CN1805965A - Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity - Google Patents
Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity Download PDFInfo
- Publication number
- CN1805965A CN1805965A CNA2004800165371A CN200480016537A CN1805965A CN 1805965 A CN1805965 A CN 1805965A CN A2004800165371 A CNA2004800165371 A CN A2004800165371A CN 200480016537 A CN200480016537 A CN 200480016537A CN 1805965 A CN1805965 A CN 1805965A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- compound
- hydroxyl
- cor
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/08—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/10—Anthelmintics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/08—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Abstract
The present invention is directed to compounds of formula (I), wherein R1, R2, R3, R4, R5, R6, R7, R8, R9, X, n and the broken lines are as defined herein which are useful as modulators of chemokine receptor activity. In particular, these compounds are useful as modulators of the chemokine receptor CCR-2.
Description
Background of invention
Chemokine belongs to (70-120 amino acid) pro-inflammatory cytokine of the small molecular weight of a family, has effective chemotactic activity.Chemokine is the chemotactic cytokine that many different cells discharge, with attract various cells (for example monocyte, scavenger cell, T cell, eosinocyte, basophilic leukocyte and neutrophil) to the inflammation position (summary is referring to Schall,
Cytokine, 3,165-183 (1991) and Murphy,
Rev.Immun., 12,593-633 (1994)).These molecules are at first by four conservative halfcystines definition, are divided into two subfamilies according to the arrangement of two halfcystines of first pair.In CXC chemokine family (comprising IL-8, GRO α, NAP-2 and IP-10), these two halfcystines are separated by an amino acid, and in CC-chemokine family (comprising RANTES, MCP-1, MCP-2, MCP-3, MIP-1 α, MIP-1 β and eosinophil chemotactic protein), these two residues are adjacent.
The main chemotactic neutrophil of α-chemokine (for example interleukin-8 (IL-8), neutrophil activator-2 (NAP-2) and melanoma growth-stimulating activity albumen (MGSA)), and beta-chemokine (for example RANTES, MIP-1 α, MIP-1 β, MCP-1 (MCP-1), MCP-2, MCP-3 and eosinophil chemotactic protein) chemotactic scavenger cell, monocyte, T cell, eosinocyte and basophilic leukocyte (Deng etc.
Nature, 381,661-666 (1996)).
Chemokine is by many dissimilar emiocytosises, in conjunction with the specificity g protein coupled receptor (GPCR) of white corpuscle and other cell (summary is referring to Horuk,
Trends Pharm. Sci., 15,159-165 (1994)).These Chemokine Receptors constitute the subfamily of GPCR, and at present, this subfamily comprises member and many orphans of 15 signs.Different with the acceptor that mixes chemoattractant (for example C5a, fMLP, PAF and LTB4), Chemokine Receptors is expressed on the white corpuscle subclass higher selectivity.Therefore, the generation of specificity chemokine provide raise the particular leukocyte subclass mechanism.
In conjunction with their cognate ligand the time, Chemokine Receptors by relevant trimerization G protein transduction, causes intracellular calcium concentration to improve fast signal in the cell.Have 7 kinds at least in conjunction with beta-chemokine or to its responsive people's acceptor, concrete characteristic formp is as follows: CCR-1 (or " CKR-1 " or " CC-CKR-1 ") [MIP-1 α, MIP-1 β, MCP-3, RANTES] (Ben-Barruch etc.,
J.Biol.Chem., 270,22123-22128 (1995); Beote etc.,
Cell, 72,415-425 (1993)); CCR-2A and CCR-2B (or " CKR-2A "/" CKR-2A " or " CC-CKR-2A " or " CC-CKR-2A ") [MCP-1, MCP-2, MCP-3, MCP-4]; CCR-3 (or " CKR-3 " or " CC-CKR-3 ") [Eotaxin, Eotaxin 2, RANTES, MCP-2, MCP-3] (Rollins etc.,
Blood, 90,908-928 (1997)); CCR-4 (or " CKR-4 " or " CC-CKR-4 ") [MIP-1 α, RANTES, MCP-1] (Rollins etc.,
Blood, 90,908-928 (1997)); CCR-5 (or " CKR-5 " or " CC-CKR-5 ") [MIP-1 α, RANTES, MIP-1 β] (Sanson etc.,
Biochemistry, 35,3362-3367 (1996)); Duffy blood group antigen [RANTES, MCP-1] (Chaudhun etc.,
J.Biol.Chem., 269,7835-7838 (1994)).Beta-chemokine also comprises eosinophil chemotactic protein, MIP (" macrophage inflammatory protein "), MCP (" MCP ") and RANTES (" regulate and activate the normal T cell expressing and the excretory factor ").
Chemokine Receptors (for example CCR-1, CCR-2, CCR-2A, CCR-2B, CCR-3, CCR-4, CCR-5, CXCR-3, CXCR-4) relates to as inflammatory immunomodulatory illness and disease (comprising asthma, rhinitis and anaphylactic disease) and autoimmunity pathology (for example rheumatoid arthritis) and atherosclerotic important medium.In the CCR-5 gene people of 32 base pair homozygosity disappearance as if to the susceptibility of rheumatoid arthritis less (Gomez etc.,
Arthritis ﹠amp; Rheumatism, 42,989-992 (1999)).Relevant eosinocyte in the summary of the effect of alterative inflammation referring to Kita, H. etc.,
J.Exp.Med.183,2421-2426 (1996).Relevant chemokine in the summary of the effect of alterative inflammation referring to Lustger, A.D.
New England J.Med., 338 (7), 426-445 (1998).
A chemokine subclass is effective chemoattractant of monocyte and scavenger cell.What characterize fullest is MCP-1 (MCP-1), and its principal recipient is CCR2.The different cell type response inflammatory stimulus things of the various species of MCP-1 (comprising rodent and people) produce, and stimulate the chemotaxis of monocyte, lymphocyte subclass.Specifically, the generation of MCP-1 is soaked into relevant with monocyte and scavenger cell in the inflammation position.Carry out homologous recombination mouse and make MCP-1 or CCR2 disappearance, cause responding the monocyte recruitement that thioglycollate injection and monocytosis Lee Salmonella (Listeria monocytogenes) inject significantly weaken (Lu etc.,
J.Exp.Med., 187,601-608 (1998); Kurihara etc.,
J.Exp.Med., 186,1757-1762 (1997); Boring etc.,
J.Clin.Invest., 100,2552-2561 (1997); Kuziel etc.,
Proc.Natl.Acad.Sci., 94,12053-12058 (1997)).In addition, monocyte infiltration reduces to the quantity of granulomatous lesion locations in these mouse, described pathology bring out by injecting schistosomicide (schistosomal) or antigen of mycobacterium (Boring etc.,
J.Clin.Invest., 100,2552-2561 (1997); Warmington etc.,
Am J.Path., 154,1407-1416 (1999)).These situations show that MCP-1 inducibility CCR2 activation plays a major role monocyte recruitement in the inflammation position, to produce enough restraining effect to immune response to this active antagonistic action, thereby provide useful treatment immunity inflammatory disease and autoimmune disorder.
So the medicine of regulating Chemokine Receptors (for example CCR-2 acceptor) can be used for such illness and disease.
In addition, raising monocyte to the inflammatory lesion position of vessel wall is to cause the major cause that atheromatous plaques forms.Behind the vascular damaged of hypercholesterolemia, MCP-1 is produced, is secreted by endotheliocyte and intimal smooth muscle cells.Raise the MCP-1 of the monocyte response release of damage position, be divided into foam cell behind the infiltration vessel wall.Several research groups verified the APO-E-that backcrosses/-, LDL-R-/-or the MCP-1-of Apo B transgenic mice/-or CCR2-/-mouse (supply food rich in fat) in aortic disease size, macrophage content and downright bad the minimizing (Boring etc.,
Nature, 394,894-897 (1998); Gosling etc.,
J.Clin.Invest., 103,773-778 (1999)).Therefore, the CCR2 antagonist can be raised and breaks up at arterial wall by reducing monocyte, thereby suppresses that atherosclerotic lesion forms and the pathology progress.
Summary of the invention
The invention further relates to such compound: they are modulators of chemokine receptor activity, can be used for prevention or treat some inflammatory immunomodulatory illness and disease, anaphylactic disease, atopy illness (comprising rhinallergosis, dermatitis, conjunctivitis and asthma) and autoimmunity pathology (for example rheumatoid arthritis) and atherosclerosis.The invention still further relates to the medicinal compositions that comprises these compounds and and these compounds and composition relate to purposes in the disease of Chemokine Receptors in prevention or treatment.
Detailed Description Of The Invention
The present invention relates to following formula I compound or its pharmacy acceptable salt or independent diastereomer:
Formula I
X is selected from C, N, O, S and SO
2
Y is N or C;
R
1Be selected from following group:
Hydrogen ,-C
1-6Alkyl ,-C
0-6Alkyl-O-C
1-6Alkyl ,-C
0-6Alkyl-S-C
1-6Alkyl ,-(C
0-6Alkyl)-(C
3-7Cycloalkyl)-(C
0-6Alkyl), hydroxyl, heterocycle ,-CN ,-NR
12R
12,-NR
12COR
13,-NR
12SO
2R
14,-COR
11,-CONR
12R
12And phenyl,
R wherein
11Independently be selected from following group: hydroxyl, hydrogen, C
1-6Alkyl ,-O-C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
R wherein
12Be selected from following group: hydrogen, C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
R wherein
13Be selected from following group: hydrogen, C
1-6Alkyl ,-O-C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
R wherein
14Be selected from following group: hydroxyl, C
1-6Alkyl ,-O-C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
Wherein said alkyl and cycloalkyl are not substituted or independently are selected from following substituting group by 1-7 and replace:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(f) C
1-3Alkyl,
(g)-O-C
1-3Alkyl,
(h)-COR
11,
(i)-SO
2R
14,
(j)-NHCOCH
3,
(k)-NHSO
2CH
3,
(l)-heterocycle,
(m)=O,
(n)-CN,
Wherein said phenyl and heterocycle are not substituted or independently are selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group and trifluoromethyl;
R
2Be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c) halogen,
(d) C
1-3Alkyl, wherein said alkyl are not substituted or are replaced by 1-6 substituting group that independently is selected from fluorine and hydroxyl,
(e)-NR
12R
12,
(f)-COR
11,
(g)-CONR
12R
12,
(h)-NR
12COR
13,
(i)-OCONR
12R
12,
(j)-NR
12CONR
12R
12,
(k)-heterocycle,
(l)-CN,
(m)-NR
12-SO
2-NR
12R
12,
(n)-NR
12-SO
2-R
14,
(o)-SO
2-NR
12R
12,
(p)=O, wherein R
2Be connected with ring by two keys;
When Y is N, R
3For oxygen or do not exist;
When Y is C, R
3Be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c) halogen,
(d) C
1-3Alkyl, wherein said alkyl are not substituted or independently are selected from following substituting group by 1-6 and replace: fluorine, hydroxyl and-COR
11,
(e)-NR
12R
12,
(f)-COR
11,
(g)-CONR
12R
12,
(h)-NR
12COR
13,
(i)-OCONR
12R
12,
(j)-NR
12CONR
12R
12,
(k)-heterocycle,
(l)-CN,
(m)-NR
12-SO
2-NR
12R
12,
(n)-NR
12-SO
2-R
14,
(o)-SO
2-NR
12R
12,
(p) nitro;
R
4Be selected from following group:
(a) hydrogen,
(b) C
1-6Alkyl,
(c) trifluoromethyl,
(d) trifluoromethoxy,
(e) chlorine,
(f) fluorine,
(g) bromine,
(h) phenyl;
R
5Be selected from following group:
(a) C
1-6Alkyl, wherein alkyl can be unsubstituted, perhaps replaced, and optionally replaced by hydroxyl by 1-6 fluorine,
(b)-O-C
2-6Alkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(c)-CO-C
1-6Alkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(d)-S-C
1-6Alkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(e)-and pyridyl, it can be unsubstituted or is selected from following substituting group and replace by one or more: halogen, trifluoromethyl, C
1-4Alkyl and COR
11,
(f) fluorine,
(g) chlorine,
(h) bromine,
(i)-C
4-6Cycloalkyl,
(j)-O-C
4-6Cycloalkyl,
(k) phenyl, it can be unsubstituted or is selected from following substituting group and replace by one or more: halogen, trifluoromethyl, C
1-4Alkyl and COR
11,
(l)-and the O-phenyl, it can be unsubstituted or is selected from following substituting group and replace by one or more: halogen, trifluoromethyl, C
1-4Alkyl and COR
11,
(m)-C
3-6Cycloalkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(n)-O-C
3-6Cycloalkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(o)-heterocycle,
(p)-CN,
(q)-COR
11;
R
6Be selected from following group:
(a) hydrogen,
(b) C
1-6Alkyl,
(c) trifluoromethyl,
(d) fluorine,
(e) chlorine,
(f) bromine;
R
7Be selected from following group:
There are not (during X=O), hydrogen, (C
0-6Alkyl)-phenyl, (C
0-6Alkyl)-heterocycle, (C
0-6Alkyl)-C
3-7Cycloalkyl, (C
0-6Alkyl)-COR
11, (C
0-6Alkyl)-(thiazolinyl)-COR
11, (C
0-6Alkyl)-SO
3H, (C
0-6Alkyl)-W-C
0-4Alkyl, (C
0-6Alkyl)-CONR
12-phenyl and (C
0-6Alkyl)-CONR
15-V-COR
11, be O, S or SO perhaps at X
2The time, R
7Do not exist,
Wherein V is C
1-6Alkyl or phenyl, W be selected from singly-bound ,-O-,-S-,-SO-,-SO
2-,-CO-,-CO
2-,-CONR
12-and-NR
12-, R
15Can be hydrogen, C
1-4Alkyl, perhaps R
15The carbon that joint by 1-5 carbon connects V constitutes ring, C
0-6Alkyl is not substituted or independently is selected from following substituting group by 1-5 and replaces:
(a) halogen,
(b) hydroxyl,
(c)-C
0-6Alkyl,
(d)-O-C
1-3Alkyl,
(e) trifluoromethyl,
(f)-C
0-2Alkyl-phenyl,
Wherein said phenyl, heterocycle, cycloalkyl and C
0-4Alkyl is not substituted or independently is selected from following substituting group by 1-5 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c) hydroxyl,
(d) C
1-3Alkyl,
(e)-O-C
1-3Alkyl,
(f)-C
0-3-COR
11,
(g)-CN,
(h)-NR
12R
12,
(i)-CONR
12R
12,
(j)-C
0-3-heterocycle,
Perhaps, described phenyl and heterocycle can be heterocyclic fused with another, and another heterocycle itself can be unsubstituted or independently is selected from following substituting group by 1-2 and replace: hydroxyl, halogen ,-COR
11With-C
1-3Alkyl,
Wherein thiazolinyl is not substituted or independently is selected from following substituting group by 1-3 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c) C
1-3Alkyl,
(d) phenyl,
(e) heterocycle;
R
8Be selected from following group:
(a) hydrogen,
(b) be O, S, SO at X
2Or during N or connect R respectively
7And R
10Carbon between when being two key, R
8Do not exist,
(c) hydroxyl,
(d) C
1-6Alkyl,
(e) C
1-6Alkyl-hydroxyl,
(f)-O-C
1-3Alkyl,
(g)-COR
11,
(h)-CONR
12R
12,
(i)-CN;
Perhaps R
7And R
8Be bonded together to constitute and be selected from following ring:
(a) 1H-indenes,
(b) 2,3-dihydro-1H-indenes,
(c) 2,3-dihydro-cumarone,
(d) 1,3-dihydro-isobenzofuran,
(e) 2,3-dihydro-thionaphthene,
(f) 1,3-dihydro-different thionaphthene,
(g) the 6H-cyclopenta [d] isoxazol-3-ol,
(h) pentamethylene,
(i) hexanaphthene,
Wherein the ring that is constituted can be unsubstituted or independently is selected from following substituting group by 1-5 and replace:
(a) halogen,
(b) trifluoromethyl,
(c) hydroxyl,
(d) C
1-3Alkyl,
(e)-O-C
1-3Alkyl,
(f)-C
0-3-COR
11,
(g)-CN,
(h)-NR
12R
12,
(i)-CONR
12R
12,
(j)-C
0-3-heterocycle,
Perhaps, R
7With R
9Or R
8With R
10Can combine constitutes phenyl ring or heterocycle, and wherein said ring is not substituted or independently is selected from following substituting group by 1-7 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c) hydroxyl,
(d) C
1-3Alkyl,
(e)-O-C
1-3Alkyl,
(f)-COR
11,
(g)-CN,
(h)-NR
12R
12,
(i)-CONR
12R
12;
R
9And R
10Independently be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c) C
1-6Alkyl,
(d) C
1-6Alkyl-COR
11,
(e) C
1-6Alkyl-hydroxyl,
(f)-O-C
1-3Alkyl,
(g)=O, at this moment R
9Or R
10By two key shacks,
(h) halogen;
N is selected from 0,1 and 2;
Dotted line is singly-bound or two key.
Another embodiment of the present invention comprises following formula I a compound or its pharmacy acceptable salt or independent diastereomer:

R wherein
1, R
2, R
3, R
5, R
9, Y and n be definition above,
R
16And R
17Independently be selected from following group:
(a) hydrogen,
(b) halogen,
(c) trifluoromethyl,
(d) hydroxyl,
(e) C
1-3Alkyl,
(f)-O-C
1-3Alkyl,
(g)-C
0-3-CO
2H,
(h)-C
0-3-CO
2C
1-3Alkyl,
(i)-CN,
(j)-C
0-3-heterocycle,
Perhaps, R
16And R
17Being bonded together constitutes and the phenyl ring condensed heterocycle, and heterocycle itself can be unsubstituted or independently is selected from following substituting group replacement by 1-2: hydroxyl, halogen ,-COR
11With-C
1-3Alkyl.
Another embodiment of the present invention also comprises following formula I b compound or its pharmacy acceptable salt or independent diastereomer:

Wherein dotted line is singly-bound or two key, R
1, R
2, R
3, R
5, R
9, R
16, R
17, Y and n be definition above.
Yet another embodiment of the invention comprises following formula I c compound or its pharmacy acceptable salt or independent diastereomer:
R wherein
1, R
2, R
3, R
5, R
9, R
16, R
17, Y and n be definition above, H is a heterocycle.
Another embodiment of the present invention comprises following formula I d compound or its pharmacy acceptable salt or independent diastereomer:

R wherein
1, R
2, R
3, R
5, R
9, R
11, Y, W and n be definition above,
C wherein
1-4Carbochain can be unsubstituted or independently is selected from following substituting group by 1-4 and replace:
(a) halogen,
(b) hydroxyl,
(c)-C
0-6Alkyl,
(d)-O-C
1-3Alkyl,
(e) trifluoromethyl,
(f)-C
0-2Alkyl-phenyl
Perhaps C
1-4Carbochain can be included in C
3-7In the cycloalkyl ring.
An embodiment more of the present invention comprises following formula I e compound or its pharmacy acceptable salt or independent diastereomer:

R wherein
1, R
2, R
3, R
5, R
9, R
16, R
17, X, Y and n be definition above; Dotted line is singly-bound or two key; O is 1 or 2; A, B and D can independently be selected from C, N, O or S, when X, A, B, D are C and o=2, constitute phenyl ring, perhaps when one of X, A, B and D at least be not C for N, O or S, and the formation heterocycle.
Yet another embodiment of the invention comprises following formula I f compound or its pharmacy acceptable salt or independent diastereomer:

R wherein
1, R
2, R
3, R
5, R
7, R
9, R
10, Y and n be definition above; X is N or O, when X is O, and R
7Do not exist.
Another embodiment of the present invention comprises following formula I g compound or its pharmacy acceptable salt or independent diastereomer:

R wherein
1, R
5, R
9, R
16, R
17With Y be above definition,
Perhaps R
16And R
17Being bonded together constitutes and the phenyl ring condensed heterocycle, and heterocycle itself can be unsubstituted or independently is selected from following substituting group replacement by 1-2: hydroxyl, halogen ,-COR
11With-C
1-3Alkyl.
An embodiment more of the present invention comprises following formula I h compound or its pharmacy acceptable salt or independent diastereomer:
Wherein dotted line is singly-bound or two key, R
1, R
5, R
9, R
16, R
17With Y be above definition.
An embodiment more of the present invention comprises following formula I i compound or its pharmacy acceptable salt or independent diastereomer:
R wherein
1, R
5, R
9, R
16, R
17With Y be above definition, H is a heterocycle.
Yet another embodiment of the invention comprises following formula I i compound or its pharmacy acceptable salt or independent diastereomer:
R wherein
1, R
5, R
9, R
11, Y and W be definition above, C
1-4Carbochain can be unsubstituted or independently is selected from following substituting group by 1-4 and replace:
(a) halogen,
(b) hydroxyl,
(c)-C
0-6Alkyl,
(d)-O-C
1-3Alkyl,
(e) trifluoromethyl,
(f)-C
0-2Alkyl-phenyl.
Another embodiment of the present invention comprises following formula I k compound or its pharmacy acceptable salt or independent diastereomer:
Formula Ik
R wherein
1, R
5, R
9, R
10With Y be above definition.
In the another embodiment of the present invention, X is C, O or N.
In another embodiment of the invention, X is C or O.
In another embodiment of the invention, R
1Following group :-C
1-6Alkyl ,-C
0-6Alkyl-O-C
1-6Alkyl and-(C
0-6Alkyl)-(C
3-7Cycloalkyl)-(C
0-6Alkyl), described alkyl and cycloalkyl are not substituted or independently are selected from following substituting group by 1-7 and replace:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(f) C
1-3Alkyl,
(g)-O-C
1-3Alkyl,
(h)-COR
11,
(i)-CN,
(j)-NR
12R
12,
(k)-CONR
12R
12。
In another aspect of this invention, R
1Be selected from following group:
(1)-C
1-6Alkyl, it is not substituted or independently is selected from following substituting group by 1-6 and replaces:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(e)-COR
11,
(2)-C
0-6Alkyl-O-C
1-6Alkyl-, it is not substituted or independently is selected from following substituting group by 1-6 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c)-COR
11,
(3)-(C
3-5Cycloalkyl)-(C
0-6Alkyl), it is not substituted or independently is selected from following substituting group by 1-7 and replaces:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(e)-COR
11。
At one side more of the present invention, R
1Be selected from following group:
(a) C
1-6Alkyl,
(b) C of hydroxyl replacement
1-6Alkyl,
(c) C that is replaced by 1-6 fluorine
1-6Alkyl.
In still another aspect of the invention, R
1Be selected from following group:
(a)-CH(CH
3)
2,
(b)-CH(OH)CH
3,
(c)-CH
2CF
3。
In another aspect of this invention, R
2Be selected from following group:
(a) hydroxyl,
(b) hydrogen,
(c)=O, wherein R
2Be connected with ring by two keys.
In another aspect of this invention, R
2Be hydrogen.
In still another aspect of the invention, when Y is N, R
3Do not exist or for O (obtaining the N-oxide compound).
In still another aspect of the invention, when Y is N, R
3Do not exist.
In still another aspect of the invention, when Y is C, R
3Be selected from following group:
(a) hydrogen,
(b) halogen,
(c) hydroxyl,
(d) C
1-3Alkyl, wherein said alkyl are not substituted or are replaced by 1-6 substituting group that independently is selected from fluorine and hydroxyl,
(e)-COR
11,
(f)-CONR
12R
12,
(g)-heterocycle,
(h)-NR
12-SO
2-NR
12R
12,
(i)-NR
12-SO
2-R
14,
(j)-SO
2-NR
12R
12,
(k)-nitro,
(l)-NR
12R
12。
In another aspect of this invention, when Y is C, R
3Be hydrogen.
In another aspect of this invention, R
4Be hydrogen.
In another aspect of this invention, R
5Be selected from following group:
(a) C that is replaced by 1-6 fluorine
1-6Alkyl,
(b) by 1-6 fluorine replace-O-C
1-6Alkyl,
(c) chlorine,
(d) bromine,
(e) phenyl.
In another aspect of this invention, R
5Be selected from following group:
(a) trifluoromethyl,
(b) trifluoromethoxy,
(c) chlorine,
(d) bromine,
(e) phenyl.
In another aspect of this invention, R
5Be trifluoromethyl.
In another aspect of this invention, R
6Be hydrogen.
In another aspect of this invention, R
7Be phenyl, heterocycle, C
3-7Cycloalkyl, C
1-6Alkyl ,-COR
11With-CONH-V-COR
11,
Wherein V is selected from C
1-6Alkyl or phenyl,
Wherein said phenyl, heterocycle, C
3-7Cycloalkyl, C
1-6Alkyl is not substituted or independently is selected from following substituting group by 1-5 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c) hydroxyl,
(d) C
1-3Alkyl,
(e)-O-C
1-3Alkyl,
(f)-COR
11,
(g)-CN,
(h)-heterocycle,
(i)-CONR
12R
12。
In another aspect of this invention, when X is not O, R
7Be phenyl, heterocycle, C
1-4Alkyl ,-COR
11With-CONH-V-COR
11, V is selected from C
1-6Alkyl or phenyl,
Wherein said phenyl, heterocycle and C
1-4Alkyl is not substituted or independently is selected from following substituting group by 1-3 and replaces:
(a) halogen,
(b) hydroxyl,
(c) C
1-3Alkyl,
(d)-O-C
1-3Alkyl,
(e)-COR
11,
(f)-heterocycle.
In another aspect of this invention, when X is C, R
7Be selected from following group:

In another aspect of this invention, when X is C, R
8Be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c)-CN,
(d)-F。
In another aspect of this invention, R
7And R
8Can combine to constitute and be selected from following ring:
(a) 1H-indenes,
(b) 2,3-dihydro-1H-indenes,
Wherein the ring that is constituted can be unsubstituted or independently is selected from following substituting group by 1-3 and replace:
(a) halogen,
(b) hydroxyl,
(c) C
1-3Alkyl,
(d)-O-C
1-3Alkyl,
(e)-COR
11,
(f)-heterocycle.
In another aspect of this invention, R
9And R
10Independently be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c)-CH
3,
(d)-O-CH
3,
(e)=O (R wherein
9And/or R
10Be connected with ring by two keys).
In another aspect of the invention, n=1 or 2.
Representative compounds of the present invention comprises compound and their pharmacy acceptable salt and the independent diastereomer of embodiment.
The compounds of this invention has two asymmetric centers at 1 and 3 of cyclopentyl at least.May have other asymmetric center, this depends on the character of different substituents on the molecule.All such asymmetric centers will independently produce two optically active isomers, and the scope of the invention comprises the form of mixtures and the pure or part neat compounds form of possible optically active isomer of institute and diastereomer.The compounds of this invention absolute configuration on the one hand is following configuration, and wherein the substituting group on the cyclopentyl (amide units and amine unit) is a cis:
The compounds of this invention absolute configuration on the other hand is the configuration of following orientation:
Wherein connect the substituent carbon of amine and be appointed as (R) absolute configuration, the carbon that connects the acid amides subunit can be according to R
1Priority be appointed as (S) or (R) absolute configuration.When for example R was sec.-propyl, the absolute stereo chemical structure that then connects the carbon of acid amides subunit will be (S), arranged because amide units and amine unit are preferably cis on cyclopentyl.
The suitable modification method disclosed herein of can passing through known in the art is synthesized diastereomer and enantiomer or chromatographic separation separately.Their absolute steric configuration can be taken a picture by the x ray crystal of crystallized product or crystallization of intermediate and be determined that if desired, the also available reagent that comprises the asymmetric center of known absolute configuration of crystallized product or crystallization of intermediate is derived.
As those skilled in the art's understanding, halogen used herein or halogen comprise chlorine, fluorine, bromine and iodine.
" alkyl " used herein is meant does not have two keys or triple-linked straight chain, side chain or ring texture.Therefore, C
1-6Alkyl is meant straight chain or the straight chain group that contains 1,2,3,4,5 or 6 carbon, such C
1-6Alkyl is particularly including methyl, ethyl, n-propyl, sec.-propyl, normal-butyl, isobutyl-, the tertiary butyl, amyl group and hexyl." cycloalkyl " is the alkyl that partly or entirely constitutes the ring of three or three above atoms.C
0Or C
0Alkyl is meant covalent linkage.
Term used herein " heterocycle " comprises following group: benzimidazolyl-; benzofuryl; benzo furazan base; the benzopyrazoles base; the benzotriazole base; benzothienyl benzoxazolyl; carbazyl; carbolinyl; cinnolinyl; furyl; imidazolyl; indolinyl; indyl; indolizine base (indolazinyl); indazolyl; isobenzofuran-base; pseudoindoyl; isoquinolyl; isothiazolyl isoxazolyl; naphtho-pyridyl oxadiazole base oxazolyl; oxetanyl; pyranyl; pyrazinyl; pyrazolyl; pyridazinyl; the pyridopyridine base; pyridazinyl; pyridyl; pyrimidyl; pyrryl; quinazolyl; quinolyl; quinoxalinyl; THP trtrahydropyranyl; tetrazyl; the tetrazolo pyridyl; thiadiazolyl group; thiazolyl; thienyl; triazolyl; azetidinyl; 1, the 4-diox; the hexahydroazepine base; piperazinyl; piperidyl; pyrrolidyl; morpholinyl; thio-morpholinyl; the dihydrobenzo imidazolyl; dihydro benzo furyl; the dihydrobenzo thienyl; Er hydrogen benzoxazolyl; the dihydrofuran base; the glyoxalidine base; indolinyl; the dihydro-isoxazole base; dihydro isothiazolyl Er Qing oxadiazole base dihydro-oxazole base; the dihydro pyrazinyl; the pyrazoline base; the dihydropyridine base; the dihydro-pyrimidin base; the pyrrolin base; the dihydroquinoline base; the dihydro tetrazyl; the thiodiazoline base; dihydro-thiazolyl; the dihydro-thiophene base; the dihydro triazolyl; the dihydro azetidinyl; the methylene-dioxy benzoyl; tetrahydrofuran base and tetrahydro-thienyl and their N-oxide compound.
Term used herein " pharmaceutically acceptable " is meant such compound, raw material, composition and/or formulation: in rational medical judgment, be fit to contact with the tissue of people and animal, and do not have over-drastic toxicity, stimulation, anaphylaxis or other problem or complication, have reasonably and be benefited/the risk ratio.
" pharmacy acceptable salt " used herein is meant parent compound modified the derivative that its hydrochlorate of preparation or alkali salt obtain.The example of pharmacy acceptable salt includes but not limited to the inorganic acid salt or the organic acid salt of alkaline residue (for example amine); The alkali salt of acidic residues (for example carboxylic acid) or organic salt etc.Pharmacy acceptable salt comprises the conventional non-toxic salt or the quaternary ammonium salt of the parent compound that forms with for example nontoxic mineral acid or organic acid.By way of example, so conventional non-toxic salt comprises mineral acid deutero-salt, for example hydrochloride, hydrobromate, vitriol, sulfamate, phosphoric acid salt, nitrate etc.; With the salt of organic acid preparation, for example acetate, propionic salt, succinate, glycollate, stearate, lactic acid salt, malate, tartrate, Citrate trianion, ascorbate salt, pamoate, maleate, hydroxymaleic acid salt, phenylacetic acid salt, glutaminate, benzoate, salicylate, sulfanilate, 2-acetoxy-benzoic acid salt, fumarate, tosylate, mesylate, ethane disulfonate, oxalate, isethionate etc.
Pharmacy acceptable salt of the present invention can be with the parent compound that comprises alkalescence or acidic moiety by the preparation of conventional chemical method.Usually, such salt can be prepared as follows: the free acid of above-claimed cpd or the suitable alkali or the acid of free alkali form and stoichiometry are reacted in water, organic solvent or both mixtures, usually use non-aqueous media, for example ether, ethyl acetate, ethanol, Virahol or acetonitrile.Suitable salt can be referring to for example Remington ' sPharmaceutical Sciences, and the 17th edition, Mack Publishing Company, Easton, PA, 1985, p.1418.
Utilize embodiment and compound disclosed herein to illustrate the present invention.
Particular compound of the present invention comprises title compound and their pharmacy acceptable salt and the independent diastereomer that is selected from following compound: embodiment.
Motif compound can be used for regulating the patient's of this adjusting of needs the method for chemokine receptor activity, and this method comprises the compound that gives significant quantity.
The present invention relates to the purposes of aforesaid compound as modulators of chemokine receptor activity.Specifically, these compounds can be used as the conditioning agent of Chemokine Receptors (particularly CCR-2).
The compounds of this invention can confirm by methods known in the art as the effectiveness of modulators of chemokine receptor activity, the disclosed chemokine binding analysis of for example following document method: VanRiper etc.,
J.Exp.Med.,
177, 851-856 (1993) is easy to improvement and is used to measure the CCR-2 keying action.
Receptor affinity is following in the CCR-2 binding analysis determines: detect
125I-MCP-1 is attached to the restraining effect of the endogenous CCR-2 acceptor of different cell types (comprising monocyte, THP-1 cell), perhaps clones acceptor and detects behind the eukaryotic cell heterogenous expression
125I-MCP-1 bonded restraining effect.(50mM HEPES, pH 7.2,5mMMgCl in binding buffer liquid with cell suspension
2, 1mM CaCl
2And 0.50%BSA), room temperature add test-compound or DMSO and
125I-MCP-1 places 1h and allows its combination.Use GFB filter membrane collecting cell then, with the 25mM HEPES damping fluid washing that contains 500mM NaCl, quantitatively combination
125The cell of I-MCP-1.
In chemotaxis assay, carry out chemotaxis with the PBMC of disappearance T cell, described PBMC from vein whole blood or white corpuscle separate blood system from, the centrifugal purification of Ficoll-Hypaque forms rosette with the sheep red blood cell (SRBC) that neuraminidase is handled then.After separation is finished, cell is washed with the HBSS that contains 0.1mg/ml BSA, with 1 * 10
7Cell/ml suspends.In the dark, under 37 ℃, with cell with 2 μ M Calcien-AM (Molecular Probes) fluorescent mark 30min.With the cell washing twice of mark, and with 5 * 10
6Cell/ml is suspended in RPMI1640 (contain L-glutaminate, 0.1mg/ml BSA, do not contain phenol red).With the 10ng/ml MCP-1 (Peprotech) of same medium dilution or only be the hole (27 μ l) that substratum adds the bottom.Behind the test-compound preincubation 15min with DMSO or different concns, monocyte (150,000 cell) is added above the filter membrane (30 μ l).The test-compound or the DMSO of same concentrations are added base apertures to prevent diffusion dilution.At 37 ℃, 5%CO
2Behind the incubation 60min, remove filter membrane, the cell of not moving to filter membrane is removed with the HBSS washing that contains 0.1mg/ml BSA in the top.Determine not have the spontaneous transporting action (chemokinesis) of chemoattractant under existing.
Specifically, hereinafter the compound of embodiment has the activity in conjunction with the CCR-2 acceptor in above-mentioned analysis, usually IC
50Less than about 1 μ M.Such result shows that compound has the intrinsic activity as modulators of chemokine receptor activity.
The mammalian chemokines acceptor provides and has disturbed or the eosinocyte of promotion Mammals (for example people) and/or the target of lymphocyte function.The compound of inhibition or promotion chemokine receptor function especially can be used for therapeutic regulation eosinocyte and/or lymphocyte function.Therefore, the compound of inhibition or promotion chemokine receptor function can be used for treatment, prevention, improves, controls multiple following disease or reduce its ill risk: inflammatory immunomodulatory illness and disease, anaphylactic disease, atopy illness (comprising rhinallergosis, dermatitis, conjunctivitis and asthma) and autoimmunity pathology (for example rheumatoid arthritis) and atherosclerosis.
For example, can suppress the chemical combination of the present invention of one or more functions of mammalian chemokines acceptor (for example human chemokine receptor) to suppress (promptly alleviate or prevent) inflammation.Thus, one or more inflammatory processes (for example swim out of, chemotaxis, exocytosis (for example exocytosis of enzyme, histamine) or inflammatory mediator discharge and be suppressed by white corpuscle.
Except Primates (for example people), many other Mammalss also can be treated according to the inventive method.For example, also can treat the Mammals that includes but not limited to ox, sheep, goat, horse, dog, cat, cavy, rat or other Bovidae, sheep section, equine, Canidae, cat family, rodent or murine species.Yet described method also can be used for other species, for example birds (for example chicken).
Can treat with The compounds of this invention with inflammation and infection diseases associated and illness.In one embodiment, described disease or illness are to need inhibition or promote lymphocyte to do in order to regulate the disease or the illness of inflammatory reaction.
Can include but not limited to the people of chemokine receptor function inhibitor for treating or the disease or the illness of other species: inflammatory or anaphylactic disease and illness comprise for example asthma (especially bronchial asthma) of respiratory tract anaphylaxis disease, rhinallergosis, supersensitivity tuberculosis, hypersensitivity pneumonitis, eosinocyte pneumonia (Loeffler syndromes for example, chronic eosinocyte pneumonia), delayed-type hypersensitivity, interstitial pneumonia (ILD) (for example idiopathic pulmonary fibrosis or and rheumatoid arthritis, systemic lupus erythematous, ankylosing spondylitis, systemic sclerosis, the Sjogren syndromes, the ILD that polymyositis or dermatomyositis are relevant); General allergy or allergy, the drug allergy transformation reactions of penicillin, cynnematin (for example to), insect bite thorn property allergy; Autoimmune disorder, for example rheumatoid arthritis, psoriatic arthritis, multiple sclerosis, systemic lupus erythematous, myasthenia gravis, juvenile diabetes; Glomerulonephritis, autoimmune thyroiditis, Behcet; Transplant rejection (for example repulsion in the transplantation) comprises the repulsion or the graft versus host disease (GVH disease) of allograft; Inflammatory bowel, for example Crohn disease and ulcerative colitis; SpA; Scleroderma; Psoriasis (comprising the cell-mediated property of T psoriasis) and inflammatory dermatosis (for example dermatitis, eczema, atopic dermatitis, contact dermatitis, urticaria); Vasculitis (for example necrotizing vasculitis, cutaneous vasculitis and allergic angiitis); Eosinocyte myositis, eosinocyte fascitis; The cancer of leukocyte infiltration skin or organ.Other disease or the illness that need to suppress bad Inflammatory response be can also treat, reperfusion injury, atherosclerosis, some hematologic malignancies, cytokine induction toxicity (for example septic shock, endotoxin shock), polymyositis, dermatomyositis included but not limited to.
Can include but not limited to the people of chemokine receptor function modulators for treatment or the disease or the illness of other species: immunosuppression (immunosuppression of for example suffering from the individuality of acquired immunodeficiency syndrome such as AIDS or other virus infection, the individual immunity that the treatment of radiotherapy, chemotherapy, autoimmune disorder or pharmacological agent (for example reflunomide therapy) cause suppresses; The immunosuppression of function of receptors birth defects or other reason; Catch, parasitosis for example, include but not limited to helminth infection, for example nematode (trichuriasis, oxyuriasis, ascariasis, uncinariasis, strongyloidiasis, trichonematosis, filaricide), fluke (schistosomicide, clonorchiasis), tapeworm (hydatidosis, teniasis, cysticercosis), the worm of internal organ, visceral larva migrans (visceral larva migraines) (for example bending roundworm), eosinocyte gastroenteritis (Anisaki sp. for example, Phocanema sp.) and cutaneous larva migrans (cutaneous larvamigraines) (ancylostoma braziliense, the dog hookworm).In addition, if estimate to give enough compounds, by inducing the Chemokine Receptors internalization to make expression of receptor on the cell surface reduce or giving compound, then can consider to treat above-mentioned inflammatory, supersensitivity and autoimmune disorder by strengthening chemokine receptor function in the mode that causes the cell migration anisotropy.
Therefore, The compounds of this invention can be used for treatment, prevention, improves, controls multiple following disease or reduce its ill risk: inflammatory immunomodulatory illness and disease, anaphylactic disease, atopy illness and autoimmunity pathology.In a special embodiment, the present invention relates to use motif compound treatment, prevention, improve, control autoimmune disorder (for example rheumatoid arthritis or psoriatic arthritis) or reduce its ill risk.
On the other hand, the present invention can be used to estimate the concrete agonist or the antagonist of inferring of Chemokine Receptors (comprising CCR-2).In view of the above, the present invention relates to above-claimed cpd in the preparation of screening analysis and the purposes in the enforcement, described analysis is used to estimate the compound of regulating chemokine receptor activity.For example, The compounds of this invention can be used for separating acceptor mutant, and acceptor mutant is to seek the more excellent screening implement of active compound.In addition, The compounds of this invention can confirm or determine the binding site of other compound and Chemokine Receptors by for example competitive inhibition.The compounds of this invention also can be used for estimating the concrete conditioning agent of inferring of Chemokine Receptors (comprising CCR-2).As this area was understood, concrete agonist and the antagonist of thoroughly estimating above Chemokine Receptors were limited by the shortage non-peptide acyl of available (antimetabolic) compound, and this compounds has high binding affinity to these acceptors.Therefore, The compounds of this invention is the commodity that will be used for these purposes future.
The invention further relates to the method for preparing medicine, described medicine comprises The compounds of this invention and pharmaceutical carrier or thinner, is used to regulate the chemokine receptor activity of humans and animals.
The invention further relates to and use The compounds of this invention treatment, prevention, improve, control retrovirus (especially simplexvirus or human immunodeficiency virus (HIV)) infects or reduce its infection risk and treatment subsequently pathologic conditions (for example AIDS) or postpone its outbreak.Treatment AIDS or prevention or treatment HIV infection are defined as and include but not limited to treat various HIV Infection Status: AIDS, have symptom or asymptomatic ARC (AIDS related syndromes) and actual or potential contact HIV.For example, suspecting in the past that perhaps when surgical operation after the contact blood samples of patients, available The compounds of this invention is treated the HIV infection because for example blood transfusion, organ transplantation, body fluid exchanges, bite, accidental needle sticks contacts HTV.
In one aspect of the invention, motif compound can be used for chemokine inhibiting and target cell Chemokine Receptors (for example CCR-2) bonded method, this method comprises makes target cell contact with a certain amount of compound, and the compound of described amount is combining of chemokine inhibiting and Chemokine Receptors effectively.
The patient of above method treatment is the Mammals people for example who needs to regulate chemokine receptor activity, comprises masculinity and femininity." adjusting " used herein comprises antagonism, excitement, part antagonism, oppositely exciting and/or part excitement.In one aspect of the present invention, regulate being meant the antagonism chemokine receptor activity.Term " treatment significant quantity " is meant that the consumption of motif compound will cause tissue, system, animal or human's biology or medical response, and this reaction is that investigator, animal doctor, doctor or other clinicists seek.
Term used herein " composition " comprises product that contains the specific composition of specified quantitative and any product that directly or indirectly obtains with the combination of specified quantitative special component." pharmaceutically acceptable " is meant that carrier, thinner or vehicle must be compatible with other composition of preparation, and can not endanger its recipient.
Term " gives " compound and is construed as the individuality that The compounds of this invention is offered the needs treatment.
Term used herein " treatment " is meant therapeutic and the above-mentioned disease of prophylactic treatment.
Comprise that appointed substituting group list replaces and polysubstituted about the term " replacement " of metalepsy on alkyl, cycloalkyl, phenyl, heterocycle or other chemical group of part, and the described single replacement on any specified chemical group or polysubstituted should be chemically to allow.
Should be understood that the substituent definition of certain specific position in the molecule is independent of the definition of this substituting group other position in molecule.So, for example work as R
3=1-5 R
12During alkyl that (definition) elsewhere replaces, each R
12Independently be selected from the group that it may be selected; Be each R
12Can with any other R
12Identical or inequality.
Term " optional replacement " comprises replacement and unsubstituted.Therefore, for example optional aryl that replaces may be pentafluorophenyl group or phenyl.
The conjoint therapy of regulating chemokine receptor activity and treating, prevent, improve, control inflammatory immunomodulatory illness and disease (comprising asthma and anaphylactic disease), autoimmunity pathology (for example rheumatoid arthritis) and atherosclerosis and above-mentioned illness thus or reduce its ill risk is by The compounds of this invention and known combination medicine explanation with other compound of such effectiveness.
For example in treatment, prevention, improve, control inflammation or reduce and suffer from the inflammation risk, The compounds of this invention can with antiphlogiston or anodyne combined utilization, opioid agonist for example, lipoxidase inhibitor (for example 5-lipoxidase inhibitor), cyclooxygenase inhibitors (for example cyclooxygenase-2 inhibitor), interleukin inhibitor (for example interleukin-1 inhibitor), nmda antagonist, nitric oxide inhibitor, the nitrogen protoxide synthetic inhibitor, nonsteroidal anti-inflammatory or cell factor inhibiting anti-inflammatory agent, for example following compound of associating: acetaminophen, Asprin, morphine monomethyl ether, etanercept (embrel), fentanyl, Ibuprofen BP/EP, INDOMETHACIN, ketorolac, morphine, Naproxen Base, phenacetin, piroxicam, the steroid anodyne, sufentanil, sulindac, tenidap etc.Similarly, The compounds of this invention can with following medication combined application: pain relief agents; Synergistic agent, for example caffeine, H2-antagonist, dimethyl silicone oil, aluminium hydroxide or magnesium hydroxide; Decongestant, for example phyenlephrinium, Phenylpropanolamine, pseudoephedrine, oxymetazoline, suprarenin (ephinephrine), naphazoline, xylometazoline, propylhexedrine (propylhexedrine) or left-handed desoxyephedrine; Cough medicine, for example morphine monomethyl ether, hydrocodone, caramiphen, pentoxiverin or Dextromethorphane Hbr; Hydragog(ue); Calmness or non-sedating antihistaminic.
Similarly, The compounds of this invention can also be united use with other medicines, and described other medicines are used for the treatment of/prevent/suppress or improve the disease or the illness of available The compounds of this invention treatment.Such other medicines can its routine administration approach and consumption and The compounds of this invention while or sequential administration.When The compounds of this invention and one or more other medicines use simultaneously, use usually to comprise the such other medicines and the composition of The compounds of this invention.Therefore, medicinal compositions of the present invention comprises such composition: except The compounds of this invention, also contain one or more other activeconstituentss.
Can include but not limited to the example that The compounds of this invention is united other activeconstituents (can separately or with same medicinal compositions administration) of use: (a) VLA-4 antagonist, US5 for example, 510,332, the antagonist of WO95/15973, WO96/01644, WO96/06108, WO96/20216, WO96/22966, WO96/31206, WO96/40781, WO97/03094, WO97/02289, WO98/42656, WO98/53814, WO98/53817, WO98/53818, WO98/54207 and WO98/58902 introduction; (b) steroide, for example beclometasone, methyl meticortelone, Betamethasone Valerate, prednisone, dexamethasone and hydrocortisone; (c) immunosuppressor, for example S-Neoral, tacrolimus, rapamycin and other FK-506 type immunosuppressor; (d) antihistaminic (H1-histamine antagonist), for example Parabromdylamine, Cholrtrimeton, dexchlorpheniramine, triprolidine, clemastine, diphenhydramine, diphenylpyraline, tripelennamine, hydroxyzine, methdilazine, promethazine, temaril, azatadine, Cyproheptadine, antazoline, pheniramine, pyrilamine, astemizole, terfenadine, Loratadine, delotadine, alerlisin, fexofenadine, decarburization oxyethyl group Loratadine etc.; (e) on-steroidal antasthmatic, for example β 2-agonist (terbutaline, Orciprenaline, Partusisten, dilabron, salbutamol, bitolterol and pirbuterol), theophylline, Sodium Cromoglicate, coromegine, ipratropium bromide, leukotriene antagonist (Lu Site, Singulair, pranlukast, iralukast, Pobilukast, SKB-106,203), leukotrienes biosynthesis inhibitor (zileuton, BAY-1005); (f) nonsteroidal anti-inflammatory (NSAID), for example propanoic derivatives (alminoprofen Compd 90459, the bucloxonic acid, carprofen, Naponol, fenoprofen, R.D. 17345, flurbiprofen, Ibuprofen BP/EP, indoprofen, Ketoprofen, Miroprofen, Naproxen Base, Taisho), pirprofen, Y-8004, sutoprofen, tiaprofenic acid is with tioxaprofen), acetogenin (INDOMETHACIN, acemetacin, Warner-Lambert), clidanac, diclofenac, Fenclofenac, fenclozic acid, Fentiazac, Furofenac, ibufenac, Isoxepac, oxpinac, sulindac, tiopinac, tolmetin, zidometacin and zomepirac), fenamic acid derivative (Flufenamic Acid, meclofenamic acid, mefenamic acid, niflumic acid and tolfenamic acid), biphenyl acid derivative (diflunisal and flufenisal), former times health class medicine (isoxicam, piroxicam, sudoxicam and tenoxicam), salicylate (acetylsalicylic acid, sulfasalazine) and pyrazoline ketone (Azapropazone, Reublonil (bezpiperylon), Zentinic, mofebutazone, crovaril, BUTE); (g) cyclooxygenase-2 (COX-2) inhibitor; (h) phosphodiesterase IN type (PDE-IV) inhibitor; (i) other antagonist of Chemokine Receptors (especially CCR-1, CCR-2, CCR-3, CXCR-3 and CCR-5); (j) anticholesteremic agent, for example HMG-CoA reductase inhibitor (lovastatin, Simvastatin and Pravastatin, fluvastatin, atorvastatin, superstatin and other he spit of fland), sequestrant (QUESTRAN and colestipol), cholesterol absorption inhibitor (ezetimibe), nicotinic acid, Fenofibric Acid derivative (gemfibrozil, clofibrate, fenofibrate and bezafibrate) and probucol; (k) antidiabetic drug, for example Regular Insulin, sulfonylurea, biguanides (metformin), alpha-glucosidase inhibitor (acarbose) and glitazone (troglitazone and pioglitazone); (l) interferon-preparation (β-1 interferon-alpha, β-1 interferon-); (m) the cell toxicant disposition chemotherapeutics of other compound (for example 5-aminosalicylic acid and prodrug thereof), antimetabolite (for example azathioprine and Ismipur) and cancer.
The part by weight of The compounds of this invention and second kind of activeconstituents can change, and depends on the effective dose of each composition.Usually, use the effective dose of each activeconstituents.Thus by way of example, when The compounds of this invention and NSAID unite when using, the part by weight of The compounds of this invention and NSAID is generally about 1000: 1 to about 1: 1000, or is about 200: 1 to about 1: 200.The combination of The compounds of this invention and other activeconstituents still in all cases, should be used the effective dose of each activeconstituents usually in above-mentioned scope.
In such combined utilization, The compounds of this invention and other active medicine can be separately or combination give.In addition, a kind of composition give can be before other medicines, simultaneously or give afterwards.
The compounds of this invention can give by following approach: oral, parenteral (for example intramuscular, intraperitoneal, intravenously, ICV, intracisternal injection or infusion, subcutaneous injection or transplant), suck in spraying, the nose, vagina, rectum, hypogloeeis or part, and can be separately or be formulated as the preparation of the suitable dose unit that comprises carrier, adjuvant and solvent jointly, described carrier, adjuvant and solvent are conventional nontoxic, pharmaceutically acceptable, and are fit to each route of administration.Except treatment warm-blooded animal (for example mouse, rat, horse, ox, sheep, dog, cat, monkey etc.), The compounds of this invention also can be effective to the people.
The medicinal compositions that gives The compounds of this invention can be rendered as dosage unit forms easily, can be by the known any method preparation of pharmaceutical field.All methods comprise activeconstituents are combined with the carrier that one or more ancillary components constitute.Usually, be prepared as follows medicinal compositions: with activeconstituents and liquid vehicle, micro-solid carrier or the two evenly, closely combine, if desired, be required preparation with product shaping again.In medicinal compositions, the content of active target compound is enough to the process of disease or illness are produced required effect.Term used herein " composition " comprises product that contains the specific composition of specified quantitative and any product that directly or indirectly obtains with the combination of specified quantitative special component.
The medicinal compositions that comprises activeconstituents can for example be tablet, lozenge, lozenge, water-based or oiliness suspensoid, dispersible powder or granule, emulsion, hard or soft balsam wafer or syrup or elixir for being fit to oral formulation.Oral compositions can be according to any currently known methods preparation in preparation medicinal compositions field, in order to obtain pharmaceutically attractive in appearance, agreeable to the taste preparation, such composition can comprise one or more and be selected from following reagent: sweeting agent, seasonings, tinting material and sanitas.Tablet comprises activeconstituents and pharmaceutically acceptable non-toxic excipients, and described vehicle is the vehicle that is fit to the preparation tablet.These vehicle can be for example inert diluent, for example lime carbonate, yellow soda ash, lactose, calcium phosphate or sodium phosphate; Granulating agent and disintegrating agent, for example W-Gum or Lalgine; Tackiness agent, for example starch, gelatin or Sudan Gum-arabic; Lubricant, for example Magnesium Stearate, stearic acid or talcum powder.Tablet can be uncoated, and perhaps they can be by known technology coated with postponing to provide secular continuous action thus in gi tract disintegration, absorption.For example can postpone raw material (for example glyceryl monostearate or distearin) duration of service.They also can apply by the technology that United States Patent (USP) 4,256,108,4,166,452 and 4,265,874 is introduced, and are formed for the osmotic therapeutic tablets of sustained release.
Oral preparations can also be the hard gelatine capsule agent, and wherein activeconstituents mixes with inert solid diluent (for example lime carbonate, calcium phosphate or kaolin); Perhaps be the soft gelatin capsule agent, wherein activeconstituents mixes with water or oil medium (for example peanut oil, whiteruss or sweet oil).
Aqueous suspension comprises activated feedstock and is fit to the vehicle of preparation aqueous suspension.Such vehicle has suspension agent, for example Xylo-Mucine, methylcellulose gum, hydroxyl-propyl methocel, sodium alginate, polyvinylpyrrolidone, tragacanth gum and Sudan Gum-arabic; Dispersion agent or wetting agent can be naturally occurring phosphatide, for example Yelkin TTS; The condensation product of alkylene oxide and lipid acid, for example stearic acid polyoxyethylene; The condensation product of oxyethane and long chain aliphatic alcohol, for example 17 vinyloxy group hexadecanols (heptadecaethyleneoxycetanol); The perhaps condensation product of oxyethane and partial ester (deriving), for example polyoxyethylene sorbitol monoleate by lipid acid and hexitol; The condensation product of oxyethane and partial ester (deriving), for example polyethylene sorbitan monooleate by lipid acid and hexitan.Aqueous suspension also can comprise one or more sanitass (for example ethyl p-hydroxybenzoate or P-hydroxybenzoic acid n-propyl), one or more tinting materials, one or more seasoningss and one or more sweeting agents (for example sucrose or asccharin).
Activeconstituents can be suspended in vegetables oil (for example peanut oil, sweet oil, sesame oil or Oleum Cocois) or mineral oil (for example whiteruss) preparation oiliness suspensoid.The oiliness suspensoid can comprise thickening material, for example beeswax, solid paraffin or hexadecanol.Can add sweeting agent (for example above-mentioned sweeting agent) and seasonings and obtain agreeable to the taste oral preparations.These compositions can be anticorrosion by adding oxidation inhibitor (for example xitix).
Dispersible powder and granule are fit to prepare aqueous suspension by adding entry, obtain the mixture of activeconstituents and dispersion agent or wetting agent, suspension agent and one or more sanitass.Suitable dispersion agent or wetting agent and suspension agent illustrate hereinbefore.Can also comprise other vehicle, for example sweeting agent, seasonings and tinting material.
Medicinal compositions of the present invention can also be oil-in-water emulsion form.Oil phase can be vegetables oil (for example sweet oil or peanut oil) or mineral oil (for example whiteruss) or their mixture.Suitable emulsifying agent can be for naturally occurring natural gum (for example Sudan Gum-arabic or tragacanth gum), naturally occurring phosphatide (for example soybean, Yelkin TTS), derived from the ester of lipid acid and hexitan or the condensation product (for example polyoxyethylene sorbitan monooleate) of partial ester (for example sorbitan monooleate) and described partial ester and oxyethane.Emulsion can also comprise sweeting agent and seasonings.
Syrup and elixir can be used sweeting agent (for example glycerine, propylene glycol, Sorbitol Powder or sucrose) preparation.Such preparation can also comprise analgesic agent, sanitas, seasonings and tinting material.
Medicinal compositions can be aseptic injection water or oiliness suspensoid.This suspensoid can be prepared according to already known processes with above-mentioned suitable dispersion agent or wetting agent and suspension agent.Aseptic injection preparation can also be aseptic injectable solution agent or the suspensoid in nontoxic parenteral acceptable diluent or the solvent, for example solution in 1,3 butylene glycol.In acceptable solvent and solvent, can make water, Ringer's solution and isotonic sodium chlorrde solution.In addition, aseptic expressed oil is usually as solvent or suspension medium.When being used for this purpose, the expressed oil of any gentleness be can use, synthetic monoglyceride or triglyceride comprised.In addition, lipid acid (for example oleic acid) can be used for injection formulations.
The compounds of this invention can also the suppository form rectal administration.These compositions can be prepared as follows: with medicine and suitable non-stimulated excipient mixture, described vehicle is solid when normal temperature, is liquid in rectal temperature, therefore will discharge medicine in the rectum fusing.Such raw material has theobroma oil or polyoxyethylene glycol.
For local application, use ointment, ointment, gelifying agent, solution or the suspensoid etc. that comprise The compounds of this invention.(for this application, topical application should comprise mouth wash shua and gargle)
Medicinal compositions of the present invention and method can further comprise above-mentioned other therapeutical active compound, and these compounds are generally used for treating above-mentioned pathologic conditions.
When treatment, prevention, improvement, control needed to regulate the disease of Chemokine Receptors or reduces its risk, suitable dosage level was generally about 0.01-500mg/kg weight in patients every day, can be divided into single agent or multi-agent administration.Dosage level is that every day about 0.1 is to about 250mg/kg; Or be that every day about 0.5 is to about 100mg/kg.The proper dosage level can be about 0.01-250mg/kg/ days, about 0.05-100mg/kg/ days or about 0.1-50mg/kg/ days.In this scope, dosage can be 0.05-0.5,0.5-5 or 5-50mg/kg/ days.For oral administration, composition can be used for the patient of needs treatment is regulated symptom for comprising 1.0-1000mg activeconstituents, 2.0-500,3.0-200 or 1,5,10,15,20,25,30,50,75,100,125,150,175,200,250,300,400,500,600,750,800,900 and/or the tablet of 1000mg activeconstituents.The usage of compound is every day 1-4 time, preferred every day 1-2 time.
But, should be understood that the concrete dosage level for any particular patient may be different with administration frequency, to depend on various factors, comprise used particular compound activity, this compound metabolic stability and action time length, patient's age, body weight, general health situation, sex and the severity and the ongoing treatment of patient of diet, mode of administration and administration time, discharge rate, drug regimen, disease specific.
Several methods that prepare The compounds of this invention illustrate in following flow process and embodiment.Initial feed be commercially available, by method preparation currently known methods preparation or that introduce according to this paper.
Flow process 1 illustrates to be used for preparing and contains 1,1, the main path of the The compounds of this invention 1-5 of the trisubstituted pentamethylene skeleton of 3-.According to this approach, make ketone acid 1-1 (according to flow process 2A, 2B, 2C and 2D preparation) and amine 1-2 (according to flow process 3A-G preparation) coupling.This reaction can be finished by different modes, comprise at first with reagent for example oxalyl chloride acid is converted into its acyl chlorides, then with amine 1-2 chemical combination in the presence of alkali (for example triethylamine).With for example NaB (OAc)
3H or NaBH
3CN makes 1-3 and amine 1-4 carry out the reductive amination reaction as reductive agent, obtains chemokine receptor modulators 1-5.Compound 1-9 (can be synthetic according to the chemical process that flow process 1 is introduced) is stereoisomer mixture (Eliel, E.E., Wilen, S.H., Stereochemistryof Organic Compounds, John Wiley ﹠amp; Sons, Inc., New York).Specifically, the compound 1-5 of acquisition is generally the mixture of cis-isomeride and trans-isomer(ide).When 1-1 is single stereoisomers (1-1a), can only obtain 2 kinds of possible isomer (cis and trans) of 1-5; They can separate by several different methods, comprise preparation type TLC, flash chromatography, MPLC or HPLC (using the chromatographic column of chiral stationary phase).When 1-1 is racemize, can obtain whole 4 kinds of possibility isomer of 1-5.The combination that they also can pass through HPLC (using the chromatographic column of chiral stationary phase) or aforesaid method separates.The synthetic method of racemic 1-1 is introduced in flow process 2A in detail, and the synthetic method of chirality 1-1a is introduced in flow process 2B and 2C.
In addition, compound 1-5 itself can be obtained new chemokine receptor modulators 1-5.1 by modification.For example the ester functional group of compound 1-5 can be hydrolyzed to corresponding carboxylic acid, and corresponding carboxylic acid also can be used as chemokine receptor modulators.
Flow process 1

Shown in flow process 1A, can under multiple condition, (comprise sodium triacetoxy borohydride or sodium cyanoborohydride), ketone-ester 1-6 is formed amino ester 1-7 with amine 1-4 reductive amination.Ester 1-7 with alkylating agent (for example alkyl chloride, alkyl bromide, alkyl iodide) alkylation in the presence of appropriate base (for example two (three silyls) Lithamide), is obtained intermediate ester 1-8.Usually, the ester that above-mentioned conversion forms is 1, and 3-is suitable-and 1,3-is anti--and the mixture of diastereomer, can be separated into two corresponding diastereomers with column chromatography.Similarly diastereomeric separation also can realize afterwards, obtains carrying out behind the corresponding sour 1-9 in the water-disintegrable cracking of ester 1-8.Hydrolysis is easy to finish under in room temperature to high temperature in normal condition (comprising lithium hydroxide, sodium hydroxide or potassium hydroxide), and this depends on ester group and substituent R
1Character.Can utilize cis-diastereomer acid littler, separate these diastereomers with different solvent crystallizations than the solvability of its trans epimer.
Under standard amide key formation property reaction conditions, comprise carbodiimide reagent (for example DCC, EDC) and catalyzer (for example DMAP, HOAT or HOBT), then with sour 1-9 and tetrahydro isoquinoline derivative 1-2 preparation formula 1-5 compound.
Flow process 1A
Intermediate 1-3 can also split by chirality HPLC and obtain 1-3a and 1-3b (flow process 1B).Can prepare suitable/trans isomer 1-5a and 1-5b then.
Flow process 1B
Flow process 2A illustrates a main path that is used to prepare intermediate 1-1 and intermediate 1-6.According to this approach, esterification 3-oxo-cyclopentane formic acid (2-1) under standard conditions (can be synthetic) (Stetter, H., Kuhlman, H., Liebigs Ann.Chim., 1979,944) with following currently known methods).R
18During for the tertiary butyl, can be prepared as follows corresponding ester 1-6: making suitable alcohol, is the trimethyl carbinol in this example, reacts in the presence of sulfuric acid with sour 2-1.Can realize protection (Greene, T., Wuts, P.G.M., ProtectiveGroups in Organic Chemistry, the John Wiley ﹠amp of oxo group among the 2-1 by many modes; Sons, Inc., New York, NY 1991).The available reagent trimethyl orthoformate is introduced specially suitable dimethylacetal blocking group in the presence of an acidic catalyst in suitable solvent (for example methylene dichloride and methyl alcohol).Perhaps, R
18During for methyl, can sour 2-1 be converted into 2-3 with trimethyl orthoformate and an acidic catalyst (for example tosic acid).In the presence of appropriate base (for example lithium diisopropylamine), alkylation ester 2-3 obtains intermediate 2-4 with alkylating agent (for example alkyl chloride, alkyl bromide or alkyl iodide).The ester protecting group of 2-4 can be sloughed in many ways, and this depends on the character of ester.Methyl esters (R
18=methyl) can be in room temperature or pyrohydrolysis in the presence of acid or alkali, and the tert-butyl ester (R
18=the tertiary butyl) under acidic conditions, is easy to cracking.Under these conditions, the dimethylacetal protection is sloughed simultaneously and is obtained 1-1.
Flow process 2A

Intermediate 1-1 can be prepared as single steric isomer (1-1a) by diverse ways (comprising the method that flow process 2B and 2C introduce).According to flow process 2B, racemic 1-1 can be converted into its benzyl ester.Have many methods can realize this esterification, wherein a kind of is with for example oxalyl chloride 1-1 to be converted into corresponding acyl chlorides earlier, uses benzyl alcohol to handle in the presence of alkali (for example triethylamine) then.Racemic benzyl ester 2-5 can obtain single stereoisomers 2-5a by the separation of chirality preparation HPLC.Can slough benzyl by several different methods and obtain chirality ketone acid 1-1a.A kind of method easily is for example to carry out hydrogenation in the presence of the Pd/C at catalyzer.
Flow process 2B

According to flow process 2C, chirality ketone acid intermediate 1-1a can prepare with commercially available optical purity amino acid 2-6.The protection of hydroxy-acid group can be passed through accomplished in many ways.R
18During for methyl, with the methyl alcohol esterification of in the presence of acid catalyst (for example HCl), finishing dealing with.Use Boc
2O handles the amine groups with protection 2-7.Stereoselective alkylation ester 2-8 in the presence of suitable alkali (for example two (three silyls) Lithamide) obtains intermediate 2-9 with alkylating agent (for example alkyl chloride, alkyl bromide or alkyl iodide).Hydrogenation obtains 2-10 in the presence of catalyzer (for example Pd/C).According to R
18Group ester hydrolysis under standard conditions obtains 2-11.R for example
18During for methyl (methyl esters), can heat or not heat with alkali (for example sodium hydroxide, lithium hydroxide or the potassium hydroxide) hydrolysis of finishing dealing with.The Boc blocking group can be sloughed under standard acidic condition (with containing the solvent (for example diox) of HCl or using TFA).Can obtain 1-1a (if the R that forms by several different methods oxidation 2-12
1Be achirality, then 1-1a is a single stereoisomers, if the R that forms
1A chiral centre is arranged, and then 1-1a is the mixture of steric isomer), comprise the method for handling with NBS, sodium methylate successively.
Flow process 2C

According to flow process shown in the 2D, in the presence of highly basic (for example lithium diisopropylamine), use ester 2-3 (R
18Be the benzyl or the tertiary butyl) enolate that obtains can with aldehyde (R
1aCHO) or ketone (R
1aR
2aCO) reaction obtains the intermediate 2-4.1 that suitable hydroxyalkyl replaces.The gained hydroxyl can be protected with different methods, comprises handling in the presence of alkali (for example triethylamine) with acetic anhydride obtaining intermediate 2-4.2.Under the condition that is fit to concrete blocking group, slough ester protecting group once more.Be the tert-butyl ester (R
18Be the tertiary butyl) time, under acidic conditions, slough protection.The latter also causes the cracking of acetal blocking group usually, and ketone acid 1-1.1 can prepare thus with one kettle way.Can be converted into final chemokine active regulator 1-9 according to preceding method (varied slightly is to adapt to the hydroxyl of protecting among the 1-1.1).
Flow process 2D
Amine 1-2 can prepare with the several different methods shown in the flow process 3A-3G.5-azepine tetrahydroisoquinoline fragment can be according to MarCoux, (J.Chem.Lett., 2000,2 (15), literature method preparation 2339-2341) such as J-F..Perhaps, it can method preparation shown in flow process 3A.With compound 3-1 (buy from market usually and obtain) bromination (Br
2, AcOH) obtain 3-2.Handle with DMF metal halogen exchange (NaH, tert-butyl lithium) back, obtains aldehyde 3-3.Available sodium formiate, hydroxylamine hydrochloride and formic acid are converted into nitrile with aldehyde radical.Gained nitrile 3-4 can handle with phosphoryl chloride and obtain 2-chloropyridine 3-5.The sodium salt displacement chloro of available dialkyl malonate.Itrile group with hydrogen and Raney Ni reduction 3-6 is followed cyclisation, obtains compound 3-7.Carry out decarboxylic reaction according to different esters with diverse ways.Under the situation of flow process 3A, with TFA the tert-butyl ester is carried out decarboxylation and obtain 3-8.Reduction reaction (BH
3), use Boc then
2O protection gained amine obtains 3-9, and 3-9 easily can purify.Can slough the Boc blocking group with several different methods and obtain 1-2a, comprise the method for handling with in anhydrous HCl Zai diox or other solvent of part.
Flow process 3A

The 1-2a compounds can also prepare according to flow process 3B.Commercially available 3-10 can be with methyl-iodide at alkali (K for example
2CO
3) methylating under existing obtains 3-11.With the protection piperidone at NH
3/ methyl alcohol carries out cycloaddition reaction under existing, and obtains 5-azepine tetrahydroisoquinoline 3-12 (R
10cCan be different blocking groups, for example benzyl or benzoyl).Nitro with hydrogen and catalyzer (for example Pd/C) hydrogenated compound 3-12 obtains 3-13.Heat with sulfuric acid after forming diazonium salt, obtain 5-azepine-7-hydroxy tetrahydro isoquinoline 99.9 3-14.According to R
10cDifferent properties and slough blocking group R with diverse ways
10cAt R
10cDuring for benzyl, can hydrogenation in the presence of HCl and catalyzer (for example Pd/C).At R
10cDuring for benzoyl, can be by the hydrolysis of the dense HCl solution of heating.Can use Boc
2O introduces the Boc blocking group easily and obtains 3-16 on 3-15.Then can be in conjunction with different R
10dObtain ether compound (referring to flow process 3C).Finally, the Boc blocking group with HCl or TFA slough gained compound 3-17 obtains 1-2b.Perhaps, compound 3-14 itself can be converted into ether compound (according to flow process 3C).Can slough the R of gained ether 3-18 as mentioned above
10c, be converted into compound 3-19.
Flow process 3B
5-azepine among the flow process 3B-7-hydroxy tetrahydro isoquinoline 99.9 3-14 can be converted into different ether compound (referring to flow process 3C) with 3-16.Can be with alkylogen and alkali (K for example
2CO
3, NaOH or NaH) the preparation alkyl oxide, obtain compound 3-19 and 3-22.Trifluoromethyl ethers can be prepared as follows: begin to generate methyl xanthate (NaH, CS
2MeI), use 1 successively then, 3-two bromo-5,5-T10 (or NBS) and HF/ pyridine solution are handled and are obtained 3-20.Aryl ethers can prepare in many ways, comprises the method for reacting in the presence of neutralized verdigris (II) and triethylamine with aryl boric acid, obtains compound 3-21.
Flow process 3C

Compound 1-2c (R
5Be halogenide (IVc)) can prepare according to flow process 3D.Compound 3-13 can be converted into halogenide 3-22 by diazonium salt according to typical method.Perhaps, can use the known cycloaddition reaction of the piperidone of due care.Slough blocking group R
10cCan finish by preceding method.
Flow process 3D
In addition, after being combined into senior intermediate, fragment 1-2c can further be modified the analogue that contains 7-aryl-5-azepine tetrahydroisoquinoline with preparation.This can followingly finish: make 5-azepine-7-halogen tetrahydroisoquinoline intermediate and aryl boric acid (or aryl stannane) coupling, by transition-metal catalyst Pd (OAc) for example
2Regulate.
The preparation method of flow process 3E-3G diagram tetrahydroisoquinoline amine component.The tetrahydroisoquinoline that is combined into the 1-5 amide moieties comprises one or two substituting group at different positions usually.These compound major parts can not be bought and obtain, but can obtain by synthetic, and flow process 3F and 3G have showed representational synthetic example.
Flow process 3E illustrates the example of synthetic simple tetrahydroisoquinoline (1-2d).According to this flow process, commercially available 4-trifluoromethyl acetonitrile (3-23) hydrogenation in the presence of Ra-Ni is obtained corresponding amine (3-24), then with trifluoro-acetic anhydride and the reaction of described amine.Gained acid amides (3-25) is handled in the presence of sulfuric acid with formaldehyde and is obtained ring compound (3-26), and it further is converted into tetrahydroisoquinoline (1-2d).
Flow process 3E

The also available 3-26 preparation of the tetrahydroisoquinoline that many 5-replace (flow process 3F).Obtain intermediate 3-28 5 iodate.After being converted into cyano compound (3-29) under palladium (0) catalytic condition, acid amides is cracked into amine 3-30,3-30 can be converted into amino ester 1-2e by two steps with high yield.Iodine compound 3-28 also can be converted into other compound shown in the experiment part.Can also after assembling final skeleton (1-5), modify these substituting groups to prepare new 1-5.1 class chemokine conditioning agent (referring to flow process 1).One is modified example is the corresponding formic acid of hydrolysis methyl esters (1-2e) preparation (referring to embodiment).
Flow process 3F

Heterocyclic 7-substituted tetrahydroisoquinolicompounds can utilize commercially available tetrahydroisoquinoline to obtain.Shown in flow process 3G, that tetrahydroisoquinoline (3-32) is nitrated in the presence of the vitriol oil with saltpetre.7-nitro-tetrahydroisoquinoline 3-33 is handled with protection amine with trifluoro-acetic anhydride, then the gained acid amides is obtained anils 3-34 with the hydrogenation under the 50psi hydrogen pressure of 10% palladium carbon.Basic hydrolysis obtains the amino tetrahydroisoquinoline (3-35) that replaces of 7-, and it can be used for the synthetic of other CCR2 antagonist or tetrahydro isoquinoline derivative.3-35 is protected in the presence of organic bases (for example triethylamine or diisopropylethylamine) with benzyl chloroformate, obtain carbamate 3-37.This intermediate can be used to prepare the tetrazolium (for example 1-2g) of tetrazolium and replacement.Intermediate 3-37 handle is generated the acid amides of trifluoroacetyl group protection with trifluoro-acetic anhydride, then it followingly is converted into the tetrazolium that trifluoromethyl replaces: make itself and triphenylphosphine under reflux, react 15h, then with sodiumazide in DMF in room temperature reaction.Obtain the tetrahydroisoquinoline 1-2g of heterocyclic substituted with the hydrogenation under nitrogen atmosphere of 10% palladium carbon.
Perhaps, 3-34 can followingly directly derive and be triazole 3-36: with N, dinethylformamide azine (N, N-dimethylformamide azine) is heated to backflow 24-48h in the presence of catalytic amount acid (for example toluenesulphonic acids).The alkaline hydrolysis of this intermediate obtains amine component 1-2f.
Flow process 3G

Being attached to other example of tetrahydroisoquinoline of The compounds of this invention amide moieties and their synthetic method further introduces at experimental section.
Amine 1-4 is obtained by different sources.Major part is commercially available, and part is that document is known and can prepare according to disclosed method, the method preparation that part is introduced according to this paper.Because their structure and preparation method are diversified, only introduce two flow processs in this part; Can find the different synthetic methods of amine 1-4 at experimental section.Flow process 4A has showed the method for the piperidines that a kind of synthetic 4-aryl replaces.The method coupling that enol trifluoromethane sulfonic acid ester 4-1 (according to Wustrow, D.J., Wise, L.D., Synthesis, (1991), 993-995 preparation) can introduce according to Wustrow and Wise with boric acid 4-2.Hydrogenation of olefin can be with hydrogen at catalyzer (Pd (OH) for example among the 4-3
2/ C) existence realizes down.Slough the Boc blocking group and can use standard acidic condition (for example HCl De dioxane solution or TFA/DCM) to realize, obtain piperidinyl-1-4.1.
Flow process 4A
Flow process 4B has showed another example of synthetic amine 1-4.At first commercially available alcohol (4-5) is obtained intermediate 4-6 with the sulfonyl methane chlorosulfonylation, then with the direct substitution heteroaryl piperidine 4-7 that obtains of tetrazolium.Under standard conditions, slough the Boc blocking group, obtain amine hydrochlorate 4-8.
Flow process 4B
Flow process 5 illustrates the another kind of main path of synthetic chemokines receptor modulators.According to this approach, use peptide coupling reagent (for example EDC) to make intermediate 2-11 (flow process 2C introduces) and amine 1-2 (flow process 1 is introduced) condensation obtain 5-1.With Boc blocking group (solvent that for example contains HCl under standard conditions; diox for example) sloughs, in the presence of reductive agent (for example sodium triacetoxy borohydride), handle gained amine 5-2 then with dialdehyde 5-3; cause dual standard reductive alkylation, follow cyclisation to obtain 1-5.2.The same with flow process 1, further modify (for example ester group of hydrolysis 1-5.2) and can obtain new chemokine receptor modulators 1-5.3.
Flow process 5
Flow process 6 illustrates a kind of method for preparing dialdehyde 5-3.According to this approach, handle with for example ozone, dimethyl sulfide successively and make the cracking of cyclenes 6-1 oxidisability, obtain dialdehyde.Perhaps, dialdehyde 5-3 substitutes with intermediate ozonide 6-2, and 6-2 can be directly used in dual reductive amination reaction, obtains 1-5.2.
Flow process 6

In some cases, in order to promote reaction or to avoid unwanted reaction product, can adjust the order of above-mentioned reaction process.Following examples only are used for further illustrating, and are not the restriction to the disclosure of invention.
Usually with rotatory evaporator concentrated solution under reduced pressure.Flash chromatography uses silica gel (230-400 order).MPLC is meant the medium pressure liquid chromatography method, except as otherwise noted, otherwise uses the silica stationary phase.Except as otherwise noted, otherwise NMR spectrum CDCl
3Solution obtains.Coupling constant (J) unit is hertz (Hz).Abbreviation: triethylamine (TEA), N, N-diisopropylethylamine (DIEA), saturated aqueous solution (sat ' d), room temperature (rt), hour (h), minute (min).
Following exemplary process is used for preparing the compound that embodiment hereinafter uses, and what can substitute that embodiment hereinafter uses can not buy the compound that obtains.
In some cases, in order to promote reaction or to avoid unwanted reaction product, can adjust the order of above-mentioned reaction process.Following examples only are used for further illustrating, and are not the restriction to the disclosure of invention.
Usually with rotatory evaporator concentrated solution under reduced pressure.Flash chromatography uses silica gel (230-400 order).Except as otherwise noted, otherwise NMR spectrum CDCl
3Solution obtains.Coupling constant (J) unit is hertz (Hz).Abbreviation: triethylamine (TEA), N, N-diisopropylethylamine (DIEA), saturated aqueous solution (sat ' d), room temperature (rt), hour (h), minute (min).
Following exemplary process is used for preparing the compound that embodiment hereinafter uses, and what can substitute that embodiment hereinafter uses can not buy the compound that obtains.
Intermediate 1
Steps A:

With 4-trifluoromethyl acetonitrile (10g, ethanol 49mmol) (100mL) and ammonium hydroxide (20mL 29.3% aqueous solution) solution Raney nickel (1g) hydrogenation 16h.Remove catalyzer by diatomite filtration, filtrate is evaporated to dried.Pure resistates is added drop-wise to cold (0 ℃) trifluoro-acetic anhydride, and (25mL 180mmol), stirs gained mixture 30min at 0 ℃.Reaction mixture is poured on the ice (250mL), stirs gained mixture 30min, filter then and shift out precipitation, the air-dry white solid product (13.4g, 90%) that obtains.
Step B:

The steps A product (13.4g, 44.0mmol) and Paraformaldehyde 96 (2g, the mixture of the disposable adding vitriol oil (90mL) and Glacial acetic acid (60mL) in mixture 50mmol) is at stirring at room gained mixture 16h.Reaction mixture is poured on the mixture of ice and water (1L), with ethyl acetate (3 * 150mL) extractions; The ethyl acetate layer water that merges (3 * 500mL), saturated sodium bicarbonate (200mL) and saturated NaCl (100mL) washing, use dried over mgso, the filtration final vacuum evaporates.Resistates (is used 10%Et with the silica column chromatographic purification
2O/ hexane wash-out) obtains product (8.29g, 60%).
Step C:

(8.29g, (20g, water 150mmol) (50mL) solution reflux and stir gained mixture 1h the trifluoroacetamide of step B preparation to add salt of wormwood in ethanol 26.0mmol) (200mL) solution.Rotary evaporation is removed ethanol, and water (150mL) is added resistates.Use CH
2Cl
2(3 * 100mL) extractions, the dichloromethane layer of merging is used dried over sodium sulfate with saturated sodium-chloride (100mL) washing, filters the final vacuum evaporation and obtains product (5.2g, 91%);
1H NMR 500MHz (CDCl
3) δ=1.81 (1H, br s), 2.84 (2H, d, J=6.0Hz), 3.15 (2H, t, J=6.0Hz), 4.05 (2H, s), 7.19 (1H, d, J=8.0Hz), 7.27 (1H, s), 7.37 (1H, d, J=8.0Hz).
Intermediate 2
Steps A:
5-trifluoromethyl-2-pyridine alcohol (51.0g, 307mmol) and sodium acetate (26.2g, add in Glacial acetic acid 319mmol) (200mL) solution bromine (16.7mL, 325mmol), with the gained mixture 80 ℃ the heating 2.5h.Allow reactant be cooled to room temperature, then reduction vaporization.Resistates neutralizes with saturated sodium bicarbonate solution, with ethyl acetate (3 * 200mL) extractions.Merge organic layer, use dried over mgso, filter, vacuum-evaporation obtains 74.45g (98.7%) crude product.
1H?NMR(400MHz,CDCl
3)δ8.04(d,J=2.6Hz,1H),7.89(m,1H)。
Step B:
Under nitrogen atmosphere, (steps A, 48.8g 202mmol) are divided into short run and add NaH (8.9g, anhydrous THF (500mL) suspension 220mmol) with the pyridine that replaces.Intermediate add finish after, reaction mixture is to-78 ℃, with syringe drip tert-butyl lithium (260mL, 444mmol).After stirring 5min, (50mL 707mmol) is lower than-50 ℃ with holding temperature slowly to add DMF.Then the gained mixture is stirred 10h, allow it rise to room temperature.Mixture is used ethyl acetate (1000mL) dilution then with 2N HCl quencher.Isolate organic layer, use the salt water washing, use dried over mgso, vacuum-evaporation.Required product is separated out from ethyl acetate and hexane, filters to obtain light brown solid (28.55g, 73.8%).
1H?NMR(500MHz,CD
3OD)δ10.13(s,1H),8.21(s,2H)。
Step C:

With the intermediate of step B (18g, 95mmol), sodium formiate (7.1g, 105mmol), hydroxylamine hydrochloride (7.3g, 110mmol) and the mixture of formic acid (150mL) at stirring at room 2h, reflux then and spend the night.Reaction mixture left standstill 7 days in room temperature.In reactant impouring water, with ethyl acetate (3x) extraction.The organic layer water (2x), the saturated NaHCO that merge
3With the salt water washing, use dried over sodium sulfate, filter, vacuum concentration obtains required brown powder shape product (17.84g, 89.8%).
1H?NMR(400MHz,CD
3OD)δ8.37(d,J=2.7Hz,1H),8.19(q,J=0.7Hz,0.3??/Hz,1H)。
Step D:
Phosphoryl chloride (13.4mL, 144mmol) and benzoquinones (8.7mL, add in mixture 73.4mmol) step C product (24.6g, 131mmol), with gained mixture backflow 3h.Cooling reactant to 100 ℃ slowly adds entry (70mL) then.Further cooling mixture carefully neutralizes with saturated sodium bicarbonate solution to room temperature.Water layer merges organic layer with ethyl acetate (3x) extraction, uses dried over mgso, filters vacuum-evaporation.Crude product is purified with flash chromatography and is obtained (23.5g, 87.0%) required compound.
1H?NMR(500MHz,CDCl
3)δ8.88(d,J=2.0Hz,1H),8.26(d,J=2.5Hz,1H)。
Step e:

Under nitrogen atmosphere, (7.8g, THF 200mmol) (100mL) suspension drip propanedioic acid tert-butyl ester methyl esters (20mL, anhydrous THF (100mL) solution 120mmol) to NaH with syringe.Stirred reaction mixture 0.5h slowly adds intermediate (20.1g, THF 97.6mmol) (200mL) solution that step D prepares with syringe then.Spend the night at the stirring at room reactant, use the quencher of ammonium chloride saturated solution then.Isolate organic layer, water layer extracts with ethyl acetate (3x).Dried over sodium sulfate is used in organic layer water (3x) washing that merges, and filters vacuum-evaporation.Flash chromatography obtains the required clean product of 31.76g (94.6%).
1H?NMR(500MHz,CDCl
3)δ9.03(d,J=1.5Hz,1H),8.25(d,J=2.0Hz,1H),5.25(s,1H),3.86(s,3H),1.52(s,9H)。
Step F:

(18.2g, ethanol 52.9mmol) (130mL) suspension places the Parr device, spends the night in 40psi hydrogenation with Raney Ni (1g) and step e product.Suspension is by diatomite filtration, and vacuum-evaporation filtrate obtains 16.35g (97.8%) crude product.
1H?NMR(500MHz,CDCl
3)δ8.83(s,1H),7.89(s,1H),7.82(s,1H),4.83(d,J=16Hz,1H),4.72(s,1H),4.49(d,J=16Hz,1H),1.45(s,9H)。
Step G:

(16g adds TFA (30mL) in DCM 51mmol) (60mL) mixture to the step F product, at stirring at room gained mixture 0.5h.Reduction vaporization solution is dissolved in DCM with resistates.Slowly add the saturated sodium bicarbonate solution neutralise mixt, shift out organic layer.The aqueous solution merges all organic layers then with DCM (4x) extraction, uses dried over sodium sulfate, filters, and vacuum-evaporation obtains the required product of 10.42g (95.2%).
1H?NMR(400MHz,CDCl
3)δ8.81(s,1H),7.78(s,1H),7.30(s,1H),4.63(s,2H),3.90(s,2H)。
Step H:

(18.0g, (417mL's THF solution of adding 1.0M borane 420mmol), spends the night at stirring at room gained solution step G product in THF 83.3mmol) (50mL) solution.Reduction vaporization solution is used resistates the 1%HCl/MeOH solution-treated then, with the gained mixture 50 ℃ of heated overnight, to decompose the borane complex compound.With twice of the methanol solution re-treatment of acid to guarantee to eliminate the borane complex compound.This reacting coarse product is used for next reaction immediately.
(43mL, (the gained mixture is in stirred overnight at room temperature for 36.4g, 167mmol) processing with tert-Butyl dicarbonate for DCM solution 250mmol) with above crude product (83.3mmol, suppose 100% transform) and DIEA.Solution saturated sodium bicarbonate solution, water and salt water washing.Combining water layer is used DCM (2x) washing again.The organic layer that merges with dried over sodium sulfate filters then, is evaporated to dried.Crude product (11.89g, 47.2%, the latter two steps) yellow solid of purifying with flash chromatography and MPLC and obtain.
1H?NMR(500MHz,CDCl
3)δ8.69(s,1H),7.66(s,1H),4.67(s,2H),3.79(t,J=6.0Hz,2H),3.08(t,J=5.5Hz,2H),1.51(s,9H)。
Step I:
The product (11.89g) of step H is handled with 4M HCl De dioxane solution.At stirring at room solution 2h, vacuum-evaporation obtains yellow powder shape intermediate 2 (10.85g, 99%) then.LC-MS C
9H
10F
3N
2[M
+H
+] calculated value 202.07, measured value 203.0.
Intermediate 3
Steps A:

With 3-oxo-cyclopentane-methyl-formiate (20g, 160mmol) and trimethyl orthoformate (85mL, (3.00g 15.6mmol) handles, at stirring at room gained solution 4h with the catalytic amount tosic acid for 780mmol) methanol solution.Solvent evaporated under reduced pressure is dissolved in resistates ether (600mL) then.Solution with saturated sodium bicarbonate (2 * 200mL), the washing of water (150mL) and salt solution (200mL), use anhydrous sodium sulfate drying, filtration, evaporating solvent as previously mentioned then.Flash column chromatography purification (eluent: 25% ether/pentane) obtain the required clarification oily of 21.52g (73%) product.
1H?NMR(500MHz,CDCl
3)δ3.68(s,3H),3.21(d,J=9.9Hz,6H),2.89(p,J=8.5Hz,1H),2.14-2.05(m,2H),2.02-1.80(m,4H)。
Step B:
The anhydrous THF of 150mL that packs in the flame-dried 500mL round-bottomed flask is cooled to-78 ℃ with acetone/the dry ice bath then under nitrogen atmosphere.With syringe with diisopropylamine (19.2mL 137mmol) adds the refrigerative solvent, slowly add then the 2.5M n-Butyl Lithium hexane solution (55mL, 140mmol).After stirring 5min, (21.52g, 50mL THF solution 114.4mmol) stir gained mixture 2h at-78 ℃ to drip the methyl ketal intermediate 3 of steps A with syringe.(34.3mL 343mmol), stirs the gained mixture overnight, allows it slowly rise to room temperature to drip 2-iodopropane with syringe then.With 10% citric acid solution quencher reactant, separation of organic substances.(3 * 150mL) extractions merge all organism to water layer, use anhydrous sodium sulfate drying, filter reduction vaporization with ether.Crude product is with quick post (flask the column) (eluent: 20% ether/pentane) obtain the required product of 16.74g (64%) of purifying.
1H?NMR(400MHz,CDCl
3)δ3.69(s,3H),3.18(d,J=20.5Hz,6H),2.57(d,J=13.9Hz,1H),2.29-2.20(m,1H),1.90(p,J=6.8Hz,1H),1.88-1.80(m,2H),1.69-1.61(m,2H),0.89(dd,J=11.9Hz,6.8Hz,6H)。
Step C:
With ester (intermediate 3 step B introduce, 16.7g, ethanol 72.7mmol) (30mL) solution is handled with 5M NaOH (55mL), with the gained mixture heating up to refluxing 3 days.Cooling mixture is used the concentrated hydrochloric acid acidifying to room temperature then.The reduction vaporization organic solvent, (5 * 100mL) extract water layer with DCM.Merge organic extract liquid, use anhydrous magnesium sulfate drying, filter, vacuum-evaporation obtains rough yellow oily 3-oxo-cyclopentane formic acid (11.07g, 90%).
1HNMR(500MHz,CDCl
3)δ2.70(d,J=18.1Hz,1H),2.44-2.39(m,1H),2.30-2.15(m,2H),2.14(dd,J=18.1,1.0Hz,1H),2.06(p,J=6.9Hz,1H),1.98(m,1H),0.98(dd,J=11.4,6.9Hz,6H)。
Step D:
Method A:
Acid (intermediate 3 step C introduce, 2.00g, and (1.54mL 17.6mmol), adds 2 DMF then to add oxalyl chloride in DCM 11.8mmol) (50mL) solution.At stirring at room solution 80min, reduction vaporization then.Resistates is dissolved in DCM (2mL), with syringe join preparation intermediate 1 (2.36g, 11.8mmol) and triethylamine (2.13mL, DCM 15.3mmol) (40mL) solution.At stirring at room gained mixture 18h, water (25mL) quencher then.Separation of organic substances with 1N HCl, saturated sodium bicarbonate and salt water washing, is used anhydrous magnesium sulfate drying, filters, then evaporation.Crude product is with the MPLC (eluent: 60% ethyl acetate/hexane) obtain intermediate 3 (3.18g, 77%) of purifying.
1H?NMR(500MHz,CDCl
3)δ7.46(d,J=7.3Hz,1H),7.39(s,1H),7.29(d,J=7.7Hz,1H),4.81(m,2H),3.93(m,1H),3.82(m,1H),2.94(m,3H),2.54(m,1H),2.43(d,J=8.5Hz,1H),2.32(m,2H),2.26(p,J=6.6Hz,1H),2.16(m,1H),0.93(dd,J=19.7Hz,6.8Hz,6H)。LC-MS C
19H
23F
3NO
2Calculated value 353.16, measured value: [M+H
+] 354.25.
Method B:
Sour intermediate 3 (1.0g with step C preparation, 5.9mmol), intermediate 1 (1.18g, 5.88mmol), DMAP (71mg, 0.59mmol), N, the N-diisopropylethylamine (1.02mL, 5.88mmol) and the mixture of methylene dichloride (20mL) with 1-[3-(dimethylamino) propyl group]-3-ethyl-carbodiimide hydrochloride (EDC, 2.25g, 11.7mmol) handle, in stirred overnight at room temperature.Reaction mixture with methylene dichloride (30mL) dilution, water (2 * 20mL), salt solution (1 * 30mL) washing, use anhydrous sodium sulfate drying, evaporating solvent.MPLC purification (eluent: 60% ethyl acetate/hexane) obtain neat compounds 1.08g (52%).LC-MS C
19H
23F
3NO
2Calculated value 353.16, measured value [M+H
+] 354.25.
Intermediate 4
Acid (intermediate 3 step C introduce, 540mg, and (0.834mL 9.60mmol), adds 2 DMF then to add oxalyl chloride in DCM 3.20mmol) (50mL) solution.At stirring at room solution 80min, reduction vaporization then.Resistates is dissolved in DCM (2mL), and the intermediate of add preparing with syringe 2 (880mg, 3.20mmol) and triethylamine (0.820mL, DCM 6.50mmol) (20mL) solution.At stirring at room gained mixture 18h, water (25mL) quencher then.Separation of organic substances with saturated sodium bicarbonate and salt water washing, is used anhydrous sodium sulfate drying, filters, then evaporation.Crude product is purified with MPLC and (is used the stage gradient eluent: the 0-70% ethyl acetate/hexane) obtain intermediate 2 (720mg, 64%).ESI-MS calculated value C
18H
21F
3N
2O
2: 354.16; Measured value 355 (M+H)
Intermediate 5
By chiral separation intermediate 4 is split as its independent enantiomer, adopts the HPLC that is equipped with preparation type ChiralPak AD post.Followingly finish separation: per injection 100mg, eluent adopt 25% Virahol and 75% heptane, flow velocity 9mL/min.
Intermediate 6

Steps A:
Method A:
With 3-oxo-cyclopentane-carboxylic acid (Stetter, H., Kuhlmann, H.Liebigs Ann.Chem., 1979,7,944-9) (5.72g, methylene dichloride 44.6mmol) (30mL) solution N, the N '-di-isopropyl-O-tertiary butyl-isourea (21.2mL, 89.3mmol) handle, spend the night at the stirring at room reaction mixture.Filtering precipitation N, N '-di-isopropyl urea, vacuum concentrated filtrate, distillation purification resistates (bp:125-129 ℃ 18mmHg) obtains 4.74g (58%) clean product.
1H?NMR(500MHz,CDCl
3):δ3.02(p,J=7.8Hz,1H),2.05-2.50(m,6H),1.45(s,9H)。
13C?NMR(125MHz,CDCl
3):δ217.00,173.47,80.99,41.88,41.14,27.94,26.57。
Method B:
Packing among the 2L round bottom RBF, (113g 940mmol), adds methylene dichloride (940mL) to anhydrous magnesium sulfate.Under agitation, with suspension with the vitriol oil (12.5mL 235mmol) handles, in 15min, add then 3-oxo-cyclopentane-carboxylic acid (30.1g, 235mmol).After stirring 15min, and the adding trimethyl carbinol (87g, 1.2mol).With the plug closes reaction vessel to help the reservation iso-butylene, at stirring at room 72h.Leach solid by Celite pad, filtrate volume is reduced to about 500mL, with saturated sodium bicarbonate solution (2 * 150mL) washings.Use the anhydrous magnesium sulfate drying organic phase, filter, underpressure distillation removes desolvate (180mmHg).Crude product is purified by distillation and is obtained 39.12g (90%) clean product.
Step B:
(11.54g, (41.4mL 251mmol) handles in the presence of tosic acid (400mg), at stirring at room 48h methylene dichloride 62.64mmol) (200mL) solution with trimethyl orthoformate with 3-oxo-cyclopentane t-butyl formate.The black reaction mixture is poured on the saturated sodium bicarbonate solution crude product dichloromethane extraction.With the organic extract liquid that anhydrous magnesium sulfate drying merges, solvent removed in vacuo, distillation purification crude product (bp.:104 ℃ 4mmHg) obtains the required product of 12.32g (85%).
1H?NMR(500MHz,CDCl
3):δ3.21(s,3H),3.20(s,3H),2.80(m,1H),2.10-1.80(bm,6H),1.46(s,9H)。
13C?NMR(125MHz,CDCl
3):δ174.9,111.2,80.3,67.8,49.2,42.5,37.4,33.8,28.3,22.0。
Step C:
The anhydrous THF of 100mL packs in the flame-dried 500mL round-bottomed flask, under nitrogen atmosphere, be cooled to-78 ℃ then with acetone/the dry ice bath, with syringe with diisopropylamine (7.9mL, 56mmol) add the refrigerative solvent, slowly add then the 2.5M n-Butyl Lithium hexane solution (22.6mL, 56.45mmol).After stirring 5min, with syringe drip acetal (intermediate 6 step B introduce, 10.0g, 50mL THF 43.4mmol) stirs gained mixture 2h at-78 ℃.(7.3mL 130mmol), stirs gained mixture 2h at-78 ℃ to drip acetaldehyde with syringe then.With quencher reactant in mixture impouring 10% citric acid solution (300mL), use methylene dichloride (2 * 150mL) extractions then.Merge organism, use anhydrous magnesium sulfate drying, filter reduction vaporization.When reaction or subsequent disposal, the part acetal is hydrolyzed to ketone, therefore, rough mixture is directly used in next step need not to purify again.
Step D:
Rough intermediate (intermediate 6 step C introduce, and 56.45mmol supposes step C transformation efficiency 100%) is handled with the dichloromethane solution of 10% trifluoroacetic acid, in stirring at room gained mixture overnight.The vacuum concentration reactant, dilute with water is used dichloromethane extraction then.Merge organism, use anhydrous magnesium sulfate drying, filter, reduction vaporization obtains 8.04g (83%) crude product, directly uses to need not to purify again.
Step e:

(intermediate 6 step D introduce in acid, 300mg, 1.74mmol), intermediate 2 (486,1.74mmol), HOAt (237mg, 1.74mmol), N, N-diisopropylethylamine (0.606mL, 3.48mmol) and the mixture of methylene dichloride (15mL) with 1-[3-(dimethylamino) propyl group]-3-ethyl-carbodiimide hydrochloride (EDC, 667mg 3.48mmol) handled, stirring at room 5 days.Reaction mixture dilutes with methylene dichloride (30mL), and anhydrous sodium sulfate drying is used in water (20mL), salt solution (20mL) washing, filters vacuum evaporating solvent.(eluent: 100% ethyl acetate) purification obtains intermediate 6260mg (42%) by the preparation template.
Intermediate 7
Steps A

With 2-fluoro-4-trifluoromethyl acetonitrile (10g, ethanol 49mmol) (100mL) and ammonium hydroxide (20mL 29.3% aqueous solution) soln using Raney nickel (1g) hydrogenation 16h.Remove catalyzer by diatomite filtration, filtrate is evaporated to dried.Pure resistates is added drop-wise to cold (0 ℃) trifluoro-acetic anhydride, and (25mL 180mmol), stirs gained mixture 30min at 0 ℃.Reaction mixture is poured on the ice (250g), stirs gained mixture 30min, filter then and shift out precipitation, the air-dry white solid product (13.4g, 90%) that obtains;
1H NMR 500MHz (CDCl
3) δ=3.02 (2H, t, J=7.0Hz), 3.66 (2H, q, J=6.6Hz), 6.44 (1H, brs), 7.34 (2H, m), 7.41 (1H, d, J=7.8Hz).
Step B

The steps A product (13g, 44mmol) and Paraformaldehyde 96 (2.0g, the mixture of the disposable adding vitriol oil (90mL) and Glacial acetic acid (60mL) in mixture 48mmol) is at stirring at room gained mixture 16h.Reaction mixture is poured on the mixture of ice and water (1L), with ethyl acetate (3 * 150mL) extractions; The ethyl acetate layer water that merges (3 * 500mL), saturated sodium bicarbonate (200mL) and saturated NaCl (100mL) washing, use dried over mgso, the filtration final vacuum evaporates.Resistates (is used 10%Et with the silica column chromatographic purification
2O/ hexane wash-out) obtains product (8.29g, 60%);
1H NMR 500MHz (CDCl
3) δ=3.01 (2H, m), 3.91 and 3.97 (2H, t, J=6.2Hz), 4.83 and 4.88 (2H, s), 7.21-7.28 (3H, m).
Step C

(8.3g, (20g, water 150mmol) (50mL) solution reflux and stir gained mixture 1h the trifluoroacetamide of step B preparation to add salt of wormwood in ethanol 26mmol) (200mL) solution.Ethanol is removed in decompression, and water (150mL) is added resistates, uses CH
2Cl
2(3 * 100mL) extractions.The dichloromethane layer that merges is used dried over sodium sulfate with saturated NaCl (100mL) washing, filters the final vacuum evaporation, obtains product (5.2g, 91%);
1H NMR 500MHz (CDCl
3) δ=1.74 (1H, br s), 2.78 (2H, d, J=6.0Hz), 3.17 (2H, t, J=6.0Hz), 4.05 (2H, s), 7.04-7.14 (3H, m).
Intermediate 8

Steps A

Three neck round-bottomed flasks are loaded onto addition funnel and condenser, and (2.45g 37.4mmol), uses the flame drying to add zinc powder.After cooling, use the nitrogen purge system, add 6mL THF, add then glycol dibromide (0.298mL, 3.46mmol).Mixture is heated to vigorous reflux with heating gun, and the stirring~30s (observing gas evolution) that refluxes is cooled to room temperature then.Repeat twice of heating, cooling.Add then the chloro trimethyl silane (0.402mL, 3.17mmol), at stirring at room mixture 20min.Tert-butoxycarbonyl-4-iodo-piperidines is (known: Billotte, S.Synlett (1998), 379., 8.97g, 15mL THF solution 28.8mmol) to add N-in about 1min.At 50 ℃ of stirred reaction mixture 1.5h, be cooled to room temperature then.Therebetween, (267mg, 1.15mmol) (298mg, mixture 0.288mmol) is dissolved in 6mLTHF, at stirring at room 15min, adds organic zinc solution with three (dibenzalacetone)-two palladium (0) chloroform adducts with three-2-furyl phosphine under nitrogen atmosphere.Add 2-bromo pyrimi piperidine (5.50g, 58mL THF 34.6mmol) and 20mL N,N-dimethylacetamide solution then.Reaction mixture is heated to 80 ℃, stirs 3.5h, be cooled to room temperature then, stir 36h.By the diatomite filtration reaction mixture, filter cake washs with ethyl acetate.Filtrate further with the ethyl acetate dilution, is washed with saturated sodium bicarbonate solution.Water layer is used ethyl acetate extraction once more, merges organic layer, washes twice with water, with the salt water washing once.The organic phase anhydrous magnesium sulfate drying filters, and concentrates then.With flash chromatography purification (silicon-dioxide, stage gradient: 25% ethyl acetate/hexane, 40% ethyl acetate/hexane, 60% ethyl acetate/hexane, 80% ethyl acetate/hexane, 100% ethyl acetate) the acquisition pure 4-of 4.92g (2-pyrimidyl)-piperidines product (65%).
1H?NMR(500MHz,CDCl
3):δ8.70(d,J=5.0Hz,2H),7.16(app?t,J=4.5Hz,1H),4.24(br?s,2H),3.05(m,1H),2.89(br?m,2H),2.01(br?d,J=13Hz,2H),1.84(dq,J=4.5,12.5Hz,2H),1.49(s,9H)。
Step B
(4.64g 17.6mmol) is dissolved in 4NHCl De dioxane solution (50mL), at stirring at room 2.25h to the N-tert-butoxycarbonyl piperidines that steps A is prepared.Concentrated reaction mixture obtains 4.16g piperidine hydrochlorate (100%), does not need to purify again.
1H NMR (500MHz, CD
3OD): δ 8.95 (d, J=5.5Hz, 2H), 7.60 (t, J=5.0Hz, 1H), 3.53 (dt, J=13,3.5Hz, 2H), 3.35 (tt, J=4.0,11.0Hz, 1H), 3.20 (brt, J=13.8Hz, 2H), 2.30 (br d, J=14.0Hz, 2H), 2.11-2.20 (m, 2H); ESI-MS C
9H
13N
3Calculated value: 163; Measured value: 164 (M+H).
Intermediate 9

This intermediate still substitutes the 2-bromo pyrimi piperidine with the 4-bromo pyrimi piperidine according to the method preparation that is used for intermediate 8.LC-MS C
9H
13N
3Calculated value 163.28, measured value [M+H]
+64.
Intermediate 10
4-(1H-1,2,4-triazol-1-yl) piperidine hydrochlorate

Steps A
4-hydroxy piperidine-1-t-butyl formate
Methylene dichloride (500mL) stirred solution to 4-hydroxy piperidine (60.8g) adds tert-Butyl dicarbonate (19g, methylene dichloride 0.55mol) (500mL) solution very lentamente.After adding, add 1h consuming time, at stirring at room gained mixture 5h.Then mixture is used saturated NaHCO
3, 3N HCl and salt water washing, drying, evaporation obtains viscous crude shape 4-hydroxy piperidine-1-t-butyl formate (90g) then.
Step B:4-[(methyl sulphonyl) oxygen base] piperidines-1-t-butyl formate
At 0 ℃, to 4-hydroxy piperidine-1-t-butyl formate (21.1g, slowly add 100mmol) and in methylene dichloride (250mL) stirred solution of triethylamine (22mL) methane sulfonyl chloride (9.0mL, 1.1eq.).Restir gained mixture 1h forms white solid during this period.Then mixture is washed with 3N HCl, use Na
2SO
4Drying, evaporation obtains white solid 4-[(methyl sulphonyl then) the oxygen base] piperidines-1-t-butyl formate (29.2g).
1H?NMR(400MHz,CDCl
3):δ4.92-4.87(m,1H),3.75-3.69(m,2H),3.34-3.28(m,2H),3.05(s,3H),2.01-1.94(m,2H),1.87-1.78(m,2H)。
Step C:4-(1H-1,2,4-triazol-1-yl) piperidine hydrochlorate
In room temperature to the 4-[(methyl sulphonyl) the oxygen base] piperidines-1-t-butyl formate (5.9g, 21mmol) and 1,2, add in the DMF stirred solution of 4-triazole (1.8g, 25mmol eq.) sodium hydride (60% is scattered in mineral oil, 1.0g, 25mmol).Stirred the mixture 5 days at 60 ℃, TLC confirms not remaining initial methanesulfonates.In mixture impouring frozen water, with ethyl acetate (3x) extraction.Dry organic layer, evaporation obtains white solid 4-(1H-1,2,4-triazol-1-yl) piperidines-1-t-butyl formate with the quick post purification of silicon-dioxide (with 0-10% methanol/ethyl acetate wash-out).(4N 10mL) handles 2h with hydrogenchloride De diox with solid then.Then most of diox is removed in the mixture evaporation and obtained white solid, it is obtained required 4-(1H-1,2,4-triazol-1-yl) piperidine hydrochlorate (5.55g) with the ethyl acetate washing.
1H?NMR(300MHz,CD
3OD):δ10.00(s,1H),8.97(s,1H),5.10-5.00(m,1H),3.63-3.58(br.d,2H),3.33-3.26(br.d,2H),2.50-2.30(m,4H)。
Following intermediate 10-16 uses the 4-[(methyl sulphonyl according to the method preparation that is similar to intermediate 10) the oxygen base] piperidines-1-t-butyl formate and suitable heterocycle.
Intermediate 11
4-(1H-pyrazol-1-yl) piperidine hydrochlorate
With the method preparation of pyrazoles according to intermediate 10.
Intermediate 12
4-(1H-imidazoles-1-yl) piperidine hydrochlorate

Prepare with the method for imidazoles according to intermediate 10:
1H NMR (400MHz, CD
3OD): (s, 1H), 7.86 (s, 1H), 7.65 (s, 1H), 4.9-4.8 (is hidden in CD to δ 9.18
3Under the OD peak, 1H), 3.61-3.61 (br.d., 2H), 3.33-3.26 (m, 2H), 2.49-2.45 (br.d, 2H), 2.39-2.28 (m, 2H).
Intermediate 13
4-(1H-1,2,3-triazol-1-yl) piperidine hydrochlorate

With the method preparation of 1,2,3-triazoles according to intermediate 10.
4-(1H-1,2,3-triazol-1-yl) piperidine hydrochlorate:
1H NMR (400MHz, CD
3OD): δ 8.77 (s, 1H), 8.54 (s, 1H), 5.26-5.19 (m, 1H), 3.65-3.59 (m, 2H), 3.37-3.29 (m, 2H), 2.60-2.54 (m, 2H), 2.50-2.39 (m, 2H).
Intermediate 14
4-(2H-1,2,3-triazole-2-yl) piperidine hydrochlorate
With the method preparation of 1,2,3-triazoles according to intermediate 10.
4-(2H-1,2,3-triazole-2-yl) piperidine hydrochlorate:
1H NMR (400MHz, CD
3OD): δ 7.72 (s, 2H), 4.94-4.87 (m, 1H), 3.54-3.48 (m, 2H), 3.28-3.22 (m, 2H), 2.46-2.32 (m, 4H).
Intermediate 15
4-(1H-tetrazolium-1-yl) piperidine hydrochlorate

With the method preparation of tetrazolium according to intermediate 10.
4-(1H-tetrazolium-1-yl) piperidine hydrochlorate:
1H NMR (400MHz, CD
3OD): δ 8.77 (s, 1H), 5.30-5.23 (m, 1H), 3.58-3.53 (m, 2H), 3.35-3.29 (m, 2H), 2.58-2.2.52 (m, 2H), 2.48-2.38 (m, 2H).
Intermediate 16
4-(2H-tetrazolium-2-yl) piperidine hydrochlorate

With the method preparation of tetrazolium according to intermediate 10.
4-(2H-tetrazolium-2-yl) piperidine hydrochlorate:
1H NMR (400MHz, CD
3OD): δ 9.32 (s, 1H), 5.08-5.00 (m, 1H), 3.61-3.57 (m, 2H), 3.33-3.28 (m, 2H), 2.52-2.47 (m, 2H), 2.42-2.32 (m, 2H).
Intermediate 17

With of the method preparation of 5-methyl tetrazolium according to intermediate 10.
1H?NMR(400MHz,CD
3OD):δ5.08-5.00(m,1H),3.61-3.57(m,2H),3.33-3.28(m,2H),2.52-2.47(m,2H),2.42-2.32(m,2H),1.68(s,3H)。
Intermediate 18
Steps A
With hydroxy amine hydrochloric acid salt (8.26g, 119mmol) and triethylamine (16.6mL 119mmol) mixes in 50mL DMSO.Filtering suspension liquid is to remove triethylamine hydrochloride, and filter cake washs with THF.The filtrate partial concentration is removed THF.(5.0g 24mmol) adds above DMSO solution, stirs gained reaction mixture 3h at 75 ℃, then in stirred overnight at room temperature with commercially available 1-tert-butoxycarbonyl-4-cyano group piperidines then.Reaction mixture dilutes with ethyl acetate, washes with water.Water layer is used ethyl acetate extraction once more, and the organic layer of merging washes with water four times, with the salt water washing once.Use the anhydrous magnesium sulfate drying organic layer, filter, concentrate then and obtain the 3.51g product.
Step B
(1.02g, (897mg 5.03mmol) handles 20mL THF solution 4.19mmol), observes gas release and heat release subsequently with thiocarbonyldiimidazole with the steps A intermediate.At stirring at room reaction mixture 1h, transfer to the 180mL 5 of silica gel #60 (20g): 1CHCl then
3In/the methanol suspension.Stirring at room reaction mixture 5 days, filter then, concentrate.MPLC purification (silicon-dioxide, 50% ethyl acetate/hexane) obtains 143mg thiadiazoles ketone.
The Boc intermediate
1H NMR (500MHz, CD
3OD): δ 4.16 (m, 2H), 2.86 (t, J=11.5Hz, 2H), 2.77 (tt, J=4.0,11.0Hz, 1H), 1.98 (dd, J=2.0,13.0Hz, 2H), 1.73 (dq, J=4.5,12.0Hz, 2H), 1.47 (s, 9H).
(139mg 0.487mmol) is dissolved in 4N HCl/ diox (5mL), at stirring at room 1.5h with the Boc intermediate.Concentrated reaction mixture obtains 94.3mg piperidine hydrochlorate product.
Intermediate 19
4-(1H-pyrazole-3-yl) piperidines
Steps A: 4-(1H-pyrazole-3-yl) pyridine
(75mL 0.68mol) and in dry-out benzene (1L) mixture of ethyl formate (109mL) adds sodium methylate (73g), with gained mixture backflow 18h to the 4-acetylpyridine.Cooling mixture goes out benzene from viscous solid (forming) decant reaction.With crude product water-soluble (700mL), add the hydrazine dihydrochloride, at stirring at room gained mixture 2h.Add 5N NaOH dissolving mixt.Precipitation is shifted out in filtration, and drying obtains 4-(1H-pyrazole-3-yl) pyridine (35g).
Step B:1-benzyl-4-(1H-pyrazole-3-yl)-1,2,3, the 6-tetrahydropyridine
Add in the 2-propyl alcohol (60mL) of 4-(1H-pyrazole-3-yl) pyridine (9.6g) heat (80 ℃) solution bromotoluene (20mL, 2.5eq.), reflux gained mixture 10min.After the cooling, filtering-depositing washs with the 2-propyl alcohol again, and is air-dry in ice bath.0 ℃ with solid suspension in ethanol, in 30min, divide several to add sodium borohydrides (13g), 30min once more stirs the mixture.Carefully add entry quencher reactant, ethanol is removed in evaporation, and resistates distributes between methylene dichloride and water.Use the dried over mgso organic layer, filter, evaporation obtains 1-benzyl-4-(1H-pyrazole-3-yl)-1,2,3,6-tetrahydropyridine (16g) then.
Step C:4-(1H-pyrazole-3-yl) piperidines
With 1-benzyl-4-(1H-pyrazole-3-yl)-1,2,3, (10%, 1g) spend the night under 40psi by hydrogenation with palladium carbon for 6-tetrahydropyridine solution (16g).Remove catalyzer by diatomite filtration, evaporated filtrate.NMR confirms that product is 1-benzyl-4-(1H-pyrazole-3-yl)-piperidines (16g).
Adding palladium carbon in ethanol (400mL) solution of 1-benzyl-4-(1H-pyrazole-3-yl)-piperidines (16g) and formic acid (30mL) (10%, 2g), in stirring at room gained mixture overnight.Remove by filter catalyzer, evaporated filtrate.By adding the dichloromethane solution purified product of tert-Butyl dicarbonate (2eq.) and triethylamine (1.5eq.), obtain the intermediate of Boc protection.The evaporation back obtains pure 4-(1H-pyrazole-3-yl) piperidines-1-t-butyl formate by silica column chromatographic purification (with 20-40% ethyl acetate/hexane wash-out).Then the Boc intermediate is handled with the methanol solution of HCl, obtained 4-(1H-pyrazole-3-yl) piperidine hydrochlorate (3.5g).Lose raw material because forming two-Boc product, do not collect two-Boc product.
1H?NMR(400MHz,CDCl
3):δ8.00(s,2H),3.48(br.d,J=13Hz,2H),3.28-3.20(m,1H),3.13(br.t,J=13Hz,2H),2.23(br.d,J=14Hz,2H),1.97-1.85(m,2H)。
Intermediate 20

Steps A:
To 4-Bretylium Tosylate hydrobromate (25g, 89mmol), pyridine (36mL, 445mmol) and CH
2Cl
2Cold (0 ℃) mixture dropping trifluoro-acetic anhydride (100mL) (18.8mL, 133mmol).After adding finishes,, be poured on then on the ice (500g) at stirring at room mixture 48h.Mixture CH
2Cl
2(4 * 100mL) extractions, the dichloromethane layer of merging with 1N HCl (4 * 100mL), saturated NaCl (100mL) washing, use dried over mgso, filter final vacuum and evaporate, obtain product (26.13g, 100%);
1H NMR 500MHz (CDCl
3) δ=2.86 (2H, t, J=7.1Hz), 3.59 (2H, q, J=6.6Hz), 6.57 (1H, br s), 7.09 (2H, d, J=8.5Hz), 7.43 (2H, d, J=8.5Hz).
Step B:

The steps A product (26g, 88mmol) and Paraformaldehyde 96 (5.6g, the mixture of the disposable adding vitriol oil (130mL) and Glacial acetic acid (195mL) in mixture 130mmol) is at stirring at room gained mixture 17h.Reaction mixture is poured on ice/water (1.5L), with ethyl acetate (3 * 300mL) extraction, the ethyl acetate layer water of merging (2 * 600mL), saturated NaHCO
3(300mL), use dried over mgso, filter the final vacuum evaporation with saturated NaCl (150mL) washing.Resistates is dissolved in ethanol (450mL), adds salt of wormwood (60g, water 434mmol) (150ml) solution.Mixture heating up to the 1h that refluxes, is cooled off vacuum-evaporation then.Water (500mL) is added resistates, use CH
2Cl
2(3 * 300mL) extractions; Dried over sodium sulfate is used in the dichloromethane layer water (500mL) that merges, saturated sodium-chloride (150mL) washing, filters vacuum concentration.Resistates with the silica column chromatographic purification (with containing 0.5%NH
4The 5%CH of OH
3OH/CH
2Cl
2Wash-out) obtains product (10g, 54%);
1H NMR 500MHz (CDCl
3) δ=1.77 (1H, br s), 2.77 (2H, d, J=6.0Hz), 3.11 (2H, t, J=6.0Hz), 3.97 (2H, s), 6.95 (1H, d, J=8.0Hz), 7.15 (1H, s) 7.23 (1H, dd, J=1.2 and 8.2Hz).
Intermediate 21
The cis racemoid
Steps A
Under nitrogen atmosphere, 3-cyclopentenes-1-formic acid (Org.Synth.75, p195-200,1998) (31.5g, anhydrous N 281mmol), add in dinethylformamide (300mL) solution salt of wormwood (97g, 710mmol) and methyl iodide (35mL, 560mmol).At stirring at room gained mixture 16h, then in the impouring water (1L), with ether (3 * 400mL) extractions.The ether layer water that merges (3 * 500mL), saturated NaCl (200mL) washing, use dried over mgso, filtration, vacuum concentration obtains 34g (96%).
1H NMR (CDCl
3, 500MHz): δ 5.64 (s, 2H), 3.68 (s, 3H), 3.11 (quintet, J=8.5Hz, 1H), 2.63 (d, J=8.3Hz, 4H).
Step B
Under nitrogen atmosphere, (34.4mL, (hexane solution of 100mL 2.5M 250mmol), stirs gained mixture 10min at-78 ℃ slowly to add n-Butyl Lithium in anhydrous tetrahydro furan 250mmol) (250mL) cold (78 ℃) solution to diisopropylamine.Add in this mixture 3-aleprolic acid methyl esters (25.8g, 200mmol), behind the restir 15min, add 2-iodopropane (41mL, 410mmol), at-78 ℃ of 30min that stir the mixture, allow then its rise to+4 ℃, leave standstill 72h in this temperature.In reaction mixture impouring 5% citric acid (700mL), with ether (3 * 300mL) extractions.The ether layer water that merges (2 * 500mL), (dried over mgso use in 1 * 100mL) washing to saturated NaCl, filtration, vacuum concentration.Resistates is by (50 ℃ of vacuum distillings; 5mmHg) purification obtains 28.9g (86%) product.
1H?NMR(CDCl
3,500MHz):δ5.54(s,2H),3.67(s,3H),2.85(d,J=15.1Hz,2H),2.30(dd,J?14.9Hz,2H),2.07(t,J=6.6Hz,1H),0.82(d,J=6.6Hz,6H)。
Step C
Under nitrogen atmosphere, (20mL, anhydrous tetrahydro furan 200mmol) (100mL) cold (0 ℃) solution adds cyclopentenes ester (28.9g, 172mmol) solution of step B preparation to borine-dimethyl sulfide with double-ended needle.After adding finishes, at stirring at room reaction mixture 20h.Cooling mixture in ice bath, (60mL 3N solution 181mmol), adds 30% hydrogen peroxide (65mL) to dropping sodium then, stirs gained mixture 1h at 40 ℃.In mixture impouring water (600mL), with ether (3 * 200mL) extractions, the ether layer water of merging (3 * 500mL), saturated NaCl (100mL) washing, use dried over mgso, filtration, vacuum concentration.Resistates obtains 18.5g (58%) product with silica column chromatographic purification (with 20%EtOAc/ hexane wash-out).
Step D

Under nitrogen atmosphere, (55mL, anhydrous methylene chloride 110mmol) (300mL) (78 ℃) solution drip methyl-sulphoxide, and (15.5mL 219mmol), stirs gained mixture 10min at-78 ℃ to oxalyl chloride.Add step C product (18.5g, anhydrous methylene chloride 100mmol) (100mL) solution with double-ended needle to this mixture.At-78 ℃ of stirred reaction mixture 15min, (69mL 500mmol), allows the gained mixture rise to room temperature in 2h to add triethylamine then.Dried over mgso is used in reaction mixture water (500mL), saturated NaCl (150mL) washing, filters, and vacuum concentration obtains the 18g product, and product is directly used in next step and need not to purify again.
Step e
Under nitrogen atmosphere, cyclopentanone (the 18g of step D preparation, 98mmol) anhydrous 1, add 4-(4-fluorophenyl) piperidine hydrochlorate (25g in 2-ethylene dichloride (500mL) solution, 120mmol), diisopropylethylamine (20.4mL, 116mmol), (112g is 531mmol) with 4 molecular sieves (Powdered 10g) for sodium triacetoxy borohydride.At stirring at room mixture 48h, use methylene dichloride (500mL) dilution then, pass through diatomite filtration.Filtrate is used dried over sodium sulfate with saturated sodium bicarbonate solution (500mL), water (500mL), saturated NaCl (200mL) washing, filters, and vacuum concentration obtains 28g (82%) product.Product is directly used in next step and need not to purify again.
Step F
1-sec.-propyl-3-(4-(4-fluorophenyl) piperidines-1-yl) cyclopentane-carboxylic acid

(28g adds potassium hydroxide (30g, water 540mmol) (100mL) solution, reflux gained mixture 18h to the pentamethylene methyl esters of step e preparation in ethanol 81mmol) (500mL) solution.Vacuum concentration refrigerative mixture adds resistates to remove ethanol with water (200mL).(3 * 200mL) extractions add in the concentrated hydrochloric acid and water layer mixture with ether.(3 * 150mL) mixture extraction, the organic extract liquid dried over sodium sulfate of merging are filtered vacuum concentration to mixture with 9/1 chloroform/2-propyl alcohol.Add acetone (70mL) in the resistates, be heated to backflow with mixture is of short duration, leave standstill 16h at+5 ℃ then.Acetone is removed from the white solid decant, and dry remaining solid obtains 11.5g (43%) product, and it is the mixture (10: 1) of cis and trans-isomer(ide).
The calculated value of ESI-MS C20H28FNO2: 333; Measured value: 334 (M+H).
Intermediate 22

With 2, (3.128g, (8.97mL 64.4mmol), is cooled to 0 ℃ to 5-dimethyl-3-pyrroline 32.19mmol) to be dissolved in triethylamine.Drip carbobenzoxy chloride (10.11mL, the dichloromethane solution of minimum 70.83mmol).Slowly the reacting by heating mixture stirs 48h to room temperature.Reactant is with saturated sodium bicarbonate solution (150mL) quencher, then with organic layer with saturated sodium bicarbonate solution (2 * 100mL) and salt solution (dried over mgso use in 1 * 100mL) washing, and filtration is concentrated.Purify (with the gradient solvent system of 5%EtOAc/ hexane) by the silica gel flash column chromatography, obtain intermediate 1 (3.844g) with 52% yield to the 10%EtOAc/ hexane.ESI-MSC
14H
17NO
2Calculated value: 231.29, measured value 232 (M+H).
Intermediate 23
Method A:
Steps A
With (1S)-(+)-2-azabicyclic [2.2.1] heptan-5-alkene-3-ketone (10.3g, 94.4mmol), the mixture of ethyl acetate (200mL) and 10%Pd/C (0.5g) is in room temperature hydrogenation.Behind 24h, filter reaction mixture, remaining 10.4g (100%) product of evaporation, with its be dissolved in 250mL methyl alcohol and HCl (12M, 6mL).At stirring at room gained mixture, finish (72h) up to reaction.Evaporation methyl alcohol, high vacuum dry obtains Off-white solid title compound (16.0g, 96%) then.
1H?NMR(500MHz,D
2O):δ3.70(s,3H),3.01(m,1H),2.38(m,1H),2.16-1.73(m,6H)。
Step B
(10.2g, anhydrous methylene chloride 56.8mmol) (200mL) suspension add the diphenylmethyl imines, and (10.2g 56.8mmol), stirs gained mixture 24h to the steps A intermediate in room temperature.Filter reaction mixture, evaporated filtrate is left yellow oil, grinds with ether (100mL), filters the back evaporation.This operation is repeated twice to guarantee that product does not have ammonium chloride impurity.The abundant vacuum-drying of gained oily matter is obtained title compound (18.03g,>100%), do not need to purify again.
1H?NMR(500MHz,CDCl
3):δ7.5-7.18(m,10H),3.75(m,1H),3.7(s,3H),2.78(m,1H),2.26-1.71(m,6H)。
Step C
Lithium diisopropylamine solution (at-78 ℃, with diisopropylamine (7.7g, the ester of adding step B 76mmol) and in the n-Butyl Lithium (30.4mL, the hexane solution of 2.5M 76mmol) prepare in tetrahydrofuran (THF) (120mL) solution) (18.0g, 58.6mmol).Stir gained dark red solution 20min, use 2-iodopropane (14.9g, 88.0mmol) quencher then.Reaction mixture progressively is heated to 0 ℃ in 3h, keeps 3h again in this temperature.Ethyl acetate extraction is used in the quencher of reactant water.Organic layer water, salt water washing, dry (anhydrous magnesium sulfate) concentrates and obtains oily matter.Add in tetrahydrofuran (THF) (100mL) solution of rough Schiff alkali (20.0g) HCl (5.0mL, 12M).At stirring at room gained reaction mixture 3h.After removing all volatile matters, hydrochloride is dissolved in methylene dichloride (250mL), adding saturated sodium bicarbonate solution (250mL) and tert-Butyl dicarbonate (26.0g, 1.4Eq.).The gained mixture is spent the night in the room temperature vigorous stirring.Isolate organic layer, water, salt water washing, dry (anhydrous magnesium sulfate) concentrates and obtains oily matter.Flash column chromatography purification (eluent: hexane/ethyl acetate 19: 1) obtain required product (4.91g, 30%).
1H?NMR(500MHz,CDCl
3):4.79(br,1H),4.01(m,1H),3.71(s,3H),2.18-1.60(m,6H),1.44(s,9H),0.87(d,J=6.9Hz,3H),0.86(d,J=6.9Hz,3H)。
Step D

(4.91g adds LiOH (3.6g, water 85mmol) (20mL) and tetrahydrofuran (THF) (10mL) solution to the ester of step C in methyl alcohol 17.2mmol) (100mL) solution.The gained mixture is finished (18h) 80 ℃ of heating up to reaction.Vacuum is removed methyl alcohol, with crude product water/ethyl acetate (200mL, 1: 4) dissolving, is cooled to 0 ℃.With the mixture acidity adjustment to pH 6.Isolate ethyl acetate layer, water, salt water washing, dry (anhydrous magnesium sulfate) concentrates and obtains oily matter.Flash column chromatography purification (eluent: hexane/ethyl acetate 1: 1+2%AcOH) obtain intermediate 11 (3.9g, 84%).
1H?NMR(500MHz,CDCl
3):11.36(br,1H),6.49(br,1H),4.83(m,1H),3.71(s,3H),2.30-1.55(m,6H),1.46(s,9H),0.94(d,J=6.9Hz,3H),0.933(d,J=6.9Hz,3H)。
Method B:
Steps A:

By typical method with commercially available (1R, 4S)-4-amino cyclopentyl-2-alkene-1-formic acid is converted into its methyl ester hydrochloride.
Step B:

(6.31g adds solid NaHCO in acetone 35.5mmol) (40mL) and water (20mL) suspension to the amine of steps A in batches
3(6.6g, 78mmol).Behind 5min, (8.5g, acetone 39mmol) (60mL) solution is at the stirring at room reaction mixture to add tert-Butyl dicarbonate.Behind 3h, vacuum is removed acetone, and resistates distributes between ether (500mL) and saturated sodium bicarbonate aqueous solution (120mL).Ether layer is further used NaHCO
3The aqueous solution (1 * 100mL), salt solution (1 * 100mL) washing, use anhydrous Na
2SO
4Drying concentrates, and obtains product (7.25g, 85%) with flash chromatography purification (15% ethyl acetate/hexane).
Step C:

At-78 ℃, (10.4g, tetrahydrofuran (THF) 62.1mmol) (100mL) solution add intermediate (6.71g, tetrahydrofuran (THF) 27.8mmol) (10mL) solution of step B in 10min to two (three silyls) Lithamide.Stir gained solution 30min at-78 ℃, and disposable then adding isopropyl iodide (3.3mL, 33mmol).Allow reactant rise to-25 ℃, keep spending the night in this temperature.Then with reactant saturated aqueous ammonium chloride (250mL) quencher.Isolate organic layer, water layer is further used ether (3 * 100mL) extractions.(anhydrous Na is used in 1 * 100mL) washing to the organic layer that merges with salt solution
2SO
4Drying is filtered, and concentrates, and obtains clarifying oily product (5.66g, 72%) (suitable/anti-=4.3/1) with flash chromatography purification (5-10% ethyl acetate/hexane).
1H NMR (500MHz, CDCl
3) cis-isomeride: δ 5.79 (s, 2H), 4.75 (m, 1H), 3.72 (s, 3H), 2.28-2.20 (m, 2H), 2.0 (dd, J=15,4Hz, 1H), 1.45 (s, 9H), 0.85 (d, J=6.6Hz, 3H), 0.81 (d, J=7Hz, 3H).
Step D:

(1.6g adds LiOH monohydrate (400mg) to step C product in tetrahydrofuran (THF) 5.7mmol) (50mL), methyl alcohol (50mL) and water (10mL) solution, reactant is heated to reflux spends the night, till TLC shows complete reaction.Vacuum is removed organic solvent, and water layer reaches 4 with the slow acidifying of dense HCl up to pH then with ether (1x) washing.Gained suspension CH
2Cl
2(3x) extraction.Organic layer with anhydrous magnesium sulfate drying merges filters, and concentrates and obtains yellow spumescence solid product (mixtures of two kinds of suitable/trans isomers) (1.5g).Under heating, this solid is dissolved in ethyl acetate (2mL), obtains settled solution with hexane (50mL) dilution.Allow this solution slowly cool to room temperature 1h, keep-25 ℃ then at refrigerator and spend the night.Trans-isomer(ide) is with the required cis-isomeride crystallization of part (500mg altogether).Collect mother liquor, concentrate and obtain title compound (1g, 66%, cis-isomeride is only arranged).
1H NMR (500MHz, CDCl
3) cis-isomeride: δ 5.80 (m, 2H), 4.80 (m, 1H), 2.40-2.20 (m, 2H), 2.15-2.0 (m, 1H), 1.5 (m, 9H), 1.0-0.8 (m, 3H).
Step e:

Add 10%Pd/C (100mg) in ethanol (30mL) solution of step D product (1g), the gained mixture stirs under 50 lb hydrogen pressures with the Parr device and spends the night.Mixture is by diatomite filtration, and vacuum concentration obtains title compound (1g, 99%).
1H?NMR(500MHz,CDCl
3):11.36(br,1H),6.49(br,1H),4.83(m,1H),3.71(s,3H),2.30-1.55(m,6H),1.46(s,9H),0.94(d,J=6.9Hz,3H),0.933(d,J=6.9Hz,3H)。
Intermediate 24

Steps A

(4.6g, 16mmol) (4.0g, 14mmol) at first (3 * 50mL) azeotropic distillation dryings place 30min under the high vacuum with toluene with intermediate 23 with intermediate 2.Under nitrogen atmosphere, add successively 4-dimethylaminopyridine (1.08g, 8.60mmol), anhydrous methylene chloride (40mL) and diisopropylethylamine (7.0mL, 40mmol).After intermediate dissolving, add bromo-three-pyrrolidyl-phosphorus hexafluorophosphate (6.80g, 14.3mmol), immediately add diisopropylethylamine (7.0mL, 40mmol).Spend the night at the stirring at room reaction mixture, use saturated NaHCO then
3Quencher.Water layer is used methylene dichloride once more, and (3 * 50mL) washings merge organic layer, use dried over sodium sulfate, filter vacuum-evaporation.Crude product obtains yellow spumescence product (4.80g, 74%) with flash chromatography purification (stage gradient 0-60%, ethyl acetate/hexane).
1H?NMR(500MHz,CDCl
3)δ8.72(s,1H),7.70(s,1H),4.88(br?d,J=17.0Hz,1H),4.78(d,J=17.6Hz,1H),4.04-3.84(m,2H),3.52(br?s,1H),3.12(br?t,J=5.6Hz,1H),2.32-2.06(m,3H),1.98-1.70(m,4H),1.64-1.54(m,1H),1.44(s,9H),0.92-0.82(m,6H)。LC-MS C
23H
32F
3N
3O
3Calculated value 455.24, measured value [M+H]
+456.2.
Step B
(1.2g 2.6mmol) is dissolved in 4M HCl/ diox (50mL), at stirring at room gained solution 1h with the steps A product.The vacuum-evaporation reaction mixture obtains white powder product (904mg, 97%).LC-MS C
18H
24F
3N
3O calculated value 355.20, measured value [M+H]
+356.2.
Intermediate 25
Steps A

Add intermediate 23 (1.1g in the flask, 4.0mmol), intermediate 1 (0.944g, 4.00mmol), bromo-three-pyrrolidyl-phosphorus hexafluorophosphate (1.85g, 4.00mmol), DMAP (0.29g, 2.4mmol), DIEA (2.77mL, 16mmol) and DCM (20mL).Under nitrogen atmosphere, stir gained mixture 36h.Whole mixtures are applied on the silicagel column, with 20%EtOAc/ hexane wash-out.The required Boc-acid amides that obtains is gumminess solid (1.5g, 82%).The calculated value of ESI-MS C24H33F3N2O3: 454; Measured value: 455 (M+H).
Step B

The Boc amino amides of steps A is handled 1h with 10mL 4N HCl/ diox.Evaporation reaction mixture, the vacuum-drying product.Obtain yellow solid intermediate 25 (1.2g).The calculated value of ESI-MSC19H25F3N2O: 354; Measured value: 355 (M+H).
Intermediate 26

Steps A
Intermediate 1 is housed, and (10g adds 30mL 70% nitric acid in flask 50mmol).At 0 ℃ of cooling mixture, in 30min, add the 30mL vitriol oil.Spend the night at stirring at room gained solution, in the impouring ice-water mixture, use solid LiOH-H at 0 ℃
2O is adjusted to pH>10.Under vigorous stirring, add tert-Butyl dicarbonate (21.8g, 500mLDCM solution 100mmol).The 30min that stirs the mixture isolates organic layer, and water layer is with DCM (2 * 200mL) extractions.Extraction liquid water (500mL) washing that merges is used dried over sodium sulfate, then evaporation.Crude product obtains white solid title compound (17.0g, 98%) with flash chromatography purification (silica gel, 20%EtOAc/ hexane).
1H?NMR(400MHz,CDCl
3)δ8.05(s,1H),7.62(s,1H),4.72(s,2H),3.67(t,J=6.0Hz,2H),3.13(t,J=6.0Hz,2H),1.49(s,9H)。
Step B
The intermediate (17.0g) of above steps A is dissolved in 100mL 4M HCl/ diox, stirs 1h, evaporation, vacuum-drying.Obtain white solid intermediate 26.
1H?NMR(400MHz,CD
3OD)δ8.75(s,1H),8.00(s,1H),2.58(s,2H),3.57(t,J=6.0Hz,2H),3.42(t,J=6.0Hz,2H)。
Intermediate 27

Steps A

Add intermediate 27 (1.10g in the flask, 4.00mmol), intermediate 23 (1.15g, 4.00mmol), bromo-three-pyrrolidyl-phosphorus hexafluorophosphate (1.85g, 4.00mmol), DMAP (0.29g, 2.4mmol), DIEA (2.7mL, 16mmol) and DCM (20mL).Under nitrogen atmosphere, stir gained mixture 36h.Whole mixtures are applied on the silicagel column, obtain gumminess solid title compound (1.5g, 75%) with 20%EtOAc/ hexane wash-out.ESI-MSC24H32F3N3O5 calculated value: 499; Measured value: 500 (M+H).
Step B

The coupling product (1.5g) of above step is handled 1h with 10mL 4N HCl/ diox, evaporation, high vacuum dry obtains yellow solid title compound (1.2g).
1H NMR (400MHz, CD
3OD) δ 8.20 (s, 1H), 7.95 (broad peak, 1H), 4.98 (s, 2H), 4.00 (dd, 2H), 3.90 (t, 2H), 3.68 (m, 1H), 3.45 (m, 3H), 3.20 (s, 2H), 2.15-2.50 (m, 3H), 1.80-2.10 (m, 2H), 1.80 (m, 2H), 0.90 (m, 6H).The calculated value C19H24F3N3O3:399 of ESI-MS; Measured value: 400 (M+H).
Intermediate 28

Steps A

To N-TFA base-7-Trifluoromethyl-1,2,3, the 4-tetrahydroisoquinoline (intermediate 1 step B, 6.0g, 20mmol), NIS (6.9g, 30mmol) and stirring the mixture of TFA (15mL) drip the vitriol oil (1.5mL).Form a large amount of solids.In the stirring at room mixture overnight, in the impouring ice-water mixture, with ethyl acetate (3x) extraction.Dried over sodium sulfate is used in the organic phase water and the salt water washing that merge, evaporation.Resistates is with silica gel purify (with 10%EtOAc/ hexane wash-out).The saturated NaHSO of part that merges
3Dried over sodium sulfate is used in washing, evaporation, and vacuum-drying obtains white solid title compound (5.0g).
1H?NMR(400MHz,CDCl
3)δ8.02(d,J=2.5Hz,1H),7.42(d,j=3.0Hz,1H),4.85,4.79(ss,2H),3.95,3.90(tt,J=1.5,1.5Hz,2H),2.97(m,2H)。ESI-MSC12H8F6INO calculated value: 423; Measured value: 424 (M+H).
Step B

Iodine compound (steps A, 4.2g, 10mmol), zinc cyanide (2.3g, 20mmol), the mixture of four-triphenylphosphine (phosphene) palladium (0) complex compound (0.4g) and 50mL DMF with nitrogen purge several times, heated overnight under 85 ℃, nitrogen atmosphere then.LC-MS confirms to transform fully.Remove by filter insoluble substance.With the filtrate water dilution, with ethyl acetate (3x) extraction.The combined ethyl acetate layer by diatomite filtration, washes with water then, uses dried over sodium sulfate, then evaporation.Resistates obtains white solid title compound (2.5g) with silica gel purification (using the 10%EtOAc/Hex wash-out).
1H?NMR(400MHz,CDCl
3)δ7.85(d,J=2.1Hz,1H),7.65(d,J=2.6Hz,1H),4.91,4.86(ss,2H),4.00(m,2H),3.25(m,2H)。The calculated value of ESI-MS C13H8F6N2O: 323; Measured value: 323 (M+H).
Step C

With the amide intermediate of step B (500mg, 1.55mmol), the mixture of salt of wormwood (1.5g), ethanol (20mL) and water (0.5mL) confirms complete cracking 80 ℃ of heating up to TLC.Evaporating solvent, dilute with water with DCM (3x) extraction, is used dried over sodium sulfate, evaporation, vacuum-drying then.Obtain white solid title product (0.41g).ESI-MS C11H9F3N2 calculated value: 226; Measured value: 227 (M+H).
Step D
(1.9g is 8.5mmol) with the dense HCl aqueous solution of 50mL backflow 48h with the cyano intermediate of above step C.LC-MS confirms complete hydrolysis.Cooling mixture filters and collects the gained precipitation to room temperature, with dense aq.HCl washing.After high vacuum dry, obtain the hydrochloride (1.75g, 73%) of required product.
1H?NMR(CD
3OD,400MHz):δ8.20(s,1H),7.80(s,1H),4.51(s,2H),3.55(m,4H)。LC-MS C11H10F3NO2 calculated value 245, measured value [M+H]
+246.
Step e
(1.75g slowly adds the pure solution of Acetyl Chloride 98Min. (5mL) to the amino acid salts hydrochlorate of above step D in 50mL methanol suspension 6.25mmol).With the gained mixture reflux up to LC-MS confirm complete esterification (~3h), evaporating solvent then, high vacuum dry obtains white solid title compound (1.85g, 100%).
1H?NMR(CD
3OD,400MHz):8.19(s,1H),7.82(s,1H),4.50(s,2H),3.94(s,3H),3.53(s,4H)。
LC-MS C12H12F3NO2 calculated value 259, measured value [M+H]
+260.
Intermediate 29

Method A:
Steps A:

With H
2SO
4(8.30mL 156mmol) is added drop-wise to sal epsom (75g, DCM 620mmol) (650mL) vigorous stirring suspension for conc., 15.3g.The 0.5h that stirs the mixture, add then known cyclopentanone-3-manthanoate (20.0g, 156mmol), then add the trimethyl carbinol (58g, 780mmol).Reaction vessel is closely sealed, in the stirring at room mixture overnight.Add the 30mL trimethyl carbinol second day morning again.Once more reaction vessel is closely sealed, it's weekend is past stirred reaction mixture.Reaction mixture passes through diatomite filtration.Filtrate is washed with 2N NaOH.Water layer washs with DCM once more.Merge organic layer, anhydrous magnesium sulfate drying is used in water, salt water washing successively, filters, and concentrates then to obtain 19.9g (69%) 3-oxo-cyclopentane t-butyl formate.Reaction process is monitored by TLC, adopts 50% ethyl acetate/hexane, with aubepine stain dyeing (SM and product are caught purple).
1H?NMR(500MHz,CDCl
3):3.02(p,J=7.8Hz,1H),2.05-2.50(m,6H),1.45(s,9H)。
13C?NMR(125MHz,CDCl
3):217.00,173.47,80.99,41.88,41.14,27.94,26.57。
Step B:

3-oxo-cyclopentane t-butyl formate (19.8g, 107mmol) 1: add successively in 1DCM/ methyl alcohol (150mL) solution trimethyl orthoformate (46.8mL, 428mmol), TsOHH
2O (~0.5g).At stirring at room reaction mixture 2h.And then adding TsOHH
2O (~0.25g), stirred reaction mixture spends the night.At the room temperature concentrated reaction mixture, the gained resistates is dissolved in ether, use saturated sodium bicarbonate solution, salt water washing successively.Use the anhydrous magnesium sulfate drying ether layer, filter, concentrate then.Obtain 22.2g (90%) 3, the 3-dimethoxy cyclopentane-carboxylic acid tert-butyl ester with flash chromatography purification (silicon-dioxide, 15% ethyl acetate/hexane).
1H?NMR(500MHz,CDCl
3):3.21(s,3H),3.20(s,3H),2.80(m,1H),2.10-1.80(bm,6H),1.46(s,9H)。
13C?NMR(125MHz,CDCl
3):174.9,111.2,80.3,67.8,49.2,42.5,37.4,33.8,28.3,22.0。
Step C:
In 10min, (THF 61mmol) (150mL) cold (78 ℃) solution drips 3, the 3-dimethoxy cyclopentane-carboxylic acid tert-butyl ester (9.37g, 25mL THF solution 40.7mmol) for the cyclohexane solution of 1.5M, 41mL to LDA.Stir gained mixture 30min at-78 ℃, drip then 2-iodopropane (16.3mL, 163mmol).After stirring 10min, allow reaction mixture rise to room temperature.After stirring is spent the night, reaction mixture is diluted with ether, use the salt water washing.Use the anhydrous magnesium sulfate drying ether layer, filter, concentrate then.Store crude product in vacuum and spend the night, obtain 8.32g 1-sec.-propyl-3, the 3-dimethoxy cyclopentane-carboxylic acid tert-butyl ester (75%) with MPLC purification (silicon-dioxide, 20% ethyl acetate/hexane).
1H?NMR(500MHz,CDCl
3)δ3.21(s,3H),3.18(s,3H),2.56(appd,J=14Hz,1H),2.26(m,1H),1.78-1.89(m,3
Step D:

With 1-sec.-propyl-3, (8.32g 30.5mmol) is dissolved in the anhydrous HCl/ diox of 4N (50mL) to the 3-dimethoxy cyclopentane-carboxylic acid tert-butyl ester, adds entry (10mL).Spend the night at the stirring at room reaction mixture, concentrate then.Resistates is dissolved in DCM, uses anhydrous magnesium sulfate drying, filter, concentrate then and obtain 5.44g 1-sec.-propyl-3-oxo-cyclopentane formic acid (directly use and need not to purify).
1H?NMR(500MHz,CDCl
3)δ2.70(d,J=18.1Hz,1H),2.44-2.39(m,1H),2.30-2.15(m,2H),2.14(dd,J=18.1,1.0Hz,1H),2.06(p,J=6.9Hz,1H),1.98(m,1H),0.98(dd,J=11.4,6.9Hz,6H)。
Step e:
With 1-sec.-propyl-3-oxo-cyclopentane formic acid (5.44g, DCM 32.0mmol) (75mL) cold (0 ℃) solution use successively oxalyl chloride (8.36mL, 95.9mmol), 3 DMF handle.Allow reaction mixture rise to room temperature, stir 1.75h.Concentrated reaction mixture then, vacuum stores 30min.The gained acyl chlorides is dissolved in DCM (75mL), is cooled to 0 ℃, use successively benzyl alcohol (8.28mL, 80.0mmol), triethylamine (8.92mL, 64.0mmol, drip) handles.Add about 100mg DMAP then, the reacting by heating mixture stirs 2h to room temperature.Reaction mixture dilutes with DCM, with 1N HCl solution, saturated sodium bicarbonate solution and salt water washing.Use the anhydrous magnesium sulfate drying organic layer, filter, concentrate then.MPLC purification (silicon-dioxide, 50% ethyl acetate/hexane) obtains 6.11g (73%) 1-sec.-propyl-3-oxo-cyclopentane formic acid benzyl ester.
1H?NMR(CDCl
3,500MHz):δ7.36(m,5H),5.17(d,J=2.5Hz,2H),2.85(d,J=18.5Hz,1H),2.48(m,1H),2.29(dd,J=10.0,3.0Hz,1H),1.98-2.23(m,3H),1.93(m,1H),0.95(m,6H)。By chirality HPLC resolution of racemic product: with chiralcel OD post, with (the 100mg/ injection of 15%2-propyl alcohol/hexane wash-out; Gilson HPLC system with programming finishes).Obtain isomer (the 1S)-1-sec.-propyl-3-oxo-cyclopentane formic acid benzyl ester of the required very fast wash-out of 2.11g.
Step F:

(1.27g, 4.88mmol) (10%Degussa 500mg) mixes in 20mL methyl alcohol, and (gas tank) stirs 2h under nitrogen atmosphere with Pd/C with (1S)-1-sec.-propyl-3-oxo-cyclopentane formic acid benzyl ester.A part (~30% transforms) has only been carried out in reaction, so filter reaction mixture, adds Pd/C (500mg) in addition, and 5h stirs the mixture under nitrogen atmosphere.Because reaction now near finishing, by the diatomite filtration reaction mixture, concentrates and obtains 704mg (1S)-1-sec.-propyl-3-oxo-cyclopentane formic acid, need not further purification.Note because the ester that obtains after chiral separation is poisoned because of impurity certainly, so used a large amount of catalyzer.This is the Special Circumstances of this specific sample.Usually catalyst consumption is a lot of less.
1H NMR consistent with above racemic acid (step D).
Method B:

(1S, 3R)-the 3-[(tert-butoxycarbonyl) amino]-(7.46g 27.5mmol) adds 4N HCl De dioxane solution (30mL) to 1-sec.-propyl cyclopentane-carboxylic acid in De diox (10mL) solution.At stirring at room reaction mixture 2h, vacuum concentration obtains corresponding amino acid salts white solid then.Then this solid is dissolved in CH
2Cl
2(100mL), add solid NaHCO
3(7.0g, 82.5mmol).After being cooled to 0 ℃, with NBS (20.0g, CH 110mmol)
2Cl
2(200mL) solution slowly adds reactant in 4h.After adding, the reactant vacuum concentration to doing, is dissolved in ethanol (100mL) then.(4.45g 82.5mmol), is heated to backflow with reactant to add NaOMe in the ethanolic soln.Behind the backflow 1h, reactant is cooled to 0 ℃, adds 2N aqueous sulfuric acid (50mL).At stirring at room mixture 1h, vacuum concentration is to about 60mL then.Remaining mixture distributes between water (150mL) and ethyl acetate (100mL).Water layer is further used twice of ethyl acetate extraction.Merge organic layer, use anhydrous magnesium sulfate drying, concentrate, obtain (1S)-1-sec.-propyl-3-oxo-cyclopentane formic acid (3.00g, 64%) with flash chromatography purification (silicon-dioxide, ethyl acetate/hexane).
Intermediate 30
Trans racemoid
Steps A:
4-[3-(ethoxy carbonyl) phenyl] piperidines-1-t-butyl formate (48g, add ruthenium oxide (IV) hydrate (6.0g in chloroform 220mmol) (900mL) stirred solution, 45mmol), add sodium periodate (150g, water 700mmol) (900mL) solution then.The inhomogeneous reaction mixture of stirring at room gained 11 days, filter by the diatomite short column then.Organic layer is shifted out water layer DCM extracting twice.The organic layer that merges is used the salt water washing once with the solution washing twice of 10% Sulfothiorine.The solution dried over mgso is filtered concentrating under reduced pressure.Product obtains 22.5g (64.8mmol) 4-[3-(ethoxy carbonyl) phenyl with flash chromatography purification (silica gel, 20%EA/ hexane)]-2-oxo-piperidine-1-t-butyl formate (29%).
ESI-MS C19H25NO5 calculated value: 347.17; Measured value 370.1 (M+Na)
Step B:
(14g 71mmol) mixes in the flame-dried round-bottomed flask of 1000mL with 300mL THF, and the gained mixture is cooled to-78 ℃ with two (three silyls) potassium amides.To be dissolved in 4-[3-(ethoxy carbonyl) phenyl of 150mL THF]-(22.5g 64.8mmol) slowly adds mixture by addition funnel to 2-oxo-piperidine-1-t-butyl formate, stirs gained reaction mixture 30min at-78 ℃.(12.1mL 195mmol), stirs 4h with reaction mixture at-78 ℃, is heated to ambient temperature overnight then to drip methyl-iodide then.With saturated ammonium chloride quencher reactant, use extracted with diethyl ether 3 times.Dried over mgso is used in the ether layer salt water washing that merges, and filters, then concentrating under reduced pressure.Product obtains the trans racemoid 4-[3-of 6.1g (ethoxy carbonyl) phenyl with flash chromatography purification (10-20%EA/ hexane)]-3-methyl-2-oxo-piperidine-1-t-butyl formate (26%).
ESI-MS C20H27NO5 calculated value: 361.19; Measured value 384.25 (M+Na).
Step C:
(6.1g 17mmol) is dissolved in 4.0M HCl/ diox, and at stirring at room 2h, concentrating under reduced pressure obtains required orange solids product then, directly uses to need not to purify in next step again with the product of step B.
ESI-MS C15H19NO3 calculated value: 261.14; Measured value 262.1 (M+H).
Step D:
Trans racemoid
With the product of above step (all amts~17mmol) be dissolved in THF (100mL), drip 2.0M borane-dimethyl sulfide THF solution (31mL, 62mmol).At stirring at room gained solution 4h, store 72h at 4 ℃ then.Removal of solvent under reduced pressure is dissolved in the gained resistates ethanolic soln of 0.5M HCl (aqueous solution~38%).Solution is heated to 50 ℃, stirs 4h.Remove and desolvate, repeat above operation and decompose to guarantee borane complex.Remove and to desolvate, product is with the MPLC (0-15% (10%NH that purifies
4OH/MeOH)/DCM) obtain required product, purity is 80%.Crude product is dissolved in DCM (100mL), with tert-Butyl dicarbonate (2.95g, 13.5mmol), diisopropylethylamine (2.30mL, 13.5mmol) and DMAP (10mg) processing.Spend the night at stirring at room gained reaction mixture, then with the DCM dilution, with 1N saturated sodium bicarbonate aqueous solution and salt water washing.Use the dried over mgso organic layer, filter concentrating under reduced pressure.Intermediate is with MPLC purify (0-40%EA/ hexane).The gained colorless oil is dissolved in 4.0M HCl/ diox, at stirring at room gained reaction mixture 1.5h.Concentrated reaction mixture is to the dried required hydrochloride of 2.13g (7.52mmol) that obtains.
ESI-MS C15H21NO2 calculated value: 247.16; Measured value 248.15 (M+H)
Embodiment 1
Intermediate 3 (150mg, 0.425mmol), 4-ethoxycarbonyl phenylpiperidines (125mg, 0.425mmol) and add molecular sieve (4 ) and NaBH (OAc) in the solution of DCM (25mL)
3(450mg, 2.12mmol).At stirring at room reaction mixture 18h, by diatomite filtration, use saturated NaHCO then
3Dilution is with DCM (3x) extraction.With the organic layer that dried over sodium sulfate merges, use preparation type TLC (3/96.7/0.3, MeOH/DCM/NH
4OH) purification obtains embodiment 1 (220mg, 97.8%).LC-MS C
27H
38F
3N
2O
3[M+H
+] calculated value 495.28, measured value 495.25.
Chemical compound lot prepares with the method for different amine according to embodiment 1.In following table, sum up these compounds.
Table 1 (embodiment 2-6)
Embodiment 7

With embodiment 1 product (185mg, 0.349mmol), 5N NaOH (200 μ L, 1.04mmol) and the mixture of MeOH (5mL) 60 ℃ the heating 3h, add 4N HCl/ dioxane solution neutralization bases then.Concentration response thing solution obtains embodiment 7 (115mg, 65.7%) with the reversed-phase HPLC purification.LC-MS C
25H
34F
3N
2O
3[M+H
+] calculated value 467.24, measured value 467.35.
Embodiment 8-12 prepares as the method for initial feed according to embodiment 7 with embodiment 2-6.In following table, sum up these compounds.
Table 2 (embodiment 8-12)

Embodiment 13
Intermediate 4 (50mg, and adding 1-ethoxy carbonyl piperazine in methylene dichloride 0.14mmol) (20mL) solution (23mg, 0.14mmol).After adding Powdered 4 molecular sieves (25mg), (180mg, 0.84mmol), stirred reaction mixture spends the night to add sodium triacetoxy borohydride.Mixture dilutes with methylene dichloride, with the saturated sodium bicarbonate aqueous solution washing, uses dried over sodium sulfate, vacuum concentration.Crude product is purified with preparation type TLC (7/92.3/.7, ethanol/methylene/ammonium hydroxide) and is obtained embodiment 13, is the mixture (55mg, 79%) of 4 kinds of diastereomers.LC-MS:MW calculated value 496.27, measured value 497.3.
Chemical compound lot wherein substitutes 1-ethoxy carbonyl piperazine with different piperazines according to the method preparation of embodiment 13.These compounds (all being prepared as the mixture of 4 kinds of diastereomers) are summed up in following table.
Table 3 (embodiment 14-16)
| Embodiment | R | Molecular formula | Calculated value [M+H +] | Measured value [M+H +] |
Embodiment 17

Intermediate 4 (50mg, add in methylene dichloride 0.14mmol) (20mL) solution 4-benzoyl piperidine hydrochlorate (32mg, 0.14mmol) and N, the N-diisopropylethylamine (73 μ L, 0.42mmol).After adding the Powdered molecular sieves of 4 (25mg), (180mg, 0.84mmol), stirred reaction mixture spends the night to add sodium triacetoxy borohydride.The mixture dichloromethane extraction with the sodium bicarbonate washing, is used dried over sodium sulfate, vacuum concentration.Crude product is purified with preparation type TLC (7/92.3/.7, ethanol/methylene/ammonium hydroxide) and is obtained embodiment 17 (30mg, 43%), is the mixture of 4 kinds of diastereomers.
ESI-MS: calculated value MW:527.28, measured value 528.25.
Embodiment 18

With intermediate 4 (176mg, 0.5mmol), the piperidines that is spirally connected (for HCl salt, 115mg, 0.6mmol), DIEA (100mg, 0.8mmol), molecular sieve (4 , 200mg), sodium triacetoxy borohydride (212mg, 1.0mmol) and the mixture of methylene dichloride (10mL) stir and to spend the night.With saturated aqueous sodium carbonate quencher reactant.Remove solid by diatomite filtration.Crude product is extracted in the methylene dichloride, and usefulness preparation type TLC (1000 microns, 10%[aq.NH
4OH/MeOH 1/9]/DCM) purify.Obtain title compound (155mg, 63%) for cis and trans racemic isomer mixture.LC-MS C26H35F3N4O2 calculated value: 492; Measured value: 493 (M+H).
C.ZHOU
Embodiment 19

Intermediate 5 (50mg, 0.14mmol) and piperidines (28 μ L, add in DCM 0.28mmol) (10mL) stirred solution Powdered molecular sieves of 4 (50mg) and sodium triacetoxy borohydride (150mg, 0.71mmol).Stirring at room gained solution 3 days, then by diatomite filtration, with saturated sodium bicarbonate aqueous solution and salt water washing.Use the dried over sodium sulfate organic layer, filter, concentrating under reduced pressure obtains rough oily matter then, with preparation type TLC (0.5%NH
4OH/4.5%MeOH/95%DCM) purification obtains the 16mg colorless solid, is the mixture of 2 kinds of diastereomers.To the diethyl ether solution of small portion free alkali adding 2N HCl, be converted into its hydrochloride.
ESI-MS C23H32F3N3O calculated value: 423.25; Measured value 424 (M+H).
The method preparation that several other embodiment introduce according to embodiment 19, but piperidines substituted with the different piperidines that replace as amine component.Table 4 has been listed these embodiment.
Table 4 (embodiment 20-28)

Embodiment 29 and embodiment 30
The free alkali product (55mg) of embodiment 22 preparation is split as its independent diastereomer, adopts the HPLC of configuration ChiralCel OD post, with 25% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Reclaim the diastereomer (embodiment 29) of the very fast wash-out of 27mg and the diastereomer (embodiment 30) of the slow wash-out of 20mg.
The calculated value of embodiment 29:ESI-MS C26H36F3N3O3: 495.27; Measured value 496 (M+H).
The calculated value of embodiment 30:ESI-MS C26H36F3N3O3: 495.27; Measured value 496 (M+H).
Embodiment 31
(10mg 0.018mmol) is dissolved in the mixture of methyl alcohol (1mL) and THF (1mL), with lithium hydroxide monohydrate (5mg, water 0.12mmol) (1mL) solution-treated with embodiment 29.At stirring at room gained solution 18h, concentrating under reduced pressure then.Product reversed-phase HPLC (C18,20-100%MeCN/H
2O) purify, be converted into hydrochloride, obtain 6.8mg product (70%) by the diethyl ether solution that adds 2N HCl.
The calculated value of ESI-MS C24H32F3N3O3: 467.24; Measured value 468 (M+H).
Embodiment 32
(10mg 0.018mmol) is dissolved in the mixture of methyl alcohol (1mL) and THF (1mL), with lithium hydroxide monohydrate (5mg, water 0.12mmol) (1mL) solution-treated with embodiment 30.At stirring at room gained solution 18h, concentrating under reduced pressure then.Product is with reversed-phase HPLC purify (C18,20-100%MeCN/H
2O), be converted into hydrochloride, obtain 3.8mg product (39%) by the diethyl ether solution that adds 2N HCl.
The calculated value of ESI-MS C24H32F3N3O3: 467.24; Measured value 468 (M+H).
Embodiment 33
Embodiment 30 (15mg, add in THF 0.026mmol) (2mL) solution lithium triethylborohydride (the THF solution of 1.0M, 150 μ L, 0.15mmol).Behind room temperature 18h, add lithium triethylborohydride (100 μ L) once more, stir gained mixture 24h, then concentrating under reduced pressure.The gained resistates is dissolved in DCM, uses saturated sodium bicarbonate aqueous solution, the 1N HCl aqueous solution and salt water washing successively.Use Na
2SO
4Dry organic layer filters, and handles with 2N HCl/ ether, hexane successively, and concentrating under reduced pressure obtains the hydrochloride (11%) of the required product of 1.5mg.
The calculated value of ESI-MS C24H34F3N3O2: 453.26; Measured value 454 (M+H).
Embodiment 34 and embodiment 35

The free alkali product (60mg) of embodiment 20 preparation is split as its independent diastereomer, adopts the HPLC of configuration ChiralCel OD post, with 25% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Reclaim the diastereomer (embodiment 34) of the very fast wash-out of 26mg and the diastereomer (embodiment 35) of the slow wash-out of 17mg.
The calculated value of embodiment 34:ESI-MS C25H35F3N4O2: 480.27; Measured value 481 (M+H).
The calculated value of embodiment 35:ESI-MS C25H35F3N4O2: 480.27; Measured value 481 (M+H).
Embodiment 36 and embodiment 37
The free alkali product (54mg) of embodiment 22 preparations is split as 2 mixtures that contain two kinds of diastereomers, adopts the HPLC of configuration ChiralCel OD post, with 13% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Reclaim the diastereomer (embodiment 36) of the very fast wash-out of 33mg and the diastereomer (embodiment 37) of the slow wash-out of 10mg.
The calculated value of embodiment 36:ESI-MS C26H36F3N3O3: 495.27; Measured value 496 (M+H).
The calculated value of embodiment 37:ESI-MS C26H36F3N3O3: 495.27; Measured value 496 (M+H).
Embodiment 38-41

The free alkali product (40mg) of embodiment 24 preparation is split as its independent diastereomer, adopts the HPLC of configuration ChiralCel OD post, with 20% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Diastereomer (embodiment 38), 1.5mg diastereomer 2 (embodiment 39), 7mg diastereomer 3 (embodiment 40) and the 6mg that reclaims the very fast wash-out of 15mg be the diastereomer (embodiment 41) of slow wash-out.
The calculated value of embodiment 38:ESI-MS C24H34F3N3O: 437.27; Measured value 438 (M+H).
The calculated value of embodiment 39:ESI-MS C24H34F3N3O: 437.27; Measured value 438 (M+H).
The calculated value of embodiment 40:ESI-MS C24H34F3N3O: 437.27; Measured value 438 (M+H).
The calculated value of embodiment 41:ESI-MS C24H34F3N3O: 437.27; Measured value 438 (M+H).
Embodiment 42-46

The free alkali product (40mg) of embodiment 25 preparation is split as its independent diastereomer, adopts the HPLC of configuration ChiralCel OD post, with 20% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Split all 6 kinds of diastereomers, but overlapping effusive peak 2 and peak 6 are mixed, obtain 4 kinds of pure diastereomers and a mixture that contains two kinds of diastereomers.
Embodiment 42: the calculated value of peak 1:ESI-MS C25H36F3N3O: 451.28; Measured value 452 (M+H).
Embodiment 43: the calculated value of peak 2/6:ESI-MS C25H36F3N3O: 451.28; Measured value 452 (M+H).
Embodiment 44: the calculated value of peak 3:ESI-MS C25H36F3N3O: 451.28; Measured value 452 (M+H).
Embodiment 45: the calculated value of peak 4:ESI-MS C25H36F3N3O: 451.28; Measured value 452 (M+H).
Embodiment 46: the calculated value of peak 5:ESI-MS C25H36F3N3O: 451.28; Measured value 452 (M+H).
Embodiment 47 and embodiment 48
The free alkali product (20mg) of embodiment 27 preparation is split as its independent diastereomer, adopts the HPLC of configuration ChiralCel OD post, with 25% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Reclaim the diastereomer (embodiment 47) of the very fast wash-out of 7mg and the diastereomer (embodiment 48) of the slow wash-out of 6mg.
The calculated value of embodiment 47:ESI-MS C23H32F3N3O2: 439.24; Measured value 440 (M+H).
The calculated value of embodiment 48:ESI-MS C23H32F3N3O2: 439.24; Measured value 440 (M+H).
Embodiment 49

Intermediate 5 (50mg, 0.14mmol) and morpholine (25 μ L, add in DCM 0.28mmol) (10mL) stirred solution Powdered molecular sieves of 4 (50mg) and sodium triacetoxy borohydride (150mg, 0.71mmol).Stirring at room gained solution 3 days, then by diatomite filtration, with saturated sodium bicarbonate aqueous solution and salt water washing.Use the dried over sodium sulfate organic layer, filter, concentrating under reduced pressure obtains rough oily matter then, with preparation type TLC (0.5%NH
4OH/4.5%MeOH/95%DCM) purification obtains the 57mg colorless solid, is the mixture of 2 kinds of diastereomers.To the diethyl ether solution of small portion free alkali adding 2N HCl, be converted into its hydrochloride.
The calculated value of ESI-MS C22H30F3N3O2: 425.23; Measured value 426 (M+H).
The method preparation that several other embodiment introduce according to embodiment 49, but morpholine substituted with different replacement morpholines as amine component.Table 5 has been listed embodiment.
Table 5 (embodiment 50-53)
Embodiment 53 and embodiment 54
The free alkali product (60mg) of embodiment 50 preparation is split as its independent diastereomer, adopts the HPLC of configuration ChiralCel OD post, with 20% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Reclaim the diastereomer (embodiment 53) of the very fast wash-out of 26mg and the diastereomer (embodiment 54) of the slow wash-out of 27mg.
The calculated value of embodiment 53:ESI-MS C24H34F3N3O2: 453.26; Measured value 454 (M+H).
The calculated value of embodiment 54:ESI-MS C24H34F3N3O2: 453.26; Measured value 454 (M+H).
Embodiment 55 and embodiment 56

The free alkali product (48mg) of embodiment 52 preparation is split as its independent diastereomer, adopts the HPLC of configuration ChiralCel OD post, with 25% ethanol/hexane wash-out.By adding the diethyl ether solution of 2N HCl, each compound is converted into corresponding hydrochloride.Reclaim the diastereomer (embodiment 53) of the very fast wash-out of 18mg and the diastereomer (embodiment 54) of the slow wash-out of 23mg.
The calculated value of embodiment 55:ESI-MS C23H30F3N3O2: 437.23; Measured value 438 (M+H).
The calculated value of embodiment 56:ESI-MS C23H30F3N3O2: 437.23; Measured value 438 (M+H).
Embodiment 57
Intermediate 5 (50mg, 0.14mmol) and tetramethyleneimine (23 μ L, add in DCM 0.28mmol) (10mL) stirred solution Powdered molecular sieves of 4 (50mg) and sodium triacetoxy borohydride (150mg, 0.71mmol).Stirring at room gained solution 3 days, then by diatomite filtration, with saturated sodium bicarbonate aqueous solution and salt water washing.Use the dried over sodium sulfate organic layer, filter, concentrating under reduced pressure obtains rough oily matter then, with preparation type TLC purification (0.5%NH
4OH/4.5%MeOH/95%DCM) obtain very fast effusive diastereomer of 15mg and the slower effusive isomer of 35.5mg, two kinds of isomer are colorless solid.To the diethyl ether solution of small portion free alkali adding 2N HCl, be converted into its hydrochloride.
Top: the calculated value of ESI-MS C22H30F3N3O: 409.23; Measured value 410 (M+H).
Bottom: the calculated value of ESI-MS C22H30F3N3O: 409.23; Measured value 410 (M+H).
The method preparation that several other embodiment introduce according to embodiment 57, but tetramethyleneimine substituted with different substituted pyrrolidins as amine component.Table 6 has been listed embodiment.
Table 6 (embodiment 58-62)
Embodiment 63
With intermediate 3 (55mg, 0.15mmol), (1S, 4S)-(-)-2-(4-fluorophenyl)-2,5-diazabicyclo [2.2.1] heptane (is a HBr salt, 85mg, 0.3mmol), DIEA (65mg, 0.5mmol), molecular sieve (4 , 250mg), sodium triacetoxy borohydride (212mg, 1.0mmol) and the mixture of methylene dichloride (5mL) stir and to spend the night.With saturated aqueous sodium carbonate quencher reactant.Remove solid by diatomite filtration.Crude product is extracted in the methylene dichloride, and usefulness preparation type TLC (1000 microns, 5%[aq.NH
4OH/MeOH 1/9]/DCM) purify.Obtain title compound (75mg, 95%) for cis and trans racemic isomer mixture.The calculated value of LC-MSC30H35F4N3O: 529; Measured value: 530 (M+H).
Embodiment 64
With intermediate 3 (55mg, 0.15mmol), (1S, 4S)-(-)-2-(2-methyl)-2,5-diazabicyclo [2.2.1] heptane (is a maleate, 100mg, 0.3mmol), DIEA (65mg, 0.5mmol), molecular sieve (4 , 250mg) and sodium triacetoxy borohydride (212mg, methylene dichloride 1.0mmol) (5mL) mixture stir and to spend the night.With saturated aqueous sodium carbonate quencher reactant.Remove solid by diatomite filtration.Crude product is extracted in the methylene dichloride, and usefulness preparation type TLC (1000 microns, 5%[aq.NH
4OH/MeOH 1/9]/DCM) purify.Obtain title compound (52mg, 66%) for cis and trans racemic isomer mixture.The calculated value of LC-MS C31H38F3N3O: 525; Measured value: 526 (M+H).
Embodiment 65
Steps A:
The pure mixture of 54g (0.29mol) (thiazolamine-4-yl) ethyl acetate and 50g (0.276mol) diphenylmethyl imines is stirred 5h at 190 ℃, then in the room temperature cooling, with the dilution of 100mL methylene dichloride.Whole mixtures are transferred on the silicagel column, with 20%EtOAc/ hexane wash-out.Obtain light yellow solid title compound (70g, 69% yield).
1H MR (300MHz, CDCl
3): δ 1.26 (t, 3H), 3.74 (s, 2H), 4.15 (q, 2H), 6.87 (s, 1H), 77.25-7.86 (m, 10H); Mass spectrum (NH
3-CI): m/z 351 (M+1).
Step B:
In room temperature, to the 35g of steps A (0.10Mol) Schiff alkali ester, suitable-1,3-two chloro-2-butylene (13mL, 0.11Mol) and add in the mixture of 500mL DME in batches solid NaH (60% oil, 10.0g, 0.25Mol).Stir the gained mixture 2 days, and in the impouring 2000mL ice-water, used the 1500mL extracted with diethyl ether.(dried over sodium sulfate is used in 3 * 500mL) washings to the ether layer water, then evaporation.FC (silica gel, 5%EtOAc/ hexane) obtains oily title compound (24g, 59%).
1H?NMR(300MHz,CDCl
3):1.20(t,3H),2.87(d,2H),3.19(d,2H),4.14(q,2H),5.29(s,2H),6.71(s,1H),7.26-7.81(m,10H)。Mass spectrum (NH
3-CI): m/z 403 (M+1).
Step C:
The cyclopentenes Schiff alkali of 24.0g (0.059mol) step B is dissolved in 100mL 4NHCl/ diox.Behind 1h, add 1.8mL water.The 3h that stirs the mixture is evaporated to dried.Resistates is dissolved in 100mL CH
2Cl
2, add 15mL DIEA.Whole mixtures are poured on the silicagel column, remove benzophenone, obtain light yellow solid title compound (12.0g, 85%) with 40%EtOAc/ hexane wash-out then with 20%EtOAc/ hexane wash-out.
1H NMR (300MHz, CDCl
3): δ 1.19 (t, 3H), 2.79 (d, 12H), 3.15 (d, 2H), 4.13 (q, 2H), 5.66 (s, 2H), 5.82 (broad peak, 2H), 6.19 (s, 1H).
Step D:
The mixture of aminothiazole, 28g (130mmol) tert-Butyl dicarbonate, 0.6g DMAP and the 250mL DCM of 12g (50mmol) step C stirred spend the night evaporation then.After the flash chromatography on silica gel method is purified (10%EtOAc/ hexane), obtain yellow oily title compound (21.0g, 96%).
1H?NMR(300MHz,CDCl
3):δ1.18(t,3H),1.49(d,18H),2.88(d,2H),3.18(d,2H),4.13(q,2H),5.65(s,2H),6.83(s,1H)。Mass spectrum (NH
3-CI): m/z 439 (M+1).
Step e:
At-78 ℃, to the 50mL anhydrous ether solution of the ester of 13.1g (30.0mmol) step D drip BH3.DMS THF solution (14mL, 24mmol).Remove cooling bath,,, add 25g sodium acetate and 55gPCC with 250mL DCM dilution at stirring at room mixture 3h.Stir the mixture and spend the night.Whole mixtures are applied to silicagel column, use 10%EtOAc/ hexane, 30%EtOAc/ hexane wash-out successively.Obtain two kinds of components.(yellow oil 6.0g) is accredited as title compound to the isomer of very fast wash-out.
1H?NMR(300MHz,CDCl
3):1.21(t,3H),1.50(s,18H),2.33(t,2H),2.42-2.70(m,2H),2.78-3.10(dd,2H),4.18(q,3H),6.88(s,1H)。Mass spectrum (NH3-CI): m/z 455 (M+1).
Step F:
The component of above slow wash-out be proved to be title compound (resinoid, 1.80g).
1H?NMR(300MHz,CDCl
3):(t,3H),1.46(s,9H),2.27(3,2H),2.38-2.62(m,2H),2.64-3.00(dd,2H),4.11(q,2H),6.66(s,1H)。Mass spectrum (NH
3-CI): m/z 355 (M+1).
Step G:
In room temperature, 1.40g (4.00mmol) ketone ester and the mixture stirring of 0.82g (13mmol) lithium hydroxide monohydrate in the 20mL MeOH and the 2mL aqueous solution of step F are spent the night.Whole mixtures are applied to silicagel column, use 10%MeOH/CH
2Cl
2Wash-out.Vacuum-evaporation obtains light yellow solid.Obtain the fluffy solid title product of 1.30g.
1H?NMR(300MHz,CDCl
3):δ(t,9H),2.10-3.20(m,8H),6.60(s,1H)。
Step H:

With the ketone acid of step G preparation (1.0g, 3.0mmol), intermediate 1 (be HCl salt, 0.66g, 3.0mmol), EDC (0.95g, 5.0mmol) and the mixture of methylene dichloride (10mL) stirring 2 days.Mixture dilutes with methylene dichloride, with 1N aq.HCl washing, uses dried over sodium sulfate, then evaporation.Resistates preparation type TLC purification (1500 microns, 10%[aq.NH
4OH/MeOH 1/9]/DCM), obtain yellow solid title product (0.52g, 34%).
The calculated value of LC-MS C25H27F3N2O4S: 508; Measured value: 509 (M+H).
Step I:
(510mg 1.0mmol) mixes 30min with TFA (10mL) with step H product.Remove TFA, resistates preparation type TLC purification (10%[aq.NH
4OH/MeOH1/9]/DCM) obtain required white solid product (223mg, 55%).The calculated value of LC-MSC20H19F3N2O2S: 408; Measured value: 409 (M+H).
Step J:
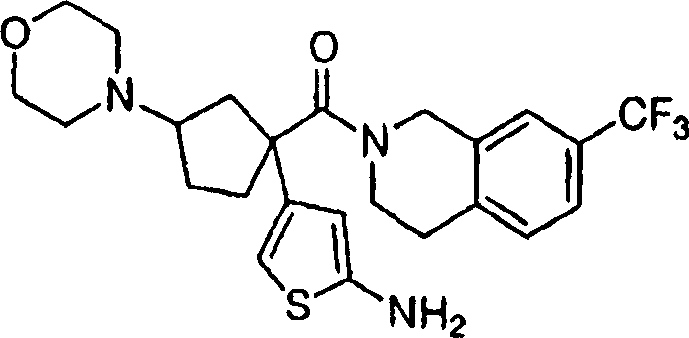
With the product of step I (210mg, 0.50mmol), morpholine (440mg, 5.0mmol), molecular sieve (4 , 500mg) and sodium triacetoxy borohydride (420mg, the mixture of methylene dichloride 2.0mmol) (15mL) stir and spend the night.With saturated aqueous sodium carbonate quencher reactant.Remove solid by diatomite filtration.Crude product is extracted in the methylene dichloride, and usefulness preparation type TLC purification (1000 microns, 10%[aq.NH
4OH/MeOH 1/9]/DCM).Obtain title compound (200mg, 84%) for cis and trans racemic isomer mixture.The calculated value of LC-MSC24H28F3N3O2S: 479; Measured value: 480 (M+H).
Embodiment 66
With intermediate 3 (70mg, 0.20mmol), suitable-2, the 6-thebaine (23 μ L, 0.20mmol), molecular sieve (4 μ L, 200mg), sodium triacetoxy borohydride (210mg, 0.99mmol) and the mixture of DCM stirred 5 days.Reaction mixture dilutes with DCM, filters, and uses sat.aq.NaHCO
3, water and salt water washing.DCM layer dried over sodium sulfate filtered, and concentrates.Resistates (is used 3%[aq.NH with preparation type TLC purification (1000 microns)
4OH/MeOH1/9]/DCM launches) to obtain be the final title compound of free alkali.Handle generation corresponding hydrochloride (62.2mg) with 4N HCl/ diox.The calculated value of ESI-MS C25H35F3N2O2: 452.27; Measured value: 453 (M+H).
Is mixture and two the single diastereomers that contain two kinds of cis diastereomers with HPLC (AD post, 5%EtOH/ hexane) with diastereomeric separation.
Embodiment 67

Embodiment 68 usefulness intermediates 3 and (1S, 4S)-(+)-2-azepine-5-oxabicyclo [2.2.1] heptane hydrochloride is according to the method preparation of embodiment 66.Cis and trans-isomerism body and function are prepared type TLC fractionation (4/95.6/0.4, MeOH/DCM/NH
4OH).The calculated value of top spot: ESI-MSC24H31F3N2O2: 436.23; Measured value: 437 (M+H).Bottom spot: the calculated value of ESI-MS C24H31F3N2O2: 436.23; Measured value: 437 (M+H).
Embodiment 68

Embodiment 68 usefulness piperidines and intermediate 3 are according to the method preparation of embodiment 49.The calculated value of ESI-MSC24H33F3N2O: 422.25; Measured value: 423 (M+H).
Embodiment 69

(563mg 2.43mmol) is dissolved in methylene dichloride, is cooled to-78 ℃ with intermediate 22.Ozone is fed in the solution up to the blueness that occurs continuing.Nitrogen is disappeared up to blueness by solution.With Na
2SO
4Add above stirred solution.Reaction mixture is filled into the flask that intermediate 23 is housed.With methylene dichloride (20mL), triethylamine (136 μ L, 0.974mmol) and NaB (OAc)
3(929mg 4.38mmol) adds reaction flask to H.Stirred reaction mixture 2h under room temperature, nitrogen atmosphere, then with methylene dichloride dilution, with saturated sodium bicarbonate solution (2 * 100mL) and salt solution (1 * 100mL) washs.The organic layer dried over mgso is filtered, and concentrates then.Crude product is divided into three batches, is loaded into three ion exchange columns.Remove impurity with 40%MeOH/ hexane (100mL) flushing.Then with required product 30%2N NH
3MeOH solution from the chromatographic column wash-out, dilute with methylene dichloride again.Embodiment 69 (131mg, 0.223mmol, 46% yield) preparation type TLC purification (using 32%EtOAc/hex).Then diastereomer is separated (using 45%EtOAc/hex) (top isomer: 20mg, 0.034mmol, 7% yield by preparation type TLC; Middle part isomer: 45mg, 0.076mmol, 16% yield; Bottom isomer: 54mg, 0.092mmol, 19% yield).The ESI-MSC32H41F3N4O3 calculated value: 586.69, measured value 587 (M+H).
Several other piperazine and high piperazine according to the preparation of the same procedure of embodiment 69.Table 7 has been summed up these compounds.
Table 7 (embodiment 70-72)

Embodiment 73

Above compound prepares according to the method for embodiment 49 with intermediate 5 and 4,5,6,7-tetrahydrochysene-1H-pyrazolo [4,3-] pyridine.The calculated value of ESI-MS C24H30F3N5O: 461.24; Measured value 462 (M+H).
Embodiment 74
Above compound prepares according to the method for embodiment 49 with intermediate 5 and 1,4-diaza ring-5-in heptan ketone.The calculated value of ESI-MS C23H31F3N4O2: 452.24; Measured value 453 (M+H).
Embodiment 75
Above compound intermediate 5 and 2,3,4,5-tetrahydrochysene-1, the 4-benzazepines is according to the method preparation of embodiment 57.Top spot: the calculated value of ESI-MS C27H32F3N3O2: 487.24; Measured value 488 (M+H).Bottom spot: the calculated value of ESI-MS C27H32F3N3O2: 487.24; Measured value 488 (M+H).
Embodiment 76
With carbon-containing palladium catalyst (top: 6mg, middle part: 18mg, bottom: 20mg) add three flasks (top: 29.6mg, middle part: 89.9mg, the bottom: 102.3mg) that one of 70 3 kinds of diastereomers of embodiment are housed respectively.Mixture is dissolved in MeOH (3-6mL), feeds hydrogen in the reaction vessel repeatedly.Reaction stirred 4.5h under room temperature, nitrogen atmosphere is then by Acrodisc syringe filter (with 0.45 μ m PTFE film).With compound 76 (top isomer: 16.0mg, 0.0354mmol, 71% yield; Middle part isomer: 42.7mg, 0.0943mmol, 62% yield; Bottom isomer: 34.5mg, 0.0762mmol, 44% yield) (use 10%[NH with preparation type TLC separation
4OH/MeOH (1: 9)]/the dichloromethane solvent system).ESI-MS C
24H
35F
3N
4The O calculated value: 452.56, measured value 453 (M+H).
Embodiment 77

(the middle part steric isomer of TLC, 11mg 0.024mmol) are dissolved in methylene dichloride (3mL), are cooled to 0 ℃ with embodiment 76.With Vinyl chloroformate (5 μ L, 0.05mmol), triethylamine (10 μ L, 0.07mmol) and catalytic amount DMAP (1-2mg) add reaction flask.Reactant is heated to room temperature, under nitrogen atmosphere, stirs 2h.After this, with DCM diluting reaction thing, with saturated sodium bicarbonate solution (1 * 25mL) and salt solution (1 * 25mL) washs.Use the dried over mgso organic layer, filter, concentrate then.Embodiment 77 (4.2mg, 0.0080mmol, 33% yield) purifies by preparation type TLC and (uses 2%[NH
4OH/MeOH (1: 9)]/the dichloromethane solvent system).ESI-MS C
27H
39F
3N
4O
3Calculated value: 524.62, measured value: 525 (M+H).
Embodiment 78

(the middle part steric isomer of TLC, 11mg 0.024mmol) are dissolved in methylene dichloride (3mL), are cooled to 0 ℃ with embodiment 76.With acetic anhydride (11 μ L, 0.12mmol), triethylamine (23 μ L, 0.17mmol) and catalytic amount DMAP (1-2mg) add reaction flask.Under nitrogen atmosphere, reactant is heated to room temperature, stir 2.5h.Then reactant is diluted with DCM, with saturated sodium bicarbonate solution (1 * 25mL) and salt solution (1 * 25mL) washs.Use the dried over mgso organic layer, filter, concentrate then.Embodiment 78 (5.3mg, 0.011mmol, 45% yield) purifies by preparation type TLC and (uses 2%[NH
4OH/MeOH (1: 9)]/the dichloromethane solvent system).ESI-MS C
26H
37F
3N
4O
2Calculated value: 494.59, measured value: 495 (M+H).
Embodiment 79

With methylene dichloride (2-3mL/ flask) add three flasks that three kinds of diastereomers of embodiment 77 are housed respectively (top: 16mg, 0.035mmol, middle part: 11mg, 0.023mmol, bottom: 14mg, 0.031mmol).With methyl isocyanate (top: 21 μ L, 0.35mmol, the middle part: 14 μ L, 0.23mmol, the bottom: 19 μ L 0.31mmol) add each reaction vessel.Each reactant is stirred 3h, concentrate back preparation type TLC (2%[NH
4OH/MeOH (1: 9)]/the dichloromethane solvent system) separation embodiment 79 (top isomer: 12.8mg, 0.0251mmol, 72% yield; Middle part isomer: 10.4mg, 0.0204mmol, 89% yield; Bottom isomer: 11.1mg, 0.0218mmol, 70% yield).ESI-MS C
26H
38F
3N
5O
2Calculated value: 509.61, measured value: 510 (M+H).
Embodiment 80
Steps A
(2.98g, (1.78mL 24.4mmol) handles, at stirring at room 1h methyl alcohol 22.7mmol) (20mL) solution with thionyl chloride with suitable-hydroxyl-D-proline(Pro).Reaction mixture being heated to 65 ℃ then spends the night.The evaporation volatile matter obtains rough methyl esters (4.0816g), and it is dissolved in methylene dichloride (50mL), and the adding diisopropylethylamine (9.59mL, 55.1mmol).Reaction mixture to 0 ℃, with syringe add pure benzyl chloroformate (3.71mL, 26.0mmol).Behind 0 ℃ of stirring 30min, remove cooling bath.(10%, 50mL) middle quencher is extracted into methylene dichloride with product with reactant impouring aqueous citric acid solution.The organic phase that merges is used the salt water washing once more, uses anhydrous sodium sulfate drying, solvent removed in vacuo.Crude product is with flash chromatography purify (silica gel, ethyl acetate: hexane/4: 6) obtain 4.0717g (69%) clean product.
1H?NMR(500MHz,CDCl
3):δ7.35m(5H),5.21(d,J=12.6Hz,2H),5.10(d,J=13Hz,1H),5.06(d,J=12.35Hz,1H),4.45(m,2H),3.80(s,3H),2.35(m,1H),2.15(m,1H)。
Step B
With the alcohol of previous step (2.63g, 9.42mmol) and diisopropylethylamine (3.28mL, methylene dichloride 40mL solution 18.8mmol) is cooled to-78 ℃, with syringe add trifluoromethanesulfonic acid tertiary butyl dimethylsilane ester (2.596mL, 11.30mmol).At-78 ℃ of stirred reaction mixture 2h, be poured on quencher on the 50mL saturated sodium bicarbonate solution.Isolate organic phase, use dried over mgso, vacuum is removed volatile matter.Resistates (6.0g) is further with column chromatography purification (silica gel, ethyl acetate: hexane/1: 4) obtain 2.8355g (76%) clean product.
1HNMR(500MHz,CDCl
3):7.35m(5H),5.20(m=2H),4.45(m,2H),3.70(m,4H),3.45(m,1H),2.23(m,2H)0.87(bs,9H),0.05(m,6H)。
Step C
At 0 ℃, (2.83g, (250mg 11.5mmol) handles, at stirring at room reaction mixture 72h THF 7.19mmol) (10mL) solution with lithium borohydride with the ester of previous step.With the ether dilution, water and 1M phosphoric acid solution wash then.The dry organic extract liquid (sodium sulfate) that merges, solvent removed in vacuo.Crude product is further with column chromatography purification (silica gel, ethyl acetate: hexane/3: 7) obtain 1.5636g (60%) clean product.LC MS:C
19H
31NO
4Si calculated value 365.20, measured value 366.25[M+H]
+
Step D

With the alcohol of previous step (1.56g, 4.27mmol) and diisopropylethylamine (1.49mL, (496 μ L are 6.41mmol) 0 ℃ of processing with methane sulfonyl chloride for methylene dichloride 8.53mmol) (20mL) solution.Stir cold reaction mixture 15min, then at stirring at room 30min.With the methylene dichloride dilution, wash with water, use dried over mgso.Solvent removed in vacuo, the crude product of acquisition is used for next step immediately.LC MS:C
20H
33NSO
6Si calculated value 443.18, measured value 444.25[M+H]
+
Step e
TBS-ether (1.82g, THF 4.10mmol) (50mL) solution tetrabutyl ammonium fluoride (4.51mL, 1M THF solution) processing with previous step.Analyze demonstration at the stirring at room reaction mixture up to LC MS and remove TBS group (about 15min) fully.Reaction mixture to 0 ℃ then, add sodium hydride (180mg, 60% suspension, 45.1mmol).At room temperature continuously stirring 24h.Reactant is poured on quencher waterborne, and crude product is extracted into ethyl acetate.Solvent removed in vacuo, crude product is with the flash chromatography (methylene dichloride: ether/7: 3) obtain the required product of 595mg (59%) of purifying.
1H?NMR(500MHz,CDCl
3):7.38(m,5H),5.30(m,2H),4.52(m,2H),3.80(m,2H),3.40(m,2H),1.70(m,2H)。
Step F

(590mg, ethanol 2.53mmol) (30mL) solution exists down in the hydrogenation of gas tank pressure at Pd/C (142mg, 10%) with the CBZ-amine of previous step.The disappearance of the disappearance of initial feed and toluene in the LC monitoring reaction.Filtration catalizer, evaporated filtrate is to doing the remaining required product of 321mg (94%).
Step G

With intermediate 3 (120mg, 0.340mmol), the amine hydrochlorate (46mg of previous step, 0.34mmol), diisopropylethylamine (60 μ L, 0.34mmol) and 4 molecular sieves (pulverize, ethylene dichloride 360mg) (10mL) solution is handled with sodium triacetoxy borohydride, at stirring at room 24h.Reactant is poured on quencher on the saturated sodium bicarbonate solution, the crude product dichloromethane extraction.Vacuum is removed volatile matter, and (ethyl acetate: ethanol: ammonium hydroxide 90: 8: 2) processing obtains clean product (118mg, 80%) to preparation type TLC.LC MS:C
24H
31N
2F
3O
2Calculated value 436.23, measured value 437.20[M+H]
+
Embodiment 81
Intermediate 21 (150mg, add in methylene dichloride 0.43mmol) (10mL) solution EDC (414mg, 2.16mmol), HOAt (59mg, 0.43mmol) and intermediate 1 (87mg, 0.43mmol), in stirring at room gained mixture 4 days.Water quencher reactant is with the dilution of 20mL methylene dichloride.Isolate organic layer, (2 * 20mL) extract water layer with DCM.Merge organism, use dried over sodium sulfate, filter the back reduction vaporization.Resistates preparation type TLC purification (eluent: 10% methyl alcohol: 89.5% methylene dichloride: 0.5%NH
4OH) obtaining the purified required end product of 40mg (17%), is the mixture of two kinds of cis-isomerides.LC-MS C
30H
36N
2OF
4Calculated value 516.28, measured value [M+H]
+517.
Embodiment 82

With pyrimidyl piperidine hydrochlorate (intermediate 8) (67mg, 0.34mmol) with intermediate 4 (100mg, 0.28mmol), triethylamine (46 μ L, 0.35mmol), the Powdered molecular sieve of 4 (100mg) mixes in DCM.Behind room temperature 15min, (240mg 1.13mmol), stirred the gained mixture 3 days, then by diatomite filtration, with the DCM dilution, with saturated sodium bicarbonate and salt water washing to add sodium triacetoxy borohydride.Use the dried over mgso organic layer, filter, concentrating under reduced pressure obtains rough oily matter, with preparation type TLC purification (silica gel, 0.3%NH
4OH/2.7%MeOH/97%DCM) obtain the 110mg colorless oil.Split each diastereomer by ChiralPak AD post HPLC (with 30% isopropanol/hexane wash-out) and obtain 2 kinds of single diastereomers and a mixture that contains 2 kinds of other diastereomers.
The calculated value of first peak 10mg:ESI-MS C28H35F3N4O: 500.28; Measured value 504 (M+H).
The calculated value of second peak 11mg:ESI-MS C28H35F3N4O: 500.28; Measured value 504 (M+H).
The calculated value of the 3rd peak 7.0mg ESI-MS C28H35F3N4O: 500.28; Measured value 504 (M+H).
Embodiment 83-91
Several other embodiment use different piperidines intermediates according to the similar approach preparation of embodiment 82.Below be these embodiment (83-91).
Embodiment 92

This product is according to the similar approach preparation of embodiment 81, but the commercially available 4-cyano group of intermediate 21 usefulness-4-Phenylpiperidine substitutes.Crude product preparation type TLC purification (eluent: 5% methyl alcohol: 94.5%DCM: 0.5%NH
4OH) obtaining embodiment 92, is 4 kinds of mixture of isomers.LC-MS C
31H
36F
3N
3O calculated value 523.28, measured value [M+H]
+524.
Embodiment 93

This product is according to the similar approach preparation of embodiment 81, but the commercially available 4-Phenylpiperidine of intermediate 21 usefulness substitutes.Crude product preparation type TLC purification (eluent: 10% methyl alcohol: 89.5%DCM: 0.5%NH
4OH) obtaining embodiment 93, is 4 kinds of mixture of isomers.LC-MSC
30H
37F
3N
2O calculated value 498.29, measured value [M+H]
+499.
Embodiment 94
Steps A
(2.3g, (1.8mL 21mmol), adds 2 DMF to cyclopentanone formic acid (intermediate 3 step C's) then to add oxalyl chloride in methylene dichloride 14mmol) (70mL) solution.At stirring at room solution 80min, reduction vaporization then.Resistates is dissolved in DCM (20mL), with syringe add ready intermediate 3 (3.0g, 14mmol) and triethylamine (2.1mL, DCM 15mmol) (50mL) solution.At stirring at room gained mixture 18h, water (25mL) quencher then.Separation of organic substances with 1N HCl, saturated sodium bicarbonate solution and salt water washing, is used anhydrous magnesium sulfate drying, filters, then evaporation.Resistates Biotage Flash40 purification (eluent: 40% ethyl acetate/60% hexane) obtain 2.2g (43%) title compound.
Step B
Product (200mg with steps A, 0.54mmol), commercially available 4-fluorophenyl piperidine hydrochlorate (120mg, 0.54mmol), diisopropylethylamine (94 μ L, 0.54mmol) and molecular sieve (4 that pulverize, methylene dichloride 100mg) (10mL) solution sodium triacetoxy borohydride (343mg, 1.60mmol) handle, in stirred overnight at room temperature.With saturated sodium bicarbonate (10mL) quencher reactant, with 10mL DCM dilution.Isolate organic layer, (2 * 5mL) wash the aqueous solution with methylene dichloride.Merge organism, use anhydrous sodium sulfate drying, filter the back evaporation.Resistates preparation type TLC purification (eluent: 8% ethanol: 90% ethyl acetate: 2%NH
4OH) obtaining two kinds of isomer, is respectively the isomer (25mg, 5%) of the higher wash-out of unknown absolute steric configuration and the isomer (37.2mg, 7.6%) of low wash-out.LC-MS C
30H
35N
2OF
5Calculated value 534.28, measured value [M+H]
+535.
Embodiment 95

This product is according to the similar approach preparation of embodiment 94, but 4-fluorophenyl piperidine hydrochlorate substitutes with intermediate 9.Crude product preparation type TLC purification (eluent: 90% ethyl acetate: 8% ethanol: 2%NH
4OH) obtaining 450mg (66%) title product, is 4 kinds of mixture of isomers.LC-MS C
28H
34N
4OF
4Calculated value 518.27, measured value [M+H]
+519.
Several other embodiment wherein adopt different piperidines intermediates according to the similar approach preparation of embodiment 94.Below be these embodiment (96-104).
Embodiment 105
Intermediate 21 (150mg, add in methylene dichloride 0.43mmol) (10mL) solution EDC (170mg, 0.86mmol), HOAt (59mg, 0.43mmol) and intermediate 21 (91mg, 0.43mmol), in stirring at room gained mixture 3 days.Water quencher reactant is with the dilution of 20mL methylene dichloride.Isolate organic layer, (2 * 20mL) extract water layer with DCM.Merge organism, use dried over sodium sulfate, filter the back reduction vaporization.Resistates preparation type TLC purification (eluent: 7% ethanol: 92% methylene dichloride: 1.0%NH
4OH) obtaining the required end product of 121mg (50%), is the mixture of two kinds of cis-isomerides.LC-MS C
29H
36BrFN
2O calculated value 526.28, measured value [M+H]
+527 and [(M+2)+H]
+529.
Embodiment 106
Embodiment 105 (100mg, 0.210mmol), phenyl-boron dihydroxide (30mg, 0.23mmol), add Na in toluene (1.4mL) and MeOH (0.6mL) solution
2CO
3(80mg, 0.74mmol) and Pd (PPh
3)
2Cl
2(8mg, H 0.01mmol)
2O (0.4mL) solution.With reaction mixture in high pressure pipe in 80 ℃ of heating 12h, then by diatomite filtration, be concentrated into dried.Enriched material dilutes with DCM, with 1N NaOH solution (3x) washing, uses dried over sodium sulfate, vacuum concentration.Crude product preparation type TLC purification (eluent: 5/94.5/0.5, MeOH/DCM/NH
4OH) obtaining embodiment 106 (63.3mg, 63.3%), is 2 kinds of cis mixtures of isomers.LC-MS C
35H
41FN
2O calculated value 524.29, measured value [M+H]
+525.4.
Embodiment 107
This product is according to the similar approach preparation of embodiment 106, but phenyl-boron dihydroxide is with 4.Pyridyl boric acid substitutes.Crude product preparation type TLC purification (eluent: 10/89/1.0, MeOH/DCM/NH
4OH) obtaining embodiment 107, is the mixture of two kinds of cis-isomerides.LC-MS C
34H
40FN
3O calculated value 525.26, measured value [M+H]
+526.3.
Embodiment 108

This product is according to the similar approach preparation of embodiment 106, but phenyl-boron dihydroxide substitutes with 3-pyridyl boric acid.Crude product preparation type TLC purification (eluent: 10/89/1.0, MeOH/DCM/NH
4OH) obtaining embodiment 108, is the mixture of two kinds of cis-isomerides.LC-MS C
34H
40FN
3O calculated value 525.26, measured value [M+H]
+526.3.
Embodiment 109
This product is according to the similar approach preparation of embodiment 106, but phenyl-boron dihydroxide substitutes with 2-pyridyl boric acid.Crude product preparation type TLC purification (eluent: 10/89/1.0, MeOH/DCM/NH
4OH) obtaining embodiment 109, is the mixture of two kinds of cis-isomerides.LC-MS C
34H
40FN
3O calculated value 525.26, measured value [M+H]
+526.3.
Embodiment 110
Steps A
Drip 1,2,3 in the ice-cold vitriol oil, the 4-tetrahydroisoquinoline (23mL, 170mmol), then with temperature rise the speed be no more than 5 ℃ add saltpetre (18.8g, 186mmol).After adding finishes,, be poured on ice (700g) and NH then at stirring at room mixture 18h
4On the stirring the mixture of OH (150mL).Mixture CHCl
3(3 * 300mL) extractions.The CHCl that merges
3Layer is used dried over sodium sulfate with saturated NaCl (200mL) washing, filters vacuum concentration.Resistates is dissolved in ethanol (130mL), under the ice bath cooling, adds concentrated hydrochloric acid (22mL).Remove by filter precipitation, obtain product (12.45g, 34%) with recrystallizing methanol;
1H NMR500MHz (DMSO-d
6) δ=3.13 (2H, t, J=6.2Hz), 3.35 (2H, t, J=6.2Hz), 4.35 (2H, s), 7.50 (1H, d, J=8.5Hz), 8.07 (1H, dd, J=2.3 and 8.5Hz), 8.19 (1H, d, J=2.3Hz) 10.02 (2H, brs).
Step B

The steps A product (12g, 58mmol), pyridine (23.7mL, anhydrous CH 293mmol)
2Cl
2(50mL) drip in cold (0 ℃) suspension trifluoro-acetic anhydride (12mL, 87mmol), at stirring at room gained mixture 18h.Reaction mixture is poured on the ice (500g), uses CH
2Cl
2(4 * 150mL) extractions.The dichloromethane layer that merges with 1N HCl (4 * 100mL), saturated NaCl (100mL) washing, use dried over sodium sulfate, filter final vacuum and evaporate and obtain product (15.92g, 89%);
1H NMR 500MHz (CDCl
3) δ=3.07 (2H, m), 3.91 and 3.94 (2H, t, J=6.2Hz), 4.85 and 4.88 (2H, s), 7.36 (1H, dd, J=8.7 and 11.9Hz), 8.07 (1H, dd, J=2.3 and 8.5Hz), 8.01-8.08 (2H m).
Step C
Under 50psi, (16g, ethanol 58mmol) (200mL) solution passes through PtO in the Parr device with step B product
2Hydrogenation is up to stopping to absorb hydrogen.Remove catalyzer by diatomite filtration, vacuum concentrated filtrate obtains product (14.2g, 100%);
1H NMR 500MHz (CDCl
3) δ=2.84 (2H, t, J=5.7Hz), 3.35 (2H, br s), 3.80 and 3.84 (2H, t, J=6.0Hz), 4.64 and 4.69 (2H, s), 6.45 (1H, d, J=10.0Hz), 6.57 (1H, td, J=2.4 and 8.5Hz), 6.95 (1H, d, J=8.5Hz).
Step D

Step C product (2.0g adds N in toluene 8.7mmol) (50mL) solution, and the dinethylformamide azine (2.3g, 16mmol) and the toluenesulphonic acids of a spatula end, reflux gained mixture 24h.Vacuum concentrated mixture, resistates is at CH
2Cl
2(50mL) and between water (50mL) distribute.Isolate organic layer,, use dried over mgso, filter vacuum concentration with saturated sodium bicarbonate (25mL) and saturated NaCl (25mL) washing.Resistates CH
2Cl
2(4mL) grind, filter, drying obtains product (1.0g, 39%);
1H NMR 500MHz (CDCl
3)=3.05 (2H, t, J=5.7Hz), 3.90 and 3.95 (2H, t, J=6.0Hz), 4.83 and 4.89 (2H, s), 7.20 (1H, d), 7.26 (1H, t), 7.38 (1H, and t) 8.45 (2H, s).
Step e

(1.0g adds salt of wormwood (2.3g, water 17mmol) (10mL) solution, reflux gained mixture 90min to step D product in ethanol 3.4mmol) (50mL) solution.The vacuum concentration cold reaction mixture, solid residue CH
2Cl
2(3 * 10mL) extractions.The CH that vacuum-evaporation merges
2Cl
2The layer, resistates with the silica column chromatographic purification (with containing 0.5%NH
4The 10%CH of OH
3OH/CH
2Cl
2Wash-out) obtains product (550mg, 82%);
1H NMR500MHz (CDCl
3) δ=2.69 (1H, br s), 2.85 (2H, t, J=5.9Hz), 3.17 (2H, t, J=6.0Hz), 4.07 (2H, s), 7.20 (1H, d, J=1.8Hz), 7.14 (1H, dd, J=1.8 and 8.0Hz), 7.23 (1H, d, J=8.0Hz), 8.43 (2H, s).
Step F
This product is according to the similar approach preparation of embodiment 81, but intermediate 1 usefulness step e product substitutes.Crude product preparation type TLC purification (eluent: 10% methyl alcohol: 89.5%DCM: 0.5%NH
4OH) obtaining embodiment 30, is the mixture of two kinds of cis-isomerides.LC-MSC
31H
38FN
5O calculated value 515.31, measured value [M+H]
+516.
Embodiment 111

Steps A

(7.5g adds salt of wormwood (17g, water 120mmol) (50mL) solution, reflux gained mixture 90min to the trifluoroacetamide of embodiment 110 step C preparation in ethanol 31mmol) (200mL) solution.The vacuum concentration cold reaction mixture dilutes resistates water (200mL).Mixture CH
2Cl
2(3 * 100mL) extractions.The CH that merges
2Cl
2Layer dried over sodium sulfate filtered, and concentrates and obtains product (3.76g, 83%);
1H NMR 500MHz (CDCl
3) δ=2.69 (2H, t, J=6.0Hz), 3.11 (2H, t, J=6.0Hz), 3.45 (2H, br s), 3.92 (2H, s), 6.36 (1H, d, J=2.3Hz), 6.52 (1H, dd, J=2.3 and 8.0Hz), 6.89 (1H, d, J=8.0Hz).
Step B
The steps A product (3.76g, add in tetrahydrofuran (THF) 25.4mmol) (100mL) solution triethylamine (4.24mL, 30.5mmol), benzyl chloroformate (4.0mL, 28mmol), at stirring at room gained mixture 4h.Ethyl acetate (100mL) is added reaction mixture, dried over mgso is used in whole water (250mL), 5% citric acid solution (150mL), saturated sodium bicarbonate (150mL) and saturated NaCl (100mL) washing, filters, vacuum concentration obtains product (7.2g, 100%).
Step C:

Under ice bath cooling, and step B product (7.2g, 25mmol) and pyridine (5.1mL, CH 64mmol)
2Cl
2(150mL) add in the solution trifluoro-acetic anhydride (5.38mL, 38.1mmol), at stirring at room gained mixture 5h.Mixture is poured on the ice (150g), uses CH
2Cl
2(4 * 100mL) extractions.The dichloromethane layer that merges with 1N HCl (4 * 75mL), saturated NaCl (100mL) washing, use dried over mgso, filter the back and evaporate.Resistates is dissolved in CCl
4(200mL), (10g, 38mmol), reflux gained mixture 15h cools off vacuum concentration to add triphenylphosphine.Resistates is dissolved in anhydrous N, dinethylformamide (150mL), this solution is added sodiumazide, and (dinethylformamide (75mL) solution is at stirring at room gained mixture 3h for 1.65g, anhydrous N 25.4mmol).In reaction mixture impouring water (500mL), use Et
2O (3 * 100mL) extractions.The Et that merges
2O layer water (2 * 250mL), saturated NaCl (100mL) washing, use dried over mgso, filtration, vacuum concentration.Resistates obtains product (3.4g, 33%) with silica column chromatographic purification (with the 10%EtOAc/ hexane to 20%EtOAc/ hexane gradient wash-out);
1H NMR 500MHz (CDCl
3) δ=2.99 (2H, br m), 3.80 (2H, t, J=6.0Hz), 4.75 (2H, s), 5.21 (2H, s), 7.20-7.45 (8H, m).
Step D:
Feeding under the nitrogen, adding 10% palladium carbon (500mg) in the step C product (3.4g, methyl alcohol 8.4mmol) (100mL) solution), under hydrogen gas tank, stirring gained mixture 5h.Remove catalyzer by diatomite filtration, vacuum-evaporation filtrate obtains product (2.2g, 97%);
1H NMR500MHz (CDCl
3) δ=2.33 (1H, brs), 2.91 (2H, t, J=6.0Hz), 3.19 (2H, t, J=6.0Hz), 4.08 (2H, s), 7.14 (1H, d, 1.8Hz), 7.22 (1H, dd, J=1.8 and 8.2Hz), 7.31 (1H, d, J=8.2Hz).
Step e:
This product is according to the similar approach preparation of embodiment 81, but intermediate 1 usefulness step D product substitutes.Crude product preparation type TLC purification (eluent: 10% methyl alcohol: 89.5%DCM:0.5%NH
4OH) obtaining embodiment 31, is the mixture of two kinds of cis-isomerides.LC-MSC
31H
36F
3N
6O calculated value 584.29, measured value [M+H]
+585.
Embodiment 112

This product is according to the preparation of the similar approach of embodiment 81, but intermediate 1 usefulness is commercially available 6, and 7-diethoxy-tetrahydroisoquinoline is alternative.Crude product preparation type TLC purification (eluent: 5% ethanol: 94%DCM: 1.0%NH
4OH) obtaining embodiment 32, is the mixture of two kinds of cis-isomerides.LC-MS C
33H
45FN
2O
3Calculated value 536.34, measured value [M+H]
+537.
Embodiment 113

Steps A

This product is according to the preparation of the similar approach of intermediate 1, but 2-fluoro-4-trifluoromethyl acetonitrile with 3,4-two-methyl fluoride phenylacetonitrile is alternative.LC-MS C
9H
9F
2N calculated value 169.07, measured value [M+H]
+170.1
Step B

This product is according to the similar approach preparation of embodiment 81, but intermediate 1 usefulness steps A product substitutes.Crude product preparation type TLC purification (eluent: 6% ethanol: 93%DCM: 1.0%NH
4OH) obtaining embodiment 33, is the mixture of two kinds of cis-isomerides.LC-MSC
29H
35F
3N
2O calculated value 484.27, measured value [M+H]
+485.
Embodiment 114

This product is according to the similar approach preparation of embodiment 82, but the commercially available 4-hydroxy-4-phenyl piperidine of intermediate 8 usefulness substitutes.Crude product preparation type TLC purification (eluent: 10% methyl alcohol: 89%DCM: 1.0%NH
4OH) obtaining embodiment 114, is 4 kinds of mixture of isomers.LC-MS C
30H
37F
3N
2O
2Calculated value 514.28, measured value [M+H]
+515.
Embodiment 115 and 116

Embodiment 115 embodiment 116
With intermediate 5 (100mg, 0.22mmol), 4-fluorophenyl piperidine hydrochlorate (57mg, 0.26mmol), diisopropylethylamine (45 μ L, 0.26mmol) and broken molecular sieve (4 , ethylene dichloride 50mg) (5mL) solution sodium triacetoxy borohydride (233mg, 1.10mmol) handle, in stirred overnight at room temperature.With saturated sodium bicarbonate (10mL) quencher reactant, dilute with 10mLDCE.Isolate organic layer, (2 * 5mL) wash water layer with methylene dichloride.Merge organism, use anhydrous sodium sulfate drying, filter the back evaporation.Resistates preparation type TLC purification (eluent: 0.5%NH
4OH: 5%MeOH: 94.5%CH
2Cl
2) obtain 72.3mg (63%) end product, be the mixture of two kinds of diastereomers.HPLC finishes separation with preparation type ChiralCel OD post, utilizes 15% ethanol and 85% hexane eluent wash-out, flow velocity 9mL/min.Obtain unwanted trans-isomer(ide) (35mg, 50%) and required cis-isomeride (25mg, 36%).Total recovery 86%.The LC-MS:C of two kinds of products
29H
35N
3OF
4Calculated value 517.28, measured value [M+H]
+518.3.
Embodiment 115:
1H NMR (the 1st kind of isomer do not need) (500MHz, CDCl
3) δ 8.73 (s, 1H), 7.69 (s, 1H), 7.20 (app dd, J=5.5,8.6Hz, 2H) 6.98 (app t, J=8.6Hz, 2H), [4.88 (d, J=17.5Hz, 1H), 4.80 (d, J=17.5Hz, 1H) (ABx q)] and 4.05-3.96 (m, 1H), 3.88 (app dt, J=5.9,13.1Hz, 1H), 3.27 (br d, J=11.4Hz, 1H), 3.12 (brt, J=5.6Hz, 3H), 2.83 (dd, J=5.7,11.9Hz, 1H), and 2.52-2.44 (m, 1H), 2.43-2.34 (m, 1H), 2.16-1.95 (m, 6H), 1.86-1.74 (m, 4H), 1.45 (br t, J=10.0,3H), 1.03 (d, J=6.6Hz, 3H), 0.83 (d, J=6.6Hz, 3H).
Embodiment 116:
1H NMR (the 2nd kind of isomer, required) (500MHz, CDCl
3) δ 8.72 (s, 1H), 7.69 (s, 1H), 7.19 (app dd, J=5.5,8.6Hz, 2H) 6.98 (app t, J=8.6Hz, 2H), [4.94 (br s, 1H), 4.69 (br d, J=17.6Hz, 1H) (ABx q)] 4.05-3.80 (m, 1H), and 3.20-3.08 (m, 4H), 2.68 (dd, J=6.6,12.8Hz, 1H), and 2.52-2.43 (m, 2H), 2.28 (dd, J=7.3,12.8Hz, 1H), 2.14-2.00 (m, 3H), 1.97-1.87 (m, 2H), 1.83 (br d, J=12.8Hz, 2H) 1.75 (br d, 12.4Hz, 2H), and 1.56-1.48 (m, 1H), 1.42-1.34 (m, 1H), 1.01 (d, J=6.5Hz, 3H), 0.80 (d, J=6.6Hz, 3H).
Embodiment 117
With the pyrimidyl piperidine hydrochlorate (intermediate 8,133mg, 0.564mmol) with intermediate 5 (100mg, 0.282mmol), DIEA (240 μ L, 1.40mmol) and the Powdered molecular sieve of 4 (200mg) in DCM, mix.Behind room temperature 15min, (300mg 1.41mmol), stirred the gained mixture 3 days, then by diatomite filtration, with the DCM dilution, with saturated sodium bicarbonate and salt water washing to add sodium triacetoxy borohydride.Use the dried over sodium sulfate organic layer, filter, concentrating under reduced pressure obtains rough oily matter, with preparation type TLC purification (silica gel, 0.5%NH
4OH/4.5%MeOH/95%DCM) obtain the 126mg colorless oil.Split suitable/trans isomer (adopting ChiralPak OD post) by HPLC and obtain 57mg trans-isomer(ide) and 45mg cis-isomeride with 20% ethanol/hexane wash-out.
The calculated value of first peak 57mg:ESI-MS C27H34F3N5O: 501.27; Measured value 502 (M+H).
The calculated value of second peak 45mg:ESI-MS C27H34F3N5O: 501.27; Measured value 502 (M+H).
Embodiment 118-129

Several other embodiment wherein adopt different piperidines intermediates according to the similar approach preparation of embodiment 117.Below be these embodiment (118-129).
Embodiment 130
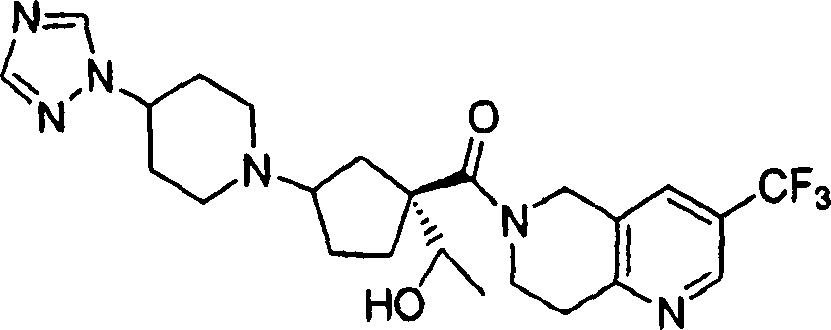
Steps A

This ketone obtains by the intermediate 6 of resolution of racemic, wherein adopts ChiralCel OD preparative column, with 15% ethanol/hexane wash-out, 9.0mL/min.Under the similar analysis condition (1.0mL/min), the retention time of the enantiomer of very fast wash-out is 11.25min.LC MSC
17H
19F
3N
2O
3Calculated value 356.13, measured value 357.05[M+H]
+
Step B

Final compound is synthetic according to the method for embodiment 19 with the ketone and the intermediate 10 of very fast wash-out in the present embodiment steps A.Corresponding cis-separate by preparation type TLC with the mixture of trans-diastereomer.LC MS C
24H
31F
3N
6O
2Calculated value 492.25, measured value 493.30[M+H]
+
Embodiment 131

This compound is synthetic according to the method for embodiment 19 with the ketone and the intermediate 9 of the preparation of embodiment 130 steps A.LC MS C
26H
32F
3N
5O
2Calculated value 503.25, measured value 504.25[M+H]
+
Embodiment 132

Steps A
The method that this ketone is introduced according to intermediate 6 is synthetic, but substitutes acetaldehyde with methoxymethyl chlorine in intermediate 6 step C.Corresponding enantiomer is by ChiralCel OD preparative column HPLC (eluent: hexane: ethanol/85: 15,9.0mL/min) separation.LC MSC
26H
32F
3N
5O
2Calculated value 503.25, measured value 504.25[M+H]
+
Step B
The ketone of final compound usefulness steps A and 4-(4-fluorophenyl) piperidines are according to the method preparation of embodiment 19.Corresponding isomer is by preparation type TLC (eluent: ethyl acetate: ethanol: ammonium hydroxide/90: 9: 1) obtain.LC MS C
26H
32F
3N
5O
2Calculated value 503.25, measured value 504.70[M+H]
+
Embodiment 133
Steps A

2-(trifluoromethyl) vinylformic acid (20.0g, 143mmol) and benzyl alcohol (14.8mL, add in DCM 142mmol) (150mL) mixture in batches EDC (40.93g, 214.2mmol).Stirred reaction mixture 2h, with the DCM dilution, dried over sodium sulfate is used in water and salt water washing, filters, and concentrates.Resistates obtains viscosity oily product (13.7g, 42%) with flash column chromatography purification (silica gel, 5%EtOAc/ hexane).
1H-NMR(400MHz,CDCl
3)δ7.36-7.43(m,5H),6.78(d,1H),6.48(d,1H),5.32(s,2H)。
Step B

Under nitrogen atmosphere, add acetate 2-[(three silyls in the flame-dried flask) methyl]-2-propylene-1-base ester (12.18mL, 57.35mmol) and the intermediate (13.2g of embodiment 52 steps A, 57.4mmol) and tetrakis triphenylphosphine palladium (0) (13.3g, THF 11.5mmol) (200mL).Reaction mixture refluxed is spent the night,, filter, concentrate then with DCM (150mL) dilution.Resistates obtains product (11.68g, 71.7%) with flash column chromatography purification (silica gel, 100% hexane-2.5%EtOAc/ hexane-5%EtOAc/ hexane).
1H-NMR(400MHz,CDCl
3)δ7.33-7.42(m,5H),5.23(s,2H),4.93(m,2H),3.03(m,1H),2.78(m,1H),2.36-2.52(m,3H),2.12-2.24(m,1H)。
Step C
(11.6g, DCM 40.8mmol) (150mL) solution is cooled to-78 ℃, feeds nitrogen to saturated with step B product.Ozone is fed reaction mixture up to the solution becomes au bleu, and (12.8g 49.0mmol) adds mixture with triphenylphosphine then.Stirred reaction mixture spends the night, then reduction vaporization.Resistates obtains title compound (8.49g, 72.7%) with flash column chromatography purification (silica gel, 30%EtOAc/ hexane).
1H-NMR(400MHz,CDCl
3)δ7.33-7.42(m,5H),5.26(s,2H),2.92(m,1H),2.65(m,1H),2.35-2.54(m,4H)。
Step D

Step C intermediate (1.00g, add in ethanol 3.49mmol) (60mL) solution Pd-C (10%, 100mg).Reaction mixture is added the Parr vibrator, at 50lb H
2Pressure is vibration 1.5h down.Solution dilutes with methyl alcohol, and by diatomite filtration, vacuum-evaporation obtains yellow oily acid (692mg, 100%) then.LC-MS C7H7F3O3 calculated value: 196.03; Measured value: 197 (M+H).
Step e

Step D product (684mg, 3.49mmol), EDC (2.01g, 10.5mmol), intermediate 2 (916mg, 3.84mmol) and add DIEA in the mixture of DCM (30mL) (670 μ L 3.84mmol), spend the night at stirring at room gained solution.Reaction mixture dilutes with DCM, and dried over sodium sulfate is used in water and salt water washing, filters the final vacuum evaporation.Resistates obtains title compound with column chromatography purification (silica gel, 50%EtOAc/ hexane).
1H-NMR(400MHz,CDCl
3)δ8.75(s,1H),7.71(s,1H),4.86(d,J=5.5Hz,2H),3.91-4.08(m,2H),3.18(t,J=5.0Hz,2H),2.98(s,2H),2.64-2.85(m,2H),2.43-2.56(m,2H)。The calculated value of LC-MS C16H14F6N2O2: 380.10; Measured value: 381 (M+H).
Step F

Step e compound (100mg, 0.263mmol), 4-Phenylpiperidine hydrochloride (52mg, 0.26mmol), molecular sieve (4 , 180mg), DIEA (46 μ L, 0.26mmol) and the mixture of DCM (5mL) in add sodium triacetoxy borohydride (167mg, 0.789mmol), in stirring at room gained mixture overnight.With DCM diluting reaction thing, by diatomite filtration, reduction vaporization.Resistates preparation type TLC purification (1000 microns, eluent: 0.4%NH
4The OH aqueous solution: 4%MeOH: 95.6%DCM) obtain for the cis of free alkali-with trans-mixture of isomers.Cis-(chirality OD post, eluent: the 5%EtOH/ hexane) separation obtains end product, is the title compound of required cis-isomeride by preparation type chirality HPLC with trans-isomer.Handle generation hydrochloride (20.4mg) with 4N HCl/ diox.
1H-NMR(400MHz,CDCl
3)δ8.74(s,1H),7.70(s,1H),7.20-7.35(m,5H),4.85(m,2H),3.99(m,2H),3.14-3.24(m,5H),2.46-2.56(m,3H),2.02-2.22(m,5H),1.70-1.91(m,5H)。The calculated value of LC-MS C27H29F6N3O: 525.22; Measured value: 526 (M+H).
Embodiment 134

Steps A
At-78 ℃, light blue up to occurring to the 100mL methylene dichloride stirred solution feeding ozone of cyclohexane (15mL).Remove excessive ozone by nitrogen gas stream, add the 60mL dimethyl sulfide then.Allow the mixture standing over night, use dried over sodium sulfate.Rough vacuum is removed and is desolvated and DMS.Crude product is directly used in next step and need not to purify again.
Step B
With intermediate 27 (135mg, 0.3mmol), steps A 1,6-hexane-dialdehyde (~100mg), molecular sieve (4 , 50mg), (130mg, the mixture of DCM 1.0mmol) (10mL crude product) stirs 5min to DIEA.Add then sodium triacetoxy borohydride (424mg, 2.0mmol).Stir gained mixture 2h, use Na
2CO
3The saturated aqueous solution quencher is filtered, and washs with DCM.Separating filtrate, the aqueous solution extracts with DCM.The DCM layer dried over sodium sulfate that merges, evaporation.Resistates (is used 10%[aq.NH with preparation type TLC purification (1000 microns)
4OH/MeOH 1/9]/DCM launches) obtain to be the title compound end product of free alkali.Handle formation hydrochloride (42mg) with 4M HCl/ diox.The calculated value of ESI-MS C25H34F3N3O3: 481; Measured value: 482 (M+H).
Embodiment 135

With embodiment 134 (35mg), Pd/C (5%, 5mg) and the mixture of methyl alcohol (20mL) hydrogenation 1h on the Parr device.Remove by filter catalyzer.Evaporated filtrate obtains white solid title product (32mg).The calculated value of ESI-MS C25H36F3N3O: 451; Measured value: 452 (M+H).
Embodiment 136

Steps A
At-78 ℃, light blue up to occurring to the 50mL methylene dichloride stirred solution feeding ozone of suberene (5mL).Remove excessive ozone by nitrogen gas stream, add the 20mL dimethyl sulfide then.Allow the mixture standing over night, use dried over sodium sulfate.Rough vacuum is removed and is desolvated and DMS.Crude product is directly used in next step and need not to purify again.
Step B
With intermediate 27 (135mg, 0.3mmol), steps A 1,7-heptane-dialdehyde (~100mg), molecular sieve (4 , 50mg), DIEA (130mg, 1.0mmol) and the mixture of DCM (10mL crude product) stir 5min.Add then sodium triacetoxy borohydride (424mg, 2.0mmol).Stir gained mixture 2h, use Na
2CO
3The saturated aqueous solution quencher is filtered, and washs with DCM.Separating filtrate, the aqueous solution extracts with DCM.The DCM layer dried over sodium sulfate that merges, evaporation.Resistates (is used 10%[aq.NH with preparation type TLC (1000 microns)
4OH/MeOH 1/9]/the DCM expansion) purifying obtains to be the title compound end product of free alkali.Handle formation hydrochloride (50mg) with 4M HCl/ diox.The calculated value of ESI-MSC26H36F3N3O3: 485; Measured value: 486 (M+H).
Embodiment 137

Steps A

With 5-norbornylene-2-formic acid (5.4g, 40mmol), benzyl alcohol (4.3g, 40mmol), EDAC.HCl (9.5g, 50mmol), (5.2g, 40mmol) weighing adds flask to DIEA.Add the 50mL methylene dichloride.Stir the mixture and spend the night,, use dried over sodium sulfate, evaporation with 2M aq.HCl, water and the washing of yellow soda ash saturated aqueous solution.Resistates is purified with MPLC (10%EtOAc/ hexane).Obtain title compound (5.2g) for external form and endo isomer mixture.
Step B

At-78 ℃, (it is light blue up to occurring that 1.2g, 50mL methylene dichloride stirred solution 5mmol) feed ozone to 5-norbornylene-2-formic acid benzyl ester.Remove excessive ozone by nitrogen gas stream, add the 20mL dimethyl sulfide then.Allow the mixture standing over night, use dried over sodium sulfate.Rough vacuum is removed and is desolvated and DMS.Crude product is directly used in next step and need not to purify again.
Step C

Intermediate 27 (86mg, 0.2mmol), two-aldehydo-ester of step B (~200mg), molecular sieve (4 , 500mg), DIEA (130mg, 1.0mmol) and the mixture of DCM (the rough thing of 10mL) stir 5min.Add then sodium triacetoxy borohydride (420mg, 2.0mmol).Stir gained mixture 2h, use Na
2CO
3The saturated aqueous solution quencher is filtered, and washs with DCM.Separating filtrate, the aqueous solution extracts with DCM.The DCM layer dried over sodium sulfate that merges, evaporation.Resistates (is used 10%[aq.NH with preparation type TLC (1000 microns)
4OH/MeOH 1/9]/the DCM expansion) purifying obtains to be the title compound end product of free alkali.Handle generation corresponding hydrochloride (62mg) with 4N HCl/ diox.The calculated value of ESI-MSC34H40F3N3O5: 627; Measured value: 428 (M+H).
Step D

With step C amino ester (60mg), Pd/C (5%, 100mg) and the mixture of methyl alcohol (20mL) with Parr device hydrogenation 1h under the 50lb nitrogen atmosphere.Remove by filter catalyzer.Evaporated filtrate, resistates are purified with preparation type TLC (launching with methyl alcohol) and are obtained white solid title product (18mg).The calculated value of ESI-MS C27H36F3N3O: 507; Measured value: 508 (M+H).
Embodiment 138
Steps A
With intermediate 26 (be hydrochloride, 1.2g, 4.0mmol), intermediate 23 (1.1g, 4.0mmol), PyBrOP (1.9g, 4.0mmol), DMAP (0.29g, 2.4mmol), DIEA (2.8mL, 16mmol) and the mixture of 10mL methylene dichloride in stirred overnight at room temperature.Whole mixtures are applied to silicagel column, with 20% ethyl acetate/hexane wash-out.Obtain white solid title compound (1.7g, 83%).LC-MS C26H35F3N2O5 calculated value 512, measured value [M+H-100]
+413.
Step B
(1.7g 3.3mmol) mixes with 20mL 4M HCl/ diox with the acid amides of above steps A.Stir gained solution 1h, evaporation, high vacuum dry obtains white solid title product (1.45g, 100%).LC-MS C21H27F3N2O3 calculated value 412, measured value [M+H]
+413.
Step C
With the intermediate of above step B (140mg, 0.30mmol), glutaraldehyde (50%H
2O, 120mg, 0.60mmol), molecular sieve (4 , 1500mg), DIEA (52mg, 0.40mmol) and the mixture of DCM (10mL) stir 5min.Add then sodium triacetoxy borohydride (212mg, 1.00mmol).Stir gained mixture 1h, use Na
2CO
3The saturated aqueous solution quencher is filtered, and washs with DCM.Separating filtrate, the aqueous solution extracts with DCM.The DCM layer dried over sodium sulfate that merges, evaporation then.Resistates (is used 10%[aq.NH with preparation type TLC (1000 microns)
4OH/MeOH 1/9]/the DCM expansion) purifying obtains to be the title compound end product of free alkali.Handle generation corresponding hydrochloride (80mg) with 4N HCl/ diox.The calculated value of ESI-MS C26H35F3N2O3: 480; Measured value: 481 (M+H).
Embodiment 139

(45mg, 0.090mmol), lithium hydroxide monohydrate (50mg), the mixture of water (0.1mL) and methyl alcohol (1.0mL) is loaded into whole mixtures on the preparation type TLC in stirred overnight at room temperature, launches with methyl alcohol with embodiment 138.Obtain white solid title compound (34mg).LC-MS C25H33F3N2O3 calculated value 466, measured value [M+H]
+467.
Embodiment 140

With the intermediate of embodiment 138 step B (140mg, 0.30mmol), embodiment 136 steps A 1,6-hexane dialdehyde (~100mg), molecular sieve (4 , 500mg), DIEA (130mg, 1.00mmol) and the mixture of DCM (20mL) stir 5min.Add then sodium triacetoxy borohydride (424mg, 2.00mmol).Stir gained mixture 1h, use Na
2CO
3The saturated aqueous solution quencher is filtered, and washs with DCM.Separating filtrate, the aqueous solution extracts with DCM.The DCM layer dried over sodium sulfate that merges, evaporation.Resistates (is used 10%[aq.NH with preparation type TLC (1000 microns)
4OH/MeOH 1/9]/the DCM expansion) purifying obtains to be the title compound end product of free alkali.Handle generation corresponding hydrochloride (75mg) with 4M HCl/ diox.The calculated value of ESI-MS C27H37F3N2O3: 494; Measured value: 495 (M+H).
Embodiment 141

(20mg 0.04mmol), the mixture of lithium hydroxide monohydrate (50mg), water (0.1mL) and methyl alcohol (1.0mL) is in stirred overnight at room temperature, and whole mixtures are loaded on the preparation type TLC, launches with methyl alcohol with embodiment 140.Obtain white solid title compound (15mg).LC-MS C26H35F3N2O3 calculated value 480, measured value [M+H]
+481.
Embodiment 142
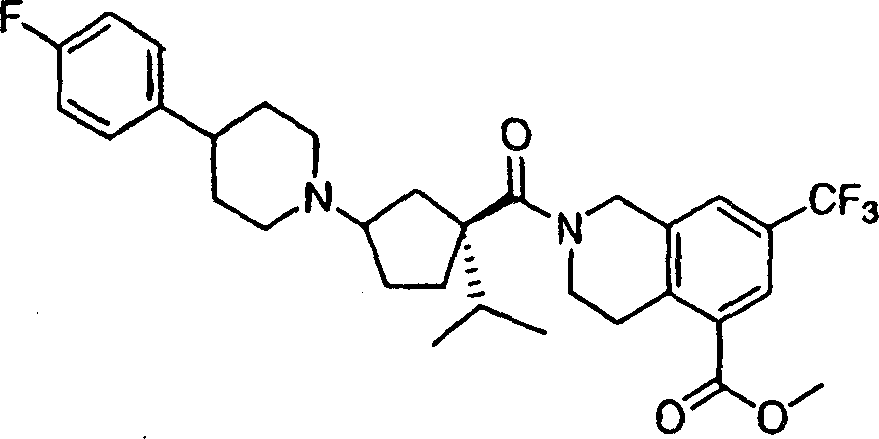
Steps A

(190mg, (dichloromethane solution 1.4mmol) adds constant DMF to intermediate 29 then for 2M, 0.70mL to add oxalyl chloride in 1mL methylene dichloride stirred solution 1.1mmol).At stirring at room mixture 30min.Vacuum-evaporation removes and desolvates and excess reagent then.Resistates is dissolved in the 1mL methylene dichloride, add intermediate 2 (295mg, 1.00mmol) and DIEA (260mg is in methylene dichloride 2.0mmol) (2mL) solution.Reaction stirred 2h..Whole mixtures are loaded into preparation type TLC plate (1000 microns), launch with 10%MeOH/DCM.Obtain yellow solid title compound (300mg).LC-MSC21H24F3NO4 calculated value 411, measured value [M+H]
+412.
Step B
Ketone (300mg with above steps A, 0.73mmol), 4-fluorophenyl piperidine hydrochlorate (214mg, 1.00mmol), DIEA (129mg, 1.00mmol), molecular sieve (4 , 500mg), sodium triacetoxy borohydride (212mg, 1.00mmol) and the mixture of 10mL methylene dichloride spend weekend in stirring at room, with the quencher of yellow soda ash saturated aqueous solution, use dichloromethane extraction, purify with preparation type TLC (1000 microns are used the 10%MeOH/DCM wash-out).Obtain the cis and the trans-isomer(ide) mixture (270mg) of title compound.LC-MSC32H38F4N2O3 calculated value 574, measured value [M+H]
+575.
Embodiment 143

With embodiment 142 (22mg, 0.04mmol), lithium hydroxide monohydrate (30mg) and MeOH/H
2(9/1, mixture 0.5mL) stirs 2h at 60 ℃ to O, and whole mixtures are loaded into preparation type TLC plate, launches with methyl alcohol.Obtain white solid title compound (12mg).LC-MS C31H34F4N2O3 calculated value 560, measured value [M+H]
+561.
Embodiment 144

Steps A:
With intermediate 29 (100mg 0.588mmol) is dissolved in DCM (20mL), use successively oxalyl chloride (153 μ L, 1.76mmol) and DMF (1) handle.At stirring at room gained solution 2h, concentrate then as for, high vacuum dry 30min.The gained resistates is dissolved in DCM (5mL), and (177mg is in DCM 0.882mmol) (5mL) and triethylamine (5mL) stirred solution to be added drop-wise to intermediate 1.Spend the night at stirring at room gained reaction mixture, then with the DCM dilution, with sodium bicarbonate, the 1N HCl aqueous solution and salt water washing.Use the dried over mgso organic layer, filter, concentrating under reduced pressure obtains the required crude product of 230mg then, is directly used in next step and need not to purify again.
Step B:
Product (115mg with previous step, 0.326mmol), 3-piperidin-4-yl ethyl benzoate hydrochloride (131mg, 0.489mmol), DIEA (83 μ L, 0.49mmol), molecular sieve (4 , 100mg), sodium triacetoxy borohydride (346mg, 1.63mmol) and the mixture of 10mL methylene dichloride spend weekend in stirring at room, use the saturated aqueous solution of sodium bicarbonate quencher, use dichloromethane extraction, with preparation type TLC purification (silica gel is with 40%THF/ hexane wash-out).Obtain the cis and the trans-isomer(ide) mixture (200mg) of title compound.The calculated value of LC-MSC33H41F3N2O3: 570.3; Measured value [M+H]
+571.6.Split each steric isomer with ChiralCelOD post (with 10% ethanol/hexane wash-out):
The calculated value of peak 1:LC-MS C33H41F3N2O3: 570.3; Measured value [M+H]
+571.6.
The calculated value of peak 2:LC-MS C33H41F3N2O3: 570.3; Measured value [M+H]
+571.6.
Embodiment 145

With embodiment 144 peaks 2 (80mg), lithium hydroxide monohydrate (30mg) and EtOH/H
2(4/1, mixture 4mL) is in stirred overnight at room temperature for O.Product is purified with reversed-phase HPLC, is converted into hydrochloride with common methods.Obtain white solid title compound (45mg).LC-MS C31H37F3N2O3 calculated value: 542.28; Measured value [M+H]
+543.
Embodiment 146

With embodiment 144 peaks 1 (78mg), lithium hydroxide monohydrate (30mg) and EtOH/H
2(4/1, mixture 4mL) is in stirred overnight at room temperature for O.Product is purified with reversed-phase HPLC, is converted into hydrochloride with common methods.Obtain white solid title compound (49mg).LC-MS C31H37F3N2O3 calculated value: 542.28; Measured value [M+H]
+543.
Embodiment 147
Embodiment 147 still substitutes 3-piperidin-4-yl ethyl benzoate hydrochlorides with intermediate 30 according to the method preparation of embodiment 144.4 kinds of stereoisomerism body and functions of gained chiral chromatography separates.
Embodiment 148

Embodiment 148 still uses a kind of product of a kind of steric isomer alternate embodiment 144 of embodiment 147 according to the method preparation of embodiment 145.
Embodiment 149

Embodiment 149 still uses a kind of product of a kind of steric isomer alternate embodiment 144 of embodiment 147 according to the method preparation of embodiment 145.
Embodiment 150

Embodiment 150 still uses a kind of product of a kind of steric isomer alternate embodiment 144 of embodiment 147 according to the method preparation of embodiment 145.
Embodiment 151
Embodiment 151 still uses a kind of product of a kind of steric isomer alternate embodiment 144 of embodiment 147 according to the method preparation of embodiment 145.
Claims (20)
1. the compound of a following formula I or its pharmacy acceptable salt or independent diastereomer:

Formula I
Wherein:
X is selected from C, N, O, S and SO
2
Y is N or C;
R
1Be selected from following group:
Hydrogen ,-C
1-6Alkyl ,-C
0-6Alkyl-O-C
1-6Alkyl ,-C
0-6Alkyl-S-C
1-6Alkyl ,-(C
0-6Alkyl)-(C
3-7Cycloalkyl)-(C
0-6Alkyl), hydroxyl, heterocycle ,-CN ,-NR
12R
12,-NR
12COR
13,-NR
12SO
2R
14,-COR
11,-CONR
12R
12And phenyl,
R wherein
11Independently be selected from following group: hydroxyl, hydrogen, C
1-6Alkyl ,-O-C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
R wherein
12Be selected from following group: hydrogen, C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
R wherein
13Be selected from following group: hydrogen, C
1-6Alkyl ,-O-C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
R wherein
14Be selected from following group: hydroxyl, C
1-6Alkyl ,-O-C
1-6Alkyl, benzyl, phenyl and C
3-6Cycloalkyl, described alkyl, phenyl, benzyl and cycloalkyl can be unsubstituted or independently be selected from following substituting group by 1-3 and replace: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group ,-CO
2H ,-CO
2-C
1-6Alkyl and trifluoromethyl,
Wherein said alkyl and cycloalkyl are not substituted or independently are selected from following substituting group by 1-7 and replace:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(f) C
1-3Alkyl,
(g)-O-C
1-3Alkyl,
(h)-COR
11,
(i)-SO
2R
14,
(j)-NHCOCH
3,
(k)-NHSO
2CH
3,
(l)-heterocycle,
(m)=O,
(n)-CN,
Wherein said phenyl and heterocycle are not substituted or independently are selected from following replacement by 1-3
Base replaces: halogen, hydroxyl, C
1-3Alkyl, C
1-3Alkoxyl group and trifluoromethyl; R
2Be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c) halogen,
(d) C
1-3Alkyl, wherein said alkyl are not substituted or are replaced by 1-6 substituting group that independently is selected from fluorine and hydroxyl,
(e)-NR
12R
12
(f)-COR
11,
(g)-CONR
12R
12,
(h)-NR
12COR
13,
(i)-OCONR
12R
12,
(j)-NR
12CONR
12R
12,
(k)-heterocycle,
(l)-CN,
(m)-NR
12-SO
2-NR
12R
12,
(n)-NR
12-SO
2-R
14,
(o)-SO
2-NR
12R
12,
(p)=O, wherein R
2Be connected with ring by two keys;
When Y is N, R
3For oxygen or do not exist;
When Y is C, R
3Be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c) halogen,
(d) C
1-3Alkyl, wherein said alkyl are not substituted or independently are selected from following substituting group by 1-6 and replace: fluorine, hydroxyl and-COR
11,
(e)-NR
12R
12,
(f)-COR
11,
(g)-CONR
12R
12,
(h)-NR
12COR
13,
(i)-OCONR
12R
12,
(j)-NR
12CONR
12R
12,
(k)-heterocycle,
(l)-CN,
(m)-NR
12-SO
2-NR
12R
12,
(n)-NR
12-SO
2-R
14,
(o)-SO
2-NR
12R
12,
(p) nitro;
R
4Be selected from following group:
(a) hydrogen,
(b) C
1-6Alkyl,
(c) trifluoromethyl,
(d) trifluoromethoxy,
(e) chlorine,
(f) fluorine,
(g) bromine,
(h) phenyl;
R
5Be selected from following group:
(a) C
1-6Alkyl, wherein alkyl can be unsubstituted, perhaps replaced, and optionally replaced by hydroxyl by 1-6 fluorine,
(b)-O-C
1-6Alkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(c)-CO-C
1-6Alkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(d)-S-C
1-6Alkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(e)-pyridyl, it can be unsubstituted or by one or more be selected from substituting group replace: halogen, trifluoromethyl, C
1-4Alkyl and COR
11,
(f) fluorine,
(g) chlorine,
(h) bromine,
(i)-C
4-6Cycloalkyl,
(j)-O-C
4-6Cycloalkyl,
(k) phenyl, it can be unsubstituted or is selected from following substituting group and replace by one or more: halogen, trifluoromethyl, C
1-4Alkyl and COR
11,
(l)-and the O-phenyl, it can be unsubstituted or is selected from following substituting group and replace by one or more: halogen, trifluoromethyl, C
1-4Alkyl and COR
11,
(m)-C
3-6Cycloalkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(n)-O-C
3-6Cycloalkyl, wherein alkyl can be unsubstituted or be replaced by 1-6 fluorine,
(o)-heterocycle,
(p)-CN,
(q)-COR
11;
R
6Be selected from following group:
(a) hydrogen,
(b) C
1-6Alkyl,
(c) trifluoromethyl,
(d) fluorine,
(e) chlorine,
(f) bromine;
R
7Be selected from following group:
Hydrogen, (C
0-6Alkyl)-phenyl, (C
0-6Alkyl)-heterocycle, (C
0-6Alkyl)-C
3-7Cycloalkyl, (C
0-6Alkyl)-COR
11, (C
0-6Alkyl)-(thiazolinyl)-COR
11, (C
0-6Alkyl)-SO
3H, (C
0-6Alkyl)-W-C
0-4Alkyl, (C
0-6Alkyl)-CONR
12-phenyl and (C
0-6Alkyl)-CONR
15-V-COR
11, be O, S or SO perhaps at X
2The time, R
7Do not exist,
Wherein V is C
1-6Alkyl or phenyl,
Wherein W be selected from singly-bound ,-O-,-S-,-SO-,-SO
2-,-CO-,-CO
2-,-CONR
12-and-NR
12-,
R wherein
15Can be hydrogen, C
1-4Alkyl, perhaps R
15The carbon that joint by 1-5 carbon connects V constitutes ring,
Wherein said C
0-6Alkyl is not substituted or independently is selected from following substituting group by 1-5 and replaces:
(a) halogen,
(b) hydroxyl,
(c)-C
0-6Alkyl,
(d)-O-C
1-3Alkyl,
(e) trifluoromethyl,
(f)-C
0-2Alkyl-phenyl,
Wherein said phenyl, heterocycle, cycloalkyl and C
0-4Alkyl is not substituted or independently is selected from following substituting group by 1-5 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c) hydroxyl,
(d) C
1-3Alkyl,
(e)-O-C
1-3Alkyl,
(f)-C
0-3-COR
11,
(g)-CN,
(h)-NR
12R
12,
(i)-CONR
12R
12,
(j)-C
0-3-heterocycle,
Perhaps, described phenyl and heterocycle can be heterocyclic fused with another, and another heterocycle itself can be unsubstituted or independently is selected from following substituting group by 1-2 and replace: hydroxyl, halogen ,-COR
11With-C
1-3Alkyl,
Wherein thiazolinyl is not substituted or independently is selected from following substituting group by 1-3 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c) C
1-3Alkyl,
(d) phenyl,
(e) heterocycle;
R
8Be selected from following group:
(a) hydrogen,
(b) be O, S, SO at X
2Or during N or connect R respectively
7And R
10Carbon between when being two key, R
8Do not exist,
(c) hydroxyl,
(d) C
1-6Alkyl,
(e) C
1-6Alkyl-hydroxyl,
(f)-O-C
1-3Alkyl,
(g)-COR
11,
(h)-CONR
12R
12,
(i)-CN;
Perhaps R
7And R
8Be bonded together to constitute and be selected from following ring:
(a) 1H-indenes,
(b) 2,3-dihydro-1H-indenes,
(c) 2,3-dihydro-cumarone,
(d) 1,3-dihydro-isobenzofuran,
(e) 2,3-dihydro-thionaphthene,
(f) 1,3-dihydro-different thionaphthene,
(g) the 6H-cyclopenta [d] isoxazol-3-ol,
(h) pentamethylene,
(i) hexanaphthene,
Wherein the ring that is constituted can be unsubstituted or independently is selected from following substituting group by 1-5 and replace:
(a) halogen,
(b) trifluoromethyl,
(c) hydroxyl,
(d) C
1-3Alkyl,
(e)-O-C
1-3Alkyl,
(f)-C
0-3-COR
11,
(g)-CN,
(h)-NR
12R
12,
(i)-CONR
12R
12,
(j)-C
0-3-heterocycle,
Perhaps, R
7With R
9Or R
8With R
10Can combine constitutes phenyl ring or heterocycle, and wherein said ring is not substituted or independently is selected from following substituting group by 1-7 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c) hydroxyl,
(d) C
1-3Alkyl,
(e)-O-C
1-3Alkyl,
(f)-COR
11,
(g)-CN,
(h)-NR
12R
12,
(i)-CONR
12R
12;
R
9And R
10Independently be selected from following group:
(a) hydrogen,
(b) hydroxyl,
(c) C
1-6Alkyl,
(d) C
1-6Alkyl-COR
11,
(e) C
1-6Alkyl-hydroxyl,
(f)-O-C
1-3Alkyl,
(g)=O, at this moment R
9Or R
10Be connected with ring by two keys,
(h) halogen;
N is selected from 0,1 and 2;
Dotted line is singly-bound or two key.
2. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ia structure:

R wherein
1, R
2, R
3, R
5, R
9, Y and n be the definition in the claim 1, R
16And R
17Independently be selected from following group:
(a) hydrogen,
(b) halogen,
(c) trifluoromethyl,
(d) hydroxyl,
(e) C
1-3Alkyl,
(f)-O-C
1-3Alkyl,
(g)-C
0-3-CO
2H,
(h)-C
0-3-CO
2C
1-3Alkyl,
(i)-CN,
(j)-C
0-3-heterocycle,
Perhaps, R
16And R
17Being bonded together constitutes and the phenyl ring condensed heterocycle, and heterocycle itself can be unsubstituted or independently is selected from following substituting group replacement by 1-2: hydroxyl, halogen ,-COR
11With-C
1-3Alkyl.
3. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ib structure::

Wherein dotted line is singly-bound or two key, R
1, R
2, R
3, R
5, R
9, R
16, R
17, Y and n be the definition in the claim 1.
4. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ic structure::
R wherein
1, R
2, R
3, R
5, R
9, R
16, R
17, Y and n be the definition in the claim 1, H is a heterocycle.
5. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Id structure:

R wherein
1, R
2, R
3, R
5, R
9, R
11, Y, W and n be the definition in the claim 1,
Wherein said C
1-4Carbochain can be unsubstituted or independently is selected from following substituting group by 1-4 and replace:
(a) halogen,
(b) hydroxyl,
(c)-C
0-6Alkyl,
(d)-O-C
1-3Alkyl,
(e) trifluoromethyl,
(f)-C
0-2Alkyl-phenyl
Perhaps described C
1-4Carbochain can be included in C
3-7In the cycloalkyl ring.
6. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ie structure:

R wherein
1, R
2, R
3, R
5, R
9, R
16, R
17, X, Y and n be the definition in the claim 1; Dotted line is singly-bound or two key; O is 1 or 2; A, B and D can independently be selected from C, N, O or S, and when X, A, B, D were C and o=2, they constituted phenyl ring, and perhaps when one of X, A, B and D at least were not C for N, O or S, they constituted heterocycle.
7. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula If structure:

R wherein
1, R
2, R
3, R
5, R
7, R
9, R
10, Y and n be the definition in the claim 1; X is N or O, when X is O, and R
7Do not exist.
8. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ig structure:

R wherein
1, R
5, R
9, R
16, R
17With Y be definition in the claim 1,
Perhaps R
16And R
17Being bonded together constitutes and the phenyl ring condensed heterocycle, and heterocycle itself can be unsubstituted or independently is selected from following substituting group replacement by 1-2: hydroxyl, halogen ,-COR
11With-C
1-3Alkyl.
9. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ih structure:

Wherein dotted line is singly-bound or two key, R
1, R
5, R
9, R
16, R
17With Y be above definition.
10. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ii structure:
R wherein
1, R
5, R
9, R
16, R
17With Y be above definition, H is a heterocycle.
10. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ij structure:
R wherein
1, R
5, R
9, R
11, Y and W be definition above, wherein said C
1-4Carbochain can be unsubstituted or independently is selected from following substituting group by 1-4 and replace:
(a) halogen,
(b) hydroxyl,
(c)-C
0-6Alkyl,
(d)-O-C
1-3Alkyl,
(e) trifluoromethyl,
(f)-C
0-2Alkyl-phenyl.
11. the compound of claim 1, it is compound or its pharmacy acceptable salt or the independent diastereomer with formula Ik structure:

Formula Ik
R wherein
1, R
5, R
9, R
10With Y be above definition.
12. the compound of claim 1, wherein R
1Be selected from following group :-C
1-6Alkyl ,-C
0-6Alkyl-O-C
1-6Alkyl and-(C
0-6Alkyl)-(C
3-7Cycloalkyl)-(C
0-6Alkyl), described alkyl and cycloalkyl are not substituted or independently are selected from following substituting group by 1-7 and replace:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(f) C
1-3Alkyl,
(g)-O-C
1-3Alkyl,
(h)-COR
11,
(i)-CN,
(j)-NR
12R
12,
(k)-CONR
12R
12。
13. the compound of claim 1, wherein R
1Be selected from following group:
(l)-C
1-6Alkyl, it is not substituted or independently is selected from following substituting group by 1-6 and replaces:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(e)-COR
11,
(2)-C
0-6Alkyl-O-C
1-6Alkyl-, it is not substituted or independently is selected from following substituting group by 1-6 and replaces:
(a) halogen,
(b) trifluoromethyl,
(c)-COR
11,
(3)-(C
3-5Cycloalkyl)-(C
0-6Alkyl), it is not substituted or independently is selected from following substituting group by 1-7 and replaces:
(a) halogen,
(b) hydroxyl,
(c)-O-C
1-3Alkyl,
(d) trifluoromethyl,
(e)-COR
11。
14. the compound of claim 13, wherein R
1Be selected from following group:
(a) C
1-6Alkyl,
(b) C of hydroxyl replacement
1-6Alkyl,
(c) C that is replaced by 1-6 fluorine
1-6Alkyl.
15. the compound of claim 14, wherein R
1Be selected from following group:
(a)-CH(CH
3)
2,
(b)-CH(OH)CH
3,
(c)-CH
2CF
3。
16. a medicinal compositions, said composition comprises the compound of inert support and claim 1.
17. a method of regulating the mammalian chemokines receptor active, this method comprises the compound of the claim 1 that gives significant quantity.
18. a method for the treatment of, improve, control or alleviate inflammatory immunomodulatory illness or disease, this method comprises the compound of the claim 1 that gives patient's significant quantity.
19. a method for the treatment of, improve, control or alleviate rheumatoid arthritis, this method comprises the compound of the claim 1 that gives patient's significant quantity.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US46367303P | 2003-04-17 | 2003-04-17 | |
| US60/463,673 | 2003-04-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1805965A true CN1805965A (en) | 2006-07-19 |
| CN100384856C CN100384856C (en) | 2008-04-30 |
Family
ID=33310806
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2004800165371A Expired - Fee Related CN100384856C (en) | 2003-04-17 | 2004-04-14 | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1622916A4 (en) |
| JP (1) | JP2006523704A (en) |
| CN (1) | CN100384856C (en) |
| AU (1) | AU2004232939A1 (en) |
| CA (1) | CA2521625A1 (en) |
| WO (1) | WO2004094371A2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103649056A (en) * | 2011-05-10 | 2014-03-19 | 吉利德科学公司 | Fused heterocyclic compounds as sodium channel modulators |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2004232939A1 (en) * | 2003-04-17 | 2004-11-04 | Merck Sharp & Dohme Corp. | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity |
| US7598243B2 (en) | 2003-04-17 | 2009-10-06 | Merck & Co., Inc. | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity |
| TWI350168B (en) | 2004-05-07 | 2011-10-11 | Incyte Corp | Amido compounds and their use as pharmaceuticals |
| US8067415B2 (en) | 2005-11-01 | 2011-11-29 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of CCR2 |
| US8067457B2 (en) | 2005-11-01 | 2011-11-29 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of CCR2 |
| WO2007122103A1 (en) * | 2006-04-20 | 2007-11-01 | F. Hoffmann-La Roche Ag | Diazepan derivatives modulators of chemokine receptors |
| JP5497429B2 (en) | 2007-03-07 | 2014-05-21 | 武田薬品工業株式会社 | Benzoxazepine derivatives and uses thereof |
| AU2010276537B2 (en) | 2009-07-27 | 2015-04-16 | Gilead Sciences, Inc. | Fused heterocyclic compounds as ion channel modulators |
| AR080374A1 (en) | 2010-03-05 | 2012-04-04 | Sanofi Aventis | PROCEDURE FOR THE PREPARATION OF 2- (CYCLOHEXILMETIL) -N- (2 - ((2S) -1-METHYLPIRROLIDIN-2-IL) ETIL) - 1,2,3,4-TETRAHYDROISOQUINOLIN-7- SULFONAMIDE |
| EP2588197B1 (en) | 2010-07-02 | 2014-11-05 | Gilead Sciences, Inc. | Fused heterocyclic compounds as ion channel modulators |
| NO3175985T3 (en) | 2011-07-01 | 2018-04-28 | ||
| TW201837023A (en) | 2011-07-01 | 2018-10-16 | 美商基利科學股份有限公司 | Fused heterocyclic compounds as ion channel modulators |
| AU2016263579B2 (en) | 2015-05-21 | 2020-12-03 | Chemocentryx, Inc. | CCR2 modulators |
| WO2018026763A1 (en) | 2016-08-01 | 2018-02-08 | Aptinyx Inc. | Spiro-lactam nmda receptor modulators and uses thereof |
| EP3490990B1 (en) | 2016-08-01 | 2023-12-06 | Tenacia Biotechnology (Hong Kong) Co., Limited | Spiro-lactam nmda modulators and methods of using same |
| PE20190503A1 (en) | 2016-08-01 | 2019-04-10 | Aptinyx Inc | NMDA ESPIRO-LACTAM RECEIVER MODULATORS AND USE OF THE SAME |
| CN111712242B (en) | 2017-09-25 | 2023-11-24 | 凯莫森特里克斯股份有限公司 | Combination therapy using chemokine receptor 2 (CCR 2) antagonists and PD-1/PD-L1 inhibitors |
| WO2019136368A2 (en) | 2018-01-08 | 2019-07-11 | Chemocentryx, Inc. | Methods of treating solid tumors with ccr2 antagonists |
| PE20210455A1 (en) | 2018-01-31 | 2021-03-08 | Aptinyx Inc | SPIRO-LACTAMA NMDA RECEIVER MODULATORS AND USES OF THEM |
| CN111056955B (en) * | 2019-12-16 | 2021-05-25 | 中国科学院大连化学物理研究所 | Method for preparing hexamethylene diamine from cyclohexene |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU8334501A (en) * | 2000-08-17 | 2002-02-25 | Merck & Co Inc | Cyclopentyl modulators of chemokine receptor activity |
| NZ536477A (en) * | 2002-04-29 | 2005-05-27 | Merck & Co Inc | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity |
| AU2003223755B2 (en) * | 2002-04-29 | 2008-08-21 | Merck Sharp & Dohme Corp. | Tetrahydropyranyl cyclopentyl tetrahydroisoquinoline modulators of chemokine receptor activity |
| AU2004232939A1 (en) * | 2003-04-17 | 2004-11-04 | Merck Sharp & Dohme Corp. | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity |
-
2004
- 2004-04-14 AU AU2004232939A patent/AU2004232939A1/en not_active Abandoned
- 2004-04-14 WO PCT/US2004/011463 patent/WO2004094371A2/en active Application Filing
- 2004-04-14 JP JP2006510018A patent/JP2006523704A/en active Pending
- 2004-04-14 EP EP04750112A patent/EP1622916A4/en not_active Withdrawn
- 2004-04-14 CA CA002521625A patent/CA2521625A1/en not_active Abandoned
- 2004-04-14 CN CNB2004800165371A patent/CN100384856C/en not_active Expired - Fee Related
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103649056A (en) * | 2011-05-10 | 2014-03-19 | 吉利德科学公司 | Fused heterocyclic compounds as sodium channel modulators |
| CN103649056B (en) * | 2011-05-10 | 2016-04-27 | 吉利德科学公司 | As the condensed heterocyclic compouds of ion channel modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2004232939A1 (en) | 2004-11-04 |
| WO2004094371A2 (en) | 2004-11-04 |
| EP1622916A4 (en) | 2008-11-05 |
| CA2521625A1 (en) | 2004-11-04 |
| CN100384856C (en) | 2008-04-30 |
| WO2004094371A3 (en) | 2005-03-24 |
| JP2006523704A (en) | 2006-10-19 |
| EP1622916A2 (en) | 2006-02-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1805965A (en) | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity | |
| CN1956975A (en) | Amino cyclopentyl heterocyclic and carbocyclic modulators of chemokine receptor activity | |
| CN1913778A (en) | Aminocyclopentyl pyridopyrazinone modulators of chemokine receptor activity | |
| CN1802187A (en) | Benzoxazinyl-amidocyclopentyl-heterocyclic modulators of chemokine receptors | |
| US6248755B1 (en) | Pyrrolidine modulators of chemokine receptor activity | |
| CN1254474C (en) | Tetrahydroazepine derivatives useful as dopamine d3 receptor modulators (antipsychotic agents) | |
| CN1189467C (en) | Pharmaceutically active sulfonamide derivatives | |
| CN1909906A (en) | 2,6-disubstituted piperidines as modulators of chemokine receptor activity | |
| US7491737B2 (en) | Heterarylpiperidine modulators of chemokine receptor activity | |
| CN1918145A (en) | Amino heterocyclic modulators of chemokine receptor activity | |
| CN1902171A (en) | Benzylether amine compounds useful as CCR-5 antagonists | |
| CN1826334A (en) | Tetrahydropyranyl cyclopentyl heterocyclic amide modulators of chemokine receptor activity | |
| CN1787818A (en) | Amino cyclobutylamide modulators of chemokine receptor activity | |
| CN1897941A (en) | Alkylamino, arylamino, and sulfonamido cyclopentyl amide modulators of chemokine receptor activity | |
| US20070238723A1 (en) | Benzoxazinyl-amidocyclopentyl-heterocyclic modulators of chemokine receptors | |
| CN1946696A (en) | Tetrahydropyranyl cyclopentyl 1-substituted and 1,1-disubstituted tetrahydroisoquinoline modulators of chemokine receptor activity | |
| US7598243B2 (en) | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity | |
| US7091211B2 (en) | Cyclopentyl modulators of chemokine receptor activity | |
| US20060183731A1 (en) | 7 and 8 membered heterocyclic cyclopentyl benzylamide modulators of chemokine receptor activity | |
| US7230008B2 (en) | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity | |
| US7589085B2 (en) | Tetrahydropyran heterocyclic cyclopentyl heteroaryl modulators of chemokine receptor activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20080430 Termination date: 20100414 |