above-mentioned collapsible particulate composition (ODP) for oral administration,
(I) coating the organic acid-containing layer outside the inert core to form organic acid-containing core particles;
(Ii) a step of coating a sustained-release film on the outside of the first organic acid-containing core particle;
(Iii) a step of coating a drug-containing layer on the outside of the sustained-release coating layer;
(Iv) coating the outside of the active ingredient-containing layer with a gastric soluble coating to form a controlled release particulate pharmaceutical composition for oral administration; and (v) the controlled release particulate pharmaceutical composition and rapid dissolution. There is provided a method for producing a particulate composition for oral administration, which comprises a step of mixing particles.
これらの工程(i)〜(v)の内、(i)〜(iv)工程は、前記放出制御型粒子状医薬組成物(MRP)の製造方法に於ける工程(i)〜(iv)と同一であり、経口投与用粒子状組成物(ODP)の製造方法においては、更に(v)工程を含む点に特徴がある。
即ち、経口投与用崩壊性粒子状組成物(ODP)の製造方法は、(i)〜(iv)工程を含む製造方法によって得られた放出制御型粒子状医薬組成物(MRP)に速溶粒子を混合する工程(v)を更に含むことを特徴とする。そこで、以下では、工程(v)について詳しく説明する。
Among these steps (i) to (v), steps (i) to (iv) are steps (i) to (iv) in the method for producing a controlled release particulate pharmaceutical composition (MRP). The method for producing a particulate composition for oral administration (ODP) is characterized in that it further comprises step (v).
That is, the method for producing a disintegrating particulate composition for oral administration (ODP) is obtained by applying fast-dissolving particles to a controlled release particulate pharmaceutical composition (MRP) obtained by a production method including steps (i) to (iv). The method further includes a step (v) of mixing. Therefore, in the following, step (v) will be described in detail.
工程(v)では、(i)〜(iv)工程で得られた放出制御型粒子状医薬組成物(MRP)と別途調製した速溶粒子とを混合して、経口投与用崩壊性粒子状組成物(ODP)を得る。速溶粒子は、糖アルコール(例えば、エリスリトール)および結合剤(例えば、ポビドン)を、水・エタノール等を溶媒として、高速攪拌混合機(High shear mixer)等を用いて混練した後、円筒造粒機(Cylindeical granulator)等を用いて造粒し、乾燥、篩分けして製造する。更に、流動化剤ないし滑沢剤(例えば、軽質無水ケイ酸)、帯電抑制剤(例えば、タルク)、必要に応じて香料も添加することができる。 In the step (v), the controlled release particulate pharmaceutical composition (MRP) obtained in the steps (i) to (iv) and the fast dissolving particles separately prepared are mixed, and the collapsible particulate composition for oral administration. (ODP) is obtained. The fast dissolving particles are prepared by kneading a sugar alcohol (for example, erythritol) and a binder (for example, povidone) with water / ethanol as a solvent using a high shear mixer or the like, and then a cylindrical granulator (Cylindical granulator) or the like is used for granulation, drying and sieving. Furthermore, a fluidizing agent or a lubricant (for example, light anhydrous silicic acid), a charge suppressing agent (for example, talc), and a fragrance as necessary can be added.
放出制御型粒子状医薬組成物(MRP)と速溶粒子との混合比は、通常、1:1〜20、好ましくは、1:2〜10の範囲、例えば1:6であってよい。混合は、例えば、交叉回転式混合機(Cross rotary mixer)等を用いて行うことができる。混合によって経口投与用崩壊性粒子状組成物(ODP)を得る際に、軽質無水ケイ酸、ステアリン酸マグネシウム、タルク、フマル酸ステアリルナトリウム等の流動化剤ないし滑沢剤、並びにL−メントール等の香料ないし矯味剤等の助剤を加えることができる。特に、流動化剤ないし滑沢剤としての軽質無水ケイ酸及び香料としてのL−メントールの添加が好ましい。流動化剤ないし滑沢剤の添加量は、ODP全量の0.05〜5重量%、好ましくは0.1〜1重量%の範囲であり、例えば0.5重量%である。香料ないし矯味剤の添加量は、ODP全量の0.005〜0.5重量%、好ましくは0.01〜0.1重量%の範囲であり、例えば0.05重量%である。 The mixing ratio of the controlled release particulate pharmaceutical composition (MRP) and fast dissolving particles may be usually in the range of 1: 1 to 20, preferably 1: 2 to 10, for example 1: 6. Mixing can be performed using, for example, a cross-rotating mixer. When obtaining a disintegrating particulate composition (ODP) for oral administration by mixing, fluidizing agents or lubricants such as light anhydrous silicic acid, magnesium stearate, talc and sodium stearyl fumarate, and L-menthol, etc. Auxiliaries such as fragrances and flavoring agents can be added. In particular, light silicic acid anhydride as a fluidizing agent or lubricant and addition of L-menthol as a fragrance are preferred. The amount of the fluidizing agent or lubricant added is in the range of 0.05 to 5% by weight, preferably 0.1 to 1% by weight, for example 0.5% by weight of the total amount of ODP. The addition amount of the fragrance or flavoring agent is in the range of 0.005 to 0.5% by weight, preferably 0.01 to 0.1% by weight of the total amount of ODP, for example 0.05% by weight.
本発明はまた、有機酸含有核粒子および徐放性皮膜層の間、徐放性皮膜層および有効成分含有層の間、ならびに有効成分含有層および胃溶性皮膜層の間の少なくとも1つにシールコート皮膜層を設ける工程を含む、前記経口投与用崩壊性粒子状組成物(ODP)の製造方法を提供する。好ましくは、有機酸含有核粒子および徐放性皮膜層の間、徐放性皮膜層および有効成分を含有する皮膜層の間、ならびに、有効成分を含有する皮膜層および胃溶性皮膜層の間の全てにシールコート皮膜層を設ける工程を含む、前記経口投与用粒子状組成物の製造方法を提供する。本発明は更に、胃溶性皮膜層の上にシールコート皮膜層を設ける工程を含む、前記経口投与用粒子状組成物の製造方法を提供する。 The present invention also provides a seal between at least one of the organic acid-containing core particles and the sustained-release coating layer, between the sustained-release coating layer and the active ingredient-containing layer, and between the active ingredient-containing layer and the gastric soluble coating layer. The manufacturing method of the said collapsible particulate composition for oral administration (ODP) including the process of providing a coat film layer is provided. Preferably, between the organic acid-containing core particles and the sustained-release coating layer, between the sustained-release coating layer and the coating layer containing the active ingredient, and between the coating layer containing the active ingredient and the gastric soluble coating layer Provided is a method for producing the particulate composition for oral administration, which comprises the step of providing a seal coat film layer on all of them. The present invention further provides a method for producing the particulate composition for oral administration, comprising the step of providing a seal coat film layer on the gastric soluble film layer.
シールコート皮膜の存在によって、本発明の目的である(1)口腔内での初期有効成分溶出の抑制、(2)その後の上部GI管での速やかな有効成分放出の開始、並びに(3)ラグタイム後の溶出速度のコントロール、をより一層精度よく達成することができる。 Due to the presence of the seal coat film, the object of the present invention is (1) suppression of elution of the initial active ingredient in the oral cavity, (2) subsequent rapid start of active ingredient release in the upper GI tract, and (3) lag Control of the elution rate after time can be achieved with higher accuracy.
シールコート皮膜層を形成する工程を、例えば、有機酸含有核粒子と徐放性皮膜層との間にシールコート皮膜層を形成する場合について説明すると、有機酸含有核粒子を転動流動層造粒機中で流動させながら、皮膜形成性の親水性ポリマー(例えば、ヒプロメロース2910とPEG−6000の組み合わせ)の水溶液をノズルからスプレーすることによって、有機酸含有核粒子上にシールコート皮膜層を形成することができる。皮膜形成性の親水性ポリマーの量は、有機酸含有核粒子100重量部に対して0.2〜20重量部、好ましくは、0.5〜10重量部の範囲、例えば、2重量部であってよい。 The step of forming the seal coat film layer will be described, for example, when the seal coat film layer is formed between the organic acid-containing core particles and the sustained-release film layer. A seal coat film layer is formed on the organic acid-containing core particles by spraying an aqueous solution of a film-forming hydrophilic polymer (for example, a combination of hypromellose 2910 and PEG-6000) from a nozzle while flowing in a granulator. can do. The amount of the film-forming hydrophilic polymer is 0.2 to 20 parts by weight, preferably 0.5 to 10 parts by weight, for example, 2 parts by weight with respect to 100 parts by weight of the organic acid-containing core particles. It's okay.
本発明の製造方法は、前記の別途調製した速溶粒子の製造方法として、糖類と結合剤を混合し、造粒後乾燥させる工程を含んでなる。即ち、糖類としての糖アルコール(例えば、エリスリトール)および結合剤(例えば、ポビドン)を、水・エタノール等を溶媒として、高速攪拌混合機(High shear mixer)等を用いて混練した後、円筒造粒機(Cylinderical granulator)等を用いて造粒し、棚式乾燥機(Bed dryer)等を用いて乾燥後、篩分けして粒径を揃え、速溶粒子とすることができる。速溶粒子の粒径は、0.1〜1.0mm、好ましくは、0.1〜0.3mmであってよい。 The production method of the present invention includes a step of mixing saccharides and a binder, and drying after granulation as a method of producing the separately prepared fast dissolving particles. That is, after kneading a sugar alcohol (for example, erythritol) as a saccharide and a binder (for example, povidone) using water, ethanol or the like as a solvent using a high-speed stirring mixer (High shear mixer) or the like, cylindrical granulation is performed. It can be granulated using a machine (Cylinder granulator) or the like, dried using a shelf type dryer (Bed dryer) or the like, and then sieved to make the particle size uniform to obtain fast dissolving particles. The particle size of the fast dissolving particles may be 0.1 to 1.0 mm, preferably 0.1 to 0.3 mm.
本発明はまた、前述のとおり、経口投与用錠剤であって、前記放出制御型粒子状医薬組成物(MRP)と速溶粒子とを含有する原料組成物を打錠することにより得られる口腔内崩壊錠(ODT)を提供する。錠剤の成形に当たっては、ODPをそのまま、あるいは賦形剤、結合剤、崩壊剤、滑沢剤等の通常用いられる添加剤を追加することができる。要すれば、更に糖アルコール(例えば、エリスリトールおよびマンニトール等)を別途加えてもよい。打錠は、例えば単発打錠機やロータリー打錠機を用いて行うことができる。 As described above, the present invention also provides a tablet for oral administration, which is obtained by tableting a raw material composition containing the controlled release particulate pharmaceutical composition (MRP) and fast dissolving particles. Provide a lock (ODT). In tablet formation, ODP can be used as it is, or commonly used additives such as excipients, binders, disintegrants and lubricants can be added. If necessary, sugar alcohols (for example, erythritol and mannitol) may be added separately. Tableting can be performed using, for example, a single tableting machine or a rotary tableting machine.
本発明の放出制御型粒子状医薬組成物(MRP)には、当該分野において慣用される公知の配合処方が利用でき、前記必須成分以外の医薬添加剤としては、医薬的に許容され、添加物として使用される各種添加剤であれば特に制限されない。かかる医薬添加剤としては、例えば、結合剤、崩壊剤、滑沢剤、矯味剤、甘味剤、清涼化剤、着香剤・香料、芳香剤、着色剤、発泡剤、安定(化)剤、抗酸化剤、保存剤、pH調節剤、可溶化剤、溶解補助剤、流動化剤、緩衝剤、基剤、賦形剤などが挙げられるが、これらに限定されるものではない。
以下に実施例を挙げて本発明を具体的に説明するが、これらにより本発明の範囲が限定されるものではない。
For the controlled release particulate pharmaceutical composition (MRP) of the present invention, a known formulation commonly used in the field can be used, and pharmaceutical additives other than the essential components are pharmaceutically acceptable and additives. If it is various additives used as, it will not restrict | limit in particular. Examples of such pharmaceutical additives include binders, disintegrants, lubricants, flavoring agents, sweeteners, refreshing agents, flavoring agents / fragrances, fragrances, coloring agents, foaming agents, stabilizing (stabilizing) agents, Examples include, but are not limited to, antioxidants, preservatives, pH adjusters, solubilizers, solubilizers, fluidizers, buffers, bases, and excipients.
EXAMPLES The present invention will be specifically described below with reference to examples, but the scope of the present invention is not limited by these examples.
実施例1[徐放性皮膜層(SR)のない放出制御型粒子状医薬組成物(MRP)の調製](製剤例4)
<1>[(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルセルロース10gを含む水溶液を撹拌下、フマル酸100g、およびタルク5gを添加して分散させた。この分散液を結晶セルロース(粒)(Celphere(登録商標)CP−102)1000gに対して、転動流動層造粒機を用いて、吸気温度約60℃、排気温度約28℃で側方噴霧し、[(1)有機酸含有層被覆粒子]を得た。フマル酸の量は、セルロース核粒子100重量部に対して10重量部であった。(計1115g)
Example 1 [Preparation of controlled release particulate pharmaceutical composition (MRP) without sustained-release coating layer (SR)] (Formulation Example 4)
<1> [(1) Preparation of organic acid-containing layer-coated particles]
Under stirring, an aqueous solution containing 10 g of hydroxypropylcellulose was added and dispersed with 100 g of fumaric acid and 5 g of talc. This dispersion is laterally sprayed on 1000 g of crystalline cellulose (grains) (Celphere® CP-102) using a rolling fluidized bed granulator at an intake temperature of about 60 ° C. and an exhaust temperature of about 28 ° C. To obtain [(1) organic acid-containing layer-coated particles]. The amount of fumaric acid was 10 parts by weight with respect to 100 parts by weight of the cellulose core particles. (Total 1115g)
<2>[(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルメチルセルロース(ヒプロメロース)20g、およびポリエチレングリコール(マクロゴール6000)5gを含む水溶液を、上記で得た(1)有機酸含有層被覆粒子に対して、転動流動層造粒機を用いて、吸気温度約85℃、排気温度約42℃で側方噴霧し、[(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。(計1140g)
<2> [(2) Seal coat coating / (1) Preparation of organic acid-containing layer coated particles]
An aqueous solution containing 20 g of hydroxypropylmethylcellulose (hypromellose) and 5 g of polyethylene glycol (Macrogol 6000) was used for the (1) organic acid-containing layer-coated particles obtained above using a rolling fluidized bed granulator. Side spraying was performed at an intake temperature of about 85 ° C. and an exhaust temperature of about 42 ° C. to obtain [(2) seal coat coating / (1) organic acid-containing layer coated particles]. (Total 1140g)
<3>[(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルセルロース32gを含む水溶液を撹拌下、バルデナフィル塩酸塩・3水和物(BayerHC社製)320g、およびタルク16gを添加して分散させた。この分散液を、上記で得た[(2)シールコート被覆/(1)有機酸含有層被覆粒子]456gに対して、転動流動層造粒機を用いて、吸気温度約55℃、排気温度約28℃で側方噴霧し、[(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。(計824g)
<3> [(3) Active ingredient-containing layer coating / (2) Seal coat coating / (1) Preparation of organic acid-containing layer-coated particles]
While stirring, an aqueous solution containing 32 g of hydroxypropylcellulose was added and dispersed with 320 g of vardenafil hydrochloride trihydrate (manufactured by BayerHC) and 16 g of talc. This dispersion was applied to 456 g of [(2) seal coat coating / (1) organic acid-containing layer coated particles] obtained above, using a rolling fluidized bed granulator, with an intake air temperature of about 55 ° C., exhaust gas Side spraying was performed at a temperature of about 28 ° C. to obtain [(3) active ingredient-containing layer coating / (2) seal coat coating / (1) organic acid-containing layer coated particles]. (Total 824g)
<4>[(4)シールコート被覆/(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルメチルセルロース(ヒプロメロース)16g、およびポリエチレングリコール(マクロゴール6000)4gを含む水溶液を、上記で得た[(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]に対して、転動流動層造粒機を用いて、吸気温度約85℃、排気温度約40℃で側方噴霧し、[(4)シールコート被覆/(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。(計844g)
<4> [(4) Seal coat coating / (3) Active ingredient-containing layer coating / (2) Seal coat coating / (1) Preparation of organic acid-containing layer-coated particles]
An aqueous solution containing 16 g of hydroxypropylmethylcellulose (hypromellose) and 4 g of polyethylene glycol (Macrogol 6000) was obtained above [(3) Active ingredient-containing layer coating / (2) Seal coat coating / (1) Organic acid-containing layer [Coated particles] are sprayed laterally at an intake air temperature of about 85 ° C. and an exhaust temperature of about 40 ° C. using a rolling fluidized bed granulator, [(4) seal coat coating / (3) active ingredient containing layer Coating / (2) Seal coat coating / (1) Organic acid-containing layer coated particles]. (Total 844g)
<5>[(5)胃溶性皮膜被覆/(4)シールコート被覆/(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ラウリル硫酸ナトリウム20gを含む水溶液を撹拌下、アミノアルキルメタクリレートコポリマーE(オイドラギット(登録商標)EPO)200g、およびステアリン酸30gを添加して分散させた。続いて、タルク70gを撹拌下に加えて分散液を調製した。この分散液を、上記で得た[(4)シールコート被覆/(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]320gに対して、転動流動層造粒機を用いて、吸気温度約45℃、排気温度約28℃で側方噴霧し、本発明の放出制御型粒子状医薬組成物(MRP)である、[(5)胃溶性皮膜被覆/(4)シールコート被覆/(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子](製剤例4:表2参照)を得た。胃溶性皮膜の被覆量は、[(4)シールコート被覆/(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]100重量部に対して100重量部であった。(計640g)。得られたMRP粒子の平均粒径は267μm、有効成分バルデナフィルの含有量は約16重量%であった。
徐放性皮膜層(SR)のない製剤例1、2及び3も上記実施例1と同様にして製造することができる。
<5> [(5) Gastric soluble coating / (4) Seal coat coating / (3) Active ingredient-containing layer coating / (2) Seal coat coating / (1) Preparation of organic acid-containing layer coated particles]
Under stirring, an aqueous solution containing 20 g of sodium lauryl sulfate was added and dispersed with 200 g of aminoalkyl methacrylate copolymer E (Eudragit (registered trademark) EPO) and 30 g of stearic acid. Subsequently, 70 g of talc was added with stirring to prepare a dispersion. This dispersion was rolled against 320 g of [(4) seal coat coating / (3) active ingredient-containing layer coating / (2) seal coat coating / (1) organic acid-containing layer coated particles] obtained above. Using a fluidized bed granulator, the spray is laterally sprayed at an intake temperature of about 45 ° C. and an exhaust temperature of about 28 ° C., which is the controlled release particulate pharmaceutical composition (MRP) of the present invention [(5) gastric soluble film Coating / (4) Seal coat coating / (3) Active ingredient-containing layer coating / (2) Seal coat coating / (1) Organic acid-containing layer coated particles] (formulation example 4: see Table 2). The coating amount of the gastric film is 100 weight parts per 100 weight parts of [(4) seal coat coating / (3) active ingredient-containing layer coating / (2) seal coat coating / (1) organic acid-containing layer coated particles]. Was part. (Total 640 g). The average particle size of the obtained MRP particles was 267 μm, and the content of the active ingredient vardenafil was about 16% by weight.
Formulation examples 1, 2, and 3 having no sustained release coating layer (SR) can also be produced in the same manner as in Example 1.
実施例2[徐放性皮膜層(SR)を有する放出制御型粒子状医薬組成物(MRP)の調製](製剤例10)
<1>[(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルセルロース20gを含む水溶液を撹拌下、フマル酸200g、およびタルク10gを添加して分散させた。この分散液を結晶セルロース(粒)(Celphere(登録商標)CP−102)1000gに対して、転動流動層造粒機を用いて、吸気温度約65℃、排気温度約30℃で側方噴霧し、[(1)有機酸含有層被覆粒子]を得た。フマル酸の量は、セルロース核粒子100重量部に対して20重量部であった。(計1230g)
Example 2 [Preparation of controlled release particulate pharmaceutical composition (MRP) having sustained-release coating layer (SR)] (Formulation Example 10)
<1> [(1) Preparation of organic acid-containing layer-coated particles]
Under stirring, an aqueous solution containing 20 g of hydroxypropylcellulose was added and dispersed with 200 g of fumaric acid and 10 g of talc. This dispersion is laterally sprayed on 1000 g of crystalline cellulose (grain) (Celphere® CP-102) using a tumbling fluidized bed granulator at an intake temperature of about 65 ° C. and an exhaust temperature of about 30 ° C. To obtain [(1) organic acid-containing layer-coated particles]. The amount of fumaric acid was 20 parts by weight with respect to 100 parts by weight of the cellulose core particles. (Total 1230g)
<2>[(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルメチルセルロース(ヒプロメロース)24g、およびポリエチレングリコール(マクロゴール6000)6gを含む水溶液を、上記で得た(1)有機酸含有層被覆粒子に対して、転動流動層造粒機を用いて、吸気温度約80℃、排気温度約42℃で側方噴霧し、[(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。(計1260g)
<2> [(2) Seal coat coating / (1) Preparation of organic acid-containing layer coated particles]
An aqueous solution containing 24 g of hydroxypropylmethylcellulose (hypromellose) and 6 g of polyethylene glycol (Macrogol 6000) was used for (1) organic acid-containing layer-coated particles obtained above using a rolling fluidized bed granulator. Side spraying was performed at an intake temperature of about 80 ° C. and an exhaust temperature of about 42 ° C. to obtain [(2) seal coat coating / (1) organic acid-containing layer coated particles]. (Total 1260g)
<3>[(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
エチルセルロース水分散液(Aquacoat(登録商標)ECD−30)400g(固形分120g)、およびクエン酸トリエチル15gを含む水分散液を、上記で得た[(2)シールコート被覆/(1)有機酸含有被覆粒子]450gに対して、転動流動層造粒機を用いて、吸気温度約75℃、排気温度約32℃で側方噴霧し、[(3)有効成分含有層被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。徐放性皮膜の被覆量は、[(2)シールコート被覆/(1)有機酸含有被覆粒子]100重量部に対して30重量部であった。(計585g)
<3> [(3) Sustained release film coating / (2) Seal coat coating / (1) Preparation of organic acid-containing layer coated particles]
An aqueous dispersion containing 400 g of ethylcellulose aqueous dispersion (Aquacoat (registered trademark) ECD-30) (solid content 120 g) and 15 g of triethyl citrate was obtained as above [(2) Seal coat coating / (1) organic acid Contained particles] 450 g is subjected to side spraying at an intake temperature of about 75 ° C. and an exhaust temperature of about 32 ° C. using a rolling fluidized bed granulator, [(3) active ingredient containing layer coating / (2) Seal coat coating / (1) Organic acid-containing layer-coated particles] was obtained. The coating amount of the sustained release film was 30 parts by weight with respect to 100 parts by weight of [(2) Seal coat coating / (1) Organic acid-containing coated particles]. (Total 585g)
<4>[(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルメチルセルロース(ヒプロメロース)12g、およびポリエチレングリコール(マクロゴール6000)3gを含む水溶液を、上記で得た[(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]に対して、転動流動層造粒機を用いて、吸気温度約80℃、排気温度約40℃で側方噴霧した。噴霧完了後、引き続き、吸気温度85℃(排気温度は成り行き)で90分間の熱処理により皮膜を熟成させ、[(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。(計600g)
<4> [(4) Seal coat coating / (3) Sustained release film coating / (2) Seal coat coating / (1) Preparation of organic acid-containing layer coated particles]
An aqueous solution containing 12 g of hydroxypropylmethylcellulose (hypromellose) and 3 g of polyethylene glycol (Macrogol 6000) was obtained above [(3) Sustained release film coating / (2) Seal coat coating / (1) Organic acid-containing layer The coated particles were sprayed sideways at an intake temperature of about 80 ° C. and an exhaust temperature of about 40 ° C. using a rolling fluidized bed granulator. After spraying is completed, the film is matured by heat treatment for 90 minutes at an intake air temperature of 85 ° C. (exhaust temperature is expected), [(4) Seal coat coating / (3) Sustained release film coating / (2) Seal coat coating / (1) Organic acid-containing layer-coated particles]. (Total 600g)
<5>[(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルセルロース20gを含む水溶液を撹拌下、バルデナフィル塩酸塩・3水和物(BayerHC社製)200g、およびタルク10gを添加して分散させた。この分散液を、上記で得た[(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]420gに対して、転動流動層造粒機を用いて、吸気温度約50℃、排気温度約28℃で側方噴霧し、[(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。(計650g)
<5> [(5) Active ingredient-containing layer coating / (4) Seal coat coating / (3) Sustained release coating / (2) Seal coat coating / (1) Preparation of organic acid-containing layer coated particles]
While stirring, an aqueous solution containing 20 g of hydroxypropylcellulose was added and dispersed with 200 g of vardenafil hydrochloride trihydrate (manufactured by BayerHC) and 10 g of talc. This dispersion was rolled against 420 g of [(4) seal coat coating / (3) sustained release coating / (2) seal coat coating / (1) organic acid-containing layer coated particles] obtained above. Using a fluidized bed granulator, side spraying is performed at an intake temperature of about 50 ° C. and an exhaust temperature of about 28 ° C. [[5) Active ingredient-containing layer coating / (4) Seal coat coating / (3) Sustained release coating Coating / (2) Seal coat coating / (1) Organic acid-containing layer coated particles]. (Total 650 g)
<6>[(6)シールコート被覆/(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ヒドロキシプロピルメチルセルロース(ヒプロメロース)12g、およびポリエチレングリコール(マクロゴール6000)3gを含む水溶液を、上記で得た[(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]に対して、転動流動層造粒機を用いて、吸気温度約85℃、排気温度約42℃で側方噴霧し、[(6)シールコート被覆/(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]を得た。(計665g)
<6> [(6) Seal coat coating / (5) Active ingredient-containing layer coating / (4) Seal coat coating / (3) Sustained release coating / (2) Seal coat coating / (1) Organic acid-containing layer Preparation of coated particles]
An aqueous solution containing 12 g of hydroxypropylmethylcellulose (hypromellose) and 3 g of polyethylene glycol (Macrogol 6000) was obtained above [(5) Active ingredient-containing layer coating / (4) Seal coat coating / (3) Sustained release coating Coating / (2) seal coat coating / (1) organic acid-containing layer coated particles] using a tumbling fluidized bed granulator and side spraying at an intake air temperature of about 85 ° C. and an exhaust temperature of about 42 ° C. [(6) Seal coat coating / (5) Active ingredient containing layer coating / (4) Seal coat coating / (3) Sustained release coating coating / (2) Seal coat coating / (1) Organic acid containing layer coating particles ] Was obtained. (Total 665g)
<7>[(7)胃溶性皮膜被覆/(6)シールコート被覆/(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]
ラウリル硫酸ナトリウム15gを含む水溶液を撹拌下、アミノアルキルメタクリレートコポリマーE(オイドラギット(登録商標)EPO)150g、およびステアリン酸22.5gを添加して分散させた。続いて、タルク52.5gを撹拌下に加えて分散液を調製した。この分散液を、上記で得た[(6)シールコート被覆/(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]320gに対して、転動流動層造粒機を用いて、吸気温度約42℃、排気温度約27℃で側方噴霧し、本発明の放出制御型粒子状医薬組成物(MRP)である、[(7)胃溶性皮膜被覆/(6)シールコート被覆/(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子](製剤例10:表4)を得た。胃溶性皮膜の被覆量は、[(6)シールコート被覆/(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子]100重量部に対して75重量部であった。(計560g)。得られたMRP粒子の平均粒径は276μm、有効成分バルデナフィルの含有量は約14重量%であった。
徐放性皮膜層(SR)を有する後記製剤例5−9、11−18及び20も上記実施例2と同様にして製造することができる。
<7> [(7) Gastric soluble coating / (6) Seal coat coating / (5) Active ingredient-containing layer coating / (4) Seal coat coating / (3) Sustained release coating coating / (2) Seal coat coating / (1) Preparation of organic acid-containing layer-coated particles]
While stirring, an aqueous solution containing 15 g of sodium lauryl sulfate was added and dispersed by adding 150 g of aminoalkyl methacrylate copolymer E (Eudragit (registered trademark) EPO) and 22.5 g of stearic acid. Subsequently, 52.5 g of talc was added with stirring to prepare a dispersion. This dispersion was obtained as described above [(6) Seal coat coating / (5) Active ingredient-containing layer coating / (4) Seal coat coating / (3) Sustained release film coating / (2) Seal coat coating / ( 1) Organic acid-containing layer-coated particles] With respect to 320 g, using a rolling fluidized bed granulator, side spraying is performed at an intake temperature of about 42 ° C. and an exhaust temperature of about 27 ° C. It is a pharmaceutical composition (MRP) [(7) gastric film coating / (6) seal coat coating / (5) active ingredient-containing layer coating / (4) seal coat coating / (3) sustained release film coating / (2) Seal coat coating / (1) Organic acid-containing layer-coated particles] (Formulation Example 10: Table 4) were obtained. The coating amount of the gastric film was [(6) Seal coat coating / (5) Active ingredient-containing layer coating / (4) Seal coat coating / (3) Sustained release film coating / (2) Seal coat coating / (1 ) Organic acid-containing layer-coated particles] 75 parts by weight with respect to 100 parts by weight. (Total 560 g). The average particle diameter of the obtained MRP particles was 276 μm, and the content of the active ingredient vardenafil was about 14% by weight.
The following formulation examples 5-9, 11-18 and 20 having a sustained-release coating layer (SR) can also be produced in the same manner as in Example 2.
比較例1[有機酸含有層不存在の粒子製剤](比較製剤例4)
実施例1における<1>[(1)有機酸含有層被覆粒子の調製]工程、および<2>[(2)シールコート被覆/(1)有機酸含有層被覆粒子の調製]工程を省略した以外は、実施例1に準じて粒子状医薬組成物を調製した。即ち、得られた粒子状医薬組成物は、[(4)胃溶性皮膜被覆/(3)シールコート被覆/(2)有効成分含有層被覆/(1)不活性コア粒子]からなる4層構造を有しており、有機酸含有層は存在しない。なお、胃溶性皮膜の被覆量は、[(3)シールコート被覆/(2)有効成分含有層被覆/(1)不活性コア粒子]100重量部に対して32.4重量部とした。
得られた[(4)胃溶性皮膜被覆/(3)シールコート被覆/(2)有効成分含有層被覆/(1)不活性コア粒子]の平均粒径は237μm、有効成分バルデナフィルの含有量は約28重量%であった(比較製剤例4:表1参照)。
有機酸含有層不存在の粒子製剤である比較製剤例3も上記比較例1と同様にして製造することができる。また、比較製剤例1及び2も上記比較例1及び上記実施例2に準じ、(5)シールコート被覆(オーバーコート)(4)徐放性皮膜被覆/(3)シールコート被覆/(2)有効成分含有層被覆/(1)不活性コア粒子の順序で各工程を適宜選択し、製造することができる。
Comparative Example 1 [Particle preparation without organic acid-containing layer] (Comparative preparation example 4)
The <1> [(1) Preparation of organic acid-containing layer-coated particles] step and <2> [(2) Seal coat coating / (1) Preparation of organic acid-containing layer-coated particles] step in Example 1 were omitted. Except for the above, a particulate pharmaceutical composition was prepared according to Example 1. That is, the obtained particulate pharmaceutical composition has a four-layer structure consisting of [(4) gastric soluble film coating / (3) seal coat coating / (2) active ingredient-containing layer coating / (1) inert core particles]. There is no organic acid-containing layer. The coating amount of the gastric film was 32.4 parts by weight with respect to 100 parts by weight of [(3) Seal coat coating / (2) Active ingredient-containing layer coating / (1) Inactive core particles].
The average particle size of the obtained [(4) gastric film coating / (3) seal coat coating / (2) active ingredient containing layer coating / (1) inert core particles] was 237 μm, and the content of the active ingredient vardenafil was About 28% by weight (Comparative formulation example 4: see Table 1).
Comparative Formulation Example 3, which is a particle formulation without an organic acid-containing layer, can also be produced in the same manner as Comparative Example 1. Further, Comparative Formulation Examples 1 and 2 were also in accordance with Comparative Example 1 and Example 2 above, (5) Seal coat coating (overcoat) (4) Sustained release film coating / (3) Seal coat coating / (2) Each step can be appropriately selected and manufactured in the order of active ingredient containing layer coating / (1) inert core particles.
実施例3[放出制御型粒子状医薬組成物(MRP:比較製剤例1〜4並びに製剤例1〜18及び20)からの有効成分(バルデナフィル)溶出プロファイル] Example 3 [Dissolution profile of active ingredient (Vardenafil) from controlled release particulate pharmaceutical composition (MRP: Comparative Formulation Examples 1-4 and Formulation Examples 1-18 and 20)]
実施例1および2で得られた放出制御型粒子状医薬組成物(MRP)、ならびに、比較例1並びにそれらに準じて得た粒子状医薬組成物について、有効成分(バルデナフィル)の溶出プロファイルを測定した。 For the controlled release particulate pharmaceutical composition (MRP) obtained in Examples 1 and 2, and Comparative Example 1 and the particulate pharmaceutical composition obtained according to them, the dissolution profile of the active ingredient (Vardenafil) was measured. did.
溶出試験は、日本薬局方一般試験法6.10溶出試験法のパドル法(装置2)に従って行った。即ち、37℃に設定した恒温水槽中の撹拌層内に所定のpHに調製した試験液900mLを入れ、バルデナフィル10mgに相当する量のMRPを分散させる。パドルを50rpm又は100rpmで撹拌しつつ、所定時間毎にサンプリングを行い、メンブランフィルターによるろ液につき、日本薬局方一般試験法2.24紫外可視吸光度測定法又は2.01液体クロマトグラフィーによりバルデナフィルの溶出量をモニターした(液体クロマトグラフィーではフマル酸の放出量も同時にモニター)。得られた結果を図4〜15に示す。尚、図4〜15中、Controlは、市販品(レビトラ(登録商標)錠10mg)であり、Comp.1〜4並びにEx.1〜18及び20はそれぞれ、比較製剤例1〜4並びに製剤例1〜18及び20を表す。
また、表8〜15にはMRPの1%溶出時間(t1% [分](t1% [分]と表示することもある。);本発明のラグタイムの定義である「1%溶出するまでに要する時間」を表す。)並びに15分後溶出率 (d15分 [%](d15分 [%]と表示することもある。);ラグタイム後の溶出速度の指標である。)を示す。表8〜15中、Controlは、市販品(レビトラ(登録商標)錠10mg)である。
これら溶出プロファイルの結果が示唆することを以下に要約する。
比較製剤例1及び2:有効成分層の外側に徐放性皮膜を施すことにより、短時間のラグタイムが生じるが(比較製剤例1)、ラグタイムを延ばすため皮膜を厚くするほどラグタイム後の溶出は抑制される。徐放性皮膜と水溶性高分子の混合皮膜は、徐放性皮膜単味と本質的に同じ問題をかかえる(比較製剤例2)。
比較製剤例3及び4:有効成分層の外側に胃溶性皮膜を施すことにより、口腔内のpH(pH6.8)における溶出を抑制できる。ただし、皮膜が厚いと長時間経過してもほとんど溶出せず(比較製剤例4)、薄いと皮膜の不完全さに基づく溶出の加速(短時間のラグタイム)が見られるものの、その後の放出は不完全であり、再現性は全く期待できない(比較製剤例3)。
製剤例1−4(徐放性皮膜なし実施例):胃溶性皮膜の厚みでラグタイム、フマル酸の量でラグタイム後の溶出率を調整可能である(pH6.8)。また酸性では速放である。
製剤例5−10(徐放性皮膜あり実施例):徐放性皮膜の厚みでラグタイム後の溶出速度も調整可能である(pH6.8)。また酸性ではやはり速放である。
製剤例11−18及び20:徐放性皮膜がエチルセルロース水分散液(アクアコートECD−30)でもアクリル酸エチル・メタクリル酸メチルコポリマー分散液(オイドラギットNE30D)でも溶出プロファイルは精密に制御されており、すなわち半透膜であれば適用可能であることを示唆する。また、製剤例16より、要すれば放出制御型粒子状医薬組成物(MRP)の表面修飾も可能で、例えばエリスリトールを10%被覆しても溶出特性は維持されることがわかる。
The dissolution test was performed according to the paddle method (apparatus 2) of the Japanese Pharmacopoeia General Test Method 6.10 Dissolution Test Method. That is, 900 mL of a test solution adjusted to a predetermined pH is placed in a stirring layer in a constant temperature water bath set at 37 ° C., and an amount of MRP corresponding to 10 mg of vardenafil is dispersed. Sampling the paddle at 50 rpm or 100 rpm, sampling every predetermined time, and elution of vardenafil by the Japanese Pharmacopoeia General Test Method 2.24 UV-Vis Absorbance Measurement Method or 2.01 Liquid Chromatography for the filtrate through the membrane filter The amount was monitored (the amount of fumaric acid released was also monitored in liquid chromatography). The obtained results are shown in FIGS. 4 to 15, Control is a commercially available product (Levitra (registered trademark) 10 mg). 1-4 and Ex. 1 to 18 and 20 represent Comparative Formulation Examples 1 to 4 and Formulation Examples 1 to 18 and 20, respectively.
Further, in Table 8-15 also be displayed with 1% elution time MRP (t1% [min] (t 1% [min]);. To lag time is defined "1% dissolution of the present invention . it represents the required time "until) and after 15 min dissolution rate (d15 min [%] (sometimes indicated as d 15 min [%].); a is an index of the dissolution rate after lag time). Show. In Tables 8 to 15, Control is a commercially available product (Levitra (registered trademark) 10 mg).
The results of these elution profiles are summarized below.
Comparative Formulation Examples 1 and 2: By applying a sustained-release film on the outside of the active ingredient layer, a short lag time is generated (Comparative Formulation Example 1), but after the lag time is increased as the film is thickened to extend the lag time. Elution is suppressed. The mixed-release film of the sustained-release film and the water-soluble polymer has essentially the same problem as the simple-release film (Comparative Formulation Example 2).
Comparative formulation examples 3 and 4: By applying a gastric film on the outside of the active ingredient layer, elution at the pH in the oral cavity (pH 6.8) can be suppressed. However, if the film is thick, it does not dissolve almost even after a long time (Comparative Formulation Example 4). If the film is thin, acceleration of dissolution based on incomplete film (short lag time) is observed, but subsequent release Is incomplete and reproducibility cannot be expected at all (Comparative Preparation Example 3).
Formulation example 1-4 (Example without sustained release film): The elution rate after the lag time can be adjusted by adjusting the thickness of the gastric soluble film and the amount of fumaric acid (pH 6.8). In addition, it is quick release when acidic.
Formulation Example 5-10 (Example with Sustained Release Film): The dissolution rate after the lag time can be adjusted by the thickness of the sustained release film (pH 6.8). Moreover, it is an immediate release when it is acidic.
Formulation Examples 11-18 and 20: Whether the sustained-release film is an aqueous dispersion of ethyl cellulose (Aquacoat ECD-30) or an ethyl acrylate / methyl methacrylate copolymer dispersion (Eudragit NE30D), the dissolution profile is precisely controlled. That is, it is suggested that a semipermeable membrane is applicable. Moreover, it can be seen from Formulation Example 16 that if necessary, the surface of the controlled-release particulate pharmaceutical composition (MRP) can be modified. For example, even when 10% of erythritol is coated, the dissolution characteristics are maintained.
実施例4 [放出制御型粒子状医薬組成物(MRP:製剤例12)からの有効成分(バルデナフィル)およびフマル酸の溶出プロファイルの比較]
実施例2に準じて調製した放出制御型粒子状医薬組成物[(7)胃溶性皮膜被覆/(6)シールコート被覆/(5)有効成分含有層被覆/(4)シールコート被覆/(3)徐放性皮膜被覆/(2)シールコート被覆/(1)有機酸含有層被覆粒子](MRP)を用いて、実施例3に準じてバルデナフィルおよびフマル酸の溶出プロファイルを同時測定した。
Example 4 [Comparison of Elution Profiles of Active Ingredient (Vardenafil) and Fumaric Acid from Controlled Release Particulate Pharmaceutical Composition (MRP: Formulation Example 12)]
Controlled release particulate pharmaceutical composition prepared according to Example 2 [(7) gastric film coating / (6) seal coat coating / (5) active ingredient containing layer coating / (4) seal coat coating / (3 The elution profiles of vardenafil and fumaric acid were simultaneously measured according to Example 3 using)) slow release film coating / (2) seal coat coating / (1) organic acid-containing layer coated particles] (MRP).
測定に用いたMRPの層構成と各層の組成は表4中の製剤例12で、溶出プロファイルは図11のとおりである。図中、Ex.12は製剤例12のことを表し、FA at pH1.2 、FA at pH5.0及びFA at pH6.8はそれぞれ、pH1.2におけるフマル酸、pH5.0におけるフマル酸及びpH6.8におけるフマル酸を表す。 The layer structure of MRP used for the measurement and the composition of each layer are Formulation Example 12 in Table 4, and the dissolution profile is as shown in FIG. In the figure, Ex. 12 represents Formulation Example 12, and FA at pH 1.2, FA at pH 5.0 and FA at pH 6.8 are respectively fumaric acid at pH 1.2, fumaric acid at pH 5.0 and fumaric acid at pH 6.8. Represents.
その溶出プロファイルの結果から、下記のことがわかる。即ち、製剤例12は、弱酸性から中性(pH5以上)において、ラグタイムは有機酸の放出開始と一致(同期)し、有機酸は制御された放出速度に応じて最外層(オイドラギットE)の一部又は全部を溶解し、外液に曝されて表面積が増大した有効成分の溶出が始まる。また、酸性(pH1.2)では最外層から溶解が進行するので、有機酸の放出よりより先に有効成分が溶出する(速放)。
The following can be seen from the results of the elution profile. That is, in Formulation Example 12, weak acid to neutral (
実施例5
[非活性速溶粒子の調製]
エリスリトール980g及びポビドン20gを高速攪拌造粒機に入れ、75%エタノールを湿潤剤として加え、練合した。この練合物を円筒造粒機を用いて造粒し、棚式乾燥機で乾燥した。網式整粒機で粒径を調整し、非活性速溶粒子を得た。
Example 5
[Preparation of non-active fast dissolving particles]
980 g of erythritol and 20 g of povidone were put into a high-speed stirring granulator, and 75% ethanol was added as a wetting agent and kneaded. This kneaded product was granulated using a cylindrical granulator and dried with a shelf dryer. The particle size was adjusted with a net type granulator to obtain inactive fast dissolving particles.
[経口投与用粒子状組成物(ODP)の調製]
実施例2に準じて得られた[徐放性皮膜層を有する放出制御型粒子状医薬組成物(製剤例12)]69.8g及び上記で得た[非活性速溶粒子]930.2gを混合し、経口投与用粒子状組成物(ODP)1000gを調製した(製剤例19)。有効成分バルデナフィルの含有量は約1重量%であった。
製剤例22−24の経口投与用粒子状組成物(ODP)も上記実施例5と同様にして製造することができる。
[Preparation of particulate composition for oral administration (ODP)]
69.8 g of [Release Controlled Particulate Pharmaceutical Composition Having Sustained Release Film Layer (Formulation Example 12)] obtained according to Example 2 and 930.2 g of [Inactive Fast Dissolving Particles] obtained above were mixed. Then, 1000 g of an oral administration particulate composition (ODP) was prepared (Formulation Example 19). The content of the active ingredient vardenafil was about 1% by weight.
A particulate composition for oral administration (ODP) of Preparation Examples 22-24 can also be produced in the same manner as in Example 5.
[口腔内崩壊錠(ODT)の調製]
製剤例21の口腔内崩壊錠(ODT)は、実施例2で製造した放出制御型粒子状医薬組成物(MRP)(製剤例10)に流動化剤(軽質無水ケイ酸)を加え(製剤例20)、[非活性速溶粒子]及び滑沢剤(フマル酸ステアリルナトリウム)と混合後、圧縮成型し、日本特許第2919771号に記載の硬化処理を施すことにより得た。
[Preparation of Orally Disintegrating Tablets (ODT)]
In the orally disintegrating tablet (ODT) of Preparation Example 21, a fluidizing agent (light anhydrous silicic acid) was added to the controlled release particulate pharmaceutical composition (MRP) (Formulation Example 10) produced in Example 2 (Formulation Example) 20), [Inactive fast-dissolving particles] and a lubricant (sodium stearyl fumarate), and then compression-molded to obtain a curing treatment described in Japanese Patent No. 2919771.
実施例6[[経口投与用粒子状組成物からの有効成分(バルデナフィル)の溶出プロファイル]
実施例5またはそれに準じて調製した経口投与用粒子状組成物(ODP)及び口腔内崩壊錠(ODT)を用いて、実施例3と同様に、有効成分(バルデナフィル)の溶出プロファイルを測定した。得られた結果を図12〜16に示す。
Example 6 [Elution profile of active ingredient (Vardenafil) from particulate composition for oral administration]
Using the particulate composition for oral administration (ODP) and the orally disintegrating tablet (ODT) prepared according to Example 5 or according thereto, the dissolution profile of the active ingredient (Vardenafil) was measured in the same manner as in Example 3. The obtained results are shown in FIGS.
図12及び13中、Controlは、市販品(レビトラ(登録商標)錠10mg)であり、図16中、Controlは、市販品(レビトラ(登録商標)錠20mg)であり、図12〜16中、Ex.12、19、21、22、23及び24はそれぞれ、製剤例12、19、21、22、23及び24を表す。 In FIGS. 12 and 13, Control is a commercially available product (Levitra (registered trademark) 10 mg), and in FIG. 16, Control is a commercially available product (Levitra (registered trademark) 20 mg), in FIGS. Ex. 12, 19, 21, 22, 23, and 24 represent Formulation Examples 12, 19, 21, 22, 23, and 24, respectively.
図13中「Ex.19,6M」とは、Ex.19の経口投与用粒子状組成物(ODP)粒子を乾燥剤とともに包装し、加速試験条件下(40℃/75%RH(相対湿度))、6ヵ月間保存したサンプルを表す。Ex.19,6Mの経口投与用粒子状組成物(ODP)の溶出特性はEx.19の経口投与用粒子状組成物(ODP)からほとんど変化しておらず、長期的な安定性を示唆する。
また、表14〜16にはODP並びにODTの1%溶出時間(t1% [分];本発明のラグタイムの定義である「1%溶出するまでに要する時間」)並びに15分後溶出率 (d15分 [%];ラグタイム後の溶出速度の指標)を示す。表14〜15中、Controlは、市販品(レビトラ(登録商標)錠10mg)であり、表16中、Controlは、市販品(レビトラ(登録商標)錠20mg)であり、表15中「製剤例19,6M」とは、製剤例19の経口投与用粒子状組成物(ODP)粒子を乾燥剤とともに包装し、加速試験条件下(40℃/75%RH(相対湿度))、6ヵ月間保存したサンプルを表す。
“Ex. 19, 6M” in FIG. 19 represents a sample of 19 orally administered particulate composition (ODP) particles packaged with a desiccant and stored under accelerated test conditions (40 ° C./75% RH (relative humidity)) for 6 months. Ex. The dissolution characteristics of 19,6M particulate composition for oral administration (ODP) are Ex. Little change from the 19 orally administered particulate composition (ODP) suggests long-term stability.
