WO2017001907A1 - Biocatalytic processes for the preparation of vilanterol - Google Patents
Biocatalytic processes for the preparation of vilanterol Download PDFInfo
- Publication number
- WO2017001907A1 WO2017001907A1 PCT/IB2016/000615 IB2016000615W WO2017001907A1 WO 2017001907 A1 WO2017001907 A1 WO 2017001907A1 IB 2016000615 W IB2016000615 W IB 2016000615W WO 2017001907 A1 WO2017001907 A1 WO 2017001907A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- vilanterol
- compound
- process according
- salt
- tartrate
- Prior art date
Links
- 0 CC(C)(OC1)Oc2c1cc([C@@](*)CNCCCCCCOCCOCc(c(Cl)ccc1)c1Cl)cc2 Chemical compound CC(C)(OC1)Oc2c1cc([C@@](*)CNCCCCCCOCCOCc(c(Cl)ccc1)c1Cl)cc2 0.000 description 1
- OWOCWBVFWIFQCF-VWLOTQADSA-N CC(C)(OC1)Oc2c1cc([C@H](CNCCCCCCOCCOCc(c(Cl)ccc1)c1Cl)O)cc2 Chemical compound CC(C)(OC1)Oc2c1cc([C@H](CNCCCCCCOCCOCc(c(Cl)ccc1)c1Cl)O)cc2 OWOCWBVFWIFQCF-VWLOTQADSA-N 0.000 description 1
- UQBUOFMNRQTQCM-UHFFFAOYSA-O CC=C(C(COCCOCCCCCC[NH3+])=C)Cl Chemical compound CC=C(C(COCCOCCCCCC[NH3+])=C)Cl UQBUOFMNRQTQCM-UHFFFAOYSA-O 0.000 description 1
- KHDUONKKQTXFDZ-KWKHWOPJSA-N CCCOCCOC/C(/C(/Cl)=C\CC)=C(/C)\Cl Chemical compound CCCOCCOC/C(/C(/Cl)=C\CC)=C(/C)\Cl KHDUONKKQTXFDZ-KWKHWOPJSA-N 0.000 description 1
- VQIDLHATGFNDLK-NSHDSACASA-O CCNC[C@@H](c(cc1)nc2c1OC(C)(C)[OH+]C2)O Chemical compound CCNC[C@@H](c(cc1)nc2c1OC(C)(C)[OH+]C2)O VQIDLHATGFNDLK-NSHDSACASA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/02—Preparation of oxygen-containing organic compounds containing a hydroxy group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/03—Ethers having all ether-oxygen atoms bound to acyclic carbon atoms
- C07C43/14—Unsaturated ethers
- C07C43/178—Unsaturated ethers containing hydroxy or O-metal groups
- C07C43/1786—Unsaturated ethers containing hydroxy or O-metal groups containing halogen
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P41/00—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture
Definitions
- This invention relates to highly efficient biocatalytic processes for the
- WO2014041565 describes a similar process to the process described in
- WO2003024439 for the preparation of Vilanterol and pharmaceutical acceptable salts thereof, by a process comprising reduction of ketone with borane.
- WO2010/025085 describes engineered ketoreductase enzymes allegedly
- WO 2004/022547 and US 2005/0075394 describe other synthetic approaches 5 which use 2-azido- or 2-(N-Boc)-amino moieties instead of the 2-bromo moiety.
- the reduction in the described processes is done with borane dimethylsulfide complex and CBS-borane catalyst.
- Use of borane and derivatives thereof requires stoichiometric amounts and borane is known to be toxic and not environmentally friendly.
- the desired process comprises a high substrate concentration, allowing high space-time yields (defined as product amount produced per volume and per time, g/l/day or g/l/h).
- the enzyme (catalyst) concentration should be kept low to allow a commercially competitive process.
- the present invention relates to a process for preparing Vilanterol, which
- the present invention relates to methods of using polypeptides for the biocatalytic conversion of the Ketone Substrate, as described herein below, to the corresponding alcohol, preferably wherein the Ketone Substrate is 2-bromo-l-(2,2- dimethyl-4H- l,3-benzodioxin-6-yl)ethanone and it is converted to enantiopure
- the present invention provides use of compound of formula V, VI and VII and salts thereof of the following formula:
- the present invention provides preparation of Vilanterol in the form of L-tartaric acid salt.
- the present invention also encompasses the vilanterol tartrate and solid state forms 20 thereof for the preparation of other vilanterol salts, or of vilanterol, solid state
- the present invention also encompasses the vilanterol tartrate and solid state forms thereof described herein for use as medicaments, particularly sole product or as a combination therapy with an inhaled corticosteroid for COPD and asthma. 25
- the present invention also encompasses a process for the preparation of
- compositions comprising combining vilanterol tartrate and solid state forms thereof, or a pharmaceutical composition comprising said vilanterol tartrate and solid state forms thereof and at least one pharmaceutically acceptable
- the present invention also encompasses a method of treating a person suffering from COPD and asthma, comprising administering a therapeutically effective amount of any one or a combination vilanterol tartrate and solid state forms
- Figure 1 shows a powder X-ray diffraction pattern ("powder XRD” or "PXRD”) of 5 (R)- 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II).
- Figure 2 shows a powder X-ray diffraction pattern of 6-(2-((2,6- dichlorobenzyl)oxy)ethoxy)hexan-l-amine L-tartaric acid salt (V L-tartrate).
- Figure 3 shows a powder X-ray diffraction pattern of (R)-N-(2-((tert- butyldimethylsilyl)oxy)-2-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethyl)-6-(2-((2,6- 10 dichlorobenzyl)oxy)ethoxy)hexan-l-amine fumarate salt (Via fumarate).
- Figure 4 shows a powder X-ray diffraction pattern of (R)-2-((6-(2-((2,6- dichlorobenzyl)oxy)ethoxy)hexyl)amino)-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- yl)ethan-l-ol L-Tartaric acid salt (VII L-tartrate)
- Figure 5 shows a powder X-ray diffraction pattern of Vilanterol L-tartrate.
- the present invention relates to highly efficient biocatalytic processes for the preparation of Vilanterol and Vilanterol intermediates.
- the present invention provides an improved, safe process with increased
- the present invention offers a desirable, well-controlled biocatalytic process, that may be adapted to a large industrial scale, and allows high substrate loadings (e.g ., > 50 g/L), high percent conversion (e.g., > 90% in 24 h), high 25 enantiomeric excess (e.g., at least about 99% ee) and low enzyme loading (e.g., less than 5 w/w%).
- the process provided in the present invention eliminates the need for an additional co-factor regenerating enzyme other than the ketoreductase enzyme/polypeptide or engineered ketoreductase polypeptide.
- the present invention encompasses crystalline salts of compounds V, VI and VII 30 which are used as intermediates of Vilanterol synthesis.
- the present invention encompasses vilanterol tartrate, solid state forms thereof and pharmaceutical compositions comprising one or more of the vilanterol salts and/or solid state forms thereof.
- the present invention also encompasses vilanterol tartrate and solid state forms thereof for use in the preparation and purification of Vilanterol and Vilanterol salt, such as Vilanterol trifenatate.
- ketooreductase refers to an enzyme/polypeptide that can
- Ketoreductase enzymes include, for example, those classified under the E.C. (or IUBMB - International 5 Union of Biochemistry and Molecular Biology) no. 1.1.1 (i.e. 1.1.1.x) (see
- ketoreductase are given various names in addition to ketoreductase, including, but not
- NADPH-dependent ketoreductases are classified under the IUBMB number of 1.1.1.2 and the CAS number of 9028-12-0.
- ketoreductases are classified under the IUBMB number of 1.1.1.1 and the CAS number 15 of 9031 -72-5.
- the ketoreductases useful for the processes of the present invention comprise enzymes from IUBMB or E.C. 1.1.1.1 and E.C. 1.1.1.2, more
- co-factor refers to a non-protein compound that operates in 20 combination with an enzyme which catalyzes the reaction of interest.
- nicotinamide co-factors such as nicotinamide adenine dinucleotide ('TSfAD”) or a salt thereof, reduced nicotinamide adenine dinucleotide ("NADH”) or a salt thereof, nicotinamide adenine dinucleotide phosphate ("NADP ⁇ + >”), reduced
- NADPH nicotinamide adenine dinucleotide phosphate
- oxidized cofactors function as intermediate hydrogen acceptors in the catalytic mechanism of the enzymes.
- salts of the co- factors include NAD tetra(cyclohexyl ammonium) salt, NAD tetrasodium salt, NAD
- the co-factor is NADP or NADPH.
- the term “isolated” in reference to compounds described in the invention corresponds to a compound that is physically separated from the reaction mixture in which it is formed.
- the term “isolated” in reference to polypeptides/enzymes refers to polypeptides/enzymes at least partially separated from the environment in which they are formed, for example from the natural environment, e.g., from bacteria.
- use of the term “isolated” indicates that a naturally occurring or recombinant enzyme has been at least partially removed from its normal cellular or natural environment, 5 e.g . from bacterial cells.
- the isolated enzyme is in a cell-free system.
- the isolated enzyme can be crude or highly purified.
- isolated does not
- the term "synthesized” or “engineered” refers to 10 an enzyme that is prepared by chemical synthesis, recombinant means, or the
- the term "purified” refers to an enzyme that is essentially free (at least about 90-95% pure) of non- enzymatic material or other enzymes.
- the isolated enzyme can be a lysate or an
- enzyme powder obtained by lyophilization of cell lysates, which can contain isolated, 15 but unpurified enzymes.
- ketoreductase is isolated.
- the ketoreductase can be separated from any host, such as mammals, filamentous fungi, yeasts, and bacteria. The isolation, purification, and
- ketoreductase is synthesized or engineered.
- the ketoreductase can be synthesized chemically or using recombinant means.
- the chemical and recombinant production of ketoreductases is described in, 25 for example, in US2016/0083759, WO2010025085, WO2011022548, and
- the ketoreductase is purified, preferably with a purity of about 90% or more, more preferably with a purity of about 95% or more, Preferably, the ketoreductase is substantially cell-free.
- a thing e.g., a reaction mixture
- room temperature often abbreviated "RT.” This means that the temperature of the thing is close to, or the same as, that of the space, e.g ., the room or fume hood, in which the thing is located.
- room temperature is from about 20°C to about 30°C, or about 22°C to about 27°C, or about 25°C.
- a process or step may be referred to herein as being carried out “overnight.”
- This time interval is from about 8 to about 20 hours, or about 10- 18 hours, typically about 16 hours.
- reduced pressure refers to a pressure of about 10 5 mbar to about 50 mbar.
- vol. or "volume” can be used to refer to ml per gram of the corresponding Vilanterol salts.
- volume can be used to refer to ml per gram of the corresponding Vilanterol salts.
- a statement that 0.5 g of Vilanterol is d issolved in ten volumes of a Solvent X would be u nderstood to mean that the 0.5 g of Vilanterol was dissolved in 5 ml of Solvent X . 10
- a crystal form may be referred to herein as being characterized by graphical data "substantially as depicted in" a Figure.
- Such data include, for example, powder X-ray diffractograms and solid state NMR spectra.
- the graphical data potentially provides additional technical information to fu rther define the 15 respective solid state form (a so-called "fingerprint") which can not necessarily be described by reference to numerical values or peak positions alone.
- the skilled person will understand that such graphical representations of data may be
- the XRPD measurements are taken 30 using copper a radiation wavelength ( 1.5418 A).
- solvate refers to a crystal form that incorporates a solvent in the crystal structure.
- the solvent is water, the solvate is often referred to as a "hydrate.
- the solvent in a solvate may be present in either a stoichiometric or in a non-stoichiometric amount. 35
- the present invention relates to polypeptides having
- ketoreductase activity and to methods of using the polypeptides for the biocatalytic conversion of the Ketone Substrate of the following structure:
- L is a leaving group, that may be selected for example from halogen containing groups, typically a chloro, bromo or iodo group; or a sulphonate group such as an 10 alkylsulphonate (particularly d. 6 alkylsulphonates), typically methane sulphonate; or an aryl sulphonate (particularly C 6 . 10 arylsulfonate) group, typically a toluenesulphonate group (e.g. para-toluenesulphonate) ;
- G ⁇ nd G 2 may each independently be a hydroxyl- protecting group or a hydrogen.
- Suitable hydroxyl-protecting protecting groups can be silyl-type protecting 15 groups according to the formula -SiR ⁇ R 3 , wherein R 1 , R 2 and R 3 are independently selected from : a C1-C15 straight or branched alkyl group, a C1-C10 cycloalkyl (preferably C3- 10 cycloalkyl or C 5 - 8 cycloalkyl) group, an optionally substituted C 6 -Ci 0 aryl group and an optionally substituted C 7 -Ci 2 arylalkyl group.
- R 1 , R 2 and R 3 are independently selected from : a C1-C15 straight or branched alkyl group, a C1-C10 cycloalkyl (preferably C3- 10 cycloalkyl or C 5 - 8 cycloalkyl) group, an optionally substituted C 6 -Ci 0 aryl group and an optionally substituted C 7 -Ci 2 arylal
- the hydroxyl- protecting groups G 1 , G 2 may be independently selected from: ether groups (e.g. Ci- 10 alkyl or C 5 - i 0 cyclic ethers, preferably C 5 .s cyclic ethers, methyl ethers or ethyl ethers) or ester groups (e.g. Ci- i 0 alkyl esters, preferably Ch alky! esters, or Cs- io aryl esters, or C7.11 araalkyl esters).
- the hydroxyl protecting groups G 1 , G 2 may be 25 independently selected from methyl or substituted methyl groups, typically
- substituted ethyl groups typically ethoxyethyl, benzyl or tert-butyl ; or ester groups, typically acetate, or aryl substituted acetate groups, for example benzoate or
- Additional hydroxyl-protecting groups can be selected 30 from those described in Greene and Wuts "Greene's Protective Groups in Organic Synthesis", 4th Edition, publ. Wiley, 2006.
- hydroxyl-protecting groups G 1 and G 2 may together represent an group suitable for protection of 1,3 diols, for example cyclic acetal or ketal, typically methylene acetal, ethylidene acetal, isopropylidene acetal (acetonide).
- G 1 , G 2 is isopropylidene acetal (acetonide).
- G 1 and G 2 of the Ketone Substrate together represent isopropylidene acetal (acetonide), and the Ketone Substrate is 2-bromo-l-(2,2-dimethyl-4H-l,3- benzodioxin-6-yl)ethanone, which is referred to as Substrate I.
- Substrate I is converted to enantiopure alcohol : (R)-2-bromo-l-(2,2- dimethyl-4H-l,3-benzodioxin-6-yl)ethanol, which is referred to herein as Compound II.
- the compound has an optical purity of : ⁇ 85%, > 90%, > 92%, > 94%, 96%, ⁇ 98%, > 99%, > 99.5%, or > 99.8%, and more preferably an optical purity of ⁇ 99%, > 99.5%, or > 99.8%.
- Compound II may be isolated, preferably it is crystalline.
- the invention also comprises crystalline Compound II, as described herein below.
- G 1 , G 2 and L are as defined above.
- the present invention comprises a process for biocatalytic reduction of the Ketone Substrate, as described above, to the corresponding alcohol, as described above, preferably wherein the Ketone Substrate is Substrate I and it is converted to enantiomerically pure alcohol, Compound II.
- This process is done by using an isolated enzyme capable of keto-reductase activity.
- ketoreductase polypeptides such as those disclosed in US2016/0083759, 5 WO2010025085, WO2011022548, and WO2009046153, herein incorporated by
- suitable enzymes may be the commercially available enzymes such as: Codexis CDX-005 or Codexis KRED-P1-H01, or an equivalent enzyme thereof.
- equivalent enzyme refers to an enzyme with similar or identical enzymatic activity, which produces the product in the desired enantiomeric 10 access and optical purity, as described in this invention.
- the effective amount of an enzyme or
- combination of enzymes may be any amount of the enzyme that is sufficient to
- a desired degree of conversion of a substrate for example, at least 90%, preferably at least 95%, more preferably at least 98% conversion of a substrate, 15 during 24 hours of reaction time,
- the above process typically allows utilizing substrate in a concentration of at least 20 g/l, preferably at least 40 g/l, more preferably at least 100 g/l .
- NADPH NADPH
- the enzyme and substrate pair used for 20 the regeneration of the co-factor e.g. glucose dehydrogenase and glucose; formate dehydrogenase and sodium formate; phosphite dehydrogenase and sodium phosphite
- glucose dehydrogenase and glucose e.g. glucose dehydrogenase and glucose
- formate dehydrogenase and sodium formate e.g. glucose dehydrogenase and glucose
- phosphite dehydrogenase and sodium phosphite e.g. glucose dehydrogenase and glucose
- formate dehydrogenase and sodium formate phosphite dehydrogenase and sodium phosphite
- the substrate (2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- yl)ethanol
- a non- 25 ionic tensioactive agent (surfactant) (preferably Triton ® X- 100) is dissolved in the substrate
- the solution of the substrate in organic solvent may be slowly added to the aqueous solution containing the enzyme and the co-factor under vigorous stirring.
- the pH of the reaction system may be kept constant during the enzymatic reaction by
- the process may be done with a biphasic solvent system, i.e., comprising an aqueous buffer and a water immiscible solvent.
- a biphasic solvent system i.e., comprising an aqueous buffer and a water immiscible solvent.
- the enzyme is dissolved in aqueous buffer.
- aqueous buffer includes potassium or sodium phosphate, hydrochloride or sulfate salts of tertiary amines, triethanolamine, TRIS
- water immiscible organic solvents include ethers, esters, aromatic and aliphatic hydrocarbons or their mixtures, preferably diisopropyl ether, methyl tert-butyl ether, ethyl acetate, isobutyl acetate, butyl acetate, ethyl butanoate, toluene, hexane, 5 heptane or mixtures thereof.
- the process can be done with a monophasic solvent system
- water miscible solvents include alcohols, aprotic polar solvents or mixtures thereof, preferably isopropanol, ethanol, isobutanol, dimethyl sulfoxide, dimethyl formamide, dimethyl 10 acetamide, acetonitrile or mixtures thereof.
- the amount of solvent utilized is an amount necessary to dissolve the enzyme and substrate.
- the amount of water immiscible organic solvent may be from about 20 to about 75 v/v%, from about 30 to about 65v/v% or from about 45 to about 55 v/v%.
- the amount of water miscible solvent may be from about 10 to 15 about 75 v/v%, from about 25 to about 65 v/v%, or from about 50 to about 55v/v%.
- the process is typically performed in the presence of a suitable co-factor as
- the reaction mixture further comprises a co-factor regenerating/recycling system.
- the co-factor regenerating/recycling 20 system comprises a substrate and a dehydrogenase.
- the co-factor recycling system can comprise an additional enzyme and its substrate in more than 1 equivalent amount relative to Ketone Substrate I.
- the co-factor recycling system can comprise, for
- glucose dehydrogenase and glucose the following pairs of enzymes and substrates: glucose dehydrogenase and glucose, formate dehydrogenase and sodium formate or phosphite dehydrogenase and 25 sodium phosphite, alcohol dehydrogenase and a secondary alcohol (e.g. isopropanol).
- a secondary alcohol e.g. isopropanol
- ketoreductase In some of the examples subject to the scope of this invention the ketoreductase
- polypeptide (enzyme) can regenerate the co-factor itself, in the presence of
- the present invention comprises a process for
- Such process may comprise:
- step a) is preferably done by using an isolated enzyme capable of ketoreductase activity.
- an isolated enzyme capable of ketoreductase activity Such enzyme may be as described above.
- the amount of enzyme is typically as described above.
- the above process typically allows utilizing substrate in the above described concentration.
- the process is done with a biphasic solvent system, i.e., comprising an aqueous buffer and a water immiscible solvent.
- a biphasic solvent system i.e., comprising an aqueous buffer and a water immiscible solvent.
- the enzyme is dissolved in the aqueous buffer. Examples of aqueous buffers and of water immiscible solvents are described above.
- the process can be done with a monophasic solvent system comprising an aqueous buffer and a water miscible organic solvent. Examples of water miscible solvent are described above.
- the amount of solvent(s) utilized and their ratio is the amount necessary to dissolve the enzyme and substrate, as described above.
- step al) is preferably done by introduction of protecting group (PG) and preparation of O-protected compound III and compound VI.
- PG protecting group
- Suitable PGs are as described above.
- the present invention comprises crystalline Compound II.
- the crystalline form can be characterized by data selected from one or more of the following : an X-ray powder diffraction pattern having peaks at 9.8, 14.9, 15.9, 18.2 and 19.9 degrees two theta ⁇ 0.2 degrees two theta; an X-ray powder diffraction pattern substantially as depicted in Figure 1 ; and combinations thereof.
- the crystalline form of Compound II may be further characterized by an X-ray powder diffraction pattern having any one, two, three, four or five additional peaks selected from peaks at 8.0, 18.7, 21.9, 24,0 and 25.6 degrees two theta ⁇ 0.2 degrees two theta.
- Table 1 The full peak list of Compound II is presented in the following table (Table 1) :
- the crystalline form of Compound II may alternatively be characterized by the peaks presented in Table 1 above, optionally with the relative intensities.
- the present invention comprises the preparation of compound V from compound IV by reaction with ammonia.
- Compound V may be used in the process of the invention as free base or in the form of a salt.
- Suitable salts may include but are not limited to inorganic acid salts, for example hydrochloride, hydrobromide, phosphate or sulphate, or organic acid salts.
- a suitable organic acid can be selected from acetic acid and its derivatives, benzoic acid or substituted benzoic acids, methanesulfonic acid, benzenesulfonic acid or substituted benzenesulfonic acid, citric acid, maleic acid, malic acid, maleic acid, malonic acid, mandelic acid, succinic acid, fumaric acid, pyroglutamic acid, oxalic acid, tartaric acid or derivatives thereof. More preferably the suitable acid can be selected from tartaric acid, preferably in its optically pure form (preferably L-tartaric acid). 5
- the present invention comprises the preparation of compound VI by reaction of compound III and compound V.
- Compound VI may be used in the process of the invention as free base or in the form of a salt.
- Suitable salts may include but are not limited to inorganic acid salts, for example hydrochloride, hydrobromide, phosphate or sulphate, or organic acid salts.
- a suitable organic acid can 10 be selected from acetic acid and its derivatives, benzoic acid or substituted benzoic acids, methanesulfonic acid, benzenesulfonic acid or substituted benzenesulfonic acid, citric acid, maleic acid, malic acid, maleic acid, malonic acid, mandelic acid, succinic acid, fumaric acid, pyroglutamic acid, oxalic acid, tartaric acid or derivatives thereof.
- the suitable acid can be selected from fumaric acid or tartaric acid, 15 preferably in its optically pure form (preferably L-tartaric acid), more preferably fumaric acid.
- the present invention comprises the preparation of compound VII from compound VI by selective deprotection of PG.
- Compound VII may be used in the process of the invention as free base or in the form of a salt. 20
- Suitable salts may include but are not limited to inorganic acid salts, for
- a suitable organic acid can be selected from acetic acid and its derivatives, benzoic acid or substituted benzoic acids, methanesulfonic acid, benzenesulfonic acid or substituted benzenesulfonic acid, citric acid, maleic acid, malic acid, maleic acid, malonic acid, 25 mandelic acid, succinic acid, fumaric acid, pyroglutamic acid, oxalic acid, tartaric acid or derivatives thereof. More preferably the suitable acid can be selected from tartaric acid, preferably in its optically pure form (preferably L-tartaric acid).
- compound V is in the form of L-tartaric acid salt as crystalline compound
- compound VI is in the form of fumaric acid salt as crystalline 30 compound
- compound VII is in a form of L-tartaric acid salt as crystalline compound.
- the present invention provides Vilanterol tartrate and solid state forms thereof, preferably in substantially pure form.
- the present invention comprises Vilanterol tartrate salt, particularly wherein the molar ratio between Vilanterol and tartaric acid can be 1.5 : 1 to 1 : 1, 35 preferably about 1 : 1, respectively.
- the above salts can be isolated.
- the above salts can be in a solid form, more preferably in a crystalline form.
- the Vilanterol tartrate may be in a crystalline form. According to one
- the present invention comprises a crystalline form of Vilanterol
- the crystalline form of Vilanterol tartrate can be characterized by data 5 selected from one or more of the following : an X-ray powder diffraction pattern substantially as depicted in Figure 2; an X-ray powder diffraction pattern having broad peaks at 7.6, 9.8, 12.1, 20.2 and 28.9 degrees two theta ⁇ 1.0 degree two theta; and combinations thereof.
- the above described Vilanterol tartrate and solid state form thereof can be used to prepare Vilanterol or other different salts of Vilanterol, as well as solid state forms thereof and/or pharmaceutical formulations comprising one or more of the salts and /or solid state forms thereof.
- the above described Vilanterol tartrate and solid state form thereof can be used to prepare Vilanterol 20 triphenylacetate (trifenatate) or Vilanterol tosylate.
- Vilanterol tartrate and solid state forms thereof can be used to purify the API Vilanterol.
- the present invention also encompasses a process for preparing other Vilanterol salts.
- the process comprises preparing Vilanterol tartrate and solid state form
- the obtained Vilanterol comprising basifying Vilanterol tartrate and solid state form thereof, and reacting the obtained Vilanterol with a suitable acid, to obtain the corresponding salt of vilanterol .
- the obtained Vilanterol and subsequently the obtained
- Vilanterol salt such as trifenatate salt
- current invention provides a process for preparation of Vilanterol substantially free from impurity A of the following
- the obtained Vilanterol and subsequently the obtained Vilanterol salt are chemically pure, i.e. having content of impurity A at amount of not more than 0.25%, preferably not more than 0.15%, 5 more preferably not more than 0.10%.
- the Vilanterol prepared by the processes of any aspect or embodiment of the present invention can achieve these chemical purities without requiring chromatographic procedures.
- the processes for preparing the Vilanterol salt such as L-tartrate salt or trifenatate salt
- the Vilanterol tartrate and solid state form thereof of the present invention can also be used as a medicament, preferably for the treatment of a person suffering from
- COPD COPD or asthma as a sole product or in a combination therapy with an inhaled 15 corticosteroid .
- the present invention further encompasses 1) a pharmaceutical composition comprising Vilanterol tartrate and solid state form thereof, as described herein; 2) a pharmaceutical formulation comprising Vilanterol tartrate and solid state form
- Powder X-ray diffraction pattern (“powder XRD” or "PXRD”) method
- Powder X-ray Diffraction was performed on an X- ay powder diffractometer
- the study of enzymatic reactions was carried out with enzyme preparations purchased from different commercial sources.
- the enzymatic reactions were studied in biphasic systems (methyl-tert-butyl ether/water and toluene/water, tested in parallel ), in 1200 microliter reaction volumes.
- the substrate 2-bromo-l-(2,2-di methyl-4H-l,3- benzodioxin-6-yl)ethanone was d issolved in the organic solvents (methyl-tert-butyl 15 ether or toluene) in 20 g/l concentration (70.1 mmol/l).
- the aqueous phase consisted of buffered solutions of nicotinamide adenine dinucleotide (NAD, oxidized form, 5
- NADP nicotinamide adeni ne dinucleotide phosphate
- aqueous phase in 4: 1 v/v ratio.
- the enzymatic reactions were set up by mixing 600 25 microliter of substrate solution (in methyl-tert-butyl ether or toluene) with 600
- Substrate 2-brorno-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (5 .00 g), having 84 Area% chromatographic purity, was dissolved in 125 ml diisopropyl ether (DIPE), Triton ® X- 100 emulsifier was added to the solution in 625 mg amount.
- DIPE diisopropyl ether
- reaction mixture was worked up by separating the organic phase, and extracting the remaining aqueous phase two times with 50 ml of methyl-tert-butyl ether. The unified 15 organic phases were dried on anhydrous sodium sulfate and the solvent was
- Substrate 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (25 ,00 g,), having 84 Area% chromatographic purity, was dissolved in 625 ml diisopropyl ether (DIPE), Triton X-100 emulsifier was added to the solution in 3.125 g amount.
- DIPE diisopropyl ether
- Example 4 (comparative example): synthesis of (R)-2-bromo-l-(2,2-dimethyl- 10 4H-l,3-benzodioxin-6-yl)ethanol, effect of enzyme addition order on the
- Substrate 2-bromo-l-(2,2-dimethyl-4H- l,3-benzodioxin-6-yl)ethanone (1.67 g,), having 84 Area% chromatographic purity, was dissolved in 42 ml of diisopropyl ether (DIPE), Triton X- 100 emuisifier was added to the solution in 208.3 mg amount.
- DIPE diisopropyl ether
- the two solutions (in DIPE and in water) were mixed under vigorous stirring, thereafter the alcohol 20 dehydrogenase identified in Example 1 (CDX-005 enzyme, purchased from Codexis) was added (in solid form - without prior dissolution) to the formed emulsion in 166,7 mg amount.
- reaction was performed at 25 C for 24 hours, the pH was adjusted continuously to 6.75 during the reaction by using a pH-stat equipment. After 24 hours of reaction time the reaction mixture was analyzed by HPLC. Conversion of 6% was 25 obtained by the HPLC analysis of the isolated crude product, the majority of the
- IPA isopropanol
- DMSO dimethyl sulfoxide
- a buffered aqueous solution of 50 ml final volume was prepared, containing MES buffer ( 100 mM concentration, 976.2 mg), caprylic acid ( 10 m 35 concentration, 72.1 mg), beta-nicotinamide adenine dinucleotide phosphate
- IMADP + monosodium salt
- the solution was diluted to the final volume (50 ml) and 32 ml of it was added to the IPA-DMSO solution of the starting material, under stirring.
- the 5 mixture was vigorously mixed by magnetic stirrer at 25 °C for 10 minutes. A slightly yellow suspension was obtained.
- KRED-P1-H01 alcohol dehydrogenase purchased from Codexis (Ketoreductase Codex Panel variant plate 1, version 1, well H01, batch number D12071) was dissolved in 200 mg amount in 10 ml of the remained buffered aqueous solution containing 10 caprylic acid and NADP + .
- the prepared enzyme solution was added under vigorous stirring, in one portion to the suspension containing the starting material, prepared as described above. The mixture was stirred for 24 hours at 25 °C temperature.
- reaction reached 95.3% conversion and it was worked up by partial evaporation of IPA and extraction with toluene.
- the extract was dried over l ⁇ la 2 S0 4 , then the solvent was 15 evaporated.
- the crude product was obtained in 4.83 g as light yellow crystalline mass, having 80.2% assay and 99.6% optical purity.
- Example 6 Scaled up preparative reaction for the synthesis of (/?)-2-bromo-l- (2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (compound II), catalyzed by alcohol dehydrogenase KRED-P1-H01 from Codexis 20
- a buffered aqueous solution of 500 ml final volume was prepared, 25 containing MES buffer ( 100 mM concentration, 9762 mg), caprylic acid ( 10 mM
- NADP + monosodium salt
- the solution was diluted to the final volume (500 ml) and 360 30 ml of it was added to the IPA-DMSO solution of the starting material, under stirring.
- KRED-P1-H01 alcohol dehydrogenase purchased from Codexis (Ketoreductase Codex Panel variant plate 1, version 1, well H01, batch number D12071) was dissolved 35 in 2.5 g amount in 100 ml of the remained buffered aqueous solution containing
- the prepared enzyme solution was added under vigorous stirring, in one portion to the suspension containing the starting material, prepared as described above. The mixture was stirred for 24 hours at 25 °C temperature.
- reaction reached 98.5% conversion in 18 hours and >99.5% conversion after 24 hours, 5 thereafter it was worked up by partial evaporation of IPA and extraction with toluene.
- the extract was dried over Na 2 S0 4 , then the solvent was evaporated .
- a light yellow crystalline mass was obtained as crude product.
- the crude product was triturated at room temperature with 1.2 volume of toluene: n-hexane 1 : 1 v/v mixture, thereafter filtered and dried in vacuum oven.
- the product was obtained in 31 g amount as off- 10 white crystalline powder, with 89.6% assay and 99.6% optical purity (first crop) .
- the filtrate was partially evaporated under vacuum then let to cool down to room
- the reactor was sealed and the content was heated to temp. 105-110° C and stirred at 20 the same temp, for 1 hour. The mixture was then cooled down to room temp. The solvent was evaporated under reduced pressure at temp. 30-40° C. The residue was partitioned between EtOAc (400 mL) and water (400 mL). Water phase was separated and the organic phase was washed with water (200 mL). Combined water phases were treated with 20 % aqueous K 2 C0 3 solution (280 mL) followed extraction of the product 25 to EtOAc (2 x 400 mL) . The obtained extract was dried over MgS0 4 and evaporated to dryness to give the title product as slightly yellowish oil (30.28 g, 83 %).
- Example 12 (R)-N-(2-((tert-butyIdimethylsiiyi)oxy)-2-(2,2-dimethyl-4H-l,3- 10 benzodioxin-6-yl)ethyl)-6-(2-((2,6-dichlorobenzyl)oxy)ethoxy)hexan-l-amine fumarate salt (Via fumarate)
- reaction mixture was cooled down to 30°C and toluene ( 1 L) and water
- Example 77(iv)) was dissolved in EtOH (50 mL), followed by addition of 1M HCI solution (50 mL). The mixture was stirred at room temperature for 90 minutes.
- the pH of the mixture was adjusted to ⁇ 9 by addition of 20 % K 2 C0 3 25 solution (25 mL).
- the mixture was then extracted to dichloromethane ( 100 mL).
- the organic phase was washed with water (2 x 25 mL), dried over MgS0 4 and evaporated to dryness.
- the crude vilanterol base ( 14.5 g, 90.9 % purity) was dissolved in
- EtOH (700 mL) was mixed with 1 M aq. HCI acid (700 mL), the formed mixture 25 was cooled to 5 °C, followed by addition of compound VII L-tartrate ( 100 g, obtained by procedure in Example 15). The mixture was stirred at 5 °C for 15 hours. Afterwards, DCM (500 mL) was added, the mixture was cooled to 0 °C and aq. Solution of K 2 C0 3 ( 130g of K 2 C0 3 in 200 mL of water) was then added drop wise to the stirred reaction mixture until pH 9 - 9.5 was obtained. Temp, during the addition was kept below 5 °C. 30 The water phase was separated, and extracted with additional DCM (300 mL).
- Example 23 Comparison of different buffers in the enzymatic 25 enantioselective reduction of 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- y!ethanone (compound I) to (R)-2-bromo-l-(2,2-dimethyI-4H-l,3- benzodioxin-6-yl)ethanone (compound II), catalyzed by ketoreductase CDX- 005 from Codexis
- Enzyme solutions were prepared by dissolving 50-50 milligrams of CDX-005 ketoreductase enzyme (purchased from Codexis) in 2.0 milliliters of each buffered cofactor solution prepared above.
- a substrate solution containing 125 g/l 2-bromo-l-(2,2-dimethyl-4H-l,3- benzodioxin-6-yl)ethanone was prepared in a solvent mixture of isopropano dimethyl 10 sulfoxide 4: 1 v/v, by dissolving 3.125 grams of 2-bromo- l-(2,2-dimethyl-4 -/-l,3- benzodioxin-6-yl)ethanone in the solvent mixture and completing it to 25 milliliters with the same.
- Buffer solution was prepared by dissolving 1180.8 grams of 4-(2- hydroxyethyl)morpholine in 75 liters of water (purified by ion exchange) and adjusting the pH of the solution to 6.75 with nitric acid (20 m/v%).
- a buffered cofactor solution of 90 liters was prepared by dissolving 35.432 10 grams of beta-nicotinamide adenine dinucleotide phosphate disodium salt (NADP+-IMa 2 , oxidized form) in the full volume of the buffer prepared above.
- the pH of the solution was adjusted to 6.75 with the aid of 20% nitric acid, thereafter completed to 90 liters with water (purified by ion exchange).
- Enzyme solution of 18 liters containing CDX-005 (ketoreductase purchased from 15 Codexis) was prepared immediately before use, by dissolving 450 grams of CDX-005 in 18 liters of buffered cofactor solution prepared above. The remaining part of the
- buffered cofactor solution ( ⁇ 72 liters) was loaded in a tempered reactor and stored at 25°C.
- Substrate solution was prepared immediately before use, by dissolving 9 20 kilograms of 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (94% assay, 29.671 moles) in a solution prepared from 18 liters of dimethyl sulfoxide (DMSO) and 54 liters of isopropanol, by stirring at room temperature. The volume of the solution was completed to 90 liters with isopropanol .
- DMSO dimethyl sulfoxide
- the biocatalytic reaction mixture was prepared by adding the prepared 25 substrate solution (90 liters) to the tempered reactor containing ⁇ 72 liters of buffer solution, under vigorous stirring . After proper homogenization the full volume of the enzyme solution ( 18 liters) was charged in one portion to the content of the reactor.
- reaction temperature was maintained at 25°C and the reaction mixture was stirred for 24 hours. By analysis of the reaction mixture 99.7% conversion to Compound II 30 was achieved .
- the reaction mixture was filtered through Perlite and the filtering layer was washed with 54 liters of toluene. The washing containing toluene was stored at 0- 5°C and used during the extraction step. The filtrate was partially evaporated under vacuum at 35°C to 110 liter volume.
- the evaporation was performed in 3 portions.
- the respective portions were stored at 0- 5°C before or after evaporation.
- the remaining volume obtained after partial evaporation of the filtrate ( ⁇ 110 liters) was extracted with 54 liters of toluene (obtained as filtrate after the washing of 5 Perlite layer). Thereafter the remaining aqueous phase was extracted with 2x 27 liters of toluene.
- the unified organic phases were extracted with 36 liters of 5% NaHC0 3 solution, thereafter with 2x 36 liters of water.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Analytical Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
A process for preparing Vilanterol includes a biocatalytic conversion of a ketone substrate to its corresponding alcohol, and then converting the obtained alcohol into Vilanterol. Polypeptides may be used for the biocatalytic conversion of the ketone substrate, such as 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone, to an enantiopure alcohol, (R)-2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol for the preparation of Vilanterol. Also disclosed is vilanterol tartrate and solid state forms thereof for use as medicaments and for the preparation of other vilanterol salts, or of vilanterol, solid state forms and/or formulations thereof. Also disclosed is a process for the preparation of pharmaceutical formulations including vilanterol tartrate and solid state forms thereof, as well as a method of treating a person suffering from COPD and asthma by administering a therapeutically effective amount of any one or a combination of vilanterol tartrate and solid state forms thereof or a pharmaceutical composition and/or formulation comprising vilanterol tartrate and solid state forms thereof.
Description
BIOCATALYTIC PROCESSES FOR THE PREPARATION OF VILANTEROL
This application is related to, and claims the benefit or priorities of, U.S. Provisional Application No. 62/151,016, entitled BIOCATALYTIC PROCESSES FOR THE PREPARATION OF VILANTEROL, filed on 22 April 2015, and U.S. Provisional Application No. 62/186,005, entitled VILANTEROL TARTRATE, filed on 29 June 2015, all of the 5 contents of which are incorporated herein by reference in their entirety for all purposes.
Field of the Invention
This invention relates to highly efficient biocatalytic processes for the
preparation of Vilanterol and intermediates in the preparation thereof.
Background of the Invention 10
Vilanterol, 4-{( lR)-2-[(6-{2-[(2,6-Dichlorobenzyl)oxy]ethoxy}hexyl)amino]-l- hydrox

is reported to be a long-acting 2-adrenoceptor agonist, administered as a dry powder formulation. Vilanterol trifenatate is available in combination with Umeclidinium 15 bromide or Fluticasone furoate for the treatment of chronic obstructive pulmonary disease (COPD) and asthma
Vilanterol, as well as certain pharmaceutically acceptable salts thereof and
processes for the preparation thereof, are described in WO2003024439 and in J. Med.
Chem. 2010, 53 (4522-4530). 20
WO2014041565 describes a similar process to the process described in
WO2003024439 for the preparation of Vilanterol and pharmaceutical acceptable salts thereof, by a process comprising reduction of ketone with borane.
WO2010/025085 describes engineered ketoreductase enzymes allegedly
capable of reducing 5-((4S)-2-oxo-4-phenyl ( l,3-oxazolidin-3-yl))-l-(4-fluorophenyl) 25 pentane-l,5-dione to (4S)-3-[(5S)-5-(4-fluorophenyl)-5-hydroxypentanoyl]-4-phenyl- l,3-oxazolidin-2-one.
2-Bromo- l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol is an alcohol
intermediate that can be used to make Vilanterol.
Liu et al. ; Tetrahedron : Asymmetry 2008, 19, 1824-2828 discloses an 30 enantioselective catalytic hydrogenation of the ketone - 2-bromo-l-(2,2-dimethyl-4H-
l,3-benzodioxin-6-yl)ethanone via a chiral Rh-complex yielding the enantiopure
alcohol, 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol. The enantiomeric excess ("ee") of this process is 93%, and can be increased to 98% only when using
PEGylated chiral ligands.
WO 2004/022547 and US 2005/0075394 describe other synthetic approaches 5 which use 2-azido- or 2-(N-Boc)-amino moieties instead of the 2-bromo moiety. The reduction in the described processes is done with borane dimethylsulfide complex and CBS-borane catalyst. Use of borane and derivatives thereof requires stoichiometric amounts and borane is known to be toxic and not environmentally friendly.
Goswami et al.; Tetrahedron : Asymmetry 2000, 11, 3701-3709 and Goswami et 10 al.; Tetrahedron : Asymmetry 2001, 12, 3343-3348 describe a microbial process,
however, carried out under whole cell fermentation conditions. Whole cells (wet
biomass) were used in a 50-fold mass excess over the substrate, thus require a large excess of whole cell while the substrate, 2-bromo-l-(4H- l,3-benzodioxin-6- yl)ethanone was used in 1 g/l concentration, which is considered very low. In these 15 processes, a 4H-l,3-benzodioxin moiety is used, which has low sensitivity to acids;
therefore its conversion to the corresponding diol requires harsh conditions of
concentrated acids. Such conditions may result in water elimination or racemization, which adversely affects the purity and yield of the final product.
A biocatalytic preparation of other compounds such as (R)-2-azido-l-(4H-l,3- 20 benzodioxin-6-yl)ethanol by whole cell reduction was also described in Procopiou et al. ; Tetrahedron : Asymmetry 2001, 12, 2005-2008. These described reactions were also done with a 1 g/l substrate concentration and yielded only moderate R-selectivity:
enantiomeric excess of 88%.
Moreover, the above described processes utilize intermediates that have limited 25 stability and are oily substances with limited scale-up options and difficult to handle in an industrialized process. Further, the above described processes involve column
chromatography for purification of the intermediates. For at least the above reasons, there is a need to have an improved, safe process with increased efficiency, that can be utilized on an industrial scale, for preparing 2-bromo-l-(2,2-dimethyl-4H-l,3- 30 benzodioxin-6-yl)ethanol and subsequently Vilanterol in high enantiomeric excess (>
98% ee). The desired process comprises a high substrate concentration, allowing high space-time yields (defined as product amount produced per volume and per time, g/l/day or g/l/h). The enzyme (catalyst) concentration should be kept low to allow a commercially competitive process. 35
Summary of the Invention
The present invention relates to a process for preparing Vilanterol, which
comprises a biocatalytic conversion of a ketone substrate to its corresponding alcohol, and then converting the obtained alcohol into Vilanterol.
In one aspect the present invention relates to methods of using polypeptides for the biocatalytic conversion of the Ketone Substrate, as described herein below, to the corresponding alcohol, preferably wherein the Ketone Substrate is 2-bromo-l-(2,2- dimethyl-4H- l,3-benzodioxin-6-yl)ethanone and it is converted to enantiopure
alcohol, (R)-2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol. In particular, these methods may be utilized for the preparation of Vilanterol.
In a further aspect, the present invention provides use of compound of formula V, VI and VII and salts thereof of the following formula:

vi vii 15 in the preparation of Vilanterol. The compounds V, VI and VII are used in the solid form as a salt.
In a further aspect, the present invention provides preparation of Vilanterol in the form of L-tartaric acid salt.
The present invention also encompasses the vilanterol tartrate and solid state forms 20 thereof for the preparation of other vilanterol salts, or of vilanterol, solid state
forms and/or formulations thereof.
The present invention also encompasses the vilanterol tartrate and solid state forms thereof described herein for use as medicaments, particularly sole product or as a combination therapy with an inhaled corticosteroid for COPD and asthma. 25
The present invention also encompasses a process for the preparation of
pharmaceutical formulations comprising combining vilanterol tartrate and solid state forms thereof, or a pharmaceutical composition comprising said vilanterol tartrate and solid state forms thereof and at least one pharmaceutically acceptable
excipient. 30
The present invention also encompasses a method of treating a person suffering from COPD and asthma, comprising administering a therapeutically effective
amount of any one or a combination vilanterol tartrate and solid state forms
thereof, or a pharmaceutical composition and/or formulation comprising vilanterol tartrate and solid state forms thereof described herein.
Brief Description of the Drawings
Figure 1 shows a powder X-ray diffraction pattern ("powder XRD" or "PXRD") of 5 (R)- 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II).
Figure 2 shows a powder X-ray diffraction pattern of 6-(2-((2,6- dichlorobenzyl)oxy)ethoxy)hexan-l-amine L-tartaric acid salt (V L-tartrate).
Figure 3 shows a powder X-ray diffraction pattern of (R)-N-(2-((tert- butyldimethylsilyl)oxy)-2-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethyl)-6-(2-((2,6- 10 dichlorobenzyl)oxy)ethoxy)hexan-l-amine fumarate salt (Via fumarate).
Figure 4 shows a powder X-ray diffraction pattern of (R)-2-((6-(2-((2,6- dichlorobenzyl)oxy)ethoxy)hexyl)amino)-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- yl)ethan-l-ol L-Tartaric acid salt (VII L-tartrate)
Figure 5 shows a powder X-ray diffraction pattern of Vilanterol L-tartrate. 15
Detailed Description of the Invention
The present invention relates to highly efficient biocatalytic processes for the preparation of Vilanterol and Vilanterol intermediates.
The present invention provides an improved, safe process with increased
efficiency, that can be utilized on an industrial scale, for preparing 2-bromo-l-(2,2- 20 dimethyl-4H-l,3-benzodioxin-6-yl)ethanol and subsequently Vilanterol, both in high enantiomeric excess (> 98% ee) and/or optical purity ( >99%).
Particularly, the present invention offers a desirable, well-controlled biocatalytic process, that may be adapted to a large industrial scale, and allows high substrate loadings (e.g ., > 50 g/L), high percent conversion (e.g., > 90% in 24 h), high 25 enantiomeric excess (e.g., at least about 99% ee) and low enzyme loading (e.g., less than 5 w/w%). In addition, the process provided in the present invention eliminates the need for an additional co-factor regenerating enzyme other than the ketoreductase enzyme/polypeptide or engineered ketoreductase polypeptide.
The present invention encompasses crystalline salts of compounds V, VI and VII 30 which are used as intermediates of Vilanterol synthesis.
The present invention encompasses vilanterol tartrate, solid state forms thereof and pharmaceutical compositions comprising one or more of the vilanterol salts and/or solid state forms thereof. The present invention also encompasses vilanterol tartrate
and solid state forms thereof for use in the preparation and purification of Vilanterol and Vilanterol salt, such as Vilanterol trifenatate.
As used herein "ketoreductase" refers to an enzyme/polypeptide that can
catalyse the reduction of a ketone to form the corresponding alcohol. Ketoreductase enzymes include, for example, those classified under the E.C. (or IUBMB - International 5 Union of Biochemistry and Molecular Biology) no. 1.1.1 (i.e. 1.1.1.x) (see
http: //www.enzyme-database.org), as well as synthetic or engineered polypeptide variants thereof (i.e. polypeptides having ketoreductase activity. Ketoreductases
(KREDs) are given various names in addition to ketoreductase, including, but not
limited to, alcohol dehydrogenase, carbonyl reductase, lactate dehydrogenase, 10 hydroxyacid dehydrogenase, hydroxyisocaproate dehydrogenase, [beta]- hydroxybutyrate dehydrogenase, steroid dehydrogenase, sorbitol dehydrogenase, aldoreductase, and the like. NADPH-dependent ketoreductases are classified under the IUBMB number of 1.1.1.2 and the CAS number of 9028-12-0. NADH-dependent
ketoreductases are classified under the IUBMB number of 1.1.1.1 and the CAS number 15 of 9031 -72-5. Preferably, the ketoreductases useful for the processes of the present invention comprise enzymes from IUBMB or E.C. 1.1.1.1 and E.C. 1.1.1.2, more
generally all enzymes belonging to E.C. 1.1.1 and engineered polypeptide variants thereof.
As used herein, the term "co-factor" refers to a non-protein compound that operates in 20 combination with an enzyme which catalyzes the reaction of interest. Co-factors
include, for example, nicotinamide co-factors such as nicotinamide adenine dinucleotide ('TSfAD") or a salt thereof, reduced nicotinamide adenine dinucleotide ("NADH") or a salt thereof, nicotinamide adenine dinucleotide phosphate ("NADP< + >"), reduced
nicotinamide adenine dinucleotide phosphate ("NADPH"), and derivatives or analogs 25 thereof. Reduced cofactors as NADPH or NADH function as intermediate hydrogen
donors, and oxidized cofactors (NADP+ or NAD+) function as intermediate hydrogen acceptors in the catalytic mechanism of the enzymes. Examples of salts of the co- factors include NAD tetra(cyclohexyl ammonium) salt, NAD tetrasodium salt, NAD
tetrasodium hydrate, NADP< + > phosphate hydrate, NADP< + > phosphate sodium salt, 30 and NADH dipotassium salt. Preferably, in the processes of the present invention, the co-factor is NADP or NADPH.
As used herein, the term "isolated" in reference to compounds described in the invention corresponds to a compound that is physically separated from the reaction mixture in which it is formed. 35
As used herein, the term "isolated" in reference to polypeptides/enzymes refers to polypeptides/enzymes at least partially separated from the environment in which they are formed, for example from the natural environment, e.g., from bacteria. Thus, use of the term "isolated" indicates that a naturally occurring or recombinant enzyme has been at least partially removed from its normal cellular or natural environment, 5 e.g . from bacterial cells. Preferably, the isolated enzyme is in a cell-free system. The isolated enzyme can be crude or highly purified. The term "isolated" does not
necessarily imply that the enzyme is the only enzyme present, but that it is the
predominant enzyme present (at least 10-20% more than any other enzyme). As used herein, when applied to an enzyme, the term "synthesized" or "engineered" refers to 10 an enzyme that is prepared by chemical synthesis, recombinant means, or the
combination thereof. As used herein, when applied to an enzyme, the term "purified" refers to an enzyme that is essentially free (at least about 90-95% pure) of non- enzymatic material or other enzymes. The isolated enzyme can be a lysate or an
enzyme powder obtained by lyophilization of cell lysates, which can contain isolated, 15 but unpurified enzymes.
Preferably, in any aspect or embodiment of the present invention, the
ketoreductase is isolated. The ketoreductase can be separated from any host, such as mammals, filamentous fungi, yeasts, and bacteria. The isolation, purification, and
characterization of a NADH-dependent ketoreductase is described in, for example, in 20 Kosjek et al, Purification and Characterization of a Chemotolerant Alcohol
Dehydrogenase Applicable to Coupled Redox Reactions, Biotechnology and
Bioengineering, 86 : 55-62 (2004) , Preferably, the ketoreductase is synthesized or engineered. The ketoreductase can be synthesized chemically or using recombinant means. The chemical and recombinant production of ketoreductases is described in, 25 for example, in US2016/0083759, WO2010025085, WO2011022548, and
WO2009046153. Preferably, the ketoreductase is purified, preferably with a purity of about 90% or more, more preferably with a purity of about 95% or more, Preferably, the ketoreductase is substantially cell-free.
A thing, e.g., a reaction mixture, may be characterized herein as being at, or 30 allowed to come to "room temperature, often abbreviated "RT." This means that the temperature of the thing is close to, or the same as, that of the space, e.g ., the room or fume hood, in which the thing is located. Typically, room temperature is from about 20°C to about 30°C, or about 22°C to about 27°C, or about 25°C.
A process or step may be referred to herein as being carried out "overnight."
This refers to a time interval, e.g . , for the process or step, that spans the time during the night, when that process or step may not be actively observed. This time interval is from about 8 to about 20 hours, or about 10- 18 hours, typically about 16 hours.
As used herein, the term "reduced pressure" refers to a pressure of about 10 5 mbar to about 50 mbar.
As used herein, the terms "vol. " or "volume" can be used to refer to ml per gram of the corresponding Vilanterol salts. For example, a statement that 0.5 g of Vilanterol is d issolved in ten volumes of a Solvent X would be u nderstood to mean that the 0.5 g of Vilanterol was dissolved in 5 ml of Solvent X . 10
A crystal form may be referred to herein as being characterized by graphical data "substantially as depicted in" a Figure. Such data include, for example, powder X-ray diffractograms and solid state NMR spectra. As is well-known in the art, the graphical data potentially provides additional technical information to fu rther define the 15 respective solid state form (a so-called "fingerprint") which can not necessarily be described by reference to numerical values or peak positions alone. In any event, the skilled person will understand that such graphical representations of data may be
subject to small variations, e. g., in peak relative intensities and peak positions due to factors such as variations in i nstrument response and variations in sample 20 concentration and purity, which are well known to the skilled person. Nonetheless, the skilled person would readily be capable of comparing the graphical data in the Figures herein with g raphical data generated for an unknown crystal form and confirm whether the two sets of g raphical data are characterizing the same crystal form or two different crystal forms. A crystal form of a Vilanterol salt referred to herein as being 25 characterized by graphical data "substantially as depicted in" a Figure will thus be
understood to include any crystal forms of the Vilanterol salt characterized with the graphical data having such small variations, as are well known to the skilled person, in comparison with the Figure.
As used herein, unless stated otherwise, the XRPD measurements are taken 30 using copper a radiation wavelength ( 1.5418 A).
As used herein and unless indicated otherwise, the term "solvate" refers to a crystal form that incorporates a solvent in the crystal structure. When the solvent is water, the solvate is often referred to as a "hydrate. " The solvent in a solvate may be present in either a stoichiometric or in a non-stoichiometric amount. 35
In one embodiment, the present invention relates to polypeptides having
ketoreductase activity, and to methods of using the polypeptides for the biocatalytic conversion of the Ketone Substrate of the following structure:

Ketone Substrate 5 to the corresponding alcohol of the following structure:
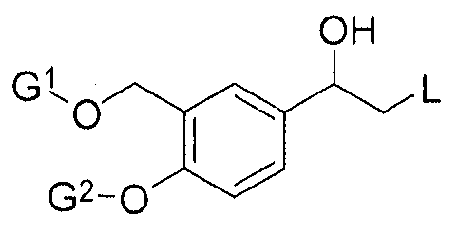
Alcohol Compound
wherein L is a leaving group, that may be selected for example from halogen containing groups, typically a chloro, bromo or iodo group; or a sulphonate group such as an 10 alkylsulphonate (particularly d.6 alkylsulphonates), typically methane sulphonate; or an aryl sulphonate (particularly C6.10 arylsulfonate) group, typically a toluenesulphonate group (e.g. para-toluenesulphonate) ; G^nd G2 may each independently be a hydroxyl- protecting group or a hydrogen.
Suitable hydroxyl-protecting protecting groups can be silyl-type protecting 15 groups according to the formula -SiR^R3, wherein R1, R2 and R3 are independently selected from : a C1-C15 straight or branched alkyl group, a C1-C10 cycloalkyl (preferably C3- 10 cycloalkyl or C5-8 cycloalkyl) group, an optionally substituted C6-Ci0 aryl group and an optionally substituted C7-Ci2 arylalkyl group. Examples of preferred silyl-type
protecting groups are t-butyldimethylsilyl (TBDMS), triethylsilyl (TES), t- 20 butyldiphenylsilyl (TBDPS), and trimethylsilyl (TMS) . Alternatively, the hydroxyl- protecting groups G1, G2 may be independently selected from: ether groups (e.g. Ci- 10 alkyl or C5- i0 cyclic ethers, preferably C5.s cyclic ethers, methyl ethers or ethyl ethers) or ester groups (e.g. Ci- i0 alkyl esters, preferably Ch alky! esters, or Cs- io aryl esters, or C7.11 araalkyl esters). Particularly, the hydroxyl protecting groups G1, G2 may be 25 independently selected from methyl or substituted methyl groups, typically
tetrahydropyranyl (ΤΉΡ), methoxymethyl (MOM), benzyloxymethyl ; or ethyl or
substituted ethyl groups, typically ethoxyethyl, benzyl or tert-butyl ; or ester groups, typically acetate, or aryl substituted acetate groups, for example benzoate or
substituted benzoate groups. Additional hydroxyl-protecting groups can be selected 30
from those described in Greene and Wuts "Greene's Protective Groups in Organic Synthesis", 4th Edition, publ. Wiley, 2006.
Preferably, hydroxyl-protecting groups G1 and G2 may together represent an group suitable for protection of 1,3 diols, for example cyclic acetal or ketal, typically methylene acetal, ethylidene acetal, isopropylidene acetal (acetonide). Preferably G1, G2 is isopropylidene acetal (acetonide).
Preferably, G1 and G2 of the Ketone Substrate together represent isopropylidene acetal (acetonide), and the Ketone Substrate is 2-bromo-l-(2,2-dimethyl-4H-l,3- benzodioxin-6-yl)ethanone, which is referred to as Substrate I.
Preferably, Substrate I is converted to enantiopure alcohol : (R)-2-bromo-l-(2,2- dimethyl-4H-l,3-benzodioxin-6-yl)ethanol, which is referred to herein as Compound II.
Preferably, by "enantiopure", it is mean that the compound has an optical purity of : ≥ 85%, > 90%, > 92%, > 94%, 96%,≥ 98%, > 99%, > 99.5%, or > 99.8%, and more preferably an optical purity of ≥ 99%, > 99.5%, or > 99.8%.
Compound II may be isolated, preferably it is crystalline. The invention also comprises crystalline Compound II, as described herein below.
In particular, the above described methods may be utilized for the preparation of Vilanterol .
The process can be illustrated by Scheme 1.
Scheme 1 :
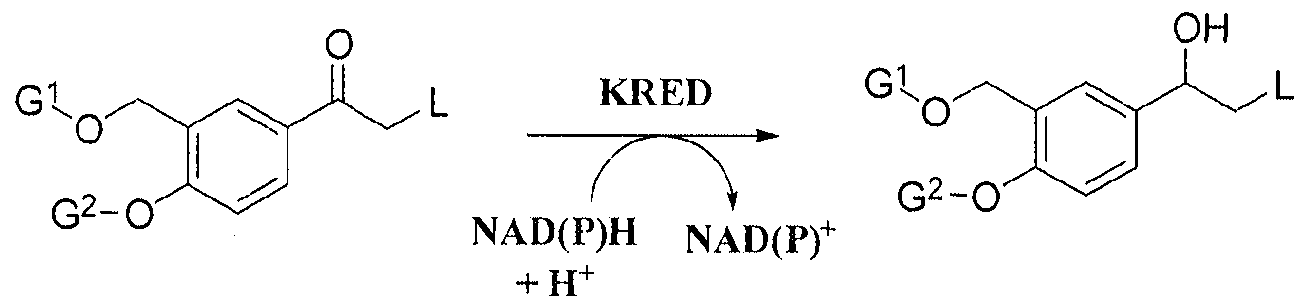
wherein G1, G2 and L are as defined above.
Particularly, when Substrate I is used to produce Compound II, as defined the process can be illustrated by the following scheme:
Scheme 2 :
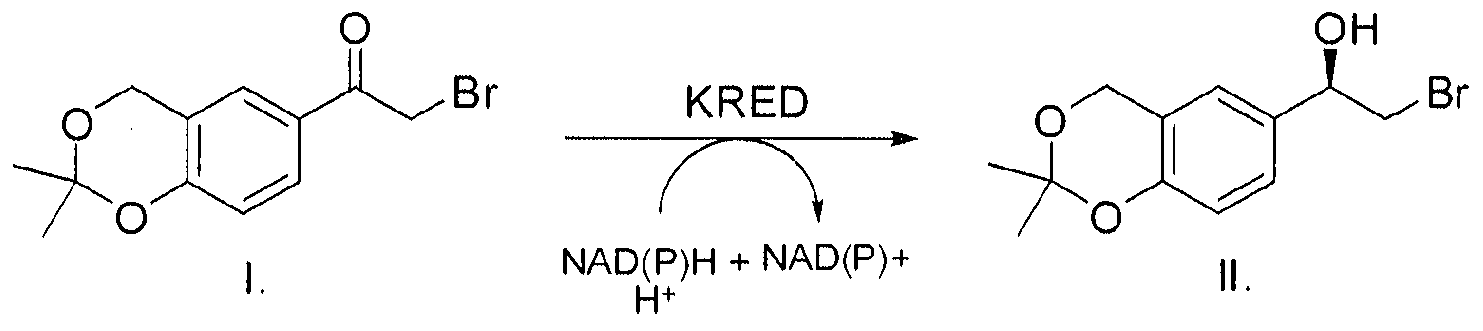
In another embodiment, the present invention comprises a process for biocatalytic reduction of the Ketone Substrate, as described above, to the
corresponding alcohol, as described above, preferably wherein the Ketone Substrate is Substrate I and it is converted to enantiomerically pure alcohol, Compound II. This process is done by using an isolated enzyme capable of keto-reductase activity.
Examples of enzymes capable of ketoreductase activity are engineered
ketoreductase polypeptides such as those disclosed in US2016/0083759, 5 WO2010025085, WO2011022548, and WO2009046153, herein incorporated by
reference. Examples of suitable enzymes may be the commercially available enzymes such as: Codexis CDX-005 or Codexis KRED-P1-H01, or an equivalent enzyme thereof.
As used herein, the term equivalent enzyme refers to an enzyme with similar or identical enzymatic activity, which produces the product in the desired enantiomeric 10 access and optical purity, as described in this invention.
According to the present invention, the effective amount of an enzyme (or
combination of enzymes) may be any amount of the enzyme that is sufficient to
achieve a desired degree of conversion of a substrate, for example, at least 90%, preferably at least 95%, more preferably at least 98% conversion of a substrate, 15 during 24 hours of reaction time,
The above process typically allows utilizing substrate in a concentration of at least 20 g/l, preferably at least 40 g/l, more preferably at least 100 g/l .
Preferably the required quantities of the enzyme and the co-factor (NADP or
NADPH) are dissolved in an aqueous buffer. The enzyme and substrate pair used for 20 the regeneration of the co-factor (e.g. glucose dehydrogenase and glucose; formate dehydrogenase and sodium formate; phosphite dehydrogenase and sodium phosphite) are dissolved in the same aqueous solution and the pH is adjusted to the required
value. Thereafter the substrate (2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- yl)ethanol) is dissolved separately in a water immiscible organic solvent, and a non- 25 ionic tensioactive agent (surfactant) (preferably Triton® X- 100) is dissolved in the
solution. The solution of the substrate in organic solvent may be slowly added to the aqueous solution containing the enzyme and the co-factor under vigorous stirring. The pH of the reaction system may be kept constant during the enzymatic reaction by
addition of an acid or a base if required. 30
The process may be done with a biphasic solvent system, i.e., comprising an aqueous buffer and a water immiscible solvent. Typically, the enzyme is dissolved in aqueous buffer. Examples of aqueous buffer includes potassium or sodium phosphate, hydrochloride or sulfate salts of tertiary amines, triethanolamine, TRIS
(tris(hydroxymethyl)aminomethane), sodium- or potassium salts of MES (2-(N- 35
morpholino)ethanesulfonic acid), HEPES (4-(2-hydroxyethyl)-l- piperazineethanesulfonic acid), MOPS (3-(N-morpholino)propanesulfonic acid), etc.
Examples of water immiscible organic solvents include ethers, esters, aromatic and aliphatic hydrocarbons or their mixtures, preferably diisopropyl ether, methyl tert-butyl ether, ethyl acetate, isobutyl acetate, butyl acetate, ethyl butanoate, toluene, hexane, 5 heptane or mixtures thereof.
Alternatively, the process can be done with a monophasic solvent system
comprising an aqueous buffer and a water miscible organic solvent. Examples of water miscible solvents include alcohols, aprotic polar solvents or mixtures thereof, preferably isopropanol, ethanol, isobutanol, dimethyl sulfoxide, dimethyl formamide, dimethyl 10 acetamide, acetonitrile or mixtures thereof.
Typically the amount of solvent utilized is an amount necessary to dissolve the enzyme and substrate. Typically, the amount of water immiscible organic solvent may be from about 20 to about 75 v/v%, from about 30 to about 65v/v% or from about 45 to about 55 v/v%. The amount of water miscible solvent may be from about 10 to 15 about 75 v/v%, from about 25 to about 65 v/v%, or from about 50 to about 55v/v%.
The process is typically performed in the presence of a suitable co-factor as
NADH or NADPH, and a co-factor regenerating system capable of converting NADP+ to NADPH, or NAD+ to NADH . Optionally, the reaction mixture further comprises a co- factor regenerating/recycling system. Typically, the co-factor regenerating/recycling 20 system comprises a substrate and a dehydrogenase. The co-factor recycling system can comprise an additional enzyme and its substrate in more than 1 equivalent amount relative to Ketone Substrate I. The co-factor recycling system can comprise, for
example, the following pairs of enzymes and substrates: glucose dehydrogenase and glucose, formate dehydrogenase and sodium formate or phosphite dehydrogenase and 25 sodium phosphite, alcohol dehydrogenase and a secondary alcohol (e.g. isopropanol).
In some of the examples subject to the scope of this invention the ketoreductase
polypeptide (enzyme) can regenerate the co-factor itself, in the presence of
isopropanol as water miscible co-solvent, or part of the water-miscible co-solvent
mixture. Reductive equivalents are transferred from the isopropanol to the oxidized co- 30 factor (NAD+ or NADP+), under the effect of the enzyme, while the co-factor is
reduced (to NADH or NADPH) and isopropanol is oxidized to acetone.
In another embodiment, the present invention comprises a process for
preparing Vilanterol, which comprises a biocatalytic conversion of a ketone substrate to its corresponding alcohol, and then converting the obtained alcohol into Vilanterol. 35
Such process may comprise:
a) converting a Ketone Substrate, such as Substrate I, into enantiomerically pure alcohol, preferably Compound II; and
al) converting the formed alcohol, preferably Compound II, to Vilanterol trifenatate, according to the below scheme:



VII L-tait rate


The process of step a) is preferably done by using an isolated enzyme capable of ketoreductase activity. Such enzyme may be as described above.
The amount of enzyme is typically as described above.
The above process typically allows utilizing substrate in the above described concentration.
The order of addition is typically as described above. Preferably, the process is done with a biphasic solvent system, i.e., comprising an aqueous buffer and a water immiscible solvent. Typically, the enzyme is dissolved in the aqueous buffer. Examples of aqueous buffers and of water immiscible solvents are described above. Alternatively the process can be done with a monophasic solvent system comprising an aqueous buffer and a water miscible organic solvent. Examples of water miscible solvent are described above.
Typically the amount of solvent(s) utilized and their ratio is the amount necessary to dissolve the enzyme and substrate, as described above.
The process of step al) is preferably done by introduction of protecting group (PG) and preparation of O-protected compound III and compound VI. Suitable PGs are as described above.
In another embodiment, the present invention comprises crystalline Compound II. The crystalline form can be characterized by data selected from one or more of the following : an X-ray powder diffraction pattern having peaks at 9.8, 14.9, 15.9, 18.2 and 19.9 degrees two theta ± 0.2 degrees two theta; an X-ray powder diffraction pattern substantially as depicted in Figure 1 ; and combinations thereof.
The crystalline form of Compound II may be further characterized by an X-ray powder diffraction pattern having any one, two, three, four or five additional peaks selected from peaks at 8.0, 18.7, 21.9, 24,0 and 25.6 degrees two theta ± 0.2 degrees two theta. The full peak list of Compound II is presented in the following table (Table 1) :
Table 1 :
Pos. Rel. Int.
[°2Θ] [%]
8.0 1
9.8 6
14.9 18
15.9 83
16.4 2
17.9 4
18.2 22
18.7 19
19.9 63
21.6 6
21.9 17
22.6 1
24.0 100
24.3 28
25.3 18
25.6 27
26.9 25
27.1 3
28.2 5
28.9 9
29.5 30
30.3 3
31.5 11
31.8 9
32.2 30
33.6 9
34.2 10
35.5 3
36.5 1
37.2 13
37.5 6
38.3 1
39.0 2
39.8 10
The crystalline form of Compound II may alternatively be characterized by the peaks presented in Table 1 above, optionally with the relative intensities.
In another embodiment, the present invention comprises the preparation of compound V from compound IV by reaction with ammonia. Compound V may be used in the process of the invention as free base or in the form of a salt. Suitable salts may include but are not limited to inorganic acid salts, for example hydrochloride, hydrobromide, phosphate or sulphate, or organic acid salts. A suitable organic acid can be selected from acetic acid and its derivatives, benzoic acid or substituted benzoic
acids, methanesulfonic acid, benzenesulfonic acid or substituted benzenesulfonic acid, citric acid, maleic acid, malic acid, maleic acid, malonic acid, mandelic acid, succinic acid, fumaric acid, pyroglutamic acid, oxalic acid, tartaric acid or derivatives thereof. More preferably the suitable acid can be selected from tartaric acid, preferably in its optically pure form (preferably L-tartaric acid). 5
In another embodiment, the present invention comprises the preparation of compound VI by reaction of compound III and compound V. Compound VI may be used in the process of the invention as free base or in the form of a salt. Suitable salts may include but are not limited to inorganic acid salts, for example hydrochloride, hydrobromide, phosphate or sulphate, or organic acid salts. A suitable organic acid can 10 be selected from acetic acid and its derivatives, benzoic acid or substituted benzoic acids, methanesulfonic acid, benzenesulfonic acid or substituted benzenesulfonic acid, citric acid, maleic acid, malic acid, maleic acid, malonic acid, mandelic acid, succinic acid, fumaric acid, pyroglutamic acid, oxalic acid, tartaric acid or derivatives thereof. Preferably the suitable acid can be selected from fumaric acid or tartaric acid, 15 preferably in its optically pure form (preferably L-tartaric acid), more preferably fumaric acid.
In another embodiment, the present invention comprises the preparation of compound VII from compound VI by selective deprotection of PG. Compound VII may be used in the process of the invention as free base or in the form of a salt. 20
Suitable salts may include but are not limited to inorganic acid salts, for
example hydrochloride, hydrobromide, phosphate or sulphate, or organic acid salts. A suitable organic acid can be selected from acetic acid and its derivatives, benzoic acid or substituted benzoic acids, methanesulfonic acid, benzenesulfonic acid or substituted benzenesulfonic acid, citric acid, maleic acid, malic acid, maleic acid, malonic acid, 25 mandelic acid, succinic acid, fumaric acid, pyroglutamic acid, oxalic acid, tartaric acid or derivatives thereof. More preferably the suitable acid can be selected from tartaric acid, preferably in its optically pure form (preferably L-tartaric acid).
In the preferred embodiment compound V is in the form of L-tartaric acid salt as crystalline compound, compound VI is in the form of fumaric acid salt as crystalline 30 compound, compound VII is in a form of L-tartaric acid salt as crystalline compound.
In another embodiment, the present invention provides Vilanterol tartrate and solid state forms thereof, preferably in substantially pure form. In specific
embodiments, the present invention comprises Vilanterol tartrate salt, particularly wherein the molar ratio between Vilanterol and tartaric acid can be 1.5 : 1 to 1 : 1, 35
preferably about 1 : 1, respectively. The above salts can be isolated. Preferably, the above salts can be in a solid form, more preferably in a crystalline form.
The Vilanterol tartrate may be in a crystalline form. According to one
embodiment, the present invention comprises a crystalline form of Vilanterol
tartrate. The crystalline form of Vilanterol tartrate can be characterized by data 5 selected from one or more of the following : an X-ray powder diffraction pattern substantially as depicted in Figure 2; an X-ray powder diffraction pattern having broad peaks at 7.6, 9.8, 12.1, 20.2 and 28.9 degrees two theta ± 1.0 degree two theta; and combinations thereof.
The above described Vilanterol tartrate and solid state form thereof is 10 particularly advantageous for the purification of the API Vilanterol. In some
embodiments the present invention comprises a process for the purification of
Vilanterol or Vilanterol salt, said process comprising preparing Vilanterol tartrate according to the process of the present invention and converting it to Vilanterol or to Vilanterol salt. 15
The above described Vilanterol tartrate and solid state form thereof can be used to prepare Vilanterol or other different salts of Vilanterol, as well as solid state forms thereof and/or pharmaceutical formulations comprising one or more of the salts and /or solid state forms thereof. Preferably, the above described Vilanterol tartrate and solid state form thereof can be used to prepare Vilanterol 20 triphenylacetate (trifenatate) or Vilanterol tosylate. Particularly, Vilanterol tartrate and solid state forms thereof can be used to purify the API Vilanterol.
The present invention also encompasses a process for preparing other Vilanterol salts. The process comprises preparing Vilanterol tartrate and solid state form
thereof by the processes of the present invention, and converting that salt to said 25 other Vilanterol salt. The conversion can be done, for example, by a process
comprising basifying Vilanterol tartrate and solid state form thereof, and reacting the obtained Vilanterol with a suitable acid, to obtain the corresponding salt of vilanterol . Preferably, the obtained Vilanterol and subsequently the obtained
Vilanterol salt, such as trifenatate salt, are chemically pure, i.e. having total 30 impurity at amount of not more than 10%, preferably not more than 5%, more preferably not more than 0.5%. Specifically, current invention provides a process for preparation of Vilanterol substantially free from impurity A of the following
formula :

Preferably, the obtained Vilanterol and subsequently the obtained Vilanterol salt, such as L-tartrate salt or trifenatate salt, are chemically pure, i.e. having content of impurity A at amount of not more than 0.25%, preferably not more than 0.15%, 5 more preferably not more than 0.10%. Advantageously, the Vilanterol prepared by the processes of any aspect or embodiment of the present invention can achieve these chemical purities without requiring chromatographic procedures. Thus, in preferred embodiments of the present invention, the processes for preparing
Vilanterol having the chemical and/or optical purities described herein, do not 10 involve a chromatographic procedure (e.g . column chromatography).
The Vilanterol tartrate and solid state form thereof of the present invention can also be used as a medicament, preferably for the treatment of a person suffering from
COPD or asthma as a sole product or in a combination therapy with an inhaled 15 corticosteroid .
The present invention further encompasses 1) a pharmaceutical composition comprising Vilanterol tartrate and solid state form thereof, as described herein; 2) a pharmaceutical formulation comprising Vilanterol tartrate and solid state form
thereof, as described herein, and at least one pharmaceutically acceptable 20 excipient; and 3) a process to prepare such formulations comprising combining
Vilanterol tartrate and solid state form thereof, or pharmaceutical compositions and at least one pharmaceutically acceptable excipient; 4) the use of Vilanterol tartrate and solid state form thereof in the manufacture of a pharmaceutical composition, and 5) a method of treating a person suffering from COPD or asthma, comprising 25 administering a therapeutically effective amount of a pharmaceutical composition or formulation comprising Vilanterol tartrate and solid state form thereof as described herein.
Having described the invention with reference to certain preferred
embodiments, other embodiments will become apparent to one skilled in the art from 30 consideration of the specification. The invention is further illustrated by reference to the
following examples describing in detail the preparation of the composition and methods of use of the invention. It will be apparent to those skilled in the art that many
modifications, both to materials and methods, may be practiced without departing from the scope of the invention.
Examples
Powder X-ray diffraction pattern ("powder XRD" or "PXRD") method
Powder X-ray Diffraction was performed on an X- ay powder diffractometer
Analytical X'pert Pro; CuKa radiation (λ = 1.541874 A?) ; X'Celerator detector with active length 2.122 degrees 2-theta; laboratory temperature 25 ± 2 °C; zero
background sample holders. Prior to analysis, the samples were gently ground using a mortar and pestle to obtain a fine powder. The ground sample was adjusted into a cavity of the sample holder and the surface of the sample was smoothed using a cover glass.
Optical purity measurement (Compound II)
Optical purity of compound II was determined on a chiral column (Chiralpak IF-3, 3.0 μιτι particle size, 250 x 4.6 mm) using the following measurement parameters:
Scan range 3 - 40 degrees 2-theta
Scan mode continuous
Step size 0.0167 degrees
Step size 42 s
Sample spin 60 rpm
Sample holder zero background silicon
20
Gradient elution :
eluent A: hexane/ethanol/methanol 98: 1 : 1 (v/v)
eluent B: hexane/ethanol 1 : 1 (v/v)
Gradient program : 25
 flow: 2 ml/min
flow: 2 ml/min
25 °C column temperature
detection : UV, 232 nm 5 injection volume : 20 μΙ_
analysis time : 40 minutes
diluent for sample preparation : hexane/ethanol 1 : 1 (v/v)
Example 1 : Screening of available Ketoreductases 10
The study of enzymatic reactions was carried out with enzyme preparations purchased from different commercial sources. The enzymatic reactions were studied in biphasic systems (methyl-tert-butyl ether/water and toluene/water, tested in parallel ), in 1200 microliter reaction volumes. The substrate 2-bromo-l-(2,2-di methyl-4H-l,3- benzodioxin-6-yl)ethanone was d issolved in the organic solvents (methyl-tert-butyl 15 ether or toluene) in 20 g/l concentration (70.1 mmol/l). The aqueous phase consisted of buffered solutions of nicotinamide adenine dinucleotide (NAD, oxidized form, 5
mmol/l), nicotinamide adeni ne dinucleotide phosphate (NADP, oxidized form, 5
mmol/l), g lucose dehyd rogenase (3 g/l), glucose (500 mmol/l), emulsifier Triton® X-
100 (0.5 w/w%) . Buffers of potassiu m hydrogen phosphate (at pH = 7.00) and 20 triethanolamine sulfate (at pH = 7.50) were used in the above reactions and tested in parallel. The enzymes tested in the form of lyophilisates were dissolved in the aqueous phase, in 2 g/l concentrations. Enzymes tested in the form of cell lysates (enzyme solutions obtained after the lysis of the biomass) were mixed with the mentioned
aqueous phase in 4: 1 v/v ratio. The enzymatic reactions were set up by mixing 600 25 microliter of substrate solution (in methyl-tert-butyl ether or toluene) with 600
microliter of enzyme solutions. The reactions were run in closed ampoules, under
shaking (350 rpm), at room temperature. The reaction mixtures were analysed by
HPLC for residual 2-bromo- l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone
(substrate) content and 2-bromo-l -(2,2-di methyl-4H-l,3-benzodioxin-6-yl)ethanol (product) content and for optical purities. CDX-005 and KRED-P1-H01 , both enzymes available from Codexis, gave the highest conversions (67.1 % and 95.9% conversions, respectively), the obtained bromoalcohol product had 98.5% and 99.2% optical purity (R enantiomer) . The high majority of the tested enzymes was S-selective, as can be 5 seen in the next table (Table 2) :Table 2 :
Optical
Sample Conversion
Enzyme Configuration purity
number (%)
(%)
07/02 CDX-005 from Codexis 67.13 R 98.5
07/31 Codexis KRED-P2-G03 89.40 S 97.9
07/05 CDX-023 from Codexis 1.67 S 72.4
07/06 CDX-025 from Codexis 73.98 s 91.2
07/15 CDX-078 from Codexis 40.65 s 97.9
07/16 CDX-051 from Codexis 99.90 s 99.7
07/17 PRZ-010 from Prozomix 13.90 s 60.2
07/42 PRZ-012 from Prozomix 14.84 s 96.5
07/19 PRZ-020 from Prozomix 37.19 s 95.5
07/44 PRZ-067 from Prozomix 54.06 s 99.0
07/45 PRZ-077 from Prozomix 38.29 s 98.6
LbADH
07/28 3.06 s 91.5
Q03TF9
Leifsonia sp,
08/34 8.44 s 95.50
Q4R1E0
Sporobolomyces
08/05 salmonicolor 19.12 s 96.80
Q9UUN9
Pseudomonas fluorescens
08/15 2.36 R 90.70
Q8RTR1
Saccharomyces
08/44 cerevisiae-YGL157W 2.44 R 92.10
P53111
23/6 Codexis KRED-P1-H01 95.94 R 99.2
Example 2: Preparative reaction for the synthesis of (R)-2-bromo-l-(2,2- dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II)
Substrate 2-brorno-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (5 .00 g), having 84 Area% chromatographic purity, was dissolved in 125 ml diisopropyl ether (DIPE), Triton® X- 100 emulsifier was added to the solution in 625 mg amount. A 5 buffered aqueous solution containing the alcohol dehydrogenase enzyme CDX-005 from Codexis, co-factor, glucose and glucose dehydrogenase was prepared separately: a 0.2 M PIPES buffer (pH = 7.0) was prepared in 100 ml amount, NADP (76.5 mg), glucose (4000 mg), glucose dehydrogenase (100 mg) and the alcohol dehydrogenase (750 mg lyophilizate) were dissolved in it and the pH was readjusted to 7.00 with potassium 10 hydroxide solution. The two solutions (in DIPE and in water) were mixed under
vigorous stirring, the reaction was performed at 25 °C for 24 hours. In the meantime the pH of the reaction mixture was controlled and readjusted manually to 7.00. The reaction mixture was worked up by separating the organic phase, and extracting the remaining aqueous phase two times with 50 ml of methyl-tert-butyl ether. The unified 15 organic phases were dried on anhydrous sodium sulfate and the solvent was
evaporated . According to the HPLC analysis of the obtained crude product, the
conversion was 99.4% during the enzymatic reaction. The crude product was purified by column chromatography over neutral silica gel (40-60 micrometer particle size) with n-hexane/ethyl acetate 3 :2 v/v as eluent. The fractions containing (R)-2-bromo-l-(2,2- 20 dimethyl-4H- l,3-benzodioxin-6-yl)ethanol were unified and the solvent was
evaporated under vacuum. In total, 3.52 g of purified (R)-2-bromo-l-(2,2-dimethyl- 4H- l,3-benzodioxin-6-yl)ethanol were obtained, having 88.2 Area% chromatographic purity and 99.0% optical purity. The yield of obtained isolated material is 73.2%
(according to chromatographic purities of substrate and product, in area%). 25 Example 3: Scaled up preparative reaction for the synthesis of (R)-2-bromo-l- (2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II)
Substrate 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (25 ,00 g,), having 84 Area% chromatographic purity, was dissolved in 625 ml diisopropyl ether (DIPE), Triton X-100 emulsifier was added to the solution in 3.125 g amount. A 30 buffered aqueous solution containing the alcohol dehydrogenase enzyme CDX-005 purchased from Codexis, co-factor, glucose and glucose dehydrogenase was prepared separately: a 0.1 M triethanolamine hydrochloride buffer (pH = 6.75) was prepared in 500 ml amount, IMADP (382.5 mg), glucose (20 g), glucose dehydrogenase (375 mg) and the alcohol dehydrogenase (2.50 g lyophilizate) were dissolved in it and the pH 35
was readjusted to 6.75 with potassium hydroxide solution. The two solutions (in DIPE and in water) were mixed under vigorous stirring, the reaction was performed at 25° C for 24 hours, the pH was adjusted continuously to 6.75 during the reaction by using a pH-stat equipment. After 24 hours of reaction time the reaction mixture was worked up similarly to Example 3, using ethyl acetate as solvent for extraction instead of methyl- 5 tert-butyl ether. Conversion of 95.6% was obtained by the HPLC analysis of the
isolated crude product, the optical purity of (R)-2-bromo-l-(2,2-dimethyl-4H-l,3- benzodioxin-6-yl)ethanol was 99.0%. The sample was analyzed by PXRD, the PXRD pattern is presented in Figure 1.
Example 4 (comparative example): synthesis of (R)-2-bromo-l-(2,2-dimethyl- 10 4H-l,3-benzodioxin-6-yl)ethanol, effect of enzyme addition order on the
conversion
Substrate 2-bromo-l-(2,2-dimethyl-4H- l,3-benzodioxin-6-yl)ethanone (1.67 g,), having 84 Area% chromatographic purity, was dissolved in 42 ml of diisopropyl ether (DIPE), Triton X- 100 emuisifier was added to the solution in 208.3 mg amount. A 15 buffered aqueous solution containing co-factor and glucose was prepared separately: a 0.1 M triethanolamine sulfate buffer (pH = 6.75) was prepared in 33 ml amount, NADP (25.5 mg), glucose (1.33 g), glucose dehydrogenase (25 mg) were dissolved in it and the pH was readjusted to 6.75 with potassium hydroxide solution. The two solutions (in DIPE and in water) were mixed under vigorous stirring, thereafter the alcohol 20 dehydrogenase identified in Example 1 (CDX-005 enzyme, purchased from Codexis) was added (in solid form - without prior dissolution) to the formed emulsion in 166,7 mg amount. The reaction was performed at 25 C for 24 hours, the pH was adjusted continuously to 6.75 during the reaction by using a pH-stat equipment. After 24 hours of reaction time the reaction mixture was analyzed by HPLC. Conversion of 6% was 25 obtained by the HPLC analysis of the isolated crude product, the majority of the
substrate remained unreacted in the reaction mixture.
Example 5: Preparative reaction for the synthesis of ( ?)-2-bromo-l-(2,2- dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (compound II), catalyzed by
alcohol dehydrogenase KRED-P1-H01 from Codexis 30
Substrate 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (starting material, 5.00 g, 15.87 mmol), having 90.5 Area% chromatographic purity, was
dissolved in a mixture of 36 ml isopropanol (IPA) and 22 ml dimethyl sulfoxide (DMSO).
Separately a buffered aqueous solution of 50 ml final volume was prepared, containing MES buffer ( 100 mM concentration, 976.2 mg), caprylic acid ( 10 m 35
concentration, 72.1 mg), beta-nicotinamide adenine dinucleotide phosphate
monosodium salt (IMADP+, oxidized form, 1 mM concentration, 38.3 mg). The pH of the aqueous solution was adjusted with NaOH 1 M solution to pH = 6.50 before the
addition of the NADP+. The solution was diluted to the final volume (50 ml) and 32 ml of it was added to the IPA-DMSO solution of the starting material, under stirring. The 5 mixture was vigorously mixed by magnetic stirrer at 25 °C for 10 minutes. A slightly yellow suspension was obtained.
KRED-P1-H01 alcohol dehydrogenase purchased from Codexis (Ketoreductase Codex Panel variant plate 1, version 1, well H01, batch number D12071) was dissolved in 200 mg amount in 10 ml of the remained buffered aqueous solution containing 10 caprylic acid and NADP+. The prepared enzyme solution was added under vigorous stirring, in one portion to the suspension containing the starting material, prepared as described above. The mixture was stirred for 24 hours at 25 °C temperature. The
reaction reached 95.3% conversion and it was worked up by partial evaporation of IPA and extraction with toluene. The extract was dried over l\la2S04, then the solvent was 15 evaporated. The crude product was obtained in 4.83 g as light yellow crystalline mass, having 80.2% assay and 99.6% optical purity.
Example 6: Scaled up preparative reaction for the synthesis of (/?)-2-bromo-l- (2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (compound II), catalyzed by alcohol dehydrogenase KRED-P1-H01 from Codexis 20
Substrate 2-bromo-l-(2,2-dimethyl-4H- l,3-benzodioxin-6-yl)ethanone (starting material, 50.00 g, 158.7 mmol), having 90.5 Area% chromatographic purity, was
dissolved in a mixture of 340 ml isopropanol (IPA) and 200 ml dimethyl sulfoxide
(DMSO).
Separately a buffered aqueous solution of 500 ml final volume was prepared, 25 containing MES buffer ( 100 mM concentration, 9762 mg), caprylic acid ( 10 mM
concentration, 721 mg), beta-nicotinamide adenine dinucleotide phosphate
monosodium salt (NADP+, oxidized form, 1 mM concentration, 383 mg). The pH of the aqueous solution was adjusted with NaOH 1 M solution to pH = 6.50 before the
addition of the NADP+. The solution was diluted to the final volume (500 ml) and 360 30 ml of it was added to the IPA-DMSO solution of the starting material, under stirring.
The mixture was vigorously mixed by magnetic stirrer at 25 °C for 10 minutes. A
slightly yellow suspension was obtained.
KRED-P1-H01 alcohol dehydrogenase purchased from Codexis (Ketoreductase Codex Panel variant plate 1, version 1, well H01, batch number D12071) was dissolved 35
in 2.5 g amount in 100 ml of the remained buffered aqueous solution containing
caprylic acid and NADP+. The prepared enzyme solution was added under vigorous stirring, in one portion to the suspension containing the starting material, prepared as described above. The mixture was stirred for 24 hours at 25 °C temperature. The
reaction reached 98.5% conversion in 18 hours and >99.5% conversion after 24 hours, 5 thereafter it was worked up by partial evaporation of IPA and extraction with toluene.
The extract was dried over Na2S04, then the solvent was evaporated . A light yellow crystalline mass was obtained as crude product. The crude product was triturated at room temperature with 1.2 volume of toluene: n-hexane 1 : 1 v/v mixture, thereafter filtered and dried in vacuum oven. The product was obtained in 31 g amount as off- 10 white crystalline powder, with 89.6% assay and 99.6% optical purity (first crop) . The filtrate was partially evaporated under vacuum then let to cool down to room
temperature. The product crystallized out and 5 grams of second crop was isolated by filtration (89.7% assay, 99.6% optical purity). Both crops contained (£)-2-chloro- l- (2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol impurity in 10.5-11 area%, as carry 15 over from the substrate.
Example 7: 6-(2-((2,6-dichlorobenzyl)oxy)ethoxy)hexan-l-amine (V)
Compound IV (44.0 g, 0.115 mol) was charged into 1L autoclave and dissolved in 40 mL of MeOH, followed by addition of ammonia (7M solution in MeOH, 800 mL).
The reactor was sealed and the content was heated to temp. 105-110° C and stirred at 20 the same temp, for 1 hour. The mixture was then cooled down to room temp. The solvent was evaporated under reduced pressure at temp. 30-40° C. The residue was partitioned between EtOAc (400 mL) and water (400 mL). Water phase was separated and the organic phase was washed with water (200 mL). Combined water phases were treated with 20 % aqueous K2C03 solution (280 mL) followed extraction of the product 25 to EtOAc (2 x 400 mL) . The obtained extract was dried over MgS04 and evaporated to dryness to give the title product as slightly yellowish oil (30.28 g, 83 %).
Example 8: 6-(2-((2,6-dichlorobenzyl)oxy)ethoxy)hexan-l-amine L-tartaric acid salt (V L-tartrate)
Compound IV (650 g) was charged into autoclave-type reactor and dissolved in 30 2-propanol (15 L). Afterwards, the reactor was sealed and ammonia gas (1400 g) was introduced while the temperature of the mixture was kept below 30 °C. The content of the reactor was then heated at temp. 85-90 °C for 3 hrs. at pressure 5-9 bars.
Afterwards, the mixture was cooled to room temp, and concentrated under reduced pressure to volume about 5L. The residue was mixed with solution of potassium 35
hydroxide in MeOH (200 mL). The mixture was stirred at room temp, for 10 min. and then inorganic precipitates were filtered off. The filtrate was concentrated to 2.5 L at temp 80-90 °C followed by addition of Toluene (0.65 L). The temp, of the mixture was set to 65 °C, followed by the addition of L-tartaric acid (254 g). The mixture was then cooled to 0 °C and filtered. The filtration cake was washed with iPrOH and dried under flow of nitrogen to give title compound as white solid (yield about 75%). The sample was analyzed by PXRD, the PXRD pattern is presented in Figure 2.
Example 9: (R)-(2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- yl)ethoxy)(tert-butyl)dimethylsilane (Ilia

Compound II ( 10.0 g, 0.035 mol) was dissolved in dry DMF (60 mL), followed by addition of t-butyldimethylsilyl chloride ( 10.5 g, 0.070 mol) and imidazole (5.0 g, 0.073 mol). The mixture was heated at 50° C and stirred under inert atm. for 2.5 hrs. Afterwards, the mixture was cooled down to room temp, and partitioned between EtOAc (240 mL) and water (240 mL). The organic phase was separated, washed with water (2 x 100 mL), brine ( 100 mL), dried over MgS0 and evaporated to dryness to give compound Ilia a yellowish oil ( 16.7 g).
Example 10: (R)-N-(2-((tert-butyldimethylsilyl)oxy)-2-(2f2-dimethyl-4H-l,3- benzodioxin-6-yl)ethyI)-6-(2-((2,6-dichlorobenzyI)oxy)ethoxy)hexan-l-amine
(V

Compound Ilia (16.4 g, obtained by procedure in Example 9) and compound V (22 g, obtained by procedure in Example 7) were mixed and heated at 85° C for 24 hours. The mixture was then cooled down to room temp, and partitioned between EtOAc ( 140 mL) and water ( 140 mL). Organic phase was separated and washed with water (140 mL), 10% aqueous HBr solution ( 120 mL), sat. NaHC03 solution (45 mL), water (45 mL), then dried over MgS04 and evaporated to dryness. The title compound Via was obtained as yellow-brownish oil (26.6 g).
Example 11: (R)-N-(2-((tert-butyldlmethylsilyl)oxy)-2-(2,2-dimethyl-4H-l,3- benzodioxin-6-yl)ethyl)-6-(2-((2,6-dichlorobenzyl)oxy)ethoxy)hexan-l-amine
(Via)
Compound Ilia (200 g, obtained by procedure in Example 9) and compound V (271 g, obtained by procedure in Example 7) were mixed and heated at 75° C for 13 hours. The mixture was then cooled down to room temp, and partitioned between
EtOAc ( 1.6 L) and water (1.2 L). Organic phase was separated and washed with water (0.8 L), 3% aqueous HBr solution (2 x 0.8 L), 8% aq. NaHC03 solution (0.8 L), 2.5 % 5 aq. NaCI solution (0.8 L), then dried over MgS04 and evaporated to dryness. The
residue was purified by column chromatography on silicagel eluting with toluene / acetone (acetone gradient from 1% to 20%). The title compound Via was obtained as yellow-brownish oil (205 g) .
Example 12: (R)-N-(2-((tert-butyIdimethylsiiyi)oxy)-2-(2,2-dimethyl-4H-l,3- 10 benzodioxin-6-yl)ethyl)-6-(2-((2,6-dichlorobenzyl)oxy)ethoxy)hexan-l-amine fumarate salt (Via fumarate)
Compound V L-tartrate (500g, obtained by procedure in Example 8) was
charged in a reactor and suspended in water ( 1.75 L). The suspension was tempered to 30°C and of aq. ammonia (25%, 375 mL) was added during 5 minutes. Afterwards, of 15 toluene (1.25 L) was added and the mixture was stirred for 5 minutes. The stirring was stopped and the phases were separated. Water phase was extracted with another portion of toluene ( 1.25 L). Organic phases were combined and heated to 90°C,
followed by evaporation of the solvent to a final volume about 2 I. Compound Ilia (155 g, obtained by procedure in Example 9) was then charged into a reactor followed by 20 addition of DMSO (0.4 L). The mixture was then heated at 75-80° C for 15 hours,
Afterwards, reaction mixture was cooled down to 30°C and toluene ( 1 L) and water
( 1L) were added with stirring. Organic phase was separated, washed by aq. HBr
solution (21 mL of cone, hydrobromic acid diluted in 0.5 L of water), aq solution of potassium carbonate (33.9 g of potassium carbonate dissolved in 0.5 L of water) and 25 finally washed by water. The organic extract was heated to 90 °C and concentrated under vacuum to final volume about 0.5 L. The solvent was then exchanged to 2- propanol by subsequent addition and evaporation of 2-propanol (4 L) in several
portions to reach final volume of the mixture about 1.6 L. The solution was then cooled down to 40 °C and fumaric acid (59.2 g) . The mixture was cooled down to 30.5 °C and 30 seeded with 2 g of the target product (Via fumarate) suspended in 2- propanol (20 mL). The mixture was cooled to 5 °C, filtered and the filtration cake was washed with 2- propanol to give the title compound (272 g, 70%). The sample was analyzed by PXRD, the PXRD pattern is presented in Figure 3.
Example 13: (R)-2-((6-(2-((2,6-dichIorobenzyl)oxy)ethoxy)hexyl)amino)-l- (2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethan-l-ol (VII)
Compound VI (26.5 g, obtained by procedure in Example 9) was dissolved in
THF (185 mL) followed by addition of tetrabutylammonium fluoride ( 100 mL, 1M
solution in THF). The mixture was stirred at room temp, for 2 hours. Afterwards, the 5 mixture was partitioned between EtOAc (300 mL) and water (300 mL). Organic phase was separated, washed with water (3 x 100 mL), dried over MgS04 and evaporated to dryness. The title compound VII was obtained as brownish oil (22.4 g).
Example 14: (R)-2-((6-(2-((2,6-dlchlorobenzyl)oxy)ethoxy)hexyl)amino)-l- (2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethan-l-ol L-Tartaric acid salt (VII L- 10 tartrate)
Compound VI (190 g, obtained by procedure in Example 11) was charged into 2L jacketed reactor and dissolved in THF (572 mL) followed by addition of
tetrabutylammonium fluoride trihydrate ( 155 g) . The mixture was stirred at room
temp, for 3 hours. Afterwards, the mixture was partitioned between EtOAc (760 mL) 15 and 2.5% aq. NaCI solution (760 mL). Organic phase was separated and mixed with solution of L-tartaric acid (47 g) in EtOH (572 mL). The mixture was cooled to 0° C and stirred overnight, then cooled to -25 0 C and stirred for additional 2 days. The formed suspension was filtered and the filtration cake was washed with MTBE. The obtained solid was dried at room temp, under inert atmosphere. The title compound VII L- 20 tartrate was obtained as white crystalline solid ( 142 g) .
Example 15: (R)-2-((6-(2-((2,6-dichlorobenzyl)oxy)ethoxy)hexyl)amino)-l- (2,2-dimethyl-4H-l,3-benzodloxln-6-yl)ethan-l-ol L-Tartaric acid salt (VII L- tartrate)
Compound Via fumarate (190 g, obtained by procedure in Example 12) was 25 charged into 2L jacketed reactor and dissolved in THF (475 mL) followed by addition of tetrabutylammonium fluoride trihydrate ( 126 g). The mixture was heated at 50 0 C for 3 hours. Afterwards, the mixture was cooled to room temp, and was partitioned
between EtOAc (760 mL) and 20% aq. potassium carbonate solution (400 mL). Organic phase was then washed with water (2 x 200 mL) and mixed with solution of L-tartaric 30 acid (39 g) in EtOH (360 mL). The mixture was then heated to 40 0 C and reputably evaporated with addition of EtOAc (800 mL) in several portions to get final volume of the mixture about 600 mL. Afterwards, additional EtOAc ( 1520 mL) was added and the mixture was cooled to 0° C and for 3hours at the same temp, then it was filtered and the filtration cake was washed with cold EtOAc. The obtained solid was dried at room 35
temp, under inert atmosphere. The title compound VII L-tartrate was obtained as white crystalline solid ( 142 g, chem. purity >99.5%, optical purity >99.85°/o). The sample was analyzed by PXRD, the PXRD pattern is presented in Figure 4.
Example 16: Vilanterol base
Compound VII (5 g, obtained by procedure in Example 10) was dissolved in 5 EtOH (50 mL), followed by addition of 1M HCI solution (50 mL). The mixture was
stirred at room temp, for 90 minutes. Afterwards, pH of the mixture was adjusted to
~9 by addition of 20 % K2C03 solution (25 mL). The mixture was then extracted to dichloromethane (100 mL). Organic phase was washed with water (2 x 25 mL), dried over MgS04 and evaporated to dryness. The residue was purified by column 10 chromatography, elution with mixture of dichloromethane/ethanol/ammonia (50/8/1 ) to give title compound as brownish slightly yellowish oil .
Example 17: Vilanterol trifenatate
Vilanterol base (0.620 g) was dissolved in EtOH (6 mL). Triphenylacetic acid
(0.370 g) was added and the mixture was heated to 50° C and stirred at the same 15 temp, for 15 min. The mixture was then cooled to room temp., followed by cooling in ice-water bath for 90 minutes. The formed suspension was filtered, the filtration cake was washed with cold EtOH and dried at room temp, overnight.
Example 18: Preparation of Vilanterol base 20
( l/ )-2-[(6-{2-[(2,6-dichlorobenzyl)oxy]ethoxy}hexyl)amino]-l-(2,2-dimethyl- 4H-l,3-benzodioxin-6-yl)ethanol (15.5 g, obtained according to the procedure in US
2005/0075394, Example 77(iv)) was dissolved in EtOH (50 mL), followed by addition of 1M HCI solution (50 mL). The mixture was stirred at room temperature for 90 minutes.
Afterwards, the pH of the mixture was adjusted to ~9 by addition of 20 % K2C03 25 solution (25 mL). The mixture was then extracted to dichloromethane ( 100 mL). The organic phase was washed with water (2 x 25 mL), dried over MgS04 and evaporated to dryness.
The crude vilanterol base ( 14.5 g, 90.9 % purity) was dissolved in
dichloromethane and the solution was loaded on a column packed with 300 g Diol-silica 30 in dichloromethane. The column was eluted with dichloromethane with gradient of ethanol (2 - 20 %) . The chromatographic fractions were monitored by TLC. The
fractions containing relatively pure vilanterol were joined and evaporated to dryness, obtaining 11.0 g of vilanterol with purity 97.1 %.
35
Examplel9: Preparation of Vilanterol L-tartrate
Crude vilanterol base ( 10.5 g, 88.9 % purity) was dissolved in ethanol ( 120 ml_) and the solution was concentrated to 37 g. Then, 3.3 g of L-( + )-tartaric acid was added and the suspension was stirred at room temperature until dissolution. 140 ml of ethyl acetate was then added to the solution stepwise and the formed crystalline suspension 5 was stirred for 1 hour at room temperature and the suspension was held 12 hours in the refrigerator. Then, the crystalline product was filtered off and the product was washed with ethyl acetate. The product was dried on filter and the mother liquors were concentrated to dryness. 11 ,2 g of crystalline vilanterol tartrate (purity was 95.7 %) and 2.4 g of dryness of mother liquors (content of vilanterol was 47.3 %) . 10
Example 20: Preparation of Vilanterol L-tartrate
Concentrated chromatographic fractions (11.0 g, 97.1 % purity) from the purification of Crude vilanterol base (90.9 % purity ) were dissolved in methanol and the solution was concentrated to 30 g. Then, 3.5 g of L-(+)-tartaric acid was added and 15 the suspension was stirred at room temperature until dissolution. 150 ml of ethyl acetate was then added to the solution stepwise and the formed crystalline suspension was stirred for 1 hour at room temperature and then the suspension was held 12 hours in the refrigerator. Then the crystalline product was filtered off and the product was washed with ethyl acetate. The product was dried on filter and the mother liquors were 20 concentrated to dryness. Obtained was 12.8 g of crystalline vilanterol tartrate (purity was 98.9 %) and 0.9 g of dryness of mother liquors (content of vilanterol was 82.3 %). The product was analyzed by PXRD; a PXRD pattern is shown in Figure 2.
Example 21: Preparation of Vilanterol L-tartrate
EtOH (700 mL) was mixed with 1 M aq. HCI acid (700 mL), the formed mixture 25 was cooled to 5 °C, followed by addition of compound VII L-tartrate ( 100 g, obtained by procedure in Example 15). The mixture was stirred at 5 °C for 15 hours. Afterwards, DCM (500 mL) was added, the mixture was cooled to 0 °C and aq. Solution of K2C03 ( 130g of K2C03 in 200 mL of water) was then added drop wise to the stirred reaction mixture until pH 9 - 9.5 was obtained. Temp, during the addition was kept below 5 °C. 30 The water phase was separated, and extracted with additional DCM (300 mL).
Combined organic extracts were warmed to temp. 20-25 °C and washed with water (2 x 500 mL), 1% brine (500 mL) and 24% brine (500 mL). Afterwards, organic extract was mixed with solution of L-Tartaric acid (26.6 g) in EtOH (210 mL). The mixture was stirred for 10 min. at temp. 20-25°C and then heated by setting the temp, of the 35
reactor jacket to 40°C. All DCM solvent was distilled off under vacuum to residual approximate 350 mL. The mixture was then cooled to 25°C, followed by addition of
EtOAc ( 1.5 L) . The mixture was stirred at 20-25 °C for 1 hour then cooled to -5 °C and stirred overnight. The product was separated by filtration, washed with cold EtOAc and dried under inert gas and room temp. Isolated yield 85%, chemical purity 99.8%, 5 optical purity 99.93%. The sample was analyzed by PXRD, the PXRD pattern is
presented in Figure 5.
Example 22: Preparation of Vilanterol trifenatate
Dichloromethane (256 mL) was mixed with water (256 mL), the formed mixture was cooled to 0 °C, followed by addition of Vilanterol L-tartrate (32 g, obtained by 10 procedure in Example 21 ) and EtOH (64 mL). Afterwards, 25% aq. solution of ammonia (34 mL) was then added drop wise to the stirred mixture. Temp, during the addition was kept below 5 °C. The water phase was separated, and extracted with additional
DCM (128 mL) . Combined organic extracts were warmed to temp. 20-25 °C mixed with MTBE (220 mL), EtOH (64 mL). The obtained mixture was then washed with water (3 x 15 220 mL). Afterwards, the obtained organic extract was mixed with triphenylacetic acid ( 14.5 g) and stirred until complete dissolution at temp. 20-25°C. Then EtOH (96 mL) was added and the mixture was heated by setting the temp, of the reactor jacket to
40°C. Part of DCM solvent was distilled off under vacuum to residual approximate volume 220 mL, The mixture was then cooled to 25°C, followed by addition of MTBE 20 (256 mL). The mixture was stirred at 20-25 °C for 1 hour then cooled to -5 °C and for additional 2 hours. The product was separated by filtration, washed with cold MTBE and dried under inert gas and room temp. Isolated yield 93%, chemical purity 99.8%, optical purity 99.93%.
Example 23 : Comparison of different buffers in the enzymatic 25 enantioselective reduction of 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- y!)ethanone (compound I) to (R)-2-bromo-l-(2,2-dimethyI-4H-l,3- benzodioxin-6-yl)ethanone (compound II), catalyzed by ketoreductase CDX- 005 from Codexis
Buffered cofactor solutions containing 1 mM beta-nicotinamide adenine 30 dinucleotide phosphate (NADP+, oxidized form) were prepared by dissolving 7.7 mg of beta-nicotinamide adenine dinucleotide phosphate monosodium salt (M = 765.39
g/mol) in 3.33 milliliters of the following buffers: triethanolamine sulfate (TEA-S04)
0.30 M, pH = 7.50, 4-(2-hydroxyethyl)morpholine sulfate (HEM-S04) 0.30 M, pH =
7.25, 4-(2-hydroxyethyl)morpholine nitrate (HEM-N03) 0.30 M, pH = 7.25. Three 35
solutions were prepared with each buffer (totally nine solutions). Water was added to each solution in 4 ml volume. The pH of the solutions were adjusted to 6.75, 7.00 and 7.25 in case of each buffer with the aid of diluted sulfuric or nitric acids or sodium
hydroxide, The final volumes of each buffered cofactor solution were adjusted to 10-10 milliliters with the aid of distilled water. 5
Enzyme solutions were prepared by dissolving 50-50 milligrams of CDX-005 ketoreductase enzyme (purchased from Codexis) in 2.0 milliliters of each buffered cofactor solution prepared above.
A substrate solution containing 125 g/l 2-bromo-l-(2,2-dimethyl-4H-l,3- benzodioxin-6-yl)ethanone was prepared in a solvent mixture of isopropano dimethyl 10 sulfoxide 4: 1 v/v, by dissolving 3.125 grams of 2-bromo- l-(2,2-dimethyl-4 -/-l,3- benzodioxin-6-yl)ethanone in the solvent mixture and completing it to 25 milliliters with the same.
The reactions were started in vials of 4 milliliter volumes. In each reaction vial was pipetted 400-500 pL of buffered cofactor solution with the desired pH and buffer 15 type, according to the tables below. Isopropanol (IPA) and/or dimethyl sulfoxide
(DMSO) were added in 0 or 100 pL volumes, according to the tables. Substrate solution was added to each vial in 490 μΙ_ volume and the content of the vial was homogenized by vigorous shaking. Finally 100 pL of enzyme solution (with buffer constituent
corresponding to the table below) was added to each vial. The vials were shaken for 24 20 hours at 22°C temperature.
Reactions with buffered cofactor solutions having pH = 6.75:

Reactions with buffered cofactor solutions having pH = 7.00:
Volume of
Volume of Conversion buffered Volume of
Reaction Tested added to
cofactor added IPA
number buffer DMSO Compound
solution (ML)
(ML) II (%)
(ML)
46/13 TEA-SO4 500 0 0 83.3
46/29 HEM-SO4 500 0 0 92.4
46/41 HEM-NO3 500 0 0 90.6
46/14 TEA-SO4 400 0 100 78.4
46/30 HEM-SO4 400 0 100 90.8
46/42 HEM- NO3 400 0 100 99.8
46/15 TEA-SO4 400 100 0 61.5
46/31 HEM-SO4 400 100 0 74.1
46/43 HEM- NO3 400 100 0 99.7
Reactions with buffered cofactor solutions having pH

The best conversions were obtained in 4-(2-hydroxyethyl)morpholine nitrate buffer. In most of the cases the difference between triethanolamine and 4-(2- 5 hydroxyethyl)morpholine buffers was also significant when both buffers were used with
sulfate as counterion, Optical purities of 98.5-99.0 % were obtained for Compound II in 4-(2-hydroxyethyl)morpholine buffer.
Example 24: Scaled up preparative reaction for the ketoreductase
catalyzed synthesis of (R)-2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6- 5 y!)ethanone
Buffer solution was prepared by dissolving 1180.8 grams of 4-(2- hydroxyethyl)morpholine in 75 liters of water (purified by ion exchange) and adjusting the pH of the solution to 6.75 with nitric acid (20 m/v%).
A buffered cofactor solution of 90 liters was prepared by dissolving 35.432 10 grams of beta-nicotinamide adenine dinucleotide phosphate disodium salt (NADP+-IMa2, oxidized form) in the full volume of the buffer prepared above. The pH of the solution was adjusted to 6.75 with the aid of 20% nitric acid, thereafter completed to 90 liters with water (purified by ion exchange).
Enzyme solution of 18 liters containing CDX-005 (ketoreductase purchased from 15 Codexis) was prepared immediately before use, by dissolving 450 grams of CDX-005 in 18 liters of buffered cofactor solution prepared above. The remaining part of the
buffered cofactor solution (~72 liters) was loaded in a tempered reactor and stored at 25°C.
Substrate solution was prepared immediately before use, by dissolving 9 20 kilograms of 2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (94% assay, 29.671 moles) in a solution prepared from 18 liters of dimethyl sulfoxide (DMSO) and 54 liters of isopropanol, by stirring at room temperature. The volume of the solution was completed to 90 liters with isopropanol .
The biocatalytic reaction mixture was prepared by adding the prepared 25 substrate solution (90 liters) to the tempered reactor containing ~72 liters of buffer solution, under vigorous stirring . After proper homogenization the full volume of the enzyme solution ( 18 liters) was charged in one portion to the content of the reactor.
The reaction temperature was maintained at 25°C and the reaction mixture was stirred for 24 hours. By analysis of the reaction mixture 99.7% conversion to Compound II 30 was achieved . The reaction mixture was filtered through Perlite and the filtering layer was washed with 54 liters of toluene. The washing containing toluene was stored at 0- 5°C and used during the extraction step.
The filtrate was partially evaporated under vacuum at 35°C to 110 liter volume.
The evaporation was performed in 3 portions. The respective portions were stored at 0- 5°C before or after evaporation.
The remaining volume obtained after partial evaporation of the filtrate (~110 liters) was extracted with 54 liters of toluene (obtained as filtrate after the washing of 5 Perlite layer). Thereafter the remaining aqueous phase was extracted with 2x 27 liters of toluene. The unified organic phases were extracted with 36 liters of 5% NaHC03 solution, thereafter with 2x 36 liters of water.
The organic phase was partially evaporated under vacuum at 35°C, to 18 kg (in 3 portions). To the concentrated organic extract at 35°C 9 liters of n-hexane (heated to 10 35°C) were added under stirring. The mixture was stirred for further 10 minutes at
35°C, thereafter cooled to 0-5°C under 5 hours, and stirred for further 2 hours. The white crystals were filtered out and flushed with nitrogen. Thereafter the crystals were suspended two times with 9 liters of n-hexane and filtered out again, flushed with nitrogen. The collected crystalline material was dried under vacuum at 35°C, for 2 15 hours, under nitrogen atmosphere. The isolated amount of (R)-2-bromo-l-(2,2- dimethyl-4H- l,3-benzodioxin-6-yl)ethanol was 7.658 kg. The assay of the prepared material was 96.5% (25.736 moles, 86.7% yield). The optical purity of the isolated material was 99.7%.
20
Claims
1. A process for the biocatalytic reduction of a ketone substrate of fo


2. A process according to Claim 1 wherein the enzyme capable of ketoreductase activity is a synthesised or engineered, and/or purified ketoreductase.
3. A process according to Claim 1 or Claim 2, wherein the enzyme capable of ketoreductase activity is an NADH-dependent ketoreductase or an NADPH-dependent ketoreductase, or an engineered polypeptide variant thereof having ketoreductase activity.
4. A process according to any of Claim 1-3 wherein the enzyme capable of ketoreductase activity is a ketoreductase or engineered polypeptide variant thereof which is based on IUBMB classification E.C. 1.1.1, more preferably IUBMB classification E.C. 1.1.1.1 and E.C. 1.1.1.2, and more preferably, wherein the enzyme capable of ketoreductase activity is an engineered ketoreductase polypeptide.
5. A process according to Claim 4, wherein the engineered ketoreductase polypeptide having ketoreductase activity is CDX-005 or KRED-P1-H01.
6. A process according to any of Claims 1 - 5, wherein the reaction is
stereoselective, and wherein the alcohol compound has the formula
 and preferably, wherei n the alcohol compound has the formula : 5
and preferably, wherei n the alcohol compound has the formula : 5

A process according to any of Claims 1-6, wherein the hydroxyl-protecting
G1 and G2 are each independently selected from the g roup consisting of: 10 a silyl protecting group, preferably having the formula -Si R1R2 3, wherein R1, R2 and R3 are independently selected from : alkyl, cycloalkyl, and optionally
substituted aryl ; more preferably a C Cis straight or branched alkyl group, a
Ci-Cio cycloalkyl (particularly C3- i0 cycloalkyl or C5-8 cycloalkyl) group; optionally 15 substituted C6-C10 aryl group and optionally substituted C7-Ci2 arylalkyl group;
particularly wherein the silyl protecting group is selected from the group
consisting of t-butyldimethylsilyl, triethylsilyl, t-butyldiphenylsilyl, and
trimethylsilyl ;
an ether protecti ng group, preferably an alkyl ether or a cyclic ether protecting 20 group; more preferably CMO alkyl ethers or C5.10 cyclic ethers; most preferably preferably C5-8 cyclic ethers, methyl ethers or ethyl ethers; or
an ester protecting grou p, preferably an alkyl ester an aryl ester or an aralkyl ester; more preferably a Ci-i0 alkyl ester (particularly Ci-6 alkyl ester), a C6.10
aryl ester, or a C7.11 aralkyl ester; and most preferably acetate, or aryl 25 substituted acetate g roups (preferably benzoate or substituted benzoate groups) a methyl or substituted methyl protecting group, an ethyl or substituted ethyl protecting group, preferably selected from the group consisti ng of:
tetrahydropyranyl, methoxymethyl, benzyloxymethyl, ethoxyethyl, benzyl or tert-butyl ; and 30
G1 and G2 together represent a group suitable for protection of 1,3-diols, preferably wherein G1 and G2 taken together represent a cyclic acetal or ketal protecting group, preferably selected from the group consisting of: methylene acetal, ethylidene acetal, isopropylidene acetal (acetonide),
8. A process according to Claim 7 wherein G1 and G2 taken together represent isopropylidene acetal (acetonide) .
9. A process according to any of Claims 1-8, wherein L is a leaving group selected from : halogen-containi ng groups (preferably halo, and more preferably chloro, bromo or iodo) or sulfonate ( preferably an alkyl sulfonate, particularly Ci_6 alkylsulphonate, and more particularly methane sulphonate), or aryl sulfonate (particularly C6.10 arylsulfonate, particularly toluene sulfonate or para-toluenesulphonate) .
10. A process according to any of Claims 1-9, wherein G1 and G2 together represent a group suitable for protection of 1,3-diols, preferably wherein G1 and G2 taken together represent a cyclic acetal or ketal protecting group, preferably selected from the group consisting of: methylene acetal, ethylidene acetal, isopropylidene acetal (acetonide), and L is halo, preferably bromo.
11. A process according to any of Claims 1- 10, wherein the ketone substrate is 2- bromo-l-( 2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanone (Substrate I) having the formula (I) :

12. A process according to any of Claims 1- 11, wherei n the alcohol compound has the formula :
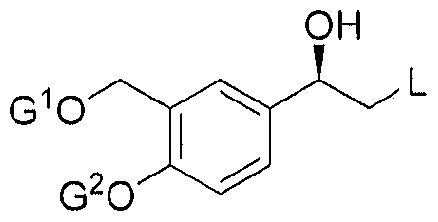

II II*
13. A process according to any of Claims 1-12, wherein the alcohol compound is enantiopure, preferably having an optical purity of : > 85%, > 90%, > 92%, > 94%, > 96%, > 98%,≥ 99%, > 99.5%, or > 99.8%, more preferably having an optical purity of > 99%,≥ 99.5%, or≥ 99.8%. 5
14. A process according to any of Claims 1-13, wherein the alcohol compound is
(R)-2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II) having the formula (II) :

15. A process according to any of Claims 1- 14, wherein the alcohol compound is enantiopure (R)-2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II) having an optical purity of ≥ 99%, > 99.5%, or > 99.8%, and preferably > 99.5% or > 99.8%.
16. A process according to any of Claims 1- 15, wherein the alcohol compound is 15 enantiopure (R)-2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound
II) having an enantiomeric excess of > 95% ee, > 96% ee, > 98% ee, or > 99% ee, preferably≥ 98% ee, or > 99% ee.
17. A process according to any of Claims 1- 16, wherein the alcohol compound is isolated, preferably wherein the alcohol compound is crystalline. 20
18. A process according to any of Claims 1- 17, comprising the steps of:
(a) providing a mixture comprising the enzyme and a co-factor and co-factor
recycling system in an aqueous buffer, optionally comprising a co-factor recycling
system;
(b) providing a solution comprising the ketone substrate in a solvent, optionally 25 comprising a non-ionic tensioactive agent;
(c) stirring the mixture (a) with solution (b); and
(d) optionally isolating the alcohol compound;
19. A process according to Claim 18, wherein the enzyme, co-factor, and co-factor recycling system are dissolved in the aqueous buffer.
20. A process according to Claim 18 or Claim 19, wherein the solvent in step (b) is a 5 water-immiscible organic solvent (preferably an ether, an ester, aromatic and aliphatic hydrocarbon or mixtures thereof, more preferably diisopropyl ether, methyl tert-butyl ether, ethyl acetate, isobutyl acetate, butyl acetate, ethyl butanoate, toluene, hexane, heptane or mixtures thereof), thereby forming a biphasic system, and preferably
wherein the solution in step (b) comprises a non-ionic tensioactive agent. 10
21. A process according to Claim 18 or Claim 19, wherein the solvent in step (b) is a water-miscible organic solvent (preferably an alcohol, aprotic polar solvent or mixtures thereof, more preferably isopropanol, ethanol, isobutanol, dimethyl sulfoxide, dimethyl formamide, dimethyl acetamide, acetonitrile or mixtures thereof.
22. A process according to any of Claims 18-21, wherein the solution formed in step 15 (b) is added slowly to the solution formed in step (a) whilst stirring the mixture, and optionally adjusting the pH during the reaction.
23. A process according to any of Claims 18-22, wherein the co-factor is NADH or NADPH, capable of converting NADP+ to NADPH, or NAD+ to NADH.
24. A process according to any of Claims 18-23, wherein the co-factor recycling 20 system comprises a dehydrogenase and a substrate, preferably wherein the co-factor recycling system comprises the following pairs: glucose dehydrogenase/glucose,
formate dehydrogenase/sodium formate or phosphite dehydrogenase/sodium
phosphite.
25. A process according to any of Claims 18-24, wherein the water-miscible organic 25 solvent comprises isopropanol as a co-solvent, whereby the enzyme capable of
ketoreductase activity can regenerate the co-factor itself.
26. A process according to any of Claims 1-17 comprising the steps of:
(a) providing a buffered aqueous solution, optionally comprising a co-factor
(b) providing a solution comprising the ketone substrate in a water miscible organic 30 solvent, optionally further comprising a tensioactive agent and a substrate for co-factor regeneration;
(c) homogenizing the buffered aqueous solution (a) with solution (b);
(d) providing an aqueous mixture comprising the keto reductase enzyme, and
optionally further comprising a co-factor recycling enzyme, and optionally comprising the co-factor;
(e) homogenizing the mixture from step (c) with the aqueous mixture (d) ;
(f) if the cofactor has not been added at step (a) or step (d), adding the co-factor 5 either in solid form or in an aqueous solution, and homogenized with the mixture
obtained at step (e) ;
(g) stirring the mixture obtained in (e) or (f); and
(h) optionally isolating the alcohol compound.
27. A process according to Claim 26, wherein the substrate for cofactor regeneration 10 is a secondary alcohol, preferably isopropanol
28. A process according to Claim 26 or Claim 27, wherein the water miscible
cosolvent comprises isopropanol , preferably in an amount of 35-50 v/v% of the
reaction mixture, and more preferably 40-45%.
29. A process according to any of Claims 26-28, wherein the solution of step (b) is 15 added slowly to buffered aqueous solution of step (a) whilst stirring.
30. A process according ot any of Claims 26-29, wherein the co-factor is added in at least in one of steps (a)-(g), and preferably at least in one of steps (a), (d) or (f).
31. A process according to any of Claims 18-30, wherein the alcohol compound is 20 (R)-2-bromo-l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II) having the formula (II) :

II
preferably having an optical purity of > 99%, > 99.5%, or > 99.8%, and preferably > 99.5% or≥ 99.8%, and preferably wherein the alcohol compound has an enantiomeric excess of > 95% ee, > 96% ee, > 98% ee, or > 99% ee, preferably≥ 98% ee, or >
99% ee.
32. A process according to any of Claims 1-31, wherein the alcohol is (R)-2-bromo- l-(2,2-dimethyl-4H-l,3-benzodioxin-6-yl)ethanol (Compound II) having the formula
(II) :

II
preferably having an optical purity of > 99%, > 99.5%, or > 99.8%, and preferably > 99.5% or≥ 99.8%, and preferably wherein the alcohol compound has an enantiomeric excess of > 95% ee, > 96% ee, > 98% ee, or > 99% ee, preferably > 98% ee, or > 99% ee, and further comprising converting the alcohol to vilanterol or a solid state form thereof, or a vilanterol salt (preferably vilanterol tartrate or vilanterol trifenatate) or a solid state form thereof.
33. A process for preparing vilanterol or a salt thereof, comprising :
(a) converting a ketone substrate of formula (I) :
 into an enantiomerically pure alcohol having the formula :
into an enantiomerically pure alcohol having the formula :


reacting the compound of formula (III) with a compound of formula V,

V
preferably wherein formula V is in the form of a salt, preferably the L-tartrate salt, to form a compou nd of formula VI*,

VI*
and optionally converting VP to a salt, preferably a fumarate salt (VI fumarate) ; 5
(d) selectively removing the protecting group PG from the compound VI* or a salt thereof, to form a compound of formula VII* :

and optionally converting VII* to a salt, preferably an L-tartrate salt (VII L-tartrate) ;
(e) removing the protecting groups G1 and G2 from VII* or a salt thereof, to form vilanterol ;
(f) optionally converting vilanterol to vilanterol L-tartrate :

Vilanterol L-tartrate ; and
optionally converting vilanterol or vilanterol L-tartrate to vilanterol trifenatate:

Vilanterol trifenatate
5 wherein step (a) is preferably carried out by a process according to any of Claims 1-32. 34. A process according to Claim 33, wherein the compound of formula V is
prepared by reacting a compound of formula IV:

35. A process according to Claim 33 or Claim 34, wherein G1 and G2 together
represent isopropylidine acetal (acetonide), and L is halo, preferably bromo.
36. A process according to any of Claims 33-35, wherein compound V is in the form of a crystalline L-tartaric acid salt, compound VI is in the form of a crystalline fumaric 15 acid salt and compound VII is in a form of a crystalline L-tartaric acid salt.
37. A process according to any of Claims 33-36 further comprising combining the vilanterol or a solid state form thereof, or a pharmaceutically acceptable salt of
vilanterol salt (preferably vilanterol trifenatate) or a solid state form thereof with at least one pharmaceutically acceptable excipient to form a pharmaceutical composition. 20
38. Use of a process according to any of Claims 1-32 for preparing vilanterol or a solid state form thereof, or a vilanterol salt (preferably vilanterol tartrate or vilanterol trifenatate) or a solid state form thereof, or a pharmaceutical composition of vilanterol or a pharmaceutically acceptable salt of vilanterol (preferably vilanterol trifenatate).
39. Vilanterol tartrate, preferably Vilanterol L-tartrate, or a solid state form thereof, 25 preferably in substantially pure form.
40. Vilanterol tartrate salt according to Claim 39, wherein the molar ratio of
vilanterol and tartaric acid is 1.5 : 1 to 1 : 1, preferably about 1 : 1.
41. Vilanterol tartrate, preferably vilanterol L-tartrate, according to Claim 39 or
Claim 40 in crystalline form, preferably characterized by data selected from one or 5 more of the following : an X-ray powder diffraction pattern substantially as depicted in Figure 2; an X-ray powder diffraction pattern having broad peaks at 7.6, 9.8, 12.1,
20.2 and 28.9 degrees two theta ± 1.0 degree two theta; and combinations thereof.
42. Use of a vilanterol tartrate salt as defined in any of Claims 39-41 for: the
purification of vilanterol, the preparation of vilanterol, the preparation of other different 10 salts of vilanterol, the purification of vilanterol API, for the preparation of solid state forms thereof, and/or for the preparation of pharmaceutical formulations comprising one or more of the salts and/or solid state forms thereof.
43. Use of a vilanterol tartrate salt as defined in any of Claims 39-41 for the
preparation of vilanterol triphenylacetate (trifenatate) or vilanterol tosylate. 15
44. A process for the purification of vilanterol or vilanterol salt comprising preparing vilanterol tartrate as defined in any of Claims 39-41 and converting the vilanterol tartrate to vilanterol or to vilanterol salt.
45. A process according to Claim 44, wherein the vilanterol or vilanterol salt is
substantially free from impurity A of the following formula: 20

46. A process according to Claim 44 or Claim 45, wherein the vilanterol or vilanterol salt (preferably L-tartrate salt or trifenatate salt) are chemically pure, preferably having content of impurity A at amount of not more than 0.25%, preferably not more than
0.15%, more preferably not more than 0.10%.
47. Vilanterol tartrate and solid state form thereof according to any of Claims 39-41, for use as a medicament, preferably for the treatment of a person suffering from COPD or asthma as a sole product or in a combination therapy with an inhaled corticosteroid.
30
48. A pharmaceutical composition comprising vilanterol tartrate or a solid state form thereof according to any of Claims 39-41.
49. A pharmaceutical formulation comprising vilanterol tartrate or a solid state form thereof according to any of Claims 39-41 and at least one pharmaceutically acceptable excipient.
50. A process for preparing a pharmaceutical composition according to Claim 48 or a formulation according to Claim 49 comprising combining the vilanterol tartrate or a solid state form thereof, or, and at least one pharmaceutically acceptable excipient.
51. Use of vilanterol tartrate or a solid state form thereof according to any of
Claims 39-41 in the manufacture of a pharmaceutical composition.
52. A method of treating a person suffering from COPD or asthma, comprising
administering a therapeutically effective amount of a pharmaceutical composition or formulation comprising Vilanterol tartrate and solid state form thereof as defined in any of Claims 39-41.
53. A crystalline form of Compound II:
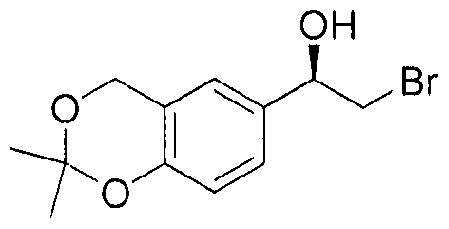
II which is preferably characterized by data selected from one or more of the following :
an X-ray powder diffraction pattern having peaks at 9.8, 14.9, 15.9, 18.2 and 19,9 20 degrees two theta ± 0.2 degrees two theta; an X-ray powder diffraction pattern
substantially as depicted in Figure 1 ; and combinations thereof.
54. A crystalline form of Compound II according to Claim 53, further characterized by an X-ray powder diffraction pattern having any one, two, three, four or five
additional peaks selected from peaks at 8.0, 18.7, 21.9, 24.0 and 25,6 degrees two 25 theta ± 0.2 degrees two theta.
55. A crystalline form of Compound II according to Claim 53 or 54, characterized by an x-ray powder diffraction pattern having peaks at 8.0, 9.8, 14.9, 15.9, 16.4, 17.9, 18.2, 18.7, 19.9, 21.6, 21.9, 22.6, 24.0, 24.3, 25.3, 25.6, 26.9, 27.1, 28.2, 28.9,
29.5, 30.3, 31.5, 31.8, 32.2, 33.6, 34.2, 35.5, 36.5, 37,2, 37.5, 38.3, 39.0 and 39.8 30 degrees two theta ± 0.2 degrees two theta.
56. A crystalline salt of:
compound V:

compound VI* :
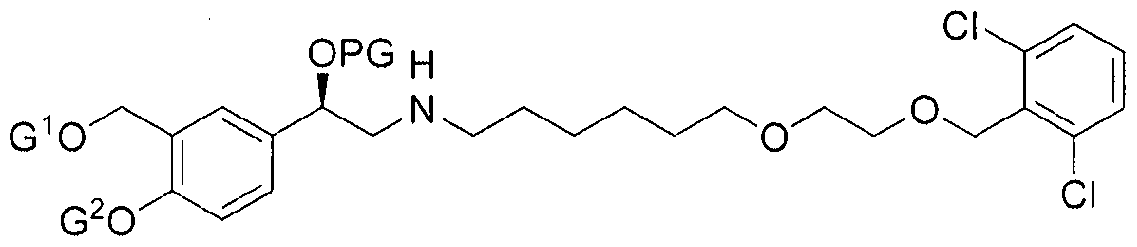
VP ; or
10 a compound VIP :

VII* preferably wherein compound V is in the form of a crystalline L-tartaric acid salt, 15 compound VI is in the form of a crystalline fumaric acid salt or compound VII is in a form of a crystalline L-tartaric acid salt.
57. Use of a crystalline salt as defined in Claim 56 as an intermediate for the
preparation of vilanterol, a vilanterol salt, or solid state forms of vilanterol or a
vilanterol salt, preferably vilanterol, vilanterol L-tartrate, or vilanterol trifenatate or 20 solid state forms thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562186005P | 2015-06-29 | 2015-06-29 | |
| US62/186,005 | 2015-06-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017001907A1 true WO2017001907A1 (en) | 2017-01-05 |
Family
ID=57607962
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2016/000615 WO2017001907A1 (en) | 2015-06-29 | 2016-04-22 | Biocatalytic processes for the preparation of vilanterol |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017001907A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019016511A2 (en) | 2017-07-19 | 2019-01-24 | Hovione Scientia Limited | New crystalline forms of vilanterol trifenatate and processes for their preparation |
| CN109574861A (en) * | 2019-01-22 | 2019-04-05 | 安徽德信佳生物医药有限公司 | A kind of method for crystallising of three phenylacetate of Vilantro |
| CN109574860A (en) * | 2019-01-22 | 2019-04-05 | 安徽德信佳生物医药有限公司 | A method of preparing Vilantro |
| CN111807973A (en) * | 2019-04-10 | 2020-10-23 | 上海谷森医药有限公司 | Preparation method of vilanterol and salt thereof |
| WO2021033198A1 (en) * | 2019-08-16 | 2021-02-25 | Melody Healthcare Pvt. Ltd | An improved process for preparation of vilanterol or a pharmaceutically acceptable salt thereof |
| US11053227B2 (en) | 2017-12-20 | 2021-07-06 | Olon S.P.A. | Process for preparing intermediates for the synthesis of optically active beta-amino alcohols by enzymatic reduction and novel synthesis intermediates |
| WO2022023291A1 (en) | 2020-07-27 | 2022-02-03 | Inke, S.A. | Method for the purification of vilanterol trifenatate |
| CN114441677A (en) * | 2022-01-25 | 2022-05-06 | 上海方予健康医药科技有限公司 | Method for simultaneously detecting multiple gene impurities of vilanterol trithionate |
| CN115745945A (en) * | 2022-11-15 | 2023-03-07 | 奥锐特药业股份有限公司 | Method for preparing vilanterol intermediate by one-pot method |
| WO2023118833A1 (en) | 2021-12-22 | 2023-06-29 | Hovione Scientia Limited | Process to prepare vilanterol trifenatate |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003024439A1 (en) | 2001-09-14 | 2003-03-27 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2004022547A1 (en) | 2002-09-06 | 2004-03-18 | Glaxo Group Limited | Phenethanolamine derivatives and their use in the treatment of respiratory diseases |
| US20040242622A1 (en) * | 2003-05-28 | 2004-12-02 | Mathai Mammen | Azabicycloalkane compounds |
| WO2008038050A2 (en) * | 2006-09-29 | 2008-04-03 | Almac Sciences Limited | Reduction of alpha-halo ketones |
| WO2009046153A1 (en) | 2007-10-01 | 2009-04-09 | Codexis, Inc. | Ketoreductase polypeptides for the production of azetidinone |
| WO2010025085A2 (en) | 2008-08-29 | 2010-03-04 | Codexis, Inc. | Ketoreductase polypeptides for the stereoselective production of (4s)-3[(5s)-5(4-fluorophenyl)-5-hydroxypentanoyl]-4-phenyl-1,3-oxazolidin-2-one |
| WO2011022548A2 (en) | 2009-08-19 | 2011-02-24 | Codexis, Inc. | Ketoreductase polypeptides for the preparation of phenylephrine |
| WO2014041565A2 (en) | 2012-09-13 | 2014-03-20 | Laurus Labs Private Limited | An improved process for the preparation of vilanterol and intermediates thereof |
| US8969571B2 (en) * | 2003-02-14 | 2015-03-03 | Theravance Respiratory Company, Llc | Biphenyl derivatives |
| US20160083759A1 (en) | 2008-08-27 | 2016-03-24 | Codexis, Inc. | Engineered ketoreductase polypeptides |
-
2016
- 2016-04-22 WO PCT/IB2016/000615 patent/WO2017001907A1/en active Application Filing
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003024439A1 (en) | 2001-09-14 | 2003-03-27 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| US20050075394A1 (en) | 2001-09-14 | 2005-04-07 | Box Philip Charles | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2004022547A1 (en) | 2002-09-06 | 2004-03-18 | Glaxo Group Limited | Phenethanolamine derivatives and their use in the treatment of respiratory diseases |
| US8969571B2 (en) * | 2003-02-14 | 2015-03-03 | Theravance Respiratory Company, Llc | Biphenyl derivatives |
| US20040242622A1 (en) * | 2003-05-28 | 2004-12-02 | Mathai Mammen | Azabicycloalkane compounds |
| WO2008038050A2 (en) * | 2006-09-29 | 2008-04-03 | Almac Sciences Limited | Reduction of alpha-halo ketones |
| WO2009046153A1 (en) | 2007-10-01 | 2009-04-09 | Codexis, Inc. | Ketoreductase polypeptides for the production of azetidinone |
| US20160083759A1 (en) | 2008-08-27 | 2016-03-24 | Codexis, Inc. | Engineered ketoreductase polypeptides |
| WO2010025085A2 (en) | 2008-08-29 | 2010-03-04 | Codexis, Inc. | Ketoreductase polypeptides for the stereoselective production of (4s)-3[(5s)-5(4-fluorophenyl)-5-hydroxypentanoyl]-4-phenyl-1,3-oxazolidin-2-one |
| WO2011022548A2 (en) | 2009-08-19 | 2011-02-24 | Codexis, Inc. | Ketoreductase polypeptides for the preparation of phenylephrine |
| WO2014041565A2 (en) | 2012-09-13 | 2014-03-20 | Laurus Labs Private Limited | An improved process for the preparation of vilanterol and intermediates thereof |
Non-Patent Citations (10)
| Title |
|---|
| GOSWAMI A ET AL: "Microbial reduction of omega-bromoacetophenones in the presence of surfactants", TETRAHEDRON ASYMMETRY, PERGAMON PRESS LTD, OXFORD, GB, vol. 11, no. 18, 22 September 2000 (2000-09-22), pages 3701 - 3709, XP004224153, ISSN: 0957-4166, DOI: 10.1016/S0957-4166(00)00341-4 * |
| GOSWAMI ET AL., TETRAHEDRON: ASYMMETRY, vol. 11, 2000, pages 3701 - 3709 |
| GOSWAMI ET AL., TETRAHEDRON: ASYMMETRY, vol. 12, 2001, pages 3343 - 3348 |
| GREENE; WUTS: "Greene's Protective Groups in Organic Synthesis, 4th Edition,", 2006, WILEY |
| J. MED. CHEM., vol. 53, 2010, pages 4522 - 4530 |
| KOSJEK ET AL.: "Purification and Characterization of a Chemotolerant Alcohol Dehydrogenase Applicable to Coupled Redox Reactions", BIOTECHNOLOGY AND BIOENGINEERING, vol. 86, 2004, pages 55 - 62, XP002315116, DOI: doi:10.1002/bit.20004 |
| LIU ET AL., TETRAHEDRON: ASYMMETRY, vol. 19, 2008, pages 1824 - 2828 |
| LIU J ET AL: "A convenient synthesis of (R)-salmeterol via Rh-catalyzed asymmetric transfer hydrogenation", TETRAHEDRON ASYMMETRY, PERGAMON PRESS LTD, OXFORD, GB, vol. 19, no. 15, 8 August 2008 (2008-08-08), pages 1824 - 1828, XP023902368, ISSN: 0957-4166, [retrieved on 20080809], DOI: 10.1016/J.TETASY.2008.07.021 * |
| PROCOPIOU ET AL., TETRAHEDRON: ASYMMETRY, vol. 12, 2001, pages 2005 - 2008 |
| PROCOPIOU P A ET AL: "Enantioselective synthesis of (S)-salmeterol via asymmetric reduction of azidoketone by Pichia angusta", TETRAHEDRON ASYMMETRY, PERGAMON PRESS LTD, OXFORD, GB, vol. 12, no. 14, 14 August 2001 (2001-08-14), pages 2005 - 2008, XP004307294, ISSN: 0957-4166, DOI: 10.1016/S0957-4166(01)00350-0 * |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111094235A (en) * | 2017-07-19 | 2020-05-01 | 好利安科技有限公司 | Amorphous form of vilanterotricin acetate and process for its preparation |
| AU2018303293B2 (en) * | 2017-07-19 | 2022-03-03 | Hovione Scientia Limited | Amorphous form of vilanterol trifenatate and processes for the preparation thereof |
| WO2019016511A3 (en) * | 2017-07-19 | 2019-03-21 | Hovione Scientia Limited | New crystalline forms of vilanterol trifenatate and processes for their preparation |
| KR102572035B1 (en) | 2017-07-19 | 2023-08-28 | 호비온 사이언티아 리미티드 | Amorphous form of vilanterol triphenatate and method for preparing the same |
| CN111108091B (en) * | 2017-07-19 | 2023-04-11 | 好利安科技有限公司 | Novel crystalline form of vilanterol triphenylacetate and process for preparing the same |
| KR20200030107A (en) * | 2017-07-19 | 2020-03-19 | 호비온 사이언티아 리미티드 | Amorphous form of bilanterol tripenatate and preparation method thereof |
| WO2019016512A1 (en) | 2017-07-19 | 2019-01-24 | Hovione Scientia Limited | Amorphous form of vilanterol trifenatate and processes for the preparation thereof |
| CN111108091A (en) * | 2017-07-19 | 2020-05-05 | 好利安科技有限公司 | Novel crystalline form of vilanterol triphenylacetate and process for preparing the same |
| US11414374B2 (en) | 2017-07-19 | 2022-08-16 | Hovione Scientia Limited | Crystalline forms of vilanterol trifenatate and processes for their preparation |
| US11434194B2 (en) | 2017-07-19 | 2022-09-06 | Hovione Scientia Limited | Amorphous form of vilanterol trifenatate and processes for the preparation thereof |
| JP2020528057A (en) * | 2017-07-19 | 2020-09-17 | ホビオネ サイエンティア リミテッド | Novel crystal forms of viranterol triphenyl acetate and their preparation process |
| AU2018303782B9 (en) * | 2017-07-19 | 2022-05-05 | Hovione Scientia Limited | New crystalline forms of vilanterol trifenatate and processes for their preparation |
| WO2019016511A2 (en) | 2017-07-19 | 2019-01-24 | Hovione Scientia Limited | New crystalline forms of vilanterol trifenatate and processes for their preparation |
| AU2018303782B2 (en) * | 2017-07-19 | 2022-03-31 | Hovione Scientia Limited | New crystalline forms of vilanterol trifenatate and processes for their preparation |
| US11053227B2 (en) | 2017-12-20 | 2021-07-06 | Olon S.P.A. | Process for preparing intermediates for the synthesis of optically active beta-amino alcohols by enzymatic reduction and novel synthesis intermediates |
| CN109574860B (en) * | 2019-01-22 | 2021-07-27 | 安徽德信佳生物医药有限公司 | Method for preparing vilanterol |
| CN109574860A (en) * | 2019-01-22 | 2019-04-05 | 安徽德信佳生物医药有限公司 | A method of preparing Vilantro |
| CN109574861A (en) * | 2019-01-22 | 2019-04-05 | 安徽德信佳生物医药有限公司 | A kind of method for crystallising of three phenylacetate of Vilantro |
| CN111807973B (en) * | 2019-04-10 | 2021-04-30 | 上海谷森医药有限公司 | Preparation method of vilanterol and salt thereof |
| CN111807973A (en) * | 2019-04-10 | 2020-10-23 | 上海谷森医药有限公司 | Preparation method of vilanterol and salt thereof |
| WO2021033198A1 (en) * | 2019-08-16 | 2021-02-25 | Melody Healthcare Pvt. Ltd | An improved process for preparation of vilanterol or a pharmaceutically acceptable salt thereof |
| WO2022023291A1 (en) | 2020-07-27 | 2022-02-03 | Inke, S.A. | Method for the purification of vilanterol trifenatate |
| WO2023118833A1 (en) | 2021-12-22 | 2023-06-29 | Hovione Scientia Limited | Process to prepare vilanterol trifenatate |
| CN114441677A (en) * | 2022-01-25 | 2022-05-06 | 上海方予健康医药科技有限公司 | Method for simultaneously detecting multiple gene impurities of vilanterol trithionate |
| CN115745945A (en) * | 2022-11-15 | 2023-03-07 | 奥锐特药业股份有限公司 | Method for preparing vilanterol intermediate by one-pot method |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2017001907A1 (en) | Biocatalytic processes for the preparation of vilanterol | |
| US20090047716A1 (en) | Reduction processes for the preparation of ezetimibe | |
| WO2008151324A1 (en) | Reduction processes for the preparation of ezetimibe | |
| US9994520B2 (en) | Enzymatic process for the preparation of (S)-5-(4-fluoro-phenyl)-5-hydroxy-1morpholin-4-yl-pentan-1-one, an intermediate of ezetimibe and further conversion to ezetimibe | |
| JP2003504070A (en) | Process for the reduction of ketocarboxylic acids and their esters | |
| An et al. | Highly α-position regioselective ring-opening of epoxides catalyzed by halohydrin dehalogenase from Ilumatobacter coccineus: a biocatalytic approach to 2-azido-2-aryl-1-ols | |
| US7622578B2 (en) | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate | |
| TWI354703B (en) | A method for the preparation of mycophenolate mofe | |
| JP2002003486A (en) | Pyranone | |
| JP7122759B2 (en) | Method for preparing purine derivatives | |
| KR100453301B1 (en) | 1-Trifluoromethyl-4-Hydroxy-7-Piperidinyl-Aminomethylchroman Derivatives | |
| SK86498A3 (en) | Process for preparing intermediates for the synthesis of antifungal agents | |
| CN1481353A (en) | Method of purifying plavastatin | |
| Kitano et al. | An efficient synthesis of optically active metabolites of platelet adhesion inhibitor OPC-29030 by lipase-catalyzed enantioselective transesterification | |
| EP4261288A1 (en) | Method for preparing alpha-branched beta -hydroxy carbonyl compounds by enzymatic-catalyzed reductive aldol reaction | |
| WO2005026373A1 (en) | Enzymatic synthesis of enatiopure intermediates by means of cholesterolesterase from yeasts | |
| WO2023158722A2 (en) | Processes for preparation of avacopan and intermediates thereof | |
| TW202233845A (en) | Method for the production of thiocarbamate derivatives a2ar inhibitors | |
| WO2009114080A2 (en) | Processes for preparing enantiomerically pure diol and dioxolane compounds | |
| CN111434671A (en) | Liver specificity AMPK agonist and preparation method and application thereof | |
| JP2001218595A (en) | Method of production for optically active cyclohexene derivative and its synthetic intermediate | |
| TW201311648A (en) | Process for production of optically active (R)-(-)-1-(2,4-dichloro-phenyl)-2-imidazole-1-yl-ethanol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16726645 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16726645 Country of ref document: EP Kind code of ref document: A1 |