WO2018066716A1 - Pattern formation method and composition - Google Patents
Pattern formation method and composition Download PDFInfo
- Publication number
- WO2018066716A1 WO2018066716A1 PCT/JP2017/036708 JP2017036708W WO2018066716A1 WO 2018066716 A1 WO2018066716 A1 WO 2018066716A1 JP 2017036708 W JP2017036708 W JP 2017036708W WO 2018066716 A1 WO2018066716 A1 WO 2018066716A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer
- pattern
- composition
- substrate
- pattern forming
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 251
- 238000000034 method Methods 0.000 title claims abstract description 134
- 230000007261 regionalization Effects 0.000 title claims abstract description 26
- 229920000642 polymer Polymers 0.000 claims abstract description 322
- 239000000758 substrate Substances 0.000 claims abstract description 121
- 239000002904 solvent Substances 0.000 claims abstract description 82
- 238000005530 etching Methods 0.000 claims abstract description 32
- 238000005191 phase separation Methods 0.000 claims abstract description 30
- 238000011049 filling Methods 0.000 claims abstract description 21
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Natural products C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 claims description 88
- 238000000576 coating method Methods 0.000 claims description 58
- 239000011248 coating agent Substances 0.000 claims description 49
- 239000000126 substance Substances 0.000 claims description 33
- 125000000524 functional group Chemical group 0.000 claims description 29
- 238000010438 heat treatment Methods 0.000 claims description 29
- 230000008569 process Effects 0.000 claims description 24
- 239000004793 Polystyrene Substances 0.000 claims description 23
- 229920002223 polystyrene Polymers 0.000 claims description 20
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 claims description 17
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 16
- 230000005855 radiation Effects 0.000 claims description 14
- 239000002253 acid Substances 0.000 claims description 13
- 238000011161 development Methods 0.000 claims description 11
- 230000018109 developmental process Effects 0.000 claims description 11
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 9
- 125000000962 organic group Chemical group 0.000 claims description 9
- 229910052710 silicon Inorganic materials 0.000 claims description 9
- 239000010703 silicon Substances 0.000 claims description 9
- 229910008051 Si-OH Inorganic materials 0.000 claims description 8
- 229910006358 Si—OH Inorganic materials 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 8
- 239000003960 organic solvent Substances 0.000 claims description 8
- 229910052760 oxygen Inorganic materials 0.000 claims description 7
- 125000003118 aryl group Chemical group 0.000 claims description 5
- 125000005843 halogen group Chemical group 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- 229910052799 carbon Chemical group 0.000 claims description 4
- 125000004432 carbon atom Chemical group C* 0.000 claims description 4
- 238000010030 laminating Methods 0.000 claims description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 3
- 239000003513 alkali Substances 0.000 claims description 3
- 125000003700 epoxy group Chemical group 0.000 claims description 3
- 238000003475 lamination Methods 0.000 claims description 3
- 125000003566 oxetanyl group Chemical group 0.000 claims description 3
- 229910004298 SiO 2 Inorganic materials 0.000 claims description 2
- 125000003011 styrenyl group Chemical class [H]\C(*)=C(/[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 147
- 239000010410 layer Substances 0.000 description 123
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 99
- 239000000243 solution Substances 0.000 description 98
- 238000006116 polymerization reaction Methods 0.000 description 94
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 92
- 230000015572 biosynthetic process Effects 0.000 description 74
- 238000006243 chemical reaction Methods 0.000 description 65
- 239000007787 solid Substances 0.000 description 64
- 238000003786 synthesis reaction Methods 0.000 description 56
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 46
- 208000005156 Dehydration Diseases 0.000 description 33
- 230000018044 dehydration Effects 0.000 description 33
- 238000006297 dehydration reaction Methods 0.000 description 33
- 238000004821 distillation Methods 0.000 description 33
- 235000006408 oxalic acid Nutrition 0.000 description 33
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 28
- 238000002360 preparation method Methods 0.000 description 27
- 229910021642 ultra pure water Inorganic materials 0.000 description 26
- 239000012498 ultrapure water Substances 0.000 description 26
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 25
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 25
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 25
- 238000001914 filtration Methods 0.000 description 25
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 23
- -1 polysiloxane Polymers 0.000 description 23
- LLHKCFNBLRBOGN-UHFFFAOYSA-N propylene glycol methyl ether acetate Chemical compound COCC(C)OC(C)=O LLHKCFNBLRBOGN-UHFFFAOYSA-N 0.000 description 23
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 21
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 21
- 239000007864 aqueous solution Substances 0.000 description 20
- 239000003112 inhibitor Substances 0.000 description 20
- 239000000741 silica gel Substances 0.000 description 20
- 229910002027 silica gel Inorganic materials 0.000 description 20
- 238000001179 sorption measurement Methods 0.000 description 20
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 18
- 238000002347 injection Methods 0.000 description 18
- 239000007924 injection Substances 0.000 description 18
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 16
- 239000012299 nitrogen atmosphere Substances 0.000 description 16
- 150000003839 salts Chemical class 0.000 description 16
- 238000004458 analytical method Methods 0.000 description 15
- 238000011156 evaluation Methods 0.000 description 15
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 15
- 239000004926 polymethyl methacrylate Substances 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 238000001035 drying Methods 0.000 description 14
- 239000000178 monomer Substances 0.000 description 14
- 238000005160 1H NMR spectroscopy Methods 0.000 description 13
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 229920001400 block copolymer Polymers 0.000 description 11
- 229920000359 diblock copolymer Polymers 0.000 description 11
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000003795 chemical substances by application Substances 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 9
- 239000011148 porous material Substances 0.000 description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 8
- 239000007789 gas Substances 0.000 description 8
- 238000005227 gel permeation chromatography Methods 0.000 description 8
- 229920001519 homopolymer Polymers 0.000 description 8
- 238000007654 immersion Methods 0.000 description 8
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 8
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 8
- 230000000052 comparative effect Effects 0.000 description 7
- 238000010304 firing Methods 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 6
- GKIPXFAANLTWBM-UHFFFAOYSA-N epibromohydrin Chemical compound BrCC1CO1 GKIPXFAANLTWBM-UHFFFAOYSA-N 0.000 description 6
- 239000004210 ether based solvent Substances 0.000 description 6
- CATSNJVOTSVZJV-UHFFFAOYSA-N heptan-2-one Chemical compound CCCCCC(C)=O CATSNJVOTSVZJV-UHFFFAOYSA-N 0.000 description 6
- 125000005647 linker group Chemical group 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 238000001020 plasma etching Methods 0.000 description 6
- 150000003440 styrenes Chemical class 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- 238000001039 wet etching Methods 0.000 description 6
- 238000010539 anionic addition polymerization reaction Methods 0.000 description 5
- 239000012295 chemical reaction liquid Substances 0.000 description 5
- 229920001577 copolymer Polymers 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 238000001312 dry etching Methods 0.000 description 5
- 150000002430 hydrocarbons Chemical group 0.000 description 5
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 5
- 229940043265 methyl isobutyl ketone Drugs 0.000 description 5
- TVMXDCGIABBOFY-UHFFFAOYSA-N n-Octanol Natural products CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 5
- 229920000193 polymethacrylate Polymers 0.000 description 5
- 239000004065 semiconductor Substances 0.000 description 5
- 238000004528 spin coating Methods 0.000 description 5
- 230000003068 static effect Effects 0.000 description 5
- ZMYIIHDQURVDRB-UHFFFAOYSA-N 1-phenylethenylbenzene Chemical group C=1C=CC=CC=1C(=C)C1=CC=CC=C1 ZMYIIHDQURVDRB-UHFFFAOYSA-N 0.000 description 4
- BBBUAWSVILPJLL-UHFFFAOYSA-N 2-(2-ethylhexoxymethyl)oxirane Chemical compound CCCCC(CC)COCC1CO1 BBBUAWSVILPJLL-UHFFFAOYSA-N 0.000 description 4
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 4
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 4
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical class C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 4
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 4
- 239000005977 Ethylene Substances 0.000 description 4
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 4
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 4
- RWGFKTVRMDUZSP-UHFFFAOYSA-N cumene Chemical compound CC(C)C1=CC=CC=C1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 4
- DMEGYFMYUHOHGS-UHFFFAOYSA-N cycloheptane Chemical compound C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 4
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 4
- 229910001882 dioxygen Inorganic materials 0.000 description 4
- LZCLXQDLBQLTDK-UHFFFAOYSA-N ethyl 2-hydroxypropanoate Chemical compound CCOC(=O)C(C)O LZCLXQDLBQLTDK-UHFFFAOYSA-N 0.000 description 4
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 229960004592 isopropanol Drugs 0.000 description 4
- VMGSQCIDWAUGLQ-UHFFFAOYSA-N n',n'-bis[2-(dimethylamino)ethyl]-n,n-dimethylethane-1,2-diamine Chemical compound CN(C)CCN(CCN(C)C)CCN(C)C VMGSQCIDWAUGLQ-UHFFFAOYSA-N 0.000 description 4
- 238000001226 reprecipitation Methods 0.000 description 4
- PJANXHGTPQOBST-UHFFFAOYSA-N stilbene Chemical group C=1C=CC=CC=1C=CC1=CC=CC=C1 PJANXHGTPQOBST-UHFFFAOYSA-N 0.000 description 4
- 150000005846 sugar alcohols Polymers 0.000 description 4
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 4
- 229920000428 triblock copolymer Polymers 0.000 description 4
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 4
- JOLQKTGDSGKSKJ-UHFFFAOYSA-N 1-ethoxypropan-2-ol Chemical compound CCOCC(C)O JOLQKTGDSGKSKJ-UHFFFAOYSA-N 0.000 description 3
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 3
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- 239000004593 Epoxy Substances 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- MKYBYDHXWVHEJW-UHFFFAOYSA-N N-[1-oxo-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propan-2-yl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(C(C)NC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 MKYBYDHXWVHEJW-UHFFFAOYSA-N 0.000 description 3
- 229940022663 acetate Drugs 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 150000001335 aliphatic alkanes Chemical class 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 238000000137 annealing Methods 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 150000004699 copper complex Chemical class 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- AFABGHUZZDYHJO-UHFFFAOYSA-N dimethyl butane Natural products CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 3
- 239000003759 ester based solvent Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000005429 filling process Methods 0.000 description 3
- XPFVYQJUAUNWIW-UHFFFAOYSA-N furfuryl alcohol Chemical compound OCC1=CC=CO1 XPFVYQJUAUNWIW-UHFFFAOYSA-N 0.000 description 3
- 239000003999 initiator Substances 0.000 description 3
- 238000010884 ion-beam technique Methods 0.000 description 3
- 239000005453 ketone based solvent Substances 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000004973 liquid crystal related substance Substances 0.000 description 3
- 238000001459 lithography Methods 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 229920000058 polyacrylate Polymers 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- 230000000379 polymerizing effect Effects 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- FYGHSUNMUKGBRK-UHFFFAOYSA-N 1,2,3-trimethylbenzene Chemical compound CC1=CC=CC(C)=C1C FYGHSUNMUKGBRK-UHFFFAOYSA-N 0.000 description 2
- RBACIKXCRWGCBB-UHFFFAOYSA-N 1,2-Epoxybutane Chemical compound CCC1CO1 RBACIKXCRWGCBB-UHFFFAOYSA-N 0.000 description 2
- KVNYFPKFSJIPBJ-UHFFFAOYSA-N 1,2-diethylbenzene Chemical compound CCC1=CC=CC=C1CC KVNYFPKFSJIPBJ-UHFFFAOYSA-N 0.000 description 2
- UAJRSHJHFRVGMG-UHFFFAOYSA-N 1-ethenyl-4-methoxybenzene Chemical compound COC1=CC=C(C=C)C=C1 UAJRSHJHFRVGMG-UHFFFAOYSA-N 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- OJVAMHKKJGICOG-UHFFFAOYSA-N 2,5-hexanedione Chemical compound CC(=O)CCC(C)=O OJVAMHKKJGICOG-UHFFFAOYSA-N 0.000 description 2
- QQZOPKMRPOGIEB-UHFFFAOYSA-N 2-Oxohexane Chemical compound CCCCC(C)=O QQZOPKMRPOGIEB-UHFFFAOYSA-N 0.000 description 2
- YIWUKEYIRIRTPP-UHFFFAOYSA-N 2-ethylhexan-1-ol Chemical compound CCCCC(CC)CO YIWUKEYIRIRTPP-UHFFFAOYSA-N 0.000 description 2
- QPRQEDXDYOZYLA-UHFFFAOYSA-N 2-methylbutan-1-ol Chemical compound CCC(C)CO QPRQEDXDYOZYLA-UHFFFAOYSA-N 0.000 description 2
- GXDHCNNESPLIKD-UHFFFAOYSA-N 2-methylhexane Natural products CCCCC(C)C GXDHCNNESPLIKD-UHFFFAOYSA-N 0.000 description 2
- SVTBMSDMJJWYQN-UHFFFAOYSA-N 2-methylpentane-2,4-diol Chemical compound CC(O)CC(C)(C)O SVTBMSDMJJWYQN-UHFFFAOYSA-N 0.000 description 2
- MCZYEFODKAZWIH-UHFFFAOYSA-N 3-(chloromethyl)-3-methyloxetane Chemical compound ClCC1(C)COC1 MCZYEFODKAZWIH-UHFFFAOYSA-N 0.000 description 2
- WVYWICLMDOOCFB-UHFFFAOYSA-N 4-methyl-2-pentanol Chemical compound CC(C)CC(C)O WVYWICLMDOOCFB-UHFFFAOYSA-N 0.000 description 2
- OZJPLYNZGCXSJM-UHFFFAOYSA-N 5-valerolactone Chemical compound O=C1CCCCO1 OZJPLYNZGCXSJM-UHFFFAOYSA-N 0.000 description 2
- DEXFNLNNUZKHNO-UHFFFAOYSA-N 6-[3-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperidin-1-yl]-3-oxopropyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1CCN(CC1)C(CCC1=CC2=C(NC(O2)=O)C=C1)=O DEXFNLNNUZKHNO-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- KWOLFJPFCHCOCG-UHFFFAOYSA-N Acetophenone Chemical compound CC(=O)C1=CC=CC=C1 KWOLFJPFCHCOCG-UHFFFAOYSA-N 0.000 description 2
- 0 CCCC*CC(*(C)*(C)CC1([C@@](C)(CC(C)C)C1)C(*)=O)c1ccccc1 Chemical compound CCCC*CC(*(C)*(C)CC1([C@@](C)(CC(C)C)C1)C(*)=O)c1ccccc1 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- NIQCNGHVCWTJSM-UHFFFAOYSA-N Dimethyl phthalate Chemical compound COC(=O)C1=CC=CC=C1C(=O)OC NIQCNGHVCWTJSM-UHFFFAOYSA-N 0.000 description 2
- RZKSECIXORKHQS-UHFFFAOYSA-N Heptan-3-ol Chemical compound CCCCC(O)CC RZKSECIXORKHQS-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 2
- NHTMVDHEPJAVLT-UHFFFAOYSA-N Isooctane Chemical compound CC(C)CC(C)(C)C NHTMVDHEPJAVLT-UHFFFAOYSA-N 0.000 description 2
- RJUFJBKOKNCXHH-UHFFFAOYSA-N Methyl propionate Chemical compound CCC(=O)OC RJUFJBKOKNCXHH-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- PJANXHGTPQOBST-VAWYXSNFSA-N Stilbene Natural products C=1C=CC=CC=1/C=C/C1=CC=CC=C1 PJANXHGTPQOBST-VAWYXSNFSA-N 0.000 description 2
- AWMVMTVKBNGEAK-UHFFFAOYSA-N Styrene oxide Chemical compound C1OC1C1=CC=CC=C1 AWMVMTVKBNGEAK-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 239000005456 alcohol based solvent Substances 0.000 description 2
- QUKGYYKBILRGFE-UHFFFAOYSA-N benzyl acetate Chemical compound CC(=O)OCC1=CC=CC=C1 QUKGYYKBILRGFE-UHFFFAOYSA-N 0.000 description 2
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N caprylic alcohol Natural products CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 150000001733 carboxylic acid esters Chemical class 0.000 description 2
- 238000004581 coalescence Methods 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- WJTCGQSWYFHTAC-UHFFFAOYSA-N cyclooctane Chemical compound C1CCCCCCC1 WJTCGQSWYFHTAC-UHFFFAOYSA-N 0.000 description 2
- 239000004914 cyclooctane Substances 0.000 description 2
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 2
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical compound C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 description 2
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 2
- 238000007324 demetalation reaction Methods 0.000 description 2
- SWXVUIWOUIDPGS-UHFFFAOYSA-N diacetone alcohol Chemical compound CC(=O)CC(C)(C)O SWXVUIWOUIDPGS-UHFFFAOYSA-N 0.000 description 2
- FLKPEMZONWLCSK-UHFFFAOYSA-N diethyl phthalate Chemical compound CCOC(=O)C1=CC=CC=C1C(=O)OCC FLKPEMZONWLCSK-UHFFFAOYSA-N 0.000 description 2
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000010894 electron beam technology Methods 0.000 description 2
- 229940116333 ethyl lactate Drugs 0.000 description 2
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 2
- XLLIQLLCWZCATF-UHFFFAOYSA-N ethylene glycol monomethyl ether acetate Natural products COCCOC(C)=O XLLIQLLCWZCATF-UHFFFAOYSA-N 0.000 description 2
- GOQYKNQRPGWPLP-UHFFFAOYSA-N heptadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 2
- NGAZZOYFWWSOGK-UHFFFAOYSA-N heptan-3-one Chemical compound CCCCC(=O)CC NGAZZOYFWWSOGK-UHFFFAOYSA-N 0.000 description 2
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 2
- 229920001903 high density polyethylene Polymers 0.000 description 2
- 239000004700 high-density polyethylene Substances 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- XAOGXQMKWQFZEM-UHFFFAOYSA-N isoamyl propanoate Chemical compound CCC(=O)OCCC(C)C XAOGXQMKWQFZEM-UHFFFAOYSA-N 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- QWTDNUCVQCZILF-UHFFFAOYSA-N isopentane Chemical compound CCC(C)C QWTDNUCVQCZILF-UHFFFAOYSA-N 0.000 description 2
- 238000010551 living anionic polymerization reaction Methods 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 229940017219 methyl propionate Drugs 0.000 description 2
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- ZWRUINPWMLAQRD-UHFFFAOYSA-N nonan-1-ol Chemical compound CCCCCCCCCO ZWRUINPWMLAQRD-UHFFFAOYSA-N 0.000 description 2
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 2
- GJQIMXVRFNLMTB-UHFFFAOYSA-N nonyl acetate Chemical compound CCCCCCCCCOC(C)=O GJQIMXVRFNLMTB-UHFFFAOYSA-N 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- XNLICIUVMPYHGG-UHFFFAOYSA-N pentan-2-one Chemical compound CCCC(C)=O XNLICIUVMPYHGG-UHFFFAOYSA-N 0.000 description 2
- FDPIMTJIUBPUKL-UHFFFAOYSA-N pentan-3-one Chemical compound CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 2
- PGMYKACGEOXYJE-UHFFFAOYSA-N pentyl acetate Chemical compound CCCCCOC(C)=O PGMYKACGEOXYJE-UHFFFAOYSA-N 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- YKYONYBAUNKHLG-UHFFFAOYSA-N propyl acetate Chemical compound CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 2
- ODLMAHJVESYWTB-UHFFFAOYSA-N propylbenzene Chemical compound CCCC1=CC=CC=C1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000010526 radical polymerization reaction Methods 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 238000012712 reversible addition−fragmentation chain-transfer polymerization Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000000992 sputter etching Methods 0.000 description 2
- 235000021286 stilbenes Nutrition 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- WGTYBPLFGIVFAS-UHFFFAOYSA-M tetramethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)C WGTYBPLFGIVFAS-UHFFFAOYSA-M 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N 1,1-Diethoxyethane Chemical class CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- VIDOPANCAUPXNH-UHFFFAOYSA-N 1,2,3-triethylbenzene Chemical compound CCC1=CC=CC(CC)=C1CC VIDOPANCAUPXNH-UHFFFAOYSA-N 0.000 description 1
- OKIRBHVFJGXOIS-UHFFFAOYSA-N 1,2-di(propan-2-yl)benzene Chemical compound CC(C)C1=CC=CC=C1C(C)C OKIRBHVFJGXOIS-UHFFFAOYSA-N 0.000 description 1
- CORMBJOFDGICKF-UHFFFAOYSA-N 1,3,5-trimethoxy 2-vinyl benzene Natural products COC1=CC(OC)=C(C=C)C(OC)=C1 CORMBJOFDGICKF-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- WEERVPDNCOGWJF-UHFFFAOYSA-N 1,4-bis(ethenyl)benzene Chemical compound C=CC1=CC=C(C=C)C=C1 WEERVPDNCOGWJF-UHFFFAOYSA-N 0.000 description 1
- QWOZZTWBWQMEPD-UHFFFAOYSA-N 1-(2-ethoxypropoxy)propan-2-ol Chemical compound CCOC(C)COCC(C)O QWOZZTWBWQMEPD-UHFFFAOYSA-N 0.000 description 1
- YIWGJFPJRAEKMK-UHFFFAOYSA-N 1-(2H-benzotriazol-5-yl)-3-methyl-8-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carbonyl]-1,3,8-triazaspiro[4.5]decane-2,4-dione Chemical compound CN1C(=O)N(c2ccc3n[nH]nc3c2)C2(CCN(CC2)C(=O)c2cnc(NCc3cccc(OC(F)(F)F)c3)nc2)C1=O YIWGJFPJRAEKMK-UHFFFAOYSA-N 0.000 description 1
- ZRZHXNCATOYMJH-UHFFFAOYSA-N 1-(chloromethyl)-4-ethenylbenzene Chemical compound ClCC1=CC=C(C=C)C=C1 ZRZHXNCATOYMJH-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- WGGLDBIZIQMEGH-UHFFFAOYSA-N 1-bromo-4-ethenylbenzene Chemical compound BrC1=CC=C(C=C)C=C1 WGGLDBIZIQMEGH-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- RWNUSVWFHDHRCJ-UHFFFAOYSA-N 1-butoxypropan-2-ol Chemical compound CCCCOCC(C)O RWNUSVWFHDHRCJ-UHFFFAOYSA-N 0.000 description 1
- FUWDFGKRNIDKAE-UHFFFAOYSA-N 1-butoxypropan-2-yl acetate Chemical compound CCCCOCC(C)OC(C)=O FUWDFGKRNIDKAE-UHFFFAOYSA-N 0.000 description 1
- KTZVZZJJVJQZHV-UHFFFAOYSA-N 1-chloro-4-ethenylbenzene Chemical compound ClC1=CC=C(C=C)C=C1 KTZVZZJJVJQZHV-UHFFFAOYSA-N 0.000 description 1
- KWHSBYQFELZKKS-UHFFFAOYSA-N 1-ethenyl-4-iodobenzene Chemical compound IC1=CC=C(C=C)C=C1 KWHSBYQFELZKKS-UHFFFAOYSA-N 0.000 description 1
- YFZHODLXYNDBSM-UHFFFAOYSA-N 1-ethenyl-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(C=C)C=C1 YFZHODLXYNDBSM-UHFFFAOYSA-N 0.000 description 1
- HYFLWBNQFMXCPA-UHFFFAOYSA-N 1-ethyl-2-methylbenzene Chemical compound CCC1=CC=CC=C1C HYFLWBNQFMXCPA-UHFFFAOYSA-N 0.000 description 1
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 1
- DOVZUKKPYKRVIK-UHFFFAOYSA-N 1-methoxypropan-2-yl propanoate Chemical compound CCC(=O)OC(C)COC DOVZUKKPYKRVIK-UHFFFAOYSA-N 0.000 description 1
- DMFAHCVITRDZQB-UHFFFAOYSA-N 1-propoxypropan-2-yl acetate Chemical compound CCCOCC(C)OC(C)=O DMFAHCVITRDZQB-UHFFFAOYSA-N 0.000 description 1
- QEDJMOONZLUIMC-UHFFFAOYSA-N 1-tert-butyl-4-ethenylbenzene Chemical compound CC(C)(C)C1=CC=C(C=C)C=C1 QEDJMOONZLUIMC-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- VXQBJTKSVGFQOL-UHFFFAOYSA-N 2-(2-butoxyethoxy)ethyl acetate Chemical compound CCCCOCCOCCOC(C)=O VXQBJTKSVGFQOL-UHFFFAOYSA-N 0.000 description 1
- FPZWZCWUIYYYBU-UHFFFAOYSA-N 2-(2-ethoxyethoxy)ethyl acetate Chemical compound CCOCCOCCOC(C)=O FPZWZCWUIYYYBU-UHFFFAOYSA-N 0.000 description 1
- CKCGJBFTCUCBAJ-UHFFFAOYSA-N 2-(2-ethoxypropoxy)propyl acetate Chemical compound CCOC(C)COC(C)COC(C)=O CKCGJBFTCUCBAJ-UHFFFAOYSA-N 0.000 description 1
- ZKCAGDPACLOVBN-UHFFFAOYSA-N 2-(2-ethylbutoxy)ethanol Chemical compound CCC(CC)COCCO ZKCAGDPACLOVBN-UHFFFAOYSA-N 0.000 description 1
- GZMAAYIALGURDQ-UHFFFAOYSA-N 2-(2-hexoxyethoxy)ethanol Chemical compound CCCCCCOCCOCCO GZMAAYIALGURDQ-UHFFFAOYSA-N 0.000 description 1
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 1
- BJINVQNEBGOMCR-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethyl acetate Chemical compound COCCOCCOC(C)=O BJINVQNEBGOMCR-UHFFFAOYSA-N 0.000 description 1
- DRLRGHZJOQGQEC-UHFFFAOYSA-N 2-(2-methoxypropoxy)propyl acetate Chemical compound COC(C)COC(C)COC(C)=O DRLRGHZJOQGQEC-UHFFFAOYSA-N 0.000 description 1
- DJCYDDALXPHSHR-UHFFFAOYSA-N 2-(2-propoxyethoxy)ethanol Chemical compound CCCOCCOCCO DJCYDDALXPHSHR-UHFFFAOYSA-N 0.000 description 1
- XYVAYAJYLWYJJN-UHFFFAOYSA-N 2-(2-propoxypropoxy)propan-1-ol Chemical compound CCCOC(C)COC(C)CO XYVAYAJYLWYJJN-UHFFFAOYSA-N 0.000 description 1
- YSUQLAYJZDEMOT-UHFFFAOYSA-N 2-(butoxymethyl)oxirane Chemical compound CCCCOCC1CO1 YSUQLAYJZDEMOT-UHFFFAOYSA-N 0.000 description 1
- JJRUAPNVLBABCN-UHFFFAOYSA-N 2-(ethenoxymethyl)oxirane Chemical compound C=COCC1CO1 JJRUAPNVLBABCN-UHFFFAOYSA-N 0.000 description 1
- LKMJVFRMDSNFRT-UHFFFAOYSA-N 2-(methoxymethyl)oxirane Chemical compound COCC1CO1 LKMJVFRMDSNFRT-UHFFFAOYSA-N 0.000 description 1
- HQLKZWRSOHTERR-UHFFFAOYSA-N 2-Ethylbutyl acetate Chemical compound CCC(CC)COC(C)=O HQLKZWRSOHTERR-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- PTTPXKJBFFKCEK-UHFFFAOYSA-N 2-Methyl-4-heptanone Chemical compound CC(C)CC(=O)CC(C)C PTTPXKJBFFKCEK-UHFFFAOYSA-N 0.000 description 1
- LCZVSXRMYJUNFX-UHFFFAOYSA-N 2-[2-(2-hydroxypropoxy)propoxy]propan-1-ol Chemical compound CC(O)COC(C)COC(C)CO LCZVSXRMYJUNFX-UHFFFAOYSA-N 0.000 description 1
- SDHQGBWMLCBNSM-UHFFFAOYSA-N 2-[2-(2-methoxyethoxy)ethoxy]ethyl acetate Chemical compound COCCOCCOCCOC(C)=O SDHQGBWMLCBNSM-UHFFFAOYSA-N 0.000 description 1
- XXSPGBOGLXKMDU-UHFFFAOYSA-N 2-bromo-2-methylpropanoic acid Chemical compound CC(C)(Br)C(O)=O XXSPGBOGLXKMDU-UHFFFAOYSA-N 0.000 description 1
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 description 1
- KKLIEUWPBXKNFS-UHFFFAOYSA-M 2-carboxyphenolate;triphenylsulfanium Chemical compound OC1=CC=CC=C1C([O-])=O.C1=CC=CC=C1[S+](C=1C=CC=CC=1)C1=CC=CC=C1 KKLIEUWPBXKNFS-UHFFFAOYSA-M 0.000 description 1
- DZFGVGDQHQHOKZ-UHFFFAOYSA-N 2-dodecylsulfanylcarbothioylsulfanyl-2-methylpropanoic acid Chemical compound CCCCCCCCCCCCSC(=S)SC(C)(C)C(O)=O DZFGVGDQHQHOKZ-UHFFFAOYSA-N 0.000 description 1
- PDELBHCVXBSVPJ-UHFFFAOYSA-N 2-ethenyl-1,3,5-trimethylbenzene Chemical compound CC1=CC(C)=C(C=C)C(C)=C1 PDELBHCVXBSVPJ-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- SVONRAPFKPVNKG-UHFFFAOYSA-N 2-ethoxyethyl acetate Chemical compound CCOCCOC(C)=O SVONRAPFKPVNKG-UHFFFAOYSA-N 0.000 description 1
- TZYRSLHNPKPEFV-UHFFFAOYSA-N 2-ethyl-1-butanol Chemical compound CCC(CC)CO TZYRSLHNPKPEFV-UHFFFAOYSA-N 0.000 description 1
- WOYWLLHHWAMFCB-UHFFFAOYSA-N 2-ethylhexyl acetate Chemical compound CCCCC(CC)COC(C)=O WOYWLLHHWAMFCB-UHFFFAOYSA-N 0.000 description 1
- CETWDUZRCINIHU-UHFFFAOYSA-N 2-heptanol Chemical compound CCCCCC(C)O CETWDUZRCINIHU-UHFFFAOYSA-N 0.000 description 1
- UPGSWASWQBLSKZ-UHFFFAOYSA-N 2-hexoxyethanol Chemical compound CCCCCCOCCO UPGSWASWQBLSKZ-UHFFFAOYSA-N 0.000 description 1
- MHXMVFDLNGKBSR-UHFFFAOYSA-N 2-hydroxyethyl 2-bromo-2-methylpropanoate Chemical compound CC(C)(Br)C(=O)OCCO MHXMVFDLNGKBSR-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- PFNHSEQQEPMLNI-UHFFFAOYSA-N 2-methyl-1-pentanol Chemical compound CCCC(C)CO PFNHSEQQEPMLNI-UHFFFAOYSA-N 0.000 description 1
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 1
- ZPVFWPFBNIEHGJ-UHFFFAOYSA-N 2-octanone Chemical compound CCCCCCC(C)=O ZPVFWPFBNIEHGJ-UHFFFAOYSA-N 0.000 description 1
- QCDWFXQBSFUVSP-UHFFFAOYSA-N 2-phenoxyethanol Chemical compound OCCOC1=CC=CC=C1 QCDWFXQBSFUVSP-UHFFFAOYSA-N 0.000 description 1
- YEYKMVJDLWJFOA-UHFFFAOYSA-N 2-propoxyethanol Chemical compound CCCOCCO YEYKMVJDLWJFOA-UHFFFAOYSA-N 0.000 description 1
- BRRVXFOKWJKTGG-UHFFFAOYSA-N 3,3,5-trimethylcyclohexanol Chemical compound CC1CC(O)CC(C)(C)C1 BRRVXFOKWJKTGG-UHFFFAOYSA-N 0.000 description 1
- PKNKULBDCRZSBT-UHFFFAOYSA-N 3,4,5-trimethylnonan-2-one Chemical compound CCCCC(C)C(C)C(C)C(C)=O PKNKULBDCRZSBT-UHFFFAOYSA-N 0.000 description 1
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 1
- QCAHUFWKIQLBNB-UHFFFAOYSA-N 3-(3-methoxypropoxy)propan-1-ol Chemical compound COCCCOCCCO QCAHUFWKIQLBNB-UHFFFAOYSA-N 0.000 description 1
- UKLWXKWTXHHMFK-UHFFFAOYSA-N 3-(chloromethyl)-3-ethyloxetane Chemical compound CCC1(CCl)COC1 UKLWXKWTXHHMFK-UHFFFAOYSA-N 0.000 description 1
- ZSPTYLOMNJNZNG-UHFFFAOYSA-N 3-Buten-1-ol Chemical compound OCCC=C ZSPTYLOMNJNZNG-UHFFFAOYSA-N 0.000 description 1
- CQZIEDXCLQOOEH-UHFFFAOYSA-N 3-bromopropanenitrile Chemical compound BrCCC#N CQZIEDXCLQOOEH-UHFFFAOYSA-N 0.000 description 1
- JSGVZVOGOQILFM-UHFFFAOYSA-N 3-methoxy-1-butanol Chemical compound COC(C)CCO JSGVZVOGOQILFM-UHFFFAOYSA-N 0.000 description 1
- QMYGFTJCQFEDST-UHFFFAOYSA-N 3-methoxybutyl acetate Chemical compound COC(C)CCOC(C)=O QMYGFTJCQFEDST-UHFFFAOYSA-N 0.000 description 1
- LDMRLRNXHLPZJN-UHFFFAOYSA-N 3-propoxypropan-1-ol Chemical compound CCCOCCCO LDMRLRNXHLPZJN-UHFFFAOYSA-N 0.000 description 1
- UUEWCQRISZBELL-UHFFFAOYSA-N 3-trimethoxysilylpropane-1-thiol Chemical compound CO[Si](OC)(OC)CCCS UUEWCQRISZBELL-UHFFFAOYSA-N 0.000 description 1
- VFXXTYGQYWRHJP-UHFFFAOYSA-N 4,4'-azobis(4-cyanopentanoic acid) Chemical compound OC(=O)CCC(C)(C#N)N=NC(C)(CCC(O)=O)C#N VFXXTYGQYWRHJP-UHFFFAOYSA-N 0.000 description 1
- JLBJTVDPSNHSKJ-UHFFFAOYSA-N 4-Methylstyrene Chemical compound CC1=CC=C(C=C)C=C1 JLBJTVDPSNHSKJ-UHFFFAOYSA-N 0.000 description 1
- SNTUCKQYWGHZPK-UHFFFAOYSA-N 4-ethenylbenzonitrile Chemical compound C=CC1=CC=C(C#N)C=C1 SNTUCKQYWGHZPK-UHFFFAOYSA-N 0.000 description 1
- FUGYGGDSWSUORM-UHFFFAOYSA-N 4-hydroxystyrene Chemical compound OC1=CC=C(C=C)C=C1 FUGYGGDSWSUORM-UHFFFAOYSA-N 0.000 description 1
- MQWCXKGKQLNYQG-UHFFFAOYSA-N 4-methylcyclohexan-1-ol Chemical compound CC1CCC(O)CC1 MQWCXKGKQLNYQG-UHFFFAOYSA-N 0.000 description 1
- VGVHNLRUAMRIEW-UHFFFAOYSA-N 4-methylcyclohexan-1-one Chemical compound CC1CCC(=O)CC1 VGVHNLRUAMRIEW-UHFFFAOYSA-N 0.000 description 1
- LPEKGGXMPWTOCB-UHFFFAOYSA-N 8beta-(2,3-epoxy-2-methylbutyryloxy)-14-acetoxytithifolin Natural products COC(=O)C(C)O LPEKGGXMPWTOCB-UHFFFAOYSA-N 0.000 description 1
- MRABAEUHTLLEML-UHFFFAOYSA-N Butyl lactate Chemical compound CCCCOC(=O)C(C)O MRABAEUHTLLEML-UHFFFAOYSA-N 0.000 description 1
- GXJRDNVWLILVAM-UHFFFAOYSA-N CCC(C)C(C)(C)CC(C(C)(C)C(O)=O)c1ccccc1 Chemical compound CCC(C)C(C)(C)CC(C(C)(C)C(O)=O)c1ccccc1 GXJRDNVWLILVAM-UHFFFAOYSA-N 0.000 description 1
- DSDOWJOFSAGLCK-UHFFFAOYSA-N CCOC(C(C)(C)C(C)(C)CC(CC(C)(CC1(C)C(O)=[O]C1(C)Br)C(OC)=O)c1ccccc1)=O Chemical compound CCOC(C(C)(C)C(C)(C)CC(CC(C)(CC1(C)C(O)=[O]C1(C)Br)C(OC)=O)c1ccccc1)=O DSDOWJOFSAGLCK-UHFFFAOYSA-N 0.000 description 1
- YYLLIJHXUHJATK-UHFFFAOYSA-N Cyclohexyl acetate Chemical compound CC(=O)OC1CCCCC1 YYLLIJHXUHJATK-UHFFFAOYSA-N 0.000 description 1
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 1
- HXQPUEQDBSPXTE-UHFFFAOYSA-N Diisobutylcarbinol Chemical compound CC(C)CC(O)CC(C)C HXQPUEQDBSPXTE-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical class COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- WRQNANDWMGAFTP-UHFFFAOYSA-N Methylacetoacetic acid Chemical compound COC(=O)CC(C)=O WRQNANDWMGAFTP-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- SUAKHGWARZSWIH-UHFFFAOYSA-N N,N‐diethylformamide Chemical compound CCN(CC)C=O SUAKHGWARZSWIH-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 1
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 1
- VCUFZILGIRCDQQ-KRWDZBQOSA-N N-[[(5S)-2-oxo-3-(2-oxo-3H-1,3-benzoxazol-6-yl)-1,3-oxazolidin-5-yl]methyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C1O[C@H](CN1C1=CC2=C(NC(O2)=O)C=C1)CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F VCUFZILGIRCDQQ-KRWDZBQOSA-N 0.000 description 1
- OHLUUHNLEMFGTQ-UHFFFAOYSA-N N-methylacetamide Chemical compound CNC(C)=O OHLUUHNLEMFGTQ-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- JKRZOJADNVOXPM-UHFFFAOYSA-N Oxalic acid dibutyl ester Chemical compound CCCCOC(=O)C(=O)OCCCC JKRZOJADNVOXPM-UHFFFAOYSA-N 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- 229910052581 Si3N4 Inorganic materials 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- JAWMENYCRQKKJY-UHFFFAOYSA-N [3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-ylmethyl)-1-oxa-2,8-diazaspiro[4.5]dec-2-en-8-yl]-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]methanone Chemical compound N1N=NC=2CN(CCC=21)CC1=NOC2(C1)CCN(CC2)C(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F JAWMENYCRQKKJY-UHFFFAOYSA-N 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 150000004791 alkyl magnesium halides Chemical class 0.000 description 1
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 238000010560 atom transfer radical polymerization reaction Methods 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 229940007550 benzyl acetate Drugs 0.000 description 1
- 230000002902 bimodal effect Effects 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- BTMVHUNTONAYDX-UHFFFAOYSA-N butyl propionate Chemical compound CCCCOC(=O)CC BTMVHUNTONAYDX-UHFFFAOYSA-N 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- UOCJDOLVGGIYIQ-PBFPGSCMSA-N cefatrizine Chemical group S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)[C@H](N)C=2C=CC(O)=CC=2)CC=1CSC=1C=NNN=1 UOCJDOLVGGIYIQ-PBFPGSCMSA-N 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 150000003950 cyclic amides Chemical class 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 150000003997 cyclic ketones Chemical class 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 150000001925 cycloalkenes Chemical class 0.000 description 1
- CGZZMOTZOONQIA-UHFFFAOYSA-N cycloheptanone Chemical compound O=C1CCCCCC1 CGZZMOTZOONQIA-UHFFFAOYSA-N 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- IIRFCWANHMSDCG-UHFFFAOYSA-N cyclooctanone Chemical compound O=C1CCCCCCC1 IIRFCWANHMSDCG-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 150000001983 dialkylethers Chemical class 0.000 description 1
- WYACBZDAHNBPPB-UHFFFAOYSA-N diethyl oxalate Chemical compound CCOC(=O)C(=O)OCC WYACBZDAHNBPPB-UHFFFAOYSA-N 0.000 description 1
- 229940028356 diethylene glycol monobutyl ether Drugs 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- 229940075557 diethylene glycol monoethyl ether Drugs 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 1
- FBSAITBEAPNWJG-UHFFFAOYSA-N dimethyl phthalate Natural products CC(=O)OC1=CC=CC=C1OC(C)=O FBSAITBEAPNWJG-UHFFFAOYSA-N 0.000 description 1
- JVSWJIKNEAIKJW-UHFFFAOYSA-N dimethyl-hexane Natural products CCCCCC(C)C JVSWJIKNEAIKJW-UHFFFAOYSA-N 0.000 description 1
- 229960001826 dimethylphthalate Drugs 0.000 description 1
- POLCUAVZOMRGSN-UHFFFAOYSA-N dipropyl ether Chemical compound CCCOCCC POLCUAVZOMRGSN-UHFFFAOYSA-N 0.000 description 1
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- JGHKDVSIFPFNIJ-UHFFFAOYSA-N dodecylsulfanylmethanedithioic acid Chemical compound CCCCCCCCCCCCSC(S)=S JGHKDVSIFPFNIJ-UHFFFAOYSA-N 0.000 description 1
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000012156 elution solvent Substances 0.000 description 1
- HFTNNOZFRQLFQB-UHFFFAOYSA-N ethenoxy(trimethyl)silane Chemical compound C[Si](C)(C)OC=C HFTNNOZFRQLFQB-UHFFFAOYSA-N 0.000 description 1
- LDLDYFCCDKENPD-UHFFFAOYSA-N ethenylcyclohexane Chemical compound C=CC1CCCCC1 LDLDYFCCDKENPD-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- IOLQWGVDEFWYNP-UHFFFAOYSA-N ethyl 2-bromo-2-methylpropanoate Chemical compound CCOC(=O)C(C)(C)Br IOLQWGVDEFWYNP-UHFFFAOYSA-N 0.000 description 1
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 1
- 229940093858 ethyl acetoacetate Drugs 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000002366 halogen compounds Chemical class 0.000 description 1
- XVEOUOTUJBYHNL-UHFFFAOYSA-N heptane-2,4-diol Chemical compound CCCC(O)CC(C)O XVEOUOTUJBYHNL-UHFFFAOYSA-N 0.000 description 1
- QNVRIHYSUZMSGM-UHFFFAOYSA-N hexan-2-ol Chemical compound CCCCC(C)O QNVRIHYSUZMSGM-UHFFFAOYSA-N 0.000 description 1
- RXTNIJMLAQNTEG-UHFFFAOYSA-N hexan-2-yl acetate Chemical compound CCCCC(C)OC(C)=O RXTNIJMLAQNTEG-UHFFFAOYSA-N 0.000 description 1
- OHMBHFSEKCCCBW-UHFFFAOYSA-N hexane-2,5-diol Chemical compound CC(O)CCC(C)O OHMBHFSEKCCCBW-UHFFFAOYSA-N 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- MLFHJEHSLIIPHL-UHFFFAOYSA-N isoamyl acetate Chemical compound CC(C)CCOC(C)=O MLFHJEHSLIIPHL-UHFFFAOYSA-N 0.000 description 1
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 1
- KXUHSQYYJYAXGZ-UHFFFAOYSA-N isobutylbenzene Chemical compound CC(C)CC1=CC=CC=C1 KXUHSQYYJYAXGZ-UHFFFAOYSA-N 0.000 description 1
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 1
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 150000002601 lanthanoid compounds Chemical class 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- IMXBRVLCKXGWSS-UHFFFAOYSA-N methyl 2-cyclohexylacetate Chemical compound COC(=O)CC1CCCCC1 IMXBRVLCKXGWSS-UHFFFAOYSA-N 0.000 description 1
- 229940057867 methyl lactate Drugs 0.000 description 1
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- SFMJNHNUOVADRW-UHFFFAOYSA-N n-[5-[9-[4-(methanesulfonamido)phenyl]-2-oxobenzo[h][1,6]naphthyridin-1-yl]-2-methylphenyl]prop-2-enamide Chemical compound C1=C(NC(=O)C=C)C(C)=CC=C1N1C(=O)C=CC2=C1C1=CC(C=3C=CC(NS(C)(=O)=O)=CC=3)=CC=C1N=C2 SFMJNHNUOVADRW-UHFFFAOYSA-N 0.000 description 1
- 229940017144 n-butyl lactate Drugs 0.000 description 1
- QJQAMHYHNCADNR-UHFFFAOYSA-N n-methylpropanamide Chemical compound CCC(=O)NC QJQAMHYHNCADNR-UHFFFAOYSA-N 0.000 description 1
- URXNVXOMQQCBHS-UHFFFAOYSA-N naphthalene;sodium Chemical compound [Na].C1=CC=CC2=CC=CC=C21 URXNVXOMQQCBHS-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- SJWFXCIHNDVPSH-UHFFFAOYSA-N octan-2-ol Chemical compound CCCCCCC(C)O SJWFXCIHNDVPSH-UHFFFAOYSA-N 0.000 description 1
- WYNVIVRXHYGNRT-UHFFFAOYSA-N octane-3,5-diol Chemical compound CCCC(O)CC(O)CC WYNVIVRXHYGNRT-UHFFFAOYSA-N 0.000 description 1
- 150000002921 oxetanes Chemical class 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- JCGNDDUYTRNOFT-UHFFFAOYSA-N oxolane-2,4-dione Chemical compound O=C1COC(=O)C1 JCGNDDUYTRNOFT-UHFFFAOYSA-N 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 238000000059 patterning Methods 0.000 description 1
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 1
- JYVLIDXNZAXMDK-UHFFFAOYSA-N pentan-2-ol Chemical compound CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 1
- GTCCGKPBSJZVRZ-UHFFFAOYSA-N pentane-2,4-diol Chemical compound CC(O)CC(C)O GTCCGKPBSJZVRZ-UHFFFAOYSA-N 0.000 description 1
- YWAKXRMUMFPDSH-UHFFFAOYSA-N pentene Chemical compound CCCC=C YWAKXRMUMFPDSH-UHFFFAOYSA-N 0.000 description 1
- GXOHBWLPQHTYPF-UHFFFAOYSA-N pentyl 2-hydroxypropanoate Chemical compound CCCCCOC(=O)C(C)O GXOHBWLPQHTYPF-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 229960005323 phenoxyethanol Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 229920002120 photoresistant polymer Polymers 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229920001281 polyalkylene Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920006294 polydialkylsiloxane Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 238000001955 polymer synthesis method Methods 0.000 description 1
- 239000003505 polymerization initiator Substances 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000010421 standard material Substances 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- BRGJIIMZXMWMCC-UHFFFAOYSA-N tetradecan-2-ol Chemical compound CCCCCCCCCCCCC(C)O BRGJIIMZXMWMCC-UHFFFAOYSA-N 0.000 description 1
- LFQCEHFDDXELDD-UHFFFAOYSA-N tetramethyl orthosilicate Chemical compound CO[Si](OC)(OC)OC LFQCEHFDDXELDD-UHFFFAOYSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- WLOQLWBIJZDHET-UHFFFAOYSA-N triphenylsulfonium Chemical compound C1=CC=CC=C1[S+](C=1C=CC=CC=1)C1=CC=CC=C1 WLOQLWBIJZDHET-UHFFFAOYSA-N 0.000 description 1
- 239000012953 triphenylsulfonium Substances 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- XMUJIPOFTAHSOK-UHFFFAOYSA-N undecan-2-ol Chemical compound CCCCCCCCCC(C)O XMUJIPOFTAHSOK-UHFFFAOYSA-N 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
Images





Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/09—Photosensitive materials characterised by structural details, e.g. supports, auxiliary layers
- G03F7/11—Photosensitive materials characterised by structural details, e.g. supports, auxiliary layers having cover layers or intermediate layers, e.g. subbing layers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F112/00—Homopolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F112/02—Monomers containing only one unsaturated aliphatic radical
- C08F112/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F112/06—Hydrocarbons
- C08F112/08—Styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F212/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F212/02—Monomers containing only one unsaturated aliphatic radical
- C08F212/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F212/06—Hydrocarbons
- C08F212/08—Styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F297/00—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer
- C08F297/02—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F297/00—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer
- C08F297/02—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type
- C08F297/026—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type polymerising acrylic acid, methacrylic acid or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/02—Compositions of unspecified macromolecular compounds characterised by the presence of specified groups, e.g. terminal or pendant functional groups
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D153/00—Coating compositions based on block copolymers containing at least one sequence of a polymer obtained by reactions only involving carbon-to-carbon unsaturated bonds; Coating compositions based on derivatives of such polymers
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/0002—Lithographic processes using patterning methods other than those involving the exposure to radiation, e.g. by stamping
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/038—Macromolecular compounds which are rendered insoluble or differentially wettable
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/039—Macromolecular compounds which are photodegradable, e.g. positive electron resists
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/039—Macromolecular compounds which are photodegradable, e.g. positive electron resists
- G03F7/0392—Macromolecular compounds which are photodegradable, e.g. positive electron resists the macromolecular compound being present in a chemically amplified positive photoresist composition
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/075—Silicon-containing compounds
- G03F7/0752—Silicon-containing compounds in non photosensitive layers or as additives, e.g. for dry lithography
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/16—Coating processes; Apparatus therefor
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/20—Exposure; Apparatus therefor
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/30—Imagewise removal using liquid means
- G03F7/32—Liquid compositions therefor, e.g. developers
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/30—Imagewise removal using liquid means
- G03F7/32—Liquid compositions therefor, e.g. developers
- G03F7/322—Aqueous alkaline compositions
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/30—Imagewise removal using liquid means
- G03F7/32—Liquid compositions therefor, e.g. developers
- G03F7/325—Non-aqueous compositions
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/40—Treatment after imagewise removal, e.g. baking
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/40—Treatment after imagewise removal, e.g. baking
- G03F7/405—Treatment with inorganic or organometallic reagents after imagewise removal
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/027—Making masks on semiconductor bodies for further photolithographic processing not provided for in group H01L21/18 or H01L21/34
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/027—Making masks on semiconductor bodies for further photolithographic processing not provided for in group H01L21/18 or H01L21/34
- H01L21/0271—Making masks on semiconductor bodies for further photolithographic processing not provided for in group H01L21/18 or H01L21/34 comprising organic layers
- H01L21/0273—Making masks on semiconductor bodies for further photolithographic processing not provided for in group H01L21/18 or H01L21/34 comprising organic layers characterised by the treatment of photoresist layers
- H01L21/0274—Photolithographic processes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/04—Manufacture or treatment of semiconductor devices or of parts thereof the devices having potential barriers, e.g. a PN junction, depletion layer or carrier concentration layer
- H01L21/18—Manufacture or treatment of semiconductor devices or of parts thereof the devices having potential barriers, e.g. a PN junction, depletion layer or carrier concentration layer the devices having semiconductor bodies comprising elements of Group IV of the Periodic Table or AIIIBV compounds with or without impurities, e.g. doping materials
- H01L21/30—Treatment of semiconductor bodies using processes or apparatus not provided for in groups H01L21/20 - H01L21/26
- H01L21/302—Treatment of semiconductor bodies using processes or apparatus not provided for in groups H01L21/20 - H01L21/26 to change their surface-physical characteristics or shape, e.g. etching, polishing, cutting
- H01L21/306—Chemical or electrical treatment, e.g. electrolytic etching
- H01L21/308—Chemical or electrical treatment, e.g. electrolytic etching using masks
- H01L21/3083—Chemical or electrical treatment, e.g. electrolytic etching using masks characterised by their size, orientation, disposition, behaviour, shape, in horizontal or vertical plane
- H01L21/3086—Chemical or electrical treatment, e.g. electrolytic etching using masks characterised by their size, orientation, disposition, behaviour, shape, in horizontal or vertical plane characterised by the process involved to create the mask, e.g. lift-off masks, sidewalls, or to modify the mask, e.g. pre-treatment, post-treatment
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/027—Making masks on semiconductor bodies for further photolithographic processing not provided for in group H01L21/18 or H01L21/34
- H01L21/0271—Making masks on semiconductor bodies for further photolithographic processing not provided for in group H01L21/18 or H01L21/34 comprising organic layers
Definitions
- the present invention relates to a pattern forming method and a composition.
- the present invention has been made based on the above-described circumstances, and an object thereof is to provide a pattern forming method and a composition capable of forming a pattern having a good shape and arrangement on a substrate.
- the invention made in order to solve the above problems includes a step of forming a basic pattern on the surface side of the substrate directly or via another layer (hereinafter also referred to as a “basic pattern forming step”), and a concave portion of the basic pattern. And a first composition (hereinafter referred to as “[A] polymer”) having at least two blocks and a solvent (hereinafter also referred to as “[S] solvent”).
- a step of filling the “composition (I)” hereinafter also referred to as “filling step” and a step of phase-separating the packed bed formed by the filling step (hereinafter also referred to as “phase separation step”).
- Step of etching the substrate The basic pattern forming step is a step of forming a resist pattern on the surface side of the substrate (hereinafter also referred to as “resist pattern forming step”), and at least a side surface of the resist pattern.
- the layer of the second polymer hereinafter also referred to as “[B] polymer” (hereinafter also referred to as “[B] polymer layer”) is placed on the surface of at least the substrate or other layer.
- a pattern forming method hereinafter also referred to as “pattern forming method (A)”.
- a first functional group (hereinafter referred to as “functional group (1) that binds to at least one terminal of the main chain and forms a chemical bond with at least one of —COOH and —OH”. ) ”) And Si—H, Si—OH, Si ⁇ O and Si—NR 2 (R each independently represents a carbon number of 1 to A polymer having a second functional group (hereinafter also referred to as “functional group (2)”) that forms a chemical bond with at least one of 20 monovalent organic groups) and a solvent. .
- Still another invention made in order to solve the above problems includes a step of laminating a basic pattern on the surface side of a substrate directly or via another layer (hereinafter also referred to as “basic pattern laminating step”), and the above basic A step of applying a fifth composition (hereinafter, also referred to as “composition (V)”) to the side and bottom surfaces of the concave portions of the pattern (hereinafter, also referred to as “coating step”) and the above-described coating step.
- a step of phase-separating the packed bed formed by the filling step (phase separation step), and a step of removing at least a part of the phase of the packed bed after the phase separation step.
- a pattern forming method comprising a step (etching step) of etching the substrate one or more times using a thinned pattern, wherein the basic pattern comprises a polymer having an aromatic ring content of 50% by mass or more as a main component And a fourth polymer (hereinafter also referred to as “[A ′] polymer”) having the first structural unit (hereinafter also referred to as “structural unit (I)”) and a solvent.
- a first block comprising the second structural unit (hereinafter also referred to as “structural unit (II)”) and the structural unit (II).
- block (B) Having a second block (hereinafter also referred to as “block (B)”) composed of a third structural unit (hereinafter also referred to as “structural unit (III)”) having a higher polarity than that of [B ′] polymer ”) and a solvent.
- structural unit (III) having a higher polarity than that of [B ′] polymer ”
- pattern forming method hereinafter also referred to as “pattern forming method (B)”.
- chemical bond is a concept including electrostatic attraction and hydrogen bond between molecules in addition to covalent bond, ionic bond, metal bond and coordination bond.
- the pattern forming methods (A) and (B) include a step of forming a basic pattern on the surface side of the substrate directly or via another layer, and a first having at least two blocks in the concave portion of the basic pattern.
- a step of etching the substrate directly or indirectly using the miniaturized pattern formed by the removing step wherein the basic pattern has a first layer on at least a side surface of the recess, and the recess
- the pattern forming method has a second layer having a contact angle different from that of the first layer on the surface side of at least the substrate or other layer.
- a pattern having a good shape and arrangement can be formed on a substrate. Therefore, these can be suitably used for lithography processes in manufacturing various electronic devices such as semiconductor devices and liquid crystal devices that are required to be further miniaturized.
- FIG. 2 is a schematic cross-sectional view showing an example of a state after forming a [B] polymer layer and a [C] polymer layer on the side surface of the resist pattern and the surface of the substrate in FIG. 1. It is typical sectional drawing which shows an example after filling the recessed part of the basic pattern in FIG. 2 with composition (I).
- FIG. 4 is a schematic cross-sectional view illustrating an example of a state after phase separation of the packed bed in FIG. 3.
- FIG. 5 is a schematic cross-sectional view showing an example of a state after removing a part of the phase of the packed bed after the phase separation step in FIG. 4.
- the pattern forming method includes a pattern forming method (A) and a pattern forming method (B). According to the pattern formation method (A), even when the pattern is fine, it is possible to form a fine pattern in which placement errors are suppressed and bottom residue is reduced. According to the pattern forming method (B), it is possible to form a fine pattern having excellent pattern size uniformity. According to the pattern forming method, a pattern having a good shape and arrangement can be formed on the substrate by using these excellent miniaturized patterns.
- the pattern formation method (A) and the pattern formation method (B) will be described.
- the pattern forming method (A) includes a basic pattern forming step, a filling step, a phase separation step, a removing step, and an etching step.
- the basic pattern forming step includes a resist pattern forming step and a polymer layer forming step.
- the pattern forming method (A) includes the above steps, and has a resist pattern forming step and a polymer layer forming step in the basic pattern forming step, and at least a side surface of the resist pattern in the polymer layer forming step.
- the [B] polymer layer is placed on the side surface of the resist pattern, that is, the side surface of the concave portion of the basic pattern, and the surface of the substrate or other layer, that is, the basic pattern.
- the [C] polymer layer is formed on the bottom surface of the recess, different layers are formed on the side surface and the bottom surface of the recess of the basic pattern. It is considered that the layer formed on the side surface and the bottom surface of the recess respectively causes the [A] polymer filling layer formed in the recess to be more appropriately phase-separated, and as a result, the pattern formed is fine. Even in this case, it is possible to form a pattern in which placement errors are suppressed and bottom residue is reduced.
- each step will be described.
- a basic pattern (hereinafter also referred to as “basic pattern (I)”) is formed directly on the surface side of the substrate or via another layer.
- the basic pattern forming step includes a resist pattern forming step and a polymer layer forming step.
- FIG. 1 shows a case where a resist pattern 2 is formed directly on the surface side of the substrate 1.
- FIG. 2 shows a case where the polymer layer 3 is formed in the concave portion of the resist pattern 2.
- the [B] polymer layer is formed on the side surface of the concave portion of the resist pattern, and the [C] polymer layer is formed on the bottom surface of the concave portion.
- a conventionally known substrate such as a silicon (Bare-Si) wafer, a silicon substrate such as silicon nitride, or a wafer coated with aluminum can be used.
- a silicon substrate is preferable, and a silicon wafer is more preferable.
- examples of other layers include a resist underlayer film, an SOG (Spin On Glass) film, and the like.
- the SOG film is a silicon-containing film.
- composition for forming the resist underlayer film examples include conventionally known organic underlayer film forming materials, and the like, for example, an underlayer film forming composition containing a crosslinking agent.
- composition for forming the SOG film a conventionally known SOG composition or the like can be used, and examples thereof include a composition containing an organic polysiloxane. Of these, the SOG film is preferable.
- the method for forming the SOG film is not particularly limited.
- the SOG composition is applied to one surface of the substrate or the surface of the lower layer opposite to the substrate 1 by a known method such as spin coating, and PB
- curing the coating film obtained by performing irradiation of a radiation and / or heating after performing is mentioned.
- radiation to be irradiated include electromagnetic waves such as visible light, ultraviolet light, far ultraviolet light, X-rays, and ⁇ rays; particle beams such as electron beams, molecular beams, and ion beams.
- 100 ° C is preferred, 150 ° C is more preferred, and 180 ° C is still more preferred.
- the lower limit of the heating time is preferably 5 seconds, more preferably 10 seconds, and even more preferably 20 seconds.
- the upper limit of the heating time is preferably 1,200 seconds, more preferably 600 seconds, and even more preferably 300 seconds.
- the lower limit of the average thickness of the SOG film is preferably 10 nm, more preferably 15 nm, and still more preferably 20 nm.
- the upper limit of the average thickness is preferably 1,000 nm, more preferably 500 nm, and still more preferably 100 nm.
- the substrate and other layers preferably have at least one of Si—H, Si—OH, Si ⁇ O, and Si—NR 2 on the top surface.
- Each R is independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms.
- Organic group refers to a group containing at least one carbon atom. Examples of the monovalent organic group include alkyl groups such as a methyl group and an ethyl group.
- R is preferably a hydrogen atom. That is, the upper part of the substrate and the other layers is preferably made of SiO 2 or SiN. When other layers are used, silicon-containing films are preferable as the other layers.
- the resist pattern 2 may be directly formed on the surface side of the substrate 1, or another layer on the surface side of the substrate 1, for example, a lower layer film, an SOG film or the like on the substrate 1. These films may be formed on the surface opposite to the substrate 1 of these films. Of these, the resist pattern 2 is preferably formed directly on the surface side of the substrate because the pattern can be easily formed on the substrate by etching using the formed resist pattern as a mask.
- Examples of the method of forming the resist pattern 2 include a method of forming the resist pattern by coating, exposing and developing a resist composition.
- the resist composition examples include a resist composition containing a polymer having an acid dissociable group and a radiation sensitive acid generator, a polymer having a crosslinkable group, and a negative resist composition containing a radiation sensitive acid generator. Conventional resist compositions such as those may be mentioned.
- the “acid-dissociable group” refers to a group that replaces a hydrogen atom such as —COOH or —OH and is dissociated by the action of an acid.
- Examples of the acid dissociable group that substitutes a hydrogen atom of —COOH include a tertiary hydrocarbon group, a t-alkyl group such as a t-butyl group and a t-amyl group, and 1-ethylcyclopentane-1- And 2-alkylbicycloalkane-2-yl group such as 1-alkylcycloalkane-1-yl group such as yl group and 2-methyladamantan-2-yl group.
- a resist composition is applied to the surface of the substrate 1 or the surface of another layer, and then pre-baked (PB) to form a resist film.
- exposure is performed through a mask pattern for forming a resist pattern 2 having a desired shape.
- radiation used for exposure include ultraviolet rays, far ultraviolet rays, extreme ultraviolet rays (EUV), electromagnetic waves such as X-rays, and charged particle beams such as electron beams and ⁇ rays.
- far ultraviolet rays are preferable, ArF excimer laser light and KrF excimer laser light are more preferable, and ArF excimer laser light is more preferable.
- immersion exposure can also be performed.
- PEB post-exposure baking
- a developer such as an alkali developer such as an aqueous 2.38 mass% tetramethylammonium hydroxide solution or an aqueous tetrabutylammonium hydroxide solution, or an organic solvent developer such as butyl acetate or anisole.
- the lower limit of the average thickness of the resist film is preferably 10 nm, more preferably 30 nm, and even more preferably 50 nm.
- the upper limit of the average thickness is preferably 1,000 nm, more preferably 500 nm, and even more preferably 200 nm.
- the shape of the resist pattern 2 can be appropriately selected according to the shape of the pattern finally formed on the substrate.
- the shape is a circle (substantially perfect circle), an ellipse (oval), or a square. , Rectangular shape, bowl shape, trapezoidal shape, triangular shape and the like. Among these, a circular shape is preferable.
- the lower limit of the average diameter is preferably 10 nm, more preferably 20 nm, still more preferably 30 nm, and particularly preferably 40 nm.
- the upper limit of the average diameter is preferably 200 nm, more preferably 100 nm, still more preferably 90 nm, and particularly preferably 80 nm.
- the lower limit of the pitch of the resist pattern 2 to be formed is preferably 30 nm, more preferably 50 nm, still more preferably 70 nm, and particularly preferably 90 nm.
- the upper limit of the pitch is preferably 1,000 nm, more preferably 500 nm, still more preferably 200 nm, and particularly preferably 150 nm.
- the resist pattern 2 preferably has at least one of —COOH and —OH on the side surface.
- Such a resist pattern 2 having at least one of —COOH and —OH on its side surface is, for example, applied to a resist composition containing a polymer having an acid-dissociable group and a radiation-sensitive acid generator, exposure, and an organic solvent. It can be formed by development with a developer. In addition, after applying a resist composition containing a polymer having an acid-dissociable group and a radiation-sensitive acid generator, exposure and development with an alkali developer, the resulting resist film is heated or exposed. Thus, it can also be formed by dissociating the acid dissociable group in the resist film.
- the [B] polymer layer is formed on at least the side surface of the resist pattern 2
- the [C] polymer layer is formed on at least the surface of the substrate or another layer.
- This step includes a step [B] forming a polymer layer (hereinafter also referred to as “[B] polymer layer forming step”) and a step [C] forming a polymer layer (hereinafter referred to as “[C] These steps may be performed separately or at the same time.
- composition (II) containing, for example, a [B] polymer and a solvent (hereinafter also referred to as “[S] solvent”) (hereinafter referred to as “B”). And a method of performing “composition (II) coating step”).
- composition (II) contains a [B] polymer and a [S] solvent.
- the composition (II) may contain other components in addition to the [B] polymer and the [S] solvent.
- the polymer preferably has a functional group (1) bonded to at least one terminal of the main chain and chemically bonded to at least one of —COOH and —OH.
- Main chain refers to the longest of the atomic chains of a polymer.
- Examples of the functional group (1) include a hydroxyl group, an epoxy group (oxiranyl group), and an oxetanyl group. These functional groups form a covalent bond with —COOH and / or —OH.
- Examples of the [B] polymer include a styrene polymer, a (meth) acrylic polymer, an ethylene polymer, and a copolymer combining these.
- the styrene polymer has a structural unit derived from substituted or unsubstituted styrene.
- substituted styrene examples include ⁇ -methylstyrene, o-, m-, p-methylstyrene, pt-butylstyrene, 2,4,6-trimethylstyrene, p-methoxystyrene, pt-butoxystyrene, o-, m-, p-vinylstyrene, o-, m-, p-hydroxystyrene, m-, p-chloromethylstyrene, p-chlorostyrene, p-bromostyrene, p-iodostyrene, p-nitrostyrene , P-cyanostyrene and the like.
- the (meth) acrylic polymer has a structural unit derived from (meth) acrylic acid or (meth) acrylic acid ester.
- (meth) acrylic acid esters examples include (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethyl (meth) acrylate, t-butyl (meth) acrylate, and 2-ethylhexyl (meth) acrylate. ; Cyclopentyl (meth) acrylate, cyclohexyl (meth) acrylate, 1-methylcyclopentyl (meth) acrylate, 2-ethyladamantyl (meth) acrylate, 2- (adamantan-1-yl) propyl (meth) acrylate, etc.
- the ethylene polymer has a structural unit derived from substituted or unsubstituted ethylene.
- substituted ethylene examples include alkenes such as propene, butene, and pentene; Vinylcycloalkanes such as vinylcyclopentane and vinylcyclohexane; Cycloalkenes such as cyclopentene and cyclohexene; Examples include 4-hydroxy-1-butene, vinyl glycidyl ether, vinyl trimethylsilyl ether, and the like.
- the lower limit of the weight average molecular weight (Mw) of the polymer is preferably 1,000, more preferably 3,000, still more preferably 5,000, and particularly preferably 5,500.
- the upper limit of Mw is preferably 100,000, more preferably 50,000, still more preferably 15,000, and particularly preferably 10,000.
- the upper limit of the ratio (dispersion degree) of the Mw to the number average molecular weight (Mn) of the polymer is preferably 5, more preferably 3, more preferably 2, and particularly preferably 1.3.
- the lower limit of the ratio is usually 1 and preferably 1.05.
- Mw and Mn in this specification are GPC columns (two "G2000HXL”, one "G3000HXL”, one “G4000HXL” from Tosoh Corporation), a flow rate of 1.0 mL / min, elution solvent: tetrahydrofuran, sample concentration : Measured by gel permeation chromatography (GPC) using a monodisperse polystyrene as a standard using a differential refractometer as a detector under the analysis conditions of 1.0 mass%, sample injection amount: 100 ⁇ L, column temperature: 40 ° C. It is a thing.
- GPC gel permeation chromatography
- the lower limit of the content of the polymer is preferably 80% by mass, more preferably 90% by mass, and still more preferably 95% by mass with respect to the total solid content in the composition (II). As an upper limit of the said content, it is 100 mass%, for example. “Total solids” refers to the sum of components other than the [S] solvent.
- the solvent is not particularly limited as long as it is a solvent that can dissolve or disperse at least the [B] polymer and other components.
- Solvents include, for example, alcohol solvents, ether solvents, ketone solvents, amide solvents, ester solvents, hydrocarbon solvents, and the like.
- alcohol solvent examples include methanol, ethanol, n-propanol, iso-propanol, n-butanol, iso-butanol, sec-butanol, tert-butanol, n-pentanol, iso-pentanol, 2-methylbutanol, sec-pentanol, tert-pentanol, 3-methoxybutanol, n-hexanol, 2-methylpentanol, sec-hexanol, 2-ethylbutanol, sec-heptanol, 3-heptanol, n-octanol, 2-ethylhexanol , Sec-octanol, n-nonyl alcohol, 2,6-dimethyl-4-heptanol, n-decanol, sec-undecyl alcohol, trimethylnonyl alcohol, sec-tetradecyl alcohol, sec -und
- ether solvents examples include dialkyl ether solvents such as diethyl ether, dipropyl ether, and dibutyl ether; Cyclic ether solvents such as tetrahydrofuran and tetrahydropyran; And aromatic ring-containing ether solvents such as diphenyl ether and anisole.
- ketone solvent examples include acetone, methyl ethyl ketone, methyl-n-propyl ketone, methyl-n-butyl ketone, diethyl ketone, methyl-iso-butyl ketone, 2-heptanone, ethyl-n-butyl ketone, methyl-n-hexyl ketone, Linear ketone solvents such as di-iso-butyl ketone and trimethylnonanone; Cyclic ketone solvents such as cyclopentanone, cyclohexanone, cycloheptanone, cyclooctanone, methylcyclohexanone; Examples include 2,4-pentanedione, acetonylacetone, acetophenone, and the like.
- amide solvent examples include cyclic amide solvents such as N, N′-dimethylimidazolidinone and N-methylpyrrolidone; Examples thereof include chain amide solvents such as N-methylformamide, N, N-dimethylformamide, N, N-diethylformamide, acetamide, N-methylacetamide, N, N-dimethylacetamide, and N-methylpropionamide.
- cyclic amide solvents such as N, N′-dimethylimidazolidinone and N-methylpyrrolidone
- chain amide solvents such as N-methylformamide, N, N-dimethylformamide, N, N-diethylformamide, acetamide, N-methylacetamide, N, N-dimethylacetamide, and N-methylpropionamide.
- ester solvents include methyl acetate, ethyl acetate, n-propyl acetate, iso-propyl acetate, n-butyl acetate, iso-butyl acetate, sec-butyl acetate, n-pentyl acetate, i-pentyl acetate, sec Acetate solvents such as pentyl, 3-methoxybutyl acetate, methylpentyl acetate, 2-ethylbutyl acetate, 2-ethylhexyl acetate, benzyl acetate, cyclohexyl acetate, methylcyclohexyl acetate, n-nonyl acetate; Ethylene glycol monomethyl ether acetate, ethylene glycol monoethyl ether acetate, diethylene glycol monomethyl ether acetate, diethylene glycol monoethyl ether acetate, diethylene glycol mono-l
- hydrocarbon solvents examples include n-pentane, iso-pentane, n-hexane, iso-hexane, n-heptane, iso-heptane, 2,2,4-trimethylpentane, n-octane, iso-octane, cyclohexane , Aliphatic hydrocarbon solvents such as methylcyclohexane; Fragrances such as benzene, toluene, xylene, mesitylene, ethylbenzene, trimethylbenzene, methylethylbenzene, n-propylbenzene, iso-propylbenzene, diethylbenzene, iso-butylbenzene, triethylbenzene, di-iso-propylbenzene, n-amylnaphthalene Group hydrocarbon solvents and the like.
- Composition (II) may contain one or more [S] solvents.
- composition (II) may contain, for example, a surfactant as other components.
- Composition (II) can improve the coating property to the resist pattern 2 by containing surfactant.
- Examples of the coating method of the composition (II) include a spin coating method.
- composition (III) coating step As a method for forming a polymer layer, for example, a step of applying a composition (III) containing a [C] polymer and a [S] solvent (hereinafter referred to as “composition (III) coating step”). And the like.
- composition (III) contains a [C] polymer and a [S] solvent.
- the composition (III) may contain components other than the [C] polymer and the [S] solvent.
- Examples of the functional group (2) include a carboxy group, a hydroxyl group, and a halogen atom. These functional groups form covalent bonds with Si—H, Si—OH, Si ⁇ O and / or Si—NR 2 .
- Examples of the [C] polymer include a styrene polymer, a (meth) acrylic polymer, an ethylene polymer, and a copolymer combining these. Examples of these polymers include polymers similar to those exemplified as the [B] polymer.
- the lower limit of the weight average molecular weight (Mw) of the polymer is preferably 1,000, more preferably 3,000, still more preferably 4,000, and particularly preferably 5,000.
- the upper limit of the Mw is preferably 100,000, more preferably 50,000, still more preferably 20,000, and particularly preferably 10,000.
- the upper limit of the ratio (dispersion degree) of the Mw to the number average molecular weight (Mn) of the polymer is preferably 5, more preferably 3, more preferably 2, and particularly preferably 1.4.
- the lower limit of the ratio is usually 1 and preferably 1.1.
- the lower limit of the content of the polymer is preferably 80% by mass, more preferably 90% by mass, and still more preferably 95% by mass with respect to the total solid content in the composition (III).
- As an upper limit of the said content it is 100 mass%, for example.
- Examples of the [S] solvent and other components in the composition (III) include the same as those exemplified as the [S] solvent and other components in the composition (II).
- Examples of the coating method of the composition (III) include a spin coating method.
- composition (IV) coating step when the [B] polymer layer forming step and the [C] polymer layer forming step are simultaneously performed, the [B] polymer, the [C] polymer, and the [S] solvent are added.
- a step of applying a composition hereinafter, also referred to as “composition (IV)” (hereinafter, also referred to as “composition (IV) coating step”) may be performed.
- composition (IV) contains a [B] polymer, a [C] polymer, and a [S] solvent.
- the composition (IV) may contain components other than the [B] polymer, the [C] polymer, and the [S] solvent.
- the [B] polymer, [C] polymer, [S] solvent and other components in the composition (IV) are the same as those exemplified as the [S] solvent and other components in the composition (II). And the like.
- the lower limit of the total content of the [B] polymer and the [C] polymer is preferably 80% by mass, more preferably 90% by mass, and 95% by mass with respect to the total solid content in the composition (IV). Further preferred.
- As an upper limit of the said content it is 100 mass%, for example.
- the [B] polymer and the [C] polymer can be synthesized by polymerization such as anionic polymerization and radical polymerization, but living anionic polymerization in which any terminal structure can be easily introduced is preferable.
- living anionic polymerization for example, a monomer such as styrene is polymerized, and the polymerization terminal is treated with an arbitrary terminal treating agent to introduce a functional group that is bonded to the terminal of the main chain of the polymer. Can do.
- a polymer composed of a polystyrene block having a functional group (1) or a functional group (2) at the terminal it is first activated by polymerizing styrene in an appropriate solvent using an anionic polymerization initiator. A terminal styrene polymer is formed. Then, the polymer by which the functional group was introduce
- examples of the terminal treatment method include a method as shown in the following scheme. That is, a terminal treatment agent such as 1,2-butylene oxide is added to the active terminal during polymerization to modify the terminal to obtain a polymer having a functional group bonded to the terminal of the main chain.
- a terminal treatment agent such as 1,2-butylene oxide
- n and m are integers of 10 to 5,000.
- Examples of the terminal treatment agent that gives a hydroxyl group include epoxy compounds such as 1,2-butylene oxide, butyl glycidyl ether, 2-ethylhexyl glycidyl ether, propylene oxide, ethylene oxide, styrene oxide, and epoxyamine.
- Examples of the end treatment agent that gives a carboxy group include carbon dioxide.
- Examples of the end treatment agent that gives an epoxy group include halogen atom-containing epoxy compounds such as epibromohydrin and epichlorohydrin.
- Examples of the end treating agent that gives an oxetanyl group include halogen atom-containing oxetane compounds such as 3-chloromethyl-3-methyloxetane and 3-chloromethyl-3-ethyloxetane.
- a polymer having a carboxy group at the terminal is, for example, a monomer such as styrene and methyl 4-cyano-4-[(dodecylsulfanylthiocarbonylsulfane) pentanoate or the like in a solvent such as anisole. It can be obtained by performing polymerization (Reversible Addition Fragmentation Chain Transfer Polymerization, reversible addition-fragmentation chain transfer polymerization).
- the polymer having a halogen atom at the terminal is prepared by charging a monomer such as styrene, an amine compound such as copper (II) bromide and tris [(2-dimethylamino) ethyl] amine, and a solvent such as anisole. Further, it can be synthesized by atom transfer radical polymerization by adding a halogen compound such as 2-hydroxyethyl 2-bromoisobutyrate to this.
- the [B] polymer and [C] polymer obtained above are preferably recovered by a reprecipitation method. That is, after completion of the terminal treatment reaction, the target copolymer is recovered as a powder by introducing the reaction solution into a reprecipitation solvent.
- a reprecipitation solvent alcohols or alkanes can be used alone or in admixture of two or more.
- the polymer can be recovered by removing low-molecular components such as monomers and oligomers by a liquid separation operation, a column operation, an ultrafiltration operation, or the like.
- the [B] polymer and the [C] polymer are preferably subjected to purification such as demetallation with an acid or the like.
- compositions (II) to (IV) for example, the [B] polymer and / or the [C] polymer, the [S] solvent and other components as necessary are mixed at a predetermined ratio, and preferably the pore size is 0. It can be prepared by filtering with a high-density polyethylene filter of about 45 ⁇ m.
- the lower limit of the solid content concentration of the compositions (II) to (IV) is preferably 0.1% by mass, more preferably 0.5% by mass, and even more preferably 0.7% by mass.
- the upper limit of the solid content concentration is preferably 30% by mass, more preferably 10% by mass, and still more preferably 3% by mass.
- the basic pattern forming step includes a resist pattern forming step, a [B] polymer layer forming step, and a [C] polymer layer forming step
- these steps can be performed in various orders as described below.
- the basic pattern forming step includes, for example: (1) resist pattern forming step, [B] polymer layer forming step, [C] polymer layer forming step in order (2) resist pattern forming step, [C] polymer layer (3) [C] Polymer layer forming step, resist pattern forming step, [B] polymer layer forming step, etc.
- the above (1) and (3) are preferable from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer.
- the order of performing these steps is, for example, (A) Resist pattern forming step The order of the composition (II) coating process, the composition (III) coating process (B) The resist pattern formation process, the composition (III) coating process, the order of the composition (II) coating process (C) The composition (III) coating step, the resist pattern forming step, the composition (II) coating step, and the like can be performed in this order.
- the above (A) and (C) are preferable from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer.
- the resist pattern forming step when performing the composition (IV) coating step, is performed from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer. It is preferable to perform a composition (IV) coating process later.
- the concave portion of the basic pattern (I) is filled with the composition (I) containing [A] a polymer and a solvent.
- composition (I) contains a [A] polymer and a solvent.
- the composition (I) can contain other components in addition to the [A] polymer and the solvent.
- the polymer is a polymer having at least two blocks. That is, the [A] polymer is a block copolymer.
- the polymer may be a diblock copolymer, a triblock copolymer, or a copolymer having four or more blocks. Among these, from the viewpoint of easy formation of the phase separation structure, a diblock copolymer and a triblock copolymer are preferable, and a diblock copolymer is more preferable.
- the polymer may have a linking group between adjacent blocks.
- the polymer When the polymer is a diblock copolymer, it has a block (I) and a block (II). In the block (II), the polarity of the structural unit constituting the block (I) is higher.
- Block (I) examples of the block (I) include a polystyrene block.
- the polystyrene block is a block containing a structural unit derived from substituted or unsubstituted styrene. Among these, a block containing a structural unit derived from styrene is preferable.
- Block (II) examples of the block (II) include a poly (meth) acrylic acid ester block, a polyalkylene glycol block, a polyester block, a polyalkylene carbonate block, a polydialkylsiloxane block, and a poly (meth) acrylic acid alkylsilyl ester block.
- the poly (meth) acrylate block is a block including a structural unit derived from (meth) acrylate. Among these, a poly (meth) acrylate block is preferable, a poly (meth) acrylate block is more preferable, a poly (meth) acrylate block is more preferable, and a polymethyl methacrylate block is particularly preferable.
- linking group examples include a divalent organic group having 1 to 50 carbon atoms.
- examples of the monomer that gives a linking group include diphenylethylene and stilbene.
- diphenylethylene and stilbene synthesize the [A] polymer by anionic polymerization, the anion terminal produced in the middle can be stabilized. Thereby, the degree of dispersion (Mw / Mn ratio) of the obtained [A] polymer becomes smaller, and as a result, the variation in the dimension of the formed pattern can be made smaller.
- the polymer may have one or more linking groups depending on the number of blocks, the target pattern shape, and the like.
- the lower limit of the weight average molecular weight (Mw) of the polymer by gel permeation chromatography (GPC) is preferably 1,000, more preferably 10,000, and even more preferably 50,000.
- the upper limit of Mw is preferably 300,000, more preferably 200,000, and even more preferably 100,000.
- the upper limit of the ratio (Mw / Mn, dispersity) between the polymer Mw and the number average molecular weight (Mn) is preferably 5, more preferably 3, more preferably 2, and particularly preferably 1.5. 1.1 is more particularly preferable.
- the lower limit of the above ratio is usually 1 and preferably 1.01.
- the lower limit of the content of the polymer is preferably 80% by mass, more preferably 90% by mass, still more preferably 95% by mass, and 99% by mass with respect to the total solid content in the composition (I). Particularly preferred.
- the “total solid content” of the composition (I) refers to the sum of components other than the solvent.
- the lower limit of the concentration of the [A] polymer in the composition (I) is preferably 0.3% by mass, more preferably 0.7% by mass, further preferably 1.0% by mass, and 1.3% by mass. Is particularly preferred.
- the upper limit of the concentration is preferably 5% by mass, more preferably 3% by mass, still more preferably 2% by mass, and particularly preferably 1.7% by mass.
- the polymer can be synthesized by living anion polymerization, living radical polymerization or the like, but living anion polymerization is more preferable.
- a polystyrene block, a polymethyl methacrylate block, and other blocks other than these can be linked while polymerizing in a desired order, and the polymerization terminal can be synthesized by treating with an arbitrary terminal treating agent.
- Examples of the solvent used for the polymerization include alkanes such as n-pentane, n-hexane, n-heptane, n-octane, n-nonane, and n-decane; Cycloaliphatic hydrocarbons such as cyclohexane, cycloheptane, cyclooctane, decalin, norbornane; Aromatic hydrocarbons such as benzene, toluene, xylene, ethylbenzene, cumene; Saturated carboxylic acid esters such as ethyl acetate, n-butyl acetate, i-butyl acetate and methyl propionate; Ketones such as acetone, methyl ethyl ketone, 4-methyl-2-pentanone, 2-heptanone; Ethers such as tetrahydrofuran, dimethoxyethanes, diethoxye
- the reaction temperature in the above polymerization may be appropriately determined according to the type of initiator, but the lower limit of the reaction temperature is preferably ⁇ 150 ° C., more preferably ⁇ 80 ° C. As an upper limit of the said reaction temperature, 50 degreeC is preferable and 40 degreeC is more preferable. As a minimum of reaction time, 5 minutes are preferred and 20 minutes are more preferred. The upper limit of the reaction time is preferably 24 hours, and more preferably 12 hours.
- Examples of the initiator used for the polymerization include alkyl lithium, alkyl magnesium halide, sodium naphthalene, alkylated lanthanoid compounds, and the like. Among these, when polymerizing using styrene, methyl methacrylate or the like as a monomer, it is preferable to use an alkyl lithium compound.
- an end treatment agent such as an alcohol such as methanol or an epoxy compound such as 2-ethylhexyl glycidyl ether is added to modify the end of the chain during the polymerization reaction. Coalescence can be obtained.
- the obtained block copolymer is preferably subjected to purification such as demetallation.
- ⁇ Phase separation process> the packed bed 4 formed by the above filling step is phase-separated. As a result, as shown in FIG. 4, the packed bed 4 is phase-separated into a block ( ⁇ ) phase 4a and a block ( ⁇ ) phase 4b.
- an annealing method or the like can be given as a method for phase-separating the packed bed 4.
- Examples of the annealing method include a heating method.
- Examples of the heating means include an oven and a hot plate.
- As a minimum of the temperature of heating 80 ° C is preferred, 100 ° C is more preferred, and 150 ° C is still more preferred.
- As an upper limit of the temperature of heating 400 degreeC is preferable, 350 degreeC is more preferable, and 300 degreeC is further more preferable.
- the lower limit of the heating time is preferably 10 seconds, more preferably 1 minute, and even more preferably 10 minutes.
- the upper limit of the heating time is preferably 120 minutes, more preferably 60 minutes, and even more preferably 30 minutes.
- a part of the block ( ⁇ ) phase 4b in the phase separation structure of the packed bed 4 is removed.
- the polymethyl methacrylate block phase 4b can be removed by etching using the difference in the etching rate of each phase separated.
- FIG. 5 shows a state after the polymethyl methacrylate block phase 4b in the phase separation structure is removed.
- the radiation when the phase to be removed by etching is a polymethyl methacrylate block phase, 172 nm radiation can be used. Since the polymethyl methacrylate block phase is decomposed by the radiation irradiation, etching becomes easier.
- Examples of a method for removing the polymethyl methacrylate block phase 4b in the phase separation structure include reactive ion etching (RIE) such as chemical dry etching and chemical wet etching; physical etching such as sputter etching and ion beam etching.
- RIE reactive ion etching
- physical etching such as sputter etching and ion beam etching.
- reactive ion etching is preferable, chemical dry etching using CF 4 , O 2 gas, etc., and chemical wet etching (wet development) using a liquid etching solution such as an organic solvent or hydrofluoric acid is more preferable.
- chemical wet etching is more preferable.
- organic solvents used for chemical wet etching include alkanes such as n-pentane, n-hexane, and n-heptane; Cycloalkanes such as cyclohexane, cycloheptane, cyclooctane; Saturated carboxylic acid esters such as ethyl acetate, n-butyl acetate, i-butyl acetate and methyl propionate; Ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone (MIBK), methyl n-pentyl ketone; Examples include alcohols such as methanol, ethanol, 1-propanol, isopropanol (IPA), and 4-methyl-2-pentanol. These solvents may be used alone or in combination of two or more. Among these, a mixed solvent with MIBK and IPA is preferable.
- the substrate is etched by directly or indirectly using the miniaturized pattern formed in the removing step.
- the miniaturized pattern is formed of the basic pattern (I) 2, the polymer layer 3, and the block ( ⁇ ) phase that has not been removed in the removing step.
- a substrate pattern is formed.
- the pattern formation method (A) it is possible to obtain a substrate pattern in which a placement error is suppressed and the shape is good.
- the fine pattern is usually used directly, that is, the etching is performed once to form the substrate pattern. obtain.
- the fine pattern is usually used indirectly, that is, the fine pattern is used to etch other layers, Etching is sequentially performed using the other layers after etching as a mask, that is, etching is performed a plurality of times to obtain a substrate pattern.
- Examples of the substrate pattern to be obtained include a hole pattern.
- etching method for example, CF 4 , O 2 gas or the like is used, chemical dry etching using a difference in etching rate of each layer or the like, chemical wet etching using a liquid etching solution such as an organic solvent or hydrofluoric acid (wet type).
- a liquid etching solution such as an organic solvent or hydrofluoric acid (wet type).
- RIE reactive ion etching
- physical etching such as sputter etching and ion beam etching.
- reactive ion etching is preferable, chemical dry etching and chemical wet etching are more preferable, and chemical dry etching is more preferable.
- the portion used as a mask is removed from the surface side of the substrate by dissolution treatment or the like, and a substrate on which a pattern is finally formed can be obtained.
- the substrate obtained by the pattern forming method (A) is suitably used for a semiconductor element and the like, and this semiconductor element is widely used for LEDs, solar cells and the like.
- the pattern forming method (B) includes a basic pattern lamination step, a coating step, a heating step, a filling step, a phase separation step, a removal step, and an etching step.
- the pattern formation method (B) comprises the above steps, and the pattern size is uniform by setting the difference in the contact angle between the side surface and the bottom surface of the concave portion of the basic pattern in the coating film after the heating step to the above value or more.
- a fine pattern having excellent properties can be formed, and by using such an excellent fine pattern, a pattern having a favorable shape and arrangement can be formed on the substrate.
- a basic pattern (hereinafter also referred to as “basic pattern (II)”) is laminated directly or via another layer on the surface side of the substrate.
- basic pattern (II) what is directly laminated
- This basic pattern (II) is mainly composed of a polymer having an aromatic ring content of 50% by mass or more. As a thing which has as a main component the polymer whose content rate of an aromatic ring is 50 mass% or more, a resist underlayer film etc. are mentioned, for example.
- Examples of the method of laminating the basic pattern (II) include the following methods.
- a resist underlayer film is formed on the surface side of the substrate directly or via another layer.
- an SOG film may be formed on the surface of the resist underlayer film opposite to the substrate.
- a resist pattern is formed on the surface of the resist underlayer film or SOG film opposite to the substrate.
- the SOG film and / or the resist underlayer film are sequentially etched using the resist pattern as a mask.
- Examples of methods for forming the substrate, other layers, the resist underlayer film, the SOG film, and the resist pattern include those similar to those exemplified in the basic pattern forming step of the pattern forming method (A).
- the substrate is preferably made of silicon.
- composition (V) is applied to the side surface and the bottom surface of the concave portion of the basic pattern (II).
- composition (V) contains a [A ′] polymer and a solvent ([S] solvent).
- the composition (V) may contain other components such as a surfactant in addition to the [A ′] polymer and the [S] solvent.
- the polymer has the structural unit (I).
- the structural unit (I) for example, a structural unit derived from substituted or unsubstituted styrene, a structural unit derived from (meth) acrylic acid or (meth) acrylic acid ester, or a structural unit derived from substituted or unsubstituted ethylene Etc.
- the substituted or unsubstituted styrene, (meth) acrylic acid ester, and substituted or unsubstituted ethylene include the same as those exemplified in the [B] polymer of the pattern forming method (A).
- the polymer preferably has a functional group that is bonded to at least one end of the main chain and chemically bonds to at least one of —COOH and —OH.
- functional groups include the functional groups exemplified as the functional group (1) of the [B] polymer in the pattern forming method (A).
- [A ′] Mw and Mw / Mn of the polymer, and a preferable range of the content of [A ′] polymer in the composition (V) include the [B] polymer and the composition in the pattern forming method (A). Same as (II).
- Examples of the [S] solvent include the same solvents as those exemplified as the [S] solvent of the composition (II) in the pattern formation method (A).
- composition (V) is prepared, for example, by mixing the [A ′] polymer, the [S] solvent, and other components as necessary at a predetermined ratio, and preferably filtering through a membrane filter having a pore diameter of about 200 nm. can do.
- a minimum of solid content concentration of composition (V) 0.1 mass% is preferred, 0.5 mass% is more preferred, and 0.7 mass% is still more preferred.
- the upper limit of the solid content concentration is preferably 30% by mass, more preferably 10% by mass, and still more preferably 3% by mass.
- Examples of the coating method of the composition (V) include a spin coating method.
- the coating film formed by the coating step is heated.
- the lower limit of the difference between ⁇ 1 and ⁇ 2 is preferably 7 ° and more preferably 10 °.
- the upper limit of the difference between ⁇ 1 and ⁇ 2 is, for example, 30 °.
- the lower limit of ⁇ 1 is preferably 50 °, more preferably 80 °.
- the upper limit of ⁇ 1 is preferably 90 ° and more preferably 88 °.
- the lower limit of ⁇ 2 is preferably 60 °, and more preferably 70 °.
- the upper limit of ⁇ 2 is preferably 85 ° and more preferably 80 °.
- Examples of the heating means include an oven and a hot plate.
- the temperature or time is controlled so that the difference between ⁇ 1 and ⁇ 2 is greater than or equal to the above value.
- the temperature and time in the heating step vary depending on, for example, the type of functional group of the [A ′] polymer of the composition (V).
- the lower limit of the heating temperature is preferably 50 ° C, more preferably 100 ° C, further preferably 120 ° C, and particularly preferably 140 ° C.
- 250 degreeC is preferable
- 200 degreeC is more preferable
- 180 degreeC is further more preferable
- 160 degreeC is especially preferable.
- the lower limit of the heating time is preferably 10 seconds, more preferably 1 minute, further preferably 5 minutes, and particularly preferably 10 minutes.
- the upper limit of the time is preferably 10 hours, more preferably 1 hour, still more preferably 30 minutes, and particularly preferably 20 minutes.
- composition (VI) contains a [B ′] polymer and a solvent ([S] solvent).
- the composition (VI) can contain other components in addition to the [B ′] polymer and the [S] solvent.
- the polymer has a block (A) and a block (B). That is, the [B ′] polymer is a block copolymer.
- the block (A) is composed of the structural unit (II)
- the block (B) is composed of the structural unit (III) having a higher polarity than the structural unit (II).
- the polymer may be a diblock copolymer, a triblock copolymer, or a copolymer having four or more blocks. Among these, from the viewpoint of easy formation of the phase separation structure, a diblock copolymer and a triblock copolymer are preferable, and a diblock copolymer is more preferable.
- the [B ′] polymer may have a linking group between adjacent blocks.
- the block (A) is preferably a polystyrene block exemplified as the block (I) of the [A] polymer in the pattern formation method (A). That is, as the structural unit (II), a structural unit derived from substituted or unsubstituted styrene is preferable.
- the block (B) include the blocks exemplified as the block (II) of the [A] polymer.
- a poly (meth) acrylate block is preferable, and a polymethyl methacrylate block is more preferable.
- the structural unit (III) a structural unit derived from a (meth) acrylic acid ester is preferable.
- [B ′] Mw and Mw / Mn of the polymer, and the content and concentration of the [B ′] polymer in the composition (VI) are the [A] polymer and the composition of the pattern formation method (A) ( A range similar to that in I) is preferred.
- Phase separation process In this step, the packed bed formed by the above filling step is phase-separated. This step is the same as the phase separation step in the pattern forming method (A).
- composition includes a polymer having a functional group bonded to at least one end of the main chain and forming a chemical bond with at least one of —COOH and —OH, and bonded to at least one end of the main chain. , Si—OH, Si ⁇ O, and Si—NR 2 (wherein R is each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms) that forms a chemical bond.
- R is each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms
- composition is described as the composition (II) used in the pattern formation method (A).
- Mw and Mn of the polymer are measured by gel permeation chromatography (GPC) using Tosoh's GPC columns ("G2000HXL", “G3000HXL” and “G4000HXL”) under the following conditions. did.
- Eluent Tetrahydrofuran (Wako Pure Chemical Industries)
- Flow rate 1.0 mL / min
- Sample concentration 1.0% by mass
- Sample injection volume 100 ⁇ L
- Detector Differential refractometer Standard material: Monodisperse polystyrene
- This polymer (a-1) had Mw of 79,000, Mn of 77,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (a-1) contained 65 mol% and 35 mol% of structural units derived from styrene and structural units derived from methyl methacrylate, respectively. .
- the polymer (a-1) is a diblock copolymer.
- This polymer (a-2) had Mw of 78,000, Mn of 76,000, and Mw / Mn of 1.03.
- the polymer (A-2) contained 65 mol% and 35 mol% of structural units derived from styrene and structural units derived from methyl methacrylate, respectively. .
- the polymer (a-2) is a diblock copolymer.
- reaction was continued for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed.
- This polymer (a-3) had Mw of 77,000, Mn of 75,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (a-3) was found to contain 70 mol% and 30 mol% of structural units derived from styrene and methyl methacrylate, respectively. .
- the block copolymer (a-3) is a diblock copolymer.
- This polymer (b-1) had Mw of 5,600, Mn of 5,300, and Mw / Mn of 1.06.
- This block copolymer (b-2) had Mw of 5,500, Mn of 5,100, and Mw / Mn of 1.08.
- This polymer (b-3) had Mw of 5,200, Mn of 5,000, and Mw / Mn of 1.04.
- This polymer (b-4) had Mw of 5,700, Mn of 5,200, and Mw / Mn of 1.10.
- This polymer (b-5) had Mw of 3,300, Mn of 3,100, and Mw / Mn of 1.06.
- This block copolymer (b-6) had an Mw of 11,600, an Mn of 10,400, and an Mw / Mn of 1.12.
- This block copolymer (b-7) had Mw of 5,900, Mn of 5,300, and Mw / Mn of 1.11.
- the resulting 4-t-butylstyrene 21.2 mL (0.115 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.19 mL (2.30 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed.
- This block copolymer (b-8) had Mw of 6,000, Mn of 5,400, and Mw / Mn of 1.11.
- This polymer (c-1) had Mw of 5,600, Mn of 4,600, and Mw / Mn of 1.22.
- the content ratios of structural units derived from styrene, methyl methacrylate and 2-hydroxyethyl methacrylate were 80 mol%, 15 mol% and 5 mol%, respectively.
- This polymer (c-2) had Mw of 5,500, Mn of 4,400, and Mw / Mn of 1.25.
- the content ratios of structural units derived from styrene, methyl methacrylate and methacrylic acid were 80 mol%, 15 mol% and 5 mol%, respectively.
- the obtained polymerization reaction liquid was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid which was precipitated. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.5 g of a yellow polymer (c-3).
- This polymer (c-3) had Mw of 8,400, Mn of 6,600, and Mw / Mn of 1.27.
- the content ratios of structural units derived from styrene and methyl methacrylate were 70 mol% and 30 mol%, respectively.
- the obtained polymerization reaction liquid was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid which was precipitated. This solid was recovered with a Buchner funnel and washed with 50 g of methanol.
- the obtained yellow polymer had Mw of 8,600, Mn of 6,500, and Mw / Mn of 1.32.
- This polymer (c-4) had an Mw of 8,400, an Mn of 6,200, and an Mw / Mn of 1.35.
- the content ratios of structural units derived from styrene and methyl methacrylate were 70 mol% and 30 mol%, respectively.
- the mixture was heated and stirred for 8 hours under a nitrogen flow.
- the obtained polymerization reaction liquid was filtered through Celite to remove the copper complex, and washing with 500 g of ultrapure water was repeated three times.
- the organic layer was collected, it was concentrated, and a solid obtained by adding 50 g of THF to the resulting concentrate was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) and purified by precipitation. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.5 g of a white polymer (c-5).
- This polymer (c-5) had Mw of 6,700, Mn of 5,800, and Mw / Mn of 1.16.
- the structural units derived from styrene and methyl methacrylate were 70 mol% and 30 mol%, respectively.
- composition (I) [Preparation of Composition (I)] [Preparation Example 1-1] To 1.5 g of the synthesized [A] polymer (a-1), 69 g of propylene glycol monomethyl ether acetate and 29.5 g of ethyl lactate as the [S] solvent were added, stirred, and then 0.45 ⁇ m The mixture was filtered through a high-density polyethylene filter having pores to prepare composition (A-1).
- compositions (D-2) to (D-12) were prepared in the same manner as in Preparation Example 1-17, except that the respective polymers shown in Table 5 were used.
- the polymerization start was carried out for 6 hours with the start of dropping as the polymerization start time.
- the polymerization reaction solution was cooled with water, cooled to 30 ° C. or lower, poured into 600 g of methanol, and the precipitated white powder was separated by filtration.
- the white powder separated by filtration was washed with 150 g of methanol twice in a slurry state, then filtered again and dried at 50 ° C. for 17 hours to obtain a white powder polymer (a1-1).
- Yield 80% Mw of the polymer (a1-1) was 6,900, and Mw / Mn was 1.35.
- the content ratios of structural units derived from (M-1) and (M-2) in the polymer (a1-1) were 49 mol% and 51 mol%, respectively. .
- the resist composition (J-1) prepared above is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, exposed to ArF immersion exposure, developed using butyl acetate, A resist hole pattern having a size of 60 nm and a pitch of 150 nm was formed.
- the obtained resist hole pattern was spin-coated with the composition (II) (compositions (B-1) to (B-8)) shown in Table 1 below at 1,500 rpm, and heated at 220 ° C. for 60 seconds. Baked. The substrate after firing was rinsed with propylene glycol monomethyl ether acetate (PGMEA) for 4 seconds to remove unreacted substances and the like, and basic patterns (P-1) to (P-12) were formed.
- composition (II) compositions (B-1) to (B-8)
- Table 1 below at 1,500 rpm
- PMEA propylene glycol monomethyl ether acetate
- the composition (B-2) is used as the composition (III), and the composition (C-1) is used as the composition (II). It was formed by using.
- the basic pattern (P-14) was formed by using the composition (B-2) as the composition (III) and the composition (II) in the formation of the basic pattern (1).
- the basic pattern (P-15) was formed by using the composition (C-1) as the composition (III) and the composition (II) in the basic pattern formation (1).
- the basic pattern (P-16) was formed without applying the composition (III) and the composition (II) in the formation of the basic pattern (1).
- a resist composition (“AIM5484JN” from JSR) is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, ArF immersion exposure, and 2.38 mass% tetrabutylammonium hydroxy
- the resist hole pattern was developed with an aqueous solution and baked at 180 ° C. to form a hole pattern with a hole size of 60 nm and a pitch of 150 nm.
- the obtained resist hole pattern was spin-coated with a composition (II) (composition (B-4)) shown in Table 2 below at 1,500 rpm and baked at 220 ° C. for 60 seconds.
- the substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like, and basic patterns (P-17) and (P-18) were formed.
- a resist composition (“AIM5484JN” from JSR) is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, ArF immersion exposure, and 2.38 mass% tetrabutylammonium hydroxy Development was performed using an aqueous solution of AlF, and the entire surface of ArF was exposed to form a resist hole pattern having a hole size of 60 nm and a pitch of 150 nm.
- the obtained resist hole pattern was spin-coated with a composition (II) (composition (B-)) shown in Table 3 below at 1,500 rpm and baked at 220 ° C. for 60 seconds.
- the substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like, and basic patterns (P-19) and (P-20) were formed.
- an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) was used to form an SOG film having an average thickness of 30 nm.
- An 85 nm resist film is formed by applying the prepared resist composition (J-1) to the obtained substrate, exposed to ArF immersion, developed using butyl acetate, and has a hole size of 60 nm and a pitch of 150 nm. A resist hole pattern was formed.
- the obtained resist hole pattern was spin-coated at 1,500 rpm with the composition (II) (compositions ((B-1) to (B-8)) shown in Table 4 below, The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted materials, etc.
- the composition (III) shown in Table 4 below (composition (C-1)) To (C-5)) were spin-coated at 1,500 rpm and baked for 60 seconds at 220 ° C. The substrate after baking was rinsed for 4 seconds using PGMEA to remove unreacted substances, etc.
- Base patterns (P-21) to (P-32) were formed.
- the composition (C-1) is used as the composition (II), and the composition (B-2) is used as the composition (III). It was formed by using.
- an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) was used to form an SOG film having an average thickness of 30 nm.
- An 85 nm resist film is formed by applying the prepared resist composition (J-1) to the obtained substrate, exposed to ArF immersion, developed using butyl acetate, and has a hole size of 60 nm and a pitch of 150 nm. A resist hole pattern was formed.
- the obtained resist hole pattern was spin-coated with the composition (IV) (compositions (D-1) to (D-12)) shown in Table 5 below at 1,500 rpm, and heated at 220 ° C. for 60 seconds. Baked. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like, and basic patterns (P-34) to (P-45) were formed.
- composition (IV) compositions (D-1) to (D-12) shown in Table 5 below at 1,500 rpm, and heated at 220 ° C. for 60 seconds. Baked.
- the substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like, and basic patterns (P-34) to (P-45) were formed.
- the basic pattern (P-46) was formed by using the composition (B-2) as the composition (IV) in the formation of the basic pattern (5).
- the basic pattern (P-47) was formed by using the composition (C-1) as the composition (IV) in the basic pattern formation (5). “-” In Table 5 indicates that the composition used does not contain both the [B] polymer and the [C] polymer.
- compositions (A-1) to (A-3)) are spin-coated with the composition (I) prepared above (compositions (A-1) to (A-3)) at 1,500 rpm. It was coated by. The substrate was phase-separated by thermal annealing at 220 ° C. for 60 seconds under nitrogen.
- the substrate after the above phase separation step was irradiated with 172 nm radiation at an intensity of 300 mJ / cm 2 , and then in a mixed solution of methyl isobutyl ketone (MIBK) / 2-propanol (IPA) (2/8 (mass ratio)). For 5 minutes to dissolve and remove the phase composed of the poly (methyl methacrylate) block in the polymers (A-1) to (A-3), thereby forming a miniaturized contact hole pattern.
- MIBK methyl isobutyl ketone
- IPA 2-propanol
- ⁇ Evaluation> About the formed miniaturized contact hole pattern, a high-magnification (300K) image is acquired using a scanning electron microscope (“CG4000” of Hitachi High-Technology Co., Ltd.), and a periodicity using a calculation tool (Hitachi High-Technology Co., Ltd.). By analyzing, the average diameter (unit: nm) and the placement error (x direction and y direction, unit: nm) of the contact hole pattern were evaluated. The placement error (x direction, y direction) can be evaluated as good when it is 3.5 nm or less, and poor when it exceeds 3.5 nm.
- a photosensitive SOG composition (“DS492Y” manufactured by JSR) was spin-coated at 1,500 rpm on the substrate after the development, and then soft-baked at 80 ° C. for 30 seconds, and then 200 mJ / second with a KrF exposure machine. After irradiation with cm 2 , a sol-gel curing reaction was performed at 80 ° C. for 120 seconds. Thus, after embedding the miniaturized contact hole pattern, the average thickness (unit: nm) of the bottom residue was measured by cross-sectional SEM observation using a length measurement SEM.
- Table 6 below shows the evaluation results of the average diameter of the miniaturized contact hole pattern, the placement error, and the average thickness of the bottom residue.
- This polymer (A-5) had Mn of 5,200 and Mw / Mn of 1.17.
- the content ratios of structural units derived from styrene and structural units derived from methyl methacrylate were 49 mol% and 51 mol%, respectively.
- the reaction was allowed to proceed for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal.
- the polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 1,000 g of a 2% by mass oxalic acid aqueous solution was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed.
- This polymer (Aa) had Mw of 79,000, Mn of 77,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (Aa) was found to have a content of 60.0% by mass (60.0 mol) of structural units derived from styrene and structural units derived from methyl methacrylate. %) And 40.0 mass% (40.0 mol%) of diblock copolymer.
- composition (V) ⁇ Preparation of composition (V)> Components other than the [A ′] polymer used for the preparation of the composition (V) are shown below.
- composition (S-1)) [A ′] 100 parts by mass of (A-1) as a polymer and 9,900 parts by weight of (S1) as a [S] solvent are mixed, and the obtained mixed solution is filtered through a membrane filter having a pore size of 200 nm. A composition (S-1) was prepared.
- compositions (S-2) to (S-6) and (Sa) were prepared in the same manner as in Preparation Example 2-1, except that the components having the types and contents shown in Table 1 were used. .
- the SOG film was etched using a mixed gas of CF 4 / O 2 / Air.
- the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern.
- a coating film is formed on the pre-pattern using the composition (V-1) shown in Table 8 below as the composition (V) and the temperature and time shown in Table 8 below. It baked on baking conditions and rinsed with PGMEA. “-” In Comparative Example 2-1 in Table 8 indicates that the composition (V) was not applied.
- a coating film was formed using (Sa) as the composition (VI) and baked at 220 ° C. for 20 minutes, and then the phase composed of the PMMA block in the polymer (Aa) with oxygen gas was formed. It removed and the refinement
- the substrate surface selectivity is considered good. As shown below, each of (S-1) to (S-6) was confirmed to have good substrate surface selectivity.
- the contact angle in the substrate (A) is considered to indicate the contact angle ( ⁇ 1) of water on the side surface side of the concave portion of the basic pattern in the coating film after the heating step.
- the difference in contact angle between the substrate (A) and the substrate (B) in Comparative Example 2-1 was less than 5 °.
- Examples 2-7 and 2-8 and Comparative Examples 2-3 and 2-4 On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) was used to form an SOG film having an average thickness of 30 nm.
- a positive resist composition (“AIM5484JN” manufactured by JSR) was applied to the obtained substrate to form a resist film having a thickness of 85 nm, ArF immersion exposure, organic solvent development, and a resist pattern were formed.
- the SOG film was etched using a mixed gas of CF 4 / O 2 / Air.
- the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern.
- a coating film of (S-1) or (S-5) as a composition (V) was formed on the pre-pattern, baked under the baking conditions shown in Table 9 below, and rinsed with PGMEA. “ ⁇ ” In Comparative Example 2-4 of Table 9 below indicates that the composition (V) was not applied.
- a coating film of (Sa) was formed as a composition (VI) and baked at 220 ° C. for 20 minutes, and then the phase consisting of the PMMA block in the polymer (Aa) was removed with oxygen gas, A refined pattern was formed.
- Example 2-9 and Comparative Example 2-5 On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) was used to form an SOG film having an average thickness of 30 nm.
- a positive resist composition (“EUVJ2121” from JSR) is applied to the obtained substrate to form a 50 nm resist film, exposed to EUV, and developed using an aqueous 2.38 mass% tetrabutylammonium hydroxide solution. Then, a resist pattern was formed.
- the SOG film was etched using a mixed gas of CF 4 / O 2 / Air.
- the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern.
- a coating film of (S-1) as a composition (V) was formed on the prepattern, baked under the baking conditions shown in Table 10 below, and rinsed with PGMEA.
- a coating film of (Sa) was formed as a composition (VI), baked at 220 ° C. for 20 minutes, and then the phase consisting of the PMMA block in the polymer (Aa) was removed with oxygen gas to obtain a fine Pattern was formed.
- CDU pattern size uniformity
- a pattern having a good shape and arrangement can be formed on a substrate. Therefore, these can be suitably used for lithography processes in manufacturing various electronic devices such as semiconductor devices and liquid crystal devices that are required to be further miniaturized.
Landscapes
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Inorganic Chemistry (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Power Engineering (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Manufacturing & Machinery (AREA)
- Computer Hardware Design (AREA)
- Architecture (AREA)
- Structural Engineering (AREA)
- Wood Science & Technology (AREA)
- Materials Engineering (AREA)
- Life Sciences & Earth Sciences (AREA)
- Photosensitive Polymer And Photoresist Processing (AREA)
- Materials For Photolithography (AREA)
- Exposure And Positioning Against Photoresist Photosensitive Materials (AREA)
- Graft Or Block Polymers (AREA)
Abstract
The present invention pertains to a pattern formation method comprising: a step for forming a basic pattern on a front face side of a substrate; a step for filling, in a recess of the basic pattern, a first composition containing a solvent and a first polymer having at least two blocks; a step for subjecting a filling layer formed in the filling step to phase-separation; a step for partially removing a phase of the filling layer after having undergone phase separation; and a step for etching the substrate using, directly or indirectly, a fine pattern formed in the removal step, wherein the basic pattern formation step comprises a step for forming a resist pattern on the front face side of the substrate, and a step for forming a second polymer layer on at least a lateral surface of the resist pattern and forming a third polymer layer different from the second polymer on at least the front surface of the substrate or the front surface of another layer.
Description
本発明は、パターン形成方法及び組成物に関する。
The present invention relates to a pattern forming method and a composition.
今日では、半導体デバイス、液晶デバイス等の各種電子デバイス構造の微細化に伴い、リソグラフィー工程におけるパターンの微細化が要求されている。このような要求に対し、一の性質を有する単量体とこの単量体とは性質の異なる単量体とが共重合してなるブロック共重合体が自己組織化により形成する相分離構造を利用して、より微細なパターンを形成する方法が提案されている(特開2008-149447号公報、特表2002-519728号公報及び特開2003-218383号公報参照)。
Today, with the miniaturization of various electronic device structures such as semiconductor devices and liquid crystal devices, there is a demand for pattern miniaturization in the lithography process. In response to such requirements, a phase separation structure formed by self-organization of a block copolymer formed by copolymerization of a monomer having one property and a monomer having a different property from this monomer. A method for forming a finer pattern by using the method has been proposed (see Japanese Patent Application Laid-Open Nos. 2008-149447, 2002-519728, and 2003-218383).
かかる方法を利用して、ホールパターンが形成された膜にブロック共重合体を含有する組成物を塗布した後、同心円柱状の相分離構造を形成させ、この相分離構造の中央の相を除去することにより、上記ホールパターンよりも小さいホール径のホールパターンを形成する方法が検討されている(米国特許出願公開第2010/0297847号明細書参照)。
Using such a method, a composition containing a block copolymer is applied to a film on which a hole pattern is formed, and then a concentric cylindrical phase separation structure is formed, and a central phase of the phase separation structure is removed. Therefore, a method for forming a hole pattern having a smaller hole diameter than the above hole pattern has been studied (see US Patent Application Publication No. 2010/0297847).
しかし、上記従来のホールパターンの形成方法では、パターンが微細化し、形成するホール径が小さくなると、ホールパターンの中心の位置がバラつくプレイスメントエラーが顕著になり、また、ホールパターンの底部における残渣の発生が顕著になるため、又はCritical Dimension Uniformity(CDU)等で示されるパターンサイズの均一性が低いため、このホールパターンからエッチング等により、基板に良好な形状及び配列のホールパターンを形成することが困難になるという不都合がある。
However, in the above conventional hole pattern forming method, when the pattern is miniaturized and the hole diameter to be formed becomes small, a placement error in which the center position of the hole pattern varies becomes significant, and the residue at the bottom of the hole pattern The formation of a hole pattern with a good shape and arrangement on the substrate by etching or the like from this hole pattern because of the occurrence of defects or the low uniformity of the pattern size indicated by Critical Dimension Uniformity (CDU), etc. Has the disadvantage of becoming difficult.
本発明は、上述のような事情に基づいてなされたものであり、その目的は、基板に良好な形状及び配列のパターンを形成することができるパターン形成方法及び組成物を提供することにある。
The present invention has been made based on the above-described circumstances, and an object thereof is to provide a pattern forming method and a composition capable of forming a pattern having a good shape and arrangement on a substrate.
上記課題を解決するためになされた発明は、基板の表面側に直接又は他の層を介して基礎パターンを形成する工程(以下、「基礎パターン形成工程」ともいう)と、上記基礎パターンの凹部に、少なくとも2つのブロックを有する第1重合体(以下、「[A]重合体」ともいう)と溶媒(以下、「[S]溶媒」ともいう)とを含有する第1組成物(以下、「組成物(I)」ともいう)を充填する工程(以下、「充填工程」ともいう)と、上記充填工程により形成された充填層を相分離させる工程(以下、「相分離工程」ともいう)と、上記相分離工程後の充填層の一部の相を除去する工程(以下、「除去工程」ともいう)と、上記除去工程により形成された微細化パターンを直接的又は間接的に用いて上記基板をエッチングする工程(以下、「エッチング工程」ともいう)とを備え、上記基礎パターン形成工程が、上記基板の表面側にレジストパターンを形成する工程(以下、「レジストパターン形成工程」ともいう)と、上記レジストパターンの少なくとも側面に第2重合体(以下、「[B]重合体」ともいう)の層(以下、「[B]重合体層」ともいう)を、少なくとも基板又は他の層の表面に上記[B]重合体とは異なる第3重合体(以下、「[C]重合体」ともいう)の層(以下、「[C]重合体層」ともいう)を形成する工程(以下、「重合体層形成工程」ともいう)とを有するパターン形成方法(以下、「パターン形成方法(A)」ともいう)である。
The invention made in order to solve the above problems includes a step of forming a basic pattern on the surface side of the substrate directly or via another layer (hereinafter also referred to as a “basic pattern forming step”), and a concave portion of the basic pattern. And a first composition (hereinafter referred to as “[A] polymer”) having at least two blocks and a solvent (hereinafter also referred to as “[S] solvent”). A step of filling the “composition (I)” (hereinafter also referred to as “filling step”) and a step of phase-separating the packed bed formed by the filling step (hereinafter also referred to as “phase separation step”). ), A step of removing a part of the packed bed after the phase separation step (hereinafter also referred to as “removal step”), and a fine pattern formed by the removal step is used directly or indirectly. Step of etching the substrate The basic pattern forming step is a step of forming a resist pattern on the surface side of the substrate (hereinafter also referred to as “resist pattern forming step”), and at least a side surface of the resist pattern. The layer of the second polymer (hereinafter also referred to as “[B] polymer”) (hereinafter also referred to as “[B] polymer layer”) is placed on the surface of at least the substrate or other layer. A step of forming a layer (hereinafter also referred to as “[C] polymer layer”) of a third polymer (hereinafter also referred to as “[C] polymer”) different from the coalescence (hereinafter referred to as “polymer layer forming step”). A pattern forming method (hereinafter also referred to as “pattern forming method (A)”).
上記課題を解決するためになされた別の発明は、主鎖の少なくとも一方の末端に結合し-COOH及び-OHの少なくとも一方と化学結合を形成する第1官能基(以下、「官能基(1)」ともいう)を有する重合体と、主鎖の少なくとも一方の末端に結合しSi-H、Si-OH、Si=O及びSi-NR2(Rは、それぞれ独立して、炭素数1~20の1価の有機基である)の少なくとも一方と化学結合を形成する第2官能基(以下「官能基(2)」ともいう)を有する重合体と、溶媒とを含有する組成物である。
Another invention made in order to solve the above-described problem is that a first functional group (hereinafter referred to as “functional group (1) that binds to at least one terminal of the main chain and forms a chemical bond with at least one of —COOH and —OH”. ) ”) And Si—H, Si—OH, Si═O and Si—NR 2 (R each independently represents a carbon number of 1 to A polymer having a second functional group (hereinafter also referred to as “functional group (2)”) that forms a chemical bond with at least one of 20 monovalent organic groups) and a solvent. .
上記課題を解決するためになされたさらに別の発明は、基板の表面側に直接又は他の層を介して基礎パターンを積層する工程(以下、「基礎パターン積層工程」ともいう)と、上記基礎パターンの凹部の側面及び底面に第5組成物(以下、「組成物(V)」ともいう)を塗工する工程(以下、「塗工工程」ともいう)と、上記塗工工程により形成された塗工膜を加熱する工程(以下、「加熱工程」ともいう)と、上記塗工膜が積層された上記基礎パターンの凹部に第6組成物(以下、「組成物(VI)」ともいう)を充填する工程(充填工程)と、上記充填工程により形成された充填層を相分離させる工程(相分離工程)と、上記相分離工程後の充填層の少なくとも一部の相を除去する工程(除去工程)と、上記除去工程により形成された微細化パターンを用いて上記基板を1又は複数回エッチングする工程(エッチング工程)とを備えるパターン形成方法であって、上記基礎パターンが芳香環の含有割合が50質量%以上の重合体を主成分とし、上記組成物(V)が第1構造単位(以下、「構造単位(I)」ともいう)を有する第4重合体(以下、「[A’]重合体」ともいう)と溶媒とを含有し、上記組成物(VI)が第2構造単位(以下、「構造単位(II)」ともいう)からなる第1ブロック(以下、「ブロック(A)」ともいう)及び上記構造単位(II)よりも極性が高い第3構造単位(以下、「構造単位(III)」ともいう)からなる第2ブロック(以下、「ブロック(B)」ともいう)を有する第5重合体(以下、「[B’]重合体」ともいう)と溶媒とを含有し、上記加熱工程後の塗工膜における基礎パターンの凹部の側面側の水の接触角θ1と底面側の水の接触角θ2との差(│θ1-θ2│)が5°以上になることを特徴とするパターン形成方法(以下、「パターン形成方法(B)」ともいう)である。
Still another invention made in order to solve the above problems includes a step of laminating a basic pattern on the surface side of a substrate directly or via another layer (hereinafter also referred to as “basic pattern laminating step”), and the above basic A step of applying a fifth composition (hereinafter, also referred to as “composition (V)”) to the side and bottom surfaces of the concave portions of the pattern (hereinafter, also referred to as “coating step”) and the above-described coating step. A step of heating the coated film (hereinafter also referred to as “heating step”) and a sixth composition (hereinafter also referred to as “composition (VI)”) in the concave portion of the basic pattern on which the coating film is laminated. ), A step of phase-separating the packed bed formed by the filling step (phase separation step), and a step of removing at least a part of the phase of the packed bed after the phase separation step. (Removal step) and formed by the above removal step A pattern forming method comprising a step (etching step) of etching the substrate one or more times using a thinned pattern, wherein the basic pattern comprises a polymer having an aromatic ring content of 50% by mass or more as a main component And a fourth polymer (hereinafter also referred to as “[A ′] polymer”) having the first structural unit (hereinafter also referred to as “structural unit (I)”) and a solvent. A first block (hereinafter also referred to as “block (A)”) comprising the second structural unit (hereinafter also referred to as “structural unit (II)”) and the structural unit (II). ) Having a second block (hereinafter also referred to as “block (B)”) composed of a third structural unit (hereinafter also referred to as “structural unit (III)”) having a higher polarity than that of [B ′] polymer ”) and a solvent. And the difference (| θ1-θ2 |) between the water contact angle θ1 on the side surface of the concave portion of the basic pattern and the water contact angle θ2 on the bottom surface in the coating film after the heating step is 5 ° or more. Is a pattern forming method (hereinafter also referred to as “pattern forming method (B)”).
ここで、「化学結合」とは、共有結合、イオン結合、金属結合及び配位結合の他、分子間の静電引力及び水素結合を含む概念である。
Here, “chemical bond” is a concept including electrostatic attraction and hydrogen bond between molecules in addition to covalent bond, ionic bond, metal bond and coordination bond.
すなわち、パターン形成方法(A)及び(B)は、基板の表面側に直接又は他の層を介して基礎パターンを形成する工程と、上記基礎パターンの凹部に、少なくとも2つのブロックを有する第1重合体と溶媒とを含有する第1組成物を充填する工程と、上記充填工程により形成された充填層を相分離させる工程と、上記相分離工程後の充填層の一部の相を除去する工程と、上記除去工程により形成された微細化パターンを直接的又は間接的に用いて上記基板をエッチングする工程とを備え、上記基礎パターンが、凹部の少なくとも側面に第1層を有し、凹部の少なくとも基板又は他の層の表面側に上記第1層とは表面の接触角が異なる第2層を有するパターン形成方法である。
That is, the pattern forming methods (A) and (B) include a step of forming a basic pattern on the surface side of the substrate directly or via another layer, and a first having at least two blocks in the concave portion of the basic pattern. A step of filling a first composition containing a polymer and a solvent, a step of phase-separating the packed bed formed by the filling step, and removing a part of the phase of the packed bed after the phase separation step. And a step of etching the substrate directly or indirectly using the miniaturized pattern formed by the removing step, wherein the basic pattern has a first layer on at least a side surface of the recess, and the recess The pattern forming method has a second layer having a contact angle different from that of the first layer on the surface side of at least the substrate or other layer.
本発明のパターン形成方法及び組成物によれば、基板に良好な形状及び配列のパターンを形成することができる。従って、これらはさらなる微細化が要求されている半導体デバイス、液晶デバイス等の各種電子デバイス製造におけるリソグラフィー工程に好適に用いることができる。
According to the pattern forming method and composition of the present invention, a pattern having a good shape and arrangement can be formed on a substrate. Therefore, these can be suitably used for lithography processes in manufacturing various electronic devices such as semiconductor devices and liquid crystal devices that are required to be further miniaturized.
以下、本発明のパターン形成方法の実施の形態について、図1~5を参照しつつ詳説する。
Hereinafter, embodiments of the pattern forming method of the present invention will be described in detail with reference to FIGS.
当該パターン形成方法としては、パターン形成方法(A)及びパターン形成方法(B)が挙げられる。パターン形成方法(A)によれば、パターンが微細な場合でも、プレイスメントエラーが抑制され、かつ底部残渣が低減された微細化パターンを形成することができる。パターン形成方法(B)によれば、パターンサイズ均一性に優れる微細化パターンを形成することができる。当該パターン形成方法によれば、これらの優れた微細化パターンを用いることにより、基板に良好な形状及び配列のパターンを形成することができる。以下、パターン形成方法(A)及びパターン形成方法(B)について説明する。
The pattern forming method includes a pattern forming method (A) and a pattern forming method (B). According to the pattern formation method (A), even when the pattern is fine, it is possible to form a fine pattern in which placement errors are suppressed and bottom residue is reduced. According to the pattern forming method (B), it is possible to form a fine pattern having excellent pattern size uniformity. According to the pattern forming method, a pattern having a good shape and arrangement can be formed on the substrate by using these excellent miniaturized patterns. Hereinafter, the pattern formation method (A) and the pattern formation method (B) will be described.
<パターン形成方法(A)>
パターン形成方法(A)は、基礎パターン形成工程と、充填工程と、相分離工程と、除去工程と、エッチング工程とを備える。パターン形成方法(A)において、基礎パターン形成工程は、レジストパターン形成工程と、重合体層形成工程とを有する。 <Pattern formation method (A)>
The pattern forming method (A) includes a basic pattern forming step, a filling step, a phase separation step, a removing step, and an etching step. In the pattern forming method (A), the basic pattern forming step includes a resist pattern forming step and a polymer layer forming step.
パターン形成方法(A)は、基礎パターン形成工程と、充填工程と、相分離工程と、除去工程と、エッチング工程とを備える。パターン形成方法(A)において、基礎パターン形成工程は、レジストパターン形成工程と、重合体層形成工程とを有する。 <Pattern formation method (A)>
The pattern forming method (A) includes a basic pattern forming step, a filling step, a phase separation step, a removing step, and an etching step. In the pattern forming method (A), the basic pattern forming step includes a resist pattern forming step and a polymer layer forming step.
パターン形成方法(A)は、上記各工程を備え、かつ基礎パターン形成工程において、レジストパターン形成工程と、重合体層形成工程とを有し、この重合体層形成工程において、レジストパターンの少なくとも側面に[B]重合体層を、少なくとも基板又は他の層の表面に[C]重合体層を形成することで、パターンが微細な場合でも、プレイスメントエラーが抑制され、かつ底部残渣が低減された微細化パターンを形成することができ、このような優れた微細化パターンを用いることにより、基板に良好な形状及び配列のパターンを形成することができる。パターン形成方法(A)が上記構成を備えることで、上記効果を奏する理由については必ずしも明確ではないが、例えば以下のように推察することができる。すなわち、レジストパターン形成工程と重合体層形成工程を有することで、レジストパターンの側面、つまり基礎パターンの凹部の側面に[B]重合体層を、基板又は他の層の表面、つまり基礎パターンの凹部の底面に[C]重合体層を形成することにより、基礎パターンの凹部の側面及び底面に異なる層が形成される。この凹部の側面及び底面にそれぞれ形成される層によって、その後にこの凹部に形成される[A]重合体の充填層がより適切に相分離すると考えられ、その結果、形成されるパターンが微細な場合であっても、プレイスメントエラーが抑制され、かつ底部残渣が低減されたパターンを形成することができる。以下、各工程について説明する。
The pattern forming method (A) includes the above steps, and has a resist pattern forming step and a polymer layer forming step in the basic pattern forming step, and at least a side surface of the resist pattern in the polymer layer forming step. By forming the [B] polymer layer and the [C] polymer layer on at least the surface of the substrate or other layer, even if the pattern is fine, placement errors are suppressed and the bottom residue is reduced. A fine pattern can be formed, and by using such an excellent fine pattern, a pattern having a good shape and arrangement can be formed on the substrate. Although the reason why the pattern forming method (A) has the above-described configuration provides the above-described effect is not necessarily clear, for example, it can be inferred as follows. That is, by having a resist pattern forming step and a polymer layer forming step, the [B] polymer layer is placed on the side surface of the resist pattern, that is, the side surface of the concave portion of the basic pattern, and the surface of the substrate or other layer, that is, the basic pattern. By forming the [C] polymer layer on the bottom surface of the recess, different layers are formed on the side surface and the bottom surface of the recess of the basic pattern. It is considered that the layer formed on the side surface and the bottom surface of the recess respectively causes the [A] polymer filling layer formed in the recess to be more appropriately phase-separated, and as a result, the pattern formed is fine. Even in this case, it is possible to form a pattern in which placement errors are suppressed and bottom residue is reduced. Hereinafter, each step will be described.
[基礎パターン形成工程]
本工程では、基板の表面側に直接又は他の層を介して基礎パターン(以下、「基礎パターン(I)」ともいう)を形成する。基礎パターン形成工程は、レジストパターン形成工程と、重合体層形成工程とを有する。図1は、基板1の表面側に直接、レジストパターン2を形成した場合を示す。図2は、このレジストパターン2の凹部に重合体層3を形成した場合を示す。図2の重合体層3のうち、レジストパターンの凹部の側面には[B]重合体層が形成されており、凹部の底面には[C]重合体層が形成されている。以下、工程について説明する。 [Basic pattern formation process]
In this step, a basic pattern (hereinafter also referred to as “basic pattern (I)”) is formed directly on the surface side of the substrate or via another layer. The basic pattern forming step includes a resist pattern forming step and a polymer layer forming step. FIG. 1 shows a case where aresist pattern 2 is formed directly on the surface side of the substrate 1. FIG. 2 shows a case where the polymer layer 3 is formed in the concave portion of the resist pattern 2. In the polymer layer 3 of FIG. 2, the [B] polymer layer is formed on the side surface of the concave portion of the resist pattern, and the [C] polymer layer is formed on the bottom surface of the concave portion. Hereinafter, the process will be described.
本工程では、基板の表面側に直接又は他の層を介して基礎パターン(以下、「基礎パターン(I)」ともいう)を形成する。基礎パターン形成工程は、レジストパターン形成工程と、重合体層形成工程とを有する。図1は、基板1の表面側に直接、レジストパターン2を形成した場合を示す。図2は、このレジストパターン2の凹部に重合体層3を形成した場合を示す。図2の重合体層3のうち、レジストパターンの凹部の側面には[B]重合体層が形成されており、凹部の底面には[C]重合体層が形成されている。以下、工程について説明する。 [Basic pattern formation process]
In this step, a basic pattern (hereinafter also referred to as “basic pattern (I)”) is formed directly on the surface side of the substrate or via another layer. The basic pattern forming step includes a resist pattern forming step and a polymer layer forming step. FIG. 1 shows a case where a
基板1としては、例えばシリコン(Bare-Si)ウェハ、窒化シリコン等のシリコン製基板、アルミニウムで被覆されたウェハ等の従来公知の基板を使用できる。これらの中で、シリコン製基板が好ましく、シリコンウェハがより好ましい。
As the substrate 1, for example, a conventionally known substrate such as a silicon (Bare-Si) wafer, a silicon substrate such as silicon nitride, or a wafer coated with aluminum can be used. Among these, a silicon substrate is preferable, and a silicon wafer is more preferable.
また、他の層としては、レジスト下層膜、SOG(Spin On Glass)膜等が挙げられる。SOG膜は、シリコン含有膜である。
In addition, examples of other layers include a resist underlayer film, an SOG (Spin On Glass) film, and the like. The SOG film is a silicon-containing film.
レジスト下層膜を形成する組成物としては、従来公知の有機下層膜形成材料等が挙げられ、例えば架橋剤を含む下層膜形成用組成物等が挙げられる。
Examples of the composition for forming the resist underlayer film include conventionally known organic underlayer film forming materials, and the like, for example, an underlayer film forming composition containing a crosslinking agent.
SOG膜を形成する組成物としては、従来公知のSOG組成物等を用いることができ、例えば有機ポリシロキサンを含有する組成物等が挙げられる。これらの中で、SOG膜が好ましい。
As the composition for forming the SOG film, a conventionally known SOG composition or the like can be used, and examples thereof include a composition containing an organic polysiloxane. Of these, the SOG film is preferable.
SOG膜の形成方法は特に限定されないが、例えば基板の一方の面又は上記下層膜の基板1とは反対側の面に、SOG組成物をスピンコート法等の公知の方法により塗工し、PBを行った後、得られる塗膜を放射線の照射及び/又は加熱を行うことにより硬化する方法等が挙げられる。照射する放射線としては、例えば可視光線、紫外線、遠紫外線、X線、γ線等の電磁波;電子線、分子線、イオンビーム等の粒子線などが挙げられる。加熱の温度の下限としては、100℃が好ましく、150℃がより好ましく、180℃がさらに好ましい。加熱の温度の上限としては、450℃が好ましく、400℃がより好ましく、350℃がさらに好ましい。加熱の時間の下限としては、5秒が好ましく、10秒がより好ましく、20秒がさらに好ましい。加熱の時間の上限としては、1,200秒が好ましく、600秒がより好ましく、300秒がさらに好ましい。SOG膜の平均厚みの下限としては、10nmが好ましく、15nmがより好ましく、20nmがさらに好ましい。上記平均厚みの上限としては、1,000nmが好ましく、500nmがより好ましく、100nmがさらに好ましい。
The method for forming the SOG film is not particularly limited. For example, the SOG composition is applied to one surface of the substrate or the surface of the lower layer opposite to the substrate 1 by a known method such as spin coating, and PB The method of hardening | curing the coating film obtained by performing irradiation of a radiation and / or heating after performing is mentioned. Examples of radiation to be irradiated include electromagnetic waves such as visible light, ultraviolet light, far ultraviolet light, X-rays, and γ rays; particle beams such as electron beams, molecular beams, and ion beams. As a minimum of temperature of heating, 100 ° C is preferred, 150 ° C is more preferred, and 180 ° C is still more preferred. As an upper limit of the temperature of heating, 450 degreeC is preferable, 400 degreeC is more preferable, and 350 degreeC is further more preferable. The lower limit of the heating time is preferably 5 seconds, more preferably 10 seconds, and even more preferably 20 seconds. The upper limit of the heating time is preferably 1,200 seconds, more preferably 600 seconds, and even more preferably 300 seconds. The lower limit of the average thickness of the SOG film is preferably 10 nm, more preferably 15 nm, and still more preferably 20 nm. The upper limit of the average thickness is preferably 1,000 nm, more preferably 500 nm, and still more preferably 100 nm.
基板及び他の層は、Si-H、Si-OH、Si=O及びSi-NR2の少なくとも1種を上面に有することが好ましい。Rは、それぞれ独立して、水素原子又は炭素数1~20の1価の有機基である。「有機基」とは、少なくとも1個の炭素原子を含む基をいう。1価の有機基としては、例えばメチル基、エチル基等のアルキル基等が挙げられる。Rとしては、水素原子が好ましい。すなわち、基板及び他の層のうち上方が、SiO2製又はSiN製であることが好ましい。他の層を用いる場合、他の層としては、シリコン含有膜が好ましい。
The substrate and other layers preferably have at least one of Si—H, Si—OH, Si═O, and Si—NR 2 on the top surface. Each R is independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms. “Organic group” refers to a group containing at least one carbon atom. Examples of the monovalent organic group include alkyl groups such as a methyl group and an ethyl group. R is preferably a hydrogen atom. That is, the upper part of the substrate and the other layers is preferably made of SiO 2 or SiN. When other layers are used, silicon-containing films are preferable as the other layers.
(レジストパターン形成工程)
レジストパターン2は、図1に示すように、基板1の表面側に直接形成してもよく、又は基板1の表面側に他の層を介して、例えば基板1上に下層膜、SOG膜等を形成し、これらの膜の基板1と反対側の面に形成してもよい。これらのうち、形成されるレジストパターンをマスクとするエッチングにより簡便に基板にパターンを形成できる点から、レジストパターン2は、基板の表面側に直接形成することが好ましい。 (Resist pattern formation process)
As shown in FIG. 1, the resistpattern 2 may be directly formed on the surface side of the substrate 1, or another layer on the surface side of the substrate 1, for example, a lower layer film, an SOG film or the like on the substrate 1. These films may be formed on the surface opposite to the substrate 1 of these films. Of these, the resist pattern 2 is preferably formed directly on the surface side of the substrate because the pattern can be easily formed on the substrate by etching using the formed resist pattern as a mask.
レジストパターン2は、図1に示すように、基板1の表面側に直接形成してもよく、又は基板1の表面側に他の層を介して、例えば基板1上に下層膜、SOG膜等を形成し、これらの膜の基板1と反対側の面に形成してもよい。これらのうち、形成されるレジストパターンをマスクとするエッチングにより簡便に基板にパターンを形成できる点から、レジストパターン2は、基板の表面側に直接形成することが好ましい。 (Resist pattern formation process)
As shown in FIG. 1, the resist
レジストパターン2の形成方法としては、例えばレジスト組成物の塗工、露光及び現像により形成する方法等が挙げられる。
Examples of the method of forming the resist pattern 2 include a method of forming the resist pattern by coating, exposing and developing a resist composition.
レジスト組成物としては、例えば酸解離性基を有する重合体及び感放射線性酸発生体を含有するレジスト組成物、架橋性基を有する重合体及び感放射線性酸発生体を含有するネガ型レジスト組成物等の従来のレジスト組成物などが挙げられる。「酸解離性基」とは、-COOH、-OH等の水素原子を置換する基であって、酸の作用により解離する基をいう。-COOHの水素原子を置換する酸解離性基としては、例えば3級炭化水素基等が挙げられ、t-ブチル基、t-アミル基等のt-アルキル基、1-エチルシクロペンタン-1-イル基等の1-アルキルシクロアルカン-1-イル基及び2-メチルアダマンタン-2-イル基等の2-アルキルビシクロアルカン-2-イル基等が挙げられる。
Examples of the resist composition include a resist composition containing a polymer having an acid dissociable group and a radiation sensitive acid generator, a polymer having a crosslinkable group, and a negative resist composition containing a radiation sensitive acid generator. Conventional resist compositions such as those may be mentioned. The “acid-dissociable group” refers to a group that replaces a hydrogen atom such as —COOH or —OH and is dissociated by the action of an acid. Examples of the acid dissociable group that substitutes a hydrogen atom of —COOH include a tertiary hydrocarbon group, a t-alkyl group such as a t-butyl group and a t-amyl group, and 1-ethylcyclopentane-1- And 2-alkylbicycloalkane-2-yl group such as 1-alkylcycloalkane-1-yl group such as yl group and 2-methyladamantan-2-yl group.
レジストパターン2の形成方法としては、まず、レジスト組成物を基板1の表面又は他の層の表面に塗工した後、プレベーク(PB)を行うことによりレジスト膜を形成する。次に、所望の形状のレジストパターン2を形成するためのマスクパターンを介して露光を行う。露光に用いる放射線としては、例えば紫外線、遠紫外線、極端紫外線(EUV)、X線等の電磁波;電子線、α線等の荷電粒子線などが挙げられる。これらの中で、遠紫外線が好ましく、ArFエキシマレーザー光及びKrFエキシマレーザー光がより好ましく、ArFエキシマレーザー光がさらに好ましい。露光方法としては液浸露光を行うこともできる。露光の後、ポストエクスポージャーベーク(PEB)を行うことが好ましい。次いで、2.38質量%テトラメチルアンモニウムヒドロキシド水溶液、テトラブチルアンモニウムヒドロキシド水溶液等のアルカリ現像液、酢酸ブチル、アニソール等の有機溶媒現像液などの現像液を用いて現像を行う。
As a method for forming the resist pattern 2, first, a resist composition is applied to the surface of the substrate 1 or the surface of another layer, and then pre-baked (PB) to form a resist film. Next, exposure is performed through a mask pattern for forming a resist pattern 2 having a desired shape. Examples of radiation used for exposure include ultraviolet rays, far ultraviolet rays, extreme ultraviolet rays (EUV), electromagnetic waves such as X-rays, and charged particle beams such as electron beams and α rays. Among these, far ultraviolet rays are preferable, ArF excimer laser light and KrF excimer laser light are more preferable, and ArF excimer laser light is more preferable. As an exposure method, immersion exposure can also be performed. It is preferable to perform post-exposure baking (PEB) after exposure. Subsequently, development is performed using a developer such as an alkali developer such as an aqueous 2.38 mass% tetramethylammonium hydroxide solution or an aqueous tetrabutylammonium hydroxide solution, or an organic solvent developer such as butyl acetate or anisole.
レジスト膜の平均厚みの下限としては、10nmが好ましく、30nmがより好ましく、50nmがさらに好ましい。上記平均厚みの上限としては、1,000nmが好ましく、500nmがより好ましく、200nmがさらに好ましい。
The lower limit of the average thickness of the resist film is preferably 10 nm, more preferably 30 nm, and even more preferably 50 nm. The upper limit of the average thickness is preferably 1,000 nm, more preferably 500 nm, and even more preferably 200 nm.
レジストパターン2の形状としては、最終的に基板に形成するパターンの形状に合わせて適宜選択することができ、例えば平面視において、円形状(略真円形状)、楕円形状(oval)、正方形状、長方形状、鈎形状、台形状、三角形状などが挙げられる。これらの中で、円形状が好ましい。
The shape of the resist pattern 2 can be appropriately selected according to the shape of the pattern finally formed on the substrate. For example, in a plan view, the shape is a circle (substantially perfect circle), an ellipse (oval), or a square. , Rectangular shape, bowl shape, trapezoidal shape, triangular shape and the like. Among these, a circular shape is preferable.
形成されるレジストパターン2が円形状の場合、その平均径の下限としては、10nmが好ましく、20nmがより好ましく、30nmがさらに好ましく、40nmが特に好ましい。上記平均径の上限としては、200nmが好ましく、100nmがより好ましく、90nmがさらに好ましく、80nmが特に好ましい。
When the resist pattern 2 to be formed is circular, the lower limit of the average diameter is preferably 10 nm, more preferably 20 nm, still more preferably 30 nm, and particularly preferably 40 nm. The upper limit of the average diameter is preferably 200 nm, more preferably 100 nm, still more preferably 90 nm, and particularly preferably 80 nm.
形成されるレジストパターン2のピッチの下限としては、30nmが好ましく、50nmがより好ましく、70nmがさらに好ましく、90nmが特に好ましい。上記ピッチの上限としては、1,000nmが好ましく、500nmがより好ましく、200nmがさらに好ましく、150nmが特に好ましい。
The lower limit of the pitch of the resist pattern 2 to be formed is preferably 30 nm, more preferably 50 nm, still more preferably 70 nm, and particularly preferably 90 nm. The upper limit of the pitch is preferably 1,000 nm, more preferably 500 nm, still more preferably 200 nm, and particularly preferably 150 nm.
レジストパターン2は、-COOH及び-OHの少なくとも一方を側面に有することが好ましい。
The resist pattern 2 preferably has at least one of —COOH and —OH on the side surface.
このような-COOH及び-OHの少なくとも一方を側面に有するレジストパターン2は、例えば酸解離性基を有する重合体及び感放射線性酸発生体を含有するレジスト組成物の塗工、露光及び有機溶媒現像液による現像により形成することができる。また、酸解離性基を有する重合体及び感放射線性酸発生体を含有するレジスト組成物の塗工、露光及びアルカリ現像液による現像を行った後、得られたレジスト膜を加熱又は露光することにより、レジスト膜中の酸解離性基を解離させることによっても形成することができる。
Such a resist pattern 2 having at least one of —COOH and —OH on its side surface is, for example, applied to a resist composition containing a polymer having an acid-dissociable group and a radiation-sensitive acid generator, exposure, and an organic solvent. It can be formed by development with a developer. In addition, after applying a resist composition containing a polymer having an acid-dissociable group and a radiation-sensitive acid generator, exposure and development with an alkali developer, the resulting resist film is heated or exposed. Thus, it can also be formed by dissociating the acid dissociable group in the resist film.
[重合体層形成工程]
本工程では、図2に示すように、レジストパターン2の少なくとも側面に[B]重合体層を、少なくとも基板又は他の層の表面に[C]重合体層を形成する。本工程は、[B]重合体層を形成する工程(以下、「[B]重合体層形成工程」ともいう)と、[C]重合体層を形成する工程(以下、「[C]重合体層形成工程」ともいう)とを有し、これらの工程を別々に行ってもよいし、同時に行ってもよい。 [Polymer layer forming step]
In this step, as shown in FIG. 2, the [B] polymer layer is formed on at least the side surface of the resistpattern 2, and the [C] polymer layer is formed on at least the surface of the substrate or another layer. This step includes a step [B] forming a polymer layer (hereinafter also referred to as “[B] polymer layer forming step”) and a step [C] forming a polymer layer (hereinafter referred to as “[C] These steps may be performed separately or at the same time.
本工程では、図2に示すように、レジストパターン2の少なくとも側面に[B]重合体層を、少なくとも基板又は他の層の表面に[C]重合体層を形成する。本工程は、[B]重合体層を形成する工程(以下、「[B]重合体層形成工程」ともいう)と、[C]重合体層を形成する工程(以下、「[C]重合体層形成工程」ともいう)とを有し、これらの工程を別々に行ってもよいし、同時に行ってもよい。 [Polymer layer forming step]
In this step, as shown in FIG. 2, the [B] polymer layer is formed on at least the side surface of the resist
([B]重合体層形成工程)
本工程では、レジストパターン2の少なくとも側面に[B]重合体層を形成する。 ([B] polymer layer forming step)
In this step, a [B] polymer layer is formed on at least the side surface of the resistpattern 2.
本工程では、レジストパターン2の少なくとも側面に[B]重合体層を形成する。 ([B] polymer layer forming step)
In this step, a [B] polymer layer is formed on at least the side surface of the resist
[B]重合体層を形成する方法としては、例えば[B]重合体と溶媒(以下、「[S]溶媒」ともいう)とを含有する組成物(II)を塗工する工程(以下、「組成物(II)塗工工程」ともいう)を行う方法等が挙げられる。
[B] As a method for forming a polymer layer, for example, a step of applying a composition (II) containing, for example, a [B] polymer and a solvent (hereinafter also referred to as “[S] solvent”) (hereinafter referred to as “B”). And a method of performing “composition (II) coating step”).
(組成物(II))
組成物(II)は、[B]重合体と[S]溶媒とを含有する。組成物(II)は、[B]重合体及び[S]溶媒以外にも他の成分を含有していてもよい。 (Composition (II))
The composition (II) contains a [B] polymer and a [S] solvent. The composition (II) may contain other components in addition to the [B] polymer and the [S] solvent.
組成物(II)は、[B]重合体と[S]溶媒とを含有する。組成物(II)は、[B]重合体及び[S]溶媒以外にも他の成分を含有していてもよい。 (Composition (II))
The composition (II) contains a [B] polymer and a [S] solvent. The composition (II) may contain other components in addition to the [B] polymer and the [S] solvent.
([B]重合体)
[B]重合体としては、主鎖の少なくとも一方の末端に結合し、-COOH及び-OHの少なくとも一方と化学結合する官能基(1)を有することが好ましい。「主鎖」とは、重合体の原子鎖のうち最も長いものをいう。 ([B] polymer)
[B] The polymer preferably has a functional group (1) bonded to at least one terminal of the main chain and chemically bonded to at least one of —COOH and —OH. “Main chain” refers to the longest of the atomic chains of a polymer.
[B]重合体としては、主鎖の少なくとも一方の末端に結合し、-COOH及び-OHの少なくとも一方と化学結合する官能基(1)を有することが好ましい。「主鎖」とは、重合体の原子鎖のうち最も長いものをいう。 ([B] polymer)
[B] The polymer preferably has a functional group (1) bonded to at least one terminal of the main chain and chemically bonded to at least one of —COOH and —OH. “Main chain” refers to the longest of the atomic chains of a polymer.
官能基(1)としては、例えば水酸基、エポキシ基(オキシラニル基)、オキセタニル基等が挙げられる。これらの官能基は、-COOH及び/又は-OHと共有結合を形成する。
Examples of the functional group (1) include a hydroxyl group, an epoxy group (oxiranyl group), and an oxetanyl group. These functional groups form a covalent bond with —COOH and / or —OH.
[B]重合体としては、例えばスチレン系重合体、(メタ)アクリル系重合体、エチレン系重合体、これらを組み合わせた共重合体等が挙げられる。
Examples of the [B] polymer include a styrene polymer, a (meth) acrylic polymer, an ethylene polymer, and a copolymer combining these.
スチレン系重合体は、置換又は非置換のスチレンに由来する構造単位を有する。
The styrene polymer has a structural unit derived from substituted or unsubstituted styrene.
置換スチレンとしては、例えばα-メチルスチレン、o-、m-、p-メチルスチレン、p-t-ブチルスチレン、2,4,6-トリメチルスチレン、p-メトキシスチレン、p-t-ブトキシスチレン、o-、m-、p-ビニルスチレン、o-、m-、p-ヒドロキシスチレン、m-、p-クロロメチルスチレン、p-クロロスチレン、p-ブロモスチレン、p-ヨードスチレン、p-ニトロスチレン、p-シアノスチレン等が挙げられる。
Examples of the substituted styrene include α-methylstyrene, o-, m-, p-methylstyrene, pt-butylstyrene, 2,4,6-trimethylstyrene, p-methoxystyrene, pt-butoxystyrene, o-, m-, p-vinylstyrene, o-, m-, p-hydroxystyrene, m-, p-chloromethylstyrene, p-chlorostyrene, p-bromostyrene, p-iodostyrene, p-nitrostyrene , P-cyanostyrene and the like.
(メタ)アクリル系重合体は、(メタ)アクリル酸又は(メタ)アクリル酸エステルに由来する構造単位を有する。
The (meth) acrylic polymer has a structural unit derived from (meth) acrylic acid or (meth) acrylic acid ester.
(メタ)アクリル酸エステルとしては、例えば
(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸t-ブチル、(メタ)アクリル酸2-エチルヘキシル等の(メタ)アクリル酸アルキルエステル;
(メタ)アクリル酸シクロペンチル、(メタ)アクリル酸シクロヘキシル、(メタ)アクリル酸1-メチルシクロペンチル、(メタ)アクリル酸2-エチルアダマンチル、(メタ)アクリル酸2-(アダマンタン-1-イル)プロピル等の(メタ)アクリル酸シクロアルキルエステル;
(メタ)アクリル酸フェニル、(メタ)アクリル酸ナフチル等の(メタ)アクリル酸アリールエステル;
(メタ)アクリル酸2-ヒドロキシエチル、(メタ)アクリル酸3-ヒドロキシアダマンチル、(メタ)アクリル酸3-グリシジルプロピル、(メタ)アクリル酸3-トリメチルシリルプロピル等の(メタ)アクリル酸置換アルキルエステルなどが挙げられる。 Examples of (meth) acrylic acid esters include (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethyl (meth) acrylate, t-butyl (meth) acrylate, and 2-ethylhexyl (meth) acrylate. ;
Cyclopentyl (meth) acrylate, cyclohexyl (meth) acrylate, 1-methylcyclopentyl (meth) acrylate, 2-ethyladamantyl (meth) acrylate, 2- (adamantan-1-yl) propyl (meth) acrylate, etc. (Meth) acrylic acid cycloalkyl ester of
(Meth) acrylic acid aryl esters such as phenyl (meth) acrylate and naphthyl (meth) acrylate;
(Meth) acrylic acid-substituted alkyl esters such as 2-hydroxyethyl (meth) acrylate, 3-hydroxyadamantyl (meth) acrylate, 3-glycidylpropyl (meth) acrylate, 3-trimethylsilylpropyl (meth) acrylate, etc. Is mentioned.
(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸t-ブチル、(メタ)アクリル酸2-エチルヘキシル等の(メタ)アクリル酸アルキルエステル;
(メタ)アクリル酸シクロペンチル、(メタ)アクリル酸シクロヘキシル、(メタ)アクリル酸1-メチルシクロペンチル、(メタ)アクリル酸2-エチルアダマンチル、(メタ)アクリル酸2-(アダマンタン-1-イル)プロピル等の(メタ)アクリル酸シクロアルキルエステル;
(メタ)アクリル酸フェニル、(メタ)アクリル酸ナフチル等の(メタ)アクリル酸アリールエステル;
(メタ)アクリル酸2-ヒドロキシエチル、(メタ)アクリル酸3-ヒドロキシアダマンチル、(メタ)アクリル酸3-グリシジルプロピル、(メタ)アクリル酸3-トリメチルシリルプロピル等の(メタ)アクリル酸置換アルキルエステルなどが挙げられる。 Examples of (meth) acrylic acid esters include (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethyl (meth) acrylate, t-butyl (meth) acrylate, and 2-ethylhexyl (meth) acrylate. ;
Cyclopentyl (meth) acrylate, cyclohexyl (meth) acrylate, 1-methylcyclopentyl (meth) acrylate, 2-ethyladamantyl (meth) acrylate, 2- (adamantan-1-yl) propyl (meth) acrylate, etc. (Meth) acrylic acid cycloalkyl ester of
(Meth) acrylic acid aryl esters such as phenyl (meth) acrylate and naphthyl (meth) acrylate;
(Meth) acrylic acid-substituted alkyl esters such as 2-hydroxyethyl (meth) acrylate, 3-hydroxyadamantyl (meth) acrylate, 3-glycidylpropyl (meth) acrylate, 3-trimethylsilylpropyl (meth) acrylate, etc. Is mentioned.
エチレン系重合体は、置換又は非置換のエチレンに由来する構造単位を有する。
The ethylene polymer has a structural unit derived from substituted or unsubstituted ethylene.
置換エチレンとしては、例えば
プロペン、ブテン、ペンテン等のアルケン;
ビニルシクロペンタン、ビニルシクロヘキサン等のビニルシクロアルカン;
シクロペンテン、シクロヘキセン等のシクロアルケン;
4-ヒドロキシ-1-ブテン、ビニルグリシジルエーテル、ビニルトリメチルシリルエーテル等が挙げられる。 Examples of the substituted ethylene include alkenes such as propene, butene, and pentene;
Vinylcycloalkanes such as vinylcyclopentane and vinylcyclohexane;
Cycloalkenes such as cyclopentene and cyclohexene;
Examples include 4-hydroxy-1-butene, vinyl glycidyl ether, vinyl trimethylsilyl ether, and the like.
プロペン、ブテン、ペンテン等のアルケン;
ビニルシクロペンタン、ビニルシクロヘキサン等のビニルシクロアルカン;
シクロペンテン、シクロヘキセン等のシクロアルケン;
4-ヒドロキシ-1-ブテン、ビニルグリシジルエーテル、ビニルトリメチルシリルエーテル等が挙げられる。 Examples of the substituted ethylene include alkenes such as propene, butene, and pentene;
Vinylcycloalkanes such as vinylcyclopentane and vinylcyclohexane;
Cycloalkenes such as cyclopentene and cyclohexene;
Examples include 4-hydroxy-1-butene, vinyl glycidyl ether, vinyl trimethylsilyl ether, and the like.
[B]重合体の重量平均分子量(Mw)の下限としては、1,000が好ましく、3,000がより好ましく、5,000がさらに好ましく、5,500が特に好ましい。上記Mwの上限としては、100,000が好ましく、50,000がより好ましく、15,000がさらに好ましく、10,000が特に好ましい。
[B] The lower limit of the weight average molecular weight (Mw) of the polymer is preferably 1,000, more preferably 3,000, still more preferably 5,000, and particularly preferably 5,500. The upper limit of Mw is preferably 100,000, more preferably 50,000, still more preferably 15,000, and particularly preferably 10,000.
[B]重合体のMwの数平均分子量(Mn)に対する比(分散度)の上限としては、5が好ましく、3がより好ましく、2がより好ましく、1.3が特に好ましい。上記比の下限としては、通常1であり、1.05が好ましい。
[B] The upper limit of the ratio (dispersion degree) of the Mw to the number average molecular weight (Mn) of the polymer is preferably 5, more preferably 3, more preferably 2, and particularly preferably 1.3. The lower limit of the ratio is usually 1 and preferably 1.05.
本明細書におけるMw及びMnは、GPCカラム(東ソー社の「G2000HXL」2本、「G3000HXL」1本、「G4000HXL」1本)を用い、流量1.0mL/分、溶出溶媒:テトラヒドロフラン、試料濃度:1.0質量%、試料注入量:100μL、カラム温度:40℃の分析条件で、検出器として示差屈折計を使用し、単分散ポリスチレンを標準とするゲルパーミエーションクロマトグラフィー(GPC)により測定したものである。
Mw and Mn in this specification are GPC columns (two "G2000HXL", one "G3000HXL", one "G4000HXL" from Tosoh Corporation), a flow rate of 1.0 mL / min, elution solvent: tetrahydrofuran, sample concentration : Measured by gel permeation chromatography (GPC) using a monodisperse polystyrene as a standard using a differential refractometer as a detector under the analysis conditions of 1.0 mass%, sample injection amount: 100 μL, column temperature: 40 ° C. It is a thing.
[B]重合体の含有量の下限としては、組成物(II)における全固形分に対して、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましい。上記含有量の上限としては、例えば100質量%である。「全固形分」とは、[S]溶媒以外の成分の総和をいう。
[B] The lower limit of the content of the polymer is preferably 80% by mass, more preferably 90% by mass, and still more preferably 95% by mass with respect to the total solid content in the composition (II). As an upper limit of the said content, it is 100 mass%, for example. “Total solids” refers to the sum of components other than the [S] solvent.
([S]溶媒)
[S]溶媒としては、少なくとも[B]重合体及び他の成分を溶解又は分散可能な溶媒であれば特に限定されない。 ([S] solvent)
[S] The solvent is not particularly limited as long as it is a solvent that can dissolve or disperse at least the [B] polymer and other components.
[S]溶媒としては、少なくとも[B]重合体及び他の成分を溶解又は分散可能な溶媒であれば特に限定されない。 ([S] solvent)
[S] The solvent is not particularly limited as long as it is a solvent that can dissolve or disperse at least the [B] polymer and other components.
[S]溶媒としては、例えばアルコール系溶媒、エーテル系溶媒、ケトン系溶媒、アミド系溶媒、エステル系溶媒、炭化水素系溶媒等が挙げられる。
[S] Solvents include, for example, alcohol solvents, ether solvents, ketone solvents, amide solvents, ester solvents, hydrocarbon solvents, and the like.
アルコール系溶媒としては、例えば
メタノール、エタノール、n-プロパノール、iso-プロパノール、n-ブタノール、iso-ブタノール、sec-ブタノール、tert-ブタノール、n-ペンタノール、iso-ペンタノール、2-メチルブタノール、sec-ペンタノール、tert-ペンタノール、3-メトキシブタノール、n-ヘキサノール、2-メチルペンタノール、sec-ヘキサノール、2-エチルブタノール、sec-ヘプタノール、3-ヘプタノール、n-オクタノール、2-エチルヘキサノール、sec-オクタノール、n-ノニルアルコール、2,6-ジメチル-4-ヘプタノール、n-デカノール、sec-ウンデシルアルコール、トリメチルノニルアルコール、sec-テトラデシルアルコール、sec-ヘプタデシルアルコール、フルフリルアルコール、フェノール、シクロヘキサノール、メチルシクロヘキサノール、3,3,5-トリメチルシクロヘキサノール、ベンジルアルコール、ジアセトンアルコール等のモノアルコール系溶媒;
エチレングリコール、1,2-プロピレングリコール、1,3-ブチレングリコール、2,4-ペンタンジオール、2-メチル-2,4-ペンタンジオール、2,5-ヘキサンジオール、2,4-ヘプタンジオール、2-エチル-1,3-ヘキサンジオール、ジエチレングリコール、ジプロピレングリコール、トリエチレングリコール、トリプロピレングリコール等の多価アルコール系溶媒;
エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールモノプロピルエーテル、エチレングリコールモノブチルエーテル、エチレングリコールモノヘキシルエーテル、エチレングリコールモノフェニルエーテル、エチレングリコールモノ-2-エチルブチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノプロピルエーテル、ジエチレングリコールモノブチルエーテル、ジエチレングリコールモノヘキシルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールモノプロピルエーテル、プロピレングリコールモノブチルエーテル、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールモノエチルエーテル、ジプロピレングリコールモノプロピルエーテル等の多価アルコール部分エーテル系溶媒などが挙げられる。 Examples of the alcohol solvent include methanol, ethanol, n-propanol, iso-propanol, n-butanol, iso-butanol, sec-butanol, tert-butanol, n-pentanol, iso-pentanol, 2-methylbutanol, sec-pentanol, tert-pentanol, 3-methoxybutanol, n-hexanol, 2-methylpentanol, sec-hexanol, 2-ethylbutanol, sec-heptanol, 3-heptanol, n-octanol, 2-ethylhexanol , Sec-octanol, n-nonyl alcohol, 2,6-dimethyl-4-heptanol, n-decanol, sec-undecyl alcohol, trimethylnonyl alcohol, sec-tetradecyl alcohol, sec -Monoalcohol solvents such as heptadecyl alcohol, furfuryl alcohol, phenol, cyclohexanol, methylcyclohexanol, 3,3,5-trimethylcyclohexanol, benzyl alcohol, diacetone alcohol;
Ethylene glycol, 1,2-propylene glycol, 1,3-butylene glycol, 2,4-pentanediol, 2-methyl-2,4-pentanediol, 2,5-hexanediol, 2,4-heptanediol, 2 Polyhydric alcohol solvents such as ethyl-1,3-hexanediol, diethylene glycol, dipropylene glycol, triethylene glycol, tripropylene glycol;
Ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monopropyl ether, ethylene glycol monobutyl ether, ethylene glycol monohexyl ether, ethylene glycol monophenyl ether, ethylene glycol mono-2-ethylbutyl ether, diethylene glycol monomethyl ether, diethylene glycol monoethyl Ether, diethylene glycol monopropyl ether, diethylene glycol monobutyl ether, diethylene glycol monohexyl ether, propylene glycol monomethyl ether, propylene glycol monoethyl ether, propylene glycol monopropyl ether, propylene glycol monobutyl ether, dipropylene glycol Monomethyl ether, dipropylene glycol monoethyl ether, and polyhydric alcohol partial ether solvents such as dipropylene glycol monopropyl ether.
メタノール、エタノール、n-プロパノール、iso-プロパノール、n-ブタノール、iso-ブタノール、sec-ブタノール、tert-ブタノール、n-ペンタノール、iso-ペンタノール、2-メチルブタノール、sec-ペンタノール、tert-ペンタノール、3-メトキシブタノール、n-ヘキサノール、2-メチルペンタノール、sec-ヘキサノール、2-エチルブタノール、sec-ヘプタノール、3-ヘプタノール、n-オクタノール、2-エチルヘキサノール、sec-オクタノール、n-ノニルアルコール、2,6-ジメチル-4-ヘプタノール、n-デカノール、sec-ウンデシルアルコール、トリメチルノニルアルコール、sec-テトラデシルアルコール、sec-ヘプタデシルアルコール、フルフリルアルコール、フェノール、シクロヘキサノール、メチルシクロヘキサノール、3,3,5-トリメチルシクロヘキサノール、ベンジルアルコール、ジアセトンアルコール等のモノアルコール系溶媒;
エチレングリコール、1,2-プロピレングリコール、1,3-ブチレングリコール、2,4-ペンタンジオール、2-メチル-2,4-ペンタンジオール、2,5-ヘキサンジオール、2,4-ヘプタンジオール、2-エチル-1,3-ヘキサンジオール、ジエチレングリコール、ジプロピレングリコール、トリエチレングリコール、トリプロピレングリコール等の多価アルコール系溶媒;
エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールモノプロピルエーテル、エチレングリコールモノブチルエーテル、エチレングリコールモノヘキシルエーテル、エチレングリコールモノフェニルエーテル、エチレングリコールモノ-2-エチルブチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノプロピルエーテル、ジエチレングリコールモノブチルエーテル、ジエチレングリコールモノヘキシルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールモノプロピルエーテル、プロピレングリコールモノブチルエーテル、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールモノエチルエーテル、ジプロピレングリコールモノプロピルエーテル等の多価アルコール部分エーテル系溶媒などが挙げられる。 Examples of the alcohol solvent include methanol, ethanol, n-propanol, iso-propanol, n-butanol, iso-butanol, sec-butanol, tert-butanol, n-pentanol, iso-pentanol, 2-methylbutanol, sec-pentanol, tert-pentanol, 3-methoxybutanol, n-hexanol, 2-methylpentanol, sec-hexanol, 2-ethylbutanol, sec-heptanol, 3-heptanol, n-octanol, 2-ethylhexanol , Sec-octanol, n-nonyl alcohol, 2,6-dimethyl-4-heptanol, n-decanol, sec-undecyl alcohol, trimethylnonyl alcohol, sec-tetradecyl alcohol, sec -Monoalcohol solvents such as heptadecyl alcohol, furfuryl alcohol, phenol, cyclohexanol, methylcyclohexanol, 3,3,5-trimethylcyclohexanol, benzyl alcohol, diacetone alcohol;
Ethylene glycol, 1,2-propylene glycol, 1,3-butylene glycol, 2,4-pentanediol, 2-methyl-2,4-pentanediol, 2,5-hexanediol, 2,4-heptanediol, 2 Polyhydric alcohol solvents such as ethyl-1,3-hexanediol, diethylene glycol, dipropylene glycol, triethylene glycol, tripropylene glycol;
Ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monopropyl ether, ethylene glycol monobutyl ether, ethylene glycol monohexyl ether, ethylene glycol monophenyl ether, ethylene glycol mono-2-ethylbutyl ether, diethylene glycol monomethyl ether, diethylene glycol monoethyl Ether, diethylene glycol monopropyl ether, diethylene glycol monobutyl ether, diethylene glycol monohexyl ether, propylene glycol monomethyl ether, propylene glycol monoethyl ether, propylene glycol monopropyl ether, propylene glycol monobutyl ether, dipropylene glycol Monomethyl ether, dipropylene glycol monoethyl ether, and polyhydric alcohol partial ether solvents such as dipropylene glycol monopropyl ether.
エーテル系溶媒としては、例えば
ジエチルエーテル、ジプロピルエーテル、ジブチルエーテル等のジアルキルエーテル系溶媒;
テトラヒドロフラン、テトラヒドロピラン等の環状エーテル系溶媒;
ジフェニルエーテル、アニソール等の芳香環含有エーテル系溶媒などが挙げられる。 Examples of ether solvents include dialkyl ether solvents such as diethyl ether, dipropyl ether, and dibutyl ether;
Cyclic ether solvents such as tetrahydrofuran and tetrahydropyran;
And aromatic ring-containing ether solvents such as diphenyl ether and anisole.
ジエチルエーテル、ジプロピルエーテル、ジブチルエーテル等のジアルキルエーテル系溶媒;
テトラヒドロフラン、テトラヒドロピラン等の環状エーテル系溶媒;
ジフェニルエーテル、アニソール等の芳香環含有エーテル系溶媒などが挙げられる。 Examples of ether solvents include dialkyl ether solvents such as diethyl ether, dipropyl ether, and dibutyl ether;
Cyclic ether solvents such as tetrahydrofuran and tetrahydropyran;
And aromatic ring-containing ether solvents such as diphenyl ether and anisole.
ケトン系溶媒としては、例えば
アセトン、メチルエチルケトン、メチル-n-プロピルケトン、メチル-n-ブチルケトン、ジエチルケトン、メチル-iso-ブチルケトン、2-ヘプタノン、エチル-n-ブチルケトン、メチル-n-ヘキシルケトン、ジ-iso-ブチルケトン、トリメチルノナノン等の鎖状ケトン系溶媒;
シクロペンタノン、シクロヘキサノン、シクロヘプタノン、シクロオクタノン、メチルシクロヘキサノン等の環状ケトン系溶媒;
2,4-ペンタンジオン、アセトニルアセトン、アセトフェノン等が挙げられる。 Examples of the ketone solvent include acetone, methyl ethyl ketone, methyl-n-propyl ketone, methyl-n-butyl ketone, diethyl ketone, methyl-iso-butyl ketone, 2-heptanone, ethyl-n-butyl ketone, methyl-n-hexyl ketone, Linear ketone solvents such as di-iso-butyl ketone and trimethylnonanone;
Cyclic ketone solvents such as cyclopentanone, cyclohexanone, cycloheptanone, cyclooctanone, methylcyclohexanone;
Examples include 2,4-pentanedione, acetonylacetone, acetophenone, and the like.
アセトン、メチルエチルケトン、メチル-n-プロピルケトン、メチル-n-ブチルケトン、ジエチルケトン、メチル-iso-ブチルケトン、2-ヘプタノン、エチル-n-ブチルケトン、メチル-n-ヘキシルケトン、ジ-iso-ブチルケトン、トリメチルノナノン等の鎖状ケトン系溶媒;
シクロペンタノン、シクロヘキサノン、シクロヘプタノン、シクロオクタノン、メチルシクロヘキサノン等の環状ケトン系溶媒;
2,4-ペンタンジオン、アセトニルアセトン、アセトフェノン等が挙げられる。 Examples of the ketone solvent include acetone, methyl ethyl ketone, methyl-n-propyl ketone, methyl-n-butyl ketone, diethyl ketone, methyl-iso-butyl ketone, 2-heptanone, ethyl-n-butyl ketone, methyl-n-hexyl ketone, Linear ketone solvents such as di-iso-butyl ketone and trimethylnonanone;
Cyclic ketone solvents such as cyclopentanone, cyclohexanone, cycloheptanone, cyclooctanone, methylcyclohexanone;
Examples include 2,4-pentanedione, acetonylacetone, acetophenone, and the like.
アミド系溶媒としては、例えば
N,N’-ジメチルイミダゾリジノン、N-メチルピロリドン等の環状アミド系溶媒;
N-メチルホルムアミド、N,N-ジメチルホルムアミド、N,N-ジエチルホルムアミド、アセトアミド、N-メチルアセトアミド、N,N-ジメチルアセトアミド、N-メチルプロピオンアミド等の鎖状アミド系溶媒などが挙げられる。 Examples of the amide solvent include cyclic amide solvents such as N, N′-dimethylimidazolidinone and N-methylpyrrolidone;
Examples thereof include chain amide solvents such as N-methylformamide, N, N-dimethylformamide, N, N-diethylformamide, acetamide, N-methylacetamide, N, N-dimethylacetamide, and N-methylpropionamide.
N,N’-ジメチルイミダゾリジノン、N-メチルピロリドン等の環状アミド系溶媒;
N-メチルホルムアミド、N,N-ジメチルホルムアミド、N,N-ジエチルホルムアミド、アセトアミド、N-メチルアセトアミド、N,N-ジメチルアセトアミド、N-メチルプロピオンアミド等の鎖状アミド系溶媒などが挙げられる。 Examples of the amide solvent include cyclic amide solvents such as N, N′-dimethylimidazolidinone and N-methylpyrrolidone;
Examples thereof include chain amide solvents such as N-methylformamide, N, N-dimethylformamide, N, N-diethylformamide, acetamide, N-methylacetamide, N, N-dimethylacetamide, and N-methylpropionamide.
エステル系溶媒としては、例えば
酢酸メチル、酢酸エチル、酢酸n-プロピル、酢酸iso-プロピル、酢酸n-ブチル、酢酸iso-ブチル、酢酸sec-ブチル、酢酸n-ペンチル、酢酸i-ペンチル、酢酸sec-ペンチル、酢酸3-メトキシブチル、酢酸メチルペンチル、酢酸2-エチルブチル、酢酸2-エチルヘキシル、酢酸ベンジル、酢酸シクロヘキシル、酢酸メチルシクロヘキシル、酢酸n-ノニル等の酢酸エステル系溶媒;
エチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノメチルエーテルアセテート、ジエチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノ-n-ブチルエーテルアセテート、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノメチルエーテルプロピオネート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート、プロピレングリコールモノブチルエーテルアセテート、ジプロピレングリコールモノメチルエーテルアセテート、ジプロピレングリコールモノエチルエーテルアセテート等の多価アルコール部分エーテルカルボキレート系溶媒;
γ-ブチロラクトン、バレロラクトン等のラクトン系溶媒;
ジメチルカーボネート、ジエチルカーボネート、エチレンカーボネート、プロピレンカーボネート等のカーボネート系溶媒;
ジ酢酸グリコール、酢酸メトキシトリグリコール、プロピオン酸エチル、プロピオン酸n-ブチル、プロピオン酸iso-アミル、シュウ酸ジエチル、シュウ酸ジ-n-ブチル、アセト酢酸メチル、アセト酢酸エチル、乳酸メチル、乳酸エチル、乳酸n-ブチル、乳酸n-アミル、マロン酸ジエチル、フタル酸ジメチル、フタル酸ジエチルなどが挙げられる。 Examples of ester solvents include methyl acetate, ethyl acetate, n-propyl acetate, iso-propyl acetate, n-butyl acetate, iso-butyl acetate, sec-butyl acetate, n-pentyl acetate, i-pentyl acetate, sec Acetate solvents such as pentyl, 3-methoxybutyl acetate, methylpentyl acetate, 2-ethylbutyl acetate, 2-ethylhexyl acetate, benzyl acetate, cyclohexyl acetate, methylcyclohexyl acetate, n-nonyl acetate;
Ethylene glycol monomethyl ether acetate, ethylene glycol monoethyl ether acetate, diethylene glycol monomethyl ether acetate, diethylene glycol monoethyl ether acetate, diethylene glycol mono-n-butyl ether acetate, propylene glycol monomethyl ether acetate, propylene glycol monomethyl ether propionate, propylene glycol monoethyl Polyhydric alcohol partial ether carbochelate solvents such as ether acetate, propylene glycol monopropyl ether acetate, propylene glycol monobutyl ether acetate, dipropylene glycol monomethyl ether acetate, dipropylene glycol monoethyl ether acetate;
Lactone solvents such as γ-butyrolactone and valerolactone;
Carbonate solvents such as dimethyl carbonate, diethyl carbonate, ethylene carbonate, propylene carbonate;
Diethyl acetate, methoxytriglycol acetate, ethyl propionate, n-butyl propionate, iso-amyl propionate, diethyl oxalate, di-n-butyl oxalate, methyl acetoacetate, ethyl acetoacetate, methyl lactate, ethyl lactate N-butyl lactate, n-amyl lactate, diethyl malonate, dimethyl phthalate, diethyl phthalate and the like.
酢酸メチル、酢酸エチル、酢酸n-プロピル、酢酸iso-プロピル、酢酸n-ブチル、酢酸iso-ブチル、酢酸sec-ブチル、酢酸n-ペンチル、酢酸i-ペンチル、酢酸sec-ペンチル、酢酸3-メトキシブチル、酢酸メチルペンチル、酢酸2-エチルブチル、酢酸2-エチルヘキシル、酢酸ベンジル、酢酸シクロヘキシル、酢酸メチルシクロヘキシル、酢酸n-ノニル等の酢酸エステル系溶媒;
エチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノメチルエーテルアセテート、ジエチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノ-n-ブチルエーテルアセテート、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノメチルエーテルプロピオネート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート、プロピレングリコールモノブチルエーテルアセテート、ジプロピレングリコールモノメチルエーテルアセテート、ジプロピレングリコールモノエチルエーテルアセテート等の多価アルコール部分エーテルカルボキレート系溶媒;
γ-ブチロラクトン、バレロラクトン等のラクトン系溶媒;
ジメチルカーボネート、ジエチルカーボネート、エチレンカーボネート、プロピレンカーボネート等のカーボネート系溶媒;
ジ酢酸グリコール、酢酸メトキシトリグリコール、プロピオン酸エチル、プロピオン酸n-ブチル、プロピオン酸iso-アミル、シュウ酸ジエチル、シュウ酸ジ-n-ブチル、アセト酢酸メチル、アセト酢酸エチル、乳酸メチル、乳酸エチル、乳酸n-ブチル、乳酸n-アミル、マロン酸ジエチル、フタル酸ジメチル、フタル酸ジエチルなどが挙げられる。 Examples of ester solvents include methyl acetate, ethyl acetate, n-propyl acetate, iso-propyl acetate, n-butyl acetate, iso-butyl acetate, sec-butyl acetate, n-pentyl acetate, i-pentyl acetate, sec Acetate solvents such as pentyl, 3-methoxybutyl acetate, methylpentyl acetate, 2-ethylbutyl acetate, 2-ethylhexyl acetate, benzyl acetate, cyclohexyl acetate, methylcyclohexyl acetate, n-nonyl acetate;
Ethylene glycol monomethyl ether acetate, ethylene glycol monoethyl ether acetate, diethylene glycol monomethyl ether acetate, diethylene glycol monoethyl ether acetate, diethylene glycol mono-n-butyl ether acetate, propylene glycol monomethyl ether acetate, propylene glycol monomethyl ether propionate, propylene glycol monoethyl Polyhydric alcohol partial ether carbochelate solvents such as ether acetate, propylene glycol monopropyl ether acetate, propylene glycol monobutyl ether acetate, dipropylene glycol monomethyl ether acetate, dipropylene glycol monoethyl ether acetate;
Lactone solvents such as γ-butyrolactone and valerolactone;
Carbonate solvents such as dimethyl carbonate, diethyl carbonate, ethylene carbonate, propylene carbonate;
Diethyl acetate, methoxytriglycol acetate, ethyl propionate, n-butyl propionate, iso-amyl propionate, diethyl oxalate, di-n-butyl oxalate, methyl acetoacetate, ethyl acetoacetate, methyl lactate, ethyl lactate N-butyl lactate, n-amyl lactate, diethyl malonate, dimethyl phthalate, diethyl phthalate and the like.
炭化水素系溶媒としては、例えば
n-ペンタン、iso-ペンタン、n-ヘキサン、iso-ヘキサン、n-ヘプタン、iso-ヘプタン、2,2,4-トリメチルペンタン、n-オクタン、iso-オクタン、シクロヘキサン、メチルシクロヘキサン等の脂肪族炭化水素系溶媒;
ベンゼン、トルエン、キシレン、メシチレン、エチルベンゼン、トリメチルベンゼン、メチルエチルベンゼン、n-プロピルベンゼン、iso-プロピルベンゼン、ジエチルベンゼン、iso-ブチルベンゼン、トリエチルベンゼン、ジ-iso-プロピルベンセン、n-アミルナフタレン等の芳香族炭化水素系溶媒などが挙げられる。 Examples of hydrocarbon solvents include n-pentane, iso-pentane, n-hexane, iso-hexane, n-heptane, iso-heptane, 2,2,4-trimethylpentane, n-octane, iso-octane, cyclohexane , Aliphatic hydrocarbon solvents such as methylcyclohexane;
Fragrances such as benzene, toluene, xylene, mesitylene, ethylbenzene, trimethylbenzene, methylethylbenzene, n-propylbenzene, iso-propylbenzene, diethylbenzene, iso-butylbenzene, triethylbenzene, di-iso-propylbenzene, n-amylnaphthalene Group hydrocarbon solvents and the like.
n-ペンタン、iso-ペンタン、n-ヘキサン、iso-ヘキサン、n-ヘプタン、iso-ヘプタン、2,2,4-トリメチルペンタン、n-オクタン、iso-オクタン、シクロヘキサン、メチルシクロヘキサン等の脂肪族炭化水素系溶媒;
ベンゼン、トルエン、キシレン、メシチレン、エチルベンゼン、トリメチルベンゼン、メチルエチルベンゼン、n-プロピルベンゼン、iso-プロピルベンゼン、ジエチルベンゼン、iso-ブチルベンゼン、トリエチルベンゼン、ジ-iso-プロピルベンセン、n-アミルナフタレン等の芳香族炭化水素系溶媒などが挙げられる。 Examples of hydrocarbon solvents include n-pentane, iso-pentane, n-hexane, iso-hexane, n-heptane, iso-heptane, 2,2,4-trimethylpentane, n-octane, iso-octane, cyclohexane , Aliphatic hydrocarbon solvents such as methylcyclohexane;
Fragrances such as benzene, toluene, xylene, mesitylene, ethylbenzene, trimethylbenzene, methylethylbenzene, n-propylbenzene, iso-propylbenzene, diethylbenzene, iso-butylbenzene, triethylbenzene, di-iso-propylbenzene, n-amylnaphthalene Group hydrocarbon solvents and the like.
これらの中で、エステル系溶媒が好ましく、多価アルコール部分エーテルカルボキシレート系溶媒がより好ましく、プロピレングリコールモノメチルエーテルアセテートがさらに好ましい。組成物(II)は、[S]溶媒を1種又は2種以上含有していてもよい。
Of these, ester solvents are preferred, polyhydric alcohol partial ether carboxylate solvents are more preferred, and propylene glycol monomethyl ether acetate is even more preferred. Composition (II) may contain one or more [S] solvents.
(他の成分)
組成物(II)は、他の成分として、例えば界面活性剤等を含有していてもよい。組成物(II)は、界面活性剤を含有することで、レジストパターン2への塗工性を向上させることができる。 (Other ingredients)
The composition (II) may contain, for example, a surfactant as other components. Composition (II) can improve the coating property to the resistpattern 2 by containing surfactant.
組成物(II)は、他の成分として、例えば界面活性剤等を含有していてもよい。組成物(II)は、界面活性剤を含有することで、レジストパターン2への塗工性を向上させることができる。 (Other ingredients)
The composition (II) may contain, for example, a surfactant as other components. Composition (II) can improve the coating property to the resist
組成物(II)の塗工方法としては、例えばスピンコート法等が挙げられる。
Examples of the coating method of the composition (II) include a spin coating method.
([C]重合体層形成工程)
本工程では、レジストパターンの少なくとも基板又は他の層の表面に[C]重合体層を形成する。 ([C] polymer layer forming step)
In this step, a [C] polymer layer is formed on the surface of at least the substrate or other layer of the resist pattern.
本工程では、レジストパターンの少なくとも基板又は他の層の表面に[C]重合体層を形成する。 ([C] polymer layer forming step)
In this step, a [C] polymer layer is formed on the surface of at least the substrate or other layer of the resist pattern.
[C]重合体層を形成する方法としては、例えば[C]重合体と[S]溶媒とを含有する組成物(III)を塗工する工程(以下、「組成物(III)塗工工程」ともいう)を行う方法等が挙げられる。
[C] As a method for forming a polymer layer, for example, a step of applying a composition (III) containing a [C] polymer and a [S] solvent (hereinafter referred to as “composition (III) coating step”). And the like.
(組成物(III))
組成物(III)は、[C]重合体と[S]溶媒とを含有する。組成物(III)は、[C]重合体及び[S]溶媒以外の他の成分を含有してもよい。 (Composition (III))
The composition (III) contains a [C] polymer and a [S] solvent. The composition (III) may contain components other than the [C] polymer and the [S] solvent.
組成物(III)は、[C]重合体と[S]溶媒とを含有する。組成物(III)は、[C]重合体及び[S]溶媒以外の他の成分を含有してもよい。 (Composition (III))
The composition (III) contains a [C] polymer and a [S] solvent. The composition (III) may contain components other than the [C] polymer and the [S] solvent.
([C]重合体)
[C]重合体としては、主鎖の少なくとも一方の末端に結合しSi-H、Si-OH、Si=O及びSi-NR2(Rは、それぞれ独立して、水素原子又は炭素数1~20の1価の有機基である)の少なくとも1種と化学結合を形成する官能基(2)を有することが好ましい。 ([C] polymer)
[C] As the polymer, Si—H, Si—OH, Si═O and Si—NR 2 bonded to at least one terminal of the main chain (R each independently represents a hydrogen atom or a carbon number of 1 to It is preferable to have a functional group (2) that forms a chemical bond with at least one of 20 monovalent organic groups.
[C]重合体としては、主鎖の少なくとも一方の末端に結合しSi-H、Si-OH、Si=O及びSi-NR2(Rは、それぞれ独立して、水素原子又は炭素数1~20の1価の有機基である)の少なくとも1種と化学結合を形成する官能基(2)を有することが好ましい。 ([C] polymer)
[C] As the polymer, Si—H, Si—OH, Si═O and Si—NR 2 bonded to at least one terminal of the main chain (R each independently represents a hydrogen atom or a carbon number of 1 to It is preferable to have a functional group (2) that forms a chemical bond with at least one of 20 monovalent organic groups.
官能基(2)としては、例えばカルボキシ基、水酸基、ハロゲン原子等が挙げられる。これらの官能基は、Si-H、Si-OH、Si=O及び/又はSi-NR2と共有結合を形成する。
Examples of the functional group (2) include a carboxy group, a hydroxyl group, and a halogen atom. These functional groups form covalent bonds with Si—H, Si—OH, Si═O and / or Si—NR 2 .
[C]重合体としては、例えばスチレン系重合体、(メタ)アクリル系重合体、エチレン系重合体、これらを組み合わせた共重合体等が挙げられる。これらの重合体としては、例えば[B]重合体として例示したものと同様の重合体等が挙げられる。
Examples of the [C] polymer include a styrene polymer, a (meth) acrylic polymer, an ethylene polymer, and a copolymer combining these. Examples of these polymers include polymers similar to those exemplified as the [B] polymer.
[C]重合体の重量平均分子量(Mw)の下限としては、1,000が好ましく、3,000がより好ましく、4,000がさらに好ましく、5,000が特に好ましい。上記Mwの上限としては、100,000が好ましく、50,000がより好ましく、20,000がさらに好ましく、10,000が特に好ましい。
[C] The lower limit of the weight average molecular weight (Mw) of the polymer is preferably 1,000, more preferably 3,000, still more preferably 4,000, and particularly preferably 5,000. The upper limit of the Mw is preferably 100,000, more preferably 50,000, still more preferably 20,000, and particularly preferably 10,000.
[C]重合体のMwの数平均分子量(Mn)に対する比(分散度)の上限としては、5が好ましく、3がより好ましく、2がより好ましく、1.4が特に好ましい。上記比の下限としては、通常1であり、1.1が好ましい。
[C] The upper limit of the ratio (dispersion degree) of the Mw to the number average molecular weight (Mn) of the polymer is preferably 5, more preferably 3, more preferably 2, and particularly preferably 1.4. The lower limit of the ratio is usually 1 and preferably 1.1.
[C]重合体の含有量の下限としては、組成物(III)における全固形分に対して、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましい。上記含有量の上限としては、例えば100質量%である。
[C] The lower limit of the content of the polymer is preferably 80% by mass, more preferably 90% by mass, and still more preferably 95% by mass with respect to the total solid content in the composition (III). As an upper limit of the said content, it is 100 mass%, for example.
組成物(III)における[S]溶媒及び他の成分としては、上記組成物(II)の[S]溶媒及び他の成分として例示したのと同様のもの等が挙げられる。
Examples of the [S] solvent and other components in the composition (III) include the same as those exemplified as the [S] solvent and other components in the composition (II).
組成物(III)の塗工方法としては、例えばスピンコート法等が挙げられる。
Examples of the coating method of the composition (III) include a spin coating method.
また、重合体層形成工程において、[B]重合体層形成工程と[C]重合体層形成工程とを同時に行う場合、[B]重合体と[C]重合体と[S]溶媒とを含有する組成物(以下、「組成物(IV)」ともいう)を塗工する工程(以下、「組成物(IV)塗工工程」ともいう)を行うこともできる。
In the polymer layer forming step, when the [B] polymer layer forming step and the [C] polymer layer forming step are simultaneously performed, the [B] polymer, the [C] polymer, and the [S] solvent are added. A step of applying a composition (hereinafter, also referred to as “composition (IV)”) (hereinafter, also referred to as “composition (IV) coating step”) may be performed.
(組成物(IV))
組成物(IV)は、[B]重合体と[C]重合体と[S]溶媒とを含有する。組成物(IV)は[B]重合体、[C]重合体及び[S]溶媒以外の他の成分を含有してもよい。 (Composition (IV))
The composition (IV) contains a [B] polymer, a [C] polymer, and a [S] solvent. The composition (IV) may contain components other than the [B] polymer, the [C] polymer, and the [S] solvent.
組成物(IV)は、[B]重合体と[C]重合体と[S]溶媒とを含有する。組成物(IV)は[B]重合体、[C]重合体及び[S]溶媒以外の他の成分を含有してもよい。 (Composition (IV))
The composition (IV) contains a [B] polymer, a [C] polymer, and a [S] solvent. The composition (IV) may contain components other than the [B] polymer, the [C] polymer, and the [S] solvent.
組成物(IV)における[B]重合体、[C]重合体、[S]溶媒及び他の成分としては、上記組成物(II)の[S]溶媒及び他の成分として例示したのと同様のもの等が挙げられる。
The [B] polymer, [C] polymer, [S] solvent and other components in the composition (IV) are the same as those exemplified as the [S] solvent and other components in the composition (II). And the like.
[B]重合体及び[C]重合体の合計含有量の下限としては、組成物(IV)における全固形分に対して、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましい。上記含有量の上限としては、例えば100質量%である。
The lower limit of the total content of the [B] polymer and the [C] polymer is preferably 80% by mass, more preferably 90% by mass, and 95% by mass with respect to the total solid content in the composition (IV). Further preferred. As an upper limit of the said content, it is 100 mass%, for example.
[[B]重合体及び[C]重合体の合成方法]
[B]重合体及び[C]重合体は、アニオン重合、ラジカル重合等の重合によって合成することができるが、任意の末端構造を容易に導入することができるリビングアニオン重合が好ましい。リビングアニオン重合によれば、例えばスチレン等の単量体を重合し、その重合末端を、任意の末端処理剤で処理することにより、重合体の主鎖の末端に結合する官能基を導入することができる。 [Method for synthesizing [B] polymer and [C] polymer]
The [B] polymer and the [C] polymer can be synthesized by polymerization such as anionic polymerization and radical polymerization, but living anionic polymerization in which any terminal structure can be easily introduced is preferable. According to living anionic polymerization, for example, a monomer such as styrene is polymerized, and the polymerization terminal is treated with an arbitrary terminal treating agent to introduce a functional group that is bonded to the terminal of the main chain of the polymer. Can do.
[B]重合体及び[C]重合体は、アニオン重合、ラジカル重合等の重合によって合成することができるが、任意の末端構造を容易に導入することができるリビングアニオン重合が好ましい。リビングアニオン重合によれば、例えばスチレン等の単量体を重合し、その重合末端を、任意の末端処理剤で処理することにより、重合体の主鎖の末端に結合する官能基を導入することができる。 [Method for synthesizing [B] polymer and [C] polymer]
The [B] polymer and the [C] polymer can be synthesized by polymerization such as anionic polymerization and radical polymerization, but living anionic polymerization in which any terminal structure can be easily introduced is preferable. According to living anionic polymerization, for example, a monomer such as styrene is polymerized, and the polymerization terminal is treated with an arbitrary terminal treating agent to introduce a functional group that is bonded to the terminal of the main chain of the polymer. Can do.
例えば末端に官能基(1)又は官能基(2)を有するポリスチレンブロックからなる重合体を合成する場合は、まずアニオン重合開始剤を使用して、適当な溶媒中でスチレンを重合することにより活性末端を有するスチレンの重合体を形成する。その後、官能基(1)又は官能基(2)を与える末端処理剤で処理することにより、ポリスチレンの主鎖の末端に官能基を導入された重合体を得ることができる。
For example, when synthesizing a polymer composed of a polystyrene block having a functional group (1) or a functional group (2) at the terminal, it is first activated by polymerizing styrene in an appropriate solvent using an anionic polymerization initiator. A terminal styrene polymer is formed. Then, the polymer by which the functional group was introduce | transduced into the terminal of the principal chain of a polystyrene can be obtained by processing with the terminal processing agent which gives a functional group (1) or a functional group (2).
すなわち、上記末端処理の方法としては、例えば下記スキームに示すような方法等が挙げられる。すなわち、重合中の活性末端に、例えば1,2-ブチレンオキシド等の末端処理剤を添加して末端を変性し、主鎖の末端に結合する官能基を有する重合体を得る。
That is, examples of the terminal treatment method include a method as shown in the following scheme. That is, a terminal treatment agent such as 1,2-butylene oxide is added to the active terminal during polymerization to modify the terminal to obtain a polymer having a functional group bonded to the terminal of the main chain.

上記スキーム中、n及びmは、10~5,000の整数である。
In the above scheme, n and m are integers of 10 to 5,000.
水酸基を与える末端処理剤としては、例えば
1,2-ブチレンオキサイド、ブチルグリシジルエーテル、2-エチルヘキシルグリシジルエーテル、プロピレンオキサイド、エチレンオキサイド、スチレンオキサイド、エポキシアミン等のエポキシ化合物等が挙げられる。 Examples of the terminal treatment agent that gives a hydroxyl group include epoxy compounds such as 1,2-butylene oxide, butyl glycidyl ether, 2-ethylhexyl glycidyl ether, propylene oxide, ethylene oxide, styrene oxide, and epoxyamine.
1,2-ブチレンオキサイド、ブチルグリシジルエーテル、2-エチルヘキシルグリシジルエーテル、プロピレンオキサイド、エチレンオキサイド、スチレンオキサイド、エポキシアミン等のエポキシ化合物等が挙げられる。 Examples of the terminal treatment agent that gives a hydroxyl group include epoxy compounds such as 1,2-butylene oxide, butyl glycidyl ether, 2-ethylhexyl glycidyl ether, propylene oxide, ethylene oxide, styrene oxide, and epoxyamine.
カルボキシ基を与える末端処理剤としては、例えば二酸化炭素等が挙げられる。
Examples of the end treatment agent that gives a carboxy group include carbon dioxide.
エポキシ基を与える末端処理剤としては、例えばエピブロモヒドリン、エピクロロヒドリン等のハロゲン原子含有エポキシ化合物等が挙げられる。
Examples of the end treatment agent that gives an epoxy group include halogen atom-containing epoxy compounds such as epibromohydrin and epichlorohydrin.
オキセタニル基を与える末端処理剤としては、例えば3-クロロメチル-3-メチルオキセタン、3-クロロメチル-3-エチルオキセタン等のハロゲン原子含有オキセタン化合物等が挙げられる。
Examples of the end treating agent that gives an oxetanyl group include halogen atom-containing oxetane compounds such as 3-chloromethyl-3-methyloxetane and 3-chloromethyl-3-ethyloxetane.
また、末端にカルボキシ基を有する重合体は、例えばスチレン等の単量体と、4-シアノ-4-[(ドデシルスルファニルチオカルボニルスルファン]ペンタン酸メチル等とを、アニソール等の溶媒中でRAFT重合(Reversible Addition Fragmentation Chain Transfer重合、可逆的付加開裂連鎖移動重合)を行うことにより得ることができる。
In addition, a polymer having a carboxy group at the terminal is, for example, a monomer such as styrene and methyl 4-cyano-4-[(dodecylsulfanylthiocarbonylsulfane) pentanoate or the like in a solvent such as anisole. It can be obtained by performing polymerization (Reversible Addition Fragmentation Chain Transfer Polymerization, reversible addition-fragmentation chain transfer polymerization).
末端にハロゲン原子を有する重合体は、例えばスチレン等の単量体と、臭化銅(II)及びトリス[(2-ジメチルアミノ)エチル]アミン等のアミン化合物と、アニソール等の溶媒とを仕込み、これに、2-ブロモイソ酪酸2-ヒドロキシエチル等のハロゲン化合物を加える方法により、原子移動ラジカル重合によって合成することができる。
The polymer having a halogen atom at the terminal is prepared by charging a monomer such as styrene, an amine compound such as copper (II) bromide and tris [(2-dimethylamino) ethyl] amine, and a solvent such as anisole. Further, it can be synthesized by atom transfer radical polymerization by adding a halogen compound such as 2-hydroxyethyl 2-bromoisobutyrate to this.
上記得られた[B]重合体及び[C]重合体は、再沈殿法により回収することが好ましい。すなわち、末端処理反応終了後、反応液を再沈溶媒に投入することにより、目的の共重合体を粉体として回収する。再沈溶媒としては、アルコール類やアルカン類等を単独で又は2種以上を混合して使用することができる。再沈殿法の他に、分液操作やカラム操作、限外ろ過操作等により、単量体、オリゴマー等の低分子成分を除去して、重合体を回収することもできる。また、[B]重合体及び[C]重合体は、酸等による脱メタル処理などの精製を行うことが好ましい。
The [B] polymer and [C] polymer obtained above are preferably recovered by a reprecipitation method. That is, after completion of the terminal treatment reaction, the target copolymer is recovered as a powder by introducing the reaction solution into a reprecipitation solvent. As the reprecipitation solvent, alcohols or alkanes can be used alone or in admixture of two or more. In addition to the reprecipitation method, the polymer can be recovered by removing low-molecular components such as monomers and oligomers by a liquid separation operation, a column operation, an ultrafiltration operation, or the like. The [B] polymer and the [C] polymer are preferably subjected to purification such as demetallation with an acid or the like.
[組成物(II)~(IV)の調製方法]
組成物(II)~(IV)は、例えば[B]重合体及び/又は[C]重合体、[S]溶媒及び必要に応じて他の成分を所定の割合で混合し、好ましくは孔径0.45μm程度の高密度ポリエチレンフィルター等で濾過することにより調製することができる。組成物(II)~(IV)の固形分濃度の下限としては、0.1質量%が好ましく、0.5質量%がより好ましく、0.7質量%がさらに好ましい。上記固形分濃度の上限としては、30質量%が好ましく、10質量%がより好ましく、3質量%がさらに好ましい。 [Method for Preparing Compositions (II) to (IV)]
In the compositions (II) to (IV), for example, the [B] polymer and / or the [C] polymer, the [S] solvent and other components as necessary are mixed at a predetermined ratio, and preferably the pore size is 0. It can be prepared by filtering with a high-density polyethylene filter of about 45 μm. The lower limit of the solid content concentration of the compositions (II) to (IV) is preferably 0.1% by mass, more preferably 0.5% by mass, and even more preferably 0.7% by mass. The upper limit of the solid content concentration is preferably 30% by mass, more preferably 10% by mass, and still more preferably 3% by mass.
組成物(II)~(IV)は、例えば[B]重合体及び/又は[C]重合体、[S]溶媒及び必要に応じて他の成分を所定の割合で混合し、好ましくは孔径0.45μm程度の高密度ポリエチレンフィルター等で濾過することにより調製することができる。組成物(II)~(IV)の固形分濃度の下限としては、0.1質量%が好ましく、0.5質量%がより好ましく、0.7質量%がさらに好ましい。上記固形分濃度の上限としては、30質量%が好ましく、10質量%がより好ましく、3質量%がさらに好ましい。 [Method for Preparing Compositions (II) to (IV)]
In the compositions (II) to (IV), for example, the [B] polymer and / or the [C] polymer, the [S] solvent and other components as necessary are mixed at a predetermined ratio, and preferably the pore size is 0. It can be prepared by filtering with a high-density polyethylene filter of about 45 μm. The lower limit of the solid content concentration of the compositions (II) to (IV) is preferably 0.1% by mass, more preferably 0.5% by mass, and even more preferably 0.7% by mass. The upper limit of the solid content concentration is preferably 30% by mass, more preferably 10% by mass, and still more preferably 3% by mass.
(基礎パターン形成工程における各工程の順序)
基礎パターン形成工程が、レジストパターン形成工程、[B]重合体層形成工程及び[C]重合体層形成工程を有する場合、これらの各工程は、下記のように種々の順序で行うことができる。すなわち、基礎パターン形成工程は、例えば
(1)レジストパターン形成工程、[B]重合体層形成工程、[C]重合体層形成工程の順
(2)レジストパターン形成工程、[C]重合体層形成工程、[B]重合体層形成工程の順
(3)[C]重合体層形成工程、レジストパターン形成工程、[B]重合体層形成工程の順
等で行うことができる。これらの中で、[B]重合体層及び[C]重合体層をより確実に形成できる観点から、上記(1)及び(3)が好ましい。 (Order of each process in the basic pattern formation process)
When the basic pattern forming step includes a resist pattern forming step, a [B] polymer layer forming step, and a [C] polymer layer forming step, these steps can be performed in various orders as described below. . That is, the basic pattern forming step includes, for example: (1) resist pattern forming step, [B] polymer layer forming step, [C] polymer layer forming step in order (2) resist pattern forming step, [C] polymer layer (3) [C] Polymer layer forming step, resist pattern forming step, [B] polymer layer forming step, etc. Among these, the above (1) and (3) are preferable from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer.
基礎パターン形成工程が、レジストパターン形成工程、[B]重合体層形成工程及び[C]重合体層形成工程を有する場合、これらの各工程は、下記のように種々の順序で行うことができる。すなわち、基礎パターン形成工程は、例えば
(1)レジストパターン形成工程、[B]重合体層形成工程、[C]重合体層形成工程の順
(2)レジストパターン形成工程、[C]重合体層形成工程、[B]重合体層形成工程の順
(3)[C]重合体層形成工程、レジストパターン形成工程、[B]重合体層形成工程の順
等で行うことができる。これらの中で、[B]重合体層及び[C]重合体層をより確実に形成できる観点から、上記(1)及び(3)が好ましい。 (Order of each process in the basic pattern formation process)
When the basic pattern forming step includes a resist pattern forming step, a [B] polymer layer forming step, and a [C] polymer layer forming step, these steps can be performed in various orders as described below. . That is, the basic pattern forming step includes, for example: (1) resist pattern forming step, [B] polymer layer forming step, [C] polymer layer forming step in order (2) resist pattern forming step, [C] polymer layer (3) [C] Polymer layer forming step, resist pattern forming step, [B] polymer layer forming step, etc. Among these, the above (1) and (3) are preferable from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer.
基礎パターン形成工程が、レジストパターン形成工程、組成物(II)塗工工程及び組成物(III)塗工工程を有する場合、これらの各工程を行う順序としては、例えば
(A)レジストパターン形成工程、組成物(II)塗工工程、組成物(III)塗工工程の順
(B)レジストパターン形成工程、組成物(III)塗工工程、組成物(II)塗工工程の順
(C)組成物(III)塗工工程、レジストパターン形成工程、組成物(II)塗工工程の順
等で行うことができる。これらの中で、[B]重合体層及び[C]重合体層をより確実に形成できる観点から、上記(A)及び(C)が好ましい。 When the basic pattern forming step includes a resist pattern forming step, a composition (II) coating step, and a composition (III) coating step, the order of performing these steps is, for example, (A) Resist pattern forming step The order of the composition (II) coating process, the composition (III) coating process (B) The resist pattern formation process, the composition (III) coating process, the order of the composition (II) coating process (C) The composition (III) coating step, the resist pattern forming step, the composition (II) coating step, and the like can be performed in this order. Among these, the above (A) and (C) are preferable from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer.
(A)レジストパターン形成工程、組成物(II)塗工工程、組成物(III)塗工工程の順
(B)レジストパターン形成工程、組成物(III)塗工工程、組成物(II)塗工工程の順
(C)組成物(III)塗工工程、レジストパターン形成工程、組成物(II)塗工工程の順
等で行うことができる。これらの中で、[B]重合体層及び[C]重合体層をより確実に形成できる観点から、上記(A)及び(C)が好ましい。 When the basic pattern forming step includes a resist pattern forming step, a composition (II) coating step, and a composition (III) coating step, the order of performing these steps is, for example, (A) Resist pattern forming step The order of the composition (II) coating process, the composition (III) coating process (B) The resist pattern formation process, the composition (III) coating process, the order of the composition (II) coating process (C) The composition (III) coating step, the resist pattern forming step, the composition (II) coating step, and the like can be performed in this order. Among these, the above (A) and (C) are preferable from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer.
基礎パターン形成工程において、組成物(IV)塗工工程を行う場合、[B]重合体層及び[C]重合体層をより確実に形成できる観点から、レジストパターン形成工程を行い、この工程の後に、組成物(IV)塗工工程を行うことが好ましい。
In the basic pattern forming step, when performing the composition (IV) coating step, the resist pattern forming step is performed from the viewpoint of more reliably forming the [B] polymer layer and the [C] polymer layer. It is preferable to perform a composition (IV) coating process later.
[充填工程]
本工程では、上記基礎パターン(I)の凹部に、[A]重合体と溶媒とを含有する組成物(I)を充填する。 [Filling process]
In this step, the concave portion of the basic pattern (I) is filled with the composition (I) containing [A] a polymer and a solvent.
本工程では、上記基礎パターン(I)の凹部に、[A]重合体と溶媒とを含有する組成物(I)を充填する。 [Filling process]
In this step, the concave portion of the basic pattern (I) is filled with the composition (I) containing [A] a polymer and a solvent.
(組成物(I))
組成物(I)は、[A]重合体と溶媒とを含有する。組成物(I)は、[A]重合体及び溶媒以外にも、その他の成分を含有することができる。 (Composition (I))
The composition (I) contains a [A] polymer and a solvent. The composition (I) can contain other components in addition to the [A] polymer and the solvent.
組成物(I)は、[A]重合体と溶媒とを含有する。組成物(I)は、[A]重合体及び溶媒以外にも、その他の成分を含有することができる。 (Composition (I))
The composition (I) contains a [A] polymer and a solvent. The composition (I) can contain other components in addition to the [A] polymer and the solvent.
([A]重合体)
[A]重合体は、少なくとも2つのブロックを有する重合体である。すなわち、[A]重合体は、ブロック共重合体である。 ([A] polymer)
[A] The polymer is a polymer having at least two blocks. That is, the [A] polymer is a block copolymer.
[A]重合体は、少なくとも2つのブロックを有する重合体である。すなわち、[A]重合体は、ブロック共重合体である。 ([A] polymer)
[A] The polymer is a polymer having at least two blocks. That is, the [A] polymer is a block copolymer.
[A]重合体は、ジブロック共重合体でも、トリブロック共重合体でも、4つ以上のブロックを有する共重合体でもよい。これらの中で、相分離構造の形成のし易さの観点から、ジブロック共重合体及びトリブロック共重合体が好ましく、ジブロック共重合体がより好ましい。また、[A]重合体は、隣接するブロック間に連結基を有していてもよい。
[A] The polymer may be a diblock copolymer, a triblock copolymer, or a copolymer having four or more blocks. Among these, from the viewpoint of easy formation of the phase separation structure, a diblock copolymer and a triblock copolymer are preferable, and a diblock copolymer is more preferable. [A] The polymer may have a linking group between adjacent blocks.
[A]重合体は、ジブロック共重合体である場合、ブロック(I)及びブロック(II)を有する。ブロック(II)はブロック(I)よりも構成する構造単位の極性が高い。
[A] When the polymer is a diblock copolymer, it has a block (I) and a block (II). In the block (II), the polarity of the structural unit constituting the block (I) is higher.
(ブロック(I))
ブロック(I)としては、例えばポリスチレンブロック等が挙げられる。ポリスチレンブロックは、置換又は非置換のスチレンに由来する構造単位を含むブロックである。これらの中で、スチレンに由来する構造単位を含むブロックが好ましい。 (Block (I))
Examples of the block (I) include a polystyrene block. The polystyrene block is a block containing a structural unit derived from substituted or unsubstituted styrene. Among these, a block containing a structural unit derived from styrene is preferable.
ブロック(I)としては、例えばポリスチレンブロック等が挙げられる。ポリスチレンブロックは、置換又は非置換のスチレンに由来する構造単位を含むブロックである。これらの中で、スチレンに由来する構造単位を含むブロックが好ましい。 (Block (I))
Examples of the block (I) include a polystyrene block. The polystyrene block is a block containing a structural unit derived from substituted or unsubstituted styrene. Among these, a block containing a structural unit derived from styrene is preferable.
(ブロック(II))
ブロック(II)としては、例えばポリ(メタ)アクリル酸エステルブロック、ポリアルキレングリコールブロック、ポリエステルブロック、ポリアルキレンカーボナートブロック、ポリジアルキルシロキサンブロック及びポリ(メタ)アクリル酸アルキルシリルエステルブロック等が挙げられる。ポリ(メタ)アクリル酸エステルブロックは、(メタ)アクリル酸エステルに由来する構造単位を含むブロックである。これらの中で、ポリ(メタ)アクリル酸エステルブロックが好ましく、ポリ(メタ)アクリル酸アルキルブロックがより好ましく、ポリ(メタ)アクリル酸メチルブロックがさらに好ましく、ポリメタクリル酸メチルブロックが特に好ましい。 (Block (II))
Examples of the block (II) include a poly (meth) acrylic acid ester block, a polyalkylene glycol block, a polyester block, a polyalkylene carbonate block, a polydialkylsiloxane block, and a poly (meth) acrylic acid alkylsilyl ester block. . The poly (meth) acrylate block is a block including a structural unit derived from (meth) acrylate. Among these, a poly (meth) acrylate block is preferable, a poly (meth) acrylate block is more preferable, a poly (meth) acrylate block is more preferable, and a polymethyl methacrylate block is particularly preferable.
ブロック(II)としては、例えばポリ(メタ)アクリル酸エステルブロック、ポリアルキレングリコールブロック、ポリエステルブロック、ポリアルキレンカーボナートブロック、ポリジアルキルシロキサンブロック及びポリ(メタ)アクリル酸アルキルシリルエステルブロック等が挙げられる。ポリ(メタ)アクリル酸エステルブロックは、(メタ)アクリル酸エステルに由来する構造単位を含むブロックである。これらの中で、ポリ(メタ)アクリル酸エステルブロックが好ましく、ポリ(メタ)アクリル酸アルキルブロックがより好ましく、ポリ(メタ)アクリル酸メチルブロックがさらに好ましく、ポリメタクリル酸メチルブロックが特に好ましい。 (Block (II))
Examples of the block (II) include a poly (meth) acrylic acid ester block, a polyalkylene glycol block, a polyester block, a polyalkylene carbonate block, a polydialkylsiloxane block, and a poly (meth) acrylic acid alkylsilyl ester block. . The poly (meth) acrylate block is a block including a structural unit derived from (meth) acrylate. Among these, a poly (meth) acrylate block is preferable, a poly (meth) acrylate block is more preferable, a poly (meth) acrylate block is more preferable, and a polymethyl methacrylate block is particularly preferable.
(連結基)
連結基としては、例えば炭素数1~50の2価の有機基等が挙げられる。連結基を与える単量体としては、例えばジフェニルエチレン、スチルベン等が挙げられる。ジフェニルエチレン及びスチルベンは、[A]重合体をアニオン重合で合成する際に、途中で生成するアニオン末端を安定化させることができる。それにより、得られる[A]重合体の分散度(Mw/Mn比)がより小さくなり、その結果、形成されるパターンの寸法のばらつきをより小さくすることができる。[A]重合体は、ブロック数や目的とするパターン形状等に応じて連結基を1種又は2種以上有していてもよい。 (Linking group)
Examples of the linking group include a divalent organic group having 1 to 50 carbon atoms. Examples of the monomer that gives a linking group include diphenylethylene and stilbene. When diphenylethylene and stilbene synthesize the [A] polymer by anionic polymerization, the anion terminal produced in the middle can be stabilized. Thereby, the degree of dispersion (Mw / Mn ratio) of the obtained [A] polymer becomes smaller, and as a result, the variation in the dimension of the formed pattern can be made smaller. [A] The polymer may have one or more linking groups depending on the number of blocks, the target pattern shape, and the like.
連結基としては、例えば炭素数1~50の2価の有機基等が挙げられる。連結基を与える単量体としては、例えばジフェニルエチレン、スチルベン等が挙げられる。ジフェニルエチレン及びスチルベンは、[A]重合体をアニオン重合で合成する際に、途中で生成するアニオン末端を安定化させることができる。それにより、得られる[A]重合体の分散度(Mw/Mn比)がより小さくなり、その結果、形成されるパターンの寸法のばらつきをより小さくすることができる。[A]重合体は、ブロック数や目的とするパターン形状等に応じて連結基を1種又は2種以上有していてもよい。 (Linking group)
Examples of the linking group include a divalent organic group having 1 to 50 carbon atoms. Examples of the monomer that gives a linking group include diphenylethylene and stilbene. When diphenylethylene and stilbene synthesize the [A] polymer by anionic polymerization, the anion terminal produced in the middle can be stabilized. Thereby, the degree of dispersion (Mw / Mn ratio) of the obtained [A] polymer becomes smaller, and as a result, the variation in the dimension of the formed pattern can be made smaller. [A] The polymer may have one or more linking groups depending on the number of blocks, the target pattern shape, and the like.
[A]重合体のゲルパーミエーションクロマトグラフィー(GPC)による重量平均分子量(Mw)の下限としては、1,000が好ましく、10,000がより好ましく、50,000がさらに好ましい。上記Mwの上限としては、300,000が好ましく、200,000がより好ましく、100,000がさらに好ましい。[A]重合体のMwを上記範囲とすることで、プレイスメントエラーがより抑制され、かつ底部残渣がより低減されたコンタクトホールパターンを形成することができる。
[A] The lower limit of the weight average molecular weight (Mw) of the polymer by gel permeation chromatography (GPC) is preferably 1,000, more preferably 10,000, and even more preferably 50,000. The upper limit of Mw is preferably 300,000, more preferably 200,000, and even more preferably 100,000. [A] By setting the Mw of the polymer in the above range, a contact hole pattern in which placement errors are further suppressed and bottom residue is further reduced can be formed.
[A]重合体のMwと数平均分子量(Mn)との比(Mw/Mn、分散度)の上限としては、5が好ましく、3がより好ましく、2がさらに好ましく、1.5が特に好ましく、1.1がさらに特に好ましい。上記比の下限としては、通常1であり、1.01が好ましい。Mw/Mn比を上記範囲とすることで、プレイスメントエラーがより抑制され、かつ底部残渣がより低減されたコンタクトホールパターンを形成することができる。
[A] The upper limit of the ratio (Mw / Mn, dispersity) between the polymer Mw and the number average molecular weight (Mn) is preferably 5, more preferably 3, more preferably 2, and particularly preferably 1.5. 1.1 is more particularly preferable. The lower limit of the above ratio is usually 1 and preferably 1.01. By setting the Mw / Mn ratio in the above range, it is possible to form a contact hole pattern in which placement errors are further suppressed and bottom residue is further reduced.
[A]重合体の含有量の下限としては、組成物(I)における全固形分に対して、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましく、99質量%が特に好ましい。組成物(I)の「全固形分」とは、溶媒以外の成分の総和をいう。
[A] The lower limit of the content of the polymer is preferably 80% by mass, more preferably 90% by mass, still more preferably 95% by mass, and 99% by mass with respect to the total solid content in the composition (I). Particularly preferred. The “total solid content” of the composition (I) refers to the sum of components other than the solvent.
組成物(I)中の[A]重合体の濃度の下限としては、0.3質量%が好ましく、0.7質量%がより好ましく、1.0質量%がさらに好ましく、1.3質量%が特に好ましい。一方、上記濃度の上限としては、5質量%が好ましく、3質量%がより好ましく、2質量%がさらに好ましく、1.7質量%が特に好ましい。
The lower limit of the concentration of the [A] polymer in the composition (I) is preferably 0.3% by mass, more preferably 0.7% by mass, further preferably 1.0% by mass, and 1.3% by mass. Is particularly preferred. On the other hand, the upper limit of the concentration is preferably 5% by mass, more preferably 3% by mass, still more preferably 2% by mass, and particularly preferably 1.7% by mass.
<[A]重合体の合成方法>
[A]重合体は、リビングアニオン重合、リビングラジカル重合等によって合成することが出来るが、リビングアニオン重合がより好ましい。例えばポリスチレンブロック、ポリメタクリル酸メチルブロック及びこれら以外の他のブロックを所望の順で重合しながら連結し、その重合末端を任意の末端処理剤で処理することにより合成することができる。 <[A] Polymer Synthesis Method>
[A] The polymer can be synthesized by living anion polymerization, living radical polymerization or the like, but living anion polymerization is more preferable. For example, a polystyrene block, a polymethyl methacrylate block, and other blocks other than these can be linked while polymerizing in a desired order, and the polymerization terminal can be synthesized by treating with an arbitrary terminal treating agent.
[A]重合体は、リビングアニオン重合、リビングラジカル重合等によって合成することが出来るが、リビングアニオン重合がより好ましい。例えばポリスチレンブロック、ポリメタクリル酸メチルブロック及びこれら以外の他のブロックを所望の順で重合しながら連結し、その重合末端を任意の末端処理剤で処理することにより合成することができる。 <[A] Polymer Synthesis Method>
[A] The polymer can be synthesized by living anion polymerization, living radical polymerization or the like, but living anion polymerization is more preferable. For example, a polystyrene block, a polymethyl methacrylate block, and other blocks other than these can be linked while polymerizing in a desired order, and the polymerization terminal can be synthesized by treating with an arbitrary terminal treating agent.
上記重合に使用される溶媒としては、例えば
n-ペンタン、n-ヘキサン、n-ヘプタン、n-オクタン、n-ノナン、n-デカン等のアルカン;
シクロヘキサン、シクロヘプタン、シクロオクタン、デカリン、ノルボルナン等の脂環式炭化水素;
ベンゼン、トルエン、キシレン、エチルベンゼン、クメン等の芳香族炭化水素;
酢酸エチル、酢酸n-ブチル、酢酸i-ブチル、プロピオン酸メチル等の飽和カルボン酸エステル;
アセトン、メチルエチルケトン、4-メチル-2-ペンタノン、2-ヘプタノン等のケトン;
テトラヒドロフラン、ジメトキシエタン類、ジエトキシエタン類等のエーテル;
メタノール、エタノール、1-プロパノール、2-プロパノール、4-メチル-2-ペンタノール等のアルコールなどが挙げられる。これらの溶媒は、単独で使用してもよく2種以上を併用してもよい。 Examples of the solvent used for the polymerization include alkanes such as n-pentane, n-hexane, n-heptane, n-octane, n-nonane, and n-decane;
Cycloaliphatic hydrocarbons such as cyclohexane, cycloheptane, cyclooctane, decalin, norbornane;
Aromatic hydrocarbons such as benzene, toluene, xylene, ethylbenzene, cumene;
Saturated carboxylic acid esters such as ethyl acetate, n-butyl acetate, i-butyl acetate and methyl propionate;
Ketones such as acetone, methyl ethyl ketone, 4-methyl-2-pentanone, 2-heptanone;
Ethers such as tetrahydrofuran, dimethoxyethanes, diethoxyethanes;
Examples thereof include alcohols such as methanol, ethanol, 1-propanol, 2-propanol and 4-methyl-2-pentanol. These solvents may be used alone or in combination of two or more.
n-ペンタン、n-ヘキサン、n-ヘプタン、n-オクタン、n-ノナン、n-デカン等のアルカン;
シクロヘキサン、シクロヘプタン、シクロオクタン、デカリン、ノルボルナン等の脂環式炭化水素;
ベンゼン、トルエン、キシレン、エチルベンゼン、クメン等の芳香族炭化水素;
酢酸エチル、酢酸n-ブチル、酢酸i-ブチル、プロピオン酸メチル等の飽和カルボン酸エステル;
アセトン、メチルエチルケトン、4-メチル-2-ペンタノン、2-ヘプタノン等のケトン;
テトラヒドロフラン、ジメトキシエタン類、ジエトキシエタン類等のエーテル;
メタノール、エタノール、1-プロパノール、2-プロパノール、4-メチル-2-ペンタノール等のアルコールなどが挙げられる。これらの溶媒は、単独で使用してもよく2種以上を併用してもよい。 Examples of the solvent used for the polymerization include alkanes such as n-pentane, n-hexane, n-heptane, n-octane, n-nonane, and n-decane;
Cycloaliphatic hydrocarbons such as cyclohexane, cycloheptane, cyclooctane, decalin, norbornane;
Aromatic hydrocarbons such as benzene, toluene, xylene, ethylbenzene, cumene;
Saturated carboxylic acid esters such as ethyl acetate, n-butyl acetate, i-butyl acetate and methyl propionate;
Ketones such as acetone, methyl ethyl ketone, 4-methyl-2-pentanone, 2-heptanone;
Ethers such as tetrahydrofuran, dimethoxyethanes, diethoxyethanes;
Examples thereof include alcohols such as methanol, ethanol, 1-propanol, 2-propanol and 4-methyl-2-pentanol. These solvents may be used alone or in combination of two or more.
上記重合における反応温度は、開始剤の種類に応じて適宜決定すればよいが、反応温度の下限としては、-150℃が好ましく、-80℃がより好ましい。上記反応温度の上限としては、50℃が好ましく、40℃がより好ましい。反応時間の下限としては、5分が好ましく、20分がより好ましい。上記反応時間の上限としては、24時間が好ましく、12時間がより好ましい。
The reaction temperature in the above polymerization may be appropriately determined according to the type of initiator, but the lower limit of the reaction temperature is preferably −150 ° C., more preferably −80 ° C. As an upper limit of the said reaction temperature, 50 degreeC is preferable and 40 degreeC is more preferable. As a minimum of reaction time, 5 minutes are preferred and 20 minutes are more preferred. The upper limit of the reaction time is preferably 24 hours, and more preferably 12 hours.
上記重合に使用される開始剤としては、例えばアルキルリチウム、アルキルマグネシウムハライド、ナフタレンナトリウム、アルキル化ランタノイド系化合物等が挙げられる。これらのうち、モノマーとしてスチレン、メタクリル酸メチル等を使用して重合する場合には、アルキルリチウム化合物を用いることが好ましい。
Examples of the initiator used for the polymerization include alkyl lithium, alkyl magnesium halide, sodium naphthalene, alkylated lanthanoid compounds, and the like. Among these, when polymerizing using styrene, methyl methacrylate or the like as a monomer, it is preferable to use an alkyl lithium compound.
上記末端処理の方法としては、メタノール等のアルコール、2-エチルヘキシルグリシジルエーテル等のエポキシ化合物などの末端処理剤を添加して、重合反応中の鎖の末端を変性することにより、所定のブロック共重合体を得ることができる。得られたブロック共重合体は、脱メタル処理等の精製を行うことが好ましい。
As the above-mentioned end treatment method, an end treatment agent such as an alcohol such as methanol or an epoxy compound such as 2-ethylhexyl glycidyl ether is added to modify the end of the chain during the polymerization reaction. Coalescence can be obtained. The obtained block copolymer is preferably subjected to purification such as demetallation.
<相分離工程>
本工程では、上記充填工程により形成された充填層4を相分離させる。これにより、図4に示すように、充填層4が、ブロック(α)相4aと、ブロック(β)相4bとに相分離する。 <Phase separation process>
In this step, the packedbed 4 formed by the above filling step is phase-separated. As a result, as shown in FIG. 4, the packed bed 4 is phase-separated into a block (α) phase 4a and a block (β) phase 4b.
本工程では、上記充填工程により形成された充填層4を相分離させる。これにより、図4に示すように、充填層4が、ブロック(α)相4aと、ブロック(β)相4bとに相分離する。 <Phase separation process>
In this step, the packed
充填層4を相分離させる方法としては、例えばアニーリングする方法等が挙げられる。
As a method for phase-separating the packed bed 4, for example, an annealing method or the like can be given.
アニーリングする方法としては、例えば加熱する方法等が挙げられる。加熱の手段としては、例えばオーブン、ホットプレート等が挙げられる。加熱の温度の下限としては、80℃が好ましく、100℃がより好ましく、150℃がさらに好ましい。加熱の温度の上限としては、400℃が好ましく、350℃がより好ましく、300℃がさらに好ましい。加熱の時間の下限としては、10秒が好ましく、1分がより好ましく、10分がさらに好ましい。加熱の時間の上限としては、120分が好ましく、60分がより好ましく、30分がさらに好ましい。
Examples of the annealing method include a heating method. Examples of the heating means include an oven and a hot plate. As a minimum of the temperature of heating, 80 ° C is preferred, 100 ° C is more preferred, and 150 ° C is still more preferred. As an upper limit of the temperature of heating, 400 degreeC is preferable, 350 degreeC is more preferable, and 300 degreeC is further more preferable. The lower limit of the heating time is preferably 10 seconds, more preferably 1 minute, and even more preferably 10 minutes. The upper limit of the heating time is preferably 120 minutes, more preferably 60 minutes, and even more preferably 30 minutes.
[除去工程]
本工程では、上記相分離工程後の充填層の一部の相を除去する。例えば図4における相分離工程後の充填層の一方の相であるブロック(β)相4bが除去され、図5に示すように、レジストパターン2、重合体層3及び除去されなかった充填層4aからなる微細化パターンが形成される。 [Removal process]
In this step, a part of the phase of the packed bed after the phase separation step is removed. For example, the block (β) phase 4b, which is one phase of the packed layer after the phase separation step in FIG. 4, is removed, and as shown in FIG. 5, the resistpattern 2, the polymer layer 3, and the packed layer 4a that has not been removed. A miniaturized pattern is formed.
本工程では、上記相分離工程後の充填層の一部の相を除去する。例えば図4における相分離工程後の充填層の一方の相であるブロック(β)相4bが除去され、図5に示すように、レジストパターン2、重合体層3及び除去されなかった充填層4aからなる微細化パターンが形成される。 [Removal process]
In this step, a part of the phase of the packed bed after the phase separation step is removed. For example, the block (β) phase 4b, which is one phase of the packed layer after the phase separation step in FIG. 4, is removed, and as shown in FIG. 5, the resist
除去工程において、例えば図4に示すように、充填層4が有する相分離構造のうちの一部のブロック(β)相4bを除去する。例えば相分離した各相のエッチングレートの差を用いて、ポリメタクリル酸メチルブロック相4bをエッチング処理により除去することができる。相分離構造のうちのポリメタクリル酸メチルブロック相4bを除去した後の状態を図5に示す。なお、上記エッチング処理の前に、必要に応じて放射線を照射してもよい。上記放射線としては、エッチングにより除去する相がポリメタクリル酸メチルブロック相である場合には、172nmの放射線を用いることができる。上記放射線照射により、ポリメタクリル酸メチルブロック相が分解されるため、よりエッチングされ易くなる。
In the removing step, for example, as shown in FIG. 4, a part of the block (β) phase 4b in the phase separation structure of the packed bed 4 is removed. For example, the polymethyl methacrylate block phase 4b can be removed by etching using the difference in the etching rate of each phase separated. FIG. 5 shows a state after the polymethyl methacrylate block phase 4b in the phase separation structure is removed. In addition, you may irradiate a radiation before the said etching process as needed. As the radiation, when the phase to be removed by etching is a polymethyl methacrylate block phase, 172 nm radiation can be used. Since the polymethyl methacrylate block phase is decomposed by the radiation irradiation, etching becomes easier.
相分離構造のうちのポリメタクリル酸メチルブロック相4bの除去の方法としては、例えばケミカルドライエッチング、ケミカルウェットエッチング等の反応性イオンエッチング(RIE);スパッタエッチング、イオンビームエッチング等の物理的エッチング等の公知の方法が挙げられる。これらの中で、反応性イオンエッチングが好ましく、CF4、O2ガス等を用いたケミカルドライエッチング、有機溶媒、フッ酸等の液体のエッチング溶液を用いたケミカルウェットエッチング(湿式現像)がより好ましく、ケミカルウェットエッチングがさらに好ましい。
Examples of a method for removing the polymethyl methacrylate block phase 4b in the phase separation structure include reactive ion etching (RIE) such as chemical dry etching and chemical wet etching; physical etching such as sputter etching and ion beam etching. There are known methods. Among these, reactive ion etching is preferable, chemical dry etching using CF 4 , O 2 gas, etc., and chemical wet etching (wet development) using a liquid etching solution such as an organic solvent or hydrofluoric acid is more preferable. Chemical wet etching is more preferable.
ケミカルウェットエッチングに用いられる有機溶媒としては、例えば
n-ペンタン、n-ヘキサン、n-ヘプタン等のアルカン;
シクロヘキサン、シクロヘプタン、シクロオクタン等のシクロアルカン;
酢酸エチル、酢酸n-ブチル、酢酸i-ブチル、プロピオン酸メチル等の飽和カルボン酸エステル;
アセトン、メチルエチルケトン、メチルイソブチルケトン(MIBK)、メチルn-ペンチルケトン等のケトン;
メタノール、エタノール、1-プロパノール、イソプロパノール(IPA)、4-メチル-2-ペンタノール等のアルコールなどが挙げられる。これらの溶媒は、単独で使用してもよく2種以上を併用してもよい。これらの中で、MIBK及びIPAとの混合溶媒が好ましい。 Examples of organic solvents used for chemical wet etching include alkanes such as n-pentane, n-hexane, and n-heptane;
Cycloalkanes such as cyclohexane, cycloheptane, cyclooctane;
Saturated carboxylic acid esters such as ethyl acetate, n-butyl acetate, i-butyl acetate and methyl propionate;
Ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone (MIBK), methyl n-pentyl ketone;
Examples include alcohols such as methanol, ethanol, 1-propanol, isopropanol (IPA), and 4-methyl-2-pentanol. These solvents may be used alone or in combination of two or more. Among these, a mixed solvent with MIBK and IPA is preferable.
n-ペンタン、n-ヘキサン、n-ヘプタン等のアルカン;
シクロヘキサン、シクロヘプタン、シクロオクタン等のシクロアルカン;
酢酸エチル、酢酸n-ブチル、酢酸i-ブチル、プロピオン酸メチル等の飽和カルボン酸エステル;
アセトン、メチルエチルケトン、メチルイソブチルケトン(MIBK)、メチルn-ペンチルケトン等のケトン;
メタノール、エタノール、1-プロパノール、イソプロパノール(IPA)、4-メチル-2-ペンタノール等のアルコールなどが挙げられる。これらの溶媒は、単独で使用してもよく2種以上を併用してもよい。これらの中で、MIBK及びIPAとの混合溶媒が好ましい。 Examples of organic solvents used for chemical wet etching include alkanes such as n-pentane, n-hexane, and n-heptane;
Cycloalkanes such as cyclohexane, cycloheptane, cyclooctane;
Saturated carboxylic acid esters such as ethyl acetate, n-butyl acetate, i-butyl acetate and methyl propionate;
Ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone (MIBK), methyl n-pentyl ketone;
Examples include alcohols such as methanol, ethanol, 1-propanol, isopropanol (IPA), and 4-methyl-2-pentanol. These solvents may be used alone or in combination of two or more. Among these, a mixed solvent with MIBK and IPA is preferable.
[エッチング工程]
本工程では、上記除去工程により形成された微細化パターンを直接的又は間接的に用いて上記基板をエッチングする。微細化パターンは、図5に示すように、基礎パターン(I)2、重合体層3及び除去工程で除去されなかったブロック(α)相から形成されている。この工程により、基板パターンが形成される。パターン形成方法(A)によれば、プレイスメントエラーが抑制され、かつ形状が良好な基板パターンを得ることができる。 [Etching process]
In this step, the substrate is etched by directly or indirectly using the miniaturized pattern formed in the removing step. As shown in FIG. 5, the miniaturized pattern is formed of the basic pattern (I) 2, thepolymer layer 3, and the block (α) phase that has not been removed in the removing step. By this step, a substrate pattern is formed. According to the pattern formation method (A), it is possible to obtain a substrate pattern in which a placement error is suppressed and the shape is good.
本工程では、上記除去工程により形成された微細化パターンを直接的又は間接的に用いて上記基板をエッチングする。微細化パターンは、図5に示すように、基礎パターン(I)2、重合体層3及び除去工程で除去されなかったブロック(α)相から形成されている。この工程により、基板パターンが形成される。パターン形成方法(A)によれば、プレイスメントエラーが抑制され、かつ形状が良好な基板パターンを得ることができる。 [Etching process]
In this step, the substrate is etched by directly or indirectly using the miniaturized pattern formed in the removing step. As shown in FIG. 5, the miniaturized pattern is formed of the basic pattern (I) 2, the
このエッチングは、基礎パターン形成工程において、基板1の表面側に直接基礎パターン(I)2を形成した場合は、通常、微細パターンを直接的に用い、すなわち、エッチングを1回行い、基板パターンを得る。基板1の表面側に他の層を介して基礎パターン(I)を形成した場合は、通常、微細化パターンを間接的に用い、すなわち、微細化パターンを用いて、他の層をエッチングし、エッチング後の他の層をマスクとして、順次エッチングを行い、すなわち、エッチングを複数回行って、基板パターンを得る。
In this etching, when the basic pattern (I) 2 is directly formed on the surface side of the substrate 1 in the basic pattern forming step, the fine pattern is usually used directly, that is, the etching is performed once to form the substrate pattern. obtain. When the basic pattern (I) is formed on the surface side of the substrate 1 through another layer, the fine pattern is usually used indirectly, that is, the fine pattern is used to etch other layers, Etching is sequentially performed using the other layers after etching as a mask, that is, etching is performed a plurality of times to obtain a substrate pattern.
得られる基板パターンとしては、例えばホールパターン等が挙げられる。
Examples of the substrate pattern to be obtained include a hole pattern.
エッチングの方法としては、例えばCF4、O2ガス等を用い、各層のエッチングレートの差等を利用するケミカルドライエッチング、有機溶媒、フッ酸等の液体のエッチング液を用いたケミカルウェットエッチング(湿式現像)等の反応性イオンエッチング(RIE);スパッタエッチング、イオンビームエッチング等の物理的エッチングなどの公知の方法が挙げられる。これらの中で、反応性イオンエッチングが好ましく、ケミカルドライエッチング及びケミカルウェットエッチングがより好ましく、ケミカルドライエッチングがさらに好ましい。
As an etching method, for example, CF 4 , O 2 gas or the like is used, chemical dry etching using a difference in etching rate of each layer or the like, chemical wet etching using a liquid etching solution such as an organic solvent or hydrofluoric acid (wet type). There are known methods such as reactive ion etching (RIE) such as development) and physical etching such as sputter etching and ion beam etching. Among these, reactive ion etching is preferable, chemical dry etching and chemical wet etching are more preferable, and chemical dry etching is more preferable.
基板のパターニング完了後、マスクとして使用された部分は溶解処理等により基板の表面側から除去され、最終的にパターンが形成された基板を得ることができる。パターン形成方法(A)により得られる基板は半導体素子等に好適に用いられ、この半導体素子はLED、太陽電池等に広く用いられる。
After completion of the patterning of the substrate, the portion used as a mask is removed from the surface side of the substrate by dissolution treatment or the like, and a substrate on which a pattern is finally formed can be obtained. The substrate obtained by the pattern forming method (A) is suitably used for a semiconductor element and the like, and this semiconductor element is widely used for LEDs, solar cells and the like.
<パターン形成方法(B)>
パターン形成方法(B)は、基礎パターン積層工程と、塗工工程と、加熱工程と、充填工程と、相分離工程と、除去工程と、エッチング工程とを備える。 <Pattern formation method (B)>
The pattern forming method (B) includes a basic pattern lamination step, a coating step, a heating step, a filling step, a phase separation step, a removal step, and an etching step.
パターン形成方法(B)は、基礎パターン積層工程と、塗工工程と、加熱工程と、充填工程と、相分離工程と、除去工程と、エッチング工程とを備える。 <Pattern formation method (B)>
The pattern forming method (B) includes a basic pattern lamination step, a coating step, a heating step, a filling step, a phase separation step, a removal step, and an etching step.
パターン形成方法(B)は、上記各工程を備え、加熱工程後の塗工膜における基礎パターンの凹部の側面側と底面側との接触角の差を上記値以上とすることで、パターンサイズ均一性に優れる微細化パターンを形成することができ、このような優れた微細化パターンを用いることにより、基板に良好な形状及び配列のパターンを形成することができる。パターン形成方法(B)が上記構成を備えることで、上記効果を奏する理由については必ずしも明確ではないが、例えば以下のように推察することができる。すなわち、加熱工程後における基礎パターンの凹部の側面側と底面側とが上記値以上の接触角の差を有することで、その後にこの凹部に形成される[A’]重合体の充填層がより適切に相分離すると考えられ、その結果、パターンサイズ均一性に優れる微細化パターンを形成することができる。以下、各工程について説明する。
The pattern formation method (B) comprises the above steps, and the pattern size is uniform by setting the difference in the contact angle between the side surface and the bottom surface of the concave portion of the basic pattern in the coating film after the heating step to the above value or more. A fine pattern having excellent properties can be formed, and by using such an excellent fine pattern, a pattern having a favorable shape and arrangement can be formed on the substrate. Although the reason why the pattern forming method (B) has the above-described configuration provides the above effect is not necessarily clear, it can be inferred as follows, for example. That is, since the side surface side and the bottom surface side of the concave portion of the basic pattern after the heating step have a difference in contact angle of the above value or more, the packed layer of [A ′] polymer formed in the concave portion after that is more It is considered that the phases are appropriately separated, and as a result, a miniaturized pattern having excellent pattern size uniformity can be formed. Hereinafter, each step will be described.
[基礎パターン積層工程]
本工程では、基板の表面側に直接又は他の層を介して基礎パターン(以下、「基礎パターン(II)」ともいう)を積層する。基礎パターン(II)としては、基板に直接積層されるものが好ましい。この基礎パターン(II)は、芳香環の含有割合が50質量%以上の重合体を主成分とする。芳香環の含有割合が50質量%以上の重合体を主成分とするものとして、例えばレジスト下層膜等が挙げられる。 [Basic pattern lamination process]
In this step, a basic pattern (hereinafter also referred to as “basic pattern (II)”) is laminated directly or via another layer on the surface side of the substrate. As basic pattern (II), what is directly laminated | stacked on a board | substrate is preferable. This basic pattern (II) is mainly composed of a polymer having an aromatic ring content of 50% by mass or more. As a thing which has as a main component the polymer whose content rate of an aromatic ring is 50 mass% or more, a resist underlayer film etc. are mentioned, for example.
本工程では、基板の表面側に直接又は他の層を介して基礎パターン(以下、「基礎パターン(II)」ともいう)を積層する。基礎パターン(II)としては、基板に直接積層されるものが好ましい。この基礎パターン(II)は、芳香環の含有割合が50質量%以上の重合体を主成分とする。芳香環の含有割合が50質量%以上の重合体を主成分とするものとして、例えばレジスト下層膜等が挙げられる。 [Basic pattern lamination process]
In this step, a basic pattern (hereinafter also referred to as “basic pattern (II)”) is laminated directly or via another layer on the surface side of the substrate. As basic pattern (II), what is directly laminated | stacked on a board | substrate is preferable. This basic pattern (II) is mainly composed of a polymer having an aromatic ring content of 50% by mass or more. As a thing which has as a main component the polymer whose content rate of an aromatic ring is 50 mass% or more, a resist underlayer film etc. are mentioned, for example.
基礎パターン(II)を積層する方法としては、例えば以下の方法が挙げられる。基板の表面側に直接又は他の層を介してレジスト下層膜を形成する。次に、必要に応じて、上記レジスト下層膜の基板と反対側の面に、SOG膜を形成してもよい。上記レジスト下層膜又はSOG膜の基板と反対側の面にレジストパターンを形成する。次いで、このレジストパターンをマスクとして、上記SOG膜及び/又は上記レジスト下層膜を順次エッチングする。
Examples of the method of laminating the basic pattern (II) include the following methods. A resist underlayer film is formed on the surface side of the substrate directly or via another layer. Next, if necessary, an SOG film may be formed on the surface of the resist underlayer film opposite to the substrate. A resist pattern is formed on the surface of the resist underlayer film or SOG film opposite to the substrate. Next, the SOG film and / or the resist underlayer film are sequentially etched using the resist pattern as a mask.
基板、他の層並びにレジスト下層膜、SOG膜及びレジストパターンを形成方法としては、例えばパターン形成方法(A)の基礎パターン形成工程において例示したのと同様のもの等が挙げられる。基板としては、シリコン製が好ましい。
Examples of methods for forming the substrate, other layers, the resist underlayer film, the SOG film, and the resist pattern include those similar to those exemplified in the basic pattern forming step of the pattern forming method (A). The substrate is preferably made of silicon.
[塗工工程]
本工程では、上記基礎パターン(II)の凹部の側面及び底面に組成物(V)を塗工する。 [Coating process]
In this step, the composition (V) is applied to the side surface and the bottom surface of the concave portion of the basic pattern (II).
本工程では、上記基礎パターン(II)の凹部の側面及び底面に組成物(V)を塗工する。 [Coating process]
In this step, the composition (V) is applied to the side surface and the bottom surface of the concave portion of the basic pattern (II).
(組成物(V))
組成物(V)は、[A’]重合体と溶媒([S]溶媒)とを含有する。組成物(V)は、[A’]重合体と[S]溶媒以外にも界面活性剤等の他の成分を含有していてもよい。 (Composition (V))
The composition (V) contains a [A ′] polymer and a solvent ([S] solvent). The composition (V) may contain other components such as a surfactant in addition to the [A ′] polymer and the [S] solvent.
組成物(V)は、[A’]重合体と溶媒([S]溶媒)とを含有する。組成物(V)は、[A’]重合体と[S]溶媒以外にも界面活性剤等の他の成分を含有していてもよい。 (Composition (V))
The composition (V) contains a [A ′] polymer and a solvent ([S] solvent). The composition (V) may contain other components such as a surfactant in addition to the [A ′] polymer and the [S] solvent.
([A’]重合体)
[A’]重合体は、構造単位(I)を有する。構造単位(I)としては、例えば置換又は非置換のスチレンに由来する構造単位、(メタ)アクリル酸又は(メタ)アクリル酸エステルに由来する構造単位、置換又は非置換のエチレンに由来する構造単位等が挙げられる。置換又は非置換のスチレン、(メタ)アクリル酸エステル及び置換又は非置換のエチレンとしては、例えばパターン形成方法(A)の[B]重合体において例示したのと同様のもの等が挙げられる。 ([A '] polymer)
[A ′] The polymer has the structural unit (I). As the structural unit (I), for example, a structural unit derived from substituted or unsubstituted styrene, a structural unit derived from (meth) acrylic acid or (meth) acrylic acid ester, or a structural unit derived from substituted or unsubstituted ethylene Etc. Examples of the substituted or unsubstituted styrene, (meth) acrylic acid ester, and substituted or unsubstituted ethylene include the same as those exemplified in the [B] polymer of the pattern forming method (A).
[A’]重合体は、構造単位(I)を有する。構造単位(I)としては、例えば置換又は非置換のスチレンに由来する構造単位、(メタ)アクリル酸又は(メタ)アクリル酸エステルに由来する構造単位、置換又は非置換のエチレンに由来する構造単位等が挙げられる。置換又は非置換のスチレン、(メタ)アクリル酸エステル及び置換又は非置換のエチレンとしては、例えばパターン形成方法(A)の[B]重合体において例示したのと同様のもの等が挙げられる。 ([A '] polymer)
[A ′] The polymer has the structural unit (I). As the structural unit (I), for example, a structural unit derived from substituted or unsubstituted styrene, a structural unit derived from (meth) acrylic acid or (meth) acrylic acid ester, or a structural unit derived from substituted or unsubstituted ethylene Etc. Examples of the substituted or unsubstituted styrene, (meth) acrylic acid ester, and substituted or unsubstituted ethylene include the same as those exemplified in the [B] polymer of the pattern forming method (A).
[A’]重合体は、主鎖の少なくとも一方の末端に結合し、-COOH及び-OHの少なくとも一方と化学結合する官能基を有することが好ましい。このような官能基としては、例えばパターン形成方法(A)における[B]重合体の官能基(1)として例示した官能基等が挙げられる。
[A ′] The polymer preferably has a functional group that is bonded to at least one end of the main chain and chemically bonds to at least one of —COOH and —OH. Examples of such functional groups include the functional groups exemplified as the functional group (1) of the [B] polymer in the pattern forming method (A).
[A’]重合体のMw及びMw/Mn、並びに組成物(V)における[A’]重合体の含有量の好ましい範囲としては、パターン形成方法(A)における[B]重合体及び組成物(II)と同様である。
[A ′] Mw and Mw / Mn of the polymer, and a preferable range of the content of [A ′] polymer in the composition (V) include the [B] polymer and the composition in the pattern forming method (A). Same as (II).
[S]溶媒としては、例えばパターン形成方法(A)における組成物(II)の[S]溶媒として例示したものと同様の溶媒等が挙げられる。
Examples of the [S] solvent include the same solvents as those exemplified as the [S] solvent of the composition (II) in the pattern formation method (A).
[組成物(V)の調製方法]
組成物(V)は、例えば[A’]重合体、[S]溶媒及び必要に応じて他の成分を所定の割合で混合し、好ましくは孔径200nm程度のメンブランフィルター等で濾過することにより調製することができる。組成物(V)の固形分濃度の下限としては、0.1質量%が好ましく、0.5質量%がより好ましく、0.7質量%がさらに好ましい。上記固形分濃度の上限としては、30質量%が好ましく、10質量%がより好ましく、3質量%がさらに好ましい。 [Method for Preparing Composition (V)]
The composition (V) is prepared, for example, by mixing the [A ′] polymer, the [S] solvent, and other components as necessary at a predetermined ratio, and preferably filtering through a membrane filter having a pore diameter of about 200 nm. can do. As a minimum of solid content concentration of composition (V), 0.1 mass% is preferred, 0.5 mass% is more preferred, and 0.7 mass% is still more preferred. The upper limit of the solid content concentration is preferably 30% by mass, more preferably 10% by mass, and still more preferably 3% by mass.
組成物(V)は、例えば[A’]重合体、[S]溶媒及び必要に応じて他の成分を所定の割合で混合し、好ましくは孔径200nm程度のメンブランフィルター等で濾過することにより調製することができる。組成物(V)の固形分濃度の下限としては、0.1質量%が好ましく、0.5質量%がより好ましく、0.7質量%がさらに好ましい。上記固形分濃度の上限としては、30質量%が好ましく、10質量%がより好ましく、3質量%がさらに好ましい。 [Method for Preparing Composition (V)]
The composition (V) is prepared, for example, by mixing the [A ′] polymer, the [S] solvent, and other components as necessary at a predetermined ratio, and preferably filtering through a membrane filter having a pore diameter of about 200 nm. can do. As a minimum of solid content concentration of composition (V), 0.1 mass% is preferred, 0.5 mass% is more preferred, and 0.7 mass% is still more preferred. The upper limit of the solid content concentration is preferably 30% by mass, more preferably 10% by mass, and still more preferably 3% by mass.
組成物(V)の塗工方法としては、例えばスピンコート法等が挙げられる。
Examples of the coating method of the composition (V) include a spin coating method.
[加熱工程]
本工程では、上記塗工工程により形成された塗工膜を加熱する。加熱工程後の塗工膜における基礎パターンの凹部の側面側の水の接触角θ1と底面側の水の接触角θ2との差(|θ1-θ2|、(θ1-θ2)の絶対値を意味する)は5°以上となる。この際、θ1とθ2の値はどちらが大きくてもよい。 [Heating process]
In this step, the coating film formed by the coating step is heated. Means the absolute value of the difference (| θ1−θ2 |, (θ1−θ2)) between the water contact angle θ1 on the side surface of the concave portion of the basic pattern in the coated film after the heating step and the water contact angle θ2 on the bottom surface side. Is 5 ° or more. At this time, either of the values of θ1 and θ2 may be larger.
本工程では、上記塗工工程により形成された塗工膜を加熱する。加熱工程後の塗工膜における基礎パターンの凹部の側面側の水の接触角θ1と底面側の水の接触角θ2との差(|θ1-θ2|、(θ1-θ2)の絶対値を意味する)は5°以上となる。この際、θ1とθ2の値はどちらが大きくてもよい。 [Heating process]
In this step, the coating film formed by the coating step is heated. Means the absolute value of the difference (| θ1−θ2 |, (θ1−θ2)) between the water contact angle θ1 on the side surface of the concave portion of the basic pattern in the coated film after the heating step and the water contact angle θ2 on the bottom surface side. Is 5 ° or more. At this time, either of the values of θ1 and θ2 may be larger.
θ1とθ2との差の下限としては、7°が好ましく、10°がより好ましい。θ1とθ2との差の上限としては、例えば30°である。
The lower limit of the difference between θ1 and θ2 is preferably 7 ° and more preferably 10 °. The upper limit of the difference between θ1 and θ2 is, for example, 30 °.
θ1の下限としては、50°が好ましく、80°がより好ましい。θ1の上限としては、90°が好ましく、88°がより好ましい。θ2の下限としては、60°が好ましく、70°がより好ましい。θ2の上限としては、85°が好ましく、80°がより好ましい。
The lower limit of θ1 is preferably 50 °, more preferably 80 °. The upper limit of θ1 is preferably 90 ° and more preferably 88 °. The lower limit of θ2 is preferably 60 °, and more preferably 70 °. The upper limit of θ2 is preferably 85 ° and more preferably 80 °.
加熱の手段としては、例えばオーブン、ホットプレート等が挙げられる。
Examples of the heating means include an oven and a hot plate.
加熱工程において、θ1とθ2との差が上記値以上となるよう、温度又は時間を制御する。加熱工程における温度及び時間は、例えば組成物(V)の[A’]重合体が有する官能基の種類等により変わってくる。
In the heating step, the temperature or time is controlled so that the difference between θ1 and θ2 is greater than or equal to the above value. The temperature and time in the heating step vary depending on, for example, the type of functional group of the [A ′] polymer of the composition (V).
加熱の温度の下限としては、50℃が好ましく、100℃がより好ましく、120℃がさらに好ましく、140℃が特に好ましい。上記温度の上限としては、250℃が好ましく、200℃がより好ましく、180℃がさらに好ましく、160℃が特に好ましい。
The lower limit of the heating temperature is preferably 50 ° C, more preferably 100 ° C, further preferably 120 ° C, and particularly preferably 140 ° C. As an upper limit of the said temperature, 250 degreeC is preferable, 200 degreeC is more preferable, 180 degreeC is further more preferable, and 160 degreeC is especially preferable.
加熱の時間の下限としては、10秒が好ましく、1分がより好ましく、5分がさらに好ましく、10分が特に好ましい。上記時間の上限としては、10時間が好ましく、1時間がより好ましく、30分がさらに好ましく、20分が特に好ましい。
The lower limit of the heating time is preferably 10 seconds, more preferably 1 minute, further preferably 5 minutes, and particularly preferably 10 minutes. The upper limit of the time is preferably 10 hours, more preferably 1 hour, still more preferably 30 minutes, and particularly preferably 20 minutes.
[充填工程]
本工程では、上記塗工膜が積層された上記基礎パターンの凹部に組成物(VI)を充填する。 [Filling process]
In this step, the concave portion of the basic pattern on which the coating film is laminated is filled with the composition (VI).
本工程では、上記塗工膜が積層された上記基礎パターンの凹部に組成物(VI)を充填する。 [Filling process]
In this step, the concave portion of the basic pattern on which the coating film is laminated is filled with the composition (VI).
(組成物(VI))
組成物(VI)は、[B’]重合体と溶媒([S]溶媒)とを含有する。組成物(VI)は、[B’]重合体及び[S]溶媒以外にも、その他の成分を含有することができる。 (Composition (VI))
The composition (VI) contains a [B ′] polymer and a solvent ([S] solvent). The composition (VI) can contain other components in addition to the [B ′] polymer and the [S] solvent.
組成物(VI)は、[B’]重合体と溶媒([S]溶媒)とを含有する。組成物(VI)は、[B’]重合体及び[S]溶媒以外にも、その他の成分を含有することができる。 (Composition (VI))
The composition (VI) contains a [B ′] polymer and a solvent ([S] solvent). The composition (VI) can contain other components in addition to the [B ′] polymer and the [S] solvent.
([B’]重合体)
[B’]重合体は、ブロック(A)及びブロック(B)を有する。すなわち、[B’]重合体は、ブロック共重合体である。ブロック(A)は構造単位(II)からなり、ブロック(B)は構造単位(II)よりも極性が高い構造単位(III)からなる。[B’]重合体は、ジブロック共重合体でも、トリブロック共重合体でも、4つ以上のブロックを有する共重合体でもよい。これらの中で、相分離構造の形成のし易さの観点から、ジブロック共重合体及びトリブロック共重合体が好ましく、ジブロック共重合体がより好ましい。また、[B’]重合体は、隣接するブロック間に連結基を有していてもよい。 ([B ′] polymer)
[B ′] The polymer has a block (A) and a block (B). That is, the [B ′] polymer is a block copolymer. The block (A) is composed of the structural unit (II), and the block (B) is composed of the structural unit (III) having a higher polarity than the structural unit (II). [B ′] The polymer may be a diblock copolymer, a triblock copolymer, or a copolymer having four or more blocks. Among these, from the viewpoint of easy formation of the phase separation structure, a diblock copolymer and a triblock copolymer are preferable, and a diblock copolymer is more preferable. Moreover, the [B ′] polymer may have a linking group between adjacent blocks.
[B’]重合体は、ブロック(A)及びブロック(B)を有する。すなわち、[B’]重合体は、ブロック共重合体である。ブロック(A)は構造単位(II)からなり、ブロック(B)は構造単位(II)よりも極性が高い構造単位(III)からなる。[B’]重合体は、ジブロック共重合体でも、トリブロック共重合体でも、4つ以上のブロックを有する共重合体でもよい。これらの中で、相分離構造の形成のし易さの観点から、ジブロック共重合体及びトリブロック共重合体が好ましく、ジブロック共重合体がより好ましい。また、[B’]重合体は、隣接するブロック間に連結基を有していてもよい。 ([B ′] polymer)
[B ′] The polymer has a block (A) and a block (B). That is, the [B ′] polymer is a block copolymer. The block (A) is composed of the structural unit (II), and the block (B) is composed of the structural unit (III) having a higher polarity than the structural unit (II). [B ′] The polymer may be a diblock copolymer, a triblock copolymer, or a copolymer having four or more blocks. Among these, from the viewpoint of easy formation of the phase separation structure, a diblock copolymer and a triblock copolymer are preferable, and a diblock copolymer is more preferable. Moreover, the [B ′] polymer may have a linking group between adjacent blocks.
ブロック(A)としては、パターン形成方法(A)における[A]重合体のブロック(I)として例示したポリスチレンブロックが好ましい。すなわち、構造単位(II)としては、置換又は非置換のスチレンに由来する構造単位が好ましい。ブロック(B)としては、例えば[A]重合体のブロック(II)として例示したブロック等が挙げられる。ブロック(B)としては、ポリ(メタ)アクリル酸エステルブロックが好ましく、ポリメタクリル酸メチルブロックがより好ましい。構造単位(III)としては、(メタ)アクリル酸エステルに由来する構造単位が好ましい。
The block (A) is preferably a polystyrene block exemplified as the block (I) of the [A] polymer in the pattern formation method (A). That is, as the structural unit (II), a structural unit derived from substituted or unsubstituted styrene is preferable. Examples of the block (B) include the blocks exemplified as the block (II) of the [A] polymer. As the block (B), a poly (meth) acrylate block is preferable, and a polymethyl methacrylate block is more preferable. As the structural unit (III), a structural unit derived from a (meth) acrylic acid ester is preferable.
[B’]重合体のMw及びMw/Mn、並びに組成物(VI)における[B’]重合体の含有量及び濃度としては、パターン形成方法(A)の[A]重合体及び組成物(I)におけるものと同様の範囲が好ましい。
[B ′] Mw and Mw / Mn of the polymer, and the content and concentration of the [B ′] polymer in the composition (VI) are the [A] polymer and the composition of the pattern formation method (A) ( A range similar to that in I) is preferred.
[相分離工程]
本工程では、上記充填工程により形成された充填層を相分離させる。この工程は、パターン形成方法(A)における相分離工程と同様である。 [Phase separation process]
In this step, the packed bed formed by the above filling step is phase-separated. This step is the same as the phase separation step in the pattern forming method (A).
本工程では、上記充填工程により形成された充填層を相分離させる。この工程は、パターン形成方法(A)における相分離工程と同様である。 [Phase separation process]
In this step, the packed bed formed by the above filling step is phase-separated. This step is the same as the phase separation step in the pattern forming method (A).
[除去工程]
本工程では、上記相分離工程後の充填層の少なくとも一部の相を除去する。この工程は、パターン形成方法(A)における除去工程と同様である。 [Removal process]
In this step, at least a part of the phase of the packed bed after the phase separation step is removed. This step is the same as the removing step in the pattern forming method (A).
本工程では、上記相分離工程後の充填層の少なくとも一部の相を除去する。この工程は、パターン形成方法(A)における除去工程と同様である。 [Removal process]
In this step, at least a part of the phase of the packed bed after the phase separation step is removed. This step is the same as the removing step in the pattern forming method (A).
[エッチング工程]
本工程では、上記除去工程により形成された微細化パターンを用いて上記基板を1又は複数回エッチングする。この工程は、パターン形成方法(A)におけるエッチング工程と同様である。 [Etching process]
In this step, the substrate is etched one or more times using the miniaturized pattern formed in the removing step. This step is the same as the etching step in the pattern forming method (A).
本工程では、上記除去工程により形成された微細化パターンを用いて上記基板を1又は複数回エッチングする。この工程は、パターン形成方法(A)におけるエッチング工程と同様である。 [Etching process]
In this step, the substrate is etched one or more times using the miniaturized pattern formed in the removing step. This step is the same as the etching step in the pattern forming method (A).
<組成物>
当該組成物は、主鎖の少なくとも一方の末端に結合し-COOH及び-OHの少なくとも一方と化学結合を形成する官能基を有する重合体と、主鎖の少なくとも一方の末端に結合しSi-H、Si-OH、Si=O及びSi-NR2(Rは、それぞれ独立して、水素原子又は炭素数1~20の1価の有機基である)の少なくとも1種と化学結合を形成する官能基を有する重合体と、溶媒とを含有する。 <Composition>
The composition includes a polymer having a functional group bonded to at least one end of the main chain and forming a chemical bond with at least one of —COOH and —OH, and bonded to at least one end of the main chain. , Si—OH, Si═O, and Si—NR 2 (wherein R is each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms) that forms a chemical bond. A polymer having a group and a solvent are contained.
当該組成物は、主鎖の少なくとも一方の末端に結合し-COOH及び-OHの少なくとも一方と化学結合を形成する官能基を有する重合体と、主鎖の少なくとも一方の末端に結合しSi-H、Si-OH、Si=O及びSi-NR2(Rは、それぞれ独立して、水素原子又は炭素数1~20の1価の有機基である)の少なくとも1種と化学結合を形成する官能基を有する重合体と、溶媒とを含有する。 <Composition>
The composition includes a polymer having a functional group bonded to at least one end of the main chain and forming a chemical bond with at least one of —COOH and —OH, and bonded to at least one end of the main chain. , Si—OH, Si═O, and Si—NR 2 (wherein R is each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms) that forms a chemical bond. A polymer having a group and a solvent are contained.
当該組成物についてはパターン形成方法(A)で用いる組成物(II)として説明している。
The composition is described as the composition (II) used in the pattern formation method (A).
以下、本発明を実施例に基づいて具体的に説明するが、本発明はこれらの実施例に限定されるものではない。各物性値の測定方法を下記に示す。
Hereinafter, the present invention will be specifically described based on examples, but the present invention is not limited to these examples. The measuring method of each physical property value is shown below.
[Mw及びMn]
重合体のMw及びMnは、ゲルパーミエーションクロマトグラフィー(GPC)により東ソー社のGPCカラム(「G2000HXL」2本、「G3000HXL」1本及び「G4000HXL」1本)を使用し、以下の条件により測定した。
溶離液:テトラヒドロフラン(和光純薬工業社)
流量:1.0mL/分
試料濃度:1.0質量%
試料注入量:100μL
カラム温度:40℃
検出器:示差屈折計
標準物質:単分散ポリスチレン [Mw and Mn]
Mw and Mn of the polymer are measured by gel permeation chromatography (GPC) using Tosoh's GPC columns ("G2000HXL", "G3000HXL" and "G4000HXL") under the following conditions. did.
Eluent: Tetrahydrofuran (Wako Pure Chemical Industries)
Flow rate: 1.0 mL / min Sample concentration: 1.0% by mass
Sample injection volume: 100 μL
Column temperature: 40 ° C
Detector: Differential refractometer Standard material: Monodisperse polystyrene
重合体のMw及びMnは、ゲルパーミエーションクロマトグラフィー(GPC)により東ソー社のGPCカラム(「G2000HXL」2本、「G3000HXL」1本及び「G4000HXL」1本)を使用し、以下の条件により測定した。
溶離液:テトラヒドロフラン(和光純薬工業社)
流量:1.0mL/分
試料濃度:1.0質量%
試料注入量:100μL
カラム温度:40℃
検出器:示差屈折計
標準物質:単分散ポリスチレン [Mw and Mn]
Mw and Mn of the polymer are measured by gel permeation chromatography (GPC) using Tosoh's GPC columns ("G2000HXL", "G3000HXL" and "G4000HXL") under the following conditions. did.
Eluent: Tetrahydrofuran (Wako Pure Chemical Industries)
Flow rate: 1.0 mL / min Sample concentration: 1.0% by mass
Sample injection volume: 100 μL
Column temperature: 40 ° C
Detector: Differential refractometer Standard material: Monodisperse polystyrene
[1H-NMR分析]
1H-NMR分析は、核磁気共鳴装置(日本電子社の「JNM-EX400」)を使用し、測定溶媒としてDMSO-d6を用いて行った。重合体における各構造単位の含有割合は、1H-NMRで得られたスペクトルにおける各構造単位に対応するピークの面積比から算出した。 [ 1 H-NMR analysis]
1 H-NMR analysis was performed using a nuclear magnetic resonance apparatus (“JNM-EX400” manufactured by JEOL Ltd.) and DMSO-d 6 as a measurement solvent. The content ratio of each structural unit in the polymer was calculated from the area ratio of the peak corresponding to each structural unit in the spectrum obtained by 1 H-NMR.
1H-NMR分析は、核磁気共鳴装置(日本電子社の「JNM-EX400」)を使用し、測定溶媒としてDMSO-d6を用いて行った。重合体における各構造単位の含有割合は、1H-NMRで得られたスペクトルにおける各構造単位に対応するピークの面積比から算出した。 [ 1 H-NMR analysis]
1 H-NMR analysis was performed using a nuclear magnetic resonance apparatus (“JNM-EX400” manufactured by JEOL Ltd.) and DMSO-d 6 as a measurement solvent. The content ratio of each structural unit in the polymer was calculated from the area ratio of the peak corresponding to each structural unit in the spectrum obtained by 1 H-NMR.
<パターン形成方法(A)>
<[A]重合体の合成>
[合成例1-1](重合体(a-1)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン(THF)200gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を0.29mL(0.256mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン22.7mL(0.197mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00077mol)及び塩化リチウムの0.5N THF溶液1.02mL(0.0005mol)を加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル10.6mL(0.100mol)をこの溶液に30分かけて滴下注入して重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤としてのメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してメチルイソブチルケトン(MIBK)で置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次にポリスチレンのホモポリマーを除去するためシクロヘキサン500gを注ぎ、ポリスチレンホモポリマーをシクロヘキサンへ溶解させて重合体を洗浄した。なお、この洗浄は4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(a-1)24.1gを得た。 <Pattern formation method (A)>
<[A] Synthesis of polymer>
[Synthesis Example 1-1] (Synthesis of Polymer (a-1))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of tetrahydrofuran (THF) that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to -78 ° C. Thereafter, 0.29 mL (0.256 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and thereafter, adsorption filtration with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 22.7 mL (0.197 mol) of styrene was added dropwise over 30 minutes, and the polymerization system was confirmed to be orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00077 mol) of 1,1-diphenylethylene and 1.02 mL (0.0005 mol) of 0.5N THF solution of lithium chloride were added to make the polymerization system dark red. I confirmed. Further, 10.6 mL (0.100 mol) of methyl methacrylate, which has been subjected to adsorption filtration with silica gel for removal of the polymerization inhibitor and distilled and dehydrated, is dropped into this solution over 30 minutes to make the polymerization system light yellow. After confirming that the reaction had occurred, the reaction was continued for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with methyl isobutyl ketone (MIBK). Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of cyclohexane was poured to remove the polystyrene homopolymer, and the polymer was washed by dissolving the polystyrene homopolymer in cyclohexane. This washing was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 24.1 g of a white polymer (a-1).
<[A]重合体の合成>
[合成例1-1](重合体(a-1)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン(THF)200gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を0.29mL(0.256mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン22.7mL(0.197mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00077mol)及び塩化リチウムの0.5N THF溶液1.02mL(0.0005mol)を加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル10.6mL(0.100mol)をこの溶液に30分かけて滴下注入して重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤としてのメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してメチルイソブチルケトン(MIBK)で置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次にポリスチレンのホモポリマーを除去するためシクロヘキサン500gを注ぎ、ポリスチレンホモポリマーをシクロヘキサンへ溶解させて重合体を洗浄した。なお、この洗浄は4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(a-1)24.1gを得た。 <Pattern formation method (A)>
<[A] Synthesis of polymer>
[Synthesis Example 1-1] (Synthesis of Polymer (a-1))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of tetrahydrofuran (THF) that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to -78 ° C. Thereafter, 0.29 mL (0.256 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and thereafter, adsorption filtration with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 22.7 mL (0.197 mol) of styrene was added dropwise over 30 minutes, and the polymerization system was confirmed to be orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00077 mol) of 1,1-diphenylethylene and 1.02 mL (0.0005 mol) of 0.5N THF solution of lithium chloride were added to make the polymerization system dark red. I confirmed. Further, 10.6 mL (0.100 mol) of methyl methacrylate, which has been subjected to adsorption filtration with silica gel for removal of the polymerization inhibitor and distilled and dehydrated, is dropped into this solution over 30 minutes to make the polymerization system light yellow. After confirming that the reaction had occurred, the reaction was continued for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with methyl isobutyl ketone (MIBK). Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of cyclohexane was poured to remove the polystyrene homopolymer, and the polymer was washed by dissolving the polystyrene homopolymer in cyclohexane. This washing was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 24.1 g of a white polymer (a-1).
この重合体(a-1)は、Mwが79,000、Mnが77,000、Mw/Mnが1.03であった。また、1H-NMR分析の結果、重合体(a-1)は、スチレンに由来する構造単位及びメタクリル酸メチルに由来する構造単位の含有割合が、それぞれ65モル%及び35モル%であった。なお、重合体(a-1)はジブロック共重合体である。
This polymer (a-1) had Mw of 79,000, Mn of 77,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (a-1) contained 65 mol% and 35 mol% of structural units derived from styrene and structural units derived from methyl methacrylate, respectively. . The polymer (a-1) is a diblock copolymer.

[合成例1-2](重合体(a-2)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF200gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を0.29mL(0.256mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン22.7mL(0.197mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00077mol)及び塩化リチウムの0.5Nテトラヒドロフラン溶液1.02mL(0.0005mol)を加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル10.6mL(0.100mol)をこの溶液に30分かけて滴下注入して重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤として2-エチルヘキシルグリシジルエーテル0.053mL(0.256mmol)を加えたのち、メタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次にポリスチレンのホモポリマーを除去するためシクロヘキサン500gを注ぎ、ポリスチレンホモポリマーをシクロヘキサンへ溶解させて重合体を洗浄した。なお、この洗浄は4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(a-2)24.1gを得た。 [Synthesis Example 1-2] (Synthesis of Polymer (a-2))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 0.29 mL (0.256 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and thereafter, adsorption filtration with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 22.7 mL (0.197 mol) of styrene was added dropwise over 30 minutes, and the polymerization system was confirmed to be orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00077 mol) of 1,1-diphenylethylene and 1.02 mL (0.0005 mol) of 0.5N tetrahydrofuran solution of lithium chloride were added to make the polymerization system dark red. I confirmed. Further, 10.6 mL (0.100 mol) of methyl methacrylate, which has been subjected to adsorption filtration with silica gel for removal of the polymerization inhibitor and distilled and dehydrated, is dropped into this solution over 30 minutes to make the polymerization system light yellow. After confirming that the reaction had occurred, the reaction was continued for 120 minutes. Thereafter, 0.053 mL (0.256 mmol) of 2-ethylhexyl glycidyl ether was added as a terminal terminator, and then 1 mL of methanol was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of cyclohexane was poured to remove the polystyrene homopolymer, and the polymer was washed by dissolving the polystyrene homopolymer in cyclohexane. This washing was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 24.1 g of a white polymer (a-2).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF200gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を0.29mL(0.256mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン22.7mL(0.197mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00077mol)及び塩化リチウムの0.5Nテトラヒドロフラン溶液1.02mL(0.0005mol)を加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル10.6mL(0.100mol)をこの溶液に30分かけて滴下注入して重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤として2-エチルヘキシルグリシジルエーテル0.053mL(0.256mmol)を加えたのち、メタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次にポリスチレンのホモポリマーを除去するためシクロヘキサン500gを注ぎ、ポリスチレンホモポリマーをシクロヘキサンへ溶解させて重合体を洗浄した。なお、この洗浄は4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(a-2)24.1gを得た。 [Synthesis Example 1-2] (Synthesis of Polymer (a-2))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 0.29 mL (0.256 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and thereafter, adsorption filtration with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 22.7 mL (0.197 mol) of styrene was added dropwise over 30 minutes, and the polymerization system was confirmed to be orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00077 mol) of 1,1-diphenylethylene and 1.02 mL (0.0005 mol) of 0.5N tetrahydrofuran solution of lithium chloride were added to make the polymerization system dark red. I confirmed. Further, 10.6 mL (0.100 mol) of methyl methacrylate, which has been subjected to adsorption filtration with silica gel for removal of the polymerization inhibitor and distilled and dehydrated, is dropped into this solution over 30 minutes to make the polymerization system light yellow. After confirming that the reaction had occurred, the reaction was continued for 120 minutes. Thereafter, 0.053 mL (0.256 mmol) of 2-ethylhexyl glycidyl ether was added as a terminal terminator, and then 1 mL of methanol was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of cyclohexane was poured to remove the polystyrene homopolymer, and the polymer was washed by dissolving the polystyrene homopolymer in cyclohexane. This washing was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 24.1 g of a white polymer (a-2).
この重合体(a-2)は、Mwが78,000、Mnが76,000、Mw/Mnが1.03であった。また、1H-NMR分析の結果、重合体(A-2)は、スチレンに由来する構造単位及びメタクリル酸メチルに由来する構造単位の含有割合が、それぞれ65モル%及び35モル%であった。なお、重合体(a-2)はジブロック共重合体である。
This polymer (a-2) had Mw of 78,000, Mn of 76,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (A-2) contained 65 mol% and 35 mol% of structural units derived from styrene and structural units derived from methyl methacrylate, respectively. . The polymer (a-2) is a diblock copolymer.

[合成例1-3](重合体(a-3)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF200gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を0.28mL(0.256mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン26.0mL(0.226mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00077mol)、及び塩化リチウムの0.5Nテトラヒドロフラン溶液1.02mL(0.0005mol)を加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル9.5mL(0.899mol)をこの溶液に30分かけて滴下注入して重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤としてのメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次にポリスチレンのホモポリマーを除去するためシクロヘキサン500gを注ぎ、ポリスチレンホモポリマーをシクロヘキサンへ溶解させて重合体を洗浄した。なお、この洗浄は4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(a-3)23.8gを得た。 [Synthesis Example 1-3] (Synthesis of Polymer (a-3))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 0.28 mL (0.256 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then, adsorption removal with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. Styrene 26.0 mL (0.226 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00077 mol) of 1,1-diphenylethylene and 1.02 mL (0.0005 mol) of 0.5N tetrahydrofuran solution of lithium chloride were added, and the polymerization system was dark red It was confirmed that it became. Further, 9.5 mL (0.899 mol) of methyl methacrylate, which has been subjected to adsorption filtration with silica gel and distillation dehydration treatment for removing the polymerization inhibitor, was dropped into this solution over 30 minutes to make the polymerization system light yellow. After confirming that the reaction had occurred, the reaction was continued for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of cyclohexane was poured to remove the polystyrene homopolymer, and the polymer was washed by dissolving the polystyrene homopolymer in cyclohexane. This washing was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 23.8 g of a white polymer (a-3).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF200gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を0.28mL(0.256mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン26.0mL(0.226mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00077mol)、及び塩化リチウムの0.5Nテトラヒドロフラン溶液1.02mL(0.0005mol)を加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル9.5mL(0.899mol)をこの溶液に30分かけて滴下注入して重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤としてのメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次にポリスチレンのホモポリマーを除去するためシクロヘキサン500gを注ぎ、ポリスチレンホモポリマーをシクロヘキサンへ溶解させて重合体を洗浄した。なお、この洗浄は4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(a-3)23.8gを得た。 [Synthesis Example 1-3] (Synthesis of Polymer (a-3))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 0.28 mL (0.256 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then, adsorption removal with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. Styrene 26.0 mL (0.226 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00077 mol) of 1,1-diphenylethylene and 1.02 mL (0.0005 mol) of 0.5N tetrahydrofuran solution of lithium chloride were added, and the polymerization system was dark red It was confirmed that it became. Further, 9.5 mL (0.899 mol) of methyl methacrylate, which has been subjected to adsorption filtration with silica gel and distillation dehydration treatment for removing the polymerization inhibitor, was dropped into this solution over 30 minutes to make the polymerization system light yellow. After confirming that the reaction had occurred, the reaction was continued for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of cyclohexane was poured to remove the polystyrene homopolymer, and the polymer was washed by dissolving the polystyrene homopolymer in cyclohexane. This washing was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 23.8 g of a white polymer (a-3).
この重合体(a-3)は、Mwが77,000、Mnが75,000、Mw/Mnが1.03であった。また、1H-NMR分析の結果、重合体(a-3)は、スチレンに由来する構造単位及びメタクリル酸メチルに由来する構造単位の含有割合が、それぞれ70モル%及び30モル%であった。なお、ブロック共重合体(a-3)はジブロック共重合体である。
This polymer (a-3) had Mw of 77,000, Mn of 75,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (a-3) was found to contain 70 mol% and 30 mol% of structural units derived from styrene and methyl methacrylate, respectively. . The block copolymer (a-3) is a diblock copolymer.

<[B]重合体の合成>
[合成例1-4](重合体(b-1)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.31mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤としてメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-1)11.7gを得た。 <[B] Synthesis of polymer>
[Synthesis Example 1-4] (Synthesis of polymer (b-1))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.31 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and thereafter, adsorption filtration with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 1 mL of methanol was injected as a terminal terminator to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.7 g of a white polymer (b-1).
[合成例1-4](重合体(b-1)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.31mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤としてメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-1)11.7gを得た。 <[B] Synthesis of polymer>
[Synthesis Example 1-4] (Synthesis of polymer (b-1))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.31 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and thereafter, adsorption filtration with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 1 mL of methanol was injected as a terminal terminator to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.7 g of a white polymer (b-1).
この重合体(b-1)は、Mwが5,600、Mnが5,300、Mw/Mnが1.06であった。
This polymer (b-1) had Mw of 5,600, Mn of 5,300, and Mw / Mn of 1.06.

[合成例1-5](重合体(b-2)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤としてスチレンオキサイド0.27mL(2.30mmol)、続いてメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-2)11.7gを得た。 [Synthesis Example 1-5] (Synthesis of Polymer (b-2))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. After that, 2.38 mL (2.30 mmol) of 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then subjected to adsorption filtration with silica gel and distillation dehydration treatment to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.27 mL (2.30 mmol) of styrene oxide as a terminal terminator and then 1 mL of methanol were injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.7 g of a white polymer (b-2).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤としてスチレンオキサイド0.27mL(2.30mmol)、続いてメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-2)11.7gを得た。 [Synthesis Example 1-5] (Synthesis of Polymer (b-2))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. After that, 2.38 mL (2.30 mmol) of 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then subjected to adsorption filtration with silica gel and distillation dehydration treatment to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.27 mL (2.30 mmol) of styrene oxide as a terminal terminator and then 1 mL of methanol were injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.7 g of a white polymer (b-2).
このブロック共重合体(b-2)は、Mwが5,500、Mnが5,100、Mw/Mnが1.08であった。
This block copolymer (b-2) had Mw of 5,500, Mn of 5,100, and Mw / Mn of 1.08.

[合成例1-6](重合体(b-3)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として二酸化炭素バブリングし、続いてメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-3)11.3gを得た。 [Synthesis Example 1-6] (Synthesis of Polymer (b-3))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. After that, 2.38 mL (2.30 mmol) of 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then subjected to adsorption filtration with silica gel and distillation dehydration treatment to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, carbon dioxide was bubbled as a terminal terminator, and then 1 mL of methanol was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.3 g of a white polymer (b-3).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として二酸化炭素バブリングし、続いてメタノール1mLを注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-3)11.3gを得た。 [Synthesis Example 1-6] (Synthesis of Polymer (b-3))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. After that, 2.38 mL (2.30 mmol) of 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then subjected to adsorption filtration with silica gel and distillation dehydration treatment to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, carbon dioxide was bubbled as a terminal terminator, and then 1 mL of methanol was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.3 g of a white polymer (b-3).
この重合体(b-3)は、Mwが5,200、Mnが5,000、Mw/Mnが1.04であった。
This polymer (b-3) had Mw of 5,200, Mn of 5,000, and Mw / Mn of 1.04.

[合成例1-7](重合体(b-4)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.19mL(2.30mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-4)11.1gを得た。 [Synthesis Example 1-7] (Synthesis of Polymer (b-4))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.30 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into this THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.19 mL (2.30 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.1 g of a white polymer (b-4).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.19mL(2.30mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-4)11.1gを得た。 [Synthesis Example 1-7] (Synthesis of Polymer (b-4))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.30 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into this THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.19 mL (2.30 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.1 g of a white polymer (b-4).
この重合体(b-4)は、Mwが5,700、Mnが5,200、Mw/Mnが1.10であった。
This polymer (b-4) had Mw of 5,700, Mn of 5,200, and Mw / Mn of 1.10.

[合成例1-8](重合体(b-5)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を3.95mL(3.84mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.32mL(3.84mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-5)11.1gを得た。 [Synthesis Example 1-8] (Synthesis of polymer (b-5))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.95 mL (3.84 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then subjected to adsorption filtration with silica gel and distillation dehydration to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.32 mL (3.84 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.1 g of a white polymer (b-5).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を3.95mL(3.84mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.32mL(3.84mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-5)11.1gを得た。 [Synthesis Example 1-8] (Synthesis of polymer (b-5))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.95 mL (3.84 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then subjected to adsorption filtration with silica gel and distillation dehydration to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.32 mL (3.84 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.1 g of a white polymer (b-5).
この重合体(b-5)は、Mwが3,300、Mnが3,100、Mw/Mnが1.06であった。
This polymer (b-5) had Mw of 3,300, Mn of 3,100, and Mw / Mn of 1.06.

[合成例1-9](重合体(b-6)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を1.19mL(1.15mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.10mL(1.15mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-6)11.5gを得た。 [Synthesis Example 1-9] (Synthesis of polymer (b-6))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 1.19 mL (1.15 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then, adsorption removal with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.10 mL (1.15 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.5 g of a white polymer (b-6).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を1.19mL(1.15mmol)注入し、その後、重合禁止剤除去のためシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.10mL(1.15mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-6)11.5gを得た。 [Synthesis Example 1-9] (Synthesis of polymer (b-6))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 1.19 mL (1.15 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) was injected into this THF, and then, adsorption removal with silica gel and distillation dehydration were performed to remove the polymerization inhibitor. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.10 mL (1.15 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.5 g of a white polymer (b-6).
このブロック共重合体(b-6)は、Mwが11,600、Mnが10,400、Mw/Mnが1.12であった。
This block copolymer (b-6) had an Mw of 11,600, an Mn of 10,400, and an Mw / Mn of 1.12.

[合成例1-10](重合体(b-7)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し6、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、3-クロロメチル-3-メチルオキセタンを0.25mL(2.30mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色のブロック共重合体(b-7)11.1gを得た。 [Synthesis Example 1-10] (Synthesis of Polymer (b-7))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.30 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into this THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes 6 and it was confirmed that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.25 mL (2.30 mmol) of 3-chloromethyl-3-methyloxetane was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.1 g of a white block copolymer (b-7).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン13.3mL(0.115mol)を30分かけて滴下注入し6、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、3-クロロメチル-3-メチルオキセタンを0.25mL(2.30mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色のブロック共重合体(b-7)11.1gを得た。 [Synthesis Example 1-10] (Synthesis of Polymer (b-7))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.30 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into this THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. 13.3 mL (0.115 mol) of styrene was added dropwise over 30 minutes 6 and it was confirmed that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.25 mL (2.30 mmol) of 3-chloromethyl-3-methyloxetane was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 11.1 g of a white block copolymer (b-7).
このブロック共重合体(b-7)は、Mwが5,900、Mnが5,300、Mw/Mnが1.11であった。
This block copolymer (b-7) had Mw of 5,900, Mn of 5,300, and Mw / Mn of 1.11.

[合成例1-11](重合体(b-8)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行った4-t-ブチルスチレン21.2mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.19mL(2.30mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-8)17.7gを得た。 [Synthesis Example 1-11] (Synthesis of polymer (b-8))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.30 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into this THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. The resulting 4-t-butylstyrene 21.2 mL (0.115 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.19 mL (2.30 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 17.7 g of a white polymer (b-8).
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を2.38mL(2.30mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行った4-t-ブチルスチレン21.2mL(0.115mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入のとき、反応溶液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、エピブロモヒドリンを0.19mL(2.30mmol)注入し重合末端の停止反応を行った。この反応溶液を室温まで昇温し、得られた反応溶液を濃縮してMIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返しシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた固体を60℃で減圧乾燥させることで白色の重合体(b-8)17.7gを得た。 [Synthesis Example 1-11] (Synthesis of polymer (b-8))
After drying the 500 mL flask reaction vessel under reduced pressure, 120 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 2.38 mL (2.30 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into this THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. The resulting 4-t-butylstyrene 21.2 mL (0.115 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.19 mL (2.30 mmol) of epibromohydrin was injected to terminate the polymerization terminal. The reaction solution was warmed to room temperature, and the resulting reaction solution was concentrated and replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. This operation was repeated three times, and oxalic acid was removed. Then, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained solid was dried under reduced pressure at 60 ° C. to obtain 17.7 g of a white polymer (b-8).
このブロック共重合体(b-8)は、Mwが6,000、Mnが5,400、Mw/Mnが1.11であった。
This block copolymer (b-8) had Mw of 6,000, Mn of 5,400, and Mw / Mn of 1.11.

<[C]重合体の合成>
[合成例1-12](重合体(c-1)の合成)
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン16.7g(0.160mol)、メタクリル酸メチル3.00g(0.030mol)、2-ヒドロキシエチルメタクレート1.30g(0.01mol)、臭化銅(II)0.29g(2.00mmol)及びトリス[(2-ジメチルアミノ)エチル]アミン0.46g(2mmol)を加え、100℃に加熱し、2-ブロモイソ酪酸エチル0.53mL(3.6mmol)、を加え、窒素フロー下、8時間加熱撹拌した。得られた重合反応液は、セライト濾過し銅錯体を除去し、超純水500gにて洗浄を3回繰り返した。有機層を回収した後、濃縮し、得られた濃縮物にTHF50gを加えたものをメタノール/超純水(質量比=5/5)1,000gに加えて、沈殿精製させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで、白色の重合体(c-1)11.2gを得た。 <Synthesis of [C] polymer>
[Synthesis Example 1-12] (Synthesis of Polymer (c-1))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, anisole 40 g, styrene 16.7 g (0.160 mol), methyl methacrylate 3.00 g (0.030 mol), 2-hydroxyethyl methacrylate 1.30 g ( 0.01 mol), 0.29 g (2.00 mmol) of copper (II) bromide and 0.46 g (2 mmol) of tris [(2-dimethylamino) ethyl] amine were added, and the mixture was heated to 100 ° C. to give 2-bromoisobutyric acid. Ethyl 0.53 mL (3.6 mmol) was added, and the mixture was heated and stirred for 8 hours under a nitrogen flow. The obtained polymerization reaction liquid was filtered through Celite to remove the copper complex, and washing with 500 g of ultrapure water was repeated three times. The organic layer was collected and then concentrated, and 50 g of THF added to the resulting concentrate was added to 1,000 g of methanol / ultra pure water (mass ratio = 5/5) to obtain a precipitated and purified solid. . This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.2 g of a white polymer (c-1).
[合成例1-12](重合体(c-1)の合成)
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン16.7g(0.160mol)、メタクリル酸メチル3.00g(0.030mol)、2-ヒドロキシエチルメタクレート1.30g(0.01mol)、臭化銅(II)0.29g(2.00mmol)及びトリス[(2-ジメチルアミノ)エチル]アミン0.46g(2mmol)を加え、100℃に加熱し、2-ブロモイソ酪酸エチル0.53mL(3.6mmol)、を加え、窒素フロー下、8時間加熱撹拌した。得られた重合反応液は、セライト濾過し銅錯体を除去し、超純水500gにて洗浄を3回繰り返した。有機層を回収した後、濃縮し、得られた濃縮物にTHF50gを加えたものをメタノール/超純水(質量比=5/5)1,000gに加えて、沈殿精製させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで、白色の重合体(c-1)11.2gを得た。 <Synthesis of [C] polymer>
[Synthesis Example 1-12] (Synthesis of Polymer (c-1))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, anisole 40 g, styrene 16.7 g (0.160 mol), methyl methacrylate 3.00 g (0.030 mol), 2-hydroxyethyl methacrylate 1.30 g ( 0.01 mol), 0.29 g (2.00 mmol) of copper (II) bromide and 0.46 g (2 mmol) of tris [(2-dimethylamino) ethyl] amine were added, and the mixture was heated to 100 ° C. to give 2-bromoisobutyric acid. Ethyl 0.53 mL (3.6 mmol) was added, and the mixture was heated and stirred for 8 hours under a nitrogen flow. The obtained polymerization reaction liquid was filtered through Celite to remove the copper complex, and washing with 500 g of ultrapure water was repeated three times. The organic layer was collected and then concentrated, and 50 g of THF added to the resulting concentrate was added to 1,000 g of methanol / ultra pure water (mass ratio = 5/5) to obtain a precipitated and purified solid. . This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.2 g of a white polymer (c-1).
この重合体(c-1)は、Mwが5,600、Mnが4,600、Mw/Mnが1.22であった。また、1H-NMR分析の結果、スチレン、メチルメタクリレート及び2-ヒドロキシエチルメタクリレートに由来する構造単位の含有割合は、それぞれ80モル%、15モル%及び5モル%であった。
This polymer (c-1) had Mw of 5,600, Mn of 4,600, and Mw / Mn of 1.22. As a result of 1 H-NMR analysis, the content ratios of structural units derived from styrene, methyl methacrylate and 2-hydroxyethyl methacrylate were 80 mol%, 15 mol% and 5 mol%, respectively.

[合成例1-13](重合体(c-2)の合成)
冷却管、滴下ロート0及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン16.7g(0.160mol)、メタクリル酸メチル3.00g(0.030mol)、メタクリル酸0.86g(0.01mol)、臭化銅(II)0.29g(2.00mmol)及びトリス[(2-ジメチルアミノ)エチル]アミン0.46g(2mmol)を加え、100℃に加熱し、2-ブロモイソ酪酸エチル0.53mL(3.6mmol)、を加え、窒素フロー下、8時間加熱撹拌した。得られた重合反応液は、セライト濾過し銅錯体を除去し、超純水500gにて洗浄を3回繰り返した。有機層を回収した後、濃縮し、得られた濃縮物にTHF50gを加えたものをメタノール/超純水(質量比5/5)1,000gに加えて、沈殿精製させポリマーを析出させた。この白色固体をテトラヒドロフラン50gに溶解させ、1N塩酸水溶液5gを加え、80℃で2時間加熱撹拌し、加水分解反応を行った。加水分解後の反応溶液は、メタノール/超純水(質量比5/5)1,000gに加えて沈殿精製させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで、白色の重合体(c-2)11.0gを得た。 [Synthesis Example 1-13] (Synthesis of polymer (c-2))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel 0 and a thermometer, anisole 40 g, styrene 16.7 g (0.160 mol), methyl methacrylate 3.00 g (0.030 mol), methacrylic acid 0.86 g (0.01 mol) ), 0.29 g (2.00 mmol) of copper (II) bromide and 0.46 g (2 mmol) of tris [(2-dimethylamino) ethyl] amine, and heated to 100 ° C. 53 mL (3.6 mmol) was added, and the mixture was heated and stirred for 8 hours under a nitrogen flow. The obtained polymerization reaction liquid was filtered through Celite to remove the copper complex, and washing with 500 g of ultrapure water was repeated three times. After collecting the organic layer, it was concentrated, and the resulting concentrate with 50 g of THF added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) was precipitated and purified to precipitate the polymer. This white solid was dissolved in 50 g of tetrahydrofuran, 5 g of 1N hydrochloric acid aqueous solution was added, and the mixture was heated and stirred at 80 ° C. for 2 hours to conduct a hydrolysis reaction. The reaction solution after hydrolysis was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid that was purified by precipitation. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.0 g of a white polymer (c-2).
冷却管、滴下ロート0及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン16.7g(0.160mol)、メタクリル酸メチル3.00g(0.030mol)、メタクリル酸0.86g(0.01mol)、臭化銅(II)0.29g(2.00mmol)及びトリス[(2-ジメチルアミノ)エチル]アミン0.46g(2mmol)を加え、100℃に加熱し、2-ブロモイソ酪酸エチル0.53mL(3.6mmol)、を加え、窒素フロー下、8時間加熱撹拌した。得られた重合反応液は、セライト濾過し銅錯体を除去し、超純水500gにて洗浄を3回繰り返した。有機層を回収した後、濃縮し、得られた濃縮物にTHF50gを加えたものをメタノール/超純水(質量比5/5)1,000gに加えて、沈殿精製させポリマーを析出させた。この白色固体をテトラヒドロフラン50gに溶解させ、1N塩酸水溶液5gを加え、80℃で2時間加熱撹拌し、加水分解反応を行った。加水分解後の反応溶液は、メタノール/超純水(質量比5/5)1,000gに加えて沈殿精製させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで、白色の重合体(c-2)11.0gを得た。 [Synthesis Example 1-13] (Synthesis of polymer (c-2))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel 0 and a thermometer, anisole 40 g, styrene 16.7 g (0.160 mol), methyl methacrylate 3.00 g (0.030 mol), methacrylic acid 0.86 g (0.01 mol) ), 0.29 g (2.00 mmol) of copper (II) bromide and 0.46 g (2 mmol) of tris [(2-dimethylamino) ethyl] amine, and heated to 100 ° C. 53 mL (3.6 mmol) was added, and the mixture was heated and stirred for 8 hours under a nitrogen flow. The obtained polymerization reaction liquid was filtered through Celite to remove the copper complex, and washing with 500 g of ultrapure water was repeated three times. After collecting the organic layer, it was concentrated, and the resulting concentrate with 50 g of THF added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) was precipitated and purified to precipitate the polymer. This white solid was dissolved in 50 g of tetrahydrofuran, 5 g of 1N hydrochloric acid aqueous solution was added, and the mixture was heated and stirred at 80 ° C. for 2 hours to conduct a hydrolysis reaction. The reaction solution after hydrolysis was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid that was purified by precipitation. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.0 g of a white polymer (c-2).
この重合体(c-2)は、Mwが5,500、Mnが4,400、Mw/Mnが1.25であった。また、1H-NMR分析の結果、スチレン、メチルメタクリレート及びメタクリル酸に由来する構造単位の含有割合は、それぞれが80モル%、15モル%、及び5モル%であった。
This polymer (c-2) had Mw of 5,500, Mn of 4,400, and Mw / Mn of 1.25. As a result of 1 H-NMR analysis, the content ratios of structural units derived from styrene, methyl methacrylate and methacrylic acid were 80 mol%, 15 mol% and 5 mol%, respectively.

[合成例1-14](重合体(c-3)の合成)
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン14.6g(0.140mol)、メタクリル酸メチル6.00g(0.060mol)、4-シアノ-[4-ドデシル(スルファニルチオ)カルボニルスルファン]ペンタン酸0.403g(1mmol)及びアゾイソブチロニトリル0.164g(1mmol)を加え、80℃、8時間加熱撹拌した。得られた重合反応液は、メタノール/超純水(質量比5/5)1,000gに加えて沈殿生成させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで、黄色の重合体(c-3)11.5gを得た。 [Synthesis Example 1-14] (Synthesis of Polymer (c-3))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 40 g of anisole, 14.6 g (0.140 mol) of styrene, 6.00 g (0.060 mol) of methyl methacrylate, 4-cyano- [4-dodecyl (sulfanyl) Thio) carbonylsulfane] pentanoic acid 0.403 g (1 mmol) and azoisobutyronitrile 0.164 g (1 mmol) were added, and the mixture was heated and stirred at 80 ° C. for 8 hours. The obtained polymerization reaction liquid was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid which was precipitated. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.5 g of a yellow polymer (c-3).
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン14.6g(0.140mol)、メタクリル酸メチル6.00g(0.060mol)、4-シアノ-[4-ドデシル(スルファニルチオ)カルボニルスルファン]ペンタン酸0.403g(1mmol)及びアゾイソブチロニトリル0.164g(1mmol)を加え、80℃、8時間加熱撹拌した。得られた重合反応液は、メタノール/超純水(質量比5/5)1,000gに加えて沈殿生成させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで、黄色の重合体(c-3)11.5gを得た。 [Synthesis Example 1-14] (Synthesis of Polymer (c-3))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 40 g of anisole, 14.6 g (0.140 mol) of styrene, 6.00 g (0.060 mol) of methyl methacrylate, 4-cyano- [4-dodecyl (sulfanyl) Thio) carbonylsulfane] pentanoic acid 0.403 g (1 mmol) and azoisobutyronitrile 0.164 g (1 mmol) were added, and the mixture was heated and stirred at 80 ° C. for 8 hours. The obtained polymerization reaction liquid was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid which was precipitated. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.5 g of a yellow polymer (c-3).
この重合体(c-3)は、Mwが8,400、Mnが6,600、Mw/Mnが1.27であった。また、1H-NMR分析の結果、スチレン及びメチルメタクリレートに由来する構造単位の含有割合が、それぞれ70モル%及び30モル%であった。
This polymer (c-3) had Mw of 8,400, Mn of 6,600, and Mw / Mn of 1.27. As a result of 1 H-NMR analysis, the content ratios of structural units derived from styrene and methyl methacrylate were 70 mol% and 30 mol%, respectively.

[合成例1-15](重合体(c-4)の合成)
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン14.6g(0.140mol)、メタクリル酸メチル6.00g(0.006mol)及び4-シアノ-4-[(ドデシルスルファニルチオカルボニル)スルファニル]ペンタン酸メチル0.417g(1.00mmol)を加え、80℃に加熱し、8時間撹拌した。得られた重合反応液は、メタノール/超純水(質量比5/5)1,000gに加えて沈殿生成させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた黄色の重合体は、Mwが、8,600、Mnが6,500、Mw/Mnが1.32であった。 [Synthesis Example 1-15] (Synthesis of Polymer (c-4))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 40 g of anisole, 14.6 g (0.140 mol) of styrene, 6.00 g (0.006 mol) of methyl methacrylate and 4-cyano-4-[(dodecylsulfanyl) 0.417 g (1.00 mmol) of methyl thiocarbonyl) sulfanyl] pentanoate was added, heated to 80 ° C., and stirred for 8 hours. The obtained polymerization reaction liquid was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid which was precipitated. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained yellow polymer had Mw of 8,600, Mn of 6,500, and Mw / Mn of 1.32.
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン14.6g(0.140mol)、メタクリル酸メチル6.00g(0.006mol)及び4-シアノ-4-[(ドデシルスルファニルチオカルボニル)スルファニル]ペンタン酸メチル0.417g(1.00mmol)を加え、80℃に加熱し、8時間撹拌した。得られた重合反応液は、メタノール/超純水(質量比5/5)1,000gに加えて沈殿生成させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた黄色の重合体は、Mwが、8,600、Mnが6,500、Mw/Mnが1.32であった。 [Synthesis Example 1-15] (Synthesis of Polymer (c-4))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 40 g of anisole, 14.6 g (0.140 mol) of styrene, 6.00 g (0.006 mol) of methyl methacrylate and 4-cyano-4-[(dodecylsulfanyl) 0.417 g (1.00 mmol) of methyl thiocarbonyl) sulfanyl] pentanoate was added, heated to 80 ° C., and stirred for 8 hours. The obtained polymerization reaction liquid was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) to obtain a solid which was precipitated. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained yellow polymer had Mw of 8,600, Mn of 6,500, and Mw / Mn of 1.32.
次に、得られた黄色の固体を、ジメチルホルムアミドに溶解させ、4,4’-アゾビス-4-シアノ吉草酸2.80g(10mmol)を加え、80℃に加熱し、3時間撹拌した。アゾ分解終了後、メタノール/超純水(質量比5/5)1,000gに加えて沈殿生成させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで、黄色の重合体(c-4)11.0gを得た。
Next, the obtained yellow solid was dissolved in dimethylformamide, 2.80 g (10 mmol) of 4,4′-azobis-4-cyanovaleric acid was added, heated to 80 ° C., and stirred for 3 hours. After the azo decomposition was completed, 1,000 g of methanol / ultra pure water (mass ratio 5/5) was added to obtain a precipitated solid. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.0 g of a yellow polymer (c-4).
この重合体(c-4)は、Mwが8,400、Mnが6,200、Mw/Mnが1.35であった。また、1H-NMR分析の結果、スチレン及びメチルメタクリレートに由来する構造単位の含有割合が、それぞれ70モル%及び30モル%であった。
This polymer (c-4) had an Mw of 8,400, an Mn of 6,200, and an Mw / Mn of 1.35. As a result of 1 H-NMR analysis, the content ratios of structural units derived from styrene and methyl methacrylate were 70 mol% and 30 mol%, respectively.

[合成例1-16](重合体(c-5)の合成)
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン14.6g(0.140mol)、メタクリル酸メチル6.00g(0.060mol)、臭化銅(II)0.29g(2.00mmol)及びトリス[(2-ジメチルアミノ)エチル]アミン0.46g(2.0mmol)を加え、100℃に加熱し、2-ブロモイソ酪酸エチル0.53(3.6mmol)をシリンジにて加え、窒素フロー下、8時間加熱撹拌した。得られた重合反応液は、セライト濾過し銅錯体を除去し、超純水500gにて洗浄を3回繰り返した。有機層を回収した後、濃縮し、得られた濃縮物にTHF50gを加えたものをメタノール/超純水(質量比5/5)1,000gに加えて沈殿精製させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで白色の重合体(c-5)11.5gを得た。 [Synthesis Example 1-16] (Synthesis of Polymer (c-5))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 40 g of anisole, 14.6 g (0.140 mol) of styrene, 6.00 g (0.060 mol) of methyl methacrylate, 0.29 g of copper (II) bromide ( 2.00 mmol) and 0.46 g (2.0 mmol) of tris [(2-dimethylamino) ethyl] amine were added and heated to 100 ° C., and 0.53 (3.6 mmol) of ethyl 2-bromoisobutyrate was added with a syringe. In addition, the mixture was heated and stirred for 8 hours under a nitrogen flow. The obtained polymerization reaction liquid was filtered through Celite to remove the copper complex, and washing with 500 g of ultrapure water was repeated three times. After the organic layer was collected, it was concentrated, and a solid obtained by adding 50 g of THF to the resulting concentrate was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) and purified by precipitation. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.5 g of a white polymer (c-5).
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにアニソール40g、スチレン14.6g(0.140mol)、メタクリル酸メチル6.00g(0.060mol)、臭化銅(II)0.29g(2.00mmol)及びトリス[(2-ジメチルアミノ)エチル]アミン0.46g(2.0mmol)を加え、100℃に加熱し、2-ブロモイソ酪酸エチル0.53(3.6mmol)をシリンジにて加え、窒素フロー下、8時間加熱撹拌した。得られた重合反応液は、セライト濾過し銅錯体を除去し、超純水500gにて洗浄を3回繰り返した。有機層を回収した後、濃縮し、得られた濃縮物にTHF50gを加えたものをメタノール/超純水(質量比5/5)1,000gに加えて沈殿精製させた固体を得た。この固体をブフナーロートにて回収し、メタノール50gにて洗浄した。得られた固体を減圧乾燥することで白色の重合体(c-5)11.5gを得た。 [Synthesis Example 1-16] (Synthesis of Polymer (c-5))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 40 g of anisole, 14.6 g (0.140 mol) of styrene, 6.00 g (0.060 mol) of methyl methacrylate, 0.29 g of copper (II) bromide ( 2.00 mmol) and 0.46 g (2.0 mmol) of tris [(2-dimethylamino) ethyl] amine were added and heated to 100 ° C., and 0.53 (3.6 mmol) of ethyl 2-bromoisobutyrate was added with a syringe. In addition, the mixture was heated and stirred for 8 hours under a nitrogen flow. The obtained polymerization reaction liquid was filtered through Celite to remove the copper complex, and washing with 500 g of ultrapure water was repeated three times. After the organic layer was collected, it was concentrated, and a solid obtained by adding 50 g of THF to the resulting concentrate was added to 1,000 g of methanol / ultra pure water (mass ratio 5/5) and purified by precipitation. This solid was recovered with a Buchner funnel and washed with 50 g of methanol. The obtained solid was dried under reduced pressure to obtain 11.5 g of a white polymer (c-5).
この重合体(c-5)は、Mwが6,700、Mnが5,800、Mw/Mnが1.16であった。また、1H-NMR分析の結果、スチレン及びメチルメタクリレートに由来する構造単位が、それぞれ70モル%及び30モル%であった。
This polymer (c-5) had Mw of 6,700, Mn of 5,800, and Mw / Mn of 1.16. As a result of 1 H-NMR analysis, the structural units derived from styrene and methyl methacrylate were 70 mol% and 30 mol%, respectively.

<組成物の調製>
[組成物(I)の調製]
[調製例1-1]
上記合成した[A]重合体としての(a-1)1.5gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート69gと乳酸エチル29.5gとを加え、撹拌したのち、0.45μmの細孔を有する高密度ポリエチレンフィルターにて濾過し、組成物(A-1)を調製した。 <Preparation of composition>
[Preparation of Composition (I)]
[Preparation Example 1-1]
To 1.5 g of the synthesized [A] polymer (a-1), 69 g of propylene glycol monomethyl ether acetate and 29.5 g of ethyl lactate as the [S] solvent were added, stirred, and then 0.45 μm The mixture was filtered through a high-density polyethylene filter having pores to prepare composition (A-1).
[組成物(I)の調製]
[調製例1-1]
上記合成した[A]重合体としての(a-1)1.5gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート69gと乳酸エチル29.5gとを加え、撹拌したのち、0.45μmの細孔を有する高密度ポリエチレンフィルターにて濾過し、組成物(A-1)を調製した。 <Preparation of composition>
[Preparation of Composition (I)]
[Preparation Example 1-1]
To 1.5 g of the synthesized [A] polymer (a-1), 69 g of propylene glycol monomethyl ether acetate and 29.5 g of ethyl lactate as the [S] solvent were added, stirred, and then 0.45 μm The mixture was filtered through a high-density polyethylene filter having pores to prepare composition (A-1).
[調製例1-2及び1-3]
[A]重合体として、上記(a-1)の代わりに、同質量の(a-2)又は(a-3)を用いた以外は、調製例1-1と同様にして、組成物(A-2)及び(A-3)を調製した。 [Preparation Examples 1-2 and 1-3]
[A] In the same manner as in Preparation Example 1-1 except that the same mass of (a-2) or (a-3) was used instead of the above (a-1) as the polymer, the composition ( A-2) and (A-3) were prepared.
[A]重合体として、上記(a-1)の代わりに、同質量の(a-2)又は(a-3)を用いた以外は、調製例1-1と同様にして、組成物(A-2)及び(A-3)を調製した。 [Preparation Examples 1-2 and 1-3]
[A] In the same manner as in Preparation Example 1-1 except that the same mass of (a-2) or (a-3) was used instead of the above (a-1) as the polymer, the composition ( A-2) and (A-3) were prepared.
[組成物(II)の調製]
[調製例1-4]
上記合成した[B]重合体としての(b-1)1.2gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート98.8gを加え溶解させた後、0.45μmの細孔を有する高密度ポリスチレンフィルターにて濾過し、組成物(B-1)を調製した。 [Preparation of Composition (II)]
[Preparation Example 1-4]
After 98.8 g of propylene glycol monomethyl ether acetate as a [S] solvent was added to 1.2 g of (b-1) as the synthesized [B] polymer and dissolved, a high pore having 0.45 μm pores was obtained. The mixture was filtered through a density polystyrene filter to prepare a composition (B-1).
[調製例1-4]
上記合成した[B]重合体としての(b-1)1.2gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート98.8gを加え溶解させた後、0.45μmの細孔を有する高密度ポリスチレンフィルターにて濾過し、組成物(B-1)を調製した。 [Preparation of Composition (II)]
[Preparation Example 1-4]
After 98.8 g of propylene glycol monomethyl ether acetate as a [S] solvent was added to 1.2 g of (b-1) as the synthesized [B] polymer and dissolved, a high pore having 0.45 μm pores was obtained. The mixture was filtered through a density polystyrene filter to prepare a composition (B-1).
[調製例1-5~1-11]
[B]重合体として、上記(b-1)の代わりに、同質量の(b-2)~(b-8)を用いた以外は、調製例1-4と同様にして、組成物(B-2)~(B-8)を調製した。 [Preparation Examples 1-5 to 1-11]
[B] In the same manner as in Preparation Example 1-4 except that the same mass of (b-2) to (b-8) was used in place of (b-1) as a polymer, the composition ( B-2) to (B-8) were prepared.
[B]重合体として、上記(b-1)の代わりに、同質量の(b-2)~(b-8)を用いた以外は、調製例1-4と同様にして、組成物(B-2)~(B-8)を調製した。 [Preparation Examples 1-5 to 1-11]
[B] In the same manner as in Preparation Example 1-4 except that the same mass of (b-2) to (b-8) was used in place of (b-1) as a polymer, the composition ( B-2) to (B-8) were prepared.
[組成物(III)の調製]
[調製例1-12]
上記合成した[C]重合体としての(c-1)1.2gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート98.8gを加え溶解させた後、0.45μmの細孔を有する高密度ポリスチレンフィルターにて濾過し、組成物(C-1)を調製した。 [Preparation of Composition (III)]
[Preparation Example 1-12]
After 98.8 g of propylene glycol monomethyl ether acetate as the [S] solvent was added to 1.2 g of (c-1) as the synthesized [C] polymer and dissolved, a high pore having 0.45 μm pores was obtained. The mixture was filtered through a density polystyrene filter to prepare a composition (C-1).
[調製例1-12]
上記合成した[C]重合体としての(c-1)1.2gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート98.8gを加え溶解させた後、0.45μmの細孔を有する高密度ポリスチレンフィルターにて濾過し、組成物(C-1)を調製した。 [Preparation of Composition (III)]
[Preparation Example 1-12]
After 98.8 g of propylene glycol monomethyl ether acetate as the [S] solvent was added to 1.2 g of (c-1) as the synthesized [C] polymer and dissolved, a high pore having 0.45 μm pores was obtained. The mixture was filtered through a density polystyrene filter to prepare a composition (C-1).
[調製例1-13~1-16]
[C]重合体として、上記(c-1)の代わりに、同質量の(c-2)~(c-5)を用いた以外は、調製例1-12と同様にして、組成物(C-2)~(C-5)を調製した。 [Preparation Examples 1-13 to 1-16]
[C] In the same manner as in Preparation Example 1-12 except that the same mass of (c-2) to (c-5) was used instead of the above (c-1) as the polymer, the composition ( C-2) to (C-5) were prepared.
[C]重合体として、上記(c-1)の代わりに、同質量の(c-2)~(c-5)を用いた以外は、調製例1-12と同様にして、組成物(C-2)~(C-5)を調製した。 [Preparation Examples 1-13 to 1-16]
[C] In the same manner as in Preparation Example 1-12 except that the same mass of (c-2) to (c-5) was used instead of the above (c-1) as the polymer, the composition ( C-2) to (C-5) were prepared.
[組成物(IV)の調製]
[調製例1-17]
上記合成した[B]重合体としての(b-2)0.6g及び[C]重合体としての(c-5)0.6gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート98.8gを加え溶解させた後、0.45μmの細孔を有する高密度ポリスチレンフィルターにて濾過し、組成物(D-1)を調製した。 [Preparation of Composition (IV)]
[Preparation Example 1-17]
The above synthesized [B] polymer (b-2) 0.6 g and [C] polymer (c-5) 0.6 g were added to [S] solvent propylene glycol monomethyl ether acetate 98.8 g. Was added and dissolved, followed by filtration with a high-density polystyrene filter having pores of 0.45 μm to prepare a composition (D-1).
[調製例1-17]
上記合成した[B]重合体としての(b-2)0.6g及び[C]重合体としての(c-5)0.6gに、[S]溶媒としてのプロピレングリコールモノメチルエーテルアセテート98.8gを加え溶解させた後、0.45μmの細孔を有する高密度ポリスチレンフィルターにて濾過し、組成物(D-1)を調製した。 [Preparation of Composition (IV)]
[Preparation Example 1-17]
The above synthesized [B] polymer (b-2) 0.6 g and [C] polymer (c-5) 0.6 g were added to [S] solvent propylene glycol monomethyl ether acetate 98.8 g. Was added and dissolved, followed by filtration with a high-density polystyrene filter having pores of 0.45 μm to prepare a composition (D-1).
[調製例1-18~1-28]
下記表5に示す各重合体を用いた以外は、調製例1-17と同様にして、組成物(D-2)~(D-12)を調製した。 [Preparation Examples 1-18 to 1-28]
Compositions (D-2) to (D-12) were prepared in the same manner as in Preparation Example 1-17, except that the respective polymers shown in Table 5 were used.
下記表5に示す各重合体を用いた以外は、調製例1-17と同様にして、組成物(D-2)~(D-12)を調製した。 [Preparation Examples 1-18 to 1-28]
Compositions (D-2) to (D-12) were prepared in the same manner as in Preparation Example 1-17, except that the respective polymers shown in Table 5 were used.
<レジスト組成物の調製>
[重合体の合成]
重合体(a1-1)及び(a2-1)の合成に用いた単量体化合物を以下に示す。 <Preparation of resist composition>
[Synthesis of polymer]
The monomer compounds used for the synthesis of the polymers (a1-1) and (a2-1) are shown below.
[重合体の合成]
重合体(a1-1)及び(a2-1)の合成に用いた単量体化合物を以下に示す。 <Preparation of resist composition>
[Synthesis of polymer]
The monomer compounds used for the synthesis of the polymers (a1-1) and (a2-1) are shown below.

[合成例1-17](重合体(a1-1)の合成)
上記化合物(M-1)12.9g(50モル%)及び化合物(M-2)17.1g(50モル%)を、メチルエチルケトン60gに溶解し、さらに2,2’-アゾビスイソブチロニトリル(AIBN)1.77gを投入して溶解させ、単量体溶液を調製した。次に、30gのメチルエチルケトンを投入した200mLの三口フラスコを30分窒素パージした後、反応釜を攪拌しながら80℃に加熱して、上記単量体溶液を、滴下漏斗を用いて3時間かけて滴下した。滴下開始を重合開始時間とし、重合反応を6時間実施した。重合反応終了後、重合反応液を水冷し30℃以下に冷却し、600gのメタノールへ投入して、析出した白色粉末を濾別した。濾別した白色粉末をそれぞれ150gのメタノールを用い、2回、スラリー状にして洗浄した後、再度濾別し、50℃にて17時間乾燥して白色粉末の重合体(a1-1)を得た(収率80%)。重合体(a1-1)のMwは6,900、Mw/Mnは1.35であった。また、13C-NMR分析の結果、重合体(a1-1)における(M-1)及び(M-2)に由来する構造単位の含有割合は、それぞれ49モル%及び51モル%であった。 [Synthesis Example 1-17] (Synthesis of Polymer (a1-1))
12.9 g (50 mol%) of the compound (M-1) and 17.1 g (50 mol%) of the compound (M-2) were dissolved in 60 g of methyl ethyl ketone, and 2,2′-azobisisobutyronitrile was further dissolved. (AIBN) 1.77 g was added and dissolved to prepare a monomer solution. Next, a 200 mL three-necked flask charged with 30 g of methyl ethyl ketone was purged with nitrogen for 30 minutes, and then the reaction kettle was heated to 80 ° C. with stirring, and the monomer solution was added over 3 hours using a dropping funnel. It was dripped. The polymerization start was carried out for 6 hours with the start of dropping as the polymerization start time. After completion of the polymerization reaction, the polymerization reaction solution was cooled with water, cooled to 30 ° C. or lower, poured into 600 g of methanol, and the precipitated white powder was separated by filtration. The white powder separated by filtration was washed with 150 g of methanol twice in a slurry state, then filtered again and dried at 50 ° C. for 17 hours to obtain a white powder polymer (a1-1). (Yield 80%). Mw of the polymer (a1-1) was 6,900, and Mw / Mn was 1.35. As a result of 13 C-NMR analysis, the content ratios of structural units derived from (M-1) and (M-2) in the polymer (a1-1) were 49 mol% and 51 mol%, respectively. .
上記化合物(M-1)12.9g(50モル%)及び化合物(M-2)17.1g(50モル%)を、メチルエチルケトン60gに溶解し、さらに2,2’-アゾビスイソブチロニトリル(AIBN)1.77gを投入して溶解させ、単量体溶液を調製した。次に、30gのメチルエチルケトンを投入した200mLの三口フラスコを30分窒素パージした後、反応釜を攪拌しながら80℃に加熱して、上記単量体溶液を、滴下漏斗を用いて3時間かけて滴下した。滴下開始を重合開始時間とし、重合反応を6時間実施した。重合反応終了後、重合反応液を水冷し30℃以下に冷却し、600gのメタノールへ投入して、析出した白色粉末を濾別した。濾別した白色粉末をそれぞれ150gのメタノールを用い、2回、スラリー状にして洗浄した後、再度濾別し、50℃にて17時間乾燥して白色粉末の重合体(a1-1)を得た(収率80%)。重合体(a1-1)のMwは6,900、Mw/Mnは1.35であった。また、13C-NMR分析の結果、重合体(a1-1)における(M-1)及び(M-2)に由来する構造単位の含有割合は、それぞれ49モル%及び51モル%であった。 [Synthesis Example 1-17] (Synthesis of Polymer (a1-1))
12.9 g (50 mol%) of the compound (M-1) and 17.1 g (50 mol%) of the compound (M-2) were dissolved in 60 g of methyl ethyl ketone, and 2,2′-azobisisobutyronitrile was further dissolved. (AIBN) 1.77 g was added and dissolved to prepare a monomer solution. Next, a 200 mL three-necked flask charged with 30 g of methyl ethyl ketone was purged with nitrogen for 30 minutes, and then the reaction kettle was heated to 80 ° C. with stirring, and the monomer solution was added over 3 hours using a dropping funnel. It was dripped. The polymerization start was carried out for 6 hours with the start of dropping as the polymerization start time. After completion of the polymerization reaction, the polymerization reaction solution was cooled with water, cooled to 30 ° C. or lower, poured into 600 g of methanol, and the precipitated white powder was separated by filtration. The white powder separated by filtration was washed with 150 g of methanol twice in a slurry state, then filtered again and dried at 50 ° C. for 17 hours to obtain a white powder polymer (a1-1). (Yield 80%). Mw of the polymer (a1-1) was 6,900, and Mw / Mn was 1.35. As a result of 13 C-NMR analysis, the content ratios of structural units derived from (M-1) and (M-2) in the polymer (a1-1) were 49 mol% and 51 mol%, respectively. .
[合成例1-18](重合体(a2-1)の合成)
上記化合物(M-3)10.4g(30モル%)及び化合物(M-4)19.6g(70モル%)を、メチルエチルケトン60gに溶解し、さらに2,2’-アゾビス(イソブチロニトリル)0.91g(化合物の合計に対して5モル%)を投入した単量体溶液を調製した。次に、30gのメチルエチルケトンを投入した200mLの三口フラスコを30分窒素パージした後、反応釜を攪拌しながら80℃に加熱し、上記単量体溶液を、滴下漏斗を用いて3時間かけて滴下した。滴下開始を重合開始時間とし、重合反応を6時間実施した。重合反応終了後、重合反応液を水冷し30℃以下に冷却し、600gのメタノールへ投入して、析出した白色粉末を濾別した。濾別した白色粉末を150gのメタノールを用い、2回、スラリー状で洗浄した後、再度濾別し、50℃にて12時間乾燥して白色粉末の重合体(a2-1)を得た(収率68%)。重合体(a2-1)のMwは5,900、Mw/Mnは1.58であった。また、13C-NMR分析の結果、重合体(a2-1)における(M-3)及び(M-4)に由来する構造単位の含有割合は、それぞれ31モル%及び69モル%であった。 [Synthesis Example 1-18] (Synthesis of Polymer (a2-1))
10.4 g (30 mol%) of the compound (M-3) and 19.6 g (70 mol%) of the compound (M-4) are dissolved in 60 g of methyl ethyl ketone, and 2,2′-azobis (isobutyronitrile) is further dissolved. A monomer solution charged with 0.91 g (5 mol% based on the total amount of compounds) was prepared. Next, a 200 mL three-necked flask charged with 30 g of methyl ethyl ketone was purged with nitrogen for 30 minutes, and then the reaction kettle was heated to 80 ° C. with stirring, and the monomer solution was added dropwise over 3 hours using a dropping funnel. did. The polymerization start was carried out for 6 hours with the start of dropping as the polymerization start time. After completion of the polymerization reaction, the polymerization reaction solution was cooled with water, cooled to 30 ° C. or lower, poured into 600 g of methanol, and the precipitated white powder was separated by filtration. The white powder separated by filtration was washed twice with 150 g of methanol, and then filtered again and dried at 50 ° C. for 12 hours to obtain a white powder polymer (a2-1) ( Yield 68%). Mw of the polymer (a2-1) was 5,900, and Mw / Mn was 1.58. As a result of 13 C-NMR analysis, the content ratios of structural units derived from (M-3) and (M-4) in the polymer (a2-1) were 31 mol% and 69 mol%, respectively. .
上記化合物(M-3)10.4g(30モル%)及び化合物(M-4)19.6g(70モル%)を、メチルエチルケトン60gに溶解し、さらに2,2’-アゾビス(イソブチロニトリル)0.91g(化合物の合計に対して5モル%)を投入した単量体溶液を調製した。次に、30gのメチルエチルケトンを投入した200mLの三口フラスコを30分窒素パージした後、反応釜を攪拌しながら80℃に加熱し、上記単量体溶液を、滴下漏斗を用いて3時間かけて滴下した。滴下開始を重合開始時間とし、重合反応を6時間実施した。重合反応終了後、重合反応液を水冷し30℃以下に冷却し、600gのメタノールへ投入して、析出した白色粉末を濾別した。濾別した白色粉末を150gのメタノールを用い、2回、スラリー状で洗浄した後、再度濾別し、50℃にて12時間乾燥して白色粉末の重合体(a2-1)を得た(収率68%)。重合体(a2-1)のMwは5,900、Mw/Mnは1.58であった。また、13C-NMR分析の結果、重合体(a2-1)における(M-3)及び(M-4)に由来する構造単位の含有割合は、それぞれ31モル%及び69モル%であった。 [Synthesis Example 1-18] (Synthesis of Polymer (a2-1))
10.4 g (30 mol%) of the compound (M-3) and 19.6 g (70 mol%) of the compound (M-4) are dissolved in 60 g of methyl ethyl ketone, and 2,2′-azobis (isobutyronitrile) is further dissolved. A monomer solution charged with 0.91 g (5 mol% based on the total amount of compounds) was prepared. Next, a 200 mL three-necked flask charged with 30 g of methyl ethyl ketone was purged with nitrogen for 30 minutes, and then the reaction kettle was heated to 80 ° C. with stirring, and the monomer solution was added dropwise over 3 hours using a dropping funnel. did. The polymerization start was carried out for 6 hours with the start of dropping as the polymerization start time. After completion of the polymerization reaction, the polymerization reaction solution was cooled with water, cooled to 30 ° C. or lower, poured into 600 g of methanol, and the precipitated white powder was separated by filtration. The white powder separated by filtration was washed twice with 150 g of methanol, and then filtered again and dried at 50 ° C. for 12 hours to obtain a white powder polymer (a2-1) ( Yield 68%). Mw of the polymer (a2-1) was 5,900, and Mw / Mn was 1.58. As a result of 13 C-NMR analysis, the content ratios of structural units derived from (M-3) and (M-4) in the polymer (a2-1) were 31 mol% and 69 mol%, respectively. .
[調製例1-29](レジスト組成物(J-1)の調製)
上記合成した重合体(a1-1)100質量部及び重合体(a2-1)3質量部、感放射線性酸発生体としてのトリフェニルスルホニウム1,1,2,2-テトラフルオロ-6-(1-アダマンタンカルボニロキシ)ヘキサン-1-スルホネート10.8質量部、酸拡散制御剤としてのトリフェニルスルホニウムサリチレート4.3質量部、並びに溶媒としてのプロピレングリコールモノメチルエーテルアセテート2,185質量部、シクロヘキサノン935質量部及びγ-ブチロラクトン30質量部を混合してレジスト組成物(J-1)を調製した。 [Preparation Example 1-29] (Preparation of resist composition (J-1))
100 parts by mass of the polymer (a1-1) synthesized above and 3 parts by mass of the polymer (a2-1), triphenylsulfonium 1,1,2,2-tetrafluoro-6- ( 1-adamantane carbonyloxy) hexane-1-sulfonate 10.8 parts by weight, 4.3 parts by weight of triphenylsulfonium salicylate as an acid diffusion controller, and 2,185 parts by weight of propylene glycol monomethyl ether acetate as a solvent Then, 935 parts by mass of cyclohexanone and 30 parts by mass of γ-butyrolactone were mixed to prepare a resist composition (J-1).
上記合成した重合体(a1-1)100質量部及び重合体(a2-1)3質量部、感放射線性酸発生体としてのトリフェニルスルホニウム1,1,2,2-テトラフルオロ-6-(1-アダマンタンカルボニロキシ)ヘキサン-1-スルホネート10.8質量部、酸拡散制御剤としてのトリフェニルスルホニウムサリチレート4.3質量部、並びに溶媒としてのプロピレングリコールモノメチルエーテルアセテート2,185質量部、シクロヘキサノン935質量部及びγ-ブチロラクトン30質量部を混合してレジスト組成物(J-1)を調製した。 [Preparation Example 1-29] (Preparation of resist composition (J-1))
100 parts by mass of the polymer (a1-1) synthesized above and 3 parts by mass of the polymer (a2-1),
<基礎パターンの形成>
[基礎パターンの形成(1)]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に下記表1に示す組成物(III)(組成物(C-1)~(C-5))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板をプロピレングリコールモノメチルエーテルアセテート(PGMEA)を用いて4秒間リンスすることにより、未反応物等の除去を行った。次いで、未反応物等除去後の基板に上記調製したレジスト組成物(J-1)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、酢酸ブチルを用いて現像し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 <Formation of basic pattern>
[Formation of basic pattern (1)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. The obtained substrate was spin-coated with the composition (III) (compositions (C-1) to (C-5)) shown in Table 1 below at 1,500 rpm and baked at 220 ° C. for 60 seconds. The substrate after baking was rinsed with propylene glycol monomethyl ether acetate (PGMEA) for 4 seconds to remove unreacted substances and the like. Next, the resist composition (J-1) prepared above is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, exposed to ArF immersion exposure, developed using butyl acetate, A resist hole pattern having a size of 60 nm and a pitch of 150 nm was formed.
[基礎パターンの形成(1)]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に下記表1に示す組成物(III)(組成物(C-1)~(C-5))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板をプロピレングリコールモノメチルエーテルアセテート(PGMEA)を用いて4秒間リンスすることにより、未反応物等の除去を行った。次いで、未反応物等除去後の基板に上記調製したレジスト組成物(J-1)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、酢酸ブチルを用いて現像し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 <Formation of basic pattern>
[Formation of basic pattern (1)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. The obtained substrate was spin-coated with the composition (III) (compositions (C-1) to (C-5)) shown in Table 1 below at 1,500 rpm and baked at 220 ° C. for 60 seconds. The substrate after baking was rinsed with propylene glycol monomethyl ether acetate (PGMEA) for 4 seconds to remove unreacted substances and the like. Next, the resist composition (J-1) prepared above is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, exposed to ArF immersion exposure, developed using butyl acetate, A resist hole pattern having a size of 60 nm and a pitch of 150 nm was formed.
得られたレジストホールパターンに対して、下記表1に示す組成物(II)(組成物(B-1)~(B-8))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板をプロピレングリコールモノメチルエーテルアセテート(PGMEA)を用いて4秒間リンスすることにより、未反応物等の除去を行い、基礎パターン(P-1)~(P-12)を形成した。
The obtained resist hole pattern was spin-coated with the composition (II) (compositions (B-1) to (B-8)) shown in Table 1 below at 1,500 rpm, and heated at 220 ° C. for 60 seconds. Baked. The substrate after firing was rinsed with propylene glycol monomethyl ether acetate (PGMEA) for 4 seconds to remove unreacted substances and the like, and basic patterns (P-1) to (P-12) were formed.
基礎パターン(P-13)は、上記基礎パターンの形成(1)において、上記組成物(III)として組成物(B-2)を用い、上記組成物(II)として組成物(C-1)を用いることにより形成した。
基礎パターン(P-14)は、上記基礎パターンの形成(1)において、上記組成物(III)及び上記組成物(II)としていずれも組成物(B-2)を用いることにより形成した。
基礎パターン(P-15)は、上記基礎パターンの形成(1)において、上記組成物(III)及び上記組成物(II)としていずれも組成物(C-1)を用いることにより形成した。
基礎パターン(P-16)は、上記基礎パターンの形成(1)において、上記組成物(III)及び上記組成物(II)の塗布を行わないで形成した。(表1中の「-」は、組成物(II)及び(III)の塗布を行わなかったことを示す。) In the basic pattern (P-13), in the formation of the basic pattern (1), the composition (B-2) is used as the composition (III), and the composition (C-1) is used as the composition (II). It was formed by using.
The basic pattern (P-14) was formed by using the composition (B-2) as the composition (III) and the composition (II) in the formation of the basic pattern (1).
The basic pattern (P-15) was formed by using the composition (C-1) as the composition (III) and the composition (II) in the basic pattern formation (1).
The basic pattern (P-16) was formed without applying the composition (III) and the composition (II) in the formation of the basic pattern (1). ("-" In Table 1 indicates that compositions (II) and (III) were not applied.)
基礎パターン(P-14)は、上記基礎パターンの形成(1)において、上記組成物(III)及び上記組成物(II)としていずれも組成物(B-2)を用いることにより形成した。
基礎パターン(P-15)は、上記基礎パターンの形成(1)において、上記組成物(III)及び上記組成物(II)としていずれも組成物(C-1)を用いることにより形成した。
基礎パターン(P-16)は、上記基礎パターンの形成(1)において、上記組成物(III)及び上記組成物(II)の塗布を行わないで形成した。(表1中の「-」は、組成物(II)及び(III)の塗布を行わなかったことを示す。) In the basic pattern (P-13), in the formation of the basic pattern (1), the composition (B-2) is used as the composition (III), and the composition (C-1) is used as the composition (II). It was formed by using.
The basic pattern (P-14) was formed by using the composition (B-2) as the composition (III) and the composition (II) in the formation of the basic pattern (1).
The basic pattern (P-15) was formed by using the composition (C-1) as the composition (III) and the composition (II) in the basic pattern formation (1).
The basic pattern (P-16) was formed without applying the composition (III) and the composition (II) in the formation of the basic pattern (1). ("-" In Table 1 indicates that compositions (II) and (III) were not applied.)
<評価>
得られた基礎パターン(P-1)~(P-16)の凹部の底面及び側面における水の静的接触角(単位:°)及び塗工膜の平均厚み(単位:nm)を、接触角計(KRUSS社の「DSA10L2E」)及び高速分光エリプソメーター(ジェー・エー・ウーラム・ジャパン社の「M2000」)を用い、基礎パターンと同様の表面状態とした塗工膜を用いて代替評価により測定した。評価結果を下記表1に合わせて示す。 <Evaluation>
The static contact angle (unit: °) of water and the average thickness (unit: nm) of the coating film on the bottom and side surfaces of the recesses of the obtained basic patterns (P-1) to (P-16) are expressed as the contact angle. Measured by alternative evaluation using a coating film with the same surface condition as the basic pattern using a meter (“DSA10L2E” from KRUSS) and a high-speed spectroscopic ellipsometer (“M2000” from JA Woollam Japan) did. The evaluation results are shown in Table 1 below.
得られた基礎パターン(P-1)~(P-16)の凹部の底面及び側面における水の静的接触角(単位:°)及び塗工膜の平均厚み(単位:nm)を、接触角計(KRUSS社の「DSA10L2E」)及び高速分光エリプソメーター(ジェー・エー・ウーラム・ジャパン社の「M2000」)を用い、基礎パターンと同様の表面状態とした塗工膜を用いて代替評価により測定した。評価結果を下記表1に合わせて示す。 <Evaluation>
The static contact angle (unit: °) of water and the average thickness (unit: nm) of the coating film on the bottom and side surfaces of the recesses of the obtained basic patterns (P-1) to (P-16) are expressed as the contact angle. Measured by alternative evaluation using a coating film with the same surface condition as the basic pattern using a meter (“DSA10L2E” from KRUSS) and a high-speed spectroscopic ellipsometer (“M2000” from JA Woollam Japan) did. The evaluation results are shown in Table 1 below.

[基礎パターンの形成(2)]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に下記表2に示す組成物(III)(組成物(C-1)又は(C-2))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板を、PGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行った。次いで、未反応物等除去後の基板にレジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、2.38質量%テトラブチルアンモニウムヒドロキシド水溶液を用いて現像し、180℃で焼成し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (2)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. The obtained substrate was spin-coated with the composition (III) (composition (C-1) or (C-2)) shown in Table 2 below at 1,500 rpm and baked at 220 ° C. for 60 seconds. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like. Next, a resist composition (“AIM5484JN” from JSR) is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, ArF immersion exposure, and 2.38 mass% tetrabutylammonium hydroxy The resist hole pattern was developed with an aqueous solution and baked at 180 ° C. to form a hole pattern with a hole size of 60 nm and a pitch of 150 nm.
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に下記表2に示す組成物(III)(組成物(C-1)又は(C-2))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板を、PGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行った。次いで、未反応物等除去後の基板にレジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、2.38質量%テトラブチルアンモニウムヒドロキシド水溶液を用いて現像し、180℃で焼成し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (2)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. The obtained substrate was spin-coated with the composition (III) (composition (C-1) or (C-2)) shown in Table 2 below at 1,500 rpm and baked at 220 ° C. for 60 seconds. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like. Next, a resist composition (“AIM5484JN” from JSR) is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, ArF immersion exposure, and 2.38 mass% tetrabutylammonium hydroxy The resist hole pattern was developed with an aqueous solution and baked at 180 ° C. to form a hole pattern with a hole size of 60 nm and a pitch of 150 nm.
得られたレジストホールパターンに対して、下記表2に示す組成物(II)(組成物(B-4))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板を、PGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行い、基礎パターン(P-17)及び(P-18)を形成した。
The obtained resist hole pattern was spin-coated with a composition (II) (composition (B-4)) shown in Table 2 below at 1,500 rpm and baked at 220 ° C. for 60 seconds. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like, and basic patterns (P-17) and (P-18) were formed.
上記形成した基礎パターン(P-17)及び(P-18)の凹部の底面及び側面における水の静的接触角及び塗工膜の平均厚みを、上記同様の方法に従い測定した。評価結果を下記表2に合わせて示す。
The static contact angle of water and the average thickness of the coating film on the bottom and side surfaces of the recesses of the basic patterns (P-17) and (P-18) formed above were measured according to the same method as described above. The evaluation results are shown in Table 2 below.

[基礎パターンの形成(3)]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に下記表3に示す組成物(III)(組成物(C-1))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板を、PGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行った。次いで、未反応物等除去後の基板にレジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、2.38質量%テトラブチルアンモニウムヒドロキシド水溶液を用いて現像し、ArF全面露光し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (3)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. The obtained substrate was spin-coated with the composition (III) (composition (C-1)) shown in Table 3 below at 1,500 rpm, and baked at 220 ° C. for 60 seconds. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like. Next, a resist composition (“AIM5484JN” from JSR) is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, ArF immersion exposure, and 2.38 mass% tetrabutylammonium hydroxy Development was performed using an aqueous solution of AlF, and the entire surface of ArF was exposed to form a resist hole pattern having a hole size of 60 nm and a pitch of 150 nm.
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に下記表3に示す組成物(III)(組成物(C-1))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板を、PGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行った。次いで、未反応物等除去後の基板にレジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、2.38質量%テトラブチルアンモニウムヒドロキシド水溶液を用いて現像し、ArF全面露光し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (3)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. The obtained substrate was spin-coated with the composition (III) (composition (C-1)) shown in Table 3 below at 1,500 rpm, and baked at 220 ° C. for 60 seconds. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like. Next, a resist composition (“AIM5484JN” from JSR) is applied to the substrate after removal of unreacted substances, etc. to form a resist film of 85 nm, ArF immersion exposure, and 2.38 mass% tetrabutylammonium hydroxy Development was performed using an aqueous solution of AlF, and the entire surface of ArF was exposed to form a resist hole pattern having a hole size of 60 nm and a pitch of 150 nm.
得られたレジストホールパターンに対して、下記表3に示す組成物(II)(組成物(B-))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板を、PGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行い、基礎パターン(P-19)及び(P-20)を形成した。
The obtained resist hole pattern was spin-coated with a composition (II) (composition (B-)) shown in Table 3 below at 1,500 rpm and baked at 220 ° C. for 60 seconds. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like, and basic patterns (P-19) and (P-20) were formed.
上記形成した基礎パターン(P-19)及び(P-20)の凹部の底面及び側面における水の静的接触角及び塗工膜の平均厚みを、上記同様の方法に従い測定した。評価結果を下記表3に合わせて示す。
The static contact angle of water and the average thickness of the coating film on the bottom and side surfaces of the recesses of the formed basic patterns (P-19) and (P-20) were measured according to the same method as described above. The evaluation results are shown in Table 3 below.

[基礎パターンの形成(4)]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に上記調製したレジスト組成物(J-1)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、酢酸ブチルを用いて現像し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (4)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. An 85 nm resist film is formed by applying the prepared resist composition (J-1) to the obtained substrate, exposed to ArF immersion, developed using butyl acetate, and has a hole size of 60 nm and a pitch of 150 nm. A resist hole pattern was formed.
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に上記調製したレジスト組成物(J-1)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、酢酸ブチルを用いて現像し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (4)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. An 85 nm resist film is formed by applying the prepared resist composition (J-1) to the obtained substrate, exposed to ArF immersion, developed using butyl acetate, and has a hole size of 60 nm and a pitch of 150 nm. A resist hole pattern was formed.
得られたレジストホールパターンに対して、下記表4に示す組成物(II)(組成物((B-1)~(B-8))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板をPGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行った。次いで、下記表4に示す組成物(III)(組成物(C-1)~(C-5))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板をPGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行い、基礎パターン(P-21)~(P-32)を形成した。
The obtained resist hole pattern was spin-coated at 1,500 rpm with the composition (II) (compositions ((B-1) to (B-8)) shown in Table 4 below, The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted materials, etc. Next, the composition (III) shown in Table 4 below (composition (C-1)) To (C-5)) were spin-coated at 1,500 rpm and baked for 60 seconds at 220 ° C. The substrate after baking was rinsed for 4 seconds using PGMEA to remove unreacted substances, etc. Base patterns (P-21) to (P-32) were formed.
基礎パターン(P-33)は、上記基礎パターンの形成(4)において、上記組成物(II)として組成物(C-1)を用い、上記組成物(III)として組成物(B-2)を用いることにより形成した。
In the basic pattern (P-33), in the formation of the basic pattern (4), the composition (C-1) is used as the composition (II), and the composition (B-2) is used as the composition (III). It was formed by using.
上記形成した基礎パターン(P-21)~(P-33)の凹部の底面及び側面における水の静的接触角及び塗工膜の平均厚みを、上記同様の方法に従い測定した。評価結果を下記表4に合わせて示す。
The static contact angle of water and the average thickness of the coating film on the bottom and side surfaces of the recesses of the basic patterns (P-21) to (P-33) formed above were measured according to the same method as described above. The evaluation results are shown in Table 4 below.

[基礎パターンの形成(5)]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に上記調製したレジスト組成物(J-1)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、酢酸ブチルを用いて現像し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (5)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. An 85 nm resist film is formed by applying the prepared resist composition (J-1) to the obtained substrate, exposed to ArF immersion, developed using butyl acetate, and has a hole size of 60 nm and a pitch of 150 nm. A resist hole pattern was formed.
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み85nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板に上記調製したレジスト組成物(J-1)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、酢酸ブチルを用いて現像し、ホールサイズ60nm、ピッチ150nmのレジストホールパターンを形成した。 [Formation of basic pattern (5)]
On the Bare-Si substrate, an underlayer film having an average thickness of 85 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. An 85 nm resist film is formed by applying the prepared resist composition (J-1) to the obtained substrate, exposed to ArF immersion, developed using butyl acetate, and has a hole size of 60 nm and a pitch of 150 nm. A resist hole pattern was formed.
得られたレジストホールパターンに対して、下記表5に示す組成物(IV)(組成物(D-1)~(D-12))を1,500rpmにてスピンコートし、220℃で60秒間焼成した。焼成後の基板をPGMEAを用いて4秒間リンスすることにより、未反応物等の除去を行い、基礎パターン(P-34)~(P-45)を形成した。
The obtained resist hole pattern was spin-coated with the composition (IV) (compositions (D-1) to (D-12)) shown in Table 5 below at 1,500 rpm, and heated at 220 ° C. for 60 seconds. Baked. The substrate after firing was rinsed with PGMEA for 4 seconds to remove unreacted substances and the like, and basic patterns (P-34) to (P-45) were formed.
基礎パターン(P-46)は、上記基礎パターンの形成(5)において、上記組成物(IV)として組成物(B-2)を用いることにより形成した。
基礎パターン(P-47)は、上記基礎パターンの形成(5)において、上記組成物(IV)として組成物(C-1)を用いることにより形成した。
表5中の「-」は、用いた組成物が[B]重合体及び[C]重合体の両方を含有するものではないことを示す。 The basic pattern (P-46) was formed by using the composition (B-2) as the composition (IV) in the formation of the basic pattern (5).
The basic pattern (P-47) was formed by using the composition (C-1) as the composition (IV) in the basic pattern formation (5).
“-” In Table 5 indicates that the composition used does not contain both the [B] polymer and the [C] polymer.
基礎パターン(P-47)は、上記基礎パターンの形成(5)において、上記組成物(IV)として組成物(C-1)を用いることにより形成した。
表5中の「-」は、用いた組成物が[B]重合体及び[C]重合体の両方を含有するものではないことを示す。 The basic pattern (P-46) was formed by using the composition (B-2) as the composition (IV) in the formation of the basic pattern (5).
The basic pattern (P-47) was formed by using the composition (C-1) as the composition (IV) in the basic pattern formation (5).
“-” In Table 5 indicates that the composition used does not contain both the [B] polymer and the [C] polymer.
上記形成した基礎パターン(P-34)~(P-47)の凹部の底面及び側面における水の静的接触角及び塗工膜の平均厚みを、上記同様の方法に従い測定した。評価結果を下記表5に合わせて示す。
The static contact angle of water and the average thickness of the coating film on the bottom and side surfaces of the recesses of the basic patterns (P-34) to (P-47) formed above were measured according to the same method as described above. The evaluation results are shown in Table 5 below.

<微細化パターンの形成>
[実施例1-1~1-54及び比較例1-1~1-5]
上記形成した基礎パターン(P-1)~(P-47)を用い、以下の手順により充填工程、相分離工程及び除去工程を行うことにより微細化コンタクトホールパターンを形成した。 <Formation of fine pattern>
[Examples 1-1 to 1-54 and Comparative Examples 1-1 to 1-5]
Using the basic patterns (P-1) to (P-47) formed above, a miniaturized contact hole pattern was formed by performing a filling step, a phase separation step and a removal step according to the following procedure.
[実施例1-1~1-54及び比較例1-1~1-5]
上記形成した基礎パターン(P-1)~(P-47)を用い、以下の手順により充填工程、相分離工程及び除去工程を行うことにより微細化コンタクトホールパターンを形成した。 <Formation of fine pattern>
[Examples 1-1 to 1-54 and Comparative Examples 1-1 to 1-5]
Using the basic patterns (P-1) to (P-47) formed above, a miniaturized contact hole pattern was formed by performing a filling step, a phase separation step and a removal step according to the following procedure.
(充填工程及び相分離工程)
上記形成した基礎パターン(P-1)~(P-47)に対し、上記調製した組成物(I)(組成物(A-1)~(A-3))を1,500rpmにてスピンコートにより塗工した。この基板を、窒素下、220℃で60秒間、熱アニールすることで相分離させた。 (Filling process and phase separation process)
The prepared basic patterns (P-1) to (P-47) are spin-coated with the composition (I) prepared above (compositions (A-1) to (A-3)) at 1,500 rpm. It was coated by. The substrate was phase-separated by thermal annealing at 220 ° C. for 60 seconds under nitrogen.
上記形成した基礎パターン(P-1)~(P-47)に対し、上記調製した組成物(I)(組成物(A-1)~(A-3))を1,500rpmにてスピンコートにより塗工した。この基板を、窒素下、220℃で60秒間、熱アニールすることで相分離させた。 (Filling process and phase separation process)
The prepared basic patterns (P-1) to (P-47) are spin-coated with the composition (I) prepared above (compositions (A-1) to (A-3)) at 1,500 rpm. It was coated by. The substrate was phase-separated by thermal annealing at 220 ° C. for 60 seconds under nitrogen.
(除去工程)
上記相分離工程後の基板について、172nmの放射線を300mJ/cm2の強度で照射した後、メチルイソブチルケトン(MIBK)/2-プロパノール(IPA)(2/8(質量比))の混合液中に5分間浸漬させて、重合体(A-1)~(A-3)におけるポリ(メタクリル酸メチル)ブロックからなる相を溶解させて除去することにより、微細化コンタクトホールパターンを形成した。 (Removal process)
The substrate after the above phase separation step was irradiated with 172 nm radiation at an intensity of 300 mJ / cm 2 , and then in a mixed solution of methyl isobutyl ketone (MIBK) / 2-propanol (IPA) (2/8 (mass ratio)). For 5 minutes to dissolve and remove the phase composed of the poly (methyl methacrylate) block in the polymers (A-1) to (A-3), thereby forming a miniaturized contact hole pattern.
上記相分離工程後の基板について、172nmの放射線を300mJ/cm2の強度で照射した後、メチルイソブチルケトン(MIBK)/2-プロパノール(IPA)(2/8(質量比))の混合液中に5分間浸漬させて、重合体(A-1)~(A-3)におけるポリ(メタクリル酸メチル)ブロックからなる相を溶解させて除去することにより、微細化コンタクトホールパターンを形成した。 (Removal process)
The substrate after the above phase separation step was irradiated with 172 nm radiation at an intensity of 300 mJ / cm 2 , and then in a mixed solution of methyl isobutyl ketone (MIBK) / 2-propanol (IPA) (2/8 (mass ratio)). For 5 minutes to dissolve and remove the phase composed of the poly (methyl methacrylate) block in the polymers (A-1) to (A-3), thereby forming a miniaturized contact hole pattern.
<評価>
上記形成した微細化コンタクトホールパターンについて、走査型電子顕微鏡(日立ハイテクノロジー社の「CG4000」)を用いて高倍率(300K)の画像を取得し、計算ツール(日立ハイテクノロジー社)を用いて周期解析を行うことにより、コンタクトホールパターンの平均直径(単位:nm)及びプレイスメントエラー(x方向及びy方向、単位:nm)を評価した。
プレイスメントエラー(x方向、y方向)は、3.5nm以下の場合は良好と、3.5nmを超える場合は不良と評価できる。 <Evaluation>
About the formed miniaturized contact hole pattern, a high-magnification (300K) image is acquired using a scanning electron microscope (“CG4000” of Hitachi High-Technology Co., Ltd.), and a periodicity using a calculation tool (Hitachi High-Technology Co., Ltd.). By analyzing, the average diameter (unit: nm) and the placement error (x direction and y direction, unit: nm) of the contact hole pattern were evaluated.
The placement error (x direction, y direction) can be evaluated as good when it is 3.5 nm or less, and poor when it exceeds 3.5 nm.
上記形成した微細化コンタクトホールパターンについて、走査型電子顕微鏡(日立ハイテクノロジー社の「CG4000」)を用いて高倍率(300K)の画像を取得し、計算ツール(日立ハイテクノロジー社)を用いて周期解析を行うことにより、コンタクトホールパターンの平均直径(単位:nm)及びプレイスメントエラー(x方向及びy方向、単位:nm)を評価した。
プレイスメントエラー(x方向、y方向)は、3.5nm以下の場合は良好と、3.5nmを超える場合は不良と評価できる。 <Evaluation>
About the formed miniaturized contact hole pattern, a high-magnification (300K) image is acquired using a scanning electron microscope (“CG4000” of Hitachi High-Technology Co., Ltd.), and a periodicity using a calculation tool (Hitachi High-Technology Co., Ltd.). By analyzing, the average diameter (unit: nm) and the placement error (x direction and y direction, unit: nm) of the contact hole pattern were evaluated.
The placement error (x direction, y direction) can be evaluated as good when it is 3.5 nm or less, and poor when it exceeds 3.5 nm.
また、上記現像後の基板上に、感光性SOG組成物(JSR社の「DS492Y」)を1,500rpmにてスピンコートした後、80℃で30秒間ソフトベークし、KrF露光機にて200mJ/cm2で照射した後、80℃で120秒間ゾル-ゲル硬化反応させた。このようにして微細化コンタクトホールパターンを埋め込んだ上で、底部残渣の平均厚み(単位:nm)を、測長SEMを用いる断面SEM観察により測定した。
Further, a photosensitive SOG composition (“DS492Y” manufactured by JSR) was spin-coated at 1,500 rpm on the substrate after the development, and then soft-baked at 80 ° C. for 30 seconds, and then 200 mJ / second with a KrF exposure machine. After irradiation with cm 2 , a sol-gel curing reaction was performed at 80 ° C. for 120 seconds. Thus, after embedding the miniaturized contact hole pattern, the average thickness (unit: nm) of the bottom residue was measured by cross-sectional SEM observation using a length measurement SEM.
微細化コンタクトホールパターンの平均直径、プレイスメントエラー及び底部残渣の平均厚みの評価結果を下記表6に合わせて示す。
Table 6 below shows the evaluation results of the average diameter of the miniaturized contact hole pattern, the placement error, and the average thickness of the bottom residue.

表6の結果から分かるように、パターン形成方法(A)によれば、ホール径が小さい場合でも、プレイスメントエラーが抑制され、かつ底部残渣が低減されたホールパターンを形成することができる。
As can be seen from the results in Table 6, according to the pattern forming method (A), even when the hole diameter is small, it is possible to form a hole pattern in which placement errors are suppressed and bottom residue is reduced.
<パターン形成方法(B)>
<[A’]重合体の合成>
[合成例2-1](重合体(A-1)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン(THF)120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を3.10mL(3.00mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として2-エチルヘキシルグリシジルエーテル0.63mL(3.00mmol)とメタノール1mLとの混合物を注入し重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、メチルイソブチルケトン(MIBK)で置換した。その後、2質量%シュウ酸水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、得られた溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色固体の下記式(A-1)で表される重合体14.8gを得た。この重合体(A-1)は、Mwが6,100、Mnが5,700、Mw/Mnが1.07であった。 <Pattern formation method (B)>
<[A ′] Polymer Synthesis>
[Synthesis Example 2-1] (Synthesis of Polymer (A-1))
A 500 mL flask reaction vessel was dried under reduced pressure, and then 120 g of tetrahydrofuran (THF) that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.10 mL (3.00 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into the THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. 16.6 mL (0.150 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, a mixture of 0.63 mL (3.00 mmol) of 2-ethylhexyl glycidyl ether and 1 mL of methanol was injected as a terminal terminator to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with methyl isobutyl ketone (MIBK). Thereafter, 1,000 g of a 2% by mass oxalic acid aqueous solution was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the resulting solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.8 g of a polymer represented by the following formula (A-1) as a white solid. This polymer (A-1) had Mw of 6,100, Mn of 5,700, and Mw / Mn of 1.07.
<[A’]重合体の合成>
[合成例2-1](重合体(A-1)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン(THF)120gを注入し、-78℃まで冷却した。その後、このTHFにsec-ブチルリチウム(sec-BuLi)の1Nシクロヘキサン溶液を3.10mL(3.00mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として2-エチルヘキシルグリシジルエーテル0.63mL(3.00mmol)とメタノール1mLとの混合物を注入し重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、メチルイソブチルケトン(MIBK)で置換した。その後、2質量%シュウ酸水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、得られた溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色固体の下記式(A-1)で表される重合体14.8gを得た。この重合体(A-1)は、Mwが6,100、Mnが5,700、Mw/Mnが1.07であった。 <Pattern formation method (B)>
<[A ′] Polymer Synthesis>
[Synthesis Example 2-1] (Synthesis of Polymer (A-1))
A 500 mL flask reaction vessel was dried under reduced pressure, and then 120 g of tetrahydrofuran (THF) that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.10 mL (3.00 mmol) of a 1N cyclohexane solution of sec-butyllithium (sec-BuLi) is injected into the THF, and thereafter, adsorption filtration with silica gel for removing the polymerization inhibitor and distillation dehydration treatment are performed. 16.6 mL (0.150 mol) of styrene was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, a mixture of 0.63 mL (3.00 mmol) of 2-ethylhexyl glycidyl ether and 1 mL of methanol was injected as a terminal terminator to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with methyl isobutyl ketone (MIBK). Thereafter, 1,000 g of a 2% by mass oxalic acid aqueous solution was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the resulting solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.8 g of a polymer represented by the following formula (A-1) as a white solid. This polymer (A-1) had Mw of 6,100, Mn of 5,700, and Mw / Mn of 1.07.
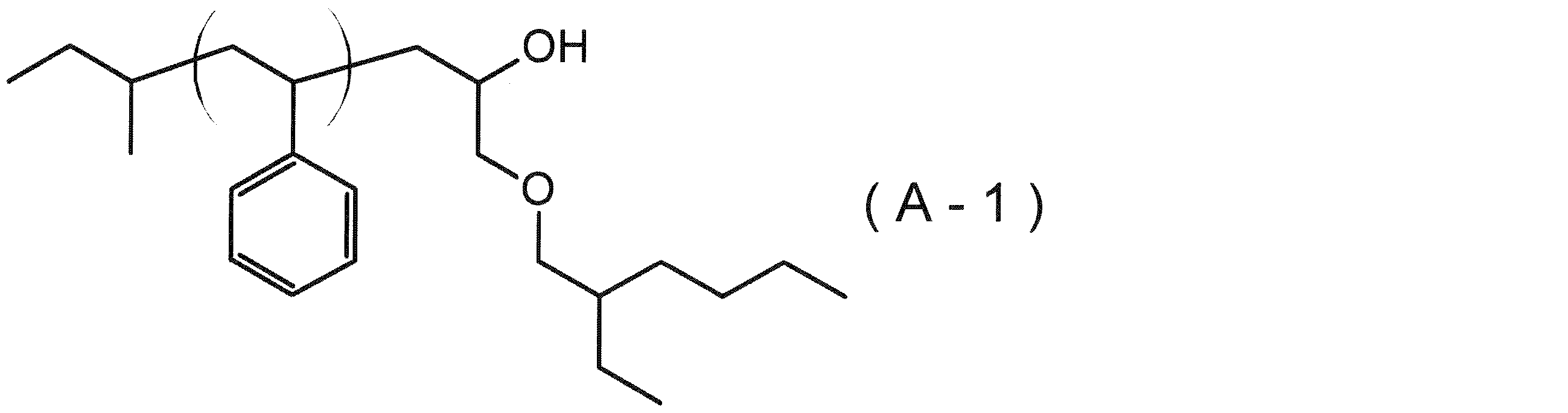
[合成例2-2](重合体(A-2)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン120gを注入し、-78℃まで冷却した。その後、このテトラヒドロフランにsec-BuLiの1Nシクロヘキサン溶液を3.10mL(3.00mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤としてメチルグリシジルエーテル0.27mL(3.00mmol)とメタノール1mLとの混合物を注入し、重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-2)で表される重合体14.6gを得た。この重合体(A-2)は、Mwが6,100、Mnが5,600、Mw/Mnが1.09であった。 [Synthesis Example 2-2] (Synthesis of Polymer (A-2))
The 500 mL flask reaction vessel was dried under reduced pressure, and then 120 g of tetrahydrofuran that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.10 mL (3.00 mmol) of a 1-N cyclohexane solution of sec-BuLi was injected into this tetrahydrofuran, and thereafter, 16.6 mL of styrene subjected to adsorption filtration with silica gel to remove the polymerization inhibitor and distillation dehydration. (0.150 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, a mixture of 0.27 mL (3.00 mmol) of methyl glycidyl ether and 1 mL of methanol was injected as an end terminator to terminate the polymerization end. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.6 g of a white polymer represented by the following formula (A-2). This polymer (A-2) had Mw of 6,100, Mn of 5,600, and Mw / Mn of 1.09.
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン120gを注入し、-78℃まで冷却した。その後、このテトラヒドロフランにsec-BuLiの1Nシクロヘキサン溶液を3.10mL(3.00mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤としてメチルグリシジルエーテル0.27mL(3.00mmol)とメタノール1mLとの混合物を注入し、重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-2)で表される重合体14.6gを得た。この重合体(A-2)は、Mwが6,100、Mnが5,600、Mw/Mnが1.09であった。 [Synthesis Example 2-2] (Synthesis of Polymer (A-2))
The 500 mL flask reaction vessel was dried under reduced pressure, and then 120 g of tetrahydrofuran that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.10 mL (3.00 mmol) of a 1-N cyclohexane solution of sec-BuLi was injected into this tetrahydrofuran, and thereafter, 16.6 mL of styrene subjected to adsorption filtration with silica gel to remove the polymerization inhibitor and distillation dehydration. (0.150 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, a mixture of 0.27 mL (3.00 mmol) of methyl glycidyl ether and 1 mL of methanol was injected as an end terminator to terminate the polymerization end. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.6 g of a white polymer represented by the following formula (A-2). This polymer (A-2) had Mw of 6,100, Mn of 5,600, and Mw / Mn of 1.09.

[合成例2-3](重合体(A-3)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン110gを注入し、-78℃まで冷却した。その後、このテトラヒドロフランにsec-BuLiの1Nシクロヘキサン溶液を3.43mL(3.33mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として3-ブロモプロピオニトリル0.27mL(3.33mmol)を注入し、重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-3)で表される重合体14.8gを得た。この重合体(A-3)は、Mwが4,900、Mnが5,600、Mw/Mnが1.14であった。 [Synthesis Example 2-3] (Synthesis of Polymer (A-3))
After drying the 500 mL flask reaction vessel under reduced pressure, 110 g of tetrahydrofuran that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.43 mL (3.33 mmol) of a 1-N cyclohexane solution of sec-BuLi was injected into this tetrahydrofuran, and thereafter, 16.6 mL of styrene subjected to adsorption filtration with silica gel to remove the polymerization inhibitor and distillation dehydration. (0.150 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.27 mL (3.33 mmol) of 3-bromopropionitrile as a terminal terminator was injected to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.8 g of a white polymer represented by the following formula (A-3). This polymer (A-3) had Mw of 4,900, Mn of 5,600, and Mw / Mn of 1.14.
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン110gを注入し、-78℃まで冷却した。その後、このテトラヒドロフランにsec-BuLiの1Nシクロヘキサン溶液を3.43mL(3.33mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として3-ブロモプロピオニトリル0.27mL(3.33mmol)を注入し、重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、シュウ酸2質量%水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-3)で表される重合体14.8gを得た。この重合体(A-3)は、Mwが4,900、Mnが5,600、Mw/Mnが1.14であった。 [Synthesis Example 2-3] (Synthesis of Polymer (A-3))
After drying the 500 mL flask reaction vessel under reduced pressure, 110 g of tetrahydrofuran that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.43 mL (3.33 mmol) of a 1-N cyclohexane solution of sec-BuLi was injected into this tetrahydrofuran, and thereafter, 16.6 mL of styrene subjected to adsorption filtration with silica gel to remove the polymerization inhibitor and distillation dehydration. (0.150 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, 0.27 mL (3.33 mmol) of 3-bromopropionitrile as a terminal terminator was injected to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 1,000 g of a 2% by mass aqueous solution of oxalic acid was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.8 g of a white polymer represented by the following formula (A-3). This polymer (A-3) had Mw of 4,900, Mn of 5,600, and Mw / Mn of 1.14.

[合成例2-4](重合体(A-4)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン120gを注入し、-78℃まで冷却した。その後、このテトラヒドロフランにsec-BuLiの1Nシクロヘキサン溶液を3.43mL(3.33mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として過剰量のエピブロモヒドリン0.54mL(6.66mmol)とメタノール1mLとの混合物を注入し、重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、シュウ酸0.2質量%水溶液500gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水500gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-4)で表される重合体14.6gを得た。この重合体(A-4)は、Mwが5,100、Mnが5,500、Mw/Mnが1.08であった。 [Synthesis Example 2-4] (Synthesis of Polymer (A-4))
The 500 mL flask reaction vessel was dried under reduced pressure, and then 120 g of tetrahydrofuran that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.43 mL (3.33 mmol) of a 1-N cyclohexane solution of sec-BuLi was injected into this tetrahydrofuran, and thereafter, 16.6 mL of styrene subjected to adsorption filtration with silica gel to remove the polymerization inhibitor and distillation dehydration. (0.150 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, a mixture of 0.54 mL (6.66 mmol) of an excess amount of epibromohydrin and 1 mL of methanol was injected as a terminal terminator to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 500 g of a 0.2% by mass aqueous solution of oxalic acid was poured and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 500 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.6 g of a white polymer represented by the following formula (A-4). This polymer (A-4) had Mw of 5,100, Mn of 5,500, and Mw / Mn of 1.08.
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったテトラヒドロフラン120gを注入し、-78℃まで冷却した。その後、このテトラヒドロフランにsec-BuLiの1Nシクロヘキサン溶液を3.43mL(3.33mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン16.6mL(0.150mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成した。この後、末端停止剤として過剰量のエピブロモヒドリン0.54mL(6.66mmol)とメタノール1mLとの混合物を注入し、重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、シュウ酸0.2質量%水溶液500gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水500gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮した後、メタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-4)で表される重合体14.6gを得た。この重合体(A-4)は、Mwが5,100、Mnが5,500、Mw/Mnが1.08であった。 [Synthesis Example 2-4] (Synthesis of Polymer (A-4))
The 500 mL flask reaction vessel was dried under reduced pressure, and then 120 g of tetrahydrofuran that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 3.43 mL (3.33 mmol) of a 1-N cyclohexane solution of sec-BuLi was injected into this tetrahydrofuran, and thereafter, 16.6 mL of styrene subjected to adsorption filtration with silica gel to remove the polymerization inhibitor and distillation dehydration. (0.150 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of dropping, the mixture was aged for 30 minutes. Thereafter, a mixture of 0.54 mL (6.66 mmol) of an excess amount of epibromohydrin and 1 mL of methanol was injected as a terminal terminator to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 500 g of a 0.2% by mass aqueous solution of oxalic acid was poured and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 500 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and then added dropwise to 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 14.6 g of a white polymer represented by the following formula (A-4). This polymer (A-4) had Mw of 5,100, Mn of 5,500, and Mw / Mn of 1.08.

[合成例2-5](重合体(A-5)の合成)
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにトルエン10g、スチレン5.60g(54mmol)及びメタクリル酸メチル4.40g(43mmol)、アゾイソブチロニトリル(AIBN)0.039g(0.24mmol)、2-メチル-2-[(ドデシルスルファニルチオカルボニル)スルファニル]プロパン酸0.088g(0.24mmol)を加え、窒素フロー下、60℃で4時間加熱撹拌した。得られた重合反応液は、メタノール100gへ沈殿精製させポリマーを析出させた。この固体をブフナーロートにて回収し、メタノール20gにて2回洗浄して開始剤由来の成分を除去した。減圧乾燥することで、白色固体の下記式(A-5)で表される重合体2.5gを得た。この重合体(A-5)はMnが5,200、Mw/Mnが1.17であった。また、1H-NMR分析の結果、スチレンに由来する構造単位及びメタクリル酸メチルに由来する構造単位の含有割合は、それぞれ49モル%及び51モル%であった。 [Synthesis Example 2-5] (Synthesis of Polymer (A-5))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 10 g of toluene, 5.60 g (54 mmol) of styrene, 4.40 g (43 mmol) of methyl methacrylate, 0.039 g of azoisobutyronitrile (AIBN) (0. 24 mmol), 0.088 g (0.24 mmol) of 2-methyl-2-[(dodecylsulfanylthiocarbonyl) sulfanyl] propanoic acid was added, and the mixture was heated and stirred at 60 ° C. for 4 hours under a nitrogen flow. The resulting polymerization reaction solution was purified by precipitation into 100 g of methanol to precipitate a polymer. This solid was recovered with a Buchner funnel and washed twice with 20 g of methanol to remove components derived from the initiator. By drying under reduced pressure, 2.5 g of a polymer represented by the following formula (A-5) as a white solid was obtained. This polymer (A-5) had Mn of 5,200 and Mw / Mn of 1.17. As a result of 1 H-NMR analysis, the content ratios of structural units derived from styrene and structural units derived from methyl methacrylate were 49 mol% and 51 mol%, respectively.
冷却管、滴下ロート及び温度計を備えた200mL三口フラスコにトルエン10g、スチレン5.60g(54mmol)及びメタクリル酸メチル4.40g(43mmol)、アゾイソブチロニトリル(AIBN)0.039g(0.24mmol)、2-メチル-2-[(ドデシルスルファニルチオカルボニル)スルファニル]プロパン酸0.088g(0.24mmol)を加え、窒素フロー下、60℃で4時間加熱撹拌した。得られた重合反応液は、メタノール100gへ沈殿精製させポリマーを析出させた。この固体をブフナーロートにて回収し、メタノール20gにて2回洗浄して開始剤由来の成分を除去した。減圧乾燥することで、白色固体の下記式(A-5)で表される重合体2.5gを得た。この重合体(A-5)はMnが5,200、Mw/Mnが1.17であった。また、1H-NMR分析の結果、スチレンに由来する構造単位及びメタクリル酸メチルに由来する構造単位の含有割合は、それぞれ49モル%及び51モル%であった。 [Synthesis Example 2-5] (Synthesis of Polymer (A-5))
In a 200 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 10 g of toluene, 5.60 g (54 mmol) of styrene, 4.40 g (43 mmol) of methyl methacrylate, 0.039 g of azoisobutyronitrile (AIBN) (0. 24 mmol), 0.088 g (0.24 mmol) of 2-methyl-2-[(dodecylsulfanylthiocarbonyl) sulfanyl] propanoic acid was added, and the mixture was heated and stirred at 60 ° C. for 4 hours under a nitrogen flow. The resulting polymerization reaction solution was purified by precipitation into 100 g of methanol to precipitate a polymer. This solid was recovered with a Buchner funnel and washed twice with 20 g of methanol to remove components derived from the initiator. By drying under reduced pressure, 2.5 g of a polymer represented by the following formula (A-5) as a white solid was obtained. This polymer (A-5) had Mn of 5,200 and Mw / Mn of 1.17. As a result of 1 H-NMR analysis, the content ratios of structural units derived from styrene and structural units derived from methyl methacrylate were 49 mol% and 51 mol%, respectively.

[合成例2-6](重合体(A-a)の合成)
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF200gを注入し、-78℃まで冷却した。その後、このTHFにsec-BuLiの1Nシクロヘキサン溶液を0.30mL(0.27mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン17.7mL(0.154mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00081mol)と、塩化リチウムの0.5NTHF溶液1.08mL(0.0005mol)とを加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル10.0mL(0.094mol)を重合反応液に30分かけて滴下注入し、重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤としてのメタノール1mLを注入し重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、2質量%シュウ酸水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次に、ポリスチレンのホモポリマーを除去するためヘプタン500gを注ぎ、重合体を洗浄し、ポリスチレンホモポリマーをヘプタンへ溶解させた。この操作を4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-a)で表される重合体(A-a)24.1gを得た。この重合体(A-a)は、Mwが79,000、Mnが77,000、Mw/Mnが1.03であった。また、1H-NMR分析の結果、重合体(A-a)は、スチレンに由来する構造単位及びメタクリル酸メチルに由来する構造単位の含有割合が、それぞれ60.0質量%(60.0モル%)及び40.0質量%(40.0モル%)のジブロック共重合体であった。 [Synthesis Example 2-6] (Synthesis of Polymer (Aa))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 0.30 mL (0.27 mmol) of a 1N cyclohexane solution of sec-BuLi was injected into this THF, and thereafter, 17.7 mL of styrene subjected to adsorption filtration with silica gel for removal of the polymerization inhibitor and distillation dehydration treatment. (0.154 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00081 mol) of 1,1-diphenylethylene and 1.08 mL (0.0005 mol) of a 0.5N THF solution of lithium chloride were added, and the polymerization system was dark red It was confirmed that it became. Further, 10.0 mL (0.094 mol) of methyl methacrylate subjected to adsorption filtration with silica gel for removing the polymerization inhibitor and distilled and dehydrated was dropped into the polymerization reaction solution over 30 minutes, and the polymerization system was thin. After confirming that the color was yellow, the reaction was allowed to proceed for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 1,000 g of a 2% by mass oxalic acid aqueous solution was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of heptane was poured to remove the polystyrene homopolymer, the polymer was washed, and the polystyrene homopolymer was dissolved in heptane. This operation was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 24.1 g of a white polymer (Aa) represented by the following formula (Aa). This polymer (Aa) had Mw of 79,000, Mn of 77,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (Aa) was found to have a content of 60.0% by mass (60.0 mol) of structural units derived from styrene and structural units derived from methyl methacrylate. %) And 40.0 mass% (40.0 mol%) of diblock copolymer.
500mLのフラスコ反応容器を減圧乾燥した後、窒素雰囲気下、蒸留脱水処理を行ったTHF200gを注入し、-78℃まで冷却した。その後、このTHFにsec-BuLiの1Nシクロヘキサン溶液を0.30mL(0.27mmol)注入し、その後、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったスチレン17.7mL(0.154mol)を30分かけて滴下注入し、重合系が橙色であることを確認した。この滴下注入の際、重合反応液の内温が-60℃以上にならないように注意した。滴下終了後に30分間熟成し、その後、1,1-ジフェニルエチレン0.11mL(0.00081mol)と、塩化リチウムの0.5NTHF溶液1.08mL(0.0005mol)とを加え、重合系が暗赤色になったことを確認した。さらに、重合禁止剤除去のためのシリカゲルによる吸着濾別と蒸留脱水処理とを行ったメタクリル酸メチル10.0mL(0.094mol)を重合反応液に30分かけて滴下注入し、重合系が薄黄色になったことを確認し、その後120分間反応させた。この後、末端停止剤としてのメタノール1mLを注入し重合末端の停止反応を行った。この重合反応液を室温まで昇温し、濃縮した後、MIBKで置換した。その後、2質量%シュウ酸水溶液1,000gを注入撹拌し、静置後、下層の水層を取り除いた。この操作を3回繰り返し、Li塩を除去した。その後、超純水1,000gを注入撹拌し、下層の水層を取り除いた。この操作を3回繰り返してシュウ酸を除去した後、溶液を濃縮してメタノール500g中に滴下することで重合体を析出させ、ブフナーロートにて固体を回収した。次に、ポリスチレンのホモポリマーを除去するためヘプタン500gを注ぎ、重合体を洗浄し、ポリスチレンホモポリマーをヘプタンへ溶解させた。この操作を4回繰り返し、再度、ブフナーロートにて固体を回収した。得られた重合体を60℃で減圧乾燥させることで白色の下記式(A-a)で表される重合体(A-a)24.1gを得た。この重合体(A-a)は、Mwが79,000、Mnが77,000、Mw/Mnが1.03であった。また、1H-NMR分析の結果、重合体(A-a)は、スチレンに由来する構造単位及びメタクリル酸メチルに由来する構造単位の含有割合が、それぞれ60.0質量%(60.0モル%)及び40.0質量%(40.0モル%)のジブロック共重合体であった。 [Synthesis Example 2-6] (Synthesis of Polymer (Aa))
After drying the 500 mL flask reaction vessel under reduced pressure, 200 g of THF that had been subjected to distillation dehydration treatment was injected under a nitrogen atmosphere and cooled to −78 ° C. Thereafter, 0.30 mL (0.27 mmol) of a 1N cyclohexane solution of sec-BuLi was injected into this THF, and thereafter, 17.7 mL of styrene subjected to adsorption filtration with silica gel for removal of the polymerization inhibitor and distillation dehydration treatment. (0.154 mol) was added dropwise over 30 minutes to confirm that the polymerization system was orange. At the time of this dropwise injection, care was taken so that the internal temperature of the polymerization reaction solution did not exceed -60 ° C. After completion of the dropwise addition, the mixture was aged for 30 minutes, and then 0.11 mL (0.00081 mol) of 1,1-diphenylethylene and 1.08 mL (0.0005 mol) of a 0.5N THF solution of lithium chloride were added, and the polymerization system was dark red It was confirmed that it became. Further, 10.0 mL (0.094 mol) of methyl methacrylate subjected to adsorption filtration with silica gel for removing the polymerization inhibitor and distilled and dehydrated was dropped into the polymerization reaction solution over 30 minutes, and the polymerization system was thin. After confirming that the color was yellow, the reaction was allowed to proceed for 120 minutes. Thereafter, 1 mL of methanol as a terminal terminator was injected to terminate the polymerization terminal. The polymerization reaction solution was warmed to room temperature, concentrated, and then replaced with MIBK. Thereafter, 1,000 g of a 2% by mass oxalic acid aqueous solution was injected and stirred, and after standing, the lower aqueous layer was removed. This operation was repeated three times to remove the Li salt. Thereafter, 1,000 g of ultrapure water was injected and stirred, and the lower aqueous layer was removed. After this operation was repeated three times to remove oxalic acid, the solution was concentrated and dropped into 500 g of methanol to precipitate a polymer, and a solid was recovered with a Buchner funnel. Next, 500 g of heptane was poured to remove the polystyrene homopolymer, the polymer was washed, and the polystyrene homopolymer was dissolved in heptane. This operation was repeated 4 times, and the solid was collected again with a Buchner funnel. The obtained polymer was dried at 60 ° C. under reduced pressure to obtain 24.1 g of a white polymer (Aa) represented by the following formula (Aa). This polymer (Aa) had Mw of 79,000, Mn of 77,000, and Mw / Mn of 1.03. As a result of 1 H-NMR analysis, the polymer (Aa) was found to have a content of 60.0% by mass (60.0 mol) of structural units derived from styrene and structural units derived from methyl methacrylate. %) And 40.0 mass% (40.0 mol%) of diblock copolymer.

[合成例2-7](重合体(A-b)の合成)
冷却管、滴下ロート及び温度計を備えた100mL三口フラスコにテトラメトキシシラン5.23g(3.44mmol)、3-メルカプトプロピルトリメトキシシラン1.12g(0.57mmol)、tert-ブチル-2-((3-トリメトキシシリルプロピル)チオ)プロピオネート9.69g(2.98mmol)、プロピレングルコールモノエチルエーテル9.20gを加え、60℃で撹拌しつつ、10%シュウ酸水溶液4.33g、超純水0.43gを滴下ロートより、20分かけて滴下し、4時間加熱撹拌した。
得られた縮合物の溶液は濃縮したのち、プロピレングリコールモノエチルエーテルを加え、固形分濃度が30質量%となるように調製した。 [Synthesis Example 2-7] (Synthesis of Polymer (Ab))
In a 100 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 5.23 g (3.44 mmol) of tetramethoxysilane, 1.12 g (0.57 mmol) of 3-mercaptopropyltrimethoxysilane, tert-butyl-2- ( 9.69 g (2.98 mmol) of (3-trimethoxysilylpropyl) thio) propionate and 9.20 g of propylene glycol monoethyl ether were added and stirred at 60 ° C., 4.33 g of 10% oxalic acid aqueous solution, ultrapure 0.43 g of water was dropped from a dropping funnel over 20 minutes, and the mixture was heated and stirred for 4 hours.
The resulting condensate solution was concentrated and then propylene glycol monoethyl ether was added to prepare a solid concentration of 30% by mass.
冷却管、滴下ロート及び温度計を備えた100mL三口フラスコにテトラメトキシシラン5.23g(3.44mmol)、3-メルカプトプロピルトリメトキシシラン1.12g(0.57mmol)、tert-ブチル-2-((3-トリメトキシシリルプロピル)チオ)プロピオネート9.69g(2.98mmol)、プロピレングルコールモノエチルエーテル9.20gを加え、60℃で撹拌しつつ、10%シュウ酸水溶液4.33g、超純水0.43gを滴下ロートより、20分かけて滴下し、4時間加熱撹拌した。
得られた縮合物の溶液は濃縮したのち、プロピレングリコールモノエチルエーテルを加え、固形分濃度が30質量%となるように調製した。 [Synthesis Example 2-7] (Synthesis of Polymer (Ab))
In a 100 mL three-necked flask equipped with a condenser, a dropping funnel and a thermometer, 5.23 g (3.44 mmol) of tetramethoxysilane, 1.12 g (0.57 mmol) of 3-mercaptopropyltrimethoxysilane, tert-butyl-2- ( 9.69 g (2.98 mmol) of (3-trimethoxysilylpropyl) thio) propionate and 9.20 g of propylene glycol monoethyl ether were added and stirred at 60 ° C., 4.33 g of 10% oxalic acid aqueous solution, ultrapure 0.43 g of water was dropped from a dropping funnel over 20 minutes, and the mixture was heated and stirred for 4 hours.
The resulting condensate solution was concentrated and then propylene glycol monoethyl ether was added to prepare a solid concentration of 30% by mass.
<組成物(V)の調製>
組成物(V)の調製に用いた[A’]重合体以外の成分について以下に示す。 <Preparation of composition (V)>
Components other than the [A ′] polymer used for the preparation of the composition (V) are shown below.
組成物(V)の調製に用いた[A’]重合体以外の成分について以下に示す。 <Preparation of composition (V)>
Components other than the [A ′] polymer used for the preparation of the composition (V) are shown below.
[[S]溶媒]
S1:プロピレングリコールモノメチルエーテルアセテート [[S] solvent]
S1: Propylene glycol monomethyl ether acetate
S1:プロピレングリコールモノメチルエーテルアセテート [[S] solvent]
S1: Propylene glycol monomethyl ether acetate
[調製例2-1](組成物(S-1)の調製)
[A’]重合体としての(A-1)100質量部、[S]溶媒としての(S1)9,900質量部を混合し、得られた混合溶液を孔径200nmのメンブランフィルターで濾過して、組成物(S-1)を調製した。 [Preparation Example 2-1] (Preparation of composition (S-1))
[A ′] 100 parts by mass of (A-1) as a polymer and 9,900 parts by weight of (S1) as a [S] solvent are mixed, and the obtained mixed solution is filtered through a membrane filter having a pore size of 200 nm. A composition (S-1) was prepared.
[A’]重合体としての(A-1)100質量部、[S]溶媒としての(S1)9,900質量部を混合し、得られた混合溶液を孔径200nmのメンブランフィルターで濾過して、組成物(S-1)を調製した。 [Preparation Example 2-1] (Preparation of composition (S-1))
[A ′] 100 parts by mass of (A-1) as a polymer and 9,900 parts by weight of (S1) as a [S] solvent are mixed, and the obtained mixed solution is filtered through a membrane filter having a pore size of 200 nm. A composition (S-1) was prepared.
[調製例2-2~2-7](組成物(S-2)~(S-6)及び(S-a)の調製)
下記表1に示す種類及び含有量の各成分を用いた以外は、調製例2-1と同様にして、組成物(S-2)~(S-6)及び(S-a)を調製した。 [Preparation Examples 2-2 to 2-7] (Preparation of compositions (S-2) to (S-6) and (Sa))
Compositions (S-2) to (S-6) and (Sa) were prepared in the same manner as in Preparation Example 2-1, except that the components having the types and contents shown in Table 1 were used. .
下記表1に示す種類及び含有量の各成分を用いた以外は、調製例2-1と同様にして、組成物(S-2)~(S-6)及び(S-a)を調製した。 [Preparation Examples 2-2 to 2-7] (Preparation of compositions (S-2) to (S-6) and (Sa))
Compositions (S-2) to (S-6) and (Sa) were prepared in the same manner as in Preparation Example 2-1, except that the components having the types and contents shown in Table 1 were used. .

<微細化パターンの形成>
[実施例2-1~2-6並びに比較例2-1及び2-2]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板にポジ型レジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、有機溶媒現像し、レジストパターンを形成した。次に、このレジストパターンをマスクとして、CF4/O2/Airの混合ガスを用いてSOG膜のエッチングを行った。次いで、得られたSOG膜パターンをマスクとして、N2/O2混合ガスにて下層膜のエッチングを行い、プレパターンを形成した。プレパターン上に組成物(V)として、(S-1)~(S-6)のうち、下記表8に示すものを用いて塗工膜を形成し、下記表8に示す温度及び時間のベーク条件でベークし、PGMEAにてリンスした。表8における比較例2-1の「-」は、組成物(V)の塗工を行わなかったことを示す。続いて組成物(VI)としての(S-a)を用いて塗工膜を形成し220℃で20分間ベークした後、酸素ガスにて重合体(A-a)におけるPMMAブロックからなる相を除去し、微細化パターンを形成した。 <Formation of fine pattern>
[Examples 2-1 to 2-6 and Comparative Examples 2-1 and 2-2]
On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. A positive resist composition (“AIM5484JN” manufactured by JSR) was applied to the obtained substrate to form a resist film having a thickness of 85 nm, ArF immersion exposure, organic solvent development, and a resist pattern were formed. Next, using this resist pattern as a mask, the SOG film was etched using a mixed gas of CF 4 / O 2 / Air. Next, using the obtained SOG film pattern as a mask, the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern. A coating film is formed on the pre-pattern using the composition (V-1) shown in Table 8 below as the composition (V) and the temperature and time shown in Table 8 below. It baked on baking conditions and rinsed with PGMEA. “-” In Comparative Example 2-1 in Table 8 indicates that the composition (V) was not applied. Subsequently, a coating film was formed using (Sa) as the composition (VI) and baked at 220 ° C. for 20 minutes, and then the phase composed of the PMMA block in the polymer (Aa) with oxygen gas was formed. It removed and the refinement | miniaturization pattern was formed.
[実施例2-1~2-6並びに比較例2-1及び2-2]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板にポジ型レジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、有機溶媒現像し、レジストパターンを形成した。次に、このレジストパターンをマスクとして、CF4/O2/Airの混合ガスを用いてSOG膜のエッチングを行った。次いで、得られたSOG膜パターンをマスクとして、N2/O2混合ガスにて下層膜のエッチングを行い、プレパターンを形成した。プレパターン上に組成物(V)として、(S-1)~(S-6)のうち、下記表8に示すものを用いて塗工膜を形成し、下記表8に示す温度及び時間のベーク条件でベークし、PGMEAにてリンスした。表8における比較例2-1の「-」は、組成物(V)の塗工を行わなかったことを示す。続いて組成物(VI)としての(S-a)を用いて塗工膜を形成し220℃で20分間ベークした後、酸素ガスにて重合体(A-a)におけるPMMAブロックからなる相を除去し、微細化パターンを形成した。 <Formation of fine pattern>
[Examples 2-1 to 2-6 and Comparative Examples 2-1 and 2-2]
On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. A positive resist composition (“AIM5484JN” manufactured by JSR) was applied to the obtained substrate to form a resist film having a thickness of 85 nm, ArF immersion exposure, organic solvent development, and a resist pattern were formed. Next, using this resist pattern as a mask, the SOG film was etched using a mixed gas of CF 4 / O 2 / Air. Next, using the obtained SOG film pattern as a mask, the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern. A coating film is formed on the pre-pattern using the composition (V-1) shown in Table 8 below as the composition (V) and the temperature and time shown in Table 8 below. It baked on baking conditions and rinsed with PGMEA. “-” In Comparative Example 2-1 in Table 8 indicates that the composition (V) was not applied. Subsequently, a coating film was formed using (Sa) as the composition (VI) and baked at 220 ° C. for 20 minutes, and then the phase composed of the PMMA block in the polymer (Aa) with oxygen gas was formed. It removed and the refinement | miniaturization pattern was formed.
(組成物(V)の基板表面選択性の評価)
Bare-Si基板上に下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、250℃で60秒間ベーク、続いて340℃で90秒間ベークした。酸素ガスにて下層膜の表層をエッチングして基板選択性評価用基板(A)を作成した。基板(A)及びBare-Si基板(以下、「基板(B)」とする)上に組成物(V)として(S-1)~(S-6)を1500rpmにてスピンコートし塗工膜を形成し、ベークし、PGMEAにてリンスした後に水接触角を測定した。ベーク条件及び接触角を下記表8に合わせて示す。なお、接触角は85°以上を親PS性表面、74°~78°をPSブロック及びPMMAブロックに対する中性表面、70°以下を親PMMA性表面とみなす。基板(A)の表面は親PS性または親PMMA性を示し、基板(B)の表面は中性を示す場合、基板表面選択性が良好とみなす。下記に示した通り(S-1)~(S-6)はいずれも基板表面選択性が良好であることが確認された。基板(A)における接触角は、加熱工程後の塗工膜における基礎パターンの凹部の側面側の水の接触角(θ1)を示すと考えられる。比較例2-1における基板(A)と基板(B)とにおける接触角の差は5°未満であった。 (Evaluation of substrate surface selectivity of composition (V))
An underlayer film having an average thickness of 100 nm was formed on a Bare-Si substrate using a composition for forming an underlayer film (“HM710” manufactured by JSR), and baked at 250 ° C. for 60 seconds and then 340 ° C. for 90 seconds. The surface layer of the lower layer film was etched with oxygen gas to prepare a substrate selectivity evaluation substrate (A). A coating film obtained by spin-coating (S-1) to (S-6) at 1500 rpm as a composition (V) on a substrate (A) and a Bare-Si substrate (hereinafter referred to as “substrate (B)”) After being formed, baked and rinsed with PGMEA, the water contact angle was measured. Bake conditions and contact angles are shown in Table 8 below. Note that a contact angle of 85 ° or more is regarded as a parent PS property surface, 74 ° to 78 ° as a neutral surface with respect to the PS block and PMMA block, and 70 ° or less as a parent PMMA surface. When the surface of the substrate (A) exhibits a PS property or a PMMA property and the surface of the substrate (B) exhibits neutrality, the substrate surface selectivity is considered good. As shown below, each of (S-1) to (S-6) was confirmed to have good substrate surface selectivity. The contact angle in the substrate (A) is considered to indicate the contact angle (θ1) of water on the side surface side of the concave portion of the basic pattern in the coating film after the heating step. The difference in contact angle between the substrate (A) and the substrate (B) in Comparative Example 2-1 was less than 5 °.
Bare-Si基板上に下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、250℃で60秒間ベーク、続いて340℃で90秒間ベークした。酸素ガスにて下層膜の表層をエッチングして基板選択性評価用基板(A)を作成した。基板(A)及びBare-Si基板(以下、「基板(B)」とする)上に組成物(V)として(S-1)~(S-6)を1500rpmにてスピンコートし塗工膜を形成し、ベークし、PGMEAにてリンスした後に水接触角を測定した。ベーク条件及び接触角を下記表8に合わせて示す。なお、接触角は85°以上を親PS性表面、74°~78°をPSブロック及びPMMAブロックに対する中性表面、70°以下を親PMMA性表面とみなす。基板(A)の表面は親PS性または親PMMA性を示し、基板(B)の表面は中性を示す場合、基板表面選択性が良好とみなす。下記に示した通り(S-1)~(S-6)はいずれも基板表面選択性が良好であることが確認された。基板(A)における接触角は、加熱工程後の塗工膜における基礎パターンの凹部の側面側の水の接触角(θ1)を示すと考えられる。比較例2-1における基板(A)と基板(B)とにおける接触角の差は5°未満であった。 (Evaluation of substrate surface selectivity of composition (V))
An underlayer film having an average thickness of 100 nm was formed on a Bare-Si substrate using a composition for forming an underlayer film (“HM710” manufactured by JSR), and baked at 250 ° C. for 60 seconds and then 340 ° C. for 90 seconds. The surface layer of the lower layer film was etched with oxygen gas to prepare a substrate selectivity evaluation substrate (A). A coating film obtained by spin-coating (S-1) to (S-6) at 1500 rpm as a composition (V) on a substrate (A) and a Bare-Si substrate (hereinafter referred to as “substrate (B)”) After being formed, baked and rinsed with PGMEA, the water contact angle was measured. Bake conditions and contact angles are shown in Table 8 below. Note that a contact angle of 85 ° or more is regarded as a parent PS property surface, 74 ° to 78 ° as a neutral surface with respect to the PS block and PMMA block, and 70 ° or less as a parent PMMA surface. When the surface of the substrate (A) exhibits a PS property or a PMMA property and the surface of the substrate (B) exhibits neutrality, the substrate surface selectivity is considered good. As shown below, each of (S-1) to (S-6) was confirmed to have good substrate surface selectivity. The contact angle in the substrate (A) is considered to indicate the contact angle (θ1) of water on the side surface side of the concave portion of the basic pattern in the coating film after the heating step. The difference in contact angle between the substrate (A) and the substrate (B) in Comparative Example 2-1 was less than 5 °.

[実施例2-7及び2-8並びに比較例2-3及び2-4]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板にポジ型レジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、有機溶媒現像し、レジストパターンを形成した。次に、このレジストパターンをマスクとして、CF4/O2/Airの混合ガスを用いてSOG膜のエッチングを行った。次いで、得られたSOG膜パターンをマスクとして、N2/O2混合ガスにて下層膜のエッチングを行いプレパターンを形成した。プレパターン上に組成物(V)として(S-1)又は(S-5)の塗工膜を形成し、下記表9に示すベーク条件でベークし、PGMEAにてリンスした。下記表9の比較例2-4における「-」は、組成物(V)の塗工を行わなかったことを示す。続いて組成物(VI)として(S-a)の塗工膜を形成し220℃で20分間ベークした後、酸素ガスにて重合体(A-a)におけるPMMAブロックからなる相を除去し、微細化パターンを形成した。 [Examples 2-7 and 2-8 and Comparative Examples 2-3 and 2-4]
On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. A positive resist composition (“AIM5484JN” manufactured by JSR) was applied to the obtained substrate to form a resist film having a thickness of 85 nm, ArF immersion exposure, organic solvent development, and a resist pattern were formed. Next, using this resist pattern as a mask, the SOG film was etched using a mixed gas of CF 4 / O 2 / Air. Next, using the obtained SOG film pattern as a mask, the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern. A coating film of (S-1) or (S-5) as a composition (V) was formed on the pre-pattern, baked under the baking conditions shown in Table 9 below, and rinsed with PGMEA. “−” In Comparative Example 2-4 of Table 9 below indicates that the composition (V) was not applied. Subsequently, a coating film of (Sa) was formed as a composition (VI) and baked at 220 ° C. for 20 minutes, and then the phase consisting of the PMMA block in the polymer (Aa) was removed with oxygen gas, A refined pattern was formed.
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板にポジ型レジスト組成物(JSR社の「AIM5484JN」)を塗布することにより85nmのレジスト膜を形成し、ArF液浸露光し、有機溶媒現像し、レジストパターンを形成した。次に、このレジストパターンをマスクとして、CF4/O2/Airの混合ガスを用いてSOG膜のエッチングを行った。次いで、得られたSOG膜パターンをマスクとして、N2/O2混合ガスにて下層膜のエッチングを行いプレパターンを形成した。プレパターン上に組成物(V)として(S-1)又は(S-5)の塗工膜を形成し、下記表9に示すベーク条件でベークし、PGMEAにてリンスした。下記表9の比較例2-4における「-」は、組成物(V)の塗工を行わなかったことを示す。続いて組成物(VI)として(S-a)の塗工膜を形成し220℃で20分間ベークした後、酸素ガスにて重合体(A-a)におけるPMMAブロックからなる相を除去し、微細化パターンを形成した。 [Examples 2-7 and 2-8 and Comparative Examples 2-3 and 2-4]
On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. A positive resist composition (“AIM5484JN” manufactured by JSR) was applied to the obtained substrate to form a resist film having a thickness of 85 nm, ArF immersion exposure, organic solvent development, and a resist pattern were formed. Next, using this resist pattern as a mask, the SOG film was etched using a mixed gas of CF 4 / O 2 / Air. Next, using the obtained SOG film pattern as a mask, the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern. A coating film of (S-1) or (S-5) as a composition (V) was formed on the pre-pattern, baked under the baking conditions shown in Table 9 below, and rinsed with PGMEA. “−” In Comparative Example 2-4 of Table 9 below indicates that the composition (V) was not applied. Subsequently, a coating film of (Sa) was formed as a composition (VI) and baked at 220 ° C. for 20 minutes, and then the phase consisting of the PMMA block in the polymer (Aa) was removed with oxygen gas, A refined pattern was formed.
(組成物(VI)の塗工膜厚均一性の評価)
AFMにてプレパターン上での組成物(VI)の塗工膜厚均一性を評価した。評価結果を下記表9に合わせて示す。組成物(V)を用いた選択修飾プロセスを適用しない場合は、塗工膜厚の二峰性分散又はアイランドパターンの形成による局所的に膜厚増加がみられた。一方で、選択修飾プロセスを適用した場合は、アイランドパターンの形成は見られず、単峰性分散を示し、良好な塗工膜厚均一性が確認された。 (Evaluation of coating film thickness uniformity of composition (VI))
The coating film thickness uniformity of the composition (VI) on the pre-pattern was evaluated by AFM. The evaluation results are shown in Table 9 below. When the selective modification process using the composition (V) was not applied, the film thickness locally increased due to the bimodal dispersion of the coating film thickness or the formation of the island pattern. On the other hand, when the selective modification process was applied, the formation of an island pattern was not observed, unimodal dispersion was exhibited, and good coating film thickness uniformity was confirmed.
AFMにてプレパターン上での組成物(VI)の塗工膜厚均一性を評価した。評価結果を下記表9に合わせて示す。組成物(V)を用いた選択修飾プロセスを適用しない場合は、塗工膜厚の二峰性分散又はアイランドパターンの形成による局所的に膜厚増加がみられた。一方で、選択修飾プロセスを適用した場合は、アイランドパターンの形成は見られず、単峰性分散を示し、良好な塗工膜厚均一性が確認された。 (Evaluation of coating film thickness uniformity of composition (VI))
The coating film thickness uniformity of the composition (VI) on the pre-pattern was evaluated by AFM. The evaluation results are shown in Table 9 below. When the selective modification process using the composition (V) was not applied, the film thickness locally increased due to the bimodal dispersion of the coating film thickness or the formation of the island pattern. On the other hand, when the selective modification process was applied, the formation of an island pattern was not observed, unimodal dispersion was exhibited, and good coating film thickness uniformity was confirmed.

[実施例2-9及び比較例2-5]
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板にポジ型レジスト組成物(JSR社の「EUVJ2121」)を塗布することにより50nmのレジスト膜を形成し、EUV露光し、2.38質量%テトラブチルアンモニウムヒドロキシド水溶液を用いて現像し、レジストパターンを形成した。次に、このレジストパターンをマスクとして、CF4/O2/Airの混合ガスを用いてSOG膜のエッチングを行った。次いで、得られたSOG膜パターンをマスクとして、N2/O2混合ガスにて下層膜のエッチングを行い、プレパターンを形成した。プレパターン上に組成物(V)として(S-1)の塗工膜を形成し、下記表10に示すベーク条件でベークし、PGMEAにてリンスした。続いて組成物(VI)として(S-a)の塗工膜を形成し220℃で20分間ベーク後、酸素ガスにて重合体(A-a)におけるPMMAブロックからなる相を除去し、微細化パターンを形成した。 [Example 2-9 and Comparative Example 2-5]
On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. A positive resist composition (“EUVJ2121” from JSR) is applied to the obtained substrate to form a 50 nm resist film, exposed to EUV, and developed using an aqueous 2.38 mass% tetrabutylammonium hydroxide solution. Then, a resist pattern was formed. Next, using this resist pattern as a mask, the SOG film was etched using a mixed gas of CF 4 / O 2 / Air. Next, using the obtained SOG film pattern as a mask, the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern. A coating film of (S-1) as a composition (V) was formed on the prepattern, baked under the baking conditions shown in Table 10 below, and rinsed with PGMEA. Subsequently, a coating film of (Sa) was formed as a composition (VI), baked at 220 ° C. for 20 minutes, and then the phase consisting of the PMMA block in the polymer (Aa) was removed with oxygen gas to obtain a fine Pattern was formed.
Bare-Si基板上に、下層膜形成用組成物(JSR社の「HM710」)を用いて平均厚み100nmの下層膜を形成し、この下層膜上に、SOG組成物(JSR社の「ISX302」)を用いて平均厚み30nmのSOG膜を形成した。得られた基板にポジ型レジスト組成物(JSR社の「EUVJ2121」)を塗布することにより50nmのレジスト膜を形成し、EUV露光し、2.38質量%テトラブチルアンモニウムヒドロキシド水溶液を用いて現像し、レジストパターンを形成した。次に、このレジストパターンをマスクとして、CF4/O2/Airの混合ガスを用いてSOG膜のエッチングを行った。次いで、得られたSOG膜パターンをマスクとして、N2/O2混合ガスにて下層膜のエッチングを行い、プレパターンを形成した。プレパターン上に組成物(V)として(S-1)の塗工膜を形成し、下記表10に示すベーク条件でベークし、PGMEAにてリンスした。続いて組成物(VI)として(S-a)の塗工膜を形成し220℃で20分間ベーク後、酸素ガスにて重合体(A-a)におけるPMMAブロックからなる相を除去し、微細化パターンを形成した。 [Example 2-9 and Comparative Example 2-5]
On the Bare-Si substrate, an underlayer film having an average thickness of 100 nm was formed using an underlayer film forming composition (“HM710” from JSR), and an SOG composition (“ISX302” from JSR) was formed on the underlayer film. ) Was used to form an SOG film having an average thickness of 30 nm. A positive resist composition (“EUVJ2121” from JSR) is applied to the obtained substrate to form a 50 nm resist film, exposed to EUV, and developed using an aqueous 2.38 mass% tetrabutylammonium hydroxide solution. Then, a resist pattern was formed. Next, using this resist pattern as a mask, the SOG film was etched using a mixed gas of CF 4 / O 2 / Air. Next, using the obtained SOG film pattern as a mask, the lower layer film was etched with a N 2 / O 2 mixed gas to form a pre-pattern. A coating film of (S-1) as a composition (V) was formed on the prepattern, baked under the baking conditions shown in Table 10 below, and rinsed with PGMEA. Subsequently, a coating film of (Sa) was formed as a composition (VI), baked at 220 ° C. for 20 minutes, and then the phase consisting of the PMMA block in the polymer (Aa) was removed with oxygen gas to obtain a fine Pattern was formed.
(パターンサイズ均一性(CDU)の評価)
上記形成した微細化ホールパターンについて、SEMにてCDU(nm)を評価した。評価結果を下記表10に合わせて示す。なお、CDUはその値が小さい程良好とみなす。組成物(V)を用いた選択修飾プロセスを適用することで、CDUで示されるパターンサイズの均一性の改良が確認された。 (Evaluation of pattern size uniformity (CDU))
CDU (nm) was evaluated by SEM for the fine hole pattern formed above. The evaluation results are shown in Table 10 below. Note that the smaller the value of CDU, the better. By applying the selective modification process using the composition (V), improvement in the uniformity of the pattern size indicated by CDU was confirmed.
上記形成した微細化ホールパターンについて、SEMにてCDU(nm)を評価した。評価結果を下記表10に合わせて示す。なお、CDUはその値が小さい程良好とみなす。組成物(V)を用いた選択修飾プロセスを適用することで、CDUで示されるパターンサイズの均一性の改良が確認された。 (Evaluation of pattern size uniformity (CDU))
CDU (nm) was evaluated by SEM for the fine hole pattern formed above. The evaluation results are shown in Table 10 below. Note that the smaller the value of CDU, the better. By applying the selective modification process using the composition (V), improvement in the uniformity of the pattern size indicated by CDU was confirmed.

表10の結果から分かるように、パターン形成方法(B)によれば、基板表面選択性に優れる組成物を用いて、膜厚均一性に優れる塗工膜を形成することにより、パターンサイズ均一性に優れるホールパターンを形成することができる。
As can be seen from the results in Table 10, according to the pattern formation method (B), by using the composition having excellent substrate surface selectivity, by forming a coating film having excellent film thickness uniformity, pattern size uniformity. Can be formed.
本発明のパターン形成方法及び組成物によれば、基板に良好な形状及び配列のパターンを形成することができる。従って、これらはさらなる微細化が要求されている半導体デバイス、液晶デバイス等の各種電子デバイス製造におけるリソグラフィー工程に好適に用いることができる。
According to the pattern forming method and composition of the present invention, a pattern having a good shape and arrangement can be formed on a substrate. Therefore, these can be suitably used for lithography processes in manufacturing various electronic devices such as semiconductor devices and liquid crystal devices that are required to be further miniaturized.
1 基板
2 レジストパターン
3 重合体層
4 充填層
4a ブロック(α)相
4b ブロック(β)相
DESCRIPTION OFSYMBOLS 1 Substrate 2 Resist pattern 3 Polymer layer 4 Packing layer 4a Block (α) phase 4b Block (β) phase
2 レジストパターン
3 重合体層
4 充填層
4a ブロック(α)相
4b ブロック(β)相
DESCRIPTION OF
Claims (21)
- 基板の表面側に直接又は他の層を介して基礎パターンを形成する工程と、
上記基礎パターンの凹部に、少なくとも2つのブロックを有する第1重合体と溶媒とを含有する第1組成物を充填する工程と、
上記充填工程により形成された充填層を相分離させる工程と、
上記相分離工程後の充填層の一部の相を除去する工程と、
上記除去工程により形成された微細化パターンを直接的又は間接的に用いて上記基板をエッチングする工程と
を備え、
上記基礎パターン形成工程が、
上記基板の表面側にレジストパターンを形成する工程と、
上記レジストパターンの少なくとも側面に第2重合体の層を、少なくとも基板又は他の層の表面に上記第2重合体とは異なる第3重合体の層を形成する工程と
を有するパターン形成方法。 Forming a basic pattern on the surface side of the substrate directly or via another layer;
Filling the concave portion of the basic pattern with a first composition containing a first polymer having at least two blocks and a solvent;
A step of phase-separating the packed bed formed by the filling step;
Removing a part of the phase of the packed bed after the phase separation step;
Etching the substrate directly or indirectly using the miniaturized pattern formed by the removing step, and
The basic pattern forming process
Forming a resist pattern on the surface side of the substrate;
Forming a second polymer layer on at least a side surface of the resist pattern, and forming a third polymer layer different from the second polymer on at least the surface of the substrate or another layer. - 上記基板及び上記他の層がSi-H、Si-OH、Si=O及びSi-NR2の少なくとも1種を上面に有し、Rは、それぞれ独立して、水素原子又は炭素数1~20の1価の有機基であり、
上記レジストパターンが-COOH及び-OHの少なくとも一方を側面に有し、
上記第2重合体が、主鎖の少なくとも一方の末端に結合し上記-COOH及び-OHの少なくとも一方と化学結合を形成する第1官能基を有し、
上記第3重合体が、主鎖の少なくとも一方の末端に結合し上記Si-H、Si-OH、Si=O及びSi-NR2の少なくとも1種と化学結合を形成する第2官能基を有するパターン形成方法。 The substrate and the other layers each have at least one of Si—H, Si—OH, Si═O, and Si—NR 2 on the upper surface, and each R is independently a hydrogen atom or a carbon number of 1 to 20 A monovalent organic group of
The resist pattern has at least one of -COOH and -OH on the side surface,
The second polymer has a first functional group bonded to at least one end of the main chain and forming a chemical bond with at least one of the -COOH and -OH;
The third polymer has a second functional group that is bonded to at least one end of the main chain and forms a chemical bond with at least one of Si—H, Si—OH, Si═O, and Si—NR 2. Pattern forming method. - 上記重合体層形成工程が、
上記第2重合体と溶媒とを含有する第2組成物を塗工する工程と、
上記第3重合体と溶媒とを含有する第3組成物を塗工する工程と
を有する請求項1又は請求項2に記載のパターン形成方法。 The polymer layer forming step is
Applying a second composition containing the second polymer and a solvent;
The pattern forming method according to claim 1, further comprising: applying a third composition containing the third polymer and a solvent. - 上記重合体層形成工程が、上記第2重合体と上記第3重合体と溶媒とを含有する第4組成物を塗工する工程を有する請求項1又は請求項2に記載のパターン形成方法。 The pattern forming method according to claim 1, wherein the polymer layer forming step includes a step of applying a fourth composition containing the second polymer, the third polymer, and a solvent.
- 上記基礎パターン形成工程を、上記レジストパターン形成工程、上記第2組成物塗工工程、上記第3組成物塗工工程の順に行う請求項3に記載のパターン形成方法。 The pattern forming method according to claim 3, wherein the basic pattern forming step is performed in the order of the resist pattern forming step, the second composition coating step, and the third composition coating step.
- 上記基礎パターン形成工程を、上記第3組成物塗工工程、上記レジストパターン形成工程、上記第2組成物塗工工程の順に行う請求項3に記載のパターン形成方法。 The pattern forming method according to claim 3, wherein the basic pattern forming step is performed in the order of the third composition coating step, the resist pattern forming step, and the second composition coating step.
- 上記基礎パターン形成工程を、上記レジストパターン形成工程を行い、この工程の後に、上記第4組成物塗工工程を行う請求項4に記載のパターン形成方法。 The pattern forming method according to claim 4, wherein the basic pattern forming step is performed by performing the resist pattern forming step, and the fourth composition coating step is performed after the step.
- 上記レジストパターンが、酸解離性基を有する重合体及び感放射線性酸発生体を含有するレジスト組成物の塗工、露光及び有機溶媒現像液による現像により形成される請求項1から請求項7のいずれか1項に記載のパターン形成方法。 The resist pattern is formed by coating a resist composition containing a polymer having an acid-dissociable group and a radiation-sensitive acid generator, exposure, and development with an organic solvent developer. The pattern formation method of any one of Claims 1.
- 上記レジストパターンが、酸解離性基を有する重合体及び感放射線性酸発生体を含有するレジスト組成物の塗工、露光、アルカリ現像液による現像及び加熱又は露光により形成される請求項1から請求項7のいずれか1項に記載のパターン形成方法。 The resist pattern is formed by coating a resist composition containing a polymer having an acid dissociable group and a radiation-sensitive acid generator, exposure, development with an alkali developer and heating or exposure. Item 8. The pattern forming method according to any one of Items 7 above.
- 上記基板又は上記他の層が、SiO2製又はSiN製である請求項1から請求項9のいずれか1項に記載のパターン形成方法。 The pattern forming method according to claim 1, wherein the substrate or the other layer is made of SiO 2 or SiN.
- 上記他の層が、シリコン含有膜である請求項1から請求項9のいずれか1項に記載のパターン形成方法。 The pattern forming method according to claim 1, wherein the other layer is a silicon-containing film.
- 上記第1官能基が、水酸基、エポキシ基又はオキセタニル基である請求項2から請求項11のいずれか1項に記載のパターン形成方法。 The pattern forming method according to any one of claims 2 to 11, wherein the first functional group is a hydroxyl group, an epoxy group, or an oxetanyl group.
- 上記第2官能基が、カルボキシ基、水酸基又はハロゲン原子である請求項2から請求項11のいずれか1項に記載のパターン形成方法。 The pattern forming method according to any one of claims 2 to 11, wherein the second functional group is a carboxy group, a hydroxyl group, or a halogen atom.
- 上記第1重合体が2つのブロックを有し、これら2つのブロックが、ポリスチレンブロック及び(メタ)アクリル酸エステルブロックである請求項1から請求項13のいずれか1項に記載のパターン形成方法。 The pattern forming method according to any one of claims 1 to 13, wherein the first polymer has two blocks, and the two blocks are a polystyrene block and a (meth) acrylate block.
- ホールパターンを形成する請求項1から請求項14のいずれか1項に記載のパターン形成方法。 The pattern formation method of any one of Claims 1-14 which forms a hole pattern.
- 主鎖の少なくとも一方の末端に結合し-COOH及び-OHの少なくとも一方と化学結合を形成する官能基を有する重合体と、
主鎖の少なくとも一方の末端に結合しSi-H、Si-OH、Si=O及びSi-NR2(Rは、それぞれ独立して、水素原子又は炭素数1~20の1価の有機基である)の少なくとも1種と化学結合を形成する官能基を有する重合体と、
溶媒と
を含有する組成物。 A polymer having a functional group bonded to at least one end of the main chain and forming a chemical bond with at least one of -COOH and -OH;
Bonded to at least one end of the main chain, Si—H, Si—OH, Si═O and Si—NR 2 (R is each independently a hydrogen atom or a monovalent organic group having 1 to 20 carbon atoms) A polymer having a functional group that forms a chemical bond with at least one of
A composition containing a solvent. - 基板の表面側に直接又は他の層を介して基礎パターンを積層する工程と、
上記基礎パターンの凹部の側面及び底面に第5組成物を塗工する工程と、
上記塗工工程により形成された塗工膜を加熱する工程と、
上記塗工膜が積層された上記基礎パターンの凹部に第6組成物を充填する工程と、
上記充填工程により形成された充填層を相分離させる工程と、
上記相分離工程後の充填層の少なくとも一部の相を除去する工程と、
上記除去工程により形成された微細化パターンを用いて上記基板を1又は複数回エッチングする工程と
を備えるパターン形成方法であって、
上記基礎パターンが芳香環の含有割合が50質量%以上の重合体を主成分とし、
上記第5組成物が第1構造単位を有する第4重合体と溶媒とを含有し、
上記第6組成物が第2構造単位からなる第1ブロック及び上記第2構造単位よりも極性が高い第3構造単位からなる第2ブロックを有する第5重合体と溶媒とを含有し、
上記加熱工程後の塗工膜における基礎パターンの凹部の側面側の水の接触角θ1と底面側の水の接触角θ2との差(|θ1-θ2|)が5°以上になることを特徴とするパターン形成方法。 A step of laminating a basic pattern directly or via another layer on the surface side of the substrate;
Applying the fifth composition to the side and bottom surfaces of the concave portion of the basic pattern;
A step of heating the coating film formed by the coating step;
Filling the sixth composition into the concave portion of the basic pattern on which the coating film is laminated;
A step of phase-separating the packed bed formed by the filling step;
Removing at least a part of the phase of the packed bed after the phase separation step;
A step of etching the substrate one or more times using the miniaturized pattern formed by the removing step,
The basic pattern is mainly composed of a polymer having an aromatic ring content of 50% by mass or more,
The fifth composition contains a fourth polymer having a first structural unit and a solvent,
The sixth composition contains a fifth polymer having a first block composed of a second structural unit and a second block composed of a third structural unit having a higher polarity than the second structural unit, and a solvent,
The difference (| θ1-θ2 |) between the water contact angle θ1 on the side surface of the concave portion of the concave portion of the basic pattern and the water contact angle θ2 on the bottom surface in the coated film after the heating step is 5 ° or more. A pattern forming method. - 上記第1構造単位と上記第2構造単位とが共に置換又は非置換のスチレンに由来する請求項17に記載のパターン形成方法。 The pattern forming method according to claim 17, wherein both the first structural unit and the second structural unit are derived from substituted or unsubstituted styrene.
- 上記積層工程で、基板の表面側に直接基礎パターンを積層し、
上記基板がシリコン製である請求項17又は請求項18に記載のパターン形成方法。 In the above lamination process, the basic pattern is laminated directly on the surface side of the substrate,
The pattern forming method according to claim 17, wherein the substrate is made of silicon. - 上記加熱工程で、温度又は時間を制御する請求項17、請求項18又は請求項19に記載のパターン形成方法。 20. The pattern forming method according to claim 17, wherein the temperature or time is controlled in the heating step.
- 上記基礎パターンがホールパターンである請求項17から請求項20のいずれか1項に記載のパターン形成方法。
The pattern formation method according to claim 17, wherein the basic pattern is a hole pattern.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018544003A JP7044976B2 (en) | 2016-10-07 | 2017-10-10 | Pattern formation method |
| US16/376,385 US10691019B2 (en) | 2016-10-07 | 2019-04-05 | Pattern-forming method and composition |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016198989 | 2016-10-07 | ||
| JP2016-198989 | 2016-10-07 | ||
| US201662417412P | 2016-11-04 | 2016-11-04 | |
| US62/417,412 | 2016-11-04 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/376,385 Continuation US10691019B2 (en) | 2016-10-07 | 2019-04-05 | Pattern-forming method and composition |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018066716A1 true WO2018066716A1 (en) | 2018-04-12 |
Family
ID=61832041
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/036708 WO2018066716A1 (en) | 2016-10-07 | 2017-10-10 | Pattern formation method and composition |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP7044976B2 (en) |
| WO (1) | WO2018066716A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11335559B2 (en) | 2016-02-08 | 2022-05-17 | Jsr Corporation | Pattern-forming method, and composition |
| TWI813048B (en) * | 2021-01-18 | 2023-08-21 | 日商國際電氣股份有限公司 | Substrate processing method, semiconductor device manufacturing method, substrate processing apparatus, and program |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014042960A (en) * | 2012-08-27 | 2014-03-13 | Tokyo Ohka Kogyo Co Ltd | Pattern forming method |
| JP2014164044A (en) * | 2013-02-22 | 2014-09-08 | Tokyo Ohka Kogyo Co Ltd | Photosensitive resin composition, pattern forming method, and production method of structure including phase separation structure |
| JP2015044186A (en) * | 2013-07-31 | 2015-03-12 | 東京応化工業株式会社 | Phase separation structure manufacturing method, pattern forming method, and fine pattern forming method |
| JP2016130844A (en) * | 2015-01-13 | 2016-07-21 | 東友ファインケム株式会社Dongwoo Fine−Chem Co., Ltd. | Photosensitive resin composition, photocurable pattern formed from the same, and image display device including the pattern |
| JP2016173415A (en) * | 2015-03-16 | 2016-09-29 | 株式会社東芝 | Pattern forming method |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5948129B2 (en) * | 2012-04-26 | 2016-07-06 | 東京応化工業株式会社 | Method for forming a pattern in which two or more isolated holes are arranged side by side |
| JP2015152702A (en) * | 2014-02-13 | 2015-08-24 | 株式会社東芝 | Pattern forming method and semiconductor device |
-
2017
- 2017-10-10 WO PCT/JP2017/036708 patent/WO2018066716A1/en active Application Filing
- 2017-10-10 JP JP2018544003A patent/JP7044976B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014042960A (en) * | 2012-08-27 | 2014-03-13 | Tokyo Ohka Kogyo Co Ltd | Pattern forming method |
| JP2014164044A (en) * | 2013-02-22 | 2014-09-08 | Tokyo Ohka Kogyo Co Ltd | Photosensitive resin composition, pattern forming method, and production method of structure including phase separation structure |
| JP2015044186A (en) * | 2013-07-31 | 2015-03-12 | 東京応化工業株式会社 | Phase separation structure manufacturing method, pattern forming method, and fine pattern forming method |
| JP2016130844A (en) * | 2015-01-13 | 2016-07-21 | 東友ファインケム株式会社Dongwoo Fine−Chem Co., Ltd. | Photosensitive resin composition, photocurable pattern formed from the same, and image display device including the pattern |
| JP2016173415A (en) * | 2015-03-16 | 2016-09-29 | 株式会社東芝 | Pattern forming method |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11335559B2 (en) | 2016-02-08 | 2022-05-17 | Jsr Corporation | Pattern-forming method, and composition |
| TWI813048B (en) * | 2021-01-18 | 2023-08-21 | 日商國際電氣股份有限公司 | Substrate processing method, semiconductor device manufacturing method, substrate processing apparatus, and program |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7044976B2 (en) | 2022-03-31 |
| JPWO2018066716A1 (en) | 2019-09-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6394042B2 (en) | Pattern forming composition and pattern forming method | |
| US9738746B2 (en) | Composition for pattern formation, pattern-forming method, and block copolymer | |
| JP2018503241A (en) | Defect reduction methods and compositions for induced self-assembly patterning | |
| JP2015170723A (en) | Patterning method and self-organization composition | |
| WO2013073505A1 (en) | Self-organizing composition for pattern formation and pattern formation method | |
| US11370872B2 (en) | Composition for pattern formation, and pattern-forming method | |
| WO2018066716A1 (en) | Pattern formation method and composition | |
| US20170255096A1 (en) | Pattern-forming method | |
| US10691019B2 (en) | Pattern-forming method and composition | |
| JP6955176B2 (en) | Film-forming compositions, film-forming methods and self-assembling lithography processes | |
| KR102353786B1 (en) | Novel compositions and processes for self-assembly of block copolymers | |
| JP6652721B2 (en) | Pattern forming composition and pattern forming method | |
| US11335559B2 (en) | Pattern-forming method, and composition | |
| JP7213495B2 (en) | Resin composition for forming phase-separated structure, method for producing structure containing phase-separated structure, and block copolymer | |
| WO2022049968A1 (en) | Self-organizing film-forming composition, and method for forming pattern | |
| US20180192524A1 (en) | Forming method of contact hole pattern | |
| WO2023083933A1 (en) | Neutral brushes with tunable polarity for self-assembly of block copolymers with poly(styrene) and poly(methyl methacrylate) containing segments | |
| JP2015139875A (en) | Self-organizing composition for pattern formation and pattern formation method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17858552 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2018544003 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17858552 Country of ref document: EP Kind code of ref document: A1 |