CN108959852B - 基于氨基酸-核苷酸成对偏好性信息的蛋白质上与rna结合模块的预测方法 - Google Patents
基于氨基酸-核苷酸成对偏好性信息的蛋白质上与rna结合模块的预测方法 Download PDFInfo
- Publication number
- CN108959852B CN108959852B CN201710374897.8A CN201710374897A CN108959852B CN 108959852 B CN108959852 B CN 108959852B CN 201710374897 A CN201710374897 A CN 201710374897A CN 108959852 B CN108959852 B CN 108959852B
- Authority
- CN
- China
- Prior art keywords
- module
- protein
- amino acid
- residues
- residue
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images




Landscapes
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
Abstract
基于氨基酸‑核苷酸成对偏好性信息的蛋白质上与RNA结合模块的预测方法,属于蛋白质‑RNA相互作用与识别技术领域。第一步,以蛋白质三维结构中的每个氨基酸残基为中心,将与之有接触的所有残基划分为一个模块;然后剔除所有的不包含任何表面残基的内部模块,保留至少含有一个表面残基的表面模块;第二步,对表面模块定义三个参数:模块的界面偏好性
Description
技术领域
本发明属于蛋白质-RNA相互作用与识别技术领域,是一种用于已知结构的非核糖体RNA结合单链蛋白质上RNA结合模块的预测方法。
背景技术
蛋白质与RNA相互作用参与生物体细胞多种重要的生理过程,如基因的表达调控、蛋白质的合成和病毒的复制等。RNA分子的重要性被人们逐渐认识,RNA既是信息分子,又是功能分子,在其众多行为的发生中,如mRNA的转录、剪切、出核、定位、翻译和降解等过程,RNA要和一系列蛋白质结合并受它们的调控。更为重要的是,一些重大疾病的发生,如肿瘤、心血管疾病、自身免疫疾病、脆性X综合症(Fragile X Syndrome)和阿尔茨海默(Alzheimer)病等都与蛋白质-RNA相互作用密切相关。因此,蛋白质-RNA相互作用的研究及结合位点的预测对理解蛋白质-RNA特异性识别的分子机制具有重要意义,并且能为分子对接和药物设计提供帮助。
由于采用实验方法预测蛋白质-RNA的结合位点耗时且费力,因此近年来,研究者们开始着力于发展能够准确识别蛋白质-RNA结合位点的理论计算方法。目前所提出的方法主要是在残基水平的结合位点预测,采用蛋白质一级序列和三维结构的信息,利用机器学习方法进行识别。
基于序列的方法是直接从蛋白质序列中提取特征,如氨基酸的进化信息、物化特性、二级结构、位置特异性打分矩阵(PSSMs)和3D基序,并采用机器学习算法,如人工神经网络(Artificial Neural Networks,ANNs)[1,2]、支持向量机(Support Vector Machines,SVMs)、贝叶斯分类器[3-5]、随机森林[6,7]或决策树方法,来预测蛋白质-RNA结合位点。基于序列的方法包括RNABindRPlus[8]和PRIPU[9]等。与基于序列的方法相比,基于结构预测蛋白质-RNA结合位点的方法是非常有限的,主要有DRNA-3D[10]和RBRDetector[11],这主要是由于蛋白质的结构相对序列来说获取较困难。
目前大部分结合位点预测方法是残基水平的,且仅考虑蛋白质表面氨基酸残基的性质,忽略了表面近邻小区域内(包括内部残基)氨基酸残基间的相互作用和协同效应。很多研究表明,蛋白质-蛋白质相互作用中,蛋白质分子是一个通过残基间各种相互作用共同维系的复杂系统,其结合界面残基具有协同效应:结合界面残基与内部残基的相互作用包含了界面区域的信息[12],界面残基往往聚集成簇[13,14]、堆积密度相对较高[15],界面结构是模块化的、模块内残基的内聚性强、模块间的耦合作用并不强[15]。鉴于以上研究结果,2012年,我们研究小组针对蛋白质-蛋白质相互作用,提出了一种能够体现残基间内聚性的蛋白质表面模块划分方案和结合模块预测方法,简称PAMA[16](Product of the solventaccessible Area Multiplied by the polyhedra contact Area)。PAMA方法是用来预测蛋白质-蛋白质复合物结合模块的方法。该方法首先将蛋白质表面模块进行划分;给每个模块一个参数,该参数是模块内残基的溶剂可及表面积与内部残基间接触面积的乘积;然后按照乘积值由大到小对模块进行排序,排在前面的模块被认为是界面模块。
在该工作中,我们给出PAMA划分蛋白质结合蛋白表面模块的方法同样适用于RNA结合蛋白表面模块的划分。在PAMA方法原理的基础上,我们对其做了进一步的改进,将课题组之前统计获得的针对蛋白质-RNA复合物结构的氨基酸-核苷酸成对偏好性信息[17]以模块的界面偏好性方式加入到模块参数中,发现模块内残基界面偏好性的平均值(模块的界面偏好性)、模块溶剂可接近表面积和内部残基间的接触面积,三者的乘积数值能够给出蛋白质-RNA相互作用的结合模块信息,为此我们提出了基于氨基酸-核苷酸成对偏好性信息的蛋白质上与RNA结合模块的预测方法。
参考文献
[1]KEIL M,EXNER T E,BRICKMANN J.Pattern recognition strategies formolecular surfaces:III.Binding site prediction with a neural network[J].JComput Chem,2004,25(6):779-789.
[2]JEONG E,CHUNG I F,MIYANO S.A neural network method foridentification of RNA-interacting residues in protein[J].Genome Inform,2004,15(1):105-116.
[3]TERRIBILINI M,SANDER J D,LEE J H,et al.RNABindR:a server foranalyzing and predicting RNA-binding sites in proteins[J].Nucleic Acids Res,2007,35(Web Server issue):W578-W584.
[4]MAETSCHKE S R,YUAN Z.Exploiting structural and topologicalinformation to improve prediction of RNA-protein binding sites[J].BMCBioinformatics,2009,10:341.
[5]TOWFIC F,CARAGEA C,GEMPERLINE D C,et al.Struct-NB:predictingprotein-RNA binding sites using structural features[J].Int J Data MinBioinform,2010,4(1):21-43.
[6]LIU Z P,WU L Y,WANG Y,et al.Prediction of protein-RNA bindingsites by a random forest method with combined features[J].Bioinformatics,2010,26(13):1616-1622.
[7]MA X,GUO J,WU J,et al.Prediction of RNA-binding residues inproteins from primary sequence using an enriched random forest model with anovel hybrid feature[J].Proteins,2011,79(4):1230-1239.
[8]WALIA R R,XUE L C,WILKINS K,et al.RNABindRPlus:a predictor thatcombines machine learning and sequence homology-based methods to improve thereliability of predicted RNA-binding residues in proteins[J].PLoS One,2014,9(5):e97725.
[9]CHENG Z,ZHOU S,GUAN J.Computationally predicting protein-RNAinteractions using only positive and unlabeled examples[J].J Bioinform ComputBiol,2015,13(3):1541005.
[10]ZHAO H,YANG Y,ZHOU Y.Structure-based prediction of RNA-bindingdomains and RNA-binding sites and application to structural genomics targets[J].Nucleic Acids Res,2011,39(8):3017-3025.
[11]YANG X X,DENG Z L,LIU R.RBRDetector:improved prediction ofbinding residues on RNA-binding protein structures using complementaryfeature-and template-based strategies[J].Proteins,2014,82(10):2455-2471.
[12]de VRIES S J,BONVIN A M.Intramolecular surface contacts containinformation about protein-protein interface regions[J].Bioinformatics,2006,22(17):2094-2098.
[13]MADABUSHI S,YAO H,MARSH M,et al.Structural clusters ofevolutionary trace residues are statistically significant and common inproteins[J].J Mol Biol,2002,316(1):139-154.
[14]GUHAROY M,CHAKRABARTI P.Conservation and relative importance ofresidues across protein-protein interfaces[J].Proc Natl Acad Sci U S A,2005,102(43):15447-15452.
[15]HINTZE A,ADAMI C.Evolution of complex modular biological networks[J].PLoS Comput Biol,2008,4(2):e23.
[16]王攀文,龚新奇,李春华,等.蛋白质表面模块划分及其在结合位点预测中的应用[J].物理化学学报,2012(11):2729-2734.
[17]LI C H,CAO L B,SU J G,et al.A new residue-nucleotide propensitypotential with structural information considered for discriminating protein-RNA docking decoys[J].Proteins,2012,80(1):14-24.
发明内容
本发明的目的是在给出非核糖体RNA结合单链蛋白结构的情况下,预测出蛋白质上RNA结合模块的位置,为蛋白质-RNA复合物结构预测和药物设计提供帮助。
基于氨基酸-核苷酸成对偏好性信息的蛋白质上与RNA结合模块的预测方法分三个步骤:一是对蛋白质进行模块划分,二是计算每个模块的自定义参数,三是将模块按照参数PPQA值由高到低进行排序(如附图1),并识别出可能的界面模块。
步骤1:蛋白质表面模块划分
以蛋白质三维结构中的每个氨基酸残基为中心,将与之有接触的所有残基(包括该中心残基、蛋白质内部残基以及表面残基)划分为一个模块(module)。传统算法是采用距离来确定残基间是否接触,本发明优选采用基于维里几何的Qcontacts算法(J StructBiol,2006,153(2):103-112)计算残基间的接触面积,来判断两残基是否接触,这种判断接触的方式比基于距离的方式更加接近真实情况。然后剔除所有的内部模块(不包含任何表面残基的模块),保留表面模块(至少有一个表面残基的模块)作为最后表面模块划分的结果(附图2)。
表面残基定义为相对溶剂可接近表面积≥15%的残基,优选溶剂可接近表面积的计算采用NACCESS算法,水分子探针半径取 另外定义界面模块为至少含有一个与RNA分子相互作用的界面残基的模块,界面模块为表面模块中的一部分。界面残基为蛋白质氨基酸中至少有一个原子与RNA中的任何一个原子间的距离小于
另外定义界面模块为至少含有一个与RNA分子相互作用的界面残基的模块,界面模块为表面模块中的一部分。界面残基为蛋白质氨基酸中至少有一个原子与RNA中的任何一个原子间的距离小于 的残基。
的残基。
步骤2:计算模块的自定义参数
对每一个表面模块计算以下各值:模块溶剂可接近表面积A、模块内部残基接触面积Q以及模块界面偏好性 如附图2所示,模块的溶剂可接近表面积A为模块中所有残基的溶剂可接近表面积之和:
如附图2所示,模块的溶剂可接近表面积A为模块中所有残基的溶剂可接近表面积之和:

其中,Ai是模块中第i个残基的溶剂可接近表面积,求和遍及模块中的所有残基。
每一个表面模块的内部接触面积Q为模块内所有残基对的接触面积之和:

其中,Qij是模块中残基i和残基j之间的接触面积,求和遍及模块中的所有残基对。
由以上两个参数的乘积可得到模块的PAMA值,即溶剂可接近表面积乘以内部接触面积PAMA=A×Q。
接下来进一步考虑模块界面偏好性,获得模块PPQA参数值。基于我们之前对非冗余非核糖体蛋白质-RNA复合物结构数据的统计分析发现,不同的氨基酸-核苷酸接触具有不同的偏好性。认为这一信息的考虑应该可以帮助识别蛋白质上结合RNA的模块,提高结合位点预测的准确性。因此在PAMA原理的基础上考虑氨基酸-核苷酸成对偏好性信息(Proteins,2012,80(1):14-24)。针对每一种氨基酸,取其与4种核苷酸的偏好性的平均值,获得该氨基酸的界面偏好性(表1中Average列)。这里针对每一个蛋白质表面模块,计算其中所有氨基酸残基界面偏好性之和的算术平均值 作为模块的界面偏好性:
作为模块的界面偏好性:

其中N表示模块中氨基酸残基的个数, 表示模块中残基i的界面偏好性,求和遍及模块中的所有残基。
表示模块中残基i的界面偏好性,求和遍及模块中的所有残基。
表1 20×4氨基酸-核苷酸成对偏好性及氨基酸界面偏好性

接着,定义模块的PPQA参数值,即模块界面偏好性、溶剂可及表面积以及其内部接触面积三者相乘:

步骤3:对模块按照PPQA值由大到小排序,预测识别界面模块
最后,根据PPQA值从大到小对模块进行排序,考察蛋白质界面模块所处的位置,从而确定前几个作为界面模块识别标准,优选根据后续的统计分析,确定前两个模块为可能的界面模块。
本发明能够比较精准地预测蛋白质上与RNA结合的模块,同时工作量不大,效率较高。该方法识别非核糖体RNA结合单链蛋白质上的结合模块有很好的效果,成功率较高。不同于传统的预测方法,它不仅考虑了受体蛋白质的表面残基,还考虑了内部残基间的相互作用,以及氨基酸-核苷酸成对偏好性信息,为蛋白质-RNA复合物结合位点的预测提供了新思路,可用于蛋白质-RNA复合物结构预测及药物设计研究。
附图说明
图1为基于氨基酸-核苷酸成对偏好性信息的蛋白质上与RNA结合模块的预测方法流程图;
图2为蛋白质表面模块的划分,0号残基与1、2、3、4、5号残基接触,是这个模块的中心残基。尽管4、5号残基是一个内部残基,但它仍然被划分为这个模块的一部分。A表示模块的溶剂可及表面积,Q表示模块内部接触面积;
图3为对蛋白质(Catalytic domain of E.coli RNase E),PPQA方法识别到的界面模块在蛋白质表面所处的位置。浅灰色是受体蛋白质,黑色和深灰色区域分别表示排在首位和次位模块所在区域,链状深灰色是该蛋白质与13-mer RNA相互作用形成复合物2C0B(PDB code)中的RNA分子;
图4为不同方法对蛋白质表面模块进行排序,前1-3位中至少含有一个界面模块的体系数目。
具体实施方式
下面结合实施例对本发明做进一步的说明,但本发明并不限于以下实施例。
实施例1
下面以一个蛋白质(Catalytic domain of E.coli RNase E)为例,它与13-merRNA相互作用形成复合物2C0B(PDB ID),来介绍PPQA方法预测蛋白质上结合RNA模块的实施过程。已知该复合物中受体蛋白质的结构,通过实施该方法获得其结合RNA的界面模块。
(1)至(4)在Linux系统下完成。
(1)蛋白质结构预处理
首先将蛋白质受体重命名为2c0b_r_b.pdb。在首次使用该方法程序包的时候,需要创建结果文件的存放目录,在本程序包的工作目录下,采用shell创建目录如下:
…]$mkdir structures
…]$mkdir data
…]$mkdir data/ReceptorModule
…]$mkdir data/Rsa
…]$mkdir data/Vor
创建完以上目录后,将预处理完的2c0b_r_b.pdb文件移动到structures目录下。
…]$mv 2c0b_r_b.pdb structures/
(2)计算蛋白质中每个氨基酸残基的溶剂可及表面积,区分出表面残基
蛋白质中每个氨基酸残基的溶剂可接近表面积通过程序NACCESS来计算(水分子探针半径取 ):
):
…]$./naccess structures/2c0b_r_b.pdb
注意:NACCESS程序要与structures在同一目录下。计算完成后每个氨基酸残基的溶剂可及表面积会存放在2c0b_r_b.rsa文件中,将2c0b_r_b.rsa文件移至data/Rsa目录下。该文件的内容如下:
表2每个氨基酸残基的溶剂可接近表面积

结果中对每个残基分别给出了五项溶剂可及表面积,即所有原子的(All-atoms)、所有侧链原子的(Total-Side)、所有主链原子的(Main-Chain)、侧链非极性原子的(Non-polar)、侧链极性原子的(All polar)溶剂可及表面积。每一项又分别给出了两个值,即绝对值(ABS)和相对值(REL)。其中All-atoms项ABS列为我们需要的每个氨基酸残基的溶剂可及表面积,会在后面求模块的溶剂可及表面积时使用。All-atoms项REL列为残基的相对溶剂可及表面积,其值≥15%的残基为表面残基。表面残基确定后,用于后面从蛋白质模块中(包括表面模块和内部模块)区分出表面模块(至少含有一个表面残基的模块)。
(3)计算氨基酸残基间的接触面积
氨基酸残基间的接触面积采用程序Qcontacts来计算:
…]$./Qcontacts.pl-i structures/2c0b_r_b.pdb-prefOut data/Vor/2c0b
程序运行完后生成2c0b_L-by-res.vor文件,其中存放氨基酸残基间的接触面积,文件如下:
表3氨基酸残基间的接触面积

Qij列为氨基酸残基间的接触面积,残基间接触面积一方面用于下一步划分蛋白质模块,另一方面用于计算模块内残基间接触面积。
(4)对蛋白质结构划分模块,计算参数PAMA值,并排序
根据上一步获得的残基间接触面积来划分蛋白质模块。模块划分完成后,根据前面(2)中获得的蛋白质的表面残基,来去除内部模块(不包含任何表面残基的模块),从而保留表面模块(至少含有一个表面残基的模块)做以下处理。
对蛋白质结构的所有表面模块,根据公式(1)计算出每个模块的溶剂可接近表面积A,即模块中每个残基的溶剂可及表面积(从前面(2)中产生的文件2c0b_r_b.rsa中获得)之和;根据公式(2)计算出模块内氨基酸残基间的接触面积Q,即模块内两两残基间的接触面积(从前面(3)产生的文件2c0b_L-by-res.vor中获得);计算每个模块的PAMA值,即A与Q的乘积。最后根据PAMA值由大到小,对表面模块进行排序。
以上对蛋白质结构划分模块,计算表面模块的参数PAMA值,并对其进行排序,三个过程可以通过运行程序getModules.pl来完成:
…]$./getModules.pl 2c0b r>data/ReceptorModule/2c0b_r.module
程序会自动按照PAMA值从大到小对模块进行排列,2c0b_r.module文件中module0是PAMA值最大的模块,以此类推,如表4。
表4用PAMA方法获得的蛋白质表面模块的各项参数

a蛋白质表面模块,其排序根据PAMA值由大到小
b模块的溶剂可接近表面积
c模块内氨基酸残基间的接触面积
d模块的溶剂可接近表面积A与模块内氨基酸残基间的接触面积Q之间的乘积
(5)考虑模块的界面偏好性,计算模块参数PPQA并排序
根据公式(3)计算每个模块的界面偏好性,该值为模块中所有氨基酸残基界面偏好性(数据来自表1中的Average列)之和的平均值。然后根据公式(4)计算模块的PPQA值。其中模块的溶剂可接近表面积A,模块内氨基酸残基间的接触面积Q,与(4)中的计算相同。
这一步在Windows系统下完成。下载Python 2.7文件包(全英文路径)。将得到的2c0b_r.module文件名改成AQ_value.txt。提取表1(20×4氨基酸-核苷酸成对偏好性及氨基酸界面偏好性)中的Average列,即每种氨基酸残基与4种核苷酸成对偏好性的平均值,存成名为aa_interface_propensity.txt的文件。根据公式(4)计算模块PPQA值的程序为PPQA.py。
启动cmd,用“cd+目录”进入结构所在目录,运行程序:
C:\Python27>PPQA.py>PPQA_out.txt
程序运算完成后,生成PPQA_out.txt文件,该文件就是加入氨基酸-核苷酸成对偏好性信息之后计算得到的蛋白质表面模块的PPQA值,这里同时也根据PPQA值由大到小的顺序对模块进行了排序。module 0是PPQA值最大的模块,以此类推,如表5。
表5用PPQA方法获得的蛋白质表面模块的各项参数

a蛋白质表面模块,其排序根据PPQA值由大到小
b模块的溶剂可接近表面积
c模块内氨基酸残基间的接触面积
d模块的界面偏好性
e模块的界面偏好性 与溶剂可及表面积A和模块内氨基酸残基间的接触面积Q之间的乘积
与溶剂可及表面积A和模块内氨基酸残基间的接触面积Q之间的乘积
选取排在前两位的表面模块作为蛋白质界面模块的预测结果。通过与复合物实验结构中真正的蛋白质界面模块(表面模块中至少含有一个界面残基的模块;界面残基为蛋白质氨基酸残基中至少有一个原子与RNA中的任何一个原子间的距离小于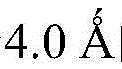 的残基。)进行比较,发现这两个预测的界面模块均是真正的界面模块。附图3给出了对蛋白质(Catalytic domain of E.coli RNase E),PPQA方法识别到的界面模块在蛋白质表面所处的位置。浅灰色是受体蛋白质,黑色和深灰色区域分别表示排在首位和次位模块所在区域,链状深灰色是该蛋白质与13-mer RNA相互作用形成复合物2C0B(PDB code)中的RNA分子。从附图3中可以看出,这两个模块都与RNA有部分接触,是真正的界面模块。
的残基。)进行比较,发现这两个预测的界面模块均是真正的界面模块。附图3给出了对蛋白质(Catalytic domain of E.coli RNase E),PPQA方法识别到的界面模块在蛋白质表面所处的位置。浅灰色是受体蛋白质,黑色和深灰色区域分别表示排在首位和次位模块所在区域,链状深灰色是该蛋白质与13-mer RNA相互作用形成复合物2C0B(PDB code)中的RNA分子。从附图3中可以看出,这两个模块都与RNA有部分接触,是真正的界面模块。
实施例2
体系来自非冗余非核糖体蛋白质-RNA复合物结构(Proteins,2012,80(1):14-24),去掉其中受体和配体是非单链的情况,最终的研究体系为69个复合物体系(如表6)。对每一个体系具体的计算过程同实施例1,这里只对结果作说明。
表6 69个非冗余非核糖体,且受体配体均是单链的蛋白质-RNA复合物


我们对使用本发明方法(以参数PPQA来命名)获得的结果,与随机挑选界面模块方法和用于蛋白质-蛋白质结合模块预测的PAMA方法获得的结果进行了比较(如表7)。
表7用不同方法获得的69个蛋白质-RNA复合物中受体蛋白界面模块最高排序结果


从界面模块最高排序来看(如表7),随机方法排序中,界面模块排在首位的有33个蛋白体系(占整个数据集的47.83%);PAMA方法排序中,界面模块排在首位的有42个体系(60.87%);PPQA方法排序中,有50个(72.46%)。从界面模块最高排序的平均值来看,随机方法、PAMA和PPQA方法分别是3.42、1.96和1.74(最小)。界面模块的排序越靠前,对预测结合模块越有利。根据PPQA方法排序后首个界面模块的最高排序的平均值为1.74,我们确定具有最大和次大PPQA值的表面模块(即排在前两位的表面模块)是界面模块。
从表面模块排序前1-3位(包括3)中含有界面模块的体系数目统计(如附图4)来看,三种方法中,PPQA方法执行的效果最好,例如前两位表面模块中至少有一个是界面模块的体系最多,有63个,占整个数据集的91.30%,与PAMA方法相比,提高了11.59%,与随机方法相比,提高了30.43%。
因此这一结果一方面说明之前用于蛋白质结合蛋白上界面模块预测PAMA中,表面模块的划分方法可以用于预测RNA结合蛋白界面模块中表面模块的划分,即蛋白质-RNA识别中蛋白质的结合界面也具有模块化的特性;另一方面说明氨基酸-核苷酸成对偏好性信息的考虑,以及其在模块参数中的考虑方式,都具有一定的合理性,可以提高蛋白质-RNA复合物界面模块的识别成功率。
Claims (3)
1.基于氨基酸-核苷酸成对偏好性信息的蛋白质上与RNA结合模块的预测方法,其特征在于,包括以下步骤:一是对蛋白质进行模块划分,二是计算每个模块的自定义参数,三是将模块按照参数PPQA值由高到低进行排序,并识别出可能的界面模块;具体如下:
步骤1:蛋白质表面模块划分
以蛋白质三维结构中的每个氨基酸残基为中心,将与之有接触的所有残基划分为一个模块,包括该中心残基、蛋白质内部残基以及表面残基;然后剔除所有的内部模块,保留表面模块作为最后表面模块划分的结果,内部模块为不包含任何表面残基的模块,表面模块为至少有一个表面残基的模块;
表面残基为相对溶剂可接近表面积≥15%的残基;界面模块为至少含有一个与RNA分子相互作用的界面残基的模块,界面模块为表面模块中的一部分;界面残基为蛋白质氨基酸中至少有一个原子与RNA中的任何一个原子间的距离小于 的残基;
的残基;
步骤2:计算模块的自定义参数
对每一个表面模块计算以下各值:模块溶剂可接近表面积A、模块内部残基接触面积Q以及模块界面偏好性
模块的溶剂可接近表面积A为模块中所有残基的溶剂可接近表面积之和:

其中,Ai是模块中第i个残基的溶剂可接近表面积,求和遍及模块中的所有残基;
模块的内部接触面积Q为模块内所有残基对的接触面积之和:

其中,Qij是模块中残基i和残基j之间的接触面积,求和遍及模块中的所有残基对;
从之前统计获得的氨基酸-核苷酸成对偏好性中,针对每一种氨基酸,取其与4种核苷酸的偏好性的平均值,获得该氨基酸的界面偏好性,表1中Average列;针对每一个蛋白质表面模块,计算其中所有氨基酸残基界面偏好性之和的算术平均值 作为模块的界面偏好性:
作为模块的界面偏好性:

其中N表示模块中氨基酸残基的个数, 表示模块中残基i的界面偏好性,求和遍及模块中的所有残基;
表示模块中残基i的界面偏好性,求和遍及模块中的所有残基;
表1 20×4氨基酸-核苷酸成对偏好性及氨基酸界面偏好性

定义模块的PPQA参数值,即模块界面偏好性、溶剂可及表面积以及其内部接触面积三者相乘:

步骤3:对模块按照PPQA值由大到小排序,预测识别界面模块
最后,根据PPQA值从大到小对模块进行排序,考察蛋白质界面模块所处的位置,从而确定前两个作为界面模块识别标准。
2.按照权利要求1所述的基于氨基酸-核苷酸成对偏好性信息的蛋白质上与RNA结合模块的预测方法,其特征在于,步骤1,采用基于维里几何的Qcontacts算法计算残基间的接触面积,来判断两残基是否接触。
3.按照权利要求1所述的基于氨基酸-核苷酸成对偏好性信息的蛋白质上与RNA结合模块的预测方法,其特征在于,溶剂可接近表面积的计算采用NACCESS算法,水分子探针半径取
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710374897.8A CN108959852B (zh) | 2017-05-24 | 2017-05-24 | 基于氨基酸-核苷酸成对偏好性信息的蛋白质上与rna结合模块的预测方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710374897.8A CN108959852B (zh) | 2017-05-24 | 2017-05-24 | 基于氨基酸-核苷酸成对偏好性信息的蛋白质上与rna结合模块的预测方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN108959852A CN108959852A (zh) | 2018-12-07 |
| CN108959852B true CN108959852B (zh) | 2021-12-24 |
Family
ID=64494263
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201710374897.8A Expired - Fee Related CN108959852B (zh) | 2017-05-24 | 2017-05-24 | 基于氨基酸-核苷酸成对偏好性信息的蛋白质上与rna结合模块的预测方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN108959852B (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111554355B (zh) * | 2020-05-05 | 2023-04-25 | 湖南大学 | 一种基于非冯诺依曼架构的分子动力学计算方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1328601A (zh) * | 1998-08-25 | 2001-12-26 | 斯克利普斯研究院 | 预测蛋白质功能的方法和系统 |
| AU2005271899A1 (en) * | 2004-07-09 | 2006-02-16 | Wyeth | Methods and systems for predicting protein-ligand coupling specificities |
| CN101146825A (zh) * | 2005-02-14 | 2008-03-19 | 阿波罗生命科学有限公司 | 分子及其嵌合分子 |
| ES2751126T3 (es) * | 2011-10-21 | 2020-03-30 | Univ Kyushu Nat Univ Corp | Método de diseño para proteína de unión a ARN usando motivo de PPR, y uso del mismo |
| CN102521527B (zh) * | 2011-12-12 | 2015-01-14 | 同济大学 | 一种根据抗体物种分类预测蛋白质抗原空间表位的方法 |
| CN103500293B (zh) * | 2013-09-05 | 2017-07-14 | 北京工业大学 | 一种非核糖体蛋白质‑rna复合物近天然结构的筛选方法 |
| CN105260626B (zh) * | 2015-09-25 | 2017-11-14 | 麦科罗医药科技(武汉)有限公司 | 蛋白质结构空间构象的全信息预测方法 |
-
2017
- 2017-05-24 CN CN201710374897.8A patent/CN108959852B/zh not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CN108959852A (zh) | 2018-12-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Giani et al. | Long walk to genomics: History and current approaches to genome sequencing and assembly | |
| EP3036344B1 (en) | Methods and systems for aligning sequences | |
| Sun et al. | Computational tools for aptamer identification and optimization | |
| NZ759659A (en) | Deep learning-based variant classifier | |
| Zhang et al. | Predicting protein-ATP binding sites from primary sequence through fusing bi-profile sampling of multi-view features | |
| JP2016540275A (ja) | 配列変異体を検出するための方法およびシステム | |
| WO2015058120A1 (en) | Methods and systems for aligning sequences in the presence of repeating elements | |
| Dobson et al. | Prediction of protein function in the absence of significant sequence similarity | |
| Fan et al. | lncLocPred: predicting LncRNA subcellular localization using multiple sequence feature information | |
| Park et al. | Comparing expression profiles of genes with similar promoter regions | |
| Yin et al. | Effective hidden Markov models for detecting splicing junction sites in DNA sequences | |
| CN108959852B (zh) | 基于氨基酸-核苷酸成对偏好性信息的蛋白质上与rna结合模块的预测方法 | |
| Gromiha | Distinct roles of conventional non-covalent and cation–π interactions in protein stability | |
| Zardoya | Quest for the best evolutionary model | |
| Cordes et al. | Initial state of an enzymic reaction. Theoretical prediction of complex formation in the active site of RNase T1 | |
| Keasar et al. | Homology as a tool in optimization problems: structure determination of 2D heteropolymers | |
| Santos et al. | Improving de novo protein structure prediction using contact maps information | |
| Chaitanya et al. | Genome sequencing, assembly, and annotation | |
| Wang et al. | How Large is the Universe of RNA-Like Motifs? A Clustering Analysis of RNA Graph Motifs Using Topological Descriptors | |
| Rafael | Quest for the Best Evolutionary Model | |
| Kinoshita et al. | Prediction of Molecular Interactions from 3D‐Structures: From Small Ligands to Large Protein Complexes | |
| Ko et al. | The development of a proteomic analyzing pipeline to identify proteins with multiple RRMs and predict their domain boundaries | |
| Tyagi | Computational molecular biology | |
| David et al. | Identification of significant descriptors for enzyme inhibition using the LASSO method and a genetic algorithm search | |
| Singh | Bioinformatics and Applications in Biotechnology |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20211224 |