ES2790586T3 - Nucleótidos modificados - Google Patents
Nucleótidos modificados Download PDFInfo
- Publication number
- ES2790586T3 ES2790586T3 ES19164666T ES19164666T ES2790586T3 ES 2790586 T3 ES2790586 T3 ES 2790586T3 ES 19164666 T ES19164666 T ES 19164666T ES 19164666 T ES19164666 T ES 19164666T ES 2790586 T3 ES2790586 T3 ES 2790586T3
- Authority
- ES
- Spain
- Prior art keywords
- nucleotide
- mmol
- nucleotides
- group
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 CC(NC(C(C#CCNC(C(F)(F)F)=O)=CN1C(C2)OC(C*)C2OC*)=NC1=O)=O Chemical compound CC(NC(C(C#CCNC(C(F)(F)F)=O)=CN1C(C2)OC(C*)C2OC*)=NC1=O)=O 0.000 description 4
Abstract
Un kit que comprende cuatro moléculas de nucleótido trifosfato modificadas, cada una de las cuales comprende una base de purina o pirimidina y una unidad estructural de azúcar desoxirribosa en el que el átomo de carbono 3' de la unidad estructural de azúcar ha unido un grupo de la estructura -O-Z en la que Z es de la fórmula -CH2N3.
Description
DESCRIPCIÓN
Nucleótidos modificados
La invención se relaciona con nucleótidos modificados. En particular, está invención divulga nucleótidos que tienen un grupo protector removible, su uso en métodos de secuenciación de polinucleótidos y un método para la desprotección química del grupo protector.
Los avances en el estudio de las moléculas han sido guiados, en parte, por la mejora en tecnologías utilizadas para caracterizar las moléculas o sus reacciones biológicas. En particular, el estudio de los ácidos nucleicos ADN y ARN se ha beneficiado de las técnicas en desarrollo utilizadas para el análisis de secuencias y el estudio de eventos de hibridación.
Un ejemplo de las tecnologías que han mejorado el estudio de los ácidos nucleicos es el desarrollo de disposiciones fabricadas de ácidos nucleicos inmovilizados. Estas disposiciones consisten típicamente de una matriz de alta densidad de polinucleótidos inmovilizados sobre un material de soporte sólido. Véase, por ejemplo, Fodor et al., Trends Biotech. 12:19-26, 1994, la cual describe maneras de ensamblar los ácidos nucleicos utilizando una superficie de vidrio sensibilizada químicamente protegida por una máscara, pero expuesta en áreas definidas para permitir la unión de fosforamiditas de nucleótidos modificadas adecuadamente. Las disposiciones fabricadas también pueden ser manufacturadas por la técnica de “manchar” polinucleótidos conocidos sobre un soporte sólido en posiciones predeterminadas (por ejemplo, Stimpson et al., Proc. Natl. Acad. Sci. USA 92: 6379-6383, 1995). La secuenciación por síntesis de ADN requiere idealmente la incorporación controlada (esto es, uno a la vez) del nucleótido complementario correcto opuesto al oligonucleótido que está siendo secuenciado. Esto permite una secuenciación precisa agregando nucleótidos en ciclos múltiples a medida que cada residuo de nucleótido es secuenciado uno a la vez, evitando así la ocurrencia de una serie no controlada de incorporaciones. El nucleótido incorporado es leído utilizando un marcador apropiado unido al mismo antes de la eliminación de la unidad estructural marcada y la subsecuente ronda siguiente de secuenciación. Con el fin de asegurar que ocurra solamente una incorporación individual, se requiere una modificación estructural (“grupo de bloqueo”) de los nucleótidos en secuenciación para asegurar una incorporación individual de nucleótido pero que evite cualquier incorporación de nucleótidos adicional en la cadena de polinucleótidos. El grupo bloqueador debe ser removible, bajo condiciones de reacción que no interfieran con la integridad del ADN que está siendo secuenciado. El ciclo de secuenciación puede continuar entonces con la incorporación del siguiente nucleótidos bloqueado, marcado. Con el fin de ser de uso práctico, el proceso completo debería consistir de etapas químicas y enzimáticas altamente específicas, de alto rendimiento para facilitar múltiples ciclos de secuenciación.
Para ser útiles en la secuenciación de ADN, el nucleótido, y más usualmente los trifosfatos de nucleótido, requieren generalmente un grupo bloqueador de 3’OH de tal manera que evite que la polimerasa utilizada para incorporarlo en una cadena de polinucleótidos continúe replicando una vez que la base sobre el nucleótido es agregada. Hay muchas limitaciones sobre la adecuabilidad de una molécula como grupo bloqueador. Debe ser tal que evite que moléculas adicionales de nucleótido se agreguen a la cadena de polinucleótidos mientras que simultáneamente sea fácil de retirar de la unidad estructural azúcar sin producir un daño a la cadena de polinucleótidos. Adicionalmente, el nucleótido modificado debe ser tolerado por la polimerasa u otras enzimas apropiadas utilizadas para incorporarlo en la cadena de polinucleótidos. El grupo bloqueador ideal por lo tanto exhibirá una estabilidad a largo plazo, será incorporado eficientemente por la enzima polimerasa, producirá un bloqueo total de incorporaciones secundarias o adicionales y tendrá la capacidad de ser retirado bajo condiciones moderadas que no produzcan daño a la estructura del polinucleótido, preferiblemente bajo condiciones acuosas. Estos requerimientos restrictivos son obstáculos formidables para el diseño y síntesis de los nucleótidos modificados requeridos.
Grupos bloqueadores reversibles para este propósito han sido descritos previamente pero ninguno de ellos satisface en general los criterios anteriores para la química de polinucleótidos, por ejemplo, que sean compatibles con ADN. Metzker et al., (Nucleic Acids Research, 22 (20): 4259-4267, 1994) divulga la síntesis y uso de ocho 5’-trifosfatos de 2-desoxirribonucleósido modificados en 3’ (dNTPs modificados en 3’) y su prueba en dos ensayos de plantilla de ADN para la actividad de incorporación. Los dNTPs modificados en 3’ incluyeron 3’alil desoxirriboadenosina 5’-trifosfato (3’-alil de ATP). Sin embargo, el compuesto bloqueado por 3’alilo no fue utilizado para demostrar un ciclo completo de terminación, desprotección y reiniciación de la síntesis de ADN: los únicos resultados de la prueba presentados fueron aquellos que mostraron la capacidad de este compuesto para terminar la síntesis de ADN en un ensayo de terminación sencillo, y de los ocho tales ensayos llevados a cabo, cada uno fue llevado a cabo con una ADN polimerasa diferente.
La WO 02/29003 (The Trustees of Columbia University en la ciudad de Nueva York) describe un método de secuenciación el cual, puede incluir el uso de un grupo protector alilo para tapar el grupo 3’-OH en una cadena creciente de ADN en una reacción con polimerasa. El grupo alilo es introducido de acuerdo con el procedimiento de Metzker (infra) y se dice que es retirado utilizando la metodología reportada por Kamal et al (Tet. Let, 40, 371-372, 1999).
La metodología de desprotección de Kamal emplea yoduro de sodio y clorotrimetilsilano de tal manera que se
genere in situ el yodotrimetilsilano, en acetonitrilo como solvente, deteniéndola con tiosulfato de sodio. Después de la extracción en acetato de etilo y secado (sulfato de sodio), la concentración posterior bajo presión reducida y cromatografía de columna (acetato de etilo: hexano; 2:3 como eluyente), se obtuvieron los alcoholes libres en un rendimiento de 90-98%.
En la WO 02/29003, se sugiere la desprotección de alilos según Kamal como directamente aplicable en el secuencialmente de ADN sin modificación, siendo las condiciones de Kamal moderadas y específicas.
Mientras que Metzker reporta la preparación de un nucleótido o nucleósido bloqueado con 3’alilo y la WO 02/29003 sugiere el uso de la funcionalidad alilo como una tapa para 3’-OH durante la secuenciación, ninguno de estos documentos enseña realmente la desprotección de un grupo hidroxilo con 3’-alilo en el contexto de un protocolo de secuenciación. Mientras que el uso de un grupo alilo como grupo protector de hidroxilo es bien conocido - es fácil de introducir y es estable a lo largo del todo el rango de pH y a temperaturas elevadas - no hay hasta la fecha una realización concreta de una escisión exitosa de un grupo 3’-alilo bajo condiciones compatibles con el ADN, esto es, condiciones bajo las cuales la integridad del ADN no es destruida total o parcialmente. En otras palabras, no ha sido posible hasta el momento llevar a cabo una secuenciación de ADN utilizando nucleótidos bloqueados con alilo en 3’OH.
La metodología de Kamal es inapropiada para ser llevada a cabo en medio acuoso puesto que el cloruro de TMS hidrolizará evitando la generación in situ de yoduro de TMS. Los intentos para llevar a cabo la desprotección de Kamal (en acetonitrilo) en secuenciación se han mostrado no exitosos en nuestras manos.
La presente invención está basada en el desarrollo sorprendente de un cierto número de grupos bloqueadores reversibles y métodos para desprotegerlos bajo condiciones compatibles con al ADN. Algunos de estos grupos bloqueadores son novedosos per se; otros han sido divulgados en la técnica anterior pero, como se anotó más arriba, no ha sido demostrada la posibilidad de utilizar estos grupos bloqueadores en la secuenciación de ADN. El documento WO91/06678 divulga métodos de secuenciación de ADN.
Nucleósidos, Nucleótidos y Ácidos Nucleicos., Vol. 19, no 10-12, páginas 1977-1991 divulga la síntesis de derivados de ribonucleósidos 2'- y 3'-O-azidometilo.
Previamente se han utilizado grupos protectores que comprenden la funcionalidad acetal como grupos bloqueadores. Sin embargo, la eliminación de tales grupos y éteres requiere desprotecciones fuertemente ácidas que son nocivas para las moléculas de ADN. Sin embargo la hidrólisis de un acetal, da como resultado la formación de un intermediario hemiacetal inestable el cual se hidroliza bajo condiciones acuosas al grupo hidroxilo natural. Los inventores han utilizado este concepto y lo han aplicado posteriormente de tal manera que esta característica de la invención reside en la utilización de grupos bloqueadores que incluyen grupos protectores para proteger moléculas intermediarias que normalmente hidrolizarían bajo condiciones acuosas. Estos grupos protectores comprenden un segundo grupo funcional que estabiliza la estructura del intermediario pero que puede ser retirado en una etapa posterior después de la incorporación en el polinucleótido. Se han utilizado grupos protectores en reacciones de síntesis orgánica para enmascarar temporalmente la química característica de un grupo funcional puesto que interfiere con otra reacción.
La invención está definida por las reivindicaciones de acuerdo con un primer aspecto en donde se provee una molécula de nucleótido o nucleósido modificada que comprende una base purina o pirimidina y una unidad estructural azúcar ribosa o desoxirribosa que tiene un grupo bloqueador 3’-OH removible unido covalentemente a la misma, de tal manera que el átomo de carbono de 3’ tiene un grupo unido de la estructura
-O-Z
en donde Z es cualquiera de -C(R')2-O-R", -C(R')2-N(R")2,-C(R')2-N(H)R", -C(R')2-S-R" y C(R')2-F,
en donde cada R" es o es parte de un grupo protector removible;
cada R' es independientemente un átomo de hidrógeno, un grupo alquilo, alquilo sustituido , arilalquilo, alquenilo, alquinilo, arilo, heteroarilo, heterocíclico, acilo, ciano, alcoxi, ariloxi, heteroariloxi o amido, o un marcador detectable unido a través de un grupo enlazante; o (R')2 representa un grupo alquilideno de fórmula =C(R"')2 en donde cada R"' puede ser el mismo o diferente y es seleccionado del grupo que comprende átomos de hidrógeno y halógeno y grupos alquilo; y
en donde dicha molécula puede hacerse reaccionar para producir un intermediario en el cual cada R" es intercambiado por H o, donde Z es -C(R')2-F, el F es intercambiado por OH, SH o NH2, preferiblemente OH, el cual de inmediato se disocia bajo condiciones acuosas para generar una molécula con un 3'OH libre;
con la condición de que donde Z es -C(R')2-S-R", ambos grupos R' no son H.
En un aspecto adicional la divulgación provee un método para controlar la incorporación de una molécula de nucleótido complementaria al nucleótido en un polinucleótido de cadena sencilla objetivo en una reacción de síntesis
o secuenciación que comprende incorporar en el polinucleótido complementario creciente una molécula de acuerdo con la divulgación, de forma que la incorporación de dicha molécula evita o bloquea la introducción de moléculas de nucleósido o nucleótido subsecuentes en dicho polinucleótido complementario creciente.
En un aspecto adicional, la divulgación provee un método para determinar la secuencia de un polinucleótido de cadena sencilla objetivo, que comprende monitorizar la incorporación secuencial de nucleótidos complementarios, en donde al menos una incorporación, y preferiblemente todas las incorporaciones son de un nucleótido de acuerdo con la invención como se describió aquí anteriormente la cual comprende preferiblemente un marcador detectable enlazado a la base del nucleósido o nucleótido mediante un enlazante escindible y en donde la identidad del nucleótido incorporado es determinada detectando el marcador, siendo removidos dicho grupo bloqueador y dicho marcador antes de la introducción del siguiente nucleótido complementario.
Desde un aspecto adicional, la divulgación provee un método para determinar la secuencia de un polinucleótido de cadena sencilla objetivo, que comprende:
(a) proveer una pluralidad de diferentes nucleótidos de acuerdo con la divulgación aquí antes descrita, nucleótidos que preferiblemente son enlazados desde la base a un marcador detectable mediante un enlazante escindible y en donde el marcador detectable enlazado a cada tipo de nucleótido puede ser distinguido por detección del marcador detectable utilizado para otros tipos de nucleótidos;
(b) incorporar el nucleótido en el complemento del polinucleótido de cadena sencilla objetivo;
(c) detectar el marcador de nucleótido de (b), determinando por lo tanto el tipo de nucleótido incorporado;
(d) eliminar el marcador de nucleótido de (b) y el grupo bloqueador; y
(e) repetir opcionalmente las etapas (b)-(d) una o más veces;
determinando por lo tanto la secuencia de un polinucleótido de cadena sencilla objetivo.
Adicionalmente, en otro aspecto, la divulgación provee un kit, que comprende:
(a) una pluralidad de nucleótidos individuales diferentes de la invención; y
(b) materiales de empaque para los mismos.
Los nucleósidos o nucleótidos de acuerdo con o usados en los métodos de la presente divulgación comprenden una base purina o pirimidina y una unidad estructural de azúcar ribosa o desoxirribosa la cual tiene un grupo bloqueador enlazado covalentemente a la misma, preferiblemente en la posición 3’O, la cual hace que las moléculas resulten útiles en técnicas que requieren el bloqueo del grupo 3’-OH para evitar la incorporación de nucleótidos adicionales, tales como por ejemplo en reacciones de secuenciación, síntesis de polinucleótidos, amplificación de ácidos nucleicos, ensayos de hibridación de ácidos nucleicos, estudios de polimorfismo de nucleótidos individuales, y otras de tales técnicas.
Cuando se utiliza aquí el término “grupo bloqueador” en el contexto de la invención, este abarca tanto los grupos bloqueadores alilo y “Z” descritos aquí. Sin embargo, será evidente que, en los métodos tal como se describen y reivindican aquí, donde se utilizan mezclas de nucleótidos, estas comprenden muy preferiblemente cada una el mismo tipo de bloqueador, esto es, bloqueado con “Z”. Cuando se utilizan nucleótidos bloqueados con “Z” cada grupo “Z” generalmente será el mismo grupo, excepto en aquellos casos en donde el marcador detectable forma parte del grupo “Z”, esto es, no está unido a la base.
Una vez que el grupo bloqueador ha sido retirado, es posible incorporar otro nucleótido al grupo 3’-OH libre.
La molécula puede ser enlazada a través de la base a un marcador detectable mediante un enlazante deseable, enlazante que puede ser un fluoróforo, por ejemplo. Si es deseable, el marcador detectable puede ser incorporado en vez de ello en los grupos bloqueadores de la fórmula “Z”. El enlazante puede ser lábil a ácidos, fotolábil o contener un enlace disulfuro. Otros enlaces, en particular los enlazantes escindibles por fosfina que contienen azida, pueden ser empleados en la invención tal como se describe en mayor detalle.
Los marcadores y enlazantes preferidos incluyen los divulgados en WO 03/048387.
Los métodos donde se incorporan los nucleótidos, por ejemplo, donde la incorporación de una molécula de nucleótido complementaria al nucleótido en un polinucleótido de cadena individual objetivo es controlada en una reacción de síntesis o secuenciación de la invención, la incorporación de la molécula puede ser lograda a través de una transferasa terminal, una polimerasa o una transcriptasa reversa.
Preferiblemente, la molécula es incorporada por una polimerasa y particularmente de Thermococcus sp., tal como una 9°N. Incluso más preferiblemente, la polimerasa es un mutante 9°N A485L y aún más preferiblemente es un doble mutante Y409V y A485L.
En los métodos para determinar la secuencia de un polinucleótido de cadena sencilla objetivo que comprende la monitorización de la incorporación secuencial de nucleótidos complementarios de la invención, se prefiere que el grupo bloqueador y el marcador puedan ser retirados en una etapa de tratamiento químico sencilla. Así, en una realización preferida de la invención, el grupo bloqueador es escindido simultáneamente con el marcador. Esto será desde luego una característica inherente a aquellos grupos bloqueadores de fórmula Z que incorporan un marcador detectable.
Adicionalmente, de manera preferible los constructos de nucleótidos bloqueados y modificados y marcados modificados de las bases nucleotídicas A, T, C y G son reconocidos como sustratos por la misma enzima polimerasa.
En los métodos descritos aquí, cada uno de los nucleótidos puede ser puesto en contacto con el objetivo secuencialmente, con eliminación de nucleótidos no incorporados antes de la adición del siguiente nucleótido, donde la detección y eliminación del marcador y el grupo bloqueador se lleva a cabo bien sea después de la adición de cada nucleótido, o después de la adición de todos los cuatro nucleótidos.
En los métodos, todos los nucleótidos pueden ser puestos en contacto con el objetivo simultáneamente, esto es, una composición que comprende todos los diferentes nucleótidos es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y subsecuentes a la eliminación del marcador y del grupo bloqueador.
Los métodos pueden comprender una primera etapa y una segunda etapa, en donde en la primera etapa, una primera composición que comprende dos de los cuatro tipos de nucleótidos modificados es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y subsecuentemente a la eliminación del marcador y del grupo bloqueador, y donde en la segunda etapa, una segunda composición que comprende los dos nucleótidos no incluidos en la primera composición es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y subsecuentemente a la eliminación del marcador y el grupo bloqueador, y en donde las primeras etapas y la segunda etapa pueden ser repetidas opcionalmente una o más veces.
En los métodos descritos aquí también pueden comprender una primera etapa y una segunda etapa, donde en la primera etapa, una composición que comprende uno de los cuatro nucleótidos es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y subsecuentemente a la eliminación del marcador y el grupo bloqueador, y donde en la segunda etapa, una segunda composición que comprende los tres nucleótidos no incluidos en la primera composición es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y subsecuentemente a la eliminación del marcador y el grupo bloqueador, y en donde las primeras etapas y la segunda etapa pueden ser repetidas opcionalmente una o más veces.
Los métodos descritos aquí también pueden comprender una primera etapa y una segunda etapa, en donde en la primera etapa, una primera composición que comprende tres de los cuatro nucleótidos es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y subsecuentemente a la eliminación del marcador y el grupo bloqueador y donde en la segunda etapa, una composición que comprende el nucleótido no incluido en la primera composición es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y subsecuentemente a la eliminación del marcador y el grupo bloqueador, y en donde las primeras etapas y la segunda etapa pueden ser repetidas opcionalmente una o más veces.
La etapa de incorporación en los métodos de la invención puede lograrse a través de una transferasa terminal, una polimerasa o una transcriptasa reversa tal como se definieron aquí más arriba. El marcador detectable y/o el enlazante escindible pueden ser de un tamaño suficiente para evitar la incorporación de un segundo nucleótido o nucleósido en la molécula de ácido nucleico.
En ciertos métodos descritos aquí para la determinación de la secuencia de un polinucleótido de cadena sencilla objetivo, cada uno de los cuatro nucleótidos, uno de los cuales será complementario a la primera base no pareada en el polinucleótido objetivo, puede ser puesto en contacto con el objetivo secuencialmente, opcionalmente con eliminación de nucleótidos no incorporados antes de la adición del siguiente nucleótido. La determinación del éxito de la incorporación puede llevarse a cabo bien sea después de la provisión de cada nucleótido, o después de la adición de todos los nucleótidos agregados. Si se determina después de la adición de menos de cuatro nucleótidos que uno de ellos ha sido incorporado, no es necesario proveer nucleótidos adicionales con el fin de detectar los nucleótidos complementarios al nucleótido incorporado.
Alternativamente, todos los nucleótidos pueden ser puestos en contacto con el objetivo simultáneamente, esto es una composición que comprende todos los diferentes nucleótidos (esto es A, T, C y G o A, U, C y G) es puesta en contacto con el objetivo, y los nucleótidos no incorporados son eliminados antes de la detección y eliminación de los marcadores. Los métodos que involucran la adhesión secuencial de nucleótidos puede comprender una primera subetapa y opcionalmente una o más subetapas subsecuentes. En la primera subetapa se provee una composición
que comprende uno, dos o tres de los cuatro nucleótidos posibles, esto es, es puesta en contacto con el objetivo. Después puede eliminarse cualquier nucleótido no incorporado y puede llevarse a cabo una etapa de detección para determinar si uno de los nucleótidos ha sido incorporado. Si uno de ellos ha sido incorporado, puede efectuarse la escisión del enlazante. De esta manera puede ser determinada la identidad de un nucleótido en el polinucleótido objetivo. El polinucleótido naciente puede ser extendido entonces para determinar la identidad del siguiente nucleótido no pareado en el oligonucleótido objetivo.
Si la primera subetapa anterior no lleva a la incorporación de un nucleótido, o si este no es conocido, puesto que la presencia de nucleótidos incorporados no es buscada inmediatamente después de la primera subetapa, pueden llevarse a cabo una o más subetapas subsecuentes en las cuales algunos o todos aquellos nucleótidos no provistos en la primera subetapa se provean bien sea, según sea apropiado, simultáneamente o subsecuentemente. Después de esto cualquier nucleótido no incorporado puede ser eliminado y puede llevarse a cabo una etapa de detección para determinar si una de las clases nucleótido ha sido incorporada. Si uno de ellos ha sido incorporado, puede efectuarse la escisión del enlazante. De esta manera, puede determinarse la identidad de un nucleótido en el polinucleótido objetivo. El polinucleótido naciente puede ser extendido entonces para determinar la identidad del siguiente nucleótido no pareado en el oligonucleótido objetivo. Si es necesario, puede efectuarse una tercera y opcionalmente una cuarta subetapa de una manera similar a la segunda subetapa. Obviamente, una vez que se han efectuado cuatro subetapas, todos los posibles nucleótidos habrán sido provistos y uno de ellos habrá sido incorporado.
Es deseable determinar si un tipo o clase de nucleótido ha sido incorporado después de que cualquier combinación particular que comprende uno, dos o tres nucleótidos haya sido provista. De esta manera se obvia el coste y tiempos innecesarios gastados en proveer los otros nucleótidos. Sin embargo, esta no es una característica requerida de la invención.
También es deseable, cuando el método de secuenciación comprende una o más subetapas, eliminar cualquier nucleótido no incorporado antes de que se provean nuevos nucleótidos. De nuevo, esta no es una característica requerida de la invención. Obviamente, es necesario que al menos algunos y preferiblemente tantos como sea practicable de los nucleótidos no incorporados sean eliminados antes de la detección del nucleótido incorporado. Los kits de la divulgación incluyen: (a) nucleótidos individuales de acuerdo con la invención aquí anteriormente descrita, en donde cada nucleótido tiene una base que está enlazada a un marcador detectable a través de un enlazante escindible, o un marcador detectable enlazado a través de un enlazante escindible a un grupo bloqueador de fórmula Z, y donde el marcador detectable enlazado a cada nucleótido puede ser distinguido por detección del marcador detectable usado para los otros tres nucleótidos; y (b) materiales de empaque para los mismos. El kit puede incluir adicionalmente una enzima para incorporar el nucleótido en la cadena de nucleótidos complementaria y reguladores apropiados para la acción e la enzima además de los agentes químicos apropiados para eliminación del grupo bloqueador y el marcador detectable, el cual puede ser preferiblemente eliminado por la misma etapa de tratamiento químico.
Los nucleótidos/nucleósidos son adecuados para uso en muchas diferentes metodologías basadas en ADN, incluyendo protocolos de síntesis de ADN y secuenciación de ADN.
La invención puede ser entendida con referencia a los dibujos anexos en los cuales:
La figura 1 muestra estructuras de nucleótidos de ejemplo. Para cada estructura, X puede ser H, fosfato, difosfato o trifosfato. R1 y R2 pueden ser el mismo o diferentes, y pueden ser seleccionados de H, OH o cualquier grupo que pueda ser transformado en un OH, incluyendo, pero no limitándose a, un carbonilo. Algunos grupos funcionales adecuados para R1 y R2 incluyen las estructuras mostradas en la figura 3 y figura 4.
La figura 2 muestra estructuras de enlazantes, incluyendo (1) enlazantes disulfuro y enlazantes lábiles a ácidos, (2) enlazantes dialcoxibencilo, (3) enlazantes de Sieber, (4) enlazantes de indol y (5) enlazantes de t-butilo de Sieber. La figura 3 muestra algunas moléculas funcionales, incluyendo algunos enlazantes escindibles y algunos grupos protectores de hidroxilo adecuados. En estas estructuras, R1 y R2 pueden ser el mismo o diferentes, y pueden ser H, OH o cualquier grupo que pueda ser transformado en un grupo Oh , incluyendo un carbonilo. R3 representa uno o más sustituyentes seleccionados independientemente de grupos alquilo, alcoxilo, amino o halógeno. R4 y R5 pueden ser H o alquilo, y R6 puede ser alquilo, cicloalquilo, alquenilo, cicloalquenilo o bencilo. X puede ser H, fosfato, difosfato o trifosfato.
La figura 4 es una ilustración esquemática de alguno de los grupos bloqueadores Z que pueden ser utilizados.
La figura 5 muestra dos ciclos de incorporación de DGTP, DCTP y dATP marcados y bloqueados respectivamente (compuestos 18, 24 y 32).
La figura 6 muestra seis ciclos de incorporación de DTTP marcado y bloqueado (compuesto 6 ).
La presenta invención se relaciona con moléculas de nucleótidos o nucleósidos que son modificados por la unión
covalente reversible de grupos bloqueadores de 3’-OH a los mismos, y moléculas que pueden ser utilizadas en reacciones en donde las moléculas de nucleótido o nucleósido bloqueadas son requeridas, tal como en reacciones de secuenciación, síntesis de polinucleótidos y similares.
Tal como se utiliza aquí el término molécula biológica se utiliza para abarcar cualquier molécula o clase de molécula que ejecute un papel biológico. Tales moléculas incluyen por ejemplo, polinucleótidos tales como ADN o ARN, oligonucleótidos y nucleótidos individuales. Además, se abarcan los péptidos e imitadores de péptidos, tales como enzimas y hormonas, etc.
Compuestos particularmente preferidos sin embargo son los polinucleótidos (incluyendo oligonucleótidos) y nucleótidos y nucleósidos, preferiblemente aquellos que contienen una base a la cual está unido un marcador detectable enlazado a través de un enlazante escindibles. Tales compuestos son útiles en la determinación de secuencias de oligonucleótidos tal como se describe aquí.
Ligandos adecuados son cualquier fosfina o ligandos mixtos nitrógeno-fosfina conocidos por los experimentados en el arte, caracterizados porque estos ligandos son derivados de tal manera que se hacen solubles en agua, por ejemplo, por introducción de uno o más residuos sulfonato, amina, hidroxilo (preferiblemente una pluralidad de hidroxilo) o carboxilato). Cuando hay presentes residuos amina, la formación de sales de amina puede ayudar en la solubilización del ligando, y así del complejo metal-alilo. Ejemplos de ligandos apropiados son triaril fosfinas, por ejemplo trifenil fosfina, derivado de tal manera que se haga soluble en agua. También se prefieren trialquil fosfinas, por ejemplo, tri-C1-6-alquil fosfinas tales como trietil fosfinas; tales trialquil fosfinas son derivadas de la misma manera de tal forma que se hagan solubles en agua. Las fosfinas que contienen sulfonato y contienen carboxilato son particularmente preferidas; un ejemplo de la primera 3,3’,3”-fosfinidintris (ácido bencenosulfónico) que está comercialmente disponible de Aldrich Chemical Company como sal trisódica; y un ejemplo preferido del último es tris (2-carboxietil)fosfina el cual está disponible de Aldrich como la sal de clorhidrato.
Las fosfinas solubles en agua derivadas y las fosfinas que contienen nitrógeno descritas aquí pueden ser utilizadas como sus sales (por ejemplo como las sales de clorhidrato o de sodio) o, por ejemplo, en el caso de las fosfinas que contienen ácido sulfónico y carboxílico descritas aquí, en forma de los ácidos libres. Así la 3,3’,3’’-fosfinidintris (ácido bencenosulfónico) y tris (2-carxyetil)fosfinas pueden ser introducidas bien sea como triácidos o las sales trisódicas. Otras sales apropiadas serán evidentes para los experimentados en el arte. La existencia en forma salina no es particularmente importante dado que las fosfinas son solubles en solución acuosa.
Otros ligandos que pueden ser usados incluyen los siguientes:

como sal de clorhidrato, compuesto como sal de clorhidrato, compuesto
neutro y sal trisódica neutro y sal trisódica


Los intermediarios producidos se disocian espontáneamente de manera ventajosa bajo condiciones acuosas para regresar a la estructura 3’ hidroxi natural, lo que permite la incorporación adicional de otro nucleótido.
Un ejemplo de grupos de estructura -O-Z en donde Z es C(R')2-N(R" )2 son aquellos en los que - N(R" )2 es azido (-N3). Uno de tal ejemplo es azidometilo en donde cada R' es H.
Los grupos "Het" son de fórmula C (R')2N3 en la que los grupos R' son hidrógeno
Cuando las moléculas contienen grupos Z de fórmula C(R’)2N3, el grupo azido puede ser convertido a amino poniendo en contacto tales moléculas con los ligandos de fosfina o de fosfina que contiene nitrógeno descritos en detalle en relación con los complejos de metales de transición que sirven para escindir los grupos alilo de los compuestos de fórmula PN-O-alilo, fórmula R-O-alilo, RaN(alilo), RMH(alilo) 2 y R-S-alilo. Cuando se transforma azido a amino, sin embargo, no es necesario un metal de transición. Alternativamente, el grupo azido en los grupos Z de fórmula C(R’)2N3 puede ser convertido en amino poniendo en contacto tales moléculas con los tioles, en particular tioles solubles en agua tales como ditiotreitol (DTT).
Como es conocido en la técnica, un "nucleótido” consiste de una base nitrogenada, un azúcar y uno o más grupos fosfato. Son unidades monoméricas de una secuencia de ácidos nucleicos. En el ARN, el azúcar es una ribosa, y en el ADN una desoxirribosa, esto es, un azúcar que carece de un grupo hidroxilo que está presente en la ribosa. La base nitrogenada es un derivado de purina o pirimidina. Las purinas son adenina (A) y guanina (G) y las pirimidinas son citosina (C) y timina (T) (en el contexto del ARN, uracilo (U)). El átomo C-1 de la desoxirribosa está enlazado a N-1 de una pirimidina o N-9 de una purina. Un nucleótido es también un éster de fosfato o un nucleósido, con esterificación presente en el grupo hidroxilo unido al C-5 del azúcar. Los nucleótidos son usualmente mono, di- o trifosfatos.
Un “nucleósido” es estructuralmente similar a un nucleótido, pero carece de las unidades estructurales fosfato. Un ejemplo de un nucleósido análogo seria uno en el cual el marcador está enlazado a la base y no hay grupo fosfato unido a la molécula de azúcar.
Aunque la base se refiere usualmente a una purina o una pirimidina, la persona experimentada sabrá que hay disponibles derivados y análogos que no alteran la capacidad del nucleótido o nucleósido para experimentar el apareamiento de bases Watson-Crick. “Derivados” o “análogos” significa un compuesto o molécula cuya estructura central es la misma que o cercanamente parecida a la de, un compuesto original, pero que tiene una modificación química o física, tal como un grupo lateral diferente o adicional, o grupos bloqueadores en 2’ y/o 3’, los cuales permiten que el nucleótido o nucleósido derivado se enlace a otra molécula. Por ejemplo, la base puede ser una desazapurina. Los derivados deberían ser capaces de experimentar el apareamiento según Watson-Crick. “Derivado” y “análogo” también significan un derivado nucleótido o nucleósido sintético que tienen unidades estructurales de base modificadas y/o unidades estructurales de azúcar modificadas. Tales derivados y análogos están discutidos, por ejemplo, en Scheit, Nucleotide Analogs (John Wiley & Son, 1980) and Uhlman et al., Chemical Reviews 90:543-584, 1990. Los análogos de nucleótidos también pueden comprender enlaces fosfodiéster modificados, incluyendo enlaces fosforotioato, fosforoditioato, alquil fosfonato, fosfaranilidato y fosforamidato. Los análogos deberían ser capaces de experimentar el apareamiento de bases según Watson-Crick. “Derivados”, “análogos” y “modificados” tal como se utilizan aquí, pueden ser utilizados de manera intercambiable, y son abarcados por los términos “nucleótidos” y “nucleósidos” definidos aquí.
En el contexto de la presente invención, el término “incorporar” significa convertirse en parte de una molécula de ácido nucleico (por ejemplo ADN) u oligonucleótido o cebador. Un oligonucleótido se refiere a una molécula sintética o natural que comprende una secuencia enlazada covalentemente de nucleótidos que está formada por un enlace fosfodiéster o fosfodiéster modificado entre la posición 3’ de la pentosa sobre un nucleótido de la posición 5’ de la pentosa en un nucleótido adyacente.
El término “alquilo” cubre grupos de cadena recta, cadena ramificada y cicloalquilo. Al menos que el contexto indique otra cosa, el término “alquilo” se refiere a grupos que tienen de 1 a 10 átomos de carbono, por ejemplo de 1 a 8 átomos de carbono, y típicamente de 1 a 6 átomos de carbono, por ejemplo de 1 a 4 átomos de carbono. Ejemplos de grupos alquilo incluyen metilo, etilo, propilo, isopropilo, n-butilo, isobutilo, tert-butilo, n-pentilo, 2-pentilo, 3-pentilo, 2-metil butilo, 3-metil butilo, y n-hexilo y sus isómeros.
Ejemplos de grupos cicloalquilo son aquellos que tienen de 3 a 10 átomos de anillo, incluyendo ejemplos particulares aquellos derivados de ciclopropano, ciclobutano, ciclopentano, ciclohexano y cicloheptano, bicicloheptano y decalina.
Cuando los grupos alquilo (incluyendo cicloalquilo) están sustituidos, particularmente cuando estos forman bien grupos R’ de las moléculas de la invención, los ejemplos de sustituyentes apropiados incluyen sustituyentes halógenos o grupos funcionales tales como hidroxilo, amino, ciano, nitro, carboxilo y similares. Tales grupos pueden ser también sustituyentes, cuando sea apropiado, de otros grupos R’ en las moléculas de la invención.
El término amino se refiere a grupos del tipo NR*R** en donde R* y R** son seleccionados independientemente de hidrógeno, un grupo alquilo C1-6 (también denominado como alquilamino C1-6 o dialquilamino C1-6).
El término “halógeno” tal como se utiliza aquí incluye flúor, cloro, bromo y yodo.
Las moléculas de nucleótidos de la presente invención son adecuadas para uso en muchos métodos diferentes donde se requiere la detección de nucleótidos.
Los métodos de secuenciación de ADN, tales como los delineados en la Patente de los Estados Unidos No.
5,302,509 pueden ser llevados a cabo utilizando los nucleótidos.
La presente invención puede hacer uso de marcadores detectables convencionales. La detección puede llevarse a cabo por cualquier método adecuado, incluyendo espectroscopía de fluorescencia o por otros medios ópticos. El marcador preferido es un fluoróforo, el cual, después de la absorción de energía emite radiación a una longitud de onda definida. Se conocen muchos marcadores fluorescentes adecuados. Por ejemplo, Welch et al. (Chem. Eur. J.
5(3): 951-960, 1999) describe unidades estructurales fluorescentes funcionalizadas con dansilo que pueden ser utilizadas en la presente invención. Zhu et al. (Cytometry 28:206-211, 1997) describe el uso de los marcadores fluorescentes Cy3 y Cy5, los cuales también pueden ser utilizados en la presente invención. También se divulgan marcadores adecuados para uso en Prober et al. (Science 238: 336-341, 1987); Connell et al. (BioTechniques 5(4): 342-384, 1987), Ansorge et al. (Nucl. Acids Res. 15(11):4593-4602, 1987) y Smith et al. (Nature 321:674, 1986). Otros marcadores fluorescentes comercialmente disponibles incluyen, pero no se limitan a fluoresceína, rodamina (incluyendo TMR, rojo de Texas y Rox), alexa, bodipy, acridina, cumarina, pireno, benzantraceno y las cianinas. También pueden utilizarse marcadores múltiples en la invención. Por ejemplo, los casetes de biofluoróforos FRET (Tet. Let. 46: 8867-8871, 2000) son bien conocidos en el arte y pueden ser utilizados en la presente invención. También pueden utilizarse sistemas dendriméricos multiflúor (J. Amer. Chem. Soc. 123: 8101-8108, 2001).
Aunque se prefieren los marcadores fluorescentes, otras formas de marcadores detectables serán evidentes como útiles para las personas de experiencia normal. Por ejemplo, las micropartículas, incluyendo puntos cuánticos (Empodocles et al., Nature 399: 126-130, 1999), nanopartículas de oro (Reichert et al., Anal. Chem. 72:6025-6029, 2000) y microperlas (Lacoste et al., Proc. Natl. Acad. Sci USA 97(17):9461-9466, 2000) pueden todos ser usados. También pueden utilizarse en la invención marcadores multicomponentes. Un marcador multicomponente es aquel que es dependiente de la interacción con un compuesto adicional para su detección. El marcador multicomponente más común usado en biología es el sistema biotina-estreptavidina. La biotina se utiliza como el marcador unido a la base de nucleótido. La estreptavidina se agrega entonces separadamente para permitir que ocurra la detección. Hay disponibles otros sistemas multicomponentes. Por ejemplo, el dinitrofenol tiene un anticuerpo fluorescente comercialmente disponible que puede ser utilizado para la detección.
La invención ha sido y será descrita con referencia a los nucleótidos. Sin embargo, a menos que se indique otra cosa, la referencia a los nucleótidos también pretende ser aplicable a los nucleósidos. La invención también será descrita adicionalmente con referencia al ADN, aunque la descripción también será aplicable a ARN, APN, y otros ácidos nucleicos, a menos que se indique otra cosa.
Los nucleótidos modificados de la invención pueden usar un enlazante escindible para unir el marcador al nucleótido. El uso de un enlazante escindible asegura que el marcador, si se requiere, puede ser eliminado después de la detección, evitando cualquier señal de interferencia con cualquier nucleótido marcado incorporado subsecuentemente.
Los experimentados en la técnica conocerán la utilidad de los didesoxinucleósidos trifosfatos en los así llamados métodos de secuenciación Sanger y protocolos relacionados (tipo Sanger), los cuales se basan en la terminación aleatorizada de cadenas en un tipo particular de nucleótido. Un ejemplo de un protocolo de secuenciación tipo Sanger es el método BASS descritos por Metzker (infra). Otros métodos de secuenciación tipo Sanger serán bien conocidos por los experimentados en el arte.
Los métodos Sanger y tipo Sanger generalmente operan mediante la ejecución de un experimento en el cual se proveen ocho tipos de nucleótidos, cuatro de los cuales contienen un grupo 3’OH; y cuatro de los cuales omiten el grupo OH y los cuales están marcados diferentemente uno de otro. Los nucleótidos usados que omiten el grupo 3’OH - didesoxinucleótido - son abreviados convencionalmente como ddNTPs. También es conocido por la persona experimentada, puesto que los ddNTPs están marcados diferentemente, al determinar las posiciones de los nucleótidos terminales incorporados, y al combinar esta información, la secuencia del oligonucleótido objetivo puede ser determinada.
Los nucleótidos de la presente invención, tal como será reconocido, pueden ser de utilidad en los métodos Sanger y protocolos relacionados puesto que el mismo efecto alcanzado utilizando ddNTPs puede ser logrado utilizando los novedosos grupos bloqueadores de 3’-OH descritos aquí: ambos previenen la incorporación de nucleótidos subsecuentes.
El uso de los nucleótidos de acuerdo con la presente invención en los métodos de secuenciación de Sanger y tipo Sanger, en donde el enlazante que conecta el marcador detectable al nucleótido puede o puede no ser escindible, forma un aspecto todavía adicional de esta invención. Vista desde este aspecto, la invención provee el uso de tales nucleótidos en un método de secuenciación de Sanger o tipo Sanger.
Cuando se utilizan los nucleótidos bloqueados con Z en 3’-OH de acuerdo con la presente invención, será evidente que los marcadores detectables unidos a los nucleótidos no necesitan estar conectados a través de enlazantes escindibles. Puesto que en cada caso cuando un nucleótido marcado de la invención es incorporado, no se necesita incorporar subsecuentemente nucleótidos y así el marcador no necesita ser eliminado del nucleótido.
Además, será evidente que la monitorización de la incorporación de los nucleótidos bloqueados en 3’OH puede ser determinada mediante el uso de 32P radioactivo en los grupos fosfato unidos. Estos pueden estar presentes bien sea en los ddNTPs mismo o en los cebadores usados para extensión. Cuando los grupos bloqueadores son de fórmula “Z”, esto representa un aspecto adicional de la invención.
Visto desde este aspecto, la invención provee el uso de un nucleótido que tiene un grupo 3’OH bloqueado con un grupo “Z” en un método de secuenciación Sanger o tipo Sanger. En esta realización, puede estar presente un marcador 32P detectable tanto en cualquiera de los ddNTPs usados en el cebador usado para la extensión.
Los enlazantes escindibles son conocidos en el arte, y puede aplicarse química convencional para unir un enlazante a una base nucleótido y un marcador. El enlazante puede ser escindido por cualquier método adecuado, incluyendo exposición a ácidos, bases, nucleófilos, electrófilos, radicales, metales, agentes reductores u oxidantes, luz, temperatura, enzimas, etc. El enlazante tal como se discute aquí también puede ser escindido con el mismo catalizador utilizado para escindir el enlace del grupo bloqueador 3’O. Enlazantes adecuados pueden ser adaptados a partir de grupos bloqueadores químicos estándar, tal como se divulga en Greene & Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons. Se divulgan enlazantes escindibles adecuados adicionales utilizados en síntesis en fase sólida en Guillier et al. (Chem. Rev. 100:2092-2157, 2000).
El uso del término “enlazante escindible” no pretende implicar que se requiere remover todo el enlazante de, por ejemplo, la base de nucleótidos. Cuando el marcador detectable está unido a la base, el sitio de escisión del nucleósido puede estar localizado en una posición del enlazante que asegure que parte del enlazante permanece unida a la base del nucleótido después de la escisión.
Cuando el marcador detectable está unido a la base, el enlazante puede estar unido en cualquier posición sobre la base de nucleótidos con la condición de que el apareamiento de bases según Watson-Crick pueda ser llevado todavía a cabo. En el contexto de bases purínicas, es preferible si el enlazante está unido a través de la posición 7 de la purina o del análogo de desazapurina preferido, a través de una purina modificada en 8, a través de una adenosina modificada en N-6 o en una guanina modificada en N-2. Para pirimidinas, la unión es preferiblemente a través de la posición 5 sobre la citosina, timidina o uracilo y la posición N-4 sobre la citosina. En la figura 1 se muestran estructuras de nucleótidos adecuadas. Para cada estructura en la figura 1 X puede ser H, fosfato, difosfato o trifosfato. R1 y R2 pueden ser el mismo o diferentes, y son seleccionados de H, OH, o la fórmula Z tal como se describe aquí o cualquier otro grupo que pueda ser transformado en un OH, incluyendo, pero no limitándose a, un carbonilo, con la condición de que al menos uno de R1 y R2 sea O-alilo o la fórmula Z tal como se describe aquí. Algunos grupos funcionales adecuados para R1 y R2 incluyen las estructuras mostradas en las figuras 3 y 4.
Enlazantes adecuados se muestran en la figura 3 e incluyen, pero no se limitan a, enlazantes disulfuro (1), enlazantes lábiles a ácidos (2, 3, 4 y 5; incluyendo enlazantes dialcoxibencilo (por ejemplo, 2), enlazantes de Sieber (por ejemplo, 3) enlazantes de indol (por ejemplo 4), enlazantes de t-butilo Sieber (por ejemplo 5)), enlazantes escindibles electrofílicamente, enlazantes escindibles nucleofilicamente, enlazantes fotoescindibles, escisión bajo condiciones reductoras, condiciones oxidativas, escisión a través del uso de enlazantes de seguridad, y escisión por mecanismos de eliminación.
A. Enlazantes escindidos electrofílicamente
Los enlazantes escindidos electrofílicamente son escindidos típicamente por protones e incluyen escisiones sensibles a ácidos. Los enlazantes adecuados incluyen sistemas bencílicos modificados tales como tritilo, ésteres de p-alcoxibencilo y amidas de p-alcoxibencilo. Otros enlazantes adecuados incluyen grupos tert-butiloxicarbonilo (Boc) y el sistema acetal.
El uso de metales tiofílicos, tales como níquel, plata o mercurio, en la escisión de tioacetal u otros grupos protectores que contienen sulfuro también puede ser considerado para la preparación de moléculas enlazantes adecuadas. B. Enlazantes escindidos nucleofílicamente.
La escisión nucleofílica también es un método bien reconocido en la preparación de moléculas enlazantes. Pueden utilizarse grupos tales como ésteres que son lábiles en agua, (esto es, pueden ser escindidos simplemente a pH básico) y grupos que son lábiles a nucleófilos no acuosos. Pueden utilizarse iones fluoruro para escindir enlaces silicio-oxígeno en grupos tales como triisopropilsilano (TIPS) o t-butildimetilsilano (TBDMS).
C. Enlazantes fotoescindibles.
Los enlazantes fotoescindibles han sido utilizados ampliamente en la química de carbohidratos. Es preferible que la luz requerida para activar la escisión no afecte los otros componentes de los nucleótidos modificados. Por ejemplo, si un fluoróforo es utilizado como marcador, es preferible que este absorba luz de una longitud de onda diferente que la requerida para escindir la molécula de enlazante. Enlazantes adecuados incluyen los basados en compuestos de O-nitrobenzol y compuestos de nitroveratril. Los enlazantes basados en la química de la benzoina también pueden ser utilizados (Lee et al., J. Org. Chem. 64:3454-3460, 1999).
D. Escisión bajo condiciones reductoras.
Hay muchos enlazantes conocidos que son susceptibles de la escisión reductora. La hidrogenación catalítica utilizando catalizadores basados en paladio ha sido utilizada para escindir grupos bencilo y benciloxicarbonilo. La reducción de enlaces disulfuro también es conocida en el arte.
E. Escisión bajo condiciones oxidativas.
Las metodologías basadas en oxidación son bien conocidas en el arte. Incluyen oxidación de grupos p-alcoxibencilo y la oxidación de los enlazantes de azufre y selenio. El uso de yodo acuoso para escindir disulfuros y otros enlazantes basados en azufre o selenio también cae dentro del alcance de la invención.
F. Enlazantes de sujeción segura.
Los enlazantes de sujeción segura son aquellos que se escinden en dos etapas. En un sistema preferido la primera etapa es la generación de un centro nucleofílico reactivo seguida por una segunda etapa que involucra una ciclización intramolecular que da como resultado la escisión. Por ejemplo, los enlazantes de éster levulínico pueden ser tratados con hidracina o fotoquímica para liberar una amina activa, la cual puede ser entonces ciclizada para escindir un éster en cualquier lugar de la molécula (Burgess et al., J. Org. Chem. 62:5165-5168, 1997).
G. Escisión por mecanismos de eliminación.
También pueden utilizarse reacciones de eliminación. Por ejemplo, pueden utilizarse la eliminación catalizada por bases de grupos tales como Fmoc y cianoetilo, y la eliminación reductora catalizada por paladio de sistemas alílicos. Así como el sitio de escisión, el enlazante puede comprender una unidad espaciadora. Las distancias espaciadoras, por ejemplo, la base de nucleótido del sitio de escisión o marcador. La longitud del enlazante no es importante dado que el marcador se mantiene a una distancia suficiente del nucleótido de tal manera que no interfiere con ninguna interacción entre el nucleótido y una enzima.
En una realización preferida el enlazante puede consistir de la misma funcionalidad que el bloque. Esto hará el proceso de desprotección y desbloqueo más eficiente, puesto que solamente se requerirá un tratamiento sencillo para remover tanto el marcador como el bloque.
Enlazantes particularmente preferidos son enlazantes que contienen azida escindible con fosfina.
Un método para determinar la secuencia de un polinucleótido objetivo puede ser llevado a cabo poniendo en contacto el polinucleótido objetivo separadamente con los diferentes nucleótidos para formar el complemento al del polinucleótido objetivo, y detectar la incorporación de los nucleótidos. Tal método hace uso de polimerización, mediante la cual una enzima polimerasa extiende la cadena complementaria incorporando el nucleótido correcto complementario al del objetivo. La reacción de polimerización también requiere de un cebador específico para iniciar la polimerización.
Para cada ciclo, la incorporación del nucleótido modificado se lleva a cabo mediante la enzima polimerasa, y se determina entonces el evento de incorporación. Existen muchas diferentes enzimas polimerasa, y será evidente para la persona de experiencia normal cual es la más apropiada para el uso. Las enzimas preferidas incluyen la ADN polimerasa I, el fragmento Klenow, la ADN polimerasa III, la T4 o T7 ADN polimerasa. La Taq polimerasa o la Vent polimerasa. También pueden utilizarse polimerasas manipuladas para tener propiedades específicas. Como se anotó anteriormente, la molécula se incorpora preferiblemente mediante una polimerasa y particularmente a partir de Thermococcus sp., tal como 9°N. Incluso más preferiblemente, la polimerasa es un mutante 9°N A485L e incluso más preferiblemente es un mutante doble Y409V y A485L. Un ejemplo de tal enzima preferida es la Thermococcus sp. 9°N exo-Y409V A485L disponible de New England Biolabs. Ejemplos de tales polimerasas apropiadas están divulgados en Proc. Natl. Acad. Sci. USA, 1996 (93), pp 5281-5285, Nucleic Acids Research, 1999(27), pp 2454 2553 y Acids Research, 2002(30), pp 605-613.
Los métodos de secuenciación se llevan a cabo preferiblemente con el polinucleótido objetivo dispuesto sobre un soporte sólido. Los polinucleótidos objetivo múltiples pueden ser inmovilizados sobre el soporte sólido a través de moléculas de enlazamiento, o pueden ser unidos a partículas, por ejemplo microesferas, que también pueden ser unidas a un material de soporte sólido. Los polinucleótidos pueden ser unidos al soporte sólido mediante un cierto número de medios, incluyendo el uso de interacciones biotina-avidina. Los métodos para inmovilizar polinucleótidos sobre un soporte sólido son bien conocidos en el arte, e incluyen técnicas litográficas y “manchado” con polinucleótidos individuales en posiciones definidas sobre un soporte sólido. Se conocen en la técnica soportes sólidos adecuados, e incluyen láminas de vidrio y perlas, superficies de cerámica y silicio y materiales plásticos. El soporte es usualmente una superficie plana aunque pueden utilizarse también perlas microscópicas (microesferas) y a su vez pueden unirse a otros soportes sólidos por medios conocidos. Las microesferas pueden ser de cualquier tamaño adecuado, típicamente en el rango de 10 nm a 100 nm de diámetro. En una realización preferida, los polinucleótidos son unidos directamente a una superficie plana, preferiblemente una superficie plana de vidrio. La unión se hará preferiblemente por medio de un enlace covalente. Preferiblemente, las disposiciones que se utilizan son disposiciones de molécula individual que comprenden polinucleótidos en áreas con resolución óptica distintas, por ejemplo, como se divulga en la Solicitud Internacional No. WO 00/06770.
El método de secuenciación puede ser llevado a cabo tanto en arreglos con una molécula de polinucleótido individual como de multipolinucleótidos, esto es, arreglos de distintas moléculas de polinucleótidos individuales y arreglos de distintas regiones que comprenden múltiples copias de una molécula de polinucleótido individual. Los arreglos de moléculas individuales permiten que cada polinucleótido individual sea resuelto separadamente. El uso de arreglo de moléculas individuales es preferido. El secuenciamiento en arreglos de moléculas individuales permite de una manera no destructiva la formación de un arreglo espacialmente direccionable.
El método hace uso de la reacción de polimerización para generar la secuencia complementaria del objetivo. Las condiciones compatibles con las reacciones de polimerización serán evidentes para la persona experimentada. Para llevar a cabo la reacción de polimerasa será necesario usualmente primero fusionar una secuencia de cebador al polinucleótido objetivo, siendo reconocida la secuencia de cebador por la enzima polimerasa y actuando como un sitio de iniciación para la extensión subsecuente de la cadena complementaria. La secuencia de cebador puede ser agregada como un componente separado con respecto al polinucleótido objetivo. Alternativamente, el cebador y el polinucleótido objetivo pueden ser cada uno parte de una molécula de cadena sencilla, formando la porción de cebador un dúplex intramolecular con una parte del objetivo, esto es, una estructura de bucle en horquilla. Esta estructura puede ser inmovilizada al soporte sólido en cualquier punto de la molécula. Otras condiciones necesarias
para llevar a cabo la reacción con polimerasa, incluyendo temperatura, pH, composiciones de los reguladores etc., serán evidentes para los experimentados en el arte.
Los nucleótidos modificados de la invención son puestos entonces en contacto con el polinucleótido objetivo, para permitir que ocurra la polimerización. Los nucleótidos pueden ser agregados secuencialmente, esto es, adición separada de cada tipo de nucleótido (A, T, G o C), o agregados juntos. Si se agregan juntos, es preferible que cada tipo de nucleótido sea marcado con un marcador diferente.
La etapa de polimerización se deja avanzar durante un tiempo suficiente para permitir la incorporación de un nucleótido.
Los nucleótidos que no son incorporados son luego eliminados, por ejemplo, sometiendo el arreglo a una etapa de lavado, y la detección de los marcadores incorporados puede ser llevada a cabo entonces.
La detección puede ser llevada a cabo por medios convencionales, por ejemplo si el marcador es una unidad estructural fluorescente, la detección de una base incorporada puede ser llevada a cabo utilizando un microscopio de barrido confocal para barrer la superficie del arreglo con un láser, para formar una imagen de un fluoróforo enlazado directamente a la base incorporada. Alternativamente, puede utilizarse un detector sensible 2-D, tal como un detector acoplado con carga (CCD) para visualizar las señales individuales generadas. Sin embargo, hay disponibles otras técnicas tales como microscopía óptica de campo cercano con barrido (SNOM) y pueden ser utilizadas cuando hay la formación de imágenes de arreglos densos. Por ejemplo, al utilizar SNOM, pueden distinguirse polinucleótidos individuales cuando están separados por una distancia de menos de 1 0 0 nm, por ejemplo 10 nm a 10 pm. Para una definición de la microscopia óptica de campo cercano con barrido véase Moyer et al., Laser Focus World 29:10, 1993. Los aparatos adecuados usados para la generación de imágenes de arreglos de polinucleótidos son conocidos y la estructuración técnica será evidente para la persona experimentada.
Después de la detección, el marcador puede ser eliminado utilizando condciones adecuadas que escindan el enlazante y el bloque en 3’OH para permitir la incorporación de nucleótidos modificados adicionales de la invención. Condiciones apropiadas pueden ser las descritas aquí para desprotecciones con grupo “Z”. Estas condiciones pueden servir para desproteger tanto el enlazante (si es escindible) y el grupo bloqueador.
Esta invención puede ser entendida adicionalmente con referencia a los siguientes ejemplos los cuales sirven para ilustrar la invención.
3’-OH protegido con un grupo azidometilo como una forma protegida de un heniaminal:

Se han sintetizado nucleótidos que portan este grupo bloqueador en la posición 3’, demostrando ser incorporados exitosamente por las ADN polimerasas, bloquear eficientemente y pueden ser eliminados subsecuentemente bajo condiciones acuosas neutras utilizando fosfinas o tioles solubles en agua que permiten una extensión posterior:

A una solución de 5-yodo-2'-desoxiuridina (1.05 g, 2.96 mmol) y Cul (114 mg, 0.60 mmol) en DMF seco (21 ml) se agregó trietilamina (0.9 ml). Después de agitar durante 5 minutos se agregaron trifluoro-N-prop-2-inil-acetamida (1.35 g, 9.0 mmol) y Pd (PPh3)4 (330 mg, 0.29 mmol) a la mezcla y la reacción se agitó a temperatura ambiente en la oscuridad durante 16 horas. Se agregaron metanol (MeOH) (40 ml) y bicarbonato Dowex a la mezcla de reacción y se agitó durante 45 minutos. La mezcla fue filtrada y el filtrado fue lavado con MeOH y el solvente fue eliminado bajo vacío. La mezcla cruda fue purificada por cromatografía sobre sílica (acetato de etilo y (EtOAc) a EtOAc:MeOH 95:5) para dar cristales ligeramente amarillos (794 mg, 71 %). 1H RMN (d6 dimetilsulfóxido (DMSO)) 6 2.13-2.17 (m, 2H, H-2'), 3.57-3.65 (m, 2H, H-5'), 3.81-3.84 (m, 1H, H-4'), 4.23-4.27 (m, 3H, H-3', CH2N) , 5.13 (t, J = 5.0 Hz, 1H, OH), 5.20 (d, J = 4.3 Hz, 1H, OH), 6.13 (t, J = 6.7 Hz, 1H, H-1'), 8.23 (s, 1H, H-6 ), 10.11 (t, J = 5.6 Hz, 1H, NH), 11.70 (br s, 1H, NH). Masa (-ve electroaspersión) calculada para C14H14F3N3O6377.08, encontrada 376.

5’-O-(tert-butidimetilsilil)-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil] -2’-desoxiuridina (2).
A una solución de (1) (656 mg, 1.74 mmol) en DMF seco (15 ml) se agregó t-butildimetilsilil cloruro (288 mg, 1.91 mmol) en pequeñas porciones, seguido por imidazol (130 mg, 1.91 mmol). La reacción fue seguida por TLC y fue completada después de agitación durante 8 horas a temperatura ambiente. La reacción fue detenida con solución acuosa saturada de NaCl. Se agregó EtOAc (25 ml) a la mezcla de reacción y la capa acuosa fue extraída con EtOAc tres veces. Después de secar las fases orgánicas combinadas (MgSO4), el solvente fue eliminado bajo vacío. La purificación por cromatografía sobre sílica (EtOAc:éter de petróleo 8:2) dio (2) en forma de cristales ligeramente amarillos (676 mg, 83 %). 1H RMN (da DMSO) 50.00 (s, 6H, CH3), 0.79 (s, 9H, tBu), 1.93 -2.00 (m, 1H, H-2’), 2.06 2.11 (m, 1H, H-2’), 3.63-3.75 (m, 2H, H-5’), 3.79-3.80 (m, 1H, H-4’), 4.12-4.14 (m, 3H, H-3’, CH2N), 5.22 (d, J = 4.1 Hz, 1H, OH), 6.03 (t, J = 6.9 Hz, 1H, H-1’), 7.86 (s, 1H, H-6), 9.95 (t, J = 5.4 Hz, 1H, NH), 11.61 (br s, 1H, NH). Masa (-ve electroaspersión)’ calculada para C20^ 8F3^ O 6Si 491.17, encontrada 490.

5’-O-(tert-Butidimetilsilil)-3’-O-metiltiometil-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2’-desoxiuridina (3).
A una solución de (2) (1.84 g, 3.7 mmol) en DMSO seco (7 ml) se agregó ácido acético (3.2 ml) y anhídrido acético (10.2 ml). La mezcla fue agitada durante 2 días a temperatura ambiente, antes de ser detenida con NaHCO3 acuosos saturado. Se agregó EtOAc (50 ml) y la capa acuosa fue extraída tres veces con acetato de etilo. Las capas orgánicas combinadas fueron lavadas con solución acuosa saturada de NaHCO3 y secada (MgSO4). Después de eliminar el solvente bajo presión reducida, el producto (3) fue purificado por cromatografía sobre sílica (EtOAc: éter de petróleo 8:2) produciendo un aceite pegajoso claro (1.83 g, 89 %), 1H RMN (d6 DMSO) : 50.00 (s, 6H, CH3), 0.79 (s, 9H, tBu), 1.96-2.06 (m, 1H, H-2’), 1.99 (s, 3H, SCH3), 2.20-2.26 (m, 1H, H-2’), 3.63-3.74 (m, 2H, H-5’), 3.92-3.95 (m, 1H, H-4’), 4.11-4.13 (m, 2H, CH2), 4.28-4.30 (m, 1H, H-3’), 4.59 (br s, 2H, CH2), 5.97 (t, J = 6.9 Hz, 1H, H-1’), 7.85 (s, 1H, H-6), 9.95 (t, J = 5.3 Hz, 1H, NH), 11.64 (s, 1H, NH). Masa (-ve electroaspersión) calculada para C22H32F3N3O6SSi 551.17, encontrada 550.

3'-O-Azidometil-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxiuridina (4).
A una solución de (3) (348 mg, 0.63 mmol) y ciclohexeno (0.32 ml, 3.2 mmol) en CH2CI2 seco (5 ml) a 4°C, se agregó cloruro de sulfurilo (1M en CH2Cl2, 0.76 ml, 0.76 mmol) gota a gota bajo N2. Después de 10 minutos la TLC indicó el consumo completo del nucleósido (3). El solvente fue evaporado y el residuo fue sometido a alto vacío durante 20 minutos. Fue redisuelto entonces en DMF seco (3 ml) y tratado con NaN3 (205 mg, 3.15 mmol). La suspensión resultante fue agitada bajo temperatura ambiente durante 2 horas. La reacción fue detenida con CH2Cl2 y las capas orgánicas fueron lavadas con solución acuosa saturada de NaCl. Después de eliminar el solvente, la goma amarilla resultante fue redisuelta en THF (2 ml) y tratada con TBAF (1 M en THF, 0.5 ml) a temperatura ambiente durante 30 minutos. El solvente fue eliminado y la reacción fue manipulada con CH2O 2 y solución acuosa saturada de NaHCO3. La capa acuosa fue extraída tres veces con CH2Cl2. La purificación por cromatografía sobre sílica (EtOAc: éter de petróleo 1:1 a EtOAc) dio (4) (100 mg, 37 %) en forma de una espuma amarilla pálida. 1H RMN (da DMSO) 8 2.15-2.26 (m, 2H, H-2'), 3.47-3.57 (m, 2H, H-5'), 3.88-3.90 (m, 1H, H-4'), 4.14 (d, J = 4.7 Hz, 2H, CH2NH), 4.24-4.27 (m, 1H, H-3'), 4.75 (s, 2H, CH2N3), 5.14 (t, J = 5.2 Hz, 1H, OH), 5.96-6.00 (m, 1H, H-1'), 8.10 (s, 1H, H-6), 10.00 (s, 1H, NHCOCF3)), 11.26 (s, 1H, NH).

Preparación de pirofosfato de bis(tri-n-butilamonio) (solución 0.5 M en DMF)
Se disolvió difosfato de tetrasodio decahidrato (1.5 g, 3.4 mmol) en agua (34 ml) y la solución fue aplicada a una columna de Dowex en la forma H+. La columna fue eluida con agua. El eluyente se hizo gotear directamente en una solución enfriada (baño de hielo) y en agitación de tri-n-butilamina (1.6 ml, 6.8 mmol) en EtOH (14 ml). La columna fue lavada hasta que el pH del eluyente se incrementó a 6. La solución en etanol acuosa fue evaporada hasta sequedad y luego coevaporada dos veces con etanol y dos veces con DMF anhidro. El residuo fue disuelto en DMF (6.7 ml). La solución amarilla pálida fue almacenada sobre tamices moleculares del 4Á.
3'-O-Azidometil-5-(3-amino-prop-1-inil)-2'-desoxiuridina 5'-O-nucleósido trifosfato (5).
El nucleósido (4) y la esponja de protones fueron secados sobre P2O5 bajo vacío durante la noche. Se agregó una solución de (4) (92 mg, 0.21 mmol) y la esponja de protones (90 mg, 0.42 mmol) en trimetilfosfato (0.5 ml) con tamices moleculares de 4Á durante 1 hora. Se agregó POCl3 recién destilado (24 pl, 0.26 mmol) y la solución fue agitada a 4°C durante 2 horas. La mezcla fue calentada lentamente hasta temperatura ambiente y se agregaron pirofosfato de bis (tri-n-butil amonio) (1.7 ml, 0.85 mmol) y de tri-n-butil amina anhidra (0.4 ml, 1.7 mmol). Después de 3 minutos, la reacción fue detenida con regulador de TEAB (bicarbonato de trietilamonio) 0.1 M (15 ml) y se agitó durante 3 horas. El agua fue eliminada bajo presión reducida y el residuo resultante fue disuelto en amoníaco concentrado (p 0.88, 15 ml) y agitado a temperatura ambiente durante 16 horas. La mezcla de reacción fue evaporada entonces hasta sequedad. El residuo fue disuelto en agua y la solución fue aplicada a una columna DEAE-Sephadex A-25. Se ejecutó MPLC con un gradiente lineal de TEAB. El trifosfato fue eluido en regulador entre 0.7 M y 0.8 M. Las fracciones que contenían el producto fueron combinadas y evaporadas hasta sequedad. El residuo fue disuelto en agua y purificado posteriormente por HPLC. HPLC: tr(5) : 18.8 min (columna preparativa Zorbax C18, gradiente: 5% a 35% B in 30 min, regulador A 0.1M TEAB, regulador B MeCN). El producto fue aislado en forma de una espuma blanca (76 O.D., 7.6 pmol, 3.8%, £280 = 10000). 1H RMN (D2O) 81.79 (s, CH2), 2.23-2.30; 2.44-2.50 (2 x m, 2H, H-2'), 3.85 (m, CH2NH), 4.10-4.18 (m, 2H, H-5'), 4.27 (br s, H-4'), 4.48-4.50 (m, H-3'), 4.70-4.77 (m, CH2N3), 6.21 (t, J = 6.6 Hz, H-1'), 8.32 (s, 1H, H-6). 31P RMN (D2O) 8 -6.6 (m, 1P, Pv), -10.3 (d, J =18.4 Hz, 1P, Pa), -21.1 (m, 1P, Pp). Masa (-ve electroaspersión) calculada para C13H19N6O14P3576.02, encontrada 575.

El disulfuro de partida (4.0 mg, 13.1 Limol) fue disuelto en DMF (300 ^L) y se agregó lentamente diisopropiletilamina (4 ^L). La mezcla fue agitada a temperatura ambiente y se agregó una solución de colorante Cy-3 (5 mg, 6.53 ^mol) en DMF (300 ^L) durante 10 minutos. Después de 3.5 horas, al terminar la reacción, los volátiles fueron evaporados bajo presión reducida y el residuo crudo fue purificado por HPLC sobre una columna analítica Zorbax SB-C18 con una rata de flujo de Iml/minuto en regulador de bicarbonato de trietilamonio 0.1 M (regulador A) y CH3CN (regulador B) utilizando el siguiente gradiente: 5 min 2% B; 31 min 55% B; 33 min 95% B; 37 min 95%; 39 min 2% B; 44 min.
2% B. El enlazante de Cy3-disulfuro esperado fue eluido con un tr: 21.8 min. en 70% de rendimiento (con base en una medición por UV; £550 150,000 cm-1 M-1 en H2O) en forma de un sólido higroscópico. 1H RMN (D2O) 51.31-1.20 (m t, J = 7.2 Hz, 5H, CH2 + CH3), 1.56-1.47 (m, 2H, CH2), 1.67 (s, 12H, 4 CH3), 1.79-1.74 (m, 2H, CH2), 2.11 (t, J = 6.9 Hz, 2H, CH2), 2.37 (t, J = 6.9 Hz, 2H, CH2), 2.60 (t, J = 6.3 Hz, 2H, CH2), 2.67 (t, J = 6.9 Hz, 2H, CH2), 3.27 (t, J = 6.1 Hz, 2H, CH2), 4.10-4.00 (m, 4H, 2 CH2), 6.29 (dd, J = 13.1, 8.1 Hz, 2H, 2 =CH), 7.29 (dd, 2H, J = 8.4, 6.1 Hz, 2 =CH), 7.75-7.71 (m, 2H, 2 =CH), 7.78 (s, 2H, =CH), 8.42 (t, J =12.8 Hz, 1H, =CH). Masa (-ve electroaspersión) calculada para C36H47K3O9S4793.22, encontrada 792 (M-H), 396 [M/2].

Una mezcla del enlazante de Cy3 disulfuro (2.5 ^mol), carbonato de disuccinimidilo (0.96 mg, 3.75 ^mol) y DMAP (0.46 mg, 3.75 ^mol) fue disuelta en DMF seco (0.5 ml) y agitada a temperatura ambiente durante 10 minutos. La reacción fue monitorizada por TLC (MeOH:CH2Cl23:7) hasta que el enlazante colorante fue consumido. Luego se agregaron una solución de (5) (7.5 ^mol) y n-Bu3N (30 ^l, 125 ^mol) en DMF (0.2 ml) a la mezcla de reacción y se agitó a temperatura ambiente durante 1 hora. La TLC (MeOH:CH2Cl24:6) mostró consumo completo del éster activado y apareció una mancha de color rojo oscuro sobre la línea base. La reacción fue detenida con regulador de TEAB (0.1M, 10 ml) y cargada sobre una columna DEAE Sephadex (2 x 5 cm). La columna fue eluida primero con regulador de TEAB 0.1 M (100 ml) para lavar los residuos orgánicos y luego regulador TEAB 1 M (100 ml). El análogo de trifosfato deseado (6) fue eluido con regulador TEAB 1 M. Las fracciones que contenían el producto fueron combinadas, evaporadas y purificadas por HPLC. Condiciones para HPLC: tr(6) : 16.1 min (Zorbax C18 preparativa columna, gradiente: 2% a 55% B in 30 min, regulador A 0.1M TEAB, regulador B MeCN). El producto fue aislado en forma de un sólido de color rojo oscuro (1.35 ^mol, 54%, e55o = 150000). 1H RMN (D2O) S 1.17-1.28 (m, 6H 3 x CH2), 1.41-1.48 (m, 3 H, CH3), 1.64 (s, 12H, 4 x CH3), 1.68-1.71 (m, 2H, CH2), 2.07-2.10 (m, 3H, H-2', CH2), 2.31-2.35 (m, 1H, H-2'), 2.50-2.54 (m, 2H, CH2), 2.65 (t, J = 5.9 Hz, 2H, CH2), 2.76 (t, J = 7.0 Hz, 2H, CH2), 3.26-3.31 (m, 2H, CH2), 3.88-3.91 (m, 2H CH2), 3.94-4.06 (m, 3H, CH2N, H-5'), 4.16 (br s, 1H, H-4'), 4.42-4.43 (m, 1H, H-3'), 4.72-4.78 (m, 2H, CH2N3), 6.24 (dd, J = 5.8, 8.2 Hz, H-1'), 6.25 (dd, J = 3.5, 8.5 Hz, 2H, HAr), 7.24, 7.25 (2d, J = 14.8 Hz, 2 x =CH), 7.69-7.86 (m, 4H, HAr, H-6), 8.42 (t, J = 13.4 Hz, =CH). 31P RMN (D2O) S -4.85 (m, 1P, PY), -9.86 (m, 1P, Pa), -20.40 (m, 1P, Pp). Masa (-ve electroaspersión) calculada para C49H64N9O22P3S4 1351.23, encontrada 1372 (M-2H+Na), 1270 [M-80], 1190 [M-160].

5-[3-(2,2,2-Trifluoroacetamido)-prop-1-inil]-2'-desoxicitidina (7).
A una solución de 5-yodo-2'-desoxicitidina (10 g, 28.32 mmol) en DMF (200 ml) en un matraz de fondo redondo protegido de la luz bajo atmósfera de argón, se agregó CuI (1.08 g, 5.67 mmol), trietilamina (7.80 ml, 55.60 mmol), 2,2,2-trifluoroN-prop-2-inil-acetamida (12.8 g, 84.76 mmol) y al final Pd(PPh)3)4 (3.27 g, 2.83 mmol). Después de 18 horas a temperatura ambiente, se agregó bicarbonato Dowex (20 mg) y la mezcla fue agitada durante 1 hora adicional. La filtración y evaporación de los volátiles bajo presión reducida dio un residuo que fue purificado por cromatografía instantánea sobre sílica gel (CH2O 2 , CH2Ch:EtOAc 1:1, EtOAc:MeOH 9:1) .El producto esperado (7) fue obtenido en forma de un sólido color beige en rendimiento cuantitativo. 1H RMN (D2O) 82.24-2.17 (m, 1H, H-2'), 2.41-2.37 (m, 1H, H-2'), 3.68 (dd, J = 12.5, 5.0 Hz, 1H, H-5'), 3.77 (dd, J = 12.5, 3.2 Hz, 1H, H-5'), 3.99 (m, 1H, H-4'), 4.27 (s, 2H, CH2N), 4.34 (m, 1H, H-3'), 6.11 (t, J = 6.3 Hz, 1H, H-1'), 8.1 (br s, 1H, NH) ; MS (ES) : m/z (%) (M-H) 375 (100).

5'-O-(tert-Butildimetilsilil)-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxicitidina (8).
A una solución del material de partida (7) (1.0 g, 2.66 mmol) e imidazol (200 mg, 2.93 mmol) en DMF (3.0 ml) a 0 °C, se agregó lentamente TBDMSC1 (442 mg, 2.93 mmol) en cuatro porciones durante 1 hora. Después de 2 horas, los volátiles fueron evaporados bajo presión reducida y el residuo fue adsorbido sobre sílica gel y purificado por cromatografía instantánea (EtOAc, EtOAc:MeOH 9.5:0.5). El producto esperado (8) fue aislado en forma de un sólido cristalino (826 mg, 64%). 1H RMN (d6 DMSO) 80.00 (s, 1H, CH3); 0.01 (s, 1H, CH3), 0.79 (s, 9 H, tBu), 1.87 1.80 (m, 1H, H-2'), 2.12 (ddd, J = 13.0, 5.8 and 3.0 Hz, 1H, H-2'), 3.65 (dd, J = 11.5, 2.9 Hz, 1H, H-5'), 3.74 (dd, J = 11.5, 2.5 Hz, 1H, H-5'), 3.81-3.80 (m, 1H, H4'), 4.10-4.09 (m, 1H, H-3'), 4.17 (d, 2H, J = 5.1 Hz, NCH2), 5.19 (d, 1H, J = 4.0 Hz, 3'-OH), 6.04 (t, J = 6.6 Hz, 1H, H1'), 6.83 (br s, 1H, NHH), 7.78 (br s, 1H, NHH), 7.90 (s, 1H, H-6), 9.86 (t, J = 5.1 Hz, 1H, -H2CNH) ; MS (ES) : m/z (%) (MH)+ 491 (40%).
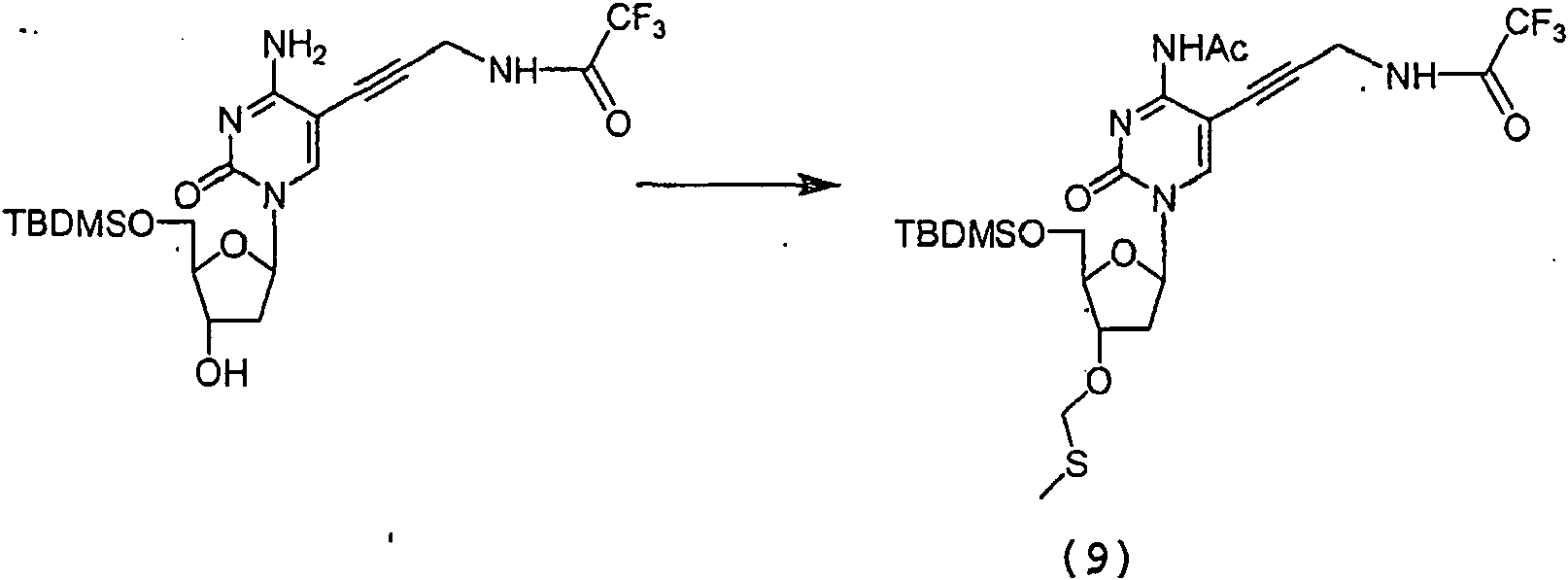
4-N-Acetil-5'-O-(tert-butildimetilsilil)-3'-O-(metiltiolmetil)-5-[3-(2,2,2-trifluoroacetamida)-prop-1-inM]2’-desoxicitidina (9).
A una solución del material de partida (8) (825 mg, 1.68 mmol) en DMSO (6.3 ml) y atmósfera de N2, se agregó lentamente ácido acético (AcOH) (1.3 ml, 23.60 mmol) seguido por anhídrido acético (Ac2O) (4.8 ml, 50.50 mmol). La solución fue agitada a temperatura ambiente durante 18 horas y detenida a 0°C mediante la adición de NaHCO3 saturado (20 ml). El producto fue extraído con EtOAc (3 x 30 ml), los extractos orgánicos se combinaron, secaron (MgSO4), filtraron, y se evaporaron los volátiles. El residuo crudo fue purificado por cromatografía instantánea sobre sílica gel (EtOAc: éter de petróleo 1:1) para dar el producto esperado en forma de un aceite incoloro (9) (573 mg, 62%). 1H RMN (da DMSO) 50.00 (s, 6H, 2 x CH3), 0.78 (s, 9H, tBu), 2.01 (s, 3H, SCH3 ), 2.19-1.97 (m, 2H, 2 x H2'), 2.25 (s, 3H, COCH3), 3.67 (dd, 1H, J = 11.5 Hz, H-5'), 3.78 (dd, 1H, J = 11.5, 3.3 Hz, H-5'), 4.06-4.05 (m, 1H, H-4'), 4.17 (d, 2H, J = 5.1 Hz, N-CH2), 4.30-4.28 (m, 1H, H-3'), 4.63 (s, 2H; CH2-S) , 5.94 (t, 1H, J = 6.5 Hz, H-1'), 8.17 (s, 1H, H-6), 9.32 (s, 1H, NHCO), 9.91 (t, 1H, J = 5.4 Hz, NHCH2) ; MS (ES) : m/z (%) (MH)+ 593.

4-N-Acet¡l-3’-O-(az¡domet¡l)-5’-O-(tert-but¡ld¡met¡ls¡l¡l)-5-[3-(2,2,2-trifluoroacetam¡da)-prop-1-¡n¡l]-2’-desox¡c¡t¡d¡na (10).
A una solución de material de partida (9) (470 mg, 0.85 mmol) en diclorometano (DCM) (8 ml) bajo atmósfera de N2 y enfriada a 0°C, se agregó ciclohexeno (430 |jl, 4.27 mmol) seguido por SO2Cl2 (1 M en DCM, 1.0 ml, 1.02 mmol). La solución fue agitada durante 30 minutos a 0°C, y los volátiles fueron evaporados. El residuo se disolvió inmediatamente en DMF (8 ml) agitado bajo N2 y se agregó lentamente azida de sodio (275 mg, 4.27 mmol). Después de 18 horas, el producto crudo fue evaporado hasta sequedad, disuelto en EtOAc (30 ml) y lavado con Na2CO3 (3 x 5 ml). Las capas orgánicas combinadas fueron mantenidas separadamente. Una segunda extracción del producto a partir de la capa acuosa fue llevada a cabo con DCM (3 x 10 ml). Todas las capas orgánicas combinadas fueron secadas (MgSO4), filtradas y los volátiles evaporados bajo presión reducida para dar aceite identificado como el producto esperado (10) (471 mg, 94 % de rendimiento). Este fue usado sin purificación posterior alguna. 1H RMN (de DMSO) 50.11 (s, 3H, CH3), 0.11 (s, 3H, CH3 ), 0.88 (s, 9H, tBu), 2.16-2.25 (m, 1H, H-2’), 2.35 (s, 3H, COCH3 ), 2.47-2.58 (m, 1H, H-2’), 3.79 (dd, J = 11.6, 3.2 Hz, 1H, H-5’), 3.90 (dd, J = 11.6, 3.0 Hz, 1H, H-5’), 4.17 4.19 (m, 1H, H-4’), 4.28 (s, 2H, NCH2), 4.32-4.35 (m, 1H, H-3’), 4.89 (dd, J = 14.4, 6.0 Hz, 2H, CH2-N3), 6.05 (t, J = 6.4 Hz, 1H, H-1’), 8.25 (s, 1H, H-6), 9.46 (br s, 1H, NHH), 10.01 (br s, 1H, NHH).

4-N-Acetil-3’-O-(azidometil)-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2’-desoxicitidina y 3’-O-(Azidometil)-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2’-desoxicitidina (11).
A una solución del material de partida (11) (440 mg, 0.75 mmol) en THF (20 ml) a 0°C y atmósfera de N2 , se agregó TBAF en THF 1.0 M (0.82 ml, 0.82 mmol). Después de 1.5 horas, los volátiles fueron evaporados bajo presión reducida y el residuo fue purificado por cromatografía instantánea sobre sílica gel (EtOAc:éter de petróleo 8:2 to EtOAc 100 % a EtOAc:MeOH 8:2). Se aislaron dos compuestos y se identificaron como se describió más arriba. El primero eluyó 4-N-acetilo (11), (53 mg, 15 %) y el segundo 4 -NH2 (12) (271 mg, 84 %). Compuesto 4-N-acetilo (11) :
1H RMN (da DMSO) 51.98 (s, 3H, CH3CO), 2.14-2.20 (m, 2H, HH-2'), 3.48-3.55 (m, 1H, H-5'), 3.57-3.63 (m, 1H, H-5'), 3.96-4.00 (m, 1H, H-4'), 4.19 (d, J = 5.3 Hz, 2H, CH2-NH), 4.23-4.28 (m, 1H, H3'), 4.77 (s, 2H, CH2-N3), 5.2 (t, 1H, J = 5.1 Hz, 5'-OH), 5.95 (t, J = 6.2 Hz, 1H, H-1'), 8.43 (s, 1H, H-6), 9.34 (s, 1H, CONH), 9.95 (t, J = 5.3 Hz, 1H, NHCH2).
Compuesto 4 -NH2 (12): 1H RMN (d6 DMSO) 51.98-2.07(2H, CHH-2'), 3.50-3.63 (m, 2H, CHH-5'), 3.96-4.00 (m, 1H, H-4'), 4.09 (d, J = 5.3 Hz, 2H, CH2-NH), 4.24-4.28 (m, 1H, H-3'), 4.76 (s, 2H, CH2-N3) 5.13 (t, J = 5.3 Hz, 1H, 5'-OH), 5.91 (br s, 1H, NHH), 6.11 (t, J = 6.4 Hz, 1H, H-1'), 8.20 (t, J = 5.3 Hz, 1H, NCH2), 8.45 (s, 1H, H-6), 11.04 (br s, 1H, NHH).

El material de partida (8) (10 g, 20.43 mmol) fue sometido a la formación de azeótropos en piridina seca (2 x 100 ml) y luego disueltos en piridina seca (160 ml) bajo atmósfera de N2. Se agregó gota a gota clorotrimetilsilano (10 ml, 79.07 mmol) a la solución y se agitó durante 2 horas a temperatura ambiente. Se agregó entonces cloruro de benzoilo (2.6 ml, 22.40 mmol) a la solución y se agitó durante 1 hora adicional. La mezcla de reacción fue enfriada a 0°C, se agregó lentamente agua destilada (50 ml) a la solución y se agitó durante 30 minutos. La piridina y el agua fueron evaporadas de la mezcla bajo alto vacío para producir un gel color marrón que fue porcionado entre 100 ml de solución de NaHCO3 acuosa saturada (100 ml) y DCM. La fase orgánica fue separada y la fase acuosa fue extraída con (2 x 100 ml) adicionales de DCM. Las capas orgánicas fueron combinadas, secadas (MgSO4), filtradas y los volátiles evaporados bajo presión reducida. El aceite marrón resultante fue purificado por cromatografía instantánea sobre sílica gel (DCM:MeOH 99:1 a 95:5) para producir un sólido cristalino amarillo claro (13) (8.92 g, 74%). 1H RMN (d6 DMSO) : 50.00 (s, 6H, CH3), 0.78 (s, 9H, tBu), 1.94 (m, 1H, H-2'), 2.27 (m, 1H, H-2'), 3.64 (d, 1H, J = 11.6 Hz, H-5'), 3.75 (d, 1H, J = 11.6 Hz, H-5'), 3.91 (m, 1H, H-4'), 4.09 (br m, 3H, CH2NH, H-3'), 5.24 (s, 1H, 3'-OH), 6.00 (m, 1H, H-1'), 7.39 (m, 2H, Ph), 7.52 (m, 2H, Ph), 7.86 (m, 1H, Ph), 8.0 (s, 1H, H-6), 9.79 (t, 1H, J = 5.4 Hz, NHCH2), 12.67 (br s, 1H, Nh ). Masa (+ve electroaspersión) calculada para C27H33F3N4O6Si 594.67, encontrada 595.

4-N-Benzoil-5'-O-(tert-butildimetilsilil)-3'-O-metiltiometil-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]2'-desoxicitidina
(14).
El material de partida (13) (2.85 g, 4.79 mmol) fue disuelto en DMSO seco (40 ml) bajo atmósfera de N2. Se agregaron ácido acético (2.7 ml, 47.9 mmol) y anhídrido acético (14.4 ml, 143.7 mmol) secuencialmente y lentamente al material de partida, lo cual fue luego agitado durante 18 horas a temperatura ambiente. Se agregó cuidadosamente solución saturada de NaHCO3 (150 ml) a la mezcla de reacción. La capa acuosa fue extraída con EtOAc (3 x 150 ml). Las capas orgánicas fueron combinadas, secadas (MgSO4), filtradas y evaporadas para producir un líquido de color naranja que fue sometido a separación de azeótropos subsecuentemente con tolueno (4 x 150 ml) hasta que el material solidificó. El residuo crudo fue purificado sobre sílica gel (éter de petróleo:EtOAc 3:1 to 2:1) para producir un sólido cristalino amarillo (14) (1.58 g, 50%). 1H RMN (d6 DMSO) : 50.00 (s, 6H, CH3), 0.78 (s, 9H, tBu), 1.99 (s, 3H, CH3), 2.09 (m, 1H, H-2'), 2.28 (m, 1H, H-2'), 3.66 (d, 1H, J = 11.5, 2.9 Hz, H-5'), 3.74 (dd, 1H, J = 11.3, 2.9 Hz, H-5'), 3.99 (m, 1H, H-4'), 4.09 (m, 1H, CH2NH), 4.29 (m, 1H, H-3'), 4.61 (s, 2H, CH2S), 6.00 (m, 1H, H-1'), 7.37 (m, 2H, Ph), 7.50 (m, 2H, Ph), 7.80 (d, 1H, J = 7.55 Hz, HAr), 7.97 (s, 1H, H-6), 9.79 (br t, 1H, NHCH2), 12.64
(br s, 1H, NH). Masa (-ve electroaspersión) calculada para C29H37F3N4O6SSÍ 654.79, encontrada 653.2.

4-N-Benzoil-5'-O-(tert-butildimetilsilil)-3'-O-azidometil-5-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxicitidina (15).
El material de partida (14) (1.65 g, 2.99 mmol) fue disuelto en DCM (18 ml) y enfriado a 0°C. Se agregó ciclohexeno (1.5 ml, 14.95 mmol) y SO2O 2 (0.72 ml, 8.97 mmol) y se agitó durante 1 hora en baño de hielo. La TLC indicó que el material de partida estaba todavía presente con lo cual se agregó una alícuota adicional de SO2O 2 (0.24 ml) y la mezcla se agitó durante 1 hora a 0°C. Los volátiles fueron eliminados por evaporación para producir un sólido de color marrón claro que fue redisuelto en 18 ml de DMF seco (18 ml) bajo N2. Se agregó entonces azida de sodio (0.97 g, 14.95 mmol) a la solución y se agitó durante 2.5 horas a temperatura ambiente. La mezcla de reacción fue pasada a través de un lecho de sílica y se eluyó con EtOAc y los volátiles fueron eliminados por evaporación a alto vacío. El gel marrón resultante fue purificado por cromatografía instantánea (éter de petróleo:EtOAc 4:1 to 2:1) para producir el producto deseado en forma de un sólido cristalino blanco (15) (0.9 g, 55%). 1H RMN (d6 DMSO); 6 0.00 (s, 6 H, CH3), 0.78 (s, 9H, tBu), 2.16 (m, 1H, H-2'), 2.22 (m, 1H, H-2'), 3.70 (d, 1H, J = 11.5 Hz, H-5'), 3.75 (d, 1H, J = 11.3 Hz, H-5'), 4.01 (m, 1H, H-4'), 4.10 (m, 1H, CH2NH), 4.23 (m, 1H, H-3'), 4.76 (s, 2H, CH2S), 5.99 (m, 1H, H-1'), 7.37 (m, 2H, Ph), 7.50 (m, 2H, Ph), 7.81 (d, 1H, J = 7.4 Hz, Ph), 7.95 (s, 1H, H-6 ), 9.78 (br s, 1H, NHCH2), 12.64 (br s, 1H, NH). Masa (-ve electroaspersión) calculada para C28H34F3NyO6Si 649.71, encontrada 648.2

El material de partida (15) (140 mg, 0.22 mmol) fue disuelto en THF (7.5 ml). Se agregó lentamente TBAF (solución 1 Molar en THF, 0.25 ml) y se agitó durante 2 horas a temperatura ambiente. El material volátil fue eliminado bajo presión reducida para producir un gel marrón y fue purificado por cromatografía instantánea (EtOAc:DCM 7:3) para dar el producto deseado (16) en forma de un sólido cristalino de color claro (0.9 g, 76%). 1H r Mn (d6 DMSO) : 6 2.16 (m, 1H, H-2'), 2.22 (m, 1H, H-2'), 3.70 (d, 1H, J = 11.5 Hz, H-5'), 3.75 (d, 1H, J = 11.3 Hz, H-5'), 4.01 (m, 1H, H-4'), 4.10 (m, 1H, CH2NH), 4.23 (m, 1H, H-3'), 4.76 (s, 2H, CH2S), 5.32 (s, 1H, 5' OH), 5.99 (m, 1H, H-1'), 7.37 (m, 2H, Ph), 7.50 (m, 2H, Ph), 7.81 (d, 1H, J = 7.35 Hz, Ph), 7.95 (s, 1H, H-6 ), 9.78 (br s, 1H, NHCH2), 12.64 (br s, 1H, NH). Masa (-ve electroaspersión) calculada para C22H20F3N7O6535.44, encontrada 534.

A una solución de (11) y (12) (290 mg, 0.67 mmol) y esponja de protones (175 mg, 0.82 mmol) (ambos previamente secados bajo P2O5 durante al menos 24 horas) en PO(OMe)3 (600 pl), a 0°C bajo atmósfera de argón, se agregó lentamente POCl3 (recién destilado) (82 pl, 0.88 mmol). La solución fue agitada vigorosamente durante 3 horas a 0°C y luego detenida mediante la adición de difosfato de tetra-tributilamonio (0.5 M) en DMF (5.2 ml, 2.60 mmol), seguido por nBu3N (1.23 ml, 5.20 mmol) y bicarbonato de trietilamonio (TEAB) 0.1 M (20 ml). Después de 1 hora a temperatura ambiente se agregó solución acuosa de amoníaco (p 0.88, 20 ml) a la mezcla. La solución se agitó a temperatura ambiente durante 15 horas, los volátiles fueron evaporados bajo presión reducida y el residuo fue purificado por MPLC con un gradiente de TEAB de 0.05 M a 0.7 M. El trifosfato esperado fue eluido de la columna a aproximadamente 0.60 M TEAB. Se realizó una segunda purificación por HPLC en una columna Zorbax SB-C18 (21.2 mm i.d. x 25 cm) eluida con TEAB 0.1 M (bomba A) y CH3CN al 30% en TEAB 0.1M (bomba B) utilizando un gradiente como sigue: 0-5 min 5% B, 0.2 ml; 5-25 min 80% B, 0.8 ml; 25-27 min 95 %B, 0.8 ml; 27-30 min 95 %B, 0.8 ml; 30-32 min 5 %B, 0.8 ml; 32-35 min 95 %B, 0,2 ml, generando el producto descrito más arriba con un rt(17) : 20.8 (14.5 pmoles, 2.5% de rendimiento); 31P RMN (D2O, 162 MHz) 5 -5.59 (d, J = 20.1 Hz, Px), -10.25 (d, J = 19.3 Hz, 1P, Pa), -20.96 (t, J = 19.5 Hz, 1P, Pp) ; 1H RMN (D2O) 52.47-2.54 (m, 1H, H-2’), 2.20-2.27 (m, 1H, H-2’), 3.88 (s, 2H, CH2 N), 4.04-4.12 (m, 1H, HH-5’), 4.16-4.22 (m, 1H, HH-5’), 4.24-4.30 (m, 1H, H-4’), 4.44-4.48 (m, 1H, H-3’), 6.13 (t, J = 6.3 Hz, 1H, H1’), 8.35 (s, 1H, H-6); MS (ES): m/z (%) (M-H) 574 (73%), 494 (100 %).20

Enlazante de disulfuro Alexa488.
El Alexa flúor 488-NHS disponible comercialmente (35 mg, 54 pmol) fue disuelto en DMF (700 pL) y, para asegurar la activación completa, se agregaron secuencialmente 4-DMAP (7 mg, 59 pmol) y carbonato de N,N’-disuccinimidilo (15 mg, 59 pmol). Después de 15 minutos en activación completa, se agregó una solución del disulfuro de inicio (32.0 mg, 108 pmol) en DMF (300 pL) que contenía diisopropiletilamina (4 pL) sobre la solución del colorante
activado. Se hizo adición posterior de diisopropiletilamina (20 pL) a la mezcla final, se sometió a ultrasonido durante 5 minutos y se hizo reaccionar durante 18 horas a temperatura ambiente en la oscuridad. Los volátiles fueron evaporados bajo presión reducida y el residuo crudo fue purificado primero pasándolo a través de una columna corta de resina de intercambio iónico Sephadex -DEAE A-25 (40-120 p), eluyendo primero con TEAB 0.1 M (25 ml) luego TEAB 1.0 M (75 ml). Este último contenía los dos compuestos finales y fue concentrado y el residuo fue purificado por HPLC en una columna Zorbax SB-C18 (21.2 mm i.d. x 25 cm) eluido con TEAB 0.1M (bomba A) y CH3CN (bomba B) utilizando un gradiente como sigue: 0-2 min 2% B, $.2 ml; 2-4 min 2% B, $.8 ml; 4-15 min 23 %B, $.8 ml; 15-24 min 23 %B, $.8 ml; 24-26 min 95 %B, $.8 ml; 26-28 min 95 %B, $.8 ml, 28-30 min 2 %B, $.8 ml, 30-33 min 2 %B, $.2 ml generando ambos compuestos detallados más arriba con tr: 19.0 (regioisómero izquierdo) y tr: 19.5 (regioisómero derecho). Ambos regioisómeros fueron pasados respectivamente a través de una columna con resina de intercambio iónico Dowex, generando respectivamente 16.2 pmol y 10.0 pmol, 62% de rendimiento total (con base en Alexa flúor 488-NHS disponible comercialmente de 76% de pureza); £493 = 71,000 cm-1 M-1 en H2O. 1H RMN (D2O) (regioisómero izquierdo) 62.51 (t, J = 6.8 Hz, 2H, CH2), 2.66 (t, J = 6.8 Hz, 2H, CH2), 2.71 (t, J = 5.8 Hz, 2H, CH2), 3.43 (t, J = 5.8 Hz, 2H, CH2), 6.64 (d, J = 9.2 Hz, 2H, HAr), 6.77 (d, J = 9.2 Hz, 2H, HAr), 7.46 (s, 1H, HAr), 7.90 (dd, J = 8.1 and 1.5 Hz, 1H, HAr), 8.20 (d, J = 8.1 Hz, 1H, HAr). 1H RMN (D2O) (regioisómero derecho) 62.67 (t, J = 6.8 Hz, 2H, CH2), 2.82 (t, J = 6.8 Hz, 2H, CH2), 2.93 (t, J = 6. 1 Hz, 2H, CH2), 3.68 (t, J = 6.1 Hz, 2H, CH2), 6.72 (d, J = 9.3 Hz, 2H, HAr), 6.90 (d, J = 9.3 Hz, 2H, HAr), 7.32 (d, J = 7.9 Hz, 1H, HAr), 8.03 (dd, J = 7.9, 1.7 Hz, 1H, HAr), 8.50 (d, J = 1.8 Hz, 1H, HAr) Masa (-ve electroaspersión) calculada para C26H23N3O12S4 697.02, encontrada 692 (M-H), 347 [M/2].

A una solución del enlazante de disulfuro Alexa flúor 488 (3.4 pmol, 2.37mg) en DMF (200 pL) fue agregado 4-DMAP (0.75 mg, 5.1 pmol) y carbonato de N,N-disuccinimidilo (1.70 mg, 5.1 pmol). La mezcla fue agitada durante 15 hasta activación completa del ácido, luego fue agregada en la solución del nucleótido (17) (3.45 mg, 6.0 pmol) en DMF (0.3 ml) que contenía nBu3N (40 pL) a 0°C. La mezcla fue sometida a sonicación durante 3 minutos y luego se agitó continuamente durante 16 horas en la ausencia de luz. Los volátiles fueron evaporados bajo presión reducida y el residuo fue purificado primeramente por filtración a través de una columna corta de resina de intercambio iónico Sephadex -DEAE A-25, eluida primero con TEAB 0.1 M (50 ml) eliminando el colorante-enlazante sin reaccionar, luego TEAB 1.0 M (100 ml) para recolectar el producto esperado (18). Después de concentración el residuo fue purificado por HPLC en una columna Zorbax SB-C18 (21.2 mm i.d. x 25 cm) eluida con TEAB 0.1M (bomba A) y CH3CN (bomba B) usando un gradiente como sigue: 0-2 min 2% B, $.2 ml; 2-4 min 2% B, $.8 ml; 4-15 min 23 %B, $.8 ml; 15-24 min 23 %B, $.8 ml; 24-26 min 95 %B, $.8 ml; 26-28 min 95 %B, $.8 ml, 28-30 min 2 %B, $.8 ml, 30 33 min 2 %B, $.2 ml generando el producto detallado más arriba con un rt(18) : 19.8 (0.26 pmols, 12% de rendimiento con base en mediciones por UV); Amax = 493 nm, e 71,000 cm-1 M-1 en H2O); 31P RMN (D2O, 162 MHz) 6 -5.06 (d, J = 20.6 Hz, 1P, Px), -10.25 (d, J = 19.3 Hz, 1P, Pa), -21.21 (t, J = 19.5 Hz, 1P, Pp) ; 1H RMN (D2O) 62.09 2.17 (m, 1H, HH-2’), 2.43-2.50 (m, 1H, HH-2’), 2.61 (t, J = 6.8 Hz, 2H, H2CS), 2.83 (2H, S-CH2), 3.68 (t, J = 6.0 Hz,
2H, ArCONCH2), 4.06 (s, 2H, CH2N), 4.08-4.17 (m, 4H, HH-5'), 4.25-4.29 (m, 1H, H-4'), 4.46-4.50 (m, 1H, H-3'), 6.09 (t, J = 6.4 Hz, 1H, H-1'), 6.88 (d, J = 9.1 Hz, 1H, HAr), 6.89 (d, J = 9.3 Hz, 1H, HAr), 7.15 (d, J = 9.3 Hz, 1H, HAr), 7.17 (d, J = 9.1 Hz, 1H, HAr), 7.64 (br s, 1H, HAr), 8.00-7.94 (m, 2H, HAr), 8.04 (s, 1H, H-6); MS (ES) : m/z (%) (M-H) 1253 (46%), (M-H+ Na)' 1275 (100 %)

7-Desaza-[3-(2,2,2-trifluoroacetamido)-prop-1 -inil]-2'-desoxiguanosina (1.9).
Bajo N2 , una suspensión de 7-desaza-7-yodo-guanosina (2 g, 2.75 mmol), Pd(PPh3)4 (582 mg, 0.55 mmol), CuI (210 mg, 1.1 mmol), Et3N (1.52 ml, 11 mmol) y la propagilamina (2.5 g, 16.5 mmol) en DMF (40 ml) fue agitada a temperatura ambiente durante 15 horas y bajo N2. La reacción fue protegida de la luz con una lámina de aluminio. Después de que la TLC indicó el consumo completo del material de partida, la mezcla de reacción fue concentrada. El residuo fue diluido con MeOH (20 ml) y tratado con Dowex-HCO3-. La mezcla fue agitada durante 30 minutos y filtrada. La solución fue concentrada y purificada por cromatografía en sílica gel (éter de petróleo:EtOAc 50:50 to éter de petróleo: EtOAc:MeOH 40: 40: 20), dando (19) en forma de un polvo amarillo (2.1 g, 92%). 1H RMN (d6 DMSO) 5 2.07-2.11 (m, 1H, H-2'), 2.31-2.33 (m, 1H, H-2'), 3.49-3.53 (m, 2H, H-5'), 3.77 (br s, 1H, H-4'), 4.25 (d, J = 4.3 Hz, 2H, ECCH2) , 4.30 (br s, 1H, H-3'), 4.95 (t, J = 5.2 Hz, 1H, 5'OH), 5.25 (d, J = 3.4 Hz, 1H, 3'-OH), 6.27-6.31 (m, 1H, H-1'), 6.37 (s, 2H, NH2), 7.31 (s, 1H, H-8), 10.10 (br s, 1H, NHCOCF3), 10.55 (s, 1H, NH). Masa (-ve electroaspersión) calculada para C16H16F3N5O5415, encontrada 414.20*53

5'-O-(tert-Butildifenil)-7-desaza-7-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxiguanosina (20).
Una solución de (19) (2.4 g, 5.8 mmol) en piridina (50 ml) fue tratada con cloruro de tert-butildifenilsililo (TBDPSCl) (1.65 ml, 6.3 mmol) gota a gota a 0°C. La mezcla de reacción fue calentada entonces hasta temperatura ambiente. Después de 4 horas, se agregó otra porción de TBDPSCl (260 ^L, 1 mmol). La reacción fue monitorizada por TLC, hasta el consumo completo del material de partida. La reacción fue detenida con MeOH (-5 ml) y evaporada hasta sequedad. El residuo fue disuelto en DCM y se agregó NaHCO3 acuoso saturado. La capa acuosa fue extraída tres veces con DCM. Los extractos orgánicos combinados fueron secados (MgSO4) y concentrados bajo vacío. La purificación por cromatografía sobre sílica (EtOAc to EtOAc:MeOH 85:15) dio (20) una espuma amarilla (3.1 g, 82%).
1H RMN (da DMSO) 5 1.07 (s, 9H, CH3), 2.19-2.23 (m, 1H, H-2'), 2.38-2.43 (m, 1H, H-2'), 3.73-3.93 (m, 2H, H-5'), 4.29 (d, J = 5.0 Hz, 2H, CH2N), 4.42-4.43 (m, 1H, H-3'), 5.41 (br s, 1H, OH), 6.37 (t, J = 6.5 Hz, H-1'), 6.45 (br s, 2H, NH2), 7.24-7.71 (m, 11H, H-8, HAr), 10.12 (t, J = 3.6 Hz, 1H, NH), 10.62 (s, 1H, H-3). Masa (+ve electroaspersión) calculada para C32H34F3N5O5Si 653, encontrada 654.

5’-O-(tert-Butildifenil)-7-desaza-3’-O-metiltiolmetil-7-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2’-desoxiguanosina (21).
Una solución de (20) (1.97 g, 3.0 mmol) en DMSO (15 ml) fue tratada con AC2O (8.5 ml, 90 mmol), y AcOH (2.4 ml, 42 mmol) y agitada a temperatura ambiente durante 15 horas, luego 2 horas a 40°C. La mezcla de reacción fue diluida con EtOAc (200 ml) y agitada con NaHCO3 acuoso saturado (200 ml) durante 1 hora. La capa acuosa fue lavada con EtOAc dos veces. La capa orgánica fue combinada, secada (MgSO4) y concentrada bajo vacío. La purificación por cromatografía sobre sílica (EtOAc:Hexano 1:1 to EtOAc:Hexano:MeOH 10:10:1) dio (21) en forma de una espuma amarilla (1.3 g, 60%). 1H RMN (CDCb) 5 1.04 (s, 9H, CH3), 2.08 (s, 3H, SCH3), 2.19-2.35 (m, 2H, H-2), 3.67-3.71 (m, 2H, H-5’), 3.97-3.99 (m, 2H, H-4’, H-3’), 4.23 (br s, 2H, CH2N), 4.58 (s, 2H, CH2S), 6.31 (dd, J = 5.7, 7.9 Hz, H-1’) 7.19-7.62 (m, 11H, H8, HAr). Masa (+ve electroaspersión) calculada para C34H38F3N5O5SSi 713, encontrada: 714.

A una solución de (21) (1.3 mg, 1.8 mmol), ciclohexeno (0.91 ml, 9 mmol) en CH2Cl2 (10 ml) a 4°C, se agregó cloruro de sulfurilo (1M en C ^C h) (1.1 ml, 1.1 mmol) gota a gota bajo N2. Después de 30 minutos, la TLC indicó el consumo completo del nucleósido (22). Después de evaporar para eliminar el solvente, el residuo fue sometido entonces a alto vacío durante 20 minutos, y luego tratado con NaN3 (585 mmol, 9 mmol) y DMF (10 ml). La suspensión resultantefue agitada bajo temperatura ambiente durante 2 horas. La extracción con C^C^/NaCl (10%) dio una goma amarilla, la cual fue tratada con TBAF en THF (1 M, 3 ml) y THF (3 ml) a temperatura ambiente durante 20 minutos. la evaporación para eliminar solventes, la extracción con EtOAc/NaHCO3 acuoso saturado, seguida por purificación por cromatografía sobre sílica (EtOAc to EtOAc:MeOH 9:1) dio (22) en forma de una espuma amarilla (420 mg, 50%). 1H RMN (d6 DMSO) : 52.36-2.42 (m, 1H, H-2’), 2.49-2.55 (m, 1H, H-2’), 3.57-3.59 (m, 2H, H-5’), 3.97-4.00 (m, 1H, H-4’), 4.29 (m, 2H, CH2N), 4.46-4.48 (m, 1H, H-3’), 4.92-4.96 (m, 2H, CH2N3), 5.14 (t, J = 5.4 Hz, 1H, 5’-OH), 5.96-6.00 (dd, J = 5.7, 8.7 Hz, 1H, H-1’), 6.46 (br s, 2H, NH2), 7.39 (s, 1H, H-6), 10.14 (s, 1H, NH), 10.63 (s, 1H, H-3).

5’-O-nucleósido trifosfato
(23).
Se disolvió difosfato de tetrasodio decahidrato (1.5 g, 3.4 mmol) en agua (34 ml) y la solución fue aplicada a una columna de Dowex 50 en la forma H+. La columna fue lavada con agua. El eluyente goteo directamente sobre una solución enfriada (baño de hielo) y agitada de tri-n-butil amina (1.6 ml, 6.8 mmol) en EtOH (14 ml). La columna fue lavada hasta que el pH del eluyente se incrementó a 6. La solución en etanol acuoso fue evaporada hasta sequedad y luego coevaporada dos veces con etanol y dos veces con DMF anhidro. El residuo fue disuelto en DMF (6.7 ml). La solución de color amarillo pálido fue almacenada sobre tamices moleculares 4Á. El nucleósido (22) y la esponja de protones fueron secados sobre P2O5 bajo vacío durante la noche. Una solución de (22) (104 mg, 0.22 mmol) y esponja de protones (71 mg, 0.33 mmol) en trimetilfosfato (0.4 ml) fue agitada con tamices moleculares 4Á durante 1 horas. Se agregó POCh recién destilado (25 ^l, 0.26 mmol) y la solución fue agitada a 4°C durante 2 horas. La mezcla fue calentada lentamente a temperatura ambiente y se agregaron pirofosfato de bis (tri-n-butil amonio) (1.76 ml, 0.88 mmol) y tri-n-butil amina anhidra (0.42 ml, 1.76 mmol). Después de 5 minutos, la reacción fue detenida con TEAB 0.1 M (bicarbonato de trietilamonio) como regulador (15 ml) y se agitó durante 3 horas. El agua fue eliminada bajo presión reducida y el residuo resultante fue disuelto en amoníaco concentrado (p 0.88, 10 ml) y agitado a temperatura ambiente durante 16 horas. La mezcla de reacción fue evaporada entonces hasta sequedad. El residuo fue disuelto en agua y la solución aplicada a una columna DEAE-Sephadex A-25. Se llevó a cabo la MPLC con un gradiente lineal de 2 L de cada uno de TEAB 0.05 M y 1 M. El trifosfato fue eluido entre regulador 0.7 M y 0.8 M. Las fracciones que contenían el producto fueron combinadas y evaporadas hasta sequedad. El residuo fue disuelto en agua y purificado posteriormente por HPLC. tr(23) = 20.5 min (columna preparativa Zorbax C18, gradiente: 5% a 35% B in 30 min, regulador A 0.1M TEAB, regulador B MeCN). El producto fue aislado en forma de una espuma blanca (225 O.D., 29.6 mmol, 13.4%, £260 = 7,600). 1H RMN (D2O) S 2.43-2.5 (m, 2H, H2’), 3.85 (m, 2H, CH2N), 3.97 4.07 (m, 2H, H-5’), 4.25 (br s, 1H, H-4’), 4.57 (br s, 1H, H-3’), 4.74-4.78 (m, 2H, CH2N3), 6.26-6.29 (m, 1H, H-1’), 7.41 (s, 1H, H-8). 31P-RMN (D2O) S -8.6 (m, 1P, PY), -10.1 (d, J =19.4 Hz, 1P, Pa), -21.8 (t, J = 19.4 Hz, 1P, Pp). Masa (-ve electroaspersión) calculada para C15H21N8O13P3614, encontrada 613.

Una mezcla de Cy3 enlazado a disulfuro (2.5 ^mol), 1-(3-dimetilaminopropil)-3-etilcarbodiimida clorhidrato (EDC) (0.95 mg, 5 ^mol), 1-hidroxibenzotriazol (HOBt) (0.68 mg, 5 ^mol) y N-metil-morfolina (0.55 ^L, 5 ^mol) en DMF (0.9 ml) fue agitada a temperatura ambiente durante 1 hora. Se agregó una solución de (23) (44 O.D., 3.75 ^mol) en 0.1 ml de agua a la mezcla de reacción a 4°C, y se dejó a temperatura ambiente durante 3 horas. La reacción fue detenida con regulador de TEAB (0.1M, 10 ml) y cargado sobre una columna DEAE Sephadex (2 x 5 cm). La columna fue eluida primero con un regulador TEAB 0.1 M (100 ml) y luego regulador TEAB 1 M (100 ml). El producto de trifosfato deseado fue eluido con regulador de TEAB 1 M. Se concentro la fracción que contenía el producto y se aplicó HPLC. tr(24) = 23.8 min (columna preparativa Zorbax C18, gradiente: 5% a 55% B in 30 min, regulador A 0.1M TEAB, regulador B MeCN). El producto fue aislado en forma de una espuma roja (0.5 ^mol, 20%, £max = 150,000). 1H RMN (D2O) 51.17-1.71 (m, 20H, 4 x CH2 , 4 x CH3), 2.07-2.15 (m, 1H, H-2'), 2.21-2.30 (m, 1H, H-2'), 2.52-2.58 (m, 2H, CH2), 2.66-2.68 (m, 2H, CH2), 2.72-2.76 (m, 2H, CH2), 3.08-3.19 (m, 2H, CH2), 3.81-3.93 (m, 6H, CH2, H-5'), 4.08-4.16 (m, 1H, H-4'), 4.45-4.47 (m, 1H, H-3'), 4.70-4.79 (m, 2H, CH2N3), 6.05-6.08 (m, 2H, HAr), 6.15-6.18 (m, 1H, H-1'), 7.11 (s, 1H, H-8), 7.09-7.18 (m, 2H, CH), 7.63-7.72 (m, 4H, HAr), 8.27-8.29 (m, 1H, CH). 31P RMN (D2O) 5 -4.7 (m, 1P, Py),-9.8 (m, 1P, Pa) , -19.7 (m, 1P, Pp). Masa (-ve electroaspersión) calculada para C51H66N11O21P3S4 1 3 8 9.2 5 , encontrada 1388 (M-H), 694 [M-2H], 462 [M-3H].

7-Desaza-7-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxiadenosina (25).
A una suspensión de 7-desaza-7-yodo-2'-desoxiadenosina (1 g, 2.65 mmol) y CuI (100 mg, 0.53 mmol) en DMF seco (20 ml) se agregó trietilamina (740 pl, 5.3 mmol). Después de agitar durante 5 minutos se agregaron trifluoro-N-prop-2-inil-acetamida (1.2 g, 7.95 mmol) y Pd(PPh3)4 (308 mg, 0.26 mmol) a la mezcla y la reacción fue agitada a temperatura ambiente en la oscuridad durante 16 horas. Se agregaron MeOH (40 ml) y bicarbonato Dowex a la mezcla de reacción y se agitó durante 45 minutos. Se filtró la mezcla. El filtrado fue lavado con MeOH y el solvente fue eliminado bajo vacío. La mezcla cruda fue purificada por cromatografía sobre sílica (EtOAc to EtOAc: MeOH 95:20) para dar un polvo ligeramente amarillo (25) (1.0 g, 95 %). 1H RMN (da DMSO) 82.11-2.19 (m, 1H, H-2'), 2.40 2.46 (m, 1H, H-2'), 3.44-3.58 (m, 2H, H-5'), 3.80 (m, 1H, H-4'), 4.29 (m, 3H, H-3', CH2N), 5.07 (t, J = 5.5 Hz, 1H, OH), 5.26 (d, J = 4.0 Hz, 1H, OH), 6.45 (dd, J = 6.1, 8.1 Hz, 1H, H-1'), 7.74 (s, 1H, H-8), 8.09 (s, 1H, H-2), 10.09 (t, J = 5.3 Hz, 1H, NH).

5'-O-(tert-Butildifenilsilil)-7-desaza-7-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxiadenosina (26).
El nucleósido (25) (1.13 g, 2.82 mmol) fue coevaporado dos veces en piridina seca (2 x 10 ml) y disuelto en piridina seca (18 ml). A esta solución se agregó cloruro de t-butildifenilsililo (748 pl, 2.87 mmol) en pequeñas porciones a 0°C. La mezcla de reacción se dejó calentar entonces a temperatura ambiente y se dejó en agitación durante la noche. La reacción fue detenida con solución acuosa saturada de NaCl. Se agregó EtOAc (25 ml) a la mezcla de reacción y la capa acuosa fue extraída con EtOAc tres veces. Después de secar los extractos orgánicos combinados (MgSO4) el solvente fue eliminado bajo vacío. La purificación por cromatografía sobre sílica (DCM y luego EtOAc a EtOAc: MeOH 85:15) dieron (26) como un polvo ligeramente amarillo (1.76 g, 97 %). 1H Rm N (d6 DMSO) 81.03 (s, 9H, tBu), 2.25-2.32 (m, 1H, H-2'), 2.06-2.47 (m, 1H, H-2'), 3.71-3.90 (m, 2H, H-5'), 3.90-3.96 (m, 1H, H-4'), 4.32 (m, 2H, CH2N), 4.46 (m, 1H, H-3'), 5.42 (br s, 1H, OH), 6.53 (t, J = 6.7 Hz, 1H, H-1'), 7.38-7.64 (m, 11H, H-8 and HAr), 8.16 (s, 1H, H-2), 10.12 (t, J = 5.3 Hz, 1H, NH).

5'-O-(tert-Butildifenilsilil)-7-desaza-4-N,N-dimetilformadin-7-[3-(2,2,2-trifluoroacetamido)-prop-1inil]-2'-desoxiadenosina (27).
Una solución del nucleósido (26) (831 mg, 1.30 mmol) fue disuelto en una mezcla de MeOH:N,N-dimetilacetal (30 ml: 3ml) y agitada a 40°C. La reacción monitorizada por TLC, se completó después de 1 hora. El solvente fue eliminado bajo vacío. La purificación por cromatografía sobre sílica (EtOAc: MeOH 95:5) dio (27) en forma de un polvo ligeramente marrón (777 mg, 86 %). 1H RMN (da DMSO) 50.99 (s, 9H, tBu), 2.22-2.29 (m, 1H, H-2'), 2.50-2.59 (m, 1H, H-2'), 3.13 (s. 3H, CH3), 3.18 (s. 3H, CH3), 3.68-3.87 (m, 2H, H-5'), 3.88-3.92 (m, 1H, H-4'), 4.25 (m, 2H, CH2N), 4.43 (m, 1H, H-3'), 6.56 (t, J = 6.6 Hz, 1H, H-1'), 7.36-7.65 (m, 10H, HAr), 7.71 (s, 1H, H-8), 8.33 (s, 1H, CH), 8.8 (s, 1H, H-2), 10.12 (t, J = 5.3 Hz, 1H, NH).
5'-O-(tert-Butildifenilsilil)-7-desaza-4-N,N-dimetilformadin-3'-O-metiltiometoxi-7-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxiadenosina (28).
A una solución de (27) (623 mg, 0.89 mmol) en DMSO seco (8 ml) se agregó ácido acético (775 |jl, 13.35 mmol) y anhídrido acético (2.54 ml, 26.7 mmol). La mezcla fue agitada durante la noche a temperatura ambiente. La reacción fue vertida entonces en EtOAc y en solución acuosa saturada de NaHCO3 (1:1) y agitada vigorosamente. La capa orgánica fue lavada una vez más con NaHCO3 acuoso saturado y secada sobre MgSO4. Después de eliminar el solvente bajo presión reducida, el producto (28) fue purificado por cromatografía sobre sílica (EtOAc: éter de petróleo 1:2, luego EtOAc) produciendo (28) (350 mg, 52 %). 1H RMN (d6 DMSO) : 5, 1.0 (s, 9H, tBu), 2.09 (s, 3H, SCH3), 2.41-2.48 (m, 1H, H-2'), 2.64-2.72 (m, 1H, H-2'), 3.12 (s, 3H, CH3), 3.17 (s, 3H, CH3), 3.66-3.89 (m, 2H, H-5'), 4.04 (m, 1H, H-4'), 4.26 (m, J = 5.6 Hz, 2H, CH2), 4.67 (m, 1H, H-3'), 4.74 (br s, 2H, CH2), 6.49 (t, J = 6.1, 8.1 Hz, 1H, H-1'), 7.37-7.48 (m, 5H, HAr), 7.58-7.67 (m, 5H, HAr), 7. 76 (s, 1H, H8), 8.30 (s, 1H, CH), 8.79 (s, 1H, H-2), 10.05 (t, J = 5.6 Hz, 1H, NH).

3'-O-Azidometil-5'-O-(tert-butildifenilsilil)-7-desaza-4-N,N-dimetilformadin-7-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2'-desoxiadenosina (29).
A una solución de (28) (200 mg, 0.26 mmol) y ciclohexeno (0.135 ml, 1.3 mmol) en CH2O 2 seco (5 ml) a 0°C, se agregó cloruro de sulfurilo (32 jl, 0.39 mmol) bajo N2. Después de 10 minutos, la TLC indicó un consumo completo
del nucleósido (28). El solvente fue evaporado y el resido fue sometido a alto vacío durante 20 minutos. Fue luego redisuelto en DMF seco (3 ml), enfriado a 0°C y tratado con NaN3 ( 8 6 mg, 1.3 mmol). La suspensión resultante fue agitada bajo temperatura ambiente durante 3 horas. La reacción fue sometida a partición entre EtOAc y agua. Las fases acuosas fueron extraídas con EtOAc. Los extractos orgánicos combinados fueron combinados y secados sobre MgSO4. Después de eliminar el solvente bajo presión reducida, la mezcla fue purificada por cromatografía sobre sílica (EtOAc) produciendo un aceite (29) (155 mg, 80 %), 1H RMN (de DMSO) : 6 0.99 (s, 9H, tBu), 2.45-2.50 (m, 1H, H-2’), 2.69-2.78 (m, 1H, H-2’), 3.12 (s, 3H, CH3), 3.17 (s, 3H, CH3), 3.67-3.88 (m, 2H, H-5’), 4.06 (m, 1H, H-4’), 4.25 (m, 2H, CH2), 4.61 (m, 1H, H-3’), 4.84-4.97 (m, 2H, CH2), 6.58 (t, J = 6 .6 Hz, 1H, H-1’), 7.35-7.47 (m, 5H, Haf), 7.58-7.65 (m, 5H, Haf), 7.77 (s, 1H, H-8 ), 8.30 (s, 1H, CH), 8.79 (s, 1H, H-2), 10.05 (brs, 1H, NH).

3’-O-Azidometil-7-desaza-4-N,N-dimetilformadin-7-[3-(2,2,2-trifluoroacetamido)-prop-1-inil]-2’-desoxiadenosina (30).
Una solución de (29) (155 mg, 0.207 mmol) en solución en tetrahidrofurano (THF) (3 ml) fue tratada con TBAF (1 M en THF, 228 pl) a 0°C. El baño de hielo fue retirado entonces y la mezcla de reacción se agitó a temperatura ambiente. Después de 2 horas la TLC indicó el consumo completo del nucleósido. Se eliminó el solvente. La purificación por cromatografía sobre sílica (EtOAc:MeOH 95:5) dio (30) ( 8 6 mg, 82 %) en forma de un aceite marrón pálido. 1H RMN (d6 DMSO) 6 2.40-2.48 (dd, J = 8.1, 13.6 Hz, 1H, H-2’), 2.59-2.68 (dd, J = 8.3, 14 Hz, 1H, H-2’), 3.12 (s, 3H, CH3), 3.17 (s, 3H, CH3) , 3.52-3.62 (m, 2H, H5’), 4.02 (m, 1H, H-4’), 4.28 (d, J = 5.6 Hz, 2H, CH2NH), 4.47 (m, 1H, H-3’), 4.89 (s, 2H, CH2N3), 5.19 (t, J = 5.6 Hz, 1H, OH), 6.49 (dd, J = 8.1, 8.7 Hz, 1H, H-1’), 7.88 (s, 1H, H-8 ), 8.34 (s, 1H, CH), 8.80 (s, 1H, H-2), 10.08 (s, 1H, NH).25*30

7-(3-Aminoprop-1-inil)-3’-O-azidometil-7-desaza-2’-desoxiadenosina 5’-O-nucleósido trifosfato (31).
El nucleósido (30) y la esponja de protones fue secada sobre P2O5 bajo vacío durante la noche. Una solución de (30) (150 mg, 0.294 mmol) y la esponja de protones (126 mg, 0.588 mmol) en fosfato de trimetilo (980 pl) fue agitada con tamices moleculares 4A durante 1 hora. Se agregó POCU recién destilado (36 pl, 0.388 mmol) y la solución fue agitada a 4°C durante 2 horas. La mezcla se dejó calentar lentamente hasta temperatura ambiente y se agregaron pirofosfato de bis (tri-n-butil amonio) 0.5 M en solución en DMF (2.35 ml, 1.17 mmol) y tri-n-butil amina anhidra (560 pl, 2.35 mmol). Después de 5 minutos, la reacción fue detenida con TEAB 0.1 M (bicarbonato de trietilamonio) como regulador (15 ml) y se agitó durante 3 horas. El agua fue eliminada bajo presión reducida y el residuo resultante fue disuelto en amoníaco concentrado (p 0.88, 15 ml) y agitado a temperatura ambiente durante 16 horas. La mezcla de reacción fue evaporada entonces hasta sequedad. El residuo fue disuelto en agua y la solución se aplico a una columna DEAE-Sephadex A-25. Se llevó a cabo la MPLC con un gradiente lineal de 0.05 M a 1 M de TEAB. Las fracciones que contenían el producto fueron combinadas y evaporadas hasta sequedad. El residuo fue disuelto en
agua y purificado posteriormente por HPLC. HPLC: tr(31) : 19.94 min (columna preparativa Zorbax C18, gradiente: 5% a 35% B in 20 min, regulador A 0.1M TEAB, regulador B MeCN). El producto (31) fue aislado en forma de una espuma blanca (17.5 pmol, 5.9%, £230 = 15000). 1H RMN (D2O) 52.67-2.84 (2m, 2H, H-2'), 4.14 (m, 2H, CH2NH), 4.17-4.36 (m, 2H, H-5'), 4.52 (br s, H-4'), 6.73 (t, J = 6.6 Hz, H-1'), 3.06 (s, 1H, H-8), 3.19 (s, 1H, H-2). 31P RMN (D2O) 5 -5.07 (d, J = 21.3 Hz, 1P, PY), -10.19 (d, J =19.3 Hz, 1P, Pa), -21.32 (t, J = 19.3 Hz, 1P, Pp). Masa (-ve electroaspersión) calculada para C15H21N3O12P3593.05, encontrada 596.

Al enlazante de disulfuro Cy3 (1.3 ^mol) en solución en DMF (450 ^l) se agregan a 0°C 50 ^l de una mezcla de 1-(3 dimetilaminopropil)-3-etilcarbodiimida clorhidrato, 1-hidroxibenzotrizol hidrato y N-metilmorfolina (26 ^M cada uno) en DMF. La mezcla de reacción fue agitada a temperatura ambiente durante 1 hora. La reacción fue monitorizada por TLC (MeOH:CH2Cl2 3:7) hasta que se consumió el enlazante del colorante. Luego se agregó DMF (400 ^l) a 0°C, seguido por el nucleótido (31) (1.2 i^mol) en solución en agua (100 i^l) y la mezcla de reacción fue agitada a temperatura ambiente durante la noche. La TLC (MeOH:CH2Cl24:6) mostró un consumo completo del éster activado y una mancha roja oscura apareció sobre la línea base. La reacción fue detenida con regulador TEAB (0.1M, 10 ml) y cargada sobre una columna DEAE Sephadex (2 x 5 cm). La columna fue eluida primero con regulador TEAB 0.1 M (100 ml) para lavar los residuos orgánicos y luego regulador TEAB 1 M (100 ml). El trifosfato deseado (32) fue eluido con regulador TEAB 1 M. Las fracciones que contenían el producto fueron combinadas, evaporadas y purificadas por HPLC. Condiciones para HPLC: tr(32) : 22.44 min (columna preparativa Zorbax C13, gradiente: 5% a 35% B in 20 min, regulador A 0.1M TEAB, regulador B MeCN). El producto fue aislado en forma de un sólido rosa oscuro (0.15 |jmol, 12.5%, £550 = 150000). 1H RMN (D2O) 52.03 (t, 2H, CH2), 2.25 (m, 1H, H-2'), 2.43 (m, 1H, H-2'), 2.50 (m, 2H, CH2), 2.66 (m, 2H, CH2), 3.79 (m, 2H CH2), 3.99 (m, 4H, CH2N, H-5'), 4.13 (br s, 1H, H-4'), 6.02, 6.17 (2d, J = 13.64 Hz, 2H, HAr), 6.30 (dd, J = 6.06, 3.53 Hz, H-1'), 7.03, 7.22 (2d, 2H, 2 x =CH), 7.53-7.32 (m, 5H, HAr, H-2, H-3), 3.29 (m, =CH). 31P RMN (D2O) 5 -4.33 (m, 1P, Py), -10.06 (m, 1P, Pa), -20.72 (m, 1P, Pp).
Incorporación de enzima de 3'-azidometil dNTPs
A un cebador de ADN/plantilla 100 nM (previamente marcado con P32 y T4 polinucleótido quinasa) en Tris HCl pH
8 .8 50 mM, Tween-20 al 0.01%, y MgSO4 4 mM, se agregó el compuesto 6 , 2 j M, y polimerasa 100 nM (Thermococcus sp. 9°N exo -Y409V A485L suministrada por New England Biolabs). La plantilla consistía de una corrida de 10 bases de adenina para mostrar el efecto del bloque. La reacción se calentó a 65°C durante 10 minutos. Para mostrar un bloqueo completo, se lleva a cabo una prueba con los cuatro nucleósido trifosfatos naturales, no bloqueados. Se pudo observar la incorporación cuantitativa de un dTTP bloqueado con azidometilo individual y así puede verse que el grupo azidometilo actúa como un bloque efectivo para incorporación posterior.
Uniendo un ADN de horquilla (cebador/plantilla autocomplementario enlazado covalentemente) a una perla de estreptavidina, la reacción puede llevarse a cabo con múltiples ciclos como se muestra en las figuras 5 y 6.
Preparación de las perlas de estreptavidina
Se retira el regulador de almacenamiento y se lavan las perlas 3 veces con regulador TE (Tris-HCl pH 8 , 10 mM y EDTA, 1 mM). Se resuspende en regulador B & W (10 mM Tris-HCl pH 7.5, 1 mM EDTA y 2.0 M NaCl), se agrega ADN de horquilla marcado con 32P biotinilado con una secuencia de plantilla colgante apropiada. Se deja en reposo a temperatura ambiente durante 15 minutos. Se elimina el regulador y se lavan las perlas 3 veces con regulador TE. Incorporación del nucleósido trifosfato completamente funcional (FFN)
A una solución de Tris-HCl pH 8 .8 50 mM, Tween-20 al 0.01%, MgSO4 4 mM, MnCh 0.4 mM (excepto ciclo 1, 0.2 mM), se agrega FFN 2 j M y polimerasa 100 nM. Esta solución se agrega entonces a las perlas y se mezcla exhaustivamente y se incuba a 65°C durante 10-15 minutos. La mezcla de reacción se retira y las perlas se lavan 3 veces con regulador TE.
Etapa de desbloqueo
Se agrega sal trisódica de Tris-(2-carboxietil) fosfina (TCEP) (0.1M) a las perlas y se mezcla exhaustivamente. La mezcla se incubó entonces a 65°C durante 15 minutos. La solución de desbloqueo se elimina y las perlas se lavan 3 veces con regulador TE.
Etapa de cubrimiento
Se agrega yodoacetamida (431 mM) en fosfato 0.1 mM a pH 6.5 a las perlas y se mezcla exhaustivamente, luego se deja a temperatura ambiente durante 5 minutos. La solución de cubrimiento se elimina y las perlas se lavan 3 veces con regulador TE.
Se repite según se requiera
Los productos de reacción pueden ser analizados colocando la solución de perlas en el pozo de un gel de secuenciación de ADN de poliacrilamida al 12% estándar en regulador de carga de formamida al 40%. Se corre el gel bajo condiciones de desnaturalización haciendo que el ADN sea liberado de las perlas sobre el gel. Los desplazamiento de la banda de ADN son afectados tanto por la presencia de colorante como por la adición de nucleótidos extra y así la escisión del colorante (y el bloque) con la fosfina producen un desplazamiento en la movilidad sobre el gel.
En las figuras 5 y 6 pueden verse dos ciclos de incorporación con los compuestos 18 (C), 24 (G) y 32 (A) y seis ciclos con el compuesto 6.
Claims (11)
1. Un kit que comprende cuatro moléculas de nucleótido trifosfato modificadas, cada una de las cuales comprende una base de purina o pirimidina y una unidad estructural de azúcar desoxirribosa en el que el átomo de carbono 3' de la unidad estructural de azúcar ha unido un grupo de la estructura
-O-Z
en la que Z es de la fórmula -CH2N3.
2. El kit de acuerdo con la reivindicación 1, que comprende los nucleótidos A, T, C y G.
3. El kit de acuerdo con la reivindicación 1 o la reivindicación 2, que comprende además una transferasa terminal, una polimerasa o una transcriptasa reversa
4. El kit de acuerdo con la reivindicación 3, que comprende una polimerasa.
5. El kit de acuerdo con la reivindicación 4, en el que la polimerasa es una Thermococcus sp.
6. Un método para controlar la incorporación de un nucleótido complementario a un segundo nucleótido en un polinucleótido de cadena sencilla objetivo en una reacción de síntesis o secuenciación, comprendiendo el método incorporar en un polinucleótido complementario creciente un nucleótido que comprende una base de purina o pirimidina y una unidad estructural de azúcar desoxirribosa en el que el átomo de carbono 3' de la unidad estructural de azúcar ha unido un grupo de la estructura
-O-Z
en la que Z es de la fórmula -CH2N3,
la incorporación de dicho nucleótido evita o bloquea la introducción de moléculas de nucleótidos subsecuentes en dicho polinucleótido complementario creciente.
7. El método de acuerdo con la reivindicación 6, en el que cuatro nucleótidos diferentes se ponen en contacto con el polinucleótido de cadena sencilla simultáneamente.
8. El método de acuerdo con la reivindicación 6 o la reivindicación 7, en el que el 3 -O-CH2N3 se elimina de la unidad estructural de azúcar desoxirribosa antes de la introducción del siguiente nucleótido complementario para generar un grupo hidroxilo 3'.
9. El método de la reivindicación 8, en el que el grupo 3 -O-CH2N3 se elimina usando una fosfina soluble en agua.
10. El método de la reivindicación 9, en el que la fosfina soluble en agua es una trialquilfosfina derivada.
11. El método de la reivindicación 10, en el que la trialquilfosfina derivatizada se deriva con una o más funcionalidades seleccionadas del grupo que comprende grupos amino, hidroxilo, carboxilo y sulfonato.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/227,131 US7057026B2 (en) | 2001-12-04 | 2002-08-23 | Labelled nucleotides |
| GB0230037A GB0230037D0 (en) | 2002-12-23 | 2002-12-23 | Modified nucleotides |
| GB0303924A GB0303924D0 (en) | 2003-02-20 | 2003-02-20 | Modified nucleotides |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2790586T3 true ES2790586T3 (es) | 2020-10-28 |
Family
ID=61953108
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19164666T Expired - Lifetime ES2790586T3 (es) | 2002-08-23 | 2003-08-22 | Nucleótidos modificados |
| ES18157128T Expired - Lifetime ES2786983T3 (es) | 2002-08-23 | 2003-08-22 | Nucleótidos modificados para secuenciación de polinucleótidos |
| ES15184520.3T Expired - Lifetime ES2664803T3 (es) | 2002-08-23 | 2003-08-22 | Nucleótidos modificados para secuenciación de polinucleótidos |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18157128T Expired - Lifetime ES2786983T3 (es) | 2002-08-23 | 2003-08-22 | Nucleótidos modificados para secuenciación de polinucleótidos |
| ES15184520.3T Expired - Lifetime ES2664803T3 (es) | 2002-08-23 | 2003-08-22 | Nucleótidos modificados para secuenciación de polinucleótidos |
Country Status (5)
| Country | Link |
|---|---|
| CY (2) | CY1123506T1 (es) |
| ES (3) | ES2790586T3 (es) |
| HK (1) | HK1253509A1 (es) |
| HU (3) | HUE038902T4 (es) |
| PT (3) | PT3002289T (es) |
-
2003
- 2003-08-22 ES ES19164666T patent/ES2790586T3/es not_active Expired - Lifetime
- 2003-08-22 HU HUE15184520A patent/HUE038902T4/hu unknown
- 2003-08-22 HU HUE19164666A patent/HUE050818T4/hu unknown
- 2003-08-22 PT PT151845203T patent/PT3002289T/pt unknown
- 2003-08-22 ES ES18157128T patent/ES2786983T3/es not_active Expired - Lifetime
- 2003-08-22 PT PT191646660T patent/PT3587433T/pt unknown
- 2003-08-22 HU HUE18157128A patent/HUE049703T4/hu unknown
- 2003-08-22 ES ES15184520.3T patent/ES2664803T3/es not_active Expired - Lifetime
- 2003-08-22 PT PT181571282T patent/PT3363809T/pt unknown
-
2018
- 2018-09-20 HK HK18112109.3A patent/HK1253509A1/zh not_active IP Right Cessation
-
2020
- 2020-04-27 CY CY20201100377T patent/CY1123506T1/el unknown
- 2020-05-11 CY CY20201100431T patent/CY1122950T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ES2664803T3 (es) | 2018-04-23 |
| HUE038902T4 (hu) | 2022-02-28 |
| PT3587433T (pt) | 2020-05-19 |
| PT3002289T (pt) | 2018-04-09 |
| PT3363809T (pt) | 2020-05-06 |
| HUE050818T4 (hu) | 2021-05-28 |
| HUE038902T2 (hu) | 2018-12-28 |
| HUE050818T2 (hu) | 2021-01-28 |
| CY1123506T1 (el) | 2022-03-24 |
| ES2786983T3 (es) | 2020-10-14 |
| HK1253509A1 (zh) | 2019-06-21 |
| CY1122950T1 (el) | 2021-10-29 |
| ES2786983T8 (es) | 2020-11-03 |
| HUE049703T4 (hu) | 2021-05-28 |
| HUE049703T2 (hu) | 2020-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2550513T3 (es) | Nucleótidos modificados para secuenciación de polinucleótidos | |
| ES2858359T3 (es) | Nucleótidos marcados | |
| ES2604951T3 (es) | Nucleótidos etiquetados | |
| US20170067104A1 (en) | Labelled nucleotides | |
| US20120156671A1 (en) | Labelled nucleotides | |
| ES2790586T3 (es) | Nucleótidos modificados |