EP2326350B1 - Compounds for killing psma-expressing, taxane-resistant cancer cells - Google Patents
Compounds for killing psma-expressing, taxane-resistant cancer cells Download PDFInfo
- Publication number
- EP2326350B1 EP2326350B1 EP09789284.8A EP09789284A EP2326350B1 EP 2326350 B1 EP2326350 B1 EP 2326350B1 EP 09789284 A EP09789284 A EP 09789284A EP 2326350 B1 EP2326350 B1 EP 2326350B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- psma
- antibody
- abgenix
- seq
- nucleic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Not-in-force
Links
Images













Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6869—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of the reproductive system: ovaria, uterus, testes, prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
Definitions
- Prostate cancer is the most common malignancy and the second leading cause of cancer death in men in the United States ( Jemal A, et al., CA Cancer J Clin 2005;55:10-30 ).
- Localized prostate cancer typically is treated with surgery or radiation, and recurrent disease can be controlled temporarily with androgen ablation ( Klein EA, et al., Urol Clin North Am 2003;30:315-30 ).
- Klein EA et al., Urol Clin North Am 2003;30:315-30
- almost all prostate carcinomas eventually become hormone refractory and then rapidly progress ( Denmeade SR, et al., Nat Rev Cancer 2002;2:389-96 ).
- Hormone-refractory or androgen-independent prostate cancer has proven to be largely resistant to conventional chemotherapy.
- MLN2704 comprises J591, a deimmunized monoclonal antibody targetingan external domain of PSMA and the maytansine analog drug maytansoid-1 (DM1).
- the present invention relates, at least in part, to the surprising discovery that antibody-drug conjugates (ADCs) comprising an antibody or antigen-binding fragment thereof that specifically binds PSMA conjugated to a dolastatin 10 derivative, in particular auristatins such as, MMAE (also referred to herein as monomethylauristatin E or monomethylauristatin norephedrine) or MMAF (also referred to herein as monomethylauristatin F or monomethylauristatin phenylalanine) can be used to kill PSMA-expressing, taxane-resistant cancer cells and to treat PSMA-expressing, taxane-resistant cancer disclosed herein, therefore, are methods for killing PSMA-expressing, taxane-resistant cancer cells.
- ADCs antibody-drug conjugates
- the methods involve killing PSMA-expressing, taxane-resistant cancer cells.
- the methods involve delaying or inhibiting progression of the cancer.
- the methods involve increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy.
- the methods involve decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level.
- CTCs circulating level of circulating tumor cells
- the methods involve decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA.
- a method for killing PSMA-expressing, taxane-resistant cancer cells comprising contacting the PSMA-expressing, taxane-resistant cancer cells with an antibody-drug conjugate in an amount effective to kill the PSMA-expressing, taxane-resistant cancer cells, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1, is disclosed.
- PSMA prostate-specific membrane antigen
- a method for treating a subject that has a PSMA-expressing, taxane-resistant cancer comprising administering to the subject that has the PSMA-expressing, taxane-resistant cancer an antibody-drug conjugate in an amount effective to treat the PSMA-expressing, taxane-resistant cancer, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1, is also disclosed.
- the method involves killing PSMA-expressing, taxane-resistant cancer cells.
- the method involves delaying or inhibiting progression of the cancer.
- the method involves increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy.
- the method involves decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level.
- the method involves decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA.
- a method for treating a subject having progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy comprising administering an effective amount of an antibody-drug conjugate, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1, is also provided.
- PSMA prostate-specific membrane antigen
- the PSMA antibody drug conjugate (also referred to herein as "PSMA ADC” or “ADC”) consists essentially of a human monoclonal antibody to PSMA conjugated to monomethylaurastatin norephedrine (MMAE) via a valine-citrulline linker.
- the method involves killing PSMA-expressing, taxane-resistant cancer cells.
- the method involves delaying or inhibiting progression of the cancer.
- the method involves increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy.
- the method involves decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level.
- the method involves decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA.
- the effective amount of the ADC is sufficient to 1) delay or inhibit progression of the cancer, 2) increase survival of the subject as compared to the median survival of subjects who have not been treated with PSMA ADC and who have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, 3) decrease a circulating level of circulating tumor cells (CTCs) compared to a baseline level and/or 4) decrease or stabilize a serum level of prostate specific antigen (PSA) compared to a baseline level of PSA.
- CTCs circulating level of circulating tumor cells
- PSA prostate specific antigen
- the killing of the PSMA-expressing, taxane-resistant cancer cells or the delay or inhibition of progression of the cancer is demonstrated by radiographic image changes in tumor burden compared to a baseline radiographic image in the subject prior to the administration of the PSMA ADC.
- the radiographic image change is a change of at least 10%.
- the radiographic image change is a change of at least 20%.
- the radiographic image change is a change of at least 30%.
- the radiographic image change is a change of at least 40%.
- the radiographic image change is a change of at least 50%.
- the radiographic image change is a change of at least 60%.
- the killing of the PSMA-expressing, taxane-resistant cancer cells or the delay or inhibition of progression of cancer is demonstrated by a change in at least one biomarker for bone metastasis and bone metabolism compared to a baseline value prior to the administration of the PSMA ADC.
- the biomarker is N-telopeptide, bone alkaline phosphatase, osteocalcin, calcitonin, calcium, pyridinoline or deoxypyridinoline.
- treatment with the PSMA ADC results in increased survival for the subject, wherein the survival is increased in comparison to the median survival time of subjects with PSMA-expressing, taxane-resistant cancer not treated with PSMA ADC.
- survival in the subject is increased by four weeks.
- survival in the subject is increased by six weeks.
- survival in the subject is increased by two months.
- survival in the subject is increased by four months.
- survival in the subject is increased by six months.
- survival in the subject is increased by eight months.
- survival in the subject is increased by ten months.
- survival in the subject is increased by twelve months.
- survival in the subject is increased by fourteen months.
- treatment with the PSMA ADC results in an improvement in the subject's quality of life as compared to the quality of life of the subject prior to treatment with the PSMA ADC.
- the PSMA-expressing, taxane-resistant cancer cells are resistant to docetaxel or paclitaxel.
- the PSMA-expressing, taxane-resistant cancer is resistant to docetaxel or paclitaxel.
- the PSMA-expressing, taxane-resistant cancer cells are prostate cancer cells or non-prostate cancer cells.
- the non-prostate cancer cells are bladder cancer cells, pancreatic cancer cells, liver cancer cells, lung cancer cells, kidney cancer cells, sarcoma cells, breast cancer cells, brain cancer cells, neuroendocrine carcinoma cells, colon cancer cells, testicular cancer cells or melanoma cells.
- the PSMA-expressing, taxane-resistant cancer is prostate cancer or non-prostate cancer.
- the non-prostate cancer is bladder cancer, pancreatic cancer, liver cancer, lung cancer, kidney cancer, sarcoma, breast cancer, brain cancer, neuroendocrine carcinoma, colon cancer, testicular cancer or melanoma.
- the PSMA-expressing, taxane-resistant cancer cells are of a tumor.
- the PSMA-expressing, taxane-resistant cancer is a tumor.
- the volume of the tumor is at least 100mm 3 prior to initiation of the method using PSMA ADC.
- the volume of the tumor is at least 200mm 3 .
- the volume of the tumor is at least 300mm 3 .
- the volume of the tumor is at least 400mm 3 .
- the volume of the tumor is at least 500mm 3 .
- the volume of the tumor is at least 600mm 3 .
- the volume of the tumor is at least 700mm 3 . In a further embodiment, the volume of the tumor is at least 800mm 3 . In yet a further embodiment, the volume of the tumor is at least 900mm 3 . In another embodiment, the volume of the tumor is at least 1000mm 3 . In yet another embodiment, the volume of the tumor is at least 1200mm 3 . In still another embodiment, the volume of the tumor is at least 1400mm 3 . In yet a further embodiment, the volume of the tumor is at least 1600mm 3 .
- treatment or contact with the PSMA ADC results in a reduction of tumor volume by at least 10% compared to the tumor volume prior to the treatment or contact with the PSMA ADC.
- the volume of the tumor is reduced by at least 20%.
- the volume of the tumor is reduced by at least 30%.
- the volume of the tumor is reduced by at least 40%.
- the volume of the tumor is reduced by at least 50%.
- the volume of the tumor is reduced by at least 60%.
- the volume of the tumor is reduced by at least 70%.
- the volume of the tumor is reduced by at least 80%.
- the volume of the tumor is reduced by at least 90%.
- the volume of the tumor is reduced by at least 95%.
- the tumor is eradicated.
- the tumor has a length of at least 5mm prior to initiation of the method using PSMA ADC. In another embodiment, the tumor has a length of at least 6mm. In still another embodiment, the tumor has a length of at least 7mm. In yet another embodiment, the tumor has a length of at least 8mm. In another embodiment, the tumor has a length of at least 9mm. In still another embodiment, the tumor has a length of at least 10mm. In yet another embodiment, the tumor has a length of at least 11mm. In a further embodiment, the tumor has a length of at least 12mm. In still a further embodiment, the tumor has a length of at least 13mm. In still a further embodiment, the tumor has a length of at least 14mm.
- the tumor has a length of at least 15mm. In yet another embodiment, the tumor has a length of at least 16mm. In still another embodiment, the tumor has a length of at least 17mm. In a further embodiment, the tumor has a length of at least 18mm. In yet a further embodiment, the tumor has a length of at least 19mm. In still a further embodiment, the tumor has a length of at least 20mm. In another embodiment, the tumor has a length of at least 21mm. In still another embodiment, the tumor has a length of at least 22mm. In yet another embodiment, the tumor has a length of at least 23mm. In a further embodiment, the tumor has a length of at least 24mm. In still a further embodiment, the tumor has a length of at least 25mm. In yet a further embodiment, the tumor has a length of at least 30mm.
- treatment or contact with the PSMA ADC results in a reduction of tumor length by at least 10% compared to the tumor length prior to the treatment or contact with the PSMA ADC.
- the length of the tumor is reduced by at least 20%.
- the length of the tumor is reduced by at least 30%.
- the length of the tumor is reduced by at least 40%.
- the length of the tumor is reduced by at least 50%.
- the length of the tumor is reduced by at least 60%.
- the length of the tumor is reduced by at least 70%.
- the length of the tumor is reduced by at least 80%.
- the length of the tumor is reduced by at least 90%.
- the length of the tumor is reduced by at least 95%.
- the length of the tumor is reduced by at least 99%.
- administration of the PSMA ADC to a subject results in the gain of body weight in the subject (as compared to the weight of the subject prior to administration of the PSMA ADC to the subject).
- the gain in body weight may result from a single administrion of the PSMA ADC or a course of treatment with the PSMA ADC (i.e., more than one administration).
- the gain is at least a 5% gain in body weight.
- the gain is at least a 10% gain.
- the gain is at least a 15% gain.
- the gain is at least a 20% gain.
- the gain is at least a 25% gain.
- the gain is at least a 30% gain. In another embodiment, the gain is a 5% gain in body weight. In another embodiment, the gain is a 10% gain. In still another embodiment, the gain is a 15% gain. In still a further embodiment, the gain is a 20% gain. In yet another embodiment, the gain a 25% gain. In a further embodiment, the gain is a 30% gain.
- the PSMA-expressing, taxane-resistant cancer cells are of a metastasis. In another embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer is metastatic.
- the antibody or antigen-binding fragment thereof is conjugated to at least 2 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules. In yet another embodiment of the methods provided, the antibody or antigen-binding fragment thereof is conjugated to at least 3 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules. In still another embodiment of the methods provided, the antibody or antigen-binding fragment thereof is conjugated to at least 4 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules.
- the monomethylauristatin norephedrine or monomethylauristatin phenylalanine is conjugated to the antibody or antigen-binding fragment thereof with a compound of the formula -A n -Y m -Z m -X n -W n -wherein, A is a carboxylic acyl unit; Y is an amino acid; Z is an amino acid; X and W are each a self-immolative spacer; n is an integer of 0 or 1; and m is an integer of 0 or 1, 2, 3, 4, 5 or 6.
- A is in which q is 1-10.
- A is 4-(N-succinimidomethyl)cyclohexane-1-carbonyl, m-succinimidobenzoyl, 4-(p-succinimidophenyl) -butyryl, 4-(2-acetamido)benzoyl, 3-thiopropionyl, 4-(1-thioethyl)-benzoyl, 6-(3-thiopropionylamido)-hexanoyl or maleimide caproyl.
- A is maleimide caproyl.
- Y is alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan or proline. In yet another embodiment, Y is valine.
- Z is lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, or citrulline. In yet a further embodiment, Z is citrulline.
- Y m -Z m is valine-citrulline. In another embodiment, Y m -Z m is a protein sequence which is selectively cleavable by a protease.
- X is a compound having the formula in which T is O, N, or S. In yet another embodiment, X is a compound having the formula -HN-R 1 -COT in which R 1 is C 1 -C 5 alkyl, T is O, N or S. In a further embodiment, X is a compound having the formula in which T is O, N, or S, R 2 is H or C 1 -C 5 alkyl. In yet a further embodiment, X is p-aminobenzylcarbamoyloxy. In still a further embodiment, X is p-aminobenzylalcohol. In another embodiment, X is p-aminobenzylcarbamate.
- X is p-aminobenzyloxycarbonyl.
- X is ⁇ -aminobutyric acid; ⁇ , ⁇ -dimethyl ⁇ -aminobutyric acid or ⁇ , ⁇ -dimethyl ⁇ -aminobutyric acid.
- W is in which T is O, S or N.
- n and n are 0.
- the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin norephedrine.
- the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin norephedrine.
- the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin phenylalanine.
- the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin phenylalanine.
- the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-monomethylauristatin phenylalanine.
- the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin norephedrine.
- the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin norephedrine.
- the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin phenylalanine.
- the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin phenylalanine.
- the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-monomethylauristatin phenylalanine.
- the monoclonal antibody or antigen-binding fragment thereof specifically binds to an extracellular domain of PSMA. In another embodiment, the monoclonal antibody or antigen-binding fragment thereof specifically binds to a conformational epitope of PSMA. In still another embodiment, the monoclonal antibody or antigen-binding fragment thereof binds live cells. In a further embodiment, the monoclonal antibody or antigen-binding fragment thereof does not require cell lysis to bind PSMA. In still a further embodiment, the monoclonal antibody or antigen-binding fragment thereof binds to cells of the neovasculature of a tumor.
- the antibody or antigen-binding fragment thereof competitively inhibits the specific binding of a second antibody to its target epitope on PSMA, wherein the second antibody is selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.7
- the antibody or antigen-binding fragment thereof binds to an epitope on PSMA defined by an antibody selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, Abgenix 4.152.1 and antibodies comprising
- the second antibody is selected from the group consisting of AB-PG1-XG1-006, AB-PG1-XG1-026 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or
- the second antibody comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 14, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 16.
- the second antibody comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 20.
- the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 90% identical to a nucleotide sequence encoding an antibody selected from the group consisting of: AB-PG1-XG1-006, AB-PG1-XG1-026 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of
- the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 95% identical. In still another embodiment, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 97% identical. In yet another embodiment, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 98% identical. In a further embodiment, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 99% identical.
- the antibody or antigen-binding fragment thereof is selected from the group consisting of antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected
- the antibody or antigen-binding fragment thereof comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 14, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 16, and antigen-binding fragments thereof.
- the antibody or antigen-binding fragment thereof comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 20, and antigen-binding fragments thereof.
- the antibody or antigen-binding fragment thereof is AB-PG1-XG1-006, AB-PG1-XG1-026 or an antigen-binding fragment thereof.
- the human antibody is an IgG1 comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8.
- an antibody-drug conjugate comprising an antibody, or antigen-binding fragment thereof, that specifically binds PSMA, conjugated to a dolastatin 10 derivative, for use in a method of treating a PSMA-expressing, taxane-resistant cancer.
- said method involves killing PSMA-expressing, taxane-resistant cancer cells.
- the method involves delaying or inhibiting progression of the cancer.
- the method involves increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy.
- the method involves decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level. In a further embodiment, the method involves decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA. It is also preferred that the PSMA-expressing, taxane-resistant cancer cells are killed by the conjugate.
- the PSMA preferably has the sequence set forth in SEQ ID NO: 1.
- the conjugate is preferably for treating progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy.
- the dolastatin 10 derivative can be an auristatin, and is preferably MMAE (also referred to herein as monomethylauristatin E or monomethylauristatin norephedrine) or MMAF (also referred to herein as monomethylauristatin F or monomethylauristatin phenylalanine).
- the conjugate can have any of the features of the conjugate employed in other aspects of the invention defined herein.
- the method involved in this further aspect moreover, can involve the administration of an effective amount of the conjugate, as defined with reference to other aspects defined herein. It can also have any of the features of any of the methods for treating a subject that are described herein.
- the present invention relates, in part, to the surprising discovery that antibody-drug conjugates (ADCs) comprising an antibody or antigen-binding fragment thereof that specifically binds PSMA conjugated to a dolastatin 10 derivative, in particular auristatins such as, MMAE (also referred to herein as monomethylauristatin E or monomethylauristatin norephedrine) or MMAF (also referred to herein as monomethylauristatin F or monomethylauristatin phenylalanine) can be used to kill PSMA-expressing, taxane-resistant cancer cells.
- ADCs antibody-drug conjugates
- MMAE also referred to herein as monomethylauristatin E or monomethylauristatin norephedrine
- MMAF also referred to herein as monomethylauristatin F or monomethylauristatin phenylalanine
- PSMA expressing cancer cells which had become resistant to the effects of an anti-cancer agent that acts by disrupting the microtubular network in cells, were sensitive to the effects of a second anti-cancer agent that also targets microtubules.
- the auristatin, MMAE, a potent tubulin inhibitor, in the form of an antibody-drug conjugate when delivered to PSMA-expressing prostate tumors that had become resistant to docetaxel was able to kill the docetaxel resistant cancer cells such that tumor volume was significantly decreased.
- PSMA-expressing cancer cells is intended to refer to cancer cells that express PSMA (e.g., human PSMA).
- PSMA is a 100 kD Type II membrane glycoprotein expressed in prostate tissues ( Horoszewicz et al., 1987, Anticancer Res. 7:927-935 ; U.S. Pat. No. 5,162,504 ).
- PSMA was characterized as a type II transmembrane protein having sequence identity with the transferrin receptor ( Israeli et al., 1994, Cancer Res. 54:1807-1811 ) and with NAALADase activity ( Carter et al., 1996, Proc. Natl. Acad. Sci. U.S.A. 93:749-753 ).
- PSMA is expressed in increased amounts in prostate cancer, and elevated levels of PSMA are also detectable in the sera of these patients (Horoszewicz et al., 1987; Rochon et al., 1994, Prostate 25:219-223 ; Murphy et al., 1995, Prostate 26:164-168 ; and Murphy et al., 1995, Anticancer Res. 15:1473-1479 ).
- PSMA expression in cancerous prostate is approximately 10-fold greater than that in normal prostate.
- Expression in normal prostate is approximately 10-fold greater than that in the brain and is 50- to 100-fold greater than that of the liver or kidney. In most normal tissues, no expression of PSMA is observed. PSMA expression increases with disease progression, becoming highest in metastatic, hormone-refractory disease.
- PSMA is also abundantly expressed on the neovasculature of a variety of non-prostate tumors, including bladder, breast, colon, pancreas, sarcoma, melanoma, renal, liver, lung (e.g., non-small cell lung carcinoma), and kidney tumors, but not on normal vasculature.
- PSMA-expressing cancer cells therefore, include, for example, prostate cancer cells as well as endothelial cells of the neovasculature of a number of non-prostate cancers.
- Tuxane-resistant cancer cells refers to cancer cells of a cancer that has been treated with a taxane but that has become resistant to treatment with the taxane (i.e., further treatment with the taxane would not slow or halt the progression of the cancer and/or would no longer provide a significant benefit to a subject with the cancer).
- a "subject with a taxane-resistant cancer” is one that has received prior treatment with a taxane but no longer experiences a significant benefit from further treatment with the taxane (e.g., no longer experiences a significant reduction in the number of cancer cells, in tumor volume, in tumor length, in the number of metastases and/or in other markers of disease progression and/or no longer experiences an improvement in quality of life and/or increased survival as compared to the expected survival time without further treatment with the taxane).
- the PSMA ADC is administered to a subject with progressive, metastatic, hormone-refractory prostate cancer resistant to taxane following prior taxane chemotherapy.
- such subject is a human.
- taxanes are anti-cancer agents that interfere with or disrupt microtubule stability, formation and/or function.
- agents include paclitaxel and docetaxel as well as derivatives thereof, wherein the derivatives function against microtubules by the same mode of action as the taxanes from which they are derived.
- the ADC can comprise an antibody or antigen-binding fragment thereof conjugated to the auristatin MMAE or MMAF.
- Auristatins are synthetic pentapeptide molecules that are structurally related to dolastatin 10, a natural product derived from a marine animal. MMAE and other auristatins act by inhibiting polymerization of tubulins, thereby preventing formation of the mitotic spindle. Apoptotic cell death is triggered when the cell cycle is arrested.
- the antibody or antigen-binding fragment thereof can be, in some embodiments, conjugated to MMAE or MMAF with a compound of the following formula (Formula 1): - A n -Y m -Z m -X n -W n -, wherein A is a carboxylic acyl unit; Y is an amino acid; Z is an amino acid; X and W are each a self-immolative spacer; n is an integer of 0 or 1; and m is an integer of 0 or 1, 2, 3, 4, 5 or 6.
- the ADC in some embodiments, is represented by the formula (Formula 2): L- ⁇ A n -Y m -Z m -X n -W n -D ⁇ p wherein L is an antibody or antigen-binding fragment thereof that binds PSMA, D is MMAE or MMAF and p is an integer of 1, 2, 3, 4, 5, 6, 7 or 8.
- L is an antibody or antigen-binding fragment thereof that binds PSMA
- D is MMAE or MMAF
- p is an integer of 1, 2, 3, 4, 5, 6, 7 or 8.
- the carboxylic unit "A n " is linked to the antibody or antigen-binding fragment via a sulfur atom derived from the antibody or antigen-binding fragment:
- A is in which q is 1-10. Therefore, in one embodiment, the conjugate is: wherein L, Y, Z, X, W, D, n, m, q and p are as previously defined.
- A is 4-(N-succinimidomethyl)cyclohexane-1-carbonyl, m-succinimidobenzoyl, 4-(p-succinimidophenyl)-butyryl, 4-(2-acetamido)benzoyl, 3-thiopropionyl, 4-(1-thioethyl)-benzoyl, 6-(3-thiopropionylamido)-hexanoyl or maleimide caproyl.
- A is maleimide caproyl.
- Representative examples of various carboxylic acyl units and methods for their synthesis and attachment are described in US Pat. No. 6,214,345 .
- Y is alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan or proline.
- Y is valine.
- Z is lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, or citrulline.
- Z is citrulline.
- Y m -Z m is valine-citrulline.
- Y m -Z m is a protein sequence which is selectively cleavable by a protease.
- X is a compound having the formula in which T is O, N, or S. In another embodiment, X is a compound having the formula -HN-R 1 -COT in which R 1 is C 1 -C 5 alkyl, T is O, N or S. In a further embodiment, X is a compound having the formula in which T is O, N, or S, R 2 is H or C 1 -C 5 alkyl. In one embodiment, X is p-aminobenzylcarbamoyloxy. In another embodiment, X is p-aminobenzylalcohol. In a further embodiment, X is p-aminobenzylcarbamate.
- X is p-aminobenzyloxycarbonyl.
- X is ⁇ -aminobutyric acid; ⁇ , ⁇ -dimethyl ⁇ -aminobutyric acid or ⁇ , ⁇ -dimethyl ⁇ -aminobutyric acid.
- W is in which T is O, S or N.
- the compound of Formula 1 is maleimidocaproyl.
- Maleimidocaproyl has been used for conjugation of two specific auristatins to an anti-CD30 mAb (AC10) ( Doronina, Svetlana et al. "Novel Linkers for Monoclonal Antibody-Mediated Delivery of Anticancer Agents", AACR, Anaheim, CA, Abstract No. 1421, April 16-20, 2005 ).
- Maleimidocaproyl reacts with thiol groups to form a thioether.
- MMAE or MMAF can be conjugated to an antibody or antigen-binding fragment thereof using methods known to those of ordinary skill in the art (e.g., See, Niemeyer, CM, Bioconjugation Protocols, Strategies and Methods, Humana Press, 2004 ) or as described herein.
- more than one MMAE or MMAF molecule is conjugated to the antibody or antigen-binding fragment thereof.
- 1, 2, 3, 4, 5, 6, 7 or 8 MMAE or MMAF molecules are conjugated to the antibody or antigen-binding fragment thereof.
- at least 2, 3, 4 or 5 MMAE or MMAF molecules are conjugated to the antibody or antigen-binding fragment thereof.
- 2, 3, 4 or 5 MMAE or MMAF molecules are conjugated to the antibody or antigen-binding fragment thereof.
- the antibodies or antigen-binding fragments thereof of the ADCs are any antibody or antigen-binding fragment thereof that specifically binds PSMA.
- the antibody or an antigen-binding fragment thereof specifically binds PSMA (e.g., specifically binds an extracellular domain of PSMA, specifically binds a conformational epitope of PSMA, etc.) and can competitively inhibit the specific binding of a second antibody to its target epitope on PSMA, wherein the second antibody is selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Ab
- the second antibody therefore, include any of the antibodies produced by the hybridomas or encoded by the plasmids shown below in Table 1.
- These hybridomas and plasmids were deposited pursuant to, and in satisfaction of, the requirements of the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedure with the American Type Culture Collection (“ATCC”) as an International Depository Authority and given the Patent Deposit Designations shown above and in Table 1.
- ATCC American Type Culture Collection
- cross-competition assays can be used to determine if an antibody or antigen-binding fragment thereof competitively inhibits binding to PSMA by another antibody or antigen-binding fragment thereof.
- cross-competition assays include cell-based methods employing flow cytometry or solid phase binding analysis.
- Other assays that evaluate the ability of antibodies or antigen-binding fragments thereof to cross-compete for PSMA molecules that are not expressed on the surface of cells, in solid phase or in solution phase, also can be used.
- the antibodies or antigen-binding fragments thereof competitively inhibit the specific binding of a second antibody to its target epitope on PSMA by at least about 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, or 99%. Inhibition can be assessed at various molar ratios or mass ratios; for example competitive binding experiments can be conducted with a 2-fold, 3-fold, 4-fold, 5-fold, 7-fold, 10-fold or more molar excess of a first antibody or antigen-binding fragment thereof over a second antibody or antigen-binding fragment thereof.
- the antibody or an antigen-binding fragment thereof specifically binds to an epitope on PSMA defined by an antibody selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, 4.248.2, 4.360.3, 4.7.1, 4.4.1, 4.177.3, 4.16.1, 4.22.3, 4.28.3, 4.40.2, 4.48.3, 4.49.1, 4.209.3, 4.219.3, 4.288.1, 4.333.1, 4.54.1, 4.153.1, 4.232.3, 4.292.3, 4.304.1, 4.78.1, and 4.152.1.
- fragments (peptides) of PSMA antigen e.g., synthetic peptides
- fragments (peptides) of PSMA antigen e.g., synthetic peptides
- a candidate antibody or antigen-binding fragment thereof binds the same epitope.
- fragments (peptides) of PSMA antigen e.g., synthetic peptides
- linear epitopes overlapping peptides of a defined length (e.g., 8 or more amino acids) are synthesized.
- the peptides can be offset by 1 amino acid, such that a series of peptides covering every 8 amino acid fragment of the PSMA protein sequence are prepared. Fewer peptides can be prepared by using larger offsets, e.g., 2 or 3 amino acids.
- longer peptides e.g., 9-, 10- or 11-mers
- binding of peptides to antibodies or antigen-binding fragments can be determined using standard methodologies including surface plasmon resonance (BIACORE) and ELISA assays.
- BIACORE surface plasmon resonance
- ELISA assays For examination of conformational epitopes, larger PSMA fragments can be used.
- Other methods that use mass spectrometry to define conformational epitopes have been described and can be used (see, e.g., Baerga-Ortiz et al., Protein Science 11:1300-1308, 2002 and references cited therein).
- Epitopes can be confirmed by introducing point mutations or deletions into a known epitope and then testing binding with one or more antibodies or antigen-binding fragments to determine which mutations reduce binding of the antibodies or antigen-binding fragments.
- the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived are those produced by hybridomas referred to herein as PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, and Abgenix 4.
- the antibodies are those encoded by the plasmids listed in Table 1.
- the antibodies are those that comprise a heavy chain encoded by a nucleic acid molecule comprising the heavy chain coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and a light chain encoded by a nucleic acid molecule comprising the light chain coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13.
- the names of the deposited hybridomas or plasmids may be used interchangeably with the names of the antibodies. It will be clear to one of ordinary skill in the art when the name is intended to refer to the antibody or when it refers to the plasmids or hybridomas that encode or produce the antibodies, respectively. Additionally, the antibody names may be an abbreviated form of the name shown in Table 1. For instance, antibody AB-PG1-XG1-006 may be referred to as AB-PG1-XG1-006, PG1-XG1-006, XG1-006, 006, etc. In another example, the antibody name PSMA 4.232.3 may be referred to as PSMA 4.232.3, 4.232.1, 4.232, etc. It is intended that all of the variations in the name of the antibody refer to the same antibody and not a different one.
- the antibodies of the ADCs include those encoded by particular sets of heavy and light chain sequences.
- the antibody (AB-PG1-XG1-006) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 2 and 8.
- the antibody (AB-PG1-XG1-026) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 3 and 9.
- the antibody (AB-PG1-XG1-051) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 4 and 10.
- the antibody (AB-PG1-XG1-069) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 5 and 11.
- the antibody (AB-PG1-XG1-077) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 6 and 12.
- the antibody is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 7 and 13.
- the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived include a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14, 18, 22, 26 and 30, and a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16, 20, 24, 28 and 32.
- the antibody includes an immunoglobulin variable sequence encoded by nucleic acid molecules which comprise the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 14 and 16.
- the antibody can be one that includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 15 and 17.
- the antibody (AB-PG1-XG1-026) includes an immunoglobulin variable sequence encoded by nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 18 and 20 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs 19 and 21.
- the antibody (AB-PG1-XG1-051) includes an immunoglobulin variable sequence encoded by the nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 22 and 24 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 23 and 25.
- the antibody (AB-PG1-XG1-069) includes an immunoglobulin variable sequence encoded by the nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 26 and 28 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 27 and 29.
- the antibody includes an immunoglobulin variable sequence encoded by the nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 30 and 32 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 31 and 33.
- the antibody includes a heavy chain variable region comprising an amino acid sequence selected from the group consisting of amino acid sequences set forth as: SEQ ID NOs: 15, 19, 23, 27 and 31, and a light chain variable region comprising an amino acid sequence selected from the group consisting of amino acid sequences set forth as: SEQ ID NOs: 17, 21, 25, 29 and 33.
- coding region refers to a region of a nucleotide sequence that encodes a polypeptide sequence. Its use herein is consistent with the recognized meaning known in the art.
- the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived are those that are encoded by nucleic acid molecules that are highly homologous to the foregoing nucleic acids.
- the homologous nucleic acid molecule can, in some embodiments, comprise a nucleotide sequence that is at least about 90% identical to the nucleotide sequence provided herein. In other embodiments, the nucleotide sequence is at least about 95% identical, at least about 97% identical, at least about 98% identical, or at least about 99% identical to a nucleotide sequence provided herein.
- the homology can be calculated using various, publicly available software tools well known to one of ordinary skill in the art. Exemplary tools include the BLAST system available from the website of the National Center for Biotechnology Information (NCBI) at the National Institutes of Health.
- the antibodies of the ADCs include antibodies having a PSMA-binding property and/or other functional properties described herein, which are encoded by nucleic acid molecules that hybridize under high stringency conditions to the foregoing nucleic acid molecules.
- Identification of related sequences can also be achieved using polymerase chain reaction (PCR) and other amplification techniques suitable for cloning related nucleic acid sequences.
- PCR primers can be selected to amplify portions of a nucleic acid sequence of interest, such as a CDR.
- high stringency conditions refers to parameters with which the art is familiar. Nucleic acid hybridization parameters may be found in references that compile such methods, e.g. Molecular Cloning: A Laboratory Manual, J. Sambrook, et al., eds., Second Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989 , or Current Protocols in Molecular Biology, F.M. Ausubel, et al., eds., John Wiley & Sons, Inc., New York .
- hybridization buffer 3.5X SSC, 0.02% Ficoll, 0.02% polyvinyl pyrrolidone, 0.02% Bovine Serum Albumin, 2.5mM NaH 2 PO 4 (pH7), 0.5% SDS, 2mM EDTA.
- SSC is 0.15M sodium chloride/0.015M sodium citrate, pH7; SDS is sodium dodecyl sulphate; and EDTA is ethylenediaminetetracetic acid.
- a membrane upon which the nucleic acid is transferred is washed, for example, in 2X SSC at room temperature and then at 0.1 - 0.5X SSC/0.1X SDS at temperatures up to 68°C.
- the term "antibody” refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds.
- Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as HCVR or V H ) and a heavy chain constant region.
- the heavy chain constant region is comprised of three domains, C H 1, C H 2 and C H 3.
- Each light chain is comprised of a light chain variable region (abbreviated herein as LCVR or V L ) and a light chain constant region.
- the light chain constant region is comprised of one domain, C L .
- V H and V L regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR).
- CDR complementarity determining regions
- FR framework regions
- Each V H and V L is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- the variable regions of the heavy and light chains contain a binding domain that interacts with an antigen.
- the constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (C1q) of the classical complement system.
- antigen-binding fragment of an antibody as used herein, refers to one or more portions of an antibody that retain the ability to specifically bind to an antigen (i.e., PSMA). It has been shown that the antigen-binding function of an antibody can be performed by fragments of a full-length antibody.
- binding fragments encompassed within the term "antigen-binding fragment" of an antibody include (i) a Fab fragment, a monovalent fragment consisting of the V L , V H , C L and C H 1 domains; (ii) a F(ab') 2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the V H and CH1 domains; (iv) a Fv fragment consisting of the V L and V H domains of a single arm of an antibody, (v) a dAb fragment ( Ward et al., (1989) Nature 341:544-546 ) which consists of a V H domain; and (vi) an isolated complementarity determining region (CDR).
- a Fab fragment a monovalent fragment consisting of the V L , V H , C L and C H 1 domains
- F(ab') 2 fragment a bivalent fragment comprising two
- the CDRs and in particular the CDR3 regions, and more particularly the heavy chain CDR3 contribute to antibody specificity. Because these CDR regions and in particular the CDR3 region confer antigen specificity on the antibody, these regions may be incorporated into other antibodies or antigen-binding fragments to confer the identical antigen specificity onto that antibody or peptide.
- the two domains of the Fv fragment, V and V H are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the V L and V H regions pair to form monovalent molecules (known as single chain Fv (scFv); see e.g., Bird et al.
- Such single chain antibodies are also intended to be encompassed within the term "antigen-binding fragment" of an antibody.
- antibody fragments are obtained using conventional procedures, such as proteolytic fragmentation procedures, as described in J. Goding, Monoclonal Antibodies: Principles and Practice, pp 98-118 (N.Y. Academic Press 1983 ), as well as by other techniques known to those with skill in the art. The fragments are screened for utility in the same manner as are intact antibodies.
- the antibodies, or antigen-binding fragments thereof, of the ADCs are, in some embodiments, isolated.
- isolated is intended to refer to an antibody (or antigen-binding fragment thereof), which is substantially free of other antibodies (or antigen-binding fragments) having different antigenic specificities (e.g., an isolated antibody that specifically binds to PSMA is substantially free of antibodies that specifically bind antigens unrelated to PSMA).
- An isolated antibody that specifically binds to an epitope, isoform or variant of PSMA may, however, have cross-reactivity to other related antigens, e.g., from other species (e.g., PSMA species homologs).
- PSMA homologs have been found in other species, such as the pig (GenBank Accession Number 077564 (amino acid)) and rat (GenBank Accession Numbers U75973 (mRNA) and AAC53423 (amino acid)).
- the antibodies or antigen-binding fragments provided herein specifically bind both human PSMA as well as a PSMA homolog from another species.
- the antibodies or antigen-binding fragments thereof specifically bind human PSMA but do not cross-react with PSMA homologs from another species.
- an isolated antibody may be substantially free of other cellular material and/or chemicals.
- substantially pure means that an antibody (or antigen-binding fragment thereof) is essentially free of other substances with which they may be found in nature or in vivo systems to an extent practical and appropriate for their intended use.
- Substantially pure antibodies or antigen-binding fragments thereof may be produced by techniques well known in the art. Because an isolated antibody (or antigen-binding fragment thereof) may be admixed with a pharmaceutically acceptable carrier in a pharmaceutical preparation, the antibody (or antigen-binding fragment thereof) may comprise only a small percentage by weight of the preparation. The antibody (or antigen-binding fragment thereof) is nonetheless isolated in that it has been separated from the substances with which it may be associated in living systems, i.e. isolated from other proteins.
- specific binding refers to antibody binding to a predetermined antigen, in this case PSMA (e.g., human PSMA).
- PSMA e.g., human PSMA
- the antibody binds with an affinity that is at least two-fold greater than its affinity for binding to a non-specific antigen (e.g., BSA, casein), which is an antigen other than PSMA, an isoform or variant of PSMA, or a closely-related antigen.
- the antibodies encompass various antibody isotypes, such as IgG1, IgG2, IgG3, IgG4, IgM, IgA1, IgA2, IgAsec, IgD, IgE.
- isotype refers to the antibody class (e.g., IgM or IgG1) that is encoded by heavy chain constant region genes.
- the antibodies can be full length or can include only an antigen-binding fragment such as the antibody constant and/or variable domain of IgG1, IgG2, IgG3, IgG4, IgM, IgA1, IgA2, IgAsec, IgD or IgE or could consist of a Fab fragment, a F(ab') 2 fragment and a Fv fragment.
- the antibodies of the ADCs are, in some embodiments monoclonal.
- the antibodies can be produced by a variety of techniques well known in the art. Monoclonal antibody production may be effected by techniques which are well known in the art.
- the term "monoclonal antibody”, as used herein, refers to a preparation of antibody molecules of single molecular composition. A monoclonal antibody displays a single binding specificity and affinity for a particular epitope.
- the process of monoclonal antibody production involves obtaining immune somatic cells with the potential for producing antibody, in particular B lymphocytes, which have been previously immunized with the antigen of interest either in vivo or in vitro and that are suitable for fusion with a B-cell myeloma line.
- Mammalian lymphocytes typically are immunized by in vivo immunization of the animal (e.g., a mouse) with the desired protein or polypeptide. Such immunizations are repeated as necessary at intervals of up to several weeks to obtain a sufficient titer of antibodies.
- animals can be used as a source of antibody-producing lymphocytes. Following the last antigen boost, the animals are sacrificed and spleen cells removed.
- Mouse lymphocytes give a higher percentage of stable fusions with the mouse myeloma lines described herein. For example, of the BALB/c mouse. However, other mouse strains, rabbit, hamster, sheep and frog may also be used as hosts for preparing antibody-producing cells.
- the ADCs comprise a fully human monoclonal antibody or an antigen-binding fragment thereof that binds PSMA.
- Somatic cells may be obtained from the lymph nodes, spleens and peripheral blood of antigen-primed animals, and the lymphatic cells of choice depend to a large extent on their empirical usefulness in the particular fusion system.
- the antibody-secreting lymphocytes are then fused with (mouse) B cell myeloma cells or transformed cells, which are capable of replicating indefinitely in cell culture, thereby producing an immortal, immunoglobulin-secreting cell line.
- the resulting fused cells, or hybridomas are cultured, and the resulting colonies screened for the production of the desired monoclonal antibodies.
- Colonies producing such antibodies are cloned, and grown either in vivo or in vitro to produce large quantities of antibody.
- a description of the theoretical basis and practical methodology of fusing such cells is set forth in Kohler and Milstein, Nature 256:495 (1975 ).
- human somatic cells capable of producing antibody are suitable for fusion with myeloma cell lines. While B lymphocytes from biopsied spleens, tonsils or lymph nodes of an individual may be used, the more easily accessible peripheral blood B lymphocytes can also be used. The lymphocytes may be derived from patients with diagnosed prostate carcinomas or another PSMA-expressing cancer. In addition, human B cells may be directly immortalized by the Epstein-Barr virus ( Cole et al., 1995, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., pp. 77-96 ). Although somatic cell hybridization procedures can be used, in principle, other techniques for producing monoclonal antibodies can be employed such as viral or oncogenic transformation of B lymphocytes.
- Myeloma cell lines suited for use in hybridoma-producing fusion procedures can be non-antibody-producing, have high fusion efficiency, and enzyme deficiencies that render them incapable of growing in certain selective media which support the growth of the desired hybridomas.
- Examples of such myeloma cell lines that may be used for the production of fused cell lines include P3-X63/Ag8, X63-Ag8.653, NS1/1.Ag 4.1, Sp2/0-Ag14, FO, NSO/U, MPC-11, MPC11-X45-GTG 1.7, S194/5XX0 Bul, all derived from mice; R210.RCY3, Y3-Ag 1.2.3, IR983F and 4B210 derived from rats and U-266, GM1500-GRG2, LICR-LON-HMy2, UC729-6, all derived from humans ( Goding, in Monoclonal Antibodies: Principles and Practice, 2d ed., pp.
- Fusion with mammalian myeloma cells or other fusion partners capable of replicating indefinitely in cell culture is effected by standard and well-known techniques, for example, by using polyethylene glycol ("PEG”) or other fusing agents (See Milstein and Kohler, Eur. J. Immunol. 6:511 (1976 ).
- PEG polyethylene glycol
- the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived are recombinant antibodies.
- the term "recombinant antibody”, as used herein, is intended to include antibodies that are prepared, expressed, created or isolated by recombinant means, such as antibodies isolated from an animal (e.g., a mouse) that is transgenic for another species' immunoglobulin genes, antibodies expressed using a recombinant expression vector transfected into a host cell, antibodies isolated from a recombinant, combinatorial antibody library, or antibodies prepared, expressed, created or isolated by any other means that involves splicing of immunoglobulin gene sequences to other DNA sequences.
- the antibodies are chimeric or humanized antibodies.
- the term “chimeric antibody” refers to an antibody, that combines the murine variable or hypervariable regions with the human constant region or constant and variable framework regions.
- the term “humanized antibody” refers to an antibody that retains only the antigen-binding CDRs from the parent antibody in association with human framework regions (see, Waldmann, 1991, Science 252:1657 ). Such chimeric or humanized antibodies retaining binding specificity of the murine antibody are expected to have reduced immunogenicity when administered in vivo as provided herein.
- the monoclonal antibodies of the ADCs can be in the form of a bispecific antibody or a multispecific antibody.
- bispecific antibody is intended to include any agent, e.g., a protein, peptide, or protein or peptide complex, which has two different binding specificities which bind to, or interact with (a) a cell surface antigen and (b) an Fc receptor on the surface of an effector cell.
- multispecific antibody is intended to include any agent, e.g., a protein, peptide, or protein or peptide complex, which has more than two different binding specificities which bind to, or interact with (a) a cell surface antigen, (b) an Fc receptor on the surface of an effector cell, and (c) at least one other component. Accordingly, the antibodies include, but are not limited to, bispecific, trispecific, tetraspecific, and other multispecific antibodies which are directed to PSMA and to Fc receptors on effector cells.
- bispecific antibodies further includes diabodies.
- Diabodies are bivalent, bispecific antibodies in which the V H and V L domains are expressed on a single polypeptide chain, but using a linker that is too short to allow for pairing between the two domains on the same chain, thereby forcing the domains to pair with complementary domains of another chain and creating two antigen-binding sites (see e.g., Holliger, P., et al. (1993) Proc. Natl. Acad. Sci. USA 90:6444-6448 ; Poijak, R.J., et al. (1994) Structure 2:1121-1123 ).
- a bispecific antibody can be formed of an antigen-binding region specific for PSMA and an antigen-binding region specific for an effector cell which has tumoricidal or tumor inhibitory activity.
- the two antigen-binding regions of the bispecific antibody are either chemically linked or can be expressed by a cell genetically engineered to produce the bispecific antibody.
- Suitable effector cells having tumoricidal activity include but are not limited to cytotoxic T-cells (primarily CD8 + cells), natural killer cells, etc.
- An effective amount of a bispecific antibody can be administered to a subject with cancer, and the bispecific antibody kills and/or inhibits proliferation of the cancer cells after localization at sites of primary or metastatic tumors bearing PSMA.
- the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived are human antibodies.
- the term "human antibody”, as used herein, is intended to include antibodies having variable and constant regions derived from human germline immunoglobulin sequences.
- the human antibodies can include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo).
- the term "human antibody”, as used herein is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences (referred to herein as "humanized antibodies”).
- Human antibodies directed against PSMA can be generated using transgenic mice carrying parts of the human immune system rather than the mouse system. Some examples of which are described elsewhere herein.
- Fully human monoclonal antibodies also can be prepared by immunizing mice transgenic for large portions of human immunoglobulin heavy and light chain loci. See, e.g., U.S. patents 5,591,669 , 5,598,369 , 5,545,806, 5,545,807 , 6,150,584 , and references cited therein. These animals have been genetically modified such that there is a functional deletion in the production of endogenous (e.g., murine) antibodies. The animals are further modified to contain all or a portion of the human germ-line immunoglobulin gene locus such that immunization of these animals results in the production of fully human antibodies to the antigen of interest.
- mice e.g., XenoMouse (Abgenix), HuMAb mice (Medarex/GenPharm)
- monoclonal antibodies can be prepared according to standard hybridoma technology. These monoclonal antibodies have human immunoglobulin amino acid sequences and, therefore, will not provoke human anti-mouse antibody (HAMA) responses when administered to humans.
- HAMA human anti-mouse antibody
- the mice are 6-16 weeks of age upon the first immunization.
- a purified or enriched preparation of PSMA antigen can be used to immunize the mice intraperitoneally (IP), although other routes of immunization known to one of ordinary skill in the art are also possible.
- PSMA antigen can be injected in combination with an adjuvant, such as complete Freund's adjuvant, and, in some embodiments, the initial injection is followed by booster immunizations with antigen in an adjuvant, such as incomplete Freund's adjuvant.
- the immune response can be monitored over the course of the immunization protocol with plasma samples obtained by, for example, retroorbital bleeds.
- the plasma can be screened by ELISA, and mice with sufficient titers of anti-PSMA human immunoglobulin can be used for fusions. Mice can be boosted intravenously with antigen 3 days before sacrifice and removal of the spleen.
- the antibody or antigen-binding fragment thereof of the ADCs can, in some embodiments, be selected for the ability to bind live PSMA-expressing cells.
- flow cytometry can be used.
- PSMA-expressing cells lines grown under standard growth conditions
- prostate cancer cells that express PSMA can be mixed with various concentrations of monoclonal antibodies in PBS containing 0.1 % Tween 80 and 20% mouse serum and incubated at 37°C for 1 hour. After washing, the cells can be reacted with fluorescein-labeled anti-human IgG secondary antibody (if human anti-PSMA antibodies were used) under the same conditions as the primary antibody staining.
- the samples can be analyzed by a fluorescence activated cell sorter (FACS) instrument using light and side scatter properties to gate on single cells.
- FACS fluorescence activated cell sorter
- An alternative assay using fluorescence microscopy can be used (in addition to or instead of) the flow cytometry assay. Cells can be stained and examined by fluorescence microscopy. This method allows visualization of individual cells but may have diminished sensitivity depending on the density of the antigen. It follows, that the ADCs, in some embodiments, bind live cells. The ADCs, in some embodiments, therefore, do not require cell lysis to bind PSMA.
- the antibodies can, in some embodiments, promote cytolysis of PSMA-expressing cells. Cytolysis can be complement-mediated or can be mediated by effector cells. In one embodiment, the cytolysis is carried out in a living organism, such as a mammal, and the live cell is a tumor cell.
- tumors which can be targeted with the antibodies or antigen-binding fragments thereof include, any tumor that expresses PSMA (this includes tumors with neovasculature expressing PSMA), such as, prostate, bladder, breast, pancreas, liver, lung (e.g., non-small cell lung carcinoma), colon and kidney tumors as well as melanomas and sarcomas.
- the tumor cell is a prostate tumor cell.
- the testing of cytolytic activity in vitro by chromium release assay can provide an initial screening prior to testing in vivo models. This testing can be carried out using standard chromium release assays. Briefly, polymorphonuclear cells (PMN), or other effector cells, from healthy donors can be purified by Ficoll Hypaque density centrifugation, followed by lysis of contaminating erythrocytes. Washed PMNs can be suspended in RPMI supplemented with 10% heat-inactivated fetal calf serum and mixed with 51 Cr labeled cells expressing PSMA, at various ratios of effector cells to tumor cells (effector cells:tumor cells). Purified anti-PSMA IgGs can then be added at various concentrations.
- PMN polymorphonuclear cells
- Irrelevant IgG can be used as a negative control. Assays can be carried out for 0-120 minutes at 37°C. Samples can be assayed for cytolysis by measuring 51 Cr release into the culture supernatant. Anti-PSMA monoclonal antibodies and/or ADCs can also be tested in combinations with each other to determine whether cytolysis is enhanced with multiple monoclonal antibodies and/or ADCs. Antibodies that bind to PSMA and/or ADCs also can be tested in an in vivo model (e.g., in mice) to determine their efficacy in mediating cytolysis and killing of cancer cells expressing PSMA, e.g., prostate tumor cells.
- an in vivo model e.g., in mice
- the antibodies of the ADCs can be selected, for example, based on the following criteria, which are not intended to be exclusive:
- the antibodies of the ADCs can meet one or more, and possibly all, of these criteria.
- the antibody or antigen-binding fragment thereof binds to a conformational epitope, such as a conformational epitope within the extracellular domain of PSMA.
- a conformational epitope such as a conformational epitope within the extracellular domain of PSMA.
- each antibody can be tested in assays using native protein (e.g., non-denaturing immunoprecipitation, flow cytometric analysis of cell surface binding) and denatured protein (e.g., Western blot, immunoprecipitation of denatured proteins). A comparison of the results will indicate whether the antibody or antigen-binding fragment thereof binds a conformational epitope.
- Antibodies or antigen-binding fragments thereof that bind to native protein but not denatured protein are, in some embodiments, those that bind conformational epitopes. It follows, that the ADCs, in some embodiments, bind comformational epitopes of PSMA.
- the antibody or antigen-binding fragment thereof binds to a dimer-specific epitope on PSMA (i.e., a conformational, dimer-specific epitope unique to the quaternary structure of dimeric PSMA).
- a dimer-specific epitope on PSMA i.e., a conformational, dimer-specific epitope unique to the quaternary structure of dimeric PSMA.
- antibodies or antigen-binding fragments thereof which bind to a dimer-specific epitope preferentially bind the PSMA dimer rather than the PSMA monomer.
- an antibody or antigen-binding fragment thereof binds preferentially (i.e., selectively and/or specifically) to a PSMA dimer
- the antibody or antigen-binding fragment thereof can be tested in assays (e.g., immunoprecipitation followed by Western blotting) using native dimeric PSMA protein and dissociated monomeric PSMA protein. A comparison of the results will indicate whether the antibody or antigen-binding fragment thereof binds preferentially to the dimer.
- the antibodies or antigen-binding fragments thereof bind to the PSMA dimer but not to the monomeric PSMA protein. It follows, that the ADCs, in some embodiments, bind to a dimer-specific epitope on PSMA.
- the ADCs provided include those that selectively bind PSMA multimers.
- selectively binds means that an antibody or antigen-binding fragment thereof of the ADCs preferentially binds to a PSMA protein multimer (e.g., with greater avidity, greater binding affinity) rather than to a PSMA protein monomer.
- the ADCs of the invention bind to a PSMA protein multimer with an avidity and/or binding affinity that is 1.1-fold, 1.2-fold, 1.3-fold, 1.4-fold, 1.5-fold, 1.6-fold, 1.7-fold, 1.8-fold, 1.9-fold, 2-fold, 3-fold, 4-fold, 5-fold, 7-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold, 70-fold, 100-fold, 200-fold, 300-fold, 500-fold, 1000-fold or more than that exhibited by the ADC for a PSMA protein monomer.
- the ADC can, in some embodiments, selectively bind a PSMA protein multimer, and not a PSMA protein monomer, i.e., exclusively binds to a PSMA protein multimer. In some embodiments, the ADC selectively binds a PSMA protein dimer.
- a PSMA protein multimer is a protein complex of at least two PSMA proteins or fragments thereof.
- the PSMA protein multimers can be composed of various combinations of full-length PSMA proteins (e.g., SEQ ID NO: 1), recombinant soluble PSMA (rsPSMA, e.g., amino acids 44-750 of SEQ ID NO: 1) and fragments of the foregoing that form multimers (i.e., that retain the protein domain required for forming dimers and/or higher order multimers of PSMA).
- at least one of the PSMA proteins forming the multimer is a recombinant, soluble PSMA (rsPSMA) polypeptide.
- the PSMA protein multimers can be dimers, such as those formed from recombinant soluble PSMA protein.
- the dimer is a rsPSMA homodimer.
- the PSMA protein multimers referred to herein are believed to assume a native conformation and can have such a conformation.
- the PSMA proteins in certain embodiments are noncovalently bound together to form the PSMA protein multimer. It has been discovered that PSMA protein noncovalently associates to form dimers under non-denaturing conditions.
- the PSMA protein multimers can retain the activities of PSMA.
- the PSMA activity may be an enzymatic activity, such as folate hydrolase activity, NAALADase activity, dipeptidyl peptidase IV activity or ⁇ -glutamyl hydrolase activity.
- the antibody or antigen-binding fragment thereof of the ADCs can bind to and be internalized with PSMA expressed on cells.
- the mechanism by which the antibody or antigen-binding fragment thereof is internalized with PSMA is not critical to the practice of the methods provided herein.
- the antibody or antigen-binding fragment thereof can induce internalization of PSMA.
- internalization of the antibody or antigen-binding fragment thereof can be the result of routine internalization of PSMA. It follows that the ADC can be internalized with PSMA expressed on cells.
- the antibodies or antigen-binding fragments thereof, and, therefore, the ADCs can, in some embodiments, specifically bind cell-surface PSMA and/or rsPSMA with sub-nanomolar affinity.
- the binding affinities can be about 1 X 10 -9 M or less, about 1 X 10 -10 M or less, or about 1 X 10 -11 M or less. In one embodiment, the binding affinity is less than about 5 X 10 -10 M.
- the antibodies or antigen-binding fragments thereof can, in some embodiments, modulate at least one enzymatic activity of PSMA.
- the activity can be selected from the group consisting of N-acetylated ⁇ -linked acidic dipeptidase (NAALADase), folate hydrolase, dipeptidyl dipeptidase IV, ⁇ -glutamyl hydrolase activity and combinations thereof in vitro or in vivo.
- NAALADase N-acetylated ⁇ -linked acidic dipeptidase
- folate hydrolase dipeptidyl dipeptidase IV
- ⁇ -glutamyl hydrolase activity ⁇ -glutamyl hydrolase activity
- Tissue levels of NAALADase activity can be determined by detergent solubilizing homogenizing tissues, pelleting the insoluble material by centrifugation and measuring the NAALADase activity in the remaining supernatant.
- the NAALADase activity in bodily fluids can also be measured by first pelleting the cellular material by centrifugation and performing a typical enzyme assay for NAALADase activity on the supernatant.
- NAALADase enzyme assays have been described by Frieden, 1959, J. Biol, Chem., 234:2891 . In this assay, the reaction product of the NAALADase enzyme is glutamic acid.
- Folate hydrolase activity of PSMA can be measured by performing enzyme assays as described by Heston and others (e.g., Clin. Cancer Res. 2(9): 1445-51,1996 ; Urology 49(3A Suppl):104-12,1997 ).
- Folate hydrolases such as PSMA remove the gamma-linked glutamates from polyglutamated folates.
- Folate hydrolase activity can be measured using substrates such as methotrexate tri-gamma glutamate (MTXGlu3), methotrexate di-gamma glutamate (MTXGlu2) or pteroylpentaglutamate (PteGlu5), for example using capillary electrophoresis (see Clin. Cancer Res. 2(9):1445-51, 1996 ). Timed incubations of PSMA with polyglutamated substrates can be followed by separation and detection of hydrolysis products.
- MTXGlu3 methotrexate tri-gamma glutamate
- MTXGlu2 methotre
- the ADCs provided have surprisingly been found to kill PSMA-expressing, taxane-resistant cancer cells and can be used to treat PSMA-expressing, taxane-resistant cancer. Methods are, therefore, provided wherein an ADC is administered to a subject with a PSMA-expressing, taxane-resistant cancer.
- the cancer cells of the PSMA-expressing, taxane-resistant cancer in some embodiments, can be cells of a tumor (e.g., primary malignant tumor) or cells of one or more metastases.
- the methods provided result in the delay or inhibition of progression of the cancer in the subject.
- delay or inhibition of progression of the cancer is intended to refer to any slowing or halting of the progression of the cancer in the subject.
- a slowing or halting of the progression of the cancer includes a reduction or stabilization in the number of cancer cells, the number of tumors, and/or the number of metastases in a subject.
- a slowing or halting is also intended to include a reduction or stabilization in the size (e.g., length or volume) of tumors and/or size of metastases in a subject.
- the delay or inhibition of progression of the cancer can be demonstrated by a change in at least one biomarker for bone metastasis or bone metabolism compared to a baseline value prior to treatment with the ADC in the subject.
- the biomarker is N-telopeptide, bone alkaline phosphatase, osteocalcin, calcitonin, calcium, pyridinoline or deoxypyridinoline.
- the delay or inhibition of progression of the cancer is demonstrated by radiographic image changes in tumor burden compared to a baseline radiographic image in the subject prior to ADC treatment. Methods for assessing radiographic changes, include, for example, bone scan, computerized axial tomography (CT) scan and magnetic resonance imaging (MRI). Other methods will be well known to those of ordinary skill in the art.
- the delay or inhibition of progression is evidenced by a radiographic image change of at least 10%, 20%, 30%, 40%, 50% or 60% or more.
- Treatment with the ADCs provided can also result in the increase in survival of subjects with a PSMA-expressing, taxane-resistant cancer.
- the survival of such a subject is increased by 4, 6, 8, 10, 12, 14, 16, 18, 20, 24, 28 or 32 weeks or more as compared to the median survival of subjects with the PSMA-expressing, taxane-resistant cancer not administered the ADC.
- the survival is increased 4, 6, 8, 10, 12, 14, 16, 18, 20, 24, 28 or 32 weeks or more as compared to the expected survival time for the subject prior to treatment with the ADC.
- the survival of the subject is increased by 10, 12, 14, 16, 18, 20, 22 or 24 months or more according to either comparison.
- the subject treated with an ADC is one that has progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, and the increase in survival of the subject is as compared to the median survival of subjects having progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy and not treated with the ADC.
- the subject treated with an ADC is one that has progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, and the increase in survival of the subject is as compared to the expected survival time for the subject prior to treatment with the ADC.
- treatment with an ADC as provided results in an increase in the quality of life for the subject as compared to the quality of life experienced by the subject prior to treatment with the ADC.
- an increase in the quality of life refers to any improvement in the subject's comfort, level of energy and/or ability to function in an activity as a result of the administration of an ADC as provided herein.
- treatment with an ADC can result in a decrease in the circulating level of circulating tumor cells (CTCs) compared to a baseline level.
- treatment with an ADC can result in a decrease or stabilization (no significant increase or decrease) in a serum level of prostate-specific antigen (PSA) compared to a baseline level of PSA.
- PSA prostate-specific antigen
- the subject has a tumor with a tumor volume that is at least 100mm 3 , 200mm 3 , 300mm 3 , 400mm 3 , 500mm 3 , 600mm 3 , 700mm 3 , 800mm 3 , 900mm 3 , 1000mm 3 , 1100mm 3 , 1200mm 3 , 1300mm 3 , 1400mm 3 , 1500mm 3 , 1600mm 3 , 1700mm 3 , 1800mm 3 , 1900mm 3 or 2000mm 3 .
- the subject has a tumor volume that is greater than 700mm 3 .
- the subject has a tumor with a length of at least 5mm, 6mm, 7mm, 8mm, 9mm, 10mm, 11 mm, 12mm, 13mm, 14mm, 15mm, 16mm, 17mm, 18mm, 19mm, 20mm, 21mm, 22mm, 23mm, 24mm, 25mm or 30mm or more.
- the tumor volume or length is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97% or 99% as a result of treatment with an ADC as provided herein.
- the tumor is eradicated. Techniques for determining the presence of a tumor and for measuring its size are well known to those of ordinary skill in the art.
- the ADCs can be used in various in vitro and in vivo methods for effecting the aforementioned therapeutic endpoints.
- the methods provided can be used to kill PSMA-expressing, taxane-resistant cancer cells in vitro or in vivo. Methods are also provided for treating any PSMA-expressing, taxane-resistant cancer.
- a cancer is, for example, prostate cancer.
- Such a cancer can also be, for example, a cancer in which PSMA is expressed on the cells of the tumor neovasculature. An exemplary list of such cancers is provided elsewhere herein.
- two or more different ADCs are used in combination.
- one or more unconjugated anti-PSMA antibodies or antigen-binding fragments thereof can be combined with one or more ADCs in a single therapy to achieve a desired therapeutic effect.
- an unconjugated anti-PSMA antibody that mediates highly effective killing of target cells in the presence of effector cells and/or that inhibits the growth of cells expressing PSMA can be used with one or more ADCs.
- the ADCs can be combined with one or more additional antitumor agents, such as corticosteroids, such as prednisone or hydrocortisone; immunostimulatory agents; immunomodulators; or some combination thereof.
- Antitumor agents include cytotoxic agents, chemotherapeutic agents and agents that act on tumor neovasculature.
- Cytotoxic agents include cytotoxic radionuclides, chemical toxins and protein toxins.
- the cytotoxic radionuclide or radiotherapeutic isotope can be an alpha-emitting isotope such as 225 Ac, 211 At, 212 Bi, 213 Bi, 212 Pb, 224 R a or 223 R a .
- the cytotoxic radionuclide can be a beta-emitting isotope such as 186 Rh, 188 Rh, 177 Lu, 90 Y, 131 I, 67 Cu, 64 Cu, 153 Sm or 166 Ho.
- the cytotoxic radionuclide can emit Auger and low energy electrons and include the isotopes 125 I, 123 I or 77 Br.
- Suitable chemical toxins or chemotherapeutic agents include members of the enediyne family of molecules, such as calicheamicin and esperamicin. Chemical toxins can also be taken from the group consisting of methotrexate, doxorubicin, melphalan, chlorambucil, ARA-C, vindesine, mitomycin C, cis-platinum, etoposide, bleomycin and 5-fluorouracil.
- Other antineoplastic agents include dolastatins ( U.S. Patent Nos. 6,034,065 and 6,239,104 ) and derivatives thereof.
- Dolastatins and derivatives thereof include dolastatin 10 (dolavaline-valine-dolaisoleuine-dolaproine-dolaphenine) and the derivatives auristatin PHE (dolavaline-valine-dolaisoleuine-dolaproine-phenylalanine-methyl ester) ( Pettit, G.R. et al., Anticancer Drug Des. 13(4):243-277, 1998 ; Woyke, T. et al., Antimicrob. Agents Chemother. 45(12):3580-3584, 2001 ), and aurastatin E and the like.
- Toxins also include poisonous lectins, plant toxins such as ricin, abrin, modeccin, botulina and diphtheria toxins. Other chemotherapeutic agents are known to those skilled in the art.
- tubulin-binding agents such as combrestatin A4 ( Griggs et al., Lancet Oncol. 2:82, 2001 ), angiostatin and endostatin (reviewed in Rosen, Oncologist 5:20, 2000 ) and interferon inducible protein 10 ( U.S. Patent No. 5,994,292 ).
- a number of other antiangiogenic agents are also contemplated and include: 2ME2, Angiostatin, Angiozyme, Anti-VEGF RhuMAb, Apra (CT-2584), Avicine, Benefin, BMS275291, Carboxyamidotriazole, CC4047, CC5013, CC7085, CDC801, CGP-41251 (PKC 412), CM101, Combretastatin A-4 Prodrug, EMD 121974, Endostatin, Flavopiridol, Genistein (GCP), Green Tea Extract, IM-862, ImmTher, Interferon alpha, Interleukin-12, Iressa (ZD1839), Marimastat, Metastat (Col-3), Neovastat , Octreotide, Paclitaxel, Penicillamine, Photofrin, Photopoint, PI-88, Prinomastat (AG-3340), PTK787 (ZK22584), RO317453, Solimastat, Squalamine, SU
- the ADCs can be administered with one or more immunostimulatory agents to induce or enhance an immune response, such as IL-2 and immunostimulatory oligonucleotides (e.g., those containing CpG motifs).
- Immunostimulatory agents can, in some embodiments, stimulate specific arms of the immune system, such as natural killer (NK) cells that mediate antibody-dependent cell cytotoxicity (ADCC).
- Immunostimulatory agents include interleukin-2, ⁇ -interferon, ⁇ -interferon, tumor necrosis factor alpha (TNF ⁇ ), immunostimulatory oligonucleotides or a combination thereof.
- Immunomodulators include cytokines, chemokines, adjuvants or a combination thereof. Chemokines useful in increasing immune responses include but are not limited to SLC, ELC, MIP3 ⁇ , MIP3 ⁇ , IP-10, MIG, and combinations thereof.
- the other therapeutic agent can also be a vaccine.
- the vaccine immunizes a subject against PSMA.
- Such vaccines include antigens, such as PSMA dimers, with, optionally, one or more adjuvants to induce or enhance an immune response.
- An adjuvant is a substance which potentiates the immune response. Adjuvants of many kinds are well known in the art.
- adjuvants include monophosphoryl lipid A (MPL, SmithKline Beecham); saponins including QS21 (SmithKline Beecham); immunostimulatory oligonucleotides (e.g., CpG oligonucleotides described by Kreig et al., Nature 374:546-9, 1995 );incomplete Freund's adjuvant; complete Freund's adjuvant; montanide; vitamin E and various water-in-oil emulsions prepared from biodegradable oils such as squalene and/or tocopherol, Quil A, Ribi Detox, CRL-1005, L-121, and combinations thereof.
- Formulations, such as those described in U.S. Application Serial No. 10/976352 are also contemplated for use as vaccines in the methods provided herein.
- the vaccines can, in some embodiments, include one or more of the isolated PSMA protein multimers described herein, such as the PSMA protein dimer.
- a PSMA protein multimer composition contains at least about 10% PSMA protein multimer (of the total amount of PSMA protein in the composition).
- the PSMA protein multimer composition contains at least about 20%, 30%, 40%, 50%, 60%, 70%, 75%, 80%, 85%, 90%, 95%, 99% or 99.5% PSMA protein multimer.
- the PSMA protein multimer composition contains substantially pure PSMA protein multimer, with substantially no PSMA protein monomer. It is understood that the list of specific percentages includes by inference all of the unnamed percentages between the recited percentages.
- Cytokines can also be used in vaccination protocols as a result of their lymphocyte regulatory properties. Many cytokines useful for such purposes will be known to one of ordinary skill in the art, including interleukin-2 (IL-2); IL-4; IL-5; IL-12, which has been shown to enhance the protective effects of vaccines ( see, e.g., Science 268: 1432-1434, 1995 ); GM-CSF; IL-15; IL-18; combinations thereof, and the like. Thus cytokines can be administered in conjunction with antigen, chemokines and/or adjuvants to increase an immune response.
- IL-2 interleukin-2
- IL-4 interleukin-5
- IL-12 which has been shown to enhance the protective effects of vaccines (see, e.g., Science 268: 1432-1434, 1995 ); GM-CSF; IL-15; IL-18; combinations thereof, and the like.
- cytokines can be administered in conjunction with antigen, chemokines and/or adjuvant
- the other therapeutic agents can be used in the methods provided in unconjugated form or in conjugated form, such as conjugated to an anti-PSMA antibody or antigen-binding fragment thereof.
- Coupling of one or more toxin molecules to the anti-PSMA antibody or antigen-binding fragment thereof can include many chemical mechanisms, for instance covalent binding, affinity binding, intercalation, coordinate binding and complexation.
- the covalent binding can be achieved either by direct condensation of existing side chains or by the incorporation of external bridging molecules.
- Many bivalent or polyvalent agents are useful in coupling protein molecules to other proteins, peptides or amine functions, etc.
- the literature is replete with coupling agents such as carbodiimides, diisocyanates, glutaraldehyde, diazobenzenes, and hexamethylene diamines. This list is not intended to be exhaustive of the various coupling agents known in the art but, rather, is exemplary of the more common coupling agents.
- Suitable crosslinking agents for use in this manner include, for example, SPDP (N-succinimidyl-3-(2-pyridyldithio)propionate), and SMPT, 4-succinimidyl-oxycarbonyl-methyl-(2-pyridyldithio)toluene.
- protein toxins can be fused to the anti-PSMA antibody or antigen-binding fragment thereof by genetic methods to form a hybrid immunotoxin fusion protein.
- the fusion proteins can include additional peptide sequences, such as peptide spacers which operatively attach, for example, the anti-PSMA antibody and toxin, as long as such additional sequences do not appreciably affect the targeting or toxin activities of the fusion protein.
- the proteins can be attached by a peptide linker or spacer, such as a glycine-serine spacer peptide, or a peptide hinge, as is well known in the art.
- the C-terminus of an anti-PSMA antibody or antigen-binding fragment thereof can be fused to the N-terminus of the protein toxin molecule to form an immunotoxin that retains the binding properties of the anti-PSMA antibody.
- Other fusion arrangements will be known to one of ordinary skill in the art.
- the nucleic acid encoding the fusion protein is inserted into an expression vector in accordance with standard methods, for stable expression of the fusion protein, such as in mammalian cells, such as CHO cells.
- the fusion protein can be isolated and purified from the cells or culture supernatant using standard methodology, such as a PSMA affinity column.
- Radionuclides typically are coupled to an antibody or antigen-binding fragment thereof by chelation.
- a bifunctional chelator is commonly used to link the isotope to the antibody or other protein of interest.
- the chelator is first attached to the antibody, and the chelator-antibody conjugate is contacted with the metallic radioisotope.
- a number of bifunctional chelators have been developed for this purpose, including the diethylenetriamine pentaacetic acid (DTPA) series of amino acids described in U.S. patents 5,124,471 , 5,286,850 and 5,434,287 .
- hydroxamic acid-based bifunctional chelating agents are described in U.S.
- DOTA 1,4,7,10-tetraazacyclododecane N,N',N",N"'-tetraacetic acid
- DOTA 1,4,7,10-tetraazacyclododecane N,N',N",N"'-tetraacetic acid
- Other therapeutic agents also include replication-selective viruses.
- Replication-competent virus such as the p53 pathway targeting adenovirus mutant dl1520, ONYX-015, kills tumor cells selectively ( Biederer, C. et al., J. Mol. Med. 80(3):163-175, 2002 ).
- the virus can, in some embodiments, be conjugated to PSMA antibodies or antigen-binding fragments thereof.
- the methods provided can further comprise the use of other therapeutic treatment modalities.
- Such other treatments include surgery, radiation, cryosurgery, thermotherapy, hormone treatment, chemotherapy, vaccines and other immunotherapies.
- the ADCs of the invention can be linked to a label.
- Labels include, for example, fluorescent labels, enzyme labels, radioactive labels, nuclear magnetic resonance active labels, luminescent labels or chromophore labels.
- compositions provided can include a physiologically or pharmaceutically acceptable carrier, excipient or stabilizer mixed with the ADC.
- a composition comprises two or more different ADCs
- each of the antibodies or antigen-binding fragments thereof of the ADCs binds to a distinct conformational epitope of PSMA.
- compositions can be administered in combination therapy, i.e., combined with other agents.
- the combination therapy can include an ADC with at least one anti-tumor agent, immunomodulator, immunostimulatory agent or other conventional therapy.
- the other agent can be conjugated to or formed as a recombinant fusion molecule with a PSMA antibody or antigen-binding fragment thereof for directed targeting of the agent to PSMA-expressing cells.
- the other therapeutic agent can be unconjugated. Additional therapeutic agents can be administered or contacted with the PSMA-expressing cells through co-administration.
- Co-administering refers to administering two or more therapeutic agents simultaneously as an admixture in a single composition, or sequentially, and close enough in time so that the compounds may exert an additive or even synergistic effect.
- an additional therapeutic agent can be administered before, during or after the administration of one or more ADCs or compositions thereof.
- pharmaceutically acceptable carrier or “physiologically acceptable carrier” includes any and all salts, solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible.
- the carrier is suitable for intravenous, intramuscular, subcutaneous, parenteral, spinal or epidermal administration (e.g., by injection or infusion).
- the active compound can be coated in a material to protect the compound from the action of acids and other natural conditions that may inactivate the compound.
- the pharmaceutical compositions When administered, the pharmaceutical compositions are applied in pharmaceutically-acceptable amounts and in pharmaceutically-acceptable compositions.
- pharmaceutically acceptable means a non-toxic material that does not interfere with the effectiveness of the biological activity of the active ingredients. Such preparations may routinely contain salts, buffering agents, preservatives, compatible carriers, and optionally other therapeutic agents, such as supplementary immune potentiating agents including adjuvants, chemokines and cytokines.
- the salts should be pharmaceutically acceptable, but non-pharmaceutically acceptable salts may conveniently be used to prepare pharmaceutically-acceptable salts thereof and are not excluded from the scope of the invention.
- a salt retains the desired biological activity of the parent compound and does not impart any undesired toxicological effects (see e.g., Berge, S.M., et al. (1977) J. Pharm. Sci. 66: 1-19 ).
- Examples of such salts include acid addition salts and base addition salts.
- Acid addition salts include those derived from nontoxic inorganic acids, such as hydrochloric, nitric, phosphoric, sulfuric, hydrobromic, hydroiodic, phosphorous and the like, as well as from nontoxic organic acids such as aliphatic mono- and dicarboxylic acids, phenyl substituted alkanoic acids, hydroxy alkanoic acids, aromatic acids, aliphatic and aromatic sulfonic acids and the like.
- Base addition salts include those derived from alkaline earth metals, such as sodium, potassium, magnesium, calcium and the like, as well as from nontoxic organic amines, such as N,N'-dibenzylethylenediamine, N-methylglucamine, chioroprocaine, choline, diethanolamine, ethylenediamine, procaine and the like.
- An ADC can be combined, if desired, with a pharmaceutically-acceptable carrier.
- pharmaceutically-acceptable carrier means one or more compatible solid or liquid fillers, diluents or encapsulating substances which are suitable for administration into a human.
- carrier denotes an organic or inorganic ingredient, natural or synthetic, with which the active ingredient is combined to facilitate the application.
- the components of the pharmaceutical compositions also are capable of being co-mingled in a manner such that there is no interaction which would substantially impair the desired pharmaceutical efficacy.
- the pharmaceutical compositions may contain suitable buffering agents, including: acetic acid in a salt; citric acid in a salt; boric acid in a salt; and phosphoric acid in a salt.
- suitable buffering agents including: acetic acid in a salt; citric acid in a salt; boric acid in a salt; and phosphoric acid in a salt.
- compositions also may contain, optionally, suitable preservatives, such as: benzalkonium chloride; chlorobutanol; and parabens.
- suitable preservatives such as: benzalkonium chloride; chlorobutanol; and parabens.
- compositions may conveniently be presented in unit dosage form and may be prepared by any of the methods well-known in the art of pharmacy. All methods include the step of bringing the active agent into association with a carrier which constitutes one or more accessory ingredients. In general, the compositions are prepared by uniformly and intimately bringing the active compound into association with a liquid carrier, a finely divided solid carrier, or both, and then, if necessary, shaping the product.
- compositions suitable for parenteral administration conveniently comprise a sterile aqueous or non-aqueous preparation of the compounds, which is, in some embodiments, isotonic with the blood of the recipient.
- This preparation may be formulated according to known methods using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation also may be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example, as a solution in 1,3-butane diol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono-or di-glycerides.
- fatty acids such as oleic acid may be used in the preparation of injectables.
- Carrier formulations suitable for oral, subcutaneous, intravenous, intramuscular, etc. administration can be found in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA .
- the active compounds can be prepared with carriers that will protect the compound against rapid release, such as a controlled release formulation, including implants, transdermal patches, and microencapsulated delivery systems.
- a controlled release formulation including implants, transdermal patches, and microencapsulated delivery systems.
- Biodegradable, biocompatible polymers can be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Many methods for the preparation of such formulations are patented or generally known to those skilled in the art. See, e.g., Sustained and Controlled Release Drug Delivery Systems, J.R. Robinson, ed., Marcel Dekker, Inc., New York, 1978 .
- the therapeutics of the invention can be administered by any conventional route, including injection or by gradual infusion over time (e.g., the ADC in saline infused over 90 minutes).
- the administration may, for example, be oral, intravenous, intraperitoneal, intramuscular, intracavity, intratumor, or transdermal.
- routes of administration include intravenous and by pulmonary aerosol. Techniques for preparing aerosol delivery systems containing antibodies are well known to those of skill in the art.
- compositions of the invention are administered in effective amounts.
- An "effective amount” is that amount of any of the ADCs provided herein that alone, or together with further doses and/or other therapeutic agents, produces the desired response. This can involve any of the therapeutic endpoints mentioned herein. In one embodiment, this involves only slowing the progression of the disease temporarily, although in some embodiments, it involves halting the progression of the disease permanently. In other embodiments, it involves eradicating the disease altogether.
- the desired therapeutic endpoint can be monitored by routine methods known to those of ordinary skill in the art.
- An amount that is effective can be the amount of an ADC alone which produces the desired therapeutic endpoint.
- An amount that is effective is also the amount of an ADC in combination with another agent that produces the desired result.
- Such amounts will depend, of course, on the particular cancer being treated, the severity of the cancer, the individual patient parameters including age, physical condition, size and weight, the duration of the treatment, the nature of concurrent therapy (if any), the specific route of administration and like factors within the knowledge and expertise of the health practitioner. These factors are well known to those of ordinary skill in the art and can be addressed with no more than routine experimentation. It is generally preferred that a maximum dose of the individual components or combinations thereof be used, that is, the highest safe dose according to sound medical judgment. It will be understood by those of ordinary skill in the art, however, that a patient may insist upon a lower dose or tolerable dose for medical reasons, psychological reasons or for virtually any other reasons.
- the pharmaceutical compositions used in the methods can be sterile and contain an effective amount of an ADC, alone or in combination with another agent, for producing the desired response in a unit of weight or volume suitable for administration to a patient.
- the response can, for example, be measured by determining the physiological effects of the ADC composition, such as a reduction in tumor volume, reduction in the size or number of metastases, an increase in survival, an improvement in quality of life and/or reduction of cancer symptoms, etc.
- Other assays will be known to one of ordinary skill in the art and can be employed for measuring the level of the response.
- the doses of ADCs administered to a subject can be chosen in accordance with different parameters, in particular in accordance with the mode of administration used and the state of the subject. Other factors include the desired period of treatment. In the event that a response in a subject is insufficient at the initial doses applied, higher doses (or effectively higher doses by a different, more localized delivery route) may be employed to the extent that patient tolerance permits.
- doses can range from about 10 ⁇ g/kg to about 100,000 ⁇ g/kg. In some embodiments, the doses can range from about 0.1 mg/kg to about 20 mg/kg. In still other embodiments, the doses range from about 0.1 mg/kg to 5 mg/kg, 0.1 mg/kg to 10 mg/kg or 0.1 mg/kg to 15 mg/kg. In yet other embodiments, the doses range from about 1 mg/kg to 5 mg/kg, 5 mg/kg to 10 mg/kg, 10 mg/kg to 15 mg/kg or 15 mg/kg to 20 mg/kg.
- the dose is about 0.1 mg/kg, 0.5 mg/kg, 1 mg/kg, 2 mg/kg, 3 mg/kg, 5 mg/kg, 7 mg/kg, 10 mg/kg, 12 mg/kg, 15 mg/kg, 17 mg/kg, 20 mg/kg, 25 mg/kg or 30 mg/kg.
- the dose is about 0.4 mg/kg, 0.7 mg/kg. 1.1 mg/kg, 1.8 mg/kg, 2.4 mg/kg, 2.9 mg/kg, 3.0 mg/kg, 3.5 mg/kg or 4.0 mg/kg.
- the dose is about 0.6 mg/kg, 0.9 mg/kg, 1.2 mg/kg or 1.5 mg/kg.
- the dose is about 1 mg/kg, 3 mg/kg, 5 mg/kg or 6 mg/kg. Based upon the composition, the dose can be delivered continuously, such as by continuous pump, or at periodic intervals. In some embodiments, when the ADC is administered intravenously, the dose is between 0.1 and 20 mg/kg or any value in between. Desired time intervals of multiple doses of a particular composition can be determined without undue experimentation by one skilled in the art. Other protocols for the administration of the compositions provided will be known to one of ordinary skill in the art, in which the dose amount, schedule of administration, site(s) of administration, mode of administration and the like vary from the foregoing. In some embodiments, subjects are administered the ADC with a dose regimen of q4d x 3 or q4d x 6. In one embodiment, the dose is administered intravenously. In another embodiment, the dose regimen is a single intravenous dose.
- Administration of an ADC or a composition comprising an ADC to mammals other than humans, e.g., for testing purposes or veterinary therapeutic purposes, etc. is carried out under substantially the same conditions as described above.
- compositions of the present invention have in vitro and in vivo therapeutic utilities.
- these molecules can be administered to cells in culture, e.g. in vitro or ex vivo, or in a subject, e.g., in vivo, to treat a variety of cancers as provided herein.
- the term "subject" is intended to include humans and non-human animals.
- Non-human animals include, e.g., dogs, cats, horses, cows, pigs, mice and rats.
- Subjects include a human patient having a PSMA-expressing, taxane-resistant cancer.
- the subject in one embodiment, has progressive, castration-resistant, metastatic prostate cancer that has been treated with taxane but has progressed.
- the subject is one that meets the entry criteria as defined in the Examples below (e.g., Examples 5, 7 and 9).
- Treatment according to the present invention is expected to be particularly effective, in some embodiments, because it can direct high levels of ADCs to the bone marrow and lymph nodes where cancer metastases, such as prostate cancer metastases, can predominate.
- Treatment in accordance with the present invention can be effectively monitored with clinical parameters such as serum prostate specific antigen and/or pathological features of a patient's cancer, including stage, Gleason score, extracapsular, seminal, vesicle or perineural invasion, positive margins, involved lymph nodes, etc. Alternatively, these parameters can be used to indicate when such treatment should be employed.
- compositions for use in the methods provided herein can be in lyophilized form or provided in an aqueous medium.
- the present invention is further illustrated by the following Examples.
- PSMA-positive and PSMA-negative cells were exposed to PSMA ADC in 96-well microplates at various concentrations. After 96-hours, percent cell survival (compared to cells in medium) was assayed using fluorescent Alamar Blue ( Fig. 5 ).
- Fig. 6 presents a table showing cytotoxicity to PSMA-expressing prostate cancer cell lines following exposure to PSMA ADC.
- the mean tumor size was 108.9 and 108.7mm 3 with a p-value of 0.452 (student t-test) for the PBS and PSMA ADC groups, respectively, at randomization. All animals then received intravenous injections of PBS or PSMA ADC (Progenics, Tarrytown, NY, toxicology lot P10306) at 6mg/kg via tail vein in a volume of 0.1 mL on day 12 through 29 twice per week for three weeks. Tumor sizes were measured twice per week or weekly for 100 days after tumor implantation, and the mean tumor volume for the two groups was plotted over time post tumor implantation ( Fig. 7 ).
- Tumor size reduction was observed in PSMA ADC treated animals.
- the experiment was followed for 100 days, and all mice were sacrificed except 4 animals in the PSMA ADC treated group.
- Fig. 7 Among the 15 animals in the PSMA ADC treated group with a mean tumor size of 72.5mm 3 at day 75 as shown in Fig. 7 , there were 4 mice (mouse #8, 11, 12 and 14) with tumors that started to grow at day 75 ( Fig. 8 ). On day 99, the average tumor volume was 688.7mm 3 (893.8, 445.3, 666.7, 749.0 mm 3 ) for the 4 mice. The animals were treated again with PSMA ADC at 6mg/kg from day 99, once a week for 13 weeks. Fig. 8 shows the tumor volume over 180 days for the 4 mice. The dotted lines indicate initial PSMA ADC treatment, and the solid lines indicate second PSMA ADC treatment. Tumor size reduction was observed immediately in all 4 mice after the second PSMA ADC treatment, and the tumor volume continued to shrink while dosing. At day 180, the tumor volume was reduced to ⁇ 100mm 3 with a mean volume of 50mm 3 .
- PSMA ADC is effective to treat large-sized tumors ( ⁇ 900mm 3 ) and also can be used to treat relapsed tumors using the same dose level from an initial PSMA ADC treatment.
- Fig. 10 An experiment ( Fig. 10 ) was designed to evaluate PSMA ADC effectiveness after docetaxel treatment failure.
- Male athymic nude mice, 6-8 weeks old, obtained from Charles River Laboratories, Inc. were implanted subcutaneously with 5 million C4-2 cells.
- the mean tumor volume was 138.0 and 138.1mm 3 for the vehicle control and docetaxel treatment groups, respectively, at randomization.
- PSMA ADC The effect of PSMA ADC treatment on tumor growth after docetaxel failure was assessed by comparing tumor reduction after the second randomization.
- the tumor volumes of animals in the two subgroups were plotted ( Fig. 12 ). The results show that tumors grew continuously while on docetaxel treatment. However, the tumor volume was significantly reduced when PSMA ADC treatment was initiated. Therefore, PSMA ADC is effective in treating animals with tumors which failed docetaxel treatment.
- Example 5 A Phase 1 Dose-Escalation Study of PSMA ADC in Subjects with Progressive, Castration-Resistant, Metastatic Prostate Cancer
- DLT dose-limiting toxicity
- NCI National Cancer Institute
- CCAE Common Terminology Criteria for Adverse Events
- toxicity grade ⁇ 3 as well as grade 2 allergic/immunologic toxicity (except isolated fever) for which a causal relationship to the study drug cannot be excluded.
- CCTCAE Common Terminology Criteria for Adverse Events
- toxicity grade ⁇ 3 as well as grade 2 allergic/immunologic toxicity (except isolated fever) for which a causal relationship to the study drug cannot be excluded.
- CCTCAE Common Terminology Criteria for Adverse Events
- Radiologic imaging will be obtained at screening and week 12. Blood samples (5 mL) for drug concentrations will be obtained prior to infusion initiation and at 90 minutes and 4, 6, 24, 48, 96, 168, 336 and 504 hours after infusion initiation at weeks 0 and 6. Serum concentrations of study drug (ADC) and total antibody (PSMA-mAb + ADC) will be measured by a fully-validated ELISA method and serum concentrations of free toxin (MMAE) will be measured by a fully-validated liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) method. Immunogenicity will be assessed by a fully validated ELISA method for day 1 (predose) and week 12 time points.
- ADC study drug
- PSMA-mAb + ADC total antibody
- MMAE serum concentrations of free toxin
- LC/MS/MS liquid chromatography/mass spectrometry/mass spectrometry
- a phase 1, open-label, dose-escalation clinical trial will include men with progressive, hormone-refractory prostate cancer, and who had prior therapy with taxane chemotherapy drugs.
- the study will investigate the duration of clinical benefit derived from PSMA ADC treatment while also assessing the investigational drug's safety and tolerability.
- the initial 12-week period will evaluate up to five intravenous doses of PSMA ADC, individually administered at three-week intervals.
- the study will include evaluations of pharmacodynamics, radiographic changes in tumor burden, and changes in prostate-specific antigen (PSA) and circulating tumor cell (CTC) values compared to baseline.
- PSA prostate-specific antigen
- CTC circulating tumor cell
- Example 7 A Phase 1 Dose-Escalation Study of PSMA ADC in Subjects with Progressive, Castration-Resistant, Metastatic Prostate Cancer
- the dose-limiting toxicity can be defined as any National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE, Version 3.0) toxicity grade ⁇ 3 with the following exception: an absolute neutrophil count (ANC) ⁇ 500mm 3 determined eight days following the first infusion of PSMA ADC.
- a DLT can be defined as a neutrophil count ⁇ 500mm 3 found on both study days 8 and 15 following the first infusion of PSMA ADC.
- a DLT can also be defined as grade ⁇ 2 allergic/immunologic toxicity (except isolated fever) for which a causal relationship to the study drug cannot be excluded. In this study, DLT was/will be determined after the first dose of each cohort.
- a dose-escalation scheme based on the DLT, was/will be employed. Once an MTD is determined or the highest dose cohort is shown safe, additional subjects will be treated. These subjects will be chosen using the same inclusion/exclusion criteria employed for the study. If two or more subjects in the lowest dose cohort experience a DLT, a cohort will be enrolled at 0.2 mg/kg of PSMA ADC.
- Radiologic imaging was/will be obtained prior to infusion and within seven days prior to the week 12 visit.
- Blood samples (5 mL) for drug concentrations was/will be obtained at designated time points in weeks 0, 3, 6, 9 and 12 (at week 12, sampling is at study day 85 ⁇ one day).
- samples were/are collected prior to infusion initiation, immediately at the end of infusion, and at four and six hours after the infusion.
- Samples collected at 24, 48, 96, 168, 336 and 504 hours after infusion initiation may be collected within ⁇ two hours of the designated time point.
- weeks 3, 6 and 9 only predose samples were/are collected.
- Serum concentrations of study drug (ADC) and total antibody (PSMA-mAb + ADC) were/will be measured by a fully-validated ELISA method and serum concentrations of free toxin (MMAE) were/will be measured by a fully-validated liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) method. Immunogenicity was/will be assessed by a fully validated ELISA method for day 1 (predose) and week 12 time points.
- a phase 1, open-label, dose-escalation clinical trial included men with progressive, hormone-refractory prostate cancer, and who had prior therapy with taxane chemotherapy drugs.
- the study investigates the duration of clinical benefit derived from PSMA ADC treatment and assesses the investigational drug's safety and tolerability.
- PSMA ADC was individually administered at three-week intervals during a 12-week period for the evaluation of at least four intravenous doses of PSMA ADC.
- the study includes evaluations of pharmacodynamics, radiographic changes in tumor burden, and changes in prostate-specific antigen (PSA) and circulating tumor cell (CTC) values compared to baseline.
- PSA prostate-specific antigen
- CTC circulating tumor cell
- IV infusions of PSMA ADC will be administered to subjects in four cycles.
- IV infusions of PSMA ADC will be administered once at the start of a cycle (duration 3 weeks or Q3W; weeks 1, 4, 7 and 10) for a total of 4 doses (4 cycles) as eight progressively enrolled treatment groups: 0.4 mg/kg, 0.7 mg/kg, 1.1 mg/kg, 1.8 mg/kg, 2.4 mg/kg, 3.0 mg/kg, 3.5 mg/kg or 4.0 mg/kg.
- IV infusions of PSMA ADC will be administered to subjects once every week for the first three weeks of a four week cycle for a total of 12 doses (4 cycles or 4QW with a dose during week 1, 2 and 3 in cycle 1; weeks, 5, 6 and 7 in cycle 2; weeks 9, 10 and 11 in cycle 3; and weeks 13, 14 and 15 in cycle 4; no doses will be administered during weeks 4, 8, 12 and 16 in cycles 1, 2, 3 and 4, respectively) as four progressively enrolled treatment groups: 0.6 mg/kg, 0.9 mg/kg, 1.2 mg/kg and 1.5 mg/kg.
- PSMA ADC will be administered in saline over approximately 90 minutes.
- the dose-limiting toxicity will be defined as any National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE, Version 3.0) toxicity grade ⁇ 3 with the following exception: an absolute neutrophil count (ANC) ⁇ 500mm 3 .
- a DLT will be defined as a neutrophil count ⁇ 500mm 3 that persist upon repeat determination 3 to 7 days following final PSMA ADC administration within the first cycle.
- an ANC ⁇ 500mm 3 prior to any dose in the first cycle is a DLT.
- a DLT is also defined as grade ⁇ 2 allergic/immunologic toxicity (except isolated fever) for which a causal relationship to the study drug cannot be excluded. In this study, DLT will be determined after the first cycle of each cohort.
- a dose-escalation scheme based on the DLT, will be employed. Determinations of DLT will occur as a separate consideration for cohorts 1-8 and 9-12. A DLT determination for cohort 1-8 will not influence enrollment in cohorts 9-12. Once an MTD is determined or the highest dose cohort is shown safe, additional subjects will be treated. These subjects will be chosen using the same inclusion/exclusion criteria employed for the study.
- radiologic imaging will be obtained prior to infusion and within seven days prior to the week 13 visit.
- Blood samples (5 mL) for drug concentrations will be obtained at time points in weeks 0, 1, 4, 7, 10 and 13 (at week 13, sampling is at study day 85 ⁇ one day).
- samples are collected prior to infusion initiation, immediately at the end of infusion, and at four and six hours after the infusion.
- Samples collected at 24, 48, 96, 168, 336 and 504 hours after infusion initiation may be collected within ⁇ two hours of the designated time point.
- At weeks 4, 7 and 10 only predose samples are collected.
- radiologic imaging will be obtained prior to the first infusion (week 1) and within seven days prior to the week 17 visit.
- Blood samples (5 mL) for drug concentrations will be obtained at designated time points: for week 1 (cycle 1) immediately prior to infusion initiation, immediately at the end of the infusion, and at four and at six hours after the infusion. Samples collected at 24, 48 and 96 hours after infusion initiation may be collected within ⁇ two hours of the designated time point. Samples collected at weeks 2, 3 and 5 are collected prior to infusion. Samples are also to be obtained at week 4.
- Serum concentrations of study drug (ADC) and total antibody (PSMA-mAb + ADC) will be measured by a fully-validated ELISA method and serum concentrations of free toxin (MMAE) will be measured by a fully-validated liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) method.
- Immunogenicity will be assessed by a fully validated ELISA method for day 1 (predose) and week 13 time points for cohorts 1-8 and study day 1 (predose) and week 17 for cohorts 9-12.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Cell Biology (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Reproductive Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Oncology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
- This application claims the benefit under 35 U.S.C. § 119 of United States provisional application
61/095,300, filed September 8, 2008 61/205,395, filed January 20, 2009 - Prostate cancer is the most common malignancy and the second leading cause of cancer death in men in the United States (Jemal A, et al., CA Cancer J Clin 2005;55:10-30). Localized prostate cancer typically is treated with surgery or radiation, and recurrent disease can be controlled temporarily with androgen ablation (Klein EA, et al., Urol Clin North Am 2003;30:315-30). However, almost all prostate carcinomas eventually become hormone refractory and then rapidly progress (Denmeade SR, et al., Nat Rev Cancer 2002;2:389-96). Hormone-refractory or androgen-independent prostate cancer has proven to be largely resistant to conventional chemotherapy. With the exception of palliative care, the only approved chemotherapy is docetaxel in combination with prednisone, which offers a modest (2.4 month) survival benefit (Gulley J, et al., Am J Ther. 2004;351:1513-20; Petrylak DP, et al., New Engl J Med 2004;351:1513-20).
- M. D GALSKY ET AL:JOURNAL OF CLINICAL ONCOLOGY, AMERICAN SOCIETY OF CLINICAL ONCOLOGY, vol. 26, no. 13, 2008 (2008-05-01) discloses the results of a clinical trial performed on the immunoconjugate MLN2704 in patients with progressive metastatic castration-resistant prostate cancer. MLN2704 comprises J591, a deimmunized monoclonal antibody targetingan external domain of PSMA and the maytansine analog drug maytansoid-1 (DM1).
- The present invention relates, at least in part, to the surprising discovery that antibody-drug conjugates (ADCs) comprising an antibody or antigen-binding fragment thereof that specifically binds PSMA conjugated to a
dolastatin 10 derivative, in particular auristatins such as, MMAE (also referred to herein as monomethylauristatin E or monomethylauristatin norephedrine) or MMAF (also referred to herein as monomethylauristatin F or monomethylauristatin phenylalanine) can be used to kill PSMA-expressing, taxane-resistant cancer cells and to treat PSMA-expressing, taxane-resistant cancer disclosed herein, therefore, are methods for killing PSMA-expressing, taxane-resistant cancer cells. Also disclosed herein, are methods for treating a subject with a PSMA-expressing, taxane-resistant cancer. In one embodiment of the latter methods, the methods involve killing PSMA-expressing, taxane-resistant cancer cells. In another embodiment of the latter methods, the methods involve delaying or inhibiting progression of the cancer. In still another embodiment of the latter methods, the methods involve increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy. In yet another embodiment of the latter methods, the methods involve decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level. In a further embodiment of the latter methods, the methods involve decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA. - In one aspect, therefore, a method for killing PSMA-expressing, taxane-resistant cancer cells comprising contacting the PSMA-expressing, taxane-resistant cancer cells with an antibody-drug conjugate in an amount effective to kill the PSMA-expressing, taxane-resistant cancer cells, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1, is disclosed.
- In another aspect, a method for treating a subject that has a PSMA-expressing, taxane-resistant cancer comprising administering to the subject that has the PSMA-expressing, taxane-resistant cancer an antibody-drug conjugate in an amount effective to treat the PSMA-expressing, taxane-resistant cancer, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1, is also disclosed. In one embodiment, the method involves killing PSMA-expressing, taxane-resistant cancer cells. In another embodiment, the method involves delaying or inhibiting progression of the cancer. In still another embodiment, the method involves increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy. In yet another embodiment, the method involves decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level. In a further embodiment, the method involves decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA.
- In yet another aspect, a method for treating a subject having progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, comprising administering an effective amount of an antibody-drug conjugate, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1, is also provided. In one embodiment, the PSMA antibody drug conjugate (also referred to herein as "PSMA ADC" or "ADC") consists essentially of a human monoclonal antibody to PSMA conjugated to monomethylaurastatin norephedrine (MMAE) via a valine-citrulline linker. In one embodiment, the method involves killing PSMA-expressing, taxane-resistant cancer cells. In another embodiment, the method involves delaying or inhibiting progression of the cancer. In still another embodiment, the method involves increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy. In yet another embodiment, the method involves decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level. In a further embodiment, the method involves decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA.
- In one embodiment of the methods provided, the effective amount of the ADC is sufficient to 1) delay or inhibit progression of the cancer, 2) increase survival of the subject as compared to the median survival of subjects who have not been treated with PSMA ADC and who have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, 3) decrease a circulating level of circulating tumor cells (CTCs) compared to a baseline level and/or 4) decrease or stabilize a serum level of prostate specific antigen (PSA) compared to a baseline level of PSA.
- In one embodiment, the killing of the PSMA-expressing, taxane-resistant cancer cells or the delay or inhibition of progression of the cancer is demonstrated by radiographic image changes in tumor burden compared to a baseline radiographic image in the subject prior to the administration of the PSMA ADC. In one embodiment, the radiographic image change is a change of at least 10%. In another embodiment, the radiographic image change is a change of at least 20%. In still another embodiment, the radiographic image change is a change of at least 30%. In yet another embodiment, the radiographic image change is a change of at least 40%. In a further embodiment, the radiographic image change is a change of at least 50%. In still a further embodiment, the radiographic image change is a change of at least 60%.
- In another embodiment of the methods provided, the killing of the PSMA-expressing, taxane-resistant cancer cells or the delay or inhibition of progression of cancer is demonstrated by a change in at least one biomarker for bone metastasis and bone metabolism compared to a baseline value prior to the administration of the PSMA ADC. In one embodiment, the biomarker is N-telopeptide, bone alkaline phosphatase, osteocalcin, calcitonin, calcium, pyridinoline or deoxypyridinoline.
- In still another embodiment of the methods provided, treatment with the PSMA ADC results in increased survival for the subject, wherein the survival is increased in comparison to the median survival time of subjects with PSMA-expressing, taxane-resistant cancer not treated with PSMA ADC. In one embodiment, survival in the subject is increased by four weeks. In another embodiment, survival in the subject is increased by six weeks. In still another embodiment, survival in the subject is increased by two months. In yet another embodiment, survival in the subject is increased by four months. In a further embodiment, survival in the subject is increased by six months. In another embodiment, survival in the subject is increased by eight months. In still another embodiment, survival in the subject is increased by ten months. In yet another embodiment, survival in the subject is increased by twelve months. In still another embodiment, survival in the subject is increased by fourteen months.
- In yet another embodiment of the methods provided, treatment with the PSMA ADC results in an improvement in the subject's quality of life as compared to the quality of life of the subject prior to treatment with the PSMA ADC.
- In a further embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer cells are resistant to docetaxel or paclitaxel. In still a further embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer is resistant to docetaxel or paclitaxel.
- In another embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer cells are prostate cancer cells or non-prostate cancer cells. In one embodiment, the non-prostate cancer cells are bladder cancer cells, pancreatic cancer cells, liver cancer cells, lung cancer cells, kidney cancer cells, sarcoma cells, breast cancer cells, brain cancer cells, neuroendocrine carcinoma cells, colon cancer cells, testicular cancer cells or melanoma cells.
- In still another embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer is prostate cancer or non-prostate cancer. In one embodiment, the non-prostate cancer is bladder cancer, pancreatic cancer, liver cancer, lung cancer, kidney cancer, sarcoma, breast cancer, brain cancer, neuroendocrine carcinoma, colon cancer, testicular cancer or melanoma.
- In a further embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer cells are of a tumor. In still a further embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer is a tumor. In one embodiment, the volume of the tumor is at least 100mm3 prior to initiation of the method using PSMA ADC. In another embodiment, the volume of the tumor is at least 200mm3. In still another embodiment, the volume of the tumor is at least 300mm3. In a further embodiment, the volume of the tumor is at least 400mm3. In still a further embodiment, the volume of the tumor is at least 500mm3. In yet another embodiment, the volume of the tumor is at least 600mm3. In still another embodiment, the volume of the tumor is at least 700mm3. In a further embodiment, the volume of the tumor is at least 800mm3. In yet a further embodiment, the volume of the tumor is at least 900mm3. In another embodiment, the volume of the tumor is at least 1000mm3. In yet another embodiment, the volume of the tumor is at least 1200mm3. In still another embodiment, the volume of the tumor is at least 1400mm3. In yet a further embodiment, the volume of the tumor is at least 1600mm3.
- In another embodiment of the methods provided, treatment or contact with the PSMA ADC results in a reduction of tumor volume by at least 10% compared to the tumor volume prior to the treatment or contact with the PSMA ADC. In yet another embodiment, the volume of the tumor is reduced by at least 20%. In still another embodiment, the volume of the tumor is reduced by at least 30%. In a further embodiment, the volume of the tumor is reduced by at least 40%. In still a further embodiment, the volume of the tumor is reduced by at least 50%. In yet a further embodiment, the volume of the tumor is reduced by at least 60%. In another embodiment, the volume of the tumor is reduced by at least 70%. In still another embodiment, the volume of the tumor is reduced by at least 80%. In yet another embodiment, the volume of the tumor is reduced by at least 90%. In a further embodiment, the volume of the tumor is reduced by at least 95%. In yet a further embodiment, the tumor is eradicated.
- In one embodiment, the tumor has a length of at least 5mm prior to initiation of the method using PSMA ADC. In another embodiment, the tumor has a length of at least 6mm. In still another embodiment, the tumor has a length of at least 7mm. In yet another embodiment, the tumor has a length of at least 8mm. In another embodiment, the tumor has a length of at least 9mm. In still another embodiment, the tumor has a length of at least 10mm. In yet another embodiment, the tumor has a length of at least 11mm. In a further embodiment, the tumor has a length of at least 12mm. In still a further embodiment, the tumor has a length of at least 13mm. In still a further embodiment, the tumor has a length of at least 14mm. In another embodiment, the tumor has a length of at least 15mm. In yet another embodiment, the tumor has a length of at least 16mm. In still another embodiment, the tumor has a length of at least 17mm. In a further embodiment, the tumor has a length of at least 18mm. In yet a further embodiment, the tumor has a length of at least 19mm. In still a further embodiment, the tumor has a length of at least 20mm. In another embodiment, the tumor has a length of at least 21mm. In still another embodiment, the tumor has a length of at least 22mm. In yet another embodiment, the tumor has a length of at least 23mm. In a further embodiment, the tumor has a length of at least 24mm. In still a further embodiment, the tumor has a length of at least 25mm. In yet a further embodiment, the tumor has a length of at least 30mm.
- In another embodiment of the methods provided, treatment or contact with the PSMA ADC results in a reduction of tumor length by at least 10% compared to the tumor length prior to the treatment or contact with the PSMA ADC. In yet another embodiment, the length of the tumor is reduced by at least 20%. In still another embodiment, the length of the tumor is reduced by at least 30%. In a further embodiment, the length of the tumor is reduced by at least 40%. In still a further embodiment, the length of the tumor is reduced by at least 50%. In yet a further embodiment, the length of the tumor is reduced by at least 60%. In another embodiment, the length of the tumor is reduced by at least 70%. In still another embodiment, the length of the tumor is reduced by at least 80%. In yet another embodiment, the length of the tumor is reduced by at least 90%. In a further embodiment, the length of the tumor is reduced by at least 95%. In yet a further embodiment, the length of the tumor is reduced by at least 99%.
- In a further embodiment of the methods provided, administration of the PSMA ADC to a subject results in the gain of body weight in the subject (as compared to the weight of the subject prior to administration of the PSMA ADC to the subject). The gain in body weight may result from a single administrion of the PSMA ADC or a course of treatment with the PSMA ADC (i.e., more than one administration). In one embodiment, the gain is at least a 5% gain in body weight. In another embodiment, the gain is at least a 10% gain. In still another embodiment, the gain is at least a 15% gain. In still a further embodiment, the gain is at least a 20% gain. In yet another embodiment, the gain is at least a 25% gain. In a further embodiment, the gain is at least a 30% gain. In another embodiment, the gain is a 5% gain in body weight. In another embodiment, the gain is a 10% gain. In still another embodiment, the gain is a 15% gain. In still a further embodiment, the gain is a 20% gain. In yet another embodiment, the gain a 25% gain. In a further embodiment, the gain is a 30% gain.
- In one embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer cells are of a metastasis. In another embodiment of the methods provided, the PSMA-expressing, taxane-resistant cancer is metastatic.
- In another embodiment of the methods provided, the antibody or antigen-binding fragment thereof is conjugated to at least 2 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules. In yet another embodiment of the methods provided, the antibody or antigen-binding fragment thereof is conjugated to at least 3 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules. In still another embodiment of the methods provided, the antibody or antigen-binding fragment thereof is conjugated to at least 4 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules.
- In a further embodiment of the methods provided, the monomethylauristatin norephedrine or monomethylauristatin phenylalanine is conjugated to the antibody or antigen-binding fragment thereof with a compound of the formula -An-Ym-Zm-Xn-Wn-wherein, A is a carboxylic acyl unit; Y is an amino acid; Z is an amino acid; X and W are each a self-immolative spacer; n is an integer of 0 or 1; and m is an integer of 0 or 1, 2, 3, 4, 5 or 6.
- In one embodiment, A is

- In another embodiment, Y is alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan or proline. In yet another embodiment, Y is valine.
- In a further embodiment, Z is lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, or citrulline. In yet a further embodiment, Z is citrulline.
- In one embodiment, Ym-Zm is valine-citrulline. In another embodiment, Ym-Zm is a protein sequence which is selectively cleavable by a protease.
- In still another embodiment, X is a compound having the formula


- In one embodiment, W is

- In another embodiment, m and n are 0.
- In still another embodiment of the methods provided, the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin norephedrine. In another embodiment of the methods provided, the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin norephedrine. In yet another embodiment, the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin phenylalanine. In another embodiment, the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin phenylalanine. In a further embodiment, the antibody-drug conjugate is AB-PG1-XG1-006-maleimide caproyl-monomethylauristatin phenylalanine. In still a further embodiment, the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin norephedrine. In another embodiment, the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin norephedrine. In yet a further embodiment, the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin phenylalanine. In yet a further embodiment, the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin phenylalanine. In another embodiment, the antibody-drug conjugate is AB-PG1-XG1-026-maleimide caproyl-monomethylauristatin phenylalanine.
- In one embodiment of the methods provided, the monoclonal antibody or antigen-binding fragment thereof specifically binds to an extracellular domain of PSMA. In another embodiment, the monoclonal antibody or antigen-binding fragment thereof specifically binds to a conformational epitope of PSMA. In still another embodiment, the monoclonal antibody or antigen-binding fragment thereof binds live cells. In a further embodiment, the monoclonal antibody or antigen-binding fragment thereof does not require cell lysis to bind PSMA. In still a further embodiment, the monoclonal antibody or antigen-binding fragment thereof binds to cells of the neovasculature of a tumor.
- In another embodiment of the methods provided, the antibody or antigen-binding fragment thereof competitively inhibits the specific binding of a second antibody to its target epitope on PSMA, wherein the second antibody is selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, Abgenix 4.152.1 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14, 18, 22, 26 and 30, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16, 20, 24, 28 and 32.
- In still another embodiment of the methods provided, the antibody or antigen-binding fragment thereof binds to an epitope on PSMA defined by an antibody selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, Abgenix 4.152.1 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14, 18, 22, 26 and 30, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16, 20, 24, 28 and 32.
- In one embodiment, the second antibody is selected from the group consisting of AB-PG1-XG1-006, AB-PG1-XG1-026 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16 and 20. In another embodiment, the second antibody comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 14, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 16. In still another embodiment, the second antibody comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 20.
- In a further embodiment of the methods provided, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 90% identical to a nucleotide sequence encoding an antibody selected from the group consisting of: AB-PG1-XG1-006, AB-PG1-XG1-026 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16 and 20. In another embodiment, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 95% identical. In still another embodiment, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 97% identical. In yet another embodiment, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 98% identical. In a further embodiment, the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 99% identical.
- In another embodiment of the methods provided, the antibody or antigen-binding fragment thereof is selected from the group consisting of antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16 and 20, and antigen-binding fragments thereof. In still another embodiment, the antibody or antigen-binding fragment thereof comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 14, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 16, and antigen-binding fragments thereof. In yet another embodiment, the antibody or antigen-binding fragment thereof comprises (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 3, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 9, or (c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 18, and d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 20, and antigen-binding fragments thereof.
- In yet a further embodiment of the methods provided, the antibody or antigen-binding fragment thereof is AB-PG1-XG1-006, AB-PG1-XG1-026 or an antigen-binding fragment thereof.
- In still a further embodiment of the methods provided, the human antibody is an IgG1 comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8.
- In a further aspect, provided is an antibody-drug conjugate comprising an antibody, or antigen-binding fragment thereof, that specifically binds PSMA, conjugated to a
dolastatin 10 derivative, for use in a method of treating a PSMA-expressing, taxane-resistant cancer. Preferably, said method involves killing PSMA-expressing, taxane-resistant cancer cells. In another embodiment, the method involves delaying or inhibiting progression of the cancer. In still another embodiment, the method involves increasing survival of the subject as compared to the median survival of subjects not been treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy. In yet another embodiment, the method involves decreasing a circulating level of circulating tumor cells (CTCs) compared to a baseline level. In a further embodiment, the method involves decreasing or stabilizing a serum level of PSA compared to a baseline level of PSA. It is also preferred that the PSMA-expressing, taxane-resistant cancer cells are killed by the conjugate. The PSMA preferably has the sequence set forth in SEQ ID NO: 1. The conjugate is preferably for treating progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy. Thedolastatin 10 derivative can be an auristatin, and is preferably MMAE (also referred to herein as monomethylauristatin E or monomethylauristatin norephedrine) or MMAF (also referred to herein as monomethylauristatin F or monomethylauristatin phenylalanine). In preferred embodiments of this further aspect, the conjugate can have any of the features of the conjugate employed in other aspects of the invention defined herein. The method involved in this further aspect, moreover, can involve the administration of an effective amount of the conjugate, as defined with reference to other aspects defined herein. It can also have any of the features of any of the methods for treating a subject that are described herein. -
-
Fig. 1 presents a schematic showing the sub-cellular localization of PSMA in normal prostate versus prostate cancer. PSMA is a 750 residue, type II transmembrane glycoprotein that is highly expressed in prostate cancer cells and has limited expression in normal non-prostatic tissues. In the normal prostate, PSMA is expressed predominantly as a cytoplasmic splice variant (PSM') that is retained in the cytoplasm. In prostate cancer, PSMA is expressed as a membrane-anchored, noncovalently associated homodimer. -
Fig. 2 presents a schematic of a number of prostate cancer treatment options. The approved chemotherapy, docetaxel (Taxotere) in combination with prednisone, for advanced prostate cancer provides a modest increase in survival (2-3 months) with significant side effects. -
Fig. 3 presents a schematic showing antibody-drug conjugate (ADC) targeted therapy. -
Fig. 4 provides the chemical structures of three examples of drug linkers that can be conjugated to an antibody or antigen-binding fragment thereof that specifically binds PSMA and form an antibody-drug conjugate. -
Fig. 5 demonstrates the potent and specific cytotoxicity of PSMA ADC to PSMA-expressing prostate cancer cells. PSMA-positive and PSMA-negative cells were exposed to PSMA ADC in 96-well microplates at various concentrations. After 96-hours, percent cell survival (compared to cells in medium) was assayed using fluorescent Alamar Blue. -
Fig. 6 presents a table that demonstrates the potent and specific cytotoxicity of PSMA ADC to PSMA-expressing prostate cancer cells. -
Fig. 7 shows tumor volume over time in nude mice treated with PSMA ADC versus control. Nude mice bearing human prostate cancer tumors were randomized atday 11 into two groups, PBS vehicle control (filled diamond, n = 15, 108.9mm3) and PSMA ADC (filled triangle, n=15, 108.7mm3), according to tumor volume. The animals received intravenous injections of PBS or PSMA ADC at 6mg/kg onday -
Fig. 8 shows the effect of PSMA ADC treatment on tumor volume with an initial and second PSMA ADC treatment. Tumors of four mice from the PSMA ADC treatment group were grown back and were treated with PSMA ADC at the same dose level of 6mg/kg. PSMA ADC effectively reduced the tumor volume to < 100mm3 from large tumor of 894mm3. The dotted lines indicate initial PSMA ADC treatment (6 dosings twice per week), and the solid lines show second PSMA ADC treatment (13 weekly dosings). -
Fig. 9 demonstrates the effects in a nude mouse having a tumor size of ∼500mm3 and treated with 12 doses of docetaxel given intraperitoneally (IP) at 12mg/kg weekly as indicated by dotted lines. The tumor responded to the docetaxel initially and then relapsed. Starting atday 100 with tumor volume of 800mm3, PSMA ADC was given intravenously (IV) at 6mg/kg for an additional 12 doses weekly (solid lines). PSMA ADC reduced the tumor volume from 800mm3 to 50mm3. -
Fig. 10 shows a scheme of randomization for testing PSMA ADC effectiveness after docetaxel treatment failure. At the first randomization, the animals were assigned to vehicle control group or docetaxel treatment group. If tumor volume exceeded 400 mm3 in the docetaxel treated group, the animals were randomized (the second randomization) at a 1:1 ratio to two subgroups: continued docetaxel treatment or PSMA ADC treatment. The tumor volumes of the animals were measured over time. -
Fig. 11 shows the tumor volume over time in nude mice treated with docetaxel versus control. Nude mice bearing human prostate cancer tumors were randomized atday 14 according to tumor volume into two groups, vehicle control (filled diamond, n = 10) and docetaxel (filled circle, n=50). The animals received intravenous injections of PBS or docetaxel at 2mg/kg/IV weekly. The average tumor volume for the two groups was plotted until the second randomization was initiated. Docetaxel treatment reduced tumor growth compared to control at least by day 21 (p=0.025) and beyond. -
Fig. 12 shows the comparison of tumor volume in two subgroups after failure of initial docetaxel treatment: (1) continued docetaxel treatment, filled triangle, (2) PSMA ADC treatment, 6mg/kg, filled squares. Since the mice were assigned to different groups at different times, time of treatment (days post second randomization) was aligned at day 0 (initiation of PSMA ADC or continuation of docetaxel post second randomization). -
Fig. 13 shows a schematic of the design of an in vivo study. -
Fig. 14 shows the tumor volume of each animal. When the tumor volume of an animal in the docetaxel-treated group exceeded 400 mm3, the animal was randomized at a 1:1 ratio into two subgroups: 1) switched to PSMA ADC treatment at 6 mg/kg/IV weekly (solid squares, mean: 515 ± 103 mm3, range 410-727 mm3) and 2) continued treatment with docetaxel at 2 mg/kg (filled triangles, mean: 495 ± 80 mm3, range: 401-650 mm3). The tumor size of each animal and time of randomization are shown. Tumor volumes at the second randomization were not significantly different between the two groups (p = 0.518, t-test). -
Fig. 15 shows the tumor size for each individual animal in two groups after second randomization. The tumor volumes of animals with PSMA ADC treatment (panel A) and continued docetaxel treatment (panel B) were plotted as a function of time following the second randomization (day 0). The tumors shrank when PSMA ADC treatment was initiated (panel A). However, even with continued docetaxel treatment, tumors continued to progress except in one animal (panel B). -
Fig. 16 shows a comparison of the mean tumor volumes in animals receiving PSMA ADC treatment (filled squares) and in animals receiving continued docetaxel treatment (filled triangles). Mean tumor volume was compared as a function of time following the second randomization (day 0). Tumor sizes of the two groups were significantly different (p < 0.007). -
Fig. 17 shows a comparison of the survival of animals over time post second randomization for animals receiving PSMA ADC treatment (solid line) and animals receiving continued docetaxel treatment (dotted line). At day 157 post second randomization, all 18 animals in the PSMA ADC treatment group were still alive. However, 16 of 18 animals in the continued docetaxel treatment group were sacrificed due to their large tumors (> 2,000 mm3) with a median survival of 45 days. PSMA ADC significantly increased the overall survival (p < 0.0001). -
Fig. 18 shows a comparison of the average body weights post second randomization in animals receiving PSMA ADC treatment (solid squares) and animals receiving continued docetaxel treatment (solid triangles). Overall, animals gained weight in the PSMA ADC treatment group, while animals lost weight in the continued docetaxel treatment group. The body weight change was significantly different (p < 0.001) between these two groups. - The present invention relates, in part, to the surprising discovery that antibody-drug conjugates (ADCs) comprising an antibody or antigen-binding fragment thereof that specifically binds PSMA conjugated to a
dolastatin 10 derivative, in particular auristatins such as, MMAE (also referred to herein as monomethylauristatin E or monomethylauristatin norephedrine) or MMAF (also referred to herein as monomethylauristatin F or monomethylauristatin phenylalanine) can be used to kill PSMA-expressing, taxane-resistant cancer cells. As described further below, it was unexpectedly found that PSMA expressing cancer cells which had become resistant to the effects of an anti-cancer agent that acts by disrupting the microtubular network in cells, were sensitive to the effects of a second anti-cancer agent that also targets microtubules. Specifically disclosed herein, the auristatin, MMAE, a potent tubulin inhibitor, in the form of an antibody-drug conjugate, when delivered to PSMA-expressing prostate tumors that had become resistant to docetaxel was able to kill the docetaxel resistant cancer cells such that tumor volume was significantly decreased. - As used herein, "PSMA-expressing cancer cells" is intended to refer to cancer cells that express PSMA (e.g., human PSMA). PSMA is a 100 kD Type II membrane glycoprotein expressed in prostate tissues (Horoszewicz et al., 1987, Anticancer Res. 7:927-935;
U.S. Pat. No. 5,162,504 ). PSMA was characterized as a type II transmembrane protein having sequence identity with the transferrin receptor (Israeli et al., 1994, Cancer Res. 54:1807-1811) and with NAALADase activity (Carter et al., 1996, Proc. Natl. Acad. Sci. U.S.A. 93:749-753). More importantly, PSMA is expressed in increased amounts in prostate cancer, and elevated levels of PSMA are also detectable in the sera of these patients (Horoszewicz et al., 1987; Rochon et al., 1994, Prostate 25:219-223; Murphy et al., 1995, Prostate 26:164-168; and Murphy et al., 1995, Anticancer Res. 15:1473-1479). PSMA expression in cancerous prostate is approximately 10-fold greater than that in normal prostate. Expression in normal prostate is approximately 10-fold greater than that in the brain and is 50- to 100-fold greater than that of the liver or kidney. In most normal tissues, no expression of PSMA is observed. PSMA expression increases with disease progression, becoming highest in metastatic, hormone-refractory disease. In addition, PSMA is also abundantly expressed on the neovasculature of a variety of non-prostate tumors, including bladder, breast, colon, pancreas, sarcoma, melanoma, renal, liver, lung (e.g., non-small cell lung carcinoma), and kidney tumors, but not on normal vasculature. PSMA-expressing cancer cells, therefore, include, for example, prostate cancer cells as well as endothelial cells of the neovasculature of a number of non-prostate cancers. - "Taxane-resistant cancer cells" refers to cancer cells of a cancer that has been treated with a taxane but that has become resistant to treatment with the taxane (i.e., further treatment with the taxane would not slow or halt the progression of the cancer and/or would no longer provide a significant benefit to a subject with the cancer). A "subject with a taxane-resistant cancer" is one that has received prior treatment with a taxane but no longer experiences a significant benefit from further treatment with the taxane (e.g., no longer experiences a significant reduction in the number of cancer cells, in tumor volume, in tumor length, in the number of metastases and/or in other markers of disease progression and/or no longer experiences an improvement in quality of life and/or increased survival as compared to the expected survival time without further treatment with the taxane).
- Therefore, in one embodiment the PSMA ADC is administered to a subject with progressive, metastatic, hormone-refractory prostate cancer resistant to taxane following prior taxane chemotherapy. In another embodiment, such subject is a human.
- As used herein, "taxanes" are anti-cancer agents that interfere with or disrupt microtubule stability, formation and/or function. Such agents include paclitaxel and docetaxel as well as derivatives thereof, wherein the derivatives function against microtubules by the same mode of action as the taxanes from which they are derived.
- The ADC can comprise an antibody or antigen-binding fragment thereof conjugated to the auristatin MMAE or MMAF. Auristatins are synthetic pentapeptide molecules that are structurally related to
dolastatin 10, a natural product derived from a marine animal. MMAE and other auristatins act by inhibiting polymerization of tubulins, thereby preventing formation of the mitotic spindle. Apoptotic cell death is triggered when the cell cycle is arrested. The antibody or antigen-binding fragment thereof can be, in some embodiments, conjugated to MMAE or MMAF with a compound of the following formula (Formula 1): - An-Ym-Zm-Xn-Wn-, wherein A is a carboxylic acyl unit; Y is an amino acid; Z is an amino acid; X and W are each a self-immolative spacer; n is an integer of 0 or 1; and m is an integer of 0 or 1, 2, 3, 4, 5 or 6. The ADC, in some embodiments, is represented by the formula (Formula 2): L-{An-Ym-Zm-Xn-Wn-D}p wherein L is an antibody or antigen-binding fragment thereof that binds PSMA, D is MMAE or MMAF and p is an integer of 1, 2, 3, 4, 5, 6, 7 or 8. The other components are as described above. In one embodiment, the carboxylic unit "An" is linked to the antibody or antigen-binding fragment via a sulfur atom derived from the antibody or antigen-binding fragment:
- In one embodiment, A is


- In another embodiment, A is 4-(N-succinimidomethyl)cyclohexane-1-carbonyl, m-succinimidobenzoyl, 4-(p-succinimidophenyl)-butyryl, 4-(2-acetamido)benzoyl, 3-thiopropionyl, 4-(1-thioethyl)-benzoyl, 6-(3-thiopropionylamido)-hexanoyl or maleimide caproyl. In a further embodiment, A is maleimide caproyl. Representative examples of various carboxylic acyl units and methods for their synthesis and attachment are described in
US Pat. No. 6,214,345 . - In another embodiment, Y is alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan or proline. In yet another embodiment, Y is valine. In a further embodiment, Z is lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, or citrulline. In still a further embodiment, Z is citrulline. In one embodiment Ym-Zm is valine-citrulline. In another embodiment, Ym-Zm is a protein sequence which is selectively cleavable by a protease.
- In a further embodiment, X is a compound having the formula


- In some embodiments, W is

- In one embodiment, the compound of
Formula 1 is maleimidocaproyl. Maleimidocaproyl has been used for conjugation of two specific auristatins to an anti-CD30 mAb (AC10) (Doronina, Svetlana et al. "Novel Linkers for Monoclonal Antibody-Mediated Delivery of Anticancer Agents", AACR, Anaheim, CA, Abstract No. 1421, April 16-20, 2005). Maleimidocaproyl reacts with thiol groups to form a thioether. - MMAE or MMAF can be conjugated to an antibody or antigen-binding fragment thereof using methods known to those of ordinary skill in the art (e.g., See, Niemeyer, CM, Bioconjugation Protocols, Strategies and Methods, Humana Press, 2004) or as described herein. In some embodiments, more than one MMAE or MMAF molecule is conjugated to the antibody or antigen-binding fragment thereof. In other embodiments, 1, 2, 3, 4, 5, 6, 7 or 8 MMAE or MMAF molecules are conjugated to the antibody or antigen-binding fragment thereof. In still other embodiments, at least 2, 3, 4 or 5 MMAE or MMAF molecules are conjugated to the antibody or antigen-binding fragment thereof. In further embodiments, 2, 3, 4 or 5 MMAE or MMAF molecules are conjugated to the antibody or antigen-binding fragment thereof.
- The antibodies or antigen-binding fragments thereof of the ADCs are any antibody or antigen-binding fragment thereof that specifically binds PSMA. In one embodiment the antibody or an antigen-binding fragment thereof specifically binds PSMA (e.g., specifically binds an extracellular domain of PSMA, specifically binds a conformational epitope of PSMA, etc.) and can competitively inhibit the specific binding of a second antibody to its target epitope on PSMA, wherein the second antibody is selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, Abgenix 4.152.1 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13. The second antibody, therefore, include any of the antibodies produced by the hybridomas or encoded by the plasmids shown below in Table 1. These hybridomas and plasmids were deposited pursuant to, and in satisfaction of, the requirements of the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedure with the American Type Culture Collection ("ATCC") as an International Depository Authority and given the Patent Deposit Designations shown above and in Table 1.
Table 1 Antibody Hybridoma/Plasmid Patent Deposit Designation Date of Deposit PSMA 3.7 PSMA 3.7 PTA-3257 April 5, 2001 PSMA 3.9 PSMA 3.9 PTA-3258 April 5, 2001 PSMA 3.11 PSMA 3.11 PTA-3269 April 10, 2001 PSMA 5.4 PSMA 5.4 PTA-3268 April 10, 2001 PSMA 7.1 PSMA 7.1 PTA-3292 April 18, 2001 PSMA 7.3 PSMA 7.3 PTA-3293 April 18, 2001 PSMA 10.3 PSMA 10.3 PTA-3347 May 1, 2001 PSMA 10.3 HC in pcDNA (SEQ ID NO: 7) PTA-4413 May 29, 2002 PSMA 10.3 Kappa in pcDNA (SEQ ID NO: 13) PTA-4414 May 29, 2002 PSMA 1.8.3 PSMA 1.8.3 PTA-3906 Dec. 5, 2001 PSMA A3.1.3 PSMA A3.1.3 PTA-3904 Dec. 5, 2001 PSMA A3.3.1 PSMA A3.3.1 PTA-3905 Dec. 5, 2001 Abgenix 4.248.2 Abgenix 4.248.2 PTA-4427 June 4, 2002 Abgenix 4.360.3 Abgenix 4.360.3 PTA-4428 June 4, 2002 Abgenix 4.7.1 Abgenix 4.7.1 PTA-4429 June 4, 2002 Abgenix 4.4.1 Abgenix 4.4.1 PTA-4556 July 18, 2002 Abgenix 4.177.3 Abgenix 4.177.3 PTA-4557 July 18, 2002 Abgenix 4.16.1 Abgenix 4.16.1 PTA-4357 May 16, 2002 Abgenix 4.22.3 Abgenix 4.22.3 PTA-4358 May 16, 2002 Abgenix 4.28.3 Abgenix 4.28.3 PTA-4359 May 16, 2002 Abgenix 4.40.2 Abgenix 4.40.2 PTA-4360 May 16, 2002 Abgenix 4.48.3 Abgenix 4.48.3 PTA-4361 May 16, 2002 Abgenix 4.49.1 Abgenix 4.49.1 PTA-4362 May 16, 2002 Abgenix 4.209.3 Abgenix 4.209.3 PTA-4365 May 16, 2002 Abgenix 4.219.3 Abgenix 4.219.3 PTA-4366 May 16, 2002 Abgenix 4.288.1 Abgenix 4.288.1 PTA-4367 May 16, 2002 Abgenix 4.333.1 Abgenix 4.333.1 PTA-4368 May 16, 2002 Abgenix 4.54.1 Abgenix 4.54.1 PTA-4363 May 16, 2002 Abgenix 4.153.1 Abgenix 4.153.1 PTA-4388 May 23, 2002 Abgenix 4.232.3 Abgenix 4.232.3 PTA-4389 May 23, 2002 Abgenix 4.292.3 Abgenix 4.292.3 PTA-4390 May 23, 2002 Abgenix 4.304.1 Abgenix 4.304.1 PTA-4391 May 23, 2002 AB-PG1-XG1-006 AB-PG1-XG1-006 Heavy Chain (SEQ ID NO: 2) PTA-4403 May 29, 2002 AB-PG1-XG1-006 Light Chain (SEQ ID NO: 8) PTA-4404 AB-PG1-XG1-026 AB-PG1-XG1-026 Heavy Chain (SEQ ID NO: 3) PTA-4405 May 29, 2002 AB-PG1-XG1-026 Light Chain (SEQ ID NO: 9) PTA-4406 AB-PG1-XG1-051 AB-PG1-XG1-051 Heavy Chain (SEQ ID NO: 4) PTA-4407 May 29, 2002 AB-PG1-XG1-051 Light Chain (SEQ ID NO: 10) PTA-4408 AB-PG1-XG1-069 AB-PG1-XG1-069 Heavy Chain (SEQ ID NO: 5) PTA-4409 May 29, 2002 AB-PG1-XG1-069 Light Chain (SEQ ID NO: 11) PTA-4410 AB-PG1-XG1-077 AB-PG1-XG1-077 Heavy Chain (SEQ ID NO: 6) PTA-4411 May 29, 2002 AB-PG1-XG1-077 Light Chain (SEQ ID NO: 12) PTA-4412 - To determine competitive inhibition, a variety of assays known to one of ordinary skill in the art can be employed. For example, cross-competition assays can be used to determine if an antibody or antigen-binding fragment thereof competitively inhibits binding to PSMA by another antibody or antigen-binding fragment thereof. These include cell-based methods employing flow cytometry or solid phase binding analysis. Other assays that evaluate the ability of antibodies or antigen-binding fragments thereof to cross-compete for PSMA molecules that are not expressed on the surface of cells, in solid phase or in solution phase, also can be used.
- In some embodiments, the antibodies or antigen-binding fragments thereof competitively inhibit the specific binding of a second antibody to its target epitope on PSMA by at least about 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, or 99%. Inhibition can be assessed at various molar ratios or mass ratios; for example competitive binding experiments can be conducted with a 2-fold, 3-fold, 4-fold, 5-fold, 7-fold, 10-fold or more molar excess of a first antibody or antigen-binding fragment thereof over a second antibody or antigen-binding fragment thereof.
- In another embodiment the antibody or an antigen-binding fragment thereof specifically binds to an epitope on PSMA defined by an antibody selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, 4.248.2, 4.360.3, 4.7.1, 4.4.1, 4.177.3, 4.16.1, 4.22.3, 4.28.3, 4.40.2, 4.48.3, 4.49.1, 4.209.3, 4.219.3, 4.288.1, 4.333.1, 4.54.1, 4.153.1, 4.232.3, 4.292.3, 4.304.1, 4.78.1, and 4.152.1. PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, Abgenix 4.152.1 and antibodies comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13. The antibodies or antigen-binding fragments of the ADCs, therefore, include those that specifically bind to an epitope on PSMA defined by the antibodies produced by the hybridomas or encoded by the plasmids provided above in Table 1.
- To determine the epitope, one can use standard epitope mapping methods known in the art. For example, fragments (peptides) of PSMA antigen (e.g., synthetic peptides) that bind the antibody can be used to determine whether a candidate antibody or antigen-binding fragment thereof binds the same epitope. For linear epitopes, overlapping peptides of a defined length (e.g., 8 or more amino acids) are synthesized. The peptides can be offset by 1 amino acid, such that a series of peptides covering every 8 amino acid fragment of the PSMA protein sequence are prepared. Fewer peptides can be prepared by using larger offsets, e.g., 2 or 3 amino acids. In addition, longer peptides (e.g., 9-, 10- or 11-mers) can be synthesized. Binding of peptides to antibodies or antigen-binding fragments can be determined using standard methodologies including surface plasmon resonance (BIACORE) and ELISA assays. For examination of conformational epitopes, larger PSMA fragments can be used. Other methods that use mass spectrometry to define conformational epitopes have been described and can be used (see, e.g., Baerga-Ortiz et al., Protein Science 11:1300-1308, 2002 and references cited therein). Still other methods for epitope determination are provided in standard laboratory reference works, such as Unit 6.8 ("Phage Display Selection and Analysis of B-cell Epitopes") and Unit 9.8 ("Identification of Antigenic Determinants Using Synthetic Peptide Combinatorial Libraries") of Current Protocols in Immunology, Coligan et al., eds., John Wiley & Sons. Epitopes can be confirmed by introducing point mutations or deletions into a known epitope and then testing binding with one or more antibodies or antigen-binding fragments to determine which mutations reduce binding of the antibodies or antigen-binding fragments.
- In particular embodiments, the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, are those produced by hybridomas referred to herein as PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, and Abgenix 4.152.1, respectively. In other embodiments, the antibodies are those encoded by the plasmids listed in Table 1. In still other embodiments, the antibodies are those that comprise a heavy chain encoded by a nucleic acid molecule comprising the heavy chain coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and a light chain encoded by a nucleic acid molecule comprising the light chain coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13.
- As used herein, the names of the deposited hybridomas or plasmids may be used interchangeably with the names of the antibodies. It will be clear to one of ordinary skill in the art when the name is intended to refer to the antibody or when it refers to the plasmids or hybridomas that encode or produce the antibodies, respectively. Additionally, the antibody names may be an abbreviated form of the name shown in Table 1. For instance, antibody AB-PG1-XG1-006 may be referred to as AB-PG1-XG1-006, PG1-XG1-006, XG1-006, 006, etc. In another example, the antibody name PSMA 4.232.3 may be referred to as PSMA 4.232.3, 4.232.1, 4.232, etc. It is intended that all of the variations in the name of the antibody refer to the same antibody and not a different one.
- The antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, include those encoded by particular sets of heavy and light chain sequences. In one embodiment, the antibody (AB-PG1-XG1-006) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 2 and 8. In another embodiment, the antibody (AB-PG1-XG1-026) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 3 and 9. In still another embodiment, the antibody (AB-PG1-XG1-051) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 4 and 10. In yet another embodiment, the antibody (AB-PG1-XG1-069) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 5 and 11. In another embodiment, the antibody (AB-PG1-XG1-077) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 6 and 12. In yet another embodiment, the antibody (PSMA 10.3) is encoded by a nucleic acid molecule which comprises the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 7 and 13. In other embodiments, the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, include a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14, 18, 22, 26 and 30, and a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16, 20, 24, 28 and 32. In one embodiment, the antibody (AB-PG1-XG1-006) includes an immunoglobulin variable sequence encoded by nucleic acid molecules which comprise the coding region or regions of the nucleic acid sequences set forth as SEQ ID NOs: 14 and 16. Likewise, the antibody can be one that includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 15 and 17. In another embodiment, the antibody (AB-PG1-XG1-026) includes an immunoglobulin variable sequence encoded by nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 18 and 20 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs 19 and 21. In still another embodiment, the antibody (AB-PG1-XG1-051) includes an immunoglobulin variable sequence encoded by the nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 22 and 24 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 23 and 25. In yet another embodiment, the antibody (AB-PG1-XG1-069) includes an immunoglobulin variable sequence encoded by the nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 26 and 28 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 27 and 29. In another embodiment, the antibody (AB-PG1-XG1-077) includes an immunoglobulin variable sequence encoded by the nucleic acid molecules comprising the coding region or regions of nucleotide sequences set forth as SEQ ID NOs: 30 and 32 or includes an immunoglobulin variable sequence which comprises the amino acid sequences set forth as SEQ ID NOs: 31 and 33. In other embodiments, the antibody includes a heavy chain variable region comprising an amino acid sequence selected from the group consisting of amino acid sequences set forth as: SEQ ID NOs: 15, 19, 23, 27 and 31, and a light chain variable region comprising an amino acid sequence selected from the group consisting of amino acid sequences set forth as: SEQ ID NOs: 17, 21, 25, 29 and 33.
- As used herein, a "coding region" refers to a region of a nucleotide sequence that encodes a polypeptide sequence. Its use herein is consistent with the recognized meaning known in the art.
- In certain embodiments, the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, are those that are encoded by nucleic acid molecules that are highly homologous to the foregoing nucleic acids. The homologous nucleic acid molecule can, in some embodiments, comprise a nucleotide sequence that is at least about 90% identical to the nucleotide sequence provided herein. In other embodiments, the nucleotide sequence is at least about 95% identical, at least about 97% identical, at least about 98% identical, or at least about 99% identical to a nucleotide sequence provided herein. The homology can be calculated using various, publicly available software tools well known to one of ordinary skill in the art. Exemplary tools include the BLAST system available from the website of the National Center for Biotechnology Information (NCBI) at the National Institutes of Health.
- One method of identifying highly homologous nucleotide sequences is via nucleic acid hybridization. Thus, the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, include antibodies having a PSMA-binding property and/or other functional properties described herein, which are encoded by nucleic acid molecules that hybridize under high stringency conditions to the foregoing nucleic acid molecules. Identification of related sequences can also be achieved using polymerase chain reaction (PCR) and other amplification techniques suitable for cloning related nucleic acid sequences. PCR primers can be selected to amplify portions of a nucleic acid sequence of interest, such as a CDR.
- The term "high stringency conditions", as used herein, refers to parameters with which the art is familiar. Nucleic acid hybridization parameters may be found in references that compile such methods, e.g. Molecular Cloning: A Laboratory Manual, J. Sambrook, et al., eds., Second Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989, or Current Protocols in Molecular Biology, F.M. Ausubel, et al., eds., John Wiley & Sons, Inc., New York. One example of high-stringency conditions is hybridization at 65°C in hybridization buffer (3.5X SSC, 0.02% Ficoll, 0.02% polyvinyl pyrrolidone, 0.02% Bovine Serum Albumin, 2.5mM NaH2PO4(pH7), 0.5% SDS, 2mM EDTA). SSC is 0.15M sodium chloride/0.015M sodium citrate, pH7; SDS is sodium dodecyl sulphate; and EDTA is ethylenediaminetetracetic acid. After hybridization, a membrane upon which the nucleic acid is transferred is washed, for example, in 2X SSC at room temperature and then at 0.1 - 0.5X SSC/0.1X SDS at temperatures up to 68°C.
- As used herein, the term "antibody" refers to a glycoprotein comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as HCVR or VH) and a heavy chain constant region. The heavy chain constant region is comprised of three domains,
C H1,C H2 andC H3. Each light chain is comprised of a light chain variable region (abbreviated herein as LCVR or VL) and a light chain constant region. The light chain constant region is comprised of one domain, CL. The VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen. The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (C1q) of the classical complement system. - The term "antigen-binding fragment" of an antibody as used herein, refers to one or more portions of an antibody that retain the ability to specifically bind to an antigen (i.e., PSMA). It has been shown that the antigen-binding function of an antibody can be performed by fragments of a full-length antibody. Examples of binding fragments encompassed within the term "antigen-binding fragment" of an antibody include (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and
C H1 domains; (ii) a F(ab')2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the VH and CH1 domains; (iv) a Fv fragment consisting of the VL and VH domains of a single arm of an antibody, (v) a dAb fragment (Ward et al., (1989) Nature 341:544-546) which consists of a VH domain; and (vi) an isolated complementarity determining region (CDR). The CDRs, and in particular the CDR3 regions, and more particularly the heavy chain CDR3 contribute to antibody specificity. Because these CDR regions and in particular the CDR3 region confer antigen specificity on the antibody, these regions may be incorporated into other antibodies or antigen-binding fragments to confer the identical antigen specificity onto that antibody or peptide. Furthermore, although the two domains of the Fv fragment, V and VH, are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the VL and VH regions pair to form monovalent molecules (known as single chain Fv (scFv); see e.g., Bird et al. (1988) Science 242:423-426; and Huston et al. (1988) Proc. Natl. Acad Sci. USA 85:5879-5883). Such single chain antibodies are also intended to be encompassed within the term "antigen-binding fragment" of an antibody. These antibody fragments are obtained using conventional procedures, such as proteolytic fragmentation procedures, as described in J. Goding, Monoclonal Antibodies: Principles and Practice, pp 98-118 (N.Y. Academic Press 1983), as well as by other techniques known to those with skill in the art. The fragments are screened for utility in the same manner as are intact antibodies. - The antibodies, or antigen-binding fragments thereof, of the ADCs are, in some embodiments, isolated. "Isolated", as used herein, is intended to refer to an antibody (or antigen-binding fragment thereof), which is substantially free of other antibodies (or antigen-binding fragments) having different antigenic specificities (e.g., an isolated antibody that specifically binds to PSMA is substantially free of antibodies that specifically bind antigens unrelated to PSMA). An isolated antibody that specifically binds to an epitope, isoform or variant of PSMA may, however, have cross-reactivity to other related antigens, e.g., from other species (e.g., PSMA species homologs). For example, PSMA homologs have been found in other species, such as the pig (GenBank Accession Number 077564 (amino acid)) and rat (GenBank Accession Numbers U75973 (mRNA) and AAC53423 (amino acid)). In some embodiments, the antibodies or antigen-binding fragments provided herein specifically bind both human PSMA as well as a PSMA homolog from another species. In other embodiments, the antibodies or antigen-binding fragments thereof specifically bind human PSMA but do not cross-react with PSMA homologs from another species.
- Moreover, an isolated antibody (or antigen-binding fragment thereof) may be substantially free of other cellular material and/or chemicals. The term "substantially pure" means that an antibody (or antigen-binding fragment thereof) is essentially free of other substances with which they may be found in nature or in vivo systems to an extent practical and appropriate for their intended use. Substantially pure antibodies or antigen-binding fragments thereof may be produced by techniques well known in the art. Because an isolated antibody (or antigen-binding fragment thereof) may be admixed with a pharmaceutically acceptable carrier in a pharmaceutical preparation, the antibody (or antigen-binding fragment thereof) may comprise only a small percentage by weight of the preparation. The antibody (or antigen-binding fragment thereof) is nonetheless isolated in that it has been separated from the substances with which it may be associated in living systems, i.e. isolated from other proteins.
- As used herein, "specific binding" refers to antibody binding to a predetermined antigen, in this case PSMA (e.g., human PSMA). Typically, the antibody binds with an affinity that is at least two-fold greater than its affinity for binding to a non-specific antigen (e.g., BSA, casein), which is an antigen other than PSMA, an isoform or variant of PSMA, or a closely-related antigen.
- The antibodies encompass various antibody isotypes, such as IgG1, IgG2, IgG3, IgG4, IgM, IgA1, IgA2, IgAsec, IgD, IgE. As used herein, "isotype" refers to the antibody class (e.g., IgM or IgG1) that is encoded by heavy chain constant region genes. The antibodies can be full length or can include only an antigen-binding fragment such as the antibody constant and/or variable domain of IgG1, IgG2, IgG3, IgG4, IgM, IgA1, IgA2, IgAsec, IgD or IgE or could consist of a Fab fragment, a F(ab')2 fragment and a Fv fragment.
- The antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, are, in some embodiments monoclonal. The antibodies can be produced by a variety of techniques well known in the art. Monoclonal antibody production may be effected by techniques which are well known in the art. The term "monoclonal antibody", as used herein, refers to a preparation of antibody molecules of single molecular composition. A monoclonal antibody displays a single binding specificity and affinity for a particular epitope. The process of monoclonal antibody production involves obtaining immune somatic cells with the potential for producing antibody, in particular B lymphocytes, which have been previously immunized with the antigen of interest either in vivo or in vitro and that are suitable for fusion with a B-cell myeloma line.
- Mammalian lymphocytes typically are immunized by in vivo immunization of the animal (e.g., a mouse) with the desired protein or polypeptide. Such immunizations are repeated as necessary at intervals of up to several weeks to obtain a sufficient titer of antibodies. Once immunized, animals can be used as a source of antibody-producing lymphocytes. Following the last antigen boost, the animals are sacrificed and spleen cells removed. Mouse lymphocytes give a higher percentage of stable fusions with the mouse myeloma lines described herein. For example, of the BALB/c mouse. However, other mouse strains, rabbit, hamster, sheep and frog may also be used as hosts for preparing antibody-producing cells. See; Goding (in Monoclonal Antibodies: Principles and Practice, 2d ed., pp. 60-61, Orlando, Fla., Academic Press, 1986). In particular, mouse strains that have human immunoglobulin genes inserted in the genome (and which cannot produce mouse immunoglobulins) can be used. Examples include the HuMAb mouse strains produced by Medarex/GenPharm International, and the XenoMouse strains produced by Abgenix. Such mice produce fully human immunoglobulin molecules in response to immunization. In some embodiments, therefore, the ADCs comprise a fully human monoclonal antibody or an antigen-binding fragment thereof that binds PSMA.
- Those antibody-producing cells that are in the dividing plasmablast stage fuse preferentially. Somatic cells may be obtained from the lymph nodes, spleens and peripheral blood of antigen-primed animals, and the lymphatic cells of choice depend to a large extent on their empirical usefulness in the particular fusion system. The antibody-secreting lymphocytes are then fused with (mouse) B cell myeloma cells or transformed cells, which are capable of replicating indefinitely in cell culture, thereby producing an immortal, immunoglobulin-secreting cell line. The resulting fused cells, or hybridomas, are cultured, and the resulting colonies screened for the production of the desired monoclonal antibodies. Colonies producing such antibodies are cloned, and grown either in vivo or in vitro to produce large quantities of antibody. A description of the theoretical basis and practical methodology of fusing such cells is set forth in Kohler and Milstein, Nature 256:495 (1975).
- Alternatively, human somatic cells capable of producing antibody, specifically B lymphocytes, are suitable for fusion with myeloma cell lines. While B lymphocytes from biopsied spleens, tonsils or lymph nodes of an individual may be used, the more easily accessible peripheral blood B lymphocytes can also be used. The lymphocytes may be derived from patients with diagnosed prostate carcinomas or another PSMA-expressing cancer. In addition, human B cells may be directly immortalized by the Epstein-Barr virus (Cole et al., 1995, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., pp. 77-96). Although somatic cell hybridization procedures can be used, in principle, other techniques for producing monoclonal antibodies can be employed such as viral or oncogenic transformation of B lymphocytes.
- Myeloma cell lines suited for use in hybridoma-producing fusion procedures can be non-antibody-producing, have high fusion efficiency, and enzyme deficiencies that render them incapable of growing in certain selective media which support the growth of the desired hybridomas. Examples of such myeloma cell lines that may be used for the production of fused cell lines include P3-X63/Ag8, X63-Ag8.653, NS1/1.Ag 4.1, Sp2/0-Ag14, FO, NSO/U, MPC-11, MPC11-X45-GTG 1.7, S194/5XX0 Bul, all derived from mice; R210.RCY3, Y3-Ag 1.2.3, IR983F and 4B210 derived from rats and U-266, GM1500-GRG2, LICR-LON-HMy2, UC729-6, all derived from humans (Goding, in Monoclonal Antibodies: Principles and Practice, 2d ed., pp. 65-66, Orlando, Fla., Academic Press, 1986; Campbell, in Monoclonal Antibody Technology, Laboratory Techniques in Biochemistry and Molecular Biology Vol. 13, Burden and Von Knippenberg, eds. pp. 75-83, Amsterdam, Elseview, 1984).
- Fusion with mammalian myeloma cells or other fusion partners capable of replicating indefinitely in cell culture is effected by standard and well-known techniques, for example, by using polyethylene glycol ("PEG") or other fusing agents (See Milstein and Kohler, Eur. J. Immunol. 6:511 (1976).
- In other embodiments, the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, are recombinant antibodies. The term "recombinant antibody", as used herein, is intended to include antibodies that are prepared, expressed, created or isolated by recombinant means, such as antibodies isolated from an animal (e.g., a mouse) that is transgenic for another species' immunoglobulin genes, antibodies expressed using a recombinant expression vector transfected into a host cell, antibodies isolated from a recombinant, combinatorial antibody library, or antibodies prepared, expressed, created or isolated by any other means that involves splicing of immunoglobulin gene sequences to other DNA sequences.
- In yet other embodiments, the antibodies are chimeric or humanized antibodies. As used herein, the term "chimeric antibody" refers to an antibody, that combines the murine variable or hypervariable regions with the human constant region or constant and variable framework regions. As used herein, the term "humanized antibody" refers to an antibody that retains only the antigen-binding CDRs from the parent antibody in association with human framework regions (see, Waldmann, 1991, Science 252:1657). Such chimeric or humanized antibodies retaining binding specificity of the murine antibody are expected to have reduced immunogenicity when administered in vivo as provided herein.
- According to an alternative embodiment, the monoclonal antibodies of the ADCs can be in the form of a bispecific antibody or a multispecific antibody. The term "bispecific antibody" is intended to include any agent, e.g., a protein, peptide, or protein or peptide complex, which has two different binding specificities which bind to, or interact with (a) a cell surface antigen and (b) an Fc receptor on the surface of an effector cell. The term "multispecific antibody" is intended to include any agent, e.g., a protein, peptide, or protein or peptide complex, which has more than two different binding specificities which bind to, or interact with (a) a cell surface antigen, (b) an Fc receptor on the surface of an effector cell, and (c) at least one other component. Accordingly, the antibodies include, but are not limited to, bispecific, trispecific, tetraspecific, and other multispecific antibodies which are directed to PSMA and to Fc receptors on effector cells. The term "bispecific antibodies" further includes diabodies. Diabodies are bivalent, bispecific antibodies in which the VH and VL domains are expressed on a single polypeptide chain, but using a linker that is too short to allow for pairing between the two domains on the same chain, thereby forcing the domains to pair with complementary domains of another chain and creating two antigen-binding sites (see e.g., Holliger, P., et al. (1993) Proc. Natl. Acad. Sci. USA 90:6444-6448; Poijak, R.J., et al. (1994) Structure 2:1121-1123).
- A bispecific antibody can be formed of an antigen-binding region specific for PSMA and an antigen-binding region specific for an effector cell which has tumoricidal or tumor inhibitory activity. The two antigen-binding regions of the bispecific antibody are either chemically linked or can be expressed by a cell genetically engineered to produce the bispecific antibody. (See generally, Fanger et al., 1995 Drug News & Perspec. 8(3):133-137). Suitable effector cells having tumoricidal activity include but are not limited to cytotoxic T-cells (primarily CD8+ cells), natural killer cells, etc. An effective amount of a bispecific antibody can be administered to a subject with cancer, and the bispecific antibody kills and/or inhibits proliferation of the cancer cells after localization at sites of primary or metastatic tumors bearing PSMA.
- In certain embodiments, the antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, are human antibodies. The term "human antibody", as used herein, is intended to include antibodies having variable and constant regions derived from human germline immunoglobulin sequences. The human antibodies can include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo). However, the term "human antibody", as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences (referred to herein as "humanized antibodies"). Human antibodies directed against PSMA can be generated using transgenic mice carrying parts of the human immune system rather than the mouse system. Some examples of which are described elsewhere herein.
- Fully human monoclonal antibodies also can be prepared by immunizing mice transgenic for large portions of human immunoglobulin heavy and light chain loci. See, e.g.,
U.S. patents 5,591,669 ,5,598,369 ,5,545,806, 5,545,807 ,6,150,584 , and references cited therein. These animals have been genetically modified such that there is a functional deletion in the production of endogenous (e.g., murine) antibodies. The animals are further modified to contain all or a portion of the human germ-line immunoglobulin gene locus such that immunization of these animals results in the production of fully human antibodies to the antigen of interest. Following immunization of these mice (e.g., XenoMouse (Abgenix), HuMAb mice (Medarex/GenPharm)), monoclonal antibodies can be prepared according to standard hybridoma technology. These monoclonal antibodies have human immunoglobulin amino acid sequences and, therefore, will not provoke human anti-mouse antibody (HAMA) responses when administered to humans. In general, but not intended to be limiting, the mice are 6-16 weeks of age upon the first immunization. For example, a purified or enriched preparation of PSMA antigen (e.g., recombinant PSMA or PSMA-expressing cells) can be used to immunize the mice intraperitoneally (IP), although other routes of immunization known to one of ordinary skill in the art are also possible. PSMA antigen can be injected in combination with an adjuvant, such as complete Freund's adjuvant, and, in some embodiments, the initial injection is followed by booster immunizations with antigen in an adjuvant, such as incomplete Freund's adjuvant. The immune response can be monitored over the course of the immunization protocol with plasma samples obtained by, for example, retroorbital bleeds. The plasma can be screened by ELISA, and mice with sufficient titers of anti-PSMA human immunoglobulin can be used for fusions. Mice can be boosted intravenously withantigen 3 days before sacrifice and removal of the spleen. - The antibody or antigen-binding fragment thereof of the ADCs can, in some embodiments, be selected for the ability to bind live PSMA-expressing cells. In order to demonstrate binding to live PSMA-expressing cells, flow cytometry can be used. For example, PSMA-expressing cells lines (grown under standard growth conditions) or prostate cancer cells that express PSMA can be mixed with various concentrations of monoclonal antibodies in PBS containing 0.1
% Tween - The antibodies can, in some embodiments, promote cytolysis of PSMA-expressing cells. Cytolysis can be complement-mediated or can be mediated by effector cells. In one embodiment, the cytolysis is carried out in a living organism, such as a mammal, and the live cell is a tumor cell. Examples of tumors which can be targeted with the antibodies or antigen-binding fragments thereof include, any tumor that expresses PSMA (this includes tumors with neovasculature expressing PSMA), such as, prostate, bladder, breast, pancreas, liver, lung (e.g., non-small cell lung carcinoma), colon and kidney tumors as well as melanomas and sarcomas. In one embodiment, the tumor cell is a prostate tumor cell.
- The testing of cytolytic activity in vitro by chromium release assay can provide an initial screening prior to testing in vivo models. This testing can be carried out using standard chromium release assays. Briefly, polymorphonuclear cells (PMN), or other effector cells, from healthy donors can be purified by Ficoll Hypaque density centrifugation, followed by lysis of contaminating erythrocytes. Washed PMNs can be suspended in RPMI supplemented with 10% heat-inactivated fetal calf serum and mixed with 51Cr labeled cells expressing PSMA, at various ratios of effector cells to tumor cells (effector cells:tumor cells). Purified anti-PSMA IgGs can then be added at various concentrations. Irrelevant IgG can be used as a negative control. Assays can be carried out for 0-120 minutes at 37°C. Samples can be assayed for cytolysis by measuring 51Cr release into the culture supernatant. Anti-PSMA monoclonal antibodies and/or ADCs can also be tested in combinations with each other to determine whether cytolysis is enhanced with multiple monoclonal antibodies and/or ADCs. Antibodies that bind to PSMA and/or ADCs also can be tested in an in vivo model (e.g., in mice) to determine their efficacy in mediating cytolysis and killing of cancer cells expressing PSMA, e.g., prostate tumor cells.
- The antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, can be selected, for example, based on the following criteria, which are not intended to be exclusive:
- 1) binding to live cells expressing PSMA;
- 2) high affinity of binding to PSMA;
- 3) binding to a unique epitope on PSMA (i.e., an epitope not recognized by a previously produced antibody);
- 4) opsonization of cells expressing PSMA;
- 5) mediation of growth inhibition, phagocytosis and/or killing of cells expressing PSMA in the presence of effector cells;
- 6) modulation (inhibition or enhancement) of NAALADase, folate hydrolase, dipeptidyl peptidase IV and/or γ-glutamyl hydrolase activities;
- 7) growth inhibition, cell cycle arrest and/or cytotoxicity in the absence of effector cells;
- 8) internalization of PSMA;
- 9) binding to a conformational epitope on PSMA;
- 10) minimal cross-reactivity with cells or tissues that do not express PSMA; and
- 11) preferential binding to dimeric forms of PSMA rather than monomeric forms of PSMA.
- The antibodies of the ADCs, or from which the antigen-binding fragments of the ADCs are derived, can meet one or more, and possibly all, of these criteria.
- In one embodiment, the antibody or antigen-binding fragment thereof binds to a conformational epitope, such as a conformational epitope within the extracellular domain of PSMA. To determine if an anti-PSMA antibody or antigen-binding fragment thereof binds to conformational epitopes, each antibody can be tested in assays using native protein (e.g., non-denaturing immunoprecipitation, flow cytometric analysis of cell surface binding) and denatured protein (e.g., Western blot, immunoprecipitation of denatured proteins). A comparison of the results will indicate whether the antibody or antigen-binding fragment thereof binds a conformational epitope. Antibodies or antigen-binding fragments thereof that bind to native protein but not denatured protein are, in some embodiments, those that bind conformational epitopes. It follows, that the ADCs, in some embodiments, bind comformational epitopes of PSMA.
- In another embodiment, the antibody or antigen-binding fragment thereof binds to a dimer-specific epitope on PSMA (i.e., a conformational, dimer-specific epitope unique to the quaternary structure of dimeric PSMA). Generally, antibodies or antigen-binding fragments thereof which bind to a dimer-specific epitope preferentially bind the PSMA dimer rather than the PSMA monomer. To determine if an antibody or antigen-binding fragment thereof binds preferentially (i.e., selectively and/or specifically) to a PSMA dimer, the antibody or antigen-binding fragment thereof can be tested in assays (e.g., immunoprecipitation followed by Western blotting) using native dimeric PSMA protein and dissociated monomeric PSMA protein. A comparison of the results will indicate whether the antibody or antigen-binding fragment thereof binds preferentially to the dimer. In some embodiments, the antibodies or antigen-binding fragments thereof bind to the PSMA dimer but not to the monomeric PSMA protein. It follows, that the ADCs, in some embodiments, bind to a dimer-specific epitope on PSMA.
- The ADCs provided include those that selectively bind PSMA multimers. As used herein, particularly with respect to the binding of PSMA multimers by the ADCs, "selectively binds" means that an antibody or antigen-binding fragment thereof of the ADCs preferentially binds to a PSMA protein multimer (e.g., with greater avidity, greater binding affinity) rather than to a PSMA protein monomer. In some embodiments, the ADCs of the invention bind to a PSMA protein multimer with an avidity and/or binding affinity that is 1.1-fold, 1.2-fold, 1.3-fold, 1.4-fold, 1.5-fold, 1.6-fold, 1.7-fold, 1.8-fold, 1.9-fold, 2-fold, 3-fold, 4-fold, 5-fold, 7-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold, 70-fold, 100-fold, 200-fold, 300-fold, 500-fold, 1000-fold or more than that exhibited by the ADC for a PSMA protein monomer. The ADC can, in some embodiments, selectively bind a PSMA protein multimer, and not a PSMA protein monomer, i.e., exclusively binds to a PSMA protein multimer. In some embodiments, the ADC selectively binds a PSMA protein dimer.
- A PSMA protein multimer, as used herein, is a protein complex of at least two PSMA proteins or fragments thereof. The PSMA protein multimers can be composed of various combinations of full-length PSMA proteins (e.g., SEQ ID NO: 1), recombinant soluble PSMA (rsPSMA, e.g., amino acids 44-750 of SEQ ID NO: 1) and fragments of the foregoing that form multimers (i.e., that retain the protein domain required for forming dimers and/or higher order multimers of PSMA). In some embodiments, at least one of the PSMA proteins forming the multimer is a recombinant, soluble PSMA (rsPSMA) polypeptide. The PSMA protein multimers can be dimers, such as those formed from recombinant soluble PSMA protein. In one embodiment, the dimer is a rsPSMA homodimer. The PSMA protein multimers referred to herein are believed to assume a native conformation and can have such a conformation. The PSMA proteins in certain embodiments are noncovalently bound together to form the PSMA protein multimer. It has been discovered that PSMA protein noncovalently associates to form dimers under non-denaturing conditions. The PSMA protein multimers can retain the activities of PSMA. The PSMA activity may be an enzymatic activity, such as folate hydrolase activity, NAALADase activity, dipeptidyl peptidase IV activity or γ-glutamyl hydrolase activity. Methods for testing the PSMA activity of multimers are well known in the art (reviewed by O'Keefe et al. in: Prostate Cancer: Biology, Genetics, and the New Therapeutics, L.W.K. Chung, W.B. Isaacs and J.W. Simons (eds.) Humana Press, Totowa, NJ, 2000, pp. 307-326).
- The antibody or antigen-binding fragment thereof of the ADCs can bind to and be internalized with PSMA expressed on cells. The mechanism by which the antibody or antigen-binding fragment thereof is internalized with PSMA is not critical to the practice of the methods provided herein. For example, the antibody or antigen-binding fragment thereof can induce internalization of PSMA. Alternatively, internalization of the antibody or antigen-binding fragment thereof can be the result of routine internalization of PSMA. It follows that the ADC can be internalized with PSMA expressed on cells.
- The antibodies or antigen-binding fragments thereof, and, therefore, the ADCs can, in some embodiments, specifically bind cell-surface PSMA and/or rsPSMA with sub-nanomolar affinity. The binding affinities can be about 1 X 10-9M or less, about 1 X 10-10M or less, or about 1 X 10-11 M or less. In one embodiment, the binding affinity is less than about 5 X 10-10M.
- The antibodies or antigen-binding fragments thereof can, in some embodiments, modulate at least one enzymatic activity of PSMA. The activity can be selected from the group consisting of N-acetylated α-linked acidic dipeptidase (NAALADase), folate hydrolase, dipeptidyl dipeptidase IV, γ-glutamyl hydrolase activity and combinations thereof in vitro or in vivo. The modulation may be enhancement or inhibition of at least one enzymatic activity of PSMA.
- Tissue levels of NAALADase activity can be determined by detergent solubilizing homogenizing tissues, pelleting the insoluble material by centrifugation and measuring the NAALADase activity in the remaining supernatant. Likewise, the NAALADase activity in bodily fluids can also be measured by first pelleting the cellular material by centrifugation and performing a typical enzyme assay for NAALADase activity on the supernatant. NAALADase enzyme assays have been described by Frieden, 1959, J. Biol, Chem., 234:2891. In this assay, the reaction product of the NAALADase enzyme is glutamic acid. This is derived from the enzyme catalyzed cleavage of N-acetylaspartylglutamate to yield N-acetylaspartic acid and glutamic acid. Glutamic acid, in a NAD(P)+ requiring step, yields 2-oxoglutarate plus NAD(P)H in a reaction catalyzed by glutamate dehydrogenase. Progress of the reaction can easily and conveniently be measured by the change in absorbance at 340nm due to the conversion of NAD(P)+ to NAD(P)H.
- Folate hydrolase activity of PSMA can be measured by performing enzyme assays as described by Heston and others (e.g., Clin. Cancer Res. 2(9): 1445-51,1996; Urology 49(3A Suppl):104-12,1997). Folate hydrolases such as PSMA remove the gamma-linked glutamates from polyglutamated folates. Folate hydrolase activity can be measured using substrates such as methotrexate tri-gamma glutamate (MTXGlu3), methotrexate di-gamma glutamate (MTXGlu2) or pteroylpentaglutamate (PteGlu5), for example using capillary electrophoresis (see Clin. Cancer Res. 2(9):1445-51, 1996). Timed incubations of PSMA with polyglutamated substrates can be followed by separation and detection of hydrolysis products.
- As mentioned above, the ADCs provided have surprisingly been found to kill PSMA-expressing, taxane-resistant cancer cells and can be used to treat PSMA-expressing, taxane-resistant cancer. Methods are, therefore, provided wherein an ADC is administered to a subject with a PSMA-expressing, taxane-resistant cancer. The cancer cells of the PSMA-expressing, taxane-resistant cancer, in some embodiments, can be cells of a tumor (e.g., primary malignant tumor) or cells of one or more metastases.
- In some embodiments, the methods provided result in the delay or inhibition of progression of the cancer in the subject. As used herein, "delay or inhibition of progression of the cancer" is intended to refer to any slowing or halting of the progression of the cancer in the subject. A slowing or halting of the progression of the cancer includes a reduction or stabilization in the number of cancer cells, the number of tumors, and/or the number of metastases in a subject. A slowing or halting is also intended to include a reduction or stabilization in the size (e.g., length or volume) of tumors and/or size of metastases in a subject. Methods for assessing the progression of cancer in a subject will be apparent to one of ordinary skill in the art. In addition, in some embodiments, the delay or inhibition of progression of the cancer can be demonstrated by a change in at least one biomarker for bone metastasis or bone metabolism compared to a baseline value prior to treatment with the ADC in the subject. In some embodiments the biomarker is N-telopeptide, bone alkaline phosphatase, osteocalcin, calcitonin, calcium, pyridinoline or deoxypyridinoline. In further embodiments, the delay or inhibition of progression of the cancer is demonstrated by radiographic image changes in tumor burden compared to a baseline radiographic image in the subject prior to ADC treatment. Methods for assessing radiographic changes, include, for example, bone scan, computerized axial tomography (CT) scan and magnetic resonance imaging (MRI). Other methods will be well known to those of ordinary skill in the art. In some embodiments, the delay or inhibition of progression is evidenced by a radiographic image change of at least 10%, 20%, 30%, 40%, 50% or 60% or more.
- Treatment with the ADCs provided can also result in the increase in survival of subjects with a PSMA-expressing, taxane-resistant cancer. In some embodiments, the survival of such a subject is increased by 4, 6, 8, 10, 12, 14, 16, 18, 20, 24, 28 or 32 weeks or more as compared to the median survival of subjects with the PSMA-expressing, taxane-resistant cancer not administered the ADC. In other embodiments, the survival is increased 4, 6, 8, 10, 12, 14, 16, 18, 20, 24, 28 or 32 weeks or more as compared to the expected survival time for the subject prior to treatment with the ADC. In other embodiments of either of the foregoing, the survival of the subject is increased by 10, 12, 14, 16, 18, 20, 22 or 24 months or more according to either comparison. In one embodiment, the subject treated with an ADC is one that has progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, and the increase in survival of the subject is as compared to the median survival of subjects having progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy and not treated with the ADC. In another embodiment, the subject treated with an ADC is one that has progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, and the increase in survival of the subject is as compared to the expected survival time for the subject prior to treatment with the ADC.
- In other embodiments, treatment with an ADC as provided results in an increase in the quality of life for the subject as compared to the quality of life experienced by the subject prior to treatment with the ADC. As used herein "an increase in the quality of life" refers to any improvement in the subject's comfort, level of energy and/or ability to function in an activity as a result of the administration of an ADC as provided herein.
- In still other embodiments, treatment with an ADC can result in a decrease in the circulating level of circulating tumor cells (CTCs) compared to a baseline level. In further embodiments, treatment with an ADC can result in a decrease or stabilization (no significant increase or decrease) in a serum level of prostate-specific antigen (PSA) compared to a baseline level of PSA. Methods for assessing the circulating level of CTCs or the serum level of PSA are well known to those of ordinary skill in the art.
- It was also surprising to discover that in subjects with a PSMA-expressing, taxane-resistant cancer, a reduction in tumor volume for even very large tumors can be achieved with the administration of an ADC. Therefore, in some embodiments, the subject has a tumor with a tumor volume that is at least 100mm3, 200mm3, 300mm3, 400mm3, 500mm3, 600mm3, 700mm3, 800mm3, 900mm3, 1000mm3, 1100mm3, 1200mm3, 1300mm3, 1400mm3, 1500mm3, 1600mm3, 1700mm3, 1800mm3, 1900mm3 or 2000mm3. In some embodiments, the subject has a tumor volume that is greater than 700mm3. In other embodiments, the subject has a tumor with a length of at least 5mm, 6mm, 7mm, 8mm, 9mm, 10mm, 11 mm, 12mm, 13mm, 14mm, 15mm, 16mm, 17mm, 18mm, 19mm, 20mm, 21mm, 22mm, 23mm, 24mm, 25mm or 30mm or more. In other embodiments, the tumor volume or length is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97% or 99% as a result of treatment with an ADC as provided herein. In a further embodiment, the tumor is eradicated. Techniques for determining the presence of a tumor and for measuring its size are well known to those of ordinary skill in the art.
- The ADCs can be used in various in vitro and in vivo methods for effecting the aforementioned therapeutic endpoints. The methods provided can be used to kill PSMA-expressing, taxane-resistant cancer cells in vitro or in vivo. Methods are also provided for treating any PSMA-expressing, taxane-resistant cancer. Such a cancer is, for example, prostate cancer. Such a cancer can also be, for example, a cancer in which PSMA is expressed on the cells of the tumor neovasculature. An exemplary list of such cancers is provided elsewhere herein.
- In some embodiments, two or more different ADCs are used in combination. In another embodiment, one or more unconjugated anti-PSMA antibodies or antigen-binding fragments thereof can be combined with one or more ADCs in a single therapy to achieve a desired therapeutic effect. As an illustration, an unconjugated anti-PSMA antibody that mediates highly effective killing of target cells in the presence of effector cells and/or that inhibits the growth of cells expressing PSMA can be used with one or more ADCs. In yet another embodiment, the ADCs can be combined with one or more additional antitumor agents, such as corticosteroids, such as prednisone or hydrocortisone; immunostimulatory agents; immunomodulators; or some combination thereof.
- Antitumor agents include cytotoxic agents, chemotherapeutic agents and agents that act on tumor neovasculature. Cytotoxic agents include cytotoxic radionuclides, chemical toxins and protein toxins. The cytotoxic radionuclide or radiotherapeutic isotope can be an alpha-emitting isotope such as 225Ac, 211At, 212Bi, 213Bi, 212Pb, 224Ra or 223Ra. Alternatively, the cytotoxic radionuclide can be a beta-emitting isotope such as 186Rh, 188Rh, 177Lu, 90Y, 131I, 67Cu, 64Cu, 153Sm or 166Ho. Further, the cytotoxic radionuclide can emit Auger and low energy electrons and include the isotopes 125I, 123I or 77Br.
- Suitable chemical toxins or chemotherapeutic agents include members of the enediyne family of molecules, such as calicheamicin and esperamicin. Chemical toxins can also be taken from the group consisting of methotrexate, doxorubicin, melphalan, chlorambucil, ARA-C, vindesine, mitomycin C, cis-platinum, etoposide, bleomycin and 5-fluorouracil. Other antineoplastic agents include dolastatins (
U.S. Patent Nos. 6,034,065 and6,239,104 ) and derivatives thereof. Dolastatins and derivatives thereof include dolastatin 10 (dolavaline-valine-dolaisoleuine-dolaproine-dolaphenine) and the derivatives auristatin PHE (dolavaline-valine-dolaisoleuine-dolaproine-phenylalanine-methyl ester) (Pettit, G.R. et al., Anticancer Drug Des. 13(4):243-277, 1998; Woyke, T. et al., Antimicrob. Agents Chemother. 45(12):3580-3584, 2001), and aurastatin E and the like. Toxins also include poisonous lectins, plant toxins such as ricin, abrin, modeccin, botulina and diphtheria toxins. Other chemotherapeutic agents are known to those skilled in the art. - Agents that act on the tumor vasculature include tubulin-binding agents such as combrestatin A4 (Griggs et al., Lancet Oncol. 2:82, 2001), angiostatin and endostatin (reviewed in Rosen, Oncologist 5:20, 2000) and interferon inducible protein 10 (
U.S. Patent No. 5,994,292 ). A number of other antiangiogenic agents are also contemplated and include: 2ME2, Angiostatin, Angiozyme, Anti-VEGF RhuMAb, Apra (CT-2584), Avicine, Benefin, BMS275291, Carboxyamidotriazole, CC4047, CC5013, CC7085, CDC801, CGP-41251 (PKC 412), CM101, Combretastatin A-4 Prodrug, EMD 121974, Endostatin, Flavopiridol, Genistein (GCP), Green Tea Extract, IM-862, ImmTher, Interferon alpha, Interleukin-12, Iressa (ZD1839), Marimastat, Metastat (Col-3), Neovastat , Octreotide, Paclitaxel, Penicillamine, Photofrin, Photopoint, PI-88, Prinomastat (AG-3340), PTK787 (ZK22584), RO317453, Solimastat, Squalamine, SU 101, SU 5416, SU-6668, Suradista (FCE 26644), Suramin (Metaret), Tetrathiomolybdate, Thalidomide, TNP-470 and Vitaxin. Additional antiangiogenic agents are described by Kerbel, J. Clin. Oncol. 19(18s):45s-51s, 2001. - The ADCs can be administered with one or more immunostimulatory agents to induce or enhance an immune response, such as IL-2 and immunostimulatory oligonucleotides (e.g., those containing CpG motifs). Immunostimulatory agents can, in some embodiments, stimulate specific arms of the immune system, such as natural killer (NK) cells that mediate antibody-dependent cell cytotoxicity (ADCC). Immunostimulatory agents include interleukin-2, α-interferon, γ-interferon, tumor necrosis factor alpha (TNFα), immunostimulatory oligonucleotides or a combination thereof. Immunomodulators include cytokines, chemokines, adjuvants or a combination thereof. Chemokines useful in increasing immune responses include but are not limited to SLC, ELC, MIP3α, MIP3β, IP-10, MIG, and combinations thereof.
- The other therapeutic agent can also be a vaccine. In some embodiments, the vaccine immunizes a subject against PSMA. Such vaccines, in some embodiments, include antigens, such as PSMA dimers, with, optionally, one or more adjuvants to induce or enhance an immune response. An adjuvant is a substance which potentiates the immune response. Adjuvants of many kinds are well known in the art. Specific examples of adjuvants include monophosphoryl lipid A (MPL, SmithKline Beecham); saponins including QS21 (SmithKline Beecham); immunostimulatory oligonucleotides (e.g., CpG oligonucleotides described by Kreig et al., Nature 374:546-9, 1995);incomplete Freund's adjuvant; complete Freund's adjuvant; montanide; vitamin E and various water-in-oil emulsions prepared from biodegradable oils such as squalene and/or tocopherol, Quil A, Ribi Detox, CRL-1005, L-121, and combinations thereof. Formulations, such as those described in
U.S. Application Serial No. 10/976352 , are also contemplated for use as vaccines in the methods provided herein. - The vaccines can, in some embodiments, include one or more of the isolated PSMA protein multimers described herein, such as the PSMA protein dimer. In some embodiments, a PSMA protein multimer composition contains at least about 10% PSMA protein multimer (of the total amount of PSMA protein in the composition). In other embodiments, the PSMA protein multimer composition contains at least about 20%, 30%, 40%, 50%, 60%, 70%, 75%, 80%, 85%, 90%, 95%, 99% or 99.5% PSMA protein multimer. In one embodiment, the PSMA protein multimer composition contains substantially pure PSMA protein multimer, with substantially no PSMA protein monomer. It is understood that the list of specific percentages includes by inference all of the unnamed percentages between the recited percentages.
- Cytokines can also be used in vaccination protocols as a result of their lymphocyte regulatory properties. Many cytokines useful for such purposes will be known to one of ordinary skill in the art, including interleukin-2 (IL-2); IL-4; IL-5; IL-12, which has been shown to enhance the protective effects of vaccines (see, e.g., Science 268: 1432-1434, 1995); GM-CSF; IL-15; IL-18; combinations thereof, and the like. Thus cytokines can be administered in conjunction with antigen, chemokines and/or adjuvants to increase an immune response.
- The other therapeutic agents can be used in the methods provided in unconjugated form or in conjugated form, such as conjugated to an anti-PSMA antibody or antigen-binding fragment thereof. Coupling of one or more toxin molecules to the anti-PSMA antibody or antigen-binding fragment thereof can include many chemical mechanisms, for instance covalent binding, affinity binding, intercalation, coordinate binding and complexation.
- The covalent binding can be achieved either by direct condensation of existing side chains or by the incorporation of external bridging molecules. Many bivalent or polyvalent agents are useful in coupling protein molecules to other proteins, peptides or amine functions, etc. For example, the literature is replete with coupling agents such as carbodiimides, diisocyanates, glutaraldehyde, diazobenzenes, and hexamethylene diamines. This list is not intended to be exhaustive of the various coupling agents known in the art but, rather, is exemplary of the more common coupling agents.
- In some embodiments, it is contemplated that one may wish to first derivatize the antibody, and then attach the therapeutic agent to the derivatized product. Suitable crosslinking agents for use in this manner include, for example, SPDP (N-succinimidyl-3-(2-pyridyldithio)propionate), and SMPT, 4-succinimidyl-oxycarbonyl-methyl-(2-pyridyldithio)toluene.
- In addition, protein toxins can be fused to the anti-PSMA antibody or antigen-binding fragment thereof by genetic methods to form a hybrid immunotoxin fusion protein. The fusion proteins can include additional peptide sequences, such as peptide spacers which operatively attach, for example, the anti-PSMA antibody and toxin, as long as such additional sequences do not appreciably affect the targeting or toxin activities of the fusion protein. The proteins can be attached by a peptide linker or spacer, such as a glycine-serine spacer peptide, or a peptide hinge, as is well known in the art. Thus, for example, the C-terminus of an anti-PSMA antibody or antigen-binding fragment thereof can be fused to the N-terminus of the protein toxin molecule to form an immunotoxin that retains the binding properties of the anti-PSMA antibody. Other fusion arrangements will be known to one of ordinary skill in the art. To express the fusion immunotoxin, the nucleic acid encoding the fusion protein is inserted into an expression vector in accordance with standard methods, for stable expression of the fusion protein, such as in mammalian cells, such as CHO cells. The fusion protein can be isolated and purified from the cells or culture supernatant using standard methodology, such as a PSMA affinity column.
- Radionuclides typically are coupled to an antibody or antigen-binding fragment thereof by chelation. For example, in the case of metallic radionuclides, a bifunctional chelator is commonly used to link the isotope to the antibody or other protein of interest. Typically, the chelator is first attached to the antibody, and the chelator-antibody conjugate is contacted with the metallic radioisotope. A number of bifunctional chelators have been developed for this purpose, including the diethylenetriamine pentaacetic acid (DTPA) series of amino acids described in
U.S. patents 5,124,471 ,5,286,850 and5,434,287 . As another example, hydroxamic acid-based bifunctional chelating agents are described inU.S. patent 5,756,82 . Another example is the chelating agent termed p-SCN-Bz-HEHA (1,4,7,10,13,16-hexaazacyclo-octadecane- N,N',N",N"',N"",N""'-hexaacetic acid) (Deal et al., J. Med. Chem. 42:2988, 1999), which is an effective chelator of radiometals such as 225Ac. Yet another example is DOTA (1,4,7,10-tetraazacyclododecane N,N',N",N"'-tetraacetic acid), which is a bifunctional chelating agent (see McDevitt et al., Science 294:1537-1540, 2001) that can be used in a two-step method for labeling followed by conjugation. - Other therapeutic agents also include replication-selective viruses. Replication-competent virus such as the p53 pathway targeting adenovirus mutant dl1520, ONYX-015, kills tumor cells selectively (Biederer, C. et al., J. Mol. Med. 80(3):163-175, 2002). The virus can, in some embodiments, be conjugated to PSMA antibodies or antigen-binding fragments thereof.
- The methods provided can further comprise the use of other therapeutic treatment modalities. Such other treatments include surgery, radiation, cryosurgery, thermotherapy, hormone treatment, chemotherapy, vaccines and other immunotherapies.
- The ADCs of the invention, such as through their antibody or antigen-binding fragment thereof, can be linked to a label. Labels include, for example, fluorescent labels, enzyme labels, radioactive labels, nuclear magnetic resonance active labels, luminescent labels or chromophore labels.
- The compositions provided can include a physiologically or pharmaceutically acceptable carrier, excipient or stabilizer mixed with the ADC. In some embodiments, when a composition comprises two or more different ADCs, each of the antibodies or antigen-binding fragments thereof of the ADCs binds to a distinct conformational epitope of PSMA.
- Pharmaceutical compositions can be administered in combination therapy, i.e., combined with other agents. For example, the combination therapy can include an ADC with at least one anti-tumor agent, immunomodulator, immunostimulatory agent or other conventional therapy. The other agent can be conjugated to or formed as a recombinant fusion molecule with a PSMA antibody or antigen-binding fragment thereof for directed targeting of the agent to PSMA-expressing cells. In another embodiment the other therapeutic agent can be unconjugated. Additional therapeutic agents can be administered or contacted with the PSMA-expressing cells through co-administration. "Co-administering," as used herein, refers to administering two or more therapeutic agents simultaneously as an admixture in a single composition, or sequentially, and close enough in time so that the compounds may exert an additive or even synergistic effect. In still other embodiments, an additional therapeutic agent can be administered before, during or after the administration of one or more ADCs or compositions thereof.
- As used herein, "pharmaceutically acceptable carrier" or "physiologically acceptable carrier" includes any and all salts, solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible. In some embodiments, the carrier is suitable for intravenous, intramuscular, subcutaneous, parenteral, spinal or epidermal administration (e.g., by injection or infusion). Depending on the route of administration, the active compound, can be coated in a material to protect the compound from the action of acids and other natural conditions that may inactivate the compound.
- When administered, the pharmaceutical compositions are applied in pharmaceutically-acceptable amounts and in pharmaceutically-acceptable compositions. The term "pharmaceutically acceptable" means a non-toxic material that does not interfere with the effectiveness of the biological activity of the active ingredients. Such preparations may routinely contain salts, buffering agents, preservatives, compatible carriers, and optionally other therapeutic agents, such as supplementary immune potentiating agents including adjuvants, chemokines and cytokines. When used in medicine, the salts should be pharmaceutically acceptable, but non-pharmaceutically acceptable salts may conveniently be used to prepare pharmaceutically-acceptable salts thereof and are not excluded from the scope of the invention.
- A salt retains the desired biological activity of the parent compound and does not impart any undesired toxicological effects (see e.g., Berge, S.M., et al. (1977) J. Pharm. Sci. 66: 1-19). Examples of such salts include acid addition salts and base addition salts. Acid addition salts include those derived from nontoxic inorganic acids, such as hydrochloric, nitric, phosphoric, sulfuric, hydrobromic, hydroiodic, phosphorous and the like, as well as from nontoxic organic acids such as aliphatic mono- and dicarboxylic acids, phenyl substituted alkanoic acids, hydroxy alkanoic acids, aromatic acids, aliphatic and aromatic sulfonic acids and the like. Base addition salts include those derived from alkaline earth metals, such as sodium, potassium, magnesium, calcium and the like, as well as from nontoxic organic amines, such as N,N'-dibenzylethylenediamine, N-methylglucamine, chioroprocaine, choline, diethanolamine, ethylenediamine, procaine and the like.
- An ADC can be combined, if desired, with a pharmaceutically-acceptable carrier. The term "pharmaceutically-acceptable carrier" as used herein means one or more compatible solid or liquid fillers, diluents or encapsulating substances which are suitable for administration into a human. The term "carrier" denotes an organic or inorganic ingredient, natural or synthetic, with which the active ingredient is combined to facilitate the application. The components of the pharmaceutical compositions also are capable of being co-mingled in a manner such that there is no interaction which would substantially impair the desired pharmaceutical efficacy.
- The pharmaceutical compositions may contain suitable buffering agents, including: acetic acid in a salt; citric acid in a salt; boric acid in a salt; and phosphoric acid in a salt.
- The pharmaceutical compositions also may contain, optionally, suitable preservatives, such as: benzalkonium chloride; chlorobutanol; and parabens.
- The pharmaceutical compositions may conveniently be presented in unit dosage form and may be prepared by any of the methods well-known in the art of pharmacy. All methods include the step of bringing the active agent into association with a carrier which constitutes one or more accessory ingredients. In general, the compositions are prepared by uniformly and intimately bringing the active compound into association with a liquid carrier, a finely divided solid carrier, or both, and then, if necessary, shaping the product.
- Compositions suitable for parenteral administration conveniently comprise a sterile aqueous or non-aqueous preparation of the compounds, which is, in some embodiments, isotonic with the blood of the recipient. This preparation may be formulated according to known methods using suitable dispersing or wetting agents and suspending agents. The sterile injectable preparation also may be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example, as a solution in 1,3-butane diol. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono-or di-glycerides. In addition, fatty acids such as oleic acid may be used in the preparation of injectables. Carrier formulations suitable for oral, subcutaneous, intravenous, intramuscular, etc. administration can be found in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA.
- The active compounds can be prepared with carriers that will protect the compound against rapid release, such as a controlled release formulation, including implants, transdermal patches, and microencapsulated delivery systems. Biodegradable, biocompatible polymers can be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Many methods for the preparation of such formulations are patented or generally known to those skilled in the art. See, e.g., Sustained and Controlled Release Drug Delivery Systems, J.R. Robinson, ed., Marcel Dekker, Inc., New York, 1978.
- The therapeutics of the invention can be administered by any conventional route, including injection or by gradual infusion over time (e.g., the ADC in saline infused over 90 minutes). The administration may, for example, be oral, intravenous, intraperitoneal, intramuscular, intracavity, intratumor, or transdermal. When compounds containing antibodies are used therapeutically, routes of administration include intravenous and by pulmonary aerosol. Techniques for preparing aerosol delivery systems containing antibodies are well known to those of skill in the art. Generally, such systems should utilize components which will not significantly impair the biological properties of the antibodies, such as the paratope binding capacity (see, for example, Sciarra and Cutie, "Aerosols," in Remington's Pharmaceutical Sciences, 18th edition, 1990, pp. 1694-1712). Those of skill in the art can readily determine the various parameters and conditions for producing antibody aerosols without resorting to undue experimentation.
- The compositions of the invention are administered in effective amounts. An "effective amount" is that amount of any of the ADCs provided herein that alone, or together with further doses and/or other therapeutic agents, produces the desired response. This can involve any of the therapeutic endpoints mentioned herein. In one embodiment, this involves only slowing the progression of the disease temporarily, although in some embodiments, it involves halting the progression of the disease permanently. In other embodiments, it involves eradicating the disease altogether. The desired therapeutic endpoint can be monitored by routine methods known to those of ordinary skill in the art. An amount that is effective can be the amount of an ADC alone which produces the desired therapeutic endpoint. An amount that is effective is also the amount of an ADC in combination with another agent that produces the desired result.
- Such amounts will depend, of course, on the particular cancer being treated, the severity of the cancer, the individual patient parameters including age, physical condition, size and weight, the duration of the treatment, the nature of concurrent therapy (if any), the specific route of administration and like factors within the knowledge and expertise of the health practitioner. These factors are well known to those of ordinary skill in the art and can be addressed with no more than routine experimentation. It is generally preferred that a maximum dose of the individual components or combinations thereof be used, that is, the highest safe dose according to sound medical judgment. It will be understood by those of ordinary skill in the art, however, that a patient may insist upon a lower dose or tolerable dose for medical reasons, psychological reasons or for virtually any other reasons.
- The pharmaceutical compositions used in the methods can be sterile and contain an effective amount of an ADC, alone or in combination with another agent, for producing the desired response in a unit of weight or volume suitable for administration to a patient. The response can, for example, be measured by determining the physiological effects of the ADC composition, such as a reduction in tumor volume, reduction in the size or number of metastases, an increase in survival, an improvement in quality of life and/or reduction of cancer symptoms, etc. Other assays will be known to one of ordinary skill in the art and can be employed for measuring the level of the response.
- The doses of ADCs administered to a subject can be chosen in accordance with different parameters, in particular in accordance with the mode of administration used and the state of the subject. Other factors include the desired period of treatment. In the event that a response in a subject is insufficient at the initial doses applied, higher doses (or effectively higher doses by a different, more localized delivery route) may be employed to the extent that patient tolerance permits.
- In general, doses can range from about 10 µg/kg to about 100,000 µg/kg. In some embodiments, the doses can range from about 0.1 mg/kg to about 20 mg/kg. In still other embodiments, the doses range from about 0.1 mg/kg to 5 mg/kg, 0.1 mg/kg to 10 mg/kg or 0.1 mg/kg to 15 mg/kg. In yet other embodiments, the doses range from about 1 mg/kg to 5 mg/kg, 5 mg/kg to 10 mg/kg, 10 mg/kg to 15 mg/kg or 15 mg/kg to 20 mg/kg. In further embodiments, the dose is about 0.1 mg/kg, 0.5 mg/kg, 1 mg/kg, 2 mg/kg, 3 mg/kg, 5 mg/kg, 7 mg/kg, 10 mg/kg, 12 mg/kg, 15 mg/kg, 17 mg/kg, 20 mg/kg, 25 mg/kg or 30 mg/kg. In a further embodiment, the dose is about 0.4 mg/kg, 0.7 mg/kg. 1.1 mg/kg, 1.8 mg/kg, 2.4 mg/kg, 2.9 mg/kg, 3.0 mg/kg, 3.5 mg/kg or 4.0 mg/kg. In a further embodiment, the dose is about 0.6 mg/kg, 0.9 mg/kg, 1.2 mg/kg or 1.5 mg/kg. In another embodiment, the dose is about 1 mg/kg, 3 mg/kg, 5 mg/kg or 6 mg/kg. Based upon the composition, the dose can be delivered continuously, such as by continuous pump, or at periodic intervals. In some embodiments, when the ADC is administered intravenously, the dose is between 0.1 and 20 mg/kg or any value in between. Desired time intervals of multiple doses of a particular composition can be determined without undue experimentation by one skilled in the art. Other protocols for the administration of the compositions provided will be known to one of ordinary skill in the art, in which the dose amount, schedule of administration, site(s) of administration, mode of administration and the like vary from the foregoing. In some embodiments, subjects are administered the ADC with a dose regimen of q4d x 3 or
q4d x 6. In one embodiment, the dose is administered intravenously. In another embodiment, the dose regimen is a single intravenous dose. - Administration of an ADC or a composition comprising an ADC to mammals other than humans, e.g., for testing purposes or veterinary therapeutic purposes, etc. is carried out under substantially the same conditions as described above.
- The compositions of the present invention have in vitro and in vivo therapeutic utilities. For example, these molecules can be administered to cells in culture, e.g. in vitro or ex vivo, or in a subject, e.g., in vivo, to treat a variety of cancers as provided herein. As used herein, the term "subject" is intended to include humans and non-human animals. Non-human animals include, e.g., dogs, cats, horses, cows, pigs, mice and rats. Subjects include a human patient having a PSMA-expressing, taxane-resistant cancer. The subject, in one embodiment, has progressive, castration-resistant, metastatic prostate cancer that has been treated with taxane but has progressed. In another embodiment, the subject is one that meets the entry criteria as defined in the Examples below (e.g., Examples 5, 7 and 9).
- Use of the methods provided has a number of benefits. Since the ADCs preferentially target PSMA e.g., on prostate cancer cells, other tissue can be spared. As a result, treatment with such biological agents is safer, particularly for elderly patients. Treatment according to the present invention is expected to be particularly effective, in some embodiments, because it can direct high levels of ADCs to the bone marrow and lymph nodes where cancer metastases, such as prostate cancer metastases, can predominate. Treatment in accordance with the present invention can be effectively monitored with clinical parameters such as serum prostate specific antigen and/or pathological features of a patient's cancer, including stage, Gleason score, extracapsular, seminal, vesicle or perineural invasion, positive margins, involved lymph nodes, etc. Alternatively, these parameters can be used to indicate when such treatment should be employed.
- The compositions for use in the methods provided herein can be in lyophilized form or provided in an aqueous medium.
- The present invention is further illustrated by the following Examples.
- PSMA-positive and PSMA-negative cells were exposed to PSMA ADC in 96-well microplates at various concentrations. After 96-hours, percent cell survival (compared to cells in medium) was assayed using fluorescent Alamar Blue (
Fig. 5 ). -
Fig. 6 presents a table showing cytotoxicity to PSMA-expressing prostate cancer cell lines following exposure to PSMA ADC. - The in vivo therapeutic efficacy of PSMA ADC in a subcutaneous xenograft model of human prostate cancer was assessed. Male athymic nude mice, 6-8 weeks old, obtained from Charles River Laboratories, Inc. (Wilmington, MA) were implanted subcutaneously with 5 million C4-2 cells (provided by Warren D.W. Heston of The Cleveland Clinic, Cleveland, OH). C4-2 is an androgen-independent human prostate cancer cell line. At
day 11, the tumor size of each mouse was measured, by length and width in millimeters, using a caliper (Mitutoyo, Aurora, IL). The tumor volume was calculated using the following formula: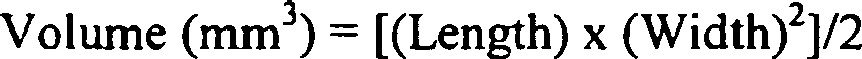
- Animals with a tumor volume between 80-140mm3 were randomized according to tumor volume into two groups for treatment with either PBS buffer (vehicle control, n = 15) or PSMA ADC (n = 15). The mean tumor size was 108.9 and 108.7mm3 with a p-value of 0.452 (student t-test) for the PBS and PSMA ADC groups, respectively, at randomization. All animals then received intravenous injections of PBS or PSMA ADC (Progenics, Tarrytown, NY, toxicology lot P10306) at 6mg/kg via tail vein in a volume of 0.1 mL on
day 12 through 29 twice per week for three weeks. Tumor sizes were measured twice per week or weekly for 100 days after tumor implantation, and the mean tumor volume for the two groups was plotted over time post tumor implantation (Fig. 7 ). - Tumor size reduction was observed in PSMA ADC treated animals. The first animal was sacrificed at day 33 in the vehicle control group due to the large size of tumor (>2000mm3); mean tumor volume for the vehicle group and the PSMA ADC treated group were 925.7mm3 and 92.5mm3 (p = 0.00005, student t-test), respectively. The experiment was followed for 100 days, and all mice were sacrificed except 4 animals in the PSMA ADC treated group.
- Among the 15 animals in the PSMA ADC treated group with a mean tumor size of 72.5mm3 at
day 75 as shown inFig. 7 , there were 4 mice (mouse # Fig. 8 ). On day 99, the average tumor volume was 688.7mm3 (893.8, 445.3, 666.7, 749.0 mm3) for the 4 mice. The animals were treated again with PSMA ADC at 6mg/kg from day 99, once a week for 13 weeks.Fig. 8 shows the tumor volume over 180 days for the 4 mice. The dotted lines indicate initial PSMA ADC treatment, and the solid lines indicate second PSMA ADC treatment. Tumor size reduction was observed immediately in all 4 mice after the second PSMA ADC treatment, and the tumor volume continued to shrink while dosing. Atday 180, the tumor volume was reduced to <100mm3 with a mean volume of 50mm3. - These results show that PSMA ADC is effective to treat large-sized tumors (∼900mm3) and also can be used to treat relapsed tumors using the same dose level from an initial PSMA ADC treatment.
- One mouse that was not included in the PSMA ADC experiment described above had a tumor size of ∼500mm3 at day 21. It was given 12 doses of docetaxel, IP, at 12mg/kg weekly. As shown in
Fig. 3 and Table 2, the animal initially responded to the docetaxel treatment, the tumor volume was reduced to ∼300mm3 atday 60, but then it progressed to ∼800mm3 atday 100. The treatment of docetaxel was stopped, and PSMA ADC was given IV at 6mg/kg for an additional 12 doses, weekly. The tumor volume was drastically reduced from 800mm3 to ∼50mm3 atday 180 when PSMA ADC was given. PSMA ADC can treat tumors that fail docetaxel treatment.Table 2. Measurement of tumor over time in an animal treated with 12 doses of 12mg/kg docetaxel followed by 12 doses of 6mg/kg PSMA ADC. The tumor responded to docetaxel treatment initially and then relapsed. The treatment was switched to PSMA ADC and then significant tumor volume reduction was observed. Days post tumor implantation Tumor Length (mm) Width (mm) Volume (mm3) 21 12.22 9.22 519.4 26 13.07 9.27 561.6 28 12.61 9.11 523.3 39 12.06 9.14 503.7 47 10.89 7.80 331.3 49 11.17 7.67 328.6 56 11.10 8.26 378.7 61 11.16 7.44 308.9 63 10.59 7.84 325.5 67 11.67 7.46 324.7 70 12.48 8.12 411.4 77 13.30 9.77 634.8 84 13.77 10.39 743.3 91 14.09 10.08 715.8 95 14.70 10.30 779.8 99 14.51 10.67 826.0 103 13.62 10.40 736.6 106 14.13 10.14 726.4 110 12.70 8.42 450.2 113 9.61 7.96 304.5 119 8.78 6.77 201.2 126 8.67 6.33 173.7 134 7.12 6.46 148.6 141 6.20 5.70 100.7 148 6.46 5.32 91.4 154 5.65 4.78 64.5 162 5.60 4.30 51.8 167 5.70 4.00 45.6 173 5.50 3.80 39.7 180 5.60 4.15 48.2 - An experiment (
Fig. 10 ) was designed to evaluate PSMA ADC effectiveness after docetaxel treatment failure. Male athymic nude mice, 6-8 weeks old, obtained from Charles River Laboratories, Inc. were implanted subcutaneously with 5 million C4-2 cells. Atday 14, animals having a tumor volume between 100-200mm3 were randomized (randomization #1) into two treatment arms: (1) vehicle control (PBS buffer, n = 10); (2) docetaxel at 2mg/kg/IV weekly (n = 50) according to tumor volume. The mean tumor volume was 138.0 and 138.1mm3 for the vehicle control and docetaxel treatment groups, respectively, at randomization. - All animals then received intravenous injections of PBS or docetaxel (Sigma, St. Louis, MO) at 2mg/kg via tail vein in a volume of 0.1 mL. Tumor sizes were measured twice per week after tumor implantation, and the average tumor volume for the two groups before the second randomization was initiated were plotted (
Fig. 11 ). Overall, docetaxel treatment reduced tumor growth compared to control. - When the tumor volume of an animal in the docetaxel treatment group exceeded 400mm3, this animal was randomized, at a 1:1 ratio, into one of the two treatment subgroups: one group continued to receive docetaxel at 2mg/kg, and the second group was switched to PSMA ADC treatment at 6mg/kg/IV weekly. Animals were randomized into these two treatment subgroups continuously over the period of 28 days to 70 days post tumor implantation. By the end of
day 70, there were 28 mice whose tumor volume had exceeded 400mm3, with 14 mice randomized into the PSMA ADC treatment subgroup (mean tumor volume of 695mm3 and 14 mice randomized into the continued docetaxel treatment subgroup (mean tumor volume of 642mm3 with a p-value of 0.28 (student t-test)). - The effect of PSMA ADC treatment on tumor growth after docetaxel failure was assessed by comparing tumor reduction after the second randomization. The tumor volumes of animals in the two subgroups were plotted (
Fig. 12 ). The results show that tumors grew continuously while on docetaxel treatment. However, the tumor volume was significantly reduced when PSMA ADC treatment was initiated. Therefore, PSMA ADC is effective in treating animals with tumors which failed docetaxel treatment. - The study design is shown in
Fig. 13 . Male athymic nude mice, 6-8 weeks old, obtained from Charles River Laboratories, Inc., were implanted with Matrigel subcutaneously into the right flank of each mouse with 5 million C4-2 cells (androgen-independent human prostate cancer cell line). Atday 14, tumor size was measured by length and width in mm (millimeter). The volume was calculated using the formula: volume (mm3) =[(length) x (width)2]/2. The animals were randomized into two groups with similar tumor volume (approximately 138 mm3): vehicle control and docetaxel at 2 mg/kg. Animals were dosed weekly through the tail vein. - When the tumor volume of an animal in the docetaxel treatment group exceeded 400 mm3, this animal was randomized, at a 1:1 ratio, into one of the two treatment subgroups: one group continued to receive docetaxel at 2 mg/kg weekly (n=18), and the second group was switched to PSMA ADC treatment at 6 mg/kg/IV weekly (n=18). Mice whose tumors durably responded to docetaxel (≤400 mm3) continued to receive docetaxel at 2 mg/kg/IV weekly. Treatment effects were assessed by measuring tumor volume and overall survival. Animal body weight was also measured. Animals with tumor size ≥2000 mm3 were sacrificed.
- Mean tumor volumes were 515 ± 103 mm3 and 495 ± 80 mm3 for the PSMA ADC and continued docetaxel treatment groups, respectively, at the second randomization (p = 0.518) (
Fig. 14 ). At end of the experiment, the survival rate was 100% for animals in the PSMA ADC treatment group (Fig. 17 ); 94% of these mice had tumor sizes <100 mm3 (Fig. 16 ). In contrast, the survival rate was 11 % in the continued docetaxel treatment group. Therefore, PSMA ADC treatment significantly shrank tumors and increased overall survival of animals compared to continued docetaxel treatment (p < 0.0001). PSMA ADC was generally well tolerated in the animal (Fig. 18 ). PSMA ADC demonstrated potent antitumor activity against large prostate tumors that had progressed following docetaxel treatment. Treatment with PSMA ADC significantly extended survival. - An open-label, dose-
escalation phase 1 study of PSMA ADC IV in subjects with progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy is initiated. Subjects will receive either 0.4mg/kg, 0.7mg/kg, 1.1 mg/kg, 1.8mg/kg or 2.9mg/kg by constant-rate 90-minute IV infusion of PSMA ADC in saline (atweeks - The dose-limiting toxicity (DLT) is defined as any National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE, Version 3.0) toxicity grade ≥ 3 as well as
grade 2 allergic/immunologic toxicity (except isolated fever) for which a causal relationship to the study drug cannot be excluded. In this study, DLT will be determined after the first dose of each cohort. A dose-escalation scheme, based on the DLT, will be employed. Once an MTD is determined or the highest dose cohort is shown safe, additional subjects will be treated. These subjects will be chosen using the same inclusion/exclusion criteria employed for the study. If two or more subjects in the lowest dose cohort experience a DLT, a cohort will be enrolled at 0.2 mg/kg of PSMA ADC. - Radiologic imaging will be obtained at screening and
week 12. Blood samples (5 mL) for drug concentrations will be obtained prior to infusion initiation and at 90 minutes and 4, 6, 24, 48, 96, 168, 336 and 504 hours after infusion initiation atweeks week 12 time points. - All subjects who have completed the 12-week study, and who, in the opinion of the investigator, are likely to benefit from continued treatment with PSMA ADC, will be offered enrollment into an extension study.
- Diagnosis and Inclusion Criteria:
- 1. Males, age ≥ 18 years (or minimum adult age as determined by local regulatory authorities)
- 2. Eastern Cooperative Oncology Group (ECOG) status of 0 or 1
- 3. Histologic confirmation of prostate cancer
- 4. A diagnosis of progressive, castration-resistant, metastatic prostate cancer based on evidence of metastatic disease on bone scan, CT scan, or MRI at any time following the initial diagnosis of prostate cancer
- 5. Prior androgen-deprivation therapy consisting of either orchiectomy or luteinizing hormone-releasing hormone (LHRH) agonists, with or without an antiandrogen and a castrate level of serum testosterone (< 50ng/mL)
- 6. Prior therapy with taxane
- 7. Lab requirements:
- White blood count (WBC) ≥ 3000/mm3
- Absolute neutrophil count (ANC) ≥ 1000/mm3
- Platelets (Plt) ≥ 100,000mm3
- Hemoglobin (Hgb) ≥ 10g/dL
- Total bilirubin ≤ 2.0mg/dL
- Serum alanine transferase/serum aspartate transaminase (ALT/AST) ≤ 2 x the upper limit of normal
- Serum creatinine ≤ 2.0mg/dL and a calculated glomerular filtration rate (GFR) of > 60mL/min
- Exclusion Criteria:
- 1. Nonprostate primary malignant neoplasm except for nonmelanoma skin cancer or low-grade papillary transitional cell carcinoma of the bladder within previous five years
- 2. Clinically significant cardiac disease (New York Heart Association Class III/IV) or severe debilitating pulmonary disease
- 3. Radiation therapy or cytotoxic chemotherapy within previous four weeks
- 4. Active central nervous system (CNS) or epidural metastatic disease
- 5. An infection requiring antibiotic treatment within seven days prior to screening and/or antibiotic treatment initiated up to the time of the first-dose
- 6. Peripheral neuropathy of
grade 2 or higher with any association to taxane therapy at the start of the study - 7. Any prior treatment with any other therapy targeting PSMA
- 8. Subjects must not have participated in any other research study within 30 days
- 9. Prior therapy with investigational or approved mAbs or Ig fusion proteins
- 10. Subjects with QTc ≥ 500 msec
- 11. Weight >225 pounds
- A
phase 1, open-label, dose-escalation clinical trial will include men with progressive, hormone-refractory prostate cancer, and who had prior therapy with taxane chemotherapy drugs. The study will investigate the duration of clinical benefit derived from PSMA ADC treatment while also assessing the investigational drug's safety and tolerability. The initial 12-week period will evaluate up to five intravenous doses of PSMA ADC, individually administered at three-week intervals. The study will include evaluations of pharmacodynamics, radiographic changes in tumor burden, and changes in prostate-specific antigen (PSA) and circulating tumor cell (CTC) values compared to baseline. - Following the 12-week period, patients will be offered, at their physician's discretion, the option to continue treatment for an additional 39 weeks with the same dose of PSMA ADC as administered in their initial cohort. Qualified subjects will receive up to 13 additional doses of PSMA ADC at three-week intervals.
- An open-label, dose-
escalation phase 1 study of PSMA ADC IV in subjects with progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy was initiated. Subjects received either 0.4mg/kg; 0.7mg/kg, 1.1 mg/kg or 1.8mg/kg or will receive either 2.4mg/kg, 3.0mg/kg, 3.5mg/kg or 4.0mg/kg by constant-rate IV infusion of PSMA ADC in saline administered over approximately 90 minutes (atweeks - The dose-limiting toxicity (DLT) can be defined as any National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE, Version 3.0) toxicity grade ≥ 3 with the following exception: an absolute neutrophil count (ANC) <500mm3 determined eight days following the first infusion of PSMA ADC. Under this circumstance, a DLT can be defined as a neutrophil count <500mm3 found on both
study days 8 and 15 following the first infusion of PSMA ADC. A DLT can also be defined as grade ≥2 allergic/immunologic toxicity (except isolated fever) for which a causal relationship to the study drug cannot be excluded. In this study, DLT was/will be determined after the first dose of each cohort. A dose-escalation scheme, based on the DLT, was/will be employed. Once an MTD is determined or the highest dose cohort is shown safe, additional subjects will be treated. These subjects will be chosen using the same inclusion/exclusion criteria employed for the study. If two or more subjects in the lowest dose cohort experience a DLT, a cohort will be enrolled at 0.2 mg/kg of PSMA ADC. - Radiologic imaging was/will be obtained prior to infusion and within seven days prior to the
week 12 visit. Blood samples (5 mL) for drug concentrations was/will be obtained at designated time points inweeks week 12, sampling is at study day 85 ± one day). Atweek 0, samples were/are collected prior to infusion initiation, immediately at the end of infusion, and at four and six hours after the infusion. Samples collected at 24, 48, 96, 168, 336 and 504 hours after infusion initiation may be collected within ± two hours of the designated time point. Atweeks week 12 time points. - All subjects who have completed the 12-week study, and who, in the opinion of the investigator, are likely to benefit from continued treatment with PSMA ADC, will be offered enrollment into an extension study.
- Diagnosis and Inclusion Criteria:
- 1. Males, age ≥18 years (or minimum adult age as determined by local regulatory authorities)
- 2. Eastern Cooperative Oncology Group (ECOG) status of 0 or 1
- 3. Histologic confirmation of prostate cancer
- 4. A diagnosis of progressive, castration-resistant, metastatic prostate cancer based on evidence of metastatic disease on bone scan, CT scan, or MRI at any time following the initial diagnosis of prostate cancer
- 5. Prior androgen-deprivation therapy consisting of either orchiectomy or luteinizing hormone-releasing hormone (LHRH) agonists, with or without an antiandrogen and a castrate level of serum testosterone (< 50ng/dL)
- 6. Prior therapy with taxane
- 7. Lab requirements:
- White blood count (WBC) ≥ 3000/mm3
- Absolute neutrophil count (ANC) ≥ 1000/mm3
- Platelets (Plt) ≥ 100,000mm3
- Hemoglobin (Hgb) ≥ 9.0g/dL
- Total bilirubin ≤ 2.0mg/dL
- Serum alanine transferase/serum aspartate transaminase (ALT/AST) ≤ 2 x the upper limit of normal (ULN)
- Serum creatinine ≤ 2.0mg/dL and a calculated glomerular filtration rate (GFR) of > 60mL/min
-
- 1. Nonprostate primary malignant neoplasm except for nonmelanoma skin cancer or low-grade papillary transitional cell carcinoma of the bladder within previous five years
- 2. Clinically significant cardiac disease (New York Heart Association Class III/IV) or severe debilitating pulmonary disease
- 3. Radiation therapy or cytotoxic chemotherapy within previous six weeks
- 4. Active central nervous system (CNS) or epidural metastatic disease
- 5. Evidence of an active infection requiring ongoing antibiotic therapy
- 6. Peripheral neuropathy of
grade 2 or higher with any association to taxane therapy at the start of the study - 7. Any prior treatment with any other therapy targeting PSMA
- 8. Subjects must not have participated in any other research study within 30 days
- 9. Prior therapy with investigational or approved mAbs or Ig fusion proteins
- 10. Subjects with QTc ≥ 500 msec
- 11. History of pancreatitis or surgical procedures to the pancreas
- 12. History of drug and/or alcohol abuse
- 13. Any medical condition that in the opinion of the investigator may interfere with a subject's participation in or compliance with the study
- A
phase 1, open-label, dose-escalation clinical trial included men with progressive, hormone-refractory prostate cancer, and who had prior therapy with taxane chemotherapy drugs. The study investigates the duration of clinical benefit derived from PSMA ADC treatment and assesses the investigational drug's safety and tolerability. PSMA ADC was individually administered at three-week intervals during a 12-week period for the evaluation of at least four intravenous doses of PSMA ADC. The study includes evaluations of pharmacodynamics, radiographic changes in tumor burden, and changes in prostate-specific antigen (PSA) and circulating tumor cell (CTC) values compared to baseline. - Following the 12-week period, patients will be offered, at their physician's discretion, the option to continue treatment for an additional 39 weeks with the same dose of PSMA ADC as administered in their initial cohort. Qualified subjects will receive up to 13 additional doses of PSMA ADC at three-week intervals.
- An open-label, dose-
escalation phase 1 study of PSMA ADC administered IV in subjects with progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy is initiated. Intravenous (IV) infusions of PSMA ADC will be administered to subjects in four cycles. In cohorts 1-8, IV infusions of PSMA ADC will be administered once at the start of a cycle (duration 3 weeks or Q3W;weeks week cycle 1; weeks, 5, 6 and 7 incycle 2;weeks cycle 3; andweeks 13, 14 and 15 in cycle 4; no doses will be administered duringweeks cycles - The dose-limiting toxicity (DLT) will be defined as any National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE, Version 3.0) toxicity grade ≥ 3 with the following exception: an absolute neutrophil count (ANC) <500mm3. For all cohorts, a DLT will be defined as a neutrophil count <500mm3 that persist upon
repeat determination 3 to 7 days following final PSMA ADC administration within the first cycle. For cohorts 9-12 an ANC <500mm3 prior to any dose in the first cycle is a DLT. A DLT is also defined as grade ≥2 allergic/immunologic toxicity (except isolated fever) for which a causal relationship to the study drug cannot be excluded. In this study, DLT will be determined after the first cycle of each cohort. A dose-escalation scheme, based on the DLT, will be employed. Determinations of DLT will occur as a separate consideration for cohorts 1-8 and 9-12. A DLT determination for cohort 1-8 will not influence enrollment in cohorts 9-12. Once an MTD is determined or the highest dose cohort is shown safe, additional subjects will be treated. These subjects will be chosen using the same inclusion/exclusion criteria employed for the study. - For subjects in cohorts 1-8, radiologic imaging will be obtained prior to infusion and within seven days prior to the week 13 visit. Blood samples (5 mL) for drug concentrations will be obtained at time points in
weeks week 1, samples are collected prior to infusion initiation, immediately at the end of infusion, and at four and six hours after the infusion. Samples collected at 24, 48, 96, 168, 336 and 504 hours after infusion initiation may be collected within ± two hours of the designated time point. Atweeks weeks - All subjects who have completed the 12-week study, and who, in the opinion of the investigator, are likely to benefit from continued treatment with PSMA ADC, will be offered enrollment into an extension study.
- Diagnosis and Inclusion Criteria:
- 1. Males, age ≥ 18 years (or minimum adult age as determined by local regulatory authorities)
- 2. Eastern Cooperative Oncology Group (ECOG) status of 0 or 1
- 3. Histologic confirmation of prostate cancer
- 4. A diagnosis of progressive, castration-resistant, metastatic prostate cancer based on evidence of metastatic disease on bone scan, CT scan, or MRI at any time following the initial diagnosis of prostate cancer
- 5. Prior androgen-deprivation therapy consisting of either orchiectomy or luteinizing hormone-releasing hormone (LHRH) agonists, with or without an antiandrogen and a castrate level of serum testosterone (< 50ng/dL)
- 6. Prior therapy with taxane
- 7. Lab requirements:
- White blood count (WBC) ≥ 3000/mm3
- Absolute neutrophil count (ANC) ≥ 1000/mm3
- Platelets (Plt) ≥ 100,000mm3
- Hemoglobin (Hgb) ≥ 9.0g/dL
- Total bilirubin ≤ 2.0mg/dL
- Serum alanine transferase/serum aspartate transaminase (ALT/AST) ≤ 2 x the upper limit of normal (ULN)
- Serum creatinine ≤ 2.0mg/dL and a calculated glomerular filtration rate (GFR) of > 60mL/min
-
- 1. Nonprostate primary malignant neoplasm except for nonmelanoma skin cancer or low-grade papillary transitional cell carcinoma of the bladder within previous five years
- 2. Clinically significant cardiac disease (New York Heart Association Class III/IV) or severe debilitating pulmonary disease
- 3. Radiation therapy or cytotoxic chemotherapy within previous six weeks
- 4. Active central nervous system (CNS) or epidural metastatic disease
- 5. Evidence of an active infection requiring ongoing antibiotic therapy
- 6. Peripheral neuropathy of
grade 2 or higher with any association to taxane therapy at the start of the study - 7. Any prior treatment with any other therapy targeting PSMA
- 8. Subjects must not have participated in any other research study within 30 days
- 9. Prior therapy with investigational or approved mAbs or Ig fusion proteins
- 10. Subjects with QTc ≥ 500 msec
- 11. History of pancreatitis or surgical procedures to the pancreas
- 12. History of drug and/or alcohol abuse
- 13. Any medical condition that in the opinion of the investigator may interfere with a subject's participation in or compliance with the study.
-
- <110> MA, Dangshe
- <120> METHODS FOR KILLING PSMA-EXPRESSING, TAXANE-RESISTANT CANCER CELLS
- <130> P0741.70011WO00
- <140>
09789284.8
<141> 2009-09-08 - <150>
61/095,300
<151> 2008-09-08 - <150>
61/205,395
<151> 2009-01-20 - <160> 33
- <170> PatentIn version 3.1
- <210> 1
<211> 750
<212> PRT
<213> Homo sapiens - <400> 1




- <210> 2
<211> 7570
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 2





- <210> 3
<211> 7597
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 3





- <210> 4
<211> 7579
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 4





- <210> 5
<211> 7558
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 5





- <210> 6
<211> 7576
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 6





- <210> 7
<211> 7561
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 7





- <210> 8
<211> 6082
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 8





- <210> 9
<211> 6082
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 9




- <210> 10
<211> 6082
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 10





- <210> 11
<211> 6085
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 11




- <210> 12
<211> 6097
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 12





- <210> 13
<211> 6094
<212> DNA
<213> Artificial Sequence - <220>
<223> Plasmid - <400> 13




- <210> 14
<211> 481
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region, portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 14


- <210> 15
<211> 142
<212> PRT
<213> Homo sapiens - <400> 15

- <210> 16
<211> 463
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 16

- <210> 17
<211> 127
<212> PRT
<213> Homo sapiens - <400> 17

- <210> 18
<211> 508
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 18

- <210> 19
<211> 143
<212> PRT
<213> Homo sapiens - <400> 19


- <210> 20
<211> 463
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 20

- <210> 21
<211> 127
<212> PRT
<213> Homo sapiens - <400> 21


- <210> 22
<211> 490
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 22

- <210> 23
<211> 145
<212> PRT
<213> Homo sapiens - <400> 23


- <210> 24
<211> 463
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 24

- <210> 25
<211> 127
<212> PRT
<213> Homo sapiens - <400> 25


- <210> 26
<211> 469
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 26

- <210> 27
<211> 138
<212> PRT
<213> Homo sapiens - <400> 27

- <210> 28
<211> 466
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 28

- <210> 29
<211> 128
<212> PRT
<213> Homo sapiens - <400> 29

- <210> 30
<211> 487
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 30


- <210> 31
<211> 144
<212> PRT
<213> Homo sapiens - <400> 31

- <210> 32
<211> 478
<212> DNA
<213> Artificial Sequence - <220>
<223> Includes BamHI/Bg1II cloning junction, signal peptide, V region,
portion of C region and 3'XbaI/NheI (heavy) or NheI (light) cloning - junction
- <400> 32


- <210> 33
<211> 132
<212> PRT
<213> Homo sapiens - <400> 33

Claims (21)
- An in vitro method for killing PSMA-expressing, taxane-resistant cancer cells comprising:contacting the PSMA-expressing, taxane-resistant cancer cells with an antibody-drug conjugate in an amount effective to kill the PSMA-expressing, taxane-resistant cancer cells, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1.
- An antibody-drug conjugate for treating a subject that has a PSMA-expressing, taxane-resistant cancer, wherein the antibody-drug conjugate comprises a monoclonal antibody or antigen-binding fragment thereof that specifically binds to prostate-specific membrane antigen (PSMA) conjugated to monomethylauristatin norephedrine or monomethylauristatin phenylalanine, and wherein the sequence of PSMA is the sequence set forth in SEQ ID NO: 1.
- An antibody-drug conjugate as claimed in claim 2, wherein survival in the subject is increased in comparison to the median survival time of subjects with PSMA-expressing, taxane-resistant cancer not treated with PSMA ADC, preferably wherein survival in the subject is increased by:a) four weeks;b) six weeks;c) two months;d) four months;e) six months;f) eight months;g) ten months;h) twelve months; and/ori) fourteen months.
- A method as claimed in claim 1, wherein the PSMA-expressing, taxane-resistant cancer cells are:a) resistant to docetaxel or paclitaxel;b) prostate cancer cells or non-prostate cancer cells, preferably wherein the non-prostate cancer cells are bladder cancer cells, pancreatic cancer cells, liver cancer cells, lung cancer cells, kidney cancer cells, sarcoma cells, breast cancer cells, brain cancer cells, neuroendocrine carcinoma cells, colon cancer cells, testicular cancer cells or melanoma cells;c) of a tumor; and/ord) of a metastasis.
- An antibody-drug conjugate as claimed in claim 2 or claim 3, wherein the PSMA-expressing, taxane-resistant cancer is:a) resistant to docetaxel or paclitaxel;b) prostate cancer or non-prostate cancer, preferably wherein the non-prostate cancer is bladder cancer, pancreatic cancer, liver cancer, lung cancer, kidney cancer, sarcoma, breast cancer, brain cancer, neuroendocrine carcinoma, colon cancer, testicular cancer or melanoma;c) a tumor; and/ord) mestastic.
- A method as claimed in claim 4 or an antibody-drug conjugate as claimed in claim 5, wherein the volume of the tumor is at least:a) 100 mm3;b) 200 mm3;c) 300 mm3;d) 400 mm3;e) 500 mm3;f) 600 mm3;g) 700 mm3;h) 800 mm3;i) 900 mm3;j) 1000 mm3;k) 1200 mm3;l) 1400 mm3; and/orm) 1600 mm3.
- A method or an antibody-drug conjugate as claimed in any of claims 4-6, wherein the volume of the tumor is reduced by at least:a) 10%;b) 20%;c) 30%;d) 40%;e) 50%;f) 60%;g) 70%;h) 80%;i) 90%; and/orj) 95%;
- A method or an antibody-drug conjugate as claimed in any of claims 1-7, wherein the antibody or antigen-binding fragment thereof is conjugated to at least 2 or 3 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules, and preferably to at least 4 monomethylauristatin norephedrine or monomethylauristatin phenylalanine molecules.
- A method or an antibody-drug conjugate as claimed in any of claims 1-7, wherein the monomethylauristatin norephedrine or monomethylauristatin phenylalanine is conjugated to the antibody or antigen-binding fragment thereof with a compound of the formula:
-An-Ym-Zm-Xn-Wn-
wherein:A is a carboxylic acyl unit;Y is an amino acid;Z is an amino acid;X and W are each a self-immolative spacer;n is an integer of 0 or 1 ; andm is an integer of 0 or 1, 2, 3, 4, 5 or 6,and preferably wherein:a) A isi) ii) 4-(N-succinimidomethyl)cyclohexane-1-carbonyl, m-succinimidobenzoyl, 4-(p-succinimidophenyl)-butyryl, 4-(2-acetamido)benzoyl, 3-thiopropionyl, 4-(1-thioethyl)-benzoyl, 6-(3-thiopropionylamido)-hexanoyl or maleimide caproyl;b) Y is alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan or proline; and/orc) Z is lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, or citrulline.
ii) 4-(N-succinimidomethyl)cyclohexane-1-carbonyl, m-succinimidobenzoyl, 4-(p-succinimidophenyl)-butyryl, 4-(2-acetamido)benzoyl, 3-thiopropionyl, 4-(1-thioethyl)-benzoyl, 6-(3-thiopropionylamido)-hexanoyl or maleimide caproyl;b) Y is alanine, valine, leucine, isoleucine, methionine, phenylalanine, tryptophan or proline; and/orc) Z is lysine, lysine protected with acetyl or formyl, arginine, arginine protected with tosyl or nitro groups, histidine, ornithine, ornithine protected with acetyl or formyl, or citrulline. - A method or an antibody-drug conjugate as claimed in claim 9, wherein:a) Ym-Zm is:i) valine-citrulline; and/orii) a protein sequence which is selectively cleavable by a protease; and/orb) X is:i) a compound having the formula:
 ii) a compound having the formula:
ii) a compound having the formula:
-HN-R1-COT
in which R1 is C1-C5 alkyl, T is O, N or S;
iii) a compound having the formula: iv) p-aminobenzylcarbamoyloxy;v) p-aminobenzylalcohol;vi) p-aminobenzylcarbamate;vii) p-aminobenzyloxycarbonyl; orviii) γ-aminobutyric acid; α,α-dimethyl γ-aminobutyric acid or β,β-dimethyl γ-aminobutyric acid.
iv) p-aminobenzylcarbamoyloxy;v) p-aminobenzylalcohol;vi) p-aminobenzylcarbamate;vii) p-aminobenzyloxycarbonyl; orviii) γ-aminobutyric acid; α,α-dimethyl γ-aminobutyric acid or β,β-dimethyl γ-aminobutyric acid. - A method or an antibody-drug conjugate as claimed in claim 9, wherein W is

- A method or an antibody-drug conjugate as claimed in any of claims 9-11, wherein m and n are 0.
- A method or an antibody-drug conjugate as claimed in claim 9, wherein the antibody-drug conjugate is:a) AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin norephedrine;b) AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin norephedrine;c) AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin phenylalanine;d) AB-PG1-XG1-006-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin phenylalanine;e) AB-PG1-XG1-006-maleimide caproyl-monomethylauristatin phenylalanine;f) AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin norephedrine;g) AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin norephedrine;h) AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzyloxycarbonyl-monomethylauristatin phenylalanine;i) AB-PG1-XG1-026-maleimide caproyl-valine-citrulline-p-aminobenzylcarbamate-monomethylauristatin phenylalanine; orj) AB-PG1-XG1-026-maleimide caproyl-monomethylauristatin phenylalanine.
- A method or an antibody-drug conjugate as claimed in any of claims 1-13, wherein the monoclonal antibody or antigen-binding fragment thereof:a) specifically binds to an extracellular domain of PSMA;b) specifically binds to a conformational epitope of PSMA;c) binds live cells;d) does not require cell lysis to bind PSMA; and/ore) binds to cells of the neovasculature of a tumor.
- A method or antibody-drug conjugate as claimed in any of claims 1-13, wherein the antibody or antigen-binding fragment thereof:I) competitively inhibits the specific binding of a second antibody to its target epitope on PSMA, wherein the second antibody is selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, Abgenix 4.152.1 and antibodies comprising:(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14, 18, 22, 26 and 30, andd) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16, 20, 24, 28 and 32,and most preferably wherein the second antibody comprises:
preferably wherein the second antibody is selected from the group consisting of AB-PG1-XG1-006, AB-PG1-XG1-026 and antibodies comprising:(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, andd) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16 and 20,i)(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 14, andd) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 16; and/orii)(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 3, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 9, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 18, andd) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 20; and/orII) binds to an epitope on PSMA defined by an antibody selected from the group consisting of PSMA 3.7, PSMA 3.8, PSMA 3.9, PSMA 3.11, PSMA 5.4, PSMA 7.1, PSMA 7.3, PSMA 10.3, PSMA 1.8.3, PSMA A3.1.3, PSMA A3.3.1, Abgenix 4.248.2, Abgenix 4.360.3, Abgenix 4.7.1, Abgenix 4.4.1, Abgenix 4.177.3, Abgenix 4.16.1, Abgenix 4.22.3, Abgenix 4.28.3, Abgenix 4.40.2, Abgenix 4.48.3, Abgenix 4.49.1, Abgenix 4.209.3, Abgenix 4.219.3, Abgenix 4.288.1, Abgenix 4.333.1, Abgenix 4.54.1, Abgenix 4.153.1, Abgenix 4.232.3, Abgenix 4.292.3, Abgenix 4.304.1, Abgenix 4.78.1, Abgenix 4.152.1 and antibodies comprising:(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2-7, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8-13, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14, 18, 22, 26 and 30, andd) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16, 20, 24, 28 and 32. - A method or an antibody-drug conjugate as claimed in any of claims 1-13, wherein the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least 90% identical to a nucleotide sequence encoding an antibody selected from the group consisting of: AB-PG1-XG1-006, AB-PG1-XG1-026 and antibodies comprising:(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, andd) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16 and 20,preferably wherein the antibody is encoded by a nucleic acid molecule comprising a nucleotide sequence that is at least:i) 95%;ii) 97%;iii) 98%; and/oriv) 99% identical.
- A method or an antibody-drug conjugate as claimed in any of claims 1-13, wherein the antibody or antigen-binding fragment thereof:I) is AB-PG1-XG1-006, AB-PG1-XG1-026 or an antigen-binding fragment thereof; and/orII) is selected from the group consisting of antibodies comprising:a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 2 and 3, andb) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 8 and 9, orc) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 14 and 18, andd) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence selected from the group consisting of nucleotide sequences set forth as SEQ ID NOs: 16 and 20, and
antigen-binding fragments thereof,
preferably wherein the antibody or antigen-binding fragment thereof comprises:i)(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 14, and(d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 16, and
antigen-binding fragments thereof; and/orii)(a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 3, and(b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 9, or(c) a heavy chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 18, and(d) a light chain variable region encoded by a nucleic acid molecule comprising the coding region or regions of the nucleotide sequence set forth as SEQ ID NO: 20, and
antigen-binding fragments thereof. - A prostate-specific membrane antigen antibody drug conjugate (PSMA ADC) for treating a subject having progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, wherein the PSMA ADC consists essentially of a human monoclonal antibody to PSMA conjugated to monomethylaurastatin norephedrine (MMAE) via a valine-citrulline linker, and is in an effective amount sufficient to 1) delay or inhibit progression of the cancer, 2) increase survival of the subject as compared to the median survival of subjects not treated with the PSMA ADC and that have progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy, 3) decrease a circulating level of circulating tumor cells (CTCs) compared to a baseline level, or 4) decrease or stabilize a serum level of PSA compared to a baseline level of PSA.
- A PSMA ADC as claimed in claim 18, wherein:i) the taxane is docetaxel;ii) the human antibody is an IgG1 comprising (a) a heavy chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 2, and (b) a light chain encoded by a nucleic acid molecule comprising the coding region or regions of a nucleotide sequence set forth as SEQ ID NO: 8;iii) the delay or inhibition of progression of the cancer is demonstrated by radiographic image changes in tumor burden compared to a baseline radiographic image in the subject prior to the administration of the PSMA ADC, preferably wherein the radiographic image change is a change of at least:a) 10%;b) 20%;c) 30%;d) 40%;e) 50%; and/orf) 60%; and/oriv) the delay or inhibition of progression of cancer is demonstrated by a change in at least one biomarker for bone metastasis and bone metabolism compared to a baseline value prior to the administration of the PSMA ADC, preferably wherein the biomarker is N-telopeptide, bone alkaline phosphatase, osteocalcin, calcitonin, calcium, pyridinoline or deoxypyridinoline.
- An antibody-drug conjugate comprising an antibody, or antigen-binding fragment thereof, that specifically binds PSMA, conjugated to a dolastatin 10 derivative, for use in a method of treating a PSMA-expressing, taxane-resistant cancer.
- An antibody-drug conjugate as claimed in claim 20, wherein:a) the method involves killing PSMA-expressing, taxane-resistant cancer cells, preferably wherein the PSMA-expressing, taxane-resistant cancer cells are killed by the conjugate;b) the PSMA has the sequence set forth in SEQ ID NO: 1;c) the cancer is progressive, castration-resistant, metastatic prostate cancer that has progressed after prior taxane therapy;d) the dolastatin 10 derivative is an auristatin; and/ore) the dolastatin is MMAE (monomethylauristatin E or monomethylauristatin norephedrine) or MMAF (monomethylauristatin F or monomethylauristatin phenylalanine).
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13182516.8A EP2727606A3 (en) | 2008-09-08 | 2009-09-08 | Compounds for killing psma-expressing, taxane-resistant cancer cells |
| PL09789284T PL2326350T3 (en) | 2008-09-08 | 2009-09-08 | Compounds for killing psma-expressing, taxane-resistant cancer cells |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9530008P | 2008-09-08 | 2008-09-08 | |
| US20539509P | 2009-01-20 | 2009-01-20 | |
| PCT/US2009/005064 WO2010027513A2 (en) | 2008-09-08 | 2009-09-08 | Methods for killing psma-expressing, taxane-resistant cancer cells |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP13182516.8A Division EP2727606A3 (en) | 2008-09-08 | 2009-09-08 | Compounds for killing psma-expressing, taxane-resistant cancer cells |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP2326350A2 EP2326350A2 (en) | 2011-06-01 |
| EP2326350B1 true EP2326350B1 (en) | 2013-09-04 |
Family
ID=41632348
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP09789284.8A Not-in-force EP2326350B1 (en) | 2008-09-08 | 2009-09-08 | Compounds for killing psma-expressing, taxane-resistant cancer cells |
| EP13182516.8A Withdrawn EP2727606A3 (en) | 2008-09-08 | 2009-09-08 | Compounds for killing psma-expressing, taxane-resistant cancer cells |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP13182516.8A Withdrawn EP2727606A3 (en) | 2008-09-08 | 2009-09-08 | Compounds for killing psma-expressing, taxane-resistant cancer cells |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9242012B2 (en) |
| EP (2) | EP2326350B1 (en) |
| DK (1) | DK2326350T3 (en) |
| ES (1) | ES2438495T3 (en) |
| HK (1) | HK1159474A1 (en) |
| PL (1) | PL2326350T3 (en) |
| PT (1) | PT2326350E (en) |
| WO (1) | WO2010027513A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9782478B1 (en) | 2011-04-22 | 2017-10-10 | Aptevo Research And Development Llc | Prostate-specific membrane antigen binding proteins and related compositions and methods |
| US11352426B2 (en) | 2015-09-21 | 2022-06-07 | Aptevo Research And Development Llc | CD3 binding polypeptides |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050215472A1 (en) | 2001-10-23 | 2005-09-29 | Psma Development Company, Llc | PSMA formulations and uses thereof |
| HUE047200T2 (en) | 2007-08-17 | 2020-04-28 | Purdue Research Foundation | Psma binding ligand-linker conjugates and methods for using |
| ES2438495T3 (en) | 2008-09-08 | 2014-01-17 | Psma Development Company, L.L.C. | Compounds to kill cancer cells expressing PSMA, resistant to taxane |
| CN102714296A (en) | 2009-05-19 | 2012-10-03 | Aic布莱博公司 | Composite current collector and methods therefor |
| US9951324B2 (en) | 2010-02-25 | 2018-04-24 | Purdue Research Foundation | PSMA binding ligand-linker conjugates and methods for using |
| US8927692B2 (en) | 2010-11-12 | 2015-01-06 | The Trustees Of The University Of Pennsylvania | Consensus prostate antigens, nucleic acid molecule encoding the same and vaccine and uses comprising the same |
| WO2012138691A2 (en) * | 2011-04-04 | 2012-10-11 | Children's Medical Center Corporation | Diagnosis and treatment of taxane-resistant cancers |
| EP3536700A1 (en) | 2012-06-07 | 2019-09-11 | Ambrx, Inc. | Prostate-specific membrane antigen antibody drug conjugates |
| MX2014015205A (en) * | 2012-06-14 | 2015-08-14 | Ambrx Inc | Anti-psma antibodies conjugated to nuclear receptor ligand polypeptides. |
| JP6892218B2 (en) | 2012-11-15 | 2021-06-23 | エンドサイト・インコーポレイテッドEndocyte, Inc. | How to treat diseases caused by drug delivery conjugates and PSMA-expressing cells |
| EP3456700A1 (en) | 2013-10-18 | 2019-03-20 | Deutsches Krebsforschungszentrum | Labeled inhibitors of prostate specific membrane antigen (psma), their use as imaging agents and pharmaceutical agents for the treatment of prostate cancer |
| WO2015057250A1 (en) * | 2013-10-18 | 2015-04-23 | Psma Development Company, Llc | Combination therapies with psma ligand conjugates |
| JP6908964B2 (en) * | 2013-10-18 | 2021-07-28 | ピーエスエムエー ディベロップメント カンパニー,エルエルシー | Combination therapy with PSMA ligand conjugate |
| US20150147339A1 (en) | 2013-11-15 | 2015-05-28 | Psma Development Company, Llc | Biomarkers for psma targeted therapy for prostate cancer |
| US10188759B2 (en) | 2015-01-07 | 2019-01-29 | Endocyte, Inc. | Conjugates for imaging |
| MX2019012017A (en) | 2017-04-07 | 2020-02-12 | Juno Therapeutics Inc | Engineered cells expressing prostate-specific membrane antigen (psma) or a modified form thereof and related methods. |
Family Cites Families (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5162504A (en) | 1988-06-03 | 1992-11-10 | Cytogen Corporation | Monoclonal antibodies to a new antigenic marker in epithelial prostatic cells and serum of prostatic cancer patients |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US5175384A (en) | 1988-12-05 | 1992-12-29 | Genpharm International | Transgenic mice depleted in mature t-cells and methods for making transgenic mice |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5124471A (en) | 1990-03-26 | 1992-06-23 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Bifunctional dtpa-type ligand |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5756825A (en) | 1992-09-08 | 1998-05-26 | Safavy; Ahmad | Hydroxamic acid-based bifunctional chelating compounds |
| EP0668777B2 (en) | 1992-11-05 | 2011-02-09 | Sloan-Kettering Institute For Cancer Research | Prostate-specific membrane antigen |
| US6953668B1 (en) | 1992-11-05 | 2005-10-11 | Sloan-Kettering Institute For Cancer Research | Prostate-specific membrane antigen |
| US7105159B1 (en) | 1992-11-05 | 2006-09-12 | Sloan-Kettering Institute For Cancer Research | Antibodies to prostate-specific membrane antigen |
| US7070782B1 (en) | 1992-11-05 | 2006-07-04 | Sloan-Kettering Institute For Cancer Research | Prostate-specific membrane antigen |
| US6034065A (en) | 1992-12-03 | 2000-03-07 | Arizona Board Of Regents | Elucidation and synthesis of antineoplastic tetrapeptide phenethylamides of dolastatin 10 |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| US6569432B1 (en) | 1995-02-24 | 2003-05-27 | Sloan-Kettering Institute For Cancer Research | Prostate-specific membrane antigen and uses thereof |
| US5935818A (en) | 1995-02-24 | 1999-08-10 | Sloan-Kettering Institute For Cancer Research | Isolated nucleic acid molecule encoding alternatively spliced prostate-specific membrane antigen and uses thereof |
| EP0690452A3 (en) | 1994-06-28 | 1999-01-07 | Advanced Micro Devices, Inc. | Electrically erasable memory and method of erasure |
| AU717937B2 (en) | 1995-02-24 | 2000-04-06 | Sloan-Kettering Institute For Cancer Research | Prostate-specific membrane antigen and uses thereof |
| US5994292A (en) | 1995-05-31 | 1999-11-30 | The United States Of America As Represented By The Department Of Health And Human Services | Interferon-inducible protein 10 is a potent inhibitor of angiogenesis |
| US5773292A (en) | 1995-06-05 | 1998-06-30 | Cornell University | Antibodies binding portions, and probes recognizing an antigen of prostate epithelial cells but not antigens circulating in the blood |
| US5788963A (en) | 1995-07-31 | 1998-08-04 | Pacific Northwest Cancer Foundation | Isolation and/or preservation of dendritic cells for prostate cancer immunotherapy |
| US20040253246A1 (en) | 1996-02-23 | 2004-12-16 | Israeli Ron S. | Prostate-specific membrane antigen and uses thereof |
| WO1997035616A1 (en) | 1996-03-25 | 1997-10-02 | Pacific Northwest Cancer Foundation | Monoclonal antibodies specific for the extracellular domain of prostate specific membrane antigen |
| US6150508A (en) | 1996-03-25 | 2000-11-21 | Northwest Biotherapeutics, Inc. | Monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen |
| US7381407B1 (en) | 1996-03-25 | 2008-06-03 | Medarex, Inc. | Monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen |
| US6962981B1 (en) | 1996-03-25 | 2005-11-08 | Medarex, Inc. | Monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen |
| US6136311A (en) * | 1996-05-06 | 2000-10-24 | Cornell Research Foundation, Inc. | Treatment and diagnosis of cancer |
| US6107090A (en) | 1996-05-06 | 2000-08-22 | Cornell Research Foundation, Inc. | Treatment and diagnosis of prostate cancer with antibodies to extracellur PSMA domains |
| US5804602A (en) | 1996-06-17 | 1998-09-08 | Guilford Pharmaceuticals Inc. | Methods of cancer treatment using naaladase inhibitors |
| US5922845A (en) | 1996-07-11 | 1999-07-13 | Medarex, Inc. | Therapeutic multispecific compounds comprised of anti-Fcα receptor antibodies |
| CA2282504A1 (en) | 1997-02-25 | 1998-08-27 | Jun-Ping Xu | Isolation and structural elucidation of the cytostatic linear and cyclo-depsipeptides dolastatin 16, dolastatin 17, and dolastatin 18 |
| US6200765B1 (en) | 1998-05-04 | 2001-03-13 | Pacific Northwest Cancer Foundation | Non-invasive methods to detect prostate cancer |
| CA2333125C (en) | 1998-05-26 | 2009-12-15 | Sloan-Kettering Institute For Cancer Research | Alpha emitting constructs and uses thereof |
| JP2002524081A (en) | 1998-09-04 | 2002-08-06 | スローン − ケッタリング インスティチュート フォー キャンサー リサーチ | Fusion receptor specific for prostate-specific membrane antigen and uses thereof |
| WO2000061605A1 (en) | 1999-04-09 | 2000-10-19 | Sloan-Kettering Institute For Cancer Research | Dna encoding the prostate-specific membrane antigen-like gene and uses thereof |
| EP1175616A4 (en) | 1999-04-13 | 2003-08-13 | Northwest Biotherapeutics Inc | Methods for the diagnosis and treatment of metastatic prostate tumors |
| EP1553414A1 (en) | 1999-04-13 | 2005-07-13 | Medarex, Inc. | Methods for the diagnosis and treatment of metastatic prostate tumors |
| CA2375228A1 (en) | 1999-06-10 | 2000-12-21 | Sloan-Kettering Institute For Cancer Research | Markers for prostate cancer |
| EP1710256A1 (en) | 1999-07-29 | 2006-10-11 | Medarex, Inc. | Human monoclonal antibodies to prostate specific membrane antigen |
| WO2001009192A1 (en) | 1999-07-29 | 2001-02-08 | Medarex, Inc. | Human monoclonal antibodies to prostate specific membrane antigen |
| US6653129B1 (en) | 1999-09-13 | 2003-11-25 | The Regents Of The University Of Colorado | Method for isolation of cells from a solution |
| US7560534B2 (en) | 2000-05-08 | 2009-07-14 | Celldex Research Corporation | Molecular conjugates comprising human monoclonal antibodies to dendritic cells |
| NZ534864A (en) | 2000-05-08 | 2006-04-28 | Celldex Therapeutics Inc | Human monoclonal antibodies to dentritic cells |
| ES2375948T3 (en) | 2000-05-12 | 2012-03-07 | Northwest Biotherapeutics, Inc. | PROCEDURE TO INCREASE THE SUBMISSION OF CLASS I OF ANTENDS? EXOGEN GENES BY CELLS DENDRÉ? TICAS HUMANAS. |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| US20040018194A1 (en) | 2000-11-28 | 2004-01-29 | Francisco Joseph A. | Recombinant anti-CD30 antibodies and uses thereof |
| US6884869B2 (en) | 2001-04-30 | 2005-04-26 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US7256257B2 (en) | 2001-04-30 | 2007-08-14 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| WO2002096460A1 (en) | 2001-05-30 | 2002-12-05 | Cornell Research Foundation, Inc. | Endopeptidase/anti-psma antibody fusion proteins for treatment of cancer |
| US7666414B2 (en) | 2001-06-01 | 2010-02-23 | Cornell Research Foundation, Inc. | Methods for treating prostate cancer using modified antibodies to prostate-specific membrane antigen |
| US7514078B2 (en) | 2001-06-01 | 2009-04-07 | Cornell Research Foundation, Inc. | Methods of treating prostate cancer with anti-prostate specific membrane antigen antibodies |
| IL158969A0 (en) | 2001-06-01 | 2004-05-12 | Cornell Res Foundation Inc | Modified antibodies to prostate-specific membrane antigen and uses thereof |
| AU2002305767B2 (en) | 2001-09-20 | 2008-04-10 | Cornell Research Foundation, Inc. | Methods and compositions for treating and preventing skin disorders using binding agents specific for PSMA |
| WO2003026577A2 (en) | 2001-09-24 | 2003-04-03 | Seattle Genetics, Inc. | P-amidobenzylethers in drug delivery agents |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| EP1448588A4 (en) | 2001-10-23 | 2006-10-25 | Psma Dev Company L L C | Psma antibodies and protein multimers |
| US20040161776A1 (en) | 2001-10-23 | 2004-08-19 | Maddon Paul J. | PSMA formulations and uses thereof |
| US20050215472A1 (en) | 2001-10-23 | 2005-09-29 | Psma Development Company, Llc | PSMA formulations and uses thereof |
| US20030157534A1 (en) | 2001-12-26 | 2003-08-21 | Manuel Engelhorn | DNA immunization with libraries of minigenes encoding degenerate variants of major histocompatability class l restricted epitopes |
| WO2003064612A2 (en) | 2002-01-28 | 2003-08-07 | Sloan-Kettering Institute For Cancer Research | Identification of mutant antigens with enhanced immunogenicity |
| AU2003224604B2 (en) | 2002-01-28 | 2007-06-14 | Medarex, Inc. | Human monoclonal antibodies to prostate specific membrane antigen (PSMA) |
| HUE027549T2 (en) | 2002-07-31 | 2016-10-28 | Seattle Genetics Inc | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| US20040136998A1 (en) | 2002-10-30 | 2004-07-15 | Bander Neil H. | Methods and compositions for treating or preventing insulin-related disorders using binding agents specific for prostate specific membrane antigen |
| AU2003293556A1 (en) | 2003-01-10 | 2004-08-10 | Millennium Pharmaceuticals, Inc. | Methods of diagnosing and treating cancer |
| EP1597356A4 (en) | 2003-02-10 | 2007-07-11 | Northwest Biotherapeutics Inc | Cultured cd14+ antigen presenting cells |
| AU2004213053C1 (en) | 2003-02-20 | 2009-07-16 | Seagen Inc. | Anti-CD70 antibody-drug conjugates and their use for the treatment of cancer and immune disorders |
| DK2489364T3 (en) | 2003-11-06 | 2015-03-02 | Seattle Genetics Inc | Monomethylvaline compounds conjugated to antibodies |
| WO2005070456A2 (en) | 2004-01-09 | 2005-08-04 | Millennium Pharmaceuticals, Inc. | Diagnosing and treating female reproductive tract or chilhood cancer with pmsa antibodies |
| WO2005084390A2 (en) | 2004-03-02 | 2005-09-15 | Seattle Genetics, Inc. | Partially loaded antibodies and methods of their conjugation |
| WO2005094882A1 (en) | 2004-03-03 | 2005-10-13 | Millennium Pharmaceuticals, Inc. | Modified antibodies to prostate-specific membrane antigen and uses thereof |
| US7378504B2 (en) | 2004-06-03 | 2008-05-27 | Medarex, Inc. | Human monoclonal antibodies to Fc gamma receptor I (CD64) |
| US20060057626A1 (en) | 2004-09-03 | 2006-03-16 | Nichol Geoffrey M | Assessment of CTLA-4 polymorphisms in CTLA-4 blockade therapy |
| EP1846454A2 (en) | 2004-09-30 | 2007-10-24 | Medarex, Inc. | Human monoclonal antibodies to fc gamma receptor ii (cd32) |
| US8288352B2 (en) | 2004-11-12 | 2012-10-16 | Seattle Genetics, Inc. | Auristatins having an aminobenzoic acid unit at the N terminus |
| EP1841467A4 (en) | 2005-01-14 | 2009-01-28 | Cytogen Corp | Combination cancer therapy with anti-psma antibodies |
| BRPI0607796A2 (en) | 2005-02-18 | 2009-06-13 | Medarex Inc | compound or a pharmaceutically acceptable salt thereof, process for preparation and use thereof, and pharmaceutical composition |
| NZ556661A (en) | 2005-02-18 | 2010-10-29 | Medarex Inc | Human monoclonal antibodies to prostate specific membrance antigen (PSMA) |
| WO2006110745A2 (en) | 2005-04-08 | 2006-10-19 | Cytogen Corporation | Conjugated anti-psma antibodies |
| WO2007002222A2 (en) * | 2005-06-20 | 2007-01-04 | Psma Development Company, Llc | Psma antibody-drug conjugates |
| US8871720B2 (en) | 2005-07-07 | 2014-10-28 | Seattle Genetics, Inc. | Monomethylvaline compounds having phenylalanine carboxy modifications at the C-terminus |
| AU2006269422B2 (en) | 2005-07-07 | 2012-12-06 | Seagen Inc. | Monomethylvaline compounds having phenylalanine side-chain modifications at the C-terminus |
| HUE035853T2 (en) | 2005-07-18 | 2018-05-28 | Seattle Genetics Inc | Beta-glucuronide-linker drug conjugates |
| US20080279868A1 (en) | 2005-09-26 | 2008-11-13 | Medarex, Inc. | Antibody-Drug Conjugates and Methods of Use |
| EP1948688A2 (en) | 2005-11-14 | 2008-07-30 | Psma Development Company, L.L.C. | Compositions of and methods of using stabilized psma dimers |
| US7750116B1 (en) | 2006-02-18 | 2010-07-06 | Seattle Genetics, Inc. | Antibody drug conjugate metabolites |
| WO2007103288A2 (en) | 2006-03-02 | 2007-09-13 | Seattle Genetics, Inc. | Engineered antibody drug conjugates |
| ES2438495T3 (en) | 2008-09-08 | 2014-01-17 | Psma Development Company, L.L.C. | Compounds to kill cancer cells expressing PSMA, resistant to taxane |
-
2009
- 2009-09-08 ES ES09789284.8T patent/ES2438495T3/en active Active
- 2009-09-08 EP EP09789284.8A patent/EP2326350B1/en not_active Not-in-force
- 2009-09-08 DK DK09789284.8T patent/DK2326350T3/en active
- 2009-09-08 PL PL09789284T patent/PL2326350T3/en unknown
- 2009-09-08 PT PT97892848T patent/PT2326350E/en unknown
- 2009-09-08 EP EP13182516.8A patent/EP2727606A3/en not_active Withdrawn
- 2009-09-08 WO PCT/US2009/005064 patent/WO2010027513A2/en active Application Filing
-
2011
- 2011-02-17 US US13/030,105 patent/US9242012B2/en active Active
- 2011-11-22 HK HK11112651.2A patent/HK1159474A1/en not_active IP Right Cessation
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9782478B1 (en) | 2011-04-22 | 2017-10-10 | Aptevo Research And Development Llc | Prostate-specific membrane antigen binding proteins and related compositions and methods |
| US11352426B2 (en) | 2015-09-21 | 2022-06-07 | Aptevo Research And Development Llc | CD3 binding polypeptides |
Also Published As
| Publication number | Publication date |
|---|---|
| DK2326350T3 (en) | 2013-12-16 |
| US20110250216A1 (en) | 2011-10-13 |
| WO2010027513A3 (en) | 2010-06-10 |
| HK1159474A1 (en) | 2012-08-03 |
| PL2326350T3 (en) | 2014-03-31 |
| PT2326350E (en) | 2013-12-10 |
| EP2727606A3 (en) | 2015-09-23 |
| ES2438495T3 (en) | 2014-01-17 |
| US9242012B2 (en) | 2016-01-26 |
| WO2010027513A2 (en) | 2010-03-11 |
| EP2727606A2 (en) | 2014-05-07 |
| EP2326350A2 (en) | 2011-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210023094A1 (en) | Psma antibody-drug conjugates | |
| EP2326350B1 (en) | Compounds for killing psma-expressing, taxane-resistant cancer cells | |
| US9695248B2 (en) | PSMA antibodies and uses thereof | |
| EP2363404B1 (en) | PSMA antibodies | |
| WO2005042029A2 (en) | Psma formulations and uses thereof | |
| US20150110814A1 (en) | Combination therapies with psma ligand conjugates | |
| AU2002356844A1 (en) | PSMA antibodies and protein multimers | |
| JP2023135666A (en) | Combination therapies with psma ligand conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20110309 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: AL BA RS |
|
| DAX | Request for extension of the european patent (deleted) | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1159474 Country of ref document: HK |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 630113 Country of ref document: AT Kind code of ref document: T Effective date: 20130915 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602009018588 Country of ref document: DE Effective date: 20131031 |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: SC4A Free format text: AVAILABILITY OF NATIONAL TRANSLATION Effective date: 20131202 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 Effective date: 20131212 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: T3 |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: TRGR |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1159474 Country of ref document: HK |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2438495 Country of ref document: ES Kind code of ref document: T3 Effective date: 20140117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130717 Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 |
|
| REG | Reference to a national code |
Ref country code: NO Ref legal event code: T2 Effective date: 20130904 |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG4D |
|
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: EP Ref document number: 20130402576 Country of ref document: GR Effective date: 20140124 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20140104 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602009018588 Country of ref document: DE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20140605 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602009018588 Country of ref document: DE Effective date: 20140605 |
|
| REG | Reference to a national code |
Ref country code: HU Ref legal event code: AG4A Ref document number: E020267 Country of ref document: HU |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130904 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 7 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 8 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R079 Ref document number: 602009018588 Country of ref document: DE Free format text: PREVIOUS MAIN CLASS: A61K0047480000 Ipc: A61K0047500000 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 9 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 20200923 Year of fee payment: 12 Ref country code: GB Payment date: 20200922 Year of fee payment: 12 Ref country code: GR Payment date: 20200918 Year of fee payment: 12 Ref country code: DK Payment date: 20200922 Year of fee payment: 12 Ref country code: FR Payment date: 20200914 Year of fee payment: 12 Ref country code: NL Payment date: 20200925 Year of fee payment: 12 Ref country code: PT Payment date: 20200827 Year of fee payment: 12 Ref country code: IE Payment date: 20200918 Year of fee payment: 12 Ref country code: FI Payment date: 20200921 Year of fee payment: 12 Ref country code: NO Payment date: 20200922 Year of fee payment: 12 Ref country code: TR Payment date: 20200907 Year of fee payment: 12 Ref country code: DE Payment date: 20200925 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20200921 Year of fee payment: 12 Ref country code: AT Payment date: 20200921 Year of fee payment: 12 Ref country code: PL Payment date: 20200901 Year of fee payment: 12 Ref country code: BE Payment date: 20200925 Year of fee payment: 12 Ref country code: SE Payment date: 20200925 Year of fee payment: 12 Ref country code: IT Payment date: 20200922 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: HU Payment date: 20201021 Year of fee payment: 12 Ref country code: ES Payment date: 20201120 Year of fee payment: 12 Ref country code: CZ Payment date: 20200908 Year of fee payment: 12 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R119 Ref document number: 602009018588 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: FI Ref legal event code: MAE |
|
| REG | Reference to a national code |
Ref country code: NO Ref legal event code: MMEP |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EBP Effective date: 20210930 |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: EUG |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: MM Effective date: 20211001 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MM01 Ref document number: 630113 Country of ref document: AT Kind code of ref document: T Effective date: 20210908 |
|
| REG | Reference to a national code |
Ref country code: BE Ref legal event code: MM Effective date: 20210930 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20210908 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220308 Ref country code: CZ Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20211001 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210909 Ref country code: NO Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210930 Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 Ref country code: HU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210909 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210930 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220401 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210930 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210930 Ref country code: GR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20220407 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210930 Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 Ref country code: DK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210930 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20221107 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210909 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20210908 |