WO2015142903A2 - Method of controlling lactate production with piperdine-dione derivatives - Google Patents
Method of controlling lactate production with piperdine-dione derivatives Download PDFInfo
- Publication number
- WO2015142903A2 WO2015142903A2 PCT/US2015/021040 US2015021040W WO2015142903A2 WO 2015142903 A2 WO2015142903 A2 WO 2015142903A2 US 2015021040 W US2015021040 W US 2015021040W WO 2015142903 A2 WO2015142903 A2 WO 2015142903A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sulfanyl
- thienyl
- dione
- piperidine
- chlorophenyl
- Prior art date
Links
- 0 *c1ccccc1OC(C(CC(c1c[s]c(*2CCC2)c1)(c1nc(*)ccc1)N1)=O)C1=O Chemical compound *c1ccccc1OC(C(CC(c1c[s]c(*2CCC2)c1)(c1nc(*)ccc1)N1)=O)C1=O 0.000 description 13
- XCMISAPCWHTVNG-UHFFFAOYSA-N Brc1c[s]cc1 Chemical compound Brc1c[s]cc1 XCMISAPCWHTVNG-UHFFFAOYSA-N 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N C1CCNCC1 Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- YNNFUGKTIJTYCF-UHFFFAOYSA-N C=[Br]c1cc(CNC(C(C(CCc2c[s]cc2)=O)Sc(cccc2)c2Cl)=O)ccc1 Chemical compound C=[Br]c1cc(CNC(C(C(CCc2c[s]cc2)=O)Sc(cccc2)c2Cl)=O)ccc1 YNNFUGKTIJTYCF-UHFFFAOYSA-N 0.000 description 1
- LFKDJXLFVYVEFG-UHFFFAOYSA-N CC(C)(C)OC(N)=O Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 1
- ZHIRQLVTKFEREF-UHFFFAOYSA-N CC(C)(C)Oc(cc1Cl)ccc1F Chemical compound CC(C)(C)Oc(cc1Cl)ccc1F ZHIRQLVTKFEREF-UHFFFAOYSA-N 0.000 description 1
- PLPLJFYYHJMSBM-CZIZESTLSA-N CC(C)(C)S(/N=C(/c1c[s]cc1)\c1cc2ccccc2cc1)=O Chemical compound CC(C)(C)S(/N=C(/c1c[s]cc1)\c1cc2ccccc2cc1)=O PLPLJFYYHJMSBM-CZIZESTLSA-N 0.000 description 1
- RFJYTFIGRIFPIH-UHFFFAOYSA-N CC(C)(C)S(NC(CC(CC(OC)=O)=O)(c1c[s]cc1)c1cc(cccc2)c2cc1)=O Chemical compound CC(C)(C)S(NC(CC(CC(OC)=O)=O)(c1c[s]cc1)c1cc(cccc2)c2cc1)=O RFJYTFIGRIFPIH-UHFFFAOYSA-N 0.000 description 1
- ZBAPTWIVXUZDIO-UHFFFAOYSA-N CC(C)Oc1nc(C(CC(C2Sc3ccccc3Cl)=O)(c3c[s]cc3)NC2=O)ccc1 Chemical compound CC(C)Oc1nc(C(CC(C2Sc3ccccc3Cl)=O)(c3c[s]cc3)NC2=O)ccc1 ZBAPTWIVXUZDIO-UHFFFAOYSA-N 0.000 description 1
- XAYUNTQABPDGOF-UHFFFAOYSA-N CC1(C)OCCN(C)C1 Chemical compound CC1(C)OCCN(C)C1 XAYUNTQABPDGOF-UHFFFAOYSA-N 0.000 description 1
- IDBNECMSCRAUNU-UHFFFAOYSA-N CCN(CC1)CC1O Chemical compound CCN(CC1)CC1O IDBNECMSCRAUNU-UHFFFAOYSA-N 0.000 description 1
- YSNHBNFKJLVKGA-UHFFFAOYSA-N CN(C(c1cc2ccccc2cc1)=O)OC Chemical compound CN(C(c1cc2ccccc2cc1)=O)OC YSNHBNFKJLVKGA-UHFFFAOYSA-N 0.000 description 1
- SZGRVVXFBLWUGL-UHFFFAOYSA-N CN(C1)CC1(OC)/[O]=C(\C1CCN(C)CC1)/O Chemical compound CN(C1)CC1(OC)/[O]=C(\C1CCN(C)CC1)/O SZGRVVXFBLWUGL-UHFFFAOYSA-N 0.000 description 1
- DUUVIWYNPHVLDS-UHFFFAOYSA-N COC(CC(CC(c1c[s]cc1)(c1cc(cccc2)c2cc1)N)=O)=O Chemical compound COC(CC(CC(c1c[s]cc1)(c1cc(cccc2)c2cc1)N)=O)=O DUUVIWYNPHVLDS-UHFFFAOYSA-N 0.000 description 1
- WCOBCKRVWREALP-UHFFFAOYSA-N COC(CC1)CCC11OCCO1 Chemical compound COC(CC1)CCC11OCCO1 WCOBCKRVWREALP-UHFFFAOYSA-N 0.000 description 1
- PFTGXSGDFZZZFY-UHFFFAOYSA-N COC(CC1)CCC1O Chemical compound COC(CC1)CCC1O PFTGXSGDFZZZFY-UHFFFAOYSA-N 0.000 description 1
- ZVARKDFRCWVEBF-UHFFFAOYSA-N COC(CCCC1)C1F Chemical compound COC(CCCC1)C1F ZVARKDFRCWVEBF-UHFFFAOYSA-N 0.000 description 1
- GPZUVOTUJWKLNB-UHFFFAOYSA-N COCC1CCOCC1 Chemical compound COCC1CCOCC1 GPZUVOTUJWKLNB-UHFFFAOYSA-N 0.000 description 1
- QKGBRANQIWBMED-UHFFFAOYSA-N COCCN(CCC1)C1=O Chemical compound COCCN(CCC1)C1=O QKGBRANQIWBMED-UHFFFAOYSA-N 0.000 description 1
- LZRCZEHBKACUNZ-UHFFFAOYSA-N COCc(cc1)ccc1F Chemical compound COCc(cc1)ccc1F LZRCZEHBKACUNZ-UHFFFAOYSA-N 0.000 description 1
- VAWHMGNMKYOSHH-UHFFFAOYSA-N CS=S1(N(C2)CC3C2COC3)=CC1 Chemical compound CS=S1(N(C2)CC3C2COC3)=CC1 VAWHMGNMKYOSHH-UHFFFAOYSA-N 0.000 description 1
- RFWVBOQCLAHRRM-UHFFFAOYSA-N C[N](C)(CC1)CCC1C#N Chemical compound C[N](C)(CC1)CCC1C#N RFWVBOQCLAHRRM-UHFFFAOYSA-N 0.000 description 1
- IBSQPLPBRSHTTG-UHFFFAOYSA-N Cc(cccc1)c1Cl Chemical compound Cc(cccc1)c1Cl IBSQPLPBRSHTTG-UHFFFAOYSA-N 0.000 description 1
- AJGOTFAQSAGFRV-UHFFFAOYSA-N Nc1cc(C(CC(C2Sc(cccc3)c3Cl)=O)(c3c[s]cc3)NC2O)ccc1 Chemical compound Nc1cc(C(CC(C2Sc(cccc3)c3Cl)=O)(c3c[s]cc3)NC2O)ccc1 AJGOTFAQSAGFRV-UHFFFAOYSA-N 0.000 description 1
- OTKNOXJYPGJJAH-UHFFFAOYSA-N O=C(CC(c1c[s]cc1)(c(cc1)nc(Oc(cc2)ccc2F)c1N1CCOCC1)NC1=O)C1Sc1ccccc1Cl Chemical compound O=C(CC(c1c[s]cc1)(c(cc1)nc(Oc(cc2)ccc2F)c1N1CCOCC1)NC1=O)C1Sc1ccccc1Cl OTKNOXJYPGJJAH-UHFFFAOYSA-N 0.000 description 1
- YBBUIMWXPCVCAU-UHFFFAOYSA-N O=C(CC(c1c[s]cc1)(c1cc(cccc2)c2cc1)N1)CC1=O Chemical compound O=C(CC(c1c[s]cc1)(c1cc(cccc2)c2cc1)N1)CC1=O YBBUIMWXPCVCAU-UHFFFAOYSA-N 0.000 description 1
- YCQDSGVAAMEUDO-UHFFFAOYSA-N O=C(CC(c1c[s]cc1)(c1cc(cccc2)c2cc1)NC1=O)C1Sc1ccccc1Cl Chemical compound O=C(CC(c1c[s]cc1)(c1cc(cccc2)c2cc1)NC1=O)C1Sc1ccccc1Cl YCQDSGVAAMEUDO-UHFFFAOYSA-N 0.000 description 1
- QOYGYMWYAFCTGL-UHFFFAOYSA-N O=C(CC(c1c[s]cc1)(c1ccc(C2CCCCC2)cc1)NC1=O)C1Sc1ccccc1Cl Chemical compound O=C(CC(c1c[s]cc1)(c1ccc(C2CCCCC2)cc1)NC1=O)C1Sc1ccccc1Cl QOYGYMWYAFCTGL-UHFFFAOYSA-N 0.000 description 1
- AZRQFCKBZNOPRG-UHFFFAOYSA-N O=C(CC(c1c[s]cc1)(c1cccc(Oc(cc2)ccc2F)n1)NC1=O)C1Sc(cccc1)c1Cl Chemical compound O=C(CC(c1c[s]cc1)(c1cccc(Oc(cc2)ccc2F)n1)NC1=O)C1Sc(cccc1)c1Cl AZRQFCKBZNOPRG-UHFFFAOYSA-N 0.000 description 1
- HABILWTZSVCETE-UHFFFAOYSA-N O=C(CC(c1c[s]cc1)(c1nc(Br)ccc1)NC1=O)C1Sc(cccc1)c1Cl Chemical compound O=C(CC(c1c[s]cc1)(c1nc(Br)ccc1)NC1=O)C1Sc(cccc1)c1Cl HABILWTZSVCETE-UHFFFAOYSA-N 0.000 description 1
- MUXWZULDSMCMLI-UHFFFAOYSA-N O=C(CC(c1c[s]cc1)(c1nc(Oc(cccc2)c2-c2c[nH]nc2)ccc1)NC1=O)C1Sc(cccc1)c1Cl Chemical compound O=C(CC(c1c[s]cc1)(c1nc(Oc(cccc2)c2-c2c[nH]nc2)ccc1)NC1=O)C1Sc(cccc1)c1Cl MUXWZULDSMCMLI-UHFFFAOYSA-N 0.000 description 1
- VVZAMWUYUAMOAI-UHFFFAOYSA-N O=C(c1c[s]cc1)c1cc2ccccc2cc1 Chemical compound O=C(c1c[s]cc1)c1cc2ccccc2cc1 VVZAMWUYUAMOAI-UHFFFAOYSA-N 0.000 description 1
- UOBYKYZJUGYBDK-UHFFFAOYSA-N OC(c1ccc(cccc2)c2c1)=O Chemical compound OC(c1ccc(cccc2)c2c1)=O UOBYKYZJUGYBDK-UHFFFAOYSA-N 0.000 description 1
- RHMPLDJJXGPMEX-UHFFFAOYSA-N Oc(cc1)ccc1F Chemical compound Oc(cc1)ccc1F RHMPLDJJXGPMEX-UHFFFAOYSA-N 0.000 description 1
- KJEJBOHUYPYKHT-UHFFFAOYSA-N SOc(cccc1)c1Br Chemical compound SOc(cccc1)c1Br KJEJBOHUYPYKHT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0018—Culture media for cell or tissue culture
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
Definitions
- the present invention relates to a method of controlling lactate production and increasing production of recombinant proteins in mammalian cell cultures.
- cell culture media and process optimization To increase product yield from cultures of mammalian cells, researchers have developed various strategies to lower lactate production. These strategies generally fall into one of two categories: (1) cell culture media and process optimization, and (2) genetic engineering of recombinant cell lines.
- one strategy is to limit the glucose supply to the cultures through dynamic glucose feeding (Gagnon et al., 2011 ; Zhou et al., 1997).
- a second strategy is to substitute or supplement glucose in the culture medium with another carbon source (Altamirano et al., 2004, 2006).
- a third strategy is to optimize the cell culture medium to enable cells to shift from net lactate production to net lactate consumption (Ma et al., 2009; Yuk et al., 2014).
- one strategy is to overexpress genes that increase carbon flux into the tricarboxylic acid cycle (Irani et al., 2002; Kim and Lee, 2007a).
- a second strategy is to manipulate sugar transport by downregulating the expression of the glucose transporter, GLUT1 (Paredes et al., 1999), or overexpressing the fructose transporter, GLUT5 (Wlaschin and Hu, 2007).
- a third strategy is to decrease the conversion of pyruvate to lactate, which is catalyzed by the lactate dehydrogenase (LDH) family of enzymes (Feron, 2009). Knocking down lactate dehydrogenase A (LDHA) gene expression by small interfering RNA lowers lactate in mammalian cultures (Kim et al., 2007b; Zhou et al., 2011).
- Figure 1 Effect of Gx onVCD for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
- Figure 2. Effect of Gx on culture viability for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
- Figure 4 Effect of Gx on culture pH for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
- Figure 7 Effect of Gx onVCD for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
- Figure 8 Effect of Gx on culture viability for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
- Figure 9 Effect of Gx on lactate for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
- Figure 10 Effect of Gx on culture pH for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
- the present invention relates to a method of reducing lactate production in cultured cells comprising growing cultured cells in a medium which comprises an effective amount of a small molecule LDHA inhibitor.
- the invention relates to a method controlling lactate production by inhibition of LDHA with compounds of Formula (I):
- the invention relates to methods of controlling lactate production using tautomers of compounds of Formula (I), such as:
- a 1 , A 2 , A 3 , A 4 , R 1 , R 4 , R 5 , R 6 , R 7 and R 8 are as defined herein.
- Compounds of Formula (I) can be useful as LDHA inhibitors.
- the term "cells in culture” or “cultured cells” refers two or more cells in a solution (e.g., a cell medium) that allows the cells to undergo one or more cell divisions.
- medium refers to is a liquid or gel designed to support the growth of microorganisms or cells.
- polynucleotide or “nucleic acid,” as used interchangeably herein, refers to polymers of nucleotides of any length, and include DNA and RNA.
- the nucleotides can be deoxyribonucleotides, ribonucleotides, modified nucleotides or bases, and/or their analogs, or any substrate that can be incorporated into a polymer by DNA or RNA polymerase.
- a polynucleotide may comprise modified nucleotides, such as methylated nucleotides and their analogs. If present, modification to the nucleotide structure may be imparted before or after assembly of the polymer.
- the sequence of nucleotides may be interrupted by non-nucleotide components.
- a polynucleotide may be further modified after polymerization, such as by conjugation with a labeling component.
- Other types of modifications include, for example, "caps", substitution of one or more of the naturally occurring nucleotides with an analog, internucleotide modifications such as, for example, those with uncharged linkages (e.g., methyl phosphonates, phosphotriesters, phosphoamidates, cabamates, etc.) and with charged linkages (e.g., phosphorothioates, phosphorodithioates, etc.), those containing pendant moieties, such as, for example, proteins (e.g., nucleases, toxins, antibodies, signal peptides, ply-L-lysine, etc.), those with intercalators (e.g., acridine, psoralen, etc.), those containing chelators (e.g
- any of the hydroxyl groups ordinarily present in the sugars may be replaced, for example, by phosphonate groups, phosphate groups, protected by standard protecting groups, or activated to prepare additional linkages to additional nucleotides, or may be conjugated to solid supports.
- the 5' and 3' terminal OH can be phosphorylated or substituted with amines or organic capping group moieties of from 1 to 20 carbon atoms.
- Other hydroxyls may also be derivatized to standard protecting groups.
- Polynucleotides can also contain analogous forms of ribose or deoxyribose sugars that are generally known in the art, including, for example, 2'-0-methyl-, 2'-0-allyl, 2'-fluoro- or 2'- azido-ribose, carbocyclic sugar analogs, a-anomeric sugars, epimeric sugars such as arabinose, xyloses or lyxoses, pyranose sugars, furanose sugars, sedoheptuloses, acyclic analogs and abasic nucleoside analogs such as methyl riboside.
- One or more phosphodiester linkages may be replaced by alternative linking groups. These alternative linking groups include, but are not limited to, embodiments wherein phosphate is replaced by
- each R or R is independently H or substituted or unsubstituted alkyl (1-20 C) optionally containing an ether (-0-) linkage, aryl, alkenyl, cycloalkyl, cycloalkenyl or araldyl. Not all linkages in a polynucleotide need be identical. The preceding description applies to all polynucleotides referred to herein, including RNA and DNA.
- heterologous nucleic acid or “heterologous polypeptide” refers to a nucleic acid or a polypeptide whose sequence is not identical to that of another nucleic acid or polypeptide naturally found in the same host cell.
- operably linked refers to a functional relationship between two or more nucleic acid (e.g., DNA) segments. Typically, it refers to the functional relationship of transcriptional regulatory sequence to a transcribed sequence.
- a promoter is operably linked to a coding sequence, such as a nucleic acid of the invention, if it stimulates or modulates the transcription of the coding sequence in an appropriate host cell or other expression system.
- promoter transcriptional regulatory sequences that are operably linked to a transcribed sequence are physically contiguous to the transcribed sequence, i.e., they are ds-acting.
- some transcriptional regulatory sequences, such as enhancers need not be physically contiguous or located in close proximity to the coding sequences whose transcription they enhance.
- promoter includes all sequences capable of driving transcription of a coding sequence in a cultured cell, e.g., a mammalian cell.
- promoters used in the constructs of the invention include ds-acting transcriptional control elements and regulatory sequences that are involved in regulating or modulating the timing and/or rate of transcription of a gene (e.g., a LDH or PDHK(s)).
- a promoter can be a exacting transcriptional control element, including an enhancer, a promoter, a transcription terminator, an origin of replication, a chromosomal integration sequence, 5' and 3' untranslated regions, or an intronic sequence, which are involved in transcriptional regulation.
- cis-acting sequences typically interact with proteins or other biomolecules to carry out (turn on/off, regulate, modulate, etc.) transcription.
- Constutive promoters are those that drive expression continuously under most environmental conditions and states of development or cell differentiation.
- Inducible or “regulatable” promoters direct expression of the nucleic acid of the invention under the influence of environmental conditions or developmental conditions. Examples of environmental conditions that may affect transcription by inducible promoters include anaerobic conditions, elevated temperature, drought, or the presence of light.
- vector means a construct, which is capable of delivering, and preferably expressing, one or more gene(s) or sequence(s) of interest (e.g., LDHa and
- PDHK(s) in a host cell.
- vectors include, but are not limited to, viral vectors, naked DNA or RNA expression vectors, plasmid, cosmid or phage vectors, DNA or RNA expression vectors associated with cationic condensing agents, DNA or RNA expression vectors encapsulated in liposomes, and certain eukaryotic cells, such as producer cells.
- Suitable vectors are those which are compatible with the host cell employed. Suitable vectors can be derived, for example, from a bacterium, a virus (such as bacteriophage T7 or a M- 13 derived phage), a cosmid, a yeast, or a plant. Protocols for obtaining and using such vectors are known to those in the art (see, for example, Sambrook et ah, Molecular Cloning: A Laboratory Manual, 2 nd ed., Cold Spring Harbor, 1989).
- Specific Productivity refers to the specific protein, e.g., antibody, production rate in pg/cell/day. Specific productivity is calculated as protein titer (pg/cell/day)/IVCC (calculate integrated viable cell count; cell/day).
- polypeptide and protein are used interchangeably herein to refer to polymers of amino acids of any length.
- the polymer may be linear or branched, it may comprise modified amino acids, and it may be interrupted by non-amino acids.
- the terms also encompass an amino acid polymer that has been modified naturally or by intervention; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation or modification, such as conjugation with a labeling component.
- polypeptides containing one or more analogs of an amino acid including, for example, unnatural amino acids, etc.
- antibody is used in the broadest sense and specifically covers monoclonal antibodies (including full length monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments.
- Antibody fragments comprise a portion of a full length antibody, generally the antigen binding or variable region thereof.
- Examples of antibody fragments include Fab, Fab', F(ab') 2 , and Fv fragments; single-chain antibody molecules; diabodies; linear antibodies; and multispecific antibodies formed from antibody fragments.
- the term "monoclonal antibody” as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical except for possible naturally occurring mutations that may be present in minor amounts. Monoclonal antibodies are highly specific, being directed against a single antigenic site. Furthermore, in contrast to conventional (polyclonal) antibody preparations which typically include different antibodies directed against different determinants (epitopes), each monoclonal antibody is directed against a single determinant on the antigen.
- the modifier "monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method.
- the monoclonal antibodies to be used in accordance with the present invention may be made by the hybridoma method first described by Kohleret al, Nature 256:495 (1975), or may be made by recombinant DNA methods (see, e.g., U.S. Pat. No. 4,816,567).
- the "monoclonal antibodies” may also be isolated from phage antibody libraries using the techniques described in Clackson et al., Nature 352:624-628 (1991) and Marks et al., J. Mol. Biol. 222:581-597 (1991), for example.
- the monoclonal antibodies herein specifically include “chimeric” antibodies
- immunoglobulins in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity (U.S. Pat. No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA 81 :6851-6855 (1984)).
- hypervariable region when used herein refers to the amino acid residues of an antibody which are responsible for antigen-binding.
- the hypervariable region comprises amino acid residues from a "complementarity determining region” or "CDR" (i.e. residues 24-34 (LI), 50-56
- Humanized forms of non-human (e.g., murine) antibodies are chimeric antibodies which contain minimal sequence derived from non-human immunoglobulin.
- humanized antibodies are human immunoglobulins (recipient antibody) in which hypervariable region residues of the recipient are replaced by hypervariable region residues from a non-human species (donor antibody) such as mouse, rat, rabbit or nonhuman primate having the desired specificity, affinity, and capacity.
- donor antibody such as mouse, rat, rabbit or nonhuman primate having the desired specificity, affinity, and capacity.
- Fv framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues.
- humanized antibodies may comprise residues which are not found in the recipient antibody or in the donor antibody. These modifications are made to further refine antibody performance.
- the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence.
- the humanized antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin.
- Fc immunoglobulin constant region
- immunoadhesin designates antibody-like molecules which combine the "binding domain" of a heterologous "adhesin” protein (e.g. a receptor, ligand or enzyme) with the effector functions of an immunoglobulin constant domain.
- adhesin protein e.g. a receptor, ligand or enzyme
- the immunoadhesins comprise a fusion of the adhesin amino acid sequence with the desired binding specificity which is other than the antigen recognition and binding site (antigen combining site) of an antibody (i.e. is "heterologous") and an immunoglobulin constant domain sequence.
- the immunoglobulin constant domain sequence in the immunoadhesin is preferably derived from ⁇ , ⁇ 2, or ⁇ 4 heavy chains since immunoadhesins comprising these regions can be purified by Protein A chromatography (Lindmark et al., J. Immunol. Meth. 62: 1-13 (1983)).
- ligand binding domain refers to any native cell-surface receptor or any region or derivative thereof retaining at least a qualitative ligand binding of a corresponding native receptor.
- the receptor is from a cell- surface polypeptide having an extracellular domain which is homologous to a member of the immunoglobulin supergenefamily.
- Other receptors which are not members of the immunoglobulin supergenefamily but are nonetheless specifically covered by this definition, are receptors for cytokines, and in particular receptors with tyrosine kinase activity (receptor tyrosine kinases), members of the hematopoietin and nerve growth factor receptor superfamilies, and cell adhesion molecules, e.g. (E-, L- and P-) selectins.
- receptor binding domain is used to designate any native ligand for a receptor, including cell adhesion molecules, or any region or derivative of such native ligand retaining at least a qualitative receptor binding ability of a corresponding native ligand. This definition, among others, specifically includes binding sequences from ligands for the above- mentioned receptors.
- an "antibody-immunoadhesin chimera” comprises a molecule which combines at least one binding domain of an antibody (as herein defined) with at least one immunoadhesin (as defined in this application).
- Exemplary antibody-immunoadhesin chimeras are the bispecific CD4-IgG chimeras described in Berg et al., PNAS (USA) 88:4723-4727 (1991) and Chamow et al., J. Immunol. 153:4268 (1994).
- osmolality refers to the number of solute particles dissolved in 1 liter of solution. Solutes which can be added to the culture medium so as to increase the osmolality thereof include proteins, peptides, amino acids, non-metabolized polymers, vitamins, ions, salts (e.g., sodium or potassium salts), sugars, metabolites, organic acids, lipids, etc.
- mOsm means “milliosmoles/Liter H 2 0."
- a "host cell” includes an individual cell, cultured cells, or cell in culture that can be or has been a recipient for vector(s) or siRNA(s) for incorporation of polynucleotide inserts to produce polypeptide.
- Host cells include progeny of a single cultured cell, and the progeny may not necessarily be completely identical (in morphology or in genomic DNA complement) to the original parent cell due to natural, accidental, or deliberate mutation.
- use of the terms "a”, “an,” and the like refers to one or more.
- references to "about” a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se. For example, description referring to "about X” includes description of "X.” Numeric ranges are inclusive of the numbers defining the range.
- alkyl refers to a saturated linear or branched-chain monovalent hydrocarbon radical of one to twelve carbon atoms (C 1 -C 12 ), wherein the alkyl radical may be optionally substituted independently with one or more substituent(s) described below.
- an alkyl radical is one to eight carbon atoms (Q-Cg), or one to six carbon atoms (Ci-C 6 ).
- alkyl groups include, but are not limited to, methyl (Me, -CH 3 ), ethyl (Et, -CH 2 CH 3 ), 1 -propyl (n-Pr, n-propyl, -CH 2 CH 2 CH 3 ), 2-propyl (i-Pr, i-propyl, -CH(CH 3 ) 2 ), 1 -butyl (n-Bu, n-butyl, -CH 2 CH 2 CH 2 CH 3 ), 2-methyl-l -propyl (i-Bu, i-butyl, -CH 2 CH(CH 3 ) 2 ), 2-butyl (s-Bu, s-butyl, -CH(CH 3 )CH 2 CH 3 ), 2-methyl-2-propyl (t-Bu, t-butyl, -C(CH 3 ) 3 ), 1-pentyl (n-pentyl, -CH 2 CH 2 CH 2 CH 3 ), 2-pentyl (n
- Ci-Ci 2 -alkoxy means a Ci-Ci 2 -alkyl group, wherein alkyl is as defined herein, that is linked to the rest of a molecule or to another group through an oxygen atom.
- alkoxy include methoxy, ethoxy, n-propoxy, isopropoxy and the different butoxy isomers and R 1 groups as exemplified therein.
- alkylene or "alkylenyl” as used herein refers to a saturated linear or branched-chain divalent hydrocarbon radical of one to twelve carbon atoms (C 1 -C 12 ), wherein the alkylene radical may be optionally substituted independently with one or more substituent(s) described below.
- an alkylene radical is one to eight carbon atoms (Q-Cg), or one to six carbon atoms (Ci-C 6 ).
- alkylene groups include, but are not limited to, methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), propylene (-CH 2 CH 2 CH 2 -), and R 1 groups as exemplified therein.
- Aryl means a monovalent aromatic hydrocarbon radical of 6-20 carbon atoms (C 6 -C 2 o) or C 6 -C 20 -aryl, derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system. Some aryl groups are represented in the exemplary structures as "Ar”.
- Aryl includes bicyclic radicals comprising an aromatic ring fused to a saturated, partially unsaturated ring, or aromatic carbocyclic ring.
- Typical aryl groups include, but are not limited to, radicals derived from benzene (phenyl), substituted benzenes, naphthalene, anthracene, biphenyl, indenyl, indanyl, 1 ,2-dihydronaphthalene, 1,2,3,4-tetrahydronaphthyl, and the like.
- Aryl groups are optionally substituted independently with one or more substituent(s) described herein. Further non limiting examples of aryl groups can be found in the definition of R 1 herein.
- aryloxy as used herein denotes an -O-aryl group, wherein aryl is as defined herein.
- Non-limiting examples of -O-aryl groups are -O-phenyl and -O-naphthyl groups.
- cyanoalkyl refers to an alky group as defined herein that is substituted by one or more cyano group, for example one cyano group.
- cyanoalkyl are Ci-Ci 2 -cyanoalkyl groups. In other embodiments "cyanoalkyl" are
- Ci-C 6 -cyanoalkyl groups for example cyanomethyl and cyanoethyl.
- Carbocycle refers to a monovalent non-aromatic, saturated or partially unsaturated ring having 3 to 12 carbon atoms (C3-C 12 ) as a monocyclic ring or 7 to 12 carbon atoms as a bicyclic ring.
- Partially unsaturated rings can also be designated as cycloalkenyl rings.
- Bicyclic carbocycles having 7 to 12 atoms can be arranged, for example, as abicyclo [4,5], [5,5], [5,6] or [6,6] system, and bicyclic carbocycles having 9 or 10 ring atoms can be arranged as a bicyclo [5,6] or [6,6] system, or as bridged systems such as bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane and bicyclo[3.2.2]nonane.
- Examples of monocyclic carbocycles or cycloalkyls include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-l-enyl, 1 -cyclopent-2-enyl, l-cyclopent-3-enyl, cyclohexyl, 1-cyclohex-l-enyl, 1 -cyclohex-2-enyl, l-cyclohex-3-enyl, cyclohexadienyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl, cyclododecyl, adamantanyl, and R 2 groups as exemplified therein.
- halo denotes chloro, iodo, fluoro and bromo, In an embodiment halo are fluoro, chloro and bromo, and yet in another embodiment fluoro and chloro.
- haloalkyl denotes an alkyl group as defined above wherein at least one of the hydrogen atoms of the alkyl group is replaced by a halogen atom, preferably fluoro or chloro, most preferably fluoro.
- haloalkyl examples include Ci-Ci 2 -haloalkyl groups, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl, sec -butyl, tert-butyl, pentyl or n-hexyl wherein one or more hydrogen atoms are replaced by CI, F, Br or I atom(s), as well as those haloalkyl groups specifically illustrated by the examples herein below.
- haloalkyl groups are monofluoro-, difluoro- or trifluoro-methyl, -ethyl or -propyl, for example 3,3,3-trifluoropropyl, 2-fluoroethyl, 2,2,2-trifluoroethyl, fluoromethyl, trifluoromethyl.
- Ci-Ci 2 -haloalkyl means a haloalkyl group having 1 to 12 carbon atoms, wherein the haloalkyl is as defined herein.
- haloalkoxy denotes a alkoxy group as defined herein wherein at least one of the hydrogen atoms of the alkoxy group is replaced by a halogen atom, preferably fluoro or chloro, most preferably fluoro.
- haloalkoxy examples include Ci-Ci 2 -haloalkoxy groups, but are not limited to, methoxy, ethoxy, propyloxy, isopropyloxy, isobutyloyx, sec-butyloxy, tert-butyloxy, pentyloxy or n-hexyloxy wherein one or more hydrogen atoms are replaced by CI, F, Br or I atom(s), as well as those haloalkoxy groups specifically illustrated by the examples herein below.
- haloalkoxy groups are monofluoro-, difluoro- or trifluoro-methoxy, -ethoxy or -propyloxy, for example 3,3,3-trifluoropropyloxy, 2-fluoroethoxy, 2,2,2-trifluoroethoxy, fluoromethoxy, trifluoromethoxy.
- groups are Ci-C 6 -haloalkoxy groups.
- heterocycle refers to a saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic radical of 3 to about 20 ring atoms in which at least one ring atom is a heteroatom selected from nitrogen, oxygen, phosphorus and sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituent(s) described below.
- heterocycly groups are 4 to 10 membered heterocyclyl, i.e.
- heterocyclyl groups comprising 2 to 9 carbon atoms and 1 , 2, 3 or 4 heteroatoms selected from N, O, P, and S.
- a heterocycle may be a monocycle having 3 to 7 ring members (2 to 6 carbon atoms and 1 to 4 heteroatoms selected from N, O, P, and S) or a bicycle having 7 to 10 ring members (4 to 9 carbon atoms and 1 to 6 heteroatoms selected from N, O, P, and S), for example: a bicyclo [4,5], [5,5], [5,6], or [6,6] system.
- Heterocycles are described in Paquette, Leo A.;
- Heterocyclyl also includes radicals where heterocycle radicals are fused with a saturated, partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring.
- heterocyclic rings include, but are not limited to, pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, piperidonyl, morpholino, thiomorpholino, thioxanyl, piperazinyl, homopiperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithiany
- heteroaryl refers to a monovalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur.
- heteroaryl groups include 5 to 10 membered heteroaryls which denotes monocyclic of bicyclic heteroaryl having 2 to 9 carbon atoms and one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur, for example, 1, 2, 3 or 4 heteroatoms independently selected from nitrogen, oxygen, and sulfur.
- heteroaryl groups include 5 or 6 membered heteroaryls which denotes monocyclic of bicyclic heteroaryl having 2 to 5 carbon atoms and one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur, for example, 1, 2, 3 or 4 heteroatoms independently selected from nitrogen, oxygen, and sulfur.
- heteroaryl groups are pyridinyl (including, for example, 2-hydroxypyridinyl), imidazolyl, imidazopyridinyl, pyrimidinyl (including, for example, 4-hydroxypyrimidinyl), pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, o
- Heteroaryl groups are optionally substituted independently with one or more substituent(s) described herein, for example alkyl, alkoxy, cyano, halo, oxo, NH 2 , OH, hydroxyalkyl, amido groups. Further examples of heteroaryl groups and of possible substituents can be found in the definition of R 2 .
- heteroaryloxy as used herein means an -O-heteroaryl, wherein heteroaryl is as defined herein.
- the heterocycle or heteroaryl groups may be carbon (carbon-linked), or nitrogen
- nitrogen bonded heterocycles or heteroaryls are bonded at position 1 of an aziridine, azetidine, pyrrole, pyrrolidine, 2-pyrroline, 3-pyrroline, imidazole, imidazolidine, 2-imidazoline, 3 -imidazoline, pyrazole, pyrazoline, 2-pyrazoline, 3-pyrazoline, piperidine, piperazine, indole, indoline, lH-indazole, benzimidazole, position 2 of a isoindole, or isoindoline, position 4 of a morpholine, and position 9 of a carbazole, or ⁇ -carboline.
- hydroxy denotes a group of formula -OH.
- hydroxyalkyl denotes an alkyl group as defined above wherein at least one of the hydrogen atoms of the alkyl group is replaced by a hydroxy group.
- hydroxyalkyl include, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl, sec-butyl, tert-butyl, pentyl or n-hexyl wherein one or more hydrogen atoms are replaced by OH, as well as those hydroxyalkyl groups specifically illustrated by the examples herein below.
- hydroxyalkyl means a hydroxyalkyl group having 1 to 12 carbon atoms, wherein hydroxyalkyl is as defined herein.
- substituent denotes a substitution by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 substituent(s) that can be independently selected from the list following this expression.
- one or more substituent(s) denotes 1, 2, 3, 4 or 5 substituents.
- one or more substituent(s) denotes 1, 2 or 3 substituents.
- beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- Those in need of treatment include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented.
- terapéuticaally effective amount means an amount of a compound of the present invention that (i) treats or prevents the particular disease, condition, or disorder, (ii) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, or disorder, or (iii) prevents or delays the onset of one or more symptoms of the particular disease, condition, or disorder described herein.
- the therapeutically effective amount of the drug may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer.
- the drug may prevent growth and/or kill existing cancer cells, it may be cytostatic and/or cytotoxic.
- efficacy can be measured, for example, by assessing the time to disease progression (TTP) and/or determining the response rate ( R).
- cancer refers to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth.
- a “tumor” comprises one or more cancerous cells. Examples of cancer include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies.
- squamous cell cancer e.g., epithelial squamous cell cancer
- lung cancer including small-cell lung cancer, non-small cell lung cancer ("NSCLC"), adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial or uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, head and neck cancer, multiple myeloma, acute myelogenous leukemia, chronic lymphoid leukemia, chronic myelogenous leukemia, lymphocytic leukemia, myeloid leukemia, oral cavity
- NSCLC non
- chiral refers to molecules which have the property of non-superimposability of the mirror image partner, while the term “achiral” refers to molecules which are superimposable on their mirror image partner.
- stereoisomers refers to compounds which have identical chemical constitution, but differ with regard to the arrangement of the atoms or groups in space. Stereoisomers include enantiomers and diastereomers.
- Diastereomer refers to a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities. Mixtures of diastereomers may separate under high resolution analytical procedures such as electrophoresis and
- Diastereomers include geometric isomers, cis/trans and E/Z isomers, and atropisomers.
- Enantiomers refer to two stereoisomers of a compound which are non-superimposable mirror images of one another.
- the compounds of the invention may contain asymmetric or chiral centers, and therefore exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of the invention, including but not limited to, diastereomers, enantiomers and atropisomers, as well as mixtures thereof such as racemic mixtures, form part of the present invention.
- optically active compounds i.e., they have the ability to rotate the plane of plane -polarized light.
- the prefixes D and L, or R and S are used to denote the absolute configuration of the molecule about its chiral center(s).
- the prefixes d and 1 or (+) and (-) are employed to designate the sign of rotation of plane-polarized light by the compound, with (-) or 1 meaning that the compound is levorotatory.
- a compound prefixed with (+) or d is dextrorotatory.
- these stereoisomers are identical except that they are mirror images of one another.
- a specific stereoisomer may also be referred to as an enantiomer, and a mixture of such isomers is often called an enantiomeric mixture.
- a 50:50 mixture of enantiomers is referred to as a racemic mixture or a racemate, which may occur where there has been no stereoselection or stereospecificity in a chemical reaction or process.
- the terms “racemic mixture” and “racemate” refer to an equimolar mixture of two enantiomeric species, devoid of optical activity.
- tautomer or “tautomeric form” refers to structural isomers of different energies which are interconvertible via a low energy barrier.
- proton tautomers also known as prototropic tautomers
- Valence tautomers include interconversions by reorganization of some of the bonding electrons.
- the compounds of Formula (I) also covers tautomers thereof, such as depicted in the following formulae:
- phrases "pharmaceutically acceptable salt” as used herein, refers to pharmaceutically acceptable organic or inorganic salts of a compound of the invention.
- Exemplary salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucuronate, saccharate, formate, benzoate, glutamate, methanesulfonate "mesylate", ethanesulfonate, benzenesulfonate, p-toluenesulfonate, and pamoate (i.e., 1,1 '-methyl ene
- a pharmaceutically acceptable salt may involve the inclusion of another molecule such as an acetate ion, a succinate ion or other counter ion.
- the counter ion may be any organic or inorganic moiety that stabilizes the charge on the parent compound.
- a pharmaceutically acceptable salt may have more than one charged atom in its structure. Instances where multiple charged atoms are part of the pharmaceutically acceptable salt can have multiple counter ions. Hence, a pharmaceutically acceptable salt can have one or more charged atoms and/or one or more counter ion.
- the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, methanesulfonic acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, trifluoroacetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-toluenesulfonic acid or
- the desired pharmaceutically acceptable salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- suitable salts include, but are not limited to, organic salts derived from amino acids, such as glycine and arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as piperidine, morpholine and piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
- phrases "pharmaceutically acceptable” indicates that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- a “solvate” refers to an association or complex of one or more solvent molecules and a compound of the invention.
- solvents that form solvates include, but are not limited to, water, isopropanol, ethanol, methanol, DMSO, ethylacetate, acetic acid, and ethanolamine.
- compound of this invention and “compounds of the present invention” and “compounds of Formula (I)” include compounds of Formulas (I), (I-a) and (I-a-1), specific compounds described herein and stereoisomers, tautomers, solvates, metabolites, and
- any formula or structure given herein, including Formula (I) compounds, is also intended to represent isotopically labeled forms of the compounds as well as unlabeled forms.
- Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as, but not limited to 2H (deuterium, D), 3H (tritium), 11C, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36C1, and 1251.
- isotopically labeled compounds of the present invention for example those into which radioactive isotopes such as 3H, 13C, and 14C are incorporated.
- Such isotopically labelled compounds may be useful in metabolic studies, reaction kinetic studies, detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients.
- Deuterium labelled or substituted therapeutic compounds of the invention may have improved DMPK (drug metabolism and pharmacokinetics) properties, relating to distribution, metabolism, and excretion (ADME).
- DMPK drug metabolism and pharmacokinetics
- substitution with heavier isotopes such as deuterium may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements.
- An 18F labeled compound may be useful for PET or SPECT studies.
- Isotopically labeled compounds of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the schemes or in the examples and preparations described below by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- substitution with heavier isotopes, particularly deuterium i.e., 2H or D
- substitution with heavier isotopes, particularly deuterium may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index.
- deuterium in this context is regarded as a substituent in the compound of the formula (I).
- concentration of such a heavier isotope, specifically deuterium may be defined by an isotopic enrichment factor.
- any atom not specifically designated as a particular isotope is meant to represent any stable isotope of that atom.
- a position is designated specifically as “H” or “hydrogen”
- the position is understood to have hydrogen at its natural abundance isotopic composition.
- any atom specifically designated as a deuterium (D) is meant to represent deuterium.
- the invention relates to methods of controlling lactate production using compounds of Formula (I):
- a 1 is O, CH 2 , or S
- a 2 is NH or N-C r C 3 -alkyl
- a 3 is N or CR 2 ;
- a 4 is N or CR 3 , provided that A 3 and A 4 are not N at the same time;
- R 1 is CI, N0 2 , or CN
- R 2 and R 6 are independently selected from the group consisting of H, halo, hydroxy,
- R 3 and R 5 are independently selected from the group consisting of:
- heterocycloalkyl • -NR a -(4 to 10 membered heterocycloalkyl), which heterocycloalkyl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C 6 -alkyl, Ci-C 6 -hydroxyalkyl, or -CO-alkyl;
- heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, -NR a R b and Ci-C 6 -alkyl;
- substituent(s) selected from the group consisting of: halo or hydroxy, Ci-C 6 -alkyl, Ci-C 6 -alkoxy, Ci-C 6 -haloalkoxy, Ci-C 6 -alkoxyaryl, Ci-C 6 -haloalkyl, Ci-C 6 -hydroxyalkyl, NR a R b , aryl, Ci-C 6 -akyl-aryl, 5 or 6 membered heteroaryl, and
- Ci-C 6 -alkyl Ci-C 6 -alkoxy, Ci-C 6 -alkyl-Ci-C 6 -alkoxy, Ci-C 6 -haloalkyl,
- Ci-C 6 -haloalkoxy Ci-C 6 -hydroxyalkyl, -S-Ci-C 6 -akyl,
- Ci-C 6 -alkyl-C 3 -C 8 -cycloalkyl C r C 6 -alkoxy-C 3 -C 8 -cycloalkyl, C r C 6 -alkyl-(4 to 10 membered heterocycloalkyl), Ci-C 6 -alkyl-(5 or 6 membered heterocycloalkyl), or 5 or 6 membered heteroaryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C 6 -alkyl, -(Ci-C 6 -alkyl)-(Ci-C 6 -alkoxy), Ci-C 6 -haloalkoxy and a Ci-C 6 -alkylene bridge;
- heteroaryl • -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, C r C 6 -alkyl, C r C 6 -hydroxyalkyl, or -NR a (CO)-C r C 6 -akyl;
- R 4 is:
- Ci-C 6 -alkoxy unsubstituted or substituted by hydroxy, Ci-C 6 -alkoxy or NR a R b ,
- substituent(s) selected from the group consisting of: halo, hydroxy, -NR a R b , Ci-C 6 -alkyl, CrC 6 -alkoxy, C r C 6 -haloalkyl, -C(0)-C r C 6 -alkyl, -C(0)-C r C 6 -cycloalkyl; -C(0)-(5 or 6 membered heterocycloalkyl);
- substituent(s) selected from the group consisting of: halo, hydroxy, -NR a R b , Ci-C 6 -alkyl, CrC 6 -alkoxy, C r C 6 -haloalkyl, -C(0)-C r C 6 -alkyl, -C(0)-C r C 6 -cycloalkyl and -C(0)-(5 or 6 membered heterocycloalkyl);
- R 7 is aryl, a 5 or 6 membered heterocycle or 5 or 6 membered heteroaryl which aryl, heterocycle or heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C 6 -alkyl, C 3 -C 8 -cycloalkyl, -O-aryl, -S-aryl, -NH-aryl, and
- R 6 and R 7 together with the carbon atoms to which they are attached form a 5 membered ring selected from a cycloalkyl or heterocycloalkyl having 5 ring members;
- R 8 is OH, -NR a R b , C r C 6 -alkoxy or -C(0)0-C r C 6 -alkyl;
- R 2 and R 3 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R 3 and R 4 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R 4 and R 5 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R 5 and R 6 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R a is H or C r C 6 -alkyl
- R b is H or C r C 6 -alkyl
- R c is H, hydroxy, halo, -NR a R b , Ci-C 6 -alkoxy, Ci-C 6 -alkenyl, 4 to 6 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C 6 -alkyl, 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C 6 -alkyl, or C 3 -C 8 -cycloalkyl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- Ci-C 6 -alkyl or Ci-C 6 -hydroxyalkyl • halo, Ci-C 6 -alkyl or Ci-C 6 -hydroxyalkyl, aryl unsubstituted or substituted by halo, 4 to 9 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C 6 -alkyl, and 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C 6 -alkyl;
- R d is H, hydroxy, Ci-C 6 -alkyl, C 3 -C 8 -cycloalkyl or aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo and -NR a -S(0)2-N(Ci-C 6 -alkyl) 2 ;
- R e is Ci-C 6 -alkyl, aryl, C 3 -C 8 -cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl and wherein said aryl, C 3 -C 8 -cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, Ci-C 6 -alkoxy, Ci-C 6 -alkyl and Ci-C 6 -haloalkyl;
- R f is H, C 3 -C 8 -cycloalkyl, 4 to 10 membered heterocycloalkyl, aryl, or 5 or 6 membered heteroaryl, which cycloalkyl, heterocycloalkyl, aryl, or heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C 6 -haloalkyl, Ci-C 6 -alkyl, Ci-C 6 -alkoxy and Ci-C 6 -hydroxyalkyl; R s is Ci-C 6 -alkoxy, C 3 -C 8 -cycloalkyl, aryl, 5 or 6 membered heteroaryl, 5 to 9 membered heterocycloalkyl, wherein said aryl, C 3 -C 8 -cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl is unsubstituted or substituted by one
- R h is aryl, 5 or 6 membered heteroaryl, 4 to 10 membered heterocycloalkyl, C 3 -C 8 -cycloalkyl, each of which is unsubstituted or substituted by halo;
- n 0 or 1.
- the invention relates to methods of controlling lactate production using compounds of F
- a 1 , A 3 , A 4 , R 1 , R 4 , R 5 , R 6 , R 8 , R 9 and R 10 are as described herein.
- the invention relates to methods of controlling lactate production using compounds of Formula (I) which are represented by the following
- the invention relates to methods of controlling lactate production using compounds of
- the invention relates to methods of controlling lactate production usin ⁇ compounds of Formula (I) which are represented by the following
- the invention relates to methods of controlling lactate production using compounds
- the invention relates to methods of controlling lactate production using compounds of owing
- the invention relates to methods of controlling lactate production using compounds of owing
- the invention relates to methods of controlling lactate production using compounds of owing
- the invention relates to methods of controlling lactate production usin ⁇ comp owing
- the invention relates to methods of controlling lactate production using comp owing
- the invention relates to methods of controlling lactate production usin ⁇ comp owing
- the compounds of Formula (I) and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, are defined as below wherein:
- a 1 is O or S
- a 2 is NH or N-C r C 3 -alkyl
- a 3 is N or CR 2 ;
- R 1 is CI, N0 2 , or CN
- R 2 and R 6 are independently selected from the group consisting of H, halo, hydroxy and NH 2 ;
- R 3 and R 5 are independently selected from the group consisting of:
- R f is 4 to 10 membered heterocycloalkyl, aryl, or 5 or 6 membered heteroaryl, which C 3 -C 8 -cycloalkyl, 5 to 9 membered heterocycloalkyl, aryl, or 5 or 6 membered heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R c is H, hydroxy, halo, -NR a R b , C r C 6 -alkoxy, C r C 6 -alkenyl, C 3 -C 8 -cycloalkyl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- Ci-C 6 -alkyl or Ci-C 6 -hydroxyalkyl aryl unsubstituted or substituted by halo, 4 to 9 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C 6 -alkyl, and 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C 6 -alkyl; • -NR a R b , wherein R a and R b are independently selected from H or Ci-C 6 -alkyl;
- R a is H or C r C 6 -alkyl and R d is H, hydroxy, C r C 6 -alkyl, C 3 -C 8 -cycloalkyl or aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R a is H or Ci-C 6 -alkyland which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- Ci-C 6 -alkoxy Ci-C 6 -haloalkyl
- Ci-C 6 -hydroxyalkyl Ci-C 6 -hydroxyalkyl
- R a is H or Ci-C 6 -alkyl and which heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- Ci-C 6 -alkyl Ci-C 6 -alkoxy
- Ci-C 6 -haloalkyl Ci-C 6 -haloalkoxy
- aryl substituted by one or more -S(0) 2 -N(alkyl) 2 ; • 4 to 10 membered heterocycloalkyl unusbstituted or substituted by one or more 5 or 6 membered heterocycloalkyl;
- R 4 is:
- Ci-C 6 -alkoxy unsubstituted or substituted by hydroxy or Ci-C 6 -alkoxy
- Ci-Ce-alkyl or -C(0)-C r C 6 -alkyl
- R 7 is 5 or 6 membered heteroaryl which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R 8 is OH, -NH 2 , C r C 6 -alkoxy, -C(0)0-C r C 6 -alkyl;
- R 2 and R 3 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R 3 and R 4 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R 4 and R 5 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- R 5 and R 6 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: • halo, hydroxy, -NH 2 , -NH(C r C 6 -alkyl), -N(C r C 6 -alkyl) 2 , C r C 6 -alkyl, C r C 6 -alkoxy and Ci-C 6 -haloalkyl;
- n 0 or 1.
- a 1 is O. In an embodiment A 1 is S. In an embodiment A 1 is CH 2 .
- a 2 is NH. In an embodiment A 2 is N-Ci-C 3 -alkyl.
- a 3 is N. In an embodiment A 3 is CR 2 .
- a 4 is N. In an embodiment A 4 is CR 3 .
- a 3 is CR 2 and A 4 is CR 3. In an embodiment, A 3 is NH and A 4 is CR 3. In one embodiment A 3 is CR 2 and A 4 is NH.
- R 1 is CI. In an embodiment R 1 is N0 2 . In an embodiment R 1 is CN.
- R 2 is H. In an embodiment R 2 is halo. In an embodiment R 2 is hydroxy. In an embodiment R 2 is Ci-C 6 -hydroxyalkyl. In an embodiment R 2 is NH 2 . In an embodiment R 2 is halo. In an embodiment R 2 is hydroxy. In an embodiment R 2 is Ci-C 6 -hydroxyalkyl.
- R 3 or R 5 is H. In an embodiment R 3 or R 5 is hydroxy. In an embodiment R 3 or R 5 is halo. In an embodiment R 3 or R 5 is -Ci-C 6 -alkyl-R f , wherein R f is as defined herein. In an embodiment R 3 or R 5 is -Ci-C 6 -alkenyl-R f , wherein R f is as defined herein. In an embodiment R 3 or R 5 is -Ci-C 6 -alkoxy-R c , wherein R c is as defined herein. In an embodiment R 3 or R 5 is -NR a R b , wherein R a and R b are as defined herein.
- R 3 or R 5 is -NR a -(Ci-C 6 -alkyl)-R d , wherein R a and R d are as defined herein.
- R 3 or R 5 is -NR a -S(0) 2 -(4 to 10 membered heterocycloalkyl), wherein R a is as defined herein.
- R 3 or R 5 is -NR a -(C 3 -C 8 -cycloalkyl), wherein R a is as defined herein and the cycloalkyl is unsubstituted or substituted by Ci-C 6 -alkyl.
- R 3 or R 5 is -NR a -aryl, wherein R a is as defined herein and the aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
- Ci-C 6 -hydroxyalkyl, Ci-C 6 -haloalkoxy and C 3 -C 8 -cycloalkyl Ci-C 6 -hydroxyalkyl, Ci-C 6 -haloalkoxy and C 3 -C 8 -cycloalkyl.
- R 3 or R 5 is -NR a -(4 to 10 membered heterocycloalkyl), wherein R a is as defined herein and the heterocycloalkyl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C 6 -alkyl, Ci-C 6 -hydroxyalkyl, or -CO-alkyl.
- R 3 or R 5 is -NR a -(5 or 6 membered heteroaryl), wherein R a is as defined herein and the heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, -NR a R b and Ci-C 6 -alkyl.
- R 3 or R 5 is -NR a (CO)-Ci-C 6 -alkyl wherein R a is as defined herein.
- R 3 or R 5 is -NR a (CO)-(aryl). In an embodiment R 3 or R 5 is -NR a (CO)-(5 or 6 membered heteroaryl).
- R 3 or R 5 is -NR a (CO)0-Ci-C 6 -alkyl wherein R a is as defined herein. In an embodiment R 3 or R 5 is -S-(alkyl) n -R h and R h is as defined herein.
- R 3 or R 5 is -S(0) 2 -aryl, which aryl is unsubstituted or substituted by one or more halo.
- R 3 or R 5 is -C(0)-R e and R e is as defined herein.
- R 3 or R 5 is -C(0)NR a -(C r C 6 -alkyl) n -R s , wherein R a and R s are as defined herein.
- R 3 or R 5 is -0-C 3 -C 8 -cycloalkyl, which cycloalkyl is unsubstituted or substituted by halo or hydroxy, Ci-C 6 -alkyl, Ci-C 6 -alkoxy, which alkoxy is unsubstituted or substituted by halo, Ci-C 6 -alkoxyaryl, Ci-C 6 -haloalkyl, aryl, Ci-C 6 -akyl-aryl, 5 or 6 membered heteroaryl, C r C 6 -haloalkoxy, C r C 6 -hydroxyalkyl, NR a R b , -(CrCs-alkylHQ-CValkoxy).
- R 3 or R 5 is -O-aryl, which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C 6 -alkyl, Ci-C 6 -alkoxy, Ci-C 6 -haloalkyl, Ci-C 6 -haloalkoxy, Ci-C 6 -hydroxyalkyl, -S-Ci-C 6 -akyl,
- R 3 or R 5 is -0-(4 to 10 membered heterocycloalkyl), which
- heterocycloalkyl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, Ci-C 6 -hydroxyalkyl and -C(0)-Ci-C 6 -alkyl.
- R 3 or R 5 is -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, or -NR a (CO)-Ci-C 6 -akyl and R a is as defined herein.
- R 3 or R 5 is C 3 -C 8 -cycloalkyl, which cycloalkyl may be fused to a phenyl.
- R 3 or R 5 is aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -C(0)OH, Ci-C 6 -hydroxyalkyl, Ci-C 6 -alkoxy, -S(0) 2 -NH(alkyl) and -S(0) 2 -N(alkyl) 2 .
- R 3 or R 5 is 4 to 10 membered heterocycloalkyl unusbstituted or substituted by one or more 5 or 6 membered heterocycloalkyl.
- R 3 or R 5 is 5 to 10 membered heteroaryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of hydroxy, -NR a R b , Ci-C 6 -alkyl, Ci-C 6 -hydroxyalkyl, and 4 to 10 membered heterocycloalkyl.
- R 3 or R 5 is -NR a -S(0) 2 -(4 to 10 membered heterocycloalkyl), for example:
- R 3 or R 5 is -S(0)2-aryl, which aryl is unsubstituted or substituted by more halo, for example:
- R 3 or R 5 is C 3 -C 8 -cycloalkyl which cycloalkyl may be fused to a phenyl, or which le:
- R 3 or R 5 is NR a -(C C 6 -alkyl)-R d , wherein R d is C 3 -C 8 -cycloalkyl, for exampl
- R 3 or R 5 is Ci-C 6 -alkenyl-R f , wherein R f is C 3 -C 8 -cycloalkyl, for example:
- R 3 or R 5 is aryl, for example phenyl unsubstituted or substituted by more halo, hydroxy, -C(0)OH, C r C 6 -hydroxyalkyl, C r C 6 -alkoxy, -S(0) 2 -NH(alkyl) and
- R 3 or R 5 is -O-aryl, for example -O-phenyl, which aryl or phenyl is unsubstituted or substituted by one or more: halo, Ci-C6-alkyl, -S-Ci-C6-akyl, Ci-C6-haloalkyl, Ci-C6-alkoxy, Ci-C6-alkoxy-C3-Cg-cycloalkyl, Ci-C6-haloalkoxy, Ci-C6-hydroxyalkyl,
- Ci-C6-alkyl-Ci-C6-alkoxy Ci-C6-alkyl-(5 or 6 membered heterocycloalkyl), 5 or 6 membered heterocycloalkyl which 5 or 6 membered heteroaryl is unsubstituted or substituted by Ci-C6-alkyl, Ci-C6-haloalkoxy, Ci-C6-alkylene bridge, naphthalene partially hydrogenated which is unsubstituted or substituted by halo for example:
- R 3 or R 5 is -NR a -(5 or 6 membered heteroaryl), which heteroaryl is unsubstituted or
- R 3 or R 5 is -NR a -(C 3 -Cg-cycloalkyl), which cycloalkyl is unsubstituted or substituted by Ci-C6-alkyl or a Ci-C 3 -alkylene bridge and R a is H or Ci-C6-alkyl, for example:
- R 3 or R 5 is halo, for example CI, F or Br.
- R 3 or R 5 is -NR a R b , wherein R a and R b are independently selected from H and Ci-Ce-alkyl, for example -NH 2 , -NHMe or -N(Me) 2 .
- R 3 or R 5 is is hydroxy
- R 3 or R 5 is -NR a (CO)0-C C 6 -alkyl, wherein R a is H or C r C 6 -alkyl, for example:
- R 3 or R 5 is -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, Ci-C 6 -alkyl, Ci-C 6 -hydroxyalkyl, or -NR a C(0) Ci-C 6 -alkyl, for example:
- R 3 or R 5 is Ci-C 6 -alkyl-R f and R f is aryl.
- R f is unsubstituted phenyl.
- R f is phenyl substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C 6 -alkoxy, Ci-C 6 -haloalkyl, and Ci-C 6 -hydroxyalkyl, for example:
- R 3 or R 5 is -Ci-C 6 -alkoxy-R c , wherein R c is hydroxy, halo, Ci-C 6 -alkoxy, Ci-C 6 -alkenyl, phenyl unsubstituted or substituted by halo, 4 to 6 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C 6 -alkyl, 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C 6 -alkyl, or C 3 -C 8 -cycloalkyl unsubstituted or substituted by halo or
- Ci-C 6 -hydroxyalkyl Ci-C 6 -alkyl, for example:
- R 3 or R 5 is Ci-C 6 -alkyl-R f and R f is 5 or 6 membered heterocycloalkyl, for example:
- R 3 or R 5 is -0-C 3 -C 6 -cycloalkyl, which cycloalkyl is unsubstituted or substituted by halo, hydroxy, Ci-C 6 -alkyl, phenyl, Ci-C 6 -alkoxy, for example:
- R 3 or R 5 is -0-(5 or 6 membered heterocycloalkyl), which
- heterocycloalkyl is unsubstituted or substituted by Ci-C 6 -alkyl or -C(0)Ci-C 6 -alkyl, for example:
- R 3 or R 5 is -NR a -C r C 6 -alkyl-R d , wherein R d is:
- R 3 or R 5 is 5 to 10 membered heteroaryl unsubstituted or substituted by -hydroxy, N -C 6 -alkyl or Ci-C 6 -hydroxyalkyl, for example:
- R 3 or R 5 is 5 or 6 membered heterocycloalkyl unusbstituted or substituted by halo, Ci- -alkyl, -C(0)-C 3 -C 8 -cycloalkyl, oxo, 5 or 6 membered heterocycloalkyl, for example:
- R 3 or R 5 is -C(0)NR a -(C r C 6 -alkyl) n -R s .
- R 3 or R 5 is -C(0)NR a -(Ci-C6-alkyl)-R s and R s is C3-C6-cycloalkyl or phenyl, which phenyl is unsubstituted or substituted by halo or R 3 or R 5 is -C(0)NR a -Ci-C6-alkoxy, for example:
- R 3 or R 5 is -S-(alkyl) n -R h .
- R 3 or R 5 is -S-phenyl and said phenyl is unsubstituted or substituted by halo, for example:
- R 3 or R 5 is -C(0)-R e and R e is phenyl which phenyl is unsubstituted or substituted by halo, for example:
- R 3 or R 5 is -NR a -S(0)2-(4 to 6 membered heterocycloalkyl), for example:
- R is H. In an embodiment R is halo. In an embodiment R is hydroxy. In an embodiment R 4 is Ci-C 6 -alkyl. In an embodiment R 4 is Ci-C 6 -haloalkyl. In an embodiment R 4 is Ci-C 6 -hydroxalkyl. In an embodiment R 4 is CN. In an embodiment R 4 is Ci-C 6 -alkoxy unsubstituted or substituted by hydroxy or Ci-C 6 -alkoxy. In an embodiment R 4 is
- R 4 is -(Ci-C 6 -alkyl) n -(C 3 -C 8 -cycloalkyl).
- R 4 is -(Ci-C 6 -alkyl) n -(C 3 -C 8 -cycloalkenyl).
- R 4 is -(Ci-C 6 -alkyl) n -(4 to 10 membered heterocycloalkyl) unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C 6 -alkyl, or -C(0)-C r C 6 -alkyl.
- R 4 is -NR a R b and R a and R b are as defined herein, for example:
- R is Ci-C6-alkoxy unsubstituted or substituted by hydroxy, Ci-C6-alkoxy or -NR a R b , wh
- R is 4 to 10 membered heterocycloalkyl unsubstituted or substituted by halo, hydroxy, cyano, oxo, Ci-C 6 -alkyl, Ci-C 6 -alkoxy, Ci-C 6 -hydroxyalkyl, -C(0)OH,
- Ci-C 4 -alkylene bridge for example:
- R 7 is 5 or 6 membered heteroaryl which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-a -aryl, -S-aryl, -NH-aryl, -(Ci-C6-alkyl) n -aryl, for example.
- R 8 is OH. In an embodiment of the present invention R 8 is -NH 2 . In an embodiment of the present invention R 8 is Ci-C 6 -alkoxy. In an embodiment of the present invention R 8 is -C(0)0-Ci-C 6 -alkyl.
- R 6 and R 7 together with the carbon atoms to which they are attached form a 5 membered ring selected from a cycloalkyl or heterocycloalkyl having 5 ring members, so that the compounds of Formula (I) are as following:
- R 2 and R 3 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -NH 2 , -NH(C r C 6 -alkyl), -N(C r C 6 -alkyl) 2 , C r C 6 -alkyl, C r C 6 -alkoxy and C r C 6 -haloalkyl.
- R 3 and R 4 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -NH 2 , -NH(C r C 6 -alkyl), -N(C r C 6 -alkyl) 2 , C r C 6 -alkyl, C r C 6 -alkoxy and C r C 6 -haloalkyl.
- R 5 and R 6 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -NH 2 , -NH(C r C 6 -alkyl), -N(C r C 6 -alkyl) 2 , C r C 6 -alkyl, C r C 6 -alkoxy and C r C 6 -haloalkyl.
- n is 0. In an embodiment of the present invention n is 1.
- R 9 is H. In an embodiment R 9 is Ci-C 6 -alkyl. In an embodiment R 9 is C 3 -C 8 -cycloalkyl. In an embodiment R 9 is halo. In an embodiment R 9 is -O-aryl, for example -O-phenyl. In an embodiment R 9 is -S-aryl, for example -S-phenyl. In an embodiment R 9 is -NH-aryl, for example -NH-phenyl. In an embodiment R 9 is -(Ci-C 6 -alkyl) n -aryl, for example
- R 10 is H. In an embodiment R 10 is Ci-C 6 -alkyl. In an embodiment R 10 is C 3 -C 8 -cycloalkyl. In an embodiment R 10 is halo. In an embodiment R 10 is -O-aryl, for example -O-phenyl. In an embodiment R 10 is -S-aryl, for example -S-phenyl. In an embodiment R 10 is -NH-aryl, for example -NH-phenyl. In an embodiment R 10 is -(Ci-C 6 -alkyl) n -aryl, for example -(C r C 6 -alkyl) n -phenyl. In one embodiment A 3 is NH.
- a 3 is CR 2 , wherein R 2 is selected from the group consisting of H, halo, hydroxy, Ci-C 6 -hydroxyalkyl, and NH.
- R 9 and R 10 are H.
- R 1 is CI.
- R 3 is NH-phenyl or NH-pyridinyl, which phenyl or pyridinyl is substituted by halo.
- R 4 , R 5 , R 6 and R 8 are H.
- a 1 is O
- a 2 is NH
- R 1 is CI
- a 3 is NH
- a 4 is CR 3 and R 3 is NH-phenyl or NH-pyridinyl, which phenyl or pyridinyl is substituted by halo
- R 4 , R 5 and R 6 are H
- R 7 is thiophenyl.
- a 1 is S
- a 2 is NH
- R 1 is halo
- a 3 is NH
- a 4 is CR 3 and R 3 is NH-phenyl or NH-pyridinyl, which phenyl or pyridinyl is substituted by halo
- R 4 ,R 5 and R 6 are H
- R 7 is thiophenyl.
- the compound of Formula (I) is selected from the compounds of the following compounds and stereoisomers, tautomers, and pharmaceutically acceptable salts thereof. These compounds can also be prepared as a racemate, mixture of diastereisomer or as single stereoisomers, all of which forms fall within the scope of the invention:
- the small molecule LDHA inhibitor is 5-(2-chlorophenyl)sulfanyl-4-hydroxy-2-(4-morpholinophenyl)-2-(3-thienyl)-l,3-dihydropyridin-6- one or a salt thereof.
- the pH value of the medium is maintained at about 7.0, 7.1, 7.2 or 7.4. In one embodiment, the temperature of the medium is maintained at 36.0, 36.5, 37.0, or 37.5 °C during the initial 2, 3, 4, 5, 6 or 7 days.
- the temperature of the medium is lowered to about 36, 35, 34, 33, 32, or 31 °C at the end of day 2, 3, 4, 5, 6, or 7.
- the concentration of the LDHA inhibitor in the medium is about 1 ⁇ , 5 ⁇ , 10 ⁇ , 15 ⁇ , 20 ⁇ , 25 ⁇ , 30 ⁇ , 35 ⁇ , 40 ⁇ , 45 ⁇ , 50 ⁇ , 60 ⁇ , 70 ⁇ , 80 ⁇ , 90 ⁇ ⁇ 100 ⁇ .
- the cultured cells produce a polypeptide.
- the polypeptide is an antibody, or a biologically functional fragment of an antibody.
- the cultured cell is a mammalian cell.
- the mammalian cell is a Chinese Hamster Ovary (CHO) cell.
- the present invention relates to a medium comprising a small molecule
- the present invention relates to a medium, wherein said LDHA inhibitor is
- LDHA inhibitors within the scope of the present invention the inventors have identified LDHA inhibitors.
- the relative efficacies of Formula (I) compounds as inhibitors of an enzyme activity (or other biological activity) can be established by determining the concentrations at which each compound inhibits the activity to a predefined extent and then comparing the results. Typically, the preferred determination is the concentration that inhibits 50% of the activity in a biochemical assay, i.e., the 50% inhibitory concentration or "IC 50 ". Determination of IC 50 values can be accomplished using conventional techniques known in the art. In general, an IC 50 can be determined by measuring the activity of a given enzyme in the presence of a range of concentrations of the inhibitor under study. The experimentally obtained values of enzyme activity then are plotted against the inhibitor concentrations used.
- IC 50 concentration of the inhibitor that shows 50%> enzyme activity (as compared to the activity in the absence of any inhibitor) is taken as the IC 50 value.
- other inhibitory concentrations can be defined through appropriate determinations of activity. For example, in some settings it can be desirable to establish a 90%> inhibitory concentration, i.e., IC 90 , etc.
- a "selective LDHA inhibitor” can be understood to refer to a compound that exhibits a 50% inhibitory concentration (IC 50 ) with respect to LDHA that is at least at least 10-fold lower than the IC 50 value with respect to any or all of the other LDHA family members.
- LDHA kinase activity of Formula (I) compounds Determination of the activity of LDHA kinase activity of Formula (I) compounds is possible by a number of direct and indirect detection methods.
- the range of IC50 values for inhibition of LDHA was less than 1 nM (nanomolar) to about 10 ⁇ (micromolar).
- Certain exemplary compounds of the invention had LDHA inhibitory IC 50 values less than 10 nM.
- Certain Formula (I) compounds may have antiproliferative properties and may be useful to treat disorders such as cancer.
- the Formula (I) compounds may inhibit LDHA in mammals and may be useful for treating human cancer patients.
- Thecompounds of Formula (I) may be synthesized by synthetic routes that include processes analogous to those well-known in the chemical arts, particularly in light of the description contained herein, and those for other heterocycles described in: Comprehensive Heterocyclic Chemistry II, Editors Katritzky and Rees, Elsevier, 1997, e.g. Volume 3; Liebigs Annalen der Chemie,
- Synthetic chemistry transformations and protecting group methodologies useful in synthesizing Formula (I) compounds and necessary reagents and intermediates are known in the art and include, for example, those described in R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T. W. Greene and P. G .M. Wuts, Protective Groups in Organic Synthesis, 3 rd Ed., John Wiley and Sons (1999); and L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) and subsequent editions thereof.
- Compounds of Formula (I) may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1,000 compounds, or 10 to 100 compounds.
- Libraries of compounds of Formula (I) may be prepared by a combinatorial 'split and mix' approach or by multiple parallel syntheses using either solution phase or solid phase chemistry, by procedures known to those skilled in the art.
- a compound library comprising at least 2 compounds, or pharmaceutically acceptable salts thereof.
- Suitable amino-protecting groups include acetyl, trifluoroacetyl, t-butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9-fluorenylmethyleneoxycarbonyl (Fmoc).
- BOC t-butoxycarbonyl
- CBz benzyloxycarbonyl
- Fmoc 9-fluorenylmethyleneoxycarbonyl
- LCMS High Pressure Liquid Chromatography / Mass Spectrometry experiments to determine retention times (R T ) and associated mass ions
- the spectrometers may have an electrospray source operating in positive and negative ion mode. Additional detection is achieved using a evaporative light scattering detector.
- Step A N,0-Dimethylhydroxylamine hydrochloride (39 g, 0.40 mol), (dimethylamino)- N, N -dimethyl(3 H -[l,2,3]triazolo[4,5- ⁇ ]pyridin-3-yloxy)-methaniminium hexafluorophosphate (152 g, 0.40 mol) and N, N -diisopropylethylamine (130.3 g, 1.01 mol) was added to a solution of 6-bromopicolinic acid (68 g, 0.34 mol) in DCM (1 L). The mixture was stirred at ambient temperature for 3 hours.
- Step B n-BuLi (158 mL, 0.4 mol) was slowly added to a solution of 3-bromothiophene (65.2 g, 0.4 mol) in isopropyl ether (1 L) at -78 °C. After stirring at -78 °C for 30 min, the reaction mixture was then slowly treated with 6-bromo-N-methoxy-N-methylpicolinamide (80 g, 0.33 mol) and stirred at -78 °C for 3 hours. The reaction mixture was quenched with saturated NH 4 C1 (300 mL), then warmed to ambient temperature.
- Step C (6-Bromopyridin-2-yl)(thiophen-3-yl)methanone (75 g, 0.28 mol) and Ti(OEt) 4 (191.5 g, 0.84 mol) was added to a solution of 2-methylpropane-2-sulfinamide (67.8 g, 0.56 mol) in THF (1 L). The mixture was heated at 70 °C for 16 hours. The suspension was allowed to cool to ambient temperature. The mixture was pour into ice water, filtered, washed with EtOAc. The filtrate was extracted with EtOAc (500 mL x 2), dried over anhydrous Na 2 S0 4 and concentrated.
- Step D Methyl 3-oxobutanoate (50.0 g, 431.2 mmol,) was added to a suspension of NaH (10.35 g, 431.2 mmol,) in THF (1 L) under 0 °C. The reaction mixture was then slowly treated with n-BuLi (172 mL, 431.2 mmol,) and stirred under 0 °C for 30 minutes,
- N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2- sulfinamide (80 g, 215.6 mmol,) was added to the mixture and stirred at 0 °C for another 2 hours.
- the reaction mixture was quenched with saturated NH 4 C1 (500 mL), then warmed to ambient temperature.
- the mixture was diluted with EtOAc (400 mL), washed with water (500 mL x 2), dried over anhydrous Na 2 S0 and concentrated to afford methyl
- Step E HCl/MeOH (150 mL) was slowly added to a solution of
- Step F Potassium carbonate (67.1 g, 485.7 mmol) was added to a solution of methyl
- Step G Potassium carbonate (36.6 g, 264.9 mmol) and l,2-bis(2-chlorophenyl)disulfane (15.2 g, 53.0 mmol) was added to a solution of
- Step A NaH (73 mg, 3.04 mmol) was added to a solution of propan-2-ol (182 mg, 3.04 mmol) in THF (10 mL) at 0 °C. After stirring 30 minutes,
- Step A 6'-Bromo-5-(2-chloro-phenylsulfanyl)-4-hydroxy-2-thiophen-3-yl-2,3- dihydro-lH-[2,2']bipyridinyl-6-one (500 mg, 1 mmol), 2-chloro-4-fluoro-phenol (178 mg, 1.2 mmol), 2-(dimethylamino)acetic acid hydrochloride (28 mg, 0.2 mmol), Cul (39 mg, 0.2 mmol) and Cs 2 C0 3 (0.99 g, 3 mmol) were combined. Dioxane (5 ml) was added, the mixture was stirred at 120 °C for 3 h under nitrogen atmosphere.
- Step A 6-(6-Bromopyridin-2-yl)-3-((2-chlorophenyl)thio)-6-(thiophen-3-yl)- piperidine-2,4-dione (300 mg, 607.5 ⁇ ), cyclohexanamine (90.4 mg, 911.3 ⁇ ), Brettphos (65.2 mg, 121.5 ⁇ ), Pd 2 (dba) 3 (55.6 mg, 60.8 ⁇ mol) and NaOiBu (116.8 mg, 1.2 mmol) were combined, dioxane (5 ml) was added. The mixture was stirred at 120 °C for 8 hours under nitrogen atmosphere.
- Step A 1 ,2-Dibromoethane (100 mg, 0.53 mmol) and l-(bromomethyl)-3-fluorobenzene (1 g, 5.3 mmol) was added to a suspension of zinc powder (345 mg, 5.3 mmol) in anhydrous THF (10 mL). The reaction mixture was stirred at room temperature for 8 hours. The resultant solution was used directly in the next step.
- Step B (3-Fluorobenzyl)zinc(II) bromide (5.7 mL, 3.04 mmol) was added to a solution of Pd(PPh 3 ) 4 (69 mg, 0.06 mmol) and
- Step A 6'-Bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro- [2,2'-bipyridin]-6(lH)-one (300 mg, 0.61 mmol) and (4-fluorophenyl)boronic acid (94 mg, 0.67 mmol) was added to a solution of K 2 C0 3 (253 mg, 0.83 mmol) and PdCl 2 (PPh 3 )2 (21 mg, 0.02 mmol) in THF (6 mL). The mixture was heated at 100 °C for 20 hours under carbon monoxide atmosphere (0.5 MPa). After cooling to room temperature, the reaction was filtered over Celite.
- Step A To a solution of 3-bromothiophene (14.43 g, 220.74 mmol) in anhydrous isopropyl ether (500 mL) was added n-BuLi (88.2 ml, 220.74 mmol) at -78 °C under nitrogen atmosphere. The reaction mixture was stirred for 1 hour. 4-Bromobenzaldehyde (100 g, 183.95 mmol) was added and the reaction mixture was stirred at -78 °C for 2 hours. The reaction was quenched with MeOH and acidified to pH 4 with 1 N HC1, extracted with DCM (100 mL x 2). The combined organic layers were dried over anhydrous Na 2 S0 4 , and concentrated.
- Step B To a solution of (4-bromophenyl)(thiophen-3-yl)methanol (100 g, 371.5 mmol) in CHC1 3 (200 ml) was added Mn0 2 (322.9 g, 3715 mmol). The reaction mixture was stirred at 60 °C for 12 hours. After cooling to room temperature, the reaction mixture was filtered over Celite and the filtrate was concentrated under vacuum. The crude residue (86 g, 86% yield) was used in the next step without further purification.
- Step C (£ ' )-N-((4-Bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2- sulfinamide was prepared in 86% yield according to the Example 1 , Step C substituting
- Step D Methyl 5-(4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5- (thiophen-3-yl) pentanoate was prepared in 85% yield according to the Example 1 , Step D:
- Step ⁇ To a solution of 6-(4-bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-6- (thiophen-3-yl)-5,6- dihydropyridin-2(lH)-one (0.25 g, 0.5 mmol) in dioxane (6 mL) was added 2-methylmorpholine (500 mg, 5 mmol), Brettphos (25 mg, 0.05 mmol), Pd 2 (dba) 3 (45 mg, 0.05 mmol) and i-BuONa (0.5 g, 5 mmol). The reaction mixture was stirred at 1 10 °C for 16 hours under nitrogen atmosphere.
- Step A To a solution of 6-(4-bromophenyl)-3-((2-chlorophenyl)thio)-6-(thiophen-3-yl) piperidine-2,4-dione (0.25 g, 0.5 mmol ) in dioxane (6 mL) and water (2 mL) was added cyclohex-l-en-l-ylboronic acid (126 mg, 1 mmol), Pd(dppf)Cl 2 (36 mg, 0.05 mmol) and K 2 C0 3 (0.27 g, 2 mmol). The reaction mixture was microwaved at 100 °C for 1 hour under nitrogen atmosphere. After cooling to room temperature, the reaction mixture was filtered though a short pad of silica gel.
- Step A To a solution of GNT C349 986 (0.8 g,1.6 mmol ) in acetic acid (20 mL) was added Pd/C (0.1 g). The reaction mixture was stirred at room temperature for 24 hours under hydrogen atmosphere (60 Psi). After relieving the pressure, the reaction mixture was filtrated over Celite and the filtrate was concentrated under vacuum. The crude residue was purified by preparative HPLC (formic acid) to afford the product (10 mg, 1.2% yield) as white solid.
- Step A Diethylzinc (40.6 ml, 40.6 mmol) and diiodomethane (9.3 g, 34.8 mmol) was added to a solution of 3-methylbut-3-en-l -ol (1 g, 11.6 mmlo) in DCM (80 mL) at -10 °C. The reaction mixture was stirred at 0 °C for 1 hour and then room temperature for additional 12 hours. The reaction was quenched with saturated NH 4 C1, extracted with DCM (50 mL x 2), dried over anhydrous Na 2 S0 4 and concentrated to afford 2-(l-methylcyclopropyl)ethanol (600 mg, 6 mmol, 52% yield) as light color oil.
- Step B 5-((2-Chlorophenyl)thio)-4-hydroxy-6'-(2-(l -methylcyclopropyl)ethoxy)-
- Step A To a stirred solution of
- Step B 5-((2-Chlorophenyl)thio)-6'-((4-fluorophenyl)amino)-4-hydroxy-l-methyl- 2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 8% yield according to the Example 4, Step A substituting cyclohexanamine for 4-fluoroaniline.
- Step C 5-((2-Chlorophenyl)thio)-6'-(4-fluorophenoxy)-4-hydroxy-l-methyl-2-
- Step A 2-Fluoro-N-methoxy-N-methylbenzamide was prepared in 73% yield according to the Example 1, Step A substituting 6-bromopicolinic acid for 2-fluorobenzoic acid.
- Step B (2-Fluorophenyl)(thiophen-3-yl)methanone was prepared in 99% yield according to the Example 1, Step B substituting 6-bromo-N-methoxy-N-methylpicolinamide for
- Step C (Z)-N-((2-Fluorophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2- sulfanamide was prepared in 46% yield according to the Example 1 , Step C substituting
- Step D Methyl 5-(l , l -dimethylethylsulfinamido)-5-(2-fluorophenyl)-3-oxo-5- (thiophen-3-yl)pentanoate was prepared in 91% yield according to the Example 1 , Step D substituting N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-
- Step E Methyl 5-amino-5-(2-fluorophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
- Step F 6-(2-Fluorophenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 89% yield according to the Example 1 , Step F substituting
- Step G 3-((2-Chlorophenyl)thio)-6-(2-fluorophenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 83% yield according to the Example 1 , Step G substituting
- Step A Chloro(methoxy)methane (19.1 g, 0.23 mol) was added to a solution of
- Step B (5-Bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanol was prepared in 77% yield according to the Example 2, Step A substituting 4-bromobenzaldehyde acid for
- Step C (5-Bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanol was prepared in 91% yield according to the Example 7, Step B substituting (4-bromophenyl)(thiophen-3-yl)methanol for (5-bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone.
- Step D A mixture of (5-bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone (10 g, 27.0 mmol), 4-fluoroaniline (10 g, 53.9 mmol), Xantphos (3.85 g, 5.39 mmol), Pd 2 (dba) 3 (3.72 g, 2.7 mmol), Cs 2 C0 3 (39.5 g, 80.9 mmol) and 1,4-dioxane (200 mL) was stirred at 110 °C for 16 hours. The reaction was cooled to room temperature, then filtered. The filtrate was concentrate under vacuum. The crude residue was purified by silica gel chromatography eluting with a gradient of 10% - 50%) EtOAc/hexanes to to afford
- Step F (Z)-tert-B ⁇ X ⁇
- Step K To a stirred solution of iert-butyl
- Step A lH-Indole-4-carbaldehyde (10 g, 69.0 mmol) was added to a suspension of NaH (2.0 g,
- Step B Thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methanol was prepared in 68% yield according to the Example 2, Step A substituting 4-bromobenzaldehyde acid for l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indole-4-carbaldehyde.
- Step C Thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methanone was prepared in 94% yield according to the Example 7, Step B substituting
- piperidine-2,4-dione was prepared in 59% yield according to the Example 1, Step G substituting 6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
- Step I To a stirred solution of
- Step A To a suspension of NaH (688 mg, 27.8 mmol) in THF (80 mL) was added diethyl malonate (7.45 g, 46.5 mmol) dropwise. Then ((2-bromoethoxy)methyl)benzene (5 g, 23.2 mmol) was added. The reaction was heated to 90 °C for 5 hours. After cooling to room temperature, the mixture was diluted with EtOAc (50 mL), washed with water (50 mL x 2), dried over anhydrous Na 2 S0 4 and concentrated under vacuum. The crude residue was purified by silica gel
- Step B To a suspension of LiAlH 4 (1.71 g, 45.0 mmol) in anhydrous THF (80 mL) was added diethyl 2-(2-(benzyloxy)ethyl)malonate (6.6 g, 22.5 mmol) dropwise in an ice bath. The reaction was warmed to room temperature and stirred for 12 hours. The reaction was quenched with water, diluted with EtOAc (50 mL), washed with water (50 mL x 2), dried over anhydrous Na 2 S0 4 and concentrated under vacuum.
- Step C To solution of 2-(2-(benzyloxy)ethyl)propane-l,3-diol (2.2 g, 10.6 mmo) in THF (20 mL) was added n-BuLi (4.2 mL, 10.6 mmol) in an ice bath. The mixture was stirred at 0 °C for 30 minutes, then TsCl (404 mg, 2.12 mmol) was added. The reaction mixture was stirred at 0 °C for 1 hour and then n-BuLi (4.2 mL, 10.6 mmol) was added. The reaction mixture was stirred at 60 °C for 6 hours, then cooled to room temperature.
- Step D A mixture of 3-(2-(benzyloxy)ethyl)oxetane (550 mg, 2.86 mmol), Pd/C (350 mg) and ethanol (5 mL) was stirred at room temperature under hydrogen atmosphere for 2 days. The mixture was filtered and the filtrate was concentrate to afford 2-(oxetan-3-yl)ethanol (200 mg, 1.96 mmol, 66% yield) as a colorless oil.
- Step A To a stirred solution of methyl 3-(bromomethyl)benzoate (5 g, 21.8 mmol) in toluene (50 mL) was added DABAL-H (43.6 ml, 43.6 mmol) in an ice bath. The reaction was stirred at 0 °C for 2 hours. The mixture was quenched with 1 N HCl, extracted with EtOAc and water. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated to afford (3-(bromomethyl)phenyl)methanol (4.0 g, 19.9 mmol, 91% yield) as a colorless oil.
- Step B A mixture of (3-(bromomethyl)phenyl)methanol (2.0 g, 10.0 mmol), 2,6-lutidine (2.13 g, 19.9 mmol), tert-butyl dimethylsilyl trifluoromethanosulfonate (3.1 g, 14.9 mmol) and DCM (30 mL) was stirred at room temperature for 2 hours. The reaction was quenched with water (20 mL), extracted with DCM. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated.
- Step C To a mixture of zinc powder (408 mg, 6.3 mol) in anhydrous THF (30 mL) was added 1 ,2-dibromoethane (107 mg, 0.57 mmol) and
- Step D To a stirred solution of
- Step E To a stirred solution of
- Step A To a stirred suspension of 2-(2-hydroxyethyl)phenol (5 g, 36.2 mmol) and CS 2 CO 3 (38.9 g, 108.7 mmol) in acetone (100 mL) was added iodomethane (6.2 g, 43.4 mmol) in an ice bath. The reaction mixture was stirred at 0 °C for 50 minutes. The mixture was filtered, the filtrate was concentrated under vacuum. The crude materials was extracted with EtOAc and water. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated to afford 2-(2-methoxyphenyl)ethanol (4.5 g, 29.6 mmol, 82% yield) as a yellow solid.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- Zoology (AREA)
- Biomedical Technology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Cell Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The invention provides novel compounds having the general formula: and tautomers and pharmaceutically acceptable salts thereof, wherein A1, A2, A3, A4, R1, R4, R5, R6, R7 and R8 are as defined herein, compositions including the compounds and methods of using the compounds.
Description
METHOD OF CONTROLLING LACTATE PRODUCTION WITH PIPERIDINE-DIONE DERIVATIVES
CROSS REFERENCE TO RELATED APPLICATIONS
This non-provisional application filed under 37 CF § 1.53(b), claims the benefit under 35 USC
§119(e) of U.S. Provisional Application Serial No. 61/954087, filed on 17 March 2014, and U.S. Provisional Application Serial No. 62/032977, filed on 4 August 2014, all of which are hereby incorporated by reference in their entirety. FIELD OF THE INVENTION
The present invention relates to a method of controlling lactate production and increasing production of recombinant proteins in mammalian cell cultures.
BACKGROUND OF THE INVENTION
Lactate correlates negatively with productivity in mammalian cell cultures used for generating recombinant proteins (Le et al., 2012; Ma et al., 2009; Pascoe et al., 2007). To increase product yield from cultures of mammalian cells, researchers have developed various strategies to lower lactate production. These strategies generally fall into one of two categories: (1) cell culture media and process optimization, and (2) genetic engineering of recombinant cell lines. Within the first category of cell culture media and process optimization, one strategy is to limit the glucose supply to the cultures through dynamic glucose feeding (Gagnon et al., 2011 ; Zhou et al., 1997). A second strategy is to substitute or supplement glucose in the culture medium with another carbon source (Altamirano et al., 2004, 2006). A third strategy is to optimize the cell culture medium to enable cells to shift from net lactate production to net lactate consumption (Ma et al., 2009; Yuk et al., 2014). Within the second category of genetic engineering of recombinant cell lines, one strategy is to overexpress genes that increase carbon flux into the tricarboxylic acid cycle (Irani et al., 2002; Kim and Lee, 2007a). A second strategy is to manipulate sugar transport by downregulating the expression of the glucose transporter, GLUT1 (Paredes et al., 1999), or overexpressing the fructose transporter, GLUT5 (Wlaschin and Hu, 2007). A third strategy is to decrease the conversion of pyruvate to lactate, which is catalyzed by the lactate dehydrogenase (LDH) family of enzymes (Feron, 2009). Knocking down lactate dehydrogenase A (LDHA) gene expression by small interfering RNA lowers lactate in mammalian cultures (Kim et al., 2007b; Zhou et al., 2011).
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1. Effect of Gx onVCD for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 2. Effect of Gx on culture viability for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 3. Effect of Gx on lactate for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 4. Effect of Gx on culture pH for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 5. Effect of Gx on product titer for CHO cell line 1 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 6. Effect of Gx on average cell-specific productivity (qp) for CHO cell line 1 cultured in duplicate shake flask cultures.
Figure 7. Effect of Gx onVCD for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 8. Effect of Gx on culture viability for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 9. Effect of Gx on lactate for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 10. Effect of Gx on culture pH for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 11. Effect of Gx on product titer for CHO cell line 2 (A) throughout the run duration, and (B) on days 7, 10, and 14.
Figure 12. Effect of Gx on average cell-specific productivity (qp) for CHO cell line 2 cultured in duplicate bioreactor cultures.
SUMMARY OF THE INVENTION
The present invention relates to a method of reducing lactate production in cultured cells comprising growing cultured cells in a medium which comprises an effective amount of a small molecule LDHA inhibitor.
In one aspect the invention relates to a method controlling lactate production by inhibition of LDHA with compounds of Formula (I):

I
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A1, A2, A3, A4, R1, R4, R5, R6, R7 and R8 are as defined herein. Compounds of Formula (I) can be useful as LDHA inhibitors.
In one aspect the invention relates to methods of controlling lactate production using tautomers of compounds of Formula (I), such as:

wherein A1, A2, A3, A4, R1, R4, R5, R6, R7 and R8 are as defined herein. Compounds of Formula (I) can be useful as LDHA inhibitors.
DETAILED DESCRIPTION OF THE INVENTION
Reference will now be made in detail to certain embodiments of the invention, examples of which are illustrated in the accompanying structures and formulas. While the invention will be described in conjunction with the enumerated embodiments, it will be understood that they are not
intended to limit the invention to those embodiments. On the contrary, the invention is intended to cover all alternatives, modifications, and equivalents which may be included within the scope of the present invention as defined by the claims. One skilled in the art will recognize many methods and materials similar or equivalent to those described herein, which could be used in the practice of the present invention. The present invention is in no way limited to the methods and materials described. In the event that one or more of the incorporated literature, patents, and similar materials differs from or contradicts this application, including but not limited to defined terms, term usage, described techniques, or the like, this application controls.
DEFINITIONS
As used herein, the term "cells in culture" or "cultured cells" refers two or more cells in a solution (e.g., a cell medium) that allows the cells to undergo one or more cell divisions.
As used herein, the term "medium", "cell medium", "cell culture medium" or "culture medium" refers to is a liquid or gel designed to support the growth of microorganisms or cells.
The term "polynucleotide" or "nucleic acid," as used interchangeably herein, refers to polymers of nucleotides of any length, and include DNA and RNA. The nucleotides can be deoxyribonucleotides, ribonucleotides, modified nucleotides or bases, and/or their analogs, or any substrate that can be incorporated into a polymer by DNA or RNA polymerase. A polynucleotide may comprise modified nucleotides, such as methylated nucleotides and their analogs. If present, modification to the nucleotide structure may be imparted before or after assembly of the polymer. The sequence of nucleotides may be interrupted by non-nucleotide components. A polynucleotide may be further modified after polymerization, such as by conjugation with a labeling component. Other types of modifications include, for example, "caps", substitution of one or more of the naturally occurring nucleotides with an analog, internucleotide modifications such as, for example, those with uncharged linkages (e.g., methyl phosphonates, phosphotriesters, phosphoamidates, cabamates, etc.) and with charged linkages (e.g., phosphorothioates, phosphorodithioates, etc.), those containing pendant moieties, such as, for example, proteins (e.g., nucleases, toxins, antibodies, signal peptides, ply-L-lysine, etc.), those with intercalators (e.g., acridine, psoralen, etc.), those containing chelators (e.g., metals, radioactive metals, boron, oxidative metals, etc.), those containing alkylators, those with modified linkages (e.g., alpha anomeric nucleic acids, etc.), as well as unmodified forms of the polynucleotide(s). Further, any of the hydroxyl groups ordinarily present in the sugars may be replaced, for example, by phosphonate groups, phosphate groups, protected by standard protecting groups, or activated to prepare additional linkages to additional nucleotides, or may be conjugated to solid supports. The 5' and 3' terminal OH can be phosphorylated or substituted with amines or organic capping group moieties of from 1 to 20 carbon atoms. Other hydroxyls may also be derivatized to standard protecting groups. Polynucleotides can also contain analogous forms
of ribose or deoxyribose sugars that are generally known in the art, including, for example, 2'-0-methyl-, 2'-0-allyl, 2'-fluoro- or 2'- azido-ribose, carbocyclic sugar analogs, a-anomeric sugars, epimeric sugars such as arabinose, xyloses or lyxoses, pyranose sugars, furanose sugars, sedoheptuloses, acyclic analogs and abasic nucleoside analogs such as methyl riboside. One or more phosphodiester linkages may be replaced by alternative linking groups. These alternative linking groups include, but are not limited to, embodiments wherein phosphate is replaced by
P(0)S("thioate"), P(S)S ("dithioate"), "(0)NR2 ("amidate"), P(0)R, P(0)OR', CO or CH2
("formacetal"), in which each R or R is independently H or substituted or unsubstituted alkyl (1-20 C) optionally containing an ether (-0-) linkage, aryl, alkenyl, cycloalkyl, cycloalkenyl or araldyl. Not all linkages in a polynucleotide need be identical. The preceding description applies to all polynucleotides referred to herein, including RNA and DNA.
The term "heterologous nucleic acid" or "heterologous polypeptide" refers to a nucleic acid or a polypeptide whose sequence is not identical to that of another nucleic acid or polypeptide naturally found in the same host cell.
As used herein, "operably linked" as used herein refers to a functional relationship between two or more nucleic acid (e.g., DNA) segments. Typically, it refers to the functional relationship of transcriptional regulatory sequence to a transcribed sequence. For example, a promoter is operably linked to a coding sequence, such as a nucleic acid of the invention, if it stimulates or modulates the transcription of the coding sequence in an appropriate host cell or other expression system.
Generally, promoter transcriptional regulatory sequences that are operably linked to a transcribed sequence are physically contiguous to the transcribed sequence, i.e., they are ds-acting. However, some transcriptional regulatory sequences, such as enhancers, need not be physically contiguous or located in close proximity to the coding sequences whose transcription they enhance.
As used herein, the term "promoter" includes all sequences capable of driving transcription of a coding sequence in a cultured cell, e.g., a mammalian cell. Thus, promoters used in the constructs of the invention include ds-acting transcriptional control elements and regulatory sequences that are involved in regulating or modulating the timing and/or rate of transcription of a gene (e.g., a LDH or PDHK(s)). For example, a promoter can be a exacting transcriptional control element, including an enhancer, a promoter, a transcription terminator, an origin of replication, a chromosomal integration sequence, 5' and 3' untranslated regions, or an intronic sequence, which are involved in transcriptional regulation. These cis-acting sequences typically interact with proteins or other biomolecules to carry out (turn on/off, regulate, modulate, etc.) transcription. "Constitutive" promoters are those that drive expression continuously under most environmental conditions and states of development or cell differentiation. "Inducible" or "regulatable" promoters direct expression of the nucleic acid of the invention under the influence of environmental conditions or developmental conditions. Examples of environmental conditions that may affect transcription by
inducible promoters include anaerobic conditions, elevated temperature, drought, or the presence of light.
As used herein, "vector" means a construct, which is capable of delivering, and preferably expressing, one or more gene(s) or sequence(s) of interest (e.g., LDHa and
PDHK(s)) in a host cell. Examples of vectors include, but are not limited to, viral vectors, naked DNA or RNA expression vectors, plasmid, cosmid or phage vectors, DNA or RNA expression vectors associated with cationic condensing agents, DNA or RNA expression vectors encapsulated in liposomes, and certain eukaryotic cells, such as producer cells. Suitable vectors are those which are compatible with the host cell employed. Suitable vectors can be derived, for example, from a bacterium, a virus (such as bacteriophage T7 or a M- 13 derived phage), a cosmid, a yeast, or a plant. Protocols for obtaining and using such vectors are known to those in the art (see, for example, Sambrook et ah, Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor, 1989).
As used herein, "Specific Productivity" or "Qp" refers to the specific protein, e.g., antibody, production rate in pg/cell/day. Specific productivity is calculated as protein titer (pg/cell/day)/IVCC (calculate integrated viable cell count; cell/day).
The terms "polypeptide" and "protein" are used interchangeably herein to refer to polymers of amino acids of any length. The polymer may be linear or branched, it may comprise modified amino acids, and it may be interrupted by non-amino acids. The terms also encompass an amino acid polymer that has been modified naturally or by intervention; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation or modification, such as conjugation with a labeling component. Also included within the definition are, for example, polypeptides containing one or more analogs of an amino acid (including, for example, unnatural amino acids, etc.), as well as other modifications known in the art. [0055] The term "antibody" is used in the broadest sense and specifically covers monoclonal antibodies (including full length monoclonal antibodies), polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments.
"Antibody fragments" comprise a portion of a full length antibody, generally the antigen binding or variable region thereof. Examples of antibody fragments include Fab, Fab', F(ab')2, and Fv fragments; single-chain antibody molecules; diabodies; linear antibodies; and multispecific antibodies formed from antibody fragments.
The term "monoclonal antibody" as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical except for possible naturally occurring mutations that may be present in minor amounts. Monoclonal antibodies are highly specific, being directed against a single antigenic site. Furthermore, in contrast to conventional (polyclonal) antibody preparations which typically include different antibodies directed against different determinants (epitopes), each monoclonal
antibody is directed against a single determinant on the antigen. The modifier "monoclonal" indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method. For example, the monoclonal antibodies to be used in accordance with the present invention may be made by the hybridoma method first described by Kohleret al, Nature 256:495 (1975), or may be made by recombinant DNA methods (see, e.g., U.S. Pat. No. 4,816,567). The "monoclonal antibodies" may also be isolated from phage antibody libraries using the techniques described in Clackson et al., Nature 352:624-628 (1991) and Marks et al., J. Mol. Biol. 222:581-597 (1991), for example.
The monoclonal antibodies herein specifically include "chimeric" antibodies
(immunoglobulins) in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity (U.S. Pat. No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA 81 :6851-6855 (1984)).
The term "hypervariable region" when used herein refers to the amino acid residues of an antibody which are responsible for antigen-binding. The hypervariable region comprises amino acid residues from a "complementarity determining region" or "CDR" (i.e. residues 24-34 (LI), 50-56
(L2) and 89-97 (L3) in the light chain variable domain and 31-35 (HI), 50-65 (H2) and 95-102 (H3) in the heavy chain variable domain; Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)) and/or those residues from a "hypervariable loop" (i.e. residues 26-32 (LI), 50-52 (L2) and 91-96 (L3) in the light chain variable domain and 26-32 (HI), 53-55 (H2) and 96-101 (H3) in the heavy chain variable domain; Chothia and Lesk J. Mol. Biol. 196:901-917 (1987)). "Framework" or "FR" residues are those variable domain residues other than the hypervariable region residues as herein defined.
"Humanized" forms of non-human (e.g., murine) antibodies are chimeric antibodies which contain minimal sequence derived from non-human immunoglobulin. For the most part, humanized antibodies are human immunoglobulins (recipient antibody) in which hypervariable region residues of the recipient are replaced by hypervariable region residues from a non-human species (donor antibody) such as mouse, rat, rabbit or nonhuman primate having the desired specificity, affinity, and capacity. In some instances, Fv framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, humanized antibodies may comprise residues which are not found in the recipient antibody or in the donor antibody. These modifications are made to further refine antibody performance. In general, the humanized antibody will comprise
substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence. The humanized antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see Jones et al., Nature 321 :522-525 (1986); Riechmann et al., Nature 332:323-329 (1988); and Presta, Curr. Op. Struct. Biol. 2:593-596 (1992).
As used herein, the term "immunoadhesin" designates antibody-like molecules which combine the "binding domain" of a heterologous "adhesin" protein (e.g. a receptor, ligand or enzyme) with the effector functions of an immunoglobulin constant domain. Structurally, the immunoadhesins comprise a fusion of the adhesin amino acid sequence with the desired binding specificity which is other than the antigen recognition and binding site (antigen combining site) of an antibody (i.e. is "heterologous") and an immunoglobulin constant domain sequence. The immunoglobulin constant domain sequence in the immunoadhesin is preferably derived from γΐ, γ2, or γ4 heavy chains since immunoadhesins comprising these regions can be purified by Protein A chromatography (Lindmark et al., J. Immunol. Meth. 62: 1-13 (1983)).
The term "ligand binding domain" as used herein refers to any native cell-surface receptor or any region or derivative thereof retaining at least a qualitative ligand binding of a corresponding native receptor. In a specific embodiment, the receptor is from a cell- surface polypeptide having an extracellular domain which is homologous to a member of the immunoglobulin supergenefamily. Other receptors, which are not members of the immunoglobulin supergenefamily but are nonetheless specifically covered by this definition, are receptors for cytokines, and in particular receptors with tyrosine kinase activity (receptor tyrosine kinases), members of the hematopoietin and nerve growth factor receptor superfamilies, and cell adhesion molecules, e.g. (E-, L- and P-) selectins.
The term "receptor binding domain" is used to designate any native ligand for a receptor, including cell adhesion molecules, or any region or derivative of such native ligand retaining at least a qualitative receptor binding ability of a corresponding native ligand. This definition, among others, specifically includes binding sequences from ligands for the above- mentioned receptors.
An "antibody-immunoadhesin chimera" comprises a molecule which combines at least one binding domain of an antibody (as herein defined) with at least one immunoadhesin (as defined in this application). Exemplary antibody-immunoadhesin chimeras are the bispecific CD4-IgG chimeras described in Berg et al., PNAS (USA) 88:4723-4727 (1991) and Chamow et al., J. Immunol. 153:4268 (1994).
The term "osmolality" refers to the number of solute particles dissolved in 1 liter of solution. Solutes which can be added to the culture medium so as to increase the osmolality thereof include proteins, peptides, amino acids, non-metabolized polymers, vitamins, ions, salts (e.g., sodium or
potassium salts), sugars, metabolites, organic acids, lipids, etc. When used herein, the abbreviation "mOsm" means "milliosmoles/Liter H20."
As used herein, a "host cell" includes an individual cell, cultured cells, or cell in culture that can be or has been a recipient for vector(s) or siRNA(s) for incorporation of polynucleotide inserts to produce polypeptide. Host cells include progeny of a single cultured cell, and the progeny may not necessarily be completely identical (in morphology or in genomic DNA complement) to the original parent cell due to natural, accidental, or deliberate mutation. For use herein, unless clearly indicated otherwise, use of the terms "a", "an," and the like refers to one or more.
Reference to "about" a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se. For example, description referring to "about X" includes description of "X." Numeric ranges are inclusive of the numbers defining the range.
The term "alkyl" as used herein refers to a saturated linear or branched-chain monovalent hydrocarbon radical of one to twelve carbon atoms (C1-C12), wherein the alkyl radical may be optionally substituted independently with one or more substituent(s) described below. In another embodiment, an alkyl radical is one to eight carbon atoms (Q-Cg), or one to six carbon atoms (Ci-C6). Examples of alkyl groups include, but are not limited to, methyl (Me, -CH3), ethyl (Et, -CH2CH3), 1 -propyl (n-Pr, n-propyl, -CH2CH2CH3), 2-propyl (i-Pr, i-propyl, -CH(CH3)2), 1 -butyl (n-Bu, n-butyl, -CH2CH2CH2CH3), 2-methyl-l -propyl (i-Bu, i-butyl, -CH2CH(CH3)2), 2-butyl (s-Bu, s-butyl, -CH(CH3)CH2CH3), 2-methyl-2-propyl (t-Bu, t-butyl, -C(CH3)3), 1-pentyl (n-pentyl, -CH2CH2CH2CH2CH3), 2-pentyl (-CH(CH3)CH2CH2CH3), 3-pentyl (-CH(CH2CH3)2),
2-methyl-2-butyl (-C(CH3)2CH2CH3), 3-methyl-2-butyl (-CH(CH3)CH(CH3)2), 3 -methyl- 1 -butyl (-CH2CH2CH(CH3)2), 2-methyl-l -butyl (-CH2CH(CH3)CH2CH3), 1-hexyl
(-CH2CH2CH2CH2CH2CH3), 2-hexyl (-CH(CH3)CH2CH2CH2CH3), 3-hexyl
(-CH(CH2CH3)(CH2CH2CH3)), 2-methyl-2-pentyl (-C(CH3)2CH2CH2CH3), 3-methyl-2-pentyl (-CH(CH3)CH(CH3)CH2CH3), 4-methyl-2-pentyl (-CH(CH3)CH2CH(CH3)2), 3-methyl-3-pentyl (-C(CH3)(CH2CH3)2), 2-methyl-3-pentyl (-CH(CH2CH3)CH(CH3)2), 2,3-dimethyl-2-butyl
(-C(CH3)2CH(CH3)2), 3,3-dimethyl-2-butyl (-CH(CH3)C(CH3)3, 1-heptyl, 1-octyl, and R2 groups as exemplified therein.
The term "Ci-Ci2-alkoxy" means a Ci-Ci2-alkyl group, wherein alkyl is as defined herein, that is linked to the rest of a molecule or to another group through an oxygen atom. Illustrative, non limiting examples of alkoxy include methoxy, ethoxy, n-propoxy, isopropoxy and the different butoxy isomers and R1 groups as exemplified therein.
The expression "(Ci-Ci2-alkylenyl)n-Ci-Ci2-alkoxy" means either a
(Ci-Ci2-alkylenyl)-Ci-Ci2-alkoxy or a Ci-Ci2-alkoxy group, wherein alkylenyl and alkoxy are as defined herein.
The term "alkylene" or "alkylenyl" as used herein refers to a saturated linear or branched-chain divalent hydrocarbon radical of one to twelve carbon atoms (C1-C12), wherein the alkylene radical may be optionally substituted independently with one or more substituent(s) described below. In another embodiment, an alkylene radical is one to eight carbon atoms (Q-Cg), or one to six carbon atoms (Ci-C6). Examples of alkylene groups include, but are not limited to, methylene (-CH2-), ethylene (-CH2CH2-), propylene (-CH2CH2CH2-), and R1 groups as exemplified therein.
"Aryl" means a monovalent aromatic hydrocarbon radical of 6-20 carbon atoms (C6-C2o) or C6-C20-aryl, derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system. Some aryl groups are represented in the exemplary structures as "Ar". Aryl includes bicyclic radicals comprising an aromatic ring fused to a saturated, partially unsaturated ring, or aromatic carbocyclic ring. Typical aryl groups include, but are not limited to, radicals derived from benzene (phenyl), substituted benzenes, naphthalene, anthracene, biphenyl, indenyl, indanyl, 1 ,2-dihydronaphthalene, 1,2,3,4-tetrahydronaphthyl, and the like. Aryl groups are optionally substituted independently with one or more substituent(s) described herein. Further non limiting examples of aryl groups can be found in the definition of R1 herein.
"aryloxy" as used herein denotes an -O-aryl group, wherein aryl is as defined herein.
Non-limiting examples of -O-aryl groups are -O-phenyl and -O-naphthyl groups..
The term "cyanoalkyl" as used herein refers to an alky group as defined herein that is substituted by one or more cyano group, for example one cyano group. In certain embodiments "cyanoalkyl" are Ci-Ci2-cyanoalkyl groups. In other embodiments "cyanoalkyl" are
Ci-C6-cyanoalkyl groups, for example cyanomethyl and cyanoethyl.
The terms "carbocycle", "carbocyclyl", "carbocyclic ring" and "cycloalkyl" refer to a monovalent non-aromatic, saturated or partially unsaturated ring having 3 to 12 carbon atoms (C3-C12) as a monocyclic ring or 7 to 12 carbon atoms as a bicyclic ring. Partially unsaturated rings can also be designated as cycloalkenyl rings. Bicyclic carbocycles having 7 to 12 atoms can be arranged, for example, as abicyclo [4,5], [5,5], [5,6] or [6,6] system, and bicyclic carbocycles having 9 or 10 ring atoms can be arranged as a bicyclo [5,6] or [6,6] system, or as bridged systems such as bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane and bicyclo[3.2.2]nonane. Examples of monocyclic carbocycles or cycloalkyls include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-l-enyl, 1 -cyclopent-2-enyl, l-cyclopent-3-enyl, cyclohexyl, 1-cyclohex-l-enyl, 1 -cyclohex-2-enyl, l-cyclohex-3-enyl, cyclohexadienyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl, cyclododecyl, adamantanyl, and R2 groups as exemplified therein.
The term "halo" denotes chloro, iodo, fluoro and bromo, In an embodiment halo are fluoro, chloro and bromo, and yet in another embodiment fluoro and chloro.
The term "haloalkyl" denotes an alkyl group as defined above wherein at least one of the hydrogen atoms of the alkyl group is replaced by a halogen atom, preferably fluoro or chloro, most preferably fluoro. Examples of haloalkyl include Ci-Ci2-haloalkyl groups, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl, sec -butyl, tert-butyl, pentyl or n-hexyl wherein one or more hydrogen atoms are replaced by CI, F, Br or I atom(s), as well as those haloalkyl groups specifically illustrated by the examples herein below. Among the preferred haloalkyl groups are monofluoro-, difluoro- or trifluoro-methyl, -ethyl or -propyl, for example 3,3,3-trifluoropropyl, 2-fluoroethyl, 2,2,2-trifluoroethyl, fluoromethyl, trifluoromethyl. The term "Ci-Ci2-haloalkyl" means a haloalkyl group having 1 to 12 carbon atoms, wherein the haloalkyl is as defined herein. The term "haloalkoxy" denotes a alkoxy group as defined herein wherein at least one of the hydrogen atoms of the alkoxy group is replaced by a halogen atom, preferably fluoro or chloro, most preferably fluoro. Examples of haloalkoxy include Ci-Ci2-haloalkoxy groups, but are not limited to, methoxy, ethoxy, propyloxy, isopropyloxy, isobutyloyx, sec-butyloxy, tert-butyloxy, pentyloxy or n-hexyloxy wherein one or more hydrogen atoms are replaced by CI, F, Br or I atom(s), as well as those haloalkoxy groups specifically illustrated by the examples herein below. Among the preferred haloalkoxy groups are monofluoro-, difluoro- or trifluoro-methoxy, -ethoxy or -propyloxy, for example 3,3,3-trifluoropropyloxy, 2-fluoroethoxy, 2,2,2-trifluoroethoxy, fluoromethoxy, trifluoromethoxy. In a certain embodiment
 groups are Ci-C6-haloalkoxy groups.
groups are Ci-C6-haloalkoxy groups.
The terms "heterocycle," "heterocyclyl" and "heterocyclic ring" are used interchangeably herein and refer to a saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic radical of 3 to about 20 ring atoms in which at least one ring atom is a heteroatom selected from nitrogen, oxygen, phosphorus and sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituent(s) described below. Examples of heterocycly groups are 4 to 10 membered heterocyclyl, i.e. heterocyclyl groups comprising 2 to 9 carbon atoms and 1 , 2, 3 or 4 heteroatoms selected from N, O, P, and S. A heterocycle may be a monocycle having 3 to 7 ring members (2 to 6 carbon atoms and 1 to 4 heteroatoms selected from N, O, P, and S) or a bicycle having 7 to 10 ring members (4 to 9 carbon atoms and 1 to 6 heteroatoms selected from N, O, P, and S), for example: a bicyclo [4,5], [5,5], [5,6], or [6,6] system. Heterocycles are described in Paquette, Leo A.;
"Principles of Modern Heterocyclic Chemistry" (W.A. Benjamin, New York, 1968), particularly Chapters 1 , 3, 4, 6, 7, and 9; "The Chemistry of Heterocyclic Compounds, A series of Monographs" (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and J. Am. Chem. Soc. (1960) 82:5566. "Heterocyclyl" also includes radicals where heterocycle radicals are fused with a saturated, partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring. Examples of heterocyclic rings include, but are not limited to, pyrrolidinyl, tetrahydrofuranyl,
dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, piperidonyl, morpholino, thiomorpholino, thioxanyl, piperazinyl, homopiperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl, dihydropyranyl, dihydrothienyl, dihydrofuranyl,
dihydroisoquinolinyl, tetrahydroisoquinolinyl, pyrazolidinylimidazolinyl, imidazolidinyl,
2- oxa-5-azabicyclo[2.2.2]octane, 3-oxa-8-azabicyclo[3.2.1]octane, 8-oxa-3-azabicyclo[3.2.1]octane, 6-oxa-3-azabicyclo[3.1.1 ]heptane, 2-oxa-5-azabicyclo[2.2.1 Jheptane, 3-azabicyco[3.1.0]hexanyl,
3- azabicyclo[4.1.0]heptanyl, azabicyclo[2.2.2]hexanyl, 3H-indolyl quinolizinyl and N-pyridyl ureas. Spiro moieties are also included within the scope of this definition. Examples of a heterocyclic group wherein 2 ring carbon atoms are substituted with oxo (=0) moieties are pyrimidinonyl and 1,1-dioxo-thiomorpholinyl. The heterocycle groups herein are optionally substituted independently with one or more substituent(s) described herein.
The term "heteroaryl" refers to a monovalent aromatic radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at least one of which is aromatic) of 5-20 atoms containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur. Examples of heteroaryl groups include 5 to 10 membered heteroaryls which denotes monocyclic of bicyclic heteroaryl having 2 to 9 carbon atoms and one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur, for example, 1, 2, 3 or 4 heteroatoms independently selected from nitrogen, oxygen, and sulfur. Examples of heteroaryl groups include 5 or 6 membered heteroaryls which denotes monocyclic of bicyclic heteroaryl having 2 to 5 carbon atoms and one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur, for example, 1, 2, 3 or 4 heteroatoms independently selected from nitrogen, oxygen, and sulfur. Non limiting examples of heteroaryl groups are pyridinyl (including, for example, 2-hydroxypyridinyl), imidazolyl, imidazopyridinyl, pyrimidinyl (including, for example, 4-hydroxypyrimidinyl), pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxadiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, triazolyl, thiadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinoxalinyl, naphthyridinyl, and furopyridinyl.
Heteroaryl groups are optionally substituted independently with one or more substituent(s) described herein, for example alkyl, alkoxy, cyano, halo, oxo, NH2, OH, hydroxyalkyl, amido groups. Further examples of heteroaryl groups and of possible substituents can be found in the definition of R2.
The term "heteroaryloxy" as used herein means an -O-heteroaryl, wherein heteroaryl is as defined herein.
The heterocycle or heteroaryl groups may be carbon (carbon-linked), or nitrogen
(nitrogen-linked) bonded where such is possible. By way of example and not limitation, carbon bonded heterocycles or heteroaryls are bonded at position 2, 3, 4, 5, or 6 of a pyridine, position 3, 4, 5, or 6 of a pyridazine, position 2, 4, 5, or 6 of a pyrimidine, position 2, 3, 5, or 6 of a pyrazine, position 2, 3, 4, or 5 of a furan, tetrahydrofuran, thiofuran, thiophene, pyrrole or tetrahydropyrrole, position 2, 4, or 5 of an oxazole, imidazole or thiazole, position 3, 4, or 5 of an isoxazole, pyrazole, or isothiazole, position 2 or 3 of an aziridine, position 2, 3, or 4 of an azetidine, position 2, 3, 4, 5, 6, 7, or 8 of a quinoline or position 1, 3, 4, 5, 6, 7, or 8 of an isoquinoline. Ring nitrogen atoms of the heterocycle or heteroaryl groups may be bonded with oxygen to form N-oxides.
By way of example and not limitation, nitrogen bonded heterocycles or heteroaryls are bonded at position 1 of an aziridine, azetidine, pyrrole, pyrrolidine, 2-pyrroline, 3-pyrroline, imidazole, imidazolidine, 2-imidazoline, 3 -imidazoline, pyrazole, pyrazoline, 2-pyrazoline, 3-pyrazoline, piperidine, piperazine, indole, indoline, lH-indazole, benzimidazole, position 2 of a isoindole, or isoindoline, position 4 of a morpholine, and position 9 of a carbazole, or β-carboline.
The term "hydroxy" denotes a group of formula -OH.
The term "hydroxyalkyl" denotes an alkyl group as defined above wherein at least one of the hydrogen atoms of the alkyl group is replaced by a hydroxy group. Examples of hydroxyalkyl include, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl, sec-butyl, tert-butyl, pentyl or n-hexyl wherein one or more hydrogen atoms are replaced by OH, as well as those hydroxyalkyl groups specifically illustrated by the examples herein below. The term

means a hydroxyalkyl group having 1 to 12 carbon atoms, wherein hydroxyalkyl is as defined herein.
Oxo denotes a group of formula =0.
The expression "one or more substituent" denotes a substitution by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 substituent(s) that can be independently selected from the list following this expression. In an embodiment, one or more substituent(s) denotes 1, 2, 3, 4 or 5 substituents. In an embodiment, one or more substituent(s) denotes 1, 2 or 3 substituents.
The terms "treat" and "treatment" refer to both therapeutic treatment and prophylactic or preventative measures, wherein the object is to prevent or slow down (lessen) an undesired physiological change or disorder, such as the development or spread of cancer. For purposes of this invention, beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable. "Treatment" can also mean prolonging survival as compared to expected survival if not receiving treatment. Those in need of
treatment include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented.
The phrase "therapeutically effective amount" means an amount of a compound of the present invention that (i) treats or prevents the particular disease, condition, or disorder, (ii) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, or disorder, or (iii) prevents or delays the onset of one or more symptoms of the particular disease, condition, or disorder described herein. In the case of cancer, the therapeutically effective amount of the drug may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer. To the extent the drug may prevent growth and/or kill existing cancer cells, it may be cytostatic and/or cytotoxic. For cancer therapy, efficacy can be measured, for example, by assessing the time to disease progression (TTP) and/or determining the response rate ( R).
The terms "cancer" refers to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth. A "tumor" comprises one or more cancerous cells. Examples of cancer include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies. More particular examples of such cancers include squamous cell cancer (e.g., epithelial squamous cell cancer), lung cancer including small-cell lung cancer, non-small cell lung cancer ("NSCLC"), adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial or uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, head and neck cancer, multiple myeloma, acute myelogenous leukemia, chronic lymphoid leukemia, chronic myelogenous leukemia, lymphocytic leukemia, myeloid leukemia, oral cavity and pharynx, non-Hodgkin lymphoma, melanoma, and villous colon adenoma.
The term "chiral" refers to molecules which have the property of non-superimposability of the mirror image partner, while the term "achiral" refers to molecules which are superimposable on their mirror image partner.
The term "stereoisomers" refers to compounds which have identical chemical constitution, but differ with regard to the arrangement of the atoms or groups in space. Stereoisomers include enantiomers and diastereomers.
"Diastereomer" refers to a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties,
e.g. melting points, boiling points, spectral properties, and reactivities. Mixtures of diastereomers may separate under high resolution analytical procedures such as electrophoresis and
chromatography. Diastereomers include geometric isomers, cis/trans and E/Z isomers, and atropisomers.
"Enantiomers" refer to two stereoisomers of a compound which are non-superimposable mirror images of one another.
Stereochemical definitions and conventions used herein generally follow S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; and Eliel, E. and Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994. The compounds of the invention may contain asymmetric or chiral centers, and therefore exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of the invention, including but not limited to, diastereomers, enantiomers and atropisomers, as well as mixtures thereof such as racemic mixtures, form part of the present invention. Many organic compounds exist in optically active forms, i.e., they have the ability to rotate the plane of plane -polarized light. In describing an optically active compound, the prefixes D and L, or R and S, are used to denote the absolute configuration of the molecule about its chiral center(s). The prefixes d and 1 or (+) and (-) are employed to designate the sign of rotation of plane-polarized light by the compound, with (-) or 1 meaning that the compound is levorotatory. A compound prefixed with (+) or d is dextrorotatory. For a given chemical structure, these stereoisomers are identical except that they are mirror images of one another. A specific stereoisomer may also be referred to as an enantiomer, and a mixture of such isomers is often called an enantiomeric mixture. A 50:50 mixture of enantiomers is referred to as a racemic mixture or a racemate, which may occur where there has been no stereoselection or stereospecificity in a chemical reaction or process. The terms "racemic mixture" and "racemate" refer to an equimolar mixture of two enantiomeric species, devoid of optical activity.
The term "tautomer" or "tautomeric form" refers to structural isomers of different energies which are interconvertible via a low energy barrier. For example, proton tautomers (also known as prototropic tautomers) include interconversions via migration of a proton, such as keto-enol and imine-enamine isomerizations. Valence tautomers include interconversions by reorganization of some of the bonding electrons. As stated above, the compounds of Formula (I) also covers tautomers thereof, such as depicted in the following formulae:

The phrase "pharmaceutically acceptable salt" as used herein, refers to pharmaceutically acceptable organic or inorganic salts of a compound of the invention. Exemplary salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucuronate, saccharate, formate, benzoate, glutamate, methanesulfonate "mesylate", ethanesulfonate, benzenesulfonate, p-toluenesulfonate, and pamoate (i.e., 1,1 '-methyl ene -bis(2-hydroxy- 3-naphthoate)) salts. A pharmaceutically acceptable salt may involve the inclusion of another molecule such as an acetate ion, a succinate ion or other counter ion. The counter ion may be any organic or inorganic moiety that stabilizes the charge on the parent compound. Furthermore, a pharmaceutically acceptable salt may have more than one charged atom in its structure. Instances where multiple charged atoms are part of the pharmaceutically acceptable salt can have multiple counter ions. Hence, a pharmaceutically acceptable salt can have one or more charged atoms and/or one or more counter ion.
If the compound of the invention is a base, the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, methanesulfonic acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, trifluoroacetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-toluenesulfonic acid or ethanesulfonic acid, or the like.
If the compound of the invention is an acid, the desired pharmaceutically acceptable salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like. Illustrative examples of suitable salts include, but are not limited
to, organic salts derived from amino acids, such as glycine and arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as piperidine, morpholine and piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
The phrase "pharmaceutically acceptable" indicates that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
A "solvate" refers to an association or complex of one or more solvent molecules and a compound of the invention. Examples of solvents that form solvates include, but are not limited to, water, isopropanol, ethanol, methanol, DMSO, ethylacetate, acetic acid, and ethanolamine.
The terms "compound of this invention," and "compounds of the present invention" and "compounds of Formula (I)" include compounds of Formulas (I), (I-a) and (I-a-1), specific compounds described herein and stereoisomers, tautomers, solvates, metabolites, and
pharmaceutically acceptable salts and prodrugs thereof. As stated above, particular tautomers of the comp

Any formula or structure given herein, including Formula (I) compounds, is also intended to represent hydrates, solvates, and polymorphs of such compounds, and mixtures thereof.
Any formula or structure given herein, including Formula (I) compounds, is also intended to represent isotopically labeled forms of the compounds as well as unlabeled forms. Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as, but not limited to 2H (deuterium, D), 3H (tritium), 11C, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36C1, and 1251. Various isotopically labeled compounds of the present invention, for example those into which radioactive isotopes such as 3H, 13C, and 14C are incorporated. Such isotopically labelled compounds may be useful in metabolic
studies, reaction kinetic studies, detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients. Deuterium labelled or substituted therapeutic compounds of the invention may have improved DMPK (drug metabolism and pharmacokinetics) properties, relating to distribution, metabolism, and excretion (ADME). Substitution with heavier isotopes such as deuterium may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements. An 18F labeled compound may be useful for PET or SPECT studies. Isotopically labeled compounds of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the schemes or in the examples and preparations described below by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent. Further, substitution with heavier isotopes, particularly deuterium (i.e., 2H or D) may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index. It is understood that deuterium in this context is regarded as a substituent in the compound of the formula (I). The concentration of such a heavier isotope, specifically deuterium, may be defined by an isotopic enrichment factor. In the compounds of this invention any atom not specifically designated as a particular isotope is meant to represent any stable isotope of that atom. Unless otherwise stated, when a position is designated specifically as "H" or "hydrogen", the position is understood to have hydrogen at its natural abundance isotopic composition. Accordingly, in the compounds of this invention any atom specifically designated as a deuterium (D) is meant to represent deuterium.
INHIBITORS OF LDHA
In one aspect, the invention relates to methods of controlling lactate production using compounds of Formula (I):

A1 is O, CH2, or S;
A2 is NH or N-CrC3-alkyl;
A3 is N or CR2;
A4 is N or CR3, provided that A3 and A4 are not N at the same time;
R1 is CI, N02, or CN;
R2 and R6 are independently selected from the group consisting of H, halo, hydroxy,
CrC6-hydroxyalkyl, and NH2;
R3 and R5 are independently selected from the group consisting of:
• H;
• hydroxy;
• halo;
· -CrC6-alkyl-Rf;
• -CrC6-alkenyl-Rf;
• -CrC6-alkoxy-Rc;
• -NRaRb;
• -NRa-(CrC6-alkyl)-Rd;
· -NRa-S(0)2-(4 to 10 membered heterocycloalkyl);
• -NRa-(C3-C8-cycloalkyl), which cycloalkyl is unsubstituted or substituted by Ci-C6-alkyl or a Ci-C3-alkylene bridge;
• -NRa-aryl, which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, hydroxy, -NH2, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-haloalkyl,
Ci-C6-hydroxyalkyl, Ci-C6-haloalkoxy and C3-C8-cycloalkyl;
• -NRa-(4 to 10 membered heterocycloalkyl), which heterocycloalkyl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C6-alkyl, Ci-C6-hydroxyalkyl, or -CO-alkyl;
· -NRa-(5 or 6 membered heteroaryl), which heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, -NRaRb and Ci-C6-alkyl;
• -NRa(CO)-CrC6-alkyl;
• -NRa(CO)-aryl;
• -NRa(CO)-(5 or 6 membered heteroaryl);
· -NRa(CO)0-CrC6-alkyl;
• -S-(alkyl)n-Rh;
• -S(0)2-aryl, which aryl is unsubstituted or substituted by one or more halo;
• -C(0)-Re;
• -C(0)NRa-(C1-C6-alkyl)n-R8;
• -C(0)NRa-CrC6-alkoxy;
• -0-C3-C8-cycloalkyl, which cycloalkyl is unsubstituted or substituted by ne or more
substituent(s) selected from the group consisting of: halo or hydroxy, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-haloalkoxy, Ci-C6-alkoxyaryl, Ci-C6-haloalkyl, Ci-C6-hydroxyalkyl, NRaRb, aryl, Ci-C6-akyl-aryl, 5 or 6 membered heteroaryl, and
-(Ci-C6-alk l)-(Ci-C6-alkoxy);
• -O-aryl, which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-alkyl-Ci-C6-alkoxy, Ci-C6-haloalkyl,
Ci-C6-haloalkoxy, Ci-C6-hydroxyalkyl, -S-Ci-C6-akyl,
-CrC6-alkyl-C3-C8-cycloalkyl, CrC6-alkoxy-C3-C8-cycloalkyl, CrC6-alkyl-(4 to 10 membered heterocycloalkyl), Ci-C6-alkyl-(5 or 6 membered heterocycloalkyl), or 5 or 6 membered heteroaryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C6-alkyl, -(Ci-C6-alkyl)-(Ci-C6-alkoxy), Ci-C6-haloalkoxy and a Ci-C6-alkylene bridge;
• -0-(4 to 10 membered heterocycloalkyl), which heterocycloalkyl is unsubstituted or
substituted by one or more substituent(s) selected from the group consisting of:
o halo, hydroxy, Ci-C6-alkyl, Ci-C6-hydroxyalkyl and -C(0)-Ci-C6-alkyl;
• -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, CrC6-alkyl, CrC6-hydroxyalkyl, or -NRa(CO)-CrC6-akyl;
• C3-C8-cycloalkyl, which cycloalkyl may be fused to a phenyl;
• aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, hydroxy, -C(0)OH, CrC6-hydroxyalkyl, CrC6-alkoxy, -S(0)2-NH(alkyl) and -S(0)2-N(alkyl)2;
• 4 to 10 membered heterocycloalkyl unusbstituted or substituted by one or more
substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkyl, -C(0)-C3-C8-cycloalkyl, oxo and 5 or 6 membered
heterocycloalkyl;
• 5 to 10 membered heteroaryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o hydroxy, -NRaRb, CrC6-alkyl, CrC6-hydroxyalkyl, and 4 to 10 membered heterocycloalkyl;
R4 is:
• H,
• cyano,
• halo,
• hydroxy,
• NRaRb,
• CrC6-alkyl,
• Ci-C6-haloalkyl,
• Ci-C6-hydroxyalkyl,
• Ci-C6-alkoxy unsubstituted or substituted by hydroxy, Ci-C6-alkoxy or NRaRb,
• -(Ci-C6-alkyl)n-(C3-C8-cycloalkyl), unsubstituted or substituted by one or more
substituent(s) selected from the group consisting of: halo, hydroxy, -NRaRb, Ci-C6-alkyl, CrC6-alkoxy, CrC6-haloalkyl, -C(0)-CrC6-alkyl, -C(0)-CrC6-cycloalkyl; -C(0)-(5 or 6 membered heterocycloalkyl);
• -(Ci-C6-alkyl)n-(C3-C8-cycloalkenyl), unsubstituted or substituted by one or more
substituent(s) selected from the group consisting of: halo, hydroxy, -NRaRb, Ci-C6-alkyl, CrC6-alkoxy, CrC6-haloalkyl, -C(0)-CrC6-alkyl, -C(0)-CrC6-cycloalkyl and -C(0)-(5 or 6 membered heterocycloalkyl);
• -(Ci-C6-alkyl)n-(5 or 6 membered heteroaryl), unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, hydroxy, -NRaRb, Ci-C6-alkyl, d-Cg-alkoxy, CrC6-haloalkyl and -C(0)-CrC6-alkyl, -C(0)-CrC6-cycloalkyl and -C(0)-(5 or 6 membered heterocycloalkyl);
• -(Ci-C6-alkyl)n-(4 to 10 membered heterocycloalkyl) unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, hydroxy, cyano, -NRaRb, CrC6-alkyl, CrC6-alkoxy, CrC6-haloalkyl, CrC6-hydroxyalkyl, -C(0)OH, a
CrC4-alkylene bridge, -C(0)-CrC6-alkyl, -C(0)-C3-C8-cycloalkyl, -C(0)-aryl, -C(0)(4 to 10 membered heterocycloalkyl) and -C(0)-(5 or 6 membered heterocycloalkyl);
R7 is aryl, a 5 or 6 membered heterocycle or 5 or 6 membered heteroaryl which aryl, heterocycle or heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-alkyl, C3-C8-cycloalkyl, -O-aryl, -S-aryl, -NH-aryl, and
-(CrC6-alkyl)n-aryl;
or R6 and R7 together with the carbon atoms to which they are attached form a 5 membered ring selected from a cycloalkyl or heterocycloalkyl having 5 ring members;
R8 is OH, -NRaRb, CrC6-alkoxy or -C(0)0-CrC6-alkyl;
or R2 and R3 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
· halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
or R3 and R4 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
or R4 and R5 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
or R5 and R6 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
Ra is H or CrC6-alkyl;
Rb is H or CrC6-alkyl;
Rc is H, hydroxy, halo, -NRaRb, Ci-C6-alkoxy, Ci-C6-alkenyl, 4 to 6 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C6-alkyl, 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C6-alkyl, or C3-C8-cycloalkyl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, Ci-C6-alkyl or Ci-C6-hydroxyalkyl, aryl unsubstituted or substituted by halo, 4 to 9 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C6-alkyl, and 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C6-alkyl;
Rd is H, hydroxy, Ci-C6-alkyl, C3-C8-cycloalkyl or aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo and -NRa-S(0)2-N(Ci-C6-alkyl)2;
Re is Ci-C6-alkyl, aryl, C3-C8-cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl and wherein said aryl, C3-C8-cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, Ci-C6-alkoxy, Ci-C6-alkyl and Ci-C6-haloalkyl;
Rf is H, C3-C8-cycloalkyl, 4 to 10 membered heterocycloalkyl, aryl, or 5 or 6 membered heteroaryl, which cycloalkyl, heterocycloalkyl, aryl, or heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-haloalkyl, Ci-C6-alkyl, Ci-C6-alkoxy and Ci-C6-hydroxyalkyl;
Rs is Ci-C6-alkoxy, C3-C8-cycloalkyl, aryl, 5 or 6 membered heteroaryl, 5 to 9 membered heterocycloalkyl, wherein said aryl, C3-C8-cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-alkoxy and Ci-C6-hydroxyalkyl;
Rh is aryl, 5 or 6 membered heteroaryl, 4 to 10 membered heterocycloalkyl, C3-C8-cycloalkyl, each of which is unsubstituted or substituted by halo;
n is 0 or 1.
In an embodiment, the invention relates to methods of controlling lactate production using compounds of F

and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A1, A3, A4, R1, R4, R5, R6, R8, R9 and R10 are as described herein.
In an embodiment, the invention relates to methods of controlling lactate production using compounds of Formula (I) which are represented by the following
formula:
 and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, A4, R1, R4, R5, R6, R7 and R8 are as described herein.
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, A4, R1, R4, R5, R6, R7 and R8 are as described herein.
In an embodiment, the invention relates to methods of controlling lactate production using compounds of
Formula (I) which are represented by the following
wherein A , A ,

In an embodiment, the invention relates to methods of controlling lactate production usin^ compounds of Formula (I) which are represented by the following
formula:

In an embodiment, the invention relates to methods of controlling lactate production using compounds
formula:
 and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, R1, R3, R4, R5 and R6 are as described herein are as described herein.
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, R1, R3, R4, R5 and R6 are as described herein are as described herein.
In an embodiment, the invention relates to methods of controlling lactate production using compounds of owing
formula:
 and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, R1 and R3 are as described herein are as described herein.
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, R1 and R3 are as described herein are as described herein.
In an embodiment, the invention relates to methods of controlling lactate production using compounds of owing
formula:
 and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, R1 and R3 are as described herein are as described herein.
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein A3, R1 and R3 are as described herein are as described herein.
In an embodiment, the invention relates to methods of controlling lactate production using compounds of owing
formula:
 and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein R1 and R3 are as described herein are as described herein.
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, wherein R1 and R3 are as described herein are as described herein.
In an embodiment, the invention relates to methods of controlling lactate production usin^ comp owing

In an embodiment, the invention relates to methods of controlling lactate production using comp owing

In an embodiment, the invention relates to methods of controlling lactate production usin^ comp owing

In an embodiment, the compounds of Formula (I) and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof, are defined as below wherein:
A1 is O or S;
A2 is NH or N-CrC3-alkyl;
A3 is N or CR2;
R1 is CI, N02, or CN;
R2 and R6 are independently selected from the group consisting of H, halo, hydroxy and NH2; R3 and R5 are independently selected from the group consisting of:
· H;
• hydroxy;
• halo;
• -Ci-C6-alkyl-Rf, wherein Rf is 4 to 10 membered heterocycloalkyl, aryl, or 5 or 6 membered heteroaryl, which C3-C8-cycloalkyl, 5 to 9 membered heterocycloalkyl, aryl, or 5 or 6 membered heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkoxy and Ci-C6-hydroxyalkyl;
• -CrC6-alkoxy-Rc, wherein Rc is H, hydroxy, halo, -NRaRb, CrC6-alkoxy, CrC6-alkenyl, C3-C8-cycloalkyl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkyl or Ci-C6-hydroxyalkyl, aryl unsubstituted or substituted by halo, 4 to 9 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C6-alkyl, and 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C6-alkyl;
• -NRaRb, wherein Ra and Rb are independently selected from H or Ci-C6-alkyl;
• -NRa-(CrC6-alkyl)-Rd, wherein Ra is H or CrC6-alkyl and Rd is H, hydroxy, CrC6-alkyl, C3-C8-cycloalkyl or aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo and -NRa-S(0)2-N(CrC6-alkyl)2;
• -NRa-S(0)2-(4 to 10 membered heterocycloalkyl), wherein Ra is H or CrC6-alkyl;
• -NRa-(C3-C8-cycloalkyl), wherein Ra is H or Ci-C6-alkyl and which cycloalkyl is
unsubstituted;
• -NRa-aryl, wherein Ra is H or Ci-C6-alkyland which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkoxy, Ci-C6-haloalkyl, and Ci-C6-hydroxyalkyl;
• -NRa-(4 to 10 membered heterocycloalkyl), wherein Ra is H or Ci-C6-alkyl;
• -NRa-(5 or 6 membered heteroaryl), wherein Ra is H or Ci-C6-alkyl and which heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, -NH2 or CrC6-alkyl;
• -NRa(CO)0-CrC6-alkyl, wherein Ra is H or CrC6-alkyl;
• -C(0)-Re, wherein Re is aryl and wherein said aryl is substituted by halo, or Ci-C6-haloalkyl;
• -C(0)NRa-(CrC6-alkyl)n-R8, wherein Ra is H or CrC6-alkyl and R8 is CrC6-alkoxy,
C3-C8-cycloalkyl;
• -0-C3-C8-cycloalkyl, which cycloalkyl is unsubstituted or substituted by halo or hydroxy, Ci-C6-alkyl, Ci-C6-alkoxy, which alkoxy is unsubstituted or substituted by Ci-C6-alkoxyaryl, Ci-C6-haloalkyl;
• -O-aryl, which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-haloalkyl, Ci-C6-haloalkoxy,
CrCg-hydroxyalkyl, -S-CrC6-akyl, -CrC6-alkyl-C3-C8-cycloalkyl, 4 to 10 membered heterocycloalkyl, 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C6-alkyl, and Ci-C6-alkylene bridge;
· -0-(4 to 10 membered heterocycloalkyl), which heterocycloalkyl is unsubstituted or
substituted by one or more substituent(s) selected from the group consisting of:
o hydroxyl, Ci-C6-hydroxyalkyl, -C(0)-Ci-C6-alkyl;
• -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, or -NRa(CO)-CrC6-akyl;
· aryl substituted by one or more -S(0)2-N(alkyl)2;
• 4 to 10 membered heterocycloalkyl unusbstituted or substituted by one or more 5 or 6 membered heterocycloalkyl;
• 5 to 10 membered heteroaryl unsubstituted or substituted by one or more4 to 10 membered heterocycloalkyl;
R4 is:
• H,
• hydroxy,
• Ci-C6-alkoxy unsubstituted or substituted by hydroxy or Ci-C6-alkoxy,
• -(CrC6-alkyl)n-(C3-C8-cycloalkyl),
• -(Ci-C6-alkyl)n-(C3-C8-cycloalkenyl),
• -(Ci-C6-alkyl)n-(4 to 10 membered heterocycloalkyl) unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, Ci-Ce-alkyl, or -C(0)-CrC6-alkyl;
R7 is 5 or 6 membered heteroaryl which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, alkyl, or -O-aryl, -S-aryl, -NH-aryl, -(Ci-C6-alkyl)n-aryl;
R8 is OH, -NH2, CrC6-alkoxy, -C(0)0-CrC6-alkyl;
or R2 and R3 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NH2, -NH(CrC6-alkyl), -N(CrC6-alkyl)2, CrC6-alkyl, CrC6-alkoxy, and Ci-C6-haloalkyl;
or R3 and R4 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NH2, -NH(CrC6-alkyl), -N(CrC6-alkyl)2, CrC6-alkyl, CrC6-alkoxy, and Ci-C6-haloalkyl;
or R4 and R5 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NH2, -NH(CrC6-alkyl), -N(CrC6-alkyl)2, CrC6-alkyl, CrC6-alkoxy, Ci-C6-haloalkyl;
or R5 and R6 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NH2, -NH(CrC6-alkyl), -N(CrC6-alkyl)2, CrC6-alkyl, CrC6-alkoxy and Ci-C6-haloalkyl;
n is 0 or 1.
Unless specifically stated otherwise herein, all of the following embodiments can be combined with one another:
In an embodiment A1 is O. In an embodiment A1 is S. In an embodiment A1 is CH2.
In an embodiment A2 is NH. In an embodiment A2 is N-Ci-C3-alkyl.
In an embodiment A3 is N. In an embodiment A3 is CR2.
In an embodiment A4 is N. In an embodiment A4 is CR3.
In an embodiment, A 3 is CR 2 and A 4 is CR 3. In an embodiment, A 3 is NH and A 4 is CR 3. In one embodiment A3 is CR2 and A4 is NH.
In an embodiment R1 is CI. In an embodiment R1 is N02. In an embodiment R1 is CN.
In an embodiment R2 is H. In an embodiment R2 is halo. In an embodiment R2 is hydroxy. In an embodiment R2 is Ci-C6-hydroxyalkyl. In an embodiment R2 is NH2. In an embodiment R2 is halo. In an embodiment R2 is hydroxy. In an embodiment R2 is Ci-C6-hydroxyalkyl.
In an embodiment R3 or R5 is H. In an embodiment R3 or R5 is hydroxy. In an embodiment R3 or R5 is halo. In an embodiment R3 or R5 is -Ci-C6-alkyl-Rf, wherein Rf is as defined herein. In an embodiment R3 or R5 is -Ci-C6-alkenyl-Rf, wherein Rf is as defined herein. In an embodiment R3 or R5 is -Ci-C6-alkoxy-Rc, wherein Rc is as defined herein. In an embodiment R3 or R5 is -NRaRb, wherein Ra and Rb are as defined herein. In an embodiment R3 or R5 is -NRa-(Ci-C6-alkyl)-Rd, wherein Ra and Rd are as defined herein. In an embodiment R3 or R5 is -NRa-S(0)2-(4 to 10 membered heterocycloalkyl), wherein Ra is as defined herein. In an embodiment R3 or R5 is -NRa-(C3-C8-cycloalkyl), wherein Ra is as defined herein and the cycloalkyl is unsubstituted or substituted by Ci-C6-alkyl. In an embodiment R3 or R5 is -NRa-aryl, wherein Ra is as defined herein and the aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, hydroxy, -NH2, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-haloalkyl,
Ci-C6-hydroxyalkyl, Ci-C6-haloalkoxy and C3-C8-cycloalkyl.
In an embodiment R3 or R5 is -NRa-(4 to 10 membered heterocycloalkyl), wherein Ra is as defined herein and the heterocycloalkyl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C6-alkyl, Ci-C6-hydroxyalkyl, or -CO-alkyl.
In an embodiment R3 or R5 is -NRa-(5 or 6 membered heteroaryl), wherein Ra is as defined herein and the heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, -NRaRb and Ci-C6-alkyl.
In an embodiment R3 or R5 is -NRa(CO)-Ci-C6-alkyl wherein Ra is as defined herein.
In an embodiment R3 or R5 is -NRa(CO)-(aryl).
In an embodiment R3 or R5 is -NRa(CO)-(5 or 6 membered heteroaryl).
In an embodiment R3 or R5 is -NRa(CO)0-Ci-C6-alkyl wherein Ra is as defined herein. In an embodiment R3 or R5 is -S-(alkyl)n-Rh and Rh is as defined herein.
In an embodiment R3 or R5 is -S(0)2-aryl, which aryl is unsubstituted or substituted by one or more halo.
In an embodiment R3 or R5 is -C(0)-Re and Re is as defined herein.
In an embodiment R3 or R5 is -C(0)NRa-(CrC6-alkyl)n-Rs, wherein Ra and Rs are as defined herein.
In an embodiment R3 or R5 is -0-C3-C8-cycloalkyl, which cycloalkyl is unsubstituted or substituted by halo or hydroxy, Ci-C6-alkyl, Ci-C6-alkoxy, which alkoxy is unsubstituted or substituted by halo, Ci-C6-alkoxyaryl, Ci-C6-haloalkyl, aryl, Ci-C6-akyl-aryl, 5 or 6 membered heteroaryl, CrC6-haloalkoxy, CrC6-hydroxyalkyl, NRaRb, -(CrCs-alkylHQ-CValkoxy).
In an embodiment R3 or R5 is -O-aryl, which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-haloalkyl, Ci-C6-haloalkoxy, Ci-C6-hydroxyalkyl, -S-Ci-C6-akyl,
-Ci-C6-alkyl-C3-C8-cycloalkyl, Ci-C6-alkyl-4 to 10 membered heterocycloalkyl, 5 or 6 membered heteroaryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C6-alkyl, -(Ci-C6-alkyl)-(Ci-C6-alkoxy), Ci-C6-haloalkoxy, Ci-C6-alkylene bridge.
In an embodiment R3 or R5 is -0-(4 to 10 membered heterocycloalkyl), which
heterocycloalkyl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, Ci-C6-hydroxyalkyl and -C(0)-Ci-C6-alkyl.
In an embodiment R3 or R5 is -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, or -NRa(CO)-Ci-C6-akyl and Ra is as defined herein.
In an embodiment R3 or R5 is C3-C8-cycloalkyl, which cycloalkyl may be fused to a phenyl. In an embodiment R3 or R5 is aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -C(0)OH, Ci-C6-hydroxyalkyl, Ci-C6-alkoxy, -S(0)2-NH(alkyl) and -S(0)2-N(alkyl)2.
In an embodiment R3 or R5 is 4 to 10 membered heterocycloalkyl unusbstituted or substituted by one or more 5 or 6 membered heterocycloalkyl.
In an embodiment R3 or R5 is 5 to 10 membered heteroaryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of hydroxy, -NRaRb, Ci-C6-alkyl, Ci-C6-hydroxyalkyl, and 4 to 10 membered heterocycloalkyl.
In an embodiment R3 or R5 is -NRa-S(0)2-(4 to 10 membered heterocycloalkyl), for example:

In an embodiment R3 or R5 is -S(0)2-aryl, which aryl is unsubstituted or substituted by more halo, for example:
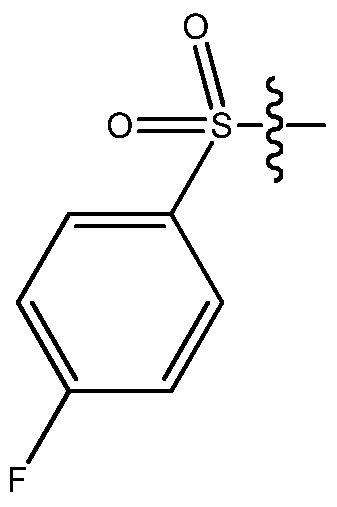
In an embodiment R3 or R5 is C3-C8-cycloalkyl which cycloalkyl may be fused to a phenyl, or which le:

In an embodiment R3 or R5 is NRa-(C C6-alkyl)-Rd , wherein Rd is C3-C8-cycloalkyl, for exampl
R3 or R5 is Ci-C6-alkenyl-Rf, wherein Rf is C3-C8-cycloalkyl, for example:

In an embodiment R3 or R5 is aryl, for example phenyl unsubstituted or substituted by more halo, hydroxy, -C(0)OH, CrC6-hydroxyalkyl, CrC6-alkoxy, -S(0)2-NH(alkyl) and
-S(0)2-N

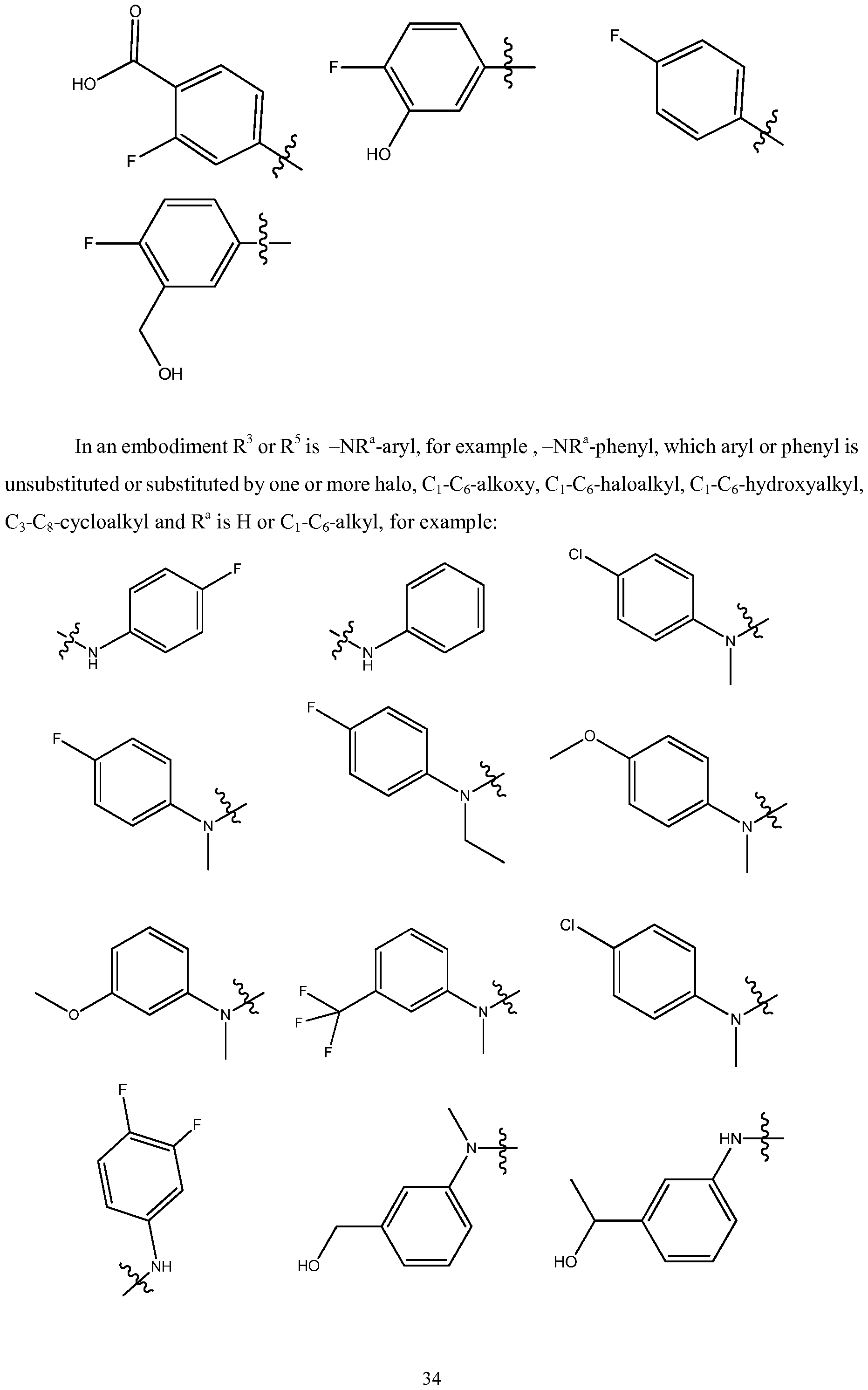

In an embodiment R3 or R5 is -O-aryl, for example -O-phenyl, which aryl or phenyl is unsubstituted or substituted by one or more: halo, Ci-C6-alkyl, -S-Ci-C6-akyl, Ci-C6-haloalkyl, Ci-C6-alkoxy, Ci-C6-alkoxy-C3-Cg-cycloalkyl, Ci-C6-haloalkoxy, Ci-C6-hydroxyalkyl,
Ci-C6-alkyl-Ci-C6-alkoxy, Ci-C6-alkyl-(5 or 6 membered heterocycloalkyl), 5 or 6 membered heterocycloalkyl which 5 or 6 membered heteroaryl is unsubstituted or substituted by Ci-C6-alkyl, Ci-C6-haloalkoxy, Ci-C6-alkylene bridge, naphthalene partially hydrogenated which is unsubstituted or substituted by halo for example:




In an embodiment R3 or R5 is -NRa-(5 or 6 membered heteroaryl), which heteroaryl is unsubstituted or


In an embodiment R3 or R5 is -NRa-(C3-Cg-cycloalkyl), which cycloalkyl is unsubstituted or substituted by Ci-C6-alkyl or a Ci-C3-alkylene bridge and Ra is H or Ci-C6-alkyl, for example:

ln an embodiment R3 or R5 is halo, for example CI, F or Br.
In an embodiment R3 or R5 is -NRaRb, wherein Ra and Rb are independently selected from H and Ci-Ce-alkyl, for example -NH2, -NHMe or -N(Me)2.
In an embodiment R3 or R5 is is hydroxy.
In an embodiment R3 or R5 is -NRa(CO)0-C C6-alkyl, wherein Ra is H or CrC6-alkyl, for example:

In an embodiment R3 or R5 is -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, Ci-C6-alkyl, Ci-C6-hydroxyalkyl, or -NRaC(0) Ci-C6-alkyl, for example:


In an embodiment R3 or R5 is Ci-C6-alkyl-Rf and Rf is aryl. In one embodiment, Rf is unsubstituted phenyl. In one embodiment, Rf is phenyl substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-alkoxy, Ci-C6-haloalkyl, and Ci-C6-hydroxyalkyl, for example:

In an embodiment R3 or R5 is -Ci-C6-alkoxy-Rc, wherein Rc is hydroxy, halo, Ci-C6-alkoxy, Ci-C6-alkenyl, phenyl unsubstituted or substituted by halo, 4 to 6 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C6-alkyl, 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C6-alkyl, or C3-C8-cycloalkyl unsubstituted or substituted by halo or
Ci-C6-hydroxyalkyl, Ci-C6-alkyl, for example:



In an embodiment R3 or R5 is Ci-C6-alkyl-Rf and Rf is 5 or 6 membered heterocycloalkyl, for example:
In an embodiment R3 or R5 is -0-C3-C6-cycloalkyl, which cycloalkyl is unsubstituted or substituted by halo, hydroxy, Ci-C6-alkyl, phenyl, Ci-C6-alkoxy, for example:



In an embodiment R3 or R5 is -0-(5 or 6 membered heterocycloalkyl), which
heterocycloalkyl is unsubstituted or substituted by Ci-C6-alkyl or -C(0)Ci-C6-alkyl, for example:

In an embodiment R3 or R5 is -NRa-CrC6-alkyl-Rd, wherein Rd is:
C3-Cg-cycloalkyl, or phenyl unsubstituted or substituted by halo, for example:


In an embodiment R3 or R5 is 5 to 10 membered heteroaryl unsubstituted or substituted by -hydroxy, N -C6-alkyl or Ci-C6-hydroxyalkyl, for example:

In an embodiment R3 or R5 is 5 or 6 membered heterocycloalkyl unusbstituted or substituted by halo, Ci- -alkyl, -C(0)-C3-C8-cycloalkyl, oxo, 5 or 6 membered heterocycloalkyl, for example:


In an embodiment R3 or R5 is -C(0)NRa-(CrC6-alkyl)n-Rs. In an embodiment R3 or R5 is -C(0)NRa-(Ci-C6-alkyl)-Rs and Rs is C3-C6-cycloalkyl or phenyl, which phenyl is unsubstituted or substituted by halo or R3 or R5 is -C(0)NRa-Ci-C6-alkoxy, for example:

In an embodiment R3 or R5 is -S-(alkyl)n-Rh. In one embodiment, R3 or R5 is -S-phenyl and said phenyl is unsubstituted or substituted by halo, for example:

In an embodiment R3 or R5 is -C(0)-Re and Re is phenyl which phenyl is unsubstituted or substituted by halo, for example:

In an embodiment R3 or R5 is -NRa-S(0)2-(4 to 6 membered heterocycloalkyl), for example:

In an embodiment R is H. In an embodiment R is halo. In an embodiment R is hydroxy. In an embodiment R4 is Ci-C6-alkyl. In an embodiment R4 is Ci-C6-haloalkyl. In an embodiment R4 is Ci-C6-hydroxalkyl. In an embodiment R4 is CN. In an embodiment R4 is Ci-C6-alkoxy unsubstituted or substituted by hydroxy or Ci-C6-alkoxy. In an embodiment R4 is
-(Ci-C6-alkyl)n-(C3-C8-cycloalkyl). In an embodiment R4 is -(Ci-C6-alkyl)n-(C3-C8-cycloalkenyl). In an embodiment R4 is -(Ci-C6-alkyl)n-(4 to 10 membered heterocycloalkyl) unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-alkyl, or -C(0)-CrC6-alkyl.
In an embodiment R4 is -NRaRb and Ra and Rb are as defined herein, for example:

In an embodiment R is Ci-C6-alkoxy unsubstituted or substituted by hydroxy, Ci-C6-alkoxy or -NRaRb, wh

embodim -C6-cycloalkyl or C3-C6-cycloalkenyl, for example

In an embodiment R is 4 to 10 membered heterocycloalkyl unsubstituted or substituted by halo, hydroxy, cyano, oxo, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-hydroxyalkyl, -C(0)OH,
-C(0)-CrC6-alkyl, -C(0)-C3-C8-cycloalkyl, -C(0)-phenyl, 4 to 10 membered heterocycloalkyl, -C(0)(5 or 6 membered heteroaryl), -C(0)(4 to 10 membered heterocycloalkyl), Ci-C4-alkylene bridge, for example:




In an embodiment of the present invention R7 is 5 or 6 membered heteroaryl which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-a -aryl, -S-aryl, -NH-aryl, -(Ci-C6-alkyl)n-aryl, for example.

In an embodiment of the present invention R8 is OH. In an embodiment of the present invention R8 is -NH2. In an embodiment of the present invention R8 is Ci-C6-alkoxy. In an embodiment of the present invention R8 is -C(0)0-Ci-C6-alkyl.
In an embodiment of the present invention or R6 and R7 together with the carbon atoms to which they are attached form a 5 membered ring selected from a cycloalkyl or heterocycloalkyl having 5 ring members, so that the compounds of Formula (I) are as following:
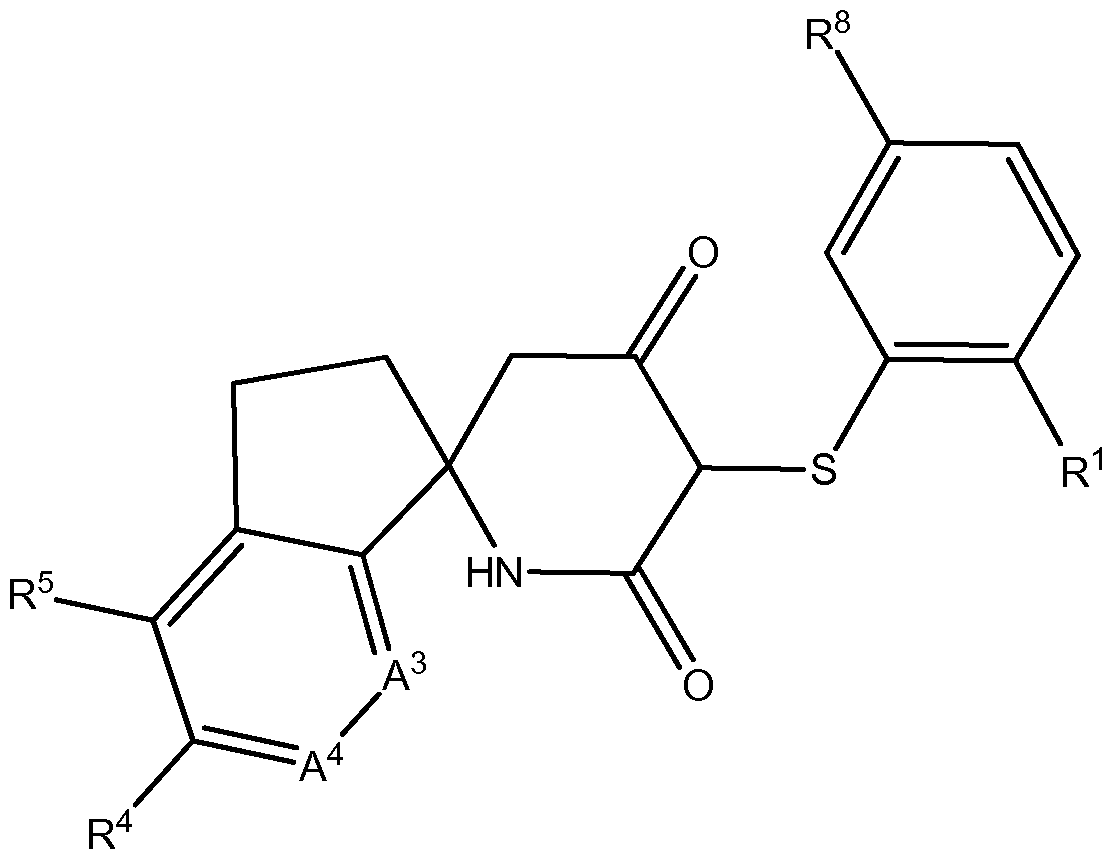
In an embodiment of the present invention R2 and R3 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -NH2, -NH(CrC6-alkyl), -N(CrC6-alkyl)2, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl.
In an embodiment of the present invention R3 and R4 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -NH2, -NH(CrC6-alkyl), -N(CrC6-alkyl)2, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl.
In an embodiment of the present invention R5 and R6 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, hydroxy, -NH2, -NH(CrC6-alkyl), -N(CrC6-alkyl)2, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl.
In an embodiment of the present invention n is 0. In an embodiment of the present invention n is 1.
In an embodiment R9 is H. In an embodiment R9 is Ci-C6-alkyl. In an embodiment R9 is C3-C8-cycloalkyl. In an embodiment R9 is halo. In an embodiment R9 is -O-aryl, for example -O-phenyl. In an embodiment R9 is -S-aryl, for example -S-phenyl. In an embodiment R9 is -NH-aryl, for example -NH-phenyl. In an embodiment R9 is -(Ci-C6-alkyl)n-aryl, for example
-(CrC6-alkyl)n-phenyl.
In an embodiment R10 is H. In an embodiment R10 is Ci-C6-alkyl. In an embodiment R10 is C3-C8-cycloalkyl. In an embodiment R10 is halo. In an embodiment R10 is -O-aryl, for example -O-phenyl. In an embodiment R10 is -S-aryl, for example -S-phenyl. In an embodiment R10 is -NH-aryl, for example -NH-phenyl. In an embodiment R10 is -(Ci-C6-alkyl)n-aryl, for example -(CrC6-alkyl)n-phenyl.
In one embodiment A3 is NH. In one embodiment A3 is CR2, wherein R2 is selected from the group consisting of H, halo, hydroxy, Ci-C6-hydroxyalkyl, and NH. In one embodiment, R9 and R10 are H. In one embodiment R1 is CI. In one embodiment R3 is NH-phenyl or NH-pyridinyl, which phenyl or pyridinyl is substituted by halo. In one embodiment R4, R5, R6 and R8 are H.
In an embodiment A1 is O, A2 is NH, R1 is CI, A3 is NH, A4 is CR3 and R3 is NH-phenyl or NH-pyridinyl, which phenyl or pyridinyl is substituted by halo, R4, R5 and R6 are H, R7 is thiophenyl.
In an embodiment A1 is S, A2 is NH, R1 is halo, A3 is NH, A4 is CR3 and R3 is NH-phenyl or NH-pyridinyl, which phenyl or pyridinyl is substituted by halo, R4,R5 and R6 are H, R7 is thiophenyl.
In an embodiment, the compound of Formula (I) is selected from the compounds of the following compounds and stereoisomers, tautomers, and pharmaceutically acceptable salts thereof. These compounds can also be prepared as a racemate, mixture of diastereisomer or as single stereoisomers, all of which forms fall within the scope of the invention:
1- [4-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]phenyl]piperidine-4-carbonitr ile;
2- [[6-(6-bromo-2-pyridyl)-2,4-dioxo-6-(3-thienyl)-3-piperidyl]sulfanyl]benzonitrile;
3- (2-chloro-5-hydroxy-phenyl)sulfanyl-6-[4-(l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione
3-(2-chlorophenoxy)-6-(4-morpholinophenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[4-(l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(2-cyclopropylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(3,4-difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-l-methyl-6-(3-tetrahydropyran-4-yloxyphenyl)-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl)sulfanyl- 1 -methyl-6- [3 -(tetrahydropyran-4-ylamino)phenyl] -6-(3-thienyl)piperid ine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(lH-indol-4-yl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-fluorophenyl)-l-methyl-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-hydroxy-4-morpholino-phenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-hydroxyphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-naphthyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-fluoro-4-morpholino-phenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-hydroxyphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-tetrahydropyran-4-yloxyphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)-6-(4-thiomorpholinophenyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 3-thienyl)-6-[6-(2,2,2-trifluoro-l-methyl-ethoxy)-2-pyridyl]piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 3-thienyl)-6-[6-(2,2,2-trifluoroethoxy)-2-pyridyl]piperidine-2,4-dion e;
3-(2-chlorophenyl)sulfanyl-6 3-thienyl)-6-[6-(4,4,4-trifluorobutoxy)-2-pyridyl]piperidine-2,4-dion e;
3- (2-chlorophenyl)sulfanyl-6 3 -thienyl)-6-[6- [3 -(trifluoromethyl)phenoxy] -2-pyridyl]piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6 3 -thienyl)-6-[6- [4-(trifluoromethoxy)phenoxy] -2-pyridyl]piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 3-thienyl)-6-[6-[4-(trifluoromethyl)cyclohexoxy]-2-pyridyl]piperidi ne -2,4-dione;
3- (2-chlorophenyl)sulfanyl-6 3- thienyl)-6-[6-[4-(trifluoromethyl)phenoxy]-2-pyridyl]piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6 4- cyclohexylphenyl)-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-cyclopropylphenyl)-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-hydroxyphenyl)-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-morpholino-3-phenyl-phenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-morpholinophenyl)-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-morpholinophenyl)-6-(5-phenyl-3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-morpholinophenyl)-6-(6-tetrahydropyran-4-yloxy-2-pyridyl)piperi dine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-morpholinophenyl)-6-thiazol-4-yl-piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4-piperazin-l-ylphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 4- pyrrolidin-l-ylphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 5- chloro-3-thienyl)-6-[6-(4-fluorophenoxy)-2-pyridyl]piperidine-2,4
-dione;
3-(2-chlorophenyl)sulfanyl-6 5- methyl-3-thienyl)-6-(4-morpholinophenyl)piperidine -2,4-dione; 3-(2-chlorophenyl)sulfanyl-6 6- chroman-4-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6 6-ethoxy-2-pyridyl)-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 6-indan-5-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 6-isobutoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 6-isopentyloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 6-isopropoxy-2-pyridyl)-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6 6-isopropoxy-5-morpholino-2-pyridyl)-6-(3-thienyl)piperidine-2,4-d ione;
3-(2-chlorophenyl)sulfanyl-6-(6-morpholino-3-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-pent-2-enoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-phenoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-phenyl-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-pyrimidin-5-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6 etrahydrofuran-3-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dio ne;
3-(2-chlorophenyl)sulfanyl-6-(6-tetralin-l-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[3-(4-fluoroanilino)phenyl]-l-methyl-6-(3-thienyl)piperidine-2,4-dio ne;
3-(2-chlorophenyl)sulfanyl-6-[3-(4-fluoroanilino)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[3-(4-fluoroanilino)phenyl]-6-phenyl-piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[3-(4-fluoro-N-methyl-anilino)phenyl]-6-phenyl-piperidine-2,4-dione
3-(2-chlorophenyl)sulfanyl-6-[3-(4-fluorophenoxy)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[3-(cyclohexylamino)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyl)sulfanyl-6-[3 -(tetrahydropyran-4-ylamino)phenyl] -6-(3 -thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[3-[(6-fluoro-5-methyl-3-pyridyl)amino]phenyl]-6-(3-thienyl)piperidi ne-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[4-(l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[4-(2,2-dimethylmo^holin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[4-(2,6-dimethylmo^holin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[4-(2-ethylmorpholin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[4-(2-hydroxyethoxy)phenyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[4-(2-methoxyethoxy)phenyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[4-(2-methylmo^holin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dion e;
3-(2-chlorophenyl)sulfanyl-6-[4-(2-oxa-5-azabicyclo[2.2.1]heptan-5-yl)phenyl]-6-(3-thienyl)piperi dine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[4-(2-oxa-6-azaspiro[3.3]heptan-6-yl)phenyl]-6-(3-thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[4-(2-oxa-7-azaspiro[3.5]nonan-7-yl)phenyl]-6-(3-thienyl)piperidine- 2,4-dione;
3 -(2-chlorophenyF )sulfanyl■6-[4 3,3-difluoroazetidin-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[4 3.3- difluoropyrrolidin-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3-fluoroazetidin-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3-fluoropyrrolidin-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyF )sulfanyl ■6-[4 3 -hydroxypropoxy)phenyl] -6-(3 -thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3-methoxypropoxy)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3 - methoxypyrrolidin- 1 -yl)phenyl] -6-(3 -thienyl)piperidine-2,4-dio ne;
3 -(2-chlorophenyF )sulfanyl ■6-[4 4.4- difluoro-l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[4 4- fluoro-l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 4-methoxy-l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 8-oxa-3-azabicyclo[3.2.1]octan-3-yl)phenyl]-6-(3-thienyl)piperidi ne-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 cyclohexen-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 dimethylamino)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 tetrahydropyran-4-ylamino)phenyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[5 4-fluoroanilino)-2-hydroxy-phenyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[5 (4-fluorophenyl)methyl]-3-thienyl]-6-(4-morpholinophenyl)piperi dine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 l,2,3,4-tetrahydroquinolin-8-yloxy)-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 l-cyclohexylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyF )sulfanyl ■6-[6 l-cyclopropylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[6 l-cyclopropylethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 lH-indazol-4-yloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyF )sulfanyl ■6-[6 2,2-difluoroethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl■6-[6 2,2-dimethylchroman-4-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine
-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2.2- dimethylpropoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[6 2.3- difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[6 2.4- difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclobutylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclohexylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclohexylethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-d ione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclopentylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclopropyl- 1 -methyl-ethoxy)-2-pyridyl] -6-(3 -thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclopropylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclopropylethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-cyclopropylpropoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dio ne;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-ethoxy- 1 -methyl-ethoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-2,4
-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-ethoxyethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-methoxy- 1 -methyl-ethoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-2
,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-methoxyphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-methylbutoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-morpholino-4-pyridyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[6 2-pyridyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 3,4-difluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3,4-difluorophenoxy)-2-pyridyl]-6-(3 hienyl)piperidine-2,4-di e;
3-(2-chlorophenyl)sulfanyl-6-[6-(3,4-difluorophenoxy)-2-pyridyl]-6-(4-morpholinophenyl)piperidi ne-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3,4-difluorophenoxy)-2-pyridyl]-6-[4-(l-piperidyl)phenyl]piperid ine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3,5-difluorophenoxy)-2-pyridyl]-6-(3 hienyl)piperidine-2,4-dion e;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-fluoro-4-methoxy-phenoxy)-2-pyridyl]-6-(3-thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-fluorophenoxy)-2-pyridyl]-6-(3 hienyl)piperidine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-(3-hydroxy-3-methyl-butoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-hydroxycyclopentoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-methoxy-3-methyl-butoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2 ,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-methoxy-N-methyl-anilino)-2-pyridyl]-6-(3-thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-methoxyphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-methoxypropoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(3-pyridyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyl)sulfanyl-6-[6-(3 -tetrahydropyran-4-ylazetidin- 1 -yl)-2-pyridyl] -6-(3 -thienyl)piper idine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4,4-difluorocyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-cyclopropyl-2-fluoro-anilino)-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-2-isopropyl-phenoxy)-2-pyridyl]-6-(3-thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-2-methoxy-phenoxy)-2-pyridyl]-6-(3-thienyl)piperidine- 2,4-dione;
(6S)-3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-2-methoxy-phenoxy)-2-pyridyl]-6-(3-thienyl)piperi dine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-2-tetrahydropyran-4-yl-phenoxy)-2-pyridyl]-6-(3-thienyl )piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl■6-[6 4-fluoro-3 -methoxy-phenyl)-2-pyridyl] -6-(3 -thienyl)piperidine-2,
4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluoro-3 -methyl-phenoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-2,
4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluoroanilino)-2-pyridyl] - 1 -methyl-6-(3 -thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 cyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluoroanilino)-2-pyridyl]-6-(4-morpholinophenyl)piperidine-2,4
-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluoroanilino)-5-morpholino-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorobenzoyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluoro-N-methyl-anilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4
-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenoxy)-2-pyridyl]-l-methyl-6-(3-thienyl)piperidine-2,4
-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenoxy)-2-pyridyl]-6-(lH-pyrazol-3-yl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenoxy)-2-pyridyl]-6-(2-hydroxyphenyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenoxy)-2-pyridyl]-6-(4-morpholinophenyl)piperidine-2
,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenoxy)-5-morpholino-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-fluorophenyl)sulfanyl-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-hydroxy-4-methyl-pentoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-iodophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 4-methoxycyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyl)sulfanyl ■6-[6
2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 4-methoxyphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 4-methylsulfanylphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6 4-pyridyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 4- pyridylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 5- fluorotetralin-l-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6 5-isoquinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 5- quinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 6- fluorotetralin-l-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6 6- quinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 7- fluorotetralin-l-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6 8- fluorochroman-4-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6 8-hydroxy-3 ,4-dihydro-2H-quinolin- 1 -yl)-2-pyridyl] -6-(3 -thienyl) piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 8-isoquinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 8-quinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclobutoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclobutylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cycloheptoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexoxy)-2-pyridyl]-6-(4-morpholinophenyl)piperidine-2,4-d ione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexoxy)-2-pyridyl]-6-[4-(l-piperidyl)phenyl]piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 cyclopentoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 cyclopentylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 cyclopentylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyl)sulfanyl -6-1 [6 -(cyclopropylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-:
e;
3 -(2-chlorophenyl sulfanyl-6-[6 dimethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3 -(2-chlorophenyl sulfanyl-6-[6 N-ethyl-4-fluoro-anilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-d ione;
3 -(2-chlorophenyl sulfanyl-6-[6 oxetan-3-ylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydrofuran-2-ylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydrofuran-3-ylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4
-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydropyran-4-ylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2, 4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydropyran-4-ylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydropyran-4-ylmethyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,
4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 thiazol-2-ylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 ,5-dimethylpyrazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidi ne -2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methyl- 1 ,2,4-triazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperi dine -2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylcyclopropyl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylimidazol-2-yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidine
-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylimidazol-2-yl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidi ne -2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylpyrazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 (2,4-difluorophenyl)methyl] -2-pyridyl] -6-(3 -thienyl)piperidine -2,
4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 (2,5-dimethylpyrazol-3-yl)amino]-2-pyridyl]-6-(3-thienyl)piperidi ne -2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 (2-methylcyclopropyl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 (2 -methylpyrazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidine-
2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(3,3-difluorocyclobutyl)methoxy]-2-pyridyl]-6-(3-thienyl)piperidi ne-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[(3,4-difluorophenyl)methyl]-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[(3,5-difluorophenyl)methyl]-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(3-ethyloxetan-3-yl)methoxy]-2-pyridyl]-6-(3 hienyl)piperidine- 2,4-dione;
5- (2-chlorophenyl)sulfanyl-4-hydroxy-2-[6-(4-methoxycyclohexoxy)-2-pyridyl]-2-(3-thienyl)-l,3-d ihydropyridin-6-one;
3-(2-chlorophenyl)sulfanyl-6-[6-[(3-fluoro-5-methoxy-phenyl)methyl]-2-pyridyl]-6-(3-thienyl)pipe ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(3-fluorophenyl)methyl]-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6-[(4-fluoro-3-methoxy-phenyl)methyl]-2-pyridyl]-6-(3-thienyl)pipe ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(4-fluorophenyl)methoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(4-fluorophenyl)methyl]-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6-[(4-fluorophenyl)methylamino]-2-pyridyl]-6-(3-thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(4-methylthiazol-2-yl)amino]-2-pyridyl]-6-(3-thienyl)piperidine-2 ,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(5-fluoro-3-pyridyl)oxy]-2-pyridyl]-6-(3 hienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6-[(5-fluoro-8-quinolyl)oxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(5-methyl-lH-imidazol-2-yl)amino]-2-pyridyl]-6-(3 hienyl)piperi dine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(5-methylthiazol-2-yl)amino]-2-pyridyl]-6-(3-thienyl)piperidine-2 ,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(5-oxotetrahydrofuran-2-yl)methoxy]-2-pyridyl]-6-(3-thienyl)pip eridine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(6-fluoro-3-pyridyl)amino]-2-pyridyl]-6-(3-thienyl)piperidine-2,4 -dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(6-fluoro-5-methyl-3-pyridyl)amino]-2-pyridyl]-6-(3 hienyl)pi^ ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[[3-(hydroxymethyl)phenyl]methyl]-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[[4-(hydroxymethyl)cyclohexyl]methoxy]-2-pyridyl]-6-(3-thienyl) piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l-(3,4-difluorophenyl)ethoxy]-2-pyridyl]-6-(3 hienyl)piperidine 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l-(3-fluorophenyl)ethoxy]-2-pyridyl]-6-(3 hienyl)piperidine-2,4- dione;
3-(2-chlorophenoxy)-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l-(4-fluorophenyl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidine -2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l-(4-fluorophenyl)ethylamino]-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[l-(4-fluorophenyl)propoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l-(4-fluorophenyl)propylamino]-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(lH-pyrazol-4-yl)phenoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2 ,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(l-methylcyclopropyl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(2,2-difluorocyclopropyl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperi dine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(2,2-dimethyl-l,3-dioxolan-4-yl)ethoxy]-2-pyridyl]-6-(3-thienyl )piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(2-oxopyrrolidin-l-yl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(3-methyltriazol-4-yl)phenoxy]-2-pyridyl]-6-(3-thienyl)piperidi ne -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(4-fluorophenyl)ethyl]-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(cyclopropylmethoxy)-4-fluoro-phenoxy]-2-pyridyl]-6-(3-thien yl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(cyclopropylmethyl)-4-fluoro-phenoxy]-2-pyridyl]-6-(3-thienyl )piperidine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[2-(methoxymethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(oxetan-3-yl)ethoxy]-2-pyridyl]-6-(3 hienyl)piperidine-2,4-dio ne;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(l-hydroxyethyl)anilino]-2-pyridyl]-6-(3 hienyl)piperidine-^ -dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(difluoromethyl)-4-fluoro-phenoxy]-2-pyridyl]-6-(3-thienyl)pip eridine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[3-(difluoromethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(hydroxymethyl)anilino]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(hydroxymethyl)-N-methyl-anilino]-2-pyridyl]-6-(3-thienyl)pip eridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-fluoro-5-(hydroxymethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)pipe ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[4-fluoro-3-(hydroxymethyl)anilino]-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[4-fluoro-3-(trifluoromethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)pip eridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[6-(hydroxymethyl)indolin-l-yl]-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyl)sulfanyl-6-[6-[N-methyl-3 -(trifluoromethyl)anilino] -2-pyridyl] -6-(3 -thienyl)pipe ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-phenyl-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-phenyl-6-thiazol-4-yl-piperidine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-thiazol-4-yl-6-(3-thienyl)piperidine-2,4-dione;
4- [3-[5-(2-chlorophenyl)sulfanyl-2-(4-morpholinophenyl)-4,6-dioxo-2-piperidyl]phenyl]-N,N-dime thyl-benzenesulfonamide;
4-[3-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]phenyl]-N,N-dimethyl-benzen esulfonamide;
4-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-2-pyridyl]-N,N-dimethyl-ben zenesulfonamide ;
3- (2-chlorophenyl)sulfanyl-6-(3 hienyl)-6-[6-[3-(trifluoromethyl)phenoxy]-2-pyridyl]piperidine-2,
4- dione;
6-(3-aminophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-anilinophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-bromo-4-mo^holino-phenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-bromophenyl)-3-(2-chlorophenyl)sulfanyl-l-methyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-bromophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(5-bromo-6-mo^holino-3-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-benzyl-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-benzyloxy-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-bromo-2-pyridyl)-3-(2-chloro-5-hydroxy-phenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-bromo-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-bromo-5-mo^holino-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[3-chloro-5-(4-fluoroanilino)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dion e;
6-[4-(l,3,3a,4,6,6a-hexahydrofuro[3,4-c]pyrrol-5-yl)phenyl^
yl)piperidine-2,4-dione;
6-[4-(2-azaspiro[3.3]heptan-2-yl)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-di one;
6-[4-(3-azabicyclo[2.1. l]hexan-3-yl)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[4-(4-acetylpiperazin-l-yl)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-N-(cyclopropylmethyl)pyridine-
2-carboxamide;
6-[6-(2-amino-5-methyl-imidazol-l-yl)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidi ne-2,4-dione;
6-[6-(2-bromophenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[6-(2-chloro-3,4-difluoro-anilino)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)piperidm^
2,4-dione;
6-[6-(2-chloro-4-fluoro-anilino)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)piperidine-2,4- dione;
6-[6-(2-chloro-4-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[6-(2-tert-butoxyethoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione
6-[6-(3-bromo-4-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[6-(3-chloro-4-fluoro-anilino)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)piperidine- dione;
6-[6-(3-chloro-4-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[6-(3-chlorophenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)piperidine-2,4-dione;
6-[6-(4-bromo-2-chloro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,
4-dione;
6-[6-(4-bromo-2-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,
4- dione;
6-[6-(4-chloro-N-methyl-anilino)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4 -dione;
6-[6-(4-chlorophenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[6-(7-bromotetralin-l-yl)oxy-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-d ione;
6-[6-[(2-chloro-6-fluoro-3-pyridyl)oxy]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidi ne-2,4-dione;
6-[6-[(4-chloro-3-fluoro-phenyl)methyl]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperid ine-2,4-dione;
6-[6-[[l-(3-chloro-4-fluoro-phenyl)-2-hydroxy-ethyl]amino]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl- 6-(3-thienyl)piperidine-2,4-dione;
6-[6-[l-(3-chloro-4-fluoro-phenyl)propylamino]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl )piperidine-2,4-dione;
6-[6-[l-(4-chlorophenyl)ethoxy]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4- dione;
N-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-2-pyridyl]azetidine-l-sulfon amide tert-butyl;
5- (2-chlorophenyl)sulfanyl-4-hydroxy-2- [4-( 1 -piperidyl)phenyl] -2-(3 -thienyl)- 1 ,3 -dihydropyridin-6 -one; and
N-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-2-pyridyl]carbamate.
In one embodiment, the small molecule LDHA inhibitor is 5-(2-chlorophenyl)sulfanyl-4-hydroxy-2-(4-morpholinophenyl)-2-(3-thienyl)-l,3-dihydropyridin-6- one or a salt thereof.
In one embodiment, the pH value of the medium is maintained at about 7.0, 7.1, 7.2 or 7.4.
In one embodiment, the temperature of the medium is maintained at 36.0, 36.5, 37.0, or 37.5 °C during the initial 2, 3, 4, 5, 6 or 7 days.
In one embodiment, the temperature of the medium is lowered to about 36, 35, 34, 33, 32, or 31 °C at the end of day 2, 3, 4, 5, 6, or 7.
In one embodiment, wherein the LDHA inhibitor is introduced into the medium at day 0, 1 ,
2, 3, 4, 5, 6, or 7, or a combination thereof.
In one embodiment, wherein the concentration of the LDHA inhibitor in the medium is about 1 μΜ, 5 μΜ, 10 μΜ, 15 μΜ, 20 μΜ, 25 μΜ, 30 μΜ, 35 μΜ, 40 μΜ, 45 μΜ, 50 μΜ, 60 μΜ, 70 μΜ, 80 μΜ, 90 μΜ θΓ 100 μΜ.
In one embodiment, the cultured cells produce a polypeptide.
In one embodiment, the polypeptide is an antibody, or a biologically functional fragment of an antibody.
In one embodiment, the cultured cell is a mammalian cell.
In one embodiment, the mammalian cell is a Chinese Hamster Ovary (CHO) cell.
In one embodiment, the present invention relates to a medium comprising a small molecule
LDHA inhibitor.
In one embodiment, the present invention relates to a medium, wherein said LDHA inhibitor is
5-(2-chlorophenyl)sulfanyl-4-hydroxy-2-(4-morpholinophenyl)-2-(3-thienyl)-l,3-dihydropyridin-6- one or a salt thereof.
BIOLOGICAL EVALUATION
Within the scope of the present invention the inventors have identified LDHA inhibitors. The relative efficacies of Formula (I) compounds as inhibitors of an enzyme activity (or other biological activity) can be established by determining the concentrations at which each compound inhibits the activity to a predefined extent and then comparing the results. Typically, the preferred determination is the concentration that inhibits 50% of the activity in a biochemical assay, i.e., the 50% inhibitory concentration or "IC50". Determination of IC50 values can be accomplished using conventional techniques known in the art. In general, an IC50 can be determined by measuring the activity of a given enzyme in the presence of a range of concentrations of the inhibitor under study. The experimentally obtained values of enzyme activity then are plotted against the inhibitor concentrations used. The concentration of the inhibitor that shows 50%> enzyme activity (as compared to the activity in the absence of any inhibitor) is taken as the IC50 value. Analogously, other inhibitory concentrations can be defined through appropriate determinations of activity. For example, in some settings it can be desirable to establish a 90%> inhibitory concentration, i.e., IC90, etc.
Accordingly, a "selective LDHA inhibitor" can be understood to refer to a compound that exhibits a 50% inhibitory concentration (IC50) with respect to LDHA that is at least at least 10-fold lower than the IC50 value with respect to any or all of the other LDHA family members.
Determination of the activity of LDHA kinase activity of Formula (I) compounds is possible by a number of direct and indirect detection methods. The range of IC50 values for inhibition of LDHA was less than 1 nM (nanomolar) to about 10 μΜ (micromolar). Certain exemplary compounds of the invention had LDHA inhibitory IC50 values less than 10 nM. Certain Formula (I) compounds may have antiproliferative properties and may be useful to treat disorders such as cancer. The Formula (I) compounds may inhibit LDHA in mammals and may be useful for treating human cancer patients.
The Example section of this patent application herein shows Formula (I) compounds that were made, characterized, and tested for inhibition of LDHA and selectivity according to the methods of this invention, and have the corresponding structures and names (ChemBioDraw Ultra, Version 11.0, CambridgeSoft Corp., Cambridge MA). PREPARATION OF FORMULA (I) COMPOUNDS
Thecompounds of Formula (I) may be synthesized by synthetic routes that include processes analogous to those well-known in the chemical arts, particularly in light of the description contained herein, and those for other heterocycles described in: Comprehensive Heterocyclic Chemistry II, Editors Katritzky and Rees, Elsevier, 1997, e.g. Volume 3; Liebigs Annalen der Chemie,
(9): 1910-16, (1985); Helvetica Chimica Acta, 41 : 1052-60, (1958); Arzneimittel-Forschung,
40(12): 1328-31, (1990), each of which are expressly incorporated by reference. Starting materials are generally available from commercial sources such as Aldrich Chemicals (Milwaukee, WI) or are readily prepared using methods well known to those skilled in the art (e.g., prepared by methods generally described in Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-23, Wiley, N.Y. (1967-2006 ed.), or Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed.
Springer- Verlag, Berlin, including supplements (also available via the Beilstein online database).
Synthetic chemistry transformations and protecting group methodologies (protection and deprotection) useful in synthesizing Formula (I) compounds and necessary reagents and intermediates are known in the art and include, for example, those described in R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T. W. Greene and P. G .M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); and L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) and subsequent editions thereof.
Compounds of Formula (I) may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1,000 compounds, or 10 to 100 compounds. Libraries of compounds of
Formula (I) may be prepared by a combinatorial 'split and mix' approach or by multiple parallel syntheses using either solution phase or solid phase chemistry, by procedures known to those skilled in the art. Thus according to a further aspect of the invention there is provided a compound library comprising at least 2 compounds, or pharmaceutically acceptable salts thereof.
In preparing compounds of Formulas I, protection of remote functionality (e.g., primary or secondary amine) of intermediates may be necessary. The need for such protection will vary depending on the nature of the remote functionality and the conditions of the preparation methods. Suitable amino-protecting groups include acetyl, trifluoroacetyl, t-butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9-fluorenylmethyleneoxycarbonyl (Fmoc). The need for such protection is readily determined by one skilled in the art. For a general description of protecting groups and their use, see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
For illustrative purposes, the following schemes show general methods for preparing compounds of Formula (I) according to the invention, as well as key intermediates. For a more detailed description of the individual reaction steps, see the Examples sections. Those skilled in the art will appreciate that other synthetic routes may be used to synthesize the inventive compounds. Although specific starting materials and reagents are depicted and discussed in the General Procedures, Examples, and schemes, other starting materials and reagents can be easily substituted to provide a variety of derivatives and/or reaction conditions. In addition, many of the exemplary compounds prepared by the described methods can be further modified in light of this disclosure using conventional chemistry well known to those skilled in the art.
EXAMPLES
The invention will be more fully understood by reference to the following examples. They should not, however, be construed as limiting the scope of the invention.
The chemical reactions described in the Examples may be readily adapted to prepare a number of other LDHA inhibitors of the invention, and alternative methods for preparing the compounds of this invention are deemed to be within the scope of this invention. For example, the synthesis of non-exemplified compounds according to the invention may be successfully performed by modifications apparent to those skilled in the art, e.g., by appropriately protecting reactive functional groups, by utilizing other suitable reagents known in the art other than those described, and/or by making routine modifications of reaction conditions. Alternatively, other reactions disclosed herein or known in the art will be recognized as having applicability for preparing other compounds of the invention.
¾ NMR spectra were recorded at ambient temperature using an NMR spectrometer, including a Varian Unity Inova (400MHz) spectrometer with a triple resonance 5mm probe.
Chemical shifts are expressed in ppm relative to tetramethylsilane. The following abbreviations have been used: br = broad signal, s = singlet, d = doublet, dd = double doublet, t = triplet, q = quartet, m = multiplet.
High Pressure Liquid Chromatography / Mass Spectrometry (LCMS) experiments to determine retention times (RT) and associated mass ions may be performed. The spectrometers may have an electrospray source operating in positive and negative ion mode. Additional detection is achieved using a evaporative light scattering detector.
Unless otherwise stated, all reactions were performed under an inert, i.e. argon or nitrogen, atmosphere.
ABBREVIATIONS
AcOH: Acetic acid; BOC: Di-tert-butyl dicarbonate; DCM: Dichloromethane; DIPEA: Diisopropylethylamine; DMAP: 4-Dimethylaminopyridine; EtOAc: Ethyl acetate; HATU:
(2-(7-Aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate); HC1:
Hydrochloric acid; MeOH: Methanol; NaBH4: Sodium borohydride, NBS: N-Bromosuccinimide; NH4CI: Ammonium chloride; NMR: Nuclear magnetic resonance; Pd(dppf)Cl2:
[l,l '-Bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane; RT: Room temperature; TFA: Trifluoroacetic acid; THF: Tetrahydrofuran.
Example 1
6-(6-bromo-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione




Scheme 1
Step A: N,0-Dimethylhydroxylamine hydrochloride (39 g, 0.40 mol), (dimethylamino)- N, N -dimethyl(3 H -[l,2,3]triazolo[4,5-^]pyridin-3-yloxy)-methaniminium hexafluorophosphate (152 g, 0.40 mol) and N, N -diisopropylethylamine (130.3 g, 1.01 mol) was added to a solution of 6-bromopicolinic acid (68 g, 0.34 mol) in DCM (1 L). The mixture was stirred at ambient temperature for 3 hours. The reaction mixture was washed with 1 Ν HQ (600 mL x 2), dried over anhydrous Na2S04 and concentrated. The crude residue was purified by silica gel chromatography eluting with a gradient of 10% - 30% EtOAc/hexanes to afford
6-bromo-N-methoxy-N-methylpicolinamide (80 g, 0.33 mol, 97% yield) as light color oil.
Step B: n-BuLi (158 mL, 0.4 mol) was slowly added to a solution of 3-bromothiophene (65.2 g, 0.4 mol) in isopropyl ether (1 L) at -78 °C. After stirring at -78 °C for 30 min, the reaction mixture was then slowly treated with 6-bromo-N-methoxy-N-methylpicolinamide (80 g, 0.33 mol) and stirred at -78 °C for 3 hours. The reaction mixture was quenched with saturated NH4C1 (300 mL), then warmed to ambient temperature. The mixture was diluted with EtOAc (400 mL), washed with water
(500 mL x 2), dried over anhydrous Na2S04 and concentrated. The crude residue was purified by silica gel chromatography eluting with a gradient of 0% -10% EtOAc/hexanes to afford
(6-bromopyridin-2-yl)(thiophen-3-yl)methanone (75 g, 0.28 mol, 86% yield) as yellow solid.
Step C: (6-Bromopyridin-2-yl)(thiophen-3-yl)methanone (75 g, 0.28 mol) and Ti(OEt)4 (191.5 g, 0.84 mol) was added to a solution of 2-methylpropane-2-sulfinamide (67.8 g, 0.56 mol) in THF (1 L). The mixture was heated at 70 °C for 16 hours. The suspension was allowed to cool to ambient temperature. The mixture was pour into ice water, filtered, washed with EtOAc. The filtrate was extracted with EtOAc (500 mL x 2), dried over anhydrous Na2S04 and concentrated. The crude was purified by silica gel chromatography eluting with a gradient of 10%> - 30%> EtOAc/hexanes to afford N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide (80 g, 215.6 mmol, 77% yield) as orange oil.
Step D: Methyl 3-oxobutanoate (50.0 g, 431.2 mmol,) was added to a suspension of NaH (10.35 g, 431.2 mmol,) in THF (1 L) under 0 °C. The reaction mixture was then slowly treated with n-BuLi (172 mL, 431.2 mmol,) and stirred under 0 °C for 30 minutes,
N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2- sulfinamide (80 g, 215.6 mmol,) was added to the mixture and stirred at 0 °C for another 2 hours. The reaction mixture was quenched with saturated NH4C1 (500 mL), then warmed to ambient temperature. The mixture was diluted with EtOAc (400 mL), washed with water (500 mL x 2), dried over anhydrous Na2S0 and concentrated to afford methyl
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen- 3-yl)pentanoate (95 g, 194.9 mol, 90% yield) as yellow oil.
Step E: HCl/MeOH (150 mL) was slowly added to a solution of
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate (95 g, 194.9 mol) in MeOH (1 L) at 0 °C. The mixture was stirred at ambient temperature for 1 hour, and then slowly acidified to pH 7 using 2 N NaOH at 0 °C. The solvent was removed under vacuum. The crude product was extracted with EtOAc (800 mL x 2), dried over anhydrous Na2S04 and concentrated to afford methyl 5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate (62 g, 161.9 mmol, 83% yield) as dark color oil.
Step F: Potassium carbonate (67.1 g, 485.7 mmol) was added to a solution of methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate (62 g, 161.9 mmol) in MeOH (800 mL). The mixture was heated at 80 °C for 2 hours. The suspension was allowed to cool to ambient temperature. The solvent was removed under vacuum, the crude product was dissolved in water (1 L), washed with EtOAc (1 L x 2). The aqueous layer was acidified to pH 4 using 3 N HC1. The mixture was extracted with EtOAc (800 mL x 2). The organic layer was dried over anhydrous Na2S0 and concentrated to afford 6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-
dihydro-[2,2'-bipyridin]-6(lH)-one (31 g, 88.3 mmol, 55% yield) as yellow solid.
Step G: Potassium carbonate (36.6 g, 264.9 mmol) and l,2-bis(2-chlorophenyl)disulfane (15.2 g, 53.0 mmol) was added to a solution of
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one (31 g, 88.3 mmol) in MeOH (800 mL). The mixture was heated at 80 °C for 2 hours. The suspension was allowed to cool to ambient temperature. The solvent was removed under vacuum, the crude product was dissolved in water (800 mL), washed with EtOAc (800 mL x 2). The aqueous layer was acidified to pH 4 using 3 N HC1. The mixture was extracted with EtOAc (800 mL x 2). The organic layer was dried over anhydrous Na2S04 and concentrated to afford
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one (38 g, 76.9 mmol, 87% yield) as light color solid.
Example 2
3-(2-chlorophenyl)sulfanyl-6-(6-isopropoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione

Step A: NaH (73 mg, 3.04 mmol) was added to a solution of propan-2-ol (182 mg, 3.04 mmol) in THF (10 mL) at 0 °C. After stirring 30 minutes,
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one (300 mg, 0.61 mmol) was added to the mixture at 0 °C, and then the mixture was refluxed for 12 hours. The suspension was cooled to 0 °C, quenched with water (10 mL), diluted with EtOAc (20mL), acidified to pH 7 using 1 N HC1, washed with brine, dried over anhydrous Na2S04 and concentrated. The crude residue was purified by preparative HPLC (formic acid) to afford
3-((2-chlorophenyl)thio)-6-(6-isopropoxy- pyridin-2-yl)-6-(thiophen-3-yl)piperidine-2,4-dione (112 mg, 0.24 mmol, 39%> yield) as white solid. Mixture of diastereoisomers: ¾ NMR (400MHz, CD3OD) δ 7.68 (dd, J= 8.4, 3.6 Hz, 1H), 7.43 (dd, J= 5.2, 2.8 Hz, 1H), 7.26 - 7.11 (m, 4H), 6.92 (dd, J= 8.0, 8.0 Hz, 1H), 6.76 - 6.67 (m, 2H), 5.97 (dd, J= 8.0, 1.2Hz, 1H), 5.39 - 5.32 (m, 1H), 3.88 (d, J= 16.4 Hz, 1H), 3.45 (d, J= 16.4 Hz, 1H), 1.31 (d, J= 6.4 Hz, 3H), 1.26 (d, J= 6.4 Hz, 3H). LCMS M+l = 472.8. Stereoisomer 1 : ¾ NMR (400MHz, CD3OD) δ 7.68 (dd, J = 8.4, 3.6 Hz, 1H), 7.43 (dd, J = 5.2, 2.8 Hz, 1H), 7.26 - 7.11 (m, 4H), 6.92 (dd, J = 8.0, 8.0 Hz, 1H), 6.76 - 6.67 (m, 2H), 5.97 (dd, J= 8.0, 1.2Hz, 1H), 5.39 - 5.32 (m, 1H), 3.89 (d, J=16.4 Hz, 1H), 3.45 (d, J = 16.4 HZ, 1H), 1.31 (d, J = 6.4 Hz, 3H), 1.26 (d, J = 6.4 Hz, 3H). LCMS M+l = 472.8. Stereoisomer 2: ¾NMR (400MHz, CD3OD) δ 7.68 (dd, J= 8.4, 3.6 Hz, 1H), 7.43 (dd, J= 5.2, 2.8 Hz, 1H), 7.26 - 7.11 (m, 4H), 6.92 (dd, J= 8.0, 8.0 Hz, 1H), 6.76 - 6.67 (m, 2H), 5.97 (dd,
J= 8.0, 1.2Hz, 1H), 5.39 - 5.32 (m, 1H), 3.89 (d, J=16.4 Hz, 1H), 3.45 (d, J= 16.4 Hz, 1H), 1.31 (d, J= 6.4 Hz, 3H), 1.26 (d, J= 6.4 Hz, 3H). LCMS M+l = 472.9.
Example 3
6-[6-(2-chloro-4-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidi ne-2,4-dione

Step A: 6'-Bromo-5-(2-chloro-phenylsulfanyl)-4-hydroxy-2-thiophen-3-yl-2,3- dihydro-lH-[2,2']bipyridinyl-6-one (500 mg, 1 mmol), 2-chloro-4-fluoro-phenol (178 mg, 1.2 mmol), 2-(dimethylamino)acetic acid hydrochloride (28 mg, 0.2 mmol), Cul (39 mg, 0.2 mmol) and Cs2C03 (0.99 g, 3 mmol) were combined. Dioxane (5 ml) was added, the mixture was stirred at 120 °C for 3 h under nitrogen atmosphere. After the suspension was cooled to ambient temperature, EtOAc (20mL) was added, and the mixture was filtered over Celite. The resulting solution was washed three times with brine, dried anhydrous Na2S04, filtered, and the solvent evaporated under reduced pressure. The crude residue was purified by preparative HPLC (formic acid) to give the product (mixture of diastereoisomers, 230 mg, 41%, 10 mg was delivered) as white solid. The mixture of diastereoisomers (220 mg) was purified by SFC (neutral) to give the isomers (stereoisomer 1, 80 mg and stereoisomer 2, 128 mg) as white solid. Mixture of diastereoisomers: 'H NMR (400MHZ, (CD3)2SO) δ 7.93 (dd, J= 8.0, 8.0 Hz, 1H), 7.60 - 7.53 (m, 1H), 7.44 (dd, J= 4.8, 2.1 Hz, 1H), 7.37 (d, J= 7.6 Hz, 1H), 7.30 - 7.23 (m, 3H), 7.18 (dd, J= 2.8, 1.2 Hz, 1H), 7.04 (d, J= 8.4 Hz, 1H), 6.98 - 6.93 (m, 1H), 6.91 (dd, J= 4.2, 1.2 Hz, 1H), 6.78 - 6.74 (m, 1H), 5.88 (dd, J= 7.6, 1.2 Hz, 1H), 3.37 (d, J = 16.4 Hz, 1H), 3.13 (d, J= 16.4 Hz, 1H). LCMS M+l = 558.7. Stereoisomer 1 : ¾ NMR (400MHz, CD3OD) δ 7.88 (dd, J= 8.0, 8.0 Hz, 1H), 7.36 (dd, J= 3.9, 2.4 Hz, 1H), 7.35 - 7.32 (m, 2H), 7.22 (dd, J= 8.0, 1.2 Hz, 1H), 7.19 - 7.09 (m, 3H), 7.07 (d, J= 8.2 Hz, 1H), 6.96 (dd, J= 3.9, 0.9 Hz, 1H), 6.94 (dd, J= 8.0, 1.2 Hz, 1H), 6.81 - 6.74 (m, 1H), 5.88 (dd, J= 8.4, 1.2 Hz, 1H), 3.48 (d, J = 16.4 Hz, 1H), 3.20 (d, J= 16.4 Hz, 1H). LCMS M+l = 558.7. Stereoisomer 2: ¾ NMR (400MHz, CD3OD) δ 7.89 (dd, J= 8.0, 8.0 Hz, 1H), 7.36 (dd, J= 3.9, 2.4 Hz, 1H), 7.35 - 7.32 (m, 2H), 7.22 (dd, J= 8.0, 1.2 Hz, 1H), 7.19 - 7.09 (m, 3H), 7.07 (d, J= 8.2 Hz, 1H), 6.96 (dd, J= 3.9, 0.9 Hz, 1H), 6.94 (dd, J= 8.0, 1.2 Hz, 1H), 6.81 - 6.74 (m, 1H), 5.97 (dd, J= 8.4, 1.2 Hz, 1H), 3.48 (d, J = 16.4 Hz, 1H), 3.20 (d, J= 16.4 Hz, 1H). LCMS M+l = 558.8.
Example 4
3-(2-chlorophenyl)sulfanyl-6-[6-(cyclohexylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-(lione
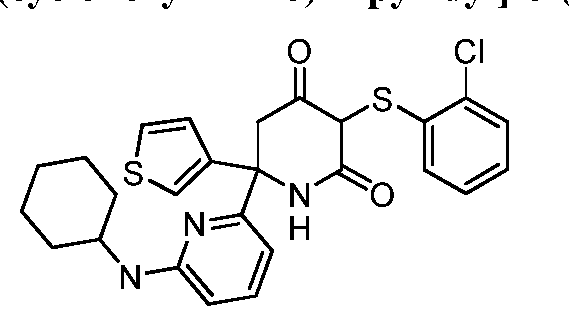
Step A: 6-(6-Bromopyridin-2-yl)-3-((2-chlorophenyl)thio)-6-(thiophen-3-yl)- piperidine-2,4-dione (300 mg, 607.5 μηιοΐ), cyclohexanamine (90.4 mg, 911.3 μηιοΐ), Brettphos (65.2 mg, 121.5 μιηοΐ), Pd2(dba)3 (55.6 mg, 60.8 μmol) and NaOiBu (116.8 mg, 1.2 mmol) were combined, dioxane (5 ml) was added. The mixture was stirred at 120 °C for 8 hours under nitrogen atmosphere. After the suspension was cooled to room temperature, ethyl acetate (15 mL) was added, and the mixture was filtered over Celite. The resulting solution was washed three times with brine, dried over sodium sulphate, filtered, and the solvent evaporated under reduced pressure. The residue was purified by preparative HPLC (formic acid) to give the desired product (mixture of diastereoisomers, 75.4 mg, 24%, 7.4 mg was delivered) as yellow solid. The mixture of
diastereoisomers (68.0 mg) was purified by SFC (neutral) to give the desired product (stereoisomer 1, 10 mg and stereoisomer 2, 5.8 mg) as yellow solid. Mixture of diastereoisomers: ¾ NMR (400MHZ, CD3OD) δ 7.88 (dd, J= 7.6, 7.6 Hz, IH), 7.69 (d, J= 2.8 Hz, IH), 7.63 (d, J= 2.8 Hz, IH), 7.28 - 7.26 (m, 2H), 7.19 (d, J = 7.6 Hz, IH), 7.10 (d, J= 7.4Hz, IH), 6.85 (d, J= 7.6 Hz, IH), 6.47 (d, J= 7.2 Hz, IH), 6.03 (d, J= 8.0 Hz, IH), 3.70 - 3.66 (m, IH), 3.88 - 3.67 (m, 5H), 3.59 (d, J= 16.0 Hz, 2H), 1.93 - 1.91 (m, 2H), 1.73 - 1.70 (m, 2H), 1.60 - 1.41 (m, 2H), 1.28 - 1.22 (m, 4H). LCMS M+l = 511.9. Stereoisomer 1 : ¾ NMR (400MHz, CD3OD) δ 7.40 (dd, J= 7.6, 7.6 Hz, IH), 7.28 (d, J= 2.8 Hz, IH), 7.18 (d, J= 2.8 Hz, IH), 7.15 (d, J= 2.8 Hz, IH), 6.91 (d, J= 7.6 Hz, IH), 6.77 (d, J= 7.4 Hz, IH), 6.64 (d, J= 7.6 Hz, IH), 6.40 (d, J= 7.2 Hz, IH), 6.06 (d, J= 8.0 Hz, IH), 3.81 - 3.77(m, IH), 3.76 (d, J= 16.0 Hz, IH), 3.41(d, J= 16.0 Hz, IH), 2.01 - 1.95 (m, 2H), 1.77 - 1.73 (m, 2H), 1.70 - 1.40 (m, 2H), 1.27 - 1.17 (m, 4H). LCMS M+l = 511.8. Stereoisomer 2: lH NMR (400MHz, CD3OD) δ 7.40 (dd, J= 7.6, 7.6 Hz, IH), 7.28 (d, J= 2.8 Hz, IH), 7.18 (d, J = 2.8 Hz, IH), 7.15 (d, J= 2.8 Hz, IH), 6.91 (d, J= 7.6 Hz, IH), 6.77 (d, J= 7.4Hz, IH), 6.62 (d, J= 7.6 Hz, IH), 6.40 (d, J= 7.2 Hz, IH), 6.06 (d, J= 8.0 Hz, IH), 3.80 - 3.78(m, IH), 3.76 (d, J= 16.0 Hz, IH), 3.42 (d, J = 16.0 Hz, IH), 2.01 - 1.95(m, 2H), 1.74 - 1.71 (m, 2H), 1.62 - 1.40 (m, 2H), 1.27 - 1.17 (m, 4H). LCMS M+l = 511.9.
Example 5
3-(2-chlorophenyl)sulfanyl-6- [6- [(3-fluorophenyl)methyl] -2-pyridyl] -6-(3-thienyl)piperidine-
2,4-dione

Step A: 1 ,2-Dibromoethane (100 mg, 0.53 mmol) and l-(bromomethyl)-3-fluorobenzene (1 g, 5.3 mmol) was added to a suspension of zinc powder (345 mg, 5.3 mmol) in anhydrous THF (10 mL). The reaction mixture was stirred at room temperature for 8 hours. The resultant solution was used directly in the next step.
Step B: (3-Fluorobenzyl)zinc(II) bromide (5.7 mL, 3.04 mmol) was added to a solution of Pd(PPh3)4 (69 mg, 0.06 mmol) and
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one (300 mg, 0.61 mmol) in anhydrous THF (5 mL). The suspension was stirred at room temperature for 12 hours, and then quenched with water, filtered over Celite. The resulting solution was dried over anhydrous Na2S04 and concentrated. The crude residue was purified by preparative HPLC (formic acid) to afford 3-(2-chlorophenyl)sulfanyl-6-[6-[(3-fluorophenyl)methyl]-2-pyridyl]- 6-(3-thienyl)piperidine-2,4 dione, (61 mg, 0.12 mmol, 20% yield) as white solid. The mixture of diastereoisomers was purified by SFC (neutral) to give the separated stereoisomers. Mixture of diastereoisomers: 'H NMR (400MHZ, CD3OD) δ 7.75 (dd, J= 8.0, 8.0 Hz, 1H), 7.41 - 7.39 (m, 2H), 7.22 - 7.17 (m, 4H), 7.10 - 7.09 (m, 2H), 7.08 (d, J= 8.0 Hz, 1H), 6.87 - 6.86 (m, 2H), 6.55 (dd, J = 8.0, 0.8 Hz, 1H), 5.82 (dd, J= 8.0, 1.6 Ηζ,ΙΗ), 4.17 (s, 2H), 3.97 (d, J= 16.8 Hz, 1H), 3.47 (d, J = 16.4 Hz, 1H). LCMS M+l = 522.9. Stereoisomer 1 : ¾ NMR (400MHz, CD3OD) δ 7.74 (dd, J= 8.0, 8.0 Hz, 1H), 7.43 - 7.41 (m, 2H), 7.24 - 7.17 (m, 4H), 7.15 - 7.09 (m, 2H), 7.07 (d, J= 8.0 Hz, 1H), 6.84 - 6.82 (m, 2H), 6.55 (dd, J= 8.0, 0.8 Hz, 1H), 5.87 (d, J= 8.4 Hz, 1H), 4.16 (s, 2H), 3.85 (d, J = 16.0 Hz, 1H), 3.45 (d, J= 16.4 Hz, 1H). LCMS M+l = 522.9. Stereoisomer 2: ¾ NMR (400MHz, CD3OD) δ 7.73 (dd, J= 8.0, 8.0 Hz, 1H), 7.42 - 7.36 (m, 2H), 7.24 - 7.21 (m, 4H), 7.09 - 7.08 (m, 2H), 7.07 (d, J= 8.0 Hz, 1H), 6.84 - 6.82 (m, 2H), 6.57 (dd, J= 8.0, 0.8 Hz, 1H), 5.90 (dd, J= 8.0, 1.6 Hz, 1H), 4.16 (s, 2H), 3.78 (d, J= 16.0 Ηζ,ΙΗ), 3.44 (d, J= 16.4 Hz, 1H). LCMS M+l = 522.9.
Example 6
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorobenzoyl)-2-pyridyl]-6-(3-thienyl)piperidine
e

Step A: 6'-Bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro- [2,2'-bipyridin]-6(lH)-one (300 mg, 0.61 mmol) and (4-fluorophenyl)boronic acid (94 mg, 0.67 mmol) was added to a solution of K2C03 (253 mg, 0.83 mmol) and PdCl2(PPh3)2 (21 mg, 0.02 mmol) in THF (6 mL). The mixture was heated at 100 °C for 20 hours under carbon monoxide atmosphere (0.5 MPa). After cooling to room temperature, the reaction was filtered over Celite. The resulting solution was dried over anhydrous Na2S04 and concentrated. The crude residue was purified by preparative HPLC (formic acid) to afford -(2-chlorophenyl)sulfanyl-6-[6-(4-fluorobenzoyl)-2- pyridyl]-6-(3-thienyl)piperidine-2,4-dione (78 mg, 0.15 mmol, 24% yield) as white solid. The mixture of diastereoisomers was purified by SFC (neutral) to give the separated stereoisomers. Mixture of diastereoisomers: mixture of diastereoisomers: lH NMR (400MHz, CD3OD) δ 7.68 (dd, J= 8.4, 3.6 Hz, 1H), 7.43 (dd, J= 5.2, 2.8 Hz, 1H), 7.26 - 7.1 1 (m, 4H), 6.92 (dd, J= 8.0, 8.0 Hz, 1H), 6.76 - 6.67 (m, 2H), 5.97 (dd, J= 8.0, 1.2Hz, 1H), 5.39 - 5.32 (m, 1H), 3.88 (d, J= 16.4 Hz, 1H), 3.45 (d, J = 16.4 Hz, 1H), 1.31 (d, J = 6.4 Hz, 3H), 1.26 (d, J = 6.4 Hz, 3H). LCMS M+l = 472.8. Stereoisomer 1 : ¾ NMR (400MHz, CD3OD) δ 7.68 (dd, J = 8.4, 3.6 Hz, 1H), 7.43 (dd, J = 5.2, 2.8 Hz, 1H), 7.26 - 7.1 1 (m, 4H), 6.92 (dd, J = 8.0, 8.0 Hz, 1H), 6.76 - 6.67 (m, 2H), 5.97 (dd, J = 8.0, 1.2Hz, 1H), 5.39 - 5.32 (m, 1H), 3.89 (d, J =16.4 Hz, 1H), 3.45 (d, J = 16.4 HZ, 1H), 1.31 (d, J = 6.4 Hz, 3H), 1.26 (d, J = 6.4 Hz, 3H). Stereoisomer 2: lH NMR (400MHz, CD3OD) δ 7.68 (dd, J= 8.4, 3.6 Hz, 1H), 7.43 (dd, J = 5.2, 2.8 Hz, 1H), 7.26 - 7.1 1 (m, 4H), 6.92 (dd, J = 8.0, 8.0 Hz, 1H), 6.76 - 6.67 (m, 2H), 5.97 (dd, J = 8.0, 1.2Hz, 1H), 5.39 - 5.32 (m, 1H), 3.89 (d, J =16.4 Hz, 1H), 3.45 (d, J = 16.4 Hz, 1H), 1.31 (d, J= 6.4 Hz, 3H), 1.26 (d, J= 6.4 Hz, 3H).
Example 7
3-(2-chlorophenyl)sulfanyl-6-[4-(2-methylmorpholin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4
-dione

7
Scheme 2
Step A: To a solution of 3-bromothiophene (14.43 g, 220.74 mmol) in anhydrous isopropyl ether (500 mL) was added n-BuLi (88.2 ml, 220.74 mmol) at -78 °C under nitrogen atmosphere. The reaction mixture was stirred for 1 hour. 4-Bromobenzaldehyde (100 g, 183.95 mmol) was added and the reaction mixture was stirred at -78 °C for 2 hours. The reaction was quenched with MeOH and acidified to pH 4 with 1 N HC1, extracted with DCM (100 mL x 2). The combined organic layers were dried over anhydrous Na2S04, and concentrated. The crude residue was purified by silica gel
chromatography (petroleum ether : EtOAc = 3 : 1) to give (4-bromophenyl)(thiophen-3-yl)methanol (100 g, 69%) as a yellow solid.
Step B: To a solution of (4-bromophenyl)(thiophen-3-yl)methanol (100 g, 371.5 mmol) in CHC13 (200 ml) was added Mn02 (322.9 g, 3715 mmol). The reaction mixture was stirred at 60 °C for 12 hours. After cooling to room temperature, the reaction mixture was filtered over Celite and the filtrate was concentrated under vacuum. The crude residue (86 g, 86% yield) was used in the next step without further purification.
Step C: (£')-N-((4-Bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2- sulfinamide was prepared in 86% yield according to the Example 1 , Step C substituting
(6-bromopyridin-2-yl)(thiophen-3-yl)methanone for (4-bromophenyl)(thiophen-3-yl) methanone.
Step D: Methyl 5-(4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5- (thiophen-3-yl) pentanoate was prepared in 85% yield according to the Example 1 , Step D:
Substituting (Z)-N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methyl- propane-2-sulfinamide for
(£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide.
Step E:
Methyl 5-amino-5-(4-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 90% yield according to the Example 1 , Step E substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- (4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl) pentanoate.
Step F:
6- (4-Bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one was prepared in 75%) yield according to the Example 1 , Step F substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for methyl
5- amino-5-(4-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step G:
6- (4-Bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH )-one was prepared in 90%> yield according to the Example 1 , Step G substituting
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3- dihydro-[2,2'-bipyridin] -6( lH)-one for 6-(4-bromophenyl)-4-hydroxy-6- (thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one.
Step Η: To a solution of 6-(4-bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-6- (thiophen-3-yl)-5,6- dihydropyridin-2(lH)-one (0.25 g, 0.5 mmol) in dioxane (6 mL) was added 2-methylmorpholine (500 mg, 5 mmol), Brettphos (25 mg, 0.05 mmol), Pd2(dba)3 (45 mg, 0.05 mmol) and i-BuONa (0.5 g, 5 mmol). The reaction mixture was stirred at 1 10 °C for 16 hours under nitrogen atmosphere. After cooling to room temperature, the reaction mixture was filtered though a
short pad of silica gel. The filtrate was concentrated under vacuum. The crude residue was purified by preparative HPLC (formic acid) to afford the product (10 mg, 3.8% yield) as white solid. lH NMR (400 MHz, (CD3)2SO) δ 8.35 (s, 1H), 7.57 (d, J= 5.2Hz, 1H), 7.32 (m, 4H), 7.16 (m, 1H), 6.98 (m, 3H), 6.73 (m, 1H), 5.92 (m, 1H), 3.93 (m, 1H), 3.64 (m, 3H), 3.58 (m, 1H), 3.37 (m, 2H), 2.69 (m, 1H), 2.34 (m, 1H), 1.15 (d, J= 6.4 Hz, 3H). LCMS M+l = 512.9.
Example 8
3-(2-chlorophenyl)sulfanyl-6-[4-(cyclohexen-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dione

Step A: To a solution of 6-(4-bromophenyl)-3-((2-chlorophenyl)thio)-6-(thiophen-3-yl) piperidine-2,4-dione (0.25 g, 0.5 mmol ) in dioxane (6 mL) and water (2 mL) was added cyclohex-l-en-l-ylboronic acid (126 mg, 1 mmol), Pd(dppf)Cl2 (36 mg, 0.05 mmol) and K2C03 (0.27 g, 2 mmol). The reaction mixture was microwaved at 100 °C for 1 hour under nitrogen atmosphere. After cooling to room temperature, the reaction mixture was filtered though a short pad of silica gel. The filtrate was concentrated under vacuum and the crude residue was purified by preparative HPLC (formic acid) to afford the product (11.7 mg, 5% yield). lH NMR (400 MHz, (CD3)2SO) δ 8.47 (s, 1H), 7.56 - 7.55 (m, 1H), 7.54 - 7.39(m, 2H), 7.32 - 7.20 (m, 3H), 7.27 (d, J= 8 Hz, 1H), 7.14 (dd, J= 5.2, 4.8 Hz, 1H), 6.93 (dd, J= 7.6, 4.8Hz, 1H), 6.15 (s, 1H), 5.85 (d, J= 8.0 Hz, 1H), 3.39 (s, 2H), 2.47 (s, 2H), 2.33 (s, 2H), 1.71 - 1.68 (m, 2H), 1.58 - 1.56 (m, 2H). LCMS M+l = 493.9; 495.9.
Example 9
3-(2-chlorophenyl)sulfanyl-6-(4-cyclohexylphenyl)-6-(3-thienyl)piperidine-2,4-dione

Step A: To a solution of GNT C349 986 (0.8 g,1.6 mmol ) in acetic acid (20 mL) was added Pd/C (0.1 g). The reaction mixture was stirred at room temperature for 24 hours under hydrogen atmosphere (60 Psi). After relieving the pressure, the reaction mixture was filtrated over Celite and the filtrate was concentrated under vacuum. The crude residue was purified by preparative HPLC (formic acid) to afford the product (10 mg, 1.2% yield) as white solid. lB NMR (400 MHz, (CD3)2SO) δ 7.49 (s, 1H), 7.35-7.32 (m, 2H), 7 '.26-7 '.25 (m, 4H), 7.19 (d, J= 8.0 Hz, 1H), 6.93 (dd, J
= 6.8, 6.8 Hz, IH), 6.72 (dd, J= 6.8, 6.8 Hz, IH), 5.98 (d, J= 6.8 Hz, IH), 3.45 (s, 2H), 1.96 - 1.74 (m, 5H), 1.48 - 1.27 (m, 5H). LCMS M+l = 495.8.
Example 10
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(l-methylcyclopropyl)ethoxy]-2-pyridyl]-6-(3-thienyl)pipe ridine-2,4-dione

10
Step A: Diethylzinc (40.6 ml, 40.6 mmol) and diiodomethane (9.3 g, 34.8 mmol) was added to a solution of 3-methylbut-3-en-l -ol (1 g, 11.6 mmlo) in DCM (80 mL) at -10 °C. The reaction mixture was stirred at 0 °C for 1 hour and then room temperature for additional 12 hours. The reaction was quenched with saturated NH4C1, extracted with DCM (50 mL x 2), dried over anhydrous Na2S04 and concentrated to afford 2-(l-methylcyclopropyl)ethanol (600 mg, 6 mmol, 52% yield) as light color oil.
Step B: 5-((2-Chlorophenyl)thio)-4-hydroxy-6'-(2-(l -methylcyclopropyl)ethoxy)-
2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 39% yield according to the Example 2, Step A substituting propan-2-ol for 2-(l-methylcyclopropyl)ethanol.
Mixture of diastereoisomers: lH NMR (400MHz, CD3OD) δ 7.74 (dd, J= 8.0, 8.0 Hz, IH), 7.47 (dd, J = 5.2, 3.2 Hz, IH), 7.30 - 7.15 (m, 4H), 6.96 (dd, J= 8.0, 8.0 Hz, IH), 6.77 - 6.75 (m, 2H), 6.01 (dd, J= 8.4, 1.6 Hz, IH), 4.49 (t, J= 7.2 Hz, 2H), 3.93 (d, J= 16.0 Ηζ,ΙΗ), 3.48 (d, J= 16.4 Ηζ,ΙΗ), 1.70 (t, J= 6.8 Hz, 2H), 1.09 (s, 3H), 0.34 - 0.23 (m, 4H). Stereoisomer 1 : lH NMR (400MHz, CD3OD) δ 7.50 (dd, J= 8.0, 8.0 Hz, IH), 7.48 (dd, J= 5.2, 3.2 Hz, IH), 7.30 - 7.22 (m, 2H), 7.06 - 7.00 (m, 2H), 6.93 (dd, J= 8.0, 8.0 Hz, IH), 6.54 - 6.52 (m, 2H), 5.79 (dd, J= 8.0, 1.6 Hz, IH), 4.26 (t, J= 6.8 Hz, 2H), 3.70 (d, J= 16.0 Ηζ,ΙΗ), 3.25 (d, J= 16.4 Ηζ,ΙΗ), 1.46 (t, J = 6.8 Hz, 2H), 0.86 (s, 3H), 0.11 - 0.00 (m, 4H). Stereoisomer 2: 1H NMR (400MHz, CD3OD) δ 7.48 (dd, J= 8.0, 8.0 Hz, lH), 7.46 (dd,
J= 5.2, 3.2 Hz, 1H), 7.21 - 7.20 (m, 2H), 7.05 - 7.03 (m, 2H), 6.93 (dd, J= 8.0, 8.0 Hz, 1H), 6.53 - 6.50 (m, 2H), 5.77 (dd, J= 8.0, 1.6 Hz, 1H), 4.24 (t, J= 6.8 Hz, 2H), 3.68 (d, J= 16.0 Ηζ,ΙΗ), 3.23 (d, J= 16.4 Ηζ,ΙΗ), 1.45 (t, J= 6.8 Hz, 2H), 0.85 (s, 3H), 0.10 - 0.01 (m, 4H).
Examples 11 and 12
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-2-pyridyl]-l-methyl-6-(3-thienyl)piperidine
-2,4-dione and
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-l-methyl-6-(3-thienyl)piperidin
-2,4-dione

Step A: To a stirred solution of
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH^ one (1 g, 2 mmol) in anhydrous THF (20 mL) at 0 °C was added NaH (288 mg, 12 mmol). The
reaction mixture was stirred at the same temperature for 0.5 hour, and then the reaction was added iodomethane (1.65 g, 12 mmol) and stirred at room temperature for 12 hours. The reaction was quenched with water, dried and concentrated. The crude residue was purified by silica gel chromatography eluting with a gradient of 10% - 50% EtOAc/hexanes to to afford
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-l-methyl-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridi n]-6(lH)-one (475 mg, 0.94 mol, 46% yield) as yellow solid.
Step B: 5-((2-Chlorophenyl)thio)-6'-((4-fluorophenyl)amino)-4-hydroxy-l-methyl- 2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 8% yield according to the Example 4, Step A substituting cyclohexanamine for 4-fluoroaniline.
Step C: 5-((2-Chlorophenyl)thio)-6'-(4-fluorophenoxy)-4-hydroxy-l-methyl-2-
(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 4% yield according to the Example 3, Step A 2-Chloro-4-fluoro-phenol for 4-fluorophenol and
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one for 6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-l-methyl-2- (thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one.
Example 11 : ¾ NMR (400MHz, CD3OD) δ 7.59 - 7.46 (m, 4H), 7.20 (dd, J= 8.0, 1.2Hz, 1H), 7.15
(dd, J= 5.2, 1.6 Hz, 1H), 7.07 (dd, J= 2.8, 1.6 Hz, 1H), 6.94 - 6.84 (m, 4H), 6.78 (dd, J= 8.4, 0.8
Hz, 1H), 6.63 (dd, J= 7.6, 0.4 Hz, 1H), 6.23 (dd, J= 7.0, 1.6 Hz, 1H), 3.88 (d, J= 16.8 Hz, 1H),
3.56 (d, J= 16.8 Hz, 1H), 2.86 (s, 3H). LCMS M+l = 537.8.
Example 12: lH NMR (400MHz, CD3OD) δ 7.88 (dd, J= 8.4, 7.6 Hz, 1H), 7.49 (dd, J= 5.2, 2.8 Hz,
1H), 7.23 (dd, J= 8.0, 1.6 Hz, 1H), 7.09 - 6.95 (m, 8 H), 6.89 - 6.83 (m, 2H), 6.04 (dd, J= 8.0, 1.2Hz,
1H), 3.57 - 3.46 (m, 2H), 2.65 (s, 3H). LCMS M+l = 538.8.
Example 13
3-(2-chlorophenyl)sulfanyl-6-(2-fluorophenyl)-l-methyl-6-(3-thienyl)piperidine-2,4-dione

13
Step A: 2-Fluoro-N-methoxy-N-methylbenzamide was prepared in 73% yield according to the Example 1, Step A substituting 6-bromopicolinic acid for 2-fluorobenzoic acid.
Step B: (2-Fluorophenyl)(thiophen-3-yl)methanone was prepared in 99% yield according to the Example 1, Step B substituting 6-bromo-N-methoxy-N-methylpicolinamide for
2-fluoro-N-methoxy-N-methyl-benzamide.
Step C: (Z)-N-((2-Fluorophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-
sulfanamide was prepared in 46% yield according to the Example 1 , Step C substituting
(6-bromopyridin-2-yl)(thiophen-3 -yl)methanone for (2-fluorophenyl)(thiophen-3 -yl)- methanone.
Step D: Methyl 5-(l , l -dimethylethylsulfinamido)-5-(2-fluorophenyl)-3-oxo-5- (thiophen-3-yl)pentanoate was prepared in 91% yield according to the Example 1 , Step D substituting N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-
2- sulfinamide for ( )-N-((2-fluorophenyl)(thiophen-3-yl)methylene)-2 -methyl- propane -2-sulfinamide.
Step E: Methyl 5-amino-5-(2-fluorophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
was prepared in 33% yield according to the Example 1 , Step E substituting methyl
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl 5-(l ,l -dimethylethylsulfinamido)-5-(2-fluorophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step F: 6-(2-Fluorophenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 89% yield according to the Example 1 , Step F substituting
methyl-5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- amino-5-(2-fluorophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step G: 3-((2-Chlorophenyl)thio)-6-(2-fluorophenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 83% yield according to the Example 1 , Step G substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
6- (2-fluorophenyl)-6-(thiophen-3-yl)piperidine-2,4-dione.
Step Η:
3- (2-chlorophenyl)sulfanyl-6-(2-fluorophenyl)-l -methyl-6-(3-thienyl)piperidine-2,4-dione was prepared in 30% yield according to the Example 1 1 , Step B substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
3-((2-chlorophenyl)thio)-6-(2-fluorophenyl)-6-(thiophen-3-yl)piperidine-2,4-dione. lH NMR (400MHz, CD3OD) δ 7.57 (d, J = 4.0 Hz, 1H), 7.50 - 7.47 (m, 1H), 7.28 - 7.16 (m, 5 H), 6.99 - 6.85 (m, 3H), 6.26 (dd, J = 8.0, 4.0 Hz, 1H), 3.76 - 3.67 (m, 2H), 2.82 (s, 3H). LCMS M+l = 445.9.
Example 14
3-(2-chlorophenyl)sulfanyl-6-[5-(4-fluoroanilino)-2-hydroxy-phenyl]-6-(3-thienyl)piperidine-
2,4-dione


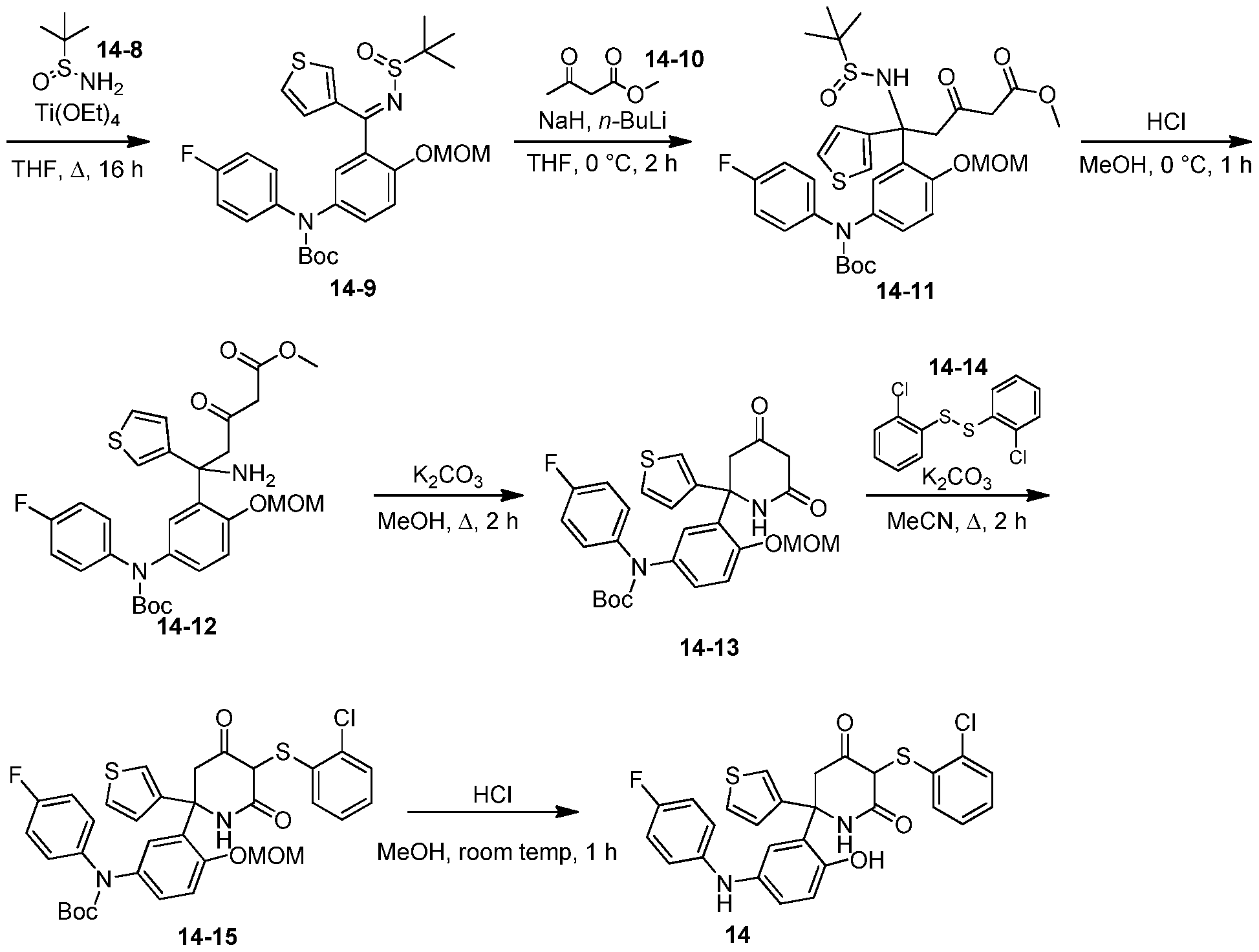
Step A: Chloro(methoxy)methane (19.1 g, 0.23 mol) was added to a solution of
5-bromo-2-hydroxybenzaldehyde (30 g, 0.15 mol) and di-z' o-propyl-ethylamine (38.5 g, 0.30 mol) at 0 °C in DCM. The mixture was warmed to ambient temperature and stirred for 18 hours. The reaction was quenched with water, dried over anhydrous Na2S04 and concentrated to afford
5-bromo-2-(methoxymethoxy)benzaldehyde (30 g, 0.12 mol, 82% yield) as light color oil.
Step B: (5-Bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanol was prepared in 77% yield according to the Example 2, Step A substituting 4-bromobenzaldehyde acid for
5-bromo-2-(methoxymethoxy)benzaldehyde.
Step C: (5-Bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanol was prepared in 91% yield according to the Example 7, Step B substituting (4-bromophenyl)(thiophen-3-yl)methanol for (5-bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone.
Step D: A mixture of (5-bromo-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone (10 g, 27.0 mmol), 4-fluoroaniline (10 g, 53.9 mmol), Xantphos (3.85 g, 5.39 mmol), Pd2(dba)3 (3.72 g, 2.7 mmol), Cs2C03 (39.5 g, 80.9 mmol) and 1,4-dioxane (200 mL) was stirred at 110 °C for 16 hours. The reaction was cooled to room temperature, then filtered. The filtrate was concentrate under vacuum. The crude residue was purified by silica gel chromatography eluting with a gradient of 10% - 50%) EtOAc/hexanes to to afford
(5-((4-fluorophenyl)amino)-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone (14 g, 39.2 mmol, 85%o yield) as yellow solid.
Step E: A mixture of
(5-((4-fluorophenyl)amino)-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone (14 g, 39.2 mol), di-tert-butyl dicarbonate (16.9 g, 78.3 mmol), 4-dimethylaminopyridine (2.37 g, 19.6 mmol) and DCM (200 mL) was stirred at room temperature for 12 hours. The mixture was diluted with DCM (200 mL), washed with water (300 mL x 2), brine, dried over Na2S04 and concentrated. The crude was purified by silica gel chromatography eluting with a gradient of 10%> - 50%>EtOAc/hexanes to afford tert-butyl (4-fluorophenyl)(4-(methoxymethoxy)-3 -(thiophene-3 -carbonyl)phenyl)carbamate (11.8 g, 25.8 mol, 91% yield) as yellow solid.
Step F: (Z)-tert-B\\X \
(3-(((tert-butylsulfinyl)imino)(thiophen-3-yl)methyl)-4-(methoxymethoxy)phenyl)(4-fluorophenyl) carbamate was prepared in 56% yield according to the Example 1, Step C substituting
(6-bromopyridin-2-yl)(thiophen-3-yl)methanone for tert-butyl
(4-fluorophenyl)(4-(methoxymethoxy)-3-(thiophene-3-carbonyl)phenyl)carbamate.
Step G: Methyl
5-(5-((iert-butoxycarbonyl)(4-fluorophenyl)amino)-2-(methoxymethoxy)phenyl)-5-( 1 , 1 -dimethylet hylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 78% yield according to the Example 1, Step D substituting
N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(Z)-tert-butyl
(3-(((tert-butylsulfinyl)imino)(thiophen-3-yl)methyl)-4-(methoxymethoxy)phenyl)(4-fluorophenyl) carbamate.
Ste H: Methyl
5-amino-5-(5-((tert-butoxycarbonyl)(4-fluorophenyl)amino)-2-(methoxymethoxy)phenyl)-3-oxo-5- (thiophen-3-yl)pentanoate was prepared in 86% yield according to the Example 1 , Step E
substituting methyl methyl
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-(5-((tert-butoxycarbonyl)(4-fluorophenyl)amino)-2-(methoxymethoxy)phenyl)-5-( 1 , 1 -dimethylet hylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step I: tert-Butyl
(3-(4,6-dioxo-2-(thiophen-3-yl)piperidin-2-yl)-4-(methoxymethoxy)phenyl)(4-fluorophenyl)carbam ate was prepared in 93% yield according to the Example 1 , Step F substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-amino-5-(5-((tert-butoxycarbonyl)(4-fluorophenyl)amino)-2-(methoxymethoxy)phenyl)-3-oxo-5- (thiophen-3 -yl)pentanoate.
Step J: tert-Butyl
(3-(5-((2-chlorophenyl)thio)-4,6-dioxo-2-(thiophen-3-yl)piperidin-2-yl)-4-(methoxymethoxy)pheny l)(4-fluorophenyl)carbamate was prepared in 70% yield according to the Example 1 , Step G
substituting 6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
tert-butyl
(3-(4,6-dioxo-2-(thiophen-3-yl)piperidin-2-yl)-4-(methoxymethoxy)phenyl)(4-fluorophenyl)carbam ate.
Step K: To a stirred solution of iert-butyl
(3-(5-((2-chlorophenyl)thio)-4,6-dioxo-2-(thiophen-3-yl)piperidin-2-yl)-4-(methoxymethoxy)phenyl)(4-flu orophenyl)carbamate (600 mg, 0.88 mmol) in methanol (10 mL) was added HCl-MeOH (10 mL) in an ice bath. The reaction was stirred at room temperature for 1 hour. The mixture was neutralized by addition of 1 N NaOH. Then the mixture was extracted with EtOAc and water. The organic layer was dried over anhydrous NaS04 and concentrated. The crude was purified by prep-HPLC (formic acid) to afford 3-((2-chlorophenyl)thio)-6-(5-((4-fluorophenyl)amino)-2-hydroxyphenyl)-6-(thiophen-3-yl)piperidine-2,4- dione (150 mg, 0.28 mmol, 32% yield) as white solid. lH NMR (400MHz, CD3OD) δ 7.41 (dd, J= 5.2, 5.2, 1H), 7.27 - 7.26 (m, 2H), 7.13 - 7.10 (m, 2H), 6.96 - 6.81 (m, 8 H), 6.25 (dd, J= 8.0, 1.6 Hz, 1H), 4.79 - 4.73 (m, 1H), 3.79 (d, J = 17.2 Hz, 1H), 3.43 (d, J = 16.8 Hz, 1H). LCMS M+l = 538.8.
Example 15
3-(2-chlorophenyl)sulfanyl-6-(lH-indol-4-yl)-6-(3-thienyl)piperidine-2,4-dione
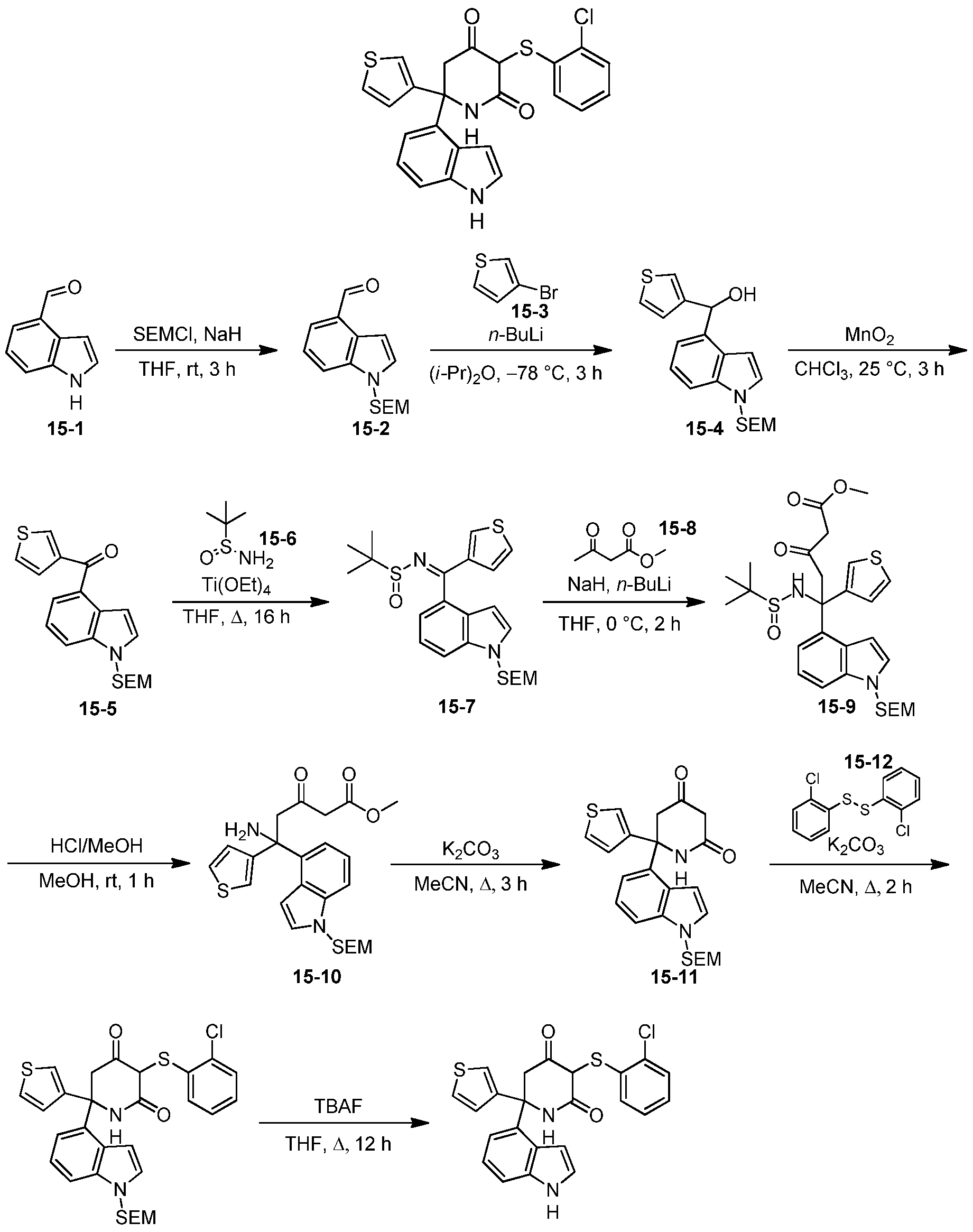
15-13 15
Step A: lH-Indole-4-carbaldehyde (10 g, 69.0 mmol) was added to a suspension of NaH (2.0 g,
82.6 mmol) in anhydrous THF (150 mL) at 0 °C. The resultant suspension was stirred at 0 °C for 30 minutes, followed by addition of 2-(trimethylsilyl) ethoxymethyl chloride (13.8 g, 82.6 mmol). The reaction mixture was stirred at room temperature for 3 hours. The reaction was quenched with water,
dried over anhydrous Na2S04 and the filtrate was concentrated under vacuum. The crude residue was purified by silica gel chromatography eluting with a gradient of 10% - 30% EtOAc/hexanes to afford l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indole-4-carbaldehyde (817 g, 61.4 mmol, 89%> yield) as dark yellow solid.
Step B: Thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methanol was prepared in 68% yield according to the Example 2, Step A substituting 4-bromobenzaldehyde acid for l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indole-4-carbaldehyde.
Step C: Thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methanone was prepared in 94% yield according to the Example 7, Step B substituting
(4-bromophenyl)(thiophen-3 -yl)methanol for
thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methanol.
Step D:
(£')-2-Methyl-N-(thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methylene)prop ane-2-sulfinamide was prepared in 64% yield according to the Example 1, Step C substituting (6-bromopyridin-2-yl)(thiophen-3 -yl)methanone for
thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methanone.
Step E: Methyl
5-(l,l-dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)-5-(l-((2-(trimethylsilyl)ethoxy)methyl)- lH-indol-4-yl)pentanoate was prepared in 88% yield according to the Example 1 , Step D substituting N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(£')-2-methyl-N-(thiophen-3-yl(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)methylene)prop ane-2-sulfinamide.
Step F: Methyl
5-amino-3-oxo-5-(thiophen-3-yl)-5-(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)pentanoate was prepared in 65% yield according to the Example 1 , Step E substituting methyl methyl 5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- (l,l-dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)-5-(l-((2-(trimethylsilyl)ethoxy)methyl)- lH-indol-4-yl)pentanoate.
Step G:
6- (Thiophen-3-yl)-6-(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)piperidine-2,4-dione was prepared in 43% yield according to the Example 1, Step F substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-amino-3-oxo-5-(thiophen-3-yl)-5-(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)-pentanoat e.
Step H:
3-((2-Chlorophenyl)thio)-6-(thiophen-3-yl)-6-^
piperidine-2,4-dione was prepared in 59% yield according to the Example 1, Step G substituting 6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
6-(thiophen-3-yl)-6-(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)piperidine-2,4-dione.
Step I: To a stirred solution of
3-((2-chlorophenyl)thio)-6-(thiophen-3-yl)-6-(l-((2-(trimethylsilyl)ethoxy)methyl)-lH-indol-4-yl)p iperidine-2,4-dione (250 mg, 0.43 mmol) in THF (4 mL) was added TBAF (4 mL, 1M in THF). The reaction was heated at 80 °C for 12 hours. After cooling to room temperature, the reaction mixture was diluted with EtOAc (20 mL), washed with water and concentrated under vacuum. The crude was purified by preparative HPLC (formic acid) to afford
3-((2-chlorophenyl)thio)-6-(lH-indol-4-yl)-6-(thiophen-3-yl)piperidine-2,4-dione (24 mg, 0.05 mmol, 12% yield) as white solid. ¾ NMR (400MHz, CD3OD) δ 7.58 (dd, J= 8.0, 8.0 Hz, 1H), 7.43 - 7.41 (m, 2H), 7.27 - 7.19 (m, 3H), 7.05 (dd, J= 8.0, 8.0 Hz, 1H), 6.91 - 6.88 (m, 1H), 6.77 - 6.75 (m, 2H), 6.53 (d, J= 8.4 Hz, 1H), 6.08 (dd, J = 8.0, 4.0 Hz, 1H), 3.88 (d, J= 16.0 Hz, 1H), 3.50 (d, J = 16.0 Hz, 1H). LCMS M+l = 452.8.
Example 16
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(oxetan-3-yl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4
-dione

Step A: To a suspension of NaH (688 mg, 27.8 mmol) in THF (80 mL) was added diethyl malonate (7.45 g, 46.5 mmol) dropwise. Then ((2-bromoethoxy)methyl)benzene (5 g, 23.2 mmol) was added. The reaction was heated to 90 °C for 5 hours. After cooling to room temperature, the mixture was diluted with EtOAc (50 mL), washed with water (50 mL x 2), dried over anhydrous Na2S04 and concentrated under vacuum. The crude residue was purified by silica gel
chromatography eluting with a gradient of 10% - 30% EtOAc/hexanes to to afford diethyl 2-(2-(benzyloxy)ethyl)malonate (6.6 g, 22.5 mmol, 81% yield) as a colorless oil.
Step B: To a suspension of LiAlH4 (1.71 g, 45.0 mmol) in anhydrous THF (80 mL) was added diethyl 2-(2-(benzyloxy)ethyl)malonate (6.6 g, 22.5 mmol) dropwise in an ice bath. The reaction was warmed to room temperature and stirred for 12 hours. The reaction was quenched with water, diluted with EtOAc (50 mL), washed with water (50 mL x 2), dried over anhydrous Na2S04 and concentrated under vacuum. The crude residue was purified by silica gel chromatography eluting
with a gradient of 10% - 30% EtOAc/hexanes to afford 2-(2-(benzyloxy)ethyl)propane-l,3-diol (2.2 g, 10.6 mmol, 47% yield) as a colorless oil.
Step C: To solution of 2-(2-(benzyloxy)ethyl)propane-l,3-diol (2.2 g, 10.6 mmo) in THF (20 mL) was added n-BuLi (4.2 mL, 10.6 mmol) in an ice bath. The mixture was stirred at 0 °C for 30 minutes, then TsCl (404 mg, 2.12 mmol) was added. The reaction mixture was stirred at 0 °C for 1 hour and then n-BuLi (4.2 mL, 10.6 mmol) was added. The reaction mixture was stirred at 60 °C for 6 hours, then cooled to room temperature. The mixture was diluted with EtOAc (30 mL), washed with water (50 mL x 2), dried over anhydrous Na2S04 and concentrated under vacuum. The crude residue was purified by silica gel chromatography eluting with a gradient of 0% - 15% EtOAc/hexanes to afford 3-(2-(benzyloxy)ethyl)oxetane (550 mg, 2.86 mmol, 27% yield) as a colorless oil.
Step D: A mixture of 3-(2-(benzyloxy)ethyl)oxetane (550 mg, 2.86 mmol), Pd/C (350 mg) and ethanol (5 mL) was stirred at room temperature under hydrogen atmosphere for 2 days. The mixture was filtered and the filtrate was concentrate to afford 2-(oxetan-3-yl)ethanol (200 mg, 1.96 mmol, 66% yield) as a colorless oil.
Step E:
5-((2-Chlorophenyl)thio)-4-hydroxy-6'-(2-(oxetan-3-yl)ethoxy)-2-(thiophen-3-yl)-2,3-dihydro-[2,2'- bipyridin]-6(lH)-one was prepared in 35% yield according to the Example 2, Step A substituting propan-2-ol for 2-(oxetan-3-yl)ethanol. lB NMR (400MHz, CD3OD) δ 7.70 (dd, J= 8.0, 8.0 Hz, 1H), 7.40 (dd, J= 2.8, 2.8 Hz, 1H), 7.27 (d, J= 2.8 Hz, 1H), 7.17 - 7.14 (m, 3H), 6.88 (dd, J= 8.0, 8.0 Hz, 1H), 6.75 - 6.73 (m, 2H), 6.00 (dd, J= 9.6, 1.6 Hz, 1H), 4.38 - 4.28 (m, 2H), 3.83 - 3.69 (m, 5 H), 3.43 - 3.41 (m, 1H), 2.71 - 2.67 (m, 1H), 2.06 - 2.02 (m, 1H), 1.75 - 1.71 (m, 1H). LCMS M+l = 514.9.
Example 17
3-(2-chlorophenyl)sulfanyl-6- [6- [ [3-(hydroxymethyl)phenyl] methyl] -2-pyridyl] -6-(3-thienyl)p iperidine-2,4-dione


17
Step A: To a stirred solution of methyl 3-(bromomethyl)benzoate (5 g, 21.8 mmol) in toluene (50 mL) was added DABAL-H (43.6 ml, 43.6 mmol) in an ice bath. The reaction was stirred at 0 °C for 2 hours. The mixture was quenched with 1 N HCl, extracted with EtOAc and water. The organic layer was dried over anhydrous Na2S04 and concentrated to afford (3-(bromomethyl)phenyl)methanol (4.0 g, 19.9 mmol, 91% yield) as a colorless oil.
Step B: A mixture of (3-(bromomethyl)phenyl)methanol (2.0 g, 10.0 mmol), 2,6-lutidine (2.13 g, 19.9 mmol), tert-butyl dimethylsilyl trifluoromethanosulfonate (3.1 g, 14.9 mmol) and DCM (30 mL) was stirred at room temperature for 2 hours. The reaction was quenched with water (20 mL), extracted with DCM. The organic layer was dried over anhydrous Na2S04 and concentrated. The crude residue was purified by chromatography on silica gel (petroleum ether / EtOAc = 20/1) to afford (3-(bromomethyl)phenyl)methanol (2.8 g, 8.9 mmol, 89% yield) as a colorless oil.
Step C: To a mixture of zinc powder (408 mg, 6.3 mol) in anhydrous THF (30 mL) was added 1 ,2-dibromoethane (107 mg, 0.57 mmol) and
((3-(bromomethyl)benzyl)oxy)(tert-butyl)dimethylsilane (1.8 g. 5.7 mmol) under nitrogen atmosphere. The mixture was stirred at room temperature for 8 hours. The reaction solution was used in next step directly.
Step D: To a stirred solution of
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one (example 1 , 300 mg, 0.61 mmol) and Pd(PPh3)4 (69 mg, 0.06 mmol) in THF (1 mL) was added
(3-(((tert-butyldimethylsilyl)oxy)methyl)benzyl)zinc(II) bromide (5.3 mL, 3.04 mmol) . The mixture was stirred at room temperature for 12 hours. The reaction was quenched with water, then filtered over Celite. The filtrate was concentrated under vacuum and the crude residue was purified by preparative HPLC (formic acid) to afford
6'-(3-(((tert-butyldimethylsilyl)oxy)methyl)benzyl)-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophe n-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one (80 mg, 0.12 mmol, 20% yield) as white solid.
Step E: To a stirred solution of
6'-(3-(((tert-butyldimethylsilyl)oxy)methyl)benzyl)-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophe n-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one (80 mg, 0.12 mmol) in MeOH (5 mL) was added HCl-MeOH (5 mL) in an ice bath. The mixture was stirred at 0 °C for 1 hour. The reaction was added water, then filtered and washed with water. The solid was dried to afford
5-((2-chlorophenyl)thio)-4-hydroxy-6'-(3-(hydroxymethyl)benzyl)-2-(thiophen-3-yl)-2,3-dihydro-[2 ,2'-bipyridin]-6(lH)-one (50 mg, 0.09 mmol, 76% yield) as a white solid. Mixture of
diastereoisomers: 'H NMR (400MHZ, CD3OD) δ 7.74 (dd, J= 8.0, 8.0 Hz, 1H), 7.42 - 7.40 (m, 2H), 7.22 - 7.17 (m, 6 H), 7.11 - 7.09 (m, 2H), 6.85 (dd, J= 8.0, 8.0 Hz, 1H), 6.51 (dd, J= 8.0, 8.0 Hz, 1H), 5.78 (dd, J= 8.4, 1.6 Hz, 1H), 4.47 (s, 2H), 4.17 (s, 2H), 4.00 (d, J= 16.4 Hz, 1H), 3.47 (d, J= 16.4 Hz, 1H). LCMS M+l = 534.9. Stereoisomer 1 : lH NMR (400MHz, CD3OD) δ 7.75 (dd, J= 8.0, 8.0 Hz, 1H), 7.44 - 7.42 (m, 2H), 7.31 - 7.14 (m, 8 H), 6.85 (dd, J= 8.0, 8.0 Hz, 1H), 6.56 (dd, J= 8.0, 8.0 Hz, 1H), 5.87 (d, J= 8.0 Ηζ,ΙΗ), 4.51 (s, 2H), 4.20 (s, 2H), 3.92 (d, J= 16.4 Hz, 1H), 3.49 (d, J= 16.4 Hz, 1H). Stereoisomer 2: lH NMR (400MHz, CD3OD) δ 7.74 (dd, J= 8.0, 8.0 Hz, 1H), 7.44 - 7.41 (m, 2H), 7.16 - 7.13 (m, 8 H), 6.87 (dd, J= 8.0, 8.0 Hz, 1H), 6.56 (dd, J= 8.0, 8.0 Hz, 1H), 5.87 (d, J = 8.0 Hz, 1H), 4.51 (s, 2H), 4.20 (s, 2H), 3.91 (d, J= 16.4 Hz, 1H), 3.49 (d, J= 16.0 Hz, 1H).
Example 18
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(lH-pyrazol-4-yl)phenoxy]-2-pyridyl]-6-(3-thienyl)piperid ine-2,4-dione

H


H
Step A: To a stirred suspension of 2-(2-hydroxyethyl)phenol (5 g, 36.2 mmol) and CS2CO3 (38.9 g, 108.7 mmol) in acetone (100 mL) was added iodomethane (6.2 g, 43.4 mmol) in an ice bath. The reaction mixture was stirred at 0 °C for 50 minutes. The mixture was filtered, the filtrate was concentrated under vacuum. The crude materials was extracted with EtOAc and water. The organic layer was dried over anhydrous Na2S04 and concentrated to afford 2-(2-methoxyphenyl)ethanol (4.5 g, 29.6 mmol, 82% yield) as a yellow solid.
Step B: To a stirred solution of 2-(2-methoxyphenyl)ethanol (4.5 g, 29.6 mmol) in DCM (80 mL) was added Dess-Martin reagent (51.1 g, 35.5 mmol) in an ice bath. The reaction mixture was stirred at 0 °C for 1 hour. The mixture was diluted with DCM (100 mL), washed with saturated NaHC03 (100 mL x 2), brine, dried over anhydrous Na2S04 and concentrated. The crude residue was purified by silica gel chromatography eluting with a gradient of 10% - 50% EtOAc/hexanes to afford 2-(2-methoxyphenyl)acetaldehyde (2.5 g, 16.7 mmol, 56% yield) as yellow oil.
Step C: A mixture of 2-(2-methoxyphenyl)acetaldehyde (2.5 g, 16.7 mmol) and
1 , 1 -dimethoxy-N,N-dimethylmethanamine (5 mL) was stirred at room temperature for 5 hours. The mixture was diluted with DCM (30 mL), washed with saturated NaHCOs (20 mL x 2), brine, dried over anhydrous Na2S04 and concentrated. The crude residue was purified by silica gel
chromatography eluting with a gradient of 10% - 50% EtOAc/hexanes to afford (£')-3-(dimethylamino)-2-(2-methoxyphenyl)acrylaldehyde (350 mg, 1.7 mmol, 12% yield) as yellow solid.
Step D: A mixture of (£')-3-(dimethylamino)-2-(2-methoxyphenyl)acrylaldehyde (350 mg, 1.7 mmol,), hydrazine hydrate (2 mL) and ethanol (5 mL) was heated to 80 °C for 30 minutes. The mixture was diluted with DCM 10 mL), washed with saturated NaHC03 (10 mL x 2), brine, dried over anhydrous Na2S04 and concentrated to afford 4-(2-methoxyphenyl)-lH-pyrazole (260 mg, 1.5 mmol, 88%) yield) as yellow solid.
Step E: To a stirred solution of 4-(2-methoxyphenyl)-lH-pyrazole (260 mg, 1.5 mmol) in DCM (5 mL) was added boron tribromide (750 mg, 3.0 mmol) in an ice bath. The reaction mixture was stirred at 0 °C for 12 hours. The mixture was diluted with DCM (20 mL), washed with saturated NaHC03 (20 mL x 2), brine, dried over anhydrous Na2S04 and concentrated to afford
2-(lH-pyrazol-4-yl)phenol (200 mg, 1.25 mmol, 84%> yield) as yellow oil.
Step F:
6'-(2-(lH-Pyrazol-4-yl)phenoxy)-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro -[2,2'-bipyridin]-6(lH)-one was prepared in 3%> yield according to the Example 3, Step A
2-Chloro-4-fluoro-phenol for 2-(lH-pyrazol-4-yl)phenol. lB NMR (400MHz, (CD3)2SO) δ 11.75 (s, 1H), 9.92 (s, 1H), 9.08 (s, 1H), 9.64 (s, 1H), 8.30 (s, 1H), 8.07 (dd, J= 8.0, 8.0 Ηζ,ΙΗ), 7.90 (d, J= 8.4 Ηζ,ΙΗ), 7.59 - 7.53 (m, 3H), 7.40 (s, 1H), 7.23 - 7.21 (m, 2H), 7.08 (dd, J= 8.0, 4.0 Hz, 1H), 6.93 - 6.84 (m, 3H), 6.60 (dd, J= 8.0, 8.0 Hz, 1H), 5.85 (d, J= 8.4 Hz, 1H), 4.04 (d, J= 16.0 Hz, 1H), 3.43 (d, J= 16.4 Hz, 1H). LCMS M+l = 573.1.
Example 19
3-(2-chlorophenyl)sulfanyl-6-(5-chloro-3-thienyl)-6-[6-(4-fluorophenoxy)-2-pyridyl]piperidin
-2,4-dione


Step A: To a solution of thiophene-3-carbaldehyde (20.0 g, 178.3 mmol) and
N-chlorosuccinimide (23.8 g, 178.3 mmol) in AcOH (180 mL) was stirred at 110 °C for 4 hours. After the completion of reaction, the solution was cooled to room temperature, and then was diluted with EtOAc (120 mL), washed with H20 (100 mL x 3), saturated NaHC03 (50 mL x 2), brine, dried over anhydrous Na2S04 and concentrated to afford 5-chlorothiophene-3-carboxylic acid (8.0 g, 54.6 mmol, 31% yield) as yellow solid, which was used directly in the next step without further purification.
Step B: (5-Chlorothiophen-3-yl)(6-(4-fluorophenoxy)pyridin-2-yl)methanol was prepared in 50% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for
5-chlorothiophene-3-carbaldehyde and 3-bromothiophene for 2-bromo-6-(4-fluorophenoxy) pyridine
Step C: (5-Chlorothiophen-3-yl)(6-(4-fluorophenoxy)pyridin-2-yl)methanone was prepared in 79%) yield according to the Example 7, Step B substituting
(4-bromophenyl)(thiophen-3-yl)methanone for (5-chlorothiophen-3-yl)(6-(4-fluorophenoxy) pyridine -2 -yl)methanone.
Step D:
(£)-N-((5-Chlorothiophen-3-yl)(6-(4-fluorophenoxy)pyridin-2-yl)methylene)-2-methylpropane-2-s ulfmamide was prepared in 74 % yield according to the Example 7, Step C substituting
(£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(£')-N-((5-chlorothiophen-3-yl)(6-(4-fluorophenoxy)pyridin-2-yl)methylene)-2-methylpropane-2-su lfinamide
Step E: Methyl 5-(5-chlorothiophen-3-yl)-5-(l ,l -dimethylethylsulfinamido)-5-(6-(4-fluoro phenoxy)pyridin-2-yl)-3-oxopentanoate was prepared in 86 % yield according to the Example 7, Step D substituting methyl
5-(4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl 5-(5-chlorothiophen-3-yl)-5-(l , 1 -dimethylethylsulfinamido)-5-(6-(4-fluoro
phenoxy)pyridin-2-yl)-3-oxopentanoate
Step F: Methyl
5-amino-5-(5-chlorothiophen-3-yl)-5-(6-(4-fluorophenoxy)pyridin-2-yl)-3-oxopentanoate was prepared in 49 % yield according to the Example 7, Step E substituting methyl
5-amino-5-(4-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- amino-5-(5-chlorothiophen-3-yl)-5-(6-(4-fluorophenoxy)pyridin-2-yl)-3-oxopentanoate
Step G: 6-(5-Chlorothiophen-3-yl)-6-(6-(4-fluorophenoxy)pyridin-2-yl)piperidine-2,4-dione was prepared in 57% yield according to the Example 7, Step F substituting
6- (4-bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one for
6-(5-chlorothiophen-3-yl)-6-(6-(4-fluorophenoxy)pyridin-2-yl)piperidine-2,4-dione.
Step Η:
5- ((2-Chlorophenyl)thio)-2-(5-chlorothiophen-3-yl)-6'-(4-fluorophenoxy)-4-hydroxy-2,3-dihydro-[ 2,2'-bipyridin]-6(lH)-one was prepared in 5.4 % yield according to the Example 7, Step G substituting
6- (4-bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH )-one for
5-((2-chlorophenyl)thio)-2-(5-chlorothiophen-3-yl)-6'-(4-fluorophenoxy)-4-hydroxy-2,3-dihydro-[2 ,2'-bipyridin]-6(lH)-one. Mixture of diastereoisomers: ¾ NMR (400MHz, CD3OD) δ 7.89 (dd, J = 7.6, 7.6 Hz, 1H), 7.31 (d, J= 8.0 Hz, 1H), 7.21 (d, J= 7.8 Hz, 1H), 7.12 - 7.08 (m, 4H), 7.01 - 6.89 (m, 3H), 6.98 - 6.89 (m, 2H), 6.07 (dd, J = 8.0, 1.2 Hz, 1H), 3.54 (d, J = 16.0 Hz, 1H), 3.25 (d, J = 16.0 Hz, 1H). LCMS M+l = 558.9. Stereoisomer 1 : lH NMR (400MHz, CD3OD) δ 7.89 (dd, J = 7.6, 7.6 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 7.10 - 7.07 (m, 4H), 7.01 - 6.89 (m, 3H), 6.89 - 6.81 (m, 2H), 6.07 (d, J = 6.8 Hz, 1H), 3.55 (d, J = 16.0 Hz, 1H), 3.28 (d, J = 16.0 Hz, 1H).
Stereoisomer 2: lH NMR (400MHz, CD3OD) δ 7.89 (dd, J= 7.6, 7.6 Hz, 1H), 7.33 (d, J= 8.0 Hz, 1H), 7.12 (d, J= 7.8 Hz, 1H), 7.10 - 7.08 (m, 4H), 7.01 - 6.99 (m, 3H), 6.89 - 6.81 (m, 2H), 6.08 (d, J= 6.8 Hz, 1H), 3.54 (d, J = 16.0 Hz, 1H), 3.28 (d, J= 16.0 Hz, 1H).
Example 20
3-(2-chlorophenyl)sulfanyl-6- [6- [2-(2,2-difluorocyclo ropyl)ethoxy] -2-pyridyl] -6-(3-thienyl)p iperidine-2,4-dione

20
Step A: Benzyl chloride (2.0 g, 14.2 mmol) was added dropwise to a solution of but-3-en-l-ol (1.2 g, 17.1 mmol) and Et3N (2.9 g, 28.5 mmol) at 0 °C in DCM (35 mL) The reaction mixture was
then warmed to ambient temperature and stirred for 3 hours. The reaction mixture was quenched with saturated aqueous NH4C1 (10 mL). The organic layer was washed with saturated NaHC03 solution (5 mL x 2), brine, dried over anhydrous Na2S04 and concentrated to afford crude product which was purified by silica gel chromatography eluting with 20% EtOAc/hexanes to afford but-3-enyl benzoate (2.3 g, 13.1 mmol, 91% yield) as yellow oil.
Step B: A mixture of but-3-enyl benzoate (500 mg, 2.8 mmol), trimethylsilyl
2,2-difluoro-2-(fluorosulfonyl)acetate (1.4 g, 5.7 mmol) andNaF (5.9 mg, 141.8 μηιτηοΐ) was heated under neat conditions at 110 °C for 2 hours. After cooling to room temperature, DCM (10 mL) and H20 (5 mL) were added, separated. The DCM extract was concentrated. The crude residue was purified by silica gel chromatography eluting with 10%> EtOAc/hexanes to afford
2-(2,2-difluorocyclopropyl)ethyl benzoate (330 mg, 1.5 mmol, 51%> yield) as yellow oil.
Step C: To a suspension of potassium hydroxide (409 mg, 7.3 mmol) in MeOH/H20 (3 :2, 5 mL) was added 2-(2,2-difluorocyclopropyl)ethyl benzoate (330 mg, 1.5 mmol) at 0 ° C, followed by stirring at room temperature for 1 hour. The reaction was quenched with saturated brine solution (5 mL), and extracted with EtOAc (10 mL x 4). The combined organic layer was dried over anhydrous Na2S04 and concentrated under vacuum to afford crude 2-(2,2-difluorocyclopropyl)ethanol (150 mg, 84%>) as colorless oil which was used directly in the next step.
Step D:
5-((2-Chlorophenyl)thio)-6'-(2-(2,2-difluorocyclopropyl)ethoxy)-4-hydroxy-2-(thiophen-3-yl)-2,3-d ihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 21%> yield according to the Example 2, Step A substituting propan-2-ol for 2-(2,2-difluorocyclopropyl)ethanol. lH NMR (400MHz, CD3OD) δ 7.73 (dd, J = 7.6, 7.6 Hz, 1H), 7.43 (d, J= 2.8 Hz, 1H), 7.27 - 7.14 (m, 4H), 7.14 (d, J= 2.8 Hz, 1H), 6.93 - 6.74 (m, 2H), 5.96 (d, J= 8.0 Hz, 1H), 4.43 (t, J= 3.6 Hz, 2H), 3.88 (d, J = 16.0 Hz, 1H), 3.47 (d, J= 16.0 Hz, 1H), 1.95 - 1.67 (m, 3H), 1.36 - 1.34 (m, 1H), 1.00 - 0.96 (m, 1H). LCMS M+l = 534.9.
Example 21
3-(2-chlorophenyl)sulfanyl-6- [6- [2-(3-methyltriazol-4-yl)phenoxy] -2-pyridyl] -6-(3-thienyl)pip
-2,4-dione

21
Step A: A solution of lH-l,2,3-triazole (1.0 g, 14.5 mmol), methyl iodide (3.1 g, 21.7 mmol) and K2CO3 (4.0 g, 28.9 mmol) in THF (15 mL) was stirred at room temperature for 3 hours. EtOAc (20 mL) and H20 (10 mL) were added, separated. The solvent was concentrated under vacuum. The crude residue was purified by silica gel chromatography eluting with 10% MeOH/DCM to afford l-methyl-lH-l,2,3-triazole (860 mg, 10.4 mmol, 71% yield) as yellow oil.
Step B: To a solution of l-methyl-lH-l,2,3-triazole (860 mg, 10.4 mmol) in THF (10 mL) at -78 °C, was added dropwise n-BuLi (5.0 mL, 12.4 mmol, 2.5 M). The mixture was stirred at -78 °C for 2 hours before addition of BusSnCl (3.7 g, 11.4 mmol). The mixture was stirred at -78 °C for 1 hour and then room temperature for 1 hour. The mixture was concentrated under vacuum and hexane was added. The insoluble material was filtered and the filtrate was concentrated under vacuum to afford l-methyl-5-(tributylstannyl)-lH-l,2,3-triazole (3.1 g, 80 %) as yellow oil which was used directly in the next step.
Step C: A solution of l-methyl-5-(tributylstannyl)-lH-l,2,3-triazole (3.1 g, 8.3 mmol), 2-bromophenol (1.7 g, 10.0 mmol), Et3N (1.7 g, 16.7 mmol) and PdCl2(PPh3)2 (l .l g, 1.7 mmol) in PhMe (16 mL) was stirred at 110 °C for 14 hours. After cooling to room temperature, DCM (25 mL) and H20 (10 mL) were added, separated. The DCM was concentrated under vacuum. The crude residue was purified by silica gel chromatography eluting with a gradient of 20% EtOAc/hexanes to 10% MeOH/DCM to afford 2-(l-methyl-lH-l,2,3-triazol-5-yl)phenol (110 mg, 8.0 %yield) as white solid.
Step D:
5-((2-Chlorophenyl)thio)-4-hydroxy-6'-(2-(l-methyl-lH-l,2,3-triazol-5-yl)phenoxy)-2-(thiophen-3- yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 8.3%> yield according to the Example 3, Step A substituting 2-chloro-4-fluoro-phenol for 2-(l-methyl-lH-l,2,3-triazol-5-yl)phenol. ¾ NMR
(400MHz, CD3OD) δ 7.80 (dd, J= 7.6, 7.6 Hz, IH), 7.47 (d, J= 2.8 Hz, IH), 7.41 - 7.39 (m, 4H), 7.30 (d, J= 2.8 Hz, IH), 7.21 (d, J= 7.6 Hz, 2H), 7.08 (s, IH), 6.94 - 6.92 (m, 3H), 6.73 (dd, J= 7.2, 7.2 Hz, IH), 5.94 (d, J= 8.0 Hz, IH), 3.87 (s, 3H), 3.50 (d, J= 16.0 Hz, IH), 3.31 (d, J= 16.0 Hz, IH). LCMS M+l = 587.8.
Example 22
3-(2-chlorophenyl)sulfanyl-6-(3-tetrahydropyran-4-yloxyphenyl)-6-(3-thienyl)piperidine-2,4-

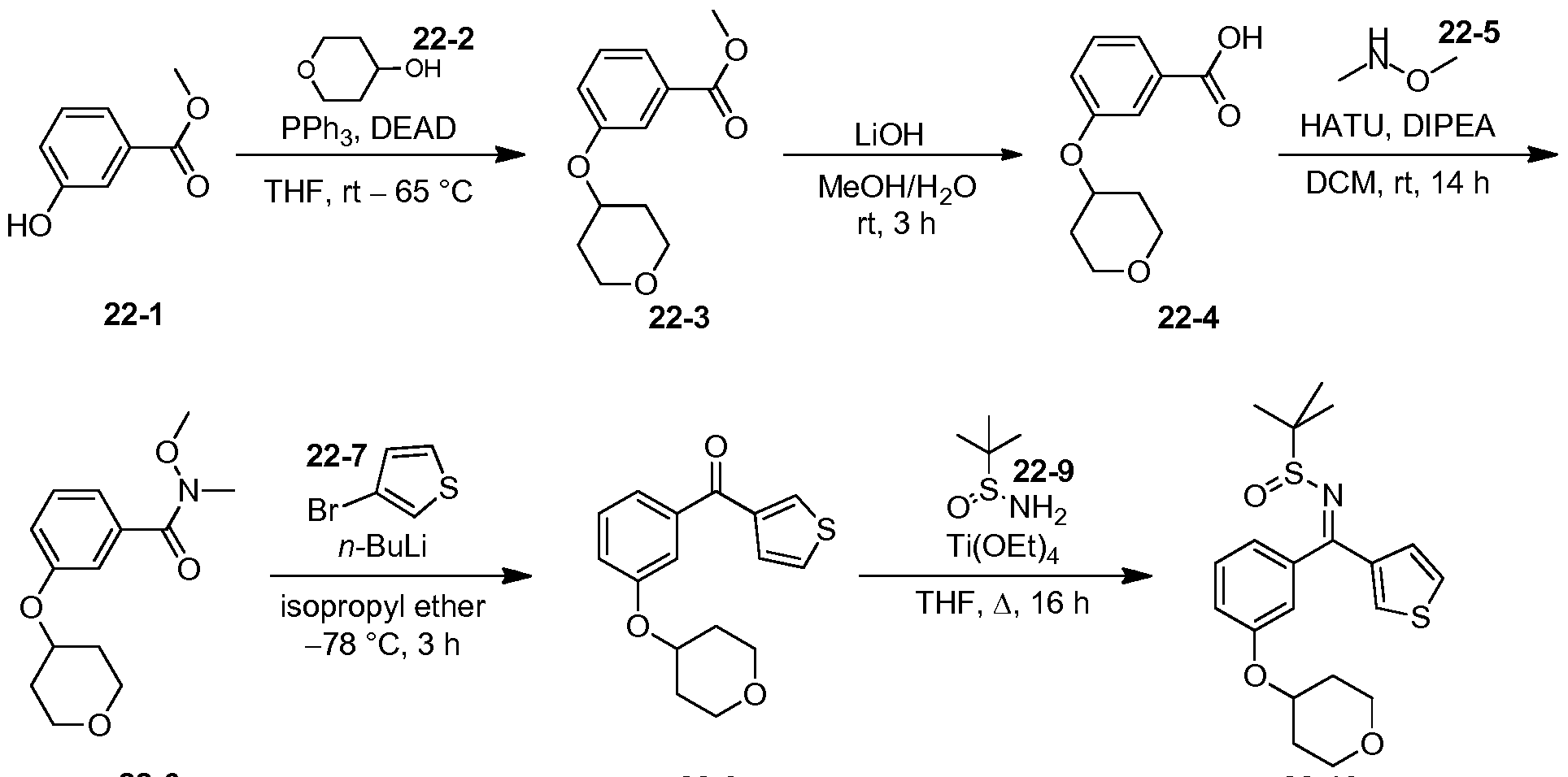

Step A: The suspension of methyl 3-hydroxybenzoate (22.0 g, 144.6 mmol),
tetrahydro-2H-pyran-4-ol (22.2 g, 216.9 mmol), PPh3 (3.8 g, 14.5 mmol) and DEAD (28.0 g, 159.1 mmol) in THF (150 ml) was refluxed for 8 hours. The reaction mixture was then cooled to room temperature, diluted with water (60 ml) and EtOAc (120 mL). The organic layer was separated and concentrated. The crude residue was purified by column chromatography on silica gel with petroleum ether:EtOAc = 3: 1 as eluent to afford methyl 3-(tetrahydro-2H-pyran-4-yloxy)benzoate (18.1 g, 53% yield) as brown oil.
Step B: A solution of methyl 3-(tetrahydro-2H-pyran-4-yloxy)benzoate (18.1 g, 76.6 mmol) and LiOH (9.2 g, 383 mmol) in methanol/H20 (80 mL/5 ml) was stirred at room temperature for 3 hours. The reaction mixture was filtered and the filtrate was adjusted to pH= 2-3 with aqueous HCl solution (1 M). The resultant solution was extracted with EtOAc (80 mL x 2), and concentaretd. The crude residue 3-(tetrahydro-2H-pyran-4-yloxy)benzoic acid (14.3 g, 84% yield) as yellow solid was used directly in the next step without further purification.
Step C: N-Methoxy-N-methyl-3-(tetrahydro-2H-pyran-4-yloxy)benzamide was prepared in 85%o yield according to the Example 1, Step A substituting 6-bromopicolinic acid for
3 -((tetrahydro-2H-pyran-4-yl)oxy)benzoic acid.
Step D: (3-(Tetrahydro-2H-pyran-4-yloxy)phenyl)(thiophen-3-yl)methanone was prepared in 67%o yield according to the Example 1 , Step B substituting
6-bromo-N-methoxy-N-methylpicolinamide for
(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)(thiophen-3-yl)methanone.
Step E:
(£')-2-Methyl-N-((3-(tetrahydro-2H-pyran-4-yloxy)phenyl)(thiophen-3-yl)methylene)propane-2-sul finamide was prepared in 63% yield according to the Example 1, Step C substituting
(Z)-N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for (£')-2-methyl-N-((3-(tetrahydro-2H-pyran-4-yloxy)phenyl)(thiophen-3-yl)methylene)propane-2-sulf inamide.
Step F:
Methyl-5-(l,l-dimethylethylsulfinamido)-3-oxo-5-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-5-(thi ophen-3-yl)pentanoate was prepared in 58% yield according to the Example 1, Step D substituting methyl
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl5-(l,l-dimethylethylsulfinamido)-3-oxo-5-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-5-(thio phen-3-yl)pentanoate.
Step G:
Methyl5-amino-3-oxo-5-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-5-(thiophen-3-yl)pentanoate was prepared in 74% yield according to the Example 1 , Step E substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for
methyl-5-amino-3-oxo-5-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-5-(thiophen-3-yl)pentanoate Step Η:
4-Hydroxy-6-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH) -one was prepared in 87% yield according to the Example 1, Step F substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
4-hydroxy-6-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH) -one
Step I:
3-(2-Chlorophenylthio)-4-hydroxy-6-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-6-(thiophen-3-yl)-5, 6-dihydropyridin-2(lH)-one was prepared in 5.0% yield according to the Example 1, Step G substituting
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one for
3-(2-chlorophenylthio)-4-hydroxy-6-(3-(tetrahydro-2H-pyran-4-yloxy)phenyl)-6-(thiophen-3-yl)-5, 6-dihydropyridin-2(lH)-one. lH NMR (400MHz, CD3OD) δ 7.48 (dd, J= 8.0, 8.0 Hz, 1H), 7.32 (dd, J= 2.0, 2.0 Hz, 1H), 7.22 (d, J= 8.0 Hz, 1H), 7.21 (d, J=8.0 Hz, 1H), 7.18 (d, J= 8.0 Hz, 1H), 7.16 - 7.14 (m, 2H), 7.15 - 7.03 (m, 2H), 6.69 (d, J= 3.2Hz, 1H), 5.92 (dd, J= 7.6, 2.4Hz, 1H), 4.54 - 4.50 (m, 1H), 3.89 (t, J= 5.6 Hz, 2H), 3.56 (d, J= 16.0 Hz, 1H), 3.54 (t, J= 5.6 Hz, 2H), 3.51 (d, J= 16.0 Hz, 1H), 1.98 - 1.92 (m, 2H), 1.70 -1.60 (m, 2H). LCMS M+l = 513.9.
Example 23
3-(2-chlorophenyl)sulfanyl-l-methyl-6-(3-tetrahydropyran-4-yloxyphenyl)-6-(3-thienyl)piper
Step A: To a suspension of NaH (60% weight, 47 mg, 1.2 mmol) in anhydrous THF (5 mL) was added dropwise methyl iodide (166 mg, 1.2 mmol) at 0 °C under nitrogen atmosphere and then the reaction was stirred for 30 minutes. The compound of example 22 (200 mg, 389 μιηοΐ,) in THF (3 mL) was added dropwise to the reaction mixture and the reaction was stirred at 0 °C for 1 hour followed by stirring at room temperature for another 1 hour. The reaction was quenched by HCl solution (1 M), and separated. The solvent was removed. The crude residue was purified by preparative HPLC (formic acid) to give the desired product (5.5 mg, 3%> yield) as white solid. lH NMR (400MHz, CD3OD) δ 7.58 (dd, J= 8.0, 8.0 Hz, 1H), 7.57 (dd, J= 2.0, 2.0 Hz, 1H), 7.37 (d, J= 8.0 Hz, 1H), 7.23 (d, J= 8.0 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 7.16 (d, J= 7.2Hz, 1H), 7.15 - 7.02(m, 2H), 6.97 - 6.79 (m, 2H), 6.10 (dd, J= 8.0, 1.2Hz, 1H), 4.51 - 4.49 (m, 1H), 3.90 (t, J= 5.6 Hz, 2H), 3.66 (d, J= 16.0 Hz, 1H), 3.55 (t, J= 5.6 Hz, 2H), 3.51 (d, J= 16.0 Hz, 1H), 2.84 (s, 3H), 1.96 - 1.92 (m, 2H), 1.69 - 1.63 (m, 2H). LCMS M+l = 527.9.
Example 24
3-(2-chlorophenyl)sulfanyl-6-[3-(4-fluorophenoxy)phenyl]-6-(3-thienyl)piperidine one

Step A:
(3-Bromophenyl)(thiophen-3-yl)methanol was prepared in 95% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for 3-bromobenzaldehyde.
Step B:
(3-Bromophenyl)(thiophen-3-yl)methanone was prepared in 95% yield according to the Example 7 Step B substituting (4-bromophenyl)(thiophen-3-yl)methanol for
(3 -bromophenyl)(thiophen-3 -yl)methanol.
Step C:
(E)-N-((3 -Bromophenyl)(thiophen-3 -yl)methylene)-2-methylpropane-2-sulfinamide was prepared in 97%) yield according to the Example 1 , Step C substituting
(6-bromopyridin-2-yl)(thiophen-3-yl)methanone for (3-bromophenyl)(thiophen-3 -yl)methanone.
Step D:
Methyl 5-(3-bromophenyl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 68% yield according to the Example 1 , Step D substituting
(Z)-N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for (£')-N-((3-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide.
Step E:
Methyl 5-amino-5-(3-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 74% yield according to the Example 1 , Step E substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- (3-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step F:
6- (4-Bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one was prepared in 80%) yield according to the Example 1 , Step F substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for methyl
5- amino-5-(3-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step G:
6- (3-Bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH )-one was prepared in 92% yield according to the Example 1 , Step G substituting
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one for 6-(3-bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one.
Step Η:
3-((2-Chlorophenyl)thio)-6-(3-(4-fluorophenoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 6.6% yield according to the Example 3, Step A substituting chloro-4-fluoro-phenol for 4-fluorophenol and
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one for
6-(3-bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH )-one. lH NMR (400MHz, CD3OD) δ 7.49 (dd, J = 4.8, 2.8 Hz, 1H), 7.29 (dd, J = 8.0, 8.0 Hz, 1H),
.30 - 7.26 (m, IH), 7.23 - 7.21 (m, 2H), 7.14 (d, J= 4.8 Hz, IH), 7.06 - 7.02 (m, 3H), 6.96 - 6.92 (m,H), 6.80 - 6.75 (m, IH), 5.98 (dd, J= 7.6 , 1.2 Hz, IH), 3.47 - 3.45 (m, 2H). LCMS M+1 = 523.8.
Example 25
-(6-bromo-5-morpholino-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine dione


Step A: Methyl 5-bromopicolinate (60.0 g, 277 mmol), morpholine (72 g, 833 mmol), Pd2(dba)3 (5.0 g, 5.55 mmol), 2,2'-bis(diphenylphosphino)-l,l '-binaphthyl (6.9 g, 11.1 mmol) and CS2CO3 (135 g, 417 mmol) were combined in a flask (2 L). Dioxane (1 L) was added, and the mixture was stirred at 120 °C for 18 h under nitrogen atmosphere. The reaction mixture was cooled to room temperature, filtered and washed with EtOAc (300 ml x 3). The filtrate was dried over anhydrous MgS04 and concentrated. Silica gel chromatography eluting with 50% EtOAc/hexanes provided methyl 5-morpholinopicolinate (25 g, 112.6 mmol, 40% yield) as yellow solid.
Step B: Methyl 5-morpholinopicolinate (25.0 g, 113 mmol) in DCM (500 ml),
N-bromosuccinimide (22 g, 123 mmol) was added. The mixture was stirred at room temperature for 16 hours. The reaction mixture was concentrated. The residue was purified by silica gel
chromatography to afford methyl 6-bromo-5-morpholinopicolinate (23 g, 69.6 mmol, 62% yield) as yellow solid.
Step C: 6-Bromo-5-morpholinopicolinate (23.0 g, 69.6 mmol) in THF (200 ml), LiOH (9.62 g, 229.1 mmol) in H20 (100 ml) was added. The reaction mixture was stirred at room temperature for 8 hours. The mixture was concentrated, the resultant aqueous solution was adjusted to pH < 4 with HC1 solution (1 M), extracted with DCM (100 ml x 3), dried with anhydrous Na2S04, and concentrated to afford 6-bromo-5-morpholinopicolinic acid (21.0 g, 73.1 mmol, 96%>) as yellow solid.
Step D: 6-Bromo-N-methoxy-N-methyl-5-morpholinopicolinamide was prepared in 75% yield according to the Example 1 , Step A substituting 6-bromopicolinic acid for
6-bromo-5-morpholinopicolinic acid.
Step E: (6-Bromo-5-morpholinopyridin-2-yl)(thiophen-3-yl)methanone was prepared in 22% yield according to the Example 1 , Step B substituting 6-bromo-N-methoxy-N-methylpicolinamide for 6-bromo-N-methoxy-N-methyl-5-morpholinopicolinamide.
Step F:
(Z)-N-((6-Bromo-5-morpholinopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfina mide was prepared in 82% yield according to the Example 1 , Step C substituting
(6-bromopyridin-2-yl)(thiophen-3-yl)methanone for (6-bromo-5
-morpholinopyridin-2-yl)(thiophen-3-yl)methanone.
Step G:
Methyl 5-(6-bromo-5-morpholinopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)- 3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 84% yield according to the Example 1 , Step D substituting (Z)-N-((6-bromopyridin-2-yl)(thiophen-3-yl)methyle
ne)-2-methylpropane-2-sulfinamide for ( )-N-((6-bromo-5-morpholinopyridin-2-yl)
-(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide.
Step H:
Methyl 5-amino-5-(6-bromo-5-morpholinopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)
pentanoate was prepared in 88% yield according to the Example 1 , Step E substituting methyl 5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for Methyl
5-(6-bromo-5-morpholinopyridin-2-yl)-5-( 1 , 1 -dimethylethylsulfinamido)- 3-oxo-5-(thiophen-3-yl)pentanoate.
Step I:
6'-Bromo-4-hydroxy-5'-mo^holino-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 89% yield according to the Example 1 , Step F substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for Methyl
5-(6-bromo-5-mo^holinopyridin-2-yl)-5-(l,l-dim
entanoate.
Step J:
6'-Bromo-5-((2-chlorophenyl)thio)-4-hydroxy-5'-mo^holino-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bi pyridin]-6(lH)-one was prepared in 36% yield according to the Example 1, Step G substituting 6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one for Methyl 5-amino5-(6-bromo-5-morpholinopyridin-2-yl)-3-oxo-5-(thiophen-3-yl) pentanoate. ¾ NMR (400MHz, (CD3)2SO) δ 11.70 (br s, IH), 8.47 (s, IH), 7.65 (d, J= 8.0 Hz, IH), 7.63 (d, J = 8.0 Hz, IH), 7.53 - 7.51 (m, IH), 7.31 - 7.27 (m, 2H), 7.12 (dd, J= 5.2, 1.6 Hz, 1H), 6.96 (d, J= 8.0 Hz, IH), 6.81 - 6.78 (m, IH), 5.95 (dd, J= 8.0, 1.2 Hz, IH), 3.80 - 3.72 (m, 5H), 3.36 (d, J= 16.4 Hz, IH), 3.01 - 2.99 (m, 4H). LCMS M+l = 579.8.
Example 26
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-5-morpholino-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione

Step A:
5-((2-Chlorophenyl)thio)-6'-((4-fluorophenyl)amino)-4-hydroxy-5'-morpholino-2-(thiophen-3-yl)-2, 3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 6% yield according to the Example 4, step A substituting cyclohexanamine for 4-fluoroaniline and
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- onel for
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-5'-morpholino-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bi pyridin]-6(lH)-one. 'H NMR (400MHZ, CD3OD) δ 7.61 - 7.58 (m, 2H), 7.50 (d, J= 8.0 Hz, IH), 7.49-7.48 (m, IH), 7.33 (s, IH), 7.28 - 7.22 (m, IH), 7.17 - 7.12 (m, IH), 7.06 -7.01 (m, 4H), 6.86 - 6.82 (m, IH), 6.25 (d, J= 8.0 Hz, IH), 3.93 (m, 4H), 3.82 (d, J= 16.4Hz, IH), 3.49 (d, J= 16.4Hz, IH), 2.98 - 2.96 (m, 4H). LCMS M+l = 608.8.
Example 27
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-5-morpholino-2-pyridyl]-6-(3-thienyl)pipe ridine-2,4-dione

5-((2-Chlorophenyl)thio)-6'-(4-fluorophenoxy)-4-hydroxy-5'-morpholino-2-(thiophen-3-yl)-2,3-dih ydro-[2,2'-bipyridin]-6(lH)-one was prepared in 3% yield according to the Example 2, Step A substituting propan-2-ol for 4-fluorophenol and
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- onel for
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-5'-morpholino-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bi pyridin]-6(lH)-one. ¾ NMR (400MHz, CD3OD) δ 7.42 (d, J = 8.4 Hz, 1H), 7.37 - 7.36 (m, 1H), 7.26 -7.22 (m, 2H), 7.12 - 7.01 (m, 5H), 6.98 - 6.94 (m, 2H), 6.82 - 6.78 (m, 1H), 6.02 (dd, J= 8.0, 1.2 Hz, 1H), 3.87 - 3.84 (m, 4H), 3.52 (d, J = 16.8 Hz, 1H), 3.24 - 3.17 (m, 5H). LCMS M+l = 609.8.
Example 28
3-(2-chlorophenyl)sulfanyl-6-[4-(3-hydroxypropoxy)phenyl]-6-(3-thienyl)piperidine


28-9 28
Step A: To a solution of 4-hydroxybenzaldehyde (25 g, 205 mmol) and
(3-bromopropoxy)(iert4outyl)dimethylsilane (57 g, 225 mmol) in MeCN (200 mL) was added K2C03 (85 g, 614 mmol). The reaction mixture was heated at 80 °C for 12 hours. After cooling to room temperation, DCM (50 mL) was added, and the mixture was filtered over Celite. Then the filtrate was concentrated, purified by silica gel column (petroleum ether / EtOAc = 20/1) to afford 4-(3-((ter/4outyldimethylsilyl)oxy)propoxy)benzaldehyde (36 g, 60% yiled) as white solid.
Step B: (4-(3-((ier/-Butyldimethylsilyl)oxy)propoxy)phenyl)(thiophen-3-yl)methanol was prepared in 84% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for 4-(3-((fert-butyldimethylsilyl)oxy)propoxy)benzaldehyde.
Step C: (4-(3-((tert-Butyldimethylsilyl)oxy)propoxy)phenyl)(thiophen-3-yl)methanone was prepared in 56% yield according to the Example 7, Step B substituting
(4-bromophenyl)(thiophen-3-yl)methanone for
(4-(3-((tert-butyldimethylsilyl)oxy)propoxy)phenyl)(thiophen-3-yl)methanol.
Step D:
(Z)-N-((4-(3-((ter^utyldimethylsilyl)oxy)propoxy)phenyl)(thiophen-3-yl)methylene)-2-methy pane-2-sulfinamide was prepared in 71% yield according to the Example 7, Step C substituting (£)-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(4-(3-((tert-butyldimethylsilyl)oxy)propoxy)phenyl)(thiophen-3-yl)methanone.
Step E: Methyl
5-(4-(3-((ter^utyldimethylsilyl)oxy)propoxy)phenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(t hiophen-3-yl)pentanoate was prepared in 82% yield according to the Example 7, Step D substituting (£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(Z)-N-((4-(3-((iert-butyldimethylsilyl)oxy)propoxy)phenyl)(thiophen-3-yl)methylene)-2-methylpro pane-2-sulfinamide.
Step F: Methyl 5-amino-5-(4-(3-hydroxypropoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 82% yield according to the Example 7, Step E substituting methyl
5-(4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl 5-(4-(3-((tert-butyldimethylsilyl)oxy)propoxy)phenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(t hiophen-3 -yl)p entanoate .
Step G: 6-(4-(3-Hydroxypropoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 90% yield according to the Example 7, Step F substituting methyl
5-amino-5-(4-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- amino-5-(4-(3-hydroxypropoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step H:
3-((2-Chlorophenyl)thio)-4-hydroxy-6-(4-(3-hydroxypropoxy)phenyl)-6-(thiophen-3-yl)-5,6-dihydr opyridin-2(lH)-one was prepared in 29% yield according to the Example 7, Step G substituting
6- (4-bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one for
6-(4-(3-hydroxypropoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione. ¾ NMR (400 MHz, (CD3)2SO) δ 1 1.41 (s, 1H), 8.41 (s, 1H), 7.55 (dd, J = 5.0, 3.0 Hz, 1H), 7.25 - 7.30 (m, 4H), 7.12 (d, J = 5.1 Hz, 1H), 6.88 - 6.96 (m, 3H), 6.72 (dd, J = 7.6, 7.6 Hz, 1H), 5.84 (dd, J = 6.0, 1.2 Hz, 1H), 4.45 (s, 1H), 4.00 (t, J= 6.3 Hz, 2H), 3.52 (t, J= 6.2 Hz, 2H), 3.39 - 3.46 (m, 2H), 1.85 - 1.79 (m, 2H). LCMS M+l = 487.9.
Example 29
3-(2-chlorophenyl)sulfanyl-6-[4-(2-hydroxyethoxy)phenyl]-6-(3-thienyl)piperidine

29-9 29
Step A: 4-(2-((feri-Butyldimethylsilyl)oxy)ethoxy)benzaldehyde was prepared in 90% yield according to the Example 27, Step A substituting (3-bromopropoxy)(ferf-butyl)dimethylsilane for (2-bromoethoxy)(fer?-butyl)dimethylsilane.
Step B: (4-(2-((tert-Butyldimethylsilyl)oxy)ethoxy)phenyl)(thiophen-3-yl)methanol was prepared in 26% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for 4-(3-((tert-butyldimethylsilyl)oxy)propoxy)benzaldehyde.
Step C: (4-(2-((tert-Butyldimethylsilyl)oxy)ethoxy)phenyl)(thiophen-3-yl)methanone was prepared in 97% yield according to the Example 7, Step B substituting
(4-bromophenyl)(thiophen-3 -yl)methanone for
(4-(2-((tert-butyldimethylsilyl)oxy)ethoxy)phenyl)(thiophen-3-yl)methanol.
Step D:
(Z)-N-((4-(2-((tert-butyldimethylsilyl)oxy)ethoxy)phenyl)(thiophen-3-yl)methylene)-2-methylprop ane-2-sulfinamide was prepared in 48% yield according to the Example 7, Step C substituting (£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(4-(2-((tert-butyldimethylsilyl)oxy)ethoxy)phenyl)(thiophen-3-yl)methanone.
Step E: Methyl
5-(4-(2-((tert-butyldimethylsilyl)oxy)ethoxy)phenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(th iophen-3-yl)pentanoate was prepared in 66%> yield according to the Example 7, Step D substituting (£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(Z -N-((4-(2-((tert-butyldimethylsilyl)oxy)ethoxy)phenyl)(thiophen-3-yl)methylene)-2-methylprop ane-2-sulfinamide.
Step F: Methyl 5-amino-5-(4-(2-hydroxyethoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 79% yield according to the Example 7, Step E substituting methyl
5-(4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl 5-(4-(2-((tert-butyldimethylsilyl)oxy)ethoxy)phenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(th iophen-3 -yl)p entanoate .
Step G: 6-(4-(2-Hydroxyethoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 93%) yield according to the Example 7, Step F substituting methyl
5-amino-5-(4-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- amino-5-(4-(2-hydroxyethoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step H:
3-((2-Chlorophenyl)thio)-6-(4-(2-hydroxyethoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 35% yield according to the Example 7, Step G substituting
6- (4-bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one for
6-(4-(2-hydroxyethoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione. lH NMR (400MHz, CD3OD) δ 8.38 (s, IH), 7.54 (dd, J= 4.8, 2.8 Hz, IH), 7.29 - 7.25 (m, 4H), 7.13(dd, J= 5.2, 1.2 Hz, IH), 6.95 - 6.89 (m, 3H), 6.74 - 6.69 (m, IH), 5.85 (dd, J = 8.0, 1.2 Hz, IH), 4.84 (s, IH), 3.96 (t, J = 4.8 Hz, 2H), 3.66 (d, J = 4.4 Hz, 2H), 3.34 (d, J = 4.4 Hz, 2H). LCMS M+l = 473.8.
Example 30
3-(2-chlorophenyl)sulfanyl-6-[4-(2-methoxyethoxy)phenyl]-6-(3-thienyl)piperidine-2,4-dione

30-9 30
Step A: 4-(2-Methoxyethoxy)benzaldehyde was prepared in 81% yield according to the Example 27, Step A substituting (3-bromopropoxy)(teri-butyl)dimethylsilane for
1 -bromo-2-methoxyethane.
Step B: (4-(2-Methoxyethoxy)phenyl)(thiophen-3-yl)methanol was prepared in 91% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for
4-(2-methoxyethoxy)benzaldehyde.
Step C: (4-(2-Methoxyethoxy)phenyl)(thiophen-3-yl)methanone was prepared in 50% yield according to the Example 7, Step B substituting (4-bromophenyl)(thiophen-3-yl)methanone for (4-(2-methoxyethoxy)phenyl)(thiophen-3-yl)methanol.
Step D:
(Z)-N-((4-(2-Methoxyethoxy)phenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide was prepared in 58% yield according to the Example 7, Step C substituting
(£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(4-(2-methoxyethoxy)phenyl)(thiophen-3-yl)methanone.
Step E: Methyl
5-(l ,l -dimethylethylsulfinamido)-5-(4-(2-methoxyethoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentano ate was prepared in 78% yield according to the Example 7, Step D substituting
(£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
( )-N-((4-(2-methoxyethoxy)phenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide.
Step F: Methyl 5-amino-5-(4-(2-methoxyethoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate in 90% yield according to the Example 7, Step E substituting methyl
5-(4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-(l ,l -dimethylethylsulfinamido)-5-(4-(2-methoxyethoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentano ate.
Step G: 6-(4-(2-Methoxyethoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 26%) yield according to the Example 7, Step F substituting methyl
5-amino-5-(4-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- amino-5-(4-(2-methoxyethoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step H:
3-((2-Chlorophenyl)thio)-6-(4-(2-methoxyethoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 30% yield according to the Example 7, Step G substituting
6- (4-bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one for
6-(4-(2-methoxyethoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione. ¾ NMR (400 MHz, (CD3)2SO) δ 8.45 (s, 1H), 7.58 (dd, J =5.0, 3.0 Hz, 1H), 7.28 - 7.33 (m, 4H), 7.16 (dd, J= 5.1 , 1.1 Hz, 1H), 6.93 - 6.98 (m, 3H), 6.72 - 6.76 (m, 1H), 5.87 (dd, J= 8.0, 1.2Hz, 1H), 4.09 - 4.1 1 (m, 2H), 3.65 - 3.67 (m, 2H), 3.42 (s, 2H), 3.31 (s, 3H). LCMS M+l = 487.9.
Example 31
3-(2-chlorophenyl)sulfanyl-6-[4-(3-methoxypropoxy)phenyl]-6-(3-thienyl)piperidine-2,4-dion e

31-9
Step A: 4-(3-Methoxypropoxy)benzaldehyde was prepared in 96% yield according to the Example 27, Step A substituting (3-bromopropoxy)(tert-butyl)dimethylsilane for
1 -bromo-3-methoxypropane.
Step B: (4-(3-Methoxypropoxy)phenyl)(thiophen-3-yl)methanol was prepared in 97% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for
4-(3-methoxypropoxy)benzaldehyde.
Step C: (4-(3-Methoxypropoxy)phenyl)(thiophen-3-yl)methanone was prepared in 68% yield according to the Example 7, Step B substituting (4-bromophenyl)(thiophen-3-yl)methanone for (4-(3-methoxypropoxy)phenyl)(thiophen-3-yl)methanol.
Step D:
(Z)-N-((4-(3-Methoxypropoxy)phenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide was prepared in 51% yield according to the Example 7, Step C substituting
(£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(4-(3-methoxypropoxy)phenyl)(thiophen-3-yl)methanone.
Step E: methyl
5-(l ,l -dimethylethylsulfinamido)-5-(4-(3-methoxypropoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentan oate was prepared in 93% yield according to the Example 7, Step D substituting
(£')-N-((4-bromophenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for
(Z)-N-((4-(3-methoxypropoxy)phenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide.
Step F: methyl 5-amino-5-(4-(3-methoxypropoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate in 90% yield according to the Example 7, Step E substituting methyl
5-(4-bromophenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-(l ,l -dimethylethylsulfinamido)-5-(4-(3-methoxypropoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentan oate.
Step G: 6-(4-(3-Methoxypropoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 33%) yield according to the Example 7, Step F substituting methyl
5-amino-5-(4-bromophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- amino-5-(4-(3-methoxypropoxy)phenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step H:
3-((2-Chlorophenyl)thio)-6-(4-(3-methoxypropoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 33% yield according to the Example 7, Step G substituting
6- (4-bromophenyl)-4-hydroxy-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one for
6-(4-(3-methoxypropoxy)phenyl)-6-(thiophen-3-yl)piperidine-2,4-dione. ¾ NMR (400 MHz, (CD3)2SO) 8 8.41 (s, 1H), 7.54 (dd, J= 5.1 , 2.9 Hz, IH), 7.25 - 7.30 (m, 4H), 7.13 (dd, J= 5.1 , 1.1 Hz, IH), 6.88 - 6.95 (m, 3H), 6.68 - 6.72 (m, IH), 5.83 (dd, J= 7.9, 1.1 Hz, IH), 4.00 (t, J= 6.4 Hz, 2H), 3.44 (t, J = 6.3 Hz, 2H), 3.21 (s, 2H), 3.25 (s, 3H), 1.90 (t, J = 6.3 Hz, 2H). LCMS M+l = 501.9.
Example 32
3-(2-chlorophenyl)sulfanyl-6-(2-naphthyl)-6-(3-thienyl)piperidine-2,4-dione

32-6 32-7 32
Step A: N-Methoxy-N-methyl-2-naphthamide was prepared in 90% yield according to the Example 1, Step A substituting 6-bromopicolinic acid for 2-naphthoic acid.
Step B: Naphthalen-2-yl(thiophen-3-yl)methanone was prepared in 25% yield according to the Example 1, Step B substituting 6-bromo-N-methoxy-N-methylpicolinamide for
N-methoxy-N-methyl-2-naphthamide.
Step C: (£')-2-Methyl-N-(naphthalen-2-yl(thiophen-3-yl)methylene)propane-2-sulfinamide was prepared in 78% yield according to the Example 1, Step C substituting
(6-bromopyridin-2-yl)(thiophen-3-yl)methanone for naphthalen-2-yl(thiophen-3-yl)methanone.
Step D: Methyl
5-(l,l-dimethylethylsulfinamido)-5-(naphthalen-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 80% yield according to the Example 1, Step D substituting
(Z)-N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for (£)-2-methyl-N-(naphthalen-2-yl(thiophen-3-yl)methylene)propane-2-sulfinamide.
Step E: Methyl 5-amino-5-(naphthalen-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 80% yield according to the Example 1 , Step E substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-(l ,l -dimethylethylsulfinamido)-5-(naphthalen-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step F: 4-Hydroxy-6-(naphthalen-2-yl)-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one was prepared in 92% yield according to the Example 1 , Step F substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for methyl
5-amino-5-(naphthalen-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step G:
3-((2-Chlorophenyl)thio)-4-hydroxy-6-(naphthalen-2-yl)-6-(thiophen-3-yl)-5,6-dihydropyridin-2(l H)-one was prepared in 9% yield according to the Example 1 , Step G substituting
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one for 4-hydroxy-6-(naphthalen-2-yl)-6-(thiophen-3-yl)-5,6-dihydropyridin-2(lH)-one. lH NMR (400MHz, (CD3)2SO) δ 1 1.53 (s, IH), 8.65 (s, IH), 7.69 - 7.62 (m, 4H), 7.61 - 7.52 (m, 4H), 7.41 (dd, J= 2.8, 1.2 Hz, IH), 7.26 - 7.22 (m, 2H), 6.88 - 6.84 (m, IH), 6.30 - 5.79 (m, IH), 5.77 (d, J= 8.0 Hz, IH), 3.61 (d, J = 16.8 Hz, IH), 3.61 (d, J = 16.8 Hz, IH). LCMS 463.8.
Example 33
3-(2-chlorophenyl)sulfanyl-6-(4-cyclopropylphenyl)-6-(3-thienyl)piperidine-2,4-dione

33-9 33
Step A: 4-Cyclopropylbenzaldehyde was prepared in 80% yield according to the Example 8, Step A substituting cyclohex-l-en-l-ylboronic acid for cyclopropylboronic acid and
6-(4-bromophenyl)-3-((2-chlorophenyl)thio)-6-(thiophen-3-yl) piperidine -2,4-dione for
4-bromobenzaldehyde.
Step B: (4-Cyclopropylphenyl)(thiophen-3-yl)methanol was prepared in 91% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for 4-cyclopropylbenzaldehyde.
Step C: (4-Cyclopropylphenyl)(thiophen-3-yl)methanone was prepared in 88% yield according to the Example 7, Step B substituting (4-bromophenyl)(thiophen-3-yl)methanol for
(4-cyclopropylphenyl)(thiophen-3-yl)methanol.
Step D:
(Z)-N-((4-Cyclopropylphenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide was prepared in 71% yield according to the Example 1 , Step C substituting
(6-bromopyridin-2-yl)(thiophen-3 -yl)methanone for
(4-cyclopropylphenyl)(thiophen-3-yl)methanone
Step E: Methyl
5-(4-cyclopropylphenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 98% yield according to the Example 1 , Step D substituting
(Z)-N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for (Z)-N-((4-cyclopropylphenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide.
Step F: Methyl 5-amino-5-(4-cyclopropylphenyl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 72% yield according to the Example 1 , Step E substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-(4-cyclopropylphenyl)-5-(l ,l -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step G: 6-(4-Cyclopropylphenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 55% yield according to the Example 1 , Step F substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for methyl
5-amino-5-(4-cyclopropylphenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step Η:
3-((2-Chlorophenyl)thio)-6-(4-cyclopropylphenyl)-6-(thiophen-3-yl)piperidine-2,4-dione was prepared in 38% yield according to the Example 1 , Step G substituting
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)- one for 6-(4-cyclopropylphenyl)-6-(thiophen-3-yl)piperidine-2,4-dione. ¾ NMR (400MHz, (CD3)2SO) δ 7.47 (dd, J = 8.0 , 8.0 Hz, 1H), 7.31 - 7.27 (m, 3H), 7.18 (dd, J= 4.0 , 4.0 Hz, 1H), 7.1 1 (dd, J = 8.0, 4.0 Hz, 1H), 7.02 (d, J= 8.0 Hz, 2H), 6.84 (dd, J= 8.0, 8.0 Hz, 1H), 6.67 (dd, J= 8.0, 8.0 Hz, 1H), 5.98 (d, J= 8.0 Hz, 1H), 3.24 (d, J = 5.2 Hz, 2H), 1.92 - 1.86 (m, 1H), 0.96 - 0.91 (m, 2H), 0.67 - 0.63 (m, 2H). LCMS M+l = 453.8.
Example 34
3-(2-chlorophenyl)sulfanyl-l-methyl-6-[3-(tetrahydropyran-4-ylamino)phenyl]-6-(3-thienyl)p iperidine-2,4-dione

24-8 34-1

Step A:
6-(3-Bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-l-methyl-6-(thiophen-3-yl)-5,6-dihy idin-2(lH)-one was prepared by Example 11 in 15% yield, step A substituting
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6 (lH)-one for 6-(3-bromophenyl)-3-((2-chlorophenyl)thio)-4-hydroxy-6- (thiophen-3-yl)-5,6- dihydropyridin- 2 (lH)-one.
Step B:
3 -(2-Chlorophenyl)sulfanyl- 1 -methyl-6- [3 -(tetrahydropyran-4-ylamino)phenyl] -6-(3 -thienyl)piperi dine-2,4-dione was prepared in 6% yield, according to Example 7. Step Η substituting
2-methylmorpholine for tetrahydro-2H-pyran-4-amine. ¾ NMR (400 MHz, (CD3)2SO) δ 11.3 (s, 1H), 7.67 (dd, J = 52, 3.2 Hz, 1H), 7.31 (dd, J= 8.0, 1.6 Hz, 1H), 7.15 - 7.10 (m, 3H), 6.98 (dd, J = 7.6, 1.2 Hz, 1H), 6.88 (dd, J= 7.6, 1.2 Hz, 1H), 6.61 (dd, J= 8.4, 1.6 Hz, 1H), 6.43 (m, 2H), 6.15 (d, J= 8.4 Hz, 1H), 3.85 (m, 2H), 3.60 (d, J= 16.8 Hz, 1H), 3.48 (d, J= 16.8 Hz, 1H), 3.44 (m, 3H), 2.69 (s, 3H), 1.82 (m, 2H), 1.34 (m, 2H). LCMS M+l = 527.0.
Example 35
-(2-chlorophenyl)sulfanyl-6-(2-hydroxy-4-morpholino-phenyl)-6-(3-thienyl)piperidine-2,4-di one
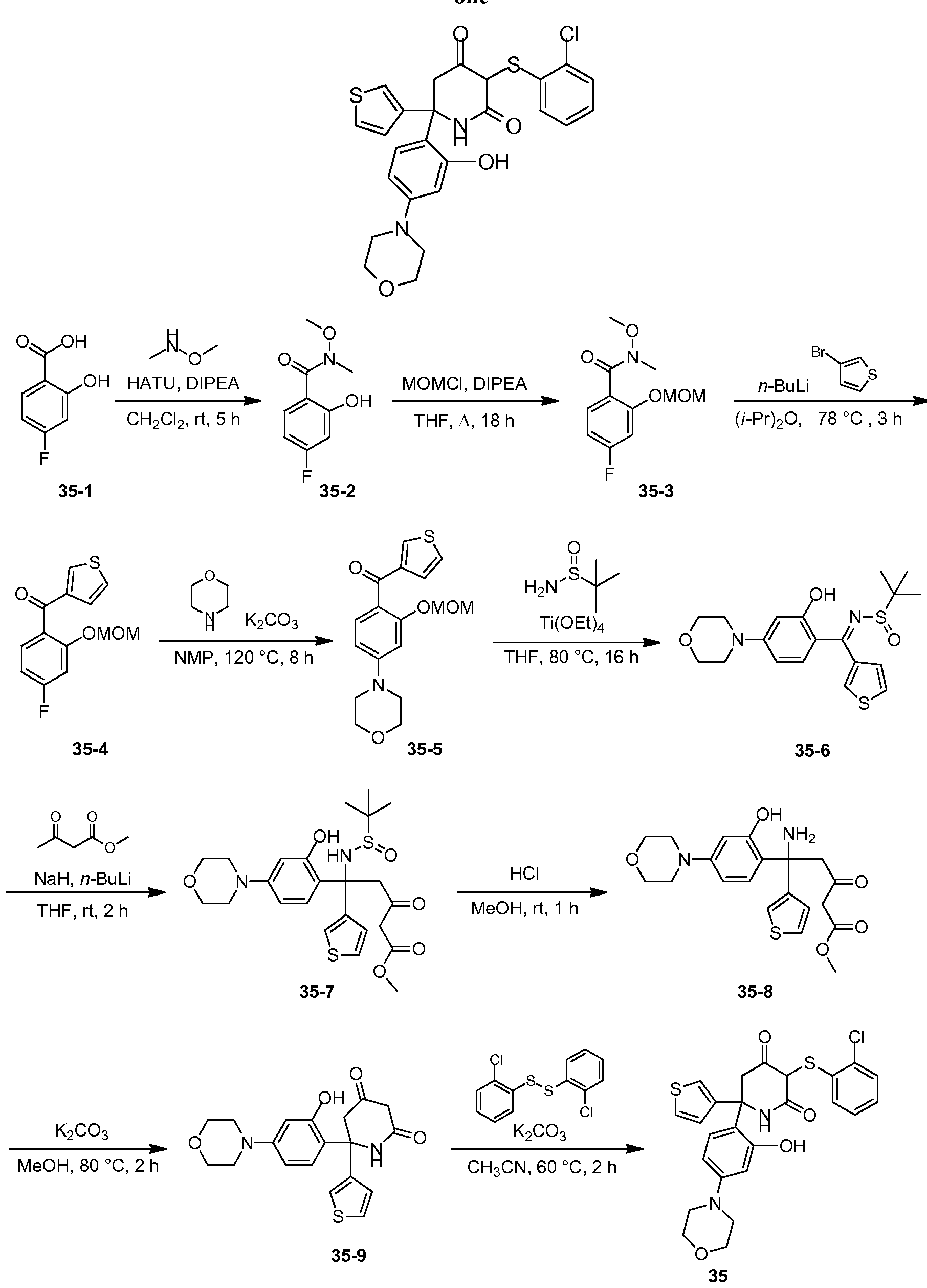
4-fluoro-2-hydroxybenzoic acid.
Step B: 4-Fluoro-N-methoxy-2-(methoxymethoxy)-N-methylbenzamide was prepared in 62% yield according to Example 13, Step A substituting 5-bromo-2-hydroxybenzaldehyde for
4- fluoro-2-hydroxy-N-methoxy-N-methylbenzamide.
Step C: (4-Fluoro-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone was prepared in 41%> yield according to the Example 1 , Step B substituting 6-bromo-N-methoxy-N-methylpicolinamide for 4-fluoro-N-methoxy-2 -(methoxymethoxy)-N-methylbenzamide.
Step D: To a solution of (4-fluoro-2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanone (10 g, 38 mmol) inNMP (50 mL) was added morpholine (16.4 g, 188 mmol) and K2C03 (10.4 g, 75 mmol). The solution was stirred at 120 °C for 8 hours. The reaction was quenched with water, adjusted to pH = 5 with HCl solution, extracted with DCM, and concentrated under vacuum. The cuude residue was purified by silica gel column to afford
(6-(4-fluorophenoxy)pyridin-2-yl)(2-(methoxymethoxy)phenyl)methanone (7.2 g, 60%> yield).
Step E: N-((2-Hydroxy-4-morpholinophenyl)(thiophen-3-yl)methylene)-2-methylpropane- 2-sulfinamide was prepared in 10%> yield according to the Example 1, Step C substituting
(6-bromopyridin-2-yl)(thiophen-3 -yl)methanone for (6-(4-fluorophenoxy)pyridin-2-yl)
(2-(methoxymethoxy)phenyl)methanone.
Step F: Methyl 5-(l,l-dimethylethylsulfinamido)-5-(2-hydroxy-4-morpholinophenyl)-3-oxo -5- (thiophen-3-yl)pentanoate was prepared in 36% yield according to the Example 1, Step D substituting N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for N-((2-hydroxy-4-morpholinophenyl)(thiophen-3 -yl)methylene)- 2-methylpropane- 2-sulfinamide.
Step G: Methyl
5- amino-5-(2-hydroxy-4-morpholinophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 40%) yield according to the Example 1 , Step E substituting methyl
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl 5-(l,l-dimethylethylsulfinamido)-5-(2-hydroxy-4-morpholinophenyl)-3-oxo -5- (thiophen-3 -yl)pentanoate.
Step H: 3-(2-Chlorophenyl) sulfanyl-6-(2-hydroxy-4-morpholino-phenyl)-6-(3-thienyl) piperidine-2,4-dione was prepared in 2% yield, according to Example 1 , Step G substituting 6'-bromo-4-hydroxy-2-(thiophen-3 -yl)-2,3 -dihydro- [2,2'-bipyridin] -6( 1 H)-one as methyl
5-amino-5-(2-hydroxy-4-morpholinophenyl)-3-oxo-5-(thiophen-3-yl)pentanoate. lH NMR (400 MHz, (CD3)2SO) δ 7.36 (d, J= 5.2 Hz, 1H), 7.20 - 7.15 (m, 3H), 7.06 (d, J= 5.2 Hz, 1H), 6.92 (dd, J = 7.6, 1.6 Hz, 1H), 6.77 (dd, J= 7.6, 1.6 Hz, 1H), 6.50 (m, 2H), 6.19 (d, J= 8.0 Hz, 1H), 3.83 (dd, J
= 4.8, 4.8 Hz, 4H), 3.68 (d, J= 16.4 Hz, 1H), 3.36 (d, J= 16.4 Hz, 1H), 3.13 (dd, J= 4.8, 4.8 Hz, 4H). LCMS M+l = 514.9.
Example 36
3-(2-chlorophenyl)sulfanyl-6-(2-hydroxyphenyl)-6-(3-thienyl)piperidine-2,4-dione

36
Step A: 2-(Methoxymethoxy)benzaldehyde was prepared in 82% according to Example 13, Step A substituting 5-bromo-2-hydroxybenzaldehyde for 2-hydroxybenzaldehyde.
Step B: (2-(Methoxymethoxy)phenyl)(thiophen-3-yl)methanol was prepared in 60% yield according to the Example 7, Step A substituting 4-bromobenzaldehyde for
2- (methoxymethoxy)benzaldehyde.
Step C: (2-(Methoxymethoxy)phenyl)(thiophen-3-yl)methanone was prepared in 71% yield according to the Example 7, Step B substituting (4-bromophenyl)(thiophen-3-yl)methanol for (2-(methoxymethoxy)phenyl)(thiophen-3-yl)methanol.
Step D:
N-((2-(Methoxymethoxy)phenyl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide was prepared in 50% yield according to the Example 1 , Step C substituting
(6-bromopyridin-2-yl)(thiophen-3-yl)methanone for (2-(methoxymethoxy)phenyl) (thiophen-
3- yl)methanone.
Step E: Methyl 5-(l ,l -dimethylethylsulfinamido)-5-(2-(methoxymethoxy)phenyl)-3-oxo-5- (thiophen-3-yl)pentanoate was prepared in 47% yield according to the Example 1 , Step D substituting N-((6-bromopyridin-2-yl)(thiophen-3-yl)methylene)-2-methylpropane-2-sulfinamide for N-((2-(methoxymethoxy)phenyl)(thiophen-3-yl)methylene)-2-methylpropane-2- sulfanamide.
Step F: Methyl 5-amino-5-(2-hydroxyphenyl)-3-oxo-5-(thiophen-3-yl)pentanoate was prepared in 51% yield according to the Example 1 , Step E substituting methyl
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl 5-(l ,l -dimethylethylsulfinamido)-5-(2-(methoxymethoxy)phenyl)-3-oxo-5- (thiophen-3 -yl)pentanoate.
Step G: 6-(2-Hydroxyphenyl)-6-(thiophen-3-yl) piperidine-2,4-dione was prepared in 60% yield according to the Example 1 , Step F substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5- amino-5-(2-hydroxyphenyl)-3-oxo-5-(thiophen-3-yl)pentanoate.
Step H:
3-((2-Chlorophenyl)thio)-4-hydroxy-6-(2-hydroxyphenyl)-6-(thiophen-3-yl)-5,6-dihydropyridin-2( lH)-one was prepared in 7% yield according to the Example 1 , Step G substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
6- (2-hydroxyphenyl)-6-(thiophen-3-yl) piperidine-2,4-dione. lH NMR (400 MHz, (CD3)2SO) δ 9.81 (s, 1H), 7.69 (s, 1H), 7.46 (d, J= 5.2 Hz, 1H), 7.29 (m, 3H), 7.26 (m, lH), 7.17 (m, 1H), 6.96 (m, 1H), 6.86 (m, 2H), 6.74 (m, 1H), 6.10 (d, J = 8.0 Hz, 1H), 3.74 (d, J = 16.4 Hz, 1H), 3.42 (d, J =\ 6A Hz, 1H). LCMS M+l = 429.8.
Example 37

37-6 37
Step A: To a stirred solution of 6-(4-bromophenyl)-6-(thiophen-3-yl)piperidine- 2,4-dione (5 g, 14.3 mmol) in DMF (50 mL) was added NBS (3.05 g, 17.8 mol) in an ice bath. The reaction was stirred at 0 °C for 30 min. The reaction mixture was used in next step directly.
Step B: The solution of 3-bromo-6-(4-bromophenyl)-6-(thiophen-3-yl) piperidine -2,4-dione (14.3 mmol) in DMF (50 mL) was added 2-chlorophenol (2.8 g, 21.5 mmol) and potassium carbonate (5.9 g, 42.9 mmol). The reaction was stirred at 80 °C for 12 hours. The reaction mixture was extracted with EtOAc and brine. The organic layer was dried and concentrated. The crude was purified by chromatography on silica gel (PE/EA = 2 IX) to afford
6-(4-bromophenyl)-3-(2-chlorophenoxy) -6-(thiophen-3-yl)piperidine-2,4-dione (2 g, 4.2 mmol, 29%) as light color solid.
Step C: In a solution of 6-(4-bromophenyl)-3-(2-chlorophenoxy)-6-(thiophen-3-yl) piperidine -2,4-dione (600 mg, 1.26 mmol ) in dioxane (10 mL) was added morpholine (328 mg, 3.77 mmol), Brettphos (65 mg, 0.13mmol), Pd2(dba)3 (64 mg, 0.07 mmol) and i-BuONa (362 mg, 3.77 mmol). The solution was stirred for 8 h at 1 10 °C under nitrogen. The solvent was removed under vacuum and the residue was purified by Prep-HPLC (FA) and SFC to afford
(6S)-3-(2-chlorophenoxy)-6-(4-morpholinophenyl) -6-(thiophen-3-yl)piperidine -2,4-dione (35 mg, 6%) as white solid.
Step D: (6S)-3-(2-chlorophenoxy)-6-(4-(piperidin-l -yl)phenyl)-6-
(thiophen-3-yl)piperidine -2,4-dione was prepared in 8% yield according to the Method 37, Step C substituting morpholine for piperidine.
Example 38
6-(6-bromopyridin-2-yl)-3-((2-chlorophenyl)thio)-6-(4-morpholinophenyl)piperidine-2,4-dion e


chromatography (PE : EA = 3 : 1) to give the desired product (6.8 g, 79%) as a yellow oil.
Step B: (6-Bromopyridin-2-yl)(4-morpholinophenyl)methanone was prepared in 69% yield according to the method 7 Step B substituting (4-bromophenyl) (thiophen-3-yl)methanol for (6-bromopyridin-2-yl) (4-morpholinophenyl)methanol.
Step C: (Z)-N-((6-Bromopyridin-2-yl)(4-morpholinophenyl) methylene)-
2-methylpropane-2-sulfinamide was prepared in 58% yield according to the Method 1 , Step C substituting (6-bromopyridin-2-yl)(thiophen-3-yl) methanone for
(6-bromopyridin-2-yl)(4-morpholinophenyl)methanone..
Step D: methyl 5-(6-bromopyridin-2-yl)-5-(l ,l -dimethylethylsulfinamido)- 5-(4-morpholinophenyl)-3-oxopentanoate was prepared in 79% yield according to the Method 1 , Step D substituting (Z)-N-((6-bromopyridin-2-yl)(thiophen-3-yl)
methylene)-2-methylpropane-2-sulfinamide for (Z)-N-((6-bromopyridin-2-yl)
(4-morpholinophenyl)methylene)-2-methylpropane-2-sulfinamide.
Step E: methyl 5-amino-5-(6-bromopyridin-2-yl)-5-(4-morpholinophenyl) -3-oxopentanoate was prepared in 67% yield according to the Method 1, Step E substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl) pentanoate for methyl
5-(6-bromopyridin-2-yl)-5-(l , 1 -dimethylethylsulfinamido)
-5-(4-morpholinophenyl)-3-oxopentanoate.
Step F: 6'-bromo-4-hydroxy-2-(4-morpholinophenyl)-2,3-dihydro- [2,2'-bipyridin]-6(lH)-one was prepared in 47% yield according to the Method 1, Step F substituting
6'-bromo-4-hydroxy-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin] -6(lH)-one for methyl
5-amino-5-(6-bromopyridin-2-yl)-5- (4-morpholinophenyl) -3-oxopentanoate.
Step G: 6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2-(4-morpholinophenyl)
-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one was prepared in 96%> yield according to the Method 1 , Step G substituting 6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy
-2-(thiophen-3-yl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for
6'-bromo-4-hydroxy-2-(4-morpholinophenyl)-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one.

Step A: 3-((2-chlorophenyl)thio)-6-(6-((4-fluorophenyl)amino)
pyridin-2-yl)-6-(4-morpholinophenyl)piperidine-2,4-dione was prepared in 42%> yield according to the Method 4, Step A substituting 6-(6-bromopyridin-2-yl)-3- ((2-chlorophenyl) thio)- 6-(thiophen-3-yl) piperidine-2,4-dione for 6-(6-bromopyridin-2-yl)
-3-((2-chlorophenyl)thio)-6-(4-morpholinophenyl) piperidine-2,4-dione and cyclohexanamine for 4-fluoroaniline. 1H NMR (400MHz, METHANOL-d4) d = 7.57 (dd, J=8.0, 8.0 Hz, IH), 7.54-7.50 (m, 2H), 7.36 (d, J=9.2, 2H), 7.20 -6.92 (m, 6H), 6.73 - 6.67 (m, 2H), 6.12 (d, J=7.2 Hz, IH), 3.84 (dd, J=9.2, 4.4 Hz, 4H), 3.75 (d, J=16.4 Hz, IH), 3.49 (d, J=16.4 Hz, IH), 3.16 (dd, J=9.2, 4.4 Hz, 4H), single stereoisomer. IH NMR (400MHz, METHANOL-d4) d = 7.51 (dd, J=8.0, 8.0 Hz, IH), 7.49-7.47 (m, 2H), 7.33 (d, J=8.8, 2H), 7.18 (d, J=8.0, IH), 6.99 -6.89 (m, 6H), 6.71 - 6.69 (m, 2H),
6.07(dd, J=6.4, 1.6 Hz, IH), 3.81 (dd, J=4.8, 4.8 Hz, 4H), 3.78 (d, J=16.8 Hz, IH), 3.47 (d, J=16.8 Hz, IH), 3.15 (dd, J=4.8, 4.8 Hz, 4H), mixture of diastereoisomers.

Step A: 3-(2-chlorophenyl)sulfanyl-6-[6-(3,4-difluorophenoxy)-2-pyridyl]
6-(4-morpholinophenyl)piperidine-2,4-dione was prepared in 47% yield according to the Method 3, Step A substituting 6-(6-bromopyridin-2-yl)-3-((2-chlorophenyl)
thio)-6-(thiophen-3-yl)piperidine-2,4-dione for 6-(6-bromopyridin-2-yl)-3-
((2-chlorophenyl)thio)-6-(4-morpholinophenyl)piperidine-2,4-dione and 2-Chloro-4-fluoro-phenol for 3,4-difluorophenol. 1H NMR (400MHz, METHANOL-d4) d = 7.88 (dd, J=8.0, 8.0 Hz, IH), 7.33 (d, J=7.2, IH), 7.20 - 7.16 (m, 4H), 7.02 -6.92 (m, 5H), 6.85 (d, J=7.2, IH), 6.72 (dd, J=8.0, 8.0, IH), 5.91(dd, J=8.0, 1.2 Hz, 1H), 3.81 (dd, J=4.8, 4.8 Hz, 4H), 3.55 (d, J=16.8 Hz, 1H), 3.31 (d, J=16.8 Hz, IH), 3.12 (dd, J=4.8, 4.8 Hz, 4H), mixture of diastereoisomers. 1H NMR (400MHz,
METHANOL-d4) d = 7.91 (dd, J=7.6, 7.6 Hz, IH), 7.37 (d, J=7.6, IH), 7.36- 7.21 (m, 4H), 7.04 -6.96 (m, 2H), 6.93 -6.85 (m, 4H), 6.75 (dd, J=7.6, 7.6, IH), 3.84 (dd, J=4.8, 4.8 Hz, 4H), 3.55 (d, J=16.8 Hz, IH), 3.33 (d, J=16.8 Hz, IH), 3.16 (dd, J=4.8, 4.8 Hz, 4H), single stereoisomer.

Step A: 3-((2-chlorophenyl)thio)-6-(6-(cyclohexyloxy)pyridin-2-yl)
-6-(4-morpholinophenyl)piperidine-2,4-dione was prepared in 11% yield according to the Method 4, Step A substituting 6-(6-bromopyridin-2-yl)-3-((2-chlorophenyl)thio)
-6-(thiophen-3-yl)piperidine-2,4-dione for 6-(6-bromopyridin-2-yl)-3- ((2-chlorophenyl) thio)-6-(4-morpholinophenyl)piperidine-2,4-dione and propan-2-ol for cyclohexanol.

42
Step A: N-methoxy-N-methylthiazole-4-carboxamide was prepared in 67% yield according to the Method 1 , Step A substituting 6-bromopicolinic acid for thiazole-4-carboxylic acid.
Step B: (4-fluorophenyl)(thiazol-4-yl)methanone was prepared in 72% yield according to the Method 36, Step A substituting 4-bromothiophene-2-carbaldehyde for
N-methoxy-N-methylthiazole-4-carboxamide.
Step C: (4-morpholinophenyl)(thiazol-4-yl)methanone was prepared in 56%> yield according to the Method 34, Step D substituting (4-fluoro-2-(methoxymethoxy)
phenyl) (thiophen-3-yl)methanone for (4-fluorophenyl)(thiazol-4-yl)methanone.
Step D: ( )-2-methyl-N-((4-morpholinophenyl)(thiazol-4-yl)methylene) propane -2-sulfinamide was prepared in 56% yield according to the Method 1, Step C substituting
(6-bromopyridin-2-yl)(thiophen-3 -yl)methanone for (4-morpholinophenyl)
(thiazol-4-yl)methanone.
Step E: methyl 5-(l,l-dimethylethylsulfinamido)-5-(4-morpholinophenyl)
-3-oxo-5-(thiazol-4-yl)pentanoate was prepared according to the Method 1, Step D substituting ( )-N-((6-bromopyridin-2-yl)(thiophen-3-yl) methylene)-2-methylpropane -2-sulfinamide for ( )-2-methyl-N-((4-morpholinophenyl) (thiazol-4-yl)methylene)propane -2-sulfinamide.
Step F: methyl 5-amino-5-(4-morpholinophenyl)-3-oxo-5-(thiazol-4-yl) pentanoate was prepared according to the Method 1 , Step E substituting methyl
5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5-(thiophen-3-yl)pentanoate for methyl
5-(l ,l -dimethylethylsulfinamido)-5-(4-morpholinophenyl)-3-oxo-5-(thiazol-4-yl)pentanoate.
Step G: 4-hydroxy-6-(4-morpholinophenyl)-6-(thiazol-4-yl)- 5,6-dihydropyridin -2(lH)-one was prepared in 18% yield over three steps according to the Method 1 , Step F substituting methyl 5-amino-5-(6-bromopyridin-2-yl)-3-oxo-5- (thiophen-3-yl)pentanoate for methyl
5-amino-5-(4-morpholinophenyl) -3-oxo- 5- (thiazol-4-yl)pentanoate.
Step Η: 3-((2-chlorophenyl)thio)-6-(4-morpholinophenyl)-6-(thiazol-4-yl) piperidine-2,4-dione was prepared in 3% yield according to the Method 1 , Step G substituting
6'-bromo-5-((2-chlorophenyl)thio)-4-hydroxy-2- (thiophen-3-yl)
-2,3-dihydro-[2,2'-bipyridin]-6(lH)-one for 4-hydroxy-6-(4-morpholinophenyl)-6- (thiazol-4-yl)-5,6-dihydropyridin-2(lH)-one.

43-1 43-2 43
Step A: 6-(4-bromophenyl)-3-((2-chloro-5-hydroxyphenyl)thio) -6-(thiophen-3-yl) piperidine-2,4-dione was prepared in 68% yield according to the method 7 Step B substituting l ,2-bis(2-chlorophenyl)disulfane for 4-chloro-3-mercaptophenol.
Step B: In a solution of 6-(4-bromophenyl)-3-((2-chloro-5-hydroxyphenyl)thio)
-6-(thiophen-3-yl)piperidine-2,4-dione (200 mg, 0.4 mmol ) in dioxane (4 mL) was added piperidine (136 mg, 1.6 mmol), Brettphos (20 mg, 0.04 mmol), Pd2(dba)3 (18 mg, 0.02 mmol) and i-BuONa (154 mg, 1.6 mmol). The solution was stirred for 8 h at 1 10 °C under nitrogen atmosphere. The solvent was removed under vacuum and the residue was purified by Prep-HPLC (FA) to afford compound 43 (12 mg, 6%>) as white solid.
The following compounds were prepared as indicated in the table, unless indicated otherwise, these compounds were prepared according to methods described herein.







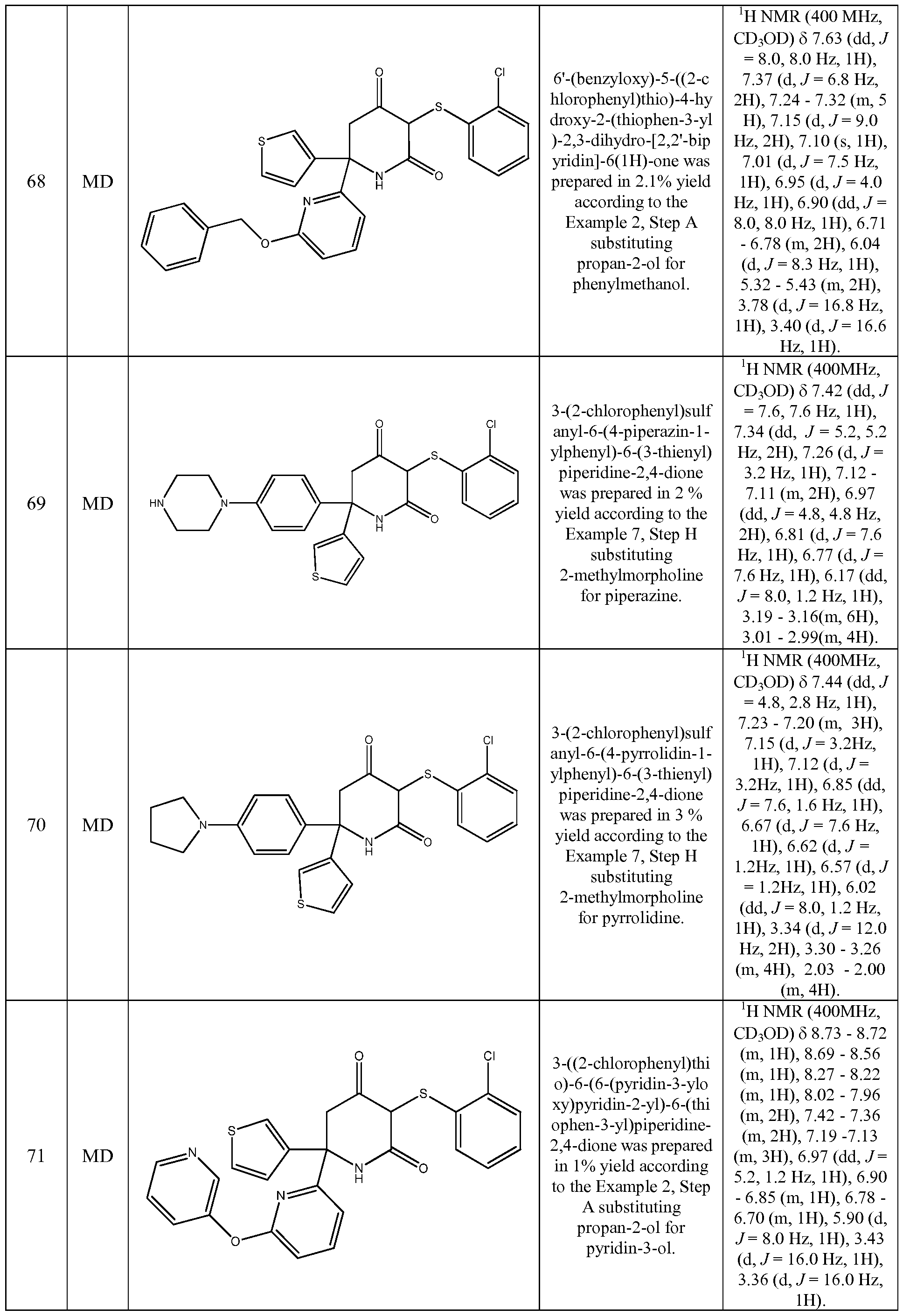







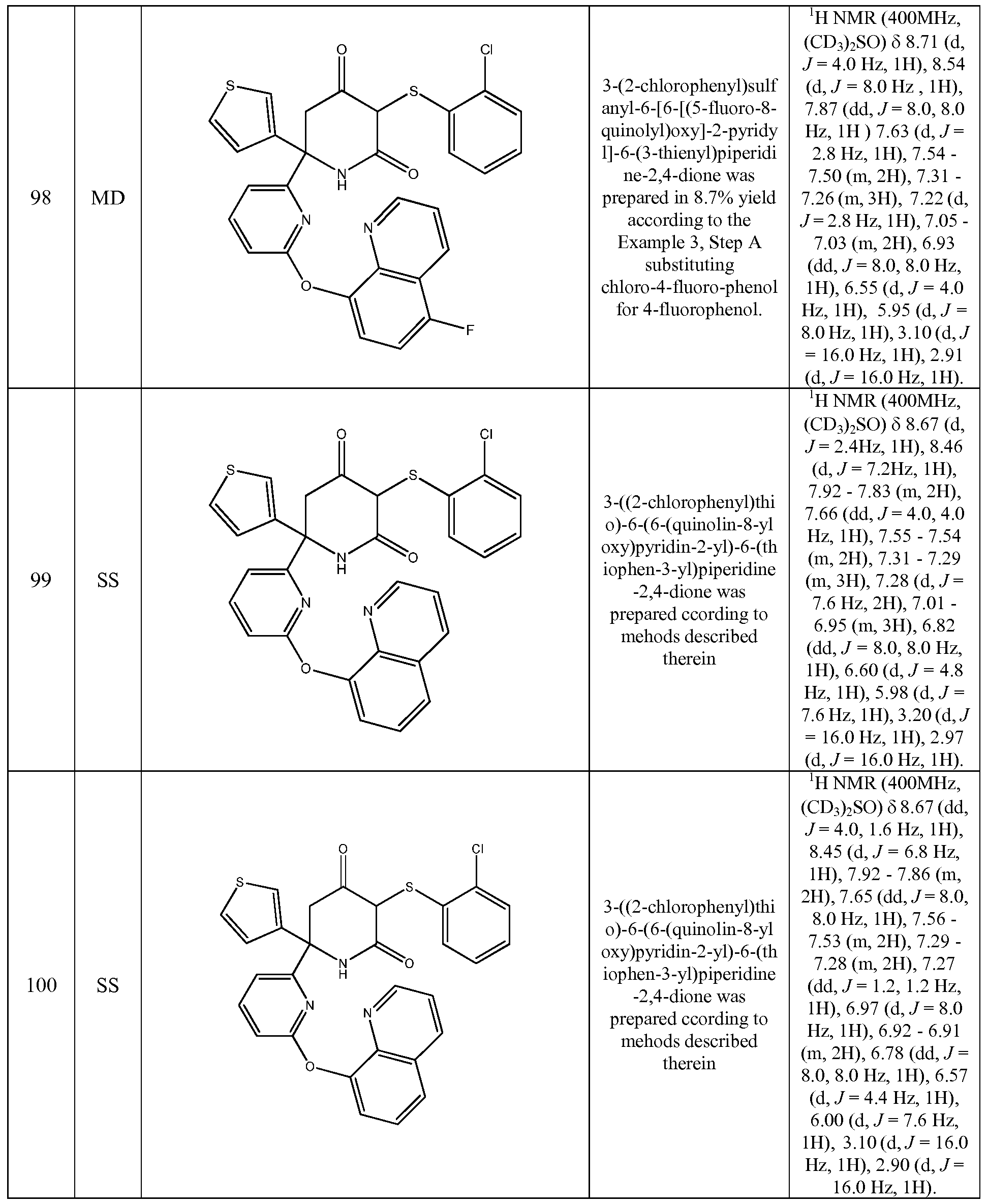









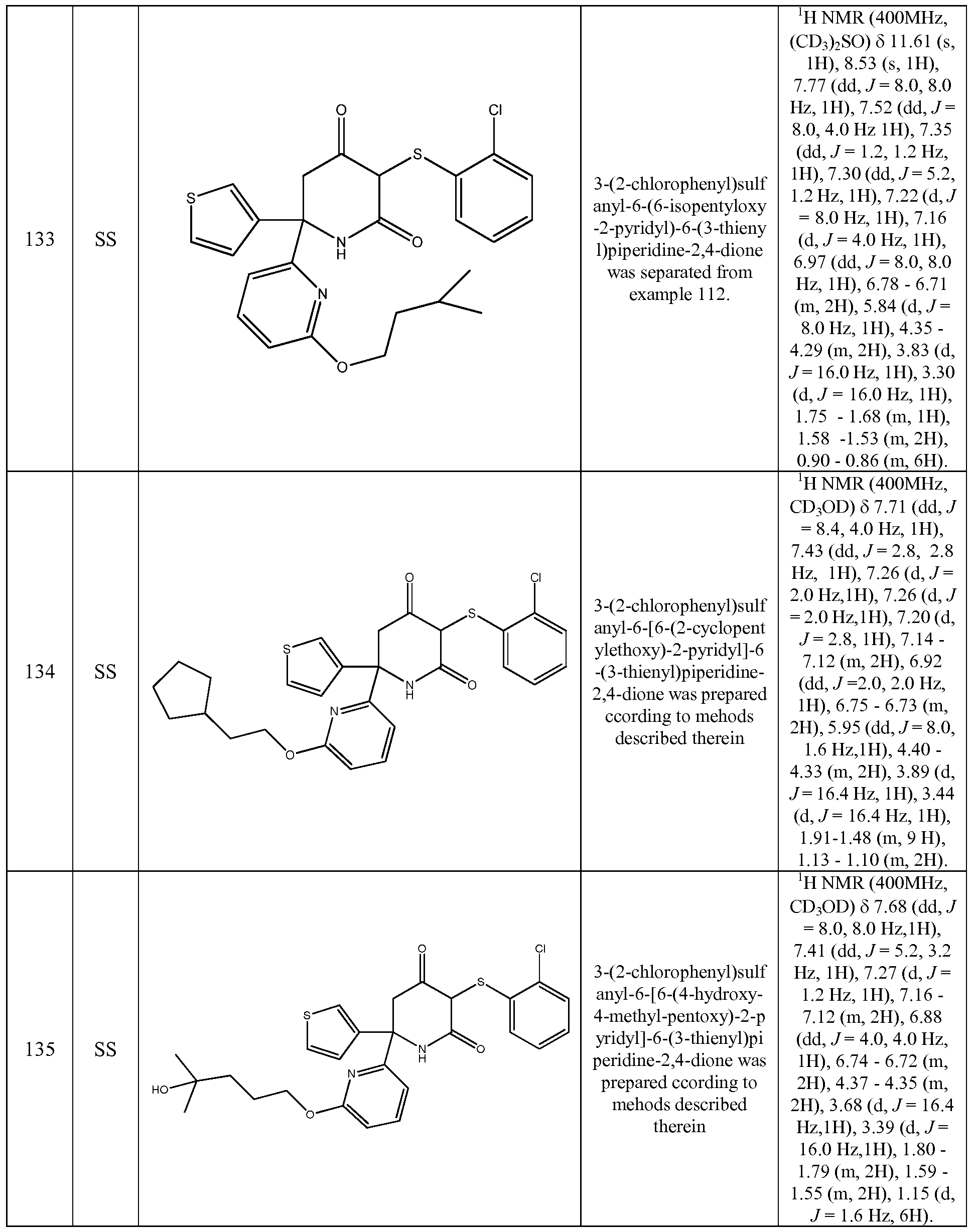






































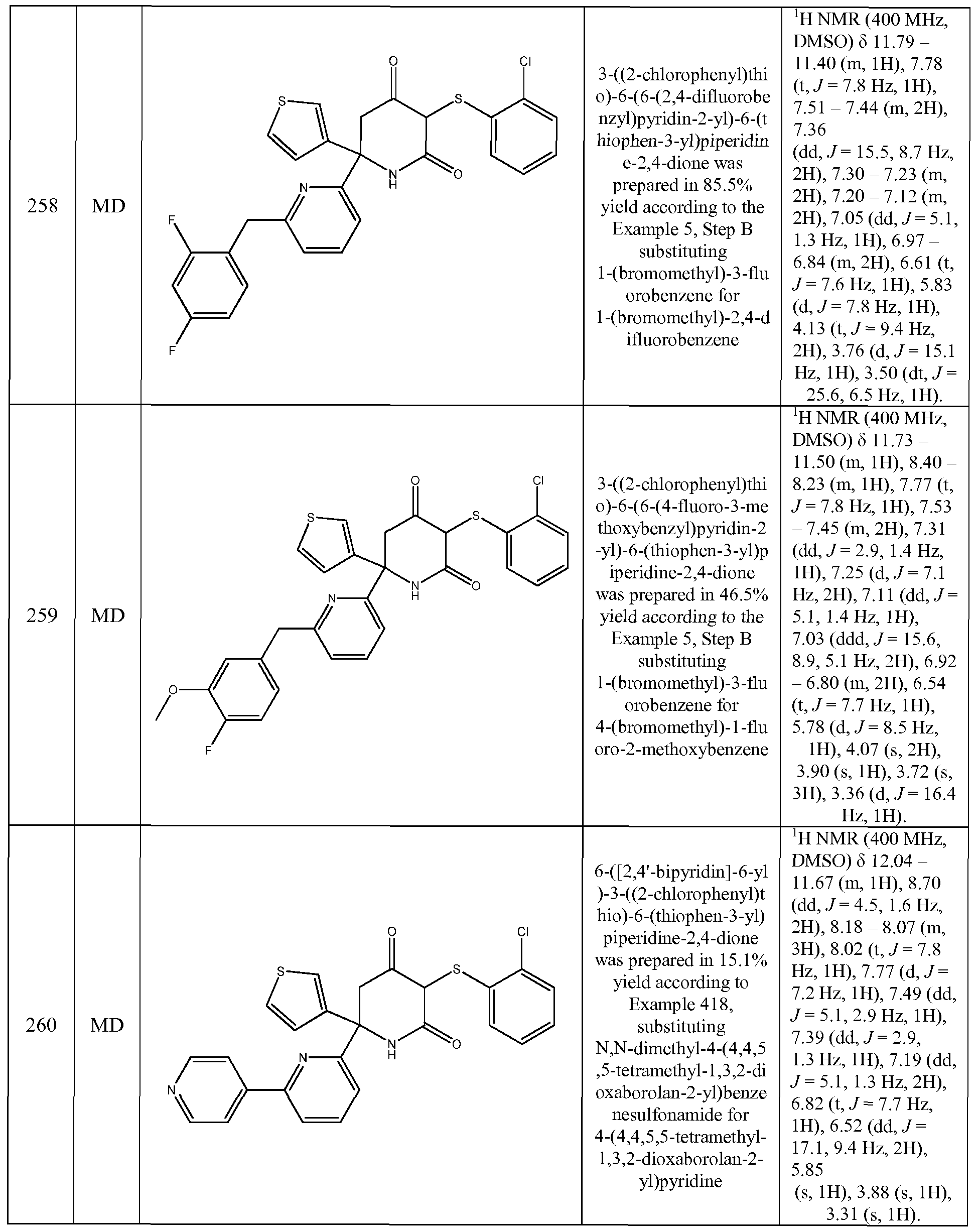


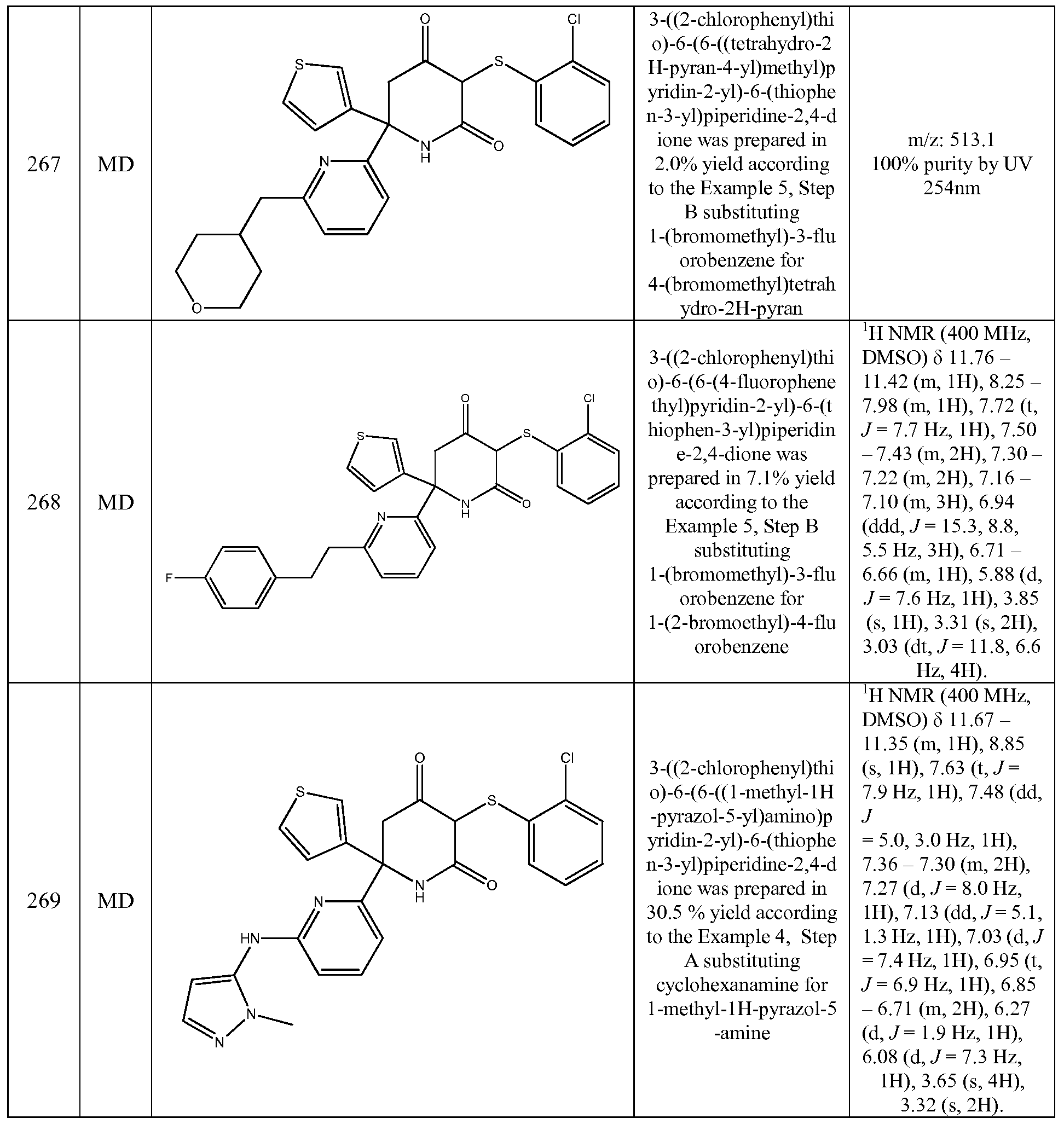







































* ST: Stereochemistry : SS = Single Stereoisomer; MD = Mixture of Diastereoisomers
Example 417
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3 hienyl)piperidine-:
(racemate)

To a solution of 2,6-dibromopyridine (5.0 g, 21 mmol) in dry THF (50 mL) cooled in dry-ice acetone bath was added 2.5 M butyl lithium (8.4 mL, 21 mmol) and the resulting mixture was stirred for 10 min. To this solution was added thiophene-3-carbaldehyde (2.4 g, 21 mmol) and the resulting mixture was stirred at -78 for 10 min. The reaction mixture was quenched with water and allowed to warm to room temperature and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The residue was purified by flash
chromatography (silica gel, 0-50 % EtO Ac/heptane) to obtain
(6-bromo-2-pyridin-2-yl)-thiphen-3-yl-methanol (2.6 g, 46 %). MS (ESI): m+H = 272. 1H NMR (400 MHz, Chloroform-d) δ 7.51 (t, J = 7.7 Hz, 1H), 7.40 (d, J = 7.8 Hz, 1H), 7.29 (dd, J = 5.0, 3.0 Hz, 1H), 7.05 - 7.01 (m, 1H), 7.26 (s, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.03 (dd, J = 5.0, 1.2 Hz, 1H), 5.84 (d, J = 4.0 Hz, 1H), 4.17 (d, J = 4.9 Hz, 1H).
A mixture of (6-bromo-2-pyridin-2-yl)-thiphen-3-yl-methanol (2.6 g, 9.6 mmol) and des martin periodinane (6.3 g, 14 mmol) in DCM (50 mLO was stirred at ambient temperature for 2 h. The solids were removed by filtration through celite and washed well with ethyl acetate. The organic layer washed with aqueous sodium bicarbonate, water, brine and dried over sodium sulfate and concentrated. The residue was purified by flash chromatography (silica gel, 0-50 % EtO Ac/heptane) to obtain (6-bromo-pyridin-2-yl)-thiophen-3-yl-methanone (1.8 g, 70%). MS (ESI): m+H = 270; H NMR (400 MHz, Chloroform-d) δ 8.92 (dd, J = 3.0, 1.1 Hz, 1H), 8.14 - 8.08 (m, 1H), 7.88 (dd, J = 5.1, 1.2 Hz, 1H), 7.75 (t, J = 7.7 Hz, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.34 (dd, J = 5.1, 3.0 Hz, 1H).
A mixture of (6-bromo-pyridin-2-yl)-thiophen-3-yl-methanone (2.5 g, 9.3 mmol),
2-Methyl-propane-2-sulfinic acid amide (1.7 g, 14 mmol) and titanium tetraethoxide (4.3 g, 19 mmol) in THF was heated at reflux for 20h. The reaction mixture was cooled, diluted with water and stirred over ethyl acetate. The solids were removed by filtration through celite. Organic layer separated, washed with brine, dried over sodium sulfate and concentrated. The residue was purified by flash chromatography (silica gel 0-100% EtO Ac/heptane) to afford the
2-Methyl-propane-2-sulfinic acid l-(6-bromo-pyridin-2-yl)-l-thiophen-3-yl-meth-(Z)-ylideneamide (2.1 g, 61%). MS (ESI): m+H = 373.
To a solution of ethyl acetate in dry THF (0.47 g, 5.4 mmol) cooled in dry-ice-acetone bath was added 2M LDA in heptane/ethylbenzene (2.7 mL, 5.4 mmol) and the mixture was stirred for 10 min and a solution of 2-methyl-propane-2-sulfinic acid
l-(6-bromo-pyridin-2-yl)-l-thiophen-3-yl-meth-(Z)-ylideneamide (1.0 g, 2.7 mmol) in THF (3 mL) was added slowly. The resulting misture was stirred for 10 min, quenched with saturated ammonium chloride, allowed to warm to ambient temperature and extracted with ethyl acetate. The organic layer separated, washed with brine, dried over sodium sulfate and concentrated. The was purified by flash chromatography (silica gel 0-100%) EtO Ac/heptane) to afford
3-(6-bromo-pyridin-2-yl)-3-(2-methyl-propane-2-sulfinylamino)-3-thiophen-3-yl-propionic acid ethyl ester (1.1 g,). MS (ESI): m+H = 461.
3-(6-Bromo-pyridin-2-yl)-3-(2 -methyl-propane -2-sulfinylamino)-3-thiophen-3-yl-propionic acid ethyl ester (1.1 g, 2.0 mmol) was dissolved in DCM ( 5 mL) and 4N HCl-l,4-dioxane (4 mL, 16 mmol) was added and the mixture stirred for 15 min. The solvents were removed and to the residue were added (2-Chloro-phenylsulfanyl)-acetic acid (0.52 g, 2.6 mmol) HATU (1.1 g, 2.8 mmol) and DMF (5 mL) followed by DIPEA (1.4 mL, 7.8 mmol). The resulting mixture was stirred 1 h and then diluted with water and extracted with ethyl acetate. The organic layer was washed with brine several times, dried over sodium sulfate and concentrated. The residue was dissolved in toluene (5 ml) and 25 %> sodium methoxide in methanol (1.5 mL, 6.5 mmol) was added and the resulting dark solution heated at 80 °C for 15 min. The reaction mixture was cooled, acidified with IN HC1 and extracted with ethyl acetate. The organic layer washed with brine, dried over sodium sulfate and concentrated.
Purification of the residue by column chromatography (silica gel, 0-100% EtO Ac/heptane) afforded 6-(64oromo-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione (0.56 g). MS (ESI): m+H = 566. 1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, lH), 8.68 (s, IH), 7.73 - 7.61 (m, 2H), 7.61 - 7.53 (m, 2H), 7.43 - 7.33 (m, 4H), 7.30 (dd, J = 8.0, 1.3 Hz, IH), 7.03 - 6.93 (m, IH), 6.77 - 6.69 (m, IH), 5.84 (dd, J = 8.0, 1.5 Hz, IH), 3.49 (s, 2H).
A mixture of
6-(6-bromo-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione (0.50 g, 1 mmol), 4-fluoroaniline (0.22 g, 2 mmol) and Brettphos-Admix (0.13 g, 0.1 mmol) and sodium tert-butoxide (0.3 g, 3 mmol) in a mixture of tert-butanol and 1,4-dioxane (1 : 1 mixture, 10 ml) was heated 105 °C in a sealed tube for lh. The reaction mixture was cooled and the solid collected by filtration. The solid was acidified with IN HCl and then dissolved in ethyl acetate. The ethyl acetate layer washed with brine, dried over sodium sulfate and concentrated. Purification by column chromatography (silica gel, 20-100% EtO Ac/heptane) afforded
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione (racemate) (0.30 g, 56%). MS(ESI): m+H = 524; IH NMR (400 MHz, DMSO-d6) δ 11.48 (s, IH), 9.12 (s, IH), 8.37 (s, IH), 7.66 - 7.57 (m, 3H), 7.53 (dd, J = 5.1, 3.0 Hz, IH), 7.39 (dd, J = 3.0, 1.4 Hz, IH), 7.30 (dd, J = 7.9, 1.3 Hz, IH), 7.18 (dd, J = 5.1, 1.4 Hz, IH), 7.11 - 7.03 (m, 3H), 6.99 - 6.92 (m, 1H), 6.81 (ddd, J = 8.5, 7.3, 1.3 Hz, 1H), 6.72 (dd, J = 8.3, 0.7 Hz, IH), 6.06 (dd, J = 8.0, 1.5 Hz, IH), 3.77 (d, J = 16.4 Hz, IH), 3.45 (d, J = 16.5 Hz, IH).
Enantiomer 1 : Chiral SFC (column: AS, EtOH w/0.1 %FA) : RT = 0.892 min. ; 1 H NMR (400
MHz, DMSO-d6) δ 11.49 (s, IH), 9.12 (s, IH), 7.67 - 7.57 (m, 3H), 7.52 (dd, J= 5.1, 2.9Hz, IH), 7.39 (dd, J= 3.0, 1.4 Hz, IH), 7.30 (dd, J= 7.9, 1.3 Hz, IH), 7.18 (dd, J= 5.1, 1.4 Hz, IH), 7.12- 7.00 (m, 3H), 6.99 - 6.92 (m, IH), 6.85 - 6.78 (m, IH), 6.72 (d, J= 8.3 Hz, IH), 6.06 (dd, J= 8.0, 1.5Hz, IH), 3.76 (d, J= 16.5 Hz, IH), 3.44 (d, J= 16.5 Hz, IH).
Entantiomer 2: Chiral SFC (column: AS, EtOH w/0.1%FA): RT = 1.276 min. IH NMR (400 MHz, DMSO-d6) δ 11.48 (s, IH), 9.12 (s, IH), 8.36 (s, IH), 7.67 - 7.55 (m, 3H), 7.53(dd, J= 5.0, 3.0 Hz, IH), 7.39 (dd, J= 3.0, 1.4 Hz, IH), 7.30 (dd, J= 7.9, 1.3 Hz, IH), 7.18 (dd, J= 5.1,1.4 Hz, IH), 7.11 - 7.02 (m, 3H), 7.00 - 6.93 (m, IH), 6.86 - 6.78 (m, IH), 6.72 (d, J= 8.3 Hz, IH), 6.06 (dd, J= 7.9, 1.5 Hz, IH), 3.77 (d, J= 16.4 Hz, IH), 3.45 (d, J= 16.4 Hz, IH).
Example 418

4-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3 hienyl)-2-piperidyl]-2-pyridyl]-N,N-dimethyl4oen zenesulfonamide (racemate)
A mixture of 6-(64oromo-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine- 2,4-dione (0.50 g, 1 mmol), 4-fluoroanili (0.06 g, 0.10 mmol),
N,N-dimethyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzenesulfonamide (0.05 g, 0.12 mmol), PdCl2(PPh3)2 (0.015 g, 0.014 mmol) and sodium carbonate (0.05 g, 0.47 mmol) in
1,4-dioxane was heated at 110 °C for 20 min in a microwave reactor. The reaction mixture was cooled, acidified by with IN HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification by column chromatography (silica gel, 0-100% EtO Ac/heptane) afforded
4-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-2-pyridyl]-N,N-dimethyl-ben zenesulfonamide (0.04 g, ): MS(ESI): m+H = 598; 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.65 (s, 1H), 8.51 - 8.43 (m, 2H), 8.16 - 8.03 (m, 2H), 7.88 - 7.81 (m, 2H), 7.74 (d, J = 7.7 Hz, 1H), 7.54 (dd, J = 5.1, 3.0 Hz, 1H), 7.38 (dd, J = 3.0, 1.4 Hz, 1H), 7.26 (dd, J = 7.9, 1.2 Hz, 1H), 7.19 (dd, J = 5.0, 1.3 Hz, lH), 6.89 (td, J = 7.7, 1.5 Hz, 1H), 6.56 (td, J = 7.7, 1.3 Hz, 1H), 5.81 (dd, J = 8.0, 1.5 Hz, 1H), 4.14 (d, J = 16.3 Hz, 1H), 3.45 (d, J = 16.2 Hz, 1H), 2.65 (s, 6H).
Example 419
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione

419-1 419-2 419
A mixture
6-(6-bromopyridin-2-yl)-3-((2-chlorophenyl)thio)-6-(thiophen-3-yl)piperidine-2,4-dione (0.50 g, 1 mmol), 4-fluorophenol (0.34 g, 3 mmol), Pd2(dba)3 (0.10 g, O.lmmol), (0.10 g, (0.10 g, 0.24 mmol) and sodium tert-butoxide (0.3 g, 3 mmol) in 1,4-dioxane (10 ml) was heated at 110 °C for 30 min in the microwave reactor. The reaction mixture was cooled and the solid collected by filtration. The solid was acidified with IN HCl and then dissolved in ethyl acetate. The ethyl acetate layer washed with brine, dried over sodium sulfate and concentrated. Purification by column chromatography (silica gel, 20-100% EtO Ac/heptane) afforded
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione racemate (0.30 g, 56%). LC/MS: m+H = 525. 1H NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H), 8.43 (s, 1H), 7.98 - 7.87 (m, 1H), 7.50 (dd, J = 5.1, 3.0 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H), 7.32 - 7.22 (m, 3H), 7.16 - 7.08 (m, 2H), 7.08 - 6.91 (m, 3H), 6.84 - 6.75 (m, 1H), 5.95 (dd, J = 8.0, 1.5 Hz, 1H), 3.57 (d, J = 16.5 Hz, 1H), 3.25 (s, 1H).
Enantiomer 1 : Chrial SFC (Column: AD; MeOH/0.1 % NH4OH): RT = 0.521
Enantiomer 2: Chrial SFC (Column: AD; MeOH/0.1 % NH4OH): RT = 0.775
Example 420
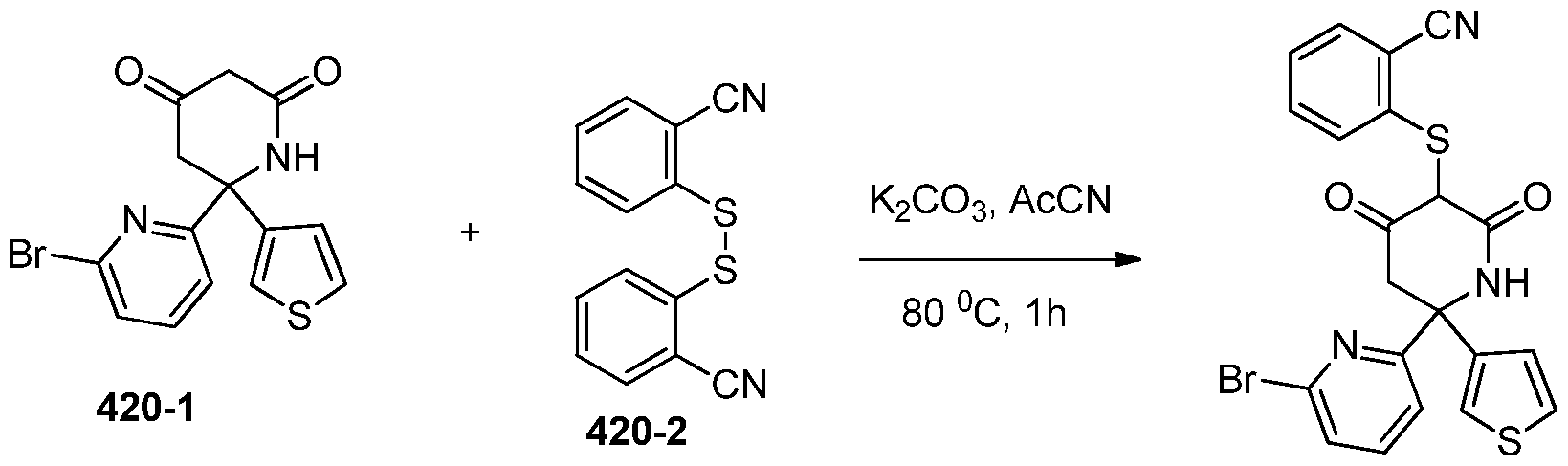
2-[[6-(6-bromo-2-pyridyl)-2,4-dioxo-6-(3-thienyl)-3-piperidyl]sulfanyl]benzonitrile (MD)
A mixture of 6-(6-bromo-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione (0.14 g, 0.40 mmol), 2-[(2-cyanophenyl)disulfanyl]benzonitrile (0.21 g, 0.80 mmol) and potassium carbonate (0.17 g, 1.20 mmol) was heated in acetonitrile (5 mL) for lh. The reaction mixture was cooled, acidified with dil HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by column chromatography (silica gel, 0-100% EtO Ac/heptane) afforded
2-[[6-(6-bromo-2-pyridyl)-2,4-dioxo-6-(3-thienyl)-3-piperidyl]sulfanyl]benzonitrile (0.04 g, 20%): MS (ESI): m+H = 484; 1H NMR (400 MHz, DMSO-d6) δ 11.94 (s, 1H), 8.58 (s, 1H), 7.86 (t, J = 7.8 Hz, 1H), 7.72 - 7.62 (m, 3H), 7.56 (dd, J = 5.1, 3.0 Hz, 1H), 7.37 - 7.30 (m, 1H), 7.15 (td, J = 5.2, 4.6, 1.7 Hz, 3H), 6.16 - 6.00 (m, 1H), 3.84 (d, J = 16.4 Hz, 1H), 3.43 (d, J = 16.4 Hz, 1H).
Example 421

A mixture of 6-(3-bromophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione (0.50 g, 1 mmol), 4-fluorophenol (0.34 g, 3 mmol), Pd2(dba)3 (0.10 g, O.lmmol), (0.10 g, (0.10 g, 0.24 mmol) and sodium tert-butoxide (0.3 g, 3 mmol) in 1,4-dioxane (10 ml) was heated at 110 °"
C for 30 min. . The reaction mixture was cooled, acidified by with IN HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification by column chromatography (silica gel, 0-100%) EtO Ac/heptane) afforded 3 -(2-chlorophenyl)sulfanyl-6-(3-hydroxyphenyl)-6-(3-thienyl)piperidine-2,4-dione (25 mg): MS (ESI): m+H = 430; DMSO-d6) δ 11.44 (s, 1H), 9.43 (s, 1H), 8.33 (s, 1H), 7.56 (dd, J =
5.1, 2.9 Hz, lH), 7.35 (dd, J = 3.0, 1.4 Hz, 1H), 7.28 (dd, J = 8.0, 1.3 Hz, 1H), 7.20 - 7.13 (m, 2H), 7.02 - 6.92 (m, 1H), 6.87 - 6.67 (m, 4H), 5.94 (dd, J = 8.0, 1.5 Hz, 1H), 3.44 - 3.36 (m, 2H).
Example 422

To a solution of 1-7 (0.21 g, 0.60 mmol) in DCM (10 mL) was added NBS ( 0.10 g, 0.60 mmol) and the resulting mixture was stirred for lh. The reaction mixture was washed with water, brine, dried over sodium sulfate and concentrated. The resulting residue was dissolved in acetonitrile (5 ml) and
2- chlorophenol (0.15 g, 1.2 mmol) and potassium carbonate (0.16 g, 1.2 mmol) were added and the mixture heated at 85 °C for 20 h. The reaction mixture was cooled, acidified with dil.HCl and extracted with ethyl acetate. Organic layer washed with brine, dried over sodium sulfate and concentrated. Purification by column chromatography (silica gel, 0-100% EtO Ac/heptane) afforded 6-(6-bromo-2-pyridyl)-3-(2-chlorophenoxy)-6-(3-thienyl)piperidine-2,4-dione (0.20 g, 70 %): MS (ESI): m+H = 479; 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.33 (s, 1H), 7.89 - 7.81 (m, 1H), 7.70 - 7.61 (m, 2H), 7.58 - 7.51 (m, 1H), 7.43 - 7.29 (m, 2H), 7.15 (dd, J = 5.1, 1.4 Hz, 1H), 6.99 - 6.83 (m, 2H), 6.13 (dd, J = 8.2, 1.5 Hz, 1H), 3.74 - 3.66 (m, 1H), 3.36 (d, J = 16.2 Hz, 1H).
6-(6-bromo-2-pyridyl)-3-(2-chlorophenoxy)-6-(3-thienyl)piperidine-2,4-dione (0.05 g, 0.10 mmol) was converted to
3- (2-chlorophenoxy)-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione (0.42 mg, 80%) as described previously: MS(ESI): m+H = 508. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.04 (s, 1H), 7.66 - 7.57 (m, 3H), 10.87 - 10.41 (m, 1H), 7.49 (dd, J = 5.1, 3.0 Hz, 1H), 7.40 (dd, J = 3.0, 1.4 Hz, 1H), 7.34 (dd, J = 7.8, 1.7 Hz, 1H), 7.17 (dd, J = 5.1, 1.4 Hz, 1H), 7.10 - 6.97 (m, 3H), 6.97 - 6.83 (m, 2H), 6.70 (d, J = 8.2 Hz, 1H), 6.17 (dd, J = 8.2, 1.6 Hz, 1H), 3.63 (d, J = 16.2 Hz, 1H), 3.36 (s, 1H).
Example 423

423
423-1
A mixture of
6-(3-bromophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione (0.07 g, 0.14 mmol), tert-butyl carbamate (0.05 g, 0.43 mmol), Brettphos-Admix (0.02 g, 0.02 mmol) and sodium tert-butoxide (0.04 mg, 0.43 mmol) in tert-butanol was heated at 120 °C for lh. The reaction mixture was cooled, acidified with dil HCl and extracted with ethyl acetate. The organic layer was washed with brine dried over sodium sulfate and concentrated. The residue was dissolved in DCM (2 mL) and 4N-HCl-l,4-dioxane was added and stirred for 30 min. The reaction mixture was concentrated, treated with sodium bicarbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate. Purification by column chromatography (silica gel, 0-100% iPrAc/heptane) afforded
6-(3-aminophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione (0.02 g, 32%)> MS(ESI): m+H = 429; 1H NMR (400 MHz, DMSO-d6) δ 8.29 (s, 1H), 7.56 (dd, J = 5.0, 3.0 Hz, 1H), 7.34 (dd, J = 2.9, 1.4 Hz, 1H), 7.29 (dd, J = 7.9, 1.3 Hz, 1H), 7.16 (dd, J = 5.1, 1.4 Hz, 1H), 7.05 - 6.95 (m, 2H), 6.85 - 6.75 (m, 1H), 6.61 (t, J = 2.0 Hz, 1H), 6.56 - 6.48 (m, 2H), 5.96 (dd, J = 7.9, 1.5 Hz, 1H), 3.42 - 3.33 (m, 2H).
Example 424

424-13
All steps and conditions are described in examples hereinabove.
Example 425
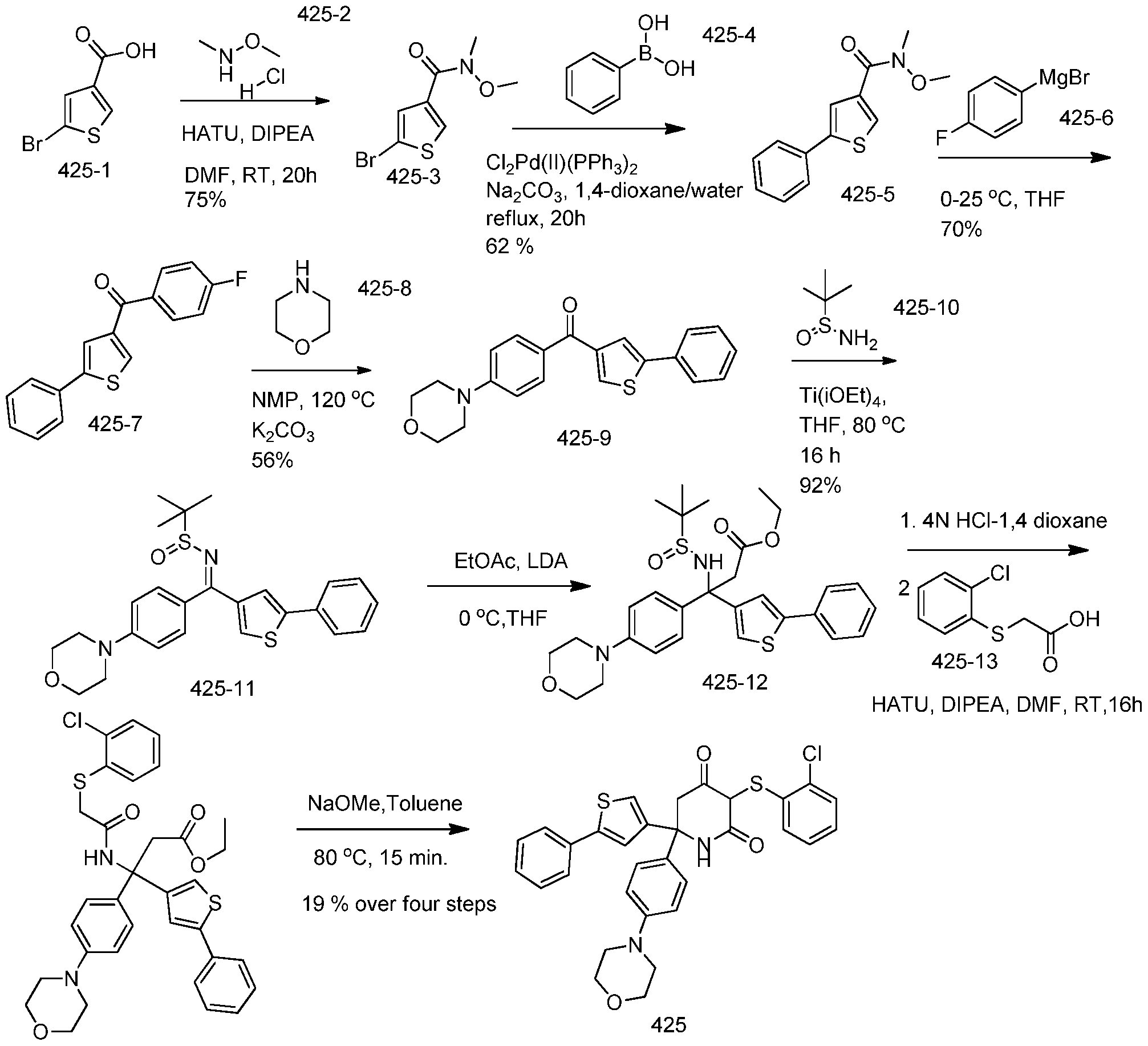
425-14
All steps and conditions are described in examples hereinabove.






example 417

example 417

ST: Stereochemistry : SS = Single Stereoisomer; MD = Mixture of Diastereoisomers
LDHA Enzyme Inhibition Assay Protocol
Human recombinant carboxy-terminal his-tagged LDHA (amino acids 2-332) was expressed and purified from E. coli. The enzyme assay was performed in uClear low volume 384-well plates
(Greiner #788092), 10 uL volume with the following final enzyme and buffer conditions: 50 mM Hepes (pH 7.2), 0.01% (v/v) TritonX-100, 0.01% (0.1 mg/mL) Bovine Gamma Globulin, 2 mM DTT, 1 nM LDHA, 50 uM NADH , and 50 uM pyruvate. Test compounds were diluted in 100% DMSO with 1 : 3 serial dilutions. Oxamate (Sigma #02751 ) was used as a positive control and was diluted in H20 (10-point 1 :3 serial dilutions, final DMSO 1%>). For the enzyme reaction, serially diluted compounds were added to a mixture of enzyme and NADH. The assay plates were then incubated at room temperature for 10 minutes and a baseline read was conducted on the FDSS700 (Hamamatsu) with excitation at 340 nm and emission at 480 nm for 12.5 seconds to identify any compounds which interfere with NADH fluorescence. Following the baseline read, pyruvate was added to the assay plates and the plates were read with excitation 340 nm and emission 480 nm for 10 minutes every 2.5 seconds. A suitable linear timeframe was selected (150-400 s) to calculate the slope of each concentration tested. The raw data were fitted to 4-parameter dose-response curves using the following equation:
fit = (A+((B-A)/(l+((C/x) D))))
inv = (C/((((B-A)/(y-A))-l)-(l/D)))
where A = minimum y, B = maximum y, C = 50% y max, and D = slope factor. The curve bottom was set to the background rate (initial 5 second recording prior to addition of pyruvate) and curve top was set to no inhibitor (DMSO only) control wells rate. Oxamate was used as a positive control and exhibited a mean IC50 value of 57.2 uM ± 13.1 uM (n = 202). For previous descriptions of LDH enzyme assays, see: Rossmann, M. G. et al. Evolutionary and structural relationships among dehydrogenases. In: Boyer, P. D. Ed., The Enzymes, vol. XL New York: Academic Press, 1975; pp61-102. See also the Supplementary Material section of: Moorhouse, A. D. et a. Chem. Commun. 2011, 47, 230.
The compounds of the present invention were tested for their capacity to inhibit LDHA activity and activation as described in the enzyme inhibition assay described above. The following table summarizes the results of this assay by reference to the exemplified compounds of the invention:

Mixture of Diastereomers 0.028
Mixture of Diastereomers 0.009
Mixture of Diastereomers 0.066
Single Stereoisomer 0.008
Single Stereoisomer 0.002
Mixture of Diastereomers 0.165
Mixture of Diastereomers 0.009
Mixture of Diastereomers 0.004
Single Stereoisomer 0.026
Mixture of Diastereomers 0.013
Mixture of Diastereomers 0.042
Single Stereoisomer 0.004
Single Stereoisomer 0.002
Mixture of Diastereomers 0.027
Mixture of Diastereomers 0.058
Mixture of Diastereomers 0.098
Mixture of Diastereomers 0.004
Mixture of Diastereomers 0.063
Mixture of Diastereomers 0.016
Mixture of Diastereomers 0.092
Mixture of Diastereomers 0.022
Mixture of Diastereomers 0.043
Mixture of Diastereomers 0.008
Mixture of Diastereomers 0.105
Mixture of Diastereomers 0.076
Mixture of Diastereomers 0.114
Mixture of Diastereomers 0.023
Mixture of Diastereomers 1.270
Mixture of Diastereomers 0.012
Single Stereoisomer 0.006
Single Stereoisomer 0.203
Mixture of Diastereomers 0.005
Mixture of Diastereomers 0.039
Mixture of Diastereomers 0.119
Mixture of Diastereomers 0.086
Mixture of Diastereomers 0.026
Mixture of Diastereomers 0.018
Mixture of Diastereomers 0.023
Mixture of Diastereomers 0.038
Mixture of Diastereomers 0.050
Mixture of Diastereomers 0.031
Mixture of Diastereomers 0.009
Mixture of Diastereomers 0.053
Mixture of Diastereomers 0.008
Mixture of Diastereomers 0.010
Mixture of Diastereomers 0.007
Mixture of Diastereomers 0.033
Mixture of Diastereomers 0.007
Mixture of Diastereomers 0.021
Mixture of Diastereomers 0.008
Mixture of Diastereomers 0.025
Mixture of Diastereomers 0.034
Mixture of Diastereomers 0.029
Mixture of Diastereomers 0.040
Mixture of Diastereomers 0.004
Mixture of Diastereomers 0.003
Mixture of Diastereomers 0.012
Mixture of Diastereomers 0.036
Mixture of Diastereomers 0.016
Mixture of Diastereomers 0.005
Mixture of Diastereomers 0.010
Mixture of Diastereomers 0.017
Mixture of Diastereomers 0.017
Mixture of Diastereomers 0.022
Mixture of Diastereomers 0.069
Mixture of Diastereomers 0.005
Mixture of Diastereomers 0.120
Mixture of Diastereomers 0.052
Mixture of Diastereomers 0.025
Mixture of Diastereomers 0.022
Mixture of Diastereomers 0.149
Mixture of Diastereomers 0.045
Mixture of Diastereomers 0.011
Mixture of Diastereomers 0.201
Mixture of Diastereomers 0.029
Mixture of Diastereomers 0.007
Mixture of Diastereomers 0.086
Mixture of Diastereomers 0.010
Mixture of Diastereomers 0.056
Mixture of Diastereomers 0.009
Mixture of Diastereomers 0.020
Mixture of Diastereomers 0.036
Mixture of Diastereomers 0.037
Mixture of Diastereomers 0.126
Mixture of Diastereomers 0.056
Mixture of Diastereomers 0.089
Mixture of Diastereomers 0.224
Mixture of Diastereomers 0.008
Single Stereoisomer 0.004
Single Stereoisomer 0.104
Mixture of Diastereomers 0.056
Mixture of Diastereomers 0.025
Single Stereoisomer 0.111
Single Stereoisomer 0.014
Mixture of Diastereomers 0.070
Mixture of Diastereomers 0.015
Mixture of Diastereomers 0.031
Single Stereoisomer 0.023
Single Stereoisomer 0.199
Mixture of Diastereomers 0.056
Mixture of Diastereomers 0.229
Mixture of Diastereomers 0.049
Mixture of Diastereomers 0.013
Mixture of Diastereomers 0.008
Mixture of Diastereomers 0.018
Mixture of Diastereomers 0.022
Mixture of Diastereomers 0.010
Mixture of Diastereomers 0.017
Mixture of Diastereomers 0.018
Mixture of Diastereomers 0.013
Mixture of Diastereomers 0.011
Single Stereoisomer 0.007
Single Stereoisomer 0.319
Single Stereoisomer 0.023
Single Stereoisomer 0.290
Mixture of Diastereomers 0.019
Mixture of Diastereomers 0.030
Mixture of Diastereomers 0.167
Mixture of Diastereomers 0.006
Single Stereoisomer 0.046
Mixture of Diastereomers 0.018
Mixture of Diastereomers 0.018
Mixture of Diastereomers 0.141
Mixture of Diastereomers 0.118
Single Stereoisomer 0.125
Single Stereoisomer 0.732
Mixture of Diastereomers 0.131
Mixture of Diastereomers 0.108
Mixture of Diastereomers 0.010
Mixture of Diastereomers 0.018
Mixture of Diastereomers 0.012
Mixture of Diastereomers 0.010
Single Stereoisomer 0.014
Single Stereoisomer 0.053
115 Single Stereoisomer 0.031
116 Single Stereoisomer 0.376
117 Mixture of Diastereomers 0.076
118 Single Stereoisomer 0.622
119 Single Stereoisomer 0.293
120 Mixture of Diastereomers 0.016
121 Mixture of Diastereomers 0.008
122 Single Stereoisomer 0.022
123 Single Stereoisomer 0.006
124 Mixture of Diastereomers 0.003
125 Mixture of Diastereomers 0.003
126 Mixture of Diastereomers 0.238
127 Mixture of Diastereomers 0.008
128 Mixture of Diastereomers 0.024
129 Mixture of Diastereomers 0.020
130 Mixture of Diastereomers 0.005
131 Mixture of Diastereomers 0.046
132 Mixture of Diastereomers 0.009
133 Mixture of Diastereomers 0.089
134 Single Stereoisomer 0.008
135 Mixture of Diastereomers 0.040
136 Single Stereoisomer 0.222
137 Mixture of Diastereomers 0.073
138 Mixture of Diastereomers 0.025
139 Mixture of Diastereomers 0.009
140 Mixture of Diastereomers 0.012
141 Single Stereoisomer 0.003
142 Single Stereoisomer 0.031
143 Mixture of Diastereomers 0.033
144 Single Stereoisomer 0.143
145 Single Stereoisomer 0.088
146 Single Stereoisomer 0.189
147 Single Stereoisomer 0.008
148 Mixture of Diastereomers 0.014
149 Mixture of Diastereomers 0.069
150 Mixture of Diastereomers 0.019
151 Mixture of Diastereomers 0.015
152 Mixture of Diastereomers 0.006
153 Single Stereoisomer 0.012
154 Mixture of Diastereomers 0.027
155 Mixture of Diastereomers 0.025
156 Single Stereoisomer 0.072
157 Mixture of Diastereomers 0.030
158 Mixture of Diastereomers 0.018
159 Mixture of Diastereomers 0.004
160 Mixture of Diastereomers 0.008
161 Single Stereoisomer 0.019
162 Single Stereoisomer 0.004
163 Single Stereoisomer 0.194
164 Single Stereoisomer 0.009
165 Single Stereoisomer 0.004
166 Mixture of Diastereomers 0.004
167 Mixture of Diastereomers 0.006
168 Single Stereoisomer 0.190
169 Mixture of Diastereomers 0.010
170 Single Stereoisomer 0.145
171 Single Stereoisomer 0.101
172 Single Stereoisomer 0.056
173 Single Stereoisomer 0.183
174 Single Stereoisomer 0.068
175 Single Stereoisomer 0.013
176 Mixture of Diastereomers 0.010
177 Mixture of Diastereomers 0.016
178 Single Stereoisomer 0.155
179 Single Stereoisomer 0.014
180 Single Stereoisomer 0.002
181 Single Stereoisomer 0.006
182 Mixture of Diastereomers 0.056
183 Single Stereoisomer 0.005
184 Single Stereoisomer 0.002
185 Mixture of Diastereomers 0.055
186 Mixture of Diastereomers 0.021
187 Mixture of Diastereomers 0.025
188 Mixture of Diastereomers 0.015
189 Single Stereoisomer 0.150
190 Single Stereoisomer 0.092
191 Single Stereoisomer 0.158
192 Single Stereoisomer 0.232
193 Single Stereoisomer 0.059
194 Single Stereoisomer 0.005
195 Single Stereoisomer 0.143
196 Single Stereoisomer 0.036
197 Single Stereoisomer 0.010
198 Single Stereoisomer 0.177
199 Single Stereoisomer 0.012
200 Mixture of Diastereomers 0.229
201 Single Stereoisomer 0.037
202 Single Stereoisomer 0.018
203 Single Stereoisomer 0.044
204 Mixture of Diastereomers 0.007
205 Mixture of Diastereomers 0.058
206 Mixture of Diastereomers 0.027
207 Mixture of Diastereomers 0.020
208 Mixture of Diastereomers 0.149
209 Mixture of Diastereomers 0.006
210 Mixture of Diastereomers 0.008
211 Mixture of Diastereomers 0.183
212 Mixture of Diastereomers 0.030
213 Mixture of Diastereomers 0.034
214 Mixture of Diastereomers 0.017
215 Mixture of Diastereomers 0.014
216 Single Stereoisomer 0.002
217 Single Stereoisomer 0.010
218 Single Stereoisomer 0.117
219 Single Stereoisomer 0.240
220 Single Stereoisomer 0.162
221 Single Stereoisomer 0.320
222 Single Stereoisomer 0.322
223 Single Stereoisomer 0.057
224 Mixture of Diastereomers 0.020
225 Mixture of Diastereomers 0.103
226 Single Stereoisomer 0.020
227 Single Stereoisomer 0.015
228 Single Stereoisomer 0.419
229 Mixture of Diastereomers 0.011
230 Mixture of Diastereomers 0.022
231 Mixture of Diastereomers 0.009
232 Mixture of Diastereomers 0.009
233 Mixture of Diastereomers 0.017
234 Mixture of Diastereomers 0.004
235 Mixture of Diastereomers 0.021
236 Mixture of Diastereomers 0.025
237 Single Stereoisomer 0.094
238 Mixture of Diastereomers 0.018
239 Single Stereoisomer 0.015
240 Single Stereoisomer 0.003
241 Single Stereoisomer 0.003
242 Mixture of Diastereomers 0.004
243 Single Stereoisomer 0.001
244 Single Stereoisomer 0.023
245 Mixture of Diastereomers 0.018
246 Single Stereoisomer 0.010
247 Single Stereoisomer 0.010
248 Single Stereoisomer 0.005
249 Mixture of Diastereomers 0.099
250 Single Stereoisomer 0.036
251 Single Stereoisomer 0.019
252 Mixture of Diastereomers 0.079
253 Mixture of Diastereomers 0.081
254 Single Stereoisomer 0.047
255 Mixture of Diastereomers 0.102
256 Single Stereoisomer 0.062
257 Mixture of Diastereomers 0.016
258 Mixture of Diastereomers 0.087
259 Single Stereoisomer 0.011
260 Single Stereoisomer 0.006
261 Single Stereoisomer 0.039
262 Mixture of Diastereomers 0.104
263 Mixture of Diastereomers 0.086
264 Mixture of Diastereomers 0.077
265 Mixture of Diastereomers 0.022
266 Single Stereoisomer 0.099
267 Single Stereoisomer 1.640
268 Single Stereoisomer 0.402
269 Single Stereoisomer 0.073
270 Single Stereoisomer 0.099
271 Single Stereoisomer 0.355
272 Single Stereoisomer 0.459
273 Single Stereoisomer 0.072
274 Single Stereoisomer 0.157
275 Single Stereoisomer 0.233
276 Single Stereoisomer 0.175
277 Single Stereoisomer 0.337
278 Single Stereoisomer 0.339
279 Single Stereoisomer 0.272
280 Single Stereoisomer 0.311
281 Single Stereoisomer 0.189
282 Mixture of Diastereomers 0.012
283 Mixture of Diastereomers 0.037
284 Mixture of Diastereomers 0.041
285 Single Stereoisomer 0.010
286 Mixture of Diastereomers 0.057
287 Mixture of Diastereomers 0.010
288 Mixture of Diastereomers 0.141
289 Mixture of Diastereomers 0.022
290 Mixture of Diastereomers 0.017
291 Single Stereoisomer 0.093
292 Mixture of Diastereomers 0.037
293 Mixture of Diastereomers 0.091
294 Mixture of Diastereomers 0.036
295 Single Stereoisomer 0.081
296 Mixture of Diastereomers 0.039
297 Single Stereoisomer 0.149
298 Single Stereoisomer 0.063
300 Mixture of Diastereomers 0.144
301 Mixture of Diastereomers 0.092
302 Mixture of Diastereomers 0.030
303 Single Stereoisomer 0.434
304 Mixture of Diastereomers 0.021
305 Mixture of Diastereomers 0.025
306 Single Stereoisomer 0.129
307 Single Stereoisomer 0.095
308 Single Stereoisomer 0.825
309 Single Stereoisomer 0.408
310 Single Stereoisomer 0.365
311 Single Stereoisomer 0.590
312 Single Stereoisomer 0.119
313 Single Stereoisomer 0.105
314 Single Stereoisomer 0.082
315 Mixture of Diastereomers 0.014
316 Mixture of Diastereomers 0.044
317 Mixture of Diastereomers 0.225
318 Single Stereoisomer 0.010
319 Single Stereoisomer 0.197
320 Single Stereoisomer 0.093
321 Single Stereoisomer 0.246
322 Single Stereoisomer 0.012
323 Single Stereoisomer 0.035
324 Single Stereoisomer 0.084
325 Mixture of Diastereomers 0.162
326 Mixture of Diastereomers 0.076
327 Single Stereoisomer 0.035
328 Mixture of Diastereomers 0.054
329 Mixture of Diastereomers 0.037
330 Single Stereoisomer 0.041
331 Single Stereoisomer 0.148
332 Single Stereoisomer 0.168
333 Single Stereoisomer 0.198
334 Single Stereoisomer 0.739
335 Single Stereoisomer 0.002
336 Mixture of Diastereomers 0.368
337 Single Stereoisomer 0.021
338 Single Stereoisomer 0.009
408 Single Stereoisomer 0.005
The foregoing description is considered as illustrative only of the principles of the invention. Further, since numerous modifications and changes will be readily apparent to those skilled in the art, it is not desired to limit the invention to the exact construction and process shown as described above. Accordingly, all suitable modifications and equivalents may be considered to fall within the scope of the invention as defined by the claims that follow.
The words "comprise," "comprising," "include," "including," and "includes" when used in this specification and in the following claims are intended to specify the presence of stated features, integers, components, or steps, but they do not preclude the presence or addition of one or more other features, integers, components, steps, or groups thereof.
Cell Culture and Analyses
The CHO cell line 1 used in this work was derived from a CHO-K1 host that had been stably transfected to produce a recombinant humanized monoclonal antibody based on a previously described method (Hu et al., 2013). A vial of cryopreserved cells that was thawed and subcultured in shake flasks provided the cell source for the experiment. To inoculate the production cultures for CHO cell line 1 , cells from the common shake flask source were centrifuged (820 g, 5 min) and resuspended at a viable cell density (VCD) of ~1 x 106 cells/mL, in a proprietary, chemically-defined production medium with the target concentrations of Gx (0 μΜ, 1 μΜ, 10 μΜ, 50 μΜ, and 100 μΜ). Each of these five Gx concentrations was tested in duplicate 250 mL shake flasks (Corning) at 100 mL working volume. These ten production cultures were agitated with an orbital shaker (150 rpm, 25 mm throw) in a humidified incubator maintained at 37°C with a 5% C02 overlay. At three, seven, and ten days post-inoculation, a concentrated chemically-defined nutrient feed was added to each culture at 1 : 10 (v/v). All the production cultures were harvested fourteen days after inoculation.
The CHO cell line 2 used in this work originated from a different CHO-K1 host but was otherwise generated using largely similar methods as CHO cell line 1. Cell lines 1 and 2 produced different recombinant monoclonal antibody products, and were developed using different chemically-defined media. The preparation for initiating the production experiment with CHO cell line 2 was similar to that for CHO cell line 1. However, the production conditions for cell lines 1 and 2 were different. The most notable difference being that CHO cell line 2 was cultured in the
advanced microscale bioreactor (ambr™) system from TAP Biosystems, which we had previously determined to be a representative scale-down model for bench-top bioreactors (Hsu et al., 2012). Therefore, in contrast to cell line 1 that was cultured in shake flasks without pH or dissolved oxygen control, cell line 2 was cultured in bioreactors with online pH and dissolved oxygen monitoring and control. The production cultures for CHO cell line 2 were inoculated in ambr™ vessels at VCD of ~2 x 106 cells/mL, in a proprietary, chemically-defined production medium supplemented with the target concentrations of Gx (0 μΜ, 10 μΜ, 25 μΜ, and 50 μΜ). Each of these four Gx concentrations was tested in duplicate ambr™ vessels at ~14 mL working volume. These eight production cultures were maintained at 37°C for the first three days, and then at 35°C thereafter. The pH and dissolved oxygen were controlled at setpoints of 7.0 and 30% of air saturation as previously described (Hsu et a., 2012). At three, seven, and ten days post-inoculation, a concentrated chemically-defined nutrient feed was added to each culture at 1 : 10 (v/v). An additional amino acid feed was also supplemented to each bioreactor culture on day 7. All the production cultures were harvested fourteen days after inoculation. The production media and nutrient feed used for cell lines 1 and 2 were similar in being chemically-defined, but were different in the actual compositions.
Culture samples taken during the experiment were analyzed immediately for VCD and viability using the Vi-Cell XR (Beckman Coulter). For the shake flask samples from CHO cell line 1 , extracellular lactate and pH were determined by the Bioprofile 400 analyzer (Nova Biomedical). For the ambr™ bioreactor samples from CHO cell line 2, extracellular lactate was measured using a commercial lactate reagent kit (Cobas C(l l l) Lactate 100T, Roche Diagnostics) and spectrophotometric absorbance at 550 nm, and pH was determined by a pH probe (InLab Ultra-Micro Electrode, Mettler Toledo). Culture supematants were stored frozen at -80°C until they were analyzed for antibody titer as described previously (Hsu et al., 2012). The average cell-specific productivity, qp, was calculated as the slope of the linear regression from the plot of titer versus time integral of VCD.
Summary of Findings
Effect of Gx on CHO cell growth (Fig. 1), culture viability (Fig. 2), lactate production (Fig. 3), and product titer (Fig. 5) for cell line 1 was dose-dependent within the 1-100 μΜ range. In particular, lactate production decreased with increasing Gx concentration. However, there was no negative effect of Gx (within the 1-100 μΜ range) on cell-specific productivity (qp).
This proof-of-concept testing here demonstrates that Gx can lower lactate production in mammalian cell cultures used for generating recombinant proteins. However, the full potential of Gx is not realized in this testing because (1) the mammalian cells were cultured without pH control and (2) the cultivation and Gx feed strategies were not optimized.
CHO cells produce less lactate at lower pH (Trummer et al., 2006). It follows that in shake flask cultures without pH control or adjustment, a feedback regulation exists between culture pH and lactate production: culture pH decreases with lactate production, and the resulting decrease in culture pH causes the cells to produce less lactate. Since this negative feedback loop between lactate and pH does not apply in bioreactor cultures maintained at a constant pH, lactate levels are typically higher in bioreactor cultures than in shake flask cultures (Hsu et al., 2012; Yuk et al., 2015). Therefore, we expect Gx supplementation to have a larger effect on lowering lactate production in bioreactor cultures than in shake flask cultures.
To assess the effects of Gx on CHO cell cultures in a pH-controlled environment, we tested Gx supplementation on CHO cell line 2 in ambr™ bioreactors. CHO cell line 2 was derived from a different CHO-K1 host from the CHO cell line 1, it was also developed using different chemically-defined media. Therefore, the use of cell line 2 also assessed whether the lactate inhibition effects observed for Gx on cell line 1 would apply to other CHO cell lines. The production culture conditions used for cell line 2 also differed from cell line 1 in several aspects, including, but not limited to the following: (1) Cell line 2 was cultured in bioreactors with online pH and dissolved oxygen monitoring and control, whereas cell line 1 was cultured in shake flasks without pH and dissolved oxygen monitoring or control. (2) The production cultures for cell lines 1 and 2 used
different chemically-defined media and nutrient feeds. (3) The production culture for cell line 2 utilized a temperature shift to 35°C on day three, whereas no temperature shift was employed for cell line 1. (4) To inoculate the production vessels, cell line 2 was seeded at almost twice the VCD as cell line 1. These differences in the productions cultures for cell lines 1 and 2 should enable us to evaluate the broader applicability of the effects of Gx on CHO cell cultures.
For CHO cell line 2 cultured in ambr™ microscale bioreactors, we observed a dose-dependent effect of Gx on CHO cell growth (Fig. 7), culture viability (Fig. 8), lactate production (Fig. 9), and product titer (Fig. 11) within the 10-50 μΜ range. Consistent with the expectation that lactate production should be considerably higher in pH-controlled bioreactors than in shake flasks without pH control (Hsu et al., 2012; Yuk et al., 2015), cell line 2 bioreactor cultures showed significantly higher levels of extracellular lactate (Figure 9) compared to cell line 1 shake flask cultures (Figure 3).
For both cell lines 1 and 2, production cultures supplemented with 10 uM Gx yielded comparable cell growth (Figures 1 and 7), lower lactate production (Figures 3 and 9), and higher final product titers (Figures 5 and 11) than the control cultures (0 uM Gx).
Likewise, for both cell lines 1 and 2, production cultures supplemented with 50 uM Gx yielded notably lower growth (Figures 1 and 7) and lower lactate levels (Figures 3 and 9) than the control cultures (0 uM Gx), At the same time, when compared to the control cultures (0 uM Gx), these 50 uM Gx-supplemented cultures yielded higher final viabilities (Figures 2 and 8) and comparable final product titers (Figures 5 and 11).
Taken together, these Gx titration experiments using CHO cell lines 1 and 2 indicated that the ability of Gx to inhibit lactate production in CHO cell cultures should apply to different CHO cell lines in different culture conditions in a dose-dependent manner. For production cultures of cell lines 1 and 2 supplemented with Gx on day 0, increasing Gx levels > 10 uM decreased extracellular lactate levels (Figures 3 and 9), but Gx > 50 uM decreased cell growth (Figures 1 and 7). Therefore, an optimal initial Gx concentration between 10 to 50 uM should exist that capitalizes on the beneficial dose-dependent lactate inhibition effects of Gx while minimizing the negative growth effects
observed at Gx > 50 uM. Indeed, when cell line 2 was supplemented with 25 uM Gx on day 0 in production cultures, the final product titer was higher than for 10 and 50 uM Gx cultures (Figure 10). In particular, the day 14 product titer for the 25 uM Gx-supplemented cultures (~4.5 g/L) represented -50% titer increase over the 0 uM Gx control cultures (~3.0 g/L)
Potential Applications
There appears to be an optimal Gx concentration within the 10-50 μΜ range that can provide maximal cell culture benefit in terms of product yield by (i) minimizing negative impact of high Gx concentration (eg., 100 μΜ) on cell growth, (ii) minimizing negative impact of high Gx concentration (eg., 100 μΜ) on culture viability, (iii) maximizing the positive impact of increasing Gx concentration on lactate production.
To harness the full benefit of lactate inhibition properties exhibited by Gx, the following strategies may be applied:
(1) Production cultures of mammalian cells should be performed in bioreactors with pH and dissolved oxygen control, consistent with large-scale manufacturing practices for therapeutic products (e.g., recombinant monoclonal antibodies).
(2) The pH in these production bioreactors should be controlled at pH and temperature setpoints that provide optimal cell growth for the cell line used (e.g., pH 7.0, 7.1, 7.2, 7.3, or 7.4; temperature 36.0, 36.5, 37.0, or 37.5°C) during the initial cell growth phase (e.g., days 0-2, days 0-3, days 0-4, days 0-5, days 0-6, or days 0-7).
(3) When the bioreactor cultures approach or reach the desired high cell density at the end of the initial cell growth phase (e.g., on day 2, day 3, day 4, day 5, day 6, or day 7), the cultivation temperature may be maintained or dropped to a lower setpoint (e.g., 36, 35, 34, 33, 32 or 31°C) to slow down further cell growth during the production phase. As implied by their names, the main purpose of the initial growth phase is cell accumulation, and the main purpose of the production phase is product accumulation.
(4) To maximize the impact of Gx on lactate inhibition, while minimizing its negative impact on cell growth and viability, different Gx doses and Gx supplementation timings should be tested in combination for the cell line of choice to determine the optimal combination that generates the maximum product titer. For example, the combinations tested should include: (i) Gx supplementation in single or multiple doses (eg., 1, 2, 3, 4, 5, 6, or 7); (ii) Gx supplementation at different concentrations (e.g., 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, or 70 μΜ); (iii) Gx supplementation at different cultivation times (e.g., day 0, 1, 2, 3, 4, 5, 6, 7, 8, or 9). More specifically, one combination may comprise of a single dose of Gx supplemented to the cultures on day 3 at 25 μΜ. Another combination may comprise of two doses of Gx supplemented to the cultures at day 3 and day 6 at concentrations of 10 and 30 μΜ, respectively. Yet another combination may comprise of three doses of Gx supplemented to the cultures at day 0, day 3, and day 7 at concentrations of 5, 15, and 25 μΜ, respectively.
(5) For ease or flexibility of operations, Gx may be supplemented directly to the bioreactor cultures, or may be added to the nutrient feeds and supplemented to the bioreactor cultures as a component of the nutrient feed. The nutrient feeds containing Gx may be supplemented to the bioreactor cultures in a bolus fashion, or in a continuous fashion. When these Gx containing nutrient feeds are supplemented in a bolus fashion, they may be supplemented on a single day, or on two or more days (e.g., 3, 4, 5 days). When these Gx containing nutrient feeds are supplemented in a continuous fashion, they may be supplemented over a period of time that spans a portion of the culture duration (e.g., from days 2-6, 3-7, 4-8, 5-9, 2-10, 3-9 etc.).
Nomenclature
CHO Chinese Hamster Ovary
LDHA lactate dehydrogenase A
VCD Viable cell density
Gx: 5-(2-chlorophenyl)sulfanyl-4-hydr^
References
Altamirano C, Paredes C, Illanes A, Cairo JJ. Godia F. 2004. Strategies for fed-batch cultivation of t-PA producing CHO cells: substitution of glucose and glutamine and rational design of culture medium. J Biotechnol 110: 171-179.
Altamirano C, Illanes A, Becerra S, Cairo JJ, Godia F. 2006. Considerations on the lactate consumption by CHO cells in the presence of galactose. J Biotechnol 125:547-556.
Feron O. 2009. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol 92:329-333.
Gagnon M, Hiller G, Luan Y-T, Kittredge A, DeFelice J, Drapeau D. 2011. High-End pH-controlled delivery of glucose effectively suppresses lactate accumulation in CHO fed-batch cultures.
Biotechnol Bioeng 108: 1328-1337.
Hu Z, Guo D, Yip SSM, Zhan D, Misaghi S, Joly JC, Snedecor BR, Shen AY. 2013. Chinese hamster ovary (CHO) Kl host cell enables stable cell line development for antibody molecules which are difficult to express in DUXB 11 -derived dihydro folate reductase (DHFR) deficient host cell. Biotechnol Prog 29:980-985.
Hsu W-T, Aulakh RPS, Traul DL, Yuk IH. 2012. Advanced microscale bioreactor system: a representative scale-down model for bench-top bioreactors. Cytotechnology 64:667-678. Irani N, Beccaria AJ, Wagner R. 2002. Expression of recombinant cytoplasmic yeast pyruvate carboxylase for the improvement of the production of human erythropoietin by recombinant BHK-21 cells.
Kim SH, Lee GM. 2007a. Functional expression of human pyruvate carboxylase for reduced lactic acid formation of Chinese hamster ovary cells (DG44). Appl Microbiol Biotechnol 76:659-665. Kim SH, Lee GM. 2007b. Down-regulation of lactate dehydrogenase-A by siRNAs for reduced lactic acid formation of Chinese hamster ovary cells producing thrombopoietin. Appl Microbiol Biotechnol 74: 152-159.
Le H, Kabbur S, Pollastrini L, Sun Z, Mills K, Johnson K, Karypis G, Hu W-S. 2012. Multivariate analysis of cell culture bioprocess data— lactate consumption as a process indicator. J
Biotechnol 162:210-223.
Ma N, Ellet J, Okediadi C, Hermes P, McCormick E, Casnocha S. 2009. A single nutrient feed supports both chemically defined NS0 and CHO fed-batch cultures: improved productivity and lactate metabolism. Biotechnol Prog 25: 1353-1363.
Paredes C, Prats E, Cairo JJ, Azorin F, Cornudella L, Godia F. 1999. Modification of glucose and glutamine metabolism in hybridoma cells through metabolic engineering. Cytotechnology
30:85-93.
Pascoe DE, Arnott D, Papoutsakis ET, Miller WM, Andersen DC. 2007. Proteome analysis of antibody-producing CHO cell lines with different metabolic profiles. Biotechnol Bioeng 98:391-410.
Trummer E, Fauland, K, Seidinger S, Schriebl, K, Lattenmayer C, Kunert R, Vorauer-Uhl K, Weik
R, Borth N, Katinger H, Muller D. 2006. Process parameter shifting: Part I. Effect of DOT, pH, and temperature on the performance of Epo-Fc expressing CHO cells cultivated in controlled batch bioreactors. Biotechnol Bioeng 94: 1033-1044
Wlaschin KF, Hu W-S. 2007. Engineering cell metabolism for high-density cell culture via manipulation of sugar transport. J Biotechnol 131 : 168-176.
Yuk IH, Zhang JD, Ebeling M, Berrera M, Gomez N, Werz S, Meiringer C, Shao Z, Swanberg JC, Lee KH, Luo J, Szperalski B. 2014. Effects of copper on CHO cells: insights from gene expression analyses. Biotechnol Prog 30:429-442.
Yuk IH, Russell S, Tang Y, Hsu W-T, Mauger JB, Aulakh RPS, Luo J, Gawlitzek M, Joly JC. 2015.
Effects of copper on CHO cells: Cellular requirements and product quality considerations.
Biotechnol Prog (in press).
Zhou M, Crawford Y, Ng D, Tung J, Pynn AFJ, Meier A, Yuk IH, Vijayasankaran N, Leach K, Joly
J, Snedecor B, Shen A. 2011. Decreasing lactate level and increasing antibody production in
Chinese Hamster Ovary cells (CHO) by reducing the expression of lactate dehydrogenase and pyruvate dehydrogenase kinases. J Biotechnol 153:27-34.
Zhou W, Rehm J, Europa A, Hu WS. 1997. Alteration of mammalian cell metabolism by dynamic nutrient feeding. Cytotechnology 24:99-108.
Claims
1. A method of reducing lactate production in cultured cells comprising growing cultured cells in a medium which comprises an effective amount of a small molecule LDHA inhibitor.
2. A method of claim 1 , wherein the small molecule LDHA inhibitor is a compound of Formula (I):

and stereoisomers, tautomers, and pharmaceutically acceptable salts thereof, wherein:
A1 is O, CH2 or S;
A2 is NH or N-CrC3-alkyl;
A3 is N or CR2;
A4 is N or CR3, provided that A3 and A4 are not N at the same time;
R1 is CI, N02, or CN;
R2 and R6 are independently selected from the group consisting of H, halo, hydroxy,
Ci-C6-hydroxyalkyl, and NH2;
R3 and R5 are independently selected from the group consisting of:
H;
hydroxy;
halo;
-CrC6-alkyl-Rf;
-CrC6-alkenyl-Rf;
-CrC6-alkoxy-Rc;
-NRaRb;
• -NRa-(C1-C6-alkyl)-Rd;
• -NRa-S(0)2-(4 to 10 membered heterocycloalkyl);
• -NRa-(C3-C8-cycloalkyl), which cycloalkyl is unsubstituted or substituted by Ci-C6-alkyl or a Ci-C3-alkylene bridge;
• -NRa-aryl, which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, hydroxy, -NH2, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-haloalkyl,
Ci-C6-hydroxyalkyl, Ci-C6-haloalkoxy and C3-C8-cycloalkyl;
• -NRa-(4 to 10 membered heterocycloalkyl), which heterocycloalkyl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: Ci-C6-alkyl, Ci-C6-hydroxyalkyl, or -CO-alkyl;
• -NRa-(5 or 6 membered heteroaryl), which heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, -NRaRb and Ci-C6-alkyl;
• -NRa(CO)-CrC6-alkyl;
• -NRa(CO)-aryl;
• -NRa(CO)-(5 or 6 membered heteroaryl);
• -NRa(CO)0-CrC6-alkyl;
• -S-(alkyl)n-Rh;
• -S(0)2-aryl, which aryl is unsubstituted or substituted by one or more halo;
• -C(0)-Re;
• -C(0)NRa-(CrC6-alkyl)n-R8;
• -C(0)NRa-CrC6-alkoxy;
• -0-C3-C8-cycloalkyl, which cycloalkyl is unsubstituted or substituted by ne or more
substituent(s) selected from the group consisting of: halo or hydroxy, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-haloalkoxy, Ci-C6-alkoxyaryl, Ci-C6-haloalkyl, Ci-C6-hydroxyalkyl, NRaRb, aryl, Ci-C6-akyl-aryl, 5 or 6 membered heteroaryl, and
-(Ci-C6-alk l)-(Ci-C6-alkoxy);
• -O-aryl, which aryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkyl, Ci-C6-alkoxy, Ci-C6-alkyl-Ci-C6-alkoxy, Ci-C6-haloalkyl,
Ci-C6-haloalkoxy, Ci-C6-hydroxyalkyl, -S-Ci-C6-akyl,
-CrC6-alkyl-C3-C8-cycloalkyl, CrC6-alkoxy-C3-C8-cycloalkyl, CrC6-alkyl-(4 to 10 membered heterocycloalkyl), Ci-C6-alkyl-(5 or 6 membered heterocycloalkyl), or 5 or 6 membered heteroaryl unsubstituted or substituted by one or more substituent(s)
selected from the group consisting of: Ci-C6-alkyl, -(Ci-C6-alkyl)-(Ci-C6-alkoxy), Ci-C6-haloalkoxy and a Ci-C6-alkylene bridge;
• -0-(4 to 10 membered heterocycloalkyl), which heterocycloalkyl is unsubstituted or
substituted by one or more substituent(s) selected from the group consisting of:
o halo, hydroxy, Ci-C6-alkyl, Ci-C6-hydroxyalkyl and -C(0)-Ci-C6-alkyl;
• -0-(5 to 10 membered heteroaryl), which heteroaryl is unsubstituted or substituted by halo, CrC6-alkyl, CrC6-hydroxyalkyl, or -NRa(CO)-CrC6-akyl;
• C3-C8-cycloalkyl, which cycloalkyl may be fused to a phenyl;
• aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o halo, hydroxy, -C(0)OH, CrC6-hydroxyalkyl, CrC6-alkoxy, -S(0)2-NH(alkyl) and -S(0)2-N(alkyl)2;
• 4 to 10 membered heterocycloalkyl unusbstituted or substituted by one or more
substituent(s) selected from the group consisting of:
o halo, Ci-C6-alkyl, -C(0)-C3-C8-cycloalkyl, oxo and 5 or 6 membered
heterocycloalkyl;
• 5 to 10 membered heteroaryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
o hydroxy, -NRaRb, CrC6-alkyl, CrC6-hydroxyalkyl, and 4 to 10 membered
heterocycloalkyl;
is:
• H,
• cyano,
• halo,
• hydroxy,
• NRaRb,
• CrC6-alkyl,
• Ci-C6-haloalkyl,
• Ci-C6-hydroxyalkyl,
• Ci-C6-alkoxy unsubstituted or substituted by hydroxy, Ci-C6-alkoxy or NRaRb,
• -(Ci-C6-alkyl)n-(C3-C8-cycloalkyl), unsubstituted or substituted by one or more
substituent(s) selected from the group consisting of: halo, hydroxy, -NRaRb, Ci-C6-alkyl, CrC6-alkoxy, CrC6-haloalkyl, -C(0)-CrC6-alkyl, -C(0)-CrC6-cycloalkyl; -C(0)-(5 or 6 membered heterocycloalkyl);
• -(Ci-C6-alkyl)n-(C3-C8-cycloalkenyl), unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, hydroxy, -NRaRb, Ci-C6-alkyl, Ci-Ce-alkoxy, CrC6-haloalkyl, -C(0)-CrC6-alkyl, -C(0)-CrC6-cycloalkyl and -C(0)-(5 or 6 membered heterocycloalkyl);
• -(Ci-C6-alkyl)n-(5 or 6 membered heteroaryl), unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, hydroxy, -NRaRb, Ci-C6-alkyl, Ci-Ce-alkoxy, CrC6-haloalkyl and -C(0)-CrC6-alkyl, -C(0)-CrC6-cycloalkyl and -C(0)-(5 or 6 membered heterocycloalkyl);
• -(Ci-C6-alkyl)n-(4 to 10 membered heterocycloalkyl) unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, hydroxy, cyano, -NRaRb, CrC6-alkyl, CrC6-alkoxy, CrC6-haloalkyl, CrC6-hydroxyalkyl, -C(0)OH, a
CrC4-alkylene bridge, -C(0)-CrC6-alkyl, -C(0)-C3-C8-cycloalkyl, -C(0)-aryl, -C(0)(4 to 10 membered heterocycloalkyl) and -C(0)-(5 or 6 membered heterocycloalkyl);
R7 is aryl, a 5 or 6 membered heterocycle or 5 or 6 membered heteroaryl which aryl, heterocycle or heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-alkyl, C3-C8-cycloalkyl, -O-aryl, -S-aryl, -NH-aryl, and
-(CrC6-alkyl)n-aryl;
or R6 and R7 together with the carbon atoms to which they are attached form a 5 membered ring selected from a cycloalkyl or heterocycloalkyl having 5 ring members;
R8 is OH, -NRaRb, CrC6-alkoxy or -C(0)0-CrC6-alkyl;
or R2 and R3 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
or R3 and R4 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
or R4 and R5 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
or R5 and R6 together with the atoms to which they are attached form a naphthyl or 9 or 10 membered heteroaryl, each of which is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, hydroxy, -NRaRb, CrC6-alkyl, CrC6-alkoxy and CrC6-haloalkyl;
Ra is H or CrC6-alkyl;
Rb is H or CrC6-alkyl;
Rc is H, hydroxy, halo, -NRaRb, Ci-C6-alkoxy, Ci-C6-alkenyl, 4 to 6 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C6-alkyl, 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C6-alkyl, or C3-C8-cycloalkyl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of:
• halo, Ci-C6-alkyl or Ci-C6-hydroxyalkyl, aryl unsubstituted or substituted by halo, 4 to 9 membered heterocycloalkyl unsubstituted or substituted by oxo or Ci-C6-alkyl, and 5 or 6 membered heteroaryl unsubstituted or substituted by Ci-C6-alkyl;
Rd is H, hydroxy, Ci-C6-alkyl, C3-C8-cycloalkyl or aryl unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo and -NRa-S(0)2-N(Ci-C6-alkyl)2;
Re is Ci-C6-alkyl, aryl, C3-C8-cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl and wherein said aryl, C3-C8-cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of: halo, Ci-C6-alkoxy, Ci-C6-alkyl and Ci-C6-haloalkyl;
Rf is H, C3-C8-cycloalkyl, 4 to 10 membered heterocycloalkyl, aryl, or 5 or 6 membered heteroaryl, which cycloalkyl, heterocycloalkyl, aryl, or heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-haloalkyl, Ci-C6-alkyl, Ci-C6-alkoxy and Ci-C6-hydroxyalkyl;
Rs is Ci-C6-alkoxy, C3-C8-cycloalkyl, aryl, 5 or 6 membered heteroaryl, 5 to 9 membered heterocycloalkyl, wherein said aryl, C3-C8-cycloalkyl, 5 to 9 membered heterocycloalkyl or 5 or 6 membered heteroaryl is unsubstituted or substituted by one or more substituent(s) selected from the group consisting of halo, Ci-C6-alkoxy and Ci-C6-hydroxyalkyl;
Rh is aryl, 5 or 6 membered heteroaryl, 4 to 10 membered heterocycloalkyl, C3-C8-cycloalkyl, each of which is unsubstituted or substituted by halo;
n is 0 or 1.
3. A method of claim 1 or 2, wherein the compound has the following general Formula:

wherein A1, A2, A3, R1, R3, R4, R5, R6, R8, R9 and R10 are as defined in claim 1 or 2.
general Formula:

general Formula:

6. A method of any one of claims 1 to 5, wherein A is NH.
7. A method of any one of claims 1 to 5, wherein A3 is CR2, wherein R2 is selected from the group consisting of H, halo, hydroxy, Ci-C6-hydroxyalkyl, and NH.
8. A method of any one of claims 1 to 6, wherein R9 and R10 are H.
9. A method of any one of claims 1 to 7, wherein R1 is CI.
10. A method of any one of claims 1 to 8, wherein R3 is NH-phenyl or NH-pyridinyl, which phenyl or pyridinyl is substituted by halo.
1 1. A method of any one of claims 1 to 9, wherein R4, R5, R6 and R8 are H.
12. A method of any one of claims 1 to 10 wherein the compound is selected from the group consisting of the following compounds as racemates, single stereoisomers, tautomers and pharmaceutically acceptable salts thereof:
1 - [4-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]phenyl]piperidine-4-carbonitr ile;
2- [[6-(6-bromo-2-pyridyl)-2,4-dioxo-6-(3-thienyl)-3-piperidyl]sulfanyl]benzonitrile;
3- (2-chloro-5-hydroxy-phenyl)sulfanyl-6-[4-(l -piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione
3-(2-chlorophenoxy)-6-(4-morpholinophenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[4-(l -piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(2-cyclopropylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(3,4-difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyl)sulfanyl- 1 -methyl-6-(3 -tetrahydropyran-4-yloxyphenyl)-6-(3 -thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl)sulfanyl- 1 -methyl-6- [3 -(tetrahydropyran-4-ylamino)phenyl] -6-(3 -thienyl)piperid ine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(lH-indol-4-yl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-fluorophenyl)-l -methyl-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-hydroxy-4-morpholino-phenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-hydroxyphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(2-naphthyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-fluoro-4-morpholino-phenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-hydroxyphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-tetrahydropyran-4-yloxyphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)-6-(4-thiomorpholinophenyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)-6-[6-(2,2,2-trifluoro-l-methyl-ethoxy)-2-pyridyl]piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)-6-[6-(2,2,2-trifluoroethoxy)-2-pyridyl]piperidine-2,4-dion e;
3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)-6-[6-(4,4,4-trifluorobutoxy)-2-pyridyl]piperidine-2,4-dion e;
3- (2-chlorophenyl)sulfanyl-6-(3 hienyl)-6-[6-[3-(trifluoromethyl)phenoxy]-2-pyridyl]piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)-6-[6-[4-(trifluoromethoxy)phenoxy]-2-pyridyl]piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)-6-[6-[4-(trifluoromethyl)cyclohexoxy]-2-pyridyl]piperidi ne-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-(3 hienyl)-6-[6-[4-(trifluoromethyl)phenoxy]-2-pyridyl]piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-(4-cyclohexylphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-cyclopropylphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-hydroxyphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-mo^holino-3-phenyl-phenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-morpholinophenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-mo^holinophenyl)-6-(5-phenyl-3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-morpholinophenyl)-6-(6-tetrahydropyran-4-yloxy-2-pyridyl)piperi dine-2,4-dione;
5- (2-chlorophenyl)sulfanyl-4-hydroxy-2-[6-(4-methoxycyclohexoxy)-2-pyridyl]-2-(3-thienyl)-l,3-d ihydropyridin-6-one;
3-(2-chlorophenyl)sulfanyl-6-(4-morpholinophenyl)-6-thiazol-4-yl-piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-piperazin-l-ylphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(4-pyrrolidin-l-ylphenyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(5-chloro-3-thienyl)-6-[6-(4-fluorophenoxy)-2-pyridyl]piperidine-2,4
-dione;
3-(2-chlorophenyl)sulfanyl-6-(5-methyl-3-thienyl)-6-(4-mo^holinophenyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-chroman-4-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-ethoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-indan-5-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-isobutoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-isopentyloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-isopropoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-(6-isopropoxy-5-mo^holino-2-pyridyl)-6-(3-thienyl)piperidine-2,4-d ione;
3- (2-chlorophenyl sulfanyl- 6-morpholino-3-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3- (2-chlorophenyl sulfanyl- 6-pent-2-enoxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3- (2-chlorophenyl sulfanyl- 6-phenoxy-2-pyridyl)-6-(3-thienyl)piperidine -2,4-dione;
3- (2-chlorophenyl sulfanyl- 6-phenyl-2-pyridyl)-6-(3-thienyl)piperidine -2,4-dione;
3- (2-chlorophenyl sulfanyl- 6-pyrimidin-5-yloxy-2-pyridyl)-6-(3-thienyl)piperidine -2,4-dione;
3- (2-chlorophenyl sulfanyl- 6-tetrahydrofuran-3-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dio ne
3- (2-chlorophenyl sulfanyl- 6-tetralin-l-yloxy-2-pyridyl)-6-(3-thienyl)piperidine-2,4-dione;
3- (2-chlorophenyl sulfanyl- 3-(4-fluoroanilino)phenyl]-l-methyl-6-(3-thienyl)piperidine-2,4-dio ne
3- (2-chlorophenyl sulfanyl- 4-fluoroanilino)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3- (2-chlorophenyl sulfanyl- 4-fluoroanilino)phenyl]-6-phenyl-piperidine-2,4-dione;
3- (2-chlorophenyl sulfanyl- 4-fluoro-N-methyl-anilino)phenyl]-6-phenyl-piperidine -2,4-dione
3 -(2-chlorophenyF )sulfanyl 4-fluorophenoxy)phenyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl cyclohexylamino)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl tetrahydropyran-4-ylamino)phenyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl (6-fluoro-5-methyl-3-pyridyl)amino]phenyl]-6-(3-thienyl)piperidi ne-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6- 1- piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6- 2,2-dimethylmorpholin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl1-6- 2,6-dimethylmorpholin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl -6- 2- ethylmorpholin-4-yl)phenyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6- 2-hydroxyethoxy)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6- 2-methoxyethoxy)phenyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6- 2-methylmorpholin-4-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl 2-oxa-5-azabicyclo[2.2.1]heptan-5-yl)phenyl]-6-(3-thienyl)piperi dine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl 2-oxa-6-azaspiro[3.3]heptan-6-yl)phenyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyF )sulfanyl■6-[4 2- oxa-7-azaspiro[3.5]nonan-7-yl)phenyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3,3-difluoroazetidin-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[4 3.3- difluoropyrrolidin-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3- fluoroazetidin-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3 -fluoropyrrolidin-l-yl)phenyl]-6-(3-thienyl)piperidine -2,4-dione
3 -(2-chlorophenyF )sulfanyl ■6-[4 3 -hydroxypropoxy)phenyl] -6-(3 -thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3-methoxypropoxy)phenyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 3 - methoxypyrrolidin- 1 -yl)phenyl] -6-(3 -thienyl)piperidine-2,4-dio ne;
3 -(2-chlorophenyF )sulfanyl ■6-[4 4.4- difluoro-l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[4 4- fluoro-l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 4-methoxy-l-piperidyl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 8-oxa-3-azabicyclo[3.2.1]octan-3-yl)phenyl]-6-(3-thienyl)piperidi ne -2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 cyclohexen-l-yl)phenyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 dimethylamino)phenyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[4 tetrahydropyran-4-ylamino)phenyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[5 4-fluoroanilino)-2-hydroxy-phenyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl ■6-[5 (4-fluorophenyl)methyl]-3-thienyl]-6-(4-morpholinophenyl)piperi dine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 l,2,3,4-tetrahydroquinolin-8-yloxy)-2-pyridyl]-6-(3-thienyl)piperi dine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl ■6-[6 l-cyclohexylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione
3 -(2-chlorophenyF )sulfanyl ■6-[6 l-cyclopropylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl ■6-[6 l-cyclopropylethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl -6-1 [6-(lH-indazol-4-yloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyF )sulfanyl -6-[6 2,2-difluoroethoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2,2-dimethylchroman-4-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine
-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2.2- dimethylpropoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl -6-[6 2.3- difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl -6-[6 2.4- difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclobutylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclohexylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclohexylethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-d ione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclopentylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclopropyl- 1 -methyl-ethoxy)-2-pyridyl] -6-(3 -thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclopropylethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclopropylethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-cyclopropylpropoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dio ne;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-ethoxy- 1 -methyl-ethoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-2,4
-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-ethoxyethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-methoxy- 1 -methyl-ethoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-2
,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-methoxyphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-methylbutoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6 2-morpholino-4-pyridyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 2- pyridyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3,4-difluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3,4-difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3,4-difluorophenoxy)-2-pyridyl]-6-(4-morpholinophenyl)piperidi ne-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3.4- difluorophenoxy)-2-pyridyl]-6-[4-(l-piperidyl)phenyl]piperid ine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3.5- difluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3 - fluoro-4-methoxy-phenoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3 -hydroxy-3 -methyl-butoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-2,
4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3-hydroxycyclopentoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3 -methoxy-3 -methyl-butoxy)-2-pyridyl] -6-(3 -thienyl)piperidine-2
,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3 -methoxy-N-methyl-anilino)-2-pyridyl] -6-(3 -thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3-methoxyphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3-methoxypropoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3- pyridyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 3 -tetrahydropyran-4-ylazetidin- 1 -yl)-2-pyridyl] -6-(3 -thienyl)piper idine -2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 4,4-difluorocyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 4- cyclopropyl-2-fluoro-anilino)-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 4-fluoro-2-isopropyl-phenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyF )sulfanyl -6-[6-( 4-fluoro-2-methoxy-phenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
(6S)-3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-2-methoxy-phenoxy)-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-2-tetrahydropyran-4-yl-phenoxy)-2-pyridyl]-6-(3-thienyl )piperidine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-3-methoxy-phenyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3- (2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-3-methyl-phenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-2-pyridyl]-l-methyl-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(4-mo^holinophenyl)piperidine-2,4
-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoroanilino)-5-mo^holino-2-pyridyl]-6-(3-thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorobenzoyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluoro-N-methyl-anilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4
-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-l-methyl-6-(3-thienyl)piperidine-2,4 -dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(lH-pyrazol-3-yl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(2-hydroxyphenyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-2-pyridyl]-6-(4-morpholinophenyl)piperidine-2
,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenoxy)-5-morpholino-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3-(2-chlorophenoxy)-6-[6-(4-fluoroanilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6-(4-fluorophenyl)sulfanyl-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3- (2-chlorophenyl)sulfanyl-6-(3-thienyl)-6-[6-[3-(trifluoromethyl)phenoxy]-2-pyridyl]piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(cyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-(4-hydroxy-4-methyl-pentoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 4-iodophenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 4-methoxycyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6 4-methoxy-N-methyl-anilino)-2-pyridyl]-6-(3-thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 4-methoxyphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 4-methylsulfanylphenoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6 4-pyridyl)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 4- pyridylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 5- fluorotetralin-l-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6 5-isoquinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 5- quinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 6- fluorotetralin-l-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6 6- quinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 7- fluorotetralin-l-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6 8- fluorochroman-4-yl)oxy-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6 8-hydroxy-3 ,4-dihydro-2H-quinolin- 1 -yl)-2-pyridyl] -6-(3 -thienyl) piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 8-isoquinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 8-quinolyloxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclobutoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclobutylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cycloheptoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexoxy)-2-pyridyl]-6-(4-morpholinophenyl)piperidine-2,4-d ione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexoxy)-2-pyridyl]-6-[4-(l-piperidyl)phenyl]piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexylamino)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 cyclohexylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3-(2-chlorophenyl)sulfanyl-6-[6 cyclopentoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 cyclopentylamino)-2-pyridyl]-6-(3-thienyl)piperidine -2,4-dione; 3 -(2-chlorophenyl sulfanyl-6-[6 cyclopentylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyl sulfanyl-6-[6 cyclopropylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dion e;
3 -(2-chlorophenyl sulfanyl-6-[6 dimethylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione; 3 -(2-chlorophenyl sulfanyl-6-[6 N-ethyl-4-fluoro-anilino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-d ione;
3 -(2-chlorophenyl sulfanyl-6-[6 oxetan-3-ylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydrofuran-2-ylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydrofuran-3-ylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4
-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydropyran-4-ylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2, 4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydropyran-4-ylmethoxy)-2-pyridyl]-6-(3-thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 tetrahydropyran-4-ylmethyl)-2-pyridyl]-6-(3-thienyl)piperidine-2,
4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 thiazol-2-ylamino)-2-pyridyl]-6-(3-thienyl)piperidine-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 ,5-dimethylpyrazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidi ne -2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methyl- 1 ,2,4-triazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperi dine -2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylcyclopropyl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylimidazol-2-yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidine
-2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylimidazol-2-yl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidi ne -2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 ( 1 -methylpyrazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidine-
2,4-dione;
3 -(2-chlorophenyl sulfanyl-6-[6 (2,4-difluorophenyl)methyl] -2-pyridyl] -6-(3 -thienyl)piperidine -2,
4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 2,5-dimethylpyrazol-3-yl)amino]-2-pyridyl]-6-(3-thienyl)piperidi ne-2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 2-methylcyclopropyl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 2-methylpyrazol-3 -yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidine- 2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 3 ,3 -difluorocyclobutyl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidi ne -2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 3.4- difluorophenyl)methyl] -2-pyridyl] -6-(3 -thienyl)piperidine -2, 4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 3.5- difluorophenyl)methyl]-2-pyridyl]-6-(3-thienyl)piperidine-2, 4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 3 -ethyloxetan-3 -yl)methoxy] -2-pyridyl] -6-(3 -thienyl)piperidine- 2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 3 -fluoro-5-methoxy-phenyl)methyl] -2-pyridyl] -6-(3 -thienyl)pipe ridine-2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 3 -fluorophenyl)methyl] -2-pyridyl] -6-(3 -thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyl )sulfanyl-6-[6 4-fluoro-3 -methoxy-phenyl)methyl] -2-pyridyl] -6-(3 -thienyl)pipe ridine-2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 4-fluorophenyl)methoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 4-fluorophenyl)methyl] -2-pyridyl] -6-(3 -thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyl )sulfanyl-6-[6 4-fluorophenyl)methylamino] -2-pyridyl] -6-(3 -thienyl)piperidine- 2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 4- methylthiazol-2-yl)amino] -2-pyridyl] -6-(3 -thienyl)piperidine-2 ,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 5- fluoro-3-pyridyl)oxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3 -(2-chlorophenyl )sulfanyl-6-[6 5-fluoro-8-quinolyl)oxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 5-methyl-lH-imidazol-2-yl)amino]-2-pyridyl]-6-(3-thienyl)piperi dine -2,4-dione;
3 -(2-chlorophenyl )sulfanyl-6-[6 5-methylthiazol-2-yl)amino]-2-pyridyl]-6-(3-thienyl)piperidine-2 ,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(5-oxotetrahydrofuran-2-yl)methoxy]-2-pyridyl]-6-(3-thienyl)pip eridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(6-fluoro-3-pyri
-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[(6-fluoro-5-met^^
ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[[3-(hydroxymethyl)phenyl]methyl]-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[[4-(hydroxymethyl)cyclohexyl]methoxy]-2-pyridyl]-6-(3-thienyl) piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l- 3 ,4-difluorophenyl)ethoxy] -2-pyridyl] -6-(3 -thienyl)piperidine- 2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l- 3-fluorophenyl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l- (4-fluorophenyl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l- 4-fluorophenyl)ethylamino] -2-pyridyl] -6-(3 -thienyl)piperidine -2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[l- 4-fluorophenyl)propoxy] -2-pyridyl] -6-(3-thienyl)piperidine -2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[l- 4-fluorophenyl)propylamino] -2-pyridyl] -6-(3 -thienyl)piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2- lH-pyrazol-4-yl)phenoxy] -2-pyridyl] -6-(3-thienyl)piperidine -2 ,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2- 1- methylcyclopropyl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2- 2,2-difluorocyclopropyl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2- 2,2-dimethyl-l,3-dioxolan-4-yl)ethoxy]-2-pyridyl]-6-(3-thienyl )piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2- 2- oxopyrrolidin-l-yl)ethoxy]-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2- 3 -methyltriazol-4-yl)phenoxy] -2-pyridyl] -6-(3 -thienyl)piperidi ne-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2- 4-fluorophenyl)ethyl]-2-pyridyl]-6-(3-thienyl)piperidine-2,4-di one;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(cyclopropylmethoxy)-4-fluoro-phenoxy]-2-pyridyl]-6-(3-thien yl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(cyclopropylmethyl)-4-fluoro-phenoxy]-2-pyridyl]-6-(3-thienyl )piperidine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[2-(methoxymethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[2-(oxetan-3-yl)ethoxy]-2-pyridyl]-6-(3 hienyl)piperidine-2,4-dio ne;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(l-hydroxyethyl)anilino]-2-pyridyl]-6-(3 hienyl)piperidine-^ -dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(difluoromethyl)-4-fluoro-phenoxy]-2-pyridyl]-6-(3-thienyl)pip eridine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-[6-[3-(difluoromethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)piperidine-2,
4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(hydroxymethyl)anilino]-2-pyridyl]-6-(3-thienyl)piperidine-2,4- dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-(hydroxymethyl)-N-methyl-anilino]-2-pyridyl]-6-(3-thienyl)pip eridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[3-fluoro-5-(hydroxymethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)pipe ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[4-fluoro-3-(hydroxymethyl)anilino]-2-pyridyl]-6-(3-thienyl)piperi dine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[4-fluoro-3-(trifluoromethyl)phenoxy]-2-pyridyl]-6-(3-thienyl)pip eridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-[6-[6-(hydroxymethyl)indolin-l-yl]-2-pyridyl]-6-(3-thienyl)piperidin e-2,4-dione;
3 -(2-chlorophenyl)sulfanyl-6-[6-[N-methyl-3 -(trifluoromethyl)anilino] -2-pyridyl] -6-(3 -thienyl)pipe ridine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-phenyl-6-(3-thienyl)piperidine-2,4-dione;
3-(2-chlorophenyl)sulfanyl-6-phenyl-6-thiazol-4-yl-piperidine-2,4-dione;
3- (2-chlorophenyl)sulfanyl-6-thiazol-4-yl-6-(3-thienyl)piperidine-2,4-dione;
4- [3-[5-(2-chlorophenyl)sulfanyl-2-(4-morpholinophenyl)-4,6-dioxo-2-piperidyl]phenyl]-N,N-dime thyl-benzenesulfonamide;
4-[3-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]phenyl]-N,N-dimethyl-benzen esulfonamide;
4-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-2-pyridyl]-N,N-dimethyl-ben zenesulfonamide ;
6-(3-aminophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-anilinophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-bromo-4-mo^holino-phenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-bromophenyl)-3-(2-chlorophenyl)sulfanyl-l-methyl-6-(3-thienyl)piperidine-2,4-dione;
6-(3-bromophenyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(5-bromo-6-mo^holino-3-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-benzyl-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-benzyloxy-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-bromo-2-pyridyl)-3-(2-chloro-5-hydroxy-phenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-bromo-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-(6-bromo-5-mo^holino-2-pyridyl)-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[3-chloro-5-(4-fluoroanilino)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dion e;
6-[4-(l,3,3a,4,6,6a-hexahydrofuro[3,4-c]pyrrol-5-yl)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hie^ yl)piperidine-2,4-dione;
6-[4-(2-azaspiro[3.3]heptan-2-yl)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-di one;
6-[4-(3-azabicyclo[2.1. l]hexan-3-yl)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[4-(4-acetylpiperazin-l-yl)phenyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-N-(cyclopropylmethyl)pyridine-
2-carboxamide;
6-[6-(2-amino-5-methyl-imidazol-l-yl)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidi ne-2,4-dione;
6-[6-(2-bromophenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[6-(2-chloro-3,4-difluoro-anilino)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)piperi
2,4-dione;
6-[6-(2-chloro-4-fluoro-anilino)-2-pyridyl]-3- dione;
6-[6-(2-chloro-4-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[6-(2-tert-butoxyethoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione
6-[6-(3-bromo-4-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[6-(3-chloro-4-fluoro-anilino)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)piperidine- dione;
6-[6-(3-chloro-4-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2, 4-dione;
6-[6-(3-chlorophenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3 hienyl)piperidine-2,4-dione;
6-[6-(4-bromo-2-chloro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,
4-dione;
6-[6-(4-bromo-2-fluoro-phenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,
4- dione;
6-[6-(4-chloro-N-methyl-anilino)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4 -dione;
6-[6-(4-chlorophenoxy)-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-dione;
6-[6-(7-bromotetralin-l-yl)oxy-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4-d ione;
6-[6-[(2-chloro-6-fluoro-3-pyridyl)oxy]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidi ne-2,4-dione;
6-[6-[(4-chloro-3-fluoro-phenyl)methyl]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperid ine-2,4-dione;
6-[6-[[l-(3-chloro-4-fluoro-phenyl)-2-hydroxy-ethyl]amino]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl- 6-(3-thienyl)piperidine-2,4-dione;
6-[6-[l-(3-chloro-4-fluoro-phenyl)propylamino]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl )piperidine-2,4-dione;
6-[6-[l-(4-chlorophenyl)ethoxy]-2-pyridyl]-3-(2-chlorophenyl)sulfanyl-6-(3-thienyl)piperidine-2,4- dione;
N-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-2-pyridyl]azetidine-l-sulfon amide tert-butyl;
5- (2-chlorophenyl)sulfanyl-4-hydroxy-2- [4-( 1 -piperidyl)phenyl] -2-(3 -thienyl)- 1 ,3 -dihydropyridin-6 -one; and
N-[6-[5-(2-chlorophenyl)sulfanyl-4,6-dioxo-2-(3-thienyl)-2-piperidyl]-2-pyridyl]carbamate.
13. A method of any one of claims 1-12, wherein the medium is maintained at a pH value about 7.0, 7.1, 7.2, 7.3 or 7.4.
14. A method of any one of the claims 1-13, wherein the medium is maintained at a temperature about 36.0, 36.5, 37.0, or 37.5 °C during the initial 2, 3, 4, 5, 6 or 7 days.
15. A method of claim 14, wherein the temperature of the medium is lowered to about 36, 35, 34, 33, 32, or 31°C at the end of day 2, 3, 4, 5, 6, or 7.
16. A method of any one of claims 1 to 15, wherein the LDHA inhibitor is introduced into the medium at day 0, 1, 2, 3, 4, 5, 6, or 7, or a combination thereof.
17. A method of any one of claims 1-15, wherein the concentration of the LDHA inhibitor in the medium is about 1 μΜ, 5 μΜ, 10 μΜ, 15 μΜ, 20 μΜ, 25 μΜ, 30 μΜ, 35 μΜ, 40 μΜ, 45 μΜ, 50 μΜ, 60 μΜ, 70 μΜ, 80 μΜ, 90 μΜ or 100 μΜ.
18. A method of any one of claims 1-17, wherein the cultured cells produce a polypeptide.
19. The method of claim 18, wherein the polypeptide is an antibody, or a biologically functional fragment of an antibody.
20. The method of claim 1, wherein the cultured cell is a mammalian cell.
21. The method of claim 20, wherein the mammalian cell is a Chinese Hamster Ovary (CHO) cell.
22. A medium comprising a small molecule LDHA inhibitor.
23. A medium of claim 22 comprising a small molecule LDHA inhibitor, wherein said LDHA inhibitor is a compound of any one of claims 1-12.
24. A medium of claim 22, wherein said LDHA inhibitor is 5-(2-chlorophenyl)sulfanyl-4-hydroxy-2-(4-morpholinophenyl)-2-(3-thienyl)-l,3-dihydropyridin-6- one or a salt thereof.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461954087P | 2014-03-17 | 2014-03-17 | |
| US61/954,087 | 2014-03-17 | ||
| US201462032977P | 2014-08-04 | 2014-08-04 | |
| US62/032,977 | 2014-08-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2015142903A2 true WO2015142903A2 (en) | 2015-09-24 |
| WO2015142903A3 WO2015142903A3 (en) | 2015-12-03 |
Family
ID=52824554
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/021040 WO2015142903A2 (en) | 2014-03-17 | 2015-03-17 | Method of controlling lactate production with piperdine-dione derivatives |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015142903A2 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| US9951069B1 (en) | 2017-01-11 | 2018-04-24 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| WO2018211276A1 (en) * | 2017-05-16 | 2018-11-22 | Spermatech As | Piperidine-dione derivatives for use as contraceptives |
| WO2018211278A1 (en) * | 2017-05-16 | 2018-11-22 | Arctic Pharma As | Ldha activity inhibitors |
| WO2018211277A1 (en) * | 2017-05-16 | 2018-11-22 | Arctic Pharma As | Ldha activity inhibitors |
| CN109369631A (en) * | 2018-12-11 | 2019-02-22 | 上海皓元生物医药科技有限公司 | A kind of synthetic method of the key intermediate for synthesizing lactic acid dehydrogenase A inhibitor |
| US10421756B2 (en) | 2015-07-06 | 2019-09-24 | Rodin Therapeutics, Inc. | Heterobicyclic N-aminophenyl-amides as inhibitors of histone deacetylase |
| CN112142711A (en) * | 2019-06-28 | 2020-12-29 | 中国科学院上海药物研究所 | Substituted thiophene compound, preparation method and application thereof |
| US10919902B2 (en) | 2015-07-06 | 2021-02-16 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US11046658B2 (en) | 2018-07-02 | 2021-06-29 | Incyte Corporation | Aminopyrazine derivatives as PI3K-γ inhibitors |
| US11225475B2 (en) | 2017-08-07 | 2022-01-18 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US11926616B2 (en) | 2018-03-08 | 2024-03-12 | Incyte Corporation | Aminopyrazine diol compounds as PI3K-γ inhibitors |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100190824A1 (en) * | 2007-06-29 | 2010-07-29 | Prabhat Kumar | Novel Substituted Piperidones as HSP Inducers |
| KR101869987B1 (en) * | 2010-05-28 | 2018-07-20 | 제넨테크, 인크. | Decreasing lactate level and increasing polypeptide production by downregulating ldh and pdhk expression |
-
2015
- 2015-03-17 WO PCT/US2015/021040 patent/WO2015142903A2/en active Application Filing
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11858939B2 (en) | 2015-07-06 | 2024-01-02 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US10421756B2 (en) | 2015-07-06 | 2019-09-24 | Rodin Therapeutics, Inc. | Heterobicyclic N-aminophenyl-amides as inhibitors of histone deacetylase |
| US10919902B2 (en) | 2015-07-06 | 2021-02-16 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US10793567B2 (en) | 2017-01-11 | 2020-10-06 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US9951069B1 (en) | 2017-01-11 | 2018-04-24 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11987580B2 (en) | 2017-01-11 | 2024-05-21 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11286256B2 (en) | 2017-01-11 | 2022-03-29 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11225479B2 (en) | 2017-01-11 | 2022-01-18 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US10519149B2 (en) | 2017-01-11 | 2019-12-31 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US10696673B2 (en) | 2017-01-11 | 2020-06-30 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11142520B2 (en) | 2017-05-16 | 2021-10-12 | Arctic Pharma As | LDHA activity inhibitors |
| JP2020520388A (en) * | 2017-05-16 | 2020-07-09 | アークティック ファルマ アーエス | LDHA activity inhibitor |
| CN110709393A (en) * | 2017-05-16 | 2020-01-17 | 阿克蒂克制药股份有限公司 | Inhibitors of LDHA activity |
| WO2018211277A1 (en) * | 2017-05-16 | 2018-11-22 | Arctic Pharma As | Ldha activity inhibitors |
| WO2018211278A1 (en) * | 2017-05-16 | 2018-11-22 | Arctic Pharma As | Ldha activity inhibitors |
| WO2018211276A1 (en) * | 2017-05-16 | 2018-11-22 | Spermatech As | Piperidine-dione derivatives for use as contraceptives |
| US11225475B2 (en) | 2017-08-07 | 2022-01-18 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US11912702B2 (en) | 2017-08-07 | 2024-02-27 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| US11926616B2 (en) | 2018-03-08 | 2024-03-12 | Incyte Corporation | Aminopyrazine diol compounds as PI3K-γ inhibitors |
| US11046658B2 (en) | 2018-07-02 | 2021-06-29 | Incyte Corporation | Aminopyrazine derivatives as PI3K-γ inhibitors |
| CN109369631A (en) * | 2018-12-11 | 2019-02-22 | 上海皓元生物医药科技有限公司 | A kind of synthetic method of the key intermediate for synthesizing lactic acid dehydrogenase A inhibitor |
| CN112142711A (en) * | 2019-06-28 | 2020-12-29 | 中国科学院上海药物研究所 | Substituted thiophene compound, preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2015142903A3 (en) | 2015-12-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015142903A2 (en) | Method of controlling lactate production with piperdine-dione derivatives | |
| EP3119760A1 (en) | Piperidine-dione derivatives | |
| JP2019163290A (en) | Novel glutaminase inhibitors | |
| JP6378308B2 (en) | 3- (aryl or heteroaryl) methylene indoline-2-one derivatives as inhibitors of cancer stem cell pathway kinase for the treatment of cancer | |
| KR102093526B1 (en) | Serine/threonine kinase inhibitors | |
| JP2021036004A (en) | Inhibitors of ret | |
| US7745641B2 (en) | Nitrogen-containing heterocyclic compound | |
| JP4667537B2 (en) | Acylthiourea compounds or salts thereof, and uses thereof | |
| EP1322650B1 (en) | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinoline and quinazoline derivatives | |
| CN114144230B (en) | Macrocyclic azolopyridine derivatives as EED and PRC2 modulators | |
| JP2016513656A5 (en) | ||
| JP5670325B2 (en) | Substituted pyridazine carboxamide compounds as kinase inhibitor compounds | |
| AU2013272701A1 (en) | Imidazo[1,2-b]pyridazine derivatives as kinase inhibitors | |
| KR20110089418A (en) | Triazine, pyrimidine and pyridine analogs and their use as therapeutic agents and diagnostic probes | |
| CN112552294A (en) | Piperazine heterocyclic derivative inhibitor, preparation method and application thereof | |
| JP2013136585A (en) | Aurora kinase modulators and method of use | |
| JP2017505804A (en) | Pyrazolone compounds and uses thereof | |
| US11986471B2 (en) | Compounds and methods of use | |
| EP3842425A1 (en) | Novel quinoline derivative inhibitor | |
| AU2014347126A1 (en) | Pyrido[2,3-d]pyrimidin-4-one compounds as tankyrase inhibitors | |
| JP2023527242A (en) | Pyrimidine compounds that are AXL inhibitors | |
| CN118055933A (en) | Selective PARP1 inhibitors and uses thereof | |
| KR20220025828A (en) | Heterocyclic compound, method for preparing the same, and method for using the same | |
| CN117024363A (en) | Cycloolefin substituted heteroaromatic ring compound and application thereof | |
| EA030067B1 (en) | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)ethanone derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15715891 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15715891 Country of ref document: EP Kind code of ref document: A2 |