CN115397428A - Sequential cancer treatment using 6-thio-dG, checkpoint inhibitors and radiation therapy - Google Patents
Sequential cancer treatment using 6-thio-dG, checkpoint inhibitors and radiation therapy Download PDFInfo
- Publication number
- CN115397428A CN115397428A CN202180028952.2A CN202180028952A CN115397428A CN 115397428 A CN115397428 A CN 115397428A CN 202180028952 A CN202180028952 A CN 202180028952A CN 115397428 A CN115397428 A CN 115397428A
- Authority
- CN
- China
- Prior art keywords
- cancer
- thio
- tumor
- cells
- treatment
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images








































































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K41/00—Medicinal preparations obtained by treating materials with wave energy or particle radiation ; Therapies using these preparations
- A61K41/0038—Radiosensitizing, i.e. administration of pharmaceutical agents that enhance the effect of radiotherapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2827—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against B7 molecules, e.g. CD80, CD86
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61N—ELECTROTHERAPY; MAGNETOTHERAPY; RADIATION THERAPY; ULTRASOUND THERAPY
- A61N5/00—Radiation therapy
- A61N5/10—X-ray therapy; Gamma-ray therapy; Particle-irradiation therapy
- A61N2005/1092—Details
- A61N2005/1098—Enhancing the effect of the particle by an injected agent or implanted device
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61N—ELECTROTHERAPY; MAGNETOTHERAPY; RADIATION THERAPY; ULTRASOUND THERAPY
- A61N5/00—Radiation therapy
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
Abstract
Disclosed herein are therapeutic methods for treating cancer using the telomerase-mediated telomere-targeting drug 6-thio-2' -deoxyguanosine (6-thio-dG), checkpoint inhibitors, and/or radiation therapy that result in tumor regression in an innate and adaptive immune-dependent manner in syngeneic and humanized mouse cancer models.
Description
Priority requirement
This application claims priority to U.S. provisional application serial No. 62/989,041, filed 3/13/2020, which is incorporated herein by reference in its entirety.
Statement of federally sponsored support
The invention was made with government support under fund number 2P50CA070907-21A1 awarded by the National Cancer Institute. The government has certain rights in this invention.
Technical Field
The present disclosure relates to the fields of medicine, pharmacology, molecular biology, and oncology. More specifically, the present disclosure relates to methods and compositions for treating cancer using sequential treatments of 6-thio-dG, checkpoint inhibitors, and/or radiation therapy.
Background
Immunotherapy radically alters the treatment of many cancers in the field of immunooncology (Brahmer et al, 2012, hodi et al, 2010, ribas and wolchok, 2018. The most commonly used immunotherapy is PD-L1/PD-1 checkpoint blockade, which has been approved by the FDA for use in advanced cancers such as melanoma, non-small cell lung Cancer, breast Cancer, cervical Cancer, colon Cancer, head and neck Cancer, hodgkin lymphoma (Hodgkin lymphoma), liver Cancer, lung Cancer, renal cell carcinoma, gastric Cancer, rectal Cancer, and any solid tumor that is unable to repair errors in its DNA that occur during replication (Garon et al, 2015 ribas et al, 2016, rizvi et al, 2015b. Despite the success of immunotherapy, many patients respond poorly to these treatments due to the emergence of immunosuppressive tumor microenvironment, tumor immunogenicity, and primary and adaptive resistance (Chen and Han,2015 gide et al, 2018. Although recent studies have shown that a large number of tumor mutations and neoantigens partially determine the response of cancer patients to checkpoint blockade, there are still a considerable number of patients with high mutations and neoantigens that respond poorly (Le et al, 2017 mandal et al, 2019 rizvi et al, 2015 a), indicating that neoantigens are insufficient to elicit an anti-tumor immune response. Therefore, there is an urgent need to identify other factors for better immune response and develop new methods to improve overall survival of patients.
The generation of an effective anti-tumor adaptive immune response requires the presentation of tumor antigens by antigen presenting cells, the activation of which depends largely on sufficient intrinsic perception. Intrinsic perception is usually provided by danger signals such as high mobility group box 1 proteins, extracellular ATP and tumor DNA released from stressed tumor cells (Kroemer et al, 2013 pitt et al, 2017). Recent studies have emphasized the importance of cytoplasmic DNA perception in radiation and DNA damage treatment (Deng et al, 2014 sen et al, 2019). The presence of DNA in the cytoplasm, for example, in the form of micronuclei (small organelles containing DNA) with missing nuclear envelopes, can trigger an immune response. Micronuclei are the products of chromosomal damage during cell division due to genotoxic stress and chromosomal mis-segregation (fen ech et al, 2011). The cytosolic DNA sensor cGAS recognizes micronuclei and converts GTP (guanosine triphosphate) and ATP (adenosine triphosphate) to the second messenger cGAMP (cyclic GMP-AMP) (Wu et al, 2013). Then, a linker protein, IFN Gene stimulating factor (Stimulator of IFN Gene, STING), binds cGAMP (ablaser et al, 2013 diner et al, 2013 gao et al, 2013 zhang et al, 2013. This complex process activates TANK binding kinase 1 (TANK-binding kinase 1, tbk 1) and IFN regulatory factor 3 (IFN regulation factor 3, irf 3) (Liu et al, 2015 tanaka and Chen, 2012) and further activates downstream transcription of type I IFNs and other cytokines (reviewed in (Li and Chen, 2018)), which ultimately increases intrinsic perception.
Eukaryotic linear chromosomes are capped by a special structure called telomeres (TTAGGG), which is critical for maintaining chromosome stability (reviewed in Blackburn, 1991). Telomeres constitute the last approximately 10kb of all human chromosomes and the last 12 to 80kb of all mouse chromosomes (Lansdorp et al, 1996. In all human cells, telomeres shorten with each cell division due to end replication problems and lack of telomere maintenance mechanisms (reviewed in (Greider, 1996)). However, unicellular eukaryotes, germ cells, and immortal cancer cells almost always maintain their telomeres at constant length by activating the enzyme telomerase (Greider and Blackburn,1985, mceacher and Blackburn,1996, morin,1989, nakamura et al, 1997. Telomerase is a reverse transcriptase that extends telomeres by adding TTAGGG repeats to the ends of chromosomes and is expressed in about 90% of human tumors but not in most normal cells (Shay and Bacchetti, 1997). Therefore, telomerase is an attractive target for the development of anti-cancer therapies.
The nucleoside analog 6-thio-2' -deoxyguanosine (6-thio-dG) is a new and effective therapeutic approach in the field of cancer. Incorporation into de novo synthesized telomeres by telomerase is known to induce damage to telomeric DNA (Mender et al, 2015 a). This leads to rapid tumor shrinkage or growth arrest in xenograft models of many tumor origin with minimal side effects (Mender et al, 2018, sengutta et al, 2018, zhang et al, 2018. The most important advantage of this telomere targeted therapy over direct telomerase inhibitors is that 6-thio-dG has no long lag phase on the tumor killing effect. In addition, it does not inhibit telomerase directly, but rather is preferentially recognized by telomerase relative to other polymerases and incorporated into telomeres, resulting in immediate termination of the DNA strand. Importantly, its action produces unstable telomeres by manipulating tumor telomerase independent of initial telomere length (Mender et al, 2015 b).
Disclosure of Invention
Accordingly, in one aspect of the disclosure, a method of treating cancer in a subject is provided that involves administering to the subject an effective amount of 6-thio-2' -deoxyguanosine (6-thio-dG) per treatment cycle followed by treatment with an immune checkpoint inhibitor. In some embodiments, the cancer is selected from one or more of the following: pancreatic cancer, lung cancer, mesothelioma, stomach cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell carcinoma (Merkel cell carcinoma), myelodysplasia syndrome, myelofibrosis, and multiple myeloma.
In some embodiments, the immune checkpoint inhibitor is a PD-1 inhibitor, a PD-L1 inhibitor, or a CTLA-4 inhibitor. In one embodiment, the immune checkpoint inhibitor is one or more CTLA-4 inhibitors, one or more PD-1 inhibitors, or a combination of one or more PD-L1 inhibitors.
In some embodiments, the PD-1 inhibitor is selected from one or more of pembrolizumab, nivolumab, cimetipril mab (cemiplimab), JTx-4014, sarajimab (sasanlimab), budilizumab (budigalimab), BI 754091, slablizumab (spartalizumab), charelizumab (camrelizumab), fiducimab (sintilimab), tirelizumab (tillizumab), separemab (zimberlimab), terlipril mab (torelizab), dolastalizab (dotrlimab), INCMGA00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-a317, BCD-100, JS001, and AMP-515.
In some embodiments, the PD-L1 inhibitor is selected from one or more of alemtuzumab, avizumab, chikulizumab (cosibelilimumab), bindraful alfa, dewalimumab (durvalumab), MGD013, KNO35, KN046, AUNP12, CA-170 and BMS-9986189.
In some embodiments, the CTLA-4 inhibitor is selected from one or more of ipilimumab (ipilimumab) and tremelimumab (tremelimumab).
In some embodiments of the methods disclosed herein, 6-thio-dG is administered for about 1 to about 5 days per treatment cycle. In some embodiments, the checkpoint inhibitor is administered for about 1 to about 3 days per treatment cycle.
The term treatment cycle as used herein means about 1 to about 12 weeks between administration of treatments.
In one embodiment of the methods disclosed herein, the 6-thio-dG and the checkpoint inhibitor are administered in combination with a chemotherapeutic agent, hormonal therapy, toxin therapy or surgery.
In another embodiment, disclosed herein is a method of treating cancer in a subject in need thereof comprising administering 6-thio-dG to said subject followed by cimetipril mab Performing a treatment, wherein the cancer is selected from one or more of: pancreatic cancer, lung cancer, mesothelioma, stomach cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell cancer, myelodysplastic syndrome, myelofibrosis, and multiple myeloma. In some embodiments of the methods, 6-thio-dG is administered for about 1 to about 5 days per treatment cycle. In some embodiments of the methods, the cimetiprizumab is administered for about 1 to about 3 days per treatment cycle. In one embodiment of the method, 6-thio-dG and cimetipril mab are administered in combination with a chemotherapeutic agent, hormonal therapy, toxin therapy, or surgery.
Performing a treatment, wherein the cancer is selected from one or more of: pancreatic cancer, lung cancer, mesothelioma, stomach cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell cancer, myelodysplastic syndrome, myelofibrosis, and multiple myeloma. In some embodiments of the methods, 6-thio-dG is administered for about 1 to about 5 days per treatment cycle. In some embodiments of the methods, the cimetiprizumab is administered for about 1 to about 3 days per treatment cycle. In one embodiment of the method, 6-thio-dG and cimetipril mab are administered in combination with a chemotherapeutic agent, hormonal therapy, toxin therapy, or surgery.
In one embodiment, disclosed herein is a method of treating cancer in a subject comprising administering 6-thio-dG to the subject and subsequent treatment with atelizumab, wherein the cancer is selected from one or more of the following: pancreatic cancer, lung cancer, mesothelioma, stomach cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell cancer, myelodysplastic syndrome, myelofibrosis, and multiple myeloma. In some embodiments of the methods, 6-thio-dG is administered for about 1 to about 5 days per treatment cycle. In some embodiments of the methods, the atuzumab is administered for about 1 to about 3 days per treatment cycle. In one embodiment of the method, 6-thio-dG and astuzumab are administered in combination with a chemotherapeutic agent, hormonal therapy, toxin therapy, or surgery.
In another aspect of the disclosure, disclosed herein is a method of treating cancer in a subject comprising administering 6-thio-dG to the subject and subsequently treating with an immune checkpoint inhibitor administered in combination with radiation therapy. In some embodiments, the checkpoint inhibitor is a PD-L1, PD-1, or CTAL-4 inhibitor. In some embodiments, the PD-L1 inhibitor is selected from one or more of alemtuzumab, avizumab, chikulizumab, bindrafusalfa, devaluzumab, MGD013, KNO35, KN046, AUNP12, CA-170, and BMS-9986189. In some embodiments, the PD-L1 inhibitor is atelizumab. In some embodiments, the PD-1 inhibitor is selected from one or more of pembrolizumab, nivolumab, cimiraprizumab, JTx-4014, saralalizumab, breglizumab, BI 754091, sibatuzumab, carpriclizumab, certralizumab, tiplizumab, tereprimab, dolaprimab, INCMGA00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-A317, BCD-100, JS001, and AMP-515. In some embodiments, the PD-1 inhibitor is cimiraprizumab. In some embodiments, the CTLA-4 inhibitor is ipilimumab or tremelimumab. In some embodiments, the cancer treated is selected from one or more of the following: pancreatic cancer, lung cancer, mesothelioma, stomach cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell cancer, myelodysplastic syndrome, myelofibrosis, and multiple myeloma. In some embodiments, the cancer treated is pancreatic cancer, lung cancer, gastric cancer, liver cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, brain cancer, colon cancer, prostate cancer, ovarian cancer, cervical cancer, testicular cancer, lymphoma, leukemia, skin cancer, or breast cancer. In some embodiments, the brain cancer is an adult brain cancer. In some embodiments, radiation therapy is administered first, followed by one or more checkpoint inhibitors. In some embodiments, the radiation therapy is administered after administration of the one or more checkpoint inhibitors.
In some embodiments of the disclosed methods, the cancer treated is lung cancer, colorectal cancer, liver cancer, melanoma, pancreatic cancer, ovarian cancer, or brain cancer (adult).
In some embodiments of the disclosed methods, the cancer treated is pancreatic cancer, lung cancer, gastric cancer, liver cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, brain cancer, colon cancer, prostate cancer, ovarian cancer, cervical cancer, testicular cancer, lymphoma, leukemia, skin cancer, or breast cancer.
In other embodiments of the disclosed methods, the total dose of 6-thio-dG administered during the treatment period of from about 1 to 5 days is from about 10 to 2000mg or from about 15 to 2000mg or from about 20 to 2000mg or from about 10 to 4800mg per treatment cycle.
In one embodiment of the disclosed method, the cancer treated is metastatic.
In some embodiments of the disclosed methods, the cancer treated is recurrent or recurrent.
In some embodiments of the disclosed methods, the cancer treated is treatment resistant. In one embodiment, the treatment-resistant cancer is treatment-resistant with a checkpoint inhibitor. In another embodiment, the treatment-resistant cancer is resistant to one or more of a PD-1 inhibitor, a PD-L1 inhibitor, and/or a CTLA-4 inhibitor. In some embodiments, the cancer is resistant to tyrosine kinase inhibitors, such as, but not limited to, erlotinib.
In some embodiments of the methods disclosed herein, the subject being treated was previously treated with checkpoint inhibitor treatment. In one embodiment, the subject was previously treated with one or more of PD-1, PD-L1, or CTLA-4. In another embodiment, the subject has been previously treated with a tyrosine kinase inhibitor treatment.
In some embodiments of the methods disclosed herein, the following is repeated at least once: 6-thio-dG is administered and subsequently treated with a checkpoint inhibitor.
In some embodiments of the methods disclosed herein, the 6-thio-dG and the checkpoint inhibitor are administered systemically. In other embodiments, the 6-thio-dG and the checkpoint inhibitor are administered locally or regionally to the tumor site. In one embodiment, 6-thio-dG is administered locally or regionally to the tumor site and the checkpoint inhibitor is administered systemically.
In some embodiments of the methods disclosed herein, administration of 6-thio-dG and the checkpoint inhibitor results in inhibition of tumor growth.
In some embodiments of the methods disclosed herein, administration of 6-thio-dG and a checkpoint inhibitor results in remission of the cancer treated.
In some embodiments of the methods disclosed herein, administering 6-thio-dG and one or more checkpoint inhibitors results in reducing tumor burden.
In some embodiments of the methods disclosed herein, administering 6-thio-dG and one or more checkpoint inhibitors results in inhibiting cancer cell metastasis.
In some embodiments of the methods disclosed herein, administration of 6-thio-dG and one or more checkpoint inhibitors results in tumor eradication.
In another aspect, disclosed herein is a method of treating cancer in a subject comprising administering to the subject a therapeutically effective dose of 6-thio-dG followed by treatment with radiation therapy. In some embodiments, the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, gastric cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, renal cancer, neuroblastoma, merkel cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma. In some embodiments, the cancer treated is pancreatic cancer, lung cancer, gastric cancer, liver cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, brain cancer, colon cancer, prostate cancer, ovarian cancer, cervical cancer, testicular cancer, lymphoma, leukemia, skin cancer, or breast cancer. In some embodiments, the brain cancer is an adult brain cancer.
In another aspect, disclosed herein is a method of treating cancer in a subject comprising administering to the subject a therapeutically effective dose of 6-thio-dG, prior to treatment with radiation therapy. In some embodiments, the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, gastric cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, brain cancer (adult), colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma. In some embodiments, the cancer treated is pancreatic cancer, lung cancer, gastric cancer, liver cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, brain cancer, colon cancer, prostate cancer, ovarian cancer, cervical cancer, testicular cancer, lymphoma, leukemia, skin cancer, or breast cancer. In some embodiments, the cancer is an adult brain cancer.
In one embodiment of the methods disclosed herein, the administration of 6-thio-dG and the radiation therapy is repeated at least once.
The cancer may exhibit telomerase activity. 6-thio-dG as well as PD-1 inhibitors, PD-L1 inhibitors and CTLA-4 inhibitors such as, for example, alemtuzumab, avilumab, chixilizumab, bintrafusi alfa, dewaruzumab, MGD013, KNO35, KN046, AUNP12, CA-170, BMS-9986189 pembrolizumab, nivolumab, cimiralizumab, JTx-4014, saratrilizumab, breglizumab, BI 754091, sibradizumab, carpizumab, riduzumab, temeprizumab, certralizumab ozolomide, dolaprimab, INCMAGMCA 00012, AMP-224, REGN2810, BMS-936558, SHR1210, I308, IBPDR 001, BGB-A317, BCD-100, JS-515, illizumab and AMP can be administered in combination with chemotherapeutic agents, radiation therapy, hormone therapy, surgical toxins, surgical therapies or surgical therapies. The daily dose of 6-thio-dG administered may be from about 0.15mg/kg to about 70mg/kg. The interval between administration of 6-thio-dG and administration of PD-L1 inhibitor, PD-1 inhibitor and/or CTLA-4 inhibitor may be from about 1 to 14 days, for example from about 1 to 4 days, or from about 2 to 5 days, or from about 2 to 6 days, or from about 2 to 7 days, or from about 2 to 8 days, or from about 2 to 9 days, or from about 2 to 10 days, or from about 2 to 11 days, or from about 2 to 12 days, or from about 2 to 13 days. The method may further comprise the step of assessing telomerase activity in adult brain cancer cells from said subject. Administration of 6-thio-dG in combination with a PD-1 inhibitor, a PD-L1 inhibitor and/or a CTLA-4 inhibitor can result in inhibition of tumor growth, remission of the cancer, reduction of tumor burden, inhibition of cancer cell metastasis or tumor eradication.
The cancer may be pancreatic cancer, lung cancer, gastric cancer, liver cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, brain cancer, colon cancer, prostate cancer, ovarian cancer, cervical cancer, testicular cancer, lymphoma, leukemia or skin cancer. The cancer may be metastatic and/or recurrent and/or resistant to treatment. The treatment-resistant cancer may be treatment-resistant with a checkpoint inhibitor, e.g., PD-L1, PD-1, and/or CTLA-4 resistance. The subject may have been previously treated with checkpoint inhibitor therapy, e.g., PD-L1, PD-1, and/or CTLA-4 therapy. Repeating at least once: administering 6-thio-dG and subsequent treatment with a PD-1 inhibitor, a PD-L1 inhibitor and/or a CTLA4 inhibitor. The 6-thio-dG and the PD-1 inhibitor, PD-L1 inhibitor and/or CTLA4 inhibitor may be administered systemically or locally or regionally to the tumor site. The 6-thio-dG may be administered by the same or different route as the PD-1 inhibitor, the PD-L1 inhibitor and/or the CTLA4 inhibitor.
Other objects, features and advantages of the present disclosure will become apparent from the following detailed description. It should be understood, however, that the detailed description and the specific examples, while indicating specific embodiments of the disclosure, are given by way of illustration only, since various changes and modifications within the spirit and scope of the disclosure will become apparent to those skilled in the art from this detailed description.
Drawings
The following drawings form part of the present specification and are included to further demonstrate certain aspects of the present disclosure. The disclosure may be better understood by reference to one or more of these drawings in combination with the detailed description of specific embodiments presented herein.
FIGS. 1A to 1G. The therapeutic effect of 6-thio-dG is dependent on CD8+ T cells. (FIG. 1A) cell viability of 6-thio-dG in MC38 cells (IC) 50 ). Cells were treated with 6-thio-dG for 5 days. (FIG. 1B and FIG. 1C) colony formation assay of 6-thio-dG in MC38 cells at the indicated dose for 13 days. Cells were treated every 3 days with 6-thio-dG, then fixed and stained with crystal violet. Representative images of three biological replicates are shown in fig. 1B and quantitative data is shown in fig. 1C. (FIGS. 1D and 1E) WT (FIG. 1D) or Rag1-/- (FIG. 1E) C57BL/6 mice (n = 5) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). (FIGS. 1F and 1G) C57BL/6 mice (n = 5) were treated with 5X 10 mice 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). 200 μ G of anti-CD 4 (FIG. 1F) or anti-CD 8 (FIG. 1G) was administered one day before treatment began and then twice weekly for 3 weeks. Tumor growth was measured every 3 days. Data are shown as mean ± SEM from two to three independent experiments. P values were determined by two-tailed unpaired t-test (fig. 1C) or two-way ANOVA (fig. 1D to 1G). See also fig. 9A to 9D.
Fig. 2A to 2F. Treatment with 6-thio-dG improved the tumor specific T cell response. (fig. 2A and 2B) C57BL/6 mice (n =4 to 5) were treated with 5 × 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Six days after the last treatment, the frequency of total T cells (fig. 2A) and Ki67+ CD8+ T cells (fig. 2B) of tumor-infiltrating T cells was analyzed. (FIG. 2C) C57BL/6 mice bearing MC38-OVA tumors (n = 5) were treated with 6-thio-dG (3 mg/kg, 7 th, 8 th, v,9 days). Three days after the last treatment, with H-2K b -OVA 257-264 Tetramer analysis OVA-specific CD8+ T cells from tumor-infiltrating T cells. (FIG. 2D and FIG. 2E) same protocol as in (A), splenocytes were harvested and restimulated with irradiated MC38 tumor cells for 48 hours. IFN- γ producing cells were identified by ELISPOT assay. Representative points are shown in fig. 2D and quantitative data (n = 5) are shown in fig. 2E. (FIG. 2F) IFN-. Gamma.reporter mice (n = 3) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Eleven days after the last treatment, tumors were minced and digested for flow cytometry detection of YFP + T cells. P values were determined by two-tailed unpaired t-test (fig. 2A to 2C, fig. 2E and fig. 2F). See also fig. 102A to F.
Fig. 3A to 3F. Treatment with 6-thio-dG enhanced the cross-priming capacity of dendritic cells. (FIG. 3A) C57BL/6 mice (n = 5) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, day 7, 8, 9). 200 μ g of anti-CSF 1R was administered one day before treatment initiation and then twice weekly for 3 weeks. (FIG. 3B) Batf 3-/-mice (n = 5) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days. (fig. 3C) percentage of tumor-free mice in WT and Batf 3-/-mice (n = 5) after 6-thio-dG treatment. (FIG. 3D) BMDCs were cultured overnight with MC38 tumor cells pretreated with 200nM 6-thio-dG or vehicle, and then DCs were purified and co-cultured with naive OT-1T cells. After 48 hours, supernatants were collected and tested for IFN- γ production by Cytometric Bead Array (CBA). (FIG. 3E) BMDCs were cultured with MC38 tumor cells pretreated with 200nM 6-thio-dG or vehicle for 18 hours and supernatants collected for IFN- β ELISA. (F) Ifnar 1-/-mice (n = 5) were treated with 5 × 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days. Data are shown as mean ± SEM from two to three independent experiments. P values were determined by two-way ANOVA (fig. 3A, 3B and 3F) or two-tailed unpaired t-test (fig. 3C to 3E).
Fig. 4A to 4G.6-Intrinsic sensing of thio-dG induction requires STING signaling in the host. (FIGS. 4A and 4B) Myd88-/- (FIG. 4A) or Tmem173-/- (FIG. 4B) mice (n = 5) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days. (fig. 4C and 4D) C57BL/6 mice (n = 5) were used with 5 × 10 5 Tmem173KO (FIG. 4C) or Mb21D1KO (FIG. 4D) MC38 tumor cells were seeded and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Tumor growth was measured every 3 days. (FIGS. 4E and 4F) MC38 tumor cells were treated with 1. Mu.M 6-thio-dG for 24 hours. TIF (Telomere dysfunction Induced Foci) assay confirmed that treatment with 6-thio-dG Induced TIF in MC38 cells. n =100 (control), n =100 (6-thio-dG). (FIG. 4G) BMDCs were cultured with HCT116 human colon cancer cells pretreated with 500nM 6-thio-dG or vehicle for 4 hours, then DCs were purified and cytoplasmic DNA was extracted. The relative abundance of MT-CO1 and human 18S in the cytosol of DCs was determined by qPCR. Data are shown as mean ± SEM from two to three independent experiments. P values were determined by two-way ANOVA (a to D) or two-tailed unpaired t-test (fig. 4F and fig. 4G). See also fig. 11A to 11H.
Figures 5A to 5f.6-thio-dG overcome PD-L1 blockade resistance in the advanced tumor model. (fig. 5A) MC38 tumor bearing C57BL/6 mice (n =4 to 5) were treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). 7 days after the first treatment, PD-1+ CD8+ T cell frequency (left) and PD-1MFI (right) were tested. (FIGS. 5B and 5C) C57BL/6 mice were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, day 10, 11). 50 μ g of anti-PD-L1 antibody was administered on days 13 and 17. Tumor growth (fig. 5B) and viability (fig. 5C) are shown. (fig. 5D) MC38 tumor bearing C57BL/6 mice (n = 5) were treated with either 6-thio-dG (3 mg/kg, day 10, 11) or anti-PD-L1 (2.5 kg/mg, day 10) or a combination of both. 7 days after the first treatment, draining lymphoid tissues were harvested and stimulated with irradiated MC38 or LLC tumor cells for IFN- γ ELISPOT. (FIGS. 5E and 5F) C57BL/6 mice (n = 5) were treated with 1X 10 6 One LLC mouse lung tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 4, 5, 6 and 10, 11). On day 8 and day 8200 μ g of anti-PD-L1 antibody was administered for 13 days. Tumor growth was measured every 3 to 4 days (fig. 5E). Six weeks later, tumor-free mice (n = 4) and control mice in the sequentially treated groups were treated with 5 × 10 6 LLC (right flank) and 5X 10 6 One MC38 (left flank) tumor cell was attacked again. Tumor growth was measured every 3 to 4 days (fig. 5F). Data are shown as mean ± SEM from two independent experiments. P values were determined by two-tailed unpaired t-test (fig. 5A, 5D) or two-factor ANOVA (fig. 5B, 5E and 5F) or log rank test (fig. 5C). See also fig. 12.
Fig. 6A to 6E. 6-thio-dG reduced human colon cancer burden in a humanized mouse model. (FIG. 6A) Total survival in colorectal adenocarcinoma patients with high and low TERT (telomerase reverse transcriptase, which is the catalytic subunit of telomerase) expression from the TCGA database. (FIG. 6B) cell viability of 6-thio-dG in HCT116 human colon carcinoma cells (IC) 50 ). Cells were treated with 6-thio-dG for 5 days. (FIG. 6C) protocol for humanizing mouse tumor models. (fig. 6D and 6E) NSG-SGM3 mice (n = 5) (fig. 6D) or humanized NSG-SGM3 mice (n = 4) (fig. 6E) were treated with 1 × 10 6 One HCT116 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, day 8, 9, 10). Tumor growth was measured every 3 days. Data are shown as mean ± SEM from two independent experiments. P values were determined by log rank test (fig. 6A) or two-way ANOVA (fig. 6D and 6E). See also fig. 13A to 13F.
Fig. 7. Schematic representation of 6-thio-dG induction of c-GAS/STING/IFN.
Fig. 8A to 8B. 6-thio-dG is then evidence that PD-L1 leads to complete tumor remission and immunogenic memory.
Fig. 9A to 9D (related to fig. 1A to 1G). (FIG. 9A) cell viability (IC) of 6-thio-dG in LLC murine Lung cancer cells 50 ). Cells were treated with 6-thio-dG for 4 days. (FIG. 9B) C57BL/6 mice (n = 5) were treated with 1X 10 6 One LLC tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 4, 5, 6). Tumor growth was measured every 3 days. (FIG. 9C) IC of 6-thio-dG in CT26 murine Colon cancer cells 50 . (FIG. 9D) BALB/C mice (n = 5) were treated with 5X 10 5 Individual CT26 tumor cells were inoculated and treated with 6-thio-dG (3 mg/kg, day 5, 6, 7). Every 3 daysAnd (4) measuring tumor growth. Data are shown as mean ± SEM from two independent experiments. P values were determined by two-way ANOVA.
Fig. 10A to 10F (related to fig. 2A to 2G). (fig. 10A to 10D) C57BL/6 mice (n =4 to 5) were treated with 5 × 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). 7 days after the first treatment, tumors were analyzed for CD8+ T cells among CD45+ cells (FIG. 10A) and total tumor cells (FIG. 10B), and CD4 of tumor-infiltrating T cells was analyzed + Foxp3 + Frequency of Treg cells (fig. 10C) and NK cells (fig. 11D). (FIG. 10E) C57BL/6 mice (n = 5) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, day 7, 8, 9). 200 μ g of anti-NK 1.1 was administered one day before treatment was started and then twice weekly for 3 weeks. (FIG. 10F) IFN-. Gamma.reporter mice (n = 3) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 7, 8, 9). Eleven days after the last treatment, tumors were minced and digested for flow cytometry detection of YFP + T cells. A representative flow cytometry gating is shown. Data are shown as mean ± SEM from two independent experiments. P values were determined by two-tailed unpaired t-test in (fig. 10A to 10D) or two-way ANOVA (fig. 10E).
Fig. 11A to 11H (related to fig. 4A to 4G). (FIG. 11A) BMDCs were cultured with MC38 tumor cells pretreated with 0.2. Mu.M or 1. Mu.M 6-thio-dG for 6 hours, and then DCs were purified with magnetic beads and subjected to western blotting. (FIG. 11B) BMDCs from Wild Type (WT) or Tmem173KO mice were cultured overnight with MC38 tumor cells pretreated with 200nM 6-thio-dG, and then DCs were purified with magnetic beads and subjected to qPCR to test the relative abundance of IFN- β. (FIGS. 11C and 11D) C57BL/6 mice (n = 3) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, days 10, 11, 12). Mice were sacrificed 3 days after the last injection; tumors were harvested and fixed for TIF (telomere dysfunction-induced lesion) staining. Images were obtained by fluorescence microscopy (100 ×). Red dots indicate DNA damage (γ -H2 AX), green dots indicate telomeres and yellow dots indicate TIF (DNA damage on telomeres). Scale bar, 10 μ M. (FIGS. 11E and 11F) 6-thiodG treatment induces micronuclei in MC38 cells. (FIG. 11E) representative pictures of two daughter cells at late evening contain telomere signaling and coated and uncoated micronuclei in MC38 cells. Green dots indicate telomeric signal and red indicates lamin a/C (nuclear envelope biomarker). (FIG. 11F) quantification of micronuclei induced by 1. Mu.M 6-thio-dG treatment after 48 hours. (FIGS. 11G and 11H) 100,000 MC38 cells were seeded in 6-well plates and labeled with 25. Mu.M EdU. After 2 days, the cells were washed and incubated with 1. Mu.M of 6-thio-dG in fresh medium O/N. Cells were then washed and co-cultured with DC O/N. The next day, the DCs were purified with magnetic beads. The purified DCs were then fixed and cytospin (cytospun) was used for immuno-FISH. Telomere probe: green, edU: red, DAPI: blue in color. Images were captured at 63 Xmagnification with Axio Imager Z2 equipped with an automatic capture system and analyzed with ISIS software (camera: coolcube 1-meters). Representative imaging (fig. 11G) and quantitative data (fig. 11H) are shown, n =100. Data are shown as mean ± SEM from two to three independent experiments. P values were determined by two-tailed unpaired t-test (B, F and H).
Fig. 12 (relating to fig. 5A to 5G). C57BL/6 mice (n = 5) were treated with 5X 10 5 One MC38 tumor cell was inoculated and treated with 6-thio-dG (3 mg/kg, day 10, 11). 50 μ g of anti-PD-L1 antibody was administered on days 13 and 17. Mouse body weights were measured. Data are shown as mean ± SEM.
Fig. 13A to 13F (related to fig. 6A to 6E). (fig. 13A to 13C) mice were tested for human CD45+ cells and CD3+ T cells in peripheral blood by flow cytometry 12 weeks after reconstitution of humanized mice. A representative flow cytometry plot is shown in fig. 13A. CD45 and CD3 frequencies in the control and 6-thiodg groups before treatment are shown in fig. 13B and 13C, with n =5. (FIG. 13D) cell viability of 6-thio-dG in A375 human melanoma cancer cells (IC) 50 ). Cells were treated with 6-thio-dG for 4 days. (FIG. 13E) NSG-SGM3 mice (n = 5) were treated with 2X 10 6 Individual a375 tumor cells were inoculated and treated with 6-thio-dG (3 mg/kg, day 7 and day 8) or anti-PD-L1 plus anti-CTLA-4 (200 μ g i.p., day 10 and day 13) or a combination of 6-thio-dG plus anti-PD-L1 and anti-CTLA-4. Each timeTumor growth was measured on 3 days. (fig. 13F) humanized NSG-SGM3 mice (n =5 to 7) were treated with 2 × 10 6 Individual a375 tumor cells were inoculated and treated with 6-thio-dG (3 mg/kg, day 13 and day 14) or anti-PD-L1 plus anti-CTLA-4 (200 μ g i.p., day 16 and day 19) or a combination of 6-thio-dG plus anti-PD-L1 and anti-CTLA-4. Tumor growth was measured every 3 days. Data are shown as mean ± SEM. P values were tested by two-tailed unpaired t (fig. 13B and 13c, n.s.p.>0.05 Or two-way ANOVA (fig. 13F).
FIG. 14 shows 6-thio-dG with the anti-PD-1 agent cimetipril mab Effect on tumor volume in mice bearing tumors of LLC origin (NSCLC). The 6-thio-dG was administered at 3mg/kg (i.p) and cimetiprizumab-10 mg/kg (i.p). The different groups were dosed as indicated in the table below. Day 1 (12/31/2020): 1000K LLC cells were inoculated into
Effect on tumor volume in mice bearing tumors of LLC origin (NSCLC). The 6-thio-dG was administered at 3mg/kg (i.p) and cimetiprizumab-10 mg/kg (i.p). The different groups were dosed as indicated in the table below. Day 1 (12/31/2020): 1000K LLC cells were inoculated into 35B 6 mice. Day 11 to 13: the experiment was started. 3mg/kg 6-thio-dG and 10mg/kg Libtayo were used in this study.
TABLE A dosing regimen

FIG. 15 shows 6-thio-dG with the anti-PD-1 agent cimetipril mab Effect on tumor volume in mice bearing tumors of LLC origin (NSCLC). The 6-thio-dG was administered at 3mg/kg (i.p) and the cimetiprilinumab-10 mg/kg.p (i.p). The different groups were dosed as indicated in the table above. Day 1 (12/31/2020): 1000K LLC cells were inoculated into
Effect on tumor volume in mice bearing tumors of LLC origin (NSCLC). The 6-thio-dG was administered at 3mg/kg (i.p) and the cimetiprilinumab-10 mg/kg.p (i.p). The different groups were dosed as indicated in the table above. Day 1 (12/31/2020): 1000K LLC cells were inoculated into 35B 6 mice. Days 11 to 13: the experiment was started. 3mg/kg 6-thio-dG and 10mg/kg Libtayo were used in this study.
Figure 16 shows the effect of 6-thio-dG in combination with the PD-1 agent pembrolizumab in a Small Cell Lung Cancer (SCLC) humanized mouse model.
Figure 17 shows 6-thio-dG in combination with a PD-L1 inhibitor and radiation in a mouse model of HCC.
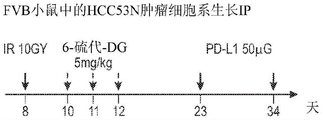
Figures 18A to 18D show 6-thio-dG in combination with PD-L1 and radiation in a mouse model of HCC. FIG. 18A: a dosing regimen. FIG. 18B: HCC53N hepatoma cells (p 53 and NRAS knockdown) were initially treated in vivo with focal IR followed by 3 doses with 6-thio-dG followed by 2 treatments with anti-PD-L1 antibody resulting in complete tumor remission. FIG. 18C: re-challenge with 10-fold more HCC53N cells, no tumor re-growth, indicating immunological memory; and FIG. 18D: when initial mice were tested, tumors grew rapidly.
Detailed Description
Telomerase is almost universally expressed in tumor cells. The telomerase-mediated telomere-targeting drug 6-thio-dG reduces the lag time between initial treatment and response to treatment by directly inducing telomere damage in telomerase positive cancer cells, but not in normal telomerase-silenced cells. In this study, the inventors aimed to explore whether 6-thio-dG inducing telomeric stress in telomerase positive cancer cells could initiate rapid DNA damage for intrinsic perception. Syngeneic wild-type mice and genetically deficient mice were used to evaluate how 6-thio-dG triggers innate perception and how it contributes to host anti-tumor immunity. Importantly, 6-thio-dG was demonstrated to overcome PD-L1 blockade resistance in advanced tumors. Unexpectedly, 6-thio-dG induces activation of DNA-mediated innate perception and immune responses in a host STING-dependent manner, resulting in increased antitumor efficacy. Furthermore, sequential 6-thio-dG followed by anti-PD-L1 treatment could completely eliminate advanced tumors. Thus, 6-thio-dG is a tumor-targeting and immune-stimulating drug that can clinically benefit telomerase-positive and PD-L1-resistant cancer patients.
These and other aspects of the disclosure are described in detail below.
I. Telomere, telomerase and telomere dysfunction
During mitosis, the cell undergoes replication of its genetic material. Half of the genetic material enters each new daughter cell. To ensure that information is successfully passed from one generation to the next, each chromosome has a special protective cap, called the telomere, located at the end of its "arm". Telomeres are controlled by the enzyme telomerase.
Telomeres are repetitive DNA sequences at the ends of the body's chromosomes (e.g., TTAGGG). Telomeres can be up to 15,000 base pairs in length. The function of telomeres is to prevent the chromosome from losing its terminal base pair sequence. It also prevents chromosomes from fusing to each other. However, each cell division, a portion of telomeres is lost (each division is typically 25 to 200 base pairs). When telomeres become too short, the chromosomes reach a "critical length" and are no longer able to replicate. This means that the cells age and die or undergo senescence through a process called apoptosis. Telomere activity is controlled by two mechanisms: erosion and growth. Erosion, as noted, occurs with each cell division because late strand DNA synthesis is not completed until finally. The increase is determined by telomerase activity.
Telomerase, also known as telomerase, is an enzyme composed of protein and RNA subunits that can extend a chromosome by adding a TTAGGG sequence to the end of an existing chromosome. Telomerase is present in fetal tissues, adult germ cells, and tumor cells. Telomerase activity is regulated during development and has very low, barely detectable activity in somatic (body) cells. Because these somatic cells do not use telomerase very often, they will age. The result of aging cells is body senescence. If telomerase is activated in a cell, the cell will continue to grow and divide. This "immortal cell" theory is in two areas of research: is important in aging and cancer.
Cellular senescence or aging is a process in which cells age and stop growing or dying. This is due to the shortening of chromosome telomeres to the point where chromosomes reach critical length. Cellular senescence is similar to a wind-up clock. If the clock remains winding, the cell becomes immortal and new cells are continuously generated. If the clock slows down (wind down), the cell stops producing new cells and undergoes so-called replicative senescence or death. The cells are constantly senescent. The ability to expand the body's cells to replicate undoubtedly results in some exciting feasibility, particularly for diseases genetically related to short telomeres (known as telomeric diseases or disorders of the telomeric lineage). Therefore, telomerase studies can lead to important findings related to the aging process.
Cancer cells evade normal short-telomeric senescence and become malignant cells. Malignant cells multiply until they form a tumor that grows uncontrollably and spreads to distant tissues throughout the body. Telomerase is detected in almost all human cancer cells. This provides a selective growth advantage for many types of tumors. If telomerase activity is turned off, telomeres in cancer cells will progressively shorten, as they do in normal somatic cells. This will prevent uncontrolled division of cancer cells at their early developmental stages. If the tumor has fully developed, it can be removed and anti-telomerase therapy administered to prevent recurrence. Essentially, preventing telomerase from performing its function would change the cancer cell from immortal to non-immortal. However, direct telomerase inhibitors require a lag phase from the start of treatment until tumor shrinkage occurs and have not progressed well in clinical development due to increased toxicity. Thus, the present invention provides a method of reducing lag phase but requiring telomerase activity to effectively and potentially reduce side effects.
Treatment of cancer
A. Therapeutic agents for sequential treatment
1. In some embodiments, the PD-L1 inhibitor is selected from one or more of alemtuzumab, avizumab, chikulizumab, bindrafusalfa, devaluzumab, MGD013, KNO35, KN046, AUNP12, CA-170, and BMS-9986189. In some embodiments, the PD-L1 inhibitor is atelizumab.
Abuzumab (trade name) ) Is a fully humanized engineered monoclonal antibody directed against the IgG1 subtype of protein programmed cell death ligand 1 (PD-L1). In 2015, it was used in clinical trials as an immunotherapy for several types of solid tumors. 2016, 5 months, approved by the FDAFor bladder cancer treatment, but in 2017, in 5 months, it failed phase III trials for second-line bladder cancer. In 10 months 2016, the FDA approved atelizumab for urothelial cancer and to treat patients with metastatic non-small cell lung cancer (NSCLC) whose disease progressed during or after platinum-containing chemotherapy. Patients with EGFR or ALK genomics tumor aberrations should have disease progression with FDA-approved treatments for these aberrations before receiving atelizumab. In 9 s 2018, atuzumab was declared to have prolonged survival in a wide-term small cell Lung Cancer treatment, based on findings demonstrated at the 19 th World Conference on Lung Cancer (WCLC), held in toronto, canada. In 2018, 10 months, the combined clinical trial of this drug with albumin-bound paclitaxel (nab-paclitaxel) was concluded for patients with advanced triple negative breast cancer. 2019, which was approved in the united states for use with protein-bound paclitaxel in adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who expressed PD-L1 (coverage of tumor-infiltrating immune cells with PD-L1 staining of any intensity ≧ 1% of the tumor area), as determined by FDA-approved testing. 2019,
) Is a fully humanized engineered monoclonal antibody directed against the IgG1 subtype of protein programmed cell death ligand 1 (PD-L1). In 2015, it was used in clinical trials as an immunotherapy for several types of solid tumors. 2016, 5 months, approved by the FDAFor bladder cancer treatment, but in 2017, in 5 months, it failed phase III trials for second-line bladder cancer. In 10 months 2016, the FDA approved atelizumab for urothelial cancer and to treat patients with metastatic non-small cell lung cancer (NSCLC) whose disease progressed during or after platinum-containing chemotherapy. Patients with EGFR or ALK genomics tumor aberrations should have disease progression with FDA-approved treatments for these aberrations before receiving atelizumab. In 9 s 2018, atuzumab was declared to have prolonged survival in a wide-term small cell Lung Cancer treatment, based on findings demonstrated at the 19 th World Conference on Lung Cancer (WCLC), held in toronto, canada. In 2018, 10 months, the combined clinical trial of this drug with albumin-bound paclitaxel (nab-paclitaxel) was concluded for patients with advanced triple negative breast cancer. 2019, which was approved in the united states for use with protein-bound paclitaxel in adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who expressed PD-L1 (coverage of tumor-infiltrating immune cells with PD-L1 staining of any intensity ≧ 1% of the tumor area), as determined by FDA-approved testing. 2019, month 3, approved in the united states for first-line treatment of adult patients with extensive small cell lung cancer (ES-SCLC) in combination with carboplatin and etoposide. The most common adverse effects in the study were fatigue, decreased appetite, nausea and infection. Urinary tract infections are the most common serious adverse effects.
Attributumab blocks the interaction of PD-L1 with programmed cell death protein 1 (PD-1) and the CD80 receptor (B7-1R). PD-L1 may be highly expressed on certain tumors, which is thought to result in reduced activation of immune cells (particularly cytotoxic T cells) that may otherwise recognize and attack cancer. Inhibition of PD-L1 by astuzumab can abrogate this inhibition and thereby generate an anti-tumor response. It is one of several approaches to block inhibitory signals associated with T cell activation, a more general strategy known as immune checkpoint inhibition. For some cancers (especially bladder cancer), the feasibility of benefit correlates with PD-L1 expression, but most cancers with PD-L1 expression still do not respond, while some (about 15%) do not respond.
Abameluumab Is a fully human IgG1 antibody developed by Merck Serono and Pfizer. Ablumumab is approved by the FDA for the treatment of metastatic merkel cell carcinoma. It failed the phase III clinical trial of gastric cancer.
Is a fully human IgG1 antibody developed by Merck Serono and Pfizer. Ablumumab is approved by the FDA for the treatment of metastatic merkel cell carcinoma. It failed the phase III clinical trial of gastric cancer.
Dewar monoclonal antibody Is a fully human IgG1 antibody developed by AstraZeneca. Dewaruzumab is approved by the FDA for the treatment of urothelial and non-resectable non-small cell lung cancer following chemoradiotherapy.
Is a fully human IgG1 antibody developed by AstraZeneca. Dewaruzumab is approved by the FDA for the treatment of urothelial and non-resectable non-small cell lung cancer following chemoradiotherapy.
KN035 is the only PD-L1 antibody with subcutaneous formulation currently being evaluated clinically in the united states, china and japan.
AUNP12 is the 29 mer peptide as the first digestible PD-1/PD-L1 inhibitor developed by Aurigene and laboratory Pierre Fabre, which is being evaluated in clinical trials for the treatment of cancer.
CA-170, discovered by Aurigene/Curis as a PD-L1 and VISTA antagonist, is currently in phase I clinical trials for the treatment of mesothelioma.
PD-1 inhibitors such as cimicipril mab, pembrolizumab, nivolumab, JTx-4014, saralalizumab, brelizumab, BI 754091, sibatuzumab, carpriclizumab, credits, tirlizumab, serralizumab, terirelizumab, dolaprimab, INCMGGA 00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-317A, BCD-100, JS001, and AMP-515. In some embodiments, the PD-1 inhibitor is cimetizumab or pembrolizumab.
Cimicpril monoclonal antibody is under the trade name It is marketed as a monoclonal antibody drug for the treatment of squamous cell skin cancer, basal cell carcinoma skin cancer and non-small cell lung cancer. The cimipipril monoclonal antibody belongs to a medicine combined with a programmed death receptor-1 (PD-1), and blocks a PD-1/PD-L1 path. In 2018, 9 months, approved by the U.S. Food and Drug Administration, FDA, for the treatment of people with metastatic squamous cell Carcinoma of Skin (CSCC) or locally advanced CSCC that are not candidates for curative surgery or curative irradiation. Cimetiprizumab is being investigated for the treatment of cervical melanoma, brain cancer, head and neck cancer, renal cell carcinoma and hodgkin's lymphoma.
It is marketed as a monoclonal antibody drug for the treatment of squamous cell skin cancer, basal cell carcinoma skin cancer and non-small cell lung cancer. The cimipipril monoclonal antibody belongs to a medicine combined with a programmed death receptor-1 (PD-1), and blocks a PD-1/PD-L1 path. In 2018, 9 months, approved by the U.S. Food and Drug Administration, FDA, for the treatment of people with metastatic squamous cell Carcinoma of Skin (CSCC) or locally advanced CSCC that are not candidates for curative surgery or curative irradiation. Cimetiprizumab is being investigated for the treatment of cervical melanoma, brain cancer, head and neck cancer, renal cell carcinoma and hodgkin's lymphoma.
Mepiquat mab (formerly lambrolizumab, under the trade name lambrolizumab) Marketed) are humanized antibodies for cancer immunotherapy. Mepiquat was approved for medical use in the united states in 2014. In 2017, the U.S. Food and Drug Administration (FDA) approved it for any unresectable or metastatic solid tumor with certain genetic abnormalities (mismatch repair defects or microsatellite instability).
Marketed) are humanized antibodies for cancer immunotherapy. Mepiquat was approved for medical use in the united states in 2014. In 2017, the U.S. Food and Drug Administration (FDA) approved it for any unresectable or metastatic solid tumor with certain genetic abnormalities (mismatch repair defects or microsatellite instability). Currently approved indications include metastatic melanoma, NSCLC, head and neck cancer, hodgkin's lymphoma, and metastatic esophageal squamous cell carcinoma. Pembrolizumab is administered by slow injection into a vein.
Currently approved indications include metastatic melanoma, NSCLC, head and neck cancer, hodgkin's lymphoma, and metastatic esophageal squamous cell carcinoma. Pembrolizumab is administered by slow injection into a vein.
3. Thiopurines, such as 6-thioguanine and 6-mercaptopurine, are currently used in clinical practice as anti-inflammatory, anti-leukemic, and immunosuppressive agents. In the activation reaction, 6-thioguanine is converted to 6-thioguanine monophosphate by hypoxanthine guanine phosphoribosyltransferase (HPRT). The monophosphate 6-thioguanosine is then further metabolized by kinases and RNA reductases into 6-thio-2 '-deoxyguanosine 5' -triphosphate, which can eventually be incorporated into the DNA strand during DNA replication. 6-thioguanine incorporated into DNA can also produce reactive oxygen species that can cause additional damage to DNA, proteins, and other cellular macromolecules, and thus block cellular replication. Although thiopurines are used clinically to treat some types of leukemia, their utility for the treatment of solid tumors is limited, in part due to increased toxicity and development of other treatments.
One particular thiopurine is 6-thio-dG. The compounds are nucleoside analogs and have been shown to be telomerase mediated telomere disrupting compounds. Thus, cancer cells are very sensitive to 6-thio-dG, where the IC observed 50 Values range from 0.7 to 2.9 μ M (depending on cell type), even including treatment resistant cancers (Mender et al, 2018). The structure is shown below:

B. treatment regimens
The present disclosure provides a sequential cancer treatment using 6-thio-dG followed by PD-L1, PD-1 and/or CTLA-4 treatment. The time period for each treatment may vary, and it is expected that short intervals between treatments will be advantageous. For example, 6-thio-dG treatment may be as short as 2 days, but may be 3, 4 or more days, including 2 to 4 days. The interval prior to PD-L1, PD-1 and/or CTLA-4 treatment should be at least 1 day and may be as long as 14 days, for example 2 to 4 days. Because of the potentially deleterious effects of 6-thio-dG on activated effector T cells, overlap between 6-thio-dG and PD-L1, PD-1 and/or CTLA-4 should be avoided.
The daily dose of 6-thio-dG will be from 0.5mg/kg to 10mg/kg, preferably intravenous or oral. The dosage of PD-L1, PD-1 and/or CTLA-4 will be in accordance with currently approved dosing regimens.
C. Telomerase positive cancers
Telomerase positive cancers are far more susceptible to the methods of the disclosure than telomerase negative cancers. Thus, it is useful, although not necessary, to test the biopsy to determine if the cancer is telomerase positive.
The most common method for detecting telomerase activity is the Telomeric Repeat Amplification Protocol (TRAP), which allows one to use some of its modifications, called ddTRAP, as a droplet digital TRAP, for semi-quantitative and quantitative analysis. Among these modifications are scintillation proximity assays, hybridization protection assays, transcription amplification assays, and magnetic bead-based extraction assays.
Telomere repeat amplification schemes can be subdivided into three major stages: primer extension, amplification of telomerase synthetic DNA and finally detection thereof. In the extension phase, telomeres are repeatedly added to the telomere mimetic oligonucleotides by telomerase present in the cell extract. PCR amplification of telomerase synthetic DNA was performed using telomere mimic and reverse primers. Different markers can be incorporated into the telomerase synthetic DNA. This stage is then followed by detection (e.g., electrophoretic separation and imaging of PCR products).
Still other methods involve quantitative isolation of telomerase and subsequent measurement of the overall activity of telomerase from a given cell mass, which can be compared to appropriate standards. Once telomerase is isolated and tested in vitro, a wide variety of labeling and detection methods can be used.
D. Drug resistant cancers
Anti-tumor resistance, often used interchangeably with chemotherapy resistance, is resistance to tumor (cancerous) cells or the ability of cancer cells to survive and grow despite anti-cancer therapy. In some cases, cancer develops resistance to multiple drugs, referred to as multidrug resistance.
There are two general reasons for the failure of anti-tumor therapy: inherent genetic characteristics (which confer resistance to cancer cells) and acquired resistance after drug exposure (which rooted in the concept of cancer cell heterogeneity). Features of resistant cells include altered membrane transport, enhanced DNA repair, defects in apoptotic pathways, altered target molecules, proteins and pathway mechanisms, such as enzyme inactivation. Since cancer is a genetic disease, two genomic events are the basis for acquired drug resistance: genomic alterations (e.g., gene amplifications and deletions) and epigenetic modifications. Cancer cells are continually using a variety of tools, including genes, proteins, and altered pathways, to ensure their survival against anti-tumor drugs.
Anti-tumor resistance is synonymous with chemotherapy resistance and refers to the ability of a cancer cell to survive and grow (i.e., its multidrug resistance) even when subjected to different anti-cancer therapies. There are two general reasons for the failure of anti-tumor therapy: (i) Intrinsic resistance (e.g., genetic trait), which is conferred resistance to cancer cells from the beginning, rooted in the concept of cancer cell heterogeneity; and (ii) acquired resistance after drug exposure.
Since cancer is a hereditary disease, two genomic events underlie these mechanisms of acquired drug resistance: genomic alterations (e.g., gene amplifications and deletions) and epigenetic modifications.
Chromosomal rearrangements due to genomic instability can lead to gene amplification and deletion. Gene amplification is an increase in the copy number of a chromosomal region. It occurs frequently in solid tumors and can promote tumor evolution by altering gene expression.
Hamster cell studies in 1993 showed that amplification of the DHFR gene involved in DNA synthesis begins with a chromosomal break below the gene and that subsequent cycles of bridge break fusion formation lead to massive intrachromosomal repeats. Over-amplification of oncogenes may occur in response to chemotherapy, and is considered a potential mechanism for several types of resistance. For example, DHFR amplification occurs in response to methotrexate, TYMS (involved in DNA synthesis) amplification occurs in response to 5-fluorouracil, and BCR-ABL amplification occurs in response to imatinib mesylate. The determination of the region of gene amplification in cells from cancer patients is of great clinical significance. Gene deletion is in contrast to gene amplification, in which a region of the chromosome is lost and drug resistance occurs through the loss of a tumor suppressor gene (e.g., TP 53).
Genomic instability can occur when replication forks are disturbed or arrested in their migration. This can occur with replication fork disorders, proteins (e.g., PTIP, CHD4, and PARP 1), which are normally cleared by the DNA damage sensors, detectors, and responders BRCA1 and BRCA2 of the cell.
Epigenetic modification of resistance to antineoplastic drugs plays an important role in carcinogenesis and drug resistance, as it contributes to the regulation of gene expression. Two major types of epigenetic control are DNA methylation and histone methylation/acetylation. DNA methylation is the process of adding a methyl group to DNA, usually in the upstream promoter region, which stops DNA transcription and effectively silences a single gene in that region. Histone modifications (e.g., deacetylation) alter chromatin formation and silence large chromosomal regions. In cancer cells in which normal regulation of gene expression is disrupted, oncogenes are activated by hypomethylation, while tumor suppressors are silenced by hypermethylation. Similarly, in drug resistance development, it has been suggested that epigenetic modifications can lead to activation and overexpression of prodrug resistance genes.
Studies with cancer cell lines have shown that hypomethylation (loss of methylation) of the MDR1 gene promoter leads to overexpression and multiple drug resistance.
In the methotrexate-resistant breast cancer cell lines without drug uptake and folate vector expression, DAC (DNA methylation inhibitor) administration increased drug uptake and folate vector expression.
Acquired resistance of melanoma cells to the alkylating drug fotemustine (fotemustine) showed high MGMT activity associated with hypermethylation of MGMT gene exons.
In imatinib Silencing of the SOCS-3 gene by methylation in resistant cell lines has been shown to result in STAT3 protein activation, leading to uncontrolled proliferation.
Silencing of the SOCS-3 gene by methylation in resistant cell lines has been shown to result in STAT3 protein activation, leading to uncontrolled proliferation.
Cancer cells can develop resistance to a variety of drugs by altering membrane transport, enhancing DNA repair, defective apoptotic pathways, altering target molecules, proteins and pathway mechanisms (e.g., enzyme inactivation).
Many classes of antineoplastic drugs act on intracellular components and pathways, such as DNA, nuclear components, which means that they need to enter cancer cells. P-glycoprotein (P-gp) or multidrug resistance protein is a phosphorylated and glycosylated membrane transporter that transports drugs out of cells, thereby reducing or eliminating drug potency. This transporter protein is encoded by the MDR1 gene and is also known as an ATP-binding cassette (ABC) protein. MDR1 has a promiscuous substrate specificity, allowing it to transport a number of structurally different compounds, primarily hydrophobic compounds, across cell membranes. It has been found that the MDR1 gene can be activated and overexpressed in response to pharmaceutical drugs, forming the basis of resistance to many drugs. Overexpression of the MDR1 gene in cancer cells is used to keep intracellular levels of antitumor drugs below cell killing levels.
For example, the antibiotic rifampicin has been found to induce MDR1 expression. Experiments in different drug resistant cell lines and patient DNA revealed gene rearrangements that initiate activation or overexpression of MDR 1. The C3435T polymorphism in exon 226 of MDR1 is also strongly associated with p-glycoprotein activity.
MDR1 is activated by NF-. Kappa.B, a protein complex that acts as a transcription factor. In rats, the NF-. Kappa.B binding site is adjacent to the mdr1B gene, and NF-. Kappa.B can be active in tumor cells because its mutated NF-. Kappa.B gene or its inhibitory Ikappa.B gene is mutated under chemotherapy. In colorectal cancer cells, inhibition of NF-. Kappa.B or MDR1 results in increased apoptosis in response to chemotherapeutic agents.
Enhanced DNA repair plays an important role in conferring cancer cells the ability to overcome drug-induced DNA damage.
Platinum-based (e.g., cisplatin) chemotherapy targets tumor cells by cross-linking their DNA strands, resulting in mutations and damage. Such damage will trigger programmed cell death (e.g., apoptosis) in the cancer cells. Cisplatin resistance occurs when cancer cells develop an enhanced ability to reverse any damage caused by cisplatin by removing it from the DNA and repairing such damage. Cisplatin-resistant cells up-regulate the expression of excision repair cross-complementing (ERCC 1) genes and proteins.
Some chemotherapeutic agents are alkylating agents, which means that they link an alkyl group to DNA to prevent it from being read. O6-methylguanine DNA methyltransferase (MGMT) is a DNA repair enzyme that removes alkyl groups from DNA. MGMT expression is upregulated in many cancer cells, which protects it from alkylating agents. Increased MGMT expression is found in colon cancer, lung cancer, non-hodgkin's lymphoma, breast cancer, glioma, myeloma and pancreatic cancer.
TP53 is a tumor suppressor gene encoding a p53 protein that responds to DNA damage by DNA repair, cell cycle arrest, or apoptosis. Loss of TP53 through gene deletion allows the cell to continue replication despite DNA damage. Tolerance to DNA damage can confer resistance to cancer cells to those drugs that typically induce apoptosis through DNA damage.
Other genes involved in resistance to apoptosis pathway-associated drugs include h-ras and bcl-2/bax. Oncogenic h-ras has been found to increase the expression of ERCC1, thereby enhancing DNA repair (see above). Inhibition of h-ras was found to increase cisplatin sensitivity in glioblastoma cells. Upregulated expression of Bcl-2 in leukemic cells (non-hodgkin's lymphoma) results in decreased levels of apoptosis in response to chemotherapeutic agents, as Bcl-2 is a pro-survival oncogene.
During targeted therapy, the target typically self-modifies and reduces its expression to the point where the therapy is no longer effective. An example of this is the loss of Estrogen Receptor (ER) and Progestin Receptor (PR) following anti-estrogen treatment of breast cancer. Tumors that lose ER and PR no longer respond to tamoxifen or other anti-estrogen treatments and, while cancer cells retain responses to estrogen synthesis inhibitors to some extent, eventually become unresponsive to endocrine manipulations and no longer grow estrogen-dependent.
Another therapeutic line for the treatment of breast cancer is a targeted kinase, such as human epidermal growth factor receptor 2 (herr 2) from the EGFR family. Mutations often occur in the HER2 gene following treatment with inhibitors, with about 50% of patients with lung cancer being found to have the EGFR-T790M gatekeeper (gatekeeper) mutation.
The treatment of Chronic Myelogenous Leukemia (CML) involves a tyrosine kinase inhibitor, called imatinib, targeting the BCR/ABL fusion gene. In some people who are resistant to imatinib, the BCR/ABL gene is reactivated or amplified, or a single point mutation occurs in the gene. These point mutations enhance the autophosphorylation of the BCR-ABL protein, resulting in the stabilization of the ATP binding site into its active form, rendering it incapable of being bound by imatinib for appropriate drug activation.
Because of its key role in DNA replication as an enzyme, topoisomerase is an advantageous target for cancer therapy, and a number of topoisomerase inhibitors have been prepared. Resistance occurs when the level of topoisomerase is reduced, or when different isomers of topoisomerase are differentially distributed within the cell. Mutant enzymes have also been reported in patients with leukemia cells and in other cancers in mutations conferring resistance to topoisomerase inhibitors.
One of the mechanisms of antitumor resistance is the overexpression of drug metabolizing enzymes or carrier molecules. By increasing the expression of metabolic enzymes, the drug is more quickly converted to the drug conjugate or inactive form, which can then be excreted. For example, increased expression of glutathione promotes drug resistance because the electrophilic nature of glutathione allows it to react with cytotoxic agents, rendering it inactive. In some cases, resistance is conferred by reduced expression or loss of expression of a drug-metabolizing enzyme, as the enzyme is required to process the drug from an inactive form to an active form. Arabinoside (a common chemotherapeutic agent for leukemia and lymphoma) is converted to cytosine arabinoside triphosphate by deoxycytidine kinase. Mutation or loss of expression of deoxycytidine kinase results in resistance to arabinoside. This is the enzyme inactive form.
Growth factor expression levels may also promote resistance to anti-tumor therapy. In breast cancer, drug resistant cells are found to express high levels of IL-6, while sensitive cells do not express significant levels of growth factors. IL-6 activates the CCAAT enhancer binding protein transcription factor (which activates MDR1 gene expression).
Another type of anti-tumor resistance is resistance to checkpoint inhibitors. Primary resistance to immune checkpoint blockade occurs in about 40% to 65% of patients with melanoma treated with anti-PD-1 based treatment. This clinical problem arises when an effective anti-tumor immune response is not induced at any of the three stages of the cancer immune cycle. Factors associated with primary resistance to date include elevated levels of baseline serum LDH, elevated baseline tumor burden, lack of PD-L1 expression in baseline melanoma tissue samples, lack of T cell infiltration, absence of PD-1T cells and PD-L1 macrophages in melanoma biopsies obtained early in the treatment, insufficient neo-antigens and low mutation burden, presence of intrinsic anti-PD-1 resistance signature (ipres) transcriptional characteristics, or absence of interferon characteristics.
Acquired resistance to immunotherapy can occur when a subpopulation of tumor cells with genetic and epigenetic characteristics is selected to evade the immune system. One example is the report of loss of B2M expression in melanoma cell lines from patients who have been treated with immunotherapy and cytokine gene therapy. This results in a loss of MHC class I expression and thus a subsequent reduction in expression by CD8 + T cell recognition. The JAK1/2 mutation has also recently been identified as a genetic marker for acquired resistance to immunotherapy in melanoma. These mutations in tumor cells result in reduced sensitivity to IFN- γ, ultimately preventing IFN- γ -induced cell growth arrest. Loss of functional mutations in the genes encoding JAK1 or JAK2 was found in recurrent tumors after whole exome sequencing of baseline and progressive biopsies; all patients had objective responses to treatment with pembrolizumab and then progressed. In addition, acquired resistance can also occur at the single cell level, where tumor cells change their gene expression in response to immune molecules in the tumor microenvironment. For example, PD-L1 may be upregulated by tumor cells in response to immune cytokines (e.g., IFN- γ released by T cells), thus limiting T cell function, and may occur in both primary and acquired resistance.
Pharmaceutical formulations and routes of administration
In case of clinical application, a pharmaceutical composition is prepared in a form suitable for the intended use. Generally, this will require the preparation of a composition that is substantially free of pyrogens (pyrogens) and other impurities that may be harmful to humans or animals.
One would typically desire to use appropriate salts and buffers to stabilize the drug and allow for uptake by the target cells. The aqueous compositions of the present disclosure comprise an effective amount of the drug dissolved or dispersed in a pharmaceutically acceptable carrier or aqueous medium. The phrase "pharmaceutically or pharmacologically acceptable" refers to molecular entities and compositions that do not produce adverse, allergic, or other untoward reactions when administered to an animal or human. As used herein, "pharmaceutically acceptable carrier" includes acceptable solvents, buffers, solutions, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like, used to formulate medicaments (e.g., medicaments suitable for administration to humans). The use of such media and agents for pharmaceutically active substances is well known in the art. Except to the extent that any conventional media or agent is incompatible with the active ingredients of the present disclosure, its use in therapeutic compositions is contemplated. Supplemental active ingredients may also be incorporated into the composition, provided they do not inactivate the agents of the composition.
The active compositions of the present disclosure may comprise classical pharmaceutical formulations. Administration of these compositions according to the present disclosure can be by any common route, so long as the target tissue is accessible by that route, but generally includes systemic administration. This includes oral, nasal or buccal (buccal). Alternatively, administration can be by intradermal, subcutaneous, intramuscular, intraperitoneal, or intravenous injection, or by intratumoral or tumoral area (e.g., in the tumor vasculature). As noted above, such compositions are typically administered as pharmaceutically acceptable compositions.
The active compounds can also be administered parenterally or intraperitoneally. By way of illustration, solutions of the active compounds as free bases or pharmacologically acceptable salts may be prepared in water suitably mixed with a surfactant, such as hydroxypropylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof, and in oils. Under ordinary conditions of storage and use, these preparations usually contain a preservative to prevent the growth of microorganisms.
The pharmaceutical forms suitable for injectable use include, for example, sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. Typically, these preparations are sterile and present as a fluid to the extent that easy syringability is achieved. The preparation should be stable under the conditions of manufacture and storage and should be preserved against the contaminating action of microorganisms, such as bacteria and fungi. Suitable solvents or dispersion media may include, for example, water, ethanol, polyols (e.g., glycerol, propylene glycol, and liquid polyethylene glycols, and the like), suitable mixtures thereof, and vegetable oils. For example, proper fluidity can be maintained, for example, by the use of a coating (e.g., lecithin), by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants. Prevention of the action of microorganisms can be achieved by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like. In many cases, it will be preferred to include isotonic agents, for example sugars or sodium chloride. Prolonged absorption of the injectable compositions can be brought about by the use in the compositions of agents delaying absorption, for example, aluminum monostearate and gelatin.
Sterile injectable solutions can be prepared by incorporating the active compound in the appropriate amount in a solvent with any other ingredient desired (e.g., as set forth above) followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterile active ingredients into a sterile vehicle which contains a base dispersion medium and the desired other ingredients (for example, as set forth above). In the case of sterile powders for the preparation of sterile injectable solutions, preferred methods of preparation include vacuum drying and freeze-drying techniques which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
The compositions of the present disclosure may be formulated in neutral or salt form. Pharmaceutically acceptable salts include, for example, acid addition salts (formed with free amino groups of the protein) derived from inorganic acids (e.g., hydrochloric or phosphoric acids), or from organic acids (e.g., acetic, oxalic, tartaric, mandelic, and the like). Salts formed with the free carboxyl groups of proteins can also be derived from inorganic bases (e.g., sodium hydroxide, potassium hydroxide, ammonium hydroxide, calcium hydroxide, or ferric hydroxide) or from organic bases (e.g., isopropylamine, trimethylamine, histidine, procaine, and the like).
At the time of formulation, the solution is preferably administered in a manner compatible with the dosage formulation and in a therapeutically effective amount. The formulations can be readily administered in a variety of dosage forms, such as injectable solutions, drug-releasing capsules, and the like. For parenteral administration in aqueous solution, for example, the solution is typically suitably buffered and the liquid diluent is first made isotonic, for example, with sufficient saline or glucose. Such aqueous solutions may be used, for example, for intravenous, intramuscular, subcutaneous, and intraperitoneal administration. Preferably, sterile aqueous media known to those skilled in the art are used, particularly in light of the present disclosure. By way of illustration, a single dose may be dissolved in 1ml of isotonic NaCl solution and added to 1000ml of subcutaneous infusion fluid or injected at the recommended infusion site (see, e.g., "Remington's Pharmaceutical Sciences" 15 th edition, pages 1035 to 1038, and pages 1570 to 1580). Some variation in dosage will necessarily occur depending upon the condition of the subject being treated. In any event, the person responsible for administration will determine the appropriate dosage for the individual subject. In addition, for human administration, the formulations should meet sterility, pyrogenicity, general safety and purity standards as required by FDA office of biologies standards.
Combination therapy
In the context of the present disclosure, 6-thio-dG/anti-PD-L1 (e.g., astuzumab) or 6-thio-dG/anti-PD-1 (e.g., altertilizumab) is also contemplated ) Or anti-CTAL-4 may be used in combination with a chemical or radiation therapeutic intervention or other treatment. It may also prove effective, in particular combining 6-thio-dG, anti-PD-L1, anti-PD-1 or anti-CTLA-4 with other therapies directed against different aspects of cancer cell function.
) Or anti-CTAL-4 may be used in combination with a chemical or radiation therapeutic intervention or other treatment. It may also prove effective, in particular combining 6-thio-dG, anti-PD-L1, anti-PD-1 or anti-CTLA-4 with other therapies directed against different aspects of cancer cell function.
To kill cells, inhibit cell growth, inhibit metastasis, inhibit angiogenesis, or otherwise reverse or reduce the malignant phenotype of tumor cells using the methods and compositions of the present disclosure, a "target" cell is typically contacted with 6-thio-dG and at least one other agent. These compositions will be provided in a sequence or in a combined amount effective to kill or inhibit cell proliferation. The process may involve contacting the cells with 6-thio-dG/anti-PD-L1, anti-PD-1 or anti-CTLA-4 and other agents or factors simultaneously. This can be accomplished by contacting the cell with a single composition or pharmacological agent comprising both agents, or by contacting the cell with two different compositions or agents simultaneously, wherein one composition comprises an interferon prodrug according to the present disclosure and the other comprises the other agent.
Alternatively, 6-thio-dG/anti-PD-L1, anti-PD-1 or anti-CTLA-4 treatment may be administered before or after the other agent treatment at intervals ranging from minutes to weeks. In some embodiments, where the additional agent and interferon prodrug are applied separately to the cell, it should generally be ensured that no significant period of time between each delivery expires, such that the agent and expression construct are still capable of exerting a favorable combined effect on the cell. In this case, it is contemplated that the cells are contacted with both forms within about 12 to 24 hours of each other, and more preferably within about 6 to 12 hours of each other, and a delay time of only about 12 hours is most preferred. In some cases, it may be desirable to significantly extend the treatment period, however, days (2, 3, 4, 5, 6, or 7 days) to weeks (1, 2, 3, 4, 5, 6, 7, or 8 weeks) are spaced between administrations.
It is also contemplated that more than one administration of an interferon prodrug or another agent is desired. Various combinations may be used, where the 6-thio-dG/anti-PD-L1, anti-PD-1 or anti-CTLA-4 treatment is "a" and the other treatment is "B", as shown below:

other combinations are contemplated. Again, to achieve cell killing, the two agents are delivered to the cells in a combined amount effective to kill the cells.
Agents or factors suitable for cancer treatment include any chemical compound or therapeutic method that induces DNA damage when applied to a cell. Such agents and factors include radiation and waves that induce DNA damage, such as radiation, microwaves, electron emissions, and the like. A variety of chemical compounds may be used, which are also described as "chemotherapeutic agents" or "genotoxic agents". This can be achieved by irradiating the local tumor site; alternatively, the tumor cells can be contacted with the agent by administering a therapeutically effective amount of the pharmaceutical composition to the subject.
Various classes of chemotherapeutic agents are contemplated for use with the present disclosure. Imetelstat is discussed below. Other chemotherapeutic agents include selective estrogen receptor antagonists ("SERMs"), such as tamoxifen, 4-hydroxytamoxifene (Afimoxfene), fulvestrant (Falsodex), raloxifene (Raloxifene), bazedoxifene (Bazedoxifene), clomiphene (Clomifene), femarelle, lasofoxifene (Lasofoxifene), oximexifene (Ormeloxifene), and Toremifene (Tormedifene). The medicaments camptothecin, actinomycin-D and mitomycin C are commonly used chemotherapeutic drugs. The present disclosure also contemplates the use of a combination of one or more DNA damaging agents (whether radiation-based or authentic compounds), for example, the use of X-rays with cisplatin or the use of cisplatin with etoposide. The agents may be prepared and used as a combined therapeutic composition.
Agents that directly cross-link DNA or form adducts are also contemplated. Agents such as cisplatin and other DNA alkylating agents can be used. Cisplatin has been widely used in the treatment of cancer, and the effective dose used in clinical practice is 20mg/m every three weeks 2 5 days, three treatment courses total. Cisplatin is not absorbed orally and therefore must be delivered by intravenous, subcutaneous, intratumoral or intraperitoneal injection.
Agents that damage DNA also include compounds that interfere with DNA replication, mitosis, and chromosome segregation. Such chemotherapeutic compounds include doxorubicin(adriamycin) (also known as doxorubicin (doxorubicin)), etoposide, verapamil (verapamil), podophyllotoxin (podophyllotoxin), and the like. When widely used in clinical settings for the treatment of neoplasms, these compounds are administered by intravenous bolus injection (bolus injection) at doses of 25 to 75mg/m every 21 days for doxorubicin 2 For etoposide 35 to 50mg/m intravenously 2 Or 2 times more orally than intravenously. Microtubule inhibitors, such as taxanes, are also contemplated. These molecules are diterpenes produced by plants of the genus Taxus (Taxus) and include paclitaxel and docetaxel.
Epidermal growth factor receptor inhibitors, such as Iressa (Iressa), the mammalian target of rapamycin mTOR (also known as FK506-binding protein 12-rapamycin associated protein 1 (FK 506-binding protein 12-rapamycin associated protein 1, frap 1)), are serine/threonine protein kinases that regulate cell growth, cell proliferation, cell motility, cell survival, protein synthesis and transcription. Thus, in accordance with the present disclosure, rapamycin and its analogs ("rapalogs") are contemplated for use in cancer therapy. Another EGFR inhibitor particularly useful herein is Gefitinib (Gefitinib).
Another possible treatment is TNF-alpha (tumor necrosis factor-alpha), a cytokine involved in systemic inflammation and a member of the group of cytokines that stimulate the acute phase response. The primary role of TNF is to regulate immune cells. TNF also induces apoptotic cell death, induces inflammation, and inhibits tumorigenesis and viral replication.
Agents that disrupt the synthesis and fidelity of nucleic acid precursors and subunits also cause DNA damage. Thus, many nucleic acid precursors have been developed. Particularly useful are agents that undergo extensive testing and are readily available. Thus, agents such as 5-fluorouracil (5-FU) are preferentially used by neoplastic tissue, making such agents particularly useful for targeting neoplastic cells. Although very toxic, 5-FU is applicable in a variety of vehicles (including surfaces), but is commonly administered intravenously at a dose of 3 to 15 mg/kg/day.
Other factors that cause DNA damage and have been widely used include gamma-rays, x-rays and/or the delivery of radioisotopes directly to tumor cells, which are commonly known. Other forms of DNA damage factors, such as microwaves and UV irradiation, are also contemplated. Most likely all of these factors cause extensive damage to DNA, DNA precursors, DNA replication and repair, and chromosome assembly and maintenance. The dose of x-rays ranges from a daily dose of 50 to 200 roentgens for an extended period of time (3 to 4 weeks) to a single dose of 2000 to 6000 roentgens. The dosage range of radioisotopes varies widely, and depends on the half-life of the isotope, the intensity and type of radiation emitted, and the uptake by neoplastic cells.
In addition, it is contemplated that different immunotherapies, hormonal treatments, toxin treatments, and/or surgeries may be used.
The skilled worker is guided by Remington's Pharmaceutical Sciences, 15 th edition, chapter 33, in particular pages 624 to 652. Some variation in dosage will necessarily occur depending on the condition of the subject being treated. In any event, the person responsible for administration will determine the appropriate dosage for the individual subject. In addition, for human administration, the formulations should meet sterility, pyrogenicity, general safety and purity standards as required by FDA office of biologies standards.
V. examples
Further details regarding examples of various embodiments are provided in the examples section below. It should be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques and/or compositions discovered by the inventors to function well. However, those of skill in the art should, in light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the present disclosure. These embodiments are illustrative of the methods and systems described herein and are not intended to limit the scope of the present disclosure. Such non-limiting examples include, but are not limited to, those given below.
Example 1 materials and methods
A mouse. In a C57BL/6J backgroundFemale C57BL/6J, BALB/C, myd88-/-, tmem173-/-, batf 3-/-and OT-1CD8+ T cell recipient transgenic mice and NSG-SMG3 mice were purchased from Jackson Laboratory (The Jackson Laboratory). Rag 1-/-mice and IFN reporter mice on C57BL/6 background (Ifng) tm3.1Lky /J) purchased from UT southwest mouse breeding center (UT southwest mouse breeding core). Ifn α r 1-/-mice were supplied by doctor Anita Chong from the University of Chicago (University of Chicago). All mice were maintained under specific pathogen free conditions. Animal care and experiments were performed according to the protocols and guidelines of the institutional and National Institutes of Health. The study was approved by the Institutional Animal Care and Use Committee (Institutional Animal Care and Use Committee) of the southwest Medical Center of Texas University (University of Texas south Medical Center).
Cell lines and reagents. MC38, CT26, LLC A375, and HCT116 cells were purchased from ATCC. MC38-OVA cells were prepared by lentiviral transduction of OVA genes. Mycoplasma contamination kit (Mycoplasma con-termination kit, R) was used for all cell lines&D) Routinely tested and cultured in Dulbecco's modified Eagle's Medium supplemented with 10% Heat-inactivated fetal bovine serum, 100U/ml penicillin and 100U/ml streptomycin at 5% CO 2 The cells were cultured at 37 ℃.
anti-CD 4 (GK 1.5), anti-NK 1.1 (PK 136), anti-CD 8 (53-5.8), and anti-CSF 1R (AFS 98) mAbs were purchased from Bioxell. anti-PD-L1 (Attributab) and anti-CTLA-4 (Epitumumab) were kindly provided by UT Southwestern Simmons Cancer Center Pharmacy (UT south western medicines Center Pharmacy). 6-thio-dG was purchased from Metkinen Oy. For in vitro studies, 6-thio-dG was dissolved in DMSO/water (1. For in vivo studies, 3mg/kg 6-thio-dG was prepared in 5% DMSO (in 1 XPBS) for intraperitoneal injection. The drug was kept frozen at-20 ℃ until use.
Cell viability assay. To determine IC using a cell proliferation assay 50 Murine and human cancer cell lines were screened with a 2-fold dilution series of 6-thio-dG in 8 different spots in a 96-well plate. Cells were plated 24 hours prior to drug addition, incubated for 4 to 5 days, and allowed to incubate according to the manufacturer's instructions (Promega)Using CellTiter 96 Aqueous single solution cell proliferation assay (CellTiter 96)
Aqueous single solution cell proliferation assay (CellTiter 96) Aqueous One Solution Cell Proliferation Assay). The number of cells per well is 1,000 to 10,000 cells per well, inversely proportional to doubling time. Dose response curves were generated and IC was calculated using Graphpad Prism 50 . All samples were analyzed in triplicate and standard deviations were from 2 to 3 independent experiments.
Aqueous One Solution Cell Proliferation Assay). The number of cells per well is 1,000 to 10,000 cells per well, inversely proportional to doubling time. Dose response curves were generated and IC was calculated using Graphpad Prism 50 . All samples were analyzed in triplicate and standard deviations were from 2 to 3 independent experiments.
Colony formation assay. MC38 cells were seeded at three different concentrations on six-well plates (1000 to 4000 cells/well) and treated with different drug concentrations every 3 to 4 days. After 13 days of treatment, cells were fixed and stained with 6% glutaraldehyde (Fisher Scientific) plus 0.5% crystal violet (Sigma) solution. After washing with tap water, the cells were air-dried and images were captured using G-BOX (Syngene, model: G-BOX F3).
Telomere dysfunction induced foci (TIF) and micronucleus assays. TIF assays use telomere sequence specific Peptide Nucleic Acid (PNA) probes based on the co-localization detection of DNA damage by antibodies to DNA damage response factors (e.g. γ -H2AX, 53BP 1) and antibodies to telomeres or telomeres (Mender and Shay, 2015). Briefly, cells were seeded into 4-well chamber slides. The following day, cells were treated with 1 μ M6-thio-dG for 24 hours (for TIF assay) or 1 to 3 μ M6-thio-dG for 48 hours (for micronucleus assay). Slides were then washed twice with PBS and fixed in 4% formaldehyde in PBS (Thermo Fisher) for 10 minutes. Cells were then washed twice with PBS and permeabilized in 0.5% Triton X-100 in PBS for 10 minutes. After permeabilization, cells were washed 3 times with PBS. Cells were blocked with 10% goat serum in 0.1% -pbst (triton x-100) for 1 hour. γ -H2AX (TIF assay, mouse, 1 1000) (Millipore) or lamin a/C (micronucleus assay, mouse, 1, 500) (Santa Cruz) was diluted in blocking solution and incubated on cells for 2 hours. In PBST (0.1% Tr)1 × PBS in iton) and washed 3 times with PBS, the cells were incubated with alexaflur 568-conjugated goat anti-mouse (1. Cells were fixed in 4% formaldehyde in PBS for 20 min at RT. Slides were dehydrated sequentially with 70%, 90%, 100% ethanol, then denatured with hybridization buffer containing the following on a heat block at 80 ℃ for 7 minutes, followed by overnight incubation at RT: FAM-conjugated telomere sequence (C-rich) specific PNA probe, 70% formamide, 30%2 XSSC, 10% (w/v) MgCl 2 .6H 2 O (Fisher Sci), 0.25% (w/v) blocking reagent for nucleic acid hybridization and detection (Roche). Slides were washed sequentially with 70% formamide (Ambion)/0.6 × SSC (Invitrogen) (2 × 1 hour), 2 × SSC (1 × 15 minutes), PBS (1 × 5 minutes), dehydrated sequentially with 70%, 90%, 100% ethanol, and then mounted with Vectashield mounting medium with DAPI (Vector Laboratory) (mount). Images were captured with a fluorescence microscope using a 100 x objective. TIF was quantified using Image J.
Detection of DNA in bone marrow-derived dendritic cells. Cells were labeled with EdU as described previously (Min et al, 2019). Briefly, 100,000 MC38 cells were seeded into 6-well plates and labeled with 25 μ M EdU. After two days, cells were washed and treated with 1. Mu.M 6-thio-dG for 24 hours. Cells were washed again and co-cultured with BMDCs overnight. The next day, the DCs were sorted with magnetic beads, washed, fixed and centrifuged on the cells. Then in fresh home-made EdU staining solution (containing 1mM CuSO) 4 2mM ascorbic acid in PBS) were stained with 6-carboxytetramethylrhodamine fluorescent azide (Invitrogen) for 30 minutes. Slides were then washed vigorously with PBS for at least 1 hour, and then telomere FISH steps were performed using FAM-TelG probe as described in the "telomere dysfunction induced foci (TIF) and micronucleus assay" methods section. Images were captured at 63 × magnification with an Axio Imager Z2 equipped with an automatic mid-capture system (Coolcube 1 camera) and analyzed with ISIS software (Metasystems).
Immune FISH. Briefly, 5 μ M tissue sections were treated with xylene (2 × 5 min), 100% ethanol (2 × 2 min), 95% ethanol (1 × 2 min), 75% ethanolThe alcohols (1 × 2 min) and 50% ethanol (1 × 2 min) were dewaxed and then washed with tap water (2 × 3 min). Deparaffinized tissue sections were incubated in sodium citrate buffer (10 mM sodium citrate, 0.05% tween 20, ph = 6.0) for 20 minutes under microwaves to recover the antigen. After the tissue sections were allowed to cool, they were rinsed with 1 × PBS for 5 minutes and then dehydrated in 95% ethanol for 3 minutes. With telomere sequence containing FITC conjugation (TTAGGG) 3 Hybridization buffer for specific PNA probes (70% formamide, 30%2 XSSC, 10% (w/v) MgCl 2 .6H 2 O (Fisher Sci), 0.25% (w/v) blocking reagent (Roche)) was denatured at 80 ℃ for 7 min on a heat block. Slides were washed sequentially with 70% formamide/0.6 × SSC (3 × 15 min), 2 × SSC (1 × 15 min), PBS (1 × 5 min), PBST (PBS +0.1% tween 20 for 1 × 5 min) and incubated with blocking buffer (4% BSA in PBST) for 30 min. Sections were incubated with the phosphohistone H2AX antibody (1. After washing with PBST for 2 × 5 min, tissue sections were incubated for 1 hour at RT in blocking buffer with alexaflur 568-conjugated goat anti-rabbit. Sections were washed sequentially with PBST (3 × 5 min) and PBS (1 × 5 min). Slides were mounted with Vectashield mounting medium with DAPI. Images were captured with a fluorescein microscope using a 100 x objective. TIF was quantified using Image J.
Tumor growth and treatment. A total of 5X 10 in 100. Mu.L of Phosphate Buffered Saline (PBS) was added 5 MC38, 5 × 10 5 CT26 or 1X 10 6 One LLC cell was inoculated subcutaneously into the right dorsal side of the mice. When the tumor grows to about 100mm 3 Tumor-bearing mice were randomly divided into treatment groups. For 6-thio-dG single treatment, 3mg/kg 6-thio-dG was administered intraperitoneally on days 7, 8 and 9 in MC38 tumors and LLC tumors and on days 5, 6, 7 for CT26 tumors. For CSF1R, NK1.1, CD4+ and CD8+ T cell depletion, 200 μ g of antibody was injected intraperitoneally 1 day before treatment initiation and then twice weekly for 2 weeks. For PD-L1 blocking combination therapy in the MC38 model, 6-thio-dG was administered on days 10 and 11 and 50 μ g PD-L1 was injected intraperitoneally on days 13 and 17. For PD-L1 resistance in LLC modelCombination therapy was discontinued with 6-thio-dG administered on days 4, 5, 6, 10 and 11 and intraperitoneal injections of 200 μ g PD-L1 on days 8 and 13. Tumor volume was measured by length (a), width (b) and height (h) and calculated as tumor volume = abh/2.
Humanized mouse tumor model. Humanized mouse recombination was previously described (Qiao et al, 2019). Briefly, 4-week-old NSG-SGM3 female mice were irradiated with 100cGy (X-ray irradiation using an X-RAD320 irradiator) one day prior to human CD34+ cell transfer. Cord blood was obtained from UT southwest Parkland Hospital (UT Southwestern Parkland Hospital). By density gradient centrifugation ( Paque Plus, GE Healthcare) human CD34+ cells were purified from cord blood, followed by positive immunomagnetic selection with anti-human CD34 microbeads (stemcells).
Paque Plus, GE Healthcare) human CD34+ cells were purified from cord blood, followed by positive immunomagnetic selection with anti-human CD34 microbeads (stemcells). 1X 10 5 Individual CD34+ cells were injected intravenously into each recipient mouse. Humanized mice with over 50% human CD45+ cell reconstitution and age and sex matched non-humanized mice were inoculated subcutaneously on the right flank with 1 x10 cells 12 weeks after transplantation 6 And HCT116 tumor cells. 3mg/kg 6-thio-dG was administered intraperitoneally on days 7, 8, and 9. Tumor volumes were measured twice weekly. The experiments were performed according to the UTSW Human Investigation Committee protocol (UTSW Human Investigation Committee protocol) and the UTSW Institutional Animal Care and Use Committee (UTSW Institutional Animal Care and Use Committee).
Tmem173 and Mb21d1 knock-out MC38 cell lines. Tmem 73 and Mb21d1 genes were knocked out in MC38 cells by CRISPR/Cas9 technology. The guide sequence 5 'CACCTAGCCTCGCACGAACT-3' (SEQ ID NO: 1) of Tmem173 and 5 'CGCAAAGGGGCTCGATCG-3' (SEQ ID NO: 2) of Mb21dl were cloned into the px458 plasmid (non-integrating plasmid with GFP selection marker) and subsequently transiently transfected into tumor cells using lipofectamine 2000 (Thermo Fisher). After 24 hours, GFP positive cells were sorted and cultured for another week. The sorted cells were then seeded into 96-well plates. After another week, GFP-negative clones were transferred to 12-well plates and western blots were performed to identify knockout clones. Finally, all knockout clones were pooled together for experiments.
IFN-. Gamma.ELISA spot assay (enzyme-linked immunosorbent assay, ELISPOT). MC38 tumors were injected subepithelially in the right flank of C57 BL/6. For 6-thio-dG single treatment, 3mg/kg 6-thio-dG was given intraperitoneally on days 7, 8, and 9; for the PD-L1 blocking combination therapy in the MC38 model, 3mg/kg 6-thio-dG was administered on days 10 and 11, and 50 μ g PD-L1 was injected intraperitoneally on day 11. Tumor draining lymph and spleen from tumor bearing mice were collected 7 days after the last treatment and single cell suspensions were prepared. Irradiated MC38 and control LLC tumor cells can be used to restimulate tumor-specific T cells. Mixing 1.5X 10 5 A draining lymph node cell or spleen cell and 7.5 × 10 4 The irradiated tumor cells were co-cultured for 48 hours and an ELISPOT assay was performed using IFN- γ ELISPOT kit (BD Bioscience) according to the manufacturer's instructions. Using CTL-ImmunoSpot The IFN-. Gamma.spots were counted by an S6 analyzer (Cellular Technology Limited).
The IFN-. Gamma.spots were counted by an S6 analyzer (Cellular Technology Limited).
In vitro co-culture of Bone Marrow Dendritic Cells (BMDCs) and T cells. Single cell suspensions of Bone Marrow (BM) cells were collected from the tibia and femur of C57BL/6 mice. BM cells were plated on 10cm dishes and cultured in complete RPMI 1640 medium containing 20ng/mL recombinant mouse GM-CSF (BioLegend). Fresh media was added to the culture on days 3 and 6. BMDCs were harvested on day 7. CD8+ T cells were isolated from lymph nodes and spleen of OT-1 transgenic mice using a negative CD8+ T cell isolation kit (Stemcell). MC38-OVA cells were pretreated with 200nM 6-thio-dG for 4 hours. The drug was then washed away and the tumor cells were cultured for an additional 72 hours and harvested on the same day as the BMDC harvest. MC38-OVA cells were then co-cultured with BMDCs overnight. The supernatant was collected for IFN- β ELISA testing (PBL). BMDCs were sorted with CD11c + positive selection kit (Stemcell) and co-cultured with OT-1CD8+ T cells for 48 hours. Supernatants were collected and IFN-. Gamma.was measured by flow bead array assay (BD Biosciences).
CytoplasmDNA extraction and quantitative real-time PCR. HCT116 cells were pretreated with 500nM 6-thio-dG for 4 hours. The drug was then washed away and the tumor cells were cultured for an additional 72 hours and harvested on the same day as the BMDC harvest. HCT116 cells were then incubated with 1X 10 6 Mix for 4 hours with BMDC 1. BMDC was purified and divided into two equal aliquots. One aliquot of total genomic DNA was extracted with Purelink genomic DNA kit (Invitrogen) and used as a normalization control. Another aliquot was resuspended in 100. Mu.L of cytoplasmic extraction buffer containing 150mM NaCl, 50mM HEPES and 25mg/mL digitonin (Sigma) and incubated for 10 min at RT for plasma membrane permeabilization (West et al, 2015). The cells were then centrifuged to pellet the intact cells. The cytosolic supernatant was collected and centrifuged at 12000g for 10 min to pellet the remaining cell debris. Cytoplasmic DNA was then extracted using the Purelink genomic DNA kit (Invitrogen). Quantitative PCR was performed on whole cell extracts and cytoplasmic fractions using human and mouse DNA primers (Xu et al, 2017).
And (5) digesting the tumor. Tumor tissue was excised and digested with 1mg/mL collagenase I (Sigma) and 0.5mg/mL DNase I (Roche) for 30 minutes at 37 deg.C, then the tumor was passed through a 70 μm cell filter to remove large undigested tumor masses. Tumor-infiltrating cells were washed twice with PBS containing 2mM EDTA.
Flow cytometry analysis. Single cell suspensions of cells were incubated with anti-Fc γ III/II receptor (clone 2.4g2) for 15 min to block non-specific binding, then stained with conjugated antibodies, and subsequently incubated with the indicated antibodies in the dark for 30 min at 4 ℃. Dead cells were excluded using the immobilizable viability dye, either eFlour 506 or eFlour 780 (eBioscience). Intracellular staining was performed for Foxp3 and Ki67 by using eukaryotic transcription factor buffer set (BioLegend) according to the manufacturer's instructions. Data were collected on a CytoFLEX flow cytometer (Beckman Coulter, inc) and analyzed by using FlowJo (Tree Star Inc., ashland, OR) software.
Quantitative real-time PCR. SsoAdvanced was used with different primer sets according to the manufacturer's instructions as follows TM Universal  Green Supermix (Bio-Rad) performed real-time PCR: (human MT-CO1, forward primer 5 'CGCCACACTCCACGGAAGCA-3' (SEQ ID NO: 3), reverse primer 5 'CGGGGCATTCCG GATAGGCC-3' (SEQ ID NO: 4); human 18s rRNA, forward primer 5 'ACCGAATTGGATGGTTTAGTGAG-3' (SEQ ID NO: 5), reverse primer 5 'CCTACGGAAACCTTACGAC-3' (SEQ ID NO: 6); mouse IFN- β, forward primer 5 'ATGAGTGGTTGGTTGCAGGC-3' (SEQ ID NO: 7), reverse primer 5 'TGATTCAAAGTAGATTCA-3' (SEQ ID NO: 8); mouse PDH,
Green Supermix (Bio-Rad) performed real-time PCR: (human MT-CO1, forward primer 5 'CGCCACACTCCACGGAAGCA-3' (SEQ ID NO: 3), reverse primer 5 'CGGGGCATTCCG GATAGGCC-3' (SEQ ID NO: 4); human 18s rRNA, forward primer 5 'ACCGAATTGGATGGTTTAGTGAG-3' (SEQ ID NO: 5), reverse primer 5 'CCTACGGAAACCTTACGAC-3' (SEQ ID NO: 6); mouse IFN- β, forward primer 5 'ATGAGTGGTTGGTTGCAGGC-3' (SEQ ID NO: 7), reverse primer 5 'TGATTCAAAGTAGATTCA-3' (SEQ ID NO: 8); mouse PDH, forward primer 5 GAAGGGTGAGTGAGGTGCAGTCA-3 '(SEQ ID NO: 9) and reverse primer AGGTAGCTGCAGT-10 AGCTGCT-3); mouse PDH-5 GAAGGTAGGTAGTGAGT-5 GAAGGTAGGTAGGTAGCTGCAGCTGAAGC-3' (SEQ ID NO: 10) were used as the internal control. 2 -ΔΔCt The method was used to calculate relative expression changes.
And (4) performing immunoblotting. BMDC and MC38 treatment were identical to "in vitro co-culture of bone marrow dendritic cells". After 6 hours of co-cultivation, DCs were isolated using a CD11c + positive selection kit (Stemcell). Protein sample preparation and immunoblotting procedures were performed as described previously (Liu et al, 2019). Proteins were detected using rabbit monoclonal antibodies against pSTING (Cell signaling, 72971), STING (Cell signaling, 50494), pTBK1 (Cell signaling, 5483), TBK1 (Cell signaling, 3504). The protein loading was determined using an antibody against cyclophilin a (Cell signaling, 2175). Anti-rabbit (1: 2000 in 5% BSA) was used for secondary antibody (Cell signaling, 7074). X-ray film (GeneMate, F-9024-8X 10) was used to develop the film. Clarity Max Western ECL substrate (Biorad, 1705062) or Supersignal West PicoPlus chemiluminescent substrate (Thermoscientific, 34577) were used for chemiluminescent Western blots.
Quantitative and statistical analysis. All data analyses were performed using GraphPad Prism statistical software and are shown as mean ± SEM. P-values were determined by two-way ANOVA for tumor growth or by log rank test for survival or unpaired two-tailed t-test for other analyses. Values of p < 0.05 were considered statistically significant.
Example 2 results
The therapeutic effect of 6-thio-dG is dependent on CD8+ T cells. All previous xenograft model studies have shownTumor growth in many tumor models can be partially controlled with 6-thio-dG intensive treatment for more than 10 days (Mender et al, 2015a. However, the potential role of this drug in the interaction between the tumor and the adaptive immune system is not clear. To explore whether 6-thio-dG induces telomere-based DNA perception for T cell responses, the present inventors first determined the inhibition of cell viability of telomerase positive murine colon cancer cells (MC 38) in an immunocompetent host by 6-thio-dG. MC38 tumor cells are sensitive to 6-thio-dG, IC 50 The concentration was 370nM (FIG. 1A). The sensitivity of 6-thio-dG in MC38 cells was also determined by a separate colony formation assay. MC38 cells were treated with 6-thio-dG every three days for 13 days, resulting in less than 50% cell colony formation at 0.5 μ M6-thio-dG treatment (fig. 1B and 1C). To evaluate whether 6-thio-dG reduced tumor burden in an in vivo syngeneic mouse model, the inventors inoculated MC38 cells subcutaneously into immunocompetent wild-type (WT) C57BL/6 mice. 7 days after tumor inoculation (when the tumor volume is about 100 mm) 3 Time), 3mg/kg of 6-thio-dG was administered daily for only three days, and tumor growth was significantly reduced compared to control tumors (fig. 1D). This is not a unique response to the MC38 tumor model, as the inventors also observed inhibition of cell viability in vitro and significant tumor growth delay in vivo in tumor models with only three days of treatment of telomerase positive LLC (lewis lung mouse carcinoma derived from C57BL/6 mice) and CT26 (colonic mouse carcinoma derived from BALB/C mice) (fig. 9A to 9D).
Since the present inventors given such a short duration of treatment with 6-thio-dG compared to the intensive dosing strategy (5 mg/kg daily for two weeks) in the xenograft model, and achieved a better antitumor effect in the syngeneic mouse model, it was speculated that 6-thio-dG could have an immunostimulatory effect in vivo. Therefore, tumors were inoculated in Rag1 knockout mice that were unable to generate mature T and B cells. Indeed, the therapeutic effect of 6-thio-dG was completely impaired (fig. 1E), indicating that adaptive immune cells are required for a large number of tumor controls in vivo. To find out which T cell subset contributed to the 6-thio-dG mediated anti-tumor effect, the inventors depleted CD4+ or CD8+ T cells concurrently with the administration of 6-thio-dG treatment and observed the marginal effect of CD4+ T cell depletion (fig. 1F). However, depletion of CD8+ T cells completely abolished the therapeutic effect of 6-thio-dG (fig. 1G). Together, the data may be interpreted to indicate an important role for CD8+ T cells in 6-thio-dG processing.
Treatment with 6-thio-dG improved the tumor specific T cell response. Since the therapeutic effect of 6-thio-dG is dependent on T cells, the present inventors concluded that 6-thio-dG treatment can alter immune cell expansion in the tumor microenvironment. To test this, the number of Tumor Infiltrating Lymphocytes (TILs) was analyzed 6 days after the last of the three daily doses of 6-thio-dG. The inventors found that the frequency of CD3+ T cells and CD8+ T cells in TILs was increased after 6-thio-dG treatment (FIGS. 2A, 10A and 10B). A significant upregulation of CD8+ T cell proliferation, indicated by increased Ki67 expression, was also observed (fig. 2B), but no significant change in Treg cells was observed (fig. 10C). Although tumor-infiltrating NK cells were also increased, the inventors did not find an effect of NK cell depletion on the therapeutic effect of 6-thio-dG (fig. 10D and 10E). Together with CD8 depletion experiments, this suggests that NK cells are not required in the 6-thio-dG mediated anti-tumor effect, but a CD8+ T cell response is required.
The inventors further tested antigen-specific T cell responses after 6-thio-dG treatment by using an MC38-OVA tumor model that allowed tracking of antigen-specific T cells in tumor tissue. Indeed, an increase in tumor-specific CD8+ T cells was observed in tumors 6 days after 6-thio-dG treatment (fig. 2C). Enhanced tumor-specific cytotoxic T cell responses were also observed in the MC38 tumor model by measuring IFN- γ producing T cells after 6-thio-dG treatment (fig. 2D and 2E). To directly assess the ability of T cells to produce IFN- γ in vivo, the present inventors utilized IFN- γ YFP reporter mice (Reinhardt et al, 2009) that allowed for the tracking of IFN- γ producing T cells with YFP expression. Treatment with 6-thio-dG significantly increased YFP + T cells in the tumor, indicating increased IFN- γ production capacity of the T cells (fig. 2F and 10F). The hallmark of an adaptive immune response is the development of memory, which initiates a rapid recall response when the same antigen is present. To determine whether 6-thio-dG treatment induced a memory response, mice with fully remitted tumors were allowed to rest for 5 weeks after 6-thio-dG treatment and were challenged again with the same MC38 tumor (but with 10-fold more tumor cells) on the contralateral flank (left flank) and inoculated with LLC tumor cells as a control on the right flank. When the initial mice (never exposed to MC38 cells or 6-thio-dG) were injected with the same number of MC38 cells, the tumors grew invasively. Notably, all mice cured by 6-thio-dG treatment spontaneously rejected a second challenge of MC38 tumor.
Treatment with 6-thio-dG enhances the cross-priming capability of dendritic cells. Cross-presentation of antigens by Antigen Presenting Cells (APCs), such as DCs or macrophages, is responsible for the activation of tumor-specific CD8+ T cells. To explore which APC subpopulations contribute to 6-thio-dG-induced T cell activation, the inventors first used anti-CSF 1R antibodies to deplete macrophages. It was found that 6-thio-dG exerted an even better effect in the macrophage depleted group (fig. 3A), which could be explained by the additive effect of removing immunosuppressive tumor-associated macrophages. Bat 3 (basic leucine zipper ATF-like transcription factor 3) dependent DCs are critical for priming antigen-specific CD8+ T cells (Broz et al, 2014. Treatment of 6-thio-dG in Batf3 deficient mice partially delayed tumor growth, but was significantly less effective than WT mice (fig. 3B). Notably, 60% of WT mice were completely tumor free, but none of the Batf 3-/-mice were tumor free (fig. 3C), suggesting an important role for the therapeutic role of the Batf 3-dependent DC in 6-thio-dG.
To directly demonstrate that 6-thio-dG treatment enhanced the cross-sensitizing capacity of DCs, the inventors co-cultured 6-thio-dG pretreated MC38-OVA tumor cells with bone marrow derived DCs (BMDCs) overnight. The DCs were then purified and recognized for OVA with expression specificity 257 to 264 Initial OT-1 transgenic CD8+ T cell co-culture of TCR of epitopes. A significant increase in IFN-. Gamma.production by CD8+ T cells was observed in the 6-thio-dG treated group (FIG. 3D), indicating that at 6-thio-dG there was a significant increase in IFN-. Gamma.production by CD8+ T cells (see FIG. 3D)After treatment, the cross-sensitization capacity of the DCs was improved. Because IFN-I signaling promotes the cross-sensitizing ability of DCs (Diamond et al, 2011 le Bon et al, 2003, sanchez-paule et al, 2017), the inventors tested IFN- β production by DCs after co-culturing them with 6-thio-dG treated tumor cells. In fact, IFN- β production was significantly increased in the 6-thio-dG-treated group, indicating that intrinsic perception of DC was increased (FIG. 3E). It was also explored whether the IFN-I pathway is essential for 6-thio-dG mediated anti-tumor effects. Using Ifnar 1-/-mice, the inventors showed that loss of IFN-I signaling in the host abrogated the anti-tumor effect of 6-thio-dG (FIG. 3F), suggesting an essential role for IFN-I signaling in 6-thio-dG therapy.
STING signaling in the host is required for the intrinsic perception of 6-thio-dG induction. Tumor cells under stress may release a danger-associated molecular pattern (DAMP) to participate in the TLR/Myd88 pathway in APC and initiate IFN-I signaling. Tumor-derived DNA can also trigger cytoplasmic DNA to sense the cGAS/STING pathway and activate the IFN-I pathway (Deng et al, 2014 li et al, 2019. To further describe which upstream pathway in 6-thio-dG triggered activation of IFN-I signaling is essential in host cells, the inventors inoculated MC38 tumors into Myd 88-/-and Tmem173-/- (Tmem 173 encodes STING) mice. 6-thio-dG treatment controlled tumor growth well in Myd 88-/-mice, but lost potency completely in Tmem 173-/-mice (FIGS. 4A and 4B), suggesting a critical role for host STING signaling in the intrinsic perception of 6-thio-dG triggering. It was also explored whether 6-thio-dG treatment activated the host STING/IFN-I pathway. Increased TBK1 phosphorylation in DCs and complete decrease in phosphorylation in Tmem173DC was observed after co-culture with 6-thio-dG pretreated tumor cells (figure 11A). 6-thio-dG treatment induced IFN- β production in DCs in a STING-dependent manner (FIG. 11B). Since previous studies reported that tumor-intrinsic STING signaling is critical in intrinsic perception-induced cancer treatment (Sen et al, 2019, vanouille-Box et al, 2017), the present inventors tested whether tumor-intrinsic STING signaling also contributes to 6-thio-dG therapeutic efficacy. The CRISPR/Cas9 was used to knock out Tmem173 and Mb21d1 (Mb 21d1 encodes cGAS) in MC38 tumor cells. In contrast to other studies, tumor-intrinsic STING signaling played an unnecessary role, as 6-thio-dG treatment still controlled tumor growth in mice bearing Tmem173KO and Mb21D1KO tumor cells (fig. 4C and 4D).
The inventors then attempted to determine how 6-thio-dG treated tumor cells trigger intrinsic perception in DCs. Since 6-thio-dG is a telomere targeting drug, 6-thio-dG induced telomere stress can contribute to the intrinsic perception of DCs by releasing DNA. Therefore, the present inventors first analyzed telomere stress by TIF (telomere dysfunction-induced foci) assay and showed that 6-thio-dG induced telomere damage in MC38 cells (fig. 4E and 4F). Since telomeres are only a small fraction of genomic DNA (about 1/6000), any co-localization of telomeres with DNA damage is significant. Similar increases in TIF were also observed in 6-thio-dG treated tumor tissues from MC38 tumor-bearing mice (fig. 11C and 11D). 6-thio-dG also induced an interval bridge between two daughter cells during telophase, and thus many daughter cells contained telomere sequences, which may explain why many telomere-containing micronuclei signaled when the cells re-entered the interval after mitosis (FIG. 11E). These cytoplasmic fragments form micronuclei with fragile nuclear envelopes (FIGS. 11E and 11F), which can eventually be recognized as danger signals. These DNA fragments are released from the cells and can be taken up by the DCs.
To confirm this hypothesis, the inventors treated HCT116 (human colon cancer cell line) with 6-thio-dG and co-cultured it with mouse BMDCs for 4 hours, and then isolated DCs and extracted cytoplasmic DNA. Short-time co-culture of human tumor cell lines with mouse BMDCs allowed differentiation between DNA of different origins. Human DNA (MT-CO 1 and human 18S) was found to be elevated in the mouse DC cytosol after 6-thio-dG treatment, indicating that DNA from the tumor entered the host DC (FIG. 4G). To determine whether 6-thio-dG treatment increased uptake of unique telomeric DNA by DCs, the inventors labeled tumor cells with EdU and then washed the cells. Next, the cells were treated with 6-thio-dG and then washed again. Finally, tumor cells were co-administered with DCsCultured, and then DCs were isolated for analysis. Having tumor DNA (EdU) in the cytosol + DC), the inventors observed an increase in co-localization of telomeres with EdU after 6-thio-dG treatment, indicating a significant uptake of tumor-derived telomeric DNA (fig. 11G and 11H). In summary, the inventors demonstrated that 6-thio-dG triggers intrinsic sensing by activating the host cytoplasmic DNA sensing STING/IFN-I pathway.
6-thio-dG overcomes PD-L1 blockade resistance in advanced tumors. Although the 6-thio-dG treatment activated CD8+ T cells, it also upregulated PD-1 expression in terms of frequency of total CD8+ T cells and on a per cell basis (fig. 5A). PD-1 is a co-inhibitory molecule that limits T cell activation. Increased PD-1 expression may ultimately inhibit cytotoxic CD8+ T cell function following 6-thio-dG treatment. Thus, the inventors conclude that 6-thio-dG in combination with PD-1/PD-L1 blockade can enhance the overall anti-tumor immune response, particularly in advanced tumor environments with a more immunosuppressive microenvironment comprising multiple resistance mechanisms that limit monotherapy efficacy. Since 6-thio-dG single treatment only for relatively small tumor sizes (about 100 mm) 3 ) Effective, therefore for advanced tumor treatment, the inventors reached tumor sizes of 150 to 200mm 3 And subsequently treated with 6-thio-dG and/or anti-PD-L1 therapy. In such advanced cancers, it is difficult to control tumor volume with two daily treatments with 6-thio-dG or by two treatments with anti-PD-L1 (fig. 5B). However, sequential administration of 6-thio-dG and anti-PD-L1 completely inhibited tumor growth (fig. 5B). Notably, only mice in the combination treatment group achieved 100% survival (fig. 5C), indicating a synergistic effect of 6-thio-dG treatment with PD-L1 blockade. In addition, the inventors did not observe any weight loss in mice in the combination treatment group (fig. 12). Tumor-specific T cell responses in draining lymph nodes (dLN) were also analyzed and anti-PD-L1 treatment was found to have little effect on T cell activation in advanced tumors. In contrast, the combination treatment significantly improved IFN- γ production compared to the other groups. The immune response was MC38 tumor specific, as there were few IFN- γ plaques in the control LLC tumor-stimulated groupDots (fig. 5D).
MC38 is known to be an immunogenic tumor model. To test whether combination therapy can also overcome PD-L1 blockade resistance in less immunogenic tumor models, the present inventors employed a mouse LLC tumor model that has been reported to be resistant to PD-L1 blockade (Bullock et al, 2019, li et al, 2017. Consistent with previous reports, monotherapy with anti-PD-L1 had no therapeutic effect (fig. 5E). Notably, the combination of 6-thio-dG with anti-PD-L1 significantly reduced mouse tumor burden, and 40% of mice eventually completely rejected the tumor (fig. 5E). The inventors re-challenged tumor-free mice 6 weeks after tumor regression to examine memory response. All mice treated with the combination spontaneously rejected LLC tumors but not MC38 tumors, indicating a persistent tumor-specific immunological memory (fig. 5F). Based on these results, 6-thio-dG treatment overcome PD-L1 blockade resistance in advanced tumors. This would potentially benefit patients with PD-1/PD-L1 blockade resistance in the clinic.
6-thio-dG reduced the burden of human colon cancer in a humanized mouse model. Previous studies have shown that patients with high TERT (catalytic subunit of telomerase) expression have poor clinical outcomes in a variety of cancers, such as non-small cell lung cancer and B-cell chronic lymphocytic leukemia (Terrin et al, 2007, wang et al, 2002. Thus, the inventors analyzed colorectal adenocarcinoma patients from the TCGA database and found that patients with abnormally high TERT expression had significantly worse overall survival rates than colon cancer patients with low TERT expression (fig. 6A). To directly demonstrate whether 6-thio-dG-induced telomere stress could benefit cancer patients in a more clinically relevant model, the inventors developed a humanized mouse model with NSG-SGM3 mice with human SCF-1, GM-CSF and IL-3 transgene expression, supporting better development of human myeloid cells. Human immune system was reconstituted with human CD34+ Hematopoietic Stem Cells (HSCs) in NSG-SGM3 mice. At 12 weeks after HSC transfer, the humanized mice had an average of more than 60% of human CD45+ cells and more than 20% of human T cells in circulating human CD45+ cells (fig. 13A to 13C). The inventors then sensitized HCT116 (sensitive to 6-thio-dG treatment)Human colon carcinoma cell line of infection, IC 50 0.73 μ M (FIG. 6B)) were inoculated into NSG-SGM3 control mice and humanized NSG-SGM3 mice. Prior to the start of treatment, the control group of humanized mice had a similar human immune cell composition to the 6-thio-dG treated group (fig. 13B and 13C). After three doses of 6-thio-dG treatment, the immunocompromised mice were not significantly different compared to the control group (fig. 6D). Notably, treatment with 6-thio-dG significantly delayed tumor growth in the humanized mice (fig. 6C and 6E). The inventors then tested the human melanoma cell line a375 sensitive to 6-thio-dG in vitro (fig. 13D). No effect was observed in the immunocompromised NSG-SGM3 mice, as a relatively short time of 6-thio-dG treatment was provided (fig. 13E). Notably, treatment with two doses of 6-thio-dG was found to partially delay tumor growth in humanized mice. In addition, combination with checkpoint blockers further reduced tumor burden, indicating that pretreatment with 6-thio-dG sensitized human tumors to checkpoint blockers (fig. 13F). Given that the humanized mice only partially restored human immunity due to the lack of some immune cells and the limited number of human T cells, it was not unexpected that the inventors observed complete tumor regression.
The inventors have shown (fig. 8A to 8B) that treatment with 6-thio-dG for three days followed by two days followed by treatment with anti-PD-L1 resulted in complete tumor remission of lewis lung cancer (lewis lung cancer). This is a very aggressive tumor type, as shown in fig. 8A. Tumors injected subcutaneously reach over 1000m within 20 days 3 . Tumor growth was observed as fast as treatment with anti-PD-L1 alone. However, only three treatments with 6-THIO-dG (THIO) resulted in significant tumor control for 20 days. Unexpectedly, treatment with THIO followed by anti-PDL-L1 (Attuzumab) resulted in complete tumor regression. The inventors maintained the cured mice for an additional 5 weeks and re-challenged the same mice with five times more LLC tumor, and no tumor growth was observed. However, if mice were cured by injection of LLC with MC38, tumors grew and anti-PD-L1 was unaffected (fig. 8B). In control mice that were never treated with THIO, the growth of control LLC tumors was similar to MC38. This indicates tumor specific immunityAnd (5) epidemic disease memory.
Overall, these data can be interpreted to support that 6-thio-dG induces telomerase-dependent DNA damage and increases uptake of tumor DNA by DCs. Cytoplasmic DNA elevation triggers the DC intrinsic STING/IFN-I pathway, resulting in enhanced cross-priming capability of the DC and subsequent tumor-specific T cell activation. In addition, 6-thio-dG overcomes PD-L1 blocking resistance in advanced tumors. This study identified 6-thio-dG as a new immunostimulatory drug that potentially benefited a clinically broad population of cancer patients.
Example 3 discussion
High telomerase expression in tumor cells is considered a poor prognostic factor for cancer development (Zhang et al, 2018). Here, the inventors report a previously unidentified role of telomerase-dependent telomere-targeted therapy (6-thio-dG) in inducing anti-tumor immune responses in syngeneic colon and lung mouse models and humanized mouse cancer models. This effect is mediated by triggering the cytoplasmic DNA-aware STING/IFN-I pathway in DCs, which ultimately enhances the cross-priming capability of DCs and subsequent tumor-specific T cell activation. This is a prominent finding, as telomerase is a universal tumor marker, and it can potentially be applied to many other telomerase-positive cancers. Furthermore, sequential administration of 6-thio-dG and anti-PD-L1 overcome PD-L1 resistance in PD-L1 blocking resistant tumors, suggesting that combination therapy may benefit PD-L1 resistant patients in the clinic.
The current belief is that 6-thio-dG therapy kills tumor cells primarily by damaging telomeres and inducing DNA damage. This study showed that this drug also relies primarily on DNA perception and T cell response to control tumors. Most previous studies used xenograft models without an intact immune system. In these models, only the tumor intrinsic role or part of the innate immune response can be studied. Although these may be important factors, T cells are essential for long-term tumor control. In addition, most previous studies tended to use high dose or intensive dosing strategies that are more effective in directly killing tumor cells, but actually suppress the immune response due to toxicity to immune cells or non-immunogenic death of tumor cells (Galluzzi et al, 2017 kroemer et al, 2013. In addition, these intensive administration strategies often lead to the emergence of tumor resistance mechanisms. In this study, the inventors used syngeneic mouse models with an intact immune system and humanized mouse models with more clinical relevance to fully evaluate the effect of lower doses and shorter treatment regimens of 6-thio-dG on the host immune response in tumor-bearing mice. This finding that 6-thio-dG is an immunostimulatory drug may allow better combination therapies (including immunotherapy) to be designed to broaden the initial immunity.
More and more studies indicate that tumor DNA-mediated innate perception is crucial for inducing anti-tumor immune responses, and that the STING/IFN I pathway is primarily involved in the initiation of anti-tumor immune responses, but whether host-or tumor-autonomous STING is more important depends on different treatment regimens (Deng et al, 2014 li et al, 2019 qiao et al, 2017 sen et al, 2019 vanouille-Box et al, 2017 woo et al, 2014. This difference may be explained by the relative strength of STING activation of the host and tumor cells, e.g., tumor cells may have STING pathway inhibition or low activity (Xia et al, 2016). The inventors show that the intrinsic perception of 6-thio-dG treatment triggering is host STING signaling dependent, as 6-thio-dG completely loses its potency in Tmem173 deficient mice, but not in Tmem173 deficient tumors. Since STING signaling is active in MC38 tumors, one explanation is that following 6-thio-dG treatment, internal STING is activated but most tumor cells die, and therefore very little type I IFN is produced. Another possibility is that there may be intrinsic mechanisms that limit STING activation in tumor cells, which is still definitively undefined. Recent reports indicate that STING signaling may also be involved in autophagy activation, which is unlikely to contribute to the therapeutic effect of 6-thio-dG (Gui et al, 2019 nassour et al, 2019), as the inventors did not see activation of autophagy in tumor cells after 6-thio-dG treatment (data not shown). In addition, 6-thio-dG in Ifnar1 deficient mice lost efficacy, indicating IFN I signaling is involved. However, STING activation of autophagy is independent of IFN I signaling.
One unique feature of 6-thio-dG, in contrast to general DNA damage induction methods (e.g., radiation therapy or chemotherapy which non-selectively induces DNA damage in all proliferating cells), is that it specifically induces telomere-associated DNA damage in telomerase expressing cells (primarily tumor cells), but does not affect immune cells and other telomerase silencing somatic cells. Importantly, 6-thio-dG can preferentially incorporate de novo synthesized telomeres and cause rapid tumor shrinkage. However, direct telomerase inhibitors act by inhibiting telomerase activity and rely on the progressive shortening of telomeres. In contrast, 6-thio-dG functions rapidly regardless of initial telomere length. This is crucial in reducing toxicity compared to direct telomerase inhibitors (Gryaznov et al, 2007, mender et al, 2015 b). The inventors show that 6-thio-dG induced DNA damage co-localizes significantly with telomeres, indicating the formation of telomere dysfunction induced foci (TIF). Telomeres account for only 1/6000 of genomic DNA, so any TIF is highly significant. In addition, some TIFs are taken up by the DC and further trigger STING-dependent IFN I signaling.
Although checkpoint blockade (especially PD-1/PD-L1 blockade) has overwhelming success, in the clinic, only a few patients respond well. Both primary and adaptive resistance limited the clinical benefit of PD-1/PD-L1 treatment (Chen and Han,2015 gide et al, 2018, zaretsky et al, 2016. The present inventors believe that the lack of proper innate perception may limit T cell activation within the tumor microenvironment, and thus there is an urgent need for combination therapies targeting both innate and adaptive immune cells. PD-L1 blocks reactivation of the adaptive immune response by "releasing the brake", whereas 6-thio-dG induces intrinsic perception by "refueling". The inventors hypothesized that the combination of 6-thio-dG with PD-L1 blockade would enhance the overall anti-tumor immune response. In fact, this study showed that sequential administration of 6-thio-dG and anti-PD-L1 had a synergistic effect in both advanced tumors and PD-L1 block resistant tumors. Further studies should be made for the best combination schemes.
Overall, these results reveal a previously unidentified role of 6-thio-dG (telomerase-dependent telomeres targeting small molecule drugs) in enhancing anti-tumor immune responses. Mechanistically, 6-thio-dG induces telomere dysfunction and enhances cytosolic DNA release. Importantly, these telomeric DNA fragments are taken up by the DC and activate the intrinsic STING/IFN pathway of the DC, resulting in enhanced cross-priming capability of the DC and subsequent tumor-specific T cell activation. In addition, this study showed significant efficacy of sequential administration of 6-thio-dG and anti-PD-L1 in both advanced tumors and PD-L1 block resistant tumors, providing a powerful scientific basis for driving combination therapy into clinical trials. The present inventors expect that these findings will be transformed in the near future and benefit more patients clinically.
*****
All methods disclosed and claimed herein can be made and executed without undue experimentation in light of the present disclosure. While the compositions and methods of this disclosure have been described in terms of some specific embodiments, it will be apparent to those of skill in the art that variations may be applied to the methods and in the steps or in the sequence of steps of the methods described herein without departing from the concept, spirit and scope of the disclosure. More specifically, it will be apparent that certain agents which are both chemically and physiologically related may be substituted for the agents described herein while the same or similar results would be achieved. All such similar substitutes and modifications apparent to those skilled in the art are deemed to be within the spirit, scope and concept of the disclosure as defined by the appended claims.
Reference VI
The following references are specifically incorporated by reference herein to the extent that they provide exemplary operational or other details supplementary to those set forth herein.
Ablasser,A.,Goldeck,M.,Cavlar,T.,Deimling,T.,Witte,G.,Rohl,I.,Hopfner,K.P.,Ludwig,J.,and Hornung,V.(2013).cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING.Nature498,380-384.
Blackburn,E.H.(1991).Structure and function of telomeres.Nature 350,569-573.
Brahmer,J.R.,Tykodi,S.S.,Chow,L.Q.,Hwu,W.J.,Topalian,S.L.,Hwu,P.,Drake,C.G.,Camacho,L.H.,Kauh,J.,Odunsi,K.,et al.(2012).Safety and activity of anti-PD-L1 antibody in patients with advanced cancer.N Engl J Med 366,2455-2465.
Broz,M.L.,Binnewies,M.,Boldajipour,B.,Nelson,A.E.,Pollack,J.L.,Erle,D.J.,Barczak,A.,Rosenblum,M.D.,Daud,A.,Barber,D.L.,et al.(2014).Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity.Cancer Cell 26,938.
Bullock,B.L.,Kimball,A.K.,Poczobutt,J.M.,Neuwelt,A.J.,Li,H.Y.,Johnson,A.M.,Kwak,J.W.,Kleczko,E.K.,Kaspar,R.E.,Wagner,E.K.,et al.(2019).Tumor-intrinsic response to IFNgamma shapes the tumor microenvironment and anti-PD-1 response in NSCLC.Life Sci Alliance 2.
Chen,L.,and Han,X.(2015).Anti-PD-1/PD-L1 therapy of human cancer:past,present,and future.J Clin Invest 125,3384-3391.
Deng,L.,Liang,H.,Xu,M.,Yang,X.,Burnette,B.,Arina,A.,Li,X.D.,Mauceri,H.,Beckett,M.,Darga,T.,et al.(2014).STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors.Immunity41,843-852.
Diamond,M.S.,Kinder,M.,Matsushita,H.,Mashayekhi,M.,Dunn,G.P.,Archambault,J.M.,Lee,H.,Arthur,C.D.,White,J.M.,Kalinke,U.,et al.(2011).Type I interferon is selectively required by dendritic cells for immune rejection of tumors.J Exp Med 208,1989-2003.
Diner,E.J.,Burdette,D.L.,Wilson,S.C.,Monroe,K.M.,Kellenberger,C.A.,Hyodo,M.,Hayakawa,Y.,Hammond,M.C.,and Vance,R.E.(2013).The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING.Cell reports 3,1355-1361.
Edelson,B.T.,Kc,W.,Juang,R.,Kohyama,M.,Benoit,L.A.,Klekotka,P.A.,Moon,C.,Albring,J.C.,Ise,W.,Michael,D.G.,et al.(2010).Peripheral CD103+dendritic cells form aunified subset developmentally related to CD8alpha+conventional dendritic cells.J Exp Med 207,823-836.
Fenech,M.,Kirsch-Volders,M.,Natarajan,A.T.,Surralles,J.,Crott,J.W.,Parry,J.,Norppa,H.,Eastmond,D.A.,Tucker,J.D.,and Thomas,P.(2011).Molecular mechanisms of micronucleus,nucleoplasmic bridge and nuclear bud formation in mammalian and human cells.Mutagenesis 26,125-132.
Galluzzi,L.,Buque,A.,Kepp,O.,Zitvogel,L.,and Kroemer,G.(2017).Immunogenic cell death in cancer and infectiousdisease.Nat Rev Immunol 17,97-111.
Gao,P.,Ascano,M.,Wu,Y.,Barchet,W.,Gaffney,B.L.,Zillinger,T.,Serganov,A.A.,Liu,Y.,Jones,R.A.,Hartmann,G.,et al.(2013).Cyclic[G(2′,5′)pA(3′,5′)p]is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase.Cell 153,1094-1107.
Garon,E.B.,Rizvi,N.A.,Hui,R.,Leighl,N.,Balmanoukian,A.S.,Eder,J.P.,Patnaik,A.,Aggarwal,C.,Gubens,M.,Horn,L.,et al.(2015).Pembrolizumab for the treatment of non-small-cell lung cancer.N Engl J Med 372,2018-2028.
Gide,T.N.,Wilmott,J.S.,Scolyer,R.A.,and Long,G.V.(2018).Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma.Clin Cancer Res 24,1260-1270.
Greider,C.W.(1996).Telomere length regulation.Annual review of biocheimistry 65,337-365.
Greider,C.W.,and Blackburn,E.H.(1985).Identification of a specific telomere terminal transferase activity in Tetrahymena extracts.Cell 43,405-413.
Gryaznov,S.M.,Jackson,S.,Dikmen,G.,Harley,C.,Herbert,B.S.,Wright,W.E.,and Shay,J.W.(2007).Oligonucleotide conjugate GRN 163L targeting human telomerase as potential anticancer and antimetastatic agent.Nucleosides Nucleotides Nucleic Acids 26,1577-1579.
Gui,X.,Yang,H.,Li,T.,Tan,X.,Shi,P.,Li,M.,Du,F.,and Chen,Z.J.(2019).Autophagy induction via STING trafficking is a primordial function of the cGAS pathway.Nature 567,262-266.
Hodi,F.S.,O′Day,S.J.,McDermott,D.F.,Weber,R.W.,Sosman,J.A.,Haanen,J.B.,Gonzalez,R.,Robert,C.,Schadendorf,D.,Hassel,J.C.,et al.(2010).Improved survival with ipilimumab in patients with metastatic melanoma.N Engl J Med 363,711-723.
Kroemer,G.,Galluzzi,L.,Kepp,O.,and Zitvogel,L.(2013).Immunogenic Cell Death in Cancer Therapy.Annual Review of Immunology 31,51-72.
Lansdorp,P.M.,Verwoerd,N.P.,van de Rijke,F.M.,Dragowska,V.,Little,M.T.,Dirks,R.W.,Raap,A.K.,and Tanke,H.J.(1996).Heterogeneity in telomere length of human chromosomes.Human molecular genetics 5,685-691.
Le Bon,A.,Etchart,N.,Rossmann,C.,Ashton,M.,Hou,S.,Gewert,D.,Borrow,P.,and Tough,D.F.(2003).Cross-priming of CD8+T cells stimulated by virus-induced type I interferon.Nat Immunol 4,1009-1015.
Le,D.T.,Durham,J.N.,Smith,K.N.,Wang,H.,Bartlett,B.R.,Aulakh,L.K.,Lu,S.,Kemberling,H.,Wilt,C.,Luber,B.S.,et al.(2017).Mismatch repair deficiency predicts response of solid tumors to PD-1blockade.Science 357,409-413.
Li,H.Y.,McSharry,M.,Bullock,B.,Nguyen,T.T.,Kwak,J.,Poczobutt,J.M.,Sippel,T.R.,Heasley,L.E.,Weiser-Evans,M.C.,Clambey,E.T.,and Nemenoff,R.A.(2017).The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade.Cancer Immunol Res 5,767-777.
Li,T.,and Chen,Z.J.(2018).The cGAS-cGAMP-STING pathway connects DNA damage to inflammation,senescence,and cancer.The Joumal of experimental medicine 215,1287-1299.
Li,X.,Liu,Z.,Zhang,A.,Han,C.,Shen,A.,Jiang,L.,Boothman,D.A.,Qiao,J.,Wang,Y.,Huang,X.,and Fu,Y.X.(2019).NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance.Nat Commun 10,3251.
Liu,S.,Cai,X.,Wu,J.,Cong,Q.,Chen,X.,Li,T.,Du,F.,Ren,J.,Wu,Y.T.,Grishin,N.V.,and Chen,Z.J.(2015).Phosphorylation of innate immune adaptor proteins MAVS,STING,and TRIF induces IRF3 activation.Science 347,aaa2630.
Liu,Z.,Han,C.,Dong,C.,Shen,A.,Hsu,E.,Ren,Z.,Lu,C.,Liu,L.,Zhang,A.,Timmerman,C.,et al.(2019).Hypofractionated EGFR tyrosine kinase inhibitor limits tumor relapse through triggering innate and adaptive immunity.Sci Immunol 4.
Mandal,R.,Samstein,R.M.,Lee,K.W.,Havel,J.J.,Wang,H.,Krishna,C.,Sabio,E.Y.,Makarov,V.,Kuo,F.,Blecua,P.,et al.(2019).Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response.Science 364,485-491.
McEachern,M.J.,and Blackburn,E.H.(1996).Cap-prevented recombination between terminal telomeric repeat arrays(telomere CPR)maintains telomeres in Kluyveromyces lactis lacking telomerase.Genes&development 10,1822-1834.
Mender,I.,Gryaznov,S.,Dikmen,Z.G.,Wright,W.E.,and Shay,J.W.(2015a).Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2′-deoxyguanosine.Cancer Discov 5,82-95.
Mender,I.,Gryaznov,S.,and Shay,J.W.(2015b).A novel telomerase substrate precursor rapidly induces telomere dysfunction in telomerase positive cancer cells but not telomerase silent normal cells.Oncoscience 2,693-695.
Mender,I.,LaRanger,R.,Luitel,K.,Peyton,M.,Girard,L.,Lai,T.P.,Batten,K.,Cornelius,C.,Dalvi,M.P.,Ramirez,M.,et al.(2018).Telomerase-Mediated Strategy for Overcoming Non-Small Cell Lung Cancer Targeted Therapy and Chemotherapy Resistance.Neoplasia 20,826-837.
Mender,I.,and Shay,J.W.(2015).Telomere Dysfunction Induced Foci(TIF)Analysis.Bio-protocol 5.
Min,J.,Wright,W.E.,and Shay,J.W.(2019).Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52.Genes Dev 33,814-827.
Morin,G.B.(1989).The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats.Cell 59,521-529.
Nakamura,T.M.,Morin,G.B.,Chapman,K.B.,Weinrich,S.L.,Andrews,W.H.,Lingner,J.,Harley,C.B.,and Cech,T.R.(1997).Telomerase catalytic subunit homologs from fission yeast and human.Science 277,955-959.
Nassour,J.,Radford,R.,Correia,A.,Fuste,J.M.,Schoell,B.,Jauch,A.,Shaw,R.J.,and Karlseder,J.(2019).Autophagic cell death restricts chromosomal instability during replicative crisis.Nature 565,659-663.
Pitt,J.M.,Kroemer,G.,and Zitvogel,L.(2017).Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment.Adv Exp Med Biol 1036,65-79.
Qiao,J.,Liu,Z.,Dong,C.,Luan,Y.,Zhang,A.,Moore,C.,Fu,K.,Peng,J.,Wang,Y.,Ren,Z.,et al.(2019).Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8(+)T Cell Apoptosis.Cancer Cell 35,901-915 e904.
Qiao,J.,Tang,H.,and Fu,Y.X.(2017).DNA sensing and immune responses in cancer therapy.Curr Opin Immunol 45,16-20.
Reinhardt,R.L.,Liang,H.E.,and Locksley,R.M.(2009).Cytokine-secreting follicular T cells shape the antibody repertoire.Nat Immunol 10,385-393.
Ribas,A.,Hamid,O.,Daud,A.,Hodi,F.S.,Wolchok,J.D.,Kefford,R.,Joshua,A.M.,Patnaik,A.,Hwu,W.J.,Weber,J.S.,et al.(2016).Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma.JAMA 315,1600-1609.
Ribas,A.,and Wolchok,J.D.(2018).Cancer immunotherapy using checkpoint blockade.Science 359,1350-1355.
Rizvi,N.A.,Hellmann,M.D.,Snyder,A.,Kvistborg,P.,Makarov,V.,Havel,J.J.,Lee,W.,Yuan,J.,Wong,P.,Ho,T.S.,et al.(2015a).Cancer immunology.Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer.Science 348,124-128.
Rizvi,N.A.,Mazieres,J.,Planchard,D.,Stinchcombe,T.E.,Dy,G.K.,Antonia,S.J.,Horn,L.,Lena,H.,Minenza,E.,Mennecier,B.,et al.(2015b).Activity and safety of nivolumab,an anti-PD-1 immune checkpoint inhibitor,for patients with advanced,refractory squamous non-small-cell lung cancer(CheckMate 063):a phase 2,single-arm trial.Lancet Oncol 16,257-265.
Sanchez-Paulete,A.R.,Teijeira,A.,Cueto,F.J.,Garasa,S.,Perez-Gracia,J.L.,Sanchez-Arraez,A.,Sancho,D.,and Melero,I.(2017).Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy.Ann Oncol 28,xii44-xii55.
Sen,T.,Rodriguez,B.L.,Chen,L.,Corte,C.M.D.,Morikawa,N.,Fujimoto,J.,Cristea,S.,Nguyen,T.,Diao,L.,Li,L.,et al.(2019).Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer.Cancer Discov 9,646-661.
Sengupta,S.,Sobo,M.,Lee,K.,Senthil Kumar,S.,White,A.R.,Mender,I.,Fuller,C.,Chow,L.M.L.,Fouladi,M.,Shay,J.W.,and Drissi,R.(2018).Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors.Molecular cancer therapeutics 17,1504-1514.
Shay,J.W.,and Bacchetti,S.(1997).A survey of telomerase activity in human cancer.European journal of cancer 33,787-791.
Singer,M.S.,and Gottschling,D.E.(1994).TLC1:template RNA component of Saccharomyces cerevisiae telomerase.Science 266,404-409.
Socinski,M.A.,Jotte,R.M.,Cappuzzo,F.,Orlandi,F.,Stroyakovskiy,D.,Nogami,N.,Rodriguez-Abreu,D.,Moro-Sibilot,D.,Thomas,C.A.,Barlesi,F.,et al.(2018).Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC.N Engl J Med 378,2288-2301.
Tanaka,Y.,and Chen,Z.J.(2012).STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway.Science signaling 5,ra20.
Terrin,L.,Trentin,L.,Degan,M.,Corradini,I.,Bertorelle,R.,Carli,P.,Maschio,N.,Bo,M.D.,Noventa,F.,Gattei,V.,et al.(2007).Telomerase expression in B-cell chronic lymphocytic leukemia predicts survival and delineates subgroups of patients with the same igVH mutation status and different outcome.Leukemia 21,965-972.
Topalian,S.L.,Hodi,F.S.,Brahmer,J.R.,Gettinger,S.N.,Smith,D.C.,McDermott,D.F.,Powderly,J.D.,Carvajal,R.D.,Sosman,J.A.,Atkins,M.B.,et al.(2012).Safety,activity,and immune correlates of anti-PD-1 antibody in cancer.N Engl J Med 366,2443-2454.
Vanpouille-Box,C.,Alard,A.,Aryankalayil,M.J.,Sarfraz,Y.,Diamond,J.M.,Schneider,R.J.,Inghirami,G.,Coleman,C.N.,Formenti,S.C.,and Demaria,S.(2017).DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity.Nat Commun 8,15618.
Wang,L.,Soria,J.C.,Kemp,B.L.,Liu,D.D.,Mao,L.,and Khuri,F.R.(2002).hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer.Clin Cancer Res 8,2883-2889.
West,A.P.,Khoury-Hanold,W.,Staron,M.,Tal,M.C.,Pineda,C.M.,Lang,S.M.,Bestwick,M.,Duguay,B.A.,Raimundo,N.,MacDuff,D.A.,et al.(2015).Mitochondrial DNA stress primes the antiviral innate immune response.Nature 520,553-557.
Woo,S.R.,Fuertes,M.B.,Corrales,L.,Spranger,S.,Furdyna,M.J.,Leung,M.Y.,Duggan,R.,Wang,Y.,Barber,G.N.,Fitzgerald,K.A.,et al.(2014).STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors.Immunity 41,830-842.
Wu,J.,Sun,L.,Chen,X.,Du,F.,Shi,H.,Chen,C.,and Chen,Z.J.(2013).Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA.Science 339,826-830.
Xia,T.,Konno,H.,Ahn,J.,and Barber,G.N.(2016).Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis.Cell Rep 14,282-297.
Xu,M.M.,Pu,Y.,Han,D.,Shi,Y.,Cao,X.,Liang,H.,Chen,X.,Li,X.D.,Deng,L.,Chen,Z.J.,et al.(2017).Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling.Immunity 47,363-373 e365.
Yu,G.L.,Bradley,J.D.,Attardi,L.D.,and Blackburn,E.H.(1990).In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs.Nature 344,126-132.
Zaretsky,J.M.,Garcia-Diaz,A.,Shin,D.S.,Escuin-Ordinas,H.,Hugo,W.,Hu-Lieskovan,S.,Torrejon,D.Y.,Abril-Rodriguez,G.,Sandoval,S.,Barthly,L.,et al.(2016).Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma.N Engl J Med 375,819-829.
Zhang,G.,Wu,L.W.,Mender,I.,Barzily-Rokni,M.,Hammond,M.R.,Ope,O.,Cheng,C.,Vasilopoulos,T.,Randell,S.,Sadek,N.,et al.(2018).Induction of Telomere Dysfunction Prolongs Disease Control of Therapy-Resistant Melanoma.Clin Cancer Res 24,4771-4784.
Zhang,X.,Shi,H.,Wu,J.,Zhang,X.,Sun,L.,Chen,C.,and Chen,Z.J.(2013).Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING.Molecular cell 51,226-235.
Zijlmans,J.M.,Martens,U.M.,Poon,S.S.,Raap,A.K.,Tanke,H.J.,Ward,R.K.,and Lansdorp,P.M.(1997).Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats.Proceedings of the National Academy of Sciences of the United States of America 94,7423-7428.
Zou,W.,Wolchok,J.D.,and Chen,L.(2016).PD-L1(B7-H1)and PD-1pathway blockade for cancer therapy:Mechanisms,response biomarkers,and combinations.Sci Transl Med 8,328rv324.
Sequence listing
<110> The Board of Regents of the University of Texas System
<120> sequential cancer treatment using 6-thio-dG, checkpoint inhibitors and radiation therapy
<130> UTFD.P3648WO
<140> unknown
<141> 2021-03-12
<150> 62/989,041
<151> 2020-03-13
<160> 10
<170> PatentIn version 3.5
<210> 1
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotide
<400> 1
<210> 2
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotide
<400> 2
<210> 3
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotide
<400> 3
cgccacactc cacggaagca 20
<210> 4
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotides
<400> 4
cggggcattc cggataggcc 20
<210> 5
<211> 22
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotide
<400> 5
accgattgga tggtttagtg ag 22
<210> 6
<211> 22
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotides
<400> 6
cctacggaaa ccttgttacg ac 22
<210> 7
<211> 19
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotide
<400> 7
atgagtggtg gttgcaggc 19
<210> 8
<211> 25
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotides
<400> 8
tgacctttca aatgcagtag attca 25
<210> 9
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotide
<400> 9
<210> 10
<211> 20
<212> DNA
<213> Artificial sequence
<220>
<223> Synthesis of oligonucleotide
<400> 10
Claims (37)
1. A method of treating cancer in a subject comprising administering 6-thio-2' -deoxyguanosine (6-thio-dG) to the subject followed by treatment with an immune checkpoint inhibitor, wherein the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, gastric cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
2. The method of claim 1, wherein the immune checkpoint inhibitor is a PD-1 inhibitor.
3. The method of claim 1, wherein the immune checkpoint inhibitor is a PD-L1 inhibitor.
4. The method of claim 1, wherein the immune checkpoint inhibitor is a CTLA-4 inhibitor.
5. The method of claim 1, wherein the immune checkpoint inhibitor is a combination of one or more CTLA-4 inhibitors and one or more PD-1 inhibitors.
6. The method of claim 1, wherein the immune checkpoint inhibitor is a combination of one or more CTLA-4 inhibitors and one or more PD-L1 inhibitors.
7. The method of any one of claims 1 to 6, wherein the 6-thio-dG is administered for about 1 to about 5 days per treatment cycle.
8. The method of any one of claims 1 to 6, wherein the checkpoint inhibitor is administered for about 1 to about 3 days per treatment cycle.
9. The method of any one of claims 1 to 8, wherein the 6-thio-dG and the checkpoint inhibitor are administered in combination with a chemotherapeutic agent, hormonal therapy, toxin therapy, or surgery.
10. A method of treating cancer in a subject comprising administering 6-thio-2' -deoxyguanosine (6-thio-dG) to the subject followed by cimetiprizumab <xnotran> , , , , , , , , , , , , , , , , , , , , , , , </xnotran>Cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
<xnotran> , , , , , , , , , , , , , , , , , , , , , , , </xnotran>Cancer, breast cancer, kidney cancer, neuroblastoma, merkel cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
11. The method of claim 10, wherein the 6-thio-dG is administered for from about 1 to about 5 days per treatment cycle.
12. The method of any one of claims 10 and 11, wherein cimetiprilinumab is administered per treatment cycle From about 1 to about 3 days.
From about 1 to about 3 days.
13. The method of any one of claims 10 to 12, wherein the 6-thio-dG and the cimetipril mab In combination with chemotherapeutic agents, hormonal therapy, toxin therapy or surgery.
In combination with chemotherapeutic agents, hormonal therapy, toxin therapy or surgery.
14. A method of treating cancer in a subject comprising administering 6-thio-2' -deoxyguanosine (6-thio-dG) to the subject followed by treatment with an immune checkpoint inhibitor administered in combination with radiation therapy, wherein the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, gastric cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, mercker cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
15. The method of any one of claims 1 to 14, wherein the total dose of 6-thio-dG administered during the treatment period of from about 1 to 5 days is from about 20 to 2000mg.
16. The method of any one of claims 1 to 14, wherein the cancer is lung cancer, colorectal cancer, liver cancer, melanoma, or glioblastoma.
17. The method of any one of claims 1 to 14, wherein the cancer is metastatic.
18. The method of any one of claims 1 to 14, wherein the cancer is recurrent.
19. The method of any one of claims 1 to 14, wherein the cancer is treatment resistant.
20. The method of claim 10, wherein the treatment-resistant cancer is checkpoint inhibitor treatment-resistant.
21. The method of claim 1, wherein the therapy-resistant cancer is resistant to one or more of a PD-1 inhibitor, a PD-L1 inhibitor, and a CTLA-4 inhibitor.
22. The method of any one of claims 1 to 9, wherein the subject was previously treated with checkpoint inhibitor therapy.
23. The method of claim 22, wherein the subject was previously treated with one or more of PD-1, PD-L1, and CTLA-4 treatment.
24. The method of any one of claims 1-23, wherein administration of 6-thio-2-deoxyguanosine (6-thio-dG) and subsequent treatment with the checkpoint inhibitor is repeated at least once.
25. The method of any one of claims 1 to 23, wherein the 6-thio-dG and the checkpoint inhibitor are administered systemically.
26. The method of any one of claims 1 to 23, wherein the 6-thio-dG and the checkpoint inhibitor are administered locally or regionally to a tumor site.
27. The method of any one of claims 1 to 23, wherein the 6-thio-dG is administered locally or regionally to a tumor site and the checkpoint inhibitor is administered systemically.
28. The method of any one of claims 1 to 23, wherein administration of 6-thio-dG and the checkpoint inhibitor results in inhibition of tumor growth.
29. The method of any one of claims 1 to 23, wherein administration of 6-thio-dG and the checkpoint inhibitor results in remission of the cancer.
30. The method of any one of claims 1 to 23, wherein administration of 6-thio-dG and the checkpoint inhibitor results in a reduction in tumor burden.
31. The method of any one of claims 1 to 23, wherein administration of 6-thio-dG and the checkpoint inhibitor results in inhibition of cancer cell metastasis.
32. The method of any one of claims 1 to 23, wherein administration of 6-thio-dG and the checkpoint inhibitor results in tumor eradication.
33. A method of treating cancer in a subject comprising administering 6-thio-2 '-deoxyguanosine (6-thio-dG) to the subject followed by treatment with radiation therapy, wherein the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, gastric cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, mercke's cell cancer, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
34. A method of treating cancer in a subject comprising treating with radiation therapy prior to administering 6-thio-2 '-deoxyguanosine (6-thio-dG) to the subject, wherein the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, gastric cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, kidney cancer, neuroblastoma, mercke's cell cancer, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
35. The method of claim 29 or 30, wherein the cancer is selected from pancreatic cancer, lung cancer, gastric cancer, liver cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, brain cancer, colon cancer, prostate cancer, ovarian cancer, cervical cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer.
36. The method of any one of claims 29 to 31 wherein the administration of 6-thioxo-2-deoxyguanosine (6-thioxo-dG) and the radiation therapy are repeated at least once.
37. A method of treating cancer in a subject comprising administering 6-thio-2' -deoxyguanosine (6-thio-dG) to the subject followed by treatment with an immune checkpoint inhibitor and radiation therapy. In some embodiments, the checkpoint inhibitor is a PD-L1 inhibitor, a PD-1 inhibitor, or a CTAL-4 inhibitor. In some embodiments, the PD-L1 inhibitor is selected from one or more of alemtuzumab, avizumab, chikulizumab, bindrafusalfa, devaluzumab, MGD013, KNO35, KN046, AUNP12, CA-170, and BMS-9986189. In a1In some embodiments, the PD-L1 inhibitor is atelizumab. In some embodiments, the PD-1 inhibitor is selected from one or more of pembrolizumab, nivolumab, cimetipril mab, JTx-4014, sartorimab, breglizumab, BI 754091, sibatuzumab, carpriluzumab, cedilizumab, tirezumab, cyprilizumab, tereprinizumab, dolaprimab, INCMGA00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-A317, BCD-100, JS001, and AMP-515. In some embodiments, the PD-1 inhibitor is cimetipril mab Which is administered in combination with radiation therapy, wherein the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, stomach cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, renal cancer, neuroblastoma, merkel cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
Which is administered in combination with radiation therapy, wherein the cancer is selected from pancreatic cancer, lung cancer, mesothelioma, stomach cancer, esophageal cancer, liver cancer, biliary tract cancer, bladder cancer, head and neck cancer, oral cancer, nasopharyngeal cancer, adult brain cancer, colon cancer, rectal cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, uterine cancer, testicular cancer, lymphoma, leukemia, skin cancer, breast cancer, renal cancer, neuroblastoma, merkel cell carcinoma, myelodysplastic syndrome, myelofibrosis, and multiple myeloma.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202062989041P | 2020-03-13 | 2020-03-13 | |
| US62/989,041 | 2020-03-13 | ||
| PCT/US2021/022090 WO2021183873A1 (en) | 2020-03-13 | 2021-03-12 | Sequential treatment of cancers using 6-thio-dg, checkpoint inhibitors and radiation therapy |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN115397428A true CN115397428A (en) | 2022-11-25 |
Family
ID=77671975
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202180028952.2A Pending CN115397428A (en) | 2020-03-13 | 2021-03-12 | Sequential cancer treatment using 6-thio-dG, checkpoint inhibitors and radiation therapy |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20210290652A1 (en) |
| EP (1) | EP4117667A4 (en) |
| JP (1) | JP2023517671A (en) |
| KR (1) | KR20220154134A (en) |
| CN (1) | CN115397428A (en) |
| AU (1) | AU2021233023A1 (en) |
| BR (1) | BR112022018238A2 (en) |
| CA (1) | CA3170930A1 (en) |
| IL (1) | IL296403A (en) |
| MX (1) | MX2022011331A (en) |
| WO (1) | WO2021183873A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108136025A (en) * | 2015-07-16 | 2018-06-08 | 比奥克斯塞尔医疗股份有限公司 | A kind of novel method using immune modulating treatment cancer |
| CN109789206A (en) * | 2016-09-16 | 2019-05-21 | 生态有限公司 | The combination treatment of antibody and checkpoint inhibitor |
| WO2019183482A1 (en) * | 2018-03-22 | 2019-09-26 | The Board Of Regents Of The University Of Texas System | USE OF 6-THIO-dG TO TREAT THERAPY-RESISTANT TELOMERASEPOSITIVE PEDIATRIC BRAIN TUMORS |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103180463B (en) * | 2010-10-22 | 2014-12-31 | 霍夫曼-拉罗奇有限公司 | A conjugate between a thiophilic solid phase and an oligonucleotide comprising a thiooxonucleotide |
| US20180036331A1 (en) * | 2016-03-24 | 2018-02-08 | The Board Of Regents Of The University Of Texas System | Treatment of drug resistant proliferative diseases with telomerase mediated telomere altering compounds |
| CA3025522A1 (en) * | 2016-05-27 | 2017-11-30 | The Board Of Regents Of The University Of Texas System | 6-thio-2'-deoxyguanosine (6-thio-dg) results in telomerase dependent telomere dysfunction and cell death in various models of therapy-resistant cancer cells |
| KR20240011262A (en) * | 2017-02-21 | 2024-01-25 | 리제너론 파아마슈티컬스, 인크. | Anti-pd-1 antibodies for treatment of lung cancer |
| WO2019152574A1 (en) * | 2018-02-01 | 2019-08-08 | Merck Sharp & Dohme Corp. | Methods for treating cancer or infection using a combination of an anti-pd-1 antibody, an anti-lag3 antibody, and an anti-tigit antibody |
-
2021
- 2021-03-12 KR KR1020227033906A patent/KR20220154134A/en active Search and Examination
- 2021-03-12 WO PCT/US2021/022090 patent/WO2021183873A1/en unknown
- 2021-03-12 EP EP21767125.4A patent/EP4117667A4/en active Pending
- 2021-03-12 IL IL296403A patent/IL296403A/en unknown
- 2021-03-12 US US17/200,539 patent/US20210290652A1/en active Pending
- 2021-03-12 BR BR112022018238A patent/BR112022018238A2/en unknown
- 2021-03-12 JP JP2022554769A patent/JP2023517671A/en active Pending
- 2021-03-12 CA CA3170930A patent/CA3170930A1/en active Pending
- 2021-03-12 MX MX2022011331A patent/MX2022011331A/en unknown
- 2021-03-12 AU AU2021233023A patent/AU2021233023A1/en active Pending
- 2021-03-12 CN CN202180028952.2A patent/CN115397428A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108136025A (en) * | 2015-07-16 | 2018-06-08 | 比奥克斯塞尔医疗股份有限公司 | A kind of novel method using immune modulating treatment cancer |
| CN109789206A (en) * | 2016-09-16 | 2019-05-21 | 生态有限公司 | The combination treatment of antibody and checkpoint inhibitor |
| WO2019183482A1 (en) * | 2018-03-22 | 2019-09-26 | The Board Of Regents Of The University Of Texas System | USE OF 6-THIO-dG TO TREAT THERAPY-RESISTANT TELOMERASEPOSITIVE PEDIATRIC BRAIN TUMORS |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2023517671A (en) | 2023-04-26 |
| CA3170930A1 (en) | 2021-09-16 |
| EP4117667A1 (en) | 2023-01-18 |
| WO2021183873A1 (en) | 2021-09-16 |
| BR112022018238A2 (en) | 2022-10-25 |
| KR20220154134A (en) | 2022-11-21 |
| EP4117667A4 (en) | 2023-11-15 |
| MX2022011331A (en) | 2022-10-27 |
| IL296403A (en) | 2022-11-01 |
| US20210290652A1 (en) | 2021-09-23 |
| AU2021233023A1 (en) | 2022-11-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Mender et al. | Telomere stress potentiates STING-dependent anti-tumor immunity | |
| Ye et al. | Advancements in clinical aspects of targeted therapy and immunotherapy in breast cancer | |
| US20190077856A1 (en) | Method of treating diseases using kinase modulators | |
| US20200253979A1 (en) | Therapeutic methods relating to hsp90 inhibitors | |
| US20240009223A1 (en) | 6-thio-2'-deoxyguanosine (6-thio-dg) results in telomerase dependent telomere dysfunction and cell death in various models of therapy-resistant cancer cells | |
| WO2019108589A1 (en) | Compositions and methods for treating cancer | |
| Franzese et al. | Tumor immunotherapy: drug-induced neoantigens (xenogenization) and immune checkpoint inhibitors | |
| KR20230059792A (en) | Combinations for Cancer Treatment | |
| Luo et al. | Necroptosis-dependent immunogenicity of cisplatin: implications for enhancing the radiation-induced abscopal effect | |
| Concannon et al. | Combining targeted DNA repair inhibition and immune-oncology approaches for enhanced tumor control | |
| CN111208283B (en) | Synergistic tumor inhibiting composition and application thereof | |
| CN111671904A (en) | Medicine containing endonuclease inhibiting function and anti-tumor application thereof | |
| Mender et al. | Activating an adaptive immune response with a telomerase-mediated telomere targeting therapeutic in hepatocellular carcinoma | |
| JP2022523571A (en) | 3'3'-Cyclic dinucleotide and prodrugs thereof | |
| US20210290652A1 (en) | SEQUENTIAL TREATMENT OF CANCERS USING 6-THIO-dG, CHECKPOINT INHIBITORS AND RADIATION THERAPY | |
| WO2019232533A1 (en) | Combination treatments of hsp90 inhibitors for enhancing tumor immunogenicity and methods of use thereof | |
| JP2023531522A (en) | Combination therapy with a deoxyuridine triphosphatase inhibitor | |
| US20230338374A1 (en) | Mdm2 inhibitors for use in the treatment or prevention of hematologic neoplasm relapse after hematopoietic cell transplantation | |
| Clancy | Potentiation of Cancer Therapy by Novel Pharmacological Inhibitors of DNA Repair | |
| Ravelli | Novel therapeutic approaches for the treatment of solid tumors | |
| Bunz | Cancer Therapy | |
| WO2024052674A1 (en) | Modified t-cells for use in the treatment of bladder cancer | |
| Raab | CDX2 as a Predictive Biomarker of Drug Response in Colon Cancer | |
| Link et al. | Cancer Therapy | |
| CN117042767A (en) | Methods of treating cancer using STING agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |