CN111239318A - Method for determining urea content in biological sample based on combination of GC-MS and enzymatic chemical method - Google Patents
Method for determining urea content in biological sample based on combination of GC-MS and enzymatic chemical method Download PDFInfo
- Publication number
- CN111239318A CN111239318A CN202010250083.5A CN202010250083A CN111239318A CN 111239318 A CN111239318 A CN 111239318A CN 202010250083 A CN202010250083 A CN 202010250083A CN 111239318 A CN111239318 A CN 111239318A
- Authority
- CN
- China
- Prior art keywords
- urea
- sample
- content
- temperature
- urease
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images







Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/88—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/88—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86
- G01N2030/8809—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86 analysis specially adapted for the sample
- G01N2030/884—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86 analysis specially adapted for the sample organic compounds
Landscapes
- Physics & Mathematics (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Analytical Chemistry (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- General Physics & Mathematics (AREA)
- Immunology (AREA)
- Pathology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
Description
技术领域technical field
本发明属于生物材料检测领域,具体涉及一种基于GC-MS与酶化学法检测生物样品中尿素含量的方法。The invention belongs to the field of biological material detection, in particular to a method for detecting urea content in biological samples based on GC-MS and an enzymatic chemical method.
背景技术Background technique
尿素别名碳酰二胺、碳酰胺、脲。是由碳、氮、氧和氢组成的有机化合物。其化学公式为CON2H4、CO(NH2)2或CN2H4O,国际非专利药品名称为Carbamide。外观是白色晶体或粉末。尿素在肝合成,是哺乳类动物排出的体内含氮代谢物。这代谢过程称为尿素循环。尿素是第一种以人工合成无机物质而得到的有机化合物。化学式:CO(NH2)2,分子质量60.06,无色或白色针状或棒状结晶体,工业或农业品为白色略带微红色固体颗粒,无臭无味。含氮量约为46.67%,密度1.335g/cm3。溶于水、醇,不溶于乙醚、氯仿,呈弱碱性。CAS No.:57-13-6,分子量:60.05;熔点:131-135℃;沸点:196.6,折射率:n20/D 1.40;闪光点:72.7℃,密度:1.335;水溶性:1080g/L(20℃)。化学性质:可与酸作用生成盐。有水解作用。在高温下可进行缩合反应,生成缩二脲、缩三脲和三聚氰酸。Urea alias carbonic diamide, carbonamide, urea. It is an organic compound composed of carbon, nitrogen, oxygen and hydrogen. Its chemical formula is CON 2 H 4 , CO(NH 2 ) 2 or CN 2 H 4 O, and the international generic drug name is Carbamide. Appearance is white crystal or powder. Urea is synthesized in the liver and is a nitrogen-containing metabolite excreted by mammals. This metabolic process is called the urea cycle. Urea is the first organic compound obtained by artificially synthesizing inorganic substances. Chemical formula: CO(NH 2 ) 2 , molecular weight 60.06, colorless or white needle-like or rod-like crystals, industrial or agricultural products are white slightly reddish solid particles, odorless and tasteless. The nitrogen content is about 46.67%, and the density is 1.335g/cm3. Soluble in water, alcohol, insoluble in ether, chloroform, weakly alkaline. CAS No.: 57-13-6, molecular weight: 60.05; melting point: 131-135 ℃; boiling point: 196.6, refractive index: n20/D 1.40; flash point: 72.7 ℃, density: 1.335; water solubility: 1080g/L ( 20°C). Chemical properties: It can react with acids to form salts. There is hydrolysis. The condensation reaction can be carried out at high temperature to generate biuret, triuret and cyanuric acid.
尿素于1828年被德国人Wohler首次合成,并于20世纪50年代被广泛使用,作为反刍动物的非蛋白氮源。它为低毒物质,大鼠经口LD50 15g/kg,小鼠经口LD50为11.5g/kg。反刍动物由于其独特的消化系统结构,使得它们能够利用尿素来合成蛋白质。在牛的瘤胃中存在着大量具有脲酶活性的微生物,当牛进食含有尿素的饲料时可以将尿素分解为氨,这些微生物能够通过氨化反应或转氨反应将氨与碳水化合物合成菌体蛋白(MCP),随后MCP在牛的真胃和小肠消化吸收。虽然尿素为低毒物质,但正因牛瘤胃中微生物能够分解尿素产氨这个特性,不恰当的使用反而会造成牛中毒死亡。主要原因如下:尿素管理不当,造成尿素被牛偷食或误食;饲料中尿素含量过高或尿素与饲料未混合均匀;投喂含尿素的饲料时同时饲喂含尿素酶的大豆饼或蚕豆;牛在食用含有尿素的饲料后大量饮水。以上均能导致尿素迅速分解,产生大量氨,导致牛瘤胃中的微生物无法将氨全部利用,以至于氨经瘤胃壁吸收入血导致中毒。在司法鉴定工作中时常会遇到牛尿素中毒的案件,而以现有技术报道方法无法很好地解决这类问题。因此,研制开发一种可快速准确的检测生物样品中尿素的方法一直是亟待解决的新课题。Urea was first synthesized by the German Wohler in 1828 and was widely used in the 1950s as a non-protein nitrogen source for ruminants. It is a low-toxic substance, with an oral LD 50 of 15g/kg for rats and an oral LD 50 of 11.5g/kg for mice. Ruminants can use urea to synthesize protein due to their unique digestive system structure. There are a large number of microorganisms with urease activity in the rumen of cattle. When cattle eat feed containing urea, urea can be decomposed into ammonia. These microorganisms can synthesize bacterial protein from ammonia and carbohydrates through ammoniation reaction or transamination reaction ( MCP), and then MCP is digested and absorbed in the bovine true stomach and small intestine. Although urea is a low-toxic substance, due to the fact that microorganisms in the rumen of cattle can decompose urea to produce ammonia, improper use will cause cattle poisoning and death. The main reasons are as follows: improper management of urea, causing urea to be stolen or ingested by cattle; excessive urea content in feed or urea and feed are not mixed evenly; feeding urease-containing feed at the same time feeding soybean cake or broad bean containing urease ; Cattle drink a lot of water after eating feed containing urea. All of the above can lead to the rapid decomposition of urea and the production of a large amount of ammonia, so that the microorganisms in the rumen of cattle cannot fully utilize the ammonia, so that the ammonia is absorbed into the blood through the rumen wall and causes poisoning. Cases of bovine urea poisoning are often encountered in forensic identification work, and such problems cannot be well resolved by the prior art reporting methods. Therefore, research and development of a rapid and accurate method for detecting urea in biological samples has always been a new topic to be solved.
发明内容SUMMARY OF THE INVENTION
尿素由于分子极性很大,且很难气化,无法直接利用气相色谱-质谱联用技术(GC-MS)检测。鉴于现有技术存在的问题,本发明的目的在于提供一种准确、简便的检测生物样品中尿素的方法,通过脲酶和衍生化试剂对尿素处理后,使用GC-MS来间接检测尿素的方法,能够有针对性的检测出生物样品中的尿素含量,可排除许多外界因素的干扰,提高了检测的灵敏度,同时大大简化了检测步骤,节省了时间和成本。Urea cannot be directly detected by gas chromatography-mass spectrometry (GC-MS) due to its high molecular polarity and difficulty in gasification. In view of the problems existing in the prior art, the object of the present invention is to provide an accurate and simple method for detecting urea in biological samples, after the urea is treated with urease and a derivatizing reagent, and a method for indirectly detecting urea using GC-MS, The urea content in the biological sample can be detected in a targeted manner, the interference of many external factors can be eliminated, the detection sensitivity is improved, the detection steps are greatly simplified, and time and cost are saved.
为了实现上述目的,本发明采用以下技术方案。In order to achieve the above objects, the present invention adopts the following technical solutions.
一种基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法,具体包括以下步骤。A method for determining urea content in a biological sample based on the combination of GC-MS and enzymatic chemistry, specifically comprising the following steps.
(1)检测条件。(1) Detection conditions.
气质联用仪:采用气相色谱-质谱联用仪(GC-MS)。Gas chromatography-mass spectrometry: gas chromatography-mass spectrometry (GC-MS) was used.
色谱柱:HP-5MS毛细管柱,规格:30m×0.25mm,0.25μm。Chromatographic column: HP-5MS capillary column, specification: 30m×0.25mm, 0.25μm.
前进样口温度、EI离子源温度、四极杆温度和接口温度分别为280℃、150℃、 230℃、280℃,质谱仪采用单离子检测扫描(SIM)模式,电子能量为70eV。The front inlet temperature, EI ion source temperature, quadrupole temperature and interface temperature were 280 °C, 150 °C, 230 °C, and 280 °C, respectively.
载气为高纯氦气(99.999%),流速为1.0ml/min,不分流进样1μL。The carrier gas was high-purity helium (99.999%), the flow rate was 1.0 ml/min, and 1 μL was injected in a splitless manner.
升温程序:初始柱温60℃,保持1min,随后以20℃/min的升温速率升至280℃,保持11min。Heating program: the initial column temperature was 60°C, held for 1 min, then increased to 280°C at a heating rate of 20°C/min, and held for 11 min.
(2)样品处理。(2) Sample processing.
取2份待检血样或其他生物样本0.5mL分别置于10mL离心管中,加入200μg/mL 内标10μL,加入PBS缓冲液1mL后再加入22U/mL尿素酶0.5mL,另一份添加0.5mL PBS缓冲液,密封后放入45℃水浴锅中温育30min。此时尿素可全部被尿素酶分解为氨,待样品冷却后,加入3mol/L NaOH溶液1mL,再加入50μL衍生化试剂七氟丁酰氯并在水浴锅中40℃水浴10min,以对氨和内标进行衍生化。水浴结束后加入2mL乙酸乙酯:正己烷=9:1,随后在涡旋仪上震荡5min以萃取,8000rpm/min离心5min。取上层有机溶剂 1mL,干燥氮气吹至近干后取1μL于GC-MS进样分析。Take 0.5mL of 2 blood samples or other biological samples to be tested and put them in 10mL centrifuge tubes respectively, add 10μL of 200μg/mL internal standard, add 1mL of PBS buffer, then add 0.5mL of 22U/mL urease, and add 0.5mL to the other part PBS buffer, sealed and placed in a water bath at 45°C for 30 min. At this point, urea can be completely decomposed into ammonia by urease. After the sample is cooled, add 1 mL of 3 mol/L NaOH solution, then add 50 μL of derivatization reagent heptafluorobutyryl chloride, and bathe it in a water bath at 40 °C for 10 min. Derivatization of the target. After the water bath, 2 mL of ethyl acetate: n-hexane = 9:1 was added, followed by shaking on a vortex for 5 min to extract, and centrifuged at 8000 rpm/min for 5 min. Take 1 mL of the organic solvent in the upper layer, blow dry nitrogen to near dryness, and then take 1 μL for injection and analysis by GC-MS.
(3)图谱分析。(3) Atlas analysis.
衍生化产物七氟丁酰胺,保留时间3.0min,特征质量数为m/z 214(分子离子峰)、m/z194、m/z 166、m/z 146、m/z 100、m/z 69和m/z 44(基峰)。Derivatized product heptafluorobutanamide, retention time 3.0min, characteristic mass numbers are m/z 214 (molecular ion peak), m/z194, m/z 166, m/z 146, m/
(4)标准工作曲线样品溶液的制备。(4) Preparation of standard working curve sample solution.
尿素酶溶液配置:将尿素酶溶于PH7.4磷酸盐缓冲液(PBS)中,配置成22U/ml的尿素酶溶液(由于尿素酶的活性会随时间降低,尽量现用现配)。Urease solution configuration: Dissolve urease in PH7.4 Phosphate Buffered Saline (PBS) and configure it into a 22U/ml urease solution (because the activity of urease will decrease with time, try to use it as needed).
内标配置:取适量1,6-己二胺,用0.1mol/L HCL稀释至200μg/ml,4℃冷藏储存。Internal standard configuration: take an appropriate amount of 1,6-hexanediamine, dilute it with 0.1mol/L HCL to 200μg/ml, and store at 4°C.
尿素工作液配置:根据所需线性范围将尿素稀释成不同浓度尿素标准液,超声波清洗机超声除菌10min,-20℃冰冻储存。Urea working solution configuration: Dilute urea into standard solution of urea with different concentrations according to the required linear range, sterilize by ultrasonic cleaning machine for 10min, and store in -20℃.
(5)含量计算。(5) Content calculation.
将步骤(4)中制成的标准工作曲线样品溶液按照步骤(2)中样品处理的方法进行处理后进样分析。选择检测m/z 100的选择离子色谱图,以衍生化产物七氟丁酰胺的峰面积为纵坐标,以血中添加尿素的质量浓度为横坐标,计算回归方程,计算含量时,将待测样品中七氟丁酰胺的峰面积代入回归方程即得到待测样品含量。The standard working curve sample solution prepared in step (4) is processed according to the sample processing method in step (2), and then injected for analysis. Select the selected ion chromatogram of detection m/
所述的生物样本为尿液、胃内容物或血浆样品。The biological samples are urine, gastric contents or plasma samples.
该方法的最低检出限以S/N=3进行计算,对于血中尿素的最低检出限为0.05μ g/ml,定量限为0.10μg/ml。The minimum detection limit of this method was calculated with S/N=3, the minimum detection limit for blood urea was 0.05 μg/ml, and the quantification limit was 0.10 μg/ml.
与现有技术相比,本发明的有益效果如下。Compared with the prior art, the beneficial effects of the present invention are as follows.
1、本发明提供的基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法检测原理为间接测定生物样品中的尿素。1. The detection principle of the method for determining the urea content in a biological sample based on the combination of GC-MS and an enzymatic chemical method provided by the present invention is to indirectly determine the urea in the biological sample.
2、本发明提供的基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法采用的是衍生化后的质谱法,而非传统的光谱法。可将血液凝固状态、溶血、试剂干扰等一些外界影响因素有效降低。2. The method for determining urea content in biological samples based on the combination of GC-MS and enzymatic chemistry provided by the present invention adopts derivatized mass spectrometry instead of traditional spectroscopy. It can effectively reduce some external factors such as blood coagulation state, hemolysis, and reagent interference.
3、本发明提供的基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法所用到的试剂种类更少,操作简单快速。3. The method for determining the urea content in a biological sample based on a combination of GC-MS and an enzymatic chemical method provided by the present invention uses fewer types of reagents, and the operation is simple and fast.
4、本发明提供的基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法可检测的生物材料包括尿液、胃内容物或血浆样品。4. The method for determining urea content in a biological sample based on a combination of GC-MS and an enzymatic chemical method provided by the present invention can detect biological materials including urine, gastric contents or plasma samples.
5、本发明提供的基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法用GC- MS检测尿素的方法,该方法利用尿素酶能专一地分解尿素地特性,将尿素转化为氨,再利用七氟丁酰氯与之较为迅速、完全地反应,使不能被GC-MS检测地尿素得以被检测与定量。该方法灵敏度较光谱法更高,线性关系较好。5. The method for detecting urea content in biological samples based on the combination of GC-MS and enzymatic chemical method provided by the present invention is a method for detecting urea by GC-MS, the method utilizes the characteristic that urease can specifically decompose urea, and converts urea into Ammonia, and then use heptafluorobutyryl chloride to react with it relatively quickly and completely, so that urea, which cannot be detected by GC-MS, can be detected and quantified. The sensitivity of this method is higher than that of spectroscopy, and the linear relationship is better.
附图说明Description of drawings
图1是尿素酶分解尿素化学式。Figure 1 is the chemical formula of urease decomposing urea.
图2是衍氨与七氟丁酰氯衍生化反应(主反应)。Figure 2 is the derivatization reaction of derivatized ammonia and heptafluorobutyryl chloride (main reaction).
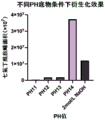
图3是不同碱性体系对衍生化效果的影响。Figure 3 shows the effect of different alkaline systems on the derivatization effect.
图4是不同温度对衍生化效果的影响。Figure 4 is the effect of different temperatures on the derivatization effect.
图5是不同反应时间对衍生化效果的影响。Figure 5 is the effect of different reaction times on the derivatization effect.
图6是尿素经气相色谱质谱联用仪测定(典型的色谱图)。Figure 6 is the determination of urea by gas chromatography-mass spectrometry (a typical chromatogram).
图7是尿素经气相色谱质谱联用仪测定(典型的质谱图)。Figure 7 is the determination of urea by gas chromatography mass spectrometry (typical mass spectrum).
图8是血中添加尿素线性关系图。Fig. 8 is a linear relationship diagram of urea addition in blood.
具体实施方式Detailed ways
下面结合附图和实施例对本发明的具体实施方式作进一步详细描述。以下实施例详细说明了本发明,但不用于限制本发明的范围。本实施例采用常规实验技术,这些均是本技术领域人员所熟悉的,可以按照本实施例使用材料厂商所提供的说明书即可进行。The specific embodiments of the present invention will be described in further detail below with reference to the accompanying drawings and examples. The following examples illustrate the invention in detail, but are not intended to limit the scope of the invention. This embodiment adopts conventional experimental techniques, which are familiar to those skilled in the art, and can be performed according to the description provided by the material manufacturer according to this embodiment.
实施例1基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法相关反应过程和条件的优化。Example 1 Optimization of the method-related reaction process and conditions for the determination of urea content in biological samples based on the combination of GC-MS and enzymatic chemistry.
尿素酶是一种含镍的寡聚酶,最适PH值为7.4,具有绝对专一性,能够特异性地催化尿素分解为氨和二氧化碳,反应过程如图1所示。由尿素酶分解尿素产生的氨随后与衍生化试剂七氟丁酰氯反应,生成七氟丁酰胺,从而被气相色谱-质谱检出。反应方程式见图 2。在此反应体系中,有两步平衡需要控制。第一,反应体系中PBS缓冲溶液用量,在尿素酶分解尿素过程中,一分子尿素生成两分子氨,并与水分子结合生成氨水,使尿素酶反应环境变为碱性,而尿素酶在PH约9的条件下失活。当样本中尿素含量过高时,尿素酶还未将尿素全部分解就已失活,在这种状况下可以适当增加PBS缓冲液容量。在本发明反应体系中共加入2mL PBS缓冲液,最大能完全分解尿素浓度约6mg/mL的样本;第二,七氟丁酰氯用量和碱液使用量,七氟丁酰氯与氨、水反应产酸,反应产物七氟丁酰胺在酸性条件下以离子形式存在,而无法被有机溶剂提取。基于此原因,在进行衍生化之前应加入适量碱液,使反应结束后下层水溶液恰好变为弱碱性,使待检物质七氟丁酰胺游离出来,加入碱液使酸碱中和,也起到了促使反应向正方向进行的作用。在第二步衍生化步骤中我们考察了不同的碱性体系、不同温度、不同反应时间对整个检测过程的影响,结果如图3-图5所示,最终选择了当PH为14,40摄氏度下反应5分钟这样的衍生化条件。Urease is a nickel-containing oligomerase with an optimum pH of 7.4, with absolute specificity, which can specifically catalyze the decomposition of urea into ammonia and carbon dioxide. The reaction process is shown in Figure 1. Ammonia produced by the decomposition of urea by urease then reacts with the derivatizing reagent heptafluorobutyryl chloride to produce heptafluorobutanamide, which is detected by gas chromatography-mass spectrometry. The reaction equation is shown in Figure 2. In this reaction system, there are two steps of equilibrium that need to be controlled. First, the amount of PBS buffer solution in the reaction system, in the process of urease decomposing urea, one molecule of urea generates two molecules of ammonia, and combines with water molecules to generate ammonia water, so that the urease reaction environment becomes alkaline, and urease is in pH. Inactivated under conditions of about 9. When the urea content in the sample is too high, the urease will be inactivated before all the urea is decomposed. In this case, the volume of PBS buffer can be appropriately increased. A total of 2 mL of PBS buffer is added to the reaction system of the present invention, which can completely decompose a sample with a urea concentration of about 6 mg/mL; second, the amount of heptafluorobutyryl chloride and the amount of lye used, heptafluorobutyryl chloride reacts with ammonia and water to produce acid , the reaction product heptafluorobutanamide exists in the form of ions under acidic conditions and cannot be extracted by organic solvents. For this reason, an appropriate amount of lye should be added before the derivatization, so that the lower aqueous solution just becomes weakly alkaline after the reaction, so that the substance to be tested heptafluorobutyramide is freed, and the lye is added to neutralize the acid and alkali. to the effect of prompting the reaction to proceed in the positive direction. In the second derivatization step, we investigated the effects of different alkaline systems, different temperatures, and different reaction times on the entire detection process. The results are shown in Figures 3-5. Finally, when the pH was 14, 40 degrees Celsius was selected. React under such derivatization conditions for 5 min.
此外,在使用有机溶剂萃取时,由于乙酸乙酯与水密度接近且反应体系为碱性,若单用乙酸乙酯提取容易导致乳化,使用乙酸乙酯:正己烷=9:1作为萃取液时能很好地避免乳化现象。In addition, when using an organic solvent for extraction, since ethyl acetate and water have close densities and the reaction system is alkaline, if ethyl acetate alone is used for extraction, it is easy to cause emulsification. When using ethyl acetate: n-hexane = 9:1 as the extract Emulsification can be well avoided.
2.色谱-质谱图(最低检测限)。2. Chromatogram-mass spectrogram (minimum detection limit).
尿素经过酶解后,与七氟丁酰氯反应,得到的典型产物质谱图见图6-7所示。After enzymatic hydrolysis of urea, it reacts with heptafluorobutyryl chloride, and the mass spectrum of the typical product obtained is shown in Figure 6-7.
3.标准曲线(以血中添加为例)。3. Standard curve (taking blood as an example).
准确吸取尿素标准工作液适量,分别加入0.5mL空白血浆中,配置成尿素浓度为0.1 μg/mL、10μg/mL、200μg/mL、1mg/mL、3mg/mL、5mg/mL的添加血浆样本,按照上述操作步骤对血样进行前处理,减去添加尿素酶的空白血浆中总氨后,以浓度为横坐标(x),氨的衍生化产物(七氟丁酰胺)与内标衍生化产物峰之比为纵坐标(y),线性关系见图8所示,得出一元线性方程为y=0.0793x+1.9703(R2=0.9994,P<0.0001)。Accurately draw an appropriate amount of urea standard working solution, add it to 0.5mL blank plasma, and configure the urea concentration as 0.1 μg/mL, 10 μg/mL, 200 μg/mL, 1 mg/mL, 3 mg/mL, 5 mg/mL. The blood samples were pretreated according to the above operation steps. After subtracting the total ammonia in the blank plasma added with urease, taking the concentration as the abscissa (x), the difference between the derivatized product of ammonia (heptafluorobutanamide) and the peak of the derivatized product of the internal standard The ratio is the ordinate (y), and the linear relationship is shown in Fig. 8, and the one-variable linear equation is obtained as y=0.0793x+1.9703 (R 2 =0.9994, P<0.0001).
4、方法学考察。4. Methodological investigation.
4.1回收率与精密度。4.1 Recovery and precision.
分别对三份样本进行日内和日间精密度考察,计算七氟丁酰胺峰面积与内标峰面积之间的比值。测得日内精密度RSD=3.7-6.2%;日间精密度RSD=4.0%-6.8%。在0.5mL空白样本中分别添加尿素10μg/mL、1mg/mL、5mg/mL,衍生化后减去空白血样中原本存在的尿素以计算回收率,平行测量三次。低、中、高三个浓度组平均回收率在90.3%-97.5%之间。The intra-day and inter-day precisions were investigated for the three samples, respectively, and the ratio between the peak area of heptafluorobutyramide and the peak area of the internal standard was calculated. The intra-day precision RSD=3.7-6.2%; the inter-day precision RSD=4.0%-6.8%. 10μg/mL, 1mg/mL, and 5mg/mL of urea were added to the 0.5mL blank sample, and the urea originally present in the blank blood sample was subtracted after derivatization to calculate the recovery rate, and the measurements were performed in parallel three times. The average recovery rates of low, medium and high concentration groups were between 90.3% and 97.5%.
4.2稳定性实验。4.2 Stability test.
基于七氟丁酰氯极其活泼的化学性质,氨与之发生的亲核取代反应所形成的共价键十分稳定,在一般外界条件下不易断裂。在对放置0h、12h、24h、48h和96h经前处理后的样本中七氟丁酰胺与内标衍生化产物峰面积比值进行比较后发现,在0-96h内该比值较为稳定P=0.8621(P>0.05),RSD=1.65%。Based on the extremely active chemical properties of heptafluorobutyryl chloride, the covalent bond formed by the nucleophilic substitution reaction between ammonia and it is very stable and is not easy to break under normal external conditions. After comparing the peak area ratio of heptafluorobutyramide to the internal standard derivatized product in the samples after pretreatment at 0h, 12h, 24h, 48h and 96h, it was found that the ratio was relatively stable within 0-96h P=0.8621 ( P>0.05), RSD=1.65%.
实施例2实际案例应用。Example 2 Practical case application.
一种基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法,具体包括以下步骤。A method for determining urea content in a biological sample based on the combination of GC-MS and enzymatic chemistry, specifically comprising the following steps.
(1)检测条件。(1) Detection conditions.
气质联用仪:采用气相色谱-质谱联用仪(GC-MS)。Gas chromatography-mass spectrometry: gas chromatography-mass spectrometry (GC-MS) was used.
色谱柱:HP-5ms毛细管柱,规格:30m×0.25mm,0.25μm。Chromatographic column: HP-5ms capillary column, specification: 30m×0.25mm, 0.25μm.
前进样口温度、EI离子源温度、四极杆温度和接口温度分别为280℃、150℃、 230℃、280℃,质谱仪采用单离子检测扫描(SIM)模式,电子能量为70eV。The front inlet temperature, EI ion source temperature, quadrupole temperature and interface temperature were 280 °C, 150 °C, 230 °C, and 280 °C, respectively.
载气为高纯氦气(99.999%),流速为1.0ml/min,不分流进样1μL。The carrier gas was high-purity helium (99.999%), the flow rate was 1.0 ml/min, and 1 μL was injected in a splitless manner.
升温程序:初始柱温60℃,保持1min,随后以20℃/min的升温速率升至280℃,保持11min。Heating program: the initial column temperature was 60°C, held for 1 min, then increased to 280°C at a heating rate of 20°C/min, and held for 11 min.
(2)样品处理。(2) Sample processing.
取待检牛血样0.5mL分别置于10mL离心管中,加入200μg/mL内标10μL,加入 PBS缓冲液1mL后再加入22U/mL尿素酶0.5mL(另一份添加0.5mL PBS缓冲液),密封后放入45℃水浴锅中温育30min。此时尿素可全部被尿素酶分解为氨,待样品冷却后,加入 3mol/L NaOH溶液1mL,再加入50μL衍生化试剂七氟丁酰氯并在水浴锅中40℃水浴 10min,以对氨和内标进行衍生化。水浴结束后加入2mL乙酸乙酯:正己烷=9:1,随后在涡旋仪上震荡5min以萃取,8000rpm/min离心5min。取上层有机溶剂1mL,干燥氮气吹至近干后取1μL于GC-MS进样分析。Take 0.5 mL of the bovine blood sample to be tested and put it into a 10 mL centrifuge tube, add 10 μL of 200 μg/mL internal standard, add 1 mL of PBS buffer, and then add 0.5 mL of 22 U/mL urease (add 0.5 mL of PBS buffer for the other), After sealing, they were placed in a 45°C water bath and incubated for 30 min. At this point, urea can be completely decomposed into ammonia by urease. After the sample is cooled, add 1 mL of 3 mol/L NaOH solution, then add 50 μL of derivatization reagent heptafluorobutyryl chloride, and bathe it in a water bath at 40 °C for 10 min. Derivatization of the target. After the water bath, 2 mL of ethyl acetate: n-hexane = 9:1 was added, followed by shaking on a vortex for 5 min to extract, and centrifuged at 8000 rpm/min for 5 min. Take 1 mL of the organic solvent in the upper layer, blow dry nitrogen to near dryness, and then take 1 μL to inject and analyze by GC-MS.
(3)图谱分析。(3) Atlas analysis.
衍生化产物七氟丁酰胺,保留时间3.0min,特征质量数为m/z 214(分子离子峰)、m/z 194、m/z 166、m/z 146、m/z 100、m/z 69和m/z 44(基峰)。Derivatized product heptafluorobutanamide, retention time 3.0min, characteristic mass number is m/z 214 (molecular ion peak), m/z 194, m/z 166, m/z 146, m/
(4)标准工作曲线样品溶液的制备。(4) Preparation of standard working curve sample solution.
尿素酶溶液配置:将尿素酶溶于PH7.4磷酸盐缓冲液(PBS)中,配置成22U/ml的尿素酶溶液(由于尿素酶的活性会随时间降低,尽量现用现配)。Urease solution configuration: Dissolve urease in PH7.4 Phosphate Buffered Saline (PBS) and configure it into a 22U/ml urease solution (because the activity of urease will decrease with time, try to use it as needed).
内标配置:取适量1,6-己二胺,用0.1mol/L HCL稀释至200μg/ml,4℃冷藏储存。Internal standard configuration: take an appropriate amount of 1,6-hexanediamine, dilute it with 0.1mol/L HCL to 200μg/ml, and store at 4°C.
尿素工作液配置:根据所需线性范围将尿素稀释成不同浓度尿素标准液,具体浓度分别为0.1μg/ml、10μg/ml、200μg/ml、2000μg/ml、5000μg/ml,超声波清洗机超声除菌10min,-20℃冰冻储存。Urea working solution configuration: Dilute urea into urea standard solutions of different concentrations according to the required linear range, the specific concentrations are 0.1μg/ml, 10μg/ml, 200μg/ml, 2000μg/ml, 5000μg/ml, ultrasonic cleaning machine ultrasonic removal. Bacteria were stored at -20°C for 10 min.
(5)含量计算。(5) Content calculation.
将步骤(4)中制成的标准工作曲线样品溶液按照步骤(2)中样品处理的方法进行处理后进样分析。选择检测m/z100的选择离子色谱图,以衍生化产物七氟丁酰胺的峰面积为纵坐标,以血中添加尿素的质量浓度为横坐标,计算回归方程得到y=0.0793x+1.9703(R2=0.9994)。计算含量时,将待测样品中七氟丁酰胺的峰面积代入回归方程即得到待测样品含量。The standard working curve sample solution prepared in step (4) is processed according to the sample processing method in step (2), and then injected for analysis. Select the selected ion chromatogram to detect m/z100, take the peak area of the derivatized product heptafluorobutanamide as the ordinate, and take the mass concentration of urea added in the blood as the abscissa, calculate the regression equation to obtain y=0.0793x+1.9703(R 2 = 0.9994). When calculating the content, the peak area of heptafluorobutanamide in the sample to be tested is substituted into the regression equation to obtain the content of the sample to be tested.
采用上述提供的基于GC-MS与酶化学法结合测定生物样本中尿素含量的方法测定了 7个案例中的牛血中尿素的含量,具体尿素含量见表1。实验结果表明该测定方法有很强的应用性。The urea content in bovine blood in 7 cases was determined by the method based on the combination of GC-MS and enzymatic chemistry method provided above for the determination of urea content in biological samples. The specific urea content is shown in Table 1. The experimental results show that the determination method has strong applicability.
表1.7个案例中的牛血中尿素的含量。
Claims (3)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010250083.5A CN111239318A (en) | 2020-04-01 | 2020-04-01 | Method for determining urea content in biological sample based on combination of GC-MS and enzymatic chemical method |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010250083.5A CN111239318A (en) | 2020-04-01 | 2020-04-01 | Method for determining urea content in biological sample based on combination of GC-MS and enzymatic chemical method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111239318A true CN111239318A (en) | 2020-06-05 |
Family
ID=70873693
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010250083.5A Pending CN111239318A (en) | 2020-04-01 | 2020-04-01 | Method for determining urea content in biological sample based on combination of GC-MS and enzymatic chemical method |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111239318A (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115078564A (en) * | 2022-05-06 | 2022-09-20 | 沈阳药科大学 | Method for ultra-fast quantifying amine substances in urine |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104764850A (en) * | 2015-04-28 | 2015-07-08 | 江南大学 | Method for rapidly determining amount of urea in white spirit by means of gas chromatogram-mass spectrum |
| CN108414661A (en) * | 2018-03-13 | 2018-08-17 | 中国医科大学 | Derivative gas chromatography-mass spectrometry method of ammonia content in a kind of detection biological sample |
-
2020
- 2020-04-01 CN CN202010250083.5A patent/CN111239318A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104764850A (en) * | 2015-04-28 | 2015-07-08 | 江南大学 | Method for rapidly determining amount of urea in white spirit by means of gas chromatogram-mass spectrum |
| CN108414661A (en) * | 2018-03-13 | 2018-08-17 | 中国医科大学 | Derivative gas chromatography-mass spectrometry method of ammonia content in a kind of detection biological sample |
Non-Patent Citations (8)
| Title |
|---|
| DONALD G. SAUNDERS 等: "Derivatization and Gas Chromatographic Measurement of Some Thermally Unstable Ureas", 《ANALYTICAL CHEMISTRY》 * |
| SCOTT,S: "Determination of derivatized urea herbicides in water by solid-phase extraction, methylation and gas chromatography with a nitrogen–phosphorus detector", 《ANALYST》 * |
| 张天娇 等: "血清尿素同位素稀释气相色谱质谱法的建立和研究", 《中华检验医学杂志》 * |
| 张新春 等: "《临床检验技术与临床应用》", 30 June 2018, 上海交通大学出版社 * |
| 李国辉 等: "气相色谱-燃烧-同位素比值质谱法测定发酵液中尿素δ~(13)C和δ~(15)N", 《质谱学报》 * |
| 王晶 等: "适用于代谢组学研究的人体尿液保存与气相色谱-质谱联用分析方法", 《环境与健康杂志》 * |
| 申世刚 等: "高效液相色谱-荧光检测器法测定白酒中尿素含量方法研究", 《酿酒科技》 * |
| 袁慧雅 等: "GC-MS衍生化方法快速检测生物样品中的氨", 《法医学杂志》 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115078564A (en) * | 2022-05-06 | 2022-09-20 | 沈阳药科大学 | Method for ultra-fast quantifying amine substances in urine |
| CN115078564B (en) * | 2022-05-06 | 2024-05-31 | 沈阳药科大学 | Method for quantifying amine substances in urine at ultra-fast speed |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113049699B (en) | Method for detecting biphenyl anhydride and related substances thereof and application | |
| CN101458238B (en) | Method for detecting Clenbuterol residual quantity in hair | |
| CN102012409A (en) | Analysis method for trace tobacco specific N-nitrosamine (TSNAs) in animal blood sample | |
| CN111239318A (en) | Method for determining urea content in biological sample based on combination of GC-MS and enzymatic chemical method | |
| US12117457B2 (en) | Method for simultaneous analysis of neurotransmitters and their metabolites based on derivatization | |
| CN106442840B (en) | The assay method of biogenic amine in a kind of cigarette mainstream flue gas | |
| CN112946099A (en) | Method for detecting related substances in amino acid glucose injection | |
| RICHARD | Determination of histidine decarboxylase activity | |
| CN102928544A (en) | Method for analyzing ammonia in cigarette mainstream smoke through gas chromatography-mass spectrometry | |
| Zhao et al. | Highly sensitive determination of putrescine in pork by an electrochemical biosensor constructed with unfolded hemoglobin and diamine oxidase | |
| CN106290598B (en) | The high efficient liquid phase analysis method of impurity in a kind of Gadoversetamide | |
| CN108414661B (en) | A Derivatized Gas Chromatography-Mass Spectrometry Method for Detecting Ammonia Content in Biological Samples | |
| CN113607854A (en) | Method and detection kit for simultaneously detecting multiple vitamins | |
| CN102495148A (en) | Method for determining polyamines in urine | |
| CN101852737A (en) | Method for determining tryptophan content in food through photometry | |
| CN103675145B (en) | Method for detecting residual quantity of cimaterol in pig hairs | |
| CN103278586A (en) | Extracting and detecting method for dicyandiamide component in dairy products | |
| CN108169362B (en) | Method for separating carbamazepine and related substances by liquid chromatography | |
| CN117723688A (en) | Pretreatment method and detection method for indole substances in aquatic products | |
| CN101852720B (en) | Method for biosensor to detect ractopamine in pork | |
| CN106770740A (en) | The detection method of N nitrosodimethylamines in a kind of malt or beer | |
| CN105842260B (en) | A kind of discrimination method of natural taurine and synthesizing taurine | |
| Bai et al. | Mechanisms of ammonia-like off-flavors formation in dried shrimp: Contribution of spoilage microbiota and their metabolism | |
| CN103822981B (en) | A kind of LC-MS measures the method for Endogenous Amino Acids asparagine and glutamine concentration | |
| CN107228906A (en) | Utilize N in gas chromatograph for determination Acotiamide bulk drug, the method for N- diisopropyl ethylenediamine residual quantities |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20200605 |
|
| RJ01 | Rejection of invention patent application after publication |