CN102976948A - Method for preparing nitisinone - Google Patents
Method for preparing nitisinone Download PDFInfo
- Publication number
- CN102976948A CN102976948A CN2012104920812A CN201210492081A CN102976948A CN 102976948 A CN102976948 A CN 102976948A CN 2012104920812 A CN2012104920812 A CN 2012104920812A CN 201210492081 A CN201210492081 A CN 201210492081A CN 102976948 A CN102976948 A CN 102976948A
- Authority
- CN
- China
- Prior art keywords
- nitro
- nitisinone
- ethyl acetate
- benzoyl chloride
- trifluoromethyl benzoyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images



Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention discloses a method for preparing nitisinone, comprising the following steps of: preparing 2-nitryl-4-trifluoromethyl-benzoyl chloride from 2-nitryl-4-trifluoromethyl-benzoic acid and thionyl chloride as raw materials; adopting 2-nitryl-4-trifluoromethyl benzoyl chloride, 1,3-cyclohexanedione, ethyl gallate and anhydrous sodium carbonate as raw materials to react; layering, extracting, washing, drying and filtering in sequence; adding triethylamine and aluminum trichloride into the obtained filtrate for reaction; adding hydrochloric acid into the solution of reaction; standing for layering, washing by water, decolorizing and filtering in sequence to obtain the crude nitisinone; and recrystallizing and refining the crude nitisinone by ethyl acetate to obtain the product which is the nitisinone. Because the ethyl acetate is used as a solvent, the aluminum trichloride is used as a catalyst, and the anhydrous sodium carbonate is used as an acid binding agent, the toxicity of the product which is the nitisinone is reduced greatly, the side effect is reduced significantly, and high product purity of more than 99.5% is achieved.
Description
Technical field
The present invention relates to a kind of preparation method of medicine, belong to the pharmaceutical chemistry field, particularly relate to a kind of preparation method of nitisinone.
Background technology
Nitisinone is developed by Sweden Swedish Orphan company, goes on the market in the U.S. first in April, 2002.This product mainly is applicable to the treatment of rare paediatrics I type hereditary tyrosinemia (HT-I), as the adjuvant drug of tyrosine and phenylalanine dietary restrictions.
At present, also synthesize the bibliographical information of aspect both at home and abroad relevant for nitisinone, but disclosed document has only simply been introduced the preparation method of nitisinone, there is no detailed description.The synthetic method of disclosed nitisinone roughly situation is as follows:
At first 2-nitro-4-5-trifluoromethylaniline is produced 2-nitro-4-trifluoromethyl cyanobenzene under acidic conditions, then under the effect of sulfuric acid, generate 2-nitro-4-trifluoromethylbenzoic acid, in sour environment, generate 2-nitro-4-trifluoromethyl benzoic acid methyl ester with methyl alcohol again, nitisinone is produced in last and hydroresorcinol reaction.Its reaction equation is:
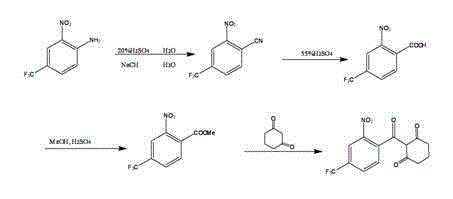
The rearrangement of final step in existing synthetic nitisinone process, what document was taked is that prussiate is catalyzer, however prussiate has higher toxicity, makes its product toxicity residual quantity larger, and side effect is larger when taking.
Summary of the invention
The technical problem to be solved in the present invention is: the preparation method that a kind of nitisinone is provided.The present invention adopts ethyl acetate in the nitisinone building-up process be that catalyzer (the available technology adopting prussiate is catalyzer), anhydrous sodium carbonate are as acid binding agent as solvent (available technology adopting methylene dichloride, acetonitrile are as solvent), aluminum chloride, the toxicity of the product nitisinone of its preparation is reduced greatly, and side effect significantly reduces.
In order to address the above problem, the technical solution used in the present invention is:
The invention provides a kind of preparation method of nitisinone, described preparation method may further comprise the steps:
The preparation of a, 2-nitro-4-trifluoromethyl benzoyl chloride:
Take 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride as raw material, 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride between the two the mol ratio of add-on be 1:2~3, take by weighing two kinds of raw materials according to the proportioning ratio between the two, the raw material 2-nitro that takes by weighing-4-trifluoromethylbenzoic acid and sulfur oxychloride are added in the reactor, be heated to 70~79 ℃ and carry out back flow reaction 3h, after finishing, reaction steams except excessive sulfur oxychloride, then collect 158 ℃ cut under vacuum tightness is the condition of 0.095mPa, the cut of collecting is 2-nitro-4-trifluoromethyl benzoyl chloride;
The preparation of b, nitisinone crude product:
With the 2-nitro of step a preparation-4-trifluoromethyl benzoyl chloride, 1, hydroresorcinol, solvent ethyl acetate and acid binding agent anhydrous sodium carbonate are raw material, described 2-nitro-4-trifluoromethyl benzoyl chloride and 1, the mol ratio of hydroresorcinol between the two is 1:1.0~1.5,2-nitro-4-trifluoromethyl benzoyl chloride and the anhydrous sodium carbonate mol ratio between the two is 1:0.5~1.0,2-nitro-4-trifluoromethyl benzoyl chloride and the ethyl acetate overall proportion between the two is 1g:5.0~12.0ml, prepare various raw materials according to proportioning ratio between each raw material, at first with the raw material 1 for preparing, hydroresorcinol, 60~80% and anhydrous sodium carbonate of ethyl acetate total amount adds in the reaction vessel, be cooled to 0 ℃, drip with this understanding the two mixed solution of 2-nitro-4-trifluoromethyl benzoyl chloride and remaining ethyl acetate, 2h dropwises mixed solution, then be warming up under 25~30 ℃ of conditions and react 2.5h, add entry after the reaction and carry out layering, the add-on of water is 0.25~0.3 times of ethyl acetate add-on, get organic phase after the layering, employing and the isopyknic mass percentage concentration of organic phase are 5% sodium carbonate solution extracting and washing organic phase, then adopt anhydrous sodium sulphate to carry out drying the organic phase after the extracting and washing, filter after the drying; Get its filtrate, be cooled to 0 ℃, then add triethylamine and aluminum chloride, described 2-nitro-4-trifluoromethyl benzoyl chloride and the triethylamine mol ratio between the two is 1:1.5~1.7,2-nitro-4-trifluoromethyl benzoyl chloride and the aluminum chloride mol ratio between the two is 1:0.05~0.07, then be warming up to 25 ℃ of reaction 12h, obtain reaction solution;
Adding concentration in reaction solution is the hydrochloric acid HCl stirring 30min of 2mol/L, described hydrochloric acid and the triethylamine mol ratio between the two is 1:1.2~1.5, standing demix, then get organic phase, organic phase is washed 2~3 times, add gac reflux decolour 30min in the organic phase after the washing, the mass ratio that described 2-nitro-4-trifluoromethyl benzoyl chloride and gac add between the two is 1:0.01~0.02, filter after the decolouring, gained filtrate steams ethyl acetate under 80 ℃ of conditions, the liquid that steams after the ethyl acetate naturally cools to room temperature, filters, and the gained filter cake is the nitisinone crude product;
The refining purification of c, nitisinone crude product: the filter cake that step b is obtained is that the nitisinone crude product adds in the container, and the adding ethyl acetate is carried out recrystallization, described filter cake and the ethyl acetate ratio between the two is 1g:3~5ml, cool off recrystallization after being heated to 70~78 ℃ of backflows, filter behind the recrystallization, the gained filter cake adopts 0 ℃ of ethyl acetate to wash, and carries out drying after the washing, obtains the product nitisinone after the drying.
