CN114634412A - High-purity gentisic acid and application thereof - Google Patents
High-purity gentisic acid and application thereof Download PDFInfo
- Publication number
- CN114634412A CN114634412A CN202210537742.2A CN202210537742A CN114634412A CN 114634412 A CN114634412 A CN 114634412A CN 202210537742 A CN202210537742 A CN 202210537742A CN 114634412 A CN114634412 A CN 114634412A
- Authority
- CN
- China
- Prior art keywords
- gentisic acid
- acid
- purity
- filtering
- gentisic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C65/00—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C65/01—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing hydroxy or O-metal groups
- C07C65/03—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing hydroxy or O-metal groups monocyclic and having all hydroxy or O-metal groups bound to the ring
- C07C65/05—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing hydroxy or O-metal groups monocyclic and having all hydroxy or O-metal groups bound to the ring o-Hydroxy carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/06—Free radical scavengers or antioxidants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/02—Preparation of carboxylic acids or their salts, halides or anhydrides from salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/41—Preparation of salts of carboxylic acids
- C07C51/412—Preparation of salts of carboxylic acids by conversion of the acids, their salts, esters or anhydrides with the same carboxylic acid part
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/43—Separation; Purification; Stabilisation; Use of additives by change of the physical state, e.g. crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/47—Separation; Purification; Stabilisation; Use of additives by solid-liquid treatment; by chemisorption
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/48—Separation; Purification; Stabilisation; Use of additives by liquid-liquid treatment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/487—Separation; Purification; Stabilisation; Use of additives by treatment giving rise to chemical modification
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Pain & Pain Management (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Toxicology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The application discloses high-purity gentisic acid, the purity of which is more than or equal to 99.50 percent, comprises: gentisic acid, salicylic acid and impurity A, wherein the structural formula of the impurity A is shown in the specification
Description
Technical Field
The present application relates to the field of pharmaceutical chemistry. In particular, the application relates to high-purity gentisic acid and application thereof.
Background
Gentisic acid, chemically known by the name 2, 5-dihydroxybenzoic acid, also known as 5-hydroxy salicylic acid, is a secondary product of salicylic acid after renal metabolism, and is commonly used clinically as antiviral, antibacterial and analgesic agents (sodium salt). Gentisic acid is a hydroquinone compound, is easy to generate oxidation reaction, and is also used as an antioxidant excipient in the pharmaceutical industry. Gentisic acid has been used as a pharmaceutical adjuvant abroad.
In the prior art, benzoic acid is adopted as a raw material to synthesize products through chlorination, hydrolysis and other steps, but five main products synthesized by the method are difficult to separate and purify, so that the final yield is only about 5%, and industrial production is difficult to realize.
In the prior art, the gentisic acid is mainly prepared by the kebel-schmitt reaction of hydroquinone, but the product prepared by the method contains a large amount of isomers of 2, 4-dihydroxybenzoic acid and 2, 6-dihydroxybenzoic acid, not only has lower product purity, but also has darker color and luster, is yellow powder crystal, can obtain a white gentisic acid product meeting the specification by repeated recrystallization purification, and has low yield and high cost.
Patent CN100363322C discloses a method for producing hydroxybenzoic acids, which comprises preparing alkali metal salts of phenolic compounds from phenolic compounds, which are reacted with carbon dioxide, wherein the preparation of alkali metal salts of phenolic compounds from phenolic compounds comprises the following steps: a) reacting the alkali metal alkoxide with an excess of a phenolic compound, the phenolic compound being in excess of the alkali metal alkoxide, to form an alkali metal salt of the phenolic compound; and b) distilling off the alcohol formed from the reaction while carrying out a). The process does not employ aprotic polar organic solvents.
Patent CN1684935A discloses a process for producing hydroxybenzoic acids by dehydrating phenols with an alkali metal compound to obtain an alkali metal salt of phenols, and then reacting the alkali metal salt of phenols with carbon dioxide, wherein the alkali metal compound and the phenol in excess of the alkali metal compound are dehydrated at 160 ℃ or higher. The method does not use aprotic polar organic solvent, and has simple process and low cost.
Patent CN102766042A discloses a method for preparing high-purity gentisic acid, comprising: adding salicylic acid and glacial acetic acid into a reaction container, and uniformly stirring; heating, dropwise adding bromine, reacting to obtain bromosalicylic acid, and distilling under reduced pressure to recover glacial acetic acid; adding liquid alkali in the presence of cuprous chloride, heating, and reacting to obtain hydroxy salicylic acid; cooling to normal temperature, filtering to remove cuprous chloride, and adjusting the pH value to 2; filtering out the mixture of the hydroxyl salicylic acid, adding the mixture into water, adjusting the pH value to 6.9-7.1, and cooling and crystallizing to obtain high-purity sodium cholate; adding the obtained sodium gentisate salt into water, adjusting the pH value to 2, and heating and refluxing in the presence of activated carbon and oxalic acid; filtering, cooling and crystallizing the filtrate to obtain the gentisic acid.
The gentisic acid products obtained by the methods in the prior art have poor purity and low yield.
Disclosure of Invention
In order to overcome the defects in the prior art, the method screens the catalyst and the alkali, optimizes and researches the reaction temperature and the like on the basis of the prior art, develops subsequent salifying, dissociating and decoloring crystallization processes under the conditions of low product purity and more impurity content, and finally obtains the qualified high-purity gentisic acid.
The specific technical scheme of the application is as follows:
1. a high-purity gentisic acid, characterized in that the purity thereof is not less than 99.50%, comprising: gentisic acid, salicylic acid and impurity A, wherein the impurity A has a structure shown in formula (I):
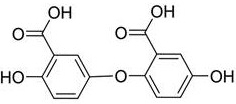
formula (I);
preferably, in the high-purity gentisic acid, the mass percent of the salicylic acid is less than or equal to 0.1 percent, and the mass percent of the impurity A is less than or equal to 0.1 percent;
preferably, the high-purity gentisic acid further comprises an impurity B, wherein the impurity B has a structure represented by formula (II):
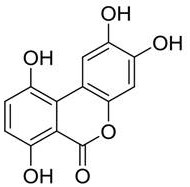
formula (II);
preferably, in the high-purity gentisic acid, the mass percentage of the impurity B is less than or equal to 0.1%.
2. The high purity gentisic acid of item 1, which is prepared by a method comprising the steps of:
feeding and reacting: adding 5-bromosalicylic acid into an inorganic alkali solution for reaction;
acidifying: adding concentrated hydrochloric acid to adjust pH, precipitating solid, and filtering;
and (3) extraction: adding an extracting agent I into the filtrate obtained in the acidification step, extracting, and standing to obtain floccules;
and (3) filtering: filtering and separating the solution to obtain an organic phase I and a water phase, adding an extracting agent II into the water phase, extracting, filtering and separating the solution to obtain an organic phase II, and combining the organic phase II and the organic phase I to obtain an organic phase III;
and (3) crystallization: concentrating the organic phase III, separating out solids, and adding the organic solvent I to obtain a crude gentisic acid product;
salifying: adding the crude gentisic acid into a second organic solvent, and dropwise adding a solution of the second organic solvent containing organic base to obtain a wet gentisic acid organic base salt;
dissociating: adding the wet gentisic acid organic base salt into water, dropwise adding concentrated hydrochloric acid to adjust the pH value, and filtering to obtain a refined gentisic acid wet product;
decoloring and crystallizing: adding the refined product wet product of gentisic acid and a decoloring agent into water, and filtering to obtain gentisic acid crystals;
and (3) filtering and drying: and leaching the gentisic acid crystal with water, then cleaning with an organic solvent I, and drying to obtain the high-purity gentisic acid.
3. The high-purity gentisic acid according to the item 2, characterized in that in the feeding reaction step, 5-bromosalicylic acid is added into an inorganic alkali solution in the presence of a copper catalyst, stirred, heated to react the 5-bromosalicylic acid with the inorganic alkali solution, cooled again, and added with water;
preferably, the inorganic alkali solution is an aqueous solution of sodium hydroxide or an aqueous solution of potassium hydroxide, preferably an aqueous solution of potassium hydroxide;
preferably, the concentration of the inorganic alkali solution is 4-8 mol/L, and more preferably 5-6 mol/L;
preferably, the mass ratio of the inorganic base to the 5-bromosalicylic acid is 4-7: 1, more preferably 5-6: 1;
preferably, the temperature is raised to 90-114 ℃, and more preferably to 105-114 ℃;
preferably, the temperature is increased to enable the 5-bromosalicylic acid to react with the inorganic alkali solution, the content of the 5-bromosalicylic acid is controlled to be less than 1.5% in HPLC, the reaction time is 20-48 h, and more preferably 20-30 h;
preferably, the temperature is reduced to 20-30 ℃;
preferably, 5-bromosalicylic acid is added into the inorganic alkali solution in the presence of a copper catalyst and oxalic acid; more preferably, the mass ratio of the oxalic acid to the 5-bromosalicylic acid is 0.05-0.15: 1;
preferably, the copper catalyst is copper sulfate pentahydrate or cuprous oxide, more preferably copper sulfate pentahydrate; preferably, the mass ratio of the copper sulfate pentahydrate to the 5-bromosalicylic acid is 0.05-0.15: 1; preferably, the mass ratio of the cuprous oxide to the 5-bromosalicylic acid is 0.025-0.075: 1.
Preferably, the mass ratio of the water to the 5-bromosalicylic acid is 40-70: 1, more preferably 50-60: 1.
4. The high-purity gentisic acid according to item 2 or 3, wherein in the acidification step, the pH is adjusted to 2.3 to 2.5;
preferably, concentrated hydrochloric acid is added at the temperature of 20-30 ℃ to adjust the pH.
5. The high-purity gentisic acid as set forth in any one of claims 2 to 4, wherein in the extraction step, the first extraction agent is one or more selected from dichloromethane, ethyl acetate, isopropyl acetate, toluene and methyl t-butyl ether;
preferably, the first extractant is methyl tert-butyl ether;
preferably, in the extraction step, an extraction agent I and an extraction aid are added into the filtrate obtained in the acidification step, wherein the extraction aid is preferably sodium chloride;
preferably, the mass ratio of the methyl tert-butyl ether to the 5-bromosalicylic acid is 4-6: 1, preferably 5-5.5: 1;
preferably, the mass ratio of the sodium chloride to the 5-bromosalicylic acid is 1.0-1.5: 1;
preferably, in the filtering step, the second extracting agent is selected from any one or more of dichloromethane, ethyl acetate, isopropyl acetate, toluene and methyl tert-butyl ether;
preferably, the mass ratio of the second extracting agent to the 5-bromosalicylic acid is 4-6: 1, preferably 5-5.5: 1;
preferably, adding a filter aid into the funnel, filtering and separating liquid to obtain an organic phase I and an aqueous phase; more preferably, the filter aid is diatomaceous earth.
6. The high-purity gentisic acid according to any one of items 2 to 5, characterized in that in the crystallization step, the organic phase III is concentrated, a solid is precipitated, the organic solvent I is added, stirring is carried out, filtering is carried out, and a filter cake is dried to obtain a gentisic acid crude product;
preferably, the organic solvent I is one or more selected from n-heptane, acetone, ethyl acetate, petroleum ether and chloroform;
preferably, the organic phase three is concentrated to precipitate a large amount of solids;
preferably, concentrating the organic phase III under reduced pressure at 40-60 ℃, more preferably at 40-50 ℃;
preferably, the mass ratio of the organic solvent I to the 5-bromosalicylic acid is 4-5.5: 1, preferably 4.5-5: 1;
preferably, the stirring time is 1-2 h;
preferably, filtering at 20-30 ℃;
preferably, the filter cake is dried in vacuum for 2-4 hours at 40-50 ℃ to obtain a rough gentisic acid product.
7. The high-purity gentisic acid according to any one of items 2 to 6, is characterized in that in the salifying step, gentisic acid crude product is added into a second organic solvent, stirred, dropwise added with a solution of the second organic solvent containing organic base, cooled and filtered to obtain a wet gentisic acid organic base salt;
preferably, under the condition of 10-35 ℃, dropwise adding a solution of an organic solvent II containing organic alkali;
preferably, the organic base is diethylamine;
preferably, the mass ratio of the organic base to the 5-bromosalicylic acid is 0.75-0.85: 1;
preferably, the mass ratio of the organic solvent II to the crude gentisic acid is 8.6-10: 1, and preferably 9-9.8: 1;
preferably, the organic solvent II is one or more selected from acetone, isopropanol and tetrahydrofuran;
preferably, the temperature is reduced to 2-8 ℃.
8. The high-purity gentisic acid as claimed in any one of items 2 to 7, wherein in the dissociating step, the gentisic acid organic base salt wet product is added into water, stirred, dropwise added with concentrated hydrochloric acid to adjust the pH value, cooled and filtered to obtain a gentisic acid refined product wet product;
preferably, concentrated hydrochloric acid is added dropwise at the temperature of 20-30 ℃ to adjust the pH value;
preferably, the pH is adjusted to 1-2;
preferably, the mass ratio of the water to the wet product of the gentisic acid organic base salt is 4-6: 1;
preferably, the temperature is reduced to 0-10 ℃.
9. The high-purity gentisic acid according to any one of items 2 to 8, wherein in the decoloring and crystallization steps, a refined gentisic acid wet product and a decoloring agent are added into water, stirred, heated, filtered, and the filtrate is cooled to obtain gentisic acid crystals;
preferably, the refined product wet product of the gentisic acid, the decolorant and the oxalic acid are added into water;
preferably, the decolorant is selected from one or more of activated carbon, alumina and clay;
preferably, the temperature is kept for 0.5 to 6 hours after the temperature is raised, preferably 0.5 to 4 hours, and more preferably 0.5 to 1.5 hours;
preferably, the temperature is increased to 65-75 ℃;
preferably, the mass ratio of the activated carbon to the refined gentisic acid wet product is 0.05-0.1: 1;
preferably, the mass ratio of the oxalic acid to the refined gentisic acid wet product is 0.01-0.03: 1;
preferably, the mass ratio of the water to the refined gentisic acid wet product is 4-6: 1;
preferably, filtering and cooling the filtrate to 0-10 ℃ to obtain the gentisic acid crystal.
10. The high-purity gentisic acid according to any one of claims 2 to 9, wherein in the filtering and drying step, the drying conditions are as follows: and (4) drying for 10-15 h in vacuum at 40-55 ℃.
11. Use of the high purity gentisic acid according to any one of claims 1 to 10 in the preparation of an antiviral agent, an antibacterial agent, an analgesic agent or an antioxidant excipient.
ADVANTAGEOUS EFFECTS OF INVENTION
The purity of the high-purity gentisic acid is controlled to be more than 99.5%, various organic impurities can be controlled to be less than 0.1%, the high-purity gentisic acid can be used as a pharmaceutical adjuvant, the high-purity gentisic acid is prepared by a specific preparation method, the yield of the high-purity gentisic acid can reach 18-22%, and the high-purity gentisic acid is suitable for large-scale industrial production.
Detailed Description
The following description of the exemplary embodiments of the present application, including various details of the embodiments of the present application to assist in understanding, should be taken as exemplary only. Accordingly, those of ordinary skill in the art will recognize that various changes and modifications of the embodiments described herein can be made without departing from the scope and spirit of the present application. Also, descriptions of well-known functions, structures and reagents are omitted in the following description for clarity and conciseness.
The application provides a high-purity gentisic acid, which is characterized in that the purity is more than or equal to 99.5%, and the high-purity gentisic acid comprises: gentisic acid, salicylic acid and impurity A. Wherein the impurity A has a structure represented by formula (I):
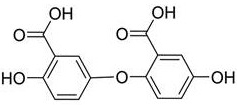
formula (I).
In one embodiment, the percentage by mass of salicylic acid in the high-purity gentisic acid is 0.1% or less, for example, 0.01%, 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0.09%, 0.1%, etc., and the percentage by mass of impurity a is 0.1% or less, for example, 0.01%, 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0.09%, 0.1%, etc.
In one embodiment, the high purity gentisic acid comprises: gentisic acid, salicylic acid, an impurity A and an impurity B, wherein the impurity B has a structure shown in a formula (II):
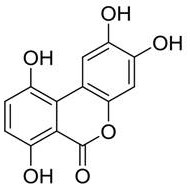
formula (II).
In one embodiment, the impurity B in the high-purity gentisic acid is 0.1% by mass or less, and may be, for example, 0.01%, 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0.09%, 0.1%, or the like.
In one embodiment, the high purity gentisic acid comprises: gentisic acid, salicylic acid and impurity A, but not impurity B.
In one embodiment, the high-purity gentisic acid contains an impurity of C (2, 3-dihydroxybenzoic acid), and the mass percentage of the 2, 3-dihydroxybenzoic acid in the high-purity gentisic acid is 0.1% or less, and may be, for example, 0.01%, 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0.09%, 0.1%, or the like.
In this application"gentisic acid", its english name is: gentisic Acid, chemical name: 2, 5-dihydroxybenzoic acid, Chemical Abstracts (CAS) No.: 490-79-9, other names: DHB, 5-hydroxy salicylic acid, molecular formula: c7H6O4The molecular weight is: 154.12, respectively; the solid is white to light yellow powder, is easily soluble in methanol, is dissolved in acetonitrile, is slightly soluble in water, and has a melting point of 207-210 ℃. The gentisic acid is used as a pharmaceutic adjuvant and used as a free radical quencher in preparation, and the radiation self-decomposition effect of a radionuclide on a prodrug is reduced.
In one embodiment, the high purity gentisic acid of the present application is prepared by a method comprising the steps of:
(1) feeding and reacting: adding 5-bromosalicylic acid into an inorganic base and a catalyst solution for reaction;
(2) acidifying: adding concentrated hydrochloric acid to adjust pH, precipitating solid, and filtering;
(3) and (3) extraction: adding an extracting agent I into the filtrate obtained in the acidification step, extracting, and standing to obtain floccules;
(4) and (3) filtering: filtering and separating the solution to obtain an organic phase I and a water phase, adding an extracting agent II into the water phase, extracting, filtering and separating the solution to obtain an organic phase II, and combining the organic phase II and the organic phase I to obtain an organic phase III;
(5) and (3) crystallization: concentrating the organic phase III, separating out a solid, and adding the organic solvent I to obtain a crude product of gentisic acid;
(6) salifying: adding the crude gentisic acid into a second organic solvent, and dropwise adding a solution of the second organic solvent containing organic base to obtain a wet gentisic acid organic base salt;
(7) dissociating: adding the wet gentisic acid organic base salt into water, dropwise adding concentrated hydrochloric acid to adjust the pH value, and filtering to obtain a refined gentisic acid wet product;
(8) decoloring and crystallizing: adding the refined gentisic acid wet product and a decoloring agent into crystallization solvent water, and filtering to obtain a refined gentisic acid product;
(9) and (3) filtering and drying: and leaching the refined gentisic acid product with water, cleaning with the organic solvent I, and drying to obtain high-purity gentisic acid.
"5-Bromofed Populus" (R) Rehd. in the present applicationAcid ", its english name is: 5-Bromosalicylic acid, other chemical names: 5-bromo-2-hydroxybenzoic acid, Chemical Abstracts (CAS) no: 89-55-4, the molecular formula is: c7H5BrO3The molecular weight is: 217.02, respectively; calculated as anhydrate, it contains C7H5BrO3Not less than 98.0 percent; it is a white or off-white crystalline powder.
In a specific embodiment, in the feeding reaction step, under the condition of the existence of a copper catalyst, 5-bromosalicylic acid is added into an inorganic alkali solution, stirred, heated to enable the 5-bromosalicylic acid to react with the inorganic alkali solution, cooled, and added with water.
In one embodiment, in the feeding reaction step, the inorganic alkali solution is an aqueous solution of sodium hydroxide or an aqueous solution of potassium hydroxide, preferably an aqueous solution of potassium hydroxide; the concentration of the inorganic alkali solution is 4 to 8mol/L, for example, 4mol/L, 4.5mol/L, 5mol/L, 5.5mol/L, 6mol/L, 6.5mol/L, 7mol/L, 7.5mol/L, 8mol/L, etc., and the lower the concentration of the inorganic alkali solution is, the slower the reaction is, and the preferable inorganic alkali solution is 5 to 6mol/L in view of cost and ease of operation.
In one embodiment, in the feeding reaction step, the ratio of the amount of the inorganic base to the 5-bromosalicylic acid, i.e., the molar ratio, is 4 to 7:1, and may be, for example, 4:1, 4.5:1, 5:1, 5.3:1, 5.5:1, 6:1, 6.5:1, 7:1, and preferably 5 to 6: 1.
In one embodiment, in the feeding reaction step, the temperature rise is raised to 0 to 114 ℃, for example, 90 ℃, 100 ℃, 101 ℃, 102 ℃, 103 ℃, 104 ℃, 105 ℃, 106 ℃, 107 ℃, 108 ℃, 109 ℃, 110 ℃, 111 ℃, 112 ℃, 113 ℃, 114 ℃ and the like, preferably to 105 to 114 ℃. 114 ℃ is the boiling point of the reaction system, the temperature cannot be increased to higher temperature, the temperature is controlled in the range, the reaction time is short, and the conversion of raw materials is complete.
In a specific embodiment, in the feeding reaction step, the temperature is raised to react the 5-bromosalicylic acid with the inorganic alkali solution, the content of the 5-bromosalicylic acid in HPLC is controlled to be less than 1.5%, and the reaction time is 20-48 h, for example, 20h, 22h, 24h, 26h, 28h, 30h, 32h, 34h, 36h, 38h, 40h, 42h, 44h, 46h, 48h, and the like, preferably 20-30 h. The reaction time is controlled within this range, and the reaction is complete with less impurities.
In one embodiment, the method for controlling the content of 5-bromosalicylic acid in HPLC in the feeding reaction step comprises the following steps:
5-Bromoesin acid positioning solution: taking a proper amount of 5-bromosalicylic acid reference substance, adding a solvent to dissolve and dilute the reference substance to prepare a solution containing about 6 micrograms of the reference substance in each 1mL, and shaking up;
test solution: taking a proper amount of reaction liquid, diluting with a solvent by 400 times (for example, diluting 0.025mL to 10 mL), shaking up, filtering, and taking the filtrate to obtain the final product;
chromatographic conditions are as follows: octadecylsilane bonded silica gel (Thermo Acclaim 120A C18, 4.6 mm. times.250 mm, 5 μm, or a column of comparable performance) was used as the filler; taking 0.05% phosphoric acid water solution as a mobile phase A and acetonitrile as a mobile phase B; a trapping column: welch Ghost-Buster column II 4.0 x 50mm or other suitable trapping column; the flow rate is 1.0mL per minute, and linear gradient elution is carried out; the column temperature was 40 ℃; the detection wavelength is 220 nm; the sample injection volume is 10 mu L;
the determination method comprises the following steps: precisely measuring a test solution, injecting the test solution into a liquid chromatograph, and recording a chromatogram;
in the limit sample solution chromatogram, the peak area of 5-bromosalicylic acid is not more than 1.5% of the total peak area calculated by an area normalization method.
In one embodiment, in the feeding reaction step, the temperature is reduced to 20-30 ℃, for example, 20 ℃, 21 ℃, 22 ℃, 23 ℃, 24 ℃, 25 ℃, 26 ℃, 27 ℃, 28 ℃, 29 ℃, 30 ℃ and the like.
In one embodiment, in the feeding reaction step, 5-bromosalicylic acid is added to the inorganic base solution in the presence of a copper catalyst and oxalic acid. Oxalic acid is used as an antioxidant and can be easily removed with water.
In one embodiment, in the feeding reaction step, the ratio of the amount of oxalic acid to 5-bromosalicylic acid, i.e., the molar ratio, is 0.05 to 0.15:1, and may be, for example, 0.05:1, 0.06:1, 0.07:1, 0.08:1, 0.09:1, 0.1:1, 0.11:1, 0.12:1, 0.13:1, 0.14:1, 0.15:1, or the like. The oxalic acid content is controlled within the range, and the product yield is high.
In one embodiment, the copper catalyst is copper sulfate pentahydrate or cuprous oxide in the feed reaction step, and is most preferably copper sulfate pentahydrate since copper sulfate is soluble in water, post-treatment is convenient, and the reaction can be more complete.
In one embodiment, in the feeding reaction step, the copper catalyst is copper sulfate pentahydrate, and the amount ratio of the copper sulfate pentahydrate to the 5-bromosalicylic acid is 0.05 to 0.15:1, such as 0.05:1, 0.06:1, 0.07:1, 0.08:1, 0.09:1, 0.1:1, 0.11:1, 0.12:1, 0.13:1, 0.14:1, 0.15: 1. The content of the copper sulfate pentahydrate is controlled within the range, the reaction time is shorter, the conversion rate is higher, and the copper sulfate pentahydrate is completely dissolved.
In one embodiment, in the feeding reaction step, the copper catalyst is cuprous oxide, and the amount ratio, i.e., molar ratio, of the cuprous oxide to the 5-bromosalicylic acid is 0.025 to 0.075:1, for example, 0.025:1, 0.03:1, 0.035:1, 0.04:1, 0.045:1, 0.05:1, 0.055:1, 0.06:1, 0.065:1, 0.07:1, 0.075:1, and the like.
In one embodiment, in the feeding reaction step, the amount ratio, i.e. the molar ratio, of the water to the 5-bromosalicylic acid is 40 to 70:1, for example, 40:1, 41.2:1, 42:1, 44:1, 46:1, 48:1, 50:1, 52:1, 54:1, 54.9:1, 55:1, 56:1, 58:1, 60:1, 62:1, 64:1, 66:1, 68:1, 68.6:1, 70:1, and the like, preferably 50 to 60: 1.
In one embodiment, in the acidification step, the pH is adjusted to 2.3 to 2.5, which may be, for example, 2.3, 2.35, 2.4, 2.45, 2.5, etc. The pH value is controlled within the range, the salicylic acid can be effectively removed, the yield is higher, pigment impurities in subsequent methyl tert-butyl ether extraction are least, and the product color is lighter.
In one embodiment, in the acidification step, concentrated hydrochloric acid is added at 20-30 deg.C to adjust pH, for example, concentrated hydrochloric acid is added at 20 deg.C, 21 deg.C, 22 deg.C, 23 deg.C, 24 deg.C, 25 deg.C, 26 deg.C, 27 deg.C, 28 deg.C, 29 deg.C, 30 deg.C to adjust pH.
In a specific embodiment, in the extraction step, the extractant one is selected from any one or more of dichloromethane, ethyl acetate, isopropyl acetate, toluene and methyl tert-butyl ether; methyl tert-butyl ether is preferred.
In one embodiment, in the extraction step, the mass ratio of the methyl tert-butyl ether to the 5-bromosalicylic acid is 4 to 6:1, for example, 4:1, 4.1:1, 4.2:1, 4.3:1, 4.4:1, 4.44:1, 4.5:1, 4.6:1, 4.7:1, 4.8:1, 4.9:1, 5:1, 5.1:1, 5.18:1, 5.2:1, 5.3:1, 5.4:1, 5.5:1, 5.6:1, 5.7:1, 5.8:1, 5.92:1, 6:1, and the like, and preferably 5 to 5.5: 1.
In one embodiment, in the extraction step, the mass ratio of the sodium chloride to the 5-bromosalicylic acid is 1.0 to 1.5:1, and may be, for example, 1.0:1, 1.1:1, 1.2:1, 1.3:1, 1.4:1, 1.5:1, and the like.
In a specific embodiment, in the filtering step, the second extractant is selected from any one or more of dichloromethane, ethyl acetate, isopropyl acetate, toluene and methyl tert-butyl ether.
In one embodiment, in the filtering step, the mass ratio of the second extractant to the 5-bromosalicylic acid is 4 to 6:1, for example, 4:1, 4.1:1, 4.2:1, 4.3:1, 4.4:1, 4.44:1, 4.5:1, 4.6:1, 4.7:1, 4.8:1, 4.9:1, 5:1, 5.1:1, 5.18:1, 5.2:1, 5.3:1, 5.4:1, 5.5:1, 5.6:1, 5.7:1, 5.8:1, 5.92:1, 6:1, and the like, and preferably 5 to 5.5: 1.
In one embodiment, in the filtration step, diatomaceous earth is added to a buchner funnel, filtered, and separated to provide an organic phase one and an aqueous phase.
In a specific embodiment, in the crystallization step, concentrating the organic phase three, separating out a solid, adding the organic solvent I, stirring, filtering, and drying a filter cake to obtain a crude gentisic acid product; the organic solvent I is one or more than two selected from n-heptane, acetone, ethyl acetate, petroleum ether and dichloromethane.
In one embodiment, in the crystallization step, the organic phase is concentrated under reduced pressure at 40 to 60 ℃, for example, 40 ℃, 42 ℃, 44 ℃, 46 ℃, 48 ℃, 50 ℃, 52 ℃, 54 ℃, 56 ℃, 58 ℃ and 60 ℃, preferably at 40 to 50 ℃ until a large amount of solids are precipitated. The temperature is controlled within the range, the product purity is high, and the concentration time is shorter.
In one embodiment, in the crystallization step, the mass ratio of the organic solvent one to the 5-bromosalicylic acid is 4 to 5.5:1, for example, 4:1, 4.08:1, 4.1:1, 4.2:1, 4.3:1, 4.4:1, 4.5:1, 4.6:1, 4.7:1, 4.8:1, 4.9:1, 5:1, 5.1:1, 5.2:1, 5.3:1, 5.44:1, 5.5:1, and the like, preferably 4.5 to 5: 1.
In a specific embodiment, in the crystallization step, the stirring time is 1 to 2 hours, and may be, for example, 1 hour, 1.1 hour, 1.2 hours, 1.3 hours, 1.4 hours, 1.5 hours, 1.6 hours, 1.7 hours, 1.8 hours, 1.9 hours, 2 hours, or the like; the filtration is carried out at 20 to 30 ℃ and may be carried out, for example, at 20 ℃, 21 ℃, 22 ℃, 23 ℃, 24 ℃, 25 ℃, 26 ℃, 27 ℃, 28 ℃, 29 ℃, 30 ℃ or the like.
In a specific embodiment, in the crystallization step, the filter cake is vacuum-dried at 40 to 50 ℃ for 2 to 4 hours to obtain a crude gentisic acid, for example, the crude gentisic acid can be vacuum-dried at 40 ℃, 41 ℃, 42 ℃, 43 ℃, 44 ℃, 45 ℃, 46 ℃, 47 ℃, 48 ℃, 49 ℃ and 50 ℃ for 2 hours, 2.2 hours, 2.4 hours, 2.6 hours, 2.8 hours, 3 hours, 3.2 hours, 3.4 hours, 3.6 hours, 3.8 hours, 4 hours and the like.
The purification cannot be effectively carried out by the solvent crystallization method alone, but gentisic acid has a carboxylic acid structure, so the salification step and the dissociation step are developed in the application.
In a specific embodiment, in the step of salifying, the crude gentisic acid is added into a second organic solvent, stirring is carried out, a solution of the second organic solvent containing an organic base is dropwise added, the temperature is reduced, and filtering is carried out, so that a salified wet gentisic acid organic base product is obtained.
In one embodiment, in the salt formation step, a solution of the organic solvent II containing the organic base is added dropwise at 10 to 35 ℃, for example, 10 ℃, 12 ℃, 14 ℃, 15 ℃, 18 ℃, 20 ℃, 22 ℃, 25 ℃, 27 ℃, 30 ℃, 32 ℃, 34 ℃, 35 ℃. The temperature is controlled within the range, so that the crystals are not easy to explode out when being cooled and separated out subsequently, and the impurities are less.
In a specific embodiment, in the salt formation step, the organic base is diethylamine, and the organic solvent is one or more selected from acetone, isopropanol and tetrahydrofuran.
In one embodiment, in the salt formation step, the amount ratio, i.e., the molar ratio, of the organic base to the 5-bromosalicylic acid is 0.75 to 0.85:1, and may be, for example, 0.75:1, 0.76:1, 0.77:1, 0.78:1, 0.79:1, 0.8:1, 0.81:1, 0.82:1, 0.83:1, 0.84:1, 0.85:1, and preferably 0.75 to 0.81: 1. By controlling the content of the organic base within the above range, salt formation can be sufficiently achieved with less impurities.
In a specific embodiment, in the salt formation step, the mass ratio of the organic solvent ii to the gentisic acid crude product is 8.6-10: 1, for example, 8.6:1, 8.7:1, 8.8:1, 8.9:1, 9:1, 9.1:1, 9.2:1, 9.3:1, 9.4:1, 9.42:1, 9.5:1, 9.6:1, 9.7:1, 9.8:1, 9.9:1, 10:1, and the like, preferably 9-9.8: 1. Controlling acetone within the above range can result in higher yield and more complete dissolution of the gentisic acid crude product.
In a specific embodiment, in the salifying step, the temperature is reduced to 2-8 ℃, for example, 2 ℃, 2.5 ℃,3 ℃, 3.5 ℃,4 ℃, 4.5 ℃,5 ℃, 5.5 ℃,6 ℃, 6.5 ℃, 7 ℃, 7.5 ℃, 8 ℃ and the like. By controlling the temperature within the above range, higher purity and yield can be obtained.
In a specific embodiment, in the step of salifying, after the dropwise addition of the organic solvent ii solution containing the organic base is completed, the temperature is maintained for 0.3 to 0.8 hour, and then the temperature is reduced, for example, the temperature may be maintained for 0.3 hour, 0.4 hour, 0.5 hour, 0.6 hour, 0.7 hour, 0.8 hour, and the like.
In a specific embodiment, in the salifying step, the temperature reduction gradient is 15-25 ℃/h, and may be, for example, 15 ℃/h, 17 ℃/h, 19 ℃/h, 20 ℃/h, 22 ℃/h, 23 ℃/h, 25 ℃/h, and the like.
In a specific embodiment, in the salifying step, the temperature is reduced and then is kept for 0.3 to 0.8h, and the filtration is carried out, for example, the temperature can be kept for 0.3h, 0.4h, 0.5h, 0.6h, 0.7h, 0.8h and the like.
In one specific embodiment, in the dissociating step, the wet gentisic acid organic base salt is added into water, stirred, dropwise added with concentrated hydrochloric acid to adjust the pH value, cooled and filtered to obtain a refined gentisic acid wet product.
In one embodiment, in the dissociation step, concentrated hydrochloric acid is added dropwise at 20-30 deg.C to adjust pH, for example, concentrated hydrochloric acid is added dropwise at 20 deg.C, 21 deg.C, 22 deg.C, 23 deg.C, 24 deg.C, 25 deg.C, 26 deg.C, 27 deg.C, 28 deg.C, 29 deg.C, 30 deg.C to adjust pH.
In one embodiment, in the liberating step, the pH is adjusted to 1-2, and can be, for example, 1, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2, and the like. The pH value is controlled within the range, so that the gentisic acid can be more completely dissociated, and the product yield is higher.
In one embodiment, in the dissociating step, the mass ratio of the water to the wet gentisic acid organic base salt is 4 to 6:1, and may be, for example, 4:1, 4.2:1, 4.4:1, 4.6:1, 4.8:1, 5:1, 5.2:1, 5.4:1, 5.6:1, 5.8:1, 6:1, and the like. Controlling the water in the range can completely dissolve the wet gentisic acid organic base salt and improve the yield of the product.
In a specific embodiment, in the dissociating step, the temperature is decreased to 0 to 10 ℃, for example, 0 ℃, 1 ℃, 2 ℃,3 ℃,4 ℃,5 ℃,6 ℃, 7 ℃, 8 ℃, 9 ℃, 10 ℃ and the like, and the temperature gradient is 15 to 25 ℃/h, for example, 15 ℃/h, 17 ℃/h, 19 ℃/h, 20 ℃/h, 22 ℃/h, 23 ℃/h, 25 ℃/h and the like.
In one embodiment, in the dissociating step, the temperature is reduced and then maintained for 0.3 to 0.8h, and the filtration is performed, for example, the temperature can be maintained for 0.3h, 0.4h, 0.5h, 0.6h, 0.7h, 0.8h, and the like.
In a specific embodiment, in the decoloring and crystallizing steps, adding a wet product of the refined gentisic acid and a decoloring agent into water, stirring, heating, filtering, and cooling the filtrate to obtain gentisic acid crystals; the decolorant is not limited to one or more of activated carbon, alumina, and clay.
In one embodiment, in the decolorizing and crystallizing step, the refined wet gentisic acid, decolorizing agent and oxalic acid are added to water, the oxalic acid acting as an antioxidant.
In one embodiment, in the decoloring and crystallizing steps, the temperature is raised and then maintained for 0.5 to 6 hours, for example, 0.5 hour, 0.6 hour, 0.7 hour, 0.8 hour, 0.9 hour, 1 hour, 1.2 hour, 1.5 hour, 2 hours, 2.5 hours, 3 hours, 3.5 hours, 4 hours, 4.5 hours, 5 hours, 5.5 hours, 6 hours, and the like, preferably 0.5 to 1.5 hours. The heat preservation time is controlled within the range, so that the wet product of the refined gentisic acid can be completely dissolved, and the content of the impurity B is further controlled.
In one embodiment, the temperature is raised to 65-75 ℃ in the decoloring and crystallizing steps, and may be, for example, 65 ℃, 66 ℃, 67 ℃, 68 ℃, 69 ℃, 70 ℃, 71 ℃, 72 ℃, 73 ℃, 74 ℃, 75 ℃ or the like. Controlling the temperature within the range can completely dissolve the refined gentisic acid wet product and improve the purity of the final product.
In one embodiment, in the decoloring and crystallizing step, the mass ratio of the activated carbon to the wet refined gentisic acid product is 0.05 to 0.1:1, and may be, for example, 0.05:1, 0.06:1, 0.07:1, 0.08:1, 0.09:1, 0.1:1, or the like. The mass ratio of the active carbon to the refined gentisic acid wet product is controlled within the range, so that the pigment removal efficiency can be improved.
In one embodiment, in the decoloring and crystallizing step, the mass ratio of the oxalic acid to the wet gentisic acid refined product is 0.01 to 0.03:1, and may be, for example, 0.01:1, 0.015:1, 0.02:1, 0.025:1, 0.03:1, or the like.
In one embodiment, in the decoloring and crystallizing step, the mass ratio of the water to the wet refined gentisic acid product is 4 to 6:1, and may be, for example, 4:1, 4.2:1, 4.4:1, 4.6:1, 4.8:1, 5:1, 5.2:1, 5.4:1, 5.6:1, 5.8:1, 6:1, or the like. The mass ratio of the water to the refined gentisic acid wet product is controlled within the range, so that the refined gentisic acid wet product can be obtained, and the product yield can be further improved.
In one embodiment, in the decoloring and crystallizing steps, filtering and cooling the filtrate to 0 to 10 ℃, for example, 0 ℃, 1 ℃, 2 ℃,3 ℃,4 ℃,5 ℃,6 ℃, 7 ℃, 8 ℃, 9 ℃, 10 ℃ and the like, and keeping the temperature for 0.3 to 0.8h, for example, 0.3h, 0.4h, 0.5h, 0.6h, 0.7h, 0.8h and the like, to obtain gentisic acid crystals.
In one embodiment, in the filtering and drying step, the drying conditions are: vacuum drying at 40-55 deg.C for 10-15 hr, such as vacuum drying at 40 deg.C, 42 deg.C, 44 deg.C, 46 deg.C, 48 deg.C, 50 deg.C, 52 deg.C, 54 deg.C, 55 deg.C for 10 hr, 11 hr, 12 hr, 13 hr, 14 hr, 15 hr, etc.
In one embodiment, the stirring in the present application is carried out under nitrogen.
In one embodiment, the water is purified water.
The present application also provides a use of the high purity gentisic acid as described in any of the above for the preparation of an antiviral agent, an antibacterial agent, an analgesic agent or an antioxidant excipient.
Examples
The materials used in the tests and the methods of the tests are generally and/or specifically described in the present application, and in the following examples, the materials or the instruments used are not indicated by the manufacturer, but are all conventional materials or instruments commercially available.
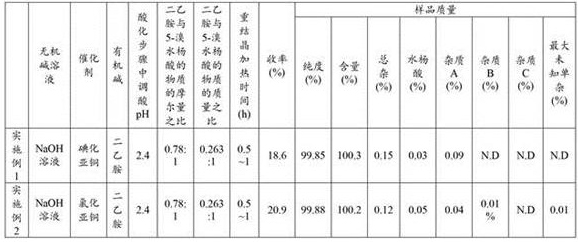
Example 1
Feeding and reacting: adding 5.0mol/L NaOH solution into a reaction kettle, and adding 50g of 5-bromosalicylic acid, 0.5g of cuprous iodide and 0.5g of oxalic acid; under the protection of nitrogen, starting stirring; heating to 100-120 ℃ for reaction for 24h, and sampling to control HPLC (5-bromosalicylic acid) to be less than or equal to 1.5%; cooling to 20-30 ℃, and adding water;
acidifying: controlling the temperature to be 20-30 ℃, adding concentrated hydrochloric acid to adjust the pH of the reaction solution to (2.4), separating out solids, and filtering;
and (3) extraction: adding 50g of sodium chloride and 250g of methyl tert-butyl ether into the filtrate, stirring and extracting, and standing to generate floccules;
and (3) filtering: adding 5g of diatomite into a Buchner funnel, filtering, and separating liquid to obtain an organic phase I and a water phase; adding 125g of methyl tert-butyl ether into the water phase, filtering, separating liquid to obtain an organic phase II, and combining the organic phase II and the organic phase I to obtain an organic phase III;
and (3) crystallization: concentrating the organic phase III at 40-50 ℃ under reduced pressure until a large amount of solid is separated out, and adding 360g of n-heptane; stirring for 1-2 h, and filtering at room temperature of 20-30 ℃; vacuum drying the filter cake for 2-4 h at 40-50 ℃ to obtain 16g of crude gentisic acid;
salifying: adding 250g of acetone into a reaction kettle, adding a rough gentisic acid product, and starting stirring under the protection of nitrogen; controlling the temperature to be 10-35 ℃, dropwise adding 36g of acetone solution containing 13.15g of diethylamine, and keeping the temperature for 0.5h after dropwise adding; closing heating, cooling (15-25 ℃/h) to 2-8 ℃, preserving heat for 0.5h, and filtering to obtain about 25g of wet gentisic acid diethylamine salt;
dissociating: adding 200g of water into a reaction kettle, and adding a wet product of diethylamine gentisate; under the protection of nitrogen, starting stirring, controlling the temperature to be 20-30 ℃, and dropwise adding concentrated hydrochloric acid to adjust the pH value; cooling to 0-10 ℃, preserving heat for 0.5h, and filtering to obtain a refined gentisic acid wet product;
decoloring and crystallizing: adding 250g of water into a reaction kettle, adding a refined product wet product of gentisic acid, adding 0.2g of activated carbon, and adding 0.1g of oxalic acid; under the protection of nitrogen, starting stirring, heating to 65-75 ℃, preserving heat for 0.5-1 h, and filtering while hot; cooling the filtrate by 0-10 ℃, and preserving the heat for 0.5 h;
and (3) filtering and drying: and (3) leaching with water, washing a filter cake with 22g of n-heptane, and vacuum-drying the filter cake at 40-55 ℃ for 12h to obtain 11g of high-purity gentisic acid.
Example 2
Feeding and reacting: adding 5.0mol/L NaOH solution into a reaction kettle, and adding 200g of 5-bromosalicylic acid, 1.8g of cuprous chloride and 2g of oxalic acid; starting stirring under the protection of nitrogen; heating to 100-120 ℃ for reaction for 24h, and sampling to control HPLC (5-bromosalicylic acid) to be less than or equal to 1.5%; cooling to room temperature (20-30 ℃), and adding water;
acidifying: controlling the temperature to be 20-30 ℃, adding concentrated hydrochloric acid to adjust the pH of the reaction solution to 2.4, separating out solids, and filtering;
and (3) extraction: adding 200g of sodium chloride and 1000g of methyl tert-butyl ether into the filtrate, stirring and extracting, and standing to generate floccules;
and (3) filtering: adding 50g of diatomite into a Buchner funnel, filtering, and separating liquid to obtain an organic phase I and a water phase; adding 500g of methyl tert-butyl ether into the water phase, filtering, separating liquid to obtain an organic phase II, and combining the organic phase II and the organic phase I to obtain an organic phase III;
and (3) crystallization: concentrating the organic phase III at 40-50 ℃ under reduced pressure until a large amount of solid is separated out, and adding 500g of n-heptane; stirring for 1-2 h, and filtering at room temperature of 20-30 ℃; vacuum drying the filter cake for 2-4 h at 40-50 ℃ to obtain 68g of crude gentisic acid;
salifying: adding 450g of acetone into a reaction kettle, adding a rough gentisic acid product, and starting stirring under the protection of nitrogen; controlling the temperature to be 10-35 ℃, dropwise adding 150g of acetone solution with 52.60g of diethylamine, and keeping the temperature for 0.5h after dropwise adding; turning off heating, cooling (15-25 ℃/h) to 2-8 ℃, preserving heat for 0.5h, and filtering to obtain 96g of wet gentisic acid diethylamine salt;
dissociating: adding water into a reaction kettle, and adding a wet product of the gentisic acid diethylamine salt; under the protection of nitrogen, starting stirring, controlling the temperature to be 20-30 ℃, and dropwise adding concentrated hydrochloric acid to adjust the pH; cooling to 0-10 ℃, preserving heat for 0.5h, and filtering to obtain a refined gentisic acid wet product;
decoloring and crystallizing: adding 1000g of water into a reaction kettle, adding a refined product wet product of gentisic acid, adding 1g of active carbon, and adding 0.5g of oxalic acid; under the protection of nitrogen, starting stirring, heating to 65-75 ℃, preserving heat for 0.5-1 h, and filtering while hot; cooling the filtrate by 0-10 ℃, and preserving the heat for 0.5 h;
and (3) filtering and drying: and (3) leaching with water, washing a filter cake with 200g of n-heptane, and vacuum-drying the filter cake at 40-55 ℃ for 12.0h to obtain 48g of high-purity gentisic acid.
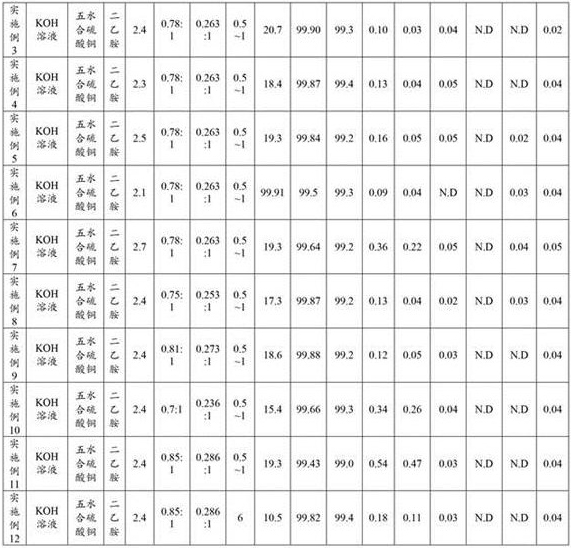
Example 3
Feeding and reacting: adding 5.0mol/L KOH solution into a reaction kettle, and adding 500g of 5-bromosalicylic acid, 5.0g of copper sulfate pentahydrate and 5g of oxalic acid; starting stirring under the protection of nitrogen; heating to 100-120 ℃ for reaction for 24h, and sampling to control HPLC (5-bromosalicylic acid) to be less than or equal to 1.5%; cooling to room temperature (20-30 ℃), and adding water;
acidifying: controlling the temperature to be 20-30 ℃, adding concentrated hydrochloric acid to adjust the pH of the reaction solution to 2.4, separating out solids, and filtering;
and (3) extraction: adding 500g of sodium chloride and 250g of methyl tert-butyl ether into the filtrate, stirring and extracting, and standing to obtain floccules;
and (3) filtering: adding 50g of diatomite into a Buchner funnel, filtering, and separating liquid to obtain an organic phase I and a water phase; adding 125g of methyl tert-butyl ether into the water phase, filtering, separating liquid to obtain an organic phase II, and combining the organic phase II and the organic phase I to obtain an organic phase III;
and (3) crystallization: concentrating the organic phase at 40-50 ℃ under reduced pressure until a large amount of solid is separated out, and adding 250g of n-heptane; stirring for 1-2 h, and filtering at room temperature of 20-30 ℃; vacuum drying the filter cake for 2-4 h at 40-50 ℃ to obtain 160g of crude gentisic acid;
salifying: adding 1200g of acetone into a reaction kettle, adding a crude gentisic acid product, and starting stirring under the protection of nitrogen; controlling the temperature to be 10-35 ℃, dropwise adding 280g of diethylamine-131.50 g of acetone solution, and keeping the temperature for 0.5h after dropwise adding; closing heating, cooling (15-25 ℃/h) to 2-8 ℃, preserving heat for 0.5h, and filtering to obtain a wet gentisic acid diethylamine salt product;
dissociating: adding 2000g of water into a reaction kettle, and adding a wet product of the gentisic acid diethylamine salt; under the protection of nitrogen, starting stirring, controlling the temperature to be 20-30 ℃, and dropwise adding concentrated hydrochloric acid to adjust the pH; cooling to 0-10 ℃, preserving heat for 0.5h, and filtering to obtain a refined gentisic acid wet product;
decoloring and crystallizing: adding 2000g of water into a reaction kettle, adding a refined product wet product of gentisic acid, adding 3g of active carbon, and adding 1.5g of oxalic acid; under the protection of nitrogen, starting stirring, heating to 65-75 ℃, preserving heat for 0.5-1 h, and filtering while hot; cooling the filtrate by 0-10 ℃, and preserving the heat for 0.5 h;
and (3) filtering and drying: and (3) leaching with water, washing a filter cake with 400g of n-heptane, and vacuum-drying the filter cake at 40-55 ℃ for 12h to obtain 110g of high-purity gentisic acid.
Example 4
This example is different from example 3 in that in the acidification step, the reaction solution pH was adjusted to 2.3.
Example 5
This example is different from example 3 in that in the acidification step, the reaction solution pH was adjusted to 2.5.
Example 6
This example is different from example 3 in that in the acidification step, the reaction solution pH was adjusted to 2.1.
Example 7
This example is different from example 3 in that in the acidification step, the reaction solution pH was adjusted to 2.7.
Example 8
This example differs from example 3 in that the ratio of the amounts of the species of diethylamine and 5-bromosalicylic acid in the salt formation step is 0.75: 1.
Example 9
This example differs from example 3 in that the ratio of the amounts of the species of diethylamine and 5-bromosalicylic acid in the salt formation step is 0.81: 1.
Example 10
This example differs from example 3 in that the ratio of the amounts of the species of diethylamine and 5-bromosalicylic acid in the salt formation step is 0.7: 1.
Example 11
This example differs from example 3 in that the ratio of the amounts of the species of diethylamine and 5-bromosalicylic acid in the salt formation step is 0.85: 1.
Example 12
The difference between this example and example 3 is that in the decolorization and crystallization step, the temperature was raised and then kept for 6 hours, and the filtrate was filtered while it was hot.
The raw materials, reagents, reaction conditions and the results of sample detection for each example are shown in table 1 below.
TABLE 1
The foregoing is directed to preferred embodiments of the present application, other than the limiting examples of the present application, and variations of the present application may be made by those skilled in the art using the foregoing teachings. However, any simple modification, equivalent change and modification of the above embodiments according to the technical essence of the present application still belong to the protection scope of the technical solution of the present application.
Claims (15)
1. A high-purity gentisic acid is characterized in that the purity is more than or equal to 99.50%, and comprises: gentisic acid, salicylic acid and impurity A, wherein the impurity A has a structure shown in formula (I):

formula (I).
2. The high purity gentisic acid as claimed in claim 1, wherein the salicylic acid is present in an amount of 0.1% by mass or less and the impurity a is present in an amount of 0.1% by mass or less.
3. The high purity gentisic acid of claim 1, further comprising impurity B, wherein the impurity B has a structure represented by formula (II):
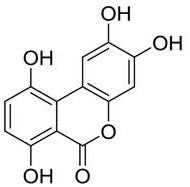
formula (II).
4. The high-purity gentisic acid as claimed in claim 3, wherein the impurity B is present in the high-purity gentisic acid in an amount of 0.1% by mass or less.
5. The high purity gentisic acid according to any one of claims 1 to 4, characterized in that it is prepared by a method comprising the steps of:
feeding and reacting: adding 5-bromosalicylic acid into an inorganic alkali solution for reaction;
acidifying: adding concentrated hydrochloric acid to adjust pH, separating out solid, and filtering;
and (3) extraction: adding an extracting agent I into the filtrate obtained in the acidification step, extracting, and standing to obtain floccules;
and (3) filtering: filtering and separating the solution to obtain an organic phase I and a water phase, adding an extracting agent II into the water phase, extracting, filtering and separating the solution to obtain an organic phase II, and combining the organic phase II and the organic phase I to obtain an organic phase III;
and (3) crystallization: concentrating the organic phase III, separating out solids, and adding the organic solvent I to obtain a crude gentisic acid product;
salifying: adding the crude gentisic acid into a second organic solvent, and dropwise adding a solution of the second organic solvent containing organic base to obtain a wet gentisic acid organic base salt;
dissociating: adding the wet gentisic acid organic base salt into water, dropwise adding concentrated hydrochloric acid to adjust the pH value, and filtering to obtain a refined gentisic acid wet product;
decoloring and crystallizing: adding refined wet gentisic acid and a decolorant into water, and filtering to obtain gentisic acid crystals;
and (3) filtering and drying: and leaching the gentisic acid crystal with water, then cleaning with an organic solvent I, and drying to obtain the high-purity gentisic acid.
6. The high-purity gentisic acid as claimed in claim 5, wherein in the acidification step, the pH is adjusted to 2.3 to 2.5.
7. The highly pure gentisic acid as claimed in claim 5, wherein in the extraction step, the extractant one is selected from any one or more of dichloromethane, ethyl acetate, isopropyl acetate, toluene and methyl t-butyl ether.
8. The highly pure gentisic acid as claimed in claim 5, wherein in the filtration step, the second extractant is selected from any one or more of dichloromethane, ethyl acetate, isopropyl acetate, toluene and methyl t-butyl ether.
9. The high-purity gentisic acid as claimed in claim 5, wherein in the crystallization step, the organic solvent one is one or more selected from n-heptane, acetone, ethyl acetate, petroleum ether and chloroform.
10. The high-purity gentisic acid as claimed in claim 5, wherein the mass ratio of the organic base to 5-bromosalicylic acid is 0.75-0.85: 1.
11. The high purity gentisic acid of claim 5 wherein the organic base is diethylamine.
12. The high purity gentisic acid as claimed in claim 5, wherein in the salt formation step, the organic solvent II is one or more selected from acetone, isopropanol and tetrahydrofuran.
13. The high-purity gentisic acid as claimed in claim 5, wherein in the decoloring and crystallizing steps, the gentisic acid refined product wet product and the decoloring agent are added into water, stirred, heated, filtered and the filtrate is cooled to obtain gentisic acid crystals.
14. The high-purity gentisic acid as claimed in claim 13, wherein in the decoloring and crystallizing steps, the temperature is raised and then maintained for 0.5-6 h.
15. Use of high purity gentisic acid as claimed in any one of claims 1 to 14 in the manufacture of an antiviral, antibacterial, analgesic or antioxidant excipient.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210537742.2A CN114634412A (en) | 2022-05-18 | 2022-05-18 | High-purity gentisic acid and application thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202210537742.2A CN114634412A (en) | 2022-05-18 | 2022-05-18 | High-purity gentisic acid and application thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN114634412A true CN114634412A (en) | 2022-06-17 |
Family
ID=81953163
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202210537742.2A Pending CN114634412A (en) | 2022-05-18 | 2022-05-18 | High-purity gentisic acid and application thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114634412A (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114716308A (en) * | 2022-05-18 | 2022-07-08 | 北京先通国际医药科技股份有限公司 | Preparation method and application of high-purity gentisic acid |
| CN117024292A (en) * | 2023-08-08 | 2023-11-10 | 深圳杉海创新技术有限公司 | Gentisic acid ion salt, preparation method thereof and daily chemical product or food or medicine |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2588679A (en) * | 1950-08-17 | 1952-03-11 | Dow Chemical Co | Purification of gentisic acid |
| JP2004189608A (en) * | 2002-12-06 | 2004-07-08 | Sumikin Air Water Chemical Inc | Method for purifying 2,5-dihydroxybenzoic acid |
| CN102766042A (en) * | 2012-07-04 | 2012-11-07 | 天长市禾益化学药品有限公司 | Method for preparing high purity gentianic acid |
-
2022
- 2022-05-18 CN CN202210537742.2A patent/CN114634412A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2588679A (en) * | 1950-08-17 | 1952-03-11 | Dow Chemical Co | Purification of gentisic acid |
| JP2004189608A (en) * | 2002-12-06 | 2004-07-08 | Sumikin Air Water Chemical Inc | Method for purifying 2,5-dihydroxybenzoic acid |
| CN102766042A (en) * | 2012-07-04 | 2012-11-07 | 天长市禾益化学药品有限公司 | Method for preparing high purity gentianic acid |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114716308A (en) * | 2022-05-18 | 2022-07-08 | 北京先通国际医药科技股份有限公司 | Preparation method and application of high-purity gentisic acid |
| CN117024292A (en) * | 2023-08-08 | 2023-11-10 | 深圳杉海创新技术有限公司 | Gentisic acid ion salt, preparation method thereof and daily chemical product or food or medicine |
| CN117024292B (en) * | 2023-08-08 | 2024-09-24 | 深圳杉海创新技术有限公司 | Gentisic acid ion salt, preparation method thereof and daily chemical product or food or medicine |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114634412A (en) | High-purity gentisic acid and application thereof | |
| US8153842B2 (en) | Method for producing 3-(2,2,2-trimethyl-hydrazinium) propionate dihydrate | |
| US10793532B2 (en) | Method for producing the crystalline form of modification a of calcobutrol | |
| CN108623486A (en) | A kind of preparation method of V hydrochloride of salbutamol intermediate | |
| CZ18597A3 (en) | Process for preparing 5-amino-2,4,6-triiodine-1,3-benzenedicarboxylic acid | |
| CN109336833B (en) | Preparation method of 1,4,7, 10-tetraazacyclododecane-1, 4, 7-triacetic acid | |
| CN101270124B (en) | Novel method for purifying and preparing high-purity fluorandiol and fluorandiol salt | |
| ES2503541T3 (en) | Procedure to produce toluidine compounds | |
| CN114716308A (en) | Preparation method and application of high-purity gentisic acid | |
| CN116554014A (en) | Preparation method of fenofibrate and impurities thereof | |
| CN110437125A (en) | A kind of preparation method of Tezacaftor intermediate II | |
| CN115784875A (en) | Preparation process of bis (2-acetoxybenzoic acid) calcium urea | |
| KR920005816B1 (en) | Process for producing aromatic hydroxycarboxylic acid | |
| CN110256434A (en) | A method of preparing high-purity diprophylline | |
| KR100651071B1 (en) | A process for the preparation of 4-carboxy-5,8,11-triscarboxymethyl-1-phenyl-2-oxa-5,8,11-triazatridecan-13-oic acid | |
| KR100235808B1 (en) | Method of producing keto acids | |
| EP0053919B1 (en) | Cephamycin c derivative and its preparation | |
| CN101492388A (en) | Method for synthesis of Miqujing medicament material | |
| CN114907207A (en) | Compound separated from gentisic acid and separation method and application thereof | |
| RU2786716C2 (en) | Method for production of crystal form of calcobutrol a modification | |
| CN111808021B (en) | Preparation method of indacaterol and salt thereof | |
| CN109851659A (en) | A kind of preparation method of lisinopril | |
| CN117362228A (en) | Roflumilast hydrate crystal and preparation method thereof | |
| EP0161499B1 (en) | Process for preparation of aminophthalideisoquinolines | |
| KR800000569B1 (en) | Process for preparing cephalosporin c derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |