JP2012184408A - Modified diene rubber, method for production thereof, and rubber composition using the rubber - Google Patents
Modified diene rubber, method for production thereof, and rubber composition using the rubber Download PDFInfo
- Publication number
- JP2012184408A JP2012184408A JP2012027339A JP2012027339A JP2012184408A JP 2012184408 A JP2012184408 A JP 2012184408A JP 2012027339 A JP2012027339 A JP 2012027339A JP 2012027339 A JP2012027339 A JP 2012027339A JP 2012184408 A JP2012184408 A JP 2012184408A
- Authority
- JP
- Japan
- Prior art keywords
- rubber
- acid
- diene rubber
- epoxidized
- modified diene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Compositions Of Macromolecular Compounds (AREA)
- Epoxy Resins (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
Description
本発明は、変性ジエン系ゴム及びその製造方法並びにそれを用いたゴム組成物に関する。 The present invention relates to a modified diene rubber, a method for producing the same, and a rubber composition using the same.
近年、環境問題への意識が高まり、自動車の燃費向上を目的とした低燃費タイヤ用ゴム材料の開発が盛んに行われている。このような低燃費を目的としたタイヤ用ゴム材料は、機械的性質、低エネルギーロス性などに優れていることが望まれており、その実現のため、フィラーであるシリカの分散性を向上させ、ゴム分子間の摩擦やシリカとゴム分子間の摩擦を低減させて発熱を減らす技術が知られている。そして、シリカの分散性を向上させるために、その材料となるゴムをエポキシ化変性させる技術が多数開発されている(特許文献1)。 In recent years, awareness of environmental issues has increased, and rubber materials for fuel-efficient tires have been actively developed for the purpose of improving the fuel efficiency of automobiles. Such rubber materials for tires intended for low fuel consumption are desired to be excellent in mechanical properties, low energy loss, etc., and to achieve this, the dispersibility of silica as a filler is improved. A technique for reducing heat generation by reducing friction between rubber molecules and friction between silica and rubber molecules is known. And in order to improve the dispersibility of a silica, many techniques which epoxidize-modify the rubber | gum used as the material are developed (patent document 1).
しかしながら、特許文献1に記載されたタイヤトレッドゴム組成物は、機械的性質、低エネルギーロス性などが必ずしも十分でない。そこで、本発明は、特に低エネルギーロス性及び反発弾性が優れた変性ジエン系ゴム及びその製造方法並びにそれを用いたゴム組成物を提供することを目的とする。 However, the tire tread rubber composition described in Patent Document 1 does not necessarily have sufficient mechanical properties, low energy loss, and the like. Accordingly, an object of the present invention is to provide a modified diene rubber particularly excellent in low energy loss and impact resilience, a method for producing the same, and a rubber composition using the same.
本発明者らは、以上の目的を達成するために、鋭意検討した結果、エポキシ化率を限定したエポキシ化ジエン系ゴムとアルコキシシラン及びカルボン酸とを反応させることによって、アルコキシシランが多数付加した変性ジエン系ゴムを製造でき、それを用いたゴム組成物は、特に低エネルギーロス性及び反発弾性に優れていることを見出し、本発明に至った。すなわち本発明は、エポキシ化率が0.1%以上15%未満のエポキシ化ジエン系ゴムと、エポキシ基と反応する官能基を有するアルコキシシラン及びカルボン酸とを反応させた変性ジエン系ゴムに関する。 As a result of intensive studies to achieve the above object, the present inventors have added a large number of alkoxysilanes by reacting an epoxidized diene rubber with a limited epoxidation rate with alkoxysilanes and carboxylic acids. A modified diene rubber can be produced, and a rubber composition using the rubber has been found to be particularly excellent in low energy loss and rebound resilience, leading to the present invention. That is, the present invention relates to a modified diene rubber obtained by reacting an epoxidized diene rubber having an epoxidation rate of 0.1% or more and less than 15%, an alkoxysilane having a functional group that reacts with an epoxy group, and a carboxylic acid.
また、本発明は、エポキシ化率が0.1%以上15%未満のエポキシ化ジエン系ゴムと、エポキシ基と反応する官能基を有するアルコキシシラン及びカルボン酸とを機械的混練により反応させることを特徴とする変性ジエン系ゴムの製造方法に関する。 In the present invention, an epoxidized diene rubber having an epoxidation rate of 0.1% or more and less than 15% is reacted with an alkoxysilane having a functional group that reacts with an epoxy group and a carboxylic acid by mechanical kneading. The present invention relates to a method for producing a modified diene rubber.
さらに、本発明は、上記変性ジエン系ゴムとシリカとを含有することを特徴とする変性ジエン系ゴム組成物に関する。 Furthermore, the present invention relates to a modified diene rubber composition comprising the modified diene rubber and silica.
以上のように、本発明によれば、特に低エネルギーロス性及び反発弾性に優れた変性ジエン系ゴム及びその製造方法並びにそれを用いたゴム組成物を提供することができる。 As described above, according to the present invention, it is possible to provide a modified diene rubber particularly excellent in low energy loss and impact resilience, a method for producing the same, and a rubber composition using the same.
本発明に係る変性ジエン系ゴムにおいて、原料となるジエン系ゴムは、特に制限はなく、公知のものを使用することができる。例えば、天然ゴム、ブタジエンゴム(BR)、シンジオタクチック−1,2−ポリブタジエン含有のブタジエンゴム(VCR)、イソプレンゴム、ブチルゴム、クロロプレンゴムなどである。この中でも、特にブタジエンゴムが好ましい。上記ジエン系ゴムは、1種類単独、または2種類以上を組み合わせて使用してもよいが、2種類以上の組合せでは、そのうち1種類のみに変性が偏る可能性がある点から1種類の単独重合体が好ましい。 In the modified diene rubber according to the present invention, the diene rubber used as a raw material is not particularly limited, and a known rubber can be used. For example, natural rubber, butadiene rubber (BR), syndiotactic-1,2-polybutadiene-containing butadiene rubber (VCR), isoprene rubber, butyl rubber, chloroprene rubber and the like. Among these, butadiene rubber is particularly preferable. The diene rubber may be used alone or in combination of two or more. However, in the combination of two or more, one type of single weight may be used because the modification may be biased to only one of them. Coalescence is preferred.
上記ジエン系ゴムは、GPC(ゲルパーミエーションクロマトグラフ法)による重量平均分子量(Mw)が5万〜200万、さらに20万〜100万、特には40万〜80万であることが好ましい。重量平均分子量(Mw)が5万未満になると、機械強度が低くなり、200万を超えると、加工性が低下するので好ましくない。 The diene rubber preferably has a weight average molecular weight (Mw) by GPC (gel permeation chromatography) of 50,000 to 2,000,000, further 200,000 to 1,000,000, particularly 400,000 to 800,000. When the weight average molecular weight (Mw) is less than 50,000, the mechanical strength is lowered, and when it exceeds 2 million, the workability is deteriorated.
また、上記ジエン系ゴムは、エポキシ化率が0.1%以上15%未満でエポキシ化されており、より好ましくは0.2%〜5%、特に好ましくは0.5%〜2.5%である。 The diene rubber is epoxidized at an epoxidation rate of 0.1% or more and less than 15%, more preferably 0.2% to 5%, particularly preferably 0.5% to 2.5%. It is.
上記ジエン系ゴムのエポキシ化の方法としては、特に制限はなく、例えば、モノ過フタル酸を用いる方法(特開昭51−060292号公報)、有機過酸またはカルボン酸その無水物とともに過酸化水素水を用いる方法(特開平05−214014号公報)、タングステン酸系触媒とリン酸化合物および相間移動触媒と過酸化水素水を用いる方法(特許第3942927号公報)等に記載の公知の方法が用いられる。 The method for epoxidizing the diene rubber is not particularly limited, and examples thereof include a method using monoperphthalic acid (Japanese Patent Laid-Open No. 51-060292), hydrogen peroxide together with organic peracid or carboxylic acid anhydride. Known methods described in, for example, a method using water (Japanese Patent Laid-Open No. 05-214141), a method using a tungstic acid catalyst and a phosphoric acid compound, a phase transfer catalyst and hydrogen peroxide (Japanese Patent No. 3944927) are used. It is done.
また、上記エポキシ化の際に、エポキシ化率を調整する方法としては、例えば過酸化水素水の添加量を調整する、反応温度を調整する、反応時間を調整する等の通常の方法が挙げられる。エポキシ化率が0.1%未満では、エポキシ基と反応する官能基を有するアルコキシシランと反応するエポキシ基の数が不足する点で好ましくない。また、エポキシ化率が15%以上では、不飽和結合が少なくなってゴム弾性が低下する点や、硫黄加硫を用いた場合に加硫戻りを生じやすく、機械的性質が低下する点で好ましくない。 In addition, examples of the method for adjusting the epoxidation rate during the epoxidation include ordinary methods such as adjusting the amount of hydrogen peroxide added, adjusting the reaction temperature, and adjusting the reaction time. . An epoxidation rate of less than 0.1% is not preferable in that the number of epoxy groups that react with an alkoxysilane having a functional group that reacts with an epoxy group is insufficient. In addition, when the epoxidation rate is 15% or more, it is preferable from the viewpoint that unsaturated bonds are reduced and rubber elasticity is lowered, and that when sulfur vulcanization is used, reversion is likely to occur and mechanical properties are lowered. Absent.
本発明に係る変性ジエン系ゴムにおいて、エポキシ基と反応する官能基を有するアルコキシシランとしては、特に制限はなく、エポキシ化ジエン系ゴム中のエポキシ基に付加反応の可能なものであれば良い。 In the modified diene rubber according to the present invention, the alkoxysilane having a functional group that reacts with an epoxy group is not particularly limited as long as it can undergo an addition reaction with the epoxy group in the epoxidized diene rubber.
上記エポキシ基と反応する官能基としては、例えば、アミノ基、メルカプト基、ビニル基、ウレイド基、アクリロキシ基、イソシアネート基、スチリル基、メタクリロキシ基、スルフィド基、ニトロ基、ハロゲン基、エポキシ基またはグリシジル基などが挙げられ、さらに、チオカルバモイルテトラスルフィド構造、ベンゾチアゾールテトラスルフィド構造、メタクリレートモノスルフィド構造を有するものなどが挙げられる。 Examples of the functional group that reacts with the epoxy group include amino group, mercapto group, vinyl group, ureido group, acryloxy group, isocyanate group, styryl group, methacryloxy group, sulfide group, nitro group, halogen group, epoxy group or glycidyl. A group having a thiocarbamoyl tetrasulfide structure, a benzothiazole tetrasulfide structure, or a methacrylate monosulfide structure.
エポキシ基と反応する官能基を有するアルコキシシランとしては、例えば、3−アミノプロピルジメチルメトキシシラン、3−アミノプロピルメチルジメトキシシラン、3−アミノプロピルエチルジメトキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルジメチルエトキシシラン、3−アミノプロピルメチルジエトキシシラン、3−アミノプロピルトリエトキシシラン、3−アミノプロピルジメチルブトキシシラン、3−アミノプロピルメチルジブトキシシラン、3−(2−アミノエチルアミノプロピル)ジメトキシメチルシラン、3−(2−アミノエチルアミノプロピル)ジメトキシエチルシラン、3−(2−アミノエチルアミノプロピル)ジメトキシプロピルシラン、3−(2−アミノエチルアミノプロピル)ジメトキシブチルシラン、3−(2−アミノエチルアミノプロピル)ジエトキシメチルシラン、3−(2−アミノエチルアミノプロピル)トリメトキシシラン、3−(2−アミノエチルアミノプロピル)トリエトキシシラン、3−(2−アミノエチルアミノプロピル)トリブトキシシランなどのアミノ基を有するものが挙げられる。これらの化合物の中では、特に変性効果の点から、3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、3−(2−アミノエチルアミノプロピル)ジメトキシメチルシラン、3−(2−アミノエチルアミノプロピル)トリメトキシシランが好適に使用される。 Examples of the alkoxysilane having a functional group that reacts with an epoxy group include 3-aminopropyldimethylmethoxysilane, 3-aminopropylmethyldimethoxysilane, 3-aminopropylethyldimethoxysilane, 3-aminopropyltrimethoxysilane, 3- Aminopropyldimethylethoxysilane, 3-aminopropylmethyldiethoxysilane, 3-aminopropyltriethoxysilane, 3-aminopropyldimethylbutoxysilane, 3-aminopropylmethyldibutoxysilane, 3- (2-aminoethylaminopropyl) Dimethoxymethylsilane, 3- (2-aminoethylaminopropyl) dimethoxyethylsilane, 3- (2-aminoethylaminopropyl) dimethoxypropylsilane, 3- (2-aminoethylaminopropyl) di Toxibutylsilane, 3- (2-aminoethylaminopropyl) diethoxymethylsilane, 3- (2-aminoethylaminopropyl) trimethoxysilane, 3- (2-aminoethylaminopropyl) triethoxysilane, 3- ( Those having an amino group, such as 2-aminoethylaminopropyl) tributoxysilane. Among these compounds, 3-aminopropyltrimethoxysilane, 3-aminopropyltriethoxysilane, 3- (2-aminoethylaminopropyl) dimethoxymethylsilane, 3- (2-amino) are particularly preferred from the viewpoint of modification effects. Ethylaminopropyl) trimethoxysilane is preferably used.
さらに、エポキシ基と反応する官能基を有するアルコキシシランとして、例えば、3−メルカプトプロピルトリメトキシシラン、3−メルカプトプロピルトリエトキシシラン、2−メルカプトエチルトリメトキシシラン、2−メルカプトエチルトリエトキシシランなどのメルカプト基を有するもの、ビニルトリメトキシシラン、ビニルトリエトキシシランなどのビニル基を有するもの、3−ウレイドプロピルトリエトキシシランなどのウレイド基を有するもの、3−アクリロキシプロピルトリメトキシシランなどのアクリロキシ基を有するもの、3−イソシアネートプロピル、トリエトキシシランなどのイソシアネート基を有するもの、p−スチリルトリメトキシシランなどのスチリル基を有するもの、3−メタクリロキシプロピルメチルジメトキシシラン、3−メタクリロキシプロピルトリメトキシシラン、3−メタクリロキシプロピルメチルジエトキシシラン、3−メタクリロキシプロピルトリエトキシシランなどのメタクリロキシ基を有するもの、ビス(3−トリエトキシシリルプロピル)テトラスルフィド、ビス(2−トリエトキシシリルエチル)テトラスルフィド、ビス(3−トリメトキシシリルプロピル)テトラスルフィド、ビス(2−トリメトキシシリルエチル)テトラスルフィドなどのスルフィド基を有するもの、3−ニトロプロピルトリメトキシシラン、3−ニトロプロピルトリエトキシシランなどのニトロ基を有するもの、3−クロロプロピルトリメトキシシラン、3−クロロプロピルトリエトキシシラン、2−クロロエチルトリメトキシシラン、2−クロロエチルトリエトキシシランなどのハロゲン基を有するもの、3−トリメトキシシリルプロピル−N,N−ジメチルチオカルバモイルテトラスルフィド、3−トリエトキシシリルプロピル−N,N−ジメチルチオカルバモイルテトラスルフィド、2−トリエトキシシリルエチル−N,N−ジメチルチオカルバモイルテトラスルフィドなどのチオカルバモイルテトラスルフィド構造を有するもの、3−トリメトキシシリルプロピルベンゾチアゾールテトラスルフィド、3−トリエトキシシリルプロピルベンゾチアゾールテトラスルフィドなどのベンゾチアゾールテトラスルフィド構造を有するもの、3−トリエトキシシリルプロピルメタクリレートモノスルフィド、3−トリメトキシシリルプロピルメタクリレートモノスルフィドなどのメタクリレートモノスルフィド構造を有するもの、2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、3−グリシジルオキシプロピルトリメトキシシラン、3−グリシドキシプロピルトリエトキシシラン、ジエトキシ(3−グリシジルオキシプロピル)メチルシラン、3−グリシジルオキシプロピル(ジメトキシ)メチルシランなどのエポキシ基またはグリシジル基を有するもの等が挙げられる。これらの化合物の中では、特に反応後の性能が優れる点から、3−グリシジルオキシプロピルトリメトキシシラン等のグリシジル基またはエポキシ基を持つアルコキシシランが好適に使用される。 Furthermore, as an alkoxysilane having a functional group that reacts with an epoxy group, for example, 3-mercaptopropyltrimethoxysilane, 3-mercaptopropyltriethoxysilane, 2-mercaptoethyltrimethoxysilane, 2-mercaptoethyltriethoxysilane, etc. Those having a mercapto group, those having a vinyl group such as vinyltrimethoxysilane and vinyltriethoxysilane, those having a ureido group such as 3-ureidopropyltriethoxysilane, and acryloxy groups such as 3-acryloxypropyltrimethoxysilane Those having an isocyanate group such as 3-isocyanatopropyl and triethoxysilane, those having a styryl group such as p-styryltrimethoxysilane, and 3-methacryloxypropylmethyldi Those having a methacryloxy group such as toxisilane, 3-methacryloxypropyltrimethoxysilane, 3-methacryloxypropylmethyldiethoxysilane, 3-methacryloxypropyltriethoxysilane, bis (3-triethoxysilylpropyl) tetrasulfide, bis Those having a sulfide group such as (2-triethoxysilylethyl) tetrasulfide, bis (3-trimethoxysilylpropyl) tetrasulfide, bis (2-trimethoxysilylethyl) tetrasulfide, 3-nitropropyltrimethoxysilane, Those having a nitro group such as 3-nitropropyltriethoxysilane, 3-chloropropyltrimethoxysilane, 3-chloropropyltriethoxysilane, 2-chloroethyltrimethoxysilane, 2-chloro One having a halogen group such as tilt triethoxysilane, 3-trimethoxysilylpropyl-N, N-dimethylthiocarbamoyl tetrasulfide, 3-triethoxysilylpropyl-N, N-dimethylthiocarbamoyl tetrasulfide, 2-triethoxy Those having a thiocarbamoyl tetrasulfide structure such as silylethyl-N, N-dimethylthiocarbamoyl tetrasulfide, benzothiazole tetrasulfides such as 3-trimethoxysilylpropylbenzothiazole tetrasulfide, 3-triethoxysilylpropylbenzothiazole tetrasulfide Methacrylates such as those having a structure, 3-triethoxysilylpropyl methacrylate monosulfide, 3-trimethoxysilylpropyl methacrylate monosulfide 2- (3,4-epoxycyclohexyl) ethyltrimethoxysilane, 3-glycidyloxypropyltrimethoxysilane, 3-glycidoxypropyltriethoxysilane, diethoxy (3-glycidyloxypropyl) The thing etc. which have epoxy groups or glycidyl groups, such as methylsilane and 3-glycidyloxypropyl (dimethoxy) methylsilane, are mentioned. Among these compounds, alkoxysilanes having a glycidyl group or an epoxy group such as 3-glycidyloxypropyltrimethoxysilane are preferably used because the performance after reaction is particularly excellent.
さらにまた、エポキシ基と反応する官能基を有するアルコキシシランとして、例えば、テトラメトキシシラン、メチルトリメトキシシラン、ジメチルジメトキシシラン、トリメチルメトキシシラン、エチルトリメトキシシラン、ジエチルジメトキシシラン、トリエチルメトキシシラン、テトラエトキシシラン、メチルトリエトキシシラン、ジメチルジエトキシシラン、トリメチルエトキシシラン、エチルトリエトキシシラン、ジエチルジエトキシシラン、トリエチルエトキシシラン、テトラブトキシシラン、メチルトリブトキシシラン、ジメチルジブトキシシラン、トリメチルブトキシシラン、エチルトリブトキシシラン、ジエチルジブトキシシラン、トリエチルブトキシシラン、トリメトキシシラン、メチルジメトキシシラン、ジメチルメトキシシラン、エチルジメトキシシラン、ジエチルメトキシシラン、トリエトキシシラン、メチルジエトキシシラン、ジメチルエトキシシラン、エチルジエトキシシラン、ジエチルエトキシシラン、トリブトキシシラン、メチルジブトキシシラン、ジメチルブトキシシラン、エチルジブトキシシラン、ジエチルブトキシシラン、フェニルトリメトキシシラン、ジフェニルジメトキシシラン、フェニルトリエトキシシラン、ジフェニルジエトキシシラン、ヘキシルトリメトキシシラン、デシルトリメトキシシラン、テトラプロポキシシラン、メチルトリプロポキシシラン、ジメチルジプロポキシシラン、エチルトリプロポキシシラン、ジエチルジプロポキシシランなどが挙げられる。これらの化合物の中では、特に添加効果とコストの両立の点から、テトラメトキシシラン、テトラエトキシシランが好適に使用される。上記エポキシ基と反応する官能基を有するアルコキシシランは、1種類単独、または2種類以上を組み合わせて使用してもよい。 Furthermore, examples of the alkoxysilane having a functional group that reacts with an epoxy group include tetramethoxysilane, methyltrimethoxysilane, dimethyldimethoxysilane, trimethylmethoxysilane, ethyltrimethoxysilane, diethyldimethoxysilane, triethylmethoxysilane, and tetraethoxy. Silane, methyltriethoxysilane, dimethyldiethoxysilane, trimethylethoxysilane, ethyltriethoxysilane, diethyldiethoxysilane, triethylethoxysilane, tetrabutoxysilane, methyltributoxysilane, dimethyldibutoxysilane, trimethylbutoxysilane, ethyltri Butoxysilane, diethyldibutoxysilane, triethylbutoxysilane, trimethoxysilane, methyldimethoxysilane, dimethylmethan Xysilane, ethyldimethoxysilane, diethylmethoxysilane, triethoxysilane, methyldiethoxysilane, dimethylethoxysilane, ethyldiethoxysilane, diethylethoxysilane, tributoxysilane, methyldibutoxysilane, dimethylbutoxysilane, ethyldibutoxysilane, Diethylbutoxysilane, phenyltrimethoxysilane, diphenyldimethoxysilane, phenyltriethoxysilane, diphenyldiethoxysilane, hexyltrimethoxysilane, decyltrimethoxysilane, tetrapropoxysilane, methyltripropoxysilane, dimethyldipropoxysilane, ethyltripropoxy Examples include silane and diethyldipropoxysilane. Among these compounds, tetramethoxysilane and tetraethoxysilane are preferably used particularly from the viewpoint of both the effect of addition and cost. The alkoxysilane having a functional group that reacts with the epoxy group may be used alone or in combination of two or more.
これらのエポキシ基と反応する官能基を有するアルコキシシランの使用量は、好ましくはエポキシ化ジエン系ゴム100gに対して0.001mol〜0.3mol、より好ましくは0.002mol〜0.25mol、特に好ましくは0.003mol〜0.1molである。アルコキシシランの使用量が0.001mol未満では、エポキシ化ジエン系ゴム中に導入されるアルコキシシランの量が少なくなり、満足すべき変性効果が現れない。一方、アルコキシシランの使用量が0.3molを超えると、エポキシ化ジエン系ゴム中に未反応のアルコキシシランが残存し、その除去に手間がかかり、アルコキシシランの無駄にもなり、さらに顕著な物性の改善効果が現れにくい。 The amount of alkoxysilane having a functional group that reacts with these epoxy groups is preferably 0.001 mol to 0.3 mol, more preferably 0.002 mol to 0.25 mol, particularly preferably 100 g per 100 g of epoxidized diene rubber. Is 0.003 mol to 0.1 mol. When the amount of alkoxysilane used is less than 0.001 mol, the amount of alkoxysilane introduced into the epoxidized diene rubber is reduced, and a satisfactory modification effect does not appear. On the other hand, if the amount of alkoxysilane used exceeds 0.3 mol, unreacted alkoxysilane remains in the epoxidized diene rubber, and it takes time to remove it, resulting in waste of alkoxysilane and further remarkable physical properties. The improvement effect is difficult to appear.
本発明に係る変性ジエン系ゴムにおいて、アルコキシシランとともにエポキシ基と反応させるカルボン酸としては、特に制限はなく、飽和モノカルボン酸、飽和ジカルボン酸、芳香族モノカルボン酸、芳香族ジカルボン酸、不飽和モノカルボン酸、不飽和ジカルボン酸、また、3つ以上のカルボキシル基を持つ不飽和、飽和カルボン酸が挙げられる。飽和モノカルボン酸としては、例えば、蟻酸、酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、ラウリン酸、ミスチリン酸、パルチミン酸、マルガリン酸、ステアリン酸などが挙げられる。飽和ジカルボン酸としては、例えば、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸などが挙げられる。芳香族モノカルボン酸としては、例えば、安息香酸、m−トルイル酸、o−トルイル酸、p−トルイル酸、ケイ皮酸、メリト酸などが挙げられる。芳香族ジカルボン酸としては、例えば、イソフタル酸、テレフタル酸、フタル酸などが挙げられる。不飽和モノカルボン酸としては、例えば、α−エレオステアリン酸、α−リノレン酸、γ−リノレン酸、アラキドン酸、アラキドン酸、イワシ酸、エイコサジエン酸、エイコサテトラエン酸、エイコサトリエン酸、エイコサペンタエン酸、エイコセン酸、エライジン酸、エルカ酸、オズボンド酸、オレイン酸、ガドレイン酸、クロトン酸、ジホモ−γ−リノレン酸、テトラコサペンタエン酸、ドコサヘキサエン酸、ニシン酸、ネルボン酸、バクセン酸、パルミトレイン酸、ボセオペンタエン酸、ミード酸、ミリストレイン酸、リノール酸などが挙げられる。不飽和ジカルボン酸としては、例えば、フマル酸、マレイン酸などが挙げられる。これらの化合物の中で、カルボキシル基以外が化学的に安定で、沸点が高く揮発しにくい性質を持ち、シラン変性剤を付加する際の効果が高い点から、飽和モノカルボン酸または飽和ジカルボン酸が好ましく、飽和ジカルボン酸が特に好ましい。中でも具体的には、ステアリン酸、マロン酸がより好ましく、マロン酸が特に好ましい。上記カルボン酸は、1種類単独、または2種類以上を組み合わせて使用してもよい。 In the modified diene rubber according to the present invention, the carboxylic acid to be reacted with the epoxy group together with the alkoxysilane is not particularly limited, and is saturated monocarboxylic acid, saturated dicarboxylic acid, aromatic monocarboxylic acid, aromatic dicarboxylic acid, unsaturated Examples thereof include monocarboxylic acids, unsaturated dicarboxylic acids, and unsaturated and saturated carboxylic acids having three or more carboxyl groups. Examples of saturated monocarboxylic acids include formic acid, acetic acid, propionic acid, butyric acid, valeric acid, caproic acid, enanthic acid, caprylic acid, pelargonic acid, capric acid, lauric acid, myristylic acid, palmitic acid, margaric acid, stearic acid Etc. Examples of the saturated dicarboxylic acid include oxalic acid, malonic acid, succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid and the like. Examples of the aromatic monocarboxylic acid include benzoic acid, m-toluic acid, o-toluic acid, p-toluic acid, cinnamic acid, and mellitic acid. Examples of the aromatic dicarboxylic acid include isophthalic acid, terephthalic acid, and phthalic acid. Examples of unsaturated monocarboxylic acids include α-eleostearic acid, α-linolenic acid, γ-linolenic acid, arachidonic acid, arachidonic acid, sardine acid, eicosadienoic acid, eicosatetraenoic acid, eicosatrienoic acid, Eicosapentaenoic acid, eicosenoic acid, elaidic acid, erucic acid, ozbond acid, oleic acid, gadoleic acid, crotonic acid, dihomo-γ-linolenic acid, tetracosapentaenoic acid, docosahexaenoic acid, nisic acid, nervonic acid, vaccenic acid Examples include palmitoleic acid, boseopentaenoic acid, mead acid, myristoleic acid, and linoleic acid. Examples of the unsaturated dicarboxylic acid include fumaric acid and maleic acid. Among these compounds, saturated monocarboxylic acids or saturated dicarboxylic acids are used because they are chemically stable except for the carboxyl group, have a high boiling point and are difficult to volatilize, and have a high effect when a silane modifier is added. Saturated dicarboxylic acids are preferred and particularly preferred. Specifically, stearic acid and malonic acid are more preferable, and malonic acid is particularly preferable. You may use the said carboxylic acid individually by 1 type or in combination of 2 or more types.
これらのカルボン酸の使用量は、好ましくはエポキシ化ジエン系ゴムに対して、10μmol/g以上5000μmol/g以下であり、より好ましくは50μmol/g以上1000μmol/g以下、特に好ましくは80μmol/g以上500μmol/g以下である。カルボン酸の使用量が10μmol/g未満では、シラン変性剤が充分付加しない。一方、カルボン酸の使用量がエポキシ化ジエン系ゴムに対して5000μmol/gを超えると、ゴム組成物とした時、エポキシ化ジエン系ゴムに付加した変性基と無機フィラーの反応が妨害されるので好ましくない。 The amount of these carboxylic acids used is preferably 10 μmol / g or more and 5000 μmol / g or less, more preferably 50 μmol / g or more and 1000 μmol / g or less, particularly preferably 80 μmol / g or more, relative to the epoxidized diene rubber. 500 μmol / g or less. When the amount of carboxylic acid used is less than 10 μmol / g, the silane modifier is not sufficiently added. On the other hand, if the amount of carboxylic acid used exceeds 5000 μmol / g with respect to the epoxidized diene rubber, the reaction between the modified group added to the epoxidized diene rubber and the inorganic filler is hindered when the rubber composition is formed. It is not preferable.
本発明に係る変性ジエン系ゴムは、上記エポキシ化率が0.1%以上15%未満のエポキシ化ジエン系ゴムを、カルボン酸存在下でエポキシ基と反応する官能基を有するアルコキシシランと機械的混練により反応させることを特徴とする。機械的混練することで、低コストで生産効率がよく、廃液等を発生させずに変性ジエン系ゴムを製造することができる。本発明においては、例えば、あらかじめ加熱した混練機を用いて原料となるエポキシ化ジエン系ゴムにアルコキシシラン及びカルボン酸を練り込むことによって反応させても良いし、製造時にエポキシ化触媒として用いられたカルボン酸を通常行われる中和工程を省略して、そのまま用いても良い。 The modified diene rubber according to the present invention is obtained by mechanically treating an epoxidized diene rubber having an epoxidation rate of 0.1% or more and less than 15% with an alkoxysilane having a functional group that reacts with an epoxy group in the presence of a carboxylic acid. The reaction is characterized by kneading. By mechanically kneading, a modified diene rubber can be produced at low cost with good production efficiency and without generating waste liquid or the like. In the present invention, for example, the reaction may be carried out by kneading alkoxysilane and carboxylic acid into a raw material epoxidized diene rubber using a preheated kneader, or used as an epoxidation catalyst during production. The neutralization step in which carboxylic acid is usually performed may be omitted and used as it is.
上記反応の反応温度としては、20℃〜140℃が好ましく、より好ましくは40℃〜120℃である。20℃未満ではエポキシ基とアルコキシシランの官能基の反応が起きず、140℃を超えるとジエン系ゴムの熱劣化が起きるため、好ましくない。 As reaction temperature of the said reaction, 20 to 140 degreeC is preferable, More preferably, it is 40 to 120 degreeC. If it is less than 20 ° C., the reaction of the functional group of the epoxy group and alkoxysilane does not occur, and if it exceeds 140 ° C., thermal degradation of the diene rubber occurs.
本発明に係る変性ジエン系ゴムの製造方法において、機械的混練とは、機械的せん断力を与えて溶融樹脂を混練することであり、その混練に用いられる混練機は、機械的せん断力を与えて溶融樹脂を混練できる装置であれば特に制限はなく、ロール混練機、バンバリーミキサー、加圧ニーダー、一軸押出機、二軸押出機、2軸テーパー押出機等、樹脂加工に用いられる一般的な混練機械を用いることができる。 In the method for producing a modified diene rubber according to the present invention, mechanical kneading means kneading a molten resin by applying mechanical shearing force, and the kneader used for kneading gives mechanical shearing force. There is no particular limitation as long as it is an apparatus capable of kneading a molten resin, and a roll kneader, a Banbury mixer, a pressure kneader, a single screw extruder, a twin screw extruder, a twin screw taper extruder, etc., which are commonly used for resin processing A kneading machine can be used.
本発明に係る変性ジエン系ゴムの製造方法によって得られた変性ジエン系ゴムによれば、低エネルギーロス性及び反発弾性に優れた変性ジエン系ゴム組成物を得ることができる。 According to the modified diene rubber obtained by the method for producing a modified diene rubber according to the present invention, a modified diene rubber composition excellent in low energy loss and rebound resilience can be obtained.
次に、本発明に係る変性ジエン系ゴムを用いたゴム組成物について説明する。本発明に係るゴム組成物は、上記変性ジエン系ゴムと、シリカとを含有することを特徴とする。 Next, the rubber composition using the modified diene rubber according to the present invention will be described. The rubber composition according to the present invention is characterized by containing the modified diene rubber and silica.
本発明に係るゴム組成物においては、上記変性ジエン系ゴムの他にそれ以外のゴムを加えて、ゴム組成物として使用することもできる。それ以外の加えられるゴムとしては、特に制限はなく、公知のものを使用することができる。例えば、天然ゴム、ブタジエンゴム(BR)、シンジオタクチック−1,2−ポリブタジエン含有のブタジエンゴム(VCR)、イソプレンゴム、ブチルゴム、クロロプレンゴムなどのジエン系単量体の重合体;アクリロニトリルブタジエンゴム(NBR)、ニトリルクロロプレンゴム、ニトリルイソプレンゴムなどのアクリロニトリル−ジエン共重合ゴム;スチレンブタジエンゴム(SBR)、スチレンクロロプレンゴム、スチレンイソプレンゴムなどのスチレン−ジエン共重合ゴム;エチレンプロピレンジエンゴム(EPDM)などが挙げられる。この中で、ブタジエンゴム、シンジオタクチック−1,2−ポリブタジエン含有のブタジエンゴム、アクリロニトリルブタジエンゴム、スチレンブタジエンゴム、天然ゴム、イソプレンゴムが好ましい。これらは単独で、または2種類以上を組み合わせて使用することができる。 In the rubber composition according to the present invention, in addition to the modified diene rubber, other rubbers may be added and used as a rubber composition. There is no restriction | limiting in particular as rubber | gum other than that added, A well-known thing can be used. For example, polymers of diene monomers such as natural rubber, butadiene rubber (BR), syndiotactic-1,2-polybutadiene-containing butadiene rubber (VCR), isoprene rubber, butyl rubber, chloroprene rubber; acrylonitrile butadiene rubber ( NBR), acrylonitrile-diene copolymer rubber such as nitrile chloroprene rubber, nitrile isoprene rubber; styrene-diene copolymer rubber such as styrene butadiene rubber (SBR), styrene chloroprene rubber, styrene isoprene rubber; ethylene propylene diene rubber (EPDM), etc. Is mentioned. Of these, butadiene rubber, syndiotactic-1,2-polybutadiene-containing butadiene rubber, acrylonitrile butadiene rubber, styrene butadiene rubber, natural rubber, and isoprene rubber are preferable. These can be used alone or in combination of two or more.
また、本発明に係るゴム組成物において、シリカの含有量は、上記変性ジエン系ゴムとそれ以外のゴム成分100重量部に対して10重量部〜120重量部、より好ましくは30重量部〜90重量部、特に好ましくは50重量部〜80重量部である。シリカが10重量部より少ないと、本発明の変性ジエン系ゴムを用いなくても充分なシリカの分散が得られるため、本発明の効果がなく、120重量部より多いと加工性が著しく悪くなり、かつ耐摩耗性も低下し、好ましくない。 In the rubber composition according to the present invention, the content of silica is 10 to 120 parts by weight, more preferably 30 to 90 parts by weight with respect to 100 parts by weight of the modified diene rubber and other rubber components. Part by weight, particularly preferably 50 to 80 parts by weight. If the amount of silica is less than 10 parts by weight, sufficient silica dispersion can be obtained without using the modified diene rubber of the present invention. Therefore, the effect of the present invention is not obtained. In addition, wear resistance is also lowered, which is not preferable.
また、本発明に係るゴム組成物は、ゴム補強剤を加えることができる。ゴム補強剤としては、上記シリカの他、各種のカーボンブラック、ホワイトカーボン、活性化炭酸カルシウム、超微粒子珪酸マグネシウム等などが挙げられる。なかでも、好ましくは、粒子径が90nm以下、ジブチルフタレート(DBP)吸油量が70ml/100g以上のカーボンブラックであり、例えば、FEF、FF、GPF、SAF、ISAF、SRF、HAF等が用いられ、特に好ましくは、低発熱性や低燃費性の観点から粒子径の小さいISAFである。 Moreover, a rubber reinforcing agent can be added to the rubber composition according to the present invention. Examples of the rubber reinforcing agent include various types of carbon black, white carbon, activated calcium carbonate, ultrafine magnesium silicate and the like in addition to the above silica. Among them, preferably, carbon black having a particle size of 90 nm or less and dibutyl phthalate (DBP) oil absorption of 70 ml / 100 g or more, for example, FEF, FF, GPF, SAF, ISAF, SRF, HAF and the like are used. Particularly preferred is ISAF having a small particle size from the viewpoint of low heat build-up and low fuel consumption.
ゴム補強剤に用いるカーボンブラックとシリカは混合するとより加工性と低エネルギーロス性や摩耗性などの両立が可能となる。特に、両者の重量比がカーボンブラック/シリカが90/10〜10/90が良く、より好ましくは、80/20〜20/80、特に好ましくは70/30〜30/70である。シリカが10%より少ないと、エネルギーロスが大きくなり、90%より多いと加工性や摩耗性が悪くなる欠点がある。 When carbon black and silica used for the rubber reinforcing agent are mixed, it becomes possible to achieve both workability, low energy loss and wear. In particular, the weight ratio of carbon black / silica is preferably 90/10 to 10/90, more preferably 80/20 to 20/80, and particularly preferably 70/30 to 30/70. If the amount of silica is less than 10%, energy loss increases, and if it is more than 90%, there is a drawback that workability and wear resistance are deteriorated.
また、本発明に係るゴム組成物は、更に、加硫剤、加硫促進剤を添加することができる。 Moreover, a vulcanizing agent and a vulcanization accelerator can be further added to the rubber composition according to the present invention.
加硫剤としては、硫黄、加熱により硫黄を生成させる化合物、有機過酸化物、酸化マグネシウム等の金属酸化物、多官能性モノマー、シラノール化合物等が挙げられる。加熱により硫黄を生成させる化合物としては、テトラメチルチウラムジスルフィド、テトラエチルチウラムジスルフィド等が挙げられる。 Examples of the vulcanizing agent include sulfur, a compound that generates sulfur by heating, a metal oxide such as an organic peroxide and magnesium oxide, a polyfunctional monomer, and a silanol compound. Examples of the compound that generates sulfur by heating include tetramethyl thiuram disulfide and tetraethyl thiuram disulfide.
加硫促進剤としては、例えばアルデヒド類、アンモニア類、アミン類、グアニジン類、チオウレア類、チアゾール類、チウラム類、ジチオカーバメイト類、キサンテート類等が挙げられ、より具体的には、テトラメチルチウラムジスルフィド(TMTD)、N−オキシジエチレン−2−ベンゾチアゾリルスルフェンアミド(OBS)、N−シクロヘキシル−2−ベンゾチアジルスルフェンアミド(CBS)、ジベンゾチアジルジスルフィド(MBTS)、2−メルカプトベンゾチアゾール(MBT)、ジンクジ−n−ブチルジチオカーバイト(ZnBDC)、ジンクジメチルジチオカーバイト(ZnMDC)等が挙げられる。 Examples of the vulcanization accelerator include aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, xanthates, and more specifically, tetramethylthiuram disulfide. (TMTD), N-oxydiethylene-2-benzothiazolylsulfenamide (OBS), N-cyclohexyl-2-benzothiazylsulfenamide (CBS), dibenzothiazyl disulfide (MBTS), 2-mercaptobenzothiazole (MBT), zinc di-n-butyldithiocarbite (ZnBDC), zinc dimethyldithiocarbide (ZnMDC) and the like.
また、本発明に係るゴム組成物は、その他、必要に応じて、老化防止剤、充填剤、プロセスオイル等、通常ゴム組成物に用いられる公知の添加剤を添加することができる。 Moreover, the rubber composition which concerns on this invention can add well-known additives normally used for rubber compositions, such as anti-aging agent, a filler, a process oil, as needed.
老化防止剤としては、アミン・ケトン系、イミダゾール系、アミン系、フェノール系、硫黄系及び燐系等の老化防止剤が挙げられる。より具体的には、老化防止剤としてはフェノール系の2,6−ジ−t−ブチル−p−クレゾール(BHT)、リン系のトリノニルフェニルフォスファイト(TNP)、硫黄系の4,6−ビス(オクチルチオメチル)−o−クレゾール、ジラウリル−3,3’−チオジプロピオネート(TPL)等が挙げられる。 Anti-aging agents include amine / ketone-based, imidazole-based, amine-based, phenol-based, sulfur-based and phosphorus-based anti-aging agents. More specifically, as the anti-aging agent, phenol-based 2,6-di-t-butyl-p-cresol (BHT), phosphorus-based trinonylphenyl phosphite (TNP), sulfur-based 4,6- Bis (octylthiomethyl) -o-cresol, dilauryl-3,3′-thiodipropionate (TPL) and the like can be mentioned.
充填剤としては、炭酸カルシウム、塩基性炭酸マグネシウム、クレー、リサージュ、珪藻土等の無機充填剤、再生ゴム、粉末ゴム等の有磯充填剤が挙げられ、プロセスオイルとしては、アロマティック系、ナフテン系、パラフィン系のプロセスオイルが挙げられる。 Examples of fillers include inorganic fillers such as calcium carbonate, basic magnesium carbonate, clay, Lissajous, diatomaceous earth, and other fillers such as recycled rubber and powder rubber. Process oils include aromatic and naphthenic fillers. And paraffinic process oil.
さらに、本発明に係るゴム組成物は、シランカップリング剤を添加してもよい。シランカップリング剤としては特にエポキシ基と反応可能な官能基を有するシランカップリング剤が好ましい。 Furthermore, a silane coupling agent may be added to the rubber composition according to the present invention. As the silane coupling agent, a silane coupling agent having a functional group capable of reacting with an epoxy group is particularly preferable.
エポキシ基と反応可能な官能基を有するシランカップリング剤として、市販で利用できるものは、例えば、以下のものが含まれるが、決してこれらに限定されるものではない。3−アミノプロピルジメチルメトキシシラン、3−アミノプロピルメチルジメトキシシラン、3−アミノプロピルエチルジメトキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルジメチルエトキシシラン、3−アミノプロピルメチルジエトキシシラン、3−アミノプロピルトリエトキシシラン、3−アミノプロピルジメチルブトキシシラン、3−アミノプロピルメチルジブトキシシラン、3−(2−アミノエチルアミノプロピル)ジメトキシメチルシラン、3−(2−アミノエチルアミノプロピル)ジメトキシエチルシラン、3−(2−アミノエチルアミノプロピル)ジメトキシプロピルシラン、3−(2−アミノエチルアミノプロピル)ジメトキシブチルシラン、3−(2−アミノエチルアミノプロピル)ジエトキシメチルシラン、3−(2−アミノエチルアミノプロピル)トリメトキシシラン、3−(2−アミノエチルアミノプロピル)トリエトキシシラン、3−(2−アミノエチルアミノプロピル)トリブトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、3−グリシドキシプロピルジメトキシメチルシラン、3−グリシドキシプロピルトリエトキシシラン、3−グリシドキシプロピルトリメトキシシラン、3−グリシドキシプロピルメチルジエトキシシラン、(3−メルカプトプロピル)トリエトキシシラン、(3−メルカプトプロピル)トリメトキシシラン、(3−メルカプトプロピル)メチルジメトキシシラン、2−メルカプトエチルトリメトキシシラン、2−メルカプトエチルトリエトキシシラン、11−メルカプトウンデシルトリメトキシシラン、11−メルカプトウンデシルトリエトキシシランなどがある。この中でも特に、(3−メルカプトプロピル)トリエトキシシランが好ましい。 Examples of commercially available silane coupling agents having a functional group capable of reacting with an epoxy group include, but are not limited to, the following. 3-aminopropyldimethylmethoxysilane, 3-aminopropylmethyldimethoxysilane, 3-aminopropylethyldimethoxysilane, 3-aminopropyltrimethoxysilane, 3-aminopropyldimethylethoxysilane, 3-aminopropylmethyldiethoxysilane, 3 -Aminopropyltriethoxysilane, 3-aminopropyldimethylbutoxysilane, 3-aminopropylmethyldibutoxysilane, 3- (2-aminoethylaminopropyl) dimethoxymethylsilane, 3- (2-aminoethylaminopropyl) dimethoxyethyl Silane, 3- (2-aminoethylaminopropyl) dimethoxypropylsilane, 3- (2-aminoethylaminopropyl) dimethoxybutylsilane, 3- (2-aminoethylaminopropyl) diethoxy Methylsilane, 3- (2-aminoethylaminopropyl) trimethoxysilane, 3- (2-aminoethylaminopropyl) triethoxysilane, 3- (2-aminoethylaminopropyl) tributoxysilane, 2- (3,4 -Epoxycyclohexyl) ethyltrimethoxysilane, 3-glycidoxypropyldimethoxymethylsilane, 3-glycidoxypropyltriethoxysilane, 3-glycidoxypropyltrimethoxysilane, 3-glycidoxypropylmethyldiethoxysilane, (3-mercaptopropyl) triethoxysilane, (3-mercaptopropyl) trimethoxysilane, (3-mercaptopropyl) methyldimethoxysilane, 2-mercaptoethyltrimethoxysilane, 2-mercaptoethyltriethoxysilane, 11- Mercaptoethyloleates undecyl trimethoxysilane, and the like 11 mercaptoundecyl triethoxysilane. Among these, (3-mercaptopropyl) triethoxysilane is particularly preferable.
エポキシ基と反応可能な官能基を有するシランカップリング剤の添加量は、エポキシ化ジエン系ゴムのもつエポキシ基数に対し0.1〜1モル当量が好ましい。当該シランカップリング剤の添加量が0.1モル当量未満では、tanδおよび耐摩耗性の改善効果が少なくなる傾向がある。また、当該シランカップリング剤の添加量が1モル当量を超えると、経済的に好ましくない傾向がある。 The addition amount of the silane coupling agent having a functional group capable of reacting with an epoxy group is preferably 0.1 to 1 molar equivalent with respect to the number of epoxy groups of the epoxidized diene rubber. When the addition amount of the silane coupling agent is less than 0.1 molar equivalent, the effect of improving tan δ and wear resistance tends to be reduced. Moreover, when the addition amount of the said silane coupling agent exceeds 1 molar equivalent, there exists a tendency which is not economically preferable.
シランカップリング剤を用いたゴム組成物は、シリカなどのゴム補強剤との混合により、ゴム補強剤のゴムマトリクス中での分散性を向上させる働きがある。その結果、低燃費性などの効果にもつながる。 A rubber composition using a silane coupling agent functions to improve the dispersibility of the rubber reinforcing agent in the rubber matrix by mixing with a rubber reinforcing agent such as silica. As a result, it also leads to effects such as low fuel consumption.
本発明に係るゴム組成物は、上記各成分を通常行われているバンバリー、オープンロール混練機、ニーダー、二軸混練り機などを用いて混練時の最高温度がシランカップリング剤とエポキシ基の反応温度以上となる条件で混練りすることで得られる。 In the rubber composition according to the present invention, the maximum temperature during kneading using the banbury, open roll kneader, kneader, biaxial kneader, etc., in which each of the above components is usually performed, is the silane coupling agent and the epoxy group. It can be obtained by kneading under the condition of the reaction temperature or higher.
以下、本発明を実施例に基づいて具体的に説明するが、これらは本発明の目的を限定するものではない。 EXAMPLES Hereinafter, although this invention is demonstrated concretely based on an Example, these do not limit the objective of this invention.
(参考例1)
先ず、参考例1として、実施例1乃至4、比較例2及び3に用いるエポキシ化ポリブタジエンゴムを作製した。具体的には、ポリブタジエン(宇部興産(株)製:UBEPOL BR150L)を100g取り、セパラブルフラスコのシクロへキサン1000ml中に投入して攪拌し、1晩(約8時間)をかけて溶解した。その後、ウォーターバスでセパラブルフラスコの温度を50℃とし、非イオン性界面活性剤テリック320(HUNTSMAN社製)1gおよび蟻酸4.25g投入して攪拌、続いて過酸化水素水(30%)10.5gを滴下ロートで投入して2時間攪拌を続けた。その後に加熱を止め、酸化防止剤(イルガノックス1520L)を0.1g投入し、ウォーターバス中に氷を投入して温度を室温まで下げた。次に、メタノール500mlを投入し、エポキシ化ポリブタジエンゴムを溶液から析出させた。溶媒を取り除いた後、2.5%炭酸ナトリウムを500ml投入し、10分間攪拌後、洗浄液を除去した。さらに、水500mlを投入し、10分間攪拌後、洗浄液を除去した。この水洗作業を3度行った。その後、反応液をテフロン(登録商標)コーティングしたバットに取り出し、90℃の真空乾燥機中に1時間置いて、乾燥させることで、エポキシ化ポリブタジエンゴムを得た。
(Reference Example 1)
First, as Reference Example 1, epoxidized polybutadiene rubber used in Examples 1 to 4 and Comparative Examples 2 and 3 was prepared. Specifically, 100 g of polybutadiene (manufactured by Ube Industries, Ltd .: UBEPOL BR150L) was taken, put into 1000 ml of cyclohexane in a separable flask and stirred, and dissolved overnight (about 8 hours). Thereafter, the temperature of the separable flask was set to 50 ° C. in a water bath, and 1 g of nonionic surfactant Telic 320 (manufactured by HUNTSMAN) and 4.25 g of formic acid were added and stirred, followed by hydrogen peroxide solution (30%) 10 0.5 g was added with a dropping funnel and stirring was continued for 2 hours. Thereafter, the heating was stopped, 0.1 g of an antioxidant (Irganox 1520L) was added, and ice was added into the water bath to lower the temperature to room temperature. Next, 500 ml of methanol was added to precipitate epoxidized polybutadiene rubber from the solution. After removing the solvent, 500 ml of 2.5% sodium carbonate was added, and after stirring for 10 minutes, the washing solution was removed. Furthermore, 500 ml of water was added, and after stirring for 10 minutes, the washing solution was removed. This washing operation was performed three times. Thereafter, the reaction liquid was taken out into a vat coated with Teflon (registered trademark), placed in a vacuum dryer at 90 ° C. for 1 hour, and dried to obtain an epoxidized polybutadiene rubber.
得られたエポキシ化ポリブタジエンゴムのエポキシ化率を測定したところ、2.2%であった。 When the epoxidation rate of the obtained epoxidized polybutadiene rubber was measured, it was 2.2%.
エポキシ化率は試料の工程終了後、3時間以上貯蔵した後、JIS K7236に準じて測定した。なお、JIS K7236と異なる点は、エポキシ化ゴムの量を0.6g〜0.9gとしたこと、エポキシ化ゴムの溶解時に用いるクロロホルムをシクロへキサンに変更したことである。また、JIS K7236では測定直前に20mlの酢酸を加えることとなっているが、臭化テトラエチルアンモニウム酢酸溶液に含まれる酢酸以外に加えなかったことである。エポキシ化率の低いゴムは規格量の酢酸を加えると塊状に析出し、滴定できなかった。酢酸量を減じた場合、当量点が分かりにくくなるが、析出した試料が油膜状に測定液上に広がり時間をかければ滴定可能となった。その他の試薬調整等はJIS K7236に述べられている通りである。 The epoxidation rate was measured according to JIS K7236 after storing for 3 hours or more after the completion of the sample process. The difference from JIS K7236 is that the amount of the epoxidized rubber was 0.6 g to 0.9 g, and that chloroform used for dissolving the epoxidized rubber was changed to cyclohexane. According to JIS K7236, 20 ml of acetic acid is added immediately before the measurement, but no acetic acid other than acetic acid contained in the tetraethylammonium bromide acetic acid solution is added. When a standard amount of acetic acid was added, a rubber having a low epoxidation rate was precipitated in a lump and could not be titrated. When the amount of acetic acid was reduced, the equivalence point became difficult to understand, but the precipitated sample spread on the measurement liquid in the form of an oil film, and titration became possible if time was taken. Other reagent adjustments are as described in JIS K7236.
また、ここでエポキシ化率は、下記計算式数1を用いて計算した。エポキシ当量とはエポキシ基1モルに相当するエポキシ化樹脂の質量(g)であり、JIS K7236に述べられている方法で求められる。100%エポキシ化ポリブタジエンの場合はブタジエン分子量+酸素1原子量である。 Moreover, the epoxidation rate was computed using the following formula 1 here. The epoxy equivalent is the mass (g) of the epoxidized resin corresponding to 1 mol of the epoxy group, and is determined by the method described in JIS K7236. In the case of 100% epoxidized polybutadiene, it is butadiene molecular weight + oxygen 1 atomic weight.
![]()
![]()
(実施例1)
次に、表1に示すとおり、参考例1において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、ステアリン酸(花王社製)2.5g、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、実施例1に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
Example 1
Next, as shown in Table 1, 100 g of the epoxidized polybutadiene rubber produced in Reference Example 1 was taken, wound around a roll (temperature: 60 ° C., roll interval: 0.8 mm), and kneaded. As the roll, a 6-inch roll kneader manufactured by Yasuda Seiki was used. Next, 2.5 g of stearic acid (manufactured by Kao) and 1 g of GOPTMS (3-glycidyloxypropyltrimethoxysilane) (manufactured by Tokyo Kasei Co., Ltd .: reagent) are dropped on the epoxidized polybutadiene rubber on the roll with a pipette and further kneaded for 5 minutes. It was crowded. Then, the modified polybutadiene rubber which concerns on Example 1 was obtained by heat-processing at 90 degreeC under vacuum with a vacuum dryer for 1 hour. The modified polybutadiene rubber was allowed to stand for 24 hours, and about 5 g was taken, dissolved in 100 ml of cyclohexane and precipitated with ethanol twice. The obtained sample was again dried at 90 ° C. for 1 hour, and the Si content was measured by ICP analysis. The results are shown in Table 3.
(実施例2)
実施例1において、ステアリン酸(花王社製)2.5gの代わりにマロン酸(和光純薬社製:試薬)1gを用いたこと以外は実施例1と同様にして、実施例2に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例1と同様にICP分析によりSiの含量を測定した。結果を表3に示す。
(Example 2)
In Example 1, the modification according to Example 2 was performed in the same manner as in Example 1 except that 1 g of malonic acid (manufactured by Wako Pure Chemical Industries, Ltd .: reagent) was used instead of 2.5 g of stearic acid (manufactured by Kao Corporation). A polybutadiene rubber was obtained. About the obtained sample, the content of Si was measured by ICP analysis in the same manner as in Example 1. The results are shown in Table 3.
(実施例3)
実施例1において、ステアリン酸(花王社製)2.5gの代わりにマロン酸(和光純薬社製:試薬)2.5gを用いたこと以外は実施例1と同様にして、実施例3に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例1と同様にICP分析によりSiの含量を測定した。結果を表3に示す。
(Example 3)
In Example 1, in the same manner as in Example 1, except that 2.5 g of malonic acid (manufactured by Wako Pure Chemical Industries, Ltd .: reagent) was used instead of 2.5 g of stearic acid (manufactured by Kao Corporation) Such modified polybutadiene rubber was obtained. About the obtained sample, the content of Si was measured by ICP analysis in the same manner as in Example 1. The results are shown in Table 3.
(実施例4)
実施例3において、変性ポリブタジエンゴムをシクロヘキサン100mlに溶解し、エタノールで析出させる操作を1回にしたこと以外は実施例3と同様にして、実施例4に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例3と同様にICP分析によりSiの含量を測定した。結果を表3に示す。
Example 4
In Example 3, the modified polybutadiene rubber according to Example 4 was obtained in the same manner as in Example 3 except that the modified polybutadiene rubber was dissolved in 100 ml of cyclohexane and precipitated with ethanol once. About the obtained sample, the content of Si was measured by ICP analysis in the same manner as in Example 3. The results are shown in Table 3.
(比較例1)
ポリブタジエン(宇部興産(株)製:UBEPOL BR150L)を24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
(Comparative Example 1)
After allowing polybutadiene (manufactured by Ube Industries Ltd .: UBEPOL BR150L) to stand for 24 hours, about 5 g was taken, dissolved in 100 ml of cyclohexane, and precipitated with ethanol twice. The obtained sample was again dried at 90 ° C. for 1 hour, and the Si content was measured by ICP analysis. The results are shown in Table 3.
(比較例2)
参考例1において作製したエポキシ化ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
(Comparative Example 2)
After leaving the epoxidized polybutadiene rubber produced in Reference Example 1 for 24 hours, about 5 g was taken, dissolved in 100 ml of cyclohexane, and precipitated with ethanol twice. The obtained sample was again dried at 90 ° C. for 1 hour, and the Si content was measured by ICP analysis. The results are shown in Table 3.
(比較例3)
参考例1において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、比較例3に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
(Comparative Example 3)
100 g of the epoxidized polybutadiene rubber produced in Reference Example 1 was taken, wound around a roll (temperature: 60 ° C., roll interval: 0.8 mm), and kneaded. As the roll, a 6-inch roll kneader manufactured by Yasuda Seiki was used. Next, 1 g of GOPTMS (3-glycidyloxypropyltrimethoxysilane) (manufactured by Tokyo Kasei Co., Ltd .: reagent) was dropped on the epoxidized polybutadiene rubber on the roll with a pipette and further kneaded for 5 minutes. Then, the modified polybutadiene rubber which concerns on the comparative example 3 was obtained by heat-processing at 90 degreeC for 1 hour under vacuum with a vacuum dryer. The modified polybutadiene rubber was allowed to stand for 24 hours, and about 5 g was taken, dissolved in 100 ml of cyclohexane and precipitated with ethanol twice. The obtained sample was again dried at 90 ° C. for 1 hour, and the Si content was measured by ICP analysis. The results are shown in Table 3.
得られた実施例1乃至4及び比較例1乃至3に係る試料30重量部と、スチレン−ブタジエンゴム(スチレン含量が23%、ML1+4、100℃が70)70重量部を、予め90℃に加温した180ccのバンバリータイプのプラストミルに投入して30秒混練した。続けてシリカ(Ultrasil 5000GR)75重量部にシランカップリング剤(Si69)を6重量部、プロセスオイル(サンセンオイル4240)21.5重量部、亜鉛華3重量部、老化防止剤(住友化学製:アンチゲン6C)を1重量部、ステアリン酸1重量部を混合し、そのうち半分をプラストミルに投入し、1分間混練した。次に、残り半分を投入し約3分30秒混練した。混練を開始してから合計で5分間経過した後、混練物をプラストミルより取り出した。次に、6インチロールに取り出した混合物を巻きつけてロール混練しながら、加硫剤である粉末硫黄を1.4重量部と加硫促進剤ノクセラーCZ1.7重量部およびノクセラーD2重量部を添加した。ロールの温度は60℃とし、約5分間の間に粉末硫黄と加硫促進剤を混合した。次に、下記に示す試験に必要な加硫成型体を得るため、加硫成型を行った。熱プレスにセットした金型を用い、金型内に混合物を入れて温度160℃、約20〜25分間加熱加圧することで加硫成型を行い、実施例1乃至4及び比較例1乃至3に係るゴム組成物を得た。用いた薬品および配合比(重量部)を表2に示す。 30 parts by weight of the samples according to Examples 1 to 4 and Comparative Examples 1 to 3 obtained and 70 parts by weight of styrene-butadiene rubber (styrene content 23%, ML 1 + 4 , 100 ° C. is 70) were previously added to 90 ° C. It was put into a heated 180 cc Banbury type plast mill and kneaded for 30 seconds. Next, 75 parts by weight of silica (Ultrasil 5000GR), 6 parts by weight of a silane coupling agent (Si69), 21.5 parts by weight of process oil (Sansen Oil 4240), 3 parts by weight of zinc oxide, an anti-aging agent (manufactured by Sumitomo Chemical: 1 part by weight of Antigen 6C) and 1 part by weight of stearic acid were mixed, half of which was put into a plastmill and kneaded for 1 minute. Next, the remaining half was charged and kneaded for about 3 minutes 30 seconds. After a total of 5 minutes from the start of kneading, the kneaded product was taken out from the plast mill. Next, while winding the mixture taken out on a 6-inch roll and kneading the roll, 1.4 parts by weight of powdered sulfur as a vulcanizing agent, 1.7 parts by weight of a vulcanization accelerator Noxeller CZ and 2 parts by weight of Noxeller D are added. did. The roll temperature was 60 ° C., and powdered sulfur and a vulcanization accelerator were mixed for about 5 minutes. Next, vulcanization molding was performed in order to obtain a vulcanization molding necessary for the test described below. Using a mold set in a hot press, the mixture was placed in the mold, and vulcanization molding was performed by heating and pressurizing at a temperature of 160 ° C. for about 20 to 25 minutes, to Examples 1 to 4 and Comparative Examples 1 to 3 Such a rubber composition was obtained. Table 2 shows the chemicals used and the mixing ratio (parts by weight).
得られた実施例1乃至4及び比較例1乃至3に係るゴム組成物について、ゴム組成物の引張強度、破壊伸び、反発弾性、加硫物tanδを以下の方法により測定した。結果を表3に示す。 For the rubber compositions according to Examples 1 to 4 and Comparative Examples 1 to 3 obtained, the tensile strength, fracture elongation, rebound resilience, and vulcanized product tan δ of the rubber composition were measured by the following methods. The results are shown in Table 3.
(引張試験)
JIS K6251に準拠して測定し、引張強度(TB)、破壊伸度(EB)、(TB×EB)/2を下記計算式数2で指数表示した。(TB×EB)/2は破断までに費やされたエネルギーのおおよその大きさを示す。TB、EB、(TB×EB)/2は一般的に指数が大きい程、ゴム組成物として有利である。
(Tensile test)
Measurement was performed according to JIS K6251 and tensile strength (TB), elongation at break (EB), and (TB × EB) / 2 were expressed as an index using the following formula 2. (TB × EB) / 2 indicates the approximate amount of energy expended before breaking. TB, EB, (TB × EB) / 2 is generally more advantageous as a rubber composition as the index is larger.
(反発弾性試験)
反発弾性は、JIS K6251に規定されている測定法に従って、トリプソ式で測定し、下記計算式数2で指数表示した。一般的に指数が大きい程、ゴム組成物として有利である。
(Rebound resilience test)
The impact resilience was measured by a trypso method according to the measurement method defined in JIS K6251 and expressed as an index using the following formula 2. Generally, the larger the index, the more advantageous as a rubber composition.
![]()
![]()
(加硫物tanδ)
EPLEXOR 100N(GABO社製)を用いて、初期歪み10%、動歪み0.3%、周波数16Hz、温度50℃の測定条件で各配合物のtanδを測定し、比較例1のtanδを100とし、下記計算式数3で指数表示した。指数が大きいほど、転がり抵抗特性が優れることを示す。
(Vulcanized product tan δ)
Using EPLEXOR 100N (manufactured by GABO), tan δ of each formulation was measured under the measurement conditions of initial strain 10%, dynamic strain 0.3%, frequency 16 Hz, temperature 50 ° C., and tan δ of Comparative Example 1 was set to 100. The index is expressed by the following formula 3. The larger the index, the better the rolling resistance characteristics.
![]()
![]()
以上より、実施例に係る変性ポリブタジエンゴムは、エポキシ基と反応したSiの量が多いため、シリカの分散性が向上し、結果ゴム組成物のtanδ及び反発弾性が向上したことが分かる。 From the above, it can be seen that the modified polybutadiene rubber according to the example has a large amount of Si reacted with the epoxy group, so that the dispersibility of silica is improved, and as a result, the tan δ and rebound resilience of the rubber composition are improved.
(参考例2)
次に、参考例2として、実施例5乃至10、比較例4に用いるエポキシ化ポリブタジエンゴムを作製した。具体的には、ポリブタジエン(宇部興産(株)製:UBEPOL BR150L)を100g取り、セパラブルフラスコのシクロへキサン1000ml中に投入して攪拌し、室温(25℃)で、1晩(約8時間)をかけて溶解した。その後、ウォーターバスでセパラブルフラスコの温度を40℃とし、非イオン性界面活性剤テリック320(HUNTSMAN社製)1gおよび蟻酸2.13g投入して攪拌、続いてセパラブルフラスコの温度を50℃まで上昇させ、過酸化水素水(30%)5.24gを滴下ロートで投入して2時間攪拌を続けた。その後に加熱を止め、酸化防止剤(イルガノックス1520L)を0.2g投入し、ウォーターバス中に氷を投入して温度を室温まで下げた。次に、エタノール500mlを投入し、エポキシ化ポリブタジエンゴムを溶液から析出させた。溶媒を取り除いた後、1.0%炭酸ナトリウムを500ml投入し、10分間攪拌後、洗浄液を除去した。さらに、水(pH=7.0)500mlを投入し、10分間攪拌後、洗浄液を除去した。この水洗作業を3度行った。その後、反応液をテフロン(登録商標)コーティングしたバットに取り出し、90℃の真空乾燥機中に3時間置いて、乾燥させることで、エポキシ化ポリブタジエンゴムを得た。
(Reference Example 2)
Next, as Reference Example 2, epoxidized polybutadiene rubber used in Examples 5 to 10 and Comparative Example 4 was produced. Specifically, 100 g of polybutadiene (manufactured by Ube Industries, Ltd .: UBEPOL BR150L) is taken, put into 1000 ml of cyclohexane in a separable flask and stirred, and then overnight (about 8 hours at room temperature (25 ° C.)). ) To dissolve. Thereafter, the temperature of the separable flask was adjusted to 40 ° C. in a water bath, 1 g of nonionic surfactant terrick 320 (manufactured by HUNTSMAN) and 2.13 g of formic acid were added and stirred, and then the temperature of the separable flask was increased to 50 ° C. Then, 5.24 g of hydrogen peroxide solution (30%) was added through a dropping funnel and stirring was continued for 2 hours. Thereafter, the heating was stopped, 0.2 g of an antioxidant (Irganox 1520L) was added, and ice was added into the water bath to lower the temperature to room temperature. Next, 500 ml of ethanol was added and epoxidized polybutadiene rubber was precipitated from the solution. After removing the solvent, 500 ml of 1.0% sodium carbonate was added, and after stirring for 10 minutes, the washing solution was removed. Furthermore, 500 ml of water (pH = 7.0) was added, and after stirring for 10 minutes, the washing solution was removed. This washing operation was performed three times. Thereafter, the reaction solution was taken out into a Teflon (registered trademark) -coated vat, placed in a vacuum dryer at 90 ° C. for 3 hours, and dried to obtain an epoxidized polybutadiene rubber.
得られたエポキシ化ポリブタジエンゴムのエポキシ化率を参考例1と同様にして測定したところ、2.5%であった。 When the epoxidation rate of the obtained epoxidized polybutadiene rubber was measured in the same manner as in Reference Example 1, it was 2.5%.
(実施例5)
次に、表4に示すとおり、参考例2において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、ステアリン酸(花王社製)1g、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、実施例5に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン120mlに溶解し、メタノール400mlで30分洗浄した。沈殿した試料を再びシクロヘキサン120mlに溶解し、老化防止剤(Irganox 1520L)0.05mlを加え、上記と同様にメタノールで洗浄することで、再度試料を析出させた。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表6に示す。
(Example 5)
Next, as shown in Table 4, 100 g of the epoxidized polybutadiene rubber produced in Reference Example 2 was taken, wound around a roll (temperature: 60 ° C., roll interval: 0.8 mm), and kneaded. As the roll, a 6-inch roll kneader manufactured by Yasuda Seiki was used. Next, 1 g of stearic acid (manufactured by Kao Corporation) and 1 g of GOPTMS (3-glycidyloxypropyltrimethoxysilane) (manufactured by Tokyo Chemical Industry: Reagent) were hung on the epoxidized polybutadiene rubber on the roll with a pipette and further kneaded for 5 minutes. . Then, the modified polybutadiene rubber which concerns on Example 5 was obtained by heat-processing at 90 degreeC under vacuum with a vacuum dryer for 1 hour. After this modified polybutadiene rubber was allowed to stand for 24 hours, about 5 g was taken and dissolved in 120 ml of cyclohexane and washed with 400 ml of methanol for 30 minutes. The precipitated sample was dissolved again in 120 ml of cyclohexane, 0.05 ml of an anti-aging agent (Irganox 1520L) was added, and the sample was precipitated again by washing with methanol in the same manner as described above. The obtained sample was again dried at 90 ° C. for 1 hour, and the Si content was measured by ICP analysis. The results are shown in Table 6.
(実施例6)
実施例5において、ステアリン酸(花王社製)1gの代わりにマロン酸(和光純薬社製:試薬)1gを用いたこと以外は実施例5と同様にして、実施例6に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例5と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
(Example 6)
A modified polybutadiene rubber according to Example 6 in the same manner as in Example 5 except that 1 g of malonic acid (manufactured by Wako Pure Chemical Industries, Ltd .: reagent) was used instead of 1 g of stearic acid (manufactured by Kao Corporation) in Example 5. Got. About the obtained sample, the content of Si was measured by ICP analysis in the same manner as in Example 5. The results are shown in Table 6.
(実施例7)
実施例5において、ステアリン酸(花王社製)1gの代わりにマロン酸(和光純薬社製:試薬)2.5gを用いたこと以外は実施例5と同様にして、実施例7に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例5と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
(Example 7)
In Example 5, the modification according to Example 7 was performed in the same manner as in Example 5 except that 2.5 g of malonic acid (manufactured by Wako Pure Chemical Industries, Ltd .: reagent) was used instead of 1 g of stearic acid (manufactured by Kao Corporation). A polybutadiene rubber was obtained. About the obtained sample, the content of Si was measured by ICP analysis in the same manner as in Example 5. The results are shown in Table 6.
(比較例4)
参考例2において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、比較例4に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン120mlに溶解し、メタノール400mlで30分洗浄した。沈殿した試料を再びシクロヘキサン120mlに溶解し、老化防止剤(Irganox 1520L)0.05mlを加え、上記と同様にメタノールで洗浄することで、再度試料を析出させた。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表6に示す。
(Comparative Example 4)
100 g of the epoxidized polybutadiene rubber produced in Reference Example 2 was taken, wound around a roll (temperature: 60 ° C., roll interval: 0.8 mm), and kneaded. As the roll, a 6-inch roll kneader manufactured by Yasuda Seiki was used. Next, 1 g of GOPTMS (3-glycidyloxypropyltrimethoxysilane) (manufactured by Tokyo Kasei Co., Ltd .: reagent) was dropped on the epoxidized polybutadiene rubber on the roll with a pipette and further kneaded for 5 minutes. Then, the modified polybutadiene rubber which concerns on the comparative example 4 was obtained by heat-processing at 90 degreeC under vacuum with a vacuum dryer for 1 hour. After this modified polybutadiene rubber was allowed to stand for 24 hours, about 5 g was taken and dissolved in 120 ml of cyclohexane and washed with 400 ml of methanol for 30 minutes. The precipitated sample was dissolved again in 120 ml of cyclohexane, 0.05 ml of an anti-aging agent (Irganox 1520L) was added, and the sample was precipitated again by washing with methanol in the same manner as described above. The obtained sample was again dried at 90 ° C. for 1 hour, and the Si content was measured by ICP analysis. The results are shown in Table 6.
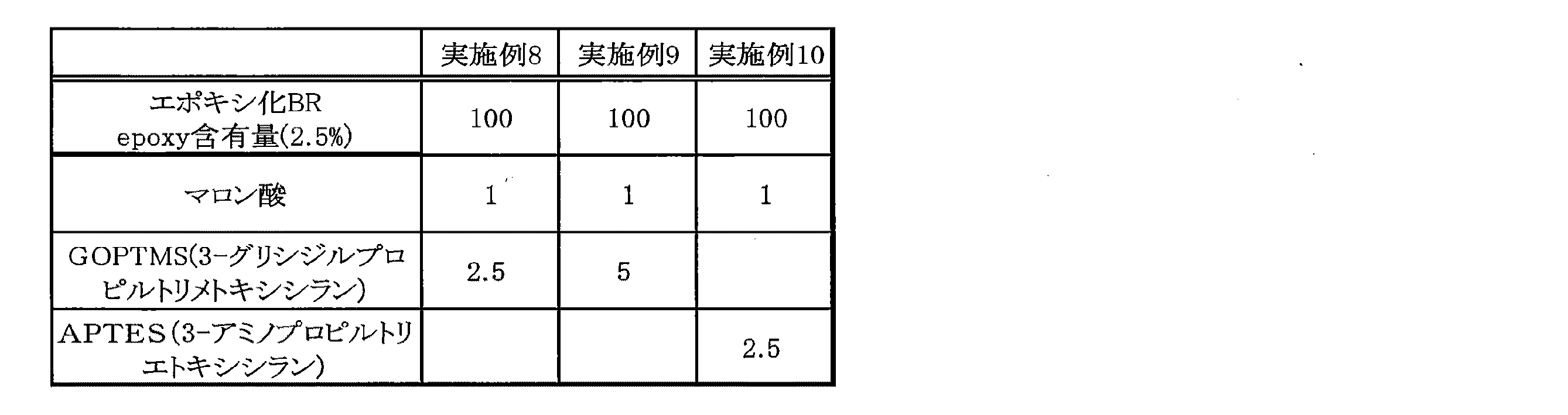
(実施例8)
次に、表5に示すとおり、参考例2において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、マロン酸(花王社製)1g、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)2.5gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、実施例8に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン120mlに溶解し、メタノール400mlで30分洗浄した。沈殿した試料を再びシクロヘキサン120mlに溶解し、老化防止剤(Irganox 1520L)0.05mlを加え、上記と同様にメタノールで洗浄することで、再度試料を析出させた。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表6に示す。
(Example 8)
Next, as shown in Table 5, 100 g of the epoxidized polybutadiene rubber produced in Reference Example 2 was taken, wound around a roll (temperature: 60 ° C., roll interval: 0.8 mm), and kneaded. As the roll, a 6-inch roll kneader manufactured by Yasuda Seiki was used. Next, 1 g of malonic acid (manufactured by Kao Co., Ltd.) and 2.5 g of GOPTMS (3-glycidyloxypropyltrimethoxysilane) (manufactured by Tokyo Kasei Co., Ltd .: reagent) are pipetted onto the epoxidized polybutadiene rubber on the roll and kneaded for 5 minutes. It was crowded. Then, the modified polybutadiene rubber which concerns on Example 8 was obtained by heat-processing at 90 degreeC under vacuum with a vacuum dryer for 1 hour. After this modified polybutadiene rubber was allowed to stand for 24 hours, about 5 g was taken and dissolved in 120 ml of cyclohexane and washed with 400 ml of methanol for 30 minutes. The precipitated sample was dissolved again in 120 ml of cyclohexane, 0.05 ml of an anti-aging agent (Irganox 1520L) was added, and the sample was precipitated again by washing with methanol in the same manner as described above. The obtained sample was again dried at 90 ° C. for 1 hour, and the Si content was measured by ICP analysis. The results are shown in Table 6.
(実施例9)
実施例8において、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)を5g用いたこと以外は実施例8と同様にして、実施例9に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例8と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
Example 9
A modified polybutadiene rubber according to Example 9 was obtained in the same manner as in Example 8, except that 5 g of GOPTMS (3-glycidyloxypropyltrimethoxysilane) (manufactured by Tokyo Chemical Industry: Reagent) was used. About the obtained sample, the content of Si was measured by ICP analysis in the same manner as in Example 8. The results are shown in Table 6.
(実施例10)
実施例8において、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)2.5gの代わりにAPTES(3−アミノプロピルトリエトキシシラン)(東京化成製:試薬)2.5gを用いたこと以外は実施例8と同様にして、実施例10に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例8と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
(Example 10)
In Example 8, 2.5 g of APTES (3-aminopropyltriethoxysilane) (manufactured by Tokyo Chemical Industry: reagent) instead of 2.5 g of GOPTMS (3-glycidyloxypropyltrimethoxysilane) (manufactured by Tokyo Chemical Industry: reagent) A modified polybutadiene rubber according to Example 10 was obtained in the same manner as Example 8 except that it was used. About the obtained sample, the content of Si was measured by ICP analysis in the same manner as in Example 8. The results are shown in Table 6.
得られた実施例5乃至10及び比較例4に係る試料30重量部と、スチレン−ブタジエンゴム(スチレン含量が23%、ML1+4、100℃が70)70重量部を、予め90℃に加温した180ccのバンバリータイプのプラストミルに投入して30秒混練した。続けてシリカ(ニプシルAQ)75重量部にシランカップリング剤(Evonik Degussa Japan社製:ニボニックデグザSi69)を6重量部、プロセスオイル(サンセンオイル4240)21.5重量部、亜鉛華3重量部、老化防止剤(住友化学製:アンチゲン6C)を1重量部、ステアリン酸(花王社製)1重量部を混合し、そのうち半分をプラストミルに投入し、1分間混練した。次に、残り半分を投入し約3分30秒混練した。混練を開始してから合計で5分間経過した後、混練物をプラストミルより取り出した。次に、6インチロールに取り出した混合物を巻きつけてロール混練しながら、加硫剤である粉末硫黄を1.4重量部と加硫促進剤ノクセラーCZ(CBS)1.7重量部およびノクセラーD(DPG)2重量部を添加した。ロールの温度は55〜65℃とし、約5分間の間に粉末硫黄と加硫促進剤を混合した。次に、下記に示す試験に必要な加硫成型体を得るため、加硫成型を行った。熱プレスにセットした金型を用い、金型内に混合物を入れて温度160℃、約20〜25分間加熱加圧することで加硫成型を行い、実施例5乃至10及び比較例4に係るゴム組成物を得た。 30 parts by weight of the samples according to Examples 5 to 10 and Comparative Example 4 and 70 parts by weight of styrene-butadiene rubber (styrene content is 23%, ML 1 + 4 , 100 ° C. is 70) are heated to 90 ° C. in advance. It was put into a 180 cc Banbury type plast mill and kneaded for 30 seconds. Next, 75 parts by weight of silica (Nipsil AQ), 6 parts by weight of a silane coupling agent (Evonik Degussa Japan: Nibonic Degussa Si69), 21.5 parts by weight of process oil (Sansen Oil 4240), 3 parts by weight of zinc oxide, aging 1 part by weight of an inhibitor (manufactured by Sumitomo Chemical Co., Ltd .: Antigen 6C) and 1 part by weight of stearic acid (manufactured by Kao Corporation) were mixed, half of which was put into a plastmill and kneaded for 1 minute. Next, the remaining half was charged and kneaded for about 3 minutes 30 seconds. After a total of 5 minutes from the start of kneading, the kneaded product was taken out from the plast mill. Next, while winding the mixture taken out on a 6-inch roll and roll kneading, 1.4 parts by weight of powdered sulfur as a vulcanizing agent, 1.7 parts by weight of a vulcanization accelerator Noxeller CZ (CBS) and Noxeller D 2 parts by weight of (DPG) was added. The temperature of the roll was 55 to 65 ° C., and powdered sulfur and a vulcanization accelerator were mixed for about 5 minutes. Next, vulcanization molding was performed in order to obtain a vulcanization molding necessary for the test described below. Rubbers according to Examples 5 to 10 and Comparative Example 4 were performed by using a mold set in a hot press, and performing a vulcanization molding by putting the mixture in the mold and heating and pressing at a temperature of 160 ° C. for about 20 to 25 minutes. A composition was obtained.
得られた5乃至10及び比較例4に係るゴム組成物について、ゴム組成物の引張強度、破壊伸び、反発弾性、加硫物tanδを上記実施例1乃至4及び比較例1乃至3と同様の方法により測定した。結果を表6に示す。 For the rubber compositions according to 5 to 10 and Comparative Example 4 obtained, the tensile strength, fracture elongation, impact resilience, and vulcanized product tan δ of the rubber composition were the same as those in Examples 1 to 4 and Comparative Examples 1 to 3. Measured by the method. The results are shown in Table 6.
以上より、実施例に係る変性ポリブタジエンゴムは、エポキシ基と反応したSiの量が多いため、シリカの分散性が向上し、結果ゴム組成物のtanδ及び反発弾性が向上したことが分かる。
From the above, it can be seen that the modified polybutadiene rubber according to the example has a large amount of Si reacted with the epoxy group, so that the dispersibility of silica is improved, and as a result, the tan δ and rebound resilience of the rubber composition are improved.
Claims (4)
A modified diene rubber composition comprising the modified diene rubber according to claim 1 or 2 and silica.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012027339A JP5998505B2 (en) | 2011-02-14 | 2012-02-10 | Method for producing modified diene rubber and method for producing rubber composition |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011028590 | 2011-02-14 | ||
| JP2011028590 | 2011-02-14 | ||
| JP2012027339A JP5998505B2 (en) | 2011-02-14 | 2012-02-10 | Method for producing modified diene rubber and method for producing rubber composition |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012184408A true JP2012184408A (en) | 2012-09-27 |
| JP5998505B2 JP5998505B2 (en) | 2016-09-28 |
Family
ID=47014744
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012027339A Expired - Fee Related JP5998505B2 (en) | 2011-02-14 | 2012-02-10 | Method for producing modified diene rubber and method for producing rubber composition |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5998505B2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017154405A1 (en) * | 2016-03-10 | 2017-09-14 | 信越化学工業株式会社 | Organosilicon compound and production process therefor |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004359773A (en) * | 2003-06-03 | 2004-12-24 | Sumitomo Rubber Ind Ltd | Rubber composition for tread, and pneumatic tire obtained using the same |
| JP2005041960A (en) * | 2003-07-25 | 2005-02-17 | Sumitomo Rubber Ind Ltd | Rubber composition for tire tread and pneumatic tire using the same |
| JP2011006548A (en) * | 2009-06-24 | 2011-01-13 | Bridgestone Corp | Polymer composition, rubber composition, and tire using the rubber composition |
| JP2011012161A (en) * | 2009-07-01 | 2011-01-20 | Sumitomo Rubber Ind Ltd | Rubber composition for tire and pneumatic tire |
-
2012
- 2012-02-10 JP JP2012027339A patent/JP5998505B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004359773A (en) * | 2003-06-03 | 2004-12-24 | Sumitomo Rubber Ind Ltd | Rubber composition for tread, and pneumatic tire obtained using the same |
| JP2005041960A (en) * | 2003-07-25 | 2005-02-17 | Sumitomo Rubber Ind Ltd | Rubber composition for tire tread and pneumatic tire using the same |
| JP2011006548A (en) * | 2009-06-24 | 2011-01-13 | Bridgestone Corp | Polymer composition, rubber composition, and tire using the rubber composition |
| JP2011012161A (en) * | 2009-07-01 | 2011-01-20 | Sumitomo Rubber Ind Ltd | Rubber composition for tire and pneumatic tire |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017154405A1 (en) * | 2016-03-10 | 2017-09-14 | 信越化学工業株式会社 | Organosilicon compound and production process therefor |
| US10323048B2 (en) | 2016-03-10 | 2019-06-18 | Shin-Etsu Chemical Co., Ltd. | Organosilicon compound and production process therefor |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5998505B2 (en) | 2016-09-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4308292B2 (en) | Rubber composition for inner liner and tire having inner liner comprising the same | |
| KR101695039B1 (en) | Natural rubber and manufacturing method thereof, rubber composition and pneumatic tire utilizing the same, modified natural rubber and manufacturing method thereof, and rubber composite for covering treads or carcass cords and pneumatic tire utilizing the same | |
| JP5924562B1 (en) | Modified rubber, tire rubber composition and tire using the same | |
| JP5904430B1 (en) | Modified rubber and method for producing the same, rubber composition, and tire | |
| JP2012012457A (en) | Method for using s-(3-aminopropyl)thiosulfuric acid and/or metal salt thereof, and method for suppressing generation of heat in vulcanized rubber composition | |
| JP2011079978A (en) | Modified diene rubber and rubber composition | |
| JP2011057922A (en) | Tire rubber composition and pneumatic tire | |
| JP5598365B2 (en) | Process for producing modified diene rubber | |
| JP5488797B2 (en) | Process for producing epoxidized diene rubber and rubber composition | |
| JP6958013B2 (en) | Method for manufacturing rubber composition | |
| JP5998505B2 (en) | Method for producing modified diene rubber and method for producing rubber composition | |
| JP5920107B2 (en) | Modified diene rubber composition and process for producing the same | |
| EP3842486A1 (en) | Rubber composition and tire | |
| JP5962121B2 (en) | Modified diene rubber, process for producing the same and rubber composition using the same | |
| JP2018035326A (en) | Rubber composition | |
| JP5541125B2 (en) | Process for producing epoxidized diene rubber and rubber composition containing epoxidized diene rubber | |
| JP5682273B2 (en) | Modified homopolymer diene rubber, process for producing the same and rubber composition using the same | |
| JP2013177520A (en) | Rubber composition for tire and pneumatic tire using the composition | |
| JP5912934B2 (en) | Rubber composition for tire and pneumatic tire | |
| JP5895517B2 (en) | Rubber composition for tire tread and pneumatic tire | |
| JP2009263446A (en) | Rubber composition for tire | |
| JP2014201651A (en) | Modified diene-based rubber, method for producing the same and rubber composition using the same | |
| JP5898007B2 (en) | Rubber composition for bead apex and pneumatic tire | |
| JP6497115B2 (en) | Rubber composition for tire | |
| JP2021195522A (en) | Rubber composition and tire |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20141226 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20151216 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160105 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160303 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20160802 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160815 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5998505 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |