WO2012059797A1 - Process for synthesis of (s) - pregabalin - Google Patents
Process for synthesis of (s) - pregabalin Download PDFInfo
- Publication number
- WO2012059797A1 WO2012059797A1 PCT/IB2011/000480 IB2011000480W WO2012059797A1 WO 2012059797 A1 WO2012059797 A1 WO 2012059797A1 IB 2011000480 W IB2011000480 W IB 2011000480W WO 2012059797 A1 WO2012059797 A1 WO 2012059797A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cyano
- methyl
- cinchonidine
- compound
- acid
- Prior art date
Links
- 0 CC(C)CC(C*=O)(C(O)=O)C#N Chemical compound CC(C)CC(C*=O)(C(O)=O)C#N 0.000 description 3
- MGWZYUMZVZMKTN-UHFFFAOYSA-N CC(C)CC(CC(O)=O)C#N Chemical compound CC(C)CC(CC(O)=O)C#N MGWZYUMZVZMKTN-UHFFFAOYSA-N 0.000 description 2
- KMPWYEUPVWOPIM-NAHPKXJFSA-N C=CC1C(CC2)CC([C@@H](c3ccnc4ccccc34)O)N2C1 Chemical compound C=CC1C(CC2)CC([C@@H](c3ccnc4ccccc34)O)N2C1 KMPWYEUPVWOPIM-NAHPKXJFSA-N 0.000 description 1
- HLOMRLLHWGUSOI-UHFFFAOYSA-O CCC(CC[NH+](C1)C(c2ccnc3ccccc23)O)C1C=C Chemical compound CCC(CC[NH+](C1)C(c2ccnc3ccccc23)O)C1C=C HLOMRLLHWGUSOI-UHFFFAOYSA-O 0.000 description 1
- XOHIDSSTBKHCPY-UHFFFAOYSA-N CCOC(C(CC(C)C)C#N)=O Chemical compound CCOC(C(CC(C)C)C#N)=O XOHIDSSTBKHCPY-UHFFFAOYSA-N 0.000 description 1
- YVGIRGNEPUPXCO-UHFFFAOYSA-N CCOC(CC(C(C)(C)C(C)C)(C(OCC)=O)C#N)=O Chemical compound CCOC(CC(C(C)(C)C(C)C)(C(OCC)=O)C#N)=O YVGIRGNEPUPXCO-UHFFFAOYSA-N 0.000 description 1
- NENOICKNJDFYMH-UHFFFAOYSA-O CCOC(CC(CC(C)C)(C(OCC)=[OH+])C#N)=O Chemical compound CCOC(CC(CC(C)C)(C(OCC)=[OH+])C#N)=O NENOICKNJDFYMH-UHFFFAOYSA-O 0.000 description 1
- CGVIWHWBHANMPJ-WOANRRIASA-N CCOC1OC1CC([C@@H](C)C(C)C)([C@H](C)C#N)C(OC)=O Chemical compound CCOC1OC1CC([C@@H](C)C(C)C)([C@H](C)C#N)C(OC)=O CGVIWHWBHANMPJ-WOANRRIASA-N 0.000 description 1
- LVLCOXXUHBMOOM-LICQEQMYSA-N CC[C@@H](C(CC(C)C)([C@H](C(C)C)C#N)C(O)=O)C([O](C)C)=O Chemical compound CC[C@@H](C(CC(C)C)([C@H](C(C)C)C#N)C(O)=O)C([O](C)C)=O LVLCOXXUHBMOOM-LICQEQMYSA-N 0.000 description 1
- LIHUTSFYFGCWQP-UHFFFAOYSA-O CNC(CC#N)=[OH+] Chemical compound CNC(CC#N)=[OH+] LIHUTSFYFGCWQP-UHFFFAOYSA-O 0.000 description 1
- ANGDWNBGPBMQHW-UHFFFAOYSA-N COC(CC#N)=O Chemical compound COC(CC#N)=O ANGDWNBGPBMQHW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/04—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C229/06—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one amino and one carboxyl group bound to the carbon skeleton
- C07C229/08—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one amino and one carboxyl group bound to the carbon skeleton the nitrogen atom of the amino group being further bound to hydrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B57/00—Separation of optically-active compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/04—Formation of amino groups in compounds containing carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/30—Preparation of optical isomers
- C07C227/34—Preparation of optical isomers by separation of optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/30—Preparation of carboxylic acid nitriles by reactions not involving the formation of cyano groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/32—Separation; Purification; Stabilisation; Use of additives
- C07C253/34—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Definitions
- the invention relates to novel, cost effective, green and industrial process for synthesis of (SJ-pregabalin. Background of the Invention:
- pregabalin (I) is a potent anticonvulsant.
- pregabalin exhibits anti-seizure activity and is found to be useful for treatment of various other conditions, like pain, fibromyalgia, physiological conditions associated with psychomotor stimulants, inflammation, gastrointestinal damage, insomnia, alcoholism and various psychiatric disorders, including mania and bipolar disorder.
- (S)-3-Cyano-5-methyl-hexanoic acid (II) is one of the key intermediates for the synthesis of (S)-pregabalin.
- a number of approaches for synthesis of racemic as well as enatiomerically pure. compound (II) are reported in the literature.
- majority of processes suffer from the drawback of using potassium cyanide or its equivalent during synthesis, thus rendering the process not eco-friendly and environmentally benign.
- (RS) - 3-cyano-5-methyl- hexanoic acid ethyl ester was resolved through lipase catalyzed kinetic resolution to obtain (S) - 3-cyano-5-methyl-hexanoic acid ethyl ester, which was hydrolyzed to obtain (S) - 3- cyano-5-methyl-hexanoic acid and subsequently converted to S-pregabalin, disclosers of which, including prior art are incorporated herein by reference.
- This invention relates to;
- the object of this invention is to provide resolution of (RS) - 3-cyano-5-methyl-hexanoic acid (XII) through diastereomeric salt formation with cinchonidine (XIII) to obtain optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II), having excellent yield and high optical purity (99 % ee) and further conversion of it to (S)-pregabalin (I) in high yield and high optical purity (>99 % ee).
- Yet another object of the present invention is synthesis of the novel compound 4-ethyl 1 - methyl 2-cyano-2-isobutylsuccinate (VII) through a novel method and further conversion of it to (S)-pregabalin (I).
- Yet another object of the present invention is synthesis of diethyl 2-cyano-2- isobutylsuccinate (X) through solvent-free, green, eco-friendly and novel method.
- Yet another object of the present invention is enhancement in the rate of decarboxylation of diethyl 2-cyano-2-isobutylsuccinate (X) to obtain (f?S)-3-cyano-5-methyl hexanoic acid ethyl ester (XI).
- Yet another object of the present invention is to investigate the effect of carbon chain length of the substrates on the decarboxylation.
- Decarboxylation of 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII) to Obtain (RS)-3-cyano-5-methyl hexanoic acid ethyl ester (XI) requires the reaction temperature around 140 °C whereas, for decarboxylation of diethyl 2- cyano-2-isobutylsuccinate (X) to obtain (RS)-3-cyano-5-methyl hexanoic acid ethyl ester (XI) requires the reaction temperature around 170 °C.
- Yet another object of the present invention is synthesis of the novel compound 2-cyano-2- isobutylsuccinic acid (XVI) through a novel method and further conversion of it to (S) - 3- cyano-5-methyl-hexanoic acid (II)
- An also important object of the present invention is to provide a process for recovery of resolving agent i.e. cinchonidine (XIII) through basification and reusability of recovered cinchonidine (XIII), thereby improving the atom economy, process efficiency and hence cost.
- resolving agent i.e. cinchonidine (XIII)
- basification and reusability of recovered cinchonidine (XIII) thereby improving the atom economy, process efficiency and hence cost.
- the present invention is directed towards synthesis of (S)-Pregabaliri.
- the invention is summarized below in scheme A and scheme B.
- scheme A detailed schematic representations of improved process for synthesis of (RS)- 3-cyano-5-methyl hexanoic acid (XII) and further conversion to (S)-pregabalin (I) are given.
- Route I Compound (V) was prepared by condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid methyl ester (III) in presence of cesium acetate, followed by hydrogenation using palladium on charcoal catalyst. Compound (V) was further reacted with haloacetic acid ethyl ester (VI) in presence of cesium carbonate without solvent or optionally in an organic solvent such as dimethyl sulfoxide to give 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII).
- compound (VII) was converted into compound (XVI) through base catalyzed hydrolysis and which was further decarboxylated to give (RS)-3-cyano-5-methyl-hexanoic acid (XII) in presence of mineral acid such as sulfuric add in organic solvent such as ethyl acetate.
- Racemic 3-cyano-5-methyl-hexanoic acid (XII) was resolved through diastereomeric salt formation with cinchonidine (XIII) to obtain (S)-3-cyano-5-methyl-hexanoic acid (II).
- Compound (II) was converted to compound (I) through hydrogenation in presence of Raney Nickel.
- compound (X) was also converted into compound (XVI) through base catalyzed hydrolysis, which was further decarboxylated to give (RS)-3-cyano-5-methyl- hexanoic acid (XII) in presence of mineral acid and in organic solvent such as ethyl acetate.
- (S)-3-cyano-5-methyl-hexanoic acid (II) was obtained from (S)-3- cyano-5-methyl-hexanoic acid salt of cinchonidine (XIV) via decomposition in biphasic mixture of aqueous mineral acid such as dilute aqueous hydrochloric acid and organic solvent such as ethyl acetate, dichloromethane, methyl tert-butyl ether; preferably ethyl acetate.
- Cinchonidine (XIII) from aqueous acid solution was also recovered through basification and reused to improve the over all atom economy and process efficiency.
- First aspect of the invention is a process for synthesis of pregabalin (I)
- halo group includes chloro, bromo and iodo, in presence of base such as sodium carbonate, potassium carbonate, cesium carbonate, preferably cesium carbonate without solvent or in an organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4- dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N, " /V-dimethyl formamide and dimethyl sulfoxide at temperature of about 10 to 90 °C to give 4-ethyl 1-alkyl-2-
- Another aspect of the invention is a process for synthesis of pregabalin (I)
- halo group includes chloro, bromo and iodo, in presence of base such as sodium carbonate, potassium carbonate, cesium carbonate, preferably cesium carbonate without solvent or in an organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4-dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N, /V-dimethyl formamide and dimethyl sulfoxide at temperature of about 10 to 90 °C to give 4-ethyl 1-alkyl-2-cyano-2- isobutylsuccinate (C);
- compound (XVI) was decarboxylated in presence of mineral acid such as sulfuric acid in organic solvent such as ethyl acetate to obtain compound (XII) at about temperature 70 to
- Yet another aspect of the invention is a process for synthesis of pregabalin (I)
- This invention provides i) Resolution via diastereomeric salt formation between (RS) - 3-cyano-5- methyl-hexanoic acid (XII) and cinchonidine (XIII) to obtain optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) in excellent yield and high optical purity (>99 % ee) and further conversion to (S)-pregabalin (I) in high yield and high optically purity (>99 % ee).
- halo group includes chloro, bromo and iodo, in presence of base such as sodium carbonate, potassium carbonate, cesium carbonate, preferably cesium carbonate without solvent or optionally in a organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4- dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N, A/-dimethyl formamide and dimethyl sulfoxide, more preferably dimethyl sulfoxide at temperature of about 10 to 90 °C, preferably 50 to 60 °C to give 4-ethyl 1-methyl 2-cyano-2-isobutylsuccinate (VII).
- base such as sodium carbonate, potassium carbonate, cesium carbonate, preferably cesium carbonate without solvent or optionally in a organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4- dioxane, dimethyl sul
- compound (XVI) was decarboxylated in presence of mineral acid such as sulfuric acid in organic solvent such as ethyl acetate to obtain compound (XII) at about temperature 70 to 80 °C; or decarboxylated through reported methods such as base catalyzed decarboxylation (J.Org. Chem, 1961 , 83, 2354)
- react erein halo group include chloro, bromo and iodo, in presence of base such as potassium carbonate, sodium carbonate and cesium carbonate preferably cesium carbonate without solvent or optionally in a organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4- dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N, /V-dimethyl formamide and dimethyl sulfoxide, more preferably dimethyl sulfoxide at temperature of about 10 to 80 °C, preferably at 50 to 60 °C to give diethyl 2-cyano-2-isobutylsuccinate (X).
- base such as potassium carbonate, sodium carbonate and cesium carbonate preferably cesium carbonate without solvent or optionally in a organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4- dioxane, dimethyl sul
- compound (XVI) was decarboxylated in presence of mineral acid such as sulfuric acid in organic solvent such as ethyl acetate to obtain compound (XII) at about temperature 70-80 °C or decarboxylated through reported methods such as base catalyzed decarboxylation (J.Org. Chem, 1961 , 83, 2354)
- resolution through diastereomeric salt between (RS)-3-cyano-5-methylhexanoic acid (XII) with cinchonidine (XIII) comprised of the following steps: a) compound (XII) was treated with cinchonidine(XIII) in presence of organic solvent such as methanol, ethanol, 1 ,4-dioxane, ethyl acetate, tetrahydrofuran, 2-methyl tetrahydrofuran, dimethoxy ethane and diglyme, preferably ethyl acetate at temperature of about 20°C to 80°C, preferably at 70°C to 80 °C to precipitate out (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV), followed by separation of compound (XIV) through known separation techniques such as filtration, centrifugation, sedimentation, etc
- ethyl acetate layer which contains the ⁇ R)-3- cyano-5-methylhexanoic acid salt of cinchonidine (XV) was treated with aqueous dilute mineral acid such as hydrochloric acid to recovered cinchonidihe.
- the enantiomeric excess (ee) for pregabalin is determined by HPLC using a Shimadzu LC 2010 system equipped with a chiral column (Purosphere star RP-18e (4.6 x 150mm), 5pm), column oven temperature 25 °C and UV visible detector (UV at 340nm). Mobile phase is buffer: acetonitrile (55:45) with flow rate 1.0 mL/min, injection volume 20 ⁇ .
- the enantiomeric excess (ee) is determined by derivatized by reacting with Marfey's reagent.
- the enantiomeric excess (ee) for (S) - 3-cyano-5-methyl-hexanoic acid ethyl ester is determined by Gas-Liquid chromatography using a Shimadzu GC 2010 system equipped with a chiral column (Chiraledex (20m x 0.25mm x 0.12mm)), and FID detector.
- the enantiomeric excess (ee) for (S) or (R) - 3-cyano-5-methyl-hexanoic acid is determined via converting into corresponding ester and analyzed on Gas-Liquid chromatography using a Shimadzu GC 2010 system equipped with a chiral column (Chiraledex (20m x 0.25mm x 0.12mm)), and FID detector.
- Example 1 Synthesis of ethyl 2-cyano-4-methylpentanoate (IX) from condensation of ethyl cyano acetate (VIII) with /so-butyraldehdye (IV) in presence of piperidine / acetic
- Reactor was purged with hydrogen gas two times and charged with hydrogen, 3 kg/cm 2 pressure was maintained in the Parr autoclave until hydrogen consumption ceases. Reaction was monitored by TLC. After completion of reaction, reaction mixture was filtered through Celite bed to remove Pd/C and filtrate was concentrated under reduced pressure to remove solvent. Residue was suspended in 100 mL water. Organic layer was separated to obtain ethyl 2-cyano-4-methylpentanoate (IX) (80 g, 95 % yield) as light yellow oil.
- IX ethyl 2-cyano-4-methylpentanoate
- Example 3 Synthesis of ethyl 2-cyano-4-methylpentanoate (IX) from condensation of ethyl cyano acetate (VIII) with /so-butyraldehdye (IV) in presence of cesium acetate /water
- Example 5 Synthesis of diethyl 2-cyano-2-isobutylsuccinate (X) from ethyl 2-cyano-4- methylpentanoate (IX) and ethyl chloro acetate (VI) in presence of potassium carbonate.
- Example 7 Synthesis of methyl-2-cyano-4-methylpentanoate (V) from condensation of methyl cyano acetate (III) with / ' so-butyraldehdye (IV) in presence of piperidine / acetic acid .
- Reactor was purged with hydrogen gas two times and charged with hydrogen, 3 kg/cm 2 pressure was maintained in the Parr autoclave until hydrogen consumption ceases. Reaction was monitored by TLC. After completion of reaction, reaction mixture was filtered through Celite bed to remove Pd/C and filtrate was concentrated under reduced pressure to remove solvent. Residue was suspended in 100 mL water. Organic layer was separated to obtain methyl-2-cyano-4- methylpentanoate (V) (170 g, 90 % yield) as light yellow oil.
- V methyl-2-cyano-4- methylpentanoate
- Example 8 Synthesis of methyl-2-cyano-4-methylpentanoate (V) from condensation of methyl cyano acetate (III) with / ' so-butyraldehdye (IV) in presence of cesium acetate in methanol
- Example 9 Synthesis of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) from methyl-2 cyano-4-methylpentanoate (V) and ethyl chloro acetate (VI) in presence of cesium carbonate.
- Example 10 Synthesis of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) from methyl- 2-cyano-4-methylpentanoate (V) and ethyl chloro acetate (VI) in presence of potassium carbonate.
- Example 11 Synthesis of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) from methyl- 2-cyano-4-methylpentanoate (V) and ethyl chloro acetate (VI) in presence of sodium carbonate.
- Example 2 Decarboxylation of diethyl 2-cyano-2-isobutylsuccinate (X) to obtain (RS) - 3- cyano-5-methylhexanoic acid ethyl ester (XI) in presence of KCI/DMSO
- a 1 L reactor was charged with diethyl 2-cyano-2-isobutylsuccinate (X) (102 g), potassium chloride (32.5 g), dimethyl sulphoxide (500 mL) and water (7.5 mL). The resulting reaction mixture was heated at 170 °C and maintained at that temperature for 4 h. Reaction was monitored by TLC for complete consumption of starting material. The reaction mixture was cooled to 40 to 50 °C and treated with methyl terf-butyl ether (200 mL). The mixture was further cooled to 0 to 5 °C and treated with water (1 L) in small portions to maintain the temperature below 40 °C. After stirring for 30 min the phases were separated.
- X diethyl 2-cyano-2-isobutylsuccinate
- potassium chloride 32.5 g
- dimethyl sulphoxide 500 mL
- water 7.5 mL
- reaction mixture was heated at 140 °C and maintained at that temperature for 4 h. Reaction was monitored by TLC for complete consumption of starting material.
- the reaction mixture was cooled to 40 to 50 °C and treated with methyl terf-butyl ether (200 mL). The mixture was further cooled to 0 to 5°C and treated with water (1 L) in small portions to maintain the temperature below 40 °C. After stirring for 30 min the phases are separated. The aqueous phase was extracted with methyl te/f-butyl ether (3 x 800 mL).
- Example 19 Decarboxylation of diethyl-2-cyano-2-isobutylsuccinate (X) to obtain ⁇ RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of diethyl amine ethane thiol/cesium carbonate
- Example 20 One pot synthesis of (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) from methyl-2-cyano-4-methylpentanoate (V)
- reaction mixture was neated to 135- 140 °C and stirred further at that temperature for 4 h.
- the reaction mixture was cooled to 40 to 50 °C and treated with methyl terf-butyl ether (200 mL).
- the mixture was further cooled to 0 to 5°C and treated with water (1 L) in small portions to maintain the temperature below 40 °C.
- the aqueous phase was extracted with methyl tert-butyl ether (3 x 800 mL); Organic phases were combined and washed twice with 100 mL water.
- the organic layer was decolorized by treating with 7.0 g of activated charcoal.
- the resultant mixture was filtered to remove charcoal and filtrate was evaporated to give (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) (39.1 g) as light brown color oil.
- Example 21 Synthesis of (RS) - 3-cyano-5-methylhexanoic acid (XII) through hydrolysis of (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of lithium hydroxide.
- Example 22 Synthesis of ⁇ RS) - 3-cyano-5-methylhexanoic acid (XII) through hydrolysis of ( ?S) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of NaOH
- Example 31 Resolution of (RS) - 3-cyano-5-methylhexanoic acid (XII) through diastereomeric salt formation with cinchonidine (1 : 0.5).
- Example 32 Resolution (RS) - 3-cyano-5-methylhexanoic acid (XII) through diastereomeric salt formation with cinehonidine (XIII) (1 : 1 mol ratio) in ethyl acetate
- Cinchonidine (14.5gm) was obtained through basification of aqueous layer of example 28 which contain the hydrochloric acid salt of cinhonidine.
- Example 39 Synthesis of (S)-Pregabalin from (S)-3-cyano-5-methyl hexanoic acid.
- reaction mixture was filtered through a Celite pad and solvent from filtrate was evaporated under reduced pressure to leave a semi-solid material, which was re- crystallized from /so-propyl alcohol: water mixture (94:06, 25 cm 3 ) to obtain (S)-pregabalin (10.0 g, 48 % and 99 % ee as per chiral HPLC analysis), as a white solid.
- Example 40 Synthesis of (S)-Pregabalin from (S)-3-cyano-5-methyl hexanoic acid and isolation by using dimethoxy ethane.
- a solution of (S)-3-cyano-5-methyl hexanoic acid (II) (20.0 g, 0.13 mol) in methanol: water (70:30) (100 cm 3 ) was added into a solution of potassium carbonate (7.2 g, 0.13 mol) in water (20 cm 3 ) at 25 °C and was stirred at room temperature for 2 h. The mixture was then transferred into a Parr autoclave reactor and carefully raney nickel (10.0 g) was added. Reactor was purged with hydrogen gas twice and then 0 atm.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Improved process for the synthesis of (S)-pregabalin having more than 99% ee through (S) 3-cyano-5-methyl-hexanoic acid has been developed. In addition to above, a novel process for resolution of (RS) - 3-cyano-5-methyl-hexanoic acid through diastereomeric salt formation with cinchonidine to obtain (S) - 3-cyano-5-methyl-hexanoic acid in high yield and high optical purity has been developed and furthermore process for recovery/ reuse of cinchonidine is also developed to improve the overall process efficiency.
Description
PROCESS FOR SYNTHESIS OF (S) - PREGABALIN
Field of the Invention:
The invention relates to novel, cost effective, green and industrial process for synthesis of (SJ-pregabalin. Background of the Invention:
(S)-3-(Aminomethyl)-5-methylhexanoic acid [CAS No. 148553-50-8], which is also known as β-isobutyl-v- aminobutyric acid, isobutyl-GABA, or pregabalin (I) is a potent anticonvulsant. As discussed in U.S. Patent No. 5,563,175, pregabalin exhibits anti-seizure activity and is found to be useful for treatment of various other conditions, like pain, fibromyalgia, physiological conditions associated with psychomotor stimulants, inflammation, gastrointestinal damage, insomnia, alcoholism and various psychiatric disorders, including mania and bipolar disorder. (U.S. Patent No. 6,242,488; U.S. Patent No. 6,326,374; U.S. Patent No. 6,001 ,876; U.S. Patent No. 6,194,459; U.S. Patent No. 6, 329, 429; U.S. Patent No. 6, 127,418; U.S. Patent No. 6,426, 368; U.S. Patent No. 6,306,910; U.S. Patent No. 6,359,005).

(S)-3-Cyano-5-methyl-hexanoic acid (II) is one of the key intermediates for the synthesis of (S)-pregabalin. A number of approaches for synthesis of racemic as well as enatiomerically pure. compound (II) are reported in the literature. However, majority of processes suffer from the drawback of using potassium cyanide or its equivalent during synthesis, thus rendering the process not eco-friendly and environmentally benign.

Our previous PCT application number PCT/IN2010/000440 dated 28 June 2010 entitled "Improved synthesis of optically pure (S) - 3-cyan0-5-methyl-hexanoic acid alkyl ester, an intermediate of (S)-pregabalin", emphasized on the novel, cost effective, eco-friendly, industrial process for synthesis of (RS) - 3-cyano-5-methyl-hexanoic acid ethyl ester without using any harmful, hazardous and poisonous chemicals. Further, (RS) - 3-cyano-5-methyl- hexanoic acid ethyl ester was resolved through lipase catalyzed kinetic resolution to obtain (S) - 3-cyano-5-methyl-hexanoic acid ethyl ester, which was hydrolyzed to obtain (S) - 3- cyano-5-methyl-hexanoic acid and subsequently converted to S-pregabalin, disclosers of which, including prior art are incorporated herein by reference.
It is evident from prior art published before our PCT application PCT/IN2010/000440 dated 28 June 2010 that the crucial feature in the manufacture of pregabalin is the synthesis of the key intermediate "(S) - 3-cyano-5-methyl-hexanoic acid" and the processes reported in the literature for its synthesis are not very attractive in view of cost, use of undesirable toxic reagents and eco-hazardous operations.
In the present invention, further improvement in the process disclosed in PCT application number PCT/IN2010/000440 dated 28 June 2010 for synthesis of (RS) - 3-cyano-5-methyl- hexanoic acid ethyl ester has been carried out with the object of improving the "greenness" of the process, energy consumption, and solvent usage, all of which have direct implications on the economic aspect of the process by reducing its over all cost.
In addition to above, a novel process for resolution of (RS) - 3-cyano-5-methyl-hexanoic acid through diastereomeric salt formation to obtain (S) - 3-cyano-5-methyl-hexanoic acid is developed. Furthermore, recovery/reuse of chiral resolving agent was developed to improve the process efficiency, atom economy, carbon efficiency, and E-factor, thereby significantly reducing the overall cost for the synthesis of the title compound.
Thus, this invention provides an improved, highly cost effective, operation friendly, "green" process for the title compound, thus satisfying almost all the criteria outlined for "green chemistry" (Green Chemistry: Theory and Practice Paul T. Anastas and John C. Warner)
This invention relates to;
1) Synthesis of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII); and diethyl 2-cyano- 2-isobutylsuccinate (X) in presence of cesium carbonate and without solvent, thus improving the "greenness" and efficiency of the process for synthesis of title compound.
2) Improved, cost effective and user friendly process for synthesis of (f?S)-3-cyano-5- methyl hexanoic acid ethyl ester (XI) from diethyl 2-cyano-2-isobutylsuccinate (X) in presence of cesium chloride and dimethyl sulfoxide, thus improving the "greenness" and efficiency of the process for synthesis of title compound.
3) Improved, cost effective and user friendly process for synthesis of (RS)-3-cyano-5- methyl hexanoic acid ethyl ester (XI) from diethyl 2-cyano-2-isobutylsuccinate (X) in presence of cesium carbonate/thiol and dimethylformamide, thus improving efficiency of the process for synthesis of title compound.
4) Easy to operate at industrial scale process for synthesis of (RS)-3-cyano-5-methyl hexanoic acid ethyl ester (XI) from 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII), thus improving the "energy consumption" and "atom economy" of the process for synthesis of title compound.
5) Synthesis of a novel compound 2-cyano-2-isobutylsuccinic add (XVI) from 4-ethyl 1- methyl 2-cyano-2-isobutylsuccinate (VII) or from diethyl 2-cyano-2-isobutylsuccinate (X) though a base catalyzed hydrolysis.
6) Efficient process for conversion of (RS)-2-cyano-2-isobutylsuccinic acid (XVI) to (RS)-3-cyano-5-methyl hexanoic acid (XII) by acid catalyzed decarboxylation.
Resolution of (RS)-3-cyano-5-methyl hexanoic acid (XII) to obtain enantiomerically pure (S)-3-cyano-5-methyl hexanoic acid (II) through diastereomeric salt formation with cinchonidine (XIII).
Synthesis of (S)-Pregabalin from (S)-3-cyano-5-methyl hexanoic acid (II) via hydrogenation in presence of metal catalysts from group VIII such as Nickel and Platinum.
9) Recovery and reusability of resolving agent i.e. cinchonidine (XIII) Objects of the Invention:
The object of this invention is to provide resolution of (RS) - 3-cyano-5-methyl-hexanoic acid (XII) through diastereomeric salt formation with cinchonidine (XIII) to obtain optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II), having excellent yield and high optical purity (99 % ee) and further conversion of it to (S)-pregabalin (I) in high yield and high optical purity (>99 % ee).
Yet another object of the present invention is synthesis of the novel compound 4-ethyl 1 - methyl 2-cyano-2-isobutylsuccinate (VII) through a novel method and further conversion of it to (S)-pregabalin (I).
Yet another object of the present invention is synthesis of diethyl 2-cyano-2- isobutylsuccinate (X) through solvent-free, green, eco-friendly and novel method.
Yet another object of the present invention is enhancement in the rate of decarboxylation of diethyl 2-cyano-2-isobutylsuccinate (X) to obtain (f?S)-3-cyano-5-methyl hexanoic acid ethyl ester (XI).
Yet another object of the present invention is to investigate the effect of carbon chain length of the substrates on the decarboxylation. Decarboxylation of 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII) to Obtain (RS)-3-cyano-5-methyl hexanoic acid ethyl ester (XI) requires the reaction temperature around 140 °C whereas, for decarboxylation of diethyl 2-
cyano-2-isobutylsuccinate (X) to obtain (RS)-3-cyano-5-methyl hexanoic acid ethyl ester (XI) requires the reaction temperature around 170 °C.
Yet another object of the present invention is synthesis of the novel compound 2-cyano-2- isobutylsuccinic acid (XVI) through a novel method and further conversion of it to (S) - 3- cyano-5-methyl-hexanoic acid (II)
An also important object of the present invention is to provide a process for recovery of resolving agent i.e. cinchonidine (XIII) through basification and reusability of recovered cinchonidine (XIII), thereby improving the atom economy, process efficiency and hence cost.
Summary of the invention:
The present invention is directed towards synthesis of (S)-Pregabaliri. The invention is summarized below in scheme A and scheme B. In scheme A, detailed schematic representations of improved process for synthesis of (RS)- 3-cyano-5-methyl hexanoic acid (XII) and further conversion to (S)-pregabalin (I) are given.
In scheme B, detailed schematic representations of resolution of ( S)-3-cyano-5-methyl hexanoic acid (XII) through diastereomeric salt formation with cinchonidine (XIII) to obtain (S)-3-cyano-5-methyl hexanoic acid (II) and method for recovery & reusability of cinchonidine (XIII) are given.

(I)
Scheme A

Scheme B
A) The processes for preparation of (S)-pregabalin (I) from (S)-3-cyano-5-methyl hexanoic acid (II).
This has been achieved through two routes:
Route I: Compound (V) was prepared by condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid methyl ester (III) in presence of cesium acetate, followed by hydrogenation using palladium on charcoal catalyst. Compound (V) was further reacted with haloacetic acid ethyl ester (VI) in presence of cesium carbonate without solvent or optionally in an organic solvent such as dimethyl sulfoxide to give 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII). Compound (VII) was then treated with cesium chloride in organic solvent such as dimethyl sulfoxide to get 3-cyano-5-methyl-hexanoic acid ethyl ester (XI), which was further converted to (f?S)-3-cyano-5-methyl-hexanoic acid (XII) through hydrolysis with lithium hydroxide.
Alternatively, compound (VII) was converted into compound (XVI) through base catalyzed hydrolysis and which was further decarboxylated to give (RS)-3-cyano-5-methyl-hexanoic acid (XII) in presence of mineral acid such as sulfuric add in organic solvent such as ethyl acetate.
Racemic 3-cyano-5-methyl-hexanoic acid (XII) was resolved through diastereomeric salt formation with cinchonidine (XIII) to obtain (S)-3-cyano-5-methyl-hexanoic acid (II). Compound (II) was converted to compound (I) through hydrogenation in presence of Raney Nickel.
Route II: Compound (IX) was prepared by condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid ethyl ester (VIII) in presence of cesium acetate, followed by hydrogenation using palladium on charcoal catalyst. Compound (IX) was further reacted with halo acetic acid ethyl ester (VI) in presence of cesium carbonate without any solvent or optionally in an organic solvent such as dimethyl sulfoxide to give diethyl 2-cyano-2- isobutylsuccinate (X). Compound (X) was then treated with cesium carbonate and thiophenol in organic solvent such as Λ/,/V-dimethylformamide to get 3-cyano-5-methyl- hexanoic acid ethyl ester (XI) or alternatively, compound (XI) was also obtained through
decarboxylation in organic solvent such as dimethylsulfoxide of diethyl 2-cyano-2- isobutylsuccinate (X) in presence of cesium chloride, which was further converted to 3- cyano-5-methyl-hexanoic acid (XII) through base catalyzed hydrolysis.
Alternatively, compound (X) was also converted into compound (XVI) through base catalyzed hydrolysis, which was further decarboxylated to give (RS)-3-cyano-5-methyl- hexanoic acid (XII) in presence of mineral acid and in organic solvent such as ethyl acetate.
(ftS)-3-cyano-5-methyl-hexanoic acid (XII) was resolved through diastereomeric salt formation with cinchonidine (XIII) to obtain (S) 3-cyano-5-methyl-hexanoic acid (II). Compound (II) was converted to compound (I) through hydrogenation in presence of Raney Nickel.
B) The processes for resolution of (RS)-3-cyano-5-methyl-hexanoic acid (XII) to obtain (S)-3-cyano-5-methyl-hexanoic acid (II) through diastereomeric salt formation with cinchonidine (XIII).
Resolution of racemic 3-cyano-5-methyl-hexanoic acid (XII) through diastereomeric salt formation with cinchonidine (XIII) is depicted in scheme B. (f?S)-3-cyano-5-methyl-hexanoic acid (XII) was refluxed with cinchonidine (XIII) in organic solvents such as ethyl acetate. During the reaction (S)-3-cyano-5-methyl-hexanoic acid salt of cinchonidine (XIV) precipitated out and (R)-3-cyano-5-methyl-hexanoic acid salt of cinchonidine (XV) remained soluble in ethyl acetate. (S)-3-cyano-5-methyl-hexanoic acid (II) was obtained from (S)-3- cyano-5-methyl-hexanoic acid salt of cinchonidine (XIV) via decomposition in biphasic mixture of aqueous mineral acid such as dilute aqueous hydrochloric acid and organic solvent such as ethyl acetate, dichloromethane, methyl tert-butyl ether; preferably ethyl acetate. Cinchonidine (XIII) from aqueous acid solution was also recovered through basification and reused to improve the over all atom economy and process efficiency.
The invention can be briefly described as follows:
First aspect of the invention is a process for synthesis of pregabalin (I)

from cyano acetic acid alkyl ester of formula (A)

(A)
Wherein,
R = CH3 : Compound III
R = C2H5 : Compound VIII
comprising,
a) condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid alkyl ester (A) in presence of organic or inorganic base such as piperidinium acetate, cesium acetate, and further hydrogenation using noble metal catalyst such as platinum oxide, palladium on carbon palladium hydroxide on carbon and also with Raney nickel in polar solvent such as methanol, ethanol, water, 1 ,4-dioxane, tetrahydrofuran, dimethoxy ethane and diglyme under hydrogen pressure of about 1 kg/cm2 to 5 kg/cm2 with subsequent isolation of the product 2-cyano-4-methyl-pentanoic acid alkyl ester (B) in solution form from the catalyst by filtration;

loride, potassium

chloride or sodium chloride in an organic solvent such as dimethyl sulfoxide at temperature of about 130°C to 180°C OR reaction of compound of formula (C) with cesium carbonate alongwith thiol at temperature of about 130 - 150°C to get (RS)-3- cyano-5-

d) hydrolysis of compound (XI) in presence of base such as lithium hydroxide, potassium hydroxide or sodium hydroxide, preferably with lithium hydroxide at temperature of °C to get (RS)-3-cyano-5-methyl-hexanoic acid (XII);


treatment of (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) with biphasic mixture of ethyl acetate: dilute hydrochloric acid (1 :1) at room temperature to obtain (S)-3-cyano-5-methylhexanoic acid (II) from ethyl acetate layer optionally accompanied with recovery of cinchonidine (XIII) from aqueous phase through basification with sodium hydroxide, potassium hydroxide;

(XIV)
g) hydrogenation of optically pure (S) - 3-cyano-5-methyl-hexa oic acid (II) in presence of Raney Nickel.

Another aspect of the invention is a process for synthesis of pregabalin (I)

from cyano acetic acid alkyl ester of formula (A)
O
O N
(A)
Wherein,
R = CH3 : Compound III
R = C2H5 : Compound VIII
comprising,
a) condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid alkyl ester (A) in presence of organic or inorganic base such as piperidinium acetate, cesium acetate, and further hydrogenation using noble metal catalyst such as platinum oxide, palladium on carbon, palladium hydroxide on carbon and also with Raney nickel in polar solvent such as
methanol, ethanol, water, 1 ,4-dioxane, tetrahydrofuran, dimethoxy ethane and diglyme under hydrogen pressure of about 1 kg/cm2 to 5 kg/cm2 with subsequent isolation of the product 2-cyano-4-methyl-pentanoic acid alkyl ester (B) in solution form from the catalyst by filtration;

b) reaction of compound of formula (B) with halo acetic acid ethyl ester (VI), wherein halo group includes chloro, bromo and iodo, in presence of base such as sodium carbonate, potassium carbonate, cesium carbonate, preferably cesium carbonate without solvent or in an organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4-dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N, /V-dimethyl formamide and dimethyl sulfoxide at temperature of about 10 to 90 °C to give 4-ethyl 1-alkyl-2-cyano-2- isobutylsuccinate (C);


x ' (XVI)
d) compound (XVI) was decarboxylated in presence of mineral acid such as sulfuric acid in organic solvent such as ethyl acetate to obtain compound (XII) at about temperature 70 to
80 °C.

(XVI) (XII)
e) treatment of compound (XII) with cinchonidine(XIII) in presence of organic solvent such as methanol, ethanol, 1 ,4-dioxane, ethyl acetate, tetrahydrofuran, 2-methyl tetrahydrofuran, dimethoxy ethane and diglyme at temperature of about 20°C to 80°C to precipitate out (S) 3- cyano-5-methylhexanoic acid salt of cinchonidine (XIV), followed by separation of compound (XIV) through known separation techniques such as filtration, centrifugation, sedimentation, followed by optional purification of (S)-3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) in ethyl acetate or through re-salt formation;


g) hydrogenation of optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) in presence of Raney Nickel.

such that at each step the intermediates were optionally isolated and purified with suitable processes. Yet another aspect of the invention is a process for synthesis of pregabalin (I)

from ( ?S)-3-cyano-5-methyl-hexanoic acid (XII);

(XII)
comprising;
a) treatment of compound (XII) with cinchonidine(XIII) in presence of organic solvent such as methanol, ethanol, 1 ,4-dioxane, ethyl acetate, tetrahydiOfuran, 2-methyl tetrahydrofuran, dimethoxy ethane and diglyme at temperature of about 20°C to 80°C to precipitate out (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV), followed by separation of compound (XIV) through known separation techniques such as filtration, centrifugation, sedimentation, followed by optional purification of (S)-3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) in ethyl acetate or through re-salt formation;

b) treatment of (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) with biphasic mixture of ethyl acetate: dilute hydrochloric acid (1 :1) at room temperature to obtain (S)-3- cyano-5-methylhexanoic acid (II) from ethyl acetate layer optionally accompanied with recovery of cinchonidine (XIII) from aqueous phase through basification with sodium hydroxide, potassium hydroxide;

(XIV)
c) hydrogenation of optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) in presence of Raney Nickel.

such that at each step the intermediates were optionally isolated and purified with suitable processes.
Detailed Description of the Invention;
This invention provides i) Resolution via diastereomeric salt formation between (RS) - 3-cyano-5- methyl-hexanoic acid (XII) and cinchonidine (XIII) to obtain optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) in excellent yield and high optical purity (>99 % ee) and further conversion to (S)-pregabalin (I) in high yield and high optically purity (>99 % ee). ϋ) Synthesis of the novel compound 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII) through a novel method as an intermediate for the title compound. iii) A novel method for decarboxylation of 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate [VII] and diethyl 2-cyano-2-isobutylsuccinate [X] in presence of thiol/cesium carbonate. iv) Green, eco-friendly, solvent free process for the synthesis of diethyl 2- cyano-2-isobutylsuccinate (X). v) Synthesis of the novel compound 2-cyano-2-isobutylsuccinic acid (XVI) through a novel method as an intermediate for the title compound.
vi) A novel method for decarboxylation of 2-cyano-2-ispbutylsuccinic acid
(XVI) in presence of mineral acid such as sulfuric acid and in organic solvent such as ethyl acetate.
vii) A method for recovery of cinchonidine (XIII) through basification and utilization of recovered cinchonidine (XIII) for resolution of (RS) - 3-cyano- 5-methyl-hexahoic acid (XII), thereby improving the process efficiency and hence cost.
A) Process for synthesis of (RS) 3-cyano-5-methylhexanoic acid of formula (XII) Route I:
1) A process for synthesis of (RS) 3-cyano-5-methylhexanoic acid of formula (XII)


a) condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid methyl ester (III) in presence of organic or inorganic base such as piperidinium acetate, cesium acetate, and further hydrogenation using noble metal catalyst such as platinum oxide, palladium on carbon, palladium hydroxide on carbon and also with Raney nickel preferably palladium on carbon and palladium hydroxide on carbon in polar solvent such as methanol, ethanol, water, 1 ,4-dioxane, tetrahydrofuran, dimethoxy ethane and diglyme, preferably methanol under hydrogen pressure of about 1 kg/cm2 to 5 kg/cm2, preferably about 2 kg/cm2, with subsequent isolation of the product in solution form from the catalyst by filtration;
0480
21


IB2011/000480
22


2) A process for synthesis of (RS) 3-cyano-5-methylhexanoic acid of formula (XII)


(III)
comprising,
a) compound (VII) was obtained as per the process described in step 'a' and 'b' in Part of route I.
b) hydrolysis of compound (VII) in presence of base such as lithium hydroxide, potassium hydroxide or sodium hydroxide, preferably with lithium hydroxide at temperature of about 20 to 80 °C, preferably at 65 to 70 °C to get 2-cyano-2- isobutylsuccinic acid (XVI)

c) compound (XVI) was decarboxylated in presence of mineral acid such as sulfuric acid in organic solvent such as ethyl acetate to obtain compound (XII) at about temperature 70 to 80 °C; or decarboxylated through reported methods such as base catalyzed decarboxylation (J.Org. Chem, 1961 , 83, 2354)

Route II:
1) A process for synthesis of {RS) 3-cyano-5-methylhexanoic acid of formula (XII)
 from cyano acetic acid ethyl ester of formula (VIII)
from cyano acetic acid ethyl ester of formula (VIII)

(VIII)
comprising,
a) condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid ethyl ester (VIII) in presence of organic or inorganic base such as piperidinium acetate, cesium acetate and further hydrogenation using noble metal catalyst such as platinum oxide, palladium on carbon, palladium hydroxide on carbon and also with Raney nickel preferably palladium on carbon and palladium hydroxide on carbon in polar solvent such as methanol, ethanol, 1 ,4- dioxane, tetrahydrofuran, dimethoxy ethane and diglyme, preferably methanol under hydrogen pressure of about 1 kg/cm2 to 5 kg/cm2, preferably about 2 kg/cm2, with subseq
b) react
 erein halo group include chloro, bromo and iodo, in presence of base such as potassium carbonate, sodium carbonate and cesium carbonate preferably cesium carbonate without solvent or optionally in a organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4- dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N, /V-dimethyl formamide and dimethyl sulfoxide, more preferably dimethyl sulfoxide at temperature of about 10 to 80 °C, preferably at 50 to 60 °C to give diethyl 2-cyano-2-isobutylsuccinate (X).
erein halo group include chloro, bromo and iodo, in presence of base such as potassium carbonate, sodium carbonate and cesium carbonate preferably cesium carbonate without solvent or optionally in a organic solvent selected from N, /V-dimethyl formamide, tetrahydrofuran, 1 ,4- dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N, /V-dimethyl formamide and dimethyl sulfoxide, more preferably dimethyl sulfoxide at temperature of about 10 to 80 °C, preferably at 50 to 60 °C to give diethyl 2-cyano-2-isobutylsuccinate (X).

(IX) (X)
It was observed that rate of reaction was faster in presence of cesium carbonate as compared to potassium carbonate and sodium carbonate and also it was worthwhile to note that reaction temperature with cesium carbonate was much lower as compared to potassium carbonate and sodium carbonate; details are summarized in Table 1. Thus, use of cesium carbonate makes the process more eco-friendly and increases the process efficiency; moreover reaction was carried out without using any organic solvent, which resulted in overall decrease in the process cost for the synthesis of title compound.
Table 1 : Effect of alkali metal carbonates on synthesis of compound [X]


Or
reaction of compound (X) with thiol/cesium carbonate in an organic solvent such as N,N- djmethylformamide at temperature of about 130°C to 150°C, preferably at 130°C to 140 °C to get 3-cyano-5-methyl-hexanoic acid ethyl ester (XI), thiol being obtained from thiophenol or diethyl amine ethane thiol; preferably thiophenol.

