WO2013029338A1 - Novel compounds - Google Patents
Novel compounds Download PDFInfo
- Publication number
- WO2013029338A1 WO2013029338A1 PCT/CN2012/001221 CN2012001221W WO2013029338A1 WO 2013029338 A1 WO2013029338 A1 WO 2013029338A1 CN 2012001221 W CN2012001221 W CN 2012001221W WO 2013029338 A1 WO2013029338 A1 WO 2013029338A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- mixture
- acetamide
- ethylsulfonyl
- water
- Prior art date
Links
- 0 CC1C(c(cc2)ccc2N*(Cc(cc2)ccc2S(*)(=O)=O)=O)=CC=CC1 Chemical compound CC1C(c(cc2)ccc2N*(Cc(cc2)ccc2S(*)(=O)=O)=O)=CC=CC1 0.000 description 3
- YZOZLABRHJTSHU-UHFFFAOYSA-N CC(C)N(CC1)CCC1=Cc(ccc(-c(cc1)c(C)cc1NC(Cc(cc1)ccc1S(C)(=O)=O)=O)c1)c1C#N Chemical compound CC(C)N(CC1)CCC1=Cc(ccc(-c(cc1)c(C)cc1NC(Cc(cc1)ccc1S(C)(=O)=O)=O)c1)c1C#N YZOZLABRHJTSHU-UHFFFAOYSA-N 0.000 description 1
- ACEKJYPWFHXQDC-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(c2c3[nH]cc2)ccc3-c(cccc2)c2OC(F)(F)F)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(c2c3[nH]cc2)ccc3-c(cccc2)c2OC(F)(F)F)=O)cc1)(=O)=O ACEKJYPWFHXQDC-UHFFFAOYSA-N 0.000 description 1
- PWJLYQQGCQHIKY-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(c2c3cc[nH]2)ccc3-c(cccc2)c2OC(F)(F)F)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(c2c3cc[nH]2)ccc3-c(cccc2)c2OC(F)(F)F)=O)cc1)(=O)=O PWJLYQQGCQHIKY-UHFFFAOYSA-N 0.000 description 1
- DBYCANNVXKNHGW-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(-c(cc3)ccc3C#N)c2C(c(cccc2)c2Cl)=O)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(-c(cc3)ccc3C#N)c2C(c(cccc2)c2Cl)=O)=O)cc1)(=O)=O DBYCANNVXKNHGW-UHFFFAOYSA-N 0.000 description 1
- YDCWYZVCWOWTIT-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(-c3ccccn3)c2-c(cccc2)c2OC(F)(F)F)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(-c3ccccn3)c2-c(cccc2)c2OC(F)(F)F)=O)cc1)(=O)=O YDCWYZVCWOWTIT-UHFFFAOYSA-N 0.000 description 1
- KLKBJOMAKCCZSL-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(C(F)(F)F)c2-c(cccc2)c2Cl)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(C(F)(F)F)c2-c(cccc2)c2Cl)=O)cc1)(=O)=O KLKBJOMAKCCZSL-UHFFFAOYSA-N 0.000 description 1
- RJXLYJKPTGSBPU-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(C(N(C)C)=O)c2-c2ccccc2OC(F)(F)F)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(C(N(C)C)=O)c2-c2ccccc2OC(F)(F)F)=O)cc1)(=O)=O RJXLYJKPTGSBPU-UHFFFAOYSA-N 0.000 description 1
- PMYXIPVHHVQYKU-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(C(c3ccccc3)=O)c2-c2cccc(C#N)c2)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(C(c3ccccc3)=O)c2-c2cccc(C#N)c2)=O)cc1)(=O)=O PMYXIPVHHVQYKU-UHFFFAOYSA-N 0.000 description 1
- SRTNLRQYXRVXBX-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(Cl)c2-c(cccc2)c2N(C2)CC2OC)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(Cl)c2-c(cccc2)c2N(C2)CC2OC)=O)cc1)(=O)=O SRTNLRQYXRVXBX-UHFFFAOYSA-N 0.000 description 1
- QKFVMGVKZSEFEE-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(Cl)c2-c(cccc2)c2Oc(cc2)ccc2Cl)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(Cl)c2-c(cccc2)c2Oc(cc2)ccc2Cl)=O)cc1)(=O)=O QKFVMGVKZSEFEE-UHFFFAOYSA-N 0.000 description 1
- BXHNCGYEVGEWMD-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)cc(Cl)c2-c(cccc2)c2Oc2ccccc2Cl)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)cc(Cl)c2-c(cccc2)c2Oc2ccccc2Cl)=O)cc1)(=O)=O BXHNCGYEVGEWMD-UHFFFAOYSA-N 0.000 description 1
- XQDKDPQSRFWIEI-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)ccc2-c2c(C(F)(F)F)cc(CC(CC3)CCN3C(C)=O)cc2)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)ccc2-c2c(C(F)(F)F)cc(CC(CC3)CCN3C(C)=O)cc2)=O)cc1)(=O)=O XQDKDPQSRFWIEI-UHFFFAOYSA-N 0.000 description 1
- ZRSJSRWQMBNGNB-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2)nc(-c3cc(Cl)ccc3)c2OC(C)C)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2)nc(-c3cc(Cl)ccc3)c2OC(C)C)=O)cc1)(=O)=O ZRSJSRWQMBNGNB-UHFFFAOYSA-N 0.000 description 1
- FRRLHLJLGQMPGG-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2-c3c[n](C)nc3)cc(Cl)c2OCC(C)C)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2-c3c[n](C)nc3)cc(Cl)c2OCC(C)C)=O)cc1)(=O)=O FRRLHLJLGQMPGG-UHFFFAOYSA-N 0.000 description 1
- KMKVLWBEGHQWBB-UHFFFAOYSA-N CCS(c1ccc(CC(Nc(cc2Cl)cc(-c3c[n](C)nc3)c2Cl)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc(cc2Cl)cc(-c3c[n](C)nc3)c2Cl)=O)cc1)(=O)=O KMKVLWBEGHQWBB-UHFFFAOYSA-N 0.000 description 1
- GWVKDEZKOSDCOC-UHFFFAOYSA-N CCS(c1ccc(CC(Nc2cc(C)c(-c(cccc3)c3OC(F)(F)F)nc2)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc2cc(C)c(-c(cccc3)c3OC(F)(F)F)nc2)=O)cc1)(=O)=O GWVKDEZKOSDCOC-UHFFFAOYSA-N 0.000 description 1
- HSMRIKYXWHIFAO-UHFFFAOYSA-N CCS(c1ccc(CC(Nc2cc(Cl)c(-c(cccc3)c3C#N)c(Cl)c2)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc2cc(Cl)c(-c(cccc3)c3C#N)c(Cl)c2)=O)cc1)(=O)=O HSMRIKYXWHIFAO-UHFFFAOYSA-N 0.000 description 1
- SHAXPZNJCXGLJI-UHFFFAOYSA-N CCS(c1ccc(CC(Nc2nc(C)c(-c3ccccc3OC(F)(F)F)nc2)=O)cc1)(=O)=O Chemical compound CCS(c1ccc(CC(Nc2nc(C)c(-c3ccccc3OC(F)(F)F)nc2)=O)cc1)(=O)=O SHAXPZNJCXGLJI-UHFFFAOYSA-N 0.000 description 1
- BSSXAWVCEBLCBX-UHFFFAOYSA-N CC[n](cc1)c(c(-c2c[n](C)nc2)c2)c1cc2NC(Cc(cc1)ccc1S(CC)(=O)=O)=O Chemical compound CC[n](cc1)c(c(-c2c[n](C)nc2)c2)c1cc2NC(Cc(cc1)ccc1S(CC)(=O)=O)=O BSSXAWVCEBLCBX-UHFFFAOYSA-N 0.000 description 1
- ZPMZTBQLEBQMQC-UHFFFAOYSA-N CS(c1ccc(CC(Nc2ccc(-c(c(C(F)(F)F)c3)ccc3F)c(Cl)c2)=O)cc1)(=O)=O Chemical compound CS(c1ccc(CC(Nc2ccc(-c(c(C(F)(F)F)c3)ccc3F)c(Cl)c2)=O)cc1)(=O)=O ZPMZTBQLEBQMQC-UHFFFAOYSA-N 0.000 description 1
- OQBXCJKABFKBNA-UHFFFAOYSA-N Cc(cc(cc1)NC(Cc(cc2)ccc2S(NC)(=O)=O)=O)c1-[n]1c2cc(C#N)ccc2cc1 Chemical compound Cc(cc(cc1)NC(Cc(cc2)ccc2S(NC)(=O)=O)=O)c1-[n]1c2cc(C#N)ccc2cc1 OQBXCJKABFKBNA-UHFFFAOYSA-N 0.000 description 1
- HGJFNRPNMNVBSR-UHFFFAOYSA-N NS(c1ccc(CC(Nc2cc(Cl)c(-c3ccccc3OC(F)F)c(Cl)c2)=O)cc1)(=O)=O Chemical compound NS(c1ccc(CC(Nc2cc(Cl)c(-c3ccccc3OC(F)F)c(Cl)c2)=O)cc1)(=O)=O HGJFNRPNMNVBSR-UHFFFAOYSA-N 0.000 description 1
- LQQJBZBNRAGAKO-UHFFFAOYSA-N O=C(Cc(cc1)ccc1S=O)Nc1cc(Cl)c(-c(cccc2)c2OC(F)(F)F)c(Cl)c1 Chemical compound O=C(Cc(cc1)ccc1S=O)Nc1cc(Cl)c(-c(cccc2)c2OC(F)(F)F)c(Cl)c1 LQQJBZBNRAGAKO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/70—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/29—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/50—Compounds containing any of the groups, X being a hetero atom, Y being any atom
- C07C311/51—Y being a hydrogen or a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
- C07C317/46—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton the carbon skeleton being further substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/32—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D207/325—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms with substituted hydrocarbon radicals directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/64—One oxygen atom attached in position 2 or 6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/70—Sulfur atoms
- C07D213/71—Sulfur atoms to which a second hetero atom is attached
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/84—Nitriles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/18—One oxygen or sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
- C07D233/61—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms not forming part of a nitro radical, attached to ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/08—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/20—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/26—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/38—One sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/20—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
- C07D249/06—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles with aryl radicals directly attached to ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/12—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms
- C07D295/135—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms with the ring nitrogen atoms and the substituent nitrogen atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/10—Oxygen atoms
- C07D309/12—Oxygen atoms only hydrogen atoms and one oxygen atom directly attached to ring carbon atoms, e.g. tetrahydropyranyl ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/14—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems
- C07D319/16—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D319/18—Ethylenedioxybenzenes, not substituted on the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Definitions
- the present invention relates to novel retinoid-related orphan receptor gamma (RORy) modulators and their use in the treatment of diseases mediated by RORy.
- RORy retinoid-related orphan receptor gamma
- RORs Retinoid-related orphan receptors
- the ROR family consists of three members, ROR alpha (RORa), ROR beta (RORp) and ROR gamma (RORy), each encoded by a separate gene (RORA, RORB and RORC, respectively).
- RORs contain four principal domains shared by the majority of nuclear receptors: an N-terminal A/B domain, a DNA-binding domain, a hinge domain, and a ligand binding domain. Each ROR gene generates several isoforms which differ only in their N-terminal A/B domain. Two isoforms of RORy have been identified: RORyl and RORyt (also known as RORy2).
- RORy is a term used to describe both RORyl and/or RORyt.
- Thl7 cells are a subset of T helper cells which produce IL- 17 and other proinflammatory cytokines. Thl7 cells have been shown to have key functions in several mouse autoimmune disease models including experimental autoimmune encephalomyelitis (EAE) and collagen-induced artliritis (CIA).
- EAE experimental autoimmune encephalomyelitis
- CIA collagen-induced artliritis
- Thl7 cells or their products have been shown to be associated with the pathology of a variety of human inflammatory and autoimmune disorders including multiple sclerosis, rheumatoid artliritis, psoriasis, Crohn's disease and asthma (Jetten (2009) Nucl. RecepL Signal. 7: e003; Manel et al. (2008) Nat. Immunol. 9:641-649).
- the pathogenesis of chronic autoimmune diseases including multiple sclerosis and rheumatoid artliritis arises from the break in tolerance towards self-antigens and the development of auto-aggressive effector T cells infiltrating the target tissues.
- Thl7 cells are one of the important drivers of the inflammatory process in tissue-specific autoimmunity (Steinman (2008) J. Exp. Med. 205: 1517- 1522; Leung et al. (2010) Cell. Mol. Immunol 7: 182-189). There is evidence that Thl7 cells are activated during the disease process and are responsible for recruiting other inflammatory cells types, especially neutrophils, to mediate pathology in the target tissues (Korn et al. (2009) Annu. Rev.
- RORyt plays a critical role in the pathogenic responses of Thl7 cells (Ivanov et al. (2006) Cell 126: 1121 -1 133). RORyt deficient mice show very little Thl7 cells. In addition, RORyt deficiency resulted in amelioration of EAE. Further support for the role of RORyt in the pathogensis of autoimmune or inflammatory diseases can be found in the following references: Jetten & Joo (2006) Adv.Dev.Biol 16:313-355; Meier et al. (2007) Immunity 26:643-654; Aloisi & Pujol-Borrell (2006) Nat. Rev. Immunol 6:205-217; Jager et al. (200 ) J. Immunol. 183:7169-7177; Serafini et al. (2004) Brain ⁇ /.14:164-174; Magliozzi et al. (2007) Brain 130: 1089-1104; Barnes (2008)
- the invention is directed to novel RORy modulators and their use in the treatment of diseases mediated by RORy. Specifically, the invention is directed to compounds according to Formula I.
- A, B, C, Rl , R2, R3, R4, R5, m, r, s and t are defined below, and to pharmaceutically- acceptable salts thereof.
- this invention provides for the use of the compounds of Formula I for the treatment of diseases mediated by RORy.
- diseases include autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid artliritis, psoriasis, Crohn's disease and asthma.
- the invention is directed to methods of treating such diseases.
- Alkosy refers to the group -O-R where R is alkyl having the specified number of member atoms. Alkoxy includes methoxy, ethoxy and propoxy. "Alkyl” refers to a monovalent saturated hydrocarbon chain having the specified number of member atoms. For example, C1-C6 alkyl refers to an alkyl group having from 1 to 6 member atoms. Alkyl groups may be optionally substituted with one or more substituent as defined herein. Alkyl groups may be straight or branched. Representative branched alkyl groups have one, two, or three branches.
- Alkyl includes methyl, ethyl, propyl (n-propyl and isopropyl), butyl (n-butyl, isobutyl, and t-butyl), pentyl (n-pentyl, isopentyl, and neopentyl), and hexyl.
- Alkenyl refers to an unsaturated hydrocarbon chain having the specified number of member atoms and having one or more carbon-carbon double bond within the chain.
- C2-C6 alkenyl refers to an alkenyl group having from 2 to 6 member atoms.
- alkenyl groups have one carbon-carbon double bond within the chain.
- alkenyl groups have more than one carbon-carbon double bond within the chain.
- Alkenyl groups may be optionally substituted with one or more substituent as defined herein.
- Alkenyl groups may be straiglit or branched. Representative branched alkenyl groups have one, two, or three branches.
- Alkenyl includes ethylenyl, propenyl, butenyl, pentenyl, and hexenyl.
- Cycloalkyl refers to a saturated hydrocarbon ring having the specified number of member atoms. Cycloalkyl groups are monocyclic ring systems. For example, C3-C6 cycloalkyl refers to a cycloalkyl group having from 3 to 6 member atoms. Cycloalkyl groups may be optionally substituted with one or more substituent as defined herein. Cycloalkyl includes cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- Enantiomerically enriched refers to products whose enantiomeric excess is greater than zero.
- enantiomerically enriched refers to products whose enantiomeric excess is greater than 50% ee, greater than 75% ee, and greater than 90% ee.
- Enantiomeric excess or "ee” is the excess of one enantiomer over the odier expressed as a percentage. As a result, since both enantiomers are present in equal amounts in a racemic mixture, the enantiomeric excess is zero (0% ee). However, if one enantiomer was enriched such that it constitutes 95% of the product, then the enantiomeric excess would be 90% ee (the amount of the enriched enantiomer, 95%, minus the amount of the other enantiomer, 5%).
- Enantiomerically pure refers to products whose enantiomeric excess is 99% ee or greater.
- Half-life refers to the time required for half of a quantity of a substance to be converted to another chemically distinct species in vitro or in vivo.
- Heteroaryl refers to an aromatic ring containing from 1 to 4 heteroatoms as member atoms in the ring. Heteroaryl groups containing more than one heteroatom may contain different heteroatoms. Heteroaryl groups may be optionally substituted with one or more substituent as defined herein. Heteroaryl groups are monocyclic ring systems or are fused or bridged bicyclic ring systems. Monocyclic heteroaryl rings have from 5 to 7 member atoms. Bicyclic heteroaryl rings have from 7 to 11 member atoms.
- Bicyclic heteroaryl rings include those rings wherein phenyl and a monocyclic heterocycloalkyl ring are attached forming a fused, spiro, or bridged bicyclic ring system, and those rings wherein a monocyclic heteroaryl ring and a monocyclic cycloalkyl, cycloalkenyl, heterocycloalkyl, or heteroaryl ring are attached forming a fused, spiro, or bridged bicyclic ring system.
- Heteroaryl includes pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, furanyl, furazanyl, thienyl, triazolyl, pyridinyl, pyrimidinyl, pyridazinyl, pyrazinyl, triazinyl, tetrazinyl, tetrazolyl, indolyl, isoindolyl, indolizinyl, indazolyl, purinyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl, pteridinyl, cinnolinyl, benzimidazolyl, benzopyranyl, benzoxazolyl, benzisoxazolyl, benzofuranyl, iso
- Heteroatom refers to a nitrogen, sulphur, or oxygen atom.
- Heterocycloalkyl refers to a saturated or unsaturated ring containing from 1 to 4 heteroatoms as member atoms in the ring. However, heterocycloalkyl rings are not aromatic.
- Heterocycloalkyl groups containing more than one heteroatom may contain different heteroatoms. Heterocycloalkyl groups may be optionally substituted with one or more substituent as defined herein. Heterocycloalkyl groups are monocyclic ring systems or are fused, spiro, or bridged bicyclic ring systems. Monocyclic heterocycloalkyl rings have from 5 to 7 member atoms. Bicyclic
- heterocycloalkyl rings have from 7 to 11 member atoms.
- heterocycloalkyl is saturated.
- heterocycloalkyl is unsaturated but not aromatic.
- Heterocycloalkyl includes pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, pyranyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothienyl, pyrazolidinyl, oxazolidinyl, thiazolidinyl, piperidinyl, homopiperidinyl, piperazinyl, morpholinyl, thiamorpholinyl, azepinyl, 1,3-dioxolanyl, 1,3-dioxanyl, 1 ,4-dioxanyl, 1 ,3-oxathiolanyl, 1,3-oxathianyl, 1,3-dithianyl, azetidin
- Member atoms refers to the atom or atoms that form a chain or ring. Where more than one member atom is present in a chain and within a ring, each member atom is covalently bound to an adjacent member atom in the chain or ring. Atoms that make up a substituent group on a chain or ring are not member atoms in the chain or ring. "Optionally substituted” indicates that a group, such as alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heteroaryl, may be unsubstituted, or the group may be substituted with one or more substituent as defined.
- RORy refers to all isoforms encoded by the RORC gene which include RORyl and RORyt.
- RORy modulator refers to a chemical compound that inhibits, either directly or indirectly, the activity of RORy.
- RORy modulators include antagonists and inverse agonists of RORy.
- “Pharmaceutically acceptable” refers to those compounds, materials, compositions, and dosage fonns which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- Substituted in reference to a group indicates that one or more hydrogen atom attached to a member atom within the group is replaced with a substituent selected from the group of defined substituents. It should be understood that die term “substituted” includes the implicit provision that such substitution be in accordance widi the permitted valence of the substituted atom and the substituent and that the substitution results in a stable compound (i.e. one that does not spontaneously undergo transformation such as by rearrangement, cyclization, or elimination and that is sufficiently robust to survive isolation from a reaction mixture). When it is stated that a group may contain one or more substituent, one or more (as appropriate) member atom within the group may be substituted. In addition, a single member atom within the group may be substituted with more than one substituent as long as such substitution is in accordance with die permitted valence of the atom. Suitable substituents are defined herein for each substituted or optionally substituted group.
- the present invention provides, in a first aspect, a compound of Formula I or a
- A is phenyl or lieteroaryl
- B is phenyl or lieteroaryl
- C is phenyl or monocyclic lieteroaryl
- each Rl is independently selected from the group consisting of:
- Rb is selected from the group consisting of:
- C1-C4 alkyl optionally substituted with CF 3 , CHF 2 , NRaRa, phenyl or cycloalkyl, 2) cycloalkyl,
- each R2 is independently selected from the group consisting of:
- each R3 is selected from the group consisting of H, C1-C3 alkyl and OH;
- each R4 is selected from the group consisting of halo, CN and C(0)NH 2 ;
- R5 is selected from the group consisting of C1-C3 alkyl, (CH 2 ) n NRaRa, (CH2) n OH, and NHC(0)CH 3 ;
- n 0, 1 or 2
- r 0, 1 or 2
- s is 1 or 2
- t is 0, 1 or 2
- each Ra is H or C1 -C3 alkyl.
- the present invention relates to compounds of Formula I wherein A is phenyl. In another embodiment, the present invention relates to compounds of Formula I wherein A is selected from the group consisting of pyridinyl, pyrazol, indolyl, indazolyl and imidazol pyridinyl.
- the present invention relates to compounds according to Formula I or any of the above embodiments wherein m is 1 or 2.
- the present invention relates to compounds according to Formula I or any of the above embodiments wherein Rl is selected from the group consisting of: OCF 3 , CFj, F, CI, CN, C3 ⁇ 4, OCH3 and OCHF 2 .
- the present invention relates to compounds according to Formula I or any of the above embodiments wherein m is 0.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein B is phenyl.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein B is selected from the group consisting of pyridinyl, pyrazinyl, pyrimidinyl and indolyl.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein r is 1. In another embodiment, the present invention also relates to compounds according to Formula I or any of the above embodiments wherein R2 is CI or methyl. The present invention also relates to compounds according to Formula I or any of the above embodiments wherein r is 2 and R2 is CI or F. The present invention also relates to compounds according to Formula I or any of the above embodiments wherein r is 0.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein C is phenyl.
- the present invention also relates to compounds according to Formula II or any of the above embodiments wherein C is pyridinyl or pyrazol.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein R3 is H.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein R3 is OH.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein s is 1.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein t is 0.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein t is 1 and R4 is halo, CN or C(0)NH 3 .
- the present invention relates to compounds according to Formula I or any of the above embodiments wherein R5 is methyl, ethyl, (CH ⁇ n NRaRa or NHC(0)CH 3 wherein n is 0, 1 or 2.
- the present invention provides, in another aspect, a compound of Formula 1(A) or a pharmaceutically acceptable salt thereof.
- A is phenyl or heteroaryl
- B is phenyl or heteroaryl
- C is phenyl or monocyclic heteroaryl
- each Rl is independently selected from the group consisting of:
- Rb is selected from the group consisting of:
- each R2 is independently selected from the group consisting of:
- R3 is H or Cl-C3 alkyl
- R4 is halo
- R5 is C1-C3 alkyl, NRaRa or (CH2) confrontOH;
- n 0, 1 or 2
- r 0, 1 or 2
- s is 1 or 2
- t 0, 1 or 2
- each Ra is H or C1-C3 alkyl.
- the present invention relates to compounds of Formula 1(A) wherein A is phenyl. In another embodiment, the present invention relates to compounds of Formula 1(A) wherein A is selected from the group consisting of pyridinyl, pyrazol, indolyl, indazolyl and imidazol pyridinyl.
- the present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein m is 1 or 2. In another embodiment, the present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein m is 0.
- the present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein Rl is selected from the group consisting of: OCF , CF 3 , F, CI, CN, CH 3j OCH 3 and OCHF 2 .
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein B is phenyl.
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein B is selected from the group consisting of pyridinyl, pyrazinyl, pyrimidinyl and indolyl.
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein r is 1 or 2.
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein r is 0.
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein R2 is selected from the group consisting of CI, F, CN, methyl and CF 3 .
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein C is phenyl.
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein C is pyridinyl or pyrazol.
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein R3 is H,
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein s is 1.
- the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein t is 0.
- the present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein R5 is methyl, ethyl or NH 2 .
- the present invention provides, in another aspect, a compound of Formula II or a pharmaceutically acceptable salt thereof.
- each Rl is independently selected from the group consisting of
- - C1-C3 alkyl optionally substituted with C1-C3 alkoxy, - piperidinylmethyl or piperidinylidenemethyl or piperazinylmethyl, wherein said piperidinyl or piperazinyl is substituted with C(0)Ra or C1-C3 alkyl,
- Rb is selected from the group consisting of:
- each R2 is independently selected from the group consisting of:
- R3 is selected from the group consisting of H, C1-C3 alkyl and OH;
- each R4 is selected from the group consisting of halo, CN and C(0)NH 2 ;
- R5 is selected from the group consisting of C1-C3 alkyl, (CH 2 ) n NRaRa, (CH2) n OH, and NHC(0)C3 ⁇ 4;
- n 0, 1 or 2
- r 0, 1 or 2
- t 0, 1 or 2and each Ra is H or C1-C3 alkyl.
- the present invention relates to compounds of Formula II wherein m is 1, and Rl is OCF 3 ,OCHF 2 , or CF 3 .
- the present invention relates to compounds of Formula II wherein m is 2, one Rl is OCF 3 or OCHF 2 , and the other Rl is CI or F.
- the present invention also relates to compounds according to Formula II or any of the above embodiments wherein r is 1, and R2 is CI or methyl at position 2 of die phenyl ring.
- the present invention also relates to compounds according to Formula II or any of the above embodiments wherein r is 2, and both R2 are CI or both R2 are F.
- the present invention also relates to compounds according to Formula II or any of the above embodiments wherein R3 is H.
- the present invention also relates to compounds according to Formula II or any of the above embodiments wherein R3 is OH.
- the present invention also relates to compounds according to Formula I or any of the above embodiments wherein t is 0.
- the present invention also relates to compounds according to Formula II or any of the above embodiments wherein t is 1 and R4 is halo, CN or C(0)NH 2 .
- the present invention relates to compounds according to Formula II or any of the above embodiments wherein R5 is methyl or ethyl.
- the present invention provides, in another aspect, a compound of Formula 11(A) or a pharmaceutically acceptable salt thereof.
- each Rl is independently selected from the group consisting of:
- Rb is selected from the group consisting of:
- each R2 is independently selected from the group consisting of:
- R5 is C1-C3 alkyl, NRaRa or (CH2) n OH;
- n 0, 1 or 2
- r 0, 1 or 2
- each Ra is H or C1-C3 alkyl
- the present invention relates to compounds of Formula 11(A) wherein m is 1, and Rl is OCF 3 or CF 3 at position 2 of the phenyl ring.
- the present invention relates to compounds of Fonnula 11(A) wherein m is 2, one Rl is OCF 3 or OCHF 2 at position 2 of the phenyl ring, and the other Rl is CI or F at position 4 of the phenyl ring.
- the present invention also relates to compounds according to Formula 11(A) or any of the above embodiments wherein r is 1, and R2 is CI or methyl at position 2 of the phenyl ring.
- the present invention also relates to compounds according to Formula 11(A) or any of the above embodiments wherein r is 2, one R2 is CI or F at position 2 of the phenyl ring, and the other R2 is CI or F at position 6 of the phenyl ring.
- the present invention relates to compounds according to Fonnula 11(A) or any of the above embodiments wherein R5 is methyl or ethyl.
- the compounds according to Formula I may contain one or more asymmetric center (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereomers, or other stereoisomeric forms, or as mixtures thereof.
- Chiral centers such as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the stereochemistry of a chiral center present in Fonnula I, or in any chemical structure illustrated herein, is not specified the structure is intended to encompass all individual stereoisomers and all mixtures thereof.
- compounds according to Fonnula I containing one or more chiral center may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers.
- Individual stereoisomers of a compound according to Fonnula I which contain one or more asymmetric center may be resolved by methods known to those skilled in the art.
- such resolution may be carried out (1) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer-specific reagent, for example by enzamatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral envionce, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- a further step is required to liberate the desired form.
- specific stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
- the compounds according to Formula I may also contain double bonds or other centers of geometric asymmetry. Where the stereochemistry of a center of geometric asymmetry present in Fonnula I, or in any chemical structure illustrated herein, is not specified, the structure is intended to encompass the trans (E) geometric isomer, the cis (Z) geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included in Formula I whether such tautomers exist in equilibrium or predominately in one form.
- compounds according to Formula I may contain an acidic functional group. In certain other embodiments, compounds according to Formula I may contain a basic functional group.
- phannaceutically-acceptable salts of the compounds according to Formula I may be prepared. Indeed, in certain embodiments of the invention, phannaceutically-acceptable salts of the compounds according to Fonnula I may be preferred over the respective free base or free acid because such salts may impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage fonn. Accordingly, the invention is further directed to the use of phannaceutically-acceptable salts of the compounds according to Formula I.
- pharmaceutically-acceptable salts refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically-acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
- the term “compounds of the invention” means both the compounds according to Formula I and the pharmaceutically-acceptable salts thereof.
- a compound of the invention also appears herein and refers to both a compound according to Formula I and its pharmaceutically-acceptable salts .
- the invention also includes various deuterated forms of the compounds of Fonnula (I). Each available hydrogen atom attached to a carbon atom may be independently replaced with a deuterium atom. A person of ordinary skill in the art will know how to syndiesize deuterated forms of the compounds of Formula (I). Commercially available deuterated starting materials may be employed in the preparation of deuterated forms of the compounds of Formula (I), or they may be synthesized using conventional techniques employing deuterated reagents (e.g. lithium aluminum deuteride).
- deuterated reagents e.g. lithium aluminum deuteride
- the compounds of the invention may exist in solid or liquid form. In the solid state, the compounds of the invention may exist in crystalline or noncrystalline form, or as a mixture thereof.
- pharmaceutically-acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization. Solvates may involve nonaqueous solvents such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice.
- Hydrates wherein water is the solvent that is incorporated into the crystalline lattice are typically referred to as "hydrates.” Hydrates include stoichiometric hydrates as well as compositions containing vaiable amounts of water. The invention includes all such solvates.
- polymorphs may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs.”
- the invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification.
- polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymorphs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
- a substituent described herein is not compatible with the synthetic methods described herein, the substituent may be protected with a suitable protecting group that is stable to the reaction conditions.
- the protecting group may be removed at a suitable point in the reaction sequence to provide a desired intermediate or target compound.
- Suitable protecting groups and tlie methods for protecting and de-protecting different substituents using such suitable protecting groups are well known to those skilled in tlie art; examples of which may be found in T. Greene and P. Wuts, Protecting Groups in Chemical Synthesis (3rd ed.), John Wiley & Sons, NY (1999).
- a substituent may be specifically selected to be reactive under the reaction conditions used. Under these circumstances, the reaction conditions convert the selected substituent into another substituent that is either useful as an intermediate compound or is a desired substituent in a target compound.
- Scheme 1 represents a general reaction scheme for preparing certain compounds of Formula I.
- Starting material substituted bromophenyl anilines 1.1 can be coupled with an appropriate reagent 1 to form amides 1.2.
- Amides 1.2 are transferred to die desired compounds of Formular I either by direct Suzuki coupling with boronic acids 1.3, or by converting to boronic esters 1.4 under catalysis of PdCl 2 (dppf) or Pd(dba) 3 first, then Suzuki coupling with bromides 1.5.
- Scheme 2 represents a general reaction scheme for preparing certain compounds of Formula I.
- Starting material bromides 2.1 can be coupled with an appropriate substituted boronic acids 2.2 to form anilines 2.3.
- Anilines 2.3 are transferred to the desired compounds of Formular I by direct coupling with reagent 1.
- Scheme 3 represents a general reaction scheme for preparing certain compounds of Formula I.
- Starting material substituted bromonitrobenzene 3.1 can be coupled with substituted boronic acids 3.2 to form phenols 3.3 under Suzuki coupling condition.
- Phenols 3.3 are transferred to difluoromethyl ethers 3.4 and then reduced to form substituted phenyl anilines 3.5.
- Phenyl anilines 3.5 can be converted to the desired compounds of Formular I by coupling with an appropriate reagent 1.
- Scheme 4 represents a general reaction scheme for preparing certain compounds of Formula I.
- Starting material substituted bromocholoronitrobenzene 4.1 can be coupled with boronic acids 4.2 to form biphenyl chlorides 4.3.
- Biphenyl chlorides 4.3 are transferred to intermediates 4.5 either by direct Suzuki coupling with boronic acids, or by converting to boronic esters 4.4 under catalysis of Pd(3 ⁇ 4(dppf) or Pd 2 (dba) 3 first, then Suzuki coupling with bromides.
- Intermediates 4,5 can be reduced to form substituted anilines 4.6.
- Anilines 4.6 can be converted to the desired compounds of Formula I by coupling with an appropriate reagent 1.
- Mobile phase water containing 0.05% TFA / acetonitrile.
- Mobile phase water containing 0.04% ammonia/ acetonitrile.
- Step 1 A solution of sodium nitrite (] 8.4 g) in 133 mL of water was added dropwise at 0 °C, while stirring, to a suspension of (4-aminophenyl)acetic acid (40.2 g) in 133 mL of water and 54 mL of concentrated hydrochloric acid. After the addition was complete, the reaction mixture was stirred at the same temperature for 45 minutes. This solution of cold diazonium salt was then added dropwise at room temperature to a mixture of potassium ethylxanthate (49.4 g), 80 mL of water and 200 mL of 2 M sodium carbonate solution. The mixture was heated to 45 °C and stirred at this temperature until gas evolution stops.
- Step 2 (4- ⁇ [(Ethyloxy)carbonothioyl]thio ⁇ phenyl)acetic acid (90 g) was taken up in 340 mL of ethanol, and a solution of 70 g of potassium hydroxide in 340 mL of water was added. Boiling at reflux was effected for 20 hours. The major portion of ethanol was subsquently removed by the distillation under reduced pressure. The aqueous phase was cooled with ice, and acidified with concentrated hydrochloric acid while stirring. The obtained solution was extracted with diethyl ether (500 mL).
- Step 3 To a solution of (4-mercaptophenyl)acetic acid (33 g) in DMF (240 mL) was added 2CO3 (108 g) and bromoethane (64.1 g). The reaction mixture was stirred at room temperature. After 2.5 hours, the starting material was totally consumed. The reaction mixture was partitioned between ethyl acetate (300 mL) and water (300 mL). The organic phase was washed with water (300 mL x 4) and brine (200 mL), dried over sodium sulphate, filtered, and concentrated to give the desired product ethyl [4-(ethylthio)phenyl] acetate (34 g) as a pale yellow solid.
- Step 4 A solution of ethyl [4-(ethylthio)phenyl] acetate (34 g) in DCM (500 mL) was cooled to 0°C with an ice bath. mCPBA (78 g) was added in portions, and the reaction mixture was stirred at RT overnight. The obtained suspension was filtered. The filtrate was washed with sat. sodium carbonate solution (400 mL x 2), water (500 mL), then brine (250 mL). The obtained solution was dried over sodium sulphate, filtered, and concentrated.
- Intennediate 6a ⁇ (4- ⁇ ⁇ - - ⁇ 1 ⁇ 1 ⁇ 1 ⁇ 6 ⁇ -2- ⁇ 4- 6& ⁇ 5 ⁇ 1 ⁇ 1 ⁇ € ⁇ 13 ⁇ 4 ⁇ 6 ⁇ 6
- Intennediate 6b N-r3-chloro-4-( , 4,4.5.5-tetrainethyl-l 1 3,2-dioxaborolan-2-yl ' )phenyll-2-
- Step 1 A mixture of [2-(trifluoromethyl)phenyl]boronic acid (0.767 g), methyl 2-bromo-5- nitrobenzoate (1 g), PdCI 2 (dppf)-CH 2 Cl 2 adduct (0.157 g) and Cs 2 C0 3 (1.504 g) in acetonitrile (12 mL) and water (4.00 mL) was bubbled with N 2 .
- the reaction vessel was sealed and heated in the microwave at 100°C for 1 hour.
- the mixture was filtered through celite and silica gel, and then washed with acetonitrile, DCM and EtOAc. The filtrate was concentrated under reduced pressure and the residue was partitoned between EtOAc and water.
- Step 2 To a solution of methyl 4-nitro-2'-(trifluoromethyl)-2-biphenylcarboxylate (300 mg) in tetrahydrofuran (THF) (8 mL) cooled at 0°C was added LAH (140 mg) slowly. The resulting mixture was stirred at room temperature overnight. The reaction was quenched by the sequential treatment with water (0.15 mL), 15% NaOH (0.15 mL) and water (0.3 mL). The mixture was then filtered and the filtrate was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na 2 S0 4 .
- THF tetrahydrofuran
- Step 3 A mixture of [4-amino-2'-(trifluoromethyl)-2-biphenylyl]metlianol (60 mg), [4- (ethyIsulfonyl)phenyl]acetic acid (intermediate la, 53.8 mg), EDC (56.0 mg) and HOBt (39.4 mg) in DCM (3 mL) was stirred at room temperature over 3 days.
- Step 1 To a solution of l-bromo-2-chloro-4-nitro-benzene (2.0 g) and 2-acetylphenyl boronic acid (1.53 g) in DMF (20 mL) was added K 2 C0 3 (3. 1 g) and PdCl 2 (dppf) (622 mg) under N 2 . The mixture was stirred for 4 hours at 100°C. The mixture was filtered and the filtrate was diluted with water (100 mL). The solution was extracted with EtOAc (100 mL x 3). The organic layers were dried over anhydrous Na 2 S0 4 and concentrated.
- Step 2 A solution of l-(2'-chloro-4'-nitro-biphenyl-2-yl)-ethanone (920 mg) and SnCl 2 '2H 2 0 in cone. HCl (20 mL) was stirred for 2 hours at 70°C. The solution was diluted with EtOAc (30 mL), The mixture was adjusted to pH ⁇ 9 with NaOH solution and filtered. The filtrate was extracted with EtOAc (50 mL x 3). The organic layers were dried over anhydrous Na 2 S0 4 and concentrated.
- Step 3 A mixture of l-(4'-amino-2'-chloro-biphenyl-2-yl)-ethanone (60 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 61.3 mg), EDC (60.9 mg) and HOBt (42.9 mg) in DCM (4 mL) was stirred at room temperature overnight. The mixture was concentrated under reduced pressure and the residue was purified by MDAP to afford N-(2'-acetyl-2-chloro-4-biphenylyl)-2-[4- (ethylsulfonyl)phenyI]acetamide (85 mg) as a light pink solid.
- Step 1 A mixture of 4-bromo-3-fiuoroaniline (400 mg), [4-(ethylsulfonyl)phenyl]acetic acid
- Step 2 A mixture of ⁇ 2-[(trifluoromethyl)oxy]phenyl ⁇ boronic acid (81 mg), N-(4-bromo-3- fluorophenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide (150 mg), PdCl 2 (dppf)-C3 ⁇ 4Cl 2 adduct (20 mg) and Cs 2 C0 3 (147 mg) in acetonitrile (2.1 mL) and water (0.700 mL) was bubbled with N 2 . The vessel was then sealed and heated in the microwave at 100°C for 30 minutes. The mixture was filtered through silica gel and celite, washed with acetonitrile, DCM and EtOAc.
- Step 1 BF 3 -Et 2 0 (6.17 g) was placed in a three-necked flask. 2-Chloro-5-nitro-phenylamine (5.0 g) in THF (40 mL) and DCM (80 lnL) was added. /-BuONO (3.6 g) was added dropwise at - 10°C, The mixture was stirred for 1 hour at 0°C. n-Hexane (200 mL) was added. The solution was filtered to give 2-chloro-5-nitro-benzenediazonium tetrafluoroborate (7.6 g) as a yellow solid.
- Step 3 A solution of 2-bromo-l -chloro-4-nitro-benzene (2 g) and Pd(PPh 3 ) 4 (980 mg) in toluene (50 mL) and EtOH (8 mL) was stirred for 20 mins at 0 °C. Na 2 C0 3 (2.96 g) in 10 mL water and 2-(trifluoromethoxy)phenylboronic acid (1.92 g) were added. The mixture was stirred for 16 hours at 100°C under N 2 . TLC indicated the reaction completed.
- Step 5 A mixture of 6-chloro-2'-trifluoromethoxy-biphenyl-3-ylamine (87 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intennediate la, 76 mg), EDC (75 mg) and HOBt (53.1 mg) in DCM (2 mL) was stirred at room temperature overnight. Solvent was removed under reduced pressure. The residue was purified by MDAP to afford N- ⁇ 6-chloro-2'-[(trifluoromethyl)oxy]-3- biphenylyl ⁇ -2-[4-(ethyIsulfonyl)phenyl]acetamide (54 mg) as a white solid.
- Step 1 A mixture of 3-bromo-4-methylbenzenamine (232 mg), phenylboronic acid (243 mg),
- Step 2 A mixture of [4-(ethylsulfonyl)phenyl] acetic acid (intermediate la, 350 mg), HOBt
- Step 2 A mixture of 2-bromo-l -isopropyl-4-nitrobenzene (1.6 g), phenylboromc acid (1.6 g), Pd(dppf)Cl 2 (0.3 g) and cesium carbonate (4.3 g) in NN-dimeihylformamide (6.0 mL) and water (3 mL) was stirred at room temperature overnight. The mixture was filtered through celite and silica gel. The filtrate was concentrated, and the residue was purified by flash chromatography (PE) to afford 2- isopropyl-5-nitrobiphenyl (1 ,2 g) as a colorless liquid.
- PE flash chromatography
- Step 1 To a solution of l-fluoro-3-(trifluoromethoxy)benzene (2 g) in THF (20 mL) at -78°C was added BuLi (4.89 mL) dropwise. The solution was stirred at -78°C for 30 mins. Iodine (4.23 g) in THF (10 mL) was added slowly. The reaction mixture was warmed to room temperature and then quenched with a solution of Na 2 COj in saturated Na 2 S 2 0 3 solution (50 mL). The mixture was extracted with ethyl acetate (40 mL).
- Step 2 A mixture ofN-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 260 mg), l-fluoro-2-iodo-3- (trifluoromethoxy)benzene (172 mg), Pd(Ph 3 P) 4 (64.8 mg) and potassium carbonate (155 mg) in 1 ,4- dioxane (5 mL) and water (1 mL) was sealed in a vessel and heated in the microwave at 100°C for 1 hour.
- Step 1 To a suspension of indoIin-2-one (5 g) in water (50 mL) was added NaOH (6 g). The mixture was stirred overnight at 100°C. The pH was adjusted to 1 with concentrated hydrochloric acid to give a solution of 2-(2-aminophenyl)acetic acid in water. MS(ES + ) m/z 152 (MH + ).
- Step 2 A solution of sodium nitrite (2.56 g) in 20 mL of water was added dropwise to a suspension of the above solution of 2-(2-aminophenyl)acetic acid in 20 mL of water and 2.7 mL of concentrated hydrochloric acid cooled at 0°C. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise at room temperature to a mixture of potassium O-ethyl carbonodithioate (6.88 g), 20 mL of water and 28 mL of 2 M sodium carbonate solution. Heating was effected to 45 °C until gas evolution stops.
- Step 3 2-(2-(Ethoxycarbonothioylthio)phenyl)acetic acid (9.5 g) was taken up in 40 mL of ethanol. A solution of KOH (6.24 g) in 30 mL of water was added. Boiling at reflux was effected for 20 hours. The major portion of ethanol was subsequently removed by distillation at reduced pressure. The aqueous phase was cooled with ice, and was rendered acid with concentrated hydrochloric acid while stirring. The solution was extracted with diethyl ether (100 mL x 5). The combined organic phases were washed with brine, dried over anhydrous sodium sulphate, filtered, and concentrated to afford 2-(2-mercaptophenyl)acetic acid (6 g). MS(ES + ) m/z 169 (MH + ).
- Step 4 To a solution of 2-(2-mercaptophenyl)acetic acid (6 g) in DMF (80 mL) was added K 2 C0 3 (34.5 g) and bromoethane (19.43 g). The reaction mixture was stirred at 30°C overnight. The reaction mixture was partitioned between ethyl acetate (300 mL) and water (100 mL). The organic phase was washed with water (100 mL x 4), brine (200 mL), dried over sodium sulfate, filtered, and concentrated to give ethyl 2-(2-(etliylthio)phenyl)acetate (6 g) as a brown solid.
- Step 5 Ethyl 2-(2-(efhylthio)phenyl)acetate (6 g) was dissolved in DCM (50 mL), and the solution was cooled to 0°C with an ice bath. mCPBA (13.85 g) was added in portions. After stirring at room temperature overnight, the reaction mixture was filtered to remove the solid. The filtrate was washed with sat. sodium carbonate solution (50 mL x 2), water (50 mL), brine (150 mL), dried over sodium sulfate, filtered, and concentrated.
- Step 1 A solution of sodium nitrite (0.916 g) in 20 mL of water was added dropwise to a suspension of 2-(3-aminophenyl)acetic acid (2 g) in 20 mL of water and 2.7 mL of concentrated hydrochloric acid cooled at 0 °C. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise to a mixture of potassium 0-ethyl carbonoditliioate (2.456 g), 20 mL of water and 10 mL of a 2 M sodium carbonate solution at room temperature. The reaction mixture was heated at 45°C until gas evolution stops.
- Step 3 To a solution of 2-(3-mercaptophenyl)acetic acid (3.3 g) in DMF (50 mL) was added 2C0 3 (10.85 g) and bromoethane (4.39 mL). The reaction mixture was stirred at room temperature for 2.5 hours. The reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL). The organic was washed with water (100 mL x 4) and brine (200 mL), dried with sodium sulfate, filtered, and concentrated to give ethyl 2-(3-(ethylthio)phenyl)acetate (2.76 g) as a pale yellow solid.
- N-f2-chlQro-6'-( , trifluoromethoxy ⁇ biphenyl-4-ylV2-f3-( ' ethylsulfonyl)phenyl)acetarnide A mixture of 2-chloro-6'-(trifluoromethoxy)biphenyI-4 -amine (intermediate 15b, 300 mg), 2-(3- (ethyIsulfonyl)phenyI)acetic acid (intermediate 16a, 309 mg), HOBt (192 mg), EDC (600 mg) and DIPEA (1.10 mL) in DCM (20 mL) was refluxed overnight.
- Intennediate 17a 3-(4-(ethylsulfonyl ' )phenyl ' )propanoic acid
- Step 1 A solution of NaN0 2 (0.42 g) in 6 mL of water was added dropwise at 0°C, while stirring, to a suspension of 3-(4-aminophenyl)propanoic acid (1.0 g) in 3 mL of water and 3 mL of concentrated hydrochloric acid. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise at room temperature to a mixture of potassium Oethyl carbonodithioate (1.16 g), 2 mL of water and 5 mL of 2 M sodium carbonate solution. The mixture was stirred at 45 °C until gas evolution stopped.
- Step 2 A mixture of 3-(4-(ethoxycarbonothioylthio)phenyl)propanoic acid (1.2 g) and potassium hydroxide (1.85 g) in ethanol (20 mL) and water (20 mL) was refluxed overnight. Ethanol was removed. The aqueous phase was cooled with ice, and acidified with concentrated hydrochloric acid while well stirring. The mixture was extracted with DCM (40 mL x 2). The combined organic phases were washed with brine, dried over anhydrous sodium sulphate, filtered, and concentrated to afford 3-(4-mercaptophenyl)propanoic acid (1.0 g) as a yellow solid. MS(ES + ) m/z 183 (MH ⁇ ).
- Step 3 A mixture of 3-(4-mercaptophenyl)propanoic acid (1.0 g), K 2 C0 3 (3.03 g) and bromoethane (1.23 mL) in DMF (10 mL) was stirred at RT for 2.5 hours. The reaction mixture was partitioned between ethyl acetate (50 mL) and water (100 mL). The organic phase was washed with water (50 mL x 3) and brine (50 mL), dried with sodium sulphate, filtered, and concentrated to give ethyl 3-(4-(ethyltmo)phenyl)propanoate (1.2 g) as a yellow oil. MS(ES + ) m/z 239 (MH + ).
- Step 4 To a solution of ethyl 3-(4-(ethylthio)phenyl)propanoate (1.2 g) in DCM (30 mL) was added mCPBA (2.61 g). The reaction mixture was stirred at RT overnight. Solid was removed by filtration. The filtrate was washed with sat. sodium carbonate solution (40 mL x 2), water (40 mL), brine (50 mL), dried over sodium sulphate, filtered, and concentrated.
- Step 1 Formic acid (1 mL) was added dropwise to acetic anhydride (2 mL) in an ice bath. After the addition, the ice bath was removed and the solution was stirred at 50°C for 30 minutes. The mixture was then cooled in an ice bath, to which was added dropwise a cold solution of 2-chloro-6'- (trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 400 mg) in formic acid (1 mL). The reaction mixture was stirred for 40 minutes at this temperature. The mixture was then warmed to room temperature and stirred for additional 80 minutes. Solvent was removed, and DCM (10 mL) was added. Solid was removed by filtration.
- Step 2 Borane-tetrahydrofuran complex (20 mL) was added dropwise to a solution of N-(2- chloro-6 , -(trifluoromethoxy)biphenyI-4-yl)formamide (480 mg) i THF (20 mL) at 0°C in 30 mins. The reaction mixture was warmed to 80°C and stirred overnight. The mixture was cooled to 0°C and quenched with brine. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate (100 mL x 2).
- Step 3 A mixture of 2-chloro-N-methyl-6 , -(trifluoromethoxy)biphenyI-4-amine (500 mg), [4- (ethylsulfonyl)phenyl] acetic acid (intermediate la, 454 mg), EDC (953 mg), HOBt (279 mg) and DIPEA (2 mL) in DCM (40 mL) was stirred at room temperature overnight. The organic solution was washed with water (50 mL) and brine (80 mL), dried and concentrated.
- Step 2 A mixture of 2-chloro-2'-phenoxybiphenyl-4-amine (200 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 232 mg), HOBt (104 mg), DIPEA (0.354 mL) and EDC (389 mg) in tetrahydrofuran (THF) (10 mL) was stirred at 60°C overnight. The mixture was concentrated under reduced pressure, and the residue was purified by prep-HPLC to afford N-(2- chloro-2'-phenoxybiphenyI-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (67 mg) as a white solid.
- Step 1 To a solution of 2-bromophenol (0.2 g) and l,l,l-trifluoro-2-iodoethane (0,267 g) in DMF (6 mL) was added K 2 C0 3 (0.320 g). The reaction mixture was heated to 110°C overnight. The reaction mixture was partitioned between ethyl acetate (30 mL) and water (20 mL). The organic phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and concentrated to give l-bromo-2-(2,2,2-trifluoroethoxy)benzene (0.25 g) as a yellow oil.
- Step 2 A mixture ofN-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsuIfonyl)phenyl]acetamide (intermediate 6b, 218 mg), l-bromo-2-(2,2,2- trifluoroethoxy)benzene (120 mg), K 2 C0 3 (163 mg) and Pd(Ph 3 P) 4 (54.4 mg) in 1 ,4-dioxane (5 mL) and water (1 mL) was heated at reflux overnight. The reaction mixture was filtered through celite and silica gel.
- Step 1 A mixture of 2-bromophenol (100 mg), 1-bromobutane (87 mg) and K 2 C0 3 (240 mg) in DMF (30 mL) was stirred at 80°C overnight. EtOAc (50 mL) was added, and the mixture was washed with brine (50 mL ⁇ 3). The combined organic phases were dried and concentrated to afford l-bromo-2-butoxybenzene (45 mg) as a colourless oil.
- Step 2 A mixture of l-bromo-2-butoxybenzene (45 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethyIsulfonyl)phenyl]acetamide (intermediate 6b, 91 mg), K 2 C0 3 (27.1 mg) and Pd(Ph 3 P) (227 mg) in 1,4-dioxane (10 mL) and water (2 mL) was stirred at 100°C overnight. The mixture was filtered tlirougli celite and silica gel.
- Step 1 To a solution of 2-bromophenol (1.0 g) in toluene (30 mL) cooled at 0°C was added 2- (dimethylamino)ethanol (0.515 g), triphenylp osphine (1.819 g) and D1AD (1.349 mL). The reaction mixture was stirred at room temperature overnight. The solid was removed by filtration, and the filtrate was concentrated to give 2-(2-bromophenoxy)-N,N-dimethylethanamine (1.5 g) as a colorless oil. SiES*) m/z 245 (Mtf).
- Step 2 A mixture of 2-(2-bromophenoxy)-N s N-dimethylethanamine (200 mg), N-[3-chloro-4- (4,4,5, -tetramethyl-l,3,2-dioxaborolan-2-yl)phenyI]-2-[4-(ethylsulfonyl)phenyl]acetamide
- Step 1 To a solution of 2-morpholinobenzenainine (500 mg) in acetonitrile (30 mL) was added /er/-butyl nitrite (579 mg) and copper(II) bromide (752 mg). The mixture was stirred at room temperature for 2 hours. The mixture was partitioned between water (60 mL) and EtOAc (60 mL). The aqueous phase was extracted with EtOAc (60 mL x 2). The combined organic phases were dried and concentrated. The residue was purified by flash chromatography (EtOAc : PE - 1 : 10) to give 4- (2-bromophenyl)morpholine (300 mg) as a brown oil. MSfES m/z 242 (MH + ).
- Step 2 A mixture of 4-(2-bromophenyl)morpholine (200 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 383 mg), Pd(Ph 3 P) 4 (95 mg) and 2 C0 3 (285 mg) in 1 ,4-dioxane (20 mL) and water (3 mL) was stirred at 90°C under N 2 for 1 hour. After removal of solvent, the residue was partitioned between EtOAc (60 mL) and water (60 mL).
- Step 1 To a solution of 2-bromophenol (1 g) and (bromomethyl)cyclopropane (0.78 g) in acetone (6 mL) was added 2 C0 3 (1.60 g). The reaction mixture was heated to reflux overnight. The reaction mixture was partitioned between ethyl acetate (40 mL) and water (20 mL). The organic phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and concentrated to give l-bromo-2-(cyclopropylmethoxy)benzene (0.95 g) as a yellow oil.
- Step 2 A mixture of N-[3-chloro-4-(4,4,5,5-tetramethyI-l,3,2-dioxaboroIan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 204 mg), l-bromo-2- (cyclopropylmethoxy)benzene (100 mg), K 2 C0 3 (152 mg), and Pd(Ph 3 P) 4 (51 mg) in 1,4-dioxane (5 mL) and water (1 mL) was heated at reflux overnight. The mixture was filtered through celite and silica gel.
- Step 1 To a solution of 2-(piperidin-l -yl)phenol (0.5 g) and trifiuoro-N-phenyl-N- (trifluoromethylsulfonyl)methanesulfonamide (1.1 g) in dichloromethane (20 mL) was added NEt 3 (0.40 mL) dropwise at 0 °C. The reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was partitioned between dichloromethane (40 mL) and water (20 mL). The organic phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and concentrated.
- Step 2 A mixture of N-[3-chloro-4-(4 ⁇ 4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 194 mg), 2-(piperidin-l -yl)phenyl
- Step 1 C lorosulfonic acid (70 mL) was added into phenylacetic acid (10 g) dropwise at 0 °C. The reaction mixture was stirred at 0°C for 1 hour, and then warmed to room temperature and stirred overnight. The reaction mixture was poured onto ice and left until all ice melted. The suspension was filtered. The obtained solid was washed with cold water and dried in vacuo to afford a white solid (10 g), which contains isomers. The mixture was recrystallized from chloroform twice to give 2-(4-
- Step 2 A mixture of 2-(4-(chlorosulfonyl)phenyl)acetic acid (200 mg) and ammonia solution
- Step 1 To a solution of ⁇ 2-bromophenyI)methanol (2 g) in DCM (30 mL) was added dropwise PBr 3 (1.01 mL) at 0 °C. The reaction mixture was stirred at room temperature overnight. Ice water (20 mL) was added, and the solution was extracted with DCM (40 mL). The organic phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and
- Step 2 To a solution of isopropanol (6 mL) was added sodium (27.6 mg). The mixture was heated to reflux until Na was completely dissolved. The mixture was cooled to RT and solvent was removed under reduced pressure. To the residue was added DMF (10 mL), followed by a solution of l-bromo-2-(bromomethyl)benzene (300 mg) in DMF (2 mL) dropwise. After stirring at RT overnight, the reaction mixture was added to ice water (10 mL), and then extrated with DCM (30 mL).
- Step 3 To a solution of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2- [4-(ethyIsulfonyl)phenyl]acetamide (intermediate 6a, 202 mg), l-bromo-2-
- Step 2 To a solution of methyl [4-(ethyIsulfonyl)phenyl] acetate (303 mg) in tetrahydrofuran
- Step 3 The intermediate methyl 2-[4-(ethylsulfonyl)phenyl]propanoate was dissolved in EtOH (2 mL). 2 M NaOH (2 mL) was added. The mixture was stirred at RT for 2 hours. 1 M HCl was used to adjust pH to about 5. The aqueous phase was extracted with EtOAc for 3 times. The combined organics were dried over Na 2 S0 4 . After removal of solvent, 2-(4-(ethylsulfonyl)phenyl)propanoic acid (202.7 mg) was obtained. MS(ES + ) m/z 243 (MH + ).
- Step 1 To a solution of 2-bromophenol (2.0 g) in toluene (30 mL) cooled at 0°C was added tetrahydro-2H-pyran-4-ol (1.81 g), triphenylphosphine (3.64 g) and DIAD (2.70 mL). The reaction mixture was stirred at RT overnight. Solid was removed by filtration, and the filtrate was
- Step 2 A mixture of 4-(2-bromophenoxy)-tetrahydro-2H-pyran (50 mg), N-[3-chloro-4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsuIfonyl)phenyl]acetamide
- Step 1 A mixture of 2-bromophenol (3.4 g), (bromomethyl)benzene (4.0 g) and K 2 C0 3 (5.4 g) in acetone (10 mL) was stirred at 50°C overnight. Solvent was removed. The residue was partitioned between water and ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by flash chromatography (PE) to give 1 - (benzyloxy)-2-bromobenzene (3.6 g) as a colorless oil.
- PE flash chromatography
- Step 2 A mixture of l-(benzyloxy)-2-bromobenzene (99 mg), N-[3-chIoro-4-(4,4,5,5- tetramethyl-1 ⁇ -dioxaborolan ⁇ -yliphenyll ⁇ - ⁇ -tethylsulfony ⁇ phenyljacetamide (intermediate 6b, 192 mg), Pd(Ph 3 P) 4 (20 mg) and K 2 C0 3 (104 mg) in 1 ,4-dioxane (20 mL) and water (3 mL) was stirred at 90°C for 3 hours. The mixture was extracted with ethyl acetate (100 mL). The organic phase was washed with brine, dried and concentrated.
- Step 2 A mixture of 3-fluoro-6'-(trifluoromethoxy)biphenyl-4-amine (200 mg), [4- (ethylsulfonyl)phenyljacetic acid (intermediate la, 202 mg), HOBt (113 mg), DIPEA (0.386 mL) and EDC (424 mg) in tetrahydrofuran (THF) (20 mL) was stirred at 60°C overnight.
- THF tetrahydrofuran
- Step 1 A mixture of 3-bromoaniline (3.0 g), (2,5-dichlorophenyl)boronic acid (3.9 g), 2 C0 3
- Step 2 To a solution of (2' ! 5'-dichloro-3-biphenylyl)amine (238 mg), EDC (0.230 g) and HOBt
- Step 1 A mixture of (2,5-dichlorophenyl)boronic acid (572 mg), 6-bromo-2-pyridinamine (519 mg), Cs 2 C0 3 (1172 mg) and PdCl 2 (dppf)-CH 2 Cl 2 adduct (196 mg) in acetonitrile (12 niL) and water (4 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times.
- Step 2 A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 100 mg), EDC (104 mg) and HOBt (73.5 mg) in DCM (2 mL) was stirred at room temperature under nitrogen for 10 mins. A solution of 6-(2,5-dichlorophenyl)-2-pyridinamine (100 mg) in DCM (1 mL) was then added. After stirring at RT overnight, the reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were dried over anliydrous Na 2 S0 4 . After filtration, solvent was removed under reduced pressure and the residue was purified by MDAP to afford N-[6-(2,5-dichlorophenyl)-2-pyridinyl]-2-[4-
- Step 1 A mixture of (2,5-diclriorophenyl)boronic acid (190.82 mg), 3-bromo-4-methylaniIine ( 86 mg), Cs 2 C0 3 (391 mg) and PdCl2(dppf)-CH 2 Cl 2 adduct (65.3 mg) in acetonitrile (3 mL) and water (1 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was partition between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anydrous Na 2 SO. t .
- Step 2 A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 95 mg), EDC (99 mg) and HOBt (69.7 mg) in DCM (3 mL) was stirred at room temperature for 15 mins. Then 2',5'- dichIoro-6-metliyl-3-biphenylamine (100 mg) was added. After stirring at room temperature overnight, the reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na 2 S0 4 . After filtration, solvent was removed under reduced pressure. The residue was purified by MDAP to afford N-(2',5 , -dichloro-6-methyl-3-biphenylyl)-2-[4-[4-
- Step 1 A mixture of (2,4-dichlorophenyl)boronic acid (572 mg), 3-bromo-4-methylaniline (558 mg), Cs 2 C0 3 (1172 mg) and PdCl 2 (dppf)-CH 2 Cl 2 adduct (196 mg) in acetonitrile (12 mL) and water (4 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anydrous Na 2 S0 4 .
- Step 2 A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate l a, 95 mg), EDC (99 mg) and HOBt (69.7 mg) in DCM (3 mL) was stirred at room temperature for 15 mins. Then 2',4'- dicliloro-6-methyl-3-biphenyIamine (100 mg) was added. After stirring at room temperature overnight, the reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were dried over anhydrous Na 2 S0 4 .
- Step 1 A mixture of (2,5-dichlorophenyl)boronic acid (333 mg), 4-bromoaniline (300 mg), Cs 2 C0 3 (682 mg) and PdCl 2 (dppf)-CH 2 Cl 2 adduct (114 mg) in acetonitrile (9 mL) and water (3 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anydrous Na 2 S0 4 .
- Step 2 A mixture of [4-(ethylsulfonyl)phenyl] acetic acid (intermediate la, 101 mg), EDC
- Step 1 A mixture of N-(4-bromophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate lb, 500 mg), 4 ) 4,4' ) 4',5,5,5',5'-octamethyl-2,2 , -bi-l t 3,2-dioxaborolane (598 mg), PdCl 2 (dppf)-CH 2 Cl2 adduct (107 mg) and potassium acetate (513 mg) in DMF (8 mL) was stirred at 100°C under nitrogen for 1.5 hours. After cooling to room temperature, the mixture was filtered through celite. The filtrate was partitioned between EtOAc and water.
- Step 2 A mixture of 2-bromo-3-(trifluoromethyl)pyridine (50 mg), 2-[4- (ethylsulfonyl)phenyl]-N-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yI)phenyl]acetamide (104 mg), Cs 2 C0 3 (87 mg) and PdCl 2 (dppf)-CH 2 Cl 2 adduct (10 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite.
- Step 1 BF 3 -Et 2 0 (1.03 g) was placed into a three necked flask. 2,6-Dichloro-4-nitro- phenylamine (1.0 g) i THF (8 mL) and DCM (16 inL) was added. Then iBuONO (598 mg) was added dropwise at -10 °C. The mixture was stirred for 1 hour at 0 °C. «-Hexane(50 mL) was added. The mixture was filtered to give 2,6-dichloro-4-nitro-benzenediazonium tetrafhioroborate (696 n g) as a yellow solid.
- Step 2 To a stirred solution of CuBr (995 mg) in H 0 (15 mL) was added 2,6-dichloro-4- nitro-benzenediazonium tetrafluoroborate (696 mg) portionwise. The mixture was stirred for 4 hours. The solution was extracted with EtOAc (30 mL x 3). The organic layers were dried over anliydrous Na 2 S0 and concentrated to give 2-bromo-l,3-dichloro-5-nitro-benzene (230 mg) as a yellow oil.
- Step 4 A solution of 2,6-dichloro-4-nitro-biphenyl (2.88 g) and SnCl 2 -2H 2 0 (12.2 g) in
- Step 5 A mixture of 2,6-dichIoro-[l,l '-biphenyl]-4-amine (80 mg), [4- (ethylsulfonyl)phenyl] acetic acid (intermediate la, 84 mg), HOBt (59.0 mg) and EDC (84 mg) in DCM (2 mL) was stirred at room temperature for 1.5 days. Solvent was removed under reduced pressure, and the residue was purified by MDAP to afford N-(2,6-dichloro-4-biphenylyl)-2-[4- (ethylsulfonyl)phenyl]acetamide (95 mg) as a white solid.
- Step 1 A mixture of 4-bromo-3-(mefhylox.y)aniline (400 mg), [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 497 mg), HOBt (348 mg) and EDC (493 mg) in DCM (8 mL) was stirred at room temperature overnight. The mixture was washed with 2M HC1 (3 times), sat. NaHC0 3 (3 times) and brine (3 times) successively. The organic layer was dried over anhydrous Na 2 S0 4 .
- Step 2 A mixture of N-[4-bromo-3-(methyloxy)phenyl]-2-[4- (ethylsulfonyl)pheny]]acetamide (60 mg), ⁇ 2-[(trifluoromethyl)oxy]phenyl ⁇ boronic acid (33.0 mg), PdCl 2 (dppf)-CH 2 Cl 2 adduct (9.51 mg) and Cs 2 C0 3 (56.9 mg) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel.
- Step 2 To a solution of 2-(2-chloro-4-nitro-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (2.0 g) and (2-bromo-phenyl)-dimethyl-amine (1.42 g) in DMF (20 mL) was added K 2 C0 3 (2.94 g) and PdCl 2 (dppf) (512 mg). The mixture was stirred for 4 hours at 100°C under N 2 . The mixture was diluted with water (200 mL). The solution was extracted with EtOAc (50 mL x 3). The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The residue was purified by column
- Step 4 A mixture of (2'-chloro-4 , -amino-biphenyl-2-yl)-dimethyI-amine (80 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 74.0 mg), HOBt (43.8 mg) and EDC (62.2 mg) in DCM (2 mL) was stirred at room temperature overnight.
- Step 1 DIAD (1.349 inL) was added into a solution of 2-bromophenoI (0.8 g), cyclopentanol (0.597 g) and triphenylphosphine (1.819 g) in tetrahydrofuran (THF) (20 mL) at 0°C dropwise. After the addition was complete, the reaction mixture was stirred at 0°C for 1 hour, then left at room temperature overnight. The reaction mixture was washed with brine. The organic layer was dried over anhydrous Na 2 SC> 4 .
- Step 2 A mixture of l-bromo-2-(cyclopentyloxy)benzene (45 mg), N[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaboi lan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 95 mg), PdCl 2 (dppf)-CH 2 Ci2 adduct (12.19 mg) and Cs 2 C0 3 (73.0 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins.
- Step 1 A mixture of 2-bromophenol (0.671 rnL), l -bromo-2-met ylpropane (1.257 mL), K 2 COj (1.598 g) and KI (0.1 g) in DMF (10 mL) was stirred at 60°C for 4.5 hours. The reaction mixture was poured into water, and extracted with EtOAc for three times. The combined organic layers were dried over anhydrous Na 2 S0 4 . After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by chromatography (PE) to afford l-bromo-2-isobutoxybenzene (1.2 g) as a colorless oil.
- PE chromatography
- Step 2 A mixture of l-bromo-2-isobutoxybenzene (45 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intennediate 6b, 100 mg), PdC3 ⁇ 4(dppf)-CH 2 CI 2 adduct (12.83 mg) and Cs 2 C0 3 (77 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins.
- Step 1 To a solution of 3-amino-benzonitrile (1.0 g) in DMF (20 mL) was added n- bromosuccinimide (1.51 g) portionwise at 0 °C. The mixture was stirred for 4 hours at room temperature.
- Step 3 A mixture of 4-amino-2'-trifliioromethoxy-biphenyl-2-carbonitrile (70 mg), [4- (ethylsulfonyI)phenyl]acetic acid (intermediate la, 63.2 mg), HOBt (44.2 mg) and EDC (62.7 mg) in DCM (2 mL) was stirred at room temperature for 1 day. Solvent was removed under reduced pressure, and the residue was purified by MDAP to afford N- ⁇ 2-cyano-2'-[(trifluoromethyl)oxy]-4-biphenylyl ⁇ - 2-[4-(ethylsulfonyl)phenyl]acetamide (39 mg) as a white solid.
- Step 1 To a solution of 2-chloro-3-nitro-benzoic acid (5.0 g) in CC1 4 (100 mL) was added yellow HgO (8.1 g) at room temperature. The mixture was heated to reflux and irradiated with light. Br 2 (1.92 mL) was added dropwise. The reaction solution was kept under reflux for 4 hours. Then the reaction was quenched with sat. NaHC0 3 solution. The mixture was filtered and the filtrate was extracted with EtOAc (50 mL x 3). The combined organic layers were dried over anhydrous Na 2 S0 4 and concentrated.
- Step 2 A solution of l-bromo-2-chloro-3-nitro-benzene (2.03 g) and Pd(PPh 3 ) 4 (1 g) in toluene (50 mL) and EtOH (8 mL) was stirred for 20 mins at 0 °C. Na 2 C0 3 (2.74 g) and 2- (trifluoromethoxy) phenylboronic acid (1.77 g) were added. The mixture was stirred for 4 hours at 100°C under N 2 . The solution was concentrated and the residue was purified by column
- Step 4 Oxalyl chloride (0.153 mL) was added into a solution of [4- (etliyIsulfonyl)phenyI]acetic acid (intermediate la, 80 mg) in DCM (2 mL) dropwise. DMF (0.054 mL) was then added. The resulting mixture was stirred at room temperature for 3 hours. After removal of solvent, the residue was dissolved in DCM (1 mL). The obtained solution was added into a solution of 2-chIoro-2'-trifluoroinethoxy-biphenyl-3-ylamine (111 mg) and pyridine (0.085 mL) in DCM (1 mL) at 0°C dropwise.
- Step 1 DIAD (1.349 mL) was added into a solution of 2-bromophenol (0.8 g),
- Step 2 A mixture of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2- [4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 90 mg), 2-bromophenyl cyclopentylmethyl ether (45 mg), PdCl 2 (dppf)-CH 2 Cl 2 adduct (11.52 mg) and Cs 2 C0 3 (69.0 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins.
- reaction mixture was filtered tlirougl'i celite and silica gel.
- the filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N- ⁇ 2-chIoro-2'-[(cyclopentylmethyl)oxy]- 4-biphenylyl ⁇ -2-[4-(ethylsulfonyl)phenyl]acetamide (32 mg) as a yellow solid.
- Step 1 To a mixture of 2-bromophenol (0.4 g), 1,1-dimetliylethyl 4-hydroxy-l- piperidinecarboxylate (0.698 g) and Ph 3 P (0.910 g) in tetrahydrofuran (THF) (10 mL) cooled at 0 °C, was added DIAD (0.674 mL) dropwise. The mixture was stirred at this temperature for 1 hour, and further stirred at room temperature overnight. The mixture was concentrated under reduced pressure.
- THF tetrahydrofuran
- Step 2 To a solution of 1,1-dimethylethyl 4-[(2-bromophenyl)oxy]-l-piperidinecarboxylate (600 mg) in DCM (6 mL) was added TFA (0.779 mL) dropwise. The reaction was stirred at room temperature for 3 days. The mixture was concentrated under reduced pressure, and the residue was neutralized with sat. Na 2 C0 3 , and then extracted with DCM. The organic layer was washed with brine and dried over anhydrous Na 2 S0 4 . After filtration, solvent was removed in vacuo to afford 4-[(2- bromophenyl)oxy]piperidine (468 mg) as a brown oil. MSfES m/z 256 (MH + ).
- Step 3 4-[(2-Bromophenyl)oxy]piperidine (110 mg) was dissolved in acetonitrile (6 mL) and acetic acid (1 mL). The mixture was cooled to 0 °C. Then 37% aqueous formaldehyde (1 mL) and sodium cyanoborohydride (135 mg) were added. The resulting mixture was allowed to room temperature and stirred for about 4 hours. The mixture was neutralized with sat. Na 2 C0 3 , and then extracted with DCM. The organic phase was dried over anhydrous Na 2 S0 4 . After filtration, solvent was removed in vacuo to afford 4-[(2-bromophenyl)oxy]-l -methylpiperidine (106 mg) as a yellow oil. MS(ES + ) m/z 270 (MH + ).
- Step 4 A mixture of 4-[(2-bromophenyl)oxy]-l-methylpiperidine (100 mg), N-[3-chloro-4- (4,4,5,5-tetramethyl-l,3,2-dioxaboroIan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetainide
- Step 1 To a solution of dimethylamine hydrochloride (0.220 g) in ethanol (20 mL) was added Et 3 N (0.377 mL), 2-bromobenzaIdehyde (0.5 g) successively. The mixture was stirred at room temperature overnight. Sodium cyanoborohydride (0.340 g) was added in one portion. The reaction mixture was stirred at room temperature for 1 day. The solid was filtered off. The filtrate was concentrated under reduced pressure. The residue was redissolved in DCM, washed with sat. Na 2 C0 3 solution, dried over anhydrous Na 2 S0 4 .
- Step 2 A mixture of 1 -(2-bromophenyl)-N,N-dimethylmethanamine (80 mg), N-[3-chloro-4-
- Step 1 A mixture of 5-bromo-4-methyl-2-pyridinamine (500 mg), [4- (ethylsulfonyl)phenyl] acetic acid (intermediate la, 671 mg), HOBt (470 mg) and EDC (666 mg) in DCM (10 mL) was stirred at room temperature overnight. The reaction mixture was washed with brine and sat. NaHC0 3 solution. The organic phase was dried over anliydrous Na 2 S0 4 .
- Step 2 A mixture of N-(5-bromo-4-methyl-2-pyridinyl)-2 ⁇ [4-(ethylsulfonyl)phenyl]acetamide
- Step 1 A mixture of ⁇ 2-[(trifluoromethyI)oxy]phenyl ⁇ boronic acid (1.097 g), l-bromo-2- chloro-4-nitrobenzene (1.2 g), PdCI 2 (dppf)-CH 2 Cl 2 adduct (100 mg) and Cs 2 C0 3 (1.984 g) in acetonitrile (13 mL) and water (4.33 mL) was bubbled with N 2 , then sealed in a reaction vessel and heated in the microwave at 100°C for 1 hour. The mixture was filtered through celite, washed with ACN and EtOAc. The filtrate was concentrated under reduced pressure, and the residue was partitioned between water and EtOAc.
- Step 1 A mixture of 4,4,5,5-tetramethyI-2-(4-nitro-2'-(trifluoromethoxy)-[l ,l '-biphenyl]-2- yI)-l ,3,2-dioxaborolane (intermediate 69a, 200 mg), 5-bromopyrimidine (93 mg), Pd(Ph 3 P) 4 (45.2 mg) and 2 M aqueous Na 2 C0 3 (0,978 mL) in 1,4-dioxane (4 mL) was bubbled with N 2) and then sealed in a reaction vessel. The mixture was heated in the microwave at 100°C for 30 mins. The mixture was partitioned between water and EtOAc.
- Step 2 To a solution of 5- ⁇ 4-nitro-2'-[(trifluoromethyl)oxy]-2-biphenylyl ⁇ pyrimidine (125 mg) in ethanol (5 mL) and tetrahydrofuran (THF) (1 mL) was added HC1 (0.6 mL), followed with addition of tin(II) chloride (394 mg) at room temperature. The resulting mixture was stirred at 50°C for 2 hours. After cooling to room temperature, the mixture was neutralized with sat. Na 2 C0 3 , and then extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na 2 S04.
- THF tetrahydrofuran
- Step 3 A mixture of 2-(5-pyrimidinyl)-2'-[(trifluoromethyl)oxy]-4-biphenyl amine (111 mg), [4-(ethylsulfonyl)phenyl]acetic acid (intermediate l a, 84 mg), EDC (77 mg) and HOBt (54.3 mg) in DCM (6 mL) was stirred at room temperature ovemiglit.
- the mixture was heated in the microwave at 100°C for 30 mins.
- the mixture was filtered through celite, washed with ACN, EtOAc and DCM.
- the combined filtrates were concentraed under reduced pressure, and the residue was parti oned between EtOAc and water.
- the organic layer was washed with brine and dried over anhydrous Na 2 S0 .
- the residue was dissolved in DCM (5 mL), to which TFA (0.173 mL) was added dropwise. The resulting mixture was stirred at room temperature for 4 hours.
- Step 1 A mixture of N- ⁇ 2-chloro-2'-[(trifluoromethyl)oxy]-4-biphenylyl ⁇ -2-[4-
- Step 2 A mixture of l-methyl-l,2,3,6-tetrahydro-4-pyridinyl trifluoromethanesulfonate (intermediate 75a, 618 mg), 2-[4-(ethylsulfonyl)phenyl]-N- ⁇ 2-(4 J 4 ) 5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-2'-[(trifluoromethyl)oxy]-4-biphenylyl ⁇ acetamide (260 mg), Pd(Ph 3 P) 4 (102 mg) and 2 M aqueous Na 2 C ( 3 ⁇ 4 (1.323 mL) in 1,4-dioxane (4 mL) was bubbled with N 2 , and then sealed in a reaction vessel.
- the mixture was heated in the microwave at 100°C for 1 hour. As most of the starting material remained, more l-methyl-l,2 ; 3,6-tetrahydro-4-pyridinyl trifluoromethanesulfonate (intermediate 75a, 464 mg) and Pd(PhjP)4 (25.5 mg) were added. The mixture was heated in the microwave for another 1.5 hours. As some of the starting material remained, more 1 -methyl -1, ,3,6- tetrahydro-4-pyridinyl trifluoromethanesulfonate (intermediate 75a, 464 mg) and Pd(Ph 3 P) (25.5 mg) were added. The mixture was heated in the microwave for an additional 1.5 hours. The mixture was partioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na 2 S0 4 . After filtration and concentration, the residue was purified by flash
- Step 2 A mixture of 1 -(2 -bromophenyl) -3-methoxyazetidine (250 mg), N-[3-chloro-4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide
- Intennediate 79a 2-f4-(propyIsulfonyl ' )phenyl ' )acetic acid
- Step 1 A mixture of (4-mercaptophenyl)acetic acid (18 g, see step 2 for synthesis of intennediate la), K 2 COj (59.2 g) and 1-bromopropane (39.5 g) in DMF (100 mL) was stirred at RT overnight. The mixture was partitioned between ethyl acetate (200 mL) and water (800 mL). The organic phase was washed with water (200 mL x 2) and brine (200 mL), dried and concentrated to give propyl 2-(4-(propylthio)phenyl) acetate (28 g) as a red liquid. MS(ES + ) m/z 253 (MH + ).
- Step 2 To a solution of propyl 2-(4-(propylthio)phenyl) acetate (27 g) in DCM (250 mL) was added wCPBA (66.5 g). The reaction mixture was stirred at RT overnight. Solid was removed by filtration. The filtrate was concentrated. The residue was purified by flash chromatography
- Step 1 A mixture of 2-bromophenoI (862 mg), 4-chloroplienylboronic acid (948 mg), Cu(OAc) 2 (996 mg) and Et 3 N (2.6 g) in DCM (10 mL) was stirred at room temperature overnight under N 2 . The mixture was washed with 1 M HCl, sat. NaHC0 3 and brine successively. The organic phase was dried over anhydrous Na 2 SC>4 and concentrated. The residue was purified by flash chromatography (PE) to give l-bromo-2-(4-chIorophenoxy)benzene (320 mg) as a colorless liquid.
- PE flash chromatography
- Step 2 A mixture of l-bromo-2-(4-chlorophenoxy)benzene (221 mg), N-[3-chloro-4-(4 f 4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 190 mg), Pd(PPh 3 ) 4 (25 mg) and Cs 2 COj (158 mg) in 1,4-dioxane (10 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 85 °C for 130 mins.
- Step 1 3- " Nitrobenzoic acid (10 g) was taken up into conc. H 2 S0 4 (30 mL). The mixture was heated to 60 °C. To this mixture was added NBS (12.78 g) in three portions during 15 mins. After stirring at this temperature for 3 hours, the reaction mixture was poured into ice-water slowly. The precipitate was collected by filtration. This solid was dissolved in EtOAc. The obtained solution was washed with brine, dried over anhydrous Na 2 S0 4 . After filtration, the solution was concentrated in vacuo to afford 3-bromo-5-nitrobenzoic acid (12 g) as a white solid.
- Step 2 A mixture of 3-bromo-5-nitrobenzoic acid (3 g), SOCl 2 (1.780 mL) and DMF (0.019 mL) in DCM (20 mL) was heated under reflux for 3 hours. After concentration, the crude benzoyl chloride was dissolved into benzene (8 mL), and then added into a suspension of aluminium chloride (1.951 g) in benzene (8 mL). The resulting mixture was heated to reflux for 4 hours, and quenched with 3 M HC1. The mixture was extracted with water. The organic layer was dried over anhydrous Na 2 SC>4. After filtration, the solution was concentrated.
- Step 3 A solution of dichlorostannane (1725 mg) in cone. HC1 (3.8 mL) was slowly added to a stirred suspension of (3-bromo-5-nitrophenyI)(phenyl)methanone (585 mg) in ethanol (8 mL). The reaction mixture was heated to 60°C for 30 mins. The obtained clear solution was treated with 30% aqueous NaOH until pH was strongly basic, and then extracted with DCM for 3 times. The combined organic layers were washed with brine and dried over anydrous Na 2 SO, 3 ⁇ 4 .
- Step 4 A mixture of (2,4-dichlorophenyl)boronic acid (207 mg), (3-amino-5- bromophenyl)(phenyl)methanone (300 mg), Cs 2 C0 3 (531 mg) and PdCl 2 (dppf)-CH 2 Cl 2 adduct (71.0 mg) in acetonitrile (6 mL) and water (2 mL) was sealed in a reaction vessel, and heated in the microwave at 120°C for 30 mins. As the starting material remained, more (2,4- dichlorophenyl)boronic acid (104 mg) and PdCl 2 (dppf)-CH 2 Cl 2 adduct (35.0 mg) were added.
- Step 5 A mixture of [4-(etliylsulfonyl)phenyl]acetic acid (intermediate la, 70.0 mg), EDC (72.8 mg) and HOBt (51.3 mg) in DCM (2 mL) was stirred at room temperature under nitrogen for 10 mins. (5-Amino-2',4 f -dichloro-3-biphenylyl)(phenyl)methanone (100 mg) was added. The reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na 2 S0 4 . After filtration, solvent was removed under reduced pressure.
- Step 1 Borane-tetrahydrofuran complex (20.41 mL) was added dropwise to a solution of 2- bromo-5-nitrobenzoic acid (2 g) in THF (100 mL) cooled at 0°C in 30 mins. The reaction mixture was warmed to room temperature and stirred overnight. The reaction mixture was cooled to 0°C and quenched with brine. The organic layer was separated. The aqueous layer was extracted with ethyl acetate (100 mL x 2). The combined organic phases were dried over anhydrous Na 2 S0 4 . After filtration, the filtrate was concentrated in vacuo to afford (2-bromo-5-nitrophenyl)methanol (2.0 g) as a yellow solid. MS(ES + ) m/z 232 (MH + ).
- Step 3 To a solution of 2-bromo-5-nitrobenzaldehyde (1.9 g) in tetrahydrofuran (THF) (50 mL) cooled at 0°C was added phenylmagnesium bromide ( 1.80 g) dropwise over 30 mins. The reaction mixture was stirred at 0°C for 2 hours, and then quenched with saturated NH 4 CI solution. The mixture was extracted with EtOAc (150 mL x 3). The combined organic phases were dried over anhydrous Na 2 S0 4 , filtered, and concentrated.
- THF tetrahydrofuran
Abstract
Disclosed are novel retinoid-related orphan receptor gamma (RORy) modulators and their use in the treatment of diseases mediated by RORy.
Description
NOVEL COMPOUNDS
The present invention relates to novel retinoid-related orphan receptor gamma (RORy) modulators and their use in the treatment of diseases mediated by RORy.
Background of the Invention
Retinoid-related orphan receptors (RORs) are transcription factors which belong to the steroid hormone nuclear receptor superfamily (Jetten & Joo (2006) Adv. Dev. Biol. 16:313-355). The ROR family consists of three members, ROR alpha (RORa), ROR beta (RORp) and ROR gamma (RORy), each encoded by a separate gene (RORA, RORB and RORC, respectively). RORs contain four principal domains shared by the majority of nuclear receptors: an N-terminal A/B domain, a DNA-binding domain, a hinge domain, and a ligand binding domain. Each ROR gene generates several isoforms which differ only in their N-terminal A/B domain. Two isoforms of RORy have been identified: RORyl and RORyt (also known as RORy2). RORy is a term used to describe both RORyl and/or RORyt.
While RORyl is expressed in a variety of tissues including thymus, muscle, kidney and liver, RORyt is exclusively expressed in the cells of the immune system. RORyt has been identified as a key regulator of Thl7 cell differentiation. Thl7 cells are a subset of T helper cells which produce IL- 17 and other proinflammatory cytokines. Thl7 cells have been shown to have key functions in several mouse autoimmune disease models including experimental autoimmune encephalomyelitis (EAE) and collagen-induced artliritis (CIA). In addition, Thl7 cells or their products have been shown to be associated with the pathology of a variety of human inflammatory and autoimmune disorders including multiple sclerosis, rheumatoid artliritis, psoriasis, Crohn's disease and asthma (Jetten (2009) Nucl. RecepL Signal. 7: e003; Manel et al. (2008) Nat. Immunol. 9:641-649). The pathogenesis of chronic autoimmune diseases including multiple sclerosis and rheumatoid artliritis arises from the break in tolerance towards self-antigens and the development of auto-aggressive effector T cells infiltrating the target tissues. Studies have shown that Thl7 cells are one of the important drivers of the inflammatory process in tissue-specific autoimmunity (Steinman (2008) J. Exp. Med. 205: 1517- 1522; Leung et al. (2010) Cell. Mol. Immunol 7: 182-189). There is evidence that Thl7 cells are activated during the disease process and are responsible for recruiting other inflammatory cells types, especially neutrophils, to mediate pathology in the target tissues (Korn et al. (2009) Annu. Rev.
Immunol. 27:485-517).
RORyt plays a critical role in the pathogenic responses of Thl7 cells (Ivanov et al. (2006) Cell 126: 1121 -1 133). RORyt deficient mice show very little Thl7 cells. In addition, RORyt
deficiency resulted in amelioration of EAE. Further support for the role of RORyt in the pathogensis of autoimmune or inflammatory diseases can be found in the following references: Jetten & Joo (2006) Adv.Dev.Biol 16:313-355; Meier et al. (2007) Immunity 26:643-654; Aloisi & Pujol-Borrell (2006) Nat. Rev. Immunol 6:205-217; Jager et al. (200 ) J. Immunol. 183:7169-7177; Serafini et al. (2004) Brain ΡσίΛο/.14:164-174; Magliozzi et al. (2007) Brain 130: 1089-1104; Barnes (2008)
Nat.Rev.Immunol. 8:183-192.
In light of the role RORy plays in the pathogenesis of diseases, it is desirable to prepare compounds that modulate RORy activity, which can be used in the treatment of diseases mediated by RORy. Summary of the Invention
The invention is directed to novel RORy modulators and their use in the treatment of diseases mediated by RORy. Specifically, the invention is directed to compounds according to Formula I.

Formula I
wherein A, B, C, Rl , R2, R3, R4, R5, m, r, s and t are defined below, and to pharmaceutically- acceptable salts thereof.
In another aspect, this invention provides for the use of the compounds of Formula I for the treatment of diseases mediated by RORy. Examples of such diseases include autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid artliritis, psoriasis, Crohn's disease and asthma. In yet another aspect, the invention is directed to methods of treating such diseases.
Detailed Description of the Invention
Terms and Definitions
In describing the invention, chemical elements are identified in accordance with the Periodic Table of the Elements.
"Alkosy" refers to the group -O-R where R is alkyl having the specified number of member atoms. Alkoxy includes methoxy, ethoxy and propoxy.
"Alkyl" refers to a monovalent saturated hydrocarbon chain having the specified number of member atoms. For example, C1-C6 alkyl refers to an alkyl group having from 1 to 6 member atoms. Alkyl groups may be optionally substituted with one or more substituent as defined herein. Alkyl groups may be straight or branched. Representative branched alkyl groups have one, two, or three branches. Alkyl includes methyl, ethyl, propyl (n-propyl and isopropyl), butyl (n-butyl, isobutyl, and t-butyl), pentyl (n-pentyl, isopentyl, and neopentyl), and hexyl.
"Alkenyl" refers to an unsaturated hydrocarbon chain having the specified number of member atoms and having one or more carbon-carbon double bond within the chain. For example, C2-C6 alkenyl refers to an alkenyl group having from 2 to 6 member atoms. In certain embodiments alkenyl groups have one carbon-carbon double bond within the chain. In other embodiments, alkenyl groups have more than one carbon-carbon double bond within the chain. Alkenyl groups may be optionally substituted with one or more substituent as defined herein. Alkenyl groups may be straiglit or branched. Representative branched alkenyl groups have one, two, or three branches. Alkenyl includes ethylenyl, propenyl, butenyl, pentenyl, and hexenyl.
"Cycloalkyl" refers to a saturated hydrocarbon ring having the specified number of member atoms. Cycloalkyl groups are monocyclic ring systems. For example, C3-C6 cycloalkyl refers to a cycloalkyl group having from 3 to 6 member atoms. Cycloalkyl groups may be optionally substituted with one or more substituent as defined herein. Cycloalkyl includes cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
"Enantiomerically enriched" refers to products whose enantiomeric excess is greater than zero. For example, enantiomerically enriched refers to products whose enantiomeric excess is greater than 50% ee, greater than 75% ee, and greater than 90% ee.
"Enantiomeric excess" or "ee" is the excess of one enantiomer over the odier expressed as a percentage. As a result, since both enantiomers are present in equal amounts in a racemic mixture, the enantiomeric excess is zero (0% ee). However, if one enantiomer was enriched such that it constitutes 95% of the product, then the enantiomeric excess would be 90% ee (the amount of the enriched enantiomer, 95%, minus the amount of the other enantiomer, 5%).
"Enantiomerically pure" refers to products whose enantiomeric excess is 99% ee or greater.
"Half-life" refers to the time required for half of a quantity of a substance to be converted to another chemically distinct species in vitro or in vivo.
"Halo" refers to the halogen radicals fluoro, chloro, bromo, and iodo.
"Heteroaryl" refers to an aromatic ring containing from 1 to 4 heteroatoms as member atoms in the ring. Heteroaryl groups containing more than one heteroatom may contain different heteroatoms. Heteroaryl groups may be optionally substituted with one or more substituent as defined herein. Heteroaryl groups are monocyclic ring systems or are fused or bridged bicyclic ring systems. Monocyclic heteroaryl rings have from 5 to 7 member atoms. Bicyclic heteroaryl rings have from 7 to 11 member atoms. Bicyclic heteroaryl rings include those rings wherein phenyl and a monocyclic heterocycloalkyl ring are attached forming a fused, spiro, or bridged bicyclic ring system, and those rings wherein a monocyclic heteroaryl ring and a monocyclic cycloalkyl, cycloalkenyl, heterocycloalkyl, or heteroaryl ring are attached forming a fused, spiro, or bridged bicyclic ring system. Heteroaryl includes pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, furanyl, furazanyl, thienyl, triazolyl, pyridinyl, pyrimidinyl, pyridazinyl, pyrazinyl, triazinyl, tetrazinyl, tetrazolyl, indolyl, isoindolyl, indolizinyl, indazolyl, purinyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl, pteridinyl, cinnolinyl, benzimidazolyl, benzopyranyl, benzoxazolyl, benzisoxazolyl, benzofuranyl, isobenzofuranyl, benzothiazolyl, benzisothiazolyl, benzothienyl, furopyridinyl, and naphthyridinyl.
"Heteroatom" refers to a nitrogen, sulphur, or oxygen atom.
"Heterocycloalkyl" refers to a saturated or unsaturated ring containing from 1 to 4 heteroatoms as member atoms in the ring. However, heterocycloalkyl rings are not aromatic.
Heterocycloalkyl groups containing more than one heteroatom may contain different heteroatoms. Heterocycloalkyl groups may be optionally substituted with one or more substituent as defined herein. Heterocycloalkyl groups are monocyclic ring systems or are fused, spiro, or bridged bicyclic ring systems. Monocyclic heterocycloalkyl rings have from 5 to 7 member atoms. Bicyclic
heterocycloalkyl rings have from 7 to 11 member atoms. In certain embodiments, heterocycloalkyl is saturated. In other embodiments, heterocycloalkyl is unsaturated but not aromatic. Heterocycloalkyl includes pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, pyranyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothienyl, pyrazolidinyl, oxazolidinyl, thiazolidinyl, piperidinyl, homopiperidinyl, piperazinyl, morpholinyl, thiamorpholinyl, azepinyl, 1,3-dioxolanyl, 1,3-dioxanyl, 1 ,4-dioxanyl, 1 ,3-oxathiolanyl, 1,3-oxathianyl, 1,3-dithianyl, azetidinyl, azabicylo[3.2.1]octyl, azabicyIo[3.3.1]nonyl,
azabicylo[4.3.0]nonyl, and oxabicylo[2.2.1]heptyl.
"Member atoms" refers to the atom or atoms that form a chain or ring. Where more than one member atom is present in a chain and within a ring, each member atom is covalently bound to an adjacent member atom in the chain or ring. Atoms that make up a substituent group on a chain or ring are not member atoms in the chain or ring.
"Optionally substituted" indicates that a group, such as alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, or heteroaryl, may be unsubstituted, or the group may be substituted with one or more substituent as defined.
"RORy" refers to all isoforms encoded by the RORC gene which include RORyl and RORyt.
"RORy modulator" refers to a chemical compound that inhibits, either directly or indirectly, the activity of RORy. RORy modulators include antagonists and inverse agonists of RORy.
"Pharmaceutically acceptable" refers to those compounds, materials, compositions, and dosage fonns which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
"Substituted" in reference to a group indicates that one or more hydrogen atom attached to a member atom within the group is replaced with a substituent selected from the group of defined substituents. It should be understood that die term "substituted" includes the implicit provision that such substitution be in accordance widi the permitted valence of the substituted atom and the substituent and that the substitution results in a stable compound (i.e. one that does not spontaneously undergo transformation such as by rearrangement, cyclization, or elimination and that is sufficiently robust to survive isolation from a reaction mixture). When it is stated that a group may contain one or more substituent, one or more (as appropriate) member atom within the group may be substituted. In addition, a single member atom within the group may be substituted with more than one substituent as long as such substitution is in accordance with die permitted valence of the atom. Suitable substituents are defined herein for each substituted or optionally substituted group.
Compounds
The present invention provides, in a first aspect, a compound of Formula I or a
pharmaceutically acceptable salt thereof.

A is phenyl or lieteroaryl;
B is phenyl or lieteroaryl;
C is phenyl or monocyclic lieteroaryl;
each Rl is independently selected from the group consisting of:
- OCF3,
- OCHF2,
- CF3,
- halo,
- CN,
- C1-C3 alkyl optionally substituted with C1-C3 alkoxy,
- piperidinylmethyl or piperidinylidenemetliyl or piperazinylmethyl, wherein said piperidinyl or piperazinyl is substituted with C(0)Ra or C1-C3 alkyl,
- (CH2)nOH,
- C(0)Ra,
- (CH2)nNRaRa ,
- (CH2)nC(0)ORa ,
- heterocycloalkyl optionally substituted with C1 -C3 alkoxy,
- phenyl,
- monocyclic lieteroaryl optionally substituted with C1-C3 alkyl, and
- ORb wherein Rb is selected from the group consisting of:
1) C1-C4 alkyl optionally substituted with CF3, CHF2, NRaRa, phenyl or cycloalkyl, 2) cycloalkyl,
3) phenyl optionally substituted with halo,
4) heterocycloalkyl optionally substituted with C1-C3 alkyl, and
5) monocyclic lieteroaryl, each R2 is independently selected from the group consisting of:
- halo,
- C1 -C6 alkyl optionally substituted with C(0)ORa or cycloalkyl,
- C2-C6 alkenyl,
- OH,
- OCF3,
- OCH2CF3
- C1-C4 alkoxy,
- CN,
- C(0)Ra,
- C(0)NRaRa,
- NRaRa,
- ORb wherein Rb is moncyclic heteroaryl or C1-C6 alkyl,
- phenylcarbonyl optionally substituted with halo,
- heterocycloalkyl, and
- monocyclic heteroaryl optionally substituted with one or more substituents selected from the group consisting of C1-C3 alkoxy, C1-C3 alkyl, COOH and CN, each R3 is selected from the group consisting of H, C1-C3 alkyl and OH;
each R4 is selected from the group consisting of halo, CN and C(0)NH2;
R5 is selected from the group consisting of C1-C3 alkyl, (CH2)nNRaRa, (CH2)nOH, and NHC(0)CH3;
m is 0, 1 or 2; each n is 0, 1 or 2; r is 0, 1 or 2; s is 1 or 2; t is 0, 1 or 2; and each Ra is H or C1 -C3 alkyl.
In one embodiment, the present invention relates to compounds of Formula I wherein A is phenyl. In another embodiment, the present invention relates to compounds of Formula I wherein A is selected from the group consisting of pyridinyl, pyrazol, indolyl, indazolyl and imidazol pyridinyl.
In one embodiment, the present invention relates to compounds according to Formula I or any of the above embodiments wherein m is 1 or 2. The present invention relates to compounds according to Formula I or any of the above embodiments wherein Rl is selected from the group consisting of: OCF3, CFj, F, CI, CN, C¾, OCH3 and OCHF2. In another embodiment, the present invention relates to compounds according to Formula I or any of the above embodiments wherein m is 0.
The present invention also relates to compounds according to Formula I or any of the above embodiments wherein B is phenyl. The present invention also relates to compounds according to
Formula I or any of the above embodiments wherein B is selected from the group consisting of pyridinyl, pyrazinyl, pyrimidinyl and indolyl.
In one embodiment, the present invention also relates to compounds according to Formula I or any of the above embodiments wherein r is 1. In another embodiment, the present invention also relates to compounds according to Formula I or any of the above embodiments wherein R2 is CI or methyl. The present invention also relates to compounds according to Formula I or any of the above embodiments wherein r is 2 and R2 is CI or F. The present invention also relates to compounds according to Formula I or any of the above embodiments wherein r is 0.
The present invention also relates to compounds according to Formula I or any of the above embodiments wherein C is phenyl. The present invention also relates to compounds according to Formula II or any of the above embodiments wherein C is pyridinyl or pyrazol.
The present invention also relates to compounds according to Formula I or any of the above embodiments wherein R3 is H. The present invention also relates to compounds according to Formula I or any of the above embodiments wherein R3 is OH. The present invention also relates to compounds according to Formula I or any of the above embodiments wherein s is 1.
The present invention also relates to compounds according to Formula I or any of the above embodiments wherein t is 0. The present invention also relates to compounds according to Formula I or any of the above embodiments wherein t is 1 and R4 is halo, CN or C(0)NH3.
In one embodiment, the present invention relates to compounds according to Formula I or any of the above embodiments wherein R5 is methyl, ethyl, (CH^nNRaRa or NHC(0)CH3 wherein n is 0, 1 or 2.
The present invention provides, in another aspect, a compound of Formula 1(A) or a pharmaceutically acceptable salt thereof.

A is phenyl or heteroaryl; B is phenyl or heteroaryl;
C is phenyl or monocyclic heteroaryl;
each Rl is independently selected from the group consisting of:
- OCF3>
- OCHF2,
- CF3,
- halo,
- CN,
- C1-C3 alkyl optionally substituted with CI -C3 alkoxy,
- piperidinylmethyl or piperidinylidenemethyl wherein said piperidinyl is substituted with
C(0)Ra or Cl-C3 alkyl,
- (CH2)nOH,
- C(0)Ra,
- (CH2)nNRaRa,
- (CH2)nC(0)ORa,
- heterocycloalkyl optionally substituted with C1-C3 alkoxy,
- phenyl,
- monocyclic heteroaryl optionally substituted with C1-C3 alkyl, and
- ORb wherein Rb is selected from the group consisting of:
1) C1-C4 alkyl optionally substituted with CF3, CHF2j NRaRa, phenyl or cycloalkyl,
2) cycloalkyl,
3) phenyl optionally substituted with halo,
4) heterocycloalkyl optionally substituted with C1-C3 alkyl, and
5) monocyclic heteroaryl, each R2 is independently selected from the group consisting of:
- halo,
- C1-C6 alkyl optionally substituted with C(0)ORa,
- OH,
- C1-C4 alkoxy,
- CF3,
- CN,
- C(0)Ra,
- C(0)NRaRa,
- NRaRa,
- ORb wherein Rb is moncyclic heteroaryl,
- phenylcarbonyl optionally substituted with halo,
- heterocycloalkyl, and
- monocyclic heteroaryl optionally substituted with one or more substituents selected from the group consisting of C1-C3 alkoxy, C1-C3 alkyl, COOH, CN and OH,
R3 is H or Cl-C3 alkyl;
R4 is halo;
R5 is C1-C3 alkyl, NRaRa or (CH2)„OH;
m is 0, 1 or 2; each n is 0, 1 or 2; r is 0, 1 or 2; s is 1 or 2; t is 0, 1 or 2; and each Ra is H or C1-C3 alkyl.
In one embodiment, the present invention relates to compounds of Formula 1(A) wherein A is phenyl. In another embodiment, the present invention relates to compounds of Formula 1(A) wherein A is selected from the group consisting of pyridinyl, pyrazol, indolyl, indazolyl and imidazol pyridinyl.
In one embodiment, the present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein m is 1 or 2. In another embodiment, the present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein m is 0.
The present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein Rl is selected from the group consisting of: OCF , CF3, F, CI, CN, CH3j OCH3 and OCHF2.
The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein B is phenyl. The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein B is selected from the group consisting of pyridinyl, pyrazinyl, pyrimidinyl and indolyl.
In one embodiment, the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein r is 1 or 2. In another embodiment, the present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein r is 0.
The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein R2 is selected from the group consisting of CI, F, CN, methyl and CF3.
The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein C is phenyl. The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein C is pyridinyl or pyrazol.
The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein R3 is H,
The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein s is 1. The present invention also relates to compounds according to Formula 1(A) or any of the above embodiments wherein t is 0.
In one embodiment, the present invention relates to compounds according to Formula 1(A) or any of the above embodiments wherein R5 is methyl, ethyl or NH2.
The present invention provides, in another aspect, a compound of Formula II or a pharmaceutically acceptable salt thereof.

each Rl is independently selected from the group consisting
- OCF3;
- OCHF2,
- CF3,
- halo,
- CN,
- C1-C3 alkyl optionally substituted with C1-C3 alkoxy,
- piperidinylmethyl or piperidinylidenemethyl or piperazinylmethyl, wherein said piperidinyl or piperazinyl is substituted with C(0)Ra or C1-C3 alkyl,
- (CH2)nOH,
- C(0)Ra,
- (CH2)„NRaRa,
- (CH2)nC(0)ORa,
- heterocycloalkyl optionally substituted with C1-C3 alkoxy,
- phenyl,
- monocyclic heteroaryl optionally substituted with C1-C3 alkyl, and
- ORb wherein Rb is selected from the group consisting of:
1) C1-C4 alkyl optionally substituted with CF3, CHF2> NRaRa, phenyl or cycloalkyl,
2) cycloalkyl,
3) phenyl optionally substituted with halo,
4) heterocycloalkyl optionally substituted with C1-C3 alkyl, and
5) monocyclic heteroaryl, each R2 is independently selected from the group consisting of:
- halo,
- C1-C6 alkyl optionally substituted with C(0)ORa or cycloalkyl,
- C2-C6 alkenyl,
- OH,
- OCF3,
- OCH2CF3
- C1-C4 alkoxy,
- CF3,
- CN,
- C(0)Ra,
- C{0)NRaRa,
- NRaRa,
- ORb wherein Rb is moncyclic heteroaryl or CI -C 6 alkyl,
- phenylcarbonyl optionally substituted with halo,
- heterocycloalkyl, and
- monocyclic heteroaryl optionally substituted with one or more substituents selected from the group consisting of C1-C3 alkoxy, C1-C3 alkyl, COOH and CN;
R3 is selected from the group consisting of H, C1-C3 alkyl and OH;
each R4 is selected from the group consisting of halo, CN and C(0)NH2;
R5 is selected from the group consisting of C1-C3 alkyl, (CH2)nNRaRa, (CH2)nOH, and NHC(0)C¾;
m is 0, 1 or 2; each n is 0, 1 or 2; r is 0, 1 or 2; t is 0, 1 or 2and each Ra is H or C1-C3 alkyl.
In one embodiment, the present invention relates to compounds of Formula II wherein m is 1, and Rl is OCF3,OCHF2, or CF3.
In another embodiment, the present invention relates to compounds of Formula II wherein m is 2, one Rl is OCF3 or OCHF2, and the other Rl is CI or F.
The present invention also relates to compounds according to Formula II or any of the above embodiments wherein r is 1, and R2 is CI or methyl at position 2 of die phenyl ring.
The present invention also relates to compounds according to Formula II or any of the above embodiments wherein r is 2, and both R2 are CI or both R2 are F.
The present invention also relates to compounds according to Formula II or any of the above embodiments wherein R3 is H. The present invention also relates to compounds according to Formula II or any of the above embodiments wherein R3 is OH.
The present invention also relates to compounds according to Formula I or any of the above embodiments wherein t is 0. The present invention also relates to compounds according to Formula II or any of the above embodiments wherein t is 1 and R4 is halo, CN or C(0)NH2.
In one embodiment, the present invention relates to compounds according to Formula II or any of the above embodiments wherein R5 is methyl or ethyl.
The present invention provides, in another aspect, a compound of Formula 11(A) or a pharmaceutically acceptable salt thereof.

Formula 11(A)
each Rl is independently selected from the group consisting of:
- OCF3,
- OCHF2,
- CF3,
- halo,
- CN,
- C1-C3 alkyl optionally substituted with C1-C3 alkoxy,
- piperidmylmethyl or piperidinylidenemethyl wherein said piperidinyl is substituted with
C(0)Ra or Cl-C3 alkyl,
- (CH2)nOH,
- C(0)Ra,
- (CH2)nNRaRa,
- (CH2)nC(0)ORa,
- heterocycloalkyl optionally substituted with C1-C3 alkoxy,
- phenyl,
- monocyclic heteroaryl optionally substituted with C1-C3 alkyl, and
- ORb wherein Rb is selected from the group consisting of:
1) C1-C4 alkyl optionally substituted with CF3, CHF2, NRaRa, phenyl or cycloalkyl,
2) cycloalkyl,
3) phenyl optionally substituted with halo,
4) heterocycloalkyl optionally substituted with C1-C3 alkyl, and
5) monocyclic heteroaryl, each R2 is independently selected from the group consisting of:
- halo,
- C1-C6 alkyl optionally substituted with C(0)ORa,
- OH,
- C1-C4 alkoxy,
- CN,
- C(0)Ra,
- C(0)NRaRa,
- NRaRa,
- ORb wherein Rb is moncyclic heteroaryl,
- phenylcarbonyl optionally substituted with halo,
- heterocycloalkyl, and
- monocyclic heteroaryl optionally substituted with one or more substituents selected from the group consisting of C1-C3 alkoxy, C1-C3 alkyl, COOH and CN,
R5 is C1-C3 alkyl, NRaRa or (CH2)nOH;
m is 0, 1 or 2; each n is 0, 1 or 2; r is 0, 1 or 2; each Ra is H or C1-C3 alkyl.
In one embodiment, the present invention relates to compounds of Formula 11(A) wherein m is 1, and Rl is OCF3 or CF3 at position 2 of the phenyl ring.
In another embodiment, the present invention relates to compounds of Fonnula 11(A) wherein m is 2, one Rl is OCF3 or OCHF2 at position 2 of the phenyl ring, and the other Rl is CI or F at position 4 of the phenyl ring.
The present invention also relates to compounds according to Formula 11(A) or any of the above embodiments wherein r is 1, and R2 is CI or methyl at position 2 of the phenyl ring.
The present invention also relates to compounds according to Formula 11(A) or any of the above embodiments wherein r is 2, one R2 is CI or F at position 2 of the phenyl ring, and the other R2 is CI or F at position 6 of the phenyl ring.
In one embodiment, the present invention relates to compounds according to Fonnula 11(A) or any of the above embodiments wherein R5 is methyl or ethyl.
The meaning of any functional group or substituent thereon at any one occurrence in Formula I, or any subformula thereof, is independent of its meaning, or any other functional group's or substituent's meaning, at any other occurrence, unless stated otherwise.
The compounds according to Formula I may contain one or more asymmetric center (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereomers, or other stereoisomeric forms, or as mixtures thereof. Chiral centers, such as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the stereochemistry of a chiral center present in Fonnula I, or in any chemical structure illustrated herein, is not specified the structure is intended to encompass all individual stereoisomers and all mixtures thereof. Thus, compounds according to Fonnula I containing one or more chiral center may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers.
Individual stereoisomers of a compound according to Fonnula I which contain one or more asymmetric center may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer-specific reagent, for example by enzamatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral enviomment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent. The skilled artisan will appreciate that where the desired stereoisomer is converted into another chemical entity by one of the separation procedures described above, a further step is required to liberate the desired form. Alternatively, specific stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
The compounds according to Formula I may also contain double bonds or other centers of geometric asymmetry. Where the stereochemistry of a center of geometric asymmetry present in Fonnula I, or in any chemical structure illustrated herein, is not specified, the structure is intended to encompass the trans (E) geometric isomer, the cis (Z) geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included in Formula I whether such tautomers exist in equilibrium or predominately in one form.
In certain embodiments, compounds according to Formula I may contain an acidic functional group. In certain other embodiments, compounds according to Formula I may contain a basic functional group. Thus, the skilled artisan will appreciate that phannaceutically-acceptable salts of the compounds according to Formula I may be prepared. Indeed, in certain embodiments of the invention, phannaceutically-acceptable salts of the compounds according to Fonnula I may be preferred over the respective free base or free acid because such salts may impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage fonn. Accordingly, the invention is further directed to the use of phannaceutically-acceptable salts of the compounds according to Formula I.
As used herein, the term "pharmaceutically-acceptable salts" refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically-acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
As used herein, the term "compounds of the invention" means both the compounds according to Formula I and the pharmaceutically-acceptable salts thereof. The term "a compound of the
invention" also appears herein and refers to both a compound according to Formula I and its pharmaceutically-acceptable salts .
The invention also includes various deuterated forms of the compounds of Fonnula (I). Each available hydrogen atom attached to a carbon atom may be independently replaced with a deuterium atom. A person of ordinary skill in the art will know how to syndiesize deuterated forms of the compounds of Formula (I). Commercially available deuterated starting materials may be employed in the preparation of deuterated forms of the compounds of Formula (I), or they may be synthesized using conventional techniques employing deuterated reagents (e.g. lithium aluminum deuteride).
The compounds of the invention may exist in solid or liquid form. In the solid state, the compounds of the invention may exist in crystalline or noncrystalline form, or as a mixture thereof. For compounds of the invention that are in crystalline form, the skilled artisan will appreciate that pharmaceutically-acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization. Solvates may involve nonaqueous solvents such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent that is incorporated into the crystalline lattice are typically referred to as "hydrates." Hydrates include stoichiometric hydrates as well as compositions containing vaiable amounts of water. The invention includes all such solvates.
The skilled artisan will further appreciate that certain compounds of the invention that exist in crystalline form, including the various solvates thereof, may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs." The invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification. The skilled artisan will appreciate that different polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymorphs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
Compound Preparation
The compounds according to Formula I are prepared using conventional organic syntheses. Suitable synthetic routes are depicted below in the following general reaction scheme.
The skilled artisan will appreciate that if a substituent described herein is not compatible with the synthetic methods described herein, the substituent may be protected with a suitable protecting group that is stable to the reaction conditions. The protecting group may be removed at a suitable point in the reaction sequence to provide a desired intermediate or target compound. Suitable protecting groups and tlie methods for protecting and de-protecting different substituents using such suitable protecting groups are well known to those skilled in tlie art; examples of which may be found in T. Greene and P. Wuts, Protecting Groups in Chemical Synthesis (3rd ed.), John Wiley & Sons, NY (1999). In some instances, a substituent may be specifically selected to be reactive under the reaction conditions used. Under these circumstances, the reaction conditions convert the selected substituent into another substituent that is either useful as an intermediate compound or is a desired substituent in a target compound.
Scheme 1

[Conditions: a) EDC, HOBt, DCM; or HATU, DIPEA, DMF; b) PdCl2(dppf), Cs2C03, C¾CN, water, 100°C, microwave; or Pd(PPh3}4, Na2C03, dioxane, water, 100°C, microwave; c) 4,4,4')4,,5,5,5',5'-octamethyl-2,2,-bi- 1,3,2-dioxaborolane, PdCl2(dppf), KOAc, DMF, 100°C; or 4,4,4,,4')5,5,5,,5'-octamethyI-2,2,-bi- 1,3,2- dioxaborolane, Pd2(dba)3, tricyclohexylphosphine, KOAc, dioxane, 90°C]
Scheme 1 represents a general reaction scheme for preparing certain compounds of Formula I. Starting material substituted bromophenyl anilines 1.1 can be coupled with an appropriate reagent 1 to form amides 1.2. Amides 1.2 are transferred to die desired compounds of Formular I either by
direct Suzuki coupling with boronic acids 1.3, or by converting to boronic esters 1.4 under catalysis of PdCl2(dppf) or Pd(dba)3 first, then Suzuki coupling with bromides 1.5.
Scheme 2

[Conditions: a) PdCl2(dppf), Cs2C03, CH3CN, water, 100°C, microwave; or Pd(PPh3)4, Na2C03, dioxane, water, 100°C, microwave; b) EDC, HOBt, DCM; or HATU, DIPEA, DMF]
Scheme 2 represents a general reaction scheme for preparing certain compounds of Formula I. Starting material bromides 2.1 can be coupled with an appropriate substituted boronic acids 2.2 to form anilines 2.3. Anilines 2.3 are transferred to the desired compounds of Formular I by direct coupling with reagent 1.
Scheme 3

[Conditions: a) tri-ieri-butylphosphine (tetrafluoroboric acid salt), Pd2(dba)3, Na2C03, dioxane, 100°C, microwave; b) KOH, diethyl (bromodifluoromethyl)phosphonate, acetonitrile, water, -78°C to RT; c) tin(II) chloride dihydrate, EtOH; d) EDC, HOBt, DCM; or HATU, DIPEA, DMF]
Scheme 3 represents a general reaction scheme for preparing certain compounds of Formula I. Starting material substituted bromonitrobenzene 3.1 can be coupled with substituted boronic acids 3.2
to form phenols 3.3 under Suzuki coupling condition. Phenols 3.3 are transferred to difluoromethyl ethers 3.4 and then reduced to form substituted phenyl anilines 3.5. Phenyl anilines 3.5 can be converted to the desired compounds of Formular I by coupling with an appropriate reagent 1.
Scheme 4

[Conditions: a) PdCl2(dppf), Cs2C03, CH3CN, water, 100CC, microwave; or Pd(PPh3)4, Na2C03, dioxane, water, 100°C, microwave; b) 4,4,4',4, J5,5!5',5'-octamethyl-2,2'-bi-l ,3,2-dioxaborolane, PdCl2(dppf), KOAc, DMF, 100°C; or 4J4,4',4',5,5,5',5'-octainethy!-2;2'-bi-l ,3,2-dioxaborolaiie, Pd2(dba)3, tricyclohexylphosphine, KOAc, dioxane, 90°C; c) tin(H) chloride dihydrate, EtOH; d) EDC, HOBt, DCM; or HATU, DIPEA, DMF]
Scheme 4 represents a general reaction scheme for preparing certain compounds of Formula I. Starting material substituted bromocholoronitrobenzene 4.1 can be coupled with boronic acids 4.2 to form biphenyl chlorides 4.3. Biphenyl chlorides 4.3 are transferred to intermediates 4.5 either by direct Suzuki coupling with boronic acids, or by converting to boronic esters 4.4 under catalysis of Pd(¾(dppf) or Pd2(dba)3 first, then Suzuki coupling with bromides. Intermediates 4,5 can be reduced to form substituted anilines 4.6. Anilines 4.6 can be converted to the desired compounds of Formula I by coupling with an appropriate reagent 1.
Examples
The following examples illustrate the invention. These examples are not intended to limit the scope of the present invention, but ratlier to provide guidance to the skilled artisan to prepare and use the compounds, compositions, and methods of the present invention. While particular
embodiments of the present invention are described, the skilled artisan will appreciate that various changes and modifications can be made without departing from the spirit and scope of the invention.
Abbreviations
ACN acetonitrile
AIBN 2,2'-azo teisobutyronitrile
BINAP 2,2' -£w(diphenylphosphino)- 1 , -binaphthyl
BOP benzotriazole-l-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate
BuLi butyl lithium
cone. concentrated
DCE 1,2-dichloroethane
DCM dichloromethane
DIAD diisopropyl azodicarboxylate
DIBAL-H diisobutylaluminum hydride
DIPEA dii sopropyl ethylamine
DMAP NN-4-dimethylaminopyridine
DMF N,N-dimethylfonnamide
DMF-DMA N,N-dimethylformamide dimethyl acetal
DMSO dimethylsulphoxide
EA ethyl acetate
EDC N-(3 -dimethylaminopropylJ-N-ethylcarbodiimide hydrochloride
ES electrospray
HATU 0-(7-azabenzotriazol-l -yl)-N^V,N',N'-tetramethyluronium hexafluorophosphate
HOBt hydroxybenzotriazole hydrate
hr hour
LAH lithium aluminum hydride
LCMS Liquid Chromatography Mass Spectrometry
MDAP mass directed automated preparative liquid chromatography
Ms mesyl (methanesulfonyl)
MS mass spectrometry
min minute
NCS N-chlorosuccinimide
NBS N-bromosucctnimide
NMP N-methyl -2-pyrrolidone
PCC pyridinium chlorochromate
Pin pinacolato
PE petroleum ether
Py pyridine
RT room temperature
sat. saturated
TBAF tetra-H-butylammonium fluoride
TEA triethylainine
Tf trifluoromethanesulfonyl
TEA trifluoroacetic acid
TFAA trifluoroacetic anhydride
THF tetrahydrofuran
TIPS triisopropylsilyl
TLC thin layer chromatography
TMSC1 trimethylchlorosilane
Chromatography
Unless stated otlierwise, all chromatography was carried out using silica columns. LCMS
1) acidic condition:
Mobile phase: water containing 0.05 % TFA/acetonitrile
Column: XBridgeTM C 18 30 x 100 mm - 5 microns
Detection: MS and photodiode array detector (PDA)
2) basic condition:
Mobile phase: water containing 0.08 % NH4HC03 / acetonitrile
Column: XBridgeTM C 18 30 x 100 mm - 5 microns;
Detection: MS and photodiode array detector (PDA)
MDAP
1) acidic condition 1 :
Instrument: Waters instrument
Column: Sunfire Prep C18 column (5 um, 19 x 50 mm)
Mobile phase: water containing 0.05% TFA / acetonitrile.
2) acidic condition 2:
Instrument: Gilson GX-281
Column: Sunfire prep C18 OBD; 5 um, 100 mm * 30 mm;
Mobile phase: A: 0.05% TFA/¾0; B: MeCN;
3) basic condition 1:
Instrumnet: Waters instrument
Column: Xbridge Prep CI 8 column (5 um, 19 x 50 mm)
Mobile phase: water containing 0.04% ammonia/ acetonitrile.
4) basic condition 2:
Instrumnet: Gilson 281(PHG-005);
Column: Shimadzii PRC-ODS 20 x250 mm, 15um two connected in series; Mobile phase: A: 10 mM NH4HC03 B:MeCN;
5) basic condition 3:
Instrumnet: Gilson GX-281 ;
Column: Agela Durashell RP 21.5*250 mm 10 μιη;
Mobile phase: A: 0.04% NH3 H20/water; B: CH3CN;
Example 1
2-[4-(ethylsulfonyI)phenyl]-iV-[2,-(trifluoromethyl)-4-biphenylyl]acetamide
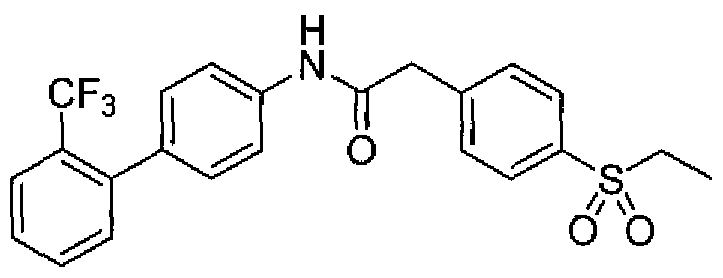
Intermediate la: [4-(ethylsulfonyl phenyl1acetic acid
Step 1: A solution of sodium nitrite (] 8.4 g) in 133 mL of water was added dropwise at 0 °C, while stirring, to a suspension of (4-aminophenyl)acetic acid (40.2 g) in 133 mL of water and 54 mL of concentrated hydrochloric acid. After the addition was complete, the reaction mixture was stirred at the same temperature for 45 minutes. This solution of cold diazonium salt was then added dropwise at room temperature to a mixture of potassium ethylxanthate (49.4 g), 80 mL of water and 200 mL of 2 M sodium carbonate solution. The mixture was heated to 45 °C and stirred at this temperature until gas evolution stops. After cooling to room temperature, pH was adjusted to 1 with concentrated hydrochloric acid and the oiled xanthogenate ester was extracted with ether. Solvent was evaporated to give (4-{[(ethyloxy)carbonothioy]]thio}phenyl)acetic acid (90 g) as a dark red liquid. MSfES m/z 257 (MH+).
Step 2: (4-{[(Ethyloxy)carbonothioyl]thio}phenyl)acetic acid (90 g) was taken up in 340 mL of ethanol, and a solution of 70 g of potassium hydroxide in 340 mL of water was added. Boiling at reflux was effected for 20 hours. The major portion of ethanol was subsquently removed by the distillation under reduced pressure. The aqueous phase was cooled with ice, and acidified with concentrated hydrochloric acid while stirring. The obtained solution was extracted with diethyl ether (500 mL). The organic phase was washed with brine, dried over anhydrous sodium sulphate, filtered, and concentrated to afford (4-mercaptophenyl)acetic acid (33 g) as a yellow solid. MS(ES ) m/z 169 (MH+).
Step 3: To a solution of (4-mercaptophenyl)acetic acid (33 g) in DMF (240 mL) was added 2CO3 (108 g) and bromoethane (64.1 g). The reaction mixture was stirred at room temperature. After 2.5 hours, the starting material was totally consumed. The reaction mixture was partitioned between ethyl acetate (300 mL) and water (300 mL). The organic phase was washed with water (300 mL x 4) and brine (200 mL), dried over sodium sulphate, filtered, and concentrated to give the desired product ethyl [4-(ethylthio)phenyl] acetate (34 g) as a pale yellow solid. MS(ES+) m/z 225 (MH+).
Step 4: A solution of ethyl [4-(ethylthio)phenyl] acetate (34 g) in DCM (500 mL) was cooled to 0°C with an ice bath. mCPBA (78 g) was added in portions, and the reaction mixture was stirred at RT overnight. The obtained suspension was filtered. The filtrate was washed with sat. sodium carbonate solution (400 mL x 2), water (500 mL), then brine (250 mL). The obtained solution was dried over sodium sulphate, filtered, and concentrated. The residue was purified by column chromatography (silica gel, EtOAc : PE = 0:1 to 1 :1) to afford ethyl [4-(ethylsulfonyl)phenyl]acetate (25 g) as a yellow liquid. MS(ES+) m/z 257 (MH4).
Step 5: To a solution of ethyl [4-(ethylsulfonyl)phenyI]acetate (25 g) in ethanol (180 mL) was added a solution of NaOH (14.28 g) in water (180 mL). The reaction mixture was stirred at room temperature overnight. Ethanol was removed under reduced pressure, and 150 mL of water was added. The aqueous phase was washed with dichloromethane (100 mL x 2), and then acidified with 6 M HCl to pH = 1. This solution was extracted widi ethyl acetate (200 mL x 2). The combined organic phases were washed with brine (200 mL), dried over sodium sulphate, filtered, and concentrated to give the desired product as a dark red oil, which slowly solidified to give [4-(ethylsulfonyl)phenyl]acetic acid (20 g) as a yellow solid. Ή-NMR (400 MHz, DMSO-i¾ 5 ppm 1.07 (t, J= 9.6 Hz, 3H), 3.26 (q, J= 9.6 Hz, 2H), 3.72 (s, 2H), 7.53 (d, J= 11.2 Hz, 2H), 7.81 (d, J= 11.2 Hz, 2H), 12.53 (s, 1H); MS(ES+) m/z 229 (MH4).
Intermediate lb: N-(4-bromophenylV2-f4-fethylsulfonyl')phenyllacetamide
A mixture of 4-bromoaniline (400 mg), [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 557 mg), EDC (579 mg) and HOBt (408 mg) in DCM (10 mL) was stirred at room temperature overnight. The mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM. The combined organic layers were washed with diluted HCl, sat. NaHC(¾ and brine. The obtained solution was dried over anliydrous Na2S04. After filtration, solvent was removed in vacuo to afford N-(4-bromophenyl)-2-[4-(ethyIsulfonyl)phenyl]acetamide (754 mg) as a yellow solid. MS(ES+) m/z 382 (MH4).
Preparation of 2 4-fethylsulfonyl)phenyl1-N-r2'- trifluoromethyl)-4-biphenylyl]acetamide
A mixture of [2-(trifluoromethyl)phenyl]boronic acid (38.3 mg), N-(4-bromophenyl)-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate lb, 70 mg), PdCl2(dppf)-CH2CI2 adduct (12.0 mg), Cs2C03 (71.6 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was bubbled with N2. The vessel was then sealed and heated in the microwave at 100°C for 30 mins. The mixture was filtered through silica gel. After removal of solvent, the residue was redissolved with DMF and purified by MDAP to afford 2-[4-(ethylsuIfonyl)phenyl]-N-[2'-(trifluorometliyl)-4-biphenylyl]acetamide (53 mg) as a white solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 1.10 (t, J= 7.4 Hz, 3H), 3.28 (q, J= 7.3 Hz, 2H), 3.83 (s, 2 H), 7.25 (d, J= 8.4 Hz, 2H), 7.38 (d, J= 7.6 Hz, 1H), 7.64 (m, 6H), 7.81 (d, J= 7.7 Hz, 1H), 7.86
(d, J= 8.3 Hz, 2H), 10.41 (s, 1H); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -55.36; MSfES ) m/z 448 (MH+).
Example 2
2-[4-(ethylsulfonyl)phenyl]-jV-[2'-(methyIoxy)-4-biphenylyl]acetamide

A mixture of [2-(methyloxy)phenyl]boronic acid (30.6 mg), N-(4-bromophenyl)-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate lb, 70 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (71.6 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was bubbled with N2. The vessel was then sealed and heated in the microwave at 100°C for 30 minutes. The mixture was filtered through silica gel. After removal of solvent, the residue was redissolved with DMF and purified by MDAP to afford 2-[4~(ethylsulfonyl)phenyl]-N-[2'-(methyloxy)-4-biphenylyl]acetamide (44 mg) as a white solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 1.10 (t, /= 7.3 Hz, 3H), 3.28 (q, J= 7.4 Hz, 2H), 3.75 (s, 3H), 3.82 (s, 2H), 7.00 (dt, J= 0.8 Hz, 7.5 Hz, 1H), 7.08 (d, J= 7.9 Hz, 1H), 7.26 (dd, J- 1.7 Hz, 7.5 Hz, 1H), 7.23 (m, 1H), 7.42 (d, J= 8.6 Hz, 2H), 7.62 (m, 4H), 7.85 (d, J= 8.3 Hz, 2H), 10.33 (s, 1H); MS(ES+) m/z 410 (MH+).
Example 3
N-(2 '-cyano-4-biphenyIyI)-2- [4-(ethyls ulf 6nyl)phenyl] acetamide

A mixture of (2-cyanophenyl)boronic acid (29.6 mg), N-(4-bromopheny])-2-[4- (e ylsulfonyl)phenyl]acetamide (intermediate lb,70 mg), PdCl2(dppf)-CH2CI2 adduct (10 mg) and Cs2C(¾ (71.6 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was bubbled with N2. The vessel was then sealed and heated in the microwave at 100°C for 30 minutes. After cooling to room temperature, the mixture was filtered through silica gel. The solvent was removed. The residue was redissolved with DMF and purified by MDAP to afford N-(2'-cyano-4-biphenylyl)-2-[4- (ethylsulfonyl)phenyl]acetamide (26 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm
1.10 (t, J= 7.4 Hz, 3H), 3.28 (q, J= 7.4 Hz, 2H), 3.85 (s, 2H), 7.58 (m, 6H), 7.76 (m, 3H), 7.86 (m, 2H), 7.93 (dd, J= 1.0 Hz, 7.8 Hz, 1H), 10.49 (s, 1H); MS(ES+) m z 405 (MH+).
Example 4
2- [4-(ethyl sulf onyl) phenyl]-/Y- {2 '- [(triflu or omethyl)oxy] -4-biphenylyl} acetamide

A mixture of {2-[(trifluoromethyI)oxy]phenyl}boronic acid (41.5 mg), N-(4-bromophenyl)-2- [4-(ethylsulfonyl)phenyl]acetamide (intermediate lb, 70 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (71.6 mg) in acetonitrile (1.5 lnL) and water (0.5 mL) was bubbled with N2. The vessel was then sealed and heated in the microwave at 100°C for 30 minutes. The mixture was filtered through silica gel and celite, washed with DCM and EtOAc. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-{2'- [(trifluoromethyl)oxy]-4-biphenylyl}acetamide (41 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 3.28 (q, J= 7.4 Hz, 2H), 3.83 (s, 2H), 7.47 (m, 6H), 7.63 (ds J= 8.3 Hz, 2H), 7.70 (d, J= 8.6 Hz, 2H), 7.86 (d, J= 8.3 Hz, 2H), 10.42 (s, 1H); l9F-NMR (376 MHz, DMSO-t/e) δ ppm -56.21 ; MS(ES+) m/z 464 (MH+).
Example 5
A'-[4,~chloro-2'-(trifluoromethyl)-4-biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide

A mixture of [4-chloro-2-(trifluoromethyl)phenyl]boronic acid (45.2 mg), N-(4-bromophenyl)- 2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate lb, 70 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (71.6 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was bubbled with N . The vessel was then sealed and heated in the microwave at 100°C for 30 minutes. The mixture was filtered through silica gel and celite, washed with DCM and EtOAc. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford N-^'-chloro^'-itrifluoromethyl)^- biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (48 mg) as a white solid. Ή-NMR (400 MHz,
DMSO-i/6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.83 (s, 2H), 7.26 (d, J= 8.4 Hz, 2H), 7.42 (d, J= 8.0 Hz, 1H), 7.63 (d, J= 8.0 Hz, 2H), 7.67 (d, J= 8.8 Hz, 2H), 7.79 {dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.86 (d, J= 8.0 Hz, 2H), 7.88 (d, J= 2.0 Hz, 1H), 10.43 (s, 1H); 19F-NMR (376 MHz, DMSO-4 δ ppm -55.90; MSiES^) m/z 482 (MH+). Example 6
iV-IZ-chloroJ'-ihydroxymethylJ^-biphenylyll-Z-^-iethylsuIfonylJphenylJacetamide

Intennediate 6a: ^(4-^οι ο- -ο1ΐ1θΓο 1ΐ6ηνΠ-2-Γ4- 6&νΐ5υ1ΓοηνΠ 1ΐ€ην1¾θ6ίαηΐίά6
A mixture of 4-bromo-3-chloroaniIine (3.3 g), [4-(ethylsulfonyl)phenyl]acetic acid
(intennediate la, 4.01 g), HOBt (3.24 g) and EDC (4.60 g) in DCM (70 lnL) was stirred at room temperature under N2 overnight. The reaction mixture was washed with 2M HC1, sat. NaHC03 solution and brine successively. The organic layer was dried over anliydrous Na2S04. After filtration, the filtrate was concentrated in vacuo to afford N-(4-bromo-3-chlorophenyl)-2-[4- (ethylsulfonyl)phenyI]acetamide (6 g) as a yellow solid. MS(ES ) m/z 416 (MH ).
Intennediate 6b: N-r3-chloro-4-(,4,4.5.5-tetrainethyl-l13,2-dioxaborolan-2-yl')phenyll-2-|"4- (ethylsulfonyllphenyllacetamide
A mixture of N-(4-bromo-3-chIoTophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (intennediate 6a, 1.5 g), 4,4,4',4',5,5,5',5,-octamethyl-2,2'-bi-lJ3,2-dioxaborolane (7.31 g), PdCI2(dppf)-CH2Cl2 adduct (0.588 g) and potassium acetate (2.12 g) in DMF (20 mL) was heated to 100°C for 6.5 hrs under nitrogen. The reaction mixture was poured into water, extracted with EtOAc for three times. The combined organic layers were filtered through celite and silica gel. The filtrate was dried over anliydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography (EtOAc : PE = 0: 100 to 40:60) to afford N-[3- chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(etliylsulfonyl)phenyl]acetamide (1.2 g) as a yellow solid. MS(ES+) m/z 464 (MH+).
Preparation of N-r2-chloro-2'-fliydroxyinethyl')-4-biphenylyll-2-r4-iethylsulfonyl phenyllacetamide
A mixture of (2-bromophenyl)methanol (50 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl-l ,3,2- dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intennediate 6b, 136 mg),
PdCl2(dppf)-CH2Cl2 adduct (15 mg) and Cs2C03 (105 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was bubbled with N2. The reaction vessel was sealed and heated in the microwave at 100°C for 30 mins. The mixture was filtered through celite and silica gel, and then washed with acetonitrile, DCM and EtOAc. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford the crude product (41 mg), which was further purified by flash chromatography (MeOH : DCM = 0: 1 to 3:97). After removal of solvent, the residue was redissolved in acetonitrile, THF and water. The mixture was freeze dried to afford N-[2-chloro-2'-(hydroxymethyl)-4- biphenylyl]-2-[4-(ethylsuIfonyl)phenyl]acetamide (25 mg) as a white solid.Ή-NMR (400 MHz, DMSO-</6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 3.28 (q, J= 7.3 Hz, 2H), 3.84 (s, 2H), 4.18 (dd, J= 5.5 Hz, 13.9 Hz, 1H), 4.26 (dd, J = 5.4 Hz, 13.9 Hz, 1H), 5.09 (t, J= 5.4 Hz, 1H), 7.07 (d, /= 6.9 Hz, 1H), 7.24 (d, J= 8.3 Hz, 1H), 7.30 (t, J = 7.3 Hz, 1H), 7.40 (t, J= 7.5 Hz, 1H), 7.51 (dd, J~ 2.0 Hz, 8.4 Hz, 1H), 7.57 (d, J= 7.6 Hz, 1H), 7.62 (d, J= 8.3 Hz, 2H), 7.86 (d, J= 8.3 Hz, 2H), 7.92 (d, J= 2.0 Hz, 1H), 10.54 (s, 1H); MS(ES+) m/z 426 (MH+-18).
Example 7
2-[4-(ethylsulfonyI)phenyl]-iV-[2-(hydroxymethy])-2'-(trifluoromethyI)-4-biphenylyl]acetamide

Step 1: A mixture of [2-(trifluoromethyl)phenyl]boronic acid (0.767 g), methyl 2-bromo-5- nitrobenzoate (1 g), PdCI2(dppf)-CH2Cl2 adduct (0.157 g) and Cs2C03 (1.504 g) in acetonitrile (12 mL) and water (4.00 mL) was bubbled with N2. The reaction vessel was sealed and heated in the microwave at 100°C for 1 hour. The mixture was filtered through celite and silica gel, and then washed with acetonitrile, DCM and EtOAc. The filtrate was concentrated under reduced pressure and the residue was partitoned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with water and dried over anhydrous Na2S04. After filtration, solvent was removed and the residue was purified by flash chromatography (EtOAc :
PE = 0:1 to 1:9) to afford methyl 4-nitro-2'-(trifluoromemyl)-2-biphenylcarboxy]ate (750 mg) as an off-white solid.Ή-NMR (400 MHz, CDC13) δ ppm 3.70 (s, 3H), 7.22 (d, J= 7.4 Hz, 1H), 7.49 (d, J= 8.4 Hz, 1H), 7.55 (m, 2H), 7.77 (d, J= 7.7 Hz, 1H), 8.39 (dd, J= 2.4 Hz, 8.4 Hz, 1H), 8.91 (d, J= 2.4 Hz, 1H); l9F-NMR (376 MHz, CDC13) 5 ppm -58.41.
Step 2: To a solution of methyl 4-nitro-2'-(trifluoromethyl)-2-biphenylcarboxylate (300 mg) in tetrahydrofuran (THF) (8 mL) cooled at 0°C was added LAH (140 mg) slowly. The resulting mixture was stirred at room temperature overnight. The reaction was quenched by the sequential treatment with water (0.15 mL), 15% NaOH (0.15 mL) and water (0.3 mL). The mixture was then filtered and the filtrate was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After filtration and removal of solvent, the residue was dissolved in methanol (8 mL), to which 10% Pd/C (30 mg) was added. The resulting mixtue was stirred under H2 (excess) at room temperature overnight. The mixture was filtered. The filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography (MeOH : DCM = 0:1 to 3:97) to afford [4-amino-2'- (trifluoromethyl)-2-biphenylyl]methanol (114 mg) as a yellow solid. MS(ES+) m/z 268 (MH+).
Step 3: A mixture of [4-amino-2'-(trifluoromethyl)-2-biphenylyl]metlianol (60 mg), [4- (ethyIsulfonyl)phenyl]acetic acid (intermediate la, 53.8 mg), EDC (56.0 mg) and HOBt (39.4 mg) in DCM (3 mL) was stirred at room temperature over 3 days. The mixture was concentrated under reduced pressure and the residue was purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-[2- (hydroxyinethyl)-2'-(trifluoromethyl)-4-biphenylyl]acetarnide (22 mg) as a white solid.Ή-NMR (400 MHz, DMSO-i/6) 5 ppm 1.10 (t, J= 7.4 Hz, 3H), 3.28 (q, = 7.4 Hz, 2H), 3.82 (s, 2H), 4.03 (dd, J= 5.2 Hz, 14.1 Hz, 1H), 4.13 (dd, J= 5.2 Hz, 14.1 Hz, 1H), 5.13 {t, J= 5.3 Hz, 1H), 7.00 (d, J= 8.2 Hz, 1H), 7.30 (d, J= 7.5 Hz, 1H), 7.61 (m, 4H), 7.69 (t, J= 7.4 Hz, 1H), 7.76 (d, J= 1.8 Hz, 1H), 7.81 (d, J= 7.7 Hz, 1H), 7.86 (d, J= 8.3 Hz, 2H), 10.38 (s, 1H); 19F-NMR (376 MHz, DMSO-< ) δ ppm - 57.39; MSfES") m/z 460 (MH+-18).
Example 8
N-(2'-acetyl-2-chloro-4-biphenylyl)-2-[4-(ethylsulfonyl)phenyl]acetamide

Step 1: To a solution of l-bromo-2-chloro-4-nitro-benzene (2.0 g) and 2-acetylphenyl boronic acid (1.53 g) in DMF (20 mL) was added K2C03 (3. 1 g) and PdCl2(dppf) (622 mg) under N2. The mixture was stirred for 4 hours at 100°C. The mixture was filtered and the filtrate was diluted with water (100 mL). The solution was extracted with EtOAc (100 mL x 3). The organic layers were dried over anhydrous Na2S04 and concentrated. The residue was purified by column chromatography on
silica gel (EtOAc : PE = 1:20) to afford l-(2'-chloro-4'-niiro-biphenyI-2-yl)-ethanone (1.1 g) as a yellow oil.
Step 2: A solution of l-(2'-chloro-4'-nitro-biphenyl-2-yl)-ethanone (920 mg) and SnCl2'2H20 in cone. HCl (20 mL) was stirred for 2 hours at 70°C. The solution was diluted with EtOAc (30 mL), The mixture was adjusted to pH ~ 9 with NaOH solution and filtered. The filtrate was extracted with EtOAc (50 mL x 3). The organic layers were dried over anhydrous Na2S04 and concentrated. The residue was purified by column chromatography on silica gel (EtOAc : PE = 1 :5) to give l-(4'-amino- 2'-chloro-biphenyl-2-yl)-etlianone as a brown oil (530 mg). MS(ES+) m/z 246 (MH+).
Step 3: A mixture of l-(4'-amino-2'-chloro-biphenyl-2-yl)-ethanone (60 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 61.3 mg), EDC (60.9 mg) and HOBt (42.9 mg) in DCM (4 mL) was stirred at room temperature overnight. The mixture was concentrated under reduced pressure and the residue was purified by MDAP to afford N-(2'-acetyl-2-chloro-4-biphenylyl)-2-[4- (ethylsulfonyl)phenyI]acetamide (85 mg) as a light pink solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 2.29 (s, 3H), 3.28 (q, J= 7.4 Hz, 2H), 3.84 (s, 2H), 7.24 (d, J= 8,4 Hz, 1H), 7.27 (dd, J= 1.0 Hz, 7.6 Hz, 1H), 7.53 (m, 2H), 7.62 (m, 3H), 7.82 (dd, J= 1.2 Hz, 7.7 Hz, 1H), 7.86 (m, 3H), 10.52 (s, 1H); MS(ES+) m/z 456 (MH÷).
Example 9
2-[4-(ethyIsulfonyl)phenyl]-iV-{2-fluoro-2,-[(trifluoromethyI)oxy]-4-biphenylyl}acetamide

(intermediate la, 505 mg), EDC (525 mg) and HOBt (370 mg) in DCM (10 mL) was stirred at room temperature overnight. The mixture was diluted with DCM, washed sequentially with water, 2M HCl, sat. NaHC03 and brine. The solution was dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford N-(4-bromo-3-fiuorophenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide (827 mg) as a yellow solid. MS(ES+) m/z 400 (MH+).
Step 2: A mixture of {2-[(trifluoromethyl)oxy]phenyl}boronic acid (81 mg), N-(4-bromo-3- fluorophenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide (150 mg), PdCl2(dppf)-C¾Cl2 adduct (20 mg) and Cs2C03 (147 mg) in acetonitrile (2.1 mL) and water (0.700 mL) was bubbled with N2. The vessel was then sealed and heated in the microwave at 100°C for 30 minutes. The mixture was filtered
through silica gel and celite, washed with acetonitrile, DCM and EtOAc. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford 2-[4- (ethylsulfonyl)phenyl]-N-{2-fluoro-2'-[(trifluoromethyl)oxy]-4-biphenylyl}acetamide (96 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i¾ δ ppm 1.10 (t, J= 7.3 Hz, 3H), 3.28 (q, J= 7.3 Hz, 2H), 3.85 (s, 2H), 7.35 (t, J= 8.3 Hz, 1H), 7.41 (dd, J= 1.9 Hz, 8.4 Hz, 1H), 7.53 (m, 4H), 7.63 (d, J= 8.3 Hz, 2H), 7.71 (dd, J= 1.8 Hz, 12.6 Hz, 1H), 7.86 (d, J= 8.3 Hz, 2H), 10.61 (s, 1H); 19F-NMR (376 MHz, DMSO-i/6) δ ppm -56.38, -113.54; MS(ES+) m/z 482 (MH+).
Example 10
N-ie-chloro-l'-Iitrifluoromethy^oxyj-S-biphenylylJ^-^-iethylsulfonylJpheny^acetamide

Step 1: BF3-Et20 (6.17 g) was placed in a three-necked flask. 2-Chloro-5-nitro-phenylamine (5.0 g) in THF (40 mL) and DCM (80 lnL) was added. /-BuONO (3.6 g) was added dropwise at - 10°C, The mixture was stirred for 1 hour at 0°C. n-Hexane (200 mL) was added. The solution was filtered to give 2-chloro-5-nitro-benzenediazonium tetrafluoroborate (7.6 g) as a yellow solid.
Step 2: To a stirred solution of CuBr (20 g) in H20 (100 mL) was added 2-chIoro-5-nitro- benzenediazonium tetrafluoroborate (7.6 g) portionwise. The mixture was stirred overnight at room temperature. The mixture was extracted with EtOAc (300 mL x 2). The organic layers were dried over anhydrous Na2S04 and concentrated. The residue was purified by column chromatography on silica gel (EtOAc : PE = 1 :50) to give 2-bromo-l-chloro-4-nitro-benzene (2.2 g).
Step 3: A solution of 2-bromo-l -chloro-4-nitro-benzene (2 g) and Pd(PPh3)4 (980 mg) in toluene (50 mL) and EtOH (8 mL) was stirred for 20 mins at 0 °C. Na2C03 (2.96 g) in 10 mL water and 2-(trifluoromethoxy)phenylboronic acid (1.92 g) were added. The mixture was stirred for 16 hours at 100°C under N2. TLC indicated the reaction completed. Then the mixture was concentrated and the residue was purified by column chromatography on silica gel (EtOAc:PE = 1:20) to give 2- chloro-5-nitro-21-trifluoromethoxy-biphenyl (2.1 g) as a yellow oil.
Step 4: A mixture of 2-chloro-5-nitro-2'-trifluoromethoxy-biphenyI (2.1 g) and SnCl2-2H20 (7.46 g) in MeOH (30 mL) was heated at 60°C overnight. The solution was concentrated. The residue was diluted with EtOAc (50 mL) and water (30 mL). The solution was adjusted to pH = 9 with NaOH solution. Then the mixture was filtered and the filtrate was extracted with EtOAc (50 mL x 3). The
combined organic layers were dried over anhydrous Na2S04 and concentrated. The residue was purified by column chromatography on silica gel (EtOAc.PE = 1:10) to give 6-chloro-2'- trifluoromethoxy-biphenyI-3-ylamine (1.15 g) as a brown oil. MS(ES+) m/z 288 (MH+).
Step 5: A mixture of 6-chloro-2'-trifluoromethoxy-biphenyl-3-ylamine (87 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intennediate la, 76 mg), EDC (75 mg) and HOBt (53.1 mg) in DCM (2 mL) was stirred at room temperature overnight. Solvent was removed under reduced pressure. The residue was purified by MDAP to afford N-{6-chloro-2'-[(trifluoromethyl)oxy]-3- biphenylyl}-2-[4-(ethyIsulfonyl)phenyl]acetamide (54 mg) as a white solid. 'H-NMR (600 MHz, DMSO-£¼) 8 ppm 1.08 (t, J= 7.2 Hz, 3H), 3.26 (q, J= 7.2 Hz, 2H), 3.82 (s, 2H), 7.41 (m, 1H), 7.50 (m, 3H)} 7.62 (m, 5H), 7.83 (d, J= 8.4 Hz, 2H), 10.50 (s, 1H); 19F-NMR (376 MHz, DMSO-iQ δ ppm -56.16; MS(ES+) m/z 498 (MH+).
Example 11
2-(4-(ethylsulfonyl)p enyl)-JV-(6-methylbip enyl-3-yl)acetamide

Pd(Ph3P)4 (86 mg) and Na2C03 (529 mg) in ethanol (10 mL), toluene (10 mL) and water (10 mL) was stirred at 80°C overnight. Solvent was removed, and etliyl acetate (50 mL) was added. The organic phase was washed with brine, dried and concentrated to afford 6-methylbiphenyl-3-amine (236 mg) as a yellow solid. MS(ES+) m/z 184 (MH+).
Step 2: A mixture of [4-(ethylsulfonyl)phenyl] acetic acid (intermediate la, 350 mg), HOBt
(391 mg), EDC (490 mg) and 6-methylbiphenyl-3 -amine (234 mg) in DCM (6 mL) was stirred at room temperature overnight. Solvent was removed, and water (30 mL) was added. The mixture was extracted with ethyl acetate (30 mL), and the organic phase was washed with brine, dried, and concentrated. Purification by flash chromatography (EtOAc:PE = 1 :4) afforded 2-(4- (ethylsulfonyl)phenyl)-N-(6-methylbiphenyl-3-yl)acetamide (450 mg) as a yellow solid. !H-NMR
(400 MHz, CDC13) δ ppm 1.10 (t, J= 7.6 Hz, 3H), 2.21 (s, 3H), 3.10 (q, J= 7.6 Hz, 2H), 3.79 (s, 2H), 7.21 (d, J= 8.4 Hz, 1H), 7.36 (m, 7H), 7.53 (d, J= 8.4 Hz, 2H), 7.88 (d, J= 8.4 Hz, 2H); MS(ES÷) m/z 394 (MH .
Example 12
2-(4-(ethylsulfonyl)phenyl)-jV-(6-isopropylbiphenyl-3-yl)acetamide

Step 1: Bromine (1.4 g) was added dropwise to a mixture of l-isopropyl-4-nitrobenzene (1.4 g), silver sulfate (1.3 g) and concentrated sulfuric acid (2 mL) in water (2 mL) at room tem erature over 5 mins. The reaction mixture was stirred for 4 hours, and then poured into ethyl acetate (80 mL). The organic layer was washed with brine, dried and concentrated. The residue was purified by flash chromatography (EtOAc:PE = 3: 100) to give 2-bromo-l-isopropyl-4-nitrobenzene (1.8 g) as a pale yellow liquid.
Step 2: A mixture of 2-bromo-l -isopropyl-4-nitrobenzene (1.6 g), phenylboromc acid (1.6 g), Pd(dppf)Cl2 (0.3 g) and cesium carbonate (4.3 g) in NN-dimeihylformamide (6.0 mL) and water (3 mL) was stirred at room temperature overnight. The mixture was filtered through celite and silica gel. The filtrate was concentrated, and the residue was purified by flash chromatography (PE) to afford 2- isopropyl-5-nitrobiphenyl (1 ,2 g) as a colorless liquid.
Step 3: To a solution of 2-isopropyl-5-nitrobiphenyl (108 mg) in methanol (15 mL) was added ammonium chloride (239 mg) in water (15 mL). The mixture was heated to 60°C, and zinc (117 mg) was added portionwise. The reaction mixture was stirred for 3 hours. Solid was removed by filtration. The filtrate was concentrated. The residue was dissolved in ethyl acetate. The obtained solution was washed with brine, dried and concentrated. The crude product was purified by reverse phase flash chromatography (MeCN : H20 = 65:35) to afford 6-isopropylbiphenyl-3-amine (80 mg) as a colorless liquid. MS(ES+) m/z 212 (MH÷).
Step 4: A mixture of [4 -(ethylsulfonyl)phenyl] acetic acid (intermediate la, 240 mg), HOBt (322 mg), EDC (403 mg) and 6-isopropylbiphenyl-3-amine (222 mg) in DCM (6 mL) was stirred at room temperature overnight. Solvent was removed, and the residue was dissolved in ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by flash chromatography (EtOAc:PE = 1 :4) to afford 2-(4-
(ethylsuIfonyl)phenyl)-N-(6-isopropylbiphenyl-3-yI)acetamide (402 mg) as a pale yellow solid. Ή- NM (400 MHz, DMSO- ) δ ppm 1.09 (m, 9H), 2.90 (m, 1H), 3.28 (q, J= 7.2 Hz, 2H), 3.78 (s, 2H), 7.25 (d, J= 7.2 Hz, 2H), 7.37 (m, 2H), 7.44 (m, 3H)} 7.53 (dd, J= 1.2 Hz, 8.4 Hz, 1H), 7.59 (d, J= 8.4 Hz, 2H), 7.83 (d, J= 8.4 Hz, 2H), 10.25 (s, 1H); MS(ES^) m/z 422 (MH+).
Example 13
2-(4-(ethylsulfonyl)phenyl)-iV-(3'-hydroxybiphenyI-4-yl)acetamide

A mixture ofN-(4-bromophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate lb, 330 mg), 3-hydroxyphenylboronic acid (119 mg), Pd(dppf)Cl2-CH2Cl2 (31.6 mg) and sodium carbonate (274 mg) in 1,4-dioxane (20 mL) and water (20 mL) was stirred at 80°C overnight. The mixture was filtered. The filtrate was partitioned between EtOAc (40 mL) and water (40 mL). The organic phase was washed with water (40 mL x 2), dried and concentrated. The residue was purified by flash chromatography (EtOAc.PE = 1:4) to give 2-(4-(ethylsulfonyl)phenyl)-N-(3'-hydroxybiphenyl-4- yl)acetamide (70 mg). Ή-NMR (400 MHz, OMSO-d6) δ ppm 1.19 (t, J= 7.6 Hz, 3H), 3.30 (q, /= 7.6 Hz, 2H), 3.84 (s, 2H), 6.74 (d, J= 8 Hz, 1H), 7.05 (d, J~ 8.0 Hz, 1H), 7.24 (t, J= 8.0 Hz, IH), 7.57 (d, J= 8.0 Hz, 2H), 7.64 (d, J= 8.0 Hz, 2H), 7.69 (d, J= 8.0 Hz, 2H), 7.87 (d, J= 8.0 Hz, 2H), 9.51 (s, IH), 10.38 (s, 1H); MS(ES^ m/z 396 (MH+).
Example 14
Ar-(2-chIoro-2,-fluoro-6'-(trifluoromethoxy)biphenyI-4-yl)-2-(4-(ethyIsulfonyl)phenyl)acetamide

Step 1: To a solution of l-fluoro-3-(trifluoromethoxy)benzene (2 g) in THF (20 mL) at -78°C was added BuLi (4.89 mL) dropwise. The solution was stirred at -78°C for 30 mins. Iodine (4.23 g) in THF (10 mL) was added slowly. The reaction mixture was warmed to room temperature and then quenched with a solution of Na2COj in saturated Na2S203 solution (50 mL). The mixture was extracted with ethyl acetate (40 mL). The organic phase was dried over Na2S04, filtered through a short silica gel column, washed with petroleum ether, and concentrated to give l-fluoro-2-iodo-3- (trifluoromethoxy)benzene (1.1 g) as a colorless oil. Ή-NMR (400 MHz, CDC13) 6 ppm 7.05 (m, IH), 7.12 (m, lH), 7.39 (m, IH).
Step 2: A mixture ofN-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 260 mg), l-fluoro-2-iodo-3- (trifluoromethoxy)benzene (172 mg), Pd(Ph3P)4 (64.8 mg) and potassium carbonate (155 mg) in 1 ,4- dioxane (5 mL) and water (1 mL) was sealed in a vessel and heated in the microwave at 100°C for 1 hour. Solvent was removed, and the residue was purified by MDAP to give N-(2-chloro-2'-fluoro-6'- (trifluorometlioxy)biphenyl-4-yl)-2-(4-(emylsulfonyI)phenyl)acetamide (52 mg) as a white solid. *H MR (400 MHz, DMSO-rf6) δ ppm 1.11 (t, J~ 7.2 Hz, 3H), 3.28 (q, J = 1.2 Hz, 2H), 3.86 (s, 2H), 7.41 (m, 3H), 7.61 (m, 4H), 7.86 (d, 7= 8.0 Hz, 2H), 8.00 (d, J= 1.6 Hz, 1H), 10.65 (s, 1H); 19F- NMR (376 MHz, DMSO-t¾ δ ppm -110.79, -56.51; MSfES^ m/z 516 (MH+). Example 15
iV-(2-chIoro-6'-(trifluoromethoxy)biphenyI-4-yl)-2-(2-(ethyJsulfonyl)phenyl)acetamide

Intermediate 15a: 2-(2-fethylsulfonyl')phenyl'¾acetic acid
Step 1: To a suspension of indoIin-2-one (5 g) in water (50 mL) was added NaOH (6 g). The mixture was stirred overnight at 100°C. The pH was adjusted to 1 with concentrated hydrochloric acid to give a solution of 2-(2-aminophenyl)acetic acid in water. MS(ES+) m/z 152 (MH+).
Step 2: A solution of sodium nitrite (2.56 g) in 20 mL of water was added dropwise to a suspension of the above solution of 2-(2-aminophenyl)acetic acid in 20 mL of water and 2.7 mL of concentrated hydrochloric acid cooled at 0°C. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise at room temperature to a mixture of potassium O-ethyl carbonodithioate (6.88 g), 20 mL of water and 28 mL of 2 M sodium carbonate solution. Heating was effected to 45 °C until gas evolution stops. The mixture was subsequently cooled to room temperature, and the pH was adjusted to 1 with concentrated hydrochloric acid. The oiled xanthogenate ester was extracted with ether, Removal of solvent gave 2- (2-(ethoxycarbonothioylthio)phenyl) acetic acid (9.5 g) as a dark red liquid. MS(ES÷) m/z 257 (MH+).
Step 3: 2-(2-(Ethoxycarbonothioylthio)phenyl)acetic acid (9.5 g) was taken up in 40 mL of ethanol. A solution of KOH (6.24 g) in 30 mL of water was added. Boiling at reflux was effected for
20 hours. The major portion of ethanol was subsequently removed by distillation at reduced pressure. The aqueous phase was cooled with ice, and was rendered acid with concentrated hydrochloric acid while stirring. The solution was extracted with diethyl ether (100 mL x 5). The combined organic phases were washed with brine, dried over anhydrous sodium sulphate, filtered, and concentrated to afford 2-(2-mercaptophenyl)acetic acid (6 g). MS(ES+) m/z 169 (MH+).
Step 4: To a solution of 2-(2-mercaptophenyl)acetic acid (6 g) in DMF (80 mL) was added K2C03 (34.5 g) and bromoethane (19.43 g). The reaction mixture was stirred at 30°C overnight. The reaction mixture was partitioned between ethyl acetate (300 mL) and water (100 mL). The organic phase was washed with water (100 mL x 4), brine (200 mL), dried over sodium sulfate, filtered, and concentrated to give ethyl 2-(2-(etliylthio)phenyl)acetate (6 g) as a brown solid. Ή-NMR (400 MHz, DMSO-i4) 5 ppm 1.16 (t, J= 9.6 Hz, 3H), 2.91 (q, J- 9.6 Hz, 2H), 3.78 (s, 2H), 4.08 (q, J= 9.6 Hz, 2H), 7.18 (m, 1H), 7.29 (m, 2H), 7.41 (m, 1H); MS(ES+) m/z 225 (MH+).
Step 5: Ethyl 2-(2-(efhylthio)phenyl)acetate (6 g) was dissolved in DCM (50 mL), and the solution was cooled to 0°C with an ice bath. mCPBA (13.85 g) was added in portions. After stirring at room temperature overnight, the reaction mixture was filtered to remove the solid. The filtrate was washed with sat. sodium carbonate solution (50 mL x 2), water (50 mL), brine (150 mL), dried over sodium sulfate, filtered, and concentrated. The residue was purified by column chromatography (silica gel; EtOAc:PE = 1:6) to afford ethyl 2-(2-(ethylsulfonyl)phenyl)acetate (2.5 g) as a yellow solid. 1 H-NMR (400 MHz, DMSO-rf6) 5 ppm 1.10 (t, J= 7.2 Hz, 3H), 1.18 (t, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 4.09 (q, J= 7.2 Hz, 2H), 4.12 (s, 2H), 7.53 (dd, J= 1.2 Hz,7.6 Hz, 1H), 7.58 (td, J= 1.2 Hz, 7.6 Hz, 1H), 7.71 (td, J= 1.2 Hz, 7.6 Hz, 1H), 7.91 (dd, J= 1.2 Hz, 7.6 Hz, 1H); MS(ES+) m/z 257 (MH .
Step 6: To a solution of ethyl 2-(2-(ethylsulfonyl)phenyl)acetate (3.4 g) in ethanol (20 mL) was added a solution of NaOH (2, 122 g) in water (20 mL). The reaction mixture was stirred at room temperature overnight. Ethanol was removed under reduced pressure, and 10 mL of water was added to the aqueous solution. The aqueous phase was washed with dichloromethane (30 mL x 3), and then acidified with 6 M HC1 to pH = 1. The solution was extracted with ethyl acetate (1000 mL x 3). The combined organic phases were washed with brine (50 mL), dried over sodium sulphate, filtered, and concentrated to give 2-(2-(ethylsulfonyl)phenyl)acetic acid (2 g) as a light yellow solid. Ή-NMR (400 MHz, D SO-i¾) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 4.06 (s, 2H), 7.55 (m, 2H), 7.70 (td, J= 1.2Hz, 7.6 Hz, 2H), 7.90 (dd, J~ 1.2 Hz, 8.0 Hz, 2H), 12.48 (s, 1H); MStES") m/z 229 (MH+).
Intermediate 15b: 2-chloro-6'-(trifluoromethoxy)biphenYl-4-amine
A mixture of 4-bromo-3-chlorobenzenamine (2 g), 2-(trifluoromethoxy)phenylboronic acid (5.98 g), Pd(PhjP)4 (1.12 g) and potassium carbonate (4.02 g) in 1,4-dioxane (50 mL) and water (50 mL) was refluxed overnight. The mixture was filtered, and to the filtrate was added EtOAc (150 mL). The organic phase was washed with water (100 mL x 2) and brine, dried and concentrated. The residue was purified by flash chromatography (EtOAc:PE = 1 :7) to give 2-chloro-6'- (trifluoromethoxy)biphenyl-4-amine (1.2 g) as a yellow oil. MSiES ) m/z 288 (MH+).
Preparation of N-f2-chloro-6'-('trifluoromethoxy'lbiphenyl-4-yl')-2-f2-fethylsulfonyl')phenyl')acetamide A mixture of 2-chloro-6'-(trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 300 mg), 2-(2- (ethylsulfonyl)phenyl)acetic acid (intermediate 15a, 309 mg), HOBt (192 mg), EDC (600 mg) and DIPEA (0.55 mL) in DCM (20 mL) was refluxed overnight. The mixture was washed with water (50 mL x 3) and brine, dried and concentrated. The residue was purified by prep-HPLC to give N-(2- chloro-6,-(trifluoromethoxy)biphenyl^-yl)-2-(2-(ethylsulfonyI)phenyl)acetamide (100 mg) as a white solid. Ή NMR (400 MHz, DMSO-if6) δ ppm 1.13 (t, J= 7.2 Hz. 3H), 3.35 (q, J= 7.2 Hz, 2H), 4.26 (s, 2H), 7.31 (d, J= 8.0 Hz, 1H), 7.36 (d, J= 6.8 Hz, 1H), 7.48 (m, 2H), 7.57 (m, 4H), 7.73 (t, J— 7.2 Hz, 1H), 7.93 (m, 2H), 10.56 (s, 1H); MS(ES+) m/z 498 (MH+).
Example 16
Ar-(2-chloro-6'-(trifluoromethoxy)biphenyl-4-yl)-2-(3-(ethyls lfonyl)phenyl)acetamide

Intermediate 16a: 2- 3- 6ίηνΐ5υΙ¾ην1) 1ΐ6ην1¼ΰ£ύο acid
Step 1: A solution of sodium nitrite (0.916 g) in 20 mL of water was added dropwise to a suspension of 2-(3-aminophenyl)acetic acid (2 g) in 20 mL of water and 2.7 mL of concentrated hydrochloric acid cooled at 0 °C. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise to a mixture of potassium 0-ethyl carbonoditliioate (2.456 g), 20 mL of water and 10 mL of a 2 M sodium carbonate solution at room temperature. The reaction mixture was heated at 45°C until gas evolution stops. After cooling to RT, pH was adjusted to 1 with concentrated hydrochloric acid. The oiled xanthogenate ester was extracted with diethyl ether. Solvent was evaporated to give 2-(3- (ethoxycarbonothioylthio)phenyl)acetic acid (4.8 g) as a dark red liquid. MS(ES+) m/z 257 (Mtf").
Step 2: 2-(3-(Ethoxycarbonomioylthio)phenyl)acetic acid (4.8 g) was taken up in 50 mL of ethanol, and a solution of KOH (1.051 g) in 50 mL of water was added. The reaction mixture was heated at reflux for 20 hours. The major portion of ethanol was subsequently removed by distillation at reduced pressure. The aqueous phase was cooled with ice, and was rendered acid with concentrated hydrochloric acid while stirring. The solution was extracted with diethyl ether (100 mL). The organic phase was washed with brine, dried over anhydrous sodium sulphate, filtered, and concentrated to afford 2-(3-mercaptophenyl)acetic acid (3.3 g) as a brown solid. MS(ES+) m/z 169 (MH+).
Step 3: To a solution of 2-(3-mercaptophenyl)acetic acid (3.3 g) in DMF (50 mL) was added 2C03 (10.85 g) and bromoethane (4.39 mL). The reaction mixture was stirred at room temperature for 2.5 hours. The reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL). The organic was washed with water (100 mL x 4) and brine (200 mL), dried with sodium sulfate, filtered, and concentrated to give ethyl 2-(3-(ethylthio)phenyl)acetate (2.76 g) as a pale yellow solid. Ή-NMR (400 MHz, DMSO-i¾) δ ppm 1.16 (t, 7= 9.6 Hz, 3H), 1.23 (t, J= 9.6 Hz, 3H), 2.97 (q, J= 9.6 Hz, 2H), 3.65 (s, 2H), 4.08 (q, J= 9.6 Hz, 2H), 7.07 (d, J= 9.6 Hz, 1H), 7.24 (m, 3H); MS(ES+) m/z 225 (MH+).
Step 4: Ethyl 2-(3-(ethylthio)phenyl)acetate (2.76 g) was dissolved in DCM (50 mL), and the solution was cooled to 0°C with an ice bath. mCPBA (6.37 g) was added in portions, and the reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the solid. The filtrate was washed with sat. sodium carbonate solution (50 mL x 2), water (50 mL), brine (150 mL), dried over sodium sulfate, filtered, and concentrated. The residue was purified by column chromatography (EtOAc : PE = 1 :6) to afford the target compound ethyl 2-(3-
(ethylsulfonyl)phenyl)acetate (2.2 g) as a yellow oil. Ή-NMR (400 MHz, DMSO-rffi) δ ppm 1.10 (t, J = 7.2 Hz, 3H), 1.19 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 4.10 (q, J= 7.2 Hz, 2H), 7.63 (m, 2H), 7.80 (m, 2H); MS(ES^) m/z 257 (MH+).
Step 5: To a solution of ethyl 2-(3-(ethylsulfonyl)phenyl)acetate (2.2 g) in ethanol (20 mL) was added a solution of NaOH (1.373 g) in water (20 mL). The reaction mixture was stirred at room temperature overnight, Ethanol was removed under reduced pressure, and 10 mL of water was added to the aqueous solution. The aqueous phase was washed with dichloromethane (30 mL x 3), and then acidified with 6 M HC1 to pH = 1. The solution was extracted with ethyl acetate (50 mL x 3). The combined organic phases were washed with brine (50 mL), dried over sodium sulphate, filtered, and concentrated. The residue was purified by column chromatography (MeOH : DCM = 1:20) to give 2- (3-(ethylsulfonyl)phenyl)acetic acid (1.9 g) as a light yellow oil. Ή-NMR (400 MHz, DMSO-c/6) δ ppm 1.10 (t, J = 7.2 Hz, 3H), 3.29 (q, J= 7.2 Hz, 2H), 3.76 (s, 2H), 7.63 (m, 2H), 7.78 (m, 2H), 12.49 (s, 1H); MSfES" m/z 229 (MH ).
Preparation of N-f2-chlQro-6'-(,trifluoromethoxy^biphenyl-4-ylV2-f3-('ethylsulfonyl)phenyl)acetarnide A mixture of 2-chloro-6'-(trifluoromethoxy)biphenyI-4 -amine (intermediate 15b, 300 mg), 2-(3- (ethyIsulfonyl)phenyI)acetic acid (intermediate 16a, 309 mg), HOBt (192 mg), EDC (600 mg) and DIPEA (1.10 mL) in DCM (20 mL) was refluxed overnight. The mixture was washed with water (30 mL x 3) and brine (30 mL), dried and concentrated. The residue was purified by prep-HPLC to give N-(2-chloro-6'-(trifluoromethoxy)biphenyI-4-yl)-2-(3-(ethylsulfonyl)phenyl)acetamide (30 mg) as a white solid. Ή NM (400 MHz, CDC13) δ ppm 1.21 (t, J= 7.2 Hz, 3H), 3.08 (q, J= 1.2 Hz, 2H), 3.71 (s, 2H), 7.10 (d, J= 8.4 Hz, IH), 7.22 (m, 3H), 7.36 (m, 2H), 7.47 (t, J= 7.6 Hz, IH), 7.62 (d, J = 7.6 Hz, IH), 7.69 (s, IH), 7.75 (d, J= 8.0 Hz, IH), 7.83 (s, IH), 8.16 (s, IH); MS(ES^) m/z 498 (MH+).
Example 17
N-(2-chloro-6'-(trifluoromethoxy)bipheny]-4-yl)-3-(4-(ethylsulfonyl)phenyl)propanamide

Intennediate 17a: 3-(4-(ethylsulfonyl')phenyl')propanoic acid
Step 1 : A solution of NaN02 (0.42 g) in 6 mL of water was added dropwise at 0°C, while stirring, to a suspension of 3-(4-aminophenyl)propanoic acid (1.0 g) in 3 mL of water and 3 mL of concentrated hydrochloric acid. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise at room temperature to a mixture of potassium Oethyl carbonodithioate (1.16 g), 2 mL of water and 5 mL of 2 M sodium carbonate solution. The mixture was stirred at 45 °C until gas evolution stopped. The mixture was subsequently cooled to room temperature, and pH was adjusted to 1 with concentrated hydrochloric acid. The oiled xanthogenate ester was extracted with EtOAc. Solvent was evaporated to give 3-(4-(ethoxycarbonothioylthio)phenyl)propanoic acid (1.2 g) as a dark yellow liquid. MS(ES^) m/z 271 (MH+).
Step 2: A mixture of 3-(4-(ethoxycarbonothioylthio)phenyl)propanoic acid (1.2 g) and potassium hydroxide (1.85 g) in ethanol (20 mL) and water (20 mL) was refluxed overnight. Ethanol was removed. The aqueous phase was cooled with ice, and acidified with concentrated hydrochloric acid while well stirring. The mixture was extracted with DCM (40 mL x 2). The combined organic
phases were washed with brine, dried over anhydrous sodium sulphate, filtered, and concentrated to afford 3-(4-mercaptophenyl)propanoic acid (1.0 g) as a yellow solid. MS(ES+) m/z 183 (MH^).
Step 3: A mixture of 3-(4-mercaptophenyl)propanoic acid (1.0 g), K2C03 (3.03 g) and bromoethane (1.23 mL) in DMF (10 mL) was stirred at RT for 2.5 hours. The reaction mixture was partitioned between ethyl acetate (50 mL) and water (100 mL). The organic phase was washed with water (50 mL x 3) and brine (50 mL), dried with sodium sulphate, filtered, and concentrated to give ethyl 3-(4-(ethyltmo)phenyl)propanoate (1.2 g) as a yellow oil. MS(ES+) m/z 239 (MH+).
Step 4: To a solution of ethyl 3-(4-(ethylthio)phenyl)propanoate (1.2 g) in DCM (30 mL) was added mCPBA (2.61 g). The reaction mixture was stirred at RT overnight. Solid was removed by filtration. The filtrate was washed with sat. sodium carbonate solution (40 mL x 2), water (40 mL), brine (50 mL), dried over sodium sulphate, filtered, and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1: 1) to afford ethyl 3-(4- (ethylsulfonyl)phenyl)propanoate (400 mg) as a white solid. MS(ES^ m/z 271 (MH+).
Step 5: A mixture of ethyl 3-(4-(ethylsulfonyl)phenyl)propanoate (400 mg) and sodium hydroxide (60 mg) in ethanol (20 mL) and water (20 mL) was stirred at RT for 1 hour. Ethanol was removed, and the aqueous solution was acidified to pH = 1 with cone. HC1. The mixture was extracted with EtOAc (40 mL x 3). The combined organic phases were dried over anhydrous sodium sulfate, filtered, and concentrated to yield 3-(4-(ethylsulfonyl)phenyl)propanoic acid (250 mg) as a white solid. MS(ES+) m/z 243 (MH+).
Preparation of N-(2-chloro-6'-(trifluoromethoxy)biphenyl-4-yl)-3 -(4- (ethylsulfonvPphenvDpropanamide
A mixture of 3-(4-(emylsulfonyl)phenyl)propanoic acid (intermediate 17a, 200 mg), 2-chloro-
6r-(trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 237 mg), HOBt (152 mg), EDC (475 mg) and DIPEA (0.87 mL) in DCM (20 mL) was refluxed overnight. The mixture was washed with water (30 mL x 3) and brine (30 mL), dried and concentrated. The residue was purified by prep-HPLC to afford N-(2-chloro-6'-(trifluoromethoxy)biphenyl-4-yl)-3-(4-(ethylsulfonyl)phenyl)propanamide (50 mg) as a white solid. Ή NMR (400 MHz, CDC13) δ ppm 1.27 (t, J= 7.2 Hz, 3H), 2.71 (t, /= 7.6 Hz,
2H), 3.12 (m, 4H), 7.21 (d, J= 8.0 Hz, 1H), 7.39 (m, 7H), 7.77 (m, 4H); MS(ES+) m/z 512 (MH+).
Example 18
7V-(2-chloro-2'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)- V-methylacetamide

Step 1: Formic acid (1 mL) was added dropwise to acetic anhydride (2 mL) in an ice bath. After the addition, the ice bath was removed and the solution was stirred at 50°C for 30 minutes. The mixture was then cooled in an ice bath, to which was added dropwise a cold solution of 2-chloro-6'- (trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 400 mg) in formic acid (1 mL). The reaction mixture was stirred for 40 minutes at this temperature. The mixture was then wanned to room temperature and stirred for additional 80 minutes. Solvent was removed, and DCM (10 mL) was added. Solid was removed by filtration. The filtrate was dried and concentrated to give N-(2-chloro- 6'-(trifluorometlioxy)biphenyI-4-yl)fonnamide (380 mg) as a brown oil. MS(ES+) m/z 316 (MIT).
Step 2: Borane-tetrahydrofuran complex (20 mL) was added dropwise to a solution of N-(2- chloro-6,-(trifluoromethoxy)biphenyI-4-yl)formamide (480 mg) i THF (20 mL) at 0°C in 30 mins. The reaction mixture was warmed to 80°C and stirred overnight. The mixture was cooled to 0°C and quenched with brine. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate (100 mL x 2). The combined organic phases were dried and concentrated to give 2- chloro-N-methyl-6'-(trifiuoromethoxy)biphenyl-4-amine (500 mg) as a brown oil. MS(ES^) m/z 302 (MH+).
Step 3: A mixture of 2-chloro-N-methyl-6,-(trifluoromethoxy)biphenyI-4-amine (500 mg), [4- (ethylsulfonyl)phenyl] acetic acid (intermediate la, 454 mg), EDC (953 mg), HOBt (279 mg) and DIPEA (2 mL) in DCM (40 mL) was stirred at room temperature overnight. The organic solution was washed with water (50 mL) and brine (80 mL), dried and concentrated. The residue was purified by prep-HPLC to give N-(2-chloro-2,-(trifluoromethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)-N- methylacetamide (100 mg) as a pale yellow oil. Ή-NMR (400 MHz, CDC13) δ ppm 1.27 (t, J= 7.2 Hz, 3H), 3.10 (q, J= 7.2 Hz, 2H), 3.33 (s, 3H), 3.63 (s, 2H), 7.12 (d, J= 7.6 Hz, 1H), 7.21 (s, 1H), 7.30 (m, 2H), 7.35 (d, J= 8.0 Hz, 2H), 7.40 (m, 2H), 7.50 (m, 1H), 7.81 (d, J= 8.0 Hz, 2H); MS(ES ) m/z 512 (MH4 .
Example 19
iV-(2-chloro-2'-phenoxybiphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyI)acetamide

Step 1: A mixture of 2-phenoxyphenylboronic acid (1.0 g), 4-bromo-3-chlorobenzenamine (1.16 g), Na2C03 (0.99 g) and Pd(Ph3P) (0.54 g) in 1,4-dioxane (20 mL) and water (20 mL) was stirred at 100°C overnight. The reaction mixture was filtered througli celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by flash chromatography (EtOAc : PE = 1:4) to afford 2-chloro-2'-phenoxybiphenyl-4-amine (450 mg) as a yellow solid.

Step 2: A mixture of 2-chloro-2'-phenoxybiphenyl-4-amine (200 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 232 mg), HOBt (104 mg), DIPEA (0.354 mL) and EDC (389 mg) in tetrahydrofuran (THF) (10 mL) was stirred at 60°C overnight. The mixture was concentrated under reduced pressure, and the residue was purified by prep-HPLC to afford N-(2- chloro-2'-phenoxybiphenyI-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (67 mg) as a white solid. Ή- NMR (400 MHz, DMSO-i¾ 6 ppm 1.09 (t, J= 7.6 Hz, 3H), 3.27 (q, J= 7.6 Hz, 2H), 3.82 (s, 2H), 6.88 (d, J= 8.4 Hz, 2H), 6.95 (d, J= 8.4 Hz, 1H), 7.05 (t, J= 7.6 Hz, 1H), 7.28 (m, 5H), 7.40 (t, J= 7.6 Hz, 1H), 7.46 (d, J= 8.0 Hz, 1H), 7.60 (d, /= 8.0 Hz, 2H), 7.84 (m, 3H), 10.05 (s, 1H); MS(ES+) mfz 506 (MH4).
Example 20
jV-(2-chloro-2'-(2,2,2-trifluoroethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyI)acetamide

Step 1: To a solution of 2-bromophenol (0.2 g) and l,l,l-trifluoro-2-iodoethane (0,267 g) in DMF (6 mL) was added K2C03 (0.320 g). The reaction mixture was heated to 110°C overnight. The reaction mixture was partitioned between ethyl acetate (30 mL) and water (20 mL). The organic phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and concentrated to give l-bromo-2-(2,2,2-trifluoroethoxy)benzene (0.25 g) as a yellow oil.
Step 2: A mixture ofN-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsuIfonyl)phenyl]acetamide (intermediate 6b, 218 mg), l-bromo-2-(2,2,2- trifluoroethoxy)benzene (120 mg), K2C03 (163 mg) and Pd(Ph3P)4 (54.4 mg) in 1 ,4-dioxane (5 mL) and water (1 mL) was heated at reflux overnight. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (EtOAc : PE = 1:2) to give the crude product. Further purification by MDAP afforded N-(2-chloro-2'-(2,2,2-triflnoroethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (45 mg) as a white solid. Ή- MR (400 MHz, DMSO^) 5 ppm 1.11 (t, J= 7.2 Hz, 3H), 3.26 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 4.70 (q, J= 8.4 Hz, 2H), 7.11 (m, 1H), 7.22 (m, 3H), 7.40 (m, 1H), 7.50 (d, J= 8.4 Hz, 1H), 7.62 (d, J= 8.0 Hz, 2H), 7.85 (d, J= 8.0 Hz, 2H), 7.89 (s, 1H), 10.50 (s, 1H);
MS(ES+) m/z 512 (MH+).
Example 21
JV-(2'-butoxy-2-chlorobiphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 2: A mixture of l-bromo-2-butoxybenzene (45 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethyIsulfonyl)phenyl]acetamide (intermediate 6b, 91 mg), K2C03 (27.1 mg) and Pd(Ph3P) (227 mg) in 1,4-dioxane (10 mL) and water (2 mL) was stirred at 100°C overnight. The mixture was filtered tlirougli celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by prep-HPLC to give N-(2'-butoxy-2- chlorobiphenyl-4-yl)-2-(4-(etliylsulfonyl)phenyI)acetamide (68 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-c/6) 6 ppm 0.82 (t, J= 7.2 Hz, 3H), 1.11 (t, J= 7.2 Hz, 3H), 1.28 (m, 2H), 1.54 (m, 2H), 3.29 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 3.94 (t, 6.0 Hz, 2H), 6.99 (t, J= 7.4 Hz, 1H), 7.07 (d, J=
8.4 Hz, 1H), 7.12 (d, J= 6.4 Hz, 1H), 7.24 (d, J= 8.4 Hz, 1H), 7.35 (t, J- 7.6 Hz, 1H), 7.49 (dd, J= 1.2 Hz, 8.4 Hz, 1H), 7.63 (d, /= 8.0 Hz, 2H), 7.87 (m, 3H), 10.49 (s, 1H); MS(ES^| m/z 486 (MH+).
Example 22
Ar-(2-chloro-2'-(2-(dimethylamino)ethoxy)biphenyl-4-yl)-2-(4-(ethyIsuIfonyl)phenyl)acetamide

Step 1: To a solution of 2-bromophenol (1.0 g) in toluene (30 mL) cooled at 0°C was added 2- (dimethylamino)ethanol (0.515 g), triphenylp osphine (1.819 g) and D1AD (1.349 mL). The reaction mixture was stirred at room temperature overnight. The solid was removed by filtration, and the filtrate was concentrated to give 2-(2-bromophenoxy)-N,N-dimethylethanamine (1.5 g) as a colorless oil. SiES*) m/z 245 (Mtf).
Step 2: A mixture of 2-(2-bromophenoxy)-NsN-dimethylethanamine (200 mg), N-[3-chloro-4- (4,4,5, -tetramethyl-l,3,2-dioxaborolan-2-yl)phenyI]-2-[4-(ethylsulfonyl)phenyl]acetamide
(intennediate 6b, 380 mg), Pd(Ph3P)4 (95 mg) and K2C03 (283 mg) in 1,4-dioxane (20 mL) and water (3 mL) was stirred at 100°C under N2 overnight. After concentration, the residue was partitioned between EtOAc (60 mL) and water (60 mL). The aqueous phase was extracted with EtOAc (60 mL x 2). The combined organic phases were washed with brine (60 mL), dried over anhydrous Na2S04, and concentrated. The residue was purified by prep-HPLC to give N-(2-chloro-2'-(2- (dimethylamino)ethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (36 mg) as a light yellow solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.31 (t, J= 7.2 Hz, 3H), 2.26 (s, 6H), 2.70 (t, J= 5.2 Hz, 2H), 3.14 (q, J= 7.2 Hz, 2H), 3.83 (s, 2H), 4.11 (t, /= 5.2 Hz, 2H), 6.97 (d, 8.4 Hz, 1H), 6.98 (m, 1H), 7.16 (d, J= 8.4 Hz, 1H), 7.23 (d, J= 8.4 Hz, 1H), 7.33 (m, 2H), 7.42 (br s, 1H), 7.58 (d, J= 8.4 Hz, 2H), 7.72 (s, 1H), 7.93 (d, J= 8.4 Hz, 2H); MS(ES+) m/z 501 (MH+).
Example 23
N-(2-chloro-2'-morpholinobiphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 1: To a solution of 2-morpholinobenzenainine (500 mg) in acetonitrile (30 mL) was added /er/-butyl nitrite (579 mg) and copper(II) bromide (752 mg). The mixture was stirred at room temperature for 2 hours. The mixture was partitioned between water (60 mL) and EtOAc (60 mL). The aqueous phase was extracted with EtOAc (60 mL x 2). The combined organic phases were dried and concentrated. The residue was purified by flash chromatography (EtOAc : PE - 1 : 10) to give 4- (2-bromophenyl)morpholine (300 mg) as a brown oil. MSfES m/z 242 (MH+).
Step 2: A mixture of 4-(2-bromophenyl)morpholine (200 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 383 mg), Pd(Ph3P)4 (95 mg) and 2C03 (285 mg) in 1 ,4-dioxane (20 mL) and water (3 mL) was stirred at 90°C under N2 for 1 hour. After removal of solvent, the residue was partitioned between EtOAc (60 mL) and water (60 mL). The aqueous phase was extracted with EtOAc (60 mL x 2). The combined organic phases were washed with brine (60 mL x 2), dried and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 1 :3 to 1: 1) to give the crude product, which was triturated with PE and EA (100: 1, 20 mL x 2) to give N-(2-chloro-2'-morpholinobiphenyl-4-yI)-2-(4- (ethylsulfonyl)phenyl)acetamide (60 mg) as a white solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.31 (t, /= 7.6 Hz, 3H), 2.81 (m, 4H), 3.14 (q, J= 7.6 Hz, 2H), 3.51 (m, 4H), 3.84 (s, 2H), 7.10 (m, 2H), 7.17 (d, J = 7.2 Hz, 1H), 7.38 (m, 4H), 7.58 (d, J= 8.0 Hz, 2H), 7.74 (s, 1H), 7.92 (d, J= 8.2 Hz, 2H); MS(ES+) m/z 499 (MH+). Example 24
N-(2-chloro-2'-(cyclopropylmethoxy)biphenyI-4-yl)-2-(4-(ethy]suIfonyl)phenyI)acetamide

Step 1: To a solution of 2-bromophenol (1 g) and (bromomethyl)cyclopropane (0.78 g) in acetone (6 mL) was added 2C03 (1.60 g). The reaction mixture was heated to reflux overnight. The reaction mixture was partitioned between ethyl acetate (40 mL) and water (20 mL). The organic
phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and concentrated to give l-bromo-2-(cyclopropylmethoxy)benzene (0.95 g) as a yellow oil.
Step 2: A mixture of N-[3-chloro-4-(4,4,5,5-tetramethyI-l,3,2-dioxaboroIan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 204 mg), l-bromo-2- (cyclopropylmethoxy)benzene (100 mg), K2C03 (152 mg), and Pd(Ph3P)4 (51 mg) in 1,4-dioxane (5 mL) and water (1 mL) was heated at reflux overnight. The mixture was filtered through celite and silica gel. The filtrate was concentrated, and the residue was purified by flash chromatography (EtOAc : PE = 1:2) to give the crude product. Further purification by MDAP afforded N-(2-chloro-21- (cyclopropylmethoxy)biphenyl-4-yl)-2-(4-(etliylsulfonyl)phenyl)acetamide (57 mg) as a white solid. Ή-NMR (400 MHz, DMSO-<¾) 6 ppm 0.20 (m, 2Η), 0.43 (m, 2H), 1.05 (m, IH), 1.10 (t, J= 7.2 Hz, 3H), 3.26 (q, J= 7.2 Hz, 2H), 3.81 (d, /= 6.8 Hz, 2H), 3.84 (s, 2Η), 7.04 (m, 3Η), 7.25 (d, J= 8.4 Hz, IH), 7.32 (m, IH), 7.50 (d, J= 8.4 Hz, IH), 7.62 (d, J= 8.0 Hz, 2H), 7.85 (d, J= 8.0 Hz, 2H), 7.89 (s, IH), 10.50 (s, IH); MSfES*) m z 484 (MH+).
Example 25
N-{2-chloro-2'-[(trifluoromethyl)oxy]-4-biphenyIyl}-2-[4-(ethylsulfonyl)phenyl]acetamide
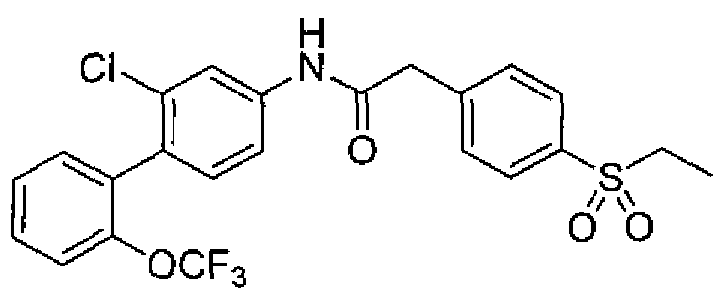
A mixture of N-(4-bromo-3-chlorophenyl)-2-[4-(ethylsulfonyI)phenyl]acetamide (intermediate 6a, 60 mg), {2-[(trifluorometliyl)oxy]phenyl}boronic acid (32.6 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2CC>3 (56.3 mg) in acetonitrile (1.5 mL)/water (0.500 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford N-{2-chloro-2'-[(trifluoromethyl)oxy]-4-biphenyIyl}-2-[4- (ethylsulfonyl)phenyl]acetamide (20 mg) as a white solid. 'H-NMR (400 MHz, DMSO-i ) 5 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, /= 7.2 Hz, 2H), 3.85 (s, 2H), 7.32 (d, J= 8.4 Hz, IH), 7.41 (m, 1H)S 7.48 (m, 2H), 7.56 (m, 2H), 7.63 (d, J= 8.4 Hz, 2H), 7.86 (d, J= 8.4 Hz, 2H), 7.94 (d, J= 2.0 Hz, IH), 10.57 (s, IH); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -56.29; MS(ES+) m/z 498 (MH+).
Example 26
AHl-chlor o-2'-(piperidin- l-yl)bip henyl-4-y I)-2-(4- (ethylsulf ony I) phenyl) ac etamide

Step 1: To a solution of 2-(piperidin-l -yl)phenol (0.5 g) and trifiuoro-N-phenyl-N- (trifluoromethylsulfonyl)methanesulfonamide (1.1 g) in dichloromethane (20 mL) was added NEt3 (0.40 mL) dropwise at 0 °C. The reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was partitioned between dichloromethane (40 mL) and water (20 mL). The organic phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 1 :10) to give 2- (piperidin-l-yl)phenyl trifluoromethanesulfonate (0.3 g). MS(ES+) m/z 310 (MH4).
Step 2: A mixture of N-[3-chloro-4-(4}4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 194 mg), 2-(piperidin-l -yl)phenyl
trifluoromethanesulfonate (130 mg), K2C03 (145 mg), and Pd(Ph3P)4 (49 mg) in 1,4-dioxane (5 mL) and water (1 mL) was stirred at reflux overniglit. The reaction mixture was filtered tlirougli celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by flash chromatography (EtOAc : PE = 1 : 1) to give the crude product. Further purification by MDAP afforded N-(2-chloro-2'-(piperidin-l -yl)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (25 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm 1.11 (t, J= 7.2 Hz, 3H), 1.28 (m, 6H), 2.68 (m, 4H), 3.28 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 7.05 (m, 3H), 7.31 (m, 2H), 7.51 (d, J= 8.4 Hz, lH), 7.62 (d, J= 8.0 Hz, 2H), 7.86 (d, J= 8.0 Hz, 2H), 7.91 (s, 1H), 10.48 (s, 1H); MS(ES+) m/z 497 (MH+). Example 27
N-(2,6-difluorobiphenyl-4-yl)-2-(4-(ethylsuIfonyl)phenyl)acetamide

Intermediate 27a: N-f4-bromo-3.5-difluorophenyl')-2-(4-(,ethylsulfonvI)phenyl')acetamide
A mixture of 4-bromo-3,5-difluorobenzenamine (2 g) in DCM (100 mL), [4- (ethylsulfonyl)phenyl] acetic acid (intermediate la, 2.63 g), HOBt (1.767 g), EDC (5.53 g) and
DIPEA (10.08 mL) was refluxed overnight. Solvent was removed, and the residue was purified by flash chromatography (DCM) to give N-(4-bromo-3,5-difluorophenyl)-2-(4- (etliyIsulfonyl)phenyl)acetamide (3 g) as a yellow solid. MS(ES+) m/z 417 (MH+).
Preparation of N-('2.6-difluorobiphenyl-4-yl')-2-<'4-(ethylsulfonyl)phenyl')acetamide
A mixture of N-(4-bromo-3,5-difluorophenyI)-2-(4-(ethylsulfonyl)phenyI)acetamide
(intermediate 27a, 200 mg), phenylboronic acid (117 mg), Pd(Ph3P)4 (27.6 mg) and potassium carbonate (132 mg) in 1,4-dioxane (20 mL) and water (20 mL) was refluxed overnight. The mixture was filtered, and to the filtrate was added water (50 mL) and EtOAc (50 mL). The organic phase was washed with water and brine, dried and concentrated. The residue was purified by prep-HPLC to give N~(2,6-difluorobiphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (23 mg) as a white solid. Ή- NMR (400 MHz, CDCI3) δ ppm 1.32 (t, J= 7.2 Hz, 3H), 3.15 (d, J= 7.2 Hz, 2H), 3.85 (s, 2H), 7.27 (m, 2H), 7.48 (m, 7H), 7.92 (d, J= 8.0 Hz, 2H); MS(ES+) m z 416 (MH+).
Example 28
iV-(2-chloro-2'-hydroxybiphenyl-4-yI)-2-(4-(ethylsulfonyl)phenyl)acetamide

A mixture of N-(4-bromo-3-chlorophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6a, 300 mg), 2-hydroxyphenylboronic acid (199 mg), K2C03 (298 mg) and Pd(Ph3P)4 (83 mg) in 1 ,4- dioxane (10 mL) and water (2 mL) was stirred at 100°C overnight. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by prep-HPLC to give N-(2-chloro-2'-hydroxybiphenyl-4-yl)-2-(4-
(ethylsulfonyI)phenyl)acetamide (65 mg) as a white solid. Ή-NMR (400 MHz, DMSO-c4) δ ppm 1.1 1 (t, J= 7.2 Hz, 3H), 3.29 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 6.83 (t, J= 7.2 Hz, 1H), 6.90 (d, J= 8.4 Hz, 1H), 7.05 (dd, J= 1.6 Hz, 7.6 Hz, 1H), 7.18 (td, J= 1.6 Hz, 8.0 Hz, 1H), 7.25 (d, J= 8.4 Hz, 1H), 7.48 (dd, J= 8.0 Hz, 8.0 Hz, 1H), 7.63 (d, J= 8.4 Hz, 2H), 7.87 (m, 3H), 9.49 (br s, 1H), 10.50 (s, 1H); MS(ES+) m z 430 (MH+).
Example 29
Ar-(2-chloro-2'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-suIfamoylphenyl)acetamide
 intermediate 29a: 2-(4-sulfamoylphenyI')acetic acid
intermediate 29a: 2-(4-sulfamoylphenyI')acetic acid
Step 1: C lorosulfonic acid (70 mL) was added into phenylacetic acid (10 g) dropwise at 0 °C. The reaction mixture was stirred at 0°C for 1 hour, and then wanned to room temperature and stirred overnight. The reaction mixture was poured onto ice and left until all ice melted. The suspension was filtered. The obtained solid was washed with cold water and dried in vacuo to afford a white solid (10 g), which contains isomers. The mixture was recrystallized from chloroform twice to give 2-(4-
(chlorosulfonyl)phenyl)acetic acid (2.2 g). Ή-NMR (400 MHz, DMSO-<¾ δ ppm 3.57 (s, 2H), 7.21 (d, J= 8.0 Hz, 2H), 7.54 (d, J= 8.0 Hz, 2H), 10.52 (s, 1H); MSiES*) m/z 235 (MH+);
Step 2: A mixture of 2-(4-(chlorosulfonyl)phenyl)acetic acid (200 mg) and ammonia solution
(7 M in methanol, 1.2 mL) in methanol (4 mL) was stirred at room temperature overnight. Solvent was removed. To the residue was added water (10 mL), and the solution was acidified with 4 M HC1 to pH = 2-3. The solid was collected by filtration, washed with water, and dried in air to give 2-(4- sulfamoylphenyI)acetic acid (120 mg) as a yellow solid. MSiES^) m/z 216 (ivfiT*).
Preparation of N-(2-chloro-2'-(trifluoroi-nethoxy')biphenyl-4-yl)-2-(4-sulfamoylphenyl')acetamide
A mixture of 2-chloro-6'-(trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 107 mg), 2-(4- suIfamoylphenyl)acetic acid (intermediate 29a, 80 mg), HOBt (68 mg) and EDC (86 mg) in DCM (5 mL) was stirred at reflux overnight. The mixture was washed with 1 M HC1, sat. NaHC03 and brine successively. The organic phase was dried over anliydrous a2S04 and concentrated. The residue was purified by MDAP to afford N-(2-chloro-2,-(trifluoromethoxy)biphenyl-4-yl)-2-(4- sulfamoylphenyl)acetamide (24 mg) as a white solid.Ή-NMR (400 MHz, DMSO-i/6) δ ppm 3.79 (s, 2H), 7.36 (m, 4H), 7.51 (m, 6H), 7.79 (d, J= 8.0 Hz, 2H), 7.94 (s, 1H), 10.59 (s, 1H); 19F-NMR (376 MHz, DMSO-<¾ δ ppm -56.27; MS(ES+) m/z 485 (MH ).
Example 30
Ar-(2-chloro-2'-(isopropoxymethyI)biphenyl-4-yl)-2-(4-(ethylsulfonyI)phenyl)acetamide

concentrated to give l-bromo-2-(bromomefhyl)benzene (1.8 g) as a yellow oil.
Step 2: To a solution of isopropanol (6 mL) was added sodium (27.6 mg). The mixture was heated to reflux until Na was completely dissolved. The mixture was cooled to RT and solvent was removed under reduced pressure. To the residue was added DMF (10 mL), followed by a solution of l-bromo-2-(bromomethyl)benzene (300 mg) in DMF (2 mL) dropwise. After stirring at RT overnight, the reaction mixture was added to ice water (10 mL), and then extrated with DCM (30 mL). The organic phase was washed with water (20 mL x 2), brine (20 mL), dried over anhydrous sodium sulfate, and concentrated to give l-bromo-2-(isopropoxymethyl)benzene (1.8 g) as a yellow oil.Ή- NMR (400 MHz, CDC13) δ ppm 1.10 (d, J = 6.0 Hz, 6H), 3.76 (m, 1H), 4.58 (s, 2H), 7.25 (m, 4H).
Step 3: To a solution of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2- [4-(ethyIsulfonyl)phenyl]acetamide (intermediate 6a, 202 mg), l-bromo-2-
(isopropoxymethyl)benzene (100 mg) and K2C03 (151 mg) in 1,4-dioxane (8 mL) and water (1 mL) was added Pd(Ph3P)4 (50.4 mg) under N2. After stirring under reflux overnight, the reaction mixture was filtered tlirough celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (EtOAc : PE = 1 : 1) to give the crude product, which was further purified by MDAP to afford N(2-chloro-21-(isopropoxymethyl)biphenyl-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (23 mg) as a white solid. Ή-NMR (400 MHz, DMSO-c¾ δ ppm 0.96 (m, 6H), 1.10 (t, J= 7.2 Hz, 3H), 3.22 (q, J= 7.2 Hz, 2H), 3.37 (m, 1H), 3.84 (s, 2H), 4.18 (m, 2H), 7.11 (d, /= 7.6 Hz, 1H)} 7.25 (d, J= 7.6 Hz, 1H), 7.37 (m, 2H), 7.49 (m, 2H), 7.62 (d, J= 8.0 Hz, 2H), 7.85 (d, J= 8.0 Hz, 2H), 7.92 (s, 1H), 10.50 (s, 1H); MS(ES+) m/z A (MH+). Example 31
Ar-(2-chloro-6'-(trifluoromethoxy)biphenyI-4-yl)-2-(4-(ethylsuIfonyl)phenyI)propanamide

Intermediate 31a: 2-f4-(ethylsulfonyl')phenyl')propanoic acid
Step 1: To a solution of [4-(ethyIsulfonyl)phenyI]acetic acid (intermediate la, 760 mg) in methanol (5 mL) was added SOC¾ (0.267 mL) at RT. The reaction mixture was heated to reflux (oil bath temperature: 80 °C) and stirred overnight. Solvent was removed under vacuum to afford methyl [4-(ethylsulfonyl)phenyl]acetate (790 mg) as a light yellow oil. MS^S ") m/z 243 (MH+),
Step 2: To a solution of methyl [4-(ethyIsulfonyl)phenyl] acetate (303 mg) in tetrahydrofuran
(THF) (1 mL) was added sodium hydride (75 mg) slowly. The vial was sealed and stirred at RT. After 30 mins, a solution of methyl iodide (0.070 mL) in tetrahydrofuran (THF) (1 mL) was carefully added at 0 °C. After stirring at this temperature for 3 hours, the reaction mixture was quenched with water. 1 M HCl was used to adjust pH to about 5. The aqueous phase was extracted with EtOAc for 3 times. The combined organics were dried over Na2S04. After removal of solvent, the obtained crude was purified by chromatography eluting with EtOAc : PE = 0: 1 to 1:2 to afford the intermediate methyl 2- [4-(ethylsulfonyI)phenyl]propanoate. MSfES4 m/z 257 (MH .
Step 3: The intermediate methyl 2-[4-(ethylsulfonyl)phenyl]propanoate was dissolved in EtOH (2 mL). 2 M NaOH (2 mL) was added. The mixture was stirred at RT for 2 hours. 1 M HCl was used to adjust pH to about 5. The aqueous phase was extracted with EtOAc for 3 times. The combined organics were dried over Na2S04. After removal of solvent, 2-(4-(ethylsulfonyl)phenyl)propanoic acid (202.7 mg) was obtained. MS(ES+) m/z 243 (MH+).
Preparation of N-( 2-chloro-6'-f trifluoromethoxy')biphenyl-4-yl -2-f4- f ethylsul fonvDphenvDpropanami de
A mixture of 2-(4-(ethylsulfonyl)phenyl)propanoic acid (intermediate 31 a, 286 mg), 2-chloro-
6'-(trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 170 mg), HOBt (109 mg), EDC (340 mg) and DIPEA (0.62 mL) in DCM (10 mL) was refluxed overnight. Solvent was removed, and the residue was purified by flash chromatography (EtOAc : PE = 1 :2) to give the crude product. Further purification by prep-HPLC afforded N-(2-chloro-6'-(trifluoromethoxy)biphenyl-4'yl)-2-(4- (ethylsulfonyl)phenyl)propanamide (20 mg) as a white solid. lH-NMR (400 MHz, DMSO-^) δ ppm 1.09 (t, J= 7.2 Hz, 3H), 1.48 (d, J= 6.8 Hz, 3H), 3.29 (q, J~ 7.2 Hz, 2H), 3.99 (q, J= 6.8 Hz, 1H), 7.30 (d, J= 8.8 Hz, 1H), 7.39 (d, J= 8.8 Hz, 1H), 7.48 (m, 2H), 7.56 (m, 2H), 7.68 (d, J= 8.0 Hz, 2H), 7.87 (d, J= 8.0 Hz, 2H), 7.95 (s, 1H), 10.49 (s, 1H); I9F-NMR (376 MHz, DMSO-i¾ δ ppm - 56.29; MS ES4) m/z 512 (Mrf). Example 32
Ar-(2-chloro-6'-(tetrahydro-2H-pyran-4-yloxy)biphenyl-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide
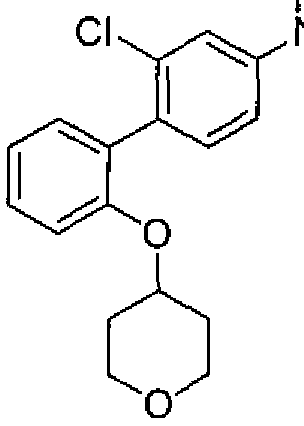
Step 1: To a solution of 2-bromophenol (2.0 g) in toluene (30 mL) cooled at 0°C was added tetrahydro-2H-pyran-4-ol (1.81 g), triphenylphosphine (3.64 g) and DIAD (2.70 mL). The reaction mixture was stirred at RT overnight. Solid was removed by filtration, and the filtrate was
concentrated. Purification by flash chromatography (EtOAc : PE = 1 : 10 to 1:1) gave 4-(2- bromophenoxy)-tetrahydro-2H-pyran (1.5 g) as a colorless oil. MS(ES+) m/z 257 (MH+).
Step 2: A mixture of 4-(2-bromophenoxy)-tetrahydro-2H-pyran (50 mg), N-[3-chloro-4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsuIfonyl)phenyl]acetamide
(intermediate 6b, 271 mg), Pd(Ph3P)4 (22.47 mg) and 2C03 (67.2 mg) in toluene (15 mL) and water (3 mL) was stirred at 90°C under N2 overnight. Solvent was removed, and the residue was partitioned between ethyl acetate (30 mL) and water (30 mL). The organic phase was washed with brine (30 mL x 2), dried and concentrated. The residue was purified by prep-HPLC to give N-(2-chloro-6'- (tetrahydro-2ii-pyran-4-yloxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (20 mg) as a white solid. Ή-NMR (400 MHz, CDCl3) δ ppm 1.31 (t, J= 7.6 Hz, 3H), 1.64 (m, 2H), 1.87 (m, 2H), 3.14 (q, J= 7.6 Hz, 2H), 3.45 (m, 2H), 3.64 (m, 2H), 3.84 (s, 2H), 4.42 (m, 1H), 7.0 (m, 2H), 7.20 (d, J= 7.2 Hz, lH), 7.24 (m, 1H), 7.33 (m, 1H), 7.39 (d, J= 8.0 Hz, 1H), 7.59 (d, J= 7.6 Hz, 2H), 7.71 (s, 1H), 7.94 (d, J= 7.6 Hz, 2H); MS(ES+) m/z 514 (MH+).
Example 33
7Ar-(6'-(benzyloxy)-2-chlorobiphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 1: A mixture of 2-bromophenol (3.4 g), (bromomethyl)benzene (4.0 g) and K2C03 (5.4 g) in acetone (10 mL) was stirred at 50°C overnight. Solvent was removed. The residue was partitioned
between water and ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by flash chromatography (PE) to give 1 - (benzyloxy)-2-bromobenzene (3.6 g) as a colorless oil.
Step 2: A mixture of l-(benzyloxy)-2-bromobenzene (99 mg), N-[3-chIoro-4-(4,4,5,5- tetramethyl-1 ^^-dioxaborolan^-yliphenyll^-^-tethylsulfony^phenyljacetamide (intermediate 6b, 192 mg), Pd(Ph3P)4 (20 mg) and K2C03 (104 mg) in 1 ,4-dioxane (20 mL) and water (3 mL) was stirred at 90°C for 3 hours. The mixture was extracted with ethyl acetate (100 mL). The organic phase was washed with brine, dried and concentrated. The residue was purified by flash chromatography (EtOAc-.PE = 1:1) to give the crude product. Further purification by prep-HPLC afforded N-(6'- (benzyloxy)-2-chlorobiphenyl-4-yl)-2-(4-(etliylsulfonyl)phenyl)acetamide (80 mg) as a white solid. Ή NMR (400 MHz, DMSO-i¾ δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.29 (q, J= 12 Hz, 2H), 3.83 (s, 2H), 5.09 (s, 2H), 7.02 (t, J= 7.2 Hz, IH), 7.15 (m, 2H), 7.31 (m, 7H), 7.49 (dd, J= 2.0 Hz, 8.8 Hz, IH), 7.61 (d, J= 8.4 Hz, 2H), 7.85 (d, J= 8.4 Hz, 2H), 7.88 (d, J= 2.0 Hz, IH), 10.51 (s, IH); MS(ES+) ;n/z 37 (MH Example 34
Ar-(2-chIoro-6'-(tri-luoromethoxy)biphenyl-4-yl)-2-(4-(methylsu]fonyl)phenyl)acetamide

A mixture of 2-chloro-61-(trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 200 mg), 2- (4-(methylsulfonyl)phenyl)acetic acid (179 mg), HOBt (128 mg), EDC (200 mg) and DIPEA (0.73 mL) in DCM (20 mL) was refluxed overnight. Solvent was removed, and the residue was purified by flash chromatography (EtOAc : PE = 1:2) to give the crude product. Further purification by prep- HPLC afforded N-(2-chloro-6'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-
(methylsulfonyl)phenyl)acetamide (130 mg) as a white solid. Ή-NMR (400 MHz,
 δ ppm 3.20 (s, 3H), 3.84 (s, 2H), 7.31 (d, J= 8.0 Hz, IH), 7.40 (d, J= 8.0 Hz, IH), 7.47 (m, 2H), 7.56 (m, 2H), 7.62 (d, J= 8.0 Hz, 2H), 7.90 (d, J= 8.0 Hz, 2H), 7.94 (s, IH), 10.56 (s, IH); l9F-NMR (376 MHz, DMSO-£¾) δ ppm -56.27; MS(ES+) mfz 484 (MH+).
δ ppm 3.20 (s, 3H), 3.84 (s, 2H), 7.31 (d, J= 8.0 Hz, IH), 7.40 (d, J= 8.0 Hz, IH), 7.47 (m, 2H), 7.56 (m, 2H), 7.62 (d, J= 8.0 Hz, 2H), 7.90 (d, J= 8.0 Hz, 2H), 7.94 (s, IH), 10.56 (s, IH); l9F-NMR (376 MHz, DMSO-£¾) δ ppm -56.27; MS(ES+) mfz 484 (MH+).
Example 35
Ar-(2-c loro-6'-(trifluoromethoxy)biphenyl-4-yI)-2-(4-(N-ethylsulfamoyl)phenyl)acetami^

Intermediate 35a: 2-f4-('N-ethylsuIfamoyl)phenyl')acetic acid
2-(4-(Chlorosulfonyl)phenyl)acetic acid (500 mg, see step 1 for synthesis of intermediate 29a) was added to a solution of ethanamine (961 mg) in methanol (50 mL), and the reaction mixture was stirred at RT overnight. Solvent was removed, and ethyl acetate was added. The solution was washed with HCl (1 M) and brine, dried and concentrated to give 2-(4-(N-ethylsulfamoyI)phenyl)acetic acid
(500 mg) as a light yellow solid. MS(ES+) m/z 244 (MH+).
Preparati on of N-(2-chloro- 6'-(trifluoromethoxyYbiphenyl -4-yl)-2 -(4-(N- ethylsulfamoyllphenvDacetamide
A mixture of 2-chloro-6'-(trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 200 mg), 2-
(4-(N-ethylsulfamoyl)phenyl)acetic acid (intermediate 35a, 203 mg), HOBt (128 mg), EDC (200 mg) and DIPEA (0.729 mL) in DCM (20 mL) was refluxed overnight. Solvent was removed, and the residue was purified by flash chromatography (EtOAc : PE = 1 :3) to give the crude product. Further purification by prep-HPLC gave N-(2-chloro-6'-(trifluoromethoxy)biphenyl-4-yI)-2-(4-(N- ethylsulfamoyl)phenyl)acetamide (70 mg) as a white solid. Ή-NMR (400 MHz, DMSO-rf6) δ ppm 0.98 (t, J= 7.2 Hz, 3H), 2.78 (q, J= 7.2 Hz, 2H), 3.81 (s, 2H), 7.31 (d, J= 8.4 Hz, 1H), 7.41 (d, J= 7.2 Hz, 1H), 7.47 (m, 7H), 7.76 (d, J= 8.0 Hz, 2H), 7.95 (s, 1H), 10.54 (s, 1H); 19F-NMR (376 MHz, DMSO--4) δ ppm -56.25; MS(ES*) m/z 513 (MH+).
Example 36
2-(4-(ethylsulfonyl)phenyl)-A'-(3-fluoro-2,-(trifluoromethoxy)biphenyl-4-yI)acetamide

Step 1: A mixture of 2-(trifluoromethoxy)phenylboronic acid (650 mg), 4-bromo-2- fluorobenzenamine (400 mg), K2C03 (873 mg) and Pd(Ph3P)4 (243 mg) in 1,4-dioxane (50 mL) and water (10.00 mL) was stirred at 100°C overnight. The mixture was filtered tlirough celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by flash
chromatography (EtOAc : PE = 1 :4) to afford 3-fluoro-6'-(trifluoromethoxy)biphenyl-4-amine (400 mg) as a yellow solid. MS(ES+) m/z 272 (MH+).
Step 2: A mixture of 3-fluoro-6'-(trifluoromethoxy)biphenyl-4-amine (200 mg), [4- (ethylsulfonyl)phenyljacetic acid (intermediate la, 202 mg), HOBt (113 mg), DIPEA (0.386 mL) and EDC (424 mg) in tetrahydrofuran (THF) (20 mL) was stirred at 60°C overnight. The mixture was concentrated under reduced pressure, and the residue was purified by prep-HPLC to give 2-(4- (ethylsulfonyl)phenyl)-N-(3-fluoro-2'-(trifluoromethoxy)biphenyl-4-yl)acetamide (60 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i¾) δ ppm 1.11 (t, J= 7.2 Hz, 3H), 3.29 (q, J= 7.2 Hz, 2H), 3.94 (s, 2H), 7.28 (dd, J~ 1.6 Hz, 8.4 Hz, 1H), 7.41 (dd, J= 1.6 Hz, 12.0 Hz, 1H), 7.52 (m, 4H), 7.65 (d, J = 8.4 Hz, 2H), 7.87 (d, J= 8.4 Hz, 2H), 8.04 (t, J= 8.4 Hz, 1H), 10.21 (s, 1H); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -56.31, -124.73; MS(ES+) m/z 482 (MH+).
Example 37
JV-(2' ,5' -dichlor o-3-biphenylyl)-2- [4-(ethylsuIf onyI)phenyll acetamide

(7.2 g) and Pd(Ph3P)4 (2.0 g) in 1,4-dioxane (150 mL) and water (15 mL) was stirred at 80°C under nitrogen for 2 hours. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (EtOAc : PE = 1 :20) to give (2',5'-dichloro-3-biphenyIyI)amine (1.9 g) as an oil. MS(ES+) m/z 238 (NOT*).
Step 2: To a solution of (2'!5'-dichloro-3-biphenylyl)amine (238 mg), EDC (0.230 g) and HOBt
(0.184 g) in DCM (15 mL) was added [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 233 mg). The reaction mixture was stirred at room temperature until no starting material left. The mixture was poured into water, and then extracted with DCM. The organic phase was washed with water, and brine. The obtained solution was dried over anhydrous sodium sulfate, filtered and concentrated to give the crude product. Further purification by MDAP afforded N-(2',5'-dichloro-3-biphenylyl)-2-[4- (ethylsuIfonyl)phenyl]acetamide (260 mg). Ή-NMR (600 MHz, DMSO-rf6) δ ppm 1.10 (t, J = 7.8 Hz, 3H), 3.27 (q, J= 7.8 Hz, 2H), 3.82 (s, 2H), 7.13 (d, /= 7.8 Hz, 1H), 7.42 (t, J= 7.8 Hz, 1H), 7.46 (d, J- 2.4 Hz, 1H), 7.48 (dd, = 2.4 Hz, 8.4 Hz, 1H), 7.61 (m, 4H), 7.72 (s, 1H), 7.85 (d, J= 8.4 Hz, 2H), 10.42 (s, 1H); MSfES4) m/z 448 (MH+).
Example 38
iV-t6-(2,5-dichioroplienyl)-2-pyridinyl]-2-[4-(ethylsulfonyI)phenyl]acetamide

Step 1: A mixture of (2,5-dichlorophenyl)boronic acid (572 mg), 6-bromo-2-pyridinamine (519 mg), Cs2C03 (1172 mg) and PdCl2(dppf)-CH2Cl2 adduct (196 mg) in acetonitrile (12 niL) and water (4 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anliydrous Na2SCv After filtration, solvent was removed in vacuo and the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1 :4) to afford 6-(2,5-dichlorophenyl)-2-pyridinamine (456 mg) as a yellow oil. MS(ES+) m/z 239 (MH+).
Step 2: A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 100 mg), EDC (104 mg) and HOBt (73.5 mg) in DCM (2 mL) was stirred at room temperature under nitrogen for 10 mins. A solution of 6-(2,5-dichlorophenyl)-2-pyridinamine (100 mg) in DCM (1 mL) was then added. After stirring at RT overnight, the reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were dried over anliydrous Na2S04. After filtration, solvent was removed under reduced pressure and the residue was purified by MDAP to afford N-[6-(2,5-dichlorophenyl)-2-pyridinyl]-2-[4-
(ethylsulfonyl)phenyl]acetamide (56 mg) as a light yellow solid. Ή-NMR (400 MHz, DMSO-< ) δ ppm 1.09 (t, J= 7.2 Hz, 3H)} 3.27 (q, J= 7.2 Hz, 2H), 3.90 (s, 2H), 7.40 (dd, J= 0.4 Hz, 7.6 Hz, 1H), 7.55 (dd, J= 2.4 Hz, 8.4 Hz, 1H), 7.63 (m} 3H), 7.66 (d, J= 2.8 Hz, 1H), 7.85 (m, 2H), 7.90 (t, J- 8.4 Hz, 1H), 8.10 (d, J= 8.4 Hz, 1H), 10.99 (s, 1H); MS(ES+) m/z 449 (MH+).
Example 39
iV-(2',5'-dichloro-6-methyI-3-biphenylyl)-2-[4-(ethylsulfonyl)phenyl3acetamide

Step 2: A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 95 mg), EDC (99 mg) and HOBt (69.7 mg) in DCM (3 mL) was stirred at room temperature for 15 mins. Then 2',5'- dichIoro-6-metliyl-3-biphenylamine (100 mg) was added. After stirring at room temperature overnight, the reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed under reduced pressure. The residue was purified by MDAP to afford N-(2',5,-dichloro-6-methyl-3-biphenylyl)-2-[4-
(ethylsulfonyl)phenyl]acetamide (82 mg) as a white solid. 'H- MR (400 MHz, DMSO-i¾ δ ppm 1.09 (t, J= 7.2 Hz, 3H), 1.99 (s, 3H), 3.26 (q, J= 7.2 Hz, 2H), 3.78 (s, 2H), 7.25 (d, J= 8.4 Hz, 1H), 7.38 (d, J~ 2.4 Hz, 1H), 7.44 (d, J= 2.0 Hz, 1H), 7.50 (dd, J= 2.4 Hz, 8.8 Hz, 2H), 7.59 (m, 3H), 7.84 (d, J= 8.4 Hz, 2H), 10.30 (s, 1H); MS(ES+) m/z 462 (MH4). Example 40
iV-(2',4,-dichloro-6-methyl-3-biphenyIyl)-2-[4-(ethylsuIfonyl)phenyl]acetamide

Step 1: A mixture of (2,4-dichlorophenyl)boronic acid (572 mg), 3-bromo-4-methylaniline (558 mg), Cs2C03 (1172 mg) and PdCl2(dppf)-CH2Cl2 adduct (196 mg) in acetonitrile (12 mL) and water (4 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anydrous Na2S04. After filtration, solvent was removed in vacuo and the residue was purified by flash chromatography
(EtOAcrPE = 0: 1 to 1:4) to afford 2',4'-dichloro-6-methyl-3-biphenylamine (423 mg) as a yellow oil. MS(ES+) m/z 252 (MH^).
Step 2: A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate l a, 95 mg), EDC (99 mg) and HOBt (69.7 mg) in DCM (3 mL) was stirred at room temperature for 15 mins. Then 2',4'- dicliloro-6-methyl-3-biphenyIamine (100 mg) was added. After stirring at room temperature overnight, the reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were dried over anhydrous Na2S04. After filtration, solvent was removed under reduced pressure and the residue was purified by MDAP to afford N-(2',4'-dichloro-6-methyl-3-biphenylyl)-2-[4-(ethylsulfonyl)phenyl3acetamide (78 mg) as a white solid. Ή-NMR (400 MHz, DMSO-<¾ δ ppm .09 (t, J= 7.2 Hz, 3H), 1.98 (s, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.78 (s, 2H), 7.25 (d, J= 8.4 Hz, 1H), 7.31 (d, J= 8.0 Hz, 1H), 7.41 (d, J= 2.0 Hz, 1H), 7.50 (m} 2H), 7.59 (d, J= 8.0 Hz, 2H), 7.75 (d, J= 2.4 Hz, 1H), 7.84 (d, J= 8.4 Hz, 2H), 10.29 (s, 1H); MSiES4) m z 462 (MH+).
Example 41
N-ii^S'-dichloro^-biphenyly - -^-tethylsulfonyliphenylJacetamidc

Step 1: A mixture of (2,5-dichlorophenyl)boronic acid (333 mg), 4-bromoaniline (300 mg), Cs2C03 (682 mg) and PdCl2(dppf)-CH2Cl2 adduct (114 mg) in acetonitrile (9 mL) and water (3 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anydrous Na2S04. After filtration, solvent was removed in vacuo and the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 15:85) to afford 2r,5'-diCmoro-4-biphenylamine ( 130 mg) as a yellow oil. MS(ES+) m/z 238 (MH+).
Step 2: A mixture of [4-(ethylsulfonyl)phenyl] acetic acid (intermediate la, 101 mg), EDC
(105 mg), and HOBt (73.8 mg) in DCM (2 mL) was stirred at room temperature under nitrogen for 10 mins. Then 2',5'-dichloro-4-biphenylamine (100 mg) was added. After stirring at room temperature overnight, the reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were washed with brine and dried over
anliydrous Na2S04. After filtration, solvent was removed under reduced pressure and the residue was purified by MDAP to afford N-(2',5'-dichloro-4-biphenylyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (68 mg) as a white solid. Ή-NMR (400 MHz, DMSO-c¾) 8 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 7.42 (m, 4H), 7.61 (m, 3H), 7.69 (d, J= 8.4 Hz, 2H), 7.86 (d, J= 8.4 Hz, 2H), 10.43 (s, 1H); MS(ES") m/z 448 (MH+).
Example 42
2-[4-(et ylsulfonyJ)phenylJ-iV-[2-(rrifluoromethyl)-4-biphenylyI]acetamide

Intermediate 42a: N-r4-bromo-3-(-rifluoromethyl'lphenyl1-2-[4-(ethylsulfonyl')pherivI]acetamide A mixture of [4-bromo-3-(trifluoromemyl)phenyl]amine (200 mg), [4-
(ethyIsulfonyl)phenyl]acetic acid (intermediate la, 209 mg), HOBt (169 mg) and EDC (240 mg) in DCM (4 niL) was stirred at room temperature overnight under N2. The mixture was washed with 1 M HC1, sat. NaHCC>3 and brine successively, dried over anhydrous Na2SC>4. After filtration, the filtrate was concentrated in vacuo to afford N-[4-bromo-3-(trifmoromethyI)phenyl]-2-[4- (ethylsulfonyi)phenyl]acetamide (330 mg) as a yellow solid. MS(ES+) m/z 450 (MH+).
Preparation of 2-r4-(ethylsulfonyl)phenyl1-N-r2-(trifluorometliyl')-4-biphenylyl1acetamide
A mixture of phenylboronic acid (20.85 mg), N-[4-bromo-3-(trifluoromethyl)phenyl]-2-[4- (ethyl sulfonyi)phenyl]acetamide (intermediate 42a, 70 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C0 (60.8 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was filtered through silica gel. The filtrate was dilute with water, and extracted with EtOAc for 3 times. The combined organic layers were dried over Na2S04. After filtration, the filtrate was concentrated under vacuum, and the residue was purified by MDAP to give 2-[4-(ethylsulfonyl)phenylJ-N-[2-(trifluoromethyl)-4- biphenylyl]acetamide (15 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 7.29 (m, 2H), 7.36 (d, J= 8.4 Hz, 1H), 7.41 (m, 3H), 7.63 (d, J- 8.0 Hz, 2H), 7.85 (m, 3H), 8.16 (d, J= 3.6 Hz, 1H), 10.68 (s, 1H); i9F-NMR (376 MHz, DMSO- ) 5 ppm -55.73; MS(ES+) m/z 448 (MH+).
Example 43
V-(2-chloro-4-biphenylyl)-2-[4-(ethylsuIfonyl)phenyI]acetamide

A mixture of phenylboronic acid (22.53 mg), N-(4-bromo-3-chlorophenyl)-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6a, 70 mg), PdC]2(dppf)-CH2Cl2 adduct (10 mg), Cs2C03 (65.7 mg) in acetonitrile (1.5 lnL) and water (0.5 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. The reaction mixture was filtered through silica gel and celite. The filtrate was dried over anhydrous Na2S0 . After filtration, the filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-(2-chloro-4-biphenylyI)- 2-[4-(ethylsulfonyl)phenyl]acetamide (21 mg) as a white solid. Ή-N R (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 7.41 (m, 6H), 7.55 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.62 (d, /- 8.4 Hz, 2H), 7.86 (d, J- 8.4 Hz, 2H), 7.93 (d, J= 2.0 Hz, 1H), 10.54 (s, 1H); MS(ES+) m/z 414 (MH+).
Example 44
2-[4-(ethylsulfonyl)phenyl]-iV-(2,2, J5'-trichloro-4-biphenyIyl)acetamide

A mixture of (2,5-dichlorophenyl)boronic acid (35.3 mg), N-(4-bromo-3-chlorophenyl
(ethylsulfonyl)pheny]]acetamide (intermediate 6a, 70 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (65.7 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a reaction vessel and heated in the microwave at 120°C for 30 mins. As there was still some starting material left, more PdCl2(dppf)-CH2Cl2 adduct (10 mg) and (2,5-dichIorophenyl)boronic acid (16.03 mg) were added. The resulting mixture was sealed again in the reaction vessel and heated in the microwave at 120°C for 30 mins. The mixture was filtered through silica gel. The filtrate was dilute with water, and extracted with EtOAc for 3 times. The combined organic layers were dried over Na2S04. After filtration, the filtrate was concentrated under vacuum, and the residue was purified by MDAP to give 2-[4-(ethylsulfonyl)phenyl]-N-(2,2',5'-trichloro-4-biphenylyl)acetamide (27 mg) as a white solid. Ή- NMR (400 MHz, DMSO-c/fi) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H),
7.32 (d, J= 8.4 Hz, 1H), 7.45 (d, J= 2.4 Hz, 1H), 7.53 (m, 2H), 7.61 (m, 3H), 7.86 (d, J= 8.4 Hz, 2H), 7.94 (d, J= 2.0 Hz, 1H), 10.59 (s, 1H); MS(ES" m z 482 (MH*).
Example 45
Ar-[2-ch[oro-2'-(trifluoromethyI)-4-biphenylyl]-2-[4-(ethylsuIfonyl)p enyl]acetamide

A mixture of [2-(trifluoromethyl)phenyl]boronic acid (30.1 mg), N-(4~bromo-3-chlorophenyi)- 2-[4-(emylsulfonyl)phenyl]acetamide (intermediate 6a, 60 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (56.3 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silice gel. The filtrate was diluted with water, and extracted with EtOAc for 3 times. The combined organic layers were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-[2-chloro-2'-(trifluoromethyl)-4-biphenylyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (26 mg) as a white solid. Ή-NM (400 MHz, DMSO-c?6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 7.27 (d, J= 8.0 Hz, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7. 1 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.64 (m, 3H), 7.73 (t, J= 7.6 Hz, 1H), 7.85 (m, 3H), 7.93 (d, 7= 2.0 Hz, 1H), 10.57 (s, 1H); 19F-NMR (376 MHz, DMSO-i/6) δ ppm -57.71; MS(ES") m/z 482 (MH+).
Example 46
3-[4'-({[4-(ethy]sulfonyl)phenyl]acetyl}amino)-2'-(trifluoromethyl)-4-biphenylyl]propanoic acid

A mixture of 3-[4-(dihydroxyboTanyl)phenyl]propanoic acid (28.4 mg), N-[4-bromo-3- (trifluoromethyl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intennediate 42a, 60 mg),
PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (52.1 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture
was filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford 3-[4'-({[4-(ethylsulfonyl)plienyl]acetyl}amino)-2'-(trifluoromethyl)-4- biphenylyljpropanoic acid (26 mg) as a white solid. Ή-NMR (400 MHz, DMSO-rf6) δ ppm 1.10 (t, J - 7.2 Hz, 3H), 2.56 (t, J= 7.6 Hz, 2H), 2.86 (t, J= 7.6 Hz, 2H), 3.28 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 7.19 (d, J= 8.0 Hz, 2H), 7.28 (d, J= 8.0 Hz, 2H), 7.34 (d, J= 8.0 Hz, 1H), 7.63 (d, J= 8.4 Hz, 2H), 7.84 (m, 3H), 8.15 (d, J= 2.0 Hz, 1H), 10.67 (s, 1H); 19F-NMR (376 MHz, DMSO-ii0) δ ppm -55.70; MS(ES+) m/z 520 (MH+).
Example 47
2- [4-(e thylsulfonyI)phenyl]-iV- [4-(4-pyridinyl)-3-(trifluoromethyl)phenyl] acctamide,
trifluoroacetic acid salt

A mixture of N-[4-bromo-3-(trifluoromethyl)phenyl]-2-[4-(ethylsulfonyI)phenyl]acetamide (intennediate 42a, 60 mg), 4-pyridinylboronic acid (18.02 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and CS2CO3 (52.1 mg) in acetonitrile (1.5 mL) and water (0.5 lnL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure to afford 2-[4-(ethylsulfonyl)phenyl]-N-[4-(4- pyridinyl)-3-(trifluoromethyl)phenyl]acetamide, trifluoroacetic acid salt (15.3 mg) as a white solid. Ή-NMR (400 MHz, DMS W6) 5 ppm 1.10 (t, 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.88 (s, 2H), 7.45 (d, J- 8.4 Hz, 1H), 7.52 (m, 2H), 7.63 (d, J= 8.4 Hz, 2H), 7.86 (d, J= 8.4 Hz, 2H), 7.92 (dd, J =1.6 Hz, 8.4 Hz, 1H), 8.23 (d, J= 2.0 Hz, 1H), 8.73 (d, J= 4.4 Hz, 2H), 10.78 (s, 1H); l9F-NMR (376 MHz, DMSO-i/e) δ ppm -55.60, -74. 1 ; MSfES^) m/z 447 (MH+).
Example 48
2-[4-(ethylsulfonyl)phenyl]-/Y-[4-(3-pyridinyl)-3-(irifIuoromethyI)phenyI]acetaniide,
trifluoroacetic acid salt

TFA
A mixture of N-[4-bromo-3-(rjifluoromethyl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetainide (intermediate 42a, 60 mg), 3-pyridinylboronic acid (18.02 mg), PdCl2(dppf)-CH2Cl2 adduct (13 mg) and Cs2C03 (52.1 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered tlirough celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-[4-(3-pyridinyl)-3-(trifluoromethyl)phenyl]acetamide, trifluoroacetic acid salt (17.4 mg) as a white solid. ¾-NMR (400 MHz, DMSO- ) δ ppm 1.10 (t, J = 7.2 Hz, 3H), 3.28 (q, /= 7.2 Hz, 2H), 3.88 (s, 2H), 7.45 (d, J= 8.4 Hz, 1H), 7.61 (m, 3H), 7.88 (m, 4H), 8.22 (s, 1H), 8.59 (s, 1H), 8.68 (d, J= 4.4 Hz, 1H), 10.75 (s, 1H); l9F-NMR (376 MHz, DMSO- <¾) δ ppm -55.84, -74.69; MS(ES+) m/z 449 (MH+).
Example 49
A'-(2,2,-dichloro-4-biphenylyl)-2-[4-(ethylsuIfonyl)phenyl]acetamide

A mixture of N-(4-bromo-3-chlorophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6a, 60 mg), (2-chlorophenyl)boronic acid (24.77 mg), Cs2C03 (56.3 mg) and PdCI2(dppf)-C¾Cl2 adduct (10 mg) in acetonitrile (1,5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-(2,2'- dichloro-4-biphenylyl)-2-[4-(ethyIsulfonyl)phenyl]acetamide (29 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-i¾ 5 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 7.28 (d, J= 8.4 Hz, 1H), 7.31 (m, 1H), 7.43 (m, 2H), 7.55 (m, 2H), 7.62 (d, J= 8.0 Hz, 2H), 7.86 (d, J= 8.4 Hz, 2H), 7.93 (d, J= 2.0 Hz, 1H), 10.57 (s, 1H); MS(ES+) m/z 448 (MH+).
Example 50
2-[4-(ethylsulfonyl)phenyl]-N-{4-[3-(trifluoromethyl)-2-pyridinyl]phenyl}acetamide

Step 2: A mixture of 2-bromo-3-(trifluoromethyl)pyridine (50 mg), 2-[4- (ethylsulfonyl)phenyl]-N-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yI)phenyl]acetamide (104 mg), Cs2C03 (87 mg) and PdCl2(dppf)-CH2Cl2 adduct (10 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-{4-[3-(trifluoromethyl)-2- pyridinyl]phenyl}acetamide (32 mg) as a white solid. 'H-NMR (400 MHz, DMSO-rf6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 7.42 (d, J= 8.4 Hz, 2H), 7.62 (m, 3H), 7.70 (d, J= 8.4 Hz, 2H), 7.86 (d, J= 8.4 Hz, 2H), 8.28 (dd, = 1.2 Hz, 8.0 Hz, 1H), 8.89 (d, J= 4.4 Hz, 1H), 10.46 (s, 1H); 19F-NMR (376 MHz, DMSO-t¼) δ ppm -55.98; MS(ES+) m/z 449 (MH+).
Example 51
N-{2-chloro-2'-[(l-methyIethyl)oxy]-4-biphenylyI}-2-[4~(ethylsuIfonyI)phenyl]acetamide

A mixture of l-bromo-2-[(l-methylethyl)oxy]benzene (38 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l}3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 90 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (69.1 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in die microwave at 100°C for 30 mins. The reaction mixture was filtered through celite. The filtrate was concentrated under reduced pressure, and the
residue was purified by MDAP to afford N-{2-chloro-2'-[(l-methylethyl)oxy]-4-biphenylyl}-2-[4- (etliylsulfonyl)phenyl]acetamide (12 mg) as a yellow solid. Ή-ΝΜΚ (600 MHz, DMSO-c ) δ ppm 1.11 (m, 9H), 3.28 (q, J= 7.2 Hz, 2H), 3.83 (s, 2H), 4.51 (m, 1H), 6.96 (t, J= 7.8 Hz, 1H), 7.09 (m, 2H), 7.22 (d, J= 6.4 Hz, 1H), 7.33 (m, 1H), 7.48 (m, 1H), 7.63 (d, J= 7.8 Hz, 2H), 7.86 (d, J= 7.8 Hz, 2H), 10.49 (s, 1H); MS(ES+) m/z Ml (MH^>.
Example 52
2-[4-(ethylsuJfonyl)phenyl]-iV-[2-methyl-2'-(trifluoromethyl)-4-biphenyly.]acetamide

Intermediate 52a: N-f4-bromo-3-methylprienvn-2-(4-(ethylsulfonvnphenyl¼cetamide
A mixture of 4-bromo-3-methylaniline (300 mg), [4-(ethylsulfonyl)phenyl]acetic acid
(inten-nediate la, 405 mg), HOBt (327 mg) and EDC (464 mg) in DCM (4 mL) was stirred at room temperature overnight. The reaction mixture was washed with 2M HCI, sat. NaHC03 solution and brine successively. The organic layer was dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure to afford N-(4-bromo-3-methylphenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide (540 mg) as a yellow solid. MS(ES+) m/z 396 (MH+).
Preparation of 2-Γ4-Γ ethyl sulfonvDphenyll -N- [2 -methyl-2'- f trifluorometh yl) -4-biphenylyllacetamide
A mixture ofN-(4-bromo-3-methylphenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide
(intennediate 52a, 70 mg), [2-(trifluoromethyl)phenyl]boronic acid (36.9 mg), PdCl2(dppf)-CH2Cl2 adduct (12.4 mg) and Cs2C03 (69.1 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl] -N-[2-methyl-2'-(trifluoromethyl)-4-biphenylyl] acetami de (41 mg) as a yellow solid. Ή-NMR (600 MHz, DMSO-i¾ δ ppm 1.10 (t, J= 7.2 Hz, 3H), 1.92 (s, 3H), 3.28 (q, J = 7.2 Hz, 2H), 3.81 (s, 2H), 7.02 (d, J= 8.4 Hz, 1H), 7.29 (d, J= 7.8 Hz, 1H), 7.44 (d, J= 7.8 Hz, 1H), 7.53 (s, 1H), 7.61 (m, 3H), 7.70 (t, J= 12 Hz, 1H), 7.84 (m, 3H), 10.32 (s, 1H); I9F- NMR (376 MHz, DMSO-rf6) δ ppm -57.68; MSiES^ m/z 462 (MH ).
Example 53
2-[4-(ethylsulfonyl)phenyl]-N-{2-(trifluoromethyl)-2t-[(trifluoromethyl)oxy]-4- biphenylyl} ac etamide

A mixture of N-[4-bromo-3-(trifluoromethyl)pheny]]-2-[4-(ethylsulfonyl)phenyl]acetamide (intennediate 42a, 70 mg), {2-[(trifluoromethyl)oxy]plienyl}boronic acid (35.2 mg), PdCI2(dppf)- CH2C12 adduct (12.7 mg) and Cs2C03 (60.8 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The mixture was filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-{2-(trifluoromethyl)-2'-[(trifluoromethyl)oxy] - biphenylyl}acetamide (21 mg) as a yellow solid. Ή-NMR (600 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.87 (s, 2H)} 7.36 (m, 2H), 7.45 (m, 2H), 7.56 (m, 1H), 7.63 (d, J= 8.4 Hz, 2H), 7.86 (m, 3H), 8.19 (s, 1H), 10.73 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm - 56.01 , -57.93; MS(ES+) m/z 532 (MH+).
Example 54
7V-[2-chloro-3'-fluoro-2,-(trifluoromethyl)-4-biphenyIyl]-2-[4-(ethylsulfonyl)phenyl]acetainide

A mixture of l-bromo-3-fluoro-2-(trifluoroinethyl)benzene (170 mg), N-[3-chloro-4-(4, 4,5,5 - tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 357 mg), PdCl2(dppf)-CH2Cl2 adduct (45.7 mg) and Cs2C03 (274 mg) in acetonitrile (4.5 mL) and water (1.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The mixture was filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was purified by chromatography on silica gel (EtOAc : PE = 0:1 to 3:7) to afford the crude product (330 mg), part of which (80 mg) was purified by MDAP to afford N-[2-chloro-3'-fluoro-2'-
(trifluoromethyl)-4-biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (13 mg) as a white solid. ¾- NMR (400 MHz, DMSO-c 6) δ ρρπΐ 1.10 (t, J- 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 7.16 (d, J= 7.6 Hz, 1H), 7.30 (d, J= 8.4 Hz, 1H), 7.57 (m, 4H), 7.78 (m, 1H), 7.86 (d, J= 8.0 Hz,
2H), 7.93 (d, J= 2.0 Hz, 1H), 10.60 (s, 1H); 19F-NMR (376 MHz, DMSO-i¾) δ ppm -54.84, -1 13.42; MSfES m/z 500 (MH+).
Example 55
A'-(2,6-dichIoro-4-biphenylyl)-2-[4-(ethylsulfonyl)phenyI]acetamide

Step 1: BF3-Et20 (1.03 g) was placed into a three necked flask. 2,6-Dichloro-4-nitro- phenylamine (1.0 g) i THF (8 mL) and DCM (16 inL) was added. Then iBuONO (598 mg) was added dropwise at -10 °C. The mixture was stirred for 1 hour at 0 °C. «-Hexane(50 mL) was added. The mixture was filtered to give 2,6-dichloro-4-nitro-benzenediazonium tetrafhioroborate (696 n g) as a yellow solid.
Step 2: To a stirred solution of CuBr (995 mg) in H 0 (15 mL) was added 2,6-dichloro-4- nitro-benzenediazonium tetrafluoroborate (696 mg) portionwise. The mixture was stirred for 4 hours. The solution was extracted with EtOAc (30 mL x 3). The organic layers were dried over anliydrous Na2S0 and concentrated to give 2-bromo-l,3-dichloro-5-nitro-benzene (230 mg) as a yellow oil.
Step 3: A solution of 2-bromo- 1 ,3-dichloro-5-nitro-benzene (3.57 g) and Pd(PPh3)4 (1 g) in toluene (100 mL) and EtOH (16 mL) was stirred for 20 mins at 0 °C. Na2C03 (5.47 g) in 20 mL water and phenylboronic (1.77 g) was added. The mixture was stirred for 4 hours at 90°C under N2. The mixture was concentrated and the residue was purified by column chromatography on silica gel (EtOAc : PE = 1:50) to give 2,6-dichloro-4-nitro-biphenyl (2.88 g).
Step 4: A solution of 2,6-dichloro-4-nitro-biphenyl (2.88 g) and SnCl2-2H20 (12.2 g) in
MeOH (50 mL) was heated to reflux for 16 hours. The solution was concentrated. The residue was diluted with EtOAc (50 mL) and water (50 mL). The solution was adjusted to pH 9 with NaOH solution. The mixture was filtered and the filtrate was extracted with EtOAc (50 mL x 3). The organic layer was dried over anhydrous Na2S04 and concentrated. The residue was purified by column chromatography on silica gel (EtOAc : PE = 1: 1) to give 2,6-dichloro-[l ,l'-biphenyI]-4-amine as a white solid (2.39 g). MSfES"* m/z 238 (MH+).
Step 5: A mixture of 2,6-dichIoro-[l,l '-biphenyl]-4-amine (80 mg), [4- (ethylsulfonyl)phenyl] acetic acid (intermediate la, 84 mg), HOBt (59.0 mg) and EDC (84 mg) in DCM (2 mL) was stirred at room temperature for 1.5 days. Solvent was removed under reduced pressure, and the residue was purified by MDAP to afford N-(2,6-dichloro-4-biphenylyl)-2-[4-
(ethylsulfonyl)phenyl]acetamide (95 mg) as a white solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.29 (q, /= 7.2 Hz, 2H), 3.85 (s, 2H), 7.24 (ra, 2H), 7.44 (m, 3H), 7.62 (d, J = 8.0 Hz, 2H), 7.81 (s, 2H), 7.86 (d, J= 8.0 Hz, 2H), 10.66 (s, 1H); MS(ES+) m/z 448 (MH .
Example 56
2-[4-(ethylsuIfonyl)phenyI]-iV-{2-(niethyIoxy)-2'-[(trifluoromethyl)oxy]-4-biphenylyl}acetamide

Step 1: A mixture of 4-bromo-3-(mefhylox.y)aniline (400 mg), [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 497 mg), HOBt (348 mg) and EDC (493 mg) in DCM (8 mL) was stirred at room temperature overnight. The mixture was washed with 2M HC1 (3 times), sat. NaHC03 (3 times) and brine (3 times) successively. The organic layer was dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure to afford N-[4-bromo-3- (methyloxy)phenyI]-2-[4-(ethylsulfonyl)phenyl]acetamide (730 mg) as a yellow solid. MS(ES+) m/z 412 (MH4).
Step 2: A mixture of N-[4-bromo-3-(methyloxy)phenyl]-2-[4- (ethylsulfonyl)pheny]]acetamide (60 mg), {2-[(trifluoromethyl)oxy]phenyl}boronic acid (33.0 mg), PdCl2(dppf)-CH2Cl2 adduct (9.51 mg) and Cs2C03 (56.9 mg) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford 2- [4-(ethylsulfonyl)phenyl]-N-{2-(methyloxy)-2,-[(trifluorometliyl)oxy]-4-biphenylyl}acetamide (38 mg) as a white solid. lH-NMR (400 MHz, DMSO-<¾) 5 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.67 (s, 3H), 3.83 (s, 2H), 7.11 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.4 Hz, 1.6 Hz, 1H), 7.36 (m, 5H), 7.63 (d, J = 8.4 Hz, 2H), 7.86 (d, J- 8.4 Hz, 2H), 10.41 (s, 1H); 19F-NMR (376 MHz, DMSO- δ ppm -55.90; MSiES4) m/z 494 (MH+).
Example 57
N-[2-chloro-2'-(dimethylamino)-4-biphenylyl]-2-[4-(ethylsulfony])phenyl]acetamide, trifluoroacetic acid salt

Step 1: To a solution of l-bromo-2-chloro-4-nitro-benzene (500 mg) and Pin2B2 (592 mg) in 1,4-dioxane (5 mL) was added KOAc (621 mg) and PdCl2(dppf) (200 mg). The mixture was stirred for 4 hours at 100°C under N2. The mixture was filtered and concentrated. The residue was purified by column chromatography on silica gel (EtOAc : PE = 1:30) to give 2-(2-chloro-4-nitro-phenyI)- 4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (215 mg). MS(ES+) m/z 202 (M-QHn+H"*).
Step 2: To a solution of 2-(2-chloro-4-nitro-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (2.0 g) and (2-bromo-phenyl)-dimethyl-amine (1.42 g) in DMF (20 mL) was added K2C03 (2.94 g) and PdCl2(dppf) (512 mg). The mixture was stirred for 4 hours at 100°C under N2. The mixture was diluted with water (200 mL). The solution was extracted with EtOAc (50 mL x 3). The organic layer was dried over anhydrous Na2S04 and concentrated. The residue was purified by column
chromatography on silica gel (EtOAc : PE = 1 :50) to give (2'-chloro-4'-nitro-biphenyl-2-yl)-dimethyl- amine as a yellow oil (1.18 g). MS(ES+) m/z 277 (MH4 .
Step 3: To a solution of (2,-chloro-4'-nitro-biphenyl-2-yl)-dimethyl-amine (350 mg) in MeOH (10 mL) was added dry Pd/C (70 mg). The mixture was stirred for 4 hours at room temperature under ¾ (1 atm). The mixture was filtered and the filtrate was concentrated. The residue was purified by column chromatography on silica gel (EtOAc : PE = 1: 10) to give (2'-chIoro-4'-amino-biphenyl-2-yl)- dimethyl-amine (200 mg) as a yellow solid. MSiES*) m/z 247 (MH^.
Step 4: A mixture of (2'-chloro-4,-amino-biphenyl-2-yl)-dimethyI-amine (80 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 74.0 mg), HOBt (43.8 mg) and EDC (62.2 mg) in DCM (2 mL) was stirred at room temperature overnight. Solvent was removed under reduced pressure, and the residue was purified by MDAP to afford N-[2-chloro-2'-(dimethylamino)-4- biphenylyl]-2-[4-(etliylsulfonyl)phenyl]acetamide, trifluoroacetic acid salt (43 mg) as a yellow solid. 1 H-NMR (400 MHz, MeOD-c/4) δ ppm 1.13 (t, J= 7.4 Hz, 3H), 3.05 (s, 6H), 3.12 (q, J= 7.3 Hz, 2H), 3.79 (s, 2H), 7.21 (m, 1H), 7.32 (d, J= 8.5 Hz, 1H), 7.45 (t, J= 7.4 Hz, 1H), 7.55 (m, 4H), 7.72 (d, J = 8.0 Hz, 1H), 7.81 (d, J= 8.3 Hz, 2H), 7.92 (m, 1H), 10.35 (s, 1H); ,9F-NMR (376 MHz, MeOD-i 4) δ ppm -77.27; MS(ES+) m/z 457 (MH+).
Example 58
N-[2-chloro-2'-(cyclopentyloxy)-4-biphenylyl]-2-[4-(ethylsulfonyi)phenyl]acetamide

Step 1: DIAD (1.349 inL) was added into a solution of 2-bromophenoI (0.8 g), cyclopentanol (0.597 g) and triphenylphosphine (1.819 g) in tetrahydrofuran (THF) (20 mL) at 0°C dropwise. After the addition was complete, the reaction mixture was stirred at 0°C for 1 hour, then left at room temperature overnight. The reaction mixture was washed with brine. The organic layer was dried over anhydrous Na2SC>4. After filtration, the filtrate was purified by chromatography (PE) to afford 1- bromo-2-(cyclopentyloxy)benzene (0.83 g) as a colorless oil.Ή-NMR (400 MHz, CDC13) δ ppm 1.64 (m, 2H), 1.88 (m, 6H), 4.81 (m, 1H), 6.79 (m, 1H), 6.90 (dd, J= 1.2 Hz, 8.4 Hz, 1H), 7.23 (m, 1H), 7.52 (dd, J= 1.6 Hz, 8.0 Hz, 1H).
Step 2: A mixture of l-bromo-2-(cyclopentyloxy)benzene (45 mg), N[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaboi lan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 95 mg), PdCl2(dppf)-CH2Ci2 adduct (12.19 mg) and Cs2C03 (73.0 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered tlirough celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-[2-chloro-2'-(cyclopentyloxy)-4- biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (37 mg) as a white solid. 1 H-NMR (600 MHz, DMSO--¾ 5 ppm 1.10 (d, J= 8.4 Hz, 3H), 1.53 (m, 6H), 1.78 (s, 2H), 3.28 (q, J= 7.2 Hz, 2H), 3.83 (s, 2H), 4.78 (s, 1H), 6.96 (t, J = 7.8 Hz, 1H), 7.05 (d, J= 8.4 Hz, 1Η), 7.Π (d, J= 7.8 Hz, 1H), 7.21 (d, J = 8.4 Hz, 1H), 7.33 (t, J= 7.2 Hz, 1H), 7.47 (d, J= 8.4 Hz, 1H), 7.62 (d, J= 7.8 Hz, 2H), 7.85 (m, 3H), 10.48 (s, 1H); MS(ES+) m z 498 (MH .
Example 59
N-{2-chloro-2'-[(2-methylpropyl)oxy]-4-biphenylyl}-2-[4-(ethylsulfonyI)phenyl]acetamide

Step 2: A mixture of l-bromo-2-isobutoxybenzene (45 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intennediate 6b, 100 mg), PdC¾(dppf)-CH2CI2 adduct (12.83 mg) and Cs2C03 (77 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered tlirougli celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-{2-chloro-2'-[(2-methylpropyl)oxy]^I- bipheny]yl}-2-[4-(ethylsulfonyl)phenyI]acetamide (46 mg) as a white solid. ¾-NMR (400 MHz,
DMSO-£¾ S ppm 0.81 (d5 J= 6.8 Hz, 6H), 1.11 (t, J= 7.2 Hz, 3H), 1.83 (m, 1H), 3.28 (q, J= 7.2 Hz, 2H), 3.70 (d, J= 6.4 Hz, 2H), 3.84 (s, 2H), 6.98 (m, 1H), 7.05 (d, J= 8.4 Hz, 1H), 7.12 (dd, J =2.0 Hz, 7.2 Hz, 1H), 7.23 (d, J= 8.4 Hz, 1H), 7.34 (m, 1H), 7.49 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.62 (d, J= 8.4 Hz, 2H), 7.86 (m, 3H), 10.45 (s, 1H); MSCES"* m/z 486 (MH+). Example 60
N-{2-cyano-2,-[(trifluoromethyl)oxy]-4-biphenylyl}-2-[4-(ethylsulfonyl)phenyl]acetamide

Step 1: To a solution of 3-amino-benzonitrile (1.0 g) in DMF (20 mL) was added n- bromosuccinimide (1.51 g) portionwise at 0 °C. The mixture was stirred for 4 hours at room temperature. The solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc : PE = 1: 100) to give 5-amino-2-bromo-benzonitrile (1.4 g).Ή-NMR (400 MHz, CDC13) δ ppm 3.90 (s, 2H), 6.71 (dd, J= 8.8 Hz, 2.8 Hz, 1H), 6.89 (d, J= 2.8 Hz, 1 H), 7.35 d, J= 8.7 Hz, 1H); MS(ES+) m/z 197 (MH+).
Step 2: A solution of 5-amino-2-bromo-benzonitrile (1.18 g) and Pd(PPh3)4 (690 mg) was stirred for 20 mins at 0 °C. Na2C03 (1.9 g) in 10 mL water and 2-(trifluoromethoxy) phenylboronic acid (1.24 g) were added. The mixture was stirred overnight at 100°C under N2. Then the solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc : PE = 1: 10) to give 4-amino-2'-trifluoromethoxy-biphenyI-2-carbonitrile as an off white solid (910 mg). MS(ES+) m z 279 (MH*).
Step 3: A mixture of 4-amino-2'-trifliioromethoxy-biphenyl-2-carbonitrile (70 mg), [4- (ethylsulfonyI)phenyl]acetic acid (intermediate la, 63.2 mg), HOBt (44.2 mg) and EDC (62.7 mg) in DCM (2 mL) was stirred at room temperature for 1 day. Solvent was removed under reduced pressure, and the residue was purified by MDAP to afford N- {2-cyano-2'-[(trifluoromethyl)oxy]-4-biphenylyl} - 2-[4-(ethylsulfonyl)phenyl]acetamide (39 mg) as a white solid. Ή-NMR (600 MHz, DMSO- ) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.88 (s, 2H). 7.53 (m, 4H), 7.63 (m, 3H), 7.88 (m, 3H), 8.21 (d, J= 1.8 Hz, 1H), 10.75 (s, 1H); i9F-NMR (376 MHz, DMSO-c?6) δ ppm -56.67;
MS(ES+) m/s 489 (MH+). Example 1
7V-{2-chloro-2'-[(trifluoromethyl)oxy]-3-biphenylyl}-2-[4-(ethylsuIfonyI)phenyl]acetarnide

Step 1: To a solution of 2-chloro-3-nitro-benzoic acid (5.0 g) in CC14 (100 mL) was added yellow HgO (8.1 g) at room temperature. The mixture was heated to reflux and irradiated with light. Br2 (1.92 mL) was added dropwise. The reaction solution was kept under reflux for 4 hours. Then the reaction was quenched with sat. NaHC03 solution. The mixture was filtered and the filtrate was extracted with EtOAc (50 mL x 3). The combined organic layers were dried over anhydrous Na2S04 and concentrated. The residue was purified by column chromatography (EtOAc : PE = 1 :500) to give l-bromo-2-chloro-3-nitro-benzene (2.27 g) as an off white solid. :H-NMR (400 MHz, CDC13) δ ppm
7.22 (dd, J= 6.5 Hz, 16.3 Hz, 1H), 7.66 (dd, J= 1.4 Hz, 8.0 Hz, 1H), 7.79 (dd, J~ 1.5 Hz, 8.2 Hz,
1H).
Step 2: A solution of l-bromo-2-chloro-3-nitro-benzene (2.03 g) and Pd(PPh3)4 (1 g) in toluene (50 mL) and EtOH (8 mL) was stirred for 20 mins at 0 °C. Na2C03 (2.74 g) and 2- (trifluoromethoxy) phenylboronic acid (1.77 g) were added. The mixture was stirred for 4 hours at
100°C under N2. The solution was concentrated and the residue was purified by column
chromatography (EtOAc : PE = 1:30) to give 2-chloro-3-nitro-2'-trifluoromethoxy-biphenyl (1.8 g) as a yellow oil.
Step 3: A solution of 2-chloro-3-nitro-2'-trifluoromethoxy-biphenyl (1.33 g) and SnCl2-2H20 (4.72 g) in MeOH (20 mL) was heated under reflux for 4 hours. The reaction mixture was concentrated. The residue was diluted with EtOAc (50 mL) and water (50 mL). The solution was adjusted to pH = 9 with NaOH solution. The mixture was filtered and the filtrated was extracted with EtOAc (50 mL x 3). The combined organic layers were dried over anhydrous NazSC^ and concentrated. The residue was purified by column chromatography on silica gel (EtOAc : PE = 1: 10) to afford 2-chloro-2'-trifluoromethoxy-biphenyl-3-ylamine (1.12 g) as an off white solid. MSiES^ m/∑ 288 (MH*).
Step 4: Oxalyl chloride (0.153 mL) was added into a solution of [4- (etliyIsulfonyl)phenyI]acetic acid (intermediate la, 80 mg) in DCM (2 mL) dropwise. DMF (0.054 mL) was then added. The resulting mixture was stirred at room temperature for 3 hours. After removal of solvent, the residue was dissolved in DCM (1 mL). The obtained solution was added into a solution of 2-chIoro-2'-trifluoroinethoxy-biphenyl-3-ylamine (111 mg) and pyridine (0.085 mL) in DCM (1 mL) at 0°C dropwise. After the addition, the reaction mixture was stirred at room temperature overnight. Solvent was removed under reduced pressure, and the residue was purifed by MDAP to afford N-{2-chloro-2'-[(trifluoromethy])oxy]-3-biphenylyI}-2-[4- (ethyIsulfonyl)phenyl]acetamide (63 mg) as a white solid. Ή-NMR (600 MHz, DMSO- ) δ ppm
1.10 (t, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.92 (s, 2H), 7.17 (dd, J= 7.6 Hz, 1.2 Hz, 1H), 7.40 (m, 2H), 7.50 (m, 2H), 7.61 (m, 3H), 7.78 (dd, J- 8.4 Hz, 1.2 Hz, 1H), 7.85 (d, J= 8.4 Hz, 2H), 9. 1 (s, 1H); 19F-NMR (376 MHz, DMSO-i/6) δ ppm -56.13; MS(ES+) m/z 498 (MH*).
Example 62
N-{2-chloro-2'-[(cyclopentylmethyl)oxy]-4-biphenyIyl}-2-[4-(ethyIsulfonyl)phenyI)acetamide

Step 1: DIAD (1.349 mL) was added into a solution of 2-bromophenol (0.8 g),
cyclopentylmethanol (0.695 g) and triphenylphosphine (1.819 g) in tetrahydrofuran (THF) (20 mL) at 0°C dropwise. The resulting reaction mixture was stirred at 0°C for 1 hour, then at room temperature
overnight. Solvent was removed under reduced pressure. The residue was purified by chromatography (PE) to afford 2-bromophenyl cyclopentylmethyl ether (760 mg) as a colorless oil. Ή-NMR (600 MHz, CDC13) δ ρριη 1.41 (m, 2H), 1.61 (m, 2H), 1.67 (m, 2H), 1.86 (m, 2H), 2.43 (m, 1H), 3.90 (d, J= 6.6 Hz, 2H), 6.81 (t, J= 7.2 Hz, 1H), 6.88 (d, J= 8.4 Hz, 1H), 7.24 (t, J= 7.2 Hz, 1H), 7.52 (m, 1H).
Step 2: A mixture of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2- [4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 90 mg), 2-bromophenyl cyclopentylmethyl ether (45 mg), PdCl2(dppf)-CH2Cl2 adduct (11.52 mg) and Cs2C03 (69.0 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered tlirougl'i celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-{2-chIoro-2'-[(cyclopentylmethyl)oxy]- 4-biphenylyl}-2-[4-(ethylsulfonyl)phenyl]acetamide (32 mg) as a yellow solid. 'H-NMR (400 MHz, DMSO-i¾) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 1.20 (m, 2H), 1.42 (m, 4H), 1.57 (m, 2H), 2.11 (m, 1H), 3.28 (q, 7.2 Hz, 2H), 3.80 (d, J= 6.4 Hz, 2H)} 3.84 (s, 2H), 6.98 (t, J= 7.6 Hz, 1H), 7.06 (d, J= 8.4 Hz, 1H), 7.11 (dd, J= 7.6 Hz, 1.6 Hz, 1H), 7.23 (d, J= 8.4 Hz, 1H), 7.34 (m, 1 H), 7.49 (dd, J= 8.4 Hz, 2.0 Hz, 1H), 7.62 (d, J- 8.4 Hz, 2H), 7.86 (m, 3H), 10.48 (s, 1H); MS(ES+) m/z 512 (MH+).
Example 63
Ar-{2-chloro-2'-[(l-methyl-4-piperidinyl)oxy]-4-biphenyIyl}-2-[4- (ethylsulfonyl)phenyl]acetamide, trifluoroacetic acid salt

Step 1: To a mixture of 2-bromophenol (0.4 g), 1,1-dimetliylethyl 4-hydroxy-l- piperidinecarboxylate (0.698 g) and Ph3P (0.910 g) in tetrahydrofuran (THF) (10 mL) cooled at 0 °C, was added DIAD (0.674 mL) dropwise. The mixture was stirred at this temperature for 1 hour, and further stirred at room temperature overnight. The mixture was concentrated under reduced pressure. The residue was purified directly by flash chromatography (EtOAc:PE = 0: 1 to 1:9) to afford 1,1- dimethylethyl 4-[(2-bromophenyl)oxy]-l-piperidinecarboxylate (767 mg) as a yellow oil. Ή-NMR (400 MHz, CDC13) δ ppm 1.47 (s, 9H), 1.86 (m, 4H), 3.49 (m, 2H), 3.64 (m, 2H), 4.57 (m, 1H), 6.84
(dt, J= I A Hz, 7.8 Hz, 1H), 6.92 (dd, J= 1.2 Hz, 8.2 Hz, 1H), 7.23 (m, 1 H), 7.54 (dd, J= 1.6 Hz, 7.9 Hz, 1H).
Step 2: To a solution of 1,1-dimethylethyl 4-[(2-bromophenyl)oxy]-l-piperidinecarboxylate (600 mg) in DCM (6 mL) was added TFA (0.779 mL) dropwise. The reaction was stirred at room temperature for 3 days. The mixture was concentrated under reduced pressure, and the residue was neutralized with sat. Na2C03, and then extracted with DCM. The organic layer was washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford 4-[(2- bromophenyl)oxy]piperidine (468 mg) as a brown oil. MSfES m/z 256 (MH+).
Step 3: 4-[(2-Bromophenyl)oxy]piperidine (110 mg) was dissolved in acetonitrile (6 mL) and acetic acid (1 mL). The mixture was cooled to 0 °C. Then 37% aqueous formaldehyde (1 mL) and sodium cyanoborohydride (135 mg) were added. The resulting mixture was allowed to room temperature and stirred for about 4 hours. The mixture was neutralized with sat. Na2C03, and then extracted with DCM. The organic phase was dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford 4-[(2-bromophenyl)oxy]-l -methylpiperidine (106 mg) as a yellow oil. MS(ES+) m/z 270 (MH+).
Step 4: A mixture of 4-[(2-bromophenyl)oxy]-l-methylpiperidine (100 mg), N-[3-chloro-4- (4,4,5,5-tetramethyl-l,3,2-dioxaboroIan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetainide
(intermediate 6b, 189 mg), PdCl2(dppf)-CH2Cl2 adduct (15 mg) and Cs2C03 (145 mg) in acetonitrile (1.8 mL) and water (0.6 mL) was bubbled with N2, and then sealed in a reaction vessel. The mixture was heated in the microwave at 100°C for 30 mins. The mixture was filtered through silica and celite, washed with ACN, DCM and EtOAc. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-{2-chloro-2'-[(l-methyl-4-piperidinyl)oxy]-4- biphenylyl}-2-[4-(ethylsulfonyI)phenyl]acetamide, trifluoroacetic acid salt (64 mg) as off-white solid. Ή-NMR (400 MHz, DMSO-i 6, after neutralized with K2C03) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 1.48 (m, 2H), 1.75 (br, 2H), 2.06 (m, 5H), 2.27 (br, 2H), 3.28 (q, J= 7.3 Hz, 2H), 3.84 (s, 2H), 4.31 (s, 1H), 6.98 (t, J= 7.4 Hz, 1H), 7.11 (m, 2H), 7.23 (d, J= 8.3 Hz, 1H), 7.32 (t, J- 7.4 Hz, 1H), 7.50 (dd, J= 1.2 Hz, 8.2 Hz, 1H), 7.62 (d, J= 8.1 Hz, 2H)} 7.86 (m, 3H), 10.56 (br, 1H); ,9F-NMR (376 MHz, DMSO-4 5 ppm -74.04; MS(ES^) m/z 527 (MH+).
Example 64
N-{3-chloro-4- [3-(trifluoromethyl)-2-pyridinyI] phenyl}-2- [4-(ethyIsulfonyl)phenyl] acetamide

A mixture of 2-bromo-3-(trifluoromethyl)pyridine (50.9 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyI]-2-[4-(ethylsulfonyl)phenyl]acetainide (intermediate 6b, 95 mg), PdCl2(dppf)-CH2Cl2 adduct (13.38 mg) and Cs2C03 (80 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered tlirougli celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-{3-chloro-4-[3-(trifluoromethy])-2- pyridinyl]phenyl}-2-[4-(ethylsulfonyl)phenyl]acetamide (42 mg) as a white solid. ]H-NMR (400 MHz, DMSO-c/e) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, /= 7.2 Hz, 2H), 3.86 (s, 2H), 7.34 (d, J= 8.4 Hz, 1H), 7.55 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.63 (d, J = 8.4 Hz, 2H), 7.70 (m, 1H), 7.86 (d, J= 8.4 Hz, 2H)5 7.94 (d, J= 2.4 Hz, 1H), 8.33 (d, J= 8.0 Hz, 1H), 8.92 (d, J= 4.4 Hz, 1H), 10.61 (s, 1H); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -58.57; MS(ES+) m/z 483 (MH÷).
Example 65
7V-{2-chloro-2'-[(methyloxy)methyI]-4-biphenylyl}-2~[4-(ethyIsulfonyl)phenyl]acetamide

A mixture of (2-bromophenyl)methyl methyl ether (45.3 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyI]-2-[4-(ethylsulfonyl)phenyl]acetamide (intennediate 6b, 95 mg), PdC¾(dppf)-CH2Cl2 adduct (13.38 mg), and Cs2C03 (80 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-{2-chloro-2'-[(methyloxy)methyl]-4- biphenylyl}-2-[4-(ethylsuIfonyl)phenyl]acetamide (31 mg) as a white solid.Ή-NMR (600 MHz, DMSO-£/6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.13 (s, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 4.09 (d, J= 8.4 Hz, 1H), 4.18 (d, J= 8.0 Hz, 1H), 7.13 (d, /= 7.2 Hz, 1H), 7.25 (d, J= 8.4 Hz, 1H), 7.35 (t, J = 7.2 Hz, 1H), 7.40 (t, J= 7.2 Hz, 1H), 7.48 (d, J= 7.2 Hz, 1H), 7.52 (dd, 7= 1.2 Hz, 8.4 Hz, 1H),
7.62 (d, J= 7.8 Hz, 2H), 7.86 (d, J- 7.8 Hz, 2H), 7.93 (d, J= 1.8 Hz, 1H), 10.56 (s, 1H); MS(ES+) m/z 426 ((M-MeOH+H)+).
Example 66
N-{2-chloro-2,-[(dimethylamino)methyll-4-biphenylyl}-2-[4-(ethyIsuIfonyl)phenyl)acetamide, trifluoroacetic acid salt

Step 1: To a solution of dimethylamine hydrochloride (0.220 g) in ethanol (20 mL) was added Et3N (0.377 mL), 2-bromobenzaIdehyde (0.5 g) successively. The mixture was stirred at room temperature overnight. Sodium cyanoborohydride (0.340 g) was added in one portion. The reaction mixture was stirred at room temperature for 1 day. The solid was filtered off. The filtrate was concentrated under reduced pressure. The residue was redissolved in DCM, washed with sat. Na2C03 solution, dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure, and the residue was purified by cliromatography (EtOAc : PE= 0:1 to 12:88) to afford l-(2- bromophenyl)-N,N-dimethylmethanamine (440 mg) as a yellow oil. MSiES^ m/z 214 (MH+).
Step 2: A mixture of 1 -(2-bromophenyl)-N,N-dimethylmethanamine (80 mg), N-[3-chloro-4-
(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(etliylsulfonyl)phenyl]acetamide
(intermediate 6b, 191 mg), Pd(Ph3P)4 (43.2 mg) and Na2C03 (1.868 mL) (2 M aqueous solution) in 1,4-dioxane (4 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture was filtered through celite and silica gel. The residue was purified by MDAP to afford N-{2-chIoro-2'-[(dimethylamino)metliyl]-4-biphenyIyl}-2-[4-(ethylsulfonyl)phenyl]acetamide, trifluoroacetic acid salt (13 mg) as a white solid. Ή-NMR (600 MHz, MeOD-c/4) δ ppm 1.22 (t, J= 7.2 Hz, 3H), 2.58 (s, 3H), 2.75 (s, 3H), 3.21 (q, J= 7.2 Hz, 2H), 3.87 (s, 2H), 4.02 (d, J= 8.8 Hz, 1H), 4.34 (d, J= 8.8 Hz, 1H), 7.32 (m, 2H), 7.60 (m, 6H), 7.90 (d, /= 8.4 Hz, 2H), 7.95 (d, J= 1.8 Hz, 1H); 19F-NM (376 MHz, MeOD-i¾ δ ppm -76.98; MSfES4) m/z 471 (MH+).
Example 67
2-[4-(ethylsulfonyl)phenyl]-N-(6-methyl-5-{2-[(trifluoromethyl)oxy]phenyl}-2- pyridinyl)acetamide, trifluoroacetic acid salt

Intermediate 67a: N-(5-bromo-6-meu lpyridm-2-vD-2-(4-( ethylsulfonvnphenyltacetamide
A mixture of 5-bromo-6-methyl-2-pyridinamine (500 mg), [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 641 mg), EDC (615 mg) and HOBt (433 mg) in DCM (12 mL) was stirred at room temperature overnight. The mixture was diluted with DCM, washed with water, sat. Na2COj and brine. The organic solution was dried over anhydrous Na2S04. After concentration, the residue was titurated from EtOAc to afford N-(5-bromo-6-methylpyridin-2-yl)-2-(4- (ethyIsulfonyl)phenyl)acetamide (395 mg) as an off-white solid. MS(ES÷) m/z 397 (MH4).
Preparation of 2-r4-(ethylsulfonyl)phenyl1-N-(6-methyl-5- j2-r(trifluoromethyl')oxylphenyl}-2- pyridinvDacetamide. trifluoroacetic acid salt
A mixture of {2-[(trifluoromethyI)oxy]phenyl}boronic acid (54.4 mg), N-(5-bromo-6- methylpyridin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (intermediate 67a, 100 mg), PdC^Cdppf)-
CH2C12 adduct (10 mg) and Cs2C03 (98 mg) in acetonitrile (1.2 mL) and water (0.4 mL) was bubbled with N2, then sealed in a reaction vessel and heated in the microwave at 100°C for 30 mins. After cooling to room temperature, the mixture was filtered through silica gel and celite, washed with ACN, EtOAc and DCM. The combined filtrates were concentrated under reduced pressure, and the residue was purified directly by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-(6-methyl-5-{2- [(trifluoromethyl)oxy]phenyl}-2-pyridinyl)acetamide, trifluoroacetic acid salt (58 mg) as a white solid. Ή-ΝΜΡν (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 2.22 (s, 3H), 3.28 (q, J= 7.3 Hz, 2H), 3.89 (s, 2H), 7.51 (m, 5H), 7.63 (d, J= 8.2 Hz, 2H), 7.85 (d, J= 8.2 Hz, 2H), 7.97 (d, J= 8.4 Hz, IH), 10.96 (s, IH); I9F-NMR (376 MHz, DMSO-J6) δ ppm -56.45, -74.96; MS(ES ) m/z 479 (MH+).
Example 68
2-[4-(ethylsulfonyl)phenyl]-iV-(4-methyI-5-{2-[(trifluoromethyl)oxy]pheiiyl}-2- pyridiny])acetamide

Step 1: A mixture of 5-bromo-4-methyl-2-pyridinamine (500 mg), [4- (ethylsulfonyl)phenyl] acetic acid (intermediate la, 671 mg), HOBt (470 mg) and EDC (666 mg) in
DCM (10 mL) was stirred at room temperature overnight. The reaction mixture was washed with brine and sat. NaHC03 solution. The organic phase was dried over anliydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure to afford N-(5-bromo-4-methyl-2-pyridinyl)-2- [4-(ethyIsulfonyl)phenyl]acetamide (700 mg) as a yellow solid. MSiJES ) m/z 397 (MH+).
Step 2: A mixture of N-(5-bromo-4-methyl-2-pyridinyl)-2~[4-(ethylsulfonyl)phenyl]acetamide
(70 mg), {2-[(trifluoromethyl)oxy]phenyl}boronic acid (39.9 mg), PdCl2(dppf)-CH2Cl2 adduct (11.51 mg) and Cs2C03 (68.9 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure. The residue was purified by MDAP to afford 2-[4-(etliylsulfonyl)phenyl]-N-(4-methyl-5- {2-[(trifluoromethyl)oxy]phenyl} -2- pyridiny])acetamide (43 mg) as a white solid.Ή-NMR (400 MHz, DMSO-i¾) δ ppm 1.10 (t, J = 7.6 Hz, 3H), 2.08 (s, 3H), 3.28 (q, J= 7.6 Hz, 2H), 3.90 (s, 2H), 7.45 (m, 1H), 7.51 (m, 1H), 7.58 (m, 1H), 7.63 (d, J= 8.0 Hz, 2H), 7.85 (d, J= 8.0 Hz, 2H), 8.04 (s, 1H), 8.09 (s, 1H), 10.89 (s, 1H); 19F-NMR (376 MHz, DMSO-c/fs) δ ppm -56.47; MS(ES+) m/z 479 (MH*). Example 69
2-[4-(ethyIsulfonyl)phenyI]-iV-{2-(5-pyrimidinyl)-2'-[(trifIuoromethyl)oxy]-4- biphenylyl}acetamide, trifluoroacetic acid salt

Intermediate 69a: 414,515 etramethyl-2-(4-nitro-2'-(trifluoromethoxy -[l,l l-bipheriyll-2-yl')-L3.2- dioxaborolane
Step 1: A mixture of {2-[(trifluoromethyI)oxy]phenyl}boronic acid (1.097 g), l-bromo-2- chloro-4-nitrobenzene (1.2 g), PdCI2(dppf)-CH2Cl2 adduct (100 mg) and Cs2C03 (1.984 g) in acetonitrile (13 mL) and water (4.33 mL) was bubbled with N2, then sealed in a reaction vessel and heated in the microwave at 100°C for 1 hour. The mixture was filtered through celite, washed with ACN and EtOAc. The filtrate was concentrated under reduced pressure, and the residue was partitioned between water and EtOAc. The organic layer was washed with brine and dried over anliydrous Na2S04. After filtration, solvent was removed in vacuo to afford 2-chloro-4-nitro-2'- (trifluoromethoxy)-l,l'-biphenyl (1.655 g) as a brown oil. MSfES^ m/z 318 (MH").
Step 2: A mixture of 2-chloro -nitro-2'-(trifluorometlioxy)-l, -biphenyI (1.3 g),
dicyclohexyl[2',4',6'-tris(l-met ylethyl)-2-biphenylyl]phosphane (0.624 g), Pd2(dba)3 (0.300 g), bis(pinacolato)diboron (3.12 g) and potassium acetate (1.205 g) in 1 ,4-dioxane (15 mL) was bubbled with N2, and then sealed in a reaction vessel. The mixture was heated in the microwave at 90°C for 1 hour. The mixture was filtered through celite, and washed with EtOAc. The filtrate was washed with brine, then dried over anhydrous Na2S04. After concentration, the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1 :9) to afford 4,4,5,5-tetramethyl-2-(4-nitro-2'- (trifluoromethoxy)-[l, -biphenyI]-2-yl) -1,3,2-dioxaborolane (788 mg). MS(ES^) m/z 410 (ΜΗ+).
Preparation of 2-r4-('ethylsulfonyl)phenyl1-N--[2-(5-pyrimidinyl")-2'-r(trifluoromethvnoxyl-4- biphenylvDacetamide. trifluoroacetic acid salt
Step 1: A mixture of 4,4,5,5-tetramethyI-2-(4-nitro-2'-(trifluoromethoxy)-[l ,l '-biphenyl]-2- yI)-l ,3,2-dioxaborolane (intermediate 69a, 200 mg), 5-bromopyrimidine (93 mg), Pd(Ph3P)4 (45.2 mg) and 2 M aqueous Na2C03 (0,978 mL) in 1,4-dioxane (4 mL) was bubbled with N2) and then sealed in a reaction vessel. The mixture was heated in the microwave at 100°C for 30 mins. The mixture was partitioned between water and EtOAc. The organic layer was washed with brine and dried over anhydrous Na2S04. After concentration, the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 3:7) to afford 5- {4-nitro-2'-[(trifluoromethyl)oxy]-2-biphenylyl}pyrimidine (125 mg) as a yellow oil. MSfES"1) mlz 362 (MH*).
Step 2: To a solution of 5-{4-nitro-2'-[(trifluoromethyl)oxy]-2-biphenylyl}pyrimidine (125 mg) in ethanol (5 mL) and tetrahydrofuran (THF) (1 mL) was added HC1 (0.6 mL), followed with addition of tin(II) chloride (394 mg) at room temperature. The resulting mixture was stirred at 50°C for 2 hours. After cooling to room temperature, the mixture was neutralized with sat. Na2C03, and then extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford 2-(5- pyrimidinyl)-2'-[(trifluoromethyl)oxy]-4-biphenylamine (111 mg) as a yellow oil. MSiES ) m/z 332 (MH4).
Step 3: A mixture of 2-(5-pyrimidinyl)-2'-[(trifluoromethyl)oxy]-4-biphenyl amine (111 mg), [4-(ethylsulfonyl)phenyl]acetic acid (intermediate l a, 84 mg), EDC (77 mg) and HOBt (54.3 mg) in DCM (6 mL) was stirred at room temperature ovemiglit. The mixture was concentrated in vacuo, and the residue was directly purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-{2-(5- pyrimidinyl)-2'-[(trifluoromethyl)oxy]-4-biphenylyl}acetamide, trifluoroacetic acid salt (54 mg) as a light yellow solid. 'H-NMR (400 MHz, MeOD-t¾) δ ppm 1.21 (t, J= 7.4 Hz, 3H), 3.20 (q, J= 7.4 Hz, 2H), 3.90 (s, 2H)5 7.12 (m, 1H), 7.42 (m, 3H), 7.60 (m, 3H), 7.77 (dd, J= 1.8 Hz, 8.4 Hz, 1H), 7.87
(m, 3H), 8.52 (s, 2H), 9.00 (s, IH); F-NMR (376 MHz, MeOD-^4) δ ppm -58.84, -77.45; MS(ES+) m/z 542 (MH4).
Example 70
AL[2 5'-dichloro-2-(trifluoromethy])-4-biphenylyl]-2-[4-(ethyIsuIfonyl)phenyl]acetamide

A mixture of (2,5-dichlorophenyl)boronic acid (34.6 mg), N-[4-bromo-3- (trifluoromethyl)phenyI]-2-[4-(ethy]sulfonyI)phenyl]acetamide (intennediate 42a, 74.2 mg),
PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (81 mg) in acetonitrile (1.5 inL) and water (0.5 mL) was sealed in a reaction vessel, and heated in the microwave at 100°C for 30 mins. As the starting material remained, more (2,5-dichIorophenyl)boronic acid (15.72 mg) and PdCI2(dppf)-CH2Cl2 adduct (10 mg) were added. The resulting mixture was heated again in the microwave at 100°C for 30 mins. The mixture was filtered through silica gel. The filtrate was diluted with water, and extracted with EtOAc for 3 times. The combined organic layers were dried over Na2S04. After filtration, the filtrate was concentrated under vacuum. The residue was purified by MDAP to give N-[2',5'-dichloro- 2-(trifluoromethyl)-4-biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (6 mg) as a white solid. Ή- NMR (400 MHz, DMSO-rf6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, /= 7.2 Hz, 2H), 3.87 (s, 2H), 7.36 (d, J= 8.4 Hz, IH), 7.43 (d, J- 2.4 Hz, IH), 7.53 (m, IH), 7.62 (m, 3H), 7.86 (m, 3H), 8.19 (d, J = 2.0 Hz, IH), 10.74 (s, IH); 19F-NMR (376 MHz, DMSO-i 6) 5 ppm -58.14; MStES*) m/z 516 (MH*).
Example 71
iV-(2' ^'-dichlor o-4-bip henylyl) -2- [4-(ethylsulfonyI)phenyl] ac etamide
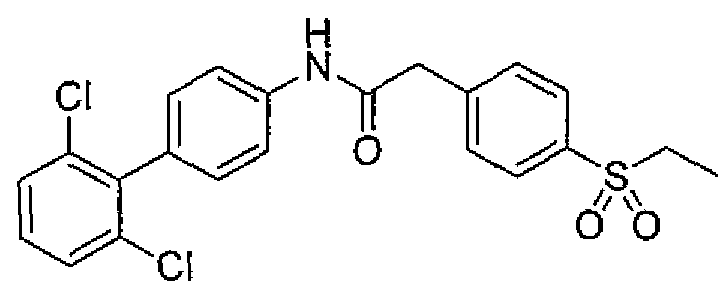
A mixture of (2,6-dichlorophenyl)boronic acid (32.9 mg), N-(4-bromophenyI)-2-[4- (ethylsulfonyl)phenyl]acetamide (intennediate lb, 60 mg), PdC]2(dppf)-CH2CI2 adduct (12.8 mg) and Cs2C03 ( 1.4 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 mins. The mixture was filtered through celite. The filtrate was
concentrated under reduced pressure, and the residue was purified by MDAP to afford N-(2',6'- dichloro-4-biphenylyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (8 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-.4) 5 ppm 1.10 (t, J= 7.2 Hz, 3Η), 3.28 (q, J= 7.2 Hz, 2Η), 3.83 (s, 2H), 7.20 (d, J= 8.4 Hz, 2H), 7.41 (t, J= 8.40 Hz, 1H)} 7.57 (d, J= 8.0 Hz, 2H), 7.63 (d, /= 8.4 Hz, 2H), 7.80 (d, J= 8.4 Hz, 2H), 7.86 (d, J= 8.0 Hz, 2H), 10.43 (s, 1H); MSiES4) m/z 448 (MH^).
Example 72
jV-(2-chIoro-2'-ethyl-4-biphenylyl)-2-[4-(ethylsuIfonyl)phenyl]acetamide
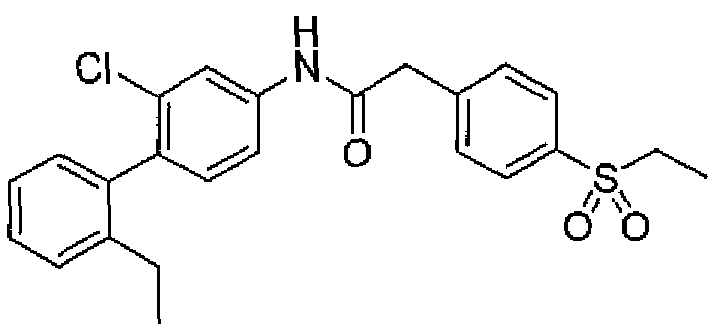
A mixture of l-bromo-2-ethylbenzene (33 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 91 mg),
PdCl2(dppf>CH2Cl2 adduct (10 mg) and Cs2C03 (69.7 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered tlirough celite. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-(2-chIoro-2'-ethyl-4-biphenylyl)-2-[4-(etJiylsulfonyl)phenyl]acetamide (3 mg) as a yellow solid. ]H-NMR (600 MHz, DMSO-i 6) δ ppm 0.09 (t, J= 4.8 Hz, 3H), 1.03 (t, J= 7.2 Hz, 3H), 2.31 (m, 2H), 3.22 (q, J- 7.2 Hz, 2H), 3.77 (s, 2 H), 6.98 (d, J= 7.8 Hz, 1H), 7.18 (d, J = 8.4 Hz, 2H), 7.26 (s, 2H), 7.45 (d, J=> 8.4 Hz, 1H), 7.56 (d, J= 7.8 Hz, 2H), 7.79 (d, J= 7.8 Hz, 2H), 7.86 (s, 1H), 10.48 (s, 1H); MSfES^ m z 442 (MH+).
Example 73
N-(2-chIoro-4,-fluoro-2'-(trifluoromethyl)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

A mixture of N-[3-chloro-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenylj-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 100 mg), 1 -bromo-4-fluoro-2- (trifluoromethyl)benzene (62.9 mg), potassium carbonate (89 mg), and Pd(Ph3P)4 (12.5 mg) in
toluene (5 mL) and water (5 mL) was refluxed overnight. The mixture was partitioned between EtOAc (20 mL) and water (20 mL). The organic phase was washed with water (10 mL x 2) and brine (10 mL), dried and concentrated. The residue was purified by prep-HPLC to give N-(2-chloro-4'- fluoro-2'-(trifluoromethyI)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (18 mg) as a light yellow solid. 'H-NMR (400 MHz, CDC13) δ ppm 1.29 (t, J= 7.2 Hz, 3H), 3.13 (q, J= 7.2 Hz, 2H), 3.82 (s, 2H), 7.17 (d, J= 8.4 Hz, 1H), 7.26 (m, 2H), 7.43 (m, 2H), 7.53 (d, J= 8.0 Hz, 2H), 7.77 (m, 4H); ,9F- MR (376 MHz, CDC13) δ ppm -59.56, -111.88; MSiES m/z 500 (MH4).
Example 74
V-[2-chloro-2'-(4-piperidinyloxy)-4-biphenyIyIJ-2-[4-(ethylsuIfonyI)phenyI]acetamide, trifluoroacetic acid salt

A mixture of 1 , 1 -dimethylethyl 4-[(2-bromophenyI)oxy]-l-piperidinecarboxylate (80 mg, see step 1 for synthesis of example 63), N-[3'Chloro-4-(4,4,5,5-tetrametliyl-l,3,2-dioxaboroIan-2- yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 108 mg), Cs2C03 (88 mg) and PdCl2(dppf)-CH2Cl2 adduct (10 mg) in acetonitrile (1.3 mL) and water (0.433 mL) was bubbled with N2, and then sealed in a reaction vessel. The mixture was heated in the microwave at 100°C for 30 mins. The mixture was filtered through celite, washed with ACN, EtOAc and DCM. The combined filtrates were concentraed under reduced pressure, and the residue was parti oned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2S0 . After filtration and concentration, the residue was dissolved in DCM (5 mL), to which TFA (0.173 mL) was added dropwise. The resulting mixture was stirred at room temperature for 4 hours. The mixture was concentrated under reduced pressure and the residue was purified by MDAP to afford N-[2-chloro-2'- (4-piperidinyloxy)-4-biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide, trifluoroacetic acid salt (17 mg) as an off-white solid. Ή-NMR (400 MHz, DMSO-i¾) δ ppm 1.11 (t, /= 7.3 Hz, 3H), 1.70 (br, 2H), 1.95 (br, 2H), 2.89 (br, 2H), 3.00 (br, 2H), 3.28 (q, J= 7.3 Hz, 2H), 3.84 (s, 2H), 4.61 (m, 1H), 7.03 (t, J= 7.5 Hz, 1H), 7.16 (m, 2H), 7.26 (d, J= 8.4 Hz, 1H), 7.37 (m, 1H), 7.52 (dd, J= 2.1 Hz,
8.4 Hz, 1H), 7.63 (d, J= 8.3 Hz, 2H), 7.87 (d, J= 8.3 Hz, 2H), 7.92 (d, J- 2.0 Hz, 1H), 10.54 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -74.43; MS(ES+) m/z 513 (MH+).
Example 75
2-[4-(ethylsulfonyI)phenyl]-N-{2-(l-methyH,2,3,6-tetrahydro-4-pyridinyI)-2'- [(trifluoromethyI)oxy]-4-biphenylyl}acetamide, trifluoroacetic acid salt

Intermediate 75a; l-metliyl-1.2.3,6-tetrahydro-4-pyridinyl trifluoromethanesulfonate
To a solution of l-methyl-4-piperidinone (1.2 g) in tetrahydrofuran (THF) (25 mL) cooled at - 78 °C was added a solution of LiHMDS (1 M in THF, 13.79 mL). The resulting mixture was stirred at this temperature for 30 mins. Then 1,1,1-trifluoro-N-phenyl-N-
[(trifluoromethyl)sulfonyl]methanesulfonamide (5.68 g) was added. The reaction mixture was stirred at this temperature for 1 hour, then gradually warmed to room temperature, and stirred for another 5 hours. The mixture was quenched with sat. NH4C1, extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After filtration and concentration, the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 4: 1) to afford l-methyl-l,2,3,6-tetrahydro-4-pyridinyl trifluoromethanesulfonate (1.76 g) as a yellow sticky oil. Ή- NMR (400 MHz, CDC13) δ ppm 2.42 (s, 3H), 2.48 (m, 2H), 2.73 (t, J= 5.8 Hz, 2H), 3.13 (m, 2H), 5.73 (m, 1H).
Preparation of 2-r4-(ethylsulfonyl)phenyll-N- (2-f 1 -methyl- 1.2.3.6-tetrahydro4-pyridinyl)-2'- f(trifluoromethyl oxyl-4-biphenylvU cetamide. trifluoroacetic acid salt
Step 1: A mixture of N-{2-chloro-2'-[(trifluoromethyl)oxy]-4-biphenylyl}-2-[4-
(ethylsulfonyl)phenyl]acetamide (Example 25, 900 mg), Pd2(dba)3 (331 mg), tricyclohexylphosphine
(406 mg), potassium acetate (532 mg) and bis(pinacolato)diboron (1836 mg) in N,N- DMF (9 mL) was bubbled with N2, and then sealed in a reaction vessel. The mixture was heated in the microwave at 120°C for 2.5 hours. The mixture was filtered through celite, and partioned between EtOAc and water. The aqueous layer was furtlier extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous a2S04. After filtration and concentration, the residue was purified with flash chromatography (EtOAc : PE = 0:1 to 3:7) to afford 2-[4-
(ethylsulfonyl)phenyl]-N-{2-(4,4,5,5 etramethyl-l,3,2-dioxaborolan-2-yl)-2,-[(trifluoromethyl)oxy]- 4-bip enylyl}acetamide (439 mg) as a yellow sticky solid. MSiES^) m/z 590 (MH+).
Step 2: A mixture of l-methyl-l,2,3,6-tetrahydro-4-pyridinyl trifluoromethanesulfonate (intermediate 75a, 618 mg), 2-[4-(ethylsulfonyl)phenyl]-N-{2-(4J4)5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-2'-[(trifluoromethyl)oxy]-4-biphenylyl}acetamide (260 mg), Pd(Ph3P)4 (102 mg) and 2 M aqueous Na2C(¾ (1.323 mL) in 1,4-dioxane (4 mL) was bubbled with N2, and then sealed in a reaction vessel. The mixture was heated in the microwave at 100°C for 1 hour. As most of the starting material remained, more l-methyl-l,2;3,6-tetrahydro-4-pyridinyl trifluoromethanesulfonate (intermediate 75a, 464 mg) and Pd(PhjP)4 (25.5 mg) were added. The mixture was heated in the microwave for another 1.5 hours. As some of the starting material remained, more 1 -methyl -1, ,3,6- tetrahydro-4-pyridinyl trifluoromethanesulfonate (intermediate 75a, 464 mg) and Pd(Ph3P) (25.5 mg) were added. The mixture was heated in the microwave for an additional 1.5 hours. The mixture was partioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2S04. After filtration and concentration, the residue was purified by flash
chromatography (EtOAc:PE— 0: 1 to 1 :0) to afford the crude product (51 mg). This crude material was further purified by MDAP to afford 2-[4-(etliylsulfonyl)phenyl]-N-{2-(l-methyl-l,2,3,6- tetrahydro-4-pyridinyl)-2'-[(1rifluoromedayl)oxy]-4-biphenylyl}acetamide, trifluoroacetic acid salt (6 mg) as a white solid. Ή-NMR (400 MHz, MeOD-ά,) δ ppm 1.21 (t, J= 7.4 Hz, 3H), 2.31 (d, J= 18.1 Hz, IH), 2.85 (s, 3H), 3.07 (m, IH), 3.20 (m, 3H), 3.48 (m, 2H), 3.83 (m, 3H), 5.43 (s, IH), 7.26 (d, / = 8.4 Hz, IH), 7.43 (m, 5H), 7.64 (d, J= 8.3 Hz, 2H), 7.76 (d, J= 2.1 Hz, IH), 7.89 (d, J= 8.3 Hz, 2H); MSiES4) m/z 559 (MH4).
Example 76
7V-(2,6-difluoro-2'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

A mixture of N-(4-bromo-3,5-difluorophenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide
(intermediate 27a, 200 mg), 2-(trifluoromethoxy)phenylboronic acid (197 mg), Pd(Ph3P)4 (27.6 mg) and potassium carbonate (132 mg) in 1,4-dioxane (20 mL) and water (20 mL) was refluxed overnight. Solid was removed by filtration. To the filtrate was added water (50 mL) and EtOAc (50 mL). The organic phase was washed with water and brine, dried and concentrated. The residue was purified by
prep-HPLC to afford N-(2)6-difluoro-2'-(trifluoromethoxy)biphenyl-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (8 mg) as a white solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.32 (t, J= 7.2 Hz, 3H), 3.16 (q, J~ 7.2 Hz, 2H), 3.85 (s, 2H), 7.29 (m, 1H), 7.39 (m, 3H), 7.48 (m, 1H), 7.55 (d, J= 8.0 Hz, 2H), 7.68 (s, 1H), 7.89 (d, J= 8.0 Hz, 2H); MS(ES ) /z 500 (MH*). Example 77
Ar-(2-chloro-2'-(3-inethoxyazet-din-l-yl)biphenyI-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 1: A mixture of 1,2-dibromobenzene (298 mg), 3-methoxyazetidine (100 mg), Pd2(dba)3 (53 mg), BINAP (72 mg) and Cs2C03 (374 mg) in 1,4-dioxane (50 mL) was refluxed under N2 overnight. Solvent was removed, and the residue was purified by flash chromatography (EtOAc : PE = 1 : 19) to give l-(2-bromophenyl)-3-methoxyazetidine (200 mg). MS(ES+) m/z 242 (MH+).
Step 2: A mixture of 1 -(2 -bromophenyl) -3-methoxyazetidine (250 mg), N-[3-chloro-4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide
(intennediate 6b, 718 mg), Pd(Ph3P)4 (60 mg) and potassium carbonate (285 mg) in water (20 mL) and 1,4-dioxane (20 mL) was refluxed under N2 overnight. Solid was removed by filtration, and the filtrate was partitioned between water (50 mL) and EtOAc (50 mL). The organic phase was dried and concentrated. The residue was purified by flash chromatography (EtOAc : PE - 2:3) to give the crude product. Further purification by prep-HPLC afforded N-(2-chloro-2'-(3 -methoxyazeti din- 1- yl)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (20 mg) as a white solid. Ή-NMR (400 MHz, DMSO-c¾ δ ppm 1.10 (t, J- 7.2 Hz, 3H), 3.08 (s , 3H), 3.20 (m, 2H), 3.28 (q, 7= 7.2 Hz, 2H), 3.49 (m, 1H), 3.58 (m, 1H), 3.85 (s, 2H), 4.03 (in, 1H), 6-53 (d, /= 8.2 Hz, 1H), 6.79 (t, J= 7.2 Hz, 1H), 6.92 (d, J= 7.2 Hz, 1H), 7.22 (m, 2H), 7.50 (dd, J~ 8.0 Hz, 2.0 Hz, 1H), 7.63 (d, J= 8.0 Hz, 2H), 7.86 (d, J= 8.0 Hz, 2H), 7.91 (d, J= 2.0 Hz, 1H), 10.52 (s, 1H); MSfES ) m/z 499 (MH+).
Example 78
iV-(2,6-dichloro-2'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyI)acetamide

Intermediate 78a: N-(4-bromo-3,5-dichlorophenyl)-2-(4-( ethylsulfonyl)phenyl)acetainide
A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 569 mg), 4-bromo-3,5- dichlorobenzenamine (500 mg), HOBt (381 mg), EDC (597 mg) and DIPEA (2.18 mL) in DCM (50 mL) was refluxed overnight. Solvent was removed. The residue was purified by flash
chromatography (EtOAc : PE = 2: 1) to give N-(4-bromo-3,5-dichlorophenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide (500 mg) as a yellow oil. MSfES ) m/z 449 (MH+).
Preparation of N-(2,6-dichloro-2'-Ctrifluoromethoxy)biphenyl-4-yl)-2-(4- fethylsulfonvDphenyllacet amide
A mixture of N-(4-bromo-3 ,5-dichlorophenyl)-2-(4-(etliylsulfonyl)phenyl)acetamide
(intennediate 78a, 300 mg), 2-(trifluoromethoxy)phenylboronic acid (274 mg), Pd(Ph3P)4 (77 mg) and potassium carbonate (184 mg) in 1,4-dioxane (20.00 mL) was refluxed under N2 overnight.
Solvent was removed, and the residue was partitioned between water (50 mL) and EtOAc (50 mL). The organic phase was washed with water and brine, dried and concentrated. The residue was purified by prep-HPLC to afford N-(2,6-dichloro-2'-(trifiuoromethoxy)biphenyl-4-yl)-2-(4-
(ethylsulfonyl)phenyl)acetamide (70 mg ) as a white solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.31 (t, J= 7.2 Hz, 3H), 3.14 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 7.25 (m, 1H), 7.36 (m, 3H), 7.48(m, 1H), 7.56 (d, J= 8.0 Hz, 2H), 7.65 (s, 2H), 7.92 (d, J= 8.0 Hz, 2H); 19F-NMR (376 MHz, CDC13) δ ppm -57.17; MS(ES+) m/z 532 (MH4). Example 79
7Y-(2-chIoro-6'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-(propyIsulfonyl)phenyl)acetamide

Intennediate 79a: 2-f4-(propyIsulfonyl')phenyl')acetic acid
Step 1: A mixture of (4-mercaptophenyl)acetic acid (18 g, see step 2 for synthesis of intennediate la), K2COj (59.2 g) and 1-bromopropane (39.5 g) in DMF (100 mL) was stirred at RT overnight. The mixture was partitioned between ethyl acetate (200 mL) and water (800 mL). The
organic phase was washed with water (200 mL x 2) and brine (200 mL), dried and concentrated to give propyl 2-(4-(propylthio)phenyl) acetate (28 g) as a red liquid. MS(ES+) m/z 253 (MH+).
Step 2: To a solution of propyl 2-(4-(propylthio)phenyl) acetate (27 g) in DCM (250 mL) was added wCPBA (66.5 g). The reaction mixture was stirred at RT overnight. Solid was removed by filtration. The filtrate was concentrated. The residue was purified by flash chromatography
(EtOAc:PE = 1:4) to afford propyl 2-(4-(propylsulfonyl)phenyl)acetate (20 g) as a light yellow solid. MS(ES+) m/z 285 (MH ).
Step 3: A mixture of propyl 2-(4-(propyIsulfonyl)phenyI)acetate (20 g) and NaOH (8.44 g) in ethanol (100 mL) and water (100 mL) was stirred at RT overnight. Ethanol was removed under reduced pressure. The aqueous phase was washed with dichloromethane (100 mL x 2), and then acidified to pH = 1 with 6 M HC1. The mixture was extracted with ethyl acetate (200 mL x 2). The combined organic phases were washed with brine, dried over anhydrous sodium sulphate, and concentrated to give 2-(4-(propylsulfonyl)phenyl)acetic acid (12 g) as a light yellow solid. MS(ES+) m/z 242 (MH .
Preparation of N-(2-chIoro-6'-( trifluoromethoxy)biphenyl-4-yl)-2-(4- (propylsulfonyllphenvDacetamide
A mixture of 2-(4-(propylsulfonyl)phenyl)acetic acid (intermediate 79a, 606 mg), 2-chloro-6'-
(trifluoromethoxy)biphenyl-4-amine (intermediate 15b, 600 mg), HOBt (383 mg), EDC (600 mg) and
DIPEA (1.093 mL) in DCM (40 mL) was refluxed overnight. Solvent was removed, and the residue was purified by flash chromatography (EtOAc : PE = 1:2) to afford the crude product. Further purification by prep-HPLC afforded N-(2-chloro-6'-(trifluoromethoxy)biphenyl-4-yl)-2-(4- (propylsuIfonyl)phenyl)acetamide (90 mg) as a white solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.01 (t, J= 7.6 Hz, 3H), 1.77 (m, 2H), 3.08 (m, 2H), 3.84 (s, 2H), 7.29 (m, 4H), 7.42 (m, 2H), 7.56 (d, J= 8.0 Hz, 2H), 7.74 (d, J= 1.6 Hz, 1H), 7.92 (d, J= 8.0 Hz, 2H); 19F-NMR (376 MHz, CDC13) δ ppm - 57.24; MSiES ) m/z 512 (MH4).
Example 80
N-(2-chloro-2T-(4-chlorophenoxy)biphenyl-4-yl)-2-(4-(ethyIsulfonyl)phenyl)acetamide
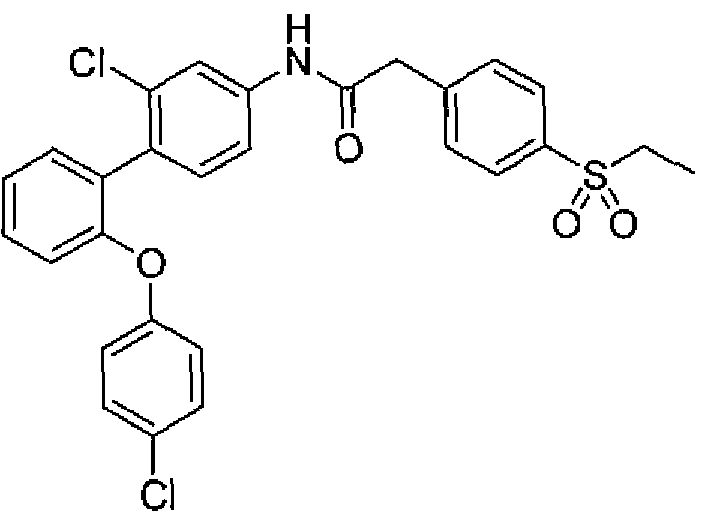
Step 1: A mixture of 2-bromophenoI (862 mg), 4-chloroplienylboronic acid (948 mg), Cu(OAc)2 (996 mg) and Et3N (2.6 g) in DCM (10 mL) was stirred at room temperature overnight under N2. The mixture was washed with 1 M HCl, sat. NaHC03 and brine successively. The organic phase was dried over anhydrous Na2SC>4 and concentrated. The residue was purified by flash chromatography (PE) to give l-bromo-2-(4-chIorophenoxy)benzene (320 mg) as a colorless liquid.
Step 2: A mixture of l-bromo-2-(4-chlorophenoxy)benzene (221 mg), N-[3-chloro-4-(4f4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 190 mg), Pd(PPh3)4 (25 mg) and Cs2COj (158 mg) in 1,4-dioxane (10 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 85 °C for 130 mins. The reaction mixture was filtered tlirougli celite and silica gel. The filtrate was concentrated. The residue was purified by prep- HPLC to afford N-(2-chloro-2'-(4-chlorophenoxy)biphenyl-4-yl)-2-(4-
(ethylsulfonyl)pheny])acetamide (60 mg) as a white solid.Ή-NMR (400 MHz, DMSO-<¾ δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.81 (s, 2H), 6.88 (d, J= 7.6 Hz, 2H), 7.02 (d, J= 8.0 Hz, 1H), 7.30 (m, 5H), 7.44 (m, 2H), 7.60 (d, J= 8.4 Hz, 2H), 7.84 (m, 3H), 10.48 (s, 1H); MS(ES+) m z 540 (ΜίΓ).
Example 81
Ar-[2',4'-dichloro-5-(phenylcarbonyl)-3-biphenyIyl]-2-[4-(ethylsulfonyl)phenyl]acetamide

Step 1: 3-"Nitrobenzoic acid (10 g) was taken up into conc. H2S04 (30 mL). The mixture was heated to 60 °C. To this mixture was added NBS (12.78 g) in three portions during 15 mins. After
stirring at this temperature for 3 hours, the reaction mixture was poured into ice-water slowly. The precipitate was collected by filtration. This solid was dissolved in EtOAc. The obtained solution was washed with brine, dried over anhydrous Na2S04. After filtration, the solution was concentrated in vacuo to afford 3-bromo-5-nitrobenzoic acid (12 g) as a white solid. 'H-NMR (400 MHz, DMSO-i¾ δ ppm 8.41 (t, J= 2.0 Hz, 1H), 8.54-8.55 (m, 1H), 8.62 (t, 2.0 Hz, 1H); MS(ES+) m/z 246 (MH+).
Step 2: A mixture of 3-bromo-5-nitrobenzoic acid (3 g), SOCl2 (1.780 mL) and DMF (0.019 mL) in DCM (20 mL) was heated under reflux for 3 hours. After concentration, the crude benzoyl chloride was dissolved into benzene (8 mL), and then added into a suspension of aluminium chloride (1.951 g) in benzene (8 mL). The resulting mixture was heated to reflux for 4 hours, and quenched with 3 M HC1. The mixture was extracted with water. The organic layer was dried over anhydrous Na2SC>4. After filtration, the solution was concentrated. The residue was purified by chromatography (EtOAc : PE = 0: 1 to 15:85) to afford (3-bromo-5-nitrophenyl)(phenyl)metlianone (1.8 g) as a yellow oil.Ή-NMR (400 MHz, CDC13) δ ppm 7.56 (t, J= 7.6 Hz, 2H), 7.71 (t, J= 1.2 Hz, 1H), 7.79 (m, 2H), 8.25 (t, J= 1.6 Hz, 1H), 8.53 (t, J= 1.6 Hz, 1H), 8.58 (t, J= 2.0 Hz, 1H); MSiES4) m/z 306 (MH4).
Step 3: A solution of dichlorostannane (1725 mg) in cone. HC1 (3.8 mL) was slowly added to a stirred suspension of (3-bromo-5-nitrophenyI)(phenyl)methanone (585 mg) in ethanol (8 mL). The reaction mixture was heated to 60°C for 30 mins. The obtained clear solution was treated with 30% aqueous NaOH until pH was strongly basic, and then extracted with DCM for 3 times. The combined organic layers were washed with brine and dried over anydrous Na2SO,¾. After filtration, solvent was removed under reduced pressure, and the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1 :4) to afford (3-amino-5-bromophenyl)(phenyl)methanone (300 mg) as a yellow solid.
MS(ES+) m/z 342 (MH÷).
Step 4: A mixture of (2,4-dichlorophenyl)boronic acid (207 mg), (3-amino-5- bromophenyl)(phenyl)methanone (300 mg), Cs2C03 (531 mg) and PdCl2(dppf)-CH2Cl2 adduct (71.0 mg) in acetonitrile (6 mL) and water (2 mL) was sealed in a reaction vessel, and heated in the microwave at 120°C for 30 mins. As the starting material remained, more (2,4- dichlorophenyl)boronic acid (104 mg) and PdCl2(dppf)-CH2Cl2 adduct (35.0 mg) were added. The reaction vessel was heated again in the microwave at 120°C for 30 mins. The mixture was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anydrous Na2S04. After filtration, solvent was removed in vacuo, and the residue was purified by flash chromatography (EtOAc:PE = 0:1 to 1:3) to afford (5-amino-2',4'-dichloro-3-biphenylyl)(phenyl)methanone (290 mg) as a yellow solid. MS(ES^) m/z 342 (MH").
Step 5: A mixture of [4-(etliylsulfonyl)phenyl]acetic acid (intermediate la, 70.0 mg), EDC (72.8 mg) and HOBt (51.3 mg) in DCM (2 mL) was stirred at room temperature under nitrogen for 10 mins. (5-Amino-2',4f-dichloro-3-biphenylyl)(phenyl)methanone (100 mg) was added. The reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed under reduced pressure. The residue was purified by MDAP to afford N-[2',4'-dichloro-5- (phenylcarbonyl)-3-biphenylyl]-2-[4-(etliyIsulfonyl)phenyl]acetamide (42 mg) as a white solid,Ή- NMR (400 MHz, DMSOtfc) δ ppm 1.09 (t, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 7.43 (t, 3= 1.6 Hz, 1H), 7.56 (m, 6H), 7.69 (m, 1H), 7.78 (m, 3H), 7.84 (d, J= 8.0 Hz, 2H), 8.03 (m, 2H), 10.66 (s, 1H); MS(ES^) m/z 553 (MH+).
Example 82
2-[4-(ethylsulfonyl)phenyll-JV-[2-(phenykarbonyl)-4-biphenylyl]acetamide

Step 2: PCC (3.34 g) was added to a mixture of (2-bromo-5-nitrophenyl)methanol (3 g) in DCM (40 mL). The reaction mixture was stirred at RT overnight. Solvent was removed in vacuo. The residue was purified by flash chromatography (EtOAc : PE = 1: 10) to give 2-bromo-5- nitrobenzaldehyde (1.9 g) as a light yellow solid.
Step 3: To a solution of 2-bromo-5-nitrobenzaldehyde (1.9 g) in tetrahydrofuran (THF) (50 mL) cooled at 0°C was added phenylmagnesium bromide ( 1.80 g) dropwise over 30 mins. The reaction mixture was stirred at 0°C for 2 hours, and then quenched with saturated NH4CI solution. The mixture
was extracted with EtOAc (150 mL x 3). The combined organic phases were dried over anhydrous Na2S04, filtered, and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 1:20) to give (2-bromo-5-nitrophenyl)(phenyl)methanol (2 g) as a white solid. (ES+) m/z 330 (MNa4).
Step 4: Dess-Martin periodinane (3.03 g) was added to a solution of (2-bromo-5- nitrophenyl)(phenyl)methanol (2.0 g) in DCM (40 mL). The reaction mixture was stirred at T overnight. Solid was removed by filtration, and the filtrate was washed with water (40 mL). The organic phase was dried over anhydrous Na2S0 , filtered, and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 1 :20) to give (2-bromo-5-nitrophenyl)(phenyl)methanone (1.9 g) as a white solid. MS(ES+) m/z 306 (MtT).
Step 5: To a solution of (2-bromo-5-nitrophenyl)(phenyl)methanone (1.9 g) in methanol (15 mL) cooled to 0°C was added iron (1.39 g), followed by the dropwise addition of 2M HCI (18.62 mL). The reaction mixture was stirred at 85 °C for 2 hours. Solid was removed by filtration, and the filtrate was concentrated. The residue was suspended in 50 mL of water, and the solution was basified to pH = 10 with aqueous NaOH. The mixture was extracted with EtOAc (100 mL x 3). The combined organic phases were dried over anhydrous Na2S04, filtered, and concentrated to afford (5-amino-2- bromophenyI)(phenyl)methanone (1.5 g) as a brown solid. MSfES ) m/z 276 (MH4).
Step 6: A mixture of (5-amino-2-bromophenyl)(phenyl)methanone (200 mg), [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 182 mg), HOBt (147 mg) and EDC (208 mg) in DCM (5 mL) was stirred at room temperature overnight. The mixture was washed with 1 M HCl, sat. NaHC03 and brine successively. The organic phase was dried over anhydrous Na2S04. After filtration, the filtrate was concentrated in vacuo to afford N-[4-bromo-3-(phenylcarbonyl)phenyl]-2- [4-(ethylsulfonyI)phenyl]acetamide (290 mg) as a yellow solid. MS(ES+) m/z 486 (MH+).
Step 7: A mixture of phenylboronic acid (19.30 mg), N-[4-bromo-3-(phenylcarbonyl)phenyl]- 2-[4-(etliylsuIfonyl)phenyl]acetamide (70 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (56.3 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a reaction vessel, and heated in the microwave at 100°C for 30 mins. The mixture was filtered through celite and silica gel. The filtrate was diluted with water, and extracted with EtOAc for 3 times. The combined organic layers were concentrated under reduced pressure. The residue was purified by MDAP to afford 2-[4- (ethylsulfonyl)phenyl]-N-[2-(phenylcarbonyl)-4-biphenylyl]acetamide (15.8 mg) as a white solid.Ή- NMR (400 MHz, DMSO-^) δ ppm 1.09 (t, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.83 (s, 2H), 7.20 (m, 5H), 7.39 (t, J= 8.0 Hz, 2H), 7.52 (m, 2H), 7.61 (m, 4H), 7.74 (d, J= 2.0 Hz, 1H), 7.85 (m, 3H), 10.55 (s, 1H); MS(ES^) m/z 484 (MH+).
Example 83
Ar-[3'-cyano-2-(phenylcarbonyl)-4-biphcnyiylJ-2-[4-(ethylsulfonyl)phenyl]acetamide

A mixture of N-[4-bromo-3-(phenyIcarbonyI)phenyl]-2-[4-(ethylsulfonyI)phenyIJacetamide (70 mg, see step 6 for synthesis of Example 82), (3-cyanophenyl)boronic acid (23.26 mg), PdCl2(dppf)- CH2C12 adduct (10 mg) and CS2CO3 (56.3 mg) in acetonitrile (1.5 lnL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 rnins. The reaction mixture was filtered tlirough celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-[3'-cyano-2-(phenylcarbonyl)-4-biphenylyl]-2-[4- (ethylsuIfonyl)phenyl]acetamide (33 mg) as a white solid.Ή-NMR (400 MHz, DMSO-c/6) δ ppm 1.09 (t, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.76 (s, 2H), 7.45 (m, 4H), 7.61 (m, 8H), 7.79 (d, J= 2.0 Hz, 1H), 7.84 (m, 2H), 7.90 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 10.61 (s, 1H); MS(ES+) m/z 509 (MH+ .
Example 84
2-[4-(ethylsulfonyl)phenyl]-.V-[6-(phenylcarbonyl)-3-biphenylyl]acetamide

Step 1: Borane-tetrahydrofuran complex (102 mL) was dropwise added to a solution of 2- bromo-4-nitrobenzoic acid (10 g) in THF (100 mL) at 0°C in 30 mins. The reaction mixture was wanned to room temperature and stirred overnight. The reaction mixture was cooled to 0°C and quenched with brine. The organic layer was separated. The aqueous layer was extracted with ethyl acetate (100 mL x 2). The combined organic phases were dried over anhydrous Na2SC>4. After filtration, the filtrate was concentrated in vacuo to afford (2-bromo-4-nitrophenyl)methanol (13 g) as a yellow solid. MS(ES+) m/z 232 (MH+).
Step 2: PCC (5.24 g) was added to a mixture of (2-bromo-4-nitrophenyl)methanol (4.7 g) in DCM (40 mL). The mixture was stirred at RT overnight. Solvent was removed in vacuo. The residue was purified by flash chromatography (EtOAc : PE = 1: 10) to give 2-bromo-4-nitrobenzaldehyde (3.3 g) as a yellow solid.
Step 3: Phenylmagnesium bromide (4.54 g) was added dropwise to a solution of 2-bromo-4- nitrobenzaldehyde (4.8 g) in tetrahydrofuran (THF) (50 mL) cooled at 0°C over 30 mins. The reaction mixture was stirred at 0°C for 2 hours, and then quenched with saturated NH4C1 solution. The mixture was extracted with EtOAc (50 mL x 3). The combined organic phases were dried and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 1:20) to give (2-bromo-4- nitrophenyl)(phenyl)methanol (3.9 g) as a yellow solid.
Step 4: Dess-Martin periodinane (5.91 g) was added to a solution of (2-bromo-4- nitrophenyl)(phenyI)methanol (3.9 g) in DCM (40 mL). The reaction mixture was stirred at RT overnight. Solid was removed by filtration, and the filtrate was washed with water (40 mL). The organic phase was dried and concentrated to afford (2-bromo-4-nitrophenyl)(phenyl)methanone (3.3 g) as a yellow solid. MS(ES+) m/z 306 (MH*).
Step 5: A mixture of (2-bromo-4-nitrophenyl)(pheny])methanone (3.29 g), phenylboronic acid (1.31 g), Cs2C03 (7.0 g) and Pd(Pl¾P)40-24 g) in 1 ,4-dioxane (50 mL) and water (10 mL) was stirred at 90°C under N2 overnight. Solid was removed by filtration, and the filtrate was concentrated. The aqueous phase was extracted with EtOAc (300 mL x 3). The combined organic phases were washed with water (100 mL). The organic phase was dried and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc : PE = 1 : 10) to give (5-nitrobipheny]-2- yI)(phenyl)methanone (1.3 g) as a white solid. MSiES4) z 304 (MH+).
Step 6: To a mixture of (5-nitrobiphenyl-2-yl)(phenyl)methanone (1.3 g) in methanol (20 mL) and water (20 mL) was added zinc (2.24 g) and ammonium formate (2.66 g). The reaction mixture was stirred at 90°C for 1 hour. The solid was removed by filtration. The filtrate was concentrated to remove methanol. The aqueous solution was extracted with EtOAc (50 mL x 3). The combined organic phases were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated in vacuo to afford (5-aminobiphenyl-2-yl)(phenyl)methanone (1.1 g) as a light yellow solid. MSfES m/z 274 (MH*).
Step 7: A mixture of [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 88 mg), EDC ( 1 mg) and HOBt (64.3 mg) in DCM (2 mL) was stirred at room temperature under nitrogen for 10 mins. (5-Amino-2-biphenylyl)(phenyl)methanone (100 mg) was added. The reaction mixture was stirred at room temperature overnight. The reaction mixture was partitioned between DCM and water. The aqueous phase was extracted with DCM for 3 times. The combined organic layers were washed with
brine and dried over anhydrous Na2S04. After filtration, solvent was removed under reduced pressure, and the residue was purified by MDAP to afford 2-[4-(ethylsulfonyl)phenyl]-N-[6-(phenyIcarbonyl)- 3-biphenylyl]acetamide (47 mg) as a white solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J~ 7.2 Hz, 2H), 3.87 (s, 2H), 7.21 (m, 5H), 7.35 (t, J= 7.2 Hz, 2H), 7.48 (m, 2H), 7.56 (d, J= 8.4 Hz, 2H), 7.63 (d, J= 8.0 Hz, 2H), 7.72 (m, 1H), 7.81 (d, J= 2.0 Hz, 1H), 7.86 (d, J= 8.4 Hz, 2H), 10.64 (s, 1H); MS(ES+) «i/z 484 (MH+).
Example 85
7V-{6-[(2-chlorophenyI)carbonyl]-4'-cyano-3-biphenylyl}-2-[4-(ethylsulfonyl)phenyI]acetamide

Step 2: PCC (2.78 g) was added to a mixture of (2-bromo-4-nitrophenyl)(2- chlorophenyl)methanol (3.4 g) in DCM (40 mL). The reaction mixture was stirred at RT overnight. Solvent was removed. The residue was purified by flash chromatography (EtOAc : PE = 1:5) to give (2-bromo-4-nitrophenyl)(2-chlorophenyl)metlianone (1.7 g) as a white solid. MS(ES+) m/z 340 (MH+).
Step 3: To a solution of (2-bromo-4-nitrophenyl)(2-chlorophenyl)methanone (0.80 g) in ethanol (30 mL) and water (15 mL) cooled to 0°C was added iron (0.53 g), and then 2M HC1 solution (7.05 mL) was added dropwise. The reaction mixture was stirred at 85 °C for 2 hours. Solid was removed by filtration, and the filtrated was concentrated. The residue was suspended in 50 mL of water, and the solution was basified to pH ~ 10 with aqueous NaOH solution. The mixture was
extracted with EtOAc (100 mL x 3). The combined organic phases were dried over anhydrous Na2S04, filtered, and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 1:20) to give (4-amino-2-bromophenyl)(2-chlorophenyl)methanone (0.6 g) as a light yellow oil. (ES^) m z 310 (MH ).
Step 4: A mixture of (4-amino-2-bromophenyl)(2-chlorophenyl)methanone (224 mg), [4-
(ethylsulfonyl)phenyl] acetic acid (intermediate la, 181 mg), HOBt (146 mg) and EDC (207 mg) in DCM (10 mL) was stirred at room temperature overnight. As the starting material remained, more [4- (ethylsulfonyl)phenyl]acetic acid (intermediate la, 82 mg), HOBt (97 mg) and EDC (138 mg) was added. The resulting mixture was stirred at room temperature overnight. The reaction mixture was washed with 2M HCl, sat. aHC03 solution and brine successively. The organic layer was dried over anhydrous Na2SC>4. After filtration, the filtrate was concentrated in vacuo to afford N-{3-bromo-4-[(2- chlorophenyl)carbonyl]phenyl}-2-[4-(ethylsulfonyl)phenyl]acetamide (230 mg) as a yellow solid.

Step 5: A mixture of N-{3-bromo-4-[(2-chlorophenyl)carbonyl]phenyl}-2-[4- (ethylsulfonyl)phenyl]acetamide (70 mg), PdCl2(dppf)-CH2Cl2 adduct (10 mg), (4- cyanophenyl)boronic acid (21.72 mg) and Cs2C03 (52.5 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-{ 6- [(2-chlorophenyl)carbonyl]-4'-cyano-3- biphenylyl} -2-[4-(ethylsulfonyl)phenyl]acetamide (11 mg) as a white solid. Ή-NMR (400 MHz, DMSO-(/G) δ ppm 1.09 (t, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 7.33 (m, 2H), 7.43 (m, 4H), 7.56 (d, J= 8.4 Hz, 1H), 7.61 (d, J= 8.4 Hz, 2H), 7.74 (m, 4H), 7.84 (d, J= 8.4 Hz, 2H), 10.76 (s, 1H); MS(ES+) m/z 543 (MH+).
Example 86
N-{6-[(2-c lorophenyl)carbonyl]-3'-cyano-3-biphenylyl}-2-[4-(ethylsulfonyI)phenyl]acetamide

A mixture of N-{3-bromo-4-[(2-chlorophenyl)carbonyl]phenyl}-2-[4- (ethylsulfonyl)phenyl]acetamide (70 mg, see step 4 for synthesis of Example 85), (3-
cyanophenyl)boronic acid (21.72 mg), PdCl2(dppf>CH2Cl2 adduct (110 mg) and Cs2C03 (52.5 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N- {6-[(2- chlorophenyl)carbonyl]-3'-cyano-3-biphenylyl}-2-[4-(ethylsulfonyI)phenyl]acetamide (10 mg) as a white solid. Ή-NMR (400 MHz, DMSO-c e) δ ppm 1.09 (t, J= 7.2 Hz, 3H), 3.28 (q, J- 7.2 Hz, 2H), 3.87 (s, 2H), 7.30 (m, 2H), 7.39 (m, 2H), 7.47 (t, J= 7.6 Hz, 1H), 7.52 (m, 1H), 7.61 (m, 4H), 7.72 (m, 3H), 7.85 (d, J= 8.4 Hz, 2H), 10.75 (s, 1H); MSiES ) m/z 543 (MH+).
Example 87
Ar-[2-chloro-4'-{[l-(2,2-dimethylpropanoyl)-4-piperidinyI]methyl}-2T-(trifluoromethyl)-4- biphenylyl] -2-[4-(ethylsulf onyl)p enyl] acetamide

Intennediate 87a: diethyl i^-bromo-S-ftrifluoromethvDphenyllmethyllphosphonate
Step 1: To a solution of 4-bromo-3-(trifluoromethyl)benzoic acid (6.8 g) and triethylamine (5.28 mL) in tetrahydrofuran (THF) (150 mL) cooled at -10°C was added isobutyl cliloroformate (4.98 mL) dropwise during 15 minutes. The resulting mixture was stirred at this temperature for 1.5 hours. Water (80 mL) was added to the mixture at -10°C and NaBH4 (3.83 g) was added slowly at 0 °C. The reaction mixture was stirred at room temperature overnight. The mixture was filtered and the filtrate was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with sat. NaHC03 and brine, then dried over anhydrous Na2S04. After filtration, solvent was removed under reduced pressure and the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1 :3) to afford [4-bromo-3- (trifluoromemyl)phenyl]methanol (6.341 g) as an off-white solid. 'H-NMR (400 MHz, CDC13) δ ppm 4.72 (s, 2H), 7.38 (dd, J= 1.1 Hz, 8.1 Hz, 1H), 7.69 (m, 2H); MSfES ) m/z 237 (MH+-18).
Step 2: To a solution of [4-bromo 3-(trifluoromethyl)phenyl]methanol (3 g) in tetrahydrofuran
(THF) (25 mL) cooled at 0°C was added PBr3 (0.5 mL) dropwise and the resulting mixture was stirred at room temperature for 2 hours. The mixture was quenched with water slowly at 0 °C, and then partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc for 3 times. The combined organic layers were washed with sat. NaHC03 and brine, dried over anhydrous Na2S04. After filtration and removal of solvent, the residue was dissolved in toluene (30 mL), and triethyl
phosphite (1.793 mL) was added. After the reaction mixture was heated to reflux overnight, more triethyl phosphite (1.793 mL) was added and the reaction was heated for another day. After cooling to room temperature, solvent was removed under reduced pressure and the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 4: 1) to afford diethyl {[4-bromo-3- (trifluoromethyl)phenyl]mediyl}phosphonate (3.208 g) as a light yellow oil. MS(ES+) m/z 375 (MH+).
Preparation of N- r2-chloro-4'- 1 Γ 1 -(2, 2-dimethylpropanovn-4-piperidinyl1 methyl } -2 '- (trifluoromethylV4-biphenylvn-2-r4-fethylsulfonyl)phenyl1acetamide
Step 1: To a solution of 4-piperidinone (2 g) in DCM (25 mL) was added Et3N (6.35 mL), followed by a dropwise addition of 2,2-dimethylpropanoyl chloride (3.92 g). After stirring at room temperature overnight, the mixture was filtered. The filtrate was diluted with DCM, washed with 2M HC1, sat. NaHC(¾ and brine, and then dried over anhydrous Na2S04. After filtration and
concentration, the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1: 1) to afford l-(2,2-dimethylpropanoyl)-4-piperidinone (1.91 g) as a white solid. MSfES*) m/z 184 (MH+).
Step 2: To a solution of diethyl {[4-bromo-3-(trifluorometliyl)phenyl]methyl}phosphonate (intermediate 87a, 409 mg) in tetrahydrofuran (THF) (8 mL) cooled at 0°C was added NaH (60% in mineral oil, 131 mg). The resulting mixture was stirred at room temperature for 30 mins. l-(2,2- Dimethylpropanoyl)-4-piperidinone (200 mg) was added. After stirring at room temperature overnight, the reaction was quenched with water, and then extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford 4-{[4-bromo-3-(trifluoromethyl)phenyl]methylidene}-l-(2,2- dimethylpropanoyl)piperidine (351 mg) as a yellow oil. MS(ES+) m z 404 (MH+).
Step 3: A mixture of 4-{[4-bromo-3-(trifluoromethyl)phenyl]methylidene}-l-(2,2- dimethylpropanoyl)piperidine (150 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2- yl)phenyl]-2-[4-(ethylsuIfonyl)phenyl]acetamide (intermediate 6b, 1 1 mg), Pd(Ph3P)4 (34.3 mg) and 2 M aqueous Na2C03 (1.484 mL) in 1,4-dioxane (3 mL) was bubbled with N2. The reaction vessel was sealed and heated in the microwave at 100°C for 40 mins. The mixture was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2S04. After filtration and concentration, the residue was purified by flash chromatography (EtOAc:PE = 0:1 to 3:2, then MeOH : DCM = 0:1 to 8:92) to afford the crude intermediate (226 mg). The intennediate was dissolved in methanol (8 mL) and tetrahydrofuran (THF) (2 mL). Pd/C (35 mg) was added and the resulting mixture was stirred under ¾ (excess) at room temperature for 2 days. The mixture was filtered. The filtrate was diluted with DMF, and then further purified by MDAP to afford N-[2-chloro- 4'-{[l -(2,2-dimethylpropanoyl)-4-piperidinyl]methyl}-2'-(trif!uoromethyl)-4-biphenylyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (14 mg) as a white solid. 'H-NMR (400 MHz, DMSO-i ) δ ppm
1- 11 (m, 14H), 1.61 (m, 2H), 1.87 (m, 1H), 2.71 (m, 4H), 3.28 (q, J= 7.3 Hz, 2H), 3.85 (s, 2H), 4.26 (d, J= 13.0 Hz, 2H), 7.25 (d, J= 8.1 Hz, 2H), 7.52 (m, 2H), 7.63 (m, 3H), 7.86 (d, J= 8.2 Hz, 2H), 7.91 (d, J= 1.8 Hz, 1H), 10.54 (s, 1H); ,9F-NMR (376 MHz, DMSO-i/6) δ ppm -57.54; MS(ES") m/z 663 (MH . Example 88
N-[2-chloro-4T-{[l-(3,3-dimethylbutanoyl)-4-piperidinyl]methyl}-2l-(trifluoromethyl)-4- biphenylyI]-2-[4-(ethylsuIfonyl)phenyl]acetamide

Step 1: To a solution of 4-piperidinone (2 g) in DCM (50 mL) was added Et3N (6.35 niL), followed by a dropwise addition of 3,3-dimethylbutanoyl chloride (4.38 g). After stirring at room temperature overnight, the mixture was filtered. The filtrate was diluted with DCM, washed with 2M HC1, sat. NaHCC>3 and brine, and then dried over anhydrous Na2S04. After filtration and
concentration, the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 7:3) to afford l-(3,3-dimethylbutanoyl)-4-piperidinone (2.326 g) as an off-white solid. MSfES4) m/z 198 (MH^).
Step 2: To a solution of diethyl {[4-bromo-3-(trifluorometliyl)phenyl]methyl}phosphonate
(intermediate 87a, 570 mg) in tetrahydrofuran (THF) (12 mL) cooled at 0°C was added NaH (60% in mineral oil, 182 mg). The resulting mixture was stirred at room temperature for 30 mins. l-(3,3- Dimethylbutanoyl)-4-piperidinone (300 mg) was added. After stirring at room temperature overnight, the reaction was quenched with water, and then extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford 4-{[4-bromo-3-(trifiuoromethyl)phenyl]methylidene}-l-(3,3- dimethylbutanoyl)piperidine (529 mg) as a brown oil. MS(ES+) m z 418 (MH+).
Step 3: A mixture of 4-{[4-bromo-3-(trifluoromethyl)phenyl]methylidene}-l-(3,3- dimethylbutanoyl)piperidine (160 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 156 mg), Pd(Ph3P) (35.4 mg) and 2 M aqueous Na2C03 (1.530 mL) in 1,4-dioxane (3 mL) was bubbled with N2. The reaction vessel was sealed and heated in the microwave at 100°C for 40 mins. The mixture was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2SC>4. After filtration and concentration, the residue was purified by flash chromatography (EtOAc:PE = 0:1 to 3:2) to afford the crude intermediate (250 mg). The intermediate was dissolved in methanol (8 mL)
and tetrahydrofuran (THF) (2 mL). Pd/C (35 mg) was added and the resulting mixture was stirred under H2 (excess) at room temperature overnight. The mixture was filtered. The filtrate was diluted with DMF, and then further purified by MDAP to afford N-[2-chloro-4'-{[l-(3;3-dimethylbutanoyl)- 4-piperidinyl]metliyl}-2'-(trifluoromethyl)-4-biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (63 mg) as a white solid. Ή-N R (400 MHz, DMSO-rfe) δ ppm 1.11 (m, 14H), 1.59 (m, 2H), 1.84 (m, 1H), 2.20 (m, 2H), 2.44 ( , 1H), 2.66 (d, J= 6.8 Hz, 2H), 2.94 (t, J= 12.3 Hz, 1H), 3.28 (q, J= 7.3 Hz, 2H), 3.84 (s, 2H), 3.95 (d, J= 13.3 Hz, 1H), 4.44 (d, 7= 12.7 Hz, 1H), 7.25 (d, 7= 8.1 Hz, 2H), 7.52 (m, 2H), 7.63 (m, 3H), 7.86 (d, J= 8.3 Hz, 2H), 7.91 (d, J= 2.0 Hz, 1H), 10.53 (s, 1H); l F- NMR (376 MHz, DMSO-<¾) 5 ppm -57.54; MS(ES^) mtz 677 (MM*), Example 89
iV-[2-ch]oro-4'-{[l-(l-rnethy]ethyl)-4-piperidinylidene]rnethyl}-2'-(trifluoromethyJ)-4- biphenylyl]-2-[4-(ethylsuIfonyl)phenyI]acetamide, trifluoroacetic acid salt

Step 1: To a solution of diethyl {[4-bromo-3-(trifluorometliyl)phenyl]methyI}phosphonate (intermediate 87a, 398 mg) in tetrahydrofuran (THF) (8 mL) cooled at 0 °C, was added NaH (60% in mineral oil, 127 mg). The resulting mixture was allowed to warm to room temperature and stirred for 30 minutes. Then l-(l-methylethyl)-4-piperidinone (1 0 mg) was added. After stirring at room temperature overnight, the mixture was quenched with water and extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous Na2SC> . After filtration, solvent was removed in vacuo to afford 4-{[4-bromo-3-
(trifluoromethyl)phenyl]methylidene}-l-(l-methylethyl)piperidine (367 mg) as a brown oil. MS(ES^) m/z 362 (MH1).
Step 2: A mixture of 4-{[4-bromo-3-(trifIuoromethyI)phenyl]methylidene}-l-(l- methylethy piperidine (200 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl]-2-[4-(ethyIsulfonyl)phenyl]acetamide (intennediate 6b, 282 mg), Pd(Ph3P) (63.8 mg) and 2 M Na2C03 solution (2.209 mL) in 1,4-dioxane (5 mL) was bubbled with N2. The reaction vessel was sealed and heated in die microwave at 100°C for 40 mins. The mixture was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2S04. After filtration and concentration, the residue was purified by flash chromatography (EtOAc : PE = 0:1 to 4: 1 , then MeOH : DCM = 0: 1 to 1 :9) to afford the desired product (313 mg) as a light brown solid.
Part of this solid (130 mg) was redissolved in DMF (6 mL) and further purified by MDAP to afford N-[2-chloro-4'-{[l-(l-methylethyl)-4-pi^
[4-(ethylsulfonyl)phenyI]acetamide, trifluoroacetic acid salt (52 mg) as a white solid. Ή-NMR (400 MHz, OMSO-d6) 5 ppm 1.10 (t, /= 7.3 Hz, 3H), 1.26 (m, 6H), 2.64 (m, 3H), 2.96 (m, 3H), 3.28 (q, / = 7.3 Hz, 2H), 3.49 (m, 3H), 3.85 (s, 2H), 6.62 (s, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.35 (d, J= 7.9 Hz, 1H), 7.53 (d, J= 8.5 Hz, 1H), 7.64 (m, 4H), 7.86 (d, J= 8.3 Hz, 2H), 7.94 (s, 1H), 9.51 (br s, 1H), 10.61 (s, 1H); I9F- MR (376 MHz, DMSO-rf6) δ ppm -57.75, -73.83; MSfES m/z 619 (MH^.
Example 90
N-[2-chloro-4,-{[l-(l-methylethyl)-4-piperidinyl]methyl}-2'-(trifluoromethyl)-4-biphenylyI]-2- [4-(ethylsulfonyI)phenyl)acetamide, trifluoroacetic acid salt

To a solution ofN-[2-chloro-4'-{[l-(l-methylethyl)-4-piperidinylidene]methyl}-2'- (trifluorometliyl)-4-biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (free base of Example 89, 180 mg) in methanol (8 mL) and tetrahydrofuran (THF) (2 mL) was added 10% Pd/C (30 mg). The resulting mixture was stirred under H2 (excess) at room temperature overnight. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by MDAP to afford N-[2-chloro-4'-{[l-(l-methylethyl)-4-piperidinyl]methyl}-2'-(trifluoromethyl)-4- biphenylyl]-2-[4-(ethylsulfonyl)phenyl]acetamide, trifluoroacetic acid salt (48 mg) as a white solid. Ή-NMR (400 MHz, MeOD-</4) δ ppm 1.21 (t, J= 7.4 Hz, 3H), 1.32 (d, J= 6.7 Hz, 6H), 1.54 (m, 2H), 1.96 (m, 3H), 2.75 (d, J- 6.5 Hz, 2H), 3.00 (t, /= 12.2 Hz, 2H), 3.20 (q, J= 7.4 Hz, 2H), 3.46 (m, 3H), 3.86 (s, 2H), 7.16 (d, J= 8.4 Hz, 1H), 7.23 (d, J= 7.8 Hz, 1H), 7.48 (m, 2H), 7.63 (m, 3H), 7.88 (m, 3H); ,9F-NMR (376 MHz, MeOD-i¾ δ ppm -60.21, -77.31; MS(ES+) m/z 621 (MH+).
Example 91
N-[4'-[(l-acetyl-4-piperidinyl)methy]]-2,-(trifluoromethyl)-4-bipheny]yl]-2-[4- (ethylsulfony l)phenyl] acetamide

A mixture of N-(4-bromophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate lb, 500 mg), 4,4,4',4',5!5,5',5,-octamethyl-252'-bi-l,3,2-dioxaborolane (598 mg), PdCl2(dppf)-CH2Cl2 adduct (107 mg) and potassium acetate (513 mg) in DMF (8 niL) was stirred at 100°C under nitrogen. After cooling to room temperature, the mixture was filtered through celite. The filtrate was partitioned between EtOAc and water. The aqueous phase was extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anliydrous Na2S04. After filtration, solvent was removed under reduced pressure and the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1:1) to afford 2-[4-(ethylsulfonyl)phenyl]-N-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl]acetamide (634 mg) as a light brown solid. MSiES4) m/z 430 (MH+).
Preparation of N-r4'-r(l-acetyl-4-piperidinvnmethyll-21-itrifluoromethyl')-4-biphenylyl1-2-r4- (ethylsulfonyl)phenvHacetamide
Step 1: A solution of 4-(bromomemyl)-l-chloro-2-(trifluoromethyl)benzene (600 mg) and triethyl phosphite (0.403 mL) in toluene (10 mL) was heated to reflux overnight. After cooling to room temperature, solvent was removed in vacuo to afford diethyl {[4-chloro-3- (trifluoromemyl)phenyl]methyl}phosphonate (927 mg) as a colorless oil. MS(ES+) m/z 331 (Mtf*).
Step 2: To a solution of l-acetyl-4-piperidinone (200 mg) and diethyl {[4-chloro-3- (trifluoromethyl)phenyl]methyl}phosphonate (601 mg) in tetrahydrofuran (THF) (10 mL) was added NaH (60% in mineral oil, 85 mg) portionwise at room temperature. After stirring at room temperature for 2 hours, more NaH (60% in mineral oil, 85 mg) was added. The mixture was stirred at room temperature for 2 days. The reaction was quenched with water, extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anliydrous Na2S04. After filtration, solvent was removed under reduced pressure and the residue was purified by flash chromatography (EtOAc : PE = 0:1 to 3:2) to afford 1 -acetyl^- {[4-chloro-3-
(trifluorometliyl)phenyl]methylidene}piperidine (415 mg) as a colorless oil. MS(ES+) m/z 318 (MH*).
Step 3: A mixture of l-acetyl-4-{[4-chloro-3-(trifiuoromethyl)phenyl]methylidene}piperidine (186 mg), 2-[4-(ethylsulfonyl)phenyl]-N-[4-(4,4,5,5-tetrametliyl-l ,3,2-dioxaboroIan-2- yl)phenyl]acetamide (intermediate 91a, 276 mg), Pd(OAc)2 (52.6 mg), tricyclohexylphosphine (131 mg) and 2 M aqueous Na2C03 (2.93 rnL) in 1,4-dioxane (5 mL) was bubbled with N2. The reaction vessel was sealed and heated in the microwave at 120°C for 1 hour. The mixture was filtered through celite and silica gel, washed with acetonitrile, DCM and EtOAc. Solvent was removed under reduced pressure. The residue was diluted with water and extracted with EtOAc for 3 times. The combined organic layers were washed with brine and dried over anhydrous NajSOi. After filtration and removal
of solvent, the residue was purified by flash chromatography (EtOAc : PE = 0: 1 to 1 :0) to afford N- [4'-[(l-acety -piperidinylidene)mediyl]-2'-(triflnoromethyl)-4-biphenyIyl]-2-[4- (emylsulfonyl)phenyl]acetamide (77 mg) as an off-white solid. MS(ES+) m/z 585 (MH+).
Step 4: To a solution of N-[4'-[(l-acetyl-4-piperidinylidene)methyl]-2,-(trifluoromethyl)^- biphenylyl]-2-[4-(etliylsulfonyl)phenyl]acetamide (77 mg) in methanol (8 mL) was added 10% Pd/C (30 mg) and the resulting mixture was stirred under H2 (excess) at room temperature overnight. The mixture was filtered and the filtrate was purified directly by MDAP to afford N-[4'-[(l-acetyl-4- piperidinyl)methyl]-2'-(trifluoromethyl)-4-biphenylyI]-2-[4-(ethylsulfony])phenyl]acetamide (6 mg) as a white solid. Ή-N R (400 MHz, DMSO- ) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 1.68 (m, 5H), 1.97 (s, 3H), 2.43 (m, 1H), 2.65 (d, J= 7.0 Hz, 2H), 2.95 (t, J= 11.7 Hz, 1H), 3.28 (q, J= 7.3 Hz, 2H), 3.80 (m, 3H), 4.35 (d, J= 12.9 Hz, 1H), 7.24 (d, J= 8.4 Hz, 2H), 7.29 (d, J= 7.8 Hz, 1H), 7.50 (d, J= 7.2 Hz, 1H), 7.63 (m, 5H), 7.86 (d, J= 8.3 Hz, 2H), 10.40 (s, 1H); MS(ES+) m/z 587 (MH÷).
Example 92
iY-[2'-chloro-2-(trifluoromethyl)-4-biphenylyI]-2-[4-(ethylsulfonyI)phenyl]acetamide

A mixture of (2-chlorophenyl)boroiiic acid (22.92 mg), N-[4-bromo-3- (trifluoromethyl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 42a, 60 mg), Cs2C03 (52.1 mg) and PdCl2(dppf)-CH2Cl2 adduct (10 mg) in acetonitrile (1.5 mL) and water (0.5 mL) was sealed in a vessel, and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was purified by MDAP to afford N-[2'-chloro-2-(trifluoromethyl)-4-biphenylyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (14 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-i¾) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.87 (s, 2H), 7.32 (m, 2H), 7.42 (m, 2H), 7.56 (dd, J = 1.2 Hz, 7.6 Hz, 1H), 7.63 (d, J^ 8.4 Hz, 2H), 7.86 (m, 3H), 8.18 (d, J= 1.6 Hz, 1H), 10.72 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -58.09; MS(ES+) m/z 482 (MH .
Example 93
N-(2-chlor 0-2 '-(3-chlorophenoxy) biphenyl-4-yl)-2-(4-(ethylsulf onyI)phenyl) ac etamide

Step 1: A mixture of l-fmoro-2-nitrobenzene (3.5 g), 3-chlorophenoI (4.8 g) and Cs2C03 (16 g) in DMF (30 mL) was stirred at 90°C for 5 hours under N2. Ethyl acetate was added, and the mixture was washed with sat. Na2COj and brine successively. The organic phase was dried over anhydrous Na2S(¾ and concentrated. The residue was purified by flash chromatography (PE) to afford l-(3- chlorophenoxy)-2-nitrobenzene (4.2 g) as a yellow liquid.
Step 2: A mixture of l-(3-chlorophenoxy)-2-nitrobenzene (3.6 g), zinc dust (6.0 g) and ammonium formate (12.0 g) in methanol (40 mL) and water (40 mL) was stirred at 90°C for 4 hours. Solvent was removed. The residue was purified by flash chromatography (EtOAc : PE = 1: 1) to afford 2-(3-chlorophenoxy)benzenamine (2.8 g) as a white solid. MS(ES+) m z 220 (MH4).
Step 3: A mixture of 2-(3-chlorophenoxy)benzenamine (1.1 g), copper bromide (1,1 g) and ieri-butyl nitrite (1 mL) in acetonitrile (10 mL) was stirred at T for 4 hours. Solvent was removed, and the residue was purified by reverse-phase chromatography (MeCN : H20 = 3: 1) to afford 1- bromo-2-(3-chlorophenoxy)benzene (280 mg) as a colorless liquid.
Step 4: A mixture of l-bromo-2-(3-chlorophenoxy)benzene (270 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 438 mg), Pd(PPh3)4 (65 mg) and Cs2C03 (358 mg) in 1,4-dioxane (10 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 85 °C for 5 hours. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by prep-HPLC to afford N-(2-chloro-2'-(3-chlorophenoxy)biphenyl-4-yl)-2-(4-
(ethylsulfonyl)phenyl)acetamide (80 mg) as a white solid. Ή-NMR (400 MHz, DMSO-cQ 5 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.81 (s, 2H), 6.82 (d, J~ 8.4 Hz, IH), 6.86 (s, IH), 7.09 (d, J= 8.0 Hz, 2H), 7.32 (m, 4H), 7.45 (m, 2H), 7.60 (d, J= 8.4 Hz, 2H), 7.84 (m, 3H), 10.48 (s, IH); MSfES") m/z 540 (MET). Example 94
Ar-(2-chloro-2'-(2-chlorophenoxy)biphenyl-4-yl)-2-(4(ethyIsu)fonyl)phenyl)acetamide

Step 1: A mixture of l-fluoro-2-nitrobenzene (2.5 g), 2-chlorophenol (4.8 g) and Cs2C03 (12 g) in DMF (25 mL) was stirred at 90°C for 5 hours under N2. Ethyl acetate (100 mL) was added, and the mixture was washed with sat. NaaCOj solution and brine successively. The organic phase was dried and concentrated. The residue was purified by flash chromatography (PE) to afford 1 -(2- chIorophenoxy)-2-nitrobenzene (3.6 g) as a yellow liquid.
Step 2: A mixture of l-(2-chlorophenoxy)-2-nitrobenzene (3.6 g), zinc dust (6.0 g) and ammonium fonnate (12.0 g) in methanol (40 mL) and water (40 mL) was stirred at 90°C for 4 hours. Solvent was removed. The residue was purified by flash chromatography (EtOAc : PE = 1 : 1) to afford 2-(2-chlorophenoxy)benzenamine (2.6 g) as a white solid. MS(ES+) m/z 220 (MFf).
Step 3: A mixture of 2-(2-chlorophenoxy)benzenamine (1.0 g), CuBr2 (1.0 g) and ter/-butyl nitrite (1 mL) in MeCN (10 mL) was stirred at 50°C for 4 hours. The reaction mixture was concentrated. The residue was purified by reverse phase flash chromatography (MeCN : H20 = 3:1) to afford l-bromo-2-(2-chIorophenoxy)benzene (700 mg) as a colorless liquid.
Step 4: A mixture of 1 -bromo-2-(2-ch]orophenoxy)benzene (246 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyI)phenyl]acetamide (intermediate 6b, 338 mg), Pd(PPh3)4 (61 mg) and Cs2C03 (428 mg) in dioxane (10 mL) and water (0.5 mL) was sealed in a vessel and heated in the microwave at 85 °C for 5 hours. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure, and the residue was purified by prep-HPLC to afford N-(2-chloro-2'-(2-chlorophenoxy)biphenyl-4-yl)-2-(4-
(ethylsulfonyl)phenyl)acetamide (75 mg) as a white solid. Ή-NMR (400 MHz,
 6 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.82 (s, 2H), 6.86 (d, J= 6.4 Hz, 1H), 6.94 (dd, J= 6.4 Hz, 1.2 Hz, 1H), 7.10 (t, J- 1.2 Hz, 1H), 7.26 (m, 2H), 7.34 (m, 2H), 7.41 (m, 1H), 7.46 (m, 2H), 7.60 (d, J = 6.8 Hz, 2H), 7.85 (m, 3H), 10.48 (s, 1H); MS(ES+) m/z 540 (MH÷). Example 95
6 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.82 (s, 2H), 6.86 (d, J= 6.4 Hz, 1H), 6.94 (dd, J= 6.4 Hz, 1.2 Hz, 1H), 7.10 (t, J- 1.2 Hz, 1H), 7.26 (m, 2H), 7.34 (m, 2H), 7.41 (m, 1H), 7.46 (m, 2H), 7.60 (d, J = 6.8 Hz, 2H), 7.85 (m, 3H), 10.48 (s, 1H); MS(ES+) m/z 540 (MH÷). Example 95
iV-(4,6-dimethyl-5-(2-(triiluoroniethoxy)phenyl)pyridin-2-yl)-2-(4- (ethyIsulfonyi)phenyi)acetamide

Step 1: A mixture of 5-bromo-4,6-dimethylpyridin-2 -amine (200 mg), 2-(4-
(ethylsulfonyl)phenyl)acetic acid (intermediate la, 341 mg), EDC (286 mg) and HOBt (228 mg) in DCM (30 mL) was stirred at RT overnight. The mixture was washed with water (30 mL x 3) and brine (30 mL x 3). The organic phase was dried and concentrated to give N-(5-bromo-4,6- dimethylpyridin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (440 mg) as a brown oil. MSiES^ m/z 411 (MH+).
Step 2: A mixture of N-(5-bromo-4,6-dimethylpyridin-2-yl)-2-(4- (ethyIsulfonyl)phenyl)acetamide (440 mg), 2-(trifluoromethoxy)phenylboronic acid (220 mg), Pd(Ph3P)4 (124 mg) and potassium carbonate (296 mg) in 1,4-dioxane (60 mL) and water (3 mL) was stirred at 90°C overnight under Ν2. The mixture was filtered, and the filtrate was concentrated. The residue was partitioned between water (80 mL), and DCM (80 mL). The organic phase was dried and concentrated. The yellow oily residue was purified by flash chromatography (EtOAc : PE = 1 :2) to give the crude product. Further purification by prep-HPLC afforded N-(4,6-dimethyl-5-(2- (trifluoromethoxy)phenyl)pyridin-2-yl)-2-(4-(ediylsulfonyl)phenyl)acetamide (27 mg) as a white solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.32 (t, /= 8.0 Hz, 3H), 2.04 (s, 3H), 2.16 (s, 3H), 3.15 (q,
8.0 Hz, 2H), 3.85 (s, 2H), 7.20 (d, J= 8.0 Hz, 1H), 7.39 (m, 2H), 7.47 (m, 1H), 7.58 (d, J= 8.0 Hz, 2H), 7.93 (d, J= 8.0 Hz, 2H), 8.00 (s, 1H), 8.11 (s, 1H); 19F-NMR (376 MHz, CDC13) δ ppm -57.27; MS(ES+) m/z 493 (MH+). Example 96
iV-(2-chloro-2'-(methyl{propyl)amino)biphenyl-4-yl)-2-(4-(ethylsulfonyI)phenyl)acetamide

Step 1: To a mixture of 1-bromopropane (4.29 g) and 2-bromobenzenamine (5 g) in DMF (20 mL) was added potassium carbonate (8.03 g) in one portion. The reaction mixture was stirred at 50°C overnight. The reaction mixture was diluted with EtOAc (80 mL), and then washed with water (100
mL). The organic phase was dried and concentrated to afford 2-bromo-N-propylbenzenamine (2.0 g) as a colorless oil. MS(ES+) m/z 214 (MH*).
Step 2: Formic acid (86 mg) was added dropwise to acetic anhydride (191 mg) cooled in an ice bath. After the addition, the ice bath was removed and the solution was stirred at 50°C for 30 minutes. The mixture was cooled again in an ice bath. The cold mixture was added dropwise to a solution of 2- bromo-N-propylbenzenamine (400 mg) in formic acid (1 mL) cooled below 10 °C. The reaction mixture was stirred at T for 80 minutes. Solvent was removed, and DCM (30 mL) was added. The mixture was filtered. The filtrate was dried and concentrated to afford N-(2-bromophenyl)-N- propylfonnamide (450 mg) as a light yellow oil. MS(ES+) m/z 242 (MH+).
Step 3: Borane-tetrahydrofuran complex (15 mL) was added dropwise to a solution of N-(2- bromophenyl)-N-propylfonnamide (450 mg) in THF (20 mL) cooled at 0°C in 30 mins. The reaction mixture was wanned to 80°C and stirred overnight. The reaction mixture was then cooled to 0°C and quenched with brine. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (100 mL x 2). The combined organic phases were dried and concentrated to afford 2-bromo- N-methyl-N-propylbenzenamine (100 mg) as a brown oil. MSfES4) m/z 228 (MIL1).
Step 4: A mixture of 2-bromo-N-metliyl-N-propylbenzenamine (100 mg), N-[3-chloro-4- (4,4,5, 5-tetramethyI-l ,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethyIsulfonyl)phenyl]acetamide
(intermediate 6b, 203 mg), Pd(Ph3P)4 (50.7 mg) and 2C03 (1 1 mg) in 1,4-dioxane (20 mL) and water (3 mL) was stirred at 80°C overnight under Nj. The mixture was filtered and the filtrate was concentrated. The residue was partitioned between water (60 mL) and DCM (60 mL), The organic phase was dried and concentrated. The residue was purified by prep-HPLC to give N-(2-chloro-2'- (methyI(propyl)amino)bipiienyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (27 mg) as a white solid. Ή-NMR (400 MHz, CDC¾) δ ppm 0.63 (t, J= 7.2 Hz, 3H), 1.27 (m, 5H)f 2.55 (s, 3H), 2.67 (s, 2H), 3.14 (q, J= 7.2 Hz, 2H), 3.84 (s, 2H), 7.06 (m, 3H), 7.33 (m, 4H), 7.58 (d, J= 8.0 Hz, 2H), 7.74 (s, 1H), 7.92 (d, 8.0 Hz, 2H); MS(ES÷) m/z 485 (MH .
Example 97
iY-(2-(lH-pyrrol-l-yl)-6'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-(ethylsuIfonyl)phenyl)acetaniide
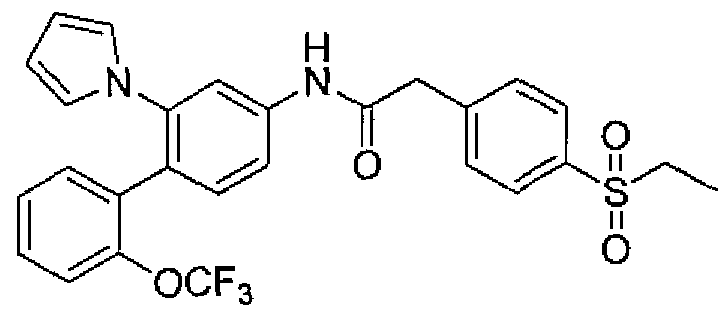
Step 2: To a mixture of 2-fluoro-4-nitro-6'-(trifluoromethoxy)biphenyl (300 mg) in DMF (30 mL) was added lH-pyrrole (134 mg) and Cs2C03 (649 mg). The reaction mixture was stirred at 120°C for 72 hours. Solvent was removed under vacuo. The residue was partitioned between water (50 mL) and EtOAc (50 mL). The aqueous phase was extracted with EtOAc (50 mL x 2). The combined organic phases were dried and concentrated to afford l-(4-nitro-6'- (trifluorornethoxy)biphenyl-2-yl)-lH-pyrrole (300 mg) as a yellow oil. MS(ES m/z 349 (MH^.
Step 3: A mixture of l-(4-nitro-6 -(trif)uoromethoxy)biphenyl-2-yl)-lH-pyrrole (300 mg), ammonium formate (543 mg) and zinc (282 mg) in methanol (30 mL) and water (10 mL) was stirred at 80°C for 3 hours. Solvent was removed, and the residue was partitioned between EtOAc (60 mL) and water (60 mL). The aqueous phase was extracted with EtOAc (60 mL x 2). The combined organic phases were dried and concentrated to afford 2-(lH-pyrrol-l-yl)-6,-(trifluoromethoxy)biphenyl-4- amine (280 mg) as a yellow solid. MS(ES*) m/z 319 (MH+).
Step 4: A mixture of 2-(lH-pyrrol-l -yl)-6'-(trifluoromethoxy)biphenyl-4-amine (280 mg), 2-
(4-(ethylsulfonyl)phenyl)acetic acid (intennediate la, 301 mg), EDC (253 mg) and HOBt (202 mg) in DCM (40 mL) was stirred at T overnight. The mixture was washed with water (30 mL x 2) and brine (30 mL). The organic phase was dried over anhydrous Na2S04 and concentrated. The residue was purified by prep-HPLC to give N-(2-(lH-pyrrol-l-yl)-6'-(trifluoromethoxy)biphenyl-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (90 mg) as a white solid. 'H-NMR (400 MHz, DMSO-c/6) δ ppm
1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 6.02 (t, J= 2.0 Hz, 2H), 6.52 (t, J= 2.0 Hz, 2H), 7.35 (m, 5H), 7.61 (d, J~ 2.0 Hz, 1H), 7.64 (d, J= 8.0 Hz, 2H), 7.81 (d, 7=2.0 Hz, 1H), 7.87 (d, J= 8.0 Hz, 2H); 19F-NMR (376 MHz, DMSO-<¾ δ ppm -56.23; MSiES") m/z 529 (MH+).
Example 98
7Y-(2-chIoro-6-fluorobiphenyI-4-yI)-2-(4-(ethy[suIfonyI)phenyI)acetamide

Step 1: A mixture of l,2-difluoro-4-nitrobenzene (8 g) and 29% ammonia (10 mL) was placed in a pressure bottle and sealed. The reaction mixture was stirred at 135 °C for 24 hours. After cooling down to room temperature, the solid was collected by filtration and washed with water followed by PE. The solid was dried to give 2-fluoro-4-nitrobenzenamine (6.4 g) as a yellow solid. MS(ES+) m/z 157 (MH+).
Step 2: To a solution of 2-fluoro-4-nitrobenzenamine (5 g) in methanol (60 mL) was added
HC1 (5.26 mL) and H202 (3.44 mL) at R.T. The reaction mixture was heated at reflux overnight. The mixture was concentrated and to the residue was added water (50 mL). The solid was collected by filtration, washed with water, and dried to give 2-chloro-6-fluoro-4-nitrobenzenamine (4.8 g) as a yellow solid. MSiES m/z 1 1 (MH÷).
Step 3: To a solution of 2-chIoro-6-fluoro-4-nitrobenzenamine (0.9 g) and 1,1-dimethyl ethyl nitrite (1.46 g) in acetonitnle (5 mL) was added copper(II) bromide (3.16 g) portionwise at 0°C under nitrogen. The reaction mixture was stirred at RT for 6 hours. The mixture was concentrated. To the residue was added water (20 mL). The mixture was extracted with EtOAc (50 mL x 2). The combined organic phases were dried and concentrated. The residue was purified by flash chromatography (PE) to give 2-bromo-l-chIoro-3-fluoro-5-nitrobenzene (300 mg) as a yellow solid.
Step 4: To a solution of 2-bromo-l-chloro-3-fluoro- -nitrobenzene (230 mg), phenylboronic acid (110 mg), and K2C03 (312 mg) in dioxane (10 mL) and water (2 mL) stirred at room temperature under nitrogen was added Pd(Ph3P)4 (104 mg). The reaction mixture was stirred at 90°C overnight.
Solvent was removed. The residue was purified by flash chromatography (PE) to give 2-chloro-6- fluoro-4-nitrobiphenyl (170 mg) as a yellow solid.
Step 5: A mixture of 2-chloro-6-fluoro-4-nitrobiphenyl (140 mg), ammonium formate (351 mg) and zinc (182 mg) in methanol (10 mL) and water (2 mL) was heated to 80°C and stirred overnight. Solvent was removed. The residue was dissolved in EtOAc (20 mL) and washed with water (10 mL) and brine (10 mL). The organic phase was dried and concentrated to give 2-chloro-6-fluorobiphenyl-
4-amine (150 mg) as a brown oil. MS(ES+) m/z 222 (MH+).
Step 6: A mixture of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 159 mg), 2- chloro-6-fluorobiphenyl-4-amine (140 mg), EDC (145 mg) and HOBt (116 mg) in dichloromethane (10 mL) was heated at reflux overnight. Solvent was removed, and the residue was purified by prep-
HPLC to give N-(2-chloro-6-fluorobiphenyl-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (45 mg) as a
white solid. Ή-NMR (400 MHz, OMSO-d6) δ ppm 1.11 (t, J= 7.2 Hz, 3H), 3.30 (q, J= 7.2 Hz, 2H), 3.87 (s, 2H), 7.34 (m, 2H), 7.45 (m, 3H), 7.61 (m, 4H), 7.87 (d, J= 7.2 Hz, 2H), 10.78 (s, 1H); 19F- NMR (376 MHz, DMSO-i¾ 5 ppm -110.42; MSiES") m/z 432 (MH ).
Example 99
N-(2-chloro-2'-(trifluoromethoxy)biphenyI-4-yl)-2-(4-(3- hydroxypropyIsulfonyl)phen I)acetamide

Step 1: To a solution of (4-mercaptophenyl)acetic acid (250 mg, see step 2 for synthesis of intermediate la) in DMF (20 mL) was added ((3-bromopropoxy)methyl)benzene (1.02 g) and potassium carbonate (822 mg). The reaction mixture was stirred at RT overnight. The mixture was partitioned between water (100 mL) and EtOAc (100 mL). The organic phase was washed with water (50 mL x 3) and brine, dried over anhydrous sodium sulphate, and concentrated. The residue was purified by flash chromatography (EtOAc : PE = 5:95) to afford 3-(benzyloxy)propyl 2-(4-(3- (benzyloxy)propyUhio)phenyl)acetate (100 mg) as a pale purple oil. MS(ES+) m z 465 (MH+).
Step 2: To a solution of 3-(benzyloxy)propyl 2-(4-(3-(benzy]oxy)propylthio)phenyl)acetate
(100 mg) in acetone (10 mL) and water (10 mL) was added oxone (179 mg). The reaction mixture was stirred at RT for 2 hours. Another portion of oxone (179 mg) was added, and the reaction mixture was stirred at RT overnight. Acetone was removed, and EtOAc (100 mL) was added. The organic phase was washed with water and brine, dried and concentrated to give 3-(benzyloxy)propyl 2-(4-(3- (benzyloxy)propylsulfonyl)phenyl)acetate (100 mg) as a colorless oil. MS(ES+) m/z 497 (MH+).
Step 3: To a solution of 3-(benzyloxy)propyl 2-(4-(3-(benzyloxy)propylsulfonyI)phenyl) acetate (100 mg) in ethanol (15 mL) was added a solution of NaOH (24.16 mg) in water (15 mL). The reaction mixture was stirred at room temperature overnight. Ethanol was removed under reduced pressure, and water (20 mL) was added. The aqueous phase was washed with dichloromethane (20 mL x 2), and then acidified to pH = 1 with 6 M HC1. The solution was extracted with ethyl acetate (30 mL x 2). The combined organic phases were washed with brine (20 mL), dried over sodium sulphate, and concentrated to give 2-(4-((3-(benzyloxy)propyl)sulfonyl)phenyl)acetic acid (80 mg) as a light yellow oil. MSfES*) m/z 349 (MH").
Step 4: A mixture of 2-(4-((3-(benzyloxy)propyl)suIfonyI)phenyl)acetic acid (80 mg), 2- chIoro-6'-(trifluoromethoxy)biphenyl-4-amine (intermediate 1 b, 79 mg), HOBt (42 mg), EDC (66
mg) and DIPEA (0.24 mL) in DCM (20 mL) was refluxed overnight. The mixture was washed with water (50 mL x 2), HCl (1 M, 20 mL), sat. NaHC03 (20 mL), and brine. The organic phase was dried and concentrated to yield 2-(4-(3-(benzyloxy)propyIsulfonyl)phenyl)-N-(2-chloro-6'- (trifluoromethoxy)biphenyl-4-yl)acetamide (87 mg) as a colorless oil. MSfES*) m/z 618 (MH+).
Step 5: To a solution of 2-(4-(3 -(benzyl oxy)propylsulfonyl)phenyl)-N-(2-chloro-6'-
(trifIuorometlioxy)biphenyl-4-yl)acetamide (87 mg) in methanol (20 mL) was added Pd/C (1.5 mg). The mixture was hydrogenated at RT overnight. Solid was removed by filtration, and the filtrate was concentrated. The residue was purified by prep-HPLC to afford N-(2-chloro-2'- (trifluoromethoxy)biphenyl-4-yl)-2-(4-(3-hydroxypropylsulfonyl)phenyl)acetamide (17 mg) as a white solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.71 (t, J= 5.2 Hz, 1H), 2.0 (m, 2H), 3.26 (t, J= 7.6 Hz, 2H), 3.75 (q, J= 5.2 Hz, 2H), 3.84 (s, 2H)5 7.23 (d, / = 8.8 Hz, 1H), 7.33 (m, 3H), 7.44 (m, 3H)} 7.56 (d, J= 8.0 Hz, 2H), 7.75 (s, 1H), 7.92 (d, J= 8.0 Hz, 2H); 19F-NMR (376 MHz, CDC13) δ ppm - 57.22; MSfES4 m/z 528 (MH+).
Example 100
N-(2-chloro-6-fluoro-2'-(trifluoromethoxy)biphenyl-4-yl)-2-(4-(ethylsulfonyI)phenyl)acetamide

Step 1: To a solution of 2-bromo-l-chloro-3-fluoro-5-nitrobenzene (280 mg) and 2- (trifluoromethoxy)phenylboronic acid (227 mg) in dioxane (10 mL) and water (2 mL) was added K2C03 (380 mg) and Pd(Ph3P)4 (127 mg) at RT under nitrogen. The reaction mixture was stirred at 100°C overnight. Solvent was removed, and the residue was purified by flash chromatography (PE) to give 2-chloro-6-fluoro-4-nitro-2'-(trifluoromethoxy)biphenyl (220 mg) as a yellow solid.
Step 2: A mixture of 2-chloro-6-fluoro-4-nitro-2'-(trifluorometlioxy)biphenyl (220 mg), ammonium fonnate (661 mg) and zinc (343 mg) in methanol (10 mL) and water (2 mL) was heated to 80°C and stirred overnight. Solvent was removed. The residue was dissolved in EtOAc (30 mL), and the solution was washed with water (10 mL) and brine (10 mL). The organic phase was concentrated to give 2-chloro-6-fluoro-2'-(trifluoromethoxy)biphenyl-4-amine (180 mg) as a brown oil. MSfES4 m/z 306 (MH4).
Step 3: A mixture of 2-chloro-6-fluoro-2'-(trifluoromethoxy)biphenyl-4-amine (150 mg), 2-(4- (ethylsulfonyl)phenyl)aceuc acid (intennediate la, 112 mg), EDC (113 mg) and HOBt (90 mg) in dichloromethane (10 mL) was stirred at reflux overnight. Solvent was removed. The residue was
purified by prep-HPLC to afford N-{2-chloro-6-fluoro-2'-(trifiuoromethoxy)biphenyl-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (55 mg) as a white solid. 'H-NMR (400 MHz, DMSO- ¾) δ ppm 1.11 (t, J= 7.2 Hz, 3H), 3.30 (q, J= 7.2 Hz, 2H), 3.87 (s, 2H), 7.50 (m, 3H), 7.59 (m, 4H), 7.70 (s, 1H), 7.87 (d, J= 8.4 Hz, 2H), 10.76 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -57.06, -109.35; MS(ES+) /JJ/Z 516 (MH+).
Example 101
2-(4-(ethyIsulfonyl)phenyl)-Ar-(2-(piperidin-l-yl)-6'-(trifluoromethoxy)biphenyl-4-yl)acetamide

Step 1: A solution of 2-fluoro-4-nitro-6'-(trifluoromethoxy)biphenyl (300 mg, see step 1 for synthesis of Example 98) in piperidine (6 mL) was stirred at 120°C for 7 days. Solvent was removed. The residue was purified by flash chromatography (EtOAc : PE - 1 :2) to give l-(4-nitro-6'- (trifluoromethoxy)biphenyl-2-yl)piperidine (400 mg) as a yellow oil. MS(ES÷) m/z 367 (MH+).
Step 2: A mixture of l-(4-nitro-6,-(trifluorometlioxy)biphenyl-2-yl)piperidine (400 mg), zinc dust (71.4 mg) and ammonium formate (67 mg) in methanol (30 mL) and water (10 mL) was heated to 80°C and stirred for 3 hours. Solvent was removed, and the residue was partitioned between water (60 mL) and EtOAc (60 mL). The aqueous phase was extracted with EtOAc (60 mL x 2), The combined organic phases were dried and concentrated to give 2-(piperidin-l-yl)-6'- (trifluoromethoxy)biphenyI-4-amine (400 mg) as a brown oil. MS(ES+) m/z 337 (MH4).
Step 3: A mixture of 2-(piperidin-l-yl)-6'-(trifluoromethoxy)biphenyl-4-amine (400 mg), 2-(4- (ethylsulfonyl)phenyI)acetic acid (intermediate la, 407 mg), EDC (342 mg) and HOBt (273 mg) in DCM (30 mL) was stirred at RT overnight. The mixture was washed with water (30 mL). The organic phase was dried over anhydrous Na2S04 and concentrated. The residue was purified by prep-HPLC to give 2-(4-(ethylsuIfonyl)phenyl)-N-(2-(piperidin-l-yl)-6,-(trifluoromethoxy)biphenyl-4-yl)acetamide (75 mg) as a light yellow solid. 1 H-NMR (400 MHz, DMSO- ) δ ppm 1.11 (t, J= 7.2 Hz, 3H), 1.31 (m, 6H), 2.63 (m, 4H), 3.29 (q, J= 7.2 Hz, 2H), 3.82 (s, 2H)} 7.08 (d, J= 8.4 Hz, 1H), 7.32 (d, J= 8.4 Hz, 1H), 7.42 (m, 4H), 7.51 (d, J- 8.4 Hz, 1H), 7.63 (d, .7= 8.4 Hz, 2H), 7.87 (d, J= 8.4 Hz, 2H), 10.32 (s, 1H); i9F-NMR (376 MHz, DMSO-i/6) δ ppm -55.55; MS(ES+) m/z 547 (MH+).
Example 102
2-(4-(ethylsulfonyl)phenyl)-N-(2-(2-methoxypyrimidin-5-yl)-2'-(trifluoromethoxy)-[l,l'- biphenyl]-4-yl)acetamide

Intermediate 102a: perfiuorophenyl 2-(4-(ethylsulfonyPphenyI)acetate
A mixture of pentafluorophenol (3.87 g), [4-(ethylsulfonyl)phenyl]acetic acid (intennediate la,
4 g), EDC (4.03 g) and HOBt (0.710 g) in DCM (35 mL) was stirred at room temperature overnight. The mixture was washed with water and brine, and then dried over anhydrous Na2S04. After concentration, the residue was purified by flash chromatography (EtOAc : PE ~ 0: 1 to 3:2) to afford a light yellow solid, which was further recrystallized from EtOAc/PE to afford pentafluorophenyl [4- (ethyIsulfonyl)phenyl]acetate (3.992 g) as a white solid with high purity. MS(ES+) m/z 395 (MH"").
Preparation of 2-f4-(,ethylsulfonyl')phenyl)-N-('2-f2-methoxypyrimidin-5-yl')-2,-('trifluoromethoxy')- Π ,1 '-biphenvfH-vnacetamide
Step 1: A mixture of 4,4,5, 5-tetramethyl-2-(4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2- yl)-l,3,2-dioxaborolane (intennediate 69a, 120 mg), 5-bromo-2-methoxypyrimidine (66.5 mg), Pd(Ph3P)4 (27.1 mg) and aq. Na2C03 solution (0.978 mL) in 1,4-dioxane (2 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by chromatography (EtOAc : PE = 0: 1 to 15:85) to afford 2-methoxy-5-(4-nitro-2'- (trifluoromethoxyi-CLl '-biphenyll^-ylipyrimidine (100 mg) as a yellow oil. MS(ES+) m/z 392 (MH ).
Step 2: Cone. HC1 (0.5 mL) was added into a mixture of 2-methoxy-5-(4-nitro-2'-
(trifluoromethoxy)-[l,l'-biphenyl]-2-yl)pyrimidine (106 mg) and tin(II) chloride dihydrate (306 mg) in ethanol (2 mL) at room temperature. The reaction mixture was heated to 60°C for 1 hour while stirring. The reaction mixture was cooled to 0°C and basified by sat. Na2C03 solution. The obtained solution was diluted with EtOAc, and then filtered. The filtrate was separated. The aqueous layer was extracted with EtOAc for 3 times. The combined organic layers were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure to afford 2-(2- metlioxypyrimidin-5-yl)-2l-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (90 mg) as a yellow oil.
MS(ES+) m/z 362 (MH+).
Step 3: A mixture of 2-(2-methoxypyrimidin-5-yl)-2'-(trifluoromethoxy)-[l, -biphenyl]-4- amine (90 mg) and perfluorophenyl 2-(4-(ethylsulfonyl)phenyl)acetate (intermediate 102a, 118 mg) in DMF (2 mL) was stirred at room temperature overnight. The reaction mixture was purified by MDAP to afford 2-(4-(ethylsulfonyl)phenyl)-N-(2-(2-methoxypyrimidin-5-yl)-2'-(trifluoromethoxy)- [l,r-biphenyl]-4-yl)acetamide (19 mg) as a yellow solid. Ή-NM (400 MHz, DMSO-^6) 5 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.86 (s, 5H), 7.26 (d, J= 6.8 Hz, 1H), 7.37 (d, J= 9.2 Hz, 1H), 7.45 (m, 3H), 7.63 (d, J= 8.4 Hz, 2H), 7.75 (m, 2H), 7.86 (d, J~ 8.4 Hz, 2H), 8.23 (s, 2H), 10.56 (s, 1H); )9F-NMR (376 MHz, DMSO-<¾) 5 ppm -56.26; MS(ES+) m/z 572 (MH* .
Example 103
Ar-(2,4,-dichloro-2,-(trinuoromethoxy)-[l,l'-biphenyl]-4-yI)-2-(4- (ethylsulfonyl)phenyl)acetamide
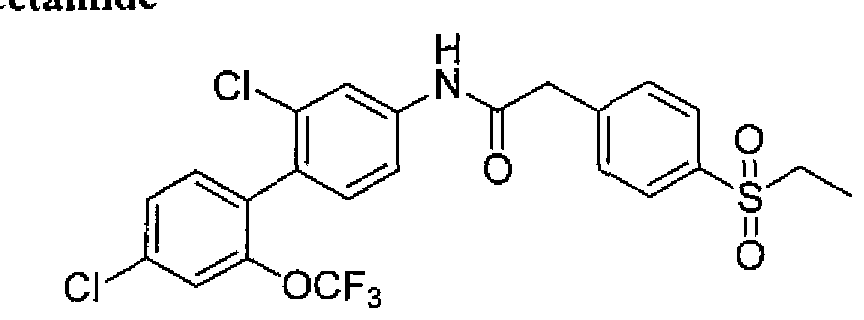
Step 1: NCS (330 mg) was added into a solution of 2-(trifluoromethoxy)phenol (440 mg) in acetic acid (5 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was poured into water, and extracted with DCM. The combined organic layers were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure to afford crude product of 4-chloro-2-(trifluoromethoxy)phenol (525 mg) as a yellow oil. MSfES ) m/z 212 (MH+).
Step 2: Tf20 (0.501 mL) was added into a solution of 4-chloro-2-(trifluoromethoxy)phenol (525 mg) and Et3N (1.033 mL) in DCM (6 mL) at 0°C dropwise. The reaction mixture was stirred at 0°C for 30 mins and then at room temperature overnight. The mixture was poured into water, and extracted with DCM. The combined organic layers were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated in vacuo and the residue was purified by column
chromatography to afford 4-chloro-2-(ti-ifluorometlioxy)phenyl trifluoromethanesulfonate (140 mg) as a yellow oil. 19F-NMR (376 MHz, CDC13) δ ppm -58.20, -73.31.
Step 3: A mixture of 4-chloro-2-(trifluoromethoxy)phenyl trifluoromethanesulfonate (70 mg), N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yi)phenyl]-2-[4-
(ethylsulfonyl)phenyl]acetamide (intennediate 6b, 104 mg), PdCl2(dppf)-CH2Cl2 adduct (13.27 mg), Cs2C03 (79 mg) in acetonitrile (1.2 mL) and water (0.4 mL) was sealed in a vessel and heated in the microwave at 100DC for 30 mins. The reaction mixture was filtered through celite and silica gel, the
filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(2,4'-dichloro- 2'-(trifluoromethoxy)-[l, -biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (42 mg) as a white solid. Ή-NMR (400 MHz, MeOD-a^) δ ppm 1.21 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.87 (s, 2H), 7.32 (m, 2H), 7.42 (m} 2H), 7.56 (dd, J= 1.2 Hz, 7.6 Hz, 1H), 7.63 (d, J= 8.4 Hz, 2H), 7.86 (m, 3H); 19F-NMR (376 MHz, MeOD-d ) 5 ppm -59.59; MSfES4 m/z 532 (MLT).
Example 104
Ar-(2-((dimethyIamino)methyl)-2,-(trifluoromethoxy)-[l.l,-biphenyI]-4-yl)-2-(4- (ethylsulfonyI)phenyl)acetamide, trifluoroacetic acid salt

Step 2: To a solution of methyl 4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2-carboxylate (400 mg) in tetrahydrofuran (THF) (10 mL) cooled at 0°C, was added LAH (133 mg) and the resulting mixture was stirred at this temperature for 2 hours. The mixture was first quenched with H20 (0.15 mL) slowly at 0 °C, followed with addition of 15% NaOH solution (0.15 mL) and H20 (0.15 mL). Then the mixture was filtered, washed with EtOAc and the filtrate was washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo. The obtained crude intermediate was dissolved in DCM (10 mL), to which Et3N (0.327 mL) and MsCl (0.137 mL) were added at 0 °C. The reaction mixture was wanned to room temperature and stirred for 1 hour. Then the mixture was quenched with brine and extracted with DCM. The combined organic layers were dried over anliydrous Na2S04 and concentrated in vacuo to afford the mesylate intennediate. The mesylate intermediate was dissolved in acetonitrile (10 mL), to which Et3N (0.490 mL) and dimethylamine, hydrochloride (115 mg) were added into the reaction mixture. The resulting solution was stirred at
room temperature overnight. The reaction mixture was concentrated and the residue was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2SC>4. After concentration, the residue was purified by column chromatography to afford N,N- dimetliyl-l -(4-nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)methanamine (47 mg) as a yellow solid. MS(ES+) m/z 341 (MH+).
Step 3: To a solution of N^-dimethyl-l^-nitro^'-itrifluoromethoxyJ-fljl'-biphenyl]^- yl)methanamine (47 mg) in ethanol (5 mL) was added HC1 (0.3 mL), followed by addition of tin(II) chloride (105 mg) and the resulting mixture was stirred at room temperature for 1 day. The mixture was basified with sat. Na2COj, and then extracted with EtOAc. The organic layer was washed with brine and dried over anhydrous Na2S04. After concentration, the residue was dissolved in DMF (5 mL), to which perfluorophenyl 2-(4-(ethyIsulfonyl)phenyl)acetate (intermediate 102a, 59.9 mg) was added and the reaction was stirred at room temperature for 4 hours. The mixture was purified by MDAP directly to afford N-(2-((dimetliylamino)methyl)-2'-(trifIuoromethoxy)-[l, -biphenyl]-4-yl)- 2-(4-(emylsulfonyl)phenyl)acetarnide, trifluoroacetic acid salt (22 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 2.41 (d, J= 4.2 Hz, 3H), 2.67 (d, J= 4.0 Hz, 3H), 3.29 (q, J= 7.3 Hz, 2H), 3.82 (dd, J= 6.5 Hz, 13.5 Hz, 1H), 3.87 (s, 2H), 4.32 (dd, J= 3.2 Hz, 13.4 Hz, 1H), 7.27 (d, J= 8.4 Hz, 1H), 7.46 (dd, J= 1.7 Hz, 7.5 Hz, 1H), 7.53 (m, 3H), 7.60 (dd, J= 1.8 Hz, 7.8 Hz, 1H), 7.64 (d, J= 8.2 Hz, 2H), 7.88 (d, J= 8.3 Hz, 2H), 8.16 (d, J= 1.9 Hz, 1H), 9.54 (br, 1H), 10.58 (s, 1H). l9F-NMR (376 MHz, OMSO-d6) δ ppm -56.12, -73.94; MS(ES+) m/z 521 (MH+).
Example 105
7Y-(2-chloro-2*-(trifluoromethoxy)-[l,l,-biphenyl]-4-yl)-2-(4-(ethylsulfon;
difluorophenyl)acetamide

Intermediate 105a: 2-(4-(ethylsulfonyI)-2,6-difluorophenyl)acetic acid
Step 1: To a suspension of NaH (2.71 g) in DMF (60 mL) was added diethyl malonate (8.14 g) at 0 °C, and the mixture was stirred at room temperature for 1.5 h under nitrogen. 1 ,2,3-Trifluoro-5- nitrobenzene (3 g) was added at 0 °C. The reaction mixture was stirred at room temperature overnight. The mixture was poured into 10% ammonium chloride solution, and then extracted with ethyl acetate (200 mL). The organic phase was washed with water (180 mL) and brine (180 mL x 3), dried over
anhydrous magnesium sulfate, filtered, and concentrated in vacuo to give diethyl 2-(2,6-difluoro-4- nitrophenyl)malonate (6 g). MSiES ) m/z 318 (MH+).
Step 2: To a solution of dietliyl 2-(2,6-difIuoro-4-nitrophenyl)malonate (8 g) in acetic acid (80 mL) and water (30 mL) was added H2S04 (20 mL). The reaction mixture was stirred at 100°C overniglit. Solvent was removed in vacuo. To the residue was added water (30 mL), and the aqueous phase was extracted with EtOAc (60 mL x 3). The organic layer was dried (Na2S04), filtered and concentrated to give 2-(2,6-difluoro-4-nitrophenyl)acetic acid (5 g) as a light yellow oil. MSiES m/z 172 (MH4).
Step 3: To a solution of 2-(2,6-difluoro-4-nitrophenyl)acetic acid (4 g) in ethanol (50 mL) was added Pd/C (1 g). The mixture was stirred under ¾ overnight. Solid was filtered, and solvent was removed in vacuo to give 2-(4-amino-2,6-difluorophenyl)acetic acid (3,4 g). MS(ES+) m/z 188 (MH+).
Step 4: A solution of sodium nitrite (0.814 g) in 40 mL of water was added dropwise at 0 °C, while stirring, to a suspension of 2-(4-amino-2,6-difIuorophenyI)acetic acid (2.2 g) in 40 mL of water and 3 mL of concentrated hydrochloric acid. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise at room temperature to a mixture of potassium <9-ethyl carbonodithioate (2.18 g), 50 mL of water and 5 mL of a 2 M sodium carbonate solution. Heating was effected to 45 °C until gas evolution stopped. The mixture was cooled to room temperature, and the pH was adjusted to 1 with concentrated hydrochloric acid. The oiled xanthogenate ester was extracted with ether. Solvent was evaporated to give a dark red liquid 2-(4-((ethoxycarbonothioyl)thio)-2,6-difluorophenyl)acetic acid (4.0 g). MSfES4) m/z 293 (MH4).
Step 5: 2-(4-({Ethoxycarbonothioyl)tI io)-2,6-difluorophenyl)acetic acid (9.5 g) was taken up in 40 mL of ethanol. A solution of OH (5.47 g) in 30 mL of water was added. Boiling at reflux was effected for 20 h. The major portion of ethanol was subsequently removed by the distillation at reduced pressure. The aqueous phase was cooled with ice, and was rendered acid with concentrated hydrochloric acid while stirring. The desired product was extracted with diethyl ether (100 mL x 5). The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to afford the desired product 2-(2,6-difluoro-4-mercaptophenyl)acetic acid (3.6 g). MS(ES+) m/z 205 (MH+).
Step 6: To a solution of 2-(2,6-difluoro-4-mercaptophenyl)acetic acid (3.6g) in DMF (40 mL) was added K2COj (17.06 g) and bromoethane (9.61 g). The reaction mixture was stirred at room temperature overniglit. The reaction mixture was partitioned between ethyl acetate (200 mL) and water (100 mL). The organic phase was washed with water (100 mL x 4) and brine (200 mL), dried
with sodium sulfate, filtered, and concentrated to give the desired product ethyl 2-(4-(ethyIthio)-2,6- difluorophenyl)acetate (3.1 g). MS(ES+) m/z 261 (MH .
Step 7: Ethyl 2-(4-(ethylthio)-2,6-difluorophenyl)acetate (3.1 g) was dissolved in DCM (30 mL), and the solution was cooled to 0°C with an ice bath. »?CPBA (6.17 g) was added in portions, and the reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the solid. The filtrate was washed with sat. sodium carbonate solution (30 mL x 2), water (30 mL), brine (30 mL), dried over sodium sulfate, and concentrated. The residue was purified by column chromatography to afford die target compound ethyl 2-(4-(ethylsulfonyl)-2,6-difluorophenyl)acetate (1.3 g). MS(ES+) m/z 293 (MH+).
Step 8: To a solution of ethyl 2-(4-(ethylsulfonyl)-2,6-difiuorophenyl)acetate (1.3 g) in ethanol
(30 mL) was added a solution of NaOH (0.534 g) in water (10 mL). The reaction mixture was stirred at room temperature overnight. Ethanol was removed under reduced pressure, and 20 mL of water was added. The aqueous phase was acidified to pH = 1 with 6 M HQ, and then extracted with ethyl acetate (50 mL x 3). The combined organic phases were washed with brine (50 mL), dried over sodium sulfate, and concentrated. The residue was purified by column chromatography to give 2-(4- (ethylsulfonyl)-2,6-difluorophenyl)acetic acid (530 mg) as a white solid. Ή-NMR (400 MHz, DMSO-4 δ ppm 1.13 (t, J= 7.6 Hz, 3H)} 3.43 (q, J= 7.6 Hz, 2H), 3.76 (s, 2H), 7.68 (m, 2H), 12.96 (s, 1 H); l9F-NMR (376 MHz, DMSO-< 6) δ ppm -110.26; MSiES m/z 265 ( H*).
Preparation of N-(2-chloro-2i-(trifluoromethoxy)-riJ'-biphenyl 4-ylV2-("4-fethylsulfonyl)-2,6- difluorophenyllacetamide
A mixture of 2-chloro-2'-[(trifluoromethyl)oxy]-4-biphenylamine (intermediate 15b, 60mg), 2-
(4-(ethylsuIfonyl)-2,6-difluorophenyl)acetic acid (intermediate 105a, 60.6 mg), HOBt (36.6 mg),
EDC (52.0 mg) in DCM (2 mL) was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-chloro-2'- (trifluoromethoxy)-[l, -biphenyl]-4-yl)-2-(4-(ethylsulfonyl)-2,6-difluorophenyl)acetamide (6.2 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-<¾) δ ppm 1.14 (t, J= 7.2 Hz, 3H), 3.45 (q, 7= 7.6 Hz,
2H), 3.94 (s, 2H), 7.32 (d, /= 8.4 Hz, 1H), 7.42 (dd, J= 2.0 Hz, 8.0 Hz 1H), 7.49 (m, 2H), 7.56 (m,
2H), 7.70 (m, 2H), 7.91 (d, J= 2.0 Hz, 1H), 10.69 (s, 1H); 19F-NMR (376 MHz, DMSO-£¾) δ ppm -
56.29, -109.95; MS(ES+) m/z 534 (MH+). Example 106
2-(4-(ethylsulfonyI)phenyI)-iV-(2-methyl-2,-(trifluorornethoxy)-[l, -biphenyl]-4-yl)acetamide

A mixture of N-(4-bromo-3-methylphenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide (intermediate 52a, 80 mg), (2-(trifluoromethoxy)phenyl)boronic acid (45.7 mg), PdCl2(dppf)-CH2Cl2 adduct (12.4 mg) and Cs2C03 (79 mg) in acetonitrile (1.5 niL)/water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by MDAP to afford 2-(4- (etliylsulfonyl)phenyl)-N-(2-mediyl-2'-(trifluoromethoxy)-[l , -biphenyl]-4-yl)acetamide (33 mg) as a yellow solid. 'H-NMR (400 MHz, DMSO-d6) δ ppm 1.10 (t, J= 7.2 Hz, 3Η), 2.03 (s, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.81 (s, 2H), 7.08 (d, J= 8.4 Hz, 1H), 7.35 (dd, J= 1.6 Hz, 8.0 Hz, 1H), 7.50 (m, 5H), 7.62 (d, J= 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 2H), 10.31 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -56.24; MS(ES+) m/z 478 (MH+).
Example 107
iV-(2'-cyano-2,6-difluoro-[l,l'-biphenyI]-4-yl)-2-(4-(ethylsulfonyl)phenyI)acetamide

(ethyIsulfonyl)phenyl)acetamide (intennediate 27a, 100 mg), Pd2(dba)3 (65.7 mg), \x -tert- butylphosphine, tetrafluoroboric acid salt (83 mg) and 2 M Na2C03 (0.478 mL) in 1,4-dioxane (2 mL) was bubbled with nitrogen, then sealed in the reaction vessel and heated in the microwave at 100°C for 1 hour. After cooled to room temperature, the mixture was filtered through celite, washed with EtOAc. The filtrate was washed with 2M HC1, sat. Na2C03 and brine, then concentrated in vacuo. The residue was purified by MDAP to afford N-(2'-cyano-2,6-difluoro-[l,l'-biphenyl]-4-yl)-2-(4- (ethyIsulfonyl)phenyl)acetamide (8 mg) as a white solid. 1 H-NMR (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 3.29 (q, J= 7.3 Hz, 2H), 3.88 (s, 2H), 7.51 (m, 2H), 7.65 (m, 4H), 7.83 (dd, J- 1.3 Hz, 7.7 Hz, 1H), 7.86 (d, 7= 8.3 Hz, 2H), 8.02 (dd, 7= 0.8 Hz, 7.7 Hz, 1H), 10.84 (s, 1H); 19F- NMR (376 MHz, DMSO-£¾ δ ppm -112.37; MStES^) m/z 441 (MH1).
Example 108
Ar-(2,6-difluoro-2'-isopropyl-[l,l'-biphenyl]-4-yl)-2-(4-(ethyIsulfonyl)phenyl)acetainide

A mixture of (2-isopropylphenyl)boronic acid (85 mg), N-(4-bromo-3,5-difluorophenyl)-2-(4- (ethyIsulfonyl)phenyl)acetamide (intermediate 27a, 120 mg), Pd2(dba)j (26.3 mg), tn-tert- butylphosphine, tetrafluoroboric acid salt (33.3 mg) and 2 M Na2C03 (0.5 mL) in 1,4-dioxane (2 mL) was bubbled with nitrogen, then sealed in the reaction vessel and heated in the microwave at 100°C for 1 hour. After cooled to room temperature, the mixture was filtered tlirough celite, washed with EtOAc. The filtrate was washed with 2M HCl, sat. Na2C03 and brine, then concentrated in vacuo. The residue was purified by MDAP to afford N-(2,6-difluoro-2'-isopropyl-[l,l'-biphenyl]-4-yI)-2-(4- (ethylsulfonyl)phenyl)acetamide (90 mg) as a white solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 1.09 (m, 9H), 2.63 (m, 1H), 3.29 (q, J= 7.3 Hz, 2H), 3.86 (s, 2H), 7.14 (d, 7= 7.0 Hz, 1H), 7.26 (dt, J = 1.6 Hz, J= 7.4 Hz, 1H), 7.43 (m, 4H), 7.62 (d, 7= 8.3 Hz, 2H), 7.86 (d, J= 8.3 Hz, 2H), 10.73 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -1 11.74; MSfES ) m/z 458 (MH+). Example 109
7V-(2'-ethyI-2,6-difluoro-[l,l'-biphenyl]-4-yl)-2-(4-(ethyIsuIfonyl)phenyI)acetamide

A mixture of (2-ethylphenyl)boronic acid (77 mg), N-(4-bromo-3,5-difluorophenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide (intermediate 27a, 120 mg), Pd2(dba)3 (26.3 mg), n-tert- butylphosphine, tetrafluoroboric acid salt (33.3 mg) and 2 M a2C03 (0.5 mL) in 1,4-dioxane (2 mL) was bubbled with N2, then sealed in the reaction vessel and heated in the microwave at 100°C for 1 hour. After cooled to room temperature, the mixture was filtered tlirough celite, washed with EtOAc. The filtrate was washed with 2M HCl, sat. Na2C03 and brine, then concentrated in vacuo. The residue was purified by MDAP to afford N-(2'-ethyl-2,6-difluoro-[l,l '-biphenyl]-4-yl)-2-(4-
(ethylsulfonyl)phenyl)acetarnide (78 mg) as a white solid. Ή-NMR (400 MHz, DMSO- 6) δ ppm 1.03 (t, J= 7.6 Hz, 3H), 1.22 (t, J= 7.4 Hz, 3H), 2.44 (q, J= 7.6 Hz, 2H), 3.20 (q, J= 7.4 Hz, 2H), 3.86 (s, 2H), 7.11 (d, J= 7.5 Hz, 1H), 7.23 (m, 1H), 7.34 (m, 4H), 7.63 (d, J~ 8.3 Hz, 2H), 7.S9 (d, J = 8.3 Hz, 2H); 19F-NMR (376 MHz, DMSO-i/6) δ ppm -113.26; MS(ES") m/z 444 (MH+). Example 110 and Example 111
2-(4-(ethylsulfonyl)phenyl)-iV-(6'-(trinuoromethoxy)-[l,l,:2,,l,,-terphenyl]-4-yl)acetarnide 2-(4-(ethylsulfonyJ)phenyl)-N-(2-(trifluoroniethoxy)-Il,l':2',l',-terphenyI]-4'-yl)acetamide

Step 1: A solution of 4,4,5,5-tetramethyl-2-(4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2-yl)- 1,3,2-dioxaborolane (intermediate 69a, 90 mg), bromobenzene (41.4 mg), Pd(Ph3P)4 (20.33 mg) and aq. Na2COj (0.440 mL) in 1,4-dioxane (4 mL) was heated to 100°C for 0.5 hour in the microwave. The resulting mixture was filtered tlirough celite, and washed by ethyl acetate. Solvent was removed and extracted with ethyl acetate, washed by saturated ammonium chloride solution and brine. The residue was purified by column chromatography and concentrated in vacuo to give 4'-nitro-2- (trifluoromethoxy)-l,l':2',l"-terphenyl (68 mg) and 4"-nitro-3'-(trifluoromethoxy)-l, :2',l"-terphenyl (68 mg) as a yellow oil. MS(ES+) m/z 360 (MH^).
Step 2: 4'-Nitro-2-(triίluoromethoxy)-l ,Γ:2',l,,-teφhen l (68 mg) and 4"-nitro-3'- (tf fluoromethoxy)-l,l':2',l"-terphenyl (68 mg) was dissolved in ethanol (10 mL). To this solution, tin(II) chloride dihydrate (410 mg) was added gradually. Then the reaction mixture was heat to 70°C for 3 hours. Solvent was removed and the sodium hydroxide solution (2 M) was added. The mixture was extracted with ethyl acetate and washed with brine. The organic layer was concentrated in vacuo to give 2-(trifluoromethoxy)-[l, :21, '-terphenyl]-4'-amine (58 mg) and 6'-(trifluoromethoxy)-
 (58 mg) as a yellow oil. MS(ES+) m/z 330 (MH4 .
(58 mg) as a yellow oil. MS(ES+) m/z 330 (MH4 .
Step 3: 2-(Trifluoromethoxy)-[l,r:2f,l"-terphenyl]-4'-amine (58 mg), 6'-(trifluoromethoxy)- [l,r:2',l"-terphenyl]-4-amine (58 mg) and perfluorophenyl 2-(4-(ethylsulfonyl)phenyl)acetate
(intennediate 102a, 83 mg) were dissolved in DCM (10 mL). Then to mis solution, DIPEA (0.055 mL) was added gradually. The reaction mixture was stirred for 3 hours. Solvent was removed. The
obtained mixture was redissolved in DMF. Solid was filtered off. The residue was purified by MDAP to give 2-(4-(ethyIsulfonyl)phenyl)-N-(2-(^
mg) as a white solid and 2-(4-(ethylsulfonyl)phenyl)-N-(6'-(trifiuorometlioxy)-[l, :2') '-terphenyl]- 4-yl)acetamide (8 mg) as a white solid.
Example 110: Ή-NMR (400 MHz, MeOD-</4) δ ppm 1.12 (t, J= 7.4 Hz, 3H), 3.11 (q, J= 7.5
Hz, 2H), 3.78 (s, 2H), 7.00 (m, 6H), 7.14 (m, 2H), 7.21 (m, 2H), 7.56 (m, 4H), 7.80 (d, J= 8.4 Hz, 2H); 19F- MR (376 MHz, DMSO-c?6) δ ppm -58.49; MS(ES^ m/z 540 (MH+).
Example 11 1: Ή-NMR (400 MHz, DMSO-c¾ δ pm 1.03 (t, J= 7.4 Hz, 3H), 3.20 (q, J~ 7.3 Hz, 2H), 3.71 (s, 2H), 6.93 (d, J= 8.8 Hz, 2H), 7.01 (d, /= 7.6 Hz, 2H), 7.14 (m, 3H), 7.37 (m, 2H), 7.41 (d, J= 8.8 Hz, 2H), 7.52 (m, 3H), 7.77 (d, J= 8.4 Hz, 2H); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -55.80; MS(ES÷) m/z 540 (MH+).
Example 112
2-(4-(ethyl sulf onyl)phenyl)-JV-(2-(pyridin-3-yl)-2'-(triflu or omethox )- [ 1, -biphenyl] -4- yl)acetamide, trifluoroacetic acid salt

Step 1:A solution of 4,4,5,5-tetramethyl-2-(4-nitro-2'-(trifluoroinethoxy)-[l,r-biphenyl]-2-yl)- 1,3,2-dioxaborolane (intermediate 69a, 150 mg), 3-bromopyridine (69.5 mg), Pd(Ph3P)4 (33.9 mg) and aq. Na2C03 (2 M, 0.733 mL) in 1,4-dioxane (5 mL) was heated to 90°C for 0.5 hour in the microwave. The resulted mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed and redissolved in ethyl acetate. The solution was washed by saturated ammonium chloride solution and brine to give 3-(4-nitro-2'-(trifluoromethoxy)-[l ,1 '-biphenyl]-2-yl)pyridine (251 mg) as a yellow oil. MS(ES÷) m/z 361 (MH4).
Step 2: 3-(4-Nitro-2'-(trifluoromethoxy)-[l,l1-biphenyl]-2-y])pyridine (251 mg) was dissolved in ethanol (15 mL). Then to this solution, tin(II) chloride dihydrate (786 mg) was added gradually. The reaction mixture was heat to 70°C for 3 hours. Solvent was removed and sodium hydroxide solution (2 M) was added. Then the mixture was extracted with ethyl acetate and washed by brine. The organic layer was concentrated in vacuo to give 2-(pyridin-3-yl)-2'-(trifluoromethoxy)-[l, - biphenyl 4-amine (178 mg) as a yellow solid. MS(ES÷) m/z 331 (MH+).
Step 3: 2-(Pyridin-3-yl)-2'-(trifluorornethoxy)-[l, -biphenyl]-4-amine (178 mg) and perfluorophenyl 2-(4-(ethylsulfonyl)phenyl)acetate (intermediate 102a, 96 mg) were dissolved in DCM (10 mL). Then to this solution, N-ethyl-N-isopropylpropan-2 -amine (31,3 mg) was added gradually. The reaction mixture was stirred for 3 hours. Solvent was removed. The mixture was redissolved in DMF. Solid was filtered off. The residue was purified by MDAP to give 2-(4- (ethylsulfonyl)phenyl)-N-(2-(pyridin-3-yl)-2'-(trifiuoromethoxy)-[l,r-biphenyl]-4-yl)acetamide, trifluoroacetic acid salt (29 mg) as a white solid. ]H-NM (400 MHz, MeOD-d4) δ ppm 1.12 (t, J= 7.4 Hz, 3H), 3.11 (q, J- 7.5 Hz, 2H), 3.80 (s, 2H), 7.06 (d, J= 7.6 Hz, 1H), 7.35 (m, 4H), 7.61 (m, 4H), 7.80 (d, J= 8.4 Hz, 2H), 7.88 (s, 1H), 8.01 (d, /= 8.4 Hz, 1H), 8.42 (s, 1H), 8.51 (d,J= 5.6 Hz, 1H); ,9F-NMR (376 MHz, MeOD-rf4) δ ppm -58.86, -77.29; MS(ES+) m/z 541 (MH+).
Example 113 and Example 114
2-(4-(ethylsulfonyl)phenyl)-N-(2-(l-methyl-lH-pyrazol-4-y])-2'-(trifluoromethoxy)-[l,l'- biphenyl]-4-yl)acetamide
2-(4-(ethylsulfonyl)phenyI)-N-(2'-(l-methyI-lH-pyrazol-4-yl)-6'-(trifluoromethoxy)-[l,l'- biphenyl]-4-yl)acetamide

Step 1: A solution of 4,4,5,5-tetramethyl-2-(4-nitro-2'-(trifiuoromethoxy)-[l, -biphenyl]-2-yl)- 1,3,2-dioxaborolane (intermediate 69a, 200 mg), 4-bromo-l-methyl-lH-pyrazole (94 mg), Pd(Ph P)4 (45.2 mg) and aq. Na2C03 (0.489 mL) in 1,4-dioxane (5 mL) was heated to 90°C for 0.5 hour in the microwave. The resulting mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed and redissolved in ethyl acetate. The solution was washed with saturated ammonium chloride solution and brine. The residue was purified by column chromatography and concentrated in vacuo to give l-methyl-4-(4-nitro-21-(trifluoromethoxy)-[l,l'-biphenyl]-2-yl)-lH-pyrazole (114 mg) and l-methyI-4-(4'-nitro-6-(trifluorometlioxy)-[l,l '-biphenyl]-2-yl)-lH-pyrazole (114 mg) as a yellow oil. MSfES4 m/z 364 (MH .
Step 2: l-Methyl-4-(4-nitro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-2-yl)-lH-pyrazole (114 mg) and l-methyl-4-(4'-nitro-6-(trifluoromethoxy)-[l ,l'-biphenyl]-2-yl)-lH-pyrazole (114.0 mg) was dissolved in ethanol (10 mL). Then to this solution, tin(II) chloride dihydrate (425 mg) was added gradually. Then the reaction mixture was heat to 70°C for 3 hours. Solvent was removed and sodium
hydroxide solution (2 M) was added. Then it was extracted with ethyl acetate and washed with brine. The organic layer was concentrated in vacuo to give 2-(l-methyHH-pyrazol-4-yl)-2'- (trifluoromethoxy)-[l, -biphenyI]-4-amine (80 mg) and 2,-(l-methyI-lH-pyrazol-4-yl)-6'- (trifluoromethoxy)-[l,r-biphenyl]-4-amine (80 mg) as a yellow oil. MS(ES+) m/z 334 (MH+).
Step 3: 2-(l-Methyl-lH-pyrazol-4-yl)-2'-(trifluoromethoxy)-[l ,l'-biphenyl]-4-amine (80 mg),
2'-(l-methyl-lH-pyrazol-4-yl)-6'-(trifluoromethoxy)-[l,l'-biphenyl]-4-amine (80 mg) and perfluorophenyl 2-(4-(ethylsulfonyl)phenyl)acetate (intermediate 102a, 88 mg) were dissolved in DCM (10 mL). Then to this solution, N-etliyl-N-isopropylpropan-2-amine (38.5 mg) was added gradually. The reaction mixture was stirred for 3 hours. Solvent was removed. The obtained mixture was redissolved in DMF and solid was filtered off. The residue was purified by MDAP to afford 2-(4- (ethylsulfonyl)phenyl)-N-(2-(l-med1yI H-pyrazol -yl)-2'-(trifluoromethoxy)-[l,r-biphenyl]-4- yl)acetamide (31 mg) as a white solid and 2-(4-(ethylsulfonyl)phenyl)-N-(2'-(l -methyl-lH-pyrazol-4- yl)-6'-(trifluoromethoxy)-[l, -biphenyl]-4-yl)acetamide (16 mg) as a white solid.
Example 113: Ή-NMR (400 MHz, MeOD-i/4) δ ppm 1.12 (t, J= 7.4 Hz, 3H), 3.11 (q, J= 7.5 Hz, 2H), 3.65 (s, 3H), 3.78 (s, 2H), 6.89 (s, 1H), 7.10 (s, 1H), 7.11 (d, J= 8.0 Hz, 1H), 7.20 (m, 2H), 7.27 (t, J= 7.4 Hz, 1H), 7.36 (d, J= 7.4 Hz, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.56 (d, J= 8.4 Hz, 2H), 7.68 (s, 1H), 7.80 (d, J= 8.4 Hz, 2H); I9F-NMR (376 MHz, MeOD-i¾) 5 ppm -58.61; MSfES ) m/z 544 (MH4).
Example 114: Ή-NMR (400 MHz, MeOD-c 4) δ ppm 1.13 (t, = 7.4 Hz, 3H), 3.11 (q, J= 7.5 Hz, 2H), 3.64 (s, 3H), 3.77 (s, 2H), 6.89 (s, 1H), 7.01 (d, J= 8.4 Hz, 2H), 7.11 (s, 1H), 7.19 (d, J= 8.0 Hz, 1H), 7.35 (t, .7= 8.0 Hz, 1H), 7.43 (d, 7= 8.0 Hz, 1H), 7.54 (m, 4H), 7.80 (d, J= 8.4 Hz, 2H); ,9F-NMR (376 MHz, MeOD-i/4) δ ppm -58.32; MS(ES+) m/z 544 (MH+).
Example 115
Ar-(2s6-difluoro-2'-methyl-[l, -biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

A mixture of Pd2(dba)3 (39.4 mg), td-teri-butylphosphine, tetrafluoroborate (41.5 mg), sat. Na2C03 solution (2 M, 0.478 mL), o-tolylboronic acid (48.8 mg) and N-(4-bromo-3,5- difluorophenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide (intennediate 27a, 100 mg) in 1,4-dioxane (2 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture
was filtered through celite and silica gel. The filtrate was washed with 2M HC1 for three times. The organic phases were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(2,6-difIuoro-2'-memyl-[l, -biphenyl]-4- yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (55 mg) as a white solid. 'H-NMR (400 MHz, DMSOi/6) δ ppm 1.10 (t, J= 7.6 Hz, 3H), 2.09 (s, 3H), 3.29 (q, J= 7.6 Hz, 2H), 3.86 (s, 2H), 7.20 (d, J= 7.6 Hz, 1H), 7.27 (m, 1H), 7.34 (m, 2H), 7.43 (m, 2H), 7.62 (d, J= 8.4 Hz, 2H), 7.86 (d, J= 8.4 Hz, 2H),10.72 (s, 1H); l9F-NMR (376 MHz, DMSO-<¾) 5 ppm -112.14; MS(ES^) z 430 (MH+).
Example 116
Ar-(2,6-difluoro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4-(/y- methylsulfamoyl)phenyl)acetamide

Intennediate 116a: 2.6-difluoro-2,-rtrifluoromethoxy')-riJ '-biphenyl1-4-amine
A mixture of (2-(trifluoromethoxy)phenyl)boronic acid (1.485 g), 4-bromo-3,5-difluoroaniline (1 g), PdCl2(dppf)-CH2Cl2 adduct (150 mg) and Cs2C03 (1.880 g) in acetonitrile (14 mL)/water (4.67 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by column chromatography to afford 2,6-difluoro-2,-(trifluoromethoxy)-[l, -biphenyl]-4- amine (0.695 g) as a yellow oil. MS(ES+) m/z 290 (Mh ).
Preparation of N-f2.6-dif1uoro-2'-ftrifluoromethoxyHl '-biphenyl -vP^-H-fN- methylsulfamoyDphenvDacetamide
A mixture of 2-(4-(N-ethylsulfamoyl)phenyl)acetic acid (intermediate 35a, 95 mg), 2,6-difluoro-
2'-(trifluoromethoxy)-[l, -biphenyl]-4-amine (intermediate 1 16a, 100 mg), EDC (99 mg) and 1H- benzo[c/][l,2,3]triazol-l-ol (37.4 mg) in DCM (1 mL) was stirred at room temperature overnight. The reactoin mixture was washed with 2M HC1 and brine. The organic phase was concentrated in vacuo and the residue was purified by MDAP to afford N-(2}6-difluoro-2'-(trifluoromethoxy)-[l,l'- biphenyl]-4-yl)-2-(4-(N-methylsulfamoyl)phenyl)acetamide (12 mg) as a yellow solid. Ή-NMR (400
MHz, DMSO-</6) 6 ppm 2.41 (d, J= 4.8 Hz, 3H), 3.83 (s, 2H), 7.44 (m, 3H), 7.54 (m, 5H), 7.62 (m,
1H), 7.75 (d, J= 8.4 Hz, 2H), 10.78 (s, 1H); ,9F-NMR (376 MHz, DMSO-i fi) 5 ppm -56.80, -112.35;
MS(ES+) m/z 501 (MH ).
Example 117
iY-(2,6-dinuoro-2'-(trifluoromethoxy)-[l,l'-bipheny[)-4-yI)-2-(4- (methylsulfonyl)phenyl)acetamide

(trifliioromethoxy)-[l,r-biphenyl]-4-amine (intennediate 1 16a, 100 mg), EDC (99 mg) and lH- benzo[i/][l ,2,3]triazol-l -ol (37.4 mg) in DCM (2 mL) was stirred at room temperature overnight. The reaction mixture was washed with 2M HC1 and brine. The organic phase was concentrated in vacuo and the residue was purified by MDAP to afford N-(2,6-difluoro-2'-(trifluoromethoxy)-[l,r- biphenyl]-4-yl)-2-(4-(methylsulfonyl)phenyl)acetamide (73 mg) as a yellow solid. Ή-NMR (400
MHz, DMSO-i¼) δ ppm 3.21 (s, 3H), 3.86 (s, 2H), 7.46 (m, 2H), 7.53 (m, 3H), 7.62 (m, 3H), 7.90 (m, 2H), 10.76 (s, 1H); 19F-NMR (376 MHz, DMSO-</6) δ ppm -56.80, -1 12.32; MS(ES+) m/z 486 (MH+).
Example 118
J/V-(2,6-difluoro-2'-(trifluoromethoxy)-[lil'-biphenyl]-4-yI)-2-(4- (methylsulfonyl)phenyl)acetamide

Step 1: To a suspension of NaH (60% in oil) (10 g) in DMF (200 mL) was added diethyl malonate (30 g) at 0°C. The mixture was stirred at room temperature for 1.5 h under nitrogen. 1 ,2- Difluoro-4-nitrobenzene (13 g) was added at 0°C. The reaction mixture was stirred at room temperature overnight. The mixture was poured into 10% ammonium chloride solution, and then extracted with ethyl acetate (200 mL). The organic phase was washed with water (180 mL) and brine (180 mL x 3), dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo to give diethyl 2-(2-fluoro-4-nitrophenyl)malonate (13 g). MS(ES+) m/z 300 (MH+).
Step 2: To a solution of diethyl 2-(2-fluoro-4-nitrophenyl)malonate (12 ) in acetic acid (80 mL) and water (30 mL) was added H2S04 (20 mL). The reaction mixture was stirred at 100°C overnight.
Solvent was removed in vacuo. To the residue was added water (100 mL), and the aqueous phase was extracted with EtOAc (200 mL x 2). The organic layers were dried over Na2S04, filtered and concentrated to give 2-(2-fluoro-4-nitrophenyl)acetic acid (7 g) as a light yellow oil.
Step 3: To a solution of 2-(2-fluoro-4-nitrophenyI)acetic acid (5 g) in ethanol (50 mL) was added Pd/C (1 g). The mixture was stirred under ¾ overnight. Solid was filtered, and solvent was removed in vacuo to give 2-(4-amino-2-fluorophenyl)acetic acid (4.6 g). MSfES^ m/z 170 (MH^.
Step 4: A solution of sodium nitrite (1.88 g) in 20 mL of water was added dropwise at 0°C, while stirring, to a suspension of 2-(4-amino-2-fluorophenyl)acetic acid (4.6 g) in 80 mL of water and 6.8 mL of concentrated hydrochloric acid. After the addition was complete, the reaction mixture was stirred at the same temperature for a further 45 minutes. This cold diazonium salt solution was then added dropwise at room temperature to a mixture of potassium O-ethyl carbonodithioate (5 g), 80 mL of water and 20 mL of a 2 M sodium carbonate solution, and heating was effected to 45°C until gas evolution stopped. The mixture was cooled to room temperature, and the pH was adjusted to 1 with concentrated hydrochloric acid. The oiled xanthogenate ester was extracted with ether. Solvent was evaporated to give a dark red liquid 2-(4-(ethoxycarbonothioyl)tliio)-2-fluorophenyl)acetic acid (5.5 g). MS(ES+) m/z 275 (MH*).
Step 5: 2-(4-(Ethoxycarbonothioy])thio)-2-fluorophenyl)acetic acid (5 g) was taken up in 80 mL of ethanol. A solution of KOH (3 g) in 60 mL of water was added, and boiling at reflux was effected for 20 h. The major portion of ethanol was subsequently removed by the distillation at reduced pressure. The aqueous phase was cooled with ice, and was rendered acid with concentrated hydrochloric acid while stirring. The desired product was extracted with diethyl ether (100 mL x 5). The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to afford the desired product 2-(2-fIuoro-4-mercaptophenyl)acetic acid (4.1 g). MS(ES+) m/z 187 (MH*).
Step 6: To a solution of 2-(2-fluoro-4-mercaptophenyl)acetic acid (3.6 g) in DMF (40 mL) was added K2C03 (18.7 g) and bromoethane (9.61 g). The reaction mixture was stirred at room temperature overnight. The reaction mixture was partitioned between ethyl acetate (80 mL) and water (60 mL). The organic was washed with water (30 mL x 3) and brine (30 mL), dried with sodium sulfate, filtered, and concentrated to give the desired product ethyl 2-(4-(ethylthio)-2- fluorophenyl)acetate (4.4 g). MS(ES+) m/z 242 (MH+).
Step 7: Ethyl 2-(4-(ethylthio)-2-fluorophenyl)acetate (3.1 g) was dissolved in DCM (50 mL). The solution was cooled to 0°C with an ice bath. wCPBA (6.6 g) was added in portions. The reaction mixture was stirred at room temperature overnight, and then filtered to remove the solid. The filtrate was washed with sat. sodium carbonate solution (30 mL x 2), water (30 mL), brine (30 mL), dried
over sodium sulfate, and concentrated. The residue was purified by column clrromatography (silica gel; EtOAc:PE = 1:1) to afford the target compound ethyl 2-(4-(etliylsulfonyl)-2-fluorophenyl)acetate (1.2 g). MS(ES+) m/z 275 (MH*).
Step 8: To a solution of ethyl 2-(4-(ethylsulfonyl)-2-fluorophenyl)acetate (0.9 g) in ethanol (20 mL) was added a solution of NaOH (0.39 g) in water (6 mL). The reaction mixture was stirred at room temperature overnight. Ethanol was removed under reduced pressure, and 20 mL of water was added. The aqueous phase was acidified to pH = 1 with 6 M HC1, and then extracted with ethyl acetate (50 mL x 3). The combined organic phases were washed with brine (50 mL), dried over sodium sulfate, and concentrated. The residue was purified by column chromatography (silica gel; MeOH : DCM = 1 :20) to give the product 2-(4-(ethylsulfonyl)-2-fluorophenyl)acetic acid (460 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) 5 ppm 1.12 (t, J= 7.6 Hz, 3H), 3.34 (q, J= 7.6 Hz, 2H), 3.78 (s, 2H), 7.69 (m, 3H), 12.70 (s, 1 H). MS(ES+) m/z 247 (MH+).
Step 9: A mixture of 2,6-difluoro-2'-(trifluoromethoxy)-[l, -biphenyl]-4-amine (intermediate 116a, 100 mg), 2-(4-(ethylsulfonyl)-2-fluorophenyl)acetic acid (94 mg), EDC (80 mg) and HOBt (23.36 mg) in DCM (3 mL) was stirred at room temperature overnight. The reaction was stirred for another 6 hours, and then concentrated in vacuo. The resulting residue was purified directly by MDAP to afford N-(256-difluoro-2'-(trifluoromedioxy)-[l,l'-biphenyl]^-yl)-2-(4-(ethylsulfonyl)-2- fluorophenyl)acetamide (79 mg) as a white solid. Ή-NMR (400 MHz, DMS W6) δ ppm 1.12 (t, J = 7.3 Hz, 3H), 3.38 (q, J= 7.3 Hz, 2H), 3.95 (s, 2H), 7.45 (m, 2H), 7.52 (m, 3H), 7.62 (m, 1H), 7.73 (m, 3H), 10.82 (s, 1H); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -56.80, -112.26, -113.65; MS(ES^) m/z 518 (MH+).
Example 119
N-(2,6-difluoro-2'-(trifIuoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyl)propanamide

Step 1: To a solution of 2-(4-nitrophenyl)propanoic acid (10 g) in methanol was added Pd/C (0.27 g). The reaction mixture was stirred at RT under ¾ for 24 hrs. The mixture was filtered, and the filtrate was concentrated to give 2-(4-aminophenyl)propanoic acid (8 g) as a brown oil.
Step 2: A solution of NaN02 (3.35 g) in 20 inL of water was added dropwise to a suspension of 2-(4-aminophenyl)propanoic acid (8.0 g) in 26 mL of water and 60 mL of concentrated hydrochloric acid cooled at 0 °C. After the addition was complete, the reaction mixture was stirred at the same temperature for another 45 mins. The cold dia2onium salt solution was then added dropwise at room temperature to a mixture of potassium O-etliyl carbonodithioate (9.0 g), 40 mL of water and 40 mL of a 2 M sodium carbonate solution. The mixture was heated at 45 °C until gas evolution stopped. The mixture was cooled to room temperature, and the pH was adjusted to 1 with concentrated hydrochloric acid. The oiled xanthogenate ester was extracted with EtOAc (100 mL x 3). The combined organic phases were dried and concentrated to give 2-(4- (ethoxycarbonothioylthio)phenyl)propanoic acid (13.0 g) as a dark red liquid. S(ES+) m/z 271 (MH+).
Step 3: 2-(4-(Ethoxycarbonothioylthio)phenyl)propanoic acid (13.0 g) was taken up in 60 mL of ethanol, and a solution of KOH (19.88 g) in 60 mL of water was added. The mixture was refluxed for 3 h. The major portion of ethanol was removed. The aqueous phase was cooled with ice, and was rendered acid with concentrated hydrochloric acid while stirring. The mixture was extracted with
DCM (50 mL x 3), and the organic phase was washed with brine, dried and concentrated to afford the desired product 2-(4-mercaptophenyl)propanoic acid (8.0 g) as a yellow solid. MS(ES+) m/z 183 (MH÷).
Step 4: To a solution of 2-(4-mercaptophenyl)propanoic acid (8.0 g) in DMF (100 mL) was added bromoethane (14.35 g) and K2C03 (24.27 g). The reaction mixture was stirred at T overnight. The mixture was partitioned between ethyl acetate (200 mL) and water (200 mL). The organic was washed with water (100 mL x 4) and brine (50 mL), dried with sodium sulphate, and concentrated to give the desired product ethyl 2-(4-(ethyIthio)phenyl)propanoate (12.0 g) as a red oil. MSfES*) m/z 239 (MET).
Step 5: To a solution of ethyl 2-(4-(ethylthio)phenyl)propanoate (12.0 g) in DCM (100 mL) was added mCPBA (26.1 g). The reaction mixture was stirred at RT overnight. The reaction mixture was filtered to remove the solid. The filtrate was washed with sat. sodium carbonate solution (200 mL x 2), water (100 mL), brine (50 mL), dried and concentrated. The residue was purified by column chromatography (silica gel; EtOAc:PE =0:1 to 1:2) to afford ethyl 2-(4- (ethylsulfonyl)phenyl)propanoate (8.0 g) as a white solid. MS(ES+) m/z 271 (MH+).
Step 6: To a solution of ethyl 2-(4-(ethylsulfonyl)phenyl)propanoate (8.0 g) in ethanol (60 mL) was added a solution of NaOH (4.73 g) in water (60 mL). The reaction mixture was stirred at RT overnight. Ethanol was removed under reduced pressure, and 100 mL of water was added to the aqueous solution. The aqueous phase was washed with dichloromethane (50 mL x 2), and then
acidified with 6 M HC1 to pH = 1. The mixture was extracted with ethyl acetate (300 mL x 2). The combined organic phases were washed with brine (50 mL), dried and concentrated to give a dark red oil, which slowly solidified to give crude 2-(4-(ethylsulfonyl)phenyl)propanoic acid (6.0 g) as a yellow solid. 3.7 g of crude was washed with ether to afford the pure product 2-(4- (ethylsuIfonyl)phenyl)propanoic acid (2.5g) as a gray solid. Ή-NMR (400 MHz, DMSO-i¾) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 1.40 (d, J= 7.2 Hz, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.85 (q, J= 7.2 Hz, 1H), 7.57 (d, J= 8.0 Hz, 2H), 7.84 (d, J = 8.0 Hz, 2H), 12.55 (s, 1H). MS(ES+) mJz 243 (MH*).
Step 7: A mixture of 2,6-difluoro-2L(trifluoromethoxy)-[l,l'-biphenyl]-4-amine (intermediate 116a, 100 mg), 2-(4-(ethylsulfonyl)phenyl)propanoic acid (92 mg), EDC (80 mg) and HOBt (23.36 mg) in DCM (3 mL) was stirred at room temperature overnight. Then more 2-(4-
(ethylsulfonyl)phenyl)propanoic acid (25.1 mg), EDC (19.89 mg) and HOBt (14.02 mg) were added to the above mixture. The reaction mixture was stirred at room temperature for 2 additional days. The mixture was concentrated in vacuo and the residue was directly purified by MDAP to afford N-(2,6- difluoro-2'-(trifluoromethoxy)-[l,r-biphenyl]^-yI)-2-(4-(ethylsulfonyI)phenyl)propanarnide (56 mg) as a white solid. Ή-NMR (400 MHz, DMSO-t¼) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 1.49 (d, J= 7.0 Hz, 3H), 3.28 (q, J= 7.4 Hz, 2H), 4.00 (q, J= 6.9 Hz, 1H), 7.49 (m, 5H), 7.60 (m, 1H), 7.67 (d, J= 8.4 Hz, 2H), 7.88 (d, J= 8.4 Hz, 2H), 10.68 (s, 1H); I9F-NMR (376 MHz, DMSO-i¾ 6 ppm -56.79, - 112.32; MS(ES+) m/z 514 (MH+).
Example 120
7Y-(2,6-dichIoro-2'-ethoxy-[l,l'-biphenyl]-4-yl)-2-(4-(ethylsuIfonyl)phenyl)acetamide

A solution of N-(4-bromo-3,5-dichlorophenyl)-2-(4-(etlrylsulfonyl)phenyl)acetamide
(intermediate 78a, 200 mg,), (2-ethoxyphenyl)boronic acid (58.9 mg), tri-ieri-butylphosphine, tetrafluoroboric acid salt (20.58 mg), Pd2(dba)3 (16.24 mg) and Na2C03 (0.355 mL) in 1,4-dioxane (4 mL) was heated to 100°C for 1 hour in the microwave. The resulting mixture was filtered through celite, and washed by ethyl acetate. Solvent was removed. The obtained mixture was redissolved in DMF. Solid was filtered off. The filtrate was purified by MDAP to afford N-(2,6-dichloro-2'-ethoxy- [l,l'-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (25 mg) as a white solid. JH-NMR (400
MHz, DMSO-4 6 ppm 1.03 (t, J = 7.4 Hz, 3H), 1.09 (t, J= 6.8 Hz, 3H); 3.20 (q, J= 7.3 Hz, 2H), 3.78 (s, 2H), 3.95 (q, J= 6.9 Hz, 2H), 6.94 (t, J= 7.4 Hz, 1H), 7.02 (m, 2H), 7.31 (t, J= 7.4 Hz, 1H), 7.55 (d, J= 8.4 Hz, 2H), 7.70 (s, 2H), 7.79 (d, J= 8.0 Hz, 2H), 10.55 (s, 1H); MS(ES+) m/z 92 (MH+). Example 121
Ar-(2,6-difluoro-2,-(trifluoromethyl)-[1 '-biphenyI]-4-yl)-2-(4-(ethylsulfonyI)phenyl)acetamide

A mixture of (2-(trifluoromethyl)phenyl)boronic acid (82 mg), N-(4-bromo-3,5-difluorophenyl)- 2-(4-(ethylsulfonyl)phenyl)acetamide (intermediate 27a, 100 mg), Pd2(dba)3 (32.8 mg), tri-/ei-f- butylphosphine, tetrafluoroborate (41.5 mg) and 2 M Na2C03 (0.478 mL) solution in 1,4-dioxane (2 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was diluted with EtOAc, washed with 2M HCl, sat. NaHC03 solution and brine successively. The organic phase was dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford N-(2,6-difluoro-2'-(trifluoromethyl)-[l , l'-biphenyl]-4-yl)-2-(4-
(ethylsulfonyl)phenyl)acetamide (64 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm
1.10 (t, J= 7.2 Hz, 3H), 3.29 (q, J= 7.6 Hz, 2H), 3.87 (s, 2H), 7.43 (m, 2H), 7.50 (d, J= 7.6 Hz, 1H), 7.62 (d, J= 8.4 Hz, 2H), 7.71 (t, /- 7.6 Hz, 1H), 7.79 (t, J= 7.6 Hz, 1H), 7.86 (m, 2H), 7.90 (d, J = 8.0 Hz, 1H), 10.76 (s, 1H); I9F-NMR (376 MHz, DMSO-i/6) S ppm -59.88, -111.35. MS(ES^) m/z 484 (MH+).
Example 122
Ar-(2'-(difIuoromethoxy)-2,6-difluoro-[l,l'-biphenyl]-4-yI)-2-(4-(ethylsuIfonyl)phenyl)acetamide

Intermediate 122a: f4-f2-('4-('ethylsulfonyl)pheny acetamido)-2.6-difluorophenvnboronic acid
A mixture of 4}4,4,,4',5,5J5',5'-octamethyl-2,2'-bi-l,3;2-dioxaborolane (601 mg), N-(4-bromo- 3}5-difluorophenyl)-2-(4-(ethyIsulfonyl)phenyI)acetamide (intermediate 27a, 300 mg), Pd2(dba)3 (59.1 mg), tricyclohexylphosphine (42.2 mg) and potassium acetate (317 mg) in 1 ,4-dioxane (6 mL) was heated to 80°C for 3 hrs. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by column chromatography to afford (4-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2,6-difluorophenyl)boronic acid (201 mg) as an off-white solid. MS(ES+) m/z 384 (MH+).
Preparation of N-^'-idifluoromethoxy'f^^-difluoro-rU'-biphenv^^-vn^- - (ethvIsulfonvDphenyDacetamide
A mixture of l-bromo-2-(difluoromethoxy)benzene (0.035 g), (4-(2-(4-
(ethylsulfonyl)phenyl)acetamido)-2,6-difluorophenyl)boronic acid (intermediate 122a, 0.066 g), Pd2(dba)3 (0.043 g),
 tetrafluoroboric acid salt (0.055 g) and 2 M Na2C03 (0.314 mL) in 1,4-dioxane (2 mL) was sealed in a vessel and heated in die microwave at 100°C for 45 rains. The reaction mixture was filtred through celite and silica gel. The Filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(2'-(difluoromethoxy)-2,6-difluoro-[l,l'- biphenyl]-4-yI)-2-(4-(ethylsulfonyl)phenyl)acetamide (9 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-i¾ δ ppm 1.11 (t, J= 7.6 Hz, 3H), 3.29 (q, J= 7.6 Hz, 2H), 3.87 (s, 2H), 7.19 (m, 3H)} 7.46 (m, 3H), 7.55 (m, 1H), 7.62 (d, J= 8.4 Hz, 2H), 7.87 (d, J= 8.4 Hz, 2H), 10.75 (s, 1H); l9F-NM (376 MHz, DMSO-<¾ 5 ppm -81.44, -111.96; MSfES*) m/z 482 (MH+). Example 123
tetrafluoroboric acid salt (0.055 g) and 2 M Na2C03 (0.314 mL) in 1,4-dioxane (2 mL) was sealed in a vessel and heated in die microwave at 100°C for 45 rains. The reaction mixture was filtred through celite and silica gel. The Filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(2'-(difluoromethoxy)-2,6-difluoro-[l,l'- biphenyl]-4-yI)-2-(4-(ethylsulfonyl)phenyl)acetamide (9 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-i¾ δ ppm 1.11 (t, J= 7.6 Hz, 3H), 3.29 (q, J= 7.6 Hz, 2H), 3.87 (s, 2H), 7.19 (m, 3H)} 7.46 (m, 3H), 7.55 (m, 1H), 7.62 (d, J= 8.4 Hz, 2H), 7.87 (d, J= 8.4 Hz, 2H), 10.75 (s, 1H); l9F-NM (376 MHz, DMSO-<¾ 5 ppm -81.44, -111.96; MSfES*) m/z 482 (MH+). Example 123
iV-(2,6-dichloro-2,-cyano-[l,l'-bipheiiyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 1: Copper(II) bromide (3.24 g) was dissolved in acetonitrile (10 mL). Tert-butyl nitrite (0.996 g) was added to the above mixture. 2,6-Dichloro-4-nitroaniline (1 g) was dissolved in tetrahydrofuran (THF) (10 mL), and then added to the reation mixture gradually. The reaction mixture was stirred at RT for 20 hours. Solvent was removed and HC1 solution (2 M) was added. Then it was extracted with ethyl acetate and washed with brine. The organic layer was concentrated vacuo to give 2-bromo-l,3-dichloro-5-nitrobenzene (1.32 g) as a yellow solid.
Step 2: 2-Bromo-l ,3-dichloro-5-nitrobenzene (1.3 g) was dissolved in ethanol (30 niL). To this solution, tin(II) chloride dihydrate (5.41 g) was added gradually. Then the reaction mixture was heated to 70°C for 3 hours. Solvent was removed and sodium hydroxide solution (2 M) was added. The mixture was extracted with ethyl acetate and washed with brine. The organic layer was concentrated in vacuo to give 4-bromo-3,5-dichloroaniline (1.0 g) as a yellow solid. MSfES ) m/z 239.9 (MH+).
Step 3: A solution of 4-bromo-3,5-dichloroaniline (200 mg), (2-cyanophenyl)boronic acid (122 mg), tri-teri-butylphosphine, tetrafluoroboric acid salt (48.2 mg), Pd2(dba)3 (38.0 mg) and Na2C03 (0.208 mL) in 1 ,4-dioxane (4 mL) was heated to 110°C for 1 hour in the microwave. The resulted mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed and extracted with ethyl acetate, washed with saturated ammonium chloride solution and brine. The crude product was purified by column choromatography and concentrated in vacuo to afford 4'-amino-2',61- dicliloro-[l ,l '-biphenyl]-2-carbonitriIe (48 mg) as a yellow oil. MS(ES+) m/z 262.9 (MH*).
Step 4: 4'-Amino-2',6'-dichloro-[l, -biphenyl]-2-carbonitrile (48 mg) and perfluoroplienyl 2-(4- (ethylsulfonyl)phenyl)acetate (intermediate 102a, 71.9 mg) were dissolved in DCM (10 mL). Then to this solution, DIPEA (0.064 mL) was added gradually. The reaction mixture was stirred for 3 hours. Solvent was removed. The obtained mixture was redissolved in DMF, and solid was filtered off. The filtrate was purified by MDAP to afford N-(2,6-dichloro-2'-cyano-[l,l'-biphenyl]-4-yl)-2-(4- (ethyIsulfonyl)phenyl)acetamide (23 mg) as a white solid. Ή-NM (400 MHz, MeOD-t¾) δ ppm 1.13 (t, J= 7.4 Hz, 3H), 3.11 (q, J- 7.5 Hz, 2H), 3.78 (s, 2H), 7.32 (d, J= 8.0 Hz, 1H), 7.52 (m, 3H), 7.68 (t, J= 7.8 Hz, 1H), 7.78 (m, 5H); MS(ES+) m/z 473 (MH+).
Example 124
N-(2-chloro-6-cyano-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide

Intermediate 124a: 4-amino-6-chloro-2'-(trifluoromethoxyV[ 1.1 '-biphenyll^-carbomtrile
Step 1: 2-Amino-3-chloro-5-nitrobenzonitrile (8 g) was dissolved in acetic acid (240 mL) and sulfuric acid (60 mL). Sodium nitrite (2.93 g) solution in water (40 mL) was added dropwise to the above mixture at 60°C during 1.5 hours. The resulted solution was stirred at 60°C for another hour.
Then the solution was added dropwise to a mixture of copper(I) bromide (8.71 g) and HBr (21.99 mL) at 00°C during 2 hous. The resulted solution was stirred for another one hour at 100°C. The reaction mixture was cooled to room temperature and quenched with water. The mixture was extracted with dichloromethane and the organic phase was washed with water, dried over Na2SC>4 an concentrated in vacuo. The residue was purified by column choromatography to afford 2-bromo-3-chloro-5- nitrobenzonitrile (6.5 g). ]H-NMR (400 MHz, CDC13) δ ppm 8.44 (d, 1H), 8.54 (d, 1H).
Step 2: HC1 (2.91 mL) was added to a solution of 2-bromo-3-chloro-5-nitrobenzonitrile (1.5 g) in ethanol (20 mL) and tetrahydrofuran (THF) (5 mL). Then the mixture was heated to 60 °C. Tin(II) chloride (3.48 g) was added to the above mixture portionwise. After the addition, the mixture was stirred at 60°C for another 4 hours. The reaction mixture was cooled to room temperature and then basified with saturated Na2C03 aqueous solution to pH around 7. Solid was filtered off and washed with ethyl acetate. The filtration was collected and concentrated in vacuo. The residue was extraced with ethyl acetate. The organic phase was washed with brine, dried over Na2S04 and concentrated in vacuo to afford 5-amino-2-bromo-3-chIorobenzonitrile (1.1 g). MS(ES+) m/z 230 (MH+).
Step 3: A mixture of tri-feri-butylphosphine, tetrafluoroboric acid salt (0.083 g), 5-amino-2- bromo-3-chlorobenzonitrile (1.1 g), (2-(trifluoromethoxy)phenyl)boronic acid (0.470 g), Pd2(dba)3 (0.087 g) and Na2C03 (0.403 g) in 1,4-dioxane (12 mL) and water (3 mL) was stirred at room temperature for 15 minutes under nitrogen. Then the mixture was heated to 110°C for 45 minutes. After cooling to RT, the mixture was extracted with ethyl acetate and washed with water and brine. The organic phase was dried over 2S04, filtered, and concentrated in vacuo. The residue was purified by column choromatography to afford 4-amino-6-chloro-2'-(trifluoromethoxy)-[l ,l '- biphenyl]-2-carbonitrile (1.3 g). MS(ES*) m/z 313 (MH+).
Preparation of N-f2-chloro-6-cyano-2'-('trifluoromethoxy')-ri .1 '-biphenyl"l-4-yl')-2-f4- (ethylsulfonyDphenyriacetamide
A mixture of 4-amino-6-chloro-2'-(triffuoromethoxy)-[l ,l'-biphenyl]-2-carbonitrile (intermediate 124a, 188 mg), 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 200 mg), HATU (329 mg) and DIPEA (0.126 mL) in dichloromethane (3 mL) was stirred at room temperature overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-chloro-6- cyano-2'-(trifluorometlioxy)-[ 1 , 1 '-biphenyl] -4-yl)-2 -(4-(ethylsulfonyl)phenyl)acetamide ( 109 mg) . 1 H- NMR (400 MHz, CDC13) δ ppm 1.31 (t, J= 7.4 Hz, 3H), 3.15 (q, J= 7.5 Hz, 2H), 3.85 (s, 2H)} 7.32 (m, 1H), 7.42 (m, 2H), 7.53 (m, 3H), 7.86 (m, 4H), 8.03 (d, J= 2.0 Hz, 1H); l9F-NMR (376 MHz, CDC13) δ ppm -57.45; MS(ES+) m/z 523 (MH+).
Example 125
JV-(2-C hloro-6-cyano-2 '-(trifiuoromethoxy)- [1,1 '-biphenyl] -4-yl)-2-(4- (methylsulfonyI)phenyl)acetamide

A mixture of 4-amino-6-chloro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-carbonitrile
(intermediate 124a, 188 mg), 2-(4-(methyIsulfonyI)pheny])acetic acid (200 mg), HATU (329 mg) and DIPEA (0.126 mL) in dichloromethane (3 mL) was stirred at room temperature overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-chloro-6- cyano-2'-(trifluoromethoxy)-[l , -biphenyl]-4-yl)-2-(4-(methylsulfonyl)phenyl)acetamide (108 mg). Ή-NMR (400 MHz, CDC13) δ ppm 3.10 (s, 3H), 3.84 (s, 2H), 7.32 (dd, J= 1.8 Hz, 7.5 Hz, 1H) 7.42 (m, 2H), 7.53 (m, 3H), 7.81 (d, J= 2.3 Hz, 1H), 7.92 (d, J= 8.3 Hz, 2H), 8.01 (s, 1H), 8.05 (d, J- 2.0 Hz, 1H); l9F-NMR (376 MHz, CDC¾) δ ppm -55.45; MS(ES^) m/∑ 509 (MH+).
Example 127
2-(4-(ethylsulfonyl)phenyl)-N-(2,4,,6-trifluoro-2'-(trifluoromethyl)-[l,l,-biphenyl]-4- yl)acetamide

A mixture of Pd2(dba)3 (32.8 mg), tri-terf-butylphosphine, tetrafluoroboric acid salt (41.6 mg), (4-fluoro-2-(trifluoromethyI)phenyl)boronic acid (74.6 mg), N-(4-bromo-3,5-difluorophenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide (intermediate 27a, 100 mg) and 2 M Na^CC^ solution (0.478 mL) in 1,4-dioxane (2 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by MDAP to afford 2-(4-(emylsulfonyl)phenyl)-N-(2,4',6-trifluoro-2'- (trifluoromethyl)-[l, -biphenyl]-4-yl)acetamide (32 mg) as a white solid. Ή-NMR (400 MHz, DMSO- 6) S ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J = 7.2 Hz, 2H), 3.86 (s, 2H), 7.43 (m, 2H), 7.60 (m, 3H), 7.78 (m, 1H), 7.84 (m, 3H), 10.77 (s, 1H); I9F- MR (376 MHz, DMSO-i 6) S ppm -60.38, - 110.45, -111.30; MSfES*) mh 502 (MH4 .
Example 128
N-(2,4'-difluoro-2'-(trifluoromethylH

A mixture of N-(4-bromo-3-fluoi phenyl)-2-(4-(ethylsulfonyl)phenyl)acetainide (150 mg, see step 1 for synthesis of Example 9), PdCl2(dppf)-CH2Cl2 adduct (20 mg), Cs2C03 (147 mg) and (4- fluoro-2-(trifluoromethyl)phenyl)boronic acid (86 mg) in acetonitrile (1.5 mL)/water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. As the reaction was not complete, more (4-fluoro-2-(trifluoromethyl)phenyl)boronic acid (86 mg) was added into the reaction mixture. The mixture was heated again in the microwave at 100°C for 30 mins. The mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(2>4,-difluoro-2'-(trifluoromethyl)-[l,l,-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (45 mg) as a yellow solid. 'H-NMR (400 MHz, DMSO-<¾) δ ppm 1.1 1 (t, J= 7.2 Hz, 3H), 3.29 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 7.27 (t, J= 8.4 Hz, 1H), 7.37 (dd, J- 1.6 Hz, 8.4 Hz, 1H), 7.50 (m, 1H), 7.63 (m, 3H), 7.69 (dd, J= 1.6 Hz, 12 Hz, 1H), 7.77 (dd, J= 2.8 Hz, 8.4 Hz, 1H), 7.87 (d, 7= 8.4 Hz, 2H), 10.61 (s, 1H); 19F-NMR (376 MHz, DMSO-c¾ δ ppm - 58.33, -111.85, -1 13.20; MS(ES+) m/z 484 (MH4).
Example 129
N-(2,4,-dichloro-2'-(trifluoromethoxy)-[l,l'-bip enyl]-4-yl)-2-{4- (methylsulfonyl)pheny])acetamide

Intermediate 129a: 2-(4-chloro-2-(trifluoromethoxy)phenyl')-4,4,515-tetramethvI-l,3,2-dioxaborolane
A mixture of bis(pinacolato)diboron (10.83 g), 4-chIoro-2-(trifluoromethoxy)phenyl trifluoromethanesulfonate (7 g, see step 2 for synthesis of Example 103), PdCl2(dppf)-CH2Cl2 adduct (1.161 g) and potassium acetate (2.79 g) in 1,4-dioxane (80 mL) was stirred under nitrogen at 100°C overnight. After cooling to room temperature, the mixture was filtered through celite, and washed
with EtOAc. The filtrate was concentrated in vacuo and the residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was purified by column chromatography to afford 2-(4-chloro-2- (trifluoromethoxy)phenyl)-4,4,5,5-tetramethyl-l,3,2'dioxaborolane (3.768 g) as a light green oil. Ή- NMR (400 MHz, DMSO-i 6) δ ppm 1.29 (s, 12H), 7.54 (m, 2H), 7.74 (d, J= 7.9 Hz, 1H).
Intermediate 129b: N-(4-bromo-3 -chlorophenyl')-2-f4-fmethylsulfonyl')phenyl')acetamide
To a solution of 4-bromo-3-chloroaniline (0.4 g), 2-(4-(methylsulfonyl)phenyl)acetic acid (0.436 g) and EDC (0.446 g) in dichloromethane (20 mL) was added HOBt (0.356 g). The reaction mixture was stirred at room temperature overnight. The mixture was diluted with DCM. The organic phase was washed with water, 2M HC1 solution (40 mL), saturated Na2C03 solution (60 mL) and brine, dried over anhydrous MgS04 and concentrated in vacuo to afford N-(4-bromo-3-chlorophenyl)-2-(4- (methylsulfonyl)phenyl)acetamide (500 mg). MS(ES+) m/z 402 (MH+).
Preparation of N-f2,4,-dichloro-2'-(trifluoromethoxy)-ri.r-biphenyl1-4-yl)-2-<'4- ( methylsulfonvDphenyl) acetami de
A mixture of 2-(4-chloro-2-(trifluoromethoxy)phenyI)-4,4,5,5-tetramethyl-l ,3,2-dioxaborolane
(intermediate 129a, 88 mg), N-(4-bromo-3-ch]orophenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (intennediate 129b, 100 mg), PdCi2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (97 mg) in acetonitrile (1,2 mL) and water (0.4 mL) was bubbled with nitrogen, then sealed and heated in the microwave at 100°C for 45 mins. The mixture was filtered through celite. The solid was washed with EtOAc. The filtrate was washed with 2M HC1, sat. Na2COj and brine, then concentrated in vacuo. The resulting residue was purified directly by MDAP to afford N-(2,4'-dichloro-2'-(trifluoromethoxy)-[l,r- biphenyl]-4-yl)-2-(4-(methylsulfonyl)phenyl)acetamide (67 mg) as an off-white solid. ¾-NMR (400 MHz, DMSO-< ) 5 ppm 3.20 (s, 3H), 3.84 (s, 2H), 7.33 (d, J= 8.4 Hz, 1H), 7.47 (d, J= 8.3 Hz, 1H), 7.60 (m, 5H), 7.90 (d, J= 8.4 Hz, 2H), 7.95 (d, J= 2.0 Hz, 1H), 10.58 (s, 1H); 19F-NMR (376 MHz, DMSO-if6) δ ppm -56.59; MS(ES÷) m/z 518 (MH+).
Example 130
N-(4'-chloro-2-fluoro-2,-(trifluoromethoxy)~[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulfonyI)phenyl)acetamide

Example 131
2-(4-(ethylsuIfonyl)phenyl)-7V-(5-(4-fIuoro-2-(trifluoromethyl)phenyl)-6-rnethylpyridin-2- yl)acetamide, trifluoroacetic acid salt
TFA

A solution of N-(5-bromo-6-methylpyridin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (intennediate 67a, 100 mg), (4-fIuoro-2-(trifluoromethyl)phenyI)boronic acid (105 mg), tri-tert- butylphosphine, tetrafluoroboric acid salt (29.2 mg), Pd2(dba)j (23.05 mg) and Na2C03 (0.378 mL) in 1,4-dioxane (3 mL) was heated to 100°C for 0.5 hour in the microwave. The resulted mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed. The residue was redissolved in DMF. Solid was filtered off. The filtrate was purified by MDAP. Solvent was evaporated in vacuo to afford 2-(4-(ethylsulfonyl)pheriyl)-N-(5-(4-fluoro-2-(trifluorometliyl)phenyI)- 6-methylpyridin-2-yl)acetamide, trifluoroacetic acid salt (112 mg) as a white solid.Ή-NMR (400 MHz, MeOD-<¾) δ ppm 1.12 (t, J= 7.4 Hz, 3H), 2.11 (s, 3H), 3.11 (q, J= 7.3 Hz, 2H), 3.85 (s, 2H), 7.29 (m, 1H), 7.38 (m, 1H), 7.52 (d, J- 8.4 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.79 (m, 3H); 19F-NMR (376 MHz, MeOD-i/4) δ ppm -60.95, -77.57, 113.59; MS(ES+) m/z 481 (MH+).
Example 132
Ar-(2,6-dichloro-2'-(trifluoromethoxy)-[l,l'-biphenyI]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide

Step 1: A solution of 4-bromo-3,5-dichloroaniline (200 mg), (2- (trifluoromethoxy)phenyl)boronic acid (342 mg), tri-ter/-butylphosphine, tetrafluoroboric acid salt (96 mg), Pd2(dba)3 (76 mg) and Na2C03 (1.245 inL) in 1,4-dioxane (4 mL) was heated to 100°C for 0.5 hour in the microwave. The resulted mixture was filtered tlirough celite, and washed with ethyl acetate. The organic solvent was removed and extracted with ethyl acetate, washed with saturated ammonium chloride solution and brine. The organic layer was concentrated in vacuo and purified by column choromatography to afford 2,6-dichloro-2'-(trifluoromethoxy)-[l, -biphenyl]-4-amine (217 mg) as a yellow oil. MS(ES+) m/z 322 (MH+).
Step 2: 2,6-DichIoro-2'-(trifluorometlioxy)-[l ,l'-biphenyl]-4-amine (100 mg), 2-(4- (methylsulfonyl)phenyl)acetic acid (80 mg) and HATU (142 mg) were dissolved in dichloromethane (10 mL). To tliis solution, DIPEA (0.108 mL) was added gradually. The reaction mixture was stirred for 15 hours. Solvent was removed. The obtained crude was redissolved in DMF. Solid was filtered off. The filtrate was submitted to MDAP for purification on Sunfire CI 8 column using
acetonitrile/water with a TFA modifier. Solvent was evaporated in vacuo to afford N-(2,6-dichloro-2'- (trifluoromethoxy)-[l, -biphenyl]-4-yl)-2-(4-(metlrylsulfonyl)phenyl)acetamide (107 mg) as a white solid. 'H-NMR (400 MHz, DMSO-t 6) δ ppm 3.22 (s, 3H), 3.86 (s, 2H), 7.41 (d, J= 8.0 Hz, 1H), 7.52 (t, = 7.4 Hz, 2H), 7.61 (m, 3H), 7.84 (s, 2H), 7.91 (d, J= 8.0 Hz, 2H), 10.69 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -56.26; MS(E8+) m/z 518 (MH+).
Example 133
7V-(4'-fluoro-2-inethyl-2'-(trifluoromethyI)-[l,l'-biphenyI]-4-yl)-2-(4- (methylsulfonyl)phenyI)acetamide

Na2C03 solution (60 mL) and brine, dried over anhydrous MgSC^, filtered and the filtrate was concentrated in vacuo to afford N-(4-bromo-3-methylphenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (540 mg). MS(ES+) mk 382 (MFf).
Step 2: To a solution of N-(4-bromo-3-methylphenyI)-2-(4-(methylsuIfonyl)phenyl)acetamide (100 mg), (4-fluoro-2-(trifluoromethyl)phenyl)boronic acid (82 mg), tri-ieri-butylphosphine, tetrafJuoroboric acid salt (45.5 mg) and Pd2(dba)3 (35.9 mg) in 1 ,4-dioxane (3 mL) was added 2 M Na2C03 solution (0.523 mL). The reaction mixture was heated to 100°C in the microwave for 45 inins. The resulted mixture was diluted with EA (45 mL) and then filtered through celite. The filtrate was washed with saturated ammonium chloride solution and brine. The organic layer was
concentrated in vacuo.The residue was purified by MDAP and evaporated in vacuo to afford N-(41- fluoro-2-methyl-2'-(trifluoromethyl)-[ 1 , 1 '-biphenyl]-4-yl)-2-(4-(methyIsulfonyl)phenyl)acetamide (50 mg). Ή-NM (400 MHz, DMSO-t/6) 5 ppm 1.92 (s, 3H), 3.20 (s, 3H), 3.80 (s, 2H), 7.03 (d, J= 8.4 Hz, 1H), 7.35 (dd, J= 5.6 Hz, 8.4 Hz, 1H), 7.44 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.53 (d, J= 2.8 Hz, 1H), 7.59 (m, 3H), 7.72 (dd, J= 2.8 Hz, 9.2 Hz, 1H), 7.90 (d, J= 8.4 Hz, 2H), 10.31 (s, 1H); 19F-NMR (376 MHz, DMSOi/e) δ ppm -58.23, -113.06; MSiES ) m/∑466 (MH+). Example 134
N-(2-chloro-4'-fluoro-2'-(trifiuoroniethyl)-[l,l'-biphenyl]-4-yl)-2-(4- (methylsulfonyl)pheny])acetamide
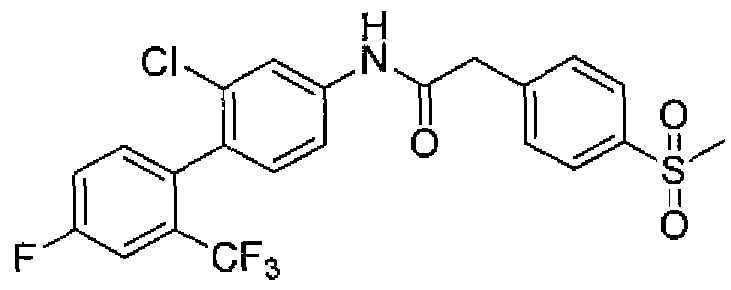
To a solution of N-(4-bromo-3-chlorophenyl)-2-(4-(metliylsulfonyl)phenyl)acetamide
(intermediate 129b, 100 mg), (4-fluoro-2-(trifluoromethyI)phenyl)boronic acid (77 mg), tri-fer/- butylphosphine, tetrafluoroboric acid salt (43.2 mg) and Pd2(dba)3 (34.1 mg) in 1,4-dioxane (3 mL) was added 2 M Na2C03 solution (0.497 mL). The reaction mixture was heated to 100°C for 45 mins in the microwave. The resulted mixture was diluted with EA (45 mL) and then filtered through celite. The filtrate was washed with saturated ammonium chloride solution and brine. The organic layer was concentrated in vacuo. The residue was purified by MDAP to afford N-(2-chloro-4'-fluoro-2'-
(trifluoromethyl)-[l, -biphenyl]-4-yl)-2-(4-(metl ylsulfonyl)phenyl)acetamide (40 mg). Ή-NMR (400 MHz, DMSO-i/e) δ ppm 3.20 (s, 3H), 3.84 (s, 2H), 7.28 (d, J= 8.4 Hz, IH), 7.42 (dd, J= 5.6 Hz, 8.4 Hz, IH), 7.52 (dd, J= 2.4 Hz, 8.4 Hz, IH), 7.61 (m, 3H), 7.74 (dd, J= 2.4 Hz, 9.2 Hz, IH), 7.90 (m, 3H), 10.56 (s, IH); 19F-NMR (376 MHz, DMSO- 6) δ ppm -58.26, -111.86; MSiES*) m/z 486 (MH÷).
Example 135
N-(2,4'-difluoro-2'-(trifluoromethyl)-[l,l,-biphenyl]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide
 Step 1: To a solution of 4-bromo-3~fluoroaniline (760 mg), 2-(4-(methylsulfonyl)phenyl)acetic acid (900 mg) and EDC (920 mg) in dichloromethane (20 mL) was added HOBt (735 mg). The reaction mixture was stirred at room temperature overnight. The mixture was diluted with additional DCM (25 mL). The organic phase was washed with water, 2M HCI solution (40 mL), saturated Na2C03 solution (60 mL) and brine, dried over anhydrous MgS04, and filtered. The filtrate was concentrated in vacuo to afford N-(4-bromo-3-fluorophenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (570 mg). MS(ES+) m/z 386 (MH ).
Step 1: To a solution of 4-bromo-3~fluoroaniline (760 mg), 2-(4-(methylsulfonyl)phenyl)acetic acid (900 mg) and EDC (920 mg) in dichloromethane (20 mL) was added HOBt (735 mg). The reaction mixture was stirred at room temperature overnight. The mixture was diluted with additional DCM (25 mL). The organic phase was washed with water, 2M HCI solution (40 mL), saturated Na2C03 solution (60 mL) and brine, dried over anhydrous MgS04, and filtered. The filtrate was concentrated in vacuo to afford N-(4-bromo-3-fluorophenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (570 mg). MS(ES+) m/z 386 (MH ).
Step 2: To a solution of N-(4-bromo-3-fIuorophenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (100 mg), (4-fluoro-2-(trifluoromethyl)phenyl)boronic acid (81 mg), tri-ier/-butylphosphine;
tetrafluoroboric acid salt (45.1 mg) and Pd2(dba)3 (35.6 mg) in 1 ,4-dioxane (3 mL) was added Na2C03 solution (0.518 mL). The reaction mixture was heated to 100°C for 45 mins in the microwave. The resulted mixture was diluted with EA (45 mL) and then filtered through celite. The filtrate was washed with saturated ammonium chloride solution and brine. The organic layer was concentrated in vacuo. The residue was purified by MDAP and evaporated in vacuo to afford N-(2,4'- difluoro-2'-(trifluoromethyl)-[l , 1 '-biphenyl]-4-yl)-2-(4-(methylsulfonyl)phenyl)acetamide (52 mg). 1 H-NMR (400 MHz, DMSO-</6) δ ppm 3.20 (s, 3H), 3.84 (s, 2H), 7.27 (t, J= 8.4 Hz, IH), 7.36 (dd, J =2.0 Hz, 8.4 Hz, IH), 7.48 (dd, J= 5.6 Hz, 8.8 Hz, IH), 7.62 (d, = 8.4 Hz, 3H), 7.68 (dd, J= 2.0 Hz, 12.0 Hz, 1H), 7.77 (dA, J= 2.8 Hz, 9.6 Hz, 1H), 7.91 ((!, /= 8.4 Hz, 2H), 10.60 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -58.33, -11 1.84, -113.18; MS(ES^) m/z 470 (MH+).
Example 136
2-(4-(methylsulfonyI)phenyl)-Ar-(2,4 6-trifluoro-2'-(trifluoromethyl)-[l,l'-biphenyl]-4- yl)acetamide
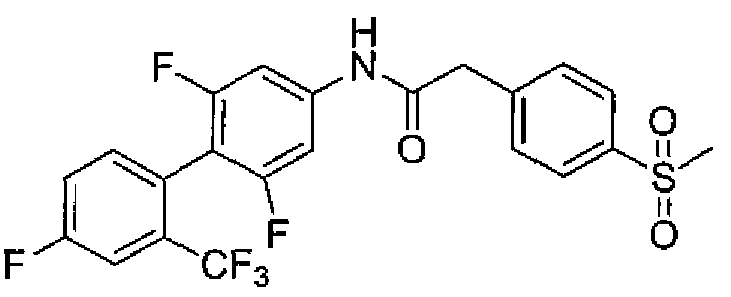
Step 1: To a solution of 4-bromo-3,5-difluoroaniline (832 mg), 2-(4- {methylsulfonyl)phenyl)acetic acid (900 mg) and EDC (920 mg) in dichloromethane (20 mL) was added HOBt (735 mg). The reaction mixture was stirred at room temperature overnight. The mixture was diluted with additional DCM (25 mL). The organic phase was washed with water, 2M HCI solution (40 mL), saturated Na2C03 solution (60 mL) and brine, dried over anhydrous MgS04, and filtered. The filtrate was concentrated in vacuo to afford N-(4-bromo-3,5-difluorophenyl)-2-(4- (methylsulfonyl)phenyl)acetamide (550 mg). MS(ES+) m/z 404 (MH+) .
Step 2: To a solution of N-(4-bromo-3,5-difluorophenyl)-2-(4- (methylsulfonyl)phenyl)acetamide ( 00 mg), (4-fluoro-2-(trifluoromethyl)phenyl)boronic acid (77 mg), trU/ert-butylphosphine, tetrafluoroboric acid salt (43.1 mg) and Pd2(dba)3 (34.0 mg) in 1,4- dioxane (3 mL) was added 2 M Na2C03 solution (0.495 mL). The reaction mixture was heated to 100°C for 45 mins in the microwave. The resulted mixture was diluted with EtOAc (45 mL) and then filtered tlirough celite. The filtrate was washed with saturated ammonium chloride solution and brine. The organic layer was concentrated in vacuo. The residue was purified by MDAP and evaporated in vacuo to afford 2-(4-(methylsulfonyl)phenyl)-N-(2,4',6-trifluoro-2'-(trifluoromethyl)-[l, -biphenyl]- 4-yl)acetamide (32 mg). 'tLNMR (400 MHz, DMSO- ) δ ppm 3.20 (s, 3H), 3.85 (s, 2H), 7.43 (m, 2H), 7.62 (d, J= 8.4 Hz, 3H), 7.68 (m, 1H), 7.83 (dd, J= 2.4 Hz, 8.8 Hz, 1H), 7.91 (d, J= 8.4 Hz, 2H), 10.75 (s, IH); 19F-NMR (376 MHz, DMSO-^) δ ppm -60.36, -1 10.45, -1 1 1.28; MS(ES+) m/z 488 (MH+).
Example 137
Ar-(5-(4-fluoro-2-(trifluoromethyl)phenyI)-6-methylpyridin-2-yl)-2-(4- (methylsulfonyl)phenyl)acetamide

Step 2: To a solution of N-(5-bromo-6-methylpyridin-2-yI)-2-(4- (methylsulfonyl)phenyl)acetamide (100 mg), (4-fluoro-2-(trifluoromethyl)phenyl)boronic acid (81 mg), tri-te^butylphosphine, tetrafluoroboric acid salt (45.4 mg) and Pd2(dba)3 (35.8 mg) in 1,4- dioxane (3 mL) was added 2 M Na2C03 solution (0.522 mL). The reaction mixture was heated to 100°C for 45 mins in the microwave. The resulted mixture was diluted with EA (45 mL) and then filtered tlirough celite. The filtrate was washed with saturated ammonium chloride solution and brine. The organic layer was concentrated in vacuo. The residue was purified by MDAP and evaporated in vacuo to afford N-(5-(4-fluoro-2-(trifluorometliyl)phenyl)-6-methylpyridin-2-yl)-2-(4-
(methylsulfonyl)phenyl)acetamide (40 mg). Ή-NMR (400 MHz, DMSO-c 6) δ ppm 2.1 1 (s, 3H); 3.20 (s, 3H), 3.88 (s, 2H), 7.50 (m, 2H), 7.64 (d, J= 8.4 Hz, 3H), 7.77 (dd, J = 2.4 Hz, 9.2 Hz, 1H), 7.92 (in, 3H), 10.96 (s, 1H); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -58.29, -112.20; MSfES4) m/z 467 (MH+). Example 138
2- (4-(ethyIsulfonyl)phenyl)-/Y-(2-(2-methoxypyr idm-4-yl)-2 '-(trifluor omethoxy)- [1,1 '-biphenyl]- 4-yl) acetamide

Step 1: To a solution of 2-methoxy-4-(4,4,5,5 -tetramethyl- l,3,2-dioxaborolan-2-yl) pyridine (200 mg) in 1,4-dioxane (9 mL) was added Cs2C03 (277 mg), 2-chloro-4-nitro-2'-(trifluoromethoxy)- Ι, -biphenyl (270 mg, see step 1 for synthesis of intermediate 69a), bis(tri-t- butylphosphine)palladium(O) (21.74 mg) and water (3 mL). The mixture was stirred under nitrogen at
120°C for 2 hours. The mixture was diluted with water, extracted with EtOAc. The organic layers were dried over Na2SO,i, concentrated and purified by column choromatography to afford 2-methoxy- 4-(4-nitro-2,-(trifluoromethoxy)-[l fl,-biphenyl]-2-yl)pyridine (200 mg). MS(ES+) m/z 391 (MH+).
Step 2: To a solution of 2-methoxy-4-(4-nitro-2'-(trifluoromethoxy) -[l,l'-bipheayl]-2- yl)pyridine (190 mg) in methanol (15 mL) was added zinc (318 mg). Then ammonium chloride (260 mg) was added to the above mixture portionwise and stirred for 1 hour. The mixture was filtered. The filtrate was diluted with water, extracted with EtOAc. The organic layers were dried over Na2S04 and concentrated to afford 2-(2-metlioxypyridin-4-yl)-2'-(trifluorometl oxy)-[l, -biphenyl]-4-amine (100 mg). MS(ES+) m/z 361 (MH* .
Step 3: To a solution of 2-(2-methoxypyridin-4-yl)-2'-(trifiuoromethoxy)-[l , 1 '-biphenyl]-4- amine (40 mg) in dichloromethane (5 mL) was added 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 27.4 mg) and triethylamine (0.031 mL). The reaction was stirred for 3 hours. The mixture was diluted witli water, extracted witli CH2CI2 for 3 times. The combined organic layers were dried over Na2S04, filtered and concentrated in vacuo. The residue was purified by MDAP to afford 2-(4-(ethylsulfonyl)phenyl)-N-(2-(2-methoxypyridin-4-yl)-2'-(trifluoromethoxy)-[l ,1 '-biphenylj-4- yljacetamide (12.1 mg) as a yellow solid. Ή-NMR (400 MHz, DMSOi¾) δ ppm 1.07 (t, 3H), 3.15 (m, 2H), 3.73 (s, 3H), 3.82 (s, 2H), 6.36 (s, 1H), 6.59 (s, 1H), 7.30 (m, 5H), 7.79 (m, 7H), 10.50 (s, 1H); MS(ES0 m/z 571 (MH1).
Example 139
2-(4-(ethylsulfonyl)phenyl)-iV-(4'-fluoro-2-methyI-2'-(trifluoromethyl)-[l}l,-biphenyl]-4- y))acetamide

A mixture of (4-fluoro-2-(trifluoromethyl)phenyl)boronic acid (86 mg), N-(4-bromo-3- metliylphenyl)-2-(4-(ethylsulfonyI)phenyl)acetamide (intermediate 52a, 150 mg), PdCl2(dppf)- CH2C12 adduct (24.73 mg) and Cs2C03 (1 7 mg) in acetonitrile (1.5 mL)/water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. As the reaction was not complete, (4- fluoro-2-(trifluoromethyl)phenyl)boronic acid (86 mg) was added to the mixture, and the mixture was heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by MDAP to afford 2- (4-(etliylsuIfonyl)phenyl)-N-(4'-fluoro-2-methyl-2'-(trifluoromethyl)-[l, -biphenyl]-4-yl)acetamide
(52 mg) as a white solid. Ή-NMR (400 MHz, DMSO- ) S ppm 1.10 (t, J= 7.2 Hz, 3H), 1.93 (s, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.81 (s, 2H), 7.03 (d, J= 8.0 Hz, 1H), 7.36 (m, 1H), 7.44 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.58 (m, 4H), 7.72 (dd, J- 2.8 Hz, 9.6 Hz, 1H), 7.85 (d, J= 8.4 Hz, 2H), 10.31 (s, 1H); I9F- NMR (376 MHz, DMSO-i/6) δ ppm -58.23, -113.07; MS(ES^) m/z 480 (MH+). Example 140
Ar-(4'-chloro-2-fluoro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (methylsulfonyI)phenyl)acetamide

A mixture of 2-(4-chloro-2-(trifluoromethoxy)phenyl)-4,4,5,5-tetrametliyl-l ,3,2-dioxaborolane (intermediate 129a, 100 mg), N-(4-bromo-3-fluorophenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (100 mg, see step 1 for synthesis of Example 135), cesium carbonate (101 mg) and PdCl2(dppf)- CH2C12 adduct (10 mg) in acetonitrile (1.5 mL)/water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaciton mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(4'-chloro- 2-fluoro-2'-(trifluoromethoxy)-[l , -biphenyl]-4-yl)-2-(4-(metliylsulfonyl)phenyl)acetamide (69 mg) as a white solid. Ή-NMR (400 MHz, DMSO-<¾) 5 ppm 3.21 (s, 3H), 3.84 (s, 2H), 7.36 (t, J= 8.0 Hz, 1H), 7.42 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.53 (d, J= 8.4 Hz, 1H), 7.63 (m, 4H), 7.71 (dd, J= 1.2 Hz, 14 Hz, 1H), 7.90 (dd, J= 1.2 Hz, 6.8 Hz, 2H), 10.62 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm - 56.68, -113.52; MS(ES+) m/z 502 (MH+). Example 141
iV-(5-(4-ch]oro-2-(trifluoromethoxy)phenyl)-6-methylpyridin-2-yl)-2-(4- (methylsulfonyl)phenyl)acetamide

Example 142
jV-(4'-ch]oro-2-methyl-2,-(trifluoromethoxy)-[l,l,-biphenyl]-4-yl)-2-(4- (methyIsulfonyl)phenyl)acetamide

A mixture of 2-(4-chloro-2-(trifluoromethoxy)phenyI)-4,4,5,5-tetramethyl-l ,3,2-dioxaborolane (intennediate 129a, 101 mg), N-(4-bromo-3-methylphenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (100 mg, see step 1 for synthesis of Example 133), PdCl2(dppf)-CH2Cl2 adduct (10 mg) and Cs2C03 (102 mg) in acetonitrile (1.5 mL)/water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 30 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(4'-chloro-2-methyl-2'- (trifluoromethoxy)-[l,r-biphenyl]-4-yl)-2-(4-(methylsulfonyl)phenyl)acetainide (74 mg) as a white solid. 'H-NMR (400 MHz, DMSO-4D δ ppm 2.03 (s, 3H), 3.20 (s, 3H), 3.81 (s, 2H), 7.08 (d, J= 8.4 Hz, 1H), 7.40 (d, J= 8.4 Hz, 1H), 7.49 (dd, J= 1.6 Hz, 8.0 Hz, 1H), 7.56 (m, 2H), 7.61 (m, 3H), 7.90 (d, J= 8.4 Hz, 2H), 10.32 (s, 1H); I9F-NMR (376 MHz, DMSO-<¾) δ ppm -56.55; MSfES^ m/z 498 (MH+).
Example 143
N-(4'-chloro-2,6-difluoro-2,-(trifluoromethoxy)-[ltl,-biphenyl]-4-yl)-2-(4- (methylsulfonyI)phenyI)acetamide

Na2CC>3 (2 M solution, 0.495 mL) was added into a mixture of 2-(4-chloro-2- (trifluoromethoxy)phenyl)-4,4,5,5-tetramethyl-l53,2-dioxaborolane (intermediate 129a, 96 mg), N-(4- bromo-3,5-difluorophenyl)-2-(4-(methylsulfony])phenyl)acetamide (100 mg, see step 1 for synthesis of Example 136) and Pd2(dba)3 (34.0 mg) in 1,4-dioxane (2 mL). The reaction mixture was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by MDAP to afford N-(4'-chloro-2J6-difluoro-2,-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide (24 mg) as a white solid. 'H-NMR (400 MHz, DMSO-cf6) δ ppm 3.21 (s, 3H), 3.87 (s, 2H), 7.47 (d, J= 10.0 Hz, 2H), 7.64 (m, 4H), 7.73 (s, 1H), 7.91 (d, J= 8.4 Hz, 2H), 10.78 (s, 1H); 19F-NMR (376 MHz, DMSO-i¾) S ppm -57.08, -112.30; MSfES^ m/z 520 (MH+).
Example 144
iV-(2,-(cyanomethyI)-2,6-difluoro-ll,l,-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

PdCl2(dppf)-CH2Cl2 adduct (2.083 g), 4,4)4,,4',5,5,5',5,-octametl yl-2,2'-bi(l,3,2-dioxaborolane) (7.12 g) and potassium acetate (5.01 g). The mixture was refluxed overnight under nitrogen. The reaction mixture was concentrated and purified by column chromatography to afford 2-(2-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)acetonitrile (700 mg) as a colorless liquid. ]H-NMR (400 MHz, DMSO-J6) δ ppm 1.02 (s, 12H), 4.14 (s, 2H), 7.36 (m, IH), 7.44 (m, IH), 7.52 (m, 1H), 7.74 (m, IH); MS(ES+) m/z 244 (MH+).
Step 2: A mixture of 2-(2-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)acetonitrile (126 mg), N-(4-bromo-3,5-difluorophenyI)-2-(4-(ethylsulfonyl)phenyl)acetamide (intermediate 27a, 120 mg), Pd2(dba)3 (26.3 mg), tri-fer/-butylphosphine, tetrafluoroboric acid salt (33.3 mg) and 2 M
aqueous Na2C03 (0.5 mL) in 1 ,4-dioxane (2 mL) was bubbled with nitrogen. The reaction mixture was sealed in a reaction vessel and heated in the microwave at 100°C for 1 hour. The mixture was filtered through celite, washed with EtOAc. The filtrate was washed with 2M HCl, sat. Na2C03 and brine, and then concentrated in vacuo. The residue was purified by MDAP to afford N-(2'- (cyanomethyl)-2,6-difluoro-[ 1 , 1 '-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (34 mg) as an off-white solid. Ή- M (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 3.29 (q, J= 7.3 Hz, 2H), 3.77 (s, 2H), 3.87 (s, 2H), 7.33 (d, J= 6.9 Hz, 1H), 7.48 (m, 4H), 7.57 (d, J- 7.4 Hz, 1H), 7.62 (d, J= 8.3 Hz, 2H), 7.87 (d, J= 8.3 Hz, 2H), 10.78 (s, 1H); 19F- MR (376 MHz, DMSO-i¾) δ ppm - 111.73; MS(ES+) m/z 455 (MH+). Example 145
Ar-(2,6-dichloro-2'-(trifluoromethoxy)-[l-l'-biphenyl]-4-yl)-2-(4-suIfamoylphenyl)acetamide

2,6-Dichloro-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (100 nig, see step 1 for synthesis of Example 132), 2-(4-sulfamoylphenyl)acetic acid (intermediate 29a, 95 mg) and HATU (142 mg) were dissolved in dichloromethane (10 mL). DIPEA (0.108 mL) was added to the above mixture gradually. The reaction mixture was stirred for 15 hours. Solvent was removed in vacuo. The mixture was redissolved in DMF, and solid was filtered off. The filtrate was purified by MDAP to afford N- (2,6-dichloro-2'-(trifluoromethoxy)-[ 1 , 1 '-biphenyl]-4-yI)-2-(4-sulfamoylphenyl)acetamide (33 mg) as a white solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 3.74 (s, 2H), 7.28 (s, 2H), 7.34 (d, J= 8.0 Hz, 1H), 7.45 (m, 4H), 7.54 (m, 1H), 7.73 (d, J= 8.0 Hz, 2H), 7.77 (s, 2H), 10.60 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -56.26; MSfES m/z 519 (MH+).
Example 146
Ar-(2,6-dic loro-2,,6'-difluoro-[l,l'-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Na2C03 (1.868 mL) solution in 1,4-dioxane (8 mL) was heated in the microwave at 110°C for 2 hours. The mixture was filtered tlirough celite, and washed with ethyl acetate. Solvent was removed and extracted with ethyl acetate, washed with saturated ammonium chloride solution and brine. The organic phase was concentrated in vacuo and purified by column choromatography to afford 2,6- dicliloro-^e'-difluoro-t^l'-bipheny^^-amine (132 mg) as a yellow oil. MS(ES÷) m/z 273.9 (MKT).
Step 2: 2,6-Dichloro-2',6'-difJuoro-[l, -biphenyl]-4-amine (132 mg) and perfluorophenyl 2-(4- (ethylsulfonyl)phenyl)acetate (intermediate 102a, 114 mg) were dissolved in dichloromethane (5 mL). Then N-ethyl-N-isopropylpropan-2-amine (37.3 mg) was added to the above solution gradually. The reaction mixture was stirred for 15 hrs. Solvent was removed. The residue was redissolved in DMF, and solid was filtered off. The filtrate was purified by MDAP to afford N-(2,6-dichloro-2',6'-difluoro- [l,l'-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (15 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm 1.03 (t, J= 7.4 Hz, 3H), 3.22 (q, J= 7.5 Hz, 2H), 3.80 (s, 2H), 7.21 (t, J= 8.0 Hz, 2H), 7.53 (m, 3H), 7.79 (d, J= 8.4 Hz, 2H), 7.81 (s, 2H), 10.70 (s, 1H); 19F-NMR (376 MHz, DMSO-<¾) δ ppm -112.13; S(ES") m/z 483.9 (MH+).
Example 147
iY-(2,6-dichloro-2'-methyl-[l,l'-biphenyl]-4-y])-2-(4-(ethylsuIfonyl)phenyl)acetamide

Step 1: A solution of 4-bromo-3,5-dichloroaniline (100 mg), o-tolylboronic acid (113 mg), tri- /.Tf-butylphosphine, tetrafluoroboric acid salt (48.2 mg), Pd2(dba)3 (38.0 mg) and Na2C03 (0.623 mL) in 1,4-dioxane solution (4 mL) was heated in the microwave at 100°C for 0.5 hour. The resulted mixture was filtered dirough celite, and washed with ethyl acetate. Solvent was removed in vacuo and extracted with ethyl acetate, washed with saturated ammonium chloride solution and brine. The organic layer was concentrated and purified by column choromatography to afford 2,6-dichloro-2'- methyI-[l,r-biphenyl]-4-amine (95 mg) as a yellow oil. MS(ES+) m z 252 (MH+).
Step 2: 2,6-Dichloro-2'-methyl-[l, -biphenyl]-4-amine (95 mg) and perfluorophenyl 2-(4- (ethylsulfonyl)phenyl)acetate (intennediate 102a, 156 mg) were dissolved in dichloromethane (10 mL). To this solution, DIPEA (0.138 mL) was added gradually. The reaction mixture was stirred for 15 hours. Solvent was removed in vacuo. The obtained mixture was redissolved in DMF, and solid
was filtered off. The filtrate was purified by MDAP to afford N-(2,6-dichIoro-2'-methyl-[l ,Γ- biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (89 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i/6) δ ppm 1.03 (t, J= 7.4 Hz, 3H), 1.92 (s, 3H), 3.22 (q, J= 7.3 Hz, 2H), 3.79 (s, 2H), 6.99 (d, J= 6.8 Hz, 1H), 7.24 (m, 3H), 7.56 (d, J= 8.4 Hz, 2H), 7.76 (s, 2H), 7.80 (d, J= 8.4 Hz, 2H), 10.60 (s, 1H); MSfES^) m/z 462 (MH+).
Example 148
2-(4-(ethylsuIfonyl)phenyl)- V-(2,2',6-trichloro-[l,l'-biphenyl]-4-yl)acetamide

Step 1: A solution of 4-bromo-3,5-dichloroaniline (100 rng), (2-chlorophenyl)boronic acid (130 mg), tri-teT-i-butylphosphine, tetrafluoroboric acid salt (48.2 mg), Pd2(dba)3 (38.0 mg) and Na2C03 solution (0.623 mL) in 1,4-dioxane (4 mL) was heated in the microwave at 100°C for 1 hour. The resulted mixture was filtered througli celite, and washed with ethyl acetate. Solvent was removed and extracted with ethyl acetate, washed with saturated ammonium chloride solution and brine. The residue was purified by column choromatography to afford 2>2',6-trichloro-[l, -biphenyl]-4-amine (100 mg) as a colorless oil. MS(ES") m/z 271.9 (MH4).
Step 2: 2,2',6-Trichloro-[lsr-biphenyl]-4-amine (100 mg) and perfluorophenyl 2-(4- (ethylsulfonyl)phenyl)acetate (intermediate 102a, 145 mg) were dissolved in dichloromethane (10 mL). To this solution, DIPEA (0.128 mL) was added gradually. The reaction mixture was stirred for 15 hours. Solvent was removed. The obtained mixture was redissolved in DMF, and solid was filtered off. The filtrate was purified by MDAP to afford 2-(4-(ethylsulfonyl)plienyl)-N-(2,2,,6-trichloro-[l,l'- biphenyl]-4-yl)acetarnide (87 mg) as a white solid. lH-NMR (400 MHz, DMSO-rf6) δ ppm 1.03 (t, J= 7.2 Hz, 3H), 3.22 (q, J= 7.3 Hz, 2H), 3.79 (s, 2H), 7.24 (d, J= 7.2 Hz, 1H), 7.40 (m, 2H), 7.55 (m, 3H), 7.76 (s, 2H), 7.80 (d, J= 8.4 Hz, 2H), 10.64 (s, lH); MS(ES ) m z 481.9 (MH+).
Example 149
7V-(4'-chloro-2-fluoro-2*-(trifluoromethyl)-[l,lt-biphenyl]-4-yl)-2-(4- (methylsulfonyI)phenyl)acetamide

To a solution of N-(4-bromo-3-fluorophenyl)-2-(4-(methylsulfonyl)phenyl)acetamide (100 mg, see step 1 for synthesis of Example 135), (4-chloro-2-(trif]uoromethyl)phenyl)boronic acid (87 mg), tri-ierZ-butylphosphine, tetrafluoroboric acid salt (45.1 mg) and Pd2(dba)3 (35.6 mg) in 1,4-dioxane (3 mL) was added 2 M Na2C03 solution (0.518 mL). The reaction mixture was heated in the microwave at 100°C for 45 mins. The resulted mixture was diluted with EA (45 mL) and then filtered through celite. The filtrate was washed with saturated ammonium chloride solution and brine. The organic layer was concentrated in vacuo.The residue was purified by MDAP and evaporated in vacuo to afford N-(4'-chloro-2-fluoro-2,-(trifiuoromethyl)-[l , 1 -biphenyl]-4-yl)-2-{4- (memylsulfonyl)phenyl)acetamide (24 mg). 'H-NMR (400 MHz, DMSO-<¾) δ ppm 3.20 (s, 3H), 3.84 (s, 2H), 7.28 (t, J= 8.8 Hz, 1H), 7.36 (dd, J= 1.6 Hz, 8.4 Hz, 1H), 7.47 (d, J= 8.4 Hz, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.68 (dd, J =2.0 Hz, 12.0 Hz, 1H), 7.83 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.91 (d, /= 8.4 Hz, 2H), 7.93 (d, J= 2.0 Hz, 1H), 10.61 (s, 1H); 19F-NMR (376 MHz, DMSO-i 6) 5 ppm -58.25, - 113.22; MS(ES+) m z 486 (MH+). Example 150
JV-(2-cyano-6-fluoro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulf ony I) phenyl) ac etamide

Intermediate 150a: 4-amino-6-fluoro-2'-(trifluoromethoxy')-ri,r-biphenyll-2-carbonitrile
Step 1: HC1 (4.13 mL) was added to a solution of 2-bromo-3-fluoro-5-nitrobenzonitrile (0.8 g) in ethanol ( 5 mL) and tetrahydrofuran (10 mL). The mixture was heated to 60°C. Tin(II) chloride dihydrate (5.89 g) was added to the above mixture portion wise. After the addition, the mixture was stirred at 60°C for another 4 hrs. The reaction was cooled to room temperature and then basified with saturated Na2COj aqueous solution to pH around 7. The solid was filtered off and washed with EA. The filtration was concentrated in vacuo. The residue was extraced with EA, The organic phase was
combined, washed with brine, dried over Na2S04 and concentrated to afford 5-amino-2-bromo-3- fluorobenzonitrile (50S mg). MSfES^ mlz 215 (MH4 .
Step 2: A mixture of tri-/er/-butyIphosphine, tetrafluoroboric acid salt (101 mg), 5-amino-2- bromo-3-fluorobenzonitrile (500 mg), (2-(trifluoromethoxy)phenyl)boronic acid (575 mg), Pd2(dba)3 (106 mg) and Na2C03 (493 mg) in 1,4-dioxane (12 mL) and water (3 liiL) was stirred at RT for 15 minutes under nitrogen. Then the mixture was heated to 110°C for 45 minutes. After cooling to RT, the mixture was extracted with EA and washed with water and brine. The organic phase was dried over Na2S04. Solvent was removed in vacuo and the residue was purified by column
choromatography to afford 4-amino-6-fluoro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-carbonitrile (520 mg). MS(ES+) mlz 297 (MH+).
Preparation of N-(2-cyano-6-fluoro-2'-(trifluoromethoxyH 1.1 '-biphenvn-4-yl)-2-(4- (e ylsulfonvDphenyl)acetamide
A mixture of 4-amino-6-fluoro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-2-carbonitrile (intermediate
150a, 75 mg), 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 63,6 mg), HATU (106 mg) and DIPEA (0.133 mL) in dichloromethane (2 mL) was stirred at RT overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-cyano-6-fluoro-2 - (trifluoromethoxy)-[l) -biphenyI]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (33 mg). Ή-NMR (400 MHz, CDClj) δ ppm 1.31 (m, 3H), 3.16 (q, J= 7.53 Hz, 2H), 3.85 (s, 2H), 7.41 (m, 3H), 7.53 (m, 3H), 7.64 (dd, J=1.00 Hz, 2.01Hz, 1H), 7.88 (m, 3H), 8.08 (s, 1H). I9F-NMR (376 MHz, CDC13) δ ppm -57.66, -107.71; MS(ES+) m/z 507 (MH").
Example 151
7V-(2-chloro-6-methyl-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-yI)-2-(4- (ethyisulfonyl)phenyl)acetamide
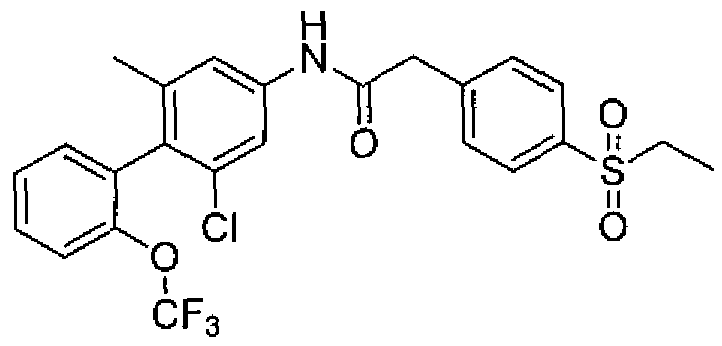
Step 1: To a solution of 2-chloro-6-methyl-4-nitroaniline (5 g) in acetonitrile (80 mL) at 0°C was added terf-butyl nitrite (3.53 mL) dropwise under nitrogen. After the addition, copper(II) bromide (6.58 g) was added to the above mixture portionwise at 0°C under nitrogen. The reaction mixture was stirred overnight at RT. The reaction mixture was concentrated and the residue was
partitioned between EA and water. The organic layers were combined, dried over Na2S04 and concentrated to afford 2-bromo-I-chloro-3-memyl-5-nitrobenzene (4.1 g). MS(ES+) m/z 250 (MH ).
Step 2: HC1 (11.05 mL) was added to a solution of 2-bromo-l-chloro-3-methyl-5-nitrobenzene (4, 1 g) in ethanol (60 mL) and tetrahydrofuran (40 mL). The mixture was heated to 60 °C. Tin(II) chloride dihydrate (18.47 g) was added to the above mixture portionwise. After the addition, the mixture was stirred at 60°C for another 4 hours. The reaction was cooled to room temperature and then basified with saturated Na2C03 aqueous solution to pH around 7. Solid was filtered off and washed with EA. The filtration was concentrated in vacuo. The residue was extraced with EA. The organic phase was washed with brine, dried over Na2S04 and concentrated to afford 4-bromo-3- chloro-5-methylaniline (2.4 g). MS(ES÷) m/z 220 (MH*).
Step 3: A mixture of tri-ter/'-butylphospliine, tetrafluoroboric acid salt (131 mg), 4-bromo-3- chloro-5-methyIaniline (661 mg), (2-(trifluoromethoxy)phenyl)boronic acid (741 mg), Pd2(dba)3 (137 mg) and Na2C03 (636 mg) was stirred at RT for 15 minutes under nitrogen. Then the mixture was heated to 110°C for 45 minutes. After cooling to RT, the mixture was extracted with EA and washed with water and brine. The organic phase was dried over Na2S04. Solvent was removed in vacuo and the residue was purified by column choromatography to afford 2-chloro-6-methyl-2'- (trifluoromethoxy)-[l,l'-biphenyI]-4-amine (520 mg). MSiES*) m/z 302 (MH+).
Preparation of N-(2-chloro-6-methyl-2,-(trifluoromethoxy - l,l'-biphenyl -ylV2-(4- fethylsulfonvDphenvDacetamide
A mixture of 2-chloro-6-methyl-2'-(trifluoromethoxy)-[l,l '-biphenyl]-4-amine (intermediate
151a, 100 mg),2-(4-(ethylsulfonyI)phenyl)acetic acid (intermediate la, 76 mg), HATU (139 mg) and DIPEA (0.174 mL) in dichloromethane (2 mL) was stirred at RT overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-chloro-6-methyl-2'- (trifluorometliiOxy)-[l,r-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (43 mg). Ή-NMR (400 MHz, CDC13) S ppm 1.30 (m, 3H), 2.00 (m, 3H), 3.14 (m, 2H), 3.83 (s, 2H), 7.19 (dd, J= 1.8 Hz, 8.0Hz, 1H), 7.35 (m, 4H), 7.44 (m, 1H), 7.56 (dd, J= 3.0 Hz, 4.5Hz, 3H), 7.90 (d, J= 8.0 Hz, 2H); "F-NMR (376 MHz, CDC13) 5 ppm -57.12; MSfES*) m/z 512 (MH*).
Example 152
iV-(2-chloro-3-fluoro-2 -(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide

Intennediate 152a: 2-chloro-3-fluoro-2'-ftrifluoromethoxyVri. -biphenyll-4-amine
A mixture of tri-fer/-butylphosphine, tetrafluoroboric acid salt (131 mg), 4-bromo-3-chloro-2- fluoroaniline (673 mg), (2-(trifluoromethoxy)phenyI)boronic acid (741 mg), Pd2(dba)3 (137 mg) and Na2COj (636 mg) in 1,4-dioxane (16 mL) and water (4 mL) was stirred at RT for 15 minutes under nitrogen. Then the mixture was heated to 1 10°C for 45 minutes. After cooling to RT, the mixture was extracted with EA and washed with water and brine. The organic phase was dried over Na^SC^ and concentrated in vacuo. The residue was purified by column choromatography to afford 2-chloro-3- fluoro-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (560 mg). MSiES*) m/z 306 (MH+).
Preparation of N-r2-chloro-3-fluoro-2'-(trifluoromethoxy)-ri .l'-biphenyl1-4-yl')-2-(4- fethylsulfonvDphenvDacetamide
A mixture of 2-chloro-3-fluoro-2'-(trifluoromethoxy)-[l, -biphenyl]-4-amine (intermediate 1 2a,
100 mg), 2-(4-(ethylsulfonyl)phenyl)acetic acid (intennediate la, 74.7 mg), HATU (137 mg) and
DIPEA (0.171 mL) in dichloromethane (2 mL) was stirred at RT overnight. The mixture was concentrated in vacuo and the residue was purified by DAP to afford N-(2-chloro-3-fluoro-2'-
(trifluoromethoxy)-[l, -biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (59 mg). Ή-NMR
(400 MHz, CDClj) δ ppm 1.30 (t, J= 7.4 Hz, 3H), 3.14 (q, J = 7.4 Hz, 2H), 3.90 (s, 2H), 7.09 (m,
1H), 7.29 (m, 1H), 7.36 (m, 2H), 7.43 (m, 1H), 7.49 (m, 1H), 7.62 (m, 2H), 7.94 (m, 2H), 8.28 (t, J-
8.0 Hz, 1H) 19F-NMR (376 MHz, CDC13) δ ppm -57.28, -130.29; MS(ES^) m/z 516 (MH^. Example 153
N-(2-chlor o-3-fluoro-2'-(trifluor omethoxy)- [1,1' -bi heny I]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide

Example 154
4-(2-(4-(ethylsulfonyl)phenyI)acetamido)-N^V-dimethyl-2,-(trifluoromethoxy)-[ltl,-biphenyl]-2- carboxamide

A mixture of 4-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2'-(trifluoromethoxy)- [l,l'-biphenyl]-2- carboxylic acid (150 mg), dimethylamine, hydrochloride (26.5 mg) and HATU (157 mg) in dichloromethane (4 mL) was stirred for 12 hrs. The mixture was concentrated and extracted with EtOAc. The organic layers were concentrated and purified by MDAP to afford 4-(2-(4- (ediylsulfonyl)phenyl)acetamido)-N,N-dimethyl-2'-(trifluoromethoxy)-[l, -biphenyl]-2-carboxamide (46 mg). 'H-NMR (400 MHz, MeOD-c/4) δ ppm 1.20 (t, J= 7.4 Hz, 3H), 2.76 (s, 3H), 2.83 (s, 3H), 3.20 (m, 2H), 3.86 (s, 2H), 7.37 (m, 5H), 7.63 (d, J= 8.4 Hz, 2H), 7.66 (d, /= 2.0 Hz, 1H), 7.69 (d, J = 2.0 Hz, 1H), 7,74 (d, J= 2.0 Hz, 1H), 7.87 (d, J= 8.4 Hz, 2H); MS(ES+) m/z 535 (MH^.
Example 155
2-(4-(ethyIsulf onyl)phenyl)-7V-(2- (py ridin-4-yl)-2' -(trifluor omethoxy)- [1,1 '-biphenyl] -4- yl)acetamide

Step 2: To a solution of 4-(4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2-yl)pyridine (150 mg) in methanol (5 mL) was added Renay Ni (14 mg). The reaction mixture was stirred under hydrogen at room temperature for 4 hrs. The mixture was filtered and the filtrate was concentrated in vacuo to afford 2-(pyridin-4-yl)-2,-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (120 mg). MSfES^ m/z 331 (MH* .
Step 3: To 2-(4-(ethylsulfonyl)phenyl)acetic acid (91 mg) was added thionyl chloride (853 mg).
The reaction mixture was stirred for 1 hr. Thionyl chloride was removed in vacuo. The residue was dissolved in dichloromethane (1 mL), and then added dropwise to a solution of 2-(pyridin-4-yi)-2'- (trifluoromethoxy)-[l, -biphenyl]-4-amine (120 mg) and triethylamine (0.152 mL) in
dichloromethane (3 mL) under nitrogen at room temperature. The reaction mixture was stirred for 4 hrs. Solvent was evaporated in vacuo and the residue was purified by MDAP to afford 2-(4-
(ethylsulfonyl)phenyl)-N-(2-(pyridin-4-yl)-2'-(trifluoromethoxy)-[l ,r-biphenyI]-4-yl)acetamide (52.1 mg). 'H-NMR (400 MHz, CDC13) δ ppm 1.21 (m, 3H), 3.01 (m, 2Η), 3.95 (s, 2Η), 6.99 (m, 1H), 7.41 (m, 4H), 7.67 (m, 2Η), 7.80 (m, 2H), 7.95 (m, 2H), 8.05 (m, 2Η), 8.41 (m, 2H), 10.35 (s, 1H);
MS(ES+) m/z 541 (MH*). Example 156
N-(2,6-dichloro-4'-fluoro-2'-methoxy-[ '-biphen^

Step 1: A solution of 4-bromo-3,5-dichloroaniline (100 mg), (4-fluoro-2- methoxyphenyl)boronic acid (106 mg), tri-ter/-butyIphosphine, tetrafluoroboric acid salt (48.2 mg),
Pd2(dba)3 (38.0 mg) and Na2C03 solution (2 M, 0.623 mL) in 1 ,4-dioxane (4 mL) was heated in the microwave at 100°C for 2 hours. The resulted mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed in vacuo and extracted with ethyl acetate. The organic layer was washed with saturated ammonium chloride solution and brine. The residue was purified by column choromatography and concentrated in vacuo to afford 2)6-dicliloro-4l-fluoro-2'-methoxy-[l , l'- biphenyl]-4-amine (83 mg) as a white solid. MS(ES ) m/z 286 (MH*).
Step 2: A solution of 2,6-dichIoro-4'-fluoro-2'-methoxy-[l,r-biphenyl]-4-amine (83 mg), perfluorophenyl 2-(4-(ethylsulfonyl)phenyl)acetate (intermediate 102a, 126 mg) and DIPEA (0.152 mL) in dichloromethane (10 mL) was stirred at RT for 18 hours. Solvent was removed in vacuo. The obtained mixture was redissolved in DMF, and solid was filtered off. The filtrate was purified by MDAP to afford N-(2,6-dichloro-4'-fluoro-2'-methoxy-[l , l'-biphenyI]-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (79 mg) as a white solid. Ή-NMR (400 MHz, DMSO-<¾) δ ppm 1.03 (t, 7= 7.2 Hz, 3H), 3.22 (q, J= 7.3 Hz, 2H), 3.66 (s, 3H), 3.78 (s, 2H), 6.80 (t, J= 8.4 Hz, 1H), 6.97 (d, J = 9.2 Hz, 1H), 7.06 (t, J - .6 Hz, 1H), 7.55 (d, 7= 8.4 Hz, 2H), 7.70 (s, 2H), 7.79 (d, J= 8.4 Hz, 2H), 10.56 (s, 1H); 19F-NMR (376 MHz, DMSO-<¾) δ ppm -110.29; MS(ES ) m/z 496 (MH4 .
Example 157
7Y-(2,6-dichloro-4'-fluoro-[l,l'-biphenyl]-4-yl)-2-(4-(ethy]sulfonyl)pheny])acetamide

Step 1: A solution of 4-bromo-3,5-dichloroaniline (100 mg), (4-fluorophenyl)boronic acid (87 mg), tri-terZ-butylphosphine, tetrafluoroboric acid salt (48.2 mg), Pd2(dba)3 (38.0 mg) and Na2C03 (2 M, 0.623 iTiL) in 1,4-dioxane (5 mL) was heated in the microwave at 120°C for 1 hour. The resulted mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed in vacuo and extracted with ethyl acetate. The organic layer was washed with saturated ammonium chloride solution and brine. The residue was purified by column choromatography and concentrated in vacuo to afford 2,6-dichloro-4'-fluoro-[l , 1 '-biphenyl]-4-amine (77 mg) as a white oil.
Step 2: A solution of 2,6-dichloro-4'-fluoro-[l,r-biphenyl]-4-amine (76 mg), perfluorophenyl 2- (4-(ethylsulfonyl)phenyl)acetate (intermediate 102a, 117 mg) and DIPEA (0.104 mL) in
dichloromethane (10 mL) was stirred at RT for 14 hours. Solvent was removed in vacuo. The obtained mixture was redissolved in DMF and solid was filtered off. The filtrate was purified by
MDAP to afford N-(2,6-dichloro-4'-fluo]O-[l , 1 '-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (37 mg) as a white solid. 'H-NMR (400 MHz, DMSO-i¾ 6 ppm 1.04 (t, J= 7.2 Hz, 3H), 3.22 (q, J= 7.5 Hz, 2H), 3.79 (s, 2H), 7.23 (d, J= 7.6 Hz, 4H), 7.55 (d, J= 8.4 Hz, 2H), 7.74 (s, 2H), 7.79 (d, J= 8.4 Hz, 2H), 10.58 (s, 1H); 19F-NMR (376 MHz, DMSO-i/6) δ ppm -113.72; MSfES4) m/z 466 (MH ). Example 158
Ar-(2,2'-dichIoro-4,-fluoro-[l,l'-biphenyl]-4-yI)-2-(4-(ethy]sulfonyl)phenyl)acetamide

A solution of N-(4-bromo-3-chlorophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6a, 100 mg), (2-chIoro-4-fluorophenyl)boronic acid (50.2 mg), tri-teri-butylphosphine,
tetrafluoroboric acid salt (27.8 mg), Pd2(dba)3 (21.97 mg,) and Na2C03 solution (2 M, 0.360 mL) in 1,4-dioxane (5 mL) was heated in the microwave at 100°C for 1 hour. The resulted mixture was filtered tlirough celite, and washed with ethyl acetate. Solvent was removed in vacuo. The obtained mixture was redissolved in DMF, and solid was filtered off. The filtrate was purified by MDAP to afford N-(2,2'-dichloro-4'-fluoro-[l}l'-biphenyl]-4-yl)-2-(4-(etliylsulfonyl)phenyl)acetamide (35 mg) as a white solid. 1 H-NMR (400 MHz, DMSO-<¾) δ ppm 1.04 (t, J= 7.4 Hz, 3H), 3.21 (q, J= 7.3 Hz, 2H), 3.78 (s, 2H), 7.24 (m, 2H), 7.32 (t, J= 7.6 Hz, 1H), 7.49 (m, 2H), 7.56 (d, J= 8.4 Hz, 2H), 7.79 (d, J= 8.4 Hz, 2H), 7.86 (s, 1H), 10.50 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -111.85; MS(ES^) m/z 466 (MH+).
Example 159
N-(2,6-dichloro-2'-fluoro- [ 1 , 1 '-biphenyl] -4-yI)-2-(4-(ethylsulf onyl)phenyl) acetamide
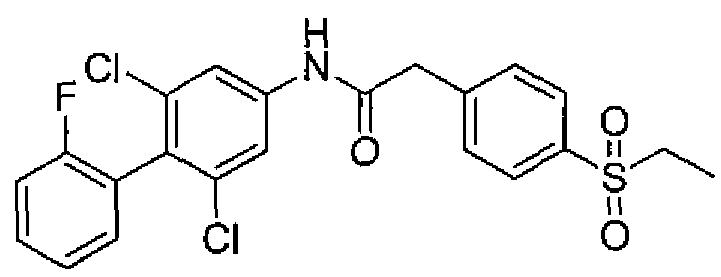
Step 1: A solution of 4-bromo-3,5-dichloroaniline (100 mg), (2-fluorophenyl)boronic acid (87 mg), tri-/er/-butylphosphine, tetrafluoroboric acid salt (48.2 mg), Pd2(dba)3 (38.0 mg) and Na2C03 solution (2 M, 0.623 mL) in 1,4-dioxane (5 mL) was heated in the microwave at 120°C for 2 hours. The resulted mixture was filtered tlirough celite, and washed with etliyl acetate. Solvent was removed
in vacuo and extracted with ethyl acetate. The organic layer was washed with saturated ammonium chloride solution and brine. The residue was purified by column choromatography and concentrated in vacuo to afford 2,6-dichloro-21-fluoro-[l,l'-biphenyl]-4-amine (75 mg) as a white solid. MS(ES+) m/z 256 (MH+).
Step 2: A solution of 2,6-dichloro-2' 1uoro-[l,l'-biphenyI]-4-amine (75 mg), perfluorophenyl 2-
(4-(ethylsulfonyl)phenyl)acetate (intermediate 102a, 115 mg) and DIPEA (0.102 mL) in
dichloromethane (10 mL) was stirred at RT for 14 hours. Solvent was removed in vacuo. The obtained mixture was redissolved in DMF and solid was filtered off. The filtrate was purified by MDAP to afford N-(2,6-dicMoro-2'-fluoro-[l, -biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (67 mg) as a light yellow solid. Ή-NMR (400 MHz, DMSO-<¾) δ ppm 1.11 (t, J= 7.4 Hz, 3H), 3.29 (q, J= 7.3 Hz, 2H), 3.87 (s, 2H), 7.33 (m, 3H), 7.51 (m, 1H), 7.63 (d, J= 8.4 Hz, 2H), 7.85 (s, 2H), 7.87 (d, J = 8.4 Hz, 2H), 10.71 (s, 1H); I9F-NMR (376 MHz, DMSO- ) δ ppm -114.63; MS(ES+) m/z 466 (MH+).
Example 160
/ -(2-cyano-6-fluoro-2'-(trifluoromethoxy)-[l,l,-biphenyl]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide

A mixture of 4-amino-6-fluoro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-carbonitrile (intermediate 150a, 100 mg), 2-(4 -(methyl sulfonyl)phenyl)acetic acid (72.3 mg), HATU (141 mg) and DIPEA (0.177 mL) in dichloromethane (2 mL) was stirred at RT overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-cyano-6-fluoro-2'-(trifluoromethoxy)- [l,r-biphenyl]-4-yl)-2-(4-(methylsulfonyl)phenyl)acetamide (34 mg). 'H-NMR (400 MHz, CDC13) δ ppm 3.09 (s, 3H), 3.85 (s, 2H), 7.42 (m, 3H), 7.53 (m, 3H), 7.62 (m, 1H), 7.86 (dd, J= 2.3 Hz, 10.8Hz, 1H), 7.91 (m, 3H); I9F- MR (376 MHz, CDC13) δ ppm -57.66, -107.66; MS(ES+) m/z 493 (MH+).
Example 161
N-(2-chIoro-6-methyI-2'-(trifluoromethoxy)-[l,l'-biphenyI]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide

A mixture of 2-chloro-6-methyl-2'-(trifluoromethoxy)-[l, -biphenyl]-4-amine {intermediate 151a, 100 mg), 2-(4-(methylsulfonyl)phenyl)acetic acid (71.0 mg), HATU (139 mg) and DIPEA (0.174 mL) in dichloromethane (2 mL) was stirred at RT overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-chloro-6-methyl-2'-(triiluoromemoxy)- [l,r-biphenyl]-4-yl)-2-(4-(methylsuIfonyl)phenyl)acetamide (42 mg). Ή-NMR (400 MHz, CDC13) δ ppm 2.02 (m, 3H), 3.07 (s, 3H), 3.82 (s, 2H), 7.19 (dd, J= 1.8 Hz, 7.8Hz, 1H), 7.36 (m, 3H), 7.44 (m, 2H), 7.55 (m, 3H), 7.93 (d, J= 8.3 Hz, 2H); 19F-NMR (376 MHz, CDC¾) 5 ppm -57.10; MSiES ) m/z 498 (MH÷). Example 162
iV-(2,6-difluoro-2'-(trjfluoromethoxy)-[l,I,-biphenyl]-4-yl)-2-(5-(ethyIsulfonyl)pyridin-2- yl)acetamide, trifluoroacetic acid salt
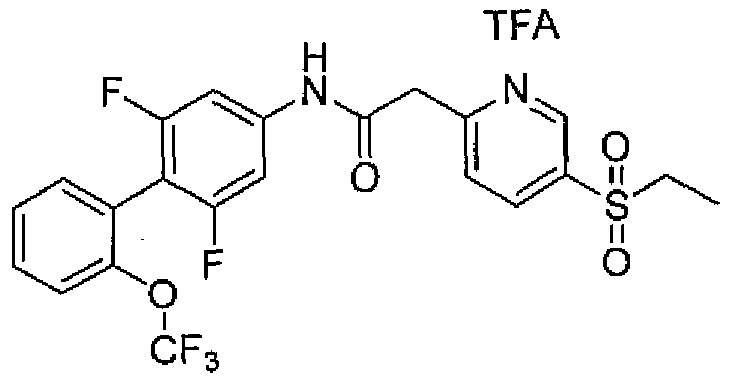
Step 1: To a mixture of 2-chloro-5-(ethylsulfonyl)pyridine (1.3 g), Pd2(dba)3 (0.289 g), tn-tert- butylphosphine, tetrafluoroboric acid salt (0.275 g) was added (2-ethoxy-2-oxoethyI)zinc(II) bromide (0.5 M in THF, 50.6 mL) under nitrogen. The resulted mixture stirred at RT for 30 minutes and then was refluxed. The mixture was stirred at 70°C overnight. The reaction was cooled to RT, queched with saturated NH C1 aqueous solution and extracted with EA. The combined organic layers were dried over Na2S04 and concentrated in vacuo. The residue was purified by column choromatography to afford ethyl 2-(5-(ethylsulfonyl)pyridin-2-yl)acetate (0.7 g). MSiES m/z 258 (MH+).
Step 2: To a solution of ethyl 2-(5-(ethylsulfonyI)pyridin-2-yl)acetate (0.6 g) in methanol (5 mL) and tetrahydrofuran (5 mL) was added a aqueous solution of NaOH (0.187 g in 5 mL water). The resulted mixture was stirred at RT for 1 hr. Most of solvent was removed in vacuo. The residue was
dissolved in water (10 mL) and extracted with EA.The aqueous phase was freeze-dried to afford 2-(5- (emylsulfonyl)pyridin-2-yl)acetic acid (40 mg). MS(ES+) m/z 230 (MH+).
Step 3: A mixture of 2-(5-(ethylsulfonyl)pyridin-2-yl)acetic acid (40 mg), 2,6-difluoro-2'- (trifluoromethoxy)-[lJl '-bip enyl]-4-amine (intennediate 116a, 50.5 mg), HATU (80 mg) and DIPEA (0.091 mL) in N^N-dimethylformamide (6 mL) was stirred at RT overnight. The reaction was quenched with water and extracted with EA. The combined organic phase was concentrated in vacuo. The residue was purified by MDAP to afford N-(2,6-difluoro-2,-(trifluorometlioxy)-[l)l'-biphenyl]-4- yl)-2-(5-(ethylsulfonyl)pyridin-2-yl)acetamide, trifluoroacetic acid salt (21 mg). Ή-NMR (400 MHz, DMSO-i ) δ ppm 1.14 (t, J= 7.4 Hz, 3H), 3.41 (q, J= 7.3 Hz, 2H), 4.11 (m, 2H), 7.47 (d, /= 9.8 Hz, 2H), 7.52 (m, 3H), 7.62 (m, 1H), 7.73 (d, J= 8.3 Hz, 1H), 8.28 (m, 1H), 8.97 (d, J= 2.5 Hz, 1H), 10.84 (s, 1H); 19F-NMR (376 MHz, DMSO-<¾ δ ppm -56.79, -75.01, -112.28; MS(ES÷) m/z 501 (MH+).
Example 163
7Y-(2-chlor o-5-fluoro-2' -(tr ifluoromethoxy)- [1,1' -biphenyl] -4-yl) -2-(4- (methylsuIfonyl)phenyl)acetamide

Step 1: To a solution of 5-chloro-2-fluoroaniline (20.0 g) in acetic acid (200 mL) at 0°C was added Br2 (3.54 mL). The reaction mixture was stirred at room temperature for 5 hrs. Solvent was removed, and the residue was basified with 50% sodium hydroxide solution (250 mL). The aqueous phase was extracted with EtOAc. The combined organic phases were dried over sodium sulfate, filtered and concentrated in vacuo. The residue was purified by column chromatography to give 4- bromo-5-chloro-2-fluoroaniline (10.11 g). Ή-NMR (400 MHz, CDC13) δ ppm 3.80 (s, 2H), 6.87 (d, J = 8.4 Hz, 1H), 7.26 (d, /= 10.4 Hz, 1H); MStES") m/z 224 (MH÷).
Step 2: A mixture of 4-bromo-5-chloro-2-fluoroani]ine (673 mg), (2- (trifluoromethoxy)phenyl)boronic acid (680 mg), Pd2(dba)3 (137 mg), tr erZ-butylphosphine, tetrafluoroboric acid salt (131 mg) and Na2C03 (636 mg) in 1,4-dioxane (12 mL) and water (3 mL) was stirred at RT for 15 minutes under nitrogen. Then the mixture was heated to 110°C and stirred for 45 minutes. The reaction was stopped and cooled to RT. The mixture was extracted with EA twice and washed with water and brine. The organic phase was collected and dried over Na2S04. Solvent
was removed and the residue was purified by chromatography to afford 2-chloro-5-fluoro-2'- (trifliioromethoxy)-[l,r-hiphenyl]-4-amine (570 mg). MS^S"") m/z 306 (MH ).
Step 3: A mixture of 2-chloro-5-fluoro-2'-(trifIuoromethoxy)-[I,I'-biphenyl]-4-amine (100 mg), 2-(4-(methylsulfonyl)phenyl)acetic acid (70.1 mg), HATU (137 mg) and DIPEA (0.171 mL) in dicliloromethane (2 mL) was stirred at RT overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-chloro-5-fluoro-2'-(trifluoromethoxy)-[l ,1 '-biphenyl]- 4-yl)-2-(4-(methylsulfonyl)phenyl)acetamide (66 mg). lH-NMR (400 MHz, CDC¾) δ ppm 3.07 (s, 3H), 3.89 (s, 2H), 7.04 (d, J= 11.0 Hz, 1H), 7.32 (m, 3H), 7.45 (m, 1H), 7.54 (br s, 1H), 7.58 (m, J= 8.3 Hz, 2H), 7.96 (m, 2H), 8.52 (m, 1H); !5F-NMR (376 MHz, CDC13) δ ppm -57.26, -133.53;
MS(ES+) m/z 502 (MH4).
Example 164
N-(2-acetyl-2,-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 1: A mixture of (2-(trifluoromethoxy)phenyl)boronic acid (1.083 g), l-(2-chloro-5- nitrophenyl)ethanone (1 g) and Cs2C03 (1,959 g) in water (5 mL) and acetonitrile (15 mL) was stirred for 10 mins under nitrogen before Pd(dppf)Cl2 (0,092 g) was added. The resulted mixture was refluxed for 50 mins. The mixture was cooled to room temperature and filtered, the filtrate was concentrated. The residue was dissolved in EtOAc and water. The EtOAc layer was washed with brine, dried over Na2S04, concentrated to give l-(4-nitro-2'-(trifluoromemoxy)-[l,r-biphenyl]-2- yl)ethanone (1 g). MS(ES÷) m/z 326 (MH* .
Step 2: A mixture of l-(4-nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)ethanone (1 g) and Pd/C (10% wt, 100 mg) in methanol (20 mL) was stirred for 2 hrs under Hydrogen (40 psi.). The residue was filtered and concentrated to give l-(4-amino-2'-(trifluorometlioxy)-[l,l,-biphenyl]-2- yl)ethanone (800 mg).
Step 3: A solution of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intennediate la, 186 mg) in SOCL
(0.5 mL) was stirred for 2 hrs at room temperature and concentrated. The residue was dissolved in CH2C12 (1 mL) and then was added to a solution of l-(4-amino-2'-(trifluoromethoxy)-[l,r-biphenyl] - 2-yl)ethanone (200 mg) and TEA (0.113 mL) in dicliloromethane (5 mL). The mixture was stirred for
30 mins, concentrated and purified by MDAP to give N-(2-acetyl-2'-(trifluoromethoxy)-[l, - biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (150 mg). Ή-NMR (400 MHz, MeOD-¾) δ ppm 1.20 (t, J= 7.4 Hz, 3H), 2.S9 (s, 3H), 3.18 (m, 2H), 3.86 (s, 2H), 7.27 (d, J= 3.6 Hz, 2H), 7.35 (m, 3H), 7.63 (d, J= 8.0 Hz, 2H), 7.77 (d, J= 2.0 Hz, 1H), 7.79 (d, J= 2.0 Hz, 2H), 8.06 (d, J= 2.0 Hz, 1H); MSfES ) m/z 506 (MH+).
Example 165
2-(4-(ethylsulfonyl)phenyl)-Ar-(5-(2-(trifluoromethoxy)phenyl)pyrazin-2-yl)acetamide, hydrochloride

Step 2: To a mixture of N-(5-bromopyrazin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (448 mg), (2-(trifiuoromethoxy)phenyI)boronic acid (200 mg) and Cs2COj (380 mg) in acetonitrile (1.5 mL) and water (0.5 mL) stirred under nitrogen at room temperature was added Pd(dppf)Cl2 (35.5 mg) in one charge. The reaction mixture was refluxed at 100°C for 1 hr. The mixture was filtered through celite, washed with CH3CN and EtOAc. The filtiate was concentrated in vacuo and the residue was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2SC>4. After filtration, solvent was removed in vacuo and the residue purified by MDAP to give 2-(4-(ethyl sulfonyl)phenyl) -N-(5 -(2-(trifluoromethoxy)phenyl)pyrazin-2-yl)acetamide, hydrochloride (10.1 mg). 'H- M (400 MHz, MeOD-<¾) δ ppm 1.21 (t, J= 3.2 Hz, 3H), 3.21 (q, J = 7.6 Hz, 2H), 3.96 (s, 2H), 7.53 (m, 3H)} 7.66 (m, 2H), 7.81 (m, 1H), 7.90 (m, 2H), 8.66 (d, J= 0.6 Hz, 1H), 9.46 (d, J= 0.8 Hz, 1H); MS(ES+) m/z 466 (MH+).
Example 166
2-(4-(ethyIsulfonyl)phenyl)-N-(6-(2-(trifluoromethoxy)phenyl)pyridin-3-yl)acetaniide, hydrochloride

Step 1: To a solution of 2-brorao-5-nitropyridine (150 mg), (2-(trifluoromemoxy)phenyl)boronic acid (183 mg) and Cs2C03 (482 mg) in acetonitrile (1.5 niL) and water (0.5 mL) stirred under nitrogen at room temperature was added Pd(dppf)Cl2 (27 mg) in one charge. The reaction mixture was stirred at 100°C for 2 hrs in the microwave. The mixture was filtered through celite, washed with EtOAc. The filtrate was concentrated in vacuo and the residue was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na2S04. Solvent was removed in vacuo to afford 5-nitro-2-(2-(trifluorometlioxy)phenyl)pyridine (250 mg). MS(ES+) m/z 285 (MH+).
Step 2: To a solution of 5-nitro-2-(2-(trifluoromethoxy)phenyl)pyridine (250 mg) in methanol (5 mL) was added Raney nickel (25.8 mg). The reaction mixture was stirred under hydrogen at room temperature for 4 hrs. The mixture was filtered and the filtrate was concentrated in vacuo to give 6- (2-(trifluoromethoxy)phenyl)pyridin-3-amine (200 mg). MSfES*) m/z 235 (MH+).
Step 3: To 2-(4-(ethylsulfonyl)phenyl)acetic acid (intennediate la, 91 mg) was added thionyl chloride (0.5 mL). After the mixture was stirred for 1 hour, thiony] chloride was removed in vacuo. The residue was dissolved in dichloromethane (1 mL) and then was added dropwise to a solution of 6-(2-(trifluoromethoxy)phenyl)pyridin-3-amine (120 mg) and triethylamine (0.197 mL) in dichloromethane (3 mL) stirred under nitrogen at room temperature. The reaction mixture was stirred for 3 hrs. Solvent was evaporated in vacuo and the residue purified by MDAP to give 2-(4- (ethylsulfonyl)phenyI)-N-(6-(2-(trifluoromethoxy)phenyI)pyridin-3-yl)acetamide, hydrochloride (86.5 mg). Ή-NMR (400 MHz, MeOD-</4) 5 ppm 1.21 (t, J= 3.2 Hz, 3H), 3.23 (q, J= 7.6 Hz, 2H), 3.98 (s, 2H), 7.64 (m, 4H), 7.88 (m, 2H), 7.90 (m, 2H), 8.17 (d, J= 9.2 Hz, 1H), 8.55 (d, J= 2.4 Hz, 1H), 9.44 (d, J = 2.4 Hz, 1H); MS(ES+) m/z 465 (MH+).
Example 167
2-(4-(ethylsulfonyl)phenyl)-JV-(2-(2-(trifluorornethoxy)phenyl)pyrimidin-5-yl)acetamide, hydrochloride

Step I: To a solution of 2-chloro-5-nitropyrimidine (500 mg) in methanol (15 mL) was added Raney nickel (92 mg). The reaction mixture was stirred under hydrogen at room temperature for 4 l rs. The mixture was filtered and the filtrate was concentrated in vacuo to give 2-chloropyrimidin-5- amine (450 mg).
Step 2: A mixture of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 291 mg) and thionyl chloride (0.5 mL) was stirred at RT for 1 hr. The excess thionyl chloride was removed in vacuo. The residue was dissolved in dichloromethane (1 mL) and then was added dropwise to a solution of 2-chloropyrimidin-5-amine (150 mg) and triethylamine (3.47 mL) in dichloromethane (2 mL). The mixture was stirred under nitrogen for 4 lirs. Solvent was evaporated in vacuo to give N-(2- chloropyrimidin-5-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (160 mg). MS(ES+) m/z 341 (MH4).
Step 3: To a solution of N-(2-chloropyrimidin-5-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (140 mg), (2-(trifluoromethoxy)phenyl)boronic acid (102 mg) and Cs2C03 (161 mg) in acetonitrile (1.5 mL) and water (0.5 mL) stirred under nitrogen at room temperature was added Pd(dppf)Cl2 (15 mg) in one charge. The reaction mixture was refluxed at 100°C for 1 hr. The mixture was filtered through celite, washed with CH3CN and EtOAc. The filtrate was concentrated in vacuo and the residue was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous After filtration, solvent was removed in vacuo and the residue was purified by MDAP to give 2-(4-(ethylsulfonyl)phenyl)-N-(2-(2-(trifluoromethoxy)phenyl)pyrimidin-5- yl)acetamide, hydrochloride (11.5 mg). Ή-NMR (400 MHz, MeOD-<¾) δ ppm 1.21 (t, 7= 3.2 Hz, 3H), 3.20 (m, 2H), 3.92 (s, 2H), 7.48 (m, 1H), 7.63 (m, 1H), 7.65 (m, 1H), 7.88 (d, J= 3.2 Hz, 2H), 7.90 (d, .7=2.0 Hz, 3H), 9.15 (s, 2H); SiES*) m z 466 (MH+).
Example 168
iV-(2,6-dichloro-2'-cyano-4'-fluoro-[l,l'-biphenyl]-4-yI)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 2: A mixture of 3,5-dichloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaboroIan-2-yl)aniline (155 mg), 2-bromo-5-fIuorobenzonitrile (215 mg), tri-/_?ri-butylphosphine, tetrafluoroboric acid salt (62.5 mg), Pd2(dba)3 (49.3 mg) and 2 M Na2C03 (0.807 mL) in 1,4-dioxane (5 mL) was heated in the microwave at 120°C for 1 hour. The resulted mixture was filtered through celite, and washed witii ethyl acetate. Solvent was removed and extracted with ethyl acetate. The organic layer was washed with saturated ammonium chloride solution and brine. The residue was purified by column chromatography and concentrated in vacuo to afford 4'-aimno-2',6'-dichloro-4-fluoro-[l,r~biphenyl]- 2-carbonitrile (39 mg) as a white solid. MS(ES+) m/z 281 (MH4 .
Step 3: A solution of 4'-amino-2',6'-dichloro-4-fluoro-[l,r-biphenyl]-2-carbonitrile (39 mg), perfluorophenyl 2-(4-(ethylsulfonyl)phenyl)acetate (intermediate 102a, 65.6 mg) and N-ethyl-N- isopropylpropan-2 -amine (53.8 mg) in dichloromethane (10 mL) was stirred at RT for 14 hours . Solvent was removed. The obtained mixture was redissolved in DMF. Solid was filtered off. The filtrate was purified by MDAP and evaporated in vacuo to afford N-(2,6-dichloro-2'-cyano-4'-fluoro- [l,r-biphenyI]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (46 mg) as a white solid. Ή-NMR (400 MHz, DMSO-4 S ppm 1.03 (t, J= 7.4 Hz, 3H), 3.22 (q, J= 7.3 Hz, 2H), 3.81 (s, 2H), 7.54 (m, 3H), 7.69 (t, 8.8 Hz, 1H), 7.79 (m, 4H), 8.00 (d, J= 8.8 Hz, 1H), 10.07 (s, 1H); l9F-NMR (376 MHz, DMSO-4,) δ ppm -110.71 ; MSiES"1) m/z 491 (MH+).
Example 169
AL(2,6-dichloro-2'-(difluoromethoxy)-[l,l'-bi henyl]-4-yl)-2-(4-(ethylsuIfonyl)phenyl)acetamide

Step 2: A mixture of 2-bromo-l ,3-dichloro-5-nitrobenzene (200 mg), (2-hydroxyphenyl)boronic acid (153 mg), tri-fer -butylphosphine, tetrafluoroboric acid salt (86 mg), Pd2(dba)3 (67.6 mg) and NaiCOs solution (2 M, 1.107 mL) in 1 ,4-dioxane (5 mL) was heated in the microwave at 100°C for 0.5 hour. The resulted mixture was filtered tlirougli celite, and washed with ethyl acetate. Solvent was removed and extracted with ethyl acetate. The organic phase was washed with saturated ammonium chloride solution and brine. The residue was purified by column chromatography and concentrated in vacuo to give 2',6'-dichloro-4'-nitro-[l,l '-biphenyl]-2-ol (180 mg) as a yellow oil. MS(ES" m/z 284 (MH+).
Step 3: To a solution of 2f,6'-dichloro-4'-nitro-[l, -biphenyl]-2-ol (180 mg) and potassium hydroxide (533 mg) in acetonitrile (5 mL) and water (5 mL), diethyl
(bromodifluoromethyl)phosphonate (254 mg) was added gradually under -78 °C. The reaction mixture was allowed to warm to room temperature and continuously stirred for 30 minutes. The resulted mixture was dissoved in ethyl acetate and washed with brine. The organic layer was concentrated in vacuo to afford 2,6-dichloro-2,-(difluoromethoxy)-4-nitro-l ,l'-biphenyl (261 mg) as a yellow oil. MSfES4 m/z 334 (MH4).
Step 4: 2,6-Dichloro-2'-(difluoromethoxy)-4-nitro-l,l '-biphenyl (261 mg) was dissolved in ethanol (15 mL). Then to this solution, tin(II) chloride dihydrate (635 mg) was added gradually. Then the reaction mixture was stirred at RT for 14 hours. The reaction mixture was heated to 70°C for 2 hours. Solvent was removed and sodium hydroxide solution (2 M) was added. Then the residue was extracted with ethyl acetate and washed with brine. The organic layer was concentrated in vacuo to afford 2,6-dichloro-2'-(difluoromethoxy)-[l, -biphenyl]-4-amine (200 mg) as a yellow solid. MS(ES+) m/z 304 (MH").
Step 5: 2,6-Dichloro-2'-(difluoromethoxy)-[l,l'-biphenyl]-4-amine (200 mg), 2-(4- (ethylsulfonyl)phenyl)acetic acid (intermediate la, 99 mg) and HATU (165 mg) were dissolved in dichloromethane (15 mL). Then to this solution, DIPEA (0.126 mL) was added gradually. The reaction mixture was stirred for 15 hours. Solvent was removed. The mixture was purified by column chromatography and concentrated in vacuo. The residue was redissolved in DMF. Solid was filtered off. The filtrate was purified by MDAP. Solvent was evaporated in vacuo to afford N-(2,6-dichloro-
2'-(difluoromethoxy)-[l,r-biphenyl]-4-yI)-2-(4-(ethylsuIfonyl)phenyl)acetarnide (57 mg) as a white solid. lH-NMR (400 MHz, DMSO- ) δ ppm 1.03 (t, J= 7.4 Hz, 3H), 3.22 (q, J- 7.3 Hz, 2H), 3.79 (s, 2H), 7.09 (t, J= 73 Hz, 1H), 7.26 (m, 3H), 7.46 (m, 1H), 7.55 (d, J= 8.4 Hz, 2H), 7.74 (s, 2H), 7.79 (d, J= 8.4 Hz, 2H), 10.61 (s, 1H); I9F-NMR (376 MHz, DMSO-i¾) δ ppm -81.00; MSfES" m/z 514 (MH+).
Example 170
2-(4-(ethyIsulfonyl)phenyl)-N-(6-methyl-5-(2-(trifluoromethoxy)phenyl)pyrazin-2-yl)acetamide

Intennediate 170a: 2-(4-(ethylsulfonvI)phenyl)acetyl chloride
To a solution of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 0.913 g) in chloroform
(16 mL) was added SOCl2 (1.168 mL) at room temperature. The mixture was refluxed for 2 hrs. The excess solvent and SOCl2 were removed in vacuo to afford 2-(4-(ethyIsulfonyl)phenyl)acetyl chloride. Intermediate 170b: 6-methyl-5-(2-(trifluoromethoxy)phenyl'>pyrazin-2-amine
Step 1: To a solution of 6-chloropyrazin-2-amine (2.59 g) in chloroform (60 mL) and acetonitrile (6 mL) at 0°C was added NBS (3.92 g) slowly. The mixture was warmed to room temperature and continuously stirred for 1 h. The excess solvent was removed in vacuo and the residue was purified by column chromatography to afford 5-bromo-6-chloropyrazin-2-amine (3.8 g). MS(ES+) m/z 208 (MH*).
Step 2: To a solution of (2-(trifluoromethoxy)phenyl)boronic acid (1.397 g), 5-bromo-6- cliIoropyrazin-2-amine (1.4 g), Pd2(dba)3 (0.492 g) and tri-teri-butylphosphine, tetrafluoroboric acid salt (0.624 g) in 1,4-dioxane (8 mL) was added Na2C03 solution (2 M, 6.72 mL). The reaction mixture was heated in the microwave at 100°C for 45 mins. The mixture was diluted with EtOAc (45 mL). The organic phase was washed with water and brine, dried over anhydrous MgS04, filtered and the filtrate was concentrated in vacuo and purified by column chromatography to afford 6-chloro-5- (2-(trifluoromethoxy)phenyl)pyrazin-2-amine (750 mg). SiES4 m/z 290 (MH+).
Step 3: To a solution of 6-cliloro-5-(2-(trifiuoromethoxy)plieny])pyrazin-2-amine (290 mg), methylboronic acid (239 mg) and Pd(Pl¾P)4 (231 mg) in 1 ,2-dimethoxyethane (6 mL) was added tripotassium phosphate (425 mg). The reaction mixture was heated in the microwave at 130°C for 30 mins. The mixture was diluted with EtOAc (45 mL). The organic phase was washed with water and brine, dried over anhydrous MgS04, filtered. The filtrate was concentrated in vacuo and purified by
column chromatography to afford 6-methyl-5-(2-(trifluoromethoxy)pheny])pyrazin-2 -amine (230 mg). S(ES+) m/z 270 (MH+).
Preparation of 2-C4-fethylsulfonyl)phenyl -N-('6-methyl-5-f2-("trifluoromethoxy')phenyl)pyrazin-2- ypacetamide
To a solution of 6-methyl-5-(2-(trifluoromethoxy)phenyI)pyrazin-2-amine (intermediate 170b,
110 mg), 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intennediate 170a, 111 mg) and pyridine (8 mL) in chloroform (2 mL) was added DMAP (24.96 mg) at room temperature. The mixture was refluxed for 40 hrs. The solvent was removed in vacuo. The residue was purified by MDAP to afford 2-(4- (ethylsulfonyl)phenyl)-N-(6-methyl-5-(2-(trifluoromedioxy)phenyl)pyrazin-2-yl)acetamide (18 mg). Ή-NMR (400 MHz, DMSO- ) δ ppm 1.11 (t, 7= 7.2 Hz, 3H), 2.29 (s, 3H), 3.28 (m, 2H), 3.94 (s, 2H), 7.54 (m, 3H), 7.62 (m, 3H), 7.87 (d, J= 8.4 Hz, 2H), 9.20 (s, 1H), 11.26 (s, 1H); 19F-NMR (376 MHz, DMSO-4 δ ppm -56.49; MS(BS+) m/∑480 (MH+).
Example 171
7V-(5-(2-chIoroph enyl)-6- (trifluoromethyl)pyridm-2- l)-2-(4-(ethyIsulfonyl)p hcny l)acetam id e

Step 1: To a solution of (2-chlorophenyl)boronic acid (144 mg), 5-bromo-6- (trifluoromethyl)pyridin-2-amine (220 mg), Pd2(dba)j (84 mg) and tri-feri-butylphosphine, tetrafluoroboric acid salt (106 mg) in 1,4-dioxane (3 mL) was added Na2C03 solution (2 M, 0.913 mL). The reaction mixture was heated in the microwave at 100°C for 45 mins. The mixture was diluted with EtOAc (40 mL). The organic phase was washed with water and brine, dried over anhydrous MgS0 , filtered and the filtrate was concentrated in vacuo and purified by column cliromatography to afford 5-(2-chlorophenyl)-6-(trifluoromethyl)pyridin-2-amine (200 mg). MSiES4 m/∑ 273 (MH .
Step 2: To a solution of 5-(2-chlorophenyl)-6-(trifluoromethyl)pyridin-2-amine (80 mg), 2-(4- (ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 80 mg) and pyridine (8 mL) in chloroform (2 mL) was added DMAP (17.92 mg) at room temperature. The mixture was refluxed for 40 hrs. The solvent was removed in vacuo and purified by MDAP to afford N-(5-(2-chlorophenyl)-6- (trifluoromethyl)pyridin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (10 mg). Ή-NMR (400 MHz, DMSO- ) δ ppm 1.11 (t, J= 7.6 Hz, 3H), 3.31 (m, 2H), 3.95 (s, 2H), 7.43 (m, 3H), 7.62 (m, 3H),
7.87 (t, J= 8.4 Hz, 3H), 8.38 (d, J= 8.4 Hz, 1H), 11.37 (s, 1H); I9F-NMR (376 MHz, DMSO-i/6) δ ppm -62.03; MS(ES+) mtz 483 (MH+).
Example 172
N-(2-chloro-5-fl oro-2,-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyI)acetamide

A mixture of 2-chloro-5-fluoro-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (100 mg, see step 2 for synthesis of Example 163), 2-(4-(ethylsulfonyl)phenyI)acetic acid (intermediate la, 74.7 mg), HATU (137 mg) and DIPEA (0.171 mL) in dichloromethane (2 mL) was stirred at room temperature overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-chloro-5-fluoro-2,-(trifluoromethoxy)-[l5l'-biphenyl]-4-yI)-2-(4-
(ethylsulfonyl)phenyl)acetamide (55 mg). Ή-NMR (400 MHz, CDCI3) 5 ppm 1.29 (t, J= 7.4 Hz, 3H), 3.13 (q, J= 7.3 Hz, 2H), 3.89 (s, 2H), 7.04 (d, J= 11.0 Hz, 1H), 7.33 (m, 3H), 7.46 (m, 2H), 7.58 (m, 2H), 7.94 (m, 2H), 8.53 (d, J= 7.3 Hz, 1H); 19F-NMR (376 MHz, CDC13) δ ppm -57.27, -133.71 ; MS(BS+) m/z 516 (MH+).
Example 173
2-(4-(ethylsulfonyI)phenyl)-A'-(2-fluoro-6-methyl-2,-(trifluoromethoxy)-[l,l'-biphenyI]-4- yl)acetamide

Step 1: A mixture of l,2-difluoro-4-nitrobenzene (16.61 mL) and ammonia (195 mL) in a sealed tube was heated to 140°C for 5 hours. The mixture was cooled to room temperature. Yellow solid was precipitated and filtered. The cake was washed with water three times. The solid was dried to afford 2-fluoro-4-nitroani]ine (19.9 g). MSCES"* m/z 157 (MH+).
Step 2: To a mixture of 2-fluoro-4-nitroaniline (20 g), ammonium acetate (0.988 g) in acetonitrile (250 mL) was added NBS (25.08 g) in portions while stirring. The resulted mixture was stirred for 6 hours. Then the mixture was concentrated in vacuo and the residue was purified by column chromatography to afford 2-bromo-6-fluoro-4-nitroaniline (26 g). MSiES"1 m/z 235 (MH+).
Step 3: A mixture of 2-bromo-6-fluoro-4-nitroaniline (0.470 g), methylboronic acid (0.144 g),
PdCl2(dppf)-CH2Cl2 adduct (0.163 g) and tripotassium phosphate (0.849 g) in 1,4-dioxane (8 mL) and water (2 mL) was sealed in a microwave tube under nitrogen. Then the reaction mixture was heated in the microwave at 120°C for 2 hours. The mixture was extracted with EA twice and washed with water and brine. The organic phase was dried over Na2S04 and concentrated in vacuo. The residue was purified by column chromatography to afford 2-fluoro-6-methyl-4-nitroaniline (0.6 g). MS(ES÷) m/z 171 (MH*).
Step 4: To a solution of 2-fluoro-6-methyl-4-nitroaniIine (1.2 g) in acetonitrile (25 mL) at 0°C was added tert-buty\ nitrite (0.93 mL) dropwise under nitrogen, Copper(II) bromide (1.733 g) was added to the above mixture in portions under nitrogen at 0 °C. The reaction mixture was warm to room temperature and stirred overnight. The reaction mixture was concentrated and water was added. The mixture was extracted with EA twice. The organic layer was dried over Na2S04 and concentrated in vacuo to afford 2-bromo-l-fluoro-3-methyl-5-nitrobenzene (1.3 g).
Step 5: HC1 (7.03 mL) was added to a solution of 2-bromo-l-fiuoro-3 -methyl -5 -nitrobenzene (1.3 g) in ethanol (30 mL) and tetrahydrofuran (10 mL). The mixture was heated to 60 °C. Tin(II) chloride dihydrate (10 g) was added to the above mixture portionwise. The mixture was stirred for another 4 hours at 60 °C. The reaction was cooled to room temperature and then basifled with saturated Na2C03 solution to pH around 7. Solid was filtered off and washed with EA. The filtration was collected and concentrated in vacuo. The residue was extraced with EA twice. The combined organic layers were washed with brine, dried over Na2S0 and concentrated in vacuo to afford 4- bromo-3-fluoro-5-methylaniline (0.9 g). MSCES") m/z 203 (MH+).
Step 6: A mixture of tri-½r/-butylphosphine, tetrafluoroboric acid salt (0.192 g), 4-bromo-3- fluoro-5-methylaniline (0.9 g), (2-(trifluoromethoxy)phenyl)boronic acid (1.090 g), Pd2(dba)j (0.202 g) and Na2C03 (0.935 g) in 1 ,4-dioxane (16 mL) and water (4 mL) was stirred at room temperature for 15 minutes under nitrogen. Then the mixture was heated to 10°C and stirred for 45 minutes. After cooling to RT, the mixture was extracted with EA twice and washed with water and brine. The organic phase was dried over Na2S04 and concentrated in vacuo. The residue was purified by column chromatography to afford 2-fluoro-6-methyI-2'-(trifluoromethoxy)-[l, r-biphenyl]-4-amine (1.0 g). MS(ES+) «^ 286 (MH+).
Preparation of 2-f4-('ethylsuIfonyl phenvIVN-('2-fluoro-6-methyl-2'-('trifl iorQmethoxyVri ,1 '- biphenyl]-4-yl)acetamide
A mixture of 2-fluoro-6-metliyl-2'-(trifluoromethoxy)-[l5 -biphenyl]-4-amine (intermediate
173a, 100 mg), 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 96 mg), HATU (173 mg) and DIPEA (0.184 mL) in dichloromethane (2 mL) was stirred at room temperature overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford 2-(4-
(emylsulfony^phenylJ-N^-fluoro-e-methyl^'-ftrifluoromethoxyJ-tLr-biphenyll^-y^acetamide (42 mg). Ή-NMR (400 MHz, CDC ) 5 ppm 1.30 (m, 3H), 2.07 (m, 3H), 3.15 (q, J= 7.3 Hz, 2H), 3.84 (s,
2H), 7.12 (d, J= 1.3 Hz, 1H), 7.25 (dd, J= 1.8 Hz, 8.0 Hz, 1H), 7.37 (m, 3H), 7.43 (m, 2H), 7.57 (m, 2H), 7.91 (m, 2H); 19F-NMR (376 MHz, CDClj) 5 ppm -57.52, -111.96; MS(ES+) m/z 496 (Mlf).
Example 174
iV-(2-fluoro-6-methyl-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (methyIsulfonyI)phenyl)acetamide

173a, 100 mg), 2-(4-(methylsuIfonyl)phenyI)acetic acid (90 mg), HATU (173 mg) and DIPEA (0.184 mL) in dichloromethane (2 mL) was stirred at room temperature overnight. The mixture was concentrated in vacuo and the residue was purified by MDAP to afford N-(2-fluoro-6-methyl-2'- (trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4-(methylsulfonyl)phenyl)acetamide (30 mg). Ή-NMR (400 MHz, CDClj) δ ppm 2.07 (s, 3H), 3.08 (m, 3H), 3.83 (s, 2H), 7.12 (m, 1H), 7.25 (dd, J= 1.9 Hz, 7.9 Hz, 1H), 7.36 (m, 3H), 7.44 (m, 2H), 7.56 (d, = 8.3 Hz, 2H), 7.95 (m, 2H); l9F-NMR (376 MHz, CDC13) δ ppm -57.52, -111.98; MS(ES+) m/z 482 (MIT).
Example 175
A42,6-dichIoro-2'-(rrifluoromethoxy)-[
yl)acetamide

Step 1 : A mixture of 5-nitropyridin-2-ol (6 g) in hydrazine (32.3 g) was heated at 1 10°C for 5 hrs. Solvent was removed to give 2-(lH-pyrazol-3-yl)acetohydrazide (6 g). MS(ES+) m/z 141 (MtT).
Step 2: To 2-(lH-pyrazol-3-yl)acetohydrazide (6 g) was added cone. HCI (120 mL). The reaction mixture was stirred at 100°C for 3 hrs. Solvent was removed in vacuo to give 2-(lH-pyrazol- 3-yl)acetic acid (5.4 g). MS(ES+) m/z 127 (MH ).
Step 3: Thionyl chloride (15.62 mL) was added dropwise to a solution of 2-(lH-pyrazol-3- yl)acetic acid (5.4 g) in methanol (100 mL). The reaction mixture was stirred at 70°C for 5 hrs. Solvent was removed. The residue was partitioned between ethyl acetate (100 mL) and sat. sodium bicarbonate (100 mL). The aqueous phase was extracted with ethyl acetate (100 mL). The combined organic phases were dried and concentrated to give methyl 2-(lH-pyrazol-3-yl)acetate (2.5 g) as a brown oil. MSfES ) m/z 141 (ΜΗ+).
Step 4: Ethanesulfonyl chloride (3.3 g) was added dropwise to a solution of methyl 2-(lH- pyrazol-3-yl)acetate (2.4 g) in pyridine (30 mL) at 0 °C. The reaction mixture was stirred at room temperature for 2 hrs. Solvent was removed in vacuo. The residue was partitioned between ethyl acetate (100 mL) and 1 M HCI (100 mL). The organic phase was washed with brine, dried and concentrated. The residue was purified by column chromatography to give methyl 2-(l - (ethylsulfonyl)-lH-pyrazol-3-yl)acetate (2.7 g) as a pale yellow oil. Ή-NM (400 MHz, CDC13) δ ppm 1.55 (t, J= 7.2 Hz, 3H), 3.48 (q, J= 7.2 Hz, 2H), 3.73 (s, 3H), 3.78 (s, 2H), 6.49 (d, J = 2.4 Hz, 1H), 7.80 (d, /= 2.4 Hz, 1H); MS(ES÷) m/z 233 (MH+).
Step 5: Lithium hydroxide monohydrate (0.47 g) was added to a solution of methyl 2-(l- (ethylsulfonyl)-lH-pyrazol-3-yl)acetate (2.6 g) in tetrahydrofuran (25 mL) and water (25 mL) . The reaction mixture was stirred at 0°C for 5 hrs. THF was removed. The aqueous phase was washed with ether (30 mL x 2), and then acidified with 2M HCI solution to pH = 2. The obtained mixture was extracted with ethyl acetate (50 mL x 2). The combined organic phases were dried and concentrated to give 2-(l -(ethylsulfonyl)-lH-pyrazol-3-yl)acetic acid (2.1 g). Ή-NMR (400 MHz, DMSO- ) 5 ppm 1.06 (t, J= 7.2 Hz, 3H), 3.64 (q, J- 7.2 Hz, 2H), 3.69 (s, 2H), 6.55 (d, J= 2.4 Hz, 1H), 8.21 (d, J= 2.4 Hz, IH), 12.59 (s, 1H); MS(ES+) m/z 219 (MH+).
Step 6: A mixture of 2,6-dichloro-2'-(trifluoromethoxy)-[l ,l'-biphenyl]-4-amine (80 mg, see step 1 for synthesis of Example 132), 2-(l-(ethylsulfonyl)-lH-pyrazol-3-yl)acetic acid (81 mg), 2-
(3H-[l,2,3]triazolo[4,5-b]pyridin-3-yl)-l,l,3,3-teta (189 mg) and N-ethyl-N-isopropylpropan-2 -amine (0.130 mL) in dichloromethane (10 mL) was stirred at RT overnight. The mixture was washed with brine. The organic layer was dried and evaporated in vacuo. The residue was purified by MDAP to afford N-(2,6-dichloro-2,-(trifluoromethoxy)-[l,l'- biphenyl]-4-yl)-2-(l-(ethylsulfonyl)-lH-pyrazol-3-yl)acetamide (31 mg). Ή-NMR (400 MHz,
DMSO- ) δ ppm 1.08 (t, J= 7.4 Hz, 3H), 3.67 (q, J= 7.2 Hz, 2H), 3.83 (s, 2H), 6.62 (d, /= 2.8 Hz, 1H), 7.41 (dd, /= 1.6 Hz, 7.9 Hz, 1H), 7.52 (m, 2H), 7.61 (m, 1H), 7.83 (s, 2H), 8.25 (d, J= 2.8 Hz, 1H), 10.70 (s, 1H); 19F-NMR (376 MHz, DMSO-</6) δ ppm -56.27; MS(ES ) m/z 522 (MH+).
Example 176
Ar-(2-chloro-2'-(difluoromethoxy)-4'-fluoro-[l5l,-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyI)acetamide

Intermediate 176a: l-bromo-2-fdifluoromethoxy)-4-fluorobenzene
To a solution of 2-bromo-5-fluorophenol (1 g) in acetonitrile (20 mL) was added KOH (5.88 g) in water (20 mL). The resulted mixture was cooled to -78 °C, and then diethyl
(bromodifluoromethyl)phosphonate (2.80 g) was added in one portion. The mixture was gradually warmed to room temperature and stirred for 30 mins. The mixture was extracted with ether. The organic layer was washed with brine and dried over anhydrous a2S04. After cocentration, the residue was purified by column chromatography to afford l-bromo-2-(difluoromethoxy)-4- fluorobenzene (1.051 g) as a colorless oil. JH-NMR (400 MHz, DMSO-i¾) δ ppm 7.14 (dt, J= 2.8 Hz, 8.4 Hz, 1H), 7.35 (dd, J= 2.8 Hz, 9.7 Hz, 1H), 7.35 (t, J= 72.9 Hz, 1H), 7.79 (dd, J= 6.1 Hz, 8.9 Hz, 1H); I9F-NMR (376 MHz, DMSOi/6) δ ppm -83.26, -111.03.
Preparation of N- -chloro^'-fdifluoroi-nethoxy'i^'-fluoro-ri J'-biphenyl -yl)^-^- (ethylsulfonyl phenyl)acetamide
A mixture of l-bromo-2-(difluoromethoxy)-4-fIuorobenzene (intermediate 176a, 70 mg), N-[3- chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide
(intermediate 6b, 148 mg), PdCl2(dppi)-CH2CI2 adduct (18.98 mg) and Cs2C03 (114 mg) in acetonitrile (1.5 mL)/water (0.5 mL) was sealed in a vessel and heated in (lie microwave at 100°C for
45 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated
under reduced pressure and the residue was purified by MDAP to afford N-(2-chloro-2'- (difluoromethoxy)-4'-fluoro-[l, -biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (22 mg) as a white solid. Ή-NMR (400 MHz, DMSO-<¾ δ ppm 1.10 (t, /= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.84 (s; 2H), 7.21 (t, J= 73.2 Hz, 1H), 7.21 (m, 2H)} 7.28 (d, J - 8.4 Hz, 1H), 7.37 (m, 1H), 7.53 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.62 (d, J= 8.0 Hz, 2H), 7.86 (d, J= 8.4 Hz, 2H), 7.91 (d, J= 2.0 Hz, 1H), 10.55 (s, 1H); 19F-NMR (376 MHz, DMSO-rf6) δ ppm -82.43, -110.64; MSiES4) m/∑ 498 (MH+).
Example 177
iV-(2,-(difluoromethoxy)-2,4',6-trifluoro-[l,l,-bipheny]]-4-yI)-2-(4- (et ylsulf onyl)phenyl) acetamide
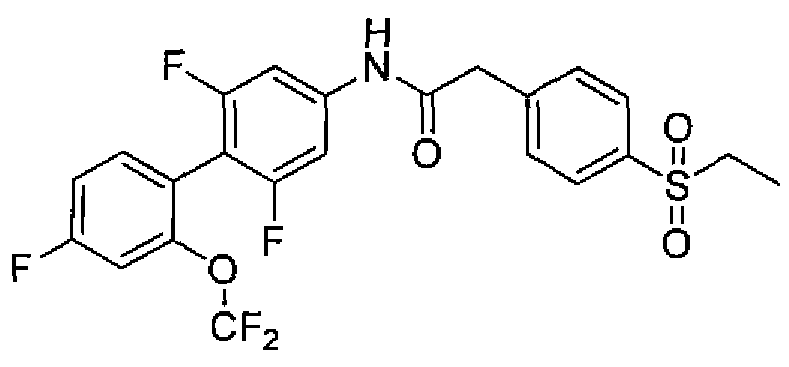
A mixture of l-bromo-2-(difluoromethoxy)-4-fluorobenzene (intermediate 176a, 150 mg), (4-(2- (4-(ethylsulfonyI)phenyl)acetamido)-2,6-difluorophenyl)boronic acid (intermediate 122a, 404 mg), tri-ierf-butylphosphine, tetrafluoroboric acid salt (217 mg), Pd2(dba)j (171 mg) and sodium carbonate solution (2 M, 1.245 mL) in 1,4-dioxane (4.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford N-(2'- (difluoromethoxy) -2,4',6-trifiuoro-[ 1 , 1 '-biphenyl] -4-yl) -2-(4-(ethylsulfonyl)phenyl) acetamide ( 133 mg) as a yellow solid. Ή-NMR (400 MHz, DMSO-<¾) 5 ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 7.25 (t, J= 73.2 Hz, 1H), 7.27 (m, 2H), 7.43 (m, 2H), 7.50 (m, 1H), 7.62 (d, J = 8.4 Hz, 2H),7.86 (d, J= 8.4 Hz, 2H), 10.74 (s, 1H); WF-NMR (376 MHz, DMSO-tf6) δ ppm -82.88, -109.41 , -111.99; MSfES^ m/z 500 (MH+).
Example 178
2-(4-(ethyIsulfonyl)phenyl)-N-(5-(4-fluoro-2-(trifluoromethoxy)phenyl)-6-methylpyridin-2- yl)acetamide, trifluoroacetic acid salt
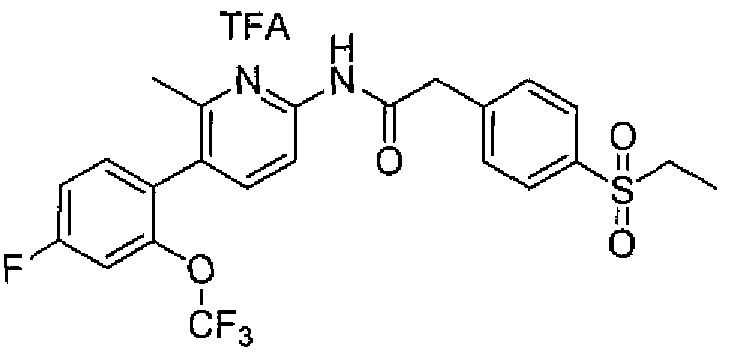
A mixture of 4,4,4, ;4, :5,5,5,,5,-octametliyl-2,2'-bi-l,3,2-dioxaborolane (5.27 g), N-(5-bromo-6- methylpyridin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (intermediate 67a, 2.5 g), Pd2(dba)3 (0.51 g), tricyclohexylphosphine (0.371 g) and potassium acetate (2.78 g) in 1 ,4-dioxane (30 l L) was heated to 80°C for 3 hrs, and then 100°C overnight. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated in vacuo and the residue was purified by column chromatography (EtOAc : PE = 0: 1 to 1: 1) to afford (6-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2- methylpyridin-3-yl)boronic acid (3.2 g) as a yellow oil. MSfES^) m/z 363 (Mtf).
Preparation of 2-f4-fethylsulfonyl')phenyl -N-f5-(4-fIuoro-2-('trifluoromethoxy^phenvn-6- methylpyridin-2-yl)acetamide. trifluoroacetic acid salt
Step 1: Acetic anhydride (1.278 lnL) was added into the solution of 3-(trifluoromethoxy)aniline
(2 g) and Et3N (2.361 mL) in dichloromethane (40 mL) at 0°C dropwise, and then tlie resulted mixture was stirred for 30 mins at 0°C and then 2 hrs at room temperature. The reaction mixture was poured into water, extracted with EtOAc. The combined organic layers were washed with sat.
NaHCOi solution, dried over anliydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure to afford N-(3-(trifluoromethoxy)phenyl)acetamide (2.3 g) as a yellow solid.
MS(ES+) m/z 220 (MH÷).
Step 2: NBS (1 ,340 g) was added to a solution of N-(3-(trifluoromethoxy)phenyl)acetamide (1.5 g) in acetic acid (20 mL) at room temperature. The mixture was stirred at room temperature for 2 days. The reaction mixture was poured into water, extracted with EtOAc. The combined organic layers were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure to afford N-(4-bromo-3-(trifluoromethoxy)phenyl)acetamide (1.8 g) as a yellow oil. MSfES m/z 298 (MH ).
Step 3: A solution of N-(4-bromo-3-(trifluoromethoxy)phenyI)acetamide (2039 mg) and cone. HC1 (3.5 mL) in ethanol (7 mL) was heated to 1 10°C for 2 hrs. The reaction mixture was basified with sat. Na2C03 solution, extracted with EtOAc. The combined organic layers were dried over anliydrous Na2S04, After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography to afford 4-bromo-3-(trifluoromedioxy)aniline (750 mg) as a yellow oil. MSfES m/z 256 (MH ).
Step 4: A mixture of 4-broino-3-(trifluoromethoxy)aniline (350 mg), (6-(2-(4-
(ethylsulfonyl)phenyl)acetamido)-2-methylpyridin-3-yl)boronic acid (intennediate 178a, 707 mg), PdCl2(dppf)-CH2Cl2 adduct (55.8 mg) and Cs2C03 (535 mg) in acetonitrile (3 mL) and water (1 mL) was bubbled with N2. The mixture was sealed in a reaction vessel and heated in the microwave at 100°C for 45 mins. As starting material still left, more (6-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2-
methylpyridin-3-yl)boronic acid (intermediate 178a, 707 mg), PdCl2(dppf)-CH2Cl2 adduct (55.8 mg) and Cs2COj (445 mg) were added to the above mixture. The reaction mixture was heated in the microwave at 100°C for another 45 mins. The mixture was filtered through celite, and washed with EtOAc. The filtrate was washed with brine and dried over anhydrous Na2S04. After concentration, the residue was purified by flash chromatography (EA : PE = 0: 1 to 1 : 1) to afford N-(5-(4-amino-2- (trifluoromethoxy)phenyl)-6-methylpyridin-2-yl)-2-(4-(ethylsnlfonyl)phenyl)acetamide (350 mg) as a yellow solid. MS(ES ) mJz 494 ( H*).
Step 5: A solution of N-(5-(4-amino-2-(trifluoromethoxy)phenyl)-6-methylpyridin-2-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (350 mg) in dichloromethane (1 mL) was added into the suspension of nitrosyl fiuoborate (249 mg) in dichloromethane (2 mL) at 0°C. The reaction mixture was stirred at 0°C for 2 hrs. Solvent was removed under reduced pressure, and the residue was dissolved into 1 ,2- dichlorobenzene (5 g). The mixture was heated to 130°C for 1 hr. The reaction mixture was cooled to 0 °C, quenched with ice-water, basified with sat. Na2CC>3 solution, extracted with DCM. The combined organic layers were dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography and then further purified by MDAP to afford 2-(4-(ethylsulfonyl)phenyl)-N-(5-(4-fluoro-2- (trifluoromethoxy)phenyl)-6-metliylpyridin-2-yl)acetamide, trifluoroacetic acid salt (93 mg) as a yellow solid. 'H-NMR (400 MHz, DMSO-d6) δ ppm 1.09 (t, J= 7.2 Hz, 3H), 2.21 (s, 3H), 3.27 (q, J = 7.2 Hz, 2H), 3.89 (s, 2H), 7.41 (m, 1H), 7.53 (m, 2H), 7.57 (d, J= 8.4 Hz, 1H), 7.63 (d, J = 8.0 Hz, 2H), 7.85 (d, J= 8.4 Hz, 2H), 7.96 (d, J = 8.4 Hz, 1H), 10.96 (s, 1H); 19F-NMR (376 MHz, DMSO- 6) δ ppm -56.73, -74.56, -1 10.39; MS(ES^| m/∑ 497 (MH+).
Example 179
N-(5-(2-(difluoromethoxy)-4-fIuorophenyI)-6-methyIpyridin-2-yI)-2-(4- (ethylsuIfonyl)phenyl)acetamide, trifluoroacetic acid salt

A mixture of l -bromo-2-(difluoromethoxy)-4-fluorobenzene (intennediate 176a, 75 mg), (6-(2- (4-(ethylsulfonyl)phenyl)acetamido)-2-methylpyridin-3-yl)boronic acid (intermediate 178a, 310 mg), PdCl2(dppf)-CH2Cl2 adduct (20.33 mg) and Na2COj (39.6 mg) in acetonitrile (2.4 mL)/water (0.8 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The mixture was filtered
through celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford N-(5-(2-(difIuoromethoxy)-4-fluorophenyl)-6-methylpyridin-2-yl)-2-(4- (ethyIsulfonyl)phenyi)acetarnide, trifluoroacetic acid salt (16 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i¾) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 2.21 (s, 3H), 3.27 (q, J= 7.2 Hz, 2H), 3.89 (s, 2H), 7.22 (t, J= 72 Hz, 1H), 7.24 (m, 2H), 7.40 (m, 1H), 7.53 (d, J= 8.4 Hz, 1H), 7.63 (d, 7= 8.4 Hz, 2H), 7.85 (d, 8.4 Hz, 2H), 7.94 (d, J= 8.4 Hz, 1H), 10.92 (s, 1H); 19F-NMR (376 MHz, DMSO-<i6) 5 ppm -73.84, -82.82, -111.03; MS(ES+) m/z 479 (ΜίΓ).
Example 180
N-(2,6-diiluoro-2,-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4-(ethylsuIfonyl)-3- fluorophenyl) acetamide

Step 1: To a suspension of NaH (60% in mineral oil, 2.7 g) in N,N-dimethylformamide (200 mL) was added benzyl ethyl malonate ( 5 g) at 0°C. The mixture was stirred at room temperature for 1 h under nitrogen. 2,4-Difluoro-l -nitrobenzene (10.7 g) was added at 0°C. The reaction mixture was stirred at room temperature overnight, and then poured into 10% ammonium chloride solution. The obtained solution was extracted with ethyl acetate (200 mL). The organic phase was washed with water and brine, dried over anhydrous magnesium sulfate, filtered, and concentrated. The residue was purified by column chromatography to give 1-benzyl 3-ethyl 2-(3-fluoro-4-nitrophenyl)malonate (1.2 g) as a yellow solid. JHNMR (400 MHz, DMSO-cQ δ ppm 1.14 (t, J= 7.2 Hz, 3H), 4.16 (m, 2H), 5.22 (m, 2H), 5.39 (s, 1H), 7.36 (m, 5H), 7.49 (d, J= 8.4 Hz, 1H), 7.62 (d, J= 12 Hz, 1H), 8.20 (m, 1H).
Step 2: To a solution of 1-benzyl 3-ethyl-2-(3-fluoro-4-nitrophenyl)malonate (1.1 g) and ammonium formate (1.2 g) in ethanol (20 mL) was added Pd/C (0.3 g). The mixture was stirred at 60°C overnight under nitrogen. The reaction mixture was filtered and the filtrate was concentrated to give ethyl 2-(4-amino-3-fluorophenyl)acetate (610 mg) as a yellow solid. MS(ES+) m/z 198 (MH*).
Step 3: A solution of sodium nitrite (158 mg) in 1 mL of water was added dropwise at 0°C to a suspension of ethyl 2-(4-amino-3-fluorophenyI)acetate (450 mg) in 5 mL of water and 1 mL of concentrated hydrochloric acid. The reaction mixture was stirred at the same temperature for another 45 minutes. This cold diazonium salt solution was then added dropwise at room temperature to a
mixture of potassium Oethyl carbonodithioate (424 mg), water (3 mL) and sodium carbonate solution (2 M, 3 mL). The mixture was heated to 45 °C until gas evolution stops. After cooling to room temperature, the pH was adjusted to 1 with concentrated hydrochloric acid. The solution was extracted with EtOAc (20 mL). Solvent was evaporated to give 2-(4-((ethoxycarbonothioyl)thio)-3- fluorophenyl)acetic acid (420 mg) as a dark red liquid. MSfES m/z 275 (MH .
Step 4: 2-(4-((Ethoxycarbonothioyl)thio)-3-fluorophenyl)acetic acid (420 mg) was taken up in 4 mL of ethanol. A solution of KOH (25S mgl) in water (3 mL) was added. The reaction was refluxed overnight. The major portion of ethanol was subsquently removed in vacuo. The aqueous phase was cooled with ice, and then acidified with concentrated hydrochloric acid. The residue was extracted with EtOAc. The organic phase was washed with brine, dried over anhydrous sodium sulphate, filtered, and concentrated to afford 2-(3-fluoro-4-mercaptophenyl)acetic acid (270 mg).
Step 5: To a solution of 2-(3-fluoro-4-mercaptophenyl)acetic acid (160 mg) in NN- dimethylformamide (5 mL) was added K2C03 (475 mg) and bromoethane (281 mg). The reaction mixture was stirred at RT overnight. The reaction mixture was partitioned between ethyl acetate (20 mL) and water (10 mL), The organic layer was washed with water and brine, dried with sodium sulphate, filtered, and concentrated to give ethyl 2-(4-(ethylthio)-3-fiuorophenyl)acetate (180 mg), MS(ES+) m/z 243 (MH*).
Step 6: Ethyl 2-(4-(ethylthio)-3-fluorophenyl)acetate (180 mg) was dissolved in
dichloromethane (10 mL). The solution was cooled to 0°C with an ice bath. mCPBA (385 mg) was added in portions, and the reaction mixture was stirred at RT overnight. DCM (10 mL) was added. The solution was washed with sat. sodium carbonate solution, water, brine, dried over sodium sulphate, filtered, and concentrated. The residue was purified by column chromatography to afford ethyl 2-(4-(ethylsulfonyl)-3-fluorophenyl)acetate (160 mg) as a yellow solid. MS(ES+) m/z 275 (MH1).
Step 7: To a solution of ethyl 2-(4-(ethylsulfonyl)-3-fIuorophenyl)acetate (130 mg) in ethanol
(6 mL) was added NaOH (2 M, 0.711 mL). The reaction mixture was stirred at room temperature overnight. Ethanol was removed under reduced pressure, and 6 mL of water was added. The aqueous phase was acidified with 6 M HCI to pH - 1 -2. The mixture was extracted with ethyl acetate for 3 times. The combined organic phases were washed with brine (10 mL), dried over sodium sulphate, filtered, and concentrated. The residue was purified by column chromatography to give 2-(4-
(ethylsulfonyl)-3-fluorophenyl)acetic acid (46 mg) as a yellow solid. 'HNMR (400 MHz, CDC¾) δ ppm 1.33 (t, J= 7.6 Hz, 3H), 3.34 (q5 J= 7.6 Hz, 2H), 3.78 (s, 2H), 7.26 (m, 2H), 7.93 (m, 1H); MS(ES+) m/z 247 (MH+).
Step 8: A mixture of 2f6-difluoro-2,-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (intermediate 116a, 50 mg), 2-(4-(ethylsulfonyI)-3-fluorophenyl)acetic acid (46.8 mg), EDC (49.7 mg) and HOBt (23.36 mg) in dichloromethane (1 mL) was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure and the residue was purified by MDAP to afford N- (2,6-difluoro-2'-(trifluoromethoxy)-[l , 1 '-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)-3- fluorophenyl)acetamide (36 mg) as a white solid.Ή-NMR (400 MHz, DMSO-£?6) δ ppm 1.15 (t, J= 7.2 Hz, 3H), 3.39 (q, J= 7.6 Hz, 2H), 3.90 (s, 2H), 7.45 (m, 3H), 7.53 (m, 4H), 7.62 (m, 1H), 7.82 (t, J= 8.0 Hz, 1H), 10.77 (s, 1H); 19F-NMR (376 MHz, DMSO- ) δ ppm -56.79, -110.50, 1 12.29; MSiES4 m/z 518 (MH+). Example 181
N-(2-chloro-4'-fluoro-2,-(trifluoromethoxy)-[l,l,-bip enyl]-4-yl)-2-(4- (ethylsulfony])phenyI)acetamide

Step 1: A mixture of 4-bromo-3-(trifluoromethoxy)aniline (300 mg), N-[3-chloro-4-(4,4,5,5- tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl]-2-[4-(ethylsulfonyl)phenyl]acetamide (intermediate 6b, 543 mg), PdC¾(dppf)-CH2Cl2 adduct (77 mg) and Cs2C03 (458 mg) in acetonitrile (4.5 mL)/water (1.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography to afford N-(4'-amino-2-chloro-2'- (trifluoromethoxy)-[l,r-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)plienyl)acetamide (164 mg) as a yellow oil. MSiES4) m/z 13 (MH4).
Step 2: A solution of N-(4'-amino-2-chloro-2'-(trifluoromemoxy)-[l, -biphenyl]-4-yl)-2-(4- (ethyIsulfonyl)phenyl)acetamide (164 mg) in dichloromethane (1 mL) was added into a suspension of nirrosyl fluoborate (112 mg) in dichloromethane (3 mL) at 0°C dropwise. The reaction mixture was stirred at 0°C for 2 hrs. DCM was removed under reduced pressure. 1 ,2-Dichlorobenzene (3 mL) was added to the residue. The mixture was stirred at 130°C for 1 hr. The reaction mixture was cooled to 0 °C, and water was added. Then the mixture was extracted with DCM. The combined organic layers were concentrated under reduced pressure. The residue was purified by short silica column: first eluted with petroleum ether to remove 1,2-dichIorobenzene, then eluted with EtOAc. The combined
EtOAc elutions were concentrated under reduced pressure and the residue was purified by MDAP to afford N-(2-clTloro-4'-flvioro-2'-(trifiuoromethoxy)-[l ,1 '-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (6.6 mg) as a yellow solid.Ή-NMR (400 MHz, OMSO-d6) δ ppm 1.10 (t, J= 7.6 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 7.32 (d, J= 8.4 Hz, 1H), 7.39 (m, 1H); 7.48 (m, 2H), 7.55 (dd, J= 2.0 Hz, 8.4 Hz, 1H), 7.62 (d, J= 8.4 Hz, 2H), 7.86 (d, J= 8.0 Hz, 2H), 7.95 (d, J= 2.0 Hz, 1H), 10.59 (s, 1H); I9F-NMR (376 MHz, DMSO-^) δ ppm -56.63, -110.02; MS(ES ;?/z 516 (MH+).
Example 183
Ar- 6-ethyl-5-(2-(trifluoromethoxy)phenyl)pyridin-2-yI)-2-(4-(ethy]sulfonyl)phenyl)acetamide

Step 1: To a solution of 6-etliylpyridin-2-amine (500 mg) in chloroform (10 mL) stirred under nitrogen at 0°C was added solid NBS (728 mg) portionwise. The reaction mixture was stirred at 0°C for 30 mins. Solvent was removed in vacuo. The residue was purified by column choromatography to give 5-bromo-6-ethylpyridin-2-amine (570 mg) as a yellow solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.21 (t, J= 7.6 Hz, 3H)} 2.76 (q, J= 7.6 Hz, 2H), 4.38 (b, 2H), 6.22 (d, J = 8.8 Hz, 1H), 7.46 (d, = 8.4 Hz, 1H); MSiES^ m/z 201 (MH+).
Step 2: A mixture of 5-bromo-6-ethylpyridin-2-amine (500 mg), Et3N (0.693 mL), EDC (572 mg), 2-(4-(ethylsuIfonyl)phenyl)acetic acid (intermediate la, 596 mg) and HOBt (457 mg) in dichloromethane (20 mL) was stirred at room temperature overnight. The mixture was diluted with DCM, washed with water, sat. Na2C03 and brine, then dried over anhydrous Na2SC>4. After concentration, the residue was triturated from EtOAc to afford N-(5-bromo-6-ethylpyridin-2-yl)-2-(4- (ethylsulfonyl)phenyl)acetamide (318 mg) as a yellow solid. 'H-NMR (400 MHz, CDC13) δ ppm 1.22 (m, 6H), 2.87 (m, 2H), 3.17 (m, 2H), 3.86 (s, 2H), 7.61 (m, 2H), 7.85 (m, 4H); MS(ES+) m/z 413 (MH+).
Step 3: A mixture of (2-(trifluoromethoxy)phenyl)boronic acid (1 15 mg), N-(5-bromo-6- ethylpyridin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (218 mg), PdCl2(dppf)-CH2Cl2 adduct (21.21 mg) and Cs2C03 (345 mg) in acetonitrile (3 mL) and water (1 mL) was heated in the microwave at 100°C for 30 mins. The mixture was filtered. The filtrate was concentrated and purified by TLC to afford 6-ethyl-5-(2-(trifluoromethoxy)phenyl)pyridin-2-amine (163 mg). The mixture of 2-(4-
(ethylsulfonyl)phenyl)acetic acid (intermediate la, 138 mg), EDC (133 mg), HOBt (106 mg), Et3N (0.161 mL) and 6-ethyl-5-(2-(trifluorome&oxy)phenyl)pyridin-2-amine (163 mg) in dichloromethane (20 mL) was stirred at room temperature overnight. The mixture was diluted with DCM, washed with water, sat. citric acid, sat. NaHC03 and brine, then dried over anliydrous Na2S04. After concentration, the residue was purified by MDAP to afford N-(6-ethyl-5-(2-(trifJuoromethoxy)phenyl)pyridin-2-yl)- 2-(4-(ethylsulfonyl)phenyl)acetamide (154.5 mg) as a white solid. Ή-NMR (400 MHz, CDC13) δ ppm 1.21 (t, J= 7.6 Hz, 3H), 1.28 (t, J= 7.6 Hz, 3H), 2.65 (m, 2H), 3.10 (q, J= 7.6 Hz, 2H), 3.94 (s, 2H), 7.23 (m, 1H), 7.38 (m, 2H), 7.49 (m, 1H), 7.63 (m, 2H), 7.70 (m, 1H), 7.91 (m, 2H), 8.25 (m, 1H); MSiES*) m/z 493 (MH+). Example 184
2-(4-(ethyIsulfonyl)phenyl)-7Y-(2-(pyridazin-3-yl)-2'-(trifluoromethoxy)-[l,l,-biphenyl]-4- yl)acetamide

Step 1: A mixture of 4J4,5,5-tetramethyl-2-(4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2-yl)- 1 ,3,2-dioxaborolane (intermediate 69a, 500 mg), 3-chloropyridazine (168 mg), Pd(Ph3P)4 (1 13 mg) and Na2C03 solution (2 M, 518 mg) in 1,4-dioxane (2 mL) was heated in the microwave at 100°C for 30 mins. The mixture was cooled to room temperature and filtered. The fitrate was concentrated in vacuo. The residue was partitioned between water and EtOAc. The aqueous layer was extracted with EtOAc. The organic layers were washed with brine, dried over Na2S(¾, and concentrated in vacuo. The residue was purified by column choromatography to afford 3-(4-nitro-2'-(trifluoromethoxy)-[l, - biphenyl]-2-yl)pyridazine (90 mg) as a red solid. Ή-NMR (400 MHz, CDC13) δ ppm 7.13 (m, 2H), 7.21 (m, 3H), 7.33 (m, 1H), 7.57 (d, J= 8.4 Hz, 1H)5 8.34 (dd, J= 2.4 Hz, 8.8 Hz, 1H), 8.64 (d, J= 2.4 Hz, 2H), 9.03 (d, J= 2.4 Hz, 1H).
Step 2: To a suspension of 3-(4-nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)pyridazine (80 mg) and zinc (145 mg) in methanol (5 mL) stirred in air at RT was added solid ammonium chloride (118 mg) in one charge. The reaction mixture was stirred at RT for 30 mins. The mixture was filtered and the filtrate was concentrated in vacuo to give 2-(pyridazin-3-yl)-2'-(trifluoromeftoxy)-[l,l'- biphenyl]-4-amine (74 mg) as a brown solid.
Step 3: A mixture of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 37.6 mg), EDC (36.1 mg), HOBt (28.8 mg), Et3N (0.044 mL) and 2-(pyridazin-3-yl)-2,-(trifluoromethoxy)-[l,l'- biphenyl]-4-amine (52 mg) in DCM (lOmL) was stirred at room temperature overnight. The mixture was diluted with DCM, washed with water, sat. citric acid, sat. Na2C03 solution and brine, then dried over anhydrous Na2S04. After concentration, the residue was purified by MDAP to afford 2-(4- (ethyl sulfonyl)phenyl) -N-(2-(p yri dazin-3 -yl) -2'-(tri fluoromethoxy)-[ 1 , 1 -biphenyl] -4-yl)acetamide (23.5 mg) as a yellow solid. lH-NMR (400 MHz, MeOD-<¾) δ ppm 1.20 (t, J= 7.6 Hz, 3H), 3.18 (q, J = 7.6 Hz, 2H), 3.89 (s, 2H), 7.13 (m, 1H), 7.43 (m, 5H), 7.64 (m, 3H), 7.86 (m, 3H), 8.03 (d, J= 2.0 Hz, 1H), 9.07 (dd, J= 1.6 Hz, 5.2 Hz, 1H); MSCES" m/z 542 (MlT). Example 185
2- (4-(ethylsuIfonyl)p henyl)-N-(5-methyl-6-(2- (trifluoromethoxy)p heny l)pyridin-3-yI)acetamide, hydrochloride

Step 1: To a solution of 2-bromo-3-methyl-5-nitropyridine (651 mg), (2- (trifluoromethoxy)phenyl)boronic acid (927 mg) and Pd(Ph3P)4 (347 mg) in 1,4-dioxane (15 mL) and water (5 mL) stirred under nitrogen at 20°C was added solid Na2C03 (954 mg) in one charge. The reaction mixture was stirred at 100°C for 16 hrs. Solvent was evaporated in vacuo and the mixture was dissolved in EA (50 mL). The organic phase was washed with water (25 mL) and saturated brine (25 mL), dried over sodium sulphate and evaporated in vacuo. The residue was purified by TLC to afford 3-methyl-5-nitro-2-(2-(trifluoromethoxy)phenyl)pyridine (534 mg) as a yellow solid. Ή-NMR (400 MHz, MeOD-<¾) δ ppm 2.22 (s, 3H), 7.44 (m, 3H), 7.55 (m, 1H), 8.51 (m, 1H), 9.18 (d, J= 2.8 Hz, 1H); MSfES^ m/z 299 (MH÷).
Step 2: To a solution of 3-methy]-5-nitro-2-(2-(trifluoromethoxy)phenyl)pyridine (298 mg) and zinc (654 mg) in methanol (10 mL) stirred in air at 20°C was added solid ammonium chloride (535 mg) in one charge. The reaction mixture was stirred at 20°C for 30 mins. Then the reaction mixture was filtered and the filterate was concentrated. The solid was dissolved in EA (60 mL) and filtered. The filtrate was concentrated to afford 5-methyl-6-(2-(trifluoromethoxy)phenyl)pyridin-3-amine (263 mg) as a colorless oil. MS(ES+) m/z 269 (MH+).
Step 3: To a solution of 5-methyl-6-(2-(trifluoromethoxy)phenyl)pyridin-3-aniine (263 mg), 2- (4-(ethylsulfonyl)phenyl)acetic acid (intermediate a, 269 mg) and PyBOP (612 mg) in Njf- dimethylformamide (5 mL) stirred under nitrogen at 20°C was added DIPEA (0.514 mL) dropwise. The reaction mixture was stirred at 20°C for 16 hrs. Then the reaction mixture was poured into water (25 mL) and extracted with EA. The organic phase was washed with saturated brine (25 mL), dried over sodium sulphate and evaporated in vacuo. The crude product was purified by MDAP to afford 2- (4-(etliylsulfonyl)phenyl)-N-(5-methyl-6-(2-(trifluoiOmethoxy)phenyl)pyridin-3-yl)acetamide, hydrochloride (213 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i¾) 6 ppm 1.10 (t, 7.6 Hz, 3H), 2.08 (d, J= 6.5 Hz, 3H), 3.29 (m, 2H), 3.94 (s, 2H), 7.55 (m, 3H), 7.66 (m, 3H), 7.86 (d, J= 4.0 Hz, 2H), 8.27 (d, J= 1.6 Hz, 1H), 8.8 (d, J= 1.6 Hz, 1H), 11.25 (br s, 1H); MS(ES+) m/z 479 (MH+).
Example 186
2-(4-(ethyIsuIfonyl)phenyI)-N-(6-(2-(trifluoromethoxy)phenyl)pyridazin-3-yI)acetamide, hydrochloride

(ethylsulfonyl)phenyl)acetamide (236 mg) as a colorlss oil. MS(ES+) m/z 340 (MH+).
Step 2: To a solution of N-(6-chloropyridazin-3-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (200 mg), (2-(trifluoromethoxy)phenyl)boronic acid (182 mg) and PdCl2(dppf) (43.1 mg) in 1 ,4-dioxane (12 mL) and water (3 mL) stirred under nitrogen at 20°C was added CS2CO3 (384 mg) in one charge. The reaction mixture was stirred at 100°C for 16 hrs. The mixture was filtered and the filtrate was concentrated. The mixture was dissolved with EA (50 mL). The organic phase was washed with water (10 mL) and saturated brine (25 mL), dried over sodium sulphate and evaporated in vacuo to give the crude product as a brown oil. The crude product was purified by prep-HPLC to obtain 2-(4- (ethylsulfonyl)phenyl)-N-(6-(2-(trifluoromethoxy)phenyl)pyridazin-3-yl)acetamide, hydrochloride
(10.2 mg) as a white solid. Ή-NMR (400 MHz, MeOD-i¾ δ ppm 1.21 (t, J= 7.6 Hz, 3H), 3.18 (d, J = 7.5 Hz, 2H), 4.03 (s, 2H), 7.54 (m, 2H), 7.63 (m, 3H), 7.81 (d, /= 3.8 Hz, 1H), 7.86 (m, 2H), 8.12 (d, J= 9.2 Hz, 1H), 8.54 (d, J= 9.2 Hz, 1H); MS(ES+) m/z 466 (MH
Example 187
2-(4-(ethylsulfonyl)phenyI)-iV-(5-methyl-6-(2-(trifluoromethoxy)phenyI)pyridazin-3- yl)acetamide

Step 1: A mixture of 3,6-dichloro-4-methylpyridazine (5 g) and ammonia hydrate (17 mL) in a sealed tube was heated to 120°C for 16 hrs. The mixture was concentrated in vacuo. The crude product was recrystallized from EtOAc to give 6-chloro-5-methylpyridazin-3-amine (2.85 g) as a gray solid. Ή-NMR (400 MHz, MeOD-i/4) δ ppm 2.26 (s, 3H), 6.87 (m, 1H); MS(ES+) m/z 144 (MH4).
Step 2: A mixture of (2-{trifluoromethoxy)phenyl)boronic acid (753 mg), 6-chloro-5- methylpyridazin-3-amine (500 mg), PdCL dppfJ-CILCL. adduct (139 mg) and Cs2C03 (2269 mg) in acetonitrile (6 mL) and water (2 rnL) was heated in the microwave at 100°C for 30 mins. The mixture was filtered and the filtrated was concentrated in vacuo. The residue was purified by TLC to afford 5- methyl-6-(2-(trifluoromethoxy)phenyl)pyridazin-3-amine (220 mg) as a brown solid. MSiES^) m/z 270 (MH+).
Step 3: A mixture of 5-methyl-6-(2-(trifluoromethoxy)phenyl)pyridazin-3-amine (220 mg), Et3N (0.228 mL) and 2-(4-(etliylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 242 mg) in dichloromethane (10 mL) was stirred for 2 hrs. The mixture was washed with 10% citric acid solution (2 mL), saturated sodium bicarbonate solution (2 mL), and brine (2 mL), dried over sodium sulphate and evaporated in vacuo. The residue was purified by MDAP to afford 2-(4-(ethylsulfonyl)phenyl)-N- (5-methyl-6-(2-(trifluoromethoxy)phenyl)pyridazin-3-yl)acetamide (70.8 mg) as a yellow solid. Ή- MR (400 MHz, MeOD-<¾) δ ppm 1.21 (t, J= 7.6 Hz, 3H), 2.25 (s, 3H), 3.21 (q, J= 7.6 Hz, 2H), 4.01 (s, 2H), 7.56 (m, 3H), 7.65 (m, 3H), 7.89 (m, 2H), 8.47 (s, 1H); MS(ES+) m/z 480 (MH+).
Example 188
2-(4-(ethyIsu]fonyl)phenyl)-N-(2-(2-methylpyrimidin-5-yl)-2,-(trifluoromethoxy)-Il,l'- biphenyl] -4-yl)acetamide, h drochloride

Step 1: A solution of Na2C03 (155 mg), 5-bromo-2-methylpyrimidine (85 mg), Pd(Ph3P)4 (1 13 mg) and 4,4,5,5-tetrame l-2-(4-nitro-2'-(trifluoromethoxy)-[l,l1-biphenyl]-2-yl)-l ,3,2- dioxaborolane (intermediate 69a, 400 mg) in 1,4-dioxane (15 mL) and water (5 mL) was added into a vessel. The reaction vessel was sealed and heated in the microwave at 100°C for 45 mins. Solvent was evaporated in vacuo. Solid was dissolved in EA (50 mL), washed with water (25 mL) and brine (25 mL), dried over sodium sulphate and evaporated in vacuo. The residue was purified by prep-TLC to give 2-methyl-5-(4-nitro-2,-(trifluoroniethoxy)-[l, -biphenyl]-2-yl)pyrimidine (176mg) as a white solid. MS(ES+) m/z 376 (MH+).
Step 2: To a solution of 2-methyl-5 -(4-nitro-2'-(trifluoromethoxy)-[ 1 , 1 '-biphenyl] -2- yl)pyrimidine (170 mg) and zinc (296 mg) in methanol (5 mL) stirred in air at 20°C was added solid ammonium chloride (242 mg) in one charge. The reaction mixture was stirred at 25 °C for 30 mins. The mixture was filtered and the filtrate was concentrated. The white solid was dissolved in EA (40 mL) and filtered. Solvent was concentrated to give 2-(2-methylpyrimidin-5-yl)-2'-(trifluoromethoxy) -[l,r-biphenyl]-4-amine (96 mg) as a colorless oil. MS(ES+) m/z 346 (MH+).
Step 3: To a solution of 2-(2-memylpyrimidin-5-yl)-2'-(trifluoromethoxy)-[l, -biphenyl]-4- amine (96 mg) and TEA (0.116 mL) in dicliloromethane (3 mL) stirred under nitrogen at 0°C was added a solution of 2-(4-(ethylsulfonyI)phenyl) acetyl chloride (intermediate 170a, 103 mg) in dicliloromethane (3 mL) dropwise. The reaction mixture was stirred at 0°C for 1 hrs. The mixture was allowed to warm to 20°C and stirred at 20°C for another 2 hrs. Solvent was evaporated in vacuo. The residue was purified by MDAP to afford 2-(4-(ethyIsulfonyl)phenyl)-N-(2-(2-metliylpyrimidin-5-yl)- 2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)acetamide, hydrochloride (12.5 mg). Ή-NMR (400 MHz, DMSO- δ ppm 1.10 (t, J- 7.6 Hz, 3H), 2.65 (s, 3H), 3.12 (q, J= 7.2 Hz, 2H), 3.89 (d, /= 1.6 Hz, 2H), 7.17 (t, J= 7.6 Hz, 1H), 7.43 (m} 4H), 7.65 (d, J= 4.0 Hz, 2H), 7.73 (d, J= 2.0 Hz, 1H), 7.87 (m, 3H), 8.48 (s, 2H); MS(ES+) m/z 556 (MH^).
Example 189
2-(4-(ethyIsulfonyl)phenyl)-Ar-(2-(isoxazol-5-yl)-2'-(trifluoromethoxy)-[l,l,-biphenyl]-4- yl)aceramide

Step 1: A solution of (2-(trifluoromethoxy)phenyl)boronic acid (1.083 g), l-(2-chloro-5- nitrophenyl)ethanone (1 g), Cs2C03 (1.959 g) and PdCl2(dppf) (0.092 g) in water (5 mL) and acetonitrile (15 mL) was refluxed for 50 mins. The mixture was filtered and the filtrate was concentrated to give l-(4-nitro-2,-(trifluoromethoxy)-[l, -biphenyI]-2-yl)ethanone (1 g). MSfES ) m/z 326 (MH4).
Step 2: A solution of l-(4-nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)ethanone (100 mg) in DMF-DMA (4] 2 μ]) was refluxed for 6 hrs. After cooling to room temperature, the mixture was dissolved in EA, washed with water and brine, dried over sodium sulphate and evaporated in vacuo to give (£)-3-(dimethylainino)-l-(4-nitro-2'-(rrifluoromethoxy)-[l ,l'-biphenyl]-2-yl)prop-2-en-l^ (100 mg). MS(ES+) m/z 381 (MH+).
Step 3: To a solution of (/¾-3-(dimethylamino)-l-(4-nitro-2'~(tiifluoromethoxy)-[l,r-biphenyl]- 2-yl)prop-2-en-l-one (100 mg) in ethanol (3 mL) was added hydroxylamine hydrochloride (18.27 mg) in one portion. The mixture was refluxed for 12 hrs, and concentrated in vacuo. The residue was partitioned between water and EA. The organic layer was washed with saturated brine, dried over sodium sulphate and evaporated in vacuo to give 5-(4-nitro-2,-(trifluoromethoxy)-[l,l'-biphenyl]-2- yI)isoxazole (80 mg). MS(ES+) m/z 351 (MH+).
Step 4: To a solution of 5-(4-nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)isoxazole (80 mg) and zinc (149 mg) in methanol (5 mL) was added ammonium chloride (48,9 mg). The mixture was stirred for 30 mins, and then filtered. The filtrate was concentrated to give 2-(isoxazol-5-yl)-2'- (trifluoromethoxy)-[l,r-biphenyl]^-amine (72 mg). MSfES ) m/z 321 (MH+).
Step 5: To a solution of 2-(isoxazol-5-yl)-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (72 mg) and DIPEA (0.079 mL) in dichloromethane (10 mL) was added 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 61.0 mg). The mixture was stirred for 2 hrs, concentrated in vacuo. The residue was purified by prep-TLC to afford 2-(4-(ethylsulfonyl)phenyl)-N-(2-(isoxazol-5-yl)-2'- (trifluoromethoxy)-[l,l'-biphenyl]-4-yl)acetamide (49 mg) as a grey solid. Ή-NMR (400 MHz,
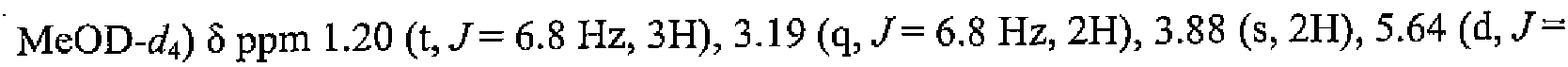 2.0 Hz, 1H), 7.33 (m, 3H), 7.40 (m, 1H), 7.42 (m, 1H), 7.49 (m, 2H) , 7.63 (d, J~ 8.4 Hz, 1H), 7.84 (m, 2H), 8.14 (d, J= 2.0 Hz, 2H), 8.18 (d, J= 2.0 Hz, 1H); MS(ES+) m/z 531 (MH+).
Example 190
2.0 Hz, 1H), 7.33 (m, 3H), 7.40 (m, 1H), 7.42 (m, 1H), 7.49 (m, 2H) , 7.63 (d, J~ 8.4 Hz, 1H), 7.84 (m, 2H), 8.14 (d, J= 2.0 Hz, 2H), 8.18 (d, J= 2.0 Hz, 1H); MS(ES+) m/z 531 (MH+).
Example 190
2-(4-(ethylsulfonyl)phenyl)-N-(2-(pyridin-2-yl)-2'-(trifluoromethox )-[l,l'-biphenyl]-4- yl)acetamide, hydrochloride

Step 2: To a solution of 2-(4-nitro-2'-(trifluorometlioxy)-[l, -biphenyl]-2-yl)pyridine (100 mg) in methanol (15 mL) was added zinc (181 mg). Ammonium chloride (148 mg) was added in portions. After 1 h, the mixture was filtered. The filtrate was diluted with water, extracted with EA, dried over Na2S04! concentrated to afford 2-(pyridin-2-yI)-2'-(trifluoromethoxy)-[l,lLbiphenyl]-4-amine (60 mg). MS(ES+) m/z 331 (MH4).
Step 3: To a solution of 2-(pyridin-2-yl)-2,-(trifluoromethoxy)-[l,l,-bipnenyl]-4-amine (40 mg) in dichloromethane (5 mL) was added 2-(4-(ethylsulfonyl)phenyl)acety] chloride (intermediate 170a, 29.9 mg), triethylamine (0.034 mL). After 3 hrs, the mixture was diluted with water, extracted with DCM (20 mL x 2), dried over Na2S04, filtered and concentrated. The residue was purified by MDAP to afford 2-(4-(etliylsulfonyl)phenyl)-N-(2-(pyridin-2-yl)-2'-(trifluoromethoxy)-[l , Γ-biphenylH- yl)acetamide, hydrochloride (24.9 mg). Ή-NMR (400 MHz, CDCL) δ ppm 1.12 (t, J = 7.6 Hz, 3H), 3.03 (q, J =7.6 Hz, 2H), 3.89 (s, 2H), 7.06 (d, J= 7.6 Hz, 1H), 7.16 (d, J= 7.6 Hz, 1H), 7.25 (m, 1H), 7.26 (m, 1H), 7.32 (m, 1H), 7.60 (d, 7= 7.6 Hz, 2H), 7.65 (m, 1H), 7.70 (d, J= 7.6 Hz, 2H), 7.90 (m, 1H), 8.14 (d, J= 7.6 Hz, 2H), 8.39 (s, 1H), 8.73 (s, 1H), 10.90 (s, 1H); MSfES"1) m/z 541 (MH+). Example 191
2-(4-(ethylsulfonyl)phenyl)-N-(2-(l-methyl-lH-pyrazol-5-yl)-2'-(trifluoromethoxy)-[l,l'- biphenyl]-4-yl)acetamide, hydrochloride

Step 1: To a solution of l-methyl-S^^jS^-tetramethyl-l^^-dioxaborolan- -ylJ-lH-pyrazole (393 mg), 2-chloro-4-nitro-2'-(trifluoromethoxy)-l,l '-biphenyl (500 mg, see step 1 for synthesis of intermediate 69a) and CS2CO3 (1026 mg) in 1,4-dioxane (15 lnL) and water (5 mL) stirred under nitrogen at room temperature was added PdCl2(dppfh (64.4mg) in one charge. The reaction vessel was sealed and heated in the microwave at 120°C for 30 mins. After cooling to room temperature, the mixture was filtered through celite, washed with C¾CN and EtOAc. The filtrate was concentrated under reduced pressure and the residue was partitioned between water and EtOAc, The organic layer was washed with brine and dried over anhydrous
 After filtration, solvent was removed in vacuo. The residue was purified by prep-TLC to afford 1 -methyl-5-(4-nitro-2'-(trifluorometlioxy)- [l,l'-biphenyl]-2-yl)-lH-pyrazole (200 mg). MS(ES+) m/z 364 (ΜΗ4).
After filtration, solvent was removed in vacuo. The residue was purified by prep-TLC to afford 1 -methyl-5-(4-nitro-2'-(trifluorometlioxy)- [l,l'-biphenyl]-2-yl)-lH-pyrazole (200 mg). MS(ES+) m/z 364 (ΜΗ4).
Step 2: To a solution of l-methyl-5-(4-nitro-2'-(trifluoromethoxy)biphenyl-2-yl)-lH-pyrazole (200 mg) in methanol (5 mL) was added Raney Ni (16.29 mg,). The reaction mixture was stirred under ¾ at room temperature for 4 hrs. The mixture was filtered and the filtrate was concentrated in vacuo to give 2-(l-methyl-lH-pyrazol-5-yl)-2'-(trifluoromethoxy)biphenyI-4-amine (150 mg).
MS(ES+) m/z 334 (MH+).
Step 3: To 2-(4-(ethylsuIfonyl)phenyl)acetic acid (intermediate la, 164 mg) was added thionyl chloride (663 mg). After stirring for 1 lir, thionyl chloride was removed under vacuum. The residue was dissolved in dichloromethane (1 mL) and added dropwise to a solution of 2-(l -methyl- lH- pyrazol-5-yl)-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (120 mg) and triethylamine (0.151 mL) in dichloromethane (3 mL). The reaction mixture was stirred for 4 hrs. Solvent was evaporated in vacuo and the residue was purified by MDAP to afford 2-(4-{ethylsulfonyl)phenyl)-N-(2-(l-methyl- lH-pyrazol-5-yl)-2'-(trifluoronietlioxy)-[l, -biphenyl]-4-yI)acetamide, hydrochloride {25.8 mg). Ή- NMR (400 MHz, MeOD-c 4) δ ppm 1.21 (t, J= 7.2 Hz, 3H), 3.18 (q, J= 7.2 Hz, 3H), 3.72 (s, 2H), 3.88 (s, 2H), 5.92 (dd, /= 0.8 Hz, 2.0 Hz, 1H), 7.19 (m, 1H), 7.34 (m, 2H), 7.38 (m, 1H), 7.42 (d, 7 = 8.4 Hz, 1H), 7.51 (d, J= 2.8 Hz, 1H), 7.64 (dd, J= 4.0 Hz, 6.0 Hz, 1H), 7.69 (d, 7= 2.4 Hz, 2H), 7.71(d, J= 2.4 Hz, 1H), 7.87 (m, 3H); MS(ES+) m/z 544 (MH+).
Example 192
2-(4-(ethyIsulfonyl)phenyl)-N-(2~(2-m^
biphenyl]-4-yl)acetamide

Step 1: A mixture of 4,4,5 ,5-tetramethyl-2-(4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2-yl)- 1,3,2-dioxaboroIane (intermediate 69a, 500 mg), 4-chloro-2-methylpvrimidine (189 mg), Pd(Ph3P)4 (113 mg) and 2 M of aqueous Na2C03 ( 18 mg) in 1,4-dioxane (10 mL) was stirred in the microwave at 100°C for 30 mins. The mixture was cooled to room tempareture and filtered. The filtrate was concentrated in vacuo. The residue was partitioned between water and EtOAc. The aqueous layer was extracted with EtOAc. The organic layers were washed with brine, dried over Na2S04 and concentrated in vacuo. The residue was purified by column choromatography to give 2-methyl-4-(4- nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)pyrimidine (90 mg) as a yellow solid. Ή-NMR (400 MHz, CDClj) δ ppm 2.66 (s, 3H), 6.75 (d, /= 7.6 Hz, 1H), 7.26 (m, 1H), 7.30 (m, 2H), 7.41 (m, 1H)} 7.58 (d, J= 8.4 Hz, 1H), 8.35 (dd, J= 2.4 Hz, 8.4 Hz, 1H), 8.43 (d, J= 5.2 Hz, 1H), 8.65 (d, /= 2.4 Hz, 1H).
Step 2: To a suspension of 2-methyl-4-(4-nitro-2'-(trifluoromethoxy)-[l,l '-biphenyl]-2- yl)pyrimidine (90 mg) and zinc (157 mg) in methanol (5 mL) stirred at RT was added ammonium chloride (128 mg) in one charge. The reaction mixture was stirred at RT for 30 mins. The mixture was filtered and the filtrate was concentrated in vacuo to give 2-(2-memylpyrimidin-4-yl)-2'- (trifluoromethoxy)-[l, -biphenyl]-4 -amine (100 mg) as a yellow solid. 'H-NMR (400 MHz, MeOD- d4) 5 ppm 2.53 (s, 3H), 6.89 (m, 2H), 7.04 (d, J= 2.4 Hz, 1H), 7.13 (m, 2H), 7.31 (m, 3H), 8.34 (d, J = 5.6 Hz, 1H).
Step 3: To a solution of 2-(4-(ethyIsulfonyl)phenyl)acetic acid (intermediate la, 83 mg) in dichloromethane (10 mL) stirred under nitrogen at 0°C was added a solution of SOCl2 (0.5 mL) dropwise. The reaction mixture was stirred at RT for 2 hrs and then concentrated in vacuo. To a solution of 2-(2-inethylpyrirnidin-4"yl)-2'-(trifIuoromethoxy)-[l, -biphenyl]-4-amine (100 mg) and Et3N (0.121 mL) in dichloromethane (10 mL) stirred under nitrogen at 0°C was added a solution of die above residue in DCM (2 mL) dropwise. The reaction mixture was stirred at 0°C for 2 hrs.
Solvent was washed with water (2 mL), 1 M HC1 (1 mL), saturated sodium bicarbonate solution (2
mL) and brine (2 mL), dried over sodium sulphate and evaporated in vacuo. The crude product was purified by MDAP to give 2-(4-(ethylsnlfonyl)phenyl)-N-(2-(2-methylpyrimidin-4-yl)-2'- (trifIuoromethoxy)-[l, -biphenyl] -yl)acetamide (30.4 mg) as a yellow solid. 'H-NMR (400 MHz, MeOD-<¾) δ ppm 1.20 (t, J= 7.6 Hz, 3H), 2.67 (s, 3H), 3,19 (q, J= 7.6 Hz, 2H), 3.90 (s, 2H), 7.22 (m, 1H), 7.44 (m, 5H), 7.64 (d, J= 8.8 Hz, 2H), 7.81 (dd, J= 2.4 Hz, 8.4 Hz, 1H), 7.89 (m, 2H), 8.33 (d, J= 2.4 Hz, 1H), 8.69 (d, J= 2.4 Hz, 1H); MStES" miz 556 (MH4).
Example 193
/y-(3-chloro-4-(lH-indol-7-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt

To a mixure of N[3-chloro-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 100 mg), 7-bromo-lH-indole (50.7 mg) and sodium carbonate solution (2 M, 0.431 mL) in DMF (3 mL) was added PdCl2(dppf)-CH2Cl2 adduct (8.80 mg) under nitrogen. The mixture was heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and extracted with EA for three times. The organic layers were dried, filtered through silica gel, and evaporated in vacuo. The residue was purified by MDAP to afford N(3-chloro-4-(lH-indol-7-yl)phenyl)-2-(4-(ethylsulfonyI)phenyl)acetamide, trifluoroacetic acid salt (18 mg). ]H-NMR (400 MHz, DMS<_W6) 5 ppm 1.04 (t, J= 7.3 Hz, 3H), 3.22 (q, J = 7.3 Hz, 2H), 3.79 (s, 2H), 6.41 (m, 1H), 6.88 (d, J= 6.3 Hz, 1H), 6.99 (t, J= 7.5 Hz, 1H), 7.19 (t, J= 2.8 Hz, 1H), 7.32 (d, J= 8.3 Hz, 1H), 7.50 (m, 2H), 7.57 (d, J= 8.3 Hz, 2H), 7.81 (d, J= 8.3 Hz, 2H), 7.92 (d, J- 2.0 Hz, 1H), 10.50 (s, 1H), 10.68 (br s, 1H); l9F-NMR (376 MHz, DMSO-<¾ δ ppm -73.96;
Example 194
JV-(3-chloro-4-(imidazo[l,2-a]pyridin-5-yI)phenyl)-2-{4-(ethylsulfonyI)phenyl)acetamide, trifluoroacetic acid salt
 To a mixture of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-y])phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 100 mg), 5-bromoimidazo[l,2-a]pyridine (51.0 mg) and sodium carbonate solution (2 M, 0.431 lnL) in DMF (3 mL) was added PdCl2(dppf)-CH2Cl2 adduct (8.80 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and then extracted with EA for three times. The organic layer was dried and filtered through silica gel, evaporated in vacuo. The residue was purified by MDAP to afford N-(3-chloro-4-(imidazo[l,2-a]pyridin-5-yl)phenyl)-2-(4- (emylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt (5.4 mg). 'H-NMR (400 MHz, DMSO-i/6) δ ppm 1.04 (t, J= 7.3 Hz, 3H), 3.23 (m, 2H), 3.83 (s, 2H), 7.34 (d, J= 7.0 Hz, 1H), 7.56 (m, 3H), 7.66 (m, 2H), 7.86 (m, 4H), 8.04 (s, 2H), 10.74 (s, 1H); 19F-NMR (376 MHz, DMSO- ) 6 ppm -73.70; MS(ES H/-: 454 (MH+).
To a mixture of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-y])phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 100 mg), 5-bromoimidazo[l,2-a]pyridine (51.0 mg) and sodium carbonate solution (2 M, 0.431 lnL) in DMF (3 mL) was added PdCl2(dppf)-CH2Cl2 adduct (8.80 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and then extracted with EA for three times. The organic layer was dried and filtered through silica gel, evaporated in vacuo. The residue was purified by MDAP to afford N-(3-chloro-4-(imidazo[l,2-a]pyridin-5-yl)phenyl)-2-(4- (emylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt (5.4 mg). 'H-NMR (400 MHz, DMSO-i/6) δ ppm 1.04 (t, J= 7.3 Hz, 3H), 3.23 (m, 2H), 3.83 (s, 2H), 7.34 (d, J= 7.0 Hz, 1H), 7.56 (m, 3H), 7.66 (m, 2H), 7.86 (m, 4H), 8.04 (s, 2H), 10.74 (s, 1H); 19F-NMR (376 MHz, DMSO- ) 6 ppm -73.70; MS(ES H/-: 454 (MH+).
Example 195
N-(3-chloro-4-(lH-indol-4-yl)phenyl)-2-(4-(ethylsulfonyI)phenyl)acetamide, trifluoroacetic acid salt

To a mixture of N-[3-chloro-4-(4,4,5,5-tetrametl-iyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 200 mg), 4-bromo-lH-indole (101 mg) and sodium carbonate solution (2 M, 0.862 mL) in DMF (3 mL) was added PdCl2(dppf)-CH2Cl2 adduct (17.61 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and extracted with EA for three times. The organic layer was dried and filtered through silica gel, evaporated in vacuo. The residue was purified by MDAP to afford N-(3-chloro-4-(lH-indol-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt (52 mg). Ή-NMR (400 MHz, DMSO-t¼) δ ppm 1.11 (t, J= 7.4 Hz, 3H), 3.28 (m, 2H), 3.86 (s, 2H), 6.11 (s, 1H), 6.93 (d, J= 7.0 Hz, 1H), 7.16 (m, 1H), 7.38 (m, 3H), 7.56 (dd, J= 8.5 Hz, 2.0 Hz, 1H), 7.64 (d, J= 8.3 Hz, 2H), 7.87 (d, J= 8.3 Hz, 2H), 7.97 (d, J- 2.0 Hz, 1H), 10.55 (s, 1H), 11.23 (br s, 1H); 19F-NMR (376 MHz, DMSO-i ) δ ppm -73.89; MS(ES+) m/z 453 (MH*).
Example 196
N-(3-chIoro-4-(imidazo[l^-a]pyridin-8-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt

To a mixture of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3;2-dioxaborolan-2-yl)phenyI]-2-[4- (ethylsulfonyl)phenyl]acetamide (intermediate 6b, 100 mg), 8-bromoimidazo[l ,2-a]pyridine (51.0 mg) and sodium carbonate solution (2 Mf 0.431 mL) in DMF (3 mL) was added PdCl (dppf)-CH2Cl2 adduct (8.80 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and extracted with EA for three times. The organic layer was dried and filtered through silica gel, evaporated in vacuo. The residue was purified by MDAP to afford N-(3-chloro -4-(imidazo[l, 2-a]pyridin-8-yl)phenyI)-2-(4- (ethylsulfonyl)phenyl)acetamide, tnfluoroacetic acid salt (4 mg). 'H-NMR (400 MHz, DMSO-i/6) δ ppm 1.1 1 (t, J= 7.3 Hz, 3H), 3.30 (m, 2H), 3.89 (s, 2H), 7.54 (d, J= 8.3 Hz, 2H), 7.65 (m} 3H), 7.81 (s, 1H), 7.87 (d, J= 8.3 Hz, 2H), 8.05 (s, 1H), 8.13 (s, 1H), 8.42 (s, 1H), 8.93 (d, J= 6.3 Hz, 1H), 10.72 (s, 1H); 19F-NMR (376 MHz, DMSO-i¾) δ ppm -73.78; MS(ES+) m/z 454 ( H+).
Example 197
l-(4-(2-(4-(ethyIsuIfonyl)phenyl)acetamido)-2'-(trifluoromethoxy)-[l,l'-biphenyl]-2-yI)-lH- pyrrole-3-carboxylic acid

Intermediate 197a: 2-fluoro-4-nitro-2'-ftrifluoromethoxy)-l .1 '-biphenyl
Step 1: To a solution of 2-fluoro-4-nitroaniline (10 g) in acetonitrile (100 mL) was added copper(I) bromide (0.919 g), tosic acid (12.18 g), tetra-ier/-butylammonium bromide (41.3 g). The mixture was stirred at 20°C for 2 hrs. The mixture was filtered. The filtrate was concentrated and purified by column chromatography to afford l-bromo-2-fluoro-4-nitrobenzene (7 g). MS(ES4) m/z 242 (MNa+).
Step 2: To a solution of 1 -bromo-2-fIuoro-4-nitrobenzene (400 mg) in 1,4-dioxane (3 mL) was added Na2C03 (385 mg), (2-(trifluorometlioxy)phenyl)boronic acid (374 mg), PdCI2(dppf) (66.5 mg) and water (1 mL). The mixture was degassed and reacted under N2 at 120°C for 2 hrs. The mixture was diluted with water, extracted with EtOAc (20 mL x 2), dried over Na2S04, concentrated and
purified by column to afford 2-fluoro-4-nitro-2'-(trifluoromethoxy)-l, -biphenyl (300 mg). MS(ES+) m/z 302 (MH+).
Preparation of l-(4-(2-(4-fethvIsulfonvnphenyl)acetamido)-2'-(trifluoromethoxy)-|"l.l'-biphenyl1-2- yI -lH-pyrrole-3-carboxylic acid
Step 1: To a solution of 2-fluoro-4-nitro-2'-(trifluoromethoxy)-l,l'-biphenyl (intermediate 197a,
0.3 g) in acetonitrile (10 mL) was added methyl lH-pyrrole-3-carboxylate (0.150 g) and Cs2C03
(0.649 g). The mixture was stirred at 100°C for 12 hrs. The mixture was filtered. The filtrate was concentrated and purified by column chromatography (PE : EA = 30: 1 to 5: 1) to afford methyl I-(4- nitro-2'-(trifluoromethoxy)-[l;r-biphenyl]-2-yl)-lH-pyrrole-3-carboxylate (150 mg). MS(BS+) m/z 407 (MH÷).
Step 2: To a solution of methyl l-(4-nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)-lH- pyrrole-3-carboxylate (150 mg) in methanol (5 mL) was added zinc (241 mg). Ammonium chloride (197 mg) was added dropwise. After 1 hr, the mixture was filtered. The filtrate was diluted with water, extracted with EtOAc (20 mL x 2), dried over Na2S04, and concentrated to afford methyl l-(4-amino- 2'-(trifluoromethoxy)-[l,l'-biphenyl]-2-yl)-lH-pyrrole-3-carboxylate (100 mg). MS(ES+) m/z 377 (ΜΗ" .
Step 3: To a solution of methyl l-(4-amino-2'-(trifluorometlioxy)-[l, -biphenyl]-2-yl)-lH- pyrrole-3-carboxylate (100 mg) in dichloromefhane (DCM) (5 mL) was added 2-(4- (ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 98 mg), triethylamine (0.111 mL). After 3 hrs, the mixture was diluted with water, extracted with CH2C12 (20 mL x 2), dried over Na2S04: and concentrated to afford a residue, which was purified by column chromatography (PE : EA = 10: 1 to 1 : 1) to afford methyl l-(4-(2-(4-(ethyIsulfonyl)phenyl)acetamido)-2'-(trifluoromethoxy)-[l,r- biphenyl]-2-yl)-lH-pyrrole-3-carboxyIate (90 mg). MS(ES+) m/z 587 (MIL*).
Step 4: To a solution of methyl l-(4-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2'- (trifIuoromethoxy)-[l, -biphenyl]-2-yl)-lH-pyrrole-3-carboxylate (80 mg) in methanol (5 mL) was added sodium hydroxide (16.36 mg) and water (1 mL). After 4 hrs, the mixture was diluted with water, washed with HC1 (1 M, 20 mL), extracted with EtOAc (20 mL x 2), dried over Na2S04, concentrated and purified by preparative HPLC to afford l-(4-(2-(4- (ethylsulfonyl)phenyl)acetamido)-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)-lH-pyrrole-3- carboxylic acid (13.4 mg) as an off white solid. Ή-NMR (400 MHz, MeOD-i/4) δ ppm 1.24 (t, J= 7.6 Hz, 3H), 3.23 (q, J= 7.6 Hz, 2H), 3.90 (s, 2H), 6.45 (m, 1H), 6.55 (m, 1H), 7.29(m, 1H), 7.34 (m, 2H), 7.37 (m, 1H), 7.45 (m, 1H), 7.68 (m, 3H), 7.86 (d, J= 2.4 Hz, 1H), 7.86 (d, J= 17.6 Hz, 2H); MS(ES+) m/z 555 ((M-OH)+).
Example 198
iV-(2-(2H-l,2,3-triazol-2-yl)-2'-(trifluoromethoxy)-[l,l'-biphenyI]-4-yl)-2-(4- (ethyIsuIfonyl)phenyI)acetamide

500 mg) in acetonitrile (10 mL) was added lH-l,2,3-triazole (138 mg), Cs2C03 (1082 mg). The mixture was stirred at 100°C for 16 hrs. The mixture was filtered. The filtrate was concentrated to afford a residue, which was purified by column chromatography (PE : EA ~ 10: 1 to 5:1) to afford 2- (4-nitro-2,-(trifiuoromethoxy)-[l,l,-biphenyl]-2-yl)-2H-l,2,3-triazole (150 mg). MS(ES+) m/z 351 ( H+).
Step 2: To a solution of 2-(4-nitro-2'-(trifluoromethoxy)-[l,l'-biphenyl] -2-yl>2H-l ,2,3-triazole (140 mg) in methanol (5 mL) was added zinc (261 mg). Ammonium chloride (214 mg) was added dropwise. After 1 hr, the mixture was filtered. The filtrate was diluted with water, and extracted with EtOAc (20 mL x 2), dried over Na2S04, concentrated to afford a residue, which was purified by column chromatography (PE : EA = 5: 1 to 1 : 1 ) to afford 2-(2H- 1 ,2,3-triazol-2-yl)-2'- (trifluoromethoxy)-[l)l '-biphenyl]-4-amine (70 mg). MSiES4 m/z 321 (MH+).
Step 3: To a solution of 2-(2H-l!2;3-triazol-2-yl)-2'-(trifluoromethoxy)- [l,l '-biphenyl]-4-amine (60 mg) in DCM (5 mL) was added 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 69.3 mg) and triethylamine (0.078 mL). After 3 hrs, the mixture was diluted with water, extracted with DCM ( 20 mL x 2), dried over Na2S04, concentrated to afford a residue, which was purified by preparative HPLC to afford N-(2-(2H-l ^.S-triazol^-yli^'-ftrifluoromethoxyi-t l'-biphe ylJ^-yl)- 2-(4-(ethylsulfonyl)phenyl)acetamide (10.1 mg) as an off-white solid. Ή-NMR (400 MHz, MeOD-i/4) δ ppm 1.24 (t, J= 7.6 Hz, 3H), 3.23 (q, J= 7.6 Hz, 2H), 3.92 (s, 2H), 7.21 (d, J= 8.0 Hz, 1H), 7.30 (m, 2H), 7.39 (m, 1H), 7.44 (d, J- 8.4 Hz, 1H), 7.68 (m, 4H), 7.82 (d, J= 2.0 Hz, 1H), 7.90 (m, 2H), 8.16 (d, J= 3.6 Hz, 1H); MS(ES+) m/z 531 (MH*).
Example 199
/Y-(2-(lH-l,2,3-triazol-l-yl)-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulfonyl) phenyl) acetamide

Step 1: To a solution of 2-fluoro-4-nitro--2'-(trifluoromethoxy)-l,r-biphenyl (intermediate 197a,
500 mg) in acetonitrile (10 mL) was added lH-l,2,3-triazole (138 mg), Cs2C03 (1082 mg). The mixture was stirred at 100°C for 16 hrs. The mixture was filtered. The filtrate was concentrated to afford a residue, which was purified by column chromatography (PE : EA = 10:1 to 1 :1) to afford 1- (4-nitro-2'-(triftuoromethoxy)-[l ,l'-biphenyl]-2-yl)-lH-l,2,3-triazole (80 mg). MS(ES+) m/z 351 (ΜΗ ).
Step 2: To a solution of l-(4-nitro-2'-(trifluoromethoxy)-[l,l'-biphenyI]-2-yl)- lH-l,2,3-triazole (80 mg) in methanol (5 mL) was added zinc (149 mg). Ammonium chloride (122 mg) was added dropwise. After 1 hr, the mixture was filtered. The filtrate was diluted with water, extracted with EtOAc (20 mL x 2), dried over Na2S04! concentrated to afford 2-(lH-l,2)3-triazol-l-yl)-2'- (trifluoromethoxy)-[l,r-biphenyl]-4-amine (40 mg). MS(ES+) m/z 321 (ΜΗ ).
Step 3: To a solution of 2-(lH-l!2,3-triazol-l-yl)-2'-(trifluoromethoxy)- [l,l'-biphenyl] -amine (30 mg) in DCM (5 mL) was added 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 34.7 mg), triethylamine (0.039 mL). After 3 hrs, the mixture was diluted with water, extracted with DCM (20 mL x 2), dried over Na2SC and concentrated to afford a residue, which was purified by preparative HPLC to afford N-(2-(lH-l,2,3 riazol-l-yl)-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)- 2-(4-(ethylsuIfonyl)phenyl)acetamide (12.8 mg) as an off-white solid. Ή-NMR (400 MHz, MeOD-c 4) δ ppm 1.24 (t, J= 7.6 Hz, 3H), 3.23 (q, J= 7.6 Hz, 2H), 3.92 (s, 2H), 7.21 (d, J= 8.4 Hz, 1H), 7.30 (m, 2H), 7.40 (m, 1H), 7.50 (d, J= 8.4 Hz, 1H), 7.67 (m, 3H), 7.74 (s, 1H), 7.82 (d, J= 2.0 Hz, 1H), 7.90 (m, 2H), 8.06 (d, J= 2.0 Hz, 1H); MS(ES÷) m/z 531 (MH4).
Example 200
/ -(2-(6-cyanopyridin-2-yI)-2,-(trifluoromethoxy)-[l,l'-biphenyl]-4-yI)-2-(4- (ethylsulfonyl)phenyl)acetamide

Step 1: To a solution of 4,4,5,5-tetramemyI-2-(4-nitro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-2- yl)-I,3,2-dioxaborolane (intermediate 69a, 1073 mg), 6-broniopicolinonitrile (400 mg) and CS2CO3 (855 mg) in 1,4-dioxane (6 mL) and water (2 mL) stirred under nitrogen at room temperature was added solid PdCl2(dppf) (80 mg) in one charge. The reaction mixture was heated in the microwave at 100°C for 20 mins. After cooling to room temperature, the mixture was filtered through celite, washed with EtOAc. The filtrate was concentrated under reduced pressure and the residue was partitioned between water and EtOAc. The organic layer was washed with brine, dried over anhydrous Na2S04, filtered and concentrated. The mixture was purified by column chromatography to give 6-(4-nitro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-2-yl)picolinonitrile (150 mg). MStES*) m/z 386 (MH+).
Step 2: To a solution of 6-(4-nitro-2'-(trifluoromethoxy)-[l,l '-bipheny]]-2-yl)picolinonitrile (150 mg) in methanol (5 mL) was added zinc (255 mg) and NH4C1 (41 mg). The reaction mixture was stirred at room temperature for 4 hrs. The mixture was filtered and the filtrate was concentrated in vacuo to give 6-(4-amino-2'-(trifluoromethoxy)-[l , l'-biphenyl]-2-yl)picolinonitrile (120 mg).
MS(ES+) m/z 356 (MH+).
Step 3: To a solution of sulfurous dichloride (603 mg) stirred under nitrogen at RT was added solid 2-(4-(ethylsulfonyl)phenyl)acetic acid (intennediate la, 216 mg) for 1 hr. Then sulfurous dichloride was removed by evaporation. The residue was dissolved in DCM (1 mL). The solution was added to a solution of 6-(4-amino-2'-(trifluorometlioxy)-[l5l'-biphenyl]-2-yl)picolinonitrile (120 mg) and triethylamine (0.197 mL) in DCM (2 mL) stirred under nitrogen at room temperature dropwise. The reaction mixture was stirred for 3 hrs. Solvent was evaporated in vacuo to give the crude product which was further purified by preparative HPLC to give a white solid N-(2-(6- cyanopyridin-2-yl)-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (19.5 mg). Ή NMR (400 MHz, MeOD-i¾ δ ppm 1.19 (t, = 3.2 Hz, 3H), 3.16 (q, J- 7.6 Hz, 2H), 3.86 (s, 2H), 7.15 (d, J= 7.2 Hz, 1H), 7.28 (d, J= 3.6 Hz ,1H), 7.35 (m, 4H), 7.41 (m, 3H), 7.65 (t, J = 2.0 Hz, 1H), 7.78 (d, J= 4.2 Hz, 1H), 7.86 (d, J= 7.2 Hz, 2H), 7.95 (s, 1H), 8.56 (s, 1 H); MSfES -) m/z 556 (MH4).
Example 201
N-(2,6-dichloro-2'-(trifluoromethoxyHl, -^
yl)acctamide, trifluoroacetic acid salt

yl)acetamide, trifluoroacetic acid salt (81 mg). Ή-NMR (400 MHz, CDC13) δ ppm 1.36 (t, J= 7.4 Hz, 3H), 3.22 (q, J= 7.4 Hz, 2H), 7.25 (dd, J= 8.0 Hz, 1.6Hz, 1H), 7.37 (m, 2H), 7.47 (dd, /= 8.0 Hz, 1.6 Hz, 1H), 7.70 (s, 2H), 7.82 (d, J= 8.0 Hz, 1H), 8.37 (dd, J= 8.0 Hz, 1.8 Hz, 1H), 9.10 (d, J= 1.8 Hz, 1H), 9.69 (s, 1H); 19F-NMR (376 MHz, DMSO-<¾ δ ppm -75.76, -57.17; MS(ES+) m/z 533 (MH+).
Example 202
7Y-(3-chloro-4-(l-methyl-lH-indazol-4-yl)phenyI)-2-(4-(ethylsulfonyl)phenyl)acetamide

To a mixture of N-[3-chloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-2-[4- (emylsulfonyl)phenyl]acetamide (intermediate 6b, 100 mg), 4-bromo-l-methyl-lH-indazole (54.6 mg) and 2 M sodium carbonate solution (0.431 mL) in DMF (3 mL) was added PdCl2(dp f)-CH2Cl2 adduct (8.80 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, extracted with EA for three times. The organic layer was dried and filtered through silica gel, and evaporated in vacuo. The residue was purified by MDAP to afford N-(3-chloro-4-(l -methyl-lH-indazoI-4-yl)phenyl)-2-(4-
(ethylsulfonyl)phenyl)acetamide (10 mg). 'H-NMR (400 MHz, DMSO-c¾ δ ppm 1.04 (t, J= 7.3 Hz,
3H), 3.21 (m, 2H), 3.80 (s, 2H), 4.01 (s, 3H), 7.02 (d, J~ 6.8 Hz, IH), 7.40 (m, 2H), 7.57 (m, 4H), 7.66 (s, IH), 7.80 (d, J= 8.3 Hz, 2H), 7.93 (d, J= 2.0 Hz, IH), 10.53 (s, IH); MS(ES+) m/z 468 (MH+).
Example 203
jV-(3,5-dichloro-4-(lH-indol-7-yl)phenyl)-2-(4-(ethyIsulfonyl)phenyl)acetamide, trifluoroacetic acid salt

Step 1: A mixture of 3,5-dichloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)aniline (800 mg, see step 1 for synthesis of Example 168), 2-(4-(ethyIsulfonyl)phenyl)acetic acid (intermediate 1 761 mg), HATU (1373 mg) and DIPEA (1.456 mL) in dichloromethane (10 mL) was stirred at RT overnight. The mixture was concentrated under rotavapor and the residue was subjected to flash chromatography to afford N-(3,5-dichloro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-2 (4-(ethylsulfonyl)phenyl)acetamide (1.2 g). MS(ES+) m/z 498 (MH+).
Step 2: To a mixture of N-(3,5-dichIoro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolaa-2-yl)phenyl)- 2-(4-(ethylsulfonyl)phenyl)acetamide (150 mg), 7-bromo-lH-indole (89 mg), sodium carbonate solution (2 M, 0.452 mL) and tri-feri-butylphosphine, tetrafluoroboric acid salt (34.9 mg) in 1 ,4- dioxane (2 mL) was added Pd2(dba)3 (27.6 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, extracted with EA for three times. The organic layer was dried, filtered through silica gel, and evaporated in vacuo. The residue was purified by MDAP to afford N-(3,5-dichloro-4-(lH-indol-7-yl)phenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt (50 mg). Ή-NMR (400 MHz, DMSO- 6) δ ppm 1.11 (t, J= 7.3 Hz, 3H), 3.29 (m, 2H), 3.88 (s, 2H), 6.48 (m, IH), 6.89 (d, J= 6.8 Hz, IH), 7.07 (t, /= 7.5 Hz, IH), 7.26 (t, J= 2.6 Hz, IH), 7.60 (d, /= 8.0 Hz, IH), 7.64 (m, 2H), 7.87 (m, 4H), 10.69 (s, IH), 10.82 (s, I H); 19F-NMR (376 MHz, DMSO-i ) 6 ppm -73.41; MS(ES+) m/z 487 (MH+).
Example 204
N-(3,5-dichloro-4-(l-methyl-lH-indazol-4-yl)phenyI)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt
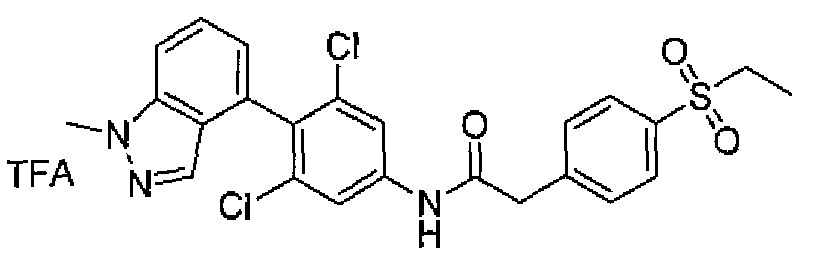
To a mixture of N-(3,5-dichloro-4-(4,4)5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyI)-2-(4- (ethylsulfonyl)phenyl)acetarnide (200 mg, see step 1 for synthesis of Example 203), 4-bromo-l- methyl-lH-indazole (127 mg), 2M sodium carbonate solution (0.602 lnL) and tri-ieri-butylphosphine, tetrafluoroboric acid salt (46.6 mg) in 1,4-dioxane (2 mL) was added Pd2(dba)3 (36.8 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and extracted with EA for three times. The organic layer was dried, filtered tlirough silica gel, and evaporated in vacuo. The residue was purified by MDAP to afford N-(3,5-dichloro-4-(l-methyl-lH-indazol-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt (43 mg). !H-NMR (400 MHz, DMSO-<¾) δ ppm 1.11 (t, J= 7.3 Hz, 3H), 3.30 (m, 2H), 3.88 (s, 2H), 4.09 (s, 3H), 7.01 (d, J= 7.0 Hz, 1H), 7.50 (m, 1H), 7.63 (m, 2H), 7.71 (d, J= 8.8 Hz, 1H), 7.88 (m, 4H), 10.72 (s; 1H); 19F-NMR (376 MHz, DMSO-4 5 ppm -74.20; MS(ES+) m/z 502 (MH+).
Example 205
Ar-(3,5-dichloro-4-(2!3-dihydrobenzo[fi] [l,4]dioxin-5-yl)phenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide

To a mixture of N-(3,5-dichloro-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide (200 mg, see step 1 for synthesis of Example 203), 5-bromo-2,3- dihydrobenzo[Z>][l,4]dioxine (129 mg), 2M sodium carbonate solution (0.602 mL) and xi-tert- butylphosphine, tetrafluoroboric acid salt (46.6 mg) in 1 ,4-dioxane (2 mL) was added Pd2(dba)3 (36.8 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and extracted with EA for three times. The organic layer was dried and filtered through silica gel, and evaporated in vacuo. The residue was purified by MDAP to afford N-(3,5-dichloro-4-(2,3-dihydrobenzo[i][l,4]dioxin-5-yl)phenyl)-2-(4-
(ethylsulfonyl)phenyl)acetamide (33 mg). Ή-NMR (400 MHz, DMSO-^) δ ppm 1.1 1 (t, J= 7.3 Hz, 3H), 3.29 (m, 2H), 3.85 (s, 2H), 4.17 (m; 2H), 4.24 (m, 2H), 6.66 (dd, 3= 6.5 Hz, 2.5 Hz, 1H), 6.91
(m, 2H), 7.62 (d, /= 8.3 Hz, 2H), 7.78 (s, 2H), 7.87 (d, J= 8.3 Hz, 2H), 10.64 (s, 1H); 19F-NM (376 MHz, DMSO- ) δ ppm -69.10, -71.10, -74.10; MS(ES+) m/z 506 (MH+).
Example 206
N-(3-chloro-4-(l-methyl-lH-indol-7-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt
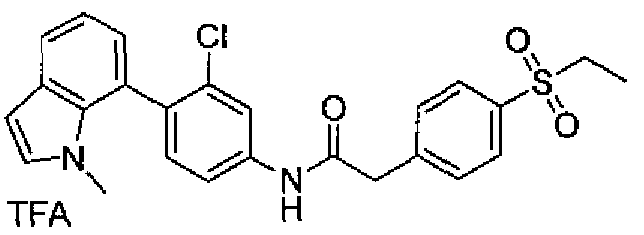
To a mixture of N-[3-chloro-4-(4,4,5,5-tetramethy]-l,3}2-dioxaborolan-2-yl)phenyl]-2-[4- (ethylsulfonyl)phenyljacetamide (intermediate 6b, 200 mg), 7-bromo-l -methyl- lH-indole (136 mg), 2M sodium carbonate solution (0.647 mL) and tri- -*ri-butylphosphine, tetrafluoroboric acid salt (50.0 mg) in 1,4-dioxane (2 mL) was added Pd2(dba)3 (39.5 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and extracted with EA for three times. The organic layer was dried, filtered through silica gel, and evaporated in vacuo. The residue was purified by MDAP to afford N-(3-chloro-4-(l -methyl -1H- indol-7-yl)phenyl)-2-(4-(etliylsulfonyI)phenyl)acetamide, trifluoroacetic acid salt (29 mg). Ή-NMR (400 MHz, DMSO-i/6) δ ppm 1.04 (t, J= 7.3 Hz, 3H), 3.18 (s, 3H), 3.23 (m, 2H), 3.79 (s, 2H), 6.41 (d, J= 3.0 Hz, 1H), 6.77 (d, J= 6.3 Hz, 1H), 6.98 (t, J= 7.5 Hz, 1H), 7.18 (d, J= 3.0 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 7.50 (m, 2H), 7.57 (d, / = 8.3 Hz, 2H), 7.80 (d, J= 8.3 Hz, 2H), 7.90 (d, J= 2.0 Hz, 1H), 10.52 (s, 1H); 19F-NMR (376 MHz, DMSO-c¾) δ ppm -73.73; MS(ES+) m/z 467 (MH+).
Example 207
Ar-(3,S-dichIoro-4-(lH-indoI-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide

To a mixture of N-(3,5-dichloro-4-(4,4,5,5-tetramediyI-l,3,2-dioxaborolan-2-yl)phenyl)-2-(4- (ethyIsulfonyl)phenyI)acetamide (200 mg, see step 1 for synthesis of Example 203), 4-bromo-lH- indole (118 mg), 2M sodium carbonate solution (0.602 mL) and tri-ier -butylphosphine,
tetrafluoroboric acid salt (46.6 mg) in 1 ,4-dioxane (2 mL) was added Pd2(dba)3 (36.8 mg) under nitrogen. The mixture was stirred and heated in the microwave at 100°C for half an hour. After cooling, the mixture was diluted with water, and extracted with EA for three times. The organic layer
was dried, filtered through silica gel, and evaporated in vacuo. The residue was purified by MDAP to afford N-(3,5-dichloro-4-(lH-indol-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide (18 mg). Ή- NMR (400 MHz, DMSO- ) δ ppm 1.11 (t, J= 7.3 Hz, 3H), 3.29 (m, 2H), 3.87 (s, 2H), 5.94 (s, 1H), 6.83 (d, /= 6.8 Hz, 1H), 7.17 (t, J= 7.7 Hz, 1H), 7.33 (t, J= 2.6 Hz, 1H), 7.44 (d, J= 8.3 Hz, 1 H), 7.64 (d, J= 8.3 Hz, 2H), 7.86 (m, 4H), 10.68 (s, 1H), 11.23 (s, 1H); MSfES"") m/z 487 (MH+).
Example 208
2-(4-(ethylsulfonyI)phenyl)-Ar-(3-(pyridin-4-yl)phenyl)acetamide, trifluoro acetic acid salt

Step 1: A solution of 3-bromoaniline (100 mg), pyridin-4-ylboronic acid (86 mg), tn-tert- butylphosphine, tetrafluoroboric acid salt (33.7 mg), Pd2(dba)3 (26.6 mg) and Na2C03 solution (2 M, 0.872 mL) was heated to 100°C for 0.5 hour in the microwave. The resulting mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed and extracted with ethyl acetate, washed with saturated ammonium chloride solution and brine to afford 3-(pyridin-4-yl)aniIine (103 mg) as a yellow solid. MS(ES+) m/z 171 (MH+).
Step 2: 3-(pyridin-4-yl)aniIine (103 mg), 2-(4-(ethylsulfonyI)phenyl)acetic acid (intermediate la,
137 mg) and HATU (228 mg) were dissovled in DCM (10 mL). To this solution, DIPEA (0.190 mL) was added gradually. The reaction mixture was stirred for 3 hours. Solvent was removed. The mixture was purified by column chromatography and concentrated in vacuo. The residue was redissolved in DMF, and solid was filtered off. The filtrate was purified by MDAP. Solvent was evaporated in vacuo to afford 2-(4-(ethylsulfonyl)phenyI)-N-(3-(pyridin-4-yl)phenyl)acetamide, trifluoroacetic acid salt (150 mg) as a white solid. Ή-NM (400 MHz, DMSO-£¾ δ ppm 1.03 (t, J= 7.4 Hz, 3H), 3.22 (q, J - 7.3 Hz, 2H), 3.80 (s, 2H), 7.48 (t, J= 8.0 Hz, 1H), 7.57 (d, /= 8.0 Hz, 3H), 7.64 (d, J= 8.0 Hz, 1H), 7.79 (d, /= 8.4 Hz, 2H), 8.00 (d, J= 6.0 Hz, 2H), 8.16 (s, 1H), 8.79 (d, 7- 6.4 Hz, 2H), 10.52 (s, 1H); MS(ES+) m/z 381 (MH+). Example 209
N-(4-chIoro-3-(pyridin-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide

Step 2: A solution of 3-bromo-4-chloroaniline (130 mg), pyridin-4-ylboronic acid (85 mg), tri- ieri-butylphosphine, tetrafluoroboric acid salt (73.1 mg), Pd2(dba)3 (57.7 mg) and Na2C03 solution (2 M, 0.944 mL) was heated to 100°C for 0.5 hour in the microwave. The resulting mixture was filtered through celite, and washed with ethyl acetate. Solvent was removed. The residue was partitioned between ethyl acetate and water. The organic phase was washed with saturated ammonium chloride solution and brine to afford 4-chloro-3-(pyridin-4-yl)aniline (151 mg) as a yellow oil. MS(ES+) m/z 205 (MH+).
Step 3: 4-Chloro-3-(pyridin-4-yl)aniline (123 mg), 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 151 mg) and HATU (251 mg) were dissovled in DCM (10 mL). To this solution, DIPEA (0.210 mL) was added gradually. The reaction mixture was stirred for 1 hour. Solvent was removed. The mixture was purified by column chromatography and concentrated in vacuo. The residue was redissolved in DMF. Solid was filtered off. The filtrate was purified by MDAP. Solvent was evaporated in vacuo to afford N-(4-chloro-3-(pyridin-4-yl)phenyI)-2-(4- (ethylsuIfonyl)phenyl)acetamide (48 mg) as a white solid. 'H-NMR (400 MHz, DMSO-c¾ δ ppm
1.02 (t, J= 7.4 Hz, 3H), 3.21 (q, J= 7.5 Hz, 2H), 3.76 (s, 2H), 7.38 (d, J= 4.4 Hz, 2H), 7.54 (m, 4H), 7.68 (s, 1H), 7.78 (d, J= 8.0 Hz, 2H), 8.61 (d, J= 6.0 Hz, 2H), 10.49 (s, 1H); MS(ES+) m/z 415 (MH^).
Example 210
N-(2,6-dichIoro-2'-(difluoromethoxy)-[l,l,-biphenyl]-4-yI)-2-(4-sulfamoylphenyl)acetamide

2,6-Dichloro-2,-(difluorometlioxy)-[l, -biphenyl]-4-amine (386 mg, see step 4 for synthesis of Example 169), 2-(4-sulfamoylphenyl)acetic acid (intermediate 29a, 355 mg) and HATU (531 mg) were dissovled in DCM (30 mL). To this solution, DIPEA (0.443 mL) was added gradually. The
reaction mixture was stirred for 24 hours. Solvent was removed. The mixture was purified by column chromatography and concentrated in vacuo. The residue was redissolved in DMF. Solid was filtered off. The filtrate was purified by MDAP. Solvent was evaporated in vacuo to afford N-(2,6-dichloro- 2'-(difIuoromethoxy)-[l,r-biphenyl]-4-yl)-2-(4-suIfamoylphenyl)acetamide (152 mg) as a white solid. JH-NMR (400 MHz, DMSO-i 6) 5 ppm 3.73 (s, 2H), 7.09 (t, J= 73.2 Hz, 1H), 7.25 (m, 5H), 7.46 (m, 3H), 7.72 (d, J= 8.4 Hz, 2H), 7.74 (s, 2H), 10.57 (s, 1H); I9F-NMR (376 MHz, DMSO- ) δ ppm - 81.00; MS(ES^) /z 501 (MH+).
Example 211
Ar-(2,6-dichloro-2'-(difluoromethoxy)-[l,l'-biphenyI]-4-yl)-2-(4- (methylsulfonyl)phenyl) ac etamide

2,6-Dichloro-2'-(difluoromethoxy)-[l, -biphenyl]-4-amine (300 mg, see step 4 for synthesis of Example 169), 2-(4-(methylsulfonyl)phenyl)acetic acid (232 mg) and HATU (413 mg) were dissovled in DCM (15 mL). To this solution, DIPEA (0.345 mL) was added gradually. The reaction mixture was stirred for 15 hours. Solvent was removed. The mixture was purified by column chromatography and concentrated in vacuo. The residue was redissolved in DMF. Solid was filtered off. The filtrate was purified by MDAP. Solvent was evaporated in vacuo to afford N-(2,6-dichloro- 2,-(difluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4-(methy]sulfonyl)phenyl)acetamide (377 mg) as a white solid. Ή-NMR (400 MHz, DMSO-i¾ δ ppm 3.22 (s, 3H), 3.86 (s, 2H), 7.16 (t, J= 73.6 Hz, 1H), 7.33 (m, 3H), 7.53 (m, 1H), 7.62 (d, J= 8.4 Hz, 2H), 7.82 (s, 2H), 7.91 (d, = 8.4 Hz, 2H), 10.68 (s, 1H); 19F-NMR (376 MHz, DMSO-< ) δ ppm -81.00; MS(ES+) m/z 500 (MH+).
Example 212
2-(4-(ethylsulfonyl)phenyl)-A-(2-(pyrimidin-5-yIoxy)-2'-(trinuorornethoxy)-[l,l'-biphenyl]-4- yl)acetamide, hydrochloride

Step 2: To a solution of 5-((4-nitro-2,-(trifluoromethoxy)-[l ,l'~biphenyl]-2-yl)oxy)pyrimidine (80 mg) in metlianol (5 mL) was added zinc (139 mg) and N¾CI (227 mg). The reaction mixture was stirred at RT for 2 hrs. The mixture was filtered and the filtrate was concentrated in vacuo to give 2- (pyrimidin-S-yloxyi^'-ttrifluoromethoxyi-lljl '-biphenyll^-amine (60 mg). MSiES*) m/z 348 (MH*).
Step 3: To a solution of sulfurous dichloride (41 1 mg) stirred under nitrogen at RT was added 2- (4-(ethylsulfonyl)phenyI)acetic acid (intermediate l a, 79 mg) for 1 hr. Then sulfurous dichloride was removed. The residue was dissolved in DCM (1 mL). The solution was added to a solution of 2- (pyriinidin-5-yIoxy)-2'-(trifluoromethoxy)-[l, -biphenyl]-4-amine (60 mg) and triethylamine (0.072 mL) in DCM (2 mL) stirred under nitrogen at room temperature dropwise. The reaction mixture was stirred for 2 hrs and evaporated in vacuo to give the crude product which was purified by Prep-HPLC to give 2-(4-(ethyIsulfonyl)phenyl)-N-(2-(pyrirnidin-5-yloxy)-2'-(trifiuoromethoxy)-[l ,l '-biphenyl]-4- yl)acetamide, hydrochloride (14.5 mg). Ή-NMR (400 MHz, MeOD- 4) δ ppm 1.20 (t, J =3.2 Hz, 3H), 3.19 (q, J = 7.6 Hz, 2H), 3.84 (s, 2H), 7.36 (m, 4H), 7.48 (d, .7=6.4 Hz, 2H), 7.59 (q, J=7.6 Hz, 3H), 7.86 (d, 7= 8.8 Hz, 2H), 8.36(s, 1H), 8.79 (s, 1H); MS(ES+) m/z 558 (MH*).
Example 213
JV-(6-(2-chlorophenyl)-5-(trifluoromethyI)pyridin-3-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide, hydrochloride

Step 3: A mixture of 5-nitro-3-(trifluoromethyl)pyridin~2(lH)-one (800 mg) and phosphoryl tribromide (2755 mg) was heated to 120°C for 8 hrs. The mixture was poured into ice/water. The mixture was neutralized with solid NaHC03 and extracted with EtOAc. The combined organic phases were dried over Na2S04, Solvents were removed under reduced pressure to give 2-bromo-5-nitro-3- (trifluorornethyl)pyridine (500 mg) . Ή-NMR (400 MHz, MeOD-i¾) S ppm 8.90 (s, 1H), 8.65 (s, 1H).
Step 4: To a solution of (2-chlorophenyl)boronic acid (190 mg), 2-bromo-5-nitro-3- (trifluoromethyl)pyridine (300 mg) and CS2CO3 (379 mg) in acetonitrile (12 mL) and water (4 mL) stirred under nitrogen at room temperature was added solid PdCl2(dppf) (19.44 mg) in one charge. The reaction mixture was heated in the microwave at 100°C for 20 mins. After cooling to RT, the mixture was filtered through celite, washed with ACN and EtOAc. The filtrate was concentrated under reduced pressure and the residue was partitioned between water and EtOAc. The organic layer was washed with brine and dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford 2-(2-chlorophenyl)-5-nitro-3-(trifluoromethyl)pyridine as a brown oil. MS(ES+) m/z 303 (MH+).
Step 5: To a solution of 2-(2-chIorophenyl)-5'nitro-3-(trifluoromethyl)pyridine (200 mg) in methanol (10 mL) was added zinc (432 mg) and NH4CI (707 mg). The reaction mixture was stirred at RT for 4 hrs. TLC (PE : EA = 1 : 1) showed the reaction was complete. The mixture was filtered and the filtrate was concentrated in vacuo to give 6-(2-chlorophenyl)-5-(trifluoromethyl)pyridin-3 -amine (150 mg).
Step 6: A mixture of HOBT (148 mg), EDC (186 mg) and 2-(4-(ethylsulfonyI)phenyl)acetic acid (intermediate la, 201 mg) in DCM (1 mL) was stirred under nitrogen at room temperature for 1 hr. Then 6-(2-chlorophenyl)-5-(trifluoromethyl)pyridin-3-amine (120 mg) was added into the mixture and stirred for 12 hrs. The mixture was washed with water, brine and dried over Na2S0 . Solvents were removed under reduced pressure. The crude product was purified by prep-HPLC to give N-(6- (2-chlorophenyl)-5-(trifluoromethyl)pyridin-3-yI)-2-(4-(ethylsulfonyl)phenyl)acetamide,
hydrochloride (55.4 mg). Ή-NMR (400 MHz, MeOD-a^) δ ppm 1.19 (t, J= 3.2 Hz, 3H), 3.16 (q, J=
7.6 Hz, 2H), 3.93 (s, 2H), 7.50 (m, 4H), 7.78 (d, J= 8.8 Hz, 2H), 7.86 (d, = 8.8 Hz, 2H), 8.65 (s, 1H), 8.90 (s, 1H); MSfJES^ m/z 483 (MH ).
Example 214
2-(4-(ethylsulfonyl)phenyl)-N-(2-(2-methylpyrid^
yl)acetamide

Step 1: A mixture of 4,4,5,5-tetramethyl-2-(4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2-yl)- 1,3,2-dioxaborolane (intermediate 69a, 600 mg), 4-bromo-2-methylpyridine (303 mg), PdC^Cdppf)- CH2C12 adduct (240 mg) and 2M of aqueous Na2COj (622 mg) in 1,4-dioxane (2 mL) was stirred in the microwave at 100°C for 30 mins. The mixture was filtered and the filtrate was concentrated. The residue was purified by TLC (PE : EA=1 : 1) to give 2-methyl-4-(4-nitro-2'-(trifluoromethoxy)-[l , 1 '- biphenyl]-2-yl)pyridine (55 mg) as a yellow oil. Ή-NMR (400 MHz, CDC13) δ ppm 2.48 (s, 3H), 6.78 (dd, J = 1.2 Hz, 5.2 Hz, 1H), 6.96 (s, 1H), 7.21-7.24 (m, 2H), 7.28-7.29 (m, 1H , 7.38-7.41 (m, 1H), 7.59 (d, / = 8.0 Hz, 1H), 8.31-8.36 (m, 3H); MS(ES+) m/z 375 (MH+).
Step 2: To a suspension of 2-methyl-4-(4-nitro-2'-{trifluoromethoxy)-[l, -biphenyl]-2- yl)pyridine (55 mg) and zinc (96 mg) in methanol (5 mL) stirred in air at RT was added solid ammonium chloride (79 mg) in one charge. The reaction mixture was stirred at RT for 30 mins. The mixture was filtered and the filtrate was concentrated in vacuo to give 2-(2-methylpyridin-4-yl)-2'- (trifluoromethoxy)-[l, -biphenyI]-4-amine (52 mg) as a brown solid. MS(ES+) m/z 345 (MH+).
Step 3: A mixture of 2-(4-(ethylsulfonyI)phenyl)acetyl chloride (intermediate 170a, 74.5 mg),
Et3N (0.074 mL) and 2-(2-methylpyridin-4-yl)-2'-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (52 mg) was stirred at RT for 1 h. TLC (PE/EA~1 :1) indicated the reaction was complete. The mixture was diluted with DCM, washed with water, sat. ctric acid, sat. Na2C03 and brine, then dried over anhydrous Na2S04. After concentration, the residue was purified by HPLC to afford 2-(4- (ethylsuIfonyl)phenyl)-N-(2-(2-methylpyridin-4-yl)-2'-(trifJuoromethoxy)biphenyI-4-yl)acetamide (21.8 mg) as a yellow solid. Ή-NMR (400 MHz, MeOD-</„) δ ppm 1.21 (t, J= 7.2 Hz, 3H), 2.66 (s, 3H), 3.19 (q, J= 7.2 Hz, 2H), 3.90 (s, 2H), 7.18-7.20 (m, 1H), 7.45-7.51 (m, 5H), 7.65-7.71 (m, 4H), 7.88 (d, J= 8.4 Hz, 2H), 8.1 1 (d, J= 2.0 Hz, 1H), 8-48 (dd, J= 0.8 Hz, 6.4 Hz, 1H); MS(ES+) m/z 555 (MH4).
Example 215
2-(4-(ethylsulfonyl)phenyl)-N-{2-(2-hydroxypyridin-4-yI)-2'-(trifluoromethoxy)-[l,l'-biphenyl]- 4-yl)acetamide

(trifluoromethoxy)phenyl)boronic acid (14.37 g), Pd(Ph3P)4 (4.03 g) and sodium carbonate (11.10 g) in 1,4-dioxane (90 mL) and water (30 mL) was refluxed for 16 hrs under N2. The mixture was washed with water and brine, dried over Na2S04 and concentrated in vacuo. The crude product was purified by column chromatography (PE : EA = 10:1 to 5:1) to obtain /ert-butyl (2-chloro-2'- (trifluoromethoxy)-[l,r-biphenyl]-4-yl)carbamate (8 g) as a colorless oil. MS(ES+) m/z 338 (MH+).
Step 2: To a solution of 2-methoxy-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridine (150 mg) in 1 ,4-dioxane (6 mL) was added CsaC03 (208 mg), ½r/-butyl (2-chloro-2'-(trifluoromethoxy)- [1 , l'-biphenyl]-4-yl)carbamate (247 mg), bis(tri-ier -butylphosphine)palladium(0) (16.30 mg) and water (2 mL). The mixture was degassed and reacted under N2 at 120°C for 2 hrs. The mixture was diluted with water, extracted with EtOAc, dried over Na2SC>4, concentrated and purified by TLC (PE : EA = 2: 1) to afford ½r/-butyl (2-(2-methoxypyridin-4-yl)-2'-(trifluoromethoxy)-[l}r-biphenyl]-4- yl)carbamate (100 mg). MS(ES+) m/z 461 (MH+).
Step 3: A mixture of tert-buty\ (2-(2-methoxypvridin-4-yl)-2'-(trifluoromethoxy)-[l, - biphenyI]-4-yl)carbamate (70 mg) in tetrahydrofuran (THF) (3 mL) was added cone. HC1 solution (0.020 mL) at 20 °C. After 3 hrs, the mixture was concentrated to afford 2-(2-methoxypyridin-4-yl)- 2,-(trifluoromethoxy)-[l,r-biphenyl]-4-amine (45 mg). MSiES*) m/z 361 (MH+).
Step 4: To a solution of 2-(2-methoxypyridin-4-yl)-2,-(trifluoromethoxy)- [l,l'-biphenyl]-4- amine (60 mg) in DCM (5 mL) was added 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 41.1 mg), triethylamine (0.046 mL). After 3 hrs, the mixture was diluted with water, extracted DCM ( 20 mL x 2), dried over Na2S04, and concentrated to afford 2-(4-(ethylsulfonyl)phenyl)-N-(2- (2-methoxypyridin^-yl)-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)acetaiTiide (100 mg). MS(ES+) m/z 571 (MH+).
Step 5: To 2-(4-(ethylsulfonyl)phenyl)-N-(2-(2-methoxypyridin-4-yl)-2'- (trifluoromethoxy)- [l, '-biphenyl]-4-yl)acetamide (80 mg) in DCM (23 mL) was added iodotrimethylsilane (0.052 mL). The mixture was stirred at 20°C for 16 hrs. The mixture was diluted with water, extracted with DCM
(20 mL x 2), dried over Na2SC>4, and concentrated to afford a residue, which was purified by preparative HPLC to afford 2-(4-(ethylsulfonyl)phenyl)-N-(2-(2-hydroxypyridin-4-yl)-2'- (trifluoromemoxy)-[l, l '-biphenyl]-4-yl)acetamide (45.5 mg). Ή-NMR (400 MHz, MeOD-d4) δ ppm 1.23 (t, J= 7.6 Hz, 3H), 3.03 (q, J= 7.6 Hz, 2H), 3.90 (s, 2H), 6.20 (d, J= 6.4 Hz, 1H), 6.38 (s, 1H), 7.24 (d, J= 6.0 Hz, 1H), 7.40 (m, 4H), 7.58 (d, J= 7.6 Hz, 1H), 7.76 (m, 1H), 7.74 (s, 1H), 7.92 (d, J = 8.0 Hz, 2H); SfES4 m/z 557 (MH+).
Example 216
N-(2-(cyanomethyI)-2'-(trifluoromethoxy)-[l,l,-biphenyl]-4-yl)-2-(4- (ethylsulfonyI)phenyl)acetamide

Step 1: To a solution of (2-(nifluoromethoxy)phenyl)boronic acid (5.67 g), CS2CO3 (10.25 g), methyl 2-bromo-5-nitrobenzoate (6.817 g) in water (18 mL) and acetonitrile (54 mL) was added PdCl2(dppf) (0.480 g) under N2. The mixture was heated to reflux for 2 hrs. TLC (PE : EA = 5: 1) showed the reaction was complete. The mixture was filtered and the filtrate was concentrated to give methyl 4-nitro-2'-(trifluoromethoxy)-[l ,1 '-biphenyI]-2-carboxylate (8.86 g).
Step 2: To a solution of methyl 4-nitro-2'-(trifiuoromethoxy)-[l,r-biphenyl]-2-carboxylate (12.26 g) in tetrahydrofuran (THF) (100 mL) was added B¾ THF (1 M in THF, 180 mL) dropwise at room temperature. The mixture was heated to reflux for 6 hrs. TLC (PE : EA = 5: 1) showed the reaction was complete. The mixture was quenched with methanol and extracted with EtOAc, dried over Na2S04 and concentrated to give (4-amino-2'-(trifluoro methoxy)-[l ,1 '-biphenyl]-2-yl)methanol
( g).
Step 3: To a solution of (4-amino-2'-(trifluoromethoxy)-[l, -biphenyI]-2-yl)methanoI (3 g), 2- (4-(ethylsulfonyl)phenyI)acetic acid (intermediate la, 3.63 g) and HATU (6.04 g) in dichloromethane (20 mL) stirred at room temperature was added DIPEA (5.55 mL). The reaction mixture was stirred at 20°C for 3 hrs. The solution was washed with water. The organic was concentrated and purified by TLC (PE : EA = 1 :3) to give 2-(4-(ethylsulfonyl)phenyl)-N-(2-(hydroxymethyl)-2'- (trifluoromethoxy)-[l ,l '-biphenyl]-4-yl)acetamide (2.5 g). MStES^ m/z 476 (MH^.
Step 4: To a solution of 2-(4-(ethylsulfonyl)phenyl)-N(2-(hydroxymethyl)-2'- (tiifluoromethoxy)-[l, -biphenyl]-4-yl)acetamide (1 g) and TEA (0.565 mL) in dichloromethane (15 mL) was added MsCl (0.237 mL) at 0°C. After stirred for 0.5 hr at this temperature, TLC (PE : EA =
1:2) showed the reaction was complete. The mixture was washed with water and extracted with DCM, The organic layer was concentrated to give (4-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2'- (trifluoromethoxy) -[l.l '-biphenyl] -2-yl)methyl metlianesulfonate (500 mg).
Step 5: A solution of (4-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2'-(trifluorornetlioxy)-[l, - biphenyl] -2-yl)methyl metlianesulfonate (100 mg), TMSCN (0.070 mL) and TBAF (137 mg) in tetrahydrofuran (THF) (3 mL) and acetonitrile (3 mL) was stirred under nitrogen at 25 °C for 2 hrs. The solution was concentrated and purifyed by prep-TLC (PE : EA = 1 : 1) to give N-(2- (cyanometliyl)-2'-(trifluoromethoxy)-[l, -biphenyl]-4-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide (53 mg) as a yellow solid. Ή-NMR {400 MHz, CDClj) 5 ppm 1.31 (t, J= 7.2 Hz, 3H), 3.13 (q, J= 7.6 Hz, 2H), 3.50 (q, J= 18.8 Hz, 2H), 3.85 (s, 2H)S 7.22 (d, J = 8.4 Hz, 1H), 7.28 (d, J= 5.6 Hz, 1H), 7.39 (m, 2H), 7.40 (t, J= 1.6 Hz, 1H),7.58 (q, J= 8.4 Hz, 4H), 7.62 (d, J= 1.6 Hz, 1H), 7.92 (d, J= 8.4 Hz, 2H); MS(ES+) m z 503 (MH+).
Example 217
2-(4-(ethyIsuIfonyl)phenyl)-iV-(2'-rnethyl-2-(l-methyl-lH-pyrazol-5-yI)-ll,l,-biphenyl]-4- yl)acetamide

Step 1: A mixture of l-bromo-2-chloro-4-nitrobenzene (1.5 g), o-tolylboronic acid (0.906 g), Cs2C<¾ (2.480 g) and PdCl2(dppf)-CH2Cl2 adduct (0.124 g) in acetonitrile (15 mL) and water (5 mL) was heated to reflux and stirred for 2 hrs. The mixture was cooled to room tempareture and filtered. The filtrate was concentrated in vacuo. The residue was dissolved in water and EtOAc. The aqueous layer was extracted with EtOAc. The organic layers were washed with brine, dried over Na2S04 and concentrated in vacuo to obtain 2-chloro-2'-methyl-4-nitro-l,r-biphenyl (1.8 g) as a brown oil. Ή- NMR (400 MHz, CDC13) δ ppm 2.04 (s, 3H), 7.03 (m, 1 H), 7.23 (m, 3H), 7.35 (d, J= 8.4 Hz, 1 H), 8.10 (dd, J= 2.4 Hz, 8.4 Hz, 1H), 8.28 (d, 2.4 Hz, 1H).
Step 2: A mixture of 2-chloro-2'-metliyl-4-nitro-l ,l'-biphenyl (300 mg), l -methyl-5-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (630 mg), Pd2(dba)3 (55.5 mg),
tricyclohexylphosphonium tetrafluoroborate (44.6 mg) and Cs2C03 (987 mg) in 1,4-dioxane (2 mL) was stirred at 120°C for 1 hr in the microwave. The mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified by prep-TLC (PE : EA = 3: 1) to obtain l-methyl-5-
(2'-methyl-4-nitro-[l,r-biphenyl]-2-yl)-lH-pyrazole (236 mg) as a yellow solid. MS^S ) m/z 294 (ΜΗ+).
Step 3: To a suspension of l-metliyl-5-(2,-methyl-4-nitro-[l,r-biphenyl]-2-yl)-lH-pyrazoIe (336 mg) and zinc (749 mg) in methanol (5 mL) stirred in air at RT was added solid ammonium chloride (613 mg) in one charge. The reaction mixture was stirred at RT for 30 mins. The mixture was filtered and the filtrate was concentrated in vacuo give 2'-methyl-2-(l-methyl-lH-pyrazol-5-yl)-[l,r- biphenyl]-4-amine (200 mg) as a yellow solid. MS(ES+) m/z 209 (MH+).
Step 4: A mixture of 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 375 mg), Et3N (0.370 mL) and 2'-med yl-2-(l-meti yl H-pyrazol-5-yl)-[l,l'-biphenyl]^-amine (200 mg) was stirred at RT for 1 nr. TLC (PE : EA = 1 : 1) indicated the reaction was complete. The mixture was diluted with DCM, washed with water, sat. citric acid, sat. Na2COj solution and brine, then dried over anhydrous Na2S04. After concentration, the residue was purified by prep-TLC (PE:EA = 1 :5) and prep-HPLC to afford 2-(4-(ethylsulfonyl)phenyl)-N-(2'-methyI-2-(l-methyl-li?-pyrazol-5-y])-[l , 1 '- biphenyl]-4-yl)acetamide (70 mg) as a pale yellow solid. Ή-NMR (400 MHz, MeOD-</4) δ ppm 1.21 (t, J= 7.2 Hz, 3H), 2.01 (s, 3H), 3.20 (q, J= 7.2 Hz, 2H), 3.52 (s, 3H), 3.87 (s, 2H), 5.95 (d, J= 2.0 Hz, 1H), 6.98 (m, 1H), 7.06 (m, 1H), 7.14 (m, 2H), 7.31 (m, 2H), 7.69 (m, 4H), 7.88 (d, /= 8.8 Hz, 2H); MSfES4 m/z 474 (MH+).
Example 218
iV-(6-(dimethylamino)-5-(2-(trifluoromethoxy)phenyl)pyridin-2-yl)-2-(4- (ethyIsuIfonyl)phenyI)acetamide, hydrochloride

Step 1: To a solution of 6-fluoropyridin-2 -amine (1 g) in DMF (5 mL) was added 1- bromopyrroIidine-2,5-dione (1.588 g) at 20 °C. After stirred for 16 hrs, the mixture was diluted with water, extracted with DCM (20 mL x 2), dried over Na2S04, and concentrated to afford a residue, which was purified by column chromatography (PE : EA = 5: 1) to afford 5-bromo-6-fluoropyridin-2- amine (1.6 g). MS(ES+) m/z 191 (MH+).
Step 2: To a solution of 5-bromo-6-fluoropyridin-2-amine (600 mg) in 1,4-dioxane (25 mL) was added Na2C03 (666 mg), (2-(trifluorometlioxy)phenyl)boronic acid (647 mg), PdCl2(dppf) (115 mg), water (8.33 mL). The mixture was degassed and reacted under N2 at 120°C for 2 hrs. The mixture was diluted with water, extracted with EtOAc (20 mL x 2), dried over Na2S04, concentrated and purified
by column chromotography (PE : EA = 5:1) to afford 6-fluoro-5-(2- (trifluoromethoxy)phenyl)pyridin-2-amine (600 mg). MSiES"*) m/z 273 (MH+).
Step 3: To a solution of 6-fluoro-5-(2-(trifluorometl oxy)phenyl)pyridin-2 -amine (0.3 g) in DMF (15 mL) was added dimethylamine (0.497 g) and 2C03 (0.914 g). The mixture was stirred at 120°C for 4 days. The mixture was diluted with water, extracted with DCM (20 mL x 2), dried over Na2S04j and concentrated to afford a residue N2,N2-dimethyl-3-(2-(trifluoromethoxy) phenyl)pyridine-2,6- diamine (120 mg). MS(ES+) m/z 298 (MH+).
Step 4: To a solution of N ,N"-dimetliyl-3-(2-(trifluoromethoxy)phenyl)pyridine -2,6-diamine (100 mg) in DCM (5 mL) was added 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 124 mg), triethylamine (0.141 mL). After stirred for 3 lirs, the mixture was diluted with water, extracted DCM (20 mL x 2), dried over Na2S04, and concentrated to afford a residue, which was purified by prep-HPLC to afford N-(6-(dimethylamino)-5-(2-(trifluoromethoxy)phenyl)pyridin-2-yl)- 2-(4-(ethyIsulfonyl)phenyl)acetamide, hydrochloride (36.5 mg) as an off white solid. Ή-NMR (400 MHz, MeOD-ά,) δ ppm 1.13 (t, 7.6 Hz, 3H), 2.90 (s, 6H), 3.23 (q, /= 7.6 Hz, 2H), 4.00 (s, 2H), 6.80 (s, IH), 7.46 (m, 4H), 7.60 (d, J= 8.4 Hz, 2H), 7.70 (m, IH), 7.90 (d, J= 8.4 Hz, 2H); MS(ES+) m/z 508 (MH+).
Example 219
2-(4-(ethylsulfonyl)phenyl)-N-(2'-fluoro-2-(l-methyl-l/r-pyra2ol-5-yl)-[l,l'-biphenyl]-4- yl)acetamide, hydrochloride

Step 1: To a solution of l-bromo-2-chloro-4-nitrobenzene (2 g) in acetonitrile (20 mL) was added Cs2C03 (3.31 g), (2-fluorophenyl)boronic acid (1.302 g), PdCl2(dppf) (0.309 g) and water (6.67 mL). The mixture was degassed and reacted under N2 at 100°C for 2 hrs. The mixture was diluted with water, extracted with EtOAc (20 mL x 2), dried over Na2S04, concentrated and purified by column chromatography (PE : EA = 20: 1) to afford 2-chloro-2'-fluoro-4-nitro-l, -biphenyl (1.5 g).

Step 2: To a solution of (l-methyl-lH-pyrazol-5-yl)boronic acid (400 mg) in 1,4-dioxane (9 mL) was added Cs2C03 (1554 mg), 2-chloro-2,-fluoro-4-nitro-l 5l,-biphenyl (400 mg), Pd2(dba)3 (72.8 mg), and tricyclohexylphosphine (44.6 mg). The mixture was degassed and reacted under N2 at 120°C for
2 hrs. The mixture was diluted with water, extracted with EtOAc (20 mL x 3), dried over Na2S04, concentrated and purified by column chromatography (PE : EA = 2: 1) to afford 5-(2'-fluoro-4-nitro- [l,r-biphenyl]-2-yl)-l-methy]-lH-pyrazole (300 mg). MSfES ) m/z 298 (MH+).
Step 3: To a solution of 5-(2,-fIuoro-4-nitro-[l,r-biphenyl]-2-yl)-l-methyl-lH-pyrazole (300 mg) in methanol (5 mL) was added zinc (660 mg). Ammonium chloride (540 mg) was added dropwise. After 1 hr, the mixture was filtered. The filtrate was diluted with water, extracted with EtOAc (20 mL x 3), dried over Na2S04, and concentrated to afford 2,-fluoro-2-(l-methyl-lH-pyrazol-5-yl)-[l,r- biphenyl]-4-amine (200 mg). MSiES'1) m/z 268 (MH+).
Step 4: To a solution of 2'-fluoro-2-(l-methyl-lH-pyrazol-5-yl)-[l, -biphenyl]-4-amine (100 mg) in DCM (5 mL) was added 2-(4-(etliylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 138 mg), triethylamine (0.156 mL). After 3 hrs, the mixture was diluted with water, extracted DCM (20 mL x 2), dried over Na2SC>4, and concentrated to afford a residue, which was purified by preparative HPLC to afford 2-(4-(ethylsulfonyl)phenyl)-N-(2'-fluoro-2-(l-methyl-lH-pyrazol-5-yl)-[l,l'- biphenylH-yl)acetamide, hydrochloride (52.9 mg). 'H-NMR (400 MHz, MeOD-i¾) δ ppm 1.23 (t, J = 7.6 Hz, 3H), 3.23 (q, /= 7.6 Hz, 2H), 3.61 (s, 3H), 3.90 (s, 2H), 6.14 (s, 1H), 7.03 (m, 1H), 7.15 (m, IH), 7.21 (m, 1H), 7.33 (m, 1H), 7.49 (t, J= 8.4 Hz, 1H), 7.68 (t, J= 8.4 Hz, 2H), 7.49 (t, J= 8.4 Hz, 1H), 7.76 (m, 1H), 7.85 (d, J= 2.4 Hz, 1H), 7.92 (d, J= 8.4 Hz, 2H); MS(ES*) m/z 478 (MH+).
Example 220
2-(4-(ethylsulfonyl)p enyl)-jV-(2*-methoxy-2-(l-methy)-lH-pyrazol-5-yl)-[l,l,-biph1
yl)acetamide

Step 1: A mixture of l-bromo-2-chloro-4-nitrobenzene (1.5 g), (2-methoxyphenyl)boronic acid (1.012 g), Cs2C03 (2.480 g) and PdCl2(dppf)-CH2Cl2 adduct (0.124 g) in acetonitrile (15 mL) and water (5 mL) was heated to reflux and stirred for 1 hr. The mixture was cooled to RT and filtered. The filtrate was concentrated in vacuo. The Tesidue was dissolved in water and EtOAc. The aqueous layer was extracted with EtOAc. The organic layers were washed with brine, dried over Na2S04 and concentrated in vacuo to give 2-chloro-2'-methoxy-4-nitro-l, -biphenyl (1.9 g) as a brown solid. Ή- NMR (400 MHz, CDC13) δ ppm 3.77 (s, 3H), 7.03 (m, 2H), 7.17 (d, J« 2.0 Hz, 1H), 7.48 (d, J= 8.4
Hz, 1H), 8.15 (dd, J= 2.0 Hz, 8.8 Hz, 1H), 8.34 (d, J= 2.4 Hz, 1H).
Step 2: A mixture of 2-chloro-2'-methoxy-4-nitro-l, l'-biphenyl (200 mg), 1-ιη€ί1ιγ1-5-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (395 mg), Pd2(dba)3 (34.7 mg), Cs2C03 (618 mg) and tricyclohexylphosphonium tetrafluoroborate (27.9 mg) in dioxane (9 inL) was stirred at 120°C for 1 hr in the microwave. The mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified by prep-TLC (PE : EA = 2:1) to obtain 5-(2'-methoxy-4-nitro-[l , 1 '-biphenyl]-2- yl)-l-methyl-lH-pyrazole (200 mg) as a yellow solid. MSiES1) m/z 310 (MH÷).
Step 3: To a suspension of 5-(2'-methoxy-4-nitro-[l; -biphenyl]-2-yl)-l-methyl-lH-pyrazole (336 mg) and zinc (710 mg) in methanol (5 inL) stirred in air at RT was added ammonium chloride (581 mg) in one charge. After stirred at RT for 30 mins, the mixture was filtered and the filtrate was concentrated in vacuo to give 2'-methoxy-2-(l-methyl-lH-pyrazol-5-yl)-[lJl,-biphenyl]-4-amine (200 mg) as a yellow solid. MS(ES > m/z 280 (Mrf
Step 4: A mixture of 2-(4-(ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 353 mg), Et3N (0.349 mL) and 2'-methoxy-2-(l-me l-lH-pyrazol-5-yl)-[l,l'-biphenyl]-4-ainine (200 mg) was stirred at RT for 1 hr. TLC (PE : EA = 1 : 1) indicated the reaction was complete. The mixture was diluted with DCM, washed with water, sat. citric acid, sat. Na2C03 solution and brine, dried over anhydrous Na2S04. After filtration and concentration, the residue was purified by TLC (PE : EA = 1:5) and HPLC to afford 2-(4-(etliylsulfonyl)phenyl)-N-(2'-methoxy-2-(l-methyl-lH-pyrazol-5-yl)- [l,l'-biphenyl]-4-yl)acetamide (72.9 mg) as a white solid. lH-NMR (400 MHz, MeOD-i¾) δ ppm 1.24 (t, J= 7.2 Hz, 3H), 3.22 (q, J= 7.2 Hz, 2H), 3.57 (s, 3H), 3.67 (s, 3H), 3.90 (s, 2H), 6.15 (d, J= 2.4 Hz, 1H), 6.93 (m, 2H), 7.12 (dd, J= 0.8 Hz, 7.6 Hz, 1H), 7.29 (t, /= 1.2 Hz, 1H), 7.41 (d, J= 8.4 Hz, 1H), 7.68 (m, 4H), 7.84 (d, J= 2.0 Hz, 1H), 7.91 (d, J= 8.4 Hz, 2H); MS(ES^) m/z 490 (MH .
Example 221
3-(4-(2-(4-(ethylsulfonyl)phenyI)acetamido)-2'-(trifluoromethoxy)-[l,l,-biphenyl]-2- yl)propanoic acid

Step 1: A solution of 2-(4-(ethylsulfonyI)phenyl)-N-(2-(hydroxymethyl)-2'-(trifluoromethoxy)- [l, -biphenyl]-4-yl)acetamide (1.5 g, see step 3 for syntliesis of Example 216) and manganese oxide (2.64 g) in CH ¾ (20 mL) was stirred at 60°C for 16 hrs. The solution was filtrated. The filtrate was
concentrated and purified by prep-TLC (PE : EA = 1 : 1) to give 2-(4-(ethylsulfonyl)phenyI)-N-(2- fomiyl-2'-(trifluorometlioxy)-[l,l'-biphenyl]^-yl)acetamide (700 mg). MS(ES+) mfz 492 (MH+).
Step 2: A solution of ethyl 2-(triphenylphosphoranylidene)acetate (595 mg) in tetrahydrofuran (15 mL) was added potassium 2-methylpropan-2-olate (192 mg) at 0°C and stirred under nitrogen at 0°C for 1 hr. To this solution was added 2-(4-(ethylsulfonyI)pheny])-N-(2-fonnyl-2'-
(trifluoromethoxy)-[l,r-biphenyl]-4-yl)acetamide (700 mg) at 0 °C. The reaction mixture was stirred at 25 °C for 3 hrs. The solution was washed with saturated NH4C1 and was extraced with DCM. The organic was washed with brine, dried over Na2S04 and concentrated to give (iT)-ethyl 3-(4-(2-(4- (ethyIsulfonyl)phenyl)acetamido)-2'-(trifluoromethoxy)-[l, -biphenyl]-2-yl)acrylate (500 mg). MS(ES+) m/z 562 (MH+).
Step 3: A solution of (£)-ethyl 3-(4-(2-(4-(ethylsulfonyl)phenyl)acetainido)-2'- (trifIuoromethoxy)-[l, -biphenyI]-2-y])acrylate (500 mg) and Pd/C (97 mg) in methanol (15 mL) was stirred under ¾ at 15 psi.. After stirring at 26 °C for 18 hrs, the mixture was filtered. The filtrate was concentrated and purified by prep-TLC (PE : EA = 1: 1) to give methyl 3-(4-(2-(4- (ethylsulfonyl)phenyl)acetamido)-2L(trifluoromethoxy)-[l ,Γ-biphenyl] -2-yl) propanoate (210 mg). MS(ES+) w/z 564 ( H+).
Step 4: A mixture of methyl 3-(4-(2-(4-(emylsulfonyl)phenyI)acetann^o)-2'-(trifluoromethoxy)- [Ι,Γ-biphenyl] -2-yl) propanoate (160 mg) and lithium hydroxide (27.2 mg) in tetrahydrofuran (THF) (6 mL) and water (2 mL) was stirred at 0°C for 16 hrs. The solution was cooled to RT, acidified to pH 5 with 1 M HC1, and extracted with DCM. The organic was concentrated and purified by prep- HPLC to give 3 -(4-(2-(4-(ethylsulfonyl)phenyl)acetamido)-2,-(trifiuoromethoxy)-[ 1 , 1 -biphenyl] -2- yl)propanoic acid (80 mg) as a white solid. Ή-NMR (400 MHz, CDC13) 5 ppm 1.27 (t, J= 7.6 Hz, 3H), 2.43 (t, J= 1.6 Hz, 2H), 2.75 (m, 2H), 3.12 (t, J= 7.6 Hz, 2H), 3.78 (s, 2H), 7.14 (d, J= 8.4 Hz, 1H), 7.24 (d, J= 12.8 Hz, 1H), 7.31 (m, 3H), 7.50 (m, 4H), 7.84 (d, J= 8.4 Hz, 2H), 7.89 (s, 1H); MSfES ) m/∑ 536 (MH+).
Example 222
Ar-(2-(l-ethyl-lH-pyrazol-4-yl)-2'-(trifl oromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyI)acetamide

Step 2: To a solution of 4i4,5i5-tetramethyl-2-(4-nitro-2'-(trifluoromethoxy)-[l, -biphenyl]-2- yl)-l,3,2-dioxaborolane (701 mg), 4-bromo-l-ethyl-lH-pyrazole (200 mg) and Cs2C03 (447 mg) in 1 ,4-dioxane (15 mL) and water (5 mL) stirred under nitrogen at RT was added PdCl2(dppf) (41.8 mg) in one charge. The reaction mixture was stirred at 100°C for 20 mins in the microwave. After cooling to room temperature, the mixture was filtered through eelite, washed with EtOAc. The filtrate was concentrated under reduced pressure and the residue was partitioned between water and EtOAc. The organic layer was washed with brine, dried over anhydrous Na2S04} and then purified by prep-TLC (PE : EA = 1: 1) to give l -ethyl-4 4-mtro-2'-(trifluoromethoxy)-[l,r-biphenyI]-2-yl)-lH-pyrazole (120 mg). MS(ES+) m/z 378 (MtT).
Step 3: To a solution of l-ethyl-4-(4-nitro-2'-(trifluoromethoxy)-[l,r-biphenyl]-2-yl)-lH- pyrazole (120 mg) in methanol (5 mL) was added zinc (208 mg) and NH4C1 (340 mg). The reaction mixture was stirred at RT for 4 hrs. The mixture was filtered and the filtrate was concentrated in vacuo to give 2-(l-ethyl-lH-pyi-azol-4-yl)-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-amine (100 mg). MS(ES+) m/z 349 (MH+).
Step 4: A mixture of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 72,3 mg) and sulfurous dichloride (819 mg) was stirred for 1 hr. Sulfurous dichloride was removed in vacuum. The residue was dissolved in DCM (3 mL) and added dropwise to a solution of 2-(l-ethyI-lH-pyrazol-4- yl)-2'-(trifluoromethoxy)-[l, -biphenyl]-4-amine (100 mg) and triethylamine (87 mg) in DCM (3 mL). The mixture was stirred under nitrogen for 4 hrs. Solvent was evaporated in vacuo. The crude product was purified by prep-HPLC to give N-(2-(l-ethyl-lH-pyrazol-4-yl)-2,-(trifluoromethoxy)- [l,r-biphenyl]-4-yl)-2-(4-(ethyIsulfonyl)phenyl)acetamide (14.1 mg). Ή-NMR (400 MHz, MeOD-t/4) δ ppm 1.20 (t, J= 3.2 Hz, 3H), 1.34 (t, J= 7.2 Hz, 3H), 3.29 (q, J= 7.6 Hz, 2H), 3.88 (s, 2H), 4.35 (q, J= 7.2 Hz, 2H), 7.16 (s, 1H), 7.20 (d, J= 4.8 Hz, 2H), 7.35 (m, 4H), 7.78 (d, J= 4.2 Hz, 1H), 7.86 (d, J= 7.2 Hz, 2H), 7.81(s, 1H), 7.88 (d, J- 4.8 Hz, 2H); MS(ES » m/z 558 (MH+).
Example 223
2-(4-(ethylsulfonyl)phenyI)-A'-(2-(l-isopropyl-lH-pyrazol-4-yl)-2'-(trifluoromethoxy)-[l,l'- biphenyl]-4-yI)acetamide

Step 1: A solution of 4-bromo-lH-pyrazole (500 mg), 2-iodopropane (694 mg) and CS2CO3 (1108 mg) in acetonitrile (10 inL) stirred under nitrogen at 80°C for 3 hrs. After cooling to room temperature, the mixture was filtered tlirough celite. The filtrate was concentrated under reduced pressure to give 4-bromo-l-isopropyl-lH-pyrazole (600 mg). MS(ES+) m/z 191 (MH+).
Step 2: To a solution of 4J4,5;5-tetramethyl-2-(4-nitro-2'-(trifluoromethoxy)-[l,r-bipheny]]-2- yl)-l,3,2-dioxaborolane (649 mg), 4-bromo-l-ethyl-lH-pyrazole (200 mg) and Cs2C03 (414 mg) in 1,4-dioxane (15 mL) and water (5 mL) stirred under nitrogen at room temperature was added PdCl2(dppf) (38.7 mg) in one charge. The reaction mixture was stirred at 100°C for 20 mins in the microwave. After cooling to room temperature, the mixture was filtered tlirough celite, and washed with EtOAc. The filtrate was concentrated under reduced pressure and the residue was partitioned between water and EtOAc. The organic layer was washed with brine and dried over anhydrous Na2S04. The residue was purified by prep-TLC (PE : EA = 1 : 1) to give l-isopropyl-4-(4-nitro-2'- (trifluoromethoxy)-[l, -biphenyl]-2-yl)-lH-pyrazoIe (120 mg). MS(ES+) m/z 392 (MEf).
Step 3: To a solution of l-isopropyl-4-(4-nitro-2,-(trifluorometlioxy)-[l,r-biphenyl]-2-yl)-lH- pyrazole (120 mg) in methanol (5 ml) was added zinc (200 mg) and NH4C1 (328 mg). The reaction mixture was stirred at T for 4 hrs. The mixture was filtered and the filtrate was concentrated in vacuo to give 2-(l -isopropyl-lH-pyrazol-4-yl)-2'-(trifiuorometlioxy)-[l ,l ,-biphenyl]-4-ainine (100 mg). MSiES m/z 363 (MH+).
Step 4: A mixture of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate l a, 83 mg) and SOCl2
(3 mL) was stirred for 1 hr. Sulfurous dichloride was removed in vacuum. The residue was dissolved in DCM (3 mL) and added dropwise to a solution of 2-(l-isopropyl-lH-pyrazol-4-yl)-2'~
(trifluoromethoxy)-[l,r-biphenyl]-4-amine (120 mg) and triethylamine (101 mg) in DCM (3 mL). The mixture was stirred under nitrogen for 4 hrs. Solvent was evaporated in vacuo. The crude product was purified by prep-HPLC to give 2-(4-(ed ylsulfonyl)phenyl)-N-(2-(l -isopropyl-lH-pyrazol-4-yl)- 2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)acetamide (17.8 mg). Ή-NMR (400 MHz, MeOD-<¾) δ ppm 1.20 (t, J- 3.2 Hz, 3H), 1.34 (d, J= 6.8 Hz, 6H), 3.29 (q, J= 7.6 Hz, 2H), 3.88 (s, 2H), 4.35 (q, J= 7.2 Hz ,1H), 7.16 (s, 1H), 7.20 (d, J= 4.8 Hz, 2H), 7.35 (m, 4H), 7.78 (d, J= 4.2 Hz, 1H), 7.81(s, 1H), 7.86 (d, J= 7.2 Hz, 2H), 7.88 (d, J= 4.8 Hz, 2H); MS(ES+) m/z 573 (MH+).
Example 224
-i^iet lsulfon lJ hen ^-^-ie-methoxy-S-i -itrifluorometho i en ^ ridin^- yl)acetamide, hydrochloride

Step 2: To a solution of 5-bromo-6-fluoropyridin-2-ainine (1 g) in methanol (20 mL) stirred under nitrogen at 25°C was added sodium methoxide (0.297 g) in one charge. The reaction mixture was stirred at 25°C for 16 hrs. Solvent was evaporated in vacuo to give the crude product. The crude product was dissolved in EtOAc (50 mL). The organic phase was washed with 10% citric acid solution (25 mL), saturated sodium bicarbonate solution (25 mL) and saturated brine (25 mL), dried over sodium sulphate and evaporated in vacuo to give 5-bromo-6-methoxypyridin-2-amine (873 mg) as a white solid. MSfES ) m/z 203 (MH .
Step 3: To a solution of 5-bromo-6-methoxypyridin-2-amine (609 mg), (2- (trifIuoromethoxy)phenyl)boronic acid (1236 mg) and sodium carbonate (954 mg) in 1,4-dioxane (15 mL) and water (5 mL) stirred under nitrogen at 25°C was added Pd(Ph3P)4 (347 mg) in one charge. The reaction mixture was stirred at 100°C for 16 hrs. EtOAc (50 mL) was added into the mixture. The organic phase was washed with water (10 mL), saturated brine (10 mL), dried over sodium sulphate and evaporated in vacuo to give the crude product. The crude product was purified by column chromatography (PE : EA = 5: 1 to 2: 1) to obtain 6-methoxy-5-(2-(trifIuoromethoxy)phenyl)pyridin- 2-amine (576 mg) as a yellow solid. MSiES ) m/z 285 (MH+).
Step 4: To a solution of 6-methoxy-5-(2-(trifluoromethoxy)phenyl)pyridin-2-amine (426 mg) and Et3N (0.627 mL) in DCM (5 mL) stirred under nitrogen at 0°C was added a solution of 2-(4- (ethylsulfonyl)phenyl)acetyl chloride (intermediate 170a, 555 mg) in DCM (5 mL) dropwise during 15 mins. The reaction mixture was stirred at 0°C for 1 hr. The mixture was allowed to warm to 25°C and stirred at 25°C for another 16 hrs. Solvent was evaporated in vacuo to give the crude product. The crude product was purified by prep-HPLC to give 2-(4-(ethylsuIfonyI)phenyl N-(6-methoxy-5-(2- (trifluoromethoxy)phenyl)pyridin-2-yl)acetamide, hydrochloride (170 mg) as a yellow solid. ]H-
N R (400 MHz, DMSO-c?6) δ ppm 1.03 (t, J= 7.6 Hz, 3H), 3.27 (q, J= 7.5 Hz, 2H), 3.85 (s, 3H), 3.93 (s, 2H), 7.43 (m, 3H), 7.49 (m} 1H), 7.63 (t, J= 6.8 Hz, 3H), 7.26 (d, J-8.0 Hz, 1H), 7.85 (d, 8.2 Hz, 2H), 10.72 (s, 1H); MSfES^I m/z 495 (MH ).
Example 225
iV-(3-ch]oro-4-isobutoxy-5-(2-methylpyridin-4-yl)phenyl)-2-(4-(ethyIsulfonyl)phenyl)acetamide

Step 1: A mixture of 2-chloro-4-nitrophenol (1.0 g) and NBS (1.026 g) in DMF (100 mL) was a stirred at 100°C for 3 hours. After cooling to room temperature, EtO Ac (100 mL) was added and the mixture was washed with brine (lOOmL x 6). The organic layer was dried over anhydrous Na2S04, filtered and concentrated to give 2-bromo-6-chloro-4-nitrophenol (1.3 g). MSfES4) m/z 252 (MH*); Ή-NMR (400 MHz, DMSO-c/6) δ ppm 7.96 (s, 1H), 8.11 (s, 1H).
Step 2: To a stirred mixture of 2-bromo-6-chloro-4-nitrophenol (1.3 g) and K2CO3 (0.854 g) in DMF (100 mL) was added l-bromo-2-methylpropane (0,672 mL) in one portion at room temperature. The mixture was heated to 100°C overnight. After cooling to room temperature, EtO Ac ( 1 OOmL) was added and the mixture was washed with brine (100 mL x 6). The organic layer was dried over anhydrous Na2S04, filtered and concentrated to give l-bromo-3-chloro-2-isobutoxy-5 -nitrobenzene (950 mg) as a yellow oil. !H-TvlMR (400 MHz, CDC13) δ ppm 1.11 (d, J= 6.8 Hz, 6H), 2.21 (m, 1H), 3.88 (d, J= 6.0 Hz, 2H), 8.24 (s, 1H), 8.36(s, 1H); MSfES4) m/z 308 (MH ).
Step 3: To a mixture of l-bromo-3-chloro-2-isobutoxy-5-nitrobenzene (950 mg) and zinc (1610 mg) in methanol (30 mL) and water (30 mL) was added ammonium formate (1553 mg) in one portion at room temperature under N2. The reaction mixture was stirred at 80°C for 2 hours. The reaction mixture was cooled to room temperature and filtered. The filtrate was evaporated under reduced pressure. The residue was dissolved in ethyl acetate (100 mL) and washed with brine (100 mL). The organic layer was concentrated to afford 3-bromo-5-chloro-4-isobutoxyaniline (790 mg) as a brown oil. MSiES4) m/z 278 (MH4 .
Step 4: To a stirred mixture of 3-bromo-5-chloro-4-isobutoxyaniline (400 mg), (2- methylpyridin-4-yl)boronic acid (197 mg) and K2C03 (198 mg) in 1,4-dioxane (50 mL) and water (10 mL) was added Ρ (Ρ1¾Ρ)4 (1659 mg) in one portion at room temperature under N2. The reaction mixture was stirred at 100°C overnight. The mixture was cooled to room temperature and filtered.
Solid was washed with EtOAc (50 mL). The combined filtrates and washings were concentrated under reduced pressure to give the crude product, which was purified by column chromatography (EA : PE = 1 :5) to afford 3-chloro^-isobutoxy-5-(2-methylpyridin^t-yI)aniline (320 mg) as a yellow oil. MSfES4) m/z 291 (MH*).
Step 5: A mixture of 3-chloro-4-isobutoxy-5-(2-methylpvridin-4-yl)aniline (320 mg), 2-(4-
(ethylsulfonyl)phenyl)acetic acid (intermediate la, 301 mg), HOBt (169 mg), EDC (633 mg) and DIPEA (0.577 mL) in tetrahydrofuran (THF) (50 mL) was stirred at 40°C overnight under N2. The mixture was cooled to room temperature and washed with brine (20 mL). The organic layer was concentrated to give the crude product, which was purified by column chromatography (DCM :
MeOH = 30: 1) to afford N-(3-chloro-4-isobutoxy-5-(2-methylpyridin-4-yl)phenyl)-2-(4-
(ethylsulfonyl)phenyl)acetamide (1 10 mg) as a white solid. 'H-NMR (400 MHz, CDC13) δ ppm 0.83 (d, J= 6.8 Hz, 6H), 1.28 (t, J= 7.2 Hz, 3H), 1.83 (t, J= 6.8 Hz, 1H), 2.58 (s, 3H), 3.1 1 (q, J= 7.2 Hz, 2H), 3.30 (d, J= 6.8 Hz, 2H), 3.79 (s, 2H), 7.26 (s, 1H), 7.35 (s, 1H), 3.39 (s, 1H), 7.50 (d, J= 7.2 Hz, 2H), 7.69 (s, 1H), 7.84 (d, J= 7.2 Hz, 2H), 7.93 (s, 1H), 8.49 (s, 1H); MS(ES+) m z 501 (MH+). Example 226
Ar-(3-chloro-5-(pyridin-4-yl)phenyl)-2-{4-(ethyIsuJfonyl)phenyI)acetamide

Step 1: A mixture of 1 ,3-dibromo-5-chlorobenzene (0.5 g), Cs2C03 (1.205 g), Pd(OAc)3 (0.041 g), diphenylmethanimine (0.335 g) and ΒΓΝΑΡ (0.067 g) in toluene (20 mL) was stirred at 70°C overnight under 2. After cooling to room temperature, the mixture was filtered and the filtrate was evaporated under reduced pressure. To the residue was added water (70 mL) and extracted with ethyl acetate (100 mL x 3). The combined organics were dried over anhydrous Na2S04, filtered and concentrated under reduced pressure to give 3-bromo-5-chloro-N-(diphenylmetliylene)aniline (500 mg) as a yellow solid. MSiES4) ni/z 370 (MIT).
Step 2: To a solution of 3-bromo-5-chloro-N-(diphenylmethylene)aniline (500 mg) in tetrahydrofuran (20 mL) was added trifluoroacetic acid (3 mL). The mixture was stirred for 2 hours at room temperature. After the reaction was complete, saturated sodium carbonate solution was added to adjust the mixture to pH 8-9. The mixture was extracted with ethyl acetate (50 mL x 2). The combined organics were washed widi brine, dried over anhydrous Na2S04, filtered and concentrated to give 3-bromo-5-chIoroaniline (500 mg) as a yellow oil. MS^S4 m/z 206 (MH4).
Step 3: A mixture of 3-bromo-5-chloroaniline (500 mg), pyridin-4-ylboronic acid (447 mg), K2C03 (1004 mg) and Pd(Ph3P)4 (280 mg) in 1,4-dioxane (20 mL) and water (4 mL) was stirred at 100°C overnight under N2. The mixture was cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure. To the residue was added water (30 mL) and adjusted to pH - 2 with 5 M HCl. The mixture was washed with DCM (30 mL x 3). The aqueous was adjusted to pH 10 with 10 M NaOH solution and then extracted with EtOAc (30 mL x 3). The combined organics were dried over anhydrous Na2S04, filtered and concentrated in vacuo to afford 3-chloro-5-(pyridin-4- yl)aniline (100 mg) as a light yellow solid. MSfES" m/z 205 (ΜΗ ).
Step 4: A mixture of 3-chloro-5-(pyridin-4-yl)aniline (170 mg), 2-(4- (ethyIsulfonyl)phenyl)acetic acid (intermediate la, 228 mg), EDC (318 mg), HOBt (127 mg) and
DIPEA (0.435 mL) in tetrahydrofuran (THF) (20 mL) was stirred overnight at room temperature. The mixture was evaporated under reduced pressure. To the residue was added EtOAc (30 mL). The resulting solid was collected and further purified by preparative HPLC to afford N-(3-chloro-5- (pyridin-4-yI)phenyl)-2-(4-(ethylsulfonyl)phenyI)acetamide (120 mg). 'H-NMR (400 MHz, DMSO- J6) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.86 (s, 2H), 7.57 (s, 1H), 7.65 (m, 4H), 7.87 (m, 4H), 8.67 (d, J= 5.6 Hz, 2H), 10.63 (s, 1H); MS(ES+) m/z 415 (MH+).
Example 227
/v"-(3-chloro-5-(l-methyl-lH-pyrazol-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide, triflu or o acetic acid salt

Ste l: Et3N (0.229 mL) was added into a solution of 2-(4-(ethylsulfonyl)phenyl)acetic acid (intennediate la, 226 mg), 3-bromo-5-chloroaniline, hydrochloride (200 mg), 1H- benzo[< l[l ,2,3]triazol-l-oI (167 mg) and EDC (237 mg) in DCM (5 mL). The mixture was stirred at room temperature overnight. As starting material still left, 2-(4-(ethylsulfonyl)phenyl)acetic acid (intermediate la, 226 mg), EDC (237 mg) was added into the mixture, and the reaction mixture was stirred at room temperature for 2 days. The mixture was washed with 2M HCI, sat. NaHCOj solution and brine successively. The organic phase was dried over anhydrous Na2S04. After filtration, the filtrate was concentrated under reduced pressure and the residue was purified by column
chromatography (EA : PE = 0: 1 to 1: 1) to afford N-(3-bromo-5-chlorophenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide (320 mg) as a yellow solid. MS(ES+) m/z 416 (MH+).
Step 2: A mixture of l-methyl-4-(4J4,5,5-tetramethyl-l,3;2-dioxaborolan-2-yl)-lH-pyrazole (65.9 mg), N-(3-bromo-5-chlorophenyl)-2-(4-(ediyIsulfonyl)phenyl)acetamide (120 mg),
PdCl2(dppf)-CH2Cl2 adduct (18.81 mg) and Cs2C03 (113 mg) in acetonitrile (1.5 mL)/water (0.5 mL) was sealed in a vessel and heated in the microwave at 100°C for 45 mins. The reaction mixture was filtered through celite and silica gel. The filtrate was concentrated under reduced pressure and the residue was purified by MDAP to afford N-(3-chloro-5-(l-methyl-lH-pyrazol-4-yl)phenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt (43 mg) as an off-white solid. Ή-ΝΜΚ (400 MHz, DMSO-i/fi) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J= 7.2 Hz, 2H), 3.82 (s, 2H), 3.86 (s, 3H), 7.35 (t, J= 1.6 Hz, 1H), 7.57 (t, J = 1.6 Hz, 1H), 7.61 (d, J= 8.4 Hz, 2H), 7.64 (t, J= 1.6 Hz, 1H), 7.82 (s, 1H), 7.85 (d, J= 8.4 Hz, 2H), 8.15 (s, 1H), 10.46 (s, 1H); MS(ES+) m/z 418 (MH+).
Example 228
2-[4-(ethylsulfonyl)phenyl]-N-(7-{2-[(trifluoromethyl)oxylphenyI}-lH-indoI-4-yl)acetamide

Step 1: A mixture of 4-chloro-3-nitroaniline (1.2 g), [4-(ethylsulfonyl)phenyl]acetic acid (intermediate la, 1.746 g), EDC (1.600 g) and HOBt (1.128 g) in DCM (15 mL) was stirred at room temperature under nitrogen. Tetrahydrofuran (THF) (5 mL) and DMF (5 mL) were added to the above mixture to increase the solubility. The resutling mixture was stirred at room temperature overnight. The mixture was diluted with DCM, washed with 1 M HC1, sat. Na2C03 and brine for several times, dried over anliydrous Na2SC>4. After filtration, solvent was removed in vacuo to afford N-(4-chloro-3-nitrophenyl)-2-[4-(ethylsulfonyl)phenyI]acetamide (2.768 g) as a brown oil. MS(ES+) m/z 383 (MH4).
Step 2: To a solution of N-(4-chloro-3-nitrophenyl)-2-[4-(ethylsulfonyl)phenyl]acetamide (1.8 g) in tetrahydrofuran (THF) (12 mL) was added a solution of vinylmagnesium bromide (1 M in THF, 19.75 mL) within 30 mins at -40 °C. The resulting mixture was stirred at this temperature for 1 hour. More vinylmagnesium bromide (1 M in THF, 9.87 mL) was added and the mixture was gradually wanned to room temperature and stirred overnight. The mixture was quenched with sat. NH4C1 solution and was stirred for 30 mins, then extracted with EtOAc. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After concentration, the residue was purified by
flash chromatography (EA : PE = 0: 1 to 4: 1) to afford N-(7-chIoro-lH-indol-4-yl)-2-[4- (ethylsulfonyl)phenyl]acetamide (1 g) as a brown solid. MSfES') m/z 377 (MH÷).
Step 3: A mixture of N-(7-chloro-lH-indol-4-yl)-2-[4-(ethylsulfonyl)phenyl]acetamide (1 g), dicyclohexyip'^'^'-tristl-methylethy ^-biphenylylJphosphane (0.406 g), Pd2(dba)3 (0.195 g), bis(pinaco!ato)diboron (1.2134 g) and potassium acetate (0.782 g) in 1 ,4-dioxane (15 lnL) was bubbled with N2. The mixture was sealed in a reaction vessel and heated in the microwave at 110°C for 1 hour. The mixture was filtered tlirough celite and the filtrate was partitioned between water and EtOAc. The organic layer was washed with brine and dried over anhydrous Na2S04. After concentration, the residue was purified by flash chromatography (EA : PE = 0:1 to 7:3) to afford 2-[4- (ethylsulfonyl)phenyl]-N-[7-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-indol-4-yl]acetamide (496 mg) as a brown solid. MSfES ) m/z 469 (MH+).
Step 4: A mixture of 2-bromophenyl trifluoromethyl ether (272 mg), 2-[4- (ethylsulfonyl)phenyl]-N-[7-(4,4,5J5-tetramethyl-l!3,2-dioxaborolan-2-yl)-lH-indol-4-yl]acetamide (480 mg), Pd(Pl¾P)4 (118 mg) and 2M aqueous Na2C03 (2.56 mL) in 1 ,4-dioxane (7.5 mL) was bubbled with N2. The mixture was sealed in a reaction vessel and heated in the microwave at 100°C for 1 hour. The mixture was partitioned between water and EtOAc. The organic layer was washed with brine and dried over anhydrous Na2S04. After concentration, the residue was purified by flash chromatography (EA : PE = 0:1 to 7:3) to afford the crude product (214 mg) as a yellow solid. Part of this crude (110 mg) was dissolved in DMF (7 mL) and further purified by MDAP to afford pure 2-[4- (ethyIsulfonyl)phenyl]-N-(7-{2-[(trifluorome-liyl)oxy]phenyl}-lH-indol-4-yl)acetamide (20 mg) as a white solid. Ή-NMR (400 MHz, MeOD-rf4) 5 ppm 1.22 (t, J= 7.4 Hz, 3H), 3.21 (q, J= 7.4 Hz, 2H), 3.99 (s, 2H), 6.64 (d, J= 3.2 Hz, 1H), 7.02 (d, J= 7.9 Hz, 1H), 7.22 (d, J= 3.2 Hz, 1H), 7.44-7.55 (m, 5H), 7.71 (d, J= 8.3 Hz, 2H), 7.91 (d, J= 8.3 Hz, 2H); 19F-NMR (376 MHz, DMSO-i/6) 5 ppm -60.53; MS(ES+) z 503 (MH+). Example 229
2-[4-(ethylsulfonyl)phenyl]-N-(4-{2-[(trifluoromethyl)oxy]phenyl}-lH-indol-7-yl)acetamide

Step 1: To a solution of 4-bromo-2-nitroaniline (5 g) and Et3N (9.63 mL) in DCM (40 mL) cooled at 0°C, was added trifluoroacetic anhydride (6.51 mL) dropwise slowly. The resulting mixture was wanned to room temperature and stirred overnight. The mixture was washed with water, 2M HCl
and brine, then dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford N-(4-bromo-2-nitrophenyI)-2,2,2-trifluoroacetamide (8.191 g) as a yellow solid. MS(ES+) m z 311 (MH .
Step 2: To a solution of N-(4-bromo-2-nifrophenyl)-2,2,2-trifluoroacetamide (4 g) in THF (30 mL) cooled at -40°C, was added a solution of vinylmagnesium bromide (1 M in THF, 77 mL) dropwise within 30 mins. The resulting mixture was stirred at this temperature for 2 hours. Upon completion, sat. NH4C1 was added. The mixture was gradually warmed to room tem erature and stirred for 30 mins. The mixture was extracted with EtOAc for several times. The combined organic layers were washed with brine and dried over anhydrous Na2S04. After concentration, the residue was purified by flash chromatography (EA : PE = 0:1 to 4: 1) to afford N-(4-bromo-lH-indol-7-yl)-2,2,2- trifluoroacetamide (1.252 g) as a brown solid. MSiES1 m/z 307 (MH+).
Step 3: A mixture of {2-[(trifluoromethyl)oxy]phenyI}boronic acid (0.823 g), N-(4-bromo-lH- indol-7-yl)-2,2}2-trifluoroacetamide (1.168 g), PdCl2(dppf)-CH2Cl2 adduct (0.250 g) and Cs2C03 (1.860 g) in acetonitrile (12 mL) and water (4 mL) was bubbled with N2. The mixture was sealed in a reaction vessel and heated in the microwave at 100°C for 1 hour. The mixture was filtered through celite, washed with ACN and EtOAc. The filtrate was concentrated in vacuo and the residue was partitioned between water and EtOAc. The organic layer was washed with brine and dried over anhydrous a2S04- After concentration, the residue was purified by flash chromatography (EA : PE = 0: 1 to 4:1) to afford 4-{2-[(trifIuoromethyl)oxy]phenyl}-lH-indol-7 -amine (533 mg) as a brown solid. MSfES" m/z 293 (MH+).
Step 4: A mixture of 4-{2-[(trifluorometliyl)oxy]phenyl}-lH-indol-7-amine (180 mg), [4- (ethyIsulfonyl)phenyl]acetic acid (intermediate la, 155 mg), EDC (142 mg) and HOBt (100 mg) in DCM (6 mL) was stirred at room temperature overnight. The mixture was diluted with DCM, washed with water, sat. Na2C03 and brine, dried over anhydrous Na2S04. After filtration, solvent was removed in vacuo to afford the crude product (299 mg) as a black solid. Part of this crude (150 mg) was dissolved in DMF (7 mL) and further purified by MDAP to afford pure 2-[4- (ethylsulfonyl)phenyl]-N-(4- {2-[(trifluoromethyl)oxy]phenyl}-lH-indol-7-yl)acetamide (32 mg) as an off-white solid. Ή-NMR (400 MHz, DMSO- ) δ ppm 1.23 (t, J= 7.4 Hz, 3H), 3.21 (q, J~ 7.4 Hz, 2H), 3.99 (s, 2H), 6.27 (d, J= 3.2 Hz, 1H), 6.99 (d, J= 7.8 Hz, 1H), 7.23 (d, J= 7.8 Hz, 1H), 7.29 (d, J= 3.2 Hz, 1H), 7.40-7.49 (m, 3H), 7.54 (dd, J= 2.0 Hz, 7.5 Hz, lH), 7.72 (d, J= 8.3 Hz, 2H), 7.91 (d, J= 8.3 Hz, 2H); 19F-NMR (376 MHz, DMSO- ) δ ppm -58.80; MSiES^ m/z 503 (MH+).
Example 230
N-(3-chloro-4-isobutoxy-5-(l-methyl-lH-pyrazol-4-yl)phenyl)-2-(4- (ethylsulfonyl)phenyl)acetamide, trifluoro acetic acid salt

The title compound was prepared using a similar procedure to that described for Example 225. 'H-NMR (400 MHz, DMSO-t?6) δ ppm 0.97 (d, J= 6.8 Hz, 6H), 1.10 (t, J= 7.4 Hz, 3H), 2.03 (m, IH), 3.28 (q, J= 7.4 Hz , 2H), 3.45 (d, J= 6.5 Hz, 2H), 3.80 (s, 2H), 3.88 (s, 3H), 7.60 (m, 3H), 7.66 (d, J= 2.5 Hz, IH), 7.75 (s, IH), 7.85 (d, J= 8.3 Hz, 2H), 8.03 (s, IH), 10.38 (s, IH); MS(ES+) m/z 490 (MH+).
Example 231
N-(2,6-dkhloro-2'-(trifluoromethoxy)-[1 '-biphenyI]-4-yl)-2-(3-(ethylsulfonyl)-lH-pyrazol-^ yl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. H-
NMR (400 MHz, CDC13) δ ppm 1.40 (t, J= 7.5 Hz, 3H), 3.32 (q, J= 7.5 Hz, 2H), 5.30 (s, 2H), 6.93 (d, J= 2.5 Hz, IH), 7.25 (m, IH), 7.38 (m, 2H), 7.48 (m, IH), 7.70 (s, 2H), 7.74 (d, J= 2.5 Hz, H), 8.96 (s, IH); MS(ES+) m/z 522 (MH+). Example 232
2-(4-(ethyIsulfonyl)phenyl)-7V-(5-fluoro-6-(2-(trifluoromethoxy)phenyl)pyridin-3-yl)acetamide, hydrochloride

The title compound was prepared using a similar procedure to that described in Scheme 2. H-
NMR (400 MHz, CDC13) δ ppm 1.28 (t, J= 7.6 Hz, 3H), 3.13 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 6.98
(d, J= 8.8 Hz, 2H), 7.43 (dd, J= 7.2 Hz, 18.4 Hz, 2H), 7.53 (m, 4H), 7-89 (d, J= 7.6 Hz, 2H),7.50 (d, J= 10.4 Hz, 2H), 8.57 (s, 1H); MSfES^ m z 483 (MH*).
Example 233
2-(4-(ethylsulfonyl)phenyI)-N-(2-(l-methyl-lH-pyrazol-3-yl)-2'-(trifluoromethoxy)biphenyl-4- yl)acetamide

The title compound was prepared using a similar procedure to that described for Example 69. 'H-NMR (400 MHz, MeOD-c¾) δ ppm 1.12 (t, J= 7.6 Hz, 3H), 3.11 (q, J= 7.6 Hz, 2H), 3.26 (s, 3H), 3.72 (s, 2H), 6.05 (s, 1H), 6.98 (d, J= 8.8 Hz, 2H), 7.33 (s, 1H), 7.34 (m, 1H), 7.43 (m, 4H),7.51 (d, J = 8.4 Hz, 2H), 7.78 (t, J= 8.4 Hz, 2H); MS(ES+) m z 544 (MH ).
Example 234
7V-(3,4-dichIoro-5-(l-methyl-lH-pyrazol-4-yI)phenyl)-2-(4-(ethylsulfonyI)phenyI)acetamide

The title compound was prepared using a similar procedure to that described for Example 227.
Ή-NMR (400 MHz, DMSO-£¾) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 3.28 (q, J= 7.4 Hz, 2H), 3.82 (s, 2H), 3.90 (s, 3H), 7.43 (d, J= 8.7 Hz, 1H), 7.49 (dd, J= 2.5 Hz, 8.7 Hz, 1H), 7.61 (d, J= 8.3 Hz, 2H), 7.72 (d, J= 0.7 Hz, 1H), 7.82 (d, J= 2.4 Hz, 1H), 7.85 (d, J- 8.4 Hz, 2H), 8.11 (s, 1H), 10.42 (s, 1H); MS(ES+) m/z 418 (MH4 . Example 235
JV-(2-(lJ/-imidazol-l-yI)-2,-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsuIfonyl)phenyl)acetamide, hydrochloride

The title compound was prepared using a similar procedure to that described for Example 198 (or state "described in Scheme 2"). 1 H-NMR (400 MHz, MeOD-<¾ 6 ppm 1.2 (t, J— 7.2 Hz, 3H), 3.2 (t, J=7.2 Hz, 2H), 7.3 (d, J= 7.6 Hz, IH), 7.5 (m, 5H), 7.6 (d, - 8.0 Hz, 2H), 7.7 (d, J= 2.4 Hz, IH), 7.9 (d, J= 8.4 Hz, IH), 8.3 (d, J= 2.0 Hz, 2H), 9.1 (s, IH); l9F-NMR (376 MHz, MeOD-rf4) δ ppm -58; MS(ES÷) m/z 530 (MH^.
Example 236
Ar-(3,4-dichloro-5-(l-methyl-lH-pyrazol-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide

The title compound was prepared using a similar procedure to that described for Example 227. ]H-NMR (400 MHz, DMSO-rf6) δ ppm 1.10 (t, J= 7.4 Hz, 3H), 3.29 (dd, J= 7.4 Hz, 14.8 Hz, 2H), 3.84 (s, 2H), 3.91 (s, 3H), 7.61 (d, J= 8.4 Hz, 2H), 7.67 (d, J= 2.5 Hz, IH), 7.74 (d, J= 0.6 Hz, IH), 7,87 (m, 3H), 8.16 (s, IH), 10.56 (s, IH); MS(ES+) m/z 452 (MH+). Example 237
JV-(4-chloro-3-(l-propyHH-pyrazol-5-yl)phenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, MeOD-rf4) δ ppm 0.76 (t, /= 7.6 Hz, 3H), 1.21 (t, /= 7.6 Hz, 3H), 1.75 (m, 2H), 3.2 (q, J= 7.2 Hz, 2H), 3.85 (s, 2H), 4.04 (t, J= 7.2 Hz, 2H), 6.5 (d, J= 2.3 Hz, IH), 7.54 (d, J= 8.8 Hz, IH), 7.62 (d, J= 8.3 Hz, 2H), 7.67 (dd, J= 2.5 Hz, 8.4 Hz, IH), 7.80 (d, J= 2.4 Hz, IH), 7.84 (d, J= 2.4 Hz, IH), 7.88 (d, J= 8.3 Hz, 2H); MSfES"") m/z 446 (MH^).
Example 238
2-(4-(ethyIsulfonyl)phenyl)-N-(2-(2-methyloxazol-5-yI)-2'-(trifluoromethoxy)-[l,l,-biphenyJ]-4- yl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή-
NMR (400 MHz, MeOD-£¾) δ ppm 1.20 (t, J= 7.6 Hz, 3H), 2.43 (s, 3H), 3.2 (t, J= 7.6 Hz, 2H), 3.89 (s, 2H)} 6.15 (s, IH), 7.2 (d, /= 8.4 Hz, IH), 7.37 (m, 2H), 7.45 (m, IH), 7.55 (m, IH), 7.65 (m, 3H), 7.90 (d, = 8.4 Hz, 2H), 8.15 (d, J= 1.6 Hz, IH); ,9F-NMR(376 MHz, MeOD-^) δ ppm -58;
MS(ES+) m/z 545 (MH+). Example 239
N-(2}6-dichloro-2'-(trifluoromethoxy)-[l,r-biphenyI]-4-yl)-3-(3-(ethylsulfonyl)-lH-pyrazol-l- yI)propanamide

The title compound was prepared using a similar procedure to that described in Scheme 2. 'H- NMR (400 MHz, DMSO-i/6) S ppm 1.06 (t, J= 7.3 Hz, 3H), 3.00 (t, J= 6.5 Hz, 3H), 3.22 (q, J= 7.4 Hz, 2H), 4.57 (t, J= 6.7 Hz, 2H), 6.78 (d, J~ 2.3 Hz, IH), 7.40 (dd, J= 1.6 Hz, 7.9 Hz, IH), 7.52 (t, J = 7.0 Hz, 2 H), 7.61 (m, IH), 7.79 (s, 2H), 8.04 (d, 2.5 Hz, IH), 10.50 (s, IH); 19F-NMR (376 MHz, DMSO-<¾) δ ppm -56; MS(ES+) m/z 536 (MH ).
Example 240
2-(4-(ethylsulfonyl)phenyl)-iV-(3-(l-meihyI-Lir-pyrazoI-3-yl)-4- (trifluoromethoxy)phenyI)acetamide

Example 241
7Y-(4,-chIoro-2,-(difluoromethoxy)-2-methyl-[l,l'-biphenyl]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide

The title compound was prepared using a similar procedure to that described for Example 176.
Ή-NMR (400 MHz, DMSO-c 6) δ ppm 2.02 (s, 3H), 3.20 (s, 3H), 3.80 (s, 2H), 7.06 (d, J= 8.4 Hz, 1H), 7.20 (t, 7= 73.6 Hz, 1H), 7.28 (d, J= 8.4 Hz, 1H), 7.38 (m, 2H), 7.47 (dd, J= 1.8 Hz, 8.3 Hz, 1H), 7.52 (d, J= 1.6 Hz, 1H), 7.61 (d, J= 8.3 Hz, 2H), 7.90 (d, J= 8.3 Hz, 2 H), 10.30 (s, 1 H);
MS(ES+) m/z 480 (MH÷). Example 242
N-(l-ethyl-7-(l-methyI-lH-pyrazol-4- !)-lH-indol-5-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή
NMR (400 MHz, CDC13) δ ppm LOO (m, 3H), 1.27 (m, 3H), 3.11 (m, 2H), 3.71 (m, 2H), 3.84 (m, 2H), 4.18 (m, 3H), 6.50 (d, J= 2.8 Hz, 1H), 7.07 (d, J= 2.8 Hz, 2H), 7.40 (m, 1H), 7.59 (m, 2H), 7.67 (m, 1H), 7.80 (m, 1H), 7.85 (m, 2H), 8.07 (m, 1H); MS(ES+) m/z 451 (MH+).
Example 243
iV-(l-(cyclopropylmethyl)-7-(l-methyl-lH-pyrazol-4-yl)-lH-indol-5-yl)-2-(4- (ethy!sulfonyl)phenyI) acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή NMR (400 MHz, DMSO-</6) δ ppm 0.10 (m, 2H), 0.28 (m, 2H), 0.80 (m, 1H), 1.10 (t, J= 7.2 Hz, 3H), 3.28 (q, J<= 7.2 Hz, 2H), 3.75 (m, 4H), 3.92 (s, 3H), 6.43 (d, J= 3.2 Hz, 1H), 7.05 (d, J= 2.0 Hz, 1H), 7.35 (d, J= 3.2 Hz, 1H), 7.55 (s, 1H), 7.62 (d, /= 8.4 Hz, 2H), 7.83 (s, 2H), 7.86 (d, = 8.4 Hz, 2H), 10.09 (s, 1H); MSiES4 m/z All (MH+).
Example 244
N-(4'-chloro-2,-(difluoromethoxy)-2-methyl-[l,l'-biphenyl]-4-yl)-2-(4- (ethylsulfonyl)phenyI)acetamide

The title compound was prepared using a similar procedure to that described in Example 176. 'H- MR (400 MHz, DMSO-</6) δ ppm 1.89 (s, 6 H), 3.20 (s, 3 H), 3.78 (s, 2 H), 7.19 (d, J= 8.4 Hz, 1 H), 7.22 (t, J= 73.6 Hz, 1 H), 7.35 (s, 2 H), 7.39 (m, 2 H), 7.61 (d, J= 8.4 Hz, 2 H), 7.89 (d, J= 8.4 Hz, 2 H), 10.21 (s, 1 H); 19F-NMR (376 MHz, DMSO-i 6) δ ppm -82; MS(ES+) m/z 494 (MH+). Example 245
r-(3'-cyano-6-(trifluoro yl)phenyl)acetamide

The title compound was prepared using a similar procedure to that described for Example 227. 'H-NMR (400 MHz, DMSO- ) δ ppm 1.09 (t, J = 1.2 Hz, 3H), 3.28 (q, J = 1.2 Hz, 2H), 3.84 (s, 2H), 7.49 (dd, J= 1.2 Hz, 7.6 Hz, 1H), 7.61 (d, J= 8.4 Hz, 2H), 7.71 (m, 2H), 7.79 (m, 1H), 7.84 (m, 3H), 7.92 (m, 2H), 10.60 (s, 1H); 19F-NMR (376 MHz, DMSO-<¾) δ ppm -57; MS(ES^) m/z 4%9 (MH+).
Example 246
N-(6-(3-chlorophenyl)-5-is henyl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή
NMR (400 MHz, DMSO-i/6) δ ppm 1.10 (t, J= 7.3 Hz, 3H), 1.26 (d, J= 6.0 Hz, 6H), 3.28 (q, J= 7.3 Hz, 2H), 3.88 (s, 2H), 4.63 (m, IH), 7.47 (m, 2H), 7.64 (m, 3H), 7.86 (d, J= 8.3 Hz, 2H), 8.00 (m, 3H)5 10.80 (s, IH); MStES*) m/z 473 (MH+).
Example 247
A"-(4-(lH-indol-4-yl)-3-methylphenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt

The title compound was prepared using a similar procedure to uiat described in Scheme 1. Ή
NMR (400 MHz, DMSO-c 6) 5 ppm 1.10 (t, J= 7.3 Hz, 3H), 2.09 (s, 3H), 3.28 (q, J= 7.3 Hz, 2H), 3.82 (s, 2H), 6.03 (s, IH), 6.82 (d, J= 6.8 Hz, IH), 7.10 (t, J= 7.7 Hz, IH), 7.18 (d, J= 8.3 Hz, IH), 7.32 (t, J=2.6 Hz, IH), 7.37 (d, J= 8.0 Hz, IH), 7.49 (dd, J= 1.9 Hz, 8.2 Hz, IH), 7.56 (d, J= 1.6 Hz, IH), 7.64 (d, J= 8.3 Hz, 2H), 7.86 (d, J= 8.3 Hz, 2H), 10.29 (s, IH), 11.18 (br s, IH); MSfES4) m/z 433 (MH+).
Example 248
2-(4-(ethyIsuIfonyl)phenyI)-7V-(3-(l-methyl-lflr-pyrazol-4-yl)-4-(2-methylprop-l-en-l- yl)phenyl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή
NMR (400 MHz, CDC13) δ ppm 1.18 (t, J= 7.6 Hz, 3H), 3.05 (q, J= 7.2 Hz, 2H), 1.66 (s, 3H), 1.81
(s, 2H), 3.73 (s, 2H), 3.89 (s, 3H), 6.07 (s, IH), 7.10 (d, J= 8.4 Hz, IH), 7.26 (d, J= 8.4 Hz, IH), 7.39 (s, IH), 7.41 (s, IH), 7.47 (d, = 8.4 Ηζ,ΙΗ), 7.52 (s, 3H), 7.82 (d, 8.0 Hz, 2H); MS(ES^) m/z 438 (Mtf).
Example 249
2-(4-(ethylsulfonyJ)phenyl)-N-(4-isobutyl-3-(l-methyl-lH-pyrazol-4-yl)phenyl)acetamide
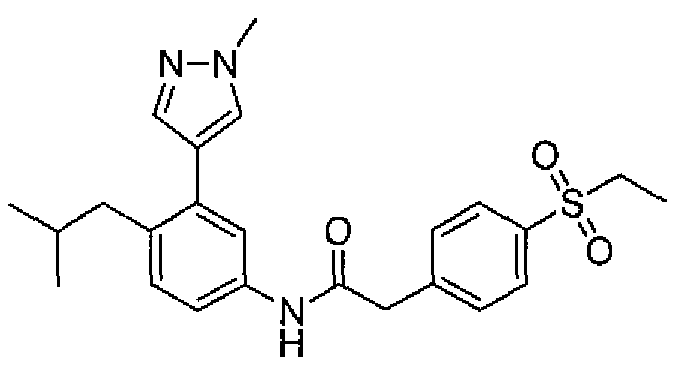
The title compound was prepared using a similar procedure to that described in Scheme 2. ]H NMR (400 MHz, CDC13) δ ppm 0.89 (d, J= 6.5 Hz, 6H), 1.31 (t, J= 7.6 Hz, 4H), 1.75 (m, IH), 2.51 (d, J= 6.8 Hz, 2H), 3.11 (q, J= 7.6 Hz, 2H), 3.80 (s, 2H), 3.94 (s, 3H), 7.19 (d, J= 8.8 Hz, IH), 7.22 (s, IH), 7.34 (m, 3H), 7.51 (s, IH), 7.54 (d, J= 8.4 Hz, 2H), 7.90 (d, J= 8.4 Hz, 2H); MS(ES+) m/z 440 (MH ).
Example 250
N-(3,5-difluoro-4-(lH-indol-4-yl)phenyl)-2-(4-(ethylsulfonyl)phenyI)acetamide

Ή NMR (400 MHz, DMSO-rf6) δ ppm 1.11 (t, J= 7.3 Hz, 3H), 3.29 (q, J= 7.4 Hz, 2 H), 3.87 (s, 2H), 6.11 (br s, IH), 6.98 (d, J= 7.0 Hz, IH), 7.17 (t, J= 7.8 Hz, IH), 7.37 (t, J= 2.8 Hz, IH), 7.45 (m, 3H), 7.63 (d, J=> 8.53 Hz, 2H), 7.87 (d, J= 8.3 Hz, 2H), 10.72 (s, IH), 11.25 (br s, IH); MS(ES^) m/z 455 (MH ). Example 251
N-(41-chloro-2'-(difluoromethoxy)-2,6-dimethyl-[l,l'-biphenyl]-4-yl)-2-(4- (methylsulfonyI)phenyI)acetamide

The title compound was prepared using a similar procedure to that described in Example 244. 'H-NMR (400 MHz, DMSO-</6) δ ppm 1.89 (s, 6H), 3.20 (s, 3H), 3.78 (s, 2H), 7.19 (d, J= 8.4 Hz, 1H), 7.22 (t, J= 36.8 Hz, 1H), 7.35 (s, 2H), 7.39 (m, 2H), 7.61 (d, J= 8.4 Hz, 2H), 7.89 (d, J= 8.4 Hz, 2H), 10.21 (s, 1H); 19F-NMR (376 MHz, DMSO-i/6) δ ppm -82; MS(ES÷) m/z 494 (MH^).
Example 252
N-(4-(3-chloro-l/T ndol-4-yl)-3-methylphenyl)-2-(4-(ethylsulfonyl)phenyl)acetamide
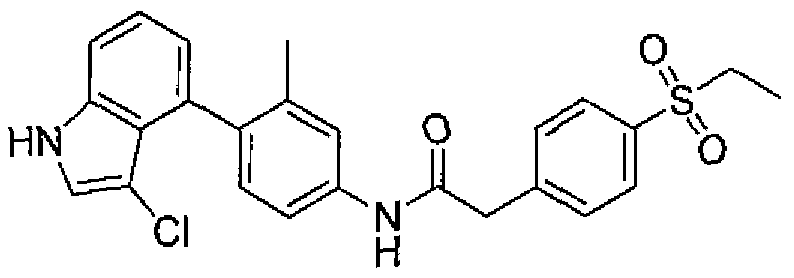
The title compound was prepared using a similar procedure to that described in Scheme 1. H NMR (400 MHz, DMSO-d6) 5 ppm 1.10 (t, J= 7.3 Hz, 3H), 1.96 (s, 3H), 3.28 (q, J= 7.4 Hz, 2H), 3.82 (s, 2H), 6.76 (d, J= 6.5 Hz, 1H), 7.08 (d, J= 8.3 Hz, 1H), 7.18 (t, .7= 7.6 Hz, 1H), 7.41 (d, J= 7.5 Hz, 1H), 7.45 (m, 2H), 7.49 (m, 1H), 7.64 (d, J= 8.3 Hz, 2H), 7.86 (d, J= 8.3 Hz, 2H), 10.26 (s, 1H), 11.47 (br s, 1H); MS(ES+) m/z 467 (MH+).
Example 253
7V-(5-(3-chIoro-lH-indoI-4-yl)-6-methylpyridin-2-yl)-2-(4-(ethylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt

The title compound was prepared using a similar procedure to that described in Scheme 1. Ή NMR (400 MHz, DMSCkfc) δ ppm 1.10 (t, = 7.3 Hz, 3H), 2.16 (s, 3H), 3.28 (q, J= 7.4 Hz, 2H), 3.89 (s, 2H), 6.84 (d, J= 7.0 Hz, 1H), 7.22 (t, J= 7.7 Hz, 1H), 7.45 (d, J= 8.3 Hz, 1H), 7.50 (d, J = 2.5 Hz, 1H), 7.54 (d, J= 8.3 Hz, 1H), 7.65 (d, J= 8.0 Hz, 2H), 7.86 (d, J= 8.3 Hz, 2H), 7.93 (d, J>= 8.3 Hz, 1H), 10.88 (s, 1H), 11.55 (br s, 1H); MS(ES+) m/z 468 (MH^.
Example 254
Ar-(3'-cyano-6-(2,2,2 phenyI)acetamide

The title compound was prepared using a similar procedure to that described for Example 227, ]H-NMR (400 MHz, MeOD-</4) δ ppm 1 .20 (t, J= 7.6 Hz, 3H), 3.1 (q, J- 7.6 Hz, 2H), 3.83 (s, 2H), 4.52 (q, J = 8.4 Hz, 2H), 7.13 (d, J= 8.8 Hz, 1H), 7.60 (m, 5H), 7.70 (m, 1H), 7.80 (m, 1 H), 7.87 (m, 3H), 10.19 (s, 1H); 19F-NMR (376 MHz, MeOO-d4) δ ppm -76; MS(ES+) m/z 503 (Μΐ ).
Example 255
iV-(3'-cyano-4'-((l-isopropylpiperidin-4-ylidene)methyI)-2-methyl-[l,l'-biphenyl]-4-yI)-2-(4- (methylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt

The title compound was prepared using a similar procedure to that described for Example 91. Ή- NMR (400 MHz, MeOD-i/4, with Na2C03 solid) δ ppm 1 .09 (m, 6H), 2.15 (br s, 1H), 2.24 (m, 3H), 2.49 (m, 1H), 2.66 (m, 4H), 3.10 (m, 5H), 3.58 (s, 1H), 3.83 (s, 2H), 5.99 (m, 1H), 7.15 (m, 1H), 7.50 (m, 4H), 7.63 (m, 3H), 7.93 (d, 2H); MS(ES+) m/z 542 (MH+).
Example 256
A'-(4,-((4-isobutj'rylpiperazin-l-yl)methy])-2-methyl-[l,l'-biphenyl]-4-yl)-2-(4- (methylsulfonyl)phenyl)acetamide, trifluoroacetic acid salt

The title compound was prepared using a similar procedure to that described in Scheme 1. Ή- NMR (400 MHz, MeOD-rf4) δ ppm 1.11 (d, J= 6.8 Hz, 6H), 2.24 (s, 3H), 2.95 (m, 1H), 3.11 (s, 3H), 3.84 (s, 2H), 4.42 (s, 2H), 7.14 (d, J- 8.3 Hz, 1H), 7.47 (m, 4H), 7.56 (d, J = 8.3 Hz, 2H), 7.63 (d, J - 8.3 Hz, 2H), 7.93 (d, J= 8.3 Hz, 2H); MS(ES+) m/z 548 (MH+).
Example 257
iV-(4'~((4-isopropylpiperazin-l-yl)methyl)-2-methyl-[l,lT-biphenyl]-4-yl)-2-(4- (methylsuIfonyl)phen

The title compound was prepared using a similar procedure to that described for Example 256.
'H-NMR (400 MHz, MeOD-dj) δ ppm 1.07 (d, J= 6.5 Hz, 6H), 2.22 (s, 3H), 2.64 (m, 9H), 3.10 (s, 3H), 3.57 (s, 2H), 3.82 (s, 2H), 7.12 (d, J= 8.3 Hz, 1H), 7.25 (d, J= 8.0 Hz, 2H), 7.37 (d, J= 7.8 Hz, 2H), 7.44 (dd, J= 2.0 Hz, 8.3 Hz, 1 H), 7.47 (s, 1 H), 7.63 (d, J= 8.3 Hz, 2H), 7.93 (d, J= 8.3 Hz, 2H); MS(ES^) m/z 520 (MH+).
Example 258
iV-(2,6-dichloro-2*-(trifluoromethoxy)-[l,l'-biphenyI]-4-yl)-2-(4-((2- (dimethylamino)ethyl)suIfonyl)phenyI)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. H-
NMR (400 MHz, CDC13) 5 ppm 2.23 (s, 6H), 2.79 (t, J= 7.6 Hz, 2H), 3.32 (t, J= 7.6 Hz, 2H), 3.86 (s, 2H), 7.40 (m, 5H), 7.56 (d, J= 8.0 Hz, 2H), 7.67 (s, 2H), 7.95 (d, = 8.4 Hz, 2H); MS(ES+) m/z 575 (MH+).
Example 259
iV-(2-chloro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yI)-2-(2-(methyIsulfonyl)pyrimidin-5- yl)acetamide

2-(2-((2-methyl-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)amino)-2-oxoethyl)-5- (methylsulfonyl)benzamide

The title compound was prepared using a similar procedure to that described in Scheme 2. JH- NMR (400 MHz, DMSO-i 6) δ ppm 2.02 (s, 3H), 3.25 (s, 3H), 4.05 (s, 2H), 7.07 (d, J= 8.3 Hz, IH), 7.35 (m, IH), 7.48 (m, 5H), 7.65 (d, J= 8.0 Hz, IH), 7.71 (s, IH), 7.96 (dd, J= 2.0 Hz, 8.0 Hz, IH), 8.00 (d, J= 1.6 Hz, IH), 8.22 (s, IH), 10.38 (s, IH); 19F-NMR (376 MHz, DMSO-c/6) 5 ppm -56; MS(ES+) m/z 507 (M^).
Example 261
2-(2-cyano-4-(methylsulfonyI)phenyl)-JV-(2-methyl-2'-(trifluoromethoxy)-[l,l'-biphi yl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, DMSO-rf6) 5 ppm 2.04 (s, 3H), 3.33 (s, 3H), 4.1 1 (s, 2H), 7.10 (d, J= 8.3 Hz, H), 7.36 (m, IH), 7.50 (m, 5H), 7.87 (d, J = 8.3 Hz, IH), 8.21 (dd, J= 1.9 Hz, 8.2 Hz, IH), 8.42 (d, J= 1.8 Hz, IH), 10.46 (s, IH). 19F-NMR (376 MHz, DMSO-i/6) -56; MS(ES+) m/z 489 (MH ).
Example 262
7V-(2,6-dichloro-2'-(trifluoromethoxy)-[l,l'-biphenyl]-4-yl)-2-(4-((2- (methylamino)ethyl)suIfonyl)phenyl)acetamide
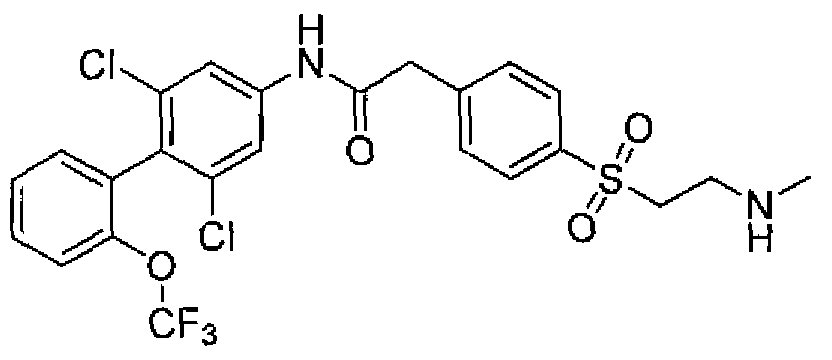
The title compound was prepared using a similar procedure to that described in Scheme 2. ¾- NMR (400 MHz, CDC13) δ ppm 2.42 (s, 3Η), 3.02 (d, J= 6.4 Hz, 2H), 3.31 (d, J— 6.4 Hz, 2H), 3.84 (S, 2H), 7.40 (m, 4H), 7.56 (d, J- 8.4 Hz, 2H), 7.64 (s, 2H), 7.94 (d, /= 8.4 Hz, 2H); MS(ES"^ m/z 561 (MH+).
Example 263
4-(2-((2,6-dichloro-2'-(trifluoromethoxy)-[l5l'-biphenyl]-4-yl)amino)-2- oxoethyl)benzenesulfonic acid

The title compound was prepared using a similar procedure to that described in Scheme 2. ¾-
NMR (400 MHz, DMSO- ) δ ppm 3.70 (s, 2H), 6.94 (s, 2H), 7.29 (d, J= 7.6 Hz, 2H), 7.41 (d, J= 8.0 Hz, 1H), 7.56 (m, 5H), 7.85 (s, 2H), 10.62 (s, 1H); MS^S^ m/z 519 (MET).
Example 264
Af-(2,6-dichloro-[l,l'-biphenyl]-4- l)-2-(4-(methylsulfonyl)phenyI)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, DMSO-<¾) δ ppm 3.21 (s, 3H), 3.85 (s, 2H), 7.24 (dd, J= 1.2 Hz, 7.2 Hz, 2H), 7.44 Cm, 3H), 7.61 (d, J= 8.4 Hz, 2H), 7.81 (s, 2H), 7.90 (d, J= 8.4 Hz, 2H), 10.65 (s, 1H); MS(ES ) m/z 434(MH
Example 265
2-(4-(iV-acetylsulfamoyl)phenyl)-N-(2,6-dimethyl-2'-(trifiuoromethoxy)-[l,l,-biphenyl]-4- yl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, DMSO-i¾ δ ppm 1.90 (s, 6H), 1.92 (s, 3H), 3.78 (s, 2Η), 7.27 (dd, J= 2.0 Hz, 8.4 Hz, IH), 7.38 (s, 2Η), 7.47 (m, 2H), 7.53 (m, IH), 7.57 (d, J= 8.4 Hz, 2H), 7.88 (d, J= 8.4 Hz, 2Η), 10.22 (s, IH), 12.07 (s, IH); 19F-NMR (376 MHz, DMSO-J6) δ ppm -56; MSfES ) m/z 521(MH+).
Example 266
iV-(3,5-dichloro-4-(2-methoxypyridin-3-yl)phenyI)-2-(4-(methylsulfonyl)phenyl)acetamide, Hydrochloride

The title compound was prepared using a similar procedure to that described for in Scheme 2. Ή-
NMR (400 MHz, DMSO-<¾ δ ppm 3.18 (s, 3H), 3.78 (s, 3H), 3.82 (s, 2H), 7.08 (dd, = 5.1 Hz, 7.3 Hz, IH), 7.58 (m, 3H)} 7.76 (s, 2H), 7.87 (d, J= 8.4 Hz, 2H), 8.23 (dd, J- 1.8 Hz, 5.1 Hz, IH), 10.64 (s, IH); MSfES*) m/z 465 (MH+).
Example 267
^-(2,6-dimethyl-2,-(trifluoromethoxy)-[l,l'-biphenyI]-4-yl)-2-hydroxy-2-(4- (methylsulfonyl)phenyI)acet

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, DMSO-<¾) δ ppm 1.90 (s, 6H), 3.21 (s, 3H), 5.27 (s, IH), 6.71 (br s, IH), 7.27 (d, J = 6.4 Hz, IH), 7.52 (m, 5H), 7.81 (d, J= 8.4 Hz, 2H), 7.94 (d, J= 8.0 Hz, 2H), 9.95 (s, IH); 19F- NMR (376 MHz, DMSO-d6) δ ppm -56; MSfES4) m/z 494 (MH^).
Example 268
JV-(4-(6-cyano-lH-indol-l-yl)^-methylphenyI)-2-(4-(N-methylsulfamoyl)phenyl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, CDC13) δ ppm 1.98 (s, 3H), 2.68 (d, J= 5.6 Hz, 3H), 3.84 (s, 2H), 4.38 (m, 1H), 6.72 (dd, J= 0.4 Hz, 3.2 Hz, 1H); 7.21 (d, J= 8.4 Hz, 1H), 7.30 (d, J= 3.2 Hz, 2H), 7.37 (d, J= 1.6 Hz, 1H), 7.45 (m, 2H), 7.53 (d, J= 8.0 Hz, 2H), 7.58 (s, 1H), 7.72 (dd, J = 0.4 Hz, 8.4 Hz, 1H), 7.87 (d, J= 8.4 Hz, 2H); MS(ES+) m/z 459 (ΜΗ^).
Example 269
2- (4-(ethyIsulfonyl)phenyl) -2-hydr oxy-N-(2-methyl-2 '-(triflu oromethoxy)-[ 1 ,Γ -biphenyl] -4- yl)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, DMSO- ) δ ppm 1.10 (t, J= 7.2 Hz, 3H), 2.02 (s, 3H), 3.27 (q, J= 7.2 Hz, 2H), 5.28 (s, 2H), 7.07 (d, J= 8.4 Hz, 1H), 7.33 (d, J= 7.2 Hz, 1H), 7.46 (t, J= 7.8 Hz, 2H), 7.52 (m, lH), 7.59 (d, J= 8.4 Hz, 1H), 7.65 (s, 1H), 7.81 (d, J= 8.4 Hz, 2H), 7.89 (d, J= 8.4 Hz, 2H), 10.02 (s, 1H); 19F- MR (376 MHz, DMSO-c/6) δ ppm -56; MS(ES+) m z 494 (MH+).
Example 270
2-hydroxy-iV-(2-methyl-2,-(trifluoromethoxy)-[l,l,-biphenyl]-4-yl)-2-(4-(N- methylsulfamoyI)phenyI)acetamide

The title compound was prepared using a similar procedure to that described in Scheme 2. Ή- NMR (400 MHz, DMSO-i/6) 5 ppm 2.03 (s, 3H), 2.41 (d, J= 5.2 Hz, 3H), 5.24 (s, 1H), 7.07 (d, J = 8.4 Hz, 1H), 7.33 (m, 1H), 7.39 (m, 1H), 7.46 (t, J= 6.8 Hz, 2H), 7.52 (m, 1H), 7.59 (dd, /= 1.6 Hz,
8.4 Hz, 1H), 7.65 (s, 1H), 7.75 (d, J= 8.4 Hz, 2H), 7.78 (d, = 8.4 Hz, 2H), 9.99 (s, 1H); I9F-NMR (376 MHz, DMSO- ) 5 ppm -56; MS(ES+) m/z 495 (MH*).
Biological Data
As stated above, tlie compounds according to Formula I are RORy modulators, and are useful in tlie treatment of diseases mediated by RORy. The biological activities of die compounds according to Formula I can be determined using any suitable assay for determining die activity of a candidate compound as a RORy modulator, as well as tissue and in vivo models.
Dual Fluorescence Energy Transfer (FRET) Assay
This assay is based on tl e knowledge that nuclear receptors interact with cofactors (transcription factors) in a ligand dependent manner. RORy is a typical nuclear receptor in that it has an AF2 domain in tlie ligand binding domain (LBD) which interacts with co-activators. The sites of interaction have been mapped to the LXXLL motifs in the co-activator SRC1(2) sequences. Short peptide sequences containing the LXXLL motif mimic the behavior of full-length co-activator.
The assay measures ligand-mediated interaction of the co-activator peptide with the purified bacterial-expressed RORy ligand binding domain (RORy-LBD) to indirectly assess ligand binding. RORy has a basal level of interaction with tlie co-activator SRC1(2) in the absence of ligand, thus it is possible to find ligands that inhibit or enhance the RORy/SRCl 2) interaction.
Materials
Generation of RORy-LBD bacterial expression plasmid
Human RORy Ligand Binding Domain (RORy-LBD) was expressed in E.coli strain
BL21(DE3) as an amino-terminal polyhistidine tagged fusion protein. DNA encoding this recombinant protein was sub-cloned into a modified pET21a expression vector (Novagen). A modified polyhistidine tag (M KHHHHHHLVPRGS) was fused in frame to residues 263-518 of the human RORy sequence.
Protein Purification
Approximately 50 g E.coli cell pellet was resuspended in 300 mL of lysis buffer (30 lnM imidazole pH 7.0 and 150 lnM NaCl). Cells were lysed by sonication and cell debris was removed by centrifugation for 30 minutes at 20,000g at 4°C. The cleared supernatant was filtered through a 0.45 uM cellulose acetate membrane filter. The clarified lysate was loaded onto a column (X -26) packed with ProBond Nickel Chelating resin (InVitrogen), pre-equilibrated with 30 inM imidazole pH 7.0 and 150 inM NaCI. After washing to baseline absorbance with the equilibration buffer, the column was developed with a gradient from 30 to 500 mM imidazole pH 7.0. Column fractions containing the
RORy-LBD protein were pooled and concentrated to a volume of 5 ml s. The concentrated protein was loaded onto a Superdex 200 column pre-equilibrated with 20 mM Tris-Cl pH 7.2 and 200 mM NaCl. The fractions containing the desired RORy-LBD protein were pooled together.
Protein Biotinylation
Purified RORy-LBD was buffer exchanged by exhaustive dialysis [3 changes of at least 20 volumes (>8000x)] against PBS [lOOmM NaPhosphate, pH 8 and 150mM NaCl]. The concentration of RORy-LBD was approximately 30uM in PBS. Five-fold molar excess of NHS-LC-Biotin (Pierce) was added in a minimal volume of PBS. This solution was incubated with occasional gentle mixing for 60 minutes at ambient room temperature. The modified RORy-LBD was dialyzed against 2 buffer changes - TBS pH 8.0 containing 5mM DTT, 2mM EDTA and 2% sucrose - each at least 20 times of the volume. The modified protein was distributed into aliquots, frozen on dry ice and stored at -80°C. The biotinylated RORy-LBD was subjected to mass spectrometric analysis to reveal the extent of modification by the biotinylation reagent. In general, approximately 95% of the protein had at least a single site of biotinylation and the overall extent of biotinylation followed a normal distribution of multiple sites ranged from one to five. A biotinylated peptide corresponding to amino acid 676 to 700 (CP S S HSSLTERHKILHRLLQEG SPS) of the co-activator steroid receptor coactivator SRC1(2) was generated using similar method.
Assay
Preparation of Europium labeled SRC 1(2) peptide: biotinylated SRC 1(2) solution was prepared by adding an appropriate amount of biotinylated SRC 1(2) from the lOOuM stock solution to a buffer containing 10 mM of freshly added DTT from solid to give a final concentration of 40 nM. An appropriate amount of Europium labeled Streptavidin was then added to the biotinylated SRC 1(2) solution in a tube to give a final concentration of 10 nM. The tube was inverted gently and incubated for 15 minutes at room temperature. Twenty-fold excess biotin from the 10 mM stock solution was added and the tube was inverted gently and incubated for 10 minutes at room temperature.
Preparation of APC labeled RORy-LBD: biotinylated RORy-LBD solution was prepared by adding an appropriate amount of biotinylated RORy-LBD from the stock solution to a buffer containing 10 mM of freshly added DTT from solid to give a final concentration of 40 nM. An appropriate amount of APC labeled Streptavidin was then added to the biotinylated RORy-LBD solution in a tube to give a final concentration of 20 nM. The tube was inverted gently and incubated for 15 minutes at room temperature. Twenty-fold excess biotin from the 10 mM stock solution was then added and the tube was inverted gently and incubated for 10 minutes at room temperature.
Equal volumes of the above-described Europium labeled SRC1(2) peptide and the APC labeled RORy-LBD were gently mixed together to give 20nM RORy-LBD, ΙΟηΜ APC-Strepavidin,
20nM SRC 1(2) and 5nM Europium-Streptavidin. The reaction mixtures were incubated for 5 minutes. Using a Thermo Combi Multidrop 384 stacker unit, 25 ul of the reaction mixtures per well was added to the 384-well assay plates containing lul of test compound per well in 100% DMSO. The plates were incubated for lhour and then read on ViewLux in Lance mode for EU/APC.
Jurkat Cell Luciferase Assay
RORy is known to bind to a CNS (conserved non-coding sequences) enhancer element in the IL17 promoter. In this assay, RORy activity is indirectly assessed using a luciferase reporter construct which contains the human IL17 promoter having the RORy-specific CNS enhancer element.
Inhibition of RORy activity by a compound will result in a decrease in luciferase activity of Jurkat cells transfected with the reporter construct.
Materials
Jurkat cell line
For the luciferase reporter plasmid, the 3 Kb human IL17 promoter containing the RORy- specific CNS enhancer element was PCR amplified from human genomic DNA and cloned into a pGL4-Luc2/hygro reporter plasmid sequencially as XhoI-HindHI (1.1 Kb) and Kpnl-Xhol (1.9 Kb) fragments. For the 1.1 Kb fragment, PCR was used to amplify human IL17 proximal promoter region from genomic DNA of 293T cells using primers as follows: forward primer, 5'- CTCGAGTAG AGCAGG ACAGGGAGGAA-3 ' (Xliol site is underlined) and reverse primer, 5'- AAGCTTGGATGGATGAGTTTGTGCCT-3 ' (Hindlll site is underlined). The 1.1 kb DNA bands were excised, purified, and inserted into pMD19-T Simple Vector (Takara). After DNA sequencing confirmation, the 1.1 kb DNA was digested with Xhol and Hindlll and inserted into Xhol/Hindlll sites of pGL4.31[Iuc2P/GAL4UAS/Hygro] (Promega) to generate the pIL17-lkb-luc reporter construct. For the 1.9 Kb fragment, PCR was used to amplify human IL17 promoter region from genomic DNA using primers as follows: forward primer,5'- GGTACCTGCCCTGCTCTATCCTGAGT-3' (Kpnl site is underlined) and reverse primer, 5'-
CTCGAGTGGTG AGTGCTG AGAG ATGG-3 ' (Xhol site is underlined). The resulting 1.9 kb DNA bands were excised, gel purified, and cloned into a pMD19-T Simple Vector (Takara). DNA sequencing analysis revealed that there were three point mutations but none of which affected RORy binding. The 1.9 kb DNA fragment was released by double digestion with Kpnl and Xhol and inserted into pIL17-lkb-luc to generate the luciferase reporter plasmid "pIL17-3kb-CNS-luc." To overexpress RORyt, the full-length cDNA of human RORyt identical to the published sequence NM_001001523 was cloned into pcDNA3.1 at the Kpnl-Notl cloning sites to generate the RORyt overexpression plasmid "CDNA3.1 DhRORy49-8".
The luciferase reporter plasmid and the RORyt overexpression plasmid were transfected into Jurkat cell line and a stable clone was identified. The stable clone was grown in 10% dialyzed FBS in RPMI (1640) with 800ug ml geneticin and 400ug ml hygromecin.
Assay
Compounds were dissolved in DMSO at three concentrations, lOmM, 400uM and 16uM, and were dispensed into 384-wells assay plate at 40nl, 12.5nl, 5nl respectively. The volume was adjusted with pure DMSO to a give a final uniform volume of 40 nl Jurkat cells described above were counted and centrifuged. The growth medium was discarded and the cells were resuspended with assay medium (phenol red free RPMI) at lE-6/ml. Cells were added to each of the compounds in the assay plates. Cells were either untreated or treated with CD3 microbeads (Miltenyi Biotec) at 1 ul beads per 500,000 cells. Cells were culture overnight and luciferase assay (Promega) was performed. Data were collected by ViewLux (using luciferase greiner 384 setting).
Thl7 ELISA/Intracellular Staining Assays
ELISA CD4+ cells were isolated from splenocytes of C57BL/6 (B6) mice (Shanghai Laboratory
Animal Resource Center) using the CD4+ T Ceil Isolation II Kit according to manufacturer's instructions (Miltenyi Biotec). 96 well plates were pre-coated with anti-CD3 antibody. CD4+ cells were resuspended in RPMI complete medium and were added to the 96-weIl plates at 3xl05 cells/well, with the total volume of each well being 90 ul. A cytokine cocktail (90 ul) was then added to stimulate Thl7 differentiation. Each compound diluted to various concentrations in DMSO (DMSO final volume 0.1%) was then added. The final concentrations of antibodies (R&D Systems) and cytokines (R&D Systems) were: anti-mCD3, 5ug/ml; anti-mCD28, 2ug ml; anti-mlFNy, lOug/ml; anti-mIL4, lOug/ml; mIL-6, 20ng/ml; mIL-23, lOng/ml; mIL-Ι β, l Ong ml; TGF-β, l Ong/ml. The cell culture was incubated at 37°C for 3 days. Supematants were collected and IL-17 concentration was determined by ELISA, performed according to manufacturer's instructions (R&D Systems). The optical density (OD) at 405 nm were measured with a microplate reader (BioRad) and the IL-17 quantity were extrapolated from the standard curve. The percentage of IL-17 inhibition was calculated by referring to the positive control (100%) and the pIC50 were determined by GraphPad.
Intracellular staining
The Thl 7 differentiation cell culture described above was maintained for 5 days instead of 3 days. The effect of compounds on the production of IL-17 and IFN-γ in the cells was determined by intracellular staining according to manufacturer's instructions (BD Biosciences).
Assay Data
It is understood that the data described below may have reasonable variation depending on the specific conditions and procedures used by the person conducting the testing.
All exemplified compounds except Examples 70-72, 74, 75, 91, 107, 110, 111, 114, 136, 138, 146, 165, 167, 171, 181, 186, 188, 194, 196, 198, 233, 239, 263 and 267 were tested in the dual FRET assay described above. All tested compounds except Examples 202 and 204 were found to have a pIC50 between 5 and 9. Examples 202 and 204 were each tested once and found to have a pIC50 below 5, the detection limit of the assay.
All exemplified compounds except Examples 2, 46, 221-224 and 268-270 were tested in the Jurkat cell Iuciferase assay described above. All tested compounds were found to have a pIC50 between 5 and 9 in at least one experiment.
All exemplified compounds except Examples 2, 3, 8, 10, 11, 13, 15-18, 22, 23, 27, 28, 32, 33, 36, 38-40, 43, 44, 46-48, 56, 61, 63, 68, 71 , 74, 75, 77, 80, 81, 83, 85, 89, 92-94, 96, 98, 104, 110-112, 114, 154, 185-188, 193, 196, 197, 202, 206, 208, 209, 215, 221 , 226, 228, 229, 233, 234, 237, 242, 255 and 257 were tested in the Thl7 ELISA assay described above. All tested compounds except Examples 6, 64, 66, 90, 137, 144, 163, 167,194, 213, 227, 249, 250, 256 and 261 were found to have a pIC50 between 5 and 9 in at least one experiment. Examples 6, 64, 66, 90, 137, 144, 163, 167, 194, 213, 227, 249, 250, 256 and 261 were each tested once and found to have a pIC50 below 5, the detection limit of the assay.
EAE Studies
Wild-type mice of the C57BL/6 (B6) strain were obtained from Shanghai Laboratory Animal
Resource Center. EAE was induced by intravenous injections of 100 ng of pertussis toxin (List Biological Laboratories) and then subcutaneous immunization with 200 μΐ of an emulsion composed of MOG3 -55 peptide (300 μg/mouse) in PBS and an equal volume of complete Freund's adjuvant containing 5 mg ml heat-killed Mycobacterium tuberculosis H37Ra (Difco Laboratories) on day 0, followed by another intravenous injections of 100 ng of pertussis toxin on day 2 as described previously (Wang et al. (2006) J. Clin. Invest. 116: 2434-2441). Each compound was given orally on day 0 at one or more of the following doses: 1, 3, 10, 30 or 100 mg/kg once or twice a day. Mice were scored for disease severity daily using a EAE scoring system (Wang et al. (2006) J. Clin. Invest. 116: 2434-2441): 0, no overt signs of disease; 1 , limp tail or hind limb weakness but not both; 2, limptail and paraparesis (weakness, incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with forelimb weakness or paralysis; and 5, moribund state or death. Clinical score data were expressed as means ± s.e.m.
CIA Studies
Collagen-induced arthritis (CIA) was induced in 8-week old male DBA 1 mice via an initial intradermal (i.d.) injection of an emulsion consisting of bovine type II collagen in CFA. Mice were intraperitoneally (i.p.) injected with bovine type II collagen 21 days later to boost the immune system, resulting in chronic inflammation in both the hind and the front paws. Each compound was given to the mice at lOOmg/kg twice a day starting from day 20 after the first immunization. Mice were examined for onset and severity of disease in a blinded manner. Arthritis symptoms were graded by the following scoring system: grade 0, normal appearance; grade 1 , slight erythema/ edema (1-3 digits); grade 2, erythema/ edema in more than 3 digits or mild swelling in ankle/wrist joint; grade 3, erythema/ edema in entire paw; grade 4, massive erythema edema of entire paw extending into proximal joints, ankylosis, loss of function. Each limb was graded, giving a maximum possible score of 16 per mouse. Clinical score data were expressed as means ± s.e.m. Foot volume of the mice was determined using a YLS-7B foot volume measuring instrument (Shandong Academy of Medical Science). Methods of Use
The compounds of the invention are modulators of RORy and can be useful in the treatment of diseases mediated by RORy, particularly autoimmune or inflammatory diseases. The Inflammatory or autoimmune diseases of the invention include multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, inflammatory bowel disease, Sjorgen's syndrome, optic neuritis, chronic obstructive pulmonary disease, asthma, type I diabetes, neuromyelitis optica, Myasthenia Gavis, uveitis, Guillain- Barre syndrome, psoriatic arthritis, Gaves' disease and allergy. Accordingly, in another aspect the invention is directed to methods of treating such diseases.
The methods of treatment of the invention comprise administering a safe and effective amount of a compound according to Formula I or a pharmaceutically-acceptable salt thereof to a patient in need thereof.
As used herein, "treat" in reference to a condition means: (1) to ameliorate or prevent the condition or one or more of the biological manifestations of the condition, (2) to interfere with (a) one or more points in the biological cascade that leads to or is responsible for the condition or (b) one or more of the biological manifestations of the condition, (3) to alleviate one or more of the symptoms or effects associated with the condition, or (4) to slow the progression of the condition or one or more of the biological manifestations of the condition.
As indicated above, "treatment" of a condition includes prevention of the condition. The skilled artisan will appreciate that "prevention" is not an absolute term. In medicine, "prevention" is understood to refer to the prophylactic administration of a drug to substantially diminish the likelihood or severity of a condition or biological manifestation thereof, or to delay the onset of such condition or biological manifestation thereof.
As used herein, "safe and effective amount" in reference to a compound of the invention or other pharmaceutically-active agent means an amount of the compound sufficient to treat the patient's condition but low enough to avoid serious side effects (at a reasonable benefit/risk ratio) within the scope of sound medical judgment, A safe and effective amount of a compound will vary with the particular compound chosen (e.g. consider the potency, efficacy, and half-life of the compound); the route of administration chosen; the condition being treated; the severity of the condition being treated; the age, size, weight, and physical condition of the patient being treated; the medical history of the patient to be treated; the duration of the treatment; the nature of concurrent therapy; the desired therapeutic effect; and like factors, but can nevertheless be routinely determined by the skilled artisan.
As used herein, "patient" refers to a human or other animal.
The compounds of the invention may be administered by any suitable route of administration, including both systemic administration and topical administration. Systemic administration includes oral administration, parenteral administration, transdermal administration, rectal administration, and administration by inhalation. Parenteral administration refers to routes of administration other dian enteral, transdermal, or by inhalation, and is typically by injection or infusion. Parenteral administration includes intravenous, intramuscular, and subcutaneous injection or infusion. Inhalation refers to administration into the patient's lungs whether inhaled tlirough the mouth or tlirough the nasal passages. Topical administration includes application to the skin as well as intraocular, otic, intravaginal, and intranasal administration.
The compounds of the invention may be administered once or according to a dosing regimen wherein a number of doses are administered at varying intervals of time for a given period of time. For example, doses may be administered one, two, three, or four times per day. Doses may be administered until the desired therapeutic effect is achieved or indefinitely to maintain the desired therapeutic effect. Suitable dosing regimens for a compound of the invention depend on the pharmacokinetic properties of that compound, such as absorption, distribution, and half-life, which can be determined by the skilled artisan. In addition, suitable dosing regimens, including the duration such regimens are administered, for a compound of the invention depend on the condition being treated, the severity of the condition being treated, the age and physical condition of the patient being
treated, the medical history of the patient to be treated, the nature of concurrent therapy, the desired therapeutic effect, and like factors within the knowledge and expertise of the skilled artisan. It will be further understood by such skilled artisans that suitable dosing regimens may require adjustment given an individual patient's response to the dosing regimen or over time as individual patient needs change.
Typical daily dosages may vary depending upon the particular route of administration chosen. Typical daily dosages for oral administration range from 0.1 mg to 1000 mg.
Additionally, the compounds of the invention may be administered as prodrugs. As used herein, a "prodrug" of a compound of the invention is a functional derivative of the compound which, upon administration to a patient, eventually liberates the compound of the invention in vivo. Administration of a compound of the invention as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome or overcome a side effect or other difficulty encountered with the compound. Typical functional derivatives used to prepare prodrugs include modifications of the compound that are chemically or enzymatically cleaved in vivo. Such modifications, which include the preparation of phosphates, amides, esters, thioesters, carbonates, and carbamates, are well known to those skilled in the art.
In one embodiment, the invention relates to the use of the compounds of the invention in the preparation of a medicament for the treatment of diseases mediated by RORy. In another embodiment, the invention relates to the compounds of the invention for use in the treatment of diseases mediated by RORy. Examples of such diseases include autoimmune or inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, psoriasis, Crohn's disease, inflammatory bowel disease, Sjorgen's syndrome, optic neuritis, chronic obstructive pulmonary disease, asthma, type I diabetes,
neuromyelitis optica, Myasthenia Gavis, uveitis, Guillain-Barre syndrome, psoriatic arthritis, Gaves' disease and allergy.
Compositions
The compounds of the invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically-acceptable excipient.
The pharmaceutical compositions of the invention may be prepared and packaged in bulk fonn wherein a safe and effective amount of a compound of the invention can be extracted and then given to the patient such as with powders or syrups. Alternatively, the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form wherein each physically discrete unit contains a safe and effective amount of a compound of the invention. When prepared in unit dosage fonn, the pharmaceutical compositions of the invention typically contain from 0.1 mg to 1000 mg.
The pharmaceutical compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. For example, in certain embodiments the pharmaceutical compositions of the invention contain two compounds of the invention. In addition, the pharmaceutical compositions of the invention may optionally further comprise one or more additional pharmaceutically active compounds.
As used herein, "pharmaceutically-acceptable excipient" means a pharmaceutically acceptable material, composition or vehicle involved in giving fonn or consistency to the pharmaceutical composition. Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a patient and interactions which would result in pharmaceutical compositions that are not pharmaceutically acceptable are avoided. In addition, each excipient must of course be of sufficiently high purity to render it pharmaceutically-acceptable.
The compound of the invention and the pharmaceutically-acceptable excipient or excipients will typically be formulated into a dosage form adapted for administration to the patient by the desired route of administration. For example, dosage forms include those adapted for (1) oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixers, suspensions, solutions, emulsions, sachets, and cachets; (2) parenteral administration such as sterile solutions, suspensions, and powders for reconstitution; (3) transdermal administration such as transdermal patches; (4) rectal administration such as suppositories; (5) inhalation such as dry powders, aerosols, suspensions, and solutions; and (6) topical administration such as creams, ointments, lotions, solutions, pastes, sprays, foams, and gels.
Suitable pharmaceutically-acceptable excipients will vary depending upon the particular dosage form chosen. In addition, suitable pharmaceutically-acceptable excipients may be chosen for a particular function that they may serve in the composition. For example, certain pharmaceutically- acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage
forms. Certain pliarmaceutically-acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pliarmaceutically-acceptable excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of the invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body. Certain phannaceutically-acceptable excipients may be chosen for their ability to enhance patient compliance.
Suitable phannaceutically-acceptable excipients include the following types of excipients: Diluents, fillers, binders, dis integrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweetners, flavoring agents, flavor masking agents, coloring agents, anticaking agents, hemectants, chelating agents, plasticizers, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents. The skilled artisan will appreciate that certain pharmaceutically-acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
Skilled artisans possess the knowledge and skill in the art to enable them to select suitable pliarmaceutically-acceptable excipients in appropriate amounts for use in the invention. In addition, there are a number of resources that are available to the skilled artisan which describe phannaceutically-acceptable excipients and may be useful in selecting suitable phannaceutically- acceptable excipients. Examples include Remington's Pliannaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Phannaceutical Excipients (the American Pharmaceutical Association and the Phannaceutical Press).
The pharmaceutical compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
In one aspect, the invention is directed to a solid oral dosage fonn such as a tablet or capsule comprising a safe and effective amount of a compound of the invention and a diluent or filler.
Suitable diluents and fillers include lactose, sucrose, dextrose, mannitol, sorbitol, starch (e.g. corn starch, potato starch, and pre-gelatinized starch), cellulose and its derivatives (e.g. microcrystalline cellulose), calcium sulfate, and dibasic calcium phosphate. The oral solid dosage fonn may further comprise a binder. Suitable binders include starch (e.g. corn starch, potato starch, and pre-gelatinized starch), gelatin, acacia, sodium alginate, alginic acid, tragacanth, guar gum, povidone, and cellulose and its derivatives (e.g. microcrystalline cellulose). The oral solid dosage form may further comprise a disintegrant. Suitable disintegrants include crospovidone, sodium starch glycolate, croscarmelose, alginic acid, and sodium carboxymethyl cellulose. The oral solid dosage form may further comprise a lubricant. Suitable lubricants include stearic acid, magnesuim stearate, calcium stearate, and talc.
Claims
WHAT IS CLAIMED IS: compound of Formula I or a harmaceutically acceptable salt thereof

Formula I
wherein:
A is phenyl or heteroaryl; B is phenyl or heteroaryl;
C is phenyl or monocyclic heteroaryl;
each Rl is independently selected from the group consisting of:
- OCF3,
- OCHF2,
- CF3,
- halo,
- C ,
- C1-C3 alkyl optionally substituted with C1-C3 alkoxy,
- piperidinylmethyl or piped dinylidenemethyl or piperazinylmethyl, wherein said piperidinyl or piperazinyl is substituted with C(0)Ra or C1-C3 alkyl,
- (CH2)„OH,
- C(0)Ra;
- (CH2)nNRaRa,
- (CH2)nC(0)ORa,
- heterocycloalkyl optionally substituted with C -C3 alkoxy,
- phenyl,
- monocyclic heteroaryl optionally substituted with C1-C3 alkyl, and
- ORb wherein Rb is selected from the group consisting of:
1) C1-C4 alkyl optionally substituted with CF3, CHF2, NRaRa, phenyl or cycloalkyl, 2) cycloalkyl,
3) phenyl optionally substituted with halo,
4) heterocycloalkyl optionally substituted with C1-C3 alkyl, and
5) monocyclic heteroaryl, each R2 is independently selected from the group consisting of:
- halo,
- C1-C6 alkyl optionally substituted with C(0)ORa or cycloalkyl,
- C2-C6 alkenyl,
- OH,
- OCF3,
- OCH2CF3
- C1-C4 alkoxy,
- CF3,
- CN,
- C(0)Ra,
- C(0)NRaRa,
- NRaRa,
- ORb wherein Rb is moncyclic heteroaryl or C1-C6 alkyl,
- phenyl carbonyl optionally substituted with halo,
- heterocycloalkyl, and
- monocyclic heteroaryl optionally substituted with one or more substituents selected from the group consisting of C1-C3 alkoxy, C1-C3 alkyl, COOH and CN, each R3 is selected from the group consisting of H, C1-C3 alkyl and OH;
each R4 is selected from the group consisting of halo, CN and C(0)NH2;
R5 is selected from the group consisting of C1-C3 alkyl, (CH2)nNRaRa, (CH2)nOH, and NHC(0)CH3;
m is 0, 1 or 2; each n is 0, 1 or 2; r is 0, 1 or 2; s is 1 or 2; t is 0, 1 or 2; and each Ra is H C1-C3 alkyl.
A compound or salt according to claim 1, wherein A is phenyl.
A compound or salt according to any of claims 1 to 2, wherein B is phenyl. A compound or salt according to any of claims 1 to 3, wherein C is phenyl. A compound of Formula II or a phannaceutically acceptable salt tliereof

Formula II
wherein:
each Rl is independently selected from the group consisting of:
- OCHF2,
- CF3,
- halo,
- CN,
- C1-C3 alkyl optionally substituted with C1-C3 alkoxy,
- piperidinylmethyl or piperidinylidenemethyl or piperazmylmethyl, wherein said piperidinyl or piperazinyl is substituted with C(0)Ra or C1-C3 alkyl,
- (CH2)nOH,
- C(0)Ra,
- (CH3)„NRaRa,
- (CH2)»C(0)ORa,
- heterocycloalkyl optionally substituted with C1-C3 alkoxy,
- phenyl,
- monocyclic heteroaryl optionally substituted with C1-C3 alkyl, and
- ORb wherein Rb is selected from the group consisting of:
1) C1-C4 alkyl optionally substituted with CF3, CHF3, NRaRa, phenyl or cycloalkyl,
2) cycloalkyl,
3) phenyl optionally substituted with halo,
4) heterocycloalkyl optionally substituted with C1-C3 alkyl, and
5) monocyclic heteroaryl, each R2 is independently selected from the group consisting of:
- halo,
- CI -C6 alkyl optionally substituted with C(0)ORa or cycloalkyl,
- C2-C6 alkenyl,
- OH,
- OCF3,
- OCH2CF3
- C1-C4 alkoxy,
- CF3,
- CN,
- C(0)Ra,
- C(0)NRaRa,
- NRaRa,
- ORb wherein Rb is moncyclic heteroaryl or C1-C6 alkyl,
- phenylcarbonyl optionally substituted with halo,
- heterocycloalkyl, and
- monocyclic heteroaryl optionally substituted with one or more substituents selected from the group consisting of C1-C3 alkoxy, C1-C3 alkyl, COOH and CN;
R3 is selected from the group consisting of H, C1-C3 alkyl and OH.
each R4 is selected from the group consisting of halo, CN and C(0)NH2.
R5 is selected from the group consisting of C1-C3 alkyl, (CH2)nNRaRa, (CH2)„OH, and NHC(0)CH3;
each m is 0, 1 or 2; each n is 0, 1 or 2; each r is 0, 1 or 2; each t is 0, 1 or 2; and each Ra is H or Cl-C3 alkyl.
6. A compound or salt according to claim 5, wherein m is 1 and Rl is OCF3, CF3, or OCHF2.
7. A compound or salt according to claim 5, wherein m is 2 and one Rl is OCF3 or OCHF2 and the other Rl is CI or F.
8. A compound or salt according to any of claims 5 to 7, wherein r is 1 and R2 is Cl or methyl.
9. A compound or salt according to any of claims 5 to 7, wherein r is 2 and R2 is Cl or F.
10. A compound or salt according to any of claims 5 to 9, wherein R3 is H and t is 0.
11. A compound or salt according to any of claims 5 to 10, wherein R5 is methyl or ethyl.
12. A compound of Formula I according to claim 1, which is a compound of Examples 1 to 270.
13. A phannaceutical composition which comprises a compound according to any of claims 1 to 11 or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
14. The use of a compound according to any of claims 1 to 11 or a phannaceutically acceptable salt thereof in the manufacture of a medicament for use in the treatment of multiple sclerosis or rheumatoid arthritis.
15. A compound according to any of claims 1 to 11 or a phannaceutically acceptable salt thereof for use in the treatment of multiple sclerosis or rheumatoid arthritis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2011001488 | 2011-09-01 | ||
| CNPCT/CN2011/001488 | 2011-09-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013029338A1 true WO2013029338A1 (en) | 2013-03-07 |
Family
ID=47755249
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2012/001221 WO2013029338A1 (en) | 2011-09-01 | 2012-08-30 | Novel compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2013029338A1 (en) |
Cited By (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013178362A1 (en) | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| WO2014008214A1 (en) * | 2012-07-02 | 2014-01-09 | Biogen Idec Ma Inc. | Biaryl-containing compounds as inverse agonists of ror-gamma receptors |
| WO2014023367A1 (en) | 2012-08-09 | 2014-02-13 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor ror gamma |
| WO2014125426A1 (en) * | 2013-02-15 | 2014-08-21 | Aurigene Discovery Technologies Limited | Trisubstituted heterocyclic derivatives as ror gamma modulators |
| WO2015006691A1 (en) | 2013-07-11 | 2015-01-15 | Evestra, Inc. | Pro-drug forming compounds |
| WO2015082533A1 (en) * | 2013-12-05 | 2015-06-11 | Lead Pharma Cel Models Ip B.V. | Ror gamma (rory) modulators |
| WO2015145371A1 (en) * | 2014-03-27 | 2015-10-01 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| WO2015159233A1 (en) * | 2014-04-16 | 2015-10-22 | Glenmark Pharmaceuticals S.A. | Aryl and heteroaryl ether compounds as ror gamma modulators |
| WO2015198232A1 (en) | 2014-06-25 | 2015-12-30 | Piramal Enterprises Limited | Fused triterpene compounds and uses thereof |
| US9266886B2 (en) | 2014-02-03 | 2016-02-23 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| WO2016039358A1 (en) * | 2014-09-10 | 2016-03-17 | 第一三共株式会社 | Carboxylic acid derivative |
| WO2016072402A1 (en) * | 2014-11-05 | 2016-05-12 | 第一三共株式会社 | Cyclic amine derivative |
| WO2016100652A3 (en) * | 2014-12-19 | 2016-08-25 | Aragon Pharmaceuticals, Inc. | Process for the preparation of a diarylthiohydantoin compound |
| CN105985248A (en) * | 2015-02-15 | 2016-10-05 | 上海泰禾国际贸易有限公司 | Method for synthesizing amino biaromatic compound |
| US9481674B1 (en) | 2016-06-10 | 2016-11-01 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| WO2016185342A1 (en) | 2015-05-15 | 2016-11-24 | Aurigene Discovery Technologies Limited | Substituted tetrahydroquinolinone compounds as ror gamma modulators |
| EP3101005A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| EP3101008A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| EP3101009A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| EP3101007A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| EP3101006A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| WO2016193894A1 (en) * | 2015-05-29 | 2016-12-08 | Glenmark Pharmaceuticals S.A. | Treatment of respiratory disorders using ror- gamma inhibitors |
| US9663515B2 (en) | 2014-11-05 | 2017-05-30 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9796710B2 (en) | 2014-10-14 | 2017-10-24 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| WO2017199103A1 (en) * | 2016-05-18 | 2017-11-23 | Glenmark Pharmaceuticals S.A. | Benzamide compounds as ror gamma modulators |
| US9845308B2 (en) | 2014-11-05 | 2017-12-19 | Vitae Pharmaceuticals, Inc. | Isoindoline inhibitors of ROR-gamma |
| CN107488113A (en) * | 2016-06-13 | 2017-12-19 | 上海泰禾国际贸易有限公司 | A kind of method for synthesizing o-aminobiphenyl class compound |
| CN107522634A (en) * | 2016-06-22 | 2017-12-29 | 复旦大学 | Biaryl ureas carboxylic acid derivates or its salt and its production and use |
| CN107522641A (en) * | 2016-06-22 | 2017-12-29 | 复旦大学 | Biaryl ureas analog derivative or its salt and its production and use |
| WO2018011747A1 (en) | 2016-07-14 | 2018-01-18 | Cadila Healthcare Limited | Polycyclic compounds as ror-gamma modulators |
| WO2018011746A1 (en) | 2016-07-14 | 2018-01-18 | Cadila Healthcare Limited | Cyclopropyl derivatives as ror-gamma modulators |
| CN107935876A (en) * | 2017-12-21 | 2018-04-20 | 金凯(辽宁)化工有限公司 | A kind of preparation method of 2 (3 amino, 4 chlorobenzoyl) benzoic acid |
| CN108026050A (en) * | 2015-08-03 | 2018-05-11 | 格兰马克药品股份有限公司 | New compound as ROR gamma modulators |
| WO2018111803A1 (en) * | 2016-12-13 | 2018-06-21 | Boehringer Ingelheim International Gmbh | Compounds as modulators of ror gamma |
| US10011566B2 (en) | 2015-12-15 | 2018-07-03 | Astrazeneca Ab | Compounds |
| WO2018145653A1 (en) * | 2017-02-09 | 2018-08-16 | 复旦大学 | Biaryl compound, preparation method and use therefor |
| WO2018181456A1 (en) * | 2017-03-29 | 2018-10-04 | 科研製薬株式会社 | Indazole derivative and pharmaceutical drug containing same |
| WO2018193297A1 (en) | 2017-04-21 | 2018-10-25 | Cadila Healthcare Limited | Novel compounds as ror-gamma modulators |
| CN108863850A (en) * | 2017-02-09 | 2018-11-23 | 复旦大学 | Biaryl base class compound and its preparation method and application |
| RU2675806C1 (en) * | 2018-07-20 | 2018-12-25 | Федеральное государственное бюджетное образовательное учреждение высшего образования "Национальный исследовательский Мордовский государственный университет им. Н.П. Огарёва" | N-(indolyl)trifluoroacetamides with antimicrobial activity production method |
| CN109134476A (en) * | 2017-06-15 | 2019-01-04 | 复旦大学 | Bridged ring piperazine derivative or its salt and its preparation method and application |
| WO2019022223A1 (en) * | 2017-07-28 | 2019-01-31 | 東レ株式会社 | Cyclic amine derivative and use thereof for medical purposes |
| US10301261B2 (en) | 2015-08-05 | 2019-05-28 | Vitae Pharmaceuticals, Llc | Substituted indoles as modulators of ROR-gamma |
| JP2019520359A (en) * | 2016-06-22 | 2019-07-18 | フーダン ユニヴァーシティFudan University | Biaryl urea derivatives or salts thereof and methods for their preparation and use |
| WO2019151270A1 (en) | 2018-01-31 | 2019-08-08 | 東レ株式会社 | Cyclic amine derivative and pharmaceutical use thereof |
| WO2019149965A1 (en) | 2018-02-05 | 2019-08-08 | Universite De Strasbourg | Compounds and compositions for the treatment of pain |
| KR20190096365A (en) * | 2016-12-05 | 2019-08-19 | 리드 파마 홀딩 비.브이. | ROR gamma (RORγ) regulator |
| WO2019213470A1 (en) | 2018-05-03 | 2019-11-07 | Eternity Bioscience Inc. | Benzimidazole derivatives as modulators of retinoid-related orphan receptor gamma (rorϒ) and pharmaceutical uses thereof |
| US20190382350A1 (en) * | 2018-06-18 | 2019-12-19 | Janssen Pharmaceutica Nv | PYRIDINYL PYRAZOLES AS MODULATORS OF RORyT |
| WO2020108538A1 (en) * | 2018-11-27 | 2020-06-04 | 正大天晴药业集团股份有限公司 | RORγ INHIBITOR HAVING SULFONYL STRUCTURE |
| WO2020140043A1 (en) * | 2018-12-28 | 2020-07-02 | Endogena Therapeutics, Inc. | Compounds for use as therapeutically active substances in the treatment and/or prevention of neuroretinal diseases |
| WO2020211836A1 (en) * | 2019-04-19 | 2020-10-22 | 北京酷瓴生物技术有限公司 | Benzene piperidine derivative, preparation method therefor, intermediate thereof and use thereof |
| US10829481B2 (en) | 2016-01-29 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Benzimidazole derivatives as modulators of ROR-gamma |
| US10913739B2 (en) | 2017-07-24 | 2021-02-09 | Vitae Pharmaceuticals, LLC (121374) | Inhibitors of RORγ |
| CN112538072A (en) * | 2019-09-21 | 2021-03-23 | 齐鲁制药有限公司 | Novel aminopyrimidine EGFR (epidermal growth factor receptor) inhibitor |
| WO2021063362A1 (en) * | 2019-09-30 | 2021-04-08 | 上海辉启生物医药科技有限公司 | Sulfo-substituted biaryl compound or salt thereof, preparation method therefor, and use thereof |
| US10975068B2 (en) | 2016-04-27 | 2021-04-13 | Janssen Pharmaceutica Nv | 6-aminopyridin-3-yl thiazoles as modulators of RORγT |
| US10975057B2 (en) | 2018-06-18 | 2021-04-13 | Janssen Pharmaceutica Nv | 6-aminopyridin-3-yl pyrazoles as modulators of RORgT |
| US10975037B2 (en) | 2018-06-18 | 2021-04-13 | Janssen Pharmaceutica Nv | Phenyl substituted pyrazoles as modulators of RORγt |
| WO2021083311A1 (en) | 2019-10-31 | 2021-05-06 | 江苏恒瑞医药股份有限公司 | ACID ADDITION SALT OF RORγ REGULATOR |
| US11008340B2 (en) | 2015-11-20 | 2021-05-18 | Vitae Pharmaceuticals, Llc | Modulators of ROR-gamma |
| US11034654B2 (en) | 2017-06-14 | 2021-06-15 | Astrazeneca Ab | 2,3-dihydroisoindole-1-carboxamides useful as ROR-gamma modulators |
| CN113072521A (en) * | 2020-01-06 | 2021-07-06 | 广东东阳光药业有限公司 | ROR gamma t inhibitor and application thereof in medicine |
| CN113072538A (en) * | 2020-01-06 | 2021-07-06 | 广东东阳光药业有限公司 | ROR gamma t inhibitor and application thereof in medicine |
| CN113072476A (en) * | 2020-01-06 | 2021-07-06 | 广东东阳光药业有限公司 | ROR gamma t inhibitor and preparation method and application thereof |
| WO2021136326A1 (en) * | 2019-12-31 | 2021-07-08 | 广东东阳光药业有限公司 | Aryl amide compound and application thereof in medicine |
| WO2021228215A1 (en) * | 2020-05-15 | 2021-11-18 | 上海辉启生物医药科技有限公司 | BIARYL COMPOUND CAPABLE OF SERVING AS RORγ REGULATOR |
| US11186573B2 (en) | 2017-07-24 | 2021-11-30 | Vitae Pharmaceuticals, Llc | Inhibitors of ROR gamma |
| WO2022007168A1 (en) * | 2020-07-10 | 2022-01-13 | 中国科学院广州生物医药与健康研究院 | Benzo five-membered nitrogen heterocyclic compound and application thereof |
| US11345666B2 (en) | 2018-06-18 | 2022-05-31 | Janssen Pharmaceutica Nv | Phenyl and pyridinyl substituted imidazoles as modulators of RORγT |
| EP4129996A4 (en) * | 2020-03-23 | 2023-07-12 | Qilu Pharmaceutical Co., Ltd. | Novel aminopyrimidine egfr inhibitor |
| WO2023240084A3 (en) * | 2022-06-06 | 2024-03-21 | Denali Therapeutics Inc. | Compounds, compositions, and methods |
| CN113072476B (en) * | 2020-01-06 | 2024-05-14 | 广东东阳光药业股份有限公司 | ROR gamma t inhibitor and preparation method and application thereof |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009144395A1 (en) * | 2008-03-21 | 2009-12-03 | Sanofi-Aventis | Polysubstituted derivatives of 2-heteroaryl-6-phenyl-imidazo[l,2- α] pyridines, and preparation and therapeutic use thereof |
| WO2010004221A2 (en) * | 2008-07-10 | 2010-01-14 | Laboratoires Fournier S.A. | Use of indole derivatives as nurr-1 activators for treating parkinson’s disease |
| WO2012028100A1 (en) * | 2010-09-01 | 2012-03-08 | Glaxo Group Limited | Novel compounds |
| WO2012027965A1 (en) * | 2010-09-01 | 2012-03-08 | Glaxo Group Limited | Novel compounds |
| WO2012100732A1 (en) * | 2011-01-24 | 2012-08-02 | Glaxo Group Limited | Retinoid-related orphan receptor gamma modulators, composition containing them and uses thereof |
| WO2012100734A1 (en) * | 2011-01-24 | 2012-08-02 | Glaxo Group Limited | Compounds useful as retinoid-related orphan receptor gamma modulators |
-
2012
- 2012-08-30 WO PCT/CN2012/001221 patent/WO2013029338A1/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009144395A1 (en) * | 2008-03-21 | 2009-12-03 | Sanofi-Aventis | Polysubstituted derivatives of 2-heteroaryl-6-phenyl-imidazo[l,2- α] pyridines, and preparation and therapeutic use thereof |
| WO2010004221A2 (en) * | 2008-07-10 | 2010-01-14 | Laboratoires Fournier S.A. | Use of indole derivatives as nurr-1 activators for treating parkinson’s disease |
| WO2012028100A1 (en) * | 2010-09-01 | 2012-03-08 | Glaxo Group Limited | Novel compounds |
| WO2012027965A1 (en) * | 2010-09-01 | 2012-03-08 | Glaxo Group Limited | Novel compounds |
| WO2012100732A1 (en) * | 2011-01-24 | 2012-08-02 | Glaxo Group Limited | Retinoid-related orphan receptor gamma modulators, composition containing them and uses thereof |
| WO2012100734A1 (en) * | 2011-01-24 | 2012-08-02 | Glaxo Group Limited | Compounds useful as retinoid-related orphan receptor gamma modulators |
Cited By (189)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10301272B2 (en) | 2012-05-31 | 2019-05-28 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ROR[γ] |
| WO2013178362A1 (en) | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| WO2014008214A1 (en) * | 2012-07-02 | 2014-01-09 | Biogen Idec Ma Inc. | Biaryl-containing compounds as inverse agonists of ror-gamma receptors |
| EP3118189A1 (en) | 2012-08-09 | 2017-01-18 | Phenex Pharmaceuticals AG | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor ror gamma |
| WO2014023367A1 (en) | 2012-08-09 | 2014-02-13 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor ror gamma |
| US9458104B2 (en) | 2012-08-09 | 2016-10-04 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted nitrogen-containing 5-membered heterocycles as modulators for the orphan nuclear receptor RORγ |
| WO2014125426A1 (en) * | 2013-02-15 | 2014-08-21 | Aurigene Discovery Technologies Limited | Trisubstituted heterocyclic derivatives as ror gamma modulators |
| WO2015006691A1 (en) | 2013-07-11 | 2015-01-15 | Evestra, Inc. | Pro-drug forming compounds |
| AU2014359324B2 (en) * | 2013-12-05 | 2019-02-21 | Lead Pharma Holding B.V. | ROR gamma (RORy) modulators |
| WO2015082533A1 (en) * | 2013-12-05 | 2015-06-11 | Lead Pharma Cel Models Ip B.V. | Ror gamma (rory) modulators |
| US9738600B2 (en) | 2013-12-05 | 2017-08-22 | Lead Pharma Cel Models Ip B.V. | ROR gamma (RORγ) modulators |
| KR102251128B1 (en) | 2013-12-05 | 2021-05-12 | 리드 파마 홀딩 비.브이. | ROR GAMMA (RORγ) MODULATORS |
| JP2017504576A (en) * | 2013-12-05 | 2017-02-09 | リード ファーマ セル モデルス アイピー ビー.ブイ. | ROR gamma (γ) modulator |
| EA030393B1 (en) * | 2013-12-05 | 2018-07-31 | ЛИД ФАРМА СЕЛ МОДЕЛЗ АйПи Б.В. | ROR GAMMA (RORγ) MODULATORS |
| KR20160105413A (en) * | 2013-12-05 | 2016-09-06 | 리드 파마 셀 모델스 아이피 비.브이. | Ror gamma (rorγ) modulators |
| CN105980353A (en) * | 2013-12-05 | 2016-09-28 | 领先制药Cel模型知识产权公司 | Ror gamma (rory) modulators |
| US9266886B2 (en) | 2014-02-03 | 2016-02-23 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10047085B2 (en) | 2014-02-03 | 2018-08-14 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US11535614B2 (en) | 2014-02-03 | 2022-12-27 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9624217B2 (en) | 2014-02-03 | 2017-04-18 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10399976B2 (en) | 2014-02-03 | 2019-09-03 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10807980B2 (en) | 2014-02-03 | 2020-10-20 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| AU2015237788B2 (en) * | 2014-03-27 | 2021-03-11 | Piramal Enterprises Limited | ROR-gamma modulators and uses thereof |
| US11084784B2 (en) | 2014-03-27 | 2021-08-10 | Piramal Enterprises Limited | ROR-gamma modulators and uses thereof |
| EP3122721A4 (en) * | 2014-03-27 | 2018-01-10 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| WO2015145371A1 (en) * | 2014-03-27 | 2015-10-01 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| WO2015159233A1 (en) * | 2014-04-16 | 2015-10-22 | Glenmark Pharmaceuticals S.A. | Aryl and heteroaryl ether compounds as ror gamma modulators |
| WO2015198232A1 (en) | 2014-06-25 | 2015-12-30 | Piramal Enterprises Limited | Fused triterpene compounds and uses thereof |
| US10105373B2 (en) | 2014-06-25 | 2018-10-23 | Piramal Enterprises Limited | Fused triterpene compounds and uses thereof |
| WO2016039358A1 (en) * | 2014-09-10 | 2016-03-17 | 第一三共株式会社 | Carboxylic acid derivative |
| US9796710B2 (en) | 2014-10-14 | 2017-10-24 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10087184B2 (en) | 2014-10-14 | 2018-10-02 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of RORγ |
| US11001583B2 (en) | 2014-11-05 | 2021-05-11 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9845308B2 (en) | 2014-11-05 | 2017-12-19 | Vitae Pharmaceuticals, Inc. | Isoindoline inhibitors of ROR-gamma |
| US9663515B2 (en) | 2014-11-05 | 2017-05-30 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| WO2016072402A1 (en) * | 2014-11-05 | 2016-05-12 | 第一三共株式会社 | Cyclic amine derivative |
| KR20170095976A (en) * | 2014-12-19 | 2017-08-23 | 아라곤 파마슈티컬스, 인코포레이티드 | Process for the preparation of a diarylthiohydantoin compound |
| CN107108507A (en) * | 2014-12-19 | 2017-08-29 | 阿拉贡药品公司 | The method for preparing Diarylthiohydantoin compounds |
| EP3372585A1 (en) * | 2014-12-19 | 2018-09-12 | Aragon Pharmaceuticals, Inc. | Process for the preparation of a diarylthiohydantoin compound |
| WO2016100652A3 (en) * | 2014-12-19 | 2016-08-25 | Aragon Pharmaceuticals, Inc. | Process for the preparation of a diarylthiohydantoin compound |
| TWI753336B (en) * | 2014-12-19 | 2022-01-21 | 美商艾瑞岡醫藥公司 | Process for the preparation of a diarylthiohydantoin compound |
| TWI703132B (en) * | 2014-12-19 | 2020-09-01 | 美商艾瑞岡醫藥公司 | Process for the preparation of a diarylthiohydantoin compound |
| US9688655B2 (en) | 2014-12-19 | 2017-06-27 | Janssen Pharmaceutica Nv | Process for the preparation of a diarylthiohydantoin compound |
| JP2020114842A (en) * | 2014-12-19 | 2020-07-30 | アラゴン ファーマシューティカルズ,インコーポレイテッド | Process for preparation of diarylthiohydantoin compound |
| KR102586059B1 (en) | 2014-12-19 | 2023-10-05 | 아라곤 파마슈티컬스, 인코포레이티드 | Process for the preparation of a diarylthiohydantoin compound |
| TWI689494B (en) * | 2014-12-19 | 2020-04-01 | 美商艾瑞岡醫藥公司 | Process for the preparation of a diarylthiohydantoin compound |
| TWI683810B (en) * | 2014-12-19 | 2020-02-01 | 美商艾瑞岡醫藥公司 | Process for the preparation of a diarylthiohydantoin compound |
| CN105985248B (en) * | 2015-02-15 | 2018-10-26 | 上海泰禾国际贸易有限公司 | A kind of method of synthesizing amino Diaromatic compound |
| CN105985248A (en) * | 2015-02-15 | 2016-10-05 | 上海泰禾国际贸易有限公司 | Method for synthesizing amino biaromatic compound |
| US11229636B2 (en) | 2015-05-15 | 2022-01-25 | Aurigene Discovery Technologies Limited | Substituted tetrahydroquinolinone compounds as ROR gamma modulators |
| WO2016185342A1 (en) | 2015-05-15 | 2016-11-24 | Aurigene Discovery Technologies Limited | Substituted tetrahydroquinolinone compounds as ror gamma modulators |
| CN113121497A (en) * | 2015-05-15 | 2021-07-16 | 奥瑞基尼探索技术有限公司 | Substituted tetrahydroquinolinone compounds as ROR gamma modulators |
| JP2020117514A (en) * | 2015-05-15 | 2020-08-06 | オーリジーン ディスカバリー テクノロジーズ リミテッドAurigene Discovery Technologies Limited | Substituted tetrahydroquinolinone compounds as ror gamma modulators |
| EP3294713A4 (en) * | 2015-05-15 | 2018-10-24 | Aurigene Discovery Technologies Limited | Substituted tetrahydroquinolinone compounds as ror gamma modulators |
| CN108026045A (en) * | 2015-05-15 | 2018-05-11 | 奥瑞基尼探索技术有限公司 | The tetrahydroquinoline ketone compound by substitution as ROR gamma modulators |
| JP2018515531A (en) * | 2015-05-15 | 2018-06-14 | オーリジーン ディスカバリー テクノロジーズ リミテッドAurigene Discovery Technologies Limited | Substituted tetrahydroquinoline compounds as ROR gamma modulators |
| EP3845526A1 (en) * | 2015-05-15 | 2021-07-07 | Aurigene Discovery Technologies Limited | Method for the preparation of tetrahydroquinolinone compounds having ror-gamma modulating activity |
| CN108026045B (en) * | 2015-05-15 | 2021-04-27 | 奥瑞基尼探索技术有限公司 | Substituted tetrahydroquinolinone compounds as ROR gamma modulators |
| WO2016193894A1 (en) * | 2015-05-29 | 2016-12-08 | Glenmark Pharmaceuticals S.A. | Treatment of respiratory disorders using ror- gamma inhibitors |
| CN107922337A (en) * | 2015-06-05 | 2018-04-17 | 领先制药控股公司 | ROR gamma modulators |
| CN108473429B (en) * | 2015-06-05 | 2021-09-10 | 领先制药控股公司 | ROR gamma modulators |
| JP2018518481A (en) * | 2015-06-05 | 2018-07-12 | リード ファーマ ホールディング ビー.ブイ. | ROR gamma (RORγ) modulator |
| WO2016193470A1 (en) * | 2015-06-05 | 2016-12-08 | Lead Pharma Cel Models Ip B.V. | Ror gamma (rory) modulators |
| WO2016193452A1 (en) * | 2015-06-05 | 2016-12-08 | Lead Pharma Cel Models Ip B.V. | ROR GAMMA (RORγ) MODULATORS |
| KR102591910B1 (en) | 2015-06-05 | 2023-10-23 | 리드 파마 홀딩 비.브이. | ROR Gamma (RORY) Adjuster |
| JP2018522837A (en) * | 2015-06-05 | 2018-08-16 | リード ファーマ ホールディング ビー.ブイ. | ROR gamma (RORγ) modulator |
| CN108430974A (en) * | 2015-06-05 | 2018-08-21 | 领先制药控股公司 | ROR gamma modulators |
| CN108430973A (en) * | 2015-06-05 | 2018-08-21 | 领先制药控股公司 | ROR gamma modulators |
| JP2018524293A (en) * | 2015-06-05 | 2018-08-30 | リード ファーマ ホールディング ビー.ブイ. | ROR gamma (RORγ) modulator |
| CN108473429A (en) * | 2015-06-05 | 2018-08-31 | 领先制药控股公司 | ROR gamma modulators |
| CN108473430A (en) * | 2015-06-05 | 2018-08-31 | 领先制药控股公司 | ROR gamma modulators |
| JP2018516938A (en) * | 2015-06-05 | 2018-06-28 | リード ファーマ ホールディング ビー.ブイ. | ROR gamma (RORγ) modulator |
| WO2016193468A1 (en) * | 2015-06-05 | 2016-12-08 | Lead Pharma Cel Models Ip B.V. | Ror gamma (rory) modulators |
| KR102576004B1 (en) | 2015-06-05 | 2023-09-06 | 리드 파마 홀딩 비.브이. | ROR gamma (RORγ) modulator |
| RU2735546C2 (en) * | 2015-06-05 | 2020-11-03 | Лид Фарма Холдинг Б.В. | ROR GAMMA MODULATORS (RORγ) |
| KR20180042213A (en) * | 2015-06-05 | 2018-04-25 | 리드 파마 홀딩 비.브이. | ROR gamma (ROR gamma) modifier |
| KR102575608B1 (en) | 2015-06-05 | 2023-09-05 | 리드 파마 홀딩 비.브이. | ROR gamma (RORγ) modulator |
| KR102546649B1 (en) | 2015-06-05 | 2023-06-21 | 리드 파마 홀딩 비.브이. | ROR Gamma (RORY) Adjuster |
| US10118895B2 (en) | 2015-06-05 | 2018-11-06 | Lead Pharma Holding B.V. | ROR gamma (RORγ) modulators |
| EP3101005A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| WO2016193461A1 (en) * | 2015-06-05 | 2016-12-08 | Lead Pharma Cel Models Ip B.V. | ROR GAMMA (RORγ) MODULATORS |
| RU2733406C2 (en) * | 2015-06-05 | 2020-10-01 | Лид Фарма Холдинг Б.В. | Ror gamma modulators (rorγ) |
| EP3101008A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| KR20180040559A (en) * | 2015-06-05 | 2018-04-20 | 리드 파마 홀딩 비.브이. | ROR gamma (ROR gamma) modifier |
| US10259782B2 (en) | 2015-06-05 | 2019-04-16 | Lead Pharma Holding B.V. | ROR gamma (RORγ) modulators |
| KR20180040560A (en) * | 2015-06-05 | 2018-04-20 | 리드 파마 홀딩 비.브이. | ROR Gamma (RORY) Regulator |
| AU2016272015B2 (en) * | 2015-06-05 | 2020-08-27 | Lead Pharma Holding B.V. | ROR gamma (RORy) modulators |
| US10315996B2 (en) | 2015-06-05 | 2019-06-11 | Lead Pharma Holding B.V. | ROR gamma (RORγ) modulators |
| AU2016273353B2 (en) * | 2015-06-05 | 2020-08-27 | Lead Pharma Holding B.V. | ROR gamma (RORy) modulators |
| EP3101009A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| CN108473430B (en) * | 2015-06-05 | 2021-05-25 | 领先制药控股公司 | ROR gamma modulators |
| CN108430973B (en) * | 2015-06-05 | 2021-09-21 | 领先制药控股公司 | ROR gamma modulators |
| JP2018518480A (en) * | 2015-06-05 | 2018-07-12 | リード ファーマ ホールディング ビー.ブイ. | ROR gamma (RORγ) modulator |
| KR20180038439A (en) * | 2015-06-05 | 2018-04-16 | 리드 파마 홀딩 비.브이. | ROR Gamma (RORY) Regulator |
| US10428018B2 (en) | 2015-06-05 | 2019-10-01 | Lead Pharma Holding B.V. | ROR gamma (RORy) modulators |
| CN108430974B (en) * | 2015-06-05 | 2021-09-17 | 领先制药控股公司 | ROR gamma modulators |
| RU2730454C2 (en) * | 2015-06-05 | 2020-08-24 | Лид Фарма Холдинг Б.В. | ROR GAMMA (RORγ) MODULATORS |
| RU2727698C2 (en) * | 2015-06-05 | 2020-07-23 | Лид Фарма Холдинг Б.В. | ROR GAMMA (RORγ) MODULATORS |
| RU2727191C2 (en) * | 2015-06-05 | 2020-07-21 | Лид Фарма Холдинг Б.В. | ROR GAMMA (RORγ) MODULATORS |
| EP3101007A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| AU2016273360B2 (en) * | 2015-06-05 | 2020-07-02 | Lead Pharma Holding B.V. | ROR gamma (RORy) modulators |
| US10556866B2 (en) | 2015-06-05 | 2020-02-11 | Lead Pharma Holding B.V. | ROR gamma (RORγ) modulators |
| EP3101006A1 (en) * | 2015-06-05 | 2016-12-07 | Lead Pharma Cel Models IP B.V. | Ror gamma (rory) modulators |
| CN107922337B (en) * | 2015-06-05 | 2021-07-02 | 领先制药控股公司 | ROR gamma modulators |
| WO2016193459A1 (en) * | 2015-06-05 | 2016-12-08 | Lead Pharma Cel Models Ip B.V. | Ror gamma (rory) modulators |
| CN108026050A (en) * | 2015-08-03 | 2018-05-11 | 格兰马克药品股份有限公司 | New compound as ROR gamma modulators |
| US10988467B2 (en) | 2015-08-03 | 2021-04-27 | Glenmark Pharmaceuticals S.A. | Compounds as ROR gamma modulators |
| JP2018522047A (en) * | 2015-08-03 | 2018-08-09 | グレンマーク・ファーマシューティカルズ・エスエー | Novel compounds as ROR gamma modulators |
| CN108026050B (en) * | 2015-08-03 | 2021-04-30 | 格兰马克药品股份有限公司 | Novel compounds as ROR gamma modulators |
| US10829448B2 (en) | 2015-08-05 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Substituted benzoimidazoles as modulators of ROR-γ |
| US10301261B2 (en) | 2015-08-05 | 2019-05-28 | Vitae Pharmaceuticals, Llc | Substituted indoles as modulators of ROR-gamma |
| US11008340B2 (en) | 2015-11-20 | 2021-05-18 | Vitae Pharmaceuticals, Llc | Modulators of ROR-gamma |
| US10988445B2 (en) | 2015-12-15 | 2021-04-27 | Astrazeneca Ab | Compounds |
| US10526286B2 (en) | 2015-12-15 | 2020-01-07 | Astrazeneca Ab | Compounds |
| US11453644B1 (en) | 2015-12-15 | 2022-09-27 | Astrazeneca, Ab | Compounds |
| US10011566B2 (en) | 2015-12-15 | 2018-07-03 | Astrazeneca Ab | Compounds |
| US10829481B2 (en) | 2016-01-29 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Benzimidazole derivatives as modulators of ROR-gamma |
| JP2021193131A (en) * | 2016-01-29 | 2021-12-23 | ヴァイティー ファーマシューティカルズ,エルエルシー | Benzimidazole derivatives as modulators of ror-gamma |
| US10975068B2 (en) | 2016-04-27 | 2021-04-13 | Janssen Pharmaceutica Nv | 6-aminopyridin-3-yl thiazoles as modulators of RORγT |
| WO2017199103A1 (en) * | 2016-05-18 | 2017-11-23 | Glenmark Pharmaceuticals S.A. | Benzamide compounds as ror gamma modulators |
| US9481674B1 (en) | 2016-06-10 | 2016-11-01 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| CN107488113A (en) * | 2016-06-13 | 2017-12-19 | 上海泰禾国际贸易有限公司 | A kind of method for synthesizing o-aminobiphenyl class compound |
| CN107522634A (en) * | 2016-06-22 | 2017-12-29 | 复旦大学 | Biaryl ureas carboxylic acid derivates or its salt and its production and use |
| JP7092356B2 (en) | 2016-06-22 | 2022-06-28 | フーダン ユニヴァーシティ | Biarylurea derivatives or salts thereof, and their preparation methods and uses |
| CN107522634B (en) * | 2016-06-22 | 2020-09-01 | 复旦大学 | Biaryl urea carboxylic acid derivative or salt thereof, and preparation method and application thereof |
| JP2019520359A (en) * | 2016-06-22 | 2019-07-18 | フーダン ユニヴァーシティFudan University | Biaryl urea derivatives or salts thereof and methods for their preparation and use |
| US10851050B2 (en) | 2016-06-22 | 2020-12-01 | Fudan University | Biaryl urea derivative or salt thereof and preparation process and use for the same |
| EP3476829A4 (en) * | 2016-06-22 | 2019-11-13 | Fudan University | Biaryl urea derivative or salt thereof, and manufacturing and application of same |
| US10647665B2 (en) | 2016-06-22 | 2020-05-12 | Fudan University | Biaryl urea derivative or salt thereof and preparation process and use for the same |
| CN107522641A (en) * | 2016-06-22 | 2017-12-29 | 复旦大学 | Biaryl ureas analog derivative or its salt and its production and use |
| WO2018011746A1 (en) | 2016-07-14 | 2018-01-18 | Cadila Healthcare Limited | Cyclopropyl derivatives as ror-gamma modulators |
| WO2018011747A1 (en) | 2016-07-14 | 2018-01-18 | Cadila Healthcare Limited | Polycyclic compounds as ror-gamma modulators |
| KR20190096365A (en) * | 2016-12-05 | 2019-08-19 | 리드 파마 홀딩 비.브이. | ROR gamma (RORγ) regulator |
| KR102554776B1 (en) | 2016-12-05 | 2023-07-11 | 리드 파마 홀딩 비.브이. | ROR gamma (RORγ) regulator |
| CN110072859A (en) * | 2016-12-13 | 2019-07-30 | 勃林格殷格翰国际有限公司 | Compound as ROR gamma modulators |
| WO2018111803A1 (en) * | 2016-12-13 | 2018-06-21 | Boehringer Ingelheim International Gmbh | Compounds as modulators of ror gamma |
| US10954215B2 (en) | 2016-12-13 | 2021-03-23 | Boehringer Ingelheim International Gmbh | Compounds as modulators of ROR gamma |
| JP2020502098A (en) * | 2016-12-13 | 2020-01-23 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Compounds as modulators of ROR gamma |
| CN110072859B (en) * | 2016-12-13 | 2022-10-11 | 勃林格殷格翰国际有限公司 | Compounds as ROR gamma modulators |
| JP7014795B2 (en) | 2016-12-13 | 2022-02-01 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Compound as a modulator of ROR gamma |
| US10844017B2 (en) | 2017-02-09 | 2020-11-24 | Fudan University | Biaryl compound, preparation method and use thereof |
| WO2018145653A1 (en) * | 2017-02-09 | 2018-08-16 | 复旦大学 | Biaryl compound, preparation method and use therefor |
| JP2020508981A (en) * | 2017-02-09 | 2020-03-26 | ▲復▼旦大学Fundan University | Biaryl compound, its production method and use |
| CN108863850A (en) * | 2017-02-09 | 2018-11-23 | 复旦大学 | Biaryl base class compound and its preparation method and application |
| WO2018181456A1 (en) * | 2017-03-29 | 2018-10-04 | 科研製薬株式会社 | Indazole derivative and pharmaceutical drug containing same |
| WO2018193297A1 (en) | 2017-04-21 | 2018-10-25 | Cadila Healthcare Limited | Novel compounds as ror-gamma modulators |
| US11034654B2 (en) | 2017-06-14 | 2021-06-15 | Astrazeneca Ab | 2,3-dihydroisoindole-1-carboxamides useful as ROR-gamma modulators |
| CN109134476A (en) * | 2017-06-15 | 2019-01-04 | 复旦大学 | Bridged ring piperazine derivative or its salt and its preparation method and application |
| CN109134476B (en) * | 2017-06-15 | 2021-03-19 | 复旦大学 | Bridged ring piperazine derivative or salt thereof, and preparation method and application thereof |
| US10913739B2 (en) | 2017-07-24 | 2021-02-09 | Vitae Pharmaceuticals, LLC (121374) | Inhibitors of RORγ |
| US11186573B2 (en) | 2017-07-24 | 2021-11-30 | Vitae Pharmaceuticals, Llc | Inhibitors of ROR gamma |
| WO2019022223A1 (en) * | 2017-07-28 | 2019-01-31 | 東レ株式会社 | Cyclic amine derivative and use thereof for medical purposes |
| CN107935876A (en) * | 2017-12-21 | 2018-04-20 | 金凯(辽宁)化工有限公司 | A kind of preparation method of 2 (3 amino, 4 chlorobenzoyl) benzoic acid |
| KR20200115491A (en) | 2018-01-31 | 2020-10-07 | 도레이 카부시키가이샤 | Cyclic amine derivatives and pharmaceutical uses thereof |
| WO2019151270A1 (en) | 2018-01-31 | 2019-08-08 | 東レ株式会社 | Cyclic amine derivative and pharmaceutical use thereof |
| WO2019149965A1 (en) | 2018-02-05 | 2019-08-08 | Universite De Strasbourg | Compounds and compositions for the treatment of pain |
| WO2019213470A1 (en) | 2018-05-03 | 2019-11-07 | Eternity Bioscience Inc. | Benzimidazole derivatives as modulators of retinoid-related orphan receptor gamma (rorϒ) and pharmaceutical uses thereof |
| US11345666B2 (en) | 2018-06-18 | 2022-05-31 | Janssen Pharmaceutica Nv | Phenyl and pyridinyl substituted imidazoles as modulators of RORγT |
| US10975057B2 (en) | 2018-06-18 | 2021-04-13 | Janssen Pharmaceutica Nv | 6-aminopyridin-3-yl pyrazoles as modulators of RORgT |
| US10975037B2 (en) | 2018-06-18 | 2021-04-13 | Janssen Pharmaceutica Nv | Phenyl substituted pyrazoles as modulators of RORγt |
| US20190382350A1 (en) * | 2018-06-18 | 2019-12-19 | Janssen Pharmaceutica Nv | PYRIDINYL PYRAZOLES AS MODULATORS OF RORyT |
| US11034658B2 (en) * | 2018-06-18 | 2021-06-15 | Janssen Pharmaceutica Nv | Pyridinyl pyrazoles as modulators of RORγT |
| RU2675806C1 (en) * | 2018-07-20 | 2018-12-25 | Федеральное государственное бюджетное образовательное учреждение высшего образования "Национальный исследовательский Мордовский государственный университет им. Н.П. Огарёва" | N-(indolyl)trifluoroacetamides with antimicrobial activity production method |
| CN113166061B (en) * | 2018-11-27 | 2023-11-21 | 正大天晴药业集团股份有限公司 | ROR gamma inhibitors containing sulfonyl structures |
| WO2020108538A1 (en) * | 2018-11-27 | 2020-06-04 | 正大天晴药业集团股份有限公司 | RORγ INHIBITOR HAVING SULFONYL STRUCTURE |
| CN113166061A (en) * | 2018-11-27 | 2021-07-23 | 正大天晴药业集团股份有限公司 | ROR gamma inhibitors containing sulfonyl structures |
| WO2020140043A1 (en) * | 2018-12-28 | 2020-07-02 | Endogena Therapeutics, Inc. | Compounds for use as therapeutically active substances in the treatment and/or prevention of neuroretinal diseases |
| CN113301958A (en) * | 2018-12-28 | 2021-08-24 | 内生疗法公司 | Compounds for use as therapeutically active substances in the treatment and/or prevention of neuroretinal diseases |
| US10752593B2 (en) | 2018-12-28 | 2020-08-25 | Endogena Therapeutics, Inc. | Compounds for use as therapeutically active substances in the treatment of retinal diseases |
| WO2020211836A1 (en) * | 2019-04-19 | 2020-10-22 | 北京酷瓴生物技术有限公司 | Benzene piperidine derivative, preparation method therefor, intermediate thereof and use thereof |
| CN112538072B (en) * | 2019-09-21 | 2024-02-06 | 齐鲁制药有限公司 | Aminopyrimidine EGFR inhibitors |
| CN112538072A (en) * | 2019-09-21 | 2021-03-23 | 齐鲁制药有限公司 | Novel aminopyrimidine EGFR (epidermal growth factor receptor) inhibitor |
| CN112851557A (en) * | 2019-09-30 | 2021-05-28 | 上海辉启生物医药科技有限公司 | Sulfo-substituted biaryl compound or salt thereof, and preparation method and application thereof |
| WO2021063362A1 (en) * | 2019-09-30 | 2021-04-08 | 上海辉启生物医药科技有限公司 | Sulfo-substituted biaryl compound or salt thereof, preparation method therefor, and use thereof |
| WO2021083311A1 (en) | 2019-10-31 | 2021-05-06 | 江苏恒瑞医药股份有限公司 | ACID ADDITION SALT OF RORγ REGULATOR |
| CN113121393A (en) * | 2019-12-31 | 2021-07-16 | 广东东阳光药业有限公司 | Aromatic amide compound and application thereof in medicines |
| WO2021136326A1 (en) * | 2019-12-31 | 2021-07-08 | 广东东阳光药业有限公司 | Aryl amide compound and application thereof in medicine |
| CN113072521A (en) * | 2020-01-06 | 2021-07-06 | 广东东阳光药业有限公司 | ROR gamma t inhibitor and application thereof in medicine |
| CN113072538A (en) * | 2020-01-06 | 2021-07-06 | 广东东阳光药业有限公司 | ROR gamma t inhibitor and application thereof in medicine |
| CN113072476A (en) * | 2020-01-06 | 2021-07-06 | 广东东阳光药业有限公司 | ROR gamma t inhibitor and preparation method and application thereof |
| WO2021139599A1 (en) * | 2020-01-06 | 2021-07-15 | 东莞市东阳光新药研发有限公司 | RORγT INHIBITOR, PREPARATION METHOD THEREFOR AND USE THEREOF |
| CN113072538B (en) * | 2020-01-06 | 2024-04-05 | 广东东阳光药业股份有限公司 | ROR gamma t inhibitor and application thereof in medicines |
| CN113072476B (en) * | 2020-01-06 | 2024-05-14 | 广东东阳光药业股份有限公司 | ROR gamma t inhibitor and preparation method and application thereof |
| CN113072521B (en) * | 2020-01-06 | 2024-04-05 | 广东东阳光药业股份有限公司 | ROR gamma t inhibitor and application thereof in medicines |
| EP4129996A4 (en) * | 2020-03-23 | 2023-07-12 | Qilu Pharmaceutical Co., Ltd. | Novel aminopyrimidine egfr inhibitor |
| WO2021228215A1 (en) * | 2020-05-15 | 2021-11-18 | 上海辉启生物医药科技有限公司 | BIARYL COMPOUND CAPABLE OF SERVING AS RORγ REGULATOR |
| CN113666853A (en) * | 2020-05-15 | 2021-11-19 | 上海辉启生物医药科技有限公司 | Biaryl compounds useful as ROR gamma modulators |
| WO2022007168A1 (en) * | 2020-07-10 | 2022-01-13 | 中国科学院广州生物医药与健康研究院 | Benzo five-membered nitrogen heterocyclic compound and application thereof |
| WO2023240084A3 (en) * | 2022-06-06 | 2024-03-21 | Denali Therapeutics Inc. | Compounds, compositions, and methods |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013029338A1 (en) | Novel compounds | |
| KR101286569B1 (en) | Novel medicaments | |
| WO2012100732A1 (en) | Retinoid-related orphan receptor gamma modulators, composition containing them and uses thereof | |
| TWI422579B (en) | New compounds | |
| WO2012028100A1 (en) | Novel compounds | |
| TWI491595B (en) | Substituted phenylureas and phenylamides as vanilloid receptor ligands | |
| AU2002212118B2 (en) | 4-phenyl-pyridine derivatives as neurokinin-1 receptor antagonists | |
| WO2012027965A1 (en) | Novel compounds | |
| WO2012100734A1 (en) | Compounds useful as retinoid-related orphan receptor gamma modulators | |
| CN101712657A (en) | heteroarylcarbamoylbenzene derivative | |
| JP2020023577A (en) | Biaryl derivative as gpr120 agonist | |
| ES2825031T3 (en) | Halogen substituted heterocyclic compound useful for the treatment of diseases caused by APL | |
| CA2938855C (en) | 1,2-substituted cyclopentanes as orexin receptor antagonists | |
| OA13247A (en) | Phenyl or pyridyl amide coumpounds as prostaglandin E2 antagonists. | |
| IL116998A (en) | Indoline compounds, processes for their preparation and pharmaceutical compositions comprising them | |
| CA2716898A1 (en) | Compound having 6-membered aromatic ring | |
| WO2005094822A1 (en) | Pyridyl derivatives and their use as mglu5 receptor antagonists | |
| KR20150108846A (en) | Azaindole derivatives as multi kinase inhibitors | |
| WO2012120397A1 (en) | Fluoro-pyridinone derivatives useful as antibacterial agents | |
| JP5552121B2 (en) | Indolizine inhibitors of leukotriene production | |
| CA2956045A1 (en) | Carboxylic acid compound, method for preparation thereof, and use thereof | |
| JP2010138073A (en) | Picolinic acid amide compound | |
| EP2205556A2 (en) | Biaryl sulfonamide derivatives | |
| BR112017010402B1 (en) | SUBSTITUTED CARBOXAMIDE BASED ON THIAZOLE AND OXAZOLE AND UREA DERIVATIVES AS VANILLOID II RECEPTOR LIGANDS | |
| CA2506799A1 (en) | Mixed lineage kinase modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12826717 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12826717 Country of ref document: EP Kind code of ref document: A1 |