Stable carteolol hydrochloride, preparation method thereof and pharmaceutical composition for eyes
Technical Field
The invention belongs to the technical field of medicines, and relates to an β -adrenergic receptor blocker, a preparation method thereof and an eye drop pharmaceutical composition containing the same, in particular to carteolol hydrochloride, a preparation method thereof and an eye drop pharmaceutical composition containing the same.
Background
Carteolol (Carteolol), which is clinically used as a hydrochloride thereof and is also called "quinalol", is a β -adrenergic receptor blocker developed by tsukamur pharmaceutical company in japan, and tsukamur pharmaceutical company in japan is marketed under the trade name of Milelan in 10 months in 1980, 1 month and 22 days in 1997, approved by the national drug administration, and tsukamur pharmaceutical company Limited in china starts to produce Carteolol hydrochloride eye drops with the trade name of "Meikaran".
The carteolol hydrochloride is a non-selective β -adrenoceptor blocking agent and has a blocking effect on both β 1 and β 2 receptors, the carteolol hydrochloride has little or no local anesthetic effect, the main difference of the carteolol hydrochloride and other β receptor blocking agents is that the carteolol hydrochloride has an intrinsic sympathomimetic activity, the carteolol hydrochloride eye drops have an intraocular pressure reducing effect on patients with high intraocular pressure and normal intraocular pressure, the intraocular pressure begins to be reduced 1 hour after the eye drops are used for glaucoma patients, the intraocular pressure is reduced maximally in 4 hours, the intraocular pressure is reduced by 5.6-9.9 mmHg, the intraocular pressure reduction rate is 7% -22%, the intraocular pressure reducing effect can last for 8-24 hours, the intraocular pressure reducing effect is kept stable for 4-32 weeks after continuous medication, 80.7% of the intraocular pressure of the patients with glaucoma is controlled below 21mmHg, the main metabolite of the carteolol hydrochloride is an intraocular β receptor blocking agent, the intraocular pressure reducing effect is also has the intraocular pressure reducing effect, the intraocular pressure reducing mechanism of the carteolol hydrochloride is mainly used for reducing the generation of aqueous humor, the atrial outflow mutation of the carteolol hydrochloride is shown by an experiment, the experiment shows that the mouse has no carcinogenic toxicity caused by a large amount of the oral administration of the rat, and the experiment of the rat has no toxicity caused by the experiment, and the experiment of the rat caused by the oral administration of the rat.
Cartinolol 2% for single eye drop of rabbit14C carteolol hydrochloride 0.01ml, the radioactivity in the aqueous humor of the instilled eye reaches the peak value 1 hour after the instillation, and most of the radioactivity is the prototype. The drug transfer to other eye tissues reaches a peak 0.5 to 1 hour after the dropping, and then rapidly disappears, and accumulation in the eye tissues is not observed. 1 hour after dropping, the ratio of radioactivity in aqueous humor, plasma and opposite aqueous humor of the eye is 200: 5: 1.1 drop of 2% of the product is dropped into each eye of a healthy person, 16% of the drop amount is discharged through urine 24 hours after the dropping, the half-life period of the excretion in urine is 5 hours, and the blood concentration of carteolol after the dropping is below a quantitative limit (5 ng/ml).
Clinically, carteolol has good intraocular pressure reducing efficacy on primary open-angle glaucoma. The eye drop can further enhance the effect of reducing intraocular pressure for certain secondary glaucoma, ocular hypertension, angle closure glaucoma which is not completely controlled after operation and other glaucoma which is not effective in medicaments and operations.
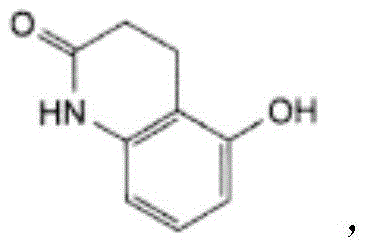
The chemical name of Carteolol Hydrochloride (Carteolol Hydrochloride) is: 5- [3- (1,1-dimethylethyl) amino ] -2-hydroxypropoxy-3, 4-dihydro-2 (1H) -quinolone hydrochloride, having the chemical name England: 5- [ (2RS) -3- [ (1, 1-dimethylthienyl) amino ] -2-hydroxyproxy ] -3,4-dihydroquinolin-2(1H) -onehydrochloride, CAS registry number 51781-21-6. The molecular formula is as follows: C16H24N2O 3. HCl, hydrochloride having a molecular weight: 328.84, having the chemical formula:
carteolol hydrochloride and its preparation, especially its eye drops, have been loaded in the chinese pharmacopoeia of 2005 edition, 2010 edition and 2015 edition, and also in the drug standards of the countries/regions such as the united states pharmacopoeia, japanese pharmacopoeia, european pharmacopoeia, british pharmacopoeia, korean pharmacopoeia, etc.
Although the clinical use history of the Chinese medicine is about 20 years and more than 10 years exist in the Chinese pharmacopoeia loaded with three versions, the raw material medicines are imported at home.
Several patent documents of carteolol hydrochloride, which were first published by tsukamur pharmaceutical company of japan in the original research, in the last 70 th century, describe a preparation process route, and a typical process route takes 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione as a starting material, and the specific process route is as follows:
however, the above-mentioned 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione is not commercially available from the market and is thus not preferable.
Other synthetic routes to carteolol hydrochloride have been reported [ Nakagawa K, Murakami N, Yoshizaki S, et al. Derivatives of 3,4-Dihydrocarbostyril as β -adrenergicg agents [ J ]. J Med Chem,1974,17 (5): 529. 533; Tamura Y, Nakagawa K, Yoshizas, et al.3,4-Dihydrocarbostyril as J.J.: J.Med.Med.Chem.Chem.J.: 1974 and a process for the synthesis of the same: US 3910924[ P ]. 1975-10-07; Luyuwa, Zhang-zhang. pharmaceutical synthesis [ M ].1 edition, Min health Press, 1989: 453. beta. 461] which uses 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone as an intermediate and makes it easier to prepare the dihydrocarbostyrol hydrochloride [ 5-Dihydrocarbostyril H-5, 5-dihydrocarbostyrol ] and easy to dehydrogenate the dihydrocarbostyrol hydrochloride [ 5-5, 5-dihydrocarbostyrol ] and 2H-5-dihydrocarbostyrol intermediate after condensation reaction with the above-5-7, easy to obtain the dihydrocarbostyrol hydrochloride-5-dihydrocarbostyrol-7-5-7-dihydrocarbostyrol-5-dihydrocarbostyrol-7-dihydrocarbostyrol-5-2-amide-ethyl-2-dihydrocarbostyrol-ethyl-2-ethyl-2-ethyl ketone (7-ethyl-7-ethyl-.
In view of the defects or shortcomings of the prior art methods for synthesizing carteolol hydrochloride, it is still very urgent for those skilled in the art to provide a method for preparing a carteolol hydrochloride drug substance meeting the pharmaceutical requirements.
Disclosure of Invention
The object of the present invention is to provide an excellent method for preparing carteolol hydrochloride, and such a method is expected to exhibit at least one aspect of excellent technical effects, such as those described in the present invention. Another object of the present invention is to provide a carteolol hydrochloride prepared by the method of the present invention. Still another object of the present invention is to use the drug substance of the present invention to prepare carteolol hydrochloride eye drops for clinical application. It has been surprisingly found that excellent technical effects as described herein can be obtained using the methods of the aspects of the invention. The present invention has been completed based on such findings.
To this end, the present invention provides, in a first aspect, a process for preparing carteolol hydrochloride comprising the steps of:
(a) the method comprises the following steps Preparation of 3-amino-2-cyclohexenone
Reacting 1, 3-cyclohexanedione with ammonium acetate under the heating condition, cooling the reactant to room temperature after the reaction is finished, adding ethyl acetate, dissolving the reactant in the ethyl acetate, cooling to 0 ℃, filtering, and drying a filter cake to obtain 3-amino-2-cyclohexenone;
step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Reacting 3-amino-2-cyclohexenone with acrylic acid under heating, cooling the reactant to room temperature after the reaction is finished, adding absolute ethyl alcohol, cooling to room temperature after heating and dissolving, filtering, and drying a filter cake to obtain 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione;
step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Dissolving 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione in a reaction solvent under stirring, then slowly dropwise adding a solution containing a dehydrogenation reagent in an ice bath under stirring, then reacting under a heating condition, adding water after the reaction is finished, standing for layering, extracting an aqueous layer with ethyl acetate, drying an organic layer, filtering, evaporating the solvent from the filtrate, and drying the residue to obtain 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone;
step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
Dissolving 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone in an aqueous alkali solution, adding epoxy chloropropane, reacting under the conditions of heating and stirring, cooling to room temperature after the reaction is finished, adding ethyl acetate, removing a lower water layer, filtering, and recrystallizing a filter cake to obtain 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone;
step e: preparation of carteolol hydrochloride
Adding a reaction solvent into a mixture of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone and tert-butylamine, reacting under the condition of adding, evaporating the solvent and unreacted amine under reduced pressure after the reaction is finished, adding acetone (the volume of the acetone is about 0.5-1 time of that of the reaction solvent), adding hydrochloric acid (the concentration of the hydrochloric acid is 1-3 mol/L, such as 1.5-2.5 mol/L, and the adding amount of the hydrochloric acid is 1-1.5 mol times of that of the 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone), stirring uniformly, filtering, evaporating the solvent from the filtrate under reduced pressure, recrystallizing the residue (such as ethanol or acetone), filtering, and drying a filter cake to obtain the carteolol hydrochloride.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (a), the molar ratio of the 1, 3-cyclohexanedione to the ammonium acetate is 1: 1-2, for example 1: 1.2 to 1.6, for example 1: 1.3 to 1.5.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (a), the heating is at a temperature of 80 to 120 ℃, for example at a temperature of 90 to 110 ℃; the reaction time is 10min to 60min, for example 15min to 45min, for example 20 min.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (b), the molar ratio of the 3-amino-2-cyclohexenone to acrylic acid is 1: 1-2, for example 1: 1.2 to 1.8, for example 1: 1.4 to 1.6.
The process according to any embodiment of the first aspect of the present invention, wherein in step (b), the heating is performed under reflux conditions of the reactants for a reaction time of 2h to 10h, such as for a reaction time of 3h to 6h, such as for a reaction time of 3h to 5 h.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (c), the reaction solvent is selected from chloroform, dichloromethane, 1, 2-dichloroethane, cyclohexane, n-hexane, cyclopentane. In one embodiment, the concentration of the 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione in the reaction solvent is 5-15% (w/v), such as 8-12% (w/v).
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (c) the dehydrogenation reagent is selected from bromine, N-bromosuccinimide. In one embodiment, the solvent in which the dehydrogenation reagent is formulated is the same as the reaction solvent. In one embodiment, the molar ratio of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione to the dehydrogenation reagent is 1: 1.5 to 2.5, for example 1: 2.
the process according to any embodiment of the first aspect of the present invention, wherein in step (c), the heating is under reflux conditions of the reactants for a reaction time of 2h to 10h, such as for a reaction time of 3h to 6h, such as for a reaction time of 3h to 5 h.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in the step (c), the amount of water added is 0.3 to 0.8 times the volume of the reactants.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (c), 2 to 4 times, for example 3 times, extraction is performed with ethyl acetate 1 to 2 times the volume of the water layer.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (c), the organic layer dries all the drying agent selected from anhydrous calcium sulfate, anhydrous magnesium sulfate, anhydrous sodium sulfate, and the like.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (d), the aqueous base is a solution selected from the group consisting of: sodium hydroxide solution, sodium carbonate solution and sodium bicarbonate solution. In one embodiment, the molar ratio of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone to the base is 1: 0.5 to 1.5, for example, in a molar ratio of 1: 0.8 to 1.2. In one embodiment, the molar concentration of the aqueous base is from 0.2 to 0.6mol/L, for example from 0.3 to 0.5 mol/L.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (d), the molar ratio of the 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone to the epichlorohydrin is 1: 2 to 4, for example in a molar ratio of 1: 3.
the method according to any embodiment of the first aspect of the present invention, wherein in the step (d), the heating is at 40 to 60 ℃, for example at 50 to 55 ℃.
The process according to any embodiment of the first aspect of the present invention, wherein in step (d), the heating reaction is performed for 3 to 10 hours, such as for 4 to 8 hours, such as for 5 to 7 hours.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in the step (d), the volume of the added ethyl acetate is 0.5 to 1 times the volume of the reactants.
The process according to any one of the embodiments of the first aspect of the present invention, wherein in step (d), the solvent used for recrystallization of the filter cake is selected from acetone, ethyl acetate.
A process according to any one of the embodiments of the first aspect of the present invention, wherein in step (e) the molar ratio of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone to tert-butylamine is 1: 3 to 6, for example, in a molar ratio of 1: 3 to 5, for example, in a molar ratio of 1: 4.
the process according to any one of the embodiments of the first aspect of the present invention, wherein in step (e), the reaction solvent is selected from the group consisting of absolute ethanol, acetone, ethyl acetate, n-hexane or a combination thereof. In one embodiment, the reaction solvent is a combination of absolute ethanol and acetone. In one embodiment, the reaction solvent is a combination of absolute ethyl alcohol and acetone, and the volume ratio of the absolute ethyl alcohol to the acetone is 8-12: 1, for example, the volume ratio is 9-11: 1, for example in a volume ratio of 10: 1.
the process according to any one of the embodiments of the first aspect of the present invention, wherein in step (e), the heating is heating under reflux conditions; the reaction time is 2 to 8 hours, for example, 3 to 6 hours, for example, 4 hours.
Further, the invention provides a carteolol hydrochloride in a second aspect.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, prepared according to a process comprising the steps of:
(a) the method comprises the following steps Preparation of 3-amino-2-cyclohexenone
Reacting 1, 3-cyclohexanedione with ammonium acetate under the heating condition, cooling the reactant to room temperature after the reaction is finished, adding ethyl acetate, dissolving the reactant in the ethyl acetate, cooling to 0 ℃, filtering, and drying a filter cake to obtain 3-amino-2-cyclohexenone;
step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Reacting 3-amino-2-cyclohexenone with acrylic acid under heating, cooling the reactant to room temperature after the reaction is finished, adding absolute ethyl alcohol, cooling to room temperature after heating and dissolving, filtering, and drying a filter cake to obtain 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione;
step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Dissolving 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione in a reaction solvent under stirring, then slowly dropwise adding a solution containing a dehydrogenation reagent in an ice bath under stirring, then reacting under a heating condition, adding water after the reaction is finished, standing for layering, extracting an aqueous layer with ethyl acetate, drying an organic layer, filtering, evaporating the solvent from the filtrate, and drying the residue to obtain 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone;
step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
Dissolving 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone in an aqueous alkali solution, adding epoxy chloropropane, reacting under the conditions of heating and stirring, cooling to room temperature after the reaction is finished, adding ethyl acetate, removing a lower water layer, filtering, and recrystallizing a filter cake to obtain 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone;
step e: preparation of carteolol hydrochloride
Adding a reaction solvent into a mixture of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone and tert-butylamine, reacting under the condition of adding, evaporating the solvent and unreacted amine under reduced pressure after the reaction is finished, adding acetone (the volume of the acetone is about 0.5-1 time of that of the reaction solvent), adding hydrochloric acid (the concentration of the hydrochloric acid is 1-3 mol/L, such as 1.5-2.5 mol/L, and the adding amount of the hydrochloric acid is 1-1.5 mol times of that of the 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone), stirring uniformly, filtering, evaporating the solvent from the filtrate under reduced pressure, recrystallizing the residue (such as ethanol or acetone), filtering, and drying a filter cake to obtain the carteolol hydrochloride.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (a), the molar ratio of the 1, 3-cyclohexanedione to ammonium acetate is 1: 1-2, for example 1: 1.2 to 1.6, for example 1: 1.3 to 1.5.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (a), the heating is at a temperature of 80 to 120 ℃, for example at a temperature of 90 to 110 ℃; the reaction time is 10min to 60min, for example 15min to 45min, for example 20 min.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (b) the molar ratio of 3-amino-2-cyclohexenone to acrylic acid is 1: 1-2, for example 1: 1.2 to 1.8, for example 1: 1.4 to 1.6.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (b), the heating is performed under reflux conditions of the reactants for a reaction time of 2h to 10h, for example for a reaction time of 3h to 6h, for example for a reaction time of 3h to 5 h.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (c) the reaction solvent is selected from chloroform, dichloromethane, 1, 2-dichloroethane, cyclohexane, n-hexane, cyclopentane. In one embodiment, the concentration of the 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione in the reaction solvent is 5-15% (w/v), such as 8-12% (w/v).
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention wherein in step (c) the dehydrogenation reagent is selected from bromine, N-bromosuccinimide. In one embodiment, the solvent in which the dehydrogenation reagent is formulated is the same as the reaction solvent.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (c), the heating is performed under reflux conditions of the reactants for a reaction time of 2h to 10h, for example for a reaction time of 3h to 6h, for example for a reaction time of 3h to 5 h.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (c), the amount of water added is 0.3 to 0.8 times the volume of the reactants.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (c), 2-4 times, for example 3 times, extraction is performed with ethyl acetate 1-2 times the volume of the water layer.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (c) the organic layer dries all the drying agents selected from anhydrous calcium sulfate, anhydrous magnesium sulfate, anhydrous sodium sulfate, and the like.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (d) the aqueous base solution is a solution selected from the group consisting of: sodium hydroxide solution, sodium carbonate solution and sodium bicarbonate solution. In one embodiment, the molar ratio of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone to the base is 1: 0.5 to 1.5, for example, in a molar ratio of 1: 0.8 to 1.2. In one embodiment, the molar concentration of the aqueous base is from 0.2 to 0.6mol/L, for example from 0.3 to 0.5 mol/L.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (d), the molar ratio of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone to epichlorohydrin is 1: 2 to 4, for example in a molar ratio of 1: 3.
carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (d), the heating is performed at 40-60 ℃, for example at 50-55 ℃.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (d), the heating reaction is performed for 3 to 10 hours, such as for 4 to 8 hours, such as for 5 to 7 hours.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (d), the volume of ethyl acetate added is 0.5 to 1 times the volume of the reactants.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (d), the solvent used for recrystallization of the filter cake is selected from acetone and ethyl acetate.
Carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (e) the molar ratio of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone to tert-butylamine is 1: 3 to 6, for example, in a molar ratio of 1: 3 to 5, for example, in a molar ratio of 1: 4.
carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (e), the reaction solvent is selected from absolute ethanol, acetone, ethyl acetate, n-hexane or a combination thereof. In one embodiment, the reaction solvent is a combination of absolute ethanol and acetone. In one embodiment, the reaction solvent is a combination of absolute ethyl alcohol and acetone, and the volume ratio of the absolute ethyl alcohol to the acetone is 8-12: 1, for example, the volume ratio is 9-11: 1, for example in a volume ratio of 10: 1.
carteolol hydrochloride according to any embodiment of the second aspect of the present invention, wherein in step (e), the heating is under reflux conditions; the reaction time is 2 to 8 hours, for example, 3 to 6 hours, for example, 4 hours.
Further, the third aspect of the present invention provides an ophthalmic pharmaceutical composition, which is in the form of eye drops, wherein the composition comprises carteolol hydrochloride and pharmaceutically acceptable excipients.
An ophthalmic pharmaceutical composition according to any embodiment of the third aspect of the present invention, having a formulation as described in any of the composition examples herein below.
An ophthalmic pharmaceutical composition according to any embodiment of the third aspect of the present invention, wherein said carteolol hydrochloride is prepared according to the method of any embodiment of the first aspect of the present invention.
An ophthalmic pharmaceutical composition according to any embodiment of the third aspect of the present invention, wherein said carteolol hydrochloride is as described in any embodiment of the second aspect of the present invention.
Further, in a fourth aspect, the present invention provides a use of the carteolol hydrochloride prepared by the method according to any one of the embodiments of the first aspect of the present invention or the carteolol hydrochloride according to any one of the embodiments of the second aspect of the present invention in the preparation of a medicament for treating or preventing glaucoma or ocular hypertension.
The use according to any of the embodiments of the fourth aspect of the invention, wherein the medicament is an ophthalmic pharmaceutical composition according to any of the embodiments of the third aspect of the invention.
In the above-described steps of the preparation method of the present invention, although the specific steps described therein are distinguished in some detail or in language description from the steps described in the preparation examples of the detailed embodiments below, those skilled in the art can fully summarize the above-described method steps in light of the detailed disclosure throughout the present disclosure.
Any embodiment of any aspect of the invention may be combined with any other embodiment of the invention, as long as they do not contradict. Furthermore, in any embodiment of any aspect of the invention, any feature may be applicable to that feature in any other embodiment of the invention, provided that they do not contradict.
The invention is further described below.
All documents cited herein are incorporated by reference in their entirety and to the extent such documents do not conform to the meaning of the present invention, the present invention shall control. Further, the various terms and phrases used herein have the ordinary meaning as is known to those skilled in the art, and even though such terms and phrases are intended to be described or explained in greater detail herein, reference is made to the term and phrase as being inconsistent with the known meaning and meaning as is accorded to such meaning throughout this disclosure.
The method provided by the invention has excellent pharmaceutical performance, and has an important effect when being used for preparing tofacitinib citrate tablets.
The carteolol hydrochloride prepared by the method is an excellent non-selective β -adrenoceptor retarder, has a retarding effect on both β 1 and β 2 receptors, and the obtained bulk drug and preparation have excellent pharmaceutical properties.
Detailed Description
The present invention will be further described by the following examples, however, the scope of the present invention is not limited to the following examples. It will be understood by those skilled in the art that various changes and modifications may be made to the invention without departing from the spirit and scope of the invention. The present invention has been described generally and/or specifically with respect to materials used in testing and testing methods. Although many materials and methods of operation are known in the art for the purpose of carrying out the invention, the invention is nevertheless described herein in as detail as possible. The following examples further illustrate the invention without limiting it.
An HPLC related substance inspection method (established by referring to a method for detecting related substances of carteolol hydrochloride bulk drug loaded in British pharmacopoeia 2013 edition, which can be abbreviated as BP2013-HPLC method in the text):
the measurement is carried out according to the specification of the high performance liquid chromatography carried in the section 0512 of page 59 of the fourth part of the Chinese pharmacopoeia 2015 edition;
test solutions: dissolving 20.0mg of a substance to be detected in a mobile phase and diluting the solution to 10.0mL by using the mobile phase;
reference solution (a): diluting 1.0mL of the test solution to 100.0mL with the mobile phase;
reference solution (b): diluting 1.0mL of the reference solution (a) to 10.0mL with the mobile phase;
reference solution (c): 10mg of system suitability carteolol was dissolved in the mobile phase and diluted to 5mL with the mobile phase;
reference solution (d): diluting 5.0mL of reference solution (b) to 10.0mL with mobile phase;
a chromatographic column: the column length is 25cm, the inner diameter is 4.6mm, the stationary phase is octadecylsilane chemically bonded silica, and the particle size is 5 mu m;
mobile phase: a mixed solution of methanol-acetonitrile-2.82 g/L sodium hexanesulfonate solution (the volume ratio of the three is 1: 20: 79);
flow rate: 1 mL/min;
ultraviolet detection, wavelength: 252 nm;
sample introduction amount: 20 mu L of the solution;
impurity identification: determining the chromatographic peak of the impurity H by using a chromatogram provided by carteolol according to the system applicability;
and (3) testing the applicability of the system: the chromatogram obtained by the reference solution (c) is similar to the chromatogram obtained by carteolol for system applicability, and the impurity H peak and the carteolol peak reach baseline separation; signal-to-noise ratio: the main peak of the chromatogram obtained by the reference solution (d) is more than 10; the number of theoretical plates: calculated by the main peak of the chromatogram obtained from the reference solution (a), is not less than 6000;
BP2013 for reference specifies impurity limits of:
-impurity H: less than 2 times (0.2%) the area of the main peak in the chromatogram obtained for reference solution (b);
-unknown impurities: for each impurity, the area of the main peak in the chromatogram obtained for reference solution (b) was lower (0.1%);
-total impurities: less than half (0.5%) of the area of the main peak in the chromatogram obtained for reference solution (a);
-ignore limit: 0.2 times (0.02%) of the area of the main peak in the chromatogram obtained for the reference solution (b);
and (3) calculating the chromatographic purity: and (4) calculating the chromatographic purity of the main component of the material by adopting an area normalization method except a solvent peak in the chromatogram of the test solution.
Possible impurities include:
impurity H:
and enantiomers thereof, chemical name: 5- [ (2RS) -3- [ (1,1-Dimethylethyl) amino group]-2-hydroxypropoxy group]Quinolin-2(1H) -one, which is an oxidation product of carteolol;
other possible impurities include:
impurity A:
4,6,7, 8-tetrahydroquinoline-2, 5(1H,3H) -dione,is a reaction intermediate;
impurity B:
5-hydroxy-3, 4-dihydroquinolin-2(1H) -one, as a reaction intermediate;
impurity C:
and its enantiomer, 5- [ [ (2RS) -oxirane2-yl]Methoxy radical]-3,4-dihydroquinolin-2(1H) -one, as reaction intermediate.
The BP2013-HPLC method can also be used for measuring the content of target objects (comprising carteolol or intermediates and the like) in various materials (comprising carteolol bulk drugs, preparations containing carteolol, bulk drugs and intermediates in the preparation process of the preparations and the like), and can also be used for measuring parameters such as the chromatographic purity of main components in the various materials.
First, examples of the preparation of the Compounds
Example 1: preparation of carteolol hydrochloride
This example employs the following reaction scheme
Step a: preparation of 3-amino-2-cyclohexenone
Adding 1, 3-cyclohexanedione (3.36g, 0.03mol) and ammonium acetate (3.23g, 0.042mol) into a 100ml three-necked bottle, stirring, reacting in an oil bath at 100 ℃ for 20min, naturally cooling after removing the oil bath, solidifying the reaction liquid, adding ethyl acetate (10ml) after cooling to room temperature, heating to dissolve, cooling to 0 ℃, filtering, and drying the filter cake to obtain a yellow crystal of 3-amino-2-cyclohexenone (3.09g, yield 93.2%), mp 132-134 ℃ (Wang GW, Miao CB, environmental impact one-pot multi-component aptamers to the synthesis of synthetic 4-arylderivatives [ J ]. Green Chem,2006, 8: 1080-.
Step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Adding 3-amino-2-cyclohexenone (2.2g, 0.02mol) and acrylic acid (2.1g, 0.03mol) into a 100ml three-necked flask, heating to reflux for 4H under stirring, cooling to room temperature, solidifying the reaction solution to obtain a red brown solid, adding absolute ethanol (5ml), heating to dissolve, cooling to room temperature, filtering, and drying the filter cake to obtain pale yellow crystals of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (2.90g, yield 88.4%), mp 194-195 ℃ (literature [ Shono T, Matsumura Y, Kashimura S.A new reactive synthesis of 5-hydroxy-3,4-dihydrocarbostyril and5-hydroxycarbostyril [ J ]. Org, 1981,46 (18: 3719): 3774%, yield 194-195 ℃).
Step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Adding 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (3.26g, 0.02mol) and chloroform (30ml) into a 100ml three-necked bottle, stirring at room temperature until the solution is dissolved, placing the bottle in an ice bath, stirring for 10min, slowly dropwise adding a chloroform (5ml) solution of bromine (1.60g, 0.02mol), heating to reflux for 4H after the dropwise addition, and cooling to room temperature after the TLC detection reaction is finished. Adding water (20ml), stirring, standing for layering, extracting the water layer with ethyl acetate (30ml × 3), combining the organic layers, drying over anhydrous magnesium sulfate, filtering, evaporating the solvent from the filtrate, and drying the residue to obtain an off-white solid 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (2.61g, yield 80.1%), mp 232-234 ℃ (literature [ Tamura Y, Terashima M, Higuchi Y, et al].Chem Ind(London),1970,7(45):1435]: the yield is 68%, mp 232-233 ℃, and the chromatographic purity of BP2013-HPLC method is 82.6%.1H-NMR(400M,DMSO-d6):δ9.93(s,1H),δ9.46(s,1H),δ6.9(t,1H),δ6.43(d,1H),δ6.31(d,1H),δ2.77(t,2H),δ2.38(t,2H)。
Step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
Dissolving 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (2.45g, 0.015mol) in 0.4mol/L sodium hydroxide solution (40ml), stirring for 10min, adding epichlorohydrin (4.14g, 0.045mol), stirring at 50-55 deg.C for about 6H to produce a large amount of white floc, detecting by TLC, cooling to room temperature, adding ethyl acetate (30ml), draining the lower water with a separating funnel, filtering, recrystallizing the filter cake with acetone to obtain a white powdery solid of 5- (2, 3-glycidoxy) -3, 4-dihydro-2 (1H) -quinolone (2.04g, yield 62.2%), mp 172-173 deg.C (Nakagawa K, Murakami N, Yoshizaki S, Medet. Derivatives of 3, 4-dihydroxybenzene β -adneragees J, 533J, 73-529J, 54 deg.C).
Step e: preparation of carteolol hydrochloride
Adding 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (4.38g, 0.02mol) and tert-butylamine (5.84g, 0.08mol) into a 100ml flask, adding absolute ethanol (40ml), stirring and heating to reflux for 4H, detecting the completion of the reaction by TLC, distilling off ethanol and unreacted tert-butylamine under reduced pressure, adding acetone (30ml) into the residue, dissolving and then adding 2mol/L hydrochloric acid (12ml), stirring for 10min, filtering, distilling off the solvent from the filtrate under reduced pressure, recrystallizing the residue with ethanol, filtering, and drying the filter cake to obtain white crystals of carteolol hydrochloride (5.28g, yield 80.5%), mp 277-278 ℃ [ Nakagawa K, Murakami N, Yoshizaki S, et al.
Purity 99.2% (BP2013-HPLC method). 1H NMR (DMSO-d6) delta: 1.31(s,9H, CH3),2.40(t, J ═ 7.6Hz,2H, CH2),2.81 to 2.89(m,2H, CH2),2.92 to 2.96(m,1H, CH2),3.10 to 3.16(m,1H, CH2),3.99(s,1H, CH),4.00(s,1H, CH2),4.24 to 4.25(m,1H, CH2),6.50(d, J ═ 7.9Hz,1H, Ar-H),6.59(d, J ═ 8.5Hz,1H, Ar-H),7.07(t, J ═ 8.2Hz,1H, Ar-H),10.06(s,1H, NH),5.98(s,1H, active hydrogen), 9.12(s,1H, active hydrogen). EI-MS (m/z): 292(M +).
Example 2: preparation of carteolol hydrochloride
Step a: preparation of 3-amino-2-cyclohexenone
Adding 1, 3-cyclohexanedione (0.03mol) and ammonium acetate (0.042mol) into a 100ml three-necked bottle, uniformly stirring, carrying out oil bath at 100 ℃ for 20min, removing the oil bath, naturally cooling, solidifying reaction liquid, cooling to room temperature, adding ethyl acetate (10ml), heating to dissolve, cooling to 0 ℃, filtering, drying filter cakes to obtain yellow crystals of 3-amino-2-cyclohexenone (the yield is 93.2%), and mp 132-133 ℃.
Step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Adding 3-amino-2-cyclohexenone (0.02mol) and acrylic acid (0.03mol) into a 100ml three-necked bottle, heating to reflux for 4H under stirring, cooling to room temperature, solidifying the reaction liquid to obtain a red brown solid, adding absolute ethyl alcohol (5ml), heating to dissolve, cooling to room temperature, filtering, and drying a filter cake to obtain a light yellow crystal of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (yield 89.1%) and mp 194-195 ℃.
Step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Adding 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (0.02mol) and cyclohexane (30ml) into a 100ml three-necked bottle, stirring at room temperature until the materials are dissolved, then placing the mixture into an ice bath, stirring for 10min, slowly dropwise adding a cyclohexane (5ml) solution of N-bromosuccinimide (0.04mol), heating and refluxing for 4H after the dropwise adding is finished, and cooling to room temperature after the TLC detection reaction is finished. Adding water (20ml), stirring, standing for layering, extracting a water layer with ethyl acetate (30ml multiplied by 3), combining organic layers, drying with anhydrous magnesium sulfate, filtering, evaporating the filtrate to remove the solvent, and drying the residue to obtain the off-white solid 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (the yield is 92.7%), mp 232-234 ℃, and the chromatographic purity of BP2013-HPLC is 94.3%.1H-NMR(400M,DMSO-d6): δ 9.93(s,1H), δ 9.46(s,1H), δ 6.9(t,1H), δ 6.43(d,1H), δ 6.31(d,1H), δ 2.77(t,2H), δ 2.38(t, 2H). It has been found that in this step, after the reaction reagent is N-bromosuccinimide and the solvent is changed to cyclohexane with low toxicity (the chloroform used in example 1 is a kind of solvent, and the toxicity is much higher than that of cyclohexane), the reaction yield of this step can be significantly improved, and the purity of the product can be significantly improved. However, in a supplementary experiment, this step c was repeated except that the cyclohexane was changed to an equivalent amount of chloroform, dichloromethane, 1, 2-dichloroethane, n-hexane, or cyclopentane, which are common solvents or even solvents having similar structure/properties, to give a productThe chromatographic purity of (a) is not more than 83%. In a further supplementary experiment, this step c was repeated, except that the N-bromosuccinimide was replaced with bromine, and the results showed a reaction yield of only 53.4% and a chromatographic purity of the product of no more than 75%, indicating that cyclohexane as a solvent with bromine as a reaction reagent was not desirable either in terms of reaction yield or product purity.
Step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
Dissolving 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (0.015mol) in 0.4mol/L sodium hydroxide solution (40ml), stirring for 10min, adding epoxy chloropropane (0.045mol), stirring at 50-55 ℃ for reacting for about 6H to generate a large amount of white floccule, cooling to room temperature after TLC detection reaction is finished, adding ethyl acetate (30ml), removing water at the lower layer by using a separating funnel, filtering, recrystallizing a filter cake by using acetone to obtain a white powdery solid which is 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (yield 63.1%), and mp 172-173 ℃.
Step e: preparation of carteolol hydrochloride
Adding 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (0.02mol) and tert-butylamine (0.08mol) into a 100ml flask, adding a mixed solution (40ml) of absolute ethanol/acetone, wherein the volume ratio of two solvents in the mixed solution is 10: 1, stirring and heating to reflux for 4 hours, detecting the reaction by TLC, removing ethanol and unreacted tert-butylamine by reduced pressure, adding acetone (30ml) into the residue, dissolving, adding 2mol/L hydrochloric acid (12ml), stirring for 10 minutes, filtering, removing the solvent by reduced pressure distillation from the filtrate, recrystallizing the residue with ethanol, filtering, and drying a filter cake to obtain white crystals of carteolol hydrochloride (5.28g, the yield of 80.5%), mp 277-278 ℃ and the purity of 99.2% (BP2013-HPLC method). 1H NMR (DMSO-d6) delta: 1.31(s,9H, CH3),2.40(t, J ═ 7.6Hz,2H, CH2),2.81 to 2.89(m,2H, CH2),2.92 to 2.96(m,1H, CH2),3.10 to 3.16(m,1H, CH2),3.99(s,1H, CH),4.00(s,1H, CH2),4.24 to 4.25(m,1H, CH2),6.50(d, J ═ 7.9Hz,1H, Ar-H),6.59(d, J ═ 8.5Hz,1H, Ar-H),7.07(t, J ═ 8.2Hz,1H, Ar-H),10.06(s,1H, NH),5.98(s,1H, active hydrogen), 9.12(s,1H, active hydrogen). EI-MS (m/z): 292(M +). It has been found that in this step, when a mixture of absolute ethanol/acetone is used as the reaction solvent, the resulting product has excellent stability, especially that characterized by the impurity H. However, when the reaction solvent is changed to absolute ethyl alcohol (without using acetone) or when the reaction solvent is changed to acetone (without using absolute ethyl alcohol) in step e of examples 2 to 6, and all the obtained bulk drugs are tested in test example 5 herein, the percentage increase of the impurity H of the bulk drugs prepared by these methods after being treated at a high temperature for 6 months is in the range of 264 to 316%; when the eye drops are prepared in composition example 1 by using the prepared crude drugs, the percentage increase of impurity H of the eye drops prepared by the methods after being treated at high temperature for 6 months is in the range of 277-353% when the eye drops prepared by the crude drugs are tested in test example 6.
Example 3: preparation of carteolol hydrochloride
Step a: preparation of 3-amino-2-cyclohexenone
Adding 1, 3-cyclohexanedione (0.03mol) and ammonium acetate (0.039mol) into a 100ml three-necked bottle, stirring uniformly, carrying out oil bath at 110 ℃ for reaction for 15min, removing the oil bath, naturally cooling, solidifying reaction liquid, cooling to room temperature, adding ethyl acetate (10ml), heating to dissolve, cooling to 0 ℃, filtering, drying filter cakes to obtain yellow crystals of 3-amino-2-cyclohexenone (the yield is 93.6%), and mp 132-133 ℃.
Step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Adding 3-amino-2-cyclohexenone (0.02mol) and acrylic acid (0.032mol) into a 100ml three-necked bottle, heating to reflux for 3H under stirring, cooling to room temperature, solidifying the reaction liquid to obtain a reddish brown solid, adding absolute ethyl alcohol (5ml), heating to dissolve, cooling to room temperature, filtering, and drying a filter cake to obtain a light yellow crystal of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (yield 89.4%), and mp 194-195 ℃.
Step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Adding 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (0.02mol) and cyclohexane (33ml) into a 100ml three-necked bottle, stirring at room temperature until the materials are dissolved, then placing the mixture into an ice bath, stirring for 10min, slowly dropwise adding a cyclohexane (5ml) solution of N-bromosuccinimide (0.04mol), heating and refluxing for 3H after the dropwise adding is finished, and cooling to room temperature after the TLC detection reaction is finished. Adding water (the amount of the water is 0.3 time of the volume of the reactant), stirring, standing for layering, extracting a water layer with ethyl acetate (the volume of the water layer is 2 times of the volume of the water layer for 2 times), combining organic layers, drying anhydrous calcium sulfate, filtering, evaporating the filtrate to remove the solvent, and drying the residue to obtain an off-white solid which is 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (the yield is 92.3%), mp 233-234 ℃, and the chromatographic purity of BP2013-HPLC is 94.7%.
Step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (0.015mol) is dissolved in 0.5mol/L sodium hydroxide solution (the amount of sodium hydroxide is 0.8 times by mol the amount of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, adding epoxy chloropropane (0.03mol), stirring at 50-55 ℃ for reaction for about 5H to generate a large amount of white floccule, cooling to room temperature after TLC detection reaction is finished, adding ethyl acetate (the amount of the ethyl acetate is 1 time of the volume of the reaction liquid), removing lower water by using a separating funnel, filtering, and recrystallizing a filter cake by using acetone to obtain a white powdery solid which is 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (the yield is 62.6 percent) and mp 172-173 ℃.
Step e: preparation of carteolol hydrochloride
Adding 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (0.02mol) and tert-butylamine (0.10mol) into a 100ml flask, adding an absolute ethanol/acetone mixed solution (40ml, the volume ratio of two solvents in the mixed solution is 9: 1), stirring and heating to reflux for 3H, detecting the reaction by TLC, evaporating ethanol and unreacted tert-butylamine under reduced pressure, adding acetone (the amount is 0.5 times of the volume of the reaction solution) into the residue, dissolving, adding 2.5mol/L hydrochloric acid (the addition amount is 1mol time of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, filtering, evaporating the filtrate under reduced pressure to remove the solvent, recrystallizing the residue with ethanol, filtering, drying a filter cake, the obtained white crystal is carteolol hydrochloride (yield is 82.3%), mp 277-278 ℃, and purity is 99.1% (BP2013-HPLC method).
Example 4: preparation of carteolol hydrochloride
Step a: preparation of 3-amino-2-cyclohexenone
Adding 1, 3-cyclohexanedione (0.03mol) and ammonium acetate (0.045mol) into a 100ml three-necked bottle, uniformly stirring, carrying out oil bath at 90 ℃ for reaction for 45min, removing the oil bath, naturally cooling, solidifying reaction liquid, cooling to room temperature, adding ethyl acetate (10ml), heating to dissolve, cooling to 0 ℃, filtering, drying filter cakes to obtain yellow crystals of 3-amino-2-cyclohexenone (yield 91.8%) and mp 131-132 ℃.
Step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Adding 3-amino-2-cyclohexenone (0.02mol) and acrylic acid (0.028mol) into a 100ml three-necked bottle, heating to reflux for 5H under stirring, cooling to room temperature, solidifying the reaction liquid to obtain a reddish brown solid, adding absolute ethyl alcohol (5ml), heating to dissolve, cooling to room temperature, filtering, and drying a filter cake to obtain a light yellow crystal of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (yield 88.7%), and mp 194-195 ℃.
Step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Adding 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (0.02mol) and cyclohexane (27ml) into a 100ml three-necked bottle, stirring at room temperature until the materials are dissolved, then placing the mixture into an ice bath, stirring for 10min, slowly dropwise adding a cyclohexane (5ml) solution of N-bromosuccinimide (0.04mol), heating and refluxing for 5H after the dropwise adding is finished, and cooling to room temperature after the TLC detection reaction is finished. Adding water (the amount of the water is 0.8 time of the volume of the reactant), stirring, standing for layering, extracting a water layer with ethyl acetate (the volume of the water layer is 1 time and 4 times of the volume of the water layer), combining organic layers, drying with anhydrous sodium sulfate, filtering, evaporating the filtrate to remove the solvent, and drying the residue to obtain an off-white solid, namely 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (the yield is 92.4%), mp 232-234 ℃, and the chromatographic purity of BP2013-HPLC is 94.1%.
Step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (0.015mol) is dissolved in 0.3mol/L sodium hydroxide solution (the amount of sodium hydroxide is 1.2 times by mol the amount of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, adding epoxy chloropropane (0.06mol), stirring at 55-60 ℃ for reaction for about 7H to generate a large amount of white floccule, cooling to room temperature after TLC detection reaction is finished, adding ethyl acetate (the amount of the ethyl acetate is 0.5 times of the volume of the reaction liquid), removing lower layer water by using a separating funnel, filtering, and recrystallizing a filter cake by using acetone to obtain a white powdery solid which is 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (the yield is 63.6 percent) and mp-174 ℃.
Step e: preparation of carteolol hydrochloride
Adding 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (0.02mol) and tert-butylamine (0.06mol) into a 100ml flask, adding an anhydrous ethanol/acetone mixed solution (40ml, the volume ratio of two solvents in the mixed solution is 11: 1), stirring and heating to reflux for 6H, detecting the reaction by TLC, evaporating ethanol and unreacted tert-butylamine under reduced pressure, adding acetone (the amount is 1 time of the volume of the reaction solution) into the residue, dissolving, adding 1.5mol/L hydrochloric acid (the addition amount is 1.5mol times of that of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, filtering, evaporating the filtrate under reduced pressure to remove the solvent, recrystallizing the residue with ethanol, filtering, drying a filter cake, the obtained white crystal is carteolol hydrochloride (yield is 84.3%), mp 277-278 ℃, and purity is 99.4% (BP2013-HPLC method).
Example 5: preparation of carteolol hydrochloride
Step a: preparation of 3-amino-2-cyclohexenone
Adding 1, 3-cyclohexanedione (0.03mol) and ammonium acetate (0.049mol) into a 100ml three-necked bottle, uniformly stirring, carrying out oil bath at 80 ℃ for reaction for 60min, removing the oil bath, naturally cooling, solidifying reaction liquid, cooling to room temperature, adding ethyl acetate (10ml), heating to dissolve, cooling to 0 ℃, filtering, drying filter cakes to obtain yellow crystals, namely 3-amino-2-cyclohexenone (the yield is 93.2%) and mp 132-133 ℃.
Step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Adding 3-amino-2-cyclohexenone (0.02mol) and acrylic acid (0.024mol) into a 100ml three-necked bottle, heating to reflux for 6H under stirring, cooling to room temperature, solidifying the reaction liquid to obtain a reddish brown solid, adding absolute ethyl alcohol (5ml), heating to dissolve, cooling to room temperature, filtering, and drying a filter cake to obtain a pale yellow crystal of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (yield 90.2%), and mp 194-195 ℃.
Step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Adding 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (0.02mol) and cyclohexane (25ml) into a 100ml three-necked bottle, stirring at room temperature until the materials are dissolved, then placing the mixture into an ice bath, stirring for 10min, slowly dropwise adding a cyclohexane (5ml) solution of N-bromosuccinimide (0.03mol), heating and refluxing for 3H after the dropwise adding is finished, and cooling to room temperature after the TLC detection reaction is finished. Adding water (the amount of the water is 0.7 time of the volume of the reactant), stirring, standing for layering, extracting a water layer with ethyl acetate (the volume of the water layer is 1 time and 4 times of the volume of the water layer), combining organic layers, drying anhydrous calcium sulfate, filtering, evaporating the filtrate to remove the solvent, and drying the residue to obtain an off-white solid which is 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (the yield is 92.7%), mp 233-234 ℃, and the chromatographic purity of BP2013-HPLC is 94.3%.
Step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (0.015mol) is dissolved in 0.2mol/L sodium hydroxide solution (the amount of sodium hydroxide is 0.5 times by mol the amount of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, adding epoxy chloropropane (0.03mol), stirring at 55-60 ℃ for reaction for about 4H to generate a large amount of white floccule, cooling to room temperature after TLC detection reaction is finished, adding ethyl acetate (the amount of the ethyl acetate is 1 time of the volume of the reaction liquid), removing lower water by using a separating funnel, filtering, and recrystallizing a filter cake by using acetone to obtain a white powdery solid which is 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (the yield is 63.6 percent) and mp 173-174 ℃.
Step e: preparation of carteolol hydrochloride
Adding 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (0.02mol) and tert-butylamine (0.06mol) into a 100ml flask, adding an anhydrous ethanol/acetone mixed solution (40ml, the volume ratio of two solvents in the mixed solution is 12: 1), stirring and heating to reflux for 8H, detecting the reaction by TLC, evaporating ethanol and unreacted tert-butylamine under reduced pressure, adding acetone (1 time of the volume of the reaction solution) into the residue, dissolving, adding 1mol/L hydrochloric acid (the adding amount of the hydrochloric acid is 1.5 times of the volume of the 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, filtering, evaporating the filtrate under reduced pressure to remove the solvent, recrystallizing the residue with ethanol, filtering, drying a filter cake, the obtained white crystal is carteolol hydrochloride (yield is 84.7%), mp 277-278 ℃, and purity is 99.6% (BP2013-HPLC method).
Example 6: preparation of carteolol hydrochloride
Step a: preparation of 3-amino-2-cyclohexenone
Adding 1, 3-cyclohexanedione (0.03mol) and ammonium acetate (0.036mol) into a 100ml three-necked bottle, stirring uniformly, carrying out oil bath at 120 ℃ for reaction for 10min, removing the oil bath, naturally cooling, solidifying reaction liquid, cooling to room temperature, adding ethyl acetate (10ml), heating to dissolve, cooling to 0 ℃, filtering, drying filter cakes to obtain yellow crystals of 3-amino-2-cyclohexenone (yield 92.3%) and mp 131-133 ℃.
Step b: preparation of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione
Adding 3-amino-2-cyclohexenone (0.02mol) and acrylic acid (0.036mol) into a 100ml three-necked bottle, heating to reflux for 3H under stirring, cooling to room temperature, solidifying the reaction liquid to obtain a reddish brown solid, adding absolute ethyl alcohol (5ml), heating to dissolve, cooling to room temperature, filtering, and drying a filter cake to obtain a light yellow crystal of 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (yield 88.3%), and mp 194-195 ℃.
Step c: preparation of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolones
Adding 3,4,7, 8-tetrahydro-2, 5(1H,6H) -quinolinedione (0.02mol) and cyclohexane (35ml) into a 100ml three-necked bottle, stirring at room temperature until the materials are dissolved, then placing the mixture into an ice bath, stirring for 10min, slowly dropwise adding a cyclohexane (5ml) solution of N-bromosuccinimide (0.05mol), heating and refluxing for 6H after the dropwise adding is finished, and cooling to room temperature after the TLC detection reaction is finished. Adding water (the amount of the water is 0.4 time of the volume of the reactant), stirring, standing for layering, extracting a water layer with ethyl acetate (the volume of the water layer is 2 times of the volume of the water layer for 2 times), combining organic layers, drying with anhydrous sodium sulfate, filtering, evaporating the filtrate to remove the solvent, and drying the residue to obtain an off-white solid which is 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (the yield is 92.2%), mp 233-234 ℃, and the chromatographic purity of BP2013-HPLC (high performance liquid chromatography) is 94.4%.
Step d: preparation of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone
5-hydroxy-3, 4-dihydro-2 (1H) -quinolone (0.015mol) is dissolved in 0.6mol/L sodium hydroxide solution (the amount of sodium hydroxide is 1.5 times by mol the amount of 5-hydroxy-3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, adding epoxy chloropropane (0.06mol), stirring at 40-45 ℃ for about 8H to generate a large amount of white floccule, cooling to room temperature after TLC detection reaction is finished, adding ethyl acetate (the amount of the ethyl acetate is 0.5 times of the volume of the reaction liquid), removing lower layer water by using a separating funnel, filtering, and recrystallizing a filter cake by using acetone to obtain a white powdery solid which is 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (the yield is 63.3 percent) and mp-173 ℃.
Step e: preparation of carteolol hydrochloride
Adding 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone (0.02mol) and tert-butylamine (0.12mol) into a 100ml flask, adding an anhydrous ethanol/acetone mixed solution (40ml, the volume ratio of two solvents in the mixed solution is 8: 1), stirring and heating to reflux for 2H, detecting the reaction by TLC, evaporating ethanol and unreacted tert-butylamine under reduced pressure, adding acetone (the amount is 0.5 times of the volume of the reaction solution) into the residue, dissolving, adding 3mol/L hydrochloric acid (the addition amount is 1 times of the mole of 5- (2, 3-epoxypropoxy) -3, 4-dihydro-2 (1H) -quinolone), stirring for 10min, filtering, evaporating the filtrate under reduced pressure to remove the solvent, recrystallizing the residue with ethanol, filtering, drying a filter cake, the obtained white crystal is carteolol hydrochloride (yield is 84.1%), mp 277-279 ℃ and purity is 99.3% (BP2013-HPLC method).
Second, test example
Test example 1: the crude drug prepared by the invention is measured according to each item in carteolol hydrochloride carried on page 903 of the second part of the 2015 version in Chinese pharmacopoeia. As a result: each batch of raw material medicines obtained in examples 1 to 6 meets the specification of the standard. This shows that the finished product of the raw material medicine prepared by different processes has no difference in quality index.
Test example 2: the content of the carteolol hydrochloride eye drops prepared by the invention is measured according to each item in carteolol hydrochloride eye drops carried on page 903 of the second part of the 2015 version in Chinese pharmacopoeia. As a result: the eye drops of composition examples 1 to 7 and 8 all met the standard, and there was no significant difference between the eye drops of composition example 8 and the corresponding eye drops of composition examples 1 to 7, which used different raw materials but had the same formulation method. This shows that the eye drop finished products prepared by using different raw material medicaments have no difference in quality index.
Test example 3: relevant substances of the bulk drugs obtained in the embodiments 1 to 6 of the invention are measured by a BP2013-HPLC method carried by the invention, and the results show that the bulk drugs have no obvious difference in three aspects of impurities H, unknown impurities and total impurities, for example, the content of the impurity H is in the range of 0.02-0.04%. This indicates that the finished product of the raw material medicine prepared by different processes has no difference in the aspect of impurity indexes.
Test example 4: according to BP2013-HPLC method carried by the invention, related substances of each batch of eye drops obtained in composition examples 1-7 and 8 are measured, and the results show that each batch of eye drops have no obvious difference in three aspects of impurities H, unknown impurities and total impurities, for example, the content of the impurity H is in the range of 0.03-0.06%. This indicates that the eye drop finished products prepared by using different raw material medicaments have no difference in the aspect of impurity indexes.
Test example 5 (stability test): the batches of the raw material medicines obtained in the embodiments 1 to 6 of the invention are respectively sealed and packaged, and are placed at the temperature of 40 ℃ for 6 months, the indexes of the raw material medicines at 0 month and 6 months are measured according to the method of the test example 1 of the invention, and the impurity indexes of the raw material medicines at 0 month and 6 months are measured according to the method of the test example 3 of the invention. As a result: for each index measured by the method of test example 1, the result at 6 months is not obviously different from the result at 0 months, and the different batches are not obviously different, for example, the detection results of the two batches of raw material medicines of example 1 and example 2 are not obviously different at 0 months and 6 months, and the detection results of the two batches of raw material medicines of example 1 and example 2 are not obviously different at the same time, for example, at 6 months; for each index measured according to test example 3, a significant difference is shown in the aspect of the impurity H (calculation results show that the total impurities may also be significantly different), specifically, the impurity H of each batch of raw material medicines obtained in examples 2 to 6 is not significantly increased at 6 months compared with 0 month, but the impurity H of the raw material medicine obtained in example 1 is significantly increased at 6 months compared with 0 month; the percentage increase of the impurity H after the raw material of example 1 is treated at a high temperature for 6 months is 283%, and the percentage increase of the impurity H after the raw material of each batch of raw material medicines obtained in examples 2 to 6 are treated at a high temperature for 6 months is 44 to 61%.
The percentage increase of the impurity H is [ (content of the impurity H at 6 months-content of the impurity H at 0 months) ÷ content of the impurity H at 0 months ]. times 100%
Test example 6 (stability test): each of the eye drops obtained in composition examples 1 to 8 of the present invention was hermetically packaged, and left at 40 ℃ for 6 months, and each of the indices at 0 month and at 6 months of each of the preparations was measured by the method of test example 2 of the present invention, and each of the indices of impurities at 0 month and at 6 months of each of the preparations was measured by the method of test example 4 of the present invention. As a result: for each index measured according to the method of test example 2, the result at 6 months is not significantly different from the result at 0 month, and there is no significant difference between different batches, for example, there is no significant difference between the test results at 0 month and 6 months between two batches of eye drops of the formulations prepared according to composition example 1 in composition example 1 and composition example 8, and there is no significant difference between the test results at the same time, for example, at 6 months, between two batches of eye drops of the formulations prepared according to composition example 1 in composition example 1 and composition example 8; for each index measured according to test example 4, a significant difference was observed in the impurity H (calculation results showed that total impurities may be significantly different from each other), specifically, the impurity H was not significantly increased at 6 months compared to 0 months in each of the eye drops obtained in composition examples 1 to 7, but was significantly increased at 6 months compared to 0 months in seven eye drops obtained in composition example 8; the percentage increase of the impurity H in the seven batches of eye drops obtained in composition example 8 after being treated at high temperature for 6 months is 276-347%, and the percentage increase of the impurity H in each batch of eye drops obtained in composition examples 1-7 after being treated at high temperature for 6 months is 58-74%. The percentage increase of impurity H was calculated in the same manner as in test example 5.
Third, composition examples section
The following composition examples 1 to 7 were prepared using carteolol hydrochloride obtained in example 2 of the present invention as a raw material drug.
Composition example 1:
preparing carteolol hydrochloride eye drops comprising the following components according to a conventional method:
carteolol hydrochloride 2.0g,
Sorbic acid 0.1g,
0.1g of sodium dihydrogen phosphate,
0.5g of sodium chloride,
0.005g of benzalkonium chloride,
Proper amount of sodium hydroxide,
Sterilizing and refining water to 100ml,
pH 7.0。
Composition example 2:
preparing carteolol hydrochloride eye drops comprising the following components according to a conventional method:
2g of carteolol hydrochloride,
Sorbic acid 0.1g,
0.2g of sodium dihydrogen phosphate,
0.43g of sodium chloride,
0.005g of benzalkonium chloride,
Proper amount of sodium hydroxide,
Sterilizing and refining water to 100ml,
pH 7.0。
Composition example 3:
preparing carteolol hydrochloride eye drops comprising the following components according to a conventional method:
1g of carteolol hydrochloride,
Sorbic acid 0.1g,
0.4g of sodium dihydrogen phosphate,
0.31g of sodium nitride,
0.005g of benzalkonium chloride,
Proper amount of sodium hydroxide,
Sterilizing and refining water to 100ml,
pH 6.5。
Composition example 4:
preparing carteolol hydrochloride eye drops comprising the following components according to a conventional method:
2g of carteolol hydrochloride,
Sorbic acid 0.15g,
0.lg of sodium dihydrogen phosphate,
0.47g of sodium chloride,
0.005g of benzalkonium chloride,
Proper amount of sodium hydroxide,
The sterilization refined water is added to 100ml,
pH 6.5。
Composition example 5:
preparing carteolol hydrochloride eye drops comprising the following components according to a conventional method:
2g of carteolol hydrochloride,
Sorbic acid 0.2g,
0.1g of sodium dihydrogen phosphate,
0.45g of sodium chloride,
0.005g of benzalkonium chloride,
Proper amount of sodium hydroxide,
Sterilizing and refining water to 100ml,
pH 7.0。
Composition example 6:
preparing carteolol hydrochloride eye drops comprising the following components according to a conventional method:
carteolol hydrochloride 0.5g,
Sorbic acid 0.17g,
0.6g of sodium dihydrogen phosphate,
0.16g of sodium chloride,
0.005g of benzalkonium chloride,
Proper amount of sodium hydroxide,
The sterilization refined water is added to 100ml,
pH 7.0。
Composition example 7:
preparing carteolol hydrochloride eye drops comprising the following components according to a conventional method:
2g of carteolol hydrochloride,
Glycerol: 1.1g
0.1g of sodium dihydrogen phosphate,
0.005g of benzalkonium chloride,
Proper amount of sodium hydroxide,
Sterilizing and refining water to 100ml,
pH 7.0。
Composition example 8:
referring to the formula and the preparation method of composition examples 1-7 respectively, except that the raw material medicines are changed into the raw material medicines obtained in example 1, 7 batches of eye drops are obtained.
The above-mentioned embodiments are merely preferred embodiments for fully illustrating the present invention, and the scope of the present invention is not limited thereto. The equivalent substitution or change made by the technical personnel in the technical field on the basis of the invention is all within the protection scope of the invention. The protection scope of the invention is subject to the claims.