FR2765221A1 - 4 - [(1H-IMIDAZOL-4-YL) PIPERIDIN-1-YL] ANILIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE - Google Patents
4 - [(1H-IMIDAZOL-4-YL) PIPERIDIN-1-YL] ANILIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE Download PDFInfo
- Publication number
- FR2765221A1 FR2765221A1 FR9707900A FR9707900A FR2765221A1 FR 2765221 A1 FR2765221 A1 FR 2765221A1 FR 9707900 A FR9707900 A FR 9707900A FR 9707900 A FR9707900 A FR 9707900A FR 2765221 A1 FR2765221 A1 FR 2765221A1
- Authority
- FR
- France
- Prior art keywords
- alkyl group
- group
- alkyl
- branched
- straight
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 C*1*CCN(C)C1 Chemical compound C*1*CCN(C)C1 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
La présente invention a pour objet des dérivés de 4- [(1H-imidazol-4-yl)pipéridin-1-yl]anilide, leur préparation et leur application en thérapeutique.The present invention relates to 4- [(1H-imidazol-4-yl) piperidin-1-yl] anilide derivatives, their preparation and their therapeutic application.
Les composés de l'invention répondent à la formule (I)
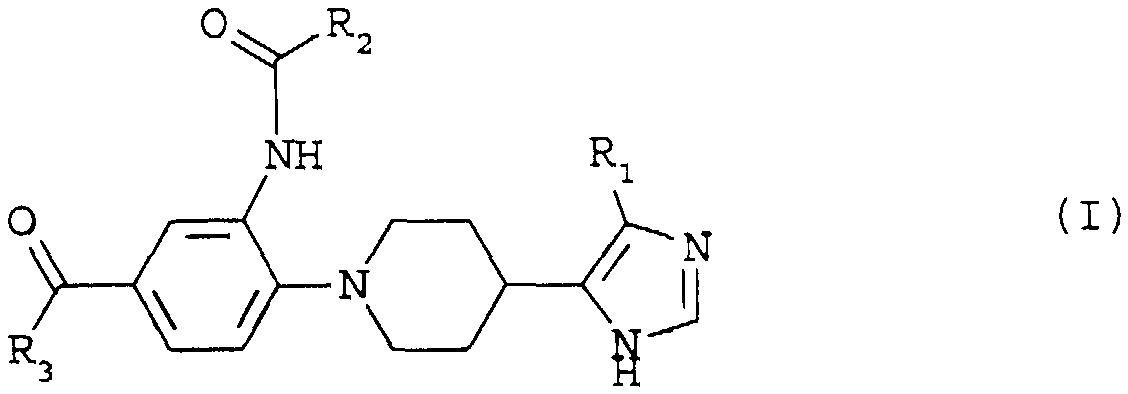
dans laquelle
R1 représente soit un atome d'hydrogène, soit un groupe (C1-C4)alkyle droit ou ramifié,
R2 représente soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle(Cl-Cs)alkyle et
R3 représente soit un groupe -OR4, soit un groupe -O(CH2)nNR5R6, soit un groupe -NHC(NH)NH2, soit un groupe -NHC(NH)N(CH3)2, soit un groupe -NR5R6, soit un groupe -MR5 (CH2) NR6R7, soit un groupe

in which
R1 represents either a hydrogen atom or a straight or branched (C1-C4) alkyl group,
R2 represents either a straight or branched (C1-C6) alkyl group, a (C3-C7) cycloalkyl group or a (C3-C7) alkyl (C1-C8) alkyl group and
R3 is either -OR4, -O (CH2) nNR5R6, -NHC (NH) NH2, -NHC (NH) N (CH3) 2, -NR5R6, or group -MR5 (CH2) NR6R7, a group
X étant choisi parmi les atomes d'oxygène et de soufre et les groupes -CHR8, -NR8, -SO- et -SO2-, R4, R51 R6, R7 et R8 étant indépendamment l'un de l'autre soit un atome d'hydrogène, soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle(Cl-Cs) alkyle, soit un groupe phényle, soit un groupe phényl(C1-C6) alkyle, soit un groupe hétéroaryle de 5 à 6 côtés, les hétéroatomes étant choisis parmi les atomes d'oxygène de soufre et d'azote, m est égal à 1 ou 2 et n est égal à 2, 3 ou 4, à l'état de bases libres ou de sels d'addition à des acides pharmaceutiquement acceptables.X being selected from oxygen and sulfur atoms and -CHR8, -NR8, -SO- and -SO2-, wherein R4, R51, R6, R7 and R8 are independently of one another; hydrogen, either a straight or branched (C1-C6) alkyl group, a (C3-C7) cycloalkyl group, a (C3-C7) alkyl (C1-C8) alkyl group, a phenyl group, or a phenyl (C1-C6) alkyl group, ie a 5- to 6-membered heteroaryl group, the heteroatoms being chosen from sulfur and nitrogen oxygen atoms, m is equal to 1 or 2 and n is equal to 2; , 3 or 4, in the form of free bases or addition salts with pharmaceutically acceptable acids.
Conformément à l'invention, on peut préparer les composés de formule (I) selon les procédés illustrés dans les schémas 1 et 2 ; dans ces schémas le groupe -C(C6H5)3 représente un groupe protecteur triphénylméthyle (groupe trityle).According to the invention, the compounds of formula (I) can be prepared according to the methods illustrated in Schemes 1 and 2; in these schemes the group -C (C6H5) 3 represents a triphenylmethyl protecting group (trityl group).
Pour préparer les composés de formule (Ia) et (Ib), on fait réagir un composé de formule (II) dans laquelle Hal représente un atome d'halogène avec un alcool de formule ROH dans laquelle R représente un groupe (C1-C6)alkyle droit ou ramifié et on obtient un composé de formule (III) que l'on fait réagir avec un composé de formule (Iv) dans laquelle R1 est tel que défini précédemment, dans un solvant aprotique comme le diméthylformamide en présence d'une base comme la
N,N-diisopropyléthylamine, pour obtenir un composé de formule (V) que l'on traite par du chlorure de triphénylméthyle dans un solvant tel que le dichlorométhane en présence d'une base comme la N-méthylmorpholine pour préparer un composé de formule (VI) que l'on soumet à une hydrogénation catalytique et on obtient un composé de formule (VII) que l'on fait réagir avec un composé de formule R2COX dans laquelle X représente un atome d'halogène et R2 est tel que défini précédemment et on obtient un composé de formule (VIII).To prepare the compounds of formula (Ia) and (Ib), a compound of formula (II) in which Hal represents a halogen atom is reacted with an alcohol of formula ROH in which R represents a group (C1-C6) straight or branched alkyl and there is obtained a compound of formula (III) which is reacted with a compound of formula (IV) in which R 1 is as defined above, in an aprotic solvent such as dimethylformamide in the presence of a base as the
N, N-diisopropylethylamine, to obtain a compound of formula (V) which is treated with triphenylmethyl chloride in a solvent such as dichloromethane in the presence of a base such as N-methylmorpholine to prepare a compound of formula ( VI) which is subjected to a catalytic hydrogenation and a compound of formula (VII) is obtained which is reacted with a compound of formula R 2 COX wherein X represents a halogen atom and R 2 is as defined above and a compound of formula (VIII) is obtained.
Ensuite si on veut préparer un composé de formule (Ia) qui correspond à un composé de formule (I) dans laquelle R3 représente un groupe -OR4, R4 étant un atome d'hydrogène ou un groupe (C1-C6)alkyle droit ou ramifié alors on déprotège le noyau imidazole du composé de formule (VIII) correspondant dans des conditions classiques connues de l'homme du métier puis on réalise éventuellement une hydrolyse de la fonction ester.Then, if it is desired to prepare a compound of formula (Ia) which corresponds to a compound of formula (I) in which R3 represents a group -OR4, R4 being a hydrogen atom or a straight or branched (C1-C6) alkyl group then the imidazole nucleus of the corresponding compound of formula (VIII) is deprotected under standard conditions known to those skilled in the art, and the ester function is optionally hydrolysed.
Si on veut préparer un composé de formule (Ib) qui correspond à un composé de formule (I) dans laquelle R3 représente soit un groupe -NHC(NH)NH2, soit un groupe -NHC(NH)N(CH3)2, alors on fait réagir le composé de formule (VIII) correspondant avec un composé de formule H2NC(NH)N(Rg) 2 dans laquelle Rg représente soit un atome d'hydrogène, soit un groupe méthyle pour obtenir un composé de formule (XI) dont on déprotège le
Schéma 1

noyau imidazole dans des conditions classiques connues de l'homme du métier.If it is desired to prepare a compound of formula (Ib) which corresponds to a compound of formula (I) in which R 3 represents either an -NHC (NH) NH 2 group or a -NHC (NH) N (CH 3) 2 group, then the corresponding compound of formula (VIII) is reacted with a compound of formula H2NC (NH) N (Rg) 2 in which Rg represents either a hydrogen atom or a methyl group in order to obtain a compound of formula (XI) whose we deprotect the
Diagram 1
imidazole nucleus under standard conditions known to those skilled in the art.
Lorsqu'on veut obtenir un composé de formule (Ic) qui correspond à un composé de formule (I) dans laquelle R3 représente soit un groupe -OR4 (R4 étant un groupe cyclo(C3-C7)alkyle, cyclo(C3-C7)alkyle(Cl-Cs)alkyle, phényle, phényl(Cl-C6)alkyle ou hétéroaryle de 5 à 6 côtés les hétéroatomes étant choisis parmi les atomes d'oxygène de soufre et d'azote), soit un groupe -O(CH2)nNR5R6, soit un groupe -NR5R6, soit un groupe -NR5(CH2)nNR6R7 (R5, R6, R7 et n étant tels que définis précédemment), soit un groupe

X étant choisi parmi les atomes d'oxygène et de soufre et les groupes -CHR8, -NR8, -So- et -SO2- et R8 et m étant tels que définis précédemment, alors on procède selon le schéma 2.X being selected from oxygen and sulfur atoms and -CHR8, -NR8, -So- and -SO2- groups and R8 and m being as defined above, then proceed as in scheme 2.
On traite le composé de formule (VIII) correspondant en milieu basique et on obtient un composé de formule (IX) que l'on fait réagir avec un composé de formule R3H (R3 étant tel que défini ci-dessus) dans un solvant aprotique comme le diméthylformamide en présence de l,l'-carbonyldiimidazole pour préparer un composé de formule (X) dont on déprotège le noyau imidazole dans des conditions classiques connues de l'homme du métier.The corresponding compound of formula (VIII) is treated in a basic medium and a compound of formula (IX) is obtained which is reacted with a compound of formula R 3 H (R 3 being as defined above) in an aprotic solvent such as dimethylformamide in the presence of 1,4-carbonyldiimidazole to prepare a compound of formula (X) which is deprotected imidazole nucleus under standard conditions known to those skilled in the art.
Dans une variante selon l'invention, on peut préparer les composés de formule (Ic) en faisant réagir les composés de formule (la) correspondants dans laquelle R3 représente un groupe -OR4, R4 étant un atome d'hydrogène, avec un compose de formule R3H dans un solvant aprotique comme le diméthylformamide en présence d'un base comme la N,N-diisopropyléthylamine et de l,l'-carbonyldiimidazole puis en réalisant une déprotection du noyau imidazole dans des conditions classiques connues de l'homme du métier. In an alternative according to the invention, the compounds of formula (Ic) can be prepared by reacting the corresponding compounds of formula (Ia) in which R3 represents a group -OR4, R4 being a hydrogen atom, with a compound of formula R3H in an aprotic solvent such as dimethylformamide in the presence of a base such as N, N-diisopropylethylamine and 1,4-carbonyldiimidazole and then carrying out a deprotection of the imidazole nucleus under standard conditions known to those skilled in the art.
Schéma 2

Les composés de départ sont décrits dans la littérature ou peuvent être préparés selon des méthodes qui y sont décrites ou qui sont connues de l'homme de métier.The starting compounds are described in the literature or may be prepared according to methods described therein or which are known to those skilled in the art.
Ainsi la préparation des composés de formule (IV) est décrite dans la demande de brevet européen EP 0507650.Thus the preparation of the compounds of formula (IV) is described in European Patent Application EP 0507650.
Les exemples qui suivent illustrent l'invention sans la limiter. Les micro-analyses et les spectres IR, RMN et de masse confirment la structure des composés obtenus. Les numéros des composés exemplifiés renvoient à ceux du tableau donné plus loin qui illustre les structures chimiques et les propriétés physiques de quelques composés selon l'invention.The following examples illustrate the invention without limiting it. Microanalyses and IR, NMR and mass spectra confirm the structure of the compounds obtained. The numbers of the exemplified compounds refer to those of the table given below which illustrates the chemical structures and the physical properties of some compounds according to the invention.
Les rapports (x:y) correspondent au rapport (acide:base).The ratios (x: y) correspond to the ratio (acid: base).
Exemple 1 (composé n0 11)
N-[2-[4-(5-méthyl- 1H- imidazol-4-yl) pipéridin-l-yl]-5-[ (4- méthylpipérazin- l-yl) carbonyl] phényl] cyclopropanecarboxamide 1.1. acide 3-[(cyclopropylcarbonyl)amino]-4-[4-f5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]
benzoate de méthyle
1.1.1 3-amino-4-[4-(5-méthyl-l-(triphénylméthyl)-lH-
imidazol-4-yl)pipéridin-l-yl]benzoate de
méthyle
a) 4-fluoro-3-nitrobenzoate de méthyle
On met 5 g (27 mmoles) d'acide 4-fluoro-3-nitrobenzoïque en solution dans 50 ml de méthanol, on ajoute à la température ambiante 7,88 ml (108 mmoles) de chlorure de thionyle et on chauffe le mélange pendant 2 heures à la température de reflux. On évapore le milieu réactionnel à sec et on reprend le résidu par 150 ml de dichlorométhane. On lave successivement par 2 fois 10 ml d'une solution aqueuse saturée d'hydrogénocarbonate de sodium puis par 50 ml d'eau et on sèche sur sulfate de magnésium.Example 1 (Compound No. 11)
N- [2- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -5 - [(4-methylpiperazin-1-yl) carbonyl] phenyl] cyclopropanecarboxamide 1.1. 3 - [(cyclopropylcarbonyl) amino] -4- [4- (5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl]
methyl benzoate
1.1.1 3-amino-4- [4- (5-methyl-1- (triphenylmethyl) -1H-
imidazol-4-yl) piperidin-1-yl] benzoate
methyl
a) methyl 4-fluoro-3-nitrobenzoate
5 g (27 mmol) of 4-fluoro-3-nitrobenzoic acid are dissolved in 50 ml of methanol, 7.88 ml (108 mmol) of thionyl chloride are added at ambient temperature and the mixture is heated for a 2 hours at reflux temperature. The reaction medium is evaporated to dryness and the residue is taken up in 150 ml of dichloromethane. Washed successively with 2 times 10 ml of a saturated aqueous solution of sodium hydrogencarbonate and then with 50 ml of water and dried over magnesium sulfate.
On obtient 5,37 g de produit.5.37 g of product is obtained.
Rendement = 91 %
Point de fusion = 70 OC
b) 4-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl]-3-
nitrobenzoate de méthyle
On met en suspension dans un mélange de 10 ml de dichlorométhane et de 2 ml de diméthylformamide, 2,38 g (10 mmoles) de dichlorhydrate de 4-(5-méthyl-lH-imidazol-4yl)pipéridine auxquels on ajoute 6,89 ml (40 mmoles) de
N,N-diisopropyléthylamine. On refroidit le mélange à - 5 OC et on ajoute goutte à goutte 1,9 g (10 mmoles) de 4-fluoro-3nitrobenzoate de méthyle en solution dans 5 ml de dichlorométhane. On laisse la température du mélange revenir à 0 "C puis on agite pendant 3 heures à cette température. On lave le milieu réactionnel avec 3 fois 10 ml d'eau, on sèche sur sulfate de magnésium et on évapore les solvants.Yield = 91%
Melting point = 70 OC
b) 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3-
methyl nitrobenzoate
10 g (10 mmol) of 4- (5-methyl-1H-imidazol-4-yl) piperidine dihydrochloride are suspended in a mixture of dichloromethane (10 ml) and dimethylformamide (2 ml) to which 6.89 g is added. ml (40 mmol)
N, N-diisopropylethylamine. The mixture is cooled to -5 ° C. and 1.9 g (10 mmol) of methyl 4-fluoro-3-nitrobenzoate dissolved in 5 ml of dichloromethane are added dropwise. The temperature of the mixture is allowed to return to 0 ° C. and then stirred for 3 hours at this temperature, the reaction mixture is washed with 3 times 10 ml of water, dried over magnesium sulphate and the solvents are evaporated off.
Après trituration dans l'éther, on obtient 2,2 g de produit sous forme d'une poudre beige.After trituration in ether, 2.2 g of product is obtained in the form of a beige powder.
Rendement = 67 8
Point de fusion = 108 OC
c) 4- [4- [5-méthyl-l- (trîphénylméthyl) -îH-imidazol-4-
yl]pipéridin-l-yl]-3-nitrobenzoate de méthyle
On met en suspension dans 190 ml de dichlorométhane à la température ambiante 20 g (58 mmoles) de 4-[4-(5-méthyl-1H- imidazol-4-yl)pipéridin-1-yl] -3-nitrobenzoate de méthyle, on ajoute 9,6 ml (87 mmoles) de N-méthylmorpholine puis 16,44 g (64 mmoles) de chlorure de trityle. On agite le mélange pendant 48 heures à la température ambiante et on ajoute 200 ml de dichlorométhane. On lave par 2 fois 150 ml d'eau.Yield = 67 8
Melting point = 108 OC
c) 4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazole-4-
methyl yl] piperidin-1-yl] -3-nitrobenzoate
190 g (58 mmol) of methyl 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3-nitrobenzoate are suspended in 190 ml of dichloromethane at room temperature. 9.6 ml (87 mmol) of N-methylmorpholine and then 16.44 g (64 mmol) of trityl chloride are added. The mixture is stirred for 48 hours at room temperature and 200 ml of dichloromethane are added. Washed with 2 times 150 ml of water.
On sèche sur sulfate de magnésium, on filtre et on évapore à sec. On cristallise le résidu obtenu dans l'éther.It is dried over magnesium sulfate, filtered and evaporated to dryness. The residue obtained is crystallized from ether.
On obtient 27 g de produit.27 g of product are obtained.
Rendement = 79 %
Point de fusion = 228 OC
d) 3-amino-4- [4- [5-méthyl-l- (triphénylméthyl) -1H-
imidazol-4-yl]pipéridin-l-yl]benzoate de méthyle
Dans 100 ml de tétrahydrofurane, on ajoute 10 g (17,02 mmoles) de 4- [4- [5-méthyl-1- (triphénylméthyl) -1H- imidazol-4-yl]pipéridin-l-yl] -3-nitrobenzoate de méthyle et une suspension de Nickel de Raney. On agite le mélange sous atmosphère d'hydrogène jusqu'à décoloration de la solution surnageante, on filtre et on évapore le solvant. On triture le résidu dans l'éther glacé et on filtre.Yield = 79%
Melting point = 228 OC
d) 3-amino-4- [4- [5-methyl-1- (triphenylmethyl) -1H-
methyl imidazol-4-yl] piperidin-1-yl] benzoate
10 g (17.02 mmol) of 4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] -3- in 4 g of tetrahydrofuran (10 g, 17.02 mmol) are added. methyl nitrobenzoate and a suspension of Raney nickel. The mixture is stirred under a hydrogen atmosphere until the supernatant solution is decolourized, filtered and the solvent evaporated. The residue is triturated in ice-cold ether and filtered.
On obtient 9,74 g de produit.9.74 g of product are obtained.
Rendement = 95 W
Point de fusion = 260 OC
1.1.2. 3-[(cyclopropylcarbonyl)amino]-4-[4-[5-méthyl-1-
(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-
yl]benzoate de méthyle
On met en suspension dans 255 ml de dichlorométhane 25 g (45 mmoles) de 3-amino-4-[4- [5-méthyl-l- (triphénylméthyl) -1H- imidazol-4-yl]pipéridin-1-yl]benzoate de méthyle et on refroidit le mélange à 0 OC sous azote. On ajoute 3,74 g (47 mmoles) de pyridine puis goutte à goutte 4,94 g (47 mmoles) de chlorure de cyclopropanecarbonyle et on agite le milieu réactionnel pendant 1 heure à cette température. On laisse le mélange revenir à la température ambiante, on ajoute 200 ml de dichlorométhane puis on lave par 2 fois 150 ml d'eau. On sèche sur sulfate de sodium, on filtre et on évapore à sec. On reprend le résidu dans l'éther, on filtre et on sèche.Yield = 95 W
Melting point = 260 OC
1.1.2. 3 - [(cyclopropylcarbonyl) amino] -4- [4- [5-methyl-1-
(Triphenylmethyl) imidazol-4-yl] piperidin-1-
yl] methyl benzoate
25 g (45 mmol) of 3-amino-4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl are suspended in 255 ml of dichloromethane. methyl benzoate and the mixture is cooled to 0 ° C. under nitrogen. 3.74 g (47 mmol) of pyridine are added, then 4.94 g (47 mmol) of cyclopropanecarbonyl chloride are added dropwise and the reaction mixture is stirred for 1 hour at this temperature. The mixture is allowed to return to room temperature, 200 ml of dichloromethane are added and the mixture is washed twice with 150 ml of water. It is dried over sodium sulphate, filtered and evaporated to dryness. The residue is taken up in ether, filtered and dried.
On obtient 25,8 g de produit.25.8 g of product are obtained.
Rendement = 91,9 %
1.1.3. acide 3-[(cyclopropylcarbonyl)amino]-4-[4-[5-
méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl]
pipéridin-l-yl]benzoïque
On met en suspension dans 150 ml de méthanol 13 g (20,8 mmoles) de 3-[(cyclopropylcarbonyl)amino]-4-[4-[5- méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl]pipéridin-l- yl]benzoate de méthyle, on ajoute 62,42 ml d'une solution aqueuse de soude 1 M (62,42 mmoles) et on chauffe à la température de reflux pendant 3,5 heures. On laisse le mélange pendant une nuit à la température ambiante, on ajoute 400 ml d'eau et on évapore le méthanol. On acidifie à pH 4 avec une solution aqueuse d'acide chlorhydrique 3 M, on filtre et on sèche le composé obtenu à 50 OC. Yield = 91.9%
1.1.3. 3 - [(cyclopropylcarbonyl) amino] -4- [4- [5-8
methyl-1- (triphenylmethyl) -1H-imidazol-4-yl]
piperidin-l-yl] benzoic acid
13 g (20.8 mmol) of 3 - [(cyclopropylcarbonyl) amino] -4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl) are suspended in 150 ml of methanol. ] piperidin-1-yl] benzoate methyl, 62.42 ml of a 1M aqueous sodium hydroxide solution (62.42 mmol) and heated at reflux temperature for 3.5 hours. The mixture is left overnight at room temperature, 400 ml of water are added and the methanol is evaporated. It is acidified to pH 4 with a 3 M aqueous hydrochloric acid solution, filtered and the compound obtained is dried at 50 ° C.
On obtient 12,1 g de produit. 12.1 g of product are obtained.
Rendement = 95 8
Point de fusion = 237 OC 1.2. N-[2-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-1-yl]-5- [(4-méthylpipérazin-1-yl)carbonyl]phényl]cyclopropane-
carboxamide
On met en suspension dans 100 ml de dichlorométhane 2,2 g (3,6 mmoles) d'acide 3-[( cyclopropylcarbonyl ) amino]-4- [4- [5- méthyl-l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1- yl]benzoïque, on ajoute 1,4 ml (8 mmoles) de
N,N-diisopropyléthylamine puis on refroidit le mélange à 0 OC. On ajoute alors 0,52 ml (4,6 mmoles) de N-méthylpipérazine en solution dans 30 ml de dichlorométhane, on poursuit l'agitation à cette température pendant 15 minutes puis on ajoute 2,4 g (4,6 mmoles) de benzotriazol-l-yl-oxytripyrrolidinophosphonium hexafluorophosphate. On laisse la température du mélange remonter à la température ambiante pendant une nuit, on ajoute 100 ml de dichlorométhane et on lave la phase organique avec successivement 50 ml d'une solution aqueuse d'hydrogénocarbonate de sodium à 8 %, 100 ml d'une solution aqueuse de chlorure de sodium à 15 % puis 50 ml d'une solution aqueuse de chlorure de sodium à 30 %. On filtre et on sèche sur sulfate de magnésium. ON filtre et on évapore à sec. On reprend le résidu dans 100 ml d'éther et on l'essorez
On obtient 2,1 g de dérivé tritylé que l'on reprend dans 57,6 ml de tétrahydrofurane, on ajoute 60 ml d'eau et 60 ml d'acide acétique puis on chauffe pendant 3 heures à 80 "C. On évapore le milieu réactionnel, on reprend le résidu dans l'éther et on essore. On purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol:ammoniaque (90:12:1).Yield = 95 8
Melting point = 237 OC 1.2. N- [2- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -5 - [(4-methylpiperazin-1-yl) carbonyl] phenyl] cyclopropane
carboxamide
2.2 g (3.6 mmol) of 3 - [(cyclopropylcarbonyl) amino] -4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol acid are suspended in 100 ml of dichloromethane. -4-yl] piperidin-1-yl] benzoic acid, 1.4 ml (8 mmol) of
N, N-diisopropylethylamine and then the mixture is cooled to 0 ° C. 0.52 ml (4.6 mmol) of N-methylpiperazine dissolved in 30 ml of dichloromethane are then added, the stirring is continued at this temperature for 15 minutes and then 2.4 g (4.6 mmol) of benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate. The temperature of the mixture is allowed to rise to ambient temperature overnight, 100 ml of dichloromethane are added and the organic phase is washed successively with 50 ml of an 8% aqueous solution of sodium hydrogencarbonate, 100 ml of 15% aqueous solution of sodium chloride and then 50 ml of a 30% aqueous solution of sodium chloride. Filtered and dried over magnesium sulfate. ON filtered and evaporated to dryness. The residue is taken up in 100 ml of ether and wrung out.
2.1 g of tritylated derivative is obtained, which is taken up in 57.6 ml of tetrahydrofuran, 60 ml of water and 60 ml of acetic acid are added and then the mixture is heated for 3 hours at 80.degree. The reaction medium is taken up in ether and the product is purified by chromatography on a column of silica gel, eluting with a dichloromethane: methanol: ammonia (90: 12: 1) mixture.
On obtient 1,12 g de produit après cristallisation dans l'acétate d'éthyle.1.12 g of product is obtained after crystallization from ethyl acetate.
Rendement = 69 %
Point de fusion = 188 OC
Exemple 2 (composé n" 2) 4-[4-(5-méthyl-lH-imldazol-4-yl)pipéridin-l-yl]-3-[(l- oxobutyl)amino]benzoate de méthyle 2.1. 4-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4-
yl]pipéridin-l-yl]-3-[(1-oxobutyl)amino]benzoate de
méthyle
Sous azote, on met 6,3 g (10,62 mmoles) de 3-amino-4-[4-[5 méthyl-l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1- yl]benzoate de méthyle en suspension dans 55 ml de dichlorométhane et on refroidit le mélange à 0-5 OC. On ajoute alors 0,95 ml (11,74 mmoles) de pyridine et 1,16 ml (11,15 mmoles) de chlorure de butyryle puis on poursuit l'agitation pendant 1 heure à 0-5 OC. On laisse revenir la température du mélange à la température ambiante et on agite pendant une nuit à cette température. On ajoute 90 ml de dichlorométhane, on lave avec 2 fois 80 ml d'eau et on sèche sur sulfate de magnésium. On filtre et on évapore à sec.Yield = 69%
Melting point = 188 OC
Example 2 (Compound No. 2) Methyl 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3 - [(1-oxobutyl) amino] benzoate 2.1. [4- [5-methyl-1- (triphenylmethyl) imidazol-4-
yl] piperidin-1-yl] -3 - [(1-oxobutyl) amino] benzoate
methyl
Under nitrogen, 6.3 g (10.62 mmol) of 3-amino-4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] benzoate are added. of methyl suspension in 55 ml of dichloromethane and the mixture is cooled to 0-5 OC. Then 0.95 ml (11.74 mmol) of pyridine and 1.16 ml (11.15 mmol) of butyryl chloride are added and stirring is continued for 1 hour at 0-5 OC. The temperature of the mixture is allowed to return to room temperature and stirred overnight at this temperature. 90 ml of dichloromethane are added, the mixture is washed with twice 80 ml of water and dried over magnesium sulphate. It is filtered and evaporated to dryness.
On obtient 6,5 g de produit sous forme d'un composé amorphe.6.5 g of product are obtained in the form of an amorphous compound.
Rendement = 98 % 2.2. 4- [4- (5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl] -3- [(I-
oxobutyl)amino]benzoate de méthyle
On solubilise 6 g (10,5 mmoles) de 4-[4-[5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]-3-[(1oxobutyl)amino]benzoate de méthyle dans 170 ml de tétrahydrofurane, on ajoute 170 ml d'eau et 340 ml d'acide acétique puis on chauffe àla température de reflux pendant 5 heures. On évapore le milieu réactionnel, on reprend le résidu dans l'éther et on essore. On purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol:ammoniaque (92:8:0,5).Yield = 98% 2.2. 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3- [(I-
methyl oxobutyl) amino] benzoate
4- [4- [5-Methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] - 3 - [(1oxobutyl) amino] 6 g (10.5 mmol) are solubilized] methyl benzoate in 170 ml of tetrahydrofuran, 170 ml of water and 340 ml of acetic acid are added and the mixture is heated at reflux temperature for 5 hours. The reaction medium is evaporated, the residue is taken up in ether and filtered. The product is purified by chromatography on a column of silica gel, eluting with a dichloromethane: methanol: ammonia mixture (92: 8: 0.5).
On obtient 3,3 g de produit après cristallisation dans l'éther.3.3 g of product are obtained after crystallization from ether.
Rendement = 82 %
Point de fusion = 166-170 OC
Exemple 3 (composé nO 1) acide 4- [4- (5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl] -3- [(1- oxobutyl)amino]benzoïque
On solubilise 2,9 g (7,87 mmoles) de 4-[4-(5-méthyl-1H- imidazol-4-yl)pipéridin-1-yl]-3-[(1-oxobutyl)amino]benzoate de méthyle dans 50 ml de méthanol, on ajoute 0,630 g (15,74 mmoles) de soude et on chauffe le mélange à la température de reflux pendant 2 heures. On évapore le méthanol, on reprend le résidu par 70 ml d'eau et on ajuste le pH à 6,5 avec une solution aqueuse d'acide chlorhydrique 3 N. On filtre et on sèche sous vide à 60 OC. Yield = 82%
Melting point = 166-170 OC
Example 3 (Compound No. 1) 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3 - [(1-oxobutyl) amino] benzoic acid
2.9 g (7.87 mmol) of 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3 - [(1-oxobutyl) amino] benzoate are solubilized. In 50 ml of methanol, 0.630 g (15.74 mmol) of sodium hydroxide are added and the mixture is heated at reflux temperature for 2 hours. The methanol is evaporated, the residue is taken up in 70 ml of water and the pH is adjusted to 6.5 with aqueous 3N hydrochloric acid solution. The mixture is filtered and dried under vacuum at 60 ° C.
On obtient 2,68 g de produit.2.68 g of product are obtained.
Rendement = 92 8
Point de fusion = 291 OC (fusion avec décomposition)
Exemple 4 (composé nO 6)
N-[2-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl]-5- (pipéridin-l-ylcarbonyl)phényl]butanamide
Méthode A A.4.1. acide 4- [4- [5-méthyl-l-(triphénylméthyl)-îH-
imidazol-4-yl]pipéridin-l-yl]-3-[(l-oxobutyl)
amino] benzoique
On met 12 g (19,46 mmoles) de 4-[4-[5-méthyl-l-(triphénylmé- thyl)-lH-imidazol-4-yl]pipéridin-l-yl]-3-[(1-oXobutyl)amino] benzoate de méthyle en suspension dans 120 ml de méthanol, on ajoute 38,93 ml (38,93 mmoles) d'une solution aqueuse de soude 0,1 N et on porte le milieu réactionnel à la température de reflux pendant 2 heures. On évapore le méthanol, on refroidit le mélange à 0-5 OC, on l'acidifie à pH 4 avec une solution aqueuse d'acide chlorhydrique 3 N et on laisse le milieu réactionnel pendant une nuit à cette température. On filtre, on essore et on sèche sur pentoxyde de phosphore sous vide à 70 OC. Yield = 92 8
Melting point = 291 OC (fusion with decomposition)
Example 4 (Compound No. 6)
N- [2- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -5- (piperidin-1-ylcarbonyl) phenyl] butanamide
Method A A.4.1. 4- [4- [5-methyl-1- (triphenylmethyl) -1H-
imidazol-4-yl] piperidin-l-yl] -3 - [(l-oxobutyl)
amino] benzoic
12 g (19.46 mmol) of 4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] -3 - [1- methyl oxo-butyl) amino] benzoate in suspension in 120 ml of methanol, 38.93 ml (38.93 mmol) of a 0.1N aqueous sodium hydroxide solution are added and the reaction medium is brought to the reflux temperature during 2 hours. The methanol is evaporated, the mixture is cooled to 0-5 ° C., acidified to pH 4 with a 3N aqueous hydrochloric acid solution and the reaction medium is left overnight at this temperature. Filtered, filtered and dried over phosphorus pentoxide under vacuum at 70 ° C.
On obtient 10,75 g de produit.10.75 g of product are obtained.
Rendement = 90 %
A.4.2. N-[2-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4- yl]pipéridin-l-yl]-5-(pipéridin-1-ylcarbonyl)phényl]
butanamide
Sous azote on met 1,5 g (2,45 mmoles) d'acide 4-[4-[5-méthyl l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-l-yl]-3-[(l- oxobutyl)amino]benzoïque en suspension dans 10 ml de diméthylformamide, on ajoute 0,44 mg (2,69 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 40 C pendant 2 heures. On ajoute ensuite 0,208 g (2,45 mmoles) de pipéridine et on laisse le mélange pendant une nuit à 80 OC. Yield = 90%
A.4.2. N- [2- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] -5- (piperidin-1-ylcarbonyl) phenyl]
butanamide
Under nitrogen, 1.5 g (2.45 mmol) of 4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] -3- [ (1-oxobutyl) amino] benzoic acid suspended in 10 ml of dimethylformamide, 0.44 mg (2.69 mmol) of 1,4-carbonyldiimidazole are added and the mixture is heated at 40 ° C. for 2 hours. Piperidine (0.208 g, 2.45 mmol) is then added and the mixture is left overnight at 80 ° C.
On évapore le diméthylformamide, on reprend le résidu par 100 ml de dichlorométhane puis on lave par 2 fois 35 ml d'eau. On sèche sur sulfate de sodium, on filtre et on évapore à sec. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (95:5).The dimethylformamide is evaporated, the residue is taken up in 100 ml of dichloromethane and then washed with twice 35 ml of water. It is dried over sodium sulphate, filtered and evaporated to dryness. The residue is purified by chromatography on a column of silica gel, eluting with a dichloromethane: methanol (95: 5) mixture.
On obtient 1,0 g de produit.1.0 g of product is obtained.
Rendement = 60 %
A.4.3. N-[2-[4-(5-méthyl-1H-imidazol-4-yl)pipéridin-1-yl] 5- (pipéridin- l-ylcarbonyl) phényl] butanamide
On solubilise 1 g (1,47 mmoles) de N-[2-[4-[5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]-5- (pipéridin-l-ylcarbonyl)phényl]butanamide dans 25 ml de tétrahydrofurane, on ajoute 25 ml d'eau et 50 ml d'acide acétique puis on chauffe à la température de reflux pendant 2 heures. On évapore le milieu réactionnel et on purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10).Yield = 60%
A.4.3. N- [2- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -5- (piperidin-1-ylcarbonyl) phenyl] butanamide
1 g (1.47 mmol) of N- [2- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] -5- (piperidine) was solubilized. 1-ylcarbonyl) phenyl] butanamide in 25 ml of tetrahydrofuran, 25 ml of water and 50 ml of acetic acid are added and the mixture is heated at reflux temperature for 2 hours. The reaction medium is evaporated and the residue is purified by chromatography on a column of silica gel, eluting with a dichloromethane: methanol (90:10) mixture.
On obtient 0,3 g de produit après cristallisation dans l'éther.0.3 g of product is obtained after crystallization from ether.
Rendement = 54 %
Point de fusion = 139-141,5 OC
Méthode B
Sous azote, on met en suspension 0,7 g (1,89 mmoles) d'acide 4-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-1-yl]-3-[(1- oxobutyl)amino]benzoïque en suspension dans 7 ml de diméthylformamide, on ajoute 0,34 g (2,08 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 60 OC pendant 2 heures. On laisse la température du milieu réactionnel redescendre à 40 OC, on ajoute 0,16 g (1,89 mmoles) de pipéridine et on chauffe le mélange à 85 OC pendant 2 heures. On évapore le diméthylformamide, on reprend le résidu dans l'éther et on le purifie par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10). On rassemble les fractions adéquates, on évapore à sec et on recristallise dans un mélange eau:méthanol (1:1). On filtre et on sèche sous vide sur pentoxyde de phosphore.Yield = 54%
Melting point = 139-141.5 OC
Method B
Under nitrogen, 0.7 g (1.89 mmol) of 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3 - [(1 (oxobutyl) amino] benzoic acid suspended in 7 ml of dimethylformamide, 0.34 g (2.08 mmol) of 1,4-carbonyldiimidazole are added and the mixture is heated at 60 ° C. for 2 hours. The temperature of the reaction medium is allowed to drop to 40 ° C., 0.16 g (1.89 mmol) of piperidine is added and the mixture is heated at 85 ° C. for 2 hours. The dimethylformamide is evaporated, the residue is taken up in ether and purified by chromatography on a column of silica gel, eluting with a dichloromethane: methanol (90:10) mixture. The appropriate fractions are pooled, evaporated to dryness and recrystallized from water / methanol (1: 1). It is filtered and dried under vacuum over phosphorus pentoxide.
On obtient 0,215 g de produit.0.215 g of product is obtained.
Rendement = 26 %
Point de fusion = 139-141,5 OC
Exemple 5 (composé nO 4) 4-[4-(5-methyl-1H- imidazol-4-yl)p ipéridin-l-yl]-3-[( I- oxobutyl) amino] -N- (phénylméthyl)benzamide 5.1. 4-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4-yl]
pipéridin-l-yl]-3-[(1-oxobutyl)amino]-N-(phénylméthyl)
benzamide
Sous azote on met 1,5 g (2,44 mmoles) d'acide 4-[4-[5-méthyl 1- (triphénylméthyl) -lH-imidazol-4-yl]pipérîdin-l-yl] -3- [(1- oxobutyl)amino]benzoïque en suspension dans 9 ml de diméthylformamide, on ajoute 0,44 g (2,69 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 60 OC pendant 2 heures.Yield = 26%
Melting point = 139-141.5 OC
Example 5 (Compound No. 4) 4- [4- (5-methyl-1H-imidazol-4-yl) -piperidin-1-yl] -3 - [(1-oxobutyl) amino] -N- (phenylmethyl) benzamide 5.1. 4- [4- [5-methyl-1- (triphenylmethyl) imidazol-4-yl]
piperidin-l-yl] -3 - [(1-oxobutyl) amino] -N- (phenylmethyl)
benzamide
Under nitrogen, 1.5 g (2.44 mmol) of 4- [4- [5-methyl-1- (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] -3- [ (1-oxobutyl) amino] benzoic acid suspended in 9 ml of dimethylformamide, 0.44 g (2.69 mmol) of 1,4-carbonyldiimidazole are added and the mixture is heated at 60 ° C. for 2 hours.
On laisse la température du milieu réactionnel revenir à 50 OC et on ajoute 0,26 g (2,44 mmoles) de phénylméthylamine puis on chauffe à 80 OC pendant une nuit. On évapore à sec, on reprend le résidu par 100 ml de dichlorométhane et on lave successivement par 40 ml d'une solution aqueuse d'acide acétique 0,1 M puis par 40 ml d'eau. On sèche sur sulfate de magnésium, on filtre et on évapore à sec. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (95:5).The temperature of the reaction medium is allowed to return to 50 ° C. and 0.26 g (2.44 mmol) of phenylmethylamine are added and the mixture is then heated at 80 ° C. overnight. It is evaporated to dryness, the residue is taken up in 100 ml of dichloromethane and washed successively with 40 ml of a 0.1M aqueous acetic acid solution and then with 40 ml of water. It is dried over magnesium sulfate, filtered and evaporated to dryness. The residue is purified by chromatography on a column of silica gel, eluting with a dichloromethane: methanol (95: 5) mixture.
On obtient 1,55 g de produit.1.55 g of product are obtained.
Rendement = 90 % 5.2. 4-[4-(5-méthyl-1H-imidazol-4-yl)pipéridin-1-yl]-3-[(1-
oxobutyl)amino]-N-(phénylméthyl)benzamide
On solubilise 1,55 g (2,21 mmoles) de 4-[4-[5-méthyl-1 (triphénylméthyl)-lH-imidazol-4-yl]pipéridln-l-yl]-3-[(loxobutyl)amino]-N-(phénylméthyl)benzamide dans 35 ml de tétrahydrofurane, on ajoute 35 ml d'eau et 70 ml d'acide acétique puis on chauffe à la température de reflux pendant 3 heures. On évapore le milieu réactionnel et on purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10). On rassemble les fractions, on évapore à sec et on laissse cristalliser dans un mélange méthanol:eau. On filtre et on sèche sous vide sur pentoxyde de phosphore à 60 OC. Yield = 90% 5.2. 4- [4- (5-methyl-1H-imidazol-4-yl) piperidin-1-yl] -3 - [(1-
oxobutyl) amino] -N- (phenylmethyl) benzamide
1.55 g (2.21 mmol) of 4- [4- [5-methyl-1 (triphenylmethyl) -1H-imidazol-4-yl] piperidin-1-yl] -3 - [(loxobutyl) amino is solubilized. ] -N- (phenylmethyl) benzamide in 35 ml of tetrahydrofuran, 35 ml of water and 70 ml of acetic acid are added and the mixture is heated at reflux temperature for 3 hours. The reaction medium is evaporated and the product is purified by chromatography on a column of silica gel, eluting with a dichloromethane: methanol (90:10) mixture. Fractions are pooled, evaporated to dryness and allowed to crystallize from methanol: water. It is filtered and dried under vacuum over phosphorus pentoxide at 60 ° C.
On obtient 0,455 g de produit.0.455 g of product is obtained.
Rendement = 45 %
Point de fusion = 233-235 OC
Légende du tableau
dans la colonne "R2", -c(C3H7) représente un groupe cyclopropyle,
dans la colonne "Sel", "fum" représente un fumarate ; les rapports entre parenthèses représentent le rapport (acide:base) ; l'absence de toute mention signifie que le composé est sous forme de base,
dans la colonne "Point de fusion", (d) coreespond à une fusion avec décomposition. Yield = 45%
Melting point = 233-235 OC
Legend of the table
in the "R2" column, -c (C3H7) represents a cyclopropyl group,
in the "Salt" column, "fum" represents a fumarate; the ratios in parentheses represent the ratio (acid: base); the absence of any mention means that the compound is in base form,
in the "Melting Point" column, (d) coresponds to a decomposition merger.
Tableau


<tb> <SEP> Point <SEP> de
<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Sel
<tb> <SEP> fusion <SEP> (0C)
<tb> 1 <SEP> -CH3 <SEP> -C3H7 <SEP> -OH <SEP> - <SEP> 291 <SEP> (d)
<tb> 2 <SEP> -CH3 <SEP> -C3H7 <SEP> -OCH3 <SEP> - <SEP> 166-170
<tb> 3 <SEP> -CH3 <SEP> -C3H7 <SEP> -NHCH3 <SEP> 138-144
<tb> <SEP> \NH
<tb> 4 <SEP> -CH, <SEP> -C,H, <SEP> 233-235
<tb> 6 <SEP> -CH3 <SEP> -C3H7 <SEP> - <SEP> NS <SEP> - <SEP> 139-141,5
<tb> 7 <SEP> /m <SEP> - <SEP> 123-129
<tb> 7 <SEP> -CH3 <SEP> -C3H7 <SEP> -N <SEP> N-CH3
<tb> <SEP> \ff
<tb> 8 <SEP> -CH3 <SEP> -CH2CH(CH3)2 <SEP> - <SEP> N3 <SEP> - <SEP> 177
<tb> 9 <SEP> -CH3 <SEP> ; <SEP> -CH2CH(CH3) <SEP> 2 <SEP> -N <SEP> N <SEP> - <SEP> - <SEP> 120 <SEP> (d)
<tb> <SEP> M
<tb>

<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Salt
<tb><SEP> merge <SEP> (0C)
<tb> 1 <SEP> -CH3 <SEP> -C3H7 <SEP> -OH <SEP> - <SEP> 291 <SEP> (d)
<tb> 2 <SEP> -CH3 <SEP> -C3H7 <SEP> -OCH3 <SEP> - <SEP> 166-170
<tb> 3 <SEP> -CH3 <SEP> -C3H7 <SEP> -NHCH3 <SEP> 138-144
<tb><SEP> \ NH
<tb> 4 <SEP> -CH, <SEP> -C, H, <SEP> 233-235
<tb> 6 <SEP> -CH3 <SEP> -C3H7 <SEP> - <SE> NS <SEP> - <SEP> 139-141.5
<tb> 7 <SEP> / m <SEP> - <SEP> 123-129
<tb> 7 <SEP> -CH3 <SEP> -C3H7 <SEP> -N <SEP> N-CH3
<tb><SEP> \ ff
<tb> 8 <SEP> -CH3 <SEP> -CH2CH (CH3) 2 <SEP> - <SEP> N3 <SEP> - <SEP> 177
<tb> 9 <SEP> -CH3 <SEP>;<SEP> -CH2CH (CH3) <SEP> 2 <SEP> -N <SEP> N <SEP> - <SEP> - <SEP> 120 <SEP> (d)
<tb><SEP> M
<Tb>
<tb> <SEP> Point <SEP> de
<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Sel
<tb> <SEP> fusion <SEP> (0C)
<tb> 10 <SEP> -CH3 <SEP> -c <SEP> (C3H7) <SEP> -É <SEP> - <SEP> 240 <SEP> (d)
<tb> 11 <SEP> -CH3 <SEP> -c(C3H7) <SEP> -N <SEP> N-cH <SEP> - <SEP> 188
<tb> <SEP> M
<tb> <SEP> fum
<tb> 12 <SEP> -CH3 <SEP> -c(C3H7) <SEP> N <SEP> Ne <SEP> < 1:1) <SEP> 130 <SEP> (d)
<tb> 13 <SEP> -CH3 <SEP> -c(C3H7) <SEP> N <SEP> N43 <SEP> ~ <SEP> 233-237
<tb> 14 <SEP> -CH3 <SEP> -c(C3H7) <SEP> -NHCH(NH)NH2 <SEP> - <SEP> 236-241,5
<tb>
Les composés de l'invention ont fait l'objet d'études pharmacologiques qui ont mis en évidence leurs propriétés inhibitrices de l'échangeur sodium/proton et leur intérêt comme substances à activité thérapeutique.<tb><SEP><SEP> Point of
<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Salt
<tb><SEP> merge <SEP> (0C)
<tb> 10 <SEP> -CH3 <SEP> -c <SEP> (C3H7) <SEP> -E <SEP> - <SEP> 240 <SEP> (d)
<tb> 11 <SEP> -CH3 <SEP> -c (C3H7) <SEP> -N <SEP> N-cH <SEP> - <SEP> 188
<tb><SEP> M
<tb><SEP> smoked
<tb> 12 <SEP> -CH3 <SEP> -c (C3H7) <SEP> N <SEP> Ne <SEP><1: 1) <SEP> 130 <SEP> (d)
<tb> 13 <SEP> -CH3 <SEP> -c (C3H7) <SEP> N <SEP> N43 <SEP> ~ <SEP> 233-237
<tb> 14 <SEP> -CH3 <SEP> -c (C3H7) <SEP> -NHCH (NH) NH2 <SEP> - <SEP> 236-241.5
<Tb>
The compounds of the invention have been the subject of pharmacological studies which have demonstrated their inhibitory properties of the sodium / proton exchanger and their interest as substances with therapeutic activity.
Ainsi, les composés de l'invention ont été soumis à un test d'inhibition du gonflement des plaquettes sanguines de lapin en milieu acide selon la méthode de Grinstein et al. (In
Methods in Enzymology, Fleisher S. And Flusher B., Vol 173, pp 777-790, Academic Press Inc., 1984).Thus, the compounds of the invention were subjected to a rabbit blood platelet swelling inhibition test in an acid medium according to the method of Grinstein et al. (In
Methods in Enzymology, Fleisher S. And Flusher B., Vol 173, pp 777-790, Academic Press Inc., 1984).
On prélève par ponction cardiaque du sang sur des lapins
Néo-Zélandais, en utilisant un anticoagulant citratedextrose. On obtient le plasma riche en plaquettes (PRP) par centrifugation à 1200 rpm pendant 20 minutes à la température ambiante. Après mesure du volume moyen plaquettaire initial on incube une fraction aliquote de PRP pendant 20 minutes dans un milieu propionate de sodium/acide propionique (140 mM) contenant du chlorure de potassium (1 mM), du chlorure de magnésium (1 mM), du glucose (10 mM) le tout tamponné par de l'Hepes (20 mM) à pH 6,7 et dont l'osmolarité est d'environ 300 mosm/l. L'acide propionique diffuse dans les plaquettes où il se dissocie, provoquant une acidification intra-cellulaire et une activation de l'antiport sodium/proton. L'influx d'ions sodium s'accompagne d'une capture d'eau qui provoque le gonflement des plaquettes. La mesure du volume moyen plaquettaire à la fin de l'incubation, diminuée du volume moyen plaquettaire initial, permet d'estimer le gonflement maximal des plaquettes. On ajoute les produits à tester au milieu d'incubation d' acide propionique aux concentrations voulues, avant l'addition de PRP. Les résultats sont exprimés en pourcentage d'inhibition du gonflement maximum permettant de calculer la CIso ou concentration inhibant de 50% le gonflement maximum.Blood is collected from rabbits by cardiac puncture
New Zealanders, using a citratedextrose anticoagulant. The platelet rich plasma (PRP) is obtained by centrifugation at 1200 rpm for 20 minutes at room temperature. After measuring the initial average platelet volume, an aliquot of PRP was incubated for 20 minutes in a propionate medium of sodium / propionic acid (140 mM) containing potassium chloride (1 mM), magnesium chloride (1 mM), glucose (10 mM) all buffered with Hepes (20 mM) at pH 6.7 and whose osmolarity is about 300 mosm / l. Propionic acid diffuses into the platelets where it dissociates, causing intracellular acidification and activation of the sodium / proton antiport. The influx of sodium ions is accompanied by a capture of water which causes the swelling of the platelets. The measurement of the mean platelet volume at the end of the incubation, minus the initial platelet mean volume, makes it possible to estimate the maximum swelling of the platelets. The test products are added to the propionic acid incubation medium at the desired concentrations prior to the addition of PRP. The results are expressed as a percentage of inhibition of maximum swelling for calculating the IC 50 or inhibiting concentration of 50% maximum swelling.
Dans ce test, les CIso des composés les plus intéressants de l'invention sont inférieures à 10 pM. In this test, the ICos of the most interesting compounds of the invention are less than 10 μM.
A ce titre ils peuvent être utilisés dans le traitement et la prévention de différentes formes de pathologies telles que l'hypertension artérielle et pulmonaire, l'arythmie cardiaque, l'ischémie cardiaque, l'infarctus cardiaque, l'insuffisance cardiaque et l'angine de poitrine, les ischémies des organes périphériques, des membres inférieurs et du système nerveux central, les néphropathies, les oedèmes, les fibroses et les cancers, ainsi que les maladies caractérisées par des hyperplasies et hypertrophies des vaisseaux ou du coeur.As such, they can be used in the treatment and prevention of various forms of pathologies such as arterial and pulmonary hypertension, cardiac arrhythmia, cardiac ischemia, cardiac infarction, heart failure and angina. chest, ischemia of peripheral organs, lower limbs and central nervous system, nephropathies, edema, fibrosis and cancers, as well as diseases characterized by hyperplasias and hypertrophies of vessels or heart.
Ils peuvent aussi être utilisés pour la protection des organes dans les opérations de chirurgie ou de transplantation d'organe.They can also be used for the protection of organs in surgery or organ transplantation operations.
Les composés de l'invention peuvent être utilisés seuls ou en association avec d'autres substances telles que les nitrates, les antagonistes du calcium, les bêta-bloquants, les antithrombotiques, les thrombolytiques, les salicylates.The compounds of the invention may be used alone or in combination with other substances such as nitrates, calcium antagonists, beta-blockers, antithrombotics, thrombolytics, salicylates.
A cet effet ils peuvent être présentés sous toutes formes appropriées à l'administration orale ou parentérale, telles que comprimés, dragées, gélules, capsules, suspensions ou solutions buvables ou injectables, etc. en association avec des excipients convenables. Ces formes sont dosées pour permettre une administration de 1 à 1000 mg/kg de 1 à 4 fois par jour.For this purpose they may be presented in any form suitable for oral or parenteral administration, such as tablets, dragees, capsules, capsules, suspensions or oral or injectable solutions, etc. in combination with suitable excipients. These forms are dosed to allow administration of 1 to 1000 mg / kg of 1 to 4 times per day.
Ils peuvent également être présentés sous toutes formes appropriées à l'administration transdermique ou sublinguale. They may also be presented in any form suitable for transdermal or sublingual administration.
Claims (6)












Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9707900A FR2765221B1 (en) | 1997-06-25 | 1997-06-25 | DERIVATIVES OF 4 - [(1H-IMIDAZOL-4-YL) PIPERIDIN-1-YL] ANILIDE, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION |
| AU82206/98A AU8220698A (en) | 1997-06-25 | 1998-06-19 | 4-{(1h-imidazol-4-yl)piperidin-1-yl}anilide derivatives, their preparation and application in therapy |
| EP98932237A EP0991639A1 (en) | 1997-06-25 | 1998-06-19 | 4- (1h-imidazol-4-yl)piperidin-1-yl]anilide derivatives, their preparation and application in therapy |
| PCT/FR1998/001288 WO1999000379A1 (en) | 1997-06-25 | 1998-06-19 | 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide derivatives, their preparation and application in therapy |
| JP50531399A JP2002506458A (en) | 1997-06-25 | 1998-06-19 | 4-[(1H-Imidazol-4-yl) piperidin-1-yl] anilide Derivatives, Their Preparation and Their Application in Therapy |
| ARP980103032A AR013127A1 (en) | 1997-06-25 | 1998-06-24 | DERIVATIVES OF 4 - [(1H-IMIDAZOL-4-IL) PIPERIDIN-1-IL] ANILIDE, ITS PREPARATION AND ITS APPLICATION IN THERAPEUTICS |
| ZA985518A ZA985518B (en) | 1997-06-25 | 1998-06-24 | 4-[(1H-imidazol-4-YL) piperidin-1-YL] anilide derivatives their preparation and their application in therapeutics |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9707900A FR2765221B1 (en) | 1997-06-25 | 1997-06-25 | DERIVATIVES OF 4 - [(1H-IMIDAZOL-4-YL) PIPERIDIN-1-YL] ANILIDE, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2765221A1 true FR2765221A1 (en) | 1998-12-31 |
| FR2765221B1 FR2765221B1 (en) | 1999-07-30 |
Family
ID=9508386
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9707900A Expired - Lifetime FR2765221B1 (en) | 1997-06-25 | 1997-06-25 | DERIVATIVES OF 4 - [(1H-IMIDAZOL-4-YL) PIPERIDIN-1-YL] ANILIDE, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP0991639A1 (en) |
| JP (1) | JP2002506458A (en) |
| AR (1) | AR013127A1 (en) |
| AU (1) | AU8220698A (en) |
| FR (1) | FR2765221B1 (en) |
| WO (1) | WO1999000379A1 (en) |
| ZA (1) | ZA985518B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001027107A2 (en) * | 1999-10-12 | 2001-04-19 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7501889B2 (en) | 2004-04-26 | 2009-03-10 | Rgb Systems, Inc. | Method and apparatus for implementing soft switching in a class D amplifier |
| JP6162694B2 (en) | 2011-07-18 | 2017-07-12 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | Benzamides |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0197840A1 (en) * | 1985-03-26 | 1986-10-15 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | (Imidazolyl-4) piperidines, their preparation and their therapeutical use |
| EP0494010A1 (en) * | 1990-12-31 | 1992-07-08 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | 1-Substituted 4-(4-imidazolyl) piperidines, process for their preparation and their therapeutic applications |
| EP0507650A1 (en) * | 1991-04-03 | 1992-10-07 | Synthelabo | Piperidine derivatives, their preparation and their therapeutic application |
| EP0591027A1 (en) * | 1992-09-28 | 1994-04-06 | Synthelabo | Piperidine derivatives, their preparation and their use as medicine |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4325822A1 (en) * | 1993-07-31 | 1995-02-02 | Hoechst Ag | Substituted benzoylguanidines, process for their preparation, their use as medicament or diagnostic agent, and medicament containing them |
-
1997
- 1997-06-25 FR FR9707900A patent/FR2765221B1/en not_active Expired - Lifetime
-
1998
- 1998-06-19 EP EP98932237A patent/EP0991639A1/en not_active Withdrawn
- 1998-06-19 JP JP50531399A patent/JP2002506458A/en active Pending
- 1998-06-19 AU AU82206/98A patent/AU8220698A/en not_active Abandoned
- 1998-06-19 WO PCT/FR1998/001288 patent/WO1999000379A1/en not_active Application Discontinuation
- 1998-06-24 AR ARP980103032A patent/AR013127A1/en unknown
- 1998-06-24 ZA ZA985518A patent/ZA985518B/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0197840A1 (en) * | 1985-03-26 | 1986-10-15 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | (Imidazolyl-4) piperidines, their preparation and their therapeutical use |
| EP0494010A1 (en) * | 1990-12-31 | 1992-07-08 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | 1-Substituted 4-(4-imidazolyl) piperidines, process for their preparation and their therapeutic applications |
| EP0507650A1 (en) * | 1991-04-03 | 1992-10-07 | Synthelabo | Piperidine derivatives, their preparation and their therapeutic application |
| EP0591027A1 (en) * | 1992-09-28 | 1994-04-06 | Synthelabo | Piperidine derivatives, their preparation and their use as medicine |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001027107A2 (en) * | 1999-10-12 | 2001-04-19 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
| WO2001027107A3 (en) * | 1999-10-12 | 2002-01-24 | Bristol Myers Squibb Co | Heterocyclic sodium/proton exchange inhibitors and method |
| US6887870B1 (en) | 1999-10-12 | 2005-05-03 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
| US7326705B2 (en) | 1999-10-12 | 2008-02-05 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
Also Published As
| Publication number | Publication date |
|---|---|
| WO1999000379A1 (en) | 1999-01-07 |
| EP0991639A1 (en) | 2000-04-12 |
| AU8220698A (en) | 1999-01-19 |
| ZA985518B (en) | 1999-01-28 |
| FR2765221B1 (en) | 1999-07-30 |
| AR013127A1 (en) | 2000-12-13 |
| JP2002506458A (en) | 2002-02-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1499589B1 (en) | Derivatives of n-phenyl(piperidin-2-yl)methyl benzamide, the preparation method thereof and application of same in therapeutics | |
| WO2005037781A2 (en) | Derivatives of n-[heteroaryl(piperidine-2-yl)methyl]benzamide, preparation method thereof and application of same in therapeutics | |
| FR2673427A1 (en) | HETEROCYCLIC DIAZOTES N - SUBSTITUTED BY BIPHENYLMETHYL GROUP, PREPARATION THEREOF, PHARMACEUTICAL COMPOSITIONS CONTAINING SAME. | |
| CZ281763B6 (en) | Indole derivatives representing 5-ht1 agonists, pharmaceutical compositions containing thereof, their use and intermediates for their preparation | |
| IL172529A (en) | Substituted diketopiperazines, process for their preparation and their use in the preparation of medicaments for antagonizing the effects of oxytocin on the oxytocin receptor | |
| EP0306375A1 (en) | 2-[(4-Piperidinyl)methyl]-1,2,3,4-tetrahydroisoquinoline derivatives, their preparation and their use in therapy | |
| FR2595352A1 (en) | INDOLE DERIVATIVES USEFUL AS MEDICAMENTS AND METHOD AND INTERMEDIATES FOR THEIR PREPARATION | |
| CA1095047A (en) | Process for the preparation of novel derivatives of 1, 4-benzodiaxane | |
| FR2696177A1 (en) | Piperidine derivatives, their preparation and their therapeutic application. | |
| FR2765221A1 (en) | 4 - [(1H-IMIDAZOL-4-YL) PIPERIDIN-1-YL] ANILIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE | |
| CA2489723C (en) | Novel aryl-{4-halo-4-[heteroaryl-methylamino)-methyl]-piperidin-1-yl}-methanone derivatives, methods for production and use thereof as medicaments | |
| FR2758329A1 (en) | New imidazole-4-butane-boronic acid derivatives | |
| EP2917204B1 (en) | Derivatives of 1h-indole-3-carboxamide and their use as p2y12 antagonists | |
| CA2258152C (en) | Novel aromatic derivatives substituted by a ribose, their method of preparation and application as medicine | |
| FR2687146A1 (en) | NOVEL PYRROLIDINE DERIVATIVES, PROCESS FOR PREPARING THEM AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. | |
| CA2166032A1 (en) | 1-oxo-2-(phenylsulfonylamino)pentylpiperidine; process for preparing them and their use as therapeutic agents | |
| FR2765580A1 (en) | (1H-IMIDAZOL-4-YL) PIPERIDINE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION | |
| FR2601951A1 (en) | NOVEL TETRAHYDROCARBAZOLONES, PHARMACEUTICAL COMPOSITION CONTAINING SAME AND PROCESS FOR PREPARING THE SAME | |
| EP0000306A1 (en) | Substituted hexahydrobenzopyrano(3,2-c)pyridines, process for their preparation and medicines containing them | |
| CA2017044A1 (en) | Pyrrolidone derivatives, process for their preparation and pharmaceutical composition containing same | |
| FR2816619A1 (en) | New tricyclic 2-(cyclic amino)-benzimidazole derivatives, are poly-(ADP-ribose) polymerase inhibitors useful e.g. for treating cardiovascular, neurodegenerative, inflammatory, immunological or tumor diseases | |
| WO1998022443A1 (en) | N-(imidazolylbutyl) benzenesulphonamide derivatives, their preparation and therapeutic application | |
| WO2000051973A1 (en) | Cyclobutene-3,4-dione derivatives, preparation method and therapeutic use | |
| EP0200583A1 (en) | 7-Hydroxy indole derivatives, process for their preparation, use as medicine, compounds containing them and their intermediates | |
| FR2761067A1 (en) | 4:substituted quinolin:2:one derivatives used as serotonin antagonists |