JP2009519945A - 非天然アミノ酸およびそれらのニューロテンシンアナログ - Google Patents
非天然アミノ酸およびそれらのニューロテンシンアナログ Download PDFInfo
- Publication number
- JP2009519945A JP2009519945A JP2008545836A JP2008545836A JP2009519945A JP 2009519945 A JP2009519945 A JP 2009519945A JP 2008545836 A JP2008545836 A JP 2008545836A JP 2008545836 A JP2008545836 A JP 2008545836A JP 2009519945 A JP2009519945 A JP 2009519945A
- Authority
- JP
- Japan
- Prior art keywords
- peptide
- group
- peptides
- compound
- amino acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CC=*CCC=C(*)N Chemical compound CC=*CCC=C(*)N 0.000 description 4
- ONJCPSHPCDMDIU-UHFFFAOYSA-N NCCCCC(C(O)=O)=N Chemical compound NCCCCC(C(O)=O)=N ONJCPSHPCDMDIU-UHFFFAOYSA-N 0.000 description 1
Images



































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C279/00—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C279/04—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of guanidine groups bound to acyclic carbon atoms of a carbon skeleton
- C07C279/14—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of guanidine groups bound to acyclic carbon atoms of a carbon skeleton being further substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/28—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/44—Nitrogen atoms not forming part of a nitro radical
- C07D233/46—Nitrogen atoms not forming part of a nitro radical with only hydrogen atoms attached to said nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/04—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C229/26—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having more than one amino group bound to the carbon skeleton, e.g. lysine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/20—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D233/24—Radicals substituted by nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/28—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/44—Nitrogen atoms not forming part of a nitro radical
- C07D233/48—Nitrogen atoms not forming part of a nitro radical with acyclic hydrocarbon or substituted acyclic hydrocarbon radicals, attached to said nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/06—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D239/08—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms directly attached in position 2
- C07D239/12—Nitrogen atoms not forming part of a nitro radical
- C07D239/14—Nitrogen atoms not forming part of a nitro radical with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached to said nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Addiction (AREA)
- Psychiatry (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Peptides Or Proteins (AREA)
- Endocrinology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Cosmetics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
たい)。NTの完全な活性はそのC末端の6アミノ酸配列(NH2−Arg(8)−Arg(9)−Pro(10)−Try(11)−Ile(12)−Leu(13)−COOH、NT[8−13]と呼称される、非特許文献4を参照されたい)に存するとは言え、該C末端の6アミノ酸配列は、血液中でのその不安定性、ならびに血液脳関門および/若しくは腸関門を横断することの不能により、IP若しくは経口で投与される場合に活性を有しない。
Can.J.Biochem.1978、56、315 Int.J.Peptide Protein Res.1977、10、240 CarrawayとLeeman、J.of Biol.Chem.248:6854(1973) CarrawayとLeeman、J.Biol.Chem.250:1907(1975)
本発明は、正に荷電した側鎖を保有することが可能であるα−デスアミノアミノ酸化合物(デスアミノアミノ酸化合物)、それらの合成、生物学的に活性のペプチドの天然のアミノ酸部分の代替物としてのそれらの応用、および、同様に、生じるペプチドに関する。とりわけ、α−デスアミノアルギニン、リシンおよびオルニチン、ならびにそれらの置換および誘導体化した側鎖アナログが本発明の好ましい態様を構成する。これらのデスアミノアミノ酸化合物は、置換されたペプチドが置換位置で切断されることができるような、いずれかの既知の生物学的に活性のペプチドのアルギニンおよび/若しくはリシン部分の代わりに用いることができる。あるいは、これらのデスアミノアミノ酸化合物は、いずれかの既知の生物学的に活性のペプチドのN末端のアミノ基にカップリングして伸長されたペプチドを生じ得る。切断型および伸長型ペプチドは、アミノペプチダーゼ分解に対するそれらの抵抗性のおかげで、有意の生物学的選択性および生物学的半減期を有する。

nは0から5まで、好ましくは2ないし5の整数であり;
mは0若しくは1の整数であり;
RはH、あるいは、C1−C6の直鎖若しくは分枝状鎖アルキル基、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基のような有機置換基であり;
R1、R2およびR3は、独立して、水素、あるいは、C1−C6の分枝状若しくは直鎖
アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、かつ、但し、R1、R2およびR3の最大2個が、芳香族、置換芳香族、ヘテロ芳香族若しくは置換ヘテロ芳香族基であるように選択されることができ、また、但し、mが0若しくは1でありかつnが0ないし5である場合は、R1、R2およびR3が全部Hではなく;
Cαは、Rの置換基が有機置換基である場合にR若しくはSいずれかの立体化学を有する炭素原子であり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せ
を有する非天然のデスアミノアミノ酸化合物に関する。
nは0から6まで、好ましくは2ないし5の整数であり;
破線aが存在しない場合、XおよびYは独立して水素、またはC1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニルであり;
破線aが存在する場合、X−Yは(CH2)Zであり、式中Zは1−8から、好ましくは2ないし4の整数であり;
Rは、H、あるいは、C1−C6の直鎖若しくは分枝状鎖アルキル基、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基のような有機置換基であり;
R4は、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、ならびに;
Cαは炭素原子であり、かつ、Rの置換基が有機置換基である場合、Cαの立体化学はR若しくはSいずれかであり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せ
の非天然のデスアミノアミノ酸化合物に関する。
nは0から5まで、好ましくは2ないし5の整数であり;
X−Yは(CH2)Zであり、式中zは0から6まで、好ましくは2ないし4の整数であり;
RはH、あるいは、C1−C6の直鎖若しくは分枝状鎖アルキル基、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基のような有機置換基であり;
R6およびR7は、独立して、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり;ならびに
Cαは炭素原子であり、かつ、Rの置換基が有機置換基である場合、Cαの立体化学はR若しくはSいずれかであり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せ
の非天然のデスアミノアミノ酸化合物に関する。
nは0から5まで、好ましくは2ないし4の整数であり;
RはH、あるいは、C1−C6の直鎖若しくは分枝状鎖アルキル基、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基のような有機置換基であり;
R9、R10およびR11は、独立して、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、かつ、但し、R9、R10およびR11の最大2個が、芳香族、置換芳香族、ヘテロ芳香族若しくは置換ヘテロ芳香族基であるように選択されることができ;ならびに
Cαは炭素原子であり、かつ、Rの置換基が有機置換基である場合、Cαの立体化学はR若しくはSいずれかであり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せ
の非天然のデスアミノアミノ酸化合物に関する。
nは0から5まで、好ましくは2ないし4の整数であり;
Rは、H、あるいは、C1−C6の直鎖若しくは分枝状鎖アルキル基、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基のような有機置換基であり;
R12、R13およびR14は、独立して、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、かつ、但し、R12、R13およびR14の最大2個が、芳香族、置換芳香族、ヘテロ芳香族若しくは置換ヘテロ芳香族基であるように選択されることができ;ならびに
Cαは炭素原子であり、かつ、Rの置換基が有機置換基である場合、Cαの立体化学はR若しくはSいずれかであり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せ
の非天然のデスアミノアミノ酸化合物に関する。
本発明の定義
本明細および付随する請求の範囲で使用されるところの単数形「ある」(「a」、「an」)および「該(the)」は、文脈が別の方法で明瞭に指図しない限り、複数の指示物を包含する。
ペプチドである。切断型ニューロテンシン(8−13)を生じるためのAA7でのその切断は選択的生物学的活性を有するペプチドを提供する。本発明により、AA8アルギニンのデスアミノアミノ酸部分への転化は、有意かつ選択的な生物学的活性もまた有するペプチドをもたらす。NTおよび転化されたバージョンの例を図1に示す。
R1、R2およびR3が、独立して、水素、またはC1−C5の低級分枝状若しくは直鎖アルキル、より好ましくは水素若しくはメチルであるが、但し、mが0若しくは1でありかつnが0ないし5である場合に、R1、R2およびR3は全部がHではないものを包含する。別の態様において、nは4である。なおさらなる一態様において、RはH、メチル、エチル若しくはプロピルである。付加的な好ましい態様は、RがH、メチル、エチル、プロピル若しくはブチルであり、ならびに
a)nが4であり、mが0であり、R1が水素であり、R2がメチルであり、式Iの化合物が酸であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
b)nが4であり、mが1であり、R1およびR2がメチルであり、R3が水素若しくはメチルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
c)nが4であり、mが1であり、R1がメチルであり、R2およびR3が水素であり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
d)nが4であり、mが1であり、R1、R2およびR3が水素であり、式Iの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
e)nが3であり、mが0であり、R1およびR2がメチルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
f)nが3であり、mが0であり、R1およびR2がエチルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
g)nが3であり、mが0であり、R1およびR2がプロピルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
h)nが3であり、mが0であり、R1およびR2がブチルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
i)nが2であり、mが0であり、R1およびR2がメチルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場
合にR若しくはSである;
j)nが2であり、mが0であり、R1およびR2がエチルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
k)nが2であり、mが0であり、R1およびR2がプロピルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
l)nが2であり、mが0であり、R1およびR2がブチルであり、式Iの化合物が酸であり、Cαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである
ものを包含する。
a)nが3であり、破線aが存在せず、式IIの化合物が酸であり、R4が水素であり、Xが水素であり、Yがメチルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
b)nが3であり、破線aが存在せず、式IIの化合物が酸であり、R4がメチルであり、Xが水素であり、Yがメチルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
c)nが3であり、破線aが存在し、式IIの化合物が酸であり、zが2であり、R4が水素であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
d)nが3であり、破線aが存在し、式IIの化合物が酸であり、zが2であり、R4がメチルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
e)nが3であり、破線aが存在せず、式IIの化合物が酸であり、R4が水素であり、Xがメチルであり、Yが水素であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
f)nが3であり、破線aが存在せず、式IIの化合物が酸であり、R4が水素であり、Xがエチルであり、Yが水素であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
g)nが2であり、破線aが存在せず、式IIの化合物が酸であり、R4が水素であり、Xが水素であり、Yがメチルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
h)nが2であり、破線aが存在せず、式IIの化合物が酸であり、R4がメチルであり、Xが水素であり、Yがプロピルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
i)nが4であり、破線aが存在し、式IIの化合物が酸であり、zが2であり、R4が水素であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブ
チルである場合にR若しくはSである;
j)nが3であり、破線aが存在し、式IIの化合物が酸であり、zが2であり、R4がメチルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
k)nが2であり、破線aが存在し、式IIの化合物が酸であり、zが3であり、R4がメチルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
l)nが3であり、破線aが存在せず、式IIの化合物が酸であり、R4がメチルであり、Xが水素であり、Yがエチルであり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
ものを包含する。
a)nが3であり、zが2であり、R6およびR7が水素であり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
b)nが3であり、zが3であり、R6およびR7が水素であり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
c)nが2であり、zが2であり、R6およびR7が水素であり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
d)nが4であり、zが2であり、R6およびR7が水素であり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
e)nが2であり、zが3であり、R6およびR7が水素であり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
f)nが4であり、zが3であり、R6およびR7が水素であり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
g)nが2であり、zが2であり、R6およびR7がメチルであり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
h)nが4であり、zが2であり、R6およびR7がメチルであり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
i)nが2であり、zが3であり、R6およびR7がメチルであり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
j)nが4であり、zが3であり、R6およびR7がメチルであり、式IIIの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである
ものを包含する。
a)nが3であり、R9およびR11が水素であり、R10がメチルであり、式IVの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
b)nが3であり、R9が水素であり、R10およびR11がメチルであり、式IVの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
c)nが3であり、R9が水素であり、R10がメチルであり、R11がエチルであり、式IVの化合物が酸であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
d)nが2であり、R9およびR11が水素であり、R10がメチルであり、式IVの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
e)nが2であり、R9が水素であり、R10およびR11がメチルであり、式IVの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
f)nが4であり、R9が水素であり、R10がメチルであり、R11がエチルであり、式IVの化合物が酸であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである
ものを包含する。
a)nが3であり、R12およびR13が水素であり、R14がメチルであり、式Vの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
b)nが3であり、R12がメチルであり、R13およびR14がメチルであり、式Vの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
c)nが3であり、R12がメチルであり、R13がメチルであり、R14がエチルであり、式Vの化合物が酸であり、およびCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
d)nが2であり、R12がメチルであり、R13が水素であり、およびR14がメチルであり、式Vの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
e)nが2であり、R12がメチルであり、R13およびR14がメチルであり、式Vの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである;
f)nが4であり、R12がメチルであり、R13がメチルであり、およびR14がエチルであり、式Vの化合物が酸であり、ならびにCαの立体化学はRの置換基がメチル、エチル、プロピル若しくはブチルである場合にR若しくはSである
ものを包含する。
本明細書で使用されるところの化合物指示子ABS201、ABS48、KH48およびペプチド28は、別の方法で示されない限り同一化合物を表す。
本発明のデスアミノアミノ酸化合物の製造法は図3に描かれる全体的合成スキームに従う。この方法の第一段階は、式IからVのnに対応するメチレン単位鎖長を有するωハロゲンカルボン酸の製造である。以下の論考および図3において、この中間体は化合物27と称される。化合物27の製造後に、そのω−ハロ基は過剰の求核剤で容易に置換されて式I〜Vのデスアミノアミノ酸化合物を生じ得る。
ならびに適切なアルキル化剤および置換剤は、“Advanced Organic Chemistry”第4版、J.March、Wiley InterScience、ニューヨーク州ニューヨーク 1992(その開示全体は引用することにより本明細書に組み込まれる)に示される教示に従う。
本発明は、それらのN末端部分として式I、II、III、IV若しくはVの化合物の残基を含有する切断型および伸長型ペプチドを包含する。これらのペプチドは、当業者にとって確立されたペプチド製造方法であるMerrifield固相法により合成し得る。Merrifield固相ペプチド合成の説明およびその条件については、R.B.Merrifield、Science、232、341−347(1986)(その開示は引用することにより本明細書に組み込まれる)を参照されたい。あるいは、N末端アミノ酸単位を除くペプチドすなわち最後から2番目のペプチドを、既知の生物学的方法により組換え発現し得、そして、式I〜Vのデスアミノアミノ酸化合物を、アミノペプチダーゼを使用する酵素的縮合によりN末端として付加し得る。ペプチドの組換え発現の説明およびその条件については、“Enzyme Structure and Mechanism”、Alan Fersht、W.H.Freeman、ニューヨーク州ニューヨーク(1985)(その開示は引用することにより本明細書に組み込まれる)を参照されたい。式I〜Vの化合物は、側鎖アミノ基で標準的保護基で適切に保護し得る。好ましい一態様において、保護基はBOCおよび/若しくはFMOCである。
は、最後から2番目のペプチドを発現するための微生物の再工作を必要とする。最後から2番目のペプチド配列をコードするDNAセグメントを、プラスミド、若しくはDNAの微生物発現を引き起こすことが可能な他のベクターに適正な読み枠で挿入し得る。ベクターはまた適切な制御、プロモーターおよび選択DNAセグメントも含有することができる。大腸菌(E.coli)若しくは枯草菌(B.subtilus)のような微生物への挿入に際して、対応する選択剤での処理により、適切なトランスフェクションについて微生物の混合物を選択し得る。典型的には、該剤は抗生物質であることができ、また、ベクターは該抗生物質の対応する解毒酵素をコードする配列を含有することができる。クロラムフェニコールおよびペニシリンはこうした剤の2種である。トランスフェクトした微生物を培養すること、および、発現されたペプチドを、培地の分泌された物質として若しくは微生物細胞を溶解することによるかのいずれかで収集することが、最終製品から2番目前の生原料ペプチドを提供することができる。最終製品から2番目前のペプチドは凍結乾燥、クロマトグラフィーなどのような既知技術により精製しうる。ペプチド発現のためのこれらの組換え技術は、“Cold Spring Harbor−Current Protocols in Molecular Biology”、Wiley Interscience、コールドスプリングハーバー(2003)(その開示は引用することにより本明細書に組み込まれる)に完全に示されている。
該ペプチドの最後から2番目の配列すなわちNT(9−13)は、p−アルコキシベンジルアルコールの固相の方法論(65)を使用して大量で合成し得、そして完全に保護された形態で保存し得る。
Chemtech(ケンタッキー州ルイビル)から購入した。PyBOP(R)はNovabiochem(カリフォルニア州サンディエゴ)から購入した。N−ヒドロキシベンゾリアゾール(N−hydroxybenzoriazole)(HOBt)、無水N,N−ジメチルホルムアミド(DMF)、N,N−ジイソプロピルエチルアミン(DIPEA)、トリイソプロピルシラン(TIS)およびトリフルオロ酢酸(TFA)はAldrich(ウィスコンシン州ミルウォーキー)から購入した。非天然アミノ酸アナログは製造したとおり使用した。略語。Fmoc、フルオレニルメトキシカルボニル;NH3、アンモニア;NH2CH3、メチルアミン;NH(CH3)2、ジメチルアミン;N(CH3)3、トリメチルアミン;EtOH、エタノール。
逆相高速液体クロマトグラフィーを使用して前述の粗ペプチドを精製し得る。例えば、Waters C18ラジアル圧縮カラムと組合せのWatersデュアルポンプ系をこの目的上使用し得る。流出液を280nmのUV吸光度によりモニターし得る。
本発明は、活性若しくは薬理学的活性についてのペプチドのスクリーニング方法を提供する。該方法は:a)選択された天然のアミノ酸配列を有するペプチドの活性若しくは薬理学的活性を測定する段階、およびb)N末端が上述された式I〜Vを有する非天然アミノ酸である、前述のペプチドと同一のアミノ酸配列に基づき伸長若しくは切断されたペプチドの同一の活性若しくは薬理学的活性を測定する段階;ならびにc)段階a)およびb)からのペプチドの測定された活性若しくは薬理学的活性を比較して、段階b)のペプチドが所望の活性若しくは薬理学的活性を有するかどうかを決定する段階を包含する。
1.受容体アゴニスト/アンタゴニスト活性:これらの活性を測定するための特異的スクリーニングの例の一覧表は、“The RBI Handbook of Receptor Classification and Signal Transduction”K.J.Watling、J.W.Kebebian、J.L.Neumeyer編
Research Biochemicals International,マサチューセッツ州ナティック、1995、およびその中の参考文献に見出し得る。分析方法は、T.Kenakin“Pharmacologic Analysis of Drug−Receptor Interactions”第2版 Raven Press、ニューヨーク、1993、およびその中の参考文献に見出し得る。
2.酵素阻害:これらの活性を測定するための特異的スクリーニングの例の一覧表は、H.Zollner“Handbook of Enzyme Inhibitors”、第2版 VCH Weinheim、FRG、1989およびその中の参考文献に見出し得る。
3.中枢神経系、自律神経系(心血管系および胃腸管)、抗ヒスタミン、抗炎症、麻酔、細胞傷害性および避妊活性:これらの活性を測定するための特異的スクリーニングの例の一覧表は、E.B.Thompson、“Drug Bioscreening:Drug Evaluation Techniques in Pharmacology”、VCH Publishers、ニューヨーク、1990、およびその中の参考文献に見出し得る。
4.抗癌活性:これらの活性を測定するための特異的スクリーニングの例の一覧表は、I.J.FidlerとR.J.White“Design of Models for Testing Cancer Therapeutic Agents”、Van Nostrand Reinhold Company、ニューヨーク、1982およびその中の参考文献に見出し得る。
5.抗生物質および抗ウイルス(とりわけ抗HIV)活性:これらの活性を測定するための特異的スクリーニングの例の一覧表は、“antibiotics in Labor
atory Medicine”、第3版、V.Lorian編 Williams and Wilkens、ボルチモア、1991、およびその中の参考文献に見出し得る。これらの活性を測定するための抗HIVスクリーニングの一覧表は、“HIV Volume 2:Biochemistry,Molecular Biology and Drug Discovery”、J.Karn編、IRL Press、オックスフォード、1995およびその中の参考文献に見出し得る。
6.免疫調節活性:これらの活性を測定するための特異的スクリーニングの例の一覧表は、V.St.Georgiev(1990)“Immunomodulatory Activity of Small Peptides”Trends Pharm.Sci.11、373−378に見出し得る。
7.薬物動態特性:該スクリーニング方法でアッセイされる薬理学的活性は、とりわけ半減期、溶解性若しくは安定性を包含する。例えば、薬物動態特性の分析および測定方法は、J.−P.Labaune“Handbook of Pharmacokinetics:Toxocity Assessment of Chemicals”、Ellis Horwood Ltd.、チチェスター、1989およびその中の参考文献に見出し得る。
本発明はさらに、式I〜Vを有するアミノ酸をそのN末端として有する伸長若しくは切断されたペプチドを被験体に投与することを含んでなる、異常状態の被験体における処置若しくは予防方法に関する。伸長若しくは切断されたペプチドがそれから形成される基礎ペプチドは、処置若しくは予防されるべき異常状態と生化学的、生理学的、薬理学的若しくは生物学的関係を有することができるか、若しくは有すると考えることができる。異常状態は、疾患、生物学的すなわち器質の機能異常、または限定されるものでないが皮膚発疹、ざ瘡などのような化粧上の異常状態を挙げることができる、通常は疾患若しくは機能異常とみなされない望ましくない生物学的状態でありうる。被験体は、ヒトのような哺乳動物、およびイヌ、ネコ、ウシ、ヒツジ、ブタのようなヒト以外の哺乳動物ならびに鳥類を包含する、医学的若しくは獣医学的患者でありうる。
安、緑内障、ヒト免疫不全ウイルス(HIV)若しくは後天的免疫不全症候群(AIDS)、神経変性(例えばアルツハイマー病およびパーキンソン病)、認知増強、クッシング症候群、アジソン病、骨粗鬆症、虚弱(frailty)、炎症性疾患(変形性関節症、関節リウマチ、喘息および鼻炎のような)、副腎機能障害、ウイルス感染症、免疫不全、免疫調節、自己免疫疾患、アレルギー、創傷治癒、強迫性行動、多剤耐性、嗜癖、精神病、食思不振、悪液質、心的外傷後ストレス症候群、術後骨折、医学的異化作用、ならびに筋虚弱の予防を処置するのに使用しうる。
and therapeutic use.Drugs.48(5):667−77、1994 Nov.で言及される)およびサイトカイン(Peters,M.;Actions of cytokines on the immune response and viral interactions:an overview.Hepatology.23(4):909−16、1996 Apr.で言及される)である。
o−Argおよび命名された他者の例は、Ojima I.;Chakravarty S.;Dong Q.Antithrombotic agents:from RGD
to peptide mimetics.Bioorganic & Medicinal Chemistry.3(4):337−60、1995に記述されている。
Pharmacological & Toxicological Methods.34(3):125−32、1995に記述されている。
して、またはアレルギー性呼吸器疾患、喘息およびアレルギー性鼻炎を処置するのに使用し得る。
は、Barker,J.N.;Adhesion molecules in cutaneous inflammation.Ciba Foundation Symposium.189:91−101に記述されている。
る。これらのペプチドの例は、Bab IA.Regulatory role of osteogenic growth peptide in proliferation,osteogenesis,and hemopoiesis.Clinical
Orthopaedics & Related Research.(313):64−8、1995に記述されている。
るのに使用し得る。
免疫系の機能障害、カルシウム恒常性および骨粗鬆症を処置するのに使用し得る。
K.Wada E.Wada K.Bombesin−like peptides:studies on food intake and social behaviour with receptor knock−out mice.Annals of Medicine.32(8):519−29、2000 Nov.;Ohki−Hamazaki H.Neuromedin B.Progress in Neurobiology.62(3):297−312、2000 Oct.;Still CD.Future trends in weight management.Jou
rnal of the American Osteophathic Association.99(10 Su Pt 2):S18−9、1999;Martinez
V.Tache Y.Bombesin and the brain−gut axis.Peptides.21(11):1617−25、2000;Afferent
signals regulating food intake.Proceedings of the Nutrition Society.59(3):373−84、2000;Takenaka Y.Nakamura F.Jinsmaa Y.Lipkowski AW.Yoshikawa M.Enterostatin(VPDPR)has anti−analgesic and anti−amnesic activities.Bioscience Biotechnology & Biochemistry.65(1):236−8、2001 Jのような例示的参考文献に記述されている。
potassium channel openers.Cardiovascular Drugs & Therapy.9 Suppl 2:185−93、1995 Mar.に記述されている。
ligands at orphan G protein−coupled receptor s.Receptor s & Channels.8(5−6):297−308、2002;Shiau AK.Coward P.Schwarz M.Lehmann JM.Orphan nuclear receptor s:from new ligand discovery technologies to nov
el signaling pathways.Current Opinion in
Drug Discovery & Development.4(5):575−90、2001;Civelli O.Nothacker HP.Saito Y.Wang Z.Lin SH.Reinscheid RK.Novel neurotransmitters as natural ligands of orphan G−protein−coupled receptor s.Trends in Neurosciences.24(4):230−7、2001;Darland T.Heinricher MM.Grandy DK.Orphan in FQ/nociceptin:a role in pain and analgesia,but so much more.Trends in Neurosciences.21(5):215−21、1998(それらの開示は引用することにより本明細書に組み込まれる)のような参考文献に記述されている。
in acute coronary syndromes.JAMA.284(12):1549−58、2000;Kereiakes DJ.Oral blockade of the platelet glycoprotein IIb/IIIa receptor:fact or fancy?.American Heart Journal.138(1 Pt 2):S39−46、1999;Bassand JP.Low−molecular−weight heparin and other antithrombotic agents in the setting of a fast−track revascularization in unstable coronary artery disease.Haemostasis.30 Suppl 2:114−21;discussion 106−7、2000を包含する。
本発明は、そのN末端として化合物式I〜Vの1残基を有する伸長若しくは切断されたペプチドの使用による、被験体の身体障壁を横断するペプチドの能力の増大方法に関する。
は血液眼を挙げることができる。好ましい一態様において、身体障壁は血液脳関門である。
本発明のある態様は、上述されたところの選ばれたペプチドの配列に基づく伸長若しくは切断されたペプチドの使用による、選ばれたペプチドの選択性の増大方法に関する。
本発明のペプチドは、対応する既知のペプチドが関連するいかなる疾患若しくは生理学的問題も処置するため、当業者に利用可能ないかなる治療手順でも使用し得る。
し750mg、最も便宜的には20ないし500mgのペプチドを含有する単位投薬形態物で便宜的に投与し得る。
メーキャップ化粧品の重要な一役割は「美化」すなわち外観をより美しくすることである。しばしば、その役割は、皮膚の粗さ、傷および色ならびに生気の補正を伴う。
油、マカダミアナッツ油、ホホバ油のような油および脂肪;イソステアリルアルコールのような高級アルコール;高級脂肪族油およびミリスチン酸イソプロピルのようなエステル油などを、本発明の外用組成物中に処方しうる。これらの油成分のなかで、とりわけ、本発明の外用組成物中で極性油を処方することは、時間の経過に伴う安定性の改良を可能にする。
ニューロテンシン(NT)は、1973年にCarrawayとLeemanにより降圧ペプチドとしてウシ視床下部から最初に単離された。それ以来、NTは中枢神経系(CNS)および末梢で多数の異なる生理学的効果を有することが示された。体温下降、抗侵害受容、d−アンフェタミン誘発性の自発運動の亢進の減弱、およびバルビツール酸誘発性鎮静の増強が、脳へのNTの直接注入により促進される。末梢では、NTは血圧低下を誘発しかつ胃酸分泌を低下させるホルモンとして作用する。構造的には、NTは以下の配列:pGlu−Leu−Tyr−Glu−Asn−Lys−Pro−Arg−Arg−Pro−Tyr−Ile−Leu−OHをもつ直鎖状トリデカペプチドである。NT研究の歴史の早期に、C末端ヘキサペプチドフラグメントArg8−Arg9−Pro10−Tyr11−Ile12−Leu13[NT(8−13)]が、NTの生理学的効果を生じることにおいてin vitroおよびin vivoで効力が等しかったことが示された。
NTR3)がヒト脳cDNAライブラリーからクローン化され、そして以前にクローン化されたgp95/ソーティリンに同一であることが見出された。NTR3は、ただ単一の膜貫通領域を有する非Gタンパク質共役型局在化タンパク質である。
いくつかの異なる系統の証拠が統合失調症の病理生理学にNTを関係させている。統合失調症のドーパミン理論の進歩が、中脳辺縁系ドーパミン系に対する多様な神経回路の集合の欠陥が該障害の発症の原因であることを裏付ける。NT系の解剖学的位置決めは、それが脳内のグルタミン作動性、ドーパミン作動性、GABA作動性およびセロトニン作動性の系と相互作用するようである。とりわけ、NTおよびドーパミン系は側坐核(妄想および幻覚の原因であると考えられる脳の領域)内で緊密に関係付けられる。NTR1受容体は腹側被蓋領域(上述された神経系と緊密に関連する脳領域)で密である。NT受容体のほぼ90%がドーパミン作動性ニューロンに位置し、また脳中のドーパミンニューロンの80%超がNTR1を発現する。統合失調症に関係している脳領域とのNT系の共局在もまたその関与を意味している。
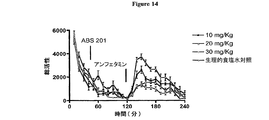
NTは「内因性神経遮断薬」として仮定され、また、NT(8−13)がその活性フラグメントと同定されて以来、NT(8−13)誘導体を潜在的抗精神病薬として開発するための努力がなされた。とりわけ2つのグループすなわちエーザイ株式会社(東京)およびRichelsonの研究グループ(Mayo Clinic、フロリダ州ジャクソンビル)が、抗精神病薬として有望性を示したNT(8−13)アナログの多数の誘導体を製造した。とりわけ、Arg8、Arg9、Tyr11およびIle12のアミノ酸置換が、末梢投与後に中枢性に活性である数種のアナログを生じた。
本発明により製造したNT(8−13)のN末端αメチル、αデスアミノホモリシルおよびオリンチルアナログ(前述の全般的論考および実施例を参照されたい)を合成し、そして抗精神病能力を予測する多数の行動アッセイで活性についてスクリーニングした。これらのペプチドは経口投与後に用量依存性の様式で体温下降を誘発した。加えて、該ペプチドの経口投与は、d−アンフェタミン誘発性の自発運動の亢進を有意に低下させた(現在の若しくは潜在的APDの治療的有効性の一尺度)。これらのアッセイで経口投与後に有意の応答を導き出すペプチドの低用量(10mg/kg)は有意である。該ペプチドは
また、反復投与後に有効性を維持する能力も示す。事実、それらは、長期にわたる最大体温下降応答を増大させる能力を示し、反復投与がそれらのCNS活動を実際に改善しうることを意味している。従って、本発明のNTペプチドは、既知の天然に存在するペプチドNTのもののような生物学的活性を有することが示され、かつ、より選択的である。これらの効果の詳細は後に続くとおりである。
CNS活性を間接的に測定するのに使用し得る。NTの体温下降効果は、NTR1(統合失調症の病理生理学で最もしばしば関係しているNTR)でのその作用に帰すことができる。IP注入後に体温下降を誘発したNT(8−13)ペプチドは従って抗精神病薬であることが示される。有意の体温下降効果は、該ペプチドが血液安定性および膜横断における顕著な改良を示したことを示すとみられる。
以下の実施例およびプロトコルは、本明細書で特許請求される化合物がどのように作成かつ評価されるかの完全な開示および記述を当業者に提供するように述べられ、そして、本発明の純粋に例示であることを意図しておりかつ発明者が彼らの発明であるとみなすものの範囲を制限することを意図していない。数(例えば量、温度など)に関する正確さを確実にするために努力がなされたが、しかし何らかの誤りおよび逸脱が説明されるべきである。別の方法で示されない限り、部分は重量部分であり、温度は℃ででありかつ室温でであり、ならびに圧力は大気圧若しくはそれ近くである。
αメチル、αデスアミノωN置換ホモリシルおよびオリンチル(8)ニューロテンシン(8−13)を合成した(図7)。α−メチルブロモ酸27aおよびcを、全般の節で概説されたとおり樹脂結合したペプチドにカップリングした。固相カップリングは後に続くとおり実施した。
Biosystems、カリフォルニア州フォスターシティ)でのMALDI−TOFMSを介して特徴付けしかつ純度について評価した。ペプチドは95%以上純度でin vivoで使用した。
化合物の構造。評価されるペプチドアナログは1個の非天然アミノ酸(スキーム1)若しくはデスアミノ酸(スキーム2)を含有する。
以前の最も活性の化合物を上回る経口活性の300%増大を有した。加えて、ABS201はIPに対して経口で投与される場合により迅速な応答を達成した。これはNT(8−13)誘導体の間で独特である。
全般的動物プロトコル。雄性Sprague Dawleyラット(250〜350g)若しくはBrattleboroラット(270〜310g)をHarlan(インジアナ州インジアナポリス)から得、そして一定温度および湿度で維持したAAALAC承認済居住室(colony room)に収容した。照明は12時間の明:暗周期で制御し、午前7時に照明を点灯した。動物はケージあたり2匹を収容し、そして実験室食および水を随意に供給した。全実験は明周期の間に実施した。
Instruments、カリフォルニア州サンディエゴ)の光ビームを1分あたり何回「切断する」かにより測定した。チャンバーは、別個の収容室とほぼ同一の周囲光および温度を伴う静寂な室中の4段のラックに配置した。実験は1日1回正午に6日間行った。各日に、6匹のナイーブなラットを各処置条件に割り当てた。用量は、異なるチャンバーの照明などの変動について制御する、装置全体に分配した12個のチャンバーでそれぞれが評価されるように割り当てた。
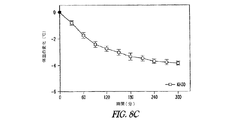
IPおよびPO投与によるABS201による体温下降誘発。ABS201は、IPで投与した場合に約1mg/kg、および経口で投与した場合に10mg/kgのED50を有する(体温下降データは示されない。下のアンフェタミン誘発性の自発運動の亢進の結果を参照されたい)。図9に見られるとおり、ABS201は、各化合物のおよそのED50の2倍で、経口で投与した場合にIPに対しより迅速な体温下降効果を実際に誘発する。
放射活性ABS201の生成のためのFmoc−プロリン−OH*の合成(図19)。L−プロリン(20.7mg、0.18mmol)(Advanced Chemtech)を450μLの10%Na2CO3溶液に溶解し、それに250μCiのL−[U−14C] egrada(Moravek、カリフォルニア州ブレア)を含有する5mLのEtOH:H2O(2:98)を添加した。3mLのジメトキシエタン(DME)中のFmoc−N−ヒドロキシスクシンイミド(Fmoc−Osu)(100mg、1.5等量)を攪拌するアミノ酸溶液に一滴ずつ添加した。反応を室温で12時間攪拌させ、そしてDMEを真空中で除去した。残存する水性溶液を10mLのH2Oで希釈し、そして飽和N−ブタノール(4×10mL)で抽出した。ブタノール抽出液を合わせかつ濃縮して青白色油状物を生じた。残余のFmoc−Osuを、MeOH:CH2Cl2(50:5
0)で溶離してシリカゲルで除去した。粗Fmoc−プロリン−OH*はさらなる精製を伴わずにペプチド合成で使用した。
受容体スクリーニング。3種の別個の濃度のABS201(10−9、10−7、10−5M)を、以下の16種の受容体、すなわち、アドレナリン(α1、α2、β)、ドーパミン、ヒスタミン(H1、H2、H3)、ムスカリン(中枢、末梢)、ニコチン、オピオイド(非選択的)、オルファニン、セロトニン(輸送体、非選択的)、モノアミンオキ
シダーゼ(A、B)に対し個別にスクリーニングした。受容体の内因性基質の置換は観察されなかった。これゆえに、ABS201はこれらの受容体のいずれとも結合すると思われない。対照的に、ABS201は標的受容体(NTR1)に対するnMの親和性を有する。
TK遺伝子座で突然変異を誘発するメタンスルホン酸ヒカントン(HYC)、および代謝活性化を伴いTK遺伝子座で突然変異を誘発する7,12−ジメチルベンズ[α]アントラセン(DMBA)を包含した。範囲発見試験を実施して0〜100%毒性を生じるABS201の濃度を同定した。陽性対照(HYCおよびDMBA)ならびに5濃度のABS201を、代謝活性化を伴うおよび伴うアッセイで使用した。細胞を各ABS用量に曝露し、そして上清を得かつ20および44時間インキュベートした。2日の発現期間後に、培養物を制限剤トリフルオロチミジン(TK−/−細胞のみの増殖を可能にする)若しくはベヒクル対照でクローン化した。突然変異の頻度および誘発された突然変異の頻度を計算した。最低1種の培養物が、対応する溶媒対照培養物の平均MFより2倍若しくはそれ以上大きかったMFを有しかつ該応答が用量依存性であった場合に、陽性の結果が得られた。変異アッセイの結果を確認するため、S−9活性化を伴わずに確認アッセイを実施した。
ents、カリフォルニア州)を使用して、ガラス製毛細管から作成した。商業的パッチクランプ増幅器を全細胞記録に使用した。デジタル化前に、電流記録をサンプリング頻度の1/5で低域フィルタリングした。
)であり、IVのABS201の急性効果は中枢神経系を介して媒介され、該化合物は血漿若しくは全血との共インキュベーションに際して代謝されるようでなく、ABS201はin vitroで血液の水性および細胞相の間で分配し;全血中のABS201のPKプロファイルは二相クリアランス過程と矛盾せず、そして観察された薬力学応答は同様に二相クリアランス過程と矛盾しない。
ArgおよびLysの一連のアミノ酸アナログを創製した。米国特許第6,043,218号;同第6,358,922号;同第6,566,330号;同第6,783,946号;同第6,858,396号明細書を参照されたい。NT(8−13)のArg8の代わりに使用される場合、抗精神病および鎮痛双方の活性を有する一連の化合物を生成した。これらの化合物の例は下の表10に見出し得る(ABS202、ABS203、ABS204、ABS206)。非天然側鎖に加え、N末端−NH2の代わりの−CH3の使用が双方の適応症に対し最も活性の化合物をもたらしたことが示された。2005年6月17日出願の第PCT/US2005/021580号、および2004年6月17日出願の米国仮出願第60/581,333号明細書を参照されたい。
3およびABS227で見られる。ABS228はABS224より良好な鎮痛活性を有し、これゆえにそれはNTR−2に対しより選択的である。ABS226は双方の潜在的適応症でより活性である。
動物。Charles River Laboratories、ノースカロライナ州ローリー(230〜260g)若しくはHarlan、アラバマ州プラットビル(240〜280g)からの実験的にナイーブな雄性Sprague−Dawleyラットを、実験前およそ1週間、任意の食餌および水を伴う規則的明/暗周期(点灯6:00〜18:00)で、動物居住区にケージあたり4匹で維持した。試験の2日の体重の範囲は230〜260グラムであった。実験は1日に行い、動物は実験を反復する前に3日間回復させた。
去までの時間を記録した。動物は10秒のカットオフ待ち時間で即座に取り除く。MPEを計算した(100%のMPEは10秒間湯浴中に留まるラットの尾に対応した)。これを3回反復した。試験の間に尾を乾燥しかつ20秒間休ませた。ラットにその後適切な量のペプチドを与え、そして実験を30および60分に反復した。処置条件は、暦時間および投与の順序のような因子について制御するように釣り合わせたデザインで試験全体に割り振った。全実験についてN=6若しくはそれ以上。
・Xf=適用した最後のvon Frey毛の値;
・k=応答パターンに基づく補正係数(較正表から)
・d=刺激間の平均距離(log単位で)]
を使用して計算する。
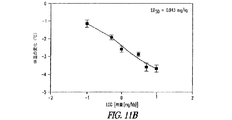
NTおよび誘導体の抗精神病および体温下降効果はNTR−1受容体活性化作用により媒介される一方、鎮痛効果はNTR−2受容体活性化作用により媒介される。実現可能な鎮痛薬であるために、NT誘導体は、in vitroでNTR−2に高親和性で結合しかつin vivoで経口で利用可能かつ安定であるべきである。ABS201はこれらの要件の全部を満たす。すなわち、表11に示されるところのマイクロモル濃度以下のNTR−2親和性延長された血漿安定性および経口生物学的利用性を有する。
201の有用性を示す。
ABS201が高親和性でNTR−1および−2双方を結合するという事実によって、NTR−1結合と関連する効果が該ペプチドを鎮痛薬としてより少なく望ましくしうる可能性が存在する。これらの効果は、ヒトで実際に非常に小さな効果である(D.Feifel、私信)ラットにおける全身体温下降の誘発、および望ましくなくないかもしれない抗精神病効力を包含する。ABS201の好都合な属性の全部を維持する、NTR−2に高度に選択的なペプチドは、「浄化体(cleaner)」鎮痛薬候補であるとみられる。
ABS201の同定の過程は、生物学的安定性および血液脳関門横断に必要な重要な構造パラメータの定義を可能にした。従って、IP若しくは経口生物学的利用性を高めうる構造要素を組み込む一群のニューロテンシン(NT)[8−13]誘導体を創製した。これらのパラメータ内で、治療薬としての潜在的開発に適切な多様な薬理学的および行動パラメータに影響を及ぼし得る他の専有の構造変化を組み込んだ。要するに、(1)それらの受容体に対する高められた結合親和性、例えば高められた結合および選択性(例えば、脳NTR−2に対する選択性をもつNT誘導体は鎮痛活性を有する一方、NTR−1に対する選択性をもつものは抗精神病活性を有する);(2)高められた生物学的障壁横断;(3)高められた安定性;ならびに(4)実行可能な合成費用に対する可能性をもつペプチドを同定した。
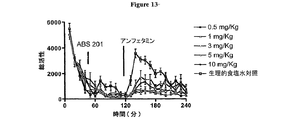
20mg/kgの用量で胃管栄養法により投与されたABS201、ABS205、ABS210、ABS212およびABS220の鎮痛効果を図28に示す。これらの結果は、ABS201、ABS205、ABS210およびABS220が経口で与えられる場合にABS212と同じくらい若しくはより活性であったことを示す。ホットプレートアッセイを使用して測定されるところのABS232およびABS239の鎮痛効果を図29AおよびBに具体的に説明する。
供する(図31A)。ホットプレートアッセイでもまた見られるとおり、ほぼ等モル濃度のモルヒネは30分後に活性を喪失する。ABS212は、しかしながら、I.P.注入により投与される場合により顕著な鎮痛効果を有する。図31BおよびCは、それぞれI.P.および経口投与についてのホルマリンアッセイを使用して測定された用量応答を示す。およそのIPおよび経口のED50は上で実施されたアッセイと同一範囲にある。鎮痛活性ABS212は、非常にわずかの体温下降(NTR−1結合の副次的効果)が治療的鎮痛用量でこのペプチドから導き出されるため、NTR−2に対し高度に選択的であるようである。(in vitro受容体結合研究は進行中である)。
ABS212の薬物動態を評価するため、血漿サンプルを、試験物品ABS212の静脈内および経口投与後の多様な時間点でSprague−Dawley(SD)ラットから収集した。これらのサンプルを、薬物動態パラメータおよび経口生物学的利用性を推定するためのLC/MS/MSによる血漿親薬物濃度の測定に使用した。ABS212のIVおよび経口投与の濃度対時間曲線を図34AおよびBに示す。
文書の以下の一覧は、背景情報、合成情報、科学的情報、プロトコルおよび関連する開示を提供する。各文書の完全な本文は、それが完全に反復されたかのように本出願の不可欠の部分として本明細書に組み込まれ、また、本明細書で引用される全部の刊行物、特許および特許出願は、引用することにより本明細書に組み込まれる。
Claims (33)
- 下記式I、II、III若しくはIVよりなる群から選択される式で表される非天然のデスアミノアルキルアミノ酸化物:
(a)式Iは
nは0から5までの整数であり;
mは0若しくは1の整数であり;
RはHであり;
R1、R2およびR3は、独立して、水素、あるいは、C1−C6の分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、かつ、但し、R1、R2およびR3の最大2個が、芳香族、置換芳香族、ヘテロ芳香族若しくは置換ヘテロ芳香族基であるように選択されることができ、また、但し、mが0若しくは1でありかつnが0ないし5である場合に、R1、R2およびR3が全部Hではなく;
そして、CαはR若しくはSいずれかの立体化学を有する炭素原子であり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せであり;
(b)式IIは
nは0から6までの整数であり;
破線aが存在しない場合、XおよびYは、独立して、水素、またはC1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニルであり;
破線aが存在する場合、X−Yは(CH2)Zであり、式中Zは1−8からの整数であり;
RはHであり;
R4は、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、そして;
Cαは炭素原子でありかつCαの立体化学はR若しくはSいずれかであり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せであり;
(c)式IIIは
nは0から5までの整数であり;
X−Yは(CH2)Zであり、式中zは0から6までの整数であり;
RはHであり;
R6およびR7は、独立して、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり;そして
Cαは炭素原子でありかつCαの立体化学はR若しくはSいずれかであり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せであり;
(d)式IVは、
nは0から5までの整数であり;
RはHであり;
R9、R10およびR11は、独立して、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、かつ、但し、R9、R10およびR11の最大2個が、芳香族、置換芳香族、ヘテロ芳香族若しくは置換ヘテロ芳香族基であるように選択されることができ;そして
Cαは炭素原子でありかつCαの立体化学はR若しくはSいずれかであり;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せであり;
さらに、
(e)式Vは:
nは0から5までの整数であり;
Rは、H、あるいは、C1−C6の直鎖若しくは分枝状鎖アルキル基、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基のような有機置換基であり;そして
R12、R13およびR14は、独立して、水素、あるいは、C1−C6の低級分枝状若しくは直鎖アルキル、アルケニル若しくはアルキニル、あるいはC6−C18の芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換芳香族基、あるいは、C4−C18ならびにいずれかの組合せの酸素、イオウおよび窒素から選択される1若しくは2個のヘテロ原子のヘテロ芳香族基、またはいずれかの組合せのハロゲン、アルキルオキシ、カルボキシ、アミド若しくはアルキルから選択される1若しくは2個の置換基をもつ対応する置換ヘテロ芳香族基であり、かつ、但し、R12、R13およびR14の最大2個が、芳香族、置換芳香族、ヘテロ芳香族若しくは置換ヘテロ芳香族基であるように選択されることができ;
あるいは、そのカルボン酸基のエステル、アミド、アルキルアミドまたは金属陽イオン若しくはアンモニウム塩、あるいはそのアミン基の有機若しくは無機酸塩、あるいはそれらのいずれかの組合せである。 - 化合物が式Vの構造を有する場合にはCαの立体化学がSである、請求項1に記載の化合物。
- R、R1、R2およびR3が、独立して、水素若しくはメチルである、請求項1に記載の化合物。
- (a)化合物が式Iのものである場合にはnが2から5までの整数であり;(b)化合物が式II若しくはIIIのものである場合には、nが2から5までの整数であり、かつ、Zが2から4までの整数であり;そして(c)化合物が式IVのものである場合には、nが2から4までの整数である、請求項1に記載の化合物。
- 側鎖アミノ基、側鎖カルボキシル基、若しくは側鎖アミノ基およびカルボキシル基双方が、アミノ基、カルボキシル基若しくは双方の基の望ましくない反応を予防しかつ他の基の切断もまた引き起こさない化学的方法により除去可能であるそれぞれの保護基により保護されている、請求項1、2、3若しくは4に記載の化合物。
- 保護基が、BOC(t−ブトキシカルボニル)、FMOC(フルオレニルメトキシカルボニル)、Alloc(アリルオキシカルボニル)、CBZ(ベンジルオキシカルボニル)、Pbf(2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル)、NO2(ニトロ)、Pmc(2,2,5,7,8−ペンタメチルクロマン−6−スルホニル)、Mtr(4−メトキシ−2,3,6−トリメチルベンゼンスルホニル)若しくはTos(トシル)である、請求項5に記載の化合物。
- 請求項1、2、3、4、5若しくは6に記載の非天然アミノ酸化合物を含んでなる半合成ペプチド。
- 非天然アミノ酸化合物が半合成ペプチドのN末端部分である、請求項7に記載の半合成ペプチド。
- 半合成ペプチドが、(i)ニューロテンシン(8−13)、(ii)転写因子、(iii)細胞受容体のリガンド、(iv)ホルモン、(v)細胞外結合ペプチド、(vi)オフェンケフリン(ofenkephlin)、(vii)LHRH若しくはそのアナログ、(viii)ニューロペプチド、(ix)グリコインクレチン、(x)インテグリン若しくはそのアナログ、(xi)グルカゴン、(xii)グルカゴン様ペプチド、(xiii)抗血栓ペプチド、(xiv)サイトカイン、(xv)インターロイキン、(xvi)トランスフェリン、(xvii)インターフェロン、(xviii)エンドセリン、(xix)ナトリウム利尿ホルモン、(xx)細胞外キナーゼリガンド、(xxi)アンジオテンシン酵素阻害剤、(xxii)ペプチド性抗ウイルス化合物、(xxiii)トロンビン、(xxiv)サブスタンスP、(xxv)サブスタンスG、(xxvi)ソマトトロピン、(xxvii)ソマトスタチン、(xxviii)GnRH若しくはそのアナログ、(xxix)セクレチン、(xxx)ブラジキニン、(xxxi)バソプレッシン若しくはそのアナログ、(xxxii)インスリン若しくはそのアナログ、(xxxiii)プロインスリン、または(xxxiv)成長因子の半合成ペプチドである、請求項8に記載の半合成ペプチド。
- ABS205、ABS207、ABS208、ABS210、ABS211、ABS212、ABS220、ABS225、ABS226、ABS227、ABS228、ABS230、ABS232、ABS234若しくはABS239である、請求項9に記載の半合成ペプチド。
- 半合成ペプチドが、該半合成ペプチドと同一のアミノ酸配列を有するがしかしそのN末端部分として置換された非天然アミノ酸化合物を有しないペプチドと比較した場合に、in vivoおよび/若しくはin vitroで延長された半減期を有する、請求項7、8、9若しくは10に記載の半合成ペプチド。
- 請求項7、8、9、10若しくは11に記載のペプチドおよび製薬学的担体を含んでなる製薬学的組成物。
- ペプチドが単位投薬形態物にある、請求項21に記載の製薬学的組成物。
- 化粧品用基剤製剤、および(a)請求項1、2、3、4、5若しくは6に記載のデスアミノアルキルアミノ酸化合物;または(b)請求項7、8、9、10若しくは11に記載のペプチドを含んでなる化粧品用製剤。
- 化粧品用基剤製剤が水性若しくは油性基剤である、請求項14に記載の化粧品用製剤。
- 医学的治療での使用のための、請求項1、2、3、4、5若しくは6に記載の化合物、または請求項7、8、9、10若しくは11に記載のペプチド。
- 哺乳動物における精神病、疼痛、癌、肥満、糖尿病若しくは覚醒剤濫用を処置するのに有用な医薬品の製造のための、請求項1、2、3、4、5若しくは6に記載の化合物または請求項7、8、9、10若しくは11に記載のペプチドの使用。
- 精神病が統合失調症である、請求項17に記載の使用。
- 患者の体温を低下させるように、(a)有効量の請求項7、8、9、10若しくは11に記載のペプチド、または(b)有効量の請求項12若しくは13に記載の組成物を患者に投与することを含んでなる、患者の体温の低下方法。
- 癌を処置するように、(a)有効量の請求項7、8、9、10若しくは11に記載のペプチド、または(b)有効量の請求項12若しくは13に記載の組成物を患者に投与することを含んでなる、癌の処置方法。
- 疼痛を処置するように、(a)有効量の請求項7、8、9、10若しくは11に記載のペプチド、または(b)有効量の請求項12若しくは13に記載の組成物を患者に投与することを含んでなる、疼痛の処置方法。
- 疼痛が神経因性疼痛である、請求項21に記載の方法。
- 精神病を処置するように、(a)有効量の請求項7、8、9、10若しくは11に記載のペプチド、または(b)有効量の請求項12若しくは13に記載の組成物を患者に投与することを含んでなる、精神病を伴う患者の処置方法。。
- 肥満を処置するように、(a)有効量の請求項7、8、9、10若しくは11に記載のペプチド、または(b)有効量の請求項12若しくは13に記載の組成物を患者に投与することを含んでなる、肥満の処置方法。
- a)既知のアミノ酸配列を有する第一のペプチドの生物学的活性を測定する段階(該第一のペプチドは非天然アミノ酸化合物を有さず);および
b)請求項7、8、9、10若しくは11に記載の半合成ペプチドの同一の生物学的活性を測定する段階(該半合成ペプチドは第一のペプチドと同一のアミノ酸配列を有するか、若しくは第一のペプチドの切断型バージョンである)
を含んでなる、非天然アミノ酸化合物を含有するペプチドの活性についてのスクリーニング方法。 - 生物学的活性が、選択性、ポトーシス(poptosis)、アポトーシス、細胞シグナリング、リガンド結合、転写、翻訳、代謝、細胞成長、細胞分化、恒常性、半減期、溶解性、輸送若しくは安定性である、請求項25に記載の方法。
- 生物学的活性が、生物学的障壁を通過する半合成ペプチドの能力の直接若しくは間接的評価を包含する、請求項25に記載の方法。
- 請求項7、8、9、10若しくは11に記載の半合成ペプチドを患者に投与することを含んでなり、該半合成ペプチドは、非天然アミノ酸化合物を除き第一のペプチドと同一の配列を有するか、若しくは非天然アミノ酸化合物を除き第一のペプチドの切断型バージョンである、既知の第一のペプチドの患者への投与により影響を及ぼされる疾患を伴う患者の処置方法。
- 疾患が脳の疾患であるか、若しくは既知の第一のペプチドが身体障壁を横断する、請求項28に記載の方法。
- 請求項7、8、9、10若しくは11に記載の半合成ペプチドを既知のペプチドの代わりに用いることを含んでなり、該半合成ペプチドは、非天然アミノ酸化合物を除き、既知のペプチドと同一の配列を有するか若しくは既知のペプチドの切断型バージョンである、生物学的障壁を横断する既知のペプチドの能力の増大、既知のペプチドの選択性の増大、若しくはペプチダーゼによる消化に対する既知のペプチドの抵抗性の増大方法。
- 障壁が、血液脳関門、細胞膜、腸上皮、皮膚若しくは血液眼を含んでなる、請求項30に記載の方法。
- 障壁が血液脳関門である、請求項31に記載の方法。
- 請求項7、8、9、10若しくは11に記載の半合成ペプチドを既知のペプチドの代わりに用いることを含んでなり、該半合成ペプチドは、非天然アミノ酸化合物を除き、第一のペプチドと同一の配列を有するか若しくは第一のペプチドの切断型バージョンである、in vivoで延長された半減期をもつ半合成ペプチドの製造方法。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US75116505P | 2005-12-16 | 2005-12-16 | |
| US81435506P | 2006-06-16 | 2006-06-16 | |
| US81424006P | 2006-06-16 | 2006-06-16 | |
| PCT/US2006/047860 WO2007070672A2 (en) | 2005-12-16 | 2006-12-15 | Non-natural amino acids and neurotensin analogues thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009519945A true JP2009519945A (ja) | 2009-05-21 |
| JP2009519945A5 JP2009519945A5 (ja) | 2011-02-24 |
Family
ID=38163538
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008545836A Pending JP2009519945A (ja) | 2005-12-16 | 2006-12-15 | 非天然アミノ酸およびそれらのニューロテンシンアナログ |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20100130432A1 (ja) |
| EP (1) | EP1962884A2 (ja) |
| JP (1) | JP2009519945A (ja) |
| KR (1) | KR20090012306A (ja) |
| AU (1) | AU2006326418A1 (ja) |
| CA (1) | CA2633224A1 (ja) |
| WO (1) | WO2007070672A2 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101035756A (zh) * | 2004-06-17 | 2007-09-12 | 南卡罗来纳州医科大学研究发展基金会 | 非天然氨基酸 |
| US20080096819A1 (en) * | 2006-05-02 | 2008-04-24 | Allozyne, Inc. | Amino acid substituted molecules |
| CA2707840A1 (en) | 2007-08-20 | 2009-02-26 | Allozyne, Inc. | Amino acid substituted molecules |
| BRPI0920209A2 (pt) | 2008-10-15 | 2015-12-22 | Angiochem Inc | conjugados de agonistas de glp-1 e usos dos mesmos |
| WO2010063122A1 (en) * | 2008-12-05 | 2010-06-10 | Angiochem Inc. | Conjugates of neurotensin or neurotensin analogs and uses thereof |
| US8476221B2 (en) | 2011-03-18 | 2013-07-02 | Halimed Pharmaceuticals, Inc. | Methods and compositions for the treatment of metabolic disorders |
| EP2896402A1 (en) | 2014-01-20 | 2015-07-22 | Vect-Horus | Activated neurotensin molecules and the uses thereof |
| JP6823055B2 (ja) | 2015-06-15 | 2021-01-27 | アンジオケム インコーポレーテッド | 軟髄膜癌腫症の治療方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4956461A (en) * | 1988-10-19 | 1990-09-11 | Dana Farber Cancer Institute, Inc. | Desamino-aminopterin and -methotrexate |
| JP2001233787A (ja) * | 2000-02-04 | 2001-08-28 | Patents Exploitation Co Bv | 小・中サイズのペプチド含有薬学的組成物 |
-
2006
- 2006-12-15 AU AU2006326418A patent/AU2006326418A1/en not_active Abandoned
- 2006-12-15 WO PCT/US2006/047860 patent/WO2007070672A2/en active Application Filing
- 2006-12-15 KR KR1020087017166A patent/KR20090012306A/ko not_active Application Discontinuation
- 2006-12-15 EP EP06848547A patent/EP1962884A2/en not_active Withdrawn
- 2006-12-15 US US12/086,528 patent/US20100130432A1/en not_active Abandoned
- 2006-12-15 JP JP2008545836A patent/JP2009519945A/ja active Pending
- 2006-12-15 CA CA002633224A patent/CA2633224A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007070672A2 (en) | 2007-06-21 |
| US20100130432A1 (en) | 2010-05-27 |
| WO2007070672A3 (en) | 2008-08-21 |
| CA2633224A1 (en) | 2007-06-21 |
| EP1962884A2 (en) | 2008-09-03 |
| AU2006326418A1 (en) | 2007-06-21 |
| KR20090012306A (ko) | 2009-02-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9359291B2 (en) | Non-natural amino acids | |
| JP2009519945A (ja) | 非天然アミノ酸およびそれらのニューロテンシンアナログ | |
| TW580501B (en) | Heterocycles as inhibitors of leucocyte adhesion and as VLA-4 antagonists | |
| US20210300932A1 (en) | Cyclic peptides targeting alpha-4-beta-7 integrin | |
| US11713338B2 (en) | Cyclic peptides multimers targeting α-4-β-7 integrin | |
| JPH09501177A (ja) | 治療効果のあるペプチド誘導体 | |
| KR20200015540A (ko) | α4β7 인테그린을 타겟팅하는 호모데틱 사이클릭 펩타이드 | |
| US6858396B2 (en) | Positively charged non-natural amino acids, methods of making and using thereof in peptides | |
| US6783946B2 (en) | Positively charged non-natural amino acids, methods of making thereof, and use thereof in peptides | |
| TWI258469B (en) | Aryl-substituted alicyclic compounds and pharmaceutical composition containing the same | |
| JP2000501083A (ja) | 改善された作用効果を有する新規lh―rh―拮抗剤 | |
| CN101472601A (zh) | 非天然氨基酸及其神经降压肽类似物 | |
| RU2773443C2 (ru) | ГОМОДЕТНЫЕ ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ИНТЕГРИН α4β7 | |
| WO2021236410A1 (en) | Antifibrotic compounds and related methods | |
| JPH05331188A (ja) | トリペプチド、その製造方法及びエンドセリン拮抗剤 | |
| AU2008200566A1 (en) | Non-natural basic amino acids, their preparation and use | |
| JPH05331187A (ja) | トリペプチド、その製造方法及びエンドセリン拮抗剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20090127 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090820 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091214 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20091214 |
|
| A072 | Dismissal of procedure [no reply to invitation to correct request for examination] |
Free format text: JAPANESE INTERMEDIATE CODE: A073 Effective date: 20110510 |