EP1584978A1 - Photothermographic material and image forming method - Google Patents
Photothermographic material and image forming method Download PDFInfo
- Publication number
- EP1584978A1 EP1584978A1 EP05006628A EP05006628A EP1584978A1 EP 1584978 A1 EP1584978 A1 EP 1584978A1 EP 05006628 A EP05006628 A EP 05006628A EP 05006628 A EP05006628 A EP 05006628A EP 1584978 A1 EP1584978 A1 EP 1584978A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- group
- photothermographic material
- mol
- weight
- silver halide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 239000000463 material Substances 0.000 title claims abstract description 166
- 238000000034 method Methods 0.000 title claims abstract description 164
- -1 silver halide Chemical class 0.000 claims abstract description 351
- 229910052709 silver Inorganic materials 0.000 claims abstract description 227
- 239000004332 silver Substances 0.000 claims abstract description 227
- 229920000642 polymer Polymers 0.000 claims abstract description 70
- 239000004816 latex Substances 0.000 claims abstract description 67
- 229920000126 latex Polymers 0.000 claims abstract description 67
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 62
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 59
- GGCZERPQGJTIQP-UHFFFAOYSA-N sodium;9,10-dioxoanthracene-2-sulfonic acid Chemical compound [Na+].C1=CC=C2C(=O)C3=CC(S(=O)(=O)O)=CC=C3C(=O)C2=C1 GGCZERPQGJTIQP-UHFFFAOYSA-N 0.000 claims abstract description 58
- 229910021612 Silver iodide Inorganic materials 0.000 claims abstract description 51
- JKFYKCYQEWQPTM-UHFFFAOYSA-N 2-azaniumyl-2-(4-fluorophenyl)acetate Chemical compound OC(=O)C(N)C1=CC=C(F)C=C1 JKFYKCYQEWQPTM-UHFFFAOYSA-N 0.000 claims abstract description 50
- 229940045105 silver iodide Drugs 0.000 claims abstract description 50
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 46
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 46
- 239000011230 binding agent Substances 0.000 claims abstract description 45
- 239000000178 monomer Substances 0.000 claims abstract description 43
- 125000005843 halogen group Chemical group 0.000 claims abstract description 26
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 9
- 150000001875 compounds Chemical class 0.000 claims description 201
- 125000001424 substituent group Chemical group 0.000 claims description 71
- 238000011161 development Methods 0.000 claims description 61
- 125000000623 heterocyclic group Chemical group 0.000 claims description 60
- 239000003795 chemical substances by application Substances 0.000 claims description 40
- 125000003118 aryl group Chemical group 0.000 claims description 34
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 claims description 32
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 25
- 125000003545 alkoxy group Chemical group 0.000 claims description 21
- 238000010438 heat treatment Methods 0.000 claims description 20
- 125000004104 aryloxy group Chemical group 0.000 claims description 18
- 125000002252 acyl group Chemical group 0.000 claims description 16
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 16
- 125000004442 acylamino group Chemical group 0.000 claims description 13
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 13
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims description 13
- 125000005647 linker group Chemical group 0.000 claims description 13
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 claims description 12
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 11
- 125000004414 alkyl thio group Chemical group 0.000 claims description 8
- 125000005110 aryl thio group Chemical group 0.000 claims description 8
- 239000007788 liquid Substances 0.000 claims description 8
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 239000012491 analyte Substances 0.000 claims description 6
- 229910052799 carbon Inorganic materials 0.000 claims description 6
- 125000000565 sulfonamide group Chemical group 0.000 claims description 6
- 125000004423 acyloxy group Chemical group 0.000 claims description 5
- 230000009477 glass transition Effects 0.000 claims description 5
- 230000002378 acidificating effect Effects 0.000 claims 1
- 239000010410 layer Substances 0.000 description 158
- 239000000243 solution Substances 0.000 description 129
- 239000006185 dispersion Substances 0.000 description 104
- 239000000839 emulsion Substances 0.000 description 98
- 238000000576 coating method Methods 0.000 description 97
- 239000011248 coating agent Substances 0.000 description 94
- 239000000126 substance Substances 0.000 description 84
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 79
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 73
- 238000002360 preparation method Methods 0.000 description 61
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 59
- 230000018109 developmental process Effects 0.000 description 58
- 239000002245 particle Substances 0.000 description 55
- 206010070834 Sensitisation Diseases 0.000 description 53
- 230000008313 sensitization Effects 0.000 description 53
- 150000003839 salts Chemical class 0.000 description 42
- 239000007864 aqueous solution Substances 0.000 description 41
- 239000000203 mixture Substances 0.000 description 40
- 239000000975 dye Substances 0.000 description 37
- 229910052757 nitrogen Inorganic materials 0.000 description 37
- 239000011241 protective layer Substances 0.000 description 35
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- 230000000274 adsorptive effect Effects 0.000 description 32
- 239000002253 acid Substances 0.000 description 31
- 238000006243 chemical reaction Methods 0.000 description 30
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 28
- 238000002156 mixing Methods 0.000 description 27
- 239000004094 surface-active agent Substances 0.000 description 27
- 230000001235 sensitizing effect Effects 0.000 description 25
- 230000015572 biosynthetic process Effects 0.000 description 24
- 229920002451 polyvinyl alcohol Polymers 0.000 description 24
- 230000008569 process Effects 0.000 description 24
- 230000035945 sensitivity Effects 0.000 description 23
- 125000003396 thiol group Chemical group [H]S* 0.000 description 22
- 229920000159 gelatin Polymers 0.000 description 21
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 21
- 238000003756 stirring Methods 0.000 description 21
- 108010010803 Gelatin Proteins 0.000 description 20
- 235000019322 gelatine Nutrition 0.000 description 20
- 235000011852 gelatine desserts Nutrition 0.000 description 20
- 229910052739 hydrogen Inorganic materials 0.000 description 20
- 125000004433 nitrogen atom Chemical group N* 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- 239000008273 gelatin Substances 0.000 description 19
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 18
- 239000004372 Polyvinyl alcohol Substances 0.000 description 17
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 17
- 229940125904 compound 1 Drugs 0.000 description 17
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 17
- 239000000047 product Substances 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- 239000001257 hydrogen Substances 0.000 description 16
- 238000007254 oxidation reaction Methods 0.000 description 16
- 239000002002 slurry Substances 0.000 description 16
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 15
- 229920001577 copolymer Polymers 0.000 description 15
- 230000003647 oxidation Effects 0.000 description 15
- 229910052714 tellurium Inorganic materials 0.000 description 15
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 14
- 235000014113 dietary fatty acids Nutrition 0.000 description 14
- 239000012153 distilled water Substances 0.000 description 14
- 239000000194 fatty acid Substances 0.000 description 14
- 229930195729 fatty acid Natural products 0.000 description 14
- 150000004665 fatty acids Chemical class 0.000 description 14
- 239000010419 fine particle Substances 0.000 description 14
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 14
- 239000012528 membrane Substances 0.000 description 14
- 229910001961 silver nitrate Inorganic materials 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 13
- 229910052751 metal Inorganic materials 0.000 description 13
- 239000002184 metal Substances 0.000 description 13
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 13
- 230000003595 spectral effect Effects 0.000 description 13
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 12
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 12
- 239000011324 bead Substances 0.000 description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 12
- NBVXSUQYWXRMNV-UHFFFAOYSA-N fluoromethane Chemical compound FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 description 12
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical compound C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 238000010521 absorption reaction Methods 0.000 description 11
- 125000003277 amino group Chemical group 0.000 description 11
- 125000004429 atom Chemical group 0.000 description 11
- 239000002738 chelating agent Substances 0.000 description 11
- 229940125782 compound 2 Drugs 0.000 description 11
- 238000006116 polymerization reaction Methods 0.000 description 11
- 239000004576 sand Substances 0.000 description 11
- 229910052717 sulfur Inorganic materials 0.000 description 11
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 11
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 10
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 10
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 10
- 241001061127 Thione Species 0.000 description 10
- 229910052798 chalcogen Inorganic materials 0.000 description 10
- 150000001787 chalcogens Chemical class 0.000 description 10
- 150000004696 coordination complex Chemical class 0.000 description 10
- 238000005336 cracking Methods 0.000 description 10
- 238000011033 desalting Methods 0.000 description 10
- 238000009826 distribution Methods 0.000 description 10
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 10
- 229910052737 gold Inorganic materials 0.000 description 10
- 239000010931 gold Substances 0.000 description 10
- 238000009607 mammography Methods 0.000 description 10
- 230000036961 partial effect Effects 0.000 description 10
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 10
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Substances [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 10
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 9
- 230000000052 comparative effect Effects 0.000 description 9
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 9
- 238000001035 drying Methods 0.000 description 9
- 238000001914 filtration Methods 0.000 description 9
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 9
- 230000005070 ripening Effects 0.000 description 9
- 239000007962 solid dispersion Substances 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- ZGOQRUPIKZGTLQ-UHFFFAOYSA-N 1,2-benzothiazole 1-oxide;sodium Chemical compound [Na].C1=CC=C2S(=O)N=CC2=C1 ZGOQRUPIKZGTLQ-UHFFFAOYSA-N 0.000 description 8
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 8
- 239000000654 additive Substances 0.000 description 8
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium peroxydisulfate Substances [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 8
- VAZSKTXWXKYQJF-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)OOS([O-])=O VAZSKTXWXKYQJF-UHFFFAOYSA-N 0.000 description 8
- 229910001870 ammonium persulfate Inorganic materials 0.000 description 8
- 229910052801 chlorine Inorganic materials 0.000 description 8
- 125000001309 chloro group Chemical group Cl* 0.000 description 8
- 238000007334 copolymerization reaction Methods 0.000 description 8
- 239000013078 crystal Substances 0.000 description 8
- 239000000428 dust Substances 0.000 description 8
- 230000003028 elevating effect Effects 0.000 description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- 150000002500 ions Chemical class 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 239000006224 matting agent Substances 0.000 description 8
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 8
- 230000003287 optical effect Effects 0.000 description 8
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical group C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 8
- XSCHRSMBECNVNS-UHFFFAOYSA-N quinoxaline Chemical compound N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 8
- ADZWSOLPGZMUMY-UHFFFAOYSA-M silver bromide Chemical compound [Ag]Br ADZWSOLPGZMUMY-UHFFFAOYSA-M 0.000 description 8
- 238000002834 transmittance Methods 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 7
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 7
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 7
- 239000004743 Polypropylene Substances 0.000 description 7
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 238000005259 measurement Methods 0.000 description 7
- 229910044991 metal oxide Inorganic materials 0.000 description 7
- 150000004706 metal oxides Chemical class 0.000 description 7
- 239000003960 organic solvent Substances 0.000 description 7
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 7
- 239000000049 pigment Substances 0.000 description 7
- 229920001155 polypropylene Polymers 0.000 description 7
- 239000011148 porous material Substances 0.000 description 7
- 238000012545 processing Methods 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 238000006722 reduction reaction Methods 0.000 description 7
- 229910052711 selenium Inorganic materials 0.000 description 7
- CVYDEWKUJFCYJO-UHFFFAOYSA-M sodium;docosanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCCCCCC([O-])=O CVYDEWKUJFCYJO-UHFFFAOYSA-M 0.000 description 7
- 239000011593 sulfur Substances 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical group C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 6
- YAJYJWXEWKRTPO-UHFFFAOYSA-N 2,3,3,4,4,5-hexamethylhexane-2-thiol Chemical compound CC(C)C(C)(C)C(C)(C)C(C)(C)S YAJYJWXEWKRTPO-UHFFFAOYSA-N 0.000 description 6
- DQYSALLXMHVJAV-UHFFFAOYSA-M 3-heptyl-2-[(3-heptyl-4-methyl-1,3-thiazol-3-ium-2-yl)methylidene]-4-methyl-1,3-thiazole;iodide Chemical compound [I-].CCCCCCCN1C(C)=CS\C1=C\C1=[N+](CCCCCCC)C(C)=CS1 DQYSALLXMHVJAV-UHFFFAOYSA-M 0.000 description 6
- IMROMDMJAWUWLK-UHFFFAOYSA-N Ethenol Chemical compound OC=C IMROMDMJAWUWLK-UHFFFAOYSA-N 0.000 description 6
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 6
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 6
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 6
- 125000003342 alkenyl group Chemical group 0.000 description 6
- 125000000304 alkynyl group Chemical group 0.000 description 6
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 6
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 6
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 6
- 235000019441 ethanol Nutrition 0.000 description 6
- 230000031700 light absorption Effects 0.000 description 6
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 6
- 229920000139 polyethylene terephthalate Polymers 0.000 description 6
- 239000005020 polyethylene terephthalate Substances 0.000 description 6
- 150000003378 silver Chemical class 0.000 description 6
- 229920003048 styrene butadiene rubber Polymers 0.000 description 6
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 6
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 5
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 5
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 5
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 5
- FOIXSVOLVBLSDH-UHFFFAOYSA-N Silver ion Chemical compound [Ag+] FOIXSVOLVBLSDH-UHFFFAOYSA-N 0.000 description 5
- 229910052771 Terbium Inorganic materials 0.000 description 5
- 125000001931 aliphatic group Chemical group 0.000 description 5
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 5
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 5
- 239000003945 anionic surfactant Substances 0.000 description 5
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 238000009835 boiling Methods 0.000 description 5
- 239000000084 colloidal system Substances 0.000 description 5
- 238000004040 coloring Methods 0.000 description 5
- 230000001276 controlling effect Effects 0.000 description 5
- 229910001873 dinitrogen Inorganic materials 0.000 description 5
- 239000003995 emulsifying agent Substances 0.000 description 5
- 229910052731 fluorine Inorganic materials 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 150000002739 metals Chemical class 0.000 description 5
- 125000005740 oxycarbonyl group Chemical group [*:1]OC([*:2])=O 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 229910052698 phosphorus Inorganic materials 0.000 description 5
- 239000004014 plasticizer Substances 0.000 description 5
- 239000003505 polymerization initiator Substances 0.000 description 5
- 235000013824 polyphenols Nutrition 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000002243 precursor Substances 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 239000011669 selenium Substances 0.000 description 5
- NHQVTOYJPBRYNG-UHFFFAOYSA-M sodium;2,4,7-tri(propan-2-yl)naphthalene-1-sulfonate Chemical compound [Na+].CC(C)C1=CC(C(C)C)=C(S([O-])(=O)=O)C2=CC(C(C)C)=CC=C21 NHQVTOYJPBRYNG-UHFFFAOYSA-M 0.000 description 5
- 238000001179 sorption measurement Methods 0.000 description 5
- 230000003068 static effect Effects 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- MRHCHKRKUVXUGE-UHFFFAOYSA-N 1-methyl-3-[2-(5-sulfanylidene-2h-tetrazol-1-yl)phenyl]urea Chemical compound CNC(=O)NC1=CC=CC=C1N1C(=S)N=NN1 MRHCHKRKUVXUGE-UHFFFAOYSA-N 0.000 description 4
- AFBBKYQYNPNMAT-UHFFFAOYSA-N 1h-1,2,4-triazol-1-ium-3-thiolate Chemical group SC=1N=CNN=1 AFBBKYQYNPNMAT-UHFFFAOYSA-N 0.000 description 4
- JAAIPIWKKXCNOC-UHFFFAOYSA-N 1h-tetrazol-1-ium-5-thiolate Chemical group SC1=NN=NN1 JAAIPIWKKXCNOC-UHFFFAOYSA-N 0.000 description 4
- PDHFSBXFZGYBIP-UHFFFAOYSA-N 2-[2-(2-hydroxyethylsulfanyl)ethylsulfanyl]ethanol Chemical compound OCCSCCSCCO PDHFSBXFZGYBIP-UHFFFAOYSA-N 0.000 description 4
- CWJJAFQCTXFSTA-UHFFFAOYSA-N 4-methylphthalic acid Chemical compound CC1=CC=C(C(O)=O)C(C(O)=O)=C1 CWJJAFQCTXFSTA-UHFFFAOYSA-N 0.000 description 4
- 235000021357 Behenic acid Nutrition 0.000 description 4
- 229910004829 CaWO4 Inorganic materials 0.000 description 4
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Substances CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- 229910021607 Silver chloride Inorganic materials 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 229910052775 Thulium Inorganic materials 0.000 description 4
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 4
- 235000010724 Wisteria floribunda Nutrition 0.000 description 4
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 4
- 229940116226 behenic acid Drugs 0.000 description 4
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 4
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical group C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 4
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 4
- 238000004061 bleaching Methods 0.000 description 4
- 238000010504 bond cleavage reaction Methods 0.000 description 4
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 150000001768 cations Chemical class 0.000 description 4
- UOCJDOLVGGIYIQ-PBFPGSCMSA-N cefatrizine Chemical group S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)[C@H](N)C=2C=CC(O)=CC=2)CC=1CSC=1C=NNN=1 UOCJDOLVGGIYIQ-PBFPGSCMSA-N 0.000 description 4
- 235000019646 color tone Nutrition 0.000 description 4
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 238000002059 diagnostic imaging Methods 0.000 description 4
- DOIRQSBPFJWKBE-UHFFFAOYSA-N dibutyl phthalate Chemical compound CCCCOC(=O)C1=CC=CC=C1C(=O)OCCCC DOIRQSBPFJWKBE-UHFFFAOYSA-N 0.000 description 4
- FLKPEMZONWLCSK-UHFFFAOYSA-N diethyl phthalate Chemical compound CCOC(=O)C1=CC=CC=C1C(=O)OCC FLKPEMZONWLCSK-UHFFFAOYSA-N 0.000 description 4
- 238000007865 diluting Methods 0.000 description 4
- 239000002612 dispersion medium Substances 0.000 description 4
- 238000010494 dissociation reaction Methods 0.000 description 4
- 230000005593 dissociations Effects 0.000 description 4
- 238000007720 emulsion polymerization reaction Methods 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 150000002367 halogens Chemical class 0.000 description 4
- 229910001385 heavy metal Inorganic materials 0.000 description 4
- 230000036571 hydration Effects 0.000 description 4
- 238000006703 hydration reaction Methods 0.000 description 4
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 4
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical group C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 4
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical group C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 125000002971 oxazolyl group Chemical group 0.000 description 4
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 4
- 150000002978 peroxides Chemical class 0.000 description 4
- 239000011574 phosphorus Substances 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 229920000728 polyester Polymers 0.000 description 4
- 239000004848 polyfunctional curative Substances 0.000 description 4
- 229910052700 potassium Inorganic materials 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 4
- ZNNZYHKDIALBAK-UHFFFAOYSA-M potassium thiocyanate Chemical compound [K+].[S-]C#N ZNNZYHKDIALBAK-UHFFFAOYSA-M 0.000 description 4
- 229940116357 potassium thiocyanate Drugs 0.000 description 4
- 238000001556 precipitation Methods 0.000 description 4
- 230000005855 radiation Effects 0.000 description 4
- 229910052761 rare earth metal Inorganic materials 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- AQRYNYUOKMNDDV-UHFFFAOYSA-M silver behenate Chemical compound [Ag+].CCCCCCCCCCCCCCCCCCCCCC([O-])=O AQRYNYUOKMNDDV-UHFFFAOYSA-M 0.000 description 4
- HKZLPVFGJNLROG-UHFFFAOYSA-M silver monochloride Chemical compound [Cl-].[Ag+] HKZLPVFGJNLROG-UHFFFAOYSA-M 0.000 description 4
- 238000007767 slide coating Methods 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 229910001220 stainless steel Inorganic materials 0.000 description 4
- 239000010935 stainless steel Substances 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- 150000003464 sulfur compounds Chemical class 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- FEJQDYXPAQVBCA-UHFFFAOYSA-J tetrasodium;ethane-1,2-diamine;tetraacetate Chemical compound [Na+].[Na+].[Na+].[Na+].CC([O-])=O.CC([O-])=O.CC([O-])=O.CC([O-])=O.NCCN FEJQDYXPAQVBCA-UHFFFAOYSA-J 0.000 description 4
- 150000003536 tetrazoles Chemical group 0.000 description 4
- 150000003567 thiocyanates Chemical class 0.000 description 4
- 125000000101 thioether group Chemical group 0.000 description 4
- 150000003585 thioureas Chemical class 0.000 description 4
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 4
- JOYRKODLDBILNP-UHFFFAOYSA-N urethane group Chemical group NC(=O)OCC JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 4
- 229920002818 (Hydroxyethyl)methacrylate Polymers 0.000 description 3
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical group C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 description 3
- WYENVTYBQKCILL-UHFFFAOYSA-N 1,2,4-triazolidine-3,5-dithione Chemical group S=C1NNC(=S)N1 WYENVTYBQKCILL-UHFFFAOYSA-N 0.000 description 3
- 229940116368 1,2-benzisothiazoline-3-one Drugs 0.000 description 3
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 3
- TUQAKXMNDMTCFO-UHFFFAOYSA-N 3-heptyl-4-phenyl-1h-1,2,4-triazole-5-thione Chemical compound CCCCCCCC1=NNC(=S)N1C1=CC=CC=C1 TUQAKXMNDMTCFO-UHFFFAOYSA-N 0.000 description 3
- CWIYBOJLSWJGKV-UHFFFAOYSA-N 5-methyl-1,3-dihydrobenzimidazole-2-thione Chemical compound CC1=CC=C2NC(S)=NC2=C1 CWIYBOJLSWJGKV-UHFFFAOYSA-N 0.000 description 3
- OVBJAABCEPSUNB-UHFFFAOYSA-N 6-propan-2-ylphthalazine Chemical compound C1=NN=CC2=CC(C(C)C)=CC=C21 OVBJAABCEPSUNB-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical group N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 229920002126 Acrylic acid copolymer Polymers 0.000 description 3
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 3
- 229930185605 Bisphenol Natural products 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 229910052688 Gadolinium Inorganic materials 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 239000002174 Styrene-butadiene Substances 0.000 description 3
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 3
- 238000000862 absorption spectrum Methods 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 150000001340 alkali metals Chemical class 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 125000000656 azaniumyl group Chemical group [H][N+]([H])([H])[*] 0.000 description 3
- DMSMPAJRVJJAGA-UHFFFAOYSA-N benzo[d]isothiazol-3-one Chemical compound C1=CC=C2C(=O)NSC2=C1 DMSMPAJRVJJAGA-UHFFFAOYSA-N 0.000 description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 3
- 229910052793 cadmium Inorganic materials 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 239000012986 chain transfer agent Substances 0.000 description 3
- 239000013522 chelant Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000003851 corona treatment Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000007547 defect Effects 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- FOBPTJZYDGNHLR-UHFFFAOYSA-N diphosphorus Chemical compound P#P FOBPTJZYDGNHLR-UHFFFAOYSA-N 0.000 description 3
- 150000002019 disulfides Chemical class 0.000 description 3
- JPIIVHIVGGOMMV-UHFFFAOYSA-N ditellurium Chemical compound [Te]=[Te] JPIIVHIVGGOMMV-UHFFFAOYSA-N 0.000 description 3
- 238000010556 emulsion polymerization method Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 3
- 238000007765 extrusion coating Methods 0.000 description 3
- 239000004744 fabric Substances 0.000 description 3
- 239000003925 fat Substances 0.000 description 3
- 238000011049 filling Methods 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 3
- 150000002391 heterocyclic compounds Chemical class 0.000 description 3
- 150000002429 hydrazines Chemical class 0.000 description 3
- 238000003384 imaging method Methods 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 125000005462 imide group Chemical group 0.000 description 3
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 229910021645 metal ion Inorganic materials 0.000 description 3
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 description 3
- 239000012046 mixed solvent Substances 0.000 description 3
- RODAXCQJQDMNSH-UHFFFAOYSA-N n-[4-(diethylamino)-6-(hydroxyamino)-1,3,5-triazin-2-yl]hydroxylamine Chemical compound CCN(CC)C1=NC(NO)=NC(NO)=N1 RODAXCQJQDMNSH-UHFFFAOYSA-N 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 3
- 150000002989 phenols Chemical class 0.000 description 3
- 235000011007 phosphoric acid Nutrition 0.000 description 3
- 150000003022 phthalic acids Chemical class 0.000 description 3
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 3
- 229920002635 polyurethane Polymers 0.000 description 3
- 239000004814 polyurethane Substances 0.000 description 3
- 238000000634 powder X-ray diffraction Methods 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229940065287 selenium compound Drugs 0.000 description 3
- 150000003343 selenium compounds Chemical class 0.000 description 3
- 150000003346 selenoethers Chemical class 0.000 description 3
- DUIOPKIIICUYRZ-UHFFFAOYSA-N semicarbazide Chemical class NNC(N)=O DUIOPKIIICUYRZ-UHFFFAOYSA-N 0.000 description 3
- 239000010944 silver (metal) Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 3
- 229910001415 sodium ion Inorganic materials 0.000 description 3
- BZHOWMPPNDKQSQ-UHFFFAOYSA-M sodium;sulfidosulfonylbenzene Chemical compound [Na+].[O-]S(=O)(=S)C1=CC=CC=C1 BZHOWMPPNDKQSQ-UHFFFAOYSA-M 0.000 description 3
- 125000003375 sulfoxide group Chemical group 0.000 description 3
- 229920003002 synthetic resin Polymers 0.000 description 3
- 239000000057 synthetic resin Substances 0.000 description 3
- GZCRRIHWUXGPOV-UHFFFAOYSA-N terbium atom Chemical compound [Tb] GZCRRIHWUXGPOV-UHFFFAOYSA-N 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 150000003852 triazoles Chemical group 0.000 description 3
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 3
- 229910052721 tungsten Inorganic materials 0.000 description 3
- 239000010937 tungsten Substances 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- LKNKAEWGISYACD-ONEGZZNKSA-N (1e)-1-bromobuta-1,3-diene Chemical compound Br\C=C\C=C LKNKAEWGISYACD-ONEGZZNKSA-N 0.000 description 2
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 2
- APXGHAWHVMPQBB-UHFFFAOYSA-N (hydroxyamino)urea Chemical class NC(=O)NNO APXGHAWHVMPQBB-UHFFFAOYSA-N 0.000 description 2
- QPFMBZIOSGYJDE-UHFFFAOYSA-N 1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)C(Cl)Cl QPFMBZIOSGYJDE-UHFFFAOYSA-N 0.000 description 2
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 description 2
- BBVIDBNAYOIXOE-UHFFFAOYSA-N 1,2,4-oxadiazole Chemical group C=1N=CON=1 BBVIDBNAYOIXOE-UHFFFAOYSA-N 0.000 description 2
- YGTAZGSLCXNBQL-UHFFFAOYSA-N 1,2,4-thiadiazole Chemical group C=1N=CSN=1 YGTAZGSLCXNBQL-UHFFFAOYSA-N 0.000 description 2
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 2
- KANAPVJGZDNSCZ-UHFFFAOYSA-N 1,2-benzothiazole 1-oxide Chemical compound C1=CC=C2S(=O)N=CC2=C1 KANAPVJGZDNSCZ-UHFFFAOYSA-N 0.000 description 2
- FKASFBLJDCHBNZ-UHFFFAOYSA-N 1,3,4-oxadiazole Chemical group C1=NN=CO1 FKASFBLJDCHBNZ-UHFFFAOYSA-N 0.000 description 2
- MBIZXFATKUQOOA-UHFFFAOYSA-N 1,3,4-thiadiazole Chemical group C1=NN=CS1 MBIZXFATKUQOOA-UHFFFAOYSA-N 0.000 description 2
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical compound C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 description 2
- WUIJCMJIYQWIMF-UHFFFAOYSA-N 1,3-benzothiazole;hydroiodide Chemical compound [I-].C1=CC=C2SC=[NH+]C2=C1 WUIJCMJIYQWIMF-UHFFFAOYSA-N 0.000 description 2
- 125000000355 1,3-benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- YHMYGUUIMTVXNW-UHFFFAOYSA-N 1,3-dihydrobenzimidazole-2-thione Chemical group C1=CC=C2NC(S)=NC2=C1 YHMYGUUIMTVXNW-UHFFFAOYSA-N 0.000 description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N 1-ethenoxybutane Chemical compound CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 2
- 125000006219 1-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- ZEQIWKHCJWRNTH-UHFFFAOYSA-N 1h-pyrimidine-2,4-dithione Chemical group S=C1C=CNC(=S)N1 ZEQIWKHCJWRNTH-UHFFFAOYSA-N 0.000 description 2
- SDJHPPZKZZWAKF-UHFFFAOYSA-N 2,3-dimethylbuta-1,3-diene Chemical compound CC(=C)C(C)=C SDJHPPZKZZWAKF-UHFFFAOYSA-N 0.000 description 2
- LCPVQAHEFVXVKT-UHFFFAOYSA-N 2-(2,4-difluorophenoxy)pyridin-3-amine Chemical compound NC1=CC=CN=C1OC1=CC=C(F)C=C1F LCPVQAHEFVXVKT-UHFFFAOYSA-N 0.000 description 2
- GOXQRTZXKQZDDN-UHFFFAOYSA-N 2-Ethylhexyl acrylate Chemical compound CCCCC(CC)COC(=O)C=C GOXQRTZXKQZDDN-UHFFFAOYSA-N 0.000 description 2
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical class C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 description 2
- KUDUQBURMYMBIJ-UHFFFAOYSA-N 2-prop-2-enoyloxyethyl prop-2-enoate Chemical compound C=CC(=O)OCCOC(=O)C=C KUDUQBURMYMBIJ-UHFFFAOYSA-N 0.000 description 2
- YNPFKIFRNDNSCG-UHFFFAOYSA-N 2-sulfanyl-1,3-dihydrotriazine-4-thione Chemical group SN1NC=CC(=S)N1 YNPFKIFRNDNSCG-UHFFFAOYSA-N 0.000 description 2
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 2
- RUBRCWOFANAOTP-UHFFFAOYSA-N 3h-1,3,4-oxadiazole-2-thione Chemical group S=C1NN=CO1 RUBRCWOFANAOTP-UHFFFAOYSA-N 0.000 description 2
- 150000000660 7-membered heterocyclic compounds Chemical class 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- SOGAXMICEFXMKE-UHFFFAOYSA-N Butylmethacrylate Chemical compound CCCCOC(=O)C(C)=C SOGAXMICEFXMKE-UHFFFAOYSA-N 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical group C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 2
- 229910052693 Europium Inorganic materials 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000004952 Polyamide Substances 0.000 description 2
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 2
- YSMRWXYRXBRSND-UHFFFAOYSA-N TOTP Chemical compound CC1=CC=CC=C1OP(=O)(OC=1C(=CC=CC=1)C)OC1=CC=CC=C1C YSMRWXYRXBRSND-UHFFFAOYSA-N 0.000 description 2
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- XEIPQVVAVOUIOP-UHFFFAOYSA-N [Au]=S Chemical compound [Au]=S XEIPQVVAVOUIOP-UHFFFAOYSA-N 0.000 description 2
- KWEGYAQDWBZXMX-UHFFFAOYSA-N [Au]=[Se] Chemical compound [Au]=[Se] KWEGYAQDWBZXMX-UHFFFAOYSA-N 0.000 description 2
- IBQKNIQGYSISEM-UHFFFAOYSA-N [Se]=[PH3] Chemical class [Se]=[PH3] IBQKNIQGYSISEM-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 229940048053 acrylate Drugs 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 2
- 125000003282 alkyl amino group Chemical group 0.000 description 2
- 125000004422 alkyl sulphonamide group Chemical group 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 230000002421 anti-septic effect Effects 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 230000003078 antioxidant effect Effects 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 2
- 125000005251 aryl acyl group Chemical group 0.000 description 2
- 125000001769 aryl amino group Chemical group 0.000 description 2
- 125000005162 aryl oxy carbonyl amino group Chemical group 0.000 description 2
- 125000004421 aryl sulphonamide group Chemical group 0.000 description 2
- 125000000732 arylene group Chemical group 0.000 description 2
- 239000007869 azo polymerization initiator Substances 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 2
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 125000005521 carbonamide group Chemical group 0.000 description 2
- 125000000473 carbonimidoyl group Chemical group [H]\N=C(/*)* 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- OIDPCXKPHYRNKH-UHFFFAOYSA-J chrome alum Chemical compound [K]OS(=O)(=O)O[Cr]1OS(=O)(=O)O1 OIDPCXKPHYRNKH-UHFFFAOYSA-J 0.000 description 2
- 239000011651 chromium Substances 0.000 description 2
- WCZVZNOTHYJIEI-UHFFFAOYSA-N cinnoline Chemical compound N1=NC=CC2=CC=CC=C21 WCZVZNOTHYJIEI-UHFFFAOYSA-N 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 125000002933 cyclohexyloxy group Chemical group C1(CCCCC1)O* 0.000 description 2
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 2
- 238000004042 decolorization Methods 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- 229960002380 dibutyl phthalate Drugs 0.000 description 2
- KZTYYGOKRVBIMI-UHFFFAOYSA-N diphenyl sulfone Chemical compound C=1C=CC=CC=1S(=O)(=O)C1=CC=CC=C1 KZTYYGOKRVBIMI-UHFFFAOYSA-N 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 125000002228 disulfide group Chemical group 0.000 description 2
- AFOSIXZFDONLBT-UHFFFAOYSA-N divinyl sulfone Chemical compound C=CS(=O)(=O)C=C AFOSIXZFDONLBT-UHFFFAOYSA-N 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 2
- 239000010946 fine silver Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 239000001087 glyceryl triacetate Substances 0.000 description 2
- 235000013773 glyceryl triacetate Nutrition 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- 229920001477 hydrophilic polymer Polymers 0.000 description 2
- 150000002443 hydroxylamines Chemical class 0.000 description 2
- VKOBVWXKNCXXDE-UHFFFAOYSA-N icosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCC(O)=O VKOBVWXKNCXXDE-UHFFFAOYSA-N 0.000 description 2
- 235000019239 indanthrene blue RS Nutrition 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 229910052741 iridium Inorganic materials 0.000 description 2
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 229940057995 liquid paraffin Drugs 0.000 description 2
- 229910001416 lithium ion Inorganic materials 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- MGFYIUFZLHCRTH-UHFFFAOYSA-N nitrilotriacetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)=O MGFYIUFZLHCRTH-UHFFFAOYSA-N 0.000 description 2
- 229910000510 noble metal Inorganic materials 0.000 description 2
- 150000002896 organic halogen compounds Chemical class 0.000 description 2
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical group C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 2
- 239000005022 packaging material Substances 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical class [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- 125000006678 phenoxycarbonyl group Chemical group 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- IJAPPYDYQCXOEF-UHFFFAOYSA-N phthalazin-1(2H)-one Chemical class C1=CC=C2C(=O)NN=CC2=C1 IJAPPYDYQCXOEF-UHFFFAOYSA-N 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 229920002647 polyamide Polymers 0.000 description 2
- 229920000768 polyamine Chemical class 0.000 description 2
- 229920000647 polyepoxide Polymers 0.000 description 2
- 239000004926 polymethyl methacrylate Substances 0.000 description 2
- 229920000098 polyolefin Polymers 0.000 description 2
- 150000008442 polyphenolic compounds Chemical class 0.000 description 2
- 229920001451 polypropylene glycol Polymers 0.000 description 2
- USHAGKDGDHPEEY-UHFFFAOYSA-L potassium persulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OOS([O-])(=O)=O USHAGKDGDHPEEY-UHFFFAOYSA-L 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- NDGRWYRVNANFNB-UHFFFAOYSA-N pyrazolidin-3-one Chemical class O=C1CCNN1 NDGRWYRVNANFNB-UHFFFAOYSA-N 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 2
- 150000005839 radical cations Chemical class 0.000 description 2
- 238000002601 radiography Methods 0.000 description 2
- 229920005604 random copolymer Polymers 0.000 description 2
- 150000002910 rare earth metals Chemical class 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000006479 redox reaction Methods 0.000 description 2
- 239000010948 rhodium Substances 0.000 description 2
- 239000005060 rubber Substances 0.000 description 2
- CRDYSYOERSZTHZ-UHFFFAOYSA-N selenocyanic acid Chemical compound [SeH]C#N CRDYSYOERSZTHZ-UHFFFAOYSA-N 0.000 description 2
- MSFPLIAKTHOCQP-UHFFFAOYSA-M silver iodide Chemical group I[Ag] MSFPLIAKTHOCQP-UHFFFAOYSA-M 0.000 description 2
- GCLGEJMYGQKIIW-UHFFFAOYSA-H sodium hexametaphosphate Chemical compound [Na]OP1(=O)OP(=O)(O[Na])OP(=O)(O[Na])OP(=O)(O[Na])OP(=O)(O[Na])OP(=O)(O[Na])O1 GCLGEJMYGQKIIW-UHFFFAOYSA-H 0.000 description 2
- 235000019982 sodium hexametaphosphate Nutrition 0.000 description 2
- CHQMHPLRPQMAMX-UHFFFAOYSA-L sodium persulfate Substances [Na+].[Na+].[O-]S(=O)(=O)OOS([O-])(=O)=O CHQMHPLRPQMAMX-UHFFFAOYSA-L 0.000 description 2
- XFTALRAZSCGSKN-UHFFFAOYSA-M sodium;4-ethenylbenzenesulfonate Chemical compound [Na+].[O-]S(=O)(=O)C1=CC=C(C=C)C=C1 XFTALRAZSCGSKN-UHFFFAOYSA-M 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 229920006132 styrene block copolymer Polymers 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- XSOKHXFFCGXDJZ-UHFFFAOYSA-N telluride(2-) Chemical compound [Te-2] XSOKHXFFCGXDJZ-UHFFFAOYSA-N 0.000 description 2
- 150000004772 tellurides Chemical class 0.000 description 2
- 150000003498 tellurium compounds Chemical class 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 239000001577 tetrasodium phosphonato phosphate Substances 0.000 description 2
- 229920002725 thermoplastic elastomer Polymers 0.000 description 2
- 150000004867 thiadiazoles Chemical group 0.000 description 2
- 150000003568 thioethers Chemical class 0.000 description 2
- 150000004764 thiosulfuric acid derivatives Chemical class 0.000 description 2
- 229960002622 triacetin Drugs 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 239000003021 water soluble solvent Substances 0.000 description 2
- 229910052984 zinc sulfide Inorganic materials 0.000 description 2
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical class C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 1
- PCPYTNCQOSFKGG-ONEGZZNKSA-N (1e)-1-chlorobuta-1,3-diene Chemical compound Cl\C=C\C=C PCPYTNCQOSFKGG-ONEGZZNKSA-N 0.000 description 1
- UZIQZDOUNBTWLH-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl)-phenyl-(2-phenylphenyl)-selanylidene-lambda5-phosphane Chemical compound FC1=C(C(=C(C(=C1P(C1=C(C=CC=C1)C1=CC=CC=C1)(C1=CC=CC=C1)=[Se])F)F)F)F UZIQZDOUNBTWLH-UHFFFAOYSA-N 0.000 description 1
- WAVDSLLYAQBITE-UHFFFAOYSA-N (4-ethenylphenyl)methanamine Chemical compound NCC1=CC=C(C=C)C=C1 WAVDSLLYAQBITE-UHFFFAOYSA-N 0.000 description 1
- CLECMSNCZUMKLM-UHFFFAOYSA-N (4-ethenylphenyl)methanol Chemical compound OCC1=CC=C(C=C)C=C1 CLECMSNCZUMKLM-UHFFFAOYSA-N 0.000 description 1
- PMJHHCWVYXUKFD-SNAWJCMRSA-N (E)-1,3-pentadiene Chemical compound C\C=C\C=C PMJHHCWVYXUKFD-SNAWJCMRSA-N 0.000 description 1
- ZFBGKBGUMMBBMY-UHFFFAOYSA-N 1,1,2-trichlorobuta-1,3-diene Chemical compound ClC(Cl)=C(Cl)C=C ZFBGKBGUMMBBMY-UHFFFAOYSA-N 0.000 description 1
- HXMRAWVFMYZQMG-UHFFFAOYSA-N 1,1,3-triethylthiourea Chemical compound CCNC(=S)N(CC)CC HXMRAWVFMYZQMG-UHFFFAOYSA-N 0.000 description 1
- KTRQRAQRHBLCSQ-UHFFFAOYSA-N 1,2,4-tris(ethenyl)cyclohexane Chemical compound C=CC1CCC(C=C)C(C=C)C1 KTRQRAQRHBLCSQ-UHFFFAOYSA-N 0.000 description 1
- UDGKZGLPXCRRAM-UHFFFAOYSA-N 1,2,5-thiadiazole Chemical group C=1C=NSN=1 UDGKZGLPXCRRAM-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical class ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- 150000005206 1,2-dihydroxybenzenes Chemical class 0.000 description 1
- 125000003363 1,3,5-triazinyl group Chemical group N1=C(N=CN=C1)* 0.000 description 1
- AIGNCQCMONAWOL-UHFFFAOYSA-N 1,3-benzoselenazole Chemical group C1=CC=C2[se]C=NC2=C1 AIGNCQCMONAWOL-UHFFFAOYSA-N 0.000 description 1
- WKKIRKUKAAAUNL-UHFFFAOYSA-N 1,3-benzotellurazole Chemical group C1=CC=C2[Te]C=NC2=C1 WKKIRKUKAAAUNL-UHFFFAOYSA-N 0.000 description 1
- YXIWHUQXZSMYRE-UHFFFAOYSA-N 1,3-benzothiazole-2-thiol Chemical group C1=CC=C2SC(S)=NC2=C1 YXIWHUQXZSMYRE-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- 150000005207 1,3-dihydroxybenzenes Chemical class 0.000 description 1
- PYWQACMPJZLKOQ-UHFFFAOYSA-N 1,3-tellurazole Chemical group [Te]1C=CN=C1 PYWQACMPJZLKOQ-UHFFFAOYSA-N 0.000 description 1
- WEERVPDNCOGWJF-UHFFFAOYSA-N 1,4-bis(ethenyl)benzene Chemical compound C=CC1=CC=C(C=C)C=C1 WEERVPDNCOGWJF-UHFFFAOYSA-N 0.000 description 1
- QIABNQHWGHBJEE-UHFFFAOYSA-N 1,4-bis(ethenyl)cyclohexane Chemical compound C=CC1CCC(C=C)CC1 QIABNQHWGHBJEE-UHFFFAOYSA-N 0.000 description 1
- 150000005208 1,4-dihydroxybenzenes Chemical class 0.000 description 1
- WSAIKWBIEKCYFN-UHFFFAOYSA-N 1,5-dimethyl-1h-1,2,4-triazol-1-ium-3-thiolate Chemical group CC1=NC(S)=NN1C WSAIKWBIEKCYFN-UHFFFAOYSA-N 0.000 description 1
- PGRNEGLBSNLPNP-UHFFFAOYSA-N 1,6-dichloro-3-methylhex-1-ene Chemical compound ClC=CC(C)CCCCl PGRNEGLBSNLPNP-UHFFFAOYSA-N 0.000 description 1
- RVXJIYJPQXRIEM-UHFFFAOYSA-N 1-$l^{1}-selanyl-n,n-dimethylmethanimidamide Chemical compound CN(C)C([Se])=N RVXJIYJPQXRIEM-UHFFFAOYSA-N 0.000 description 1
- PQUXFUBNSYCQAL-UHFFFAOYSA-N 1-(2,3-difluorophenyl)ethanone Chemical compound CC(=O)C1=CC=CC(F)=C1F PQUXFUBNSYCQAL-UHFFFAOYSA-N 0.000 description 1
- PSIFIJBZVPUWTO-UHFFFAOYSA-N 1-(4-chlorophenyl)-2-[2-(4-chlorophenyl)phenyl]sulfonylbenzene Chemical compound C1=CC(Cl)=CC=C1C1=CC=CC=C1S(=O)(=O)C1=CC=CC=C1C1=CC=C(Cl)C=C1 PSIFIJBZVPUWTO-UHFFFAOYSA-N 0.000 description 1
- ZRZHXNCATOYMJH-UHFFFAOYSA-N 1-(chloromethyl)-4-ethenylbenzene Chemical compound ClCC1=CC=C(C=C)C=C1 ZRZHXNCATOYMJH-UHFFFAOYSA-N 0.000 description 1
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical group C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 1
- PNWKMUUTDFAROK-UHFFFAOYSA-N 1-bis(4-tert-butylphenyl)phosphoryl-4-tert-butylbenzene Chemical compound C1=CC(C(C)(C)C)=CC=C1P(=O)(C=1C=CC(=CC=1)C(C)(C)C)C1=CC=C(C(C)(C)C)C=C1 PNWKMUUTDFAROK-UHFFFAOYSA-N 0.000 description 1
- QTKWLGPILKQVJB-UHFFFAOYSA-N 1-buta-1,3-dienylnaphthalene Chemical compound C1=CC=C2C(C=CC=C)=CC=CC2=C1 QTKWLGPILKQVJB-UHFFFAOYSA-N 0.000 description 1
- GXZPMXGRNUXGHN-UHFFFAOYSA-N 1-ethenoxy-2-methoxyethane Chemical compound COCCOC=C GXZPMXGRNUXGHN-UHFFFAOYSA-N 0.000 description 1
- OSSNTDFYBPYIEC-UHFFFAOYSA-N 1-ethenylimidazole Chemical compound C=CN1C=CN=C1 OSSNTDFYBPYIEC-UHFFFAOYSA-N 0.000 description 1
- AFAKZSJEQYSXTB-UHFFFAOYSA-N 1-ethyl-3-(4-methyl-1,3-thiazol-2-yl)thiourea Chemical compound CCNC(=S)NC1=NC(C)=CS1 AFAKZSJEQYSXTB-UHFFFAOYSA-N 0.000 description 1
- 125000006432 1-methyl cyclopropyl group Chemical group [H]C([H])([H])C1(*)C([H])([H])C1([H])[H] 0.000 description 1
- XLPJNCYCZORXHG-UHFFFAOYSA-N 1-morpholin-4-ylprop-2-en-1-one Chemical compound C=CC(=O)N1CCOCC1 XLPJNCYCZORXHG-UHFFFAOYSA-N 0.000 description 1
- HDPWHFLTRDUOHM-UHFFFAOYSA-N 1-naphthalen-1-ylphthalazine Chemical compound C1=CC=C2C(C=3C4=CC=CC=C4C=CC=3)=NN=CC2=C1 HDPWHFLTRDUOHM-UHFFFAOYSA-N 0.000 description 1
- CKQAOGOZKZJUGA-UHFFFAOYSA-N 1-nonyl-4-(4-nonylphenoxy)benzene Chemical compound C1=CC(CCCCCCCCC)=CC=C1OC1=CC=C(CCCCCCCCC)C=C1 CKQAOGOZKZJUGA-UHFFFAOYSA-N 0.000 description 1
- QEDJMOONZLUIMC-UHFFFAOYSA-N 1-tert-butyl-4-ethenylbenzene Chemical compound CC(C)(C)C1=CC=C(C=C)C=C1 QEDJMOONZLUIMC-UHFFFAOYSA-N 0.000 description 1
- IGGDKDTUCAWDAN-UHFFFAOYSA-N 1-vinylnaphthalene Chemical compound C1=CC=C2C(C=C)=CC=CC2=C1 IGGDKDTUCAWDAN-UHFFFAOYSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- AAILEWXSEQLMNI-UHFFFAOYSA-N 1h-pyridazin-6-one Chemical compound OC1=CC=CN=N1 AAILEWXSEQLMNI-UHFFFAOYSA-N 0.000 description 1
- GKZPEYIPJQHPNC-UHFFFAOYSA-N 2,2-bis(hydroxymethyl)propane-1,3-diol prop-2-enoic acid Chemical compound OC(=O)C=C.OC(=O)C=C.OC(=O)C=C.OC(=O)C=C.OC(=O)C=C.OC(=O)C=C.OCC(CO)(CO)CO GKZPEYIPJQHPNC-UHFFFAOYSA-N 0.000 description 1
- LIFLRQVHKGGNSG-UHFFFAOYSA-N 2,3-dichlorobuta-1,3-diene Chemical compound ClC(=C)C(Cl)=C LIFLRQVHKGGNSG-UHFFFAOYSA-N 0.000 description 1
- JNYKOGUXPNAUIB-UHFFFAOYSA-N 2,3-dihydro-1-benzofuran-5-ol Chemical class OC1=CC=C2OCCC2=C1 JNYKOGUXPNAUIB-UHFFFAOYSA-N 0.000 description 1
- SEIZZTOCUDUQNV-UHFFFAOYSA-N 2,3-dihydrophthalazine Chemical compound C1=CC=CC2=CNNC=C21 SEIZZTOCUDUQNV-UHFFFAOYSA-N 0.000 description 1
- KGLPWQKSKUVKMJ-UHFFFAOYSA-N 2,3-dihydrophthalazine-1,4-dione Chemical compound C1=CC=C2C(=O)NNC(=O)C2=C1 KGLPWQKSKUVKMJ-UHFFFAOYSA-N 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- YKUDHBLDJYZZQS-UHFFFAOYSA-N 2,6-dichloro-1h-1,3,5-triazin-4-one Chemical compound OC1=NC(Cl)=NC(Cl)=N1 YKUDHBLDJYZZQS-UHFFFAOYSA-N 0.000 description 1
- LZDYZEGISBDSDP-UHFFFAOYSA-N 2-(1-ethylaziridin-1-ium-1-yl)ethanol Chemical compound OCC[N+]1(CC)CC1 LZDYZEGISBDSDP-UHFFFAOYSA-N 0.000 description 1
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- RUYYZQCJUGZUCW-UHFFFAOYSA-N 2-(prop-2-enoylamino)ethyl 3-oxobutanoate Chemical compound CC(=O)CC(=O)OCCNC(=O)C=C RUYYZQCJUGZUCW-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- ALIOIMRELZMHLE-UHFFFAOYSA-N 2-[(3-amino-3-oxopropyl)-(carboxymethyl)amino]acetic acid Chemical compound NC(=O)CCN(CC(O)=O)CC(O)=O ALIOIMRELZMHLE-UHFFFAOYSA-N 0.000 description 1
- URDCARMUOSMFFI-UHFFFAOYSA-N 2-[2-[bis(carboxymethyl)amino]ethyl-(2-hydroxyethyl)amino]acetic acid Chemical compound OCCN(CC(O)=O)CCN(CC(O)=O)CC(O)=O URDCARMUOSMFFI-UHFFFAOYSA-N 0.000 description 1
- SJBOEHIKNDEHHO-UHFFFAOYSA-N 2-[2-aminoethyl(carboxymethyl)amino]acetic acid Chemical compound NCCN(CC(O)=O)CC(O)=O SJBOEHIKNDEHHO-UHFFFAOYSA-N 0.000 description 1
- WZFUQSJFWNHZHM-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)N1CC2=C(CC1)NN=N2 WZFUQSJFWNHZHM-UHFFFAOYSA-N 0.000 description 1
- FCKYPQBAHLOOJQ-NXEZZACHSA-N 2-[[(1r,2r)-2-[bis(carboxymethyl)amino]cyclohexyl]-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)[C@@H]1CCCC[C@H]1N(CC(O)=O)CC(O)=O FCKYPQBAHLOOJQ-NXEZZACHSA-N 0.000 description 1
- WYMDDFRYORANCC-UHFFFAOYSA-N 2-[[3-[bis(carboxymethyl)amino]-2-hydroxypropyl]-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)CN(CC(O)=O)CC(O)=O WYMDDFRYORANCC-UHFFFAOYSA-N 0.000 description 1
- TXBCBTDQIULDIA-UHFFFAOYSA-N 2-[[3-hydroxy-2,2-bis(hydroxymethyl)propoxy]methyl]-2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(CO)(CO)COCC(CO)(CO)CO TXBCBTDQIULDIA-UHFFFAOYSA-N 0.000 description 1
- QHHFAXFIUXRVSI-UHFFFAOYSA-N 2-[carboxymethyl(ethyl)amino]acetic acid Chemical compound OC(=O)CN(CC)CC(O)=O QHHFAXFIUXRVSI-UHFFFAOYSA-N 0.000 description 1
- XWSGEVNYFYKXCP-UHFFFAOYSA-N 2-[carboxymethyl(methyl)amino]acetic acid Chemical compound OC(=O)CN(C)CC(O)=O XWSGEVNYFYKXCP-UHFFFAOYSA-N 0.000 description 1
- VIBPNYGNMMRDQG-UHFFFAOYSA-N 2-[dimethylcarbamothioyl(methyl)amino]acetic acid Chemical compound CN(C)C(=S)N(C)CC(O)=O VIBPNYGNMMRDQG-UHFFFAOYSA-N 0.000 description 1
- ZTJNPDLOIVDEEL-UHFFFAOYSA-N 2-acetyloxyethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOC(C)=O ZTJNPDLOIVDEEL-UHFFFAOYSA-N 0.000 description 1
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical class NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 1
- DADINJSQBPUGEI-UHFFFAOYSA-N 2-buta-1,3-dienylnaphthalene Chemical compound C1=CC=CC2=CC(C=CC=C)=CC=C21 DADINJSQBPUGEI-UHFFFAOYSA-N 0.000 description 1
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 description 1
- WHBAYNMEIXUTJV-UHFFFAOYSA-N 2-chloroethyl prop-2-enoate Chemical compound ClCCOC(=O)C=C WHBAYNMEIXUTJV-UHFFFAOYSA-N 0.000 description 1
- AEPWOCLBLLCOGZ-UHFFFAOYSA-N 2-cyanoethyl prop-2-enoate Chemical compound C=CC(=O)OCCC#N AEPWOCLBLLCOGZ-UHFFFAOYSA-N 0.000 description 1
- BQBSIHIZDSHADD-UHFFFAOYSA-N 2-ethenyl-4,5-dihydro-1,3-oxazole Chemical compound C=CC1=NCCO1 BQBSIHIZDSHADD-UHFFFAOYSA-N 0.000 description 1
- XUDBVJCTLZTSDC-UHFFFAOYSA-N 2-ethenylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1C=C XUDBVJCTLZTSDC-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- WDQMWEYDKDCEHT-UHFFFAOYSA-N 2-ethylhexyl 2-methylprop-2-enoate Chemical compound CCCCC(CC)COC(=O)C(C)=C WDQMWEYDKDCEHT-UHFFFAOYSA-N 0.000 description 1
- BQHQZFUAEAVJRE-UHFFFAOYSA-N 2-fluorobuta-1,3-diene Chemical compound FC(=C)C=C BQHQZFUAEAVJRE-UHFFFAOYSA-N 0.000 description 1
- FLFWJIBUZQARMD-UHFFFAOYSA-N 2-mercapto-1,3-benzoxazole Chemical group C1=CC=C2OC(S)=NC2=C1 FLFWJIBUZQARMD-UHFFFAOYSA-N 0.000 description 1
- YXYJVFYWCLAXHO-UHFFFAOYSA-N 2-methoxyethyl 2-methylprop-2-enoate Chemical compound COCCOC(=O)C(C)=C YXYJVFYWCLAXHO-UHFFFAOYSA-N 0.000 description 1
- UYJCRMOPNVBSNM-UHFFFAOYSA-N 2-methyl-1-[4-(2-methylprop-2-enoyl)piperazin-1-yl]prop-2-en-1-one Chemical compound CC(=C)C(=O)N1CCN(C(=O)C(C)=C)CC1 UYJCRMOPNVBSNM-UHFFFAOYSA-N 0.000 description 1
- WPFTVIVHDBZTSZ-UHFFFAOYSA-N 2-methyl-n-(2,4,4-trimethylpentan-2-yl)prop-2-enamide Chemical compound CC(=C)C(=O)NC(C)(C)CC(C)(C)C WPFTVIVHDBZTSZ-UHFFFAOYSA-N 0.000 description 1
- LEKIODFWYFCUER-UHFFFAOYSA-N 2-methylidenebut-3-enenitrile Chemical compound C=CC(=C)C#N LEKIODFWYFCUER-UHFFFAOYSA-N 0.000 description 1
- RKOOOVKGLHCLTP-UHFFFAOYSA-N 2-methylprop-2-enoic acid;propane-1,2,3-triol Chemical compound CC(=C)C(O)=O.OCC(O)CO RKOOOVKGLHCLTP-UHFFFAOYSA-N 0.000 description 1
- NDAJNMAAXXIADY-UHFFFAOYSA-N 2-methylpropanimidamide Chemical compound CC(C)C(N)=N NDAJNMAAXXIADY-UHFFFAOYSA-N 0.000 description 1
- NSMJMUQZRGZMQC-UHFFFAOYSA-N 2-naphthalen-1-yl-1H-imidazo[4,5-f][1,10]phenanthroline Chemical compound C12=CC=CN=C2C2=NC=CC=C2C2=C1NC(C=1C3=CC=CC=C3C=CC=1)=N2 NSMJMUQZRGZMQC-UHFFFAOYSA-N 0.000 description 1
- QCDWFXQBSFUVSP-UHFFFAOYSA-N 2-phenoxyethanol Chemical compound OCCOC1=CC=CC=C1 QCDWFXQBSFUVSP-UHFFFAOYSA-N 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- SCNKFUNWPYDBQX-UHFFFAOYSA-N 2-sulfanyl-3h-thiadiazol-5-amine Chemical group NC1=CNN(S)S1 SCNKFUNWPYDBQX-UHFFFAOYSA-N 0.000 description 1
- NBNQOWVYEXFQJC-UHFFFAOYSA-N 2-sulfanyl-3h-thiadiazole Chemical group SN1NC=CS1 NBNQOWVYEXFQJC-UHFFFAOYSA-N 0.000 description 1
- GCSVNNODDIEGEX-UHFFFAOYSA-N 2-sulfanylidene-1,3-oxazolidin-4-one Chemical class O=C1COC(=S)N1 GCSVNNODDIEGEX-UHFFFAOYSA-N 0.000 description 1
- UGWULZWUXSCWPX-UHFFFAOYSA-N 2-sulfanylideneimidazolidin-4-one Chemical compound O=C1CNC(=S)N1 UGWULZWUXSCWPX-UHFFFAOYSA-N 0.000 description 1
- GPNYZBKIGXGYNU-UHFFFAOYSA-N 2-tert-butyl-6-[(3-tert-butyl-5-ethyl-2-hydroxyphenyl)methyl]-4-ethylphenol Chemical compound CC(C)(C)C1=CC(CC)=CC(CC=2C(=C(C=C(CC)C=2)C(C)(C)C)O)=C1O GPNYZBKIGXGYNU-UHFFFAOYSA-N 0.000 description 1
- DHEOBQCWCKRUKJ-UHFFFAOYSA-N 2-tert-butyl-6-[1-(3-tert-butyl-2-hydroxyphenyl)pentyl]-4-methylphenol Chemical compound C=1C(C)=CC(C(C)(C)C)=C(O)C=1C(CCCC)C1=CC=CC(C(C)(C)C)=C1O DHEOBQCWCKRUKJ-UHFFFAOYSA-N 0.000 description 1
- KFNGWPXYNSJXOP-UHFFFAOYSA-N 3-(2-methylprop-2-enoyloxy)propane-1-sulfonic acid Chemical compound CC(=C)C(=O)OCCCS(O)(=O)=O KFNGWPXYNSJXOP-UHFFFAOYSA-N 0.000 description 1
- SBWOBTUYQXLKSS-UHFFFAOYSA-N 3-(2-methylprop-2-enoyloxy)propanoic acid Chemical compound CC(=C)C(=O)OCCC(O)=O SBWOBTUYQXLKSS-UHFFFAOYSA-N 0.000 description 1
- PYSRRFNXTXNWCD-UHFFFAOYSA-N 3-(2-phenylethenyl)furan-2,5-dione Chemical compound O=C1OC(=O)C(C=CC=2C=CC=CC=2)=C1 PYSRRFNXTXNWCD-UHFFFAOYSA-N 0.000 description 1
- KWYJDIUEHHCHCZ-UHFFFAOYSA-N 3-[2-[bis(2-carboxyethyl)amino]ethyl-(2-carboxyethyl)amino]propanoic acid Chemical compound OC(=O)CCN(CCC(O)=O)CCN(CCC(O)=O)CCC(O)=O KWYJDIUEHHCHCZ-UHFFFAOYSA-N 0.000 description 1
- CWAOVKFGVZXUNW-UHFFFAOYSA-N 3-methylidenehex-1-ene Chemical compound CCCC(=C)C=C CWAOVKFGVZXUNW-UHFFFAOYSA-N 0.000 description 1
- IGLWCQMNTGCUBB-UHFFFAOYSA-N 3-methylidenepent-1-ene Chemical compound CCC(=C)C=C IGLWCQMNTGCUBB-UHFFFAOYSA-N 0.000 description 1
- XDLMVUHYZWKMMD-UHFFFAOYSA-N 3-trimethoxysilylpropyl 2-methylprop-2-enoate Chemical compound CO[Si](OC)(OC)CCCOC(=O)C(C)=C XDLMVUHYZWKMMD-UHFFFAOYSA-N 0.000 description 1
- OCVLSHAVSIYKLI-UHFFFAOYSA-N 3h-1,3-thiazole-2-thione Chemical group SC1=NC=CS1 OCVLSHAVSIYKLI-UHFFFAOYSA-N 0.000 description 1
- VPWNQTHUCYMVMZ-UHFFFAOYSA-N 4,4'-sulfonyldiphenol Chemical class C1=CC(O)=CC=C1S(=O)(=O)C1=CC=C(O)C=C1 VPWNQTHUCYMVMZ-UHFFFAOYSA-N 0.000 description 1
- RYYXDZDBXNUPOG-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine;dihydrochloride Chemical compound Cl.Cl.C1C(N)CCC2=C1SC(N)=N2 RYYXDZDBXNUPOG-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- DBCAQXHNJOFNGC-UHFFFAOYSA-N 4-bromo-1,1,1-trifluorobutane Chemical compound FC(F)(F)CCCBr DBCAQXHNJOFNGC-UHFFFAOYSA-N 0.000 description 1
- KFDVPJUYSDEJTH-UHFFFAOYSA-N 4-ethenylpyridine Chemical compound C=CC1=CC=NC=C1 KFDVPJUYSDEJTH-UHFFFAOYSA-N 0.000 description 1
- OIYTYGOUZOARSH-UHFFFAOYSA-N 4-methoxy-2-methylidene-4-oxobutanoic acid Chemical compound COC(=O)CC(=C)C(O)=O OIYTYGOUZOARSH-UHFFFAOYSA-N 0.000 description 1
- KXFRSVCWEHBKQT-UHFFFAOYSA-N 4-naphthalen-1-yl-2h-phthalazin-1-one Chemical compound C12=CC=CC=C2C(=O)NN=C1C1=CC=CC2=CC=CC=C12 KXFRSVCWEHBKQT-UHFFFAOYSA-N 0.000 description 1
- SLBQXWXKPNIVSQ-UHFFFAOYSA-N 4-nitrophthalic acid Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)C=C1C(O)=O SLBQXWXKPNIVSQ-UHFFFAOYSA-N 0.000 description 1
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 1
- HVUGMEVRSDKZHF-UHFFFAOYSA-N 5,5-diethyl-2-sulfanylidene-1,3-thiazolidin-4-one Chemical compound CCC1(CC)SC(=S)NC1=O HVUGMEVRSDKZHF-UHFFFAOYSA-N 0.000 description 1
- CFIUCOKDVARZGF-UHFFFAOYSA-N 5,7-dimethoxy-2h-phthalazin-1-one Chemical compound C1=NNC(=O)C2=CC(OC)=CC(OC)=C21 CFIUCOKDVARZGF-UHFFFAOYSA-N 0.000 description 1
- JCWOGOMMXQGTDA-UHFFFAOYSA-N 5,7-dimethoxyphthalazine Chemical compound C1=NN=CC2=CC(OC)=CC(OC)=C21 JCWOGOMMXQGTDA-UHFFFAOYSA-N 0.000 description 1
- OBDSPDZCPRBIIA-UHFFFAOYSA-N 5-sulfanyl-3h-1,3-thiazole-2-thione Chemical group SC1=CN=C(S)S1 OBDSPDZCPRBIIA-UHFFFAOYSA-N 0.000 description 1
- XDECIMXTYLBMFQ-UHFFFAOYSA-N 6-chloro-2h-phthalazin-1-one Chemical compound C1=NNC(=O)C=2C1=CC(Cl)=CC=2 XDECIMXTYLBMFQ-UHFFFAOYSA-N 0.000 description 1
- AINDGCOQTNWCCB-UHFFFAOYSA-N 6-chlorophthalazine Chemical compound C1=NN=CC2=CC(Cl)=CC=C21 AINDGCOQTNWCCB-UHFFFAOYSA-N 0.000 description 1
- HXONAWDYNNJUQI-UHFFFAOYSA-N 6-tert-butylphthalazine Chemical compound C1=NN=CC2=CC(C(C)(C)C)=CC=C21 HXONAWDYNNJUQI-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- DPUOLQHDNGRHBS-UHFFFAOYSA-N Brassidinsaeure Natural products CCCCCCCCC=CCCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-UHFFFAOYSA-N 0.000 description 1
- 239000006171 Britton–Robinson buffer Substances 0.000 description 1
- 101001123543 Caenorhabditis elegans Phosphoethanolamine N-methyltransferase 1 Proteins 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- 244000025254 Cannabis sativa Species 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical class NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- LEVWYRKDKASIDU-QWWZWVQMSA-N D-cystine Chemical compound OC(=O)[C@H](N)CSSC[C@@H](N)C(O)=O LEVWYRKDKASIDU-QWWZWVQMSA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- URXZXNYJPAJJOQ-UHFFFAOYSA-N Erucic acid Natural products CCCCCCC=CCCCCCCCCCCCC(O)=O URXZXNYJPAJJOQ-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- NVXLIZQNSVLKPO-UHFFFAOYSA-N Glucosereductone Chemical class O=CC(O)C=O NVXLIZQNSVLKPO-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 229910002226 La2O2 Inorganic materials 0.000 description 1
- 229910002420 LaOCl Inorganic materials 0.000 description 1
- 229910001477 LaPO4 Inorganic materials 0.000 description 1
- 239000002879 Lewis base Substances 0.000 description 1
- 235000021353 Lignoceric acid Nutrition 0.000 description 1
- CQXMAMUUWHYSIY-UHFFFAOYSA-N Lignoceric acid Natural products CCCCCCCCCCCCCCCCCCCCCCCC(=O)OCCC1=CC=C(O)C=C1 CQXMAMUUWHYSIY-UHFFFAOYSA-N 0.000 description 1
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-N Metaphosphoric acid Chemical compound OP(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-N 0.000 description 1
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical group SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 1
- GYCMBHHDWRMZGG-UHFFFAOYSA-N Methylacrylonitrile Chemical compound CC(=C)C#N GYCMBHHDWRMZGG-UHFFFAOYSA-N 0.000 description 1
- 229910017672 MgWO4 Inorganic materials 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- FCSHMCFRCYZTRQ-UHFFFAOYSA-N N,N'-diphenylthiourea Chemical compound C=1C=CC=CC=1NC(=S)NC1=CC=CC=C1 FCSHMCFRCYZTRQ-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- 101001123538 Nicotiana tabacum Putrescine N-methyltransferase 1 Proteins 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 229920001328 Polyvinylidene chloride Polymers 0.000 description 1
- NPYPAHLBTDXSSS-UHFFFAOYSA-N Potassium ion Chemical compound [K+] NPYPAHLBTDXSSS-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229920000147 Styrene maleic anhydride Polymers 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- MNOILHPDHOHILI-UHFFFAOYSA-N Tetramethylthiourea Chemical compound CN(C)C(=S)N(C)C MNOILHPDHOHILI-UHFFFAOYSA-N 0.000 description 1
- 241001422033 Thestylus Species 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- DAKWPKUUDNSNPN-UHFFFAOYSA-N Trimethylolpropane triacrylate Chemical compound C=CC(=O)OCC(CC)(COC(=O)C=C)COC(=O)C=C DAKWPKUUDNSNPN-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical class C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 1
- XZKRXPZXQLARHH-XVNBXDOJSA-N [(1e)-buta-1,3-dienyl]benzene Chemical compound C=C\C=C\C1=CC=CC=C1 XZKRXPZXQLARHH-XVNBXDOJSA-N 0.000 description 1
- HVVWZTWDBSEWIH-UHFFFAOYSA-N [2-(hydroxymethyl)-3-prop-2-enoyloxy-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate Chemical compound C=CC(=O)OCC(CO)(COC(=O)C=C)COC(=O)C=C HVVWZTWDBSEWIH-UHFFFAOYSA-N 0.000 description 1
- HSZUHSXXAOWGQY-UHFFFAOYSA-N [2-methyl-3-prop-2-enoyloxy-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate Chemical compound C=CC(=O)OCC(C)(COC(=O)C=C)COC(=O)C=C HSZUHSXXAOWGQY-UHFFFAOYSA-N 0.000 description 1
- RLAVJXQZTLDBRB-UHFFFAOYSA-N [S].[Se].[Au] Chemical compound [S].[Se].[Au] RLAVJXQZTLDBRB-UHFFFAOYSA-N 0.000 description 1
- YDHWWBZFRZWVHO-UHFFFAOYSA-N [hydroxy(phosphonooxy)phosphoryl] phosphono hydrogen phosphate Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(O)=O YDHWWBZFRZWVHO-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 1
- 238000010669 acid-base reaction Methods 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 150000007933 aliphatic carboxylic acids Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 description 1
- 125000005250 alkyl acrylate group Chemical group 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- AOPRFYAPABFRPU-UHFFFAOYSA-N amino(imino)methanesulfonic acid Chemical class NC(=N)S(O)(=O)=O AOPRFYAPABFRPU-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- SOIFLUNRINLCBN-UHFFFAOYSA-N ammonium thiocyanate Chemical compound [NH4+].[S-]C#N SOIFLUNRINLCBN-UHFFFAOYSA-N 0.000 description 1
- 239000002280 amphoteric surfactant Substances 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 238000010539 anionic addition polymerization reaction Methods 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 239000007798 antifreeze agent Substances 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- 239000002216 antistatic agent Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 125000005135 aryl sulfinyl group Chemical group 0.000 description 1
- 125000005200 aryloxy carbonyloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- CHCFOMQHQIQBLZ-UHFFFAOYSA-N azane;phthalic acid Chemical compound N.N.OC(=O)C1=CC=CC=C1C(O)=O CHCFOMQHQIQBLZ-UHFFFAOYSA-N 0.000 description 1
- DZGUJOWBVDZNNF-UHFFFAOYSA-N azanium;2-methylprop-2-enoate Chemical compound [NH4+].CC(=C)C([O-])=O DZGUJOWBVDZNNF-UHFFFAOYSA-N 0.000 description 1
- HEJFWWFWBQGZGQ-UHFFFAOYSA-N azuleno(2,1,8-ija)azulene Chemical compound C1=CC=C2C=C(C=CC=CC3=C4)C3=C2C4=C1 HEJFWWFWBQGZGQ-UHFFFAOYSA-N 0.000 description 1
- 229910001632 barium fluoride Inorganic materials 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000000043 benzamido group Chemical group [H]N([*])C(=O)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 150000008109 benzenetriols Chemical class 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 150000001559 benzoic acids Chemical class 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- GCTPMLUUWLLESL-UHFFFAOYSA-N benzyl prop-2-enoate Chemical compound C=CC(=O)OCC1=CC=CC=C1 GCTPMLUUWLLESL-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 150000001602 bicycloalkyls Chemical group 0.000 description 1
- 239000001045 blue dye Substances 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- UORVGPXVDQYIDP-BJUDXGSMSA-N borane Chemical class [10BH3] UORVGPXVDQYIDP-BJUDXGSMSA-N 0.000 description 1
- RJTANRZEWTUVMA-UHFFFAOYSA-N boron;n-methylmethanamine Chemical compound [B].CNC RJTANRZEWTUVMA-UHFFFAOYSA-N 0.000 description 1
- 229950005228 bromoform Drugs 0.000 description 1
- MCIQPHNFQZFKKM-UHFFFAOYSA-N bromoform;5-sulfonylcyclohexa-1,3-diene Chemical compound BrC(Br)Br.O=S(=O)=C1CC=CC=C1 MCIQPHNFQZFKKM-UHFFFAOYSA-N 0.000 description 1
- GJKZSOHUVOQISW-UHFFFAOYSA-N buta-1,3-diene;2-methylbuta-1,3-diene;styrene Chemical compound C=CC=C.CC(=C)C=C.C=CC1=CC=CC=C1.C=CC1=CC=CC=C1 GJKZSOHUVOQISW-UHFFFAOYSA-N 0.000 description 1
- RTACIUYXLGWTAE-UHFFFAOYSA-N buta-1,3-diene;2-methylbuta-1,3-diene;styrene Chemical compound C=CC=C.CC(=C)C=C.C=CC1=CC=CC=C1 RTACIUYXLGWTAE-UHFFFAOYSA-N 0.000 description 1
- UTOVMEACOLCUCK-PLNGDYQASA-N butyl maleate Chemical compound CCCCOC(=O)\C=C/C(O)=O UTOVMEACOLCUCK-PLNGDYQASA-N 0.000 description 1
- NCMHKCKGHRPLCM-UHFFFAOYSA-N caesium(1+) Chemical compound [Cs+] NCMHKCKGHRPLCM-UHFFFAOYSA-N 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000005606 carbostyryl group Chemical group 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000010538 cationic polymerization reaction Methods 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 229920006217 cellulose acetate butyrate Polymers 0.000 description 1
- ZUIVNYGZFPOXFW-UHFFFAOYSA-N chembl1717603 Chemical compound N1=C(C)C=C(O)N2N=CN=C21 ZUIVNYGZFPOXFW-UHFFFAOYSA-N 0.000 description 1
- 238000002144 chemical decomposition reaction Methods 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- YACLQRRMGMJLJV-UHFFFAOYSA-N chloroprene Chemical compound ClC(=C)C=C YACLQRRMGMJLJV-UHFFFAOYSA-N 0.000 description 1
- GZCJJOLJSBCUNR-UHFFFAOYSA-N chroman-6-ol Chemical class O1CCCC2=CC(O)=CC=C21 GZCJJOLJSBCUNR-UHFFFAOYSA-N 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 229920001940 conductive polymer Polymers 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 239000002826 coolant Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- XCJYREBRNVKWGJ-UHFFFAOYSA-N copper(II) phthalocyanine Chemical compound [Cu+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 XCJYREBRNVKWGJ-UHFFFAOYSA-N 0.000 description 1
- 239000011258 core-shell material Substances 0.000 description 1
- 125000000853 cresyl group Chemical group C1(=CC=C(C=C1)C)* 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 238000007766 curtain coating Methods 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000006639 cyclohexyl carbonyl group Chemical group 0.000 description 1
- KBLWLMPSVYBVDK-UHFFFAOYSA-N cyclohexyl prop-2-enoate Chemical compound C=CC(=O)OC1CCCCC1 KBLWLMPSVYBVDK-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- OGVXYCDTRMDYOG-UHFFFAOYSA-N dibutyl 2-methylidenebutanedioate Chemical compound CCCCOC(=O)CC(=C)C(=O)OCCCC OGVXYCDTRMDYOG-UHFFFAOYSA-N 0.000 description 1
- 125000004915 dibutylamino group Chemical group C(CCC)N(CCCC)* 0.000 description 1
- 150000001990 dicarboxylic acid derivatives Chemical class 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- ZOMNIUBKTOKEHS-UHFFFAOYSA-L dimercury dichloride Chemical class Cl[Hg][Hg]Cl ZOMNIUBKTOKEHS-UHFFFAOYSA-L 0.000 description 1
- LDCRTTXIJACKKU-ARJAWSKDSA-N dimethyl maleate Chemical compound COC(=O)\C=C/C(=O)OC LDCRTTXIJACKKU-ARJAWSKDSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Natural products C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-N diphosphoric acid Chemical compound OP(O)(=O)OP(O)(O)=O XPPKVPWEQAFLFU-UHFFFAOYSA-N 0.000 description 1
- RBYDCVNTVVKIQT-UHFFFAOYSA-L dipotassium;2-methylidenebutanedioate Chemical compound [K+].[K+].[O-]C(=O)CC(=C)C([O-])=O RBYDCVNTVVKIQT-UHFFFAOYSA-L 0.000 description 1
- GOMCKELMLXHYHH-UHFFFAOYSA-L dipotassium;phthalate Chemical compound [K+].[K+].[O-]C(=O)C1=CC=CC=C1C([O-])=O GOMCKELMLXHYHH-UHFFFAOYSA-L 0.000 description 1
- HQWKKEIVHQXCPI-UHFFFAOYSA-L disodium;phthalate Chemical compound [Na+].[Na+].[O-]C(=O)C1=CC=CC=C1C([O-])=O HQWKKEIVHQXCPI-UHFFFAOYSA-L 0.000 description 1
- 238000012674 dispersion polymerization Methods 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000012990 dithiocarbamate Substances 0.000 description 1
- WNAHIZMDSQCWRP-UHFFFAOYSA-N dodecane-1-thiol Chemical compound CCCCCCCCCCCCS WNAHIZMDSQCWRP-UHFFFAOYSA-N 0.000 description 1
- GMSCBRSQMRDRCD-UHFFFAOYSA-N dodecyl 2-methylprop-2-enoate Chemical compound CCCCCCCCCCCCOC(=O)C(C)=C GMSCBRSQMRDRCD-UHFFFAOYSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 239000003822 epoxy resin Substances 0.000 description 1
- DPUOLQHDNGRHBS-KTKRTIGZSA-N erucic acid Chemical compound CCCCCCCC\C=C/CCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-KTKRTIGZSA-N 0.000 description 1
- HDERJYVLTPVNRI-UHFFFAOYSA-N ethene;ethenyl acetate Chemical class C=C.CC(=O)OC=C HDERJYVLTPVNRI-UHFFFAOYSA-N 0.000 description 1
- XJELOQYISYPGDX-UHFFFAOYSA-N ethenyl 2-chloroacetate Chemical compound ClCC(=O)OC=C XJELOQYISYPGDX-UHFFFAOYSA-N 0.000 description 1
- CMXXMZYAYIHTBU-UHFFFAOYSA-N ethenyl 2-hydroxybenzoate Chemical compound OC1=CC=CC=C1C(=O)OC=C CMXXMZYAYIHTBU-UHFFFAOYSA-N 0.000 description 1
- UIWXSTHGICQLQT-UHFFFAOYSA-N ethenyl propanoate Chemical compound CCC(=O)OC=C UIWXSTHGICQLQT-UHFFFAOYSA-N 0.000 description 1
- NKSJNEHGWDZZQF-UHFFFAOYSA-N ethenyl(trimethoxy)silane Chemical compound CO[Si](OC)(OC)C=C NKSJNEHGWDZZQF-UHFFFAOYSA-N 0.000 description 1
- GCSJLQSCSDMKTP-UHFFFAOYSA-N ethenyl(trimethyl)silane Chemical compound C[Si](C)(C)C=C GCSJLQSCSDMKTP-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- FARYTWBWLZAXNK-WAYWQWQTSA-N ethyl (z)-3-(methylamino)but-2-enoate Chemical compound CCOC(=O)\C=C(\C)NC FARYTWBWLZAXNK-WAYWQWQTSA-N 0.000 description 1
- SUPCQIBBMFXVTL-UHFFFAOYSA-N ethyl 2-methylprop-2-enoate Chemical compound CCOC(=O)C(C)=C SUPCQIBBMFXVTL-UHFFFAOYSA-N 0.000 description 1
- CEIPQQODRKXDSB-UHFFFAOYSA-N ethyl 3-(6-hydroxynaphthalen-2-yl)-1H-indazole-5-carboximidate dihydrochloride Chemical compound Cl.Cl.C1=C(O)C=CC2=CC(C3=NNC4=CC=C(C=C43)C(=N)OCC)=CC=C21 CEIPQQODRKXDSB-UHFFFAOYSA-N 0.000 description 1
- VGEWEGHHYWGXGG-UHFFFAOYSA-N ethyl n-hydroxycarbamate Chemical class CCOC(=O)NO VGEWEGHHYWGXGG-UHFFFAOYSA-N 0.000 description 1
- 229940093476 ethylene glycol Drugs 0.000 description 1
- STVZJERGLQHEKB-UHFFFAOYSA-N ethylene glycol dimethacrylate Substances CC(=C)C(=O)OCCOC(=O)C(C)=C STVZJERGLQHEKB-UHFFFAOYSA-N 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- IFQUWYZCAGRUJN-UHFFFAOYSA-N ethylenediaminediacetic acid Chemical compound OC(=O)CNCCNCC(O)=O IFQUWYZCAGRUJN-UHFFFAOYSA-N 0.000 description 1
- 239000007888 film coating Substances 0.000 description 1
- 238000009501 film coating Methods 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 1
- JKFAIQOWCVVSKC-UHFFFAOYSA-N furazan Chemical group C=1C=NON=1 JKFAIQOWCVVSKC-UHFFFAOYSA-N 0.000 description 1
- 238000000769 gas chromatography-flame ionisation detection Methods 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 229910021397 glassy carbon Inorganic materials 0.000 description 1
- VOZRXNHHFUQHIL-UHFFFAOYSA-N glycidyl methacrylate Chemical compound CC(=C)C(=O)OCC1CO1 VOZRXNHHFUQHIL-UHFFFAOYSA-N 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 150000002344 gold compounds Chemical class 0.000 description 1
- YPDKFMYSITXPDU-UHFFFAOYSA-B hafnium(4+) tetraphosphate Chemical compound [Hf+4].[Hf+4].[Hf+4].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O YPDKFMYSITXPDU-UHFFFAOYSA-B 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- UIZVMOZAXAMASY-UHFFFAOYSA-N hex-5-en-1-ol Chemical compound OCCCCC=C UIZVMOZAXAMASY-UHFFFAOYSA-N 0.000 description 1
- 229940005740 hexametaphosphate Drugs 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- FSSWIVRJWMTXDI-UHFFFAOYSA-N hexathiocane-7-thione Chemical compound S=C1CSSSSSS1 FSSWIVRJWMTXDI-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- VKOBVWXKNCXXDE-UHFFFAOYSA-M icosanoate Chemical compound CCCCCCCCCCCCCCCCCCCC([O-])=O VKOBVWXKNCXXDE-UHFFFAOYSA-M 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- NBZBKCUXIYYUSX-UHFFFAOYSA-N iminodiacetic acid Chemical compound OC(=O)CNCC(O)=O NBZBKCUXIYYUSX-UHFFFAOYSA-N 0.000 description 1
- ICPGNGZLHITQJI-UHFFFAOYSA-N iminosilver Chemical compound [Ag]=N ICPGNGZLHITQJI-UHFFFAOYSA-N 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- UETZVSHORCDDTH-UHFFFAOYSA-N iron(2+);hexacyanide Chemical compound [Fe+2].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-] UETZVSHORCDDTH-UHFFFAOYSA-N 0.000 description 1
- 125000005929 isobutyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])OC(*)=O 0.000 description 1
- 125000002462 isocyano group Chemical group *[N+]#[C-] 0.000 description 1
- GWVMLCQWXVFZCN-UHFFFAOYSA-N isoindoline Chemical compound C1=CC=C2CNCC2=C1 GWVMLCQWXVFZCN-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- BITXABIVVURDNX-UHFFFAOYSA-N isoselenocyanic acid Chemical class N=C=[Se] BITXABIVVURDNX-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229910052746 lanthanum Inorganic materials 0.000 description 1
- 125000000400 lauroyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- PBOSTUDLECTMNL-UHFFFAOYSA-N lauryl acrylate Chemical compound CCCCCCCCCCCCOC(=O)C=C PBOSTUDLECTMNL-UHFFFAOYSA-N 0.000 description 1
- 150000007527 lewis bases Chemical class 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- CDOSHBSSFJOMGT-UHFFFAOYSA-N linalool Chemical compound CC(C)=CCCC(C)(O)C=C CDOSHBSSFJOMGT-UHFFFAOYSA-N 0.000 description 1
- 239000010808 liquid waste Substances 0.000 description 1
- 150000004668 long chain fatty acids Chemical class 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- BQPIGGFYSBELGY-UHFFFAOYSA-N mercury(2+) Chemical class [Hg+2] BQPIGGFYSBELGY-UHFFFAOYSA-N 0.000 description 1
- FQPSGWSUVKBHSU-UHFFFAOYSA-N methacrylamide Chemical compound CC(=C)C(N)=O FQPSGWSUVKBHSU-UHFFFAOYSA-N 0.000 description 1
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 1
- 229940117841 methacrylic acid copolymer Drugs 0.000 description 1
- RBQRWNWVPQDTJJ-UHFFFAOYSA-N methacryloyloxyethyl isocyanate Chemical compound CC(=C)C(=O)OCCN=C=O RBQRWNWVPQDTJJ-UHFFFAOYSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- MPHUYCIKFIKENX-UHFFFAOYSA-N methyl 2-ethenylbenzoate Chemical compound COC(=O)C1=CC=CC=C1C=C MPHUYCIKFIKENX-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- PWAJIGUTWKGVLP-UHFFFAOYSA-N methyl non-8-enoate Chemical compound COC(=O)CCCCCCC=C PWAJIGUTWKGVLP-UHFFFAOYSA-N 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- ZIUHHBKFKCYYJD-UHFFFAOYSA-N n,n'-methylenebisacrylamide Chemical compound C=CC(=O)NCNC(=O)C=C ZIUHHBKFKCYYJD-UHFFFAOYSA-N 0.000 description 1
- 229940088644 n,n-dimethylacrylamide Drugs 0.000 description 1
- YLGYACDQVQQZSW-UHFFFAOYSA-N n,n-dimethylprop-2-enamide Chemical compound CN(C)C(=O)C=C YLGYACDQVQQZSW-UHFFFAOYSA-N 0.000 description 1
- WTNTZFRNCHEDOS-UHFFFAOYSA-N n-(2-hydroxyethyl)-2-methylpropanamide Chemical compound CC(C)C(=O)NCCO WTNTZFRNCHEDOS-UHFFFAOYSA-N 0.000 description 1
- OMNKZBIFPJNNIO-UHFFFAOYSA-N n-(2-methyl-4-oxopentan-2-yl)prop-2-enamide Chemical compound CC(=O)CC(C)(C)NC(=O)C=C OMNKZBIFPJNNIO-UHFFFAOYSA-N 0.000 description 1
- YFJKOCSMGQMGNP-UHFFFAOYSA-N n-(dimethylcarbamoselenoyl)-2,2,2-trifluoro-n-methylacetamide Chemical compound CN(C)C(=[Se])N(C)C(=O)C(F)(F)F YFJKOCSMGQMGNP-UHFFFAOYSA-N 0.000 description 1
- RKISUIUJZGSLEV-UHFFFAOYSA-N n-[2-(octadecanoylamino)ethyl]octadecanamide Chemical compound CCCCCCCCCCCCCCCCCC(=O)NCCNC(=O)CCCCCCCCCCCCCCCCC RKISUIUJZGSLEV-UHFFFAOYSA-N 0.000 description 1
- PMJFVKWBSWWAKT-UHFFFAOYSA-N n-cyclohexylprop-2-enamide Chemical compound C=CC(=O)NC1CCCCC1 PMJFVKWBSWWAKT-UHFFFAOYSA-N 0.000 description 1
- 125000006610 n-decyloxy group Chemical group 0.000 description 1
- 125000001298 n-hexoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- SEEYREPSKCQBBF-UHFFFAOYSA-N n-methylmaleimide Chemical compound CN1C(=O)C=CC1=O SEEYREPSKCQBBF-UHFFFAOYSA-N 0.000 description 1
- YPHQUSNPXDGUHL-UHFFFAOYSA-N n-methylprop-2-enamide Chemical compound CNC(=O)C=C YPHQUSNPXDGUHL-UHFFFAOYSA-N 0.000 description 1
- BPCNEKWROYSOLT-UHFFFAOYSA-N n-phenylprop-2-enamide Chemical compound C=CC(=O)NC1=CC=CC=C1 BPCNEKWROYSOLT-UHFFFAOYSA-N 0.000 description 1
- SUZXWXGJCOCMHU-UHFFFAOYSA-N n-sulfonylbenzamide Chemical compound O=S(=O)=NC(=O)C1=CC=CC=C1 SUZXWXGJCOCMHU-UHFFFAOYSA-N 0.000 description 1
- XFHJDMUEHUHAJW-UHFFFAOYSA-N n-tert-butylprop-2-enamide Chemical compound CC(C)(C)NC(=O)C=C XFHJDMUEHUHAJW-UHFFFAOYSA-N 0.000 description 1
- DWJIJRSTYFPKGD-UHFFFAOYSA-N naphthalen-2-yl benzoate Chemical compound C=1C=C2C=CC=CC2=CC=1OC(=O)C1=CC=CC=C1 DWJIJRSTYFPKGD-UHFFFAOYSA-N 0.000 description 1
- 125000004957 naphthylene group Chemical group 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 239000000025 natural resin Substances 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 230000006911 nucleation Effects 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 125000002801 octanoyl group Chemical group C(CCCCCCC)(=O)* 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005447 octyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- 150000004866 oxadiazoles Chemical class 0.000 description 1
- 125000003452 oxalyl group Chemical group *C(=O)C(*)=O 0.000 description 1
- 125000003355 oxamoyl group Chemical group C(C(=O)N)(=O)* 0.000 description 1
- 125000001096 oxamoylamino group Chemical group C(C(=O)N)(=O)N* 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Chemical compound [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- JTJMJGYZQZDUJJ-UHFFFAOYSA-N phencyclidine Chemical compound C1CCCCN1C1(C=2C=CC=CC=2)CCCCC1 JTJMJGYZQZDUJJ-UHFFFAOYSA-N 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- 229920006287 phenoxy resin Polymers 0.000 description 1
- 239000013034 phenoxy resin Substances 0.000 description 1
- 229960005323 phenoxyethanol Drugs 0.000 description 1
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 125000005328 phosphinyl group Chemical group [PH2](=O)* 0.000 description 1
- 239000011941 photocatalyst Substances 0.000 description 1
- 125000005498 phthalate group Chemical group 0.000 description 1
- 150000003021 phthalic acid derivatives Chemical class 0.000 description 1
- 239000001007 phthalocyanine dye Substances 0.000 description 1
- PMJHHCWVYXUKFD-UHFFFAOYSA-N piperylene Natural products CC=CC=C PMJHHCWVYXUKFD-UHFFFAOYSA-N 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920002285 poly(styrene-co-acrylonitrile) Polymers 0.000 description 1
- 229920002037 poly(vinyl butyral) polymer Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920002857 polybutadiene Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920001225 polyester resin Polymers 0.000 description 1
- 239000004645 polyester resin Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001228 polyisocyanate Polymers 0.000 description 1
- 239000005056 polyisocyanate Substances 0.000 description 1
- 229920001195 polyisoprene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000005077 polysulfide Substances 0.000 description 1
- 229920001021 polysulfide Polymers 0.000 description 1
- 150000008117 polysulfides Polymers 0.000 description 1
- 229920005749 polyurethane resin Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- ABPDLUFRMRZSIX-UHFFFAOYSA-M potassium;4-ethenylbenzenesulfinate Chemical compound [K+].[O-]S(=O)C1=CC=C(C=C)C=C1 ABPDLUFRMRZSIX-UHFFFAOYSA-M 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- FBCQUCJYYPMKRO-UHFFFAOYSA-N prop-2-enyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC=C FBCQUCJYYPMKRO-UHFFFAOYSA-N 0.000 description 1
- HJWLCRVIBGQPNF-UHFFFAOYSA-N prop-2-enylbenzene Chemical compound C=CCC1=CC=CC=C1 HJWLCRVIBGQPNF-UHFFFAOYSA-N 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- USPWKWBDZOARPV-UHFFFAOYSA-N pyrazolidine Chemical compound C1CNNC1 USPWKWBDZOARPV-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 229940005657 pyrophosphoric acid Drugs 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- WPPDXAHGCGPUPK-UHFFFAOYSA-N red 2 Chemical compound C1=CC=CC=C1C(C1=CC=CC=C11)=C(C=2C=3C4=CC=C5C6=CC=C7C8=C(C=9C=CC=CC=9)C9=CC=CC=C9C(C=9C=CC=CC=9)=C8C8=CC=C(C6=C87)C(C=35)=CC=2)C4=C1C1=CC=CC=C1 WPPDXAHGCGPUPK-UHFFFAOYSA-N 0.000 description 1
- 238000001028 reflection method Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229910001419 rubidium ion Inorganic materials 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 150000003872 salicylic acid derivatives Chemical class 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 150000004756 silanes Chemical class 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- ZUNKMNLKJXRCDM-UHFFFAOYSA-N silver bromoiodide Chemical compound [Ag].IBr ZUNKMNLKJXRCDM-UHFFFAOYSA-N 0.000 description 1
- FJOLTQXXWSRAIX-UHFFFAOYSA-K silver phosphate Chemical compound [Ag+].[Ag+].[Ag+].[O-]P([O-])([O-])=O FJOLTQXXWSRAIX-UHFFFAOYSA-K 0.000 description 1
- 229940019931 silver phosphate Drugs 0.000 description 1
- 229910000161 silver phosphate Inorganic materials 0.000 description 1
- RHUVFRWZKMEWNS-UHFFFAOYSA-M silver thiocyanate Chemical compound [Ag+].[S-]C#N RHUVFRWZKMEWNS-UHFFFAOYSA-M 0.000 description 1
- YRSQDSCQMOUOKO-KVVVOXFISA-M silver;(z)-octadec-9-enoate Chemical compound [Ag+].CCCCCCCC\C=C/CCCCCCCC([O-])=O YRSQDSCQMOUOKO-KVVVOXFISA-M 0.000 description 1
- MNMYRUHURLPFQW-UHFFFAOYSA-M silver;dodecanoate Chemical compound [Ag+].CCCCCCCCCCCC([O-])=O MNMYRUHURLPFQW-UHFFFAOYSA-M 0.000 description 1
- LTYHQUJGIQUHMS-UHFFFAOYSA-M silver;hexadecanoate Chemical compound [Ag+].CCCCCCCCCCCCCCCC([O-])=O LTYHQUJGIQUHMS-UHFFFAOYSA-M 0.000 description 1
- ORYURPRSXLUCSS-UHFFFAOYSA-M silver;octadecanoate Chemical compound [Ag+].CCCCCCCCCCCCCCCCCC([O-])=O ORYURPRSXLUCSS-UHFFFAOYSA-M 0.000 description 1
- LPYHADGLCYWDNC-UHFFFAOYSA-M silver;tetracosanoate Chemical compound [Ag+].CCCCCCCCCCCCCCCCCCCCCCCC([O-])=O LPYHADGLCYWDNC-UHFFFAOYSA-M 0.000 description 1
- OHGHHPYRRURLHR-UHFFFAOYSA-M silver;tetradecanoate Chemical compound [Ag+].CCCCCCCCCCCCCC([O-])=O OHGHHPYRRURLHR-UHFFFAOYSA-M 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 229940047670 sodium acrylate Drugs 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- VGTPCRGMBIAPIM-UHFFFAOYSA-M sodium thiocyanate Chemical compound [Na+].[S-]C#N VGTPCRGMBIAPIM-UHFFFAOYSA-M 0.000 description 1
- PODWXQQNRWNDGD-UHFFFAOYSA-L sodium thiosulfate pentahydrate Chemical compound O.O.O.O.O.[Na+].[Na+].[O-]S([S-])(=O)=O PODWXQQNRWNDGD-UHFFFAOYSA-L 0.000 description 1
- 235000019832 sodium triphosphate Nutrition 0.000 description 1
- GGRBDFIKUKYKLY-UHFFFAOYSA-M sodium;3-(5-sulfanylidene-2h-tetrazol-1-yl)benzenesulfonate Chemical compound [Na+].[O-]S(=O)(=O)C1=CC=CC(N2C(N=NN2)=S)=C1 GGRBDFIKUKYKLY-UHFFFAOYSA-M 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000001119 stannous chloride Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 229910052712 strontium Inorganic materials 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- WGPCGCOKHWGKJJ-UHFFFAOYSA-N sulfanylidenezinc Chemical group [Zn]=S WGPCGCOKHWGKJJ-UHFFFAOYSA-N 0.000 description 1
- WSANLGASBHUYGD-UHFFFAOYSA-N sulfidophosphanium Chemical class S=[PH3] WSANLGASBHUYGD-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical class [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 238000010558 suspension polymerization method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 239000005061 synthetic rubber Substances 0.000 description 1
- 229940042055 systemic antimycotics triazole derivative Drugs 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- 238000010345 tape casting Methods 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- 125000001302 tertiary amino group Chemical group 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- AUHHYELHRWCWEZ-UHFFFAOYSA-N tetrachlorophthalic anhydride Chemical compound ClC1=C(Cl)C(Cl)=C2C(=O)OC(=O)C2=C1Cl AUHHYELHRWCWEZ-UHFFFAOYSA-N 0.000 description 1
- CBXCPBUEXACCNR-UHFFFAOYSA-N tetraethylammonium Chemical compound CC[N+](CC)(CC)CC CBXCPBUEXACCNR-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- AWDBHOZBRXWRKS-UHFFFAOYSA-N tetrapotassium;iron(6+);hexacyanide Chemical compound [K+].[K+].[K+].[K+].[Fe+6].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-] AWDBHOZBRXWRKS-UHFFFAOYSA-N 0.000 description 1
- OSBSFAARYOCBHB-UHFFFAOYSA-N tetrapropylammonium Chemical compound CCC[N+](CCC)(CCC)CCC OSBSFAARYOCBHB-UHFFFAOYSA-N 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000004149 thio group Chemical group *S* 0.000 description 1
- YUKQRDCYNOVPGJ-UHFFFAOYSA-N thioacetamide Chemical compound CC(N)=S YUKQRDCYNOVPGJ-UHFFFAOYSA-N 0.000 description 1
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 1
- 125000001391 thioamide group Chemical group 0.000 description 1
- 150000003556 thioamides Chemical class 0.000 description 1
- 125000000034 thioazolyl group Chemical group 0.000 description 1
- 230000009974 thixotropic effect Effects 0.000 description 1
- FRNOGLGSGLTDKL-UHFFFAOYSA-N thulium atom Chemical compound [Tm] FRNOGLGSGLTDKL-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- BVBALDDYDXBEKK-UHFFFAOYSA-N tributoxy(selanylidene)-$l^{5}-phosphane Chemical compound CCCCOP(=[Se])(OCCCC)OCCCC BVBALDDYDXBEKK-UHFFFAOYSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000004953 trihalomethyl group Chemical group 0.000 description 1
- KTFAZNVGJUIWJM-UHFFFAOYSA-N trimethyl(sulfanylidene)-$l^{5}-phosphane Chemical compound CP(C)(C)=S KTFAZNVGJUIWJM-UHFFFAOYSA-N 0.000 description 1
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- ZFVJLNKVUKIPPI-UHFFFAOYSA-N triphenyl(selanylidene)-$l^{5}-phosphane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=[Se])C1=CC=CC=C1 ZFVJLNKVUKIPPI-UHFFFAOYSA-N 0.000 description 1
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 1
- 229940048102 triphosphoric acid Drugs 0.000 description 1
- NZKWZUOYGAKOQC-UHFFFAOYSA-H tripotassium;hexachloroiridium(3-) Chemical compound [Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[K+].[K+].[K+].[Ir+3] NZKWZUOYGAKOQC-UHFFFAOYSA-H 0.000 description 1
- WFRMLFFVZPJQSI-UHFFFAOYSA-N tris(4-methylphenoxy)-selanylidene-$l^{5}-phosphane Chemical compound C1=CC(C)=CC=C1OP(=[Se])(OC=1C=CC(C)=CC=1)OC1=CC=C(C)C=C1 WFRMLFFVZPJQSI-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- PBYZMCDFOULPGH-UHFFFAOYSA-N tungstate Chemical compound [O-][W]([O-])(=O)=O PBYZMCDFOULPGH-UHFFFAOYSA-N 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- KOZCZZVUFDCZGG-UHFFFAOYSA-N vinyl benzoate Chemical compound C=COC(=O)C1=CC=CC=C1 KOZCZZVUFDCZGG-UHFFFAOYSA-N 0.000 description 1
- 229920001567 vinyl ester resin Polymers 0.000 description 1
- NLVXSWCKKBEXTG-UHFFFAOYSA-N vinylsulfonic acid Chemical compound OS(=O)(=O)C=C NLVXSWCKKBEXTG-UHFFFAOYSA-N 0.000 description 1
- 238000004832 voltammetry Methods 0.000 description 1
- 238000004073 vulcanization Methods 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 238000004804 winding Methods 0.000 description 1
- 125000005023 xylyl group Chemical group 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- 229910000164 yttrium(III) phosphate Inorganic materials 0.000 description 1
- 150000007934 α,β-unsaturated carboxylic acids Chemical class 0.000 description 1
Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03C—PHOTOSENSITIVE MATERIALS FOR PHOTOGRAPHIC PURPOSES; PHOTOGRAPHIC PROCESSES, e.g. CINE, X-RAY, COLOUR, STEREO-PHOTOGRAPHIC PROCESSES; AUXILIARY PROCESSES IN PHOTOGRAPHY
- G03C1/00—Photosensitive materials
- G03C1/494—Silver salt compositions other than silver halide emulsions; Photothermographic systems ; Thermographic systems using noble metal compounds
- G03C1/498—Photothermographic systems, e.g. dry silver
- G03C1/49836—Additives
- G03C1/49863—Inert additives, e.g. surfactants, binders
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03C—PHOTOSENSITIVE MATERIALS FOR PHOTOGRAPHIC PURPOSES; PHOTOGRAPHIC PROCESSES, e.g. CINE, X-RAY, COLOUR, STEREO-PHOTOGRAPHIC PROCESSES; AUXILIARY PROCESSES IN PHOTOGRAPHY
- G03C1/00—Photosensitive materials
- G03C1/494—Silver salt compositions other than silver halide emulsions; Photothermographic systems ; Thermographic systems using noble metal compounds
- G03C1/498—Photothermographic systems, e.g. dry silver
- G03C1/49818—Silver halides
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03C—PHOTOSENSITIVE MATERIALS FOR PHOTOGRAPHIC PURPOSES; PHOTOGRAPHIC PROCESSES, e.g. CINE, X-RAY, COLOUR, STEREO-PHOTOGRAPHIC PROCESSES; AUXILIARY PROCESSES IN PHOTOGRAPHY
- G03C5/00—Photographic processes or agents therefor; Regeneration of such processing agents
- G03C5/16—X-ray, infrared, or ultraviolet ray processes
- G03C5/17—X-ray, infrared, or ultraviolet ray processes using screens to intensify X-ray images
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03C—PHOTOSENSITIVE MATERIALS FOR PHOTOGRAPHIC PURPOSES; PHOTOGRAPHIC PROCESSES, e.g. CINE, X-RAY, COLOUR, STEREO-PHOTOGRAPHIC PROCESSES; AUXILIARY PROCESSES IN PHOTOGRAPHY
- G03C1/00—Photosensitive materials
- G03C1/005—Silver halide emulsions; Preparation thereof; Physical treatment thereof; Incorporation of additives therein
- G03C1/0051—Tabular grain emulsions
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03C—PHOTOSENSITIVE MATERIALS FOR PHOTOGRAPHIC PURPOSES; PHOTOGRAPHIC PROCESSES, e.g. CINE, X-RAY, COLOUR, STEREO-PHOTOGRAPHIC PROCESSES; AUXILIARY PROCESSES IN PHOTOGRAPHY
- G03C1/00—Photosensitive materials
- G03C1/005—Silver halide emulsions; Preparation thereof; Physical treatment thereof; Incorporation of additives therein
- G03C1/06—Silver halide emulsions; Preparation thereof; Physical treatment thereof; Incorporation of additives therein with non-macromolecular additives
- G03C1/061—Hydrazine compounds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03C—PHOTOSENSITIVE MATERIALS FOR PHOTOGRAPHIC PURPOSES; PHOTOGRAPHIC PROCESSES, e.g. CINE, X-RAY, COLOUR, STEREO-PHOTOGRAPHIC PROCESSES; AUXILIARY PROCESSES IN PHOTOGRAPHY
- G03C1/00—Photosensitive materials
- G03C1/494—Silver salt compositions other than silver halide emulsions; Photothermographic systems ; Thermographic systems using noble metal compounds
- G03C1/498—Photothermographic systems, e.g. dry silver
- G03C1/49836—Additives
Definitions
- the present invention relates to an improved photothermographic material and an image forming method. More specifically, the invention relates to a photothermographic material which exhibits a reduced degree of haze, high image quality, and excellent pressure resistance, and an image forming method utilizing the same.
- images for medical imaging require a particularly high image quality excellent in sharpness and granularity since fine representation is required, and are characterized in that images of blue-black tones are preferred from the viewpoint of easy diagnosis.
- various kinds of hard copy systems utilizing dyes or pigments such as ink jet printers and electrophotographic systems have been marketed as general image forming systems, but these are not satisfactory as output systems for medical images.
- Such photothermographic materials generally comprise an image forming layer in which a catalytically active amount of photocatalyst (for example, a silver halide), a reducing agent, a reducible silver salt (for example, an organic silver salt), and if necessary, a toner for controlling the color tone of silver, are dispersed in a binder.
- a photothermographic material forms a black silver image by being heated to a high temperature (for example, 80°C or higher) after imagewise exposure to cause an oxidation-reduction reaction between a silver halide or a reducible silver salt and a reducing agent.
- the oxidation-reduction reaction is accelerated by the catalytic action of a latent image, which is generated on the silver halide grain by exposure.
- the photosensitive material for photography as described herein means a photosensitive material on which images are recorded by one-shot exposing by camera work, rather than by writing the image information by scanning exposure with a laser beam or the like.
- photosensitive materials for photography are generally used in the field of wet developing photosensitive materials, and direct or indirect radiography films, mammography films, and the like are widely known as photosensitive materials for use in medical imaging.
- an X-ray photothermographic material coated on both sides using a blue fluorescent intensifying screen is described in Japanese Patent (JP) No.
- JP-A Japanese Patent Application Laid-Open
- a photosensitive material for medical use containing tabular grains that have a high content of silver chloride and have (100) major faces, and that are coated on both sides of a support is described in JP-A No. 10-282602.
- Double-sided coated photothermographic materials are disclosed in JP-A Nos. 2000-227642, 2001-22027, 2001-109101, and 2002-90941.
- developed images can be obtained from photothermographic materials by heating alone, they have many benifical features such as simple image forming operation, no need for controlling a wet processing solution or dispose thereof, and the like.
- haze turbidity of a membrane
- Another problem is an increase of fog known as print-out, which occurs because silver halide grains, unreacted organic silver salts, and reducing agents remain in the membrane.
- a first aspect of the invention provides a photothermographic material comprising, on at least one side of a support, an image forming layer containing at least a photosensitive silver halide, a non-photosensitive organic silver salt, a reducing agent, and a binder, wherein: the photosensitive silver halide has an average silver iodide content of 40 mol% or higher, and at least 50% of a projected area of total grains of the photosensitive silver halide is occupied by tabular grains having an aspect ratio of 2 or more; and 50% by weight or more of the binder is formed by a polymer latex containing the following monomer component in a range of from 10% by weight to 70% by weight:
- a second aspect of the invention provides an image forming method using the photothermographic material according to the first aspect, wherein the method comprises: (a) providing an assembly for forming an image by placing the photothermographic material between a pair of fluorescent intensifying screens; (b) putting an analyte between the assembly and an X-ray source; (c) applying exposure to the analyte using X-rays having an energy level in a range of 25 kVp to 125 kVp; (d) taking the photothermographic material out of the assembly; and (e) heating the photothermographic material at a temperature at which the material can be developed thermally.
- the present inventors found that for the above two problems of turbity of the membrane (haze) and increase of fog known as print-out, the use of tabular photosensitive silver iodide grains is useful. Moreover the present invention solves the problem of generating a break at the surface of a photothermographic material when the material comprising the grains is impressed before thermal development by using a polymer having a specific monomer component for a binder in an image forming layer. In addition, the present invention provides a method for forming a radiation image with high sensitivity.
- the photothermographic material of the invention has an image forming layer comprising at least a photosensitive silver halide, a non-photosensitive organic silver salt, a reducing agent, and a binder on at least one side of a support.
- the image forming layer may have disposed thereon a surface protective layer, or a back layer, a back protective layer, or the like may be disposed on the opposite side of the image forming layer with respect to the support.
- the image forming layer of the present invention may be disposed on one side, or may be disposed on both sides of the support. Particularly, in the case of photothermographic material used for forming a radiation image, the image forming layer is preferably coated on both sides.
- the organic silver salt which can be used in the present invention is relatively stable to light but serves as to supply silver ions and forms silver images when heated to 80°C or higher under the presence of an exposed photosensitive silver halide and a reducing agent.
- the organic silver salt may be any organic material containing a source capable of supplying silver ions that are reducible by a reducing agent.
- Such a non-photosensitive organic silver salt is disclosed, for example, in JP-A No. 10-62899 (paragraph Nos. 0048 to 0049), European Patent (EP) No. 0803764A (page 18, line 24 to page 19, line 37), EP No. 0962812A1, JP-A Nos. 11-349591, 2000-7683, and 2000-72711, and the like.
- a silver salt of an organic acid particularly, a silver salt of long chained aliphatic carboxylic acid (having 10 to 30 carbon atoms, and preferably having 15 to 28 carbon atoms) is preferable.
- Preferred examples of the silver salt of fatty acid can include, for example, silver lignocerate, silver behenate, silver arachidinate, silver stearate, silver oleate, silver laurate, silver capronate, silver myristate, silver palmitate, silver erucate and mixtures thereof.
- silver salt of fatty acid with a silver behenate content of 80 mol% to 99 mol%, more preferably, 85 mol% to 99 mol%, and further preferably, 95 mol% to 99 mol%. Further, it is preferred to use a silver salt of fatty acid with a silver erucate content of 2 mol% or less, more preferably, 1 mol% or less, and further preferably, 0.1 mol% or less.
- organic silver salt usable in the invention there is no particular restriction on the shape of the organic silver salt usable in the invention and it may be needle-like, bar-like, tabular, or flake shaped.
- the percentage for the value obtained by dividing the standard deviation for the length of minor axis and major axis by the minor axis and the major axis respectively is, preferably, 100% or less, more preferably, 80% or less and, further preferably, 50% or less.
- the shape of the organic silver salt can be measured by analyzing a dispersion of an organic silver salt as transmission type electron microscopic images.
- Another method of measuring the monodispersion is a method of determining of the standard deviation of the volume weighted mean diameter of the organic silver salt in which the percentage for the value defined by the volume weighted mean diameter (variation coefficient), is preferably, 100% or less, more preferably, 80% or less and, further preferably, 50% or less.
- a commercially available laser-beam scattering grain size analyzer can be used.
- Methods known in the art may be applied to the method for producing the organic silver salt used in the invention and to the dispersing method thereof.
- the amount of the photosensitive silver salt to be disposed in the aqueous dispersion is preferably, 1 mol% or less, more preferably, 0.1 mol% or less per 1 mol of the organic acid silver salt in the solution and, further preferably, positive addition of the photosensitive silver salt is not conducted.
- the photosensitive material can be prepared by mixing an aqueous dispersion of an organic silver salt and an aqueous dispersion of a photosensitive silver salt and the mixing ratio between the organic silver salt.
- a method of mixing two or more kinds of aqueous dispersions of organic silver salts and two or more kinds of aqueous dispersions of photosensitive silver salts upon mixing is used preferably for controlling the photographic properties.
- a total amount of coated silver including silver halide is preferably in a range from 0.1 g/m 2 to 5.0 g/m 2 , more preferably from 0.3 g/m 2 to 3.0 g/m 2 , and further preferably from 0.5 g/m 2 to 2.0 g/m 2 .
- the total amount of coated silver is preferably 1.8 mg/m 2 or less, and more preferably 1.6 mg/m 2 or less.
- the photothermographic material of the invention preferably contains a reducing agent for the organic silver salt.
- the reducing agent may be any substance (preferably, organic substance) capable of reducing silver ions into metallic silver. Examples of the reducing agent are described in JP-A No. 11-65021 (column Nos. 0043 to 0045) and EP No. 0803764A1 (page 7, line 34 to page 18, line 12).
- a so-called hindered phenolic reducing agent or a bisphenol reducing agent having a substituent at the ortho-position to the phenolic hydroxy group is preferred.
- the compound represented by the following formula (R) is more preferred.
- R 11 and R 11' each independently represent an alkyl group having 1 to 20 carbon atoms.
- R 12 and R 12' each independently represent a hydrogen atom or a substituent capable of substituting for a hydrogen atom on a benzene ring.
- L represents an -S- group or a -CHR 13 -group.
- R 13 represents a hydrogen atom or an alkyl group having 1 to 20 carbon atoms.
- X 1 and X 1' each independently represent a hydrogen atom or a group capable of substituting for a hydrogen atom on a benzene ring.
- R 12 and R 12' are, preferably, an alkyl group having 1 to 20 carbon atoms and can include, specifically, a methyl group, an ethyl group, a propyl group, a butyl group, an isopropyl group, a t-butyl group, a t-amyl group, a cyclohexyl group, a 1-methylcyclohexyl group, a benzyl group, a methoxymethyl group, a methoxyethyl group, and the like. More preferred are a methyl group, an ethyl group, a propyl group, an isopropyl group, and a t-butyl group.
- X 1 and X 1' are, preferably, a hydrogen atom, a halogen atom, or an alkyl group, and more preferably, a hydrogen atom.
- L is preferably a -CHR 13 - group.
- R 13 is preferably a hydrogen atom or an alkyl group having 1 to 15 carbon atoms.
- the alkyl group can include a methyl group, an ethyl group, a propyl group, an isopropyl group, and a 2,4,4-trimethylpentyl group.
- Particularly preferable R 13 is a hydrogen atom, a methyl group, an ethyl group, a propyl group, or an isopropyl group.
- R 12 and R 12' are preferably an alkyl group having 2 to 5 carbon atoms, more preferably an ethyl group or a propyl group, and most preferably an ethyl group.
- R 12 and R 12 are preferably a methyl group.
- the primary or secondary alkyl group having 1 to 8 carbon atoms as R 13 is preferably a methyl group, an ethyl group, a propyl group, or an isopropyl group, and more preferably a methyl group, an ethyl group, or a propyl group.
- R 13 is preferably a secondary alkyl group.
- the secondary alkyl group as R 13 is preferably an isopropyl group, an isobutyl group, or a 1-ethylpentyl group, and more preferably an isopropyl group.
- the reducing agent described above shows different thermal developing performances, color tones of developed silver images, or the like depending on the combination of R 11 , R 11' , R 12 , R 12' , and R 13 . Since these performances can be controlled by using two or more kinds of reducing agents at various mixing ratios, it is preferred to use two or more kinds of reducing agents in combination depending on the purpose.
- the addition amount of the reducing agent is, preferably, from 0.1 g/m 2 to 3.0 g/m 2 , more preferably, 0.2 g/m 2 to 1.5 g/m 2 and, further preferably 0.3 g/m 2 to 1.0 g/m 2 . It is preferably contained in a range of 5 mol% to 50 mol%, more preferably, 8 mol% to 30 mol% and, further preferably, 10 mol% to 20 mol% per 1 mol of silver in the image forming layer.
- the reducing agent of the invention is preferably contained in the image forming layer.
- the reducing agent may be incorporated into photothermographic material by being added into the coating solution, such as in the form of solution, emulsion dispersion, solid fine particle dispersion, and the like.
- emulsion dispersing method there can be mentioned a method comprising dissolving the reducing agent using an oil such as dibutyl phthalate, tricresyl phosphate, glyceryl triacetate, diethyl phthalate, or the like, as well as an auxiliary solvent such as ethyl acetate, cyclohexanone, or the like; from which an emulsion dispersion is mechanically produced.
- an oil such as dibutyl phthalate, tricresyl phosphate, glyceryl triacetate, diethyl phthalate, or the like
- an auxiliary solvent such as ethyl acetate, cyclohexanone, or the like
- solid fine particle dispersing method there can be mentioned a method comprising dispersing the powder of the reducing agent in a proper medium such as water, by means of ball mill, colloid mill, vibrating ball mill, sand mill, jet mill, roller mill, or ultrasonics, thereby obtaining solid dispersion.
- a protective colloid such as polyvinyl alcohol
- a surfactant for instance, an anionic surfactant such as sodium triisopropylnaphthalenesulfonate (a mixture of compounds having the isopropyl groups in different substitution sites).
- the dispersion media In the mills enumerated above, generally used as the dispersion media are beads made of zirconia and the like, and Zr and the like eluting from the beads may be incorporated in the dispersion. Although depending on the dispersing conditions, the amount of Zr and the like generally incorporated in the dispersion is in the range from 1 ppm to 1000 ppm. It is practically acceptable so long as Zr is incorporated in an amount of 0.5 mg or less per 1 g of silver.
- an antiseptic for instance, benzisothiazolinone sodium salt
- an antiseptic for instance, benzisothiazolinone sodium salt
- the reducing agent is used as a solid particle dispersion, and the reducing agent is added in the form of fine particles having mean particle size from 0.01 ⁇ m to 10 ⁇ m, and more preferably, from 0.05 ⁇ m to 5 ⁇ m, and further preferably, from 0.1 ⁇ m to 2 ⁇ m.
- other solid dispersions are preferably used with this particle size range.
- the development accelerator described above is used in a range from 0.1 mol% to 20 mol%, preferably, in a range from 0.5 mol% to 10 mol% and, more preferably, in a range from 1 mol% to 5 mol% with respect to the reducing agent.
- the introducing methods to the photothermographic material can include similar methods as those for the reducing agent and, it is particularly preferred to add as a solid dispersion or an emulsion dispersion.
- emulsion dispersion it is preferred to add as an emulsion dispersion dispersed by using a high boiling solvent which is solid at a normal temperature and an auxiliary solvent at a low boiling point, or to add as a so-called oilless emulsion dispersion not using the high boiling solvent.
- hydrazine compounds represented by formula (D) described in the specification of JP-A No. 2002-156727 hydrazine compounds represented by formula (D) described in the specification of JP-A No. 2002-156727
- phenolic or naphtholic compounds represented by formula (2) described in the specification of JP-A No. 2001-264929 phenolic or naphtholic compounds represented by formula (2) described in the specification of JP-A No. 2001-264929.
- Particularly preferred development accelerators of the invention are compounds represented by the following formulae (A-1) and (A-2).
- Formula (A-1) Q 1 -NHNH-Q 2 (wherein Q 1 represents an aromatic group or a heterocyclic group which bonds to -NHNH-Q 2 at a carbon atom, and Q 2 represents one selected from a carbamoyl group, an acyl group, an alkoxycarbonyl group, an aryloxycarbonyl group, a sulfonyl group, and a sulfamoyl group).
- the aromatic group or the heterocyclic group represented by Q 1 is preferably a 5 to 7-membered unsaturated ring.
- Preferred examples include a benzene ring, a pyridine ring, a pyrazine ring, a pyrimidine ring, a pyridazine ring, a 1,2,4-triazine ring, a 1,3,5-triazine ring, a pyrrole ring, an imidazole ring, a pyrazole ring, a 1,2,3-triazole ring, a 1,2,4-triazole ring, a tetrazole ring, a 1,3,4-thiadiazole ring, a 1,2,4-thiadiazole ring, a 1,2,5-thiadiazole ring, a 1,3,4-oxadiazole ring, a 1,2,4-oxadiazole ring, a 1,2,5-oxadiazole ring,
- the rings described above may have substituents and in a case where they have two or more substituents, the substituents may be identical or different from each other.
- substituents can include a halogen atom, an alkyl group, an aryl group, a carbonamide group, an alkylsulfonamide group, an arylsulfonamide group, an alkoxy group, an aryloxy group, an alkylthio group, an arylthio group, a carbamoyl group, a sulfamoyl group, a cyano group, an alkylsulfonyl group, an arylsulfonyl group, an alkoxycarbonyl group, an aryloxycarbonyl group, and an acyl group.
- substituents are groups capable of substitution, they may have further substituents and examples of preferred substituents can include a halogen atom, an alkyl group, an aryl group, a carbonamide group, an alkylsulfonamide group, an arylsulfonamide group, an alkoxy group, an aryloxy group, an alkylthio group, an arylthio group, an acyl group, an alkoxycarbonyl group, an aryloxycarbonyl group, a carbamoyl group, a cyano group, a sulfamoyl group, an alkylsulfonyl group, an arylsulfonyl group, and an acyloxy group.
- a halogen atom an alkyl group, an aryl group, a carbonamide group, an alkylsulfonamide group, an arylsulfonamide group, an alkoxy group, an aryloxy group, an alkylthi
- the carbamoyl group represented by Q 2 is a carbamoyl group preferably having 1 to 50 carbon atoms and, more preferably, having 6 to 40 carbon atoms, and examples can include unsubstituted carbamoyl, methyl carbamoyl, N-ethylcarbamoyl, N-propylcarbamoyl, N-sec-butylcarbamoyl, N-octylcarbamoyl, N-cyclohexylcarbamoyl, N-tert-butylcarbamoyl, N-dodecylcarbamoyl, N-(3-dodecyloxypropyl)carbamoyl, N-octadecylcarbamoyl, N- ⁇ 3-(2,4-tert-pentylphenoxy)propyl ⁇ carbamoyl, N-(2-hexyldecyl)carbamo
- the acyl group represented by Q 2 is an acyl group, preferably having 1 to 50 carbon atoms and, more preferably having 6 to 40 carbon atoms and can include, for example, formyl, acetyl, 2-methylpropanoyl, cyclohexylcarbonyl, octanoyl, 2-hexyldecanoyl, dodecanoyl, chloroacetyl, trifluoroacetyl, benzoyl, 4-dodecyloxybenzoyl, and 2-hydroxymethylbenzoyl.
- the alkoxycarbonyl group represented by Q 2 is an alkoxycarbonyl group, preferably having 2 to 50 carbon atoms and, more preferably, having 6 to 40 carbon atoms and can include, for example, methoxycarbonyl, ethoxycarbonyl, isobutyloxycarbonyl, cyclohexyloxycarbonyl, dodecyloxycarbonyl, and benzyloxycarbonyl.
- the aryloxy carbonyl group represented by Q 2 is an aryloxycarbonyl group, preferably having 7 to 50 carbon atoms and, more preferably having 7 to 40 carbon atoms, and can include, for example, phenoxycarbonyl, 4-octyloxyphenoxycarbonyl, 2-hydroxymethylphenoxycarbonyl, and 4-dodecyloxyphenoxycarbonyl.
- the sulfonyl group represented by Q 2 is a sulfonyl group, preferably having 1 to 50 carbon atoms and, more preferably having 6 to 40 carbon atoms, and can include, for example, methylsulfonyl, butylsulfonyl, octylsulfonyl, 2-hexadecylsulfonyl, 3-dodecyloxypropylsulfonyl, 2-octyloxy-5-tert-octylphenyl sulfonyl, and 4-dodecyloxyphenyl sulfonyl.
- the sulfamoyl group represented by Q 2 is a sulfamoyl group, preferably having 0 to 50 carbon atoms and, more preferably having 6 to 40 carbon atoms, and can include, for example, unsubstituted sulfamoyl, N-ethylsulfamoyl group, N-(2-ethylhexyl)sulfamoyl, N-decylsulfamoyl, N-hexadecylsulfamoyl, N- ⁇ 3-(2-ethylhexyloxy)propyl ⁇ sulfamoyl, N-(2-chloro-5-dodecyloxycarbonylphenyl)sulfamoyl, and N-(2-tetradecyloxyphenyl)sulfamoyl.
- the group represented by Q 2 may further have a group mentioned as the example of the substituent of 5 to 7-membered unsaturated ring represented by Q 1 at the position capable of substitution. In a case where the group has two or more substituents, such substituents may be the same or different from each other.
- a 5 or 6-membered unsaturated ring is preferred for Q 1 , and a benzene ring, a pyrimidine ring, a 1,2,3-triazole ring, a 1,2,4-triazole ring, a tetrazole ring, a 1,3,4-thiadiazole ring, a 1,2,4-thiadiazole ring, a 1,3,4-oxadiazole ring, a 1,2,4-oxadiazole ring, a thioazole ring, an oxazole ring, an isothiazole ring, an isooxazole ring, and a ring in which the ring described above is condensed with a benzene ring or unsaturated hetero ring are further preferred.
- Q 2 is preferably a carbamoyl group and, particularly, a carbamoyl group having a hydrogen atom on
- R 1 represents one selected from an alkyl group, an acyl group, an acylamino group, a sulfonamide group, an alkoxycarbonyl group, and a carbamoyl group.
- R 2 represents one selected from a hydrogen atom, a halogen atom, an alkyl group, an alkoxy group, an aryloxy group, an alkylthio group, an arylthio group, an acyloxy group, and a carbonate ester group.
- R 3 and R 4 each independently represent a group capable of substituting for a hydrpgen atom on a benzene ring which is mentioned as the example of the substituent for formula (A-1).
- R 3 and R 4 may link together to form a condensed ring.
- R 1 is preferably an alkyl group having 1 to 20 carbon atoms (for example, a methyl group, an ethyl group, an isopropyl group, a butyl group, a tert-octyl group, a cyclohexyl group, or the like), an acylamino group (for example, an acetylamino group, a benzoylamino group, a methylureido group, a 4-cyanophenylureido group, or the like), or a carbamoyl group (for example, a n-butylcarbamoyl group, an N,N-diethylcarbamoyl group, a phenylcarbamoyl group, a 2-chlorophenylcarbamoyl group, a 2,4-dichlorophenylcarbamoyl group, or the like).
- An acylamino group including a urei
- R 2 is preferably a halogen atom (more preferably, a chlorine atom or a bromine atom), an alkoxy group (for example, a methoxy group, a butoxy group, an n-hexyloxy group, an n-decyloxy group, a cyclohexyloxy group, a benzyloxy group, or the like), or an aryloxy group (for example, a phenoxy group, a naphthoxy group, or the like).
- a halogen atom more preferably, a chlorine atom or a bromine atom
- an alkoxy group for example, a methoxy group, a butoxy group, an n-hexyloxy group, an n-decyloxy group, a cyclohexyloxy group, a benzyloxy group, or the like
- an aryloxy group for example, a phenoxy group, a naphthoxy group, or the like.
- R 3 is preferably a hydrogen atom, a halogen atom, or an alkyl group having 1 to 20 carbon atoms, and most preferably a halogen atom.
- R 4 is preferably a hydrogen atom, an alkyl group, or an acylamino group, and more preferably an alkyl group or an acylamino group. Examples of the preferred substituent thereof are similar to those for R 1 . In the case where R 4 is an acylamino group, R 4 may preferably link with R 3 to form a carbostyryl ring.
- R 3 and R 4 in formula (A-2) link together to form a condensed ring
- a naphthalene ring is particularly preferred as the condensed ring.
- the same substituent as the example of the substituent referred to for formula (A-1) may bond to the naphthalene ring.
- R 1 is preferably a carbamoyl group. Among them, a benzoyl group is particularly preferred.
- R 2 is preferably an alkoxy group or an aryloxy group and, particularly preferably an alkoxy group.
- the reducing agent in the case where the reducing agent has an aromatic hydroxy group (-OH) or an amino group (-NHR, R represents a hydrogen atom or an alkyl group), particularly in the case where the reducing agent is a bisphenol described above, it is preferred to use in combination, a nonreducing compound having a group capable of reacting with these groups, and that is also capable of forming a hydrogen bond therewith.
- a group forming a hydrogen bond with a hydroxy group or an amino group there can be mentioned a phosphoryl group, a sulfoxide group, a sulfonyl group, a carbonyl group, an amide group, an ester group, a urethane group, a ureido group, a tertiary amino group, a nitrogen-containing aromatic group, and the like.
- Particularly preferred among them is a phosphoryl group, a sulfoxide group, an amide group (not having >N-H moiety but being blocked in the form of >N-Ra (where, Ra represents a substituent other than H)), a urethane group (not having >N-H moiety but being blocked in the form of >N-Ra (where, Ra represents a substituent other than H)), and a ureido group (not having >N-H moiety but being blocked in the form of >N-Ra (where, Ra represents a substituent other than H)).
- R 21 to R 23 each independently represent one selected from an alkyl group, an aryl group, an alkoxy group, an aryloxy group, an amino group, and a heterocyclic group, which may be substituted or unsubstituted.
- R 21 to R 23 contain a substituent
- substituents include a halogen atom, an alkyl group, an aryl group, an alkoxy group, an amino group, an acyl group, an acylamino group, an alkylthio group, an arylthio group, a sulfonamide group, an acyloxy group, an oxycarbonyl group, a carbamoyl group, a sulfamoyl group, a sulfonyl group, a phosphoryl group, and the like, in which preferred as the substituents are an alkyl group or an aryl group, e.g., a methyl group, an ethyl group, an isopropyl group, a t-butyl group, a t-octyl group, a phenyl group, a 4-alkoxyphenyl group, a 4-acyloxyphenyl group, and the like.
- an alkyl group expressed by R 21 to R 23 include a methyl group, an ethyl group, a butyl group, an octyl group, a dodecyl group, an isopropyl group, a t-butyl group, a t-amyl group, a t-octyl group, a cyclohexyl group, a 1-methylcyclohexyl group, a benzyl group, a phenetyl group, a 2-phenoxypropyl group, and the like.
- aryl group there can be mentioned a phenyl group, a cresyl group, a xylyl group, a naphthyl group, a 4-t-butylphenyl group, a 4-t-octylphenyl group, a 4-anisidyl group, a 3,5-dichlorophenyl group, and the like.
- alkoxyl group there can be mentioned a methoxy group, an ethoxy group, a butoxy group, an octyloxy group, a 2-ethylhexyloxy group, a 3,5,5-trimethylhexyloxy group, a dodecyloxy group, a cyclohexyloxy group, a 4-methylcyclohexyloxy group, a benzyloxy group, and the like.
- aryloxy group there can be mentioned a phenoxy group, a cresyloxy group, an isopropylphenoxy group, a 4-t-butylphenoxy group, a naphthoxy group, a biphenyloxy group, and the like.
- an amino group there can be mentioned are a dimethylamino group, a diethylamino group, a dibutylamino group, a dioctylamino group, an N-methyl-N-hexylamino group, a dicyclohexylamino group, a diphenylamino group, an N-methyl-N-phenylamino group, and the like.
- R 21 to R 23 is an alkyl group, an aryl group, an alkoxy group, or an aryloxy group. Concerning the effect of the invention, it is preferred that at least one or more of R 21 to R 23 are an alkyl group or an aryl group, and more preferably, two or more of them are an alkyl group or an aryl group. From the viewpoint of low cost availability, it is preferred that R 21 to R 23 are of the same group.
- the compound expressed by formula (D) used in the invention can be used in the photothermographic material by being incorporated into the coating solution in the form of solution, emulsion dispersion, or solid fine particle dispersion, similar to the case of reducing agent. However, it is preferred to use the compound in the form of solid dispersion.
- the compound expressed by formula (D) forms a hydrogen-bonded complex with a compound having a phenolic hydroxy group or an amino group, and can be isolated as a complex in crystalline state depending on the combination of the reducing agent and the compound expressed by formula (D).
- crystal powder thus isolated in the form of solid fine particle dispersion, because it provides stable performance. Further, it is also preferred to use a method of leading to form complex during dispersion by mixing the reducing agent and the compound expressed by formula (D) in the form of powders and dispersing them with a proper dispersion agent using sand grinder mill or the like.
- the compound expressed by formula (D) is preferably used in a range from 1 mol% to 200 mol%, more preferably from 10 mol% to 150 mol%, and further preferably, from 20 mol% to 100 mol%, with respect to the reducing agent.
- the photosensitive silver halide of the present invention has an average silver iodide content of 40 mol% or higher, and at least 50% of a projected area of total grains of the photosensitive silver halide is occupied by tabular grains having an aspect ratio of 2 or more.
- the photothermographic material contains a compound which substantially reduces visible light absorption by photosensitive silver halide after thermal development versus before thermal development.
- a silver iodide complex-forming agent is used as the compound which substantially reduces visible light absorption by photosensitive silver halide after thermal development.
- the silver iodide complex-forming agent As for the silver iodide complex-forming agent according to the present invention, at least one of a nitrogen atom or a sulfur atom in the compound can contribute to a Lewis acid-base reaction which gives an electron to a silver ion, as a ligand atom (electron donor: Lewis base).
- the stability of the complex is defined by successive stability constant or total stability constant, but it depends on the combination of silver ion, iodo ion and the silver complex forming agent. As a general guide, it is possible to obtain a large stability constant by a chelate effect from intramolecular chelate ring formation, by means of increasing the acid-base dissociation constant and the like.
- the ultra violet-visible light absorption spectrum of the photosensitive silver halide can be measured by a transmission method or a reflection method.
- the ultra violet-visible light absorption spectrum of photosensitive silver halide can be observed by using, independently or in combination, the means of difference spectrum and removal of other compounds by solvent and the like.
- a 5 to 7-membered heterocyclic compound containing at least one nitrogen atom is preferable.
- the said 5 to 7-membered nitrogen-containing heterocycle may be saturated or unsaturated, and may have another substituent.
- the substituent on a heterocycle may bind to each other to form a ring.
- pyridine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indole, isoindole, indolizine, quinoline, isoquinoline, benzimidazole, 1H-imidazole, quinoxaline, quinazoline, cinnoline, phthalazine, 1,8-naphthylizine, 1,10-phenanthroline, benzotriazole, 1,2,4-triazine, 1,3,5-triazine, and the like can be described.
- pyridine imidazole, pyrazine, pyrimidine, pyridazine, phtharazine, triazine, 1,8-naphthylizine, 1,10-phenanthroline, and the like can be described.
- a halogen atom fluorine atom, chlorine atom, bromine atom, or iodine atom
- an alkyl group a straight, a branched, a cyclic alkyl group containing a bicycloalkyl group and an active methine group
- an alkenyl group an alkynyl group, an aryl group, a heterocyclic group (substituted position is not asked)
- an acyl group an alkoxycarbonyl group, an aryloxycarbonyl group, a heterocyclic oxycarbonyl group, a carbamoyl group, an N-acylcarbamoyl group, an N-sulfonylcarbamoyl group, an N-carbamoylcarbamoyl group, an N-sulfamoylcarbamoyl group
- an active methine group means a methine group substituted by two electron-attracting groups, wherein the electron-attracting group means an acyl group, an alkoxycarbonyl group, an aryloxycarbonyl group, a carbamoyl group, an alkylsulfonyl group, an arylsulfonyl group, a sulfamoyl group, a trifluoromethyl group, a cyano group, a nitro group, a carbonimidoyl group.
- two electron-attracting groups may bind each other to form a cyclic structure.
- the salt means a salt formed with positive ion such as an alkaline metal, an alkaline earth metal, a heavy metal, or the like, or organic positive ion such as an ammonium ion, a phosphonium ion, or the like. These substituents may be further substituted by these substituents.
- heterocycles may be further condensed by another ring.
- the substituent is an anion group (e.g., -CO 2 - , -SO 3 - , -S - , or the like)
- the heterocycle containing nitrogen atom of the invention may become a positive ion (e.g., pyridinium, 1,2,4-triazolium, or the like) and may form an intramolecular salt.
- the acid dissociation constant (pKa) of a conjugated acid of nitrogen-containing heterocyclic part in acid dissociation equilibrium of the said compound is preferably 3 to 8 in the mixture solution of tetrahydrofuran/water (3/2) at 25°C, and more preferably, the pKa is 4 to 7.
- heterocyclic compound pyridine, pyridazine, or phtharazine derivative is preferable, and particularly preferable is pyridine or phthalazine derivative.
- heterocyclic compounds have a mercapto group, a sulfide group or a thione group as the substituent
- pyridine, thiazole, isothiazole, oxazole, isoxazole, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, triazine, triazole, thiadiazole, and oxadiazole derivatives are preferable
- thiazole, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, triazine, and triazole derivatives are particularly preferable.
- R 11 and R 12 each independently represent a hydrogen atom or a substituent.
- R 21 and R 22 each independently represent a hydrogen atom or a substituent. However, both of R 11 and R 12 are not hydrogen atoms simultaneously and both of R 21 and R 22 are not hydrogen atoms simultaneously.
- the substituent herein the substituent explained as the substituent of a 5 to 7-membered nitrogen-containing heterocyclic type silver iodide complex-forming agent mentioned above can be described.
- R 31 to R 35 each independently represent a hydrogen atom or a substituent.
- the substituent represented by R 31 to R 35 the substituent of a 5 to 7-membered nitrogen-containing heterocyclic type silver iodide complex-forming agent mentioned above can be used.
- preferred substituting position is R 32 to R 34 .
- R 31 to R 35 may bind each other to form a saturated or an unsaturated ring.
- a preferred substituent is a halogen atom, an alkyl group, an aryl group, a carbamoyl group, a hydroxy group, an alkoxy group, an aryloxy group, a carbamoyloxy group, an amino group, an acylamino group, a ureido group, an alkoxycarbonylamino group, an aryloxycarbonylamino group, or the like.
- the acid dissociation constant (pKa) of conjugated acid of pyridine ring part preferably is 3 to 8 in the mixed solution of tetrahydrofuran /water (3/2) at 25°C, and particularly preferably 4 to 7.
- R 41 to R 44 each independently represent a hydrogen atom or a substituent.
- R 41 to R 44 may bind each other to form a saturated or an unsaturated ring.
- the substituent represented by R 41 to R 44 the substituent of a 5 to 7-membered nitrogen-containing heterocyclic type silver iodide complex-forming agent mentioned above can be described.
- an alkyl group, an alkenyl group, an alkynyl group, an aryl group, a hydroxy group, an alkoxy group, an aryloxy group a heterocyclic oxy group, and a group which forms a phthalazine ring by benzo-condensation are described.
- a hydroxy group exists at the carbon atom adjacent to nitrogen atom of the compound represented by formula (4), there exists equilibrium between pyridazinone.
- the compound represented by formula (4) more preferably forms a phthalazine ring represented by the following formula (5), and furthermore, this phthalazine ring particularly preferably has at least one subsutituent.
- R 51 to R 56 in formula (5) the substituent of a 5 to 7-membered nitrogen-containing heterocyclic type silver iodide complex-forming agent mentioned above can be described.
- an alkyl group, an alkenyl group, an alkynyl group, an aryl group, a hydroxy group, an alkoxy group, an aryloxy group, and the like are described.
- An alkyl group, an alkenyl group, an aryl group, an alkoxy group, and an aryloxy group are preferable and an alkyl group, an alkoxy group, and an aryloxy group are more preferable.
- R 61 to R 63 each independently represent a hydrogen atom or a substituent.
- R 62 the substituent of a 5 to 7-membered nitrogen-containing heterocyclic type silver iodide complex-forming agent mentioned above can be described.
- R 71 and R 72 each independently represent a hydrogen atom or a substituent.
- L represents a divalent linking group.
- n represents 0 or 1.
- an alkyl group (containing a cycloalkyl group), an alkenyl group (containing a cycloalkenyl group), an alkynyl group, an aryl group, a heterocyclic group, an acyl group, an aryloxycarbonyl group, an alkoxycarbonyl group, a carbamoyl group, an imide group, and a complex substituent containing these groups are described as examples.
- a divalent linking group represented by L preferably has the length of 1 to 6 atoms and more preferably has the length of 1 to 3 atoms, and furthermore, may have a substituent.
- One more of the compounds preferably used is a compound represented by formula (8).
- R 81 to R 84 each independently represent a hydrogen atom or a substituent.
- substituent represented by R 81 to R 84 an alkyl group (including a cycloalkyl group), an alkenyl group (including a cycloalkenyl group), an alkynyl group, an aryl group, a heterocyclic group, an acyl group, an aryloxycarbonyl group, an alkoxycarbonyl group, a carbamoyl group, an imide group, and the like are described as examples.
- the compounds represented by formulae (3), (4), (5), (6) and (7) are more preferable and, the compounds represented by formulae (3) and (5) are particularly preferable.
- silver iodide complex-forming agent Preferable examples of silver iodide complex-forming agent are described below, however the present invention is not limited in these.
- the silver iodide complex-forming agent according to the present invention can also be a compound common to a toner, in the case where the agent achieves the function of conventionally known toner.
- the silver iodide complex-forming agent according to the present invention can be used in combination with a toner. And, two or more kinds of the silver iodide complex-forming agents may be used in combination.
- the silver iodide complex-forming agent according to the present invention preferably exists in a film under the state separated from a photosensitive silver halide, such as a solid state. It is also preferably added to the layer adjacent to the image forming layer. Concerning the silver iodide complex-forming agent according to the present invention, a melting point of the compound is preferably adjusted to a suitable range so that it can be dissolved when heated at thermal developing temperature.
- an absorption intensity of ultra violet-visible light absorption spectrum of photosensitive silver halide after thermal development preferably becomes 80% or less as compared with before thermal development, more preferably 40% or less and, particularly preferably 10% or less.
- the silver iodide complex-forming agent according to the invention may be incorporated into a photothermographic material by being added into the coating solution, such as in the form of a solution, an emulsion dispersion, a solid fine particle dispersion, or the like.
- Well known emulsion dispersing methods include a method comprising dissolving the silver iodide complex-forming agent in an oil such as dibutylphthalate, tricresylphosphate, glyceryl triacetate, diethylphthalate, or the like, and an auxiliary solvent such as ethyl acetate, cyclohexanone, or the like, followed by mechanically forming an emulsified dispersion.
- an oil such as dibutylphthalate, tricresylphosphate, glyceryl triacetate, diethylphthalate, or the like
- an auxiliary solvent such as ethyl acetate, cyclohexanone, or the like
- Solid fine particle dispersing methods include a method comprising dispersing the powder of the silver iodide complex-forming agent according to the invention in a proper solvent such as water or the like, by means of ball mill, colloid mill, vibrating ball mill, sand mill, jet mill, roller mill, or ultrasonics, thereby obtaining a solid dispersion.
- a protective colloid such as polyvinyl alcohol
- a surfactant for instance, an anionic surfactant such as sodium triisopropylnaphthalenesulfonate (a mixture of compounds having the three isopropyl groups in different substitution sites)).
- the dispersion media In the mills enumerated above, generally used as the dispersion media are beads made of zirconia and the like, and Zr and the like eluting from the beads may be incorporated in the dispersion.
- the amount of Zr and the like generally incorporated in the dispersion is in a range of from 1 ppm to 1000 ppm. It is practically acceptable so long as Zr is incorporated in the photothermographic material in an amount of 0.5 mg or less per 1 g of silver.
- an antiseptic for instance, benzisothiazolinone sodium salt
- an antiseptic for instance, benzisothiazolinone sodium salt
- the silver iodide complex-forming agent according to the invention is preferably used in the form of a solid dispersion.
- the silver iodide complex-forming agent according to the invention is preferably used in the range from 1 mol% to 5000 mol%, more preferably, from 10 mol% to 1000 mol% and, further preferably, from 50 mol% to 300 mol%, with respect to the photosensitive silver halide in each case.
- 50% by weight or more of the binder used for the image forming layer of the present invention is formed by a polymer latex containing a monomer component represented by the following formula (M) within a range of from 10% by weight to 70% by weight.
- an alkyl group having 1 to 4 carbon atoms is preferred, and more preferably an alkyl group having 1 to 2 carbon atoms is used.
- a halogen atom a fluorine atom, a chlorine atom, or a bromine atom is preferred, and more preferred is a chlorine atom.
- one of R 01 and R 02 represents a hydrogen atom and the other represents a methyl group or a chlorine atom.
- monomer represented by the above formula (M) of the present invention include 2-ethyl-1,3-butadiene, 2-n-propyl-1,3-butadiene, 2,3-dimethyl-1,3-butadiene, 2-methyl-1,3-butadiene, 2-chloro-1,3-butadiene, 1-bromo-1,3-butadiene, 2-fluoro-1,3-butadiene, 2,3-dichloro-1,3-butadiene, and 2-cyano-1,3-butadiene.
- the binder of the present invention are polymers obtained by copolymerizing monomers represented by formula (M), where the copolymerization ratio of the monomer represented by formula (M) for the polymer is in a range of from 10% by weight to 70% by weight, preferably from 15% by weight to 65% by weight, and more preferably from 20% by weight to 60% by weight.
- the other monomers which are capable to copolymerize with the monomer represented by formula (M), are not particularly restricted, and any monomers may be preferably used provided that they are polymerizable by usual radical polymerization or ion polymerization.
- the monomer which can be used preferably, it is capable to select the combination independently and freely from the monomer groups (a) to (j) described below. -Monomer groups (a) to (j)-
- Preferred examples of a polymer copolymerized with the monomer represented by formula (M) of the present invention include copolymers with styrene (for example, random copolymer, block polymer, or the like), copolymers with styrene and butadiene (for example, random copolymer, butadiene-isoprene-styrene block copolymer, styrene-butadiene-isoprene-styrene block copolymer, or the like), copolymers with ethylene and propylene, copolymers with acrylonitrile, copolymers with isobutyrene, copolymers with acrylic esters (for example, as acrylic ester, ethyl acrylate, butyl acrylate, or the like can be used), and copolymers with acrylic ester and acrylonitrile (the same acrylic esters as mentioned above can be used).
- styrene for example, random cop
- the polymer of the present invention is preferably copolymerized with a monomer having an acid group.
- a monomer having an acid group preferred are a carboxylic acid, a sulfonic acid, and a phosphoric acid.
- the copolymerization ratio of a monomer having the acid group is preferably in a range of from 1% by weight to 20% by weight, and more preferably from 1% by weight to 10% by weight.
- Examples of a monomer having the acid group include acylic acid, methacrylic acid, itaconic acid, p-styrene sulfonic acid sodium salt, isopyrene sulfonic acid, phoshoryl ethyl methacrylate, and the like. Any kind of polymer may be used in combination with the polymer copolymerized with the monomer represented by formula (M) as the binder of the invention.
- Suitable as the polymer which can be used in combination are those that are transparent or translucent, and that are generally colorless, such as natural resin or polymer and their copolymers; synthetic resin or polymer and their copolymer; or media forming a film; for example, included are gelatin, rubber, poly(vinyl alcohol), hydroxyethyl cellulose, cellulose acetate, cellulose acetate butyrate, poly(vinyl pyrrolidone), casein, starch, poly(acrylic acid), poly(methylmethacrylic acid), poly(vinyl chloride), poly(methacrylic acid), styrene-maleic anhydride copolymers, styreneacrylonitrile copolymers, styrene-butadiene copolymers, poly(vinyl acetal) (e.g., poly(vinyl formal) or poly(vinyl butyral)), polyester, polyurethane, phenoxy resin, poly(vinylidene chloride),
- the binder of the invention preferably has a grass transition temperature (Tg) in a range of -30°C to 70°C, more preferably -10°C to 50°C, and further preferably 0°C to 40°C, considering work brittleness and image storability.
- Tg grass transition temperature
- Two or more kinds of polymers can be blended for the binder, and in this case, Tg of the blended polymer as a composition weighed average preferably falls within the range above.
- Tg of each phase preferably falls within the range above.
- Tgi is the glass transition temperature (absolute temperature) of the homopolymer obtained with the ith monomer.
- Values for the glass transition temperature (Tgi) of the homopolymers derived from each of the monomers were obtained from J. Brandrup and E. H. Immergut, Polymer Handbook (3rd Edition) (Wiley-Interscience, 1989).
- the polymer used in the invention can be readily obtained by a solution polymerization method, a suspension polymerization method, an emulsion polymerization method, a dispersion polymerization method, an anionic polymerization method, a cationic polymerization method, or the like, however most preferable is an emulsion polymerization method by which polymer can be obtained as a latex.
- the polymer latex is obtained by emulsion polymerization at about 30°C to 100°C, preferably at 60°C to 90°C, for 3 hours to 24 hours with stirring using water or a mixed solvent of water and a water-miscible organic solvent (for example, methanol, ethanol, acetone, or the like) as a dispersion medium, and using a monomer mixture in an amount of 5% by weight to 150% by weight with respect to the dispersion solvent, an emulsifying agent in an amount of 0.1 % by weight to 20% by weight with respect to a total amount of monomers, and a polymerization initiator.
- a water-miscible organic solvent for example, methanol, ethanol, acetone, or the like
- Conditions such as the dispersion medium, monomer concentration, the amount of the initiator, the amount of the emulsifying agent, the amount of the dispersing agent, the reaction temperature and the addition method of the monomer may be appropriately determined considering the kind of the monomer used.
- the dispersing agent is preferably used, if necessary.
- Emulsion polymerization is usually carried out according to the following documents: "Gosei Jushi Emulsion (Synthetic Resin Emulsion)” ed. by Taira Okuda and Hiroshi Inagaki, Polymer Publishing Association (1978); “Gosei Latex no Oyo (Application of Synthetic Latex)” ed. by Taka-aki Sugimura, Yasuo Kataoka, Soichi Suzuki and Keiji Kasahara, Polymer Publishing Association (1993); and “Gosei Latex no Kagaku (Chemistry of Synthetic Latex)" by Soichi Muroi, Polymer Publishing Association (1970).
- Emulsion polymerization method for synthesizing the polymer latex of the invention may be selected from an overall polymerization method, a monomer addition (continuous or divided) method, an emulsion addition method and a seed polymerization method.
- the overall polymerization method, monomer addition (continuous or divided) method, and emulsion addition method are preferable in view of productivity of the latex.
- the polymerization initiator described above may have a radical generation ability, and examples of them available include inorganic peroxides such as persulfate salts and hydrogen peroxide, peroxides described in the catalogue of organic peroxides by Nippon Oil and Fat Co., and azo compounds described in azo polymerization initiator catalogue by Wako Pure Chemical Industries, Ltd. Among them, a water-soluble peroxides such as persulfate, and water-soluble azo compounds described in azo polymerization initiator catalogue by Wako Pure Chemical Industries, Ltd., are preferable.
- Ammonium persulfate, sodium persulfate, potassium persulfate, azobis(2-methylpropionamidine) hydrochloride, azobis(2-methyl-N-(2-hydroxyethyl)propionamide and azobiscyanovaleric acid are more preferable, and particularly, peroxides such as ammonium persulfate, sodium persulfate and potassium persulfate are preferable from the viewpoint of image storability, solubility, and cost.
- the addition amount of the polymerization initiator described above is preferably in a range from 0.3% by weight to 2.0% by weight, more preferably 0.4% by weight to 1.75% by weight, and particularly preferably 0.5% by weight to 1.5% by weight, based on a total amount of monomers.
- Image storability decreases when the amount of the polymerization initiator is less than 0.3% by weight, while the latex tends to be aggregated to deteriorate coating ability when the amount of the polymerization initiator exceeds 2.0% by weight.
- any surfactants such as an anionic surfactant, a nonionic surfactant, a cationic surfactant, or an amphoteric surfactant can be employed.
- An anionic surfactant is preferably employed from the viewpoint of dispersibility and image storability, and more preferred is a sulfonic acid-type anionic surfactant which maintains the polymerization stability even in a small amount and has a hydrolysis resistance.
- a sulfonic acid-type surfactant is preferably used in a rage of from 0.1% by weight to 10.0% by weight based on the total amount of monomers, more preferably from 0.2% by weight to 7.5% by weight, and particularly preferably from 0.3% by weight to 5.0% by weight. Stability in the emulsion polymerization process can not secure when the addition amount of the polymerization emulsifying agent is less than 0.1% by weight, while image storability decreases when the addition amount exceeds 10.0 % by weight.
- Chelating agents are preferably used for the synthesis of the polymer latex used in the invention.
- the chelating agent is a compound capable of coordinating multi-valent metal ions such as iron ion, and alkali earth metal ions such as calcium ion, and examples thereof include the compounds described in JP-B No. 6-8956; USP No. 5053322; and JP-A Nos.
- 4-73645 4-127145, 4-247073, 4-305572, 6-11805, 5-173312, 5-66527, 5-158195, 6-118580, 6-110168, 6-161054, 6-175299, 6-214352, 7-114161, 7-114154, 7-120894, 7-199433, 7-306504, 9-43792, 8-314090, 10-182571, 10-182570, and 11-190892.
- the chelating agent used in the invention is preferably an inorganic chelating compound (sodium tripolyphosphate, sodium hexametaphosphate, sodium tetrapolyphosphate, or the like), an aminopolycarboxylic acid chelating compound (nitrilotriacetic acid, ethylenediamine tetraacetic acid, or the like), an organic phosphonic acid chelating agent (compounds described in Research Disclosure No. 18170, JP-A Nos.
- an inorganic chelating compound sodium tripolyphosphate, sodium hexametaphosphate, sodium tetrapolyphosphate, or the like
- an aminopolycarboxylic acid chelating compound nitrilotriacetic acid, ethylenediamine tetraacetic acid, or the like
- organic phosphonic acid chelating agent compounds described in Research Disclosure No. 18170, JP-A Nos.
- aminopolycarboxylic acid derivative Preferable examples of the aminopolycarboxylic acid derivative are described in the supplement table of "EDTA (-Chemistry of Complexane-)", Nankodo 1977.
- a part of the carboxyl group of these compounds may be substituted by a salt of alkali metal such as sodium or potassium, or an ammonium salt.
- aminocarboxylic acid derivatives include iminodiacetic acid, N-methyliminodiacetic acid, N-(2-aminoethyl)iminodiacetic acid, N-(carbamoylethyl)iminodiacetic acid, nitrilotriacetic acid, ehylenediamine-N,N'-diacetic acid, ehylenediamine-N,N'-di- ⁇ -propionic acid, ethylenediamine-N,N'-di- ⁇ -propionic acid, N,N'-ethylene-bis( ⁇ -o-hydroxyphenyl)glycine, N,N'-di(2-hydroxybenzyl)ethylenediamine-N,N'-diacetic acid, ethylenediamine-N,N'-diacetic acid-N,N'-diacetohydroxamic acid, N-hydroxyethylethylenediamine-N,N',N'-triacetic acid,
- the addition amount of the chelating agent described above is preferable 0.01% by weight to 0.4% by weight, more preferably 0.02% by weight to 0.3% by weight, and particularly preferably 0.03% by weight to 0.15% by weight based on a total amount of monomers.
- Metal ions mingling in the production process of the polymer latex are insufficiently trapped when the amount of the chelating agent is less than 0.01% by weight to decrease stability of the latex against aggregation to deteriorate coating ability.
- the content exceeds 0.4%, on the other hand, the viscosity of the latex increases to deteriorate coating ability.
- the chain transfer agent is preferably used in the synthesis of the polymer latex used in the invention.
- the compounds described in Polymer Handbook Third Edition (Wiley-Interscience, 1989) are preferable as the chain transfer agents. Sulfur compounds are preferable since they have high chain transfer ability to make the amount of use of the reagent small.
- Particularly preferable chain reaction agents are hydrophobic mercaptan chain transfer agents such as tert-dodecylmercaptan, n-dodecylmercaptan, and the like.
- the amount of the chain transfer agent described above is preferable 0.2% by weight to 2.0% by weight, more preferably 0.3% by weight to 1.8% by weight, and particularly preferably 0.4% by weight to 1.6% by weight based on a total amount of monomers. Work brittleness is decreased when the amount of the chain transfer agent is less than 0.2% by weight, while image storability is deteriorated when the amount exceeds 2.0% by weight.
- additives such as an electrolyte, a stabilizer, a thickener, a defoaming agent, an antioxidant, a vulcanizing agent, an antifreeze agent, a gelling agent, vulcanization accelerator, and the like described in Synthetic Rubber Handbook and the like may be used in addition to the compounds above.
- x, y, z, and z' in chemical formula show the mass ratios in the polymer composition, and the sum of x, y, z, and z' is equal to 100%.
- Tg represents the glass transition temperature of a dry film obtained from the polymer.
- the mixture was heated to 90°C with stirring for 3 hours. After the reaction was completed, the inner temperature of the reaction vessel was cooled to room temperature.
- the polymer obtained was filtered through a filter cloth (mesh: 225), then 1145 g of the example compound P-1 (solid content of 45 % by weight, particle diameter of 112 nm) was obtained.
- water solvent for the solvent of a coating solution for the polymer latex, water solvent can be used and any of water-admixing organic solvents may be used in combination.
- organic solvents there can be used, for example, alcohols such as methyl alcohol, ethyl alcohol, propyl alcohol, and the like; cellosolves such as methyl cellosolve, ethyl cellosolve, butyl cellosolve, and the like; ethyl acetate, dimethylformamide, and the like.
- the addition amount of the organic solvent is 50% by weight or less, and more preferably 30% by weight or less, with respect to the solvent.
- the concentration of the polymer is preferably 10% by weight to 70% by weight, more preferably 20% by weight to 60% by weight, and particularly preferably 30% by weight to 55% by weight, with respect to the latex solution in each case.
- the equilibrium water content under 25°C and 60%RH is preferably 2% by weight or lower, but is more preferably, 0.01% by weight to 1.5% by weight, and is further preferably, 0.02% by weight to 1.0% by weight.
- polymers capable of being dispersed in an aqueous solvent are particularly preferable.
- dispersed states may include a latex, in which water-insoluble fine particles of hydrophobic polymer are dispersed, or such in which polymer molecules are dispersed in molecular states or by forming micelles, but preferred are latex-dispersed particles.
- the average particle size of the latex-dispersed particles is in a range of from 1 nm to 50,000 nm, preferably from 5 nm to 1,000 nm, more preferably 10 nm to 500 nm, and further preferably 50 nm to 200 nm.
- particle size distribution of the dispersed particles there is no particular limitation concerning particle size distribution of the dispersed particles, and they may be widely distributed or may exhibit a monodisperse particle size distribution. From the viewpoint of controlling the physical properties of the coating solution, preferred mode of usage includes mixing two or more types of particles each having monodisperse particle distribution.
- hydrophilic polymers such as gelatin, polyvinyl alcohol, methyl cellulose, hydroxypropyl cellulose, carboxymethyl cellulose, and the like.
- the hydrophilic polymers above are added at an amount of 30% by weight or less, preferably 20% by weight or less, with respect to the total weight of the binder incorporated in the image forming layer.
- the image forming layer of the present invention is preferably formed by using a polymer latex.
- the weight ratio for total binder to organic silver salt is preferably in a range of from 1/10 to 10/1, more preferably from 1/3 to 5/1, and further preferably 1/1 to 3/1.
- the weight ratio for total binder to photosensitive silver halide is preferably in a range of from 400 to 5, and more preferably, from 200 to 10.
- the total amount of binder in the image forming layer of the invention is preferably in a range from 0.2 g/m 2 to 30 g/m 2 , more preferably from 1 g/m 2 to 15 g/m 2 , and further preferably 2 g/m 2 to 10 g/m 2 .
- a crosslinking agent for crosslinking or a surfactant and the like to improve coating properties.
- antifoggant As an antifoggant, stabilizer and stabilizer precursor usable in the invention, there can be mentioned those disclosed as patents in paragraph number 0070 of JP-A No. 10-62899 and in line 57 of page 20 to line 7 of page 21 of EP-A No. 0803764A1, the compounds described in JP-A Nos. 9-281637 and 9-329864, in USP No. 6,083,681, and in EP-A No. 1048975.
- the antifoggant preferably used in the invention is an organic halogen compound, and those disclosed in paragraph Nos. 0111 to 0112 of JP-A No. 11-65021 can be enumerated as examples thereof.
- the organic halogen compound expressed by formula (P) in JP-A No. 2000-284399 the organic polyhalogen compound expressed by formula (II) in JP-A No. 10-339934, and organic polyhalogen compounds described in JP-A Nos. 2001-31644 and 2001-33911 are preferred.
- Q represents one selected from an alkyl group, an aryl group, and a heterocyclic group
- Y represents a divalent linking group
- n represents 0 or 1
- Z 1 and Z 2 each represent a halogen atom
- X represents a hydrogen atom or an electron-attracting group.
- Q is preferably an aryl group, or a heterocyclic group.
- Q is preferably a nitrogen-containing heterocyclic group having 1 or 2 nitrogen atoms, and particularly preferably a 2-pyridyl group or a 2-quinolyl group.
- Q in the case where Q is an aryl group, Q preferably is a phenyl group substituted by an electron-attracting group whose Hammett substituent coefficient ⁇ p yields a positive value.
- Hammett substituent coefficient reference can be made to Journal of Medicinal Chemistry, vol. 16, No. 11 (1973), pp. 1207 to 1216, and the like.
- examples include, halogen atoms (fluorine atom ( ⁇ p value: 0.06), chlorine atom ( ⁇ p value: 0.23), bromine atom ( ⁇ p value: 0.23), iodine atom ( ⁇ p value: 0.18)), trihalomethyl groups (tribromomethyl ⁇ p value: 0.29), trichloromethyl ( ⁇ p value: 0.33), trifluoromethyl ( ⁇ p value: 0.54)), a cyano group ( ⁇ p value: 0.66), a nitro group ( ⁇ p value: 0.78), an aliphatic aryl sulfonyl goup or a heterocyclic sulfonyl group (for example, methanesulfonyl ( ⁇ p value: 0.72)), an aliphatic aryl acyl group or a heterocyclic acyl group (for example, acetyl ( ⁇ p value: 0.50) and benzoyl ( ⁇ p value: 0.43)), an alphatic atoms (
- Preferred range of the ⁇ p value is from 0.2 to 2.0, and more preferably, from 0.4 to 1.0.
- Particularly preferred as the electron-attracting groups are a carbamoyl group, an alkoxycarbonyl group, an alkylsulfonyl group, and an alkylphosphoryl group, and most preferred among them is a carbamoyl group.
- X is preferably an electron-attracting group, and more preferably, a halogen atom, an aliphatic aryl sulfonyl group, a heterocyclic sulfonyl group, an aliphatic aryl acyl group, a heterocyclic acyl group, an aliphatic aryl oxycarbonyl group, a heterocyclic oxycarbonyl group, a carbamoyl group, or a sulfamoyl group; particularly preferred among them is a halogen atom.
- halogen atoms preferred are chlorine atom, bromine atom, and iodine atom; more preferred are chlorine atom and bromine atom; and particularly preferred is bromine atom.
- n represents 0 or 1, and preferably represents 1.
- organic polyhalogen compounds of the invention other than those above, there can be mentioned compounds disclosed in JP-A Nos. 2001-31644, 2001-56526, and 2001-209145.
- the compounds expressed by formula (H) of the invention are preferably used in an amount of from 10 -4 mol to 1 mol, more preferably, 10 -3 mol to 0.5 mol, and further preferably, 1 ⁇ 10 -2 mol to 0.2 mol, per 1 mol of non-photosensitive silver salt incorporated in the image forming layer.
- usable methods for incorporating the antifoggant into the photosensitive material are those described above in the method for incorporating the reducing agent.
- the organic polyhalogen compound is also preferably used in the form of solid fine particle dispersion.
- antifoggants there can be mentioned a mercury (II) salt described in paragraph number 0113 of JP-A No. 11-65021, benzoic acids described in paragraph number 0114 of the same literature, a salicylic acid derivative described in JP-A No. 2000-206642, a formaline scavenger compound expressed by formula (S) in JP-A No. 2000-221634, a triazine compound related to Claim 9 of JP-A No. 11-352624, a compound expressed by formula (III), 4-hydroxy-6-methyl-1,3,3a,7-tetrazaindene and the like, described in JP-A No. 6-11791.
- a mercury (II) salt described in paragraph number 0113 of JP-A No. 11-65021
- benzoic acids described in paragraph number 0114 of the same literature
- a salicylic acid derivative described in JP-A No. 2000-206642
- the photothermographic material of the invention may further contain an azolium salt in order to prevent fogging.
- Azolium salts useful in the present invention include a compound expressed by formula (XI) described in JP-A No. 59-193447, a compound described in JP-B No. 55-12581, and a compound expressed by formula (II) in JP-A No. 60-153039.
- the azolium salt may be added to any part of the photothermographic material, but as an additional layer, it is preferred to select a layer on the side having thereon the image forming layer, and more preferred is to select the image forming layer itself.
- the azolium salt may be added at any time of the process of preparing the coating solution; in the case where the azolium salt is added into the image forming layer, any time of the process may be selected, from the preparation of the organic silver salt to the preparation of the coating solution, but preferred is to add the salt after preparing the organic silver salt and just before coating.
- any method for adding the azolium salt any method using a powder, a solution, a fine-particle dispersion, and the like, may be used.
- additives such as sensitizing agents, reducing agents, toners, and the like.
- the azolium salt may be added at any amount, but preferably, it is added in a range from 1 ⁇ 10 -6 mol to 2 mol, and more preferably, from 1 ⁇ 10 -3 mol to 0.5 mol, per 1 mol of silver.
- formic acid or formates as a strong fogging agent, it is preferably incorporated into the side having thereon the image forming layer containing photosensitive silver halide, at an amount of 5 mmol or less, and preferably 1 mmol or less, per 1 mol of silver.
- Acids resulting from the hydration of diphosphorus pentaoxide, or a salt thereof include metaphosphoric acid (salt), pyrophosphoric acid (salt), orthophosphoric acid (salt), triphosphoric acid (salt), tetraphosphoric acid (salt), hexametaphosphoric acid (salt), and the like.
- Particularly preferred acids obtainable by the hydration of diphosphorus pentaoxide or salts thereof include orthophosphoric acid (salt) and hexametaphosphoric acid (salt).
- the salts are sodium orthophosphate, sodium dihydrogen orthophosphate, sodium hexametaphosphate, ammonium hexametaphosphate, and the like.
- the addition amount of the acid obtained by hydration of diphoshorus pentaoxide or the salt thereof may be set as desired depending on sensitivity and fogging, but preferred is an amount of from 0.1 mg/m 2 to 500 mg/m 2 , and more preferably, from 0.5 mg/m 2 to 100 mg/m 2 .
- the temperature for preparing the coating solution for the image forming layer of the invention is preferably from 30°C to 65°C, more preferably, from 35°C or more to less than 60°C, and further preferably, from 35°C to 55°C. Furthermore, the temperature of the coating solution for the image forming layer immediately after adding the polymer latex is preferably maintained in the temperature range from 30°C to 65°C.
- the photothermographic material of the present invention may be either "single-sided type" having an image forming layer on one side of the support, or "double-sided type” having image forming layers on both sides of the support.
- the black and white photothermographic material of the present invention is preferably applied for an image forming method to record radiation images using a fluorescent intensifying screen.
- the photothermographic material can be preferably employed as described below: where the photothermographic material is exposed with a monochromatic light having the same wavelength as the main emission peak wavelength of the fluorescent intensifying screen and having a half band width of 15 ⁇ 5 nm, and after a thermal developing process, an exposure value required for a density of fog+0.5 for an image obtained by removing the image forming layer that is disposed on the opposite side of an exposure face is 0.005 Lux ⁇ sec to 0.07 Lux ⁇ sec.
- the image forming method using the photothermographic materials described above comprises:
- the photothermographic material used for the assembly in the present invention is subjected to X-ray exposure through a step wedge tablet and thermal development.
- the thermal developed image may have the photographic characteristic curve where the average gamma ( ⁇ ) made at the points of a density of fog+0.1 and a density of fog+0.5 is from 0.5 to 0.9, and the average gamma ( ⁇ ) made at the points of a density of fog+1.2 and a density of fog+1.6 is from 3.2 to 4.0.
- the use of photothermographic material having the aforesaid photographic characteristic curve would give the radiation images with excellent photographic properties that exhibit an extended bottom portion and high gamma value at a middle density area.
- the photographic properties mentioned have the advantage of that the depiction in a low density portion on the mediastinal region and the heart shadow region having little X-ray transmittance becomes excellent, and that the density becomes easy to view, and that the contrast in the images on the lung field region having much X-ray transmittance becomes excellent.
- the photothermographic material having the preferred photographic characteristic curve mentioned above can be easily prepared, for example, by the method where each of the image forming layer of both sides may be constituted of two or more image forming layers containing silver halide and having a sensitivity different from each other.
- the aforesaid image forming layer preferably comprises an emulsion of high sensitivity for the upper layer and an emulsion with photographic properties of low sensitivity and high contrast for the lower layer.
- the sensitivity difference between the silver halide emulsion in each layer is preferably from 1.5 times to 20 times, and more preferably from 2 times to 15 times.
- the ratio of the amount of emulsion used for forming each layer may depend on the sensitivity difference between emulsions used and the covering power. Generally, as the sensitivity difference is large, the ratio of the using amount of high sensitivity emulsion is reduced. For example, if the sensitivity difference is two times, and the covering power is equal, the ratio of the amount of high sensitivity emulsion to low sensitivity emulsion would be preferably adjusted to be in a range from 1:20 to 1:50 based on silver amount.
- the techniques such as an emulsion sensitizing method, kinds of additives and constituents employed in the production of the photothermographic material of the present invention are not particularly limited.
- various kinds of techniques described in JP-A Nos. 2-68539, 2-103037 and 2-115837 can be applied.
- the radiographic intensifying screen essentially comprises a support and a fluorescent substance layer coated on one side of the support as the fundamental structure.
- the fluorescent substance layer is a layer where the fluorescent substance is dispersed in binders.
- a transparent protective layer is generally disposed to protect the fluorescent substance layer from chemical degradation and physical shock.
- Tungstate fluorescent substances (CaWO 4 MgWO 4 CaWO 4 :Pb and the like), terbium activated rare earth sulfoxide fluorescent substances (Y 2 O 2 S:Tb, Gd 2 O 2 S:Tb, La 2 O 2 S:Tb, (Y,Gd) 2 O 2 S:Tb, (Y,Gd)O 2 S:Tb, Tm, and the like), terbium activated rare earth phosphate fluorescent substances (YPO 4 :Tb, GdPO 4 :Tb, LaPO 4 :Tb, and the like), terbium activated rare earth oxyhalogen fluorescent substances (LaOBr:Tb, LaOBr:Tb, Tm, LaOCI:Tb, LaOCI:Tb, Tm, LaOBr:Tb, GdOBr:Tb, GdOCl:Tb, and the like), thulium activated rare earth oxyhalogen fluorescent substances (LaOBr:Tm
- the fluorescent substances are preferably packed in the grain size graded structure.
- fluorescent substance particles having a large particle size is preferably coated at the side of the surface protective layer and fluorescent substance particles having a small particle size is preferably coated at the side of the support.
- the small particle size of fluorescent substance is preferably in a range from 0.5 ⁇ m to 2.0 ⁇ m and the large size is preferably in a range from 10 ⁇ m to 30 ⁇ m.
- the single-sided type photothermographic material of the present invention is preferably applied for an X-ray photosensitive material used for mammography.
- the method to draw the photographic characteristic curve of the photothermographic material of the present invention is explained below.
- molybdenum target tube which emits a low pressure X-ray
- the intensifying screen comprising substantially the fluorescent substance comprising Gd 2 O 2 S:Tb
- the photographic characteristic curve obtained by changing the X-ray exposure value by the method of distance using the X-ray beam emitted by tungsten target tube as the beam source may give substantially the same result obtained above.
- X-ray emitted from tungsten target tube operated by three-phase electric power supply at 50 KVp and penetrated through an aluminum plate having a thickness of 3 mm is used.
- the commercially availabe UM-Fine screen and the photosensitive material to be measured are made contact and installed in ECMA cassette produced by Fuji Photo Film Co., Ltd.
- the photothermographic material and the screen may be set, from X-ray tube, in turn, X-ray irradiation is performed.
- the assembly is subjected to exposure with a step wedge tablet having a width of 0.15 in terms of log E.
- the exposed photothermographic material is thermally developed under the determined condition. Thereafter, density is measured, and then the photographic characteristic curve is obtained where the logarithm of radiation exposure value is plotted on abscissa axis, and the optical density is plotted on ordinate axis.
- the contrast is determined from the gradient (tan ⁇ , when the angle to the abscissa axis is ⁇ ) of the straight line connecting the points at a density of fog+0.25 and a density of fog+2.0.
- the measuring method of the sensitivity of the photosensitive material is explained.
- the light source a monochromatic light having the same wavelength as a main emission peak wavelength of the fluorescent intensifying screen is employed.
- a method using the filter system where interference filters are combined can be used. According to the aforesaid method, usually the monochromic light having a required exposure value and a half band width of 15 ⁇ 5 nm can be obtained easily, although it depends also on the combination of interference filters used.
- the monochromic light whose intensity is correctly measured by an illuminometer in advance is employed as the light source.
- the photothermographic material is subjected to exposure with a step wedge tablet through a neutral filter for one second, where the photothermographic material and the light source are one meter apart.
- the density is measured after a thermal developing process, the sensitivity can be obtained by determining the exposure value required to give a density of fog+0.5 and can be expressed by Lux • second.
- Preferred sensitivity of the photothermographic material used for mammography according to the invention is 0.01 Lux ⁇ sec to 0.07 Lux ⁇ sec.
- Preferred contrast for the photothermographic material used for mammography according to the present invention is from 3.0 to 5.0.
- the fluorescent intensifying screen for mammography used in the invention is explained in detail below.
- the fluorescent intensifying screen used for photographic assembly of mammography used in the present invention is required to have high image sharpness in comparison with the conventional chest diagnosis.
- the image sharpness of commercially available fluorescent intensifying screen used for mammography is usually enhanced by coloring the fluorescent substance layer.
- the light emitted by X-ray beam absorbed in the inner side of the fluorescent substance to the X-ray irradiation plane cannot effectively be taken out from the colored screen.
- it is required to provide the intensifying screen coated with the amount of fluorescent substances enough to absorb X-ray and having high image sharpness without coloring the fluorescent substance layer substantially.
- the particle size of fluorescent substances preferably may be below a fixed size.
- the measurement of the particle size is performed by Coulter counter or observation through electron microscope.
- the mean equivalent spherical diameter of the fluorescent substance particles is preferably in a range from 1 ⁇ m to 5 ⁇ m, and more preferably from 1 ⁇ m to 4 ⁇ m.
- the weight ratio of binder/fluorescent substance is preferably from 1/50 to 1/20, and more preferably from 1/50 to 1/25.
- the binder known substances described in JP-A No. 6-75097, from line 45 on right column at page 4 to line 10 on left column at page 5, can be employed.
- the thermoplastic elastomer having a softening temperature or a melting temperature of 30°C to 150°C can be preferably used alone or in combination with the other binder polymer.
- the proper selection of the binder used is very important to resist to the defect, because of the poor durability of the screen. It is desirable to choose entirely flexible binders as the solution for the defect. And also plasticizers and the like are preferably added in the fluorescent substance layer.
- thermoplastic elastomer polystyrenes, polyolefines, polyurethanes, polyesters, polyamides, polybutadienes, ethylene vinyl acetates, natural rubbers, fluorinated rubbers, polyisoprenes, ethylene chlorides, styrene-butadiene rubbers, silicone rubbers, and the like can be described.
- polyurethanes are particularly preferred.
- the selection of the binder for the undercoat of the fluorescent substance layer is very important. Acrylate binders are preferably employed.
- the thickness of the surface protective layer is preferably thin.
- the preferred thickness of the surface protective layer is in a range of from 2 ⁇ m to 7 ⁇ m.
- the materials for the surface protective layer of the screen films such as PET (especially, stretched type), PEN, nylon, and the like can be preferably stuck thereon.
- the surface protective layer of the screen is preferably formed by coating the fluorinated resins dissolved in a suitable solvent from the standpoint of preventing stain.
- the preferred embodiments of the fluorinated resins are described in detail in JP-A No. 6-75097, line 4 on left column at page 6 to line 43 on right column at the same page.
- the resin suited for solvent coating type to form the surface protective layer polyurethane resins, polyacrylate resins, cellulose derivatives, polymethyl methacrylates, polyester resins, epoxy resins, and the like can be mentioned other than fluorinated resins.
- the volume filling factor of the fluorescent substance is preferably from 60% to 80%, and more preferably from 65 % to 80%.
- the compression processes of fluorescent substance layer described in JP-A No. 6-75097, line 29 on right column at page 4 to line 1 on left column at page 6, are preferably applied.
- the fluorescent substance used in the present invention preferably comprises substantially Gd 2 O 2 S:Tb.
- the term "substantially” described here means that main component of the fluorescent substance is Gd 2 O 2 S:Tb, and several % of any other additives to improve the property of the screen, and silica and the like to decorate the surface can preferably be included. And also, in place of Gd, Y, La, or Lu can be possibly mixed inside the ratio of several ten %.
- fluorescent substance having a heavy density is preferred to absorb X-ray effectively.
- fluorescent substance that shows a desirable X-ray absorption ability in beam source used for mammography YTaO 4 and the one adding various kinds of activator as the emission center thereto, CaWO 4 , BaFBr:Eu, and the like can be mentioned besides Gd 2 O 2 S:Tb.
- the image forming method using photothermographic material is perfomed in combination with a fluorescent substance having a main emission peak at 400 nm or lower. And more preferably, the image forming method is performed in combination with a fluorescent substance having a main emission peak at 380 nm or lower. Either single-sided photosensitive material or double-sided photosensitive material can be applied for the assembly.
- the screen having a main emission peak at 400 nm or lower the screens described in JP-A No. 6-11804 and WO No. 93/01521 and the like are used, but the present invention is not limited to these.
- crossover cut for double-sided photosensitive material
- anti-halation for single-sided photosensitive material
- the technique described in JP-A No. 8-76307 can be applied.
- ultraviolet absorbing dyes the dye described in JP-A No. 2001-144030 is particularly preferred.
- the photothermographic material according to the invention can have a non-photosensitive layer in addition to the image forming layer.
- the non-photosensitive layers can be classified depending on the layer arrangement into (a) a surface protective layer provided on the image forming layer (on the side farther from the support), (b) an intermediate layer provided among plural image forming layers or between the image forming layer and the protective layer, (c) an undercoat layer provided between the image forming layer and the support, and (d) a back layer provided to the side opposite to the image forming layer.
- a layer that functions as an optical filter may be provided as (a) or (b) above.
- An antihalation layer may be provided as (c) or (d) to the photothermographic material.
- thermal development process is usually performed by elevating the temperature of the photothermographic material exposed imagewise.
- the temperature for development is preferably 90°C to 180°C, more preferably 95°C to 140°C, and further preferably 100°C to 130°C.
- Time period for development is generally 1 second to 60 seconds, preferably 3 seconds to 30 seconds, and more preferably 5 seconds to 15 seconds.
- a drum type heater or a plate type heater may be used as for the process for thermal development.
- a plate type heater process is preferred.
- a preferable process for thermal development by a plate type heater is a process described in JP-A No. 11-133572, which discloses a thermal developing device in which a visible image is obtained by bringing a photothermographic material with a formed latent image into contact with a heating means at a thermal developing portion, wherein the heating means comprises a plate heater, and a plurality of pressing rollers are oppositely provided along one surface of the plate heater, the thermal developing device is characterized in that thermal development is performed by passing the photothermographic material between the pressing rollers and the plate heater.
- the plate heater is divided into 2 to 6 portions, with the leading end having a lower temperature by 1°C to 10°C.
- 4 sets of plate heaters which can be independently subjected to the temperature control, are used, and are controlled so that they respectively become 112°C, 119°C, 121°C, and 120°C.
- the heater is more stably controlled, and a top part of one sheet of the photothermographic material is exposed and thermal development of the exposed portion is started before exposure of the end part of the sheet has completed,
- Preferred imagers capable of rapid processing for use in the invention are described in, for example, JP-A Nos. 2002-289804 and 2002-287668.
- thermal development within 14 seconds is possible with a plate type heater having three heating plates which are controlled, for example, at 107°C, 121°C, and 121°C, respectively.
- the output time period for the first sheet can be reduced to about 60 seconds.
- the photothermographic material and the image forming method of the invention are employed for forming black and white images by silver imaging.
- the photothermographic material and the image forming method of the invention can be employed for medical imaging.
- the product was pelletized, dried at 130°C for 4 hours, melted at 300°C. Thereafter, the mixture was extruded from a T-die and rapidly cooled to form a non-tentered film.
- the film was stretched along the longitudinal direction by 3.3 times using rollers of different peripheral speeds, and then stretched along the transverse direction by 4.5 times using a tenter machine.
- the temperatures used for these operations were 110°C and 130°C, respectively.
- the film was subjected to thermal fixation at 240°C for 20 seconds, and relaxed by 4% along the transverse direction at the same temperature. Thereafter, the chucking part was slit off, and both edges of the film were knurled. Then the film was rolled up at the tension of 4 kg/cm 2 to obtain a roll having the thickness of 175 ⁇ m.
- Both surfaces of the support were treated at room temperature at 20 m/minute using Solid State Corona Discharge Treatment Machine Model 6KVA manufactured by Piller GmbH. It was proven that treatment of 0.375 KV ⁇ A ⁇ minute ⁇ m -2 was executed, judging from the readings of current and voltage on that occasion. The frequency upon this treatment was 9.6 kHz, and the gap clearance between the electrode and dielectric roll was 1.6 mm.
- a solution was prepared by adding 4.3 mL of a 1% by weight potassium iodide solution, and then 3.5 mL of 0.5 mol/L sulfuric acid, 36.5 g of phthalated gelatin, and 160 mL of a 5% by weight methanol solution of 2,2'-(ethylene dithio)diethanol to 1421 mL of distilled water.
- the solution was kept at 75°C while stirring in a stainless steel reaction vessel, and thereto were added total amount of: solution A prepared through diluting 22.22 g of silver nitrate by adding distilled water to give the volume of 218 mL; and solution B prepared through diluting 36.6 g of potassium iodide with distilled water to give the volume of 366 mL.
- a method of controlled double jet was executed through adding total amount of the solution A at a constant flow rate over 16 minutes, accompanied by adding the solution B while maintaining the pAg at 10.2.
- Potassium hexachloroiridate (III) was added in its entirety to give 1 ⁇ 10 -4 mol per 1 mol of silver, at 10 minutes post initiation of the addition of the solution C and the solution D.
- potassium hexacyanoferrate (II) in an aqueous solution was added in its entirety to give 3 ⁇ 10 -4 mol per 1 mol of silver.
- 32.7 g of phthalated gelatin was added.
- the mixture was adjusted to the pH of 3.8 with 0.5 mol/L sulfuric acid. After stopping stirring, the mixture was subjected to precipitation/ desalting/ water washing steps.
- the mixture was adjusted to the pH of 5.9 with 1 mol/L sodium hydroxide to produce a silver halide dispersion having the pAg of 11.0.
- the silver halide emulsion A was a pure silver iodide emulsion, and the obtained silver halide grains had a mean projected area equivalent diameter of 0.93 ⁇ m, a variation coefficient of a projected area equivalent diameter distribution of 17.7%, a mean thickness of 0.057 ⁇ m and a mean aspect ratio of 16.3. Tabular grains having an aspect ratio of 2 or more occupied 80% or more of the total projected area.
- the mean equivalent spherical diameter of the grains was 0.42 ⁇ m. 90% or more of the silver iodide existed in ⁇ phase from the result of powder X-ray diffraction analysis.
- the mixture was adjusted to the pH of 3.8 with 0.5 mol/L sulfuric acid. After stopping stirring, the mixture was subjected to precipitation/ desalting/ water washing steps. The mixture was adjusted to the pH of 5.9 with 1 mol/L sodium hydroxide to produce a silver halide dispersion having the pAg of 11.0.
- the above-mentioned silver halide dispersion was kept at 38°C with stirring, and thereto was added 5 mL of a 0.34% by weight methanol solution of 1,2-benzisothiazoline-3-one, and after 40 minutes the temperature was elevated to 60°C.
- sodium benzene thiosulfonate in a methanol solution was added at 7.6 ⁇ 10 -5 mol per 1 mol of silver.
- tellurium sensitizer C in a methanol solution was added at 2.0 ⁇ 10 -5 mol per 1 mol of silver and subjected to ripening for 91 minutes.
- Preparation of silver halide emulsion C was conducted in a similar manner to the process in the preparation of the silver halide emulsion A except that adequately changing the addition amount of a 5 % by weight methanol solution of 2,2'-(ethylene dithio)diethanol, the temperature at grain formation step, and the time for adding the solution A.
- the silver halide emulsion C was a pure silver iodide emulsion.
- the obtained silver halide grains had a mean projected area equivalent diameter of 1.369 ⁇ m, a variation coefficient of a projected area equivalent diameter distribution of 19.7%, a mean thickness of 0.130 ⁇ m and a mean aspect ratio of 11.1.
- Tabular grains having an aspect ratio of 2 or more occupied 80% or more of the total projected area.
- the mean equivalent spherical diameter of the grains was 0.71 ⁇ m. 90% or more of the silver iodide existed in ⁇ phase from the result of powder X-ray diffraction analysis.
- Preparation of silver halide emulsion D was conducted in a similar manner to the process in the preparation of silver halide emulsion B, except that using silver halide emulsion C. Thereby, silver halide emulsion D containing 5 mol% of epitaxial silver bromide was prepared.
- benzothiazolium iodide 7 ⁇ 10 -3 mol per 1 mol of silver with a 1% by weight aqueous solution.
- the compounds Nos. 1, 2, and 3 were added respectively in an amount of 2 ⁇ 10 -3 mol per 1 mol of silver in silver halide.
- the compound Nos. 1 and 2 were added respectively in an amount of 8 ⁇ 10 -3 mol per 1 mol of silver halide.
- Behenic acid manufactured by Henkel Co. (trade name: Edenor C22-85R) in an amount of 100 kg was admixed with 1200 kg of isopropyl alcohol, and dissolved at 50°C.
- the mixture was filtrated through a 10 ⁇ m filter, and cooled to 30°C to allow recrystallization. Cooling speed for the recrystallization was controlled to be 3 °C/hour.
- the resulting crystal was subjected to centrifugal filtration, and washing was performed with 100 kg of isopropyl alcohol. Thereafter, the crystal was dried.
- the resulting crystal was esterified, and subjected to GC-FID analysis to give the results of the content of behenic acid being 96 mol%, lignoceric acid 2 mol%, and arachidic acid 2 mol%. In addition, erucic acid was included at 0.001 mol%.
- a reaction vessel charged with 635 L of distilled water and 30 L of t-butyl alcohol was kept at 30°C, and thereto were added the total amount of the solution of sodium behenate and the total amount of the aqueous silver nitrate solution with sufficient stirring at a constant flow rate over 93 minutes and 15 seconds, and 90 minutes, respectively.
- the added material was restricted to the aqueous silver nitrate solution alone.
- the addition of the solution of sodium behenate was thereafter started, and during 14 minutes and 15 seconds following the completion of adding the aqueous silver nitrate solution, the added material was restricted to the solution of sodium behenate alone.
- the temperature inside of the reaction vessel was then set to be 30°C, and the temperature outside was controlled so that the liquid temperature could be kept constant.
- the temperature of a pipeline for the addition system of the solution of sodium behenate was kept constant by circulation of warm water outside of a double wall pipe, so that the temperature of the liquid at an outlet in the leading edge of the nozzle for addition was adjusted to be 75°C.
- the temperature of a pipeline for the addition system of the aqueous silver nitrate solution was kept constant by circulation of cool water outside of a double wall pipe.
- Position at which the solution of sodium behenate was added and the position, at which the aqueous silver nitrate solution was added, was arranged symmetrically with a shaft for stirring located at a center. Moreover, both of the positions were adjusted to avoid contact with the reaction liquid.
- the mixture was left to stand at the temperature as it was for 20 minutes. The temperature of the mixture was then elevated to 35°C over 30 minutes followed by ripening for 210 minutes. Immediately after completing the ripening, solid matters were filtered out with centrifugal filtration. The solid matters were washed with water until the electric conductivity of the filtrated water became 30 ⁇ S/cm. A silver salt of fatty acid was thus obtained. The resulting solid matters were stored as a wet cake without drying.
- a stock liquid after the preliminary dispersion was treated three times using a dispersing machine (trade name: Microfluidizer M-610, manufactured by Microfluidex International Corporation, using Z type Interaction Chamber) with the pressure controlled to be 1150 kg/cm 2 to give a dispersion of the silver behenate.
- a dispersing machine trade name: Microfluidizer M-610, manufactured by Microfluidex International Corporation, using Z type Interaction Chamber
- the pressure controlled to be 1150 kg/cm 2 to give a dispersion of the silver behenate.
- coiled heat exchangers were equipped in front of and behind the interaction chamber respectively, and accordingly, the temperature for the dispersion was set to be 18°C by regulating the temperature of the cooling medium.
- reducing agent-1 (2,2'-methylenebis-(4-ethyl-6-tertbutylphenol)
- 16 kg of a 10% by weight aqueous solution of modified polyvinyl alcohol manufactured by Kuraray Co., Ltd., Poval MP-203
- a slurry was fed with a diaphragm pump, and was subjected to dispersion with a horizontal sand mill (UVM-2: manufactured by AIMEX Co., Ltd.) packed with zirconia beads having a mean particle diameter of 0.5 mm for 3 hours.
- UVM-2 manufactured by AIMEX Co., Ltd.
- a benzoisothiazolinone sodium salt and water were added thereto, thereby adjusting the concentration of the reducing agent to be 25% by weight.
- This dispersion was subjected to heat treatment at 60°C for 5 hours to obtain reducing agent-1 dispersion.
- Particles of the reducing agent included in the resulting reducing agent dispersion had a median diameter of 0.40 ⁇ m, and a maximum particle diameter of 1.4 ⁇ m or less.
- the resultant reducing agent dispersion was subjected to filtration with a polypropylene filter having a pore size of 3.0 ⁇ m to remove foreign substances such as dust, and stored.
- reducing agent-2 (6,6'-di-t-butyl-4,4'-dimethyl-2,2'-butylidenediphenol)
- a 10% by weight aqueous solution of modified polyvinyl alcohol manufactured by Kuraray Co., Ltd., Poval MP-203
- UVM-2 manufactured by AIMEX Co., Ltd.
- zirconia beads having a mean particle diameter of 0.5 mm for 3 hours and 30 minutes.
- 0.2 g of a benzoisothiazolinone sodium salt and water were added thereto, thereby adjusting the concentration of the reducing agent to be 25% by weight.
- This dispersion was warmed at 40°C for one hour, followed by a subsequent heat treatment at 80°C for one hour to obtain reducing agent-2 dispersion.
- Particles of the reducing agent included in the resulting reducing agent-2 dispersion had a median diameter of 0.50 ⁇ m, and a maximum particle diameter of 1.6 ⁇ m or less.
- the resultant reducing agent-2 dispersion was subjected to filtration with a polypropylene filter having a pore size of 3.0 ⁇ m to remove foreign substances such as dust, and stored.
- This dispersion was warmed at 40°C for one hour, followed by a subsequent heat treatment at 80°C for one hour to obtain hydrogen bonding compound-1 dispersion.
- Particles of the hydrogen bonding compound included in the resulting hydrogen bonding compound dispersion had a median diameter of 0.45 ⁇ m, and a maximum particle diameter of 1.3 ⁇ m or less.
- the resultant hydrogen bonding compound dispersion was subjected to filtration with a polypropylene filter having a pore size of 3.0 ⁇ m to remove foreign substances such as dust, and stored.
- Particles of the development accelerator included in the resulting development accelerator dispersion had a median diameter of 0.48 ⁇ m, and a maximum particle diameter of 1.4 ⁇ m or less.
- the resultant development accelerator dispersion was subjected to filtration with a polypropylene filter having a pore size of 3.0 ⁇ m to remove foreign substances such as dust, and stored.
- dispersion was executed similar to the development accelerator-1, and thus dispersions of 20% by weight and 15% by weight were respectively obtained.
- organic polyhalogen compound-1 tribromomethane sulfonylbenzene
- 10 kg of a 20% by weight aqueous solution of modified polyvinyl alcohol manufactured by Kuraray Co., Ltd., Poval MP-203
- 0.4 kg of a 20% by weight aqueous solution of sodium triisopropylnaphthalenesulfonate and 14 kg of water were thoroughly admixed to give a slurry.
- This slurry was fed with a diaphragm pump, and was subjected to dispersion with a horizontal sand mill (UVM-2: manufactured by AIMEX Co., Ltd.) packed with zirconia beads having a mean particle diameter of 0.5 mm for 5 hours.
- UVM-2 manufactured by AIMEX Co., Ltd.
- zirconia beads having a mean particle diameter of 0.5 mm for 5 hours.
- 0.2 g of a benzisothiazolinone sodium salt and water were added thereto, thereby
- Particles of the organic polyhalogen compound included in the resulting organic polyhalogen compound dispersion had a median diameter of 0.41 ⁇ m, and a maximum particle diameter of 2.0 ⁇ m or less.
- the resultant organic polyhalogen compound dispersion was subjected to filtration with a polypropylene filter having a pore size of 10.0 ⁇ m to remove foreign substances such as dust, and stored.
- organic polyhalogen compound-2 N-butyl-3-tribromomethane sulfonylbenzamide
- 20 kg of a 10% by weight aqueous solution of modified polyvinyl alcohol manufactured by Kuraray Co., Ltd., Poval MP-203
- 0.4 kg of a 20% by weight aqueous solution of sodium triisopropylnaphthalenesulfonate were thoroughly admixed to give a slurry.
- This slurry was fed with a diaphragm pump, and was subjected to dispersion with a horizontal sand mill (UVM-2: manufactured by AIMEX Co., Ltd.) packed with zirconia beads having a mean particle diameter of 0.5 mm for 5 hours. Thereafter, 0.2 g of a benzisothiazolinone sodium salt and water were added thereto, thereby adjusting the concentration of the organic polyhalogen compound to be 30% by weight.
- UVM-2 manufactured by AIMEX Co., Ltd.
- This fluid dispersion was heated at 40°C for 5 hours to obtain organic polyhalogen compound-2 dispersion.
- Particles of the organic polyhalogen compound included in the resulting organic polyhalogen compound dispersion had a median diameter of 0.40 ⁇ m, and a maximum particle diameter of 1.3 ⁇ m or less.
- the resultant organic polyhalogen compound dispersion was subjected to filtration with a polypropylene filter having a pore size of 3.0 ⁇ m to remove foreign substances such as dust, and stored.
- Mercapto compound-2 (1-(3-methylureidophenyl)-5-mercaptotetrazole) in an amount of 20 g was dissolved in 980 g of water to give a 2.0% by weight aqueous solution.
- C.I. Pigment Blue 60 in an amount of 64 g and 6.4 g of DEMOL N manufactured by Kao Corporation were added to 250 g of water and thoroughly mixed to give a slurry.
- Zirconia beads having the mean particle diameter of 0.5 mm were provided in an amount of 800 g, and charged in a vessel with the slurry.
- Dispersion was performed with a dispersing machine (1/4G sand grinder mill: manufactured by AIMEX Co., Ltd.) for 25 hours. Thereto was added water to adjust so that the concentration of the pigment became 5% by weight to obtain a pigment-1 dispersion.
- Particles of the pigment included in the resulting pigment dispersion had a mean particle diameter of 0.21 ⁇ m.
- SBR Latex (RP-1) was prepared as follows.
- Degassing was conducted with a vacuum pump, followed by repeating nitrogen gas replacement several times. Thereto was injected 108.75 g of 1,3-butadiene, and the inner temperature is elevated to 60°C. Thereto was added a solution of 1.875 g of ammonium persulfate dissolved in 50 mL of water, and the mixture was stirred for 5 hours as it stands.
- the temperature was further elevated to 90°C, followed by stirring for 3 hours.
- filtration with a polypropylene filter having the pore size of 1.0 ⁇ m was conducted to remove foreign substances such as dust followed by storage. Accordingly, SBR latex RP-1 was obtained in an amount of 774.7 g.
- concentration of chloride ion was revealed to be 3 ppm.
- concentration of the chelating agent by high performance liquid chromatography it was revealed to be 145 ppm.
- the aforementioned latex had a mean particle diameter of 90 nm, Tg of 17°C, solid matter concentration of 44% by weight, the equilibrium moisture content at 25°C and 60%RH of 0.6% by weight, ionic conductance of 4.80 mS/cm (measurement of the ionic conductance performed using a conductivity meter CM-30S manufactured by Toa Electronics Ltd. for the latex stock solution (44% by weight) at 25°C).
- the coating solution for the image forming layer prepared by adding the mixed emulsion for coating solution thereto followed by thorough mixing just prior to the coating was fed directly to a coating die, and was coated.
- Viscosity of the coating solution was 58 [mPa ⁇ s] which was measured with a B type viscometer at 40°C (No. 1 rotor, 60 rpm).
- Viscosity of the coating solution was 20 [mPa ⁇ s] which was measured with a B type viscometer at 40°C (No. 1 rotor, 60 rpm).
- Viscosity of the coating solution was 19 [mPa ⁇ s] which was measured with a B type viscometer at 40°C (No. 1 rotor, 60 rpm).
- Simultaneous overlaying coating by a slide bead coating method was subjected, on both sides of the support, in order of the image forming layer, intermediate layer, first layer of the surface protective layers and second layer of the surface protective layers, and thus samples of the photothermographic material were produced.
- the temperature of the coating solution was adjusted to 31°C for the image forming layer and intermediate layer, to 36°C for the first layer of the surface protective layers, and to 37°C for the second layer of the surface protective layers.
- the amount of coated silver in the image forming layer was 0.821 g/m 2 per one side, with respect to the sum of the amounts of silver salt of fatty acid and silver halide. This was coated on both sides of the support.
- each compound (g/m 2 ) for the image forming layer per one side is as follows.
- the thickness of the image forming layer was 10 ⁇ m.
- the coating amount of silver salt of fatty acid was 0.00625 mol/m 2 .
- Silver salt of fatty acid A Organic polyhalogen compound-1 0.028 Organic polyhalogen compound-2 0.094
- Silver iodide complex-forming agent 0.46 Latex binder (see Table 1) 5.20 Reducing agent-1 0.33 Reducing agent-2 0.13 Hydrogen bonding compound-1 0.15 Development accelerator-1 0.005 Development accelerator-2 0.035
- Silver halide (on the basis of Ag content) 0.146
- the support was decharged by ionic wind. Coating was performed at the speed of 160 m/min. Conditions for coating and drying were adjusted within the range described below, and conditions were set to obtain the most stable surface state.
- the clearance between the leading end of the coating die and the support was 0.10 mm to 0.30 mm.
- the pressure in the vacuum chamber was set to be lower than atmospheric pressure by 196 Pa to 882 Pa.
- the coating solution was cooled by wind having the dry-bulb temperature of 10°C to 20°C.
- the coated support was dried with an air of the dry-bulb of 23°C to 45°C and the wet-bulb of 15°C to 21°C in a helical type contactless drying apparatus.
- moisture conditioning was performed at 25°C in the humidity of 40%RH to 60%RH.
- the film surface was heated to be 70°C to 90°C, and after heating, the film surface was cooled to 25°C.
- Compound 1 that can be one-electron-oxidized to provide a one-electron oxidation product which releases one or more electrons
- Compound 2 that can be one-electron-oxidized to provide a one-electron oxidation product which releases one or more electrons
- Compound 3 that can be one-electron-oxidized to provide a one-electron oxidation product which releases one or more electrons
- the resulting sample was cut into a half-cut size, and was wrapped with the following packaging material under an environment of 25°C and 50%RH, and stored for 2 weeks at an ambient temperature.
- X-ray regular screen HI-SCREEN-B3 (CaWO 4 was used as fluorescent substance, the emission peak wavelength of 425 nm) produced by Fuji Photo Film Co., Ltd. were used, and the assembly for image formation was provided by inserting the sample between them. This assembly was subjected to X-ray exposure for 0.05 seconds, and then X-ray sensitometry was performed.
- the X-ray apparatus used was DRX-3724HD (trade name) produced by Toshiba Corp., and a tungsten target tube was used.
- the thermal developing portion of Fuji Medical Dry Laser Imager FM-DP L was modified so that it can heat from both sides, and by another modification the transportation rollers in the thermal developing portion were changed to the heating drum so that the sheet of film could be conveyed.
- the temperature of four panel heaters were set to 112°C- 118°C- 120°C-120°C, and the temperature of the heating drum was set to 120°C.
- the total time period for thermal development was set to be 14 seconds.
- Photothermogaphic material-11 to -16 were prepared as shown in Table 3, using silver halide emulsion F, H, or J instead of silver halide emulsion B, which was used in Sample Nos. 1 and 8 prepared in the Example.
- Latex P-1b was prepared in a similar manner to the preparation of latex P-1 used for Sample No.1 in Example 1 except that using "SANDET BL” (trade name, available from Sanyo Kasei K.K.) instead of the surfactant "PIONIN A-43-S” (trade name, available from Takemoto Oil & Fat Co., Ltd.) used in the synthesis of latex P-1. Chloride ion concentration of this latex was 553 ppm.
- Sample No.21 was prepared where latex P-1 used for Sample No.1 was changed for latex P-1b by same weight.
- the processed photothermographic materials were stored under an environment of 1000 Lux (25°C and 60%RH) for one day and thereafter fog was measured, in addition to the evaluation terms carried out in Example 1.
- the photothermographic materials containing the polymer latex having a high chloride ion concentration exhibit unfavorable increase in fog after exposure to light.
- the above-mentioned silver halide dispersion was kept at 38°C with stirring, and thereto was added 5 mL of a 0.34% by weight methanol solution of 1,2-benzisothiazoline-3-one, and after 40 minutes the temperature was elevated to 60°C.
- sodium benzene thiosulfonate in a methanol solution was added in an amount of 7.6 ⁇ 10 -5 mol per 1 mol of silver.
- an aqueous solution of sodium thiosulfate pentahydrate was added in an amount of 7.5 ⁇ 10 -5 mol per 1 mol of silver, and then after 10 minutes, an aqueous solution of aurichloric acid and an aqueous solution of potassium thiocyanate were added in an amount of 9 ⁇ 10 -6 mol per 1 mol of silver and 8 ⁇ 10 -3 mol per 1 mol of silver respectively, and subjected to ripening for 90 minutes.
- a silver halide dispersion containing 10 mol% of epitaxial silver bromide was prepared in a similar manner to the process in the preparation of silver halide emulsion K, except that using silver halide emulsion C of Example 1 instead of using silver halide emulsion A.
- the above-mentioned silver halide dispersion was kept at 38°C with stirring, and thereto was added 5 mL of a 0.34% by weight methanol solution of 1,2-benzisothiazoline-3-one, and after 20 minutes the temperature was elevated to 50°C. At 20 minutes after elevating the temperature, sodium benzene thiosulfonate in a methanol solution was added in an amount of 7.6 ⁇ 10 -5 mol per 1 mol of silver.
- a methanol solution of 4-oxo-3-benzyl-oxazolizine-2-thione as a sulfur sensitizer was added thereto in an amount of 4.5 ⁇ 10 -5 mol per 1 mol of silver, and after 10 minutes, an aqueous solution of aurichloric acid and an aqueous solution of potassium thiocyanate were added in an amount of 6 ⁇ 10 -6 mol per 1 mol of silver and 5 ⁇ 10 -3 mol per 1 mol of silver respectively, followed by ripening for 60 minutes.
- benzothiazolium iodide 1.5 ⁇ 10 -2 mol per 1 mol of silver with a 1% by weight aqueous solution.
- the compounds Nos. 1, 2, and 3 were added respectively in an amount of 4 ⁇ 10 -3 mol per 1 mol of silver in silver halide.
- the compound Nos. 1 and 2 were added respectively in an amount of 4 ⁇ 10 -3 mol per 1 mol of silver halide.
- Photothermographic materials were prepared similar to Example 1, except that using the mixed emulsion 2 for coating solution instead of the mixed emulsion for coating solution used in Example 1.
- the obtained samples were evaluated similar to Example 1.
- the photothermographic materials of the present invention attain excellent results in fog, haze, and membrane cracking after processing.
Landscapes
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Non-Silver Salt Photosensitive Materials And Non-Silver Salt Photography (AREA)
Abstract
CH2=CR01-CR02=CH2
Description
CH2=CR01-CR02=CH2







Q1-NHNH-Q2






















X-L2-Y



A-(W)n-B
-C≡CH group and the said hydrogen atom may be substituted.The adsorptive group described above may have any substituent.Further, as typical examples of an adsorptive group, the compounds described in pages 4 to 7 in the specification of JP-A No.11-95355 are described.As an adsorptive group represented by A in formula (I), a mercapto-substituted heterocyclic group (e.g., a 2-mercaptothiadiazole group, a 2-mercapto-5-aminothiadiazole group, a 3-mercapto-1,2,4-triazole group, a 5-mercaptotetrazole group, a 2-mercapto-1,3,4-oxadiazole group, a 2-mercaptobenzimidazole group, a 1,5-dimethyl-1,2,4-triazorium-3-thiolate group, a 2,4-dimercaptopyrimidine group, a 2,4- dimercaptotriazine group, a 3,5-dimercapto-1,2,4-triazole group, a 2,5-dimercapto-1,3-thiazole group, or the like) or a nitrogen-containing heterocyclic group having a -NH- group capable to form an imino-silver (>NAg) as a partial structure of heterocycle (e.g., a benzotriazole group, a benzimidazole group, an indazole group, or the like) is preferable, and more preferable as an adsorptive group is a 2-mercaptobenzimidazole group or a 3,5-dimercapto-1,2,4-triazole group.In formula (I), W represents a divalent linking group. The said linking group may be any divalent linking group, as far as it does not give a bad effect toward photographic properties. For example, a divalent linking group which includes a carbon atom, a hydrogen atom, an oxygen atom, a nitrogen atom, or a sulfur atom can be used. As typical examples, an alkylene group having 1 to 20 carbon atoms (e.g., a methylene group, an ethylene group, a trimethylene group, a tetramethylene group, a hexamethylene group, or the like), an alkenylene group having 2 to 20 carbon atoms, an alkynylene group having 2 to 20 carbon atoms, an arylene group having 6 to 20 carbon atoms (e.g., a phenylene group, a naphthylene group, or the like), -CO-, -SO2-, -O-, -S-, -NR1-, and the combination of these linking groups are described. Herein, R1 represents a hydrogen atom, an alkyl group, a heterocyclic group, or an aryl group.The linking group represented by W may have any substituent.In formula (I), a reducible group represented by B represents the group capable to reduce a silver ion. As the examples, a formyl group, an amino group, a triple bond group such as an acetylene group, a propargyl group and the like, a mercapto group, and residues which are obtained by removing one hydrogen atom from hydroxylamines, hydroxamic acids, hydroxyureas, hydroxyurethanes, hydroxysemicarbazides, reductones (reductone derivatives are contained), anilines, phenols (chroman-6-ols, 2,3-dihydrobenzofuran-5-ols, aminophenols, sulfonamidophenols and polyphenols such as hydroquinones, catechols, resorcinols, benzenetriols, bisphenols are contained), acylhydrazines, carbamoylhydrazines, 3-pyrazolidones, and the like can be described. They may have any substituent.The oxidation potential of a reducible group represented by B in formula (I) can be measured by using the measuring method described in Akira Fujishima, "DENKIKAGAKU SOKUTEIHO" (pages 150 to 208, GIHODO SHUPPAN), and The Chemical Society of Japan, "ZIKKEN KAGAKUKOZA", 4th ed. (vol. 9, pages 282 to 344, MARUZEN). For example, the method of rotating disc voltammetry can be used; namely the sample is dissolved in the solution (methanol : pH 6.5 Britton-Robinson buffer = 10% : 90% (% by volume)) and after deaerated by nitrogen gas for 10 minutes, the voltamograph can be measured under the condition of 1000 rotations/minute, the sweep rate 20 mV/second, at 25°C by using a rotating disc electrode (RDE) made by glassy carbon as a working electrode, a platinum electrode as a counter electrode and a saturated calomel electrode as a reference electrode. The half wave potential (E1/2) can be calculated by that obtained voltamograph.When a reducible group represented by B in the present invention is measured by the method described above, an oxidation potential is preferably in a range of about -0.3 V to about 1.0 V, more preferably about -0.1 V to about 0.8 V, and particularly preferably about 0 V to about 0.7 V.In formula (I), a reducible group represented by B is preferably a residue which is obtained by removing one hydrogen atom from hydroxylamines, hydroxamic acids, hydroxyureas, hydroxysemicarbazides, reductones, phenols, acylhydrazines, carbamoylhydrazines, 3-pyrazolidones, or the like.The compound of formula (I) in the present invention may have the ballasted group or polymer chain in it generally used in the non-moving photographic additives as a coupler. And as a polymer, for example, the polymer described in JP-A No. 1-100530 can be selected.The compound of formula (I) in the present invention may be bis or tris type of compound. The molecular weight of the compound represented by formula (I) in the present invention is preferably 100 to 10,000 and more preferably 120 to 1,000 and particularly preferably 150 to 500.The examples of the compound represented by formula (I) in the present invention are shown below, but the present invention is not limited in these.





R11―S―R12





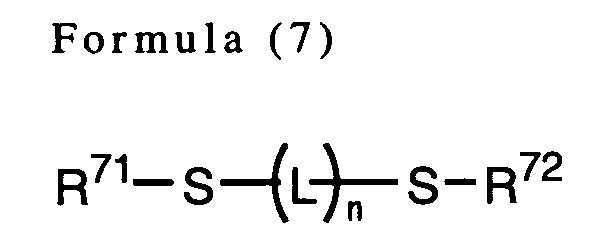














CH2=CR01-CR02 =CH2
-Monomer groups (a) to (j)-

















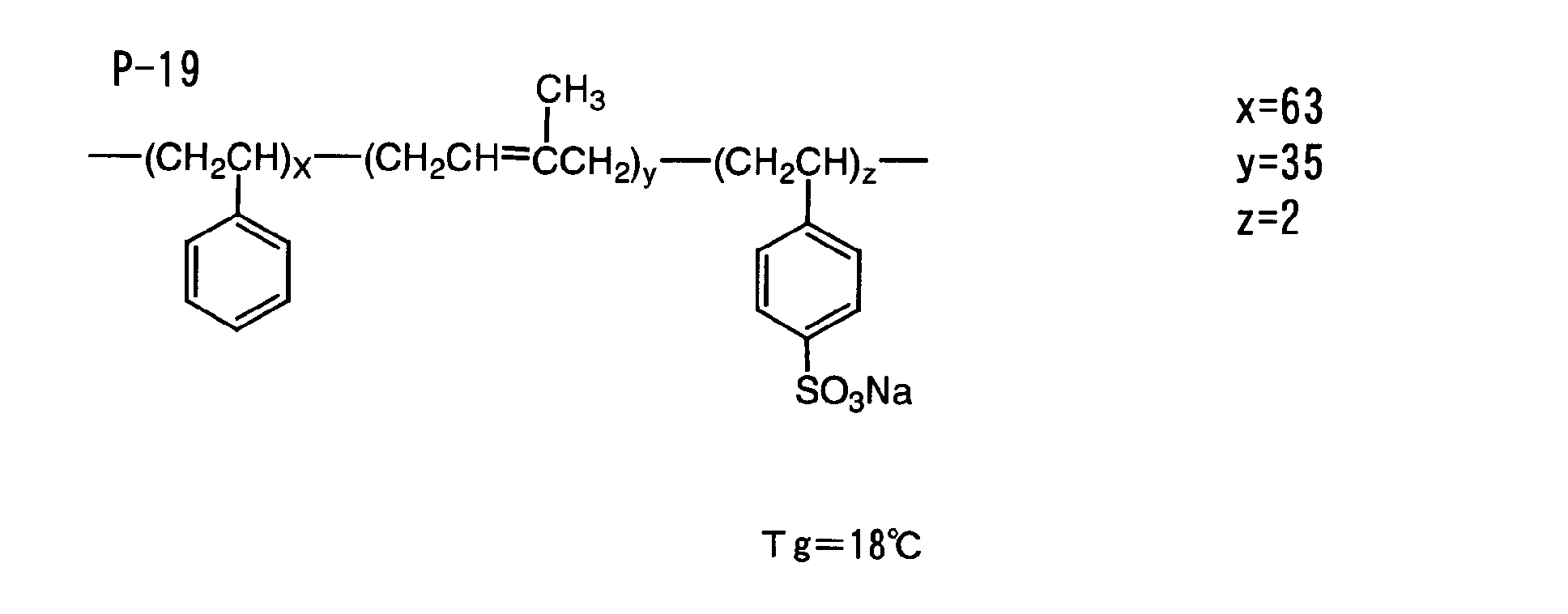










Q-(Y)n-C (Z1) (Z2)X






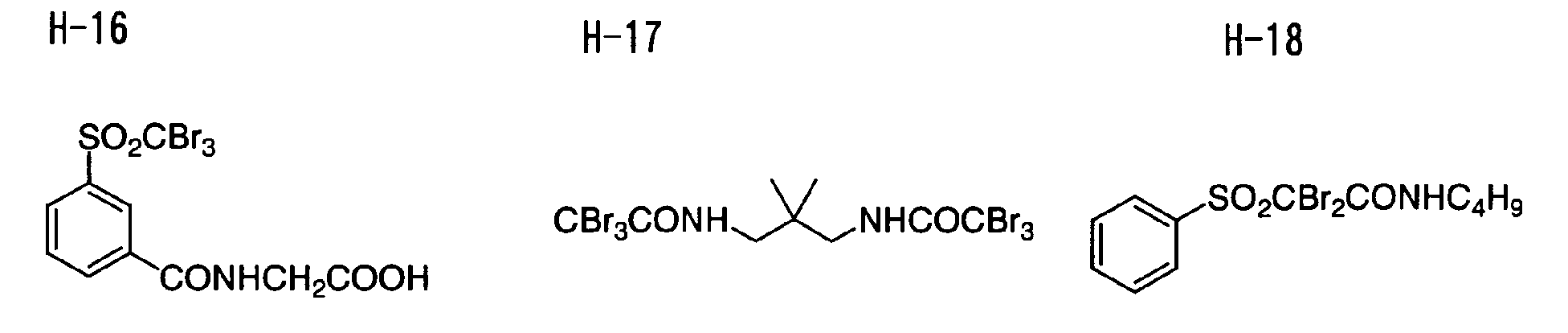

| Silver salt of fatty acid A | |
| Organic polyhalogen compound-1 | 0.028 |
| Organic polyhalogen compound-2 | 0.094 |
| Silver iodide complex-forming agent | 0.46 |
| Latex binder (see Table 1) | 5.20 |
| Reducing agent-1 | 0.33 |
| Reducing agent-2 | 0.13 |
| Hydrogen bonding compound-1 | 0.15 |
| Development accelerator-1 | 0.005 |
| Development accelerator-2 | 0.035 |
| Color-tone-adjusting agent-1 | 0.002 |
| Mercapto compound-1 | 0.001 |
| Mercapto compound-2 | 0.003 |
| Silver halide (on the basis of Ag content) | 0.146 |














- Fog: Fog is an optical density (Dmin) of an unexposed part.
- Haze after Thermal Development: Concerning haze after thermal development, an unexposed part of the sample was measured using a haze measuring apparatus Model 1001DP produced by Nippon Denshoku Co., Ltd.. The measured haze is an index which represents the transparency of the membrane and correlates with the light scattering properties caused by the incorporated fine particles and the disturbance of the boundary. The ratio of the scattered light is expressed by percent. The smaller is the value, the higher is the transparency of the film. The ratio is expressed by a relative value based on the value of Sample No.1, which was taken as 100.
- Evaluation of Membrane Cracking during Thermal Development A scratch test was carried out at a linear speed of 5 cm/second by using a stainless steel stylus having a point diameter of 0.4 mm, while applying a load which weights in a range of from 0 g to 20 g by increasing 2 grams and making the stylus perpendicular contact with the surface of the sample. Thereafter the sample was subjected to the thermal developing process. Thereby some samples may result in a membrane cracking on the surface of an image forming layer where the scratch test described above was performed. The load under which the cracking starts is an index for the pressure resistance, and the value is expressed by a relative value based on the value for Sample No.1, which was taken as 100. The bigger is the value, the superior is the cracking resistance of the material.
| Sample No. | Polymer Latex | Fog | Haze after Development | Membrane Cracking | Note |
| 1 | P-1 (Tg :17°C) | 0.16 | 100 | 100 | Invention |
| 2 | P-2 (Tg : 20°C) | 0.17 | 102 | 95 | Invention |
| 3 | P-4 (Tg : 13°C) | 0.16 | 104 | 120 | Invention |
| 4 | P-7 (Tg : 8°C) | 0.16 | 98 | 110 | Invention |
| 5 | P-19 (Tg : 18°C) | 0.17 | 99 | 100 | Invention |
| 6 | C-1 | 0.18 | 110 | 80 | Invention |
| 7 | C-2 | 0.18 | 125 | 25 | Comparative |
| 8 | RP-1 (Tg : 17°C) | 0.19 | 128 | 35 | Comparative |
| C-1 : methyl methacrylate/isoprene/itaconic acid (weight ratio: 47. 5/47. 5/5, Tg: 0°C) | |||||
| C-2 : methyl methacrylate/butadiene/itaconic acid (weight ratio: 45/45/10, Tg: -2°C) |
| Silver Halide Emulsion | Mean Projected Area Equivalent Diameter | Variation Coefficient | Mean Thickness (µm) | Mean Aspect Ratio | Equivalent Spherical Diameter (µm) |
| A | 0.93 | 17.7% | 0.057 | 16.3 | 0.42 |
| C | 1.369 | 19.7% | 0.130 | 11.1 | 0.71 |
| F | 0.621 | 18.5% | 0.062 | 10.0 | 0.33 |
| H | 0.655 | 18.0% | 0.180 | 3.6 | 0.34 |
| J | 0.602 | 17.2% | 0.330 | 1.8 | 0.30 |
| Sample No. | Polymer Latex | Silver Halide Emulsion (Aspect Ratio) | Fog | Haze after Development | Membrane Cracking | Note Note |
| 1 | P-1 | B (16.3) | 0.16 | 100 | 100 | Invention |
| 8 | RP-1 | B (16.3) | 0.19 | 128 | 35 | Comparative |
| 11 | P-1 | F (10) | 0.16 | 102 | 105 | Invention |
| 12 | RP-1 | F (10) | 0.22 | 130 | 40 | Comparative |
| 13 | P-1 | H (3.6) | 0.16 | 103 | 110 | Invention |
| 14 | RP-1 | H (3.6) | 0.22 | 131 | 39 | Comparative |
| 15 | P-1 | J (1.8) | 0.23 | 133 | 125 | Comparative |
| 16 | RP-1 | J (1.8) | 0.23 | 135 | 122 | Comparative |
| Sample No. | Polymer Latex | Fog | Haze after Development | Membrane Cracking | Note |
| 1 | P-1 | 0.16 | 100 | 100 | Invention |
| 21 | P-1b | 0.19 | 128 | 35 | Invention |
Claims (20)
- A photothermographic material comprising, on at least one side of a support, an image forming layer including at least a photosensitive silver halide, a non-photosensitive organic silver salt, a reducing agent, and a binder, wherein:the photosensitive silver halide has an average silver iodide content of 40 mol% or higher and at least 50% of a projected area of total grains of the photosensitive silver halide is occupied by tabular grains having an aspect ratio of 2 or more; and50% by weight or more of the binder is formed by a polymer latex containing a monomer component shown below within a range of from 10% by weight to 70% by weight:
CH2=CR01-CR02=CH2 - The photothermographic material according to claim 1, wherein the photosensitive silver halide has an average silver iodide content of 80 mol% or higher.
- The photothermographic material according to claim 2, wherein the photosensitive silver halide has an average silver iodide content of 90 mol% or higher.
- The photothermographic material according to claim 1, wherein the photosensitive silver halide tabular grains have a mean equivalent spherical diameter of 0.3 µm to 8.0 µm.
- The photothermographic material according to claim 1, wherein the photosensitive silver halide tabular grains have a mean thickness of 0.3 µm or less.
- The photothermographic material according to claim 1, wherein the polymer latex comprises a monomer component having an acidic group within a range of from 1% by weight to 20% by weight.
- The photothermographic material according to claim 1, wherein the polymer latex has a glass transition temperature (Tg) of -10°C to 70°C.
- The photothermographic material according to claim 1, wherein the polymer latex has a glass transition temperature (Tg) of 5°C to 35°C.
- The photothermographic material according to claim 1, wherein the polymer latex has a halogen ion content of 500 ppm or less per latex liquid.
- The photothermographic material according to claim 1, wherein the polymer latex has styrene in a range of from 30% by weight to 90% by weight as the monomer component.
- The photothermographic material according to claim 1, wherein the reducing agent is a compound represented by the following formula (R):wherein: R11 and R11' each independently represent an alkyl group having 1 to 20 carbon atoms; R12 and R12' each independently represent a hydrogen atom or a substituent capable of substituting for a hydrogen atom on a benzene ring; L represents an -S- group or a -CHR13- group; R13 represents a hydrogen atom or an alkyl group having 1 to 20 carbon atoms; and X1 and X1' each independently represent a hydrogen atom or a group capable of substituting for a hydrogen atom on a benzene ring.

- The photothermographic material according to claim 11, wherein, in the reducing agent represented by the formula (R), R11 and R11' each independently represent a secondary or tertiary alkyl group having 3 to 15 carbon atoms.
- The photothermographic material according to claim 1, further comprising a development accelerator.
- The photothermographic material according to claim 13, wherein the development accelerator is a compound represented by the following formula (A-1):
Q1-NHNH-Q2 - The photothermographic material according to claim 13, wherein the development accelerator is a compound represented by the following formula (A-2):wherein: R1 represents one selected from an alkyl group, an acyl group, an acylamino group, a sulfonamide group, an alkoxycarbonyl group, and a carbamoyl group; R2 represents one selected from a hydrogen atom, a halogen atom, an alkyl group, an alkoxy group, an aryloxy group, an alkylthio group, an arylthio group, an acyloxy group, and a carbonate ester group; R3 and R4 each independently represent a group capable of substituting for a hydrogen atom on a benzene ring; and R3 and R4 may link together to form a condensed ring.

- The photothermographic material according to claim 1, further comprising an organic polyhalogen compound as an antifoggant.
- The photothermographic material according to claim 16, wherein the organic polyhalogen compound is a compound represented by the following formula (H):
Q-(Y)n-C(Z1)(Z2)X - The photothermographic material according to claim 1, further comprising a silver iodide complex-forming agent.
- The photothermographic material according to claim 1 having the image forming layer on both sides of the support.
- An image forming method utilizing a photothermographic material according to claim 1, comprising:(a) providing an assembly for forming an image by placing the photothermographic material between a pair of fluorescent intensifying screens;(b) putting an analyte between the assembly and an X-ray source;(c) applying exposure to the analyte using X-rays having an energy level in a range of 25 kVp to 125 kVp;(d) taking the photothermographic material out of the assembly; and(e) heating the photothermographic material at a temperature at which the material can be developed thermally.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004093593 | 2004-03-26 | ||
| JP2004093593 | 2004-03-26 | ||
| JP2005008257A JP2005309381A (en) | 2004-03-26 | 2005-01-14 | Heat developable photosensitive material and image forming method |
| JP2005008257 | 2005-01-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| EP1584978A1 true EP1584978A1 (en) | 2005-10-12 |
Family
ID=34914549
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP05006628A Withdrawn EP1584978A1 (en) | 2004-03-26 | 2005-03-24 | Photothermographic material and image forming method |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP1584978A1 (en) |
| JP (1) | JP2005309381A (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4635822B2 (en) | 2004-11-09 | 2011-02-23 | パナソニック株式会社 | Modulation circuit and transmitter, receiver and communication system using the same |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6300044B1 (en) * | 1996-06-13 | 2001-10-09 | Agfa-Gevaert | Production method for a photothermographic material and a recording process |
| US20030232288A1 (en) * | 2001-11-05 | 2003-12-18 | Yutaka Oka | Photothermographic material and method of thermal development of the same |
| US20030235795A1 (en) * | 2001-08-22 | 2003-12-25 | Fuji Photo Film Co., Ltd. | Photothermographic material |
-
2005
- 2005-01-14 JP JP2005008257A patent/JP2005309381A/en active Pending
- 2005-03-24 EP EP05006628A patent/EP1584978A1/en not_active Withdrawn
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6300044B1 (en) * | 1996-06-13 | 2001-10-09 | Agfa-Gevaert | Production method for a photothermographic material and a recording process |
| US20030235795A1 (en) * | 2001-08-22 | 2003-12-25 | Fuji Photo Film Co., Ltd. | Photothermographic material |
| US20030232288A1 (en) * | 2001-11-05 | 2003-12-18 | Yutaka Oka | Photothermographic material and method of thermal development of the same |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2005309381A (en) | 2005-11-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20070026348A1 (en) | Black and white photothermographic material and image forming method | |
| US7150963B2 (en) | Silver halide emulsion, silver halide photosensitive material, and photothermographic material | |
| US20070065762A1 (en) | Black and white photothermographic material and image forming method | |
| US20060014113A1 (en) | Photothermographic material | |
| US7129032B2 (en) | Black and white photothermographic material and image forming method | |
| US7135277B2 (en) | Photothermographic material and image forming method | |
| US7429444B2 (en) | Black and white photothermographic material and image forming method | |
| US20050026093A1 (en) | Photothermographic material and image forming method | |
| EP1582918B1 (en) | Photothermographic material and image forming method | |
| US7135276B2 (en) | Photothermographic material and method for preparing photosensitive silver halide emulsion | |
| US7129033B2 (en) | Photothermographic material and image forming method | |
| US7402380B2 (en) | Photothermographic material | |
| US20060073428A1 (en) | Photothermographic material and image forming method | |
| US20060204908A1 (en) | Photothermographic material | |
| US7429447B2 (en) | Photothermographic material and image forming method | |
| US7232652B2 (en) | Photothermographic material and image forming method | |
| US7144692B2 (en) | Photothermographic material | |
| EP1584978A1 (en) | Photothermographic material and image forming method | |
| JP2007065626A (en) | Black and white heat developable photosensitive material and image forming method | |
| US20070065764A1 (en) | Black and white photothermographic material and image forming method | |
| US20070020566A1 (en) | Photothermographic material and image forming method | |
| US7384732B2 (en) | Method of manufacturing a photothermographic material by aqueous coating and a photothermographic material prepared therewith | |
| EP1637925B1 (en) | Photothermographic material | |
| EP1681593A2 (en) | Image forming method using photothermographic material | |
| JP2006308968A (en) | Heat-developable photosensitive material and image forming method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL BA HR LV MK YU |
|
| 17P | Request for examination filed |
Effective date: 20060330 |
|
| AKX | Designation fees paid |
Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU MC NL PL PT RO SE SI SK TR |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: FUJIFILM CORPORATION |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION HAS BEEN WITHDRAWN |
|
| 18W | Application withdrawn |
Effective date: 20090213 |