CN102002017A - Method for preparing febuxostat intermediate - Google Patents
Method for preparing febuxostat intermediate Download PDFInfo
- Publication number
- CN102002017A CN102002017A CN 201010534098 CN201010534098A CN102002017A CN 102002017 A CN102002017 A CN 102002017A CN 201010534098 CN201010534098 CN 201010534098 CN 201010534098 A CN201010534098 A CN 201010534098A CN 102002017 A CN102002017 A CN 102002017A
- Authority
- CN
- China
- Prior art keywords
- acid
- methylthiazol
- ethyl ester
- hydroxy phenyl
- formic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Thiazole And Isothizaole Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
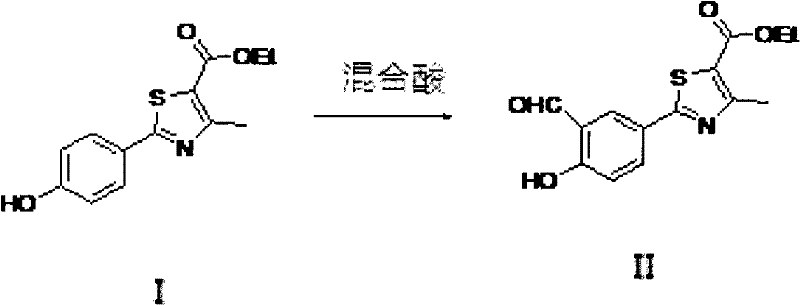
The invention relates to a method for preparing a febuxostat intermediate, which comprises the following steps of: dissolving 2-[4-hydroxyphenyl]-4-methylthiazol-5-ethyl formate in a mixed acid reaction solvent, adding a certain amount of urotropine, heating to react for 1-36 h at certain temperature, and treating the reaction liquid to obtain corresponding heterocyclic aldehyde.
Description
Technical field
The invention belongs to the pharmaceutical chemistry field, relate to febuxostat intermediate 2-[3-aldehyde radical-4-hydroxy phenyl]-preparation of 4-methylthiazol-5-formic acid ethyl ester.
Background technology
Gout has now become the especially common disease of middle-aging male of the world today, and pathogenesis is too much, the kidney removing ability drop of generation uric acid in the body, causes uric acid constantly to be put aside in vivo, forms hyperuricemia.The generation of uric acid is relevant with purine metabolism in the body, final stage at purine metabolism, xanthine generates uric acid under the effect of XOD (XO), the activity that suppresses XO can effectively reduce the generation of uric acid, and therefore the development of novel anti-gout drugs is the focus of drug research always.。
Febuxostat, chemistry 2-[3-cyano-4-isobutoxy phenyl by name]-the 4-methylthiazol-5-formic acid, be a new generation's non-purine class selectivity xanthine oxidase inhibitor, be used for the treatment of chronic hyperuricemia that urate deposition takes place (comprised once or occur now gout or urarthritis) clinically.
Synthesizing of Febuxostat, report that more route can reduce three:
(1) at document Heterocycles, 47 (2), among the 857-864 with 4-isobutoxy-1, the 3-benzene dinitrile is a raw material, obtain 3-cyano-4-isobutoxy thiobenzamide with the thioacetamide reaction, the latter again with the cyclization of 2-chloroacetyl acetacetic ester, the 2-of gained (3-cyano-4-isobutoxy phenyl)-4-methylthiazol-5-formic acid ethyl ester hydrolysis under alkaline condition obtains Febuxostat.

(2) at Chinese Journal of Pharmaceuticals 2009; 40 (10); 2-among the 726-728 (3-formyl radical-4-hydroxy phenyl)-4-methylthiazol-5-formic acid ethyl ester is used the oxammonium hydrochloride cyaniding in methyl alcohol; the 2-that obtains (3-cyano group-4-hydroxy phenyl)-4-methylthiazol-5-formic acid ethyl ester and isobutane bromide reaction make 2-(3-cyano-4-isobutoxy phenyl)-4-methylthiazol-5-formic acid ethyl ester, obtain Febuxostat after latter's hydrolysis.

(3) in the document US 5614520,2-(3-nitro-4-isobutoxy phenyl)-4-methylthiazol-5-formic acid ethyl ester hydrogenation under the catalysis of palladium carbon, the 2-that obtains (3-amino-4-isobutoxy phenyl)-4-methylthiazol-5-formic acid ethyl ester, elder generation's diazotization is reacted with cuprous cyanide again, get 2-(3-cyano-4-isobutoxy phenyl)-4-methylthiazol-5-formic acid ethyl ester, latter's hydrolysis obtains Febuxostat.

2-[3-aldehyde radical-4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is the important intermediate of synthetic Febuxostat, structure is as follows

In the existing synthetic document, the synthetic method of II formula is by the Duff-Bills reaction, uses urotropine aldehyde on three of phenyl ring, and reaction solvent has two kinds,
(1) in JP1045733, use PPA (polyphosphoric acid) as solvent, yield 70%,
(2) in CN101412699, use trifluoroacetic acid as solvent, yield 95%,
Use PPA (polyphosphoric acid) as reaction solvent, advantage is low price, but reaction generation impurity is many, difficult treatment, and product yield is low.
Use trifluoroacetic acid as reaction solvent, it is few that reaction produces impurity, and product yield height, but trifluoroacetic acid price height are difficult to reclaim, and can increase synthetic cost.
We find in synthetic Febuxostat process, if the mixing acid that uses polyphosphoric acid and other strong acid is as reaction solvent, can obtain high yield (92%), can reduce the difficulty of aftertreatment again, reducing synthetic cost, is a kind of good method of intermediate of synthetic Febuxostat, this method reaction conditions gentleness, simple to operate, be convenient to industrialization.
Summary of the invention
The invention provides compound 2-[3-aldehyde radical-4-hydroxy phenyl of a kind of structure I I]-preparation method of 4-methylthiazol-5-formic acid ethyl ester, may further comprise the steps:
2-[4-hydroxy phenyl with structure I]-4-methylthiazol-5-formic acid ethyl ester is dissolved in the mixing acid reaction solvent, adds urotropine, reacts 1-36 hour, obtains the compound of structure I I.

Wherein said mixing acid reaction solvent is selected from: the mixed solvent of polyphosphoric acid and concentrated hydrochloric acid, polyphosphoric acid and the vitriol oil, polyphosphoric acid and nitric acid or polyphosphoric acid and trifluoroacetic acid, polyphosphoric acid and other sour volume ratio are 4 in the mixing acid: 1-1: 1.
The amount that wherein adds urotropine is a substrate 2-[4-hydroxy phenyl]-0.5-10 of 4-methylthiazol-5-formic acid ethyl ester molar weight is doubly.
Temperature of reaction 40-120 ℃.
The add-on of mixing acid is a substrate 2-[4-hydroxy phenyl]-5-10 of 4-methylthiazol-5-formic acid ethyl ester charging capacity is doubly.
The advantage of the inventive method is: simple to operate, and purity height, yield height, reaction conditions gentleness.
Embodiment
Further specify the present invention by the following examples, but not as limitation of the present invention.
Embodiment 1:
26.3g 2-[4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is dissolved in 100ml polyphosphoric acid and the 40ml concentrated hydrochloric acid, adds the 13g urotropine, is heated to 80 ℃, keeps 12 hours, and reaction solution is chilled to room temperature, and processing obtains product, yield 90%.
Embodiment 2:
26.3g 2-[4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is dissolved in the 100ml polyphosphoric acid and the 50ml vitriol oil, adds the 12g urotropine, is heated to 80 ℃, keeps 12 hours, and reaction solution is chilled to room temperature, and processing obtains product, yield 93%.
Embodiment 3:
26.3g 2-[4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is dissolved in 100ml polyphosphoric acid and the 40ml phosphoric acid, adds the 10g urotropine, is heated to 120 ℃, keeps 12 hours, and reaction solution is chilled to room temperature, and processing obtains product, yield 70%.
Embodiment 4:
26.3g 2-[4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is dissolved in 150ml polyphosphoric acid and the 25ml trifluoroacetic acid, adds the 13g urotropine, is heated to 90 ℃, keeps 6 hours, and reaction solution is chilled to room temperature, and processing obtains product, yield 93%.
Embodiment 5:
26.3g 2-[4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is dissolved in 100ml polyphosphoric acid and the 40ml concentrated hydrochloric acid, adds the 13g urotropine, is heated to 60 ℃, keeps 24 hours, and reaction solution is chilled to room temperature, and processing obtains product, yield 79%.
Embodiment 6:
26.3g 2-[4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is dissolved in 100ml polyphosphoric acid and the 40ml concentrated hydrochloric acid, adds the 13g urotropine, is heated to 100 ℃, keeps 3 hours, and reaction solution is chilled to room temperature, and processing obtains product, yield 80%.
Embodiment 7:
26.3g 2-[4-hydroxy phenyl]-4-methylthiazol-5-formic acid ethyl ester is dissolved in 100ml polyphosphoric acid and the 40ml concentrated hydrochloric acid, adds the 15g urotropine, is heated to 80 ℃, keeps 12 hours, and reaction solution is chilled to room temperature, and processing obtains product, yield 93%.
Claims (6)
1. the preparation method of a structural formula II compound, it is characterized in that, may further comprise the steps: with the compound 2-[4-hydroxy phenyl of structure I]-4-methylthiazol-5-formic acid ethyl ester is dissolved in the mixing acid reaction solvent, adds urotropine, reacted 1-36 hour, and obtained the compound of structure I I.
2. according to the method for claim 1, it is characterized in that described mixing acid reaction solvent is selected from: the mixed solvent of polyphosphoric acid and concentrated hydrochloric acid, polyphosphoric acid and the vitriol oil, polyphosphoric acid and nitric acid or polyphosphoric acid and trifluoroacetic acid.
3. according to the method for claim 1, it is characterized in that the amount that adds urotropine is a substrate 2-[4-hydroxy phenyl]-0.5-10 of 4-methylthiazol-5-formic acid ethyl ester molar weight is doubly.
4. according to the method for claim 1, it is characterized in that temperature of reaction 40-120 ℃.
5. according to the method for claim 1, it is characterized in that the add-on of mixing acid is a substrate 2-[4-hydroxy phenyl]-5-10 of 4-methylthiazol-5-formic acid ethyl ester charging capacity is doubly.
6. according to the method for claim 2, it is characterized in that polyphosphoric acid and other sour volume ratio are 4 in the mixing acid: 1-1: 1.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010534098A CN102002017B (en) | 2010-11-02 | 2010-11-02 | Method for preparing febuxostat intermediate |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010534098A CN102002017B (en) | 2010-11-02 | 2010-11-02 | Method for preparing febuxostat intermediate |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102002017A true CN102002017A (en) | 2011-04-06 |
| CN102002017B CN102002017B (en) | 2012-09-26 |
Family
ID=43809751
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201010534098A Active CN102002017B (en) | 2010-11-02 | 2010-11-02 | Method for preparing febuxostat intermediate |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102002017B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103910695A (en) * | 2014-04-24 | 2014-07-09 | 重庆科瑞制药(集团)有限公司 | Synthetic method of febuxostat |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1045733A (en) * | 1996-08-01 | 1998-02-17 | Teijin Ltd | Production of 2-(4-alkoxy-3-cyanophenyl)thiazole derivative |
| CN101412699A (en) * | 2007-10-19 | 2009-04-22 | 上海医药工业研究院 | Preparation of 2-(3-carboxaldehyde-4-hydroxy phenyl)-4-methyl-5-thiazole ethyl formate |
| CN102086169A (en) * | 2009-12-04 | 2011-06-08 | 重庆医药工业研究院有限责任公司 | Preparation method of intermediates of Febuxostat |
-
2010
- 2010-11-02 CN CN201010534098A patent/CN102002017B/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1045733A (en) * | 1996-08-01 | 1998-02-17 | Teijin Ltd | Production of 2-(4-alkoxy-3-cyanophenyl)thiazole derivative |
| CN101412699A (en) * | 2007-10-19 | 2009-04-22 | 上海医药工业研究院 | Preparation of 2-(3-carboxaldehyde-4-hydroxy phenyl)-4-methyl-5-thiazole ethyl formate |
| CN102086169A (en) * | 2009-12-04 | 2011-06-08 | 重庆医药工业研究院有限责任公司 | Preparation method of intermediates of Febuxostat |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103910695A (en) * | 2014-04-24 | 2014-07-09 | 重庆科瑞制药(集团)有限公司 | Synthetic method of febuxostat |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102002017B (en) | 2012-09-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103304512A (en) | Preparation method for febuxostat | |
| CN103387500A (en) | Preparation methods for mirabegron and intermediate thereof | |
| CN103012311A (en) | Preparation method of high-purity febuxostat | |
| García-Báez et al. | Benzothiazoles from condensation of o-aminothiophenoles with carboxylic acids and their derivatives: A review | |
| CN103073519A (en) | Method for preparing dextro-pramipexole hydrochloride | |
| CN109503513B (en) | One-pot synthesis method of febuxostat intermediate | |
| CN105566162B (en) | The preparation technology of rilpivirine intermediate | |
| CN102002017B (en) | Method for preparing febuxostat intermediate | |
| CN101665471A (en) | Preparation method of febuxostat intermediate | |
| CN101723915B (en) | Method for preparing Febuxostat intermediate | |
| CN102936230A (en) | New preparation method of febuxostat | |
| CN104496936B (en) | A kind of preparation method of body of Pramipexole dihydrochloride | |
| CN103788010A (en) | Febuxostat intermediate and preparation method thereof | |
| CN103408507A (en) | Preparation method for 2-amino-1,3,4-thiadiazole compounds | |
| US20130172571A1 (en) | Process to prepare ethyl 4-methyl-2-(4-(2-methylpropyloxy)-3-cyanophenyl)-5-thiazolecarboxylate | |
| CN103073492A (en) | Synthesis method of 2-(3-(S)-(3-(2-7-chlorine-2-quinolyl) vinyl) phenyl)-3-hydroxypropyl) benzoate | |
| CN110229117A (en) | A kind of novel preparation method of Febustat | |
| CN102391170B (en) | A kind of preparation method of N, N-diallyl-5-methoxytryptamine hydrochlorides | |
| CN103396373A (en) | Preparation method of deferasirox and intermediate compound of deferasirox | |
| CN102911123A (en) | Preparation method of 2-chloro trifluoromethyl pyrimidine compound | |
| CN102875399B (en) | D-valine preparation method | |
| CN102127034B (en) | Preparation method of cefozopran side chain acid | |
| CN103073503B (en) | Preparation method of 2-arylbenzimidazole | |
| CN100448836C (en) | Method for preparing key intermediate of medication for anti AIDS | |
| CN103880859A (en) | Preparation method of (3aS, 6aR)-1,3-dibenzyltetrahydro-4H-thieno[3,4-d]imidazol-2,4-(1H)-dione |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C53 | Correction of patent for invention or patent application | ||
| CB02 | Change of applicant information |
Address after: 100021 Beijing city Chaoyang District West business center boziwan Jinhai rich 402 (A) 21 storey building Applicant after: China Resources Saike Pharmaceutical Co., Ltd. Address before: 100021 Beijing city Chaoyang District West business center boziwan Jinhai rich 402 (A) 21 storey building Applicant before: Saike Pharmaceutical Co., Ltd., Beijing |
|
| COR | Change of bibliographic data |
Free format text: CORRECT: APPLICANT; FROM: BEIJING SAIKE PHARMACEUTICAL CO., LTD. TO: CHINA RESOURCES SAIKE PHARMACEUTICAL CO., LTD. |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |