CN101298414A - Preparation of 4-hydroxy benzophenone - Google Patents
Preparation of 4-hydroxy benzophenone Download PDFInfo
- Publication number
- CN101298414A CN101298414A CNA2008100395532A CN200810039553A CN101298414A CN 101298414 A CN101298414 A CN 101298414A CN A2008100395532 A CNA2008100395532 A CN A2008100395532A CN 200810039553 A CN200810039553 A CN 200810039553A CN 101298414 A CN101298414 A CN 101298414A
- Authority
- CN
- China
- Prior art keywords
- preparation
- phenol
- dihydroxy benaophenonel
- reaction
- benzene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention relates to a preparation method of 4-hydroxybenzophenone. The preparation method mainly includes the step of: preparing a target compound by using trihalo-methyl benzene to react with phenol by Friedel-Crafts. The product yield by adopting the preparation method of the 4-hydroxybenzophenone of the invention can reach 90 percent; besides, the preparation method of the 4-hydroxybenzophenone also has the advantages of simple steps and low operation cost, etc; the invention is a method which is easy for scale commercial preparation.
Description
Technical field
The present invention relates to a kind of preparation method of 4-dihydroxy benaophenonel.
Background technology
The 4-dihydroxy benaophenonel is the key intermediate of preparation (ultraviolet) light trigger.
U.S. Pat 2,839,541 disclose the method that is made the 4-dihydroxy benaophenonel by benzophenone in the presence of special catalyzer through hydrolysis.Though the step of this method is comparatively succinct, the selectivity of reaction is bad, except the 4-dihydroxy benaophenonel, also has 2-dihydroxy benaophenonel and 3-dihydroxy benaophenonel in the product, and the separation and purification of this mixture is very difficult.
U.S. Pat 1,961,630 disclose the synthetic method of a kind of halogenated benzophenone hydrolysis system 4-dihydroxy benaophenonel under High Temperature High Pressure.This method is catalyzer with the copper oxide, and the hydrolysis under pressure of 4-chlorobenzophenone is got the 4-dihydroxy benaophenonel; U.S. Pat 2,694,729 disclose a kind of with AlCl
3Be catalyzer; method to methoxy benzophenone demethylation system dihydroxy benaophenonel; but these preparation methods' step comparatively complicated (needing preparation halogenated benzophenone or methoxy benzophenone earlier), the atom availability is not high simultaneously, is not suitable for the mass-producing commercial production.
Given this, to press for a kind of step succinct and be suitable for the preparation method of the 4-dihydroxy benaophenonel of mass-producing commercial production in this area.
Summary of the invention
The objective of the invention is to, provide a kind of step succinct and be suitable for the preparation method of the 4-dihydroxy benaophenonel of mass-producing commercial production.
The said 4-dihydroxy benaophenonel of the present invention, its structure is suc as formula shown in the I:
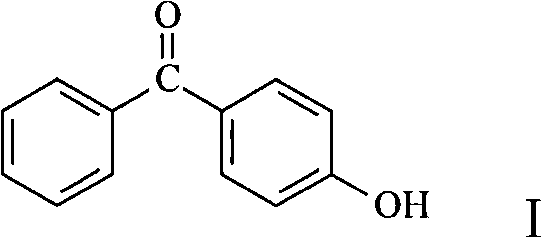
The present inventor finds after deliberation: when phenol and trihalogenmethyl benzene (as trifluoromethylbenzene, benzenyl trichloride, trisbromomethyl benzene or triiodo methylbenzene, when benzenyl trichloride especially) carrying out the Friedel-Crafts reaction, almost all generate the 4-dihydroxy benaophenonel, basically do not have isomer to produce, promptly Fan Ying selectivity is very high.
Given this, the present invention proposes to be prepared as follows the method for 4-dihydroxy benaophenonel.
The method for preparing the 4-dihydroxy benaophenonel of the present invention, its key step is: make target compound (compound shown in the formula I) by trihalogenmethyl benzene (as trifluoromethylbenzene, benzenyl trichloride, trisbromomethyl benzene or triiodo methylbenzene) and phenol by the Friedel-Crafts reaction;
In Friedel-Crafts reaction of the present invention, used catalyzer is that Louis (Lewis) acid is (as anhydrous AlCl
3, anhydrous ZnCl
2Or anhydrous FeCl
3Deng); Used reaction solvent aliphatics haloalkane.
The synthetic route of preparation 4-dihydroxy benaophenonel method of the present invention is as follows:

Wherein: X is halogen (F, Cl, Br or I), and used trihalogenmethyl benzene is commercially available chemical reagent.
In optimal technical scheme of the present invention, described trihalogenmethyl benzene is benzenyl trichloride;
In another optimal technical scheme of the present invention, the mol ratio of phenol and catalyzer (Lewis acid) is 1: (1.0~2.5), preferred mol ratio are 1: (1.1~1.5);
In another optimal technical scheme of the present invention, the mol ratio of phenol and trihalogenmethyl benzene (compound shown in the formula III) is 1: (1.0~2.0), preferred mol ratio are 1: (1.1~1.4).
Embodiment
The method for preparing the 4-dihydroxy benaophenonel of the present invention comprises following rapid:
Under 0 ℃~5 ℃ conditions, successively with 1 of catalyzer (Lewis acid), benzenyl trichloride and phenol, the 2-dichloroethane solution joins 1, in the 2-ethylene dichloride, wherein the mol ratio of phenol and catalyzer is 1: (1.1~1.5), the mol ratio of phenol and benzenyl trichloride are 1: (1.1~1.4).It is complete to feed intake, in 20 ℃~25 ℃ reactions (thin plate chromatography " tracking " reaction is reacted till the raw material point disappearance),
Under agitation condition, reaction solution is poured in ice/water, there is solids to separate out, filtration, gained solids are target compound (4-dihydroxy benaophenonel) after recrystallization and drying.Filtrate can be reclaimed reaction flux (1, the 2-ethylene dichloride) through distillation, can be recycled.
Adopt the method for preparing the 4-dihydroxy benaophenonel of the present invention, product yield can reach 90%.In addition, the present invention also has advantages such as the succinct and process cost of step is cheap, is a kind of method that is easy to the mass-producing commercial production.
Below will the invention will be further elaborated by embodiment, its purpose only is better to understand content of the present invention.Therefore, the cited case does not limit protection scope of the present invention.
Embodiment 1
Add 1 in the 250ml there-necked flask, 2-ethylene dichloride (80ml) and anhydrous AlCl
3(20g, 0.15mol), ice bath is cooled to 0 ℃-5 ℃, stirs half an hour, makes anhydrous AlCl
3Be dispersed into the fine particle shape, (15.7ml 0.11mol), finishes in 20 minutes, and insulation reaction is 20 minutes then, and this moment, reaction solution became orange red to drip benzenyl trichloride again.(9.41g 0.1mol) is dissolved in 1, in the 2-ethylene dichloride (20ml), drips this phenol solution in the time of 0 ℃-5 ℃, drips in 20 minutes and finishes with phenol.There are this moment a large amount of HCl gases to produce, absorb with sig water by gas absorbing device.In the reaction later stage, question response mixture color becomes intense violet color and is warmed up to 20 ℃-25 ℃ again, reacts 3 hours.After reacting end, stir down reaction solution is poured in ice/water, left standstill 2 hours, filter the solid of separating out, with rare Glacial acetic acid recrystallization, obtain the 4-dihydroxy benaophenonel of off-white color after the drying at 10 ℃, mp.133-135 ℃, purity 98%, yield 86%.Mother liquor washes with water to neutrality, distills after the Calcium Chloride Powder Anhydrous drying, collects 78 ℃ of-79 ℃ of cuts, and it can be recycled.
