JP2004510434A - Method for clonal expansion of hepatic progenitor cells - Google Patents
Method for clonal expansion of hepatic progenitor cells Download PDFInfo
- Publication number
- JP2004510434A JP2004510434A JP2002532583A JP2002532583A JP2004510434A JP 2004510434 A JP2004510434 A JP 2004510434A JP 2002532583 A JP2002532583 A JP 2002532583A JP 2002532583 A JP2002532583 A JP 2002532583A JP 2004510434 A JP2004510434 A JP 2004510434A
- Authority
- JP
- Japan
- Prior art keywords
- cells
- progeny
- progenitor cells
- mixture
- cell
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0607—Non-embryonic pluripotent stem cells, e.g. MASC
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/067—Hepatocytes
- C12N5/0672—Stem cells; Progenitor cells; Precursor cells; Oval cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
- C12N2500/24—Iron; Fe chelators; Transferrin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
- C12N2500/24—Iron; Fe chelators; Transferrin
- C12N2500/25—Insulin-transferrin; Insulin-transferrin-selenium
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/30—Organic components
- C12N2500/36—Lipids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/30—Organic components
- C12N2500/38—Vitamins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/90—Serum-free medium, which may still contain naturally-sourced components
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/11—Epidermal growth factor [EGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/33—Insulin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/38—Hormones with nuclear receptors
- C12N2501/39—Steroid hormones
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2502/00—Coculture with; Conditioned medium produced by
- C12N2502/13—Coculture with; Conditioned medium produced by connective tissue cells; generic mesenchyme cells, e.g. so-called "embryonic fibroblasts"
Abstract
哺乳類の前駆細胞、その子孫、またはその混合物を哺乳類の胚フィーダー細胞上で培地中で培養することを含む、哺乳類の内胚葉に由来する、肝前駆細胞のような前駆細胞、その子孫、またはその混合物の増殖方法が開発される。培地には1つまたは複数のホルモンおよび他の増殖剤が添加され得る。これらのホルモンおよび他の増殖剤には、インスリン、デキサメタゾン、トランスフェリン、ニコチンアミド、血清アルブミン、βメルカプトエタノール、遊離脂肪酸、グルタミン、CuSO4、およびH2SeO3が含まれ得る。また培地には抗体も含まれ得る。重要なことに、培地には血清が含まれない。本発明は、それぞれ上皮成長因子を添加または除去することによる、肝前駆細胞の肝実質細胞または胆管細胞への分化のような、前駆細胞のその成体の運命への分化の誘導方法も含まれる。その後、前駆細胞は、増殖および分化因子の同定、毒性試験、薬剤開発、抗菌試験、または人工肝臓のような体外臓器の調製といった過程の1つまたは複数において使用できるという点から、哺乳類前駆細胞を生産する方法は有用である。A progenitor cell, such as a hepatic progenitor cell, derived from a mammalian endoderm, or a progeny thereof, or a progeny thereof, comprising culturing the mammalian progenitor cell, its progeny, or a mixture thereof on a mammalian embryo feeder cell in a medium. A method of growing the mixture is developed. The medium may be supplemented with one or more hormones and other growth agents. These hormones and other proliferating agents can include insulin, dexamethasone, transferrin, nicotinamide, serum albumin, β-mercaptoethanol, free fatty acids, glutamine, CuSO 4 , and H 2 SeO 3 . The medium may also include antibodies. Importantly, the medium does not contain serum. The invention also includes a method of inducing the differentiation of progenitor cells into their adult fate, such as the differentiation of hepatic progenitor cells into hepatocytes or bile duct cells, by adding or removing epidermal growth factor, respectively. Mammalian progenitor cells can then be used in one or more of the following processes, such as identification of growth and differentiation factors, toxicity testing, drug development, antimicrobial testing, or preparation of extracorporeal organs such as artificial livers. The production method is useful.
Description
【0001】
1. 発明の分野
本発明は、多能性細胞、幹細胞、および他の初期肝前駆細胞を含む、哺乳類肝前駆細胞のクローン増殖のための新規の条件に関する。特に、本発明は合成培地および共培養でフィーダー細胞を使用して肝前駆細胞を増殖させる方法に関する。また、本発明はフィーダーとして使用され、肝前駆細胞の増殖を支持する能力のある細胞にも関する。
【0002】
2. 関連技術の説明
哺乳類組織の多能性前駆細胞群の同定は、臨床的および商業的に重要であり、また発生過程と組織のホメオスタシスの理解のためにも重要である。前駆細胞群は、遺伝子療法、細胞移植、および生体人工臓器の組織工学の理想的な標的である(Millar, A.D. 1992 Nature 357, 455; Langer, R.およびVacanti, J.P. 1993 Science 260, 920; Gage, F.H. 1998 Nature 392, 18)。
【0003】
増殖能および/または多能性の高い組織特異的な「運命づけられた」幹細胞または前駆細胞が存在することは、各々組織に合った特定の方法を用いてクローン同定した、造血幹細胞(Spangrude, G.J.ら、1998 Science 241, 58)、神経幹細胞(Davis, A.A.およびTemple, S. 1994 Nature 372, 263; Stemple, D.L.およびAnderson, D.J. 1992 Cell 71, 973)、および上皮幹細胞(Jones, P.H.およびWatt, F.M. 1993 Cell 73, 713)の研究から明らかである。これらの前駆細胞は、正常な造血、神経、または上皮組織のホメオスタシスを担う、および重度の傷害後に再生応答を担う細胞だと考えられている(Hall, P.A.およびWatt, F.M. 1989 Development 106, 619)。
【0004】
哺乳類の成体の肝臓は、通常は代謝回転が遅く、静止状態の組織であるにも関わらず、高度の肝毒性による傷害または部分的肝切除の後に回復する能力が非常に高い(Fishback, F.C. 1929 Arch. Pathol. 7, 955); (Higgins, G.M.およびAnderson, R.M. 1931 Arch. Pathol. 12, 186)。マウスにおける最近の試験データでは、一連の移植実験で調べたところ、成体の実質細胞は、ほぼ無限の増殖可能性を持っていることが示唆された(Overturfら、1997 Am. J. Pathol. 151, 1273); (Rhim, J.A.ら、1994 Science 263, 1149)。これらの実験では不均質の幹細胞群を利用しているため、観察された増殖可能性が成体の実質細胞に由来するのか、成体の実質細胞の亜集団に由来するのか、および/または実質細胞の未成熟期(即ち、前駆細胞)に由来するのかを証明する能力が限定されている。さらに実験では、使用された宿主がアルブミン・ウロキナーゼ導入遺伝子またはチロシン分解酵素の欠損を持っていたため、胆管上皮分化の証拠が示されていない;このいずれの宿主も、肝細胞系を選択するような特性を持つ。したがって、このアッセイ法では両能性細胞群の試験ができなかった。
【0005】
いくつかの組織学的試験により、妊娠中期の胎児の初期肝細胞が、胆管上皮および成熟幹細胞に分化する両能性の能力を持つことが確立している(Shiojiri, N. 1997 Microscopy Res. Tech. 39, 328−35)。肝臓の発生は、内胚葉の上皮が造心中胚葉と相互作用をした直後に、腹側前腸内胚葉から始まる(Douarin, N.M. 1975 Medical Biol. 53, 427); (Houssaint, E. 1980 Cell Differ. 9, 269)。この肝臓への関係づけは、マウスでは胎齢(E) 8日で起きる。肝臓発生の最初の段階は、形態学的変化の前に、内胚葉において血清アルブミンおよびαフェトプロテインのmRNAが誘導されて、明らかになる(Gualdi, Rら、1996 Genes Dev. 10, 1670)。マウス胎齢E 9.5日で、特定の細胞が増殖し、糸のような形で横中隔の間充織に侵入し、肝臓原基を形成する。その後、肝臓の質量は劇的に増加するが、質量の増加は主に造血細胞によるもので、これはマウスではE10に胎児肝にコロニーを形成し(Houssaint, E. 1981 Cell Differ. 10, 243)、肝細胞に影響を与えて極度にゆがんだ不規則な形をとらせる(Luzzatto, A.C. 1981 Cell Tissue Res. 215, 133)。興味深いことに、遺伝子ターゲティング変異マウスを用いた最近のデータは、いくつかの遺伝子の欠損により、E12からE15の間に、致死的な肝不全、実質細胞のアポトーシス、および/または壊死が引き起こされたことを示す(Gunes, C.ら、1998 EMBO J. 17, 2846)、(Hilberg, F.ら、1993 Nature 365, 1791)、(Motoyama, J.ら、1997 Mech. Dev. 66, 27)、(Schmidt, Cら、1995 Nature 373, 699)。ストレス活性化カスケード (Ganiatsas, S.ら、1998, Proc. Natl. Acad. Sci. USA 95, 6881)、(Nishina, H.ら、1999 Development 126, 505) または抗アポトーシスカスケード(Beg, A.ら、1995 Nature 376, 167)、(Li, Q.ら、1999 Science 284, 321)、(Tanaka, Mら、1999 Immunity 10, 421)の一部である遺伝子の破壊は、不活化された遺伝子が広く発現されるにも関わらず、肝発生をひどく損なうが、造血は損なわない。肝細胞が発生ストレスの刺激に本来感受性なのか、胎児肝自体の中の特定の微小環境がそのような破壊的な効果を引き起こすのかは明らかではない(Doi, T.Sら、1999 Proc. Natl. Acad. Sci. USA 96, 2994)。一方で、成体肝の基礎的構築は、門脈を取り囲む胆管上皮の最初の円柱の出現に依存する(Shiojiri, N. 1997 Microscopy Res. Tech. 39, 328)。免疫組織学的には、肝内胆管上皮細胞の分化の最初の徴候は、胆汁特異的サイトケラチン(CK)の発現である。上皮細胞の細胞質中間フィラメント(IF)タンパク質であるCKタンパク質は、多重遺伝子族によってコードされ、組織および分化に特異的に発現される(Moll, Rら、1982 Cell 31, 11)。成体の肝実質細胞はCK19を全く発現しないが、成体の胆管上皮細胞は発現するので、CK19は胆汁マーカーの中で最も目を引くものの1つである。CK8とCK18のみが初期の肝細胞から成体の肝実質細胞まで通して発現される(Moll, Rら、1982 Cell 31, 11)。マウスのE14に相当するラットの発現のE15.5において、胆管前駆細胞はCK18抗体およびCK8抗体の両方によって強く染色され、一部の胆管前駆細胞はCK19を発現する。発生が進むに連れ、成熟しつつある胆管はCK19に加えてCK7も徐々に発現し始め、ALBの発現を失う(Shiojiri, Nら、1991 Cancer Res. 51, 2611)。ラットのE13という早い時期の肝細胞は均一な細胞群と考えられているが、全ての初期の肝細胞が胆管上皮細胞系に分化することができるのかどうか、およびこのような運命がどのようにして決定されるのかはまだ分かっていない。レトロウイルスベクターを用いるような、決定的な系統マーキング試験は肝細胞については行われておらず、両能性肝前駆細胞の立証に必要なクローン培養条件は決定されていない。
【0006】
クローン増殖分析の1つの大きな障害は、造血細胞の爆発的な増加であり、これにより肝細胞のエクスビボでの増殖が観察できなくなる。したがって、肝細胞の濃縮方法を用いる必要がある。胎児肝において造血細胞を分画が必要な表面マーカーは、詳細に調べられているが(Dzierzak, Eら、1998 Immunol. Today 19, 228)、肝前駆細胞のマーカーの研究は初期段階で、これらはまだあまり決定されていない(Sigal, Sら、1994 Hepatology 19, 999)。さらに、成体肝細胞で通常使用されるエクスビボの増殖条件では、ALB発現のような組織特異的機能の損失を伴った分化が起きる(Block, G.D.ら、1996 J. Cell Biol. 132, 1133)。組織特異的mRNAを合成するやや改善した能力と、翻訳後に完全に組織特異的遺伝子を調節する修復能力とは、無血清で、ホルモン、増殖因子、および/または一部の細胞外マトリックス成分が定義されている混合物を用いて維持された肝細胞にのみ、見受けられる(Jefferson, D.M.ら、1984. Mol. Cell. Biol. 4, 1929; Enat Rら、1984 Proc. Natl. Acad. Sci., 81, 1411)。しかし、増殖する胎児肝細胞は、インビボでそのような血清タンパク質の発現を維持する。当技術分野で明らかでないのは、インビトロで肝前駆細胞を維持して増殖させるためにどうしたらよいかである。肝前駆細胞のエクスビボの増殖を維持する条件の同定には、未充足の需要がある。同様に、新しく肝組織から単離された肝前駆細胞のクローン増殖能力を決定するためのインビトロコロニー形成アッセイ(CFA)法に対する未充足の需要がある;クローン増殖は、培養液に接種された単一の細胞が、接種された細胞に由来するクローンの娘細胞群を生成する能力と定義される。高い細胞密度で接種され、肝臓培養において近接して増殖する細胞の塊からなるクローン増殖も他の研究者により記述されている(Block, G.D.らJ. Cell Biol. 1996, 132, 1133);しかし、これらの以前の研究に記述されている細胞のコロニーは、継代培養(subculture)できず、したがって、定義からしてクローンとは言えず、有用性が限られている。
【0007】
インビトロで肝実質細胞を増殖させる試みもなされてきた。ノートン(Naughton)らの米国特許第5,510,524号は、肝細胞の培養は生体適合性であるが生きていない材料の3次元フレームワークに依存すると主張している。人工的フレームワークがなく、肝前駆細胞が増殖および培養できるような培養条件には未充足の需要がある。また、胆管細胞および肝実質細胞の両方を生成する能力のある両能性分化能を持ち、ならびに特に人工肝臓の成分としての利用、肝毒素の試験、および薬剤開発に適する、クローン化肝前駆細胞には未充足の需要がある。
【0008】
ノートンらの米国特許第5,559,022号では、「予備細胞(reserve cell)」の特徴付けに使用される染料であるエオシンYに結合する肝予備細胞を特許請求しているが、肝予備細胞の同定に、確立されたマーカーを使用しておらず、また予備細胞のクローン増殖の方法も、生存能力のある肝予備細胞の単離に用いたマーカーも提供されていない。少なくとも1つの特異的マーカーの発現および肝実質細胞または胆管細胞のいずれかに分化する能力を含め、肝前駆細胞に必須の多くの特徴を持つ細胞の単離および培養法を開示している方法には、未充足の需要がある。肝前駆細胞のクローン増殖の方法にも未充足の需要がある。クローン増殖は、多能性肝前駆細胞の明確で厳密な区別および同定方法として必須である。
【0009】
ポンティング(Ponting)の米国特許第5,405,772号は、細胞増殖のための培地を特許請求している。米国特許第5,405,772号では、3〜30μg/mlコレステロール、5〜30μg/mlヌクレオシド、および2〜100μg/cm2のIV型コラーゲンまたは0.5〜100μg/cm2のフィブロネクチンのいずれかが必要とされる。これには肝前駆細胞の増殖に特異的で、かつ最適化された培地が必要である。
【0010】
クリ・ハークーチ(Kuri−Harcuch)らの米国特許第4,914,032号は、肝実質細胞を培養する過程を特許請求している。本発明とは反対に、米国特許第4,914,032号は肝前駆細胞の培養または肝細胞のクローン増殖の条件のいずれも開示していない。同様に、クリ・ハークーチらの米国特許第5,030,105号は、肝実質細胞培養を処理することによって薬剤を評価する方法を特許請求している。規定された細胞群を試験に使用するためのクローン増殖条件、および肝前駆細胞の培養のための方法には、未充足の需要がある。
【0011】
ノートンらの米国特許第5,858,721号は、間質細胞のトランスフェクションを特許請求している。しかし米国特許第5,858,721号は、生体適合性の生きていない材料のフレームワークを必要とする点で限定されている。これとは対照に、本発明では合成のメッシュワークを必要としない増殖条件といった未充足の需要がある。
【0012】
本発明者らは、はるかに有用な肝前駆細胞からではなく、肝実質細胞のような成熟した肝細胞を培養するのは不適当であると認識した。発明者らは肝前駆細胞の単離パラメーターおよびクローン増殖の要件を注意深く決定した。前駆細胞ならびに前駆細胞の選択および培養方法には、肝不全の患者の治療薬、および毒性物質の評価、および薬剤の評価を含め、多くの利用方法がある。
【0013】
リード(Reid)らの米国特許第5,576,207号および5,789,246号は、フィーダーおよびホルモン添加合成培地の必要性を開示している。これらの先行研究は、胚の肝臓の間質細胞と、規定された細胞外マトリックス基底、および無血清のホルモン添加培地を組み合せて用いて、肝前駆細胞の増殖条件としている。しかし、使用された合成培地は本発明で使用されたものよりも、複雑であり;細胞は精製されたマトリックス基底(IV型コラーゲンおよびラミニン)上に接種されたが、本明細書ではフィーダー(マトリックスを供給する)の上に直接まかれており;また、胚間質細胞は、胚肝臓からの初代培養として調製され、細胞株として確立されなかった。胚間質細胞株を使用することにより、フィーダー細胞は、はるかに簡単で、実際的で、再現性のある細胞支持手段となる。さらに、STOフィーダーは肝前駆細胞に限定して支持するのではなく、複数の種類の組織から得た前駆細胞にも使用できると考えるのは、合理的である。先行特許では、肝前駆細胞培養液は、高い細胞密度で接種され、その増殖はコロニー形成として観察されたが、これは細胞のクローンではなく、塊の増殖が誘導されたことを意味する。
【0014】
3. 発明の概要
本発明は、前駆細胞、その子孫、またはその混合物を増殖させる方法に関する。特に、本発明は内胚葉由来の前駆細胞、その子孫、またはその混合物を増殖させる方法に関する。細胞は内胚葉組織から得られる。その後、内胚葉由来の前駆細胞、その子孫、またはその混合物は、培地中で、フィーダー細胞を含む層の上で培養される。前駆細胞、その子孫、またはその混合物は、脊椎動物細胞であり得る。前駆細胞、その子孫、またはその混合物は、ICAMまたはICAM−1陽性、および古典的MHCクラスI抗原陰性の表現型を発現する。古典的MHCクラスI抗原は、MHCクラスIa抗原とも呼ばれる。
【0015】
本発明は無血清の、ホルモン添加合成培地、およびフィーダー細胞を用いた肝幹細胞および他の前駆細胞の培養方法にも関する。また、本発明は前駆細胞の子孫、または前駆細胞および前駆細胞の子孫の組み合せを培養する方法にも関する。好ましくは、前駆細胞は肝前駆細胞である。同様に、本発明は特定の培養条件を用いて肝臓の多能性前駆細胞をクローニングする方法にも関する。好ましくは、本発明は肝臓の多能性前駆細胞をクローニングする方法に関する。肝臓の多能性前駆細胞は、任意の無脊椎動物または脊椎動物種、より好ましくは哺乳類に由来できる。より好ましくは、肝臓の多能性前駆細胞は、ヒト、霊長類、ブタ、イヌ、ラット、ウサギ、またはマウスに由来する。最も好ましくは、多能性前駆細胞はヒトに由来する。本発明は肝前駆細胞およびその子孫のエクスビボの増殖に必要な特定の培養条件を開示する。本発明は肝前駆細胞のフィーダー細胞として、STOマウス胚細胞のような胚のフィーダー細胞の使用も開示する。フィーダー細胞は、本発明で開示される新規で無血清のホルモン添加合成培地(HDM)と組み合せて使用される。この組み合せによって、細胞が悪性形質転換せずに、E15ラット肝から種々のラット胎児肝細胞株を確立することができた。
【0016】
さらに、本発明は肝前駆細胞およびその子孫の増殖を支持する能力のあるフィーダー細胞のクローニング方法に関する。
【0017】
本発明はフィーダーとして使用されると肝前駆細胞の増殖を支持する、特定の細胞株にも関する。
【0018】
また、本発明は肝前駆細胞のクローニング方法にも関する。本発明は、新しく単離された肝前駆細胞のクローン増殖能力を決定するためのインビトロコロニー形成アッセイ(CFA)法の開発のための、幹細胞株およびHDM−STO共培養系の使用を開示する。CFAを、特異的な抗原プロファイルによって精製された細胞と組み合せると、両能性肝前駆細胞が明らかになる。例えば、マウスのE11.5に対応するE13ラット肝から得た増殖能力の高い前駆細胞は、古典的MHCクラスI(RT1A1)−、OX18 (汎MHCクラスI)dull、および細胞内接着分子1(ICAM−1)+という同じ表現型を持つ。
【0019】
また本発明はクローン肝細胞増殖を支持できる培地にも関する。培地は、いくつかの特定のホルモンおよび栄養を特徴とし、血清を含まない。
【0020】
さらに、本発明はフィーダー細胞の生合成産物を含む培地における肝前駆細胞の培養に関する。
【0021】
さらに本発明は肝実質細胞および胆管細胞の表現型を含む、肝細胞の分化の誘導方法に関する。本発明では、上皮成長因子(EGF)が、前駆細胞コロニーの増殖と、肝実質細胞または胆管上皮細胞としてのいずれかの運命の両方に影響を与えることが開示される。
【0022】
5. 好ましい態様の詳細な説明
本発明は幹細胞の増殖および利用の過程である。外胚葉、中胚葉、および内胚葉起源の組織を含め、種々の組織が、前駆細胞の適当な供給源である。外胚葉組織には、皮膚組織、脳組織、および他の神経組織が含まれる。中胚葉組織には、筋肉、血液、および造血系が含まれる。内胚葉組織には、消化管、胃、膵臓、甲状腺、および消化器系に伴う腺が含まれる。特に、本発明は肝幹細胞および他の肝前駆細胞の増殖の過程である。本過程には、単離した肝幹細胞および/または肝前駆細胞および/またはその子孫の細胞群を、クローン増殖すなわち非常に低い細胞密度での増殖を支持する能力のある増殖条件に暴露することを含む。好ましい態様では、この過程には、フィーダー細胞層上での肝前駆細胞の増殖を支持するため、無血清の、ホルモン添加合成培地の使用が含まれる。フィーダー細胞の機能は複数あり、栄養の供給、接着表面の提供、および肝前駆細胞の生存、増殖および/または分化に必要な特定の増殖因子および細胞外マトリックスを培地中に分泌することが含まれる。別の好ましい態様では、この過程には肝幹細胞および肝前駆細胞の増殖を支持する能力のある細胞の選択が含まれる。フィーダー細胞は、爬虫類、鳥類、甲殻類、魚類、環形動物、軟体類、線虫類、昆虫類、または哺乳類、好ましくはヒト由来でよい。好ましくは、フィーダー細胞は胚組織に由来する。また好ましくは、フィーダー細胞は胚組織に由来する。また好ましくは、フィーダー細胞は胚の肝組織に由来する。また、フィーダー細胞は遺伝子組換えされていても良い。さらに好ましい態様では、この過程には幹細胞を最適に支持するフィーダー細胞のクローニングが含まれる。
【0023】
親和性に基づく相互作用、例えば、アフィニティパニング、補体と組み合せたイムノサージャリー、フローサイトメトリー、遠心水簸、分画遠心等、肝幹細胞および肝前駆細胞を単離する任意の方法が使用できる。単離された肝幹細胞および前駆細胞は、一部または全ての表現型マーカー(古典的MHCクラスI−、ICAM−1+、OX18dull、αフェトプロテイン+、またはアルブミン+)を発現する能力を持つ。本発明の別の態様では、肝前駆細胞は、塊、コロニーまたはクラスターとして積み重なった細胞の形成で特徴付けられる増殖パターンを発現する。
【0024】
本発明の好ましい態様では、幹細胞は無血清のホルモン添加合成培地(HDM)で選択的に増殖する。
【0025】
HDMの組成には、約40 ng/mlまでのEGF、約5〜10μg/mlまでのインスリン、約10−6 Mまでのデキサメタゾンまたは他のグルココルチコイドホルモン、約10μg/mlまでの鉄飽和トランスフェリン、約5 x 10−2 Mまでのニコチンアミド、約2%までのウシ血清アルブミン、約5 x 10−4 Mまでの2−メルカプトエタノールまたは同等の還元剤、約8μeq/lまでの遊離脂肪酸、約2 x 10−2 Mまでのグルタミン、約1 x 10−6 MまでのCuSO4、約3 x 10−8 MまでのH2SeO3、および選択的に抗生物質を添加した、ダルベッコ変法イーグル培地およびハムF12の混合物を含むがこれに限定されることのない、栄養培地が含まれる。抗生物質には、ペニシリン、ストレプトマイシン、ゲンタマイシン、および他の当技術分野で一般的な抗生物質、およびその組み合せが含まれる。当業者には、DMEM/F12の代わりに、最小限の試験後に、他の栄養培地、例えば、ハムF−10、Medium 199、またはMCDB 151およびMCDB 302を含むMCDBシリーズも使用できることが理解されると思われる。細胞培養の最も最低の条件は、ホルモンなしでフィーダーを使用することであり;上述のホルモン要件の中で最も重要なのはグルココルチコイド、インスリン、トランスフェリン、およびEGFで、これは前駆細胞の増殖のためのホルモン性分裂促進剤を構成している。他のホルモン因子も添加でき、2次的な増殖効果があるかも知れないが、上述の重要な要件を置き換えるものではない。同様に、当業者が行なえるようなホルモン組成の変更は、本発明の範囲内である。
【0026】
好ましい範囲には、10〜50 ng/ml EGF、2〜10μg/mlインスリン、5 x 10−7 M〜5 x 10−6 Mデキサメタゾン(9a−フルオロ−16a−メチル−プレドニゾロン)、5〜20μg/ml鉄飽和トランスフェリン、2〜8 x 10−3 Mニコチンアミド、0.05〜0.5%血清アルブミン、2〜8 x 10−5 M 2−メルカプトエタノール、5〜10μeq遊離脂肪酸混合物、1〜3 x 10−3 Mグルタミン、0.5〜2 x 10−6 M CuSO4、1〜5 x 10−8 M H2SeO3、1〜5μMパルミチン酸、0.1〜0.4μMパルミトレイン酸、0.5〜1.2μMステアリン酸、0.5〜2μMオレイン酸、1〜5μMリノール酸、および0.2〜0.8μMリノレン酸が含まれる。
【0027】
本発明の無血清のホルモン添加合成培地は、肝細胞のクローン増殖に適している。このHDMには、ダルベッコ変法イーグル培地(DME)、ハムF12、RPMI 1640、ウィリアムズE培地等のようないくつかのオプションのうちの任意の基礎培地を含み得る。好ましい態様は、ダルベッコ変法イーグル培地とハムF12の1:1の混合物(DMEM/F12、例えばGIBCO/BRL、Grand Island, NY)である。基礎培地には、好ましい濃度10 ng/mlの上皮成長因子EGF(例えば、Collaborative Biomedical Productsより)、好ましい濃度5μg/mlのインスリン(例えばSigma)、10−6Mデキサメタゾン(例えばSigma)、10μg/ml鉄飽和トランスフェリン(Sigma)、4.4 x 10−3Mニコチンアミド(例えばSigma)、0.2%ウシ血清アルブミン(例えばSigma)、5 x 10−5M 2−メルカプトエタノール(例えばSigma)、7.6 μeq/l遊離脂肪酸混合物(2.4μMパルミチン酸、0.21μMパルミトレイン酸、0.88μMステアリン酸、1μMオレイン酸、2.7μMリノール酸、および0.43μMリノレン酸)、2 x 10−3Mグルタミン(例えばGIBCO/BRL)、1 x 10−6M CuSO4、3 x 10−8M H2SeO3および抗生物質を添加する。インスリン様成長因子(IGF)、インターロイキン(IL)−6ファミリー、肝細胞増殖因子(HGF)、および線維芽細胞成長因子(FGF)を含むがこれらに限定されないフィーダー細胞から分泌される増殖因子は、フィーダーの効果を増強するために培地に添加でき、単独または種々の組み合せで添加してもフィーダーの効果を置き換えるものではない。これは、フィーダー細胞は、単独またはこれらの増殖因子との組み合せで必要とされる他のシグナルも生産していることを意味する。
【0028】
本発明の別の態様では、肝前駆細胞は単一の前駆細胞から増殖する、すなわち、細胞がクローニングされる。コロニーで細胞を増殖させることは、単一の細胞に由来する細胞の増殖と黙示的および明示的に定義されるクローン増殖とは必ずしも同じではない。限界希釈法と呼ばれる、前駆細胞を培養プレートのウェル1つあたり1細胞またはそれ未満に希釈する方法を含め、当技術分野で周知のいくつかのクローニング方法の任意のものが適当である。同様に前駆細胞は、クローニングリング、選択的剥離、微粒子上の希釈培養、フローサイトメトリーを用いた単細胞ソート、マイクロピペットまたは光学ピンセットを用いた個々の細胞の選択、および寒天を用いてもクローニングできる。
【0029】
本発明の別の態様では、クローニングされた前駆細胞の多くが有糸分裂できる。前駆細胞は少なくとも1回の有糸分裂ができることが好ましく、少なくとも10回の分裂ができることがさらに好ましい。
【0030】
本発明のさらに別の態様では、肝前駆細胞およびその子孫は、フィーダー細胞の代謝および生合成産物を添加した培地で増殖する。添加物は、調整培地、すなわち生きたフィーダー細胞が以前にインキュベートされた培地の形でよい。好ましくは、添加は、フィーダー細胞の調整培地から、肝前駆細胞およびその子孫の増殖を維持し増加するタンパク質、ペプチド、脂質、炭化水素、および代謝調節因子を含む因子を単離する形で良い。タンパク質は、細胞外マトリックスの可溶性および不溶性成分、および上皮成長因子およびインスリン様成長因子を含む増殖因子を含み得る。
【0031】
さらに好ましくは、胚または成体細胞または他の適当な細胞であるフィーダー細胞の層を用いて、肝細胞は培養で選択的に増殖する。1つの態様では、フィーダー細胞は間質細胞または線維芽細胞である。線維芽細胞または他の適当な細胞は、例えばトランスフェクションにより、遺伝子組換えされていてもよい。好ましくは、線維芽細胞または他の適当な細胞はヒト、非ヒト霊長類、ブタ、イヌ、ウサギ、ラット、またはマウスの中胚葉細胞であるが、また他の哺乳類および鳥類の中胚葉細胞も適している。さらに、線維芽細胞は、クローニングされ肝前駆細胞を支持する能力について選択され得る。
【0032】
本発明の好ましい態様では、単離された肝前駆細胞は、上皮成長因子(EGF)または他の分化シグナルの選択的使用またはその欠如により、肝実質細胞または胆管細胞系に運命づけられている。
【0033】
本発明のさらに好ましい態様では、単離された幹細胞および他の肝前駆細胞は、体外肝補助装置として使用され得る人工肝臓の成分として使用できる。本発明のさらに好ましい態様では、単離された肝前駆細胞およびその子孫を含む人工肝臓は、肝の機能不全または不全を患う患者の生命を維持するために使用される。
【0034】
6. 実施例
以下の実施例は、本発明を例示するものであるが、本発明はこれらの具体的な実施例に限定されない。当業者はこれらの実施例から本発明を実行する手段を得るものと思われる。当業者は本発明の範囲に含まれる多くの別の態様を認識すると思われる。
【0035】
6.1 肝幹細胞および肝前駆細胞の調製および分析
ラット
チャールズ・リバー・ブリーディングラボラトリー(Charles River Breeding Laboratory)(Wilmingon, MA)から、妊娠したフィッシャー334ラットを入手する。時刻を設定した妊娠には、午後に動物を一緒にして、プラグが観察された朝を0日とする。成体の肝細胞には、オスのフィッシャー334ラット(200〜250 g)が使用される。
【0036】
幹細胞株の確立
胎齢15日の胎児肝が調製される。単細胞懸濁液は、0.05%トリプシンおよび0.5 mM EDTAまたは10ユニット/mlサーモリシン(Sigma, St. Louis, MO)および100ユニット/mlデオキシリボヌクレアーゼI(Sigma)で肝臓を37℃でインキュベートして得られる。細胞は、フィコール−パーク(Pharmacia Biotech, Uppsala, Sweden)に重ね、450gで15分間密度勾配遠心を行なう。th1120−3およびrter6またはrhel4321について、底部の画分の細胞を、それぞれ17 mg/ml IV型コラーゲン(Collaborative Biomedical Products, Bedford, MA)または12μg/mlラミニン(Collaborative Biomedical Products)でコートした組織培養ディッシュに接種する。無血清のホルモン添加培地HDMは、ダルベッコ変法イーグル培地およびハムF12の1:1の混合物(DMEM/F12、GIBCO/BRL、Grand Island, NY)に、20 ng/ml EGF(Collaborative Biomedical Products)、5μg/mlインスリン(Sigma)、10−7Mデキサメタゾン(Sigma)、10μg/ml鉄飽和トランスフェリン(Sigma)、4.4 x 10−3Mニコチンアミド(Sigma)、0.2%ウシ血清アルブミン(Sigma)、5 x 10−5M 2−メルカプトエタノール(Sigma)、7.6 μeq/l遊離脂肪酸、2 x 10−3Mグルタミン(GIBCO/BRL)、1 x 10−6M CuSO4、3 x 10−8M H2SeO3および抗生物質を添加したものである。記載の各濃度は、培地中の最終濃度である。4週間の培養後、トリプシン処理した細胞をマイトマイシンCで処理したSTOマウス胚線維芽細胞株(American Type Culture Collection, Rockville MD)のフィーダー層上で培養する。Th1120−3、rter6およびrhel4321は、胎児肝細胞の3つの独立した調製物からクローニングし、HDMを用いてSTOフィーダー細胞上で維持する。細胞株の確立後、全ての培養に関してEGFの濃度は10 ng/mlに低下させる。
【0037】
細胞接着アッセイ法
フィブロネクチン(Collaborative Biomedical Products)、ラミニン、およびIV型コラーゲンに対する細胞の接着は、これらのタンパク質を0.3〜10μg/mlでコートした96穴マイクロタイタープレート(Corning, Cambridge, MA)を用いて評価する。200gで15分間のパーコール(Pharmacia Biotech)密度勾配遠心によりSTO細胞を除去した後、各ウェル中で3 x 104細胞の肝細胞株th1120−3、rter6、およびrhel4321をHDMを用いて10時間培養する。浮遊細胞を除去するために2回すすいだ後、テトラゾリウム塩WST−1を含む新しい培地(Boehringer Mannheim, Indianapolis, IN)を添加して、生存可能な接着細胞の数を測定する。4時間後に、製造元のプロトコールにしたがって、吸光度を決定する。
【0038】
STO亜系統
ATCCから得た100個の親STO細胞は、10%の熱で不活化した胎児ウシ血清、2 x 10−3Mグルタミン、5 x 10−5M 2−メルカプトエタノール、および抗生物質を添加したDMEM/F12中で、100mm培養ディッシュで7日間培養する。細胞の形態および増殖速度によって、さらに解析するために4つのサブクローンが選択される。rter6のCFAは4つのサブクローンで行われるが、その1つであるSTO6はマイトマイシンC処理後に培養プレートに接着しなくなる。1つのサブクローンSTO5はアダムス博士(Dr. J. M. Adams, The Walter and Eliza Hall Institute of Medical Research)から提供を受けたpEF−Hlx−MC1neoまたはpEF−MC1neoによってトランスフェクトする。Nde I部位で線状にしたプラスミドを、DOSPERリポソームトランスフェクション試薬(Boehringer Mannheim)によって細胞中に導入する。G418選択後、6つのクローンが単離される。各々3つのクローンがCFAで分析される。
【0039】
コロニーの免疫組織化学染色
培養プレートはメタノール−アセトン(1:1)で室温で2分間固定し、すすぎ、20%のヤギ血清(GIBCO/BRL)を含むハンクス液(Hanks Balanced Salt Solution (HBSS))により、4℃でブロックする。αフェトプロテインとアルブミンの二重免疫組織化学のためには、プレートは抗ラットアルブミン抗体(ICN Biomedicals, Costa Mesa, CA)処理後にテキサスレッド結合抗ウサギIgG(Vector laboratories, Burlingame, CA)、およびFITC結合抗ラットαフェトプロテインポリクローナル抗体(Nordic Immunology, Tilburg, Netherlands)とインキュベートする。アルブミンとCK19の二重標識には、抗αフェトプロテイン抗体の代わりに、抗CK19モノクローナル抗体(Amersham, Buckinghamshire, England)およびFITC結合抗マウスIgG(Caltag, Burlingame, CA)が使用される。
【0040】
E13の胎児肝の解離
胎児肝は10 mM HEPES, 0.8 mM MgSO4および1mM EGTA (pH 7.4)を添加した、Ca++フリーの氷冷HBSS中に切開して入れる。肝臓は10 mM HEPES、0.8 mM MgSO4、および1 mM CaCl2で調製されたHBSS中の0.2% IV型コラゲナーゼ(Sigma)および16.5ユニット/mlサーモリシン(Sigma)を用いて、粉砕する。37℃で10分間インキュベーションした後、細胞懸濁液を0.025%トリプシンおよび2.5 mM EDTA (Sigma)で10分間消化する。その後、1 mg/mlのトリプシン阻害剤(Sigma)を添加してトリプシンを抑制する。最後に、細胞を200ユニット/mlデオキシリボヌクレアーゼI (Sigma)で処理する。全ての実験において、肝臓1つあたり3〜5 x 105細胞が得られる。
【0041】
成体肝細胞の単離
肝細胞を単離するために、2段階の肝臓潅流法が用いられる。潅流後、細胞を50gで1分間、2回遠心し、大きな実質細胞を濃縮する。生存度はトリパンブルー排除による測定によると>90%である。
【0042】
フローサイトメトリー分析
細胞はFACScan (Becton−Dickinson, Mountain View, CA)で分析し、Moflow Flow Cytometer (Cytomation, Fort Collins, CO)を用いてソートする。E13胎児肝から得た細胞懸濁液は、非特異的抗体結合を予防するために、氷上で20%ヤギ血清(GIBCO/BRL)および1%硬骨類のゼラチン(Sigma)を含むHBSSとインキュベートする。すすいだ後、細胞はFITC結合抗ラットRT1Aa,b,l抗体B5 (Pharmingen, San Diego, CA)およびPE結合抗ラットICAM−1抗体1A29(Pharmingen)と懸濁する。一部の実験では、3色染色のため、細胞はビオチン化抗ラット単形性MHCクラスI抗体OX18 (Pharmingen)で染色した後、ストレプトアビジン−レッド670 (GIBCO/BRL)による第2の染色を行なう。全ての染色は、10 mM HEPES, 0.8 mM MgSO4, 0.2 mM EGTA、および0.2% BSA (pH 7.4)を含み、Ca++フリーの氷冷HBSSを用いて行なう。確立された3つの細胞株は、トリプシン処理をして、パーコール密度勾配遠心によってフィーダー細胞を除去する。ラットヘパトーマ細胞株FTO−2Bおよびラット肝上皮細胞株WB−F344ならびに成体肝細胞は、胎児肝細胞株と比較するために染色する。細胞株は、それぞれフォアニエー博士(Dr. R.E.K. Fournier, Fred Hutchinson Cancer Research Center, Seattle, WA)、およびソウ博士(Dr. M.−S. Tsao, University of North Carolina, Chapel Hill, NC)から贈与された。細胞はブロックして、FITC結合B5, OX18, PE結合1A29、または抗FITC結合ラットインテグリンβ1抗体Ha2/5 (Pharmingen)により染色する。OX18にはFITC結合抗マウスIgGが使用される。マウス細胞群を排除するために、3つの胎児肝細胞株の細胞懸濁液は、ビオチン化抗マウスCD98で染色後、ストレプトアビジン−レッド670および抗ラットモノクローナル抗体による第2の染色を行なう。
【0043】
細胞ごとに、異なる相対数で様々な抗原が発現される。現実的な使用法では、特定の抗原の発現レベルは、発現なし、低レベルの発現、多くの抗原にとって通常の発現レベル、および高レベルの発現であり得る。この使用法では、「低い(low)」という用語は弱い(weakまたはdull)と互換的に使用される。発現レベルのより詳しい記述を行なうこともできるが、多くの目的にはこれらの4つのレベルで充分である。例えばフローサイトメトリーによる抗原発現の測定は、抗原発現について連続的な範囲を提供することは明らかである。
【0044】
肝細胞株、ソートした細胞、および成体肝細胞のCFA
肝細胞株は細胞株の維持に使用されたのと同一のHDMを用いて、マイトマイシンC処理STOフィーダー層上に、9.6 cm2あたり500細胞で、3連で播く。播く前に、細胞はトリプシン処理をして、パーコール密度勾配遠心によってフィーダー細胞を除去する。培養液は2日ごとに培地を取り換えながら10〜14日インキュベートする。その後、αフェトプロテインおよびアルブミンの二重免疫蛍光染色が行われる。各ウェル100コロニーを、コロニーの形態、PまたはF型、およびαフェトプロテインおよびアルブミンの発現について分析する。コロニーはDiff−Quick (Baxter, McGaw Park, IL)を用いて染色し、各ウェルのコロニー数を数える。初代のソートした細胞および成体肝細胞のCFAでは、記述のように播く細胞数を変更する。別のわずかな変更として、培養期間を14および17日の間に延長し、デキサメタゾンの濃度を10−6Mに上昇させる。他の手順は上述のように実行される。成体肝細胞のCFAでは、調製後、細胞懸濁液から、少数の肝細胞クランプが除去されない。したがって、クランプから不確定数のコロニーが作られる可能性がある。ソートした細胞上の胆管分化のCFAでは、コロニーのアルブミンおよびCK19の二重免疫蛍光染色がEGFの存在下または非存在下で、培養5日目に行われる。培養5日目に、CK19+細胞を1個を超えて持つコロニーは、CK19+コロニーとして数える。10日および15日目には、2個のCK19+細胞のクラスターを複数含むコロニー、または3個を超えるCK19+細胞のクラスターを1つ含むコロニーは、CK19+コロニーとして数える。各ウェルで約100のコロニーが数えられる。各点は3連の染色した培養物の平均±SDを表す。
【0045】
6.2. ホルモン添加合成培地を用いた、マウス胚細胞のフィーダーを用いた胎児ラット肝細胞株の作製および解析
どれだけの期間、胎児肝細胞が維持され、エクスビボで増殖し、子孫を産生できるかを知るために、ラットE15肝細胞の単純な長期培養を試みる。造血単核細胞を除去するために密度勾配遠心を行なった後、胎児肝細胞をIV型コラーゲンまたはラミニンでコートした培養ディッシュおよびHDM(実施例6.1参照)を用いて培養する。細胞は4週間以上、生存する。しかし、新しくIV型コラーゲンまたはラミニンでコートしたディッシュ上の2次培養では、さらには増殖しない。マイトマイシンC処理のSTO胚マウス線維芽細胞株を2次培養のフィーダー層として使用すると、細胞の多くの塊が増殖する。最終的に、4つの独立した実験から、いくつかの安定した肝細胞株が確立される。
【0046】
αフェトプロテインおよびアルブミンの免疫組織分析は、細胞株のクローニングの前に、連続的に増殖する細胞群で行われる。αフェトプロテインおよびアルブミンの両方のタンパク質は、細胞群が肝細胞系起源であることを確認するマーカーとして使用される。Pコロニーと呼ばれる、細胞の積み重なりを形成する傾向のある細胞群では、αフェトプロテインおよびアルブミンを強く発現したが、Fコロニーと呼ばれる平坦な単層では、αフェトプロテインの発現が低下し、アルブミンは発現しなかった。胚マウス線維芽細胞STOは、いずれの抗体にも反応性を示さない。さらに解析するために、P型またはF型のコロニーという形態的な基準により、独立した実験で、3つのクローニングされた肝細胞株が選択される。Rhel4321(図1A)は、主に小さな細胞の詰まったP型コロニーからなり、th1120−3(図1C)は、F型コロニーの平らな単層のみを形成する。Rter6(図1B)はこの2つの表現型の中間である。興味深いことに、rter6の不均一性は、平らなコロニーを連続3回クローニングした後でも観察される。rhel4321およびrter6の単一細胞に由来するコロニーの不均一性を見るために、9.6 cm2(6穴プレートの1つのウェル)あたり500細胞の密度で播種し、細胞をSTO線維芽細胞上で10〜14日培養した。その後、コロニーの形態およびαフェトプロテインおよびアルブミンの発現の解析をした。図2A、2B、2C、2D、2E、および2Fはその結果を表す。rhel4321(図2B)およびrter6(図2C)の細胞株、ならびにクローニング前の元の細胞群(図2A)は、ほぼ全てのP型コロニーで強くαフェトプロテインを発現するのに対して、F型コロニーでは発現しない。さらに、αフェトプロテインとアルブミンの両方の強い発現は、P型コロニーのみに観察される。クローニングされた肝細胞株の形態的違いは、P型コロニーの割合と相関している(図2Bおよび2C)。rter6およびrhel4321のCFAのP型コロニーの割合は、それぞれ33.3%(±8.6% SD)および65.7%(±4.0% SD)である。1ウェルあたりの総コロニー数を数えて、クローン増殖効率(コロニー効率)を計算する。rter6およびrhel4321の効率は、それぞれ45.7%(±1.3% SD)および36.4%(±1.1% SD)である。th1120−3細胞は、側面の境界にそって互いに強く接着しあっており、単細胞懸濁液の調製が、非常に困難である。しかし、th1120−3細胞は、重なり合ったクラスターを形成しない。
【0047】
マウス肝細胞のラミニン、IV型コラーゲン、およびフィブロネクチンのような細胞外マトリックス(ECM)タンパク質に対する接着は発生段階によって異なるため、各細胞株が特定のECM成分に接着するときの選好性を次に調べる。成体肝細胞での所見と同様に、IV型コラーゲンはth1120−3の接着に最も効果的であるが(図1C)、rter6(図1B)およびrhel4321(図1A)ではそれほどでもない。ラミニンはrhel4321の接着に最も有効な基質である(図1A)。この選好性は、マウス胎児肝細胞の初代培養と同様である。要約すると、P型コロニーにおけるαフェトプロテインおよびアルブミンの保存された発現、ならびにrhel4321によるラミニンヘの優先した接着は、P型コロニーを産生している細胞群がより厳密に肝前駆細胞と関連していることを示唆する。
【0048】
6.3 コロニー形成のためのSTOサブクローンの単離;肝前駆細胞のアッセイ法
増殖能力の高い、両能性肝前駆細胞を同定するためのCFAシステムを開発するために、培養系は、クローン播種密度で細胞の増殖を支持し、重要な元の肝機能を保存するものでなくてはならない。初期の肝臓発生に最も重要なマーカーのうちの2つがアルブミンおよびαフェトプロテインである。F型ではなくP型のコロニーがクローン増殖中にαフェトプロテインおよびアルブミンの発現を維持するので、P型コロニーを最適化する培養条件が最良である。したがって、rter6のP型コロニーを支持するSTOサブクローンの能力が比較される。1つのクローンSTO5は、他の全ての亜系統および親株以上にP型コロニー形成を支持する(図2D)。rhel4321のCFAも、STO5が親STOよりも有効なフィーダーであることを確認する(図2E)。E10.5の消化管の内側を覆っている間充織細胞で発現するマウスH1x遺伝子産物は、胎児肝細胞の増殖に必須である。H1x遺伝子のmRNA発現がSTOサブクローンで分析されても、サブクローン間での発現の有意な差はない(データは示さず)。さらに、マウスH1xのSTO5中の安定なトランスフェクタントは、コロニー形成アッセイ法を改善しない(図2F)。しかし、トランスフェクタントの1つのクローンは、比較的高密度の継代で、STO5の元の形態をより安定的に持続させるので、このクローンはさらなる実験に使用される。
【0049】
6.4 表面抗原マーカーおよびコロニー形成アッセイ法を用いた、E13胎児肝からの肝前駆細胞の同定
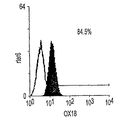
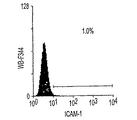
肝形成と大量の造血とが、胎児肝の中に共存する。これまで、造血前駆細胞の抗原性プロファイルは広く分析されてきたが、初期の肝前駆細胞の研究はまだ初期段階である。本研究で確立された3つの肝細胞株、成体肝癌細胞株(FTO−2B)、成体ラット肝由来の上皮細胞株(WB−F344)、および新しく単離された成体肝細胞を用いて、肝細胞の抗原性プロファイルが分析される。FTO−2B、WB−F344、および成体肝細胞と比較して、最も未熟な胎児肝細胞株rhel4321は、古典的MHCクラスI(RT1A1)の発現がないという点で、非常に独特である(図3A〜3X)。細胞株th1120−3(図3I〜3L)は、RT1A1、OX18(汎MHCクラスI)、およびICAM−1のパターンがrhel4321と類似しているが(図3A〜3D)、rter6(図3E〜3H)はRT1A1およびOX18の発現が比較的高い(図3)。さらに、異なる実験で得られた別の細胞株は、rhel4321と同一の形態を持つが、やはりRT1A1−、OX18dull、およびICAM−1+である。インテグリンb1の発現は、全ての細胞株で類似しているが、RT1Aa, b, 1およびICAM−1のパターンは細胞において独特である。成体肝細胞の抗原性プロファイルはRT1A1+、OX18+、およびICAM−1+である。成体ラット中では、赤血球を除く全ての骨髄細胞がMHCクラスI分子を強く発現するので、胎児肝細胞群は、MHCクラスI発現によって造血細胞群から分離できる。ラットE13肝臓から得られた細胞懸濁液を抗RT1A1およびICAM−1抗体で染色する。図4Aは、RT1A1およびICAM−1の2色染色パターンを示す。どの画分に肝細胞群が含まれているかを決定するために、蛍光活性化セルソートによって5つの画分を単離し、クローン増殖能力をCFAによってスクリーニングする。図4Bは、ソート後の、5つの画分の再ソート結果を表す。アルブミンおよびαフェトプロテインの発現で定義される肝細胞コロニーは、形態的にも区別可能なので、各ウェルの肝コロニーの数を数えることが可能である。肝コロニーの大部分は、RT1A1dullおよびICAM−1+のゲートで検出され(表1、図4B、ゲート2)、P型コロニーの頻度は75.6%(±4.9% SD)である。ゲート1は、はるかに少ないコロニー数を示し、他の画分のコロニー形成能力を持つ細胞の数はごくわずかだった。ゲート1および2では、全ての肝コロニーにおいて、αフェトプロテインおよびアルブミンの両方の発現が確認される。ゲート2の細胞に由来する一部のコロニーは、他よりも大きい。肝細胞上のMHCクラスI発現を詳細に調べるために、細胞分画のために、RT1A1、ICAM−1、およびOX18の3色の染色、ならびにもう1つのパラメーターとして側方散乱光(SSC)が使用される。胎児肝細胞はE11という早期の胎齢でも脂肪滴を含むので(Luzzatto, 1981)、細胞の粒状度を反映する側方散乱光(SSC)は、造血細胞から肝細胞を分離するために有用なパラメーターである。図4Cは、ゲート2に最も多数のコロニー形成細胞が含まれていることを示す。SSCに基づいてR2でゲートをかけると、ゲート2に対応する細胞群は明らかにRT1A1−およびOX18dullの表現型を示す(図4C、4D)。CFAにより、R4にはゲート2よりも多くのコロニー形成細胞が含まれることが確認される(表1)。これらの結果は、E13ラット肝から得られたRT1A1−およびOX18dull、およびICAM−1+細胞群の大部分が、αフェトプロテイン+およびアルブミン+のコロニーを生成する肝細胞であることを示唆する。これはrhel4321細胞で見られるのと同一の抗原性プロファイルである(図3)。
【0050】
【表1】RT1AおよびICAM−1の発現に基づいてソートしたE13胎児肝由来の肝コロニーの頻度

STO5hlx上でのコロニー形成培養は、E13の胎児肝の各画分から得られる、示された数の細胞を含む。肝コロニーの数を、3連の染色した培養物から確立した(平均±SD)。コロニー形成の効率は、培養液に接種した細胞のうち、16日の培養後に分析したところ、コロニーを形成するようになった割合を示す。
【0052】
6.5. E13肝細胞および成体肝細胞の異なる培養要件
E13肝臓から得られたソートされた肝細胞の成長要件は、決められたSTO5フィーダーおよびHDMを用いて調べられた。EGFは成体肝細胞の強力な増殖因子であることが以前から知られている。したがって、ソートした肝細胞のコロニー形成に対するEGFの効果を調べる。RT1A1− OX18dull、ICAM−1+肝細胞のコロニーサイズはEGFがないと大きくなるが、成体肝細胞はEGFの存在下でのみ、コロニーを生成した(図6C)。さらに、成体肝細胞に由来するコロニーの形態は、通常F型であるが、RT1A1−肝細胞はEGFなしでP型コロニーを形成する。しかし、コロニー効率はEGFがないとわずかに低下する(図6A)。興味深いことに、EGFのない培養条件は、2種類のPコロニー、P1およびP2を際立たせた。培養12日目にはコロニーの大部分はP2型であるが、図6Aのような典型的な形態を持たないコロニーもあるので、2つのタイプを完全に区別するのは困難である。これらの結果は、胎児肝細胞および成体肝細胞は、その増殖要件ならびにRT1A1発現(図3および4)およびコロニーの形態において、本質的に異なることを示唆している。
【0053】
増殖が最大に到達したと思われる培養3週間後に、RT1A1− 、OX18、およびICAM−1の発現を評価する。図5B〜5Dに示すように、RT1A1の発現は誘導されず、OX18の発現は低下している。ICAM−1のレベルは変化しない。さらに、単一コロニーの平均細胞数を、回収した細胞数、ラット肝細胞の割合、およびコロニー効率から計算する。見積もられる細胞数は3〜4 x 103(表2)に達する。これは、コロニーを形成する単一細胞が、この培養条件では平均で約11〜12回分裂したことを示す。
【0054】
【表2】単一肝コロニーにおける細胞数の計算
図4CのR4からソートした細胞は、60 mmまたは100 mmのディッシュでSTO5hlxフィーダー細胞上で培養した。示された期間培養した後、全ての細胞を回収し、総細胞数を計算した。ラット細胞の割合は、ラットICAM−1およびマウスCD98の発現に基づく、フローサイトメトリー分析による。コロニー効率は、培養に接種された細胞のうち、コロニーを形成したものの割合を示す。3連の染色した培養(平均)からのデータは、平行して行なった実験から得られた。
【0056】
単一コロニー中の平均細胞数=(回収した細胞の数 x ラット細胞のパーセンテージ/100)/接種した細胞の数 x コロニー効率/100)
【0057】
6.6 RT1A1−肝前駆細胞が両能性である証拠
ラット胎齢E13において、肝細胞は成熟した肝実質細胞および胆管上皮細胞を生み出す両能性前駆細胞だと考えられる。しかし、本発明以前では、この2つの運命が単一の細胞から生ずるのかどうかを示す直接の証拠がなかった。RT1A1− OX18dull ICAM−1+の胎児肝細胞がこの培養系で胆管細胞系に分化できるかどうかを知るために、胆管上皮細胞に特異的なマーカーとして抗CK19でコロニーの染色を行なう。CK19は、胎児肝で15.5日以降に胆管上皮前駆細胞で発現され、この時点で細胞のアルブミン発現が消失する。ソートされたRT1A1− ICAM−1+細胞をEGFの存在下または非存在下で培養し、その運命は培養5日目にCK19およびアルブミンの発現によってモニターする。最初から5日後、EGF処理した培養物ではCK19+のコロニーがほとんどないが、EGFのない培養物ではCK19+細胞を含むコロニーがいくつか見られる。CK19の強度はかなり弱いが、CK19+細胞ではアルブミン発現が低下している。培養10日目には、CK19のみまたはアルブミンのみを発現するコロニーや、両方が陽性のコロニーが存在する。単一コロニーの中でのCK19+およびアルブミン+細胞のパターンは相反している。両方陽性のコロニーおよびCK19のみ陽性のコロニーの数は、やはりEGF非存在下のほうが高い(図7A)。EGF存在下では、10日目には多くのコロニーがアルブミン+細胞のみからなる(図7B)。最終的には、両方陽性のコロニーの数は、15日目にEGF非存在下では100%近くになる(図7A)。全体では、EGFは培養を通してCK19+のコロニーの出現を劇的に抑制する(図7B)。これらの結果は、E13胎児肝から得たRT1A1−、OX18dull、およびICAM−1+細胞が、胆管細胞系に分化でき、その運命はインビトロではEGFに影響されることを示唆する(図8)。
【0058】
6.7 肝幹細胞および肝前駆細胞のクローン増殖を支持する能力のあるフィーダー細胞の単離およびクローニングのためのプロトコール
ブタ、ビーグル、ウサギ、マウス、またはサルの新鮮な胚組織または凍結組織(例えば、肝臓、肺、腎臓、筋肉、腸)を、カルシウムを含まないリン酸緩衝生理食塩水(PBS)中で細かく切り刻む。PBSで2回すすいだ後、0.25%トリプシンと37℃で10分間、または室温で60分間電磁攪拌機を用いて撹拌しながら、細胞懸濁液をインキュベートする。残った細胞の塊は、懸濁液をメッシュで濾過して除去する。その後、細胞を血清(例えば、10%胎児ウシ血清)および任意の種々の増殖補給剤(例えば、2 mMグルタミン、ピルビン酸ナトリウム、およびMEM非必須アミノ酸)を添加した基礎培地(例えば、イーグルMEM)を用いて組織培養ディッシュで培養する。プラスチックの基底および血清添加培地は、多くの場合中胚葉由来(例えば、間質細胞)である支持細胞(「フィーダー細胞」)の候補である細胞群の増殖を可能にし、かつ別の種類の細胞(例えば、前駆細胞)の生存、増殖、および/または機能を支持する因子を提供する、一般的な条件である。フィーダー細胞は、コンフルエントまたはそれに近くなったら、0.05%トリプシンを用いて継代培養する。継代培養を数回行なったら、増殖した細胞を凍結ストックとして調製し、使用まで凍結保存する。フィーダー細胞の別の供給源は、市販の初代培養フィーダー細胞またはフィーダー細胞株である。いずれの場合も、適当なフィーダー細胞を同定するためには、以下の基準が必要である。
【0059】
フィーダー細胞は以下を支持する:
1) 古典的MHCクラスI抗原陰性、ICAM−1陽性、および/または非古典的MHCクラスI抗原弱陽性という表現型マーカーを持つ肝前駆細胞のクローン増殖;
2) 古典的MHCクラスI抗原陰性、ICAM−1陽性、非古典的MHCクラスI抗原弱陽性、αフェトプロテイン陽性、アルブミン陽性、またはCK19陽性という表現型マーカーを持つ子孫を持つ前駆細胞のクローン増殖;または
3) 両能性肝前駆細胞の定義に必要な、肝細胞系および胆管細胞系の両方への誘導可能な分化。
【0060】
当技術分野では、古典的MHCクラスI抗原は、MHCクラスIa抗原としても知られる。非古典的MHCクラスI抗原は、MHCクラスIb抗原としても知られる。MHC抗原は、異なる種では異なる呼び方をされる:例えば、ラットではRT1,マウスではH−2、およびヒトではHLAである。
【0061】
上記のアッセイ法は以下に説明される:
【0062】
肝前駆細胞のクローン増殖の条件
肝前駆細胞は、増殖を停止した、すなわち増殖を予防するように処理されたフィーダー細胞上に、9.6 cm2あたり500細胞を接種する。フィーダー細胞は、マイトマイシンCまたは放射線照射(細胞の種類によって3000〜5000ラド)で処理して、増殖を停止する。増殖停止したフィーダー細胞および前駆細胞には、無血清HDMを供給する。例えば、齧歯類細胞のHDMは、ダルベッコ変法イーグル培地とハムF12の1:1混合物に10 ng/ml EGF、 5μg/mlインスリン、10−6Mデキサメタゾン、10μg/ml鉄飽和トランスフェリン、4.4 x 10−3Mニコチンアミド、0.2%ウシ血清アルブミン、5 x 10−5M 2−メルカプトエタノール、7.6 μeq/l遊離脂肪酸混合物、2 x 10−3Mグルタミン、1 x 10−6M CuSO4、3 x 10−8M H2SeO3および抗生物質を添加したものである。培養液は、2日ごとに培地を交換しながら10〜14日間インキュベートする。その後、αフェトプロテイン、アルブミン、および/またはCK19の2重免疫蛍光染色を行なって子孫の運命を同定する。αフェトプロテインおよびアルブミンの発現について、約100のコロニーが分析される。さらに、コロニーの形態、P型かF型かも適切な子孫の同定に役に立つ場合がある。
【0063】
フィーダー細胞と肝前駆細胞の理想的な組み合せは、同一の種から得られたものである。好ましくは、フィーダー細胞は肝前駆細胞と同一の組織および同一の種から得られる。しかし、1つの種からのフィーダーと別の種からの前駆細胞を混合することも可能である。例えば、齧歯類のフィーダーでさえも、ヒト肝前駆細胞に使用できる。可溶性および不溶性の因子(種および/または組織特異的な可能性がある)は、肝幹細胞または肝前駆細胞のクローン増殖を助ける。このような因子の供給源には、次のようなものがある:
1) 最適な種および組織のフィーダー細胞を培養した条件培地。フィーダー細胞は間質細胞のみならず、任意の種類の細胞で良い。
2) 重要な因子が既知の場合には、肝前駆細胞に活性を示す最適なフィーダー細胞に由来する適切な分子(シグナル)を合成するために、それぞれ転写または翻訳のためのcDNAまたはmRNAを任意の細胞に導入することによって生物活性のあるフィーダー細胞群を作製できる供給源。
3)肝前駆細胞に活性な最適フィーダー細胞から得られるシグナルが、タンパク質、ペプチド、炭化水素、脂質、糖ペプチド、糖タンパク質、リポタンパク質、糖脂質、またはその組み合せであるかに関わらず、重要な因子が既知の場合には、そのようなシグナルを培地に添加することによってフィーダー細胞を完全に置き換えることもできる供給源。
【0064】
上述の実施例は、例示のためのみに説明されており、本発明の範囲または態様を制限するものではない。上記に特に説明されていない他の態様も、当業者には明らかであると思われる。しかし、そのような他の態様も、本発明の範囲および精神に含まれると考えられる。したがって、本発明は添付の特許請求の範囲によってのみ、正しく限定される。
【0065】
本明細書で引用された全ての特許および出版物は、その全体が参照として本明細書に組み入れられる。
【図面の簡単な説明】
【図1】15日目の胎児ラットの肝臓から得た肝細胞株の特徴付けである。
【図2】線維芽細胞フィーダー細胞上のコロニー形成アッセイ法である。
【図3】成体肝細胞中の様々な細胞株上のラット細胞表面抗原の発現である。
【図4】13日目の胎児ラットの肝臓の表現型分析を現す。
【図5】EGF存在下および非存在下での肝臓コロニーの特徴付けである。
【図6】RT1A1−肝細胞上のCK19発現の誘導を表す。
【図7】STO5フィーダー細胞上の肝臓コロニー形成の模式図である。[0001]
1. Field of the invention
The present invention relates to novel conditions for clonal expansion of mammalian hepatic progenitor cells, including pluripotent cells, stem cells, and other early hepatic progenitor cells. In particular, the invention relates to a method for growing hepatic progenitor cells using feeder cells in a synthetic medium and co-culture. The invention also relates to cells used as feeders and capable of supporting the growth of hepatic progenitor cells.
[0002]
2. Description of related technology
Identification of pluripotent progenitor cells in mammalian tissues is of clinical and commercial importance, as well as for understanding developmental processes and tissue homeostasis. Progenitor cells are ideal targets for gene therapy, cell transplantation, and tissue engineering of bioartificial organs (Millar, AD 1992 Nature 357, 455; Langer, R. and Vacanti, JP 1993). Science 260, 920; Gage, FH 1998 Nature 392, 18).
[0003]
The presence of tissue-specific "fate" stem cells or progenitor cells with high proliferative and / or pluripotent properties was confirmed by hematopoietic stem cells (Spangrde, GJ, et al., 1998 Science 241, 58), neural stem cells (Davis, A.A. and Temple, S. 1994 Nature 372, 263; Temple, DL and Anderson, DJ. 1992 Cell 71, 973), and epithelial stem cells (Jones, PH and Watt, FM 1993 Cell 73, 713). These progenitor cells are thought to be responsible for normal hematopoietic, neuronal, or epithelial tissue homeostasis, and for the regenerative response after severe injury (Hall, PA and Watt, FM). 1989 Development 106, 619).
[0004]
The adult mammalian liver has a very high capacity to recover following severe hepatotoxic injury or partial hepatectomy, despite the normally slow turnover and quiescent tissue (Fishback, F. et al. C. 1929 Arch. Pathol. 7, 955); (Higgins, GM and Anderson, RM 1931 Arch. Pathol. 12, 186). Recent test data in mice have shown that adult parenchymal cells have an almost endless proliferation potential when examined in a series of transplantation experiments (Overturf et al., 1997 Am. J. Pathol. 151). (Rhim, JA, et al., 1994 Science 263, 1149). Because these experiments utilize heterogeneous populations of stem cells, the observed growth potential was derived from adult parenchymal cells, a subpopulation of adult parenchymal cells, and / or The ability to prove that it is from an immature phase (ie, a progenitor cell) is limited. Furthermore, experiments have shown no evidence of biliary epithelial differentiation because the host used had a deficiency in the albumin urokinase transgene or tyrosine degrading enzyme; neither of these hosts had the potential to select a hepatocyte lineage. Has characteristics. Therefore, this assay could not test a population of bipotent cells.
[0005]
Several histological studies have established that fetal early hepatocytes in the second trimester have a bipotential ability to differentiate into biliary epithelium and mature stem cells (Shiojiri, N. 1997 Microscopy Res. Tech.). 39, 328-35). Liver development begins in the ventral foregut endoderm immediately after the endoderm epithelium interacts with the cardiogenic mesoderm (Douarin, NM 1975 Medical Biol. 53, 427); (Houssaint, E., et al. 1980 Cell Differ. 9, 269). This link to the liver occurs at 8 days of fetal age (E) in mice. The first stages of liver development are revealed by induction of serum albumin and alpha-fetoprotein mRNA in the endoderm before morphological changes (Gualdi, R et al., 1996 Genes Dev. 10, 1670). At 9.5 days of mouse embryonic age, certain cells proliferate and invade the mesenchyme of the transverse septum in the form of threads, forming the liver primordium. Thereafter, the mass of the liver increases dramatically, but the increase in mass is mainly due to hematopoietic cells, which colonize the fetal liver at E10 in mice (Houssaint, E. 1981 Cell Differ. 10, 243). ), Causing hepatocytes to take an extremely distorted and irregular shape (Luzzatto, AC 1981 Cell Tissue Res. 215, 133). Interestingly, recent data using gene-targeting mutant mice indicate that deletion of several genes caused fatal liver failure, parenchymal apoptosis, and / or necrosis between E12 and E15 (Gunes, C. et al., 1998 EMBO J. 17, 2846), (Hilberg, F. et al., 1993 Nature 365, 1791), (Motoyama, J. et al., 1997 Mech. Dev. 66, 27), (Schmidt, C et al., 1995 Nature 373, 699). Stress activation cascade (Ganiatsas, S. et al., 1998, Proc. Natl. Acad. Sci. USA 95, 6881), (Nishina, H. et al., 1999 Development 126, 505) or anti-apoptotic cascade (Beg, A. et al.). , 1995 Nature 376, 167), (Li, Q. et al., 1999 Science 284, 321), (Tanaka, M. et al., 1999
[0006]
One major obstacle to clonal proliferation assays is the explosive proliferation of hematopoietic cells, which makes observable ex vivo proliferation of hepatocytes unobservable. Therefore, it is necessary to use a method for enriching hepatocytes. Surface markers that require fractionation of hematopoietic cells in fetal liver have been investigated in detail (Dzierzak, E. et al., 1998 Immunol. Today 19, 228), but studies of markers for hepatic progenitor cells are in the early stages. Has not yet been determined (Sigal, S et al., 1994 Hepatology 19, 999). Furthermore, ex vivo growth conditions commonly used for adult hepatocytes result in differentiation with loss of tissue-specific functions such as ALB expression (Block, GD et al., 1996 J. Cell Biol. 132, 1133). The slightly improved ability to synthesize tissue-specific mRNA and the ability to repair tissue-specific genes completely after translation are defined as serum-free, defined by hormones, growth factors, and / or some extracellular matrix components. Is found only in hepatocytes maintained using the mixture described (Jefferson, DM, et al., 1984. Mol. Cell. Biol. 4, 1929; Enat R et al., 1984 Proc. Natl. Acad. Sci. , 81, 1411). However, proliferating fetal hepatocytes maintain expression of such serum proteins in vivo. What is not clear in the art is how to maintain and grow hepatic progenitor cells in vitro. There is an unmet need for identification of conditions that maintain ex vivo expansion of hepatic progenitor cells. Similarly, there is an unmet need for an in vitro colony formation assay (CFA) method to determine the clonal expansion potential of hepatic progenitor cells isolated from fresh liver tissue; One cell is defined as the ability to generate a population of daughter cells of a clone derived from the inoculated cells. Clonal growth consisting of clumps of cells that are inoculated at high cell density and grow in close proximity in liver culture has also been described by others (Block, GD, et al., J. Cell Biol. 1996, 132, 1133). However, the colonies of cells described in these previous studies cannot be subcultured and are, by definition, not clones and have limited utility.
[0007]
Attempts have also been made to grow hepatocytes in vitro. U.S. Patent No. 5,510,524 to Naughton et al. Claims that culturing hepatocytes relies on a three-dimensional framework of biocompatible but non-living material. There is an unmet need for culture conditions that do not have an artificial framework and allow hepatic progenitor cells to grow and culture. Also, cloned hepatic progenitor cells that have the potential for generating both bile duct cells and hepatocytes, and are particularly suitable for use as a component of an artificial liver, for testing hepatotoxins, and for drug development Has unmet demand.
[0008]
US Pat. No. 5,559,022 to Norton et al. Claims hepatic reserve cells that bind to eosin Y, a dye used to characterize "reserve cells". No established markers are used for cell identification, nor is a method for clonal expansion of spare cells nor a marker used to isolate viable liver spare cells. Methods that disclose methods of isolating and culturing cells having many characteristics essential for hepatic progenitor cells, including the expression of at least one specific marker and the ability to differentiate into either hepatocytes or bile duct cells Have unsatisfied demand. There is also an unmet need for methods of clonal expansion of hepatic progenitor cells. Clonal expansion is essential as a clear and precise method of identifying and identifying pluripotent hepatic progenitor cells.
[0009]
U.S. Pat. No. 5,405,772 to Ponting claims a medium for cell growth. In U.S. Patent No. 5,405,772, 3-30 g / ml cholesterol, 5-30 g / ml nucleoside, and 2-100 g / cm.2Type IV collagen or 0.5-100 μg / cm2Any of the fibronectins is required. This requires a medium that is specific and optimized for the growth of hepatic progenitor cells.
[0010]
U.S. Pat. No. 4,914,032 to Kuri-Haruch et al. Claims a process for culturing hepatocytes. Contrary to the present invention, U.S. Pat. No. 4,914,032 does not disclose any conditions for culturing hepatic progenitor cells or clonal expansion of hepatocytes. Similarly, U.S. Pat. No. 5,030,105 to Kri Herkuch et al. Claims a method of evaluating a drug by treating a hepatocyte cell culture. There is an unmet need for clonal expansion conditions for using defined populations of cells for testing, and methods for culturing hepatic progenitor cells.
[0011]
Norton et al., US Patent No. 5,858,721, claims transfection of stromal cells. However, U.S. Pat. No. 5,858,721 is limited in that it requires a framework of biocompatible non-living materials. In contrast, there is an unmet need in the present invention for growth conditions that do not require synthetic meshwork.
[0012]
The present inventors have recognized that it is inappropriate to culture mature hepatocytes, such as hepatocytes, rather than from much more useful hepatic progenitor cells. The inventors have carefully determined the isolation parameters of hepatic progenitor cells and the requirements for clonal expansion. There are many uses for progenitor cells and methods for selecting and culturing progenitor cells, including the evaluation of drugs and toxicants in patients with liver failure, and the evaluation of drugs.
[0013]
U.S. Patent Nos. 5,576,207 and 5,789,246 to Reid et al. Disclose the need for feeder and hormone-supplemented synthetic media. These prior studies have used embryonic liver stromal cells in combination with a defined extracellular matrix basal and serum-free hormone-supplemented medium to provide conditions for hepatic progenitor cell growth. However, the synthetic medium used was more complex than that used in the present invention; the cells were inoculated on purified matrix basal (type IV collagen and laminin), but here the feeder (matrix) was used. And germ stromal cells were prepared as primary cultures from embryonic liver and were not established as a cell line. By using embryonic stromal cell lines, feeder cells provide a much simpler, practical, and reproducible means of supporting cells. Furthermore, it is reasonable to assume that the STO feeder is not limited to hepatic progenitor cells but can also be used for progenitor cells obtained from multiple types of tissues. In prior patents, hepatic progenitor cell cultures were inoculated at high cell densities and their growth was observed as colony formation, which means that clumps, rather than cell clones, were induced to grow.
[0014]
3. Summary of the Invention
The present invention relates to a method of growing a progenitor cell, its progeny, or a mixture thereof. In particular, the invention relates to a method of expanding endoderm-derived progenitor cells, progeny thereof, or mixtures thereof. Cells are obtained from endoderm tissue. The endoderm-derived progenitor cells, their progeny, or a mixture thereof are then cultured in a medium on a layer containing feeder cells. The precursor cell, its progeny, or a mixture thereof can be a vertebrate cell. The progenitor cells, their progeny, or a mixture thereof, express an ICAM or ICAM-1 positive and classical MHC class I antigen negative phenotype. Classical MHC class I antigens are also called MHC class Ia antigens.
[0015]
The present invention also relates to a serum-free, hormone-supplemented synthetic medium and a method for culturing hepatic stem cells and other progenitor cells using feeder cells. The invention also relates to a method of culturing progeny of progenitor cells, or a combination of progenitor cells and progeny of progenitor cells. Preferably, the progenitor cells are liver progenitor cells. Similarly, the invention relates to a method of cloning hepatic pluripotent progenitor cells using specific culture conditions. Preferably, the invention relates to a method for cloning pluripotent progenitor cells of the liver. Hepatic pluripotent progenitor cells can be derived from any invertebrate or vertebrate species, more preferably mammals. More preferably, the pluripotent progenitor cells of the liver are derived from humans, primates, pigs, dogs, rats, rabbits, or mice. Most preferably, the pluripotent progenitor cells are derived from a human. The present invention discloses the specific culture conditions required for the ex vivo expansion of hepatic progenitor cells and their progeny. The invention also discloses the use of embryonic feeder cells, such as STO mouse embryonic cells, as hepatic progenitor feeder cells. Feeder cells are used in combination with the novel, serum-free, hormone-supplemented synthetic medium (HDM) disclosed in the present invention. By this combination, various rat fetal liver cell lines could be established from E15 rat liver without cells undergoing malignant transformation.
[0016]
Furthermore, the present invention relates to a method for cloning feeder cells capable of supporting the growth of hepatic progenitor cells and their progeny.
[0017]
The invention also relates to certain cell lines that support the growth of hepatic progenitor cells when used as feeders.
[0018]
The present invention also relates to a method for cloning hepatic progenitor cells. The present invention discloses the use of a stem cell line and an HDM-STO co-culture system for the development of an in vitro colony formation assay (CFA) method for determining the clonal expansion potential of newly isolated hepatic progenitor cells. Combining CFA with cells purified by a specific antigen profile reveals bipotent hepatic progenitor cells. For example, highly proliferative progenitor cells obtained from E13 rat liver corresponding to mouse E11.5 are derived from classical MHC class I (RT1A1)−, OX18 (Pan MHC Class I)dull, And intracellular adhesion molecule 1 (ICAM-1)+With the same phenotype.
[0019]
The present invention also relates to a medium that can support clonal hepatocyte proliferation. The medium is characterized by some specific hormones and nutrients and does not contain serum.
[0020]
Furthermore, the present invention relates to the culture of hepatic progenitor cells in a medium containing a biosynthetic product of feeder cells.
[0021]
Further, the present invention relates to a method for inducing hepatocyte differentiation, including the phenotype of hepatocytes and bile duct cells. The present invention discloses that epidermal growth factor (EGF) affects both the growth of progenitor cell colonies and the fate of either hepatocytes or biliary epithelial cells.
[0022]
5. Detailed description of preferred embodiments
The present invention is a process of stem cell proliferation and utilization. A variety of tissues are suitable sources of progenitor cells, including tissues of ectoderm, mesoderm, and endoderm origin. Ectodermal tissue includes skin tissue, brain tissue, and other neural tissue. Mesodermal tissue includes muscle, blood, and the hematopoietic system. Endoderm tissue includes the gut, stomach, pancreas, thyroid, and glands associated with the digestive system. In particular, the invention is a process of proliferating hepatic stem cells and other hepatic progenitor cells. The process involves exposing the isolated population of hepatic stem cells and / or hepatic progenitor cells and / or progeny thereof to growth conditions capable of supporting clonal expansion, ie, growth at very low cell densities. Including. In a preferred embodiment, this process involves the use of serum-free, hormone-supplemented, synthetic media to support the growth of hepatic progenitor cells on the feeder cell layer. Feeder cells have multiple functions, including providing nutrients, providing an adherent surface, and secreting certain growth factors and extracellular matrices into the medium necessary for hepatic progenitor cell survival, growth and / or differentiation. . In another preferred embodiment, the process includes selecting cells capable of supporting the growth of hepatic stem cells and hepatic progenitor cells. The feeder cells may be from reptiles, birds, crustaceans, fish, annelids, mollusks, nematodes, insects, or mammals, preferably humans. Preferably, the feeder cells are derived from embryonic tissue. Also preferably, the feeder cells are derived from embryonic tissue. Also preferably, the feeder cells are derived from embryonic liver tissue. Further, the feeder cells may be genetically modified. In a further preferred embodiment, this process involves cloning feeder cells that optimally support the stem cells.
[0023]
Any method for isolating hepatic stem cells and hepatic progenitor cells can be used, such as affinity-based interactions, such as affinity panning, immunosurgery in combination with complement, flow cytometry, centrifugal elutriation, differential centrifugation, and the like. Isolated hepatic stem cells and progenitor cells have some or all of the phenotypic markers (classical MHC class I−, ICAM-1+, OX18dull, Α-fetoprotein+Or albumin+) With the ability to express In another aspect of the invention, the hepatic progenitor cells develop a proliferation pattern characterized by the formation of cells stacked as clumps, colonies or clusters.
[0024]
In a preferred embodiment of the present invention, the stem cells are selectively grown in serum free hormone-supplemented synthetic medium (HDM).
[0025]
The composition of HDM includes EGF up to about 40 ng / ml, insulin up to about 5-10 μg / ml,-6 M dexamethasone or other glucocorticoid hormone, up to about 10 μg / ml iron-saturated transferrin, about 5 × 10-2 Nicotinamide up to M, bovine serum albumin up to about 2%, about 5 x 10-4 M 2-mercaptoethanol or equivalent reducing agent, free fatty acids up to about 8 μeq / l, about 2 × 10-2 Glutamine up to M, about 1 x 10-6 CuSO up to M4, About 3 x 10-8 H up to M2SeO3And nutrient media, including, but not limited to, a mixture of Dulbecco's modified Eagle's medium and Ham F12, optionally with the addition of antibiotics. Antibiotics include penicillin, streptomycin, gentamicin, and other common antibiotics in the art, and combinations thereof. Those skilled in the art will understand that instead of DMEM / F12, after minimal testing, other nutrient media can also be used, such as HamDB-10, Medium199, or the MCDB series including MCDB151 and MCDB302. I think that the. The worst conditions for cell culture are to use feeders without hormones; the most important of the above hormonal requirements are glucocorticoids, insulin, transferrin, and EGF, which are It constitutes a hormonal mitogen. Other hormonal factors can also be added and may have a secondary proliferative effect, but do not replace the important requirements described above. Similarly, alterations of the hormonal composition that can be made by those skilled in the art are within the scope of the invention.
[0026]
Preferred ranges include 10-50 ng / ml EGF, 2-10 μg / ml insulin, 5 × 10-7 M-5 x 10-6 M dexamethasone (9a-fluoro-16a-methyl-prednisolone), 5-20 μg / ml iron-saturated transferrin, 2-8 × 10-3 M nicotinamide, 0.05-0.5% serum albumin, 2-8 x 10-5 M 2-mercaptoethanol, 5-10 μeq free fatty acid mixture, 1-3 x 10-3 M-glutamine, 0.5-2 x 10-6 M CuSO4, 1-5
[0027]
The serum-free synthetic medium with hormones of the present invention is suitable for clonal expansion of hepatocytes. The HDM may include any of a number of options, such as Dulbecco's Modified Eagle Medium (DME), Ham F12, RPMI 1640, Williams E Medium, and the like. A preferred embodiment is a 1: 1 mixture of Dulbecco's modified Eagle's medium and Ham F12 (DMEM / F12, such as GIBCO / BRL, Grand Island, NY). The basal medium contains a preferred concentration of 10 ng / ml of epidermal growth factor EGF (eg, from Collaborative Biomedical Products), a preferred concentration of 5 μg / ml of insulin (eg, Sigma),-6M dexamethasone (eg Sigma), 10 μg / ml iron-saturated transferrin (Sigma), 4.4 × 10-3M nicotinamide (eg, Sigma), 0.2% bovine serum albumin (eg, Sigma), 5 × 10-5M 2-mercaptoethanol (eg Sigma), 7.6 μeq / l free fatty acid mixture (2.4 μM palmitic acid, 0.21 μM palmitoleic acid, 0.88 μM stearic acid, 1 μM oleic acid, 2.7 μM linoleic acid, and 0 .43 μM linolenic acid), 2 × 10-3M glutamine (eg, GIBCO / BRL), 1 × 10-6M CuSO4, 3 x 10-8MH2SeO3And add antibiotics. Growth factors secreted from feeder cells include, but are not limited to, insulin-like growth factor (IGF), interleukin (IL) -6 family, hepatocyte growth factor (HGF), and fibroblast growth factor (FGF). Can be added to the medium to enhance the effect of the feeder, and addition alone or in various combinations does not replace the effect of the feeder. This means that feeder cells also produce other signals required alone or in combination with these growth factors.
[0028]
In another aspect of the invention, the hepatic progenitor cells are expanded from a single progenitor cell, ie, the cells are cloned. Growing cells in a colony is not necessarily the same as clonal expansion, which is defined implicitly and explicitly, by growing cells derived from a single cell. Any of a number of cloning methods known in the art are suitable, including a method of diluting progenitor cells to one cell or less per well of a culture plate, referred to as limiting dilution. Similarly, progenitor cells can be cloned using cloning rings, selective exfoliation, dilution culture on microparticles, single cell sorting using flow cytometry, selection of individual cells using micropipette or optical tweezers, and agar. .
[0029]
In another aspect of the invention, many of the cloned progenitor cells are capable of mitosis. Preferably, the progenitor cells are capable of at least one mitosis, more preferably at least ten.
[0030]
In yet another aspect of the invention, the hepatic progenitor cells and their progeny are grown in media supplemented with feeder cell metabolism and biosynthesis products. The supplement may be in the form of a conditioned medium, a medium in which live feeder cells have been previously incubated. Preferably, the addition can be in the form of isolating from the conditioned medium of the feeder cells factors including proteins, peptides, lipids, hydrocarbons, and metabolic regulators that maintain and increase the growth of hepatic progenitor cells and their progeny. Proteins can include soluble and insoluble components of the extracellular matrix, and growth factors, including epidermal growth factor and insulin-like growth factor.
[0031]
More preferably, hepatocytes are selectively grown in culture using a layer of feeder cells, which are embryonic or adult cells or other suitable cells. In one aspect, the feeder cells are stromal cells or fibroblasts. Fibroblasts or other suitable cells may be genetically modified, for example, by transfection. Preferably, the fibroblasts or other suitable cells are human, non-human primate, porcine, dog, rabbit, rat, or mouse mesodermal cells, but also other mammalian and avian mesodermal cells are suitable. ing. In addition, fibroblasts can be cloned and selected for their ability to support hepatic progenitor cells.
[0032]
In a preferred embodiment of the invention, the isolated hepatic progenitor cells are committed to a hepatic parenchymal or cholangiocyte cell line by selective use or lack of epidermal growth factor (EGF) or other differentiation signals.
[0033]
In a further preferred embodiment of the present invention, the isolated stem cells and other hepatic progenitor cells can be used as components of an artificial liver that can be used as an extracorporeal liver assist device. In a further preferred embodiment of the invention, the artificial liver comprising the isolated hepatic progenitor cells and their progeny is used to support the life of a patient suffering from hepatic dysfunction or failure.
[0034]
6. Example
The following examples illustrate the invention, but the invention is not limited to these specific examples. Those skilled in the art will obtain the means for practicing the invention from these examples. Those skilled in the art will recognize many alternative embodiments that fall within the scope of the invention.
[0035]
6.1 Preparation and analysis of hepatic stem cells and hepatic progenitor cells
Rat
Pregnant Fisher 334 rats are obtained from the Charles River Breeding Laboratory (Wilmington, MA). For timed pregnancies, the animals are brought together in the afternoon and the morning when the plug is observed is taken as
[0036]
Establishment of stem cell line
Fetal liver of 15 days of age is prepared. Single cell suspensions are incubated with the liver at 37 ° C. with 0.05% trypsin and 0.5 mM EDTA or 10 units / ml thermolysin (Sigma, St. Louis, Mo.) and 100 units / ml DNase I (Sigma). Is obtained. Cells are overlaid on Ficoll-Park (Pharmacia Biotech, Uppsala, Sweden) and subjected to density gradient centrifugation at 450 g for 15 minutes. For th1120-3 and rter6 or Rhel4321, the fraction of cells in the bottom, 17 mg / ml IV collagen respectively (Collaborative Biomedical Products, Bedford, MA) or 12 [mu] g / ml laminin (Collaborative Biomedical Products) tissue culture dishes coated with Inoculate. Serum-free hormone-supplemented medium HDM was prepared by adding 20 ng / ml EGF (Collaborative Biomedical Products) to a 1: 1 mixture of Dulbecco's modified Eagle medium and Ham F12 (DMEM / F12, GIBCO / BRL, Grand Island, NY). 5 μg / ml insulin (Sigma), 10-7M dexamethasone (Sigma), 10 μg / ml iron-saturated transferrin (Sigma), 4.4 × 10-3M nicotinamide (Sigma), 0.2% bovine serum albumin (Sigma), 5 x 10-5M 2-mercaptoethanol (Sigma), 7.6 μeq / l free fatty acid, 2 × 10-3M glutamine (GIBCO / BRL), 1 x 10-6M CuSO4, 3 x 10-8MH2SeO3And antibiotics added. Each concentration described is the final concentration in the medium. After 4 weeks of culture, trypsinized cells are cultured on the feeder layer of STO mouse embryo fibroblast cell line (American Type Culture Collection, Rockville MD) treated with mitomycin C. Th1120-3, rter6 and rhel4321 are cloned from three independent preparations of fetal hepatocytes and maintained on STO feeder cells using HDM. After cell line establishment, the concentration of EGF is reduced to 10 ng / ml for all cultures.
[0037]
Cell adhesion assay
Cell adhesion to fibronectin (Collaborative Biomedical Products), laminin, and type IV collagen is assessed using 96-well microtiter plates (Corning, Cambridge, Mass.) Coated with these proteins at 0.3-10 μg / ml. . After removing STO cells by Percoll (Pharmacia Biotech) density gradient centrifugation at 200 g for 15 minutes, 3 × 10 5 in each well.4The cell hepatocyte lines th1120-3, rter6, and rhel4321 are cultured for 10 hours using HDM. After rinsing twice to remove floating cells, a fresh medium containing the tetrazolium salt WST-1 (Boehringer Mannheim, Indianapolis, Ind.) Is added and the number of viable adherent cells is determined. After 4 hours, the absorbance is determined according to the manufacturer's protocol.
[0038]
STO substrain
100 parental STO cells obtained from the ATCC are 10% heat inactivated fetal bovine serum, 2 x 10-3M-glutamine, 5 x 10-5Cultivate for 7 days in a 100 mm culture dish in DMEM / F12 supplemented with M2-mercaptoethanol and antibiotics. Depending on cell morphology and growth rate, four subclones are selected for further analysis. CFA of rter6 is performed on four subclones, one of which, STO6, does not adhere to the culture plate after mitomycin C treatment. One subclone STO5 is transfected with pEF-Hlx-MC1neo or pEF-MC1neo provided by Dr. Adams (Dr. JM Adams, The Walter and Eliza Hall Institute of Medical Research). Plasmids linearized at the Nde I site are introduced into cells by DOPER liposome transfection reagent (Boehringer Mannheim). After G418 selection, six clones are isolated. Each of the three clones is analyzed by CFA.
[0039]
Colony immunohistochemical staining
Culture plates were fixed with methanol-acetone (1: 1) at room temperature for 2 minutes, rinsed, blocked at 4 ° C. with Hanks Balanced Salt Solution (HBSS) containing 20% goat serum (GIBCO / BRL). I do. For dual immunohistochemistry of alpha fetoprotein and albumin, plates were treated with anti-rat albumin antibody (ICN Biomedicals, Costa Mesa, Calif.) followed by Texas Red-conjugated anti-rabbit IgG (Vector laboratories, Burlingame, Calif.), and FITC binding. Incubate with anti-rat α-fetoprotein polyclonal antibody (Nordic Immunology, Tilburg, Netherlands). For double labeling of albumin and CK19, an anti-CK19 monoclonal antibody (Amersham, Buckinghamshire, England) and a FITC-conjugated anti-mouse IgG (Caltag, Burlingame, CA) are used instead of the anti-α-fetoprotein antibody.
[0040]
Fetal liver dissociation at E13
Fetal liver was 10 mM HEPES, 0.8 mM MgSO4Dissect into Ca ++-free ice-cold HBSS supplemented with 1 mM EGTA (pH 7.4). Liver was 10 mM HEPES, 0.8 mM MgSO4, And 1 mM CaCl2Mill with 0.2% type IV collagenase in HBSS (Sigma) and 16.5 units / ml thermolysin (Sigma) prepared in. After 10 minutes incubation at 37 ° C., the cell suspension is digested with 0.025% trypsin and 2.5 mM EDTA (Sigma) for 10 minutes. Thereafter, trypsin is inhibited by adding 1 mg / ml trypsin inhibitor (Sigma). Finally, the cells are treated with 200 units / ml deoxyribonuclease I (Sigma). In all experiments, 3-5 x 10 per liver5Cells are obtained.
[0041]
Isolation of adult hepatocytes
To isolate hepatocytes, a two-stage liver perfusion method is used. After perfusion, the cells are centrifuged twice at 50 g for 1 minute to concentrate large parenchymal cells. Viability is> 90% as measured by trypan blue exclusion.
[0042]
Flow cytometry analysis
Cells are analyzed on a FACScan (Becton-Dickinson, Mountain View, CA) and sorted using a FlowFlow Cytometer (Cytomation, Fort Collins, CO). Cell suspensions obtained from E13 fetal liver are incubated on ice with HBSS containing 20% goat serum (GIBCO / BRL) and 1% teleost gelatin (Sigma) to prevent non-specific antibody binding. . After rinsing, cells were FITC-conjugated anti-rat RT1Aa, b, lSuspend with antibody B5 (Pharmingen, San Diego, CA) and PE-conjugated anti-rat ICAM-1 antibody 1A29 (Pharmingen). In some experiments, cells were stained with a biotinylated anti-rat monomorphic MHC class I antibody OX18 (Pharmingen) followed by a second stain with streptavidin-red 670 (GIBCO / BRL) for three-color staining. Do. All stains were 10 mM HEPES, 0.8 mM MgSO.4, 0.2 mM EGTA, and 0.2% BSA, pH 7.4, using Ca ++ free ice-cold HBSS. The three established cell lines are trypsinized and feeder cells are removed by Percoll density gradient centrifugation. Rat hepatoma cell line FTO-2B and rat liver epithelial cell line WB-F344 and adult hepatocytes are stained for comparison with fetal hepatocyte cell lines. The cell lines were Dr. FEK Fournier, Fred Hutchinson Cancer Research Center, Seattle, WA, respectively, and Dr. Saw (Dr. M.-S. Tsao, University of California, University of California, USA). NC). Cells are blocked and stained with FITC-conjugated B5, OX18, PE-conjugated 1A29, or anti-FITC-conjugated rat integrin β1 antibody Ha2 / 5 (Pharmingen). FITC-conjugated anti-mouse IgG is used for OX18. To eliminate the mouse cell population, cell suspensions of the three fetal liver cell lines are stained with biotinylated anti-mouse CD98, followed by a second staining with streptavidin-red 670 and an anti-rat monoclonal antibody.
[0043]
Different antigens are expressed in different relative numbers for each cell. In practical use, the level of expression of a particular antigen can be no expression, low levels of expression, normal levels of expression for many antigens, and high levels of expression. In this usage, the term "low" is used interchangeably with "weak" or "dull". Although a more detailed description of the expression levels can be made, these four levels are sufficient for many purposes. Obviously, measurement of antigen expression by flow cytometry obviously provides a continuous range for antigen expression.
[0044]
CFA of hepatocyte cell lines, sorted cells, and adult hepatocytes
The hepatocyte line was 9.6 cm above the mitomycin C treated STO feeder layer using the same HDM used to maintain the cell line.2Seed in triplicate at 500 cells per cell. Prior to seeding, cells are trypsinized and feeder cells are removed by Percoll density gradient centrifugation. The culture solution is incubated for 10 to 14 days while changing the medium every two days. Thereafter, double immunofluorescence staining of α-fetoprotein and albumin is performed. One hundred colonies in each well are analyzed for colony morphology, P or F type, and expression of α-fetoprotein and albumin. Colonies are stained using Diff-Quick (Baxter, McGaw Park, IL) and the number of colonies in each well is counted. For CFA of primary sorted cells and adult hepatocytes, change the number of cells seeded as described. As another minor change, the culture period was extended between 14 and 17 days and the concentration of dexamethasone was increased to 10-6Increase to M. Other procedures are performed as described above. In adult hepatocyte CFA, a small number of hepatocyte clamps are not removed from the cell suspension after preparation. Thus, an indefinite number of colonies can be produced from the clamp. For CFA of bile duct differentiation on sorted cells, double immunofluorescence staining of colonies with albumin and CK19 is performed on
[0045]
6.2. Production and analysis of fetal rat hepatocyte cell line using mouse embryo cell feeder using hormone-supplemented synthetic medium
A simple long-term culture of rat E15 hepatocytes is attempted to see how long fetal hepatocytes can be maintained, proliferated ex vivo, and produce offspring. After density gradient centrifugation to remove hematopoietic mononuclear cells, fetal hepatocytes are cultured using a type IV collagen or laminin-coated culture dish and HDM (see Example 6.1). Cells survive for more than 4 weeks. However, secondary cultures on dishes newly coated with type IV collagen or laminin do not proliferate further. When the STO embryonic mouse fibroblast cell line treated with mitomycin C is used as a feeder layer for secondary culture, many clumps of cells proliferate. Finally, from four independent experiments, several stable hepatocyte cell lines are established.
[0046]
Immunohistochemical analysis of alpha fetoprotein and albumin is performed on a continuously growing population of cells prior to cloning of the cell line. Both alpha-fetoprotein and albumin proteins are used as markers to confirm that the cell population is of hepatocyte origin. A group of cells that tend to form cell stacks, called P colonies, strongly expressed α-fetoprotein and albumin, while a flat monolayer, called F colonies, had reduced expression of α-fetoprotein and albumin. Did not. Embryonic mouse fibroblast STO shows no reactivity with any antibody. For further analysis, three cloned hepatocyte cell lines are selected in independent experiments by morphological criteria of P-type or F-type colonies. Rhel4321 (FIG. 1A) consists mainly of P-cell colonies packed with small cells, while th1120-3 (FIG. 1C) forms only a flat monolayer of F-type colonies. Rter6 (FIG. 1B) is intermediate between the two phenotypes. Interestingly, the heterogeneity of rter6 is observed even after three consecutive clonings of flat colonies. To see the heterogeneity of the colonies from single cells of rhel4321 and rter6, 9.6 cm2Cells were seeded at a density of 500 cells (one well of a 6-well plate) and cells were cultured on STO fibroblasts for 10-14 days. Thereafter, the morphology of the colonies and the expression of α-fetoprotein and albumin were analyzed. 2A, 2B, 2C, 2D, 2E, and 2F show the results. The rhel4321 (FIG. 2B) and rter6 (FIG. 2C) cell lines and the original cell population before cloning (FIG. 2A) express α-fetoprotein strongly in almost all P-type colonies, whereas F-type colonies Does not appear. Furthermore, strong expression of both α-fetoprotein and albumin is observed only in P-type colonies. Morphological differences in cloned hepatocyte lines correlate with the percentage of P-type colonies (FIGS. 2B and 2C). The percentages of P-type colonies of CFA of rter6 and rhel4321 are 33.3% (± 8.6% SD) and 65.7% (± 4.0% SD), respectively. The total number of colonies per well is counted, and the clonal growth efficiency (colony efficiency) is calculated. The efficiencies of rter6 and rhel4321 are 45.7% (± 1.3% SD) and 36.4% (± 1.1% SD), respectively. The th1120-3 cells are strongly adhered to each other along the lateral border, making the preparation of a single cell suspension very difficult. However, th1120-3 cells do not form overlapping clusters.
[0047]
Since the adhesion of mouse hepatocytes to extracellular matrix (ECM) proteins such as laminin, type IV collagen, and fibronectin varies by developmental stage, we next examine each cell line's preference for adhering to specific ECM components. . Similar to the findings in adult hepatocytes, type IV collagen is most effective for th1120-3 adhesion (FIG. 1C), but not so much for rter6 (FIG. 1B) and rhel4321 (FIG. 1A). Laminin is the most effective substrate for rhel4321 adhesion (FIG. 1A). This preference is similar to the primary culture of fetal mouse hepatocytes. In summary, the conserved expression of α-fetoprotein and albumin in P-type colonies, and preferential adhesion of laminin by rhel4321 indicates that the population of cells producing P-type colonies is more strictly associated with hepatic progenitor cells Suggests.
[0048]
6.3 Isolation of STO subclone for colony formation; Assay for hepatic progenitor cells
To develop a CFA system to identify highly proliferative, bipotent hepatic progenitor cells, culture systems support cell growth at clonal seeding densities and preserve important original liver function. Must-have. Two of the most important markers for early liver development are albumin and alpha-fetoprotein. Culture conditions that optimize P-type colonies are best, as P-type but not F-type colonies maintain α-fetoprotein and albumin expression during clonal expansion. Therefore, the ability of STO subclones to support the rter6 P-type colonies is compared. One clone, STO5, supports P-type colony formation over all other sublines and parental strains (FIG. 2D). The CFA of rhel4321 also confirms that STO5 is a more effective feeder than the parent STO (FIG. 2E). The mouse H1x gene product expressed in the mesenchymal cells lining the gastrointestinal tract of E10.5 is essential for the growth of fetal hepatocytes. When H1x gene mRNA expression was analyzed in STO subclones, there was no significant difference in expression between subclones (data not shown). Furthermore, stable transfectants in mouse H1x STO5 do not improve the colony formation assay (FIG. 2F). However, one clone of the transfectant, at a relatively high density of passages, more stably retains the original form of STO5, so this clone is used for further experiments.
[0049]
6.4 Identification of hepatic progenitor cells from E13 fetal liver using surface antigen markers and colony formation assays
Liver formation and massive hematopoiesis coexist in fetal liver. To date, the antigenic profile of hematopoietic progenitor cells has been extensively analyzed, but studies of early hepatic progenitor cells are still in their infancy. Using the three hepatocyte lines established in this study, an adult hepatoma cell line (FTO-2B), an epithelial cell line derived from adult rat liver (WB-F344), and newly isolated adult hepatocytes, The antigenic profile of the cells is analyzed. Compared to FTO-2B, WB-F344 and adult hepatocytes, the most immature fetal hepatocyte cell line, rhel4321, is a classic MHC class I (RT1A1) Are very unique in that there is no expression (FIGS. 3A-3X). Cell line th1120-3 (FIGS. 3I-3L) contains RT1A1, OX18 (pan-MHC class I), and ICAM-1 patterns are similar to rhel4321 (FIGS. 3A-3D), but rter6 (FIGS. 3E-3H) is RT1A.1And OX18 expression is relatively high (FIG. 3). In addition, another cell line obtained in a different experiment has the same morphology as rhel4321, but also RT1A1-, OX18dull, And ICAM-1+It is. Integrin b1 expression is similar in all cell lines, but RT1Aa, b, 1And the pattern of ICAM-1 is unique in cells. The antigenic profile of adult hepatocytes is RT1A1+, OX18+, And ICAM-1+It is. In adult rats, fetal hepatocytes can be separated from hematopoietic cells by MHC class I expression, because all bone marrow cells except erythrocytes strongly express MHC class I molecules. Cell suspension obtained from rat E13 liver was used as anti-RT1A1And staining with ICAM-1 antibody. FIG. 4A shows RT1A.12 shows a two-color staining pattern of ICAM-1 and ICAM-1. To determine which fractions contain the hepatocyte population, five fractions are isolated by fluorescence activated cell sorting and screened for clonal expansion potential by CFA. FIG. 4B shows the result of re-sorting the five fractions after sorting. Hepatocyte colonies defined by the expression of albumin and α-fetoprotein are morphologically distinguishable, so that the number of liver colonies in each well can be counted. Most of the liver colonies are RT1A1 dulAnd ICAM-1+(Table 1, FIG. 4B, gate 2), and the frequency of P-type colonies is 75.6% (± 4.9% SD).
[0050]
TABLE 1 Frequency of liver colonies from E13 fetal liver sorted based on RT1A and ICAM-1 expression
Colony forming cultures on STO5hlx contain the indicated number of cells obtained from each fraction of E13 fetal liver. The number of liver colonies was established from triplicate stained cultures (mean ± SD). The efficiency of colony formation indicates the percentage of cells inoculated into the culture solution, at which the colony was formed after analysis after 16 days of culture.
[0052]
6.5. Different culture requirements for E13 hepatocytes and adult hepatocytes
The growth requirements of sorted hepatocytes obtained from E13 liver were examined using a defined STO5 feeder and HDM. EGF has long been known to be a potent growth factor for adult hepatocytes. Therefore, the effect of EGF on sorted hepatocyte colony formation is examined. RT1A1- OX18dull, ICAM-1+Hepatocyte colony size increases in the absence of EGF, but adult hepatocytes generated colonies only in the presence of EGF (FIG. 6C). Furthermore, the morphology of colonies derived from adult hepatocytes is usually F-type, but RT1A1-Hepatocytes form P-type colonies without EGF. However, colony efficiency is slightly reduced in the absence of EGF (FIG. 6A). Interestingly, culture conditions without EGF highlighted two types of P colonies, P1 and P2. At day 12 of culture, most of the colonies are of the P2 type, but it is difficult to completely distinguish the two types, as some colonies do not have the typical morphology as in FIG. 6A. These results indicate that fetal and adult hepatocytes have their growth requirements and RT1A1It suggests that the expression (FIGS. 3 and 4) and the morphology of the colonies are essentially different.
[0053]
After 3 weeks in culture when growth appears to have reached a maximum, RT1A1- , OX18 and ICAM-1 are evaluated. As shown in FIGS. 5B-5D, RT1A1Is not induced and OX18 expression is reduced. The level of ICAM-1 does not change. In addition, the average cell number of a single colony is calculated from the number of cells recovered, the percentage of rat hepatocytes, and the colony efficiency. The estimated cell number is 3-4 x 103(Table 2) is reached. This indicates that a single cell forming a colony divided about 11 to 12 times on average in this culture condition.
[0054]
Table 2: Calculation of cell number in a single liver colony
Cells sorted from R4 in FIG. 4C were cultured on STO5hlx feeder cells in 60 mm or 100 mm dishes. After culturing for the indicated period, all cells were harvested and the total cell number was calculated. The percentage of rat cells is by flow cytometry analysis based on the expression of rat ICAM-1 and mouse CD98. Colony efficiency indicates the percentage of cells that formed a colony among the cells inoculated into the culture. Data from triplicate stained cultures (average) were obtained from experiments performed in parallel.
[0056]
Average number of cells in a single colony = (number of cells recovered x percentage of rat cells / 100) / number of cells inoculated x colony efficiency / 100)
[0057]
6.6 RT1A1-Evidence that hepatic progenitor cells are bipotent
In rat embryonic age E13, hepatocytes are considered to be bipotent progenitor cells that give rise to mature hepatocytes and bile duct epithelial cells. However, prior to the present invention, there was no direct evidence to indicate whether these two fates originated from a single cell. RT1A1- OX18dull ICAM-1+Colonies are stained with anti-CK19 as a marker specific for bile duct epithelial cells to determine if the fetal hepatocytes of this can be differentiated into bile duct cell lines in this culture. CK19 is expressed in biliary epithelial progenitor cells in fetal liver after day 15.5, at which point the cells have lost albumin expression. RT1A sorted1- ICAM-1 + cells are cultured in the presence or absence of EGF, and their fate is monitored on
[0058]
6.7 Protocol for isolation and cloning of feeder cells capable of supporting clonal expansion of hepatic stem cells and hepatic progenitor cells
Slice fresh embryo or frozen tissue (eg, liver, lung, kidney, muscle, intestine) of pig, beagle, rabbit, mouse, or monkey in calcium-free phosphate buffered saline (PBS) . After rinsing twice with PBS, the cell suspension is incubated with 0.25% trypsin for 10 minutes at 37 ° C. or 60 minutes at room temperature using a magnetic stirrer. Remaining cell clumps are removed by filtering the suspension through a mesh. The cells are then placed in a basal medium (eg, Eagle MEM) supplemented with serum (eg, 10% fetal bovine serum) and any of a variety of growth supplements (eg, 2 mM glutamine, sodium pyruvate, and MEM non-essential amino acids). Culture in a tissue culture dish using. Plastic basal and serum-supplemented media allow the growth of a population of cells that are candidates for feeder cells (“feeder cells”), often derived from mesoderm (eg, stromal cells), and provide another type of cell General conditions that provide factors that support the survival, proliferation, and / or function of (eg, progenitor cells). Feeder cells are subcultured with 0.05% trypsin when it is at or near confluence. After several passages, the grown cells are prepared as frozen stocks and stored frozen until use. Another source of feeder cells is commercially available primary culture feeder cells or feeder cell lines. In any case, the following criteria are required to identify a suitable feeder cell.
[0059]
Feeder cells support:
1) clonal expansion of hepatic progenitor cells with phenotypic markers of classical MHC class I antigen negative, ICAM-1 positive, and / or non-classical MHC class I antigen weak positive;
2) clonal expansion of progenitor cells having progeny with a phenotypic marker of classical MHC class I antigen negative, ICAM-1 positive, non-classical MHC class I antigen weak positive, α-fetoprotein positive, albumin positive, or CK19 positive; Or
3) Inducible differentiation into both hepatocellular and cholangiocyte cell lines required for the definition of bipotent hepatic progenitor cells.
[0060]
In the art, classical MHC class I antigens are also known as MHC class Ia antigens. Non-classical MHC class I antigens are also known as MHC class Ib antigens. MHC antigens are called differently in different species: for example, RT1 in rats, H-2 in mice, and HLA in humans.
[0061]
The above assay is described below:
[0062]
Conditions for clonal expansion of hepatic progenitor cells
Hepatic progenitor cells were 9.6 cm above feeder cells that stopped growing, ie, were treated to prevent growth.2Inoculate 500 cells per. Feeder cells are treated with mitomycin C or irradiation (3000-5000 rads depending on cell type) to stop growth. Serum-free HDM is supplied to feeder cells and progenitor cells that have stopped growing. For example, the HDM of rodent cells can be obtained by adding 10 ng / ml EGF, 5 μg / ml insulin, 10 μg / ml to a 1: 1 mixture of Dulbecco's modified Eagle's medium and Ham F12.-6M dexamethasone, 10 μg / ml iron-saturated transferrin, 4.4 × 10-3M nicotinamide, 0.2% bovine serum albumin, 5 × 10-5M 2-mercaptoethanol, 7.6 μeq / l free fatty acid mixture, 2 × 10-3M glutamine, 1 x 10-6M CuSO4, 3 x 10-8MH2SeO3And antibiotics added. The culture is incubated for 10 to 14 days, changing the medium every two days. Subsequently, double immunofluorescence staining for α-fetoprotein, albumin, and / or CK19 is performed to identify the fate of the offspring. Approximately 100 colonies are analyzed for expression of alpha fetoprotein and albumin. In addition, the morphology of the colony, P-type or F-type, may also help identify suitable offspring.
[0063]
The ideal combination of feeder cells and hepatic progenitor cells is from the same species. Preferably, the feeder cells are obtained from the same tissue and the same species as the hepatic progenitor cells. However, it is also possible to mix feeders from one species with precursor cells from another species. For example, even rodent feeders can be used for human liver progenitor cells. Soluble and insoluble factors, which may be species and / or tissue specific, assist in clonal expansion of hepatic stem cells or hepatic progenitor cells. Sources of such factors include the following:
1) A conditioned medium in which feeder cells of the optimal species and tissue have been cultured. The feeder cells may be not only stromal cells but also any kind of cells.
2) When important factors are known, cDNA or mRNA for transcription or translation, respectively, is optionally used to synthesize an appropriate molecule (signal) derived from an optimal feeder cell that is active in hepatic progenitor cells. A source from which biologically active feeder cell groups can be produced by introducing the cells into the cells.
3) Regardless of whether the signal obtained from the optimal feeder cell active on hepatic progenitor cells is a protein, peptide, hydrocarbon, lipid, glycopeptide, glycoprotein, lipoprotein, glycolipid, or a combination thereof, A source that, if the factors are known, can also completely replace feeder cells by adding such a signal to the medium.
[0064]
The above examples are described by way of illustration only and do not limit the scope or aspects of the present invention. Other embodiments not specifically described above will be apparent to those skilled in the art. However, such other embodiments are also considered to be within the scope and spirit of the present invention. Therefore, the present invention is properly limited only by the appended claims.
[0065]
All patents and publications cited herein are hereby incorporated by reference in their entirety.
[Brief description of the drawings]
FIG. 1 is a characterization of a hepatocyte cell line obtained from fetal rat liver on
FIG. 2 is a colony formation assay on fibroblast feeder cells.
FIG. 3. Expression of rat cell surface antigen on various cell lines in adult hepatocytes.
FIG. 4 depicts phenotypic analysis of fetal rat liver on day 13.
FIG. 5 is a characterization of liver colonies in the presence and absence of EGF.
FIG. 6: RT1A1-FIG. 4 shows induction of CK19 expression on hepatocytes.
FIG. 7 is a schematic diagram of liver colony formation on STO5 feeder cells.
Claims (80)
(a) 細胞間接着分子抗原を発現し、主要組織適合性抗原を発現しない、少なくとも1つの肝前駆細胞を提供する段階、および
(b) 少なくとも1つのフィーダー細胞の生合成産物を含む培地中で肝前駆細胞を培養する段階
を含む方法。A method of expanding hepatic progenitor cells, progeny thereof, or a mixture thereof,
(A) providing at least one hepatic progenitor cell that expresses an intercellular adhesion molecule antigen and does not express a major histocompatibility antigen; and (b) in a medium containing a biosynthetic product of at least one feeder cell. A method comprising culturing hepatic progenitor cells.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2000/027428 WO2002029012A1 (en) | 2000-10-03 | 2000-10-03 | Processes for clonal growth of hepatic progenitor cells |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004510434A true JP2004510434A (en) | 2004-04-08 |
| JP2004510434A5 JP2004510434A5 (en) | 2005-11-17 |
Family
ID=21741846
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002532583A Pending JP2004510434A (en) | 2000-10-03 | 2000-10-03 | Method for clonal expansion of hepatic progenitor cells |
Country Status (9)
| Country | Link |
|---|---|
| EP (1) | EP1325111A1 (en) |
| JP (1) | JP2004510434A (en) |
| KR (1) | KR100887623B1 (en) |
| CN (1) | CN1461341A (en) |
| AU (2) | AU7753500A (en) |
| CA (1) | CA2424779C (en) |
| HK (1) | HK1054570A1 (en) |
| IL (1) | IL155387A0 (en) |
| WO (1) | WO2002029012A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010534065A (en) * | 2007-07-20 | 2010-11-04 | セルアーティス アーベー | A population of novel hepatocytes derived from human blastocyst stem cells via definitive endoderm (DE-hep) |
| JP2015006137A (en) * | 2013-06-24 | 2015-01-15 | 独立行政法人医薬基盤研究所 | Culture medium for growing hepatic precursor cells |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1117762A4 (en) | 1998-09-29 | 2004-02-25 | Gamida Cell Ltd | Methods of controlling proliferation and differentiation of stem and progenitor cells |
| US20020187133A1 (en) | 1999-10-01 | 2002-12-12 | Hiroshi Kubota | Methods of isolating bipotent hepatic progenitor cells |
| NZ528462A (en) | 2001-03-27 | 2005-11-25 | Vertex Pharma | Compositions and methods useful for HCV infection |
| IL152904A0 (en) * | 2002-01-24 | 2003-06-24 | Gamida Cell Ltd | Utilization of retinoid and vitamin d receptor antagonists for expansion of renewable stem cell populations |
| RU2327479C2 (en) * | 2002-03-15 | 2008-06-27 | Юниверсити Оф Норт Каролина Эт Чепел Хилл | Primitive and proximal stem hepatocytes |
| SG141296A1 (en) * | 2002-05-17 | 2008-04-28 | Sinai School Medicine | Mesoderm and definitive endoderm cell populations |
| US20060003446A1 (en) | 2002-05-17 | 2006-01-05 | Gordon Keller | Mesoderm and definitive endoderm cell populations |
| US8669109B2 (en) | 2003-08-19 | 2014-03-11 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Methods of producing proteins in Chinese hamster ovary (CHO) cells |
| DE10338531A1 (en) * | 2003-08-19 | 2005-04-07 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Method for recloning production cells |
| CA2457296A1 (en) | 2003-08-19 | 2005-02-19 | Takahiro Ochiya | Methods for inducing differentiation of pluripotent cells |
| JP5588587B2 (en) * | 2004-01-14 | 2014-09-10 | ノーバヘップ アーベー | Human liver progenitor cells and methods of use thereof |
| CN1914313A (en) * | 2004-02-13 | 2007-02-14 | 旭科技玻璃股份有限公司 | Medium for preparing feeder cells for embryonic stem cells and feeder cells |
| US8874380B2 (en) | 2010-12-09 | 2014-10-28 | Rutgers, The State University Of New Jersey | Method of overcoming therapeutic limitations of nonuniform distribution of radiopharmaceuticals and chemotherapy drugs |
| JP2010509316A (en) * | 2006-11-09 | 2010-03-25 | ガミダ セル リミテッド | Use of ex vivo cultured hematopoietic cells to treat peripheral vascular disease |
| CN101275121B (en) * | 2007-03-26 | 2011-05-11 | 芦银雪 | In vitro culture-amplified human liver progenitor cell and preparation thereof |
| CN101701219B (en) * | 2009-11-06 | 2013-02-20 | 哈尔滨医科大学 | cDNA for encoding rabbit hepatocyte growth factor and expression vector and application thereof |
| CN104630130A (en) * | 2014-12-05 | 2015-05-20 | 新乡医学院 | Method for separating rat hepatocytes |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08112092A (en) * | 1994-08-23 | 1996-05-07 | Res Dev Corp Of Japan | Hepatic parenchymal cell having cloning proliferation potency and methods for obtaining and subculturing the same |
| WO2000043498A2 (en) * | 1999-01-19 | 2000-07-27 | University Of North Carolina At Chapel Hill | Human liver progenitors |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4914032A (en) * | 1985-06-06 | 1990-04-03 | Centro De Investigation Y Estudios Avanzados Del Instituto Politecnico Nacional | Process for the long-term surviving culture of hepatocytes |
| EP0597964A4 (en) * | 1991-08-07 | 1994-11-30 | Einstein Coll Med | Proliferation of hepatocyte precursors. |
| JPH09505206A (en) * | 1993-11-19 | 1997-05-27 | イェシバ・ユニバーシティ | Hepatoblast and its isolation method |
| CA2146735C (en) * | 1994-04-11 | 2007-02-20 | Chise Tateno | Liver parenchymal cells having clonal growth ability, method for obtaining same, method for subculturing same, and subculturing system of primary hepatocytes |
| WO2002028997A2 (en) * | 2000-10-03 | 2002-04-11 | University Of North Carolina | Methods of isolating bipotent hepatic progenitor cells |
-
2000
- 2000-10-03 AU AU7753500A patent/AU7753500A/en active Pending
- 2000-10-03 WO PCT/US2000/027428 patent/WO2002029012A1/en active IP Right Grant
- 2000-10-03 EP EP00967317A patent/EP1325111A1/en not_active Ceased
- 2000-10-03 JP JP2002532583A patent/JP2004510434A/en active Pending
- 2000-10-03 KR KR1020037004803A patent/KR100887623B1/en not_active IP Right Cessation
- 2000-10-03 AU AU2000277535A patent/AU2000277535B2/en not_active Ceased
- 2000-10-03 IL IL15538700A patent/IL155387A0/en unknown
- 2000-10-03 CN CN00820051A patent/CN1461341A/en active Pending
- 2000-10-03 CA CA2424779A patent/CA2424779C/en not_active Expired - Fee Related
-
2003
- 2003-09-19 HK HK03106718.5A patent/HK1054570A1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08112092A (en) * | 1994-08-23 | 1996-05-07 | Res Dev Corp Of Japan | Hepatic parenchymal cell having cloning proliferation potency and methods for obtaining and subculturing the same |
| WO2000043498A2 (en) * | 1999-01-19 | 2000-07-27 | University Of North Carolina At Chapel Hill | Human liver progenitors |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010534065A (en) * | 2007-07-20 | 2010-11-04 | セルアーティス アーベー | A population of novel hepatocytes derived from human blastocyst stem cells via definitive endoderm (DE-hep) |
| US8951792B2 (en) | 2007-07-20 | 2015-02-10 | Cellartis Ab | Methods for making definitive endoderm hepatocyte (DE-hep) progenitor cells |
| JP2015006137A (en) * | 2013-06-24 | 2015-01-15 | 独立行政法人医薬基盤研究所 | Culture medium for growing hepatic precursor cells |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2424779C (en) | 2016-08-23 |
| AU2000277535B2 (en) | 2007-03-29 |
| AU7753500A (en) | 2002-04-15 |
| CN1461341A (en) | 2003-12-10 |
| KR20040047732A (en) | 2004-06-05 |
| KR100887623B1 (en) | 2009-03-11 |
| WO2002029012A1 (en) | 2002-04-11 |
| EP1325111A1 (en) | 2003-07-09 |
| IL155387A0 (en) | 2003-11-23 |
| CA2424779A1 (en) | 2002-04-11 |
| HK1054570A1 (en) | 2003-12-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090053758A1 (en) | Processes for clonal growth of hepatic progenitor cells | |
| AU2008202656B8 (en) | Primitive and proximal hepatic stem cells | |
| KR100887623B1 (en) | Processes for clonal growth of hepatic progenitor cells | |
| US9625450B2 (en) | Methods of isolating bipotent hepatic progenitor cells | |
| US10093895B2 (en) | Hepatic stellate cell precursors and methods of isolating same | |
| AU2000277535A1 (en) | Processes for clonal growth of hepatic progenitor cells | |
| AU2000278582B2 (en) | Methods of isolating bipotent hepatic progenitor cells | |
| AU2000278582A1 (en) | Methods of isolating bipotent hepatic progenitor cells | |
| US20030175255A1 (en) | Methods of isolating bipotent hepatic progenitor cells | |
| KR100845471B1 (en) | Processes for clonal growth of hepatic progenitor cells | |
| KR20070030936A (en) | Processes for clonal growth of hepatic progenitor cells | |
| AU2007202994A1 (en) | Processes for clonal growth of hepatic progenitor cells |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040406 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20050420 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20071001 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100628 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100902 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100909 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101228 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111019 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120117 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120124 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120625 |