According to the preparation method of above-mentioned nitisinone, the nitro of 2-described in the step a-4-trifluoromethylbenzoic acid and sulfur oxychloride between the two the mol ratio of add-on be 1:2.
According to the preparation method of above-mentioned nitisinone, carry out drying after the washing described in the step c, its drying is dry 1h under 50 ℃ of conditions at first, then is warming up to dry 12h under 80 ℃ of conditions.
According to the preparation method of above-mentioned nitisinone, the concentration of the nitisinone of product described in the step c is 〉=99.9%.
Positive beneficial effect of the present invention:
1, to adopt ethyl acetate in the nitisinone building-up process be that catalyzer (the available technology adopting prussiate is catalyzer), anhydrous sodium carbonate are as acid binding agent as solvent (available technology adopting methylene dichloride, acetonitrile are as solvent), aluminum chloride in the present invention, the product nitisinone toxicity of its preparation is reduced greatly, and side effect significantly reduces.
2, utilize the complete composite demand of nitisinone quality product of technical solution of the present invention preparation, and purity is higher, its purity to 99.5% above (the correlation detection data of product of the present invention see table 1 and accompanying drawing for details).
3, utilize the nitisinone product toxicity residual quantity of technical solution of the present invention preparation less, less (the rearrangement of final step in existing synthetic nitisinone process of side effect when taking, taking prussiate is catalyzer, yet prussiate has higher toxicity, make its product toxicity residual quantity larger, side effect is larger when taking.And the catalyzer that the present invention adopts is aluminum chloride, is inorganics after the hydrolysis, does not have residual after the washing.)。
The detection analytical data of table 1 product nitisinone of the present invention

Four, description of drawings:
The atlas analysis of Fig. 1 embodiment of the invention 1 products obtained therefrom nitisinone;
The atlas analysis of Fig. 2 embodiment of the invention 2 products obtained therefrom nitisinones;
The atlas analysis of Fig. 3 embodiment of the invention 3 products obtained therefrom nitisinones.
Five, embodiment:
Further set forth the present invention below in conjunction with embodiment, but do not limit content of the present invention.
Embodiment 1:
The preparation method of nitisinone of the present invention, this preparation method's detailed step is as follows:
The preparation of a, 2-nitro-4-trifluoromethyl benzoyl chloride:
Take 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride as raw material, 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride between the two the mol ratio of add-on be 1:2, take by weighing two kinds of raw materials according to the proportioning ratio between the two, the raw material 2-nitro that takes by weighing-4-trifluoromethylbenzoic acid and sulfur oxychloride are added in the reactor, be heated to 70 ℃ and carry out back flow reaction 3h, after finishing, reaction steams except excessive sulfur oxychloride, then collect 158 ℃ cut under vacuum tightness is the condition of 0.095mPa, the cut of collecting is 2-nitro-4-trifluoromethyl benzoyl chloride;
The preparation of b, nitisinone crude product:
With the 2-nitro of step a preparation-4-trifluoromethyl benzoyl chloride, 1, hydroresorcinol, solvent ethyl acetate and acid binding agent anhydrous sodium carbonate are raw material, described 2-nitro-4-trifluoromethyl benzoyl chloride and 1, the mol ratio of hydroresorcinol between the two is 1:1.0,2-nitro-4-trifluoromethyl benzoyl chloride and the anhydrous sodium carbonate mol ratio between the two is 1:0.5,2-nitro-4-trifluoromethyl benzoyl chloride and the ethyl acetate overall proportion between the two is 1g:7.0ml, prepare various raw materials according to proportioning ratio between each raw material, at first with the raw material 1 for preparing, hydroresorcinol, 60% and anhydrous sodium carbonate of ethyl acetate total amount adds in the reaction vessel, be cooled to 0 ℃, drip with this understanding the two mixed solution of 2-nitro-4-trifluoromethyl benzoyl chloride and remaining ethyl acetate, 2h dropwises mixed solution, then be warming up under 25 ℃ of conditions and react 2.5h, add entry after the reaction and carry out layering, the add-on of water is 0.25 times of ethyl acetate add-on, get organic phase after the layering, employing and the isopyknic mass percentage concentration of organic phase are 5% sodium carbonate solution extracting and washing organic phase, then adopt anhydrous sodium sulphate to carry out drying the organic phase after the extracting and washing, filter after the drying; Get its filtrate, be cooled to 0 ℃, then add triethylamine and aluminum chloride, described 2-nitro-4-trifluoromethyl benzoyl chloride and the triethylamine mol ratio between the two is 1:1.5,2-nitro-4-trifluoromethyl benzoyl chloride and the aluminum chloride mol ratio between the two is 1:0.05, then be warming up to 25 ℃ of reaction 12h, obtain reaction solution;
Adding concentration in reaction solution is the hydrochloric acid HCl stirring 30min of 2mol/L, described hydrochloric acid and the triethylamine mol ratio between the two is 1:1.2, standing demix, then get organic phase, organic phase is washed 2~3 times, add gac reflux decolour 30min in the organic phase after the washing, the mass ratio that described 2-nitro-4-trifluoromethyl benzoyl chloride and gac add between the two is 1:0.01, filter after the decolouring, gained filtrate steams ethyl acetate under 80 ℃ of conditions, the liquid that steams after the ethyl acetate naturally cools to room temperature, filters, and the gained filter cake is the nitisinone crude product;
The refining purification of c, nitisinone crude product: the filter cake that step b is obtained is that the nitisinone crude product adds in the container, and the adding ethyl acetate is carried out recrystallization, described filter cake and the ethyl acetate ratio between the two is 1g:4ml, cool off recrystallization after being heated to 70 ℃ of backflows, filter behind the recrystallization, the gained filter cake adopts 0 ℃ of ethyl acetate to wash, (described drying is dry 1h under 50 ℃ of conditions at first to carry out drying after the washing, then be warming up to dry 12h under 80 ℃ of conditions), obtain product nitisinone (the products obtained therefrom purity check sees accompanying drawing 1 for details) after the drying.
Embodiment 2:
The preparation method of nitisinone of the present invention, this preparation method's detailed step is as follows:
The preparation of a, 2-nitro-4-trifluoromethyl benzoyl chloride:
Take 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride as raw material, 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride between the two the mol ratio of add-on be 1:2.5, take by weighing two kinds of raw materials according to the proportioning ratio between the two, the raw material 2-nitro that takes by weighing-4-trifluoromethylbenzoic acid and sulfur oxychloride are added in the reactor, be heated to 75 ℃ and carry out back flow reaction 3h, after finishing, reaction steams except excessive sulfur oxychloride, then collect 158 ℃ cut under vacuum tightness is the condition of 0.095mPa, the cut of collecting is 2-nitro-4-trifluoromethyl benzoyl chloride;
The preparation of b, nitisinone crude product:
With the 2-nitro of step a preparation-4-trifluoromethyl benzoyl chloride, 1, hydroresorcinol, solvent ethyl acetate and acid binding agent anhydrous sodium carbonate are raw material, described 2-nitro-4-trifluoromethyl benzoyl chloride and 1, the mol ratio of hydroresorcinol between the two is 1:1.2,2-nitro-4-trifluoromethyl benzoyl chloride and the anhydrous sodium carbonate mol ratio between the two is 1:0.7,2-nitro-4-trifluoromethyl benzoyl chloride and the ethyl acetate overall proportion between the two is 1g:10.0ml, prepare various raw materials according to proportioning ratio between each raw material, at first with the raw material 1 for preparing, hydroresorcinol, 70% and anhydrous sodium carbonate of ethyl acetate total amount adds in the reaction vessel, be cooled to 0 ℃, drip with this understanding the two mixed solution of 2-nitro-4-trifluoromethyl benzoyl chloride and remaining ethyl acetate, 2h dropwises mixed solution, then be warming up under 28 ℃ of conditions and react 2.5h, add entry after the reaction and carry out layering, the add-on of water is 0.28 times of ethyl acetate add-on, get organic phase after the layering, employing and the isopyknic mass percentage concentration of organic phase are 5% sodium carbonate solution extracting and washing organic phase, then adopt anhydrous sodium sulphate to carry out drying the organic phase after the extracting and washing, filter after the drying; Get its filtrate, be cooled to 0 ℃, then add triethylamine and aluminum chloride, described 2-nitro-4-trifluoromethyl benzoyl chloride and the triethylamine mol ratio between the two is 1:1.6,2-nitro-4-trifluoromethyl benzoyl chloride and the aluminum chloride mol ratio between the two is 1:0.06, then be warming up to 25 ℃ of reaction 12h, obtain reaction solution;
Adding concentration in reaction solution is the hydrochloric acid HCl stirring 30min of 2mol/L, described hydrochloric acid and the triethylamine mol ratio between the two is 1:1.3, standing demix, then get organic phase, organic phase is washed 2~3 times, add gac reflux decolour 30min in the organic phase after the washing, the mass ratio that described 2-nitro-4-trifluoromethyl benzoyl chloride and gac add between the two is 1:0.015, filter after the decolouring, gained filtrate steams ethyl acetate under 80 ℃ of conditions, the liquid that steams after the ethyl acetate naturally cools to room temperature, filters, and the gained filter cake is the nitisinone crude product;
The refining purification of c, nitisinone crude product: the filter cake that step b is obtained is that the nitisinone crude product adds in the container, and the adding ethyl acetate is carried out recrystallization, described filter cake and the ethyl acetate ratio between the two is 1g:5ml, cool off recrystallization after being heated to 75 ℃ of backflows, filter behind the recrystallization, the gained filter cake adopts 0 ℃ of ethyl acetate to wash, (described drying is dry 1h under 50 ℃ of conditions at first to carry out drying after the washing, then be warming up to dry 12h under 80 ℃ of conditions), obtain product nitisinone (the products obtained therefrom purity check sees accompanying drawing 2 for details) after the drying.
Embodiment 3:
The preparation method of nitisinone of the present invention, this preparation method's detailed step is as follows:
The preparation of a, 2-nitro-4-trifluoromethyl benzoyl chloride:
Take 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride as raw material, 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride between the two the mol ratio of add-on be 1:3, take by weighing two kinds of raw materials according to the proportioning ratio between the two, the raw material 2-nitro that takes by weighing-4-trifluoromethylbenzoic acid and sulfur oxychloride are added in the reactor, be heated to 79 ℃ and carry out back flow reaction 3h, after finishing, reaction steams except excessive sulfur oxychloride, then collect 158 ℃ cut under vacuum tightness is the condition of 0.095mPa, the cut of collecting is 2-nitro-4-trifluoromethyl benzoyl chloride;
The preparation of b, nitisinone crude product:
With the 2-nitro of step a preparation-4-trifluoromethyl benzoyl chloride, 1, hydroresorcinol, solvent ethyl acetate and acid binding agent anhydrous sodium carbonate are raw material, described 2-nitro-4-trifluoromethyl benzoyl chloride and 1, the mol ratio of hydroresorcinol between the two is 1:1.5,2-nitro-4-trifluoromethyl benzoyl chloride and the anhydrous sodium carbonate mol ratio between the two is 1:1.0,2-nitro-4-trifluoromethyl benzoyl chloride and the ethyl acetate overall proportion between the two is 1g:12.0ml, prepare various raw materials according to proportioning ratio between each raw material, at first with the raw material 1 for preparing, hydroresorcinol, 80% and anhydrous sodium carbonate of ethyl acetate total amount adds in the reaction vessel, be cooled to 0 ℃, drip with this understanding the two mixed solution of 2-nitro-4-trifluoromethyl benzoyl chloride and remaining ethyl acetate, 2h dropwises mixed solution, then be warming up under 30 ℃ of conditions and react 2.5h, add entry after the reaction and carry out layering, the add-on of water is 0.3 times of ethyl acetate add-on, get organic phase after the layering, employing and the isopyknic mass percentage concentration of organic phase are 5% sodium carbonate solution extracting and washing organic phase, then adopt anhydrous sodium sulphate to carry out drying the organic phase after the extracting and washing, filter after the drying; Get its filtrate, be cooled to 0 ℃, then add triethylamine and aluminum chloride, described 2-nitro-4-trifluoromethyl benzoyl chloride and the triethylamine mol ratio between the two is 1:1.7,2-nitro-4-trifluoromethyl benzoyl chloride and the aluminum chloride mol ratio between the two is 1:0.07, then be warming up to 25 ℃ of reaction 12h, obtain reaction solution;
Adding concentration in reaction solution is the hydrochloric acid HCl stirring 30min of 2mol/L, described hydrochloric acid and the triethylamine mol ratio between the two is 1:1.5, standing demix, then get organic phase, organic phase is washed 2~3 times, add gac reflux decolour 30min in the organic phase after the washing, the mass ratio that described 2-nitro-4-trifluoromethyl benzoyl chloride and gac add between the two is 1:0.02, filter after the decolouring, gained filtrate steams ethyl acetate under 80 ℃ of conditions, the liquid that steams after the ethyl acetate naturally cools to room temperature, filters, and the gained filter cake is the nitisinone crude product;
The refining purification of c, nitisinone crude product: the filter cake that step b is obtained is that the nitisinone crude product adds in the container, and the adding ethyl acetate is carried out recrystallization, described filter cake and the ethyl acetate ratio between the two is 1g:3ml, cool off recrystallization after being heated to 78 ℃ of backflows, filter behind the recrystallization, the gained filter cake adopts 0 ℃ of ethyl acetate to wash, (described drying is dry 1h under 50 ℃ of conditions at first to carry out drying after the washing, then be warming up to dry 12h under 80 ℃ of conditions), obtain product nitisinone (the products obtained therefrom purity check sees accompanying drawing 3 for details) after the drying.
Claims (4)
1. the preparation method of a nitisinone is characterized in that, described preparation method may further comprise the steps:
The preparation of a, 2-nitro-4-trifluoromethyl benzoyl chloride:
Take 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride as raw material, 2-nitro-4-trifluoromethylbenzoic acid and sulfur oxychloride between the two the mol ratio of add-on be 1:2~3, take by weighing two kinds of raw materials according to the proportioning ratio between the two, the raw material 2-nitro that takes by weighing-4-trifluoromethylbenzoic acid and sulfur oxychloride are added in the reactor, be heated to 70~79 ℃ and carry out back flow reaction 3h, after finishing, reaction steams except excessive sulfur oxychloride, then collect 158 ℃ cut under vacuum tightness is the condition of 0.095mPa, the cut of collecting is 2-nitro-4-trifluoromethyl benzoyl chloride;
The preparation of b, nitisinone crude product:
With the 2-nitro of step a preparation-4-trifluoromethyl benzoyl chloride, 1, hydroresorcinol, solvent ethyl acetate and acid binding agent anhydrous sodium carbonate are raw material, described 2-nitro-4-trifluoromethyl benzoyl chloride and 1, the mol ratio of hydroresorcinol between the two is 1:1.0~1.5,2-nitro-4-trifluoromethyl benzoyl chloride and the anhydrous sodium carbonate mol ratio between the two is 1:0.5~1.0,2-nitro-4-trifluoromethyl benzoyl chloride and the ethyl acetate overall proportion between the two is 1g:5.0~12.0ml, prepare various raw materials according to proportioning ratio between each raw material, at first with the raw material 1 for preparing, hydroresorcinol, 60~80% and anhydrous sodium carbonate of ethyl acetate total amount adds in the reaction vessel, be cooled to 0 ℃, drip with this understanding the two mixed solution of 2-nitro-4-trifluoromethyl benzoyl chloride and remaining ethyl acetate, 2h dropwises mixed solution, then be warming up under 25~30 ℃ of conditions and react 2.5h, add entry after the reaction and carry out layering, the add-on of water is 0.25~0.3 times of ethyl acetate add-on, get organic phase after the layering, employing and the isopyknic mass percentage concentration of organic phase are 5% sodium carbonate solution extracting and washing organic phase, then adopt anhydrous sodium sulphate to carry out drying the organic phase after the extracting and washing, filter after the drying; Get its filtrate, be cooled to 0 ℃, then add triethylamine and aluminum chloride, described 2-nitro-4-trifluoromethyl benzoyl chloride and the triethylamine mol ratio between the two is 1:1.5~1.7,2-nitro-4-trifluoromethyl benzoyl chloride and the aluminum chloride mol ratio between the two is 1:0.05~0.07, then be warming up to 25 ℃ of reaction 12h, obtain reaction solution;
Adding concentration in reaction solution is the hydrochloric acid HCl stirring 30min of 2mol/L, described hydrochloric acid and the triethylamine mol ratio between the two is 1:1.2~1.5, standing demix, then get organic phase, organic phase is washed 2~3 times, add gac reflux decolour 30min in the organic phase after the washing, the mass ratio that described 2-nitro-4-trifluoromethyl benzoyl chloride and gac add between the two is 1:0.01~0.02, filter after the decolouring, gained filtrate steams ethyl acetate under 80 ℃ of conditions, the liquid that steams after the ethyl acetate naturally cools to room temperature, filters, and the gained filter cake is the nitisinone crude product;
The refining purification of c, nitisinone crude product: the filter cake that step b is obtained is that the nitisinone crude product adds in the container, and the adding ethyl acetate is carried out recrystallization, described filter cake and the ethyl acetate ratio between the two is 1g:3~5ml, cool off recrystallization after being heated to 70~78 ℃ of backflows, filter behind the recrystallization, the gained filter cake adopts 0 ℃ of ethyl acetate to wash, and carries out drying after the washing, obtains the product nitisinone after the drying.
2. the preparation method of nitisinone according to claim 1 is characterized in that: the nitro of 2-described in the step a-4-trifluoromethylbenzoic acid and sulfur oxychloride between the two the mol ratio of add-on be 1:2.
3. the preparation method of nitisinone according to claim 1 is characterized in that: carry out drying after the washing described in the step c, its drying is dry 1h under 50 ℃ of conditions at first, then is warming up to dry 12h under 80 ℃ of conditions.
4. the preparation method of nitisinone according to claim 1, it is characterized in that: the concentration of the nitisinone of product described in the step c is 〉=99.9%.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210492081.2A CN102976948B (en) | 2012-11-28 | 2012-11-28 | Method for preparing nitisinone |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210492081.2A CN102976948B (en) | 2012-11-28 | 2012-11-28 | Method for preparing nitisinone |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102976948A true CN102976948A (en) | 2013-03-20 |
| CN102976948B CN102976948B (en) | 2014-07-09 |
Family
ID=47851353
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210492081.2A Active CN102976948B (en) | 2012-11-28 | 2012-11-28 | Method for preparing nitisinone |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102976948B (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015101794A1 (en) * | 2014-01-03 | 2015-07-09 | Cycle Pharmaceuticals Ltd | Pharmaceutical composition |
| CN112107548A (en) * | 2020-10-30 | 2020-12-22 | 兆科药业(广州)有限公司 | Pharmaceutical composition containing nitisinone and preparation method thereof |
| CN113845449A (en) * | 2020-06-28 | 2021-12-28 | 沈阳中化农药化工研发有限公司 | Method for preparing mesotrione herbicide |
| WO2024047657A1 (en) * | 2022-08-28 | 2024-03-07 | Brawn Laboratories Ltd | Process for the preparation of nitisinone |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4774360A (en) * | 1987-06-29 | 1988-09-27 | Stauffer Chemical Company | Converting enol ester precursor of a benzoyl-1,3-cycloalkyldione to a benzoyl-1,3-cycloalkyldione |
| WO1993000080A1 (en) * | 1991-06-24 | 1993-01-07 | Zeneca Ltd. | Use of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione in the treatment of tyrosinaemia and pharmaceutical compositions |
-
2012
- 2012-11-28 CN CN201210492081.2A patent/CN102976948B/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4774360A (en) * | 1987-06-29 | 1988-09-27 | Stauffer Chemical Company | Converting enol ester precursor of a benzoyl-1,3-cycloalkyldione to a benzoyl-1,3-cycloalkyldione |
| WO1993000080A1 (en) * | 1991-06-24 | 1993-01-07 | Zeneca Ltd. | Use of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione in the treatment of tyrosinaemia and pharmaceutical compositions |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015101794A1 (en) * | 2014-01-03 | 2015-07-09 | Cycle Pharmaceuticals Ltd | Pharmaceutical composition |
| US10328029B2 (en) | 2014-01-03 | 2019-06-25 | Cycle Pharmaceuticals Ltd | Pharmaceutical composition |
| CN113845449A (en) * | 2020-06-28 | 2021-12-28 | 沈阳中化农药化工研发有限公司 | Method for preparing mesotrione herbicide |
| CN113845449B (en) * | 2020-06-28 | 2023-07-25 | 沈阳中化农药化工研发有限公司 | Method for preparing mesotrione herbicide |
| CN112107548A (en) * | 2020-10-30 | 2020-12-22 | 兆科药业(广州)有限公司 | Pharmaceutical composition containing nitisinone and preparation method thereof |
| WO2024047657A1 (en) * | 2022-08-28 | 2024-03-07 | Brawn Laboratories Ltd | Process for the preparation of nitisinone |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102976948B (en) | 2014-07-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102976948B (en) | Method for preparing nitisinone | |
| CN101940947B (en) | Method for preparing polystyrene resin-immobilized Salon-Co (III) catalyst | |
| CN101759528B (en) | Synthesizing method of 2-methallyl alcohol | |
| CN102702292B (en) | Preparation method of azacitidine | |
| CN106916105A (en) | A kind of method that purifying can win U.S. | |
| CN105152980A (en) | Chiral preparation method for N-t-butyloxycarboryl-(4S)-(p-phenyl phenyl methyl)-4-amino-(2R)-methylbutyric acid | |
| CN103570568A (en) | Clean production process of glycine in coproduction with ammonium chloride | |
| CN103204801A (en) | Synthesis method for N-Boc-3-piperidone | |
| CN102617374B (en) | Method for preparing betaine hydrochloride | |
| CN101486687B (en) | Preparation technique of setastine hydrochloride | |
| CN101973932B (en) | Preparation method of bisacodyl | |
| CN104231033A (en) | Preparation method of dutasteride | |
| CN103980481B (en) | The preparation method of watermiscible vitamin E | |
| CN102603603B (en) | Method for preparing (S)-oxiracetam | |
| CN104356043B (en) | One prepares the method for 5-(2-fluorophenyl)-1H-pyrroles's-3-formaldehyde | |
| CN103804234B (en) | The synthetic method of Alpha-Methyl-(3,4-Dimethoxyphenyl)-alpha-amino group propionitrile | |
| CN102212075A (en) | Preparation method for cefbuperazone | |
| CN103709039A (en) | Method for synthesizing methyl (ethyl) gallate through catalysis of Cu-mordenite | |
| CN104311471A (en) | Improved mitiglinide calcium industrialized preparation method | |
| CN102241599B (en) | Method for preparing glycine | |
| CN102093271A (en) | Preparation method of 2-hydroxy-4-methylthioalkyl butyrate | |
| CN104177271B (en) | A kind of preparation method of ALC | |
| CN103922988A (en) | Method for purifying levetiracetam crude product | |
| CN103739635B (en) | A kind of purification process of mannose triflate intermediate | |
| CN102718810B (en) | After-treatment method of benzylation reaction product |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |