JP2012193176A - Milnacipran-containing pharmaceutical composition for percutaneous absorption - Google Patents
Milnacipran-containing pharmaceutical composition for percutaneous absorption Download PDFInfo
- Publication number
- JP2012193176A JP2012193176A JP2012045291A JP2012045291A JP2012193176A JP 2012193176 A JP2012193176 A JP 2012193176A JP 2012045291 A JP2012045291 A JP 2012045291A JP 2012045291 A JP2012045291 A JP 2012045291A JP 2012193176 A JP2012193176 A JP 2012193176A
- Authority
- JP
- Japan
- Prior art keywords
- milnacipran
- composition according
- menthol
- weight
- absorption
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





Abstract
Description
本発明はミルナシプラン又はその薬学的に許容される塩を有効成分として含有する経皮吸収用医薬組成物又はこの組成物を含有する医薬に関する。 The present invention relates to a pharmaceutical composition for percutaneous absorption containing milnacipran or a pharmaceutically acceptable salt thereof as an active ingredient, or a medicine containing this composition.
うつ病は、気分障害の一種であり、抑うつ気分(不安、焦燥など)、意欲の低下(無気力、億劫など)、精神活動の低下(考えがまとまらないなど)、各種の身体症状(不眠、易疲労感、頭重・頭痛、食欲不振など)を呈する精神疾患である。うつ病は、近年罹患者が増大しており、重症化すると日常生活に支障をきたし、自殺企図など生命に係ることもあることから、社会的にも重要な疾患である。そこで、その発症機序の解明が望まれているが、うつ病の発症には精神的な要因と脳神経科学的な要因が複雑に絡んでいると考えられ、十分な解明には至っていない。このうつ病の治療は、現在、薬物療法(抗うつ剤)と心理学的なケアを組み合わせて行われている。抗うつ剤としては、イミプラミンやアミトリプチリンなどの三環系抗うつ剤(第1世代)が先ず開発され使用されてきた。しかし、三環系抗うつ剤は、シナプス後部のヒスタミンH1受容体、アセチルコリン性ムスカリンM1受容体、アドレナリン性α1受容体なども遮断するため、抗コリン作用による便秘や排尿障害などの副作用が多かった。また、大量服用では致死的になるという問題があった。そこで、より副作用の少ない四環系抗うつ剤(第2世代)が開発されたが、抗うつ効果に物足りなさがあり、抗コリン作用もまだ残っていた。1980年代に、さらに抗コリン作用による副作用の少ないセロトニン選択的再取り込み阻害剤(SSRI)(第3世代)が開発されたが、SSRIは脳内や腸管のセロトニン受容体に対する刺激作用による吐気や嘔吐、服薬中断による離脱症候群などの新たな副作用を生じさせた。その後、1990年代になって、選択的セロトニン・ノルアドレナリン再取り込み阻害剤(SNRI)が開発され、胃腸症状はなお残るものの、抗コリン作用のない薬剤として、広く臨床現場で使用されている。 Depression is a type of mood disorder, including depressed mood (anxiety, agitation, etc.), decreased motivation (such as lethargy, hunger, etc.), decreased mental activity (such as lack of thought), and various physical symptoms (such as insomnia, easy sleep) It is a mental illness that causes fatigue, headache / headache, loss of appetite, etc. Depression is a socially important disease because the number of affected individuals has increased in recent years, and when it becomes severe, it has an impact on daily life and can be life-threatening, such as suicide attempts. Therefore, elucidation of the onset mechanism is desired, but it is thought that psychological factors and neuroscience factors are complicatedly involved in the onset of depression, and it has not been fully elucidated. The treatment of depression is currently carried out by a combination of pharmacotherapy (antidepressants) and psychological care. As antidepressants, tricyclic antidepressants (first generation) such as imipramine and amitriptyline have been first developed and used. However, since tricyclic antidepressants also block histamine H 1 receptor, acetylcholine muscarinic M 1 receptor, and adrenergic α 1 receptor at the postsynaptic side, side effects such as constipation and dysuria due to anticholinergic action There were many. In addition, there is a problem that it becomes fatal in large doses. Therefore, a tetracyclic antidepressant (second generation) with fewer side effects was developed, but the antidepressant effect was insufficient and anticholinergic action still remained. In the 1980s, a serotonin-selective reuptake inhibitor (SSRI) (third generation) with fewer side effects due to anticholinergic activity was developed, but SSRIs are used to induce nausea and vomiting by stimulating serotonin receptors in the brain and intestinal tract. , Caused new side effects such as withdrawal syndrome due to medication interruption. Later, in the 1990s, selective serotonin and noradrenaline reuptake inhibitors (SNRI) were developed and are widely used in clinical settings as drugs that do not have anticholinergic effects although gastrointestinal symptoms still remain.
うつ病の薬物療法では血中の薬剤濃度を有効濃度で一定に維持することが、副作用を低減して患者への治療効果を最大限もたらすために重要である。しかし、うつ病の治療では、服薬が長期にわたることやうつ症状が寛解・増悪を繰り返すため、うつ病の治療に一般的に使用されている経口剤を患者がコンプライアンス(医師から処方された薬剤を患者が指示通りに使用すること)よく服薬することが困難で、飲み忘れによる症状の悪化や過剰摂取による副作用を起こすことがある。特に、SSRIは1日1回の服用でよい製剤があるが、SNRIとして汎用されているミルナシプランは1日2〜3回服用する必要があり、さらにコンプライアンスよく服薬することが困難であった。
このような経口剤に対し、薬剤を経皮吸収させて投与する経皮剤は、薬剤が持続的に徐々に皮膚から吸収されるため、経口剤に比べて、有効血中濃度を持続的に維持することが可能である。その結果、特に1日2回以上服用する必要のあった薬剤を1日1回使用すればよい製剤にすることが可能となり、コンプライアンスが改善され得る。加えて、経皮剤の場合、最高血中濃度を低下させることができることから、副作用を回避し易くなる。また、経皮投与は、肝臓での薬剤の代謝による初回通過効果を回避できることから、経口投与より少量の薬剤で血中有効用量を維持できる可能性がある。さらに、薬剤を支持基材に塗布して貼付剤とした場合には、患者が薬剤を使用していることを患者のみならず、医師や看護師、家族などが容易に確認でき、副作用発生時にも貼付剤を剥すことによって以降の薬剤吸収を容易に中止できるメリットがある。従って、経皮剤は患者のコンプライアンス及びクオリティー・オブ・ライフ(QOL)の改善に非常に有用である。しかし、薬剤の種類によっては経皮吸収され難いものもあり、適切な経皮吸収製剤になり得る処方を見出すことが困難な場合も多い。
In drug therapy for depression, it is important to keep the drug concentration in the blood constant at an effective concentration in order to reduce side effects and maximize the therapeutic effect on the patient. However, in the treatment of depression, patients take compliance with oral drugs commonly used to treat depression (drugs prescribed by doctors) because the medication is long-lasting and the symptoms of depression remit and repeat. (Patients should use as directed) It is difficult to take medicine well, which may cause worsening of symptoms due to forgetting to take or side effects due to overdose. In particular, there are preparations that can be taken once a day for SSRI, but milnacipran, which is widely used as SNRI, needs to be taken 2 to 3 times a day, and it is difficult to take medicine with good compliance. .
In contrast to such oral agents, transdermal agents that are administered by transdermally absorbing the drug continuously absorb the drug continuously from the skin. It is possible to maintain. As a result, it is possible to obtain a preparation that can be used only once a day, especially for a drug that has been required to be taken twice or more a day, and compliance can be improved. In addition, in the case of a transdermal agent, since the maximum blood concentration can be reduced, side effects can be easily avoided. In addition, since transdermal administration can avoid the first-pass effect due to drug metabolism in the liver, there is a possibility that an effective blood dose can be maintained with a smaller amount of drug than oral administration. In addition, when a drug is applied to a support substrate to make a patch, not only the patient but also the doctor, nurse, family, etc. can easily confirm that the patient is using the drug. There is also a merit that subsequent drug absorption can be easily stopped by removing the patch. Thus, transdermal agents are very useful for improving patient compliance and quality of life (QOL). However, some drugs are difficult to be transdermally absorbed, and it is often difficult to find a formulation that can be an appropriate transdermally absorbable preparation.
ミルナシプラン〔(±)−シス−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミド〕は代表的なSNRIの一つで、脳内神経接合部においてセロトニンとノルアドレナリンの再取り込みを選択的に阻害することにより抗うつ作用を発揮することが知られている第4世代の抗うつ剤である。ミルナシプランは、三環系や四環系抗うつ剤のように神経伝達物質の受容体に結合しないことから抗コリン作用による副作用がなく、また、第3世代の抗うつ剤であるSSRIのように脳内や腸管でセロトニン受容体を刺激することによる吐気や嘔吐の副作用も少ない。効果的にも、セロトニンに加えてノルアドレナリンの再取り込みも阻害することから、セロトニンの再取り込みを選択的に阻害するSSRIより強い抗うつ作用を発揮し、特にSSRIではあまり効果のない意欲にも効果を有する。また、ミルナシプランは、抗うつ作用のみならず疼痛にも有効であり、神経因性疼痛、線維筋痛症、緊張型頭痛等に対する治療効果が報告されている。
なお、本発明において、便宜的にミルナシプランという場合でも、ミルナシプラン塩酸塩を指すことがある。
Milnacipran [(±) -cis-2- (aminomethyl) -N, N-diethyl-1-phenylcyclopropanecarboxamide] is one of the typical SNRIs, and it contains serotonin and noradrenaline at the intracerebral nerve junction. It is a fourth generation antidepressant that is known to exert an antidepressant action by selectively inhibiting reuptake. Milnacipran has no side effects due to anticholinergic action because it does not bind to neurotransmitter receptors like tricyclic and tetracyclic antidepressants, and the third-generation antidepressant SSRI Thus, there are few side effects of nausea and vomiting by stimulating serotonin receptors in the brain and intestinal tract. Effectively, it also inhibits the reuptake of noradrenaline in addition to serotonin, so it exerts stronger antidepressant action than SSRI that selectively inhibits the reuptake of serotonin, and it is also effective for willingness that is not particularly effective in SSRI. Have Moreover, milnacipran is effective not only for antidepressive action but also for pain, and has been reported to have therapeutic effects on neuropathic pain, fibromyalgia, tension-type headache and the like.
In the present invention, even when referred to as milnacipran for convenience, milnacipran hydrochloride may be referred to.
ミルナシプランの経皮吸収剤に関しては、特許文献1の報告がある。しかしながら、これはミルナシプランの経粘膜投与剤に関すものであり、具体的にはミルナシプランの鼻腔内投与用の製剤を開示するのみのものである。粘膜が比較的薬物を透過しやすい組織であるのに対し、皮膚上皮は強力な薬物の透過阻止機能を有する。従って、経皮投与用製剤では、薬剤を経皮吸収させるための、より強力な吸収促進作用を有する処方を見出すことが必要となる。
Regarding the transdermal absorption agent of milnacipran, there is a report of
本発明の目的は、有効成分であるミルナシプラン又はその薬学的に許容される塩の皮膚透過性を亢進させ、優れた薬理効果を発揮させるための、新規な経皮吸収用医薬組成物又はこれを含有する医薬を提供することにある。 An object of the present invention is to provide a novel pharmaceutical composition for percutaneous absorption for enhancing the skin permeability of milnacipran, which is an active ingredient, or a pharmaceutically acceptable salt thereof, and exhibiting an excellent pharmacological effect. It is in providing the pharmaceutical containing this.
本発明者らは、経皮吸収が困難なミルナシプラン又はその薬学的に許容される塩の経皮吸収性を向上させることにより、経皮吸収剤として優れた薬理効果を発揮させるための製剤化について鋭意検討を行った。その結果、ミルナシプランに吸収促進剤としてメントール、シネオール又はグリセリン脂肪酸エステル或いはそれらのうちの2種以上の組合せを配合することにより、ミルナシプランの経皮吸収性が顕著に亢進されることを見出し、本発明を完成した。 The present inventors provide a preparation for exerting an excellent pharmacological effect as a transdermal absorbent by improving transdermal absorbability of milnacipran or a pharmaceutically acceptable salt thereof which is difficult to transdermally absorb. We have made an intensive study on the development. As a result, by incorporating menthol, cineol or glycerin fatty acid ester or a combination of two or more thereof as an absorption promoter in milnacipran, the transdermal absorbability of milnacipran is significantly enhanced. The headline and the present invention were completed.
本発明医薬組成物は、ミルナシプラン又はその薬学的に許容される塩を経皮吸収により効果を発揮させることができるものである。従って、本発明医薬組成物を含有する医薬は、外用剤として有用である。特に貼付剤とした場合、コンプライアンスよく長期間使用可能な経皮吸収型の抗うつ薬又は鎮痛薬として非常に有用性が高いものである。 The pharmaceutical composition of the present invention can exert an effect by transdermal absorption of milnacipran or a pharmaceutically acceptable salt thereof. Therefore, the medicine containing the pharmaceutical composition of the present invention is useful as an external preparation. In particular, when used as a patch, it is extremely useful as a transdermal antidepressant or analgesic that can be used for a long time with good compliance.
本発明は、ミルナシプラン又はその薬学的に許容される塩を有効成分とし、吸収促進剤としてメントール、シネオール又はグリセリン脂肪酸エステル或いはそれらのうちの2種以上の組合せを含有する新規な経皮吸収用医薬組成物又はこれを含有する医薬に関する。 The present invention relates to a novel transdermal absorption containing milnacipran or a pharmaceutically acceptable salt thereof as an active ingredient and containing menthol, cineol or glycerin fatty acid ester or a combination of two or more thereof as an absorption enhancer. The present invention relates to a pharmaceutical composition for use or a medicine containing the same.
本発明医薬組成物又は医薬の有効成分は、ミルナシプラン又はその薬学的に許容される塩である。塩としては、薬学的に許容される酸付加塩であれば特に制限なく使用でき、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、フッ化水素酸塩、臭化水素酸塩等の無機酸塩や酢酸、酒石酸塩、乳酸塩、クエン酸塩、フマール酸塩、マレイン酸塩、メシル酸塩、コハク酸塩、メタンスルホン酸塩、ベンゼンスルホン酸塩、トルエンスルホン酸塩、ナフタレンスルホン酸塩、カンファースルホン酸塩等の有機酸塩を挙げることができる。好ましくは、市販され広く臨床的に用いられているミルナシプラン塩酸塩が挙げられる。また、本願においては、ミルナシプランの水和物や溶媒和物も本発明の有効成分であるミルナシプランに包含される。 The active ingredient of the pharmaceutical composition or medicament of the present invention is milnacipran or a pharmaceutically acceptable salt thereof. The salt can be used without particular limitation as long as it is a pharmaceutically acceptable acid addition salt. For example, inorganic salts such as hydrochloride, sulfate, nitrate, phosphate, hydrofluoride, hydrobromide, etc. Acid salt, acetic acid, tartrate, lactate, citrate, fumarate, maleate, mesylate, succinate, methanesulfonate, benzenesulfonate, toluenesulfonate, naphthalenesulfonate And organic acid salts such as camphor sulfonate. Preferably, milnacipran hydrochloride which is commercially available and widely used clinically is used. Moreover, in this application, the hydrate and solvate of a milnacipran are also included by the milnacipran which is an active ingredient of this invention.
本発明医薬組成物又は医薬において有効成分として含有されるミルナシプラン又はその薬学的に許容される塩の重量%は、0.1乃至10重量%、好ましくは0.2乃至5重量%とすることができる。 The weight% of milnacipran or a pharmaceutically acceptable salt thereof contained as an active ingredient in the pharmaceutical composition or medicine of the present invention is 0.1 to 10% by weight, preferably 0.2 to 5% by weight. be able to.
本発明医薬組成物で使用されるメントールは、その光学異性体であるl−メントール、dl−メントールを包含する。また、メントールを主成分とするハッカ油やハッカ水も本発明のメントールに包含される。使用されるメントールの量は、後述する剤形などによって異なるが、0.75乃至10重量%、好ましくは1乃至5重量%とすることができる。 Menthol used in the pharmaceutical composition of the present invention includes l-menthol and dl-menthol which are optical isomers thereof. Mentha oil and mint water mainly composed of menthol are also included in the menthol of the present invention. The amount of menthol used varies depending on the dosage form described below, but can be 0.75 to 10% by weight, preferably 1 to 5% by weight.
本発明医薬組成物で使用されるシネオールは、1,8−シネオール、1,8−エポキシ−p−メンタン又はユーカリプトールとも呼称され、シネオールを主成分とするユーカリ精油も本発明のシネオールに包含される。使用されるシネオールの量は、後述する剤形などによって異なるが、0.75乃至10重量%、好ましくは1乃至5重量%とすることができる。 Cineol used in the pharmaceutical composition of the present invention is also called 1,8-cineol, 1,8-epoxy-p-menthane or eucalyptol, and eucalyptus essential oil mainly composed of cineol is also included in the cineol of the present invention. Is done. The amount of cineole used varies depending on the dosage form described below, but can be 0.75 to 10% by weight, preferably 1 to 5% by weight.
本発明医薬組成物においては、上記メントール及びシネオールを組合せて使用することもでき、使用されるメントール及びシネオールの量は、後述する剤形などによって異なるが、合わせて0.75乃至10重量%、好ましくは1乃至5重量%とすることができる。 In the pharmaceutical composition of the present invention, the above menthol and cineole can be used in combination, and the amount of menthol and cineole used varies depending on the dosage form and the like described later, but 0.75 to 10% by weight in total, Preferably, it can be 1 to 5% by weight.
本発明医薬組成物で利用可能なグリセリン脂肪酸エステルとしては、脂肪酸の炭素数が5乃至25であるグリセリン脂肪酸エステル等が挙げられる。より具体的には、カプリル酸グリセリン、カプリン酸グリセリン、ラウリン酸グリセリン、パルミチン酸グリセリン、オレイン酸グリセリン、ステアリン酸グリセリン、ジカプリル酸グリセリン、トリカプリル酸グリセリン等が挙げられ、オレイン酸グリセリンが好ましい。グリセリン脂肪酸エステルの量は、後述する剤形などによって異なるが、2乃至30重量%、好ましくは5乃至20重量%とすることができる。 Examples of the glycerin fatty acid ester that can be used in the pharmaceutical composition of the present invention include glycerin fatty acid esters in which the fatty acid has 5 to 25 carbon atoms. More specifically, glyceryl caprylate, glyceryl caprate, glyceryl laurate, glyceryl palmitate, glyceryl oleate, glyceryl stearate, glyceryl dicaprylate, glyceryl tricaprylate, and the like, glycerin oleate is preferred. The amount of glycerin fatty acid ester varies depending on the dosage form described later, but can be 2 to 30% by weight, preferably 5 to 20% by weight.
本発明医薬組成物で使用される低級アルコールとしては、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、イソブタノール、2−ブタノール等の炭素数1乃至4のアルコールが挙げられる。使用される低級アルコールの量は、後述する剤形などによって異なるが、5乃至40重量%、好ましくは10乃至30重量%とすることができる。 Examples of the lower alcohol used in the pharmaceutical composition of the present invention include alcohols having 1 to 4 carbon atoms such as methanol, ethanol, propanol, isopropanol, butanol, isobutanol, and 2-butanol. The amount of the lower alcohol used varies depending on the dosage form described below, but can be 5 to 40% by weight, preferably 10 to 30% by weight.
本発明に係る経皮吸収用医薬組成物を含有する医薬の具体的な剤形としては特に制限はなく、例えば、液剤、クリーム剤、軟膏剤、ゲル剤、ローション剤、スプレー剤、貼付剤、テープ剤等の外用剤として製剤化できる。製剤化に際しては、各剤形に適した添加剤や基剤を適宜使用し、日本薬局方の製剤総則などに記載される通常の方法に従って製造することができる。処方にあたっては、ミルナシプランと共に他の医薬活性成分との配合剤としてもよい。 The specific dosage form of the pharmaceutical containing the pharmaceutical composition for transdermal absorption according to the present invention is not particularly limited, and examples thereof include solutions, creams, ointments, gels, lotions, sprays, patches, It can be formulated as an external preparation such as a tape. In the formulation, additives and bases suitable for each dosage form are used as appropriate, and they can be produced according to the usual methods described in the general rules of formulation of the Japanese Pharmacopoeia. In prescription, it is good also as a compounding agent with another pharmaceutical active ingredient with a milnacipran.
貼付剤の例としては、ヒドロゲルの形態の貼付剤(パップ剤)に製剤化することが挙げられる。ヒドロゲルの作成は、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシビニルポリマー等の粘稠剤を用い、水で膨潤させて基剤とするという、通常行われている方法を利用でき、主薬であるミルナシプランや上記吸収促進剤を製剤中に均一に混合させるために、イソプロパノールやエタノール等の適当な溶剤を用いて製剤化することができる。 As an example of the patch, it can be formulated into a patch (patch) in the form of a hydrogel. Hydrogels can be made using a commonly used method that uses a viscous agent such as hydroxyethylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose, carboxyvinyl polymer, etc., and swells with water as a base. In order to uniformly mix a certain milnacipran and the above absorption enhancer into the preparation, it can be formulated using an appropriate solvent such as isopropanol or ethanol.
本発明の経皮吸収用医薬組成物及び医薬は、うつ病又は慢性疼痛等で従来経口用ミルナシプランを投与されていた患者に用いることができる。鎮痛剤としては、慢性疼痛の他或いはこれに含まれるものとして、神経因性疼痛、頭痛、偏頭痛、緊張性頭痛、慢性骨盤痛、筋肉痛、関節痛、腰痛、線維筋痛症等の種々の痛みの治療に用いられ得る。また、他にも例えば、ミルナシプランの効果が報告されている腹圧性尿失禁等の疾患の治療にも用いられ得る。 The percutaneous absorption pharmaceutical composition and medicament of the present invention can be used for patients who have been conventionally administered oral milnacipran due to depression or chronic pain. As analgesics, in addition to or included in chronic pain, various types of pain such as neuropathic pain, headache, migraine, tension headache, chronic pelvic pain, muscle pain, joint pain, low back pain, fibromyalgia, etc. Can be used to treat pain. In addition, for example, it can be used for treatment of diseases such as stress urinary incontinence for which the effect of milnacipran has been reported.
以下に、実施例により本発明を具体的に説明するが、本発明はこれらに限定されるものではない。 EXAMPLES The present invention will be specifically described below with reference to examples, but the present invention is not limited to these examples.
実施例1.吸収促進剤の検討
(1)ミルナシプラン含有ヒドロゲルの作成
吸収促進剤が異なるミルナシプラン含有ヒドロゲルを以下の方法で作成した。なお、以下の本実施例においては、ミルナシプランの塩酸塩を用い、各組成成分の表示重量%は最終の総量(100重量%)に対するものである。
ヒドロキシエチルセルロース及びヒドロキシプロピルセルロースをそれぞれ1重量%となるように水に添加し、さらにミルナシプラン塩酸塩を2重量%となるように加えて一晩放置し基剤を膨潤させた。その後、吸収促進剤として2重量%のl−メントール、2重量%のd−リモネン、2重量%の1,8−シネオール、1重量%のl−メントールと1重量%の1,8−シネオールの組合せ、10重量%のN−メチルピロリドン又は10重量%のモノオレイン酸グリセリンを添加し、さらに20重量%のイソプロピルアルコールを添加して一晩静置して均一なヒドロゲルとした。なお、成分の配合量の増減に伴って使用する水の量を加減し、総量が100重量%となるようにした。さらに、対照として、吸収促進剤を添加しないミルナシプラン含有ヒドロゲルを同様に作成した。
Example 1. Examination of absorption accelerator (1) Preparation of milnacipran-containing hydrogels Milnacipran-containing hydrogels having different absorption accelerators were prepared by the following method. In the following examples, milnacipran hydrochloride is used, and the indicated weight% of each composition component is based on the final total amount (100 weight%).
Hydroxyethyl cellulose and hydroxypropyl cellulose were each added to water so as to be 1% by weight, and milnacipran hydrochloride was further added so as to be 2% by weight, and left overnight to swell the base. Thereafter, 2% by weight of 1-menthol, 2% by weight of d-limonene, 2% by weight of 1,8-cineole, 1% by weight of 1-menthol and 1% by weight of 1,8-cineole as absorption promoters. In combination, 10% by weight of N-methylpyrrolidone or 10% by weight of glyceryl monooleate was added, and further 20% by weight of isopropyl alcohol was added and allowed to stand overnight to form a uniform hydrogel. In addition, the amount of water to be used was adjusted in accordance with the increase / decrease in the amount of the components so that the total amount became 100% by weight. Furthermore, as a control, a milnacipran-containing hydrogel without the addition of an absorption enhancer was similarly prepared.
(2)インビトロ(in vitro)皮膚透過性試験
7週齢の雄性Hos;HR−1系ヘアレスマウス摘出皮膚(登録商標:ラボスキン、株式会社星野実験動物飼育所)を縦型拡散セル(有効拡散面積:2.01cm2、レシーバーセル体積16.0 mL)に皮膚角質層がドナー側、皮膚基底膜側がレシーバー側となるように装着した。上記のヒドロゲル1.0gをドナーセル内に適用し、レシーバーセルをpH 7.4のリン酸緩衝生理食塩液(PBS)で満たした。レシーバーセルから1時間ごとに0.2mLの溶液を分取して、ミルナシプランを高速液体クロマトグラフィー(HPLC)で定量した。また、分取後直ちに、0.2mLのPBSをレシーバーセルに補充した。HPLCは、移動相がリン酸二水素カリウム−水酸化カリウム緩衝液(pH5.5、イオン強度59mM):メタノール=60:40、カラムがYCM−Pack ODS−A(150mm×44.6mm)、波長が222nm、流速1.0mL/分、カラム温度が室温、注入量が90μLの条件にて実施した。なお、試験は各々3回行い、平均値±標準偏差を求めた。
(2) In vitro skin permeability test 7-week-old male Hos; HR-1 hairless mouse isolated skin (registered trademark: Labo Skin, Hoshino Laboratory Animal Breeding) vertical diffusion cell (effective diffusion area) : 2.01 cm 2 , receiver cell volume 16.0 mL) so that the skin stratum corneum is on the donor side and the skin basement membrane side is on the receiver side. 1.0 g of the above hydrogel was applied in the donor cell and the receiver cell was filled with phosphate buffered saline (PBS) at pH 7.4. A 0.2 mL solution was taken from the receiver cell every hour, and milnacipran was quantified by high performance liquid chromatography (HPLC). Immediately after the separation, 0.2 mL of PBS was replenished to the receiver cell. For HPLC, the mobile phase was potassium dihydrogen phosphate-potassium hydroxide buffer (pH 5.5, ionic strength 59 mM): methanol = 60: 40, the column was YCM-Pack ODS-A (150 mm × 44.6 mm), wavelength Was 222 nm, the flow rate was 1.0 mL / min, the column temperature was room temperature, and the injection volume was 90 μL. Each test was performed three times, and the average value ± standard deviation was obtained.
(3)ミルナシプラン皮膚透過性試験の結果
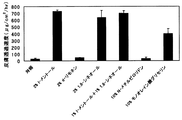
上記のインビトロ皮膚透過性試験により、各々の吸収促進剤を添加したミルナシプラン含有ヒドロゲルからのミルナシプランの単位面積当たりの累積皮膚透過量を経時的に測定した結果(累積透過量−時間曲線のグラフ)を図1に示した。また、上記測定値より求めたミルナシプランの皮膚透過速度(μg/cm2/h)〔皮膚透過速度は累積透過量−時間曲線のプロットを直線回帰して得られた回帰直線の傾きである〕を図2に示した。さらに、この回帰直線より求めた遅延時間〔皮膚透過速度が定常状態に達するまでの時間〕を図3に示した。
(3) Results of milnacipran skin permeability test As a result of the above in vitro skin permeability test, the cumulative skin permeation amount per unit area of milnacipran from the milnacipran-containing hydrogel added with each absorption enhancer was measured over time. FIG. 1 shows the results of the measurement (cumulative permeation amount-time curve graph). Further, the skin permeation rate (μg / cm 2 / h) of milnacipran determined from the above measured values [the skin permeation rate is the slope of the regression line obtained by linear regression of the cumulative permeation amount-time curve plot. 2 is shown in FIG. Furthermore, the delay time obtained from this regression line [time until the skin permeation speed reaches a steady state] is shown in FIG.
図1及び図2から明らかなように、吸収促進剤を添加しないミルナシプラン含有組成物(対照)では、ほとんど皮膚透過性が認められなかったが、l−メントール(2重量%)又は1,8−シネオール(2重量%)を含有する組成物では顕著にミルナシプランの皮膚透過性が促進され、モノオレイン酸グリセリン(10重量%)を含有する組成物も、ミルナシプランの皮膚透過性が促進されることが示された。しかし、d−リモネン又はN−メチルピロリドンを含有する組成物では皮膚透過性の促進は認められなかった。特に、d−リモネンはl−メントールや1,8−シネオールと同様にモノテルペン類に属し、化学構造が類似しているにもかかわらず、ミルナシプランの吸収促進作用が認められなかった。このことは、ミルナシプランという薬物とこれに適した吸収促進剤の組合せが経皮吸収促進に重要であることを示すものである。 As is clear from FIGS. 1 and 2, the milnacipran-containing composition to which no absorption enhancer was added (control) showed almost no skin permeability, but 1-menthol (2% by weight) or 1, The composition containing 8-cineole (2% by weight) significantly promotes the skin permeability of milnacipran, and the composition containing glyceryl monooleate (10% by weight) also has a skin permeability of milnacipran. Was shown to be promoted. However, the composition containing d-limonene or N-methylpyrrolidone did not promote skin permeability. In particular, d-limonene belongs to monoterpenes like l-menthol and 1,8-cineole, and although the chemical structure is similar, the absorption promoting action of milnacipran was not recognized. This indicates that a combination of a drug called milnacipran and an absorption enhancer suitable for it is important for promoting transdermal absorption.
また、l−メントール及び1,8−シネオールをそれぞれ1重量%ずつ含有する組成物では、l−メントール又は1,8−シネオールを各2重量%含有する組成物と同等にミルナシプランの皮膚透過性が促進された(図2)。なお、図3から明らかなように、l−メントール及び1,8−シネオールをそれぞれ1重量%ずつ配合した組成物では、l−メントール又は1,8−シネオールを2重量%含有する組成物よりもミルナシプランの皮膚透過遅延時間を短縮する効果が認められた。遅延時間が短い方が薬物の効果が現れるまでの時間が早くなることで、組成物として好ましい性質となり得る。 Further, in the composition containing 1% by weight of 1-menthol and 1,8-cineole, milnacipran penetrates the skin in the same manner as the composition containing 1% by weight of l-menthol or 2,8-cineole. Sex was promoted (FIG. 2). As is apparent from FIG. 3, the composition containing 1% by weight of l-menthol and 1,8-cineole each is more than the composition containing 2% by weight of l-menthol or 1,8-cineole. The effect of milnacipran on shortening the skin permeation delay time was recognized. The shorter the delay time, the faster the time until the drug effect appears, which can be a favorable property for the composition.
実施例2.ミルナシプランの用量検討
(1)ミルナシプラン含有ヒドロゲルの作成
表1に示す処方のヒドロゲルを次の方法で作成した。すなわち、ミルナシプラン、ヒドロキシプロピルセルロース及びヒドロキシエチルセルロースに水を添加して一晩放置し基剤を膨潤させた。その後、イソプロピルアルコール(及びl−メントール)を添加して一晩静置して均一なヒドロゲルとした。なお、ミルナシプランとその他の成分の配合量の増減に伴って使用する水の量を加減し、総量が100重量%となるようにした。
Example 2 Dose study of milnacipran (1) Preparation of milnacipran-containing hydrogel A hydrogel having the formulation shown in Table 1 was prepared by the following method. That is, water was added to milnacipran, hydroxypropylcellulose, and hydroxyethylcellulose and allowed to stand overnight to swell the base. Thereafter, isopropyl alcohol (and 1-menthol) was added and allowed to stand overnight to form a uniform hydrogel. In addition, the amount of water to be used was adjusted according to the increase / decrease in the blending amount of milnacipran and other components so that the total amount became 100% by weight.

(2)ミルナシプラン皮膚透過性試験の結果
全組成物の重量を100重量%としたときのミルナシプランの添加量を1、2又は3重量%に設定し、l−メントールを添加しない場合と2重量%のl−メントールを添加した場合のミルナシプラン皮膚透過性を比較した。実施例1のインビトロ皮膚透過性試験と同様に、単位面積当たりのミルナシプランの累積皮膚透過量を経時的に測定した累積透過量−時間曲線のグラフを図4に示した。また、上記測定値より求めたミルナシプランの皮膚透過速度(μg/cm2/h)を図5に示した。図4及び図5から明らかなように、l−メントールを含有しない処方ではミルナシプランの濃度(用量)にかかわらず、ミルナシプランがほとんど皮膚透過しなかった。他方、l−メントールを含有する組成物では、時間の経過と共にミルナシプランの皮膚透過量の上昇が認められた(図4)。また、図5に示したとおり、同じミルナシプラン含有量(1%、2%及び3%)の組成物で比較した場合、l−メントール添加によりミルナシプランの皮膚透過性が有意に促進された(n=3、***P<0.001)。さらに、l−メントールを2重量%含有する各組成物では、1重量%のミルナシプランと比較したとき、ミルナシプランの濃度(用量)に依存して線形的に促進された(n=3、**P<0.01、***P<0.001)。これにより、ミルナシプランの含量を調整することにより、皮膚吸収速度を調整することが可能であることが示された。
(2) Results of milnacipran skin permeability test When the amount of milnacipran added is set to 1, 2 or 3 wt% when the weight of all compositions is 100 wt%, and l-menthol is not added And milnacipran skin permeability when 2% by weight of 1-menthol was added. Similar to the in vitro skin permeability test of Example 1, FIG. 4 shows a graph of a cumulative permeation amount-time curve in which the cumulative skin permeation amount of milnacipran per unit area was measured over time. Further, the skin permeation rate (μg / cm 2 / h) of milnacipran determined from the above measured values is shown in FIG. As apparent from FIGS. 4 and 5, milnacipran hardly permeated through the skin in the formulation containing no l-menthol regardless of the concentration (dose) of milnacipran. On the other hand, in the composition containing l-menthol, an increase in the skin permeation amount of milnacipran was observed with time (FIG. 4). Further, as shown in FIG. 5, when compared with compositions having the same milnacipran content (1%, 2% and 3%), the skin permeability of milnacipran was significantly promoted by the addition of l-menthol. (N = 3, *** P <0.001). Furthermore, each composition containing 2% by weight l-menthol was promoted linearly (n = 3) depending on the concentration (dose) of milnacipran when compared to 1% by weight milnacipran. , ** P <0.01, *** P <0.001). Thus, it was shown that the skin absorption rate can be adjusted by adjusting the content of milnacipran.
実施例3.インビボ(in vivo)強制水泳試験
ミルナシプランが皮膚を経由して生体内に送達されて薬理効果を発揮することを確認するため、下記の強制水泳試験を行った。なお、試験は各々5回行い、平均値±標準誤差を求めた。
Example 3 In vivo forced swimming test In order to confirm that milnacipran is delivered into the living body through the skin and exerts a pharmacological effect, the following forced swimming test was performed. Each test was performed 5 times, and the average value ± standard error was obtained.
直径9.5cmのガラスシリンダーを適量の水で満たし、25℃水浴下で保温した。その後、ガラスシリンダー内で7週齢の雄性C57BL/6Jマウス(東京実験動物株式会社)を10分間強制水泳によるストレス負荷を行った。ストレス負荷12時間後に実施例1のミルナシプラン含有ヒドロゲルをマウス背部徐毛部に12時間適用した(有効拡散面積;0.16cm2)。12時間後、マウス背部よりヒドロゲルを除去し、再びガラスシリンダー内にて6分間の強制水泳を行った。このとき、終わり5分間のマウスの無動時間を測定し、無動時間の低下をミルナシプランの抗うつ作用として評価した。また、陽性対照として、ミルナシプラン60mg/kgを腹腔内(i.p.)投与したマウスにて同様に試験した。 A glass cylinder having a diameter of 9.5 cm was filled with an appropriate amount of water and kept warm in a 25 ° C. water bath. After that, a 7-week-old male C57BL / 6J mouse (Tokyo Experimental Animal Co., Ltd.) was subjected to stress loading by forced swimming for 10 minutes in a glass cylinder. After 12 hours of stress loading, the milnacipran-containing hydrogel of Example 1 was applied to the back hair of the mouse for 12 hours (effective diffusion area; 0.16 cm 2 ). After 12 hours, the hydrogel was removed from the back of the mouse, and forced swimming was performed again in a glass cylinder for 6 minutes. At this time, the immobility time of the mouse for the last 5 minutes was measured, and the decrease in the immobility time was evaluated as an antidepressant action of milnacipran. Further, as a positive control, the same test was carried out in mice administered milnacipran 60 mg / kg intraperitoneally (ip).
その結果を図6に示した。発症対照に対してミルナシプランを腹腔内投与した陽性対照の無動時間は有意に低下した(n=5、**P<0.01)。l−メントールを含まないヒドロゲルを適用した場合、ミルナシプランによる有意な効果は認められなかった。これに対して2重量%l−メントールを添加した各組成物では、2又は3重量%のミルナシプランを添加した本発明組成物の場合、l−メントールを添加しないものに比べて、ミルナシプランによる用量依存的な無動時間の低下が有意に認められ、陽性対照と同等の抗うつ作用が認められた(n=5、**P<0.01)。また、2重量%l−メントールを添加した組成物においては、ミルナシプランを添加しない組成物と比較して、2又は3重量%のミルナシプランを添加した本発明医薬組成物では、それぞれ有意な無動時間の低下が認められ(n=5、*P<0.05)、この無動時間の低下はミルナシプランの皮膚透過による薬効発現であることが示された。 The results are shown in FIG. The immobility time of the positive control in which milnacipran was intraperitoneally administered relative to the onset control was significantly reduced (n = 5, ** P <0.01). When a hydrogel containing no l-menthol was applied, no significant effect with milnacipran was observed. On the other hand, in each composition to which 2% by weight of 1-menthol was added, in the case of the composition of the present invention to which 2 or 3% by weight of milnacipran was added, mil The dose-dependent decrease in immobility time by the plan was significantly observed, and an antidepressant effect equivalent to that of the positive control was observed (n = 5, ** P <0.01). Moreover, in the composition to which 2% by weight of 1-menthol was added, the pharmaceutical composition of the present invention to which 2 or 3% by weight of milnacipran was added was significantly different from the composition to which milnacipran was not added. A decrease in the immobility time was observed (n = 5, * P <0.05), and this decrease in the immobility time was shown to be a manifestation of the medicinal effect due to skin penetration of milnacipran.
上記薬理試験の結果から明らかなように、適切な吸収促進剤を含有させることにより、ミルナシプランの経皮吸収性を促進し、顕著な薬理効果を発現させることが可能となった。吸収促進剤としては、メントール、シネオール又はグリセリン脂肪酸エステル或いはそれらのうちの2種以上の組合せが可能であり、メントール及びシネオールの組合せが好ましい。また、本発明医薬組成物においては、イソプロパノール又はエタノール等の低級アルコールをさらに添加することにより、より経皮吸収の優れた医薬製剤とすることが可能である。本発明医薬組成物含有する医薬は、うつ病治療薬として広く臨床で使用され、また鎮痛剤としての有用性も認められているミルナシプランを有効成分とするものであり、経皮吸収性に優れ、コンプライアンスよく長期間使用可能な経皮吸収型の抗うつ剤及び鎮痛剤として非常に有用性が高いものである。 As is apparent from the results of the above pharmacological test, by incorporating an appropriate absorption enhancer, it became possible to promote transdermal absorbability of milnacipran and develop a remarkable pharmacological effect. As the absorption enhancer, menthol, cineol, glycerin fatty acid ester or a combination of two or more thereof can be used, and a combination of menthol and cineol is preferable. In addition, in the pharmaceutical composition of the present invention, a pharmaceutical preparation having better percutaneous absorption can be obtained by further adding a lower alcohol such as isopropanol or ethanol. The pharmaceutical composition containing the pharmaceutical composition of the present invention comprises milnacipran, which is widely used clinically as a therapeutic drug for depression and has been recognized as being useful as an analgesic, and has a transdermal absorbability. It is highly useful as a transdermal absorption type antidepressant and analgesic that is excellent and can be used for a long time with good compliance.
Claims (20)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012045291A JP2012193176A (en) | 2011-03-03 | 2012-03-01 | Milnacipran-containing pharmaceutical composition for percutaneous absorption |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011046445 | 2011-03-03 | ||
| JP2011046445 | 2011-03-03 | ||
| JP2012045291A JP2012193176A (en) | 2011-03-03 | 2012-03-01 | Milnacipran-containing pharmaceutical composition for percutaneous absorption |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012193176A true JP2012193176A (en) | 2012-10-11 |
Family
ID=47085438
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012045291A Pending JP2012193176A (en) | 2011-03-03 | 2012-03-01 | Milnacipran-containing pharmaceutical composition for percutaneous absorption |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2012193176A (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5376481B1 (en) * | 2013-03-04 | 2013-12-25 | 日本臓器製薬株式会社 | Pharmaceutical composition for transdermal absorption |
| JP2014208623A (en) * | 2013-03-29 | 2014-11-06 | 小林製薬株式会社 | External pharmaceutical composition |
| WO2014187943A1 (en) * | 2013-05-24 | 2014-11-27 | Nestec S.A. | Treatment or prevention of depression using menthol and/or icilin |
| US20150110903A1 (en) * | 2012-05-01 | 2015-04-23 | Amberwing Solutions Inc. | Method and product for headache relief |
-
2012
- 2012-03-01 JP JP2012045291A patent/JP2012193176A/en active Pending
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150110903A1 (en) * | 2012-05-01 | 2015-04-23 | Amberwing Solutions Inc. | Method and product for headache relief |
| JP5376481B1 (en) * | 2013-03-04 | 2013-12-25 | 日本臓器製薬株式会社 | Pharmaceutical composition for transdermal absorption |
| WO2014136717A1 (en) * | 2013-03-04 | 2014-09-12 | 日本臓器製薬株式会社 | Transdermally absorbable pharmaceutical composition |
| JP2014208623A (en) * | 2013-03-29 | 2014-11-06 | 小林製薬株式会社 | External pharmaceutical composition |
| JP2019034978A (en) * | 2013-03-29 | 2019-03-07 | 小林製薬株式会社 | Pharmaceutical composition for external application |
| WO2014187943A1 (en) * | 2013-05-24 | 2014-11-27 | Nestec S.A. | Treatment or prevention of depression using menthol and/or icilin |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5021621B2 (en) | Beta-2 adrenergic receptor agonist for treating cutaneous connective tissue disease | |
| JPWO2007086493A1 (en) | Nasal medication | |
| NO20110342L (en) | Fentanyl preparation for nasal administration | |
| CN117531017A (en) | Esketamine for treating depression | |
| PT2680833E (en) | Parenteral administration of tapentadol | |
| JP2021080276A (en) | Prevention or treatment of sleep disorders using dexmedetomidine formulation | |
| KR20220044385A (en) | Methods for treating pruritus | |
| JP2012193176A (en) | Milnacipran-containing pharmaceutical composition for percutaneous absorption | |
| JP2020505322A (en) | Topical detomidine preparation | |
| JP2005531612A (en) | Nefopam formulation and its use in the treatment of pain | |
| WO2017120468A1 (en) | Therapeutic use of nalbuphine without aquaretic effects | |
| KR20090120423A (en) | A pharmaceutical composition for the treatment of premature ejaculation | |
| BR112020003025A2 (en) | methods of treating osteoarthritis with cannabidiol transdermal gel | |
| EP3432871B1 (en) | Treatment of uremic pruritus | |
| Michel | A benefit-risk assessment of extended-release oxybutynin | |
| US11147799B2 (en) | Topical pharmaceutical composition containing phenytoin and a (co-) analgesic for the treatment of chronic pain | |
| US11602521B2 (en) | N,N-dimethyltryptamine compositions and methods | |
| ITMI20100260A1 (en) | PHARMACEUTICAL COMPOSITION CONTAINING A DRUG TO REDUCE THE SIDE EFFECTS OF ANTIPSYCHOTIC DRUGS | |
| JP5376481B1 (en) | Pharmaceutical composition for transdermal absorption | |
| US20100190860A1 (en) | Methods for selectively enhancing antinociceptive potency of local anesthetics | |
| US10004734B2 (en) | Compositions and methods for treating rebound erythema associated with topical alpha-adrenergic agonists | |
| WO2006038596A1 (en) | Pharmaceutical composition for xerophthalmia and xerostomia treatment | |
| CA3074302A1 (en) | Topical compositions and methods for treatment | |
| ES2260160T3 (en) | TOPICAL COMPOSITION AND PROCEDURE TO TREAT URINARY INCONTINENCE OF EFFORT. | |
| US20160287711A1 (en) | Formulation Solution Adapted to Prolong Plasma Times of Drugs in Mammals Including Humans |