CN102850257A - Preparation method of 1,2-cyclopentane dicarboximide - Google Patents
Preparation method of 1,2-cyclopentane dicarboximide Download PDFInfo
- Publication number
- CN102850257A CN102850257A CN2012103437603A CN201210343760A CN102850257A CN 102850257 A CN102850257 A CN 102850257A CN 2012103437603 A CN2012103437603 A CN 2012103437603A CN 201210343760 A CN201210343760 A CN 201210343760A CN 102850257 A CN102850257 A CN 102850257A
- Authority
- CN
- China
- Prior art keywords
- preparation
- alcohols
- cyclopentanediformylandne
- sodium salt
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 22
- QCWDCTDYSDJKTP-UHFFFAOYSA-N 4,5,6,6a-tetrahydro-3ah-cyclopenta[c]pyrrole-1,3-dione Chemical compound C1CCC2C(=O)NC(=O)C21 QCWDCTDYSDJKTP-UHFFFAOYSA-N 0.000 title abstract 6
- -1 alcohol sodium salt Chemical class 0.000 claims abstract description 29
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 20
- 239000002904 solvent Substances 0.000 claims abstract description 19
- 239000002994 raw material Substances 0.000 claims abstract description 13
- 238000006243 chemical reaction Methods 0.000 claims abstract description 11
- 238000009833 condensation Methods 0.000 claims abstract description 6
- 230000005494 condensation Effects 0.000 claims abstract description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 31
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 18
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 claims description 15
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 15
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 claims description 12
- 230000001476 alcoholic effect Effects 0.000 claims description 10
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 10
- 150000001298 alcohols Chemical class 0.000 claims description 8
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 5
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 claims description 5
- 235000019445 benzyl alcohol Nutrition 0.000 claims description 5
- 229960004217 benzyl alcohol Drugs 0.000 claims description 5
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 claims description 5
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 claims description 5
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 claims description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 4
- 238000002425 crystallisation Methods 0.000 claims description 4
- 230000008025 crystallization Effects 0.000 claims description 4
- 239000003960 organic solvent Substances 0.000 claims description 4
- 238000010792 warming Methods 0.000 claims description 4
- 230000035484 reaction time Effects 0.000 claims description 3
- 238000004807 desolvation Methods 0.000 claims description 2
- 159000000000 sodium salts Chemical class 0.000 claims description 2
- BOVGTQGAOIONJV-BETUJISGSA-N 1-[(3ar,6as)-3,3a,4,5,6,6a-hexahydro-1h-cyclopenta[c]pyrrol-2-yl]-3-(4-methylphenyl)sulfonylurea Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1C[C@H]2CCC[C@H]2C1 BOVGTQGAOIONJV-BETUJISGSA-N 0.000 abstract description 10
- 229960000346 gliclazide Drugs 0.000 abstract description 10
- 238000000034 method Methods 0.000 abstract description 7
- 238000005265 energy consumption Methods 0.000 abstract description 3
- 239000000126 substance Substances 0.000 abstract description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 abstract 4
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 abstract 1
- 235000011114 ammonium hydroxide Nutrition 0.000 abstract 1
- 238000010438 heat treatment Methods 0.000 abstract 1
- 238000009776 industrial production Methods 0.000 abstract 1
- 239000002351 wastewater Substances 0.000 abstract 1
- 239000000047 product Substances 0.000 description 9
- 238000004821 distillation Methods 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 150000008065 acid anhydrides Chemical class 0.000 description 3
- 238000005576 amination reaction Methods 0.000 description 3
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- FJYWNYLUZBMVKI-UHFFFAOYSA-N 3,3a,4,5,6,6a-hexahydro-1h-cyclopenta[c]pyrrol-2-amine Chemical compound C1CCC2CN(N)CC21 FJYWNYLUZBMVKI-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 230000008707 rearrangement Effects 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 2
- 238000001291 vacuum drying Methods 0.000 description 2
- PVFOHMXILQEIHX-UHFFFAOYSA-N 8-[(6-bromo-1,3-benzodioxol-5-yl)sulfanyl]-9-[2-(2-bromophenyl)ethyl]purin-6-amine Chemical compound C=1C=2OCOC=2C=C(Br)C=1SC1=NC=2C(N)=NC=NC=2N1CCC1=CC=CC=C1Br PVFOHMXILQEIHX-UHFFFAOYSA-N 0.000 description 1
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 1
- MQIUGAXCHLFZKX-UHFFFAOYSA-N Di-n-octyl phthalate Natural products CCCCCCCCOC(=O)C1=CC=CC=C1C(=O)OCCCCCCCC MQIUGAXCHLFZKX-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- BOTDANWDWHJENH-UHFFFAOYSA-N Tetraethyl orthosilicate Chemical compound CCO[Si](OCC)(OCC)OCC BOTDANWDWHJENH-UHFFFAOYSA-N 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 238000005904 alkaline hydrolysis reaction Methods 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 239000003849 aromatic solvent Substances 0.000 description 1
- 238000010923 batch production Methods 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 150000001793 charged compounds Chemical class 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 229940127017 oral antidiabetic Drugs 0.000 description 1
- 239000003538 oral antidiabetic agent Substances 0.000 description 1
- SUSQOBVLVYHIEX-UHFFFAOYSA-N phenylacetonitrile Chemical compound N#CCC1=CC=CC=C1 SUSQOBVLVYHIEX-UHFFFAOYSA-N 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- YROXIXLRRCOBKF-UHFFFAOYSA-N sulfonylurea Chemical class OC(=N)N=S(=O)=O YROXIXLRRCOBKF-UHFFFAOYSA-N 0.000 description 1
- 238000007039 two-step reaction Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention discloses a preparation method of 1,2-cyclopentane dicarboximide disclosed as a chemical structural formula on the right. The preparation method comprises the following steps: reacting the raw material alpha-bromocyclohexanone formamide with alcohol and alcohol sodium salt at low temperature for 0.5-1.5 hours, wherein the mol ratio of alpha-bromocyclohexanone formamide to alcohol sodium salt is 1:(1-2); and after the reaction finishes, removing the solvent, continuing heating to 180-220 DEG C to carry out condensation, removing the alcohol solvent, and recrystallizing with a proper solvent to obtain the 1,2-cyclopentane dicarboximide. The 1,2-cyclopentane dicarboximide prepared by the method disclosed by the invention can be used as an intermediate of gliclazide; and compared with the 1,2-cyclopentane dicarboximide prepared by the traditional ammonia water technique, the 1,2-cyclopentane dicarboximide prepared by the method disclosed by the invention has the advantages of high yield, low energy consumption, less wastewater and the like, and provides a feasible technical route for industrial production.
Description
Technical field
The present invention relates to a kind of organism 1, the chemical synthesis process of 2-cyclopentanediformylandne belongs to the field of chemical synthesis.
Background technology
1,2-cyclopentanediformylandne is the important intermediate of preparation gliclazide (Gliclazide).Gliclazide is s-generation sulfonylurea oral antidiabetic drug, be used for the treatment of non insulin dependent diabetes (type ii diabetes), have hypoglycemicly and improve the dual function of coagulation function, and slight lipid metabolism improvement effect is arranged, side effect seldom is widely used clinically.
The synthetic of gliclazide can be by reaction preparations such as N-amino-3-azabicyclo [3.3.0] octane and tolysulfonyl amido ethyl formates, and 1, the 2-cyclopentanediformylandne then is the key intermediate of synthetic N-amino-3-azabicyclo [3.3.0] octane, and the yield of this intermediate is on the low side.Gliclazide usually with pimelinketone and urea as starting raw material, through condensation, hydrolysis, bromination, rearrangement, amination, reduction, nitrosification, restore, the series reaction such as condensation again, obtain the finished product.And 1,2-cyclopentanediformylandne is the key intermediate of synthetic gliclazide, if by traditional Amination Technique yield lower (~ 68%), then directly have influence on overall yield, and then promoted the overall high expensive of gliclazide.
By having technology utilization pimelinketone and urea now as starting material, synthetic 1,2-cyclopentanediformylandne, need experience condensation, hydrolysis, bromo, rearrangement and amination to amount to for five steps, wherein from the bromo-derivative to the aminate, namely 1, the 2-cyclopentanediformylandne, its yield only has 47%.

The yield that adopts synthetic 1, the 2-cyclopentanediformylandne of 1,2-pentamethylene dicarboxylic anhydride only is 51% and 67.23%(Geoffrey C.C, Barry J.S, etc.Synthetic communications, 1981,11,447-454; J.E.
Brenner, J.Org.Chem., 1961,26,22-27), and 1,2-pentamethylene dicarboxylic anhydride is made raw material with pimelinketone, then needs six-step process just can obtain, and step is longer, does not have economic worth.

Be raw material with 1,2-pentamethylene diformamide, need through alkaline hydrolysis that refining 1, the 2-pentamethylene dioctyl phthalate that obtains again through dehydration, forms acid anhydrides, again through underpressure distillation, logical ammonia is re-refined again, and yield only has 57.66%.And the preparation of 1,2-pentamethylene diformamide itself is synthetic as raw material from alpha-brominated pimelinketone methane amide, so if from alpha-brominated pimelinketone methane amide as starting raw material, then total recovery only is 46.12%.This method also exists when becoming acid anhydrides by acid in addition, and raw materials used is diacetyl oxide, and the acid anhydrides thing has the charing phenomenon when high temperature purification, and yield is lower, and raw materials cost is high.Other domestic patent (CN1876636A), reported that with 1,2-pentamethylene diformamide be raw material, through salify, cyclization, its yield is 82.15%, although yield increases, has the particular requirement of high temperature energy consumption and equipment, mention in the patent in addition 1,2-pentamethylene diformamide raw material generally is synthetic by alpha-brominated pimelinketone methane amide, and then total recovery also only has 65.72%.
 ?
?
Summary of the invention
In order to solve above-mentioned technical problem, the technical problem to be solved in the present invention provide that a kind of total recovery is high, raw materials cost is low and purifying easy 1, the preparation method of 2-cyclopentanediformylandne.
Its preparation method is to adopt one kettle way synthetic 1 take alpha-brominated pimelinketone methane amide as starting material, the 2-cyclopentanediformylandne, this product can be as the intermediate of synthetic hypoglycemic drug gliclazide, effectively improved yield, cost and energy consumption have been reduced, shorten the production cycle, reduced the discharging of the three wastes.
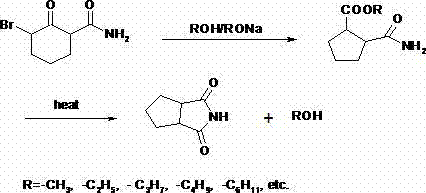
In order to solve the problems of the technologies described above, the invention provides a kind of 1, the preparation method of 2-cyclopentanediformylandne, its chemical structural formula is:
As raw material, carry out low-temp reaction in alcohols and alcohols sodium salt with alpha-brominated pimelinketone methane amide, the reaction times is 0.5 ~ 1.5 hour, alpha-brominated pimelinketone methane amide and alcohols sodium salt are with suitable mol ratio, after reaction finished, desolvation continued to be warming up to 180 ~ 220 ℃, carry out condensation, remove alcoholic solvent, the residual thing of gained adds ethyl acetate or other appropriate solvents, through activated carbon decolorizing, crystallization obtains 1,2-cyclopentanediformylandne.
Improvement as preparation method of the present invention: used alcohols and alcohols sodium salt thereof and adopted the mode that drips.
Further improvement as preparation method of the present invention: alcoholic solvent is the alcohol organic solvent such as methyl alcohol, ethanol, propyl alcohol, Virahol, propyl carbinol, isopropylcarbinol, ethylene glycol, glycol ether, hexalin, benzylalcohol; The alcohols sodium salt is the sodium salt of the corresponding alcohols such as methyl alcohol, ethanol, propyl alcohol, Virahol, propyl carbinol, isopropylcarbinol, ethylene glycol, glycol ether, hexalin, benzylalcohol.
Further improvement as preparation method of the present invention: the alcohols mass ratio of alcohols sodium salt is 10 ~ 60% alcoholic solution.
Further improvement as solvent that purification process of the present invention is used: esters solvent is the ester class organic solvents such as methyl-formiate, ethyl formate, methyl acetate, ethyl acetate, tetraethyl silicate, and aromatic solvents is benzene,toluene,xylene, oil of mirbane, benzyl cyanide etc.; Be the alcohol organic solvent such as methyl alcohol, ethanol, propyl alcohol, Virahol, propyl carbinol, isopropylcarbinol, ethylene glycol, glycol ether, hexalin, benzylalcohol as alcoholic solvent.
Reaction equation of the present invention can be expressed as:
Adopt the product of method preparation of the present invention through gas phase-spectrometer analysis, determined the molecular ion peak (M of M=139
+, 100), and 111 (M
+,-CO, 26.87), 96 (M
+,-CONH, 83.29), 68 (M
+,-CONHCO, 59.61) and fragment ion.Through infrared analysis, at 1704cm
-1(charateristic avsorption band C=O) is at 3292 cm the amidocarbonylation carbon-oxygen bond to have occurred
-1, 3172cm
-1Hydrogen bound to nitrogen (charateristic avsorption band N-H) has appearred.Fusing point conforms to the fusing point of actual product at 230-240 ℃, finally is defined as 1,2-cyclopentanediformylandne.
With of the present invention 1, the 2-cyclopentanediformylandne is for the production of gliclazide, and reaction process is as follows:

In sum, adopt of the present inventionly 1, the 2-cyclopentanediformylandne can be merged into one kettle way with original two-step reaction, can effectively improve yield, and is conducive to environmental protection and reduces production costs, and is suitable for large-scale batch production.
Embodiment
The invention will be further described below in conjunction with specific embodiment:
Embodiment 1: a kind of 1, the preparation method of 2-cyclopentanediformylandne, carry out following steps successively:
Churned mechanically 250mL there-necked flask is being housed, is adding 70ml methyl alcohol, the alpha-brominated pimelinketone methane amide of 30g is cooled to 0-5 ℃, slowly drips the 50ml methanol solution that contains 14.8 gram methyl alcohol sodium salts, after dropwising, and insulated and stirred 30 minutes.
Reaction will be installed this and be common distillation after being finished, and steam except alcoholic solvent, begin afterwards to be warming up under 180 ~ 220 ℃, carry out the solvent that generation is removed in underpressure distillation, add ethyl acetate 150ml after the cooling, add gac 5g, reflux 1 hour, filtered while hot, the crystallization of filtrate normal temperature obtains product 1, the 2-cyclopentanediformylandne, 40 ℃ of lower vacuum-dryings 1 hour, obtain product 14.58 g, yield is 76.55%.
Embodiment 2: a kind of 1, the preparation method of 2-cyclopentanediformylandne, carry out following steps successively:
Churned mechanically 250mL there-necked flask is being housed, is adding 100ml methyl alcohol, the alpha-brominated pimelinketone methane amide of 30g is cooled to 0-5 ℃, slowly drips the 20ml methanol solution that contains 7.4 gram methyl alcohol sodium salts, after dropwising, and insulated and stirred 30 minutes.
Reaction will be installed this and be common distillation after being finished, and steam except alcoholic solvent, begin afterwards to be warming up under 180 ~ 220 ℃, carry out the solvent that generation is removed in underpressure distillation, add ethyl acetate 150ml after the cooling, add gac 5g, reflux 1 hour, filtered while hot, the crystallization of filtrate normal temperature obtains product 1, the 2-cyclopentanediformylandne, 40 ℃ of lower vacuum-dryings 1 hour, obtain product 13.79g, yield is 72.41%.
Embodiment 3 ~ 11: change solvent, alcohols sodium salt concentration, reaction times in above-described embodiment 1 or 2, can obtain corresponding embodiment 3 ~ 11.Particular content sees Table 1, and the product yield of each embodiment gained sees Table 1.
The concrete data of table 1, embodiment 3-11

What enumerate in addition only is several specific embodiments of the present invention.Obviously, the invention is not restricted to above embodiment, can also other, such as all scenario that those of ordinary skill in the art can directly derive or associate from content disclosed by the invention, all should think protection scope of the present invention.
Claims (7)
1.1, the preparation method of 2-cyclopentanediformylandne, it is characterized in that: with alpha-brominated pimelinketone methane amide as raw material, in alcohols and alcohols sodium salt, carry out low-temp reaction, reaction times is 0.5 ~ 1.5 hour, alpha-brominated pimelinketone methane amide and alcohols sodium salt are with suitable mol ratio, after reaction finished, desolvation continued to be warming up to 180 ~ 220 ℃, carry out condensation, remove alcoholic solvent, the residual thing of gained adds ethyl acetate or other appropriate solvents, through activated carbon decolorizing, crystallization obtains 1,2-cyclopentanediformylandne.
2. according to 1 of claim, the preparation method of 2-cyclopentanediformylandne is characterized in that: first alpha-brominated pimelinketone methane amide is joined in the alcoholic solvent, then at low temperatures the alcoholic solution of alcohols sodium salt is dripped.
3. according to 1 of claim, the preparation method of 2-cyclopentanediformylandne is characterized in that: described solvent is the alcohol organic solvent such as methyl alcohol, ethanol, propyl alcohol, Virahol, propyl carbinol, isopropylcarbinol, ethylene glycol, glycol ether, hexalin, benzylalcohol.
4. according to 1 of claim, the preparation method of 2-cyclopentanediformylandne is characterized in that: described alcohols sodium salt is the sodium salt of the corresponding alcohols such as methyl alcohol, ethanol, propyl alcohol, Virahol, propyl carbinol, isopropylcarbinol, ethylene glycol, glycol ether, hexalin, benzylalcohol.
5. according to 1 of claim, the preparation method of 2-cyclopentanediformylandne is characterized in that: the mol ratio of alpha-brominated pimelinketone methane amide and alcohols sodium salt is 1:1 ~ 2.
6. according to claim 51, the preparation method of 2-cyclopentanediformylandne is characterized in that: the alcohols mass ratio of described alcohols sodium salt is 10 ~ 60% alcoholic solution.
7. according to the related solvent of claim aftertreatment purifying, it is characterized in that: ethyl acetate, toluene, alcohols.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210343760.3A CN102850257B (en) | 2012-09-17 | 2012-09-17 | The preparation method of 1,2-cyclopentanediformylandne |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210343760.3A CN102850257B (en) | 2012-09-17 | 2012-09-17 | The preparation method of 1,2-cyclopentanediformylandne |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102850257A true CN102850257A (en) | 2013-01-02 |
| CN102850257B CN102850257B (en) | 2015-09-30 |
Family
ID=47397293
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210343760.3A Expired - Fee Related CN102850257B (en) | 2012-09-17 | 2012-09-17 | The preparation method of 1,2-cyclopentanediformylandne |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102850257B (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108569994A (en) * | 2018-06-01 | 2018-09-25 | 滨海博大化工有限公司 | The synthetic method of 1,2- of one kind rings, penta dicarboximide |
| CN109206358A (en) * | 2018-09-14 | 2019-01-15 | 济南爱思医药科技有限公司 | The synthetic method of 1,2- of one kind ring, penta dicarboximide |
| CN119285528A (en) * | 2024-12-11 | 2025-01-10 | 济南大学 | A kind of synthetic method of 1,2-cyclopentanedicarboximide |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0132392B1 (en) * | 1983-07-22 | 1992-01-02 | Ici Australia Limited | Asymmetric synthesis of alpha-substituted-alpha-cyanomethyl alcohols |
| CN1314340A (en) * | 2000-03-21 | 2001-09-26 | 山东省医药工业研究所 | Method for preparing cyclopentane imide |
| KR20110088755A (en) * | 2010-01-29 | 2011-08-04 | 주식회사 포켐바이오제닉스 | Method for preparing imipenem intermediate |
-
2012
- 2012-09-17 CN CN201210343760.3A patent/CN102850257B/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0132392B1 (en) * | 1983-07-22 | 1992-01-02 | Ici Australia Limited | Asymmetric synthesis of alpha-substituted-alpha-cyanomethyl alcohols |
| CN1314340A (en) * | 2000-03-21 | 2001-09-26 | 山东省医药工业研究所 | Method for preparing cyclopentane imide |
| KR20110088755A (en) * | 2010-01-29 | 2011-08-04 | 주식회사 포켐바이오제닉스 | Method for preparing imipenem intermediate |
Non-Patent Citations (2)
| Title |
|---|
| I. N. TARABARA,等: "Synthesis, Structure, and Transformations of New Endic Anhydride Derivatives", 《RUSSIAN JOURNAL OF ORGANIC CHEMISTRY》 * |
| 张柯华: "格列齐特关键中间体顺式-1,2-环戊烷二甲酰亚胺的合成", 《上海医药工业研究院博士学位论文》 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108569994A (en) * | 2018-06-01 | 2018-09-25 | 滨海博大化工有限公司 | The synthetic method of 1,2- of one kind rings, penta dicarboximide |
| CN109206358A (en) * | 2018-09-14 | 2019-01-15 | 济南爱思医药科技有限公司 | The synthetic method of 1,2- of one kind ring, penta dicarboximide |
| CN109206358B (en) * | 2018-09-14 | 2021-10-29 | 济南爱思医药科技有限公司 | Synthesis method of 1, 2-cyclopentadiimide |
| CN119285528A (en) * | 2024-12-11 | 2025-01-10 | 济南大学 | A kind of synthetic method of 1,2-cyclopentanedicarboximide |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102850257B (en) | 2015-09-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102627594A (en) | Preparation method of waterless aziridine compound | |
| CN102850257B (en) | The preparation method of 1,2-cyclopentanediformylandne | |
| CN102627608B (en) | Preparation method for analgesic and antipyretic drug-analgin | |
| CN102731333B (en) | Method for preparing tetracaine | |
| CN105566138A (en) | Method for synthesizing sitagliptin intermediate | |
| CN102311394B (en) | Preparation method for 5-ethyl-5-phenyl barbituric acid | |
| CN101239919A (en) | Synthetic method of aromatic diamine monomer | |
| CN102093346A (en) | Preparation method of praziquantel | |
| CN101891645A (en) | A kind of preparation method of salicylamide | |
| CN101353446A (en) | Manufacturing method of light-stability agent | |
| CN102603533B (en) | Preparation method of 4,4'-dinitrodiphenyl ether | |
| CN101575262B (en) | Method for reducing content of 2-methylnaphthalene impurity | |
| CN102234253B (en) | Method for preparing febuxostat intermediate | |
| CN103333103B (en) | Method for preparing flupirtine maleate by one-pot method | |
| CN103664654A (en) | Industrial production method of high-purity sulfuric acid terbutaline | |
| CN114478540A (en) | DBU synthesis method | |
| CN102491902A (en) | Preparation method of isopropyl 2-(3-nitrobenzylidene)acetoacetate | |
| CN116396290B (en) | Method for preparing moxifloxacin intermediate (S, S) -2, 8-diazabicyclo [4,3,0] nonane | |
| CN102070471A (en) | Medicine intermediate 2-phenoxy aniline and preparation method thereof | |
| CN103896784A (en) | Method for reducing nitro of Fingolimod intermediate to amino | |
| CN102659626B (en) | Method for reducing waste residues of 4-Bromomethyl-2-cyanobiphenyl | |
| CN108191633A (en) | A kind of method for synthesizing homoanisic acid | |
| CN102311362A (en) | Method for preparing ethyl hydrazinoacetate hydrochloride | |
| CN102127052B (en) | Method for synthesizing 1,4-benzdioxan-2-carboxylic acid | |
| CN108947928B (en) | Nitrogen, oxygen and oxygen trisubstituted six-membered cyclic lactone compound and preparation method and use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C41 | Transfer of patent application or patent right or utility model | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20170213 Address after: 274500 Heze Chemical Industrial Park, Dongming, Shandong, China Patentee after: Shandong Hongzhi Biotechnology Co., Ltd. Address before: 274500 the Yellow River Road, Dongming, Shandong, Heze Patentee before: Shandong Fangming Pharmaceutical Group Co., Ltd. |
|
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20150930 Termination date: 20190917 |