EP4384500B1 - Phenoxy-acetyl-thioureido-benzolsulfonamid-derivate und ihre verwendungen - Google Patents
Phenoxy-acetyl-thioureido-benzolsulfonamid-derivate und ihre verwendungen Download PDFInfo
- Publication number
- EP4384500B1 EP4384500B1 EP22799854.9A EP22799854A EP4384500B1 EP 4384500 B1 EP4384500 B1 EP 4384500B1 EP 22799854 A EP22799854 A EP 22799854A EP 4384500 B1 EP4384500 B1 EP 4384500B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- compound
- mmol
- mixture
- cells
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images






















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4706—4-Aminoquinolines; 8-Aminoquinolines, e.g. chloroquine, primaquine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
- A61K31/635—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide having a heterocyclic ring, e.g. sulfadiazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C327/00—Thiocarboxylic acids
- C07C327/38—Amides of thiocarboxylic acids
- C07C327/40—Amides of thiocarboxylic acids having carbon atoms of thiocarboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C327/42—Amides of thiocarboxylic acids having carbon atoms of thiocarboxamide groups bound to hydrogen atoms or to acyclic carbon atoms to hydrogen atoms or to carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C335/00—Thioureas, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C335/04—Derivatives of thiourea
- C07C335/16—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/18—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D207/22—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/24—Oxygen or sulfur atoms
- C07D207/26—2-Pyrrolidones
- C07D207/273—2-Pyrrolidones with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to other ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/22—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with hetero atoms directly attached to ring nitrogen atoms
- C07D295/28—Nitrogen atoms
- C07D295/30—Nitrogen atoms non-acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2866—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for cytokines, lymphokines, interferons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- the invention relates to phenoxy-acetyl-thioureido-benzenesulfonamide derivatives, and their use, in particular for treating viral infections.
- the Coronaviridae family is subdivided into four genera based on phylogenetic clustering: Alpha-, Beta-, Gamma- and Delta-coronavirus.
- the main outcome of CoV-2 infection is acute respiratory distress syndrome (85%), but other clinical disorders exist, including acute cardiac and kidney injuries (31% and 23% respectively), secondary infection (31%), and shock (23%).
- CoV-2 is an enveloped positive-strand RNA virus exhibiting a • 30 kb genome enwrapped into a particle composed of a cellular lipid bilayer and four structural proteins: Spike (S), Nucleocapsid (N), Envelope (E), and Membrane (M).
- CoV2 entry is highly dependent on the angiotensin-converting enzyme 2 (ACE2), similarly to its close relative SARS-CoV.
- ACE2 angiotensin-converting enzyme 2
- CoV2 contains a furin cleavage site between S1/S2, allowing S to be processed before egress that is absent from SARS-CoV, which explains its partial dependency for TMPRSS2.
- CoV-2 Spike can be cleaved and activated directly at the cell surface, although it is still possible that the virus is internalized into endosomes before fusion. The virus is internalized in a clathrin-dependent manner and endosomal acidification is required if the target cells do not express TMPRSS2.
- RNA genome of CoV-2 Upon fusion with cellular membranes, the RNA genome of CoV-2, protected by N protein, is targeted to ER membranes where translation immediately of replication proteins occurs, initiating the formation of viral replication organelles consisting of characteristic perinuclear double-membrane vesicles (DMVs).
- DMVs characteristic perinuclear double-membrane vesicles
- the aim of the invention is to obviate the drawbacks of the prior art, and to propose a specific and efficient medicament for curing SARS-CoV-2 infection, or preventing its side effects.
- the invention relates to a compound of formula I: wherein
- the invention relates to the above defined compounds, the compound having one of the following formulas: wherein A, Y1, Y2, W, Z, X1 and X2 and R and R1 to R7 are as defined above.
- the inventor unexpectedly identified that specific phenoxy-acetyl-thioureido-benzenesulfonamide derivatives, are potent and efficient antiviral compounds, but are also efficient against coronaviruses, in particular against SARS-CoV-2 virus.
- the invention relates to the compound as defined above, having one of the following formulas: and wherein Y1, Y2, W, Z, X1, X2, R and R1 to R7 are as defined above.
- the invention relates to the compound as defined above, having one of the following formulas: and wherein Y1, Y2, W, X1, X2, R and R1 to R7 are as defined above.
- the invention relates to the compound as defined above, having one of the following formulas: and wherein Y1, Y2, X1 and X2, R and R1 to R7 are as defined above.
- X1 and X2 are O.
- R3, R4, R6 and R7 are H.
- R5 is -O-CH 3 .
- the invention relates to the compound as defined above, the compound having one of the following formulas: and
- the invention relates to the compound as defined above, the compound having one of the following formulas: and
- the invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising, as active ingredient, the compound as defined above, in association with a pharmaceutically acceptable carrier.
- compositions within the scope of the invention include all compositions wherein the compounds of the present invention are contained in an amount which is effective to achieve its intended purpose. While individual needs vary, determination of optimal ranges of effective amounts of each component is within the skill of the art.
- the compounds may be administered to mammals, e.g. humans, orally at a dose of 0.0025 to 50 mg/kg, or an equivalent amount of the pharmaceutically acceptable salt thereof, per day of the body weight of the mammal being treated for disorders responsive to induction of apoptosis. In one embodiment, about 0.01 to about 25 mg/kg is orally administered to treat, ameliorate, or prevent such disorders.
- the dose is generally about one-half of the oral dose.
- a suitable intramuscular dose would be about 0.0025 to about 25 mg/kg, or from about 0.01 to about 5 mg/kg.
- the unit oral dose may comprise from about 0.01 to about 1000 mg, for example, about 0.1 to about 100 mg of the compound.
- the unit dose may be administered one or more times daily as one or more tablets or capsules each containing from about 0.1 to about 10 mg, conveniently about 0.25 to 50 mg of the compound or its solvates.
- the compound may be present at a concentration of about 0.01 to 100 mg per gram of carrier. In a one embodiment, the compound is present at a concentration of about 0.07-1.0 mg/ml, for example, about 0.1-0.5 mg/ml, and in one embodiment, about 0.4 mg/ml.
- the compounds of the invention may be administered as part of a pharmaceutical preparation containing suitable pharmaceutically acceptable carriers comprising excipients and auxiliaries which facilitate processing of the compounds into preparations which can be used pharmaceutically.
- suitable pharmaceutically acceptable carriers comprising excipients and auxiliaries which facilitate processing of the compounds into preparations which can be used pharmaceutically.
- the preparations particularly those preparations which can be administered orally or topically and which can be used for one type of administration, such as tablets, dragees, slow release lozenges and capsules, mouth rinses and mouth washes, gels, liquid suspensions, hair rinses, hair gels, shampoos and also preparations which can be administered rectally, such as suppositories, as well as suitable solutions for administration by intravenous infusion, injection, topically or orally, contain from about 0.01 to 99 percent, in one embodiment from about 0.25 to 75 percent of active compound(s), together with the excipient.
- compositions of the invention may be administered to any patient which may experience the beneficial effects of the compounds of the invention.
- mammals e.g., humans, although the invention is not intended to be so limited.
- Other patients include veterinary animals (cows, sheep, pigs, horses, dogs, cats and the like).
- the compounds and pharmaceutical compositions thereof may be administered by any means that achieve their intended purpose.
- administration may be by parenteral, subcutaneous, intravenous, intramuscular, intraperitoneal, transdermal, buccal, intrathecal, intracranial, intranasal or topical routes.
- administration may be by the oral route.
- the dosage administered will be dependent upon the age, health, and weight of the recipient, kind of concurrent treatment, if any, frequency of treatment, and the nature of the effect desired.
- compositions of the present invention are manufactured in a manner which is itself known, for example, by means of conventional mixing, granulating, dragee making, dissolving, or lyophilizing processes.
- pharmaceutical preparations for oral use can be obtained by combining the active compounds with solid excipients, optionally grinding the resulting mixture and processing the mixture of granules, after adding suitable auxiliaries, if desired or necessary, to obtain tablets or dragee cores.
- Suitable excipients are, in particular, fillers such as saccharides, for example lactose or sucrose, mannitol or sorbitol, cellulose preparations and/or calcium phosphates, for example tricalcium phosphate or calcium hydrogen phosphate, as well as binders such as starch paste, using, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, tragacanth, methyl cellulose, hydroxypropylmethylcellulose, sodium carboxymethylcellulose, and/or polyvinyl pyrrolidone.
- fillers such as saccharides, for example lactose or sucrose, mannitol or sorbitol, cellulose preparations and/or calcium phosphates, for example tricalcium phosphate or calcium hydrogen phosphate, as well as binders such as starch paste, using, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, tragacanth, methyl cellulose,
- disintegrating agents may be added such as the above-mentioned starches and also carboxymethyl-starch, cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof, such as sodium alginate.
- Auxiliaries are, above all, flow-regulating agents and lubricants, for example, silica, talc, stearic acid or salts thereof, such as magnesium stearate or calcium stearate, and/or polyethylene glycol.
- Dragee cores are provided with suitable coatings which, if desired, are resistant to gastric juices.
- concentrated saccharide solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, polyethylene glycol and/or titanium dioxide, lacquer solutions and suitable organic solvents or solvent mixtures.
- suitable cellulose preparations such as acetylcellulose phthalate or hydroxypropylmethyl-cellulose phthalate, are used.
- Dye stuffs or pigments may be added to the tablets or dragee coatings, for example, for identification or in order to characterize combinations of active compound doses.
- Other pharmaceutical preparations which can be used orally include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer such as glycerol or sorbitol.
- the push-fit capsules can contain the active compounds in the form of granules which may be mixed with fillers such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active compounds are in one embodiment dissolved or suspended in suitable liquids, such as fatty oils, or liquid paraffin.
- stabilizers may be added.
- Possible pharmaceutical preparations which can be used rectally include, for example, suppositories, which consist of a combination of one or more of the active compounds with a suppository base.
- Suitable suppository bases are, for example, natural or synthetic triglycerides, or paraffin hydrocarbons.
- gelatin rectal capsules which consist of a combination of the active compounds with a base.
- Possible base materials include, for example, liquid triglycerides, polyethylene glycols, or paraffin hydrocarbons.
- Suitable formulations for parenteral administration include aqueous solutions of the active compounds in water-soluble form, for example, water-soluble salts and alkaline solutions.
- suspensions of the active compounds as appropriate oily injection suspensions may be administered.
- Suitable lipophilic solvents or vehicles include fatty oils, for example, sesame oil, or synthetic fatty acid esters, for example, ethyl oleate or triglycerides or polyethylene glycol-400.
- Aqueous injection suspensions may contain substances which increase the viscosity of the suspension include, for example, sodium carboxymethyl cellulose, sorbitol, and/or dextran.
- the suspension may also contain stabilizers.
- the topical compositions of this invention are formulated in one embodiment as oils, creams, lotions, ointments and the like by choice of appropriate carriers.
- Suitable carriers include vegetable or mineral oils, white petrolatum (white soft paraffin), branched chain fats or oils, animal fats and high molecular weight alcohol (greater than C12).
- the carriers may be those in which the active ingredient is soluble.
- Emulsifiers, stabilizers, humectants and antioxidants may also be included as well as agents imparting color or fragrance, if desired.
- transdermal penetration enhancers can be employed in these topical formulations. Examples of such enhancers can be found in U.S. Pat. Nos. 3,989,816 and 4,444,762 .
- Ointments may be formulated by mixing a solution of the active ingredient in a vegetable oil such as almond oil with warm soft paraffin and allowing the mixture to cool.
- a vegetable oil such as almond oil
- a typical example of such an ointment is one which includes about 30% almond oil and about 70% white soft paraffin by weight.
- Lotions may be conveniently prepared by dissolving the active ingredient, in a suitable high molecular weight alcohol such as propylene glycol or polyethylene glycol.
- the invention also relates to a compound as defined above, for its use as a medicament, medicine or drug
- the invention also relates to a compound as defined above, for its use for the treatment of viral infections or a pathology linked to the viral infection.
- the invention relates to the above compounds, wherein the viral infection is a coronavirus infection.
- the invention relates to the above compounds, for its use as defined above, wherein the viral infection is a Sars-CoV-2 infection, and the pathology liked to the viral infection is COVID-19.
- Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an enveloped, positive-sense, single-stranded RNA beta-coronavirus that emerged in Wuhan in November 2019 and rapidly developed into a global pandemic.
- the associated disease, COVID-19 has an array of symptoms, ranging from flu-like illness and gastrointestinal distress to acute respiratory distress syndrome, heart arrhythmias, strokes, and death. Drug repurposing has played an important role in the search for COVID-19 therapies.
- the invention also relates to the use of the above defined compound for the preparation of a drug for the treatment, or the prevention of viral infection, in particular for the treatment of or the prevention of SARS-CoV-2 infections, or the treatment or the prevention of COVID-19.
- the invention relates to a method for preventing or treating viral infection in a subject in a need thereof, comprising administering to the subject an effective amount of the compound as defined or an effective amount of the pharmaceutical composition as defined above.
- the invention also relates to a kit comprising
- Vaccine against a viral infection can be either an attenuated virus, i.e. a virus deficient in its replication process but liable to infect target cells, or a nucleic acid molecule of the virus liable to produce viral protein to elicit an immune response.
- Antiviral compound can be one of the followings: interferon, remdesivir, azithromycin, hydroxychloroquine, chloroquine tocilizumab and sarilumab.
- remdesivir when present, it is present in an amount between about 1 mg and about l00mg, preferably from 10mg to 50 mg, more preferably from 20 mg to 30 mg.
- Phenol (1 equiv) was dissolved in dry DMF. The clear solution was cooled to 0 °C and Sodium Hydride (60% in mineral oil, 1.5 equiv) was added portionwise. A strong gaz evolution was observed. Mixture was stirred for 45 min and 1,1-Dimethylethyl N-(2-chloroethyl)carbamate (1.1 equiv) was added. Resulting mixture was stirred at room temperature overnight. Reaction mixture was diluted in 50 mL EtOAc then washed with saturated ammonium chloride solution (2 x 25 mL), water (2 x 25 mL) and brine (25 mL). Organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated to dryness. The crude mixture was purified by flash chromatography using a DCM-EtOAc or Hexanes-EtOAc gradient mixtures as eluent.
- Boc-protected amine was dissolved in DCM and the mixture cooled to 0 °C. TFA was then added dropwise to afford a 1:2 DCM:TFA ratio. Resulting mixture is stirred for 1 h, initially at 0 °C and allowed to reach room temperature. After this time, reaction mixture was concentrated to dryness and the residue was quenched with addition of a saturated sodium bicarbonate solution (30 mL). The organic layer was extracted with butanol (3 x 30 mL) and concentrated to dryness. The crude product was purified by reverse phase chromatography using a mixture of H 2 O-MeOH.
- Arylsulfonamide (1equiv) was suspended in dry DCM (0.2 M) under inert atmosphere. Diisopropylethylamine (3 equiv) was added to the mixture, followed by thiocarbonyldiimidazole (1 equiv). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted in EtOAc (40 mL), water (2 x 20 mL) and brine (20 mL). The organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography using a mixture of EtOAc in Hexanes.
- Boc-Amino-pyrrolidine (1 equiv) and cesium carbonate (1.2 equiv) were dissolved in anhydrous Toluene (0.3 M) and arylbromide (1.15 equiv) was added.
- the resulting mixture was degassed under argon for 5 min and Pd 2 (dba) 3 (6 mol%) was added, followed by BINAP (15 mol %).
- the mixture was then stirred at 110 °C under argon atmosphere for 5 h. After this time, the reaction mixture was diluted in ethyl acetate, washed with water (3 x 20 mL) and brine (20 mL). The organic layer was separated and concentrated to dryness.
- the crude product was purified by flash chromatography using a mixture of EtOAc in Hexanes.
- Arylsulfonamide (100 mg, 0.399 mmol) was suspended in dry DCM (2 mL) under inert atmosphere. Diisopropylethylamine (151 ⁇ L) was added to the mixture, followed by carbonyldiimidazole (0.399 mmol, 65 mg). The reaction mixture was stirred at room temperature for 45 min then amine compound (101 mg, 0.399 mmol) was added and the reaction mixture was heated under microwave irradiation for 1.5 h at 100 °C. The reaction mixture was diluted in EtOAc (40 mL), water (2 x 20 mL) and brine (20 mL). The organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- Arylsulfonamide (0.435 mmol, 110 mg) was suspended in dry DCM (2 mL) under inert atmosphere. Diisopropylethylamine (166 ⁇ M) was added to the mixture, followed by thiocarbonyldiimidazole (0.435 mmol, 78 mg). The reaction mixture was stirred at room temperature for 2 h then amine compound (112 mg, 0.435 mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted in EtOAc (40 mL), water (2 x 20 mL) and brine (20 mL). The organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- Arylsulfonamide (90 mg, 0.354 mmol) was suspended in dry dichloromethane (5 mL) under inert atmosphere. Diisopropylethylamine (135 ⁇ L) was added to the mixture, followed by thiocarbonyldiimidazole (63 mg, 0.354 mmol). The reaction mixture was stirred at room temperature for 2 h. Amide compound (68 mg, 0.354 mmol) was added and the resulting mixture was stirred at room temperature for 1.5 h. The reaction mixture was diluted in EtOAc (40 mL), water (2 x 20 mL) and brine (20 mL). The organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- Arylsulfonamide (40 mg, 0.157 mmol) was suspended in dry dichloromethane (2 mL) under inert atmosphere. Diisopropylethylamine (60 ⁇ L) was added to the mixture, followed by thiocarbonyldiimidazole (28 mg, 0.157 mmol). The reaction mixture was stirred at room temperature for 3 h. Amide compound (26 mg, 0.157 mmol) was added and the resulting mixture was stirred at room temperature for 3 h. The reaction mixture was diluted in EtOAc (40 mL), water (2 x 20 mL) and brine (20 mL). The organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- Arylsulfonamide (40 mg, 0.157 mmol) was suspended in dry dichloromethane (2 mL) under inert atmosphere. Diisopropylethylamine (60 ⁇ L) was added to the mixture, followed by thiocarbonyldiimidazole (28 mg, 0.157 mmol). The reaction mixture was stirred at room temperature for 3 h. Amine compound (29 mg, 0.157 mmol) was added and the resulting mixture was stirred at room temperature for 3 h. The reaction mixture was diluted in EtOAc (40 mL), water (2 x 20 mL) and brine (20 mL). The organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- Arylsulfonamide 50 mg, 0.238 mmol was suspended in dry dimethylformamide (2 mL) under inert atmosphere.
- HATU 109 mg, 0.285 mmol was added to the mixture, followed by diisopropylethylamine (123 ⁇ L, 0.714 mmol).
- Carboxylic acid 73 mg, 0.285 mmol was added and the resulting mixture was stirred at room temperature overnight. After this time, the crude mixture was directly purified by reverse phase chromatography using a mixture of Acetonitrile in H 2 O. Compound RG30 was obtained as a white solid (79 mg, 74 % yield).
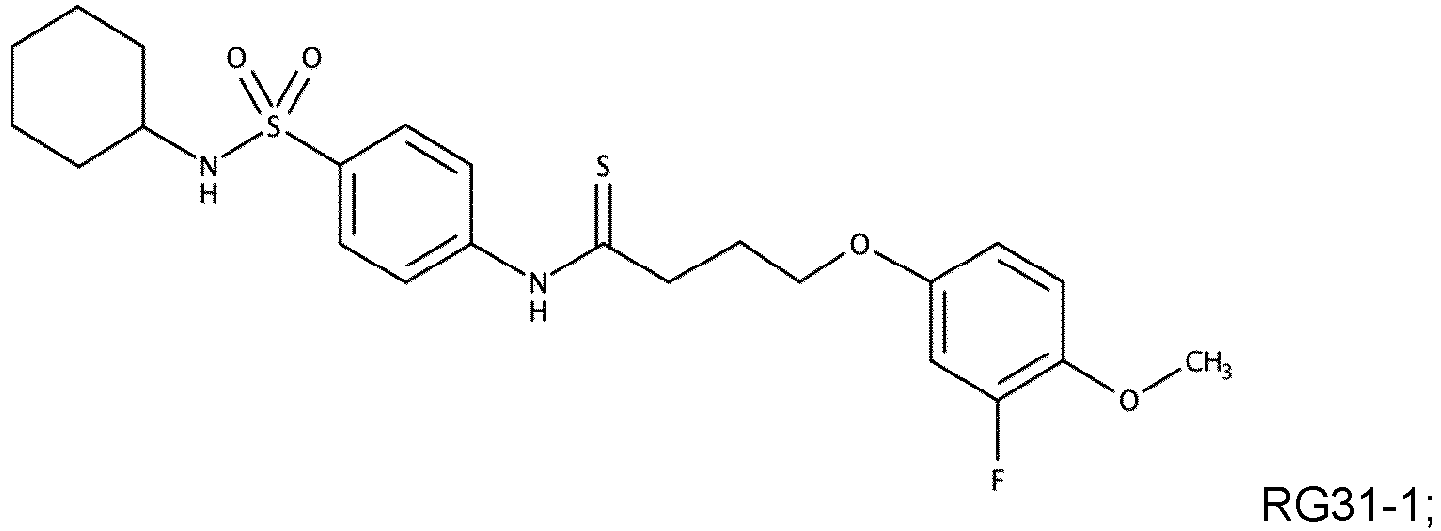
- RG30 (79 mg, 0.177 mmol) was dissolved in dry tetrahydrofuran (3 mL) under inert atmosphere. Lawesson reagent (61 mg, 0.152 mmol) was added to the mixture. The reaction mixture was stirred at 66 °C for 5 h. After this time, reaction mixture was diluted with a saturated sodium bicarbonate solution (20 mL) and organic layer was extracted with DCM (3 x 20 mL) and concentrated to dryness. The crude product was purified by flash chromatography using a mixture of EtOAc in Hexanes, followed by reverse phase chromatography using a mixture of Acetonitrile in H 2 O. Compound RG31 was obtained as a white solid (34 mg, 41 % yield).
- Arylsulfonamide 50 mg, 0.195 mmol was suspended in dry DCM (3 mL) under inert atmosphere.
- Diisopropylethylamine 74 ⁇ L, 0.429 mmol was added to the mixture, followed by thiocarbonyldiimidazole (35 mg, 0.195 mmol,).
- the reaction mixture was stirred at room temperature 3.5 h.
- Amine compound 38 mg, 0.195 mmol was added and the resulting mixture was stirred at room temperature for 1 h.
- the reaction mixture was diluted in EtOAc (50 mL), water (2 x 25 mL) and brine (25 mL).
- Arylsulfonamide 45 mg, 0.166 mmol was suspended in dry DCM (3 mL) under inert atmosphere.
- Diisopropylethylamine 63 ⁇ L, 0.366 mmol was added to the mixture, followed by thiocarbonyldiimidazole (30 mg, 0.166 mmol,).
- the reaction mixture was stirred at room temperature 3 h.
- Amine compound 32 mg, 0.166 mmol
- the reaction mixture was diluted in EtOAc (50 mL), water (2 x 25 mL) and brine (25 mL). The organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- Arylsulfonamide 50 mg, 0.197 mmol was suspended in dry DCM (3 mL) under inert atmosphere.
- Diisopropylethylamine 109 ⁇ L, 0.629 mmol was added to the mixture, followed by thiocarbonyldiimidazole (35 mg, 0. 197 mmol,).
- the reaction mixture was stirred at room temperature 3.5 h.
- Amine compound 48 mg, 0.197 mmol was added and the resulting mixture was stirred at room temperature for 1 h.
- the reaction mixture was diluted in EtOAc (50 mL), water (2 x 25 mL) and brine (25 mL).
- Arylsulfonamide (47 mg, 0.185 mmol) was suspended in dry DCM (3 mL) under inert atmosphere. Diisopropylethylamine (71 ⁇ L, 0.408 mmol) was added to the mixture, followed by thiocarbonyldiimidazole (33 mg, 0. 185 mmol,). The reaction mixture was stirred at room temperature 4.5 h. Amine compound (39 mg, 0.185 mmol) was added and the resulting mixture was stirred at room temperature for 30 min. The reaction mixture was diluted in EtOAc (50 mL), water (2 x 25 mL) and brine (25 mL).
- Compound RG27-2 Compound 11 (21 mg, 0.08 mmol) was suspended in dry dichloromethane (1 mL), N,N-diisopropylethylamine (31 ⁇ L, 0.18 mmol) was added to the mixture, followed by 1,1'-thiocarbonyldiimidazole (14 mg, 0.08 mmol). The reaction mixture was stirred at room temperature for 5h. Compound 2 (34 mg, 0.08 mmol) was added, and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted in ethyl acetate washed with water and brine. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- Compound RG27-8 Compound 13 (20 mg, 0.08 mmol) was suspended in dry dichloromethane (0.8 mL), N,N-diisopropylethylamine (42 • L, 0.24 mmol) was added to the mixture, followed by 1,1'-thiocarbonyldiimidazole (14 mg, 0.08 mmol). The reaction mixture was stirred at room temperature for 5h. Compound 22 (35 mg, 0.08 mmol) was added, and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted in ethyl acetate and washed with water and brine. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- Compound 25 1-(3-chloro-4-methoxyphenyl)pyrrolidin-3-amine .
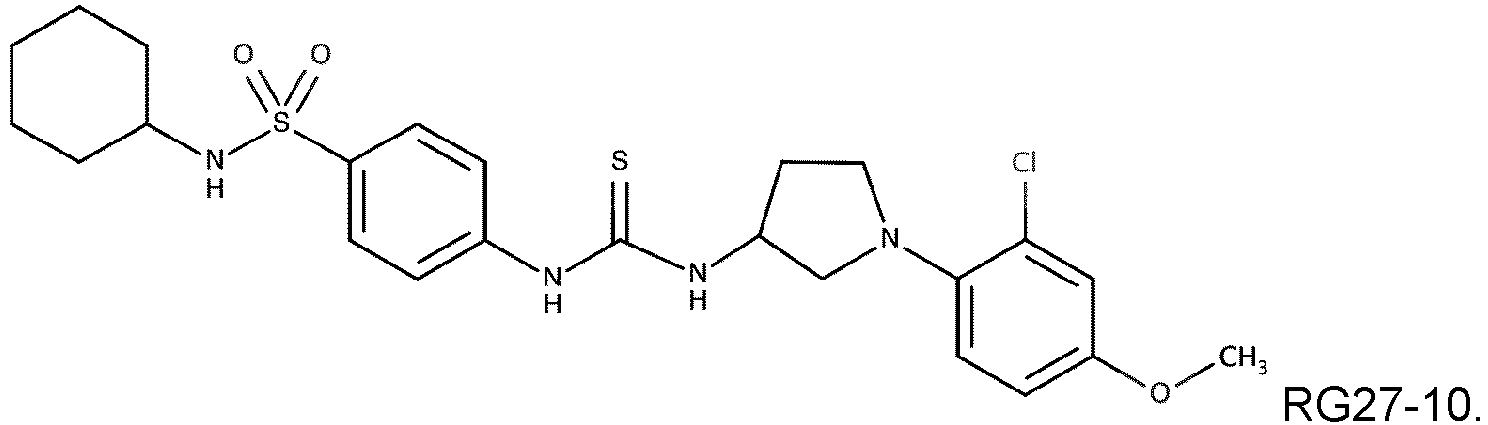
- Compound RG27-10 Compound 13 (98 mg, 0.365 mmol) was suspended in dry dichloromethane (4 mL), N,N-diisopropylethylamine (140 • L, 0.803 mmol) was added to the mixture, followed by 1,1'-thiocarbonyldiimidazole (65 mg, 0.365 mmol). The reaction mixture was stirred at room temperature for 5h. Compound 28 (166 mg, 0.365 mmol) was added, and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted in ethyl acetate and washed with water and brine. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- Example 2 Pharmacological perturbation of intracellular dynamics as a SARS-CoV-2 antiviral strategy
- SARS-CoV-2 (CoV2) is the viral agent responsible for the pandemic of the coronavirus disease 2019 (COVID-19). Vaccines are being deployed all over the world with good efficacy, but there is no approved antiviral treatment to date. This is particularly needed since the emergence of variants and the potential immune escape may prolong pandemic spreading of the infection for much longer than anticipated.
- the inventor developed a series of small molecules and identified RG10 as a potent antiviral compound against SARS-CoV-2 in cell lines and in human airway epithelia (HAE). RG10 localizes to endoplasmic reticulum (ER) membranes, perturbing ER morphology and inducing ER stress.
- ER endoplasmic reticulum
- RG10 does not associate with SARS-CoV-2 replication sites although preventing virus replication.
- the inventor developed fluorescent SARS-CoV-2 viral particles allowing them to track virus arrival to ER membranes. Live cell imaging of replication-competent virus infection revealed that RG10 of formula: stalls the intracellular dynamics of virus-ER dynamics.
- RG10b a stable version of RG10, of formula: that showed increased potency in vitro and in HAE with a pharmacokinetic half-life greater than 2 h.
- the inventor's work reports on a novel fluorescent virus model and innovative antiviral strategy consisting of the perturbation of ER/virus dynamics, highlighting the promising antiviral properties of RG10 and RG10b.
- the inventor had synthesized a series of sulfamoyl-phenyl derived compounds designed as potential antiviral agents. While most of them did not prevent CoV2 replication at 2 and 10 ⁇ M, they identified the molecule RG10, a novel small molecule of unknown target, as a potent antiviral molecule with similar activity levels as Remdesivir (Rem; Figure 1 ). RG10 did not show cytotoxicity at working concentrations ( Figure 2 ) and measurement of its potency in Huh7.5.1 cells returned an EC50 • 1.5 ⁇ M ( Figure 3 ).
- RG10 exhibited high CoV2 antiviral activity in Vero E6 cells but also in human Caco2 and Huh7.5.1 cells ( Figure 4 ) and retained potency at high multiplicity of infection (MOI) similar to Rem ( Figure 5 ).
- MOI multiplicity of infection
- S neosynthesized Spike protein
- Figure 6 Microscopy and flow cytometry analyses confirmed that both RG10 and Rem prevent viral protein synthesis ( Figures 7 and 8 ).
- immunolabeling using an antibody recognizing double stranded RNA (dsRNA), specific of viral replication complexes showed that RG10 prevents viral RNA synthesis ( Figures 7 and 8 ).
- RG10 inhibits CoV2 replication
- RG10 inhibits virus entry or post-entry events
- the inventor performed time-of-addition experiments where the molecule was added for four hours and then washed-out (Pre), or conversely; no drug was added for the first 4 h to allow early virus entry steps to occur and only then RG10 was added to the cells (Post; Figure 9 ).
- the inventor found that RG10 had no antiviral activity when incubated only during virus entry while adding RG10 post-entry significantly decreased vRNA expression levels ( Figure 10 ).
- comparing the amount of viral N protein and dsRNA levels by flow cytometry confirmed that RG10 exerts strong antiviral activity post entry, although some inhibition was also observed at the entry steps ( Figures 11 and 12 ).
- RG10 localizes to the endoplasmic reticulum
- RG10 perturbed ER morphology ( Figure 17 and Figure 18 ) and thus investigated whether RG10 could exert its antiviral activity through ER stress induction.
- the inventor measured the levels of RNAs of known ER stress mediators from the IRE1- ⁇ and PERK pathways and found that the spliced form of XBP-1 (sXBP-1) was enriched upon 50 ⁇ M RG10 and 1 mM DTT treatment ( Figure 19 ).
- 50 ⁇ M of the RG10 derivative compound RG7, which has no antiviral activity ( Figure 1 ) did not significantly increase sXBP-1 mRNA levels ( Figure 20 ).
- the increase of sXBP-1 RNA was lower at 10 ⁇ M and no difference was observed between RG10 and RG7 ( Figure 20 ), and thus, ER stress induction cannot fully explain the antiviral activity of RG10.
- the inventor generated fluorescent infectious CoV2 using a trans-complementation approach.
- the inventor generated a Vero E6 cell line stably expressing the nucleocapsid (N) from CoV2 fused to the red fluorescent protein mRuby3.
- Wild type or Vero E6 N-mRuby3 cells were infected with wild type CoV2 virus ( Figures 27 and 28 ) and the produced virions incorporated a subset of fluorescent N-mRuby3 proteins in trans (not encoded by the viral genome; Figure 21 ).
- the N-mRuby3-containing viruses were as infectious as their non-fluorescent counterparts ( Figure 22 and Figure 29 ), indicating that they represent suitable tools to study CoV2 infection.
- RG10 exhibits coronavirus specific antiviral activity
- RG10 had antiviral activity against another virus known to replicate at the ER. Therefore, the inventor compared in parallel the infection of Vero E6 cells by CoV2 or by Zika virus (ZIKV), another enveloped positive-strand RNA virus but from the Flaviviridae family. Interestingly, the inventor found that RG10 had no detectable effect on ZIKV, while in contrast, the ER stress inducer Tunicamycin was active against both viruses ( Figures 30 and 31 ). To determine whether RG10 was specific to CoV2 or can act against other coronaviruses, the inventor tested its activity against the human coronavirus 229E (HCoV-229E) and observed significant antiviral activity, although potency was lower ( Figure 32 ).
- HCV-229E human coronavirus 229E
- HAE human airway epithelia
- RG10 decreases viral infection using a fluorescent mNeonGreen replication-competent virus, and that virus protein expression was also altered ( Figures 33 and 34 ).
- HAE viability was similar, the actin network seemed to be rearranged at the apical side of RG10-treated HAE ( Figure 35 ).
- sXBP-1 was upregulated upon RG10 treatment ( Figure 36 ), indicating that RG10 mediates ER stress in this primary HAE model.
- RT-qPCR viral RNA was decreased by RG10, although the extent of the decrease was much lower than Remdesivir treatment ( Figure 37 ).
- RG10b The low potency of RG10 in HAE could be attributed to its weak stability and therefore, the inventor designed a novel RG10 derivative, which the inventor called RG10b. This derivative harbors a modification shielding a molecular weakness present in RG10. RG10b was more potent than RG10 with an EC50 ⁇ 1 ⁇ M ( Figures 38 and 39 ). To verify the stability of RG10 and RG10b in physiological conditions, the inventor performed pharmacokinetics analyses in mice and found that RG10b has a half-life of 2h19 in the blood, a value more than 3-fold higher than for RG10 ( Figure 40 ).
- the inventor has characterized a novel SARS-CoV-2 antiviral molecule and explored its mode of action. To this end, he generated novel tools to study the spatiotemporal dynamics of the virus at high resolution.
- DAA Direct-acting antiviral agents
- RNA viruses being prone to mutations (as highlighted by the emerging variants) suggest that this approach may not be sufficient by itself to counteract virus dissemination and may instead favor escape mutants in monotherapies.
- HTAs that target cellular partners important for the virus life cycle is an approach of choice, that must be considered in parallel to DAA and vaccination.
- the inventor choses a bottom-up approach, using a series of related proprietary molecules that had no known functions, and started to characterize it.
- he also generated new tools to perform live cell imaging of SARS-CoV-2 entry, offering the opportunity to learn more about the virus' life cycle, while tailoring an antiviral molecule with promising pharmacological properties.
- HTAs often have broad-spectrum antiviral activity due to the fact that viruses may exploit similar pathways.
- (hydroxy)chloroquine, ribavirin, and favipiravir are examples of broad-spectrum antivirals that are used in vitro at high concentrations to be effective.
- favipiravir shows very low potency in vitro.
- Hydroxychloroquine which has an EC50 • 2 ⁇ M in Vero E6 cells, does not decrease the viral loads of infected non-human primates.
- the heterogeneity between in vitro and in vivo potency highlights the complexity to develop HTAs.
- RG10 is likely an HTA as it does not localize at virus replication sites to exert its antiviral activity.
- the improved version of RG10, RG10b shows EC50 below 1 ⁇ M, high stability in vivo (> 2 h), with no toxicity observed ( Figures 38 - 42 ).
- the target of RG10 is unknown but the inventor identified that the molecule strongly affects virus and ER dynamics ( Figures 21 - 26 ), suggesting that it perturbs players involved in intracellular trafficking routes.
- the inventor showed that the actin cytoskeleton was disorganized by RG10 in primary HAE cells ( Figures 33 - 37 ). Preserved integrity of the actin cytoskeleton is required for SARS-CoV-2 infection.
- the FDA-approved drugs Sunitinib and BNTX inhibit SARS-CoV-2 entry and their mode of action was correlated to the disruption of actin dynamics.
- SARS-CoV-2 increases filopodia number and length, while traveling along them, further indicating that a strong interplay exists between CoV2 and the actin network.
- RG10 has very low cytotoxicity compared to Latrunculin, jasplakinolide and cytocalasin D, three well-known direct-acting actin perturbators that inhibit CoV2 infection. It is therefore likely that RG10 subtly impacts the actin cytoskeleton in a direct or indirect manner that remains to be identified.
- RG10 induces ER stress at high doses, but it was difficult to correlate this observation to the antiviral effect of RG10 because at lower doses, RG10 and RG7, two closely related molecules, similarly induced ER stress ( Figures 18 - 20 ) even though RG7 showed no antiviral activity ( Figures 1 - 8 ). Furthermore, RG10 was unable to inhibit ZIKV replication, which also occurs in the ER. The inventor used Tunicamycin as a potent ER stress inducer that inhibits infection by CoV2 and ZIKV, further suggesting that ER stress induction may not be sufficient for RG10 to exert an antiviral activity.
- RG10 is targeted to the ER and impacts ER morphology. Constant reshaping of the ER structure is a requirement for maintaining its numerous functions, and ER membranes are often targeted by pathogens. For instance, influenza A virus was recently shown to impact mitochondrial morphodynamics and endoplasmic reticulum-mitochondria contact sites, and altering mitochondrial dynamics was thus proposed as a new antiviral strategy. Similarly, one can hypothesize that ER morphodynamics can represent an antiviral strategy to be further explored. In this regard, atlastins play important roles in ER dynamics and morphology and it was recently shown that atlastin-dependent misshaped ER leads to decreased ZIKV replication.
- Mouse anti-Spike and anti-GAPDH were purchased from Genetex and Rabbit anti-N from Sino Biological (Clinisciences).
- the mouse anti-dsRNA antibody (J2) was from Jena Bioscience and rabbit anti-Calnexin from Elabscience.
- NHS-Rhodamine and all secondary Alexa Fluor antibodies were purchased from Thermo Fisher Scientific.
- Phalloidin CF-633 was purchased from Biotium and the ER Staining Kit was from Abcam. RG10 and derivatives were synthesized by AGV Discovery.
- Huh7.5.1 Zhong J. et al. (2005) Proceedings of the National Academy of Sciences of the United States of America 102: 9294-9
- Vero E6 ECACC #85020206
- Caco2 HEK 293T cells
- DMEM GlutaMAX Thermo Fisher Scientific
- HAE cells Epidermal calf serum and penicillin-streptomycin (500 • g/ml; GIBCO).
- HAE cells epihelix
- N was firstly amplified by PCR from the plasmid pLVX-EF1alpha-SARS-CoV-2-N-2xStrep-IRES-Puro (addgene#141391) using the sense primer 5' TTGCGGCCGCGCCACCATGAGCGATAACG-3' (SEQ ID NO: 1) (Notl site underlined) and the antisense primer 5'-CGCCTGAGTAGAATCGGCT-3' (SEQ ID NO: 2) (overlapped region underlined).
- the Ruby-3 fragment was amplified by PCR from a gblock (Integrated DNA-Technologies) using the sense primer 5'-AGCCGATTCTACTCAGGCGGGAGGTTCTGGTGGTTCTG-3' (SEQ ID NO: 3) (overlapped region underlined) and the antisense primer 5'- GA GGATC CTCACTTGTACAGCTCGTCCAT-3' (SEQ ID NO: 4) (BamHI site underlined).
- the individual N and Ruby3 fragments were linked by overlapping PCR of the N-CoV2 using primers 5'- TTGCGGCCGCGCCAC-3' (SEQ ID NO: 5) and 5'- GAGGATCCTCACTTGTACAGCTCGT-3' (SEQ ID NO: 6).
- the resulting PCR product was ligated into the digested Notl and BamHl lentiviral plasmid pHAGE IRES puro.
- the resulting plasmid pHAGE N-Ruby3 IRES puro is available on Addgene (#170466).
- a pHAGE N-mNeonGreen IRES puro was also generated (Addgene #170467).
- HEK 293T cells were transfected with pxPAX2, VSV-G and pHAGE N-Ruby3 IRES puro using JetPrime (Poluplus Transfection) and washed 6 h post-transfection. The supernatant was harvested 2 days later and cleared by centrifugation at 2000 x g for 5 min at 4°C. The lentivirus-containing supernatant was added to Vero E6 cells (ECACC #85020206) for 48 h. Selection of the positive cells was performed using 4 ⁇ g/ml puromycin and viable cells were expanded for flow cytometry cell sorting using a FACSMelody (BD Biosciences) equipped with a 561 nm excitation laser and 613/18 filter. Upon sorting, the cells were tested negative for mycoplasma. Vials of the Vero E6 N-Ruby3 stable cell line were conserved in liquid nitrogen in 10% DMSO.
- the SARS-CoV-2 strain BetaCoV/France/IDF0372/2020 was supplied by the National Reference Centre for Respiratory Viruses hosted by Institut Pasteur (Paris, France) and headed by Dr. Sylvie van der Werf.
- the strain was amplified through the infection of Vero E6 cells at MOI 0.0001 in DMEM GlutaMAX (Gibco) supplemented with 2% fetal bovine serum (FBS; Andrea Dutscher) and 1X penicillin-streptomycin (GIBCO).
- FBS fetal bovine serum
- GIBCO 1X penicillin-streptomycin
- the cleared virus-containing supernatant was frozen in 1 ml aliquots at -80°C.
- a vial was thawed for titration by plaque assay in Vero E6 cells to estimate plaque-forming units per mL of virus (PFU/mL) as described in Gaudin & Barteneva (Gaudin & Barteneva, 2015).
- Viral titers ranged between 3 10 6 and 2 10 7 pfu/ml.
- HCoV-229E-Renilla was a gift from Volker Thiel (van den Worm et al., 2012) and was amplified for 5-7 days at 33°C in Huh7.5.1 cells in 5% FCS-containing DMEM. Viral stocks were harvested when cells showed > 50% CPEs. Virus stock was titrated through TCID 50 in Huh7.5.1 cells used for their amplification and titer was 10 8 TCID 50 /mL.
- the ZIKV strain was obtained through BEI Resources (NIAID NR-50183, strain FLR) isolated in December 2015 from infected patient blood from Colombia.
- ZIKV was amplified in Vero cells for 72 h in DMEM GlutaMAX (Gibco) supplemented with 2% fetal bovine serum (FBS; Miguel Dutscher), 10 mM HEPES (Thermo Fisher Scientific) and 1X penicillin-streptomycin (Gibco).
- FBS fetal bovine serum
- HEPES Thermo Fisher Scientific
- the supernatant was harvested and clarified by centrifugation at 2000 g RT for 10 min.
- the virus was stored at -80°C.
- the virus titer was obtained by using standard plaque assay on Vero cells to measure plaque-forming unit per mL of virus (PFU/mL). Viral titer was 3.8 x10 6 pfu/mL.
- SARS-CoV-2 viral RNA was detected using the N1 probe that targets specifically the N protein.
- primers targeting Rpl27 or GAPDH were utilized.
- ER stress Primers Forward Reverse sXBP-1 5' -CTGAGTCCGAATCAGGTGCAG- 3' (SEQ ID NO: 7) 5' -ATCCATGGGGAGATGTTCTGG- 3' (SEQ ID NO: 8) usXBP-1 5' -CAGCACTCAGACTACGTGCA- 3' (SEQ ID NO: 9) 5' -ATCCATGGGGAGATGTTCTGG- 3' (SEQ ID NO: 10) tXBP-1 5' -TGGCCGGGTCTGCTGAGTCCG- 3' (SEQ ID NO: 11) 5' -ATCCATGGGGAGATGTTCTGG- 3' (SEQ ID NO: 12) ATF4 5' -GTTCTCCAGCGACAAGGCTA- 3' (SEQ ID NO: 13) 5' -AT
- Image acquisition was performed on an AxioObserver.Z1 inverted microscope (Zeiss) mounted with a CSU-X1 spinning disk head (Yokogawa), a back-illuminated EMCCD camera (Evolve, Photometrics) and a X100, 1.4 NA oil objective (Zeiss) controlled by MetaMoprh.
- the microscope chamber was heated at 37°C and the automated multiposition stage maintained in 95% humidity at 37°C and 5% CO2. Images were processed using FiJi (ImageJ software version 1.51h) and quantitative analyses were performed using Bitplane Imaris x64 version 9.2. Analyses of the movies consisted on the segmentation of the single viral particles based on intensity and size. Then, tracking was performed for each individual particle and mean speed and track duration were extracted.
- Antiviral activity was assessed as explained in Example 2. Vero E6 cells were treated with the indicated RG derivatives or Remdesivir at indicated concentrations and subsequently infected with SARS-CoV-2 at MOI 0.01 for 48 h in the presence of the compounds.
- RG38 and RG41 exhibit similar range of antiviral activity against SARS-CoV-2 that the one of RG27, suggesting that they could be good alternative molecules for future antiviral development.
- RG27 derivatives namely RG27-1, RG27-2, RG27-8 and RG27-10 of formula:
- Antiviral activity was assessed as explained in Example 2. Human Huh 7.5.1 cells were used instead of Vero cells. Cells were treated with the indicated RG derivatives or Remdesivir at indicated concentrations and subsequently infected with SARS-CoV-2 at MOI 0.01 for 48 h in the presence of the compounds.
- Compounds RG27-2 and RG27-10 harbor a similar antiviral activity compared to RG27 compound, at 10 ⁇ M. Moreover, at 25 ⁇ M, compounds RG27-1 and RG27-8 harbor also an antiviral activity that cannot be observed at 10 ⁇ M.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Claims (15)
- Eine Verbindung der Formel I
 - R ist H oder ein C1-C4-Alkyl;- A ist NH oder CH2;- Y1 ist CH oder N;- Y2 ist O, N oder CH- Z ist NH oder O;- G1 ist einer der folgenden Fälle:-
- R ist H oder ein C1-C4-Alkyl;- A ist NH oder CH2;- Y1 ist CH oder N;- Y2 ist O, N oder CH- Z ist NH oder O;- G1 ist einer der folgenden Fälle:-

 - X1 und X2 sind, unabhängig voneinander, O, NH, N-R9, wobei R9 ein C1-C3-Alkyl, linear oder verzweigt, eine -CN-Gruppe oder eine -CF3-Gruppe ist;- R3 bis R7 sind, unabhängig voneinander, H oder eine funktionelle Gruppe aus der Liste bestehend aus Fluor, Chlor, Brom, Nitro, Cyano, Amino, Aminoalkyl wie Aminomethyl und Aminoethyl, Methyl, Hydroxylmethyl, Trifluormethyl, Methoxy, Trifluormethyloxy, Ethyl, Trifluorethyl, Ethoxy und Trifluorethyloxyoder ein Salz oder ein Solvat davon.
- X1 und X2 sind, unabhängig voneinander, O, NH, N-R9, wobei R9 ein C1-C3-Alkyl, linear oder verzweigt, eine -CN-Gruppe oder eine -CF3-Gruppe ist;- R3 bis R7 sind, unabhängig voneinander, H oder eine funktionelle Gruppe aus der Liste bestehend aus Fluor, Chlor, Brom, Nitro, Cyano, Amino, Aminoalkyl wie Aminomethyl und Aminoethyl, Methyl, Hydroxylmethyl, Trifluormethyl, Methoxy, Trifluormethyloxy, Ethyl, Trifluorethyl, Ethoxy und Trifluorethyloxyoder ein Salz oder ein Solvat davon. - Verbindung nach Anspruch 1 mit einer der folgenden Formeln


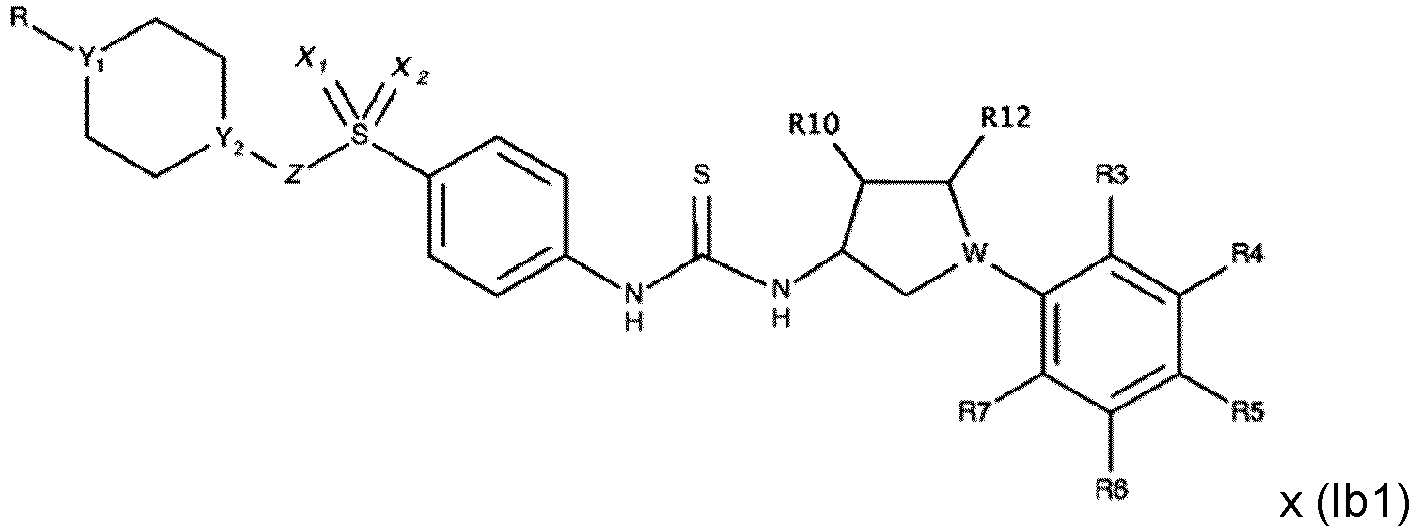



- Verbindung nach Anspruch 1 oder 2 mit einer der folgenden Formeln:

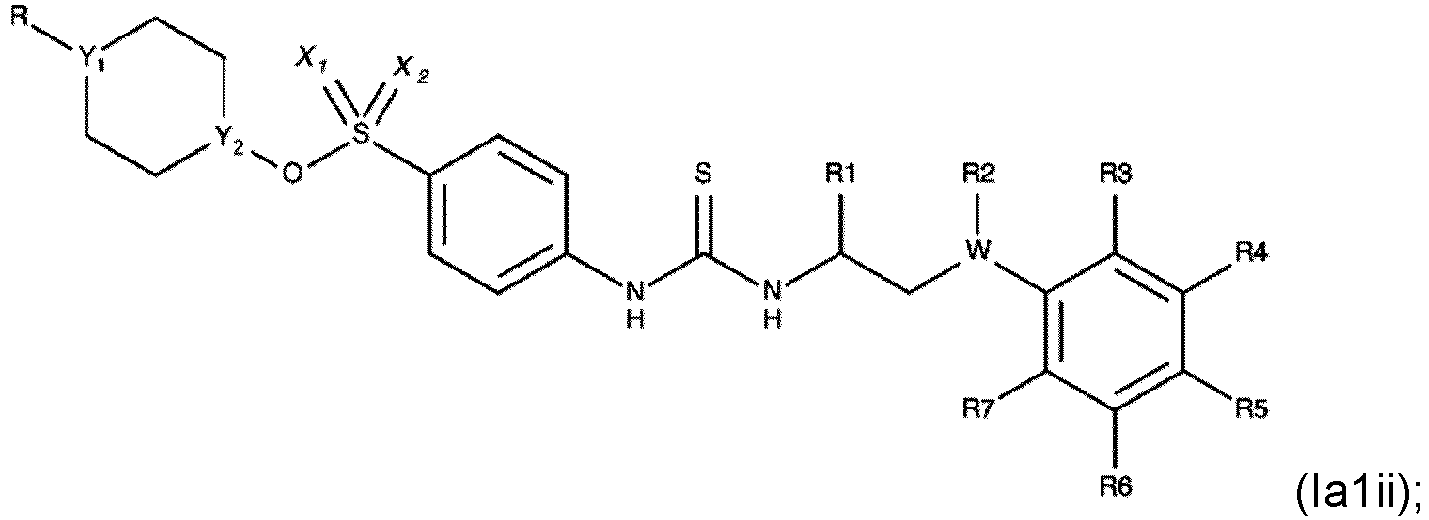









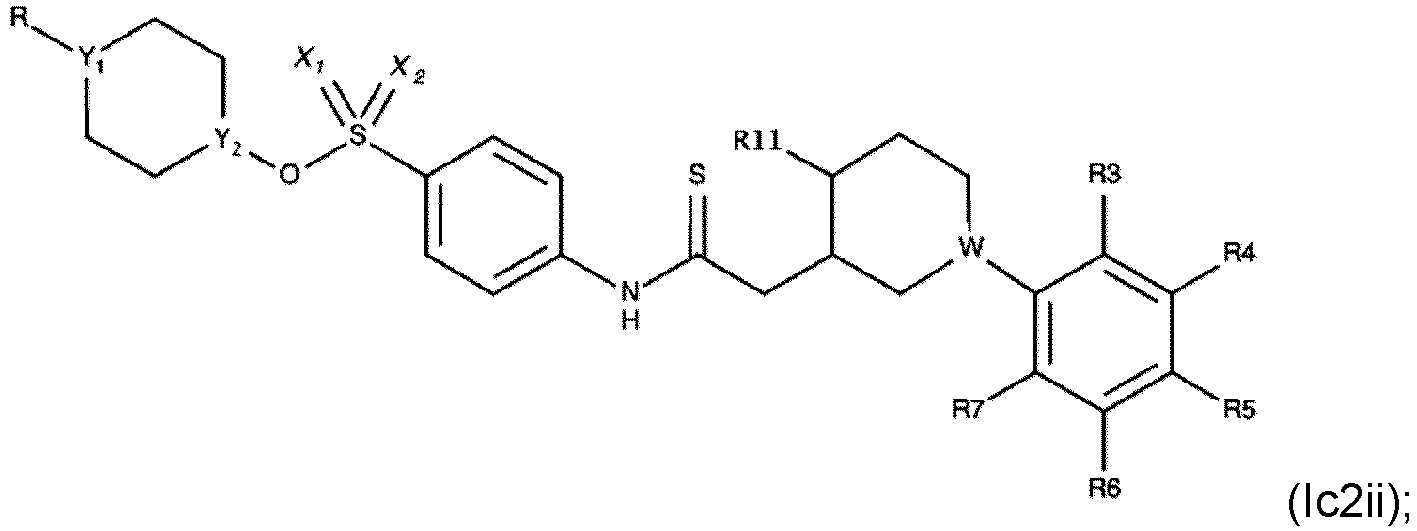
- Verbindung nach einem der Ansprüche 1 bis 3, die eine der folgenden Formeln aufweist





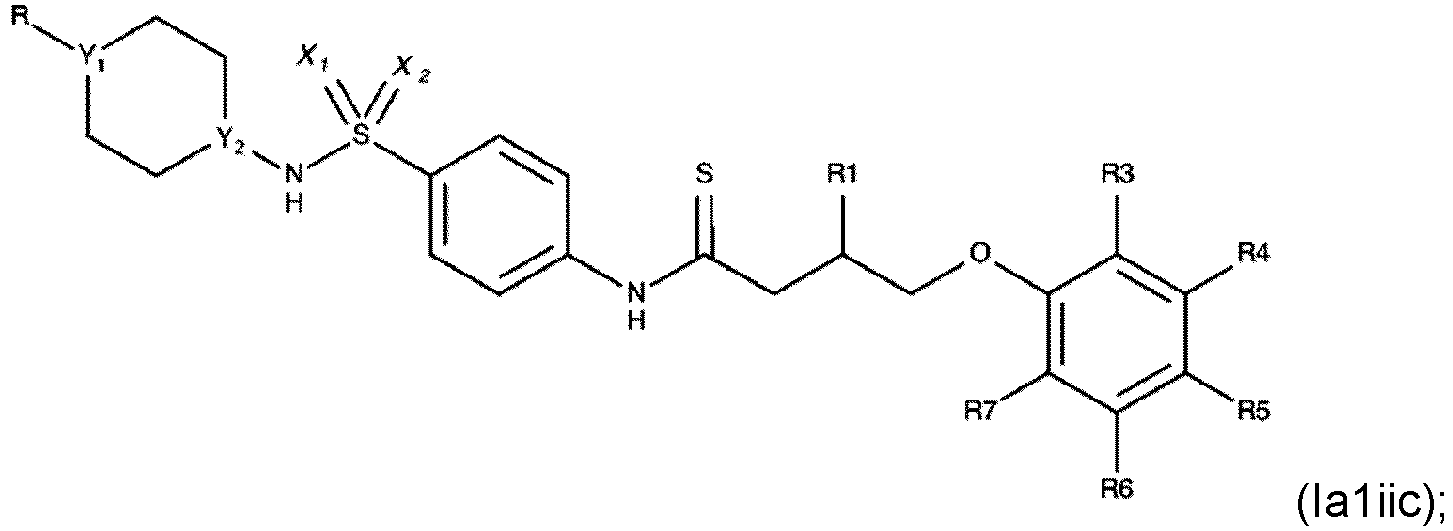




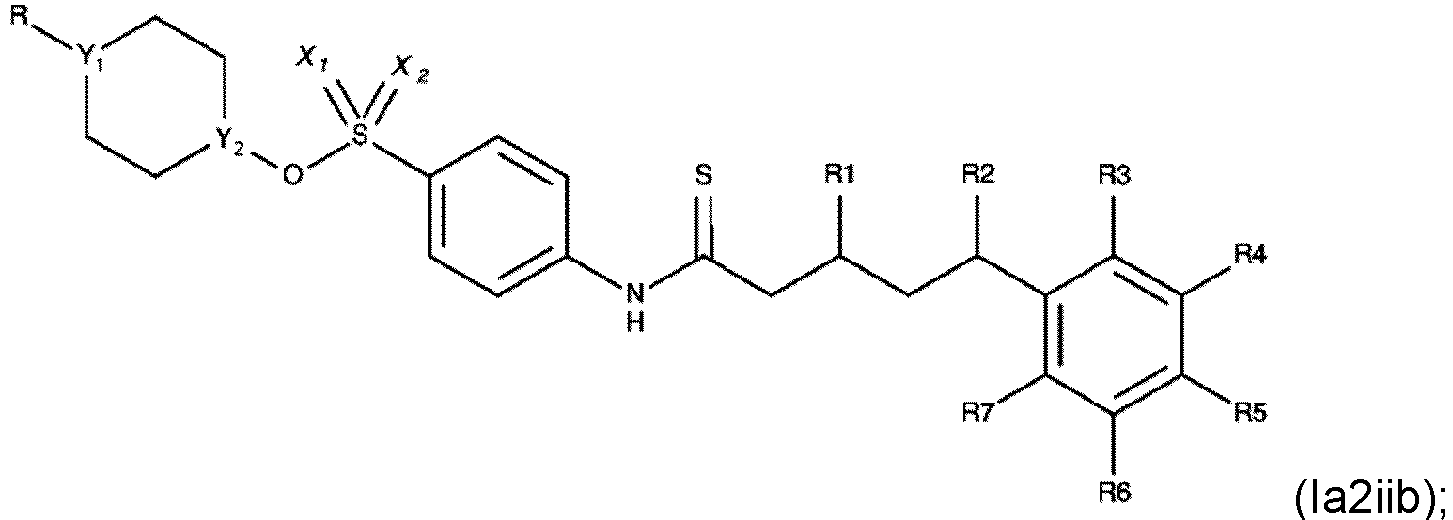


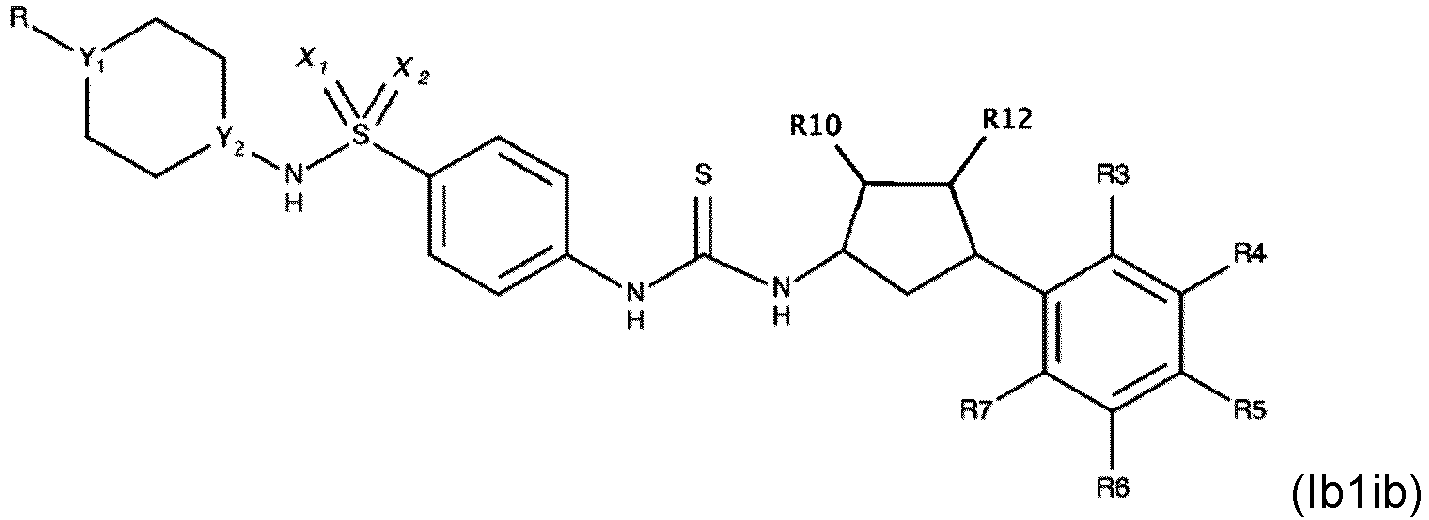








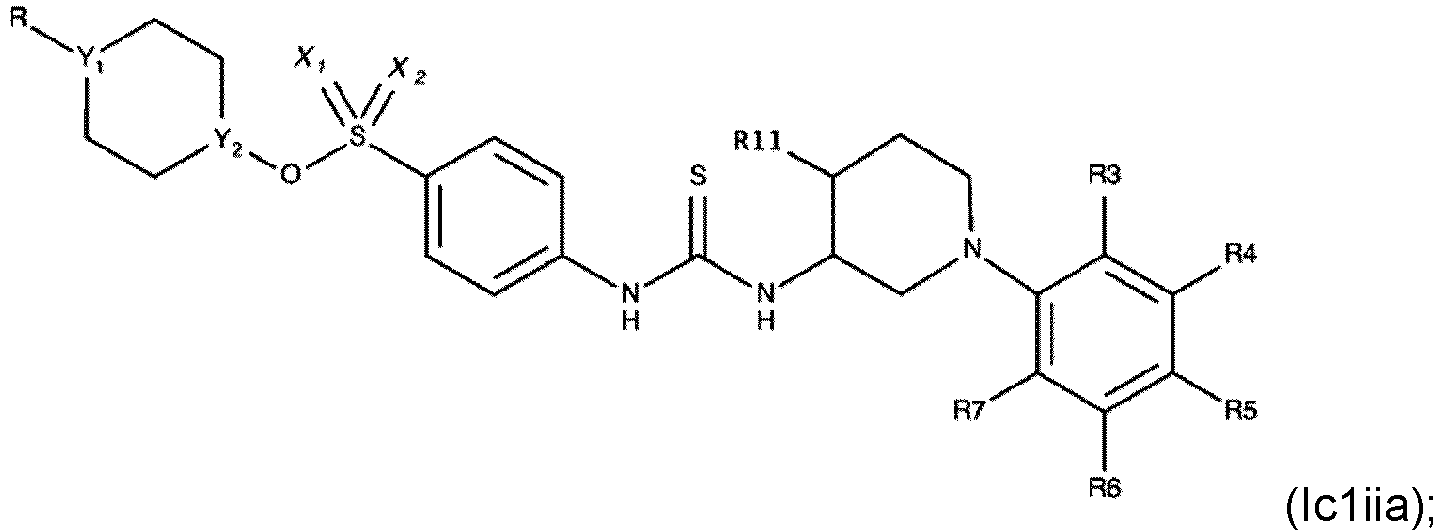


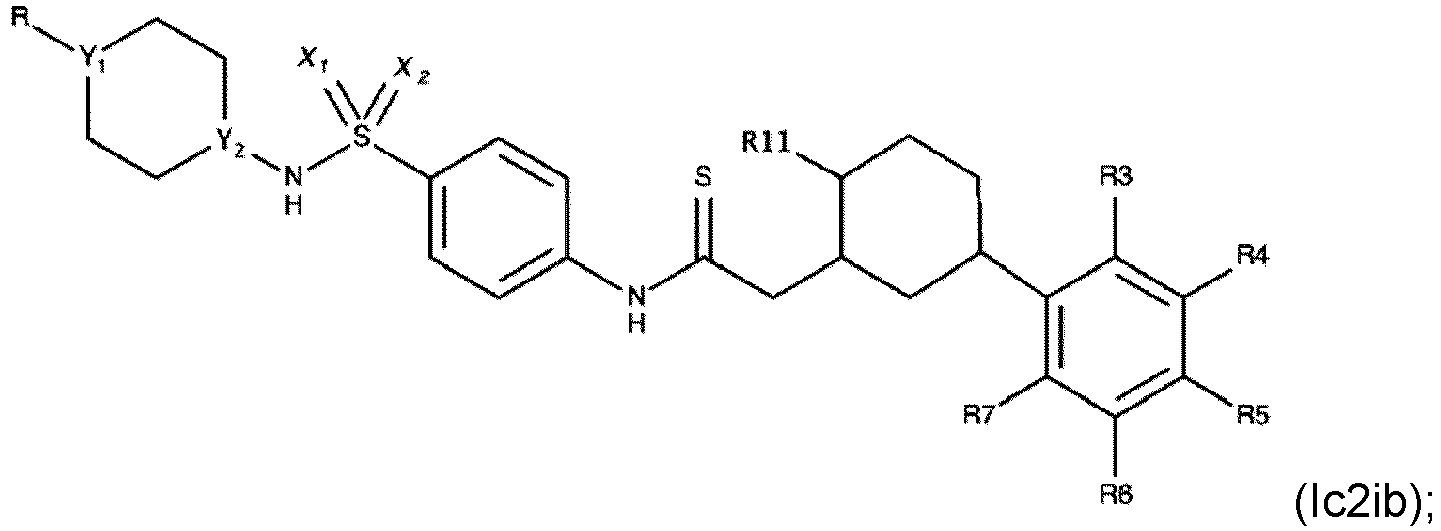

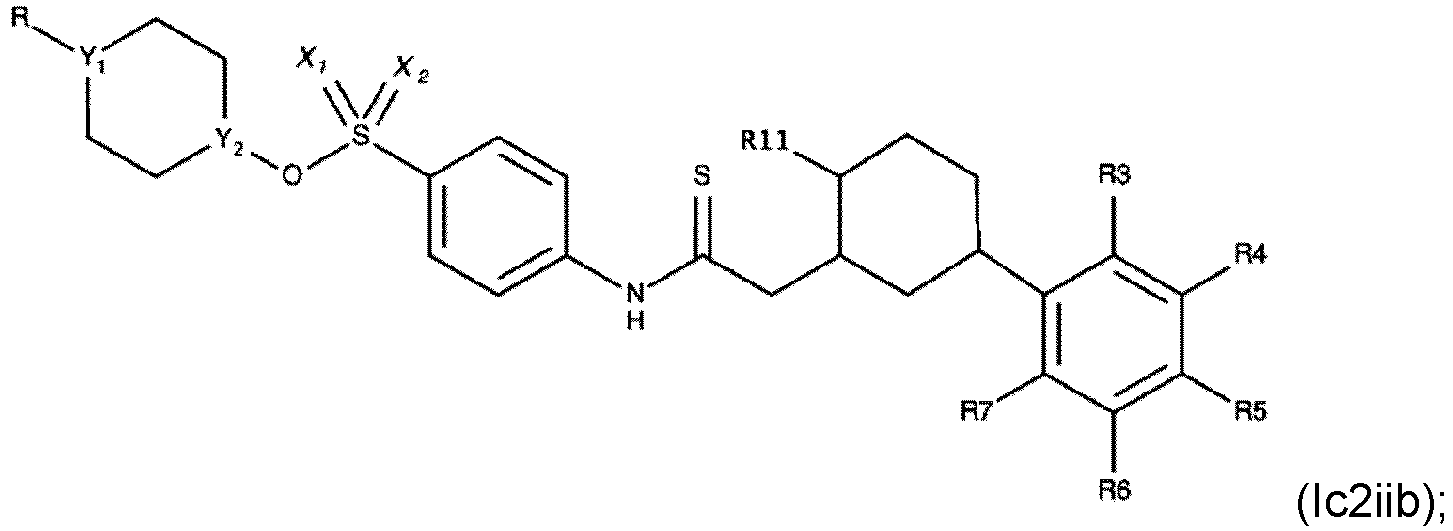
- Verbindung nach einem der Ansprüche 1 bis 4, wobei R3, R4, R6 und R7 H sind.
- Verbindung nach einem der Ansprüche 1 bis 5, wobei R5 -O-CH3 ist.
- Verbindung nach einem der Ansprüche 1 bis 6, wobei X1 und X2 O sind.
- Die Verbindung nach einem der Ansprüche 1 bis 7, wobei die Verbindung eine der folgenden Formeln aufweist:
























- Die Verbindung nach einem der Ansprüche 1 bis 4, wobei die Verbindung eine der folgenden Formeln aufweist:





- Pharmazeutische Zusammensetzung, umfassend als Wirkstoff die in einem der Ansprüche 1 bis 9 definierte Verbindung in Verbindung mit einem pharmazeutisch annehmbaren Träger.
- Verbindung nach einem der Ansprüche 1 bis 9 zu ihrer Verwendung als Medikament.
- Verbindung nach einem der Ansprüche 1 bis 9 zur Verwendung für die Behandlung von Virusinfektionen oder einer mit der Virusinfektion verbundenen Pathologie.
- Verbindung für ihre Verwendung nach Anspruch 12, wobei die Virusinfektion eine Coronavirusinfektion ist.
- Die Verbindung für ihre Verwendung gemäß Anspruch 11 oder 12, wobei die Virusinfektion eine Sars-CoV-2-Infektion ist und die mit der Virusinfektion verbundene Pathologie COVID-19 ist.
- Ein Bausatz bestehend aus- eine Verbindung nach einem der Ansprüche 1 bis 9; und- einen Impfstoff gegen eine virale Infektion oder eine antivirale Verbindung, vorzugsweise eine der folgenden: Remdesivir, Azithromycin, Hydroxychloroquin, Chloroquin, Tocilizumab und Sarilumab.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21306242.5A EP4148042A1 (de) | 2021-09-10 | 2021-09-10 | Phenoxy-acetyl-thioureido-benzolsulfonamid-derivate und ihre verwendungen |
| PCT/EP2022/075138 WO2023036945A1 (en) | 2021-09-10 | 2022-09-09 | Phenoxy-acetyl-thioureido-benzenesulfonamide derivatives, and their uses |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP4384500A1 EP4384500A1 (de) | 2024-06-19 |
| EP4384500B1 true EP4384500B1 (de) | 2025-06-11 |
| EP4384500C0 EP4384500C0 (de) | 2025-06-11 |
Family
ID=77998918
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP21306242.5A Withdrawn EP4148042A1 (de) | 2021-09-10 | 2021-09-10 | Phenoxy-acetyl-thioureido-benzolsulfonamid-derivate und ihre verwendungen |
| EP22799854.9A Active EP4384500B1 (de) | 2021-09-10 | 2022-09-09 | Phenoxy-acetyl-thioureido-benzolsulfonamid-derivate und ihre verwendungen |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP21306242.5A Withdrawn EP4148042A1 (de) | 2021-09-10 | 2021-09-10 | Phenoxy-acetyl-thioureido-benzolsulfonamid-derivate und ihre verwendungen |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20240391872A1 (de) |
| EP (2) | EP4148042A1 (de) |
| WO (1) | WO2023036945A1 (de) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7559223B2 (ja) | 2020-08-24 | 2024-10-01 | ギリアード サイエンシーズ, インコーポレイテッド | リン脂質化合物及びその使用 |
| TW202344257A (zh) | 2020-10-16 | 2023-11-16 | 美商基利科學股份有限公司 | 磷脂化合物及其用途 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3989816A (en) | 1975-06-19 | 1976-11-02 | Nelson Research & Development Company | Vehicle composition containing 1-substituted azacycloheptan-2-ones |
| US4444762A (en) | 1980-04-04 | 1984-04-24 | Nelson Research & Development Company | Vehicle composition containing 1-substituted azacyclopentan-2-ones |
| CA3182306A1 (en) * | 2020-05-05 | 2021-11-11 | The Scripps Research Institute | Methods and compositions for the treatment of sars-cov-2 |
| WO2021242850A1 (en) * | 2020-05-28 | 2021-12-02 | The Regents Of The University Of Michigan | Compositions and methods for preventing and treating sars-cov-2 infection |
-
2021
- 2021-09-10 EP EP21306242.5A patent/EP4148042A1/de not_active Withdrawn
-
2022
- 2022-09-09 EP EP22799854.9A patent/EP4384500B1/de active Active
- 2022-09-09 US US18/690,833 patent/US20240391872A1/en active Pending
- 2022-09-09 WO PCT/EP2022/075138 patent/WO2023036945A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| WO2023036945A1 (en) | 2023-03-16 |
| EP4384500A1 (de) | 2024-06-19 |
| US20240391872A1 (en) | 2024-11-28 |
| EP4148042A1 (de) | 2023-03-15 |
| EP4384500C0 (de) | 2025-06-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114057702B (zh) | 一种新型冠状病毒主蛋白酶的抑制剂及其制备方法和用途 | |
| WO2021207409A2 (en) | Small molecule inhibitors of sars-cov-2 viral replication and uses thereof | |
| EP4384500B1 (de) | Phenoxy-acetyl-thioureido-benzolsulfonamid-derivate und ihre verwendungen | |
| Xu et al. | Structure–activity relationship studies on diversified salicylamide derivatives as potent inhibitors of human adenovirus infection | |
| Pascual et al. | Structure-based drug design for envelope protein E2 uncovers a new class of bovine viral diarrhea inhibitors that block virus entry | |
| CN104744433A (zh) | Ald及其作为ev71病毒和cav16病毒抑制剂的用途 | |
| JP5894935B2 (ja) | サングリフェリン系化合物 | |
| US20060003317A1 (en) | Drug discovery method | |
| Xu et al. | Discovery of novel substituted N-(4-Amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues as potent human adenovirus inhibitors | |
| US9775835B2 (en) | Small molecule inhibitors of viral protein interactions with human t-RNA | |
| EP3056202A1 (de) | Benzopyrrolidone-derivate mit antiviralen und antitumor-eigenschaften | |
| JP2022509763A (ja) | アレナウイルス感染症の治療のための化合物 | |
| WO2022122968A1 (en) | Compounds for use in the treatment of covid-19 | |
| Ferla et al. | Rational modifications, synthesis and biological evaluation of new potential antivirals for RSV designed to target the M2-1 protein | |
| CA2975382C (en) | Design, synthesis and methods of use of acyclic fleximer nucleoside analogues having anti-coronavirus activity | |
| EP4408833A1 (de) | Inhibitoren der molluscum-antagiosum-infektion und verfahren zu ihrer verwendung | |
| Bakhache et al. | Pharmacological perturbation of intracellular dynamics as a SARS-CoV-2 antiviral strategy | |
| US11046731B2 (en) | Zinc-binder based EBNA1-specific compounds | |
| CN118955471B (zh) | 一种具有双手性中心的多取代哌嗪类化合物及其制备方法和应用 | |
| EP4052726B1 (de) | Cd4-mimic-verbindung mit anti-hiv-aktivität | |
| Wang | Development of Bioactive Peptidomimetics | |
| VanGraafeiland et al. | Approaches to the Identification of Molecules Altering Programmed Ribosomal Frameshifting in Viruses | |
| Ottavi | Efforts toward the discovery and development of novel tuberculosis therapeutics | |
| US20240425473A2 (en) | Enzyme inhibitors and viral infection therapy | |
| AU2024204967B1 (en) | Application of tetrahydroquinolone-amide-thiazole compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: UNKNOWN |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE INTERNATIONAL PUBLICATION HAS BEEN MADE |
|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: REQUEST FOR EXAMINATION WAS MADE |
|
| 17P | Request for examination filed |
Effective date: 20240311 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| DAV | Request for validation of the european patent (deleted) | ||
| DAX | Request for extension of the european patent (deleted) | ||
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: GRANT OF PATENT IS INTENDED |
|
| INTG | Intention to grant announced |
Effective date: 20250228 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE PATENT HAS BEEN GRANTED |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602022015904 Country of ref document: DE |
|
| U01 | Request for unitary effect filed |
Effective date: 20250616 |
|
| U07 | Unitary effect registered |
Designated state(s): AT BE BG DE DK EE FI FR IT LT LU LV MT NL PT RO SE SI Effective date: 20250626 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: U11 Free format text: ST27 STATUS EVENT CODE: U-0-0-U10-U11 (AS PROVIDED BY THE NATIONAL OFFICE) Effective date: 20251002 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250611 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250912 Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250911 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250611 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250911 |
|
| U20 | Renewal fee for the european patent with unitary effect paid |
Year of fee payment: 4 Effective date: 20250930 |