CN106256824B - Preparation method of high-purity delafloxacin meglumine salt - Google Patents
Preparation method of high-purity delafloxacin meglumine salt Download PDFInfo
- Publication number
- CN106256824B CN106256824B CN201510339691.2A CN201510339691A CN106256824B CN 106256824 B CN106256824 B CN 106256824B CN 201510339691 A CN201510339691 A CN 201510339691A CN 106256824 B CN106256824 B CN 106256824B
- Authority
- CN
- China
- Prior art keywords
- delafloxacin
- formula
- compound
- purity
- meglumine salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Abstract
The invention relates to a refining method of a delafloxacin intermediate compound shown in a formula 1, which comprises the steps of dissolving the compound shown in the formula 1 in a good solvent, mixing with a poor solvent, heating, stirring, cooling, crystallizing and separating crystals. Hydrolyzing the refined compound shown in the formula 1 to obtain high-purity delafloxacin, and reacting the high-purity delafloxacin with meglumine to obtain delafloxacin meglumine salt, wherein the purity of the delafloxacin meglumine salt is more than 99.5%. The method can effectively remove impurities, simultaneously introduces few solvents and reagents, has high yield, is simple in technological process operation, and effectively reduces the preparation cost.
Description
Technical Field
The invention belongs to the field of pharmaceutical chemistry, and particularly relates to a refining method of a delafloxacin intermediate and a preparation method of delafloxacin meglumine salt.
Background
Delafloxacin (Delafloxacin), chemical name: 1- (6-amino-3, 5-difluoropyridin-2-yl) -8-chloro-6-fluoro-7- (3-hydroxyazetidin-1-yl) -4-oxo-1, 4-dihydroquinoline-3-carboxylic acid, having the structural formula 2, the corresponding meglumine salt thereof having the structure of formula 3:
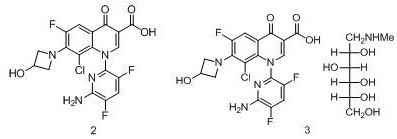
delafloxacin, a fluoroquinolone compound developed by Wakunaga pharmaceutical ltd, japan, is currently developed by Melinta therapeutic corporation to obtain the QIDP qualification of the FDA in the united states, and is in the phase III clinical study. The action mechanism of the compound is the same as other fluoroquinolones, the compound acts on bacterial DNA gyrase and topoisomerase IV, the prominent enzyme inhibiting activity can reduce the selectivity of bacterial drug resistance mutation, and the compound is expected to become a candidate drug for treating respiratory tract and urinary tract infection, Acute Bacterial Skin and Skin Structure Infection (ABSSSI) and other diseases.
WO9711068 and WO2006015194 disclose methods for the synthesis of delafloxacin, mainly two methods:
method one (WO 9711068):
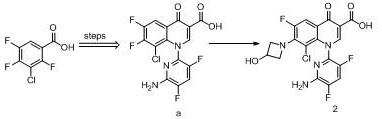
the method uses 2,4, 5-trifluoro, 2-chlorobenzoic acid as a starting material, and obtains a compound a through multi-step reaction, and the compound a is condensed with azetidine to obtain delafloxacin.
Method two (WO2006015194):

the method takes 2,4, 5-trifluorobenzoic acid as an initial material, prepares a compound 1 through multi-step reaction, hydrolyzes to obtain delafloxacin, and finally forms delafloxacin meglumine salt with meglumine.
The delafloxacin meglumine salt has long synthesis steps and more impurities, so the conventional method is difficult to meet the requirement of medicinal grade. The preparation methods of the two process routes disclosed above do not refer to the purification of intermediates and the purification of finished products, and the purity of the obtained finished products can hardly meet the requirement of pharmaceutical grade.
WO2001034595 discloses a delafloxacin refining method, which comprises reacting delafloxacin with alkali metal hydroxide such as lithium hydroxide to generate alkali metal salt, acidifying to obtain delafloxacin, and refining. CN201310021838.4 discloses an improved purification method, which is characterized in that a high-purity delafloxacin alkali metal salt solid compound is prepared by preparing an ester compound before the carboxyl of delafloxacin is not hydrolyzed, directly reacting with alkali metal, and then acidifying to prepare high-purity delafloxacin. However, the method always needs to introduce strong base and acid, the whole operation is still complicated, the risk of introducing residues is high, and meanwhile, the yield is still low due to the two acid-base processes, so that the method is not beneficial to industrial scale-up production.
CN201310124425.9 discloses another refining method of delafloxacin. The delafloxacin is added into an aprotic solvent, water is added, and the mixture is heated, stirred and recrystallized, so that the delafloxacin with high purity is obtained. The solvent introduced by the method, such as N-N dimethylacetamide, has high boiling point, is not easy to remove completely, has the risk of solvent residue, and has higher temperature, high energy consumption and low yield.
Although the above 3 methods obtain medicinal grade delafloxacin meglumine salt by refining delafloxacin, the method still has the defects of low yield, residual solvent, high energy consumption and unsuitability for industrial production, and therefore, a more optimized method is required to obtain the medicinal grade delafloxacin meglumine salt.
Disclosure of Invention
The invention aims to obtain the high-purity delafloxacin meglumine salt, the delafloxacin meglumine salt obtained by the method has high yield and high purity, meets the medicinal requirements, and the method is simple and economic and is suitable for industrial production.
The inventor of the present invention conducted research on delafloxacin, and found that delafloxacin has poor solubility, and has low solubility or insolubility in common solvents such as alcohols, esters, and aromatic alkanes except for good solubility in a few polar aprotic solvents such as DMF, NMP, and the like. Meanwhile, systematic research is also carried out on the compound of the formula 1 before hydrolysis of delafloxacin, and the compound 1 is found to have better solubility in alcohols, esters, acetonitrile, acetone and other common solvents. The inventor surprisingly discovers that the compound shown in the formula 1 is refined, hydrolyzed to obtain the delafloxacin and then converted into delafloxacin meglumine salt, wherein the delafloxacin can be used for obtaining the delafloxacin meglumine salt with high purity without purification, and the medicinal requirement is met.
To achieve the object of the present invention, the following embodiments are provided.
In one embodiment, the present invention provides a method for purifying a compound of formula 1, comprising:
comprises dissolving the compound of formula 1 in a good solvent, mixing with a poor solvent, heating and stirring, cooling and crystallizing, separating out crystals,
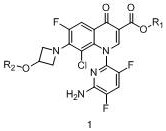
in the formula 1, R1 is alkyl, and R2 is alkanoyl.
In the above embodiments, the process of the present invention, wherein R1 is a C1 to C4 alkyl group, preferably ethyl, and R2 is a C1 to C6 alkanoyl group, preferably isobutyryl.
In the above embodiments, the good solvent of the method of the present invention is selected from one or more of methanol, ethanol, isopropanol, n-butanol, acetonitrile, tetrahydrofuran, acetone, ethyl acetate, methyl acetate, n-butyl acetate, and isopropyl acetate, preferably methanol, ethanol, or ethyl acetate; the poor solvent is selected from one or more of water, petroleum ether, n-hexane, cyclohexane and n-heptane, preferably water and petroleum ether, and the mass volume ratio of the compound shown in the formula 1 to the good solvent is 1: 1-1: 10g/ml, preferably 1: 2-1: 6 g/ml; the mass-to-volume ratio of the compound of formula 1 to the poor solvent is 1: 0-1: 10g/ml, preferably. The content of the water is 1: 2-1: 5 g/ml.
In the above embodiment, the solution system is in a clear or non-clear state during heating and stirring in the method of the present invention. The heating temperature is 30-100 ℃, preferably 40-70 ℃, and the heating and stirring time is 0.5-5 h.
In a specific embodiment, the method for purifying the compound of formula 1 of the present invention comprises adding a proper amount of a good solvent and a poor solvent to the compound of formula 1, heating and stirring for 0.5 to 5 hours, gradually cooling and crystallizing, and separating a solid.

In the compound of formula 1, R1 is alkyl, preferably C1-C4 alkyl, more preferably ethyl, R2 is alkanoyl, preferably C1-C6 alkanoyl, more preferably isobutyryl.
In the above embodiments, the good solvent is selected from one or more of methanol, ethanol, isopropanol, n-butanol, acetonitrile, tetrahydrofuran, acetone, ethyl acetate, methyl acetate, n-butyl acetate, and isopropyl acetate, preferably methanol, ethanol, or ethyl acetate; the poor solvent is selected from one or more of water, petroleum ether, n-hexane, cyclohexane and n-heptane, and is preferably water or petroleum ether. The mass-volume ratio of the compound of the formula 1 to the good solvent is 1: 1-1: 10g/ml, preferably 1: 2-1: 6 g/ml; the mass-to-volume ratio of the compound of formula 1 to the poor solvent is 1: 0-1: 10g/ml, preferably 1: 2-1: 5 g/ml. The heating temperature is 30-100 ℃, and preferably 40-70 ℃. The heating and stirring time is 1-3 h. The cooling crystallization time is 1-20 h, preferably 3-10 h.
Herein, the compound of formula 1 is a known compound, and can be prepared by the following technical scheme.
According to the method disclosed in WO2006015194 (incorporated by reference in its entirety), the compound of formula 1 can be prepared as follows:
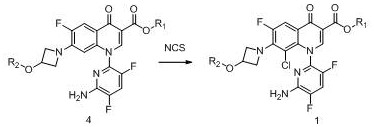
in another embodiment, the present invention provides a method for preparing delafloxacin meglumine salt comprising the steps of:
1) dissolving the compound of formula 1 in a good solvent, mixing with a poor solvent, heating, stirring, cooling, crystallizing, and separating out crystals to obtain the compound of formula 1 with high purity;
2) hydrolyzing the high-purity compound shown in the formula 1 to obtain delafloxacin;
3) and (3) reacting the delafloxacin obtained in the step (a) with meglumine to obtain delafloxacin meglumine salt.
The specific reaction formula is as follows:
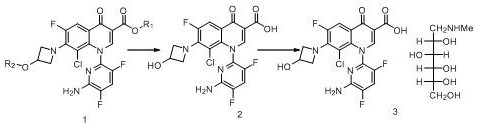
preferably, the method for preparing delafloxacin meglumine salt comprises the steps of dissolving the compound shown in the formula 1 obtained by refining by the method in the invention in an alcohol solution, adding a strong base solution, acidifying and crystallizing to obtain delafloxacin, and salifying the delafloxacin and meglumine in water to obtain the delafloxacin meglumine salt.
The invention has the following positive effects:
if the existing preparation method of delafloxacin is not refined, the prepared delafloxacin has low purity and is difficult to meet the medicinal requirements. In the disclosed refining method, the meglumine salt meets the requirement of medicinal grade mainly by purifying delafloxacin, and has the defects of low yield and the conditions and processes related to acid-base, high boiling point solvent, high-temperature recrystallization and the like. The method overcomes the defects, the compound with better solubility in the formula 1 is refined, the yield is more than 90%, the high-purity delafloxacin (the purity is more than 99.0%) is obtained by hydrolysis, the obtained delafloxacin can be directly salified with meglumine without refining to obtain the high-purity delafloxacin meglumine salt (the purity is more than 99.5%), the yield is more than 90%, the refining effect is obvious, the operation is simple, a high-boiling-point solvent and strong acid and strong base are avoided, the yield is high, and the method is suitable for large-scale industrial production.
Detailed Description
The present invention will be further described with reference to the following examples, which will enable those skilled in the art to more fully understand the present invention, but which are not intended to limit the scope of the present invention in any way.
Example 1 preparation of a compound of formula 1 wherein R1 is ethyl and R2 is isobutyryl, the reaction is as follows:
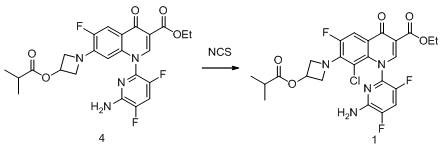
in a 100L reactor, 5.0Kg (9.9mol) of the compound of formula 4, 25Kg of ethyl acetate and 500g of sulfuric acid were added in this order, stirred at room temperature, and a solution of 1.7Kg of NCS (12.8mol) dissolved in 25Kg of methyl acetate was slowly added dropwise. After the dropwise addition, the mixture is stirred for 5 hours at room temperature, and then washed by sodium bicarbonate solution, sodium sulfite solution and saturated saline solution respectively, and the organic phase is concentrated to dryness to obtain 5.0Kg of crude compound of the formula 1 with the purity of 97 percent.
EXAMPLE 2 purification of Compound of formula 1
Adding 1Kg of the crude compound of formula 1 prepared in example 1 into a 20L reaction flask, adding 2L of methanol and 3L of water, heating to 50 ℃, stirring for 1 hour, then turning off the heating, slowly cooling to room temperature, crystallizing in ice-water bath for 5 hours, performing suction filtration, washing a filter cake with a small amount of cold methanol, draining, and drying the filter cake under reduced pressure to constant weight to obtain 930g of light yellow powder with yield of 93%, HPLC purity: 99.1 percent.
EXAMPLE 3 purification of Compound of formula 1
Adding 1Kg of the crude compound of formula 1 prepared in example 1 into a 20L reaction flask, adding 6L of ethanol and 2L of water, heating to 60 ℃, stirring for 1 hour, then turning off the heating, slowly cooling to room temperature, crystallizing in ice-water bath for 7 hours, performing suction filtration, washing a filter cake with a small amount of cold ethanol, draining, and drying the filter cake under reduced pressure to constant weight to obtain 900g of light yellow powder with yield of 90%, HPLC purity: 99.3 percent.
Example 4 purification of Compound of formula 1
Adding 1Kg of crude compound of formula 1 prepared in example 1 into a 20L reaction flask, adding 5L of ethyl acetate, heating to 60 ℃, stirring to dissolve, adding 5L of petroleum ether, closing and heating, slowly cooling the system to room temperature, crystallizing in ice-water bath for 8h, performing suction filtration, washing the filter cake with a small amount of cold ethyl acetate, performing suction drying, and drying the filter cake under reduced pressure to constant weight to obtain 840g of light yellow powder, the yield is 84%, and the HPLC purity: 99.5 percent.
Example 5 preparation of delafloxacin
500g of the refined product of the compound 1 of the formula 1 obtained in example 2 was put into a 20L reaction flask, 10L of isopropyl alcohol was added, the mixture was dissolved by stirring, 4L of a sodium hydroxide solution having a mass fraction of 4% was slowly dropped, and 5L of an acetic acid solution having a mass fraction of 12% was dropped after stirring until hydrolysis was completed. After full crystallization, the solid is filtered, washed by water and dried under reduced pressure to obtain 384g of light yellow powder, the yield is 94 percent, and the purity is 99.3 percent.
Example 6 preparation of delafloxacin
500g of the refined product of the compound of formula 1 obtained in example 4 was put into a 20L reaction flask, 10L of isopropyl alcohol was added, and dissolved by stirring, 4L of a sodium hydroxide solution having a mass fraction of 4% was slowly dropped, and 5L of an acetic acid solution having a mass fraction of 12% was dropped after stirring until hydrolysis was completed. After full crystallization, the solid is filtered, washed by water and dried under reduced pressure to obtain 380g of light yellow powder, the yield is 93 percent and the purity is 99.6 percent.
Comparative example 1 preparation of delafloxacin
500g of the crude compound of the formula 1 prepared in example 1 is added into a 20L reaction bottle, 10L of isopropanol is added, stirred and dissolved, 4L of sodium hydroxide solution with the mass fraction of 4% is slowly dripped, and 5L of acetic acid solution with the mass fraction of 12% is dripped after stirring until hydrolysis is completed. After full crystallization, the solid is filtered, washed with water and dried under reduced pressure to obtain 375g of light yellow powder, the yield is 92 percent, and the purity is 97.9 percent
Example 7 preparation of delafloxacin meglumine salt
Adding 200g of delafloxacin prepared in example 5 and 120g of meglumine into a 2L reaction bottle, adding 1L of water, stirring, heating for dissolving, slowly cooling, fully crystallizing, filtering, washing with cold water, and drying to obtain 237g of yellowish powder, wherein the yield is 82% and the HPLC purity is 99.7%.
Example 8 preparation of delafloxacin meglumine salt
200g of delafloxacin prepared in example 6 and 120g of meglumine are added into a 2L reaction bottle, 1L of water is added, stirring and heating are carried out, the temperature is slowly reduced and the mixture is cooled after the mixture is dissolved and cleared, crystallization is fully carried out, then the mixture is filtered, washed by cold water and dried to obtain 237g of yellowish powder, the yield is 82 percent, and the HPLC purity is 99.8 percent.
Comparative example 2 preparation of delafloxacin meglumine salt
Adding 200g of delafloxacin prepared in the comparative example 1 and 120g of meglumine into a 2L reaction bottle, adding 1L of water, stirring, heating for dissolving, slowly cooling, fully crystallizing, filtering, washing with cold water, and drying to obtain 240g of yellowish powder, wherein the yield is 83% and the HPLC purity is 98.6%.
Claims (7)
1. A refining method of delafloxacin intermediate compound shown in formula I comprises the steps of dissolving the compound shown in formula I in a good solvent, mixing the mixture with a poor solvent, heating, stirring, cooling, crystallizing, and separating out crystals, wherein in the formula I, R1 is ethyl, R2 is isobutyryl, wherein the good solvent is one or more selected from methanol, ethanol and ethyl acetate, the poor solvent is one or more selected from water and petroleum ether,
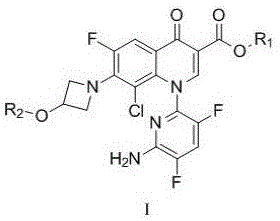
2. the method according to claim 1, wherein the solution is in a clear or non-clear state when heated and stirred.
3. The method as claimed in claim 1, wherein the mass-to-volume ratio of the compound of formula I to the good solvent is 1: 2-1: 6 g/ml.
4. The method according to claim 1, wherein the mass-to-volume ratio of the compound of formula I to the poor solvent is 1:2 to 1:5 g/m.
5. The method according to claim 1, wherein the heating temperature is 40 to 70 ℃.
6. The method of claim 1, wherein the heating and stirring time is 0.5-5 h.
7. A method of preparing delafloxacin meglumine salt comprising the steps of:
1) purifying the compound of formula I according to the process of claims 1-6 to obtain a purified compound of formula I;
2) hydrolyzing the purified compound of the formula I to obtain delafloxacin;
3) and (3) reacting the delafloxacin obtained in the previous step with meglumine to obtain delafloxacin meglumine salt.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510339691.2A CN106256824B (en) | 2015-06-18 | 2015-06-18 | Preparation method of high-purity delafloxacin meglumine salt |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510339691.2A CN106256824B (en) | 2015-06-18 | 2015-06-18 | Preparation method of high-purity delafloxacin meglumine salt |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN106256824A CN106256824A (en) | 2016-12-28 |
| CN106256824B true CN106256824B (en) | 2020-10-27 |
Family
ID=57713827
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201510339691.2A Active CN106256824B (en) | 2015-06-18 | 2015-06-18 | Preparation method of high-purity delafloxacin meglumine salt |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN106256824B (en) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106831723B (en) * | 2017-02-15 | 2020-07-28 | 鲁南制药集团股份有限公司 | Improved refining method of delafloxacin |
| CN110467600A (en) * | 2018-05-10 | 2019-11-19 | 上海度德医药科技有限公司 | A kind of De Lasha star meglumine salt crystal form L and preparation method thereof |
| CN111718330A (en) * | 2019-03-23 | 2020-09-29 | 南京海润医药有限公司 | Delafloxacin impurity III and product refining method |
| CN111718329A (en) * | 2019-03-23 | 2020-09-29 | 南京海润医药有限公司 | Delafloxacin impurity IV and product refining method |
| CN111718331A (en) * | 2019-03-23 | 2020-09-29 | 南京海润医药有限公司 | Impurity I and II of delafloxacin and product refining method |
| CN113527262B (en) * | 2021-06-22 | 2022-07-15 | 安徽普利药业有限公司 | Refining method of delafloxacin and meglumine salt thereof |
| CN116514775A (en) * | 2022-01-20 | 2023-08-01 | 海南普利制药股份有限公司 | New crystal form of meglumine salt of DELASHANXIA and preparation method thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6136823A (en) * | 1996-11-28 | 2000-10-24 | Wakunaga Pharmaceutical Co., Ltd. | Pyridonecarboxylic acid derivatives or salts thereof and drugs containing the same as the active ingredient |
| WO2006015194A2 (en) * | 2004-07-30 | 2006-02-09 | Abbott Laboratories | Preparation of pyridonecarboxylic acid antibacterials |
| CN102164912A (en) * | 2008-09-24 | 2011-08-24 | Rib-X制药有限公司 | Process for making quinolone compounds |
| CN103936717A (en) * | 2013-01-22 | 2014-07-23 | 上海医药工业研究院 | Intermediate of delafloxacin and preparation method thereof |
| CN104098548A (en) * | 2013-04-11 | 2014-10-15 | 上海医药工业研究院 | Delafloxacin purifying method |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL3056492T3 (en) * | 2004-10-08 | 2022-03-21 | Abbvie Inc. | Meglumine salt and crystalline forms thereof of a drug (delafloxacin) |
-
2015
- 2015-06-18 CN CN201510339691.2A patent/CN106256824B/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6136823A (en) * | 1996-11-28 | 2000-10-24 | Wakunaga Pharmaceutical Co., Ltd. | Pyridonecarboxylic acid derivatives or salts thereof and drugs containing the same as the active ingredient |
| WO2006015194A2 (en) * | 2004-07-30 | 2006-02-09 | Abbott Laboratories | Preparation of pyridonecarboxylic acid antibacterials |
| CN102164912A (en) * | 2008-09-24 | 2011-08-24 | Rib-X制药有限公司 | Process for making quinolone compounds |
| CN103936717A (en) * | 2013-01-22 | 2014-07-23 | 上海医药工业研究院 | Intermediate of delafloxacin and preparation method thereof |
| CN104098548A (en) * | 2013-04-11 | 2014-10-15 | 上海医药工业研究院 | Delafloxacin purifying method |
Non-Patent Citations (2)
| Title |
|---|
| Chlorination at the 8-Position of a Functionalized Quinolone and the Synthesis of Quinolone Antibiotic;Barnes, David M等;《Organic Process Research & Development》;20060621;第10卷(第4期);803-807 * |
| Synthesis of the Quinolone ABT-492: Crystallizations for Optimal Processing,Organic Process Research & Development,Synthesis of the Quinolone ABT-492: Crystallizations for Optimal Processing;Anthony R. Haight等;《Organic Process Research & Development》;20060623;第10卷(第4期);751-756 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN106256824A (en) | 2016-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN106256824B (en) | Preparation method of high-purity delafloxacin meglumine salt | |
| CN108658858B (en) | Preparation and refining method of hydroxychloroquine and preparation method of sulfate thereof | |
| CN104230803B (en) | Preparation method of hydroxychloroquine sulfate | |
| CN103724261B (en) | A kind of industrialized process for preparing of hydroxychloroquine sulfate quinoline | |
| CN101941969B (en) | Preparation method of moxifloxacin hydrochloride | |
| CN103664912B (en) | A kind of synthesis technique of prucalopride | |
| WO2023071293A1 (en) | Method for preparing molnupiravir | |
| CN102395591B (en) | Method for preparing prasugrel | |
| CN102887885B (en) | Preparation method of esomeprazole sodium | |
| WO2020182228A1 (en) | Method of refining sodium taurocholate | |
| CN101270124B (en) | Novel method for purifying and preparing high-purity fluorandiol and fluorandiol salt | |
| CN103787924A (en) | New purification method of antitumor drug Belinostat | |
| CN108383745B (en) | Preparation method of aceclofenac | |
| CN105254629A (en) | Preparation method of moxifloxacin hydrochloride | |
| CN111548310B (en) | Levosimendan sodium crystal form and preparation method thereof | |
| CN110551052A (en) | Preparation method of (R) -4-hydroxy-2-oxo-1-pyrrolidine acetate | |
| CN106565776A (en) | Separating and purifying method for 4-(methyl hydroxyl phosphoryl)-2-carbonyl butyric acid | |
| WO2016078584A1 (en) | Emtricitabine purification method | |
| CN105732547A (en) | Preparation method of dehydrated andrographolide diacid half ester basic salt | |
| CN111320622A (en) | Method for synthesizing moxifloxacin hydrochloride | |
| CN103012264A (en) | Method for resolving 3-substituted amino-hexahydro-1H-azacycloheptane | |
| WO2021056382A1 (en) | Preparation method for alpha-penta-o-acetyl mannose | |
| CN107417599B (en) | Preparation method of etoricoxib crystal form | |
| CN117624009A (en) | Continuous synthesis method of JAK inhibitor drug heterocyclic intermediate | |
| CN108069970B (en) | Preparation method of pralatrexate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |