WO2015124764A1 - Synthesis process of dabigatran etexilate mesylate, intermediates of the process and novel polymorph of dabigatran etexilate - Google Patents
Synthesis process of dabigatran etexilate mesylate, intermediates of the process and novel polymorph of dabigatran etexilate Download PDFInfo
- Publication number
- WO2015124764A1 WO2015124764A1 PCT/EP2015/053702 EP2015053702W WO2015124764A1 WO 2015124764 A1 WO2015124764 A1 WO 2015124764A1 EP 2015053702 W EP2015053702 W EP 2015053702W WO 2015124764 A1 WO2015124764 A1 WO 2015124764A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- amino
- methyl
- pyridin
- dabigatran etexilate
- Prior art date
Links
- KSGXQBZTULBEEQ-UHFFFAOYSA-N dabigatran etexilate Chemical compound C1=CC(C(N)=NC(=O)OCCCCCC)=CC=C1NCC1=NC2=CC(C(=O)N(CCC(=O)OCC)C=3N=CC=CC=3)=CC=C2N1C KSGXQBZTULBEEQ-UHFFFAOYSA-N 0.000 title claims abstract description 45
- 238000000034 method Methods 0.000 title claims abstract description 34
- 229960000288 dabigatran etexilate Drugs 0.000 title claims abstract description 21
- 229960004951 dabigatran etexilate mesylate Drugs 0.000 title claims abstract description 17
- 239000000543 intermediate Substances 0.000 title abstract description 24
- 230000015572 biosynthetic process Effects 0.000 title abstract description 9
- 238000003786 synthesis reaction Methods 0.000 title abstract description 7
- 150000001875 compounds Chemical class 0.000 claims abstract description 126
- 238000004519 manufacturing process Methods 0.000 claims abstract description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 77
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 45
- 239000000203 mixture Substances 0.000 claims description 38
- 150000003839 salts Chemical class 0.000 claims description 33
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 30
- 235000019441 ethanol Nutrition 0.000 claims description 28
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 claims description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 24
- 238000006243 chemical reaction Methods 0.000 claims description 23
- -1 4-methylamino-3-nitro-benzoyl Chemical group 0.000 claims description 21
- BGLLICFSSKPUMR-UHFFFAOYSA-N ethyl 3-[[2-[(4-carbamimidoylanilino)methyl]-1-methylbenzimidazole-5-carbonyl]-pyridin-2-ylamino]propanoate Chemical compound C=1C=CC=NC=1N(CCC(=O)OCC)C(=O)C(C=C1N=2)=CC=C1N(C)C=2CNC1=CC=C(C(N)=N)C=C1 BGLLICFSSKPUMR-UHFFFAOYSA-N 0.000 claims description 21
- 239000002904 solvent Substances 0.000 claims description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 15
- KJRQMXRCZULRHF-UHFFFAOYSA-N 2-(4-cyanoanilino)acetic acid Chemical compound OC(=O)CNC1=CC=C(C#N)C=C1 KJRQMXRCZULRHF-UHFFFAOYSA-N 0.000 claims description 10
- WBAHJRHRIPHILC-UHFFFAOYSA-N 4-[(2-imidazol-1-yl-2-oxoethyl)amino]benzonitrile Chemical compound N1(C=NC=C1)C(CNC1=CC=C(C#N)C=C1)=O WBAHJRHRIPHILC-UHFFFAOYSA-N 0.000 claims description 10
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 10
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 10
- 229910002651 NO3 Inorganic materials 0.000 claims description 9
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 claims description 9
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims description 9
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 8
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 claims description 8
- 238000002425 crystallisation Methods 0.000 claims description 8
- 230000008025 crystallization Effects 0.000 claims description 8
- 239000011976 maleic acid Substances 0.000 claims description 8
- 239000003054 catalyst Substances 0.000 claims description 7
- SNFCXIDUTQBELJ-UHFFFAOYSA-N 4-(methylamino)-3-nitrobenzoyl chloride hydrochloride Chemical compound Cl.CNC1=C(C=C(C(=O)Cl)C=C1)[N+](=O)[O-] SNFCXIDUTQBELJ-UHFFFAOYSA-N 0.000 claims description 6
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 claims description 6
- 150000002688 maleic acid derivatives Chemical class 0.000 claims description 6
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 6
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 claims description 5
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 claims description 5
- 239000012458 free base Substances 0.000 claims description 5
- KIWBRXCOTCXSSZ-UHFFFAOYSA-N hexyl carbonochloridate Chemical compound CCCCCCOC(Cl)=O KIWBRXCOTCXSSZ-UHFFFAOYSA-N 0.000 claims description 5
- 238000002955 isolation Methods 0.000 claims description 5
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 5
- 230000007935 neutral effect Effects 0.000 claims description 5
- 229910017604 nitric acid Inorganic materials 0.000 claims description 5
- KSMLIIWEQBYUKA-UHFFFAOYSA-N 4-(methylamino)-3-nitrobenzoic acid Chemical compound CNC1=CC=C(C(O)=O)C=C1[N+]([O-])=O KSMLIIWEQBYUKA-UHFFFAOYSA-N 0.000 claims description 4
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 claims description 4
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 239000001099 ammonium carbonate Substances 0.000 claims description 4
- 235000012501 ammonium carbonate Nutrition 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 101100515517 Arabidopsis thaliana XI-I gene Proteins 0.000 claims description 3
- 239000011261 inert gas Substances 0.000 claims description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 2
- 239000000460 chlorine Substances 0.000 claims description 2
- 229910052801 chlorine Inorganic materials 0.000 claims description 2
- 238000001914 filtration Methods 0.000 claims description 2
- 230000001131 transforming effect Effects 0.000 claims description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 60
- 239000000243 solution Substances 0.000 description 51
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 17
- 238000002360 preparation method Methods 0.000 description 15
- 239000007787 solid Substances 0.000 description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical group ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 7
- 238000004821 distillation Methods 0.000 description 7
- 239000012071 phase Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 238000004090 dissolution Methods 0.000 description 6
- 239000003960 organic solvent Substances 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 238000001556 precipitation Methods 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- 229960000583 acetic acid Drugs 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 235000011181 potassium carbonates Nutrition 0.000 description 5
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 230000002051 biphasic effect Effects 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 150000002823 nitrates Chemical class 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- 239000012362 glacial acetic acid Substances 0.000 description 3
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- YTAHJIFKAKIKAV-XNMGPUDCSA-N [(1R)-3-morpholin-4-yl-1-phenylpropyl] N-[(3S)-2-oxo-5-phenyl-1,3-dihydro-1,4-benzodiazepin-3-yl]carbamate Chemical compound O=C1[C@H](N=C(C2=C(N1)C=CC=C2)C1=CC=CC=C1)NC(O[C@H](CCN1CCOCC1)C1=CC=CC=C1)=O YTAHJIFKAKIKAV-XNMGPUDCSA-N 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 238000006482 condensation reaction Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- FGYADSCZTQOAFK-UHFFFAOYSA-N 1-methylbenzimidazole Chemical group C1=CC=C2N(C)C=NC2=C1 FGYADSCZTQOAFK-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 206010003658 Atrial Fibrillation Diseases 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- BXFMIILPWULKDH-UHFFFAOYSA-N CCOC(CCN(C(c1ccc2[n](C)c(CNc(cc3)ccc3C(OCC)=N)nc2c1)=O)c1ncccc1)=O Chemical compound CCOC(CCN(C(c1ccc2[n](C)c(CNc(cc3)ccc3C(OCC)=N)nc2c1)=O)c1ncccc1)=O BXFMIILPWULKDH-UHFFFAOYSA-N 0.000 description 1
- 206010051055 Deep vein thrombosis Diseases 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 206010047249 Venous thrombosis Diseases 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 238000007046 ethoxylation reaction Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000007306 functionalization reaction Methods 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000012035 limiting reagent Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229940066336 pradaxa Drugs 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000005979 thermal decomposition reaction Methods 0.000 description 1
- 238000007669 thermal treatment Methods 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to a synthesis process of Dabigatran etexilate mesylate, to two compounds that form as intermediates of the process, as well as to a novel Dabigatran etexilate polymorph, a further intermediate of the process.
- Dabigatran etexilate mesylate is an active substance developed by Boehringer
- Dabigatran etexilate mesylate acts as direct inhibitor of thrombin (Factor I la) and is used as an anticoagulant, for example, for preventing strokes in patients with atrial fibrillation or blood clots in the veins (deep vein thrombosis) that could form following surgery.
- Dabigatran etexilate mesylate is the INN name of the compound 3-( ⁇ 2-[(4- ⁇ Amino- [(E)-hexyloxycarbonylimino]-methyl ⁇ -phenylamino)-methyl]-1 -methyl-1 H- benzimidazol-5-carbonyl ⁇ -pyridin-2-yl-amino)-ethyl propanoate methanesulphonate, having the following structural formula:
- A is a chlorine or nitrate anion
- step d although reported after step c), may be carried out at any moment before step e).
- the invention relates to the compound 3-( ⁇ 2-[(4- cyano-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl ⁇ -pyridin-2-yl- amino)-ethyl propanoate nitrate, intermediate compound (XI) of the above-described process in which A is the nitrate anion.
- the present invention provides the compound 3-( ⁇ 2-[(4-cyano- phenyl-amino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl ⁇ -pyridin-2-yl-amino)- ethyl propanoate as nitrate salt and a process for the preparation thereof.
- this selected compound of the invention is directly isolated from the reaction mixture without passing from the free base, a procedure that allows compound (XI) to be obtained with a high degree of purity and good yields.
- the invention relates to a novel polymorph of Dabigatran etexilate, intermediate compound (XIV) of the above-described process, characterised by an XRPD profile comprising peaks at 4.80°, 8.78°, 15.16°, 16.90°, 22.76° and 24.30° 2 ⁇ , and by three endothermic phenomena in the DSC graph, respectively with peaks at 83.0-85.0 °C, 104.0-104.2 °C (main peak), and 129.9 °C, when scanning under inert gas at a heating rate of 10 °C/min.
- FIG. 1 is an XRPD spectrum of the novel polymorph of Dabigatran etexilate of the invention
- FIG. 2 is the graph of a DSC test on the novel polymorph of Dabigatran etexilate of the invention.
- the first aspect of the invention relates to the novel process for the synthesis of Dabigatran etexilate mesylate comprising the steps a) to k) above. These steps are indicated (and will be described below) separately for clarity of description, however in the process some of these steps are performed in sequence, without isolating the intermediates that form. In particular, steps a) and b); steps d) to f); and steps g) to i), are performed in the same container.
- step a) 4-methylamino-3-nitrobenzoic acid (I), a commercially available compound, is used as the starting reagent.
- Compound (I) is suspended in toluene, then preferably a slight stoichiometric excess (for example, 0.5%) of pyridine is added with the function of activator as it salifies the acid permitting the reaction thereof with thionyl chloride.
- Thionyl chloride is then added to this solution, typically in stoichiometric excess, for example between 5 and 10%, with respect to compound (I).
- the mixture is brought to a temperature of between 30 and 50 °C, and is maintained in this condition for several tens of minutes, for example, 30', then the mixture is distilled to obtain a dense residue, which is dissolved with toluene.
- the solution thus obtained contains the intermediate compound 4-methylamino-3-nitrobenzoyl chloride hydrochloride (II) that is not isolated.
- step b) the solution thus obtained is brought to a temperature typically within the range between 0 and 30 °C.
- Triethylamine is then added with the function of neutralising both the hydrochloric acid that salifies the intermediate, and the hydrochloric acid that is released by reaction.
- the intermediate (II) is reacted with the compound 3-(2-pyridylamino)ethyl propanoate (III), typically added in an a sub-stoichiometric amount with respect to the starting compound (I), the latter being the less costly reagent; compound (III) is thus the limiting agent of the reaction, on which the yield thereof is calculated.
- the addition of triethylamine to neutralize hydrochloric acid, mentioned above, may also be carried out in this phase, in the form of a mixture with 3-(2-pyridylamino)ethyl propanoate (III) in a suitable solvent.
- Step c) consists in the catalytic hydrogenation of compound (IV).
- the solid compound (IV) obtained in the previous step is loaded into an autoclave together with an solvent inert under reaction conditions, typically ethyl acetate, or with a mixture thereof with a second solvent, for example ethyl alcohol; compound (IV) is solubilised.
- a hydrogenation catalyst consisting of palladium on carbon support is added to the solution; the amount of palladium is preferably equal to 5% of the total weight of the catalyst. While maintaining the temperature of the mixture to values between 10 and 30 °C, hydrogen at a pressure of between 1 and 2 bar is loaded into the autoclave, and the system is left to react for a few hours.
- the product compound 3-[(3-amino-4-methylamino-benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V)
- V is recovered by filtering the solution to eliminate the catalyst, concentrating the liquid phase to cause precipitation of the product, and lastly, centrifuging and washing with a solvent the obtained solid.
- the isolation of compound V thus occurs from ethyl acetate.
- Step d) is performed separately, and can be carried out at any moment before step e).
- This step consists in the functionalization of the compound N-(4- cyanophenyl)glycine (VI) with 1 ,1 -carbonyl-diimidazole (CDI).
- Suitable solvents for the reaction are well known to those skilled in the art; the use of ethyl acetate is preferred.
- the reaction takes place at a temperature of between about 10 and 30 °C with a slight stoichiometric excess of CDI and completes in about 2 hours, and aims to activate the carboxylic group of N-(4-cyanophenyl)glycine.
- a suspension of the benzonitrile (VII) is obtained, that is not isolated.
- step e the suspension of compound (VI I) is poured into a reaction vessel containing compound (V) obtained in step c).
- the acyl group corresponding to the initial glycine forms a peptide bond with one of the two primary or secondary amino groups of compound (V), giving rise to one, or generally a mixture, of compounds (VIII) and (IX).
- a further amount of the solvent used in the previous step can be added to adjust the concentration of the solution and prevent the same from being too dense.
- the mass thus obtained is brought to a temperature of between 35 and 55 °C, which is maintained for a time period of at least 6 hours.
- reaction is quenched by addition of water, preferably in a volume equal to at least half of the total volume of solvent used.
- Compounds (VIII) and/or (IX) remain in the organic phase which is separated from the aqueous phase, which is instead eliminated.
- step f) an acid is added to the organic phase recovered at the end of the previous step, with the function of catalysing the condensation reaction between the carbonyl adjacent to one of the two amino groups on the central phenyl ring of compounds (VIII) and (IX) and the other of said two amino groups; the use of glacial acetic acid is preferred.
- This condensation reaction leads to the formation of the N- methyl-benzimidazole ring of compound (X), both starting from compound (VIII) and from compound (IX), which do not therefore require to be separated or isolated at the end of the previous step.
- the process of the invention provides that the ring-closure of the intermediates (VIII) and (IX), aimed at giving the compound (X), directly takes place by adding glacial acetic acid to the reaction mixture, without distilling the solvent used for the previous phase or without changing it and without using acetic acid in large excess as the reaction solvent.
- step f For carrying out the reaction of step f), the reagent mixture is heated to reflux for at least 6 hours, after which it is brought back to room temperature. Water and an ammonia solution are then added to the mixture and the aqueous phase is separated and eliminated, while the organic phase is concentrated by distillation until an oily residue containing compound (X) is obtained, which is not further purified.
- the oily residue is diluted with a suitable solvent, such as acetone, ethanol, ethyl acetate, N,N- dimethylformamide (DMF) or a mixture thereof, preferably a mixture DMF/ethanol; the solution obtained is brought to a temperature of between 30 and 70 °C, and an acid selected between concentrated hydrochloric acid (aqueous solution at 37% by weight) and nitric acid (aqueous solution at 65% by weight) is added; the mass is then cooled to a temperature of between 0 and 25 °C, centrifuged, and the solid obtained is washed with acetone, thus obtaining the salt (XI). Both salts (nitrate and hydrochloride) are formed by one molecule of acid per molecule of compound (X).
- a suitable solvent such as acetone, ethanol, ethyl acetate, N,N- dimethylformamide (DMF) or a mixture thereof, preferably a mixture DMF/ethanol
- Salt (XI) may be optionally recrystallized to purify it, according to general methods known in the field.
- recrystallization may be carried out dissolving raw salt (XI) in DMF at a temperature of about 50-55 °C, adding ethanol to the resulting clear solution thus obtained, cooling the mixture between 30 and 40 °C, and stirring it until crystallization occurs (with times typically around 1 hour); the obtained suspension is then cooled to a temperature in the range of 0 to 15 °C and filtered.
- the invention provides compound (XI) when this is the nitrate salt in isolated form, and a process for the isolation of compound (XI), both in case of the hydrochloride and in case of the nitrate salts, without isolating the corresponding free base (X).
- base (X) is liberated from the salt. This is obtained by treating salt (XI) with a diluted basic water solution, e.g., a solution of sodium hydroxide or ammonia, and extracting the resulting free base with an organic solvent, preferably ethyl acetate. The resulting organic phase is concentrated to a solid residue by distillation, without however isolating and/or purifying the base; the distillation residue is used as such in step g).
- a diluted basic water solution e.g., a solution of sodium hydroxide or ammonia
- organic solvent preferably ethyl acetate
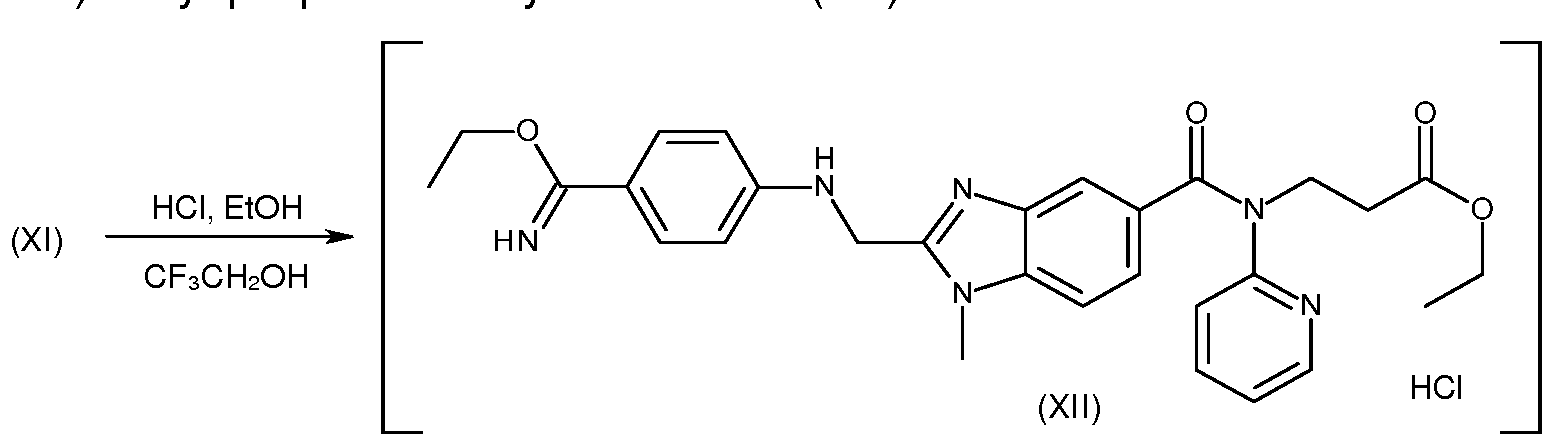
- the next step, g), consists of the ethoxylation of the cyan function of compound (XI) to give compound (XII).
- This reaction is carried out by adding ethyl alcohol to the solid residue described above; the alcohol is added in considerable excess with respect to the stoichiometric amount required, because it also acts as a solvent for the reaction; then, 2,2,2-trifluoroethanol is added and the solution is brought to a temperature of between 0 and 25 °C, gaseous hydrochloric acid being bubbled as a catalyst in the system, and the reaction is continued for at least 20 hours.
- the invention thus provides the use of 2,2,2- trifluoroethanol as co-solvent and activator that accelerates the reaction that leads from compound (XI) of the invention to compound (XII).
- Step h) consists in replacing the ethoxy group introduced in the previous step with an amino group, to obtain compound (XIII).
- the mixture obtained in the previous step is distilled in order to obtain an oily residue, then a suitable solvent is added, preferably ethyl alcohol, and brought to a temperature of between 20 and 40 °C, then adding ammonium carbonate in stoichiometric excess with respect to compound (XII).
- step i) this residue is dissolved by adding an organic solvent, for example ethanol or, preferably, ⁇ , ⁇ -dimethylformamide, then stirred and heated to 20-60 °C, preferably 30-40 °C.
- an organic solvent for example ethanol or, preferably, ⁇ , ⁇ -dimethylformamide
- maleic acid is added in an amount such that the stoichiometric ratio maleic acid:compound (XIII) is at least 0.5:1 ; maleic acid may be added in slight excess, for example in an amount such that said ratio is up to 0.8:1 .
- the amount of compound (XIII) can be calculated by assuming a yield of 100% in the previous step; alternatively, the solution of compound (XIII) may be titrated before the addition of maleic acid.
- the neutral maleic salt (XI 11 ') is obtained.
- a second organic solvent for example ethyl acetate
- a thermal treatment comprising the steps of heating to 70-80 °C, cooling to 20-25 °C, then further cooling to a temperature between -10 and 10 °C; the mixture is finally filtered off to obtain the desired compound (XI 11 ')
- step j maleate salt (XI 11 ') is transformed into Dabigatran etexilate, compound (XIV).
- the residue is dissolved in a mixture of water and a water-miscible solvent, preferably acetone, and the mass is brought to a temperature of between 0 and 25 °C.
- Potassium carbonate is then added and, while maintaining the temperature in the indicated range, hexyl chloroformate is added, in slight stoichiometric excess with respect to the theoretical amount of compound (XIII).
- the reactant mixture is maintained in said temperature range for at least two hours, after which the temperature is raised to a value between 40 and 60 °C until complete dissolution of the solids and the formation of a biphasic solution, from which the aqueous phase is eliminated by separation.
- the organic phase recovered is distilled until a paste-like residue is obtained, which is dissolved with a mixture of water and a water-immiscible solvent, preferably ethyl acetate, methylene chloride, toluene or toluene/ethanol, or possibly mixtures thereof, while adding potassium carbonate to the biphasic system.
- the system is heated to a temperature of between 30 and 75 °C to promote the solubilisation of the components, after which the aqueous phase is eliminated.
- the organic phase is distilled to a dense residue.
- the residue is dissolved with acetone and cooled to a temperature of between 20 and 40 °C to allow precipitation of the product, the desired compound (XIV).
- the solution is further cooled, after which it is centrifuged and washed with acetone, thus obtaining the crude compound (XIV).
- Compound (XIV) is then purified by means of recrystallization as known in the field, using anhydrous acetone as recrystallization solvent, initially bringing the mass to a temperature of between 40 and 60 °C to allow complete dissolution of compound (XIV), then cooling to 20-40 °C first and then to between 0 and 15 °C to completely precipitate the product, centrifuging and washing the solid obtained.
- Compound (XIV) thus produced is obtained in the form of a novel polymorph, which is described below.
- step k compound (XIV) obtained as described is salified. Salification typically takes place with methanesulfonic acid.
- the compound obtained is mono-methanesulphonate.
- compound (XIV) is dissolved in acetone at a temperature of between 40 and 60 °C to promote the complete dissolution thereof.
- the solution thus obtained is cooled to a temperature of between 28 and 32 °C and slowly added to a second, separately- prepared solution, pre-cooled at 0-10 °C, of methanesulfonic acid in an organic solvent, for example acetone, ethyl acetate, butyl acetate, ethanol, THF, toluene, or preferably acetone.
- an organic solvent for example acetone, ethyl acetate, butyl acetate, ethanol, THF, toluene, or preferably acetone.
- the mass is allowed to rest at 28-32 °C for a few hours to promote the complete precipitation of the product, after which it is centrifuged, the precipitate is washed with acetone, and dried at a temperature of between 50 and 70 °C, thus obtaining the desired salt.
- the invention relates to intermediate (XI) of the process described above, 3- ⁇ 2-[(4-cyano-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5- carbonyl ⁇ -pyridin-2-yl-amino)-ethyl propanoate nitrate, and a process for the synthesis thereof.
- the invention relates to the intermediate (XI 11 ') of the process described above, the neutral maleate of Dabigatran ethyl ester.
- a fourth aspect of the invention consists in the polymorph of Dabigatran etexilate obtained as intermediate (XIV) in the above-described process, characterised by an XRPD diffractogram showing peaks at 4.80°, 8.78°, 15.16°, 16.90°, 22.76° and 24.30° 2 ⁇ (each value of the 2 ⁇ angles of the diffractograms of powders must be understood with a degree of accuracy of ⁇ 0.2°), and by a DSC plot that, in an inert gas scan at a speed of 10 °C/min, shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0 -104.2 °C (main), and 129.9 °C.
- the DSC measurements are obtained with a Perkin-Elmer DSC 6 calorimeter.
- step a Preparation of 4-methylamino-3-nitrobenzoyl chloride hydrochloride (II) (step a).
- 1 1 .1 kg of 4-methylamino-3-nitrobenzoic acid (56.6 mol), 47.7 kg of toluene and 4.50 kg of pyridine (56.9 mol) are loaded into a reactor.
- 7.3 kg of thionyl chloride (61 .36 mol) is poured into the solution, which is maintained at a T of between 30 and 50 °C.
- the reagent solution is maintained at 30-50 °C for 30 minutes, and is then distilled to a dense residue, which is dissolved with 10.0 kg of toluene.
- the solution thus obtained contains compound (II).
- the solution prepared in step a) is brought to a T of between 0 and 30 °C; a second solution, prepared by dissolving 9.6 kg of 3-(2-pyridylamino)ethyl propanoate (49.42 mol) in 1 1 .2 kg of triethylamine and 40.0 kg of toluene, is slowly poured into said solution.
- the novel solution thus obtained is maintained at 0-30 °C for 3 hours; at the end of this period it is filtered to eliminate the salts present, and the filtered organic solution is washed with 60 kg of water and a mixture of 17.2 kg water and 2.8 kg aqueous ammonia 30%.
- the organic phase is concentrated by distillation to a dense residue, then 30.0 kg of ethanol are added.
- the residue is dissolved by heating at 60-70 °C, then the solution is cooled at 20-30 °C, stirred at this temperature until the crystallization occurs (4 hours) and finally centrifuged at 0-10 °C, washing the solid with 10.0 kg of ethanol.
- Compound (IV) obtained in step b) is loaded into a reactor along with 96 kg of ethyl acetate, 8.0 g of ethanol and 0.6 kg of Pd/C catalyst at 5%.
- Gaseous hydrogen is fed into the reactor at a pressure of 2 bar, and the mixture is maintained at 10-30 °C for 8 hours.
- the solution is filtered to eliminate the catalyst and the solution is concentrated by evaporation to reduce the volume to about one half the starting volume.
- the resulting solution is cooled to 10-25 °C, then centrifuged and the solid obtained is washed with 8.0 kg of ethyl acetate.
- step d Preparation of 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII) (step d). 6.5 kg of N-(4-cyanophenyl)glycine (36.9 mol) and 6.8 kg of 1 ,1 - carbonyldiimidazole (41 .94 mol) in 48.0 kg of ethyl acetate are loaded into a reactor. The mixture is left under stirring at 10-30 °C for 2 hours, obtaining a suspension containing compound (VII).
- Example 4 The suspension obtained in Example 4 is slowly poured into a reaction vessel containing the 35.05 moles of compound (V) obtained in Example 3 dissolved in 48.0 kg of ethyl acetate.
- the mass is heated to 35-55 °C, and maintained under these conditions for at least 6 hours, then 60 kg of water are loaded and left to interact. Then, the aqueous phase is separated and eliminated.
- the organic phase contains the two title compounds, (VIII) and (IX).
- the organic phase is distilled to an oily residue, then 6.0 kg of DMF and 72.0 kg of ethanol are added.
- the mixture is heated at 50-60 °C to obtain a clear solution, then it is cooled at 35-40 °C and 3.74 kg of nitric acid (65% aqueous solution; 38.6 moles) are added.
- the resulting mixture is stirred until crystallization occurs (which takes about 30 minutes), then it is cooled at 20-25 °C, stirred for about 2 hours and finally filtered at 0-20 °C.
- nitrate salt (XI) 12.43 kg of compound (XI) obtained in the previous example are dissolved in 12.4 kg of DMF at a temperature of about 50-55 °C. To the resulting clear solution, 49.6 kg of ethanol are added, then the mixture is cooled at about 35 °C and allowed to stir until crystallization occurs (almost 1 hour). The suspension is then cooled at 0-5 °C and filtered.
- a reactor is charged with 10.2 kg of compound (XI) (18.7 mol), 81 .6 kg of ethyl acetate and 10.2 kg of water. The mixture is stirred at 55-60 °C until complete dissolution occurs, then 2.1 kg of aqueous 30% solution of ammonia are added and the organic phase is separated, washed with water and brine and finally concentrate to obtain a solid residue. This residue is dissolved with 20.4 kg of 2,2,2-trifluoroethanol and 20.4 kg of ethanol. The mass is brought to 0-10 °C and 17.0 kg of gaseous hydrochloric acid is bubbled therein. The mass is then maintained at 20-25 °C under stirring for 20 hours, after which it is distilled under vacuum to eliminate most of the hydrochloric acid.
- the distillation residue contains compound (XII).
- Example 7 71 .4 kg of ethanol are added to the residue obtained in Example 7. The mass is brought to about 40 °C, then 8.98 kg (93.5 moles) of ammonium carbonate are added.
- the reacting mass is maintained at 30-35 °C for 16 hours, then centrifuged at the same temperature to eliminate the salts formed and then washed with 10.2 kg of ethanol.
- the washing ethanol is recovered and reunited to the centrifugal mother liquors, thus obtaining a solution in which the product of interest is present.
- the reacting mass is maintained at 20-25 °C for 2 hours and is then heated to 60 °C until complete dissolution of the reagents. Lastly, the phases are separated and the aqueous phase is eliminated.
- the organic phase is distilled to obtain a paste-like residue, then 73 kg of toluene, 7.3 kg of ethanol, 7.3 kg of distilled water and 0.45 kg of potassium carbonate are added.
- the mass is heated to 60-70 °C until complete dissolution of the reagents, after which the phases are separated and the aqueous phase is eliminated.
- the organic phase is washed with water and brine, then distilled under vacuum to obtain a paste-like residue.
- 21 .9 kg of dry acetone are added and the solvent is distilled out under vacuum.
- 29.2 kg of acetone are then added, the mixture is heated at 50-60 °C to turn into a clear solution, then it is cooled to 30 °C, and this T is maintained for two hours obtaining a complete precipitation of the solid.
- the mixture is cooled to 0-5 °C, then centrifuged and the solid is washed with 7.3 kg of cold acetone, obtaining 5.34 kg of crude Dabigatran etexilate, equal to 8.51 moles of compound, with a yield of 65% with respect to compound (XI I ⁇ ).
- the crude Dabigatran etexilate is then purified by means of recrystallization from 21 .36 kg of dry acetone.
- the mass is heated at 60 °C, until a complete solution of the product is obtained, then cooled to 25-30 °C, and maintained at this temperature for 2 hours, until complete precipitation. It is then cooled to 0-5 °C, then centrifuged, washing with 5.34 kg of cooled dry acetone. 4.7 kg (7.49 moles) of pure Dabigatran etexilate are obtained, with a yield of 88% with respect to the crude product.
- step k Preparation of Dabigatran etexilate mesylate.
- Example 1 1 All the Dabigatran etexilate obtained in Example 1 1 (4.7 kg; 7.49 moles) is loaded into a reactor along with 28.2 kg of acetone and the mass is heated at 50-60 °C until a complete solution is obtained; it is then filtered to remove suspended impurities. The filtered solution is brought to 28-32 °C.
- a second solution is prepared by dissolving 0.705 kg (7.34 moles) of methanesulfonic acid in 4.7 kg of acetone; the second solution is cooled down to 0-10 °C. The second solution is poured into the Dabigatran etexilate solution during 30 minutes, while maintaining the temperature of the resulting solution at 28-32 °C with cooling.
- the salt of the title is formed.
- the mass is maintained at 28-32 °C for 2 hours, then cooled to 18-23 °C to complete precipitation and the system is maintained at this temperature for 2 hours; lastly, centrifugation takes place, washing the precipitate with 5 kg of acetone.
- the precipitate is dried at 60 °C.
- Example 1 1 0.5 g of the crystalline compound (XIV) obtained in Example 1 1 are ground thoroughly and loaded into the sample holder of a Rigaku Miniflex diffractometer with copper anode.
- Example 1 1 0.7 g of the crystalline compound (XIV) obtained in Example 1 1 are loaded into the sample holder of a Perkin-Elmer DSC 6 calorimeter, performing a scan from ambient T to 350 °C at a rate of 10 °C/min in nitrogen atmosphere.
- the graph of the test is shown in Figure 2, and shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0-104.2 °C and 129.9 °C; events linked to the thermal decomposition of the compound are evident at about 200 °C.
Abstract
12956PTWO_eng 23 "SYNTHESIS PROCESS OF DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE" Abstract A novel process is described for the production of Dabigatran etexilate mesylate, a 5 compound having the following structural formula: and two novel intermediates of said process.
Description
"SYNTHESIS PROCESS OF DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE"
**************************
Field of the invention
The present invention relates to a synthesis process of Dabigatran etexilate mesylate, to two compounds that form as intermediates of the process, as well as to a novel Dabigatran etexilate polymorph, a further intermediate of the process.
Background of the invention
Dabigatran etexilate mesylate is an active substance developed by Boehringer
Ingelheim and marketed under the name Pradaxa® in the form of tablets for oral administration; Dabigatran etexilate mesylate acts as direct inhibitor of thrombin (Factor I la) and is used as an anticoagulant, for example, for preventing strokes in patients with atrial fibrillation or blood clots in the veins (deep vein thrombosis) that could form following surgery.
Dabigatran etexilate mesylate is the INN name of the compound 3-({2-[(4-{Amino- [(E)-hexyloxycarbonylimino]-methyl}-phenylamino)-methyl]-1 -methyl-1 H- benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate methanesulphonate, having the following structural formula:

The family of compounds to which Dabigatran etexilate belongs was described for the first time in patent US 6,087,380, which also reports possible synthesis pathways.
The preparation of polymorphs of Dabigatran etexilate or Dabigatran etexilate mesylate is described in patent applications US 2006/0276513 A1 , WO 2012/027543 A1 , WO 2008/059029 A2, WO 2013/124385 A2, WO 2013/124749 A1 , WO 2013/1 1 1 163 A2 and WO 2013/144903 A1 , while patent applications WO 2012/044595 A1 , US 2006/0247278 A1 , US 2009/0042948 A2, US 2010/0087488 A1 and WO 2012/077136 A2 describe salts of these compounds.
One of the objects of the invention is to provide an alternative process for the preparation of Dabigatran etexilate mesylate and two novel intermediates of the process.
Summary of the invention
These objects are achieved with the present invention, which, in a first aspect thereof, relates to a process for the production of Dabigatran etexilate mesylate, comprising the following steps:
a) reacting 4-methylamino-3-nitrobenzoic acid (I) with thionyl chloride to give 4- methylamino-3-nitrobenzoyl chloride hydrochloride (II):

(I) (ID
b) reacting compound (II) with 3-(2-pyridylamino) ethyl propanoate (III) to give the compound 3-[(4-methylamino-3-nitro-benzoyl)-pyridyn-2-yl-amino]-ethyl propanoate (IV):

(II) (IV)
reducing compound (IV) with hydrogen to 3-[(3-amino-4-methyl benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V):

(IV) (V)
d) reacting N-(4-cyanophenyl)glycine (VI) with 1 ,1 -carbonyldiimidazole (CDI) to give 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII):

(VI) (VII) e) reacting compound (VII) with compound (V) obtained in step c) to give one of compounds 3-({3-[2-(4-cyano-phenylamino)-acetylamino]-4-methylamino- benzoyl}-pyridin-2-yl-amino)-ethyl propanoate (VIII) and 3-[(3-amino-4-{[(2- (4-cyano-phenylamino)-acetyl]-methylamino}-benzoyl)-pyridin-2-yl- amino]ethyl propanoate (IX), or a mixture of the two compounds (VIII) and (IX):

f) transforming, through treatment with acetic acid, compounds (VIII) or (IX) or the mixture thereof into the compound 3-({2-[(4-cyano-phenylamino)-methyl]- 1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate (X), and then treating compound (X) with hydrochloric or nitric acid to form the corresponding salt (XI):
CHsCOOH
[(VIII) ; (IX)]


wherein A is a chlorine or nitrate anion;
liberating in solution compound (X) from salt (XI), and reacting compound (X) in solution with ethyl alcohol in the presence of hydrochloric acid and 2,2,2- trifluoroethanol to give the compound 3-({2-[(4-ethoxycarbonimidoyl- phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl- amino)-ethyl propanoate hydrochloride (XII):
reacting compound (XII) with ammonium carbonate to form compound Dabigatran ethyl ester (XIII):

reacting compound (XIII) with maleic acid to produce the maleate salt thereof (XI 11 ') and isolating the latter:



a gatran etex ate mesy ate
In the above formulae, the compounds indicated in brackets are not isolated during the process and are reacted, in the same reaction mixture in which they were produced, to form the next compound. Besides, step d), although reported after step c), may be carried out at any moment before step e).
In a second aspect thereof, the invention relates to the compound 3-({2-[(4- cyano-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl- amino)-ethyl propanoate nitrate, intermediate compound (XI) of the above-described process in which A is the nitrate anion.
In particular, the present invention provides the compound 3-({2-[(4-cyano- phenyl-amino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)- ethyl propanoate as nitrate salt and a process for the preparation thereof.
Specifically, this selected compound of the invention is directly isolated from the reaction mixture without passing from the free base, a procedure that allows compound (XI) to be obtained with a high degree of purity and good yields.
Lastly, the invention relates to a novel polymorph of Dabigatran etexilate, intermediate compound (XIV) of the above-described process, characterised by an XRPD profile comprising peaks at 4.80°, 8.78°, 15.16°, 16.90°, 22.76° and 24.30° 2Θ, and by three endothermic phenomena in the DSC graph, respectively with peaks at
83.0-85.0 °C, 104.0-104.2 °C (main peak), and 129.9 °C, when scanning under inert gas at a heating rate of 10 °C/min.
Brief description of the Drawings
- Figure 1 is an XRPD spectrum of the novel polymorph of Dabigatran etexilate of the invention;
- Figure 2 is the graph of a DSC test on the novel polymorph of Dabigatran etexilate of the invention.
Detailed description of the invention
The first aspect of the invention relates to the novel process for the synthesis of Dabigatran etexilate mesylate comprising the steps a) to k) above. These steps are indicated (and will be described below) separately for clarity of description, however in the process some of these steps are performed in sequence, without isolating the intermediates that form. In particular, steps a) and b); steps d) to f); and steps g) to i), are performed in the same container.
In step a) 4-methylamino-3-nitrobenzoic acid (I), a commercially available compound, is used as the starting reagent. Compound (I) is suspended in toluene, then preferably a slight stoichiometric excess (for example, 0.5%) of pyridine is added with the function of activator as it salifies the acid permitting the reaction thereof with thionyl chloride.
Thionyl chloride is then added to this solution, typically in stoichiometric excess, for example between 5 and 10%, with respect to compound (I). The mixture is brought to a temperature of between 30 and 50 °C, and is maintained in this condition for several tens of minutes, for example, 30', then the mixture is distilled to obtain a dense residue, which is dissolved with toluene. The solution thus obtained contains the intermediate compound 4-methylamino-3-nitrobenzoyl chloride hydrochloride (II) that is not isolated.
In step b), the solution thus obtained is brought to a temperature typically within the range between 0 and 30 °C. Triethylamine is then added with the function of neutralising both the hydrochloric acid that salifies the intermediate, and the hydrochloric acid that is released by reaction.
The intermediate (II) is reacted with the compound 3-(2-pyridylamino)ethyl propanoate (III), typically added in an a sub-stoichiometric amount with respect to the starting compound (I), the latter being the less costly reagent; compound (III) is thus
the limiting agent of the reaction, on which the yield thereof is calculated. The addition of triethylamine to neutralize hydrochloric acid, mentioned above, may also be carried out in this phase, in the form of a mixture with 3-(2-pyridylamino)ethyl propanoate (III) in a suitable solvent. The mixture thus obtained is left to react for a few hours, after which it is filtered to eliminate the salts that have formed, the organic phase is washed with water, concentrated by distillation to a dense residue to which ethanol is added. The residue is dissolved by heating at 60-70 °C, then the solution is cooled at 20-30 °C until the crystallization occurs and finally centrifuged at 0-10 °C. The solid obtained is washed with ethanol, thus obtaining the compound 3-[(4-methylamino-3-nitro- benzoyl)-pyridin-2-yl-amino]-ethyl propanoate (IV) in solid form. Conveniently, this phase of the process thus provides for the isolation of compound (IV) by crystallization from ethanol.
Step c) consists in the catalytic hydrogenation of compound (IV).
The solid compound (IV) obtained in the previous step is loaded into an autoclave together with an solvent inert under reaction conditions, typically ethyl acetate, or with a mixture thereof with a second solvent, for example ethyl alcohol; compound (IV) is solubilised. A hydrogenation catalyst consisting of palladium on carbon support is added to the solution; the amount of palladium is preferably equal to 5% of the total weight of the catalyst. While maintaining the temperature of the mixture to values between 10 and 30 °C, hydrogen at a pressure of between 1 and 2 bar is loaded into the autoclave, and the system is left to react for a few hours. At the end of the reaction, the product, compound 3-[(3-amino-4-methylamino-benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V), is recovered by filtering the solution to eliminate the catalyst, concentrating the liquid phase to cause precipitation of the product, and lastly, centrifuging and washing with a solvent the obtained solid. According to one embodiment of the invention, the isolation of compound V thus occurs from ethyl acetate.
Step d) is performed separately, and can be carried out at any moment before step e). This step consists in the functionalization of the compound N-(4- cyanophenyl)glycine (VI) with 1 ,1 -carbonyl-diimidazole (CDI). Suitable solvents for the reaction are well known to those skilled in the art; the use of ethyl acetate is preferred. The reaction takes place at a temperature of between about 10 and 30 °C with a slight stoichiometric excess of CDI and completes in about 2 hours, and aims to activate the
carboxylic group of N-(4-cyanophenyl)glycine. At the end of the reaction, a suspension of the benzonitrile (VII) is obtained, that is not isolated.
In the next step e), the suspension of compound (VI I) is poured into a reaction vessel containing compound (V) obtained in step c). In reaction, the acyl group corresponding to the initial glycine forms a peptide bond with one of the two primary or secondary amino groups of compound (V), giving rise to one, or generally a mixture, of compounds (VIII) and (IX). A further amount of the solvent used in the previous step can be added to adjust the concentration of the solution and prevent the same from being too dense. The mass thus obtained is brought to a temperature of between 35 and 55 °C, which is maintained for a time period of at least 6 hours. After this period, the reaction is quenched by addition of water, preferably in a volume equal to at least half of the total volume of solvent used. Compounds (VIII) and/or (IX) remain in the organic phase which is separated from the aqueous phase, which is instead eliminated.
In step f), an acid is added to the organic phase recovered at the end of the previous step, with the function of catalysing the condensation reaction between the carbonyl adjacent to one of the two amino groups on the central phenyl ring of compounds (VIII) and (IX) and the other of said two amino groups; the use of glacial acetic acid is preferred. This condensation reaction leads to the formation of the N- methyl-benzimidazole ring of compound (X), both starting from compound (VIII) and from compound (IX), which do not therefore require to be separated or isolated at the end of the previous step.
In particular, the process of the invention provides that the ring-closure of the intermediates (VIII) and (IX), aimed at giving the compound (X), directly takes place by adding glacial acetic acid to the reaction mixture, without distilling the solvent used for the previous phase or without changing it and without using acetic acid in large excess as the reaction solvent.
For carrying out the reaction of step f), the reagent mixture is heated to reflux for at least 6 hours, after which it is brought back to room temperature. Water and an ammonia solution are then added to the mixture and the aqueous phase is separated and eliminated, while the organic phase is concentrated by distillation until an oily residue containing compound (X) is obtained, which is not further purified. The oily residue is diluted with a suitable solvent, such as acetone, ethanol, ethyl acetate, N,N-
dimethylformamide (DMF) or a mixture thereof, preferably a mixture DMF/ethanol; the solution obtained is brought to a temperature of between 30 and 70 °C, and an acid selected between concentrated hydrochloric acid (aqueous solution at 37% by weight) and nitric acid (aqueous solution at 65% by weight) is added; the mass is then cooled to a temperature of between 0 and 25 °C, centrifuged, and the solid obtained is washed with acetone, thus obtaining the salt (XI). Both salts (nitrate and hydrochloride) are formed by one molecule of acid per molecule of compound (X).
Salt (XI) may be optionally recrystallized to purify it, according to general methods known in the field. For example, recrystallization may be carried out dissolving raw salt (XI) in DMF at a temperature of about 50-55 °C, adding ethanol to the resulting clear solution thus obtained, cooling the mixture between 30 and 40 °C, and stirring it until crystallization occurs (with times typically around 1 hour); the obtained suspension is then cooled to a temperature in the range of 0 to 15 °C and filtered.
In accordance with further aspects thereof, the invention provides compound (XI) when this is the nitrate salt in isolated form, and a process for the isolation of compound (XI), both in case of the hydrochloride and in case of the nitrate salts, without isolating the corresponding free base (X).
In preparation of the next step, base (X) is liberated from the salt. This is obtained by treating salt (XI) with a diluted basic water solution, e.g., a solution of sodium hydroxide or ammonia, and extracting the resulting free base with an organic solvent, preferably ethyl acetate. The resulting organic phase is concentrated to a solid residue by distillation, without however isolating and/or purifying the base; the distillation residue is used as such in step g).
The next step, g), consists of the ethoxylation of the cyan function of compound (XI) to give compound (XII). This reaction is carried out by adding ethyl alcohol to the solid residue described above; the alcohol is added in considerable excess with respect to the stoichiometric amount required, because it also acts as a solvent for the reaction; then, 2,2,2-trifluoroethanol is added and the solution is brought to a temperature of between 0 and 25 °C, gaseous hydrochloric acid being bubbled as a catalyst in the system, and the reaction is continued for at least 20 hours. At the end of the reaction, distillation takes place at reduced pressure to eliminate most of the hydrochloric acid, and the resulting reaction mixture, containing compound (XII), is used as is in the next step. The inventors have observed that the use of 2,2,2-
trifluoroethanol in the starting solution of this step significantly increases the speed of the reaction. According to some aspects, the invention thus provides the use of 2,2,2- trifluoroethanol as co-solvent and activator that accelerates the reaction that leads from compound (XI) of the invention to compound (XII).
Step h) consists in replacing the ethoxy group introduced in the previous step with an amino group, to obtain compound (XIII). For carrying out this reaction, the mixture obtained in the previous step is distilled in order to obtain an oily residue, then a suitable solvent is added, preferably ethyl alcohol, and brought to a temperature of between 20 and 40 °C, then adding ammonium carbonate in stoichiometric excess with respect to compound (XII). Reaction conditions are maintained for at least 16 hours, and at the end of this period the mass is centrifuged, typically at 20-60 °C, the precipitate washed with ethanol and the wash ethanol reunited to the liquid phase separated by centrifugation; the precipitate consists of salts that are eliminated, while compound (XIII) is found in the centrifugal mother liquors. This solution is distilled to a dense residue, after which an organic solvent and water are added in similar amounts, thus obtaining a biphasic system; the organic solvent is preferably methylene chloride. The biphasic system is stirred in the presence of a base, for example an alkali metal carbonate such as potassium carbonate, then the phases are separated. The organic phase is then distilled off obtaining an oily residue containing the desired compound (XIII).
In step i), this residue is dissolved by adding an organic solvent, for example ethanol or, preferably, Ν,Ν-dimethylformamide, then stirred and heated to 20-60 °C, preferably 30-40 °C. To this solution, maleic acid is added in an amount such that the stoichiometric ratio maleic acid:compound (XIII) is at least 0.5:1 ; maleic acid may be added in slight excess, for example in an amount such that said ratio is up to 0.8:1 . The amount of compound (XIII) can be calculated by assuming a yield of 100% in the previous step; alternatively, the solution of compound (XIII) may be titrated before the addition of maleic acid. The neutral maleic salt (XI 11 ') is obtained. After the addition of maleic acid, a second organic solvent, for example ethyl acetate, is added, then the mixture is subjected to a thermal treatment comprising the steps of heating to 70-80 °C, cooling to 20-25 °C, then further cooling to a temperature between -10 and 10 °C; the mixture is finally filtered off to obtain the desired compound (XI 11 ')
In step j), maleate salt (XI 11 ') is transformed into Dabigatran etexilate, compound
(XIV). For carrying out this reaction, the residue is dissolved in a mixture of water and a water-miscible solvent, preferably acetone, and the mass is brought to a temperature of between 0 and 25 °C. Potassium carbonate is then added and, while maintaining the temperature in the indicated range, hexyl chloroformate is added, in slight stoichiometric excess with respect to the theoretical amount of compound (XIII). The reactant mixture is maintained in said temperature range for at least two hours, after which the temperature is raised to a value between 40 and 60 °C until complete dissolution of the solids and the formation of a biphasic solution, from which the aqueous phase is eliminated by separation.
The organic phase recovered is distilled until a paste-like residue is obtained, which is dissolved with a mixture of water and a water-immiscible solvent, preferably ethyl acetate, methylene chloride, toluene or toluene/ethanol, or possibly mixtures thereof, while adding potassium carbonate to the biphasic system. The system is heated to a temperature of between 30 and 75 °C to promote the solubilisation of the components, after which the aqueous phase is eliminated. The organic phase is distilled to a dense residue. The residue is dissolved with acetone and cooled to a temperature of between 20 and 40 °C to allow precipitation of the product, the desired compound (XIV). The solution is further cooled, after which it is centrifuged and washed with acetone, thus obtaining the crude compound (XIV).
Compound (XIV) is then purified by means of recrystallization as known in the field, using anhydrous acetone as recrystallization solvent, initially bringing the mass to a temperature of between 40 and 60 °C to allow complete dissolution of compound (XIV), then cooling to 20-40 °C first and then to between 0 and 15 °C to completely precipitate the product, centrifuging and washing the solid obtained. Compound (XIV) thus produced is obtained in the form of a novel polymorph, which is described below.
Finally, in step k), compound (XIV) obtained as described is salified. Salification typically takes place with methanesulfonic acid.
The compound obtained is mono-methanesulphonate. For this purpose, compound (XIV) is dissolved in acetone at a temperature of between 40 and 60 °C to promote the complete dissolution thereof. The solution thus obtained is cooled to a temperature of between 28 and 32 °C and slowly added to a second, separately- prepared solution, pre-cooled at 0-10 °C, of methanesulfonic acid in an organic solvent, for example acetone, ethyl acetate, butyl acetate, ethanol, THF, toluene, or preferably
acetone. The mass is allowed to rest at 28-32 °C for a few hours to promote the complete precipitation of the product, after which it is centrifuged, the precipitate is washed with acetone, and dried at a temperature of between 50 and 70 °C, thus obtaining the desired salt.
In a second aspect, the invention relates to intermediate (XI) of the process described above, 3-{2-[(4-cyano-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5- carbonyl}-pyridin-2-yl-amino)-ethyl propanoate nitrate, and a process for the synthesis thereof.
In a third aspect thereof, the invention relates to the intermediate (XI 11 ') of the process described above, the neutral maleate of Dabigatran ethyl ester.
Lastly, a fourth aspect of the invention consists in the polymorph of Dabigatran etexilate obtained as intermediate (XIV) in the above-described process, characterised by an XRPD diffractogram showing peaks at 4.80°, 8.78°, 15.16°, 16.90°, 22.76° and 24.30° 2Θ (each value of the 2Θ angles of the diffractograms of powders must be understood with a degree of accuracy of ± 0.2°), and by a DSC plot that, in an inert gas scan at a speed of 10 °C/min, shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0 -104.2 °C (main), and 129.9 °C.
The invention will be further illustrated by the following examples, which are provided only for purpose of illustration and are not intended to limit the scope of the disclosure in any matter. All percentages are by weight unless specified otherwise.
The instrument used to obtain the XRPD diffractograms is a Rigaku Miniflex diffractometer, equipped with a copper anode, using the Και and Kci2 radiations of the element, having wavelengths λ = 1 .54051 A and λ = 1 .54430 A, respectively.
The DSC measurements are obtained with a Perkin-Elmer DSC 6 calorimeter.
EXAMPLE 1
Preparation of 4-methylamino-3-nitrobenzoyl chloride hydrochloride (II) (step a). 1 1 .1 kg of 4-methylamino-3-nitrobenzoic acid (56.6 mol), 47.7 kg of toluene and 4.50 kg of pyridine (56.9 mol) are loaded into a reactor. 7.3 kg of thionyl chloride (61 .36 mol) is poured into the solution, which is maintained at a T of between 30 and 50 °C.
The reagent solution is maintained at 30-50 °C for 30 minutes, and is then distilled to a dense residue, which is dissolved with 10.0 kg of toluene. The solution thus obtained contains compound (II).
EXAMPLE 2
Preparation of 3-[(4-methylamino-3-nitro-benzoyl)-pyridin-2-yl-amino]-ethyl propanoate (IV) (step b).
The solution prepared in step a) is brought to a T of between 0 and 30 °C; a second solution, prepared by dissolving 9.6 kg of 3-(2-pyridylamino)ethyl propanoate (49.42 mol) in 1 1 .2 kg of triethylamine and 40.0 kg of toluene, is slowly poured into said solution. The novel solution thus obtained is maintained at 0-30 °C for 3 hours; at the end of this period it is filtered to eliminate the salts present, and the filtered organic solution is washed with 60 kg of water and a mixture of 17.2 kg water and 2.8 kg aqueous ammonia 30%.
The organic phase is concentrated by distillation to a dense residue, then 30.0 kg of ethanol are added. The residue is dissolved by heating at 60-70 °C, then the solution is cooled at 20-30 °C, stirred at this temperature until the crystallization occurs (4 hours) and finally centrifuged at 0-10 °C, washing the solid with 10.0 kg of ethanol.
16.0 kg of 3-[(4-methylamino-3-nitrobenzoyl)-pyridin-2-yl-amino]-ethyl propanoate, equal to 42.97 moles of the compound, are obtained, with a yield of 87.0%, calculated on 3-(2-pyridylamino)ethyl propanoate, compound (IV).
EXAMPLE 3
Preparation of 3-[(3-amino-4-methylamino-benzoyl)-pyridin-2-yl-amino]-ethyl propanoate (V) (step c).
Compound (IV) obtained in step b) is loaded into a reactor along with 96 kg of ethyl acetate, 8.0 g of ethanol and 0.6 kg of Pd/C catalyst at 5%. Gaseous hydrogen is fed into the reactor at a pressure of 2 bar, and the mixture is maintained at 10-30 °C for 8 hours. At the end, the solution is filtered to eliminate the catalyst and the solution is concentrated by evaporation to reduce the volume to about one half the starting volume. The resulting solution is cooled to 10-25 °C, then centrifuged and the solid obtained is washed with 8.0 kg of ethyl acetate.
12.0 kg are obtained, equal to 35.05 moles, of 3-[(3-amino-4- methylaminobenzoyl)-pyridin-2-yl-amino]-ethyl propanoate, compound (V), with a yield in this step equal to 81 .6%.
EXAMPLE 4
Preparation of 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII) (step d). 6.5 kg of N-(4-cyanophenyl)glycine (36.9 mol) and 6.8 kg of 1 ,1 - carbonyldiimidazole (41 .94 mol) in 48.0 kg of ethyl acetate are loaded into a reactor.
The mixture is left under stirring at 10-30 °C for 2 hours, obtaining a suspension containing compound (VII).
EXAMPLE 5
Preparation of a mixture of the compounds 3-({3-[2-(4-cyano-phenylamino)- acetylamino]-4-methylamino-benzoyl}-pyridin-2-yl-amino)-ethyl propanoate (VIII) and 3-[(3-amino-4-{[(2-(4-cyano-phenylamino)-acetyl]-methylamino}-benzoyl)-pyridin-2-yl- amino]-ethyl propanoate (IX) (step e).
The suspension obtained in Example 4 is slowly poured into a reaction vessel containing the 35.05 moles of compound (V) obtained in Example 3 dissolved in 48.0 kg of ethyl acetate.
The mass is heated to 35-55 °C, and maintained under these conditions for at least 6 hours, then 60 kg of water are loaded and left to interact. Then, the aqueous phase is separated and eliminated. The organic phase contains the two title compounds, (VIII) and (IX).
EXAMPLE 6
Preparation of 3-({2-[(4-cyano-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol- 5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate nitrate (XI) (step f).
12.0 kg of glacial acetic acid is added to the organic phase obtained in Example 5 and the mixture is heated to reflux for 6 hours; the reacting mass is then cooled to 25-30 °C, after which 42.0 kg of water and 13.6 kg of an aqueous solution of ammonia at 30% by weight, are added. The phases are separated and the aqueous phase eliminated. The organic phase contains compound (X).
The organic phase is distilled to an oily residue, then 6.0 kg of DMF and 72.0 kg of ethanol are added. The mixture is heated at 50-60 °C to obtain a clear solution, then it is cooled at 35-40 °C and 3.74 kg of nitric acid (65% aqueous solution; 38.6 moles) are added. The resulting mixture is stirred until crystallization occurs (which takes about 30 minutes), then it is cooled at 20-25 °C, stirred for about 2 hours and finally filtered at 0-20 °C.
12.43 kg of 3-({2-[(4-cyano-phenylamino)-methyl]-1 -methyl-1 H-benzoimidazole- 5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate nitrate, compound (XI), equal to 22.78 moles, are obtained, with a yield in this step of 65%.
EXAMPLE 7
Purification of nitrate salt (XI).
12.43 kg of compound (XI) obtained in the previous example are dissolved in 12.4 kg of DMF at a temperature of about 50-55 °C. To the resulting clear solution, 49.6 kg of ethanol are added, then the mixture is cooled at about 35 °C and allowed to stir until crystallization occurs (almost 1 hour). The suspension is then cooled at 0-5 °C and filtered. 10.2 kg of purified 3-({2-[(4-cyano-phenylamino)-methyl]-1 -methyl-1 H- benzoimidazole-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate nitrate, compound (XI), are obtained, with a yield of 82.3%.
EXAMPLE 8
Preparation of 3-({2-[(4-ethoxycarbamimidoyl-phenylamino)-methyl]-1 -methyl- 1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate hydrochloride (XII) (step g).
A reactor is charged with 10.2 kg of compound (XI) (18.7 mol), 81 .6 kg of ethyl acetate and 10.2 kg of water. The mixture is stirred at 55-60 °C until complete dissolution occurs, then 2.1 kg of aqueous 30% solution of ammonia are added and the organic phase is separated, washed with water and brine and finally concentrate to obtain a solid residue. This residue is dissolved with 20.4 kg of 2,2,2-trifluoroethanol and 20.4 kg of ethanol. The mass is brought to 0-10 °C and 17.0 kg of gaseous hydrochloric acid is bubbled therein. The mass is then maintained at 20-25 °C under stirring for 20 hours, after which it is distilled under vacuum to eliminate most of the hydrochloric acid.
The distillation residue contains compound (XII).
EXAMPLE 9
Preparation of Dabigatran ethyl ester (XIII) (step h).
71 .4 kg of ethanol are added to the residue obtained in Example 7. The mass is brought to about 40 °C, then 8.98 kg (93.5 moles) of ammonium carbonate are added.
The reacting mass is maintained at 30-35 °C for 16 hours, then centrifuged at the same temperature to eliminate the salts formed and then washed with 10.2 kg of ethanol.
The washing ethanol is recovered and reunited to the centrifugal mother liquors, thus obtaining a solution in which the product of interest is present.
This solution is distilled to a dense residue, then 81 .6 kg of methylene chloride,
10.2 kg of water and 1 .3 kg of K2CO3 are added, the mixture is stirred at 30-35 °C and the organic phase is recovered, washed with water and distilled off to obtain an oily residue containing compound (XIII).
Yield of compound (XIII) is not calculated.
EXAMPLE 10
Preparation of Dabigatran ethyl ester neutral maleic salt (XI 11 ') (step i).
The residue obtained in example 9 is dissolved in 10.2 kg of DMF at 40-45 °C. The solution thus obtained is titrated, and the content of compound (XIII) is determined to be 16.64 moles. 0.966 kg of maleic acid (8.32 moles; 0.5:1 molar ratio with respect to compound (XIII)) are added and the resulting solution is stirred for 30', then 40.8 kg ethyl acetate are added. The mixture is stirred at 40-45 °C until crystallization occurs (adding 0.1 kg of seed), then it is heated at 70-75 °C for about 1 hour. The suspension is then cooled at 20-25 °C and finally filtered at a temperature between -5 and 0 °C. 7.3 kg of Dabigatran ethyl ester neutral maleic salt (XI 11 ') are recovered (corresponding to 6.545 moles of salt, and 13.09 moles of compound (XIII)), with a yield of 70%.
EXAMPLE 11
Preparation of crude Dabigatran etexilate (XIV) (step j).
The 7.3 kg of salt (XI I Γ) prepared in Example 10 (13.09 moles of the corresponding free base XIII) are dissolved in a mixture of 21 .9 kg of water and 36.5 kg of acetone. The mass is brought to 20-25 °C, then 5.42 kg of potassium carbonate are added and, while maintaining the temperature between 20 and 25 °C, a solution of 2.37 kg of n-hexyl chloroformate (14.4 moles) in 7.3 kg of acetone is added in about 1 hours.
The reacting mass is maintained at 20-25 °C for 2 hours and is then heated to 60 °C until complete dissolution of the reagents. Lastly, the phases are separated and the aqueous phase is eliminated.
The organic phase is distilled to obtain a paste-like residue, then 73 kg of toluene, 7.3 kg of ethanol, 7.3 kg of distilled water and 0.45 kg of potassium carbonate are added.
The mass is heated to 60-70 °C until complete dissolution of the reagents, after which the phases are separated and the aqueous phase is eliminated.
The organic phase is washed with water and brine, then distilled under vacuum to obtain a paste-like residue. 21 .9 kg of dry acetone are added and the solvent is distilled out under vacuum. 29.2 kg of acetone are then added, the mixture is heated at 50-60 °C to turn into a clear solution, then it is cooled to 30 °C, and this T is maintained for two hours obtaining a complete precipitation of the solid. The mixture is
cooled to 0-5 °C, then centrifuged and the solid is washed with 7.3 kg of cold acetone, obtaining 5.34 kg of crude Dabigatran etexilate, equal to 8.51 moles of compound, with a yield of 65% with respect to compound (XI I Γ).
The crude Dabigatran etexilate is then purified by means of recrystallization from 21 .36 kg of dry acetone. The mass is heated at 60 °C, until a complete solution of the product is obtained, then cooled to 25-30 °C, and maintained at this temperature for 2 hours, until complete precipitation. It is then cooled to 0-5 °C, then centrifuged, washing with 5.34 kg of cooled dry acetone. 4.7 kg (7.49 moles) of pure Dabigatran etexilate are obtained, with a yield of 88% with respect to the crude product.
EXAMPLE 12
Preparation of Dabigatran etexilate mesylate (step k).
All the Dabigatran etexilate obtained in Example 1 1 (4.7 kg; 7.49 moles) is loaded into a reactor along with 28.2 kg of acetone and the mass is heated at 50-60 °C until a complete solution is obtained; it is then filtered to remove suspended impurities. The filtered solution is brought to 28-32 °C. Separately, a second solution is prepared by dissolving 0.705 kg (7.34 moles) of methanesulfonic acid in 4.7 kg of acetone; the second solution is cooled down to 0-10 °C. The second solution is poured into the Dabigatran etexilate solution during 30 minutes, while maintaining the temperature of the resulting solution at 28-32 °C with cooling. The salt of the title is formed. The mass is maintained at 28-32 °C for 2 hours, then cooled to 18-23 °C to complete precipitation and the system is maintained at this temperature for 2 hours; lastly, centrifugation takes place, washing the precipitate with 5 kg of acetone. The precipitate is dried at 60 °C.
4.88 kg of Dabigatran etexilate mesylate, equal to 6.74 moles of compound, are obtained, with a yield in this step of 90%.
EXAMPLE 13
0.5 g of the crystalline compound (XIV) obtained in Example 1 1 are ground thoroughly and loaded into the sample holder of a Rigaku Miniflex diffractometer with copper anode.
The diffractogram shown in Figure 1 is obtained; a comparison with the XRPD data of the known Dabigatran etexilate polymorphs allows to verify that the polymorph of Example 1 1 is novel.
EXAMPLE 14
0.7 g of the crystalline compound (XIV) obtained in Example 1 1 are loaded into
the sample holder of a Perkin-Elmer DSC 6 calorimeter, performing a scan from ambient T to 350 °C at a rate of 10 °C/min in nitrogen atmosphere. The graph of the test is shown in Figure 2, and shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0-104.2 °C and 129.9 °C; events linked to the thermal decomposition of the compound are evident at about 200 °C.
Claims
A process for producing Dabigatran etexilate mesylate, which comprises the following steps:
a) reacting 4-methylamino-3-nitrobenzoic acid (I) with thionyl chloride to give 4- methylamino-3-nitrobenzoyl chloride hydrochloride (II):
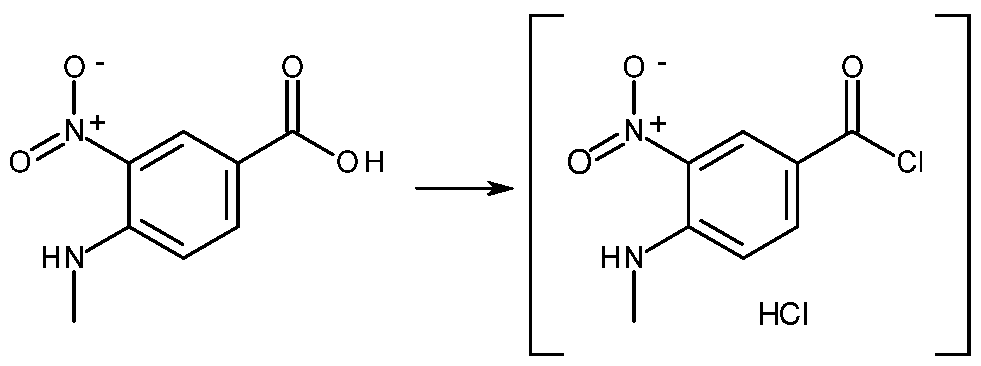
(I) (ID
b) reacting compound (II) with 3-(2-pyridylamino) ethyl propanoate (III) to give the compound 3-[(4-methylamino-3-nitro-benzoyl)-pyridyn-2-yl-amino]-ethyl propanoate (IV):

(II) (IV)
reducing compound (IV) with hydrogen to 3-[(3-amino-4-methyl benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V):

(IV) (V)
d) reacting N-(4-cyanophenyl)glycine (VI) with 1 ,1 -carbonyldiimidazole (CDI) to give 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII):

(VI) (VII)
e) reacting compound (VII) with compound (V) obtained in step c) to give one of compounds 3-({3-[2-(4-cyano-phenylamino)-acetylamino]-4-methylamino- benzoyl}-pyridin-2-yl-amino)-ethyl propanoate (VIII) and 3-[(3-amino-4-{[(2- (4-cyano-phenylamino)-acetyl]-methylamino}-benzoyl)-pyridin-2-yl- amino]ethyl propanoate (IX) or a mixture of the two compounds (VIII) and (IX):

f) transforming, through treatment with acetic acid, compounds (VIII) or (IX) or the mixture thereof into the compound 3-({2-[(4-cyano-phenylamino)-methyl]- 1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate (X), and then treating compound (X) with hydrochloric or nitric acid to form the corresponding salt (XI):

wherein A is a chlorine or nitrate anion;
g) liberating in solution compound (X) from salt (XI), and reacting compound (X) in solution with ethyl alcohol in the presence of hydrochloric acid and 2,2,
2- trifluoroethanol to give the compound 3-({2-[(4-ethoxycarbonimidoyl- phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl- amino)-ethyl propanoate hydrochloride (XII):

reacting compound (XII) with ammonium carbonate to form compound Dabigatran ethyl ester (XIII):

i) reacting compound (XIII) with maleic acid to produce the maleate salt thereof (XI 11 ') and isolating the latter:



3. Dabigatran ethyl ester neutral maleate salt, (XI I Γ).
4. A polymorph of Dabigatran etexilate, characterised by an XRPD profile comprising peaks at 4.80°, 8.78°, 15.16°, 16.90°, 22.76° and 24.30° ± 0.2°, 2Θ.
5. A polymorph according to claim 4, characterised by a DSC graph which, in a scan in inert gas at a speed of 10 °C/min, shows three endothermic phenomena with maxima at 83.0-85.0 °C, 104.0-104.2 °C and 129.9 °C.
6. A process for production of 3-({2-[(4-cyano-phenylamino)-methyl]-1 -methyl-1 H- benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate nitrate, (XI), comprising steps a) to f) of the process of claim 1 , characterized in that in step f) compound (X) is treated with nitric acid and in that said nitrate is isolated without previously isolating the corresponding free base (X).
7. A process according to claim 6 wherein the reactions in steps a) and/or b) are carried out in the presence of toluene as solvent.
8. A process according to claim 6 or claim 7 wherein step b) comprises isolation of the intermediate compound (IV) by crystallization from ethanol.
9. A process according to any one of claims 6 to 8, wherein step c) comprises isolation of compound (V) by concentration of the mixture in ethyl acetate, preferably after filtration of the catalyst.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI20140272 | 2014-02-24 | ||
| ITMI2014A000272 | 2014-02-24 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2015124764A1 true WO2015124764A1 (en) | 2015-08-27 |
| WO2015124764A8 WO2015124764A8 (en) | 2015-12-30 |
Family
ID=50693778
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2015/053702 WO2015124764A1 (en) | 2014-02-24 | 2015-02-23 | Synthesis process of dabigatran etexilate mesylate, intermediates of the process and novel polymorph of dabigatran etexilate |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015124764A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105153118A (en) * | 2015-09-24 | 2015-12-16 | 青岛黄海制药有限责任公司 | Central control method of midbody in dabigatran etexilate mesylate preparation process |
| CN105294651A (en) * | 2015-09-23 | 2016-02-03 | 烟台东诚药业集团股份有限公司 | Method for synthesizing and preparing pradaxa formamidine intermediates |
| CN105906607A (en) * | 2015-12-23 | 2016-08-31 | 嘉实(湖南)医药科技有限公司 | Dabigatran etexilate synthesis method |
| WO2017037743A3 (en) * | 2015-09-03 | 2018-09-27 | Sun Pharmaceutical Industries Limited | Dabigatran etexilate 1,4-butanedisulfonate salt and its crystal form |
| CN108658939A (en) * | 2018-07-16 | 2018-10-16 | 上海现代制药股份有限公司 | A kind of synthetic method of dabigatran etexilate methanesulfonate key intermediate |
| CN115141124A (en) * | 2022-06-16 | 2022-10-04 | 重庆华森制药股份有限公司 | Method for preparing gabexate mesylate |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6087380A (en) | 1949-11-24 | 2000-07-11 | Boehringer Ingelheim Pharma Kg | Disubstituted bicyclic heterocycles, the preparations and the use thereof as pharmaceutical compositions |
| US20060247278A1 (en) | 2005-04-27 | 2006-11-02 | Boehringer Ingelheim International Gmbh | Physiologically acceptable salts of 3-[(2-{[4-(hexyloxycarbonylamino-imino-methyl)-phenylamino]-methyl}-1-methyl-1H-benzimidazol-5-carbonyl)-pyridin-2-yl-amino]-propionic acid ethyl ester |
| US20060276513A1 (en) | 2005-06-04 | 2006-12-07 | Boehringer Ingelheim International Gmbh | Polymorphs of 3-[(2-{[4-(hexyloxycarbonylamino-imino-methyl)-phenylamino]-methyl}-1-methyl-1H- benzimidazole-5-carbonyl)-pyridin-2-yl-amino]-propionic acid ethyl ester |
| WO2008059029A2 (en) | 2006-11-16 | 2008-05-22 | Boehringer Ingelheim International Gmbh | New polymorphs of ethyl 3-[(2-{[4-(hexyloxycarbonylamino-imino-methyl)-phenylamino]-methyl}-1-methyl-1h-benzimidazole-5-carbonyl)-pyridin-2-yl-amino]-propionate |
| WO2009111997A1 (en) * | 2008-03-14 | 2009-09-17 | Zentiva, K.S. | A method for the preparation of dabigatran |
| US20100087488A1 (en) | 2006-10-10 | 2010-04-08 | Boehringer Ingelheim International Gmgh | Physiologically Acceptable Salts of 3-[(2--1-methyl-1H-benzimidazol-5-carbonyl)-pyridin-2-yl-amino]-propionic acid ethyl ester |
| WO2012004396A2 (en) * | 2010-07-09 | 2012-01-12 | Esteve Química, S.A. | Process of preparing a thrombin specific inhibitor |
| WO2012027543A1 (en) | 2010-08-25 | 2012-03-01 | Teva Pharmaceuticals Usa, Inc. | Solid state forms of dabigatran etexilate, dabigatran etexilate mesylate and processes for preparation thereof |
| WO2012044595A1 (en) | 2010-09-27 | 2012-04-05 | Ratiopharm Gmbh | Dabigatran etexilate bismesylate salt, solid state forms and process for preparation thereof |
| WO2012077136A2 (en) | 2010-12-06 | 2012-06-14 | Msn Laboratories Limited | Process for the preparation of benzimidazole derivatives and its salts |
| EP2522662A1 (en) * | 2011-05-11 | 2012-11-14 | Medichem, S.A. | Dabigatran etexilate and related substances, processes and compositions, and use of the substances as reference standards and markers |
| WO2013111163A2 (en) | 2012-01-20 | 2013-08-01 | Cadila Healthcare Limited | Process for the preparation of dabigatran etexilate mesylate and polymorphs of intermediates thereof |
| WO2013124385A2 (en) | 2012-02-23 | 2013-08-29 | Esteve Química, S.A. | Solid forms of dabigatran etexilate mesylate and processes for their preparation |
| WO2013124749A1 (en) | 2012-02-20 | 2013-08-29 | Alembic Pharmaceuticals Limited | Novel polymorph of dabigatran etexilate |
| WO2013144903A1 (en) | 2012-03-28 | 2013-10-03 | Dr. Reddy's Laboratories Limited | Processes for the preparation of dabigatran etexilate |
| WO2013150545A2 (en) * | 2012-04-02 | 2013-10-10 | Msn Laboratories Limited | Process for the preparation of benzimidazole derivatives and salts thereof |
-
2015
- 2015-02-23 WO PCT/EP2015/053702 patent/WO2015124764A1/en active Application Filing
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6087380A (en) | 1949-11-24 | 2000-07-11 | Boehringer Ingelheim Pharma Kg | Disubstituted bicyclic heterocycles, the preparations and the use thereof as pharmaceutical compositions |
| US20060247278A1 (en) | 2005-04-27 | 2006-11-02 | Boehringer Ingelheim International Gmbh | Physiologically acceptable salts of 3-[(2-{[4-(hexyloxycarbonylamino-imino-methyl)-phenylamino]-methyl}-1-methyl-1H-benzimidazol-5-carbonyl)-pyridin-2-yl-amino]-propionic acid ethyl ester |
| US20090042948A1 (en) | 2005-04-27 | 2009-02-12 | Boehringer Ingelheim International Gmbh | Physiologically acceptable salts of 3--1-methyl-1H-benzimidazol-5-carbonyl)-pyridin-2-yl-amino]-propionic acid ethyl ester |
| US20060276513A1 (en) | 2005-06-04 | 2006-12-07 | Boehringer Ingelheim International Gmbh | Polymorphs of 3-[(2-{[4-(hexyloxycarbonylamino-imino-methyl)-phenylamino]-methyl}-1-methyl-1H- benzimidazole-5-carbonyl)-pyridin-2-yl-amino]-propionic acid ethyl ester |
| US20100087488A1 (en) | 2006-10-10 | 2010-04-08 | Boehringer Ingelheim International Gmgh | Physiologically Acceptable Salts of 3-[(2--1-methyl-1H-benzimidazol-5-carbonyl)-pyridin-2-yl-amino]-propionic acid ethyl ester |
| WO2008059029A2 (en) | 2006-11-16 | 2008-05-22 | Boehringer Ingelheim International Gmbh | New polymorphs of ethyl 3-[(2-{[4-(hexyloxycarbonylamino-imino-methyl)-phenylamino]-methyl}-1-methyl-1h-benzimidazole-5-carbonyl)-pyridin-2-yl-amino]-propionate |
| WO2009111997A1 (en) * | 2008-03-14 | 2009-09-17 | Zentiva, K.S. | A method for the preparation of dabigatran |
| WO2012004396A2 (en) * | 2010-07-09 | 2012-01-12 | Esteve Química, S.A. | Process of preparing a thrombin specific inhibitor |
| WO2012027543A1 (en) | 2010-08-25 | 2012-03-01 | Teva Pharmaceuticals Usa, Inc. | Solid state forms of dabigatran etexilate, dabigatran etexilate mesylate and processes for preparation thereof |
| WO2012044595A1 (en) | 2010-09-27 | 2012-04-05 | Ratiopharm Gmbh | Dabigatran etexilate bismesylate salt, solid state forms and process for preparation thereof |
| WO2012077136A2 (en) | 2010-12-06 | 2012-06-14 | Msn Laboratories Limited | Process for the preparation of benzimidazole derivatives and its salts |
| EP2522662A1 (en) * | 2011-05-11 | 2012-11-14 | Medichem, S.A. | Dabigatran etexilate and related substances, processes and compositions, and use of the substances as reference standards and markers |
| WO2013111163A2 (en) | 2012-01-20 | 2013-08-01 | Cadila Healthcare Limited | Process for the preparation of dabigatran etexilate mesylate and polymorphs of intermediates thereof |
| WO2013124749A1 (en) | 2012-02-20 | 2013-08-29 | Alembic Pharmaceuticals Limited | Novel polymorph of dabigatran etexilate |
| WO2013124385A2 (en) | 2012-02-23 | 2013-08-29 | Esteve Química, S.A. | Solid forms of dabigatran etexilate mesylate and processes for their preparation |
| WO2013144903A1 (en) | 2012-03-28 | 2013-10-03 | Dr. Reddy's Laboratories Limited | Processes for the preparation of dabigatran etexilate |
| WO2013150545A2 (en) * | 2012-04-02 | 2013-10-10 | Msn Laboratories Limited | Process for the preparation of benzimidazole derivatives and salts thereof |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017037743A3 (en) * | 2015-09-03 | 2018-09-27 | Sun Pharmaceutical Industries Limited | Dabigatran etexilate 1,4-butanedisulfonate salt and its crystal form |
| CN105294651A (en) * | 2015-09-23 | 2016-02-03 | 烟台东诚药业集团股份有限公司 | Method for synthesizing and preparing pradaxa formamidine intermediates |
| CN105153118A (en) * | 2015-09-24 | 2015-12-16 | 青岛黄海制药有限责任公司 | Central control method of midbody in dabigatran etexilate mesylate preparation process |
| CN105906607A (en) * | 2015-12-23 | 2016-08-31 | 嘉实(湖南)医药科技有限公司 | Dabigatran etexilate synthesis method |
| CN108658939A (en) * | 2018-07-16 | 2018-10-16 | 上海现代制药股份有限公司 | A kind of synthetic method of dabigatran etexilate methanesulfonate key intermediate |
| CN108658939B (en) * | 2018-07-16 | 2021-07-20 | 上海现代制药股份有限公司 | Synthesis method of dabigatran etexilate mesylate key intermediate |
| CN115141124A (en) * | 2022-06-16 | 2022-10-04 | 重庆华森制药股份有限公司 | Method for preparing gabexate mesylate |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2015124764A8 (en) | 2015-12-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015124764A1 (en) | Synthesis process of dabigatran etexilate mesylate, intermediates of the process and novel polymorph of dabigatran etexilate | |
| EP3645518B1 (en) | Synthesis of omecamtiv mecarbil | |
| EP1926705B1 (en) | Process for preparing valsartan | |
| CN106905314A (en) | Method for preparing 5 fluorine 1H pyrazolo-pyridines of substitution | |
| CN107365275B (en) | High purity celecoxib | |
| CA2988594C (en) | Methods of making protein deacetylase inhibitors | |
| US10927095B2 (en) | Processes for the preparation of Niraparib and intermediates thereof | |
| JP5078993B2 (en) | Process for producing 1- (3,4-dichlorobenzyl) -5-octyl biguanide or a salt thereof | |
| US6495685B1 (en) | Process for preparing piperazine derivatives | |
| EP2872499A1 (en) | Process for the preparation of intermediates for the synthesis of dabigatran etexilate, and crystalline forms of said intermediates | |
| JP2013531004A (en) | Intermediates and methods for the preparation of thrombin specific inhibitors | |
| KR20150123267A (en) | Process for the preparation of enantiomerically enriched 3-aminopiperidine | |
| CN108864084B (en) | Apixaban related substances and preparation method thereof | |
| EP2928472B1 (en) | Process for making reverse transcriptase inhibitors | |
| WO2011117876A1 (en) | An improved process for the preparation of amlodipine free base and acid addition salts thereof | |
| US11932614B2 (en) | Method for preparing diazoxide | |
| JP5078211B2 (en) | Method for producing heterocyclic compound | |
| CN107722007A (en) | The preparation method of Eliquis impurity | |
| WO2006022182A1 (en) | Method for producing 2-(4-methyl-2-phenylpiperazine-1-yl)-3-cyanopiridine | |
| CN117843613A (en) | Preparation method of dabigatran etexilate intermediate | |
| WO2015162551A1 (en) | Process for the preparation of apixaban | |
| KR101266224B1 (en) | An improved process for the preparation of trityl olmesartan medoxomil | |
| WO2010010058A1 (en) | Method of preparing anhydrous alfusozin hydrochloride | |
| MXPA00001529A (en) | Crystalline 10,10-bis((2-fluoro-4-pyridinyl)methyl)-9(10h)-anthracenone and an improved process for preparing the same | |
| KR20180018697A (en) | Novel manufacturing process of terry flunomide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15711432 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15711432 Country of ref document: EP Kind code of ref document: A1 |