Embodiment 2
Add 1 in the 250ml there-necked flask, 2-ethylene dichloride (80ml) and anhydrous AlCl
3(14.69g, 0.11mol), ice bath is cooled to 0 ℃-5 ℃, stirs half an hour, makes anhydrous AlCl
3Be dispersed into the fine particle shape, drip benzenyl trichloride (15.7ml 0.11mol), finishes in 20 minutes, insulation reaction 20 minutes, this moment, reaction solution became orange red.(9.41g 0.1mol) is dissolved in 1, in the 2-ethylene dichloride (20ml), drips this phenol solution in the time of 0 ℃-5 ℃, drips in 20 minutes and finishes with phenol.There are this moment a large amount of HCl gases to produce, absorb with sig water by gas absorbing device.In the reaction later stage, question response mixture color becomes intense violet color and is warmed up to 20 ℃-25 ℃ again, reacts 3 hours.After reacting end, stir down reaction solution is poured in ice/water, left standstill 2 hours, filter the solid of separating out, with rare Glacial acetic acid recrystallization, obtain the 4-dihydroxy benaophenonel of off-white color after the drying at 10 ℃, mp.133-135 ℃, purity 98%, yield 80%.The processing of mother liquor is the same.
Embodiment 3
Add 1 in the 250ml there-necked flask, 2-ethylene dichloride (80ml) and anhydrous AlCl
3(14.69g, 0.11mol), ice bath is cooled to 0 ℃-5 ℃, stirs half an hour, makes anhydrous AlCl
3Be dispersed into the fine particle shape, drip benzenyl trichloride (19.9ml 0.14mol), finishes in 20 minutes, insulation reaction 20 minutes, this moment, reaction solution became orange red.(9.41g 0.1mol) is dissolved in 1, in the 2-ethylene dichloride (20ml), drips this phenol solution in the time of 0 ℃-5 ℃, drips in 20 minutes and finishes with phenol.There are this moment a large amount of HCl gases to produce, absorb with sig water by gas absorbing device.In the reaction later stage, question response mixture color becomes intense violet color and is warmed up to 20 ℃-25 ℃ again, reacts 3 hours.After reacting end, stir down reaction solution is poured in ice/water, left standstill 2 hours, filter the solid of separating out, with rare Glacial acetic acid recrystallization, obtain the 4-dihydroxy benaophenonel of off-white color after the drying at 10 ℃, mp.133 ℃-135 ℃, purity 98%, yield 78%.The processing of mother liquor is the same.
Embodiment 4
Add 1 in the 250ml there-necked flask, 2-ethylene dichloride (80ml) and anhydrous AlCl
3(20g, 0.15mol), ice bath is cooled to 0 ℃-5 ℃, stirs half an hour, makes anhydrous AlCl
3Be dispersed into the fine particle shape, drip again benzenyl trichloride (19.9ml 0.14mol), finishes in 20 minutes, insulation reaction 20 minutes, this moment, reaction solution became orange red.(9.41g 0.1mol) is dissolved in 1, in the 2-ethylene dichloride (20ml), drips this phenol solution in the time of 0 ℃-5 ℃, drips in 20 minutes and finishes with phenol.There are this moment a large amount of HCl gases to produce, absorb with sig water by gas absorbing device.In the reaction later stage, question response mixture color becomes intense violet color and is warmed up to 20 ℃-25 ℃ again, reacts 3 hours.After reacting end, stir down reaction solution is poured in ice/water (500g), left standstill 2 hours, filter the solid of separating out, with rare Glacial acetic acid recrystallization, obtain the 4-dihydroxy benaophenonel of off-white color after the drying at 10 ℃, mp.133-135 ℃, purity 98%, yield 85%.The processing of mother liquor is the same.
Embodiment 5
Add 1 in the 250ml there-necked flask, 2-ethylene dichloride (80ml) and anhydrous ZnCl
2(20.4g, 0.15mol), ice bath is cooled to 0 ℃-5 ℃, stirs half an hour, drip benzenyl trichloride (19.9ml 0.14mol), finishes in 20 minutes, insulation reaction 30 minutes, this moment, reaction solution became orange.(9.41g 0.1mol) is dissolved in 1, in the 2-ethylene dichloride (20ml), drips this phenol solution in the time of 0 ℃-5 ℃, drips in 30 minutes and finishes with phenol.There is this moment HCl gas to produce, absorbs with sig water by gas absorbing device.In the reaction later stage, question response mixture color becomes scarlet and is warmed up to 20 ℃-25 ℃ again, reacts 5 hours.After reacting end, stir down reaction solution is poured in ice/water, left standstill 2 hours, filter the solid of separating out, with rare Glacial acetic acid recrystallization, obtain the 4-dihydroxy benaophenonel of off-white color after the drying at 10 ℃, mp.133-135 ℃, purity 989%, yield 68%.The processing of mother liquor is the same.
Embodiment 6
Add 1 in the 250ml there-necked flask, 2-ethylene dichloride (80ml) and anhydrous FeCl
3(24.35g, 0.15mol), ice bath is cooled to 0 ℃-5 ℃, stirs half an hour, and (19.9ml 0.14mol), finishes in 20 minutes, and insulation reaction is 20 minutes then, and this moment, reaction solution became orange red to drip benzenyl trichloride again.(9.41g 0.1mol) is dissolved in 1, in the 2-ethylene dichloride (20ml), drips this phenol solution in the time of 0 ℃-5 ℃, drips in 20 minutes and finishes with phenol.There are this moment a large amount of HCl gases to produce, absorb with sig water by gas absorbing device.In the reaction later stage, question response mixture color becomes intense violet color and is warmed up to 20 ℃-25 ℃ again, reacts 3 hours.After reacting end, stir down reaction solution is poured in ice/water (500g), left standstill 2 hours, filter the solid of separating out, with rare Glacial acetic acid recrystallization, obtain the 4-dihydroxy benaophenonel of off-white color after the drying at 10 ℃, mp.133 ℃-135 ℃, purity 98%, yield 78%.The processing of mother liquor is the same.
Claims (7)
1, a kind of method for preparing the 4-dihydroxy benaophenonel is characterized in that, described preparation method's key step is: make target compound by trihalogenmethyl benzene and phenol by the Friedel-Crafts reaction;
In described Friedel-Crafts reaction, used catalyzer is a Lewis acid; Used reaction solvent is the aliphatics haloalkane.
2, preparation method as claimed in claim 1 is characterized in that, wherein the mol ratio of phenol and catalyzer is 1: (1.0~2.5).
3, preparation method as claimed in claim 2 is characterized in that, wherein the mol ratio of phenol and catalyzer is 1: (1.1~1.5).
4, preparation method as claimed in claim 1 is characterized in that, wherein the mol ratio of phenol and trihalogenmethyl benzene is 1: (1.0~2.0).
5, preparation method as claimed in claim 4 is characterized in that, wherein the mol ratio of phenol and trihalogenmethyl benzene is 1: (1.1~1.4).
6, as any described preparation method in the claim 1~5, it is characterized in that wherein used trihalogenmethyl benzene is benzenyl trichloride.
7, as any described preparation method in the claim 1~5, it is characterized in that wherein used catalyzer is anhydrous AlCl
3, anhydrous ZnCl
2Or anhydrous FeCl
3
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNA2008100395532A CN101298414A (en) | 2008-06-26 | 2008-06-26 | Preparation of 4-hydroxy benzophenone |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNA2008100395532A CN101298414A (en) | 2008-06-26 | 2008-06-26 | Preparation of 4-hydroxy benzophenone |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101298414A true CN101298414A (en) | 2008-11-05 |
Family
ID=40078391
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2008100395532A Pending CN101298414A (en) | 2008-06-26 | 2008-06-26 | Preparation of 4-hydroxy benzophenone |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101298414A (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102093190A (en) * | 2010-12-09 | 2011-06-15 | 常州花山化工有限公司 | Method for synthesizing hydroxybenzophenone compound |
| CN102585045A (en) * | 2012-01-16 | 2012-07-18 | 北京化工大学常州先进材料研究院 | Macromolecular polymerizable photoinitiator and preparation thereof |
| CN107879910A (en) * | 2017-11-02 | 2018-04-06 | 武汉科技大学 | A kind of green synthesis process of 2,4 dihydroxy benaophenonel |
| CN109574863A (en) * | 2017-09-28 | 2019-04-05 | 安徽省庆云医药股份有限公司 | A kind of synthetic method of -4 ' of 2- amino-fluoro- benzophenone |
-
2008
- 2008-06-26 CN CNA2008100395532A patent/CN101298414A/en active Pending
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102093190A (en) * | 2010-12-09 | 2011-06-15 | 常州花山化工有限公司 | Method for synthesizing hydroxybenzophenone compound |
| CN102585045A (en) * | 2012-01-16 | 2012-07-18 | 北京化工大学常州先进材料研究院 | Macromolecular polymerizable photoinitiator and preparation thereof |
| CN102585045B (en) * | 2012-01-16 | 2013-09-04 | 北京化工大学常州先进材料研究院 | Macromolecular polymerizable photoinitiator and preparation thereof |
| CN109574863A (en) * | 2017-09-28 | 2019-04-05 | 安徽省庆云医药股份有限公司 | A kind of synthetic method of -4 ' of 2- amino-fluoro- benzophenone |
| CN109574863B (en) * | 2017-09-28 | 2021-09-14 | 安徽省庆云医药股份有限公司 | Synthesis method of 2-amino-4' -fluoro-benzophenone |
| CN107879910A (en) * | 2017-11-02 | 2018-04-06 | 武汉科技大学 | A kind of green synthesis process of 2,4 dihydroxy benaophenonel |
| CN107879910B (en) * | 2017-11-02 | 2021-06-11 | 武汉科技大学 | Green synthesis process of 2, 4-dihydroxy benzophenone |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101298414A (en) | Preparation of 4-hydroxy benzophenone | |
| CN101774897B (en) | Method for preparing vanillin and analogue thereof | |
| CN111302907A (en) | Preparation method of 4,4' -dibromodiphenyl ether | |
| CN102211996A (en) | Preparation method of 2,5-dihydroxy terephthalic acid | |
| CN106278824B (en) | A method of preparing cresols using modified eutectic solvent alkaline hydrolysis | |
| WO2008086942A2 (en) | A method for preparation of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphtoic acid | |
| CN114920637A (en) | Preparation process of 4-chloro-4' -hydroxybenzophenone | |
| TWI632136B (en) | Novel bis(hydroxyphenyl) benzoxazole compound | |
| CN114805094B (en) | Preparation method of bis (3-amino-4-hydroxyphenyl) hexafluoropropane | |
| JPH0579053B2 (en) | ||
| JP4438465B2 (en) | Method for producing tetrakis (4-hydroxyphenyl) ethane compound | |
| CN112225657A (en) | Preparation method of flurbiprofen | |
| CN102531870A (en) | Preparation method of 2,4-difluoroacetophenone | |
| US6875896B2 (en) | Method for producing high purity 1,1-bis(4-hydroxyphenyl)cyclohexanes | |
| CN112479843B (en) | Preparation method of 5-methyl-3-cyclohexenone and application thereof in preparation of m-cresol | |
| CN116178114B (en) | Method for separating cresol isomers by complexation extraction method | |
| JP7476448B2 (en) | Method for producing 4-hydroxy-2-methylbenzoic acid | |
| CN115650901B (en) | Synthesis method of benzo [ b ] carbazole compound | |
| CN101723864A (en) | Method for preparing p-tert-butyl o-nitrothiophenol | |
| WO2005092839A1 (en) | A novel catalytic process for the production of 3,3', 4,4'-tetraminobiphenyl | |
| JP2008115140A (en) | Method for continuous production of 9,9-bis(4-hydroxyphenyl)fluorenes | |
| JPH0267239A (en) | Purification of 2,2-bis(4-hydroxyphenyl)hexafluoropropane | |
| CN117466726A (en) | Preparation method of 4-hydroxy phenyl ketone compound | |
| CN110615775A (en) | Synthetic method of benzofuran-2 (3H) -ketone | |
| CN116903532A (en) | Preparation method of roflumilast |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Open date: 20081105 |