JP2007308564A - Hydrorefining method - Google Patents
Hydrorefining method Download PDFInfo
- Publication number
- JP2007308564A JP2007308564A JP2006138296A JP2006138296A JP2007308564A JP 2007308564 A JP2007308564 A JP 2007308564A JP 2006138296 A JP2006138296 A JP 2006138296A JP 2006138296 A JP2006138296 A JP 2006138296A JP 2007308564 A JP2007308564 A JP 2007308564A
- Authority
- JP
- Japan
- Prior art keywords
- oil
- catalyst
- hydrogen
- oxygen
- mass
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P30/00—Technologies relating to oil refining and petrochemical industry
- Y02P30/20—Technologies relating to oil refining and petrochemical industry using bio-feedstock
Landscapes
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
- Catalysts (AREA)
Abstract
【課題】含酸素炭化水素化合物を含有する被処理油を用いた場合に、酸素分が十分に低減された水素化精製油を得ることが可能な水素化精製方法を提供すること。
【解決手段】本発明の水素化精製方法は、水素の存在下、含酸素炭化水素化合物を含有する被処理油と、アルミニウム、ケイ素、ジルコニウム、ホウ素、チタン及びマグネシウムから選ばれる2種以上の元素を含んで構成される多孔性無機酸化物並びに該多孔性無機酸化物に担持された周期律表第8族の元素から選ばれる1種以上の金属を含有する触媒とを、水素圧力2〜13MPa、液空間速度0.1〜3.0h−1、水素油比150〜1500NL/L、反応温度150〜380℃の条件下で接触させることを特徴とする。
【選択図】なしTo provide a hydrorefining method capable of obtaining a hydrorefined oil having a sufficiently reduced oxygen content when an oil to be treated containing an oxygen-containing hydrocarbon compound is used.
The hydrorefining method of the present invention comprises an oil to be treated containing an oxygen-containing hydrocarbon compound in the presence of hydrogen, and two or more elements selected from aluminum, silicon, zirconium, boron, titanium and magnesium. And a catalyst containing one or more metals selected from Group 8 elements of the periodic table supported on the porous inorganic oxide, and a hydrogen pressure of 2 to 13 MPa The liquid space velocity is 0.1 to 3.0 h −1 , the hydrogen oil ratio is 150 to 1500 NL / L, and the reaction temperature is 150 to 380 ° C.
[Selection figure] None
Description
本発明は、水素化精製方法に関し、より詳しくは、動植物油由来の油脂成分を含む被処理油の水素化精製方法に関する。 The present invention relates to a hydrorefining method, and more particularly, to a hydrotreating method for oil to be treated containing fat and oil components derived from animal and vegetable oils.
地球温暖化の防止対策として、バイオマスエネルギーの有効利用に注目が集まっている。バイオマスエネルギーの中でも植物由来のバイオマスエネルギーは、植物の成長過程で光合成により二酸化炭素から変換された炭化水素を有効利用できるため、ライフサイクルの観点からすると大気中の二酸化炭素の増加につながらない、いわゆる、カーボンニュートラルという性質を持つ。 Attention has been focused on the effective use of biomass energy as a measure to prevent global warming. Among biomass energy, biomass energy derived from plants can effectively use hydrocarbons converted from carbon dioxide by photosynthesis during the growth of plants, so it does not lead to an increase in carbon dioxide in the atmosphere from the viewpoint of life cycle, so-called, It has the property of being carbon neutral.
このようなバイオマスエネルギーの利用は、輸送用燃料の分野においても種々検討がなされている。例えば、ディーゼル燃料として動植物油由来の燃料を使用できれば、ディーゼルエンジンの高いエネルギー効率との相乗効果により二酸化炭素の排出量削減において有効な役割を果たすと期待されている。動植物油を利用したディーゼル燃料としては、脂肪酸メチルエステル油(Fatty Acid Methyl Ester)が知られている。脂肪酸メチルエステル油は、動植物油の一般的な構造であるトリグリセリド構造に対し、アルカリ等によってメタノールとのエステル交換を行うことで製造されている。しかしながら、脂肪酸メチルエステル油を製造するプロセスにおいては、以下の特許文献1に記載されている通り、副生するグリセリンの処理が必要であったり、生成油の洗浄などにコストやエネルギーがかかったりすることが指摘されている。
動植物油由来の油脂成分やこれを原料として製造される燃料を使用するには、上記のような問題に加え、以下のような問題がある。すなわち、動植物油由来の油脂成分は、一般に分子中に酸素原子を有しているため、酸素分がエンジン材質に与える悪影響が懸念されること、並びに、当該酸素分を極低濃度まで除去することが困難であることなどである。また、動植物油由来の油脂成分と石油系炭化水素留分とを混合して使用する場合には、従来の技術では、当該油脂成分中の酸素分及び石油系炭化水素留分中の硫黄分の両方を十分に低減化することができない。 In addition to the above problems, there are the following problems in order to use oil and fat components derived from animal and vegetable oils and fuels produced from these components. That is, since the oil and fat component derived from animal and vegetable oils generally has an oxygen atom in the molecule, there is a concern that the oxygen content may adversely affect the engine material, and that the oxygen content is removed to an extremely low concentration. Is difficult. In addition, when using a mixture of oil and fat components derived from animal and vegetable oils and petroleum hydrocarbon fractions, in the conventional technology, the oxygen content in the oil and fat components and the sulfur content in the petroleum hydrocarbon fractions are used. Both cannot be reduced sufficiently.
そこで、本発明は、含酸素炭化水素化合物を含有する被処理油を用いた場合に、酸素分が十分に低減された水素化精製油を得ることが可能な水素化精製方法を提供することを目的とする。 Therefore, the present invention provides a hydrorefining method capable of obtaining a hydrorefined oil having a sufficiently reduced oxygen content when an oil to be treated containing an oxygen-containing hydrocarbon compound is used. Objective.
上記課題を解決するために、本発明は、水素の存在下、含酸素炭化水素化合物を含有する被処理油と、アルミニウム、ケイ素、ジルコニウム、ホウ素、チタン及びマグネシウムから選ばれる2種以上の元素を含んで構成される多孔性無機酸化物並びに該多孔性無機酸化物に担持された周期律表第8族の元素から選ばれる1種以上の金属を含有する触媒とを、水素圧力2〜13MPa、液空間速度0.1〜3.0h−1、水素油比150〜1500NL/L、反応温度150〜380℃の条件下で接触させることを特徴とする水素化精製方法を提供する。 In order to solve the above problems, the present invention comprises an oil to be treated containing an oxygen-containing hydrocarbon compound in the presence of hydrogen, and two or more elements selected from aluminum, silicon, zirconium, boron, titanium and magnesium. A porous inorganic oxide comprising and a catalyst containing one or more metals selected from Group 8 elements supported on the porous inorganic oxide, a hydrogen pressure of 2 to 13 MPa, There is provided a hydrorefining method characterized by contacting under the conditions of a liquid space velocity of 0.1 to 3.0 h −1 , a hydrogen oil ratio of 150 to 1500 NL / L, and a reaction temperature of 150 to 380 ° C.
本発明の水素化精製方法によれば、含酸素炭化水素化合物を含有する被処理油と上記特定の触媒とを上記特定の条件下で接触させることによって、酸素分が十分に低減された水素化精製油を経済的に極めて有効に得ることができる。 According to the hydrorefining method of the present invention, hydrogenation in which the oxygen content is sufficiently reduced by contacting an oil to be treated containing an oxygen-containing hydrocarbon compound with the specific catalyst under the specific conditions. Refined oil can be obtained very effectively economically.
本発明の水素化精製方法においては、被処理油の全量を基準として、酸素分の含有量が0.1〜15質量%であることが好ましい。また、被処理油が硫黄分を含有するものである場合、その含有量は50質量ppm以下であることが好ましい。被処理油の酸素分及び硫黄分がそれぞれ上記の範囲内であると、安定した脱酸素活性を長期にわたって維持することができる。したがって、本発明によれば、含酸素炭化水素化合物及び含硫黄炭化水素化合物を含有する被処理油を用いて、酸素分及び硫黄分の両方が十分に低減された水素化精製油を得ることが可能である。 In the hydrorefining method of this invention, it is preferable that content of oxygen content is 0.1-15 mass% on the basis of the whole quantity of to-be-processed oil. Moreover, when the to-be-processed oil contains a sulfur content, it is preferable that the content is 50 mass ppm or less. When the oxygen content and sulfur content of the oil to be treated are within the above ranges, stable deoxygenation activity can be maintained over a long period of time. Therefore, according to the present invention, it is possible to obtain a hydrorefined oil in which both the oxygen content and the sulfur content are sufficiently reduced by using the oil to be treated containing the oxygen-containing hydrocarbon compound and the sulfur-containing hydrocarbon compound. Is possible.
また、本発明の水素化精製方法においては、バイオマスエネルギーの有効利用の点から、含酸素炭化水素化合物が動植物油に由来する油脂成分であることが好ましい。 Moreover, in the hydrorefining method of this invention, it is preferable that an oxygen-containing hydrocarbon compound is an oil-fat component originating in animal and vegetable oil from the point of effective utilization of biomass energy.
また、原材料の加工に必要なエネルギーを低減できることから、含酸素炭化水素化合物に占めるトリグリセリド構造を有する化合物の割合は90モル%以上であることが好ましい。 Moreover, since the energy required for processing raw materials can be reduced, the proportion of the compound having a triglyceride structure in the oxygen-containing hydrocarbon compound is preferably 90 mol% or more.
また、本発明の水素化精製方法に用いられる触媒の脱酸素活性を一層向上させる観点から、当該触媒に担持された金属は、Pd、Pt、Rh、Ir、Au及びNiから選ばれる1種以上の元素であることが好ましい。 From the viewpoint of further improving the deoxygenation activity of the catalyst used in the hydrorefining method of the present invention, the metal supported on the catalyst is one or more selected from Pd, Pt, Rh, Ir, Au and Ni. It is preferable that it is an element of these.
さらに、本発明においては、高い触媒活性を十分長期にわたり維持させる観点から、触媒に担持されている金属が還元状態にある当該触媒1g当たりの一酸化炭素吸着量が、0.003〜0.05mmolであることが好ましい。 Furthermore, in the present invention, from the viewpoint of maintaining a high catalyst activity for a sufficiently long time, the carbon monoxide adsorption amount per 1 g of the catalyst in which the metal supported on the catalyst is in a reduced state is 0.003 to 0.05 mmol. It is preferable that
本発明によれば、含酸素炭化水素化合物を含有する被処理油を用いた場合に、酸素分が十分に低減された水素化精製油を経済的に極めて有効に得ることが可能な水素化精製方法が提供される。 According to the present invention, hydrotreating capable of obtaining a hydrorefined oil having a sufficiently reduced oxygen content economically and effectively when an oil to be treated containing an oxygen-containing hydrocarbon compound is used. A method is provided.
以下、本発明の好適な実施形態について詳細に説明する。 Hereinafter, preferred embodiments of the present invention will be described in detail.
本発明においては、含酸素炭化水素化合物を含有する被処理油が用いられる。含酸素炭化水素化合物としては、動植物油由来の油脂成分が好適である。ここで、本発明における油脂成分には、天然もしくは人工的に生産、製造される動植物油脂及び動植物油成分及び/又はこれらの油脂を由来して生産、製造される成分及びこれらの油脂製品の性能を維持、向上させる目的で添加される成分が包含される。 In the present invention, an oil to be treated containing an oxygen-containing hydrocarbon compound is used. As the oxygen-containing hydrocarbon compound, oil and fat components derived from animal and vegetable oils are suitable. Here, the fats and oils component in the present invention includes natural and artificially produced and manufactured animal and vegetable oils and fats and / or animal and vegetable oil components and / or components produced and produced from these fats and oils and the performance of these fat and oil products. Components added for the purpose of maintaining and improving the above are included.
動植物油に由来する油脂成分としては、例えば、牛脂、菜種油、大豆油、パーム油などが挙げられる。本発明においては動植物油に由来する油脂成分として、いかなる油脂を用いてもよく、これら油脂を使用した後の廃油でもよい。ただし、カーボンニュートラルの観点からは植物油脂が好ましく、脂肪酸アルキル鎖炭素数及びその反応性の観点から、菜種油、大豆油及びパーム油がより好ましい。なお、上記の油脂は1種を単独で又は2種以上を混合して用いてもよい。 Examples of the oil and fat component derived from animal and vegetable oils include beef tallow, rapeseed oil, soybean oil, and palm oil. In the present invention, any fats and oils derived from animal and vegetable oils may be used, and waste oil after using these fats and oils may be used. However, vegetable oils and fats are preferable from the viewpoint of carbon neutral, and rapeseed oil, soybean oil, and palm oil are more preferable from the viewpoint of the number of fatty acid alkyl chain carbons and their reactivity. In addition, you may use said fats and oils individually by 1 type or in mixture of 2 or more types.
動植物油に由来する油脂成分は、一般に脂肪酸トリグリセリド構造を有しているが、その他の脂肪酸や脂肪酸メチルエステルなどのエステル体に加工されている油脂成分を含んでいてもよい。ただし、植物油脂から脂肪酸や脂肪酸エステルを製造する際には二酸化炭素が発生するため、二酸化炭素の排出量を低減化する観点から、植物油脂としてトリグリセリド構造を有した成分が主体であることが好ましい。本発明においては、被処理油に含まれる含酸素炭化水素化合物に占めるトリグリセリド構造を有する化合物の割合が90モル%以上であることが好ましく、92モル%以上であることがより好ましく、95モル%以上であることが更に好ましい。 Oils and fats derived from animal and vegetable oils generally have a fatty acid triglyceride structure, but may contain other oils and fats processed into esters such as fatty acids and fatty acid methyl esters. However, since carbon dioxide is generated when producing fatty acids and fatty acid esters from vegetable oils and fats, it is preferable that the components having a triglyceride structure are mainly used as vegetable oils and fats from the viewpoint of reducing carbon dioxide emissions. . In the present invention, the proportion of the compound having a triglyceride structure in the oxygenated hydrocarbon compound contained in the oil to be treated is preferably 90 mol% or more, more preferably 92 mol% or more, and 95 mol%. It is still more preferable that it is above.
なお、被処理油は、含酸素炭化水素化合物として、上記の動植物油由来の油脂成分の他、プラスチックや溶剤等の化学品由来の化合物を含んでいてもよく、一酸化炭素と水素とからなる合成ガスを原料としたフィッシャートロプシュ反応を経由して得られる合成油を含んでいてもよい。 The oil to be treated may contain, as an oxygen-containing hydrocarbon compound, a compound derived from a chemical such as a plastic or a solvent, in addition to the oil and fat component derived from the above-mentioned animal and vegetable oils, and consists of carbon monoxide and hydrogen. A synthetic oil obtained via a Fischer-Tropsch reaction using synthesis gas as a raw material may be contained.
また、被処理油は、石油系炭化水素留分を含んでいてもよく、原油の蒸留によって得られる留分や水素化脱硫、水素化分解、流動接触分解、接触改質などの反応で得られる留分を含んでいてもよい。これらの留分の混合量は、被処理油に含まれる酸素分及び硫黄分が所定の濃度範囲を満たしている限りにおいて任意に設定することができる。更に、これらの留分と、上記の化学品由来の化合物やフィッシャートロプシュ反応を経由して得られる合成油とを併せて混合してもよい。 The oil to be treated may contain a petroleum hydrocarbon fraction, and can be obtained by a fraction obtained by distillation of crude oil or a reaction such as hydrodesulfurization, hydrocracking, fluid catalytic cracking, catalytic reforming. A fraction may be included. The mixing amount of these fractions can be arbitrarily set as long as the oxygen content and sulfur content contained in the oil to be treated satisfy a predetermined concentration range. Furthermore, these fractions may be mixed together with the above-mentioned chemical-derived compounds and synthetic oils obtained via the Fischer-Tropsch reaction.
被処理油に含まれる酸素分は、被処理油全量を基準として、好ましくは0.1〜15質量%であり、より好ましくは1〜15質量%、更に好ましくは3〜14質量%、特に好ましくは5〜13質量%である。酸素分の含有量が0.1質量%未満であると、脱酸素活性及び脱硫活性を安定的に維持することが困難となる傾向にある。他方、酸素分の含有量が15質量%を超えると、副生する水の処理に要する設備が必要となることや、水と触媒担体との相互作用が過度となり活性低下したり触媒強度が低下したりする。なお、酸素分の含有量は、一般的な元素分析装置で測定することができ、例えば、試料を白金炭素上で一酸化炭素に変換し、もしくは更に二酸化炭素に変換した後に熱伝導度検出器を用いて測定することができる。 The oxygen content contained in the oil to be treated is preferably 0.1 to 15% by mass, more preferably 1 to 15% by mass, still more preferably 3 to 14% by mass, particularly preferably based on the total amount of the oil to be treated. Is 5 to 13% by mass. If the oxygen content is less than 0.1% by mass, it tends to be difficult to stably maintain the deoxygenation activity and desulfurization activity. On the other hand, if the oxygen content exceeds 15% by mass, equipment required for the treatment of by-product water is required, the interaction between water and the catalyst carrier becomes excessive, and the activity decreases or the catalyst strength decreases. To do. The oxygen content can be measured with a general elemental analyzer. For example, the sample is converted to carbon monoxide on platinum carbon, or further converted to carbon dioxide, and then a thermal conductivity detector. Can be measured.
石油系炭化水素留分としては、一般的な石油精製工程で得られる留分を用いることができる。例えば、常圧蒸留装置や減圧蒸留装置から得られる所定の沸点範囲に相当する留分、あるいは、水素化脱硫装置、水素化分解装置、残油直接脱硫装置、流動接触分解装置などから得られる、所定の沸点範囲に相当する留分を使用してもよい。なお、上記の各装置から得られる留分は1種を単独で又は2種以上を混合して用いてもよい。 As the petroleum hydrocarbon fraction, a fraction obtained in a general petroleum refining process can be used. For example, a fraction corresponding to a predetermined boiling range obtained from an atmospheric distillation apparatus or a vacuum distillation apparatus, or obtained from a hydrodesulfurization apparatus, a hydrocracking apparatus, a residual oil direct desulfurization apparatus, a fluid catalytic cracking apparatus, etc. A fraction corresponding to a predetermined boiling range may be used. In addition, you may use the fraction obtained from each said apparatus individually by 1 type or in mixture of 2 or more types.
被処理油に含まれる硫黄分は、被処理油全量を基準として、好ましくは50質量ppm以下、より好ましくは20質量ppm以下、更に好ましくは10質量ppm以下である。硫黄分の含有量が50質量ppmを超える場合、脱酸素活性を安定的に維持することが困難となる傾向にあるとともに、水素化精製油に含まれる硫黄分含有量が増加する傾向にあり、ディーゼルエンジン等の燃料として用いる場合にエンジン排ガス浄化装置への悪影響が懸念される。なお、本発明における硫黄分は、JIS K 2541「硫黄分試験方法」又はASTM−5453に記載の方法に準拠して測定される硫黄分の質量含有量を意味する。 The sulfur content contained in the oil to be treated is preferably 50 mass ppm or less, more preferably 20 mass ppm or less, still more preferably 10 mass ppm or less, based on the total amount of the oil to be treated. When the sulfur content exceeds 50 mass ppm, it tends to be difficult to stably maintain the deoxygenation activity, and the sulfur content contained in the hydrorefined oil tends to increase. When used as a fuel for a diesel engine or the like, there is a concern about an adverse effect on the engine exhaust gas purification device. In addition, the sulfur content in this invention means the mass content of the sulfur content measured based on the method of JISK2541 "Sulfur content test method" or ASTM-5453.
本発明で用いられる被処理油は、沸点300℃以上の留分を含有することが好ましく、また、沸点700℃を超える重質な留分を含んでいないことが好ましい。沸点300℃以上の留分を含有しない被処理油を用いると、過度の分解によって十分な収率を得ることが困難となる傾向にある。他方、被処理油が沸点700℃を超える重質な留分を含む場合は、重質成分によって触媒における炭素の析出が促進され、活性が低下する傾向にある。なお、本発明における沸点は、JIS K 2254「蒸留試験方法」又はASTM−D86に記載の方法に準拠して測定される値である。 The oil to be treated used in the present invention preferably contains a fraction having a boiling point of 300 ° C. or higher, and preferably does not contain a heavy fraction having a boiling point exceeding 700 ° C. When the oil to be treated that does not contain a fraction having a boiling point of 300 ° C. or higher is used, it tends to be difficult to obtain a sufficient yield due to excessive decomposition. On the other hand, when the oil to be treated contains a heavy fraction having a boiling point higher than 700 ° C., carbon deposition in the catalyst is promoted by the heavy components, and the activity tends to decrease. In addition, the boiling point in this invention is a value measured based on the method as described in JISK2254 "distillation test method" or ASTM-D86.
本発明の水素化精製方法においては、アルミニウム、ケイ素、ジルコニウム、ホウ素、チタン及びマグネシウムから選ばれる2種以上の元素を含んで構成される多孔性無機酸化物並びに該多孔性無機酸化物に担持された周期律表第8族の元素から選ばれる1種以上の金属を含有する触媒が用いられる。 In the hydrorefining method of the present invention, a porous inorganic oxide comprising two or more elements selected from aluminum, silicon, zirconium, boron, titanium, and magnesium, and the porous inorganic oxide are supported. In addition, a catalyst containing one or more metals selected from Group 8 elements of the periodic table is used.
本発明で用いられる触媒の担体としては、上述のようにアルミニウム、ケイ素、ジルコニウム、ホウ素、チタン及びマグネシウムから選ばれる2種以上を含んで構成される多孔性無機酸化物が用いられる。かかる多孔性無機酸化物としては、脱酸素活性及び脱硫活性を一層向上できる点から、アルミニウム、ケイ素、ジルコニウム、ホウ素、チタン及びマグネシウムから選ばれる2種以上であることが好ましく、アルミニウムと他の元素とを含む無機酸化物(酸化アルミニウムと他の酸化物との複合酸化物)が更に好ましい。 As the catalyst carrier used in the present invention, a porous inorganic oxide comprising two or more selected from aluminum, silicon, zirconium, boron, titanium and magnesium as described above is used. The porous inorganic oxide is preferably at least two selected from aluminum, silicon, zirconium, boron, titanium and magnesium from the viewpoint that the deoxygenation activity and desulfurization activity can be further improved. Aluminum and other elements And an inorganic oxide (a composite oxide of aluminum oxide and another oxide) is more preferable.
アルミニウム以外の担体構成元素である、ケイ素、ジルコニウム、ホウ素、チタン及びマグネシウムを担体に導入する方法は特に制限されず、これらの元素を含有する溶液などを原料として用いればよい。例えば、ケイ素については、ケイ素、水ガラス、シリカゾルなど、ホウ素についてはホウ酸など、リンについては、リン酸やリン酸のアルカリ金属塩など、チタンについては硫化チタン、四塩化チタンや各種アルコキサイド塩など、ジルコニウムについては硫酸ジルコニウムや各種アルコキサイド塩などを用いることができる。 The method for introducing silicon, zirconium, boron, titanium and magnesium, which are carrier constituent elements other than aluminum, is not particularly limited, and a solution containing these elements may be used as a raw material. For example, for silicon, silicon, water glass, silica sol, etc., for boron, boric acid, etc., for phosphorus, phosphoric acid and alkali metal salts of phosphoric acid, etc., for titanium, titanium sulfide, titanium tetrachloride and various alkoxide salts, etc. As for zirconium, zirconium sulfate and various alkoxide salts can be used.
上記の酸化アルミニウム以外の担体構成成分の原料は、担体の焼成より前の工程において添加することが好ましい。例えば、アルミニウム水溶液に予め上記原料を添加した後、これらの構成成分を含む水酸化アルミニウムゲルを調製してもよく、調合した水酸化アルミニウムゲルに対して上記原料を添加してもよい。あるいは、市販の酸化アルミニウム中間体やベーマイトパウダーに水もしくは酸性水溶液を添加して混練する工程において上記原料を添加してもよいが、水酸化アルミニウムゲルを調合する段階で共存させることがより好ましい。酸化アルミニウム以外の担体構成成分の効果発現機構は必ずしも解明されたわけではないが、アルミニウムと複合的な酸化物状態を形成していると推察され、このことが担体表面積の増加や活性金属との相互作用を生じることにより、活性に影響を及ぼしていると考えられる。 It is preferable to add the raw materials for the carrier constituents other than the above-described aluminum oxide in the step prior to the firing of the carrier. For example, after adding the said raw material previously to aluminum aqueous solution, the aluminum hydroxide gel containing these structural components may be prepared, and the said raw material may be added with respect to the prepared aluminum hydroxide gel. Alternatively, the above raw materials may be added in a step of adding water or an acidic aqueous solution to a commercially available aluminum oxide intermediate or boehmite powder and kneading them, but it is more preferable to coexist at the stage of preparing aluminum hydroxide gel. Although the mechanism of the effect of the carrier constituents other than aluminum oxide has not necessarily been elucidated, it is presumed that it forms a complex oxide state with aluminum, which increases the surface area of the carrier and the interaction with the active metal. It is considered that the activity is affected by producing the action.
担体としての上記多孔性無機酸化物には、周期律表第8族の元素から選ばれる1種以上の金属が担持される。これらの金属の中でも、Pd、Pt、Rh、Ir、Au及びNiから選ばれる1種以上の金属を用いることが好ましく、2種以上を組み合わせて用いることがより好ましい。好適な組み合わせとしては、例えば、Pd−Pt、Pd−Ir、Pd−Rh、Pd−Au、Pd−Ni、Pt−Rh、Pt−Ir、Pt−Au、Pt−Ni、Rh−Ir、Rh−Au、Rh−Ni、Ir−Au、Ir−Ni、Au−Ni、Pd−Pt−Rh、Pd−Pt−Ir、Pd−Pt−Niなどが挙げられる。このうち、Pd−Pt、Pd−Ni、Pt−Ni、Pd−Ir、Pt−Rh、Pt−Ir、Rh−Ir、Pd−Pt−Rh、Pd−Pt−Ni、Pd−Pt−Irの組み合わせがより好ましく、Pd−Pt、Pd−Ni、Pt−Ni、Pd−Ir、Pt−Ir、Pd−Pt−Ni、Pd−Pt−Irの組み合わせが更に好ましい。水素化精製に際しては、これらの金属を還元状態に変換して使用する。 The porous inorganic oxide as a carrier carries one or more metals selected from Group 8 elements of the periodic table. Among these metals, it is preferable to use one or more metals selected from Pd, Pt, Rh, Ir, Au, and Ni, and it is more preferable to use a combination of two or more. Suitable combinations include, for example, Pd—Pt, Pd—Ir, Pd—Rh, Pd—Au, Pd—Ni, Pt—Rh, Pt—Ir, Pt—Au, Pt—Ni, Rh—Ir, Rh— Au, Rh—Ni, Ir—Au, Ir—Ni, Au—Ni, Pd—Pt—Rh, Pd—Pt—Ir, Pd—Pt—Ni, and the like can be given. Of these, combinations of Pd—Pt, Pd—Ni, Pt—Ni, Pd—Ir, Pt—Rh, Pt—Ir, Rh—Ir, Pd—Pt—Rh, Pd—Pt—Ni, Pd—Pt—Ir Is more preferable, and a combination of Pd—Pt, Pd—Ni, Pt—Ni, Pd—Ir, Pt—Ir, Pd—Pt—Ni, and Pd—Pt—Ir is still more preferable. In hydrorefining, these metals are used after being converted to a reduced state.
触媒質量を基準とする活性金属の合計担持量は、金属として0.1〜2質量%が好ましく、0.2〜1.5質量%がより好ましく、0.5〜1.3質量%が更に好ましい。金属の合計担持量が0.1質量%未満であると、活性点が少なくなり、十分な活性が得られなくなる傾向がある。他方、2質量%を越えると、金属が効果的に分散せず、十分な活性が得られなくなる傾向がある。 The total supported amount of the active metal based on the catalyst mass is preferably 0.1 to 2% by mass, more preferably 0.2 to 1.5% by mass, and further 0.5 to 1.3% by mass as the metal. preferable. If the total supported amount of the metal is less than 0.1% by mass, the active sites tend to decrease and sufficient activity cannot be obtained. On the other hand, if it exceeds 2% by mass, the metal is not effectively dispersed and sufficient activity tends not to be obtained.
これらの活性金属を触媒に含有させる方法は特に限定されず、通常の水素化精製触媒を製造する際に適用される公知の方法を用いることができる。通常、活性金属の塩を含む溶液を触媒担体に含浸する方法が好ましく採用される。また、平衡吸着法、Pore−filling法、Incipient−wetness法なども好ましく採用される。例えば、Pore−filling法は、担体の細孔容積を予め測定しておき、これと同じ容積の金属塩溶液を含浸する方法である。なお、含浸方法は特に限定されるものではなく、金属担持量や触媒担体の物性に応じて適当な方法で含浸することができる。 A method for incorporating these active metals into the catalyst is not particularly limited, and a known method applied when producing an ordinary hydrorefining catalyst can be used. Usually, a method of impregnating a catalyst carrier with a solution containing a salt of an active metal is preferably employed. In addition, an equilibrium adsorption method, a pore-filling method, an incident-wetness method, and the like are also preferably employed. For example, the pore-filling method is a method in which the pore volume of the carrier is measured in advance and impregnated with a metal salt solution having the same volume. The impregnation method is not particularly limited, and it can be impregnated by an appropriate method according to the amount of metal supported and the physical properties of the catalyst carrier.
本発明において、使用する水素化精製触媒の種類数は特に限定されない。例えば、一種類の触媒を単独で使用してもよく、活性金属種や担体構成成分の異なる触媒を複数使用してもよい。 In the present invention, the number of hydrorefining catalysts to be used is not particularly limited. For example, one type of catalyst may be used alone, or a plurality of catalysts having different active metal species and carrier components may be used.
担体成分が異なる複数の触媒を組み合せる場合には、例えば、担体の総質量を基準として酸化アルミニウムの含有量が50質量%以上であり且つ97質量%未満の触媒の後段に、酸化アルミニウムの含有量が1〜30質量%の範囲にある触媒を用いればよい。 When a plurality of catalysts having different support components are combined, for example, the content of aluminum oxide is included in the subsequent stage of the catalyst having an aluminum oxide content of 50% by mass or more and less than 97% by mass based on the total mass of the support. A catalyst having an amount in the range of 1 to 30% by mass may be used.
本発明において用いられる上記触媒の活性金属が還元状態にあるときの、触媒1g当たりの一酸化炭素吸着量は0.003〜0.05mmolであることが好ましく、0.005〜0.04mmolであることがより好ましく、0.009〜0.03mmolであることが更に好ましい。当該吸着量が0.003mmol未満であると、金属が凝集した状態にあり、活性点が減少する傾向にある。他方、当該吸着量が0.05mmolを超えると、反応時間の経過とともに活性低下が促進される傾向にある。一酸化炭素吸着量の測定は、還元金属を担持した触媒に用いる一般的な測定方法を適用することができる。具体的には、水素気流下、温度350℃で一定量の触媒を還元した後に、50℃まで冷却して、パルス法や定容法によって求めることができる。 When the active metal of the catalyst used in the present invention is in a reduced state, the carbon monoxide adsorption amount per 1 g of the catalyst is preferably 0.003 to 0.05 mmol, and is preferably 0.005 to 0.04 mmol. It is more preferable, and it is still more preferable that it is 0.009-0.03 mmol. When the adsorption amount is less than 0.003 mmol, the metal is in an aggregated state, and the active sites tend to decrease. On the other hand, when the adsorption amount exceeds 0.05 mmol, the activity decrease tends to be promoted as the reaction time elapses. For the measurement of the carbon monoxide adsorption amount, a general measurement method used for a catalyst supporting a reduced metal can be applied. Specifically, after reducing a certain amount of catalyst at a temperature of 350 ° C. in a hydrogen stream, the catalyst can be cooled to 50 ° C. and obtained by a pulse method or a constant volume method.
さらに、上記の触媒(水素化精製触媒)以外に、必要に応じて被処理油に随伴して流入するスケール分をトラップしたり触媒床の区切り部分で水素化精製触媒を支持したりする目的でガード触媒、脱金属触媒、不活性充填物を用いてもよい。なお、これらは単独又は組み合せて用いることができる。 Furthermore, in addition to the above catalyst (hydrorefining catalyst), for the purpose of trapping the scale component that flows along with the oil to be treated, if necessary, or supporting the hydrorefining catalyst at the separation part of the catalyst bed. A guard catalyst, a metal removal catalyst, or an inert packing may be used. In addition, these can be used individually or in combination.
水素の存在下で上記の被処理油と触媒とを接触させる際の条件は、水素圧力2〜13MPa、液空間速度(LHSV)0.1〜3.0h−1、水素油比(水素/油比)150〜1500NL/Lである。好ましくは水素圧力2.5〜10MPa、液空間速度0.5〜2.0h−1、水素油比380〜1200NL/Lであり、より好ましくは水素圧力3〜8MPa、液空間速度0.8〜1.8h−1、水素油比350〜1000NL/Lである。これらの条件はいずれも反応活性を左右する因子であり、例えば水素圧力及び水素油比が上記の下限値に満たない場合には、反応性が低下したり活性が急速に低下したりする傾向がある。他方、水素圧力及び水素油比が上記の上限値を超える場合には、圧縮機等の過大な設備投資が必要となる傾向がある。また、液空間速度は低いほど反応に有利な傾向にあるが、上記の下限値未満の場合は、極めて大きな内容積の反応器が必要となり過大な設備投資が必要となる傾向があり、他方、液空間速度が上記の上限値を超える場合は、反応が十分に進行しなくなる傾向がある。 The conditions for contacting the oil to be treated and the catalyst in the presence of hydrogen are as follows: hydrogen pressure 2-13 MPa, liquid space velocity (LHSV) 0.1-3.0 h −1 , hydrogen oil ratio (hydrogen / oil Ratio) 150-1500 NL / L. Preferably, the hydrogen pressure is 2.5 to 10 MPa, the liquid space velocity is 0.5 to 2.0 h −1 , and the hydrogen oil ratio is 380 to 1200 NL / L, more preferably the hydrogen pressure is 3 to 8 MPa, and the liquid space velocity is 0.8 to It is 1.8h < -1 >, hydrogen oil ratio 350-1000NL / L. These conditions are factors that influence the reaction activity. For example, when the hydrogen pressure and the hydrogen oil ratio are less than the above lower limit values, the reactivity tends to decrease or the activity rapidly decreases. is there. On the other hand, when the hydrogen pressure and the hydrogen oil ratio exceed the above upper limit values, there is a tendency that excessive equipment investment such as a compressor is required. Further, the lower the liquid space velocity tends to be advantageous for the reaction, but if the liquid space velocity is less than the above lower limit value, there is a tendency that an extremely large internal volume reactor is required and excessive equipment investment is required, When the liquid space velocity exceeds the above upper limit, the reaction tends not to proceed sufficiently.
また、水素の存在下で被処理油と触媒とを接触させる際の温度条件は、150〜380℃である。好ましくは温度170〜360℃であり、より好ましくは温度220〜350℃である。当該温度が150℃未満であると、脱酸素活性が不十分となる傾向があり、他方、380℃を越えると、被処理油が過度に分解され、液体燃料の製造に有用な留分(例えば、沸点温度が250〜350℃の範囲の留分)の収率が低下する傾向がある。 Moreover, the temperature conditions at the time of making a to-be-processed oil and a catalyst contact in presence of hydrogen are 150-380 degreeC. The temperature is preferably 170 to 360 ° C, more preferably 220 to 350 ° C. If the temperature is less than 150 ° C, the deoxygenation activity tends to be insufficient. On the other hand, if it exceeds 380 ° C, the oil to be treated is excessively decomposed, and a fraction useful for the production of liquid fuel (for example, , The yield of the boiling point temperature in the range of 250 to 350 ° C.) tends to decrease.
反応器の形式としては、固定床方式を採用することができる。すなわち、水素は被処理油に対して向流又は並流のいずれの形式を採用することができる。また、複数の反応器を用いて、向流、並流を組み合せた形式としてもよい。一般的な形式としては、ダウンフローであり、気液双並流形式を採用することができる。また、反応器は単独又は複数を組み合せてもよく、一つの反応器内部を複数の触媒床に区分した構造を採用してもよい。 As the type of the reactor, a fixed bed system can be adopted. That is, hydrogen can adopt either a countercurrent or a parallel flow type with respect to the oil to be treated. Moreover, it is good also as a form which combined the countercurrent and the parallel flow using several reactors. As a general format, it is a down flow, and a gas-liquid twin parallel flow format can be adopted. Moreover, the reactor may be used alone or in combination, and a structure in which one reactor is divided into a plurality of catalyst beds may be adopted.
反応器内で水素化精製された水素化精製油は気液分離工程や精留工程等を経て所定の留分を含有する水素化精製油に分画される。例えば、軽油留分や残さ留分に分画される。さらに必要に応じてガス、ナフサ留分、灯油留分を分画することもある。生成するこのような軽質炭化水素留分の一部を、水蒸気改質装置において改質することにより水素を製造することができる。このようにして製造された水素は、水蒸気改質に用いた原料がバイオマス由来炭化水素であることから、カーボンニュートラルという特徴を有しており、環境への負荷を低減することができる。なお、被処理油に含まれている酸素分や硫黄分の反応に伴って水、一酸化炭素、二酸化炭素、硫化水素などが発生する可能性があるが、複数の反応器の間や生成物回収工程に気液分離設備やその他の副生ガス除去装置を設置してもよい。 The hydrorefined oil hydrorefined in the reactor is fractionated into a hydrorefined oil containing a predetermined fraction through a gas-liquid separation process, a rectification process, and the like. For example, it is fractionated into a light oil fraction and a residual fraction. Furthermore, the gas, naphtha fraction, and kerosene fraction may be fractionated as necessary. Hydrogen can be produced by reforming a part of the light hydrocarbon fraction produced in a steam reformer. The hydrogen produced in this way has a characteristic of carbon neutral because the raw material used for steam reforming is a biomass-derived hydrocarbon, and can reduce the burden on the environment. In addition, water, carbon monoxide, carbon dioxide, hydrogen sulfide, etc. may be generated due to the reaction of oxygen and sulfur contained in the oil to be treated. A gas-liquid separation facility or other by-product gas removal device may be installed in the recovery process.
水素ガスは加熱炉を通過前もしくは通過後の被処理油に随伴させて最初の反応器の入口から導入することが一般的であるが、これとは別に、反応器内の温度を制御するとともに、反応器内全体にわたって水素圧力を維持する目的で触媒床の間や複数の反応器の間から水素ガスを導入してもよい。このようにして導入される水素を一般にクエンチ水素と呼ぶ。被処理油に随伴して導入する水素ガスに対するクエンチ水素の割合は、10〜60容量%であることが好ましく、15〜50容量%であることがより好ましい。クエンチ水素の割合が10容量未満であると後段の反応部位での反応が十分に進行しない傾向があり、クエンチ水素の割合が60容積%を超えると反応器入口付近での反応が十分に進行しない傾向がある。 In general, hydrogen gas is introduced from the inlet of the first reactor along with the oil to be treated before or after passing through the heating furnace, but separately from this, the temperature in the reactor is controlled. In order to maintain the hydrogen pressure throughout the reactor, hydrogen gas may be introduced between the catalyst beds or between a plurality of reactors. The hydrogen thus introduced is generally called quench hydrogen. The ratio of quench hydrogen to hydrogen gas introduced along with the oil to be treated is preferably 10 to 60% by volume, and more preferably 15 to 50% by volume. If the rate of quench hydrogen is less than 10 volumes, the reaction at the subsequent reaction site tends not to proceed sufficiently. If the rate of quench hydrogen exceeds 60% by volume, the reaction near the reactor inlet does not proceed sufficiently. Tend.
本発明によって製造される水素化精製油を軽油留分基材として用いる場合は、少なくとも260〜300℃の沸点を有する留分を含有し、硫黄分の含有量が10質量ppm以下であり且つ酸素分の含有量0.5質量%以下であることが好ましく、硫黄分の含有量が7質量ppm以下であり且つ酸素分の含有量0.3質量%以下であることがより好ましく、硫黄分の含有量が3質量ppm以下であり且つ酸素分の含有量0.2質量%以下であることが更に好ましい。硫黄分及び酸素分が上記の上限値を超える場合、ディーゼルエンジンの排出ガス処理装置で使用されるフィルターや触媒、さらにエンジンその他の材質に影響を及ぼす恐れがある。 When the hydrorefined oil produced according to the present invention is used as a light oil fraction base, it contains a fraction having a boiling point of at least 260 to 300 ° C., a sulfur content of 10 mass ppm or less, and oxygen The content of min is preferably 0.5% by mass or less, more preferably the content of sulfur is 7% by mass or less, and the content of oxygen is 0.3% by mass or less. More preferably, the content is 3 mass ppm or less and the oxygen content is 0.2 mass% or less. When the sulfur content and the oxygen content exceed the above upper limit values, there is a risk of affecting the filter and catalyst used in the exhaust gas treatment device of the diesel engine, and further the engine and other materials.
本発明によって製造される水素化精製油は、特にディーゼル軽油や重油基材として好適に用いることができる。水素化精製油は単独でディーゼル軽油や重油基材として用いてもよいが、他の基材などの成分を混合したディーゼル軽油又は重質基材として用いることができる。他の基材としては、一般的な石油精製工程で得られる軽油留分及び/又は灯油留分、本発明の水素化精製方法で得られる残さ留分を混合することもできる。さらに、水素と一酸化炭素から構成される、いわゆる合成ガスを原料とし、フィッシャートロプシュ反応などを経由して得られる合成軽油もしくは合成灯油を混合することができる。これらの合成軽油や合成灯油は芳香族分をほとんど含有せず、飽和炭化水素を主成分とし、セタン価が高いことが特徴である。なお、合成ガスの製造方法としては公知の方法を用いることができ、特に限定されるものではない。 The hydrorefined oil produced by the present invention can be suitably used particularly as a diesel light oil or heavy oil base material. The hydrorefined oil may be used alone as a diesel light oil or heavy oil base material, but can be used as a diesel light oil or a heavy base material mixed with components such as other base materials. As other base materials, a light oil fraction and / or kerosene fraction obtained in a general petroleum refining process and a residual fraction obtained by the hydrorefining method of the present invention can be mixed. Furthermore, a synthetic light oil or a kerosene obtained through a Fischer-Tropsch reaction or the like using so-called synthesis gas composed of hydrogen and carbon monoxide as a raw material can be mixed. These synthetic light oils and kerosene are characterized by containing almost no aromatic content, having a saturated hydrocarbon as a main component, and a high cetane number. In addition, a well-known method can be used as a manufacturing method of synthesis gas, and it is not specifically limited.
本発明の水素化精製方法で得られる残さ留分は、硫黄分の含有量が0.1質量%以下であり、酸素分の含有量が1質量%以下であり、低硫黄重質基材として使用することができる。また、当該残さ留分は、接触分解用原料油として好適である。このように低硫黄レベルの残さ留分を接触分解装置に供することにより、硫黄分の少ないガソリン基材やその他燃料油基材を製造することができる。さらに、当該残さ留分は、水素化分解用原料油として用いることもできる。このような残さ留分を水素化分解装置に供することにより、分解活性の向上や生成油各留分性状の高品質化を達成することができる。 The residual fraction obtained by the hydrorefining method of the present invention has a sulfur content of 0.1% by mass or less, an oxygen content of 1% by mass or less, and is used as a low sulfur heavy substrate. Can be used. The residual fraction is suitable as a feedstock for catalytic cracking. Thus, a gasoline base material and other fuel oil base materials with little sulfur content can be manufactured by using the residue fraction of a low sulfur level for a catalytic cracking apparatus. Further, the residual fraction can be used as a feedstock for hydrocracking. By providing such a residual fraction to a hydrocracking apparatus, it is possible to improve the cracking activity and to improve the quality of each product oil fraction.
以下、実施例及び比較例に基づき本発明を更に具体的に説明するが、本発明は以下の実施例に何ら限定されるものではない。 EXAMPLES Hereinafter, although this invention is demonstrated more concretely based on an Example and a comparative example, this invention is not limited to a following example at all.
(触媒の調製)
<触媒A>
濃度5質量%のアルミン酸ナトリウム水溶液3000gに水ガラス3号185gを加え、65℃に保温した容器に入れた。他方、65℃に保温した別の容器において濃度2.5質量%の硫酸アルミニウム水溶液3000gを調製し、これに前述のアルミン酸ナトリウムを含む水溶液を滴下した。混合溶液のpHが7.0になる時点を終点とし、得られたスラリー状の生成物をフィルターに通して濾取し、ケーキ状のスラリーを得た。
(Preparation of catalyst)
<Catalyst A>
185 g of water glass No. 3 was added to 3000 g of an aqueous sodium aluminate solution having a concentration of 5% by mass and placed in a container kept at 65 ° C. On the other hand, 3000 g of an aluminum sulfate aqueous solution having a concentration of 2.5% by mass was prepared in another container kept at 65 ° C., and the aqueous solution containing sodium aluminate described above was added dropwise thereto. The time when the pH of the mixed solution reached 7.0 was set as the end point, and the resulting slurry product was filtered through a filter to obtain a cake slurry.
ケーキ状のスラリーを還流冷却器を取り付けた容器に移し、蒸留水150mlと27%アンモニア水溶液10gを加え、75℃で20時間加熱攪拌した。該スラリーを混練装置に入れ、80℃以上に加熱し水分を除去しながら混練し、粘土状の混練物を得た。得られた混練物を押出し成形機によって直径1.5mmシリンダーの形状に押し出し、110℃で1時間乾燥した後、550℃で焼成し、成形担体を得た。 The cake-like slurry was transferred to a container equipped with a reflux condenser, 150 ml of distilled water and 10 g of 27% aqueous ammonia solution were added, and the mixture was heated and stirred at 75 ° C. for 20 hours. The slurry was put in a kneading apparatus and heated to 80 ° C. or higher and kneaded while removing moisture to obtain a clay-like kneaded product. The obtained kneaded material was extruded into a shape of a cylinder having a diameter of 1.5 mm by an extrusion molding machine, dried at 110 ° C. for 1 hour, and then fired at 550 ° C. to obtain a molded carrier.
得られた成形担体50gをナス型フラスコに入れ、ロータリーエバポレータ−で脱気しながらテトラアンミン白金(II)クロライドとテトラアンミンパラジウム(II)クロライドの混合水溶液35mlを用いて金属を含浸させた。含浸した試料を110℃で乾燥させた後、350℃で焼成し、触媒Aを得た。触媒Aにおける白金及びパラジウムの担持量は、触媒質量を基準として、それぞれ0.5質量%及び0.7質量%であった。調製した触媒Aの物性を表1に示す。 50 g of the obtained shaped carrier was put into an eggplant-shaped flask, and impregnated with metal using 35 ml of a mixed aqueous solution of tetraammineplatinum (II) chloride and tetraamminepalladium (II) chloride while degassing with a rotary evaporator. The impregnated sample was dried at 110 ° C. and then calcined at 350 ° C. to obtain Catalyst A. The supported amounts of platinum and palladium in the catalyst A were 0.5% by mass and 0.7% by mass, respectively, based on the catalyst mass. Table 1 shows the physical properties of the prepared catalyst A.
<触媒B>
濃度5質量%のアルミン酸ナトリウム水溶液3000gを65℃に保温した容器に入れた。他方、65℃に保温した別の容器において濃度2.5質量%の硫酸アルミニウム水溶液3000gを調製し、これに前述のアルミン酸ナトリウム水溶液を滴下した。混合溶液のpHが7.0になる時点を終点とし、得られたスラリー状生成物をフィルターに通して濾取し、ケーキ状のスラリーを得た。
<Catalyst B>
A sodium aluminate aqueous solution having a concentration of 5% by mass was placed in a container kept at 65 ° C. On the other hand, 3000 g of a 2.5 mass% aluminum sulfate aqueous solution was prepared in another container kept at 65 ° C., and the above-mentioned sodium aluminate aqueous solution was added dropwise thereto. The end point was when the pH of the mixed solution reached 7.0, and the resulting slurry product was filtered through a filter to obtain a cake-like slurry.
ケーキ状スラリーを還流冷却器を取り付けた容器に移し、蒸留水150mlと27%アンモニア代水溶液10gを加え、75℃で20時間加熱撹拌した。該スラリーを混練装置に入れ、80℃以上に加熱し水分を除去しながら混練し、粘土状の混練物を得た。得られた混練物を押出し成形機によって直径1.5mmシリンダーの形状に押し出し、110℃で1時間乾燥した後、550℃で焼成し、成形担体を得た。 The cake-like slurry was transferred to a container equipped with a reflux condenser, 150 ml of distilled water and 10 g of a 27% ammonia aqueous solution were added, and the mixture was heated and stirred at 75 ° C. for 20 hours. The slurry was put in a kneading apparatus and heated to 80 ° C. or higher and kneaded while removing moisture to obtain a clay-like kneaded product. The obtained kneaded material was extruded into a shape of a cylinder having a diameter of 1.5 mm by an extrusion molding machine, dried at 110 ° C. for 1 hour, and then fired at 550 ° C. to obtain a molded carrier.
得られた成形担体50gをナス型フラスコに入れ、ロータリーエバポレータ−で脱気しながら、ジニトロアンミン白金(II)とジニトロアンミンパラジウム(II)の混合水溶液35mlを用いて金属を含浸させた。含浸した試料を110℃で乾燥させた後、350℃で焼成し、触媒Bを得た。触媒Bにおける白金及びパラジウムの担持量は、触媒質量を基準として、それぞれ0.5質量%及び0.7質量%であった。調製した触媒Bの物性を表1に示す。 50 g of the obtained shaped carrier was put into an eggplant-shaped flask, and impregnated with metal using 35 ml of a mixed aqueous solution of dinitroammineplatinum (II) and dinitroamminepalladium (II) while degassing with a rotary evaporator. The impregnated sample was dried at 110 ° C. and then calcined at 350 ° C. to obtain Catalyst B. The supported amounts of platinum and palladium in the catalyst B were 0.5 mass% and 0.7 mass%, respectively, based on the catalyst mass. The physical properties of the prepared catalyst B are shown in Table 1.
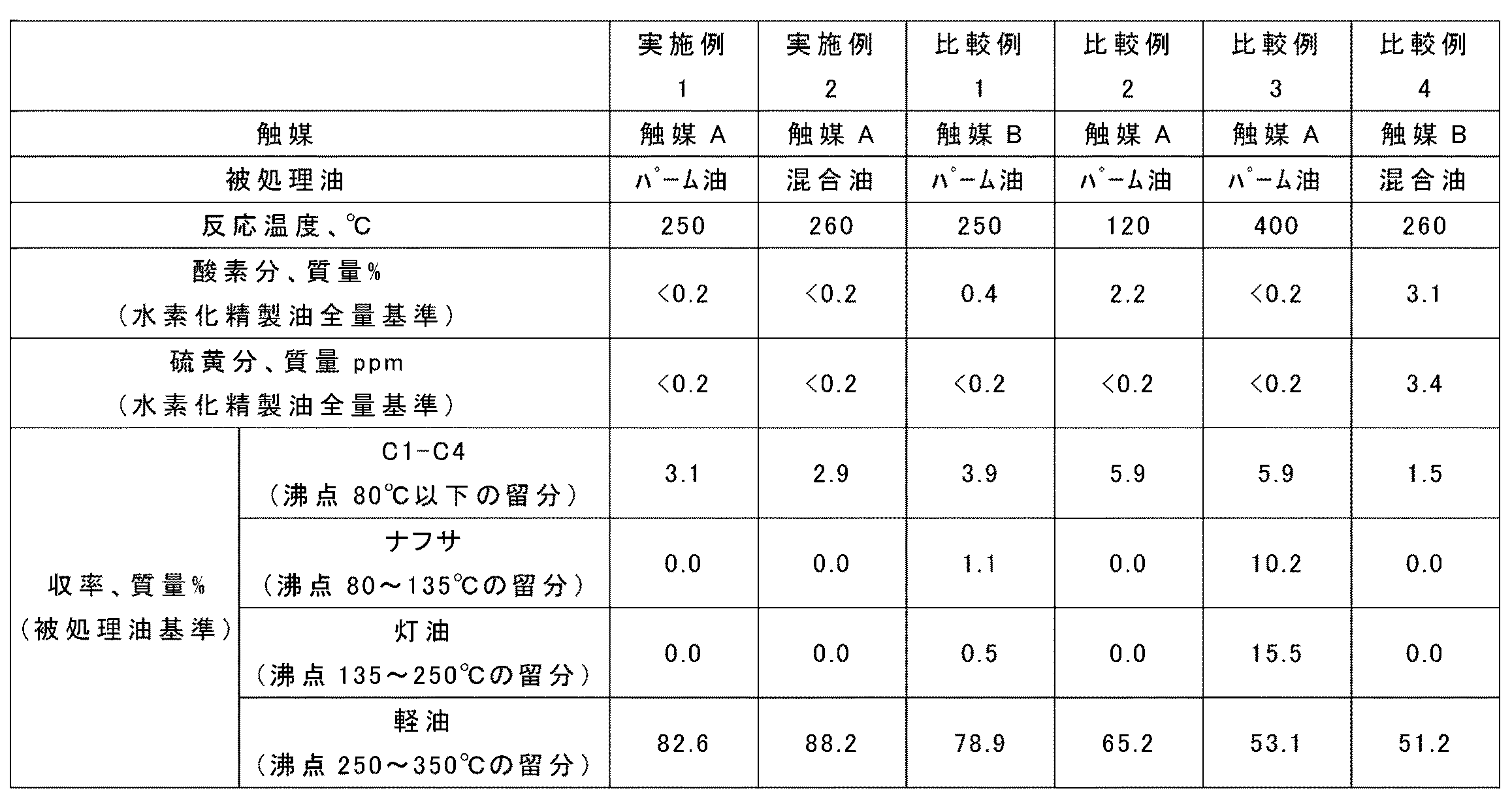
(実施例1)
触媒A(50ml)を充填した第一反応管(内径20mm)と、同じく触媒A(50ml)を充填した第二反応管(内径20mm)を直列に固定床流通式反応装置に取り付けた。その後、触媒の還元処理を、触媒層平均温度(反応温度)320℃、水素分圧5MPa、水素ガス量83ml/分の条件下で6時間行った。
Example 1
A first reaction tube (inner diameter 20 mm) filled with catalyst A (50 ml) and a second reaction tube (inner diameter 20 mm) also filled with catalyst A (50 ml) were attached in series to a fixed bed flow reactor. Thereafter, the reduction treatment of the catalyst was performed for 6 hours under the conditions of an average catalyst layer temperature (reaction temperature) of 320 ° C., a hydrogen partial pressure of 5 MPa, and a hydrogen gas amount of 83 ml / min.
触媒の還元処理を行った後、被処理油としてパーム油(含酸素炭化水素化合物に占めるトリグリセリド構造を有する化合物の割合:98モル%)を用いて、水素化精製を行った。被処理油の15℃密度は0.916g/ml、酸素分の含有量は11.4質量%であった。また、水素化精製の条件は、第一及び第二反応管の反応温度を250℃、圧力を5.5MPa、液空間速度を0.8h−1とした。なお、第一反応管と第二反応管の間で導入する水素ガスの容量比率(クエンチ水素比率)は全導入水素の20容量%とし、導入した全水素によって求めた水素/油比を600NL/Lとした。得られた結果を表2に示す。 After reducing the catalyst, hydrorefining was performed using palm oil (ratio of the compound having a triglyceride structure in the oxygen-containing hydrocarbon compound: 98 mol%) as the oil to be treated. The 15 ° C. density of the oil to be treated was 0.916 g / ml, and the oxygen content was 11.4% by mass. The hydrorefining conditions were such that the reaction temperature of the first and second reaction tubes was 250 ° C., the pressure was 5.5 MPa, and the liquid space velocity was 0.8 h −1 . The volume ratio of hydrogen gas introduced between the first reaction pipe and the second reaction pipe (quenched hydrogen ratio) is 20% by volume of the total introduced hydrogen, and the hydrogen / oil ratio obtained from the total hydrogen introduced is 600 NL / L. The obtained results are shown in Table 2.
(実施例2)
実施例1で使用したものと同一のパーム油70体積部と石油系脱硫軽油30体積部とを混合した混合油を被処理油として用い、第一及び第二反応管の反応温度を260℃としたこと以外は、実施例1と同様にして水素化精製を行った。石油系脱硫軽油の15℃密度は0.838g/ml、硫黄分の含有量は15質量ppm、初留点及び終点は、それぞれ211℃及び365℃である。被処理油の密度は0.893g/ml、酸素分の含有量は8.2質量%、硫黄分の含有量は4.2質量ppmである。
(Example 2)
A mixed oil obtained by mixing 70 parts by volume of the same palm oil and 30 parts by volume of petroleum-based desulfurized light oil used in Example 1 was used as the oil to be treated, and the reaction temperature of the first and second reaction tubes was 260 ° C. Except that, hydrorefining was performed in the same manner as in Example 1. The petroleum-based desulfurized light oil has a 15 ° C. density of 0.838 g / ml, a sulfur content of 15 ppm by mass, and an initial boiling point and an end point of 211 ° C. and 365 ° C., respectively. The density of the oil to be treated is 0.893 g / ml, the oxygen content is 8.2 mass%, and the sulfur content is 4.2 mass ppm.
(比較例1)
触媒Aの代わりに触媒Bを用いたこと以外は実施例1と同様にして水素化精製を行った。得られた結果を表2に示す。
(Comparative Example 1)
The hydrorefining was performed in the same manner as in Example 1 except that the catalyst B was used instead of the catalyst A. The obtained results are shown in Table 2.
(比較例2)
水素化精製の際に第一及び第二反応管の反応温度を120℃としたこと以外は実施例1と同様にして水素化精製を行った。得られた結果を表2に示す。
(Comparative Example 2)
Hydrorefining was carried out in the same manner as in Example 1 except that the reaction temperature of the first and second reaction tubes was 120 ° C. during the hydrorefining. The obtained results are shown in Table 2.
(比較例3)
水素化精製の際に第一及び第二反応管の反応温度を400℃としたこと以外は実施例1と同様にして水素化精製を行った。得られた結果を表2に示す。
(Comparative Example 3)
Hydrorefining was performed in the same manner as in Example 1 except that the reaction temperature of the first and second reaction tubes was set to 400 ° C. during hydrorefining. The obtained results are shown in Table 2.
(比較例4)
触媒Aの代わりに触媒Bを用いたこと以外は実施例2と同様にして水素化精製を行った。得られた結果を表2に示す。
(Comparative Example 4)
The hydrorefining was performed in the same manner as in Example 2 except that the catalyst B was used instead of the catalyst A. The obtained results are shown in Table 2.
Claims (6)
含酸素炭化水素化合物を含有する被処理油と、
アルミニウム、ケイ素、ジルコニウム、ホウ素、チタン及びマグネシウムから選ばれる2種以上の元素を含んで構成される多孔性無機酸化物並びに該多孔性無機酸化物に担持された周期律表第8族の元素から選ばれる1種以上の金属を含有する触媒とを、
水素圧力2〜13MPa、液空間速度0.1〜3.0h−1、水素油比150〜1500NL/L、反応温度150〜380℃の条件下で接触させることを特徴とする水素化精製方法。 In the presence of hydrogen,
An oil to be treated containing an oxygen-containing hydrocarbon compound;
From a porous inorganic oxide comprising two or more elements selected from aluminum, silicon, zirconium, boron, titanium and magnesium, and an element belonging to Group 8 of the periodic table carried on the porous inorganic oxide A catalyst containing one or more selected metals,
A hydrorefining method comprising contacting under conditions of a hydrogen pressure of 2 to 13 MPa, a liquid space velocity of 0.1 to 3.0 h −1 , a hydrogen oil ratio of 150 to 1500 NL / L, and a reaction temperature of 150 to 380 ° C.
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006138296A JP2007308564A (en) | 2006-05-17 | 2006-05-17 | Hydrorefining method |
| KR1020087030783A KR101362950B1 (en) | 2006-05-17 | 2007-05-15 | Hydrorefining process |
| PCT/JP2007/059972 WO2007132857A1 (en) | 2006-05-17 | 2007-05-15 | Hydrorefining process |
| CNA2007800178680A CN101448918A (en) | 2006-05-17 | 2007-05-15 | Hydrofining process |
| MYPI20084494A MY165883A (en) | 2006-05-17 | 2008-11-10 | Hydrorefining process |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006138296A JP2007308564A (en) | 2006-05-17 | 2006-05-17 | Hydrorefining method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2007308564A true JP2007308564A (en) | 2007-11-29 |
Family
ID=38841726
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006138296A Pending JP2007308564A (en) | 2006-05-17 | 2006-05-17 | Hydrorefining method |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2007308564A (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007308563A (en) * | 2006-05-17 | 2007-11-29 | Nippon Oil Corp | Hydrorefining method |
| JP2009138144A (en) * | 2007-12-07 | 2009-06-25 | Nippon Oil Corp | Method for producing hydrocarbon oil |
| JP2010084060A (en) * | 2008-10-01 | 2010-04-15 | Jfe Engineering Corp | Apparatus for manufacturing high grade hydrocarbon oil and manufacturing method therefor |
| WO2012120926A1 (en) | 2011-03-07 | 2012-09-13 | Jx日鉱日石エネルギー株式会社 | Hydrocarbon fuel production method |
| JP2012523473A (en) * | 2009-04-07 | 2012-10-04 | ガス、テクノロジー、インスティチュート | Biohydrolysis of biomass to produce high quality liquid fuel |
| JP2013035945A (en) * | 2011-08-08 | 2013-02-21 | Kitakyushu Foundation For The Advancement Of Industry Science & Technology | Catalytic cracking method for biomass and decarbonation/hydrogenation catalytic cracking catalyst used for the same |
| US8915981B2 (en) | 2009-04-07 | 2014-12-23 | Gas Technology Institute | Method for producing methane from biomass |
| JP2019171337A (en) * | 2018-03-29 | 2019-10-10 | Jxtgエネルギー株式会社 | Hydrogenation catalyst and method for producing low aromatic solvent |
| US10647933B2 (en) | 2015-11-12 | 2020-05-12 | Gas Technology Institute | Activated carbon as a high value product of hydropyrolysis |
| JP2021518859A (en) * | 2018-04-30 | 2021-08-05 | グリーン テクノロジー リサーチ シーオー.,エルティーディーGreen Technology Research Co.,Ltd | Bio-based material processing method and equipment for processing it |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59108088A (en) * | 1982-11-10 | 1984-06-22 | Honda Motor Co Ltd | Production of paraffin hydrocarbon |
| JP2003171670A (en) * | 2001-12-07 | 2003-06-20 | Kawaken Fine Chem Co Ltd | Method for producing hydrocarbons and catalyst for producing hydrocarbons |
| US20040230085A1 (en) * | 2002-09-06 | 2004-11-18 | Juha Jakkula | Process for producing a hydrocarbon component of biological origin |
-
2006
- 2006-05-17 JP JP2006138296A patent/JP2007308564A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59108088A (en) * | 1982-11-10 | 1984-06-22 | Honda Motor Co Ltd | Production of paraffin hydrocarbon |
| JP2003171670A (en) * | 2001-12-07 | 2003-06-20 | Kawaken Fine Chem Co Ltd | Method for producing hydrocarbons and catalyst for producing hydrocarbons |
| US20040230085A1 (en) * | 2002-09-06 | 2004-11-18 | Juha Jakkula | Process for producing a hydrocarbon component of biological origin |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007308563A (en) * | 2006-05-17 | 2007-11-29 | Nippon Oil Corp | Hydrorefining method |
| JP2009138144A (en) * | 2007-12-07 | 2009-06-25 | Nippon Oil Corp | Method for producing hydrocarbon oil |
| JP2010084060A (en) * | 2008-10-01 | 2010-04-15 | Jfe Engineering Corp | Apparatus for manufacturing high grade hydrocarbon oil and manufacturing method therefor |
| JP2012523473A (en) * | 2009-04-07 | 2012-10-04 | ガス、テクノロジー、インスティチュート | Biohydrolysis of biomass to produce high quality liquid fuel |
| US8915981B2 (en) | 2009-04-07 | 2014-12-23 | Gas Technology Institute | Method for producing methane from biomass |
| WO2012120926A1 (en) | 2011-03-07 | 2012-09-13 | Jx日鉱日石エネルギー株式会社 | Hydrocarbon fuel production method |
| JP2013035945A (en) * | 2011-08-08 | 2013-02-21 | Kitakyushu Foundation For The Advancement Of Industry Science & Technology | Catalytic cracking method for biomass and decarbonation/hydrogenation catalytic cracking catalyst used for the same |
| US10647933B2 (en) | 2015-11-12 | 2020-05-12 | Gas Technology Institute | Activated carbon as a high value product of hydropyrolysis |
| JP2019171337A (en) * | 2018-03-29 | 2019-10-10 | Jxtgエネルギー株式会社 | Hydrogenation catalyst and method for producing low aromatic solvent |
| JP2021518859A (en) * | 2018-04-30 | 2021-08-05 | グリーン テクノロジー リサーチ シーオー.,エルティーディーGreen Technology Research Co.,Ltd | Bio-based material processing method and equipment for processing it |
| JP2023076755A (en) * | 2018-04-30 | 2023-06-01 | グリーン テクノロジー リサーチ シーオー.,エルティーディー | Processing method of bio-based raw material and processing device thereof |
| JP7490851B2 (en) | 2018-04-30 | 2024-05-27 | グリーン テクノロジー リサーチ シーオー.,エルティーディー | Method for processing bio-based materials and device for processing same |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4832871B2 (en) | Hydrorefining method | |
| JP4878824B2 (en) | Manufacturing method of environmentally low load type fuel and environmentally low load type fuel | |
| JP5317644B2 (en) | Method for producing aviation fuel base material | |
| AU2009293731B2 (en) | Process for producing hydrocarbon oil | |
| JP5070169B2 (en) | Method for producing hydrocarbon oil | |
| JP5057954B2 (en) | Method for producing hydrocarbon oil | |
| JP5005451B2 (en) | Method for producing hydrocarbon oil | |
| JP2010121071A (en) | Aviation fuel oil base and aviation fuel oil composition | |
| KR101452793B1 (en) | Hydrogenation purification method | |
| JP5022117B2 (en) | Method for producing hydrocarbon oil | |
| KR101362950B1 (en) | Hydrorefining process | |
| JP2007308564A (en) | Hydrorefining method | |
| JP5189740B2 (en) | Hydrorefining method | |
| JP4914643B2 (en) | Hydrorefining method and environment-friendly gasoline base material | |
| JP5070168B2 (en) | Method for producing hydrocarbon oil | |
| JP4914644B2 (en) | Hydrorefining method and environment-friendly gasoline base material | |
| JP5349213B2 (en) | Aviation fuel oil base material production method and aviation fuel oil composition | |
| JP5588171B2 (en) | Method for producing hydrocarbon oil |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090224 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111004 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111130 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120626 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120918 |