EP3699173A1 - Gem-disubstituted pyrrolidines, piperazines, and diazepanes, and compositions and methods of making the same - Google Patents
Gem-disubstituted pyrrolidines, piperazines, and diazepanes, and compositions and methods of making the same Download PDFInfo
- Publication number
- EP3699173A1 EP3699173A1 EP19204164.8A EP19204164A EP3699173A1 EP 3699173 A1 EP3699173 A1 EP 3699173A1 EP 19204164 A EP19204164 A EP 19204164A EP 3699173 A1 EP3699173 A1 EP 3699173A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- alkyl
- compound
- formula
- aryl
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 238000000034 method Methods 0.000 title claims description 317
- 239000000203 mixture Substances 0.000 title claims description 94
- 150000004885 piperazines Chemical class 0.000 title description 3
- 150000003235 pyrrolidines Chemical class 0.000 title description 3
- 150000000117 diazepanes Chemical class 0.000 title description 2
- 239000003446 ligand Substances 0.000 claims abstract description 166
- 150000001875 compounds Chemical class 0.000 claims description 331
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 266
- 125000000217 alkyl group Chemical group 0.000 claims description 261
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 233
- -1 heteroaryl halide Chemical class 0.000 claims description 215
- 125000001072 heteroaryl group Chemical group 0.000 claims description 211
- 125000003118 aryl group Chemical group 0.000 claims description 200
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 196
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 155
- 229910052736 halogen Inorganic materials 0.000 claims description 146
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 122
- 125000003545 alkoxy group Chemical group 0.000 claims description 119
- 239000003054 catalyst Substances 0.000 claims description 112
- 150000002367 halogens Chemical group 0.000 claims description 109
- 238000006243 chemical reaction Methods 0.000 claims description 96
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 82
- 125000004104 aryloxy group Chemical group 0.000 claims description 75
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 73
- 239000002904 solvent Substances 0.000 claims description 71
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 70
- 125000001188 haloalkyl group Chemical group 0.000 claims description 67
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 66
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 63
- 125000003342 alkenyl group Chemical group 0.000 claims description 62
- 125000000304 alkynyl group Chemical group 0.000 claims description 62
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical group ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 60
- 150000003839 salts Chemical class 0.000 claims description 59
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 57
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 53
- 125000002252 acyl group Chemical group 0.000 claims description 50
- 125000004475 heteroaralkyl group Chemical group 0.000 claims description 48
- 229910052739 hydrogen Inorganic materials 0.000 claims description 48
- 125000005843 halogen group Chemical group 0.000 claims description 47
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 45
- 125000001424 substituent group Chemical group 0.000 claims description 42
- 125000004429 atom Chemical group 0.000 claims description 40
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 claims description 40
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 39
- 238000002360 preparation method Methods 0.000 claims description 35
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 31
- 239000001257 hydrogen Substances 0.000 claims description 30
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 28
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 24
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 23
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 claims description 23
- 125000004366 heterocycloalkenyl group Chemical group 0.000 claims description 23
- 230000029936 alkylation Effects 0.000 claims description 21
- 238000005804 alkylation reaction Methods 0.000 claims description 21
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 20
- 229910052763 palladium Inorganic materials 0.000 claims description 20
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical group C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 claims description 20
- 125000003107 substituted aryl group Chemical group 0.000 claims description 20
- 230000002194 synthesizing effect Effects 0.000 claims description 20
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 19
- 229910052801 chlorine Inorganic materials 0.000 claims description 19
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 claims description 19
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 18
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 18
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 18
- 229940126601 medicinal product Drugs 0.000 claims description 18
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 16
- 150000001502 aryl halides Chemical class 0.000 claims description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 15
- 229910001868 water Inorganic materials 0.000 claims description 15
- 125000003368 amide group Chemical group 0.000 claims description 14
- 229910052759 nickel Inorganic materials 0.000 claims description 14
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 13
- XXJGBENTLXFVFI-UHFFFAOYSA-N 1-amino-methylene Chemical compound N[CH2] XXJGBENTLXFVFI-UHFFFAOYSA-N 0.000 claims description 12
- 125000003282 alkyl amino group Chemical group 0.000 claims description 11
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 11
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 claims description 11
- JRTIUDXYIUKIIE-KZUMESAESA-N (1z,5z)-cycloocta-1,5-diene;nickel Chemical group [Ni].C\1C\C=C/CC\C=C/1.C\1C\C=C/CC\C=C/1 JRTIUDXYIUKIIE-KZUMESAESA-N 0.000 claims description 10
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 10
- 125000004414 alkyl thio group Chemical group 0.000 claims description 10
- 229910052757 nitrogen Inorganic materials 0.000 claims description 10
- 229910052760 oxygen Inorganic materials 0.000 claims description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 10
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 claims description 10
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 9
- 125000004442 acylamino group Chemical group 0.000 claims description 9
- 125000004423 acyloxy group Chemical group 0.000 claims description 9
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 9
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 claims description 9
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 8
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 8
- 125000002102 aryl alkyloxo group Chemical group 0.000 claims description 8
- 150000002148 esters Chemical class 0.000 claims description 8
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 claims description 8
- 229910052717 sulfur Inorganic materials 0.000 claims description 8
- 150000007970 thio esters Chemical class 0.000 claims description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 7
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 6
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 6
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 6
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 6
- 239000012280 lithium aluminium hydride Substances 0.000 claims description 6
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 claims description 6
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 5
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 claims description 5
- 239000004202 carbamide Substances 0.000 claims description 5
- LZWQNOHZMQIFBX-UHFFFAOYSA-N lithium;2-methylpropan-2-olate Chemical compound [Li+].CC(C)(C)[O-] LZWQNOHZMQIFBX-UHFFFAOYSA-N 0.000 claims description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 5
- 125000002577 pseudohalo group Chemical group 0.000 claims description 5
- 229940124530 sulfonamide Drugs 0.000 claims description 5
- 150000003456 sulfonamides Chemical class 0.000 claims description 5
- 150000003457 sulfones Chemical class 0.000 claims description 5
- 150000003462 sulfoxides Chemical class 0.000 claims description 5
- 125000004001 thioalkyl group Chemical group 0.000 claims description 5
- 150000003568 thioethers Chemical class 0.000 claims description 5
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 claims description 4
- QARVLSVVCXYDNA-UHFFFAOYSA-N phenyl bromide Natural products BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 claims description 4
- 229910010084 LiAlH4 Inorganic materials 0.000 claims description 3
- YCOXTKKNXUZSKD-UHFFFAOYSA-N as-o-xylenol Natural products CC1=CC=C(O)C=C1C YCOXTKKNXUZSKD-UHFFFAOYSA-N 0.000 claims description 3
- 238000001914 filtration Methods 0.000 claims description 3
- SNHMUERNLJLMHN-UHFFFAOYSA-N iodobenzene Chemical compound IC1=CC=CC=C1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 claims description 3
- GRJHONXDTNBDTC-UHFFFAOYSA-N phenyl trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=CC=CC=C1 GRJHONXDTNBDTC-UHFFFAOYSA-N 0.000 claims description 3
- RNAIPFYGOQYYTF-XRLMYZCVSA-N (2s,5s)-1-cyclopenta-2,4-dien-1-yl-2,5-di(propan-2-yl)phospholane;iron(2+) Chemical compound [Fe+2].CC(C)[C@@H]1CC[C@@H](C(C)C)P1[C-]1C=CC=C1.CC(C)[C@@H]1CC[C@@H](C(C)C)P1[C-]1C=CC=C1 RNAIPFYGOQYYTF-XRLMYZCVSA-N 0.000 claims description 2
- QXXSYUGLJIMLFH-VUTIHBPYSA-N (2s,5s)-1-cyclopenta-2,4-dien-1-yl-2,5-dimethylphospholane;iron(2+) Chemical compound [Fe+2].C[C@H]1CC[C@H](C)P1[C-]1C=CC=C1.C[C@H]1CC[C@H](C)P1[C-]1C=CC=C1 QXXSYUGLJIMLFH-VUTIHBPYSA-N 0.000 claims description 2
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 claims description 2
- HFHFWISYOSHESF-OLNLWVBHSA-N C(C)[C@@H]1P([C@H](CC1)CC)[C-]1C=CC=C1.[C-]1(C=CC=C1)P1[C@H](CC[C@@H]1CC)CC.[Fe+2] Chemical compound C(C)[C@@H]1P([C@H](CC1)CC)[C-]1C=CC=C1.[C-]1(C=CC=C1)P1[C@H](CC[C@@H]1CC)CC.[Fe+2] HFHFWISYOSHESF-OLNLWVBHSA-N 0.000 claims description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 claims description 2
- 239000012346 acetyl chloride Substances 0.000 claims description 2
- 125000001691 aryl alkyl amino group Chemical group 0.000 claims description 2
- 125000001769 aryl amino group Chemical group 0.000 claims description 2
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 claims description 2
- QARVLSVVCXYDNA-IDEBNGHGSA-N bromobenzene Chemical group Br[13C]1=[13CH][13CH]=[13CH][13CH]=[13CH]1 QARVLSVVCXYDNA-IDEBNGHGSA-N 0.000 claims description 2
- GPAYUJZHTULNBE-UHFFFAOYSA-N diphenylphosphine Chemical compound C=1C=CC=CC=1PC1=CC=CC=C1 GPAYUJZHTULNBE-UHFFFAOYSA-N 0.000 claims description 2
- 230000003301 hydrolyzing effect Effects 0.000 claims description 2
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims 4
- 150000003951 lactams Chemical class 0.000 abstract description 126
- 238000006254 arylation reaction Methods 0.000 abstract description 46
- 238000006886 vinylation reaction Methods 0.000 abstract description 42
- 238000010276 construction Methods 0.000 abstract description 4
- 239000012039 electrophile Substances 0.000 abstract description 3
- 230000007704 transition Effects 0.000 abstract description 2
- 150000003953 γ-lactams Chemical class 0.000 abstract 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 214
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 130
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 112
- 235000019439 ethyl acetate Nutrition 0.000 description 90
- 239000000243 solution Substances 0.000 description 80
- 239000000047 product Substances 0.000 description 67
- 239000000741 silica gel Substances 0.000 description 66
- 229910002027 silica gel Inorganic materials 0.000 description 66
- 238000003818 flash chromatography Methods 0.000 description 59
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 58
- 238000005160 1H NMR spectroscopy Methods 0.000 description 57
- 239000011780 sodium chloride Substances 0.000 description 56
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 53
- 239000010408 film Substances 0.000 description 52
- 238000004808 supercritical fluid chromatography Methods 0.000 description 44
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 42
- 239000000758 substrate Substances 0.000 description 38
- IWELDVXSEVIIGI-UHFFFAOYSA-N piperazin-2-one Chemical class O=C1CNCCN1 IWELDVXSEVIIGI-UHFFFAOYSA-N 0.000 description 34
- 229910052723 transition metal Inorganic materials 0.000 description 34
- 238000000746 purification Methods 0.000 description 33
- 239000011541 reaction mixture Substances 0.000 description 33
- 230000002829 reductive effect Effects 0.000 description 33
- 150000003624 transition metals Chemical class 0.000 description 33
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 31
- 239000000377 silicon dioxide Substances 0.000 description 31
- 125000000623 heterocyclic group Chemical group 0.000 description 30
- 125000001183 hydrocarbyl group Chemical group 0.000 description 30
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 25
- 239000012230 colorless oil Substances 0.000 description 24
- 229920006395 saturated elastomer Polymers 0.000 description 23
- 238000003756 stirring Methods 0.000 description 22
- 230000000694 effects Effects 0.000 description 21
- 238000006579 Tsuji-Trost allylation reaction Methods 0.000 description 20
- 239000002585 base Substances 0.000 description 20
- 229920002554 vinyl polymer Polymers 0.000 description 20
- 239000007832 Na2SO4 Substances 0.000 description 19
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 19
- 238000011156 evaluation Methods 0.000 description 19
- 229910052938 sodium sulfate Inorganic materials 0.000 description 19
- NQPJDJVGBDHCAD-UHFFFAOYSA-N 1,3-diazinan-2-one Chemical class OC1=NCCCN1 NQPJDJVGBDHCAD-UHFFFAOYSA-N 0.000 description 17
- 238000011068 loading method Methods 0.000 description 17
- LKUDPHPHKOZXCD-UHFFFAOYSA-N 1,3,5-trimethoxybenzene Chemical compound COC1=CC(OC)=CC(OC)=C1 LKUDPHPHKOZXCD-UHFFFAOYSA-N 0.000 description 16
- 150000001500 aryl chlorides Chemical class 0.000 description 16
- 239000003921 oil Substances 0.000 description 16
- 235000019198 oils Nutrition 0.000 description 16
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 15
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 15
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 14
- 125000005842 heteroatom Chemical group 0.000 description 14
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 13
- PNLQPWWBHXMFCA-UHFFFAOYSA-N 2-chloroprop-1-ene Chemical compound CC(Cl)=C PNLQPWWBHXMFCA-UHFFFAOYSA-N 0.000 description 12
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- 230000003197 catalytic effect Effects 0.000 description 12
- 229910052751 metal Chemical class 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 11
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 11
- 239000000654 additive Substances 0.000 description 11
- 229910052681 coesite Inorganic materials 0.000 description 11
- 229910052906 cristobalite Inorganic materials 0.000 description 11
- 239000012074 organic phase Substances 0.000 description 11
- 239000007858 starting material Substances 0.000 description 11
- 229910052682 stishovite Inorganic materials 0.000 description 11
- 238000006467 substitution reaction Methods 0.000 description 11
- 229910052905 tridymite Inorganic materials 0.000 description 11
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 10
- 239000008346 aqueous phase Substances 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 10
- KWYUFKZDYYNOTN-UHFFFAOYSA-M potassium hydroxide Inorganic materials [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- KWISWUFGPUHDRY-UHFFFAOYSA-N 1-Chloro-2-methylpropene Chemical compound CC(C)=CCl KWISWUFGPUHDRY-UHFFFAOYSA-N 0.000 description 9
- YUKILTJWFRTXGB-UHFFFAOYSA-N 1-chloro-3-methoxybenzene Chemical compound COC1=CC=CC(Cl)=C1 YUKILTJWFRTXGB-UHFFFAOYSA-N 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 9
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 9
- 239000010410 layer Substances 0.000 description 9
- 239000002184 metal Chemical class 0.000 description 9
- WXHIJDCHNDBCNY-UHFFFAOYSA-N palladium dihydride Chemical compound [PdH2] WXHIJDCHNDBCNY-UHFFFAOYSA-N 0.000 description 9
- 229910000104 sodium hydride Inorganic materials 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- CRUILBNAQILVHZ-UHFFFAOYSA-N 1,2,3-trimethoxybenzene Chemical compound COC1=CC=CC(OC)=C1OC CRUILBNAQILVHZ-UHFFFAOYSA-N 0.000 description 8
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 230000008878 coupling Effects 0.000 description 8
- 238000010168 coupling process Methods 0.000 description 8
- 238000005859 coupling reaction Methods 0.000 description 8
- 125000004122 cyclic group Chemical group 0.000 description 8
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 8
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 239000000376 reactant Substances 0.000 description 8
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 8
- 238000004809 thin layer chromatography Methods 0.000 description 8
- WKSFKEGGQALVAF-UHFFFAOYSA-N 1-benzyl-1,3-diazinan-2-one Chemical compound O=C1NCCCN1CC1=CC=CC=C1 WKSFKEGGQALVAF-UHFFFAOYSA-N 0.000 description 7
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 7
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 7
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 7
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 239000008194 pharmaceutical composition Substances 0.000 description 7
- 125000006239 protecting group Chemical group 0.000 description 7
- 239000012312 sodium hydride Substances 0.000 description 7
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 7
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 6
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 6
- 229910006148 NiII Inorganic materials 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 6
- 125000001246 bromo group Chemical group Br* 0.000 description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 6
- AIYUHDOJVYHVIT-UHFFFAOYSA-M caesium chloride Chemical compound [Cl-].[Cs+] AIYUHDOJVYHVIT-UHFFFAOYSA-M 0.000 description 6
- 239000006260 foam Substances 0.000 description 6
- 238000009815 homocoupling reaction Methods 0.000 description 6
- 150000002576 ketones Chemical class 0.000 description 6
- 150000002596 lactones Chemical class 0.000 description 6
- 239000012434 nucleophilic reagent Substances 0.000 description 6
- 239000003960 organic solvent Substances 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 6
- 239000012041 precatalyst Substances 0.000 description 6
- VNUGUFKJRPDTPJ-UHFFFAOYSA-N prop-2-enyl cyanoformate Chemical compound C=CCOC(=O)C#N VNUGUFKJRPDTPJ-UHFFFAOYSA-N 0.000 description 6
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 6
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 6
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 5
- MWTBZONAQZFWDV-UHFFFAOYSA-N 1-aminopiperazin-2-one Chemical compound NN1CCNCC1=O MWTBZONAQZFWDV-UHFFFAOYSA-N 0.000 description 5
- GIQYJSUDPMGEOZ-UHFFFAOYSA-N 1-methyldiazepan-3-one Chemical compound CN1CCCCC(=O)N1 GIQYJSUDPMGEOZ-UHFFFAOYSA-N 0.000 description 5
- 241000282414 Homo sapiens Species 0.000 description 5
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 229910052786 argon Inorganic materials 0.000 description 5
- 125000002619 bicyclic group Chemical group 0.000 description 5
- 210000004027 cell Anatomy 0.000 description 5
- 230000003247 decreasing effect Effects 0.000 description 5
- 230000002950 deficient Effects 0.000 description 5
- CRKULFPPCILKNJ-UHFFFAOYSA-N diazepan-3-one Chemical class O=C1CCCCNN1 CRKULFPPCILKNJ-UHFFFAOYSA-N 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 150000002170 ethers Chemical class 0.000 description 5
- 125000001153 fluoro group Chemical group F* 0.000 description 5
- 150000004702 methyl esters Chemical class 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 5
- 239000008177 pharmaceutical agent Substances 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 125000003396 thiol group Chemical group [H]S* 0.000 description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 5
- KJCIMSSFGUGTGA-UHFFFAOYSA-N 1-methylpiperazin-2-one Chemical compound CN1CCNCC1=O KJCIMSSFGUGTGA-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 4
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 4
- DMOLTNKQLUAXPI-UHFFFAOYSA-N [2-(4-tert-butyl-4,5-dihydro-1,3-oxazol-2-yl)phenyl]-diphenylphosphane Chemical compound CC(C)(C)C1COC(C=2C(=CC=CC=2)P(C=2C=CC=CC=2)C=2C=CC=CC=2)=N1 DMOLTNKQLUAXPI-UHFFFAOYSA-N 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 4
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 description 4
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 230000003292 diminished effect Effects 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 238000011065 in-situ storage Methods 0.000 description 4
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 150000002894 organic compounds Chemical class 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 4
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical compound [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 description 4
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 4
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 4
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 4
- 230000009257 reactivity Effects 0.000 description 4
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 4
- 238000004611 spectroscopical analysis Methods 0.000 description 4
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 4
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 4
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 4
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 4
- WGTYBPLFGIVFAS-UHFFFAOYSA-M tetramethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)C WGTYBPLFGIVFAS-UHFFFAOYSA-M 0.000 description 4
- 229940030010 trimethoxybenzene Drugs 0.000 description 4
- 150000003952 β-lactams Chemical class 0.000 description 4
- NMBHLXCMHMXPAX-UHFFFAOYSA-N 1-benzyldiazepan-3-one Chemical compound C(C1=CC=CC=C1)N1NC(CCCC1)=O NMBHLXCMHMXPAX-UHFFFAOYSA-N 0.000 description 3
- DBSPZYVXETVRMN-UHFFFAOYSA-N 1-ethyl-1,3-diazinan-2-one Chemical compound CCN1CCCNC1=O DBSPZYVXETVRMN-UHFFFAOYSA-N 0.000 description 3
- UZOOHZSLRFBNQD-UHFFFAOYSA-N 2-bromo-5-chloro-1,3-benzoxazole Chemical compound ClC1=CC=C2OC(Br)=NC2=C1 UZOOHZSLRFBNQD-UHFFFAOYSA-N 0.000 description 3
- SBYMUDUGTIKLCR-UHFFFAOYSA-N 2-chloroethenylbenzene Chemical compound ClC=CC1=CC=CC=C1 SBYMUDUGTIKLCR-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- YCIPQJTZJGUXND-UHFFFAOYSA-N Aglaia odorata Alkaloid Natural products C1=CC(OC)=CC=C1C1(C(C=2C(=O)N3CCCC3=NC=22)C=3C=CC=CC=3)C2(O)C2=C(OC)C=C(OC)C=C2O1 YCIPQJTZJGUXND-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- 229910021585 Nickel(II) bromide Inorganic materials 0.000 description 3
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 101150003085 Pdcl gene Proteins 0.000 description 3
- 239000004809 Teflon Substances 0.000 description 3
- 229920006362 Teflon® Polymers 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 150000001409 amidines Chemical class 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 150000002466 imines Chemical class 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- AWIJRPNMLHPLNC-UHFFFAOYSA-N methanethioic s-acid Chemical compound SC=O AWIJRPNMLHPLNC-UHFFFAOYSA-N 0.000 description 3
- GTCAXTIRRLKXRU-UHFFFAOYSA-N methyl carbamate Chemical compound COC(N)=O GTCAXTIRRLKXRU-UHFFFAOYSA-N 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 235000010446 mineral oil Nutrition 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 3
- IPLJNQFXJUCRNH-UHFFFAOYSA-L nickel(2+);dibromide Chemical compound [Ni+2].[Br-].[Br-] IPLJNQFXJUCRNH-UHFFFAOYSA-L 0.000 description 3
- 150000002825 nitriles Chemical class 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 238000005457 optimization Methods 0.000 description 3
- 125000004043 oxo group Chemical group O=* 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- TYZYRCHEVXXLSJ-UHFFFAOYSA-N phenylmethoxymethoxymethoxymethylbenzene Chemical compound C=1C=CC=CC=1COCOCOCC1=CC=CC=C1 TYZYRCHEVXXLSJ-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- FCMLWBBLOASUSO-UHFFFAOYSA-N tert-butyl 3-oxopiperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNC(=O)C1 FCMLWBBLOASUSO-UHFFFAOYSA-N 0.000 description 3
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 3
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 3
- 238000012800 visualization Methods 0.000 description 3
- 239000011592 zinc chloride Substances 0.000 description 3
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 3
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 2
- GTIXSUJKFAATAE-UHFFFAOYSA-N (r)-c3-tunephos Chemical compound C=12C(C(=CC=C3)P(C=4C=CC=CC=4)C=4C=CC=CC=4)=C3OCCCOC2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 GTIXSUJKFAATAE-UHFFFAOYSA-N 0.000 description 2
- HABIAKDIZVCTLU-UHFFFAOYSA-N 1,3-diazinan-4-one Chemical compound O=C1CCNCN1 HABIAKDIZVCTLU-UHFFFAOYSA-N 0.000 description 2
- DKYBVKMIZODYKL-UHFFFAOYSA-N 1,3-diazinane Chemical class C1CNCNC1 DKYBVKMIZODYKL-UHFFFAOYSA-N 0.000 description 2
- QPPLBCQXWDBQFS-UHFFFAOYSA-N 1,4-diazepan-5-one Chemical class O=C1CCNCCN1 QPPLBCQXWDBQFS-UHFFFAOYSA-N 0.000 description 2
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 2
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 2
- ZIQMBOJQMDWMLJ-UHFFFAOYSA-N 4,5-dihydro-1,3-oxazol-2-ylphosphane Chemical compound PC1=NCCO1 ZIQMBOJQMDWMLJ-UHFFFAOYSA-N 0.000 description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 2
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- GQIPDOCYJYGOPV-UHFFFAOYSA-N O=C1NCCNC1.CNC(O)=O Chemical compound O=C1NCCNC1.CNC(O)=O GQIPDOCYJYGOPV-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- MXGXXBYVDMVJAO-UHFFFAOYSA-N [1-[2-bis(3,5-dimethylphenyl)phosphanylnaphthalen-1-yl]naphthalen-2-yl]-bis(3,5-dimethylphenyl)phosphane Chemical compound CC1=CC(C)=CC(P(C=2C=C(C)C=C(C)C=2)C=2C(=C3C=CC=CC3=CC=2)C=2C3=CC=CC=C3C=CC=2P(C=2C=C(C)C=C(C)C=2)C=2C=C(C)C=C(C)C=2)=C1 MXGXXBYVDMVJAO-UHFFFAOYSA-N 0.000 description 2
- GLYOPNLBKCBTMI-UHFFFAOYSA-N [2-chloro-2-(1-chloro-2-phenylethoxy)ethyl]benzene Chemical compound C=1C=CC=CC=1CC(Cl)OC(Cl)CC1=CC=CC=C1 GLYOPNLBKCBTMI-UHFFFAOYSA-N 0.000 description 2
- JZOSBBLJKXSBBN-UHFFFAOYSA-N [3-(4-diphenylphosphanyl-2,6-dimethoxypyridin-3-yl)-2,6-dimethoxypyridin-4-yl]-diphenylphosphane Chemical compound COC=1N=C(OC)C=C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)C=1C=1C(OC)=NC(OC)=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 JZOSBBLJKXSBBN-UHFFFAOYSA-N 0.000 description 2
- ZNORAFJUESSLTM-UHFFFAOYSA-N [4-[5-bis(3,5-ditert-butyl-4-methoxyphenyl)phosphanyl-1,3-benzodioxol-4-yl]-1,3-benzodioxol-5-yl]-bis(3,5-ditert-butyl-4-methoxyphenyl)phosphane Chemical compound C1=C(C(C)(C)C)C(OC)=C(C(C)(C)C)C=C1P(C=1C(=C2OCOC2=CC=1)C=1C(=CC=C2OCOC2=1)P(C=1C=C(C(OC)=C(C=1)C(C)(C)C)C(C)(C)C)C=1C=C(C(OC)=C(C=1)C(C)(C)C)C(C)(C)C)C1=CC(C(C)(C)C)=C(OC)C(C(C)(C)C)=C1 ZNORAFJUESSLTM-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 125000002015 acyclic group Chemical group 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 238000012435 analytical chromatography Methods 0.000 description 2
- 230000000561 anti-psychotic effect Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 150000001499 aryl bromides Chemical class 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 229940049706 benzodiazepine Drugs 0.000 description 2
- BAHFPJFBMJTOPU-UHFFFAOYSA-N benzyl 3-oxopiperazine-1-carboxylate Chemical compound C1CNC(=O)CN1C(=O)OCC1=CC=CC=C1 BAHFPJFBMJTOPU-UHFFFAOYSA-N 0.000 description 2
- IYVSQWKHFMWCIW-UHFFFAOYSA-N benzyl N-(benzenesulfonylmethyl)carbamate Chemical compound C1(=CC=CC=C1)S(=O)(=O)CNC(OCC1=CC=CC=C1)=O IYVSQWKHFMWCIW-UHFFFAOYSA-N 0.000 description 2
- 239000003782 beta lactam antibiotic agent Substances 0.000 description 2
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- FUSUHKVFWTUUBE-UHFFFAOYSA-N buten-2-one Chemical compound CC(=O)C=C FUSUHKVFWTUUBE-UHFFFAOYSA-N 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 125000004452 carbocyclyl group Chemical group 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 2
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 description 2
- 229960004170 clozapine Drugs 0.000 description 2
- 230000000536 complexating effect Effects 0.000 description 2
- 238000010668 complexation reaction Methods 0.000 description 2
- 239000007819 coupling partner Substances 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- QPMLSUSACCOBDK-UHFFFAOYSA-N diazepane Chemical compound C1CCNNCC1 QPMLSUSACCOBDK-UHFFFAOYSA-N 0.000 description 2
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000004519 grease Substances 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 229940030980 inova Drugs 0.000 description 2
- 206010022437 insomnia Diseases 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229930014626 natural product Natural products 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 238000006464 oxidative addition reaction Methods 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 150000004686 pentahydrates Chemical class 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- IYPZRUYMFDWKSS-UHFFFAOYSA-N piperazin-1-amine Chemical compound NN1CCNCC1 IYPZRUYMFDWKSS-UHFFFAOYSA-N 0.000 description 2
- RFIOZSIHFNEKFF-UHFFFAOYSA-M piperazine-1-carboxylate Chemical compound [O-]C(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-M 0.000 description 2
- 125000003367 polycyclic group Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 2
- 229910000105 potassium hydride Inorganic materials 0.000 description 2
- 238000012746 preparative thin layer chromatography Methods 0.000 description 2
- 230000000135 prohibitive effect Effects 0.000 description 2
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 2
- 239000012264 purified product Substances 0.000 description 2
- 150000004040 pyrrolidinones Chemical class 0.000 description 2
- 239000011669 selenium Substances 0.000 description 2
- 229910000077 silane Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 230000000707 stereoselective effect Effects 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- JYTNQNCOQXFQPK-MRXNPFEDSA-N suvorexant Chemical compound C([C@H]1C)CN(C=2OC3=CC=C(Cl)C=C3N=2)CCN1C(=O)C1=CC(C)=CC=C1N1N=CC=N1 JYTNQNCOQXFQPK-MRXNPFEDSA-N 0.000 description 2
- 229960001198 suvorexant Drugs 0.000 description 2
- XNSBXNNKFMZTSW-UHFFFAOYSA-N tert-butyl n-(benzenesulfonylmethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCS(=O)(=O)C1=CC=CC=C1 XNSBXNNKFMZTSW-UHFFFAOYSA-N 0.000 description 2
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 239000002132 β-lactam antibiotic Substances 0.000 description 2
- 229940124586 β-lactam antibiotics Drugs 0.000 description 2
- 150000003954 δ-lactams Chemical class 0.000 description 2
- 150000003955 ε-lactams Chemical class 0.000 description 2
- ASXSNYYGHRKVEQ-SFHVURJKSA-N (2S)-2-benzyl-2-[[(2-methylpropan-2-yl)oxycarbonylamino]methyl]pent-4-enoic acid Chemical compound C(C1=CC=CC=C1)[C@@](C(=O)O)(CC=C)CNC(=O)OC(C)(C)C ASXSNYYGHRKVEQ-SFHVURJKSA-N 0.000 description 1
- IFUCTRCZLDIJLE-HNNXBMFYSA-N (3S)-1-benzoyl-3-methyl-3-propylpiperazin-2-one Chemical compound C(C1=CC=CC=C1)(=O)N1C([C@@](NCC1)(CCC)C)=O IFUCTRCZLDIJLE-HNNXBMFYSA-N 0.000 description 1
- VIMMECPCYZXUCI-MIMFYIINSA-N (4s,6r)-6-[(1e)-4,4-bis(4-fluorophenyl)-3-(1-methyltetrazol-5-yl)buta-1,3-dienyl]-4-hydroxyoxan-2-one Chemical compound CN1N=NN=C1C(\C=C\[C@@H]1OC(=O)C[C@@H](O)C1)=C(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 VIMMECPCYZXUCI-MIMFYIINSA-N 0.000 description 1
- RRWJVRBCODZBAN-MRXNPFEDSA-N (6R)-4-benzoyl-6-propyl-1,4-diazabicyclo[4.2.0]octane-5,8-dione Chemical compound C(C1=CC=CC=C1)(=O)N1CCN2C(C[C@@]2(C1=O)CCC)=O RRWJVRBCODZBAN-MRXNPFEDSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- OTPDWCMLUKMQNO-UHFFFAOYSA-N 1,2,3,4-tetrahydropyrimidine Chemical compound C1NCC=CN1 OTPDWCMLUKMQNO-UHFFFAOYSA-N 0.000 description 1
- DTOUUUZOYKYHEP-UHFFFAOYSA-N 1,3-bis(2-ethylhexyl)-5-methyl-1,3-diazinan-5-amine Chemical compound CCCCC(CC)CN1CN(CC(CC)CCCC)CC(C)(N)C1 DTOUUUZOYKYHEP-UHFFFAOYSA-N 0.000 description 1
- 229940005561 1,4-benzoquinone Drugs 0.000 description 1
- WJIFKOVZNJTSGO-UHFFFAOYSA-N 1-bromo-3-methylbenzene Chemical compound CC1=CC=CC(Br)=C1 WJIFKOVZNJTSGO-UHFFFAOYSA-N 0.000 description 1
- BLRHMMGNCXNXJL-UHFFFAOYSA-N 1-methylindole Chemical compound C1=CC=C2N(C)C=CC2=C1 BLRHMMGNCXNXJL-UHFFFAOYSA-N 0.000 description 1
- GEKQOEQTSMDIKA-UHFFFAOYSA-N 1-prop-2-ynyl-1,3-diazinan-2-one Chemical compound O=C1NCCCN1CC#C GEKQOEQTSMDIKA-UHFFFAOYSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- FALCMQXTWHPRIH-UHFFFAOYSA-N 2,3-dichloroprop-1-ene Chemical compound ClCC(Cl)=C FALCMQXTWHPRIH-UHFFFAOYSA-N 0.000 description 1
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical class O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- GAGJMOQGABUOBK-UHFFFAOYSA-N 3-aminopropanamide;hydrochloride Chemical compound Cl.NCCC(N)=O GAGJMOQGABUOBK-UHFFFAOYSA-N 0.000 description 1
- DGEHWJDSVVYFBP-UHFFFAOYSA-N 3-benzoyl-5-methyl-1-[(2-methylpropan-2-yl)oxycarbonyl]-4-oxo-1,3-diazinane-5-carboxylic acid Chemical compound CC1(CN(CN(C1=O)C(=O)C2=CC=CC=C2)C(=O)OC(C)(C)C)C(=O)O DGEHWJDSVVYFBP-UHFFFAOYSA-N 0.000 description 1
- QSVDFJNXDKTKTJ-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1h-indene Chemical compound C1CCCC2=C1CC=C2 QSVDFJNXDKTKTJ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QAEIIJPYZWWCNB-UHFFFAOYSA-N C(C1=CC=CC=C1)(=O)N1CN(CCC1=O)C(=O)OC(C)(C)C Chemical compound C(C1=CC=CC=C1)(=O)N1CN(CCC1=O)C(=O)OC(C)(C)C QAEIIJPYZWWCNB-UHFFFAOYSA-N 0.000 description 1
- DYRWLYGRVMHRPQ-UHFFFAOYSA-N C1CN(C(=O)C(N1C(=O)O)C(=O)O)C(=O)C2=CC=CC=C2 Chemical compound C1CN(C(=O)C(N1C(=O)O)C(=O)O)C(=O)C2=CC=CC=C2 DYRWLYGRVMHRPQ-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 101150041968 CDC13 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 238000007445 Chromatographic isolation Methods 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- PCKPVGOLPKLUHR-UHFFFAOYSA-N OH-Indolxyl Natural products C1=CC=C2C(O)=CNC2=C1 PCKPVGOLPKLUHR-UHFFFAOYSA-N 0.000 description 1
- 229910004727 OSO3H Inorganic materials 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 229910021605 Palladium(II) bromide Inorganic materials 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical class CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 229910006069 SO3H Inorganic materials 0.000 description 1
- 229910002808 Si–O–Si Inorganic materials 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 229910001508 alkali metal halide Inorganic materials 0.000 description 1
- 150000008045 alkali metal halides Chemical class 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 150000001349 alkyl fluorides Chemical class 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 125000005219 aminonitrile group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- MXMOTZIXVICDSD-UHFFFAOYSA-N anisoyl chloride Chemical compound COC1=CC=C(C(Cl)=O)C=C1 MXMOTZIXVICDSD-UHFFFAOYSA-N 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 230000000949 anxiolytic effect Effects 0.000 description 1
- 229940005530 anxiolytics Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 150000001503 aryl iodides Chemical class 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 238000000065 atmospheric pressure chemical ionisation Methods 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 210000004082 barrier epithelial cell Anatomy 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- CUCPUSVHPQCAOD-UHFFFAOYSA-N benzyl 4-benzoyl-3-oxopiperazine-1-carboxylate Chemical compound C(C1=CC=CC=C1)(=O)N1C(CN(CC1)C(=O)OCC1=CC=CC=C1)=O CUCPUSVHPQCAOD-UHFFFAOYSA-N 0.000 description 1
- 239000003781 beta lactamase inhibitor Substances 0.000 description 1
- 150000001576 beta-amino acids Chemical class 0.000 description 1
- 229940126813 beta-lactamase inhibitor Drugs 0.000 description 1
- BVCRERJDOOBZOH-UHFFFAOYSA-N bicyclo[2.2.1]heptanyl Chemical group C1C[C+]2CC[C-]1C2 BVCRERJDOOBZOH-UHFFFAOYSA-N 0.000 description 1
- JBFDZEJAJZJORO-UHFFFAOYSA-N bicyclo[4.1.0]hept-3-ene Chemical compound C1C=CCC2CC21 JBFDZEJAJZJORO-UHFFFAOYSA-N 0.000 description 1
- DCRRIOWFXXDTHV-UHFFFAOYSA-N bicyclo[4.2.0]oct-3-ene Chemical compound C1C=CCC2CCC21 DCRRIOWFXXDTHV-UHFFFAOYSA-N 0.000 description 1
- RPZUBXWEQBPUJR-UHFFFAOYSA-N bicyclo[4.2.0]octane Chemical compound C1CCCC2CCC21 RPZUBXWEQBPUJR-UHFFFAOYSA-N 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 125000005884 carbocyclylalkyl group Chemical group 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 230000006315 carbonylation Effects 0.000 description 1
- 238000005810 carbonylation reaction Methods 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- FWXAUDSWDBGCMN-ZEQRLZLVSA-N chiraphos Chemical compound C=1C=CC=CC=1P([C@@H](C)[C@H](C)P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 FWXAUDSWDBGCMN-ZEQRLZLVSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000012650 click reaction Methods 0.000 description 1
- 230000002153 concerted effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000006880 cross-coupling reaction Methods 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- XUWHAWMETYGRKB-UHFFFAOYSA-N delta-valerolactam Natural products O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000000664 diazo group Chemical group [N-]=[N+]=[*] 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- 230000004890 epithelial barrier function Effects 0.000 description 1
- OLAMWIPURJGSKE-UHFFFAOYSA-N et2o diethylether Chemical compound CCOCC.CCOCC OLAMWIPURJGSKE-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000007306 functionalization reaction Methods 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000012252 genetic analysis Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- 229960004867 hexetidine Drugs 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- ATQYNBNTEXNNIK-UHFFFAOYSA-N imidazol-2-ylidene Chemical group [C]1NC=CN1 ATQYNBNTEXNNIK-UHFFFAOYSA-N 0.000 description 1
- 150000002462 imidazolines Chemical class 0.000 description 1
- 150000007976 iminium ions Chemical class 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical compound C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 238000009434 installation Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 1
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 1
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- VASIZKWUTCETSD-UHFFFAOYSA-N manganese(II) oxide Inorganic materials [Mn]=O VASIZKWUTCETSD-UHFFFAOYSA-N 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- BCVXHSPFUWZLGQ-UHFFFAOYSA-N mecn acetonitrile Chemical compound CC#N.CC#N BCVXHSPFUWZLGQ-UHFFFAOYSA-N 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 1
- MUTCAPXLKRYEPR-ITWZMISCSA-N methyl (e,3r,5s)-7-[4-bromo-2,3-bis(4-fluorophenyl)-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyhept-6-enoate Chemical compound COC(=O)C[C@H](O)C[C@H](O)\C=C\N1C(C(C)C)=C(Br)C(C=2C=CC(F)=CC=2)=C1C1=CC=C(F)C=C1 MUTCAPXLKRYEPR-ITWZMISCSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- BMGNSKKZFQMGDH-FDGPNNRMSA-L nickel(2+);(z)-4-oxopent-2-en-2-olate Chemical compound [Ni+2].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O BMGNSKKZFQMGDH-FDGPNNRMSA-L 0.000 description 1
- QEKXARSPUFVXIX-UHFFFAOYSA-L nickel(2+);triphenylphosphane;dibromide Chemical compound [Ni+2].[Br-].[Br-].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QEKXARSPUFVXIX-UHFFFAOYSA-L 0.000 description 1
- KFBKRCXOTTUAFS-UHFFFAOYSA-N nickel;triphenylphosphane Chemical compound [Ni].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 KFBKRCXOTTUAFS-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- 125000005593 norbornanyl group Chemical group 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 150000002905 orthoesters Chemical class 0.000 description 1
- 150000003901 oxalic acid esters Chemical class 0.000 description 1
- 150000002916 oxazoles Chemical class 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- BHAAPTBBJKJZER-UHFFFAOYSA-N p-anisidine Chemical compound COC1=CC=C(N)C=C1 BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 1
- JKDRQYIYVJVOPF-FDGPNNRMSA-L palladium(ii) acetylacetonate Chemical compound [Pd+2].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O JKDRQYIYVJVOPF-FDGPNNRMSA-L 0.000 description 1
- INIOZDBICVTGEO-UHFFFAOYSA-L palladium(ii) bromide Chemical compound Br[Pd]Br INIOZDBICVTGEO-UHFFFAOYSA-L 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- KZQFPRKQBWRRHQ-UHFFFAOYSA-N phenyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OC1=CC=CC=C1 KZQFPRKQBWRRHQ-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 150000003870 salicylic acids Chemical class 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000012363 selectfluor Substances 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 125000003748 selenium group Chemical group *[Se]* 0.000 description 1
- 150000003346 selenoethers Chemical class 0.000 description 1
- 150000003962 selenoxides Chemical class 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000004426 substituted alkynyl group Chemical group 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- BTNRCYVVRFTNGI-UHFFFAOYSA-N tert-butyl 4-(4-methoxybenzoyl)-3-oxopiperazine-1-carboxylate Chemical compound COC1=CC=C(C(=O)N2C(CN(CC2)C(=O)OC(C)(C)C)=O)C=C1 BTNRCYVVRFTNGI-UHFFFAOYSA-N 0.000 description 1
- VOEATNGJULDSLO-UHFFFAOYSA-N tert-butyl 4-benzoyl-3-oxopiperazine-1-carboxylate Chemical compound C(C1=CC=CC=C1)(=O)N1C(CN(CC1)C(=O)OC(C)(C)C)=O VOEATNGJULDSLO-UHFFFAOYSA-N 0.000 description 1
- BKYGBKHDICSEDZ-UHFFFAOYSA-N tert-butyl 4-oxo-1,3-diazinane-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)NC1 BKYGBKHDICSEDZ-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 150000003628 tricarboxylic acids Chemical class 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- XLOMVQKBTHCTTD-UHFFFAOYSA-N zinc oxide Inorganic materials [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/36—Oxygen or sulfur atoms
- C07D207/38—2-Pyrrolones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/18—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D207/22—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/24—Oxygen or sulfur atoms
- C07D207/26—2-Pyrrolidones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/06—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/06—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members
- C07D241/08—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/18—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/08—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D507/00—Heterocyclic compounds containing a condensed beta-lactam ring system, not provided for by groups C07D463/00, C07D477/00 or C07D499/00 - C07D505/00; Such ring systems being further condensed
Definitions
- Nitrogen-containing heterocycles are ubiquitous structural motifs that can be found across all areas and applications of organic chemistry.
- piperazine a representative diazaheterocycle
- pyrrolidines and pyrrolidinones occur widely in nature, possess a wide range of biological and pharmacological properties, and occur readily in materials and catalysis.
- diazepane heterocycles are common structural motifs found in a variety of pharmaceuticals, including the benzodiazepine anxiolytics, the antipsychotic clozapine, and the anti-insomnia drug suvorexant.
- Incorporation of a carbonyl group in the heterocycle has been shown to be a useful means of introducing chirality.
- the oxo group when introduced on a carbon atom also bearing the nitrogen atom, may be used to selectively introduce chirality at the ⁇ position and later removed to produce the functionalized heterocycle or other downstream products.
- pyrrolidinones progress has been made toward enantioselective allylic alkylation and enantioselective ⁇ -acylation of ⁇ -butyrolactams; and in piperazines, Stoltz et al.
- the invention relates to a method for the preparation of a compound of formula (I): comprising treating a compound of formula (II): or a salt thereof; with a Pd or Ni catalyst comprising a chiral ligand; an optionally substituted aryl halide, optionally substituted aryl pseudohalide, optionally substituted heteroaryl halide, an optionally substituted heteroaryl pseudohalide, or and a base; wherein, as valence and stability permit,
- the invention relates to a compound of formula (I) made by any of the methods described herein.
- the invention relates to a method comprising preparing a compound of formula (I) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (I).
- the invention relates to a compound of formula (III) or formula (IV): wherein, as valence and stability permit,
- p is 0; the compound of formula (III) is a compound of formula (IIIa): and the compound of formula (IV) is a compound of formula (IVa): wherein, as valence and stability permit,
- p is 1; the compound of formula (III) is a compound of formula (IIIb): the compound of formula (IV) is a compound of formula (IVb): wherein, as valence and stability permit:
- the invention relates to a compound of formula (V), (VI), or (VII): wherein, as valence and stability permit,
- the invention relates to a compound selected from: and
- the invention relates to a compound of Formula (VIII): wherein, as valence and stability permit,
- the invention relates to a compound of Formula (IX) or (X): wherein, as valence and stability permit,
- the invention relates to a compound of formula (XI): wherein, as valence and stability permit,
- the invention relates to a compound of formula (XII): wherein, as valence and stability permit,
- the invention relates to a compound of formula (XIII): wherein, as valence and stability permit,
- the invention relates to a method for preparation of a compound of formula (IV): the method comprising: treating a compound of formula (III) or a salt thereof, with a Pd(0) catalyst and a ligand L under alkylation conditions, wherein, as valence and stability permit,
- the compound of formula (IV) is a compound of formula (IVa): the compound of formula (III) is a compound of formula (IIIa): or a salt thereof, with a Pd(0) catalyst and a ligand L under alkylation conditions, wherein, as valence and stability permit,
- the invention relates to a compound of formula (IVa) made by any of the methods described herein.
- the invention relates to a method comprising preparing a compound of formula (IVa) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (IVa).
- p is 1; the compound of formula (IV) is a compound of formula (IVb) the compound of formula (III) is a compound of formula (IIIb) or a salt thereof, wherein, as valence and stability permit,
- the invention relates to a compound of formula (E): or a salt thereof, wherein, as valence and stability permit,
- the invention relates to a compound of formula (IVb) made by any of the methods described herein.
- the invention relates to a method comprising preparing a compound of formula (IVb) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (IVb).
- the invention relates to a method of preparation of a compound of formula (VI): the method comprising: treating a compound of formula (V): or a salt thereof, with a Pd(0) catalyst and a ligand L, under alkylation conditions, wherein, as valence and stability permit, R 25 is C 1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen.
- the invention relates to a compound of formula (VI) made by any of the methods described herein.
- the invention relates to a method comprising preparing a compound of formula (VI) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (VI).
- the invention is directed to a method for preparation of a compound of formula (VIII): the method comprising: treating a compound of formula (IVa): with TFA in a solvent; wherein, as valence and stability permit,
- the invention relates to a compound of formula (VIII) made by any of the methods described herein.
- the invention relates to a method for the preparation of a compound of formula (IX): the method comprising: treating a compound of formula (IVa): or a salt thereof, with LiOH in a first solvent, wherein, as valence and stability permit,
- the invention relates to a compound of formula (IX) made by any of the methods described herein.
- the invention relates to a method of preparation of a compound of formula (XI): the method comprising: treating a compound of formula (IVa): or a salt thereof, with hydrogen gas in the presence of a Pd/C catalyst in a first solvent, to form a first product mixture; filtering the first product mixture, to form a crude first product; and treating the crude first product with TFA in a second solvent; wherein, as valence and stability permit,
- the invention relates to a compound of formula (XI) made by any of the methods described herein.
- the invention relates to method of preparation of a compound of formula (XIII): the method comprising: treating a compound of formula (VI): or a salt thereof, with TFA in a first solvent, to form a first product; treating the first product with lithium hydroxide in a second solvent, to form a second product; treating the second product with Boc 2 O in the presence of a base, to form a third product; treating the third product with K 2 CO 3 and R 13 -X 21 in a third solvent, to form a compound of formula (XIII); wherein, as valence and stability permit,
- the invention relates to a compound of formula (XIII) made by any of the methods described herein.
- the invention relates to any one of the compounds described in the Examples.
- FIG. 1 shows the structures of examples of enantioenriched phosphine ligands.
- acyl is art-recognized and refers to a group represented by the general formula hydrocarbyl-C(O)-, preferably alkyl-C(O)-.
- acylamino is art-recognized and refers to an amino group substituted with an acyl group and may be represented, for example, by the formula hydrocarbyl-C(O)NH-.
- acyloxy is art-recognized and refers to a group represented by the general formula hydrocarbyl-C(O)O-, preferably alkyl-C(O)O-.
- alkoxy refers to an alkyl group, preferably a lower alkyl group, having an oxygen attached thereto.
- Representative alkoxy groups include methoxy, trifluoromethoxy, ethoxy, propoxy, tert-butoxy and the like.
- alkoxyalkyl refers to an alkyl group substituted with an alkoxy group and may be represented by the general formula alkyl-O-alkyl.
- alkenyl refers to an aliphatic group containing at least one double bond and from 1 to about 20 carbon atoms, preferably from 1 to about 10 carbon atoms unless otherwise defined, and is intended to include both "unsubstituted alkenyls" and “substituted alkenyls", the latter of which refers to alkenyl moieties having substituents replacing a hydrogen on one or more carbons of the alkenyl group. Such substituents may occur on one or more carbons that are included or not included in one or more double bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed below, except where stability is prohibitive.
- alkenyl groups substitution of alkenyl groups by one or more alkyl, carbocyclyl, aryl, heterocyclyl, or heteroaryl groups is contemplated.
- a C 2 -C 6 alkenyl group is also referred to as a "lower alkenyl" group.
- alkyl group or “alkane” is a straight chained or branched non-aromatic hydrocarbon which is completely saturated. Typically, a straight chained or branched alkyl group has from 1 to about 20 carbon atoms, preferably from 1 to about 10 unless otherwise defined. Examples of straight chained and branched alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, tert-butyl, pentyl, hexyl, pentyl and octyl. A C 1 -C 6 straight chained or branched alkyl group is also referred to as a "lower alkyl" group.
- alkyl (or “lower alkyl) as used throughout the specification, examples, and claims is intended to include both “unsubstituted alkyls” and “substituted alkyls”, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- substituents can include, for example, a halogen (e.g., fluoro), a hydroxyl, an oxo, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxy, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, a halogen
- the substituent on substituted alkyls are selected from C 1 -C 6 alkyl, C 3 -C 6 cycloalkyl, halogen, carbonyl, cyano, or hydroxyl. In more preferred embodiments, the substituents on the substituted alkyls are selected from fluoro, carbonyl, cyano, or hydroxyl. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate.
- the substituents of a substituted alkyl may include substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl (including phosphonate and phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), -CF 3 , -CN and the like. Exemplary substituted alkyls are described below.
- Cycloalkyls can be further substituted with alkyls, alkenyls, alkoxys, alkylthios, aminoalkyls, carbonyl-substituted alkyls, -CF 3 , -CN, and the like.
- C x -C y when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain.
- C x -C y alkyl refers to substituted or unsubstituted saturated hydrocarbon groups, including straight-chain alkyl and branched-chain alkyl groups that contain from x to y carbons in the chain, including haloalkyl groups.
- Preferred haloalkyl groups include trifluoromethyl, difluoromethyl, 2,2,2-trifluoroethyl, and pentafluoroethyl.
- Co alkyl indicates a hydrogen where the group is in a terminal position, a bond if internal.
- the terms "C 2 -C y alkenyl” and “C 2 -C y alkynyl” refer to substituted or unsubstituted unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- alkylamino refers to an amino group substituted with at least one alkyl group.
- alkylthio refers to a thiol group substituted with an alkyl group and may be represented by the general formula alkyl-S-.
- alkynyl refers to an aliphatic group containing at least one triple bond and is intended to include both "unsubstituted alkynyls" and “substituted alkynyls", the latter of which refers to alkynyl moieties having substituents replacing a hydrogen on one or more carbons of the alkynyl group. Such substituents may occur on one or more carbons that are included or not included in one or more triple bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed above, except where stability is prohibitive. For example, substitution of alkynyl groups by one or more alkyl, carbocyclyl, aryl, heterocyclyl, or heteroaryl groups is contemplated.
- amide refers to a group wherein each R 10 independently represent a hydrogen or hydrocarbyl group, aryl, heteroaryl, acyl, or alkoxy, or two R 10 are taken together with the N atom to which they are attached complete a heterocycle having from 3 to 8 atoms in the ring structure.
- amine and “amino” are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by wherein each R 10 independently represents a hydrogen or a hydrocarbyl group, or two R 10 are taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
- aminoalkyl refers to an alkyl group substituted with an amino group.
- aralkyl refers to an alkyl group substituted with an aryl group.
- aryl as used herein include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon.
- the ring is a 6- to 10-membered ring, more preferably a 6-membered ring.
- aryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Aryl groups include benzene, naphthalene, phenanthrene, aniline, and the like.
- Example substitutions on an aryl group include a halogen, a haloalkyl such as trifluoromethyl, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl such as an alkylC(O)), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxy, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a silyl ether, a sulfhydryl, an alkylthio, a sulfate, a sulfonate,
- carboxylate is art-recognized and refers to a group wherein R 9 and R 10 independently represent hydrogen or a hydrocarbyl group, such as an alkyl group, or R 9 and R 10 taken together with the intervening atom(s) complete a heterocycle having from 4 to 8 atoms in the ring structure.
- Carbocycle refers to a saturated or unsaturated ring in which each atom of the ring is carbon.
- carbocycle includes both aromatic carbocycles and non-aromatic carbocycles.
- Non-aromatic carbocycles include both cycloalkyl and cycloalkenyl rings.
- Carbocycle includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings.
- Carbocycle includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings.
- fused carbocycle refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring.
- Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings.
- an aromatic ring e.g., phenyl
- a saturated or unsaturated ring e.g., cyclohexane, cyclopentane, or cyclohexene.
- Exemplary "carbocycles” include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5-cyclooctadiene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]oct-3-ene, naphthalene and adamantane.
- Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4,5,6,7-tetrahydro-1H-indene and bicyclo[4.1.0]hept-3-ene.
- Carbocycles may be substituted at any one or more positions capable of bearing a hydrogen atom.
- Carbocyclylalkyl refers to an alkyl group substituted with an carbocycle group.
- carbonate is art-recognized and refers to a group -OCO 2 -R A , wherein R A represents a hydrocarbyl group.
- a “cycloalkyl” group is a cyclic hydrocarbon, which is completely saturated.
- “Cycloalkyl” includes monocyclic and bicyclic rings. Typically, a monocyclic cycloalkyl group has from 3 to about 10 carbon atoms, from 3 to 8 carbon atoms, or more typically from 3 to 6 carbon atoms, unless otherwise defined.
- the second ring of a bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings. Cycloalkyl includes bicyclic substituents in which one, two or three or more atoms are shared between the two rings (e.g., fused bicyclic substituents, bridged bicyclic substituents, and spirocyclic substituents).
- fused bicyclic substituent refers to a bicyclic substituent in which two rings share two adjacent atoms. In other words, the rings share one covalent bond, i.e., the so-called bridgehead atoms are directly connected.
- bridgehead atoms are directly connected.
- each of the rings shares two adjacent atoms with the other ring, and the second ring of a fused bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings.
- a "cycloalkenyl” group is a cyclic hydrocarbon containing one or more double bonds.
- bridged bicyclic substituent refers to a bicyclic substituent in which the two rings share three or more atoms, separating the two bridgehead atoms by a bridge containing at least one atom.
- norbornanyl also known as bicyclo[2.2.1]heptanyl
- spirocyclic substituent refers to a bicyclic substituent in which the two rings have only one single atom, the spiro atom, in common.
- dithiosulfide is art-recognized and refers to a group -S-S-R A , wherein R A represents a hydrocarbyl group.
- esters refers to a group -C(O)OR 10 wherein R 10 represents a hydrocarbyl group.
- ether refers to a hydrocarbyl group linked through an oxygen to another hydrocarbyl group. Accordingly, an ether substituent of a hydrocarbyl group may be hydrocarbyl-O-. Ethers may be either symmetrical or unsymmetrical. Examples of ethers include, but are not limited to, heterocycle-O-heterocycle and aryl-O-heterocycle. Ethers include "alkoxyalkyl” groups, which may be represented by the general formula alkyl-O-alkyl.
- heteroalkyl refers to a saturated or unsaturated chain of carbon atoms and at least one heteroatom, for example, wherein no two heteroatoms are adjacent.
- heteroarylkyl refers to an alkyl group substituted with a hetaryl group.
- heteroaryl and “hetaryl” include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heteroaryl and “hetaryl” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heteroaryl groups include 5- to 10-membered cyclic or polycyclic ring systems, such as pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, and pyrimidine, and the like.
- heteroatom as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
- heterocyclyl refers to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, preferably 3-to 7-membered rings, more preferably 5- to 6-membered rings, in some instances, most preferably a 5-membered ring, in other instances, most preferably a 6-membered ring, which ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heterocyclyl and “heterocyclic” also include 5- to 10- membered polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heterocyclyl groups include, for example, pyrrolidine, piperidine, piperazine, pyrrolidine, tetrahydropyran, tetrahydrofuran, morpholine, lactones, lactams, oxazoles, imidazolines, and the like.
- heterocyclylalkyl refers to an alkyl group substituted with a heterocycle group.
- Hydrocarbyl groups include, but are not limited to aryl, heteroaryl, carbocycle, heterocyclyl, alkyl, alkenyl, alkynyl, and combinations thereof.
- hydroxyalkyl refers to an alkyl group substituted with a hydroxy group.
- lower when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups where there are ten or fewer non-hydrogen atoms in the substituent, preferably six or fewer, more preferably three or fewer.
- acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy substituents defined herein are respectively lower acyl, lower acyloxy, lower alkyl, lower alkenyl, lower alkynyl, or lower alkoxy, whether they appear alone or in combination with other substituents, such as in the recitations hydroxyalkyl and aralkyl (in which case, for example, the atoms within the aryl group are not counted when counting the carbon atoms in the alkyl substituent).
- orthoester as used herein is art-recognized and refers to a group - C(OR A ) 3 , wherein each R A independently represents hydrogen or a hydrocarbyl, such as alkyl, or any occurrence of R A taken together with another and the intervening atom(s) complete a heterocycle having from 4 to 8 atoms in the ring structure.
- polycyclyl refers to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls) in which two or more atoms are common to two adjoining rings, e.g., the rings are "fused rings".
- Each of the rings of the polycycle can be substituted or unsubstituted.
- each ring of the polycycle contains from 3 to 10 atoms in the ring, preferably from 5 to 7.
- pseudohalide means a polyatomic analogue of a halide.
- Preferred pseudohalides include sulfonates, e.g., arylsulfonates and alkylsulfonates, such as p-toluenesulfonate and trifluoromethanesulfonate.
- selenium is equivalent to an ether, wherein the oxygen is replaced with a selenium.
- siloxane is art-recognized and refers to a group with an Si-O-Si linkage, such as the group -Si(R A ) 2 -O-Si-(R A ) 3 , wherein each R A independently represents hydrogen or hydrocarbyl, such as alkyl, or two R A taken together with the intervening atom(s) complete a heterocycle having from 4 to 8 atoms in the ring structure.
- sil refers to a silicon moiety with three hydrocarbyl moieties attached thereto.
- substituted refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds.
- the permissible substituents can be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms.
- Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, an oxo, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxy, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an alkyl, an amino, an amido, an amidine, an imine, an oxime, a cyano, a nitro, an azido, a silyl ether, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl,
- the substituents on substituted alkyls are selected from C 1 -C 6 alkyl, C 3 -C 6 cycloalkyl, halogen, carbonyl, cyano, or hydroxyl. In more preferred embodiments, the substituents on substituted alkyls are selected from fluoro, carbonyl, cyano, or hydroxyl. It will be understood by those skilled in the art that substituents can themselves be substituted, if appropriate. Unless specifically stated as "unsubstituted,” references to chemical moieties herein are understood to include substituted variants. For example, reference to an "aryl" group or moiety implicitly includes both substituted and unsubstituted variants.
- sulfate is art-recognized and refers to the group -OSO 3 H, or a pharmaceutically acceptable salt thereof.
- sulfonamide is art-recognized and refers to the group represented by the general formulae wherein R 9 and R 10 independently represents hydrogen or hydrocarbyl, such as alkyl, or R 9 and R 10 taken together with the intervening atom(s) complete a heterocycle having from 4 to 8 atoms in the ring structure.
- sulfoxide is art-recognized and refers to the group -S(O)-R 10 , wherein R 10 represents a hydrocarbyl.
- sulfonate is art-recognized and refers to the group SO 3 H, or a pharmaceutically acceptable salt thereof.
- sulfone is art-recognized and refers to the group -S(O) 2 -R 10 , wherein R 10 represents a hydrocarbyl.
- thioalkyl refers to an alkyl group substituted with a thiol group.
- thioester refers to a group -C(O)SR 10 or -SC(O)R 10 wherein R 10 represents a hydrocarbyl.
- thioether is equivalent to an ether, wherein the oxygen is replaced with a sulfur.
- urea is art-recognized and may be represented by the general formula wherein each R 9 and R 10 independently represents hydrogen or a hydrocarbyl, such as alkyl, or either occurrence of R 9 taken together with R 10 or the other R 9 and the intervening atom(s) complete a heterocycle having from 4 to 8 atoms in the ring structure.
- Protecting group refers to a group of atoms that, when attached to a reactive functional group in a molecule, mask, reduce or prevent the reactivity of the functional group. Typically, a protecting group may be selectively removed as desired during the course of a synthesis. Examples of protecting groups can be found in Greene and Wuts, Protective Groups in Organic Chemistry, 3rd Ed., 1999, John Wiley & Sons, NY and Harrison et al., Compendium of Synthetic Organic Methods, Vols. 1-8, 1971-1996, John Wiley & Sons, NY .
- nitrogen protecting groups include, but are not limited to, formyl, acetyl, trifluoroacetyl, anisoyl ("An”), benzyl (“Bn”), benzoyl (“Bz”), benzyloxycarbonyl (“CBZ”), tert-butoxycarbonyl (“Boc”), trimethylsilyl (“TMS”), 2- trimethylsilyl-ethanesulfonyl (“TES”), trityl and substituted trityl groups, allyloxycarbonyl, 9- fluorenylmethyloxycarbonyl (“FMOC”), nitro-veratryloxycarbonyl (“NVOC”) and the like.
- hydroxyl protecting groups include, but are not limited to, those where the hydroxyl group is either acylated (esterified) or alkylated such as benzyl and trityl ethers, as well as alkyl ethers, tetrahydropyranyl ethers, trialkylsilyl ethers (e.g., TMS or TIPS groups), glycol ethers, such as ethylene glycol and propylene glycol derivatives and allyl ethers.
- compositions, excipients, adjuvants, polymers and other materials and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- “Pharmaceutically acceptable salt” or “salt” is used herein to refer to an acid addition salt or a basic addition salt that is suitable for or compatible with the treatment of patients.
- pharmaceutically acceptable acid addition salt means any non-toxic organic or inorganic salt of any base compounds disclosed herein.
- Illustrative inorganic acids that form suitable salts include hydrochloric, hydrobromic, sulfuric and phosphoric acids, as well as metal salts such as sodium monohydrogen orthophosphate and potassium hydrogen sulfate.
- Illustrative organic acids that form suitable salts include mono-, di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic, phenylacetic, cinnamic and salicylic acids, as well as sulfonic acids such as p-toluene sulfonic and methanesulfonic acids. Either the mono or di-acid salts can be formed, and such salts may exist in either a hydrated, solvated or substantially anhydrous form.
- mono-, di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic, phenylacetic, cinnamic and salicylic acids, as well as sul
- the acid addition salts of compounds disclosed herein are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms.
- the selection of the appropriate salt will be known to one skilled in the art.
- Other non-pharmaceutically acceptable salts e.g., oxalates, may be used, for example, in the isolation of compounds of the invention for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
- pharmaceutically acceptable basic addition salt means any non-toxic organic or inorganic base addition salt of any acid compounds of the invention, or any of their intermediates.
- Illustrative inorganic bases that form suitable salts include lithium, sodium, potassium, calcium, magnesium, or barium hydroxide.
- Illustrative organic bases which form suitable salts include aliphatic, alicyclic, or aromatic organic amines such as methylamine, trimethylamine and picoline or ammonia. The selection of the appropriate salt will be known to a person skilled in the art.
- stereogenic center in their structure.
- This stereogenic center may be present in a R or a S configuration, said R and S notation is used in correspondence with the rules described in Pure Appl. Chem. (1976), 45, 11-30 .
- the disclosure contemplates all stereoisomeric forms such as enantiomeric and diastereoisomeric forms of the compounds, salts, prodrugs or mixtures thereof (including all possible mixtures of stereoisomers). See, e.g., WO 01/062726 .
- This disclosure is based on the discovery of novel C-arylation and novel C-vinylation reactions that generate an ⁇ -quaternary substituted lactam.
- the methods comprise treating a lactam with a chiral nickel (Ni) or palladium (Pd) catalyst, and an aryl halide, heteroaryl halide or a vinyl halide.
- a ligand preferably a chiral ligand, enables the construction of quaternary stereocenters on ⁇ -substituted lactams to form ⁇ -aryl or ⁇ -vinyl lactams.
- a wide range of structurally-diverse, functionalized products are prepared by a stereoselective method of nickel or palladium-catalyzed enantioselective arylation or vinylation. This chemistry is useful in the synthesis of lactams, and for the construction of novel building blocks for medicinal and polymer chemistry.
- This disclosure is also based, in part, on the discovery of novel decarboxylative asymmetric allylic alkylation reactions that generate ⁇ -quatemary substituted piperazinones, tetrahydropyrimidinones, and diazepanones.
- the methods comprise enolate functionalization of protected 3-(allyloxycarbonyl)piperazine-2-ones, 5-(allyloxycarbonyl)tetrahydropyrimidin-4-one, and 6-(allyloxycarbonyl)-1,4-diazepan-5-ones, followed by decarboxylative asymmetric allylic alkylation.
- This chemistry is useful in the construction of novel building blocks for medicinal chemistry, such as in the synthesis of ⁇ -lactams (such as in ⁇ -lactam antibiotics), hexahydropyrimidines, and benzodiazepines.
- ⁇ -lactams such as in ⁇ -lactam antibiotics
- hexahydropyrimidines and benzodiazepines.
- tetrahydropyrimidinones may be reductively transformed into hexahydropyrimidines, which are found in bioactive molecules such as the antibiotic hexetidine and the macrocyclic natural product verbamethine.
- bioactive molecules such as the antibiotic hexetidine and the macrocyclic natural product verbamethine.
- diazepane heterocycles are also found in the antipsychotic clozapine and the anti-insomnia drug suvorexant.
- Also provided herein are methods of synthesizing a pharmaceutical agent and/or composition comprising preparing a compound of formula (I), (Ia), (Ib), (IVa), or (IVb) as defined below, according to a method as described herein and synthesizing the pharmaceutical agent and/or composition from the compound of formula (I), (Ia), (Ib), (IVa), or (IVb), e.g., by carrying out one or more chemical reactions on the compound of formula (I), (Ia), (Ib), (IVa), or (IVb) and/or combining the pharmaceutical agent with one or more pharmaceutically acceptable carriers and/or excipients.
- the pharmaceutical agent and/or composition prepared from the compound of formula (I), (Ia), (Ib), (IVa), or (IVb) may be utilized to treat an individual in need thereof.
- the individual is a mammal such as a human, or a non-human mammal.
- the composition or the compound is preferably administered as a pharmaceutical composition comprising, for example, a compound of the invention and a pharmaceutically acceptable carrier.
- Pharmaceutically acceptable carriers are well known in the art and include, for example, aqueous solutions such as water or physiologically buffered saline or other solvents or vehicles such as glycols, glycerol, oils such as olive oil, or injectable organic esters.
- the aqueous solution is pyrogen-free, or substantially pyrogen-free.
- the excipients can be chosen, for example, to effect delayed release of an agent or to selectively target one or more cells, tissues or organs.
- the pharmaceutical composition can be in dosage unit form such as tablet, capsule (including sprinkle capsule and gelatin capsule), granule, lyophile for reconstitution, powder, solution, syrup, suppository, injection or the like.
- the composition can also be present in a transdermal delivery system, e.g., a skin patch.
- the composition can also be present in a solution suitable for topical administration, such as a lotion, cream, or ointment.
- Methods of preparing these formulations or compositions include the step of bringing into association an active compound, such as a compound of the invention, with the carrier and, optionally, one or more accessory ingredients.
- an active compound such as a compound of the invention
- the formulations are prepared by uniformly and intimately bringing into association a compound of the present invention with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- the present disclosure provides for the preparation of a compound of formula (I):
- the invention relates to a method for the preparation of a compound of formula (I): comprising treating a compound of formula (II): or a salt thereof; with a Pd or Ni catalyst comprising a chiral ligand; an optionally substituted aryl halide, optionally substituted aryl pseudohalide, optionally substituted heteroaryl halide, an optionally substituted heteroaryl pseudohalide, or and a base; wherein, as valence and stability permit,
- the compound of formula (I) is represented by formula (Ia): and the compound of formula (II) is represented by formula (IIa): wherein:
- the sum of m and n is 0, 1, 2, or 3.
- each occurrence of B, D, and E is independently -CR 5 R 6 -, -CR 5 -, or -C(O)-.
- E is -CR 5 -; and the sum of m and n is 0.
- R 1 is selected from optionally substituted alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, -C(O)alkyl, -C(O)aryl, -C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), and -S(O) 2 (aryl); and R 5 is selected from hydrogen, hydroxyl, halogen, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl,
- R 1 is selected from optionally substituted alkyl, aryl, aralkyl, alkenyl, -C(O)alkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), and -S(O) 2 (aryl), for example, substituted aryl.
- R 1 and the occurrence of R 5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group, for example, R 1 and the occurrence of R 5 on E are taken together to form an optionally substituted heterocycloalkyl, or heterocycloalkenyl group.
- the compound of formula (I) is represented by formula (Ib): and the compound of formula (II) is represented by formula (IIb): wherein:
- R 1 is selected from optionally substituted alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, -C(O)alkyl, -C(O)aryl, -C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), and -S(O) 2 (aryl); and R 5 is selected from hydrogen, hydroxyl, halogen, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl,
- R 1 is selected from optionally substituted alkyl, aryl, aralkyl, alkenyl, -C(O)alkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), and -S(O) 2 (aryl), for example, substituted aryl.
- R 1 and the occurrence of R 5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group.
- R 2 represents substituted or unsubstituted alkyl, alkenyl, alkynyl, aralkyl, aralkenyl, aryl, heteroaralkyl, heteroaralkenyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, or halo.
- R 2 is selected from alkyl, alkenyl, aryl, aralkyl, aralkenyl, and heteroaralkenyl, optionally substituted with halo, alkyl, haloalkyl, hydroxy, alkoxy, aryloxy, arylalkoxy, cyano, nitro, azido, -CO 2 H, -C(O)O(alkyl), amino, alkylamino, arylamino, aralkylamino, or amido, for example, R 2 is selected from alkyl, alkenyl, aryl, aralkyl, aralkenyl, and heteroaralkenyl, optionally substituted with halo, alkyl, haloalkyl, alkoxy, aryloxy, or arylalkoxy.
- the Pd catalyst is Pd(dmdba) 2 or Pd 2 (dmdba) 3 .
- the Ni(0) catalyst is Ni(COD) 2 .
- the Pd or Ni catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (II), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (II), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (II).
- the catalyst is a Pd catalyst and the chiral ligand is an enantioenriched ferrocelane ligand, or the catalyst is a Ni catalyst and the chiral ligand is an enantioenriched Josiphos ligand;
- the enantioenriched ferrocelane ligand is 1,1'-bis[(2 S ,5 S )-2,5-diethyl-1-phospholanyl]ferrocene, 1,1'-bis[(2 S ,5 S )-2,5-dimethyl-1-phospholanyl]ferrocene, or 1,1'-bis[(2 S ,5 S )-2,5-diisopropyl-1-phospholanyl]ferrocene, preferably, the enantioenriched Josiphos ligand is ( R )-1-[( S )-2-(dicyclohexylphosphino)ferrocenylethyl]diphenylphosphin
- the chiral ligand is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (II), for example, the chiral ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (II), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (II).
- the aryl halide or aryl pseudohalide is selected from bromobenzene, chlorobenzene, iodobenzene, phenyl triflate, and chlorotoluene.
- the base is selected from hexamethyldisilazane sodium salt (NaHMDS), KHMDS, LiHMDS, and lithium tert-butoxide (tBuOLi), preferably LiHMDS, or preferably NaHMDS.
- NaHMDS hexamethyldisilazane sodium salt
- KHMDS KHMDS
- LiHMDS LiHMDS
- tBuOLi lithium tert-butoxide
- the reaction includes dioxane, for example, the dioxane is used in an amount from about 0.01 M to about 0.4 M, preferably, the dioxane is used in an amount of about 0.01 M, about 0.05 M, about 0.1 M, about 0.15 M, about 0.2 M, about 0.25 M, about 0.3 M, about 0.35 M, or about 0.4 M.
- the compound of formula (I) is enantioenriched.
- no aryl nitrile is used in the method.
- the invention relates to a compound of formula (I) made by any of the methods described herein.
- the invention relates to a method comprising preparing a compound of formula (I) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (I).
- the invention relates to a compound of formula (III) or formula (IV): wherein, as valence and stability permit,
- p is 0, the compound of formula (III) is a compound of formula (IIIa): and the compound of formula (IV) is a compound of formula (IVa): wherein, as valence and stability permit,
- R 21 is Bz or An;
- R 22 is Boc, Cbz, or Bz;
- R 23 is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , CH 2 NHCbz, or CH 2 NHBoc; and
- R 24 is H or Cl.
- R 22 is Boc or Cbz.
- R 22 is Boc
- the compound is: or
- the compound is:
- the compound is or
- p is 1, the compound of formula (III) is a compound of formula (IIIb): the compound of formula (IV) is a compound of formula (IVb): wherein, as valence and stability permit:
- R 21 is Bz, An, or pCF 3 Bz;
- R 22 is Boc; and
- the compound of formula (IIIb) is: or
- the compound of formula (IVb) is:
- the invention relates to a compound of formula (V), (VI), or (VII): wherein, as valence and stability permit,
- the compound is:
- the compound is:
- the compound is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-oxidethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the invention is directed to a compound selected from: and
- the invention relates to compound of Formula (VIII): wherein, as valence and stability permit,
- R 21 is Bz or An
- R 23b is H, CH 3 , CH 2 Ph, CH 2 NHCbz, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , CH 2 NH 2
- R 24 is H or Cl.
- R 21 is Bz; R 23b is CH 2 NH 2 ; and R 24 is H.
- the invention relates to a compound of Formula (IX) or (X): wherein, as valence and stability permit,
- R 23d is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 OH, CH 2 CH 2 C(O)CH 3 , CH 2 CH 2 CH(OH)CH 3 , CH 2 NHCBz, or CH 2 NHBoc; and R 24 is H or Cl.
- R 23d is CH 2 NHBoc; and R 24 is H.
- the invention relates to a compound of formula (XI): wherein, as valence and stability permit,
- R 21 is Bz or An;
- R 24 is H or Cl; and
- R 23e is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , or CH 2 NH 2 .
- R 21 is Bz; R 23e is CH 3 ; and R 24 is H.
- the invention relates to a compound of formula (XII): wherein, as valence and stability permit,
- R 21 is Bz or An; and R 24 is H or Cl.
- R 21 is Bz and R 24 is H.
- the invention relates to a compound of formula (XIII): wherein, as valence and stability permit,
- R 25a is CH 2 Ph and R 13 is Bn.
- the invention relates to a method for preparation of a compound of formula (IV): the method comprising: treating a compound of formula (III) or a salt thereof, with a Pd(0) catalyst and a ligand L under alkylation conditions, wherein, as valence and stability permit,
- p is 0, the compound of formula (IV) is a compound of formula (IVa): and the compound of formula (III) is a compound of formula (IIIa): or a salt thereof, wherein, as valence and stability permit,
- R 21 is Bz or An;
- R 22 is Boc, Cbz, or Bz;
- R 23 is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , CH 2 NHCbz, or CH 2 NHBoc; and
- R 24 is H or Cl.
- R 22 is Boc or Cbz.
- the compound of formula (IVa) is: or
- the alkylation conditions include reaction in toluene, THF, MTBE, or hexanes, or a combination thereof.
- the Pd(0) catalyst is Pd 2 (pmdba) 3 or Pd 2 (dba) 3 .
- the Pd(0) catalyst is Pd 2 (dba) 3 .
- the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol % relative to the compound of formula (IIIa). In certain embodiments, the Pd catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (III), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (III), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (III).
- the Pd(0) catalyst is used in an amount of about 4 mol % relative to the compound of formula (IIIa).
- the ligand L is: or In certain such embodiments, the ligand L is (S)-L1.
- the ligand L is used in an amount from about 0.1 mol % to about 100 mol % relative to the compound of formula (IIIa).
- the chiral ligand is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (IIIa), for example, the chiral ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (IIIa), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (IIIa).
- the chiral ligand is used in
- the compound of formula (IVa) is enantioenriched.
- the invention relates to a method of synthesizing a medicinal product, the method comprising:
- p is 1; the compound of formula (IV) is a compound of formula (IVb) the compound of formula (III) is a compound of formula (IIIb) or a salt thereof, wherein, as valence and stability permit,
- R 21 is Bz, An, or pCF 3 Bz;
- R 22 is Boc; and
- the compound of formula (IIIb) is: or
- the compound of formula (IVb) is:
- the alkylation conditions include reaction in THF, 1,4-dioxane, MTBE, toluene, cyclohexane, or methylcyclohexane, or a combination thereof. In certain preferred embodiments, the alkylation conditions include reaction in methylcyclohexane.
- the Pd(0) catalyst is Pd 2 (pmdba) 3 or Pd 2 (dba) 3 . In certain preferred embodiments, the Pd(0) catalyst is Pd 2 (pmdba) 3 .
- the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol % relative to the compound of formula (IIIb). In certain embodiments, the Pd(0) catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (IIIb), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (IIIb), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (IIIb). In certain preferred embodiments, the Pd(0) catalyst is used in an amount of about 4 mol % relative to the compound of formula (IIIb).
- the ligand L is:
- L is (S)-(CF 3 ) 3 -tBuPHOX.
- the ligand L is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (IIIb), for example, the chiral ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (IIIb), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (IIIb). In certain embodiments, the ligand L is used in an amount of about 4 mol % relative to the compound of formula (IIIb).
- the compound of formula (IVb) is enantioenriched.
- the invention is directed to a method of synthesizing a medicinal product, the method comprising:
- the synthesizing the medicinal product from the compound of formula (IVb) comprises: treating the compound of formula (IVb) with lithium hydroxide and subsequently with lithium aluminum hydride, to form a product of formula (A) treating the compound of formula (A) with a compound of formula (B) and subsequently with acetyl chloride in methanol, to form a product of formula (C) treating the compound of formula (C) with a compound of formula (D) to form a compound of formula (E)
- the invention relates to a compound of formula (E): or a salt thereof, wherein, as valence and stability permit,
- the invention relates to a method for preparation of a compound of formula (VI): the method comprising: treating a compound of formula (V): or a salt thereof, with a Pd(0) catalyst and a ligand L, under alkylation conditions, wherein, as valence and stability permit, R 25 is C 1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen.
- the compound of formula (VI) is:
- alkylation conditions include reaction in hexanes, toluene, or a mixture thereof.
- the Pd(0) catalyst is Pd 2 (dba) 3 or Pd 2 (pmdba) 3 . In certain preferred embodiments, the Pd(0) catalyst is Pd 2 (pmdba) 3 .
- the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol %. In certain embodiments, the Pd catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (V), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (V), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (V). In certain preferred embodiments, the Pd(0) catalyst is used in an amount of about 4 mol %.
- L is: In certain preferred embodiments, L is (S)-L1.
- L is used in an amount of from about 0.1 mol % to about 100 mol %.
- the ligand is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (V), for example, the ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (V), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (V).
- L is used in an amount of about 10 mol %.
- the compound of formula (VI) is enantioenriched.
- the method further comprises hydrolyzing the compound of formula (VI), to form a compound of formula (VII): or a salt thereof.
- the invention relates to a method for preparation of a compound of formula (VIII): the method comprising: treating a compound of formula (IVa): with TFA in a solvent; wherein, as valence and stability permit,
- R 21 is Bz or An;
- R 22 is Boc;
- R 23 is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , CH 2 NHCbz, or CH 2 NHBoc;
- R 24 is H or Cl; and
- R 23b is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , CH 2 NHCbz, or CH 2 NH 2 .
- R 21 is Bz; R 22 is Boc; R 23 is CH 2 NHBoc; R 24 is H; and R 23b is CH 2 NH 2 .
- the solvent is CH 2 Cl 2 .
- the invention relates to a method for the preparation of a compound of formula (IX): the method comprising: treating a compound of formula (IVa): or a salt thereof, with LiOH in a first solvent, wherein, as valence and stability permit,
- R 21 is Bz or An
- R 23d is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 OH, CH 2 CH 2 C(O)CH 3 , CH 2 CH 2 CH(OH)CH 3 , CH 2 NHCbz, CH 2 NHBoc
- R 24 is H or Cl.
- R 21 is Bz; R 22 is Boc; R 23 is CH 2 NHBoc; R 23d is CH 2 NHBoc; and R 24 is H.
- the first solvent comprises methanol, water, or a mixture thereof. In certain such embodiments, the first solvent comprises methanol and water.
- the method further comprises preparing a compound of formula (X): the method comprising: treating the compound of formula (IX) with LiAlH 4 in a second solvent.
- the second solvent comprises THF.
- the invention relates to a method of preparation of a compound of formula (XI): the method comprising: treating a compound of formula (IVa): or a salt thereof, with hydrogen gas in the presence of a Pd/C catalyst in a first solvent, to form a first product mixture; filtering the first product mixture, to form a crude first product; and treating the crude first product with TFA in a second solvent; wherein, as valence and stability permit,
- R 21 is Bz or An;
- R 22 is Boc;
- R 23 is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , or CH 2 NHBoc;
- R 23e is H, CH 3 , CH 2 Ph, CH 2 OBn, CH 2 CH 2 CN, CH 2 CH 2 C(O)CH 3 , or CH 2 NH 2 ;
- R 24 is H or Cl.
- R 21 is Bz; R 23 is CH 3 ; R 23e is CH 3 ; and R 24 is H.
- the first solvent comprises methanol.
- the second solvent comprises CH 2 Cl 2 , ethyl acetate, or a mixture thereof.
- the method further comprises preparation of a compound of formula (XII): the method comprising: treating a compound of formula (XI): or a salt thereof, with Pd(OPiv) 2 , xantphos, AgOPiv, and benzoquinone in the presence of CO (g) in a third solvent; wherein, as valence and stability permit,
- R 24 is H.
- R 21 is Bz or An.
- R 21 is Bz; R 23e is CH 3 ; and R 24 is H.
- the third solvent comprises toluene.
- the invention relates to a method of preparation of a compound of formula (XIII): the method comprising: treating a compound of formula (VI): or a salt thereof, with TFA in a first solvent, to form a first product; treating the first product with lithium hydroxide in a second solvent, to form a second product; treating the second product with Boc 2 O in the presence of a base, to form a third product; treating the third product with K 2 CO 3 and R 13 -X 21 in a third solvent, to form a compound of formula (XIII); wherein, as valence and stability permit,
- R 13 is Bn.
- the first solvent comprises dichloromethane.
- the second solvent comprises methanol.
- the third solvent comprises DMF.
- the invention relates to a method as described in Table 1. In certain embodiments, the invention relates to a method as described in Table 2. In certain embodiments, the invention relates to a method as described in Table 3. In certain embodiments, the invention relates to a method as described in Table 4. In certain embodiments, the invention relates to a method as described in Table 5. In certain embodiments, the invention relates to a method as described in Table 6. In certain embodiments, the invention relates to a method as described in Table 7. In certain embodiments, the invention relates to a method as described in Table 8. In certain embodiments, the invention relates to a method as described in Table 9. In certain embodiments, the invention relates to a method as described in Table 10.
- the invention relates to a method as described in Table 11. In certain embodiments, the invention relates to a method as described in Table 12. In certain embodiments, the invention relates to a method as described in Table 13. In certain embodiments, the invention relates to a method as described in Table 14. In certain embodiments, the invention relates to a method as described in Table 15. In certain embodiments, the invention relates to a method as described in Table 16. In certain embodiments, the invention relates to a method as described in Table 17. In certain embodiments, the invention relates to a method as described in Table 18. In certain embodiments, the invention relates to a method as described in Table 19. In certain embodiments, the invention relates to a method as described in Table 20.
- the invention relates to a method as described in Table 21. In certain embodiments, the invention relates to a method as described in Table 22. In certain embodiments, the invention relates to a method as described in Table 23. In certain embodiments, the invention relates to a method as described in Table 24. In certain embodiments, the invention relates to a method as described in Table 25. In certain embodiments, the invention relates to a method as described in Table 26. In certain embodiments, the invention relates to a method as described in Table 27. In certain embodiments, the invention relates to a method as described in Table 28. In certain embodiments, the invention relates to a method as described in Table 29. In certain embodiments, the invention relates to a method as described in Table 30.
- the invention relates to a method as described in Table 31. In certain embodiments, the invention relates to a method as described in Table 32. In certain embodiments, the invention relates to a method as described in Table 33. In certain embodiments, the invention relates to a method as described in Table 34. In certain embodiments, the invention relates to a method as described in Table 35. In certain embodiments, the invention relates to a method as described in Table 36. In certain embodiments, the invention relates to a method as described in Table 37. In certain embodiments, the invention relates to a method as described in the table in Example 35. In certain embodiments, the invention relates to a method as described in Table 38. In certain embodiments, the invention relates to a method as described in Table 39.
- Preferred transition metal catalysts of the disclosure are complexes of nickel or palladium comprising a chiral ligand.
- the complex is of Ni(0) comprising a chiral ligand.
- the complex is of Ni(II) comprising a chiral ligand.
- the complex is of Pd(0) comprising a chiral ligand.
- the complex is of Pd(II) comprising a chiral ligand.
- transition metal catalysts having a low oxidation state suffer from air- and moisture-sensitivity, such that these complexes of transition metals necessitate appropriate handling precautions. This may include the following precautions without limitation: minimizing exposure of the reactants to air and water prior to reaction; maintaining an inert atmosphere within the reaction vessel; properly purifying all reagents; and removing water from reaction vessels prior to use.
- the Ni(0) catalyst is a precatalyst.
- the Pd(0) catalyst is a precatalyst.
- the Pd(II) catalyst is a precatalyst.
- Example Ni(0) catalysts that may be used in the methods of the disclosure include Ni[(1,5-cyclooctadiene) 2 ], which is also referred to herein as Ni(COD) 2 .
- the transition metal catalysts of the disclosure are complexes of Ni(0) or Ni(II), such as Ni(COD) 2 , NiCl, and NiBr 2 .
- the transition metal catalysts of the disclosure are complexes of Pd(0) or Pd(II).
- Pd(II) catalysts are typically robust, and are less sensitive to air and moisture than their lower-oxidation state counterparts.
- Pd(0) catalysts examples include Pd(PPh 3 ) 4 , Pd(dm-dba) 2 , Pd(dba) 2 , Pd 2 (dba) 3 •CHCl 3 , Pd 2 (dba) 3 , and Pd 2 (pm-dba) 3 .
- the transition metal catalyst is Pd(PPh 3 ) 4 .
- the transition metal catalyst is Pd(dm-dba) 2 .
- the transition metal catalyst is Pd(dba) 2 .
- the transition metal catalyst is Pd 2 (dba) 3 •CHCl 3 .
- the transition metal catalyst is Pd 2 (dba) 3 .
- the transition metal catalyst is Pd 2 (pm-dba) 3 .
- Pd(II) catalysts examples include Pd(OC(O)R c ) 2 , wherein R c is optionally substituted alkyl, alkenyl, alkynyl, aryl, heteroaryl, aralkyl, heteroaralkyl, cycloalkyl, heterocycloalkyl, (cycloalkyl)alkyl, or (heterocycloalkyl)alkyl.
- the transition metal catalyst is Pd(OC(O)R c ) 2 , wherein R c is defined above, For example, R c may be alkyl, substituted by one or more halo or cyano groups.
- the transition metal catalyst is Pd(OAc) 2 .
- the transition metal catalyst is [Pd(cinnamyl)Cl] 2 .
- additives include salts. These additives are preferably used in an amount that is in the range of about 0.1 equivalents to about 15 equivalents relative to the amount of the reactant, more preferably in the range of about 0.5 equivalents to about 10 equivalents relative to the reactant, and most preferably in the range of about 2 equivalents to about 7 equivalents relative to the reactant.
- additives include one or more of Mn 0 , Zn 0 , LiCl, LiBr, NaCl, NaBr, NaI, KCl, KBr, CsCl, ZnCl 2 , CuI, AlCl 3 , HMDS, COD, LiO t -Bu, NaO t -Bu, and KO t- Bu. These additives are preferably used in an amount that is in the range of about 1 equivalent to about 5 equivalents relative to the amount of the catalyst.
- a low oxidation state of a transition metal i.e., an oxidation state sufficiently low to undergo oxidative addition, can be obtained in situ, by the reduction of transition metal complexes that have a high oxidation state.
- Reduction of the transition metal complex can optionally be achieved by adding nucleophilic reagents including, without limitation, tetrabutyl ammonium hydroxide, tetrabutyl ammonium difluorotriphenylsilicate (TBAT), tetrabutylammonium fluoride (TBAF), 4-dimethylaminopyridine (DMAP), tetramethylammonium hydroxide (e.g., as the pentahydrate), KOH/1,4,7,10,13,16-hexaoxacyclooctadecane, sodium ethoxide, TBAT/trimethyl-(2-methyl-cyclohex-1-enyloxy)-silane, and combinations thereof.
- the nucleophilic reagent is used in an amount in the range of about 1 mol % to about 20 mol % relative to the reactant, more preferably in the range of about 1 mol % to about 10 mol % relative to the substrate, and most preferably in the range of about 5 mol % to about 8 mol % relative to the substrate.
- a Pd(II) complex can be reduced in situ to form a Pd(0) catalyst.
- exemplary transition metal complexes that may be reduced in situ include, without limitation, allylchloro[1 ,3-bis(2,6-di-iso-propylphenyl)imidazol-2-ylidene]palladium(II), ([2 S ,3 S ]-bis[diphenylphosphino]butane)( ⁇ 3 -allyl)palladium(II) perchlorate, [ S ]-4-tert-butyl-2-(2-diphenylphosphanyl-phenyl)-4,5-dihydro-oxazole( ⁇ 3 -allyl)palladium(II) hexafluorophosphate (i.e., [Pd( S -tBu-PHOX)(allyl)]PF 6 ), and cyclopentadienyl( ⁇ 3 -allyl) palladium
- nucleophilic reagents including, without limitation, NaHMDS, KHMDS, LiHMDS, tBuOLi, tetrabutyl ammonium hydroxide, tetrabutyl ammonium difluorotriphenylsilicate (TBAT), tetrabutylammonium fluoride (TBAF), 4-dimethylaminopyridine (DMAP), tetramethylammonium hydroxide (e.g., as the pentahydrate), KOH/1,4,7,10,13,16-hexaoxacyclooctadecane, sodium ethoxide, TBAT/trimethyl-(2-methyl-cyclohex-1-enyloxy)-silane, and combinations thereof.
- nucleophilic reagents including, without limitation, NaHMDS, KHMDS, LiHMDS, tBuOLi, tetrabutyl ammonium hydroxide, tetrabutyl ammonium difluorotriphen
- the nucleophilic reagent When a nucleophilic reagent is added, the nucleophilic reagent is used in an amount in the range of about range of about 0.1 equivalents to about 10 equivalents relative to the amount of the reactant, more preferably in the range of about 0.1 equivalents to about 5 equivalents relative to the reactant, and most preferably in the range of about 0.5 equivalents to about 2 equivalents relative to the reactant.
- Catalyst loading may be expressed as a percentage that is calculated by dividing the moles of catalyst complex by the moles of the substrate present in a given reaction.
- Catalyst loading is alternatively expressed as a percentage that is calculated by dividing the moles of total transition metal (for example, nickel) by the moles of the substrate present in a given reaction.
- the transition metal catalyst is present under the conditions of the reaction from an amount of about 0.1 mol% to about 20 mol% total transition metal relative to the substrate, which is the compound of formula (II), (IIIa), (IIIb), or (V). In certain embodiments, the catalyst loading is from about 1 mol% to about 15 mol% total transition metal relative to the substrate.
- the catalyst loading is from about 1 mol% to about 14 mol%, about 1 mol% to about 12%, about 1 mol% to about 10%, about 2 mol% to about 9 mol%, about 2.5 mol% to about 8 mol%, about 3 mol% to about 7 mol%, about 3.5 mol% to about 6.5 mol%, or about 4 mol% to about 6 mol% total transition metal relative to the substrate.
- the catalyst loading is about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% total transition metal.
- the catalyst loading is about 2 mol%, about 2.5 mol%, about 3 mol%, about 3.5 mol%, about 4 mol%, about 4.25 mol%, about 4.5 mol%, about 4.75 mol%, about 5 mol%, about 5.25 mol%, about 5.5 mol%, about 5.75 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, or about 9% total transition metal.
- the methods disclosed herein use a Ni(0) catalyst comprising a chiral ligand. In certain embodiments, the methods disclosed herein use a Pd(0) catalyst comprising a chiral ligand. In certain embodiments, the methods disclosed herein use a Pd(II) catalyst comprising a chiral ligand.
- Enantioselectivity results from the use of chiral ligands during the arylation or vinylation reaction.
- the asymmetric environment that is created around the metal center by the presence of chiral ligands produces an enantioselective reaction.
- the chiral ligand forms a complex with the transition metal (e.g., nickel), thereby occupying one or more of the coordination sites on the metal and creating an asymmetric environment around the metal center. This complexation may or may not involve the displacement of achiral ligands already complexed to the metal.
- the displacement may proceed in a concerted fashion, i.e., with both the achiral ligand decomplexing from the metal and the chiral ligand complexing to the metal in a single step.
- the displacement may proceed in a stepwise fashion, i.e., with decomplexing of the achiral ligand and complexing of the chiral ligand occurring in distinct steps.
- Complexation of the chiral ligand to the transition metal may be allowed to occur in situ, i.e., by admixing the ligand and metal before adding the substrate.
- the ligand-metal complex can be formed separately, and the complex isolated before use in the arylation or vinylation reactions of the present disclosure.
- the chiral ligand influences the orientation of other molecules as they interact with the transition metal catalyst. Coordination of the metal center with an aryl halide and reaction of the substrate with the aryl halide-metal complex are dictated by the presence of the chiral ligand. The orientation of the reacting species determines the stereochemistry of the products.
- Chiral ligands of the disclosure may be bidentate or monodentate or, alternatively, ligands with higher denticity (e.g., tridentate, tetradentate, etc.) can be used.
- the ligand will be substantially enantiopure.
- enantiopure is meant that only a single enantiomer is present.
- substantially enantiopure ligands e.g., ee >99%, preferably >99.5%, even more preferably >99.9%
- substantially enantiopure ligands can be purchased from commercial sources, obtained by successive recrystallizations of an enantioenriched substance, or by other suitable means for separating enantiomers.
- the chiral ligand is an enantioenriched phosphine ligand.
- the enantioenriched phosphine ligand is a ferrocenyl ligand such as a Mandyphos-type ligand, a Josiphos-type ligand, a Taniaphos-type ligand, or a Walphos-type ligand.
- Preferred chiral ligands of the disclosure include a Mandyphos-type ligand a Josiphos-type ligand, or a ferrocenyl-type ligand.
- the Mandyphos-type ligand or the Josiphos-type ligand is selected from SL-M001-2, SL-M003-2, SL-M004-1, SL-M004-2, SL-M009-1, SL-M009-2, SL-J001-1, SL-J002-1, SL-J003-1, SL-J004-1, SL-J006-1, SL-J007-1, SL-J013-1, SL-J212-1, and SL-J418-1.
- the ferrocenyl-type ligand is selected from (2 S ,5 S )-Me-ferocelane, (2 S ,5 S )-Et-ferocelane, and (2 S ,5 S )-iPr-ferocelane.
- the enantioenriched phosphine ligand is selected from ( R )-BINAP, ( R )-DM-BINAP, ( S )-DTBM-SEGPHOS, ( R )-BTFM-Garphos, ( S )-C3-TunePhos, ( R )-P-Phos, (2 S ,5 S )-Me-ferocelane, (2 S ,5 S )-Et-ferocelane, (2 S ,5 S )-iPr-ferocelane, (2 S ,5 S )-Me-f-Ketalphos, SL-M001-2, SL-M003-2, SL-M004-1, SL-M004-2, SL-M009-1, SL-M009-2, SL-J001-1, SL-J002-1, SL-J003-1, SL-J004-1, SL-J006-1, SL-J007-1,
- the enantioenriched phosphine ligand is selected from ( R )-BINAP, ( R )-DM-BINAP, ( S )-C3-TunePhos, SL-M001-2, SL-M003-2, SL-M004-1, SL- M004-2, SL-M009-1, SL-M009-2, SL-J001-1, SL-J002-1, SL-J003-1, SL-J004-1, SL-J006-1, SL-J013-1, SL-J212-1, SL-W001-1, SL-W002-1, SL-W005-1, SL-W006-1, SL-W00S-1, and SL-W009-1.
- the enantioenriched phosphine ligand is selected from (S)- DTBM-SEGPHOS, ( R )-BTFM-Garphos, ( R )-P-Phos, (2 S ,5 S )-Me-ferocelane, (2 S ,5 S )-Et-ferocelane, (2 S ,5 S )-Me-f-Ketalphos, SL-J007-1, SL-J418-1, and SL-W022-1.
- Exemplary ligand structures are depicted in Figure 1 .
- the chiral ligand is present in an amount in the range of about 0.1 equivalents to about 10 equivalents relative to the amount of total metal from the catalyst, preferably in the range of about 0.1 to about 6 equivalents relative to the amount of total metal from the catalyst, and most preferably in the range of about 0.5 to about 4.5 equivalents relative to the amount of total metal from the catalyst.
- the amount of the chiral ligand can be measured relative to the amount of the substrate.
- the ligand is present under the conditions of the reaction from an amount of about 0.1 mol% to about 100 mol% relative to the substrate, which is the compound of formula (II), (IIIa), (IIIb), or (V).
- the amount of ligand present in the reaction is alternatively referred to herein as "ligand loading" and is expressed as a percentage that is calculated by dividing the moles of ligand by the moles of the substrate present in a given reaction.
- the ligand loading is from about 0.5 mol% to about 50 mol%.
- the ligand loading is about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol%, or about 15 mol%.
- the ligand is in excess of the transition metal catalyst. In certain embodiments, the ligand loading is about 10 times the transition metal catalyst loading.
- the reactions of the disclosure may enrich the stereocenter bearing R 2 in the product relative to the enrichment at this center, if any, of the starting material.
- the chiral ligand used in the methods of the disclosure yields a compound of formula (I) that is enantioenriched.
- the level of enantioenrichment of a compound may be expressed as enantiomeric excess (ee).
- the ee of a compound may be measured by dividing the difference in the fractions of the enantiomers by the sum of the fractions of the enantiomers.
- the ee of the compound is (98-2)/(98+2), or 96%.
- the compound of formula (I) has about 5% ee or greater, 10% ee or greater, 15% ee or greater, 20% ee or greater, 25% ee or greater, 30% ee or greater, 40% ee or greater, 50% ee or greater, 60% ee or greater, 70% ee or greater, about 80% ee, about 85% ee, about 88% ee, about 90% ee, about 91% ee, about 92% ee, about 93% ee, about 94% ee, about 95% ee, about 96% ee, about 97% ee, about 98% ee, about 99% ee, or above about 99% ee, even where this % ee is greater than the
- the compound of formula (I) is enantioenriched. In certain embodiments, the compound of formula (I) is enantiopure. In embodiments where the starting material has more than one stereocenter, reactions of the disclosure may enrich the stereocenter bearing R 2 relative to the enrichment at this center, if any, of the starting material, and substantially independently of the stereochemical disposition/enrichment (de) of any other stereocenters of the molecule.
- a product of the methods described herein may have 5% de or greater, 10% de or greater, 15% de or greater, 20% de or greater, 25% de or greater, 30% de or greater, 40% de or greater, 50% de or greater, 60% de or greater, 70% de or greater, 80% de or greater, 90% de or greater, 95% de or greater, or even 98% de or greater at the stereocenter of the product bearing R 2 .
- the methods of the disclosure include treating a compound of formula (II) with a palladium or nickel catalyst comprising a chiral ligand, and an aryl halide or a heteroaryl halide under arylation conditions.
- the methods of the disclosure include treating a compound of formula (II) with a palladium or nickel catalyst comprising a chiral ligand, and a vinyl halide under vinylation conditions.
- the palladium catalyst under arylation or vinylation conditions is a Pd(0) catalyst comprising a chiral ligand.
- the palladium catalyst under arylation or vinylation conditions is a Pd(II) catalyst comprising a chiral ligand.
- the nickel catalyst under arylation or vinylation conditions is a Ni(0) catalyst comprising a chiral ligand.
- the palladium catalyst under arylation or vinylation conditions is a Ni(II) catalyst comprising a chiral ligand.
- arylation and vinylation conditions further comprise a base, such as NaHMDS, KHMDS, and LiHMDS.
- the base is LiHMDS.
- the base is NaHMDS.
- arylation and vinylation conditions of the reaction include one or more additives, such as a salt.
- the salt is one or more of LiCl, NaCl, NaBr, NaI, KCl, KBr, CsCl, CuI, LiO t -Bu, and KO t -Bu.
- arylation, vinylation, and alkylation conditions of the reaction include one or more organic solvents.
- organic solvents include aromatic or non-aromatic hydrocarbons, ethers, alkylacetates, nitriles, or combinations thereof.
- organic solvents include hexane, pentane, benzene, toluene, xylene, cyclohexane, methylcyclohexane, cyclic ethers such as optionally substituted tetrahydrofuran and dioxane, acyclic ethers such as dimethoxyethane, diethyl ether, methyl tertbutyl ether, and cyclopentyl methyl ether, acetonitrile, isobutyl acetate, ethyl acetate, isopropyl acetate, or combinations thereof.
- the solvent is toluene, tetrahydrofuran, dioxane, methyl tert-butyl ether, cyclohexane, methylcyclohexane, dimethoxyethane, or a mixture of toluene and tetrahydrofuran.
- the solvent is a mixture of two organic solvents.
- the solvent is a mixture of two organic solvents in a ratio of 1:5, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, or 15:1.
- the solvent is dioxane.
- the solvent is a mixture of toluene and tetrahydrofuran.
- the mixture of toluene and tetrahydrofuran is in a ratio of 1:5, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, or 15:1.
- the mixture of toluene and tetrahydrofuran is in a ratio of 5:1 or 10:1.
- the solvent is a mixture of toluene and hexanes.
- the mixture of toluene and hexanes is in a ratio of 1:5, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, or 15:1. In certain embodiments, the mixture of toluene and hexanes is in a ratio of 5:1 or 10:1. In certain embodiments, the solvent is methylcyclohexane.
- arylation, vinylation, and alkylation conditions of the reaction include a reaction temperature.
- the reaction temperature is ambient temperature (about 20 °C to about 26 °C). In preferred embodiments, the reaction temperature is about 60 °C to about 100 °C. In preferred embodiments, the reaction temperature is higher than ambient temperature, such as, for example, about 30 °C, about 35 °C, about 40 °C, about 45 °C, about 50 °C, about 55 °C, about 60 °C, about 65 °C, about 70 °C, about 75 °C, about 80 °C, about 85 °C, about 90 °C, about 95 °C, or about 100 °C. Reaction temperature may be optimized per each substrate.
- Example 1 Initial evaluation of ligands and bases for Ni-catalyzed lactam ⁇ -arylation
- N-protecting groups were evaluated. Generally low yields were observed for N -aryl and N -benzyl lactams (Table 8, 15a-15f ). Little to no improvement in yield was observed for benzoyl or tosyl protected substrates (15g, 15h), an oxindole (15i), a ⁇ -lactam (15j), and an ⁇ -lactam (15k). Both an ⁇ -Et-substituted (151) and an unsubstituted lactam (15m) resulted in low product to internal standard ratios. Table 8 Effect of N-protecting groups Substrate Conv. (%) Yield (%) Substrate Conv.
- Nitrile-/Josiphos-ligated Ni complexes have been demonstrated to be particularly reactive toward oxidative addition of aryl halides.
- Complex 20 was synthesized according to a literature procedure (Scheme 1) and used as a precatalyst in the coupling of lactam 15 with 3-chloroanisole (17). Complex 20 did not result in improved reactivity as compared to Ni(COD) 2 (Table 12).
- Table 12 Use of a Ni 0 (PhCN) complex as a precatalyst entry mol% Ni temp. % added L5 % Yield ( 16 ) 1 5 80 °C - 14 2 10 80 °C - 13 3 5 50 °C 5 12 4 10 50 °C 10 15
- Ni-catalyzed ⁇ -arylation of lactam 15 with an aryl chloride may be achieved with high enantioselectivity but low yield. Both lower yields and levels of enantioselectivity were observed for an aryl bromide or an aryl iodide. Product formation was found to terminate within 1 hour, and increasing the Ni loading decreased yield. The stoichiometric reaction of an isolated Ni II ArCl complex and the lithium enolate derived from 15 resulted in predominant aryl chloride homocoupling.
- Example 8 Evaluation of ligands and bases for Pd-catalyzed lactam ⁇ -arylation with 3-chloroanisole and chlorobenzene
- Example 10 Evaluation of reaction time, temperature and Pd sources for Pd-catalyzed lactam ⁇ -arylation
- Example 11 Scope of aryl halides and lactams for Pd-catalyzed lactam ⁇ -arylation
- Aryl bromides and aryl chlorides with a variety of substitution patterns are accommodated in the arylation (Table 20).
- Aryl halides possessing electron-deficient (see products 3ab, 3ad, 3ah, 3ae) and electron-rich (3af, 3ag) substituents at the para position led to products with excellent enantioselectivites using Method A and B, respectively.
- Aryl halides possessing substituents at the meta position are also permissible in both Method A and B, although slightly diminished enantioselectivity is observed when 3-chloroanisole is used as the electrophile (3ai). Trace product is observed when ortho -substituted aryl halides are exposed to the reaction conditions. An N -methyl indole was also well tolerated. 5-indolyl lactam 3al was obtained in moderate yield and excellent enantioselectivity.
- Example 15 Evaluation of temperature for Pd-catalyzed lactam a-vinylation
- Example 16 Evaluation of solvents and solvent concentration for Pd-catalyzed lactam ⁇ -vinylation
- Example 17 Evaluation of stoichiometry for Pd-catalyzed lactam a-vinylation
- Example 19 Scope of the vinyl halide for Pd-catalyzed lactam a-vinylation
- Analytical chiral HPLC was performed with an Agilent 1100 Series HPLC utilizing a Chiralcel OD-H column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd. with visualization at 254 nm.
- Analytical SFC was performed with a Mettler SFC supercritical CO 2 analytical chromatography system utilizing a Chiralcel AD column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd. with visualization at 254 nm.
- 1 H-NMR spectroscopic data were collected on a Varian 500 MHz spectrometer at ambient temperature.
- GC analyses were obtained on an Agilent 6890 Series GC system with a DB-1 column (length 30 m, internal diameter 0.25 mm).
- THF, Et 2 O, CH 2 Cl 2 , toluene, CH 3 CN, TBME and dioxane were dried by passage through an activated alumina column under argon.
- Purified water was obtained using a Barnstead NANOpure Infinity UV/UF system. Commercially available reagents were purchased.
- N- aryl lactam substrates Ni benzonitrile complex 20, and Ni II ArCl complex 21 were prepared according to previously reported procedures.
- the Schlenk-tube was closed with a Teflon screw-cap and the reaction mixture was heated to 80 °C for 20 h. After 20 h, the reaction was allowed to cool to room temperature. After quenching with MeOH, followed by the addition of aqueous, saturated NH 4 Cl-solution and vigorous stirring, the layers were separated and the aqueous phase extracted with ethyl acetate (3 x 20 mL). The combined organic extracts were dried over Na 2 SO 4 and the volatiles were removed under reduced pressure. Chromatography of the crude mixture on silica eluting with 50% ethyl acetate/hexane yielded the title compound as an off-white solid.
- Example 21 Representative procedure for evaluating ligands in the Ni-catalyzed a-arylation of lactam with aryl halide
- n the number of reactions in the screen.
- Ni(COD) 2 (1.38 n mg, 0.005 n mmol, 0.100 equiv), toluene (0.25 n mL), and a stir bar were added to a 4-mL vial A.
- LiHMDS 8.37 n mg, 0.050 n mmol, 1.00 equiv
- lactam (10.3 n mg, 0.050 n mmol, 1.00 equiv)
- toluene (0.25 n mL)
- the contents of A and B were stirred for ⁇ 2 minutes.
- Example 22 Representative procedure for evaluating ligands in the Pd-catalyzed ⁇ -arylation of lactam with aryl halide
- n the number of reactions in the screen.
- Pd(OAc) 2 (1.10 n mg, 0.005 n mmol, 0.100 equiv), toluene (0.25 n mL), and a stir bar were added to a 4-mL vial A.
- LiHMDS 8.37 n mg, 0.050 n mmol, 1.00 equiv
- lactam (12.3 n mg, 0.060 n mmol, 1.20 equiv)
- toluene (0.25 n mL)
- the contents of A and B were stirred for ⁇ 2 minutes.
- Example 25 Scope of ⁇ -substituents for Pd-catalyzed decarboxylative alkylation of tetrahydropyrimidin-4-ones
- the optimized conditions in 2:1 hexanes-toluene furnished the decarboxylative alkylation product 6a in high yield and ee.
- the reaction proved amenable to a variety of ⁇ -substituents, including ethyl ( 6b ), methyl ester ( 6c ), 2-chloroallyl ( 6d ), and benzyl ( 6e ).
- the reaction is scalable, as shown for the benzyl substrate 5e in a 1 gram reaction.
- the nitrile and benzyloxymethyl ether products 6f and 6g were isolated with reduced enantioselectivities.
- the fluorine and propargyl substrates 5h and 5i were also able to be used, and may serve as a precursor to a novel fluorinated ⁇ 2,2 -amino acid, or for biorthogonal click reactions, respectively.
- Both Boc groups of 4m could be removed by treatment with excess TFA to give aminopiperazinone 8 (Table 36, row a). With LiOH, the benzoyl group could be orthogonally removed to provide the free amide 9 (Table 36, row b). The amide 9 could then be reduced with LiAlH 4 to the corresponding chiral aminopiperazine 10.
- Boc-deprotection of N4 we hydrogenated the allyl olefin of methyl piperazinone 4e before cleaving the Boc group to obtain 11.
- novel unprotected ⁇ 2,2 -amino acids 14-16 bearing pendant carboxylic acid, chloroallyl, and fluorine atom functional groups could be obtained directly by purification with reverse phase preparatory HPLC following the two-step deprotection sequence (Table 36, row e).
- reaction were performed in flame-dried glassware under an argon or nitrogen atmosphere using dry, deoxygenated solvents. Solvents were dried by passage through an activated alumina column under argon. tert-Butyl 3-oxopiperazine-1-carboxylate 1 was obtained from Combi-Blocks. Commercially obtained reagents were used as received. Chemicals were purchased from Sigma Aldrich/Strem/Alfa Aesar/Combi-Blocks and used as received. Reaction temperatures were controlled by an IKAmag temperature modulator. Glove box manipulations were performed under a nitrogen atmosphere. Thin-layer chromatography (TLC) was performed using E.
- TLC Thin-layer chromatography
- High resolution mass spectra were obtained from an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI+), atmospheric pressure chemical ionization (APCI+), or mixed ionization mode (MM: ESI-APCI+).
- ESI+ electrospray ionization
- APCI+ atmospheric pressure chemical ionization
- MM mixed ionization mode
- Optical rotations were measured with a Jasco P-2000 polarimeter operating on the sodium D-line (589 nm), using a 100 mm pathlength cell and are reported as: [a]D T (concentration in g/100 mL, solvent). Stereochemistry is assigned by analogy to previous results. 1 (a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc.
- Allyl cyanoformate was prepared according to the method of Weber. [ M. E. Childs, W . P. Weber, J. Org. Chem. 1976, 41, 3486-3487 .] Phosphinooxazoline (PHOX) ligands (S)-L1 , (S)-L2 , and achiral GlyPhox were prepared by methods described in our previous work. [(a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126, 15044-15045 . (b) K. Tani, D. C. Behenna, R. M. McFadden, B. M. Stoltz, Org. Lett.
- PHOX Phosphinooxazoline
- Di-benzoylated allylic alkylation substrate 3g was prepared according to the method of Korch. [ K. M. Korch, C. Eidamshaus, D. C. Behenna, S. Nam, D. Home, B. M. Stoltz, Angew. Chem. Int. Ed.
- Tris(4,4'- methoxydibenzylideneacetone)dipalladium(0) [Pd 2 (pmdba) 3 ] was prepared according to the method of Ibers [ J. Organomet. Chem. 1999, 65, 253-266 ] or Fairlamb [ Org. Lett. 2004, 6, 4435-4438 ]. AgOPiv was prepared using Grubbs' procedure [ J. Am. Chem. Soc. 2011, 133, 8525-8527 ].
- tert -Butyl ((phenylsulfonyl)methyl)carbamate and benzyl ((phenylsulfonyl)methyl)carbamate were prepared according to the method of Zwierzak or Dikshit [(a) Klepacz, A.; Zwierzak, A. Tetrahedron Lett. 2002, 43, 1079 .
- Benzyl 3-oxopiperazine-1-carboxylate was prepared according to the method of Batey. [ Tetrahedron 2010, 66, 3370-3377 ].
- 2-chloroallyl chloroformate was prepared according to the method of Stoltz. [ J. Am. Chem. Soc. 2008, 130, 810-811 .]
- tert-butyl 4-benzoyl-3-oxopiperazine-1-carboxylate SI-1
- THF 250 mL
- nBuLi 11.4 mL, 2.4M solution in hexane, 27.5 mmol, 1.1 equiv
- the resulting yellow solution was stirred for 10 min at -78 °C.
- Benzoyl chloride (3.48 mL, 30.0 mmol, 1.2 equiv) was then added dropwise at -78 °C, giving an orange solution.
- the reaction was stirred for 2.5 h at -78 °C, quenched by addition of saturated aqueous NH 4 Cl (100 mL), and diluted with ethyl acetate (100 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (3 ⁇ 100 mL). The combined organic layers were dried over anhydrous Na 2 SO 4 , decanted, and concentrated under reduced pressure onto silica gel.
- tert-butyl 4-(4-methoxybenzoyl)-3-oxopiperazine-1-carboxylate SI-2.
- THF 250 mL
- n BuLi 11.4 mL, 2.4M solution in hexane, 27.5 mmol, 1.1 equiv
- the resulting yellow solution was stirred for 10 min at -78 °C.
- the silica-loaded crude reaction mixture was filtered through a plug of silica (1% ⁇ 2% MeOH/CH 2 Cl 2 ) to give crude anisoyl protected ketopiperazine SI-2 as a white foam, which was directly used without further purification in subsequent acylation reactions with allyl cyanoformate.
- benzyl 4-benzoyl-3-oxopiperazine-1-carboxylate (SI-3).
- benzyl 3-oxopiperazine-1-carboxylate [ Tetrahedron 2010, 66, 3370-3377 ] (1.0 g, 4.3 mmol, 1.0 equiv) in THF (42 mL) at -78 °C was added dropwise n BuLi (1.96 mL, 2.4M solution in hexane, 4.7 mmol, 1.1 equiv) over 20 minutes. The solution was stirred for 10 min at -78 °C.
- Benzoyl chloride (0.595 mL, 5.1 mmol, 1.2 equiv) was then added dropwise at -78 °C, giving a light yellow solution.
- the reaction was stirred for 1 h at -78 °C, quenched by addition of saturated aqueous NH 4 Cl (50 mL), and diluted with ethyl acetate (50 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (3 ⁇ 50 mL). The combined organic layers were dried over anhydrous Na 2 SO 4 , decanted, and concentrated under reduced pressure onto silica gel.
- reaction was quenched with aqueous NH4Cl (10 mL) and extracted with EtOAc (3 x 5 mL). The combined organic phases were dried over anhydrous Na 2 SO 4 , decanted, and concentrated under reduced pressure onto silica gel.
- reaction solution was stirred at -78 °C for 40 min, allowed to warm up to room temperature, and was then quenched with saturated aqueous NH 4 Cl (50 mL).
- the mixture was diluted with EtOAc (100 mL) and the aqueous phase was extracted with EtOAc (3 x 80 mL).
- the combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure onto silica gel.
- reaction mixture was stirred for 3 h at 0 °C and diluted with saturated aqueous NH 4 Cl (2 mL) and EtOAc (2 mL).
- the aqueous phase was extracted with EtOAc (4 x 3 mL) and the combined organic phases were dried over anhydrous Na 2 SO 4 , decanted, and concentrated under reduced pressure onto silica gel.
- reaction was quenched with aqueous NH 4 Cl (2 mL) and diluted with EtOAc (2 mL).
- the aqueous phase was extracted with EtOAc (3 x 3mL) and the combined organic phases were dried over anhydrous Na 2 SO 4 , decanted, and concentrated under reduced pressure onto silica gel.
- the vial was sealed with a teflon cap and heated to 40 °C.
- the reaction mixture was removed from the glovebox and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography to afford the desired oxopiperazine.
- tert-Bbtyl (R)-2-allyl-4-benzoyl-2-(((tert-butoxycarbonyl)amino)methyl)-3-oxopiperazine-1-carboxylate (4m).
- allyl ester 3m 100 mg, 0.19 mmol, 1.0 equiv
- Pd 2 (pmdba) 3 8.5 mg, 0.0077 mmol, 4 mol %)
- S )-(CF 3 ) 3 - t Bu-PHOX (11.4 mg, 0.019 mmol, 10 mol %) in hexanes/toluene (2:1, 4.0 mL).
- methylated pyrimidinone 5a (15 mg, 0.037 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd 2 (pmdba) 3 (1.6 mg, 0.0015 mmol, 4 mol %) and ( S )-(CF 3 ) 3 - t Bu-PHOX (2.2 mg, 0.0037 mmol, 10 mol %) in hexanes/toluene (2:1, 1.7 mL).
- cyanoethylated tetrahydropyrimidinone 5f (15 mg, 0.034 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd 2 (pmdba) 3 (1.2 mg, 0.0014 mmol, 4 mol %) and ( S )-(CF 3 ) 3 - t Bu-PHOX (2.0 mg, 0.0034 mmol, 10 mol %) in hexanes/toluene (2:1, 1.4 mL).
- the flask was stirred at ambient glovebox temperature (27 °C) for 30 min and then 5e (lg, 2.1 mmol, 1.0 equiv) was added as a solution in hexane/toluene (2:1, 100 mL, total concentration 0.014M).
- the flask was sealed with a Kontes valve, removed from the glovebox, and heated to 40 °C for 16 h.
- the solution was concentrated under reduced pressure and purified by silica gel flash chromatography (15% EtOAc/hexanes) to give benzyl tetrahydropyrimidinone 6e as a yellow oil (780 mg, 87% yield, 95% ee); spectroscopic data vide supra.
- tert-butyl (R)-((2-allyl-4-benzoyl-3-oxopiperazin-2-yl)methyl)carbamate (8) Trifluoroacetic acid (114 ⁇ L, 1.5 mmol, 20 equiv) was added dropwise to a solution of methylcarbamate oxopiperazine 4m (35 mg, 0.07 mmol, 1.0 equiv) in CH 2 Cl 2 (0.74 mL) at 0 °C. The reaction mixture was allowed to warm up to room temperature, stirred for 3 h and concentrated under reduced pressure. The residue was repeatedly taken up in CH 2 Cl 2 (1.0 mL) and concentrated, four times.
- tert-butyl (R)-2-allyl-2-(((tert-butoxycarbonyl)amino)methyl)-3-oxopiperazine-1-carboxylate (9).
- LiOH monohydrate (2.5 mg, 0.06 mmol, 1.4 equiv) was added in one portion to a solution of methylcarbamate oxopiperazine 4m (20 mg, 0.04 mmol, 1.0 equiv) in methanol/water (1:1, 1.8 mL) at room temperature.
- the reaction mixture was stirred for 1 h, diluted with EtOAc (2 mL) and washed with saturated aqueous NaHCO 3 (2 mL).
- H 2 O 40 ⁇ L
- 15% aqueous NaOH 40 ⁇ L
- H 2 O 120 ⁇ L
- MgSO 4 was added.
- the mixture was stirred for another 5 minutes at room temperature and then filtered over a pad of celite, rinsing with EtOAc.
- Racemic products were synthesized according to the general procedure, using achiral GlyPHOX ligand instead of (S)-(CF 3 ) 3 -tBu-PHOX.
- Diazepanone 4a was subjected to selective debenzoylation under basic conditions, followed by reduction with LiAlH 4 to yield diazepane 5 bearing a free secondary amine. Then, nucleophilic aromatic substitution of aryl bromide 6 with 5 [ Mangion, I. K.; Sherry, B. D.; Yin, J.; Fleitz, F. J. Enantioselective Synthesis of a Dual Orexin Receptor Antagonist. Org. Lett. 2012, 14, 3458-3461 ], followed by Boc deprotection, with in situ generated HCl, furnished secondary amine 7. A final coupling with the benzoyl chloride derived from carboxylic acid 8 [ Mangion, I.
- NMR spectra are complicated by rotational isomerism about amide bonds. This behavior is illustrated by variable-temperature NMR spectra of compound 4e in DMSO (p. S102).
- IR spectra were obtained by use of a Perkin Elmer Spectrum BXII spectrometer or Nicolet 6700 FTIR spectrometer using thin films deposited on NaCl plates and reported in frequency of absorption (cm -1 ).
- Optical rotations were measured with a Jasco P-2000 polarimeter operating on the sodium D-line (589 nm), using a 100 mm path-length cell.
- allyl ester 3l 53 mg, 0.0997 mmol, 1.0 equiv
- Pd 2 (dba) 3 Purification by automated silica gel flash chromatography (0 ⁇ 50% EtOAc/hexanes) provided carbamate 4l as a white foam (37 mg, 0.0759 mmol, 76% yield, 93% ee).
- tert-butyl 4-benzoyl-5-oxo-1,4-diazepane-1-carboxylate (SI1).
- SI1 tert-butyl 4-benzoyl-5-oxo-1,4-diazepane-1-carboxylate
- THF 230 mL, 0.1 M
- n-BuLi 2.18 M in hexanes
- tert-butyl S -oxo-4-(4-(trifluoromethyl)benzoyl)-1,4-diazepane-1-carboxylate SI2
- THF 25 mL, 0.1 M
- n -BuLi 2.5 M in hexanes, 1.02 mL, 2.56 mmol, 1.1 equiv
- tert -butyl 4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1-carboxylate SI3
- n -BuLi 2.5 M in hexanes, 1.64 mL, 4.1 mmol, 1.1 equiv
- diisopropylamine (266 ⁇ L, 1.88 mmol, 1.2 equiv) in THF (10 mL) at -78 °C in a flame-dried round-bottom flask was added n -BuLi (2.5 M in hexanes, 792 ⁇ L, 1.73 mmol, 1.1 equiv), the resulting solution was stirred at -78 °C for 45 min.
- allyl cyanoformate (166 ⁇ L, 1.55 mmol, 1.2 equiv) was added dropwise at -78 °C, after which the solution slowly became colorless. After stirring for 1 h at -78 °C, the reaction was poured into 2 M HCl (20 mL) and extracted with ethyl acetate (4 x 20 mL). The combined organic extracts were dried over anhydrous Na 2 SO 4 and NaHCO 3 , passed through filter paper, and concentrated under reduced pressure.
- allyl ester 2a (1.00 g, 2.49 mmol, 1.0 equiv) in THF (25 mL, 0.1 M) at 0 °C was added NaH (60% dispersion in mineral oil, 107 mg, 2.74 mmol, 1.1 equiv) and the mixture was stirred at 0 °C for 30 min.
- allyl ester 2a 240 mg, 0.596 mmol, 1.0 equiv
- THF 6 mL, 0.1 M
- 60 % NaH 26 mg, 0.657 mmol, 1.1 equiv
- MeI 186 ⁇ L, 2.98 mmol, 5.0 equiv
- the reaction was heated to 45 °C and stirred for 16 h, cooled to 23 °C, poured into saturated aqueous NH 4 Cl (5 mL), and extracted with EtOAc (3 x 3 mL). The combined organic extracts were dried over Na 2 SO 4 and concentrated onto silica gel.
- the silica-adsorbed crude product was purified by silica gel flash chromatography (20% EtOAc/hexanes) to afford the title compound as a light yellow oil (200 mg, 0.480 mmol, 81% yield).
- allyl ester 2b 150 mg, 0.319 mmol, 1.0 equiv
- Cs 2 CO 3 208 mg, 0.638 mmol, 2.0 equiv
- MeI 99 ⁇ L, 1.59 mmol, 5.0 equiv
- the reaction was heated to 45 °C and stirred for 5 h, then cooled to 23 °C, poured into saturated aqueous NH 4 Cl (6 mL), and extracted with EtOAc (3 x 3 mL). The combined organic extracts were dried over anhydrous Na 2 SO 4 , filtered, and concentrated under reduced pressure.
- Cs 2 CO 3 301 mg, 0.925 mmol, 2.0 equiv
- MeI 143 ⁇ L, 2.31 mmol, 5.0 equiv
- the reaction was heated to 45 °C and stirred for 40 min, then cooled to 23 °C, poured into saturated aqueous NH 4 Cl (10 mL), and extracted with EtOAc (3 x 5 mL). The combined organic extracts were dried over anhydrous Na 2 SO 4 , filtered, and concentrated under reduced pressure.
- the crude product was purified by automated silica gel flash chromatography (Teledyne ISCO, 0 ⁇ 90% EtOAc/hexanes) to provide the title compound as a colorless oil (70 mg, 0.157 mmol, 34% yield).
- a solution of allyl ester 2c (300 mg, 0.694 mmol, 1.0 equiv) in THF (7 mL, 0.1 M) at 0 °C was added NaH (60% dispersion in mineral oil, 38 mg, 0.972 mmol, 1.4 equiv) and the mixture was stirred at 0 °C for 15 min and then allowed to warm to 23 °C over 15 min.
- allyl ester 2c 300 mg, 0.694 mmol, 1.0 equiv
- Cs 2 CO 3 453 mg, 1.39 mmol, 2.0 equiv
- 2,3-dichloropropene 320 ⁇ L, 3.47 mmol, 5.0 equiv
- 6-allyl 1-( tert -butyl) 4-benzoyl-6-fluoro-5-oxo-1,4-diazepane-1,6-dicarboxylate (3g).
- allyl ester 2a 250 mg, 0.621 mmol, 1.0 equiv
- THF 7.4 mL, 0.1 M
- allyl ester 2c 250 mg, 0.578 mmol, 1.0 equiv
- THF 5.8 mL, 0.1 M
- NaH 50% dispersion in mineral oil, 25 mg, 0.636 mmol, 1.1 equiv
- the crude product was purified by automated silica gel flash chromatography (Teledyne ISCO, 0 ⁇ 50% acetone/hexanes) to provide propargyl allyl ester 3i as a colorless oil (220 mg, 0.468 mmol, 81% yield).
- allyl ester 2c 300 mg, 0.694 mmol, 1.0 equiv
- K 2 CO 3 480 mg, 3.47 mmol, 5.0 equiv
- acetone 2.8 mL, 0.25 M
- methyl acrylate 126 ⁇ L, 1.39 mmol, 2.0 equiv
- allyl ester 2c 300 mg, 0.694 mmol, 1.0 equiv
- K 2 CO 3 480 mg, 3.47 mmol, 5.0 equiv
- acetone 2.8 mL, 0.25 M
- acrylonitrile 182 ⁇ L, 2.78 mmol, 4.0 equiv
- 6-allyl 1-( tert -butyl) 4-benzoyl-6-(((tert-butoxycarbonyl)amino)methyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (3l).
- reaction mixture was then diluted with diethyl ether (10 mL) and water (300 ⁇ L) was added. After gas generation subsided, 15% aqueous NaOH (300 ⁇ L) was added, followed by additional water (900 ⁇ L). After stirring at 0 °C for 15 min, anhydrous MgSO 4 was added, and the mixture was stirred for an additional 10 min, whereafter it was filtered through celite and concentrated under reduced pressure.
Abstract
Description
- This application claims the benefit of priority to
U.S. Patent Application No. 62/792,811 filed January 15, 2019 U.S. Patent Application No. 62/749,591 filed October 23, 2018 U.S. Patent Application No. 62/747,511 filed October 18, 2018 - Nitrogen-containing heterocycles are ubiquitous structural motifs that can be found across all areas and applications of organic chemistry. For example, piperazine, a representative diazaheterocycle, is the third most prevalent heterocycle in small molecule pharmaceuticals. As another example, pyrrolidines and pyrrolidinones occur widely in nature, possess a wide range of biological and pharmacological properties, and occur readily in materials and catalysis. As a further example, diazepane heterocycles are common structural motifs found in a variety of pharmaceuticals, including the benzodiazepine anxiolytics, the antipsychotic clozapine, and the anti-insomnia drug suvorexant. For these reasons, the development of stereoselective approaches to functionalized five-, six-, and seven-membered nitrogen-containing heterocycles is a topic of great interest in the synthesis of small molecules and natural products. Chiral gem-disubstituted nitrogen-containing heterocycles are targets of particular interest in pharmaceuticals, because of their increased molecular complexity and correspondingly enhanced binding affinity and specificity for a target, as receptor-ligand binding is often defined by three-dimensional contacts. However, installation of fully substituted chiral centers into azaheterocycles, especially diazaheterocycles, remains a challenge.
- Incorporation of a carbonyl group in the heterocycle has been shown to be a useful means of introducing chirality. The oxo group, when introduced on a carbon atom also bearing the nitrogen atom, may be used to selectively introduce chirality at the α position and later removed to produce the functionalized heterocycle or other downstream products. In pyrrolidinones, progress has been made toward enantioselective allylic alkylation and enantioselective α-acylation of γ-butyrolactams; and in piperazines, Stoltz et al. reported in 2015 a palladium-catalyzed decarboxylative asymmetric allylic alkylation reaction to synthesize chiral α,α-disubstituted N4-benzylated piperazin-2-ones. However, α-arylation of γ-butyrolactams has remained elusive, as the use of monocyclic lactams in transition metal-catalyzed α-arylation risks the generation of unwanted aryne intermediates and catalyst decomposition. Likewise, the 2015 Stoltz chiral α,α-disubstituted N4-benzylated piperazin-2-one chemistry is plagued by low product yields owing to side reactions of the benzyl-protected N4, and other protecting groups such as PMB proved recalcitrant to deprotection.
- Accordingly, improved methods are needed for generation of gem-disubstituted pyrrolidines, piperazines, and diazepanes.
- In certain embodiments, the invention relates to a method for the preparation of a compound of formula (I):


with a Pd or Ni catalyst comprising a chiral ligand;
an optionally substituted aryl halide, optionally substituted aryl pseudohalide, optionally substituted heteroaryl halide, an optionally substituted heteroaryl pseudohalide, or
a base;
wherein, as valence and stability permit, - ring A represents an optionally substituted heterocycloalkyl, or heterocycloalkenyl group;
- R in each occurrence independently represents optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, -OR10, -SR10, or -NR10R11;
- R1 represents hydrogen or optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, alkynyl, -C(O)alkyl, -C(O)aryl, - C(O)aralkyl,
-C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), -S(O)2(aryl), -S(O)2(alkyl), -S(O)2(haloalkyl), -OR10, -SR10, or -NR10R11; - or R1 taken together with a substituent on ring A and the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;
- or a substituent on ring A taken together with another substituent on ring A and the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;
- R2 represents substituted or unsubstituted alkyl, alkenyl, alkynyl, aralkyl, aralkenyl, aryl, heteroaralkyl, heteroaralkenyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkoxy, amino, or halo;
- R10 and R11 are independently selected for each occurrence from hydrogen or substituted or unsubstituted alkyl, aralkyl, aryl, heteroaralkyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, and alkynyl;
- X is a halogen or pseudohalide; and
- Y represents optionally substituted aryl, optionally substituted heteroaryl, or

- In certain embodiments, the invention relates to a compound of formula (I) made by any of the methods described herein.
- In certain embodiments, the invention relates to a method comprising
preparing a compound of formula (I) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (I). - In certain aspects, the invention relates to a compound of formula (III) or formula (IV):


- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R23 is H, halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, alkoxycarbonyl, halogen, aryl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R24 is H or halogen; and
- p is 0 or 1.
- In certain embodiments, p is 0; the compound of formula (III) is a compound of formula (IIIa):


- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R23 is H, or C1-6 alkyl optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain embodiments, p is 1; the compound of formula (III) is a compound of formula (IIIb):


- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a;
- R22a is C1-6 alkyl;
- R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with OH, halogen, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl; and
- R24 is H.
- In certain aspects, the invention relates to a compound of formula (V), (VI), or (VII):



- R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, alkynyl, or -NHC(O)OR25b; halogen; or allyl, optionally substituted with halogen;
- R25b is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl.
- In certain aspects, the invention relates to a compound selected from:



- In certain aspects, the invention relates to a compound of Formula (VIII):

- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R23b is H, or C1-6 alkyl optionally substituted with OH, CN, alkoxy, aryloxy, amino, acyl, or -NHC(O)R23c;
- R23c is C1-6 alkyl, (C6-10 aryl)alkyl or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain aspects, the invention relates to a compound of Formula (IX) or (X):


- R23d is H, or is C1-6 alkyl optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, - NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain aspects, the invention relates to a compound of formula (XI):

- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R24 is H or halogen; and
- R23e is H, or is C1-6 alkyl optionally substituted with halogen, OH, aryl, aryloxy, CN, acyl, or amino.
- In certain aspects, the invention relates to a compound of formula (XII):

- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl; and
- R24 is H or halogen.
- In certain aspects, the invention relates to a compound of formula (XIII):

- R25a is C1-6 alkyl, optionally substituted with CN, OH, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen; and
- R13 is aralkyl or hetaralkyl.
- In certain aspects, the invention relates to a method for preparation of a compound of formula (IV):

treating a compound of formula (III)
with a Pd(0) catalyst and a ligand L under alkylation conditions,
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl is optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl;
- R23 is H, halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, alkoxycarbonyl, halogen, aryl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R24 is H or halogen; and
- p is 0 or 1.
- In certain embodiments, p is 0, the compound of formula (IV) is a compound of formula (IVa):


with a Pd(0) catalyst and a ligand L under alkylation conditions,
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl is optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl;
- R23 is H, or C1-6 alkyl optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain embodiments, the invention relates to a compound of formula (IVa) made by any of the methods described herein.
- In certain embodiments, the invention relates to a method comprising preparing a compound of formula (IVa) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (IVa).
- In certain embodiments, p is 1; the compound of formula (IV) is a compound of formula (IVb)


wherein, as valence and stability permit, - R22 is -C(O)OR22a;
- R22a is C1-6 alkyl;
- R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl; and
- R24 is H.
- In certain aspects, the invention relates to a compound of formula (E):

wherein, as valence and stability permit, - R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl; and
- R24 is H.
- In certain embodiments, the invention relates to a compound of formula (IVb) made by any of the methods described herein.
- In certain embodiments, the invention relates to a method comprising preparing a compound of formula (IVb) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (IVb).
- In certain embodiments, the invention relates to a method of preparation of a compound of formula (VI):

treating a compound of formula (V):
with a Pd(0) catalyst and a ligand L, under alkylation conditions,
wherein, as valence and stability permit,
R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen. - In certain embodiments, the invention relates to a compound of formula (VI) made by any of the methods described herein.
- In certain embodiments, the invention relates to a method comprising preparing a compound of formula (VI) according to any of the methods described herein; and synthesizing a medicinal product from the compound of formula (VI).
- In certain embodiments, the invention is directed to a method for preparation of a compound of formula (VIII):

treating a compound of formula (IVa):
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is Boc;
- R23 is H, or is C1-6 alkyl optionally substituted with CN, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R24 is H or halogen;
- R23b is H; or is C1-6 alkyl optionally substituted with halogen, OH, CN, alkoxy, aryloxy, amino, acyl, or
-NHC(O)OR23c; and - R23c is (C6-10 aryl)alkyl or (C5-9 heteroaryl)alkyl.
- In certain embodiments, the invention relates to a compound of formula (VIII) made by any of the methods described herein.
- In certain embodiments, the invention relates to a method for the preparation of a compound of formula (IX):

treating a compound of formula (IVa):
with LiOH in a first solvent,
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is Boc;
- R23 is H, or C1-6 alkyl optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, acyl, or
-NHC(O)OR23a; - R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R23d is H, or is C1-6 alkyl optionally substituted with halogen, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a; and
- R24 is H or halogen.
- In certain embodiments, the invention relates to a compound of formula (IX) made by any of the methods described herein.
- In certain embodiments, the invention relates to a method of preparation of a compound of formula (XI):

treating a compound of formula (IVa):
with hydrogen gas in the presence of a Pd/C catalyst in a first solvent, to form a first product mixture;
filtering the first product mixture, to form a crude first product; and
treating the crude first product with TFA in a second solvent;
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is Boc;
- R23 is H, or is C1-6 alkyl optionally substituted with halogen, OH, alkoxy, aryloxy, CN, aryl, or
-NHC(O)OR23a; - R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R23e is H, or is C1-6 alkyl optionally substituted with halogen, OH, aryl, aryloxy, CN, acyl, or amino; and
- R24 is H or halogen.
- In certain embodiments, the invention relates to a compound of formula (XI) made by any of the methods described herein.
- In certain embodiments, the invention relates to method of preparation of a compound of formula (XIII):

treating a compound of formula (VI):
with TFA in a first solvent, to form a first product;
treating the first product with lithium hydroxide in a second solvent, to form a second product;
treating the second product with Boc2O in the presence of a base, to form a third product;
treating the third product with K2CO3 and R13-X21 in a third solvent, to form a compound of formula (XIII);
wherein, as valence and stability permit, - R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen;
- R25a is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen;
- R13 is aralkyl or hetaralkyl; and
- X21 is halogen.
- In certain embodiments, the invention relates to a compound of formula (XIII) made by any of the methods described herein.
- In certain embodiments, the invention relates to any one of the compounds described in the Examples.
-
FIG. 1 shows the structures of examples of enantioenriched phosphine ligands. - Unless otherwise defined herein, scientific and technical terms used in this application shall have the meanings that are commonly understood by those of ordinary skill in the art. Generally, nomenclature used in connection with, and techniques of, chemistry, cell and tissue culture, molecular biology, cell and cancer biology, neurobiology, neurochemistry, virology, immunology, microbiology, pharmacology, genetics and protein and nucleic acid chemistry, described herein, are those well-known and commonly used in the art.
- The methods and techniques of the present disclosure are generally performed, unless otherwise indicated, according to conventional methods well known in the art and as described in various general and more specific references that are cited and discussed throughout this specification. See, e.g., "Principles of Neural Science", McGraw-Hill Medical, New York, N.Y. (2000); Motulsky, "Intuitive Biostatistics", Oxford University Press, Inc. (1995); Lodish et al., "Molecular Cell Biology, 4th ed.", W. H. Freeman & Co., New York (2000); Griffiths et al., "Introduction to Genetic Analysis, 7th ed.", W. H. Freeman & Co., N.Y. (1999); and Gilbert et al., "Developmental Biology, 6th ed.", Sinauer Associates, Inc., Sunderland, MA (2000).
- Chemistry terms used herein, unless otherwise defined herein, are used according to conventional usage in the art, as exemplified by "The McGraw-Hill Dictionary of Chemical Terms", Parker S., Ed., McGraw-Hill, San Francisco, C.A. (1985).
- All of the above, and any other publications, patents and published patent applications referred to in this application are specifically incorporated by reference herein. In case of conflict, the present specification, including its specific definitions, will control.
- The definitions for the terms described below are applicable to the use of the term by itself or in combination with another term.
- The term "acyl" is art-recognized and refers to a group represented by the general formula hydrocarbyl-C(O)-, preferably alkyl-C(O)-.
- The term "acylamino" is art-recognized and refers to an amino group substituted with an acyl group and may be represented, for example, by the formula hydrocarbyl-C(O)NH-.
- The term "acyloxy" is art-recognized and refers to a group represented by the general formula hydrocarbyl-C(O)O-, preferably alkyl-C(O)O-.
- The term "alkoxy" refers to an alkyl group, preferably a lower alkyl group, having an oxygen attached thereto. Representative alkoxy groups include methoxy, trifluoromethoxy, ethoxy, propoxy, tert-butoxy and the like.
- The term "alkoxyalkyl" refers to an alkyl group substituted with an alkoxy group and may be represented by the general formula alkyl-O-alkyl.
- The term "alkenyl", as used herein, refers to an aliphatic group containing at least one double bond and from 1 to about 20 carbon atoms, preferably from 1 to about 10 carbon atoms unless otherwise defined, and is intended to include both "unsubstituted alkenyls" and "substituted alkenyls", the latter of which refers to alkenyl moieties having substituents replacing a hydrogen on one or more carbons of the alkenyl group. Such substituents may occur on one or more carbons that are included or not included in one or more double bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed below, except where stability is prohibitive. For example, substitution of alkenyl groups by one or more alkyl, carbocyclyl, aryl, heterocyclyl, or heteroaryl groups is contemplated. A C2-C6 alkenyl group is also referred to as a "lower alkenyl" group.
- An "alkyl" group or "alkane" is a straight chained or branched non-aromatic hydrocarbon which is completely saturated. Typically, a straight chained or branched alkyl group has from 1 to about 20 carbon atoms, preferably from 1 to about 10 unless otherwise defined. Examples of straight chained and branched alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, tert-butyl, pentyl, hexyl, pentyl and octyl. A C1-C6 straight chained or branched alkyl group is also referred to as a "lower alkyl" group.
- Moreover, the term "alkyl" (or "lower alkyl") as used throughout the specification, examples, and claims is intended to include both "unsubstituted alkyls" and "substituted alkyls", the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone. Such substituents, if not otherwise specified, can include, for example, a halogen (e.g., fluoro), a hydroxyl, an oxo, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxy, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. In preferred embodiments, the substituent on substituted alkyls are selected from C1-C6 alkyl, C3-C6 cycloalkyl, halogen, carbonyl, cyano, or hydroxyl. In more preferred embodiments, the substituents on the substituted alkyls are selected from fluoro, carbonyl, cyano, or hydroxyl. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate. For instance, the substituents of a substituted alkyl may include substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl (including phosphonate and phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), -CF3, -CN and the like. Exemplary substituted alkyls are described below. Cycloalkyls can be further substituted with alkyls, alkenyls, alkoxys, alkylthios, aminoalkyls, carbonyl-substituted alkyls, -CF3, -CN, and the like.
- The term "Cx-Cy", when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain. For example, the term "Cx-Cy alkyl" refers to substituted or unsubstituted saturated hydrocarbon groups, including straight-chain alkyl and branched-chain alkyl groups that contain from x to y carbons in the chain, including haloalkyl groups. Preferred haloalkyl groups include trifluoromethyl, difluoromethyl, 2,2,2-trifluoroethyl, and pentafluoroethyl. Co alkyl indicates a hydrogen where the group is in a terminal position, a bond if internal. The terms "C2-Cyalkenyl" and "C2-Cyalkynyl" refer to substituted or unsubstituted unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- The term "alkylamino", as used herein, refers to an amino group substituted with at least one alkyl group.
- The term "alkylthio", as used herein, refers to a thiol group substituted with an alkyl group and may be represented by the general formula alkyl-S-.
- The term "alkynyl", as used herein, refers to an aliphatic group containing at least one triple bond and is intended to include both "unsubstituted alkynyls" and "substituted alkynyls", the latter of which refers to alkynyl moieties having substituents replacing a hydrogen on one or more carbons of the alkynyl group. Such substituents may occur on one or more carbons that are included or not included in one or more triple bonds. Moreover, such substituents include all those contemplated for alkyl groups, as discussed above, except where stability is prohibitive. For example, substitution of alkynyl groups by one or more alkyl, carbocyclyl, aryl, heterocyclyl, or heteroaryl groups is contemplated.
- The term "amide", as used herein, refers to a group

- The terms "amine" and "amino" are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by

- The term "aminoalkyl", as used herein, refers to an alkyl group substituted with an amino group.
- The term "aralkyl", as used herein, refers to an alkyl group substituted with an aryl group.
- The term "aryl" as used herein include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon. Preferably the ring is a 6- to 10-membered ring, more preferably a 6-membered ring. The term "aryl" also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Aryl groups include benzene, naphthalene, phenanthrene, aniline, and the like. Example substitutions on an aryl group include a halogen, a haloalkyl such as trifluoromethyl, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl such as an alkylC(O)), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxy, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a silyl ether, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety.
- The term "carbamate" is art-recognized and refers to a group

- The term "carbocycle" refers to a saturated or unsaturated ring in which each atom of the ring is carbon. The term carbocycle includes both aromatic carbocycles and non-aromatic carbocycles. Non-aromatic carbocycles include both cycloalkyl and cycloalkenyl rings. "Carbocycle" includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings. Carbocycle includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings. The term "fused carbocycle" refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring. Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings. In an exemplary embodiment, an aromatic ring, e.g., phenyl, may be fused to a saturated or unsaturated ring, e.g., cyclohexane, cyclopentane, or cyclohexene. Any combination of saturated, unsaturated and aromatic bicyclic rings, as valence permits, is included in the definition of carbocyclic. Exemplary "carbocycles" include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5-cyclooctadiene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]oct-3-ene, naphthalene and adamantane. Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4,5,6,7-tetrahydro-1H-indene and bicyclo[4.1.0]hept-3-ene. "Carbocycles" may be substituted at any one or more positions capable of bearing a hydrogen atom.
- The term "carbocyclylalkyl", as used herein, refers to an alkyl group substituted with an carbocycle group.
- The term "carbonate" is art-recognized and refers to a group -OCO2-RA, wherein RA represents a hydrocarbyl group.
- The term "carboxy", as used herein, refers to a group represented by the formula-CO2H.
- A "cycloalkyl" group is a cyclic hydrocarbon, which is completely saturated. "Cycloalkyl" includes monocyclic and bicyclic rings. Typically, a monocyclic cycloalkyl group has from 3 to about 10 carbon atoms, from 3 to 8 carbon atoms, or more typically from 3 to 6 carbon atoms, unless otherwise defined. The second ring of a bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings. Cycloalkyl includes bicyclic substituents in which one, two or three or more atoms are shared between the two rings (e.g., fused bicyclic substituents, bridged bicyclic substituents, and spirocyclic substituents).
- The term "fused bicyclic substituent" refers to a bicyclic substituent in which two rings share two adjacent atoms. In other words, the rings share one covalent bond, i.e., the so-called bridgehead atoms are directly connected. For example, in a fused cycloalkyl, each of the rings shares two adjacent atoms with the other ring, and the second ring of a fused bicyclic cycloalkyl may be selected from saturated, unsaturated and aromatic rings.
- A "cycloalkenyl" group is a cyclic hydrocarbon containing one or more double bonds.
- The term "bridged bicyclic substituent" refers to a bicyclic substituent in which the two rings share three or more atoms, separating the two bridgehead atoms by a bridge containing at least one atom. For example, norbornanyl, also known as bicyclo[2.2.1]heptanyl, can be thought of as a pair of cyclopentane rings each sharing three of their five carbon atoms.The term "spirocyclic substituent" refers to a bicyclic substituent in which the two rings have only one single atom, the spiro atom, in common.
- The term "diazo", as used herein, refers to a group represented by the formula =N=N.
- The term "disulfide" is art-recognized and refers to a group -S-S-RA, wherein RA represents a hydrocarbyl group.
- The term "enol ester", as used herein, refers to a group -C(O)O-C(RA)=C(RA)2 wherein RA represents a hydrocarbyl group.
- The term "ester", as used herein, refers to a group -C(O)OR10 wherein R10 represents a hydrocarbyl group.
- The term "ether", as used herein, refers to a hydrocarbyl group linked through an oxygen to another hydrocarbyl group. Accordingly, an ether substituent of a hydrocarbyl group may be hydrocarbyl-O-. Ethers may be either symmetrical or unsymmetrical. Examples of ethers include, but are not limited to, heterocycle-O-heterocycle and aryl-O-heterocycle. Ethers include "alkoxyalkyl" groups, which may be represented by the general formula alkyl-O-alkyl.
- The terms "halo" and "halogen" as used herein means halogen and includes chloro, fluoro, bromo, and iodo.
- The term "heteroalkyl", as used herein, refers to a saturated or unsaturated chain of carbon atoms and at least one heteroatom, for example, wherein no two heteroatoms are adjacent.
- The terms "heteroaralkyl" and "hetaralkyl", as used herein, refers to an alkyl group substituted with a hetaryl group.
- The terms "heteroaryl" and "hetaryl" include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms "heteroaryl" and "hetaryl" also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heteroaryl groups include 5- to 10-membered cyclic or polycyclic ring systems, such as pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, and pyrimidine, and the like.
- The term "heteroatom" as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
- The terms "heterocyclyl", "heterocycle", and "heterocyclic" refer to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, preferably 3-to 7-membered rings, more preferably 5- to 6-membered rings, in some instances, most preferably a 5-membered ring, in other instances, most preferably a 6-membered ring, which ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms "heterocyclyl" and "heterocyclic" also include 5- to 10- membered polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heterocyclyl groups include, for example, pyrrolidine, piperidine, piperazine, pyrrolidine, tetrahydropyran, tetrahydrofuran, morpholine, lactones, lactams, oxazoles, imidazolines, and the like.
- The term "heterocyclylalkyl", as used herein, refers to an alkyl group substituted with a heterocycle group.
- The term "hydrocarbyl", as used herein, refers to a group that is bonded through a carbon atom that does not have a =O or =S substituent, and typically has at least one carbon-hydrogen bond and a primarily carbon backbone, but may optionally include heteroatoms. Thus, groups like methyl, ethoxyethyl, 2-pyridyl, and trifluoromethyl are considered to be hydrocarbyl for the purposes of this application, but substituents such as acetyl (which has a =O substituent on the linking carbon) and ethoxy (which is linked through oxygen, not carbon) are not. Hydrocarbyl groups include, but are not limited to aryl, heteroaryl, carbocycle, heterocyclyl, alkyl, alkenyl, alkynyl, and combinations thereof.
- The term "hydroxyalkyl", as used herein, refers to an alkyl group substituted with a hydroxy group.
- The term "lower" when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups where there are ten or fewer non-hydrogen atoms in the substituent, preferably six or fewer, more preferably three or fewer. A "lower alkyl", for example, refers to an alkyl group that contains ten or fewer carbon atoms, preferably six or fewer, more preferably three or fewer. In certain embodiments, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy substituents defined herein are respectively lower acyl, lower acyloxy, lower alkyl, lower alkenyl, lower alkynyl, or lower alkoxy, whether they appear alone or in combination with other substituents, such as in the recitations hydroxyalkyl and aralkyl (in which case, for example, the atoms within the aryl group are not counted when counting the carbon atoms in the alkyl substituent).
- The term "orthoester" as used herein is art-recognized and refers to a group - C(ORA)3, wherein each RA independently represents hydrogen or a hydrocarbyl, such as alkyl, or any occurrence of RA taken together with another and the intervening atom(s) complete a heterocycle having from 4 to 8 atoms in the ring structure.
- The term "phosphoester", as used herein, refers to a group -OP(=O)(OH)2.
- The term "phosphodiester", as used herein, refers to a group -OP(=O)(ORA)2 wherein RA represents a hydrocarbyl group.
- The terms "polycyclyl", "polycycle", and "polycyclic" refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls) in which two or more atoms are common to two adjoining rings, e.g., the rings are "fused rings". Each of the rings of the polycycle can be substituted or unsubstituted. In certain embodiments, each ring of the polycycle contains from 3 to 10 atoms in the ring, preferably from 5 to 7.
- The term "pseudohalide," as used herein, means a polyatomic analogue of a halide. Preferred pseudohalides include sulfonates, e.g., arylsulfonates and alkylsulfonates, such as p-toluenesulfonate and trifluoromethanesulfonate.
- The term "selenide", as used herein, is equivalent to an ether, wherein the oxygen is replaced with a selenium.
- The term "selenoxide" is art-recognized and refers to the group -Se(O)-RA, wherein RA represents a hydrocarbyl.
- The term "siloxane" is art-recognized and refers to a group with an Si-O-Si linkage, such as the group -Si(RA)2-O-Si-(RA)3, wherein each RA independently represents hydrogen or hydrocarbyl, such as alkyl, or two RA taken together with the intervening atom(s) complete a heterocycle having from 4 to 8 atoms in the ring structure.
- The term "silyl" refers to a silicon moiety with three hydrocarbyl moieties attached thereto.
- The term "substituted" refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that "substitution" or "substituted with" includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term "substituted" is contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds. The permissible substituents can be one or more and the same or different for appropriate organic compounds. For purposes of this disclosure, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, an oxo, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxy, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an alkyl, an amino, an amido, an amidine, an imine, an oxime, a cyano, a nitro, an azido, a silyl ether, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. In preferred embodiments, the substituents on substituted alkyls are selected from C1-C6 alkyl, C3-C6 cycloalkyl, halogen, carbonyl, cyano, or hydroxyl. In more preferred embodiments, the substituents on substituted alkyls are selected from fluoro, carbonyl, cyano, or hydroxyl. It will be understood by those skilled in the art that substituents can themselves be substituted, if appropriate. Unless specifically stated as "unsubstituted," references to chemical moieties herein are understood to include substituted variants. For example, reference to an "aryl" group or moiety implicitly includes both substituted and unsubstituted variants.
- The term "sulfate" is art-recognized and refers to the group -OSO3H, or a pharmaceutically acceptable salt thereof.
- The term "sulfonamide" is art-recognized and refers to the group represented by the general formulae

- The term "sulfoxide" is art-recognized and refers to the group -S(O)-R10, wherein R10 represents a hydrocarbyl.
- The term "sulfonate" is art-recognized and refers to the group SO3H, or a pharmaceutically acceptable salt thereof.
- The term "sulfone" is art-recognized and refers to the group -S(O)2-R10, wherein R10 represents a hydrocarbyl.
- The term "thioalkyl", as used herein, refers to an alkyl group substituted with a thiol group.
- The term "thioester", as used herein, refers to a group -C(O)SR10 or -SC(O)R10 wherein R10 represents a hydrocarbyl.
- The term "thioether", as used herein, is equivalent to an ether, wherein the oxygen is replaced with a sulfur.
- The term "urea" is art-recognized and may be represented by the general formula

- "Protecting group" refers to a group of atoms that, when attached to a reactive functional group in a molecule, mask, reduce or prevent the reactivity of the functional group. Typically, a protecting group may be selectively removed as desired during the course of a synthesis. Examples of protecting groups can be found in Greene and Wuts, Protective Groups in Organic Chemistry, 3rd Ed., 1999, John Wiley & Sons, NY and Harrison et al., Compendium of Synthetic Organic Methods, Vols. 1-8, 1971-1996, John Wiley & Sons, NY. Representative nitrogen protecting groups include, but are not limited to, formyl, acetyl, trifluoroacetyl, anisoyl ("An"), benzyl ("Bn"), benzoyl ("Bz"), benzyloxycarbonyl ("CBZ"), tert-butoxycarbonyl ("Boc"), trimethylsilyl ("TMS"), 2- trimethylsilyl-ethanesulfonyl ("TES"), trityl and substituted trityl groups, allyloxycarbonyl, 9- fluorenylmethyloxycarbonyl ("FMOC"), nitro-veratryloxycarbonyl ("NVOC") and the like. Representative hydroxyl protecting groups include, but are not limited to, those where the hydroxyl group is either acylated (esterified) or alkylated such as benzyl and trityl ethers, as well as alkyl ethers, tetrahydropyranyl ethers, trialkylsilyl ethers (e.g., TMS or TIPS groups), glycol ethers, such as ethylene glycol and propylene glycol derivatives and allyl ethers.
- The phrase "pharmaceutically acceptable" is art-recognized. In certain embodiments, the term includes compositions, excipients, adjuvants, polymers and other materials and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- "Pharmaceutically acceptable salt" or "salt" is used herein to refer to an acid addition salt or a basic addition salt that is suitable for or compatible with the treatment of patients.
- The term "pharmaceutically acceptable acid addition salt" as used herein means any non-toxic organic or inorganic salt of any base compounds disclosed herein. Illustrative inorganic acids that form suitable salts include hydrochloric, hydrobromic, sulfuric and phosphoric acids, as well as metal salts such as sodium monohydrogen orthophosphate and potassium hydrogen sulfate. Illustrative organic acids that form suitable salts include mono-, di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic, phenylacetic, cinnamic and salicylic acids, as well as sulfonic acids such as p-toluene sulfonic and methanesulfonic acids. Either the mono or di-acid salts can be formed, and such salts may exist in either a hydrated, solvated or substantially anhydrous form. In general, the acid addition salts of compounds disclosed herein are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms. The selection of the appropriate salt will be known to one skilled in the art. Other non-pharmaceutically acceptable salts, e.g., oxalates, may be used, for example, in the isolation of compounds of the invention for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
- The term "pharmaceutically acceptable basic addition salt" as used herein means any non-toxic organic or inorganic base addition salt of any acid compounds of the invention, or any of their intermediates. Illustrative inorganic bases that form suitable salts include lithium, sodium, potassium, calcium, magnesium, or barium hydroxide. Illustrative organic bases which form suitable salts include aliphatic, alicyclic, or aromatic organic amines such as methylamine, trimethylamine and picoline or ammonia. The selection of the appropriate salt will be known to a person skilled in the art.
- Many of the compounds useful in the methods and compositions of this disclosure have at least one stereogenic center in their structure. This stereogenic center may be present in a R or a S configuration, said R and S notation is used in correspondence with the rules described in Pure Appl. Chem. (1976), 45, 11-30. The disclosure contemplates all stereoisomeric forms such as enantiomeric and diastereoisomeric forms of the compounds, salts, prodrugs or mixtures thereof (including all possible mixtures of stereoisomers). See, e.g.,
WO 01/062726 - Furthermore, certain compounds which contain alkenyl groups may exist as Z (zusammen) or E (entgegen) isomers. In each instance, the disclosure includes both mixtures and separate individual isomers.
- Some of the compounds may also exist in tautomeric forms. Such forms, although not explicitly indicated in the formulae described herein, are intended to be included within the scope of the present disclosure.
- This disclosure is based on the discovery of novel C-arylation and novel C-vinylation reactions that generate an α-quaternary substituted lactam. The methods comprise treating a lactam with a chiral nickel (Ni) or palladium (Pd) catalyst, and an aryl halide, heteroaryl halide or a vinyl halide. Use of a ligand, preferably a chiral ligand, enables the construction of quaternary stereocenters on α-substituted lactams to form β-aryl or β-vinyl lactams.
- According to some embodiments of the present disclosure, a wide range of structurally-diverse, functionalized products are prepared by a stereoselective method of nickel or palladium-catalyzed enantioselective arylation or vinylation. This chemistry is useful in the synthesis of lactams, and for the construction of novel building blocks for medicinal and polymer chemistry.
- This disclosure is also based, in part, on the discovery of novel decarboxylative asymmetric allylic alkylation reactions that generate α-quatemary substituted piperazinones, tetrahydropyrimidinones, and diazepanones. The methods comprise enolate functionalization of protected 3-(allyloxycarbonyl)piperazine-2-ones, 5-(allyloxycarbonyl)tetrahydropyrimidin-4-one, and 6-(allyloxycarbonyl)-1,4-diazepan-5-ones, followed by decarboxylative asymmetric allylic alkylation. This chemistry is useful in the construction of novel building blocks for medicinal chemistry, such as in the synthesis of β-lactams (such as in β-lactam antibiotics), hexahydropyrimidines, and benzodiazepines. For example, tetrahydropyrimidinones may be reductively transformed into hexahydropyrimidines, which are found in bioactive molecules such as the antibiotic hexetidine and the macrocyclic natural product verbamethine. [A. Guggisberg, K. Drandarov, M. Hesse, Helvetica Chimica Acta 2000, 83, 3035-3042; K. Drandarov, A. Guggisberg, M. Hesse, Helvetica Chimica Acta 2002, 85, 979-989.] In addition, diazepane heterocycles are also found in the antipsychotic clozapine and the anti-insomnia drug suvorexant.
- Also provided herein are methods of synthesizing a pharmaceutical agent and/or composition, comprising preparing a compound of formula (I), (Ia), (Ib), (IVa), or (IVb) as defined below, according to a method as described herein and synthesizing the pharmaceutical agent and/or composition from the compound of formula (I), (Ia), (Ib), (IVa), or (IVb), e.g., by carrying out one or more chemical reactions on the compound of formula (I), (Ia), (Ib), (IVa), or (IVb) and/or combining the pharmaceutical agent with one or more pharmaceutically acceptable carriers and/or excipients.
- The pharmaceutical agent and/or composition prepared from the compound of formula (I), (Ia), (Ib), (IVa), or (IVb) may be utilized to treat an individual in need thereof. In certain embodiments, the individual is a mammal such as a human, or a non-human mammal. When administered to an animal, such as a human, the composition or the compound is preferably administered as a pharmaceutical composition comprising, for example, a compound of the invention and a pharmaceutically acceptable carrier. Pharmaceutically acceptable carriers are well known in the art and include, for example, aqueous solutions such as water or physiologically buffered saline or other solvents or vehicles such as glycols, glycerol, oils such as olive oil, or injectable organic esters. In preferred embodiments, when such pharmaceutical compositions are for human administration, particularly for invasive routes of administration (i.e., routes, such as injection or implantation, that circumvent transport or diffusion through an epithelial barrier), the aqueous solution is pyrogen-free, or substantially pyrogen-free. The excipients can be chosen, for example, to effect delayed release of an agent or to selectively target one or more cells, tissues or organs. The pharmaceutical composition can be in dosage unit form such as tablet, capsule (including sprinkle capsule and gelatin capsule), granule, lyophile for reconstitution, powder, solution, syrup, suppository, injection or the like. The composition can also be present in a transdermal delivery system, e.g., a skin patch. The composition can also be present in a solution suitable for topical administration, such as a lotion, cream, or ointment.
- Methods of preparing these formulations or compositions include the step of bringing into association an active compound, such as a compound of the invention, with the carrier and, optionally, one or more accessory ingredients. In general, the formulations are prepared by uniformly and intimately bringing into association a compound of the present invention with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- In certain aspects, the present disclosure provides for the preparation of a compound of formula (I):
In certain embodiments, the invention relates to a method for the preparation of a compound of formula (I):

with a Pd or Ni catalyst comprising a chiral ligand;
an optionally substituted aryl halide, optionally substituted aryl pseudohalide, optionally substituted heteroaryl halide, an optionally substituted heteroaryl pseudohalide, or
a base;
wherein, as valence and stability permit, - ring A represents an optionally substituted heterocycloalkyl, or heterocycloalkenyl group;
- R in each occurrence independently represents optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, -OR10, -SR10, or -NR10R11;
- R1 represents hydrogen or optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, alkynyl, -C(O)alkyl, -C(O)aryl,-C(O)aralkyl,
-C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), -S(O)2(aryl), -S(O)2(alkyl), -S(O)2(haloalkyl), -OR10, -SR10, or -NR10R11; - or R1 taken together with a substituent on ring A and the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;
- or a substituent on ring A taken together with another substituent on ring A and the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;
- R2 represents substituted or unsubstituted alkyl, alkenyl, alkynyl, aralkyl, aralkenyl, aryl, heteroaralkyl, heteroaralkenyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkoxy, amino, or halo;
- R10 and R11 are independently selected for each occurrence from hydrogen or substituted or unsubstituted alkyl, aralkyl, aryl, heteroaralkyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, and alkynyl;
- X is a halogen or pseudohalide; and
- Y represents optionally substituted aryl, optionally substituted heteroaryl, or

- In certain embodiments, the compound of formula (I) is represented by formula (Ia):


- B, D, and E independently for each occurrence represent, as valence permits, O, S, NR4, CR5R6, C(O), CR5, or N; provided that no two adjacent occurrences of N, B, D, and E are NR4, O, S, or N;
- R4 represents hydrogen or optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, alkynyl, -C(O)alkyl, -C(O)aryl,-C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl),-C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), -S(O)2(aryl),-S(O)2(alkyl), -S(O)2(haloalkyl), -OR10, -SR10, or -NR10R11;
- R5 and R6 each independently represent hydrogen, hydroxyl, halogen, nitro, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, cyano, carboxyl, sulfate, amino, alkoxy, aryloxy, arylalkoxy, alkylamino, alkylthio, hydroxyalkyl, alkoxyalkyl, aminoalkyl, thioalkyl, haloalkyl, ether, thioether, ester, amido, thioester, carbonate, carbamate, urea, sulfonate, sulfone, sulfoxide, sulfonamide, acyl, acyloxy, or acylamino;
- or any two occurrences of R1, R4, R5, and R6 on adjacent N, B, D, or E groups, taken together with the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;
- each occurrence of independently represents a double bond or a single bond as permitted by valence; and

- m and n are integers each independently selected from 0, 1, and 2.
- In some embodiments, the sum of m and n is 0, 1, 2, or 3.
- In exemplary embodiments, each occurrence of B, D, and E is independently -CR5R6-, -CR5-, or -C(O)-.
- In certain embodiments, E is -CR5-; and the sum of m and n is 0.
- In certain embodiments,
R1 is selected from optionally substituted alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl,
-C(O)alkyl, -C(O)aryl, -C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), and -S(O)2(aryl); and
R5 is selected from hydrogen, hydroxyl, halogen, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, amino, alkoxy, aryloxy, alkylamino, amido, and acylamino; or
R1 and the occurrence of R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group. - In certain embodiments, R1 is selected from optionally substituted alkyl, aryl, aralkyl, alkenyl, -C(O)alkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), and -S(O)2(aryl), for example, substituted aryl.
- In some embodiments, R1 and the occurrence of R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group, for example, R1 and the occurrence of R5 on E are taken together to form an optionally substituted heterocycloalkyl, or heterocycloalkenyl group.
- In some embodiments, the compound of formula (I) is represented by formula (Ib):


- R5 and R6 each independently represent hydrogen, hydroxyl, halogen, nitro, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, cyano, carboxyl, sulfate, amino, alkoxy, aryloxy, arylalkoxy, alkylamino, alkylthio, hydroxyalkyl, alkoxyalkyl, aminoalkyl, thioalkyl, haloalkyl, ether, thioether, ester, amido, thioester, carbonate, carbamate, urea, sulfonate, sulfone, sulfoxide, sulfonamide, acyl, acyloxy, or acylamino;
- or any two occurrences of R1, R5, and R6, taken together with the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group.
- In certain embodiments,
R1 is selected from optionally substituted alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl,
-C(O)alkyl, -C(O)aryl, -C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), and -S(O)2(aryl); and
R5 is selected from hydrogen, hydroxyl, halogen, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, amino, alkoxy, aryloxy, alkylamino, amido, and acylamino; or
R1 and R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group. - In certain embodiments, R1 is selected from optionally substituted alkyl, aryl, aralkyl, alkenyl, -C(O)alkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), and -S(O)2(aryl), for example, substituted aryl.
- In some embodiments, R1 and the occurrence of R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group.
- In exemplary embodiments, R2 represents substituted or unsubstituted alkyl, alkenyl, alkynyl, aralkyl, aralkenyl, aryl, heteroaralkyl, heteroaralkenyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, or halo.
- In certain embodiments, R2 is selected from alkyl, alkenyl, aryl, aralkyl, aralkenyl, and heteroaralkenyl, optionally substituted with halo, alkyl, haloalkyl, hydroxy, alkoxy, aryloxy, arylalkoxy, cyano, nitro, azido, -CO2H, -C(O)O(alkyl), amino, alkylamino, arylamino, aralkylamino, or amido, for example, R2 is selected from alkyl, alkenyl, aryl, aralkyl, aralkenyl, and heteroaralkenyl, optionally substituted with halo, alkyl, haloalkyl, alkoxy, aryloxy, or arylalkoxy.
- In some embodiments, the Pd catalyst is Pd(dmdba)2 or Pd2(dmdba)3.
- In other embodiments, the Ni(0) catalyst is Ni(COD)2.
- In certain embodiments, the Pd or Ni catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (II), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (II), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (II).
- In some embodiments, the catalyst is a Pd catalyst and the chiral ligand is an enantioenriched ferrocelane ligand, or the catalyst is a Ni catalyst and the chiral ligand is an enantioenriched Josiphos ligand; for example, the enantioenriched ferrocelane ligand is 1,1'-bis[(2S,5S)-2,5-diethyl-1-phospholanyl]ferrocene, 1,1'-bis[(2S,5S)-2,5-dimethyl-1-phospholanyl]ferrocene, or 1,1'-bis[(2S,5S)-2,5-diisopropyl-1-phospholanyl]ferrocene, preferably, the enantioenriched Josiphos ligand is (R)-1-[(S)-2-(dicyclohexylphosphino)ferrocenylethyl]diphenylphosphine.
- In certain embodiments, the chiral ligand is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (II), for example, the chiral ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (II), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (II).
- In some embodiments, the aryl halide or aryl pseudohalide is selected from bromobenzene, chlorobenzene, iodobenzene, phenyl triflate, and chlorotoluene.
- In certain embodiments, the base is selected from hexamethyldisilazane sodium salt (NaHMDS), KHMDS, LiHMDS, and lithium tert-butoxide (tBuOLi), preferably LiHMDS, or preferably NaHMDS.
- In certain embodiments, the reaction includes dioxane, for example, the dioxane is used in an amount from about 0.01 M to about 0.4 M, preferably, the dioxane is used in an amount of about 0.01 M, about 0.05 M, about 0.1 M, about 0.15 M, about 0.2 M, about 0.25 M, about 0.3 M, about 0.35 M, or about 0.4 M.
- In preferred embodiments, the compound of formula (I) is enantioenriched.
- In certain preferred embodiments, no aryl nitrile is used in the method.
- In certain embodiments, the invention relates to a compound of formula (I) made by any of the methods described herein.
- In some embodiments, the invention relates to a method comprising
preparing a compound of formula (I) according to any of the methods described herein; and
synthesizing a medicinal product from the compound of formula (I). - In certain aspects, the invention relates to a compound of formula (III) or formula (IV):


- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R23 is H, halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, alkoxycarbonyl, halogen, aryl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R24 is H or halogen; and
- p is 0 or 1.
- In certain embodiments, p is 0, the compound of formula (III) is a compound of formula (IIIa):


- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;
- R23 is H, or C1-6 alkyl optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain embodiments,
R21 is Bz or An;
R22 is Boc, Cbz, or Bz;
R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, or CH2NHBoc; and
R24 is H or Cl. - In certain embodiments, R22 is Boc or Cbz.
- In some such embodiments, R22 is Boc.
- In certain preferred embodiments, the compound is:







- In other preferred embodiments, the compound is:






- In certain other preferred embodiments, the compound is


- In certain embodiments, p is 1, the compound of formula (III) is a compound of formula (IIIb):


- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a;
- R22a is C1-6 alkyl;
- R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl; and
- R24 is H.
- In certain embodiments,
R21 is Bz, An, or pCF3Bz;
R22 is Boc; and
R23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc. - In certain embodiments, the compound of formula (IIIb) is:




- In certain embodiments, the compound of formula (IVb) is:



- In certain preferred embodiments, the compound of formula (IVb) is

- In certain aspects, the invention relates to a compound of formula (V), (VI), or (VII):
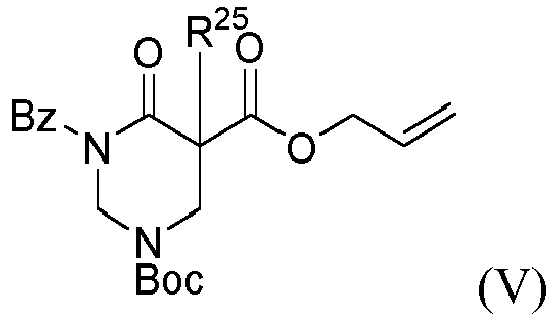


- R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, alkynyl, or -NHC(O)OR25b; halogen; or allyl, optionally substituted with halogen;
- R25b is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl.
- In certain embodiments, the compound is:




- In other embodiments, the compound is:




- In some such embodiments, the compound is

- In still other embodiments, the compound is a compound of formula (VII) wherein R25 is -CH3, -CH2CH3, -CH2CH2C(O)CH3, -CH2CCl=CH2, -CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH.
- In certain aspects, the invention is directed to a compound selected from:



- In certain aspects, the invention relates to compound of Formula (VIII):

- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R23b is H, or C1-6 alkyl optionally substituted with CN, OH, alkoxy, aryloxy, amino, acyl, or -NHC(O)R23c;
- R23c is C1-6 alkyl, (C6-10 aryl)alkyl or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain embodiments,
R21 is Bz or An;
R23b is H, CH3, CH2Ph, CH2NHCbz, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NH2; and
R24 is H or Cl. - In some such embodiments, R21 is Bz; R23b is CH2NH2; and R24 is H.
- In certain aspects, the invention relates to a compound of Formula (IX) or (X):


- R23d is H, or is C1-6 alkyl optionally substituted with cyano, OH, alkoxy, aryloxy, acyl,-NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain embodiments,
R23d is H, CH3, CH2Ph, CH2OBn, CH2CH2OH, CH2CH2C(O)CH3, CH2CH2CH(OH)CH3, CH2NHCBz, or CH2NHBoc; and
R24 is H or Cl. - In some such embodiments, R23d is CH2NHBoc; and R24 is H.
- In certain aspects, the invention relates to a compound of formula (XI):

- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;
- R24 is H or halogen; and
- R23e is H, or is C1-6 alkyl optionally substituted with halogen, OH, aryl, aryloxy, CN, acyl, or amino.
- In certain embodiments,
R21 is Bz or An;
R24 is H or Cl; and
R23e is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, or CH2NH2. - In some such embodiments, R21 is Bz; R23e is CH3; and R24 is H.
- In certain aspects, the invention relates to a compound of formula (XII):

- R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl; and
- R24 is H or halogen.
- In certain embodiments, R21 is Bz or An; and R24 is H or Cl.
- In certain preferred embodiments, R21 is Bz and R24 is H.
- In certain aspects, the invention relates to a compound of formula (XIII):

- R25a is C1-6 alkyl, optionally substituted with CN, OH, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen; and
- R13 is aralkyl or hetaralkyl.
- In certain embodiments,
R25a is -CH3, -CH2CH3, -CH2CH2C(O)CH3, -CH2CCl=CH2, -CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH; and
R13 is Bn. - In certain preferred embodiments, wherein R25a is CH2Ph and R13 is Bn.
- In certain aspects, the invention relates to a method for preparation of a compound of formula (IV):

treating a compound of formula (III)
with a Pd(0) catalyst and a ligand L under alkylation conditions,
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl is optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl;
- R23 is H, halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, alkoxycarbonyl, halogen, aryl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R24 is H or halogen; and
- p is 0 or 1.
- In certain embodiments, p is 0, the compound of formula (IV) is a compound of formula (IVa):


wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl is optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl;
- R23 is H, or C1-6 alkyl optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; and
- R24 is H or halogen.
- In certain embodiments,
R21 is Bz or An;
R22 is Boc, Cbz, or Bz;
R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, or CH2NHBoc; and
R24 is H or Cl. - In certain preferred embodiments, R22 is Boc or Cbz.
- In certain more preferred embodiments, the compound of formula (IVa) is:







- In certain embodiments, the alkylation conditions include reaction in toluene, THF, MTBE, or hexanes, or a combination thereof.
- In certain embodiments, the Pd(0) catalyst is Pd2(pmdba)3 or Pd2(dba)3.
- In certain preferred embodiments, the Pd(0) catalyst is Pd2(dba)3.
- In certain embodiments, the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol % relative to the compound of formula (IIIa). In certain embodiments, the Pd catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (III), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (III), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (III).
- . In certain preferred embodiments, the Pd(0) catalyst is used in an amount of about 4 mol % relative to the compound of formula (IIIa).
- In certain embodiments, the ligand L is:


- In certain embodiments, the ligand L is used in an amount from about 0.1 mol % to about 100 mol % relative to the compound of formula (IIIa). In certain embodiments, the chiral ligand is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (IIIa), for example, the chiral ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (IIIa), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (IIIa). In certain such embodiments, the ligand L is used in an amount of about 10 mol %.
- In certain embodiments, the compound of formula (IVa) is enantioenriched.
- In certain embodiments, the invention relates to a method of synthesizing a medicinal product, the method comprising:
- preparing a compound of formula (IVa) according to any of the methods disclosed herein; and
- synthesizing a medicinal product from the compound of formula (IVa).
- In certain embodiments,
p is 1;
the compound of formula (IV) is a compound of formula (IVb)

wherein, as valence and stability permit, - R22 is -C(O)OR22a;
- R22a is C1-6 alkyl;
- R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl; and
- R24 is H.
- In certain embodiments,
R21 is Bz, An, or pCF3Bz;
R22 is Boc; and
R23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc. - In certain embodiments, the compound of formula (IIIb) is:




- In certain embodiments, the compound of formula (IVb) is:



- In certain preferred embodiments, the compound of formula (IVb) is

- In certain embodiments, the alkylation conditions include reaction in THF, 1,4-dioxane, MTBE, toluene, cyclohexane, or methylcyclohexane, or a combination thereof. In certain preferred embodiments, the alkylation conditions include reaction in methylcyclohexane.
- In certain embodiments, the Pd(0) catalyst is Pd2(pmdba)3 or Pd2(dba)3. In certain preferred embodiments, the Pd(0) catalyst is Pd2(pmdba)3.
- In certain embodiments, the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol % relative to the compound of formula (IIIb). In certain embodiments, the Pd(0) catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (IIIb), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (IIIb), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (IIIb). In certain preferred embodiments, the Pd(0) catalyst is used in an amount of about 4 mol % relative to the compound of formula (IIIb).
- In certain embodiments, the ligand L is:


- In certain preferred embodiments, L is (S)-(CF3)3-tBuPHOX.
- In certain embodiments, the ligand L is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (IIIb), for example, the chiral ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (IIIb), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (IIIb). In certain embodiments, the ligand L is used in an amount of about 4 mol % relative to the compound of formula (IIIb).
- In certain embodiments, the compound of formula (IVb) is enantioenriched.
- In certain embodiments, the invention is directed to a method of synthesizing a medicinal product, the method comprising:
- preparing a compound of formula (IVb) according to a method as disclosed herein; and
- synthesizing a medicinal product from the compound of formula (IVb).
- In some such embodiments, the synthesizing the medicinal product from the compound of formula (IVb) comprises:
treating the compound of formula (IVb)





- In certain preferred embodiments, R23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc. In certain more preferred embodiments, R23 is CH2Ph.
- In certain aspects, the invention relates to a compound of formula (E):

wherein, as valence and stability permit, - R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl; and
- R24 is H.
- In certain embodiments, R23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc. In certain preferred embodiments, R23 is CH2Ph.
- In certain aspects, the invention relates to a method for preparation of a compound of formula (VI):

treating a compound of formula (V):
with a Pd(0) catalyst and a ligand L, under alkylation conditions,
wherein, as valence and stability permit,
R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen. - In certain embodiments, the compound of formula (VI) is:




- In certain embodiments, alkylation conditions include reaction in hexanes, toluene, or a mixture thereof.
- In certain embodiments, the Pd(0) catalyst is Pd2(dba)3 or Pd2(pmdba)3. In certain preferred embodiments, the Pd(0) catalyst is Pd2(pmdba)3.
- In certain embodiments, the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol %. In certain embodiments, the Pd catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (V), for example, an amount from about 1 mol % to about 15 mol % relative to the compound of formula (V), preferably, an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (V). In certain preferred embodiments, the Pd(0) catalyst is used in an amount of about 4 mol %.
- In certain embodiments, L is:


- In certain embodiments, L is used in an amount of from about 0.1 mol % to about 100 mol %. In certain embodiments, the ligand is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (V), for example, the ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (V), preferably, the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (V). In certain preferred embodiments, L is used in an amount of about 10 mol %.
- In certain embodiments, the compound of formula (VI) is enantioenriched.
- In certain embodiments, the method further comprises hydrolyzing the compound of formula (VI), to form a compound of formula (VII):

- In certain aspects, the invention relates to a method for preparation of a compound of formula (VIII):

treating a compound of formula (IVa):
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is Boc;
- R23 is H, or is C1-6 alkyl optionally substituted with CN, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;
- R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R24 is H or halogen;
- R23b is H; or is C1-6 alkyl optionally substituted with halogen, OH, CN, alkoxy, aryloxy, amino, acyl, or
-NHC(O)OR23c; and - R23c is (C6-10 aryl)alkyl or (C5-9 heteroaryl)alkyl.
- In certain embodiments,
R21 is Bz or An;
R22 is Boc;
R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, or CH2NHBoc;
R24 is H or Cl; and
R23b is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, or CH2NH2. - In certain such embodiments, R21 is Bz; R22 is Boc; R23 is CH2NHBoc; R24 is H; and R23b is CH2NH2.
- In certain embodiments, the solvent is CH2Cl2.
- In certain aspects, the invention relates to a method for the preparation of a compound of formula (IX):

treating a compound of formula (IVa):
with LiOH in a first solvent,
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is Boc;
- R23 is H, or C1-6 alkyl optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, acyl, or
-NHC(O)OR23a; - R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R23d is H, or is C1-6 alkyl optionally substituted with halogen, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a; and
- R24 is H or halogen.
- In certain embodiments,
R21 is Bz or An;
R23d is H, CH3, CH2Ph, CH2OBn, CH2OH, CH2CH2C(O)CH3, CH2CH2CH(OH)CH3, CH2NHCbz, CH2NHBoc; and
R24 is H or Cl. - In certain such embodiments, R21 is Bz; R22 is Boc; R23 is CH2NHBoc; R23d is CH2NHBoc; and R24 is H.
- In certain embodiments, the first solvent comprises methanol, water, or a mixture thereof. In certain such embodiments, the first solvent comprises methanol and water.
- In certain embodiments, the method further comprises preparing a compound of formula (X):

treating the compound of formula (IX) with LiAlH4 in a second solvent. - In certain embodiments, the second solvent comprises THF.
- In certain embodiments, R22 is Boc; R23d is CH2NHBoc; and R24 is H.
- In certain aspects, the invention relates to a method of preparation of a compound of formula (XI):

treating a compound of formula (IVa):
with hydrogen gas in the presence of a Pd/C catalyst in a first solvent, to form a first product mixture;
filtering the first product mixture, to form a crude first product; and
treating the crude first product with TFA in a second solvent;
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;
- R22 is Boc;
- R23 is H, or is C1-6 alkyl optionally substituted with halogen, OH, alkoxy, aryloxy, CN, aryl, or
-NHC(O)OR23a; - R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;
- R23e is H, or is C1-6 alkyl optionally substituted with halogen, OH, aryl, aryloxy, CN, acyl, or amino; and
- R24 is H or halogen.
- In certain embodiments,
R21 is Bz or An;
R22 is Boc;
R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, or CH2NHBoc;
R23e is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, or CH2NH2;
R24 is H or Cl. - In certain embodiments, R21 is Bz; R23 is CH3; R23e is CH3; and R24 is H.
- In certain embodiments, the first solvent comprises methanol.
- In certain embodiments, the second solvent comprises CH2Cl2, ethyl acetate, or a mixture thereof.
- In certain embodiments, the method further comprises preparation of a compound of formula (XII):

treating a compound of formula (XI):
with Pd(OPiv)2, xantphos, AgOPiv, and benzoquinone in the presence of CO(g) in a third solvent;
wherein, as valence and stability permit, - R21 is -C(O)aryl or -C(O)heteroaryl;
- R23e is methyl; and
- R24 is H or halogen.
- In certain embodiments, R24 is H.
- In certain preferred embodiments, R21 is Bz or An.
- In certain more preferred embodiments, R21 is Bz; R23e is CH3; and R24 is H.
- In certain embodiments, the third solvent comprises toluene.
- In certain aspects, the invention relates to a method of preparation of a compound of formula (XIII):

treating a compound of formula (VI):
with TFA in a first solvent, to form a first product;
treating the first product with lithium hydroxide in a second solvent, to form a second product;
treating the second product with Boc2O in the presence of a base, to form a third product;
treating the third product with K2CO3 and R13-X21 in a third solvent, to form a compound of formula (XIII);
wherein, as valence and stability permit, - R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen;
- R25a is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen;
- R13 is aralkyl or hetaralkyl; and
- X21 is halogen.
- In certain embodiments,
R25 is CH3, CH2CH3, CH2CH2CO2CH3, CH2CCl=CH2, CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH;
R25a is CH3, CH2CH3, CH2CH2CO2CH3, CH2CCl=CH2, CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH; and
R13 is Bn. - In certain embodiments, R25 is CH2Ph; R25a is CH2Ph; and X21 is Br.
- In certain embodiments, the first solvent comprises dichloromethane.
- In certain embodiments, the second solvent comprises methanol.
- In certain embodiments, the third solvent comprises DMF.
- In certain embodiments, the invention relates to a method as described in Table 1. In certain embodiments, the invention relates to a method as described in Table 2. In certain embodiments, the invention relates to a method as described in Table 3. In certain embodiments, the invention relates to a method as described in Table 4. In certain embodiments, the invention relates to a method as described in Table 5. In certain embodiments, the invention relates to a method as described in Table 6. In certain embodiments, the invention relates to a method as described in Table 7. In certain embodiments, the invention relates to a method as described in Table 8. In certain embodiments, the invention relates to a method as described in Table 9. In certain embodiments, the invention relates to a method as described in Table 10. In certain embodiments, the invention relates to a method as described in Table 11. In certain embodiments, the invention relates to a method as described in Table 12. In certain embodiments, the invention relates to a method as described in Table 13. In certain embodiments, the invention relates to a method as described in Table 14. In certain embodiments, the invention relates to a method as described in Table 15. In certain embodiments, the invention relates to a method as described in Table 16. In certain embodiments, the invention relates to a method as described in Table 17. In certain embodiments, the invention relates to a method as described in Table 18. In certain embodiments, the invention relates to a method as described in Table 19. In certain embodiments, the invention relates to a method as described in Table 20. In certain embodiments, the invention relates to a method as described in Table 21. In certain embodiments, the invention relates to a method as described in Table 22. In certain embodiments, the invention relates to a method as described in Table 23. In certain embodiments, the invention relates to a method as described in Table 24. In certain embodiments, the invention relates to a method as described in Table 25. In certain embodiments, the invention relates to a method as described in Table 26. In certain embodiments, the invention relates to a method as described in Table 27. In certain embodiments, the invention relates to a method as described in Table 28. In certain embodiments, the invention relates to a method as described in Table 29. In certain embodiments, the invention relates to a method as described in Table 30. In certain embodiments, the invention relates to a method as described in Table 31. In certain embodiments, the invention relates to a method as described in Table 32. In certain embodiments, the invention relates to a method as described in Table 33. In certain embodiments, the invention relates to a method as described in Table 34. In certain embodiments, the invention relates to a method as described in Table 35. In certain embodiments, the invention relates to a method as described in Table 36. In certain embodiments, the invention relates to a method as described in Table 37. In certain embodiments, the invention relates to a method as described in the table in Example 35. In certain embodiments, the invention relates to a method as described in Table 38. In certain embodiments, the invention relates to a method as described in Table 39.
- Preferred transition metal catalysts of the disclosure are complexes of nickel or palladium comprising a chiral ligand. In certain embodiments, the complex is of Ni(0) comprising a chiral ligand. In other embodiments, the complex is of Ni(II) comprising a chiral ligand. In other embodiments, the complex is of Pd(0) comprising a chiral ligand. In yet other embodiments, the complex is of Pd(II) comprising a chiral ligand.
- It should be appreciated that typical transition metal catalysts having a low oxidation state (e.g., (0) or (I)) suffer from air- and moisture-sensitivity, such that these complexes of transition metals necessitate appropriate handling precautions. This may include the following precautions without limitation: minimizing exposure of the reactants to air and water prior to reaction; maintaining an inert atmosphere within the reaction vessel; properly purifying all reagents; and removing water from reaction vessels prior to use. In certain embodiments, the Ni(0) catalyst is a precatalyst. In other embodiments, the Pd(0) catalyst is a precatalyst. In yet other embodiments, the Pd(II) catalyst is a precatalyst.
- Example Ni(0) catalysts that may be used in the methods of the disclosure include Ni[(1,5-cyclooctadiene)2], which is also referred to herein as Ni(COD)2.
- In certain embodiments, the transition metal catalysts of the disclosure are complexes of Ni(0) or Ni(II), such as Ni(COD)2, NiCl, and NiBr2.
- In certain embodiments, the transition metal catalysts of the disclosure are complexes of Pd(0) or Pd(II). In certain embodiments, Pd(II) catalysts are typically robust, and are less sensitive to air and moisture than their lower-oxidation state counterparts.
- Examples of Pd(0) catalysts that may be used in the methods of the invention include Pd(PPh3)4, Pd(dm-dba)2, Pd(dba)2, Pd2(dba)3•CHCl3, Pd2(dba)3, and Pd2(pm-dba)3. In preferred embodiments, the transition metal catalyst is Pd(PPh3)4. In other preferred embodiments, the transition metal catalyst is Pd(dm-dba)2. In other preferred embodiments, the transition metal catalyst is Pd(dba)2. In other preferred embodiments, the transition metal catalyst is Pd2(dba)3•CHCl3. In yet other preferred embodiments, the transition metal catalyst is Pd2(dba)3. In other preferred embodiments, the transition metal catalyst is Pd2(pm-dba)3.
- Examples of Pd(II) catalysts that may be used in the methods of the invention include Pd(OC(O)Rc)2, wherein Rc is optionally substituted alkyl, alkenyl, alkynyl, aryl, heteroaryl, aralkyl, heteroaralkyl, cycloalkyl, heterocycloalkyl, (cycloalkyl)alkyl, or (heterocycloalkyl)alkyl. Further examples of Pd(II) catalysts include Pd(OC(O)Rc)2, Pd(OC(=O)CH3)2 (i.e., Pd(OAc)2), Pd(TFA)2, Pd(acac)2, PdCl, PdBr2, PdCl (R23CN)2 (e.g., Pd(PhCN)2Cl and Pd(CH3CN)2Cl), PdCl(PR24R25R26)2, [Pd(η3-allyl)Cl]2, and pre-formed Pd(II)-ligand complex, wherein R23, R24, R25, and R26 are independently selected from hydrocarbyl, substituted hydrocarbyl, heteroatom- containing hydrocarbyl, and substituted heteroatom-containing hydrocarbyl. Alternatively, the transition metal catalyst is Pd(OC(O)Rc)2, wherein Rc is defined above, For example, Rc may be alkyl, substituted by one or more halo or cyano groups. In preferred embodiments, the transition metal catalyst is Pd(OAc)2. In other preferred embodiments, the transition metal catalyst is [Pd(cinnamyl)Cl]2.
- To improve the effectiveness of the catalysts discussed herein, additional reagents may be employed in the methods of the invention, including, without limitation, salts, solvents, and other small molecules, such as a chiral ligand (see below). In some preferred embodiments, additives include salts. These additives are preferably used in an amount that is in the range of about 0.1 equivalents to about 15 equivalents relative to the amount of the reactant, more preferably in the range of about 0.5 equivalents to about 10 equivalents relative to the reactant, and most preferably in the range of about 2 equivalents to about 7 equivalents relative to the reactant.
- In certain embodiments, additives include one or more of Mn0, Zn0, LiCl, LiBr, NaCl, NaBr, NaI, KCl, KBr, CsCl, ZnCl2, CuI, AlCl3, HMDS, COD, LiOt-Bu, NaOt-Bu, and KOt-Bu. These additives are preferably used in an amount that is in the range of about 1 equivalent to about 5 equivalents relative to the amount of the catalyst.
- A low oxidation state of a transition metal, i.e., an oxidation state sufficiently low to undergo oxidative addition, can be obtained in situ, by the reduction of transition metal complexes that have a high oxidation state. Reduction of the transition metal complex can optionally be achieved by adding nucleophilic reagents including, without limitation, tetrabutyl ammonium hydroxide, tetrabutyl ammonium difluorotriphenylsilicate (TBAT), tetrabutylammonium fluoride (TBAF), 4-dimethylaminopyridine (DMAP), tetramethylammonium hydroxide (e.g., as the pentahydrate), KOH/1,4,7,10,13,16-hexaoxacyclooctadecane, sodium ethoxide, TBAT/trimethyl-(2-methyl-cyclohex-1-enyloxy)-silane, and combinations thereof. When a nucleophilic reagent is needed for the reduction of the metal complex, the nucleophilic reagent is used in an amount in the range of about 1 mol % to about 20 mol % relative to the reactant, more preferably in the range of about 1 mol % to about 10 mol % relative to the substrate, and most preferably in the range of about 5 mol % to about 8 mol % relative to the substrate.
- For example, a Pd(II) complex can be reduced in situ to form a Pd(0) catalyst. Exemplary transition metal complexes that may be reduced in situ, include, without limitation, allylchloro[1 ,3-bis(2,6-di-iso-propylphenyl)imidazol-2-ylidene]palladium(II), ([2S,3S]-bis[diphenylphosphino]butane)(η3-allyl)palladium(II) perchlorate, [S]-4-tert-butyl-2-(2-diphenylphosphanyl-phenyl)-4,5-dihydro-oxazole(η3-allyl)palladium(II) hexafluorophosphate (i.e., [Pd(S-tBu-PHOX)(allyl)]PF6), and cyclopentadienyl(η3-allyl) palladium(II).
- The effectiveness of the catalysts discussed herein can be improved by adding nucleophilic reagents including, without limitation, NaHMDS, KHMDS, LiHMDS, tBuOLi, tetrabutyl ammonium hydroxide, tetrabutyl ammonium difluorotriphenylsilicate (TBAT), tetrabutylammonium fluoride (TBAF), 4-dimethylaminopyridine (DMAP), tetramethylammonium hydroxide (e.g., as the pentahydrate), KOH/1,4,7,10,13,16-hexaoxacyclooctadecane, sodium ethoxide, TBAT/trimethyl-(2-methyl-cyclohex-1-enyloxy)-silane, and combinations thereof. When a nucleophilic reagent is added, the nucleophilic reagent is used in an amount in the range of about range of about 0.1 equivalents to about 10 equivalents relative to the amount of the reactant, more preferably in the range of about 0.1 equivalents to about 5 equivalents relative to the reactant, and most preferably in the range of about 0.5 equivalents to about 2 equivalents relative to the reactant.
- Accordingly, when describing the amount of transition metal catalyst used in the methods of the disclosure, the following terminology applies. The amount of transition metal catalyst present in a reaction is alternatively referred to herein as "catalyst loading". Catalyst loading may be expressed as a percentage that is calculated by dividing the moles of catalyst complex by the moles of the substrate present in a given reaction. Catalyst loading is alternatively expressed as a percentage that is calculated by dividing the moles of total transition metal (for example, nickel) by the moles of the substrate present in a given reaction.
- In certain embodiments, the transition metal catalyst is present under the conditions of the reaction from an amount of about 0.1 mol% to about 20 mol% total transition metal relative to the substrate, which is the compound of formula (II), (IIIa), (IIIb), or (V). In certain embodiments, the catalyst loading is from about 1 mol% to about 15 mol% total transition metal relative to the substrate. In certain embodiments, the catalyst loading is from about 1 mol% to about 14 mol%, about 1 mol% to about 12%, about 1 mol% to about 10%, about 2 mol% to about 9 mol%, about 2.5 mol% to about 8 mol%, about 3 mol% to about 7 mol%, about 3.5 mol% to about 6.5 mol%, or about 4 mol% to about 6 mol% total transition metal relative to the substrate. For example, in certain embodiments, the catalyst loading is about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% total transition metal. In certain embodiments, the catalyst loading is about 2 mol%, about 2.5 mol%, about 3 mol%, about 3.5 mol%, about 4 mol%, about 4.25 mol%, about 4.5 mol%, about 4.75 mol%, about 5 mol%, about 5.25 mol%, about 5.5 mol%, about 5.75 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, or about 9% total transition metal.
- In certain embodiments, the methods disclosed herein use a Ni(0) catalyst comprising a chiral ligand. In certain embodiments, the methods disclosed herein use a Pd(0) catalyst comprising a chiral ligand. In certain embodiments, the methods disclosed herein use a Pd(II) catalyst comprising a chiral ligand.
- One aspect of the disclosure relates to the enantioselectivity of the methods. Enantioselectivity results from the use of chiral ligands during the arylation or vinylation reaction. Without being bound by theory, the asymmetric environment that is created around the metal center by the presence of chiral ligands produces an enantioselective reaction. The chiral ligand forms a complex with the transition metal (e.g., nickel), thereby occupying one or more of the coordination sites on the metal and creating an asymmetric environment around the metal center. This complexation may or may not involve the displacement of achiral ligands already complexed to the metal. When displacement of one or more achiral ligands occurs, the displacement may proceed in a concerted fashion, i.e., with both the achiral ligand decomplexing from the metal and the chiral ligand complexing to the metal in a single step. Alternatively, the displacement may proceed in a stepwise fashion, i.e., with decomplexing of the achiral ligand and complexing of the chiral ligand occurring in distinct steps. Complexation of the chiral ligand to the transition metal may be allowed to occur in situ, i.e., by admixing the ligand and metal before adding the substrate. Alternatively, the ligand-metal complex can be formed separately, and the complex isolated before use in the arylation or vinylation reactions of the present disclosure.
- Once coordinated to the transition metal center, the chiral ligand influences the orientation of other molecules as they interact with the transition metal catalyst. Coordination of the metal center with an aryl halide and reaction of the substrate with the aryl halide-metal complex are dictated by the presence of the chiral ligand. The orientation of the reacting species determines the stereochemistry of the products.
- Chiral ligands of the disclosure may be bidentate or monodentate or, alternatively, ligands with higher denticity (e.g., tridentate, tetradentate, etc.) can be used. Preferably, the ligand will be substantially enantiopure. By "enantiopure" is meant that only a single enantiomer is present. In many cases, substantially enantiopure ligands (e.g., ee >99%, preferably >99.5%, even more preferably >99.9%) can be purchased from commercial sources, obtained by successive recrystallizations of an enantioenriched substance, or by other suitable means for separating enantiomers.
- Examples of chiral ligands used in certain embodiments may be found in
U.S. Patent No. 10,040,784 Figure 1 . - Generally, the chiral ligand is present in an amount in the range of about 0.1 equivalents to about 10 equivalents relative to the amount of total metal from the catalyst, preferably in the range of about 0.1 to about 6 equivalents relative to the amount of total metal from the catalyst, and most preferably in the range of about 0.5 to about 4.5 equivalents relative to the amount of total metal from the catalyst. Alternatively, the amount of the chiral ligand can be measured relative to the amount of the substrate.
- In certain embodiments, the ligand is present under the conditions of the reaction from an amount of about 0.1 mol% to about 100 mol% relative to the substrate, which is the compound of formula (II), (IIIa), (IIIb), or (V). The amount of ligand present in the reaction is alternatively referred to herein as "ligand loading" and is expressed as a percentage that is calculated by dividing the moles of ligand by the moles of the substrate present in a given reaction. In certain embodiments, the ligand loading is from about 0.5 mol% to about 50 mol%. For example, in certain embodiments, the ligand loading is about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol%, or about 15 mol%. In certain embodiments, the ligand is in excess of the transition metal catalyst. In certain embodiments, the ligand loading is about 10 times the transition metal catalyst loading.
- Where a chiral ligand is used, the reactions of the disclosure may enrich the stereocenter bearing R2 in the product relative to the enrichment at this center, if any, of the starting material. In certain embodiments, the chiral ligand used in the methods of the disclosure yields a compound of formula (I) that is enantioenriched. The level of enantioenrichment of a compound may be expressed as enantiomeric excess (ee). The ee of a compound may be measured by dividing the difference in the fractions of the enantiomers by the sum of the fractions of the enantiomers. For example, if a compound is found to comprise 98% (S)-enantiomer, and 2% (R)-enantiomer, then the ee of the compound is (98-2)/(98+2), or 96%. In certain embodiments, the compound of formula (I) has about 5% ee or greater, 10% ee or greater, 15% ee or greater, 20% ee or greater, 25% ee or greater, 30% ee or greater, 40% ee or greater, 50% ee or greater, 60% ee or greater, 70% ee or greater, about 80% ee, about 85% ee, about 88% ee, about 90% ee, about 91% ee, about 92% ee, about 93% ee, about 94% ee, about 95% ee, about 96% ee, about 97% ee, about 98% ee, about 99% ee, or above about 99% ee, even where this % ee is greater than the % ee of the starting material, such as 0% ee (racemic). In certain embodiments, the compound of formula (I) is enantioenriched. In certain embodiments, the compound of formula (I) is enantiopure. In embodiments where the starting material has more than one stereocenter, reactions of the disclosure may enrich the stereocenter bearing R2 relative to the enrichment at this center, if any, of the starting material, and substantially independently of the stereochemical disposition/enrichment (de) of any other stereocenters of the molecule. For example, a product of the methods described herein may have 5% de or greater, 10% de or greater, 15% de or greater, 20% de or greater, 25% de or greater, 30% de or greater, 40% de or greater, 50% de or greater, 60% de or greater, 70% de or greater, 80% de or greater, 90% de or greater, 95% de or greater, or even 98% de or greater at the stereocenter of the product bearing R2.
- In certain embodiments, the methods of the disclosure include treating a compound of formula (II) with a palladium or nickel catalyst comprising a chiral ligand, and an aryl halide or a heteroaryl halide under arylation conditions. In certain embodiments, the methods of the disclosure include treating a compound of formula (II) with a palladium or nickel catalyst comprising a chiral ligand, and a vinyl halide under vinylation conditions. In certain embodiments, the palladium catalyst under arylation or vinylation conditions is a Pd(0) catalyst comprising a chiral ligand. In certain embodiments, the palladium catalyst under arylation or vinylation conditions is a Pd(II) catalyst comprising a chiral ligand. In certain embodiments, the nickel catalyst under arylation or vinylation conditions is a Ni(0) catalyst comprising a chiral ligand. In certain embodiments, the palladium catalyst under arylation or vinylation conditions is a Ni(II) catalyst comprising a chiral ligand. In certain embodiments, arylation and vinylation conditions further comprise a base, such as NaHMDS, KHMDS, and LiHMDS. In certain embodiments, the base is LiHMDS. In certain embodiments, the base is NaHMDS. In certain embodiments, arylation and vinylation conditions of the reaction include one or more additives, such as a salt. In certain embodiments, the salt is one or more of LiCl, NaCl, NaBr, NaI, KCl, KBr, CsCl, CuI, LiOt-Bu, and KOt-Bu.
- In certain embodiments, arylation, vinylation, and alkylation conditions of the reaction include one or more organic solvents. In certain embodiments, organic solvents include aromatic or non-aromatic hydrocarbons, ethers, alkylacetates, nitriles, or combinations thereof. In certain embodiments, organic solvents include hexane, pentane, benzene, toluene, xylene, cyclohexane, methylcyclohexane, cyclic ethers such as optionally substituted tetrahydrofuran and dioxane, acyclic ethers such as dimethoxyethane, diethyl ether, methyl tertbutyl ether, and cyclopentyl methyl ether, acetonitrile, isobutyl acetate, ethyl acetate, isopropyl acetate, or combinations thereof. In certain preferred embodiments, the solvent is toluene, tetrahydrofuran, dioxane, methyl tert-butyl ether, cyclohexane, methylcyclohexane, dimethoxyethane, or a mixture of toluene and tetrahydrofuran. In some embodiments, the solvent is a mixture of two organic solvents. In some such embodiments, the solvent is a mixture of two organic solvents in a ratio of 1:5, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, or 15:1. In preferred embodiments, the solvent is dioxane. In certain other embodiments, the solvent is a mixture of toluene and tetrahydrofuran. In certain embodiments, the mixture of toluene and tetrahydrofuran is in a ratio of 1:5, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, or 15:1. In certain embodiments, the mixture of toluene and tetrahydrofuran is in a ratio of 5:1 or 10:1. In certain other embodiments, the solvent is a mixture of toluene and hexanes. In certain embodiments, the mixture of toluene and hexanes is in a ratio of 1:5, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 11:1, 12:1, 13:1, 14:1, or 15:1. In certain embodiments, the mixture of toluene and hexanes is in a ratio of 5:1 or 10:1. In certain embodiments, the solvent is methylcyclohexane.
- In certain embodiments, arylation, vinylation, and alkylation conditions of the reaction include a reaction temperature. In certain embodiments, the reaction temperature is ambient temperature (about 20 °C to about 26 °C). In preferred embodiments, the reaction temperature is about 60 °C to about 100 °C. In preferred embodiments, the reaction temperature is higher than ambient temperature, such as, for example, about 30 °C, about 35 °C, about 40 °C, about 45 °C, about 50 °C, about 55 °C, about 60 °C, about 65 °C, about 70 °C, about 75 °C, about 80 °C, about 85 °C, about 90 °C, about 95 °C, or about 100 °C. Reaction temperature may be optimized per each substrate.
- The disclosure described generally herein will be more readily understood by reference to the following examples, which are included merely for purposes of illustration of certain aspects and embodiments of the present disclosure, and are not intended to limit the disclosure.
- Chlorobenzene and the lithium enolate derived from lactam 15 generated a small amount of arylated product 16 in high enantioselectivity in the presence of Ni(COD)2 and a Josiphos ligand (L5) (Table 1, entry 5). A variety of other ligand classes resulted in unreacted aryl chloride.
Table 1 Initial evaluation of ligands in Ni-catalyzed coupling of lactam 15 with chlorobenzene 
entry ligand Conv. (%) ee (%) biphenyl (%) 1 L1 <5 - 14 2 L2 <5 - 25 3 L3 16 66 9 4 L4 <5 - 9 5 L5 11 96 9 

- Reactions in the absence of Ni and ligand revealed a significant background reaction resulting from the direct reaction of the enolate with chlorobenzene. Direct reaction was observed in the presence of LDA, NaHMDS, or KHMDS but not in the presence of LiHMDS (Table 2). Weaker bases resulted in little to no product formation. When bromo- or iodobenzene was used instead of chlorobenzene, enantioselectivity decreased significantly (Table 3,
entries 2 and 3). Phenyl triflate and phenyl tosylate were unreactive under the reaction conditions (entries 4 and 5).Table 2 Initial evaluation of bases in the Ni-catalyzed coupling of lactam 15 with chlorobenzene 
entry base temp. arylation 1 LDA 23 °C 86% 2 LiHMDS 23 °C not observed 3 LiHMDS 60 °C not observed 4 NaHMDS 23 °C not observed 5 NaHMDS 60 °C observed 6 KHMDS 23 °C observed 7 KHMDS 60 °C observed Table 3 Effect of aryl (pseudo)halide leaving group on conversion and enantioselectivity 
entry X Conv. (%) ee (%) biphenyl (%) 1 Cl 50 94 5 2 Br 27 55 <5 3 I 29 5 <5 4 OTf <5 - <5 5 OTs <5 - <5 - Although a number of Josiphos ligands were evaluated, only L5 resulted in >5% α-arylation (Table 4, entry 1). Anisole (18) and homocoupled electrophile (19) were consistently formed under the reaction conditions.
Table 4 Evaluation of Josiphos ligands 
entry ligand Conv. (%) % yield (16) ee (%) % anisole (18) % homocoupling (19) 1 L5 75 21 97 26 5 2 L4 66 <5 - 31 <5 3 L6 61 <5 - 30 9 4 L7 51 <5 - 28 <5 5 L8 66 <5 - 35 10 6 L9 39 <5 - 12 10 7 L10 65 <5 - 29 <5 8 L11 62 <5 - 41 <5 9 L12 40 <5 - 40 <5 10 L13 74 5 - 32 <5 

- A variety of reaction parameters were evaluated in the α-arylation of lactam 15 with 3-chloroanisole (17). Neither an excess of enolate relative to aryl chloride nor an excess of aryl chloride relative to enolate provided an improvement in yield or enantioselectivity relative to a 1:1 ratio (Table 5, entries 1-3). Ethereal solvents provided equivalent or slightly diminished yields as compared to toluene (Table 6). Ni(COD)2 was found to be a preferred Ni source, as other Ni0 or NiII salts resulted in decreased yields (Table 7).
Table 5 Effect of stoichiometry on yield, enantioselectivity, and side product formation 
entry Equiv. lactam Equiv. base Equiv. ArCl Conv. (%) % Yield (16) ee (%) % anisole (18) % homocoupling (19) 1 1.5 1.5 1.0 58 18 95 18 <5 2 1.0 1.0 1.0 36 17 97 12 <5 3 1.0 1.0 1.5 35 18 95 12 <5 4 1.0 2.0 2.0 90 8 - 65 8 5 1.0 2.0 1.0 79 5 - 53 <5 Table 6 Effect of solvent 
entry Solvent Conv. (%) % Yield (16) ee (%) % anisole (18) % homocoupling (19) 1 toluene 70 23 98 21 <5 2 THF 72 13 88 32 6 3 1,4-dioxane 76 18 99 28 5 4 DME 59 8 - 22 8 5 toluene/THF (1:1) 73 15 - 30 8 Table 7 Effect of Ni source 
entry Ni source Additive Conv. (%) % Yield (16) % anisole (18) % homocoupling (19) 1a Ni(COD)2 Mn0 38 14 10 <5 2a Ni(COD)2 Zn0 29 3 9 <5 3 NiCl2•glyme - 33 9 14 <5 4a NiCl2•glyme Mn0 <5 <5 <5 <5 5a NiCl2•glyme Zn0 9 5 <5 <5 6 NiBr2•glyme - 22 <5 7 <5 7 NiBr2•diglyme - 18 6 6 <5 8 Ni(acac)2 - 51 <5 4 <5 9 NiBr2(PPh3)2 - 53 5 27 <5 10 Ni(PPh3)4 - 60 11 33 <5 aThe reaction was run for 2 h instead of 20 h - A number of N-protecting groups were evaluated. Generally low yields were observed for N-aryl and N-benzyl lactams (Table 8, 15a-15f). Little to no improvement in yield was observed for benzoyl or tosyl protected substrates (15g, 15h), an oxindole (15i), a δ-lactam (15j), and an ε-lactam (15k). Both an α-Et-substituted (151) and an unsubstituted lactam (15m) resulted in low product to internal standard ratios.
Table 8 Effect of N-protecting groups 
Substrate Conv. (%) Yield (%) Substrate Conv. (%) Yield (%) 
63 22 
17 <5 61 19 28 9 
73 <5 
3 <5 
54 12 
33 <5 
84 25 
57 <5 
85 <5 
79 P/IS = 0.32 
14 <5 
33 P/IS = 0.21 - Although additives have been beneficial in several examples of Ni-catalyzed cross-coupling, neither alkali metal halides (Table 9, entries 2-10) nor Lewis acids (entries 11-16) provided a boost in yield. The expected byproducts of this reaction, HMDS and COD, did not have a significant detrimental effect when added to the reaction (entries 17-18).
Table 9 Effect of additives 
entry additive mol % % conv % Yield (16) 1 none 100 58 31 2 LiCl 100 40 16 3 LiCl 10 68 21 4 LiBr 100 33 7 5 NaCl 100 55 25 6 NaBr 100 60 22 7 NaI 100 45 15 8 KCl 100 nd 24 9 KBr 100 26 20 10 CsCl 100 32 21 11 ZnCl2 100 27 5 12 ZnCl2 10 56 17 13 CuI 100 10 <5 14 CuI 10 57 21 15 AlCl3 100 16 <5 16 AlCl3 10 50 13 17 HMDS 100 63 17 18 COD 100 69 20 - Monitoring the reaction over time revealed that a low terminal yield of α-arylation product 16 is generated within 1 h at 80 °C (Table 10). At 40 °C or 60 °C, lower terminal yields were obtained. Increasing the Ni loading to 20% led to a somewhat decreased yield (Table 11, entries 7 and 8).


Table 11 Effect of Ni loading 
entry mol% Ni mol% L5 time (h) % conv % Yield (16) % anisole (18) % homocoupling (19) 1 2.5 3 1 17 5 1 <5 2 2.5 3 16 40 12 14 <5 3 5 6 1 29 9 4 <5 4 5 6 16 57 13 16 <5 5 10 12 1 48 14 9 <5 6 10 12 16 70 22 14 <5 7 20 40 1 68 11 12 <5 8 20 40 26 83 11 17 <5 - Nitrile-/Josiphos-ligated Ni complexes have been demonstrated to be particularly reactive toward oxidative addition of aryl halides. Complex 20 was synthesized according to a literature procedure (Scheme 1) and used as a precatalyst in the coupling of lactam 15 with 3-chloroanisole (17). Complex 20 did not result in improved reactivity as compared to Ni(COD)2 (Table 12).

Table 12 Use of a Ni0(PhCN) complex as a precatalyst 
entry mol% Ni temp. % added L5 % Yield (16) 1 5 80 °C - 14 2 10 80 °C - 13 3 5 50 °C 5 12 4 10 50 °C 10 15 - Complex 20 was subjected to literature conditions for the synthesis of a Josiphos-ligated NiIIArCl species (21, Scheme 2). When a 1:1 ratio of NiII complex 21, lactam 15, and LiHMDS was reacted in THF at 80 °C, a 50% yield of homocoupled aryl chloride (19) and only 5% product 16 were obtained (Scheme 3).


- The Ni-catalyzed α-arylation of lactam 15 with an aryl chloride may be achieved with high enantioselectivity but low yield. Both lower yields and levels of enantioselectivity were observed for an aryl bromide or an aryl iodide. Product formation was found to terminate within 1 hour, and increasing the Ni loading decreased yield. The stoichiometric reaction of an isolated NiIIArCl complex and the lithium enolate derived from 15 resulted in predominant aryl chloride homocoupling.
- α-Arylation of lactam 15 with 3-chloroanisole (17) was achieved in the presence of LiHMDS and catalytic Pd(OAc)2. In contrast to the Ni-catalyzed process, α-arylation was observed with several ferrocene-based ligands, including Josiphos and Mandyphos ligand classes. Moderate yields of product were obtained with Mandyphos ligands L17 and L18 (Table 13, entries 8 and 9). Moderate levels of enantioselectivity were achieved with a Josiphos ligand (L5, entry 1) and several Mandyphos ligands (L18, entry 9; L19, entry 10; L21, entry 12).
Table 13 Effect of ligands on Pd-catalyzed coupling of lactam 15 and 3-chloroanisole 
Entry Ligand Conv. (%) % Yield (16) % anisole (18) ee (%) 1 L5 61 27 5 45 2 L6 37 <5 28 - 3 L8 88 8 40 - 4 L4 62 16 6 18 5 L15 41 <5 25 - 6 L13 24 20 5 -10 7 L16 66 24 19 -8 8 L17 65 47 12 -34 9 L18 59 43 12 -48 10 L19 14 8 19 -55 11 L20 59 <5 35 - 12 L21 35 13 13 54 13 L22 4 <5 4 - 14 L23 45 18 13 2 15 L24 7 <5 4 - 16 L25 46 21 19 16 17 L26 75 38 9 -12 18 L1 8 <5 3 - 19 L3 <5 <5 5 - 20 L27 <5 <5 <5 - 21 L28 8 7 <5 - 22 L29 58 6 10 - 


- In an investigation of various lactam substrates, an N-benzyl protected lactam (22) provided the highest levels of enantioselectivity (Table 14, entries 1 and 4). However, lower yields were observed compared to those observed for the N-PMP protected lactam 15.
Table 14 Effect of bases in combination with Josiphos and Mandyphos ligandsa 
Entry Ligand Base Conv. (%) % Yield (XX) % anisole (XX) ee (%) 1 L18 LiHMDS 60 19 22 -80 2 L18 NaHMDS 57 7 13 nd 3 L18 KHMDS 81 13 9 4 4 L17 LiHMDS 73 20 18 -76 5 L17 NaHMDS 67 9 15 nd 6 L17 KHMDS 79 13 11 17 7 L5 LiHMDS 59 8 9 nd 8 L5 NaHMDS 70 23 12 59 9 L5 KHMDS 87 27 14 28 a Ligand numbers refer to those in Table 14 - α-Arylation of lactam 1a with chlorobenzene was achieved in the presence of LiHMDS and catalytic Pd(dmdba)2 using various ligands (Table 15).
Table 15 Effect of ligands on Pd-catalyzed coupling of lactam 15 and chlorobenzene 
entry ligand yield(%)b ee(%)c 1 L1 - - 2 L2 29 5 3 L3 65 93 4 L4 69 94 5 L5 19 2 6 L6 6 0 
a Conditions: 0.1 mmol scale, la (1.5 equiv), 2a (1.0 equiv), LiHMDS (1.5 equiv), 10 mol % Pd(dmdba)2, 12 mol % ligand, 0.5 mL dioxane. b Determined by LC-MS analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as a standard. cDetermined by chiral SFC analysis of the isolated product. PMP = p-methoxyphenyl. dmdba = 3,5,3',5'-dimethoxydibenzylideneacetone. - Changing the Pd source produced a more active pre-catalyst that allowed for a lower catalyst loading and shorter reaction time with L3 (Table 16, entry 1). By employing the less hindered ligand L4, we were able to isolate 3aa in 86% yield and 92% ee after 12 hours (Table 16, entry 7). With this set of conditions, the reaction proceeds at a lower temperature; the desired product is obtained in 84% yield and 92% ee (Table 16, entry 9) after 15 h at 80 °C. Moreover, at 54 °C the product is still obtained in good yield and high enantiomeric excess (Table 16, entry 10).
Table 16 Optimization of reaction conditions for arylation of lactam 1a 
entry Ph-X base ligand time (h) yield (%)b ee (%)c 1 Ph-Cl LiHMDS L3 6 65 94 2 Ph-I LiHMDS L3 20 - - 3 Ph-OTf LiHMDS L3 20 - - 4 Ph-Cl NaHMDS L3 20 39 55 5 Ph-Cl KHMDS L3 20 7 6d Ph-Br NaHMDS L3 16 64 96 7 Ph-Br NaHMDS L4 12 86 92 8 Ph-Br LiHMDS L4 15 31 72 9d Ph-Br NaHMDS L4 15 84 92 10e Ph-Br NaHMDS L4 15 72 92 aConditions: 0.1 mmol scale, la (1.5 equiv), Ph-X (1.0 equiv), base 1.5 equiv), Pd2(pmdba)3 (2.5 mol %), ligand (7.5 mol %), 0.5 mL dioxane. byield determined by LCMS analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as a standard. cDetermined by chiral SFC analysis of the isolated product. dReaction performed at 80 °C. eReaction performed at 54 °C. PMP = p-methoxyphenyl. pmdba = 4,4' - dimethoxydibenzylideneacetone. - A number of different N-protecting groups were tolerated (Table 17). Bis-methoxyphenyl lactam 1b performs just as well as 1a with Method B, but a slight decrease in yield and enantioselectivity is observed when subjected to Method A (3ba). Switching to ortho-methoxy phenyl substituted 1c or electron-deficient trifluoromethylphenyl 1e led to diminished yield and enantioselectivity (3ca and 3ea). Although N-phenyl 1d does not outperform 1a in Method A, it does exhibit higher reactivity and enantioselectivity when exposed to Method B, furnishing the desired product in 91% yield and 93% ee (3da). Benzyl-protected lactam 1f affords α-quaternary lactam 3fa in high levels of enantiomeric excess across both Methods.

- When the reaction was monitored over time, the yield did not significantly increase between 30 minutes and 8 hours (Table 18). At 60 °C, a lower yield was obtained. Other Pd sources resulted in similarly low yields (Table 19).
Table 18 Effect of time on Pd-catalyzed lactam α-arylation 
Entry Ligand Reaction time Conv. (%) % Yield (22) % anisole (18) ee (%) 1 L18 30 min 36 24 19 -79 2 L18 1 h 64 23 17 -79 3 L18 8 h 76 33 25 -79 4 L17 30 min 65 23 16 -79 5 L17 1 h 71 24 17 -78 6 L17 8 h 81 28 26 Table 19 Effect of Pd sources on Pd-catalyzed lactam α-arylation 
Entry [Pd] Additive Conv. (%) % Yield (22) % anisole (18) 1 [Pd(cinnamyl)Cl]3 - 55 27 24 2 [Pd(cinnamyl)Cl]2 LiOt-Bu 59 20 23 3 [Pd(cinnamyl)Cl]2 NaOt-Bu 76 12 18 4 [Pd(cinnamyl)Cl]2 KOt-Bu 93 25 14 5 Pd(PPh3)4 - 52 16 5 6 Pd(Cp)(allyl) - 40 13 17 - Aryl bromides and aryl chlorides with a variety of substitution patterns are accommodated in the arylation (Table 20). Aryl halides possessing electron-deficient (see products 3ab, 3ad, 3ah, 3ae) and electron-rich (3af, 3ag) substituents at the para position led to products with excellent enantioselectivites using Method A and B, respectively. Aryl halides possessing substituents at the meta position are also permissible in both Method A and B, although slightly diminished enantioselectivity is observed when 3-chloroanisole is used as the electrophile (3ai). Trace product is observed when ortho-substituted aryl halides are exposed to the reaction conditions. An N-methyl indole was also well tolerated. 5-indolyl lactam 3al was obtained in moderate yield and excellent enantioselectivity.

- Sterically demanding α-substituents are well tolerated in both Methods (Table 21). Although the yields are slightly diminished, the high levels of enantioselectivity are retained. Examples having ethyl (3ha, 3hb), benzyl (3ga, 3gb), propyl (3ia), phenethyl (3ja), and 2-naphthylmethyl (3ka) substitution all furnish the α-arylated products in good enantioselectivity. α-Benzyl substituted lactam 1g was also employed in the reaction with a number of different electrophilic coupling partners using both Methods. Even with a more hindered substrate, similar patterns of reactivity and selectivity to α-methyl substituted 1a were observed. When an electron-deficient aryl chloride coupling partner is used in Method A or m-bromotoluene is used in Method B with 1g, the desired product is formed in high enantioselectivity and good yield (3gc and 3gk).

- The arylation reaction was used in the synthesis of different five-membered heterocycles (Scheme 4). α-Quaternary lactam 3aa can be swiftly deprotected with ceric ammonium nitrate (CAN) providing unprotected lactam 4 in 73% yield. Reduction of the lactam carbonyl with lithium aluminum hydride provides the corresponding medicinally valuable pyrrolidine (5). Partial reduction of the lactam with lithium triethylborohydride and trapping of the resulting iminium ion with potassium cyanide yields chiral aminonitrile 6 in moderate yield, but with high diastereoselectivity.

- α-Vinylation of lactam 15 with 2-chloroprop-1-ene (25) was achieved in the presence of LiHMDS and various palladium sources (Table 22).
Table 22 Effect of various palladium sources on α-vinylation of lactam 15 with vinyl 25 
Entry [Pd] (x) % Yield (26) ee (%) 1 Pd2(pm-dba)3 (2.5) 21 91 2 Pd2(dba)3 (2.5) 34 95 3 Pd(OAc)2 (5) trace - - α-Vinylation of lactam 15 with 1-chloro-2-methylprop-1-ene (27) was achieved in the presence of LiHMDS and various palladium sources (Table 23).
Table 23 Effect of various palladium sources on a-vinylation of lactam 15 with vinyl 27 
Entry [Pd] : % Yield (28) % ee 1 Pd(dba)2 (5 mol%) 40 94 2 Pd(dba)2 (10 mol%) 24 - 3 Pd(dm-dba)2 (5 mol%) 8 - - α-vinylation of lactam 15 with vinyl 25 was achieved in the presence of LiHMDS and catalytic Pd2(pm-dba)3 using various ligands (Table 24).

- α-vinylation of lactam 15 with vinyl 27 was achieved in the presence of LiHMDS and catalytic Pd(dba)2 using various ligands (Table 25).

- α-Vinylation of lactam 15 with vinyl 25 was achieved in the presence of LiHMDS and catalytic Pd2(pm-dba)3 at various temperatures (Table 26).
Table 26 Effect of temperature on α-vinylation of lactam 15 with vinyl 25 
Entry T % RSMa % Yield ( 26 )a % ee 1 60 23 37 88 2 50 31 42 86 3 40 67 8 - 4 60 - 36 b 86 a Internal standard trimethoxybenzene (TMB) was used for measurements.
b Isolated yield. - α-Vinylation of lactam 15 with vinyl 27 was achieved in the presence of LiHMDS and catalytic Pd2(dba)3 at various temperatures (Table 27).
Table 27 Effect of temperature on α-vinylation of lactam 15 with vinyl 27 
Entry X T % yield (28) 1 a Cl 50 N.D. 2 60 2 3 70 12 4 80 16 5 b 80 43 6 b 100 13 7 a Br r.t. trace 8 a 40 7 9 a 50 8 10 a 80 13 a (S,S)-Me-ferrocelane
b 24 h - α-Vinylation of lactam 15 with vinyl 25 was achieved in the presence of LiHMDS and catalytic Pd2(pm-dba)3 at various concentrations of dioxane (Table 28).
Table 28 Effect of Dioxane concentration on α-vinylation of lactam 15 with vinyl 25 
Entry x % yield ( 26 )a % ee 1 0.20 29 90 2 0.33 23 90 3 0.10 27 90 a Internal standard trimethoxybenzene (TMB) was used for measurements. - α-Vinylation of lactam 15 with vinyl 27 was achieved in the presence of LiHMDS and catalytic Pd2(dba)3 with a solvent mixture or Toluene alone (Table 29).
Table 29 Effect of Dioxane/Toluene mixture or Toluene alone on α-vinylation of lactam 15 with vinyl 27 
Entry Solvent % yield % ee 1 Dioxane /Toluene (1:1) 23 83 2 Toluene 30 84 - α-Vinylation of lactam 15 with vinyl 25 was achieved in the presence of LiHMDS and catalytic Pd2(pm-dba)3 with a variation of the stoichiometry of 15 and 25 (Table 30, entry 1), and α-vinylation of lactam 15 with vinyl 27 was achieved in the presence of LiHMDS and catalytic Pd(dba)2 with several variations in the stoichiometry of 15 and 27 (Table 30, entries 2-6).

- α-Vinylation of lactam 15 with various N-protecting groups was achieved in the presence of LiHMDS and catalytic Pd2(dba)3 (Table 31) using vinyl 27.

- α-Vinylation of lactam 15 with 2-chlorovinylbenzene (29) was achieved in the presence of LiHMDS and catalytic Pd(dba)2 (Table 32).
Table 32 Scope of vinyl halide for α-vinylation of lactam 15 
Entry Temp., Time % RSM (15) % yield (30) % ee 1 80 °C, 18 h 0 73 25 2 80 °C, 24 h 0 75 - 3 50 °C, 24 h 24 72 6 - Unless otherwise stated, reactions were performed in flame-dried or oven-dried glassware under an argon or nitrogen atmosphere using dry, deoxygenated solvents. Reaction temperatures were controlled by an IKAmag temperature modulator unless otherwise indicated. Glovebox manipulations were performed under a N2 atmosphere. TLC was performed using E. Merck silica gel 60 F254 precoated glass plates (0.25 mm) and visualized by UV fluorescence quenching or KMNO3 staining. Silicycle SiliaFlash P60 Academic Silica gel (particle size 0.040-0.064 mm) was used for flash column chromatography. Analytical chiral HPLC was performed with an Agilent 1100 Series HPLC utilizing a Chiralcel OD-H column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd. with visualization at 254 nm. Analytical SFC was performed with a Mettler SFC supercritical CO2 analytical chromatography system utilizing a Chiralcel AD column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd. with visualization at 254 nm. 1H-NMR spectroscopic data were collected on a Varian 500 MHz spectrometer at ambient temperature. GC analyses were obtained on an Agilent 6890 Series GC system with a DB-1 column (length 30 m, internal diameter 0.25 mm).
- THF, Et2O, CH2Cl2, toluene, CH3CN, TBME and dioxane were dried by passage through an activated alumina column under argon. Purified water was obtained using a Barnstead NANOpure Infinity UV/UF system. Commercially available reagents were purchased.
- N-aryl lactam substrates, Ni benzonitrile complex 20, and NiIIArCl complex 21 were prepared according to previously reported procedures.
-

- This procedure was adapted from a previously reported procedure. In a Schlenk-tube, to a solution of p-anisidine (219 mg, 1.78 mmol, 3.07 equiv) in CH2Cl2 (2.0 mL) cooled to 0 °C was added dropwise via syringe a trimethylalumnium-solution (2.0 M in heptane, 0.330 mL, 0.660 mmol, 1.13 equiv). The solution was allowed to warm to room temperature. Lactone 24 (121 mg, 0.580 mmol, 1.00 equiv) was added in CH2Cl2 (1.9 mL). The Schlenk-tube was closed with a Teflon screw-cap and the reaction mixture was heated to 80 °C for 20 h. After 20 h, the reaction was allowed to cool to room temperature. After quenching with MeOH, followed by the addition of aqueous, saturated NH4Cl-solution and vigorous stirring, the layers were separated and the aqueous phase extracted with ethyl acetate (3 x 20 mL). The combined organic extracts were dried over Na2SO4 and the volatiles were removed under reduced pressure. Chromatography of the crude mixture on silica eluting with 50% ethyl acetate/hexane yielded the title compound as an off-white solid. 1H-NMR (CDCl3, 500 MHz): δ 7.59 - 7.55 (m, 2H), 7.27-7.23 (m, 1H), 7.03 - 6.99 (m, 2H), 6.93 - 6.89 (m, 2H), 6.79 (m, 1H), 3.81 (s, 3H), 3.80 (s, 3H), 2.57 (ddd, 1H, J = 12.6, 6.6, 4.1 Hz), 2.26 (dt, 1H, J = 12.7, 8.0 Hz), 1.62 (s, 3H).
- n = the number of reactions in the screen. In a glovebox, Ni(COD)2 (1.38n mg, 0.005n mmol, 0.100 equiv), toluene (0.25n mL), and a stir bar were added to a 4-mL vial A. To a separate 4-mL vial B was added LiHMDS (8.37n mg, 0.050n mmol, 1.00 equiv), lactam (10.3n mg, 0.050n mmol, 1.00 equiv), toluene (0.25n mL), and a stir bar. The contents of A and B were stirred for ∼2 minutes. To a separate vial was added the ligand (0.006 mmol, 0.120 equiv) and a stir bar followed by 0.25 mL of solution A. The ligand/Ni mixture was then stirred for 10 minutes and aryl halide (7.1 mg, 6.1 µL, 0.050 mmol, 1.00 equiv) and 0.25 mL of solution B were added. The vial was then sealed with a PTFE-lined cap and removed from the glovebox. The reaction mixture was then heated at and stirred for 16-20 h. The vial was then cooled to room temperature and opened to air. A solution of 1,3,5-trimethoxybenzene (8.41 mg, 0.050 mmol, 1.00 equiv) in Et2O (0.1 mL) was added as an internal standard. The reaction mixture was then filtered through a short silica plug, eluting with Et2O (2 mL). An aliquot of the eluate was then removed for GC determination of aryl halide conversion with respect to 1,3,5-trimethoxybenzene. The remaining eluate was then concentrated by rotary evaporator and dissolved in CDCl3 to determine an 1HNMR yield with respect to 1,3,5-trimethoxybenzene. Then, the sample was concentrated and purified by preparative TLC (EtOAc/hexanes). The purified product was then dissolved in hexanes for HPLC analysis on a Chiralcel OD column (20% IPA/hexanes, 1.0 mL/min).
- n = the number of reactions in the screen. In a glovebox, Pd(OAc)2 (1.10n mg, 0.005n mmol, 0.100 equiv), toluene (0.25n mL), and a stir bar were added to a 4-mL vial A. To a separate 4-mL vial B was added LiHMDS (8.37n mg, 0.050n mmol, 1.00 equiv), lactam (12.3n mg, 0.060n mmol, 1.20 equiv), toluene (0.25n mL), and a stir bar. The contents of A and B were stirred for ∼2 minutes. To a separate vial was added the ligand (0.006 mmol, 0.120 equiv) and a stir bar followed by 0.25 mL of solution A. The ligand/Pd mixture was then stirred for 10 minutes and aryl halide (7.1 mg, 6.1 µL, 0.050 mmol, 1.00 equiv) and 0.25 mL of solution B were added. The vial was then sealed with a PTFE-lined cap and removed from the glovebox. The reaction mixture was then heated at 80 °C and stirred for 16-20 h. The vial was then cooled to room temperature and opened to air. A solution of 1,3,5-trimethoxybenzene (8.41 mg, 0.050 mmol, 1.00 equiv) in Et2O (0.1 mL) was added as an internal standard. The reaction mixture was then filtered through a short silica plug, eluting with Et2O (2 mL). An aliquot of the eluate was then removed for GC determination of aryl halide conversion with respect to 1,3,5-trimethoxybenzene. The remaining eluate was then concentrated by rotary evaporator and dissolved in CDCl3 to determine an 1HNMR yield with respect to 1,3,5-trimethoxybenzene. Then, the sample was concentrated and purified by preparative TLC (EtOAc/hexanes). The purified product was then dissolved in hexanes for SFC analysis on a Chiralcel AD column (40% IPA/hexanes, 2.5 mL/min).
- With model substrate 3m, conditions based on our previous report (Korch et al., Angew. Chem. Int. Ed., 2015, 54, 179-183) using 10 mol % (S)-(CF3)3-t-BuPHOX ligand and 4 mol % Pd2(pmdba)3 in 0.014 M toluene gave the product 3m in only 76% ee (Table 33, entry 1). Other commonly used allylic alkylation ligands such as (S)-t-BuPHOX (L2) and (S,S)-ANDEN-Ph Trost (L3) were also tested (entries 2-3). We then examined the effect of solvent on the enantioselectivity (entries 4-6), ultimately finding that 2:1 hexanes-toluene provided high yield and ee.

- The substrate scope of the decarboxylative alkylation of various N4-Boc protected piperazinones was also explored (Table 34). We first tested the α-monosubstituted piperazinone 3a, finding that typical conditions in a 0.033 M solution of toluene afforded the product 4a in high yield and enantioselectivity. Another monosubstituted piperazinone 4b with a N1-anisoyl protecting group, was also obtained in high yield and ee. Furthermore, replacement of the N4-Boc protecting group with Cbz (3c), and 2-chloro substitution of the allyl group (3d) provided similar results.
- Next, we examined α,α-disubstituted substrates, finding that for the simple cases of methyl substitution (3e, 3f), performing the reaction at 0.033 M concentration in toluene gave high yields and ee, with no improvement with the use of 2:1 hexanes-toluene at 0.014 M concentration. The exception was N4-benzoyl protected substrate 3g, which in our previous work gave 52% ee in toluene (Korch et al., Angew. Chem. Int. Ed., 2015, 54, 179-183); under our optimized conditions in 2:1 hexanes-toluene, the substrate gave an improved albeit modest ee of 70%. Larger substituents such as α-benzylated compound 3h or benzyloxymethyl ether 3i required the use of 2:1 hexanes-toluene to achieve high enantioselectivity. This unique mixed-solvent requirement was observed in all substituents other than methyl. A wide range of functional groups was tolerated: notably, the nitrile, ketone, and methylcarbamate substrates, which could not be accessed in our previously described efforts, gave high ee and yields (3j-m).

- If decarboxylative alkylation could be achieved with the versatile tetrahydropyrimidin-2-one scaffold, a broader range of chiral β2,2-amino acids might be accessible:

- Beginning the investigations with methyl substrate 5a (Table 35), the optimized conditions in 2:1 hexanes-toluene furnished the decarboxylative alkylation product 6a in high yield and ee. The reaction proved amenable to a variety of α-substituents, including ethyl (6b), methyl ester (6c), 2-chloroallyl (6d), and benzyl (6e). Furthermore, the reaction is scalable, as shown for the benzyl substrate 5e in a 1 gram reaction. In contrast, the nitrile and benzyloxymethyl ether products 6f and 6g were isolated with reduced enantioselectivities. The fluorine and propargyl substrates 5h and 5i were also able to be used, and may serve as a precursor to a novel fluorinated β2,2-amino acid, or for biorthogonal click reactions, respectively.

- Both Boc groups of 4m could be removed by treatment with excess TFA to give aminopiperazinone 8 (Table 36, row a). With LiOH, the benzoyl group could be orthogonally removed to provide the free amide 9 (Table 36, row b). The amide 9 could then be reduced with LiAlH4 to the corresponding chiral aminopiperazine 10. To further illustrate the synthetic versatility realized by Boc-deprotection of N4, we hydrogenated the allyl olefin of methyl piperazinone 4e before cleaving the Boc group to obtain 11. We then applied Gaunt's method of C-H carbonylation on the aliphatic amine of 11 to forge the fused β-lactam 12, which bears resemblance to the core of various β-lactam antibiotics and β-lactamase inhibitors (Table 36, row c). [J. R. Cabrera-Pardo, A. Trowbridge, M. Nappi, K. Ozaki, M. J. Gaunt, Angew. Chem. Int. Ed. 2017, 56, 11958-11962.] Lastly, we transformed four tetrahydropyrimidinone substrates, 6c-e and 6h, into their corresponding acyclic forms (Table 36, rows d-e). Using a two-step protocol involving TFA-mediated Boc cleavage followed by saponification with LiOH, we successfully obtained the crude β2,2-amino acid (Table 36, row d); to facilitate silica gel chromatographic isolation, we chose to mask the free amine and carboxylic acid with Boc and benzyl groups, respectively, resulting in protected β2,2-amino acid 13. We note that in this four step sequence, we only performed one chromatographic separation to isolate the protected β-amino acid. In contrast, novel unprotected β2,2-amino acids 14-16 bearing pendant carboxylic acid, chloroallyl, and fluorine atom functional groups could be obtained directly by purification with reverse phase preparatory HPLC following the two-step deprotection sequence (Table 36, row e).

- Unless otherwise stated, reactions were performed in flame-dried glassware under an argon or nitrogen atmosphere using dry, deoxygenated solvents. Solvents were dried by passage through an activated alumina column under argon. tert-Butyl 3-oxopiperazine-1-carboxylate 1 was obtained from Combi-Blocks. Commercially obtained reagents were used as received. Chemicals were purchased from Sigma Aldrich/Strem/Alfa Aesar/Combi-Blocks and used as received. Reaction temperatures were controlled by an IKAmag temperature modulator. Glove box manipulations were performed under a nitrogen atmosphere. Thin-layer chromatography (TLC) was performed using E. Merck silica gel 60 F254 precoated plates (0.25 mm) and visualized by UV fluorescence quenching, iodine on silica, or KMnO4 staining. SiliaFlash P60 Academic Silica gel (particle size 0.040-0.063 mm) was used for flash chromatography. Analytical SFC was performed with a Mettler SFC supercritical CO2 analytical chromatography system utilizing a Chiralpak IC column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd. with visualization at 254 nm. 1H NMR spectra were recorded on a Varian Inova 500 MHz spectrometer or a Bruker Avance HD 400 MHz spectrometer and are reported relative to residual CHCl3 (δ 7.26 ppm). 13C NMR spectra were recorded on a Varian Inova 500 MHz spectrometer or a Bruker Avance HD 400 MHz spectrometer and are reported relative to residual CDCl3 (δ 77.16 ppm). Data for 1H NMR are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sept = septuplet, m = multiplet, br s = broad singlet. Data for 13C NMR are reported in terms of chemical shifts (δ ppm). Some reported spectra include minor solvent impurities of water (δ 1.56 ppm), ethyl acetate (δ 4.12, 2.05, 1.26 ppm), methylene chloride (δ 5.30 ppm), acetone (δ 2.17 ppm), grease (δ 1.26, 0.86 ppm), and/or silicon grease (δ 0.07 ppm), which do not impact product assignments. IR spectra were obtained using a Perkin Elmer Paragon 1000 spectrometer using thin films deposited on NaCl plates and reported in frequency of absorption (cm-1). High resolution mass spectra (HRMS) were obtained from an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI+), atmospheric pressure chemical ionization (APCI+), or mixed ionization mode (MM: ESI-APCI+). Optical rotations were measured with a Jasco P-2000 polarimeter operating on the sodium D-line (589 nm), using a 100 mm pathlength cell and are reported as: [a]DT (concentration in g/100 mL, solvent). Stereochemistry is assigned by analogy to previous results.1 (a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126, 15044-15045; b) J. T. Mohr, D. C. Behenna, A. M. Harned, B. M. Stoltz, Angew. Chem. Int. Ed. 2005, 44, 6924-6927; Angew. Chem. 2005, 117, 7084-7087; c) M. Seto, J. L. Roizen, B. M. Stoltz, Angew. Chem. Int. Ed. 2008, 47, 6873-6876; Angew. Chem. 2008, 120, 6979-6982; d) J. Streuff, D. E. White, S. C. Virgil, B. M. Stoltz, Nat. Chem. 2010, 2, 192-196; e) D. C. Behenna, Y. Liu, T. Yurino, J. Kim, D. E. White, S. C. Virgil, B. M. Stoltz, Nat. Chem. 2012, 4, 130-133; f) C. M. Reeves, C. Eidamshaus, J. Kim, B. M. Stoltz, Angew. Chem. Int. Ed. 2013, 52, 6718-6721; Angew. Chem. 2013, 125, 6850-6853.)
- List of Abbreviations: An - Anisoyl, Boc - tert-Butyloxycarbonyl, BRSM - based on recovered starting material, Bz - benzoyl, CH2Cl2 - methylene chloride, Cbz - carboxybenzyl, ee - enantiomeric excess, Et2O - diethyl ether, EtOAc - ethyl acetate, IPA - isopropanol, LHMDS - lithium hexamethyldisilazide, MeCN - acetonitrile, MeOH - methanol, SFC - supercritical fluid chromatography, THF - tetrahydrofuran., TLC - thin-layer chromatography.
- Allyl cyanoformate was prepared according to the method of Weber. [M. E. Childs, W . P. Weber, J. Org. Chem. 1976, 41, 3486-3487.] Phosphinooxazoline (PHOX) ligands (S)-L1, (S)-L2, and achiral GlyPhox were prepared by methods described in our previous work. [(a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126, 15044-15045. (b) K. Tani, D. C. Behenna, R. M. McFadden, B. M. Stoltz, Org. Lett. 2007, 9, 2529-2531. (c) M. R. Krout, J. T. Mohr, B. M. Stoltz, Org. Synth. 2009, 86, 181-193.] Di-benzoylated allylic alkylation substrate 3g was prepared according to the method of Korch. [K. M. Korch, C. Eidamshaus, D. C. Behenna, S. Nam, D. Home, B. M. Stoltz, Angew. Chem. Int. Ed. 2015, 54, 179-183.] Tris(4,4'- methoxydibenzylideneacetone)dipalladium(0) [Pd2(pmdba)3] was prepared according to the method of Ibers [ J. Organomet. Chem. 1999, 65, 253-266] or Fairlamb [Org. Lett. 2004, 6, 4435-4438]. AgOPiv was prepared using Grubbs' procedure [ J. Am. Chem. Soc. 2011, 133, 8525-8527]. tert-Butyl ((phenylsulfonyl)methyl)carbamate and benzyl ((phenylsulfonyl)methyl)carbamate were prepared according to the method of Zwierzak or Dikshit [(a) Klepacz, A.; Zwierzak, A. Tetrahedron Lett. 2002, 43, 1079. (b) Sikriwal, D.; Kant, R.; Maulik, P. R.; Dikshit, D. K. Tetrahedron 2010, 66, 6167]. Benzyl 3-oxopiperazine-1-carboxylate was prepared according to the method of Batey. [Tetrahedron 2010, 66, 3370-3377]. 2-chloroallyl chloroformate was prepared according to the method of Stoltz. [J. Am. Chem. Soc. 2008, 130, 810-811.]
-

- tert-butyl 4-benzoyl-3-oxopiperazine-1-carboxylate (SI-1). To a solution of tert-butyl 3-oxopiperazine-1-carboxylate 1 (5.0 g, 24.9 mmol, 1 equiv) in THF (250 mL) at -78 °C was added dropwise nBuLi (11.4 mL, 2.4M solution in hexane, 27.5 mmol, 1.1 equiv) over 20 minutes. The resulting yellow solution was stirred for 10 min at -78 °C. Benzoyl chloride (3.48 mL, 30.0 mmol, 1.2 equiv) was then added dropwise at -78 °C, giving an orange solution. The reaction was stirred for 2.5 h at -78 °C, quenched by addition of saturated aqueous NH4Cl (100 mL), and diluted with ethyl acetate (100 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded crude reaction mixture was purified by silica gel flash column chromatography (10% → 15% → 20% EtOAc/hexanes) to give protected ketopiperazine SI-1 as a white solid (3.2 g, 42.1% yield). Product identity was confirmed by comparison to previously reported characterization data. [A. Chollet, G. Mori, C. Menendez, F. Rodriguez, I. Fabing, M. R. Pasca, J. Madacki, J. Korduláková, P. Constant, A. Quémard, et al., Eur. J. Med. Chem. 2015, 101, 218-235.]

- tert-butyl 4-(4-methoxybenzoyl)-3-oxopiperazine-1-carboxylate (SI-2). To a solution of tert-Butyl 3-oxopiperazine-1-carboxylate (5.0 g, 24.9 mmol, 1 equiv) in THF (250 mL) at -78 °C was added dropwise nBuLi (11.4 mL, 2.4M solution in hexane, 27.5 mmol, 1.1 equiv) over 20 minutes. The resulting yellow solution was stirred for 10 min at -78 °C. Anisoyl chloride (4.1 mL, 30.0 mmol, 1.2 equiv) was then added dropwise at -78 °C, giving a bright orange solution. The reaction was stirred for 2 h at -78 °C, quenched by addition of saturated aqueous NH4Cl (100 mL), and diluted with ethyl acetate (100 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded crude reaction mixture was filtered through a plug of silica (1% → 2% MeOH/CH2Cl2) to give crude anisoyl protected ketopiperazine SI-2 as a white foam, which was directly used without further purification in subsequent acylation reactions with allyl cyanoformate.

- benzyl 4-benzoyl-3-oxopiperazine-1-carboxylate (SI-3). To a solution of benzyl 3-oxopiperazine-1-carboxylate [Tetrahedron 2010, 66, 3370-3377] (1.0 g, 4.3 mmol, 1.0 equiv) in THF (42 mL) at -78 °C was added dropwise nBuLi (1.96 mL, 2.4M solution in hexane, 4.7 mmol, 1.1 equiv) over 20 minutes. The solution was stirred for 10 min at -78 °C. Benzoyl chloride (0.595 mL, 5.1 mmol, 1.2 equiv) was then added dropwise at -78 °C, giving a light yellow solution. The reaction was stirred for 1 h at -78 °C, quenched by addition of saturated aqueous NH4Cl (50 mL), and diluted with ethyl acetate (50 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded crude reaction mixture was purified by silica gel flash column chromatography (33% EtOAc/hexanes) to give Bz-Cbz-protected ketopiperazine SI-3 as a white solid (1.0 g, 70.0% yield); 1H NMR (500 MHz, CDCl3) δ 7.61 - 7.56 (m, 2H), 7.54 - 7.49 (m, 1H), 7.44 - 7.31 (m, 7H), 5.21 (s, 2H), 4.30 (s, 2H), 3.95 (dd, J= 6.8, 4.4 Hz, 2H), 3.83 (dd, J = 6.9, 4.3 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ (a mixture of two rotamers) 172.8, 168.0, 167.6, 154.5, 136.0, 135.0, 132.3, 128.7, 128.5, 128.4, 128.3, 128.3, 68.0, 48.5, 43.3, 41.7, 41.4; IR (Neat Film, NaCl) 3386, 3063, 3033, 2954, 2894, 1706, 1600, 1584, 1498, 1449, 1422, 1394, 1367, 1302, 1231, 1177, 1162, 1123, 1060, 1028, 1010, 944, 857, 796, 765, 731, 699, 639, 612 cm-1; HRMS (MM: ESI-APCI) m/z calc'd for C19H19N2O4 [M+H]+: 339.1339, found 339.1338.

- 2-allyl 1-(tert-butyl) 4-benzoyl-3-oxopiperazine-1,2-dicarboxylate. To a solution of Bz-protected oxopiperazine SI-1 (1.5 g, 4.9 mmol, 1.0 equiv) in THF (40 mL) at -78 °C was added LiHMDS (907 mg, 5.42 mmol, 1.10 equiv.) in THF (10mL) dropwise. The resulting orange reaction mixture was stirred for 15 min at -78 °C. Then, allyl cyanoformate (590 µL, 5.2 mmol, 1.05 equiv) was added dropwise at -78 °C, giving a yellow solution. After stirring for 1.5 h at -78 °C, the reaction was quenched with saturated aqueous NH4Cl (20 mL) and diluted with ethyl acetate (50 mL). The aqueous phase was extracted with ethyl acetate (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica. The silica-loaded crude mixture was purified by silica gel flash chromatography (10% → 20% EtOAc/hexanes) to give the allyl ester 2 as a white solid (1.3 g, 68% yield): 1H NMR (500 MHz, CDCl3) δ (a mixture of two rotamers) 7.59 (d, J = 7.7 Hz, 2H), 7.52 (t, J = 7.6 Hz, 1H), 7.40 (t, J = 7.6 Hz, 2H), 5.92 (m, 1H), 5.44 - 5.07 (m, 3H), 4.84 - 4.57 (m, 2H), 4.28 - 3.61 (m, 4H), 1.43 (br s, 9H); 13C NMR (126 MHz, CDCl3) δ (a mixture of two rotamers) 172.7, 166.9, 164.4, 154.1, 153.5, 134.6, 132.6, 131.0, 128.6, 128.4, 119.9, 119.4, 82.2, 82.0, z67.1, 62.7, 61.7, 43.2, 42.6, 41.5, 40.2, 29.4, 28.3; IR (Neat Film, NaCl) 2978 1746 1695 1600 1450 1393 1367 1310 1277 1231 1177 1158 1127 1088 1059 1009 958 861 794 770 728 694 623 cm-1; HRMS (MM: ESI-APCI) m/z calc'd for C16H17N2O6 [(M-tBu)+H]+: 333.1081, found 333.1075.

- 2-allyl 1-(tert-butyl) 4-(4-methoxybenzoyl)-3-oxopiperazine-1,2-dicarboxylate (3b). Following the procedure described for the preparation of 2, anisoyl protected oxopiperazine SI-2 (1.0 g, 3.0 mmol, 1.0 equiv) was treated with LiHMDS (550 mg, 3.3 mmol, 1.1 equiv) and acylated with allyl cyanoformate (336 µL, 3.1 mmol, 1.05 equiv) to give, after purification by silica gel flash chromatography (Dry load SiO2, 18% EtOAc/hexanes), allyl ester 3b as a white solid (566 mg, 45% yield): 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.61 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.91 (ddt, J = 16.4, 10.8, 5.8 Hz, 1H), 5.46-5.12 (m, 3H), 4.70 (s, 2H), 4.13-3.65 (m, 4H), 3.81 (s, 3H), 1.46 (m, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 171.9, 171.8, 167.0, 164.4, 164.2, 163.4, 154.0, 153.4, 131.4, 131.0, 126.3, 119.6, 119.2, 113.6, 82.0, 81.8, 66.9, 62.5, 61.6, 55.5, 43.3, 42.8, 41.5, 40.1, 28.2; IR (Neat Film, NaCl) 3384, 3060, 2978, 2843, 2568, 2049, 1732, 1605, 1580, 1513, 1456, 1372, 1258, 1088, 1059, 959, 844, 769, 736, 706, 634 cm-1; HRMS (MM: ESI-APCI) m/z calc'd for C21H27N2O7 [M+H]+: 419.1813, found 419.1815.

- 2-allyl 1-benzyl 4-benzoyl-3-oxopiperazine-1,2-dicarboxylate (3c). Following the procedure described for the preparation of 2, Cbz protected oxopiperazine SI-3 (1.0 g, 3.0 mmol, 1.0 equiv) was treated with LiHMDS (544 mg, 3.3 mmol, 1.1 equiv) and acylated with allyl cyanoformate (331 µL, 3.1 mmol, 1.05 equiv) to give, after purification by silica gel flash chromatography (dry load SiO2, 20 → 25% EtOAc/hexanes), allyl ester 3a as a white solid (720 mg, 58% yield): 1H NMR (500 MHz, CDCl3) δ (a mixture of two rotamers) 7.65 - 7.56 (m, 2H), 7.56 - 7.46 (m, 1H), 7.46 - 7.28 (m, 7H), 5.87 (dtd, J = 47.5, 10.7, 5.4 Hz, 1H), 5.55 - 5.04 (m, 5H), 4.84 - 4.48 (m, 2H), 4.24 - 3.99 (m, 2H), 3.99 - 3.66 (m, 2H); 13C NMR (126 MHz, CDCl3) δ (a mixture of two rotamers) 172.4, 172.3, 166.5, 164.0, 163.8, 154.8, 154.2, 135.7, 135.6, 134.4, 132.5, 130.9, 130.8, 128.7, 128.6, 128.5, 128.5, 128.4, 128.3, 128.2, 128.1, 119.6, 119.5, 68.3, 68.2, 67.2, 67.1, 62.1, 61.9, 42.8, 42.3, 41.3, 40.8; IR (Neat Film, NaCl) 3386, 3064, 3033, 2955, 2897, 1746, 1713, 1694, 1651, 1600, 1584, 1498, 1450, 1417, 1368, 1304, 1278, 1229, 1195, 1178, 1160, 1124, 1088, 1061, 1014, 985, 951, 860, 794, 768, 729, 696, 675, 623 cm-1; HRMS (MM: ESI-APCI) m/z calc'd for C23H23N2O6 [M+H]+: 423.1551, found 423.1547.

- 1-(tert-butyl) 2-(2-chloroallyl) 4-benzoyl-3-oxopiperazine-1,2-dicarboxylate (3d). Following the procedure described for the preparation of 2, benzoyl protected oxopiperazine SI-1 (440 mg, 1.5 mmol, 1.0 equiv) was treated with LiHMDS (267 mg, 1.6 mmol, 1.1 equiv) and acylated with 2-chloroallyl chloroformate (235 mg, 1.5 mmol, 1.05 equiv) [J. Am. Chem. Soc. 2008, 130, 810-811] to give, after purification by silica gel flash chromatography (Dry load SiO2, 15% EtOAc/hexanes), 2-chloroallyl ester 3d as an off-white solid (431 mg, 70% yield): 1HNMR (500 MHz, CDCl3): δ (a mixture of two rotamers) 7.59-7.54 (m, 2H), 7.50-7.44 (m, 1H), 7.43-7.37 (m, 2H), 6.67 (m, 1H), 5.42-5.33 (m, 2H), 4.54 (m, 2H), 3.95-3.89 (m, 2H), 3.72 (t, J = 5.2 Hz, 2H), 1.51 (s, 9H); 13C NMR (101 MHz, CDCl3): δ (a mixture of two rotamers) 167.5, 167.3, 152.4, 152.2, 151.1, 151.0, 135.1, 134.9, 134.3, 131.3, 131.2, 128.5, 128.4, 128.0, 127.9, 126.7, 115.9, 115.7, 107.9, 107.6, 82.3, 82.2, 69.9, 69.8, 44.2, 43.1, 42.3, 41.7, 28.3; IR (Neat Film, NaCl) 3396, 3129, 3062, 2979, 2936, 2253, 1770, 1691, 1372, 1242, 1050, 987, 950, 922, 859, 839, 764, 731, 707, 647 cm-1; HRMS (MM: ESI-APCI) m/z calculated for C20H24ClN2O6 [M+H]+: 423.1317, found 423.1316.

- 2-allyl 1-(tert-butyl) 4-(4-methoxybenzoyl)-2-methyl-3-oxopiperazine-1,2-dicarboxylate (3e). Sodium hydride (60% in mineral oil, 25 mg, 0.62 mmol, 1.2 equiv) was added to a solution of allyl ester 2 (200 mg, 0.52 mmol, 1.0 equiv) in THF (5 mL) at 0 °C. After stirring for 30 min at 0 °C, Mel (160 µL, 2.57 mmol, 5.0 equiv) was added. The reaction mixture was allowed to warm to room temperature and stirred for 16 h. The reaction was quenched with aqueous NH4Cl (10 mL) and extracted with EtOAc (3 x 5 mL). The combined organic phases were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded residue was purified by silica gel flash chromatography (20% EtOAc/hexanes) to give methylated allyl ester 3e as a colorless oil (180 mg, 85% yield): 1H NMR (400 MHz, CDCl3): δ 7.59 - 7.46 (m, 3H), 7.46 - 7.32 (m, 2H), 5.92 (ddt, J= 17.2, 10.4, 5.8 Hz, 1H), 5.41 - 5.19 (m, 2H), 4.66 (d, J= 5.8 Hz, 2H), 4.24 - 4.09 (m, 1H), 4.05 - 3.90 (m, 2H), 3.81 - 3.64 (m, 1H), 1.84 (s, 3H), 1.46 (s, 9H); 13C NMR (101 MHz, CDCl3): 8 (a mixture of two rotamers) 172.5, 169.2, 168.0, 153.2, 134.8, 132.4, 131.5, 128.4, 128.0, 119.1, 82.7, 68.7, 66.9, 43.7, 40.9, 28.3, 21.5; IR (Neat Film, NaCl) 3384, 3064, 2980, 2939, 2876, 1962, 1766, 1694, 1600, 1584, 1451, 1394, 1368, 1306, 1270, 1232, 1206, 1164, 1096, 1060, 1032, 1016, 993, 968, 936, 854, 795, 770, 727, 696, 675, 633, 618 cm-1; HRMS (MM: ESI-APCI) m/z calculated for C16H19N2O4 [(M-Boc)+H]+: 303.1341, found 303.1339.

- 2-allyl 1-(tert-butyl) 4-(4-methoxybenzoyl)-2-methyl-3-oxopiperazine-1,2-dicarboxylate (3f). Following the procedure described for the preparation of 3e, anisoyl protected oxopiperazine 3b (120 mg, 0.29 mmol, 1.0 equiv) was treated with NaH (60% in mineral oil, 13 mg, 0.32 mmol, 1.1 equiv) and methylated with MeI (90 µL, 1.43 mmol, 5.0 equiv) to give, after purification by silica gel flash chromatography (dry load SiO2, 15% - 20% - 25% - 30% EtOAc/hexanes), methylated allyl ester 3f as a colorless oil (110 mg, 89% yield): 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 5.92 (ddt, J = 17.2, 10.4, 5.8 Hz, 1H), 5.40 - 5.17 (m, 2H), 4.66 (d, J = 5.8 Hz, 2H), 4.15 - 4.02 (m, 1H), 4.02 - 3.87 (m, 2H), 3.83 (s, 3H), 3.80 - 3.67 (m, 1H), 1.84 (s, 3H), 1.46 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.8, 169.0, 168.1, 163.4, 153.2, 131.5, 131.0, 126.6, 119.0, 113.7, 82.6, 68.5, 66.8, 55.5, 43.8, 41.0, 28.3, 21.6; IR (Neat Film, NaCl) 3081, 2978, 2938, 2842, 1766, 1702, 1604, 1579, 1512, 1460, 1394, 1368, 1308, 1287, 1259, 1235, 1208, 1169, 1114, 1095, 1060, 1021, 1004, 969, 933, 845, 789, 770, 733, 706, 648, 634 cm-1; HRMS (MM: ESI-APCI) m/z calculated for C18H21N2O7 [(M-tBu)+H]+: 377.1343, found 377.1339.

- 2-allyl 1-(tert-butyl) 2-benzyl-4-(4-methoxybenzoyl)-3-oxopiperazine-1,2-dicarboxylate (3h). Following the procedure described for the preparation of 3e, anisoyl-protected allyl ester 3b (200 mg, 0.48 mmol, 1.0 equiv) was treated with potassium hydride (23 mg, 0.57 mmol, 1.2 equiv) and alkylated with benzyl bromide (170 µL, 1.43 mmol, 3.0 equiv) to give, after purification by silica gel flash chromatography (Dry load SiO2, 20% - 25% EtOAc/hexanes), the benzyl ester 3h as a colorless oil (100 mg, 41% yield) (Note: attempts using sodium hydride as a base failed to give conversion of starting material. Instead, potassium hydride resulted in conversion to the desired product.): 1H NMR (400 MHz, CDCl3) ö (a mixture of two rotamers) 7.52 (m, 2H), 7.42 - 7.23 (m, 3H), 7.11 (m, 2H), 6.92 - 6.75 (m, 2H), 5.97 (ddt, J = 17.2, 10.4, 5.8 Hz, 1H), 5.50 - 5.17 (m, 2H), 4.87 - 4.57 (m, 2H), 3.86 (s, 3H), 3.79 - 3.56 (m, 4H), 2.95 (ddd, J = 13.3, 7.4, 3.0 Hz, 1H), 2.86 (br s, 1H), 1.55 (s, 9H); 13C NMR (101 MHz, CDCl3) ö (a mixture of two rotamers) 172.1, 168.0, 167.4, 166.9, 163.4, 153.6, 153.4, 136.0, 135.3, 131.8, 131.3, 130.7, 128.8, 128.7, 128.4, 128.2, 127.8, 127.7, 127.5, 126.6, 119.4, 118.9, 113.7, 82.9, 81.8, 72.7, 67.0, 66.5, 55.6, 44.5, 43.0, 42.2, 41.4, 40.8, 40.0, 38.8, 28.5; IR (Neat Film, NaCl) 2978, 1764, 1698, 1604, 1512, 1496, 1455, 1394, 1366, 1307, 1282, 1256, 1196, 1155, 1078, 1020, 1000, 921, 844, 768 733, 704 cm-1; HRMS (MM: ESI-APCI) m/z calculated for C28H33N2O7 [M+H]+: 509.2282, found 509.2285.

- 2-allyl 1-(tert-butyl) 4-benzoyl-2-((benzyloxy)methyl)-3-oxopiperazine-1,2-dicarboxylate (3i). Following the procedure described for the preparation of 3e, allyl ester 2 (150 mg, 0.39 mmol, 1.00 equiv) was treated with sodium hydride (17 mg, 0.42 mmol, 1.1 equiv) and alkylated with benzyl chloromethyl ether (107 µL, 0.77 mmol, 2.0 equiv) to give, after purification by silica gel flash chromatography (Dry load SiO2, 10% → 15% EtOAc/hexanes), benzyloxy methyl ether 3i as a colorless oil (50 mg, 55% yield BRSM): 1H NMR (500 MHz, CDCl3) δ (a mixture of two rotamers) 7.64 - 7.55 (m, 2H), 7.51 - 7.44 (m, 1H), 7.41 - 7.27 (m, 7H), 5.88 (ddt, J = 17.3, 10.4, 5.8 Hz, 1H), 5.40 - 5.17 (m, 2H), 4.74 - 4.47 (m, 4H), 4.38 (m, 1H), 4.17 (m, 1H), 4.08 - 3.84 (m, 4H), 1.42 (m, 9H); 13C NMR (126 MHz, CDCl3) δ (a mixture of two rotamers) 173.0, 167.4, 167.2, 166.5, 153.6, 153.1, 141.0, 137.9, 137.5, 134.8, 132.4, 131.6, 131.1, 128.7, 128.7, 128.6, 128.4, 128.3, 128.0, 127.7, 127.6, 127.1, 119.5, 118.9, 82.7, 81.9, 73.9, 73.3, 72.6, 71.2, 71.1, 66.8, 65.4, 43.6, 43.4, 42.5, 41.7, 28.4, 28.3; IR (Neat Film, NaCl) 3528, 3064, 3031, 2978, 2934, 1762, 1694, 1600, 1496, 1453, 1394, 1366, 1314, 1270, 1231, 1205, 1161, 1094, 1055, 1014, 972, 941, 854, 795, 770, 730, 696, 674, 624 cm-1; HRMS (MM: ESI-APCI) m/z calculated for C24H25N2O7 [(M-tBu)+H]+: 453.1656, found 453.1649.

- 2-allyl 1-(tert-butyl) 2-(2-cyanoethyl)-4-(4-methoxybenzoyl)-3-oxopiperazine-1,2-dicarboxylate (3j). DBU (7.1 µL, 0.048 mmol, 0.10 equiv) was added to a solution of allyl ester 3b (200 mg, 0.478 mmol, 1.0 equiv) and acrylonitrile (94 µL, 1.43 mmol, 3.0 equiv) in DMF (2.4 mL) at room temperature. After stirring for 2 h at 70 °C and 24 h at 55 °C, additional DBU (14 µL, 0.1 mmol, 0.2 equiv) was added and the orange solution was maintained at 70 °C for 4 h. Then, additional DBU (21 µL, 0.143 mmol, 0.30 equiv) was added and the mixture was stirred at 70 °C for 3 h. After allowing the reaction mixture to cool to room temperature, the reaction was quenched with saturated aqueous NH4Cl (2 mL) and diluted with EtOAc (6 mL). The aqueous phase was extracted with EtOAc (3 x 5 mL). The combined organic layers were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded residue was purified by flash chromatography (20% EtOAc/hexanes) to give the α-cyanoethylated allyl ester 3j as a pale yellow oil (77.6 mg, 34% yield): 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 2H), 6.85 (d, J = 8.9 Hz, 2H), 5.92 (ddt, J = 17.2, 10.4 Hz, 5.9 Hz, 1H), 5.36 (dd, J = 17.2, 1.4 Hz, 1H), 5.29 (d, J = 10.5 Hz, 1H), 4.69 (d, J = 5.8 Hz, 2H), 4.25 (s, 1H), 4.06 (ddd, J = 13.1, 5.7, 3.1 Hz, 1H), 3.96 (ddd, J = 13.1, 8.6, 3.4 Hz, 1H), 3.83 (s, 3H), 3.64 - 3.49 (m, 1H), 2.77 (s, 2H), 2.49 (m, 1H), 2.44 - 2.31 (m, 1H), 1.47 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 171.7, 167.24, 166.3, 163.5, 153.4, 131.1, 126.2, 119.6, 119.1, 113.8, 82.9, 70.5, 67.3, 55.5, 43.9, 42.3, 29.8, 28.2, 12.8; IR (Neat Film, NaCl) 2978, 2249, 1760, 1694, 1604, 1579, 1512, 1462, 1394, 1368, 1311, 1258, 1160, 1018, 933, 846, 769, 737, 704 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C24H30N3O7 [M+H]+: 472.2078, found 472.2078.

- 2-allyl 1-(tert-butyl) 4-(4-methoxybenzoyl)-3-oxo-2-(3-oxobutyl)piperazine-1,2-dicarboxylate (3k). To a solution of allyl ester 3b (200 mg, 0.5 mmol, 1.0 equiv) and methyl vinyl ketone (80 µL, 0.96 mmol, 2.0 equiv) in acetone (2 mL) at room temperature was added DBU (7.1 µL, 0.05 mmol, 0.1 equiv). After stirring for 24 h at 55 °C, additional DBU (7.1 µL, 0.05 mmol, 0.1 equiv) was added and the orange solution was maintained at 55 °C for an additional 24 h. The reaction mixture was allowed to cool to ambient temperature and concentrated under reduced pressure onto silica gel. The silica-loaded crude reaction mixture was purified by silica gel flash column chromatography (33% EtOAc/hexanes) to afford the ketone 3k as a pale yellow oil (90 mg, 39% yield): 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 5.92 (ddt, J = 17.2, 10.4, 5.8 Hz, 1H), 5.35 (dd, J = 17.2, 1.5 Hz, 1H), 5.26 (d, J = 10.4 Hz, 1H), 4.71 - 4.60 (m, 2H), 4.11 - 3.93 (m, 3H), 3.82 (s, 3H), 3.71 - 3.59 (m, 1H), 2.72 (q, J = 9.3, 8.1 Hz, 1H), 2.60 (dt, J = 9.6, 6.2 Hz, 3H), 2.10 (s, 3H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 207.8, 172.0, 167.9, 167.4, 163.4, 153.5, 131.5, 131.1, 126.6, 119.2, 113.7, 82.4, 70.6, 66.9, 55.5, 43.5, 42.1, 38.8, 29.8, 28.7, 28.3; IR (Neat Film, NaCl): 2977, 2934, 1761, 1704, 1604, 1512, 1456, 1394, 1367, 1312, 1257, 1199, 1168, 1090, 1018, 845, 768 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C25H33N2O8 [M+H]+: 489.2231, found: 489.2228.

- 2-allyl 1-(tert-butyl) 2-((((benzyloxy)carbonyl)amino)methyl)-4-(4-methoxybenzoyl)-3-oxopiperazine-1,2-dicarboxylate (3l). Following the procedure described for the preparation of 3m, anisoyl-protected allyl ester 3b (400 mg, 0.96 mmol, 1.0 equiv) was treated with Cs2CO3 (779 mg, 02.39 mmol, 2.5 equiv) and alkylated with benzyl ((phenylsulfonyl)methyl)carbamate8 (350 mg, 1.15 mmol, 2.5 equiv) to give, after purification by silica gel flash chromatography (Dry load SiO2, 10% - 15% - 20% EtOAc/hexanes), the aminomethyl allyl ester 3l as a white foam (300 mg, 54% yield): 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.69 - 7.50 (m, 2H), 7.41 - 7.25 (m, 5H), 6.95 - 6.72 (m, 2H), 5.92 (ddt, J = 17.3, 10.4, 5.9 Hz, 1H), 5.49 - 4.96 (m, 5H), 4.85 - 4.53 (m, 2H), 4.27 - 3.87 (m, 5H), 3.83 (d, J = 14.6 Hz, 3H), 3.60 (d, J = 22.7 Hz, 1H), 1.47 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 171.7, 167.3, 166.5, 163.4, 156.8, 153.7, 152.7, 136.4, 134.21, 132.5, 131.5, 131.2, 129.4, 129.3, 128.9, 128.6, 128.3, 126.5, 119.8, 119.3, 113.7, 83.1, 82.1, 71.4, 70.6, 68.3, 67.8, 67.0, 55.5, 45.0, 44.2, 43.7, 42.9, 41.9, 28.3; IR (Neat Film, NaCl) 3366, 2977, 1704, 1604, 1513, 1456, 1394, 1367, 1313, 1257, 1232, 1169, 1091, 1018, 1002, 845, 767, 698 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C30H36N3O9 [M+H]+: 582.2446, found 582.2438.

- 2-allyl 1-(tert-butyl) 4-benzoyl-2-(((tert-butoxycarbonyl)amino)methyl)-3-oxopiperazine-1,2-dicarboxylate (3m). To a suspension of allyl ester 2 (200 mg, 0.5 mmol, 1.0 equiv) and tert-butyl ((phenylsulfonyl)methyl)carbamate (168 mg, 0.6 mmol, 1.2 equiv), in dichloromethane (2.5 mL) at room temperature was added Cs2CO3 (419 mg, 1.3 mmol, 2.5 equiv). [(a) Klepacz, A.; Zwierzak, A. Tetrahedron Lett. 2002, 43, 1079. (b) Sikriwal, D.; Kant, R.; Maulik, P. R.; Dikshit, D. K. Tetrahedron 2010, 66, 6167.] After stirring for 3 h, saturated aqueous NH4Cl (1 mL) was added and the biphasic mixture was vigorously stirred for 20 min. The aqueous phase was extracted with dichloromethane (3 x 3 mL). The combined organic phases were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded crude reaction mixture was purified by silica gel flash column chromatography (15% EtOAc/hexanes) to give methylcarbamate allyl ester 3m as a white foam (202 mg, 76% yield): 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.55 (d, J = 7.5 Hz, 2H), 7.49 (t, J = 7.5 Hz, 1H), 7.36 (t, J = 7.7 Hz, 2H), 5.91 (ddt, J = 16.5, 10.4, 5.8 Hz, 1H), 5.34 - 5.27 (m, 2H), 4.96 (m, 1H), 4.66 (br s, 2H), 4.19 - 3.85 (m, 5H), 3.79 - 3.64 (m, 1H), 1.48 (s, 9H), 1.40 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 172.4, 167.3, 166.8, 156.1, 153.6, 152.9, 134.9, 132.4, 131.7, 131.1, 128.3, 128.3, 119.6, 119.0, 83.1, 82.1, 79.9, 71.4, 70.6, 67.0, 44.3, 43.8, 43.2, 42.2, 41.6, 28.4, 28.3; IR (Neat Film, NaCl) 3386, 2978, 1760, 1698, 1601, 1505, 1451, 1394, 1367, 1314, 1232, 1203, 1164, 1092, 1067, 1012, 915, 854, 766, 730, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C26H36N3O8 [M+H]+: 518.2497, found 518.2496.
-

- tert-Butyl 4-oxotetrahydropyrimidine-1(2H)-carboxylate (SI-4). A solution of 3-aminopropanamide hydrochloride (5 g, 40.1 mmol, 1.0 equiv), potassium hydroxide (3.38 g, 60.2 mmol, 1.5 equiv), and formaldehyde (37% in water, 5.97 mL, 80.3 mmol, 2.0 equiv) in ethanol (13 mL) was stirred at reflux for 4 h. The suspension was then maintained at 55 °C while triethylamine (5.5 mL, 40.1 mmol, 1 equiv) and di-tert-butyl dicarbonate (9.2 g, 42.1 mmol, 1.05 equiv) were added successively. The reaction was stirred for 2 h at 55 °C and then allowed to cool to ambient temperature. The precipitate was filtered off, the filtrate was concentrated under reduced pressure, and was then purified by silica gel flash chromatography (1 → 3% MeOH/CH2Cl2) to afford Boc-protected tetrahydropyrimidinone SI-4 as a white solid (4.09 g, 51% yield over two steps): 1H NMR (500 MHz, CDCl3) δ 8.14 - 7.60 (m, 1H), 4.77 - 4.61 (m, 2H), 3.58 (t, J = 6.5 Hz, 2H), 2.41 (t, J = 6.5 Hz, 2H), 1.41 (s, 9H); 13C NMR (126 MHz, CDCl3) δ (a mixture oftwo rotamers) 171.5, 153.8, 153.4, 81.1, 54.8, 54.0, 40.4, 39.4, 31.4, 28.3; IR (Neat Film, NaCl) 3193, 2970, 1710, 1643, 1488, 1404, 1366, 1326, 1276, 1245, 1210, 1159, 1136, 1020, 949, 776 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C9H17N2O3 [M+H]+: 201.1234, found 201.1231.

- tert-Butyl 3-benzoyl-4-oxotetrahydropyrimidine-1(2H)-carboxylate (SI-5). To a solution of tetrahydropyrimidinone SI-4 (2.07 g, 10.4 mmol, 1.0 equiv) in THF (100 mL) at -78 °C was added n-butyllithium (2.2 M in hexanes, 4.94 mL, 10.9 mmol, 1.05 equiv) dropwise over 10 min. After stirring the solution at -78 °C for 20 min, benzoyl chloride (1.43 mL, 12.4 mmol, 1.2 equiv) was added dropwise at -78 °C. The reaction solution was stirred at -78 °C for 40 min, allowed to warm up to room temperature, and was then quenched with saturated aqueous NH4Cl (50 mL). The mixture was diluted with EtOAc (100 mL) and the aqueous phase was extracted with EtOAc (3 x 80 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure onto silica gel. The silica-loaded residue was purified by silica gel flash chromatography (30% EtOAc/hexanes) to give Bz-protected tetrahydropyrimidinone SI-5 as a white solid (2.72 g, 86% yield): 1H NMR (400 MHz, Chloroform-d) δ 7.55 (dt, J = 8.3, 1.4 Hz, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.39 (t, J = 7.6 Hz, 2H), 5.28 (s, 2H), 3.74 (t, J = 6.6 Hz, 2H), 2.68 (t, J = 6.6 Hz, 2H), 1.49 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.3, 170.7, 153.8, 135.0, 132.2, 128.4, 128.2, 81.7, 57.0, 40.4, 33.4, 28.3; IR (Neat Film, NaCl) 2978, 1698, 1480, 1450, 1408, 1367, 1304, 1266, 1239, 1141, 1015, 936, 863, 797, 700, 618 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C16H21N2O4 [M+H]+: 305.1496, found 305.1500.

- 5-Allyl 1-(tert-butyl) 3-benzoyl-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (SI-6). To a solution of diisopropylamine (224 µL, 1.59 mmol, 1.2 equiv) in THF (3 mL) at -78 °C was added n-butyllithium (2.2 M in hexanes, 664 µL, 1.46 mmol, 1.1 equiv). The solution was maintained at -78 °C for 15 min and then cannulated over 10 min into a solution of Bz-protected tetrahydropyrimidine SI-5 (404 mg, 1.33 mmol, 1.0 equiv) in THF (10 mL) at -78 °C. After stirring the solution at -78 °C for 25 min, allyl cyanoformate (156 µL, 1.46 mmol, 1.1 equiv) was added dropwise at -78 °C. The reaction mixture was maintained at -78 °C for 50 min and was then quenched with saturated aqueous NH4Cl (10 mL). The reaction mixture was diluted with EtOAc (10mL) and allowed to warm to room temperature. The aqueous phase was extracted with EtOAc (3 x 15 mL) and the combined organic phases were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded residue was purified by silica gel flash chromatography (18 → 20 → 30% EtOAc/hexanes) to give re-isolated starting material SI-5 (154 mg, 38% yield) and allyl ester SI-6 as a white crystalline solid (310 mg, 60% yield, 97% yield based on recovered starting material): 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.2 Hz, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.40 (t, J = 7.6 Hz, 2H), 5.95 (ddt, J = 17.2, 10.4, 5.9 Hz, 1H), 5.37 (dq, J = 17.2, 1.5 Hz, 1H), 5.36 (m, 1H), 5.30 (dd, J = 10.4, 1.3 Hz, 1H), 5.17 (d, J = 12.6 Hz, 1H), 4.69 (d, J = 6.0 Hz, 2H), 4.27 (ddd, J = 13.7, 5.4, 1.2 Hz, 1H), 3.85 (dd, J = 13.7, 6.2 Hz, 1H), 3.63 (t, J = 5.7 Hz, 1H), 1.49 (s, 9H); 13C NMR (101 MHz, CDCl3) 0 172.9, 167.8, 167.0, 153.4, 134.6, 132.5, 131.3, 128.7, 128.3, 119.7, 82.3, 67.0, 58.5, 50.1, 43.9, 28.3; IR (Neat Film, NaCl) 3406, 3064, 2978, 2935, 1714, 1601, 1480, 1450, 1416, 1369, 1287, 1148, 1072, 1017, 934 , 859, 796, 768, 736, 703, 626 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C20H25N2O6 [M+H]+: 389.1707, found 389.1708.

- 1-(tert-butyl) 3-benzoyl-5-methyl-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5a). To a solution of allyl ester SI-6 (100 mg, 0.26 mmol, 1.0 equiv) in acetonitrile (2.6 mL) at 0 °C was added cesium carbonate (168 mg, 0.52 mmol, 2.0 equiv). After stirring the suspension for 30 min at 0 °C, methyl iodide (48 µL, 0.77 mmol, 3.0 equiv) was added. The reaction mixture was stirred for 3 h at 0 °C and diluted with saturated aqueous NH4Cl (2 mL) and EtOAc (2 mL). The aqueous phase was extracted with EtOAc (4 x 3 mL) and the combined organic phases were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The residue was purified by silica gel flash chromatography (15% EtOAc/hexanes) to give methylated allyl ester 5a as a colorless oil (100 mg, 95% yield): 1H NMR (500 MHz, CDCl3) δ (a mixture of two rotamers) 7.71 (d, J = 15.3 Hz, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.39 (t, J = 6.8 Hz, 2H), 5.96 (ddt, J = 16.6, 10.4, 6.0 Hz, 1H), 5.48 - 5.17 (m, 4H), 4.71 (d, J = 6.1 Hz, 2H), 4.43 (m, 1H), 3.38 (m, 1H), 1.49 (s, 9H), 1.47 (s, 3H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 173.2, 170.9, 170.7, 153.2, 135.0, 132.3, 131.3, 128.4, 128.2, 119.7, 82.1, 67.1, 58.9, 58.1, 52.8, 50.6, 50.3, 28.3, 19.3; IR (Neat Film, NaCl) 3406, 3065, 2980, 2939, 1714, 1602, 1450, 1423, 1369, 1288, 1251, 1162, 1134, 1104, 1028, 984, 938, 902, 857, 804, 765, 732, 697, 657, 635 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C21H27N2O6 [M+H]+: 403.1864, found 403.1868.

- 5-allyl 1-(tert-butyl) 3-benzoyl-5-ethyl-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5b). Following the procedure described for the preparation of 5a, allyl ester SI-6 (200 mg, 0.52 mmol, 1.0 equiv) was treated with cesium carbonate (336 mg, 1.03 mmol, 2.0 equiv) and alkylated with ethyl iodide (124 µL, 1.54 mmol, 3.0 equiv) to give, after purification by silica gel flash chromatography (dry load SiO2, 15% EtOAc/hexanes), ethylated allyl ester 5b as a colorless oil (182 mg, 85% yield): 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.70 (d, J = 8.0 Hz, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 7.7 Hz, 2H), 5.95 (ddt, J = 16.6, 10.4, 6.0 Hz, 1H), 5.38 (dd, J = 17.2, 1.5 Hz, 1H), 5.31 (d, J = 10.3 Hz, 2H), 5.20 (d, J = 12.5 Hz, 1H), 4.70 (t, J = 5.5 Hz, 2H), 4.47 - 4.26 (m, 1H), 3.49 (d, J = 13.5 Hz, 1H), 1.96 (q, J = 7.5 Hz, 2H), 1.49 (s, 9H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 172.9, 170.4, 170.1, 169.8, 153.2, 135.0, 132.3, 131.2, 128.4, 128.2, 119.8, 81.9, 66.9, 57.9, 57.4, 56.7, 47.8, 47.4, 28.3, 26.4, 9.1; IR (Neat Film, NaCl) 2976, 1703, 1450, 1422, 1367, 1288, 1247, 1156, 1134, 1015, 942, 894, 802, 766, 718, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H29N2O6 [M+H]+: 417.2020, found 417.2019.

- 5-allyl 1-(tert-butyl) 3-benzoyl-5-(3-methoxy-3-oxopropyl)-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5c). To a suspension of allyl ester SI-6 (100 mg, 0.26 mmol, 1.0 equiv) and potassium carbonate (178 mg, 1.29 mmol, 5.0 equiv) in acetone (1.0 mL) at room temperature was added methyl acrylate (47 µL, 0.52 mmol, 2.0 equiv). The reaction mixture was stirred for 3.5 h at 55 °C, allowed to cool to room temperature, and filtered through a cotton plug. The filter cake was washed with acetone (3 x 1 mL) and the combined organic phases were concentrated by under reduced pressure onto silica gel. The silica-loaded residue was purified by silica gel flash chromatography (19% EtOAc/hexanes) to give pyrimidinone 5c as a colorless oil (101 mg, 83% yield): 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.70 (br s, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 7.7 Hz, 2H), 5.95 (ddt, J = 16.6, 10.3, 6.0 Hz, 1H), 5.38 (dd, J = 17.1, 1.6 Hz, 1H), 5.32 (d, J = 10.4 Hz, 1H), 5.25 (m, 2H), 4.70 (d, J = 6.0 Hz, 2H), 4.41 (m, 1H), 3.62 (s, 3H), 3.47 (m, 1H), 2.58 (ddd, J= 16.0, 9.5, 6.4 Hz, 1H), 2.46 - 2.33 (m, 1H), 2.21 (ddd, J = 10.0, 6.3, 3.2 Hz, 2H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 173.0, 172.9, 169.7, 153.1, 134.9, 132.4, 131.1, 128.4, 128.3, 120.1, 82.2, 67.2, 58.3, 57.6, 55.6, 51.9, 48.7, 48.2, 29.7, 28.3; IR (Neat Film, NaCl) 2978, 1704, 1423, 1368, 1248, 1153, 987, 803, 722, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C24H31N2O8 [M+H]+: 475.2075, found 475.2074.

- 5-allyl 1-(tert-butyl) 3-benzoyl-5-(2-chloroallyl)-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5d). To a suspension of allyl ester SI-6 (200 mg, 0.51 mmol, 1.0 equiv) and tetrabutylammonium iodide (17 mg, 0.05 mmol, 0.1 equiv), in THF (5.1 mL) at 0 °C was added NaH (60% in mineral oil, 25 mg, 0.62 mmol, 1.2 equiv). After stirring for 30 min at 0 °C, 2,3-dichloro-1-propene (95 µL, 1.02 mmol, 2 equiv) was added and the reaction mixture heated at 40 °C for 16 h. The reaction was quenched with aqueous NH4Cl (10 mL) and extracted with EtOAc (3 x 5 mL). The combined organic phases were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded residue was purified by silica gel flash chromatography (10% → 15% EtOAc/hexanes) to give 2-chloro-allyl allyl ester 5d as a colorless oil (140 mg, 59% yield): 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 8.6 Hz, 2H), 7.51 (s, 1H), 7.39 (t, J = 7.5 Hz, 2H), 6.07 - 5.90 (m, 1H), 5.71 - 5.48 (m, 1H), 5.33 (m, 4H), 5.03 (d, J = 12.5 Hz, 1H), 4.85 - 4.43 (m, 3H), 3.58 (mz, 1H), 3.32 (d, J = 15.1 Hz, 1H), 3.03 (d, J = 15f.1 Hz, 1H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 173.3, 169.9, 168.7, 153.4, 136.3, 134.9, 132.4, 131.2, 128.7, 128.2, 120.1, 118.8, 82.1, 67.7, 58.5, 57.8, 55.1, 47.7, 47.2,41.0, 28.3; IR (Neat Film, NaCl) 2978, 1703, 1632, 1478, 1450, 1423, 1368, 1289, 1246, 1137, 902, 803, 721, 695, 633 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C23H28ClN2O6 [M+H]+: 463.1630, found 463.1641.

- 5-Allyl 1-(tert-butyl) 3-benzoyl-5-benzyl-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5e). To a solution of allyl ester SI-6 (100 mg, 0.26 mmol, 1.0 equiv) in THF (2.6 mL) at room temperature was added sodium hydride (11 mg, 0.28 mmol, 1.1 equiv). After stirring for 15 min, benzyl bromide (37 µL, 0.31 mmol, 1.2 equiv) was added. The reaction mixture was maintained at room temperature for 22 h and at 55 °C for 24 h. The reaction was quenched with aqueous NH4Cl (2 mL) and diluted with EtOAc (2 mL). The aqueous phase was extracted with EtOAc (3 x 3mL) and the combined organic phases were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded residue was purified by silica gel flash chromatography (15% EtOAc/hexanes) to give benzylated allyl ester 5e as a colorless oil (94 mg, 76% yield): 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 7.6 Hz, 2H), 7.52 (t, J = 7.5 Hz, 1H), 7.39 (t, J = 7.5 Hz, 2H), 7.29 - 7.19 (m, 3H), 7.14 (dd, J = 7.4, 2.2 Hz, 2H), 5.95 (ddt, J = 16.6, 10.4, 6.0 Hz, 1H), 5.44 - 5.19 (m, 3H), 4.73 (m, 3H), 4.56 - 4.37 (m, 1H), 3.50 (d, J = 14.0 Hz, 1H), 3.33 (d, J = 13.9 Hz, 1H), 3.14 (d, J = 14.0 Hz, 1H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 173.1, 170.4, 169.3, 153.2, 135.0, 134.9, 132.4, 131.3, 130.9, 128.7, 128.7, 128.2, 127.5, 119.9, 81.9, 67.3, 58.0, 57.5, 57.3, 47.8, 47.2, 38.1, 28.3; IR (Neat Film, NaCl) 3063, 2978, 2935, 1704, 1602, 1479, 1451, 1418, 1368, 1287, 1251, 1152, 1093, 1002, 927, 902, 857, 804, 764, 727, 697, 635 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C27H31N2O6 [M+H]+: 479.2177, found 479.2180.

- 5-allyl 1-(tert-butyl) 3-benzoyl-5-(2-cyanoethyl)-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5f). To a solution of allyl ester SI-6 (100 mg, 0.26 mmol, 1.0 equiv) and acrylonitrile (34 µL, 0.52 mmol, 2.0 equiv) in acetonitrile (1.3 mL) at room temperature was added DBU (1.9 µL, 0.013 mmol, 0.05 equiv). After 22 h at room temperature, the reaction mixture was heated to 70 °C for 32 h, allowed to cool to room temperature, and treated with additional DBU (1.9 µL, 0.013 mmol, 0.05 equiv). After 2 h at 70 °C, the reaction mixture was allowed to cool to room temperature, directly concentrated onto silica gel, and purified by silica gel flash chromatography (22% EtOAc/hexanes) to give cyanoethylated pyrimidinone 5f as a colorless oil (71.5 mg, 63% yield): 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 7.7 Hz, 2H), 7.53 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.6 Hz, 2H), 5.97 (ddt, J = 16.7, 10.3, 6.1 Hz, 1H), 5.41 (dd, J = 17.4, 1.5 Hz, 1H), 5.36 (d, J = 10.5 Hz, 1H), 5.28 (m, 2H), 4.74 (m, 2H), 4.41 (d, J = 13.8 Hz, 1H), 3.49 (d, J = 13.8 Hz, 1H), 2.72 (dt, J = 16.2, 7.9 Hz, 1H), 2.50 (dt, J = 16.6, 7.8 Hz, 1H), 2.20 (t, J = 7.9 Hz, 2H), 1.49 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 173.0, 169.4, 169.0, 153.1, 134.6, 132.7, 130.8, 128.4, 128.4, 120.6, 118.9, 82.6, 67.6, 67.6, 58.8, 57.9, 55.1, 49.0, 48.4, 29.2, 28.4, 13.6; IR (Neat Film, NaCl) 2979, 2250, 1698, 1450, 1423, 1369, 1286, 1250, 1155, 1030, 939, 857, 803, 718, 696, 635 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C23H31N4O6 [M+NH4]+: 459.2238, found 459.2243.

- 5-allyl 1-(tert-butyl) 3-benzoyl-5-((benzyloxy)methyl)-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5g). Following the procedure described for the preparation of 5a, allyl ester SI-6 (200 mg, 0.52 mmol, 1.0 equiv) was treated with sodium hydride (29 mg, 0.72 mmol, 1.4 equiv) and alkylated with benzyl chloromethyl ether (127 µL, 0.93 mmol, 1.8 equiv) to give, after two rounds of purification by silica gel flash chromatography (dry load SiO2, 16 → 25% EtOAc/hexanes), BOM-alkylated allyl ester 5g as a colorless oil (57 mg, 22% yield): 1H NMR (400 MHz, CDCl3) δ 7.73 (t, J = 8.3 Hz, 2H), 7.50 (t, J = 7.9 Hz, 1H), 7.41 - 7.24 (m, 7H), 5.94 (ddt, J = 16.5, 10.3, 6.0 Hz, 1H), 5.63 (d, J = 12.2 Hz, 1H), 5.43 - 5.25 (m, 2H), 4.95 (d, J = 12.4 Hz, 1H), 4.74 (dd, J = 13.0, 5.9 Hz, 1H), 4.70 - 4.36 (m, 4H), 4.07 (d, J = 9.2 Hz, 1H), 3.96 - 3.80 (m, 1H), 3.74 (d, J = 9.3 Hz, 1H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 173.2, 169.0, 168.7, 168.3, 153.6, 153.3, 137.5, 134.9, 132.2, 131.3, 131.0, 128.6, 128.1, 128.0, 127.7, 119.9, 119.7, 81.9, 73.9, 70.0, 67.1, 58.7, 57.9, 57.0, 47.0, 46.7, 28.3; IR (Neat Film, NaCl) 2978, 2360, 1704, 1453, 1418, 1368, 1290, 1248, 1153, 1128, 1072, 1003, 904, 857, 803, 735, 697, 633 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C28H33N2O7 [M+H]+: 509.2282, found 509.2279.

- 5-allyl 1-(tert-butyl) 3-benzoyl-5-fluoro-4-oxotetrahydropyrimidine-1,5(2H)-dicarboxylate (5h). To a solution of allyl ester SI-6 (100 mg, 0.257 mmol, 1.0 equiv) in THF (2.6 mL) at room temperature was added sodium hydride (11 mg, 0.28 mmol, 1.1 equiv). After stirring for 15 min, Selectfluor (109 mg, 0.31 mmol, 1.2 equiv) was added and the reaction mixture was stirred for 1.5 h at room temperature. The reaction was quenched with aqueous NH4Cl (2 mL) and diluted with EtOAc (2 mL). The aqueous phase was extracted with EtOAc (3 x 3 mL) and the combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated by reduced pressure onto silica gel. The residue was purified by silica gel flash chromatography (4:1 hexanes/EtOAc) to give fluorinated allyl ester 5h as a colorless oil (92 mg, 0.226 mmol, 88%); 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.67 (s, 2H), 7.52 (t, J = 7.5 Hz, 1H), 7.39 (t, J = 7.6 Hz, 2H), 5.95 (ddt, J = 16.6, 10.3, 6.0 Hz, 1H), 5.63 - 5.07 (m, 4H), 4.88 - 4.77 (m, 1H), 4.75 (s, 1H), 4.43 (m, 1H), 3.99 (m, 1H), 1.49 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 172.4, 165.3, 164.4, 153.1, 133.8, 132.9, 130.5, 128.6, 128.5, 120.4, 89.3 (d, J CF = 192.9 Hz, appears as four peaks due to the presence of two rotamers and coupling with fluorine), 82.8, 67.8, 59.0, 58.4, 49.2, (d, J CF = 28.3 Hz), 48.4 (d, J CF = 27.3 Hz), 28.2; IR (Neat Film, NaCl) 2979, 2360, 1770, 1715, 1601, 1478, 1450, 1418, 1369, 1287, 1252, 1157, 1134, 1072, 1018, 907, 857, 829, 803, 764, 730, 695, 658, 633 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C20H27FN3O6 [M+NH4]+: 424.1878, found 424.1877.

- 5-allyl 1-(tert-butyl) 3-benzoyl-4-oxo-5-(prop-2-yn-1-yl)tetrahydropyrimidine-1,5(2H)-dicarboxylate (5i). To a solution of allyl ester SI-6 (200 mg, 0.51 mmol, 1.0 equiv) in THF (5 mL) at 0 °C was quickly added sodium hydride (23 mg, 0.57 mmol, 1.1 equiv). After stirring at 0 °C for 30 minutes, propargyl bromide (111 µL, 1.03 mmol, 2 equiv) was added and the reaction mixture was heated to 50 °C. After three hours, more propargyl bromide (111 µL, 1.03 mmol, 2 equiv) was added and the reaction was allowed to continue for 16 h at 50 °C. The reaction was quenched with aqueous NH4Cl (10 mL) and extracted with EtOAc (3 x 5 mL). The combined organic phases were dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure. The crude residue was purified by silica gel flash chromatography (50% CH2Cl2/hexanes - 70% CH2Cl2/hexanes - 20% EtOAc/hexanes) to afford the propargylated allyl ester 5i as a colorless oil (160 mg, 73% yield): 1H NMR (400 MHz, CDCl3) δ 7.74 (s, 2H), 7.50 (d, J = 7.4 Hz, 1H), 7.38 (t, J = 7.5 Hz, 2H), 5.96 (dd, J = 17.0, 10.6 Hz, 1H), 5.64 (dd, J = 12.4, 2.1 Hz, 1H), 5.39 (d, J = 17.2 Hz, 1H), 5.33 (d, J = 10.4 Hz, 1H), 5.16 - 4.88 (m, 1H), 4.82 - 4.32 (m, 3H), 4.01 - 3.63 (m, 1H), 3.03 (d, J = 18.2 Hz, 1H), 2.71 (dd, J = 17.0, 2.7 Hz, 1H), 2.09 (t, J= 2.6 Hz, 1H), 1.49 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 173.3, 169.6, 168.6, 153.5, 134.9, 132.4, 131.2, 130.9, 128.6, 128.2, 120.0, 82.2, 78.8, 72.6, 67.5, 58.9, 58.2, 55.2, 48.2, 28.3, 23.0; IR (Neat Film, NaCl) 3280, 2978, 1704, 1600, 1479, 1450, 1422, 1368, 1287, 1245, 1155, 1140, 1073, 1016, 929, 904, 856, 803, 765, 733 695, 656 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C23H27N2O6 [M+H]+: 427.1864, found 427.1859.
-

- In a nitrogen-filled glovebox, an oven-dried 1 dram vial was charged with Pd2(pmdba)3 (1.7 mg, 0.0015 mmol, 4 mol %), ligand (10 mol %), solvent (1 mL), and a magnetic stir bar. The vial was stirred at ambient glovebox temperature (27 °C) for 30 min and then substrate 3m (20 mg, 0.04 mmol, 1.0 equiv) was added as a solution in solvent (1.8 mL, total concentration 0.014 M). The vial was sealed with a teflon cap and heated to 40 °C. When complete consumption of the starting material was observed by thin layer chromatography, the reaction mixture was removed from the glovebox and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography to afford the oxopiperazine 4m.
- Absolute configuration for all other products has been inferred by analogy. [a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126, 15044-15045; b) J. T. Mohr, D. C. Behenna, A. M. Harned, B. M. Stoltz, Angew. Chem. Int. Ed. 2005, 44, 6924-6927; Angew. Chem. 2005, 117, 7084-7087; c) M. Seto, J. L. Roizen, B. M. Stoltz, Angew. Chem. Int. Ed. 2008, 47, 6873-6876; Angew. Chem. 2008, 120, 6979-6982; d) J. Streuff, D. E. White, S. C. Virgil, B. M. Stoltz, Nat. Chem. 2010, 2, 192-196; e) D. C. Behenna, Y. Liu, T. Yurino, J. Kim, D. E. White, S. C. Virgil, B. M. Stoltz, Nat. Chem. 2012, 4, 130-133; f) C. M. Reeves, C. Eidamshaus, J. Kim, B. M. Stoltz, Angew. Chem. Int. Ed. 2013, 52, 6718-6721; Angew. Chem. 2013, 125, 6850-6853.] Respective SFC conditions are described below under Determination of Enantiomeric Excess.
- In a nitrogen-filled glovebox, an oven-dried 1-dram vial or 20 mL scintillation vial was charged with Pd2(pmdba)3 or Pd2(dba)3 (4 mol %), (S)-(CF3)3-tBu-PHOX (10 mol %), hexane/toluene (2:1), and a magnetic stir bar. The vial was stirred at ambient glovebox temperature (27 °C) for 30 min and then the substrate (1.0 equiv) was added as a solution in hexane/toluene (2:1, total concentration 0.014 M or 0.033 M). The vial was sealed with a teflon cap and heated to 40 °C. When complete consumption of the starting material was observed by thin layer chromatography, the reaction mixture was removed from the glovebox and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography to afford the desired oxopiperazine.
-

- tert-butyl (S)-2-allyl-4-benzoyl-3-oxopiperazine-1-carboxylate (4a). Following the general procedure, allyl ester 3a (25 mg, 0.064 mmol, 1.0 equiv) in toluene (1.45 mL) was added to a solution of Pd2(dba)3 (2.3 mg, 0.0026 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (3.8 mg, 0.0064 mmol, 10 mol %) in toluene (0.5 mL). Purification by flash chromatography (20% EtOAc/hexanes) gave monosubstituted oxopiperazine 4a as a yellow oil (21 mg, 90% yield, 92% ee). 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.64 - 7.46 (m, 3H), 7.41 (m, 2H), 5.83 (ddt, J = 17.2, 10.0, 7.3 Hz, 1H), 5.22 - 5.07 (m, 2H), 4.70 (m, 1H), 4.45 - 3.99 (m, 1H), 3.99 - 3.70 (m, 2H), 3.42 (m, 1H), 2.90 - 2.52 (m, 2H), 1.50 (s, 9H); 13C NMR (101 MHz, CDCl3 ) δ (a mixture of two rotamers) 173.6, 170.6, 153.8, 135.3, 133.2, 132.2, 128.3, 128.3, 119.0, 81.3, 58.4, 44.5, 38.1, 37.2, 28.5. IR (Neat Film, NaCl) 2977, 2930, 1692, 1600, 1450, 1413, 1392, 1366, 1300, 1231, 1159, 1130, 1008, 973, 920, 856, 795, 762, 729, 696, 656 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C19H25N2O4 [M+H]+: 345.1809, found 345.1810; [α]D23.0 +49.5 (c 1.00, CHCl3); SFC conditions: 15% IPA, 2.5 mL/min, Chiralpak AD-H column, X = 210 nm, tR (min): major = 3.741, minor = 2.682.

- tert-butyl (S)-2-allyl-4-(4-methoxybenzoyl)-3-oxopiperazine-1-carboxylate (4b). Following the general procedure, anisoyl-protected allyl ester 3b (15 mg, 0.036 mmol, 1.0 equiv) in toluene (0.6 mL) was added to a solution of Pd2(dba)3 (1.7 mg, 0.0014 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.8 mg, 0.0036 mmol, 10 mol %) in toluene (0.5 mL). Purification by flash chromatography (20% EtOAc/hexanes) gave monosubstituted oxopiperazine 4b as a light yellow oil (12 mg, 92% yield, 96% ee): 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.60 (d, J = 8.9 Hz, 2H), 6.89 (d, J = 8.9 Hz, 2H), 5.85 (ddt, J = 17.1, 10.0, 7.3 Hz, 1H), 5.24 - 5.04 (m, 2H), 4.69 (m, 1H), 4.19 (m, 1H), 3.85 (m, 5H), 3.42 (m, 1H), 2.89 - 2.56 (m, 2H), 1.49 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.0, 170.6, 163.3, 153.9, 133.4, 131.2, 127.1, 118.9, 113.7, 81.3, 58.4, 55.6, 44.6, 38.3, 37.4, 28.5; IR (Neat Film, NaCl) 2976, 1694, 1605, 1512, 1462, 1416, 1366, 1315, 1257, 1234, 1170, 1130, 1003, 973, 842, 770 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C20H27N2O5 [M+H]+: 375.1914, found 375.1931; [α]D23.0 +41.7 (c 1.00, CHCl3); SFC conditions: 15% IPA, 2.5 mL/min, Chiralpak AD-H column, X = 210 nm, tR (min): major = 4.708, minor = 3.998.

- benzyl (S)-2-allyl-4-benzoyl-3-oxopiperazine-1-carboxylate (4c). Following the general procedure, Cbz-protected allyl ester 3c (20 mg, 0.047 mmol, 1.0 equiv) in toluene (0.9 mL) was added to a solution of Pd2(dba)3 (2.2 mg, 0.0019 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (3.5 mg, 0.0047 mmol, 10 mol %) in toluene (0.5 mL). Purification by flash chromatography (20% EtOAc/hexanes) gave monosubstituted oxopiperazine 4c as a colorless oil (15 mg, 83% yield, 99% ee): 1H NMR (500 MHz, CDC13) δ (a mixture of two rotamers) 7.62 - 7.46 (m, 3H), 7.44 - 7.29 (m, 7H), 5.79 (m, 1H), 5.16 (m, 4H), 4.83 (m, 1H), 4.26 (m, 1H), 3.97 (m, 1H), 3.87 (m, 1H), 3.49 (m, 1H), 2.89 - 2.53 (m, 2H); 13C NMR (126 MHz, CDCl3) δ (a mixture of two rotamers) 173.5, 170.2, 154.7, 136.0, 135.2, 132.9, 132.3, 128.8, 128.6, 128.6, 128.4, 128.3, 119.3, 68.0, 58.4, 44.3, 39.6, 38.9, 37.2, 36.9; IR (Neat Film, NaCl) 3065, 2951, 1702, 1600, 1449, 1427, 1395, 1362, 1302, 1228, 1163, 1127, 1015, 975, 922, 796, 761,730, 697, 656 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H23N2O4 [M+H]+: 379.1652, found 379.1659; [α]D23.0 +66.0 (c 1.0, CHCl3); SFC conditions: 30% IPA, 2.5 mL/min, Chiralpak IC column, X = 210 nm, tR (min): major = 4.873, minor = 6.748.

- tert-butyl (S)-4-benzoyl-2-(2-chloroallyl)-3-oxopiperazine-1-carboxylate (4d). Following the general procedure, 2-chloroallyl ester 3d (20 mg, 0.047 mmol, 1.0 equiv.) in hexanes/toluene (2:1, 1.9 mL) was added to a solution of Pd2(pmdba)3 (2.1 mg, 0.0019 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.8 mg, 0.0047 mmol, 10 mol %) in hexanes/toluene (2:1, 1.5 mL). Purification by silica gel flash chromatography (15% EtOAc/hexanes) gave oxopiperazine 4d as a light yellow oil (15 mg, 85% yield, 98% ee).1H NMR (400 MHz, CDCl3) δ (A mixture of two rotamers) 7.56 (d, J = 7.8 Hz, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.41 (t, J= 7.5 Hz, 2H), 5.30 (s, 1H), 5.24 (s, 1H), 5.06 - 4.88 (m, 1H), 4.40 - 4.06 (m, 1H), 3.94 (m, 1H), 3.84 (m, 1H), 3.55 - 3.29 (m, 1H), 2.95 (dd, J = 13.1, 7.4 Hz, 2H), 1.51 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.5, 169.8, 153.7, 137.5, 135.3, 132.3, 128.4, 128.3, 116.8, 81.9, 57.0, 44.6, 41.8, 37.7, 28.4. IR (Neat Film, NaCl) 2977, 2359, 1694, 1635, 1456, 1418, 1394, 1367, 1284, 1232, 1200, 1158, 1007, 971, 892, 856, 796, 730, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C19H24ClN2O4 [M+H]+: 379.1419, found 379.1416; [α]D 23.1 +33.8 (c 1.00, CHCl3); SFC conditions: 15% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 254 nm, tR (min): major = 4.482, minor = 3.224.

- tert-butyl (S)-2-allyl-4-benzoyl-2-methyl-3-oxopiperazine-1-carboxylate (4e). Following the general procedure, methylated allyl ester 3e (23 mg, 0.057 mmol, 1.0 equiv) in toluene (1.2 mL) was added to a solution of Pd2(pmdba)3 (2.7 mg, 0.0023 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (3.7 mg, 0.0057 mmol, 10 mol %) in toluene (0.5 mL). Purification by flash chromatography (15% EtOAc/hexanes) gave di-substituted oxopiperazine 4e as a light yellow oil (17 mg, 85% yield, 96% ee). 1H NMR (500 MHz, CDCl3) δ 7.57 - 7.46 (m, 3H), 7.40 (t, J = 7.6 Hz, 2H), 5.81 - 5.67 (m, 1H), 5.17 - 5.13 (m, 1H), 5.12 (d, J = 1.1 Hz, 1H), 4.16 - 4.02 (m, 1H), 4.03 - 3.91 (m, 1H), 3.79 (ddd, J = 12.9, 9.0, 3.0 Hz, 1H), 3.53 (ddd, J = 14.1, 9.0, 2.8 Hz, 1H), 3.11 (m, 1H), 2.84 - 2.70 (m, 1H), 1.77 (s, 3H), 1.53 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 174.3, 172.8, 135.8, 133.1, 131.8, 128.3, 127.6, 119.5, 81.2, 67.3, 44.2, 42.7, 41.0, 28.6, 25.5; IR (Neat Film, NaCl) 3076, 2977, 2934, 1692, 1641, 16001,102, 1450, 1392, 1366, 1301, 1230, 1166, 1106, 1047, 1016, 955, 922, 852, 790, 757, 727, 695 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C20H27N2O4 [M+H]+: 359.1965, found 359.1966; [α]D22.8 +6.5 (c 2.0, CHCl3); SFC conditions: 7% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): major = 7.208, minor = 4.714.

- tert-butyl (S)-2-allyl-4-(4-methoxybenzoyl)-2-methyl-3-oxopiperazine-1-carboxylate (4f). Following the general procedure, anisoyl-protected allyl ester 3f (25 mg, 0.058 mmol, 1.0 equiv) in toluene (1.3 mL) was added to a solution of Pd2(pmdba)3 (2.5 mg, 0.0023 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (3.4 mg, 0.0058 mmol, 10 mol %) in toluene (0.5 mL). Purification by flash chromatography (20% EtOAc/hexanes) gave di-substituted oxopiperazine 4f as a light pink oil (19 mg, 86% yield, 96% ee). 1H NMR (400 MHz, CDCl3) δ 7.70 - 7.53 (m, 2H), 6.99 - 6.82 (m, 2H), 5.86 - 5.67 (m, 1H), 5.23 - 5.04 (m, 2H), 4.01 (dddd, J = 33.5, 13.9, 5.8, 2.8 Hz, 2H), 3.85 (s, 3H), 3.74 (ddd, J = 12.7, 9.0, 2.8 Hz, 1H), 3.52 (ddd, J = 13.9, 9.0, 2.7 Hz, 1H), 3.20 (s, 1H), 2.80 (ddt, J = 14.0, 7.1, 1.2 Hz, 1H), 1.77 (s, 3H), 1.52 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.1, 172.4, 163.0, 153.9, 133.2, 130.6, 127.5, 119.4, 113.6, 81.2, 67.1, 55.5, 44.2, 42.8, 41.1, 28.6, 25.6; IR (Neat Film, NaCl) 3076, 2977, 2934, 1692, 1641, 16001,102, 1450, 1392, 1366, 1301, 1230, 1166, 1106, 1047, 1016, 955, 922, 852, 790, 757, 727, 695 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C21H29N2O5 [M+H]+: 389.2071, found 389.2083; [α]D22.0 +75.6 (c 2.9, CHCl3); SFC conditions: 10% IPA, 2.5 mL/min, Chiralpak AD-H column, X = 210 nm, tR (min): major = 5.370, minor = 4.278.

- tert-butyl (S)-2-allyl-4-(4-methoxybenzoyl)-2-methyl-3-oxopiperazine-1-carboxylate (4g). Following the general procedure, di-Bz-protected allyl ester 3g (10 mg, 0.025 mmol, 1.0 equiv) in toluene (1.3 mL) was added to a solution of Pd2(dba)3 (0.9 mg, 0.00098 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (1.5 mg, 0.0025 mmol, 10 mol %) in toluene (0.5 mL). Purification by flash chromatography (20% EtOAc/hexanes) gave di-substituted oxopiperazine 4f as a colorless oil (8 mg, 89% yield, 70% ee). Product identity matched previously reported characterization data. [K. M. Korch, C. Eidamshaus, D. C. Behenna, S. Nam, D. Home, B. M. Stoltz, Angew. Chem. Int. Ed. 2015, 54, 179-183]. SFC conditions: 10% MeOH, 2.5 mL/min, Chiralpak OJ-H column, X = 254 nm, tR (min): major = 5.574, minor = 6.659.

- tert-butyl (R)-2-allyl-2-benzyl-4-(4-methoxybenzoyl)-3-oxopiperazine-1-carboxylate (4h). Following the general procedure, benzylated allyl ester 3h (10 mg, 0.02 mmol, 1.0 equiv) in hexanes/toluene (2:1, 0.9 mL) was added to a solution of Pd2(pmdba)3 (0.86 mg, 0.00078 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (1.2 mg, 0.002 mmol, 10 mol %) in hexanes/toluene (2:1, 0.5 mL). Purification by flash chromatography (5% - 10% - 15% EtOAc/hexanes) gave di-substituted oxopiperazine 4h as a colorless oil (7 mg, 77% yield, 96% ee): 1H NMR (400 MHz, CDCl3) δ (A mixture of two rotamers) 7.57 (t, J = 8.5 Hz, 2H), 7.29 (d, J = 5.7 Hz, 3H), 7.22 - 7.07 (m, 2H), 6.91 (d, J = 8.3 Hz, 2H), 5.82 (ddt, J = 17.0, 9.6, 7.1 Hz, 1H), 5.29 - 5.13 (m, 2H), 3.88 (m, 4H), 3.74 - 3.45 (m, 2H), 3.31 (m, 1H), 3.16 (d, J = 12.0 Hz, 2H), 2.98 - 2.57 (m, 2H), 1.57 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (A mixture of two rotamers) 172.7, 172.3, 172.0, 163.2, 154.7, 153.4, 137.1, 136.4, 133.1, 132.6, 131.3, 130.4, 128.7, 128.5, 127.5, 127.2, 119.9, 119.7, 113.5, 82.2, 80.6, 71.7, 55.6, 43.6, 43.3, 42.9, 42.6, 41.8, 29.8, 28.9, 28.6; IR (Neat Film, NaCl) 2975, 1691, 1604, 1512, 1454, 1365,1309, 1282, 1258, 1167, 1104, 1077, 1021, 993, 925, 839, 768, 740, 704cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C27H33N2O5 [M+H]+: 465.2384, found 465.2390; [α]D 23.4 +31.0 (c 0.47, CHCl3); SFC conditions: 15% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 254 nm, tR (min): major = 7.471, minor = 5.802.

- tert-butyl (R)-2-allyl-4-benzoyl-2-((benzyloxy)methyl)-3-oxopiperazine-1-carboxylate (4i). (Following the general procedure, benzyloxy methyl ether allyl ester 3i (25 mg, 0.049 mmol, 1.0 equiv) in hexanes/toluene (2:1, 3.0 mL) was added to a solution of Pd2(pmdba)3 (2.2 mg, 0.0020 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.9 mg, 0.0049 mmol, 10 mol %) in hexanes/toluene (2:1, 0.5 mL). Purification by flash chromatography (15% EtOAc/hexanes) gave di-substituted oxopiperazine 4i as a colorless oil (20 mg, 87% yield, 92% ee): 1H NMR (500 MHz, CDCl3) δ (A mixture of two rotamers) 7.61 (dd, J = 8.2, 1.4 Hz, 2H), 7.51 - 7.43 (m, 1H), 7.38 - 7.26 (m, 7H), 5.72 (ddt, J = 17.6, 10.3, 7.4 Hz, 1H), 5.21 - 5.09 (m, 2H), 4.57 (t, J= 13.3 Hz, 3H), 4.32 - 3.63 (m, 5H), 3.25 - 2.93 (m, 1H), 2.60 (br s, 1H), 1.50 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 173.3, 172.5, 153.4, 135.6, 132.1, 131.9, 128.6, 128.3, 128.2, 127.9, 127.5, 120.0, 75.1, 74.2, 73.7, 70.6, 43.5, 38.5, 37.0, 28.6; IR (Neat Film, NaCl) 3064, 2976, 2931, 1692, 1601, 1474, 1452, 1392, 1365, 1317, 1286, 1251, 1232, 1164, 1102, 1062, 1016, 969, 924, 857, 730, 696, 671 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C27H33N2O5 [M+H]+: 465.2384, found 465.2388; [α]D23.8 -13.9 (c 0.33, CHCl3); SFC conditions: 7% IPA, 2.5 mL/min, Chiralpak OJ-H column, X = 254 nm, tR (min): major = 3.737, minor = 4.398.

- tert-butyl (S)-2-allyl-2-(2-cyanoethyl)-4-(4-methoxybenzoyl)-3-oxopiperazine-1-carboxylate (4j). Following the general procedure, α-cyanoethylated allyl ester 3j (25 mg, 0.053 mmol, 1.0 equiv) in hexanes/toluene (2:1, 2.8 mL) was added to a solution of Pd2(dba)3 (1.9 mg, 0.0021 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (3.1 mg, 0.0053 mmol, 10 mol %) in hexanes/toluene (2:1, 1.0 mL). Purification by silica gel flash chromatography (dry load SiO2, 3:1 hexanes/EtOAc) gave oxopiperazine 4j as a colorless oil (20 mg, 88% yield, 97% ee): 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H), 5.79 (ddt, J = 16.6, 10.4, 7.5 Hz, 1H), 5.24 (d, J = 1.7 Hz, 1H), 5.20 (dd, J = 9.9, 1.9 Hz, 1H), 3.98 - 3.87 (m, 2H), 3.86 (s, 3H), 3.84 - 3.71 (m, 2H), 3.27 (m, 1H), 2.82 (m, 1H), 2.69 (dd, J = 13.8, 7.4 Hz, 1H), 2.47 (dt, J = 14.1, 7.4 Hz, 1H), 2.32 (t, J = 7.2 Hz, 2H), 1.53 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.4, 171.5, 163.4, 153.6, 132.0, 131.2, 127.0, 120.6, 119.1, 113.7, 82.0, 69.0, 55.6, 43.8, 43.2, 42.0, 32.3, 28.5, 13.1; IR (Neat Film, NaCl) 2976, 2933, 2359, 2247, 1694, 1605, 1579, 1512, 1456, 1366, 1281, 1258 1168, 1113, 1020, 974, 928, 843, 768, 614 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C23H30N3O5 [M+H]+: 428.2180, found 428.2182; [α]D23.2 -4.3 (c 1.0, CHCl3); SFC conditions: 15% IPA, 2.5 mL/min, Chiralpak OD-H column, X = 210 nm, tR (min): major = 6.049, minor = 5.143.

- tert-butyl (S)-2-allyl-4-(4-methoxybenzoyl)-3-oxo-2-(3-oxobutyl)piperazine-1-carboxylate (4k). Following the general procedure, ketone 3k (25 mg, 0.051 mmol, 1.0 equiv) in hexanes/toluene (2:1, 2.2 mL) was added to a solution of Pd2(dba)3 (1.9 mg, .002 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (3.0 mg, 0.0051 mmol, 10 mol %) in hexanes/toluene (2:1, 1.5 mL). Purification by silica gel flash chromatography (dry load SiO2, 30% EtOAc/hexanes) gave ketone 4k as a pale yellow oil (17 mg, 73% yield, 97% ee); 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 8.9 Hz, 2H), 6.89 (d, J = 8.9 Hz, 2H), 5.79 (ddt, J = 14.6, 9.4, 7.5 Hz, 1H), 5.18 (dd, J = 13.8, 1.9 Hz, 2H), 3.92 - 3.81 (m, 6H), 3.80 - 3.66 (m, 1H), 3.22 (s, 1H), 2.74 (dd, J = 13.8, 7.3 Hz, 1H), 2.72, (m, 1H), 2.50 - 2.27 (m, 3H), 2.13 (s, 3H), 1.51 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 207.5, 172.6, 172.4, 163.2, 132.8, 131.0, 127.3, 120.0, 113.6, 81.6, 69.5, 55.6, 43.9, 43.2, 39.1, 32.3, 30.0, 28.6, 24.8; IR (Neat Film, NaCl) 2975, 1694, 1605, 1512, 1456, 1392, 1366, 1282, 1258, 1168, 1113, 1062, 1019, 974, 923, 841, 768 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C24H33N2O6 [M+H]+: 445.2333, found 445.2335; [α]D23.2 +25.1 (c 0.97, CHCl3); SFC conditions: 7% IPA, 2.5 mL/min, Chiralpak OD-H column, X = 235 nm, tR (min): major = 9.712, minor = 10.434.

- tert-butyl (R)-2-allyl-2-((((benzyloxy)carbonyl)amino)methyl)-4-(4-methoxybenzoyl)-3-oxopiperazine-1-carboxylate (41). Following the general procedure, aminomethyl allyl ester 3l (100 mg, 0.17 mmol, 1.0 equiv) in hexanes/toluene (2:1, 10 mL) was added to a solution of Pd2(dba)3 (6.3 mg, 0.0069 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (10 mg, 0.017 mmol, 10 mol %) in hexanes/toluene (2:1, 2 mL). Purification by silica gel flash chromatography (15% - 20% - 30% EtOAc/hexanes) gave di-substituted oxopiperazine 41 as a yellow oil (60 mg, 65% yield, 92% ee); 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.64 (t, J = 8.9 Hz, 2H), 7.44 - 7.23 (m, 5H), 6.89 (d, J= 8.5 Hz, 2H), 5.79 (dq, J = 16.8, 7.8 Hz, 1H), 5.32 - 4.93 (m, 5H), 4.31 - 3.97 (m, 1H), 3.95 - 3.64 (m, 7H), 3.64 - 3.45 (m, 1H), 3.45 - 2.87 (m, 1H), 2.64 (dd, J = 14.0, 7.3 Hz, 1H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.6, 172.3, 163.1, 156.4, 153.7, 136.6, 132.2, 131.1, 128.6, 128.4, 128.3, 127.3, 120.1, 113.6, 70.2, 67.0, 55.6, 46.9, 43.8, 43.2, 39.6, 28.6; IR (Neat Film, NaCl) 3357, 2975, 2361, 1694, 1605, 1512, 1456, 1366, 1317, 1283, 1255, 1169, 1094, 1061, 1020, 923, 841, 768, 699 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C29H36N3O7 [M+H]+: 538.2548, found 538.2543; [α]D22.8 +3.74 (c 2.0, CHCl3); SFC conditions: 15% IPA, 2.5 mL/min, Chiralpak OD-H column, λ = 280 nm, tR (min): major = 8.425, minor = 7.817.

- tert-Bbtyl (R)-2-allyl-4-benzoyl-2-(((tert-butoxycarbonyl)amino)methyl)-3-oxopiperazine-1-carboxylate (4m). Following the general procedure, allyl ester 3m (100 mg, 0.19 mmol, 1.0 equiv) in hexanes/toluene (2:1, 8.0 mL) was added to a solution of Pd2(pmdba)3 (8.5 mg, 0.0077 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (11.4 mg, 0.019 mmol, 10 mol %) in hexanes/toluene (2:1, 4.0 mL). Purification by flash chromatography (15% EtOAc/hexanes) gave di-substituted oxopiperazine 4m as a pale yellow foam (85 mg, 93% yield, 93% ee): 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.61 (d, J = 7.6 Hz, 2H), 7.51 (tt, J = 7.5, 2.1 Hz, 1H), 7.41 (t, J = 7.5 Hz, 2H), 5.91 - 5.66 (m, 1H), 5.19 (d, J = 15.6 Hz, 2H), 4.73 (s, 1H), 4.09 - 3.72 (m, 5H), 3.67 (dd, J = 14.0, 7.0 Hz, 1H), 3.43 - 2.97 (m, 1H), 2.72 - 2.51 (m, 1H), 1.54 (s, 9H), 1.43 (s, 9H); 13C NMR (101 MHz, CDCl3) 172.9, 172.8, 155.7, 153.6, 135.8, 132.3, 131.9, 128.2, 128.0, 120.1, 81.2, 79.7, 70.5, 46.5, 43.7, 43.2, 39.4, 28.6, 28.5; IR (Neat Film, NaCl): = 3374, 2977, 1694, 1504, 1454, 1392, 1366, 1317, 1286, 1231, 1165, 1094, 1060, 1014, 921, 855, 765, 729, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C25H36N3O6 [M+H]+: 474.2599, found: 474.2602; [α]D23.2 +2.7 (c 1.00, CH3Cl); SFC conditions: 10% IPA, 2.5 mL/min, Chiralpak OD-H column, λ = 254 nm, tR (min): major = 4.429, minor = 3.910.
-

- tert-butyl (R)-5-allyl-3-benzoyl-5-methyl-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6a). Following the general procedure, methylated pyrimidinone 5a (15 mg, 0.037 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd2(pmdba)3 (1.6 mg, 0.0015 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.2 mg, 0.0037 mmol, 10 mol %) in hexanes/toluene (2:1, 1.7 mL). Purification by silica gel flash chromatography (13% EtOAc/hexanes) gave α-methyl pyrimidinone 6a as a colorless oil (11 mg, 83% yield, 93% ee): 1H NMR (500 MHz, CDCl3) δ 7.57 - 7.46 (m, 3H), 7.40 (t, J = 7.6 Hz, 2H), 5.78 (dq, J = 16.9, 8.0 Hz, 1H), 5.31 (d, J = 8.4 Hz, 1H), 5.25 - 5.09 (m, 3H), 3.69 (d, J= 13.8 Hz, 1H), 3.55 (m, 1H), 2.52 (m, 1H), 2.31 (dd, J = 13.9, 8.0 Hz, 1H), 1.51 (s, 9H), 1.26 (s, 3H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 176.8, 176.5, 173.9, 153.7, 135.6, 132.5, 132.1, 128.3, 127.9, 120.0, 81.8, 59.4, 59.0, 50.6, 49.7, 45.2, 41.0, 28.4, 22.4; IR (Neat Film, NaCl) 2976, 2932, 1698, 1426, 1367, 1286, 1246, 1136, 1027, 924, 858, 802, 750, 719, 695, 635 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C20H27N2O4 [M+H]+: 359.1965, found 359.1963; [α]D23.2 -25.7 (c 1.0, CHCl3); SFC conditions: 7% IPA, 2.5 mL/min, Chiralpak OJ-H column, X = 254 nm, tR(min): major = 3.209, minor = 2.569.

- tert-butyl (R)-5-allyl-3-benzoyl-5-ethyl-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6b). Following the general procedure, ethylated allyl ester 5b (15 mg, 0.036 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd2(pmdba)3 (1.6 mg, 0.0014 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.1 mg, 0.0036 mmol, 10 mol %) in hexanes/toluene (2:1, 1.6 mL). Purification by silica gel flash chromatography (13% EtOAc/hexanes) gave α-ethyl tetrahydropyrimidinone 6b as a colorless oil (13 mg, 98% yield, 94% ee); 1H NMR (400 MHz, CDCl3) δ 7.57 - 7.45 (m, 3H), 7.39 (t, J= 7.5 Hz, 2H), 5.87 - 5.62 (m, 1H), 5.48 - 5.24 (m, 1H), 5.20 - 5.03 (m, 3H), 3.75 (dd, J = 14.0, 1.4 Hz, 1H), 3.65 - 3.52 (m, 1H), 2.50 (dd, J = 14.2, 6.7 Hz, 1H), 2.27 (d, J = 16.1 Hz, 1H), 1.79 (dq, J = 14.9, 7.5 Hz, 1H), 1.68 (dq, J = 14.6, 7.4 Hz, 1H), 1.51 (s, 9H), 0.95 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 175.8, 174.1, 153.7, 135.8, 132.9, 132.0, 128.3, 128.0, 119.7, 81.7, 59.2, 58.8, 48.6, 48.0, 47.6, 39.1, 38.6, 28.7, 28.4, 8.4; IR (Neat Film, NaCl) 2976, 2927, 1698, 1426, 1367, 1286, 1263, 1246, 1136, 1017, 923, 859, 801, 738, 695, 635 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C21H29N2O4 [M+H]+: 373.2122, found: 373.2122; [α]D22.2 -21.0 (c 1.0, CHCl3); SFC conditions: 7% IPA, 2.5 mL/min, Chiralpak OJ-H column, X = 254 nm, tR(min): major = 3.956, minor = 2.585.

- tert-butyl (R)-5-allyl-3-benzoyl-5-(3-methoxy-3-oxopropyl)-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6c). Following the general procedure, methyl ester pyrimidinone 5c (15 mg, 0.032 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd2(pmdba)3 (1.2 mg, 0.0013 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (1.9 mg, 0.0032 mmol, 10 mol %) in hexanes/toluene (2:1, 1.3 mL). Purification by silica gel flash chromatography (15% EtOAc/hexanes) gave methyl ester 6c as a colorless oil (9.1 mg, 67% yield, 95% ee): 1H NMR (400 MHz, CDCl3) δ 7.50 (dd, J = 13.1, 7.2 Hz, 3H), 7.38 (t, J= 7.5 Hz, 2H), 5.73 (dq, J = 16.6, 7.4 Hz, 1H), 5.47 - 5.22 (m, 1H), 5.22 - 5.04 (m, 3H), 3.66 (m, 5H), 2.49 (dt, J = 12.9, 6.4 Hz, 1H), 2.37 (m, 2H), 2.31 - 2.19 (m, 1H), 2.14 - 1.91 (m, 2H), 1.51 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 175.2, 174.9, 173.9, 173.3, 153.5, 135.6, 132.1, 132.0, 128.4, 127.9, 120.3, 82.0, 59.2, 58.8, 51.1, 48.7, 47.7, 39.1, 38.8, 30.3, 28.9, 28.3; IR (Neat Film, NaCl) 2978, 1738, 1698, 1428, 1368, 1286, 1247, 1147, 925, 856, 802, 764, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C23H31N2O6 [M+H]+: 431.2177, found 431.2173; [α]D 23.1 +5.5 (c 0.9, CHCl3); SFC conditions: 10% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 254 nm, tR (min): major = 5.591, minor = 6.372.

- tert-butyl (S)-5-allyl-3-benzoyl-5-(2-chloroallyl)-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6d). Following the general procedure, 2-chloropropenyl allyl ester 5d (20 mg, 0.049 mmol, 1.0 equiv) in hexanes/toluene (2:1, 3.0 mL) was added to a solution of Pd2(pmdba)3 (2.1 mg, 0.0020 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.9 mg, 0.0049 mmol, 10 mol %) in hexanes/toluene (2:1, 0.5 mL). Purification by flash chromatography (EtOAc/hexanes 15%) gave di-substituted oxopiperazine 6d as a yellow oil (17 mg, 94% yield, 94% ee); 1H NMR (400 MHz, CDCl3) δ (a mixture of two rotamers) 7.65 - 7.45 (m, 3H), 7.40 (t, J = 7.6 Hz, 2H), 5.95 - 5.76 (m, 1H), 5.76 - 5.47 (m, 1H), 5.31 (d, J = 1.3 Hz, 1H), 5.30 - 5.16 (m, 2H), 4.92 (s, 1H), 4.05 (s, 1H), 3.61 (d, J = 14.0 Hz, 1H), 3.06 (d, J = 14.8 Hz, 1H), 2.69 - 2.55 (m, 1H), 2.45 (d, J = 14.5 Hz, 2H), 1.52 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 174.4, 173.9, 153.6, 137.5, 135.6, 132.1, 132.0, 128.3, 128.1, 120.8, 118.1, 82.0, 59.1, 48.2, 47.6, 46.6, 42.8, 42.3, 41.7, 28.4; IR (Neat Film, NaCl) 2977, 1698, 1630, 1478, 1426, 1368, 1286, 1246, 1140, 902, 802, 765, 724, 695, 636 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H28ClN2O4 [M+H]+: 419.1732, found 419.1732; [α]D 22.6 +22.2 (c 1.0, CHCl3); SFC conditions: 10% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): major = 4.679, minor = 3.777.

- tert-butyl (S)-5-allyl-3-benzoyl-5-benzyl-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6e). Following the general procedure, benzylated allyl ester 5e (15 mg, 0.031 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd2(pmdba)3 (1.4 mg, 0.0013 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (1.9 mg, 0.0031 mmol, 10 mol %) in hexanes/toluene (2:1, 1.2 mL). Purification by silica gel flash chromatography (13% EtOAc/hexanes) gave benzyl tetrahydropyrimidinone 6e as a colorless oil (11.5 mg, 84% yield, 95% ee); 1H NMR (500 MHz, CDCl3) δ 7.58 - 7.49 (m, 3H), 7.42 (t, J= 7.7 Hz, 2H), 7.31 - 7.24 (m, 3H), 7.20 (d, J = 7.2 Hz, 2H), 5.86 (dddd, J = 16.8, 10.2, 7.9, 6.6 Hz, 1H), 5.48 - 5.25 (m, 1H), 5.22 (d, J = 10.0 Hz, 1H), 5.21 - 5.14 (m, 1H), 4.87 (d, J = 11.9 Hz, 1H), 3.89 - 3.72 (m, 1H), 3.55 (d, J = 13.7 Hz, 1H), 3.37 - 3.21 (m, 1H), 2.80 - 2.68 (m, 1H), 2.65 (dd, J= 14.0, 6.6 Hz, 1H), 2.32 - 2.19 (m, 1H), 1.52 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 175.4, 173.9, 153.6, 136.1, 135.6, 132.6, 132.1, 131.0, 128.5, 128.3, 128.1, 127.1, 120.2, 81.9, 58.8, 49.5, 47.2, 46.5, 41.1, 40.6, 28.4; IR (Neat Film, NaCl) 2978, 2930, 2360, 1698, 1424, 1368, 1288, 1245, 1142, 1029, 924, 856, 802, 718, 696, 636 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C26H31N2O4 [M+H]+: 435.2278, found: 435.2274; [α]D22.1 -5.6 (c 1.0, CHCl3); SFC conditions: 20% IPA, 2. mL/min, Chiralpak IC column, X = 254 nm, tR (min): major = 4.096, minor = 4.670.

- tert-butyl (R)-5-allyl-3-benzoyl-5-(2-cyanoethyl)-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6f). Following the general procedure, cyanoethylated tetrahydropyrimidinone 5f (15 mg, 0.034 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd2(pmdba)3 (1.2 mg, 0.0014 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.0 mg, 0.0034 mmol, 10 mol %) in hexanes/toluene (2:1, 1.4 mL). Purification by silica gel flash chromatography (20% EtOAc/hexanes) gave cyanoethylated tetrahydropyrimidinone 6f as a colorless oil (9.0 mg, 67% yield, 74% ee): 1H NMR (500 MHz, CDCl3) δ 7.56 - 7.49 (m, 3H), 7.43 (t, J = 7.6 Hz, 2H), 5.75 (dddd, J = 16.8, 10.2, 7.9, 6.6 Hz, 1H), 5.35 - 5.13 (m, 4H), 3.86 - 3.55 (m, 2H), 2.57 - 2.51 (m, 1H), 2.44 (dd, J = 10.0, 5.9 Hz, 2H), 2.32 (dd, J = 14.1, 8.0 Hz, 1H), 2.09 (dt, J = 14.6, 8.5 Hz, 1H), 1.96 (ddd, J = 14.3, 9.8, 6.2 Hz, 1H), 1.53 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.6, 173.7, 153.4, 135.4, 132.4, 131.2, 128.5, 127.8, 121.2, 119.2, 82.5, 59.4, 48.3, 47.7, 39.1, 31.0, 28.4, 12.6; IR (Neat Film, NaCl) 2977, 2931, 2248, 1694, 1601, 1478, 1427,1368, 1285, 1263, 1246, 1141, 1027, 926,901, 857, 802, 763, 721, 696, 636, cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H28N3O4 [M+H]+: 398.2074, found 398.2071; [α]D23.2 +10.0 (c 1.0, CHCl3); SFC conditions: 30% IPA, 2.5 mL/min, Chiralpak IC column, X = 254 nm, tR(min): major = 3.148, minor = 4.927.

- tert-butyl (R)-5-allyl-3-benzoyl-5-((benzyloxy)methyl)-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6g). Following the general procedure, allyl ester 5g (15 mg, 0.029 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.6 mL) was added to a solution of Pd2(pmdba)3 (1.3 mg, 0.0012 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (1.7 mg, 0.0029 mmol, 10 mol %) in hexanes/toluene (2:1, 0.5 mL). Purification by flash chromatography (10 - 15% EtOAc/hexanes) gave di-substituted oxopiperazine 6g as a yellow oil (13 mg, 87% yield, 87% ee): 1H NMR (400 MHz, CDCl3) δ 7.69 - 7.51 (m, 2H), 7.51 - 7.42 (m, 1H), 7.42 - 7.22 (m, 7H), 5.77 (ddt, J = 15.0, 10.3, 7.4 Hz, 1H), 5.54 - 5.49 (m, 1H), 5.28 - 5.10 (m, 2H), 5.06 (d, J = 12.1 Hz, 1H), 4.63 - 4.40 (m, 2H), 3.94 (m, 1H), 3.82 (d, J = 13.9 Hz, 1H), 3.75 (d, J = 8.9 Hz, 1H), 3.40 (d, J= 9.0 Hz, 1H), 2.42 (m, 2H), 1.50 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 174.7, 173.5, 153.6, 137.8, 135.5, 132.0, 128.6, 128.3, 128.2, 127.9, 127.7, 112.0, 81.8, 73.8, 58.8, 50.0, 46.9, 46.3, 38.6, 28.4; IR (Neat Film, NaCl) 2977, 2926, 2283, 1698, 1641, 1478, 1451, 1426, 1367, 1287, 1246, 1151, 1028, 906, 857, 801, 740, 697, 635 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C27H32N2O5 [M+H]+: 465.2384, found 465.2378; [α]D 23.4 +11.9 (c 0.67, CHCl3); SFC conditions: 20% IPA, 2.5 mL/min, Chiralpak IC column, λ = 210 nm, tR (min): major = 5.131, minor = 4.419.

- tert-butyl (S)-5-allyl-3-benzoyl-5-fluoro-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6h). Following the general procedure, fluorinated tetrahydropyrimidinone 5h (15 mg, 0.037 mmol, 1.0 equiv) in hexanes/toluene (2:1, 1.0 mL) was added to a solution of Pd2(pmdba)3 (1.6 mg, 0.0015 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.2 mg, 0.0037 mmol, 10 mol %) in hexanes/toluene (2:1, 1.6 mL). Purification by silica gel flash chromatography (15% EtOAc/hexanes) gave fluorinated tetrahydropyrimidinone 6h as a pale yellow oil (9.6 mg, 72% yield, 92% ee); 1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 8.3, 1.3 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.8 Hz, 2H), 5.82 (ddt, J = 14.9, 9.6, 7.2 Hz, 1H), 5.66 - 5.34 (m, 1H), 5.29 (s, 1H), 5.25 (d, J = 5.3 Hz, 1H), 5.24 - 5.13 (m, 1H), 4.13 - 3.72 (m, 2H), 2.81 (td, J = 14.3, 6.9 Hz, 1H), 2.67 (ddd, J = 22.4, 14.5, 7.6 Hz, 1H), 1.51 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 172.5, 168.23 (d, JCF = 23.6 Hz), 153.4, 134.3, 132.7, 129.7, 129.6, 128.5, 128.5, 128.3, 128.0, 121.4, 92.2 (d, JCF = 197.0 Hz, appears as four peaks due to the presence of two rotamers and coupling with fluorine), 82.4, 57.7, 49.4 (appears as four poorly resolved peaks due to the presence of two rotamers and coupling with fluorine), 38.2 (d, JCF = 22.8 Hz), 28.3; IR (Neat Film, NaCl) 2978, 1710, 1416, 1368, 1286, 1245, 1158, 1137, 906, 858, 801, 749, 723, 694, 662 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C19H24N2O4 [M+H]+: 363.1715, found: 363.1713; [α]D21.8 -28.6 (c 0.96, CHCl3); SFC conditions: 10% IPA, 2.5 mL/min, Chiralpak IC column, X = 254 nm, tR (min): major = 6.226, minor = 5.041.

- tert-butyl (S)-5-allyl-3-benzoyl-4-oxo-5-(prop-2-yn-1-yl)tetrahydropyrimidine-1(2H)-carboxylate (6i). Following the general procedure, propargylated allyl ester 5i (20 mg, 0.047 mmol, 1.0 equiv) in hexanes/toluene (2:1, 2.8 mL) was added to a solution of Pd2(pmdba)3 (2.1 mg, 0.0019 mmol, 4 mol %) and (S)-(CF3)3-tBu-PHOX (2.8 mg, 0.0047 mmol, 10 mol %) in hexanes/toluene (2:1, 0.5 mL). Purification by flash chromatography (EtOAc/hexanes 5% - 10% - 15%) gave propargyl tetrahydropyrimidinone 6i as a yellow oil (15 mg, 83% yield, 90% ee): 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.50 (t, J = 7.4 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 5.89 - 5.68 (m, 1H), 5.42 (s, 1H), 5.31 - 4.98 (m, 3H), 4.15 - 3.57 (m, 2H), 2.61 (dd, J = 16.9, 2.7 Hz, 2H), 2.42 (dd, J = 16.9, 2.7 Hz, 2H), 2.11 (t, J = 2.6 Hz, 1H), 1.54 (s, 9H); 13C NMR (101 MHz, CDCl3) δ (a mixture of two rotamers) 174.3, 173.7, 153.6, 135.4, 132.2, 131.8, 128.3, 128.2, 120.6, 82.0, 79.6, 72.4, 59.3, 58.9, 48.2, 47.8, 40.1, 39.6, 28.4, 25.1; IR (Neat Film, NaCl) 3271, 2978, 2930, 1698, 1478, 1450, 1425, 1368, 1284, 1247, 1139, 1028, 927, 854, 802, 764, 720, 695, 635 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H27N2O4 [M+H]+: 383.1965, found 383.1973; [α]D23.0 +22.1 (c 0.47, CHCl3); SFC conditions: 10% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): major = 4.769, minor = 4.399.
-

- tert-butyl (S)-5-allyl-3-benzoyl-5-benzyl-4-oxotetrahydropyrimidine-1(2H)-carboxylate (6e). In a nitrogen-filled glovebox, a 250 mL schlenk flask was charged with Pd2(pmdba)3 (69 mg, 0.063 mmol, 4 mol %), (S)-(CF3)3-tBu-PHOX (99 mg, 0.17 mmol, 8 mol %), hexane/toluene (2:1, 50 mL), and a magnetic stir bar. The flask was stirred at ambient glovebox temperature (27 °C) for 30 min and then 5e (lg, 2.1 mmol, 1.0 equiv) was added as a solution in hexane/toluene (2:1, 100 mL, total concentration 0.014M). The flask was sealed with a Kontes valve, removed from the glovebox, and heated to 40 °C for 16 h. The solution was concentrated under reduced pressure and purified by silica gel flash chromatography (15% EtOAc/hexanes) to give benzyl tetrahydropyrimidinone 6e as a yellow oil (780 mg, 87% yield, 95% ee); spectroscopic data vide supra.
-

- tert-butyl (R)-((2-allyl-4-benzoyl-3-oxopiperazin-2-yl)methyl)carbamate (8). Trifluoroacetic acid (114 µL, 1.5 mmol, 20 equiv) was added dropwise to a solution of methylcarbamate oxopiperazine 4m (35 mg, 0.07 mmol, 1.0 equiv) in CH2Cl2 (0.74 mL) at 0 °C. The reaction mixture was allowed to warm up to room temperature, stirred for 3 h and concentrated under reduced pressure. The residue was repeatedly taken up in CH2Cl2 (1.0 mL) and concentrated, four times. The crude residue was then purified by silica gel flash chromatography (10% MeOH/CH2Cl2) to yield deprotected oxopiperazine 8 as a pale yellow foam (27 mg, 73% yield); 1H NMR (400 MHz, CDCl3) δ 7.48 (t, J= 8.0 Hz, 3H), 7.37 (t, J = 7.4 Hz, 2H), 5.66 (dd, J = 16.0, 8.8 Hz, 1H), 5.30 (d, J = 9.8 Hz, 1H), 5.25 (d, J = 16.8 Hz, 1H), 3.99 - 3.79 (m, 1H), 3.73 - 3.55 (m, 1H), 3.44 - 3.16 (m, 2H), 3.05 (d, J = 13.2 Hz, 1H), 2.89 (d, J = 11.6 Hz, 1H), 2.76 (dd, J= 14.0, 7.0 Hz, 1H), 2.45 (dd, J= 14.2, 7.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 173.7, 172.4, 135.5, 132.2, 129.7, 128.4, 128.0, 122.5, 62.0, 47.0, 43.5, 39.7, 38.1; IR (Neat Film, NaCl) 2976, 1682, 1470, 1282, 1203, 1135, 926, 836, 799, 722, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C15H20N3O4 [M+H]+: 274.1550, found 274.1555; [α]D22.3 +15.1 (c 1.0, CHCl3).

- tert-butyl (R)-2-allyl-2-(((tert-butoxycarbonyl)amino)methyl)-3-oxopiperazine-1-carboxylate (9). LiOH monohydrate (2.5 mg, 0.06 mmol, 1.4 equiv) was added in one portion to a solution of methylcarbamate oxopiperazine 4m (20 mg, 0.04 mmol, 1.0 equiv) in methanol/water (1:1, 1.8 mL) at room temperature. The reaction mixture was stirred for 1 h, diluted with EtOAc (2 mL) and washed with saturated aqueous NaHCO3 (2 mL). The aqueous phase was extracted with EtOAc (3x3 mL) and the combined organic phases were washed with brine (3 mL), dried over anhydrous Na2SO4, decanted, and concentrated under reduced pressure onto silica gel. The silica-loaded residue was purified by silica gel flash chromatography (hexanes/EtOAc 1:1) to yield lactam 9 as a white foam (14 mg, 92% yield): 1H NMR (400 MHz, CDCl3) δ 6.96 (s, 1H), 5.72 (ddt, J = 17.2, 10.2, 7.4 Hz, 1H), 5.16 - 5.04 (m, 2H), 4.88 (t, J = 6.9 Hz, 1H), 4.26 - 3.91 (m, 1H), 3.82 (s, 1H), 3.60 (dd, J = 14.0, 5.9 Hz, 1H), 3.50 (s, 1H), 3.39 - 2.83 (m, 3H), 2.62 (s, 1H), 1.52 (s, 9H), 1.41 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.7, 155.7, 153.9, 132.7, 119.2, 81.2, 79.3, 69.1, 46.0, 43.1, 40.9, 38.9, 28.6, 28.5; IR (Neat Film, NaCl) 3337, 2977, 2360, 1698, 1520, 1367, 1243, 1168, 1085, 1058, 919, 866, 768, 733 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C18H32N3O5 [M+H]+: 370.2336, found 370.2337; [α]D 21.9 -5.7 (c 1.0, CHCl3).

- tert-butyl (S)-2-allyl-2-(((tert-butoxycarbonyl)amino)methyl)piperazine-1-carboxylate (10). To a solution of lactam 9 (50 mg, 0.14 mmol, 1 equiv) in THF (1.4 mL) at 0 °C was quickly added LAH (8 mg, 0.2 mmol, 1.5 equiv) in one portion. The mixture was stirred at room temperature and three portions of LAH (8 mg, 0.2 mmol, 1.5 equiv) were added over four hours until all starting material was consumed as determined by TLC analysis. The reaction mixture was then cooled to 0 °C and diluted with Et2O. H2O (40 µL), 15% aqueous NaOH (40 µL), and H2O (120 µL) were added successively at 0 °C. The mixture was stirred for 5 minutes at room temperature and then MgSO4 was added. The mixture was stirred for another 5 minutes at room temperature and then filtered over a pad of celite, rinsing with EtOAc. The solvent was concentrated under reduced pressure and the crude residue was purified by silica gel flash chromatography (MeOH/CH2Cl2, 1 - 2 - 4%) to afford piperazine 10 as a colorless oil (30 mg, 63% yield); 1H NMR (400 MHz, CDCl3) δ 5.84 - 5.66 (m, 1H), 5.28 - 5.01 (m, 3H), 3.75 (dd, J = 14.1, 5.6 Hz, 1H), 3.62 (dt, J = 13.6, 4.2 Hz, 1H), 3.35 (dd, J = 14.1, 7.3 Hz, 1H), 3.24 (ddd, J = 13.6, 9.8, 3.8 Hz, 1H), 3.02 - 2.91 (m, 1H), 2.85 (t, J = 11.1 Hz, 2H), 2.79 - 2.63 (m, 2H), 2.57 - 2.26 (m, 2H), 1.46 (s, 9H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 156.8, 156.0, 133.1, 119.1, 80.6, 79.6, 59.9, 50.8, 46.2, 45.1, 42.3, 38.1, 28.6, 28.5; IR (Neat Film, NaCl) 3789, 3662, 3451, 3341, 3074, 2976, 2930, 2284, 1693, 1641, 1502, 1453, 1391, 1365, 1298, 1249, 1169, 1085, 996, 914, 859, 771 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C18H34N3O4 [M+H]+: 356.2544, found 356.2549; [α]D22.8 +5.3 (c 0.67, CHCl3).

- (S)-1-benzoyl-3-methyl-3-propylpiperazin-2-one (11). Piperazinone 4e (80 mg, 0.22 mmol, 1 equiv) was dissolved in MeOH (2.2 mL). The reaction flask was purged with Argon before adding Pd/C (10%, 24 mg, 0.022 mmol, 0.1 equiv). The flask was evacuated and filled with H2 three times, and then sparged with H2 for 5 minutes. The reaction was stirred at room temperature for 6 hours before being filtered through a pad of silica gel while rinsing with EtOAc. The crude hydrogenated product was then dissolved in CH2Cl2 (2.2 mL) and TFA (171 µL, 2.23 mmol, 10 equiv) was added. The reaction was stirred for 16 hours and then quenched with saturated aqueous NaHCO3 (5 mL). The solution was extracted with EtOAc (3x5 mL), dried over Na2SO4, decanted, and concentrated under reduced pressure. The crude piperazinone was purified by silica gel flash chromatography (2.5 - 5% MeOH/CH2Cl2) to afford the desired product 11 (55 mg, 94% yield over two steps): 1H NMR (400 MHz, CDCl3) δ 7.58 - 7.50 (m, 2H), 7.54 - 7.46 (m, 1H), 7.46 - 7.36 (m, 2H), 3.92 (ddd, J = 12.6, 5.4, 4.6 Hz, 1H), 3.81 (ddd, J = 12.5, 6.7, 5.4 Hz, 1H), 3.32 - 3.18 (m, 2H), 1.92 (ddd, J = 13.6, 12.1, 4.7 Hz, 1H), 1.62 (ddd, J = 13.6, 12.2, 4.5 Hz, 1H), 1.42 (s, 3H), 1.54 - 1.23 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 176.7, 174.7, 136.4, 131.6, 128.3, 127.5, 61.2, 48.2, 41.2, 38.7, 25.0, 16.9, 14.5; IR (Neat Film, NaCl) 3331, 2960, 2872, 1681, 1600, 1448, 1378, 1284, 1202, 1176, 1152, 1112, 966, 794, 726, 694, 669 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C15H21N2O2 [M+H]+: 261.1598, found 261.1596; [α]D22.6 -59.6 (c 1.35, CHCl3).

- (R)-4-benzoyl-6-propyl-1,4-diazabicyclo[4.2.0]octane-5,8-dione (12). To a 10 mL roundbottom flask was added Pd(OPiv)2 (3 mg, 0.0096 mmol, 0.1 equiv), AgOPiv7 (60 mg, 0.29 mmol, 3 equiv), Xantphos (6 mg, 0.0096 mmol, 0.1 equiv), and 1,4-benzoquinone (21 mg, 0.19 mmol, 2 equiv). A solution of piperazine 11 (25 mg, 0.096 mmol, 1 equiv) in toluene (1 mL) was then added and the flask was evacuated and filled with carbon monoxide three times. The flask was then stirred at 80 °C for 16 hours. The reaction mixture was allowed to cool to room temperature, diluted with EtOAc, and filtered through celite while rinsing with additional EtOAc. The solvent was concentrated under reduced pressure onto silica gel and then purified by silica gel flash chromatography (2:1 hexanes/EtOAc) to provide the β-lactam 12 and as a light yellow oil (15 mg, 55% yield): 1H NMR (400 MHz, CDCl3) δ (5:1 ratio of desired product and an inseparable isomer resulting from insertion into another β-C-H) 7.56 - 7.51 (m, 3H), 7.46 - 7.40 (m, 2H), 4.51 (ddd, J= 14.0, 6.1, 2.9 Hz, 1H), 3.94 (ddt, J = 12.3, 10.8, 5.7 Hz, 1H), 3.78 - 3.67 (m, 1H), 3.40 (dddd, J = 12.3, 4.8, 2.8, 1.5 Hz, 1H), 3.27 - 3.10 (m, 2H), 1.98 (ddd, J= 14.2, 11.2, 5.5 Hz, 1H), 1.87 (ddd, J = 14.2, 11.0, 5.7 Hz, 1H), 1.59 - 1.40 (m, 2H), 1.00 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 174.2, 173.2, 169.1, 135.1, 132.6, 128.6, 128.3, 59.7, 46.7, 41.5, 39.7, 37.8, 17.7, 14.3; IR (Neat Film, NaCl) 3374, 2961, 1760, 1688, 1600, 1505, 1449, 1350, 1318, 1279, 1228, 1183, 1151, 1110, 963, 936, 796, 726, 694, 662 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C16H19N2O3 [M+H]+: 287.1390, found 287.1385; [α]D23.1 -12.8 (c 0.47, CHCl3).

- (S)-2-benzyl-2-(((tert-butoxycarbonyl)amino)methyl)pent-4-enoic acid (13). To a solution of benzyl tetrahydropyrimidinone 6d (200 mg, 0.46 mmol, 1 equiv) in methylene chloride (4.6 mL) was added TFA (352 µL, 4.6 mmol, 10 equiv) dropwise at room temperature. The solution was stirred for 24 hours at room temperature and then quenched with aqueous NaHCO3 (10 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (4 x 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was dissolved in MeOH/H2O (1:1, 5 mL) and LiOH monohydrate (290 mg, 6.9 mmol, 15 equiv) was added. The reaction mixture was heated to 80 °C for 60 hours and then allowed to cool to room temperature. Then, NEt3 (77 µL, 0.55 mmol, 1.2 equiv) and Boc2O (110 mg, 0.51 mmol, 1.1 equiv) were added successively at room temperature. The reaction was stirred for 1 hour at room temperature and then acidified with 1 M HCl (4 mL). The solution was extracted with EtOAc (3 x 5 mL). The combined organic layers were dried over Na2SO4. The crude Boc-protected fl-amino acid was taken up in DMF (2.3 mL). K2CO3 (95 mg, 0.69 mmol, 1.5 equiv) and BnBr (66 µL, 0.55 mmol, 1.2 equiv) were added at room temperature. The reaction was stirred at room temperature for 1 hour and then quenched with saturated aqueous NH4Cl. The solution was extracted with EtOAc (3 x 5 mL) and the combined organic layers were concentrated under reduced pressure and placed under high vacuum until trace DMF had evaporated. The residue was taken up in CH2Cl2, concentrated under reduced pressure onto silica gel and purified by silica gel flash chromatography (5 → 10% EtOAc/hexanes) to afford protected β2,2-amino acid 13 as a white solid (80 mg, 40% yield): 1H NMR (400 MHz, CDCl3) δ 7.42 - 7.27 (m, 5H), 7.20 (dd, J = 4.9, 1.9 Hz, 3H), 7.10 - 6.93 (m, 2H), 5.92 - 5.75 (m, 1H), 5.18 - 5.04 (m, 4H), 4.96 - 4.81 (m, 1H), 3.34 (qd, J = 14.0, 6.5 Hz, 2H), 2.98 (d, J = 13.8 Hz, 1H), 2.86 (d, J = 13.8 Hz, 1H), 2.49 (ddt, J = 14.2, 6.7, 1.4 Hz, 1H), 2.31 (ddt, J = 14.2, 7.9, 1.1 Hz, 1H), 1.43 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 175.1, 156.1, 136.6, 135.6, 133.4, 130.2, 128.8, 128.6, 128.5, 128.4, 126.9, 119.2, 79.4, 66.8, 51.6, 44.1, 41.0, 38.8, 28.5; IR (Neat Film, NaCl) 3452, 3065, 3031, 2977, 2930, 1721, 1640, 1604, 1503, 1454, 1391, 1365, 1245, 1169, 1094, 1030z, 994, 917, 859, 776, 741, 700 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C25H32NO4 [M+H]+: 410.2326, found 410.2324; [α]D23.4 +3.47 (c 1.0, CHCl3).
- Racemic products were synthesized according to the general procedure, using achiral GlyPHOX ligand instead of (S)-(CF3)3-tBu-PHOX. [(a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126, 15044-15045. (b) K. Tani, D. C. Behenna, R. M. McFadden, B. M. Stoltz, Org. Lett. 2007, 9, 2529- 2531. (c) M. R. Krout, J. T. Mohr, B. M. Stoltz, Org. Synth. 2009, 86, 181-193.]
Entry Product Assay Conditions Retention time of major isomer (min) Retention time of minor isomer (min) %ee 1 
SFC Chiralpak AD-H 15% iPrOH isocratic, 2.5 mL/min 3.741 2.682 92% 2 
SFC Chiralpak AD-H 15% iPrOH isocratic, 2.5 mL/min 4.708 3.998 96% 3 
SFC Chiralpak IC 30% iPrOH isocratic, 2.5 mL/min 4.873 6.748 99% 4 
SFC Chiralpak AD-H 15% iPrOH isocratic, 2.5 mL/min 4.482 3.224 98% 5 
SFC Chiralpak AD-H 7% iPrOH isocratic, 2.5 mL/min 7.295 4.714 96% 6 
SFC Chiralpak OJ-H 10% MeOH isocratic, 2.5 mL/min 5.574 6.659 70% 7 
SFC Chiralpak AD-H 10% iPrOH isocratic, 2.5 mL/min 5.370 4.278 96% 8 
SFC Chiralpak AD-H 15% iPrOH isocratic, 2.5 mL/min 7.471 5.802 96% 9 
SFC Chiralpak OJ-H 7% iPrOH isocratic, 2.5 mL/min 3.737 4.398 92% 10 
SFC Chiralpak OD-H 10% iPrOH isocratic, 2.5 mL/min 4.429 3.910 93% 11 
SFC Chiralpak OD-H 15% MeOH isocratic, 2.5 mL/min 8.425 7.817 92% 12 
SFC Chiralpak OD-H 15% iPrOH isocratic, 2.5 mL/min 6.049 5.143 97% 13 
SFC Chiralpak OD-H 7% iPrOH isocratic, 2.5 mL/min 9.712 10.434 97% - We began by examining conditions for the asymmetric allylic alkylation of diazepanone 3a (R2 = Bn), utilizing Pd2(pmdba)3 as a palladium source and (S)-(CF3)3-t-BuPHOX as a chiral ligand (Table 38). Generally, polar solvents led to only modest ee of the enantioenriched product 4a (entries 1-3). An ee of 86% was obtained for compound 4a in 2:1 hexanes/toluene (entry 6). Finally, an even more nonpolar solvent, methylcyclohexane [Can. J. Chem. 1984, 62, 2560-2565], further enhanced enantioselectivity, yielding 4a in 89% ee (entry 9). In the course of examining reaction conditions, we also discovered that use of the highly electron-deficient ligand (S)-Ty-PHOX [Chem. Sci. 2019, 10, 5996-6000] resulted in a drop in ee (entry 8). This is in contrast with previous results indicating that more electron-deficient ligands often give higher enantioselectivity in related systems. [Nature Chem. 2012, 4, 130-133; Chem. Eur. J. 2013, 19, 4414-4418; Angew. Chem. Int. Ed. 2015, 54, 179-183; Chem. Sci. 2019, 10, 788-792] Using a more electron-rich ligand, (S)-t-BuPHOX, also decreased the ee of the product (entry 11).
Table 38 Optimization of reaction conditionsa 
entry solvent ligand ee b (%) 1 THF (S)-(CF3)3-t-BuPHOX 20 2 1,4-dioxane (S)-(CF3)3-t-BuPHOX 38 3 MTBE (S)-(CF3)3-t-BuPHOX 70 4 toluene (S)-(CF3)3-t-BuPHOX 66 5 2:1 hexanes/benzene (S)-(CF3)3-t-BuPHOX 84 6 2:1 hexanes/toluene (S)-(CF3)3-t-BuPHOX 86 7 cyclohexane (S)-(CF3)3-t-BuPHOX 88 8 cyclohexane (S)-Ty-PHOX 80 9 MeCy (S)-(CF3)3-t-BuPHOX 89 10 2:1 MeCy/toluene (S)-(CF3)3-t-BuPHOX 86 11 MeCy (S)-t-BuPHOX 50 a Screening was performed on a 0.02 mmol scale. b Values determined by chiral SFC analysis. 
- Firstly, the effect of the electronics of the lactam protecting group on the reaction outcome was investigated. Switching from a benzoyl group (4b) to a more electron-poor p-CF3-benzoyl group (4c) resulted in a slight increase in enantioselectivity. The use of an electron-rich p-anisoyl group delivered product 4d in 94% ee. It is worth noting that use of the p-anisoyl protecting group was not beneficial to enantioselectivity in all cases (4a/4e, 4g/4h), and was often on par with the unsubstituted benzoyl group. A variety of functional groups at the quaternary carbon were tolerated, including groups bearing an alkenyl chloride (4f) and a tert-butyl carbamate (4l). This method also proved reliable for the formation of tertiary alkyl fluorides (4g, 4h). The propargyl lactam 4i was obtained in low yield. Additionally, this method allowed for the preparation of benzoyl lactam 4e on a 1 mmol scale, albeit in somewhat diminished yield and ee.

- Diazepanone 4a was subjected to selective debenzoylation under basic conditions, followed by reduction with LiAlH4 to yield diazepane 5 bearing a free secondary amine. Then, nucleophilic aromatic substitution of aryl bromide 6 with 5 [Mangion, I. K.; Sherry, B. D.; Yin, J.; Fleitz, F. J. Enantioselective Synthesis of a Dual Orexin Receptor Antagonist. Org. Lett. 2012, 14, 3458-3461], followed by Boc deprotection, with in situ generated HCl, furnished secondary amine 7. A final coupling with the benzoyl chloride derived from carboxylic acid 8 [Mangion, I. K.; Sherry, B. D.; Yin, J.; Fleitz, F. J. Enantioselective Synthesis of a Dual Orexin Receptor Antagonist. Org. Lett. 2012, 14, 3458-3461] in the same pot provided target compound 9, an analogue of the drug suvorexant bearing an all-carbon quaternary stereocenter.

- Unless otherwise stated, reactions were performed in flame-dried glassware under an argon or nitrogen atmosphere using dry, deoxygenated solvents. Solvents were dried by passage through an activated alumina column under argon. Reaction progress was monitored by thin-layer chromatography (TLC) or Agilent 1290 UHPLC-MS. TLC was performed using E. Merck silica gel 60 F254 precoated glass plates (0.25 mm) and visualized by UV fluorescence quenching or KMnO4 staining. Silicycle SiliaFlash® P60 Academic Silica gel (particle size 40-63 nm) was used for flash chromatography. 1H NMR spectra were recorded on Varian Inova 500 MHz, Varian 400 MHz, and Bruker 400 MHz spectrometers and are reported relative to residual CHCl3 (δ 7.26 ppm). 13C NMR spectra were recorded on a Varian Inova 500 MHz spectrometer (125 MHz), a Varian 400 MHz spectrometer (100 MHz), and Bruker 400 MHz spectrometers (100 MHz) and are reported relative to CHCl3 (δ 77.16 ppm). Data for 1H NMR are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sept = septuplet, m = multiplet, br s = broad singlet, br d = broad doublet. Data for 13C NMR are reported in terms of chemical shifts (δ ppm) Some reported spectra include minor solvent impurities of water (δ 1.56ppm), ethyl acetate (δ 4.12, 2.05, 1.26 ppm), methylene chloride (δ 5.30 ppm), acetone (δ 2.17 ppm), grease (δ 1.26, 0.86 ppm), and/or silicon grease (δ 0.07 ppm), which do not impact product assignments. Most NMR spectra are complicated by rotational isomerism about amide bonds. This behavior is illustrated by variable-temperature NMR spectra of compound 4e in DMSO (p. S102). IR spectra were obtained by use of a Perkin Elmer Spectrum BXII spectrometer or Nicolet 6700 FTIR spectrometer using thin films deposited on NaCl plates and reported in frequency of absorption (cm-1). Optical rotations were measured with a Jasco P-2000 polarimeter operating on the sodium D-line (589 nm), using a 100 mm path-length cell. Analytical SFC was performed with a Mettler SFC supercritical CO2 analytical chromatography system utilizing Chiralpak (AD-H, AS-H or IC) or Chiralcel (OD-H, OJ-H, or OB-H) columns (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd. High resolution mass spectra (HRMS) were obtained from Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI+), atmospheric pressure chemical ionization (APCI+), or mixed ionization mode (MM: ESI-APCI+). Absolute stereochemistry is assigned by analogy to previous results by our group. [J. Am. Chem. Soc. 2004, 126, 15044-15045; Angew. Chem. Int. Ed. 2005, 44, 6924-6927; Angew. Chem. Int. Ed. 2008, 47, 6873-6876; Nat. Chem. 2010, 2, 192-196; Nat. Chem. 2012, 4, 130-133; Angew. Chem. Int. Ed. 2013, 52, 6718-6721.]
- Reagents were purchased from commercial sources and used as received unless otherwise stated. Ligands (S)-(CF3)3-t-BuPHOX and (S)-Ty-PHOX were prepared according to literature procedures. [Tetrahedron Lett. 2010, 51, 5550-5554; Chem. Sci. 2019, 10, 5996-6000]
List of Abbreviations: ee - enantiomeric excess, SFC - supercritical fluid chromatography, TLC - thin-layer chromatography, IPA - isopropanol, An = 4-anisoyl, MeCy = methylcyclohexane -

- In a N2 filled glovebox, Pd2(dba)3 (4 mol %) or Pd2(pmdba)3 (4 mol %) and (S)-(CF3)3-t-BuPHOX (10 mol %) were suspended in methylcyclohexane (2 mL) in a 20 mL glass vial. After stirring for 20 minutes at 25 °C, the appropriate diazepanone (1.0 equiv) and methylcyclohexane (5.2 mL, total substrate concentration 0.014 M) were added to the pre-stirred catalyst solution. The vial was then sealed and heated to 40 °C. After full consumption of starting material, as monitored by TLC, the reaction mixture was exposed to air. The crude reaction mixture was loaded directly onto a flash column and the product was isolated by silica gel flash chromatography.

- tert-butyl (S)-6-allyl-4-benzoyl-6-benzyl-5-oxo-1,4-diazepane-1-carboxylate (4a). Prepared according to the general procedure with allyl ester 3a (51.2 mg, 0.104 mmol, 1.0 equiv), Pd2(pmdba)3 (4.4 mg, 0.004 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (5.9 mg, 0.01 mmol, 10 mol %). Purified by silica gel flash chromatography (15% EtOAc/hexanes) to provide benzyl diazepanone 4a as a colorless oil (43.4 mg, 0.0967 mmol, 93% yield, 90% ee); 1H NMR (400 MHz, CDCl3) δ 7.56 - 7.40 (m, 3H), 7.40 - 7.32 (m, 2H), 7.32 - 7.19 (m, 3H), 7.19 - 7.00 (m, 2H), 5.88 (br s, 1H), 5.26 - 5.07 (m, 2H), 4.26 - 4.03 (m, 1H), 3.94 (d, J = 15.4 Hz, 1H), 3.73 (d, J = 42.2 Hz, 1H), 3.54 (d, J = 15.3 Hz, 1H), 3.40 (s, 2H), 3.09 (dd, J = 61.0, 13.7 Hz, 1H), 2.94 - 2.34 (m, 3H), 1.48 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 179.2, 174.8, 156.0, 155.4, 136.6, 136.4, 133.0, 131.5, 130.8, 128.6, 128.4, 127.8, 127.1, 120.0, 119.7, 80.8, 54.3, 53.9, 49.1, 47.4, 46.9, 42.5, 42.0, 41.5, 40.3, 28.5; IR (Neat Film, NaCl) 3062, 2975, 2928, 1693, 1682, 1601, 1452, 1415, 1392, 1365, 1322, 1283, 1246, 1156, 1044, 978, 917, 865, 728, 697 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C27H33N2O4 [M+H]+: 449.2435, found 449.2429; [α]D 22.4+14.19 (c 0.66, CHCl3); SFC conditions: 20% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): major = 3.51, minor = 2.71.

- tert-butyl (R)-6-allyl-4-benzoyl-6-methyl-5-oxo-1,4-diazepane-1-carboxylate (4b). Prepared according to the general procedure with allyl ester 3b (39.0 mg, 0.0937 mmol, 1.0 equiv), Pd2(pmdba)3 (4.4 mg, 0.004 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (5.9 mg, 0.01 mmol, 10 mol %). Purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide methyl diazepanone 4b as a colorless, waxy solid (32.3 mg, 0.868 mmol, 93% yield, 90% ee); 1H NMR (400 MHz, CDCl3) δ 7.54 - 7.43 (m, 3H), 7.43 - 7.32 (m, 2H), 5.74 (ddt, J = 17.1, 9.9, 7.4 Hz, 1H), 5.19 - 5.06 (m, 2H), 4.30 - 3.89 (m, 3H), 3.85 - 3.69 (m, 1H), 3.66 - 3.34 (m, 2H), 2.63 - 2.20 (m, 2H), 1.50 (s, 9H), 1.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 180.8, 174.5, 155.4, 155.0, 136.3, 132.9, 131.4, 128.3, 127.5, 119.5, 80.7, 50.9, 49.9, 47.2, 46.5, 42.5, 42.1, 41.8, 28.4, 23.5, 23.1; IR (Neat Film, NaCl) 2976, 2933, 1694, 1450, 1418, 1392, 1366, 1323, 1284, 1246, 1146, 1057, 983, 917, 868, 768, 729, 696 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C21H29N2O4 [M+H]+: 373.2122, found 373.2117; [α]D 22.31-12.69 (c 1.0, CHCl3); SFC Conditions: 20% IPA, 2.5 mL/min, Chiralpak IC column, λ = 210 nm, tR (min): minor = 4.31, major = 5.68.

- tert-butyl (R)-6-allyl-6-methyl-5-oxo-4-(4-(trifluoromethyl)benzoyl)-1,4-diazepane-1-carboxylate (4c). Prepared according to the general procedure with allyl ester 3c (55.3 mg, 0.114 mmol, 1.0 equiv), Pd2(dba)3 (4.2 mg, 4.57 µmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (6.7 mg, 0.011 mmol, 10 mol %). Purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide methyl diazepanone 4c as a colorless oil (45.1 mg, 0.102 mmol, 90% yield, 92% ee); 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.2 Hz, 2H), 7.55 (d, J = 8.2 Hz, 2H), 5.71 (ddt, J = 17.2, 10.1, 7.4 Hz, 1H), 5.23 - 5.07 (m, 2H), 4.21 - 4.03 (m, 2H), 4.02 - 3.64 (m, 2H), 3.58 - 3.38 (m, 2H), 2.59 - 2.21 (m, 2H), 1.49 (s, 9H), 1.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 181.0, 180.8, 173.1, 155.4, 155.1 140.0, 132.8 (q, JC-F = 32.9 Hz), 132.7, 127.6, 125.5 (q, JC-F = 3.7 Hz), 123.7 (q, JC-F = 272.5 Hz), 119.9, 81.0, 50.9, 50.0, 47.2, 46.4, 42.4, 42.0, 41.8, 28.5, 23.6, 23.3; IR (Neat Film, NaCl) 3366, 3077, 2978, 2934, 1694, 1452, 1410, 1394, 1367, 1326, 1248, 1167, 1147, 1066, 1014, 984, 925, 852, 764 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H31F3N3O4 [M+NH4]+: 458.2261, found 458.2250; [α]D 22.6-12.32 (c 1.0, CHCl3); SFC Conditions: 5% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): minor = 4.49, major = 5.86.

- tert-butyl (R)-6-allyl-4-(4-methoxybenzoyl)-6-methyl-5-oxo-1,4-diazepane-1-carboxylate (4d). Prepared according to the general procedure with allyl ester 3d (45.8 mg, 0.103 mmol, 1.0 equiv), Pd2(dba)3 (3.7 mg, 0.004 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (5.9 mg, 0.01 mmol, 10 mol %). Purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide methyl diazepanone 4d as a colorless oil (38.9 mg, 0.0966 mmol, 94% yield, 94% ee); 1H NMR (400 MHz, CDCl3) δ 7.62 - 7.44 (m, 2H), 6.95 - 6.75 (m, 2H), 5.76 (m, 1H), 5.26 - 4.99 (m, 2H), 4.24 - 3.87 (m, 3H), 3.83 (s, 3H), 3.75 (m, 1H), 3.59 - 3.37 (m, 2H), 2.60 - 2.25 (m, 2H), 1.49 (s, 9H), 1.31 (s, 3H); 13C NMR (100 MHz, CDCl3) 8 180.7, 180.6, 174.3, 162.5, 155.4, 155.0, 133.0, 130.1, 128.1, 119.4, 113.6, 80.7, 55.4, 50.8, 49.8, 47.4, 46.6, 42.7, 42.4, 28.4, 23.5, 23.1; IR (Neat Film, NaCl) 3352, 3076, 2975, 2932, 2841, 2568, 1690, 1605, 1579, 1542, 1511, 1458, 1420, 1392, 1366, 1322, 1284, 1256, 1214, 1168, 1146, 1056, 1032, 984, 924, 868, 842, 807, 762, 743, 736, 650, 633, 621, 608 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H31N2O5 [M+H]+: 403.2227, found 403.2225; [α]D 22.45-40.51 (c 1.0, CHCl3); SFC Conditions: 20% MeOH, 2.5 mL/min, Chiralpak IC column, λ = 210 nm, tR (min): minor = 5.11, major = 5.66.

- tert-butyl (S)-6-allyl-6-benzyl-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1-carboxylate (4e). Prepared according to the general procedure with allyl ester 3e (52.3 mg, 0.100 mmol, 1.0 equiv), Pd2(dba)3 (3.7 mg, 0.004 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (5.9 mg, 0.01 mmol, 10 mol %). Purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide benzyl diazepanone 4e as a colorless oil (48.1 mg, 0.100 mmol, >99% yield, 89% ee); 1H NMR (400 MHz, CDCl3) δ 7.51 - 7.44 (m, 2H), 7.32 - 7.20 (m, 3H), 7.18 - 7.09 (m, 2H), 6.87 - 6.81 (m, 2H), 5.93 (br s, 1H), 5.26 - 5.12 (m, 2H), 4.09 - 3.88 (m, 2H), 3.84 (s, 3H), 3.78 - 2.41 (m, 8H), 1.48 (s, 9H); 13CNMR (100 MHz, CDCl3) δ 179.0, 174.6, 162.6, 156.0, 155.4, 136.7, 133.1, 130.8, 130.5, 128.5, 128.2, 127.0, 119.9, 119.6, 113.7, 80.9, 80.7, 55.5, 54.2, 53.8, 49.1, 47.5, 47.3, 42.4, 42.0, 41.2, 40.9, 40.0, 28.5; IR (Neat Film, NaCl) 3374, 2974, 2927, 1694, 1604, 1581, 1510, 1454, 1416, 1392, 1365, 1320, 1282, 1256, 1211, 1166, 1028, 979, 925, 838, 762, 742, 705, 678, 636, 610 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C28H35N2O5 [M+H]+: 479.2540, found 479.2533; [α]D 22.81 +19.02 (c 1.0, CHCl3); SFC Conditions: 20% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): minor = 3.94, major = 6.20.

- tert-butyl (S)-6-allyl-6-(2-chloroallyl)-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1-carboxylate (4f). Prepared according to the general procedure with allyl ester 3f (48.1 mg, 0.0949 mmol, 1.0 equiv), Pd2(dba)3 (3.7 mg, 0.004 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (5.9 mg, 0.01 mmol, 10 mol %). Purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide alkenyl chloride 4f as a colorless oil (36.7 mg, 0.0793 mmol, 84% yield, 90% ee); 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 5.93 - 5.71 (m, 1H), 5.36 - 5.07 (m, 4H), 4.36 - 3.84 (m, 4H), 3.83 (s, 3H), 3.82 - 3.51 (m, 2H), 3.00 - 2.33 (m, 4H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 178.2, 174.6, 162.7, 155.8, 155.2, 137.5, 132.9, 130.5, 128.3, 120.1, 118.2, 113.7, 81.1, 80.9, 55.5, 52.6, 49.2, 47.7, 47.4, 47.0, 44.5, 43.8, 42.8, 42.0, 41.8, 40.3, 28.5; IR (Neat Film, NaCl) 2976, 2930, 1694, 1631, 1604, 1580, 1510, 1456, 1421, 1393, 1366, 1320, 1282, 1256, 1212, 1167, 1150, 1030, 980, 928, 840, 765, 682, 636, 610 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C24H32ClN2O5 [M+H]+: 463.1994, found 463.2005; [α]D 22.68-24.10 (c 0.5, CHCl3); SFC Conditions: 20% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): minor = 2.52, major = 2.77.

- tert-butyl (S)-6-allyl-4-benzoyl-6-fluoro-5-oxo-1,4-diazepane-1-carboxylate (4g). Prepared according to the general procedure with allyl ester 3g (43.2 mg, 0.103 mmol, 1.0 equiv), Pd2(pmdba)3 (4.4 mg, 0.004 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (5.9 mg, 0.01 mmol, 10 mol %). Purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide alkyl fluoride 4g as a white, amorphous solid (32.6 mg, 0.0866 mmol, 84% yield, 90% ee); 1H NMR (400 MHz, CDCl3) δ 7.61 - 7.53 (m, 2H), 7.53 - 7.45 (m, 1H), 7.45 - 7.34 (m, 2H), 5.94 - 5.72 (m, 1H), 5.34 - 5.17 (m, 2H), 4.58 - 4.38 (m, 1H), 4.26 - 4.02 (m, 2H), 3.99 - 3.74 (m, 1H), 3.39 - 3.10 (m, 2H), 2.96 - 2.74 (m, 1H), 2.73 - 2.43 (m, 1H), 1.47 (s, 9H); 13CNMR (100 MHz, CDCl3) 8 173.9, 173.9 (d, JC-F = 26.3 Hz), 155.1, 135.2, 132.1, 130.3, 128.4, 128.2, 121.0, 97.7 (dd, JC-F = 193.9, 47.2 Hz), 81.1, 49.8 (dd, JC-F = 35.3, 23.1 Hz), 47.2, 46.6, 42.6, 39.7 (dd, JC-F = 27.6, 21.9 Hz), 28.3; IR (Neat Film, NaCl) 2978, 2926, 1694, 1450, 1414, 1393, 1367, 1329, 1246, 1152, 1042, 999, 979, 926, 857, 766, 724, 694, 672, 648 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C20H29FN3O4 [M+NH4]+: 438.2035, found 438.2040; [α]D 22.85+28.89 (c 1.0, CHCl3); SFC Conditions: 10% IPA, 2.5 mL/min, Chiralcel OD-H column, λ = 210 nm, tR (min): minor = 6.26, major = 4.99.

- tert-butyl (S)-6-allyl-6-fluoro-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1-carboxylate (4h). Prepared according to the general procedure with allyl ester 3h (60 mg, 0.133 mmol, 1.0 equiv), Pd2(dba)3 (4.9 mg, 0.0053 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (7.9 mg, 0.013 mmol, 10 mol %). Purified by automated silica gel flash chromatography (0→50% acetone/hexanes) to provide alkyl fluoride 4h as a colorless oil (45 mg, 0.111 mmol, 84% yield, 83% ee); 1H NMR (400 MHz, CDCl3) δ 7.61 - 7.53 (m, 2H), 6.92 - 6.84 (m, 2H), 5.93 - 5.78 (m, 1H), 5.30 - 5.21 (m, 2H), 4.35 (t, J = 16.0 Hz, 1H), 4.22 - 4.02 (m, 2H), 3.96 - 3.85 (m, 1H), 3.84 (s, 3H), 3.40 - 3.19 (m, 2H), 2.94 - 2.78 (m, 1H), 2.71 - 2.48 (m, 1H), 1.47 (s, 9H); 13CNMR (100 MHz, CDCl3) δ 173.9, 173.7, 163.1, 155.3, 131.0, 130.6, 127.1, 121.0, 113.8, 97.8 (dd, JC-F = 193.7, 52.5 Hz), 81.2, 55.5, 49.8 (dd, JC-F = 33.5, 23.3 Hz), 47.5, 46.9, 43.2, 39.8 (dd, JC-F = 32.1, 21.8 Hz), 28.3; IR (Neat Film, NaCl) 2977, 2932, 1696, 1603, 1578, 1511, 1448, 1413, 1366, 1327, 1256, 1169, 1152, 1029, 1000, 977, 923, 835, 766 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C21H28FN2O5 [M+H]+: 407.1977, found 407.1973; [α]D 22.5 +46.99 (c 1.7, CHCl3); SFC conditions: 20% IPA, 2.5 mL/min, Chiralcel OD-H column, λ = 210 nm, tR (min): major = 2.69, minor = 3.25.

- tert-butyl (S)-6-allyl-4-(4-methoxybenzoyl)-5-oxo-6-(prop-2-yn-1-yl)-1,4-diazepane-1-carboxylate (4i). Prepared according to the general procedure with allyl ester 3i (70.0 mg, 0.149 mmol, 1.0 equiv), Pd2(pmdba)3 (5.4 mg, 4.9 µmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (8.8 mg, 0.015 mmol, 10 mol %) at 50 °C. Purification by automated silica gel flash chromatography (Teledyne ISCO, 0→40% acetone/hexanes) provided alkyne 4i as a colorless oil (28.0 mg, 0.0656 mmol, 44% yield, 94% ee); 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J= 8.2 Hz, 2H), 6.89 - 6.82 (m, 2H), 5.93 - 5.63 (m, 1H), 5.30 - 5.10 (m, 2H), 4.36 - 4.15 (m, 1H), 4.09 - 3.68 (m, 4H), 3.83 (s, 3H), 3.62 - 3.37 (m, 1H), 2.83 - 2.43 (m, 4H), 2.20 - 1.99 (m, 1H), 1.51 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 177.5, 174.6, 162.9, 155.6, 155.2, 132.1, 130.7, 127.9, 120.0, 113.7, 81.1, 80.9, 80.5, 72.1, 55.6, 52.7, 49.1, 46.9, 47.7, 43.0, 42.3, 39.1, 37.5, 28.5, 26.5, 25.9; IR (Neat Film, NaCl) 3283, 2972, 2922, 1692, 1603, 1511, 1454, 1418, 1365, 1322, 1255, 1169, 1031, 980, 926, 839, 766, 670 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C24H31N2O5 [M+H]+: 427.2227, found 427.2238; [α]D 22.1-7.69 (c 1.0, CHCl3); SFC conditions: 10% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): major = 10.85, minor = 10.29.

- tert-butyl (R)-6-allyl-6-(3-methoxy-3-oxopropyl)-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1-carboxylate (4j). Prepared according to the general procedure with allyl ester 3j (51.9 mg, 0.100 mmol, 1.0 equiv), Pd2(dba)3 (3.7 mg, 0.004 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (5.9 mg, 0.01 mmol, 10 mol %). Purified by silica gel flash chromatography (33% EtOAc/hexanes) to provide methyl ester 4j as a white, amorphous solid (45.8 mg, 0.0965 mmol, 96% yield, 95% ee); 1H NMR (400 MHz, CDCl3) δ 7.57 - 7.48 (m, 2H), 6.92 - 6.83 (m, 2H), 5.80 - 5.64 (m, 1H), 5.22 - 5.09 (m, 2H), 4.21 (ddd, J = 15.7, 6.6, 2.1 Hz, 1H), 4.13 - 3.85 (m, 3H), 3.83 (s, 3H), 3.62 (s, 3H), 3.51 (d, J = 15.2 Hz, 1H), 3.42 - 3.29 (m, 1H), 2.64 - 2.20 (m, 4H), 2.20 - 1.86 (m, 2H), 1.49 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 179.2, 174.7, 173.6, 173.4, 162.7, 155.4, 155.0, 132.7, 132.5, 130.3, 128.3, 120.0, 113.8, 81.1, 80.9, 55.5, 51.8, 50.1, 48.9, 47.6, 46.8, 43.2, 42.9, 40.3, 39.7, 29.3, 28.8, 28.5; IR (Neat Film, NaCl) 2975, 2360, 1736, 1694, 1605, 1580, 1510, 1426, 1393, 1366, 1321, 1283, 1254, 1167, 1031, 980, 927, 842, 811, 762, 647, 610 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C25H35N2O7 [M+H]+: 475.2439, found 475.2438; [α]D 22.52 +7.73 (c 1.0, CHCl3); SFC conditions: 15% IPA, 2.5 mL/min, Chiralcel OD-H column, λ = 210 nm, tR (min): major = 5.27, minor = 4.84.

- tert-butyl (R)-6-allyl-6-(2-cyanoethyl)-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1-carboxylate (4k). Prepared according to the general procedure with allyl ester 3k (62.2 mg, 0.128 mmol, 1.0 equiv), Pd2(dba)3 (4.7 mg, 0.00512 mmol, 4 mol %), and (S)-(CF3)3-t-BuPHOX (7.6 mg, 0.0128 mmol, 10 mol %), using 9:1 methylcyclohexane-toluene as the reaction solvent. Purified by silica gel flash chromatography (33% EtOAc/hexanes) to provide nitrile 4j as a white, amorphous solid (48.6 mg, 0.110 mmol, 86% yield, 84% ee); 1H NMR (400 MHz, CDCl3) δ 7.59 - 7.47 (m, 2H), 6.96 - 6.85 (m, 2H), 5.82 - 5.64 (m, 1H), 5.30 - 5.11 (m, 2H), 4.09 - 3.93 (m, 2H), 3.93 - 3.72 (m, 2H), 3.85 (s, 3H), 3.70 - 3.38 (m, 2H), 2.60 - 2.25 (m, 4H), 2.22 - 1.93 (m, 2H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 178.0, 174.5, 163.0, 155.6, 154.9, 131.7, 130.3, 127.9, 120.7, 119.7, 113.9, 81.4, 55.6, 52.2, 49.6, 48.0, 47.4, 46.8, 43.0, 42.6, 39.9, 39.0, 32.1, 31.5, 28.4, 12.4; IR (Neat Film, NaCl) 2975, 2931, 2361, 2246, 1690, 1604, 1579, 1510, 1456, 1419, 1366, 1321, 1256, 1168, 1148, 1031, 980, 926, 840, 811, 766, 607 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C24H35N4O5 [M+NH4]+: 459.2602, found 459.2602; [α]D 22.4+9.31 (c 0.5, CHCl3); SFC conditions: 20% IPA, 2.5 mL/min, Chiralcel OD-H column, λ = 310 nm, tR (min): major = 8.33, minor = 6.18.

- tert-butyl (S)-6-allyl-4-benzoyl-6-(((tert-butoxycarbonyl)amino)methyl)-5-oxo-1,4-diazepane-1-carboxylate (4l). Prepared according to the general procedure with allyl ester 3l (53 mg, 0.0997 mmol, 1.0 equiv) and Pd2(dba)3. Purification by automated silica gel flash chromatography (0→50% EtOAc/hexanes) provided carbamate 4l as a white foam (37 mg, 0.0759 mmol, 76% yield, 93% ee). 1H NMR (400 MHz, CDCl3) δ 7.55 - 7.44 (m, 3H), 7.44 - 7.35 (m, 2H), 5.74 (ddt, J = 15.7, 10.5, 7.4 Hz, 1H), 5.35 (br s, 0.5H), 5.21 - 5.06 (m, 2H), 4.79 (br s, 0.5H), 4.48 - 4.28 (m, 1H), 4.14 - 3.09 (m, 7H), 2.69 - 2.18 (m, 2H), 1.50 (s, 9H), 1.44 (s, 9H); 13CNMR (100 MHz, CDCl3) δ 179.3, 178.8, 174.5, 156.3, 155.9, 155.1, 136.3, 132.5, 132.1, 131.7, 128.5, 127.7, 120.1, 81.3, 81.1, 79.5, 54.8, 48.9, 47.3, 46.8, 46.3, 42.6, 41.1, 38.7, 37.1, 28.5, 28.5; IR (Neat Film, NaCl) 2977, 1687, 1502, 1422, 1391, 1365, 1322, 1282, 1245, 1168, 978, 916, 753 HRMS (MM: ESI-APCI): m/z calc'd for C26H37N3O6 [M+H]+: 488.2755, found 488.2747; [α]D 23.2-3.70 (c 1.85, CHCl3); SFC conditions: 20% IPA, 2.5 mL/min, Chiralpak IC column, λ = 254 nm, tR (min): major = 5.85, minor = 4.65.

- To a 500 mL Schlenk flask was added Pd2(dba)3 (37 mg, 0.04 mmol, 4 mol %), (S)-(CF3)3-t-BuPHOX (59 mg, 0.1 mmol, 10 mol %), and MeCy (20 mL). After stirring for 20 minutes at 25 °C, allyl ester 3e (523 mg, 1.0 mmol, 1.0 equiv) and methylcyclohexane (52 mL, total substrate concentration 0.014 M) were added to the pre-stirred catalyst solution. After stirring for 23 h at 40 °C, the reaction mixture was directly loaded onto a flash column and purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide benzyl diazepanone 4e as a colorless oil (393 mg, 0.82 mmol, 82% yield, 83% ee); All characterization data matched those reported above for compound 4e; [α]D 21.96 +14.757 (c 1.0, CHCl3); SFC Conditions: 20% IPA, 2.5 mL/min, Chiralpak AD-H column, λ = 210 nm, tR (min): minor = 3.76, major = 5.90.
-

- tert-butyl 4-benzoyl-5-oxo-1,4-diazepane-1-carboxylate (SI1). To a solution of tert-butyl 5-oxo-1,4-diazepane-1-carboxylate (5.00 g, 23.3 mmol, 1.0 equiv) in THF (230 mL, 0.1 M) at -78 °C was slowly added n-BuLi (2.18 M in hexanes, 12.8 mL, 27.9 mmol, 1.2 equiv). The opaque mixture was allowed to warm to ambient temperature until the solution became homogeneous, at which point it was again cooled to -78 °C. Then, benzoyl chloride (3.52 mL, 30.3 mmol, 1.3 equiv) was added dropwise and the reaction turned light orange over several minutes. The reaction was stirred for 1 h at -78 °C, then poured into saturated aqueous NH4Cl (200 mL) and extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over Na2SO4 and concentrated. The crude product was purified by silica gel flash chromatography (20% acetone/hexanes) to afford benzoyl-protected lactam SI1 as a white solid (7.43 g, 23.3 mmol, >99% yield); 1H NMR (400 MHz, CDCl3) δ 7.57 - 7.49 (m, 2H), 7.49 - 7.41 (m, 1H), 7.41 - 7.32 (m, 2H), 4.03 - 3.96 (m, 2H), 3.71 (m, 4H), 2.82 - 2.75 (m, 2H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 175.6, 173.7, 154.5, 135.9, 131.7, 128.2, 127.9, 80.7, 47.8, 47.10, 45.4, 41.6, 41.0, 40.6, 28.3; IR (Neat Film, NaCl) 2976, 2932, 2251, 1682, 1599, 1582, 1450, 1422, 1392, 1366,1327, 1285, 1247, 1229, 1157, 1115, 1032, 1018, 976, 954, 915,862, 793,769, 729, 696, 647 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C17H26N3O4 [M+NH4]+: 336.1918, found 336.1912.

- tert-butyl S-oxo-4-(4-(trifluoromethyl)benzoyl)-1,4-diazepane-1-carboxylate (SI2). To a solution of tert-butyl 5-oxo-1,4-diazepane-1-carboxylate (500 mg, 2.33 mmol, 1 equiv) in THF (25 mL, 0.1 M) at -78 °C was slowly added n-BuLi (2.5 M in hexanes, 1.02 mL, 2.56 mmol, 1.1 equiv), and the reaction mixture was stirred at -78 °C for 15 min. Then, 4-trifluoromethylbenzoyl chloride (450 µL, 3.03 mmol, 1.3 equiv) was added dropwise, and the reaction was stirred for 30 min at -78 °C. The reaction mixture was then poured into saturated aqueous NH4Cl (20 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (3 x 10 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (25% EtOAc/hexanes) to afford the title compound as a white solid (698 mg, 1.81 mmol, 77% yield); 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.1 Hz, 2H), 4.14 - 4.02 (m, 2H), 3.84 - 3.66 (m, 4H), 2.91 - 2.77 (m, 2H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 175.7, 172.5, 154.6, 139.6, 133.0 (q, JC-F = 32.8 Hz), 130.6, 128.0, 125.5 (q, JC-F = 3.8 Hz), 123.7 (q, JC-F = 272.6 Hz), 81.2, 47.7 (br), 45.3, 41.4 (br), 40.8, 28.5; IR (Neat Film, NaCl) 2981, 1689, 1455, 1422, 1367, 1326, 1301, 1249, 1230, 1159, 1127, 1066, 1028, 1015, 977, 955, 852, 832, 769 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C18H25F3N3O4 [M+NH4]+: 404.1742, found 404.1797.

- tert-butyl 4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1-carboxylate (SI3). To a solution of tert-butyl 5-oxo-1,4-diazepane-1-carboxylate (800 mg, 3.73 mmol, 1 equiv) in THF (37 mL, 0.1 M) at -78 °C was slowly added n-BuLi (2.5 M in hexanes, 1.64 mL, 4.1 mmol, 1.1 equiv). The opaque mixture was allowed to warm to ambient temperature until the solution became homogeneous, at which point it was again cooled to -78 °C. Then, 4-methoxybenzoyl chloride (657 µL, 4.85 mmol, 1.3 equiv) was added dropwise and the reaction was stirred for 30 min at -78 °C. The reaction mixture was then poured into saturated aqueous NH4Cl (30 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (3 x 10 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by automated silica gel flash chromatography (Teledyne ISCO, 0→100% EtOAc/hexanes) to afford the title compound as a white solid (1.2 g, 3.44 mmol, 92% yield); 1H NMR (400 MHz, CDCl3) δ 7.62 - 7.54 (m, 2H), 6.93 - 6.84 (m, 2H), 4.00 - 3.93 (m, 2H), 3.83 (s, 3H), 3.78 - 3.69 (m, 4H), 2.85 - 2.78 (m, 2H), 1.48 (s, 9H); 13CNMR (100 MHz, CDCl3) δ 175.7, 173.4, 162.9, 154.6, 130.9, 130.6, 127.7, 113.7, 80.8, 55.5, 48.0, 47.4, 46.1, 41.9, 41.3, 40.8, 28.5; IR (Neat Film, NaCl) 2974, 2936, 1774, 1687, 1604, 1578, 1510, 1458, 1420, 1391, 1366, 1327, 1284, 1249, 1166, 1114, 1023, 977, 956, 916, 860, 842, 809, 767, 632 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C18H25N2O5 [M+H]+: 349.1758, found 349.1760.

- 6-allyl 1-(tert-butyl) 4-benzoyl-5-oxo-1,4-diazepane-1,6-dicarboxylate (2a). To a solution of diisopropylamine (266 µL, 1.88 mmol, 1.2 equiv) in THF (10 mL) at -78 °C in a flame-dried round-bottom flask was added n-BuLi (2.5 M in hexanes, 792 µL, 1.73 mmol, 1.1 equiv), the resulting solution was stirred at -78 °C for 45 min. To this solution was then added lactam SI1 (500 mg, 1.57 mmol, 1.0 equiv) in THF (6 mL, 0.1 M total concentration) dropwise while stirring at -78 °C. The reaction mixture was stirred for 75 min at -78 °C. Allyl cyanoformate (201 µL, 1.88 mmol, 1.2 equiv) was then added dropwise at -78 °C. After stirring for 3 h at -78 °C, the reaction mixture was poured into saturated aqueous NH4Cl (10 mL) and extracted with ethyl acetate (3 x 20 mL). The combined organic extracts were concentrated under reduced pressure onto silica (4 g). The silica-adsorbed crude mixture was purified by silica gel flash chromatography (20→30% EtOAc/hexanes) to provide allyl ester 2a as an off-white solid (550 mg, 1.37 mmol, 87% yield); 1H NMR (500 MHz, CDCl3) δ 7.65 - 7.57 (m, 2H), 7.53 - 7.45 (m, 1H), 7.42 - 7.34 (m, 2H), 5.92 (ddt, J = 17.2, 10.4, 5.9 Hz, 1H), 5.40 - 5.22 (m, 2H), 4.79 - 4.59 (m, 2H), 4.33 - 4.03 (m, 2H), 4.02 - 3.88 (m, 3H), 3.87 - 3.66 (m, 1H), 3.55 - 3.40 (m, 1H), 1.48 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 173.7, 171.4, 167.5, 154.7, 135.3, 132.2, 131.4, 128.4, 128.4, 119.5, 81.3, 66.6, 56.0, 46.7 (br), 44.5, 43.3 (br), 28.4; IR (Neat Film, NaCl) 3374, 3062, 2977, 2934, 1746, 1694, 1600, 1582, 1450, 1419, 1393, 1367, 1327, 1246, 1156, 1037, 1020, 995, 968, 939, 857, 792, 769, 727, 695, 616 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C21H30N3O6 [M+NH4]+: 420.2129, found 420.2109.

- 6-allyl 1-(tert-butyl) 5-oxo-4-(4-(trifluoromethyl)benzoyl)-1,4-diazepane-1,6-dicarboxylate (2b). To a solution of lactam SI2 (500 mg, 1.29 mmol, 1.0 equiv) in THF (8 mL, 0.1 M total concentration) at -78 °C was added LiHMDS (303 mg, 1.81 mmol, 1.4 equiv) in THF (5 mL) dropwise. The resulting yellow reaction mixture was stirred for 15 min at -78 °C. Then, allyl cyanoformate (166 µL, 1.55 mmol, 1.2 equiv) was added dropwise at -78 °C, after which the solution slowly became colorless. After stirring for 1 h at -78 °C, the reaction was poured into 2 M HCl (20 mL) and extracted with ethyl acetate (4 x 20 mL). The combined organic extracts were dried over anhydrous Na2SO4 and NaHCO3, passed through filter paper, and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (20→33% EtOAc/hexanes) to provide allyl ester 2b as a white solid (266 mg, 0.565 mmol, 44% yield); 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 8.3 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 5.92 (ddt, J = 17.2, 10.4, 5.9 Hz, 1H), 5.42 - 5.23 (m, 2H), 4.79 - 4.59 (m, 2H), 4.46 - 3.63 (m, 6H), 3.52 (m, 1H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3) 8 172.4, 171.4, 167.4, 154.6, 138.9, 133.3 (q, JC-F = 32.6 Hz), 131.2, 128.3, 125.4 (q, JC-F = 3.8 Hz), 123.7 (q, JC-F = 272.5 Hz), 119.8, 81.5, 66.8, 56.0, 47.4, 46.3, 44.2, 43.0, 28.4; IR (Neat Film, NaCl) 3377, 3083, 2980, 2935, 2463, 2358, 1928, 1798, 1747, 1694, 1652, 1619, 1584, 1513, 1455, 1414, 1394, 1368, 1327, 1246, 1156, 1131, 1067, 1034, 1016, 994, 970, 940, 879, 853, 824, 770, 723, 679, 639, 630, 612 HRMS (MM: ESI-APCI): m/z calc'd for C22H29F3N3O6 [M+NH4]+: 488.2003, found 488.2022.

- 6-allyl 1-(tert-butyl) 4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (2c). To a solution of lactam SI3 (1.00 g, 2.87 mmol, 1.0 equiv) in THF (20 mL, 0.1 M total concentration) at -78 °C was added LiHMDS (528 mg, 3.16 mmol, 1.1 equiv) in THF (9 mL) dropwise. The resulting pale yellow reaction mixture was stirred for 15 min at -78 °C. Allyl cyanoformate (368 µL, 3.44 mmol, 1.2 equiv) was then added dropwise at -78 °C, resulting in a clear solution. After stirring for 1.5 h at -78 °C, the reaction was poured into 1 M HCl (10 mL) and diluted with ethyl acetate (20 mL). The layers were separated and the aqueous phase was extracted with ethyl acetate (3 x 30 mL). The combined organic extracts were dried over anhydrous Na2SO4 and NaHCO3, filtered, and concentrated under reduced pressure onto silica. The silica-adsorbed crude mixture was purified by silica gel flash chromatography (10→20% EtOAc/hexanes) to provide allyl ester 2c as a colorless oil (600 mg, 1.39 mmol, 48% yield); 1H NMR (400 MHz, CDCl3) δ 7.72 - 7.60 (m, 2H), 6.91 - 6.79 (m, 2H), 5.92 (ddt, J = 17.3, 10.4, 5.9 Hz, 1H), 5.42 - 5.20 (m, 2H), 4.77 - 4.56 (m, 2H), 4.40 - 3.92 (m, 4H), 3.92 - 3.62 (m, 2H), 3.82 (s, 3H), 3.58 - 3.24 (m, 1H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) 8 173.1, 171.3, 167.6, 163.2, 154.6, 131.4, 131.2, 127.0, 119.4, 113.7, 81.1, 66.5, 55.9, 55.4, 47.7 and 46.7, 45.1, 43.3, 42.8, 28.3; IR (Neat Film, NaCl) 2977, 1746, 1693, 1603, 1578, 1511, 1454, 1419, 1392, 1366, 1324, 1255, 1168, 1025, 995, 965, 842, 766 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H28N2O7 [M+H]+: 433.1969, found 433.1966.

- 6-allyl 1-(tert-butyl) 4-benzoyl-6-benzyl-5-oxo-1,4-diazepane-1,6-dicarboxylate (3a). To a flame-dried round bottom flask containing a solution of allyl ester 2a (1.00 g, 2.49 mmol, 1.0 equiv) in THF (25 mL, 0.1 M) at 0 °C was added NaH (60% dispersion in mineral oil, 107 mg, 2.74 mmol, 1.1 equiv) and the mixture was stirred at 0 °C for 30 min. BnBr (1.50 mL, 12.45 mmol, 5.0 equiv) was then added dropwise and the reaction mixture was warmed to 45 °C. After 16 h, the temperature was further increased to 53 °C due to sluggish reactivity. After another 45 min of stirring at 53 °C, the reaction mixture was cooled to 23 °C and poured into saturated aqueous NH4Cl (25 mL), the layers were separated, and the aqueous layer was extracted with ethyl acetate (3 x 10 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide the title compound as a colorless foam (922 mg, 1.87 mmol, 75% yield); 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 7.6 Hz, 2H), 7.55 - 7.46 (m, 1H), 7.37 (t, J = 7.7 Hz, 2H), 7.31 - 7.25 (m, 3H), 7.24 - 7.12 (m, 2H), 5.86 (tq, J = 22.8, 6.5 Hz, 1H), 5.42 - 5.26 (m, 2H), 4.71 - 4.55 (m, 2H), 4.22 (dd, J = 75.3, 15.5 Hz, 1H), 4.05 - 3.57 (m, 4H), 3.57 - 3.36 (m, 2H), 3.22 (dd, J = 68.5, 13.7 Hz, 1H), 1.45 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (174.4, 174.0, 172.2, 172.0, 170.8, 170.2, 155.5, 155.0, 135.7, 135.6, 135.5, 132.0, 131.1, 130.9, 130.8, 130.6, 128.5, 128.5, 128.3, 127.5, 127.4, 120.6, 120.2, 81.2, 81.0, 67.0, 66.9, 62.6, 62.2, 47.3, 46.7, 46.2, 45.8, 42.4, 42.1, 42.0, 28.5; IR (Neat Film, NaCl) 3063, 3030, 2977, 2933, 1694, 1601, 1583, 1495, 1450, 1416, 1393, 1366, 1325, 1280, 1247, 1154, 1132, 1092, 1041, 1023, 980, 939, 868, 796, 768, 728, 703, 662 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C24H25N2O6 [M- t Bu+2H]+: 437.1707, found 437.1697.

- 6-allyl 1-(tert-butyl) 4-benzoyl-6-methyl-5-oxo-1,4-diazepane-1,6-dicarboxylate (3b). To a solution of allyl ester 2a (240 mg, 0.596 mmol, 1.0 equiv) in THF (6 mL, 0.1 M) at 0 °C was added 60 % NaH (26 mg, 0.657 mmol, 1.1 equiv). The solution was stirred at 0 °C for 40 min, after which MeI (186 µL, 2.98 mmol, 5.0 equiv) was added rapidly. The reaction was heated to 45 °C and stirred for 16 h, cooled to 23 °C, poured into saturated aqueous NH4Cl (5 mL), and extracted with EtOAc (3 x 3 mL). The combined organic extracts were dried over Na2SO4 and concentrated onto silica gel. The silica-adsorbed crude product was purified by silica gel flash chromatography (20% EtOAc/hexanes) to afford the title compound as a light yellow oil (200 mg, 0.480 mmol, 81% yield). 1H NMR (400 MHz, CDCl3) δ 7.78 - 7.63 (m, 2H), 7.58 - 7.43 (m, 1H), 7.38 (t, J = 7.6 Hz, 2H), 5.96 (ddt, J = 16.6, 10.4, 6.0 Hz, 1H), 5.49 - 5.26 (m, 2H), 4.85 - 4.64 (m, 2H), 4.46 - 4.22 (m, 1H), 4.10 (br d, J = 14.8 Hz, 1H), 3.86 - 3.42 (m, 4H), 1.57 (s, 3H), 1.45 (s, 9H); 13CNMR (100 MHz, CDCl3) δ 174.5 174.1, 173.0, 171.7, 155.1, 154.9, 135.7, 132.0, 131.2, 128.3, 128.2, 120.2, 81.1, 66.9, 57.7, 49.8, 49.0, 47.1, 46.0, 43.2, 28.4, 23.6; IR (Neat Film, NaCl) 2977, 1693, 1449, 1416, 1366, 1325, 1281, 1249, 1139, 1104, 1047, 983, 938, 768, 727, 694 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H29N2O6 [M+H]+: 417.2020, found 417.2010.

- 6-allyl 1-(tert-butyl) 6-methyl-5-oxo-4-(4-(trifluoromethyl)benzoyl)-1,4-diazepane-1,6-dicarboxylate (3c). To a suspension of allyl ester 2b (150 mg, 0.319 mmol, 1.0 equiv) and Cs2CO3 (208 mg, 0.638 mmol, 2.0 equiv) in acetonitrile (3.2 mL, 0.1 M) was added MeI (99 µL, 1.59 mmol, 5.0 equiv) at 23 °C. The reaction was heated to 45 °C and stirred for 5 h, then cooled to 23 °C, poured into saturated aqueous NH4Cl (6 mL), and extracted with EtOAc (3 x 3 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (15% EtOAc/petroleum ether) to provide the title compound as a colorless oil (146 mg, 0.301 mmol, 95% yield); 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 8.1 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H), 5.97 (ddt, J = 17.3, 10.3, 6.1 Hz, 1H), 5.48 - 5.29 (m, 2H), 4.84 - 4.68 (m, 2H), 4.49 - 4.30 (m, 1H), 4.18 - 3.98 (m, 1H), 3.90 - 3.39 (m, 4H), 1.57 (s, 3H), 1.45 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 173.2, 172.7, 171.7, 154.9, 139.2, 133.1 (q, JC-F = 32.5 Hz), 131.1, 128.2, 125.4, 123.8 (q, JC-F = 272.7 Hz), 120.5, 81.3, 67.0, 57.7, 49.7, 49.0, 46.9, 45.8, 43.1, 42.9, 28.4, 23.7; IR (Neat Film, NaCl) 3384, 3083, 2979, 2937, 1698, 1619, 1584, 1514, 1478, 1453, 1416, 1394, 1367, 1326, 1285, 1250, 1207, 1166, 1136, 1110, 1066, 1022, 1012, 985, 938, 855, 832, 817, 790, 769, 740, 722, 680 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C23H28F3N2O6 [M+H]+: 485.1894, found 485.1907.

- 6-allyl 1-(tert-butyl) 4-(4-methoxybenzoyl)-6-methyl-5-oxo-1,4-diazepane-1,6-dicarboxylate (3d). To a suspension of allyl ester 2c (200 mg, 0.462 mmol, 1.0 equiv), Cs2CO3 (301 mg, 0.925 mmol, 2.0 equiv) in acetonitrile (4.6 mL, 0.1 M) was added MeI (143 µL, 2.31 mmol, 5.0 equiv) at 23 °C. The reaction was heated to 45 °C and stirred for 40 min, then cooled to 23 °C, poured into saturated aqueous NH4Cl (10 mL), and extracted with EtOAc (3 x 5 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by automated silica gel flash chromatography (Teledyne ISCO, 0→90% EtOAc/hexanes) to provide the title compound as a colorless oil (70 mg, 0.157 mmol, 34% yield). 1H NMR (400 MHz, CDCl3) δ 7.75 - 7.65 (m, 2H), 6.90 - 6.82 (m, 2H), 5.96 (ddt, J = 17.3, 10.4, 6.0 Hz, 1H), 5.44 - 5.29 (m, 2H), 4.77 - 4.66 (m, 2H), 4.27 - 4.15 (m, 1H), 4.14 - 4.04 (m, 1H), 3.83 (s, 3H), 3.80 - 3.70 (m, 1H), 3.67 - 3.58 (m, 2H), 3.56 - 3.46 (m, 1H), 1.57 (s, 3H), 1.44 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 174.0, 173.6, 172.9, 172.0, 171.8, 162.9, 155.2, 154.9, 131.2, 131.2, 131.0, 127.6, 120.1, 120.0, 113.6, 81.0, 66.8, 57.5, 55.5, 49.6, 48.9, 47.2, 46.1, 43.7, 28.4, 23.7; IR (Neat Film, NaCl) 2974, 2937, 1698, 1604, 1578, 1511, 1453, 1416, 1392, 1366, 1324, 1280, 1256, 1169, 1139, 1103, 1031, 1001, 983, 929, 840, 768, 733 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C23H31N2O7 [M+H]+: 447.2126, found 447.2128.

- 6-allyl 1-(tert-butyl) 6-benzyl-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (3e). To a flame-dried round bottom flask containing a solution of allyl ester 2c (300 mg, 0.694 mmol, 1.0 equiv) in THF (7 mL, 0.1 M) at 0 °C was added NaH (60% dispersion in mineral oil, 38 mg, 0.972 mmol, 1.4 equiv) and the mixture was stirred at 0 °C for 15 min and then allowed to warm to 23 °C over 15 min. BnBr (412 µL, 3.47 mmol, 5.0 equiv) was then added dropwise and the reaction mixture was heated to 50 °C. After stirring for 8 h, the reaction mixture was allowed to cool to 23 °C and poured into saturated aqueous NH4Cl (5 mL), the layers were separated, and the aqueous phase was extracted with ethyl acetate (3 x 2 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (20% EtOAc/hexanes) to provide the title compound as a colorless foam (303 mg, 0.580 mmol, 84% yield); 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 8.5 Hz, 2H), 7.31 - 7.12 (m, 5H), 6.90 - 6.80 (m, 2H), 5.95 - 5.75 (m, 1H), 5.41 - 5.26 (m, 2H), 4.69 - 4.54 (m, 2H), 4.21 - 4.05 (m, 1H), 4.02 - 3.86 (m, 2H), 3.83 (s, 3H), 3.78 - 3.62 (m, 2H), 3.56 - 3.49 (m, 1H), 3.43 - 3.10 (m, 2H), 1.45 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 173.7, 173.4, 172.0, 171.7, 170.8, 170.2, 162.9, 155.4, 154.8, 135.7, 135.6, 131.1, 130.9, 130.7, 130.5, 128.5, 128.3, 128.2, 127.5, 127.3, 127.0, 120.2, 119.9, 113.5, 80.9, 80.7, 66.6, 62.4, 62.0, 55.3, 47.1, 46.8, 46.0, 45.8, 42.8, 42.5, 41.9, 28.3; IR (Neat Film, NaCl) 2976, 2359, 1698, 1604, 1512, 1455, 1416, 1366, 1324, 1258, 1155, 1028, 979, 840, 741, 703, 671, 634 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C29H35N2O7 [M+H]+: 523.2439, found 523.2446.

- 6-allyl 1-(tert-butyl) 6-(2-chloroallyl)-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (3f). To a suspension of allyl ester 2c (300 mg, 0.694 mmol, 1.0 equiv) and Cs2CO3 (453 mg, 1.39 mmol, 2.0 equiv) in acetonitrile (7 mL, 0.1 M) was added 2,3-dichloropropene (320 µL, 3.47 mmol, 5.0 equiv) at 23 °C. The reaction mixture was heated to 50 °C and stirred for 19 h, after which starting material remained as judged by TLC. Tetrabutylammonium iodide (25.6 mg, 0.0694 mmol, 0.1 equiv) was added and the reaction mixture was stirred at 50 °C for an additional 9 h, then allowed to cool to 23 °C. The mixture was filtered through a cotton plug and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (20% EtOAc/petroleum ether) to provide the title compound as a colorless oil (196 mg, 0.387 mmol, 56% yield); 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 6.12 - 5.94 (m, 1H), 5.60 - 5.25 (m, 4H), 4.78 (qdt, J = 12.8, 6.0, 1.2 Hz, 2H), 4.25 (br t, J = 13.9 Hz, 1H), 4.17 - 3.87 (m, 3H), 3.84 (s, 3H), 3.76 - 3.51 (m, 1H), 3.48 - 3.30 (m, 1H), 3.29 - 2.99 (m, 2H), 1.43 (d, J= 16.9 Hz, 9H); 13C NMR (100 MHz, CDCl3) δ 173.9, 173.3, 170.3, 163.1, 155.9, 155.1, 137.0, 136.7, 131.1, 127.2, 120.6, 120.3, 119.4, 118.4, 113.6, 81.4, 81.0, 67.5, 60.0, 55.5, 47.1, 45.9, 46.0, 45.2, 45.6, 44.7, 43.1, 42.8, 28.4; IR (Neat Film, NaCl) 3356, 3080, 2977, 2933, 2841, 2568, 2254, 1700, 1629, 1605, 1579, 1512, 1456, 1417, 1393, 1367, 1326, 1281, 1257, 1217, 1196, 1153, 1029, 988, 910, 842, 811, 780, 757, 732, 668, 634, 621 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C25H32ClN2O7 [M+H]+: 507.1893, found 507.1902.

- 6-allyl 1-(tert-butyl) 4-benzoyl-6-fluoro-5-oxo-1,4-diazepane-1,6-dicarboxylate (3g). To a 20 mL vial containing allyl ester 2a (250 mg, 0.621 mmol, 1.0 equiv) in THF (7.4 mL, 0.1 M) at 23 °C was added NaH (60% dispersion in mineral oil, 27.3 mg, 0.683 mmol, 1.1 equiv). After stirring for 12 min, Selectfluor™ (264 mg, 0.745 mmol, 1.2 equiv) was added in a single portion, and the reaction mixture was warmed to 50 °C and stirred for 24 h, after which starting material remained as judged by TLC. Additional Selectfluor™ (264 mg, 0.745 mmol, 1.2 equiv) was then added, and the reaction mixture was stirred for an additional 8 h at 50 °C. The reaction mixture was allowed to cool to 23 °C and water (5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 5 mL). The combined organic extracts were dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by silica gel flash chromatography (25% EtOAc/hexanes) to provide the title compound (167 mg, 0.397 mmol, 64% yield); 1H NMR (500 MHz, CDCl3) δ 7.64 - 7.56 (m, 2H), 7.52 - 7.43 (m, 1H), 7.36 (t, J= 7.7 Hz, 2H), 5.99 - 5.80 (m, 1H), 5.43 - 5.19 (m, 2H), 4.83 - 4.56 (m, 2H), 4.52 - 4.17 (m, 3H), 4.03 - 3.56 (m, 2H), 3.27 - 3.08 (m, 1H), 1.47 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 173.1, 169.6, 169.4, 164.8, 164.6, 154.9, 134.3, 132.5, 130.7, 128.5, 128.4, 119.7, 95.7 (d, J = 204.8 Hz), 81.5, 67.2, 47.6 (dd, JC-F = 137.5, 24.1 Hz), 47.3, 46.4, 42.8, 28.3; IR (Neat Film, NaCl) 2978, 2926, 1694, 1450, 1414, 1393, 1367, 1329, 1246, 1152, 1042, 999, 979, 926, 857, 766, 724, 694, 672, 648 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C21H29FN3O6 [M+NH4]+: 438.2035, found 438.2040.

- 6-allyl 1-(tert-butyl) 6-fluoro-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (3h). To a 20 mL vial containing allyl ester 2c (320 mg, 0.740 mmol, 1.0 equiv) and NaH (60% dispersion in mineral oil, 32.5 mg, 0.814 mmol, 1.1 equiv) was added THF (7.4 mL, 0.1 M) at 23 °C. After stirring for 30 min, Selectfluor™ (315 mg, 0.889 mmol, 1.2 equiv) was added in a single portion, and the reaction mixture was warmed to 50 °C and stirred for 5 h. The crude reaction mixture was then concentrated under reduced pressure and purified by silica gel flash chromatography (30% acetone/hexanes) to provide the title compound (290 mg, 0.644 mmol, 87% yield); 1H NMR (400 MHz, CDCl3) δ 7.66 - 7.58 (m, 2H), 6.90 - 6.82 (m, 2H), 5.91 (ddt, J = 16.3, 10.9, 5.7 Hz, 1H), 5.43 - 5.21 (m, 2H), 4.84 - 4.40 (m, 3H), 4.40 - 4.16 (m, 2H), 4.00 - 3.86 (m, 1H), 3.82 (s, 3H), 3.77 - 3.56 (m, 1H), 3.22 - 3.09 (m, 1H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 172.6, 169.6 (dd, JC-F = 48.2, 25.4 Hz), 164.9 (d, JC-F = 25.8 Hz), 163.4, 155.0, 131.3, 130.8, 126.1, 119.8, 113.9, 95.7 (dd, JC-F = 205.6, 14.6 Hz), 81.5, 67.3, 55.5, 47.5 (dd, JC-F = 109.0, 23.7 Hz), 47.2 (d, JC-F = 92.2 Hz), 43.4, 28.3; IR (Neat Film, NaCl) 2976, 2936, 2844, 1759, 1698, 1603, 1578, 1512, 1449, 1414, 1367, 1327, 1258, 1168, 1151, 1076, 1030, 997, 977, 942, 929, 841, 817, 770, 760, 730, 698 HRMS (MM: ESI-APCI): m/z calc'd for C22H28FN2O7 [M+H]+: 451.1875, found 451.1877.

- 6-allyl 1-(tert-butyl) 4-(4-methoxybenzoyl)-5-oxo-6-(prop-2-yn-1-yl)-1,4-diazepane-1,6-dicarboxylate (3i). To a solution of allyl ester 2c (250 mg, 0.578 mmol, 1.0 equiv) in THF (5.8 mL, 0.1 M) was added NaH (60% dispersion in mineral oil, 25 mg, 0.636 mmol, 1.1 equiv) at 0 °C. After stirring for 30 min at 0 °C, propargyl bromide (80% wt/wt in toluene, 125 µL, 1.16 mmol, 2.0 equiv) was added at 0 °C. The reaction mixture was heated to 50 °C and stirred for 16 h. The mixture was allowed to cool to 23 °C, quenched with aqueous NaHCO3 (10 mL) and extracted with EtOAc (3 x 5 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by automated silica gel flash chromatography (Teledyne ISCO, 0→50% acetone/hexanes) to provide propargyl allyl ester 3i as a colorless oil (220 mg, 0.468 mmol, 81% yield). 1H NMR (500 MHz, CDCl3) δ 7.73 - 7.59 (m, 2H), 6.84 (d, J = 8.6 Hz, 2H), 5.99 (ddt, J = 17.3, 10.4, 6.0 Hz, 1H), 5.54 - 5.27 (m, 2H), 4.85 - 4.69 (m, 2H), 4.33 - 3.85 (m, 4H), 3.82 (s, 3H), 3.76 - 3.56 (m, 1H), 3.56 - 3.39 (m, 1H), 3.14 - 2.90 (m, 2H), 2.08 (s, 1H), 1.42 (d, J = 14.3 Hz, 9H); 13C NMR (125 MHz, CDCl3) δ 173.7, 173.2, 170.5, 170.3, 169.7, 169.3, 163.0, 155.6, 154.9, 131.2, 131.0, 127.1, 120.3, 120.0, 113.6, 81.1, 81.0, 78.9, 78.6, 72.3, 67.3, 60.7, 60.4, 55.5, 47.1, 46.5, 46.2, 46.0, 43.0, 28.3, 27.0, 26.9; IR (Neat Film, NaCl) 3280, 2975, 2936, 1737, 1694, 1604, 1579, 1547, 1512, 1454, 1416, 1393, 1366, 1326, 1280, 1258, 1156, 1134, 1030, 994, 980, 841, 778, 770, 737, 706, 677, 634, 622 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C25H31N2O7 [M+H]+: 471. 2126, found 471.2130.

- 6-allyl 1-(tert-butyl) 6-(3-methoxy-3-oxopropyl)-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (3j). To a 20 mL vial containing allyl ester 2c (300 mg, 0.694 mmol, 1.0 equiv) and K2CO3 (480 mg, 3.47 mmol, 5.0 equiv) was added acetone (2.8 mL, 0.25 M) and methyl acrylate (126 µL, 1.39 mmol, 2.0 equiv) at 23 °C. The vessel was sealed and heated to 50 °C. After stirring for 5 h, additional methyl acrylate (126 µL, 1.39 mmol, 2.0 equiv) was added and the reaction was stirred for an additional 14 h. The reaction mixture was then filtered through a plug of cotton, concentrated under reduced pressure, and purified by silica gel flash chromatography (33% EtOAc/petroleum ether) to provide diester 3j as a colorless, waxy solid (185 mg, 0.357 mmol, 51% yield); 1H NMR (500 MHz, CDCl3) δ 7.77 - 7.65 (m, 2H), 6.92 - 6.79 (m, 2H), 5.96 (ddt, J = 17.2, 10.3, 6.1 Hz, 1H), 5.48 - 5.27 (m, 2H), 4.82 - 4.63 (m, 2H), 4.32 - 3.84 (m, 3H), 3.83 (s, 3H), 3.81 - 3.66 (m, 1H), 3.62 (s, 3H), 3.58 - 3.40 (m, 2H), 2.56 - 2.13 (m, 4H), 1.44 (s, 9H); 13CNMR (125 MHz, CDCl3) δ 173.8, 173.5, 173.0, 171.9, 170.9, 170.6, 163.0, 155.1, 154.8, 131.1, 131.0, 127.5, 120.6, 113.6, 81.2, 67.0, 60.4, 55.5, 51.7, 48.7, 47.8, 47.0, 46.0, 43.4, 31.4, 29.8, 28.3; IR (Neat Film, NaCl) 3354, 2976, 2843, 2568, 2255, 2044, 1694, 1605, 1579, 1556, 1513, 1416, 1393, 1367, 1260, 1168, 1030, 982, 916, 843, 811, 782, 766, 732, 648, 634 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C26H38N3O9 [M+NH4]+: 536.2603, found 536.2603.

- 6-allyl 1-(tert-butyl) 6-(2-cyanoethyl)-4-(4-methoxybenzoyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (3k). To a 20 mL vial containing allyl ester 2c (300 mg, 0.694 mmol, 1.0 equiv) and K2CO3 (480 mg, 3.47 mmol, 5.0 equiv) was added acetone (2.8 mL, 0.25 M) and acrylonitrile (182 µL, 2.78 mmol, 4.0 equiv) at 23 °C. The vessel was sealed and heated to 50 °C. After 17 h of stirring, the reaction mixture was filtered through a plug of cotton, concentrated under reduced pressure, and purified by silica gel flash chromatography (33% EtOAc/petroleum ether) to provide 3k as a colorless foam (176 mg, 0.362 mmol, 52% yield); 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.9 Hz, 2H), 6.01 (ddt, J = 16.7, 10.3, 6.3 Hz, 1H), 5.51 - 5.36 (m, 2H), 4.89 - 4.75 (m, 2H), 4.32 - 3.88 (m, 3H), 3.86 (s, 3H), 3.84 - 3.34 (m, 3H), 2.71 - 2.09 (m, 4H), 1.53 - 1.34 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 170.8, 170.3, 163.2, 155.3, 131.0, 130.6, 127.1, 121.4, 121.1, 119.1, 113.7, 81.4, 67.5, 60.2, 55.5, 47.5, 46.8, 43.6, 32.9, 28.3, 13.6; IR (Neat Film, NaCl) 2975, 2934, 2250, 1694, 1605, 1579, 1512, 1455, 1419, 1393, 1367, 1326, 1255, 1164, 1031, 1000, 979, 941, 916, 842, 813, 781, 762, 733, 648, 634 HRMS (MM: ESI-APCI): m/z calc'd for C25H35N4O7 [M+NH4]+: 503.2500, found 503.2505.

- 6-allyl 1-(tert-butyl) 4-benzoyl-6-(((tert-butoxycarbonyl)amino)methyl)-5-oxo-1,4-diazepane-1,6-dicarboxylate (3l). A solution of allyl ester 2a (200 mg, 0.497 mmol, 1.0 equiv) and tert-butyl ((phenylsulfonyl)methyl)carbamate (162 mg, 0.597 mmol, 1.2 equiv) in CH2Cl2 (2.5 mL, 0.2 M) at 23 °C was stirred for 5 min, after which time Cs2CO3 (405 mg, 1.24 mmol, 2.5 equiv) was added at the same temperature. [Klepacz, A.; Zwierzak, A. Tetrahedron Lett. 2002, 43, 1079-1080; Sikriwal, D.; Kant, R.; Maulik, P. R.; Dikshit, D. K. Tetrahedron 2010, 66, 6167-6173.] After an additional 30 min of stirring, saturated aqueous NH4Cl (1 mL) was added and the biphasic mixture was vigorously stirred for 20 min. The layers were separated and the aqueous phase was extracted with CH2Cl2 (3 x 3 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure onto silica gel (2 g). The silica-adsorbed crude reaction mixture was purified by automated silica gel flash chromatography (Teledyne ISCO, 10→40% acetone/hexanes) to provide carbamate 3l as a white foam (200 mg, 0.376 mmol, 76% yield): 1H NMR (400 MHz, CDCl3) δ 7.83 - 7.71 (m, 2H), 7.59 - 7.46 (m, 1H), 7.46 - 7.36 (m, 2H), 6.00 (ddt, J = 16.6, 10.3, 6.1 Hz, 1H), 5.51 - 5.25 (m, 2H), 5.17 (br s, 1H), 4.79 - 4.64 (m, 2H), 4.48 - 4.23 (m, 1H), 4.14 - 3.20 (m, 7H), 1.44 (s, 9H), 1.42 (s, 9H); 13CNMR (100 MHz, CDCl3) δ 174.1, 173.8, 172.7, 170.1, 169.6, 156.0, 155.2, 154.7, 135.5, 132.2, 131.4, 128.5, 128.4, 120.2, 81.4, 79.6, 67.4, 62.7, 47.1, 46.8, 45.9, 43.2, 28.5, 28.4; IR (Neat Film, NaCl) 3457, 2977, 2934, 2253, 1704, 1600, 1503, 1450, 1417, 1392, 1367, 1325, 1283, 1248, 1158, 1042, 980, 913,77 860, 767, 729, 693, 663 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C27H38N3O8 [M+H]+: 532.2653, found 532.2664.
-

- Prepared according to the literature procedure by Mangion and coworkers and used directly in the synthesis of 7. [Org. Lett. 2012, 14, 3458-3461].

- 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid (8). Prepared according to the literature procedure by Mangion and coworkers. [Org. Lett. 2012, 14, 3458-3461]. All characterization data matched those reported in the literature.

- tert-butyl (S)-6-allyl-6-benzyl-S-oxo-1,4-diazepane-1-carboxylate (SI4). To a flask containing benzoyl-protected diazepanone 4a (460 mg, 1.03 mmol, 1.0 equiv) was added isopropyl alcohol (100 mL, 0.01 M) and water (10 mL), followed by LiOH·H2O (61 mg, 1.45 mmol, 1.5 equiv) at 23 °C. After stirring for 4 h at 23 °C, the isopropyl alcohol was removed under reduced pressure and the resulting aqueous mixture extracted with EtOAc (4 x 50 mL). The combined organic extracts were dried with sodium sulfate, filtered, and concentrated under reduced pressure to yield a crude oil (400 mg) that was used without further purification.
- tert-butyl (R)-6-allyl-6-benzyl-1,4-diazepane-1-carboxylate (5). Crude lactam SI4 (350 mg [theoretical maximum 310 mg lactam], 0.903 mmol, 1 equiv) was dissolved in THF (10.2 mL, 0.1 M) and cooled to 0 °C. LiAlH4 (77 mg, 2.03 mmol, 2.25 equiv) was then added, and the reaction mixture was stirred at 0 °C for 4 h, over the course of which an additional 3.37 equiv (116 mg, 3.05 mmol) of LiAlH4 were added in total (77 mg, followed by 39 mg, at equal intervals). The reaction mixture was then diluted with diethyl ether (10 mL) and water (300 µL) was added. After gas generation subsided, 15% aqueous NaOH (300 µL) was added, followed by additional water (900 µL). After stirring at 0 °C for 15 min, anhydrous MgSO4 was added, and the mixture was stirred for an additional 10 min, whereafter it was filtered through celite and concentrated under reduced pressure. Purification by automated silica gel flash chromatography (Teledyne ISCO, 0→20% MeOH/CH2Cl2) provided the product as a light yellow oil (130 mg, 0.393 mmol, 44% yield); 1H NMR (400 MHz, CDCl3) δ 7.40 - 7.14 (m, 5H), 6.01 (dq, J = 17.1, 7.8 Hz, 1H), 5.18 (d, J = 15.9 Hz, 2H), 3.67 - 3.30 (m, 4H), 2.97 (m, 2H), 2.88 - 2.48 (m, 4H), 2.30 - 2.06 (m, 3H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 155.8, 138.1, 134.6, 130.7, 128.0, 126.2, 118.2, 79.8, 79.5, 57.9, 57.2, 55.3, 54.2, 50.9, 49.8, 49.3, 43.5, 41.3, 39.8, 39.4, 28.5; IR (Neat Film, NaCl) 3357, 3066, 3028, 2976, 2928, 1694, 1602, 1464, 1455, 1416, 1391, 1365, 1334, 1302, 1248, 1166, 1031, 996, 952, 912, 866, 771, 733, 703, 685, 672, 659, 644, 612 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C20H31N2O2 [M+H]+: 331.2380, found 331.2399; [α]D 22.24-6.496 (c 2.0, CHCl3).

- tert-butyl (R)-6-allyl-6-benzyl-4-(5-chlorobenzo[d]oxazol-2-yl)-1,4-diazepane-1-carboxylate (SI5). To a 1 dram vial containing diazepane 5 (9.5 mg, 0.0287 mmol, 1.0 equiv), aryl bromide 6 (10.0 mg, 0.0431 mmol, 1.5 equiv), and K2CO3 (7.9 mg, 0.0574 mmol, 2 equiv) was added MeCN (0.3 mL, 0.1 M) at 23 °C. After stirring for 24 h at 23 °C, saturated aqueous NH4Cl (1 mL) was added, and the mixture was extracted with EtOAc (3 x 1 mL). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude material (15.4 mg) thus obtained was carried forward without further purification.
- (S)-2-(6-allyl-6-benzyl-1,4-diazepan-1-yl)-5-chlorobenzo[d]oxazole (7). Crude carbamate SI5 was dissolved in MeOH (0.3 mL, 0.1 M) and AcCl (20.5 µL, 0.288 mmol, 10 equiv) was added at 23 °C. After stirring for 5 h at 23 °C, the reaction mixture was concentrated under reduced pressure and purified by silica gel flash chromatography (66% EtOAc/benzene + 1% Et3N) to provide 7 as a beige, amorphous solid (4.3 mg, 0.0113 mmol, 39% yield from 5) of sufficient purity for use in the next reaction, however, further purification was possible by silica gel flash chromatography with 2% Et3N in Et2O; 1H NMR (400 MHz, CDCl3) δ 7.34 - 7.27 (m, 3H), 7.25 - 7.17 (m, 3H), 7.12 (d, J = 8.4 Hz, 1H), 6.94 (dd, J = 8.4, 2.3 Hz, 1H), 5.99 (ddt, J = 14.5, 10.4, 7.2 Hz, 1H), 5.30 (d, J = 3.1 Hz, 1H), 5.20 - 5.10 (m, 1H), 3.87 - 3.61 (m, 3H), 3.15 - 3.00 (m, 2H), 2.90 - 2.66 (m, 3H), 2.57 (d, J = 13.9 Hz, 1H), 2.22 - 2.10 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 147.5, 137.8, 134.1, 130.8, 129.4, 128.2, 126.4, 120.1, 118.8, 116.2, 109.2, 57.8, 56.5, 53.3, 49.2, 43.8, 41.3, 39.8; IR (Neat Film, NaCl) 2922, 1638, 1570, 1458, 1249, 1167, 921, 792, 710 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C22H25ClN3O [M+H]+: 382.1681, found 382.1695; [α]D 21-89+7.524 (c 0.07, CHCl3).

- (R)-(6-allyl-6-benzyl-4-(5-chlorobenzo[d]oxazol-2-yl)-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (9). To a vial containing carboxylic acid 8 (35 mg, 0.172 mmol, 1.0 equiv) in CH2Cl2 (1.8 mL) at 23 °C was added DMF (4 µL, 0.0517 mmol, 0.3 equiv) and oxalyl chloride (18 µL, 0.207 mmol, 1.2 equiv). After stirring for 1 h, Et3N (48 µL, 0.344 mmol, 2.0 equiv) was added, followed by amine 7 (60 mg, 0.155 mmol, 0.9 equiv) in CH2Cl2 (1.8 mL, 0.05 M total concentration). After stirring for an additional 1 h at 23 °C, the reaction mixture was quenched with saturated aqueous NaHCO3 (3 mL), the layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 x 2 mL). The combined organic extracts were dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by automated silica gel flash chromatography (Teledyne ISCO, 0→40% Et2O/hexanes) to provide amide 9 as a beige oil (29.6 mg, 0.0522 mmol, 34% yield); 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1H), 7.55 (s, 1H), 7.34 - 7.27 (m, 6H), 7.20 - 7.09 (m, 3H), 7.04 - 6.92 (m, 2H), 6.11 (ddt, J = 17.6, 10.4, 7.3 Hz, 1H), 5.26 - 5.12 (m, 2H), 4.24 - 4.07 (m, 1H), 3.96 (dd, J = 17.1, 14.4 Hz, 1H), 3.90 - 3.75 (m, 1H), 3.70 - 3.40 (m, 4H), 3.38 - 3.12 (m, 2H), 2.99 - 2.80 (m, 2H), 2.43 - 2.38 (m, 2H), 2.38 - 2.33 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 170.7, 170.5, 170.0, 163.0, 147.2, 144.4, 138.7, 138.6, 138.5, 136.9, 135.9, 135.8, 135.8, 135.7, 135.7, 135.6, 135.6, 134.2, 133.9, 133.8, 133.6, 133.5, 133.2, 132.9, 130.9, 130.8, 130.6, 130.6, 130.4, 130.4, 129.9, 129.5, 129.5, 129.0, 129.0, 128.3, 128.3, 128.2, 128.2, 128.2, 128.1, 128.1, 126.6, 126.6, 122.2, 122.0, 121.9, 120.5, 120.5, 119.2, 119.1, 119.0, 116.3, 116.3, 109.3, 109.2, 56.2, 56.1, 55.5, 54.3, 52.9, 52.2, 49.5, 49.3, 49.2, 48.9, 47.6, 45.0, 43.5, 43.1, 42.4, 42.2, 42.0, 41.8, 39.5, 39.5, 37.6, 21.0, 21.0, 20.9; IR (Neat Film, NaCl) 3431, 2923, 2854, 2356, 1644, 1634, 1574, 1568, 1538, 1505, 1462, 1454, 1428, 1372, 1308, 1251, 1216, 1172, 1054, 952, 921, 852, 822, 794, 737, 704 cm-1; HRMS (MM: ESI-APCI): m/z calc'd for C32H32ClN6O2 [M+H]+: 567.2270, found 567.2294; [α]D 22.24 +41.90 (c 1.0, CHCl3).
- All publications and patents mentioned herein are hereby incorporated by reference in their entirety as if each individual publication or patent was specifically and individually indicated to be incorporated by reference. In case of conflict, the present application, including any definitions herein, will control.
- While specific embodiments of the subject disclosure have been discussed, the above specification is illustrative and not restrictive. Many variations of the disclosure will become apparent to those skilled in the art upon review of this specification and the claims below. The full scope of the disclosure should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
Claims (153)
- A method for the preparation of a compound of formula (I):


with a Pd or Ni catalyst comprising a chiral ligand;
an optionally substituted aryl halide, optionally substituted aryl pseudohalide, optionally substituted heteroaryl halide, an optionally substituted heteroaryl pseudohalide, or
wherein, as valence and stability permit,ring A represents an optionally substituted heterocycloalkyl, or heterocycloalkenyl group;R in each occurrence independently represents optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, -OR10, -SR10, or -NR10R11;R1 represents hydrogen or optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, alkynyl, -C(O)alkyl, -C(O)aryl, - C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), - C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), -S(O)2(aryl), - S(O)2(alkyl), -S(O)2(haloalkyl), -OR10, -SR10, or -NR10R11;or R1 taken together with a substituent on ring A and the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;or a substituent on ring A taken together with another substituent on ring A and the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;R2 represents substituted or unsubstituted alkyl, alkenyl, alkynyl, aralkyl, aralkenyl, aryl, heteroaralkyl, heteroaralkenyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkoxy, amino, or halo;R10 and R11 are independently selected for each occurrence from hydrogen or substituted or unsubstituted alkyl, aralkyl, aryl, heteroaralkyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, and alkynyl;X is a halogen or pseudohalide; andY represents optionally substituted aryl, optionally substituted heteroaryl, or
- The method of claim 1, wherein the compound of formula (I) is represented by formula (Ia):

the compound of formula (II) is represented by formula (IIa): B, D, and E independently for each occurrence represent, as valence permits, O, S, NR4, CR5R6, C(O), CR5, or N; provided that no two adjacent occurrences of N, B, D, and E are NR4, O, S, or N;R4 represents hydrogen or optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, alkynyl, -C(O)alkyl, -C(O)aryl, - C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), - C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), -S(O)2(aryl), - S(O)2(alkyl), -S(O)2(haloalkyl), -OR10, -SR10, or -NR10R11;R5 and R6 each independently represent hydrogen, hydroxyl, halogen, nitro, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, cyano, carboxyl, sulfate, amino, alkoxy, aryloxy, arylalkoxy, alkylamino, alkylthio, hydroxyalkyl, alkoxyalkyl, aminoalkyl, thioalkyl, haloalkyl, ether, thioether, ester, amido, thioester, carbonate, carbamate, urea, sulfonate, sulfone, sulfoxide, sulfonamide, acyl, acyloxy, or acylamino;or any two occurrences of R1, R4, R5, and R6 on adjacent N, B, D, or E groups, taken together with the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;each occurrence ofindependently represents a double bond or a single bond as permitted by valence; and
B, D, and E independently for each occurrence represent, as valence permits, O, S, NR4, CR5R6, C(O), CR5, or N; provided that no two adjacent occurrences of N, B, D, and E are NR4, O, S, or N;R4 represents hydrogen or optionally substituted alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, alkynyl, -C(O)alkyl, -C(O)aryl, - C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, -C(O)O(alkyl), -C(O)O(aryl), - C(O)O(aralkyl), -C(O)O(heteroaryl), -C(O)O(heteroaralkyl), -S(O)2(aryl), - S(O)2(alkyl), -S(O)2(haloalkyl), -OR10, -SR10, or -NR10R11;R5 and R6 each independently represent hydrogen, hydroxyl, halogen, nitro, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, cyano, carboxyl, sulfate, amino, alkoxy, aryloxy, arylalkoxy, alkylamino, alkylthio, hydroxyalkyl, alkoxyalkyl, aminoalkyl, thioalkyl, haloalkyl, ether, thioether, ester, amido, thioester, carbonate, carbamate, urea, sulfonate, sulfone, sulfoxide, sulfonamide, acyl, acyloxy, or acylamino;or any two occurrences of R1, R4, R5, and R6 on adjacent N, B, D, or E groups, taken together with the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group;each occurrence ofindependently represents a double bond or a single bond as permitted by valence; and m and n are integers each independently selected from 0, 1, and 2.
m and n are integers each independently selected from 0, 1, and 2. - The method of claim 2, wherein the sum of m and n is 0, 1, 2, or 3.
- The method of claim 2 or claim 3, wherein each occurrence of B, D, and E is independently -CR5R6-, -CR5-, or -C(O)-.
- The method of claim 2, wherein E is -CR5-; and the sum of m and n is 0.
- The method of claim 5, wherein
R1 is selected from optionally substituted alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, -C(O)alkyl, -C(O)aryl, -C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, - C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), - C(O)O(heteroaralkyl), and -S(O)2(aryl); and
R5 is selected from hydrogen, hydroxyl, halogen, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, amino, alkoxy, aryloxy, alkylamino, amido, and acylamino; or
R1 and the occurrence of R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group. - The method of claim 6, wherein R1 is selected from optionally substituted alkyl, aryl, aralkyl, alkenyl, -C(O)alkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), and -S(O)2(aryl).
- The method of claim 7, wherein R1 is substituted aryl.
- The method of claim 6, wherein R1 and the occurrence of R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group.
- The method of claim 9, wherein R1 and the occurrence of R5 on E are taken together to form an optionally substituted heterocycloalkyl, or heterocycloalkenyl group.
- The method of claim 1, wherein the compound of formula (I) is represented by formula (Ib):

the compound of formula (II) is represented by formula (IIb): R5 and R6 each independently represent hydrogen, hydroxyl, halogen, nitro, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, cyano, carboxyl, sulfate, amino, alkoxy, aryloxy, arylalkoxy, alkylamino, alkylthio, hydroxyalkyl, alkoxyalkyl, aminoalkyl, thioalkyl, haloalkyl, ether, thioether, ester, amido, thioester, carbonate, carbamate, urea, sulfonate, sulfone, sulfoxide, sulfonamide, acyl, acyloxy, or acylamino;or any two occurrences of R1, R5, and R6, taken together with the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group.
R5 and R6 each independently represent hydrogen, hydroxyl, halogen, nitro, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, cyano, carboxyl, sulfate, amino, alkoxy, aryloxy, arylalkoxy, alkylamino, alkylthio, hydroxyalkyl, alkoxyalkyl, aminoalkyl, thioalkyl, haloalkyl, ether, thioether, ester, amido, thioester, carbonate, carbamate, urea, sulfonate, sulfone, sulfoxide, sulfonamide, acyl, acyloxy, or acylamino;or any two occurrences of R1, R5, and R6, taken together with the intervening atoms, form an optionally substituted aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heterocycloalkenyl group. - The method of claim 11, wherein
R1 is selected from optionally substituted alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, alkenyl, -C(O)alkyl, -C(O)aryl, -C(O)aralkyl, -C(O)heteroaryl, -C(O)heteroaralkyl, - C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), -C(O)O(heteroaryl), - C(O)O(heteroaralkyl), and -S(O)2(aryl); and
R5 is selected from hydrogen, hydroxyl, halogen, alkyl, cycloalkyl, (cycloalkyl)alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, alkenyl, alkynyl, amino, alkoxy, aryloxy, alkylamino, amido, and acylamino; or
R1 and R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group. - The method of claim 12, wherein R1 is selected from optionally substituted alkyl, aryl, aralkyl, alkenyl, -C(O)alkyl, -C(O)O(alkyl), -C(O)O(aryl), -C(O)O(aralkyl), and - S(O)2(aryl).
- The method of claim 13, wherein R1 is substituted aryl.
- The method of claim 12, wherein R1 and the occurrence of R5 on E are taken together to form an optionally substituted heteroaryl, heterocycloalkyl, or heterocycloalkenyl group.
- The method of any one of claims 1 - 15, wherein R2 represents substituted or unsubstituted alkyl, alkenyl, alkynyl, aralkyl, aralkenyl, aryl, heteroaralkyl, heteroaralkenyl, heteroaryl, (cycloalkyl)alkyl, cycloalkyl, (heterocycloalkyl)alkyl, heterocycloalkyl, or halo.
- The method of any one of claims 1 - 16, wherein R2 is selected from alkyl, alkenyl, aryl, aralkyl, aralkenyl, and heteroaralkenyl, optionally substituted with halo, alkyl, haloalkyl, hydroxy, alkoxy, aryloxy, arylalkoxy, cyano, nitro, azido, -CO2H, -C(O)O(alkyl), amino, alkylamino, arylamino, aralkylamino, or amido.
- The method of any one of claims 1 - 17, wherein R2 is selected from alkyl, alkenyl, aryl, aralkyl, aralkenyl, and heteroaralkenyl, optionally substituted with halo, alkyl, haloalkyl, alkoxy, aryloxy, or arylalkoxy.
- The method of any one of claims 1 - 18, wherein the Pd catalyst is Pd(dmdba)2 or Pd2(dmdba)3.
- The method of any one of claims 1 - 18, wherein the Ni(0) catalyst is Ni(COD)2.
- The method of any one of claims 1 - 20, wherein the Pd or Ni catalyst is used in an amount from about 0.1 mol % to about 20 mol % relative to the compound of formula (II).
- The method of any one of claims 1 - 21, wherein the Pd or Ni catalyst is used in an amount from about 1 mol % to about 15 mol % relative to the compound of formula (II).
- The method of any one of claims 1 - 22, wherein the Pd or Ni catalyst is used in an amount of about 1 mol%, about 2 mol%, about 3 mol%, about 4 mol%, about 5 mol%, about 6 mol%, about 7 mol%, about 8 mol%, about 9 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, or about 14 mol% relative to the compound of formula (II).
- The method of any one of claims 1 - 23, wherein the catalyst is a Pd catalyst and the chiral ligand is an enantioenriched ferrocelane ligand, or the catalyst is a Ni catalyst and the chiral ligand is an enantioenriched Josiphos ligand.
- The method of claim 24, wherein the enantioenriched ferrocelane ligand is 1,1'-bis[(2S,5S)-2,5-diethyl-1-phospholanyl]ferrocene, 1,1'-bis[(2S,5S)-2,5-dimethyl-1-phospholanyl]ferrocene, or 1,1'-bis[(2S,5S)-2,5-diisopropyl-1-phospholanyl]ferrocene.
- The method of claim 24, wherein the enantioenriched Josiphos ligand is (R)-1-[(S)-2-(dicyclohexylphosphino)ferrocenylethyl]diphenylphosphine.
- The method of any one of claims 1 - 26, wherein the chiral ligand is used in an amount from about 0.5 mol % to about 50 mol % relative to the compound of formula (II).
- The method of any one of claims 1 - 27, wherein the chiral ligand is used in an amount from about 1 mol % to about 20 mol % relative to the compound of formula (II).
- The method of any one of claims 1 - 28, wherein the chiral ligand is used in an amount of about 5 mol%, about 5.5 mol%, about 6 mol%, about 6.5 mol%, about 7 mol%, about 7.5 mol%, about 8 mol%, about 8.5 mol%, about 9 mol%, about 9.5 mol%, about 10 mol%, about 11 mol%, about 12 mol%, about 13 mol%, about 14 mol %, or about 15 mol % relative to the compound of formula (II).
- The method of any one of claims 1 - 29, wherein the aryl halide or aryl pseudohalide is selected from bromobenzene, chlorobenzene, iodobenzene, phenyl triflate, and chlorotoluene.
- The method of any one of claims 1 - 30, wherein the base is selected from hexamethyldisilazane sodium salt (NaHMDS), KHMDS, LiHMDS, and lithium tert-butoxide (tBuOLi).
- The method of any one of claims 1 - 31, wherein the base is LiHMDS.
- The method of any one of claims 1 - 31, wherein the base is NaHMDS.
- The method of any one of claims 1 - 33, wherein the reaction includes dioxane.
- The method of claim 34, wherein the dioxane is used in an amount from about 0.01 M to about 0.4 M.
- The method of claim 34 or 35, wherein the dioxane is used in an amount of about 0.01 M, about 0.05 M, about 0.1 M, about 0.15 M, about 0.2 M, about 0.25 M, about 0.3 M, about 0.35 M, or about 0.4 M.
- The method of any one of claims 1 - 36, whereby the compound of formula (I) is enantioenriched.
- The method of any one of claims 1-37, wherein no aryl nitrile is used in the method.
- A compound of formula (I) made by the method of any one of claims 1-38.
- A method comprising
preparing a compound of formula (I) according to the method of any of claims 1 - 38;
and
synthesizing a medicinal product from the compound of formula (I). - A compound of formula (III) or formula (IV):

 R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R23 is H, halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, alkoxycarbonyl, halogen, aryl, or -NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;R24 is H or halogen; andp is 0 or 1.
R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R23 is H, halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, alkoxycarbonyl, halogen, aryl, or -NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;R24 is H or halogen; andp is 0 or 1. - The compound of claim 41, wherein:
p is 0;
the compound of formula (III) is a compound of formula (IIIa):
 R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R23 is H, or C1-6 alkyl optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; andR24 is H or halogen.
R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl, wherein each of aryl and heteroaryl are optionally substituted with alkoxy, alkyl, or haloalkyl;R23 is H, or C1-6 alkyl optionally substituted with OH, cyano, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; andR24 is H or halogen. - The compound of claim 41 or claim 42, wherein:R21 is Bz or An;R22 is Boc, Cbz, or Bz;R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, or CH-2NHBoc; andR24 is H or Cl.
- The compound of any one of claims 41-43, wherein R22 is Boc or Cbz.
- The compound of claim 44, wherein R22 is Boc.
- The compound of any one of claims 41-43, wherein the compound is:







- The compound of any one of claims 41-43, wherein the compound is:





- The compound of claim 47, wherein the compound is


- The compound of claim 41, wherein:
p is 1;
the compound of formula (III) is a compound of formula (IIIb):
 R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a;R22a is C1-6 alkyl;R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR21a;R23a is C1-6 alkyl; andR24 is H.
R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a;R22a is C1-6 alkyl;R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR21a;R23a is C1-6 alkyl; andR24 is H. - The compound of claim 49, wherein:R21 is Bz, An, or pCF3Bz;R22 is Boc; andR23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc.
- The compound of claim 49 or claim 50, wherein the compound of formula (IIIb) is:



- The compound of claim 49 or claim 50, wherein the compound of formula (IVb) is:


- The compound of claim 52, wherein the compound of formula (IVb) is

- A compound of formula (V), (VI), or (VII):


 R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, alkynyl, or -NHC(O)OR25b; halogen; or allyl, optionally substituted with halogen;R25b is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl.
R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, alkynyl, or -NHC(O)OR25b; halogen; or allyl, optionally substituted with halogen;R25b is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl. - The compound of claim 44, wherein the compound is:




- The compound of claim 49, wherein the compound is:



- The compound of claim 56, wherein the compound is

- The compound of claim 54, wherein the compound is a compound of formula (VII) wherein R25 is -CH3, -CH2CH3, -CH2CH2C(O)CH3, -CH2CCl=CH2, -CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH.
- A compound selected from:



- A compound of Formula (VIII):
 R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R23b is H, or C1-6 alkyl optionally substituted with CN, OH, alkoxy, aryloxy, amino, acyl, or -NHC(O)R23c;R23c is C1-6 alkyl, (C6-10 aryl)alkyl or (C5-9 heteroaryl)alkyl; andR24 is H or halogen.
R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R23b is H, or C1-6 alkyl optionally substituted with CN, OH, alkoxy, aryloxy, amino, acyl, or -NHC(O)R23c;R23c is C1-6 alkyl, (C6-10 aryl)alkyl or (C5-9 heteroaryl)alkyl; andR24 is H or halogen. - The compound of claim 60, wherein:R21 is Bz or An;R23b is H, CH3, CH2Ph, CH2NHCbz, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NH2; andR24 is H or Cl.
- The compound of claim 61, wherein:R21 is Bz;R23b is CH2NH2; andR24 is H.
- A compound of Formula (IX) or (X):

 R23d is H, or is C1-6 alkyl optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, - NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; andR24 is H or halogen.
R23d is H, or is C1-6 alkyl optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, - NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; andR24 is H or halogen. - The compound of claim 63, wherein:R23d is H, CH3, CH2Ph, CH2OBn, CH2CH2OH, CH2CH2C(O)CH3, CH2CH2CH(OH)CH3, CH2NHCBz, or CH2NHBoc; andR24 is H or Cl.
- The compound of claim 64, wherein:R23d is CH2NHBoc; andR24 is H.
- A compound of formula (XI):
 R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R24 is H or halogen; andR23e is H, or is C1-6 alkyl optionally substituted with halogen, OH, aryl, aryloxy, CN, acyl, or amino.
R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl;R24 is H or halogen; andR23e is H, or is C1-6 alkyl optionally substituted with halogen, OH, aryl, aryloxy, CN, acyl, or amino. - The compound of claim 66, wherein:R21 is Bz or An;R24 is H or Cl; andR23e is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, or CH2NH2.
- The compound of claim 67, wherein:R21 is Bz;R23e is CH3; andR24 is H.
- A compound of formula (XII):
 R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl; andR24 is H or halogen.
R21 is -C(O)aryl or -C(O)heteroaryl, each optionally substituted with alkoxy, alkyl, or haloalkyl; andR24 is H or halogen. - The compound of claim 69, wherein:R21 is Bz or An; andR24 is H or Cl.
- The compound of claim 70, wherein R21 is Bz and R24 is H.
- A compound of formula (XIII):
 R25a is C1-6 alkyl, optionally substituted with CN, OH, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen; andR13 is aralkyl or hetaralkyl.
R25a is C1-6 alkyl, optionally substituted with CN, OH, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen; andR13 is aralkyl or hetaralkyl. - The compound of claim 72, wherein:R21a is -CH3, -CH2CH3, -CH2CH2C(O)CH3, -CH2CCl=CH2, -CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH; andR13 is Bn.
- The compound of claim 73, wherein R25a is CH2Ph and R13 is Bn.
- A method for preparation of a compound of formula (IV):

treating a compound of formula (III)
with a Pd(0) catalyst and a ligand L under alkylation conditions,
wherein, as valence and stability permit,R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl is optionally substituted with alkoxy, alkyl, or haloalkyl;R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl;R23 is H, halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, alkoxycarbonyl, halogen, aryl, or -NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;R24 is H or halogen; andp is 0 or 1. - The method of claim 75, wherein:
p is 0;
the compound of formula (IV) is a compound of formula (IVa)

wherein, as valence and stability permit,R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is -C(O)OR22a, -C(O)aryl, or -C(O)heteroaryl, wherein each of aryl and heteroaryl is optionally substituted with alkoxy, alkyl, or haloalkyl;R22a is C1-6 alkyl, C6-10 aryl, or C5-9 heteroaryl;R23 is H, or C1-6 alkyl optionally substituted with cyano, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl; andR24 is H or halogen. - The method of claim 75 or claim 76, wherein:R21 is Bz or An;R22 is Boc, Cbz, or Bz;R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, or CH-2NHBoc; andR24 is H or Cl.
- The method of claim 77, wherein R22 is Boc or Cbz.
- The method of claim 77 or claim 78, wherein the compound of formula (IVa) is:






- The method of any one of claims 75-79, wherein the alkylation conditions include reaction in toluene, THF, MTBE, or hexanes, or a combination thereof.
- The method of any one of claims 75-80, wherein the Pd(0) catalyst is Pd2(pmdba)3 or Pd2(dba)3.
- The method of any one of claims 75-81, wherein the Pd(0) catalyst is Pd2(dba)3.
- The method of any one of claims 75-82, wherein the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol % relative to the compound of formula (IIIa).
- The method of claim 83, wherein the Pd(0) catalyst is used in an amount of about 4 mol % relative to the compound of formula (IIIa).
- The method of any one of claims 75-83, wherein the ligand L is:


- The method of claim 85, wherein the ligand L is (S)-L1.
- The method of any one of claims 75-86, wherein the ligand L is used in an amount from about 0.1 mol % to about 100 mol % relative to the compound of formula (IIIa).
- The method of claim 87, wherein the ligand L is used in an amount of about 10 mol %.
- The method of any one of claims 75-88, wherein the compound of formula (IVa) is enantioenriched.
- A method of synthesizing a medicinal product, the method comprising:preparing a compound of formula (IVa) according to any one of claims 75-89; andsynthesizing a medicinal product from the compound of formula (IVa).
- The method of claim 75, wherein:
p is 1;
the compound of formula (IV) is a compound of formula (IVb)

wherein, as valence and stability permit,R22 is -C(O)OR22a;R22a is C1-6 alkyl;R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR21a;R23a is C1-6 alkyl; andR24 is H. - The method of claim 91, wherein:R21 is Bz, An, or pCF3Bz;R22 is Boc; andR23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc.
- The method of claim 91 or claim 92, wherein the compound of formula (IIIb) is:



- The method of claim 91 or claim 92, wherein the compound of formula (IVb) is:


- The method of claim 94, wherein the compound of formula (IVb) is

- The method of any one of claims 91-95, wherein the alkylation conditions include reaction in THF, 1,4-dioxane, MTBE, toluene, cyclohexane, or methylcyclohexane, or a combination thereof.
- The method of claim 96, wherein the alkylation conditions include reaction in methylcyclohexane.
- The method of any one of claims 91-97, wherein the Pd(0) catalyst is Pd2(pmdba)3 or Pd2(dba)3.
- The method of claim 98, wherein the Pd(0) catalyst is Pd2(pmdba)3.
- The method of any one of claims 91-99, wherein the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol % relative to the compound of formula (IIIb).
- The method of claim 100, wherein the Pd(0) catalyst is used in an amount of about 4 mol % relative to the compound of formula (IIIb).
- The method of any one of claims 91-101, wherein the ligand L is:


- The method of claim 102, wherein L is (S)-(CF3)3-tBuPHOX.
- The method of any one of claims 91-103, wherein the compound of formula (IVb) is enantioenriched.
- A method of synthesizing a medicinal product, the method comprising:preparing a compound of formula (IVb) according to any one of claims 91-104; andsynthesizing a medicinal product from the compound of formula (IVb).
- The method of claim 105, wherein the synthesizing the medicinal product from the compound of formula (IVb) comprises:
treating the compound of formula (IVb)





- The method of claim 106, wherein R23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc.
- The method of claim 107, wherein R23 is CH2Ph.
- A compound of formula (E):

wherein, as valence and stability permit,R23 is halogen, alkynyl, alkenyl, or C1-6 alkyl, each optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, alkoxycarbonyl, or -NHC(O)OR21a;R23a is C1-6 alkyl; andR24 is H. - The compound of claim 109, wherein R23 is CH3, CH2Ph, CH2CCl=CH2, F, CH2CCH, CH2CH2CO2CH3, CH2CH2CN, or CH2NHBoc.
- The compound of claim 110, wherein R23 is CH2Ph.
- A method for preparation of a compound of formula (VI):

treating a compound of formula (V):
with a Pd(0) catalyst and a ligand L, under alkylation conditions,
wherein, as valence and stability permit,
R25 is C1-6 alkyl, optionally substituted with halogen, OH, CN, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen. - The method of claim 112, wherein the compound of formula (VI) is:



- The method of claim 112 or claim 113, wherein alkylation conditions include reaction in hexanes, toluene, or a mixture thereof.
- The method of any one of claims 112-114, wherein the Pd(0) catalyst is Pd2(dba)3 or Pd2(pmdba)3.
- The method of claim 115, wherein the Pd(0) catalyst is Pd2(pmdba)3.
- The method of any one of claims 112-116, wherein the Pd(0) catalyst is used in an amount from about 0.01 mol % to about 10 mol %.
- The method of claim 117, wherein the Pd(0) catalyst is used in an amount of about 4 mol %.
- The method of any one of claims 112-118, wherein L is:


- The method of claim 119, wherein L is (S)-L1.
- The method of any one of claims 112-120, wherein L is used in an amount of from about 0.1 mol % to about 100 mol %.
- The method of claim 121, wherein L is used in an amount of about 10 mol %.
- The method of any one of claims 112-122, wherein the compound of formula (VI) is enantioenriched.
- The method of any one of claims 112-123, wherein the method further comprises hydrolyzing the compound of formula (VI), to form a compound of formula (VII):

- A method for preparation of a compound of formula (VIII):

treating a compound of formula (IVa):
wherein, as valence and stability permit,R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is Boc;R23 is H, or is C1-6 alkyl optionally substituted with CN, alkoxy, aryloxy, acyl, or -NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;R24 is H or halogen;R23b is H; or is C1-6 alkyl optionally substituted with halogen, OH, CN, alkoxy, aryloxy, amino, acyl, or
-NHC(O)OR23c; andR23c is (C6-10 aryl)alkyl or (C5-9 heteroaryl)alkyl. - The method of claim 125, wherein:R21 is Bz or An;R22 is Boc;R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, orCH2NHBoc;R24 is H or Cl; andR23b is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, CH2NHCbz, or CH2NH2.
- The method of claim 125 or claim 126, wherein:R21 is Bz;R22 is Boc;R23 is CH2NHBoc;R24 is H; andR23b is CH2NH2.
- The method of any one of claims 125-127, wherein the solvent is CH2Cl2.
- A method for the preparation of a compound of formula (IX):

treating a compound of formula (IVa):
with LiOH in a first solvent,
wherein, as valence and stability permit,R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is Boc;R23 is H, or C1-6 alkyl optionally substituted with halogen, OH, cyano, alkoxy, aryloxy, acyl, or
-NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;R23d is H, or is C1-6 alkyl optionally substituted with halogen, OH, alkoxy, aryloxy, acyl, or -NHC(O)OR23a; andR24 is H or halogen. - The method of claim 129, wherein:R21 is Bz or An;R23d is H, CH3, CH2Ph, CH2OBn, CH2OH, CH2CH2C(O)CH3, CH2CH2CH(OH)CH3, CH2NHCbz, CH2NHBoc; andR24 is H or Cl.
- The method of claim 129 or claim 130, wherein:R21 is Bz;R22 is Boc;R23 is CH2NHBoc;R23d is CH2NHBoc; andR24 is H.
- The method of any one of claims 129-131, wherein the first solvent comprises methanol, water, or a mixture thereof.
- The method of any one of 129-132, wherein the first solvent comprises methanol and water.
- The method of any one of claims 129-133, wherein the method further comprises preparing a compound of formula (X):

treating the compound of formula (IX) with LiAlH4 in a second solvent. - The method of claim 134, wherein the second solvent comprises THF.
- The method of claim 134 or claim 135, wherein:R22 is Boc;R23d is CH2NHBoc; andR24 is H.
- A method of preparation of a compound of formula (XI):

treating a compound of formula (IVa):
with hydrogen gas in the presence of a Pd/C catalyst in a first solvent, to form a first product mixture;
filtering the first product mixture, to form a crude first product; and
treating the crude first product with TFA in a second solvent;
wherein, as valence and stability permit,R21 is -C(O)aryl or -C(O)heteroaryl, optionally substituted with alkoxy, alkyl, or haloalkyl;R22 is Boc;R23 is H, or is C1-6 alkyl optionally substituted with halogen, OH, alkoxy, aryloxy, CN, aryl, or
-NHC(O)OR23a;R23a is C1-6 alkyl, (C6-10 aryl)alkyl, or (C5-9 heteroaryl)alkyl;R23e is H, or is C1-6 alkyl optionally substituted with halogen, OH, aryl, aryloxy, CN, acyl, or amino; andR24 is H or halogen. - The method of claim 137, wherein:R21 is Bz or An;R22 is Boc;R23 is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, or CH2NHBoc;R23e is H, CH3, CH2Ph, CH2OBn, CH2CH2CN, CH2CH2C(O)CH3, or CH2NH2;R24 is H or Cl.
- The method of claim 137 or claim 138, wherein:R21 is Bz;R23 is CH3;R23e is CH3; andR24 is H.
- The method of any one of claims 137-139, wherein the first solvent comprises methanol.
- The method of any one of claims 137-140, wherein the second solvent comprises CH2Cl2, ethyl acetate, or a mixture thereof.
- The method of any one of claims 137-141, wherein the method further comprises preparation of a compound of formula (XII):

treating a compound of formula (XI):
with Pd(OPiv)2, xantphos, AgOPiv, and benzoquinone in the presence of CO(g) in a third solvent;
wherein, as valence and stability permit,R21 is -C(O)aryl or -C(O)heteroaryl;R23e is methyl; andR24 is H or halogen. - The method of claim 142, wherein R24 is H.
- The method of claim 142 or claim 143, wherein R21 is Bz or An.
- The method of any one of claims 142-144, wherein:R21 is Bz;R23e is CH3; andR24 is H.
- The method of any one of claims 142-145, wherein the third solvent comprises toluene.
- A method of preparation of a compound of formula (XIII):

treating a compound of formula (VI):
with TFA in a first solvent, to form a first product;
treating the first product with lithium hydroxide in a second solvent, to form a second product;
treating the second product with Boc2O in the presence of a base, to form a third product;
treating the third product with K2CO3 and R13-X21 in a third solvent, to form a compound of formula (XIII);
wherein, as valence and stability permit,R25 is C1-6 alkyl, optionally substituted with halogen, CN, OH, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen;R25a is C1-6 alkyl, optionally substituted with halogen, CN, OH, aryl, heteroaryl, aryloxy, or alkynyl; halogen; or allyl, optionally substituted with halogen;R13 is aralkyl or hetaralkyl; andX21 is halogen. - The method of claim 147, wherein:R25 is CH3, CH2CH3, CH2CH2CO2CH3, CH2CCl=CH2, CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH;R25a is CH3, CH2CH3, CH2CH2CO2CH3, CH2CCl=CH2, CH2Ph, CH2CH2CN, CH2OBn, F, or CH2CCH; andR13 is Bn.
- The method of claim 147 or claim 148, wherein:R25 is CH2Ph;R25a is CH2Ph; andX21 is Br.
- The method of any one of claims 147-149, wherein the first solvent comprises dichloromethane.
- The method of any one of claims 147-150, wherein the second solvent comprises methanol.
- The method of any one of claims 147-151, wherein the third solvent comprises DMF.
- A method of synthesizing a medicinal product, the method comprising:preparing a compound of formula (VI) according to any one of claims 112-123; andsynthesizing a medicinal product from the compound of formula (VI).
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862747511P | 2018-10-18 | 2018-10-18 | |
| US201862749591P | 2018-10-23 | 2018-10-23 | |
| US201962792811P | 2019-01-15 | 2019-01-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| EP3699173A1 true EP3699173A1 (en) | 2020-08-26 |
Family
ID=68296159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP19204164.8A Withdrawn EP3699173A1 (en) | 2018-10-18 | 2019-10-18 | Gem-disubstituted pyrrolidines, piperazines, and diazepanes, and compositions and methods of making the same |
Country Status (2)
| Country | Link |
|---|---|
| US (3) | US11214568B2 (en) |
| EP (1) | EP3699173A1 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130267699A1 (en) | 2011-06-24 | 2013-10-10 | California Institute Of Technology | Quaternary heteroatom containing compounds |
| US10421696B2 (en) | 2014-12-18 | 2019-09-24 | California Institute Of Technology | Enantioselective synthesis of α-quaternary mannich adducts by palladium-catalyzed allylic alkylation |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001062726A2 (en) | 2000-02-23 | 2001-08-30 | Ucb, S.A. | 2-oxo-1-pyrrolidine derivatives, processes for preparing them and their uses |
| WO2011154374A1 (en) * | 2010-06-09 | 2011-12-15 | Janssen Pharmaceutica Nv | 5-amino-3,6-dihydro-1h-pyrazin-2-one derivatives useful as inhibitors of beta-secretase (bace) |
| WO2017156239A1 (en) * | 2016-03-11 | 2017-09-14 | California Institute Of Technology | Compositions and methods for acylating lactams |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE668489C (en) | 1937-08-26 | 1938-12-05 | Hoffmann La Roche & Co Akt Ges | Process for the preparation of 2,4-dioxo-3, 3-dialkyltetrahydropyridine-6-carboxylic acid esters or the corresponding free acids |
| US2886487A (en) | 1955-08-22 | 1959-05-12 | Ortho Pharma Corp | Quaternary ammonium salt and n-thiotrichloromethyl hydantoin vaginal composition |
| DE3242252A1 (en) | 1982-11-15 | 1984-05-17 | Bayer Ag, 5090 Leverkusen | HETEROCYCLICALLY SUBSTITUTED HYDROXYALKYL-AZOLYL DERIVATIVES |
| DE4035961A1 (en) | 1990-11-02 | 1992-05-07 | Thomae Gmbh Dr K | CYCLIC IMINODERIVATES, MEDICAMENTS CONTAINING SUCH COMPOUNDS AND METHOD FOR THE PRODUCTION THEREOF |
| AU7836594A (en) | 1994-03-14 | 1995-10-03 | Merck & Co., Inc. | Pyridyl ethylation of lactam derivatives |
| TR200200118T2 (en) | 1999-07-21 | 2002-12-23 | Astrazeneca Ab | New combinations |
| US7244721B2 (en) | 2000-07-21 | 2007-07-17 | Schering Corporation | Peptides as NS3-serine protease inhibitors of hepatitis C virus |
| CA2528592A1 (en) | 2003-06-17 | 2005-02-10 | Vicuron Pharmaceuticals Inc. | Novel lincomycin derivatives possessing antimicrobial activity |
| WO2005037823A1 (en) | 2003-10-16 | 2005-04-28 | Yuhan Corporation | Process for preparing 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1h-imidazole-1-yl)methyl]-4h-carbazol-4-one or its salt |
| US7696356B2 (en) | 2004-08-17 | 2010-04-13 | Taro Pharmaceutical Industries Limited | Process for preparing 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one and ondansetron therefrom |
| US7235698B2 (en) | 2004-08-27 | 2007-06-26 | California Institute Of Technology | Enantioselective, catalytic allylation of ketones and olefins |
| DE102007028406A1 (en) | 2007-06-20 | 2008-12-24 | Bayer Healthcare Ag | Substituted oxazolidinones and their use |
| TWI457122B (en) | 2007-07-20 | 2014-10-21 | Orion Corp | 2,3-dihydrobenzo[1,4]dioxin-2-ylmethyl derivatives as alpha2c antagonists for use in the treatment of peripheric and central nervous system diseases |
| EP2297096A2 (en) | 2008-06-18 | 2011-03-23 | F. Hoffmann-La Roche AG | Aryl ketone as mri |
| JO2998B1 (en) | 2010-06-04 | 2016-09-05 | Amgen Inc | Piperidinone derivatives as mdm2 inhibitors for the treatment of cancer |
| US20130267699A1 (en) | 2011-06-24 | 2013-10-10 | California Institute Of Technology | Quaternary heteroatom containing compounds |
| US8822679B2 (en) | 2011-06-24 | 2014-09-02 | California Institute Of Technology | Quaternary heteroatom containing compounds |
| US9518034B2 (en) | 2013-10-14 | 2016-12-13 | California Institute Of Technology | Synthesis of chiral enaminones, their derivatives, and bioactivity studies thereof |
| US20160096810A1 (en) | 2014-10-07 | 2016-04-07 | Brian M. Stoltz | Enantioselective synthesis of dialkylated n,o-heterocycles by palladium-catalyzed allylic alkylation |
| US10421696B2 (en) | 2014-12-18 | 2019-09-24 | California Institute Of Technology | Enantioselective synthesis of α-quaternary mannich adducts by palladium-catalyzed allylic alkylation |
| US10106479B2 (en) | 2015-03-27 | 2018-10-23 | California Institute Of Technology | Asymmetric catalytic decarboxylative alkyl alkylation using low catalyst concentrations and a robust precatalyst |
| EP3480190B1 (en) | 2017-11-01 | 2023-01-04 | California Institute of Technology | Methods for enantioselective allylic alkylation of esters, lactones, and lactams with unactivated allylic alcohols |
-
2019
- 2019-10-18 EP EP19204164.8A patent/EP3699173A1/en not_active Withdrawn
- 2019-10-18 US US16/657,672 patent/US11214568B2/en active Active
-
2021
- 2021-12-31 US US17/566,915 patent/US20220213081A1/en active Pending
- 2021-12-31 US US17/566,919 patent/US20220119377A1/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001062726A2 (en) | 2000-02-23 | 2001-08-30 | Ucb, S.A. | 2-oxo-1-pyrrolidine derivatives, processes for preparing them and their uses |
| WO2011154374A1 (en) * | 2010-06-09 | 2011-12-15 | Janssen Pharmaceutica Nv | 5-amino-3,6-dihydro-1h-pyrazin-2-one derivatives useful as inhibitors of beta-secretase (bace) |
| WO2017156239A1 (en) * | 2016-03-11 | 2017-09-14 | California Institute Of Technology | Compositions and methods for acylating lactams |
| US10040784B2 (en) | 2016-03-11 | 2018-08-07 | California Institute Of Technology | Compositions and methods for acylating lactams |
Non-Patent Citations (41)
| Title |
|---|
| "The McGraw-Hill Dictionary of Chemical Terms", 1985, MCGRAW-HILL |
| A. CHOLLETG. MORIC. MENENDEZF. RODRIGUEZI. FABINGM. R. PASCAJ. MADACKIJ. KORDULAKOVAP. CONSTANTA. QUEMARD ET AL., EUR. J. MED. CHEM., vol. 101, 2015, pages 218 - 235 |
| A. GUGGISBERGK. DRANDAROVM. HESSE, HELVETICA CHIMICA ACTA, vol. 83, 2000, pages 3035 - 3042 |
| ALEXANDER M TAYLOR ET AL: "Palladium-Catalyzed Enantioselective alpha-Arylation and alpha-Vinylation of Oxindoles Facilitated by an Axially Chiral P-Stereogenic Ligand", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, AMERICAN CHEMICAL SOCIETY, US, vol. 131, no. 29, 29 July 2009 (2009-07-29), pages 9900 - 9901, XP008138085, ISSN: 0002-7863, [retrieved on 20090706], DOI: 10.1021/JA903880Q * |
| ANGEW. CHEM., vol. 117, 2005, pages 7084 - 7087 |
| ANGEW. CHEM., vol. 120, 2008, pages 6979 - 6982 |
| ANGEW. CHEM., vol. 125, 2013, pages 6850 - 6853 |
| C. M. REEVESC. EIDAMSHAUSJ. KIMB. M. STOLTZ, ANGEW. CHEM. INT. ED., vol. 52, 2013, pages 6718 - 6721 |
| CAN. J. CHEM., vol. 62, 1984, pages 2560 - 2565 |
| CARINA I. JETTE ET AL: "Palladium-Catalyzed Construction of Quaternary Stereocenters by Enantioselective Arylation of [gamma]-Lactams with Aryl Chlorides and Bromides", ANGEWANDTE CHEMIE, vol. 58, no. 13, 22 March 2019 (2019-03-22), DE, pages 4297 - 4301, XP055714704, ISSN: 1433-7851, DOI: 10.1002/anie.201814475 * |
| CHEM. EUR. J., vol. 19, 2013, pages 4414 - 4418 |
| CHEM. SCI., vol. 10, 2019, pages 5996 - 6000 |
| CHENG-KANG MAI ET AL: "alpha-Arylation of 3-Aryloxindoles", ORGANIC LETTERS, AMERICAN CHEMICAL SOCIETY, US, vol. 12, no. 10, 21 May 2010 (2010-05-21), pages 2306 - 2309, XP008138081, ISSN: 1523-7060, [retrieved on 20100428], DOI: 10.1021/OL100666V * |
| D. C. BEHENNAB. M. STOLTZ, J. AM. CHEM. SOC., vol. 126, 2004, pages 15044 - 15045 |
| D. C. BEHENNAY. LIUT. YURINOJ. KIMD. E. WHITES. C. VIRGILB. M. STOLTZ, NAT. CHEM., vol. 4, 2012, pages 130 - 133 |
| DOUGLAS C. BEHENNA ET AL: "Enantioselective construction of quaternary N-heterocycles by palladium-catalysed decarboxylative allylic alkylation of lactams", NATURE CHEMISTRY, vol. 4, no. 2, 18 December 2011 (2011-12-18), pages 130 - 133, XP055146417, ISSN: 1755-4330, DOI: 10.1038/nchem.1222 * |
| HARI MANGUNURU ET AL: "Enantioselective Arylation of Oxindoles Using Modified BI-DIME Ligands", SYNTHESIS, vol. 50, no. 22, 28 June 2018 (2018-06-28), STUTTGART, DE., pages 4435 - 4443, XP055714729, ISSN: 0039-7881, DOI: 10.1055/s-0036-1591590 * |
| HARRISON ET AL.: "Compendium of Synthetic Organic Methods", vol. 1-8, 1971, JOHN WILEY & SONS |
| J. AM. CHEM. SOC., vol. 130, 2008, pages 810 - 811 |
| J. AM. CHEM. SOC., vol. 133, 2011, pages 8525 - 8527 |
| J. ORGANOMET. CHEM., vol. 65, 1999, pages 253 - 266 |
| J. R. CABRERA-PARDOA. TROWBRIDGEM. NAPPIK. OZAKIM. J. GAUNT, ANGEW. CHEM. INT. ED., vol. 56, 2017, pages 11958 - 11962 |
| J. STREUFFD. E. WHITES. C. VIRGILB. M. STOLTZ, NAT. CHEM., vol. 2, 2010, pages 192 - 196 |
| J. T. MOHRD. C. BEHENNAA. M. HARNEDB. M. STOLTZ, ANGEW. CHEM. INT. ED., vol. 44, 2005, pages 6924 - 6927 |
| K. DRANDAROVA. GUGGISBERGM. HESSE, HELVETICA CHIMICA ACTA, vol. 85, 2002, pages 979 - 989 |
| K. M. KORCHC. EIDAMSHAUSD. C. BEHENNAS. NAMD. HOMEB. M. STOLTZ, ANGEW. CHEM. INT. ED., vol. 54, 2015, pages 179 - 183 |
| K. TANID. C. BEHENNAR. M. MCFADDENB. M. STOLTZ, ORG. LETT., vol. 9, 2007, pages 2529 - 2531 |
| KANKANALA KAVITHA ET AL: "Chemistry of Cyclic Imides: An Overview on the Past, Present and Future", CURRENT ORGANIC CHEMISTRY, vol. 20, no. 19, 12 July 2016 (2016-07-12), NL, pages 1955 - 2001, XP055714991, ISSN: 1385-2728, DOI: 10.2174/1385272820666160530145014 * |
| KLEPACZ, A.ZWIERZAK, A., TETRAHEDRON LETT., vol. 43, 2002, pages 1079 - 1080 |
| M. E. CHILDSW . P. WEBER, J. ORG. CHEM., vol. 41, 1976, pages 3486 - 3487 |
| M. R. KROUTJ. T. MOHRB. M. STOLTZ, ORG. SYNTH., vol. 86, 2009, pages 181 - 193 |
| M. SETOJ. L. ROIZENB. M. STOLTZ, ANGEW. CHEM. INT. ED., vol. 47, 2008, pages 6873 - 6876 |
| MANGION, I. K.SHERRY, B. D.YIN, J.FLEITZ, F. J.: "Enantioselective Synthesis of a Dual Orexin Receptor Antagonist", ORG. LETT., vol. 14, 2012, pages 3458 - 3461, XP002754730, DOI: 10.1021/ol3014123 |
| MOTULSKY: "Intuitive Biostatistics", 1995, OXFORD UNIVERSITY PRESS, INC. |
| NATURE CHEM., vol. 4, 2012, pages 130 - 133 |
| ORG. LETT., vol. 14, 2012, pages 3458 - 3461 |
| ORG. LETT., vol. 6, 2004, pages 4435 - 4438 |
| RYAN A ALTMAN ET AL: "Orthogonal Pd- and Cu-Based Catalyst Systems for C- and N-Arylation of Oxindoles", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, AMERICAN CHEMICAL SOCIETY, US, vol. 130, no. 29, 23 July 2008 (2008-07-23), pages 9613 - 9620, XP008138086, ISSN: 0002-7863, [retrieved on 20080628], DOI: 10.1021/JA803179S * |
| SIKRIWAL, D.KANT, R.MAULIK, P. R.DIKSHIT, D. K., TETRAHEDRON, vol. 66, 2010, pages 6167 - 6173 |
| TETRAHEDRON LETT., vol. 51, 2010, pages 5550 - 5554 |
| YANHUI LU ET AL: "Palladium-Catalyzed Enantioselective C sp 3 -C sp 3 Cross-Coupling for the Synthesis of (Poly)fluorinated Chiral Building Blocks", ORGANIC LETTERS, vol. 20, no. 18, 21 September 2018 (2018-09-21), US, pages 5657 - 5660, XP055714798, ISSN: 1523-7060, DOI: 10.1021/acs.orglett.8b02369 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20220119377A1 (en) | 2022-04-21 |
| US20220213081A1 (en) | 2022-07-07 |
| US20200199114A1 (en) | 2020-06-25 |
| US11214568B2 (en) | 2022-01-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220119377A1 (en) | Gem-disubstituted pyrrolidines, piperazines, and diazepanes, and compositions and methods of making the same | |
| Volkmann et al. | 2-Thioalkyl penems: an efficient synthesis of sulopenem, a (5R, 6S)-6-(1 (R)-hydroxyethyl)-2-[(cis-1-oxo-3-thiolanyl) thio]-2-penem antibacterial | |
| AU2011261263B2 (en) | Piperidinone derivatives as MDM2 inhibitors for the treatment of cancer | |
| EP3538525B1 (en) | 3-substituted propionic acids as alpha v integrin inhibitors | |
| Dener et al. | . alpha.-Acylamino radical cyclizations: application to the synthesis of (-)-swainsonine | |
| CN102348698B (en) | Inhibitors of beta-secretase | |
| CA3128062A1 (en) | Benzopyridone heterocyclic compound and use thereof | |
| TWI498115B (en) | Imidazole carbonyl compounds | |
| JPWO2008072655A1 (en) | Imidazothiazole derivatives | |
| EP3347355B1 (en) | Heterotricyclic sulfonamides as anti-cancer agents | |
| KR20060023152A (en) | 3-substituted 5,6-diaryl-pyrazine-2-carboxamide and -2-sulfonamide derivatives as cb1 modulators | |
| EP3480190B1 (en) | Methods for enantioselective allylic alkylation of esters, lactones, and lactams with unactivated allylic alcohols | |
| CN116917286A (en) | 6-carbamate substituted heteroaromatic ring derivatives | |
| WO2023284651A1 (en) | N-(2-aminophenyl)benzamide compound and application thereof | |
| Pattenden et al. | Total synthesis of (−)-ulapualide A, a novel tris-oxazole macrolide from marine nudibranchs, based on some biosynthesis speculation | |
| CN101622240B (en) | Fused substituted aminopyrrolidine derivative | |
| Muratake et al. | Synthesis of teleocidins A, B and their congeners. Part 2. Synthesis of lyngbyatoxin A (teleocidin A-1), teleocidin A-2, pendolmycin, and (R, E)-and (S, E)-7-(3, 7, 11-trimethyl-1, 6, 10-dodecatrien-3-yl)-(−)-indolactams V | |
| JP2009298713A (en) | Imidazothiazole derivative | |
| JP2022527213A (en) | Process for preparing alpha-carboxamide pyrrolidine derivatives | |
| JP3761096B2 (en) | Process for producing 2-alkyl-substituted carbapenem derivatives | |
| AU2005233648A1 (en) | Methods for minimizing thioamide impurities | |
| Golliher et al. | The synthesis and use of γ-chloro-enamides for the subsequent construction of novel enamide-containing small molecules | |
| US3711464A (en) | 1-formyl-2-alpha-(2-acyloxy-2-propylmercapto)-3 alpha-n-acyl-aminoazetidin-4-ones and process for their preparation | |
| Choi et al. | Synthesis of mutidentate imidazole-containing macrocycles | |
| CN103635474A (en) | Tricyclic antibiotics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION HAS BEEN PUBLISHED |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: BA ME |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: REQUEST FOR EXAMINATION WAS MADE |
|
| 17P | Request for examination filed |
Effective date: 20210224 |
|
| RBV | Designated contracting states (corrected) |
Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: EXAMINATION IS IN PROGRESS |
|
| 17Q | First examination report despatched |
Effective date: 20220503 |
|
| P01 | Opt-out of the competence of the unified patent court (upc) registered |
Effective date: 20230516 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION IS DEEMED TO BE WITHDRAWN |
|
| 18D | Application deemed to be withdrawn |
Effective date: 20230803 |