EP0000338A2 - Isoxazolo(5,4-c)pyridine derivatives, their preparation and pharmaceutical compositions containing them - Google Patents
Isoxazolo(5,4-c)pyridine derivatives, their preparation and pharmaceutical compositions containing them Download PDFInfo
- Publication number
- EP0000338A2 EP0000338A2 EP78100191A EP78100191A EP0000338A2 EP 0000338 A2 EP0000338 A2 EP 0000338A2 EP 78100191 A EP78100191 A EP 78100191A EP 78100191 A EP78100191 A EP 78100191A EP 0000338 A2 EP0000338 A2 EP 0000338A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- group
- hydrogen
- general formula
- compound
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 CC(C(C*=NCC1)=C1ONC)O Chemical compound CC(C(C*=NCC1)=C1ONC)O 0.000 description 2
- INECCWXQRVOTEG-UHFFFAOYSA-N Oc1n[o]c2c1CCCC2 Chemical compound Oc1n[o]c2c1CCCC2 INECCWXQRVOTEG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/72—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D211/78—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- GABA gamma-aminobutyric acid
- CNS central nervous system
- muscimol of the formula (a substance found in fly amanita (Amanita muscaria)) has various interesting pharmacological properties and especially shows an inhibition of motoric functions. Later, it was reported that muscimol is a very potent GABA agonist with respect to bicuculline--sensitive postsynaptic receptors (Johnston et al., Biochem. Pharmacol.
- agents influencing the GABA system are therefore under consideration and research for the therapeutical treatment of such GABA system malfunction-related diseases. It is also under consideration to administer agents influencing the GABA system against diseases in which malfunctions of the pituitary hormones are involved, e.g. diseases where a decreased secretion of prolactin is involved, and it is, furthermore, contemplated that such agents may be useful against artereoschlerotic diseases in the brain where a vasodilatation is desired.
- muscimol has toxic effects, such as narcotic effects (derealisation and depersonalisation), and the difference between the effective dose and the toxic dose of muscimol is very small (Arzneiffenaba, 1968, 18, 311 - 315), which may limit or prevent the therapeutic use of muscimol.
- various muscimol-analogues or muscimol-like substances have been synthesized and tested (P.
- the present invention relates to novel compounds showing GABA--related activity, to salts thereof with acids or bases, and to pharmaceutical compositions containing the novel compounds or a salt thereof as an active ingredient. Moreover, the present invention relates to methods for the preparation of the novel compounds and salts thereof and to a method for the treatment of neurological and psychiatrical disorders, such as epilepsy, parkinsonism, schizophrenia and Huntington's chorea, or diseases in which malfunctions of the pituitary hormones are involved, or artereoschlerotic diseases in the brain where a vasodilatation is desired, by administering a therapeutically active amount of the novel compound or a non-toxic salt thereof to a living animal body including human beings.
- neurological and psychiatrical disorders such as epilepsy, parkinsonism, schizophrenia and Huntington's chorea, or diseases in which malfunctions of the pituitary hormones are involved, or artereoschlerotic diseases in the brain where a vasodilatation is desired, by administer
- the novel compound of the formula Ia (4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-3-ol) is well tolerated and is a very potent GABA agonist having a very specific activity, being inactive as a GABA-uptake inhibitor. Particulars concerning the activity of this compound are given in the section "Test Results” below.
- the potent, specific GABA agonist activity of the compound Ia is especially remarkable on the background of the fact that the known very closely related compounds, that is, 5,6,7,8-tetrahydro-4H-isoxazolo-[4,5-c]-azepine-3-ol (P. Krogsgaard-Larsen, Acta Chem. Scand. B 31, 1977, 584 - 588, and P. Krogsgaard-Larsen and G.A.R. Johnston, J. Neurochem., 1978, 30, 1377 - 1382). 5,6,7,8-tetrahydro-4H-isoxazolo-[5,4-c]-azepine-3-ol (P.
- the present invention is not to be limited by any theory, it is believed that the remarkable selective activity of the compound Ia is ascribable to the particular position of the nitrogen atom in the 6-membered ring in relation to the acidic hydroxy group in the 5-membered ring.
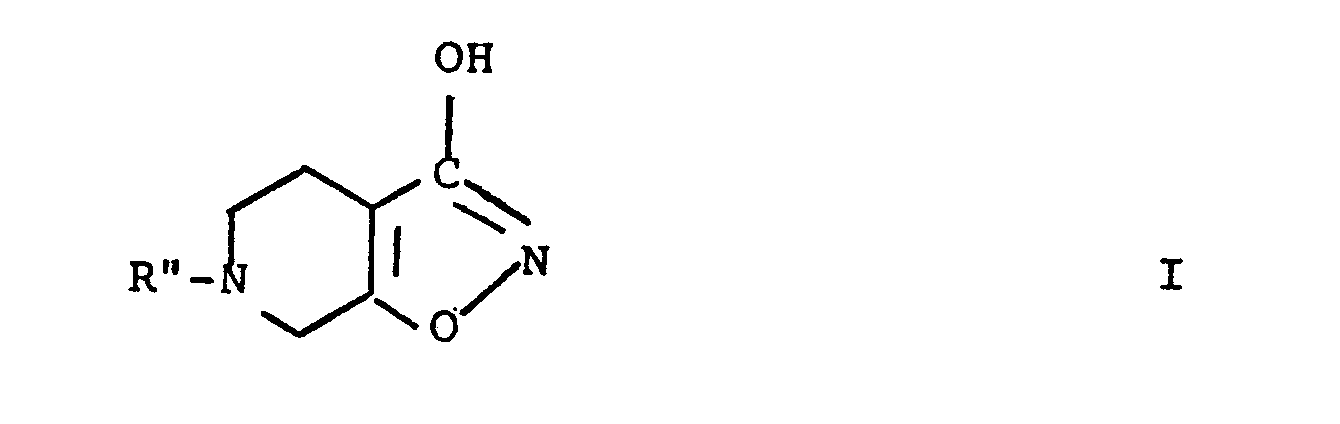
- the present invention therefore relates to the novel compound Ia and to derivatives thereof which upon administration will be decomposed in situ to yield the parent compound Ia, in particular compounds of the general formula I wherein R" is hydrogen, acetyl or a group of the general formula VII wherein R 5 is C 1-8 alkyl; phenyl; phenyl substituted in the 4- position with halogen, lower alkoxy, or lower alkyl; or phenylalkyl such as benzyl or phenylethyl in which the phenyl group may be substituted in the 4-position with halogen, lower alkoxy, or lower alkyl; and salts thereof.
- R" is hydrogen, acetyl or a group of the general formula VII wherein R 5 is C 1-8 alkyl; phenyl; phenyl substituted in the 4- position with halogen, lower alkoxy, or lower alkyl; or phenylalkyl such as benzyl or phenylethyl in which
- the compounds I the only species showing pronounced GABA agonist activity in the brain is the compound Ia.
- the groups R" which are different from hydrogen may enhance the penetration of the compounds into the brain in that they may enhance the ability of the compounds to pass the blood- brain barrier, and will thereafter be split off in situ to yield the parent compound.
- a prolonged effect of Ia may be obtained via decomposition in situ of compounds wherein R" is different from hydrogen, to yield the parent compound.
- lower alkyl and “lower alkoxy” designate such groups containing 1 - 4 carbon atoms.
- the compounds of the general formula I may exist in a tautomeric form, as shown by the formula I' and in the present specification and claims, the formula I is to be understood as covering also this tautomeric form and mixtures of the two tautomeric forms.
- salts of the compound of the formula Ia are acid addition salts thereof, such as pharmaceutically acceptable salts with inorganic acids, e.g. hydrochloric, hydrobromic, nitric, sulfuric, phosphoric acids and the like, or with organic acids, such as organic carboxylic acids, e.g. acetic, propionic, glycolic, malonic, succinic, maleic, fumaric, malic, tartaric, citric, glucuronic, benzoic, pamoic acid and the like, or organic sulfonic acids, e.g.
- salts may be prepared by procedures known per se, e.g. by adding the acid in question to the base, preferably in a solvent.
- Compounds of formula I may form pharmaceutically acceptable salts with bases, such as metal salts, e.g. sodium, potassium, calcium or aluminium salts, and ammonium and substituted ammonium salts, e.g. salts of amines such as triethylamine, triethanolamine, ethylpiperidine, procaine, dibenzylamine and the like.
- the compound Ia was tested in microelectrophoretic experiments. Experiments were performed on lumbar dorsal horn interneurones and Renshaw cells of cats anaesthetized with pentobarbitone sodium. The approximate potency of the depressant actions of the compound was assessed relative to that of GABA on the basis of electrophoretic currents required to produce equal and submaximal inhibitions of the firin of the central neurones. The inhibitory action of Ia on central neurones was antagonized by the specific GABA antagonist bicu- culline methochloride (BMC).
- BMC bicu- culline methochloride
- the compound Ia did not interact with the GABA uptake system at concentrations of 5 x 10 4 M, and it did not interact with the GABA metabolizing enzymes GABA:2-oxo-glutarate aminotransferase and L-glutamate 1-carboxylase at concentrations of 10 -3 M.
- the compound Ia is a specific and very potent GABA agonist.
- compound Ia is considerably less toxic than muscimol.
- Ia was shown to be weaker than muscimol.
- I a was found to be weaker than muscimol.
- test compound is injected i.p. in the doses 0, 1/2, 1/8 and 1/32 of the determined “i.v. LD 50 ".
- doses 0, 1/4, 1/16 and 1/64 of the determined "i.p. LD 50 " are used.
- mice are used for each dose level.
- isoniazide 300 mg/kg is injected s.c. This dose of isoniazide induces intermittent tonic clonic seizures within 60 minutes.
- compound Ia has been shown to be a potent GABA agonist.
- Compound Ia is weaker than muscimol but considerably less toxic.
- the compounds of formula I may be prepared by
- Compound IVa in reaction scheme I is a key intermediate in the above synthesis and in other syntheses of the compounds of the present invention. Similar key intermediates may contain other hydrolysable N-protecting groups and other lower alkyl groups, and hence, in its broad concept, this novel key intermediate of the present invention has the general formula IV in which Alk is a lower alkyl group and Z is hydrogen or an amino--protecting group readily removable, e.g. by hydrolysis, suitably a group R" (as defined above) or a trityl or formyl group.
- Z are the following: hydrogen, methoxycarbonyl, ethoxycarbonyl, propyloxycarbonyl, tert.butyloxycarbonyl, benzyloxycarbonyl, p-chlorobenzyloxycarbonyl, trityl, formyl, acetyl.
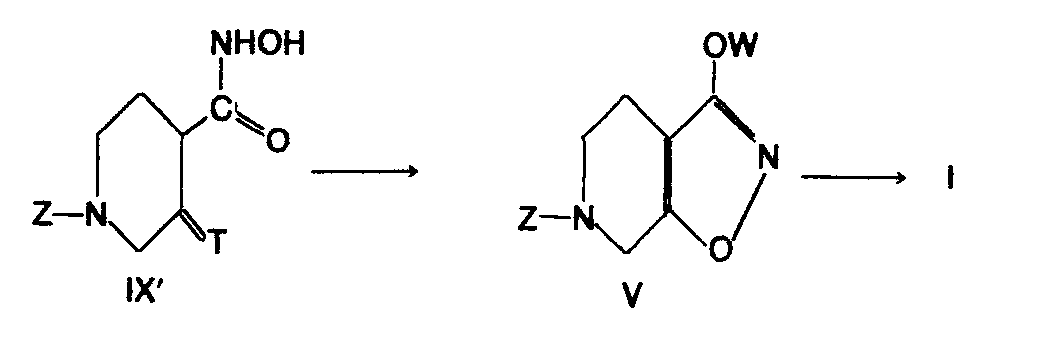
- novel intermediates according to the present invention are the compounds of the formulae VIIIa and IXa in reaction scheme I, and also the generic classes which they represent, which is, compounds of the general formula VIII' in which Z is as defined above, T is a group convertible, by hydrolysis, into an oxy group, e.g., an acetal group such as ethylene dioxy, and Q is a leaving group which, on reaction with hydroxylamine, forms a hydroxamic acid group, examples of Q being halogen, especially chlorine and bromine, hydroxy, the residue of an acid, the residue of an activated amide, the residue of an activated ester, lower alkoxy, and the like, and compounds of the general formula IX' in which Z and T are as defined above, and also, at the stage of compound Va (which is both a compound of the general formula I and an intermediate for the preparation of compounds of the general formula I), an intermediate may be used which in generalized form has the formula V above.
- T is a group convertible, by hydrolysis, into an oxy group,
- An interesting aspect of the present invention is the compound Ia as intermediate in the preparation of compounds of formula I in which R" is different from hydrogen.
- the present invention also relates to the total sequence of synthesis stages IV ⁇ VIII' ⁇ IX' ⁇ V ⁇ I and to the final stages thereof, i.e., VIII' ⁇ IX' ⁇ V ⁇ I and IX' ⁇ V ⁇ I.
- the conversion of ethyl l-benzyl-3-oxy-piperidine-4-carboxylate into the intermediate IV as exemplified by IVa is usually performed in lower alkanols, e.g. ethanol or ethanol/water.
- the removal of the N-benzyl group may be effected with gaseous hydrogen in the presence of a hydrogenation catalyst, e.g. platinum, palladium or Raney nickel.
- the alkyl 3-oxy-piperidine-4-carboxylate formed is dissolved, e.g. in water, and treated with an acid acceptor, e.g. alkali carbonate, and an ester of chloroformic acid, e.g. methyl chloroformate.
- the temperature is kept near 0°C during the reaction.
- the compound IV is isolated by extraction into an organic solvent followed by evaporation of the solvent.
- the formation of the compound of formula VIII' as exemplified by the ethylene acetal VIIIa is usually performed in a solvent, e.g. benzene, which forms an azeotropic mixture with water.
- a solvent e.g. benzene
- the reaction is preferably carried out at reflux temperature and with a strong acid, e.g. a sulfonic acid as catalyst.
- the hydroxamic acid IX' as exemplified by IXa is synthesized by reacting VIIIa with hydroxylamine, preferably in water or a lower alcohol, e.g. methanol and usually at a temperature between -20°C and room temperature, preferably at 0 - 10°C.
- the compound may be isolated and purified by a manner known per se, e.g. column chromatography.
- Q in formula VIII' is a halogen or the residue of an acid
- the reaction is effected in the presence of a base.
- a condensing agent e.g. dicyclohexyl carbodiimide or carbonyldiimidazole.
- solvent an iner
- the hydrolysis of the acetal group of IXa or, quite generally, the conversion of T in compounds of formula IX' into an oxo group, followed by cyclization to a compound of formula V as exemplified by Va may be effected by an aqueous solution of a strong acid optionally also containing acetic acid, e.g. concentrated hydrochloric acid or 70% perchloric acid at a temperature between 0°C and 100°C, preferably at 50 - 80°C.
- the compound V may be isolated by extraction with an organic solvent or by evaporation of the water.
- the compound can be purified by column chromatography or by crystallization.
- Removal of the protecting group Z and/or W in compound V may be effected with a strong inorganic acid, e.g. hydrochloric or hydrobromic acid, in a solvent, e.g. glacial acetic acid or water, or a mixture of water and glacial acetic acid.
- a strong inorganic acid e.g. hydrochloric or hydrobromic acid
- the temperature may be kept between room temperature and the boiling point of the solvent.
- the reaction time is usually short, e.g. less than 1 hour.
- the Ia salt may be isolated by evaporation of the solvent.
- the Ia salt may be transformed into Ia by treatment with a base, e.g. a tertiary amine, in a solvent, usually a mixture of water and a lower alkanol.
- Compound Ia may be transformed into another salt as described above.
- reaction of a compound of the general formula IV as exemplified by IVa with hydroxylamine may give a mixture of a compound of the general formula V and the corresponding isomeric compound V as exemplified by Va and VIa.
- the reaction may be effected at a temperature between -30°C and 50°C, preferably between -30 and -lO o C.
- the solvent is usually water or a lower alkanol or mixtures thereof.
- reaction scheme II although yielding a mixture of two isomers, is nevertheless advantageous. It is very time-saving in that it avoids the protection of the oxo group in compounds of the general formula IV and the subsequent hydroxamic acid formation.
- the compounds formed in the reaction of IV with hydroxylamine, as exemplified by Va and VIa, are easily separated by manners known per se, e.g. by column chromatography.
- the introduction of the group R" may be performed by manners known per se.
- R" is a group of the above formula VII
- the introduction may be performed by treatment of compound Ia with the appropriate. formic acid ester of the general formula X'- -OR 5 wherein X' is a leaving group, especially halogen, azido, etc., in the presence of an acid acceptor, for example an alkali carbonate.
- an acid acceptor for example an alkali carbonate.
- the BOC-derivative can be made by means of tert.butyl azidoformate.
- R" is acetyl
- a reactive derivative of acetic acid e.g. acetyl chloride or acetanhydride may be used for the introduction of the group R".
- the compounds of the formula I, and salts thereof may be formulated for administration in any convenient way by analogy with other pharmaceuticals.
- compositions comprising the compounds of the invention may be in the form of pharmaceutical preparations, e.g. in solid, semisolid or liquid form, which contain the active compound of the invention in admixture with a pharmaceutical organic or inorganic carrier or excipient suitable for enteral or parenteral application.
- the active ingredient may, e.g., be formulated with the usual carriers for tablets, pellets, capsules, suppositories, solutions, emulsions, aqueous suspensions and other suitable administration forms.
- Examples of carriers are glucose, lactose, gum acacia, gelatin, mannitol, starch paste, magnesium trisilicate, talc, corn starch, keratin, colloidal silica, potato starch, urea, and other carriers suitable for use in manufacturing compositions in solid, semisolid, or liquid form, and in addition auxiliary, stabilizing, thickening, colouring, flavouring, and preservative agents can be contained in the composition of this invention.
- the active compound is included in the compositions of the invention in an amount sufficient to produce the desired therapeutical effect upon administration.
- the dosage or therapeutically effective quantity of the compound varies and also depends upon the age and condition of each individual patient being treated.
- a preferred tablet or capsule formulation for oral administration contains 0.1 - 200 mg, preferably 1 - 100, especially 5 - 50, mg of a compound of the formula I or a salt thereof per unit dosage which may be administeret 1 - 4 times per day or as a sustained release composition.
- Injection preparations preferably contain 0.1 - 200 mg, preferably 1 - 100, especially 5 - 50, mg of a compound of the formula I or a salt thereof per unit dosage.
- a preferred injected dose is about 0.5 to 2 ml.
- the invention also relates to the use of the compounds of the general formula I and salts thereof in medicaments for treating GABA system malfunction-related diseases, and a process of treating GABA system malfunction-related diseases in human beings by administering, to the human being, an effective dose of a compound of the general formula I, or a salt thereof.
- compositions and the above-mentioned uses it may be suitable or preferred to combine the compounds of the general formula I or a salt thereof with minor tranquillizers such as benzodiazepines or neuroleptics, for example butyrophenones such as haloperidol, phenothiazines such as chloropromazine, thioxanthene, and the like.
- minor tranquillizers such as benzodiazepines or neuroleptics, for example butyrophenones such as haloperidol, phenothiazines such as chloropromazine, thioxanthene, and the like.
- the neuroleptics are suitably administered in their effective amounts or, in a preferred embodiment in lower amounts than the amounts in which they would be effective when used alone.
- IXa (1.9 g; 29%) as a crystalline and TLC-pure substance [R F : 0.23; eluent: ethyl acetate-methanol-formic acid (90:9:1)].
- An analytical sample was recrystallized (ethanol-benzene) to give IXa as colourless crystals, m.p. 150.0--152.0°C.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Cardiology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Hydrogenated Pyridines (AREA)
Abstract


Description
- GABA (gamma-aminobutyric acid) is known to be a neurotransmitter in the central nervous system (CNS) in mammals. GABA is found predominantly in the brain where it is a dominant inhibitory transmitter (Curtis, D.R. and Johnston, G.A.R., Ergebn. Physiol., 1974, 69, 97 - 188).
- It has been reported (Arzneimittelforschung, 1968, 18, 311 - 315) that muscimol of the formula

- The present invention relates to novel compounds showing GABA--related activity, to salts thereof with acids or bases, and to pharmaceutical compositions containing the novel compounds or a salt thereof as an active ingredient. Moreover, the present invention relates to methods for the preparation of the novel compounds and salts thereof and to a method for the treatment of neurological and psychiatrical disorders, such as epilepsy, parkinsonism, schizophrenia and Huntington's chorea, or diseases in which malfunctions of the pituitary hormones are involved, or artereoschlerotic diseases in the brain where a vasodilatation is desired, by administering a therapeutically active amount of the novel compound or a non-toxic salt thereof to a living animal body including human beings.
- According to the present invention, it has now been found that the novel compound of the formula Ia

- The potent, specific GABA agonist activity of the compound Ia is especially remarkable on the background of the fact that the known very closely related compounds, that is,




- Although the present invention is not to be limited by any theory, it is believed that the remarkable selective activity of the compound Ia is ascribable to the particular position of the nitrogen atom in the 6-membered ring in relation to the acidic hydroxy group in the 5-membered ring.
- The present invention therefore relates to the novel compound Ia and to derivatives thereof which upon administration will be decomposed in situ to yield the parent compound Ia, in particular compounds of the general formula I


- It is believed that among the compounds I, the only species showing pronounced GABA agonist activity in the brain is the compound Ia. However, the groups R" which are different from hydrogen may enhance the penetration of the compounds into the brain in that they may enhance the ability of the compounds to pass the blood- brain barrier, and will thereafter be split off in situ to yield the parent compound. Also, a prolonged effect of Ia may be obtained via decomposition in situ of compounds wherein R" is different from hydrogen, to yield the parent compound.
- In the present specification, "lower alkyl" and "lower alkoxy" designate such groups containing 1 - 4 carbon atoms.
- The compounds of the general formula I may exist in a tautomeric form, as shown by the formula I'

- Examples of compounds of the general formula I in which R" is different from hydrogen, are:
- 6-acetyl-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-3-ol,
- methyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-6--carboxylate,
- ethyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-6--carboxylate,
- tert.butyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine--6-carboxylate,
- phenyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-6--carboxylate,
- 4-chlorophenyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-6-carboxylate,
- 4-methoxyphenyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-6-carboxylate,
- benzyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-6--carboxylate,
- Examples of salts of the compound of the formula Ia are acid addition salts thereof, such as pharmaceutically acceptable salts with inorganic acids, e.g. hydrochloric, hydrobromic, nitric, sulfuric, phosphoric acids and the like, or with organic acids, such as organic carboxylic acids, e.g. acetic, propionic, glycolic, malonic, succinic, maleic, fumaric, malic, tartaric, citric, glucuronic, benzoic, pamoic acid and the like, or organic sulfonic acids, e.g. methane sulfonic, ethane sulfonic, benzene sulfonic, toluene sulfonic acid and the like, which salts may be prepared by procedures known per se, e.g. by adding the acid in question to the base, preferably in a solvent. Compounds of formula I may form pharmaceutically acceptable salts with bases, such as metal salts, e.g. sodium, potassium, calcium or aluminium salts, and ammonium and substituted ammonium salts, e.g. salts of amines such as triethylamine, triethanolamine, ethylpiperidine, procaine, dibenzylamine and the like.
- In order to study the interactions of the compound Ia with the central GABA receptors in vitro, the compound Ia was tested in affinity binding experiments. The affinity binding (sodium-independent binding) of GABA to membranes isolated from rat brains was studied as described by Enna, S.J. and Snyder, S.H., Brain Res., 1975, 100, 81 - 97. IC50 values, inhibitor concentrations causing 50% inhibition of GABA binding were determined.
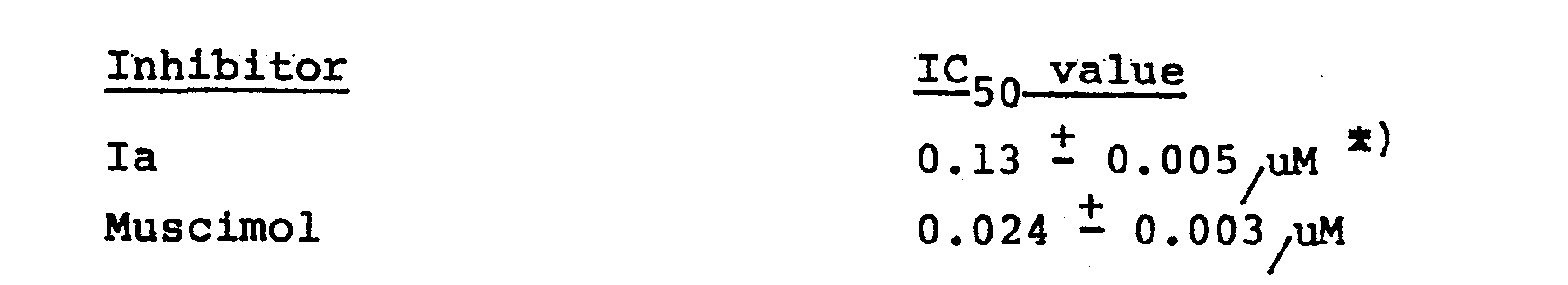
- *) In earlier studies 2.6 - 0.6/uM was found. The value stated (0.13 - 0.005/uM)is based on studies of 5 different concentrations of Ia, each determined in triplicate, and the stated IC50 value is calculated by log-probit analysis. The difference between the two IC50 values determined for Ia is the result of the development of an improved technique for the preparation of rat brain membranes.
- In order to study the interactions of the compound Ia with the central GABA receptors in vivo, the compound Ia was tested in microelectrophoretic experiments. Experiments were performed on lumbar dorsal horn interneurones and Renshaw cells of cats anaesthetized with pentobarbitone sodium. The approximate potency of the depressant actions of the compound was assessed relative to that of GABA on the basis of electrophoretic currents required to produce equal and submaximal inhibitions of the firin of the central neurones. The inhibitory action of Ia on central neurones was antagonized by the specific GABA antagonist bicu- culline methochloride (BMC).

- The compound Ia did not interact with the GABA uptake system at concentrations of 5 x 104 M, and it did not interact with the GABA metabolizing enzymes GABA:2-oxo-glutarate aminotransferase and L-glutamate 1-carboxylase at concentrations of 10-3 M.
- Based on the above-mentioned experiments, the compound Ia is a specific and very potent GABA agonist.
- Compound Ia has been compared with muscimol, the most potent GABA agonist so far known, in a series of pharmacological experiments:
- Compound Ia has been shown to be a well-tolerated substance:

- Thus, compound Ia is considerably less toxic than muscimol.
- Injections into Substantia Nigra in Rats.
- a) Bilateral injections. 0.1, 0.5, and 1.0µg of Ia,HBr have been injected. The rats showed a pronounced stereo- typic behaviour.
- Ia was shown to be weaker than muscimol.
- b) Unilateral injections. 0.1 and 0.5µg of Ia,Hbr have been injected. The rats showed a strong and prolonged contralateral turning.
- Ia was found to be weaker than muscimol.
- a) Potentiation of methylphenidate-induced gnawing (Scheel-Krüger et al.: Muscimol differentially facilitates stereotypy but antagonizes motility induced by dopaminergic drugs. A complex GABA--DOPAMINE interaction. Life Sciences, 1978, Vol. 22, 75 - 84).
- ED50 mg/kg (the dose which causes potentiation in 50% of the animals)

- b) Antagonism of morphine-induced motility (Christensen et al.: Muscimol antagonizes morphine hypermotility without potentiation of analgesia. European J. Pharmacol., 1978, 48, 459 - 462).
- MED mg/kg (minimum effective dose)

- c) Antagonism of isoniazide-induced convulsions (Modification (mice; two times lower concentration of isoniazide) of Mao et al.: Evidence for an involvement of GABA in the mediation of cerebellar c-GMP decrease and the anticonvulsant action of diazepam. Naunyn-Smiedeberg's Arch. Pharmacol. 1975, 289, 369 - 378).
- MED mg/kg (minimum effective dose)

- Conditions and procedure for isoniazide antagonism test:
- Mice, male, 20 - 25 g.
- Isoniazide 300 mg/kg s.c.
- Macrolon cages type II.
- The test compound is injected i.p. in the doses 0, 1/2, 1/8 and 1/32 of the determined "i.v. LD50". In case of insoluble substances, the doses 0, 1/4, 1/16 and 1/64 of the determined "i.p. LD50" are used. Five mice are used for each dose level. Immediately after administration of test substance, isoniazide 300 mg/kg is injected s.c. This dose of isoniazide induces intermittent tonic clonic seizures within 60 minutes.
- The calculations are performed as an "on line procedure" on the EDP-terminal. The results are recorded as % increase in time until convulsions occur and in addition the least dose (MED) which shows significant effect (minimal effective dose, calculated by means of van der Waerden-test).
- Based on these experiments, compound Ia has been shown to be a potent GABA agonist. Compound Ia is weaker than muscimol but considerably less toxic.
- The compounds of formula I may be prepared by
- a) subjecting a compound of the general formula V


- b) for the preparation of a compound of the general formula I in which R" is different from hydrogen, subjecting a compound of the general formula IX"

- An example of a full synthesis of the compound Ia from a known starting material appears from the examples and from the below Reaction Scheme I:

- Compound IVa in reaction scheme I is a key intermediate in the above synthesis and in other syntheses of the compounds of the present invention. Similar key intermediates may contain other hydrolysable N-protecting groups and other lower alkyl groups, and hence, in its broad concept, this novel key intermediate of the present invention has the general formula IV


an acetal group such as ethylene dioxy, and Q is a leaving group which, on reaction with hydroxylamine, forms a hydroxamic acid group, examples of Q being halogen, especially chlorine and bromine, hydroxy, the residue of an acid, the residue of an activated amide, the residue of an activated ester, lower alkoxy, and the like, and compounds of the general formula IX'
- An interesting aspect of the present invention is the compound Ia as intermediate in the preparation of compounds of formula I in which R" is different from hydrogen.
- The present invention also relates to the total sequence of synthesis stages IV → VIII' → IX' → V → I and to the final stages thereof, i.e., VIII'→ IX' → V → I and IX' → V → I.
- The conversion of ethyl l-benzyl-3-oxy-piperidine-4-carboxylate into the intermediate IV as exemplified by IVa, is usually performed in lower alkanols, e.g. ethanol or ethanol/water. The removal of the N-benzyl group may be effected with gaseous hydrogen in the presence of a hydrogenation catalyst, e.g. platinum, palladium or Raney nickel. The alkyl 3-oxy-piperidine-4-carboxylate formed is dissolved, e.g. in water, and treated with an acid acceptor, e.g. alkali carbonate, and an ester of chloroformic acid, e.g. methyl chloroformate. The temperature is kept near 0°C during the reaction. The compound IV is isolated by extraction into an organic solvent followed by evaporation of the solvent.
- The formation of the compound of formula VIII' as exemplified by the ethylene acetal VIIIa is usually performed in a solvent, e.g. benzene, which forms an azeotropic mixture with water. The reaction is preferably carried out at reflux temperature and with a strong acid, e.g. a sulfonic acid as catalyst.
- The hydroxamic acid IX' as exemplified by IXa is synthesized by reacting VIIIa with hydroxylamine, preferably in water or a lower alcohol, e.g. methanol and usually at a temperature between -20°C and room temperature, preferably at 0 - 10°C. The compound may be isolated and purified by a manner known per se, e.g. column chromatography. When Q in formula VIII' is a halogen or the residue of an acid, the reaction is effected in the presence of a base. Alternatively, the piperidine carboxylic acid itself (VIII', Q=OH) may be reacted with hydroxylamine in the presence of a condensing agent, e.g. dicyclohexyl carbodiimide or carbonyldiimidazole. As solvent, an inert solvent, e.g. methylene chloride or chloroform can be used.
- The hydrolysis of the acetal group of IXa or, quite generally, the conversion of T in compounds of formula IX' into an oxo group, followed by cyclization to a compound of formula V as exemplified by Va may be effected by an aqueous solution of a strong acid optionally also containing acetic acid, e.g. concentrated hydrochloric acid or 70% perchloric acid at a temperature between 0°C and 100°C, preferably at 50 - 80°C. The compound V may be isolated by extraction with an organic solvent or by evaporation of the water. The compound can be purified by column chromatography or by crystallization.
- Removal of the protecting group Z and/or W in compound V may be effected with a strong inorganic acid, e.g. hydrochloric or hydrobromic acid, in a solvent, e.g. glacial acetic acid or water, or a mixture of water and glacial acetic acid. The temperature may be kept between room temperature and the boiling point of the solvent. The reaction time is usually short, e.g. less than 1 hour. The Ia salt may be isolated by evaporation of the solvent. The Ia salt may be transformed into Ia by treatment with a base, e.g. a tertiary amine, in a solvent, usually a mixture of water and a lower alkanol.
- Compound Ia may be transformed into another salt as described above.
- An interesting synthesis is illustrated in the below reaction scheme II in which a compound of the general formula I, as exemplified by the compound Ia, is prepared:

- The reaction of a compound of the general formula IV as exemplified by IVa with hydroxylamine may give a mixture of a compound of the general formula V and the corresponding isomeric compound V as exemplified by Va and VIa. The reaction may be effected at a temperature between -30°C and 50°C, preferably between -30 and -lOoC. The solvent is usually water or a lower alkanol or mixtures thereof.
- The process illustrated in reaction scheme II, although yielding a mixture of two isomers, is nevertheless advantageous. It is very time-saving in that it avoids the protection of the oxo group in compounds of the general formula IV and the subsequent hydroxamic acid formation. The compounds formed in the reaction of IV with hydroxylamine, as exemplified by Va and VIa, are easily separated by manners known per se, e.g. by column chromatography.
- When it is desired to prepare compounds of the general formula I in which R" is different from hydrogen, one may either omit the removal of the group Z if the group Z has the same identity as the desired group R", or one may introduce such group R" into the compound of the general formula Ia.
- The introduction of the group R" may be performed by manners known per se. Thus, for example, when R" is a group of the above formula VII, the introduction may be performed by treatment of compound Ia with the appropriate. formic acid ester of the general formula X'--OR5 wherein X' is a leaving group, especially halogen, azido, etc., in the presence of an acid acceptor, for example an alkali carbonate. For example, the BOC-derivative can be made by means of tert.butyl azidoformate. When R" is acetyl, a reactive derivative of acetic acid, e.g. acetyl chloride or acetanhydride may be used for the introduction of the group R".

- The compounds of the formula I, and salts thereof may be formulated for administration in any convenient way by analogy with other pharmaceuticals.
- Thus, the composition comprising the compounds of the invention may be in the form of pharmaceutical preparations, e.g. in solid, semisolid or liquid form, which contain the active compound of the invention in admixture with a pharmaceutical organic or inorganic carrier or excipient suitable for enteral or parenteral application. The active ingredient may, e.g., be formulated with the usual carriers for tablets, pellets, capsules, suppositories, solutions, emulsions, aqueous suspensions and other suitable administration forms. Examples of carriers are glucose, lactose, gum acacia, gelatin, mannitol, starch paste, magnesium trisilicate, talc, corn starch, keratin, colloidal silica, potato starch, urea, and other carriers suitable for use in manufacturing compositions in solid, semisolid, or liquid form, and in addition auxiliary, stabilizing, thickening, colouring, flavouring, and preservative agents can be contained in the composition of this invention.
- The active compound is included in the compositions of the invention in an amount sufficient to produce the desired therapeutical effect upon administration. The dosage or therapeutically effective quantity of the compound varies and also depends upon the age and condition of each individual patient being treated.
- A preferred tablet or capsule formulation for oral administration contains 0.1 - 200 mg, preferably 1 - 100, especially 5 - 50, mg of a compound of the formula I or a salt thereof per unit dosage which may be administeret 1 - 4 times per day or as a sustained release composition.
- Injection preparations preferably contain 0.1 - 200 mg, preferably 1 - 100, especially 5 - 50, mg of a compound of the formula I or a salt thereof per unit dosage. A preferred injected dose is about 0.5 to 2 ml.
- The invention also relates to the use of the compounds of the general formula I and salts thereof in medicaments for treating GABA system malfunction-related diseases, and a process of treating GABA system malfunction-related diseases in human beings by administering, to the human being, an effective dose of a compound of the general formula I, or a salt thereof.
- In the above-mentioned compositions and the above-mentioned uses, it may be suitable or preferred to combine the compounds of the general formula I or a salt thereof with minor tranquillizers such as benzodiazepines or neuroleptics, for example butyrophenones such as haloperidol, phenothiazines such as chloropromazine, thioxanthene, and the like. In such combinations, compositions and combined usages, the neuroleptics are suitably administered in their effective amounts or, in a preferred embodiment in lower amounts than the amounts in which they would be effective when used alone.
- The invention is further illustrated by the below working examples. All compounds prepared according to the working examples have been subjected to elemental analysis for C, H, N and halogen, when present, and all agreed within ± 0.3% with the calculated values.
- A solution of ethyl l-benzyl-3-oxopiperidine-4-carboxylate (Iselin, B.M. and Hoffmann, K., Helv.Chim. Acta, 1954, 37, 178) (14.0 g; 47 mmol) in aqueous ethanol (300 ml; 50%) was hydrogenated (ca. 300 kPa) in a PARR hydrogenation apparatus by using a 10% Pd-C catalyst (1.4 g). The reaction mixture was filtered and evaporated to dryness in vacuo. To an ice cooled solution of the residue in water (50 ml) was added with stirring an iced solution of potassium carbonate (19.4 g; 140 mmol) in water (20 ml) followed by addition of methyl chloroformate (11.3 g; 120 mmol). Stirring was continued at 0°C for 30 minutes and at 25°C for 30 minutes. The mixture was extracted with three 100 ml portions of ether. The combined and dried (Na2SO4) ether phases were evaporated in vacuo to give 10.0 g of crude product. Ball--tube distillation at 40--130 Pa (oven temperature 170°C) gave IVa (9.0 g; 84%) as a colourless oil, which slowly crystallized, m.p. 36--38°C. IR (film): 2980--2850 (several bands, m-s), 1700 (s), 1655 (s), 1620 (m) cm-1. 1H NMR (CCl4): δ 12.3 (1 H, s), 4.13 (q, J 7 Hz) and 4.0--3.9 (m) (a total of 4 H), 3,62 (3 H, s), 3,43 (2 H, t, J 6 Hz), 2.4--2.1 (2 H, m), 1.30 (3 H, t, J 7 Hz).
- A mixture of ethyl 1-methoxycarbonyl-3-oxopiperidine-4-carboxylate (9.0 g; 39 mmol), ethylene glycol (100 ml), 4-toluenesulfonic acid (0.7 g), and benzene (500 ml) was refluxed for 6 days using a Dean-Stark water separator. The mixture was washed with aqueous sodium carbonate (300 ml; 1 M), water (300 ml), and saturated aqueous sodium chloride (300 ml). The organic phase was dried (K2CO3) and evaporated in vacuo to give 8.6 g of an oil. CC [silica gel (Woelm 0.063--0.1 mm): 350 g; eluents: methylene chloride to which ethyl acetate (20--35%) was added] followed by ball-tube distillation at 40 Pa (oven temperature 170°C) gave VIIIa (7.0 g; 65%) as a colourless oil. IR (film): 2970 (s), 2900 (s), 1730 (s) cm-1. 1H NMR (CCl4): δ 4.05 (q, J 7 Hz) and 3.92 (s) (a total of 6 H), 3.60 (s) and 3.7--3.0 (m) (a total of 7 H), 2.8--2.5 (1 H, t), 2.2--l.6 (2 H, m), 1.23 (3 H, t, J 7 Hz).
- To a stirred and iced solution of potassium hydroxide (7.3 g; 130 mmol) in methanol (30 ml) was added hydroxylammonium chloride (6.9 g; 100 mmol). After stirring at 0°C for further 30 minutes a solution of ethyl l-methoxycarbonyl-3-oxopiperidine-4-carboxylate ethylene acetal (6.8 g; 25 mmol) in methanol (20 ml) was added, and the mixture was left at 8°C for 8 days. Upon addition of glacial acetic acid (15 ml) and filtration the filtrate was evaporated in vacuo to give a treacly mass. CC [silica gel (Woelm 0.063--0.1 mm): 250 g; eluents: ethyl acetate to which methanol (15--26%) and formic acid (1%) was added] afforded IXa (1.9 g; 29%) as a crystalline and TLC-pure substance [RF: 0.23; eluent: ethyl acetate-methanol-formic acid (90:9:1)]. An analytical sample was recrystallized (ethanol-benzene) to give IXa as colourless crystals, m.p. 150.0--152.0°C. IR (KBr): 3700--3350 (m), 3280 (m), 3210 (s), 3055 (w), 3000--2870 (several bands, w-m), 1690 (s), 1640 (s), 1550 (w) cm-1. 1H NMR [CDCl3-DMSO-d6 (1:1): δ 10.5--10.1 (1 H, m), 4.9--4.3 (1 H, m), 3.93 (s), 3.60 (s), and 4.1--3.1 (m) (a total of 11 H), 2.8--2.6 (1 H, m), 2.2--l.8 (2 H, m).
- A solution of l-methoxycarbonyl-3-oxopiperidine-4-carbohydroxamic acid ethylene acetal (750 mg; 2.9 mmol) in concentrated hydrochloric acid (13 ml) was heated to 70°C for 10 minutes. The mixture was evaporated in vacuo to give a black oil. CC [silica gel (Woelm 0.063--0.1 mm): 60 g; eluents: benzene to which ethyl acetate (40--70%) and formic acid (1%) was added] gave crystalline and TLC-pure Va (244 mg; 43%) [RF: 0.27; eluent: benzene-ethyl acetate-formic acid (50:50:1)]. An analytical sample was recrystallized (benzene--cyclohexane) to give pure Va as colourless crystals, m.p. 136.0--138.0°C. IR (KBr): 3700--3300 (m), 3300--2500 (several bands, w-m), 1655 (s), 1525 (m), 1490 (s) cm-1.
- UV [methanol (log ε)]: 212 (3.64) nm. 1H NMR (CDC13): δ 10.6 (1 H, s), 4.43 (2 H, s), 3.70 (s) and 3.8--3.5 (t) (a total of 5 H), 2.6--2.3 (2 H, t).
- A solution of methyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]-pyridine-6-carboxylate (309 mg; 1.6 mmol) in a solution of hydrogen bromide in glacial acetic acid (3 ml; 43%) was refluxed for 15 minutes. Upon evaporation to dryness in vacuo the residue was treated with the same reagent (3 ml) for further 15 minutes. Evaporation of the reaction mixture to dryness in vacuo and recrystallization (methanol-ether) of the residue gave Ia (salt) (193 mg; 56%) as faintly reddish crystals, m.p. 162--163°C (decomp.). IR (KBr): 3700--3300 (m), 3070 (s), 3000-2300 (several bands, m-s), 1670 (m), 1580 (m), 1525 (s), 1505 (w) cm-1. UV (methanol): <210 nm. 1H NMR [D2O (sodium 3-(trimethylsilyl)-propanesulfonate was used as an internal standard)]: δ4.77 (ca. 5 H, s), 4.43 (2 H, t, J 1 Hz), 3.7--3.4 (2 H, q, J 6 and 7 Hz), 3.0--2.7 (2 H, t).
- To a solution of 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridinium bromide (77 mg; 0.35 mmol) in water (0.6 ml) was added a solution of triethylamine (39 mg; 0.39 mmol) in ethanol (0.6 ml). The mixture was left at 25°C for 2 hours. Ia (42 mg; 86%) was isolated as colourless crystals, m.p. 242--244°C (decomp.). IR (KBr): 3700--2900 (s), 2900--1900 (several bands, m-s), 1670 (s), 1625 (m) cm-1. UV [methanol (log ε)]: 212 (3.64) nm. pKA values (H20, 25°C): 4.44 ± 0.03, 8.48 ± 0.04.
- To an iced solution of sodium hydroxide (9.6 g; 0.24 mol) and hydroxylammonium chloride (8.34 g:, 0.12 mol) in water (100 ml) was added with stirring ethyl l-methoxycarbonyl-3-oxopiperidine--4-carboxylate (22.9 g; 0,1 mol). Upon standing at 5°C for 5 hours the solution was evaporated to dryness in vacuo. The residue was dissolved in concentrated hydrochlorid acid (75 ml) and heated to 70°C for 10 minutes. The mixture was evaporated in vacuo to the formation of a black residue, which was extracted with three 100 ml portions of chloroform. The combined chloroform phases were dried (Na2SO4) and evaporated in vacuo to the formation of a black semisolid residue. TLC ((silica gel F254)' eluent: benzene-ethyl acetate-formic acid (25:25:1)) showed the presence of two compounds with R- values 0.31 and 0.16 corresponding to Va and VIa, respectively. Column chromatography (silica gel: 300 g; eluent: benzene-ethyl acetate-formic acid (30:20:1)) lead to Va and VIa.
- A solution of the hydroxamic acid (IXa) (10 g) in perchloric acid (70%; 35 ml) was heated to 60°C for 30 minutes. Upon cooling, NaOH (40 ml; 28%) was added with stirring and cooling. The mixture was extracted with three 50 ml portions of chloroform. The combined and dried (MgSO4) chloroform phases were filtered and evaporated to dryness in vacuo to form a residue which was dissolved in ethyl acetate (50 ml). Upon standing and cooling, Va was isolated as crystals (6.3 g; 82%). Isolation and washing twice with 30 ml portions of ethyl acetate yielded crystalline Va, m.p. 139 - 141°C.
- Instead of heating to 60°C for 30 minutes, the same result may be achieved on standing at ambient temperature for 16 hours.
- 4,5,6,7-Tetrahydroisoxazolo[5,4-c]pyridine-3-ol zwitterion (Ia).
- A solution of methyl 3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]-pyridine-6-carboxylate (Va) (37 g) in hydrogen bromide in glacial acetic acid (33% HBr, 250 ml) was left for 16 hours at ambient temperature. Evaporation to dryness in vacuo gave the HBr salt of Ia as a yellowish crystalline material, which was dissolved in a mixture of water (100 ml) and ethanol (200 ml). Triethylamine was added until pH 6.5, which caused Ia zwitterion to crystallize. Upon standing for 3 hours at 5°C and filtration, the precipitate was washed on the filter with a mixture of water (25 ml) and ethanol (50 ml) to give the zwitterion (26 g; 95%) as a white crystalline material, m.p. 242 - 2440C (decomp.).
and salts thereof with bases.
if desired, converting a resulting compound in which R" is different from hydrogen, into a salt thereof.
Claims (9)


characterized in that it is 4,5,6,7-tetrahydroisoxazolo[5,4-c]-pyridine-3-ol, or a salt thereof.



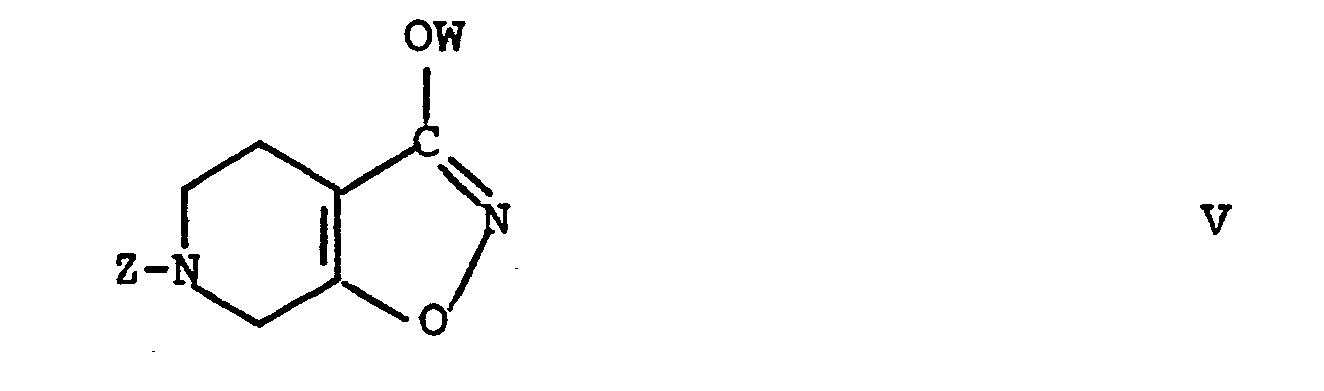

group may be substituted in the 4-position with halogen, lower alkoxy, or lower alkyl; and salts thereof,
characterized by

to removal of any group W different from hydrogen and any group Z different from R" and, if desired, removal of any group Z which falls under the definition of R"; if desired, converting the compound of formula Ia, if obtained as a salt thereof, into the zwitterion form thereof by treatment with a base or into another salt, and, if desired, converting the compound Ia, when obtained, into a compound I in which R" is different from hydrogen, by treatment with a reactive derivative of acetic acid or with an ester of the general formula


if desired, converting a resulting compound in which R" is different from hydrogen into a salt thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB2574077 | 1977-06-20 | ||
| GB2574077 | 1977-06-20 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP80106497.3 Division-Into | 1980-10-23 | ||
| EP80106498.1 Division-Into | 1980-10-23 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0000338A2 true EP0000338A2 (en) | 1979-01-24 |
| EP0000338A3 EP0000338A3 (en) | 1979-06-27 |
| EP0000338B1 EP0000338B1 (en) | 1981-11-25 |
Family
ID=10232513
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP80106497A Withdrawn EP0027279A1 (en) | 1977-06-20 | 1978-06-19 | Isoxazolo(5,4-C)pyridines |
| EP78100190A Withdrawn EP0000167A1 (en) | 1977-06-20 | 1978-06-19 | 1,2,3,6-Tetrahydroisonicotinic acid and derivatives thereof, methods and starting products for their preparation, and pharmaceutical compositions containing them. |
| EP80106498A Withdrawn EP0028017A1 (en) | 1977-06-20 | 1978-06-19 | 3-Piperidinone-4-carboxylic acid derivatives |
| EP78100191A Expired EP0000338B1 (en) | 1977-06-20 | 1978-06-19 | Isoxazolo(5,4-c)pyridine derivatives, their preparation and pharmaceutical compositions containing them |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP80106497A Withdrawn EP0027279A1 (en) | 1977-06-20 | 1978-06-19 | Isoxazolo(5,4-C)pyridines |
| EP78100190A Withdrawn EP0000167A1 (en) | 1977-06-20 | 1978-06-19 | 1,2,3,6-Tetrahydroisonicotinic acid and derivatives thereof, methods and starting products for their preparation, and pharmaceutical compositions containing them. |
| EP80106498A Withdrawn EP0028017A1 (en) | 1977-06-20 | 1978-06-19 | 3-Piperidinone-4-carboxylic acid derivatives |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US4278676A (en) |
| EP (4) | EP0027279A1 (en) |
| JP (2) | JPS5436275A (en) |
| AT (1) | AT368505B (en) |
| AU (2) | AU3724478A (en) |
| CA (1) | CA1107736A (en) |
| DK (2) | DK270278A (en) |
| ES (2) | ES470912A1 (en) |
| FI (2) | FI64376C (en) |
| IE (1) | IE47200B1 (en) |
| IT (2) | IT7868449A0 (en) |
| NO (3) | NO782128L (en) |
| NZ (1) | NZ187615A (en) |
| ZA (2) | ZA783492B (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2494691A1 (en) * | 1980-11-27 | 1982-05-28 | Kefalas As | NEWS 2-ACYL 4,5,6,7-TETRAHYDROISOXAZOLO (5,4-C) PYRIDINONES-3 USEFUL AS MEDICINAL PRODUCTS, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| FR2494692A1 (en) * | 1980-11-27 | 1982-05-28 | Kefalas As | NEW 4,5,6,7-TETRAHYDROISOXAZOLO (5,4-C) 3-SUBSTITUTED PYRIDINES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| EP0126654A1 (en) * | 1983-05-24 | 1984-11-28 | H. Lundbeck A/S | Tetrahydroisoxazolo(4,5-c)pyridine derivatives and preparation thereof |
| EP0350051A1 (en) * | 1988-07-06 | 1990-01-10 | Bristol-Myers Squibb Company | Thaz derivatives for enhancemet of cerebral function |
| WO2005023820A1 (en) * | 2003-09-05 | 2005-03-17 | H. Lundbeck A/S | Method for the manufacture of thip |
| WO2005073237A2 (en) * | 2004-01-30 | 2005-08-11 | Merck Sharp & Dohme Limited | Polymorphic forms of a gabaa agonist |
| WO2006118897A1 (en) * | 2005-04-29 | 2006-11-09 | H.Lundbeck A/S | Acid and base salt forms of gaboxadol |
| EP1848420A2 (en) * | 2005-01-28 | 2007-10-31 | Merck & Co., Inc. | Polymorphic forms of a gabaa agonist |
| EP2145620A2 (en) | 2003-06-25 | 2010-01-20 | H. Lundbeck A/S | Gaboxadol for treating depression and other affective disorders |
| US9339495B2 (en) | 2014-06-06 | 2016-05-17 | Ovid Therapeutics Inc | Methods of increasing tonic inhibition and treating secondary insomnia |
| WO2016150953A1 (en) | 2015-03-24 | 2016-09-29 | H. Lundbeck A/S | Manufacture of 4,5,6,7-tetrahydroisozaxolo[5,4-c]pyridin-3-ol |
| WO2017015049A1 (en) | 2015-07-17 | 2017-01-26 | Ovid Therapeutics Inc. | Methods of treating developmental disorders with gaboxadol |
| US9682069B2 (en) | 2015-07-17 | 2017-06-20 | Ovid Therapeutics Inc | Methods of treating Dravet syndrome |
| WO2018144827A1 (en) | 2017-02-03 | 2018-08-09 | Ovid Therapeutics Inc. | Use of gaboxadol in the treatment of tinnitus |
| US10363246B1 (en) | 2016-08-11 | 2019-07-30 | Ovid Therapeutics Inc. | Methods and compositions for treatment of epileptic disorders |
| US10765666B2 (en) | 2018-09-20 | 2020-09-08 | Ovid Therapeutics Inc | Use of gaboxadol for the treatment of Tourette syndrome, tics and stuttering |
| US10813918B2 (en) | 2017-08-04 | 2020-10-27 | Ovid Therapeutics Inc. | Use of Gaboxadol in the treatment of diabetes and related conditions |
| US11690829B2 (en) | 2018-12-17 | 2023-07-04 | Ovid Therapeutics Inc. | Use of gaboxadol for the treatment of non-24 hour sleep-wake disorder |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0083378A1 (en) * | 1981-12-22 | 1983-07-13 | Chugai Seiyaku Kabushiki Kaisha | Tetrahydronicotinamide derivatives, a process for producing the same and a pharmaceutical composition comprising the same |
| IT1228426B (en) * | 1987-07-20 | 1991-06-17 | Ausimont Spa | HETEROCYCLIC PEROXIDE |
| CN1043051C (en) * | 1994-07-22 | 1999-04-21 | 国际壳牌研究有限公司 | Process for producing a hydrowax |
| US20050234093A1 (en) * | 2003-06-25 | 2005-10-20 | H. Lundbeck A/S | Treatment of depression and other affective disorders |
| EP1691811B1 (en) | 2003-12-11 | 2014-07-23 | Sunovion Pharmaceuticals Inc. | Combination of a sedative and a neurotransmitter modulator, and methods for improving sleep quality and treating depression |
| US20050137222A1 (en) * | 2003-12-18 | 2005-06-23 | H. Lundbeck A/S | Treatment of insomnia in human patients |
| US20050215521A1 (en) * | 2003-12-22 | 2005-09-29 | Karim Lalji | Modafinil combination therapy for improving sleep quality |
| AU2004308962A1 (en) * | 2003-12-24 | 2005-07-14 | Sepracor Inc. | Melatonin combination therapy for improving sleep quality |
| DE602005018763D1 (en) * | 2004-02-18 | 2010-02-25 | Sepracor Inc | DOPAMINE AGONIST COMBINATION THERAPY WITH SEDATIVA TO IMPROVE THE SLEEP QUALITY |
| GB0417558D0 (en) * | 2004-08-06 | 2004-09-08 | Merck Sharp & Dohme | Novel combination therapy |
| JP2009506069A (en) | 2005-08-26 | 2009-02-12 | ブレインセルス,インコーポレイティド | Neurogenesis through modulation of muscarinic receptors |
| EP2258359A3 (en) | 2005-08-26 | 2011-04-06 | Braincells, Inc. | Neurogenesis by muscarinic receptor modulation with sabcomelin |
| AU2006304787A1 (en) | 2005-10-21 | 2007-04-26 | Braincells, Inc. | Modulation of neurogenesis by PDE inhibition |
| CA2625210A1 (en) * | 2005-10-31 | 2007-05-10 | Braincells, Inc. | Gaba receptor mediated modulation of neurogenesis |
| US20070203216A1 (en) * | 2006-02-14 | 2007-08-30 | Bjarke Ebert | Method of treating inflammatory diseases |
| US20100216734A1 (en) | 2006-03-08 | 2010-08-26 | Braincells, Inc. | Modulation of neurogenesis by nootropic agents |
| AU2007249399A1 (en) | 2006-05-09 | 2007-11-22 | Braincells, Inc. | Neurogenesis by modulating angiotensin |
| US7678808B2 (en) | 2006-05-09 | 2010-03-16 | Braincells, Inc. | 5 HT receptor mediated neurogenesis |
| EP2068872A1 (en) | 2006-09-08 | 2009-06-17 | Braincells, Inc. | Combinations containing a 4-acylaminopyridine derivative |
| US20100193652A1 (en) * | 2009-02-02 | 2010-08-05 | William Stajos | Wall Display System And Method Of Providing The Same |
| WO2010099217A1 (en) | 2009-02-25 | 2010-09-02 | Braincells, Inc. | Modulation of neurogenesis using d-cycloserine combinations |
| US8784835B2 (en) * | 2012-07-02 | 2014-07-22 | Trent Austin | Method for producing muscimol and/or reducing ibotenic acid from amanita tissue |
| CN104974175A (en) * | 2014-04-11 | 2015-10-14 | 天津药物研究院 | Preparation method of aminomethyl hydroxyisooxazole analogue |
| US9399034B1 (en) | 2015-08-11 | 2016-07-26 | Ovid Therapeutics Inc | Methods of sedation during critical care treatment |
| US20180338959A1 (en) | 2017-05-24 | 2018-11-29 | Ovid Therapeutics Inc. | Treatment of depressive disorders |
| WO2020106927A1 (en) | 2018-11-21 | 2020-05-28 | Certego Therapeutics | Gaboxadol for reducing risk of suicide and rapid relief of depression |
| IL298334A (en) | 2020-05-20 | 2023-01-01 | Certego Therapeutics Inc | Ring deuterated gaboxadol and its use for the treatment of psychiatric disorders |
| CN114292224B (en) * | 2022-03-07 | 2022-05-20 | 中国农业科学院农产品加工研究所 | Cannabidiol-2- (N-acetyl) piperidine acid ester and application thereof |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2784190A (en) * | 1952-03-28 | 1957-03-05 | Upjohn Co | Alkyl piperidinepropionates |
| CA762455A (en) * | 1962-03-22 | 1967-07-04 | Dr. Karl Thomae Gesellschaft Mit Beschrankter Haftung | Pyrido-pyrimidines |
| US3381016A (en) * | 1966-03-04 | 1968-04-30 | Upjohn Co | Isoxazolo[5, 4-b]pyridine derivatives and a method for their preparation |
| DE2221770A1 (en) * | 1972-05-04 | 1973-11-15 | Bayer Ag | 1,2,5,6-tetrahydropyridines - prepd from corresp 1-benzyl tetrahydropyridines and phosgene |
| DK62791A (en) * | 1991-04-09 | 1992-11-09 | Tulip Int As | PROCEDURE FOR SALTING MEAT AND PLANT FOR USE IN EXERCISE OF THE PROCEDURE |
-
1978
- 1978-06-15 DK DK270278A patent/DK270278A/en unknown
- 1978-06-15 DK DK270378A patent/DK270378A/en unknown
- 1978-06-19 FI FI781954A patent/FI64376C/en not_active IP Right Cessation
- 1978-06-19 ES ES470912A patent/ES470912A1/en not_active Expired
- 1978-06-19 FI FI781955A patent/FI781955A/en not_active Application Discontinuation
- 1978-06-19 EP EP80106497A patent/EP0027279A1/en not_active Withdrawn
- 1978-06-19 NO NO782128A patent/NO782128L/en unknown
- 1978-06-19 NZ NZ187615A patent/NZ187615A/en unknown
- 1978-06-19 IE IE1234/78A patent/IE47200B1/en unknown
- 1978-06-19 NO NO782127A patent/NO152049C/en unknown
- 1978-06-19 ES ES470913A patent/ES470913A1/en not_active Expired
- 1978-06-19 EP EP78100190A patent/EP0000167A1/en not_active Withdrawn
- 1978-06-19 ZA ZA00783492A patent/ZA783492B/en unknown
- 1978-06-19 US US05/917,118 patent/US4278676A/en not_active Expired - Lifetime
- 1978-06-19 AU AU37244/78A patent/AU3724478A/en active Pending
- 1978-06-19 ZA ZA00783493A patent/ZA783493B/en unknown
- 1978-06-19 EP EP80106498A patent/EP0028017A1/en not_active Withdrawn
- 1978-06-19 EP EP78100191A patent/EP0000338B1/en not_active Expired
- 1978-06-20 JP JP7479978A patent/JPS5436275A/en active Pending
- 1978-06-20 IT IT7868449A patent/IT7868449A0/en unknown
- 1978-06-20 AT AT0448678A patent/AT368505B/en not_active IP Right Cessation
- 1978-06-20 CA CA305,798A patent/CA1107736A/en not_active Expired
- 1978-06-20 IT IT68450/78A patent/IT1159739B/en active
- 1978-06-20 JP JP7480078A patent/JPS5436290A/en active Pending
- 1978-06-20 AU AU37298/78A patent/AU521040B2/en not_active Expired
-
1979
- 1979-09-03 NO NO792839A patent/NO792839L/en unknown
- 1979-12-17 US US06/104,080 patent/US4301287A/en not_active Expired - Lifetime
Non-Patent Citations (4)
| Title |
|---|
| ACTA CHEMICA SCANDINAVICA B28, Pages 533-38 (1974) * |
| CHEMICAL ABSTRACTS 84, 159505z (1976) * |
| CHEMICAL ABSTRACTS 88, 37672p (1978) & Acta Chemica Scandinavica B31 (7), 584-8 (1977) * |
| NATUR (London) 268 (5615) 53-5 (1977) * |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2494691A1 (en) * | 1980-11-27 | 1982-05-28 | Kefalas As | NEWS 2-ACYL 4,5,6,7-TETRAHYDROISOXAZOLO (5,4-C) PYRIDINONES-3 USEFUL AS MEDICINAL PRODUCTS, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| FR2494692A1 (en) * | 1980-11-27 | 1982-05-28 | Kefalas As | NEW 4,5,6,7-TETRAHYDROISOXAZOLO (5,4-C) 3-SUBSTITUTED PYRIDINES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| EP0126654A1 (en) * | 1983-05-24 | 1984-11-28 | H. Lundbeck A/S | Tetrahydroisoxazolo(4,5-c)pyridine derivatives and preparation thereof |
| EP0350051A1 (en) * | 1988-07-06 | 1990-01-10 | Bristol-Myers Squibb Company | Thaz derivatives for enhancemet of cerebral function |
| EP2145620A2 (en) | 2003-06-25 | 2010-01-20 | H. Lundbeck A/S | Gaboxadol for treating depression and other affective disorders |
| WO2005023820A1 (en) * | 2003-09-05 | 2005-03-17 | H. Lundbeck A/S | Method for the manufacture of thip |
| US7371863B2 (en) | 2003-09-05 | 2008-05-13 | H. Lundbeck A/S | Method for manufacture of THIP |
| AU2004270323B2 (en) * | 2003-09-05 | 2010-01-07 | H. Lundbeck A/S | Method for the manufacture of THIP |
| WO2005073237A3 (en) * | 2004-01-30 | 2005-10-20 | Merck Sharp & Dohme | Polymorphic forms of a gabaa agonist |
| CN1914212B (en) * | 2004-01-30 | 2010-10-06 | H.隆德贝克有限公司 | Polymorphic forms of a GABAA agonist |
| EA009413B1 (en) * | 2004-01-30 | 2007-12-28 | Мерк Шарп Энд Домэ Лимитед | Polymorphic forms of gaboxadol, gabaagonist |
| US7262300B2 (en) | 2004-01-30 | 2007-08-28 | Merck Sharp & Dohme Ltd. | Polymorphic forms of a GABAA agonist |
| US8236958B2 (en) | 2004-01-30 | 2012-08-07 | H. Lundbeck A/S | Polymorphic forms of a GABAA agonist |
| WO2005073237A2 (en) * | 2004-01-30 | 2005-08-11 | Merck Sharp & Dohme Limited | Polymorphic forms of a gabaa agonist |
| EP2042505A1 (en) | 2004-01-30 | 2009-04-01 | H. Lundbeck A/S | Polymorphic forms of a GABAa agonist |
| EP1848420A4 (en) * | 2005-01-28 | 2008-01-23 | Merck & Co Inc | Polymorphic forms of a gabaa agonist |
| US8022084B2 (en) | 2005-01-28 | 2011-09-20 | H. Lundbeck A/S | Polymorphic forms of a GABAA agonist |
| US8193216B2 (en) | 2005-01-28 | 2012-06-05 | H. Lundbeck A/S | Polymorphic forms of a GABAA agonist |
| EP1848420A2 (en) * | 2005-01-28 | 2007-10-31 | Merck & Co., Inc. | Polymorphic forms of a gabaa agonist |
| EP2292222A1 (en) | 2005-01-28 | 2011-03-09 | H. Lundbeck A/S | Polymorphic Forms of a GABAA Agonist |
| WO2006118897A1 (en) * | 2005-04-29 | 2006-11-09 | H.Lundbeck A/S | Acid and base salt forms of gaboxadol |
| EP1906953A1 (en) * | 2005-04-29 | 2008-04-09 | H. Lundbeck A/S | Acid and base salt forms of gaboxadol |
| EP1906953A4 (en) * | 2005-04-29 | 2009-05-20 | Lundbeck & Co As H | Acid and base salt forms of gaboxadol |
| US9801864B2 (en) | 2014-06-06 | 2017-10-31 | Ovid Therapeutics Inc | Methods of increasing tonic inhibition and treating secondary insomnia |
| US9339495B2 (en) | 2014-06-06 | 2016-05-17 | Ovid Therapeutics Inc | Methods of increasing tonic inhibition and treating secondary insomnia |
| US9446028B2 (en) | 2014-06-06 | 2016-09-20 | Ovid Therapeutics Inc | Methods of increasing tonic inhibition and treating fragile X syndrome and angelman syndrome |
| US11278529B2 (en) | 2014-06-06 | 2022-03-22 | Ovid Therapeutics Inc. | Methods for treating aggression associated with Alzheimer's disease |
| US9744159B2 (en) | 2014-06-06 | 2017-08-29 | Ovid Therapeutics Inc | Methods of treatment Rett syndrome |
| WO2016150953A1 (en) | 2015-03-24 | 2016-09-29 | H. Lundbeck A/S | Manufacture of 4,5,6,7-tetrahydroisozaxolo[5,4-c]pyridin-3-ol |
| WO2017015049A1 (en) | 2015-07-17 | 2017-01-26 | Ovid Therapeutics Inc. | Methods of treating developmental disorders with gaboxadol |
| US9682069B2 (en) | 2015-07-17 | 2017-06-20 | Ovid Therapeutics Inc | Methods of treating Dravet syndrome |
| US20230071127A1 (en) * | 2015-07-17 | 2023-03-09 | Ovid Therapeutics Inc. | Methods of treating developmental disorders with gaboxadol |
| US11096929B2 (en) | 2015-07-17 | 2021-08-24 | Ovid Therapeutics Inc. | Methods of treating developmental disorders with gaboxadol |
| EP4233861A2 (en) | 2016-08-11 | 2023-08-30 | Ovid Therapeutics, Inc. | Compositions for treatment of essential tremor |
| US10363246B1 (en) | 2016-08-11 | 2019-07-30 | Ovid Therapeutics Inc. | Methods and compositions for treatment of epileptic disorders |
| EP3586845A1 (en) | 2016-08-11 | 2020-01-01 | Ovid Therapeutics, Inc. | Compositions for treatment of essential tremor |
| WO2018144827A1 (en) | 2017-02-03 | 2018-08-09 | Ovid Therapeutics Inc. | Use of gaboxadol in the treatment of tinnitus |
| US10188635B2 (en) | 2017-02-03 | 2019-01-29 | Ovid Therapeutics Inc. | Use of gaboxadol in the treatment of tinnitus |
| US10071083B2 (en) | 2017-02-03 | 2018-09-11 | Ovid Therapeutics Inc | Use of gaboxadol in the treatment of tinnitus |
| US10813918B2 (en) | 2017-08-04 | 2020-10-27 | Ovid Therapeutics Inc. | Use of Gaboxadol in the treatment of diabetes and related conditions |
| US11291658B2 (en) | 2017-08-04 | 2022-04-05 | Ovid Therapeutics Inc. | Use of gaboxadol in the treatment of diabetes and related conditions |
| US11090293B2 (en) | 2018-09-20 | 2021-08-17 | Ovid Therapeutics Inc. | Use of gaboxadol for the treatment of Tourette syndrome, tics and stuttering |
| US10765666B2 (en) | 2018-09-20 | 2020-09-08 | Ovid Therapeutics Inc | Use of gaboxadol for the treatment of Tourette syndrome, tics and stuttering |
| US11690829B2 (en) | 2018-12-17 | 2023-07-04 | Ovid Therapeutics Inc. | Use of gaboxadol for the treatment of non-24 hour sleep-wake disorder |
Also Published As
| Publication number | Publication date |
|---|---|
| US4278676A (en) | 1981-07-14 |
| ATA448678A (en) | 1982-02-15 |
| ES470912A1 (en) | 1979-02-01 |
| ZA783492B (en) | 1979-06-27 |
| IE47200B1 (en) | 1984-01-11 |
| DK270378A (en) | 1978-12-21 |
| EP0000167A1 (en) | 1979-01-10 |
| US4301287A (en) | 1981-11-17 |
| NO152049C (en) | 1985-07-24 |
| FI64376C (en) | 1983-11-10 |
| AT368505B (en) | 1982-10-25 |
| JPS5436275A (en) | 1979-03-16 |
| NZ187615A (en) | 1981-12-15 |
| CA1107736A (en) | 1981-08-25 |
| JPS5436290A (en) | 1979-03-16 |
| NO152049B (en) | 1985-04-15 |
| FI64376B (en) | 1983-07-29 |
| NO782127L (en) | 1978-12-21 |
| IE781234L (en) | 1978-12-20 |
| FI781955A (en) | 1978-12-21 |
| IT7868450A0 (en) | 1978-06-20 |
| IT1159739B (en) | 1987-03-04 |
| ES470913A1 (en) | 1979-02-01 |
| EP0000338B1 (en) | 1981-11-25 |
| ZA783493B (en) | 1979-06-27 |
| EP0027279A1 (en) | 1981-04-22 |
| IT7868449A0 (en) | 1978-06-20 |
| EP0028017A1 (en) | 1981-05-06 |
| DK270278A (en) | 1978-12-21 |
| EP0000338A3 (en) | 1979-06-27 |
| NO792839L (en) | 1978-12-21 |
| FI781954A (en) | 1978-12-21 |
| AU521040B2 (en) | 1982-03-11 |
| AU3724478A (en) | 1980-01-03 |
| NO782128L (en) | 1978-12-21 |
| AU3729878A (en) | 1980-01-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0000338B1 (en) | Isoxazolo(5,4-c)pyridine derivatives, their preparation and pharmaceutical compositions containing them | |
| US4996210A (en) | Heterocyclic spiro compounds and methods for preparing the same | |
| KR0160979B1 (en) | N-phenyl-l-(4-piperidinyl)amides useful as analgesics | |
| US6187782B1 (en) | Morphinane derivatives and medicinal use thereof | |
| PT96405B (en) | PROCESS FOR THE PREPARATION OF 3-AMINOPIPERIDINE DERIVATIVES AND HETEROCYCLES CONTAINING NITROGEN | |
| IE850103L (en) | Xanthine derivatives | |
| SU1308196A3 (en) | Method of producing 5,11-dihydro-11-(((1-methyl-4-piperidinyl)-amino)-carbonyl)-6h-dibenz (b,e) azepine-6-one or its salts | |
| CA2158952C (en) | 2-substituted morpholine and thiomorpholine derivatives as gaba-b antagonists | |
| US3864348A (en) | 1-Oxa-3,8-diaza spiro (4,5) decane compounds | |
| US4758559A (en) | Pyrrolo[1,2-a] [4,1]benzoxazepine derivatives useful as calmodulin and histamine inhibitors | |
| US4235921A (en) | Treating muscular spasms and convulsions with 3-azabicyclo[3.1.0]hexanes | |
| CS203940B2 (en) | Process for preparing aryloktahydropyridines | |
| CA1125288A (en) | Heterocyclic compounds which are gaba-agonists and production of same | |
| CA1148150A (en) | Phenylmorphans, intermediates and method of preparation | |
| US4495178A (en) | Enkephalin analogs | |
| US4332810A (en) | N-(Substituted)-2,5-ethano-8-hydroxy (or methoxy)-1,2,3,4,5,6-hexahydro-3 (or 4)-benzazocine centrally-acting analgesics | |
| US4376779A (en) | N-(Substituted)-2-aza-2'-hydroxy-5,6-benzotricyclo[6.3.01,8.04,11 ] undecane centrally-acting analgesics | |
| Chignell et al. | Structures Related to Morphine. XXVIII. 1 Alternative Syntheses of α-and β-2, 9-Dimethyl-2'-hydroxy-5-propyl-6, 7-benzomorphan | |
| EP0020885A1 (en) | Derivatives of 2-hydroxy-6,9-methano-11-amino-5,6,7,8,9,10-hexahydro-benzocyclooctene, process for their preparation and pharmaceutical compositions containing the same | |
| US4113877A (en) | Substituted 2-aminomethylphenyl sulfamates | |
| US4341904A (en) | Derivatives of 2-hydroxy-6,9-methano-11-amino-5,6,7,8,9,10-hexahydro-benzocyclooctene | |
| DK146129B (en) | METHOD OF ANALOGY FOR THE PREPARATION OF ERGOPEPTIDALCALOID DERIVATIVES | |
| US3198800A (en) | 2, 8-(para-f-4-oxo-butyl)-diazaspiro[4, 5]-decane-1, 3-diones | |
| US4010161A (en) | Piperazinoethyl-N-(2,3-dimethyl-5-oxo-1-phenyl-3Δ-pyrazolin-4-yl)carbamates | |
| CA1267898A (en) | Substituted benzoic acid alkylene bridged piperidyl amides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB LU NL SE |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB LU NL SE |
|
| 17P | Request for examination filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB LU NL SE |
|
| REF | Corresponds to: |
Ref document number: 2861342 Country of ref document: DE Date of ref document: 19820128 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19820630 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 19830609 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19840612 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19840629 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 19840630 Year of fee payment: 7 Ref country code: BE Payment date: 19840630 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19840730 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19860630 Year of fee payment: 9 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19870620 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19870630 |
|
| BERE | Be: lapsed |
Owner name: H. LUNDBECK & CO. A/S Effective date: 19870630 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19880101 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19880226 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19880301 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19881117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Effective date: 19890630 |
|
| EUG | Se: european patent has lapsed |
Ref document number: 78100191.2 Effective date: 19880711 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |