CN115385926A - Preparation method of connection base drug conjugate and intermediate thereof - Google Patents
Preparation method of connection base drug conjugate and intermediate thereof Download PDFInfo
- Publication number
- CN115385926A CN115385926A CN202110566920.XA CN202110566920A CN115385926A CN 115385926 A CN115385926 A CN 115385926A CN 202110566920 A CN202110566920 A CN 202110566920A CN 115385926 A CN115385926 A CN 115385926A
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- preparation
- mixture
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/28—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to acyclic carbon atoms of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/188—Preparation; Treatments not provided for in C07F7/20 by reactions involving the formation of Si-O linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/1892—Preparation; Treatments not provided for in C07F7/20 by reactions not provided for in C07F7/1876 - C07F7/1888
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/021—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-(X)n-C(=0)-, n being 5 or 6; for n > 6, classification in C07K5/06 - C07K5/10, according to the moiety having normal peptide bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06043—Leu-amino acid
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Abstract
The invention disclosesA preparation method of a connection base drug conjugate and an intermediate thereof, in particular discloses a preparation method of a compound shown in a formula I, which comprises the following steps: removing R from the compound of formula II 4 Protecting groups to give the compounds of formula I; wherein R is 1 Is C 1 ~C 6 Alkyl, one or more R 1‑3 S(O) 2 -substituted C 1 ~C 6 Alkyl, or one or more N (R) 1‑1 )(R 1 ‑ 2 ) -substituted C 1 ~C 6 An alkyl group; r 2 And R 3 Each independently is C 1 ~C 6 Alkyl, one or more halogen substituted C 1 ~C 6 Alkyl, or halogen; r 1‑1 、R 1‑2 And R 1‑3 Each independently is C 1 ~C 4 An alkyl group; r 4 Is a hydroxyl protecting group. The preparation method has one or more of the following advantages: the method has the advantages of simple operation, high yield, effective avoidance of generation of special byproducts, easy control of product quality and suitability for industrial production.
Description
Technical Field
The invention belongs to the field of drug synthesis, and particularly relates to a preparation method of a connection base drug conjugate and an intermediate thereof.
Background
Antibody conjugated drugs (ADCs) have been one of the hotspots of interest to the pharmaceutical industry in recent years. Because of the unsatisfactory clinical efficacy of many antibody drugs, many industries are increasingly turning their eyes to ADC drugs. The basic module of the ADC medicine comprises an antibody, a linker and an effector molecule, and the effector molecule is transmitted to a tumor site by the antibody for enrichment, so that tumor cells are killed. Most of the traditional effector molecules are high-activity tubulin inhibitors, and generally have larger toxic and side effects, so that the application of ADC is limited. Recently, immunoledics company invented a novel ADC drug IMMU-132 (ZL 200980156218) which takes a camptothecin compound as an effector molecule and shows a better anti-tumor effect, and the first three co-invented another ADC drug DS-8201a (ZL 201380053256) which takes a camptothecin compound as an effector molecule and also shows a better anti-tumor effect.
WO2020259258A1 discloses an ADC compound using camptothecin derivative Dxd as an effector molecule, and the compound also shows a good anti-tumor effect. Among them, the target ADC compound can be obtained by coupling the camptothecin derivative represented by formula I with an antibody, wherein the linker drug conjugate represented by formula I can be prepared by the following synthetic scheme 1 or synthetic scheme 2.
Route 1:

the synthetic method of scheme 1 comprises: reacting a compound 1-1 with 4-aminobenzyl alcohol, reacting the obtained compound with bis (p-nitrophenyl) carbonate, then reacting with substituted alkylamine to obtain a compound 1-2, reacting the compound 1-2 with paraformaldehyde and trimethylchlorosilane to obtain a compound 1-3, reacting the compound 1-3 with tert-butyl hydroxyacetate, removing tert-butyl under the action of trifluoroacetic acid to obtain a compound 1-4, reacting the compound 1-4 with irinotecan mesylate to obtain a compound 1-5, removing Fmoc protection on amino under the action of DBU, and then carrying out coupling reaction with 6- (maleimide) hexanoic acid succinimidyl ester to obtain a target compound I.
Route 2:

the synthetic method of scheme 2 comprises: reacting the compound 2-1 with paraformaldehyde and trimethylchlorosilane, reacting the obtained compound with tert-butyl glycolate to obtain a compound 2-2, removing tert-butyl from the compound 2-2 under the action of trifluoroacetic acid, reacting with irinotecan mesylate to obtain a compound 2-3, reducing azide into amino by the compound 2-3 under the action of triethylphosphine to obtain a compound 2-4, and carrying out coupling reaction on the compound 2-4 and MC-V to obtain a target compound I.
However, the synthesis methods of scheme 1 and scheme 2 above have the problem of unacceptable purity of the final product during practical production scale-up, and therefore a new scheme is required to be explored to make the purity of the final product meet the requirement.
Disclosure of Invention
The invention aims to solve the technical problem that the preparation method of the connection base drug conjugate shown in the formula I in the prior art is difficult to obtain a final product with qualified purity, so that the invention provides a novel preparation method of the connection base drug conjugate shown in the formula I and an intermediate thereof. The preparation method has one or more of the following advantages: the method has the advantages of simple operation, high yield, effective avoidance of generation of special byproducts, easy control of product quality and suitability for industrial production.
The invention provides a preparation method of a compound shown in a formula I, which comprises the following steps: removing R from the compound of formula II 4 Protecting groups to obtain the compound of formula I;

wherein R is 1 Is C 1 ~C 6 Alkyl, one or more R 1-3 S(O) 2 -substituted C 1 ~C 6 Alkyl, or one or more N (R) 1-1 )(R 1-2 ) -substituted C 1 ~C 6 An alkyl group;
R 2 and R 3 Each independently is C 1 ~C 6 Alkyl, one or more halogen substituted C 1 ~C 6 Alkyl, or halogen;
R 1-1 、R 1-2 and R 1-3 Each independently is C 1 ~C 4 An alkyl group;
R 4 is a hydroxyl protecting group.
In some embodiments, at R 1 、R 2 And R 3 In the definition of (1), the C 1 ~C 6 The alkyl group may be methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl or tert-butylPreferably methyl or ethyl.
In some embodiments, at R 2 And R 3 In the definition of (1), the halogen may be fluorine, chlorine, bromine or iodine, preferably fluorine.
In some embodiments, at R 1-1 、R 1-2 And R 1-3 In the definition of (1), the C 1 ~C 4 The alkyl group may be methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl or tert-butyl, preferably methyl.
In some embodiments, R 1 Is a R 1-3 S(O) 2 -substituted C 1 ~C 6 Alkyl or a-NR 1-1 R 1-2 Substituted C 1 ~C 6 The alkyl group is preferably a methylsulfonylethyl group or an N, N-dimethylethyl group, and more preferably a methylsulfonylethyl group.
In some embodiments, R 2 Is C 1 ~C 6 Alkyl, preferably methyl.
In some embodiments, R 3 Is halogen, preferably fluorine or chlorine, more preferably fluorine.
In some embodiments, R 1 Is a R 1-3 S(O) 2 -substituted C 1 ~C 6 Alkyl groups such as methylsulfonylethyl;
R 2 is C 1 ~C 6 Alkyl groups such as methyl;
R 3 is halogen, such as fluorine;
R 1-3 is C 1 ~C 4 Alkyl groups, such as methyl.
In some embodiments, R 1 As defined in any of the preceding schemes, R 2 Is methyl, and R 3 Is fluorine.
In some embodiments, R 1 Is methylsulfonylethyl, R 2 Is methyl, and R 3 Is fluorine.
In some embodiments, at R 4 In the definition of (1), the hydroxyl protecting group is a hydroxyl protecting group conventional in the art, such as an ester protecting group (i.e., R) 4 Is acyl) or silyl ether protecting group (i.e. R) 4 Is silicon based). When the hydroxyl protecting group is an ester protecting group, R 4 May be an acetyl group, a propionyl group, a benzoyl group or a pivaloyl group, and is preferably an acetyl group or a propionyl group, and is more preferably an acetyl group. When the hydroxyl protecting group is a silyl ether protecting group, R 4 It may be trimethylsilyl, triisopropylsilyl, tert-butyldimethylsilyl, tert-butyldiphenylsilyl or tri-tert-butylsilyl, preferably tert-butyldiphenylsilyl or tri-tert-butylsilyl, and more preferably tert-butyldiphenylsilyl. Preferably, R 4 Acetyl or tert-butyl diphenyl silicon group.
In some embodiments, in the methods of making compounds of formula I, R 1 Is methylsulfonylethyl; r 2 Is methyl; r 3 Is fluorine; r 4 Is acetyl.
In some embodiments, in the methods of making compounds of formula I, R 1 Is methylsulfonylethyl; r 2 Is methyl; r 3 Is fluorine; r 4 Is tert-butyl diphenyl silicon base.
In some embodiments, the compound of formula I is prepared by a process comprising removing R 4 Reaction conditions for protecting group (e.g. solvent and amount thereof, removing R) 4 The reagents used for protecting groups and their amounts, reaction times, etc.) may be conventional in the art for such reactions, and may be adjusted depending on the type of hydroxy protecting group.
In some embodiments, when the hydroxy protecting group is an ester protecting group (e.g., R) in the process for preparing the compound of formula I 4 When acetyl is present), R is removed 4 The deprotecting agent used for the protecting group may be sodium hydroxide, potassium hydroxide, sodium carbonate, potassium bicarbonate or acetyl chloride/methanol, preferably potassium carbonate or acetyl chloride/methanol, and more preferably acetyl chloride/methanol.
In some embodiments, the process for preparing a compound of formula I, when the hydroxy protecting group is an ester protecting group (e.g., R) 4 When acetyl is present), R is removed 4 For protecting groupsThe molar ratio of deprotecting agent (e.g. acetyl chloride) to compound of formula II may be in the range of 0.5 to 2, preferably 0.5 to 1, more preferably 0.5.
In some embodiments, when the hydroxy protecting group is an ester protecting group (e.g., R) in the process for preparing the compound of formula I 4 When acetyl is present), R is removed 4 The solvent used for the protecting group may be a conventional solvent used in the art for such a reaction, such as methanol, ethanol, isopropanol, dichloromethane, or a mixture of any two or more thereof, and is preferably a "mixed solvent of methanol and dichloromethane" or a "mixed solvent of ethanol and dichloromethane", and is more preferably a mixed solvent of methanol and dichloromethane. In the mixed solvent of methanol and dichloromethane, the volume ratio of methanol to dichloromethane can be 1.
In some embodiments, when the hydroxy protecting group is an ester protecting group (e.g., R) in the process for preparing the compound of formula I 4 When acetyl is present), R is removed 4 The temperature at which the protecting groups are reacted is from 10 to 50 deg.C, for example from 20 to 30 deg.C.
In some embodiments, when the hydroxy protecting group is an ester protecting group (e.g., R) in the process for preparing the compound of formula I 4 When acetyl is present), R is removed 4 The operation of the reaction of the protecting group may be a routine operation of such a reaction in the art, and may include, for example, the following steps: adding acetyl chloride to the solution of the compound of formula II in batches (e.g. dropwise), and reacting after the addition is finished; preferably, the temperature of the system is controlled to be 0-5 ℃ during the addition of the acetyl chloride.
In some embodiments, when the hydroxy protecting group is an ester protecting group (e.g., R) in the process for preparing the compound of formula I 4 When acetyl is present), R is removed 4 The progress of the protecting group reaction can be monitored using conventional test methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula II is no longer detectable as an end point of the reaction. In some embodiments, de-R 4 The reaction time of the protecting group may be 2 to 8 hours, preferably 2 to 5 hours, and more preferably 2 to 3 hours。
In some embodiments, the process for preparing a compound of formula I, when the hydroxy protecting group is an ester protecting group (e.g., R) 4 When acetyl is present), R is removed 4 After the protecting group reaction is completed, the method can further comprise the following post-treatment steps: extracting and washing the reaction solution, and concentrating an organic phase to obtain a crude product of the compound shown in the formula I; optionally, the crude compound of the formula I is purified by silica gel column chromatography to obtain the compound of the formula I.
In some embodiments, when the hydroxy protecting group is a silyl ether protecting group (e.g., R) in the process for preparing the compound of formula I 4 In the case of tert-butyldiphenylsilyl), R is removed 4 The deprotection reagent used for the protecting group may be a reagent conventionally used in the art for such a reaction, for example, lithium hydroxide, tetrabutylammonium fluoride/acetic acid, sodium hydroxide, a pyridine hydrogen fluoride complex, tert-butylammonium fluoride or tert-butylammonium fluoride/acetic acid, preferably tert-butylammonium fluoride or tert-butylammonium fluoride/acetic acid, and more preferably tert-butylammonium fluoride/acetic acid.
In some embodiments, in the process for preparing a compound of formula I, when the hydroxy protecting group is a silyl ether protecting group (e.g., R) 4 In the case of tert-butyldiphenylsilyl), R is removed 4 The molar ratio of the deprotecting reagent used for the protecting group (e.g. tert-butylammonium fluoride or tert-butylammonium fluoride/acetic acid) to the compound of formula II may be 1.2 to 3, preferably 1.2 to 1.6, and more preferably 1.5.
In some embodiments, the process for preparing a compound of formula I, when the hydroxy protecting group is a silyl ether protecting group (e.g., R) 4 In the case of tert-butyldiphenylsilyl), R is removed 4 The solvent used for the protecting group may be a conventional solvent used in the art for such a reaction, such as methanol, ethanol, isopropanol, dichloromethane, or a mixture of any two or more thereof, and is preferably a "mixed solvent of methanol and dichloromethane" or a "mixed solvent of ethanol and dichloromethane", and is more preferably a mixed solvent of methanol and dichloromethane. In the mixed solvent of the methanol and the dichloromethane, the methanolThe volume ratio of the organic solvent to dichloromethane can be 1.
In some embodiments, when the hydroxy protecting group is a silyl ether protecting group (e.g., R) in the process for preparing the compound of formula I 4 In the case of tert-butyldiphenylsilyl), R is removed 4 The temperature at which the protecting groups are reacted is from 10 to 50 deg.C, for example from 20 to 30 deg.C.
In some embodiments, when the hydroxy protecting group is a silyl ether protecting group (e.g., R) in the process for preparing the compound of formula I 4 In the case of tert-butyldiphenylsilyl), R is removed 4 The operation of the reaction of the protecting group may be a routine operation of such a reaction in the art, and may include, for example, the following steps: adding tert-butyl ammonium fluoride and acetic acid into the solution of the compound of the formula II in batches, and carrying out reaction after the addition is finished.
In some embodiments, when the hydroxy protecting group is a silyl ether protecting group (e.g., R) in the process for preparing the compound of formula I 4 In the case of tert-butyldiphenylsilyl), R is removed 4 The progress of the reaction of the protecting group can be monitored by customary test methods in the art (e.g. TLC, GC, HPLC or NMR, etc.), generally so that the compound of formula II is no longer detectable as an end point of the reaction. In some embodiments, de-R 4 The time for the reaction of the protecting group may be 10 to 20 hours, preferably 12 to 16 hours, and more preferably 14 to 16 hours.
In some embodiments, the process for preparing a compound of formula I, when the hydroxy protecting group is a silyl ether protecting group (e.g., R) 4 In the case of tert-butyldiphenylsilyl), R is removed 4 After the protecting group reaction is completed, the method can further comprise the following post-treatment steps: and (3) carrying out solid-liquid separation on the reaction liquid to obtain a solid, namely the compound product of the formula I.
In some embodiments, R 4 Is acetyl;
preferably, de-R 4 The deprotection reagent used by the protecting group is acetyl chloride/methanol;
preferably, de-R 4 The solvent used for protecting group is a mixed solvent of methanol and dichloromethane.
In some embodiments, R 4 Is tert-butyl diphenyl silicon base;
preferably, de-R 4 The deprotection reagent used by the protecting group is tert-butyl ammonium fluoride/acetic acid;
preferably, de-R 4 The solvent used for protecting group is a mixed solvent of methanol and dichloromethane.
The process for preparing the compound of formula I may further comprise a process for preparing a compound of formula II, which may comprise the steps of: carrying out coupling reaction on a compound shown in a formula III and 6- (maleimide) hexanoic acid succinimide ester in a solvent to obtain a compound shown in a formula II;

wherein R is 1 、R 2 、R 3 And R 4 As defined above.
In some embodiments, in the method for preparing the compound of formula II, the molar ratio of the succinimidyl 6- (maleimido) hexanoate to the compound of formula III may be 1 to 5, preferably 1 to 2, more preferably 1.0 to 1.5, and still more preferably 1.0.
In some embodiments, in the preparation method of the compound of formula II, the solvent may be a solvent conventional in such reactions in the art, such as an amide solvent, a chloroalkane solvent, an ether solvent, a nitrile solvent, or a mixture of any two or more thereof, preferably an amide solvent, a chloroalkane solvent, or a mixture thereof, more preferably an amide solvent or a chloroalkane solvent, and even more preferably a chloroalkane solvent. Among them, the amide-based solvent is preferably N, N-dimethylformamide, N-dimethylacetamide, or a mixture thereof, and more preferably N, N-dimethylformamide. The chloroalkane solvent is preferably dichloromethane, chloroform, dichloroethane or a mixture of any two or more thereof, and more preferably dichloromethane. The ether solvent is preferably tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether, or a mixture of any two or more thereof, and more preferably tetrahydrofuran. The nitrile solvent is preferably acetonitrile. In some embodiments, the compound of formula II is prepared by a process wherein the solvent is a chlorinated alkane solvent, such as dichloromethane.
In some embodiments, the reaction temperature in the process for preparing the compound of formula II may be a temperature conventional in the art for such reactions, for example, from 0 to 50 ℃, preferably from 25 to 40 ℃, and more preferably 40 ℃.
In some embodiments, the coupling reaction may be performed in a manner conventional in the art for such reactions, and may, for example, comprise the steps of: stirring a mixture of the compound of formula III, 6- (maleimide) hexanoyl compound and solvent to carry out the coupling reaction.
In some embodiments, the progress of the coupling reaction in the preparation of the compound of formula II may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula III is no longer detected as an end point of the reaction. The reaction time of the coupling reaction may be 1 to 24 hours, preferably 12 to 20 hours, and more preferably 16 hours.
In some embodiments, the method for preparing the compound of formula II may further comprise a post-treatment step after the coupling reaction is completed: and removing the solvent in the reaction solution, and purifying the obtained residue to obtain the compound shown in the formula II. The purification can be carried out by conventional purification methods in the art, such as slurry, crystallization, preparative chromatography or silica gel column chromatography, etc., preferably by silica gel column chromatography, wherein the eluent is preferably a mixture of dichloromethane and methanol, and the elution gradient is preferably 50.
The process for preparing the compound of formula II may further comprise a process for preparing a compound of formula III, which may comprise the steps of: carrying out reduction reaction on a compound shown in a formula IV and a reducing agent in an organic solvent in the presence of an acid buffer solution to obtain a compound shown in a formula III;

wherein R is 1 、R 2 、R 3 And R 4 As defined above.
In some embodiments, in the preparation method of the compound of formula III, the reducing agent may be a reducing agent conventional in the art for such reactions, preferably triphenylphosphine, tri-tert-butylphosphine or trimethylphosphine, more preferably trimethylphosphine.
In some embodiments, in the method for preparing the compound of formula III, the molar ratio of the reducing agent to the compound of formula IV may be 1 to 5, preferably 1 to 2, more preferably 1.0 to 1.5, and still more preferably 1.1 to 1.3.
In some embodiments, in the preparation method of the compound of formula III, the organic solvent may be a solvent conventional in such reactions in the art, preferably an ether solvent, such as tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether, or a mixture of any two or more thereof, and further preferably tetrahydrofuran.
In some embodiments, in the preparation method of the compound of formula III, the volume/mass ratio of the organic solvent to the compound of formula IV may be 5 to 50mL/g, preferably 10 to 20mL/g, and more preferably 13 to 19mL/g.
In some embodiments, in the method for preparing the compound of formula III, the acid buffer may be an acid buffer conventional in such reactions in the art, such as an acetate buffer, a formate buffer, and preferably an acetate buffer. The pH of the acid buffer may be 4.0 to 6.0, preferably 4.5 to 5.5, and more preferably 5.0. In some embodiments, the acid buffer is an acetic acid buffer having a pH of 4.0 to 6.0 (preferably 4.5 to 5.5, and more preferably 5.0).
In some embodiments, in the preparation method of the compound of formula III, the volume ratio of the organic solvent to the acid buffer solution can be 1 to 5, preferably 1 to 2, more preferably 1.25 to 1.35, and most preferably 1.28 to 1.30.
In some embodiments, the temperature of the reduction reaction in the process for preparing the compound of formula III may be a temperature conventional in the art for such reactions, for example, 0 to 20 ℃, preferably 0 to 10 ℃, and more preferably 0 to 5 ℃.
In some embodiments, the reduction reaction may be performed in a manner conventional in the art for such reactions, and may, for example, comprise the steps of: the mixture of organic solvent, acid buffer, compound of formula IV and reducing agent is stirred to carry out the reduction reaction.
In some embodiments, the progress of the reduction reaction in the preparation of the compound of formula III may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, or the like), typically such that the compound of formula IV is no longer detectable as a reaction endpoint. The reaction time of the reduction reaction may be 1 to 24 hours, preferably 2 to 5 hours, and more preferably 2 hours.
In some embodiments, in the preparation method of the compound of formula III, after the reduction reaction is finished, a post-treatment step may be further included: and (3) extracting the reaction liquid, removing the solvent from an organic phase obtained by extraction, and purifying the obtained residue to obtain the compound shown in the formula III. The purification can be carried out by conventional purification methods in the field, such as slurry, crystallization, preparative chromatography or silica gel column chromatography, and the like, preferably by silica gel column chromatography, wherein the eluent is preferably a mixture of dichloromethane and methanol, and the elution gradient is preferably from 50.
The process for preparing the compound of formula III may further comprise a process for preparing a compound of formula IV, which may comprise the steps of: carrying out substitution reaction on a compound of a formula V and a compound of a formula VIa in a solvent in the presence of alkali to obtain a compound of a formula IV;

wherein R is 1 、R 2 、R 3 And R 4 As previously described.
In some embodiments, the molar ratio of the compound of formula V to the compound of formula VIa in the process for preparing the compound of formula IV may be 1 to 5, preferably 1 to 2, and more preferably 1.
In some embodiments, in the methods of preparing the compounds of formula IV, the base may be a base conventional in such reactions in the art, such as an organic base, an inorganic base, or a mixture thereof, preferably an organic base. The organic base is preferably tert-butyl potassium, sodium methoxide, triethylamine, DMAP, pyridine, dipyridine, or a mixture of any two or more thereof, and more preferably sodium methoxide. The inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof. In some embodiments, the compound of formula IV is prepared by a process wherein the base is sodium methoxide.
In some embodiments, the molar ratio of the base used to the compound of formula VIa in the process for preparing the compound of formula IV may be 1 to 5, preferably 1 to 2, and more preferably 1.5.
In some embodiments, in the method for preparing the compound of formula IV, the solvent may be a solvent conventional in such reactions in the art, preferably an aprotic organic solvent, such as an ether solvent, a chloroalkane solvent, a nitrile solvent, or a mixture of any two or more thereof, preferably an ether solvent or a chloroalkane solvent. The ether solvent can be tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of the above, preferably 1,4-dioxane. The chloroalkane solvent can be dichloromethane, dichloroethane, chloroform or a mixture of any two or more of the dichloromethane, and dichloromethane is preferred. The nitrile solvent may be acetonitrile. In some embodiments, when R 4 In the case of a silyl ether protecting group such as tert-butyldiphenylsilyl, the solvent may be a chloroalkane solventThe agent is preferably dichloromethane. In some embodiments, when R 4 In the case of an ester protecting group such as acetyl, the solvent may be an ether solvent, preferably 1,4-dioxane.
In some embodiments, in the methods of preparing the compounds of formula IV, the reaction temperature may be a temperature conventional in such reactions in the art, e.g., from 0 to 80 ℃. In some embodiments, when R 4 In the case of an ester-protecting group such as acetyl, the reaction temperature may be 40 to 60 ℃ and more preferably 60 ℃. In some embodiments, when R 4 In the case of a silyl ether-type protecting group such as t-butyldiphenylsilyl group, the reaction temperature may be 0 to 20 ℃ and preferably 0 to 5 ℃.
In some embodiments, the progress of the reaction in the preparation of the compound of formula IV may be monitored by conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula VIa is no longer detectable as an end point of the reaction. The reaction time for the substitution reaction may be 2 to 12 hours. In some embodiments, when R 4 In the case of silyl ether protecting groups such as t-butyldiphenylsilyl, the reaction time may be 3 to 8 hours, for example 4 to 5 hours. In some embodiments, when R 4 In the case of an ester protecting group such as acetyl, the reaction time may be 2 to 3 hours, for example 2 hours.
In some embodiments, the preparation method of the compound of formula IV may further comprise a post-treatment step after the substitution reaction is finished, and the post-treatment step may be a post-treatment step conventional in the art. In some embodiments, when R 4 Where an ester protecting group such as acetyl, the post-treatment step may comprise: the solvent in the reaction solution was removed, the organic phase was washed, the solvent in the organic phase was removed, and the obtained residue was purified. Wherein, the organic phase in the washing can adopt ethyl acetate or dichloromethane, preferably dichloromethane. The water phase in the washing can be prepared by using an aqueous acid solution, water and/or saturated salt water; the aqueous acid solution may be 0.1N hydrochloric acid, 0.05N sulfuric acid or a mixture thereof, preferably 0.1N hydrochloric acid. Preferably, the washing may comprise sequentiallyThe organic phase is washed with an aqueous acid solution, water and saturated brine, preferably with dilute 0.1N acid, water and saturated brine in that order. In some embodiments, the purification can be performed by a conventional purification method in the art, such as slurry, crystallization, preparative chromatography or silica gel column chromatography, and the like, preferably by column silica gel column chromatography, wherein the eluent used is preferably a mixture of dichloromethane and methanol, and the elution gradient is preferably from 100. In some embodiments, when R 4 In the case of silyl ether type protecting groups such as t-butyldiphenylsilyl, the post-treatment step may comprise: washing the reaction solution, and concentrating the obtained organic phase to obtain the compound shown in the formula IV.
In some embodiments, in the methods of preparing the compounds of formula IV, the substitution reaction is preferably performed under anhydrous conditions.
The process for preparing the compound of formula IV may further comprise a process for preparing a compound of formula VIa, which may comprise the steps of: carrying out condensation reaction on a compound shown in a formula VIb and bromoacetic acid in the presence of a condensing agent and alkali to obtain a compound shown in a formula VIa;

wherein R is 2 、R 3 And R 4 As defined above.
In some embodiments, the solvent for the condensation reaction in the preparation method of the compound of formula VIa may be a solvent conventional in such reactions in the art, such as dichloromethane, tetrahydrofuran, N-dimethylformamide, N-dimethylacetamide, dimethylsulfoxide, or a mixture of any two or more thereof, preferably dichloromethane, N-dimethylformamide or a mixture thereof, and more preferably dichloromethane.
In some embodiments, in the method for preparing the compound of formula VIa, the molar ratio of the bromoacetic acid to the compound of formula VIb may be 1 to 5, preferably 1.5 to 3.0, and more preferably 1.5 to 2.0.
In some embodiments, in the method for preparing the compound of formula VIa, the condensing agent may be a conventional condensing agent used in the art for condensation reactions, such as Dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIC), 1- (3-dimethylaminopropyl) -3-Ethylcarbodiimide (EDC) or salts thereof, 2- (7-azabenzotriazole) -N, N' -tetramethylurea Hexafluorophosphate (HATU), propylphosphoric anhydride (T;) and the like 3 P), benzotriazol-N, N' -tetramethyluronium Hexafluorophosphate (HBTU), diphenylphosphoryl azide (DPPA), 4- (4,6-dimethoxytriazin-2-yl) -4-methylmorpholine or a salt thereof (e.g., 4- (4,6-dimethoxytriazin-2-yl) -4-methylmorpholine hydrochloride), preferably Dicyclohexylcarbodiimide (DCC), 4- (4,6-dimethoxytriazin-2-yl) -4-methylmorpholine or a salt thereof, more preferably 4- (4,6-dimethoxytriazin-2-yl) -4-methylmorpholine or a salt thereof, and further preferably 4- (4,6-dimethoxytriazin-2-yl) -4-methylmorpholine hydrochloride.
In some embodiments, in the method for preparing the compound of formula VIa, the molar ratio of the condensing agent to the compound of formula VIb may be 1 to 5, preferably 1.5 to 3.0, and more preferably 1.5 to 2.0.
In some embodiments, the base used in the condensation reaction in the preparation method of the compound of formula VIa may be a base conventionally used in the art for condensation reactions, such as N, N-Diisopropylethylamine (DIEA), triethylamine or 1,8-diazabicycloundecen-7-ene (DBU), preferably triethylamine or N, N-diisopropylethylamine, and more preferably N, N-diisopropylethylamine.
In some embodiments, the molar ratio of the base used in the condensation reaction to the compound of formula VIb in the preparation method of the compound of formula VIa may be 1 to 5, preferably 2 to 4, more preferably 2.0 to 3.0, and still more preferably 2.0 to 2.5.
In some embodiments, the condensation reaction may be carried out at a temperature conventional to such reactions in the art, for example, from 20 to 50 deg.C, preferably from 20 to 30 deg.C, in the process for preparing the compound of formula VIa.
In some embodiments, in the preparation method of the compound of formula VIa, the condensation reaction is preferably performed under the protection of an inert gas, for example, in a nitrogen or helium environment.
In some embodiments, the progress of the condensation reaction in the preparation of the compound of formula VIa may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, or the like), typically such that the compound of formula VIb is no longer detectable as an end point of the reaction. The reaction time of the condensation reaction may be 5 to 20 hours, more preferably 13 to 15 hours, and still more preferably 13 hours.
In some embodiments, in the preparation method of the compound of formula VIa, the compound of formula VIa may be purified by a conventional purification method in the art, such as slurry, crystallization, preparative chromatography or silica gel column chromatography, etc., preferably by column chromatography, and the eluent used is preferably a mixture of dichloromethane and methanol, and the elution gradient may be 100.
The preparation method of the compound of formula VIa may further comprise a preparation method of the compound of formula VIb, which may comprise the following steps: removing 4-methoxyl triphenylmethyl connected with amino in the compound of the formula VIc to obtain the compound of the formula VIb;

wherein R is 2 、R 3 And R 4 As previously described.
In some embodiments, the solvent used in the reaction for removing the 4-methoxytriphenylmethyl group in the preparation of the compound of formula VIb is a reagent commonly used in the art for such reactions, such as chloroform, dichloromethane or mixtures thereof, preferably dichloromethane.
In some embodiments, the deprotection reagent used to remove the 4-methoxytriphenylmethyl group in the preparation method of the compound of formula VIb is a deprotection reagent conventional in the art for such reactions, such as triisopropylsilane or triethylsilane, preferably triethylsilane.
In some embodiments, the molar ratio of the deprotection reagent used to remove 4-methoxytriphenylmethyl group to the compound of formula VIc in the method for preparing the compound of formula VIb may be 1 to 5, preferably 1 to 3, and more preferably 1.2 to 2.5.
In some embodiments, the temperature for the reaction for removing the 4-methoxytriphenylmethyl group in the preparation method of the compound of formula VIb may be a temperature conventional in the art, such as-20 to 10 ℃, preferably-10 to 5 ℃, and more preferably-5 to 5 ℃.
In some embodiments, the 4-methoxytriphenylmethyl group is removed preferably under an inert gas atmosphere, for example, under a nitrogen or helium atmosphere.
In some embodiments, the process of the preparation of the compound of formula VIb, wherein the progress of the reaction to remove 4-methoxytriphenylmethyl group is monitored by conventional methods of art (e.g., TLC, GC, HPLC, NMR, etc.), is typically such that the compound of formula VIc is no longer detectable as an end point of the reaction. In some embodiments, the reaction time for removing the 4-methoxytriphenylmethyl group may be 1 to 5 hours, more preferably 1 to 2 hours, still more preferably 1 to 1.5 hours.
In some embodiments, the method for preparing the compound of formula VIb may further comprise a post-treatment step after the reaction for removing the 4-methoxytriphenylmethyl group is completed. The post-treatment step may be a post-treatment step conventional in this type of reaction in the art, and may for example comprise the steps of: after the reaction is finished, dropwise adding an ether solvent into the reaction system to separate out the compound of the formula VIb from the mixed solution, and separating the separated solid to obtain a crude product of the compound of the formula VIb; the ether solvent can be methyl tert-butyl ether, anisole, propylene glycol methyl ether, propylene glycol ethyl ether, propylene glycol butyl ether or a mixture of any two or more of them, preferably methyl tert-butyl ether or anisole, and more preferably methyl tert-butyl ether. The crude compound of formula VIb may be purified to provide a product compound of formula VIb, which may include: dissolving the crude compound of the formula VIb by using a chloroalkane solvent, then dropwise adding an ether solvent to separate out the compound of the formula VIb, and separating out the separated solid to obtain a compound product of the formula VIb; the chloroalkane solvent can be dichloromethane, dichloroethane or a mixture thereof, and is preferably dichloromethane; the ether solvent can be methyl tert-butyl ether, anisole or a mixture thereof, and is preferably methyl tert-butyl ether; the volume mol ratio of the chloroalkane solvent to the compound of formula VIc can be 10-30mL/mmol, preferably 20-25mL/mmol, and more preferably 20mL/mmol; the volume ratio of the ether solvent to the chloroalkane solvent used may be 2 to 4, preferably 2 to 3, and more preferably 2.
The process for preparing the compound of formula VIb may further comprise a process for preparing a compound of formula VIc, which may comprise the steps of: carrying out hydroxyl protection reaction on a compound shown in a formula VId and a hydroxyl protection reagent to obtain a compound shown in a formula VIc;

wherein R is 2 、R 3 And R 4 As previously described.
In some embodiments, the hydroxy protecting agent used in the preparation method of the compound of formula VIc may be a hydroxy protecting agent conventional in the art, such as acetic anhydride, propionic anhydride, acetyl chloride, propionyl chloride, tert-butyldimethylchlorosilane or tert-butyldiphenylchlorosilane, preferably acetic anhydride, acetyl chloride or tert-butyldiphenylchlorosilane, and more preferably acetic anhydride or tert-butyldiphenylchlorosilane.
In some embodiments, the molar ratio of the hydroxy protecting reagent to the compound of formula VId used in the method of preparing the compound of formula VIc may be 1 to 2, preferably 1 to 1.5, and more preferably 1.1 to 1.44.
In some embodiments, the compound of formula VIc is prepared by a method in which the hydroxyl protection reaction is carried out in the presence of a base. The base may be a base commonly used in the art for such reactions, for example triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine, 1,8-diazabicyclo-bicyclo (5,4,0) -7-undecene, 1,5-diazabicyclo [4.3.0] non-5-ene, N-methylmorpholine, tetramethylethylenediamine, pyridine or a mixture of any two or more thereof, preferably triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine or a mixture of any two or more thereof. In some embodiments, when the hydroxy protecting reagent used in the preparation of the compound of formula VIc is acetic anhydride, the base used is preferably triethylamine, 4-dimethylaminopyridine or a mixture thereof, and more preferably a mixture of triethylamine and 4-dimethylaminopyridine; in the mixing of triethylamine and 4-dimethylaminopyridine, the molar ratio of triethylamine to the compound of formula VId can be 1-5, preferably 1.5-2.0, and more preferably 1.6-1.7; the molar ratio of 4-dimethylaminopyridine to compound of formula VId may be in the range of 1 to 5, preferably 2 to 3, more preferably 2. In some embodiments, when the protecting agent used in the process for preparing the compound of formula VIc is t-butyldiphenylchlorosilane, the base used is preferably N, N-diisopropylethylamine; the molar ratio of N, N-diisopropylethylamine to the compound of formula VId may be 1 to 5, preferably 2 to 3, more preferably 2.
In some embodiments, the hydroxy-protecting reaction of the process for preparing the compound of formula VIc may be performed as is conventional in the art, and may, for example, comprise the steps of: adding (e.g., dropwise) a hydroxyl protecting agent to a solution of the compound of formula VId in portions; optionally, the temperature of the process control reaction system added in portions is from-5 to 10 ℃ (e.g., 0-10 ℃ or 0-5 ℃).
In some embodiments, the solvent for the hydroxy-protecting reaction in the process for preparing the compound of formula VIc may be a solvent conventional in the art for such reactions, such as dichloromethane, dichloroethane, N-dimethylformamide, N-dimethylacetamide, or a mixture of any two or more thereof; preferably dichloromethane or N, N-dimethylformamide. In some embodiments, when the hydroxyl protecting agent used is acetic anhydride, the solvent used may be methylene chloride. In some embodiments, when the hydroxy protecting agent used is t-butyldiphenylchlorosilane, the solvent used may be N, N-dimethylformamide.
In some embodiments, the temperature of the hydroxyl protection reaction in the process for preparing the compound of formula VIc may be a temperature conventional in the art for such reactions, for example, from 0 to 40 deg.C, preferably from 10 to 30 deg.C, and more preferably from 20 to 30 deg.C.
In some embodiments, in the preparation method of the compound of formula VIc, the hydroxyl protecting reaction is preferably performed under the protection of an inert gas, for example, under a nitrogen or helium atmosphere.
In some embodiments, the progress of the hydroxy-protecting reaction in the preparation of the compound of formula VIc may be monitored by conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula VId is no longer detectable as an end point of the reaction. The reaction time of the hydroxyl group-protecting reaction may be 2 to 10 hours, more preferably 3 to 5 hours, and still more preferably 3 hours.
The process for preparing the compound of formula VIc may further comprise a process for preparing a compound of formula VId, which may comprise the steps of:
(i) Reacting irinotecan with trimethylchlorosilane;
(ii) (ii) reacting the reaction solution obtained in the step (i) with 4-methoxytriphenylmethane chloride in the presence of alkali to obtain the compound shown in the formula VId;

wherein R is 2 And R 3 As defined above.
In some embodiments, the solvent used in the reaction of steps (i) and (ii) in the preparation of the compound of formula VId may be a solvent conventional in the art, such as dichloromethane, chloroform, dichloroethane, or a mixture of any two or more thereof, preferably dichloromethane.
In some embodiments, in the preparation method of the compound of formula VId, the molar ratio of trimethylchlorosilane to irinotecan used in the reaction of step (i) may be 1 to 3, preferably 1.2 to 2.0, and more preferably 1.2.
In some embodiments, in the method for preparing the compound of formula VId, the reaction temperature in step (i) may be 20 to 60 ℃, preferably 40 to 45 ℃, and more preferably 45 ℃.
In some embodiments, the progress of the reaction in step (i) in the process for preparing the compound of formula VId may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically with no more irinotecan being detected as an end point of the reaction. In some embodiments, the reaction time of step (i) may be 1 to 5 hours, preferably 1 to 2 hours, and further preferably 1 hour.
In some embodiments, the compound of formula VId may be prepared in a molar ratio of 4-methoxytriphenylmethane chloride of step (ii) to irinotecan of step (i) of 1 to 3, preferably 1.2 to 2.0, and more preferably 1.2.
In some embodiments, in the method for preparing the compound of formula VId, the base of step (ii) may be a commonly used base used in the art for such reactions, such as triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine, 1,8-diazabicyclo-bicyclo (5,4,0) -7-undecene, 1,5-diazabicyclo [4.3.0] non-5-ene, N-methylmorpholine, tetramethylethylenediamine, pyridine, or a mixture of any two or more thereof, preferably triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine, or a mixture of any two or more thereof, more preferably N, N-diisopropylethylamine.
In some embodiments, the compound of formula VId may be prepared in a process wherein the molar ratio of base of step (ii) to irinotecan of step (i) is from 2 to 5, preferably from 3 to 4, and more preferably 3.
In some embodiments, in the method for preparing a compound of formula VId, the step (ii) may comprise the steps of: (ii) adding 4-methoxytriphenylmethane chloride and a base to the reaction solution of the step (i), wherein the reaction system is controlled to be 0-10 ℃ (e.g. 0-5 ℃, e.g. 0 ℃).
In some embodiments, in the method for preparing the compound of formula VId, the reaction temperature in step (ii) may be a temperature conventional in such reactions in the art, for example, 0 to 40 ℃, preferably 10 to 30 ℃, and more preferably 20 to 30 ℃.
In some embodiments, in the preparation of the compound of formula VId, the progress of the reaction of step (ii) may be monitored by conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically with no further detection of the product of step (i) as an end point of the reaction. The reaction time in the step (ii) may be 12 to 24 hours, more preferably 16 to 24 hours, and still more preferably 16 to 18 hours.
The process for preparing the compound of formula IV may further include a process for preparing a compound of formula V, which may include the steps of: reacting a compound shown in a formula VII with paraformaldehyde in the presence of alkali to obtain a compound shown in a formula V;

wherein R is 1 As previously described.
In some embodiments, the molar ratio of the paraformaldehyde to the compound of formula VII in the process for preparing a compound of formula V is 1 to 3, preferably 1 to 2, and more preferably 1.5 in terms of formaldehyde.
In some embodiments, the base in the process for preparing the compound of formula V may be a base conventional in such reactions in the art, such as an organic base, an inorganic base, or a mixture thereof, preferably an inorganic base. The organic base is preferably tert-butyl potassium, sodium methoxide, triethylamine, DMAP, pyridine, dipyridine, or a mixture of any two or more thereof, and more preferably sodium methoxide. The inorganic base is preferably an alkali metal carbonate, an alkali metal hydroxide, an alkali metal phosphate, or a mixture of any two or more thereof, and more preferably sodium hydrogen carbonate, sodium carbonate, potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof. In some embodiments, the compound of formula V is prepared by a process wherein the base is sodium bicarbonate.
In some embodiments, the molar ratio of the sodium bicarbonate to the compound of formula VII in the process for preparing the compound of formula V is 1-4, preferably 1-2, and more preferably 1.4.
In some embodiments, in the preparation method of the compound of formula V, the solvent may be a solvent conventional in such reactions in the art, and is preferably a mixed system of an ether solvent and water; the ether solvent can be tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of the above, preferably 1,4-dioxane.
In some embodiments, the reaction temperature in the process for preparing the compound of formula V may be a temperature conventional in such reactions in the art, for example, from 10 to 40 ℃, preferably from 25 to 40 ℃, and more preferably from 25 to 30 ℃.
In some embodiments, the reaction of the process for preparing the compound of formula V may be conducted in a manner conventional in the art for such reactions, including, for example, the steps of: sodium bicarbonate is added in portions to a mixture of the compound of formula VII, paraformaldehyde and solvent, and the reaction is carried out with stirring.
In some embodiments, the progress of the reaction in the process for preparing the compound of formula V may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula VII is no longer detected as an end point of the reaction. The reaction time of the reaction may be 1 to 60 hours, preferably 12 to 40 hours, more preferably 16 to 35 hours, and still more preferably 24 hours.
In some embodiments, the method for preparing the compound of formula V may further comprise the following post-treatment step after the substitution reaction is completed: and (3) carrying out solid-liquid separation on the reaction liquid, removing the solvent from the obtained liquid phase, and directly using the obtained residue for the next reaction.
The process for preparing the compound of formula V may further comprise a process for preparing a compound of formula VII, which may comprise the steps of: reacting a compound shown in a formula VIII with a sulfonyl azide compound in the presence of a base and a catalyst in a solvent to obtain a compound shown in a formula VII;

wherein R is 1 As previously described.
In some embodiments, in the method of preparing the compound of formula VII, the sulfonyl azide compound may be 1H-imidazole-1-sulfonyl azide hydrochloride, 2-azido-1,3-dimethylimidazole hexafluorophosphate, trifluorosulfonyl azide, p-methylbenzenesulfonylazide, or methanesulfonylazide, preferably 1H-imidazole-1-sulfonyl azide hydrochloride.
In some embodiments, in the method for preparing the compound of formula VII, the molar ratio of the sulfonyl azide compound to the compound of formula VIII is 1.0.
In some embodiments, the base in the process for preparing the compound of formula VII may be a base conventional in such reactions in the art, such as an organic base, an inorganic base, or a mixture thereof, preferably an inorganic base; wherein the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof, and further preferably potassium carbonate; the organic base is preferably tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-lutidine, or a mixture of any two or more thereof. In some embodiments, the process for preparing the compound of formula V wherein the base is an alkali metal carbonate, such as potassium carbonate.
In some embodiments, the molar ratio of the base to the compound of formula VIII in the process for preparing the compound of formula VII is 1.5 to 3.0, preferably 2.0 to 2.5, and more preferably 2.0.
In some embodiments, the catalyst in the process for preparing the compound of formula VII may be a catalyst conventional in the art for such reactions, such as a ketone salt, preferably copper sulfate, more preferably copper sulfate pentahydrate.
In some embodiments, in the method of preparing the compound of formula VII, the molar ratio of the ketone salt to the compound of formula VIII may be 0.1 to 0.5, preferably 0.1 to 0.3, more preferably 0.1 to 0.2, and most preferably 0.1.
In some embodiments, in the preparation method of the compound of formula VII, the solvent may be a solvent conventional in the art for such reactions, preferably a mixed solvent of an organic solvent and water, and the organic solvent may be an alcohol solvent, a chloroalkane solvent, an ether solvent, or a mixture of any two or more thereof, preferably a mixture of an alcohol solvent and a chloroalkane solvent; the alcohol solvent can be methanol, ethanol, isopropanol or a mixture of any two or more of the methanol, the ethanol and the isopropanol, and preferably methanol; the chloroalkane solvent can be dichloromethane, chloroform, dichloroethane or a mixture of any two or more of the dichloromethane, and dichloromethane is preferred; the ether solvent is preferably tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of the above. In some embodiments, the solvent is a mixture of methanol, dichloromethane and water.
In some embodiments, the reaction temperature in the process for the preparation of the compound of formula VII may be a temperature conventional in such reactions in the art, e.g. from 10 to 40 ℃, preferably from 25 to 40 ℃, and more preferably from 25 to 30 ℃.
In some embodiments, the reaction of the process for preparing the compound of formula VII may be performed in a manner conventional in the art for such reactions, for example, including the steps of: adding the sulfonyl azide compound into a mixed system of the compound shown in the formula VIII, alkali, catalyst and solvent (preferably, adding the sulfonyl azide compound after the mixed system is clarified), and stirring for reacting.
In some embodiments, the progress of the reaction in the preparation of the compound of formula VII may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula VIII is no longer detectable as an end point of the reaction. The reaction time of the reaction may be 1 to 24 hours, preferably 12 to 20 hours, more preferably 16 to 20 hours, and still more preferably 16 hours.
In some embodiments, the method for preparing the compound of formula VII may further comprise the following post-treatment steps after the reaction is completed: removing the organic solvent from the reaction solution, extracting (for example, extracting with dichloromethane), and recrystallizing the organic phase obtained by extraction with ethanol and activated carbon to obtain the compound of formula VII.
The process for preparing the compound of formula VII may further comprise a process for preparing a compound of formula VIII, which may comprise the steps of: carrying out Fmoc removal reaction on a compound in a formula IX in an organic solvent in the presence of a base to obtain a compound in a formula VIII;

wherein, R 1 As previously described.
In some embodiments, in the methods of preparing the compounds of formula VIII, the base may be a base conventional in such reactions in the art, such as an organic base, an inorganic base, or a mixture thereof, preferably an organic base; wherein, the organic base is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-dimethylpyridine or a mixture of any two or more of them, and is further preferably ethylenediamine; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof. In some embodiments, in the method of making a compound of formula VIII, the base is diethylamine.
In some embodiments, in the method for preparing the compound of formula VIII, the volume ratio of the base to the organic solvent may be 0.1 to 0.5, preferably 0.2 to 0.3, and more preferably 0.2.
In some embodiments, in the method for preparing the compound of formula VIII, the organic solvent may be DMF, DMSO, tetrahydrofuran, 1,4-dioxane, or a mixture of any two or more thereof, preferably DMF.
In some embodiments, the temperature for the Fmoc removal reaction in the method for preparing the compound of formula VIII may be a temperature conventional in the art, such as 10-40 deg.C, preferably 25-40 deg.C, and more preferably 25-30 deg.C.
In some embodiments, the Fmoc removal reaction of the process for preparing the compound of formula VIII may be performed according to procedures conventional in the art, such as by stirring a mixture of the compound of formula IX, a base and an organic solvent.
In some embodiments, the progress of the Fmoc removal reaction in the process for preparing the compound of formula VIII may be monitored using assays routine in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula IX is no longer detectable as an end point of the reaction. The reaction time of the reaction may be 1 to 24 hours, preferably 4 to 12 hours, more preferably 4 to 6 hours, and still more preferably 4 hours.
In some embodiments, the method for preparing the compound of formula VIII may further include the following post-treatment steps after the reaction is completed: and (3) carrying out solid-liquid separation on the reaction liquid, and removing the solvent in the obtained liquid phase to obtain a crude product. The post-processing step may further include the steps of: pulping the crude product to obtain a solid product of the compound of formula VIII. The solvent used for pulping can be an ether solvent, such as tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of them, preferably methyl tert-butyl ether. The post-processing step may further include the steps of: and recrystallizing and purifying the solid product of the compound of the formula VIII obtained by pulping. The solvent used for recrystallization may be an alcohol solvent, such as methanol, ethanol, isopropanol, or a mixture of any two or more thereof, preferably ethanol.
The process for preparing the compound of formula VIII may further comprise a process for preparing a compound of formula IX, which may comprise the steps of: carrying out coupling reaction on a compound shown in the formula X and N-Fmoc-L-valine N-butadiene amine imino ester in a solvent to obtain a compound shown in the formula IX;

wherein R is 1 As previously described.
In some embodiments, in the method of preparing the compound of formula IX, the molar ratio of the N-Fmoc-L-valine N-butanamidine ester to the compound of formula X may be 0.8 to 5, preferably 0.8 to 1.2, and more preferably 1.
In some embodiments, in the method of preparing the compound of formula IX, the solvent may be DMF, DMSO, acetonitrile, dichloromethane, dichloroethane, or a mixture of any two or more thereof, preferably dichloromethane.
In some embodiments, the temperature of the coupling reaction in the process for preparing the compound of formula IX may be a temperature conventional in the art for such reactions, for example, from 10 to 40 ℃, preferably from 35 to 40 ℃, and more preferably 40 ℃.
In some embodiments, in the preparation method of the compound of formula IX, the coupling reaction is preferably performed under a gas protection. The gas in the gas shield does not participate in the reaction, such as argon, helium or nitrogen, and further such as nitrogen.
In some embodiments, the coupling reaction of the process for preparing the compound of formula IX may be conducted in a manner conventional in the art and may, for example, comprise stirring a mixture of the compound of formula X, N-Fmoc-L-valine N-butanamine ester, and a solvent to effect the coupling reaction.
In some embodiments, the progress of the coupling reaction in the preparation of the compound of formula IX may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula X is no longer detected as an end point of the reaction. The reaction time of the coupling reaction may be 1 to 24 hours, preferably 12 to 20 hours, more preferably 16 to 20 hours, and still more preferably 16 hours.
In some embodiments, the method for preparing the compound of formula IX may further comprise a post-treatment step after the coupling reaction, and the post-treatment step may comprise: after adding an alcoholic solvent (for example, methanol, ethanol, isopropanol, or a mixture of any two or more thereof, preferably methanol) to the reaction system, stirring (the stirring temperature may be 20 to 40 ℃, preferably 35 to 40 ℃, more preferably 40 ℃, and the stirring time may be 1 to 24 hours, preferably 4 to 12 hours, more preferably 4 to 6 hours, and even more preferably 4 hours), and separating the solid in the system to obtain the compound of formula VII.
The process for preparing the compound of formula IX may further comprise a process for preparing a compound of formula X, which may comprise the steps of: carrying out Fmoc removal reaction on a compound in a formula XI in a solvent in the presence of alkali to obtain a compound in a formula X;

wherein R is 1 As previously described.
In some embodiments, in the methods of preparing the compounds of formula X, the base may be a base conventional in such reactions in the art, such as an organic base, an inorganic base, or a mixture thereof, preferably an organic base; wherein, the organic alkali is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-dimethylpyridine or a mixture of any two or more of them, and more preferably ethylenediamine; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof. In some embodiments, the method of making a compound of formula X, wherein the base is diethylamine.
In some embodiments, in the method of preparing the compound of formula X, the solvent may be DMF, DMSO, tetrahydrofuran, 1,4-dioxane, or a mixture of any two or more thereof, preferably DMF.
In some embodiments, in the method for preparing the compound of formula X, the volume ratio of the base to the solvent may be 0.2 to 0.5, preferably 0.3 to 0.4, and more preferably 0.3.
In some embodiments, the reaction temperature for the Fmoc removal reaction in the method for preparing the compound of formula X may be a temperature conventional in the art for such reactions, such as 10-40 deg.C, preferably 25-40 deg.C, and more preferably 25-30 deg.C.
In some embodiments, the Fmoc removal reaction of the process for preparing the compound of formula X may be performed in a manner conventional in the art for such reactions, and may, for example, comprise the step of stirring a mixture of the compound of formula XI, a base and a solvent to perform the Fmoc removal reaction.
In some embodiments, the process of preparing the compound of formula X, wherein the progress of the Fmoc removal reaction is monitored using assays conventional in the art (e.g., TLC, GC, HPLC, NMR, etc.), is generally such that the compound of formula XI is no longer detectable as an end point of the reaction. The reaction time of the reaction may be 1 to 24 hours, preferably 2 to 12 hours, more preferably 2 to 6 hours, and still more preferably 2 hours.
In some embodiments, the method for preparing the compound of formula X may further include a post-treatment step after the Fmoc removal reaction is completed, and the post-treatment step may include: and removing the solvent in the reaction solution, and pulping the obtained residue to obtain a solid, namely the compound of the formula X. The solvent used for pulping can be an ether solvent, such as tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of them, preferably methyl tert-butyl ether.
The process for the preparation of the compound of formula X may further comprise a process for the preparation of a compound of formula XI, which may comprise the steps of: reacting a compound of formula XII with an amino compound R 1 NH 2 Carrying out coupling reaction in a solvent in the presence of alkali to obtain a compound shown in a formula XI;

wherein R is 1 As previously described.
In some embodiments, in the process for preparing a compound of formula XI, the amino compound R 1 NH 2 The molar ratio to the compound of the formula XII may be from 1.0 to 3.0, preferably from 1.1 to 1.5, more preferably 1.1.
In some embodiments, the base in the process for preparing the compound of formula XI may be a base conventional in such reactions in the art, such as an organic base, an inorganic base, or a mixture thereof, preferably an organic base; wherein, the organic base is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-dimethylpyridine or a mixture of any two or more of them, and is more preferably DMAP; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof. In some embodiments, the base can be DMAP in the process for preparing a compound of formula XI.
In some embodiments, the molar ratio of the base to the compound of formula XII in the process for preparing a compound of formula XI can be from 2 to 4, preferably from 2.5 to 3.0, and more preferably 2.5.
In some embodiments, in the process for preparing the compound of formula XI, the solvent may be DMF, DMSO, dichloromethane, dichloroethane, tetrahydrofuran, 1,4-dioxane, or a mixture of any two or more thereof, preferably dichloromethane.
In some embodiments, the coupling reaction temperature in the process for the preparation of the compound of formula XI may be a temperature conventional in such reactions in the art, for example, from 10 to 40 deg.C, preferably from 25 to 40 deg.C, and more preferably from 25 to 30 deg.C.
In some embodiments, the preparation of the compound of formula XIIn the process, the coupling reaction may be carried out as is conventional in the art for such reactions, and may, for example, comprise the step of stirring a compound of formula XII, an amino compound R 1 NH 2 And a mixed system of alkali and solvent to carry out coupling reaction.
In some embodiments, the progress of the coupling reaction in the preparation of the compounds of formula XI may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula XII is no longer detected as an end point of the reaction. The reaction time of the reaction may be 1 to 24 hours, preferably 12 to 20 hours, more preferably 12 to 16 hours, and still more preferably 12 hours.
In some embodiments, the method for preparing a compound of formula XI may further comprise a post-treatment step after the coupling reaction, and the post-treatment step may comprise: the reaction solution is subjected to solid-liquid separation, and the solid obtained by solid-liquid separation (the solid may be washed with an organic solvent such as diethyl ether, ethyl acetate or a mixture thereof, preferably ethyl acetate after solid-liquid separation) is a part of the product of the compound of formula XI. The post-processing step may further include: and (3) carrying out extraction washing on the liquid phase obtained by the solid-liquid separation, removing the solvent from the obtained organic phase, and pulping the obtained solid to obtain the other part of the product of the compound in the formula XI. The extraction washing may be performed by using an aqueous acid solution, an aqueous alkali solution, water and a saturated saline solution, preferably by sequentially using an aqueous acid solution, an aqueous alkali solution, water and a saturated saline solution; further preferably, washing with an aqueous acid solution 1 time, washing with an aqueous alkali solution 2 times, washing with water 1 time, and washing with saturated brine 1 time in this order; the aqueous acid solution may be an aqueous hydrochloric acid solution, an aqueous sulfuric acid solution or an aqueous phosphoric acid solution, preferably 1N hydrochloric acid, 0.5N sulfuric acid or 0.33N phosphoric acid, and more preferably 1N hydrochloric acid; the aqueous alkali solution may be an aqueous sodium hydroxide solution, an aqueous potassium hydroxide solution, or a mixture thereof, preferably 1N sodium hydroxide, 1N potassium hydroxide, or a mixture thereof, and more preferably 1N sodium hydroxide. The solvent for pulping can be dichloromethane, ethyl acetate or a mixture thereof, preferably dichloromethane; the beating temperature may be 10 to 40 ℃, preferably 25 to 40 ℃, and more preferably 25 to 30 ℃.
The process for the preparation of the compound of formula XI may further comprise a process for the preparation of a compound of formula XII, which may comprise the steps of: reacting a compound of formula XIII and a compound of formula XIV in the presence of a base and in a solvent to obtain said compound of formula XII;

in some embodiments, in the method for preparing the compound of formula XII, the molar ratio of the compound of formula XIV to the compound of formula XIII may be 3.0 to 1.2, preferably 2.0 to 1.5, and more preferably 1.5.
In some embodiments, in the process for the preparation of compounds of formula XII, the base may be a base conventional in such reactions in the art, such as an organic base, an inorganic base, or a mixture thereof, preferably an organic base; wherein, the organic base is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-dimethylpyridine or a mixture of any two or more of them, and is further preferably pyridine; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture thereof, and more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof. In some embodiments, the process for preparing a compound of formula XII wherein the base is pyridine.
In some embodiments, in the method for preparing the compound of formula XII, the molar ratio of the base to the compound of formula XIII may be 1.0 to 4.0, preferably 2.0 to 3.0, and more preferably 2.0.
In some embodiments, in the method of preparing the compound of formula XII, the solvent can be DMF, DMSO, dichloromethane, dichloroethane, tetrahydrofuran, 1,4-dioxane, or a mixture of any two or more thereof, preferably dichloromethane.
In some embodiments, the reaction temperature in the process for the preparation of the compound of formula XII may be a temperature conventional in such reactions in the art, for example, from 10 to 40 ℃, preferably from 25 to 40 ℃, and more preferably from 25 to 30 ℃.
In some embodiments, the reaction of the process for preparing the compound of formula XII may be carried out according to a conventional procedure in the art, and for example, the reaction may be carried out by adding the compound of formula XIV in portions (which may be divided into 6 to 3 portions, preferably 5 to 4 portions) to a mixed system of the compound of formula XIII, the base and the solvent (the addition may be carried out while controlling the temperature of the mixed system to 0 to 20 ℃, preferably 10 to 0 ℃, and more preferably 0 to 5 ℃) and stirring.
In some embodiments, the progress of the reaction in the process for preparing the compound of formula XII may be monitored using conventional testing methods in the art (e.g., TLC, GC, HPLC, NMR, etc.), typically such that the compound of formula XIII is no longer detectable as an end point of the reaction. The reaction time of the reaction may be 1 to 24 hours, preferably 2 to 12 hours, more preferably 4 to 8 hours, and still more preferably 4 hours.
In some embodiments, the method for preparing the compound of formula XII may further include a post-treatment step after the reaction is completed, and the post-treatment step may include: and (3) carrying out solid-liquid separation on the reaction liquid, wherein the solid obtained by the solid-liquid separation is a part of the product of the compound shown in the formula XII. The post-processing step may further include: and (3) washing the liquid phase obtained by solid-liquid separation, removing the solvent from the obtained organic phase, and pulping the obtained solid to obtain the other part of the compound shown in the formula XII. The extraction washing can use acid aqueous solution, alkali aqueous solution, water and saturated saline solution, preferably uses acid aqueous solution, alkali aqueous solution, water and saturated saline solution to carry out extraction washing in sequence; further preferably, washing with an aqueous acid solution 1 time, washing with an aqueous alkali solution 2 times, washing with water 1 time, and washing with saturated brine 1 time in this order; the aqueous acid solution may be an aqueous hydrochloric acid solution, an aqueous sulfuric acid solution or an aqueous phosphoric acid solution, preferably 1N hydrochloric acid, 0.5N sulfuric acid or 0.33N phosphoric acid, and more preferably 1N hydrochloric acid; the aqueous alkali solution may be an aqueous sodium hydroxide solution, an aqueous potassium hydroxide solution, or a mixture thereof, preferably 1N sodium hydroxide, 1N potassium hydroxide, or a mixture thereof, and more preferably 1N sodium hydroxide. The solvent for pulping can be tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of the above, preferably methyl tert-butyl ether.
The present invention also provides a compound of formula II:

wherein R is 1 、R 2 、R 3 And R 4 As defined above.
In some embodiments, the compound of formula II is

The invention also provides a preparation method of the compound shown in the formula II, which comprises the following steps: carrying out coupling reaction on a compound shown in a formula III and 6- (maleimide) hexanoic acid succinimide ester in a solvent to obtain a compound shown in a formula II;

wherein R is 1 、R 2 、R 3 And R 4 As defined above.
In the preparation method of the compound of the formula II, the reaction conditions can be as described above. The process for preparing the compound of formula II may further include the process for preparing the compound of formula III as described herein.
The present invention also provides a compound of formula III:

wherein R is 1 、R 2 、R 3 And R 4 As defined above.
In some embodiments, the compound of formula III is

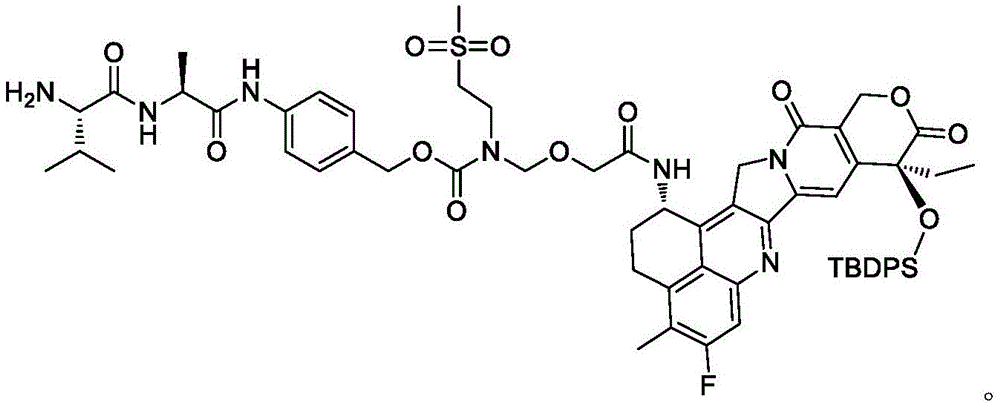
The invention also provides a preparation method of the compound shown in the formula III, which comprises the following steps: carrying out reduction reaction on a compound shown in a formula IV and a reducing agent in an organic solvent in the presence of an acid buffer solution to obtain a compound shown in a formula III;

wherein R is 1 、R 2 、R 3 And R 4 As defined above.
In the preparation method of the compound of the formula III, the reaction conditions can be as described above. The process for preparing the compound of formula III may further include the process for preparing the compound of formula IV as described herein.
The present invention also provides a compound of formula IV:

wherein R is 1 、R 2 、R 3 And R 4 As defined above.
In some embodiments, the compound of formula IV is

The invention also provides a preparation method of the compound shown in the formula IV, which comprises the following steps: carrying out substitution reaction on a compound shown in a formula V and a compound shown in a formula VIa in a solvent in the presence of alkali to obtain a compound shown in a formula IV;

wherein R is 1 、R 2 、R 3 And R 4 As previously described.
In the preparation method of the compound of formula IV, the reaction conditions can be as described above. The process for preparing the compound of formula IV may further comprise a process for preparing a compound of formula V and/or a compound of formula VIa as described herein.
The invention also provides a compound represented by formula VIa:

wherein R is 2 、R 3 And R 4 As defined above.
In some embodiments, the compound of formula VIa is

The invention also provides a preparation method of the compound of the formula VIa, which comprises the following steps: carrying out condensation reaction on a compound shown in a formula VIb and bromoacetic acid in the presence of a condensing agent and alkali to obtain a compound shown in a formula VIa;

wherein R is 2 、R 3 And R 4 As defined above.
In the preparation method of the compound of formula VIa, the reaction conditions can be as described above. The process for preparing the compound of formula VIa may further comprise a process for preparing a compound of formula VIb as described herein.
The invention also provides a compound of formula VIb:

wherein R is 2 、R 3 And R 4 As defined above.
In some embodiments, the compound of formula VIb is

The present invention also provides a process for the preparation of a compound of formula VIb, which may comprise the steps of: removing 4-methoxyl triphenylmethyl connected with amino in the compound of formula VIc to obtain the compound of formula VIb;

wherein R is 2 、R 3 And R 4 As defined above.
In the preparation method of the compound of formula VIb, the reaction conditions can be as described above. The process for preparing the compound of formula VIb may further comprise a process for preparing a compound of formula VIc as described herein.
The invention also provides a compound of formula VIc:

wherein R is 2 、R 3 And R 4 As defined above.
In some embodiments, the compound of formula VIc is

The invention also provides a preparation method of the compound of the formula VIc, which comprises the following steps: carrying out hydroxyl protection reaction on a compound shown in a formula VId and a hydroxyl protection reagent to obtain a compound shown in a formula VIc;

wherein R is 2 、R 3 And R 4 As defined above.
In the preparation method of the compound of formula VIc, the reaction conditions can be as described above. The process for preparing the compound of formula VIc may further comprise a process for preparing a compound of formula VId as described herein.
The present invention also provides a compound of formula VId:
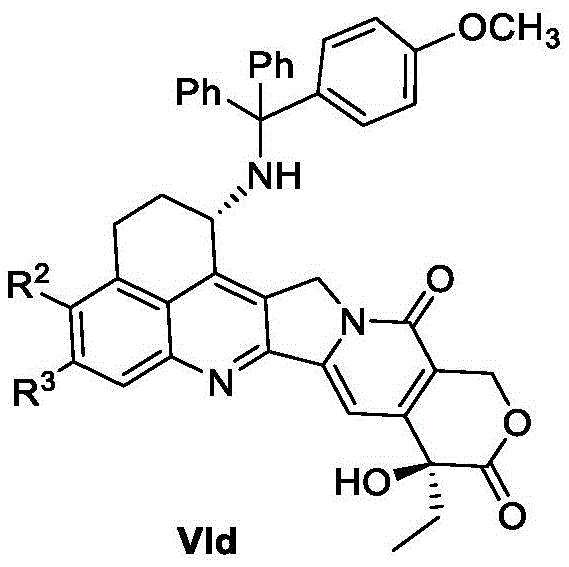
wherein R is 2 And R 3 As defined above.
The invention also provides a preparation method of the compound of the formula VId, which comprises the following steps:
(i) Reacting irinotecan with trimethylchlorosilane;
(ii) (ii) reacting the reaction solution obtained in the step (i) with 4-methoxytriphenylmethane chloride in the presence of alkali to obtain the compound shown in the formula VId;

wherein R is 2 And R 3 As defined above.
In the preparation method of the compound of formula VId, the reaction conditions can be as described above.
The invention also provides a compound, which has a structure shown in the formula V:

wherein R is 1 As defined above.
In one embodiment, the compound of formula V is

The invention also provides a preparation method of the compound of the formula V, which comprises the following steps: reacting a compound shown in a formula VII with paraformaldehyde in the presence of alkali to obtain a compound shown in a formula V;

wherein R is 1 As previously described.
In the preparation method of the compound of the formula V, the reaction conditions can be as described above.
Definition of
In the present invention, the term "C 1 -C 6 Alkyl "denotes a saturated straight-chain or branched alkyl group comprising 1 to 6, in particular 1 to 4, carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl and the like, in particular methyl or ethyl.
In the present invention, the term "halogen" denotes fluorine, chlorine, bromine or iodine, in particular fluorine or chlorine.
Table 1: abbreviations


The above preferred conditions can be arbitrarily combined to obtain preferred embodiments of the present invention without departing from the common general knowledge in the art.
The reagents and starting materials used in the present invention are commercially available.
The positive progress effects of the invention are as follows: provides a preparation method of a novel connecting base drug conjugate shown in formula I and an intermediate thereof. The preparation method is simple to operate, easy to control the product quality, high in yield and suitable for industrial production. The preparation process of the present invention is advantageous compared to the processes of synthetic routes 1 and 2 of WO2020259258A1 mentioned in the background section: the intermediate products of each step after irinotecan is introduced in the synthetic routes 1 and 2 of WO2020259258A1 are difficult to dissolve in most organic solvents, so that the purification is difficult, and the purity of the final product is influenced; it has been found through exploration that the reaction route of the invention introduces the compound with R 4 The solubility of intermediate compounds obtained after the irinotecan of the protecting group is protected is obviously improved, and key intermediates with better purity can be obtained by purification by methods such as recrystallization, column purification and the like, so that final products meeting requirements can be further obtained.
Detailed Description
The invention is further illustrated by the following examples, which are not intended to limit the invention thereto. The experimental methods without specifying specific conditions in the following examples were selected according to the conventional methods and conditions, or according to the commercial instructions.
In the following examples, the mass spectrum adopts a Waters Acquity Xevo G2-XS QTof UPLC/MS ultra-performance liquid chromatography high-resolution mass spectrometry system, 1 H-NMR was performed by using Bruker AVANCE III MHz NMR instrument or Bruker AVANCE III HD MHz NMR instrument, and HPLC was performed by Agilent 1260 HPLC.
In the following examples, room temperature means 20-30 ℃.
Example 1: synthesis method of compound 1
Step 1: synthesis of Compound 11

Compound 13 (10.0g, 32.2mmol) and compound 14 (4.0g, 32.5mmol) were dispersed in 200mL of dichloromethane at room temperature, and EEDQ (9.5g, 38.4 mmol) was added in 3 portions. After the addition, the reaction solution was mechanically stirred at room temperature for 12 hours, and a large amount of white solid was precipitated in the system. TLC monitored that the reaction of the starting material was complete, the reaction mixture was directly filtered by suction and the filter cake was dried to give Compound 11 as a white solid (11.0 g, 82% yield).
MS:m/z=417.2(M+H);
1 H NMR(400MHz,DMSO-d 6 )δppm 9.95(s,1H),7.88(t,J=9.9Hz,2H),7.80-7.65(m,3H),7.55(t,J=7.7Hz,2H),7.46-7.19(m,6H),5.20-5.02(m,1H),4.43(t,J=7.7Hz,2H),4.32-4.06(m,4H),1.41-1.21(m,3H)。
Step 2: synthesis of Compound 10

Compound 11 (11.0 g,26.4 mmol) was dispersed in 200mL of dichloromethane at room temperature, and pyridine (4.2 mL,52.8 mmol) was added. The resulting mixture was cooled to 0 ℃ in an ice bath, p-nitrophenol chloroformate (4 times, 8.0g,39.6 mmol) was added in portions while maintaining the temperature in the ice bath, and after the addition was completed, the resulting reaction mixture was stirred at room temperature for 4 hours. After TLC monitoring of the reaction of the raw materials is completed, carrying out suction filtration on the reaction solution, collecting a filter cake and drying to obtain a1 st batch of solid; the filtrate was washed with 1N hydrochloric acid, 1N sodium hydroxide (2 times), water and saturated brine in this order, dried, concentrated, slurried with methyl tert-butyl ether for 1 hour, filtered, and the filter cake was collected and dried to obtain batch 2 solid. The two batches of solids were combined to give compound 10 as a pale yellow solid (12.0 g, 78% yield).
And 3, step 3: synthesis of Compound 9

Compound 10 (12.0 g,20.6 mmol) and thiamphenyethylamine hydrochloride (3.6 g,22.6mmol,1.1 equiv) were dispersed in 200mL of dichloromethane at room temperature, and DMAP (6.3 g,51.6 mmol) was added in 3 portions. The reaction solution was further stirred at room temperature for 12 hours, and a large amount of pale yellow solid was precipitated. TLC monitors the reaction of the raw materials completely, then the mixture is filtered by suction, and the filter cake is washed 2 times with ethyl acetate (100 mL each) and dried to obtain 7.6g of a first batch of white solid. The filtrate was spin-dried, dissolved in 150mL of ethyl acetate, washed successively with 1N hydrochloric acid (50 mL), 1N sodium hydroxide (2 times 50mL each), water and saturated brine (100 mL), dried, concentrated, slurried with dichloromethane (20 mL), filtered and dried to give 2.0g of batch 2 as a white solid. The two solid batches were combined to give compound 9 (9.6 g, 82% yield) as a white solid.
MS:m/z=566.2(M+H);
1 H-NMR(400MHz,DMSO-d 6 )δppm 10.04(s,1H),7.88(t,J=10.8Hz,2H),7.79-7.66(m,3H),7.59(d,J=8.3Hz,2H),7.51-7.22(m,6H),4.97(s,2H),4.37-4.05(m,4H),3.41(dd,J=12.8,6.6Hz,2H),3.30-3.18(m,2H),2.99(s,3H),1.31(d,J=7.1Hz,3H)。
And 4, step 4: synthesis of Compound 8

Compound 9 (9.6 g,16.9 mmol) was dissolved in 50mL of DMF at room temperature, 15mL of diethylamine was added, the resulting mixture was stirred at room temperature for 2 hours and TLC monitored for completion of the starting material reaction. The reaction was directly concentrated to remove the solvent, and the resulting residue was slurried with methyl tert-butyl ether (50 mL) to give Compound 8 (5.2 g, 89% yield) as a white solid.
MS:m/z=344.1(M+H);
1 H-NMR(400MHz,DMSO-d 6 )δppm 7.63(d,J=8.5Hz,2H),7.45(t,J=5.6Hz,1H),7.28(d,J=8.4Hz,2H),4.97(s,2H),3.41(qd,J=7.0,3.2Hz,3H),3.29-3.18(m,2H),2.99(s,3H),1.20(d,J=6.9Hz,3H)。
And 5: synthesis of Compound 7

N-Fmoc-L-valine N-butanamidine ester (5.1g, 11.7mmol) and amino compound 8 (4.0g, 11.7mmol) were dispersed in 100mL of DCM at room temperature, and the reaction mixture was stirred overnight at 40 ℃ under nitrogen atmosphere, and there was always a white insoluble matter in the system. Then, 5mL of methanol was added to the reaction system, and stirring was continued at 40 ℃ for 4 hours. The resulting reaction solution was filtered with suction, and the filter cake was collected and dried to give compound 7 which was used directly in the next step.
MS:m/z=665.3(M+H);
1 H-NMR(400MHz,DMSO-d 6 )δppm 10.07(d,J=34.2Hz,1H),8.16(t,J=23.9Hz,1H),7.89(d,J=7.5Hz,2H),7.80-7.69(m,2H),7.58(d,J=8.4Hz,2H),7.52-7.37(m,3H),7.31(dd,J=17.6,8.0Hz,4H),4.97(s,2H),4.42(p,J=6.8Hz,1H),4.34-4.17(m,3H),3.98-3.83(m,1H),3.41(dd,J=13.3Hz,2H),3.29-3.19(m,2H),2.99(s,3H),1.99(dq,J=13.5Hz,1H),1.31(d,J=7.1Hz,3H),1.01-0.74(m,6H)。
And 6: synthesis of Compound 6

Compound 7 from step 5 was dissolved in 50mL DMF at room temperature, 10mL diethylamine was added, the mixture was stirred at room temperature for 4 hours and the starting material was monitored by TLC for completion. At this time, the reaction solution had white insoluble impurities. The impurities were removed by filtration, the obtained filtrate was dried by an oil pump, and the resulting pale yellow oil was sufficiently stirred and slurried with methyl tert-butyl ether (50 mL) for about 2 hours and then filtered to obtain a white solid. The obtained white solid was purified by recrystallization from ethanol to obtain Compound 6 (5.0 g, 97% yield in two steps) as a white solid.
MS:m/z=443.2(M+H)。
And 7: synthesis of Compound 5

Compound 6 (5.0 g,11.3 mmol) was dispersed in 50mL of methanol, 10mL of methylene chloride and 25mL of water at room temperature, and potassium carbonate (3.1 g,22.6 mmol) and copper sulfate pentahydrate (0.3 g,1.2 mmol) were added. After the resulting mixture was clarified by stirring, 1H-imidazole-1-sulfonyl azide hydrochloride (CAS: 952234-36-5,2.4g,11.5 mmol) was added and the resulting reaction solution was further stirred at room temperature overnight. After TLC monitoring the starting material reaction was complete, 25mL of water was added, most of the methanol was removed under reduced pressure, and the resulting mixture was extracted 2 times with dichloromethane (50 mL each). The combined organic phases were washed with saturated brine, dried and recrystallized from 40mL of ethanol and 0.3g of activated carbon to give Compound 5 (3.3 g, yield 62%) as a white solid. MS: m/z =469.2 (M + H).
And 8: synthesis of Compound 4

Compound 5 (3.3 g,6.0 mmol) synthesized in step 7, paraformaldehyde (271.0 mg, 9.0mmol in terms of formaldehyde), and sodium hydrogen carbonate (705.7 mg,8.4 mmol) were added to a mixed solution of 50mL of 1, 4-dioxane and 10mL of water, the resulting mixture was stirred at room temperature for 24 hours, after completion of the reaction of the starting materials was monitored by TLC, 30mL of water and 60mL of dichloromethane were added, the mixture was stirred and allowed to stand for phase separation, the solvent was evaporated under reduced pressure by organic phase, and vacuum was applied on a high vacuum pump for 2 hours to obtain a foamy solid compound 4 (3.3 g) (which was used directly in the next step).
Step 9a: synthesis of Compound 3a

The crude compound 4 (3.3g, 6.6 mmol) obtained in step 8 was dissolved in total in 25mL of ultra dry 1,4-dioxane, and then Dxd-a (4.0g, 6.6mmol, synthesized according to the method of example 2) and sodium methoxide (357.6mg, 9.9 mmol) were added thereto, and the resulting mixture was heated to 60 ℃ and stirred for 2 hours. The resulting reaction solution was evaporated under reduced pressure to remove the solvent, the resulting crude product was dissolved in 100mL of methylene chloride, and the crude solution was washed with 0.1mol/L dilute hydrochloric acid (100 mL), water (100 mL) and saturated brine (100 mL) in this order, and the resulting organic phase was dried over anhydrous sodium sulfate overnight. The dried solution was evaporated to remove the solvent and subjected to silica gel column chromatography eluting with dichloromethane/methanol = 60.
And step 9b: synthesis of Compound 3b

A further batch of crude compound 4 (3.3 g,6.6 mmol) obtained in step 8 was dissolved in dry dichloromethane (150 mL) and then compound Dxd-b (5.3 g,6.6mmol, synthesized as in example 3) was added and the reaction was replaced three times with nitrogen and after cooling to 0-5 ℃ with an ice bath sodium methoxide (357.6 mg,9.9 mmol) was added slowly in portions and dropwise over 2 hours. The resulting mixture was stirred at 0-5 ℃ for 4-5 hours and TLC monitored for completion of the reaction. The resulting reaction solution was washed with saturated brine (three times, 50mL each) and dried over anhydrous sodium sulfate, and then filtered and concentrated under reduced pressure to give a crude compound of compound 3b (8.0 g, used directly in the next step).
Step 10a: synthesis of Compound 2a

Compound 3a (7.1 g) from step 9a was dissolved in 35mL of tetrahydrofuran, followed by addition of 50mL of tetrahydrofuran and pH 5.0 acetic acid buffer (125mL, 12.5 mmol), followed by addition of 1M trimethylphosphine in tetrahydrofuran (7.7 mL,7.7 mmol). The resulting mixture was stirred at 0 to 5 ℃ for 2 hours, and after completion of the reaction, 200mL of saturated saline was added to the resulting reaction mixture, followed by extraction with 150mL of methylene chloride. The organic phase was dried over anhydrous sodium sulfate overnight and the solvent was evaporated under reduced pressure, and the crude product was subjected to silica gel column chromatography, eluting with dichloromethane/methanol 20.
MS:m/z=990.4(M+H)。
Step 10b: synthesis of Compound 2b

Compound 3b (8.5 g) from step 9b was dissolved in 35mL of tetrahydrofuran, then 120mL of tetrahydrofuran and pH 5.0 acetic acid buffer (125mL, 12.5 mmol) were added, followed by 1M trimethylphosphine in tetrahydrofuran (7.7 mL,7.7 mmol). The resulting mixture was stirred at 0 to 5 ℃ for 2 hours, and after completion of the reaction, 200mL of saturated saline was added to the resulting reaction mixture, followed by extraction with 150mL of methylene chloride. The organic phase was dried over anhydrous sodium sulfate overnight and the solvent was evaporated under reduced pressure, and the crude product was subjected to silica gel column chromatography, eluting with dichloromethane/methanol 20.
MS:m/z=1186.5(M+H)。
Step 11a: synthesis of Compound 1a

Compound 2a (3.7g, 3.7mmol) and EMCS (commercially available, 0.8g, 3.7mmol) were mixed and dissolved in 20mL of methylene chloride, and the resulting mixture was stirred at 40 ℃ overnight. After completion of the reaction, the solvent was evaporated under reduced pressure, and the residue was subjected to silica gel column chromatography, eluting with dichloromethane/methanol = 50.
MS:m/z=1183.4(M+H);
1 H NMR(500MHz,CDCl 3 )δ9.22(s,1H),7.61(d,J=9.2Hz,1H),7.59-7.53(m,3H),7.52(s,1H),7.44(d,J=8.4Hz,1H),7.36-7.30(m,3H),7.19(t,J=1.0Hz,1H),6.71(s,2H),5.13(s,1H),5.13-5.07(m,3H),5.05(d,J=9.5Hz,1H),4.91-4.82(m,3H),4.76(dd,J=12.5,1.1Hz,1H),4.57(dd,J=8.8,6.4Hz,1H),4.46(dq,J=8.4,5.7Hz,1H),4.15-4.09(m,2H),3.72-3.55(m,4H),3.25(td,J=8.2,1.6Hz,2H),3.03(s,3H),2.95(ddd,J=12.3,8.6,5.9Hz,1H),2.84(ddd,J=12.5,8.4,5.9Hz,1H),2.26(s,3H),2.24-2.17(m,3H),2.17-2.06(m,6H),2.00(dddd,J=12.3,Hz,1H),1.69(p,J=6.2Hz,2H),1.55-1.45(m,2H),1.41-1.37(m,1H),1.37-1.32(m,4H),1.08-1.01(m,3H),0.89(dd,J=6.5,2.1Hz,6H)。
Step 11b: synthesis of Compound 1b

Compound 2b (4.7g, 4.0mmol) and EMCS (commercially available, 0.8g, 4.0mmol) were mixed and dissolved in 60mL of dichloromethane, and the resulting mixture was stirred at 40 ℃ overnight. After completion of the reaction, the solvent was distilled off under reduced pressure from the obtained reaction solution, and the obtained residue was subjected to silica gel column chromatography (eluent dichloromethane/methanol = 50.
MS:m/z=1379.6(M+H);
1 H NMR(500MHz,CDCl 3 )δ9.22(s,1H),7.66-7.58(m,5H),7.58-7.53(m,2H),7.53(d,J=8.8Hz,1H),7.44(d,J=8.4Hz,1H),7.41-7.30(m,9H),7.18(t,J=1.0Hz,1H),6.71(s,1H),5.15-5.03(m,6H),4.91-4.83(m,3H),4.57(dd,J=8.8,6.4Hz,1H),4.42(dq,J=8.6,5.7Hz,1H),4.13(d,J=4.0Hz,2H),3.72-3.62(m,2H),3.65-3.55(m,2H),3.25(td,J=8.2,1.6Hz,2H),3.03(s,2H),2.95(ddd,J=12.4,8.6,5.9Hz,1H),2.84(ddd,J=12.4,8.6,5.9Hz,1H),2.27-2.13(m,4H),2.03-1.86(m,3H),1.69(p,J=6.2Hz,2H),1.55-1.45(m,2H),1.41-1.32(m,5H),1.07-0.98(m,9H),0.89(dd,J=6.5,2.1Hz,6H)。
Step 12a: preparation of Compound 1 starting from Compound 1a

Compound 1a (2.8g, 2.4mmol) was dissolved in a mixed solvent of methanol and methylene chloride (75 mL, volume ratio 1:1) in a 250mL three-necked flask, and after the three-necked flask reaction system was replaced with nitrogen three times, the mixture was cooled to 0 ℃ with an ice bath under nitrogen protection. Slowly adding acetyl chloride (0.1g, 1.2mmol) solution dissolved in methanol and dichloromethane mixed solvent (5 mL, the volume ratio is 1:1) into a reaction system, and maintaining the temperature of the reaction system at 0-5 ℃ in the dropping process; after the dropwise addition, the cooling device is removed to enable the reaction system to be recovered to the normal temperature, then stirring is continued for 2-3 hours, and the TLC monitors that the reaction is finished. The resulting reaction solution was washed with aqueous sodium bicarbonate solution (twice, 50mL each) and saturated brine (once, 50 mL) at pH 7 to 8, dried over anhydrous sodium sulfate and filtered, and finally the residue obtained by concentration under reduced pressure was subjected to silica gel column chromatography (eluent dichloromethane/methanol =30, 1 to 15.
MS:m/z=1141.4(M+H);
1 H NMR(500MHz,CDCl 3 )δ9.22(s,1H),7.61(d,J=9.2Hz,1H),7.59-7.53(m,2H),7.53(d,J=8.8Hz,1H),7.44(d,J=8.4Hz,1H),7.36-7.30(m,3H),7.24(t,J=1.0Hz,1H),6.71(s,1H),5.23(dd,J=12.4,1.1Hz,1H),5.15-5.10(m,3H),5.06(d,J=9.5Hz,1H),4.91-4.83(m,3H),4.77-4.70(m,2H),4.57(dd,J=8.8,6.4Hz,1H),4.46(dq,J=8.4,5.7Hz,1H),4.13(d,J=4.0Hz,2H),3.72-3.55(m,4H),3.25(td,J=8.2,1.6Hz,2H),3.03(s,2H),2.95(ddd,J=12.5,8.6,6.0Hz,1H),2.84(ddd,J=12.4,8.6,5.9Hz,1H),2.27-2.13(m,4H),2.01-1.86(m,2H),1.79(dq,J=13.7,8.0Hz,1H),1.69(p,J=6.2Hz,2H),1.55-1.45(m,2H),1.41-1.34(m,2H),1.34(d,J=5.7Hz,3H),0.97(t,J=8.0Hz,3H),0.89(dd,J=6.5,2.1Hz,6H)。
Step 12b preparation of Compound 1 starting from Compound 1b

Compound 1b (3.8g, 2.8mmol) was added to a mixed solution of dichloromethane and methanol (30 mL, volume ratio 20. The resulting mixture was stirred at room temperature overnight and the reaction was complete as detected by TLC. The reaction solution was filtered, and the obtained cake was washed with dichloromethane (twice, in amounts of 20mL and 10mL respectively), and then the residual solvent was removed under reduced pressure to obtain Compound 1 (3.0 g, purity 98%, yield 95%) as a white solid.
MS:m/z=1141.4(M+H);
1 H NMR(500MHz,CDCl 3 )δ9.22(s,1H),7.61(d,J=9.2Hz,1H),7.59-7.53(m,2H),7.53(d,J=8.8Hz,1H),7.44(d,J=8.4Hz,1H),7.36-7.30(m,3H),7.24(t,J=1.0Hz,1H),6.71(s,1H),5.23(dd,J=12.4,1.1Hz,1H),5.15-5.10(m,3H),5.06(d,J=9.5Hz,1H),4.91-4.83(m,3H),4.77-4.70(m,2H),4.57(dd,J=8.8,6.4Hz,1H),4.46(dq,J=8.4,5.7Hz,1H),4.13(d,J=4.0Hz,2H),3.72-3.55(m,4H),3.25(td,J=8.2,1.6Hz,2H),3.03(s,2H),2.95(ddd,J=12.5,8.6,6.0Hz,1H),2.84(ddd,J=12.4,8.6,5.9Hz,1H),2.27-2.13(m,4H),2.01-1.86(m,2H),1.79(dq,J=13.7,8.0Hz,1H),1.69(p,J=6.2Hz,2H),1.55-1.45(m,2H),1.41-1.34(m,2H),1.34(d,J=5.7Hz,3H),0.97(t,J=8.0Hz,3H),0.89(dd,J=6.5,2.1Hz,6H)。
Example 2: synthesis of compound Dxd-a

Step 1: synthesis of intermediate 17
In a 250mL three-necked flask, irinotecan (4.4g, 10.0mmol) and methylene chloride (66 mL) were charged, trimethylchlorosilane (1.6mL, 12.0mmol) was further added, and the resulting milky-white mixture was heated to 45 ℃ for 1 hour under reflux, and it was monitored by TLC that irinotecan had been reacted, and then the reaction solution was cooled to 0 ℃ followed by addition of N, N-diisopropylethylamine (5.0mL, 30.0mmol) and 4-methoxytriphenylmethane chloride (3.7g, 12.0mmol), and the resulting mixture was stirred at room temperature overnight and the reaction was monitored by TLC to be completed. The reaction mixture was washed with a sodium acetate buffer solution (pH = 5) (2 times, 20mL each) and a saturated saline solution (2 times, 20mL each), and the organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product of intermediate 17 (6.0 g, crude yield 85%).
MS:m/z=708.3(M+H);
1 H NMR(500MHz,CDCl 3 )δ7.34-7.26(m,11H),7.24(t,J=1.0Hz,1H),7.16-7.10(m,2H),6.89-6.84(m,2H),5.24(dd,J=12.4,1.0Hz,1H),5.11(d,J=9.5Hz,1H),5.04(d,J=9.5Hz,1H),4.88(dt,J=8.6,4.4Hz,1H),4.75(s,1H),4.70(dd,J=12.3,1.0Hz,1H),3.85(d,J=8.2Hz,1H),3.78(s,2H),2.93(dt,J=12.5,6.3Hz,1H),2.82(dt,J=12.5,6.4Hz,1H),2.08(td,J=6.3,4.4Hz,2H),2.04-1.89(m,2H),0.97(t,J=8.0Hz,3H)。
Step 2: synthesis of intermediate 16a
Intermediate 17 (2.4 g,3.0 mmol), dichloromethane (36 mL), 4-dimethylaminopyridine (1.3 g,10.0 mmol) and triethylamine (0.7 mL,5.0 mmol) were added to a 100mL three-necked flask, and the resulting reaction was cooled to 0 ℃ with an ice bath and replaced with nitrogen three times. A solution (12 mL) of acetic anhydride (0.4 g,4.0 mmol) in methylene chloride was slowly added dropwise to the reaction system, and the temperature of the system was maintained at 0-5 ℃ during the dropwise addition. After the addition was completed, the ice bath was removed to warm the reaction solution to room temperature, the resulting reaction solution was stirred for 3 hours, and the completion of the reaction was monitored by TLC. The reaction was added to a buffer of sodium acetate/acetic acid (50 mL) at pH =5.0 and the resulting mixture was extracted with dichloromethane (100 mL). The organic phase was washed with saturated brine (three times, 50mL each) and then dried over anhydrous sodium sulfate with stirring for 0.5 h, followed by filtration and concentration to give intermediate 16a as a solid (1.7 g, yield 75%).
MS:m/z=750.3(M+H);
1 H NMR(500MHz,CDCl 3 )δ7.33-7.26(m,10H),7.19(t,J=1.0Hz,1H),7.16-7.10(m,2H),6.89-6.84(m,2H),5.14-5.08(m,2H),5.05(d,J=9.3Hz,1H),4.88(dt,J=8.6,4.4Hz,1H),4.74(dd,J=12.5,0.9Hz,1H),3.85(d,J=8.2Hz,1H),3.78(s,2H),2.93(dt,J=12.5,6.3Hz,1H),2.82(dt,J=12.5,6.4Hz,1H),2.26(s,2H),2.17(dq,J=14.1,8.3Hz,1H),2.13-2.07(m,1H),2.10-2.04(m,2H),1.05(t,J=8.3Hz,3H)。
And 3, step 3: synthesis of intermediate 15a
At room temperature, adding the intermediate 16a (1.7g, 2.3mmol) and dichloromethane (40 mL) into a three-necked flask, adding triethylsilane (0.7g, 5.8mmol) after the mixture is dissolved, cooling the obtained mixture to-5-5 ℃, replacing the mixture with argon for three times, keeping the internal temperature of the reaction solution at-5-5 ℃ under the protection of argon, continuing stirring for 1-1.5 hours, and detecting by TLC to finish the reaction. To the resulting reaction solution, methyl t-butyl ether (80 mL) was added dropwise, the internal temperature was maintained at 0-5 ℃ and after completion of the addition, the supernatant was poured off, the resulting viscous solid was dissolved with methylene chloride (40 mL) under stirring, the internal temperature was maintained at 0-10 ℃ and during the addition, a solid precipitated, and the solid was collected by filtration to give intermediate 15a (0.8 g, crude yield 75%). MS: m/z =478.2 (M + H);
1 H NMR(500MHz,CDCl 3 )δ7.32(d,J=7.9Hz,1H),7.19(t,J=1.0Hz,1H),5.13-5.07(m,2H),5.05(d,J=9.5Hz,1H),4.76(dd,J=12.5,1.1Hz,1H),4.26(tdd,J=6.8Hz,1H),2.93(ddd,J=12.4Hz,1H),2.85(ddd,J=12.4Hz,1H),2.45(t,J=6.9Hz,1H),2.34(t,J=6.9Hz,1H),2.26(s,2H),2.19-2.00(m,3H),1.90(ddt,J=12.3Hz,1H),1.05(t,J=8.3Hz,3H).
and 4, step 4: synthesis of Dxd-a
To a 100mL three-necked flask, intermediate 15a (0.8g, 2.0mmol), bromoacetic acid (416.8mg, 3.0mmol) and 4- (4,6-dimethoxytriazin-2-yl) -4-methylmorpholine hydrochloride (1.2g, 4.0mmol) were charged, and methylene chloride (30 mL) was further added, and the resulting reaction system was replaced with nitrogen three times, and N, N-diisopropylethylamine (0.7g, 5.0mmol) was added. The resulting mixture was stirred at room temperature for 13 hours and the reaction was complete by TLC. The resulting reaction solution was concentrated and purified by column chromatography (using dichloromethane: methanol =50 as an eluent) to obtain compound Dxd-a (921.5 g, yield 77%). And (2) MS: m/z =598.2 (M + H);
1 H NMR(500MHz,CDCl 3 )δ7.62(d,J=9.3Hz,1H),7.32(d,J=7.9Hz,1H),7.19(t,J=1.0Hz,1H),5.15-5.08(m,2H),5.05(d,J=9.5Hz,1H),4.74(dd,J=12.4Hz,1H),3.96(d,J=5.5Hz,2H),3.86-3.80(m,1H),2.95(ddd,J=12.3Hz,1H),2.84(ddd,J=12.5Hz,1H),2.26(s,2H),2.22-2.10(m,2H),2.19(s,3H),2.13-2.04(m,1H),2.00(dddd,J=12.3Hz,1H),1.05(t,J=8.3Hz,3H)。
example 3: synthesis of compound Dxd-b

Step 1: synthesis of intermediate 16b
Adding the intermediate 17 (3.5g, 5.0mmol), N-dimethylformamide (70 mL) and N, N-diisopropylethylamine (1.1g, 10.0mmol) into a 250mL three-neck bottle, cooling the obtained reaction system to 3 ℃, replacing the reaction system with nitrogen for three times, dropwise adding tert-butyldiphenylchlorosilane (1.5g, 5.5mmol) into the reaction system, keeping the internal temperature at 0-10 ℃ in the dropwise adding process, heating the reaction solution to room temperature after the dropwise adding is finished, continuing stirring for 3 hours, and monitoring the completion of the reaction by TLC. The resulting reaction solution was poured into ice water (50 mL), followed by extraction with ethyl acetate (100 mL). The organic phase was washed with saturated brine (three times, 80mL each), dried over anhydrous sodium sulfate, stirred, filtered and concentrated to give intermediate 16b as a solid (4.4 g, 92% yield).
MS:m/z=946.4(M+H);
1 H NMR(500MHz,CDCl 3 )δ7.66-7.58(m,4H),7.41-7.34(m,6H),7.37-7.26(m,10H),7.18(t,J=1.0Hz,1H),7.16-7.10(m,2H),6.89-6.84(m,2H),5.15-5.02(m,4H),4.88(ddd,J=8.6,5.8,3.0Hz,1H),3.85(d,J=8.2Hz,1H),3.78(s,2H),2.94(ddd,J=12.6Hz,1H),2.81(ddd,J=12.4Hz,1H),2.19(dddd,J=12.5Hz,1H),2.03-1.87(m,3H),1.07-0.98(m,9H).
And 2, step: synthesis of intermediate 15b
Intermediate 16b (3.3g, 4.7 mmol), dichloromethane (60 mL) was added to a three-necked flask at room temperature, triethylsilane (0.7 g, 6.1mmol) was added, the resulting reaction system was cooled to-5-5 ℃ and replaced with argon three times, and then the internal temperature was maintained at-5-5 ℃ under argon protection and stirring was continued for 1-1.5 hours, and the reaction was detected by TLC to be complete. To the resulting reaction solution, methyl t-butyl ether (120 mL) was added dropwise, the internal temperature was maintained at 0-5 ℃ and the supernatant was poured out after completion of the addition, the resulting viscous solid was dissolved with methylene chloride (60 mL) under stirring, the internal temperature was maintained at 0-10 ℃ and a solid precipitated during the addition of methyl t-butyl ether (120 mL), and the solid was collected by filtration to give intermediate 15b (2.5 g, crude yield 78%).
MS:m/z=674.3(M+H)。
And step 3: synthesis of Compound Dxd-b
To a 250mL three-necked flask were added intermediate 15b (1.3 g, 2.0mmol), bromoacetic acid (416.8mg, 3.0mmol), and 4- (4,6-dimethoxytriazin-2-yl) -4-methylmorpholine hydrochloride (1.2g, 4.0mmol). DCM (55 mL) was added thereto, the resulting reaction system was replaced with nitrogen three times, N-diisopropylethylamine (0.7g, 5.0mmol) was added thereto, the resulting mixture was stirred at room temperature for 13 hours, and the completion of the reaction was monitored by TLC. The resulting reaction was concentrated and purified by column chromatography (using DCM: meOH =50 as eluent) to give compound Dxd-b (1.2 g, 73% yield).
MS:m/z=794.3(M+H);
1 H NMR(500MHz,CDCl 3 )δ7.65-7.58(m,5H),7.41-7.30(m,7H),7.18(t,J=1.0Hz,1H),5.15-5.09(m,2H),5.11-5.03(m,2H),4.89(ddd,J=9.3Hz,1H),3.83(t,J=5.5Hz,1H),2.95(ddd,J=12.4Hz,1H),2.84(ddd,J=12.5Hz,1H),2.39(d,J=5.5Hz,2H),2.21(d,J=5.8Hz,2H),2.2(s,3H),2.18(dddd,J=12.3Hz,1H),2.03-1.86(m,3H),1.07-0.98(m,10H)。
Example 4: comparison of end product purity for several process routes:
the final product of compound 1 obtained according to scheme 1 (i.e. the synthesis of LE14 in example 7) and scheme 2 (i.e. the synthesis of LE14 in example 10) disclosed in WO2020259258A1 was compared in purity by high performance liquid phase with the final product of compound 1 obtained according to the synthesis method of steps 12a and 12b of example 1 of the present invention, and the results are shown in table 3 below.
The liquid phase conditions used were that phase A was 0.1% formic acid aqueous solution, phase B was 0.1% formic acid acetonitrile solution, the detection wavelength was 370nm, the apparatus was Agilent 1260, and the column chromatography was ZORBAX Eclipse Plus C18,3.5 μm, 4.6X 150mm. The gradient settings are as follows in table 2.
TABLE 2 gradient setup of mobile phase
| Time (min) | Mobile phase A% | Mobile phase B% |
| 0.00 | 80.0 | 20.0 |
| 10.00 | 60.0 | 40.0 |
| 25.00 | 60.0 | 40.0 |
| 35.00 | 30.0 | 70.0 |
| 41.00 | 30.0 | 70.0 |
| 41.10 | 80.0 | 20.0 |
| 43.00 | 80.0 | 20.0 |
TABLE 3 comparison of end product purities for different process routes
| Route 1 | Route 2 | EXAMPLE 1 step 12a | EXAMPLE 1 step 12b | |
| Purity of final product | 95% | 96% | 97% | 98% |
| Maximum single hetero | 3% | 2% | <1% | <1.5% |
Claims (14)
1. A process for the preparation of a compound of formula IV comprising the steps of: carrying out substitution reaction on a compound of a formula V and a compound of a formula VIa in a solvent in the presence of alkali to obtain a compound of a formula IV;

wherein R is 1 Is C 1 ~C 6 Alkyl, one or more R 1-3 S(O) 2 -substituted C 1 ~C 6 Alkyl, or one or more N (R) 1-1 )(R 1-2 ) -substituted C 1 ~C 6 An alkyl group;
R 2 and R 3 Each independently is C 1 ~C 6 Alkyl, one or more halogen substituted C 1 ~C 6 Alkyl, or halogen;
R 1-1 、R 1-2 and R 1-3 Each independently is C 1 ~C 4 An alkyl group;
R 4 is a hydroxyl protecting group.
2. The method of claim 1, wherein R is 1 Is a R 1-3 S(O) 2 -substituted C 1 ~C 6 Alkyl groups such as methylsulphonylethyl;
and/or, R 2 Is C 1 ~C 6 Alkyl groups such as methyl;
and/or, R 3 Is halogen, such as fluorine;
and/or, R 1-3 Is C 1 ~C 4 Alkyl groups such as methyl;
and/or, the hydroxyl protecting group is an ester protecting group or a silicon ether protecting group.
3. The process of claim 1, wherein the compound of formula IV is prepared in a molar ratio of the compound of formula V to the compound of formula VIa of from 1 to 5, preferably from 1 to 2;
and/or, in the preparation method of the compound shown in the formula IV, the base is organic base, inorganic base or a mixture thereof; wherein, the organic base is preferably tert-butyl potassium, sodium methoxide, triethylamine, DMAP, pyridine, dipyridine or a mixture of any two or more of the above, and is further preferably sodium methoxide; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and is more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof;
and/or, in the preparation method of the compound of the formula IV, the molar ratio of the base to the compound of the formula VIa is 1-5, preferably 1-2, and more preferably 1.5;
and/or in the preparation method of the compound shown in the formula IV, the solvent is an ether solvent, a chloroalkane solvent, a nitrile solvent or a mixture of any two or more of the solvents; the ether solvent can be tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of the above, preferably 1,4-dioxane; the chloroalkane solvent can be dichloromethane, dichloroethane, chloroform or a mixture of any two or more of the dichloromethane, and dichloromethane is preferred; the nitrile solvent may be acetonitrile;
and/or, in the preparation method of the compound shown in the formula IV, the reaction temperature is 0-80 ℃; when R is 4 When the protecting group is an ester protecting group, the reaction temperature can be 40-60 ℃; when R is 4 In the case of the silyl ether protecting group, the reaction temperature may be 0 to 20 ℃.
4. The process of claim 1, wherein the process for preparing the compound of formula IV further comprises a process for preparing a compound of formula VIa and/or a process for preparing a compound of formula V;
preferably, the preparation method of the compound of formula VIa comprises the following steps: carrying out condensation reaction on a compound shown in the formula VIb and bromoacetic acid in the presence of a condensing agent and alkali to obtain a compound shown in the formula VIa;

wherein R is 2 、R 3 And R 4 As defined in claim 1;
further preferably, in the preparation method of the compound of formula VIa, the preparation method of the compound of formula VIb preferably comprises the following steps: removing 4-methoxyl triphenylmethyl connected with amino in the compound of formula VIc to obtain the compound of formula VIb;

wherein R is 2 、R 3 And R 4 As claimed in claim 1;
further preferably, in the preparation method of the compound of formula VIb, the preparation method of the compound of formula VIc preferably comprises the following steps: carrying out hydroxyl protection reaction on a compound shown in a formula VId and a hydroxyl protection reagent to obtain a compound shown in a formula VIc;

wherein R is 2 、R 3 And R 4 As claimed in claim 1;
further preferably, in the preparation method of the compound of formula VIc, the preparation method of the compound of formula VId comprises the following steps:
(i) Reacting irinotecan with trimethylchlorosilane;
(ii) (ii) reacting the reaction solution obtained in the step (i) with 4-methoxytriphenylmethane chloride in the presence of alkali to obtain the compound shown in the formula VId;

wherein R is 2 And R 3 As defined in claim 1;
preferably, the preparation method of the compound of formula V comprises the following steps: reacting a compound shown in a formula VII with paraformaldehyde in the presence of alkali to obtain a compound shown in a formula V;

wherein R is 1 As defined in claim 1;
further preferably, in the preparation method of the compound of formula V, the preparation method of the compound of formula VII comprises the following steps: reacting a compound shown in a formula VIII with a sulfonyl azide compound in the presence of a base and a catalyst in a solvent to obtain a compound shown in a formula VII;

wherein R is 1 As defined in claim 1;
further preferably, in the preparation method of the compound of formula VII, the preparation method of the compound of formula VIII comprises the following steps: carrying out Fmoc removal reaction on a compound in a formula IX in an organic solvent in the presence of a base to obtain a compound in a formula VIII;

wherein R is 1 As defined in claim 1;
further preferably, in the preparation method of the compound of formula VIII, the preparation method of the compound of formula IX comprises the following steps: carrying out coupling reaction on a compound shown in a formula X and N-Fmoc-L-valine N-butadiene amine imine ester in a solvent to obtain a compound shown in a formula IX;

wherein R is 1 As defined in claim 1;
further preferably, in the preparation method of the compound of formula IX, the preparation method of the compound of formula X comprises the following steps: carrying out Fmoc removal reaction on a compound in a formula XI in a solvent in the presence of alkali to obtain a compound in a formula X;

wherein R is 1 As defined in claim 1;
further preferably, in the preparation method of the compound of formula X, the preparation method of the compound of formula XI comprises the following steps: reacting a compound of formula XII with an amino compound R 1 NH 2 Carrying out coupling reaction in a solvent in the presence of alkali to obtain a compound shown in a formula XI;

wherein R is 1 As defined in claim 1;
further preferably, in the preparation method of the compound of formula XI, the preparation method of the compound of formula XII comprises the following steps: reacting a compound of formula XIII and a compound of formula XIV in the presence of a base and in a solvent to obtain said compound of formula XII;

5. the process according to claim 4, wherein the solvent for the condensation reaction in the process for preparing the compound of formula VIa is dichloromethane, tetrahydrofuran, N-dimethylformamide, N-dimethylacetamide, dimethylsulfoxide, or a mixture of any two or more thereof, preferably dichloromethane, N-dimethylformamide or a mixture thereof, more preferably dichloromethane;
and/or, in the preparation method of the compound of formula VIa, the molar ratio of the bromoacetic acid to the compound of formula VIb is 1 to 5, preferably 1.5 to 3, and more preferably 1.5 to 2;
and/or in the preparation method of the compound shown in the formula VIa, the condensing agent is dicyclohexylcarbodiimide, diisopropylcarbodiimide, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide or salts thereof, 2- (7-azabenzotriazole) -N, N '-tetramethylurea hexafluorophosphate, propyl phosphoric anhydride, benzotriazol-N, N' -tetramethylurea hexafluorophosphate, diphenylphosphoric acid azide, 4- (4,6-dimethoxytriazine-2-yl) -4-methylmorpholine or salts thereof, preferably Dicyclohexylcarbodiimide (DCC), 4- (4,6-dimethoxytriazine-2-yl) -4-methylmorpholine or salts thereof, further preferably 4- (4,6-dimethoxytriazine-2-yl) -4-methylmorpholine or salts thereof, further preferably 4- (4,6-dimethoxytriazine-2-yl) -4-methylmorpholine hydrochloride;
and/or, in the preparation method of the compound of formula VIa, the molar ratio of the condensing agent to the compound of formula VIb is 1-5, preferably 1.5-3.0, and more preferably 1.5-2.0;
and/or, in the preparation method of the compound shown in the formula VIa, the base used in the condensation reaction is N, N-Diisopropylethylamine (DIEA), triethylamine, 1,8-diazabicycloundecen-7-ene (DBU) or a mixture of any two or more of them, preferably triethylamine and/or N, N-diisopropylethylamine, and more preferably N, N-diisopropylethylamine;
and/or, in the preparation method of the compound of formula VIa, the molar ratio of the base used in the condensation reaction to the compound of formula VIb is 1-5, preferably 2-4, more preferably 2.0-3.0, and even more preferably 2.0-2.5;
and/or, in the preparation method of the compound of the formula VIa, the condensation reaction temperature is 20-50 ℃, preferably 20-30 ℃;
and/or, in the preparation method of the compound of formula VIa, the condensation reaction is carried out under the protection of inert gas, for example, in a nitrogen or helium environment;
and/or, in the preparation method of the compound shown in the formula VIb, the solvent used for the reaction of removing the 4-methoxyl triphenylmethyl is chloroform, dichloromethane or a mixture thereof, preferably dichloromethane;
and/or in the preparation method of the compound shown in the formula VIb, a deprotection reagent used for removing the 4-methoxytriphenylmethyl is triisopropylsilane, triethylsilane or a mixture thereof, and preferably triethylsilane;
and/or, in the preparation method of the compound of formula VIb, the molar ratio of the deprotection reagent used for removing the 4-methoxytriphenylmethyl group to the compound of formula VIc is 1-5, preferably 1-3, and more preferably 1.3-2.5;
and/or, in the preparation method of the compound of the formula VIb, the temperature of the reaction for removing the 4-methoxyl triphenylmethyl is-20 to 10 ℃, preferably-10 to 5 ℃, and further preferably-5 to 5 ℃;
and/or, in the preparation method of the compound of formula VIb, the 4-methoxyl triphenylmethyl removing reaction is carried out under the protection of inert gas, such as nitrogen or helium atmosphere;
and/or, in the preparation method of the compound of formula VIc, the hydroxyl protecting reagent used is acetic anhydride, propionic anhydride, acetyl chloride, propionyl chloride, tert-butyldimethylchlorosilane or tert-butyldiphenylchlorosilane, preferably acetic anhydride, acetyl chloride or tert-butyldiphenylchlorosilane, and more preferably acetic anhydride or tert-butyldiphenylchlorosilane;
and/or, in the preparation method of the compound of formula VIc, the molar ratio of the hydroxyl protecting reagent to the compound of formula VId is 1-2, preferably 1-1.5, and more preferably 1.1-1.44;
and/or, in the preparation method of the compound of the formula VIc, the hydroxyl protection reaction is carried out in the presence of alkali; the base is preferably triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine, 1,8-diazabicyclo-bicyclo (5,4,0) -7-undecene, 1,5-diazabicyclo [4.3.0] non-5-ene, N-methylmorpholine, tetramethylethylenediamine, pyridine or a mixture of any two or more thereof, preferably triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine or a mixture of any two or more thereof;
and/or in the preparation method of the compound shown in the formula VIc, the solvent for hydroxyl protection reaction is dichloromethane, dichloroethane, N-dimethylformamide, N-dimethylacetamide or the mixture of any two or more of the dichloromethane, the dichloroethane, the N, N-dimethylformamide and the N, N-dimethylacetamide; preferably dichloromethane, N-dimethylformamide or mixtures thereof;
and/or, in the preparation method of the compound of formula VIc, the temperature of hydroxyl protection reaction is 0-40 ℃, preferably 10-30 ℃, and more preferably 20-30 ℃;
and/or, in the preparation method of the compound of formula VIc, the hydroxyl protection reaction is carried out under the protection of inert gas, such as nitrogen or helium atmosphere;
and/or, in the preparation method of the compound shown in the formula VId, the solvent used in the reaction of the step (i) and the step (ii) is dichloromethane, chloroform, dichloroethane or a mixture of any two or more of the dichloromethane, preferably dichloromethane;
and/or, in the preparation method of the compound shown in the formula VId, in the step (i), the molar ratio of the trimethylchlorosilane to the irinotecan used in the reaction is 1-3, preferably 1.2-2.0, and more preferably 1.2;
and/or, in the preparation method of the compound of formula VId, the reaction temperature of step (i) is 20-60 ℃, preferably 40-45 ℃, and more preferably 45 ℃;
and/or, in the preparation method of the compound shown in the formula VId, the molar ratio of the 4-methoxytriphenylmethane chloride in the step (ii) to the irinotecan in the step (i) is 1-3, preferably 1.2-2.0, and more preferably 1.2;
and/or, in the preparation method of the compound of formula VId, the base in step (ii) is triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine, 1,8-diazabicyclo-bicyclo (5,4,0) -7-undecene, 1,5-diazabicyclo [4.3.0] non-5-ene, N-methylmorpholine, tetramethylethylenediamine, pyridine or a mixture of any two or more thereof, preferably triethylamine, 4-dimethylaminopyridine, N-diisopropylethylamine or a mixture of any two or more thereof, and further preferably N, N-diisopropylethylamine;
and/or, in the preparation method of the compound shown in the formula VId, the molar ratio of the base in the step (ii) to the irinotecan in the step (i) is 2-5, preferably 3-4, and more preferably 3;
and/or, in the preparation method of the compound of formula VId, the reaction temperature in the step (ii) is 0-40 ℃, preferably 10-30 ℃, and more preferably 20-30 ℃;
and/or, in the preparation method of the compound of the formula V, the molar ratio of the paraformaldehyde to the compound of the formula VII in terms of formaldehyde is 1-3, preferably 1-2, and more preferably 1.5;
and/or, in the preparation method of the compound of the formula V, the base is an organic base, an inorganic base or a mixture thereof, preferably an inorganic base; wherein, the organic base is preferably tert-butyl potassium, sodium methoxide, triethylamine, DMAP, pyridine, dipyridine or a mixture of any two or more of the above, and is further preferably sodium methoxide; the inorganic base is preferably an alkali metal carbonate, an alkali metal hydroxide, an alkali metal phosphate, or a mixture of any two or more thereof, and is more preferably sodium bicarbonate, sodium carbonate, potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof;
and/or, in the preparation method of the compound of the formula V, the molar ratio of the sodium bicarbonate to the compound of the formula VII is 1-4, preferably 1-2, and more preferably 1.4;
and/or in the preparation method of the compound shown in the formula V, the solvent is a mixed system of an ether solvent and water; the ether solvent can be tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of the above, preferably 1,4-dioxane;
and/or, in the preparation method of the compound shown in the formula V, the reaction temperature is 10-40 ℃, preferably 25-40 ℃, and further preferably 25-30 ℃;
and/or, in the preparation method of the compound shown in the formula VII, the sulfonyl azide compound is 1H-imidazole-1-sulfonyl azide hydrochloride, 2-azido-1,3-dimethyl imidazole hexafluorophosphate, trifluorosulfonyl azide, p-methyl benzenesulfonyl azide or methylsulfonyl azide, preferably 1H-imidazole-1-sulfonyl azide hydrochloride;
and/or, in the preparation method of the compound of the formula VII, the molar ratio of the sulfonyl azide compound to the compound of the formula VIII is 1.0-1.5, preferably 1.0-1.2, and more preferably 1.02;
and/or, in the preparation method of the compound shown in the formula VII, the base is organic base, inorganic base or a mixture thereof; wherein the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof, and more preferably potassium carbonate; the organic base is preferably tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-lutidine or a mixture of any two or more of the tert-butyl potassium, the triethylamine, the DMAP, the pyridine, the dipyridine and the 2,6-lutidine;
and/or, in the preparation method of the compound of the formula VII, the molar ratio of the base to the compound of the formula VIII is 1.5-3.0, preferably 2.0-2.5, and more preferably 2.0;
and/or, in the preparation method of the compound shown in the formula VII, the catalyst is a ketone salt, preferably copper sulfate, and more preferably copper sulfate pentahydrate;
and/or, in the preparation method of the compound of the formula VII, the molar ratio of the ketone salt to the compound of the formula VIII is 0.1-0.5, preferably 0.1-0.3, and more preferably 0.1-0.2;
and/or in the preparation method of the compound of the formula VII, the solvent is a mixed solvent of an organic solvent and water, the organic solvent is preferably an alcohol solvent, a chloroalkane solvent, an ether solvent or a mixture of any two or more of the alcohol solvent, the chloroalkane solvent and the chloroalkane solvent; the alcohol solvent is preferably methanol, ethanol, isopropanol or a mixture of any two or more of the methanol, the ethanol and the isopropanol, and is preferably methanol; the chloroalkane solvent is preferably dichloromethane, chloroform, dichloroethane or a mixture of any two or more of the dichloromethane, preferably dichloromethane; the ether solvent is preferably tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of the tetrahydrofuran, the diethyl ether, the 1,4-dioxane, the anisole and the methyl tert-butyl ether;
and/or in the preparation method of the compound shown in the formula VII, the reaction temperature is 10-40 ℃, preferably 25-40 ℃, and further preferably 25-30 ℃;
and/or, in the preparation method of the compound of the formula VIII, the base is an organic base, an inorganic base or a mixture thereof, and preferably an organic base; wherein, the organic base is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-dimethylpyridine or a mixture of any two or more of them, and is further preferably ethylenediamine; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and is more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof;
and/or, in the preparation method of the compound of the formula VIII, the volume ratio of the alkali to the organic solvent is 0.1-0.5, preferably 0.2-0.3, and more preferably 0.2;
and/or, in the preparation method of the compound of formula VIII, the organic solvent is DMF, DMSO, tetrahydrofuran, 1,4-dioxane or a mixture of any two or more of the DMF, the DMSO, the tetrahydrofuran, the 1,4-dioxane or the mixture of any two or more of the DMF and the DMF;
and/or in the preparation method of the compound shown in the formula VIII, the temperature of Fmoc removal reaction is 10-40 ℃, preferably 25-40 ℃, and further preferably 25-30 ℃;
and/or, in the preparation method of the compound of formula IX, the molar ratio of the N-Fmoc-L-valine N-butanamidine ester to the compound of formula X is 0.8-5, preferably 0.8-1.2, and more preferably 1;
and/or, in the preparation method of the compound of formula IX, the solvent is DMF, DMSO, acetonitrile, dichloromethane, dichloroethane or a mixture of any two or more thereof, preferably dichloromethane;
and/or, in the preparation method of the compound of the formula IX, the temperature of the coupling reaction is 10-40 ℃, preferably 35-40 ℃;
and/or, in the preparation method of the compound of the formula X, the base is organic base, inorganic base or a mixture thereof; wherein, the organic base is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-dimethylpyridine or a mixture of any two or more of them, and is further preferably ethylenediamine; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof;
and/or, in the preparation method of the compound of the formula X, the solvent is DMF, DMSO, tetrahydrofuran, 1,4-dioxane or a mixture of any two or more of the DMF, the DMSO, the tetrahydrofuran, the 1,4-dioxane or the mixture of any two or more of the DMF and the DMF;
and/or, in the preparation method of the compound of the formula X, the volume ratio of the base to the solvent is 0.2-0.5, preferably 0.3-0.4, and more preferably 0.3;
and/or, in the preparation method of the compound of the formula X, the reaction temperature of Fmoc removal reaction is 10-40 ℃, preferably 25-40 ℃, and further preferably 25-30 ℃;
and/or, in the process for the preparation of the compounds of the formula XI, the amino compounds R 1 NH 2 The molar ratio to the compound of the formula XII is from 1.0 to 3.0, preferably from 1.1 to 1.5;
and/or, in the preparation method of the compound shown in the formula XI, the base is organic base, inorganic base or a mixture thereof; wherein the organic base is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridamole, 2,6-lutidine or a mixture of any two or more thereof, and more preferably DMAP; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate, or a mixture of any two or more thereof, and is more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate, or a mixture of any two or more thereof;
and/or, in the preparation method of the compound of the formula XI, the molar ratio of the base to the compound of the formula XII is 2-4, preferably 2.5-3.0, and more preferably 2.5;
and/or, in the preparation method of the compound in the formula XI, the solvent can be DMF, DMSO, dichloromethane, dichloroethane, tetrahydrofuran, 1,4-dioxane or a mixture of any two or more of the DMF, the DMSO, the dichloromethane, the dichloroethane, the tetrahydrofuran, the 1,4-dioxane or the mixture of any two or more of the DMF, the DMSO, the dichloromethane and the mixture of the dichloromethane;
and/or, in the preparation method of the compound of the formula XI, the coupling reaction temperature is 10-40 ℃, preferably 25-40 ℃, and further preferably 25-30 ℃;
and/or, in the preparation method of the compound of the formula XII, the molar ratio of the compound of the formula XIV to the compound of the formula XIII is 3.0 to 1.2, preferably 2.0 to 1.5, and more preferably 1.5;
and/or, in the preparation method of the compound shown in the formula XII, the alkali is organic alkali, inorganic alkali or a mixture thereof; wherein, the organic base is preferably diethylamine, tert-butyl potassium, triethylamine, DMAP, pyridine, dipyridine, 2,6-dimethylpyridine or a mixture of any two or more of them, and is further preferably pyridine; the inorganic base is preferably an alkali metal hydroxide, an alkali metal carbonate, an alkali metal phosphate or a mixture thereof, and is more preferably potassium phosphate, potassium carbonate, potassium hydroxide, cesium carbonate or a mixture of any two or more thereof;
and/or, in the preparation method of the compound shown in the formula XII, the molar ratio of the base to the compound shown in the formula XIII is 1.0-4.0, preferably 2.0-3.0, and more preferably 2.0;
and/or, in the preparation method of the compound shown in the formula XII, the solvent is DMF, DMSO, dichloromethane, dichloroethane, tetrahydrofuran, 1,4-dioxane or a mixture of any two or more of the solvents, preferably dichloromethane;
and/or, in the preparation method of the compound shown in the formula XII, the reaction temperature is 10-40 ℃, preferably 25-40 ℃, and more preferably 25-30 ℃.
6. A process for preparing a compound of formula III, said process comprising the steps of:
the method comprises the following steps: preparing a compound of formula IV according to the process for the preparation of a compound of formula IV as claimed in any one of claims 1 to 5;
step two: carrying out reduction reaction on a compound shown in a formula IV and a reducing agent in an organic solvent in the presence of an acid buffer solution to obtain a compound shown in a formula III;

wherein R is 1 、R 2 、R 3 And R 4 Is as defined in claim 1.
7. The process according to claim 6, wherein the reducing agent is triphenylphosphine, tri-tert-butylphosphine or trimethylphosphine, preferably trimethylphosphine;
and/or, in the preparation method of the compound of the formula III, the molar ratio of the reducing agent to the compound of the formula IV is 1 to 5, preferably 1 to 2, more preferably 1.0 to 1.5, and still more preferably 1.1 to 1.3;
and/or, in the preparation method of the compound of formula III, the organic solvent is an ether solvent, such as tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether, or a mixture of any two or more thereof, and further preferably tetrahydrofuran;
and/or, in the preparation method of the compound of the formula III, the acid buffer solution is an acetic acid buffer solution and a formic acid buffer solution, preferably an acetic acid buffer solution; the pH of the acid buffer is preferably 4.0 to 6.0, more preferably 4.5 to 5.5, and still more preferably 5.0;
and/or, in the preparation method of the compound of the formula III, the temperature of the reduction reaction is 0-20 ℃, preferably 0-10 ℃, and more preferably 0-5 ℃.
8. A process for preparing a compound of formula II, said process comprising the steps of:
the method comprises the following steps: preparing a compound of formula III according to the process for the preparation of a compound of formula III as claimed in claim 6 or 7;
step two: carrying out coupling reaction on a compound shown in a formula III and 6- (maleimide) hexanoic acid succinimide ester in a solvent to obtain a compound shown in a formula II;

wherein R is 1 、R 2 、R 3 And R 4 Is as defined in claim 1.
9. The process of claim 8, wherein in the process of preparing the compound of formula II, the molar ratio of the succinimidyl 6- (maleimido) hexanoate to the compound of formula III is 1 to 5, preferably 1 to 2, more preferably 1.0 to 1.5;
and/or in the preparation method of the compound shown in the formula II, the solvent is an amide solvent, a chloroalkane solvent, an ether solvent, a nitrile solvent or a mixture of any two or more of the amide solvent, the chloroalkane solvent, the ether solvent and the nitrile solvent; wherein the amide solvent is preferably N, N-dimethylformamide, N-dimethylacetamide, or a mixture thereof, and more preferably N, N-dimethylformamide; the chloroalkane solvent is preferably dichloromethane, chloroform, dichloroethane or a mixture of any two or more of the dichloromethane, and is further preferably dichloromethane; the ether solvent is preferably tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, methyl tert-butyl ether or a mixture of any two or more of them, and more preferably tetrahydrofuran; the nitrile solvent is preferably acetonitrile;
and/or, in the preparation method of the compound of the formula II, the reaction temperature is 0-50 ℃, preferably 25-40 ℃.
10. A process for the preparation of a compound of formula I, comprising the steps of:
the method comprises the following steps: preparing a compound of formula II according to the process for the preparation of a compound of formula II as claimed in claim 8 or 9;
step two: removing R from the compound of formula II 4 Protecting groups to obtain the compound of formula I;

wherein R is 1 、R 2 、R 3 And R 4 Is as defined in claim1, the preparation method is as follows.
11. The method of claim 10, wherein R is R when the hydroxyl protecting group is an ester protecting group 4 Acetyl, propionyl, benzoyl or pivaloyl, preferably acetyl;
and/or, when the hydroxyl protecting group is a silyl ether protecting group, R 4 Is trimethylsilyl, triisopropylsilyl, tert-butyldimethylsilyl, tert-butyldiphenylsilyl or tri-tert-butylsilyl, preferably tert-butyldiphenylsilyl;
and/or, when the hydroxyl protecting group is an ester protecting group, removing R 4 The deprotection reagent used by the protecting group is sodium hydroxide, potassium hydroxide, sodium carbonate, potassium bicarbonate or acetyl chloride/methanol, preferably acetyl chloride/methanol;
and/or, when the hydroxyl protecting group is an ester protecting group, removing R 4 The molar ratio of the deprotection reagent used for protecting groups to the compound of the formula II is 0.5-2, preferably 0.5-1;
and/or, when the hydroxyl protecting group is an ester protecting group, removing R 4 The solvent used for the protecting group is methanol, ethanol, isopropanol, dichloromethane or a mixture of any two or more of the methanol, the ethanol, the isopropanol and the dichloromethane, and preferably the mixed solvent of methanol and dichloromethane or the mixed solvent of ethanol and dichloromethane;
and/or, when the hydroxyl protecting group is an ester protecting group, removing R 4 The temperature of the protecting group reaction is 10-50 ℃, for example 20-30 ℃;
and/or, when the hydroxyl protecting group is a silyl ether protecting group, removing R 4 The deprotection reagent used for the protecting group is lithium hydroxide, tetrabutylammonium fluoride/acetic acid, sodium hydroxide, pyridine hydrogen fluoride complex, tert-butylammonium fluoride or tert-butylammonium fluoride/acetic acid, preferably tert-butylammonium fluoride or tert-butylammonium fluoride/acetic acid;
and/or, when the hydroxyl protecting group is a silyl ether protecting group, removing R 4 The molar ratio of the deprotection reagent used for protecting groups to the compound of formula II is 1.2-3, preferably 1.2-1.6, more preferably 1.5;
and/or, when the hydroxyl protecting group is a silyl ether protecting group, removing R 4 The solvent used for the protecting group is methanol, ethanol, isopropanol, dichloromethane or a mixture of any two or more of the methanol, the ethanol, the isopropanol and the dichloromethane, and preferably the mixed solvent of methanol and dichloromethane or the mixed solvent of ethanol and dichloromethane;
and/or, when the hydroxyl protecting group is a silyl ether protecting group, removing R 4 The temperature at which the protecting group is reacted is from 10 to 50 deg.C, for example from 20 to 30 deg.C.
12. A compound, the structure of which is shown as a formula VIa or V;

13. The compound of claim 12, wherein R is 1 Is methylsulfonylethyl; r 2 Is methyl; r 3 Is fluorine; r 4 Acetyl or tert-butyl diphenyl silicon group.
14. A process for the preparation of a compound according to claim 12 or 13,
the process for the preparation of the compound of formula VIa comprises the steps of: carrying out condensation reaction on a compound shown in a formula VIb and bromoacetic acid in the presence of a condensing agent and alkali to obtain a compound shown in a formula VIa; the reaction conditions are as defined in claim 4 or 5;

wherein R is 2 、R 3 And R 4 As defined in claim 1;
the preparation method of the compound of the formula V comprises the following steps: reacting a compound shown in a formula VII with paraformaldehyde in the presence of alkali to obtain a compound shown in a formula V; the reaction conditions are as defined in claim 4 or 5;

wherein R is 1 Is as defined in claim 1.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110566920.XA CN115385926A (en) | 2021-05-24 | 2021-05-24 | Preparation method of connection base drug conjugate and intermediate thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110566920.XA CN115385926A (en) | 2021-05-24 | 2021-05-24 | Preparation method of connection base drug conjugate and intermediate thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN115385926A true CN115385926A (en) | 2022-11-25 |
Family
ID=84114364
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110566920.XA Pending CN115385926A (en) | 2021-05-24 | 2021-05-24 | Preparation method of connection base drug conjugate and intermediate thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN115385926A (en) |
-
2021
- 2021-05-24 CN CN202110566920.XA patent/CN115385926A/en active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107660206A (en) | For preparing the method and intermediate of { base of 1 (ethylsulfonyl) 3 [base of 4 (base of 7H pyrrolo-es [2,3 d] pyrimidine 4) 1H pyrazoles 1] azetidine 3 } acetonitrile | |
| WO2018137679A1 (en) | Process for the Preparation of (10R) -7- (2-aminoacetyl) amino-12-fluoro-2, 10, 16-trimethyl-15-oxo-10, 15, 16, 17-tetrahydro-2H-8, 4- (metheno) pyrazolo [4, 3-h] [2, 5, 11] -benzoxadiazacyclotetradecine-3-carbonitrile | |
| CN114957247A (en) | Synthesis method of Rimegepant and intermediate thereof | |
| CN115385926A (en) | Preparation method of connection base drug conjugate and intermediate thereof | |
| CN115215921A (en) | Preparation method of connection base drug conjugate and intermediate thereof | |
| EP1501799A1 (en) | Compounds useful in preparing camptothecin derivatives | |
| EP2714691B1 (en) | Process for the preparation of 2-amino-9-((2-phenyl-1,3-dioxan-5-yloxy)methyl)-1h-purin-6(9h)-one compound useful in the preparation of valganciclovir | |
| CN111574463B (en) | Rivastigmine intermediate compound IV | |
| JPH10114788A (en) | Block for constituting dna/pna co-oligomer | |
| CN111574520B (en) | Riagliptin intermediate compound V | |
| JPH03209393A (en) | Synthesis of 1-(3-azide-2, 3-dideoxy-beta- d-erythropentfuranosil) thymine and its related compound | |
| WO2022204947A1 (en) | Preparation method for linker drug conjugate and intermediate thereof | |
| CN112272665A (en) | Process for preparing sitagliptin | |
| CN114539288B (en) | Preparation method of everolimus | |
| RU2304583C1 (en) | Method for synthesis di- and triaminochlorines | |
| JP3511788B2 (en) | 7-Amino-2,3-dihydro-2-oxo-pyrido [2,3-d] pyrimidine and method for producing the same | |
| WO2022205111A1 (en) | Method for preparing exatecan derivative, and intermediate thereof | |
| US6426417B1 (en) | Processes and intermediates useful to make antifolates | |
| CN108864113B (en) | MDM2-HDAC double-target inhibitor, pharmaceutical composition, preparation and application thereof | |
| JPS61263995A (en) | Production of cytosine nucleoside | |
| CN116217654A (en) | Preparation method of linker drug conjugate and intermediate thereof | |
| KR100531668B1 (en) | 4-Hydroxyphenylglycine derivatives and processes for the preparation thereof | |
| WO2022032644A1 (en) | Method for preparing substituted imidazo[1,2-a]pyridin-2-ylamide compound, and intermediate thereof | |
| NZ529168A (en) | Preparation and isolation of indolocarbazole glycosides | |
| CN115703712A (en) | Process for the synthesis of 5,8-diamino-3,4-dihydro-2H-1-naphthalenone and intermediate compounds useful therein |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication |