JP2009512705A - Ccr5アンタゴニストとして有用なピペラジン誘導体 - Google Patents
Ccr5アンタゴニストとして有用なピペラジン誘導体 Download PDFInfo
- Publication number
- JP2009512705A JP2009512705A JP2008536749A JP2008536749A JP2009512705A JP 2009512705 A JP2009512705 A JP 2009512705A JP 2008536749 A JP2008536749 A JP 2008536749A JP 2008536749 A JP2008536749 A JP 2008536749A JP 2009512705 A JP2009512705 A JP 2009512705A
- Authority
- JP
- Japan
- Prior art keywords
- mmol
- solution
- product
- added
- etoac
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CCCN(CC1(CC1)N)C(CC1)(CCN1C(c1c(C)ncnc1C)=O)C=* Chemical compound CCCN(CC1(CC1)N)C(CC1)(CCN1C(c1c(C)ncnc1C)=O)C=* 0.000 description 14
- RNHFORFHXASUHN-BVZXCIRXSA-N C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)ncnc2C)=O)N1[C@@H](COC(C(C(C1O)O)O)OC1C(O)=O)c1ccc(C(F)(F)F)cc1 Chemical compound C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)ncnc2C)=O)N1[C@@H](COC(C(C(C1O)O)O)OC1C(O)=O)c1ccc(C(F)(F)F)cc1 RNHFORFHXASUHN-BVZXCIRXSA-N 0.000 description 2
- ZISJWLFZQNHTTB-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CC2(CC2)N1C(c1ccc(C(F)(F)F)cc1)C#N)=O Chemical compound CC(C)(C)OC(N(CC1)CC2(CC2)N1C(c1ccc(C(F)(F)F)cc1)C#N)=O ZISJWLFZQNHTTB-UHFFFAOYSA-N 0.000 description 1
- PKVQJGAKQUHNRK-HSZRJFAPSA-N CC(CC1)(CCN1C(c1c(C)ncnc1C)=O)N(CC1)CC2(CC2)N1[C@@H](C(O)=O)c1ccc(C(F)(F)F)cc1 Chemical compound CC(CC1)(CCN1C(c1c(C)ncnc1C)=O)N(CC1)CC2(CC2)N1[C@@H](C(O)=O)c1ccc(C(F)(F)F)cc1 PKVQJGAKQUHNRK-HSZRJFAPSA-N 0.000 description 1
- WLNOFRXLATWAAY-ZDUSSCGKSA-N C[C@@H](C1)NCCN1C(C)(CC1)CCN1C(OC(C)(C)C)=O Chemical compound C[C@@H](C1)NCCN1C(C)(CC1)CCN1C(OC(C)(C)C)=O WLNOFRXLATWAAY-ZDUSSCGKSA-N 0.000 description 1
- SXQRWLSMVOKZLP-GAJHUEQPSA-N C[C@@H](CN(CC1)C(C)(CC2)CCN2C(C2=C(C)NCN=C2C)=O)N1[C@@H](C(O)=O)c1ccc(C(F)(F)F)cc1 Chemical compound C[C@@H](CN(CC1)C(C)(CC2)CCN2C(C2=C(C)NCN=C2C)=O)N1[C@@H](C(O)=O)c1ccc(C(F)(F)F)cc1 SXQRWLSMVOKZLP-GAJHUEQPSA-N 0.000 description 1
- MBERILMVQOVKQU-CYFREDJKSA-N C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)nc[n+](O)c2C)=O)N1[C@@H](COC)c1ccc(C(F)(F)F)cc1 Chemical compound C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)nc[n+](O)c2C)=O)N1[C@@H](COC)c1ccc(C(F)(F)F)cc1 MBERILMVQOVKQU-CYFREDJKSA-N 0.000 description 1
- KKISTQPVRGXETN-MBSDFSHPSA-N C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)ncnc2C)=O)N1[C@@H](CO)c1ccc(C(F)(F)F)cc1 Chemical compound C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)ncnc2C)=O)N1[C@@H](CO)c1ccc(C(F)(F)F)cc1 KKISTQPVRGXETN-MBSDFSHPSA-N 0.000 description 1
- IGSPKXXKILIWGL-ITWWAKKDSA-N C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)ncnc2C)=O)N1[C@@H](COC(C(C1O)O)OC(C2OCC2O)C1O)c1ccc(C(F)(F)F)cc1 Chemical compound C[C@@H](CN(CC1)C(C)(CC2)CCN2C(c2c(C)ncnc2C)=O)N1[C@@H](COC(C(C1O)O)OC(C2OCC2O)C1O)c1ccc(C(F)(F)F)cc1 IGSPKXXKILIWGL-ITWWAKKDSA-N 0.000 description 1
- PQITVSZLERYIHP-OALUTQOASA-N C[C@@H](c(cc1)ccc1C(C)=O)N(CC1)[C@@H](C)CN1C(CC1)CCN1C(c(c(N)ccc1)c1Cl)=O Chemical compound C[C@@H](c(cc1)ccc1C(C)=O)N(CC1)[C@@H](C)CN1C(CC1)CCN1C(c(c(N)ccc1)c1Cl)=O PQITVSZLERYIHP-OALUTQOASA-N 0.000 description 1
- DTKUANPECHGGBY-UNMCSNQZSA-N C[C@@H](c1ccc(C(F)(F)F)cc1)N(CC1)[C@@H](C)CN1C(C)(CC1)CCN1C(c1c(C)nccc1C)=O Chemical compound C[C@@H](c1ccc(C(F)(F)F)cc1)N(CC1)[C@@H](C)CN1C(C)(CC1)CCN1C(c1c(C)nccc1C)=O DTKUANPECHGGBY-UNMCSNQZSA-N 0.000 description 1
- XQNVDQZWOBPLQZ-UHFFFAOYSA-N O=Cc(cc1)ccc1OC(F)(F)F Chemical compound O=Cc(cc1)ccc1OC(F)(F)F XQNVDQZWOBPLQZ-UHFFFAOYSA-N 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- Biotechnology (AREA)
- Virology (AREA)
- Neurosurgery (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Neurology (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Transplantation (AREA)
- Oncology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Communicable Diseases (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
本発明は、選択的CCR5拮抗薬として有用なピペラジン誘導体、該化合物を含む医薬品組成物、および該化合物を用いる治療方法に関する。本発明は、本発明のCCR5拮抗薬と、ヒト免疫不全ウイルス(HIV)の治療において有用な1つ以上の抗ウイルス剤またはその他の剤との組み合わせの使用にも関する。さらに、本発明は、固形臓器移植拒絶、移植片対宿主疾患、関節炎、関節リウマチ、炎症性腸疾患、アトピー性皮膚炎、乾癬、喘息、アレルギーまたは多発性硬化症の治療における単独または別の剤と組み合わせた本発明のCCR‐5拮抗薬の使用に関する。
本発明は、HIVの治療に関し、構造式I

Rは、R8‐フェニル、R8‐ピリジル、R8‐チオフェニルまたはR8‐ナフチルであり、
R1は、水素またはC1〜C6アルキルであり、
R2は、R9、R10、R11‐フェニル、R9、R10、R11‐置換6員ヘテロアリール、R9、R10、R11‐置換6員ヘテロアリールN‐オキシド、R12、R13‐置換5員ヘテロアリール、ナフチル、フルオレニル、ジフェニルメチル、
R3は、水素、C1〜C6アルキル、(C1〜C6)アルコキシ(C1〜C6)アルキル、C3〜C10シクロアルキル、C3〜C10シクロアルキル(C1〜C6)アルキル、R8‐フェニル、R8‐フェニル(C1〜C6)アルキル、R8‐ナフチル、R8‐ナフチル(C1〜C6)アルキル、R8‐ヘテロアリールまたはR8‐ヘテロアリール(C1〜C6)アルキルであり、
R4、R5、R7およびR13は、独立に、水素および(C1〜C6)‐アルキルからなる群から選ばれ、
R6は、水素、C1〜C6アルキルまたはC2〜C6アルケニルであり、
R8は、水素、ハロゲン、C1〜C6アルキル、C1〜C6アルコキシ、‐CF3、CF3O‐、CH3C(O)‐、‐CN、CH3SO2‐、CF3SO2‐、R14‐フェニル、R14‐ベンジル、CH3C(=NOCH3)、CH3C(=NOCH2CH3)、
R9およびR10は、独立に、(C1〜C6)アルキル、ハロゲン、‐NR17R18、‐OH、−CF3、‐OCH3、‐O‐アシル、‐OCF3および‐Si(CH3)3からなる群から選ばれ、
R11は、R9、水素、フェニル、‐NO2、‐CN、‐CH2F、‐CHF2、‐CHO、‐CH=NOR17、ピリジル、ピリジルN‐オキシド、ピリミジニル、ピラジニル、‐N(R17)CONR18R19、‐NHCONH(クロロ‐(C1〜C6)アルキル)、‐NHCONH((C3−C1)シクロアルキル(C1〜C6)アルキル)、‐NHCO(C1〜C6)アルキル、‐NHCOCF3、‐NHSO2N((C1〜C6)アルキル)2、‐NHSO2(C1〜C6)アルキル、‐N(SO2CF3)2、‐NHCO2(C1〜C6)アルキル、C3〜C10シクロアルキル、‐SR20、‐SOR20、‐SO2R20、−SO2NH(C1〜C6アルキル)、‐OSO2(C1〜C6)アルキル、‐OSO2CF3、ヒドロキシ(C1〜C6)アルキル、‐CONR17R18、‐CON(CH2CH2‐O‐CH3)2、‐OCONH(C1〜C6)アルキル、‐CO2R17、‐Si(CH3)3または‐B(OC(CH3)2)2であり、
R12は、(C1〜C6)アルキル、‐NH2またはR14‐フェニルであり、
R14は、水素、(C1〜C6)アルキル、‐CF3、‐CO2R17、‐CN、(C1〜C6)アルコキシおよびハロゲンからなる群から独立に選択される1から3個の置換基であり、
R15およびR16は、水素およびC1〜C6アルキルからなる群から独立に選ばれるか、または、R15とR16は一緒になってC2〜C5アルキレン基であり、それらが結合する炭素と3から6個の炭素原子のスピロ環を形成し、
R17、R18およびR19は、独立に、HおよびC1〜C6アルキルからなる群から選ばれ、
R20は、C1〜C6アルキルまたはフェニルである。
(1)Raは、R8a‐フェニル、R8b‐ピリジル、R8b‐チオフェニルまたはR8‐ナフチルであり、
R1は、水素またはC1〜C6アルキルであり、
R2は、R9、R10、R11‐フェニル、R9、R10、R11‐置換6員ヘテロアリール、R9、R10、R11‐置換6員ヘテロアリールN‐オキシド、R12、R13‐置換5員ヘテロアリール、ナフチル、フルオレニル、ジフェニルメチル、
R3は、水素、C1〜C6アルキル、(C1〜C6)アルコキシ(C1〜C6)アルキル、C3〜C10シクロアルキル、C3〜C10シクロアルキル(C1〜C6)アルキル、R8‐フェニル、R8‐フェニル(C1〜C6)アルキル、R8‐ナフチル、R8‐ナフチル(C1〜C6)アルキル、R8‐ヘテロアリールまたはR8‐ヘテロアリール(C1〜C6)アルキルであり、
R4、R5、R7およびR13は、独立に、水素および(C1〜C6)‐アルキルからなる群から選ばれ、
R6は、水素、C1〜C6アルキルまたはC2〜C6アルケニルであり、
R8は、水素、ハロゲン、C1〜C6アルキル、C1〜C6アルコキシ、‐CF3、CF3O‐、CH3C(O)‐、‐CN、CH3SO2‐、CF3SO2‐、R14‐フェニル、R14‐ベンジル、CH3C(=NOCH3)、CH3C(=NOCH2CH3)、
R8aは、水素、ハロゲン、‐CF3、CF3O‐、‐CN、CF3SO2‐、R14‐フェニル、‐NHCOCF3、5員ヘテロアリールおよび
R8bは、水素、ハロゲン、‐CF3、CF3O‐、CH3C(O)‐、‐CN、CF3SO2‐、R14‐ベンジル、CH3C(=NOCH3)、CH3C(=NOCH2CH3)、
R9およびR10は、独立に、(C1〜C6)アルキル、ハロゲン、‐NR17R18、‐OH、−CF3、‐OCH3、‐O‐アシル、‐OCF3および‐Si(CH3)3からなる群から選ばれ、
R11は、R9、水素、フェニル、‐NO2、‐CN、‐CH2F、‐CHF2、‐CHO、‐CH=NOR17、ピリジル、ピリジルN‐オキシド、ピリミジニル、ピラジニル、‐N(R17)CONR18R19、‐NHCONH(クロロ‐(C1〜C6)アルキル)、‐NHCONH((C3〜C1)シクロアルキル(C1〜C6)アルキル)、‐NHCO(C1〜C6)アルキル、‐NHCOCF3、‐NHSO2N((C1〜C6)アルキル)2、‐NHSO2(C1〜C6)アルキル、‐N(SO2CF3)2、‐NHCO2(C1〜C6)アルキル、C3〜C10シクロアルキル、‐SR20、‐SOR20、‐SO2R20、‐SO2NH(C1〜C6アルキル)、‐OSO2(C1〜C6)アルキル、‐OSO2CF3、ヒドロキシ(C1〜C6)アルキル、‐CONR17R18、‐CON(CH2CH2‐O‐CH3)2、‐OCONH(C1〜C6)アルキル、‐CO2R17、‐Si(CH3)3または‐B(OC(CH3)2)2であり、
R12は、(C1〜C6)アルキル、‐NH2またはR14‐フェニルであり、
R14は、水素、(C1〜C6)アルキル、‐CF3、‐CO2R17、‐CN、(C1〜C6)アルコキシおよびハロゲンからなる群から独立に選択される1から3個の置換基であり、
R15およびR16は、水素およびC1〜C6アルキルからなる群から独立に選ばれるか、またはR15とR16は一緒になってC2〜C5アルキレン基であり、それらが結合している炭素と3から6個の炭素原子のスピロ環を形成し、
R17、R18およびR19は、HおよびC1〜C6アルキルからなる群から独立に選ばれ、
R20は、C1〜C6アルキルまたはフェニルであり、あるいは、
(2)Raは、R8‐フェニル、R8‐ピリジルまたはR8‐チオフェニルであり、
R2は、フルオレニル、ジフェニルメチル、
R1、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、R16、R17、R18、R19およびR20は、(1)で定義したと同じである。
本明細書で用いられる以下の用語は、他の定義を示さない限り、下記の定義に従って用いられる。
(a)2つのNRTIと1つのPIと、または(b)2つのNRTIと1つのNNRTIと、などの三剤併用治療、および(c)2つのNRTI、1つのPIおよび第2のPIまたは1つのNNRTIなどの四剤併用治療を含む。薬物未使用患者の治療においては、三剤併用治療による抗HIV‐1治療を開始するのが好ましい。PIに対する不耐性がなければ、2つのNRTIと1つのPIとの使用が好ましい。服薬遵守は不可欠である。3〜6ヶ月ごとにCD4+およびHIV‐1‐RNA血漿レベルをモニターする必要がある。ウイルス量が一定になる場合、第4の薬物、例えば1つのPIまたは1つのNNRTIを加えられ得る。一般的な治療をさらに記載した下の表を参照すること。
A.三剤併用療法
1. 2つのNRTI1+1つのPI2
2. 2つのNRTI1+1つのNNRTI3
B.四剤併用治療4
2つのNRTI+1つのPI+第2のPIまたは1つのNNRTI
C.代替法5
2つのNRTI1
1つのNRTI5+1つのPI2
2つのPl6±1つのNRTI7またはNNRTI3
1つのPI2+1つのNRTI7+1つのNNRTI3
表の脚注
1. 以下、すなわちジドブジン+ラミブジン、ジドブジン+ジダノシン、スタブジン+ラミブジン、スタブジン+ジダノシン、ジドブジン+ザルシタビンのうちの1つ。
2. インジナビル、ネルフィナビル、リトナビルまたはサキナビルの軟質ゲルカプセル。
3. ネビラピンまたはデラビルジン。
4. A−M. Vandamne et al Antiviral Chemistry & Chemotherapy 9: 187のp 193−197および図1+2を参照すること。
5. コンプライアンスの問題または毒性が原因となって、推薦した治療を受けることができない患者および推薦したレジメンでは失敗または再発する患者のための代替投与計画がある。多くの患者において二剤のヌクレオシド併用法はHIV抵抗性および臨床失敗に至ることがある。
6. 大部分のデータは、サキナビルとリトナビルとで(一日二回各400mg)得た。
7. ジドブジン、スタブジンまたはジダノシン。
炎症性腸疾患:IL‐10(米国特許第5,368,854参照)、ステロイドおよびアザルフィジン、
関節リウマチ:メトトレキサート、アザチオプリン、シクロホスファミド、ステロイドおよびミコフェノール酸モフェチル、
多発性硬化症:インターフェロン‐ベータ、インターフェロン‐アルファおよびステロイド。
パラ位にアルキルまたはアリールスルホニルR8基を有する化合物の合成を、対応するパラ‐置換アセトフェノンで開始した。パラ‐置換アセトフェノンを実施例4、工程1〜6と同じように処理し、式
5%のCH3OH‐CH2Cl2中のTLCのRf=0.45。
*HPLC VYDAC218TP5405カラム、グラジエント5〜95%B、10分間、2分間一定、溶液A 0.1%TFA/H2O、溶液B 0.1%TFA/CH3CN、245nm。
工程1 ベンゼン中の実施例11、工程4のN‐BOC保護生成物(2.5g、5.8mmol)の溶液に、フェニルホウ酸(1.68g、13.8mmol)、2M Na2CO3(14ml)およびテトラキス(トリ‐フェニルホスフィン)パラジウム(0.67g、0.58mmol)を加えた。この混合物を還流下で一夜撹拌した。EtOAcを用いて抽出により後処理した後、シリカゲルカラムクロマトグラフィーに付し、フェニル誘導体(1.37g、62%収率、Rf=0.5、ヘキサン/EtOAc、5:1)をシロップ状物として得た。
元素分析C27H36N5OF3・2HCl・0.5H2O、計算値C55.38%、H6.71%、N11.96%、Cl12.11%、実測値C55.19%、H6.69%、N11.75%、Cl11.45%。
アリールシクロプロピルアミド
方法A
工程3
(S,S)‐ジアステレオマー(A)、TLC Rf=0.6(25%、Et2O‐ヘキサン)。0.9gの無色のガム状物。
(R,S)‐ジアステレオマー(B)、TLC Rf=0.5(25%、Et2O‐ヘキサン)。0.8gの無色のガム状物。
A Rf=0.5(ヘキサン中50%のEt2O)。淡黄色のガム状物(0.9g、42%)
B Rf=0.4(ヘキサン中50%のEt2O)。コハク色のガム状物(1.13g、44%)
工程4 実施例1、工程4に記載したと同じように、Aから誘導された遊離ピペラジン(0.9g、2.2mmol)のN‐BOC‐ピペリジン‐4‐オンによる還元アミノ化およびイプソ‐メチル基の導入を実行し、BOC‐保護ピペリジニル化合物(0.87g、92%)を得た。Rf=0.3(ヘキサン中50%のEtOAc)。
化学薬品 14C‐ビクリビロック(Vicriviroc)[1‐[(4,6‐ジメチル‐5‐ピリミジニル)カルボニル]‐4‐[4‐[2‐メトキシ‐1(R)‐[4‐(トリフルオロメチル)フェニル]エチル]‐3(S)‐メチル‐l‐ピペラジニル]‐4‐メチルピペリジン、すなわち、下記
に示す14C‐化合物29A]は、Schering−Plough Research Institute(Kenilworth,NJ)において合成し、>97%の放射性化学物質純度を有していた。他のすべての化合物/標準品は、Schering−Plough Research InstituteのChemical Research Departmentから入手した。HPLC用アセトニトリルおよびメタノールは、BurdickおよびJackson(Muskegon,MI)から供給された。水は、Millipore Milli−QPlus水精製システム(Bedford,MA)を用いて精製した。
試料集積
被検者集団/動物集団ごとに時点別にそれぞれの種の血漿試料を集積した。残りのマトリックスはすべて、所望の採集間隔で最初にそれぞれの被検者/動物ごとに、次に被検者集団/動物集団ごとに集積し、排出された放射能の>90%をそれぞれのマトリックス中に含む複合試料を得た。
LC‐MSおよびLC‐MSn実験すべてについて、HPLCカラムは室温に保持した。5%のアセトニトリルを含む95%の10mM酢酸アンモニウム(pH6.0)(A)と、95%のアセトニトリルおよび5%の水(B)からなる移動相を一定の流速(1mL/分)に保持した。すべてのLC‐MS実験について、カラム溶出液を分流し、20〜25%をTSQ Quantum(ThermoElectron,San Jose,CA)質量分析計に流し、残りをFlow Scintillation Analyzer(FSA)アナライザーに流した。
次の表に要約する移動相組成のプログラム線形変化を用いて代謝産物の分離を達成した。
下記に列挙する条件下で作動させたTSQ質量分析計(ThermoElectron,San Jose,CA)を用いて、すべてのLC‐MSおよびLC‐MS/MS実験を実行した。
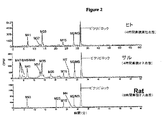
表1に示すように、健康な男性志願者へのVICの50mg単回経口投与の後、投与物は、尿および糞便にほぼ等しく排出されていた。これに対して、調査したすべての非臨床種からは投与物の過半数(53〜71%)は糞便中に回収された。図1に示すように、VICは、50mg、6mg/kgおよび5mg/kgの14C‐VICの単回経口投与後、ヒト、サルおよびラットにおいてそれぞれ急速かつ広範に代謝された。VICの代謝に性別関連の定性的差異はなかったため、図2〜4にはオスのラットおよびサルからの各マトリックスのプロフィルだけを示す。
図2は、健康な男性志願者、オスのサルおよびオスのラットへのビクリビロックの単回経口投与後の集積した血漿抽出物の代表的なラジオクロマトグラフィーのプロフィルの比較を示す。
・ヒト、サルおよびラットからの血漿中では定性的に類似のプロフィルが観測された。
・ヒト、サルおよびラットにおける主な循環薬物誘導成分はVICであった。
・ヒトおよびサル血漿中ではO‐デスメチル‐VIC(M35)のグルクロニド抱合体が目立った循環代謝産物であったが、ラットではこの代謝産物は極微量しか検出されなかった。
・ヒトに特異的な循環代謝産物は検出されなかった。
・ヒト、サルおよびラットにおいて、主な尿代謝物N‐デスアルキル‐VIC(M41)およびO‐デスメチル‐VIC‐グルクロニド(M35)は全体としてそれぞれ、投与量の21%、8%および4%を占めた。
・M35は、尿中では投与量の11%および3%の寄与を示したが、この代謝産物は、ラットからの尿中では1%しか占めなかった。
・ヒト、サルおよびラットでは、全体として投与した用量の16〜35%を占める主要な糞便代謝物は、N‐デスアルキル‐VIC(M41)、O‐デメチル‐VIC(M15)、およびM15の酸化生成物(m/z534のM20/M20a)を含む。
・健康な志願者への単回経口50mg投与の後、ビクリビロック(VIC、化合物29A)およびその代謝産物は、糞便および尿中にほぼ等しく排出された。これに対して、ラットおよびサルへのVICのそれぞれ単回5mg/kgおよび6mg/kg経口投与の後、放射能は大部分糞便中に排出された。
・調査したすべての種で、VICの一次生体内変換は、O‐脱メチル化、N‐脱アルキル化、酸化およびグルクロン酸化を含んでいた。
CCR5膜結合アッセイを利用する高スループットスクリーニングによって、RANTES結合の阻害剤を同定する。このアッセイは、ヒトCCR5ケモカイン受容体を発現するNIH 3T3細胞から調製した膜を利用する。この膜には、この受容体の天然リガンドであるRANTESに結合する能力がある。96ウェルプレートフォーマットを用いて、化合物の存在下または非存在下に膜調製物を125I‐RANTESと1時間インキュベートする。化合物を0.001μg/mlから1μg/mlの広い範囲に連続希釈し、3連で試験する。ガラスファイバーフィルタによって反応カクテルを集め、徹底的に洗う。反復についての全計数値を平均し、総125I‐RANTES結合の50パーセントを阻害するために必要な濃度としてデータを報告する。膜結合アッセイにおいて強力な活性を有する化合物を、二次細胞(secondary cell)ベースのHIV‐1進入および複製アッセイにおいてさらに特徴付ける。
HIV‐1のNL4‐3株をコードするプラスミド(エンベロープ遺伝子の変異およびルシフェラーゼレポータープラスミドの導入によって改変されている)を、Connor et al, Virology, 206(1995), p. 935−944に記載されているいくつかのHIV‐1エンベロープ遺伝子の1つをコードするプラスミドとともに同時トランスフェクションして、複製欠損HIV‐1レポータービリオンを生成する。リン酸カルシウム沈殿法によるこれら2つのプラスミドのトランスフェクションの後、第3日にウイルス上清を集め、機能的なウイルス力価を測定する。次に、これらのストックを用いて、試験化合物の存在下または非存在下で予めインキュベートした、CD4とケモカイン受容体CCR5とを安定に発現するU87細胞を感染させる。37℃で2時間感染を行い、細胞を洗い、培地を、化合物を含む新しい培地に換える。細胞を3日間インキュベートし、溶解させ、ルシフェラーゼ活性を測定する。結果を対照培養物中のルシフェラーゼ活性の50%を阻害するために必要な化合物の濃度として報告する。
このアッセイは、初代末梢血単核細胞または安定なU87‐CCR5細胞株を用いて初代HIV‐1株の感染を阻止する抗CCR5化合物の効果を測定する。正常かつ健康なドナーからの初代リンパ球を精製し、感染させる3日前にPHAおよびIL‐2でインビトロ刺激する。96ウェルプレートフォーマットを用いて、37℃で1時間細胞を薬物で予備処理し、続いて向M性HIV‐1分離株で感染させる。感染後、細胞を洗って残留接種物を除去し、化合物の存在下で4日間培養する。培養上清を集め、ウイルスp24抗原濃度の測定によってウイルス複製を測定する。
HIV共受容体CCR5を発現する細胞にカルシウム感受性染料を負荷させた後、化合物または天然CCR5リガンドを加える。作動薬特性を有する化合物は、細胞内にカルシウムフラックスシグナルを誘導し、一方、CCR5拮抗薬はそれ自体ではシグナル伝達を誘導しないが、天然リガンドRANTESによるシグナル伝達を遮断することができる化合物として同定される。
GTPγS結合アッセイは、CCR5リガンドによる受容体活性化を測定する。このアッセイは、適切なリガンドによる受容体活性化の結果として起こる、受容体結合型G蛋白質への35S標識化GTPの結合を測定する。このアッセイでは、CCR5リガンドRANTESをCCR5発現細胞由来の膜とともにインキュベートし、結合した35S標識をアッセイして受容体活性化(または結合)に対する結合を測定する。このアッセイは、化合物が受容体の活性化を誘導して作動薬特性を示すか、あるいは競合または非競合的な様式でのRANTES結合の阻害を測定して、拮抗薬特性を示すかを定量的に測定する。
走化性アッセイは、試験化合物の作動薬対拮抗薬特性を特徴付ける機能的なアッセイである。このアッセイは、ヒトCCR5(BaF‐550)を発現する非接着性マウス細胞株が、試験化合物または天然リガンド(すなわちRANTES、MIP‐1β)のどちらかへの応答として膜を通って移行する能力を測定する。細胞は、浸透性の膜を通って作動薬活性を有する化合物の方へ移行する。拮抗薬である化合物は、走化性を誘導することができないだけでなく、既知のCCR5リガンドへの応答として細胞移行を抑制することもできる。
Claims (15)
- 純粋な単離された形の化合物であって、
- 請求項1に記載の化合物であって、
- 請求項1に記載の化合物であって、
- 請求項1に記載の化合物であって、
- 請求項1に記載の化合物であって、
- 請求項1に記載の化合物であって、
- 請求項1に記載の化合物、またはその薬学的に許容される塩、溶媒和物またはエステルの有効な量を薬学的に許容されるキャリアと組み合わせて含む医薬品組成物。
- ヒト免疫不全ウイルスを治療する方法であって、該方法は、請求項1に記載の化合物、またはその薬学的に許容される塩、溶媒和物またはエステルの治療有効量をそのような治療を必要とするヒトに投与することを含む、方法。
- 請求項1に記載の化合物、またはその薬学的に許容される塩、溶媒和物またはエステルと組み合わせて、ヒト免疫不全ウイルスの治療において有用な1つ以上の抗ウイルス剤またはその他の剤を投与することをさらに含む、請求項8に記載の方法。
- 前記抗ウイルス剤が、ヌクレオシド逆転写酵素阻害剤、非ヌクレオシド逆転写酵素阻害剤およびプロテアーゼ阻害剤からなる群から選択される、請求項9に記載の方法。
- 前記抗ウイルス剤が、ジドブジン、ラミブジン、ザルシタビン、ジダノシン、スタブジン、アバカビル、アデフォビルジピボキシル、ロブカビル、BCH‐10652、エミトリシタビン、ベータ‐L‐FD4、DAPD、ロデノシン、ネビラピン、デラビルジン、エファビレンツ、PNU‐142721、AG‐1549、MKC‐442、(+)‐カラノライドAおよびB、サキナビル、インジナビル、リトナビル、ネルフィナビル、ラシナビル、DMP‐450、BMS‐2322623、ABT‐378、アンプレナビル、ヒドロキシ尿素、リバビリン、IL‐2、IL‐12、ペンタフシド、イッサム第11607号およびAG‐1549からなる群から選択される、請求項10に記載の方法。
- 固形臓器移植拒絶、移植片対宿主疾患、関節炎、関節リウマチ、炎症性腸疾患、アトピー性皮膚炎、乾癬、喘息、アレルギーまたは多発性硬化症を治療する方法であって、該方法は、請求項1に記載の化合物、またはその薬学的に許容される塩、溶媒和物またはエステルの治療有効量を、そのような治療を必要とするヒトに投与することを含む、方法。
- 固形臓器移植拒絶、移植片対宿主疾患、関節炎、関節リウマチ、炎症性腸疾患、アトピー性皮膚炎、乾癬、喘息、アレルギーまたは多発性硬化症の治療のための請求項12に記載の方法であって、該方法は、該疾患の治療において有用な1つ以上の他の剤をさらに含む、方法。
- 単一のパッケージ中の別々の容器内に、ヒト免疫不全ウイルスを治療するために組み合わせて使用するための複数の医薬品組成物を含むキットであって、該キットは、薬学的に許容されるキャリア中の請求項1に記載の化合物またはその薬学的に許容される塩、溶媒和物またはエステルの有効な量を含む医薬品組成物を1つの容器の中に含み、薬学的に許容されるキャリア中のヒト免疫不全ウイルスの治療において有用な抗ウイルス剤または他の剤の有効な量を含む1つ以上の医薬品組成物を別の容器の中に含む、キット。
- 患者が、式
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/255,643 US7825121B2 (en) | 1999-05-04 | 2005-10-21 | Piperazine derivatives useful as CCR5 antagonists |
| PCT/US2006/040636 WO2007050375A2 (en) | 2005-10-21 | 2006-10-18 | Piperazine derivatives useful as ccr5 antagonists |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012007201A Division JP2012072195A (ja) | 2005-10-21 | 2012-01-17 | Ccr5アンタゴニストとして有用なピペラジン誘導体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009512705A true JP2009512705A (ja) | 2009-03-26 |
| JP2009512705A5 JP2009512705A5 (ja) | 2012-03-22 |
Family
ID=37872167
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008536749A Pending JP2009512705A (ja) | 2005-10-21 | 2006-10-18 | Ccr5アンタゴニストとして有用なピペラジン誘導体 |
| JP2012007201A Pending JP2012072195A (ja) | 2005-10-21 | 2012-01-17 | Ccr5アンタゴニストとして有用なピペラジン誘導体 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012007201A Pending JP2012072195A (ja) | 2005-10-21 | 2012-01-17 | Ccr5アンタゴニストとして有用なピペラジン誘導体 |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US7825121B2 (ja) |
| EP (1) | EP1951708B1 (ja) |
| JP (2) | JP2009512705A (ja) |
| KR (1) | KR20080058486A (ja) |
| CN (1) | CN101326178A (ja) |
| AR (1) | AR058105A1 (ja) |
| AU (1) | AU2006306491B2 (ja) |
| BR (1) | BRPI0617731A2 (ja) |
| CA (1) | CA2626565A1 (ja) |
| EC (1) | ECSP088378A (ja) |
| IL (1) | IL190902A0 (ja) |
| NO (1) | NO20082297L (ja) |
| PE (1) | PE20070711A1 (ja) |
| RU (1) | RU2008119652A (ja) |
| TW (1) | TW200800234A (ja) |
| WO (1) | WO2007050375A2 (ja) |
| ZA (1) | ZA200803454B (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015518957A (ja) * | 2012-05-14 | 2015-07-06 | リチャード ジー. ペステル | がんの治療のためのccr5の修飾薬の使用 |
| US10952415B2 (en) | 2011-03-09 | 2021-03-23 | Richard G. Pestell | Prostate cancer cell lines, gene signatures and uses thereof |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1734966B1 (en) * | 2004-04-13 | 2013-07-31 | Incyte Corporation | Piperazinylpiperidine derivatives as chemokine receptor antagonists |
| US20080280892A1 (en) * | 2005-10-21 | 2008-11-13 | Nathalie Cailleau | Compounds |
| WO2008030853A2 (en) * | 2006-09-06 | 2008-03-13 | Incyte Corporation | Combination therapy for human immunodeficiency virus infection |
| WO2010081851A1 (en) | 2009-01-14 | 2010-07-22 | Genoscience Pharma | Piperidin-4-ylpiperazine compounds for the treatment of hcv infection |
| GB201118876D0 (en) | 2011-11-01 | 2011-12-14 | Astex Therapeutics Ltd | Pharmaceutical compounds |
| WO2016094011A1 (en) * | 2014-12-11 | 2016-06-16 | Merck Sharp & Dohme Corp. | Crystal forms of a ccr5 antagonist |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002543185A (ja) * | 1999-05-04 | 2002-12-17 | シェーリング コーポレイション | Ccr5アンタゴニストとして有用なピペラジン誘導体 |
| JP2004524360A (ja) * | 2001-03-29 | 2004-08-12 | シェーリング コーポレイション | Ccr5アンタゴニストとして有用なアリールオキシム−ピペラジン |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6391865B1 (en) * | 1999-05-04 | 2002-05-21 | Schering Corporation | Piperazine derivatives useful as CCR5 antagonists |
-
2005
- 2005-10-21 US US11/255,643 patent/US7825121B2/en not_active Expired - Fee Related
-
2006
- 2006-10-18 EP EP06826151.0A patent/EP1951708B1/en active Active
- 2006-10-18 BR BRPI0617731-0A patent/BRPI0617731A2/pt not_active IP Right Cessation
- 2006-10-18 CA CA002626565A patent/CA2626565A1/en not_active Abandoned
- 2006-10-18 WO PCT/US2006/040636 patent/WO2007050375A2/en active Application Filing
- 2006-10-18 AU AU2006306491A patent/AU2006306491B2/en not_active Ceased
- 2006-10-18 RU RU2008119652/04A patent/RU2008119652A/ru not_active Application Discontinuation
- 2006-10-18 CN CNA2006800462485A patent/CN101326178A/zh active Pending
- 2006-10-18 KR KR1020087011479A patent/KR20080058486A/ko not_active Application Discontinuation
- 2006-10-18 JP JP2008536749A patent/JP2009512705A/ja active Pending
- 2006-10-19 AR ARP060104564A patent/AR058105A1/es not_active Application Discontinuation
- 2006-10-19 PE PE2006001267A patent/PE20070711A1/es not_active Application Discontinuation
- 2006-10-20 TW TW095138842A patent/TW200800234A/zh unknown
-
2008
- 2008-04-15 IL IL190902A patent/IL190902A0/en unknown
- 2008-04-17 EC EC2008008378A patent/ECSP088378A/es unknown
- 2008-04-18 ZA ZA200803454A patent/ZA200803454B/xx unknown
- 2008-05-20 NO NO20082297A patent/NO20082297L/no not_active Application Discontinuation
-
2012
- 2012-01-17 JP JP2012007201A patent/JP2012072195A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002543185A (ja) * | 1999-05-04 | 2002-12-17 | シェーリング コーポレイション | Ccr5アンタゴニストとして有用なピペラジン誘導体 |
| JP2004524360A (ja) * | 2001-03-29 | 2004-08-12 | シェーリング コーポレイション | Ccr5アンタゴニストとして有用なアリールオキシム−ピペラジン |
Non-Patent Citations (4)
| Title |
|---|
| JPN6011056927; 久保田和彦他: 薬理学テキスト 第2版1刷, 19870525, p.445-456, 460-463, 廣川書店 * |
| JPN6012022933; Jayaram R. Tagat et al: 'Piperazine-Based CCR5 Antagonists as HIV-1 Inhibitors. IV. Discovery of 1-[(4,6-Dimethyl-5-pyrimidin' Journal of Medicinal Chemistry Vol.47, No.10, 2004, p.2405-2408 * |
| JPN6012022934; 医薬品の臨床薬物動態試験について , 20010601 * |
| JPN6012022935; 非臨床薬物動態試験ガイドラインについて , 19980626 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10952415B2 (en) | 2011-03-09 | 2021-03-23 | Richard G. Pestell | Prostate cancer cell lines, gene signatures and uses thereof |
| JP2015518957A (ja) * | 2012-05-14 | 2015-07-06 | リチャード ジー. ペステル | がんの治療のためのccr5の修飾薬の使用 |
| US11633397B2 (en) | 2012-05-14 | 2023-04-25 | Richard G. Pestell | Use of modulators of CCR5 in the treatment of cancer and cancer metastasis |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2626565A1 (en) | 2007-05-03 |
| NO20082297L (no) | 2008-07-21 |
| IL190902A0 (en) | 2008-11-03 |
| BRPI0617731A2 (pt) | 2011-08-02 |
| ECSP088378A (es) | 2008-05-30 |
| WO2007050375A3 (en) | 2007-06-14 |
| WO2007050375A2 (en) | 2007-05-03 |
| PE20070711A1 (es) | 2007-08-11 |
| KR20080058486A (ko) | 2008-06-25 |
| JP2012072195A (ja) | 2012-04-12 |
| ZA200803454B (en) | 2009-04-29 |
| EP1951708A2 (en) | 2008-08-06 |
| US20060105964A1 (en) | 2006-05-18 |
| AU2006306491A1 (en) | 2007-05-03 |
| AR058105A1 (es) | 2008-01-23 |
| TW200800234A (en) | 2008-01-01 |
| AU2006306491B2 (en) | 2012-12-13 |
| US7825121B2 (en) | 2010-11-02 |
| EP1951708B1 (en) | 2013-11-20 |
| CN101326178A (zh) | 2008-12-17 |
| RU2008119652A (ru) | 2009-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1175401B1 (en) | Piperazine derivatives useful as ccr5 antagonists | |
| US6391865B1 (en) | Piperazine derivatives useful as CCR5 antagonists | |
| EP1175402B1 (en) | Piperidine derivatives useful as ccr5 antagonists | |
| US8114879B2 (en) | Piperazine derivatives useful as CCR5 antagonists | |
| EP1421075B1 (en) | Piperidine derivatives useful as ccr5 antagonists for the treatment of hiv | |
| JP4248251B2 (ja) | Aidsの処置に有用なccr5アンタゴニスト | |
| JP2012072195A (ja) | Ccr5アンタゴニストとして有用なピペラジン誘導体 | |
| JP4589728B2 (ja) | Ccr5アンタゴニストとして有用なピペリジン誘導体 | |
| CA2643323A1 (en) | Ccr5 antagonists useful for treating hiv | |
| MXPA01011185A (en) | Piperazine derivatives useful as ccr5 antagonists | |
| AU2002329889A1 (en) | Piperidine derivatives useful as CCR5 antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090330 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111031 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20120117 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120507 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120801 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20121120 |