DE102007049039A1 - Process for the preparation of 8-hydrazino-8-aryl-octanoyl derivatives and their use - Google Patents
Process for the preparation of 8-hydrazino-8-aryl-octanoyl derivatives and their use Download PDFInfo
- Publication number
- DE102007049039A1 DE102007049039A1 DE102007049039A DE102007049039A DE102007049039A1 DE 102007049039 A1 DE102007049039 A1 DE 102007049039A1 DE 102007049039 A DE102007049039 A DE 102007049039A DE 102007049039 A DE102007049039 A DE 102007049039A DE 102007049039 A1 DE102007049039 A1 DE 102007049039A1
- Authority
- DE
- Germany
- Prior art keywords
- arylalkyl
- branched
- hydrogen
- alkyl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 B*(C(C[C@@]1C(C)C)[C@](C[C@]2C(C)C)OC2=O)C1c(cc1)cc(O*)c1O* Chemical compound B*(C(C[C@@]1C(C)C)[C@](C[C@]2C(C)C)OC2=O)C1c(cc1)cc(O*)c1O* 0.000 description 6
- OFDPCWVLRYMCLQ-XTDUXTJISA-O CCN(CC)C([C@@H](C/C=C/C[C@@H](C(C)C)C(c(cc1)cc(OCCCOC)c1OC)=[NH2+])C(C)C)=O Chemical compound CCN(CC)C([C@@H](C/C=C/C[C@@H](C(C)C)C(c(cc1)cc(OCCCOC)c1OC)=[NH2+])C(C)C)=O OFDPCWVLRYMCLQ-XTDUXTJISA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/72—Hydrazones
- C07C251/86—Hydrazones having doubly-bound carbon atoms of hydrazone groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/02—Formation of carboxyl groups in compounds containing amino groups, e.g. by oxidation of amino alcohols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/10—Hydrazines
- C07C243/12—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms
- C07C243/16—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms of an unsaturated carbon skeleton
- C07C243/18—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C281/00—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group
- C07C281/02—Compounds containing any of the groups, e.g. carbazates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C281/00—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group
- C07C281/02—Compounds containing any of the groups, e.g. carbazates
- C07C281/04—Compounds containing any of the groups, e.g. carbazates the other nitrogen atom being further doubly-bound to a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C281/00—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group
- C07C281/06—Compounds containing any of the groups, e.g. semicarbazides
- C07C281/08—Compounds containing any of the groups, e.g. semicarbazides the other nitrogen atom being further doubly-bound to a carbon atom, e.g. semicarbazones
- C07C281/14—Compounds containing any of the groups, e.g. semicarbazides the other nitrogen atom being further doubly-bound to a carbon atom, e.g. semicarbazones the carbon atom being further bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
- C07D207/50—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/04—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having less than three double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/22—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with hetero atoms directly attached to ring nitrogen atoms
- C07D295/28—Nitrogen atoms
- C07D295/30—Nitrogen atoms non-acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/30—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/32—Oxygen atoms
- C07D307/33—Oxygen atoms in position 2, the oxygen atom being in its keto or unsubstituted enol form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Die Erfindung betrifft ein Verfahren zur Herstellung von 8-Aryl-Octanoylderivaten, insbesondere chiralen 8-Hydrazino-8-Aryl-Octanoylamiden, sowie neue Zwischenprodukte, die in dem Verfahren zur Herstellung der genannten Octanoylderivate eingesetzt werden, und deren Verwendung.The invention relates to a process for the preparation of 8-aryl-octanoyl derivatives, in particular chiral 8-hydrazino-8-aryl-octanoylamides, and to novel intermediates which are used in the process for the preparation of said octanoyl derivatives, and to their use.
Description
Die Erfindung betrifft ein Verfahren zur Herstellung von 8-Aryl-Octanoylderivaten, insbesondere chiralen 8-Hydrazinn-8-Aryl-Octanoylderivaten, sowie neue Zwischenprodukte, die in dem Verfahren zur Herstellung der genannten Octanoylderivate eingesetzt werden und deren Verwendung.The Invention relates to a process for the preparation of 8-aryl-octanoyl derivatives, in particular chiral 8-hydrazine-8-aryl-octanoyl derivatives, as well as new intermediates used in the process for preparing the mentioned Octanoylderivate be used and their use.
Chirale
8-Aryl-Octanoylderivate der allgemeinen Formel (X) 
In
den Dokumenten
Die
Die
Nachteilig an den bekannten Verfahren ist vor allem der hohe verfahrenstechnische Aufwand zur gezielten Konfigurierung stereochemischer Zentren, welche nur unter hohem technischen und Kostenaufwand durchführbar ist.adversely in the known method is mainly the high procedural Effort for the specific configuration of stereochemical centers, which only with high technical and cost feasible is.
Die Aufgabe der vorliegenden Erfindung bestand somit in der Bereitstellung eines vereinfachten Herstellungsverfahrens für 8-Aryl-Octanoylderivate der allgemeinen Formel (X).The Object of the present invention was therefore in the provision a simplified production process for 8-aryl-octanoyl derivatives the general formula (X).
Die
gestellte Aufgabe wird gelöst mit einem Verfahren zur Herstellung
von 8-Aryl-Octanoylderivaten der allgemeinen Formel (X) oder deren
Salzen 
R1 und
R2 unabhängig voneinander für
Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl,
bevorzugt Benzyl, Alkoxyalkyl oder Alkoxyaryl stehen;
und
X
für Wasserstoff, Halogen, O–,
OR12 steht, worin R12 Wasserstoff,
verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, bevorzugt
Benzyl, oder M bedeutet, wobei M für Alkalimetall oder
ein Äquivalent eines Erdalkalimetalls steht,
oder
für
NR8R9 steht, worin
R8 und R9 unabhängig
voneinander für Wasserstoff, verzweigtes oder unverzweigtes
Alkyl, Arylalkyl, bevorzugt Benzyl, Hydroxyalkyl, Alkoxyalkyl, Alkanoyloxyalkyl,
HO(O)C-Alkyl, NH2C(O)-Alkyl, Alkyl-NHC(O)Alkyl,
(Alkyl)N-Alkyl oder CH2C(CH3)2CONH2 stehen;
oder
deren Stereoisomere oder Gemische davon, wobei man mindestens einen
der folgenden Verfahrensschritte durchführt
- A) Umsetzung einer Verbindung der Formel (IV) worin R1 und R2 die oben angegebene Bedeutung haben und E für eine Gruppe der folgenden Formeln steht
 worin in Formel a A für N steht, und B für NR4R5 steht, worin R4 und R5 unabhängig voneinander für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes, gegebenenfalls durch Halogen substituiertes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, stehen; oder R4 und R5 gemeinsam mit dem Stickstoffatom ein heterocyclisches Ringsystem bilden; und in Formel b b1) die einfach punktierte Linie eine Einfachbindung darstellt; und A für NR3 steht, worin R3 für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, steht; und B für NR4 steht, worin R4 für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, steht; oder b2) die einfach punktierte Linie keine Bindung darstellt; und A für die Gruppe NR3-NR4R5 steht, worin R3, R4 und R5 unabhängig voneinander für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, stehen oder R4 und R5 gemeinsam mit dem Stickstoffatom ein heterocyclisches Ringsystem bilden; und B für eine Stickstoff-Funktionalität, wie Azid, NR6R7 oder NH-NR6R7, steht, worin R6 und R7 unabhängig Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, bevorzugt Benzyl oder Trialkylsilyl bedeuten oder R6 und R7 gemeinsam mit dem Stickstoffatom ein heterocyclisches Ringsystem, wie Phthalimid, bilden, oder deren Stereoisomeren oder Gemischen davon oder deren delactonisierter Derivate in einem oder mehreren Schritten mit einem geeigneten Reduktionsmittel unter Entfernung der Stickstoff-Funktionalität in C8-Position und unter Bildung einer Amingruppe in C5-Position direkt zu einer Verbindung der Formel (X) oder zu einer Verbindung der Formel (IX)
worin in Formel a A für N steht, und B für NR4R5 steht, worin R4 und R5 unabhängig voneinander für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes, gegebenenfalls durch Halogen substituiertes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, stehen; oder R4 und R5 gemeinsam mit dem Stickstoffatom ein heterocyclisches Ringsystem bilden; und in Formel b b1) die einfach punktierte Linie eine Einfachbindung darstellt; und A für NR3 steht, worin R3 für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, steht; und B für NR4 steht, worin R4 für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, steht; oder b2) die einfach punktierte Linie keine Bindung darstellt; und A für die Gruppe NR3-NR4R5 steht, worin R3, R4 und R5 unabhängig voneinander für Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, C(O)R10, CO2R10, C(O)NR10R11, worin R10, R11 jeweils unabhängig voneinander Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Arylalkyl, bevorzugt Benzyl, bedeuten, stehen oder R4 und R5 gemeinsam mit dem Stickstoffatom ein heterocyclisches Ringsystem bilden; und B für eine Stickstoff-Funktionalität, wie Azid, NR6R7 oder NH-NR6R7, steht, worin R6 und R7 unabhängig Wasserstoff, verzweigtes oder unverzweigtes Alkyl, Aryl, Arylalkyl, bevorzugt Benzyl oder Trialkylsilyl bedeuten oder R6 und R7 gemeinsam mit dem Stickstoffatom ein heterocyclisches Ringsystem, wie Phthalimid, bilden, oder deren Stereoisomeren oder Gemischen davon oder deren delactonisierter Derivate in einem oder mehreren Schritten mit einem geeigneten Reduktionsmittel unter Entfernung der Stickstoff-Funktionalität in C8-Position und unter Bildung einer Amingruppe in C5-Position direkt zu einer Verbindung der Formel (X) oder zu einer Verbindung der Formel (IX)

- B) Öffnung des Lactonrings bei einer Verbindung der Formel (IV) oder (IX),
- C) ggfs. Überführung einer Verbindung der Formel (X) oder eines delactonisierten Derivats einer Verbindung der Formel (IV) in ein weiteres geeignetes Derivat, bevorzugt für eine der oben angegebenen Bedeutungen von X.
R 1 and R 2 are independently hydrogen, branched or unbranched alkyl, aryl, arylalkyl, preferably benzyl, alkoxyalkyl or alkoxyaryl;
and
X is hydrogen, halogen, O - , OR 12 , in which R 12 is hydrogen, branched or unbranched alkyl, aryl, arylalkyl, preferably benzyl, or M, where M is alkali metal or one equivalent of an alkaline earth metal,
or
is NR 8 R 9 , wherein R 8 and R 9 are independently hydrogen, branched or unbranched alkyl, arylalkyl, preferably benzyl, hydroxyalkyl, alkoxyalkyl, alkanoyloxyalkyl, HO (O) C-alkyl, NH 2 C (O) -alkyl , Alkyl-NHC (O) alkyl, (alkyl) N-alkyl or CH 2 C (CH 3 ) 2 CONH 2 ;
or their stereoisomers or mixtures thereof, wherein at least one of the following process steps is carried out
- A) Reaction of a compound of the formula (IV) wherein R 1 and R 2 are as defined above and E is a group of the following formulasin formula a A is N, and B is NR 4 R 5 , in which R 4 and R 5 independently of one another represent hydrogen, branched or unbranched alkyl, aryl, arylalkyl, C (O) R 10 , CO 2 R 10 , C (O) NR 10 R 11 , wherein R 10 , R 11 are each independently of one another hydrogen, branched or unbranched, optionally halogen-substituted alkyl, arylalkyl, preferably benzyl; or R 4 and R 5 together with the nitrogen atom form a heterocyclic ring system; and in formula b b1) the single-dotted line represents a single bond; and A is NR 3 wherein R 3 is hydrogen, branched or unbranched alkyl, aryl, arylalkyl, C (O) R 10 , CO 2 R 10 , C (O) NR 10 R 11 , wherein R 10 , R 11 each independently of one another are hydrogen, branched or unbranched alkyl, arylalkyl, preferably benzyl, and B is NR 4 wherein R 4 is hydrogen, branched or unbranched alkyl, aryl, arylalkyl, C (O) R 10 , CO 2 R 10 , C (O) NR 10 R 11 , wherein R 10 , R 11 each independently of one another are hydrogen, branched or unbranched alkyl, arylalkyl, preferably benzyl, or b2) the single-dotted line represents no bond; and A is the group NR 3 -NR 4 R 5 wherein R 3 , R 4 and R 5 are independently hydrogen, branched or unbranched alkyl, aryl, arylalkyl, C (O) R 10 , CO 2 R 10 , C (O) NR 10 R 11 wherein R 10 , R 11 are each independently of one another hydrogen, branched or unbranched alkyl, arylalkyl, preferably benzyl, or R 4 and R 5 together with the nitrogen atom form a heterocyclic ring system; and B is a nitrogen functionality such as azide, NR 6 R 7 or NH-NR 6 R 7 , wherein R 6 and R 7 are independently hydrogen, branched or unbranched alkyl, aryl, arylalkyl, preferably benzyl or trialkylsilyl, or R 6 and R 7 together with the nitrogen atom form a heterocyclic ring system, such as phthalimide, or their stereoisomers or mixtures thereof or their delactonized derivatives in one or more steps with a suitable reducing agent with removal of the nitrogen functionality in the C8 position and with formation of an amine group in the C5 position directly to a compound of the formula (X) or to a compound of the formula (IX)
- B) opening of the lactone ring in a compound of formula (IV) or (IX),
- C) optionally, converting a compound of the formula (X) or a delactonated derivative of a compound of the formula (IV) into another suitable derivative, preferably one of the meanings of X given above.
Die Reihenfolge der Verfahrensschritte A), B) und C) kann je nach den Verfahrensbedingungen gewählt werden, wobei Schritt C) vorzugsweise optional ist und im Anschluß an Schritt A) und/oder Schritt B) durchgeführt wird.The The sequence of process steps A), B) and C) can vary depending on the Process conditions are selected, wherein step C) is preferably optional and following step A) and / or step B) is performed.
In einer bevorzugten Variante des erfindungsgemäßen Verfahrens werden Verbindungen der Formel (IV) zunächst gemäß Verfahrensschritt A) reduktiv zu einer Verbindung der Formel (IX) umgesetzt und anschließend einer Lactonringöffnung gemäß Verfahrensschritt B) unterworfen, wobei nachfolgend optional gemäß Verfahrensschritt C) derivatisiert wird.In a preferred variant of the invention Process are compounds of formula (IV) first according to process step A) reductive to a compound of the formula (IX) and then a Lactonringöffnung subjected to process step B), wherein subsequently optional according to process step C) is derivatized.
In einer weiteren bevorzugten Variante des erfindungsgemäßen Verfahrens werden Verbindungen der Formel (IV) zunächst gemäß Verfahrensschritt B) einer Lactonringöffnung unterworfen, wobei nachfolgend optional gemäß Verfahrensschritt C) derivatisiert wird, und die erhaltenen delactonisierten Derivate anschließend gemäß Verfahrensschritt A) reduktiv zu einer Verbindung der Formel (X) umgesetzt, wobei nachfolgend optional gemäß Verfahrensschritt C) derivatisiert wird.In a further preferred variant of the invention Process are compounds of formula (IV) first according to process step B) of a lactone ring opening subjected, optionally subsequently according to method step C) is derivatized, and the resulting delactonated derivatives subsequently according to the method step A) reductively converted to a compound of formula (X), wherein subsequently optional according to process step C) is derivatized.
Der
Reduktionsschritt A) kann in einem oder mehreren Schritten durch
dem Fachmann an sich bekannte Methoden erfolgen, wie sie beispielsweise
in
Der Reduktionsschritt A) kann mit Wasserstoff bevorzugt in Gegenwart eines üblichen homogenen (z. B. Wilkinson-Katalysator) oder heterogenen Katalysators durchgeführt werden. Bevorzugt werden Metallkatalysatoren wie Pt oder Pd oder Raney-Ni, Ru, Rh oder Ir gegebenenfalls an einem Träger oder komplexiert mit einem N,O,P-haltigen Liganden eingesetzt. Die Reaktion kann unter Normal-Druck oder Überdruck bis zu 100 bar, bevorzugt bis zu 50 bar, bei Temperaturen von –20 bis 150°C, vorzugsweise 10°C bis 150°C, durchgeführt werden. Vorteilhaft wird die Reaktion in einem Lösungsmittel durchgeführt. Als Lösungsmittel können polare protische oder aprotische Lösungsmittel sowie apolare Lösungsmittel wie Alkohole oder AcOH, THF, DMF, Methylenchlorid, Ether oder aliphatische oder aromatische Kohlenwasserstoffe wie z. B. Toluol, Hexan oder Heptan usw. angewendet werden.Of the Reduction step A) may be preferred with hydrogen in the presence a conventional homogeneous (eg Wilkinson catalyst) or heterogeneous catalyst. Prefers are metal catalysts such as Pt or Pd or Raney-Ni, Ru, Rh or Ir optionally on a support or complexed used with an N, O, P-containing ligands. The reaction can under normal pressure or overpressure up to 100 bar, preferably up to 50 bar, at temperatures from -20 to 150 ° C, preferably 10 ° C to 150 ° C, are performed. Advantageously, the reaction is carried out in a solvent. As a solvent, polar protic or aprotic solvents and apolar solvents such as alcohols or AcOH, THF, DMF, methylene chloride, ether or aliphatic or aromatic hydrocarbons such as. As toluene, hexane or Heptane, etc. are used.
Der Reduktionsschritt kann auch in mehreren Schritten erfolgen, so dass beispielsweise zunächst die Sauerstoffunktion A in C8-Position reduktiv entfernt und anschließend die Stickstoffunktion B in C5-Position in eine Aminogruppe umgewandelt wird. Ein umgekehrter Reaktionsablauf ist ebenfalls möglich.Of the Reduction step can also be done in several steps, so that For example, first the oxygen function A in C8 position reductively removed and then the nitrogen function B is converted into an amino group in C5 position. A reverse one Reaction process is also possible.
Für
die Spaltung der N-N-Bindung können insbesondere auch,
nach an sich bekannter Methoden (s.
Insbesondere im Falle mehrerer Reduktionsschritte können die jeweiligen Schritte mit unterschiedlichen Reduktionsmitteln und in mehreren Schritten durchgeführt werden.Especially in the case of several reduction steps, the respective Steps with different reducing agents and in several Steps are performed.
Als
weitere mögliche Reduktionsmittel können Metallhydride,
bevorzugt LiAlH4, Redal, NaBH4 oder
DIBAH usw., oder Metalle wie Alkalimetalle, Erdalkalimetalle oder
Al, Fe, Zn usw. vorteilhaft jeweils wie üblich in protischen
oder aprotischen Lösungsmitteln wie z. B. Alkoholen, flüssigem
Ammoniak, niederen Carbonsäuren wie AcOH usw. verwendet
werden. Auch die als Birch-Reduktion bekannten Bedingungen mit diversen
Metallen in flüssigem Ammoniak oder Aminen können
verwendet werden (siehe z. B.
In den Fällen a) und b1), in welchen die Gruppe E einen stickstoffhaltigen Heterocyclus darstellt, erfolgt im Reduktionsschritt A) eine Ringöffnung, bevorzugt selektiv an der C8-A-Bindung, ggfs. unter Bruch der Bindung A-B. Je nach den gewählten Bedingungen ist auch ein umgekehrter Reaktionsablauf möglich, wobei zunächst die A-B Bindung gespalten und danach die A-Funktion in der Position C8 reduktiv entfernt wird.In Cases a) and b1), in which the group E is a nitrogen-containing Represents heterocycle, takes place in the reduction step A) a ring opening, preferably selectively at the C8-A bond, possibly with breakage of the bond FROM. Depending on the chosen conditions is also a reverse Reaction process possible, initially the A-B Binding split and then the A-function in position C8 reductive Will get removed.
In Abhängigkeit von der gewählten Reaktionsroute wird entweder A oder B an C5-Position erhalten und in eine Aminofunktion umgewandelt.In Dependence on the chosen reaction route is obtained either A or B at C5 position and in an amino function transformed.
Wie oben beschrieben erfolgt die Reihenfolge der Reduktion (Spaltung der A-B Bindung und reduktive Entfernung der A-Funktion in der C8-Position sowie die Bildung der Aminofunktion in der C5-Position) abhängig von den verwendeten Reduktionsmitteln und gewählten Bedingungen. Wird z. B. der Reduktionsschritt mit Wasserstoff ausgeführt wird bevorzugt die A-B Bindung zunächst gespalten unter gleichzeitiger oder nachfolgender Bildung der Aminofunktion in der Position C5 und anschließender reduktiver Entfernung der A-Funktion aus der Position C8.As described above, the order of reduction (cleavage the A-B bond and reductive removal of the A function in the C8 position and the formation of the amino function in the C5 position) of the reducing agents used and the conditions chosen. If z. B. carried out the reduction step with hydrogen Preferably, the A-B bond is initially cleaved under simultaneous or subsequent formation of the amino function in the Position C5 and subsequent reductive removal of the A function from position C8.
Die
Lactonöffnung gemäß Schritt B) erfolgt
ebenfalls auf an sich bekannte Weise in einem oder mehreren Schritten
durch Umsetzung z. B. mit Wasser, einem Alkohol oder Aminen in die
entsprechende Carbonsäure, Ester oder Amid (siehe z. B.
Die Lactonöffnung kann sowohl während des Reduktionsschritts A) als auch nach der Reduktion erfolgen. Wird z. B. als Lösungsmittel Alkohol für den Reduktionsschritt verwendet, erfolgt die Öffnung des Lactons unter gleichzeitiger Bildung eines entsprechenden Esters.The Lactone opening can occur during both the reduction step A) and after the reduction. If z. B. as a solvent Alcohol used for the reduction step, the opening takes place of the lactone with simultaneous formation of a corresponding ester.
Wie
oben erwähnt, kann die Reihenfolge der Schritte A), B)
und C) geändert werden. Es kann somit vor dem Reduktionsschritt
A) das Lacton zunächst mit Wasser und/oder Alkohol oder
direkt mit einem Amin unter gleichzeitiger Bildung der Carbonsäure,
des Esters oder des Amids geöffnet werden (Schritt B und
ggfs. C) gefolgt durch einen Reduktionsschritt A) an einem entsprechenden
delactonisierten Derivat der Carbonsäure. So kann z. B.
das Lacton zunächst mit einem Amin in ein Amid überführt
werden, unter Bedingungen, wie z. B. in der
Bevorzugt kann die Lactonöffnung gleichzeitig mit dem Reduktionsschritt A) erfolgen.Prefers For example, the lactone opening may be simultaneously with the reduction step A).
Die
ggfs. zusätzliche Derivatisierung, z. B. des delactonisierten
Derivats, gemäß Verfahrensschritt C) erfolgt wiederum
nach für den Fachmann an sich bekannten Methoden (siehe
z. B.
Beispielsweise
kann aus einem Carbonsäureester durch Umsetzung mit Aminen
in Gegenwart von Trialkylaluminium oder Dialkylaluminiumhalogenid
oder einer Lewis Säure oder Base das Carbonsäureamid
erhalten werden (siehe z. B.
Das
Säurehalogenid wird auf bekannte Weise durch Umsetzung
der freien Säure oder des Säuresalzes mit einem
Halogenierungsmittel, z. B. Thionylchlorid, gewünschtenfalls
lösungsmittelfrei oder in einem inerten Lösungsmittel,
z. B. einem Kohlenwasserstoff, wie Toluol oder Hexan ggfs. in Gegenwart
eines Katalysators, z. B. Zinkchlorid oder Dimethylformamid bei
Temperaturen bevorzugt zwischen 20 und 120°C erhalten (siehe
z. B.
Ein Carbonsäureester oder ein Carbonsäureamid kann beispielsweise durch alkalische Verseifung in die freie Säure überführt werden.One Carboxylic acid ester or a carboxylic acid amide can For example, converted by alkaline saponification in the free acid become.
Unter
Derivatisierung C) wird im Rahmen der vorliegenden Erfindung auch
die Überführung einer erhaltenen Verbindung mit
mindestens einer salzbildenden Gruppe in ihr Salz, die Überführung
eines Salzes in die freie Verbindung oder in ein anderes Salz verstanden,
wie dies z. B. in der
Die Ausgangsverbindungen der Formel (IV) können erfindungsgemäß entsprechend den nachfolgenden Synthesewegen erhalten werden, wobei die einzelnen Umsetzungsschritte auf dem Fachmann an sich bekannte Weise erfolgen. Dabei können verschiedene Reaktionsfolgen durchgeführt werden. Über die Wahl der Reste R3, R4 und R5, sowie die Stereochemie an der C8-Position kann die Diastereomerenselektivität der Zyklisierungen gesteuert werden. Dabei kann, ausgehend von Verbindungen der allgemeinen Formel (II) je nach Reaktionsführung und nach Art der Reste R3, R4, R5 und X zunächst ein Lacton der allgemeinen Formel (III) oder ein Stickstoffheterocyclus (Verbindungen Va, Vb) gebildet werden. Die entsprechenden Verbindungen Va und Vb können nur gebildet werden, wenn das cyclusbildende Stickstoffatom ein freies Proton aufweist, d. h. R3 für Wasserstoff steht und/oder R4 bzw. R5 für Wasserstoff stehen.According to the invention, the starting compounds of the formula (IV) can be obtained in accordance with the following synthesis routes, with the individual reaction steps being carried out in a manner known per se to one skilled in the art. Different reaction sequences can be carried out. The choice of R 3 , R 4 and R 5 and the stereochemistry at the C 8 position can be used to control the diastereomer selectivity of the cyclizations. Depending on the reaction procedure and on the nature of the radicals R 3 , R 4 , R 5 and X, starting from compounds of the general formula (II), it is possible initially to form a lactone of the general formula (III) or a nitrogen heterocycle (compounds Va, Vb) , The corresponding compounds Va and Vb can only be formed if the cyclus-forming nitrogen atom has a free proton, ie R 3 is hydrogen and / or R 4 or R 5 are hydrogen.
Eine
bevorzugte Variante des erfindungsgemäßen Verfahren,
wie in Schema 2 angegeben, geht von Verbindungen der Formel (IVa)
oder (IVb) aus 
R1, R2 die oben angegebene
Bedeutung haben,
A und B die oben unter a) und b1) angegebene
Bedeutung haben,
oder deren Stereoisomere oder Gemische davon,
die
erhalten werden durch Umsetzung einer Verbindung der Formel (II) 
R1,
R2, R3, R4 und R5 und X die
oben angegebene Bedeutung haben, wobei R3,
R4 und R5 bevorzugt
nicht für Wasserstoff stehen,
oder deren Stereoisomeren
oder Gemischen davon
mit einem Halogenierungsmittel, wie z.
B. Chlor, Brom, NCS, NBS, Iod, I-Cl, I-Br, I-OAc oder Bispyridin-Iodonium-tetrafluorborat,
einem Oxidationsmittel wie Osmiumtetroxid, Wasserstoffperoxid oder
meta-Chlorperbenzoesäure, ggfs. unter Zugabe von chiralen
Auxiliaren, oder einer Quecksilberverbindung, wie Hg(OAc)2 oder Hg(O2CCF3)2, oder einer Selenverbindung,
wie PhSeCl oder ArSeOTf, und ggfs. nachfolgende Behandlung mit einer
Base, wie LiOH in Wasser, sowie ggfs. Einführung einer
Schutzgruppe, wie Mesylat, Tosylat oder Triflat, unter Lactonisierung
zu einer Verbindung der Formel (III) 
R1,
R2, R3, R4 und R5 die oben
angegebene Bedeutung haben und Y für Brom, Chlor, Iod,
OH oder OR18 steht, worin OR18 für
eine Abgangsgruppe, wie Mesylat, Tosylat oder Triflat steht,
oder
deren Stereoisomeren oder Gemischen davon,
und
anschließende Überführung
der Verbindung der Formel (III) durch intramolekulare Cyclisierung
in die Zielverbindung der Formel (IVa) und/oder (IVb).A preferred variant of the process according to the invention, as indicated in Scheme 2, is based on compounds of the formula (IVa) or (IVb)
R 1 , R 2 have the abovementioned meaning,
A and B have the meaning given above under a) and b1),
or their stereoisomers or mixtures thereof,
which are obtained by reacting a compound of the formula (II)
R 1 , R 2 , R 3 , R 4 and R 5 and X have the abovementioned meaning, where R 3 , R 4 and R 5 are preferably not hydrogen,
or their stereoisomers or mixtures thereof
with a halogenating agent, such as. For example, chlorine, bromine, NCS, NBS, iodine, I-Cl, I-Br, I-OAc or bispyridine-iodonium tetrafluoroborate, an oxidizing agent such as osmium tetroxide, hydrogen peroxide or meta-chloroperbenzoic acid, if necessary, with the addition of chiral auxiliaries, or a mercury compound, such as Hg (OAc) 2 or Hg (O 2 CCF 3 ) 2 , or a selenium compound, such as PhSeCl or ArSeOTf, and optionally subsequent treatment with a base, such as LiOH in water, and, if appropriate, introduction of a protective group, such as Mesylate, tosylate or triflate, with lactonization to give a compound of the formula (III)
R 1 , R 2 , R 3 , R 4 and R 5 have the abovementioned meaning and Y is bromine, chlorine, iodine, OH or OR 18 , in which OR 18 is a leaving group, such as mesylate, tosylate or triflate,
or their stereoisomers or mixtures thereof,
and
subsequent conversion of the compound of the formula (III) by intramolecular cyclization into the target compound of the formula (IVa) and / or (IVb).
Die
Cyclisierung zum Halo-Lacton (Verbindung III, mit Y = Br, Cl; I)
erfolgt dabei nach an sich bekannten Methoden, wie beispielsweise
in der
Optional
kann die Halogenverbindung im Anschluss nach an sich bekannten Methoden,
z. B.: mit einer Base wie z. B.: LiOH in Wasser, wie beispielsweise
in
Alternativ
können Verbindungen der allgemeinen Formel (II) auch durch
Epoxidierung, nach an sich bekannter Weise zu den entsprechenden
Verbindungen der allgemeinen Formel III (mit Y = OH) umgesetzt werden.
Als Epoxidierungsmittel dienen z. B. Persäure, Peroxid,
ggfs. in Gegenwart eines üblichen Katalysators, bspw. auf
Basis von Übergangsmetallen, wie z. B. Ti-Alkoxide, V,
Mo, W, deren Salze oder Komplexe mit anorganischen oder organischen
Liganden, z. B. unter Bedingungen beschrieben als "Sharpless Epoxidation" (siehe
Vor
der Bildung des zweiten Cyclus unter Erhalt von Verbindungen der
allgemeinen Formel (IVa) und/oder (IVb) werden ausgehend von Verbindungen
der allgemeinen Formel (III), worin R3,
R4, R5 alle ≠ H, zunächst
eine oder mehrere der Stickstoffschutzgruppen entfernt. Bei Verbindungen
der allgemeinen Formel (III), mit R3, R4 und/oder R5 = H
können optional weitere Schutzgruppen entfernt werden.
Hierzu werden jeweils die an sich bekannten Methoden zur Abspaltung
von Stickstoff-Schutzgruppen verwendet, wie sie z. B.: in Standardwerken,
wie
Die intramolekulare Cyclisierung sowie die Bildung der Produkte IVa und IVb kann gewünschtenfalls in polaren protischen oder aprotischen sowie apolaren Lösungsmitteln durchgeführt werden. Bevorzugt werden Lösungsmittel wie Acetonitril, DMF, Wasser, N-Methylpyrrolidon (NMP) usw., insbesondere DMF, NMP, Acetonitril oder Toluol, verwendet, die für eine SN2-Substitution üblicherweise verwendet werden. Die Reaktion kann außerdem mit Basen wie z. B. Alkalimetallhydroxiden, Alkoholaten oder Metallhydriden oder auch Aminbasen wie Alkylaminen, wie z. B. organische Trialkylamine, bevorzugt Triethylamin katalysiert werden. Auch Säuren, besonders Lewis-Säuren können verwendet werden. Die Reaktionstemperatur liegt bevorzugt zwischen –20°C und dem Siedepunkt des jeweiligen Lösungsmittels.If desired, the intramolecular cyclization and the formation of the products IVa and IVb can be carried out in polar protic or aprotic as well as apolar solvents. Preference is given to using solvents such as acetonitrile, DMF, water, N-methylpyrrolidone (NMP), etc., in particular DMF, NMP, acetonitrile or toluene, which are usually used for SN 2 substitution. The reaction can also with bases such. As alkali metal hydroxides, alcoholates or metal hydrides or amine bases such as alkylamines, such as. As organic trialkylamines, preferably triethylamine are catalysed. Also acids, especially Lewis acids can be used. The reaction temperature is preferably between -20 ° C and the boiling point of the respective solvent.
In
einer weiteren bevorzugten Variante des erfindungsgemäßen
Verfahren, wie in Schema 2 angegeben, werden die Verbindungen der
Formel (IVa) oder (IVb) erhalten durch Umsetzung einer Verbindung
der Formel (II), wobei mindestens einer der Reste R3,
R4 und R5 für
Wasserstoff steht, oder deren Stereoisomeren oder Gemischen davon
mit einem Halogenierungsmittel, wie Chlor, Brom, NCS, NBS, Brom,
Iod, I-Cl, I-Br, I-OAc oder Bispyridin-Iodonium-tetrafluorborat,
einem Oxidationsmittel wie Osmiumtetroxid, Wasserstoffperoxid oder
meta-Chlorperbenzoesäure, ggfs. unter Zugabe von chiralen
Auxiliaren, oder einer Quecksilberverbindung, wie Hg(OAc)2 oder Hg(O2CCF3)2, oder einer Selenverbindung,
wie PhSeCl oder ArSeOTf zu einer Verbindung der Formel 
R1, R2, R3,
R4 und R5 die oben
angegebene Bedeutung haben und
Y für Brom, Chlor,
Iod, OH oder OR18 steht, worin OR18 für eine Abgangsgruppe, wie Mesylat,
Tosylat oder Triflat steht,
anschließende Überführung
der erhaltenen Verbindung der Formel (Va) oder (Vb) durch Lactonisierung
in die Zielverbindung der Formel (IVa) und/oder (IVb). Das Lacton
der allgemeinen Formel (IVa) bzw. (IVb) wird je nach Definition
der Reste X und Y unter saurer oder basischer Aktivierung gebildet;
nach an sich bekannten Methoden (siehe z. B.:
R 1 , R 2 , R 3 , R 4 and R 5 have the abovementioned meaning and
Y is bromine, chlorine, iodine, OH or OR 18 , wherein OR 18 is a leaving group such as mesylate, tosylate or triflate,
subsequent conversion of the resulting compound of the formula (Va) or (Vb) by lactonization into the target compound of the formula (IVa) and / or (IVb). The lactone of the general formula (IVa) or (IVb) is formed depending on the definition of the radicals X and Y with acidic or basic activation; according to methods known per se (see, for example:
Für die saure Aktivierung werden Lewissäuren oder Brönstedt-Säuren, bevorzugt Essigsäure, Trifluoressigsäure, p-Toluolsulfonsäure zweckmäßig in polaren protischen oder aprotischen Lösungsmitteln verwendet. Die Reaktion kann außerdem mit Basen wie z. B. Alkalimetallhydroxiden, Alkalimetallcarbonaten, Alkoholaten oder Metallhydriden oder Aminbasen wie Alkylaminen, bevorzugt Triethylamin, katalysiert werden. Die bevorzugte Reaktionstemperatur liegt zwischen –20°C und dem Siedepunkt des jeweiligen Lösungsmittels. Bevorzugt werden Lösungsmittel wie Acetonitril, DMF, Wasser, N-Methylpyrrolidon (NMP), usw. verwendet, die für eine SN2-Substitution üblicherweise verwendet werden.For acidic activation, Lewis acids or Bronsted acids, preferably acetic acid, trifluoroacetic acid, p-toluenesulfonic acid are advantageously used in polar protic or aprotic solvents. The reaction can also be carried out with bases such. As alkali metal hydroxides, alkali metal carbonates, alcoholates or metal hydrides or amine bases such as alkylamines, preferably triethylamine, are catalyzed. The preferred reaction temperature is between -20 ° C and the boiling point of the respective solvent. It is preferable to use solvents such as acetonitrile, DMF, water, N-methylpyrrolidone (NMP), etc., which are commonly used for SN 2 substitution.
In
einer weiteren bevorzugten Verfahrensvariante werden beide Cyclen
ohne Isolierung der Monocyclen gebildet. Dazu werden Verbindungen
der Formel (II) bevorzugt mit Reagenzien umgesetzt, die direkt zu zweifach
cyclisierten Verbindungen der allgemeinen Formeln (IVa) und (IVb)
führen, z. B. hypervalente Iodverbindungen, wie PhI(OAc)2, PhI(O2CCF3)2, oder (Hydroxy-(tosyloxy)-iod)-benzol
(
In
einer weiteren bevorzugten Verfahrensvariante geht das Verfahren
von Verbindungen der Formel (IVc) 
R1,
R2 die oben angegebene Bedeutung haben,
A
und B die oben unter b2) angegebene Bedeutung haben,
oder deren
Stereoisomere oder Gemische davon,
aus, die erhalten werden
aus einer Verbindung der Formel (III) 
R1,
R2, R3, R4 und R5 die oben
angegebene Bedeutung haben und
Y für Brom, Chlor,
Iod, OH oder OR18 steht, worin OR18 für eine Abgangsgruppe, wie Mesylat,
Tosylat oder Triflat steht,
oder deren Stereoisomeren oder
Gemischen davon,
durch Umsetzung mit einem Stickstoff-Nukleophil
in die Zielverbindung (IVc). In diesem Falle erfolgt eine nucleophile
Substitution der Abgangsgruppe Y durch die Stickstoffnucleophile,
wie z. B. Azid, Aminen, Ammoniak, Cyanamiden, etc. Hierzu werden
die Verbindungen der Formel (III) mit einem stickstoffhaltigen Reagenz,
wie Ammoniak, Cyanamiden, Aziden oder Aminen, unter an sich bekannten
Bedingungen, die zur SN2-Substitution angewendet
werden, so z. B. DMF, Acetonitril oder NMP als Lösungsmittel,
gegebenenfalls in Gegenwart einer Base wie z. B. Alkalimetallhydroxide,
Alkoxide oder org. Aminbasen, zur Verbindung der Formel (IVc) umgesetzt.In a further preferred process variant, the process of compounds of the formula (IVc)
R 1 , R 2 have the abovementioned meaning,
A and B have the meaning given above under b2),
or their stereoisomers or mixtures thereof,
which are obtained from a compound of the formula (III)
R 1 , R 2 , R 3 , R 4 and R 5 have the abovementioned meaning and
Y is bromine, chlorine, iodine, OH or OR 18 , wherein OR 18 is a leaving group such as mesylate, tosylate or triflate,
or their stereoisomers or mixtures thereof,
by reaction with a nitrogen nucleophile in the target compound (IVc). In this case, a nucleophilic substitution of the leaving group Y by the nitrogen nucleophiles, such as. Azide, amines, ammonia, cyanamides, etc. For this purpose, the compounds of the formula (III) are reacted with a nitrogen-containing reagent, such as ammonia, cyanamides, azides or amines, under conditions known per se, which are used for SN 2 substitution. so z. As DMF, acetonitrile or NMP as a solvent, optionally in the presence of a base such as. As alkali metal hydroxides, alkoxides or org. Amine bases, converted to the compound of formula (IVc).
Die
erfindungsgemäß eingesetzten Verbindungen der
Formel (II) können erhalten werden durch Umsetzung einer
Verbindung der allgemeinen Formel (VII) 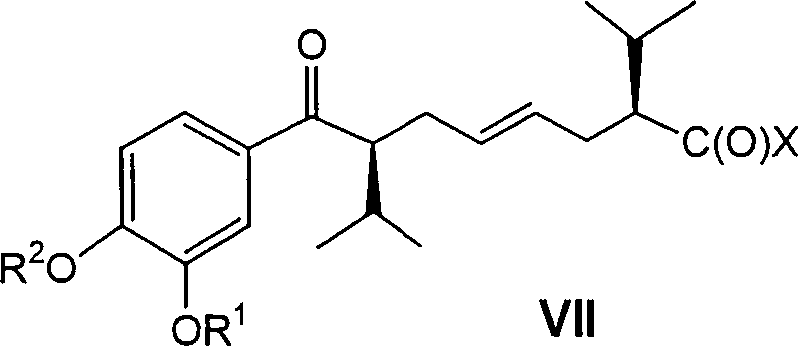
mit
einem substituierten Hydrazinderivat, zweckmäßig
der allgemeinen Formel
zu einer Verbindung der allgemeinen
Formel (I) 
with a substituted hydrazine derivative, suitably the general formula
to a compound of general formula (I)
So
können direkt derivatisierte Hydrazone der allgemeinen
Formel (I) mit entsprechenden Substituenten R4 und R5 erhalten werden.
Dazu wird z. B. nach einer an sich bekannten Methode (siehe
Vorteilhaft
kann man die Verbindung der allgemeinen Formel (VII) mit Hydrazin
der Formel H2N-NH2 zu
einer Verbindung der allgemeinen Formel (Ia) 
Die Umsetzung mit Hydrazin zum Hydrazon (Ia) erfolgt auf an sich bekannte Weise. Dabei können wasserentziehende Mittel, wie Molsieb, Wasserabscheider, Orthoester, etc. eingesetzt werden. Die Reaktion kann unter Aktivierung mit Lewissäure, wie z. B. Titanisopropoxid, oder unter saurer Katalyse, z. B. Essigsäure, Trifluoressigsäure oder p-Toluolsulfonsäure durchgeführt werden. Bevorzugt wird wasserfreies Hydrazin verwendet. Die Reaktion wird in den üblichen protischen oder aprotischen Lösungsmitteln, bevorzugt Tetrahydrofuran oder halogenierte Kohlenwasserstoffe, wie z. B. Dichlorethan, bei Temperaturen von Raumtemperatur bis zur Siedetemperatur des jeweiligen Lösungsmittels durchgeführt.The Reaction with hydrazine to give the hydrazone (Ia) is carried out in known manner Wise. In this case, dehydrating agents, such as molecular sieve, Water separator, Orthoester, etc. are used. The reaction can under activation with Lewis acid, such as. For example, titanium isopropoxide, or under acid catalysis, e.g. As acetic acid, trifluoroacetic acid or p-toluenesulfonic acid. Anhydrous hydrazine is preferably used. The reaction will in the usual protic or aprotic solvents, preferably tetrahydrofuran or halogenated hydrocarbons, such as As dichloroethane, at temperatures from room temperature to to the boiling temperature of the particular solvent.
Je
nach gewünschtem Substitutionsmuster wird das Hydrazon
(Ia) dann einfach oder mehrfach substituiert, wie in Schema 1 beispielhaft
dargestellt. Mehrfachsubstitutionen können in einer Stufe
oder in zwei aufeinanderfolgenden Syntheseschritten eingeführt
werden. Hierzu können die an sich bekannten Methoden der
Schätzung von Stickstofffunktionen verwendet werden, wie
z. B. in Standardwerken, wie
Die Hydrazine der allgemeinen Formel (II) werden durch Reduktion in an sich bekannter Weise aus den entsprechenden Hydrazonen der allgemeinen Formel (I) hergestellt. Dabei kann die Bildung des neuen Stereozentrums in Position C-8 je nach Reduktionsbedingungen diastereoselektiv erfolgen.The Hydrazines of the general formula (II) are obtained by reduction in in a known manner from the corresponding hydrazones of the general Formula (I) prepared. This can be the formation of the new stereo center in position C-8, depending on the reduction conditions, diastereoselective respectively.
Als
Reduktionsmittel dienen hierbei z. B. komplexe Metallhydride, wie
LiAlH4 im Neutralen, Metallborhydride, bevorzugt
NaBH4, BH3·NMe3, NaBH3(OAc), NaBH(OAc)3 oder NaBH3CN, wie
auch Siliziumhydride wie z. B. Et3SiH jeweils
unter Aktivierung durch Lewis-Säuren oder Brönstedt-Säuren,
z. B.: Trifluoressigsäure, Essigsäure, p-Toluolsulfonsäure,
Salzsäure, etc. Des weiteren können Metallamalgame,
wie Na(Hg), Zn(Hg) oder Al(Hg), oder Metallsalze, wie z. B. SnCl2 verwendet werden. Alternativ kann Wasserstoff
unter homogener oder heterogener Katalyse mit geeigneten desaktivierten
Katalysatoren, sowie die unter dem Namen „Birch-Reduktion"
bekannten Bedingungen, wie z. B.: Natrium, Lithium in Ammoniak,
verwendet werden (s. z. B.
Dabei werden jeweils die üblichen protischen oder aprotischen Lösungsmitteln, bei Temperaturen von –78°C bis zur Siedetemperatur des jeweiligen Lösungsmittels verwendet.there each become the usual protic or aprotic Solvents, at temperatures of -78 ° C used up to the boiling temperature of the particular solvent.
Besonders bevorzugt ist hierbei die Verwendung von Metallborhydriden unter Säureaktivierung.Especially preferred here is the use of metal borohydrides under Acid activation.
Anschließend kann das erhaltene Hydrazin (II) je nach gewünschtem Schutzgruppenmuster weiter derivatisiert werden. Dazu können die bereits oben beschriebenen Methoden zur Schätzung von Aminfunktionen verwendet werden.Subsequently For example, the resulting hydrazine (II) can vary depending on the desired protective group pattern be further derivatized. This can already be done above described methods for estimating amine functions become.
Alternativ ist es auch möglich, ungeschützte Hydrazone (Ia) zu reduzieren und anschließend nach den oben beschriebenen Methoden die Stickstoff-Atome zu derivatisieren.alternative it is also possible to use unprotected hydrazones (Ia) reduce and then after the above described Methods to derivatize the nitrogen atoms.
Mit den beispielhaft in Schema 1 beschriebenen Wegen lässt sich so das gewünschte Substitutionsmuster an den Stickstoffatomen darstellen. Dabei beeinflusst die Wahl der Reste R3, R4 und R5 sowie die Stereochemie an der C8-Position die Diastereomerenselektivität der nachfolgenden Reaktionen. Damit können die neu zu generierenden Stereozentren in C4 und C5-Position mit hoher Diastereoselektivität eingeführt werden.By way of example, the routes described in Scheme 1 can be used to represent the desired substitution pattern on the nitrogen atoms. The choice of the radicals R 3 , R 4 and R 5 as well as the stereochemistry at the C8 position influences the diastereomer selectivity of the subsequent reactions. This allows the new stereogenic centers to be generated in C4 and C5 positions with high diastereoselectivity.
Die 8-Hydrazinn-8-Aryl-Octanoylderivate der allgemeinen Formel (II) können zur Herstellung von Reninhemmern der allgemeinen Formel (X) verwendet werden. (siehe Schema 2).The 8-hydrazine-8-aryl-octanoyl derivatives of the general formula (II) can be used for the preparation of renin inhibitors of the general Formula (X) can be used. (see Scheme 2).
Die
erfindungsgemäß eingesetzten Verbindungen der
Formel (VII) können nach 
an eine chirale
Verbindung der Formel (2) 
W für
z. B. für Wasserstoff oder Halogen steht, wobei die Gruppe
C(O)W und C(O)OR21 jeweils auch durch Nitril
ersetzt sein kann, und
R21 für
verzweigtes oder unverzweigtes Alkyl steht, wobei die Gruppe OR21 auch für Halogen stehen kann,
ggfs.
unter nachfolgender Derivatisierung, d. h. einer nachgeordneten
Manipulation der funktionellen Gruppe in Abhängigkeit der
Bedeutung des Restes X der Verbindung der Formel (VII). Die Reaktion
kann auch mit anderen als den angegebenen Isomeren der jeweiligen
Verbindungen oder deren Mischungen durchgeführt werden,
was zu entsprechenden Isomeren und/oder Mischungen der Verbindung
der Formel (VII) führt.The compounds of the formula (VII) used according to the invention can according to
to a chiral compound of the formula (2)
W for z. B. is hydrogen or halogen, wherein the group C (O) W and C (O) OR 21 may each be replaced by nitrile, and
R 21 is branched or unbranched alkyl, where the group OR 21 may also be halogen,
if appropriate with subsequent derivatization, ie a downstream manipulation of the functional group as a function of the meaning of the radical X of the compound of the formula (VII). The reaction can also be carried out with other than the specified isomers of the respective compounds or mixtures thereof, which leads to corresponding isomers and / or mixtures of the compound of formula (VII).
Die Reaktionstemperatur kann zwischen –78°C und Rückflusstemperatur des Lösungsmittels liegen, bevorzugt ist THF bei 0°C oder RT.The Reaction temperature can be between -78 ° C and reflux temperature of the solvent, preferably THF is at 0 ° C. or RT.
Überraschend
wurde nun gefunden, daß die Kupplung vorteilhaft in Gegenwart
eines Metallkatalysators aus der Gruppe Fe(III), z. B.: Fe(acac)3, Ni(0), Pd(0) und V(III), z. B. VCl3 (siehe dazu auch,
Die
zur Kupplung benötigte chirale Verbindung 2, vorzugsweise
die Disäure mit W = OH; R21 = H,
wird bevorzugt als (S,S)-Verbindung eingesetzt. Zur Isolierung des
gewünschten Stereoisomers aus der Diastereomeren Mischung
wird nach der
Überraschend
wurde nun gefunden, daß es bei der vorgenannten meso/rac-Trennung
vorteilhaft ist die kinetische Kristallisation zu stabilisieren,
bevorzugt durch Addition von Keimbildungsinhibitoren, wie z. B. Tensiden,
oder durch Emulsionskristallisation (s. dazu auch:
Dabei werden ausgehend von ca. 50:50 Mischungen der meso/rac Disäure bevorzugt 1–2 Äquivalente (bzgl. meso-Verbindung) des Amins, bevorzugt Piperazin, in einem organischen Lösungsmittel, z. B. Ethanol oder Isopropylacetat, zugegeben. Das als Feststoff erhaltene Salz ist stark mit der meso-Verbindung angereichert, die Mutterlauge enthält überwiegend die racemische Verbindung. Die vorangereicherten meso- bzw. racemischen-Verbindungen können jeweils, gegebenenfalls nach Salzspaltung und Entfernung des Amins mit den üblichen Methoden wie z. B. Extraktion mit organischen Lösungsmitteln aus einer sauren wässrigen Lösung, durch einfache Umkristallisierung aus, bzw. „Slurry-Wäsche" in organischen Lösungsmitteln weiter angereichert werden. Die jeweils erhaltenen Mutterlaugen können in den Prozess zurückgeführt werden, so dass die Gesamtausbeute nahezu quantitativ ist.there are starting from about 50:50 mixtures of meso / rac diacid preferably 1-2 equivalents (with respect to meso compound) of the amine, preferably piperazine, in an organic solvent, z. For example, ethanol or isopropyl acetate. That as a solid salt obtained is strongly enriched with the meso compound, which Mother liquor contains predominantly the racemic Connection. The pre-enriched meso or racemic compounds can each, optionally after salt splitting and removal of the amine with the usual methods such. B. Extraction with organic solvents from an acidic aqueous Solution, by simple recrystallization or "slurry washing" be further enriched in organic solvents. The mother liquors obtained in each case can be in the process be returned, so that the overall yield is almost quantitative.
Im Rahmen der vorliegenden Erfindung bezieht sich der Ausdruck "Halogen" auf Fluor, Chlor, Brom, Jod, bevorzugt Chlor und Brom.in the For the purposes of the present invention, the term "halogen" to fluorine, chlorine, bromine, iodine, preferably chlorine and bromine.
"Alkyl" bezieht sich, sofern nicht anders angegeben, auf geradkettige oder verzweigte oder cyclische gesättigte Kohlenwasserstoffe oder deren Kombinationen mit vorzugsweise 1 bis 20 Kohlenstoffatomen, insbesondere 1 bis 10, besonders bevorzugt 1 bis 5 Kohlenstoffatomen. Beispiele solcher Alkylgruppen (vorausgesetzt, die bezeichnete Länge umfasst das spezielle Beispiel) sind Methyl, Ethyl, Propyl, Isopropyl, Butyl, sek.-Butyl, tert.-Butyl, Pentyl, Isopentyl, Neopentyl, tert.-Pentyl, 1-Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, Hexyl, Isohexyl, Heptyl und Octyl, oder Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl, Cycloheptyl, Cyclopropenyl, Cyclobutenyl, Cyclopentenyl, Cyclohexenyl, Cycloheptenyl oder 1,3-Cyclobutadienyl."Alkyl" Unless otherwise indicated, refers to straight-chain or straight chain branched or cyclic saturated hydrocarbons or combinations thereof preferably having 1 to 20 carbon atoms, in particular 1 to 10, particularly preferably 1 to 5 carbon atoms. Examples of such alkyl groups (provided the designated length includes the specific example) are methyl, ethyl, propyl, isopropyl, Butyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, tert-pentyl, 1-methylbutyl, 2-methylbutyl, 3-methylbutyl, hexyl, isohexyl, heptyl and octyl, or cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, Cycloheptyl, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, Cycloheptenyl or 1,3-cyclobutadienyl.
"Alkoxy" bezieht sich auf über Sauerstoff gebundenes geradkettiges oder verzweigtes gesättigtes Alkyl mit vorzugsweise 1 bis 20 Kohlenstoffatomen, insbesondere 1 bis 10, besonders bevorzugt 1 bis 5, ganz besonders bevorzugt 1 bis 2 Kohlenstoffatomen. Beispiele solcher Alkoxygruppen (vorausgesetzt, die bezeichnete Länge umfasst das spezielle Beispiel) sind Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy und tert.-Butoxy."Alkoxy" refers to oxygen linked straight chain or branched saturated alkyl having preferably 1 to 20 carbon atoms, in particular 1 to 10, particularly preferred 1 to 5, most preferably 1 to 2 carbon atoms. Examples such alkoxy groups (provided the designated length includes the specific example) are methoxy, ethoxy, propoxy, isopropoxy, Butoxy, isobutoxy and tert-butoxy.
Die Alkyl- und Alkoxygruppen können substituiert sein durch eine oder mehrere der folgenden Gruppen ausgewählt aus Halogen, Hydroxy, Cyano, C1-C6-Alkoxy, Nitro, Amino, C1-C6-Alkylamino, Di-C1-C6-Alkylamino, Carboxy, C1-C6-Alkoxycarbonyl, Aminocarbonyl, Halogenmethyl, Dihalogenmethyl, Trihalogenmethyl, Halogenethyl, Dihalogenethyl, Trihalogenethyl, Tetrahalogenethyl, Pentahalogenethyl.The alkyl and alkoxy groups may be substituted by one or more of the following groups selected from halogen, hydroxy, cyano, C 1 -C 6 alkoxy, nitro, amino, C 1 -C 6 alkylamino, diC 1 -C 6 Alkylamino, carboxy, C 1 -C 6 alkoxycarbonyl, aminocarbonyl, halomethyl, dihalomethyl, trihalomethyl, haloethyl, dihaloethyl, trihaloethyl, tetrahaloethyl, pentahalogenethyl.
Der Ausdruck „Aryl" bedeutet einen cyclischen oder polycyclischen Ring, bestehend aus vorzugsweise 6 bis 12 Kohlenstoffatomen, der unsubstituiert sein kann oder substituiert ist durch eine oder mehrere Substituentengruppen, die oben für die Alkyl- und Alkoxygruppen angegeben sind. Beispiele für Arylgruppen sind Phenyl, 2,6-Dichlorphenyl, 2- oder 3- oder 4-Methoxyphenyl, Naphthyl, 4-Thionaphthyl, Tetralinyl, Anthracinyl, Phenanthrenyl, Benzonaphthenyl, Fluorenyl, 2-Acetamidofluoren-9-yl und 4'-Brombiphenyl.Of the The term "aryl" means a cyclic or polycyclic one Ring consisting of preferably 6 to 12 carbon atoms, the may be unsubstituted or substituted by one or more Substituent groups above for the alkyl and alkoxy groups are indicated. Examples of aryl groups are phenyl, 2,6-dichlorophenyl, 2- or 3- or 4-methoxyphenyl, naphthyl, 4-thionaphthyl, Tetralinyl, anthracinyl, phenanthrenyl, benzonaphthenyl, fluorenyl, 2-acetamidofluoren-9-yl and 4'-bromobiphenyl.
Der Ausdruck „heterocyclisch" bedeutet ein mono- oder bicyclisches, heterocyclisches Ringsystem. Monocyclische heterocyclische Ringe bestehen aus etwa 3 bis 7 Ringatomen mit 1 bis 5 Heteroatomen, ausgewählt aus N, O oder S und vorzugsweise 3 bis 7 Atomen im Ring. Bicyclische Heterocyclen bestehen aus etwa 5 bis 17 Ringatomen, bevorzugt 5 bis 12 Ringatome. Beispiele für heterocyclische Ringsysteme sind Phthalimido, Morpholino, 1,3,5-Dioxacinyl, 2,3-Diphenylmaleoyl, und dergleichen.Of the Term "heterocyclic" means a mono- or bicyclic, heterocyclic ring system. Monocyclic heterocyclic rings consist of about 3 to 7 ring atoms with 1 to 5 heteroatoms selected from N, O or S and preferably 3 to 7 atoms in the ring. bicyclic Heterocycles consist of about 5 to 17 ring atoms, preferably 5 up to 12 ring atoms. Examples of heterocyclic ring systems are phthalimido, morpholino, 1,3,5-dioxacinyl, 2,3-diphenylmaleoyl, and the same.
Der Ausdruck "Salze" bezieht sich bevorzugt auf Säureadditionssalze, Salze mit Basen und Metallsalze, insbesondere Alkalimetallsalze.Of the Term "salts" refers preferably to acid addition salts, Salts with bases and metal salts, in particular alkali metal salts.
Salze, Hydrate und Solvate der erfindungsgemäßen Verbindungen sind ebenfalls mit eingeschlossen. Im erfindungsgemäßen Verfahren können die Verbindungen ggfs. als Salz, Hydrat oder Solvat eingesetzt oder erhalten werden.salts Hydrates and solvates of the compounds of the invention are also included. In the invention If necessary, the compounds can be used as salt, hydrate or solvate can be used or obtained.
Der Ausdruck "delactonisiertes Derivat" bedeutet im Rahmen der vorliegenden Erfindung ein von einem Lacton unter Öffnung des Lactonrings abgeleitetes Derivat, wie z. B. die freie Carbonsäure oder entsprechende Carbonsäurehalogenide, Carbonsäureamide, Carbonsäureester, etc.Of the Term "delactonized derivative" means within the scope of the present invention Invention of a lactone with opening of the lactone ring derived derivative, such as. As the free carboxylic acid or corresponding carboxylic acid halides, carboxylic acid amides, Carboxylic esters, etc.
Die erfindungsgemäßen Verbindungen der Formel (II), (III), (IV), (V) und (VI) sowie die Verbindungen der Formel (I), (VII), (IX) und (X) besitzen chirale Zentren und können in jeder stereoisomeren Form vorliegen. Dies beinhaltet auch E/Z-Isomere bei Verbindungen der Formel (I). Die vorliegende Erfindung umfasst jegliche stereoisomere Formen oder deren Mischungen einer erfindungsgemäßen Verbindung oder Zielverbindung, wobei es bekannt ist, wie die optisch aktiven Formen (beispielsweise durch Auftrennung der racemischen Form durch Rekristallisationsverfahren, durch Synthese aus optisch aktiven Ausgangsmaterialien, durch chirale bzw. asymmetrische Synthese oder durch chromatographische Trennung mittels einer chiralen stationären Phase) erhalten werden können.The compounds of the formula (II) according to the invention, (III), (IV), (V) and (VI) and the compounds of the formula (I), (VII), (IX) and (X) have chiral centers and can in any stereoisomeric form. This also includes E / Z isomers in the case of compounds of the formula (I). The present invention includes any stereoisomeric forms or mixtures thereof of an inventive Compound or target compound, it being known how the optically active forms (for example, by resolution of the racemic Form by recrystallization process, by synthesis from optical active starting materials, by chiral or asymmetric synthesis or by chromatographic separation using a chiral stationary phase) can be obtained.
Die
in den jeweiligen Verbindungen vorhandenen funktionellen Gruppen,
beispielsweise Carboxy, Amino oder Hydroxy, können statt
in freier auch in geschützter Form vorliegen. Entsprechende
geeignete Schutzgruppen und ihre Einführung und Abspaltung
sind z. B. in Standardwerken, wie
Das erfindungsgemäße Verfahren umfaßt auch in diejenigen Ausführungsformen, bei denen Zwischenprodukte isoliert, Ausgangsstoffe und Reagenzien in situ hergestellt werden und/oder Zwischen- und Endprodukte ohne Isolierung weiterverarbeitet werden.The inventive method also includes in those embodiments where intermediates isolated, starting materials and reagents are prepared in situ and / or intermediate and final products processed without isolation become.
Die einzelnen Verfahrensstufen werden nach Standardmethoden, wie beispielsweise in den oben angegebenen Literaturstellen beschrieben, ggfs. in Gegenwart von Lösungsmitteln bei niedrigen Temperaturen, Raumtemperatur oder erhöhten Temperaturen, bevorzugt im Bereich des Siedepunkts des jeweiligen Lösungsmittels, bei Atmosphärendruck oder Überdruck, ggf. unter Inertgasatmosphäre durchgeführt.The individual process steps are determined by standard methods, such as in the references given above, if necessary. In the presence of solvents at low temperatures, room temperature or elevated temperatures, preferably in the range of the boiling point of the respective solvent, at atmospheric pressure or overpressure, if appropriate under an inert gas atmosphere carried out.
Geeignete Lösungsmittel sind Wasser und organische Lösungsmittel, die auch als Gemische von mindestens zwei Lösungsmitteln eingesetzt werden können.suitable Solvents are water and organic solvents, also as mixtures of at least two solvents can be used.
Beispiele für geeignete Lösungsmittel sind ggfs. halogenierte Kohlenwasserstoffe, wie Pentan, Hexan, Cyclohexan, Benzol, Toluol, Methylenchlorid, Chloroform, Tetrachlorethan oder Chlorbenzol; Ether, wie Diethylether, Dioxan oder Tetrahydrofuran; Carbonsäureester und Lactone, wie Essigsäuremethylester, Essigsäureethylester oder Valerolacton; N,N-substituierte Carbonsäureamide und Lactame, wie Dimethylformamid, Dimethylacetamid oder N-Methylpyrrolidon; Ketone, wie Aceton oder Cyclohexanon; Sulfoxide und Sulfone, wie Dimethylsulfoxid oder Dimethylsulfon; Alkohole, wie Methanol, Ethanol, Hexanol, Cyclohexanol, Benzylalkohol, Ethylenglycol, oder Propandiol; Nitrile, wie Acetonitril oder Propionitril; tertiäre Amine, wie Triethylamin, Diethylamin, Pyridin, N-Methylpyrrolidin oder N-Methylmorpholin; und organische Säuren, wie Essigsäure oder Ameisensäure.Examples for suitable solvents are optionally halogenated Hydrocarbons, such as pentane, hexane, cyclohexane, benzene, toluene, Methylene chloride, chloroform, tetrachloroethane or chlorobenzene; ethers, such as diethyl ether, dioxane or tetrahydrofuran; Carbonsäureester and lactones, such as methyl acetate, ethyl acetate or valerolactone; N, N-substituted carboxylic acid amides and Lactams, such as dimethylformamide, dimethylacetamide or N-methylpyrrolidone; Ketones, such as acetone or cyclohexanone; Sulfoxides and sulfones, such as dimethyl sulfoxide or dimethylsulfone; Alcohols, such as methanol, ethanol, hexanol, cyclohexanol, Benzyl alcohol, ethylene glycol, or propanediol; Nitriles, such as acetonitrile or propionitrile; tertiary amines, such as triethylamine, diethylamine, Pyridine, N-methylpyrrolidine or N-methylmorpholine; and organic Acids, such as acetic acid or formic acid.
Die Zielverbindungen können mittels bekannter Methoden isoliert werden, wie z. B. Extraktion, Kristallisation oder Filtration und deren Kombinationen.The Target compounds can be isolated by known methods be such. As extraction, crystallization or filtration and their combinations.
Die
nachfolgenden Beispiele dienen zur Illustration der Erfindung. Beispiel
1 
Zu
einer Lösung von N,N-Dimethylhydrazin (180 mg, 3.0 mmol)
in trockenem Toluol (1.5 ml) wird Trimethylaluminium (2 M in Toluol;
1.5 ml; 3.0 mmol) zugegeben und die Mischung wird bei 70°C
für 2 h gerührt. Dann wird trans-2-Isopropyl-7-[4-methoxy-3-(3-methoxy-propoxy)-benzoyl]-8-methyl-non-4-ensäure
diethylamid (476 mg; 0.97 mmol) zugegeben und die Mischung wird
für 12 h bei 90°C gerührt. Die Mischung
wird auf Raumtemperatur gebracht und eine gesättigte wässrige
Kochsalzlösung (10 ml) wird zugegeben. Die wässrige Phase
wird mit Toluol extrahiert. Die vereinigten organischen Phasen werden über
Na2SO4 getrocknet
und im Vakuum eingeengt. Der Rückstand wird über
Flash-Chromatographie an SiO2 (MTBE/Heptan
3:2 + 1% DEA) gereinigt. Es werden 454 mg (0.85 mmol; 88%) eines
grünen durchsichtigen Öls erhalten.
1H-NMR (250 MHz, CDCl3, Überschussisomer):
0.81-1.11 (m, 18H), 1.75-1.90 (m, 2H), 2.01-2.55 (m, 7H), 2.39 (s,
6H), 3.20-3.28 (m, 4H), 3.30-3.36 (m, 1H), 3.31 (s, 3H), 3.54 (app.
t, 2H, J = 6.2 Hz), 3.81 (s, 3H), 4.08 (app.t, 2H, J = 6.4 Hz),
5.26-5.40 (m, 2H), 6.78 (s, 1H), 7.01 (d, 1H, J = 2.0 Hz), 7.07
(d, 1H, J = 2.0 Hz) ppm. Ausgewählte Daten für
das Unterschussisomer: 2.30 (s, 6H), 3.53 (t, 1H, J = 6.2 Hz), 3.82
(s, 3H), 4.04 (t, 1H, J = 6.4 Hz), 6.75 (s, 1H), 6.85-6.87 (m, 1H),
7.01 (d, 1H, J = 2.0 Hz) ppm. Beispiel
2 
1 H-NMR (250 MHz, CDCl 3 , excess isomer): 0.81-1.11 (m, 18H), 1.75-1.90 (m, 2H), 2.01-2.55 (m, 7H), 2.39 (s, 6H), 3.20- 3.28 (m, 4H), 3.30-3.36 (m, 1H), 3.31 (s, 3H), 3.54 (app.t, 2H, J = 6.2 Hz), 3.81 (s, 3H), 4.08 (app.t, 2H, J = 6.4 Hz), 5.26-5.40 (m, 2H), 6.78 (s, 1H), 7.01 (d, 1H, J = 2.0 Hz), 7.07 (d, 1H, J = 2.0 Hz) ppm. Selected data for the deficient isomer: 2.30 (s, 6H), 3.53 (t, 1H, J = 6.2 Hz), 3.82 (s, 3H), 4.04 (t, 1H, J = 6.4 Hz), 6.75 (s, 1H) , 6.85-6.87 (m, 1H), 7.01 (d, 1H, J = 2.0 Hz) ppm. Example 2
Zu
einer Lösung von trans-2-Isopropyl-7-[4-methoxy-3-(3-methoxy-propoxy)-benzoyl]-8-methyl-non-4-ensäurediethylamid
(790 mg; 1.61 mmol) in trockenem THF (8 mL) wird wasserfreies Hydrazin
(8 mL; 1 M in THF) und Titan-tetraisopropoxid (2.76 g) zugegeben
und für 12 h bei 80°C gerührt. Die Mischung wird
auf Raumtemperatur gebracht, mit Dichlormethan (30 mL) verdünnt
und mit Wasser (4.82 g) versetzt. Der Niederschlag wird abfiltriert
und die erhaltene Mutterlauge wird im Vakuum eingeengt. Man erhält
die Titelverbindung als farbloses Öl (810 mg; quant.).
DC:
Rf (MTBE 1 + 1% DEA) = 0.32 und 0.37. 1H-NMR (250 MHz, CDCl3, Überschussisomer):
0.78-0.99 (m, 12H), 0.78-0.99 (m, 12H), 1.00-1.15 (m, 6H), 1.69-1.93
(m, 2H), 2.00-2.56 (m, 7H), 3.18-3.43 (m, 5H), 3.30 (s, 3H), 3.53
(app. t, 1H, J = 6.2 Hz), 3.83 (s, 3H), 4.07 (app. t, 1H, J = 6.6
Hz), 5.00 (bs, 2H), 5.25-5.53 (m, 2H), 6.69-7.11 (m, 3H) ppm. Ausgewählte
Daten für das Unterschussisomer: 3.30 (s, 3H), 3.52 (app.
t, 1H, J = 6.2 Hz), 3.82 (s, 3H), 4.04 (app. t, 1H, J = 6.5 Hz)
ppm. Beispiel
3 
DC: R f (MTBE 1 + 1% DEA) = 0.32 and 0.37. 1 H-NMR (250 MHz, CDCl 3 , excess isomer): 0.78-0.99 (m, 12H), 0.78-0.99 (m, 12H), 1.00-1.15 (m, 6H), 1.69-1.93 (m, 2H), 2.00-2.56 (m, 7H), 3.18-3.43 (m, 5H), 3.30 (s, 3H), 3.53 (app.t, 1H, J = 6.2 Hz), 3.83 (s, 3H), 4.07 (app. t, 1H, J = 6.6 Hz), 5.00 (bs, 2H), 5.25-5.53 (m, 2H), 6.69-7.11 (m, 3H) ppm. Selected data for the deficient isomer: 3.30 (s, 3H), 3.52 (app.t, 1H, J = 6.2 Hz), 3.82 (s, 3H), 4.04 (app.t, 1H, J = 6.5 Hz) ppm. Example 3
50
mg trans-7-(hydrazono-[4-methoxy-3-(3-methoxy-propoxy)-phenyl]-methyl)-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid (0.10 mmol) werden in 1 ml Methylenchlorid gelöst
und mit 24 mg (0.2 mmol) Phenylisocyanat versetzt und für
1 h bei RT gerührt. Die Mischung wird direkt durch Chromatographie
an SiO2 (Heptan/MTBE 1:2 + 1% DEA) gereinigt.
Man erhält das Harnstoffderivat als klares Öl.
1H-NMR (250 MHz, CDCl3,
ca. eine 1:1 Mischung von E/Z Isomeren und eine ca. 1:1 Mischung
von Rotameren): 0.78-1.21 (m, 36H), 1.71-1.93 (m, 4H), 1.95-2.61
(m, 14H), 3.17-3.44 (m, 10H), 3.29 (s, 1.5H), 3.31 (s, 1.5H), 3.32
(s, 1.5H), 3.34 (s, 1.5H), 3.53 (app. t, 2H, J = 6.1 Hz), 3.54 (app.
t, 2H, J = 6.1 Hz), 3.85 (s, 6H), 4.05 (app. t, 2H, J = 6.4 Hz),
4.13 (app. t, 2H, J = 6.5 Hz), 5.23-5.56 (m, 4H), 6.29 (bs, 2H),
6.64 (d, 1H, J = 1.8 Hz), 6.70 (dd, 1H, J = 1.9 Hz, J = 8.2 Hz),
6.83 (m, 1H), 6.89 (d, 1H, J = 8.2 Hz), 6.93-7.01 (m, 2H), 7.06
(dd, 1H, J = 1.9 Hz, J = 8.5 Hz), 7.15 (d, 1H, J = 2.0 Hz), 7.20-7.37
(m, 6H), 7.45 (m, 2H), 7.61 (bs, 0.5 H), 8.04 (bs, 0.5H), 8.18 (bs,
0.5H), 8.28 (bs, 0.5H) ppm Beispiel
4 
1 H-NMR (250 MHz, CDCl 3 , ca. 1: 1 mixture of E / Z isomers and ca. 1: 1 mixture of rotamers): 0.78-1.21 (m, 36H), 1.71-1.93 (m, 4H), 1.95-2.61 (m, 14H), 3.17-3.44 (m, 10H), 3.29 (s, 1.5H), 3.31 (s, 1.5H), 3.32 (s, 1.5H), 3.34 (s, 1.5 H), 3.53 (app.t, 2H, J = 6.1 Hz), 3.54 (app.t, 2H, J = 6.1 Hz), 3.85 (s, 6H), 4.05 (app.t, 2H, J = 6.4 Hz ), 4.13 (app.t, 2H, J = 6.5 Hz), 5.23-5.56 (m, 4H), 6.29 (bs, 2H), 6.64 (d, 1H, J = 1.8 Hz), 6.70 (dd, 1H, J = 1.9 Hz, J = 8.2 Hz), 6.83 (m, 1H), 6.89 (d, 1H, J = 8.2 Hz), 6.93-7.01 (m, 2H), 7.06 ( dd, 1H, J = 1.9 Hz, J = 8.5 Hz), 7.15 (d, 1H, J = 2.0 Hz), 7.20-7.37 (m, 6H), 7.45 (m, 2H), 7.61 (bs, 0.5 H) , 8.04 (bs, 0.5H), 8.18 (bs, 0.5H), 8.28 (bs, 0.5H) ppm Example 4
25
mg trans-7-(Hydrazono-[4-methoxy-3-(3-methoxy-propoxy)-phenyl]-methyl)-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid (50 μmol) werden in 125 μl Pyridin
gelöst und bei 0°C mit 15 mg Essigsäureanhydrid
versetzt. Die Mischung wird für 1 h gerührt und
danach direkt durch Chromatographie an SiO2 (Heptan/MTBE
1:2 + 1% DEA) gereinigt. Man erhält die isomeren Hydrazone
als farblose zähe Öle.
1H-NMR
(250 MHz, CDCl3, Überschussisomer):
0.76-0.93 (m, 12H), 0.99-1.20 (m, 6H), 1.68-1.88 (m, 2H), 2.00-2.40
(m, 7H), 2.24 (s, 3H), 3.17-3.29 (m, 4H), 3.31 (s, 3H), 3.34-3.44
(m, 1H), 3.53 (app. t, 2H, J = 6.1 Hz), 3.84 (s, 3H), 4.03 (app.
t, 2H, J = 6.4 Hz), 5.19-5.50 (m, 2H), 6.57 (d, 1H, J = 1.8 Hz),
6.63 (dd, 1H, J = 1.8 Hz, J = 8.2 Hz), 6.86 (d, 1H, J = 8.2 Hz),
8.29 (bs, 1H) ppm. Ausgewählte Daten für das Unterschussisomer: 2.31
(s, 3H), 3.54 (app. t, 2H, J = 6.1 Hz), 4.10 (app. t, 2 H, J = 6.5
Hz), 6.77 (d, 1H, J = 8.5 Hz), 7.08 (dd, 1H, J = 2.1 Hz, J = 8.5
Hz), 7.20 (d, 1H, J = 2.1 Hz), 8.58 (bs, 1H) ppm. Beispiel
5 
1 H-NMR (250 MHz, CDCl 3 , excess isomer): 0.76-0.93 (m, 12H), 0.99-1.20 (m, 6H), 1.68-1.88 (m, 2H), 2.00-2.40 (m, 7H), 2.24 (s, 3H), 3.17-3.29 (m, 4H), 3.31 (s, 3H), 3.34-3.44 (m, 1H), 3.53 (app.t, 2H, J = 6.1 Hz), 3.84 (s, 3H), 4.03 (app.t, 2H, J = 6.4 Hz), 5.19-5.50 (m, 2H), 6.57 (d, 1H, J = 1.8 Hz), 6.63 (dd, 1H, J = 1.8 Hz, J = 8.2 Hz), 6.86 (d, 1H, J = 8.2 Hz), 8.29 (bs, 1H) ppm. Selected data for the deficient isomer: 2.31 (s, 3H), 3.54 (app.t, 2H, J = 6.1 Hz), 4.10 (app.t, 2H, J = 6.5 Hz), 6.77 (d, 1H, J = 8.5 Hz), 7.08 (dd, 1H, J = 2.1 Hz, J = 8.5 Hz), 7.20 (d, 1H, J = 2.1 Hz), 8.58 (bs, 1H) ppm. Example 5
1.2
g trans-7-(Hydrazono-[4-methoxy-3-(3-methoxy-propoxy)-phenyl]-methyl)-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid wurden in 8 ml wasserfreiem Pyridin gelöst
und unter Eis/Salz-Kühlung langsam mit 512 mg Cbz-Cl versetzt.
Es wurde noch eine Stunde bei 0°C gerührt. Die
Lösung wurde mit 20 ml MTBE verdünnt und mit 9
ml konzentrierter Salzsäure und 36 ml Wasser hydrolisiert.
Die wässrige Phase wurde 2 mal mit 10 ml Toluol extrahiert
und die vereinigten organischen Phasen wurden über Na2SO4 getrocknet. Das
LM wurde unter Vakuum entfernt und der Rückstand durch
Chromatographie an SiO2 (MTBE/Heptan 2:1) gereinigt.
Es werden 1.25 g der geschützten Hydrazone als schwach
grüne Öle erhalten.
DC: Rf (MTBE/Heptan
2:1) = 0.19 und 0.14. 1H-NMR (250 MHz, CDCl3, Überschussisomer): 0.77-1.20
(m, 18H), 1.70-1.92 (m, 2H), 2.08-2.37 (m, 7H), 3.16-3.44 (m, 5H),
3.29 (s, 3H), 3.52 (app. t, 2H, J = 6.2 Hz), 3.83 (s, 3H), 4.02
(app. t, 2H, J = 6.5 Hz), 5.23-5.53 (m, 2H), 5.13 (bm, 2H), 6.58
(d, 1H, J = 1.8 Hz), 6.63 (dd, 1H, J = 1.8 Hz, J = 8.2 Hz), 6.87
(d, 1H, J = 8. 2 Hz), 7.25-7.34 (m, 5H), 7.70 (bs, 1H) ppm. Ausgewählte
Daten für das Unterschussisomer: 3.30 (s, 3H), 3.52 (app.
t, 2H, J = 6.3 Hz), 3.82 (s, 3H), 4.08 (app. t, 2H, J = 6.4 Hz), 5.22
(bm, 2 H), 6.74 (d, 1H, J = 8.4 Hz), 7.07 (dd, 1H, J = 2.0 Hz, J
= 8.4 Hz), 7.23 (d, 1H, J = 2.0 Hz), 8.05 (bs, 1H) ppm. HPLC (ODS-2
250/4.6 150/5 C18; ACN-Wasser; ACN:0-1 min 50%; 30 min 100%) Rt = 23.89 (35%) und 26.65 (65%). Beispiel
6 
TLC: R f (MTBE / heptane 2: 1) = 0.19 and 0.14. 1 H-NMR (250 MHz, CDCl 3 , excess isomer): 0.77-1.20 (m, 18H), 1.70-1.92 (m, 2H), 2.08-2.37 (m, 7H), 3.16-3.44 (m, 5H), 3.29 (s, 3H), 3.52 (app.t, 2H, J = 6.2 Hz), 3.83 (s, 3H), 4.02 (app.t, 2H, J = 6.5 Hz), 5.23-5.53 (m, 2H) , 5.13 (bm, 2H), 6.58 (d, 1H, J = 1.8 Hz), 6.63 (dd, 1H, J = 1.8 Hz, J = 8.2 Hz), 6.87 (d, 1H, J = 8. 2 Hz) , 7.25-7.34 (m, 5H), 7.70 (bs, 1H) ppm. Selected data for the deficient isomer: 3.30 (s, 3H), 3.52 (app.t, 2H, J = 6.3 Hz), 3.82 (s, 3H), 4.08 (app.t, 2H, J = 6.4 Hz), 5.22 (app. bm, 2 H), 6.74 (d, 1H, J = 8.4 Hz), 7.07 (dd, 1H, J = 2.0 Hz, J = 8.4 Hz), 7.23 (d, 1H, J = 2.0 Hz), 8.05 (bs, 1H) ppm. HPLC (ODS-2 250 / 4.6 150/5 C18; ACN water; ACN: 0-1 min 50%; 30 min 100%) R t = 23.89 (35%) and 26.65 (65%). Example 6
100
mg trans-7-(Hydrazono-[4-methoxy-3-(3-methoxy-propoxy)-phenyl]-methyl)-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid (0.2 mmol) in 220 mg Boc2O (0.5
mmol, 5 eq) werden zusammen geschmolzen und für 30 min
bei rt gerührt. Diese Mischung wird direkt durch Chromatographie
an SiO2 (Heptan/MTBE 1:2 + 1% DEA) gereinigt.
Man erhält 116 mg an den beiden geschützten Hydrazonen
als schwach grüne klare Öle.
DC: Rf (MTBE 1 + 1% DEA) = 0.60 und 0.52. 1H-NMR (250 MHz, CDCl3,
54:46 Mischung der Isomere, nicht zugeordnet): 0.75-1.20 (m, 18H),
1.48 (s, 9H), 1.69-1.86 (m, 2H), 1.98-2.45 (m, 7H), 3.18-3.42 (m,
5H), 3.30 (s, 3H), 3.52 (app. t, 2 H, J = 6.1 Hz), 3.84 (s, 3H),
4.08 (app. t, 2H, J = 6.4 Hz), 5.26-5.52 (m, 2H), 6.60 (d, 1H, J
= 1.7 Hz), 6.64 (dd, 1H, J = 1.7 Hz, J = 8.1 Hz), 6.88 (d, 1H, J
= 8.1 Hz), 7.85 (bs, 1H) ppm. Ausgewählte Daten für
das andere Isomer: 1.39 (s, 9H), 3.29 (s, 3H), 3.51 (app. t, 2H,
J = 6.3 Hz), 3.80 (s, 3H), 4.03 (app. t, 2H, J = 6.2 Hz), 7.05 (dd,
1H, J = 1.6 Hz, J = 8.4 Hz), 7.26 (d, 1H, J = 1.6 Hz), 6.71 (d,
1H, J = 8.4 Hz), 7.50 (bs, 1H) ppm. HPLC (ODS-2 250/4.6 150/5 C18;
ACN-Wasser; ACN:0-1 min 50%; 30 min 100%): Rt =
23.90 (46%) und 26.21 (54%). Beispiel
7 
DC: R f (MTBE 1 + 1% DEA) = 0.60 and 0.52. 1 H-NMR (250 MHz, CDCl 3 , 54:46 mixture of isomers, not assigned): 0.75-1.20 (m, 18H), 1.48 (s, 9H), 1.69-1.86 (m, 2H), 1.98-2.45 (m, 7H), 3.18-3.42 (m, 5H), 3.30 (s, 3H), 3.52 (app.t, 2H, J = 6.1 Hz), 3.84 (s, 3H), 4.08 (app. 2H, J = 6.4 Hz), 5.26-5.52 (m, 2H), 6.60 (d, 1H, J = 1.7 Hz), 6.64 (dd, 1H, J = 1.7 Hz, J = 8.1 Hz), 6.88 (d, 1H, J = 8.1 Hz), 7.85 (bs, 1H) ppm. Selected data for the other isomer: 1.39 (s, 9H), 3.29 (s, 3H), 3.51 (app.t, 2H, J = 6.3 Hz), 3.80 (s, 3H), 4.03 (app. T, 2H, J = 6.2 Hz), 7.05 (dd, 1H, J = 1.6 Hz, J = 8.4 Hz), 7.26 (d, 1H, J = 1.6 Hz), 6.71 (d, 1H, J = 8.4 Hz), 7.50 (bs , 1H) ppm. HPLC (ODS-2 250 / 4.6 150/5 C18; ACN water; ACN: 0-1 min 50%; 30 min 100%): R t = 23.90 (46%) and 26.21 (54%). Example 7
232
mg (400 μmol) des CBZ-geschützten Hydrazons (Verbindung
Ib, mit R4 = CO2tBu;
X = NEt2; R2 = Me;
R1 = (CH2)3OMe) wurden in 8 g F3COOH/THF
(1:1) vorgelegt und mittels Eis/Salz-Kühlung auf –20°C
gekühlt. Zu der tiefgrünen Lösung wurden
unter starkem Rühren 76 mg (2 mmol; 5 eq) NaBH4 portionsweise
gegeben. Die nun wieder farblose Lösung wurde noch für
20 min bei RT gerührt und dann unter Kühlen auf
20 ml einer 10%igen wässrigen NaOH-Lösung gegossen.
Es wurde 3-mal mit je 5 ml MTBE extrahiert, die organische Phase über
Na2SO4 getrocknet
und das Lösungsmittel im Vakuum entfernt. Durch Chromatographie
an SiO2 (MTBE/Heptan 1:1) wurden 245 mg
(96%) an den diastereomeren Hydrazinen als farblose hochviskose Öle
erhalten.
DC: Rf (MTBE/Heptan 2:1 +
0.5% DEA) = 0.25. 1H-NMR (250 MHz, CDCl3): 0.78-1.10 (m, 18H), 1.71-1.92 (m, 2H),
1.20-1.27 (m, 7H), 3.16-3.37 (m, 5H), 3.28 (s, 3H), 3.50 (app. t,
2H, J = 6.1 Hz), 3.78 (s, 3H), 3.88-3.97 (m, 1H), 4.01 (app. t,
2H, J = 6.4 Hz), 4.22-4.50 (bs, 1H), 4.95-5.50 (m, 4H), 6.03 (bs,
1H), 6.72 (m, 2H), 6.79 (m, 1H), 7.22-7.30 (m, 5H) ppm. Ms (FAB)
m/z = 640 (17) [M+H+], 474 (100) [M-C8H9N2O2]. HPLC (ODS-2 250/4.6 150/5 C18; ACN-Wasser;
ACN: 0–1 min 50%; 30 min 100%): Rt =
26.1 (68%) and 27.4 (32%). Beispiel
8 
TLC: R f (MTBE / heptane 2: 1 + 0.5% DEA) = 0.25. 1 H-NMR (250 MHz, CDCl 3 ): 0.78-1.10 (m, 18H), 1.71-1.92 (m, 2H), 1.20-1.27 (m, 7H), 3.16-3.37 (m, 5H), 3.28 ( s, 3H), 3.50 (app.t, 2H, J = 6.1 Hz), 3.78 (s, 3H), 3.88-3.97 (m, 1H), 4.01 (app.t, 2H, J = 6.4 Hz), 4.22 -4.50 (bs, 1H), 4.95-5.50 (m, 4H), 6.03 (bs, 1H), 6.72 (m, 2H), 6.79 (m, 1H), 7.22-7.30 (m, 5H) ppm. Ms (FAB) m / z = 640 (17) [M + H + ], 474 (100) [MC 8 H 9 N 2 O 2 ]. HPLC (ODS-2 250 / 4.6 150/5 C18; ACN water; ACN: 0-1 min 50%; 30 min 100%): R t = 26.1 (68%) and 27.4 (32%). Example 8
Zu
einer Lösung von 53 mg (100 μmol) trans-7-{N,N-Dimethyl-hydrazono-[4-methoxy-3-(3-methoxy-propoxy)-phenyl]-methyl}-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid in 1 g THF wird langsam 1 g F3CCOOH
zugetropft. Die nun tief violette Lösung wird auf –20°C
gekühlt und unter heftigem Rühren mit 19 mg (500 μmol)
NaBH4 versetzt. Die nun farblose Lösung
wird noch eine halbe Stunde bei 0°C gerührt. Die
Lösung wird vorsichtig mit 5 ml 10%ige NaOH-Lösung
hydrolisiert und mit MTBE extrahiert. Nach dem Trocknen (Na2SO4) wird das LM im
Vakuum entfernt und der Rückstand durch Chromatographie
an SiO2 (MTBE/Heptan 1:1 + 0.5% DEA) gereinigt.
Man erhielt 39 mg (73%) an dem Hydrazinderivat als klares zähes Öl.
DC:
Rf (MTBE/Heptan 1:1 + 0.5% DEA) = 0.34. 1H-NMR (250 MHz, CDCl3, Überschussisomer:
0.84-1.16 (m, 18H), 1.78-1.92 (m, 2H), 2.04-2.31 (m, 7H), 2.44 (s,
6H), 3.24-3.46 (m, 6H), 3.36 (s, 3H), 3.59 (app. t, 2H, J = 6.2
Hz), 3.70 (bs, 1H), 3.86 (s, 3H), 4.12 (app.t, 2H, J = 6.4 Hz),
5.31-5.41 (m, 2H), 6.78-7.05 (m, 3H) ppm. Ausgewählte Daten
für das Unterschussisomer: 3.58 (t, 1H, J = 6.2 Hz), 3.84
(s, 3H), 4.08 (t, 1H, J = 7.0 Hz). Beispiel
9 
TLC: R f (MTBE / heptane 1: 1 + 0.5% DEA) = 0.34. 1 H-NMR (250 MHz, CDCl 3 , excess isomer: 0.84-1.16 (m, 18H), 1.78-1.92 (m, 2H), 2.04-2.31 (m, 7H), 2.44 (s, 6H), 3.24-3.46 (m, 6H), 3.36 (s, 3H), 3.59 (app.t, 2H, J = 6.2 Hz), 3.70 (bs, 1H), 3.86 (s, 3H), 4.12 (app.t, 2H, J = 6.4 Hz), 5.31-5.41 (m, 2H), 6.78-7.05 (m, 3H) ppm Selected data for the deficient isomer: 3.58 (t, 1H, J = 6.2 Hz), 3.84 (s, 3H), 4.08 (t, 1H, J = 7.0 Hz) Example 9
120
mg (200 μmol) des mono-Boc-geschützten Hydrazons
(Verbindung Ib, mit R4 = CO2tBu;
X = NEt2; R2 = Me;
R1 = (CH2)3OMe) werden in 1 ml THF gelöst;
und mit 16 mg (240 μmol; 1.2 eq) NaCNBH3 versetzt. Nun
wird die Mischung unter starkem Rühren portionsweise mit
38.5 mg (240 μmol, 1.2 eq) p-TsOH·H2O
innerhalb einer Stunde versetzt. Es wird noch eine weitere Stunde
bei RT gerührt. Die Suspension wird mit 1 ml wässriger
NaOH-Lösung (1%ig) versetzt und 3 mal mit MTBE extrahiert.
Das LM wird getrocknet (Na2SO4)
und unter Vakuum entfernt. Der Rückstand wird durch Chromatographie
an SiO2 (MTBE/Heptan 2:1 + 0.5% DEA) gereinigt.
Man erhält das Boc-Hydrazin als Mischung der Diastereomere. Beispiel
10 
50
mg (100 μmol) des ungeschützten Hydrazons (Verbindung
Ia, mit X = NEt2; R2 =
Me; R1 = (CH2)3OMe) werden in 1 ml THF gelöst
und mit 16 mg (240 μmol; 2.4 eq) NaCNBH3 versetzt.
Nun wird unter starker Rühren portionsweise 38.5 mg (240 μmol,
1.2 eq) p-TsOH·H2O innerhalb einer
Stunde zugegeben. Es wird noch eine weitere Stunde bei RT gerührt.
Die Suspension wird mit 1 ml NaOH-Lösung (1%ig) versetzt
und 3 mal mit MTBE (2 ml) extrahiert. Das LM wird getrocknet (Na2SO4) und unter Vakuum
entfernt. Der Rückstand wird durch Chromatographie an SiO2 (MTBE/Heptan 2:1 + 0.5% DEA) gereinigt.
Man erhält das Hydrazin als Mischung der Diastereomeren. Beispiel
11 
60
mg (100 μmol) des mono-Boc-geschützten Hydrazons
(Verbindung Ib, mit R4 = CO2tBu;
X = NEt2; R2 = Me;
R1 = (CH2)3OMe), 15 mg Dimethylaminopyridin (120 μmol)
und 218 mg (1.0 mmol) Boc2O werden in 500 μl
Triethylamin gelöst und bei 40°C für
24 h gerührt. Die Lösung wird mit gesättigter
NH4Cl-Lösung (2 ml) versetzt und
mit MTBE extrahiert (3 mal 2 ml). Die organische Phase wird getrocknet
(Na2SO4) und das
LM im Vakuum entfernt. Nach Reinigung des Rückstandes durch
Chromatographie (SiO2, MTBE/Heptan 1:1)
wird das doppelt geschützte Hydrazon erhalten. Beispiel
12 
107
mg (200 μmol) des Dimethyl-Hydrazins (Verbindung II, mit
R3 = H; R4 = R5 = Me, X = NEt2;
R2 = Me; R1 = (CH2)3OMe), 1.5 mg Dimethylaminopyridin
(cat.) und 109 mg (500 μmol) Boc2O
werden in 1 ml Triethylamin gelöst und für 12
h bei RT gerührt. Die Lösung wird mit 2 ml ges.
NH4Cl-Lösung versetzt und mit MTBE (3
mal 5 ml) extrahiert. Nach dem Trocknen über Na2SO4 wird das LM
unter Vakuum entfernt und der Rückstand durch Chromatographie
(SiO2, Heptan/MTBE 1:1 + 0.5 DEA) gereinigt.
Man erhält das vierfachsubstituierte Hydrazin als Mischung
der Diastereomeren. Beispiel
13 
41
mg von trans-7-{bis-N',N'-Dimethyl-N-tert-butyloxycarbonyl-hydrazino-[4-methoxy-3-(3-methoxy-propoxy)-phenyl]-methyl}-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid wird zweiphasig mit 2 ml DCM/H2O
4:1 stark gerührt. Unter Kühlung (Eis/Salz) wird
portionsweise 14 mg NBS dazugegeben und für 2 h gerührt.
Die Lösung wird mit gesättigter Bisulfit-Lösung
(1 ml) versetzt und extrahiert (3 mal 2 ml MTBE). Die organische
Phase wird über Natriumsulfat getrocknet und im Vakuum
eingeengt. Nach Reinigung durch Säulenchromatographie wird
die Titelverbindung als Gemisch der Diastereomeren erhalten. Beispiel
14 
Das
Bromolacton (Verbindung III, mit R3 = CO2tBu; R4 = R5 = Me; Y = Br; R2 =
Me; R1 = (CH2)3OMe) wird in Essigsäureethylester/3
M HCl (1:1) zweiphasig für 10 min gerührt und
dann wird die wässrige Phase mit NaHCO3 auf
pH = 9 eingestellt. Die Lösung wird für weitere
12 h zweiphasig gerührt; dann wird mit MTBE extrahiert.
Die organischen Phasen werden über Na2SO4 getrocknet und das LM unter Vakuum entfernt.
Nach Chromatographie (SiO2) wird die Titelverbindung
erhalten. Beispiel
15 
20
mg des Pyrrolidinderivats (Verbindung IVa, mit R4 =
R5 = Me; R2 = Me;
R1 = (CH2)3OMe) wird in 1 ml EtOAc mit 10 mg Pd/C und
H2 in hydriert. Nach beendeter Wasserstoffaufnahme
wird vom Katalysator abfiltriert und das LM unter Vakuum entfernt.
Nach Chromatographie an SiO2 wird das Amin
erhalten. Beispiel
16 
52
mg 3-Isopropyl-5-(1-bromo-3-isopropyl-4-N',N'-Dimethyl-N-tert-butyloxycarbonyl-hydrazino-4-[4-methoxy-3-(3-methoxypropoxy)phenyl]-butyl)-dihydrofuran-2(3H)-on
(Verbindung III, mit R3 = CO2tBu;
R4 = R5 = Me; Y
= Br; R2 = Me; R1 =
(CH2)3OMe) in 0.5
ml DMPU und 20 mg NaN3 werden für
5 d bei 40°C gerührt. Es wird mit Wasser verdünnt
(20 ml) und mit MTBE extrahiert. Die organischen Phasen werden über
Na2SO4 getrocknet
und das LM unter Vakuum entfernt. Nach Chromatographie wird die
Titelverbindung erhalten. Beispiel
17 
40
mg 3-Isopropyl-5-(1-azido-3-isopropyl-4-N',N'-Dimethyl-N-tert-butyloxycarbonyl-hydrazino-4-[4-methoxy-3-(3-methoxypropoxy)phenyl]-butyl)-dihydrofuran-2(3H)-on
(Verbindung VI, mit R3 = CO2tBu;
R4 = R5 = Me; Y'
= N3; R2 = Me; R1 = (CH2)3OMe) wird in Ethanol (1 ml) mit einer Spatelspitze
Rainey-Nickel und H2 bei RT für
12 h hydriert. Nach beendeter Reaktion wird mit EtOH verdünnt,
vom Katalysator abfiltriert und unter Vakuum aufkonzentriert. Nach
säulenchromatographischer Reinigung wird die Titelverbindung
erhalten. Beispiel
18 
60
mg trans-7-{N,N-bis-tert-butyloxycarbonyl-hydrazono-[4-methoxy-3-(3-methoxypropoxy)-phenyl]-methyl}-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid (Verbindung I, mit R4 = R5= CO2tBu; X = NEt2; R2 = Me; R1 = (CH2)3OMe) werden in 1 ml THF gelöst
und mit 8 mg (120 μmol) NaCNBH3 versetzt.
Nun wird die Mischung unter starkem Rühren portionsweise
mit 19 mg (120 μmol, 1.2 eq) p-Toluolsulfonsäure·H2O innerhalb einer Stunde versetzt. Es wird
noch eine weitere Stunde bei RT gerührt. Die Suspension
wird mit 1 ml wässriger NaOH-Lösung (1%ig) versetzt
und 3 mal mit MTBE extrahiert. Die organische Phase wird getrocknet
(Na2SO4) und das
LM unter Vakuum entfernt. Der Rückstand wird durch Chromatographie
an SiO2 (MTBE/Heptan 2:1 + 0.5% DEA) gereinigt.
Man erhält die Titelverbindung als Mischung der Diastereomere. Beispiel
19 
61
mg trans-7-{N,N-bis-tert-butyloxycarbonyl-hydrazino-[4-methoxy-3-(3-methoxypropoxy)-phenyl]-methyl}-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid (Verbindung II, mit R3 = H;
R4 = R5 = CO2tBu; X = NEt2; R2 = Me; R1 = (CH2)3OMe) werden mit
34 mg Benzylbromid und einer Spatelspitze NaI in 500 μl
Aceton bei 40°C für 24 h gerührt. Die
Mischung wird mit ges. NaHCO3-Lösung
(2 ml) versetzt und extrahiert (MTBE, 3 mal 2 ml). Die organischen
Phasen werden über Na2SO4 getrocknet und unter Vakuum aufkonzentriert.
Nach chromatographischer Reinigung (SiO2)
wird das vierfachsubstituierte Hydrazinderivat erhalten. Beispiel
20 
45
mg trans-7-{N-Benzyl-N',N'-bis-tert-butyloxycarbonyl-hydrazino-[4-methoxy-3-(3-methoxy-propoxy)-phenyl]-methyl}-2-Isopropyl-8-methyl-non-4-ensäure
diethylamid (Verbindung II, mit R3 = CH2Ph; R4 = R5 = CO2tBu; X = NEt2; R2 = Me; R1 = (CH2)3OMe) wird zweiphasig mit 2 ml DCM/H2O (4:1) stark gerührt. Unter Kühlung
wird portionsweise NBS dazugegeben und für weitere 12 h
gerührt. Die Lösung wird mit gesättigter
wässriger Bisulfit-Lösung (1 ml) versetzt und
extrahiert (3 mal 2 ml MTBE). Die organischen Phasen werden über
Na2SO4 getrocknet
und unter Vakuum aufkonzentriert. Nach chromatographischer Reinigung
(SiO2) wird die Titelverbindung erhalten. Beispiel
21 
34
mg 3-Isopropyl-5-(1-bromo-3-isopropyl-4-(N-benzyl-N',N'-bis-tert-butyloxycarbonyl)-hydrazino-4-[4-methoxy-3-(3-methoxypropoxy)phenyl]-butyl)-dihydrofuran-2(3H)-on
(Verbindung III, mit R3 = Bn; R4 =
R5 = CO2tBu; Y =
Br; R2 = Me; R1 =
(CH2)3OMe) wird
in 2 ml CH2Cl2/F3COOH (10:1) gelöst und für
2 h bei RT gerührt. Das LM wird im Vakuum entfernt und
der Rückstand in THF (1 ml) aufgenommen. Es wird Triethylamin
(0.2 ml) hinzugegeben und für 3 h bei RT gerührt.
Die Lösung wird mit Wasser (2 ml) gewaschen, über
Na2SO4 getrocknet
und im Vakuum aufkonzentriert. Nach Chromatographie (SiO2) wird die Titelverbindung erhalten. Beispiel
22 
22
mg 5-(1-benzyl-5-isopropyl-6-[4-methoxy-3-(3-methoxypropoxy)phenyl]-hexahydropyridazin-3-yl)-3-isopropyl-dihydrofuran-2(3H)-on.
(Verbindung IVb, mit R3 = Bn; R4 =
H; R2 = Me; R1 =
(CH2)3OMe) wird
in EtOH (1 ml) mit einer Spatelspitze Rainey-Nickel und H2 bei RT für 16 h hydriert. Nach
beendeter Reaktion wird mit EtOH verdünnt, vom Katalysator
abfiltriert und unter Vakuum aufkonzentriert. Nach säulenchromatographischer
Reinigung wird die Titelverbindung erhalten. Beispiel
23 
Eine
Lösung aus 107 mg (200 μmol) des Dimethyl-Hydrazins
(Verbindung II, mit R3 = H; R4 =
R5 = Me; X = NEt2;
R2 = Me; R1 = (CH2)3OMe), 1.5 mg Dimethylaminopyridin (cat.)
und 42 mg Trifluoressigsäureanhydrid in 500 μl
Pyridin werden für 12 h unter Stickstoffatmosphäre
gerührt. Es wird mit gesättigter Kochsalzlösung
(2 ml) versetzt und mit MTBE extrahiert. Die vereinigten organischen
Phasen werden getrocknet (Na2SO4)
und das LM im Vakuum entfernt. Nach chromatographischer Reinigung
wird die Titelverbindung erhalten. Beispiel
24 
Zu
einer Lösung von 3-Isopropyl-5-(1-bromo-3-isopropyl-4-N',N'-Dimethyl-N-tert-butyloxycarbonyl-hydrazino-4-[4-methoxy-3-(3-methoxypropoxy)phenyl]-butyl)-dihydrofuran-2(3H)-on
(56 mg) in 1 ml Isopropanol wird tropfenweise 5 mg LiOH gelöst
in 100 μl H2O gegeben. Die Lösung
wird für 2 h gerührt und direkt weiterverwendet.
Zu dieser Lösung wird bei 0°C 300 μl
einer 1 N HCl-Lösung tropfenweise gegeben. Die Lösung
wird für 4 h gerührt, mit H2O
verdünnt, mit TBME extrahiert und über Na2SO4 getrocknet.
Nach Einengen im Vakuum erhält man die Titelverbindung. Beispiel
25 
Es
wird 19 mg m-Chlorperbenzoesäure auf einmal dazugegeben.
Es wird noch 30 min bei 0°C gerührt, mit ges.
NaHCO3-Lösung (1 ml) gewaschen, über
Na2SO4 getrocknet,
und im Vakuum aufkonzentriert. Nach Chromatographie kann unter anderem
die gewünschte Verbindung isoliert werden. Beispiel
26 
Es werden 20 mg Methansulfonamid und 280 mg AD-Mix-alpha in tert-Butanol (1 ml) und Wasser (1 ml) gelöst. Es wird auf 0°C gekühlt, dann 107 mg des Hydrazinderivates (Verbindung II, mit R3 = Boc; R4 = R5 = Me, X = NEt2; R2 = Me; R1 = (CH2)3OMe) dazugegeben und solange gerührt, bis das Startmaterial vollständig abreagiert hat (DC). Nach Chromatographie an SiO2 erhält man das Diol.Dissolve 20 mg of methanesulfonamide and 280 mg of AD-Mix-alpha in tert-butanol (1 ml) and water (1 ml). It is cooled to 0 ° C., then 107 mg of the hydrazine derivative (compound II, where R 3 = Boc; R 4 = R 5 = Me, X = NEt 2 ; R 2 = Me; R 1 = (CH 2 ) 3 OMe) was added and stirred until the starting material has completely reacted (TLC). After chromatography on SiO 2 , the diol is obtained.
Beispiel 27Example 27
(2S,7S)-trans-2-Isopropyl-7-[4-methoxy-3-(3-methoxy-propoxy)-benzoyl]-8-methylnon-4-ensäure (VII mit R1 = (CH2)3OMe und R2 = Me und X = OH)(2S, 7S) -trans-2-isopropyl-7- [4-methoxy-3- (3-methoxy-propoxy) -benzoyl] -8-methylnon-4-enoic acid (VII with R 1 = (CH 2 ) 3 OMe and R 2 = Me and X = OH)
Zu
einer gekühlten (–78°C) Lösung
von 4-Brom-1-methoxy-2-(3-methoxypropyloxy)-benzol (2.75 g; 10 mmol)
in trockenem THF (15 ml) wurde n-BuLi (4.5 ml; 12.5 M in Hexan)
zugetropft und die Reaktionsmischung für 30 min bei –78°C
gerührt. Danach wurde eine MgCl2-Lösung
(22.5 ml; 0.50 M in THF, frisch hergestellt) zugegeben, die Reaktionsmischung
für 20 min bei –78°C gerührt,
auf RT erwärmt und für weitere 30 min gerührt.
Diese Reaktionsmischung wurde innerhalb von 2 min zu einer –78°C
gekühlten Suspension von (2S,7S)-trans-2,7- Diisopropyl-oct-4-endionsäurechlorid
(2.93 g; 10 mmol) und Fe(acac)3 (128 mg,
360 μmol) in trockenem THF (15 ml) zugegeben. Die Reaktionsmischung
wurde bei –78°C für 5 min gerührt
und mit 10 ml Wasser und 2 ml HCl versetzt und auf RT auftauen lassen.
Die wässrige Schicht wurde 3× mit tert.-Butylmethylether
extrahiert und die organische Schicht über MgSO4 getrocknet und unter reduziertem Druck
aufkonzentriert. Der rohe Rückstand wurde durch Säulenchromatographie
an SiO2 gereinigt (Heptan, MTBE 2:1 + 0.5%
Essigsäure) unter Erhalt der Titelverbindung (IV) (2.0
g; 46% Ausbeute) als ein blassgelbem Öl. (2S,7S)-trans-2-Isopropyl-7-[4-methoxy-3-(3-methoxy-propoxy)-benzoyl]-8-methyl-non-4-ensäure
DC:
(Hexan/MTBE 1:1 + 0,5% AcOH): Rf = 0.30;
1H-NMR
(CDCl3, 400 MHz): δ = 0.90 (m,
12H); 1.81 (m, 1H); 1.98-2.29 (m, 6H); 2.45 (m, 1H); 3.21 (m, 1H); 3.38
(s, 3H); 3.59 (dd, J1 = J2 =
7Hz, 2H); 3.92 (s, 3 H); 4.18 (dd, J1 =
J2 = 7Hz, 2H); 5.38 (m, 2H); 6.89 (d, J
= 9Hz, 1H); 7.04 (m, 2H); 7.75 (bs, 1H).
13C-NMR
(CDCl3, 100,6 MHz): δ = 19.68;
19.75; 20.02; 21.19; 29.39; 29.56; 29.69; 30.49; 32.20; 32.39; 52.30; 56.01;
58.54; 66.18; 69.26; 110.46; 112.50; 122.67; 128.92; 130.10; 131.67;
148.48; 153.57; 179.60; 202.64.To a cooled (-78 ° C) solution of 4-bromo-1-methoxy-2- (3-methoxypropyloxy) benzene (2.75 g, 10 mmol) in dry THF (15 mL) was added n-BuLi (4.5 mL; 12.5 M in hexane) was added dropwise and the reaction mixture for 30 min at -78 ° C stirred. Thereafter, a MgCl 2 solution (22.5 ml, 0.50 M in THF, freshly prepared) was added, the reaction mixture stirred for 20 min at -78 ° C, warmed to RT and stirred for a further 30 min. This reaction mixture was added within 2 min to a -78 ° C cooled suspension of (2S, 7S) -trans-2,7-diisopropyl-oct-4-endioic acid chloride (2.93 g, 10 mmol) and Fe (acac) 3 (128 mg, 360 μmol) in dry THF (15 ml). The reaction mixture was stirred at -78 ° C for 5 min and treated with 10 ml of water and 2 ml of HCl and thawed to RT. The aqueous layer was extracted 3x with tert-butyl methyl ether and the organic layer was dried over MgSO 4 and concentrated under reduced pressure. The crude residue was purified by column chromatography on SiO 2 (heptane, MTBE 2: 1 + 0.5% acetic acid) to give the title compound (IV) (2.0 g, 46% yield) as a pale yellow oil. (2S, 7S) -trans-2-isopropyl-7- [4-methoxy-3- (3-methoxy-propoxy) -benzoyl] -8-methyl-non-4-enoic acid
TLC: (hexane / MTBE 1: 1 + 0.5% AcOH): Rf = 0.30;
1 H-NMR (CDCl3, 400 MHz): δ = 0.90 (m, 12H); 1.81 (m, 1H); 1.98-2.29 (m, 6H); 2.45 (m, 1H); 3.21 (m, 1H); 3.38 (s, 3H); 3.59 (dd, J 1 = J 2 = 7Hz, 2H); 3.92 (s, 3H); 4.18 (dd, J 1 = J 2 = 7Hz, 2H); 5.38 (m, 2H); 6.89 (d, J = 9Hz, 1H); 7.04 (m, 2H); 7.75 (bs, 1H).
13 C-NMR (CDCl 3 , 100.6 MHz): δ = 19.68; 19.75; 2.20; 21:19; 29.39; 29.56; 29.69; 30.49; 32.20; 32.39; 52.30; 56.01; 58.54; 66.18; 69.26; 110.46; 112.50; 122.67; 128.92; 130.10; 131.67; 148.48; 153.57; 179.60; 202.64.
Beispiel 28Example 28
Trennung meso/rac-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure mit PiperazinSeparation of meso / rac-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid with piperazine
Eine Mischung von meso/rac trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (59,6 g; meso/rac 54:46) und Piperazin (14.35 g) in EtOH (277 g) wird 4 h stark gerührt. Die erhaltenen Kristalle (meso-angereichertes Piperazinsalz) werden abfiltriert. Die Mutterlauge wird mit 100 ml 5%iger Salzsäure zweiphasig gerührt und 3 mal mit jeweils 100 ml MTBE extrahiert. Die vereinigten organischen Phasen werden über Na2SO4 getrocknet und das LM im Vakuum entfernt. Der Rückstand wird aus Isopropylacetat umkristallisiert (Umkristallisierungsschritt 1). Es werden 16,5 g an rac-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (HPLC: > 98.5%) als farblose Kristalle erhalten.A mixture of meso / rac trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (59.6 g; meso / rac 54:46) and piperazine (14.35 g) in EtOH (277 g). is stirred vigorously for 4 h. The resulting crystals (meso-enriched piperazine salt) are filtered off. The mother liquor is stirred in two phases with 100 ml of 5% strength hydrochloric acid and extracted 3 times with in each case 100 ml of MTBE. The combined organic phases are dried over Na 2 SO 4 and the LM removed in vacuo. The residue is recrystallized from isopropyl acetate (recrystallization step 1). There are obtained 16.5 g of rac-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (HPLC:> 98.5%) as colorless crystals.
Das meso-angereicherte Piperazinsalz wird zweiphasig mit 5%iger HCl und MTBE gerührt. Die organische Phase wird abgetrennt und über Na2SO4 getrocknet. Nach dem Entfernen des LM unter Vakuum wird der Rückstand und die Mutterlauge aus dem Umkristallisierungsschritt 1 (42 g gesamt) in 174 g Isopropylacetat suspendiert und für 12 h stark gerührt (Kristallisationsschritt 2). Der Feststoff (23,1 g; meso-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure, HPLC: > 98.5%) wird abfiltriert. Aus der Mutterlauge des Kristallisationsschritts 2 werden durch Entfernen des LM unter reduziertem Druck 19,3 g an meso/rac, trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (meso/rac 45:55) isoliert, welche erneut nach dem selben Prinzip aufgetrennt werden können.The meso-enriched piperazine salt is stirred in two phases with 5% HCl and MTBE. The organic phase is separated and dried over Na 2 SO 4 . After removal of the LM under vacuum, the residue and the mother liquor from the recrystallization step 1 (42 g total) are suspended in 174 g of isopropyl acetate and stirred vigorously for 12 h (crystallization step 2). The solid (23.1 g, meso-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid, HPLC:> 98.5%) is filtered off. 19.3 g of meso / rac, trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (meso / rac 45:55) are removed from the mother liquor of crystallization step 2 by removing the LM under reduced pressure. isolated, which can be separated again according to the same principle.
Beispiel 29Example 29
Trennung meso/rac-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure mittels EmulsionskristallisationSeparation of meso / rac-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid by emulsion crystallization
Eine Mischung von meso/rac trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (10 g; meso/rac 44:56) in Isobutanol/NMP/Wasser/NP10 (58:11:14:17; 10,4 g) wird bei 65°C homogenisiert. Nach Abkühlen auf 25°C werden Impfkristalle von rac-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (11 mg) zugegeben und die Mischung wird bei Raumtemperatur für 1 h gerührt. Die Kristalle werden abfiltriert, mit Isobutanol gewaschen und getrocknet. Man erhält 1,34 g rac-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (rac/meso: 94:6).A Mixture of meso / rac trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (10g, meso / rac 44:56) in isobutanol / NMP / water / NP10 (58: 11: 14: 17; 10.4 g) is homogenized at 65 ° C. After cooling At 25 ° C, seed crystals of rac-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (11 mg) is added and the mixture is stirred at room temperature for Stirred for 1 h. The crystals are filtered off, with isobutanol washed and dried. This gives 1.34 g of rac-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (rac / meso: 94: 6).
Zur Mutterlauge wird weiteres Ausgangsmaterial von meso/rac trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (1,3 g; meso/rac 44:56) zugegeben und die Mischung wird bei 65°C homogenisiert. Nach Abkühlen auf 25°C werden Impfkristalle von meso-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (10 mg) zugegeben und die Mischung wird bei Raumtemperatur für 1 h gerührt. Die Kristalle werden abfiltriert, mit Isobutanol gewaschen und getrocknet. Man erhält 1,5 g meso-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (rac/meso: 34:66). Die erhaltene Mutterlauge ist dann wieder leicht mit rac-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure angereichert und kann erneut im 1. Schritt eingesetzt werden. Die meso-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (1,5 g; rac/meso: 34:66) wird in 6g Isopropylacetat für 16h bei RT gerührt. Die Kristalle werden abfiltriert, mit Isopropylacetat gewaschen und getrocknet. Man erhält 0,48 g meso-trans-2,7-Diisopropyl-oct-4-en-1,8-dionsäure (rac/meso: 6:94). Aus der Mutterlauge wird nach Einengen im Vakuum 1,0 g der Disäure (rac/meso: 47:53) isoliert, welche in Schritt 1 wieder verwendet werden kann.To the mother liquor is added additional starting material of meso / rac trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (1.3 g, meso / rac 44:56) and the mixture becomes 65 ° C homogenized. After cooling At 25 ° C, seed crystals of meso-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (10 mg) are added and the mixture is stirred at room temperature for 1 h. The crystals are filtered off, washed with isobutanol and dried. This gives 1.5 g of meso-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (rac / meso: 34:66). The resulting mother liquor is then again slightly enriched with rac-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid and can be used again in the 1st step. The meso-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (1.5 g, rac / meso: 34:66) is stirred in 6 g of isopropyl acetate for 16 h at RT. The crystals are filtered off, washed with isopropyl acetate and dried. This gives 0.48 g of meso-trans-2,7-diisopropyl-oct-4-ene-1,8-dionic acid (rac / meso: 6:94). After concentration in vacuo, 1.0 g of the diacid (rac / meso: 47:53) is isolated from the mother liquor, which can be reused in step 1.
ZITATE ENTHALTEN IN DER BESCHREIBUNGQUOTES INCLUDE IN THE DESCRIPTION
Diese Liste der vom Anmelder aufgeführten Dokumente wurde automatisiert erzeugt und ist ausschließlich zur besseren Information des Lesers aufgenommen. Die Liste ist nicht Bestandteil der deutschen Patent- bzw. Gebrauchsmusteranmeldung. Das DPMA übernimmt keinerlei Haftung für etwaige Fehler oder Auslassungen.This list The documents listed by the applicant have been automated generated and is solely for better information recorded by the reader. The list is not part of the German Patent or utility model application. The DPMA takes over no liability for any errors or omissions.
Zitierte PatentliteraturCited patent literature
- - WO 02/02508 [0003] - WO 02/02508 [0003]
- - WO 02/08172 [0003] WO 02/08172 [0003]
- - WO 01/09083 [0003] - WO 01/09083 [0003]
- - WO 02/02487 [0003] - WO 02/02487 [0003]
- - WO 02/02500 [0003] - WO 02/02500 [0003]
- - WO 01/09079 [0003] WO 01/09079 [0003]
- - WO 02/092828 [0003] WO 02/092828 [0003]
- - WO 2006/024501 [0004] WO 2006/024501 [0004]
- - EP 0678503 [0005, 0021, 0023, 0025, 0029] EP 0678503 [0005, 0021, 0023, 0025, 0029]
- - WO 2006/131304 [0023] WO 2006/131304 [0023]
- - EP 0258183 [0027, 0029, 0032] EP 0258183 [0027, 0029, 0032]
- - US 6730798 B2 [0033] - US 6730798 B2 [0033]
- - WO 2007/048620 [0054, 0057] - WO 2007/048620 [0054, 0057]
- - US 6428583 [0058] - US 6428583 [0058]
Zitierte Nicht-PatentliteraturCited non-patent literature
- - J. March, Advanced Organic Chemistry, John Wiley & Sons, 1992 oder W. Carruthers, Some modern methods of organic synthesis, Cambridge University Press, 3rd edition, 1986 [0012] - J. March, Advanced Organic Chemistry, John Wiley & Sons, 1992 or W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd edition, 1986 [0012]
- - Newbold, B.T. in Patai The Chemistry of the Hydrazo, Azo and Azoxy Groups, pt. 1, Wiley: NY, 1975, 2–629 [0015] Newbold, BT in Patai The Chemistry of the Hydrazo, Azo and Azoxy Groups, pt. 1, Wiley: NY, 1975, 2-629 [0015]
- - W. Carruthers, Some modern methods of organic synthesis, Cambridge University Press, 3rd edition, 1986, Seiten 440–450 [0017] - W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd edition, 1986, pp. 440-450 [0017]
- - H.P. Latscha, H.A. Klein "Organische Chemie", 4. Auflage, Springer-Verlag, Berlin, 1997 [0021] - HP Latscha, HA Klein "Organic Chemistry", 4th edition, Springer-Verlag, Berlin, 1997 [0021]
- - S. Weinreb, Org. Synthesis, VI, S. 49, 1988 [0026] - S. Weinreb, Org. Synthesis, VI, p. 49, 1988 [0026]
- - W. Carruthers, Some modern methods of organic synthesis, Cambridge University Press, 3rd edition, 1986, Seiten 374–377 [0034] - W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd edition, 1986, pp. 374-377 [0034]
- - Shi et al. J. Am. Chem. Soc. 1997, 199, 11224–11235 [0034] - Shi et al. J. Am. Chem. Soc. 1997, 199, 11224-11235 [0034]
- - Sharpless et al. Chem. Rev. 1994, 94, 2483–2547 [0034] Sharpless et al. Chem. Rev. 1994, 94, 2483-2547 [0034]
- - J.F.W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London und New York 1973 [0035] - JFW McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973 [0035]
- - Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981 [0035] Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981. [0035]
- - "The Peptides"; Band 3 (Herausg. E. Gross und J. Meienhofer), Academic Press, London und New York 1981 [0035] - "The Peptides"; Volume 3 (eds. E. Gross and J. Meienhofer), Academic Press, London and New York 1981. [0035]
- - "Methoden der organischen Chemie", Houben-Weyl, 4. Auflage, Bd. 15/I, Georg Thieme Verlag, Stuttgart 1974 [0035] - "Methods of Organic Chemistry", Houben-Weyl, 4th Edition, Vol. 15 / I, Georg Thieme Verlag, Stuttgart 1974 [0035]
- - Acct. Chem. Res 14, 95 (1981); J. Org Chem. 55, 5867 (1990) [0037] - Acct. Chem. Res. 14, 95 (1981); J. Org Chem. 55, 5867 (1990) [0037]
- - Kosers Reagenz; siehe dazu Koser, G.F., Aldrichimica Acta, Vol. 34, No.3 (2001) [0039] Koser's reagent; see Koser, GF, Aldrichimica Acta, Vol. 34, No.3 (2001) [0039]
- - S.91; Moriarty et al., Synlett 1990, 365 [0039] - p.91; Moriarty et al., Synlett 1990, 365 [0039]
- - Zhdankin, V.V. et al., Chem. Rev. 2002, 102, 2523–84 [0039] - Zhdankin, VV et al., Chem. Rev. 2002, 102, 2523-84 [0039]
- - B.Bildstein et al., Synthesis, 1994, 158–160 [0042] B.Bildstein et al., Synthesis, 1994, 158-160 [0042]
- - J.F.W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London und New York 1973 [0045] - JFW McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973 [0045]
- - in Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981 [0045] Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981. [0045]
- - "The Peptides"; Band 3 (Herausg. E. Gross und J. Meienhofer), Academic Press, London und New York 1981 [0045] - "The Peptides"; Volume 3 (eds. E. Gross and J. Meienhofer), Academic Press, London and New York 1981. [0045]
- - "Methoden der organischen Chemie", Houben-Weyl, 4. Auflage, Bd. 15/I, Georg Thieme Verlag, Stuttgart 1974 [0045] - "Methods of Organic Chemistry", Houben-Weyl, 4th Edition, Vol. 15 / I, Georg Thieme Verlag, Stuttgart 1974 [0045]
- - R.Calabretta et al. Synthesis 1991, 536–539 [0047] R. Calabretta et al. Synthesis 1991, 536-539 [0047]
- - R. Karl Dieter, Tetrahedron 55 (1999) 4177–4236 [0056] R. Karl Dieter, Tetrahedron 55 (1999) 4177-4236 [0056]
- - B. Scheiper et al, J. Org. Chem. 2004, 69, 3943–3949 [0056] Scheiper, et al, J. Org. Chem. 2004, 69, 3943-3949. [0056]
- - J.F.W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London und New York 1973 [0070] - JFW McOmie, "Protective Groups in Organic Chemistry", Plenary Press, London and New York 1973 [0070]
- - Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981 [0070] Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981. [0070]
- - "The Peptides"; Band 3 (Herausg. E. Gross und J. Meienhofer), Academic Press, London und New York 1981 [0070] - "The Peptides"; Volume 3 (eds. E. Gross and J. Meienhofer), Academic Press, London and New York 1981 [0070]
- - "Methoden der organischen Chemie", Houben-Weyl, 4. Auflage, Bd. 15/I, Georg Thieme Verlag, Stuttgart 1974 [0070] - "Methods of Organic Chemistry", Houben-Weyl, 4th Edition, Vol. 15 / I, Georg Thieme Verlag, Stuttgart 1974 [0070]
Claims (18)






























Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102007049039A DE102007049039A1 (en) | 2007-10-11 | 2007-10-11 | Process for the preparation of 8-hydrazino-8-aryl-octanoyl derivatives and their use |
| PCT/EP2008/008597 WO2009049837A1 (en) | 2007-10-11 | 2008-10-10 | Process for preparing 8-hydrazino-8-aryloctanoyl derivatives and their use |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102007049039A DE102007049039A1 (en) | 2007-10-11 | 2007-10-11 | Process for the preparation of 8-hydrazino-8-aryl-octanoyl derivatives and their use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| DE102007049039A1 true DE102007049039A1 (en) | 2009-04-16 |
Family
ID=40340645
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE102007049039A Withdrawn DE102007049039A1 (en) | 2007-10-11 | 2007-10-11 | Process for the preparation of 8-hydrazino-8-aryl-octanoyl derivatives and their use |
Country Status (2)
| Country | Link |
|---|---|
| DE (1) | DE102007049039A1 (en) |
| WO (1) | WO2009049837A1 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011051853A1 (en) * | 2009-10-29 | 2011-05-05 | CarboDesign LLC | Manufacturing process for preparing enaniomerically pure 8- aryloctanoic acid derivatives such as aliskiren |
| US8703976B2 (en) | 2011-10-02 | 2014-04-22 | Milan Soukup | Manufacturing process for 8-aryloctanoic acids such as Aliskiren |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0258183A2 (en) | 1986-08-13 | 1988-03-02 | Ciba-Geigy Ag | Process for the preparation of 5-amino-4-hydroxyvaleric-acid derivatives |
| EP0678503A1 (en) | 1994-04-18 | 1995-10-25 | Ciba-Geigy Ag | Delta-amino-gamma-hydroxy-omega-aryl alkanoic acid amides with enzyme especially renin inhibiting activities |
| WO2001009079A1 (en) | 1999-07-29 | 2001-02-08 | Speedel Pharma Ag | 2-alkyl-5-halogen-pent-4-ene carboxylic acids and their production |
| WO2002002500A1 (en) | 2000-07-03 | 2002-01-10 | Speedel Pharma Ag | Preparation of (r)-2-alkyl-3-phenylpropionic acids |
| WO2002002487A1 (en) | 2000-07-03 | 2002-01-10 | Speedel Pharma Ag | Process for the preparation of (r)-2-alkyl-3-phenyl-1-propanols |
| WO2002002508A1 (en) | 2000-07-05 | 2002-01-10 | Speedel Pharma Ag | Process for the preparation of substituted octanoyl amides |
| WO2002008172A1 (en) | 2000-07-25 | 2002-01-31 | Speedel Pharma Ag | Process for the preparation of substituted octanoyl amides |
| US6428583B1 (en) | 1997-09-06 | 2002-08-06 | Reuter Chemische Apparatebau Kg | Separation process |
| WO2002092828A2 (en) | 2001-05-15 | 2002-11-21 | Speedel Pharma Ag | Process for the preparation of substituted carboxylic acid estersby enzymatic hydrolysis |
| WO2006024501A1 (en) | 2004-08-31 | 2006-03-09 | Novartis Ag | Alternative synthesis of renin inhibitors and intermediates thereof |
| WO2006131304A2 (en) | 2005-06-08 | 2006-12-14 | Novartis Ag | Process for preparing renin inhibitors |
| WO2007048620A1 (en) | 2005-10-28 | 2007-05-03 | Reuter Chemischer Apparatebau Kg | Process for preparing octenoic acid derivatives |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0521083D0 (en) * | 2005-10-17 | 2005-11-23 | Novartis Ag | Organic compounds |
| DE102006017681A1 (en) * | 2006-04-12 | 2007-10-18 | Reuter Chemischer Apparatebau Kg | Process for the preparation of 8-aryl-octanoyl derivatives |
-
2007
- 2007-10-11 DE DE102007049039A patent/DE102007049039A1/en not_active Withdrawn
-
2008
- 2008-10-10 WO PCT/EP2008/008597 patent/WO2009049837A1/en active Application Filing
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0258183A2 (en) | 1986-08-13 | 1988-03-02 | Ciba-Geigy Ag | Process for the preparation of 5-amino-4-hydroxyvaleric-acid derivatives |
| EP0678503A1 (en) | 1994-04-18 | 1995-10-25 | Ciba-Geigy Ag | Delta-amino-gamma-hydroxy-omega-aryl alkanoic acid amides with enzyme especially renin inhibiting activities |
| US6428583B1 (en) | 1997-09-06 | 2002-08-06 | Reuter Chemische Apparatebau Kg | Separation process |
| WO2001009079A1 (en) | 1999-07-29 | 2001-02-08 | Speedel Pharma Ag | 2-alkyl-5-halogen-pent-4-ene carboxylic acids and their production |
| WO2001009083A1 (en) | 1999-07-29 | 2001-02-08 | Speedel Pharma Ag | Production of n-substituted 2,7-dialkyl-4-hydroxy-5-amino-8-aryl-octanoylamides |
| WO2002002500A1 (en) | 2000-07-03 | 2002-01-10 | Speedel Pharma Ag | Preparation of (r)-2-alkyl-3-phenylpropionic acids |
| WO2002002487A1 (en) | 2000-07-03 | 2002-01-10 | Speedel Pharma Ag | Process for the preparation of (r)-2-alkyl-3-phenyl-1-propanols |
| WO2002002508A1 (en) | 2000-07-05 | 2002-01-10 | Speedel Pharma Ag | Process for the preparation of substituted octanoyl amides |
| US6730798B2 (en) | 2000-07-05 | 2004-05-04 | Speedel Pharma Ag | Process for the preparation of substituted octanoyl amides |
| WO2002008172A1 (en) | 2000-07-25 | 2002-01-31 | Speedel Pharma Ag | Process for the preparation of substituted octanoyl amides |
| WO2002092828A2 (en) | 2001-05-15 | 2002-11-21 | Speedel Pharma Ag | Process for the preparation of substituted carboxylic acid estersby enzymatic hydrolysis |
| WO2006024501A1 (en) | 2004-08-31 | 2006-03-09 | Novartis Ag | Alternative synthesis of renin inhibitors and intermediates thereof |
| WO2006131304A2 (en) | 2005-06-08 | 2006-12-14 | Novartis Ag | Process for preparing renin inhibitors |
| WO2007048620A1 (en) | 2005-10-28 | 2007-05-03 | Reuter Chemischer Apparatebau Kg | Process for preparing octenoic acid derivatives |
Non-Patent Citations (21)
| Title |
|---|
| "Methoden der organischen Chemie", Houben-Weyl, 4. Auflage, Bd. 15/I, Georg Thieme Verlag, Stuttgart 1974 |
| "The Peptides"; Band 3 (Herausg. E. Gross und J. Meienhofer), Academic Press, London und New York 1981 |
| Acct. Chem. Res 14, 95 (1981); J. Org Chem. 55, 5867 (1990) |
| B. Scheiper et al, J. Org. Chem. 2004, 69, 3943-3949 |
| B.Bildstein et al., Synthesis, 1994, 158-160 |
| H.P. Latscha, H.A. Klein "Organische Chemie", 4. Auflage, Springer-Verlag, Berlin, 1997 |
| in Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981 |
| J. March, Advanced Organic Chemistry, John Wiley & Sons, 1992 oder W. Carruthers, Some modern methods of organic synthesis, Cambridge University Press, 3rd edition, 1986 |
| J.F.W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London und New York 1973 |
| Kosers Reagenz; siehe dazu Koser, G.F., Aldrichimica Acta, Vol. 34, No.3 (2001) |
| Newbold, B.T. in Patai The Chemistry of the Hydrazo, Azo and Azoxy Groups, pt. 1, Wiley: NY, 1975, 2-629 |
| R. Karl Dieter, Tetrahedron 55 (1999) 4177-4236 |
| R.Calabretta et al. Synthesis 1991, 536-539 |
| S. Weinreb, Org. Synthesis, VI, S. 49, 1988 |
| S.91; Moriarty et al., Synlett 1990, 365 |
| Sharpless et al. Chem. Rev. 1994, 94, 2483-2547 |
| Shi et al. J. Am. Chem. Soc. 1997, 199, 11224-11235 |
| Th. W. Greene, "Protective Groups in Organic Synthesis", Wiley, New York 1981 |
| W. Carruthers, Some modern methods of organic synthesis, Cambridge University Press, 3rd edition, 1986, Seiten 374-377 |
| W. Carruthers, Some modern methods of organic synthesis, Cambridge University Press, 3rd edition, 1986, Seiten 440-450 |
| Zhdankin, V.V. et al., Chem. Rev. 2002, 102, 2523-84 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009049837A1 (en) | 2009-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6061158B2 (en) | Synthesis intermediate of 6- (7-((1-aminocyclopropyl) methoxy) -6-methoxyquinolin-4-yloxy) -N-methyl-1-naphthamide, or a pharmaceutically acceptable salt thereof, and its use | |
| EP3421470A1 (en) | Substituted 5-fluoro-1h-pyrazolopyridines in crystalline form | |
| BR112020001457A2 (en) | intermediates useful for the synthesis of a selective inhibitor against protein kinase and processes for preparing them | |
| EP1669346A1 (en) | Process for the preparation of carbamic acid derivatives | |
| DE102004041531A1 (en) | Process for the preparation of biphenylamines | |
| DE102007049039A1 (en) | Process for the preparation of 8-hydrazino-8-aryl-octanoyl derivatives and their use | |
| NZ231108A (en) | Preparation of 1,4-diazabicyclo(3.2.2)nonane and intermediate compounds used therein | |
| DE60307109T2 (en) | PROCESS FOR THE PRODUCTION OF ZOLMITRIPTAN COMPOUNDS | |
| HUE033534T2 (en) | Asymmetric synthesis of a substituted pyrrolidine-2-carboxamide | |
| EP3617181A1 (en) | Synthesis of trans-2-phenylcyclopropylamine or a salt or solvate thereof | |
| WO2007118681A1 (en) | Method for the production of 8-aryl-octanoyl derivatives | |
| JP2009242244A (en) | Method for producing pyrazine derivative and intermediate of the same | |
| CN108147996B (en) | Synthetic method of arylmethylene bispyrazole ester monopotassium salt | |
| DE60031299T2 (en) | Process for the industrial production of (aminomethyl) trifluoromethylcarbinol derivatives | |
| DE2263527B2 (en) | 2,2-Disubstituted phenylacetonitrile derivatives, processes for their preparation and their use | |
| EP1833814B1 (en) | Method for the production of substituted 2-alkoxycarbonyl-3-aminothiophenes | |
| EP1347977B1 (en) | Method for the production of 2-(2-ethoxyphenyl)-substituted imidazotriazinones | |
| EP0440582A1 (en) | Process for preparing O-substituted hydroxylamines | |
| DE60308170T2 (en) | PROCESS FOR THE PREPARATION OF CHINOLINE DERIVATIVES | |
| DE2351556C2 (en) | Process for the preparation of N-haloformyl-carbamic acid halides | |
| DE69106316T2 (en) | Process for the preparation of an aliphatic amide and salts thereof. | |
| TW201700458A (en) | Method for producing dicarboxylic acid compound | |
| KR100488444B1 (en) | Quinolone derivatives and a preparation method thereof | |
| DE3815046A1 (en) | 3-CHLORINE-2-METHYLPHENETHYLAMINE DERIVATIVES | |
| WO2022218733A1 (en) | A process for preparation of substituted enamine compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| R119 | Application deemed withdrawn, or ip right lapsed, due to non-payment of renewal fee |
Effective date: 20110502 |