EP0248523A1 - Pyrazoles - Google Patents
Pyrazoles Download PDFInfo
- Publication number
- EP0248523A1 EP0248523A1 EP87303666A EP87303666A EP0248523A1 EP 0248523 A1 EP0248523 A1 EP 0248523A1 EP 87303666 A EP87303666 A EP 87303666A EP 87303666 A EP87303666 A EP 87303666A EP 0248523 A1 EP0248523 A1 EP 0248523A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- compound
- formula
- pyrazol
- alkyl
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 150000003217 pyrazoles Chemical class 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 84
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 41
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 35
- 239000001257 hydrogen Substances 0.000 claims abstract description 33
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 28
- 239000002253 acid Substances 0.000 claims abstract description 26
- 238000000034 method Methods 0.000 claims abstract description 24
- 150000003839 salts Chemical class 0.000 claims abstract description 22
- 150000002431 hydrogen Chemical group 0.000 claims abstract description 19
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 12
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 11
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 10
- 150000002367 halogens Chemical class 0.000 claims abstract description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 9
- 125000001589 carboacyl group Chemical group 0.000 claims abstract description 8
- 125000006239 protecting group Chemical group 0.000 claims description 10
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 claims description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 230000004968 inflammatory condition Effects 0.000 claims description 4
- YLOCHNXWGDQLIH-UHFFFAOYSA-N 2-(1h-pyrazol-5-ylamino)phenol Chemical compound OC1=CC=CC=C1NC1=NNC=C1 YLOCHNXWGDQLIH-UHFFFAOYSA-N 0.000 claims description 3
- 239000002671 adjuvant Substances 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- 125000004464 hydroxyphenyl group Chemical group 0.000 claims description 3
- 238000011282 treatment Methods 0.000 claims description 3
- YNLDOCYALPXWOV-UHFFFAOYSA-N 2-[(1-methylpyrazol-3-yl)amino]phenol Chemical compound CN1C=CC(NC=2C(=CC=CC=2)O)=N1 YNLDOCYALPXWOV-UHFFFAOYSA-N 0.000 claims description 2
- QGBMZPFZRRUQGV-UHFFFAOYSA-N 4-(1h-pyrazol-5-ylamino)phenol Chemical compound C1=CC(O)=CC=C1NC1=NNC=C1 QGBMZPFZRRUQGV-UHFFFAOYSA-N 0.000 claims description 2
- FGPOTFQRHSVAPE-UHFFFAOYSA-N 4-[(1-benzylpyrazol-3-yl)amino]phenol Chemical compound C1=CC(O)=CC=C1NC1=NN(CC=2C=CC=CC=2)C=C1 FGPOTFQRHSVAPE-UHFFFAOYSA-N 0.000 claims description 2
- ANDSTFHJMKJIKO-UHFFFAOYSA-N 4-[(1-methylpyrazol-3-yl)amino]phenol Chemical compound CN1C=CC(NC=2C=CC(O)=CC=2)=N1 ANDSTFHJMKJIKO-UHFFFAOYSA-N 0.000 claims description 2
- NKRXUTNZNMPPTN-UHFFFAOYSA-N 4-[(5-phenyl-1h-pyrazol-3-yl)amino]phenol Chemical compound C1=CC(O)=CC=C1NC1=NNC(C=2C=CC=CC=2)=C1 NKRXUTNZNMPPTN-UHFFFAOYSA-N 0.000 claims description 2
- SGHPAUUSPSGRQB-UHFFFAOYSA-N 4-[[1-[(4-methoxyphenyl)methyl]pyrazol-3-yl]amino]phenol Chemical compound C1=CC(OC)=CC=C1CN1N=C(NC=2C=CC(O)=CC=2)C=C1 SGHPAUUSPSGRQB-UHFFFAOYSA-N 0.000 claims description 2
- 201000004681 Psoriasis Diseases 0.000 claims description 2
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- XYJWVEMIGXHRAA-UHFFFAOYSA-N n-(4-methoxyphenyl)-4,5-diphenyl-1h-pyrazol-3-amine Chemical compound C1=CC(OC)=CC=C1NC1=NNC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 XYJWVEMIGXHRAA-UHFFFAOYSA-N 0.000 claims description 2
- XNKUXEFTNCRWAX-UHFFFAOYSA-N n-(4-methoxyphenyl)-5-methyl-1h-pyrazol-3-amine Chemical compound C1=CC(OC)=CC=C1NC1=NNC(C)=C1 XNKUXEFTNCRWAX-UHFFFAOYSA-N 0.000 claims description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims description 2
- 238000011321 prophylaxis Methods 0.000 claims description 2
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 2
- CPVDCIXTOJGKCD-UHFFFAOYSA-N 4-[3-(4-methoxyanilino)-1H-pyrazol-5-yl]phenol Chemical compound C1=CC(OC)=CC=C1NC1=NNC(C=2C=CC(O)=CC=2)=C1 CPVDCIXTOJGKCD-UHFFFAOYSA-N 0.000 claims 1
- 239000003814 drug Substances 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 19
- 238000002360 preparation method Methods 0.000 abstract description 4
- 229940121363 anti-inflammatory agent Drugs 0.000 abstract description 2
- 239000002260 anti-inflammatory agent Substances 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 27
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 9
- -1 for example Chemical group 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 238000005899 aromatization reaction Methods 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- 229940114079 arachidonic acid Drugs 0.000 description 4
- 235000021342 arachidonic acid Nutrition 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012362 glacial acetic acid Substances 0.000 description 3
- 150000008282 halocarbons Chemical class 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- JVVRJMXHNUAPHW-UHFFFAOYSA-N 1h-pyrazol-5-amine Chemical class NC=1C=CNN=1 JVVRJMXHNUAPHW-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 102000001381 Arachidonate 5-Lipoxygenase Human genes 0.000 description 2
- 108010093579 Arachidonate 5-lipoxygenase Proteins 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 102000003820 Lipoxygenases Human genes 0.000 description 2
- 108090000128 Lipoxygenases Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 102000004005 Prostaglandin-endoperoxide synthases Human genes 0.000 description 2
- 108090000459 Prostaglandin-endoperoxide synthases Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 235000010418 carrageenan Nutrition 0.000 description 2
- 229920001525 carrageenan Polymers 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 210000002429 large intestine Anatomy 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 125000003884 phenylalkyl group Chemical group 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- ZTBQVCJFZOKYJV-UHFFFAOYSA-N 1-benzyl-5-methyl-n-phenylpyrazol-3-amine Chemical compound N=1N(CC=2C=CC=CC=2)C(C)=CC=1NC1=CC=CC=C1 ZTBQVCJFZOKYJV-UHFFFAOYSA-N 0.000 description 1
- XITDNKSONMUDPZ-UHFFFAOYSA-N 1-benzyl-n-(4-methoxyphenyl)pyrazol-3-amine Chemical compound C1=CC(OC)=CC=C1NC1=NN(CC=2C=CC=CC=2)C=C1 XITDNKSONMUDPZ-UHFFFAOYSA-N 0.000 description 1
- ZXSJPHOSHCYSHT-UHFFFAOYSA-N 2-(4-methylphenyl)sulfonyl-3,4-dihydropyrazol-5-amine Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1N=C(N)CC1 ZXSJPHOSHCYSHT-UHFFFAOYSA-N 0.000 description 1
- UVQQWQCXQCZJQF-UHFFFAOYSA-N 2-[(1-methylpyrazol-3-yl)amino]phenol;hydrochloride Chemical compound Cl.CN1C=CC(NC=2C(=CC=CC=2)O)=N1 UVQQWQCXQCZJQF-UHFFFAOYSA-N 0.000 description 1
- OSUKHONFUUPPTH-UHFFFAOYSA-N 2-benzyl-3,4-dihydropyrazol-5-amine Chemical compound C1CC(N)=NN1CC1=CC=CC=C1 OSUKHONFUUPPTH-UHFFFAOYSA-N 0.000 description 1
- WTYPGBGRYCNDGE-UHFFFAOYSA-N 2-benzyl-n-(4-methoxyphenyl)-3,4-dihydropyrazol-5-amine Chemical compound C1=CC(OC)=CC=C1NC(CC1)=NN1CC1=CC=CC=C1 WTYPGBGRYCNDGE-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- QISOBCMNUJQOJU-UHFFFAOYSA-N 4-bromo-1h-pyrazole-5-carboxylic acid Chemical compound OC(=O)C=1NN=CC=1Br QISOBCMNUJQOJU-UHFFFAOYSA-N 0.000 description 1
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 1
- AFMSYOHKVDOEFY-UHFFFAOYSA-N 5-methyl-n-phenyl-1h-pyrazol-3-amine Chemical compound N1C(C)=CC(NC=2C=CC=CC=2)=N1 AFMSYOHKVDOEFY-UHFFFAOYSA-N 0.000 description 1
- FDQGNLOWMMVRQL-UHFFFAOYSA-N Allobarbital Chemical compound C=CCC1(CC=C)C(=O)NC(=O)NC1=O FDQGNLOWMMVRQL-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000035939 Alveolitis allergic Diseases 0.000 description 1
- 206010002198 Anaphylactic reaction Diseases 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 102000011730 Arachidonate 12-Lipoxygenase Human genes 0.000 description 1
- 108010076676 Arachidonate 12-lipoxygenase Proteins 0.000 description 1
- 102000009515 Arachidonate 15-Lipoxygenase Human genes 0.000 description 1
- 108010048907 Arachidonate 15-lipoxygenase Proteins 0.000 description 1
- 208000004300 Atrophic Gastritis Diseases 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- HIYAVKIYRIFSCZ-CVXKHCKVSA-N Calcimycin Chemical compound CC([C@H]1OC2([C@@H](C[C@H]1C)C)O[C@H]([C@H](CC2)C)CC=1OC2=CC=C(C(=C2N=1)C(O)=O)NC)C(=O)C1=CC=CN1 HIYAVKIYRIFSCZ-CVXKHCKVSA-N 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 241000272201 Columbiformes Species 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 1
- 208000027445 Farmer Lung Diseases 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Natural products OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 208000007882 Gastritis Diseases 0.000 description 1
- 208000036495 Gastritis atrophic Diseases 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 206010018634 Gouty Arthritis Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 239000002879 Lewis base Substances 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 208000008469 Peptic Ulcer Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 206010042496 Sunburn Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- GPWHDDKQSYOYBF-UHFFFAOYSA-N ac1l2u0q Chemical compound Br[Br-]Br GPWHDDKQSYOYBF-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical compound [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000036783 anaphylactic response Effects 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 208000002399 aphthous stomatitis Diseases 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 230000002917 arthritic effect Effects 0.000 description 1
- 125000005002 aryl methyl group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FGNLEIGUMSBZQP-UHFFFAOYSA-N cadaverine dihydrochloride Chemical compound Cl.Cl.NCCCCCN FGNLEIGUMSBZQP-UHFFFAOYSA-N 0.000 description 1
- HIYAVKIYRIFSCZ-UHFFFAOYSA-N calcium ionophore A23187 Natural products N=1C2=C(C(O)=O)C(NC)=CC=C2OC=1CC(C(CC1)C)OC1(C(CC1C)C)OC1C(C)C(=O)C1=CC=CN1 HIYAVKIYRIFSCZ-UHFFFAOYSA-N 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 208000016644 chronic atrophic gastritis Diseases 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 235000019441 ethanol Nutrition 0.000 description 1
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethyl cyclohexane Natural products CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 208000022195 farmer lung disease Diseases 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 208000007565 gingivitis Diseases 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- 210000003405 ileum Anatomy 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical class CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 150000007527 lewis bases Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 210000001589 microsome Anatomy 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- DAIALAXTILYAHE-UHFFFAOYSA-N n-(2-methoxyphenyl)-1-methylpyrazol-3-amine Chemical compound COC1=CC=CC=C1NC1=NN(C)C=C1 DAIALAXTILYAHE-UHFFFAOYSA-N 0.000 description 1
- IIMMPVUXXYHFFW-UHFFFAOYSA-N n-(2-methoxyphenyl)-1h-pyrazol-5-amine Chemical compound COC1=CC=CC=C1NC1=CC=NN1 IIMMPVUXXYHFFW-UHFFFAOYSA-N 0.000 description 1
- BMBPISKCYNCYMW-UHFFFAOYSA-N n-(2-methoxyphenyl)-2-(4-methylphenyl)sulfonyl-3,4-dihydropyrazol-5-amine Chemical compound COC1=CC=CC=C1NC1=NN(S(=O)(=O)C=2C=CC(C)=CC=2)CC1 BMBPISKCYNCYMW-UHFFFAOYSA-N 0.000 description 1
- DZODIUGJYAUAEH-UHFFFAOYSA-N n-(4-methoxyphenyl)-1-methyl-5-phenylpyrazol-3-amine Chemical compound C1=CC(OC)=CC=C1NC1=NN(C)C(C=2C=CC=CC=2)=C1 DZODIUGJYAUAEH-UHFFFAOYSA-N 0.000 description 1
- PVWSVMVCDCGOLW-UHFFFAOYSA-N n-(4-methoxyphenyl)-1-methylpyrazol-3-amine Chemical compound C1=CC(OC)=CC=C1NC1=NN(C)C=C1 PVWSVMVCDCGOLW-UHFFFAOYSA-N 0.000 description 1
- SBBYLGZCLMFIJA-UHFFFAOYSA-N n-(4-methoxyphenyl)-1h-pyrazol-5-amine Chemical compound C1=CC(OC)=CC=C1NC1=NNC=C1 SBBYLGZCLMFIJA-UHFFFAOYSA-N 0.000 description 1
- HVAMZIRCNOJSMJ-UHFFFAOYSA-N n-(4-methoxyphenyl)-3-phenylprop-2-ynethioamide Chemical compound C1=CC(OC)=CC=C1NC(=S)C#CC1=CC=CC=C1 HVAMZIRCNOJSMJ-UHFFFAOYSA-N 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- VMPITZXILSNTON-UHFFFAOYSA-N o-anisidine Chemical compound COC1=CC=CC=C1N VMPITZXILSNTON-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- BHAAPTBBJKJZER-UHFFFAOYSA-N p-anisidine Chemical compound COC1=CC=C(N)C=C1 BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 210000003200 peritoneal cavity Anatomy 0.000 description 1
- 210000003024 peritoneal macrophage Anatomy 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000003127 radioimmunoassay Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 210000001625 seminal vesicle Anatomy 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000001117 sulphuric acid Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 201000010653 vesiculitis Diseases 0.000 description 1
- 239000007762 w/o emulsion Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/38—Nitrogen atoms
Definitions
- This invention relates to new heterocyclic compounds, processes for their preparation and compositions containing them.
- the cyclisation reaction (a) is preferably carried out in a polar solvent, eg ethanol, at a temperature of from about 25 to 75°C.
- a polar solvent eg ethanol
- the process of reaction (b) is preferably carried out in a solvent.
- the solvent is preferably inert to the reaction conditions, eg a polar, aprotic solvent such as 1,4-dioxan, dimethylformamide or N-methylpyrrolidone.
- the reaction may, if desired, be carried out in the presence of a non-nucleophilic base, eg sodium carbonate, sodium hydride or potassium t-butoxide.
- Good leaving groups that X may represent include tosylate and halide, eg chloride, bromide and iodide.
- the reaction may be carried out at a temperature of from about 0 to 150°C.
- the aromatisation of process (c) may be carried out in the presence of an agent which is capable of accepting hydrogen, for example palladium or charcoal.
- the aromatisation may be carried out by bubbling air or oxygen through a solution of the compound of formula V in an inert solvent, eg an halogenated hydrocarbon.
- an oxidising agent eg a metal oxide such as manganese dioxide.
- the oxidation is preferably carried out in a solvent which is inert to the reaction conditions, eg in an halogenated hydrocarbon, at a temperature of from about -70 to I50°C.
- Leaving groups that L may represent in the aromatisation of process (d) include halogen, Oacetyl and especially arylsulphonyl, for example, tosyl.
- the aromatisation may be effected by heating. However we prefer to carry out the reaction in the presence of a base, eg a metal alkoxide in an alcohol. Sodium ethoxide in ethanol is particularly suitable. The reaction may be carried out at a temperature of from 40 to 150 o C .
- Protected hydroxy groups that ArIa, Ar 2 a and Ar 3 a may bear include alkyloxy, eg methoxy, acetoxy, trifluoroacetoxy and arylmethoxy, eg phenylmethoxy.
- Other protecting groups are well known and include those described in Protective Groups in Organic Chemistry, ed JWF McOmie, Plenum Press (1973) and Protective Groups in Organic Synthesis, TW Greene, Wiley-Interscience (1981). Removal of the protecting group depends on the nature of the protecting group; conventional techniques may generally be employed, including acidic or basic cleavage or hydrogenolysis.
- protecting alkyl or phenylalkyl groups may be removed using a protic acid, eg hydrochloric acid or hydrobromic acid at a temperature of from 0 to 150°C, or a Lewis acid, eg by reacting with boron trihalide in a halocarbon solvent.
- a protic acid eg hydrochloric acid or hydrobromic acid at a temperature of from 0 to 150°C
- a Lewis acid eg by reacting with boron trihalide in a halocarbon solvent.
- cleavage may be effected using a base, eg sodium hydroxide, in a suitable solvent, eg aqueous ethanol.
- Lewis bases eg pyridine hydrochloride, may be used to cleave alkyl or phenylalkyl groups.
- Arylmethyl groups eg benzyl
- Compounds of formula V may be prepared by reacting a compound of formula VIII, in which L 2 represents a leaving group and R 1 , R 4 and R 5 are as defined above, with a compound of formula IX, in which R 3 and Ar 2 are as defined above.
- the reaction may be carried out by heating the two reactants with or without a solvent, eg at a temperature of 50 to 250°C.
- Suitable leaving groups that L 2 may represent include halogen, eg chlorine, NH 2 and hydroxy.
- Compounds of formula VI may be prepared from compounds of formula V in which R 1 represents hydrogen by conventional techniques, for example when L represents p-toluenesulphonyl, compounds of formula VI may be prepared by reacting a compound of formula V in which R 1 is hydrogen with p-toluenesulphonyl chloride in the presence of a base.
- the acid addition salts of the compounds of formula I may be prepared by reaction of the free base with an appropriate acid.
- the acid addition salts may be converted to the corresponding free base by the action of a stronger base.
- the process as described above may produce the compound of formula I or an acid addition, salt thereof. It is also within the scope of this invention to treat any derivatives so produced to liberate the free compound of formula I, or to convert one derivative into another.
- Pharmaceutically acceptable acid addition salts include, eg salts of mineral acids, such as hydrohalic acids, eg hydrochloric acid or hydrobromic acid, or organic acids such as formic, acetic or lactic acids.
- the acid may be polybasic, for example sulphuric, fumaric or citric acid.
- the compounds of formula I, and pharmaceutically acceptable acid addition salts thereof, are useful because they possess pharmacological activity in animals.
- the compounds are useful as broad spectrum anti-inflammatory agents as indicated in one or more of the following assay systems:
- the compounds are indicated for use in the treatment or prophylaxis of inflammatory conditions in mammals, including man. Conditions that may be specifically mentioned are: rheumatoid arthritis, rheumatoid spondylitis, osteoarthritis, gouty arthritis, and other arthritic conditions, inflamed joints;
- the dosage administered will, of course, vary with the compound employed, the mode of administration and the treatment desired. However, in general satisfactory results are obtained when the compounds are administered at a daily dosage of from about O.lmg to about 20mg per kg of animal body weight, preferably given in divided doses 1 to 4 times ,a day or in sustained release form.
- the total daily dose is in the range of from 7.0mg to 1,400mg and unit dosage forms suitable for oral administration comprise from 2.0mg to 1,400mg of the compound admixed with a solid or liquid pharmaceutical carrier or diluent.
- the compounds of formula I, and pharmaceutically acceptable acid addition salts thereof, may be used on their own or in the form of appropriate medicinal preparations for enteral, parenteral or topical administration.
- a pharmaceutical composition comprising preferably less than 80% and more preferably less than 50% by weight of a compound of formula I, or a pharmaceutically acceptable acid addition salt thereof, in admixture with a pharmaceutically acceptable adjuvant, diluent or carrier.
- adjuvants, diluents and carriers are:- for tablets and dragees: lactose, starch, talc, stearic acid; for capsules: tartaric acid or lactose; for injectable solutions: water, alcohols, glycerin, vegetable oils; for suppositories: natural or hardened oils or waxes.
- compositions in a form suitable for oral, ie oesophageal administration include tablets, capsules and dragees;
- the compounds of formula I and pharmaceutically acceptable acid addition salts thereof have the advantage that they are less toxic, more efficacious, are longer acting, have a broader range of activity, are more potent, produce fewer side effects, are more easily absorbed, eg orally or have other useful pharmacological properties, than compounds of similar structure.
- R 1 , R 3' R 4 , R 5 , R 6 or R 7 represent alkyl or alkanoyl
- the group preferably contains up to 10, more preferably up to six carbon atoms.
- Particular alkyl groups that R 1 , R 3 , R 4 , R 5 , R 6 and R 7 may represent include saturated and unsaturated alkyl groups, for example, methyl, ethyl, 2-propenyl and butyl.
- Particular NR 6 R 7 groups that may substitute the N-phenyl include -NH 2 , -NHCH 3 and -N(CH 3 ) 2 .
- R 1 may represent include hydrogen, acetyl, benzyl, 4-alkoxybenzyl, methyl and isopropyl.
- R 1 may represent CH2Arl, alkyl or hydrogen.
- R 1 may represent hydrogen when R 5 represents Ar 3 .
- Ar 1 may represent include phenyl and substituted by hydroxy or alkoxy.
- phenyl group of Ar 2 to bear a substituent in the 2- or 4- position.
- phenyl group to bear an OH, alkoxy, eg methoxy or dialkylamino group in the 2- or 4- position.
- An especially favoured group that Ar 2 may represent is hydroxyphenyl, in particular 2- or 4-hydroxyphenyl.
- Ar 2 may be further substituted by groups selected from halogen, alkoxy and especially alkyl, eg methyl.
- R 3 to represent alkyl and particularly hydrogen.
- R 4 to represent phenyl and particularly hydrogen.
- R 5 may represent include hydrogen, methyl and Ar 3 .
- Ar 3 may represent include phenyl and hydroxyphenyl, eg 4-hydroxyphenyl.
- Certain of the compounds of formula I possess one or more chiral centres and the invention also provides the compounds in the form of their individual optical isomers or as racemic or other mixtures thereof. Certain of the compounds of formula I may also exist as stereoisomers and in these cases the invention provides all stereoisomeric forms. The various isomers may be prepared and/or separated using conventional processes known per se.
- Example 2 The compound of Example 1 (1.0g), acetyl chloride (0.43g) and pyridine (0.43g) were stirred together at room temperature for 0.5 hr. The mixture was partitioned between ethyl acetate and aqueous brine. The organic layer was extracted, dried and chromatographed on silica eluting with 5% ethyl acetate/95% dichloromethane, to give the title produced as colourless crystals, 280 mg, mp 123-124°.
- step a) To a stirred solution of the product of step a) (13.3g) in dichloromethane (250ml) was added active manganese dioxide in portions until all traces of starting material had disappeared. The reaction mix was filtered, evaporated to a gum and purified by chromatography on silica, eluting with ether, to give the product, mp 68-71°.
- Example 3 The title pyrazole of Example 3 (6.3g) was heated on a steam bath in a mixture of aqueous HBr (48%, 200ml) and glacial acetic acid (25ml) for 6 hours. The reaction mixture was cooled, poured into water, neutralised with sodium bicarbonate, extracted with ethyl acetate, washed with brine, dried and evaporated to give after chromatography the title compound, l.Og, mp 114-116°
- step a) The product of step a) (7.5g) was added to a solution of sodium ethoxide (60ml, prepared from 0.6g sodium), at a temperature of 60°. The mixture was refluxed for 10 minutes, cooled, added to water, extracted with ethyl acetate, dried and evaporated to give the sub-title compound as a purple oil, ms 189 (M + ).
- step b) The product of step b) (4.6g) was dissolved in 48% aqueous HBr (100ml) and refluxed for 8 hours. The mixture was poured onto water (750ml), neutralised with solid sodium bicarbonate, extracted with ethyl acetate, washed with brine and evaporated to give the title compound as a white crystalline solid, 0.49g, mp 163-4° (from ethyl acetate/cyclohexane).
- the pyrazole from Example 5 b) (4.0g) was added to a mixture of crushed potassium hydroxide pellets (4.7g) in dry dimethylsulphoxide, which had previously been stirred for 5 minutes under nitrogen at room temperature. After stirring for 0.75 hours, methyl iodide (3.1g) was added to the red solution and stirring was maintained for a further 0.75 hours. The reaction mix was poured into water, extracted with ethyl acetate, the organic layer washed, dried, evaporated and chromatographed to give a light brown oil.
- Example 2 The title pyrazole from Example 1 (3g) was heated in a mixture of aqueous HBr (30%, 10ml) and glacial acetic acid (2ml) on a steam bath for 72 hours. The mixture was cooled, filtered and neutralised with dilute sodium bicarbonate to give the title compound as a colourless solid, 2.2g, mp 215-17° (from ethanol).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract

- R1 represents hydrogen, alkyl, alkanoyl or alkyl substituted by Ar1,
- R3, R4 and R5, which may be the same or different, each independently represent hydrogen, alkyl or Ar3,
- Ar,, Ar2 and Ar3, which may be the same or different, independently represent phenyl or phenyl substituted by one or more of alkyl, hydroxy, alkoxy, NR6R7 or halogen,
- R6 and R7, which may be the same or different, independently represent hydrogen or alkyl,
- and pharmaceutically acceptable acid addition salts thereof.
Description
- This invention relates to new heterocyclic compounds, processes for their preparation and compositions containing them.
- Chemical Abstracts, Vol 71, 1969, 22058Z describes several 3-aryl-5-methylpyrazol-3-amines but does not disclose any pharmaceutical uses of these compounds.
- The Indian Journal of Chemistry, Section B, 1985 24(B), 472-6 describes several 3,5-diarylpyrazol-3-amines but does not disclose any pharmaceutical uses of these compounds.
- Pyrazole-3-amines are also described in Chemische Berichte, 1962 95 937-43, Chemische Berichte 1969, 100, 2577-2584, and in East German Patent 211343.
- We have now found that certain pyrazol-3-amines have useful pharmacological properties.
- According to the invention there are provided the compounds of formula I,

- R l-represents hydrogen, alkyl, alkanoyl or alkyl substituted by Arl,
- R 3, R4 and R5, which may be the same or different, each independently represent hydrogen, alkyl or Ar3,
- Ar1, Ar2 and,Ar3, which may be the same or different, independently represent phenyl or phenyl substituted by one or more of alkyl, hydroxy, alkoxy, NR6R7 or halogen,
- R6 and R7, which may be the same or different, independently represent hydrogen or alkyl,
- and pharmaceutically acceptable acid addition salts thereof,
- for use as a pharmaceutical.
- According to the invention there are also provided the novel compounds of formula I, as hereinbefore defined, provided that
- i) when R1, R3 and R4 each represent hydrogen, and RS represents methyl, then Ar2 does not represent phenyl or phenyl substituted by alkyl, alkoxy or halogen and
- ii) when R1, R3 and R4 each represent hydrogen and Ar2 represents phenyl or phenyl substituted by alkyl, then R5 does not represent phenyl or phenyl substituted by alkyl, alkoxy or halogen and
- iii) when R1 and R3 represent hydrogen, and Ar2 represents phenyl, then
- (a) R4 and R5 do not both represent phenyl and
- (b) R5 does not represent hydrogen when R4 represents alkyl,
- and pharmaceutically acceptable acid addition salts thereof.
- According to the invention there is further provided a process for the preparation of compounds of formula I or pharmaceutically acceptable acid addition salts thereof, which comprises
- (a) reacting a compound of formula II,
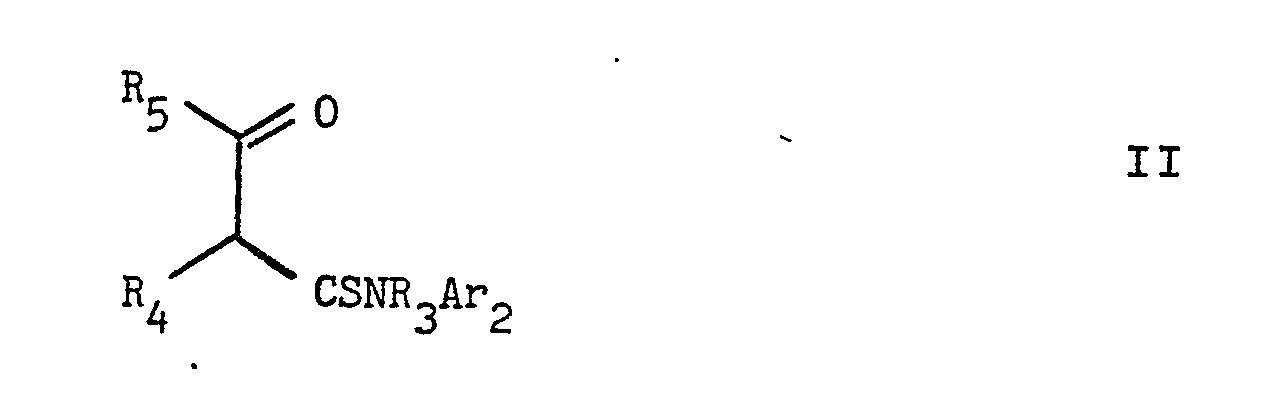

- (b) producing a compound of formula I in which R1 represents alkyl, alkanoyl or alkyl substituted by Ar1, by reacting a corresponding compound of formula I in which R1 represents hydrogen, with a compound of formula IV,

- (c) aromatising a compound of formula V,

- (d) producing a compound of formula I in which R1 represents hydrogen, by aromatising a compound of formula VI,

- (e) producing a compound of formula I in which one or more of Ar1, Ar2 and Ar3 is substituted by at least one -OH, by removing one or more protecting groups from a corresponding compound of formula VII,

- and where desired or necessary converting the resulting compound of formula I into a pharmaceutically acceptable acid addition salt thereof or vice versa.
- The cyclisation reaction (a) is preferably carried out in a polar solvent, eg ethanol, at a temperature of from about 25 to 75°C.
- The process of reaction (b) is preferably carried out in a solvent. The solvent is preferably inert to the reaction conditions, eg a polar, aprotic solvent such as 1,4-dioxan, dimethylformamide or N-methylpyrrolidone. The reaction may, if desired, be carried out in the presence of a non-nucleophilic base, eg sodium carbonate, sodium hydride or potassium t-butoxide. Good leaving groups that X may represent include tosylate and halide, eg chloride, bromide and iodide. The reaction may be carried out at a temperature of from about 0 to 150°C.
- The aromatisation of process (c) may be carried out in the presence of an agent which is capable of accepting hydrogen, for example palladium or charcoal. The aromatisation may be carried out by bubbling air or oxygen through a solution of the compound of formula V in an inert solvent, eg an halogenated hydrocarbon. However we prefer to carry out the aromatisation using an oxidising agent, eg a metal oxide such as manganese dioxide. The oxidation is preferably carried out in a solvent which is inert to the reaction conditions, eg in an halogenated hydrocarbon, at a temperature of from about -70 to I50°C.
- Leaving groups that L may represent in the aromatisation of process (d) include halogen, Oacetyl and especially arylsulphonyl, for example, tosyl. The aromatisation may be effected by heating. However we prefer to carry out the reaction in the presence of a base, eg a metal alkoxide in an alcohol. Sodium ethoxide in ethanol is particularly suitable. The reaction may be carried out at a temperature of from 40 to 150o C.
- Protected hydroxy groups that ArIa, Ar2a and Ar3a may bear include alkyloxy, eg methoxy, acetoxy, trifluoroacetoxy and arylmethoxy, eg phenylmethoxy. Other protecting groups are well known and include those described in Protective Groups in Organic Chemistry, ed JWF McOmie, Plenum Press (1973) and Protective Groups in Organic Synthesis, TW Greene, Wiley-Interscience (1981). Removal of the protecting group depends on the nature of the protecting group; conventional techniques may generally be employed, including acidic or basic cleavage or hydrogenolysis. For example protecting alkyl or phenylalkyl groups may be removed using a protic acid, eg hydrochloric acid or hydrobromic acid at a temperature of from 0 to 150°C, or a Lewis acid, eg by reacting with boron trihalide in a halocarbon solvent. When the protecting group is alkanoyl or haloalkanoyl, cleavage may be effected using a base, eg sodium hydroxide, in a suitable solvent, eg aqueous ethanol. Lewis bases, eg pyridine hydrochloride, may be used to cleave alkyl or phenylalkyl groups. Arylmethyl groups, eg benzyl, may be removed by catalytic hydrogenation using a suitable catalyst, eg palladium, in a suitable solvent, eg ethanol or acetic acid. Further methods for the removal of protecting groups are described in both McOmie and Greene, loc. cit. Both McOmie and Greene also describe numerous methods for the application of protecting groups.
- Compounds of formula V may be prepared by reacting a compound of formula VIII,


- The reaction may be carried out by heating the two reactants with or without a solvent, eg at a temperature of 50 to 250°C. Suitable leaving groups that L2 may represent include halogen, eg chlorine, NH2 and hydroxy.
- Compounds of formula VI may be prepared from compounds of formula V in which R1 represents hydrogen by conventional techniques, for example when L represents p-toluenesulphonyl, compounds of formula VI may be prepared by reacting a compound of formula V in which R1 is hydrogen with p-toluenesulphonyl chloride in the presence of a base.
- Compounds of formula VII may be prepared by processes analogous to those described in process a), b), c) or d).
- Compounds of formulae II, III, IV, VIII and IX are either known, or may be made from known compounds using conventional techniques known per se.
- The acid addition salts of the compounds of formula I may be prepared by reaction of the free base with an appropriate acid. The acid addition salts may be converted to the corresponding free base by the action of a stronger base.
- The process as described above may produce the compound of formula I or an acid addition, salt thereof. It is also within the scope of this invention to treat any derivatives so produced to liberate the free compound of formula I, or to convert one derivative into another.
- The compounds of formula I and the intermediates thereto may be isolated from their reaction mixtures using conventional techniques.
- Pharmaceutically acceptable acid addition salts include, eg salts of mineral acids, such as hydrohalic acids, eg hydrochloric acid or hydrobromic acid, or organic acids such as formic, acetic or lactic acids. The acid may be polybasic, for example sulphuric, fumaric or citric acid.
- When R represents hydrogen, compounds of formula I may exist in a tautomeric form, formula A,

- These compounds are included in the scope of the definition of the compounds of formula I.
- The compounds of formula I, and pharmaceutically acceptable acid addition salts thereof, are useful because they possess pharmacological activity in animals. In particular, the compounds are useful as broad spectrum anti-inflammatory agents as indicated in one or more of the following assay systems:
- (a) Inhibition of lipoxygenases, e.g. 5, 12 and 15 lipoxygenase, in the presence of exogenous arachidonic acid and measurement of the enzyme activity by either a modification of B A Jakschik et al, Biochemical and Biophysical Research Communications, 95(1), 103, (1980) using reverse phase HPLC to quantify the products or by a modification of the method of F F Sun et al, Prostaglandins 21 (2) 333 (1981) using uv absorption to quantify product formation.
- (b) Inhibition of prostaglandin synthetase, utilising bovine seminal vesicle microsomes as the enzyme source after the method of Egan et al Biochemistry 17, 2230 (1978) using either radiolabelled arachidonic acid as substrate and product separation by thin layer chromatography and quantification by scintillation counting or unlabelled arachidonic acid as substrate and a specific radioimmunoassay kit (New England Nuclear) to measure prostaglandin E2 produced.
- (c) Inhibition of 5 lipoxygenase activity in intact human neutrophils stimulated by ionophore A23187 and supplemented with exogenous arachidonic acid after the method of P Borgeat and B Samuelsson, Proceedings New York Academy of Science 70 2148 (1979) using reverse phase HPLC to measure the products.
- (d) Inhibition of formation of arachidonic acid metabolites by mouse peritoneal macrophages challenged in vitro with immune complexes by the method of Blackham et al, J. Pharm. Pharmac. (1985).
- (e) Inhibition of PGE2 formation and cell infiltration in the carrageenin sponge model by the method of Higgs et al, Eur. J. Pharmac. 66 81 (1980).
- (f) Inhibition of immune complex mediated inflammation in the mouse peritoneal cavity by the method of Blackham et al, J. Pharmac. Methods (1985).
- (g) Inhibition of carrageenin oedema in the rat by the method of Winter et al, Proc. Soc. Exp. Biol. 111 544 (1962).
- (h) Inhibition of bronchial anaphylaxis in guinea pigs by the method of Anderson, Br. J. Pharmac. 77 301 (1982).
- The compounds are indicated for use in the treatment or prophylaxis of inflammatory conditions in mammals, including man. Conditions that may be specifically mentioned are: rheumatoid arthritis, rheumatoid spondylitis, osteoarthritis, gouty arthritis, and other arthritic conditions, inflamed joints;
- eczema, psoriasis or other inflammatory skin conditions such as sunburn;
- inflammatory eye conditions including conjunctivitis;
- lung disorders in which inflammation is involved, eg asthma, bronchitis, pigeon fancier's disease and farmer's lung;
- conditions of the gastrointestinal tract including aphthous ulcers, gingivitis, Crohn's disease (a condition of the small, and sometimes also of the large intestine), atrophic gastritis and gastritis varialoforme (conditions of the stomach), ulcerative colitis (a condition of the large intestine and sometimes the small intestine) coeliac disease (a condition of the small intestine), regional ileitis (a regional inflammatory condition of the terminal ileum), peptic ulceration (a condition of the stomach and duodenum) and irritable bowel syndrome; pyresis, pain;
- and other conditions associated with inflammation, particularly those in which lipoxygenase and cyclooxygenase products are a factor.
- For the above mentioned uses the dosage administered will, of course, vary with the compound employed, the mode of administration and the treatment desired. However, in general satisfactory results are obtained when the compounds are administered at a daily dosage of from about O.lmg to about 20mg per kg of animal body weight, preferably given in divided doses 1 to 4 times ,a day or in sustained release form. For man the total daily dose is in the range of from 7.0mg to 1,400mg and unit dosage forms suitable for oral administration comprise from 2.0mg to 1,400mg of the compound admixed with a solid or liquid pharmaceutical carrier or diluent.
- The compounds of formula I, and pharmaceutically acceptable acid addition salts thereof, may be used on their own or in the form of appropriate medicinal preparations for enteral, parenteral or topical administration.
- According to our invention we also provide a pharmaceutical composition comprising preferably less than 80% and more preferably less than 50% by weight of a compound of formula I, or a pharmaceutically acceptable acid addition salt thereof, in admixture with a pharmaceutically acceptable adjuvant, diluent or carrier. Examples of such adjuvants, diluents and carriers are:- for tablets and dragees: lactose, starch, talc, stearic acid; for capsules: tartaric acid or lactose; for injectable solutions: water, alcohols, glycerin, vegetable oils; for suppositories: natural or hardened oils or waxes.
- Compositions in a form suitable for oral, ie oesophageal administration include tablets, capsules and dragees;
- compositions in a form suitable for administration to the lung include aerosols, particularly pressurised aerosols;
- compositions in a form suitable for administration to the skin include creams, eg oil-in-water emulsions or water-in-oil emulsion;
- compositions in a form suitable for administration to the eye include drops and ointments.
- We prefer the composition to contain up to 50% and more preferably up to 25% by weight of the compound of formula I, or of the pharmaceutically acceptable acid addition salt thereof.
- The compounds of formula I and pharmaceutically acceptable acid addition salts thereof have the advantage that they are less toxic, more efficacious, are longer acting, have a broader range of activity, are more potent, produce fewer side effects, are more easily absorbed, eg orally or have other useful pharmacological properties, than compounds of similar structure.
- When R1, R 3' R 4, R5, R6 or R7 represent alkyl or alkanoyl, the group preferably contains up to 10, more preferably up to six carbon atoms. Particular alkyl groups that R1, R3, R4, R 5, R6 and R7 may represent include saturated and unsaturated alkyl groups, for example, methyl, ethyl, 2-propenyl and butyl.
- Particular NR6R7 groups that may substitute the N-phenyl include -NH2, -NHCH3 and -N(CH3)2.
- Particular groups that R1 may represent include hydrogen, acetyl, benzyl, 4-alkoxybenzyl, methyl and isopropyl. We prefer R1 to represent CH2Arl, alkyl or hydrogen. We particularly prefer R1 to represent hydrogen when R5 represents Ar3. Specific groups that Ar1 may represent include phenyl and substituted by hydroxy or alkoxy.
- We prefer the phenyl group of Ar2 to bear a substituent in the 2- or 4- position. We particularly prefer the phenyl group to bear an OH, alkoxy, eg methoxy or dialkylamino group in the 2- or 4- position. An especially favoured group that Ar2 may represent is hydroxyphenyl, in particular 2- or 4-hydroxyphenyl. Ar2 may be further substituted by groups selected from halogen, alkoxy and especially alkyl, eg methyl.
- We prefer R3 to represent alkyl and particularly hydrogen.
- We prefer R4 to represent phenyl and particularly hydrogen.
- Particular groups that R5 may represent include hydrogen, methyl and Ar3. Particular groups that Ar3 may represent include phenyl and hydroxyphenyl, eg 4-hydroxyphenyl.
- Certain of the compounds of formula I possess one or more chiral centres and the invention also provides the compounds in the form of their individual optical isomers or as racemic or other mixtures thereof. Certain of the compounds of formula I may also exist as stereoisomers and in these cases the invention provides all stereoisomeric forms. The various isomers may be prepared and/or separated using conventional processes known per se.
- The invention will now be illustrated by the following Examples, in which temperatures are in degrees centigrade.
- Hydrazine hydrate (0.55ml) was added to a suspension of N-(4-methoxyphenyl)-3-phenyl-thiopropiolamide (2.7g) in ethanol (20ml) stirred at room temperature. After 2 hours, the mixture was heated to 60° for 2 hours then cooled to room temperature. The mixture was partitioned between ethyl acetate and dilute aqueous brine. The organic layer was collected and dried. The solvent was evaporated and the residue was recrystallised from ethanol to give the title compound, l.lg m.p. 155-157°. C16H15N3O:
- Requires: C = 72.45%, H = 5.66%, N = 15.85%
- Found: C = 72.36%, H = 5.82%, N = 15.80%
- The compound of Example 1 (1.0g), acetyl chloride (0.43g) and pyridine (0.43g) were stirred together at room temperature for 0.5 hr. The mixture was partitioned between ethyl acetate and aqueous brine. The organic layer was extracted, dried and chromatographed on silica eluting with 5% ethyl acetate/95% dichloromethane, to give the title produced as colourless crystals, 280 mg, mp 123-124°.
- 4,5-dihydro-1-phenylmethyl-1H-pyrazol-3-amine (24g) (prepared by the method of H. Dorm, A. Otto, Chem Ber. 103 2505 (1970), p-anisidine (17.5g) and p-toluenesulphonic acid (100mg) was heated at 1700 under dry nitrogen until evolution of ammonia ceased. The reaction mix was cooled, purified by chromatography on SiO2 eluting with ether:pentane 1:1 to give the sub-title compound as colourless cubes (13.3.g).
- To a stirred solution of the product of step a) (13.3g) in dichloromethane (250ml) was added active manganese dioxide in portions until all traces of starting material had disappeared. The reaction mix was filtered, evaporated to a gum and purified by chromatography on silica, eluting with ether, to give the product, mp 68-71°.
- The title pyrazole of Example 3 (6.3g) was heated on a steam bath in a mixture of aqueous HBr (48%, 200ml) and glacial acetic acid (25ml) for 6 hours. The reaction mixture was cooled, poured into water, neutralised with sodium bicarbonate, extracted with ethyl acetate, washed with brine, dried and evaporated to give after chromatography the title compound, l.Og, mp 114-116°
- A mixture of 4,5-dihydro-l-(4-toluenesulphonyl)-lH- pyrazol-3-amine (8.08g) (Organic Synthesis 48,8, (1968)), o-anisidine (8.0g) and glacial acetic acid (80ml) was heated on a steam bath for 4 hours. The reaction mixture was cooled, filtered and washed with ether to give the sub-title product, 7.6g, mp 176-9°.
- The product of step a) (7.5g) was added to a solution of sodium ethoxide (60ml, prepared from 0.6g sodium), at a temperature of 60°. The mixture was refluxed for 10 minutes, cooled, added to water, extracted with ethyl acetate, dried and evaporated to give the sub-title compound as a purple oil, ms 189 (M+).
- The product of step b) (4.6g) was dissolved in 48% aqueous HBr (100ml) and refluxed for 8 hours. The mixture was poured onto water (750ml), neutralised with solid sodium bicarbonate, extracted with ethyl acetate, washed with brine and evaporated to give the title compound as a white crystalline solid, 0.49g, mp 163-4° (from ethyl acetate/cyclohexane).
- The pyrazole from Example 5 b) (4.0g) was added to a mixture of crushed potassium hydroxide pellets (4.7g) in dry dimethylsulphoxide, which had previously been stirred for 5 minutes under nitrogen at room temperature. After stirring for 0.75 hours, methyl iodide (3.1g) was added to the red solution and stirring was maintained for a further 0.75 hours. The reaction mix was poured into water, extracted with ethyl acetate, the organic layer washed, dried, evaporated and chromatographed to give a light brown oil.
- The pyrazole from step a) (1.7g) was dissolved in dichloromethane (30ml) under an atmosphere of nitrogen. Brown tribromide (8ml) in dichloromethane was added dropwise and the mixture stirred at room temperature for 24 hours. After addition of dilute sodium bicarbonate, the organic phase was removed, dried and evaporated to give a pale oil, which was converted to the hydrochloride salt of the title compound, by trituration with ethereal HC1, to give an off-white solid, 0.75g, mp 162-1640.
- The title pyrazole from Example 1 (3g) was heated in a mixture of aqueous HBr (30%, 10ml) and glacial acetic acid (2ml) on a steam bath for 72 hours. The mixture was cooled, filtered and neutralised with dilute sodium bicarbonate to give the title compound as a colourless solid, 2.2g, mp 215-17° (from ethanol).
- The following compounds were prepared by methods analogous to those described in Examples 1 to 7 above:
- (a) N-[4-Methoxyphenyl]-1-[1-methyl]ethyl-1H-pyrazol-3- amine, oil;
- (b) N-[4-Methoxyphenyl]-1-methyl-lH-pyrazol-3-amine, mp 64-66°;
- (c) 5-Methyl-N-phenyl-l-phenylmethyl-lH-pyrazol-3-amine, mp 97-98°;
- (d) N-[4-Methoxyphenyl]-lH-pyrazol-3-amine, mp 95-96°;
- (e) 5-Methyl-N-phenyl-lH-pyrazole-3-amine, mp 130-131°;
- (f) N-[4-Methoxyphenyl]-5-methyl-lH-pyrazol-3-amine, mp 132-133°;
- (g) N-[4-Methoxyphenyl]-1-methyl-5-phenyl-1H-pyrazol-3- amine, mp 107-108°;
- (h) N-[4-Methoxyphenyl]-4,5-diphenyl-1H-pyrazol-3-amine, mp 190-191 0.
- (i) 4-[3-(4-Methoxyphenylamino)-1H-pyrazol-5-yl)phenol, mp 170-172°.
- (j) N-(4-Hydroxyphenyl)-lH-pyrazol-3-amine, mp 190-192°. (k) 4-(1-methylpyrazol-3-yl)aminophenol, mp 183-185°.
- (1) 1-Methyl-N-(4-dimethylaminophenyl)-1H-pyrazol-3-amino, mp 84-85°.
- (m) 4-(1-(4-Methoxyphenyl)methylpyrazol-3-yl)aminophenol, mp 123-124 0.
Claims (13)







Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT87303666T ATE68485T1 (en) | 1986-05-07 | 1987-04-27 | PYRAZOLE. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB8611102 | 1986-05-07 | ||
| GB868611102A GB8611102D0 (en) | 1986-05-07 | 1986-05-07 | Heterocyclic compounds |
| GB8630906 | 1986-12-24 | ||
| GB868630906A GB8630906D0 (en) | 1986-12-24 | 1986-12-24 | Heterocyclic compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0248523A1 true EP0248523A1 (en) | 1987-12-09 |
| EP0248523B1 EP0248523B1 (en) | 1991-10-16 |
Family
ID=26290729
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP87303666A Expired - Lifetime EP0248523B1 (en) | 1986-05-07 | 1987-04-27 | Pyrazoles |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US4803216A (en) |
| EP (1) | EP0248523B1 (en) |
| KR (1) | KR870011101A (en) |
| AT (1) | AT390732B (en) |
| AU (1) | AU595602B2 (en) |
| CA (1) | CA1305157C (en) |
| DE (1) | DE3773746D1 (en) |
| DK (1) | DK231687A (en) |
| FI (1) | FI871974A (en) |
| IL (1) | IL82421A0 (en) |
| NO (1) | NO871879L (en) |
| NZ (1) | NZ220214A (en) |
| PT (1) | PT84831B (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990014338A1 (en) * | 1989-05-20 | 1990-11-29 | Fisons Plc | Anti-inflammatory 4-aminophenol derivatives |
| WO2005047273A1 (en) * | 2003-11-14 | 2005-05-26 | Novartis Ag | Thiazole and pyrazole derivatives as flt-3 kinase inhibitors |
| WO2009135944A1 (en) | 2008-05-09 | 2009-11-12 | Janssen Pharmaceutica Nv | Trisubstituted pyrazoles as acetylcholine receptor modulators |
| WO2011033018A1 (en) | 2009-09-17 | 2011-03-24 | Janssen Pharmaceutica Nv | Substituted n-phenyl-1-(4-pyridinyl)-1h-pyrazol-3-amines |
| CN111836801A (en) * | 2018-02-07 | 2020-10-27 | 韩国化学硏究院 | Compound for inhibiting TNIK and medical application thereof |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4234886A1 (en) * | 1992-10-16 | 1994-04-21 | Wella Ag | New N-phenylaminopyrazole derivatives and agents and processes for coloring hair |
| GB9312853D0 (en) * | 1993-06-22 | 1993-08-04 | Euro Celtique Sa | Chemical compounds |
| US5500439A (en) * | 1993-12-09 | 1996-03-19 | Alteon Inc. | Aminopyrazoles |
| US5591776A (en) * | 1994-06-24 | 1997-01-07 | Euro-Celtique, S.A. | Pheynl or benzyl-substituted rolipram-based compounds for and method of inhibiting phosphodiesterase IV |
| US5922751A (en) * | 1994-06-24 | 1999-07-13 | Euro-Celtique, S.A. | Aryl pyrazole compound for inhibiting phosphodiesterase IV and methods of using same |
| EP0766676B1 (en) * | 1994-06-24 | 2002-05-22 | Euroceltique S.A. | Compounds for inhibiting phosphodiesterase iv |
| AU4527996A (en) * | 1994-12-13 | 1996-07-03 | Euro-Celtique S.A. | Trisubstituted thioxanthines |
| US6025361A (en) * | 1994-12-13 | 2000-02-15 | Euro-Celtique, S.A. | Trisubstituted thioxanthines |
| WO1996018399A1 (en) * | 1994-12-13 | 1996-06-20 | Euro-Celtique, S.A. | Aryl thioxanthines |
| US6166041A (en) * | 1995-10-11 | 2000-12-26 | Euro-Celtique, S.A. | 2-heteroaryl and 2-heterocyclic benzoxazoles as PDE IV inhibitors for the treatment of asthma |
| US6075016A (en) * | 1996-04-10 | 2000-06-13 | Euro-Celtique S.A. | 6,5-fused aromatic ring systems having enhanced phosphodiesterase IV inhibitory activity |
| US5864037A (en) | 1996-06-06 | 1999-01-26 | Euro-Celtique, S.A. | Methods for the synthesis of chemical compounds having PDE-IV inhibitory activity |
| US5744473A (en) * | 1996-09-16 | 1998-04-28 | Euro-Celtique, S.A. | PDE IV inhibitors: "bis-compounds" |
| DE60234057D1 (en) | 2001-07-25 | 2009-11-26 | Raptor Pharmaceutical Inc | COMPOSITIONS AND METHODS FOR MODULATING TRANSPORT BY THE BLOOD-BRAIN BARRIER |
| ATE410415T1 (en) * | 2003-02-27 | 2008-10-15 | Smithkline Beecham Corp | NEW CONNECTIONS |
| CN103259027A (en) | 2005-04-28 | 2013-08-21 | 普罗透斯数字保健公司 | Pharma-informatics system |
| JO3019B1 (en) | 2006-04-19 | 2016-09-05 | Janssen Pharmaceutica Nv | Trisubstituted 1,2,4-Triazoles |
| KR20090071598A (en) | 2006-09-18 | 2009-07-01 | 랩터 파마슈티컬 인코포레이티드 | Treatment of liver disorders by administration of receptor-associated protein(rap)-conjugates |
| MX2010004177A (en) | 2007-10-18 | 2010-05-03 | Janssen Pharmaceutica Nv | Trisubstituted 1,2,4-triazoles. |
| JO2784B1 (en) | 2007-10-18 | 2014-03-15 | شركة جانسين فارماسوتيكا ان. في | 1,3,5-trisubstitued triazole derivative |
| AR070936A1 (en) | 2008-03-19 | 2010-05-12 | Janssen Pharmaceutica Nv | 1,2,4-TRISUSTITUTED TRIAZOLS |
| PT2398500T (en) | 2009-02-20 | 2019-06-14 | 2 Bbb Medicines B V | Glutathione-based drug delivery system |
| TWI556839B (en) | 2009-05-06 | 2016-11-11 | 研究室護膚股份有限公司 | Dermal delivery compositions comprising active agent-calcium phosphate particle complexes and methods of using the same |
| US20120077778A1 (en) | 2010-09-29 | 2012-03-29 | Andrea Bourdelais | Ladder-Frame Polyether Conjugates |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0178035A1 (en) * | 1984-05-12 | 1986-04-16 | FISONS plc | Anti-inflammatory 1,N-diarylpyrazol-3-amines, compositions containing them and processes for their preparation |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DD211343A1 (en) * | 1982-11-08 | 1984-07-11 | Univ Ernst Moritz Arndt | PROCESS FOR SYNTHESIS OF PYRAZOLES |
-
1987
- 1987-04-27 DE DE8787303666T patent/DE3773746D1/en not_active Expired - Fee Related
- 1987-04-27 EP EP87303666A patent/EP0248523B1/en not_active Expired - Lifetime
- 1987-05-05 IL IL82421A patent/IL82421A0/en unknown
- 1987-05-05 FI FI871974A patent/FI871974A/en not_active IP Right Cessation
- 1987-05-05 US US07/046,656 patent/US4803216A/en not_active Expired - Fee Related
- 1987-05-06 CA CA000536506A patent/CA1305157C/en not_active Expired - Fee Related
- 1987-05-06 NO NO871879A patent/NO871879L/en unknown
- 1987-05-06 DK DK231687A patent/DK231687A/en not_active Application Discontinuation
- 1987-05-06 NZ NZ220214A patent/NZ220214A/en unknown
- 1987-05-07 AU AU72584/87A patent/AU595602B2/en not_active Ceased
- 1987-05-07 PT PT84831A patent/PT84831B/en not_active IP Right Cessation
- 1987-05-07 KR KR870004448A patent/KR870011101A/en not_active Application Discontinuation
- 1987-11-03 AT AT0290487A patent/AT390732B/en not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0178035A1 (en) * | 1984-05-12 | 1986-04-16 | FISONS plc | Anti-inflammatory 1,N-diarylpyrazol-3-amines, compositions containing them and processes for their preparation |
Non-Patent Citations (5)
| Title |
|---|
| CHEMICAL ABSTRACT, vol. 88, no. 7, 13th February 1978, page 164, abstract no. 46216v, Columbus, Ohio, US; A.N. BORISEVICH et al.: "Synthesis and growth regulating effect of substituted 1-acylpyrazole", & FIZIOL. AKT. VESHCHESTVA 1977, 9, 47-52 * |
| CHEMICAL ABSTRACTS, vol. 104, no. 19, 12th May 1986, page 667, abstract no. 168404z, Columbus, Ohio, US; J.N. VISHWAKARMA et al.: "Reactions of polarized ketene S,N-acetals with hydrazine: a facile general route to 3(5)-sbstituted amino-4,5(3)-substituted pyrazoles. Part XLV.", & INDIAN J. CHEM. SECT. B 1985, 24B(5), 472-6 * |
| CHEMICAL ABSTRACTS, vol. 71, no. 5, 4th August 1969, page 323, abstract no. 22058t, Columbus, Ohio, US; A.N. BORISEVICH et al.: "Arylamides of substituted thionoacetic acid. IV. Interaction of arylamides of monoacetylthionoacetic acid with nucleophilic reagents", & KHIM. GETEROTSIKL. SOEDIN. 1969, (2), 312-15 * |
| CHEMISCHE BERICHTE, vol. 100, no. 8, 1967, pages 2577-2584, Weinheim, DE; A. DORNOW et al.: "Synthesen von Pyrazolo-pyrimidinen aus neuen Pyrazol- und Pyrimidin-Derivaten" * |
| CHEMISCHE BERICHTE, vol. 95, no. 4, 1962, pages 937-943, Weinheim, DE; S. HÜNIG et al.: "Synthesen mit Enaminen, VIII. Heterocyclen aus Enamin-Isothiocyanat-Addukten" * |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990014338A1 (en) * | 1989-05-20 | 1990-11-29 | Fisons Plc | Anti-inflammatory 4-aminophenol derivatives |
| US5428044A (en) * | 1989-05-20 | 1995-06-27 | Fisons Plc | Anti-inflammatory 4-aminophenyl derivatives |
| WO2005047273A1 (en) * | 2003-11-14 | 2005-05-26 | Novartis Ag | Thiazole and pyrazole derivatives as flt-3 kinase inhibitors |
| EA018187B1 (en) * | 2008-05-09 | 2013-06-28 | Янссен Фармацевтика Нв | Trisubstituted pyrazoles as acetylcholine receptor modulators |
| US20110065683A1 (en) * | 2008-05-09 | 2011-03-17 | Thuring Johannes Wilhelmus John F | Trisubstituted pyrazoles as acetylcholine receptor modulators |
| WO2009135944A1 (en) | 2008-05-09 | 2009-11-12 | Janssen Pharmaceutica Nv | Trisubstituted pyrazoles as acetylcholine receptor modulators |
| US8779158B2 (en) * | 2008-05-09 | 2014-07-15 | Janssen Pharmaceutica Nv | Trisubstituted pyrazoles as acetylcholine receptor modulators |
| WO2011033018A1 (en) | 2009-09-17 | 2011-03-24 | Janssen Pharmaceutica Nv | Substituted n-phenyl-1-(4-pyridinyl)-1h-pyrazol-3-amines |
| CN111836801A (en) * | 2018-02-07 | 2020-10-27 | 韩国化学硏究院 | Compound for inhibiting TNIK and medical application thereof |
| EP3749648A4 (en) * | 2018-02-07 | 2021-11-24 | Korea Research Institute of Chemical Technology | Compounds for inhibiting tnik and medical uses thereof |
| US11485711B2 (en) | 2018-02-07 | 2022-11-01 | Korea Research Institute Of Chemical Technology | Compounds for inhibiting TNIK and medical uses thereof |
| AU2019217094B2 (en) * | 2018-02-07 | 2023-04-06 | Industry-Academic Cooperation Foundation, Yonsei University | Compounds for inhibiting TNIK and medical uses thereof |
| US11767297B2 (en) | 2018-02-07 | 2023-09-26 | Korea Research Institute Of Chemical Technology | Compounds for inhibiting TNIK and medical uses thereof |
| CN111836801B (en) * | 2018-02-07 | 2024-02-02 | 韩国化学硏究院 | Compounds for inhibiting TNIK and medical use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| FI871974A0 (en) | 1987-05-05 |
| PT84831B (en) | 1990-02-08 |
| NO871879D0 (en) | 1987-05-06 |
| NZ220214A (en) | 1990-06-26 |
| FI871974A (en) | 1987-11-08 |
| IL82421A0 (en) | 1987-11-30 |
| DK231687A (en) | 1987-11-08 |
| PT84831A (en) | 1987-06-01 |
| AU595602B2 (en) | 1990-04-05 |
| DK231687D0 (en) | 1987-05-06 |
| KR870011101A (en) | 1987-12-21 |
| DE3773746D1 (en) | 1991-11-21 |
| EP0248523B1 (en) | 1991-10-16 |
| CA1305157C (en) | 1992-07-14 |
| AU7258487A (en) | 1987-11-12 |
| NO871879L (en) | 1987-11-09 |
| ATA290487A (en) | 1989-12-15 |
| US4803216A (en) | 1989-02-07 |
| AT390732B (en) | 1990-06-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0248523B1 (en) | Pyrazoles | |
| US4810719A (en) | Anti-inflammator 1,n-diarylpyrazol-3-amines | |
| US5459151A (en) | N-acyl substituted phenyl piperidines as bronchodilators and antiinflammatory agents | |
| US4216220A (en) | Platelet aggregation inhibiting 2-oxyindoles, their compositions and method of use | |
| US5428044A (en) | Anti-inflammatory 4-aminophenyl derivatives | |
| US4313947A (en) | Platelet aggregation inhibiting 2-oxyindoles, their compositions and method of use | |
| US4294841A (en) | Derivatives of 1-phenyl 3-(4-piperidyl) 1-propanone usable as drugs | |
| US5110831A (en) | Vinylogous hydroxamic acids and derivatives thereof as 5-lipoxygenase inhibitors | |
| Child et al. | Fenbufen, a new anti-inflammatory analgesic: synthesis and structure-activity relationships of analogs | |
| EP0485984B1 (en) | Diarylmethoxypiperidine derivatives | |
| US4009166A (en) | Pyrido(2,3-d) pyrimidinones | |
| US5051430A (en) | 3-(1H-indazol-3-yl)-4-pyridinamines | |
| US4757078A (en) | Cyclic aryl hydroxamic acids, derivatives thereof and method of use as anti-allergy agents | |
| EP0127371B1 (en) | 1,n-diaryl-4,5-dihydro-1h-pyrazol-3-amines, compositions containing them and methods for their preparation | |
| US4411907A (en) | Antiinflammatory 3H-naphtho(1,2-d)imidazoles | |
| US4243665A (en) | 2-Heterocyclylalkyl-6-methoxy-naphthalenes | |
| US4454136A (en) | Substituted benzopyranotriazoles and antiallergic use | |
| EP0287971B1 (en) | Benzimidazole derivatives and process for their preparations | |
| JPS62267269A (en) | Pyrazole-3-amines | |
| EP0121806A1 (en) | Pyrazolo(1,5-a)pyridine derivatives and compositions containing them | |
| US3972883A (en) | 1,6-Dimethyl-8β-[2' or 3'-pyrroyloxyethyl or substituted pyrroyloxyethyl]-10α-methoxyergolene compounds | |
| US4026899A (en) | Benzo and benzothiopyranoindazole N-oxides | |
| KOMESHIMA et al. | Synthesis and anti-inflammatory activity of antioxidants, 4-alkylthio-o-anisidine derivatives | |
| US4473570A (en) | Imidazo[1,5-c]pyrimidin-5-ones | |
| US5166163A (en) | 3-(1H-indazol-3-yl)-4-pyridinamines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 19870506 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| 17Q | First examination report despatched |
Effective date: 19890614 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 19911016 Ref country code: LI Effective date: 19911016 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 19911016 Ref country code: AT Effective date: 19911016 Ref country code: CH Effective date: 19911016 Ref country code: NL Effective date: 19911016 Ref country code: BE Effective date: 19911016 Ref country code: SE Effective date: 19911016 |
|
| REF | Corresponds to: |
Ref document number: 68485 Country of ref document: AT Date of ref document: 19911115 Kind code of ref document: T |
|
| REF | Corresponds to: |
Ref document number: 3773746 Country of ref document: DE Date of ref document: 19911121 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 19920127 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| EN | Fr: translation not filed | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Effective date: 19920306 |
|
| NLV1 | Nl: lapsed or annulled due to failure to fulfill the requirements of art. 29p and 29m of the patents act | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19920430 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 19940419 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19940426 Year of fee payment: 8 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19950427 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19950427 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19960103 |