EP0101951A1 - 1,3-Dioxolo(4,5-g)quinolines and preparation thereof - Google Patents
1,3-Dioxolo(4,5-g)quinolines and preparation thereof Download PDFInfo
- Publication number
- EP0101951A1 EP0101951A1 EP83107464A EP83107464A EP0101951A1 EP 0101951 A1 EP0101951 A1 EP 0101951A1 EP 83107464 A EP83107464 A EP 83107464A EP 83107464 A EP83107464 A EP 83107464A EP 0101951 A1 EP0101951 A1 EP 0101951A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- compound
- hydrogen
- formula
- alkyl
- formyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 C*c(cnc1c2cc3OCOc3c1)c2O Chemical compound C*c(cnc1c2cc3OCOc3c1)c2O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/056—Ortho-condensed systems with two or more oxygen atoms as ring hetero atoms in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Definitions
- the present invention relates to compounds having the 1,3-dioxolo[4,5-g]quinoline ring system: which compounds are useful as intermediates and antibacterial agents, and to methods for the preparation thereof.
- U.S. Patent 4,284,629 discloses certain 4-quinolone-3-carboxylic acids having a tertiary-amino substituent in the 1-position, stated to possess antibacterial activity.
- Exemplary of the compounds disclosed are 1-dimethylamino-6-nitro-2-methyl-4-quinolone-3-carboxylic acid, methyl ester (Example 4); 1-dimethylamino-7-chloro-6-nitro-2-methyl-4-quinolone-3-carboxylic acid, methyl ester (Example 5); and 1-dimethylamino-7-(n-butylmercapto)-6-nitro-2-methyl-4-quinolone-3-carboxylic acid, methyl ester (Example 17).
- the present invention relates to compounds of the formula where R is hydrogen or alkyl or 1-3 carbon atoms; R' is hydrogen, formyl, alkyl or 1-3 carbon atoms, or alkoxycarbonyl where the alkyl moiety of the latter group is identical with R; and R" is hydrogen or lower-alkyl; and alkali metal or amine salts of compounds where R" is hydrogen.
- a composition for treatment of bacterial infections in animals, including humans comprises an antibacterially effective amount of a compound of Formula I where R is methyl or ethyl, and R' and R" are hydrogen, together with one or more pharmaceutically acceptable excipients.
- lower-alkyl in defining R' stands for alkyl preferably having from one to four carbon atoms.
- the amino function is introduced into the 5-position of the 1,3-dioxolo[4,5-g]quinoline ring by treatment of the known (U.S. Pat. 3,287,458) starting material of the formula wherein R" is hydrogen or lower-alkyl, with an aminating agent, e.g. an O - arylhydroxylamine such as O-(2,4-dinitrophenyl)-hydroxylamine [2,4-(O 2 N) 2 C 6 H 3 ONH 2 ]; cf. Tamura et al., J. Org. Chem. 38, 1239 (1973).
- the reaction takes place at ambient temperature in an inert organic solvent in the presence of a base such as potassium carbonate, resulting in the formation of the N-amino compound of Formula II below.
- the N-formyl derivative (III) is prepared from the primary amino compound (II) by a conventional formylation procedure using the mixed anhydride derived from formic acid and acetic anhydride.
- the alkylation procedure is carried out either on the primary amino compound (II) or the N-formyl derivative (III).
- the alkylation is carried out with an alkyl halide, preferably bromide or iodide, where alkyl has from 1 to 3 carbon atoms, in the presence of an alkali metal carbonate, preferably potassium carbonate, to yield as the product an N-alkyl-N-(alkoxycarbonyl) compound (IV).
- the alkylation is carried out with an alkyl halide in the presence of any strong base, e.g.
- alkali metal hydroxide, carbonate or alkoxide the product being an N-alkyl-N-formyl compound (V), where alkyl is methyl, ethyl, propyl or isopropyl.
- V N-alkyl-N-formyl compound
- the alkylation reactions take place at a temperature between about 25°C. and 100°C. in an inert organic solvent.
- the alkaline hydrolysis of a compound of Formula IV or V to form an N-alkyl compound of Formula VI is preferably carried out in aqueous alcoholic solution at a temperature between about 50°C. and 100°C.
- R" in compounds of Formula IV or V is lower-alkyl
- hydrolysis using two or more molar equivalents of base produces a compound of Formula VI where R" is hydrogen.
- selective hydrolysis of the alkoxycarbonyl or formyl group attached to nitrogen is realized, affording a compound of Formula VI where R" is lower-alkyl.
- the compounds of Formula I where both R and R' are alkyl of 1-3 carbon atoms can be prepared by reacting a compound of Formula II with a dialkyl sulfate. The reactants are heated together at a temperature between about 75°C. and 150°C.
- the compounds of Formula I where R" is hydrogen can also be prepared and used in the form of their alkali metal or amine salts, preferably the sodium, potassium or N-methylglucamine salts.
- the chloroform layer was dried over anhydrous magnesium sulfate, decolorized with charcoal and concentrated to give 3.1 g of solid residue. The latter was recrystallized from acetonitrile to give 2.5 g of ethyl 5,8-dihydro-5-[(formyl)methylamino]-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylate, colorless crystals, m.p. 242-244°C.
- Ethyl 5-(ethyl(formyl)amino]-5,8-dihydro-8-oxo-1,3-dioxolo-[4,5-g]quinoline-7-carboxylate [V; R and R" are C 2 H 5 ] was prepared from 5 g of ethyl 5-(formylamino)-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 3), 4.5 g of potassium carbonate and 3.7 ml of ethyl iodide according to the procedure of Example 6.
- reaction mixture was allowed to stand overnight, then chilled in an ice-bath, and the solid product was collected by filtration and washed with absolute alcohol and ether to give 2 g of 5-(ethylamino)-5,8-dihydro-8-oxo- i,3-dioxolo[4,5-g]quinoline-7-carboxylic acid in the form of its potassium salt hemihydrate m.p. 242°C. (decompn.) after being dried for several days at 75°C. (0.1 mm).
- the final products of the invention have been found to possess antibacterial activity.
- the in vitro antibacterial activity was determined by conventional serial dilution procedures. Bacterial cultures were grown in tryptose phosphate broth or brain heart infusion broth (containing heatinactivated normal horse serum for tests with S. pyogenes) overnight at 37°C. and subsequently diluted in double strength broth to provide bacterial inocula of about 2 x 10 S cells/ml.
- Aqueous solutions of the compounds of the invention were prepared by dissolving the free acid form in 0.5N sodium hydroxide. The solutions were diluted with sterile distilled water to 1000 mcg/ml of compound in terms of the free acid.
- mice infected with E. coli were medicated once (0.5 ml) one-half hour post infection, the test compound being administered by the subcutaneous (s.c.) route. Deaths were recorded daily for seven days.
- mice infected with K. pneumoniae were medicated at the following times: seventeen hours and one hour pre- infection, six hours postinfection and twice a day for the next three days.
- the test compound was administered by the subcutaneous (0.2 ml) route. Deaths were recorded daily for fourteen days.
- the antibacterial activity of the compounds of Formula VI (R" decreases as the carbon content of the group R increases.
- the preferred compound is that of Example 9, namely 5,8-dihydro-5-(methylamino)-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylic acid.
- the intermediates for the compounds of Formula VI are mostly inactive as antibacterial agents.
- the compound of Example 2 had some in vitro activity (MIC 7.8, 3.9 and 3.9 against E. coli, P. mirab. and P. vulg., respectively); and the compound of Example 7 was active in vitro against E. coli, K. pneum., P. mirab. and P. vulg. (MIC 31.3, 15.6, 7.8 and 15.6, respectively) and had a PD 50 value of 40 in vivo against E. coli.
- the antibacterially active compounds of the invention can be prepared for use by conventional pharmaceutical procedures: that is, by dissolving or suspending them in a pharmaceutically acceptable vehicle, e.g., water, aqueous alcohol, glycol, oil solution or oil-water emulsion, for parenteral or oral administration; or by incorporating them in unit dosage form as capsules or tablets for oral administration either alone or in combination with conventional adjuvants or excipients, e.g., calcium carbonate, starch, lactose, talc, magnesium stearate, gum acacia, and the like.
- a pharmaceutically acceptable vehicle e.g., water, aqueous alcohol, glycol, oil solution or oil-water emulsion
- conventional adjuvants or excipients e.g., calcium carbonate, starch, lactose, talc, magnesium stearate, gum acacia, and the like.
Abstract
Compounds of formula
<CHEM>
where R is methyl or ethyl, useful as antibacterial agents, are prepared from 8-hydroxy-1,3-dioxolo (4,5-g) quinoline-7-carboxylic acid or a lower-alkyl ester thereof via a series of novel intermediates.
Description
- The present invention relates to compounds having the 1,3-dioxolo[4,5-g]quinoline ring system:

- Certain substituted 1,3-dioxolo[4,5-g]quinolines are known to possess antibacterial activity. Illustrative of these compounds are those of the formula
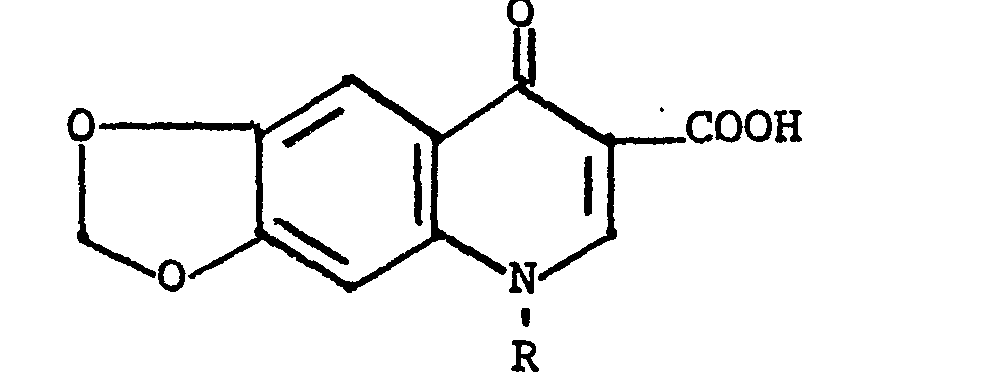
- U.S. Patent 4,284,629 discloses certain 4-quinolone-3-carboxylic acids having a tertiary-amino substituent in the 1-position, stated to possess antibacterial activity. Exemplary of the compounds disclosed are 1-dimethylamino-6-nitro-2-methyl-4-quinolone-3-carboxylic acid, methyl ester (Example 4); 1-dimethylamino-7-chloro-6-nitro-2-methyl-4-quinolone-3-carboxylic acid, methyl ester (Example 5); and 1-dimethylamino-7-(n-butylmercapto)-6-nitro-2-methyl-4-quinolone-3-carboxylic acid, methyl ester (Example 17). There is no disclosure of compounds having a methylenedioxy group attached to the phenyl ring.
- The present invention relates to compounds of the formula
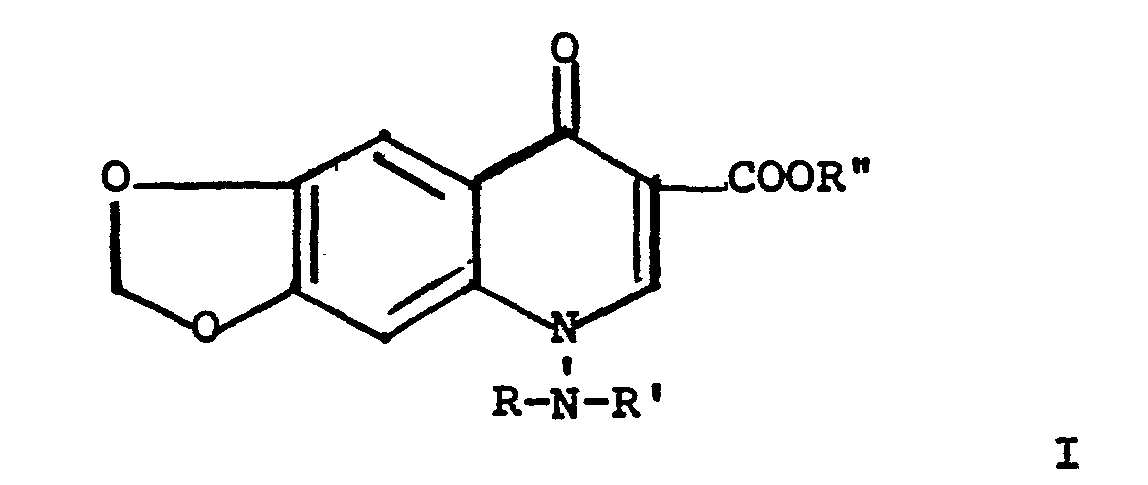
- A composition for treatment of bacterial infections in animals, including humans, comprises an antibacterially effective amount of a compound of Formula I where R is methyl or ethyl, and R' and R" are hydrogen, together with one or more pharmaceutically acceptable excipients.
- One can prepare a compound of the Formula I by treating a compound of the Formula VII (herein) with an aminating agent, wherein R" is hydrogen or lower-alkyl, to prepare the compound wherein R and R' are each hydrogens;
- if desired, formylating a compound of Formula I obtained where R and R' are each hydrogens to prepare the compound where R' is formyl and R" is hydrogen;
- if desired, alkylating a compound of the Formula I obtained wherein R' is formyl or hydrogen with an alkyl halide of 1-3 carbon atoms in the presence of a base when R' is formyl or an alkali metal carbonate when R' is hydrogen to give a compound of the Formula I wherein R" is lower-alkyl, R is alkyl of 1-3 carbon atoms and R' is formyl or alkoxycarbonyl, respectively;
- if desired, subjecting a compound of the Formula I obtained to hydrolysis with one or more molar equivalents of an alkali metal base to prepare a compound of Formula I wherein R' is hydrogen, said hydrolysis producing an alkali metal salt of a compound wherein R" is hydrogen when two or more molar equivalents of the alkali metal base are used;
- if desired, converting an alkali metal salt to the free base (R" is hydrogen);
- if desired, converting a compound of the Formula I obtained wherein R" is lower-alkyl to the corresponding compound wherein R" is hydrogen;
- if desired, reacting a compound of the Formula I obtained wherein R and R' are both hydrogens with dialkyl sulfate to obtain the corresponding compound wherein both Rand R' are alkyl of 1-3 carbon atoms;
- One can treat bacterial infections in animals, including humans, by administering to an animal or human so infected a composition comprising an antibacterially effective amount of a compound of Formula I where R is methyl or ethyl, and R and R" are hydrogen.
- In the definition of the variables in Formula I above the term "lower-alkyl" in defining R' stands for alkyl preferably having from one to four carbon atoms.
- The amino function is introduced into the 5-position of the 1,3-dioxolo[4,5-g]quinoline ring by treatment of the known (U.S. Pat. 3,287,458) starting material of the formula

- The synthesis of the compounds of the invention proceeds in accordance with the following flow-sheet:

- The N-formyl derivative (III) is prepared from the primary amino compound (II) by a conventional formylation procedure using the mixed anhydride derived from formic acid and acetic anhydride.
- The alkylation procedure is carried out either on the primary amino compound (II) or the N-formyl derivative (III). In the case of the former, the alkylation is carried out with an alkyl halide, preferably bromide or iodide, where alkyl has from 1 to 3 carbon atoms, in the presence of an alkali metal carbonate, preferably potassium carbonate, to yield as the product an N-alkyl-N-(alkoxycarbonyl) compound (IV). In the case of the N-formyl derivative, the alkylation is carried out with an alkyl halide in the presence of any strong base, e.g. alkali metal hydroxide, carbonate or alkoxide, the product being an N-alkyl-N-formyl compound (V), where alkyl is methyl, ethyl, propyl or isopropyl. The alkylation reactions take place at a temperature between about 25°C. and 100°C. in an inert organic solvent.
- The alkaline hydrolysis of a compound of Formula IV or V to form an N-alkyl compound of Formula VI is preferably carried out in aqueous alcoholic solution at a temperature between about 50°C. and 100°C. In the case where R" in compounds of Formula IV or V is lower-alkyl, hydrolysis using two or more molar equivalents of base produces a compound of Formula VI where R" is hydrogen. On the other hand, if only one molar equivalent of base is used, selective hydrolysis of the alkoxycarbonyl or formyl group attached to nitrogen is realized, affording a compound of Formula VI where R" is lower-alkyl.
- The compounds of Formulas III, IV and V where R" is lower-alkyl can be readily converted to the respective free acids (R" is hydrogen) by heating with formic acid to effect an ester exchange.
- Alkylation of compounds of Formulas II and III where R" is hydrogen will also cause esterification of the carboxyl group to form compounds of Formulas IV and V, respectively, where R" is lower-alkyl.
- The compounds of Formula I where both R and R' are alkyl of 1-3 carbon atoms can be prepared by reacting a compound of Formula II with a dialkyl sulfate. The reactants are heated together at a temperature between about 75°C. and 150°C.
- The compounds of Formula I where R" is hydrogen can also be prepared and used in the form of their alkali metal or amine salts, preferably the sodium, potassium or N-methylglucamine salts.
- The following examples will further illustrate the invention.
- To a stirred mixture of 12.8 g (0.049 mole) of ethyl 8-hydroxy-l,3-dioxolo[4,5-g]quinoline-7-carboxylate and 13.5 g (0.098 mole) of potassium carbonate in 500 ml of dimethylformamide was added 11.7 g (0.059 mole) of 0-(2,4-dinitrophenyl)hydroxylamine. The reaction mixture was stirred overnight at room temperature. An additional 5.0 g of 0-(2,4-dinitrophenyl)hydroxylamine and 300 ml of dimethylformamide were then added and stirring was continued again overnight. The solvent was removed in vacuo, the residue stirred in 500 ml of water, and the product was collected by filtration and recrystallized from dimethylformamide to give 7.6 g of ethyl 5-amino-5,8-dihydro-8-oxo-1,3-dioxolo[4,5-g]quinoline-7-carboxylate, pale orange crystals, m.p. 251-253°C.
- To a solution of 4.4 g (0.11 mole) of sodium hydroxide in 350 ml of water was added 3.0 g (0.047 mole) of ethyl 5-amino-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g] quinoline-7-carboxylate (Example 1). The resulting slurry was stirred at about 90°C. for three hours, then cooled to room temperature and neutralized with 9.3 ml of acetic acid. The solid product was collected by filtration and recrystallized from dimethylformamide to give 10.3 g of 5-amino-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylic acid, colorless crystals, m.p. 309-313°C. (decompn.).
- Formic acid (95.7%) (17.35 g, 0.36 mole) was added dropwise to 37.0 g (0.36 mole) of acetic anhydride at 0°C. The mixture was stirred 15 min. at 0°C., 15 min. at 50°C, and again cooled to 0°C. There were then added 10.0 g (0.036 mole) of ethyl 5-amino-5,8-dihydro-8-oxo- l,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 1) and 10 ml of formic acid. The resulting solution was stirred one hour at 0°C. and at room temperature overnight. The reaction mixture was poured into 400 ml of ice-water, stirred for one hour, and the solid product was collected by filtration and recrystallized from dimethylformamide to give 7 g of product, m.p. 252-254°C. (decompn.). Further recrystallizations from dimethylformamide and dimethylformamide- ethanol gave a sample of ethyl 5-(formylamino-5,8-dihydro-8-oxo-1,3-dioxolo[4,5-g]quinoline-7-carboxylate, colorless crystals, m.p. 255-257°C. (decompn.).
- A mixture of 5.8 g of ethyl 5-(formylamino)-5,8-dihydro-8-oxo-1,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 3) and 100 ml of 97% formic acid was heated at reflux under a Dean-Stark separator for about eight hours. The reaction mixture was then distilled to remove 30 ml of volatiles, cooled and stirred to induce crystallization of product. The latter was collected by filtration and washed with absolute alcohol and ether to give 2.3 g of 5-(formylamino)-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g] quinoline-7-carboxylic acid, tan crystals, m.p. 302°C. (decompn.).
- A mixture of 6.0 g (0.0217 mole) of ethyl 5-amino-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 1), 30.9 g (0.217 mole) of methyl iodide and 30.0 g 0.217 mole) of potassium carbonate in 90 ml of dimethylformamide was stirred at room temperature for 24 hours. The reaction was still incomplete, so an additional 30.9 g of methyl iodide was added and the mixture heated at reflux for 18 hours. The solvent was removed in vacuo and the residue partitioned between chloroform and water. The chloroform layer was dried over anhydrous magnesium sulfate, concentrated and chromatographed on a Waters Prep. 500 HPLC apparatus. There was obtained 6.5 g of product which was recrystallized from ethyl acetate-hexane to give 6 g of ethyl 5,8-dihydro-5-[(methoxycarbonyl)methylamino]-8-oxo- l,3-dioxolo[4,5-g]quinoline-7-carboxylate, colorless crystals, m.p. 142-145°C.
- A mixture of 3.0 g (0.01 mole) of ethyl 5-(formyl- amino)-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 3) and 2.76 g (0.02 mole) of potassium carbonate in 50 ml of dimethylformamide (distilled from calcium hydride) was stirred at room temperature for six hours. Methyl iodide (2.84 g, 0.02 mole) was then added and the resulting slurry was stirred overnight. The solvent was removed in vacuo and the residue partitioned between chloroform and water. The chloroform layer was dried over anhydrous magnesium sulfate, decolorized with charcoal and concentrated to give 3.1 g of solid residue. The latter was recrystallized from acetonitrile to give 2.5 g of ethyl 5,8-dihydro-5-[(formyl)methylamino]-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylate, colorless crystals, m.p. 242-244°C.
- S[(Formyl)methylamino]-5,8-dihydro-8-oxo-1,3-dioxolo[4,5-g]-quinoline-7-carboxylic acid [V; R is CH3, R" is H] was prepared by heating ethyl 5,8-dihydro-5[(formyl)methylamino]-8-oxo-1,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 6) with formic acid according to the procedure of Example 4, and was obtained in the form of colorless plates, m.p. above 330°C. when precipitated from a solution of its potassium salt by addition of acetic acid.
- Ethyl 5-(ethyl(formyl)amino]-5,8-dihydro-8-oxo-1,3-dioxolo-[4,5-g]quinoline-7-carboxylate [V; R and R" are C2H5] was prepared from 5 g of ethyl 5-(formylamino)-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 3), 4.5 g of potassium carbonate and 3.7 ml of ethyl iodide according to the procedure of Example 6. There was obtained 3.2 g of ethyl 5-[ethyl(formyl)amino]-5,8-dihydro-8-oxo-1,3-dioxolo[4,5-g]quinoline-7-carboxylate, colorless granules, m.p. 218-221°C. when recrystallized from ethanol.
- A mixture of 4.6 g (0.0132 mole) of ethyl 5,8-dihydro-5-[(methoxycarbonyl)methylamino]-8-oxo-1,3-dioxolo [4,5-g]-quinoline-7-carboxylate (Example 5), 1.6 g (0.0396 mole) of sodium hydroxide and about 100 ml of water was stirred at 95°C. for 4.5 hours. The reaction mixture was cooled, acidified with 5 ml of acetic acid with stirring, and kept in a refrigerator overnight. The solid product was collected by filtration, washed with water and recrystallized from 70 ml of boiling dimethylformamide. There was obtained 3 g of 5,8-dihydro-5-(methylamino)-8-oxo-1,3-dioxolo[4,5-g]quinoline-7-carboxylic acid, colorless crystals, m.p. above 300°C.
- A mixture of 0.6 g (0.0019 mole) of ethyl 5,8-dihydro-5[(formyl)methylamino]-8-oxo-l,3-dioxolo[4,5-g] quinoline-7-carboxylate (Example 6), 0.30 g (0.0075 mole) of sodium hydroxide and 20 ml of water was heated on a steam bath for two hours. The reaction mixture was cooled to room temperature and 0.6 g of acetic acid was added. The resulting precipitated product was collected by filtration, washed with 100 ml of water and dried in vacuo at 60°C. for one day to give 0.45 g of colorless solid, determined by thin layer chromatography to be identical with compound obtained in Example 9, namely 5,8-dihydro-5-(methylamino)-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylic acid.
- To a suspension of 4.1 g (0.013 mole) of ethyl 5,8-dihydro-5-[(formyl)methylamino]-8-oxo-1,3-dioxolo[4,5-g]-quinoline-7-carboxylate (Example 6) in 40 ml of hot absolute ethanol was added a solution of 0.8 g (0.013 mole) of potassium hydroxide (85%) in 2.6 ml of water. The mixture was heated at near the boiling point of the ethanol for about 10 minutes and then chilled in an ice bath. The solid product was collected by filtration and washed with absolute ethanol and ether. After a further washing with water, tetrahydrofuran and ether and drying at 0.1 mm overnight there was obtained 3.1 g of ethyl 5,8-dihydro-5-(methylamino)-8-oxo-l,3-dioxolo [4,5-g]-quinoline-7-carboxylate, colorless crystals, m.p. 211-218°C.
- To a stirred suspension of 2 g (0.006 mole) of ethyl 5-[ethyl(formyl)amino]-5,8-dihydro-8-oxo-l,3-dioxolo [4,5-g]-quinoline-7-carboxylate (Example 8) in 53.8 ml of refluxing ethanol was added a solution of 0.9 g (0.014 mole) of potassium hydroxide in 3 ml of water over a period of about two minutes. After a few more minutes of reflux, 30 ml of additional ethanol was added and reflux continued for one hour. The reaction mixture was allowed to stand overnight, then chilled in an ice-bath, and the solid product was collected by filtration and washed with absolute alcohol and ether to give 2 g of 5-(ethylamino)-5,8-dihydro-8-oxo- i,3-dioxolo[4,5-g]quinoline-7-carboxylic acid in the form of its potassium salt hemihydrate m.p. 242°C. (decompn.) after being dried for several days at 75°C. (0.1 mm).
- To a stirred mixture of 22.7 g (0.151 mole) of potassium carbonate in 113 ml of dimethylformamide was added 14.7 g (0.048 mole) of ethyl 5-(formylamino)-5,8-dihydro-8-oxo-1,2-dioxolo[4,5-g]quinoline-7-carboxylate (Example 3). The mixture was stirred for 30 minutes, then a few crystals of potassium iodide were added followed by 33.2 ml (0.36 mole) of propyl bromide. The reaction mixture was stirred in a hot water bath for five hours, then at room temperature overnight, and poured into ice-water. The mixture was extracted three times with chloroform, and the chloroform extracts were washed with water and sodium chloride solution and dried over anhydrous magnesium sulfate. The solution was concentrated in vacuo and the solid residue (16.4 g) was slurried in tetrahydrofuran, filtered and washed with tetrahydrofuran and ether. The product was recrystallized from absolute ethanol to give 7.8 g of ethyl 5-[formyl(propyl) amino]-5,8-dihydro-8-oxo-1,3-dioxolo-[4,5-g]quinoline-7-carboxylate, colorless crystals, m.p. 188-192°C.
- 5,8-Dihdro-8-oxo-5-(proplamino)-1,3-dioxolo[4,5-]quinoline-7-carboxylic acid [VI; R is CH3CH2CH2, R' is H], was prepared from 4 g (0.012 mole) of ethyl 5-[formyl(propyl)amino]-5,8-dihydro-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 13) and 1.8 g (0.027 mole) of potassium hydroxide in aqueous ethanol, heated at reflux for one hour. The crude product was acidified with acetic acid and the solid product collected and dried to give 3.2 g of 5,8-dihydro-8-oxo-5-(propylamino)-1,3-dioxolol4,5-glquinoline-7-carboxylic acid, colorless crystals, m.p. 258-262°C.
- A mixture of 0.5 g of ethyl 5-amino-5,8-dihydro-8-oxo-1,3-dioxolo[4,5-g]quinoline-7-carboxylate (Example 1) and 10 ml of dimethyl sulfate was heated with stirring at 100°C. for 18 hours. The reaction mixture was poured into ice-water and stirred while solid potassium carbonate was added until the mixture was basic. The organic product was extracted with chloroform, and the extracts were dried over anhydrous magnesium sulfate and concentrated. The residue was separated by thin layer chromatography into two fractions which were identified by mass spectroscopy. One fraction was determined to be the mono-methyl compound of Example 11 and the other fraction was identified as the above-entitled dimethyl compound.
- The final products of the invention have been found to possess antibacterial activity. The in vitro antibacterial activity was determined by conventional serial dilution procedures. Bacterial cultures were grown in tryptose phosphate broth or brain heart infusion broth (containing heatinactivated normal horse serum for tests with S. pyogenes) overnight at 37°C. and subsequently diluted in double strength broth to provide bacterial inocula of about 2 x 10S cells/ml. Aqueous solutions of the compounds of the invention were prepared by dissolving the free acid form in 0.5N sodium hydroxide. The solutions were diluted with sterile distilled water to 1000 mcg/ml of compound in terms of the free acid. Serial two-fold dilutions of the compound stock solutions were prepared in water and 0.5 ml of each dilution was transferred to sets of tubes, one set for each bacterial inoculum. Each tube was then inoculated with 0.5 ml of the appropriate culture, resulting in a final bacterial concentration of about 1 x 105 cells/ml. The minimal inhibitory concentration (MIC), defined as the lowest concentration of the test compound to inhibit visible bacterial growth, was recorded after 18-20 hours of static incubation at 37°C. The results are recorded in Table I:

- The in vivo antibacterial activity of the compounds of the invention was determined in female mice, 18-20 grams each, by the following procedure:
- Aqueous solutions of the compounds to be tested were prepared by dissolving the free acid form in dilute sodium hydroxide and diluting the solution with distilled water to the desired volume.
- Cultures of Escherichia coli Vogel prepared in brain heart infusion broth, cultures of Klebsiella pneumoniae 39645 grown in tryptose phosphate broth with 5% rabbit serum ) diluted in the same broth were used to infect the mice as follows:
- E. coli: mice were inoculated intraperitoneally with 0.5 ml of the bacterial test inoculum (1.87 x 107 and 5 x 106 cells/ml respectively).
- 5 K. pneumoniae: mice were inoculated intramuscularly in the right hind leg with 0.2 ml of the bacterial test inoculum (2.05 x 104 cells/ml).
- Mice infected with E. coli were medicated once (0.5 ml) one-half hour post infection, the test compound being administered by the subcutaneous (s.c.) route. Deaths were recorded daily for seven days.
- Mice infected with K. pneumoniae were medicated at the following times: seventeen hours and one hour pre- infection, six hours postinfection and twice a day for the next three days. The test compound was administered by the subcutaneous (0.2 ml) route. Deaths were recorded daily for fourteen days.
- Groups of ten animals each for four or five dose levels were thus treated and the number of survivors in each group recorded. The fifty percent protective dose values (PD50) were then calculated. The results obtained are given in Table II:

- The antibacterial activity of the compounds of Formula VI (R" is H) decreases as the carbon content of the group R increases. Thus the preferred compound is that of Example 9, namely 5,8-dihydro-5-(methylamino)-8-oxo-l,3-dioxolo[4,5-g]quinoline-7-carboxylic acid.
- The intermediates for the compounds of Formula VI are mostly inactive as antibacterial agents. The compound of Example 2 had some in vitro activity (MIC 7.8, 3.9 and 3.9 against E. coli, P. mirab. and P. vulg., respectively); and the compound of Example 7 was active in vitro against E. coli, K. pneum., P. mirab. and P. vulg. (MIC 31.3, 15.6, 7.8 and 15.6, respectively) and had a PD50 value of 40 in vivo against E. coli.
- The antibacterially active compounds of the invention can be prepared for use by conventional pharmaceutical procedures: that is, by dissolving or suspending them in a pharmaceutically acceptable vehicle, e.g., water, aqueous alcohol, glycol, oil solution or oil-water emulsion, for parenteral or oral administration; or by incorporating them in unit dosage form as capsules or tablets for oral administration either alone or in combination with conventional adjuvants or excipients, e.g., calcium carbonate, starch, lactose, talc, magnesium stearate, gum acacia, and the like.
and if desired, converting a compound of the Formula I obtained wherein R" is hydrogen into an alkali metal or amine salt thereof.
Claims (7)
1. A compound of Formula I (herein) where R is hydrogen or alkyl of 1-3 carbon atoms; R' is hydrogen, formyl, alkyl of 1-3 carbon atoms, or alkoxycarbonyl where the alkyl moiety of the latter group is identical with R; and R" is hydrogen or lower-alkyl; or an alkali metal salt or amine salt of a compound where R" is hydrogen.
2. A compound according to claim 1, where R' and R" are hydrogen.
3. 5,8-Dihydro-5-(methylamino)-8-oxo-l,3-dioxolo-[4,5-g]quinoline-7-carboxylic acid.
4. A compound according to claim 1, where R' is formyl.
5. A process for preparing a compound according to claim 1, which comprises treating a compound of the Formula VII (herein) with an aminating agent, wherein R" is hydrogen or lower-alkyl, to prepare the compound wherein R and R' are each hydrogens;
and if desired, converting a compound of the Formula I obtained wherein R" is hydrogen into an alkali metal or amine salt thereof.
if desired, formylating a compound of Formula I obtained where R and R' are each hydrogens to prepare the compound where R' is formyl and R" is hydrogen;
if desired, alkylating a compound of the Formula I obtained wherein R' is formyl or hydrogen with an alkyl halide of 1-3 carbon atoms in the presence of a base when R' is formyl or an alkali metal carbonate when R' is hydrogen to give a compound of the Formula I wherein R" is lower-alkyl, R is alkyl of 1-3 carbon-atoms and R' is formyl or alkoxycarbonyl, respectively;
if desired, subjecting a compound of the Formula I obtained to hydrolysis with one or more molar equivalents of an alkali metal base to prepare a compound of Formula I wherein R' is hydrogen, said hydrolysis producing an alkali metal salt of a compound wherein R" is hydrogen when two or more molar equivalents of the alkali metal base are used;
if desired, converting an alkali metal salt to the free base (R" is hydrogen);
if desired, converting a compound of the Formula I obtained wherein R" is lower-alkyl to the corresponding compound wherein R" is hydrogen;
if desired, reacting a compound of the Formula I obtained wherein R and R' are both hydrogens with dialkyl sulfate to obtain the corresponding compound wherein both R and R' are alkyl of 1-3 carbon atoms;
and if desired, converting a compound of the Formula I obtained wherein R" is hydrogen into an alkali metal or amine salt thereof.
6. A composition for treatment of bacterial infections in animals, including humans, which comprises an antibacterial effective amount of a compound according to any one of claims 1-5, wherein R is methyl or ethyl, and R' and R" are hydrogen, together with one or more pharmaceutically acceptable excipients.
7. A method for treatment of bacterial infections in animals, including humans, which comprises administering to an animal or a human so infected an antibacterially effective amount of a composition according to claim 6.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US06/405,263 US4439436A (en) | 1982-08-04 | 1982-08-04 | 1,3-Dioxolo(4,5-G)quinoline compounds |
| US405263 | 1982-08-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| EP0101951A1 true EP0101951A1 (en) | 1984-03-07 |
Family
ID=23602957
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP83107464A Withdrawn EP0101951A1 (en) | 1982-08-04 | 1983-07-28 | 1,3-Dioxolo(4,5-g)quinolines and preparation thereof |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US4439436A (en) |
| EP (1) | EP0101951A1 (en) |
| JP (1) | JPS5946283A (en) |
| KR (1) | KR840005813A (en) |
| AU (1) | AU1689683A (en) |
| CA (1) | CA1218659A (en) |
| DK (1) | DK353983A (en) |
| FI (1) | FI832780A (en) |
| GR (1) | GR78962B (en) |
| IL (1) | IL69267A0 (en) |
| NO (1) | NO832810L (en) |
| NZ (1) | NZ204900A (en) |
| PH (1) | PH19689A (en) |
| PT (1) | PT77133B (en) |
| ZA (1) | ZA835239B (en) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4730000A (en) * | 1984-04-09 | 1988-03-08 | Abbott Laboratories | Quinoline antibacterial compounds |
| US4774246A (en) * | 1984-01-26 | 1988-09-27 | Abbott Laboratories | Quinoline antibacterial compounds |
| US4528285A (en) * | 1984-04-26 | 1985-07-09 | Abbott Laboratories | Methylenedioxy quino-benzothiazine derivatives |
| US4607032A (en) * | 1984-04-26 | 1986-08-19 | Abbott Laboratories | Quino-benoxazine antibacterial compounds |
| US4542133A (en) * | 1984-04-26 | 1985-09-17 | Abbott Laboratories | Methylenedioxy quino-benoxazine derivatives and antibacterial use |
| US4687770A (en) * | 1985-12-23 | 1987-08-18 | Abbott Laboratories | Isoxazolo-pyrido-benzoxazine and isothiazolo-pyrido-benzoxazine derivatives |
| US4689325A (en) * | 1985-12-23 | 1987-08-25 | Abbott Laboratories | Isoxazolo-pyrido-phenoxazine and isothiazolo-pyrido-phenoxazine derivatives |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1338961A (en) * | 1970-07-09 | 1973-11-28 | Sumitomo Chemical Co | Alkylenedioxy quinoline-3-carboxylic acid derivatives |
| EP0004279A1 (en) * | 1978-02-24 | 1979-10-03 | Bayer Ag | Process for the preparation of 4-pyridone-3-carboxylic acids, 1-cyclopropyl-4-pyridone-3-carboxylic acid derivatives and medicines containing them |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1076828A (en) * | 1963-12-12 | 1967-07-26 | Warner Lambert Pharmaceutical | Methylenedioxy quinoline derivatives |
| DE2103805C3 (en) * | 1970-01-28 | 1980-03-20 | Sumitomo Chemical Co., Ltd., Osaka (Japan) | Process for the preparation of N-substituted 6,7-methylenedioxy-4-quinolones |
-
1982
- 1982-08-04 US US06/405,263 patent/US4439436A/en not_active Expired - Fee Related
-
1983
- 1983-07-14 NZ NZ204900A patent/NZ204900A/en unknown
- 1983-07-15 AU AU16896/83A patent/AU1689683A/en not_active Abandoned
- 1983-07-18 IL IL69267A patent/IL69267A0/en unknown
- 1983-07-19 ZA ZA835239A patent/ZA835239B/en unknown
- 1983-07-20 PH PH29257A patent/PH19689A/en unknown
- 1983-07-28 GR GR72072A patent/GR78962B/el unknown
- 1983-07-28 EP EP83107464A patent/EP0101951A1/en not_active Withdrawn
- 1983-07-29 PT PT77133A patent/PT77133B/en unknown
- 1983-08-02 FI FI832780A patent/FI832780A/en not_active Application Discontinuation
- 1983-08-03 DK DK353983A patent/DK353983A/en not_active Application Discontinuation
- 1983-08-03 KR KR1019830003631A patent/KR840005813A/en not_active Application Discontinuation
- 1983-08-03 NO NO832810A patent/NO832810L/en unknown
- 1983-08-03 CA CA000433752A patent/CA1218659A/en not_active Expired
- 1983-08-04 JP JP58143086A patent/JPS5946283A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1338961A (en) * | 1970-07-09 | 1973-11-28 | Sumitomo Chemical Co | Alkylenedioxy quinoline-3-carboxylic acid derivatives |
| EP0004279A1 (en) * | 1978-02-24 | 1979-10-03 | Bayer Ag | Process for the preparation of 4-pyridone-3-carboxylic acids, 1-cyclopropyl-4-pyridone-3-carboxylic acid derivatives and medicines containing them |
Also Published As
| Publication number | Publication date |
|---|---|
| PT77133B (en) | 1986-01-24 |
| NO832810L (en) | 1984-02-06 |
| CA1218659A (en) | 1987-03-03 |
| ZA835239B (en) | 1984-03-28 |
| DK353983D0 (en) | 1983-08-03 |
| FI832780A0 (en) | 1983-08-02 |
| PH19689A (en) | 1986-06-13 |
| NZ204900A (en) | 1986-11-12 |
| JPS5946283A (en) | 1984-03-15 |
| DK353983A (en) | 1984-02-05 |
| FI832780A (en) | 1984-02-05 |
| KR840005813A (en) | 1984-11-19 |
| AU1689683A (en) | 1984-02-09 |
| IL69267A0 (en) | 1983-11-30 |
| PT77133A (en) | 1983-08-01 |
| US4439436A (en) | 1984-03-27 |
| GR78962B (en) | 1984-10-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0009425B1 (en) | Novel naphtyridine derivatives and pharmaceutical compositions containing them | |
| EP0049355B1 (en) | 7-Amino-1-cyclopropyl-4-oxo-1,4-dihydro-(naphthyridine or quinoline)-3-carboxylic acids, process for their preparation and pharmaceutical compositions containing them | |
| US4499091A (en) | 1-Amino (or substituted amino)-1,4-dihydro-4-oxo-6-fluoro-7-heterylquinoline-3-carboxylic acids and their use as antibacterial agents | |
| FR2555584A1 (en) | DERIVATIVES OF FLUORO-6-DIHYDRO-1,4-OXO-4-PIPERAZINYL SUBSTITUTED-7-QUINOLINE-CARBOXYLIC-3 ACID AND PROCESS FOR THEIR PREPARATION AND ANTI-BACTERIAL ACTION-BASED PHARMACEUTICAL COMPOSITIONS BASED ON SAID DERIVATIVES | |
| EP0109284B1 (en) | 6,7-dihydro-5,8-dimethyl-9-fluoro-1-oxo-1h,5h-benzo(ij)quinolizine-2-carboxylic acid and derivatives | |
| EP0090424B1 (en) | New quinolone compounds and preparation thereof | |
| US4439436A (en) | 1,3-Dioxolo(4,5-G)quinoline compounds | |
| EP0028698B1 (en) | Quinoline compounds, process for their preparation, and pharmaceutical compositions | |
| EP0119779A1 (en) | 8-Alkoxy-6,7-dihydro-5-methyl-9-fluoro-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylic acids | |
| US4539326A (en) | 5-Oxo-5H-(1)benzopyrano(2,3-b)pyridine derivatives, their production and use as anti-inflammatory agents | |
| US4390541A (en) | Quinolone derivatives and their use in a method of controlling an immediate hypersensitivity disease | |
| US4496566A (en) | Naphthyridine derivatives | |
| US2772280A (en) | Synthesis of 4-amino-3-isoxazolidone and its derivatives | |
| US6545149B2 (en) | Synthesis and crystallization of piperazine ring-containing compounds | |
| EP0021000A2 (en) | Substituted acetophenones, their preparation and pharmaceutical compositions containing them | |
| US4720495A (en) | Benzo[ij]quinolizine-2-carboxylic acids useful for treating bacterial infection | |
| US3673177A (en) | Substituted 4-(anilinomethylene)-3-galanthamaninones | |
| US4517191A (en) | 1-Amino-1,8-naphthyridine compounds useful as bactericides | |
| US4497816A (en) | 7-(4-Pyridyl)-1,8-naphthyridine derivatives and their antibacterial compositions | |
| US4060527A (en) | Pyrido[2,3-c]-acridine-1-hydroxy-2-carboxylic acid derivatives | |
| RU2068415C1 (en) | 5,7-dihydroxy-2-methyl-8-[4-(3-hydroxy-1)-(1-propyl)piperidinyl]-4h-1-benzopyran-4-one or its stereoisomers or pharmacologically acceptable acid-additive salts and a method of their synthesis | |
| US4057544A (en) | α-Alkylsulfobenzyl penicillins | |
| US3594413A (en) | Cyclopentene derivatives | |
| US4008237A (en) | 2,3,5,8-Tetrahydro-5-alkoxy-8-oxofuro(2,3-g)quinoline-7-carboxylic acid derivatives | |
| US4330555A (en) | Indanyloxamic derivatives, their preparation and pharmaceutical compositions containing them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): AT BE CH DE FR GB IT LI NL SE |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION IS DEEMED TO BE WITHDRAWN |
|
| 18D | Application deemed to be withdrawn |
Effective date: 19841107 |
|
| RIN1 | Information on inventor provided before grant (corrected) |
Inventor name: BAILEY, DENIS MAHLON Inventor name: WENTLAND, MARK PHILIP |