Tables 14 to 16 show 1% elution time of ODP and ODT (t1% [min]; “time required for 1% elution” as defined by the lag time of the present invention) and elution rate after 15 minutes ( d15 minutes [%]; index of elution rate after lag time). In Tables 14 to 15, Control is a commercially available product (Levitra (registered trademark) 10 mg), and in Table 16, Control is a commercially available product (Levitra (registered trademark) 20 mg). "19,6M" means that the particulate composition for oral administration (ODP) particles of Preparation Example 19 are packaged with a desiccant and stored for 6 months under accelerated test conditions (40 ° C / 75% RH (relative humidity)). Represents the sample.
製剤例19、21、22、23及び24の経口投与用粒子状組成物(ODP)又は口腔内崩壊錠(ODT)のように、唾液の分泌を促す甘味成分(別途調製した速溶顆粒)、必要に応じて流動化剤ないし滑沢剤や香料等を加えて分包化ないし錠剤化すると、服用や持ち運びに便利であると同時に、溶出特性は維持され、安定性も良好(6ヵ月間の加速試験)であった。 Sweet ingredients that promote the secretion of saliva (fast-dissolving granules prepared separately), such as the particulate compositions for oral administration (ODP) or orally disintegrating tablets (ODT) of Preparation Examples 19, 21, 22, 23 and 24, necessary Depending on the condition, adding a fluidizing agent, lubricant or fragrance, etc. to sachet or tablet, it is convenient for taking and carrying, while maintaining dissolution properties and good stability (acceleration for 6 months) Test).
比較製剤例1〜4及び製剤例1〜24の処方を以下の表1〜7に示し、1%溶出時間(t1% [分])並びに15分後溶出率 (d15分 [%])を以下の表8〜16に示す。 The formulations of Comparative Preparation Examples 1 to 4 and Preparation Examples 1 to 24 are shown in Tables 1 to 7 below, and the 1% elution time (t1% [min]) and the elution rate after 15 minutes (d15 minutes [%]) are shown below. Are shown in Tables 8-16.
表1[有機酸含有層不存在の粒子製剤:比較製剤例1〜4] Table 1 [Particle preparation without organic acid-containing layer: Comparative preparation examples 1 to 4]

表2[徐放性皮膜層(SR)のない放出制御型粒子状医薬組成物(MRP):製剤例1〜4] Table 2 [Release Controlled Particulate Pharmaceutical Composition (MRP) Without Sustained Release Film Layer (SR): Formulation Examples 1-4]
表3[徐放性皮膜層(SR)を有する放出制御型粒子状医薬組成物(MRP):製剤例5〜8] Table 3 [Release Controlled Particulate Pharmaceutical Composition (MRP) Having Sustained Release Coating Layer (SR): Formulation Examples 5 to 8]
表4[徐放性皮膜層(SR)を有する放出制御型粒子状医薬組成物(MRP):製剤例9〜12] Table 4 [Release Controlled Particulate Pharmaceutical Composition (MRP) Having Sustained Release Coating Layer (SR): Formulation Examples 9-12]
表5[徐放性皮膜層(SR)を有する放出制御型粒子状医薬組成物(MRP):製剤例13〜16] Table 5 [Release Controlled Particulate Pharmaceutical Composition (MRP) Having Sustained Release Coating Layer (SR): Formulation Examples 13 to 16]
表6[徐放性皮膜層(SR)を有する放出制御型粒子状医薬組成物(MRP)又はMRPを用いた経口投与用粒子状組成物(ODP):製造例17〜20] Table 6 [Release Controlled Particulate Pharmaceutical Composition (MRP) Having Sustained Release Coating Layer (SR) or Particulate Composition for Oral Administration (ODP) Using MRP: Production Examples 17 to 20]
表7[徐放性皮膜層(SR)を有する放出制御型粒子状医薬組成物(MRP)を用いた経口投与用粒子状組成物(ODP)又は口腔内崩壊錠(ODT):製造例21〜24] Table 7 [Particulate composition for oral administration (ODP) or orally disintegrating tablet (ODT) using controlled release particulate pharmaceutical composition (MRP) having sustained-release coating layer (SR): Production Examples 21 to 21] 24]
表8[Control(市販品レビトラ(登録商標)錠10mg)及びMRPのpH1.2における1%溶出時間(t1% [分])及び15分後溶出率 (d15分 [%]):バルデナフィル10mg相当量分散、パドルを50rpmで攪拌、900ml試験液使用:図4に対応する。]
Table 8 [Control (commercially available Levitra (registered trademark)
表9[Control(市販品レビトラ(登録商標)錠10mg)及びMRPのpH1.2における1%溶出時間(t1% [分])及び15分後溶出率 (d15分 [%]):バルデナフィル10mg相当量分散、パドルを50rpmで攪拌、900ml試験液使用:図6及び8に対応する。]
Table 9 [Control (commercially available Levitra (registered trademark)
表10[Control(市販品レビトラ(登録商標)錠10mg)及びMRPのpH6.8における1%溶出時間(t1% [分])及び15分後溶出率 (d15分[%]):バルデナフィル10mg相当量分散、パドルを50rpmで攪拌、900ml試験液使用:図5に対応する。]
Table 10 [Control (commercial product Levitra (registered trademark)
表11[Control(市販品レビトラ(登録商標)錠10mg)及びMRPのpH6.8における1%溶出時間(t1% [分])及び15分後溶出率 (d15分[%]):バルデナフィル10mg相当量分散、パドルを50rpmで攪拌、900ml試験液使用:図7及び9に対応する。]
Table 11 [Control (commercially available Levitra (registered trademark)
表12[Control(市販品レビトラ(登録商標)錠10mg)及びMRPのpH6.8における1%溶出時間(t1% [分])及び15分後溶出率 (d15分[%]):バルデナフィル10mg相当量分散、パドルを100rpmで攪拌、900ml試験液使用:図10に対応する。]
Table 12 [Control (commercially available Levitra (registered trademark)
表13[MRPのpH1.2、5.0及び6.8における1%溶出時間(t1% [分])及び15分後溶出率 (d15分[%]):バルデナフィル10mg相当量分散、パドルを50rpmで攪拌、900ml試験液使用:図11に対応する。] Table 13 [MRP pH 1.2, 5.0 and 6.8 at 1% elution time (t1% [min]) and elution rate after 15 min (d15 min [%]): Vardenafil equivalent to 10 mg dispersion, paddle Stirring at 50 rpm, using 900 ml test solution: corresponding to FIG. ]
表14[Control(市販品レビトラ(登録商標)錠10mg)、MRP、ODP及びODTのpH1.2における1%溶出時間(t1% [分])及び15分後溶出率 (d15分 [%]):バルデナフィル10mg相当量分散、パドルを50rpmで攪拌、900ml試験液使用:図12及び14に対応する。]
Table 14 [Control (commercially available Levitra (registered trademark)
表15[Control(市販品レビトラ(登録商標)錠10mg)、MRP、ODP及びODTのpH6.8における1%溶出時間(t1% [分])及び15分後溶出率 (d15分 [%]):バルデナフィル10mg相当量分散、パドルを50rpmで攪拌、900ml試験液使用:図13及び15に対応する。]
Table 15 [Control (commercially available Levitra (registered trademark)
表16[Control(市販品レビトラ(登録商標)錠20mg)及びODPのpH6.8における1%溶出時間(t1% [分])及び15分後溶出率 (d15分 [%]):バルデナフィル20mg相当量分散、パドルを100rpmで攪拌、900ml試験液使用:図16に対応する。]
Table 16 [Control (commercially available Levitra (registered trademark)
本発明は、バルデナフィルに代表される弱酸性ないし塩基性の有効成分において、十分な長さのラグタイムを有し、ラグタイム後に速やかに有効成分を放出でき、かつ有効成分・製剤の特性・目的に応じて、ラグタイムの長さおよびその後の溶出速度を任意にコントロール可能な放出制御型粒子状医薬組成物ならびに該組成物を含有する口腔内速崩壊粒子剤もしくは錠剤を提供する。 The present invention is a weakly acidic or basic active ingredient typified by vardenafil, has a sufficiently long lag time, and can release the active ingredient promptly after the lag time, and the characteristics / objectives of the active ingredient / formulation Accordingly, the present invention provides a controlled-release particulate pharmaceutical composition capable of arbitrarily controlling the length of lag time and the subsequent dissolution rate, and an intraoral rapidly disintegrating particle or tablet containing the composition.
Claims (28)
(i)不活性コアの外側に有機酸含有層を被覆し有機酸含有核粒子を形成する工程、
(ii)該有機酸含有核粒子の外側に徐放性皮膜を被覆する工程
(iii)該徐放性皮膜層の外側に有効成分を含有する層を被覆する工程、および
(iv)該有効成分含有層の外側を胃溶性皮膜で被覆して経口投与用の放出制御型粒子状医薬組成物を形成する工程
を含むことを特徴とする、放出制御型粒子状医薬組成物の製造方法。 A method for producing a controlled release particulate pharmaceutical composition according to any one of claims 1 to 14,
(I) coating the organic acid-containing layer outside the inert core to form organic acid-containing core particles;
(Ii) a step of coating the organic acid-containing core particles with a sustained-release coating (iii) a step of coating a layer containing an active ingredient on the outside of the sustained-release coating layer, and (iv) the active component A method for producing a controlled release particulate pharmaceutical composition comprising the step of coating the outer side of a containing layer with a gastric soluble film to form a controlled release particulate pharmaceutical composition for oral administration.
(i)不活性コアの外側に有機酸含有層を被覆し有機酸含有核粒子を形成する工程、
(ii)該有機酸含有核粒子の外側に徐放性皮膜を被覆する工程
(iii)該徐放性皮膜層の外側に有効成分を含有する層を被覆する工程、
(iv)該有効成分含有層の外側を胃溶性皮膜で被覆して経口投与用の放出制御型粒子状医薬組成物を形成する工程、および
(v)該放出制御型粒子状医薬組成物および速溶粒子、ならびに、要すれば流動化剤もしくは滑沢剤および/または香料を混合する工程
を含むことを特徴とする、経口投与用粒子状組成物の製造方法。 A method for producing a disintegrating particulate composition for oral administration according to any one of claims 17-21,
(I) coating the organic acid-containing layer outside the inert core to form organic acid-containing core particles;
(Ii) a step of coating a sustained-release coating on the outside of the organic acid-containing core particles (iii) a step of coating a layer containing an active ingredient on the outside of the sustained-release coating layer;
(Iv) coating the outside of the active ingredient-containing layer with a gastric soluble coating to form a controlled release particulate pharmaceutical composition for oral administration; and (v) the controlled release particulate pharmaceutical composition and fast dissolution A method for producing a particulate composition for oral administration, comprising a step of mixing particles and, if necessary, a fluidizing agent or lubricant and / or a fragrance.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011119340A JP2012246252A (en) | 2011-05-27 | 2011-05-27 | Modified-release type particle pharmaceutical composition for oral administration, method for producing the same, and orally disintegrating tablet including the composition |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011119340A JP2012246252A (en) | 2011-05-27 | 2011-05-27 | Modified-release type particle pharmaceutical composition for oral administration, method for producing the same, and orally disintegrating tablet including the composition |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012246252A true JP2012246252A (en) | 2012-12-13 |
Family
ID=47467077
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011119340A Withdrawn JP2012246252A (en) | 2011-05-27 | 2011-05-27 | Modified-release type particle pharmaceutical composition for oral administration, method for producing the same, and orally disintegrating tablet including the composition |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2012246252A (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107693361A (en) * | 2017-09-13 | 2018-02-16 | 北京化工大学 | A kind of preparation facilities of high-efficiency sustained-release Nano medication |
| CN109963554A (en) * | 2016-09-15 | 2019-07-02 | 优尼特尔制药公司 | Non-agglomerated solid particulate form includes the quick intake of two kinds of different type particles and is convenient for the solid composite swallowed |
| WO2021091188A1 (en) * | 2019-11-05 | 2021-05-14 | 아주대학교산학협력단 | Sustained-release pharmaceutical composition for oral administration, containing rebamipide or pharmaceutically acceptable salt thereof |
| CN114652684A (en) * | 2022-02-21 | 2022-06-24 | 北京罗诺强施医药技术研发中心有限公司 | Solid pharmaceutical composition and process for preparing the same |
| CN107693361B (en) * | 2017-09-13 | 2024-04-26 | 北京化工大学 | Preparation facilities of high-efficient slowly-releasing nano-drug |
-
2011
- 2011-05-27 JP JP2011119340A patent/JP2012246252A/en not_active Withdrawn
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109963554A (en) * | 2016-09-15 | 2019-07-02 | 优尼特尔制药公司 | Non-agglomerated solid particulate form includes the quick intake of two kinds of different type particles and is convenient for the solid composite swallowed |
| JP2019534239A (en) * | 2016-09-15 | 2019-11-28 | ユニザー ファーマシューティカルズUnither Pharmaceuticals | Solid composition with rapid ingestion and easy swallowing, comprising two different types of particles and in the form of non-aggregated solid particles |
| JP7035020B2 (en) | 2016-09-15 | 2022-03-14 | ユニザー ファーマシューティカルズ | A solid composition containing two different types of particles, in the form of non-aggregating solid particles, with rapid ingestion and easy swallowing. |
| US11304905B2 (en) | 2016-09-15 | 2022-04-19 | Unither Pharmaceuticals | Solid composition for quick ingestion with facilitated swallowing, in the form of solid, non-agglomerated particles, comprising two different types of particles |
| AU2017327889B2 (en) * | 2016-09-15 | 2023-04-13 | Unither Pharmaceuticals | Solid composition for quick ingestion with facilitated swallowing, in the form of solid, non-agglomerated particles, comprising two different types of particles |
| CN107693361A (en) * | 2017-09-13 | 2018-02-16 | 北京化工大学 | A kind of preparation facilities of high-efficiency sustained-release Nano medication |
| CN107693361B (en) * | 2017-09-13 | 2024-04-26 | 北京化工大学 | Preparation facilities of high-efficient slowly-releasing nano-drug |
| WO2021091188A1 (en) * | 2019-11-05 | 2021-05-14 | 아주대학교산학협력단 | Sustained-release pharmaceutical composition for oral administration, containing rebamipide or pharmaceutically acceptable salt thereof |
| CN114652684A (en) * | 2022-02-21 | 2022-06-24 | 北京罗诺强施医药技术研发中心有限公司 | Solid pharmaceutical composition and process for preparing the same |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5433236B2 (en) | Drug delivery system comprising a weakly basic selective serotonin 5-HT3 blocker and an organic acid | |
| USRE42096E1 (en) | Oral pulsed dose drug delivery system | |
| KR101752014B1 (en) | Orally disintegrating tablet compositions comprising combinations of high and low-dose drugs | |
| RU2504362C2 (en) | Systems of delivering medical substances, including weakly basic medicinal substances and organic acids | |
| JP5604304B2 (en) | Orally disintegrating solid preparation | |
| US8747895B2 (en) | Orally disintegrating tablets of atomoxetine | |
| JP5366558B2 (en) | Orally disintegrating solid preparation | |
| JP2014501224A (en) | Orally disintegrating tablets | |
| KR20090029830A (en) | Pharmaceutical preparation for oral administration with controlled active ingredient release in the small intestine and methods for its production | |
| CA2994073C (en) | Tablet comprising a core of acetylsalicylic acid with enteric coating and an outer layer with a potassium-competitive acid blocker | |
| US20090202630A1 (en) | Orally disintegrating tablet compositions of ranitidine and methods of manufacture | |
| EP3513784A1 (en) | Esomeprazole-containing complex capsule and preparation method therefor | |
| EP3981390A1 (en) | Lacosamide pharmaceutical composition and pharmaceutical preparation thereof | |
| JP2012246252A (en) | Modified-release type particle pharmaceutical composition for oral administration, method for producing the same, and orally disintegrating tablet including the composition | |
| US20220401373A1 (en) | Timed-elution masking particles and oral pharmaceutical composition containing the same | |
| EP3796908B1 (en) | Controlled release propiverine formulations | |
| AU2006236052B2 (en) | Oral pulsed dose drug delivery system |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20140326 |
|
| A300 | Withdrawal of application because of no request for examination |
Free format text: JAPANESE INTERMEDIATE CODE: A300 Effective date: 20140805 |