In literature, decarboxylation of activated esters in presence of thiol/cesium carbonate (J. Org. Chem. 1986, 51, 3165-31369) is reported for benzylic substances but there is no report of decarboxylation of compound (X) or similar substances in presence of thiolate/cesium carbonate.
The decarboxylation of compound (X) in presence of thiol/cesium carbonate was carried out at lower temperature as compared to decarboxylation in presence of alkali metal chloride/DMSO, which results in overall decrease in the energy consumption, hence reducing the overall process cost for synthesis of title compound.
It was also observed that decarboxylation of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) occurs at lower temperature as compared to decarboxylation of diethyl 2-cyano-2- isobutylsuccinate (X). Comparison of different methods for decarboxylation is given in Table
2. ■■
Table 2: Comparison of different methods for decarboxylation

2) A process for synthesis of (RS) 3-cyano-5-methylhexanoic acid of formula (XII)
 from cyano acetic acid ethyl ester of formula (VIII)
from cyano acetic acid ethyl ester of formula (VIII)
O
N
(VIII)
comprising,
a) compound (X) was obtained as per the process described in step a and b in Part 1 of route II.
b) hydrolysis of compound (X) in presence of base such as lithium hydroxide, potassium hydroxide or sodium hydroxide, preferably with lithium hydroxide at temperature of about 20 to 80 °C, preferably at 65 to 70 °C to get 2-cyano-2- isobutylsuccinic acid (XVI)


B) Resolution of (/?S)-3-cyano-5-methyl-hexanoic acid (XII) to obtain optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) via diastereomeric salt formation with cinchonidine (XIII).
WO2007/143152 A2, reports the optical resolution of (/?S)-3-cyano-5-methylhexanoic acid (XII) through diastereomeric salt formation with (S^-phenyl ethyl amine to obtain (S)-3 - cyano -5-methyl hexanoic acid (II) in acetone. Following the experimental conditions reported in the said patent, it has not been possible to obtain precipitate of the desired diastereomeric salt crystallizing out, and in spite of varying the experimental conditions such as solvent, reaction temperature and ratio between reactants, no precipitation of desired diastereomeric salt was obtained. Further other chiral amines have also been suggested in the said patent for resolution of (f?S)-3-cyano-5- methylhexanoic acid (XII) to obtain (S)-3-cyano-5-methylhexanoic acid (II) through diastereomeric salt formation but no enablement whatsoever has been reported i.e. one does not know how to perform these speculative experiments.
Hence there was need for identifying suitable resolving agents, which could give the excellent separation to obtain optically pure compound (II) in high % ee and yield. Further, the process must be easy to operate in industrial scale i.e. one could separate the desired diastereomeric salt through efficient crystallization in high optical purity and yield. Furthermore, to improve the process efficiency, the recovery of the resolving agent along with its reuse should be easy and efficient.
After a series of experimentation, it was observed that resolution of (f?S)-3-cyano-5- methylhexanoic acid (XII) via diastereomeric salt with cinchonidine (XIII) provides the desired separation in high optical purity and yield.
Thus, resolution through diastereomeric salt between (RS)-3-cyano-5-methylhexanoic acid (XII) with cinchonidine (XIII) comprised of the following steps: a) compound (XII) was treated with cinchonidine(XIII) in presence of organic solvent such as methanol, ethanol, 1 ,4-dioxane, ethyl acetate, tetrahydrofuran, 2-methyl tetrahydrofuran, dimethoxy ethane and diglyme, preferably ethyl acetate at temperature of about 20°C to 80°C, preferably at 70°C to 80 °C to precipitate out (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV), followed by separation of compound (XIV) through known separation techniques such as filtration, centrifugation, sedimentation, etc
b) (S)-3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) was further purified through reflux in ethyl acetate or through re-salt formation in order to dissolve occluded (f?)-3-cyano-5-methylhexanoic acid salt of cinchonidine (XV) thereby improving enantiomeric purity of (S)-3-cyano-5-methylhexanoic acid (II).
(S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) was characterized by the powder X-ray diffraction peaks at the 2-theta values 5.84, 7.27, 7.69, 10.72, 11.65, 13.79, 14.92, 15.39,15.73, 16.69, 17.31 , 17.41 , 17.58, 17.99, 19.48, 20.03,
20.71 , 21.18, 21.92, 23.18, 24.93, 25.29, 25.95, 26.38, 27.07, 27.91 , 28.79, 31.06, 31.65, 35.36, 38.00 and 39.35; specific optical rotation value -54.29 0 (c=1 in DMSO at 25 °C); DSC (differential scanning calorimetry) peak at 152.49 ° C (onset =149.86 ° C).
c) (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) was decomposed in a biphasic mixture of ethyl acetate: dilute hydrochloric acid (1 :1 ) at room temperature. (S)-3-cyano-5-methylhexanoic acid (II) was obtained from ethyl acetate layer and cinchonidine (XIII) was recovered from aqueous phase through basification with sodium hydroxide, potassium hydroxide and was reused for resolution.
d) Mother liquor of salt formation i.e. ethyl acetate layer, which contains the {R)-3- cyano-5-methylhexanoic acid salt of cinchonidine (XV) was treated with aqueous dilute mineral acid such as hydrochloric acid to recovered cinchonidihe.
e) Optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) was converted into S- pregabalin (I) by hydrogenation in presence of Raney Nickel.

Nomenclatures used for the compounds mentioned herein are as understood from the CambridgeSoft® ChemOffice software ChemDraw Ultra version 6.0.1.
Analytical Methods:
The enantiomeric excess (ee) for pregabalin is determined by HPLC using a Shimadzu LC 2010 system equipped with a chiral column (Purosphere star RP-18e (4.6 x 150mm), 5pm), column oven temperature 25 °C and UV visible detector (UV at 340nm). Mobile phase is buffer: acetonitrile (55:45) with flow rate 1.0 mL/min, injection volume 20 μΙ. The enantiomeric excess (ee) is determined by derivatized by reacting with Marfey's reagent.
The enantiomeric excess (ee) for (S) - 3-cyano-5-methyl-hexanoic acid ethyl ester is determined by Gas-Liquid chromatography using a Shimadzu GC 2010 system equipped with a chiral column (Chiraledex (20m x 0.25mm x 0.12mm)), and FID detector.
The enantiomeric excess (ee) for (S) or (R) - 3-cyano-5-methyl-hexanoic acid is determined via converting into corresponding ester and analyzed on Gas-Liquid chromatography using a Shimadzu GC 2010 system equipped with a chiral column (Chiraledex (20m x 0.25mm x 0.12mm)), and FID detector.
NMR spectra are obtained at 200 and 400 MHz Bruker instruments, with CDCI3 as solvent unless otherwise stated. Chemical shifts (<5) are given in ppm relative to tetramethylsilane (<5
= 0 ppm). IR spectra are recorded on Perkin Elmer Spectrum (Model: Spectrum 100) and absorption bands are given in cm"1. Mass analyses are performed on Shimadzu LCMS 201 OA instrument. Powder X-ray diffraction is recorded on PANalytical B. V. Netherlands model PN3040/60X'Part Pro. DSC is recorded on Perkin Elmer model Diamond DSC at the rate of 10 °C/min, and endothermic peak is recorded in °C.
Brief Description of Accompanying Drawings
Figure 1: PXRD of (S)-3-cyano-5-methyl hexanoic acid salt of cinchonidine (XIV)
Figure 2: DSC of (S)-3-cyano-5-methyl hexanoic acid salt of cinchonidine (XIV)
· .
Example 1 : Synthesis of ethyl 2-cyano-4-methylpentanoate (IX) from condensation of ethyl cyano acetate (VIII) with /so-butyraldehdye (IV) in presence of piperidine / acetic

Ethyl cyano acetate (VIII) (56.5 g, 0.5 mol) was dissolved in methanol (100 mL) and iso- butyraldehyde (IV) (43.2 g, 0.6 mol) was added to it at room temperature. The mixture was cooled to 4 °C and a solution of acetic acid (12 mL) and piperidine (2 mL) in 50 mL of methnaol was added slowly over a period of 20 min by maintaining temperature below 20 °C. The reaction mixture was transferred into a Parr autoclave reactor followed by addition of 2 % catalyst palladium on carbon (50 % wet (10% Pd loading)). Reactor was purged with hydrogen gas two times and charged with hydrogen, 3 kg/cm2 pressure was maintained in the Parr autoclave until hydrogen consumption ceases. Reaction was monitored by TLC. After completion of reaction, reaction mixture was filtered through Celite bed to remove Pd/C and filtrate was concentrated under reduced pressure to remove solvent. Residue was suspended in 100 mL water. Organic layer was separated to obtain ethyl 2-cyano-4-methylpentanoate (IX) (80 g, 95 % yield) as light yellow oil.
P T/IB2011/000480
33
FTIR (neat): 2962, 2249, 1746, 1469, 1186 cm 1.
1H NMR (CDCIj, 200 MHz): δ 0.95 (d, 3H), 0.96 (d, 3H), 1.28 (t, 3H), 1.17-1.87 (m, 3H), 3.49 (q, 1H), 4.22 (q, 2H).
MS (El): C9H15N02: 169.0; [M+H20] +: 186.85 and [M] : 167.80
Example 2: Synthesis of ethyl 2-cyano-4-methylpentanoate (IX) from condensation of ethyl cyano acetate (VIII) with so-butyraldehdye (IV) in presence of cesium acetate /methanol
eact
acid

Example 3: Synthesis of ethyl 2-cyano-4-methylpentanoate (IX) from condensation of ethyl cyano acetate (VIII) with /so-butyraldehdye (IV) in presence of cesium acetate /water
eacti
acid
 P T/IB2011/000480
P T/IB2011/000480
34 -4-

A reactor was charged with ethyl 2-cyano-4-methylpentanoate (IX) (103.0 g, 609 mmol), ethyl chloro acetate (VI) ( (82.1 , 670 mmol) and benzyl triethyl ammonium chloride (1.4 g, 6,09 mmol) and resulting reaction mixture was stirred for 15-20 min at room temperature. To above reaction mixture activated fine powder of cesium carbonate (198.0 g, 609 mmol) was added slowly in small portions while stirring over a period of 10-15 min, addition of cesium carbonate result into rise in the reaction temperature upto 60 to 65 °C. After complete addition of cesium carbonate, reaction mixture was stirred further for 1 h at 60 °C. Reaction was monitored by TLG for complete consumption of starting materials and after completion of reaction; it was quenched by adding 100 mL water and organic layer was separated to obtain diethyl 2-cyano-2- isobutylsuccinate (X) (155.0 g, 88% yield) as yellow oil.
FTIR (neat): 2963, 2248, 1743, 1469, 1195, 1025 cm-1.
1H NMR (CDCI3, 200 MHz): 50.95 (d, 3H), 0.96 (d, 3H), 1.23 (t, 3H), 1.28 (t, 3H), 1.70-1.89 (m, 3H), 2.80 (d, 1H), 3.02 (d, 1 H), 4.16 (q, 2H), 4.28 (q, 2H),
MS (El): CgH15N02: 255; [M+H20] +: 273.05.
Example 5: Synthesis of diethyl 2-cyano-2-isobutylsuccinate (X) from ethyl 2-cyano-4- methylpentanoate (IX) and ethyl chloro acetate (VI) in presence of potassium carbonate.

Reaction was carried out as per the procedure described in example 4 with activated fine powder of potassium carbonate at temperature 90 °C for 120 min to obtain diethyl 2-cyano-2- isobutylsuccinate (X) as dark brown oil. Example 6: Synthesis of diethyl 2-cyano-2-isobutylsuccinate (X) from ethyl 2-cyano-4- methylpentanoate (IX) and ethyl chloro acetate (VI) in presence of sodium carbonate.

Reaction was carried out as per the procedure described in example 4 with activated fine powder of sodium carbonate at temperature 90 °C for 180 min to obtain diethyl 2-cyano-2- isobutylsuccinate (X).
Example 7: Synthesis of methyl-2-cyano-4-methylpentanoate (V) from condensation of methyl cyano acetate (III) with /'so-butyraldehdye (IV) in presence of piperidine / acetic acid .

Methyl cyano acetate (III) (113.0 g, 1.14mol) was dissolved in methanol (125 mL), iso- butyraldehyde (IV) (98.0 g, 1.36 mol) and glacial acetic acid (12 mL) was added to it at room temperature. The mixture was cooled to 4 °C and a solution of acetic acid (12 mL) and piperidine (4 mL) in 50 mL of methanol was added slowly over a period of 20 min by maintaining temperature below 20 °C. The reaction mixture was transferred into a Parr autoclave reactor followed by addition of 2 % catalyst palladium on carbon (50 % wet (10% Pd loading)). Reactor was purged with hydrogen gas two times and charged with hydrogen, 3 kg/cm2 pressure was maintained in the Parr autoclave until hydrogen consumption ceases. Reaction was monitored by TLC. After completion of reaction, reaction mixture was filtered through Celite bed to remove Pd/C and filtrate was concentrated under reduced pressure to remove solvent. Residue was suspended in 100 mL water. Organic layer was separated to obtain methyl-2-cyano-4- methylpentanoate (V) (170 g, 90 % yield) as light yellow oil.
FTIR (neat): 2958, 2872, 2642, 2250, 1751 , 1468, 1185, 1131 , 1010 cm 1
1H NMR (CDCI3, 200 MHz): δ 0.92 (d, 3H), 0.99 (d, 3H), 1.74-1.98 (m, 3H), 3.53 (t, 1H) 3.81 (s, 3H)
MS (El): C9H15N02: 169.11 ; [M+H] +: 170.15
Example 8: Synthesis of methyl-2-cyano-4-methylpentanoate (V) from condensation of methyl cyano acetate (III) with /'so-butyraldehdye (IV) in presence of cesium acetate in methanol

ion was carried out as per process described in example 7 by replacing piperidine/ acetic acid with cesium acetate to obtain methyl-2-cyano-4-methylpentanoate (V.
Example 9: Synthesis of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) from methyl-2 cyano-4-methylpentanoate (V) and ethyl chloro acetate (VI) in presence of cesium carbonate.

A reactor was charged with methyl 2-cyano-4-methylpentanoate (V) (41.0 g, 265.0 mmol), ethyl chloro acetate (VI) (35.7, 291 mmol) and benzyl triethyl ammonium chloride (0.6 g) and resulting reaction mixture was stirred for 15-20 min at room temperature. To above reaction mixture activated fine powder of cesium carbonate (47.3 g, 145,5 mmol) was added slowly in small portions whil stirring over a period of 10-15 min, addition of cesium carbonate result into rise in the reaction temperature upto 65 to 70 °C. After complete addition of cesium carbonate, reaction mixture was stirred further for 1 h at 60 °C. Reaction was monitored by TLC for complete consumption of starting materials and after completion of reaction; it was quenched by adding
100 mL water and organic layer was separated to obtain 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII) (57.5 g, 90% yeild) as light yellow oil.
FTIR (neat): 2958, 2248, 1741 , 1637, 1467, 1199, 1025 cm"1.
1H NMR (CDCI3, 200 MHz): 50.88 (d, 3H), 0.92 (d, 3H), 1.05 (t, 3H), 1.70-1.89 (m, 3H), 2.79 (d, 1 H), 3.03 (d, 1 H), 3.84 (s, 3H), 4.18 (q, 2H).
MS (El): C9H15N02: 241 ; [M+H20] +: 259.05.
Example 10: Synthesis of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) from methyl- 2-cyano-4-methylpentanoate (V) and ethyl chloro acetate (VI) in presence of potassium carbonate.
Reaction was carried out as per the procedure described in example 9 with activated fine powder of potassium carbonate at temperature 90 °C to obtain 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII) as light brown oil.
Example 11 : Synthesis of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) from methyl- 2-cyano-4-methylpentanoate (V) and ethyl chloro acetate (VI) in presence of sodium carbonate.
Reaction was carried out as per the procedure described in example 9 with activated fine powder of sodium carbonate at temperature 90 °C to obtain 4-ethyl 1 -methyl 2-cyano-2- isobutylsuccinate (VII) as light brown oil.
Example 2: Decarboxylation of diethyl 2-cyano-2-isobutylsuccinate (X) to obtain (RS) - 3- cyano-5-methylhexanoic acid ethyl ester (XI) in presence of KCI/DMSO

FTIR (neat): 2961 , 2242, 1738, 1469, 1182, 1023 cm"1.
1H NMR (CDCI3) 200 MHz): δ 0.95 (d, 3H), 0.96 (d, 3H), 1.22-1.24 (m, 4H), 1.58 (m, 1 H), 1.83 (m, 1 H), 2.49 (dd, 1 H), 2.65 (dd, 1 H), 2.98-3.06 (m, 1 H), 4.17 (q, 2H). 13C NMR (CDCI3) 50 MHz): 14.1 , 21.2, 22.8, 25.8, 26.0, 37.1 , 40.7, 61.4, 121.1 , 169.7.
MS (El): C,oH17N02: 183; [M+H20] +: 201.05. Example 13: Decarboxylation of diethyl 2-cyano-2-isobutylsuccinate (X) to obtain (RS) - 3- cyano-5-methylhexanoic acid ethyl ester (XI) in presence of CsCI/DMSO
Reaction was carried out as per procedure described in example 12 by replacing potassium chloride with cesium chloride to obtain (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI). Example 14: Decarboxylation of diethyl 2-cyano-2-isobutylsuccinate (X) to obtain (RS) - 3- cyano-5-methylhexanoic acid ethyl ester (XI) in presence of NaCI/DMSO
Reaction was carried out as per procedure described in example 12 by replacing sodium chloride with cesium chloride to obtain (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI). Example 15: Decarboxyiation of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) to obtain (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of KCI/DMSO

FTIR (neat): 2961 , 2242, 1738, 1469, 1182, 1023 cm 1.
*H NMR (CDCI3, 200 MHz): δ 0.95 (d, 3H), 0.96 (d, 3H), 1.22-1.24 (m, 4H), 1.58 (m, 1 H), 1.83 (m, 1 H), 2.49 (dd, 1 H), 2.65 (dd, 1 H), 2.98-3.06 (m, 1 H), 4.17 (q, 2H). 13C NMR (CDCI3, 50 MHz): 14.1 , 21.2, 22.8, 25.8, 26.0, 37.1 , 40.7, 61.4, 121.1 , 169.7.
MS (El): C10H17NO2: 183; [M+H20] +: 201.05. Example 16: Decarboxylation of 4-ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) to obtain (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of CsCI/DMSO
Reaction was carried out as per procedure described in example 15 with cesium chloride instead of potassium chloride to obtain (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI).
Example 17: Decarboxylation of 4-ethyl -methyl 2-cyano-2-isobutylsuccinate (VII) to obtain (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of NaCI/DMSO
Reaction was carried out as per procedure described in example 15 with sodium chloride instead of potassium chloride to obtain {RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI). Example 18: Decarboxylation of diethyl 2-cyano-2-isobutylsuccinate (X) to obtain {RS) - 3- cyano-5-methylhexanoic acid ethyl ester (XI) in presence of thiophenol/cesium carbonate in dimethylformamide.

A 250 mL reactor was charged with diethyl 2-cyano-2-isobutylsuccinate (13.1 g, 51.3 mmol), thiophenol (8.47, 77.0mmol), cesium carbonate (5.0 g, 15.4 mmol) and A/,A/-dimethylformamide (40 mL). The resulting reaction mixture was heated at 130 °C and maintained at that temperature for 4 h. Reaction was monitored by GC for conversion of (RS) - 3-cyano-5- methylhexanoic acid ethyl ester (XI) .
Example 19: Decarboxylation of diethyl-2-cyano-2-isobutylsuccinate (X) to obtain {RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of diethyl amine ethane thiol/cesium carbonate
Rea
thiol

Example 20: One pot synthesis of (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) from methyl-2-cyano-4-methylpentanoate (V)

A reactor was charged with methyl-2-cyano-4-methylpentanoate (V) (41.0 g, 265.0 mmol), ethyl chloro acetate (VI) (35.7 g, 291 mmol), benzyl triethyl ammonium chloride (0.6 g, 2.64 mmol) and dimethyl sulfoxide (63 mL). The resulting reaction mixture was stirred for 15 -20 min at room temperature. Activated powder of cesium carbonate was added slowly in small portions to the above reaction mixture while stirring. Addition of cesium carbonate result into increase in the reaction temperature upto 60 to 65 °C. The reaction mixture was stirred further for 2 h at 60 °C. Reaction was monitored by TLC for complete consumption of starting materials. After which reaction mixture was neated to 135- 140 °C and stirred further at that temperature for 4 h. The reaction mixture was cooled to 40 to 50 °C and treated with methyl terf-butyl ether (200 mL). The mixture was further cooled to 0 to 5°C and treated with water (1 L) in small portions to maintain the temperature below 40 °C. After stirring for 30 min the phases were separated. The aqueous phase was extracted with methyl tert-butyl ether (3 x 800 mL); Organic phases were combined and washed twice with 100 mL water. The organic layer was decolorized by treating with 7.0 g of activated charcoal. The resultant mixture was filtered to remove charcoal and filtrate was evaporated to give (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) (39.1 g) as light brown color oil.
Example 21 : Synthesis of (RS) - 3-cyano-5-methylhexanoic acid (XII) through hydrolysis of (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of lithium hydroxide.

A reactor was charged with (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) (52.0 g,) and water (250 mL). A solution of lithium hydroxide (15.0 gm) in water (25 mL) was added slowly while stirring. The reaction mixture was stirred further for 12 h at 60 °G. After which reaction mixture was cooled to room temperature and un-reacted (RS) - 3-cyano-5-methylhexanoic acid ethyl ester, if any was extracted with di-/'so propyl ether. Aqueous layer was acidified with dilute hydrochloric acid upto pH 2 and extracted with dichloromethane (3 *150 mL). Combined organic layer was dried over sodium sulfate and solvent was evaporated under reduced pressure to obtain (RS) - 3-cyano-5-methylhexanoic acid (XII) (31.1 gm) as yellow oil.
■ ' · - -
FTIR (neat): 31 18, 2961 , 2935, 2875, 2642, 2244, 1715, 1470, 1174, 1113 cm'1
1H NMR (CDCI3, 200 MHz): δ 0.95 (d, 3H), 0.96 (d, 3H), 1.36-1.38 (d, 1 H), 1.59-1.66 (m, 1 H), 1.79-1.85 (m, 1 H), 2.59-2.61 (dd, 1 H), 2.69-2.75 (dd, 1 H), 2.98-3.04 (m, 1 H).
MS (El): C8H13N02: 155.19; [M-H] ": 154.00; [M+H] +: 156.15
Example 22: Synthesis of {RS) - 3-cyano-5-methylhexanoic acid (XII) through hydrolysis of ( ?S) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of NaOH
Reaction was carried our as per process given in example 21 by replacing lithium hydroxide with sodium hydroxide.
Example 23: Synthesis of {RS) - 3-cyano-5-methylhexanoic acid (XII) through hydrolysis of (RS) - 3-cyano-5-methylhexanoic acid ethyl ester (XI) in presence of KOH
Reaction was carried our as per process given in example 21 by replacing lithium hydroxide with Potassium hydroxide.
Example 24: Synthesis of 2-cyano-2-isobutylsuccinic acid (XVI) through hydrolysis

A reactor was charged with diethyl 2-cyano-2-isobutylsuccinate (X) (200.0 g,) and water (300 mL). A solution of lithium hydroxide (72.3 gm) in water (350 mL) was added slowly while stirring. The reaction mixture was stirred further for 12 h at 70 °C. After which reaction mixture was cooled to room temperature and un-reacted diethyl 2-cyano-2-isobutylsuccinate, if any was extracted with di-/so propyl ether. Aqueous layer was acidified with dilute hydrochloric acid upto pH 2 and extracted with ethyl acetate (3 *350 mL). Combined organic layer was dried over sodium sulfate and solvent was evaporated under reduced pressure to obtain, 2-cyano-2- isobutylsuccinic acid (XVI) (140.1 gm) as off white solid.
FTIR (KBr): cm"1 3429, 2962, 2271 , 1742, 1703, 1469, 1438, 1402, 1214, 910, 844 and 642 1H NMR (DMSO-de, 200 MHz): δ 0.82-0.89 (d, 3H), 0.96-0.97 (d, 3H), 1.64-1.75 (m, 3H), 2.86 (s, 2H), 13.15 (s, 2H); 13C NMR (DMSO-d6, 50 MHz): 23.4, 23.6, 25.6, 41.7, 45.1, 120.6, 170.4, 171.1
MS (El): C9H13N04: 199.2; [M-H]": 197.80; [M+HzO] +: 216.90
Example 25: Synthesis of 2-cyano-2-isobutylsuccinic acid (XVI) through hydrolysis of diethyl 2-cyano-2-isobutylsuccinate (X) in presence of KOH
Reaction was carried our as per process given in example 24 by replacing lithium hydroxide with Potassium hydroxide to obtain 2-cyano-2-isobutylsuccinic acid (XVI).
Example 26: Synthesis of 2-cyano-2-isobutylsuccinic acid (XVI) through hydrolysis of diethyl 2-cyano-2-isobutylsuccinate (X) in presence of NaOH
Reaction was carried our as per process given in example 24 by replacing lithium hydroxide with Potassium hydroxide to obtain 2-cyano-2-isobutylsuccinic acid (XVI). Example 27: Synthesis of 2-cyano-2-isobutylsuccinic acid (XVI) through hydrolysis of 4- ethyl 1-methyl 2-cyano-2-isobutylsuccinate (VII) in presence of Lithium hydroxide.

A reactor was charged with 4-ethyl 1-methyl 2-cyano-2-isobutylsuccinate (VII) (100.0 g,) and water (150 mL). A solution of lithium hydroxide (36.5 gm) in water (150 mL) was added slowly while stirring. The reaction mixture was stirred further for 12 h at 70 °C. After which reaction mixture was cooled to room temperature and un-reacted diethyl 2-cyano-2-isobutylsuccinate, if any was extracted with di-/so propyl ether. Aqueous layer was acidified with dilute hydrochloric acid upto pH 2 and extracted with ethyl acetate (3 *350 mL). Combined organic layer was dried over sodium sulfate and solvent was evaporated under reduced pressure to obtain 2-cyano-2- isobutylsuccinic acid (XVI) (72.1 gm) as off white solid.
FTIR (KBr): cm" 3429, 2962, 2271, 1742, 1703, 1469, 1438, 1402, 1214, 910, 844 and 642 1H MR (DMSO-d6> 200 MHz): δ 0.82-0.89 (d, 3H), 0.96-0.97 (d, 3H), 1.64-1.75 (m, 3H), 2.86 (s, 2H), 13.15 (s, 2H); 13C NMR (DMSO-d6, 50 MHz): 23.4, 23.6, 25.6, 41.7, 45.1, 120.6, 170.4,171.1
MS (El): C9H13N04: 199.2; [M-H] ": 197.80; [M+H20] +: 216.90
Example 28: Synthesis of 2-cyano-2-isobutylsuccinic acid (XVI) through hydrolysis of 4- ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) in presence of potassium hydroxide
Reaction was carried our as per process given in example 27 by replacing lithium hydroxide with Potassium hydroxide to obtain 2-cyano-2-isobutylsuccinic acid (XVI) Example 29: Synthesis of 2-cyano-2-isobutylsuccinic acid (XVI) through hydrolysis of 4- ethyl 1 -methyl 2-cyano-2-isobutylsuccinate (VII) in presence of sodium hydroxide
Reaction was carried our as per process given in example 27 by replacing lithium hydroxide withsodium hydroxide to obtain 2-cyano-2-isobutylsuccinic acid (XVI) Example 30: Synthesis of (RS) - 3-cyano-5-methylhexanoic acid (XII) through decarboxylation of 2-cyano-2-isobutylsuccinic acid (XVI)

Example 31 : Resolution of (RS) - 3-cyano-5-methylhexanoic acid (XII) through diastereomeric salt formation with cinchonidine (1 : 0.5).

(Precipitate)
A reactor was charged with cinehonidine (XIII) (18.95, 64.5 mmol) and ethyl acetate (500 ml) and resulting reaction mixture was heating to 70 °C. A solution of (RS) - 3-cyano-5- methylhexanoic acid (XII) (20.0 g, 129.0 mmol) in ethyl acetate (200 mL) was added into above reaction mixture over a period of 15- 20 min and reaction mixture was further stirred for 5 h at reflux temperature. After which reaction mixture was cooled to room temperature and stirred further for 12 h. (S)-3-cynao-5-methylhexanoic acid salt of cinehonidine precipitate out. The resultant mixture was filtered to give (S) - 3-cyano-5-methylhexanoic acid salt of cinehonidine as a white solid (23.0 g, 95% ee for (S)-3-cynao 5-methylhexanoic acid as GC analysis).
Example 32: Resolution (RS) - 3-cyano-5-methylhexanoic acid (XII) through diastereomeric salt formation with cinehonidine (XIII) (1 : 1 mol ratio) in ethyl acetate

(Precipitate)
A reactor was charged with cinchonidine (XIII) (37.9, 129 mmol) and ethyl acetate (500 ml) and resulting reaction mixture was heating to 70 °C. A solution of (RS) - 3-cyano-5-methylhexanoic acid (XII) (20.0 g, 129.0 mmol) in ethyl acetate (200 mL) was added into above reaction mixture over a period of 15- 20 min and reaction mixture was further stirred for 5 h at reflux temperature. After which reaction mixture was cooled to room temperature and stirred further for 12 h. (S)-3- cynao-5-methylhexanoic acid salt of cinchonidine precipitated out. The resultant mixture was filtered to give (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) as a white solid (28.3 g, 97% ee for (S)-3-cynao 5-methylhexanoic acid ethyl ester by GC area %). Example 33: Resolution (RS) - 3-cyano-5-methylhexanoic acid (XII) through diastereomeric salt formation with cinchonidine (XIII) (1 : 1 mol ratio) in 2-methyl tetrahydrofuran

(Precipitate)
A reactor was charged with cinchonidine (XIII) (11.2 g) and 2-methyl tetrahydrofuran (196 ml) and resulting reaction mixture was heating to 70 °C. A solution of (RS) - 3-cyano-5- methylhexanoic acid (XII) (5.9 g,) in 2-methyl tetrahydrofuran (45 mL) was added into above reaction mixture over a period of 15- 20 min and reaction mixture was further stirred: for 1 h at reflux temperature. After which reaction mixture was cooled to 0°C and stirred further for 2 h. (S)-3-cynao-5-methylhexanoic acid salt of cinchonidine precipitated out. The resultant mixture was filtered to give (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) as a white solid (7.1 g, 93% ee for (S)-3-cynao 5-methylhexanoic acid ethyl ester by GC area %). Example 34: Resolution (RS) - 3-cyano-5-methylhexanoic acid (XII) through diastereomeric salt formation with cinchonidine (XIII) (1 : 1 mol ratio) in dimethoxy ethane
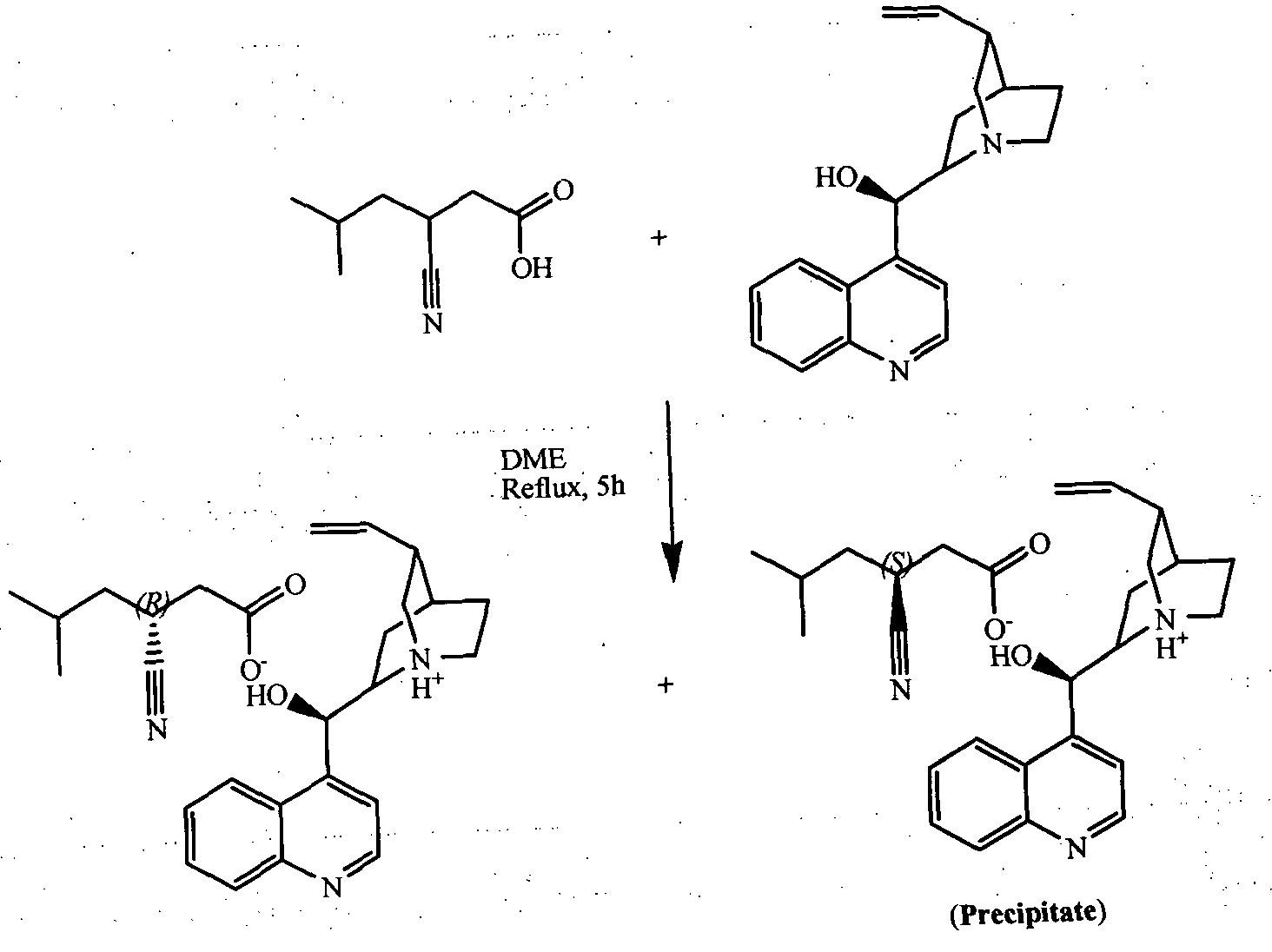
A reactor was charged with cinchonidine (XIII) (11.2 g) and dimethoxy ethane (196 ml) and resulting reaction mixture was heating to 70 °C. A solution of (RS) - 3-cyano-5-methylhexanoic acid (XII) (5.9 g) in dimethoxy ethane (45 mL) was added into above reaction mixture over a period of 15- 20 min and reaction mixture was further stirred for 2 h at reflux temperature. After which reaction mixture was cooled to room temperature and stirred further for 5 h. (S)-3-cynao- 5-methylhexanoic acid salt of cinchonidine precipitated out. The resultant mixture was filtered to give (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) as a white solid (28.3 g, 92% ee for (S)-3-cynao 5-methylhexanoic acid ethyl ester by GC area %). Example 35: Purification (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine (through ref I uxing in ethyl acetate).
A reactor was charged with (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine as a white solid (28.3 g, 97% ee for (S)-3-cynao 5-methylhexanoic acid) and ethyl acetate (200 ml) and resulting reaction mixture was heating for 5 h at reflux temperature. After which reaction mixture was cooled to room temperature and stirred further for 12 h. (S)-3-cynao-5-methylhexanoic acid salt of cinchonidine precipitate out. The resultant mixture was filtered to give (S) - 3-cyano-5- methylhexanoic acid salt of cinchonidine as a white solid (24.5 g, 98.5% ee for (S)-3-cynao 5- methylhexanoic acid ethyl ester by GC area %).
Example 36: Purification (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine (resalt formation)
A reactor was charged (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine, (23.0 g) dichlormethane (100 mL) and aqueous dilute hydrochloric acid ( OOmL). Resulting heterogeneous mixture was stirred for 1 hr. Organic layer was separated and aqueous layer was washed with dichloromethane. Combined organic layer was dried over sodium sulfate and solvent was evaporated under reduced pressure to (S) - 3-cyano-5-methylhexanoic acid (7.5 g). Cinchonidine was recovered from aqueous layer through basification (14.5gm).
A reactor was charged with recovered cinchonidine (14.01) and ethyl acetate (200 ml) and resulting reaction mixture was heating to 70 °C. A solution of enantiomeric excess (S) - 3-cyano- 5-methylhexanoic acid (7.5 g,) in ethyl acetate (50 mL) was added into above reaction mixture over a period of 15- 20 min and reaction mixture was further stirred for 5 h at reflux temperature. After which reaction mixture was cooled to room temperature and stirred further for 12 h. (S)-3- cynao-5-methylhexanoic acid salt of cinchonidine precipitate out. The resultant mixture was filtered to give (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine as a white solid (20.3 g, 99.82 % ee for (S)-3-cynao 5-methylhexanoic acid ethyl ester by GC area %).
FTIR (KBr): 3413, 3071 , 2955, 2234, 1639, 1595, 1508, 1394, 1102, 915, 785, 759, 619 cm"1. H NMR (DMSO-D6, 200 MHz): δ 0.89 (d, 3H), 0.91 (d, 3H), 1.30-1.45 (m, 1 H), 1.52-1.59 (m, 3H), 1.69-1.82 (m, 4H), 2.38 (bs, 1 H), 2.43-2.55 (dd, 3H), 2.60-2.68 (m, 2H), 2.97-3.07 (m, 2H), 3.25 (s, 1 H), 3.49 (s, 1 H), 4.93 (d, 1 H), 4.99 (d, 1 H), 5.64 (d, 1 H), 5.78-5.87 (m, 1H), 7.60-7.64 (m, 2H), 7.75 (t, 1 H), 8.04 (d, 1 H), 8.36 (d, 1 H), 8.86 (d, 1 H). 13C NMR (DMSO-D6, 50 MHz): 21.6, 22.1 , 23.2, 26.2, 26.3, 26.4, 27.6, 38.3, 38.9, 42.3, 55.1 , 60.5, 69.4, 115.3, 119.4, 123.0, 124.4, 125.9, 126.9, 129.3, 130.1 , 141.6, 148.2, 149.6, 150.5, 172.9.
Powder x*ray diffraction pattern PXRD [2Θ] (Cu Kal = 1.54060 A, Kct2 = 1.54443 A, Κβ = 1.39225 A; 40 mA, 45 kV): 5.84, 7.27, 7.69, 10.72, 11.65, 13.79, 14.92, 15.39,15.73, 16.69, 17.31 , 17.41 , 17.58, 17.99, 19.48, 20.03, 20.71 , 21.18, 21.92, 23.18, 24.93, 25.29, 25.95, 26.38, 27.07, 27.91 , 28.79, 31.06, 31.65, 35.36, 38.00 and 39.35
DSC Value: Peak = 152.49 °C Onset = 149.86°C.
Example 37: Decomposition of (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine to obtain (S) - 3-cyano-5-methylhexanoic acid
A reactor was charged with (S) - 3-cyano-5-methylhexanoic acid salt of cinchonidine, (20.0g) dichlormethane (100 mL) and aqueous dilute hydrochloric acid (lOOmL). Resulting heterogeneous mixture was stirred for 1 hr. Organic layer was separated and aqueous layer was washed with dichloromethane. Combined organic layer was dried over sodium sulfate and solvent was evaporated under reduced pressure to give (S) - 3-cyano-5-methylhexanoic acid (7.5 g).
Example 38: Recovery of Cinchonidine through basification
Cinchonidine (14.5gm) was obtained through basification of aqueous layer of example 28 which contain the hydrochloric acid salt of cinhonidine.
Example 39: Synthesis of (S)-Pregabalin from (S)-3-cyano-5-methyl hexanoic acid.
A solution of (S)-3-cyano-5-methyl hexanoic acid (II) (20.0 g, 0.13 mol) in methanol: water (50:50) (100 cm3) was added into a solution of potassium carbonate (9.8 g, 0.071 mol) in water (20 cm3) at 25 °C and was stirred at room temperature for 2 h. The mixture was then transferred into a Parr autoclave reactor and carefully raney nickel (10.0 g) was added. Reactor was purged with hydrogen gas twice and then 10 atm. hydrogen pressure was maintained for 24 h. The reaction mixture was filtered through a Celite pad and solvent from filtrate was evaporated under reduced pressure to leave a semi-solid material, which was re- crystallized from /so-propyl alcohol: water mixture (94:06, 25 cm3) to obtain (S)-pregabalin (10.0 g, 48 % and 99 % ee as per chiral HPLC analysis), as a white solid.
FTIR (KBr): 3400, 2956, 1645, 1551 , 1489, 1278 cm"1
'H NMR (CD3OD, 200 MHz): 0.91-0.96 (m, 6H), 1.22-1.23 (q, 2H), 1.64-1.74 (q, 1 H), 2.20-2-48 (m, 3H), 2.79-3.00 (m, 2H).
13C NMR (CDCI3> 50 MHz): 21.4, 21.9, 24.3, 31.6, 40.5, 40.6, 43.5, 181,1.
MS (El): C8 H17N02: 159.13; [M+H] += 159.96.
Example 40: Synthesis of (S)-Pregabalin from (S)-3-cyano-5-methyl hexanoic acid and isolation by using dimethoxy ethane.
A solution of (S)-3-cyano-5-methyl hexanoic acid (II) (20.0 g, 0.13 mol) in methanol: water (70:30) (100 cm3) was added into a solution of potassium carbonate (7.2 g, 0.13 mol) in water (20 cm3) at 25 °C and was stirred at room temperature for 2 h. The mixture was then transferred into a Parr autoclave reactor and carefully raney nickel (10.0 g) was added. Reactor was purged with hydrogen gas twice and then 0 atm. hydrogen pressure was maintained for 24 h. The reaction mixture was filtered through a Celite pad and resulting filtrate was acidified with acetic acid (8.52 mL, 0.013 mol) and stirred for 10 min. solvent from resulting reaction mixture was evaporated under reduced pressure to leave a semisolid material, which further suspended in dimethoxy ethane (100 mL) and stirred for 2 h at 70 °C. After which reaction mixture was allow to cool at 5 °C and stirred for 5 h at 5 °C to obtain (S)-pregabalin (12.0 g, 60 % and 99.81 % ee as per chiral HPLC analysis), as a white solid.
Claims
1) A process for synthesis of pregabalin (I)

from cyano acetic acid alkyl ester of formula (A) 
(A)
Wherein,
R = CH3 : Compound III
R = C2H5 : Compound VIII
comprising,
a) condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid alkyl ester (A) in presence of organic or inorganic base such as piperidinium acetate, cesium acetate, and further hydrogenation using noble metal catalyst such as platinum oxide, palladium on carbon palladium hydroxide on carbon and also with Raney nickel in polar solvent such as methanol, ethanol, water, 1 ,4-dioxane, tetrahydrofuran, dimethoxy ethane and diglyme under hydrogen pressure of about 1 kg/cm2 to 5 kg/cm2 with subsequent isolation of the product 2-cyano-4-methyl-pentanoic acid alkyl ester (B) in solution form from the catalyst by filtration;
55
b) reaction of compound of formula (B) with halo acetic acid ethyl ester (VI), wherein halo group includes chloro, bromo and iodo, in presence of base such as sodium carbonate, potassium carbonate, cesium carbonate, preferably cesium carbonate without solvent or in an organic solvent selected from N, N-dimethyl formamide, tetrahydrofuran, 1 ,4-dioxane, dimethyl sulfoxide, and dimethoxy ethane, preferably N,r /V-dimethyl formamide and dimethyl sulfoxide at
isobutylsuccin
c) reaction of

chloride or sodium chloride in an organic solvent such as dimethyl sulfoxide at temperature of about 130°C to 180°C OR reaction of compound of formula (C) with cesium carbonate alongwith thiol at temperature of about 130 - 150°C to get (RS)-3-cyano-5-methyl-hexanoic acid ethyl ester (XI);

d) hydrolysis of compound (XI) in presence of base such as lithium hydroxide, potassium hydroxide or sodium hydroxide, preferably with lithium hydroxide at temperature of about 20 to 80 °C to get (RS)-3-cyano-5-methyl-hexanoic acid (XII);


mixture of ethyl acetate: dilute hydrochloric acid (1 :1) at room temperature to obtain (S)-3- cyano-5-methylhexanoic acid (II) from ethyl acetate layer optionally accompanied with recovery of cinchonidine (XIII) from aqueous phase through basification with sodium hydroxide, potassium hydroxide; 
(XIV) g) hydrogenation of optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) in presence Raney Nickel.

such that at each step the intermediates were optionally isolated and purified with suita e processes. 2) A process for synthesis of pregabaline (I)

from cyano acetic acid alkyl ester of formula (A)  58 Wherein,
58 Wherein,
R = CH3 : Compound III
R = C2H5 : Compound VIII
comprising,
a) condensation of 2-methyl-propionaldehyde (IV) with cyano acetic acid alkyl ester (A) in presence of organic or inorganic base such as piperidinium acetate, cesium acetate, and further hydrogenation using noble metal catalyst such as platinum oxide, palladium on carbon, palladium hydroxide on carbon and also with Raney nickel in polar solvent such as methanol, ethanol, water, 1 ,4-dioxane, tetrahydrofuran, dimethoxy ethane and diglyme under hydrogen pressure of about 1 kg/cm2 to 5 kg/cm2 with subsequent isolation of the product 2-cyano-4-methyl-pentanoic acid alkyl ester (B) in solution form from the catalyst by filtratio

59 c) hydrolysis of compound (C) in presence of base such as lithium hydroxide, potassium hydroxide or sodium hydroxide, preferably with lithium hydroxide at temperature of about 20 to 80 °C, preferably at 65 to 70 °C to get 2-cyano-2-isobutylsuccinic acid (XVI)

d) compound (XVI) was decarboxylated in presence of mineral acid such as sulfuric acid in organic solvent such as ethyl acetate to obtain compound (XII) at about temperature 70 to 80 °C.

e) treatment of compound (XII) with cinchonidine(XIII) in presence of organic solvent such as methanol, ethanol, 1 ,4-dioxane, ethyl acetate, tetrahydrofuran,
2-methyl tetrahydrofuran, dimethoxy ethane and diglyme at temperature of about 20°C to 80°C to precipitate out (S) 3- cyano-5-methylhexanoic acid salt of cinchonidine (XIV), followed by separation of compound (XIV) through known separation techniques such as filtration, centrifugation, sedimentation, followed by optional purification of (S)-3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) in ethyl acetate or through re-salt formation; 60

(ΧΙΠ)
(XIV)
f) treatment of (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) with Diphasic mixture of ethyl acetate: dilute hydrochloric acid (1 :1) at room temperature to obtain (S)-3- cyano-5-methylhexanoic acid (II) from ethyl acetate layer optionally accompanied with recovery of cinchonidine (XIII) from aqueous phase through basification with sodium

(XIV) g) hydrogenation of optically pure (S) - 3-cyano-5-methyl-hexanoic acid (II) in presence of Raney Nickel.

such that at each step the intermediates were optionally isolated and purified with suitable processes. 61
3. A process for synthesis of pregabaline (I)
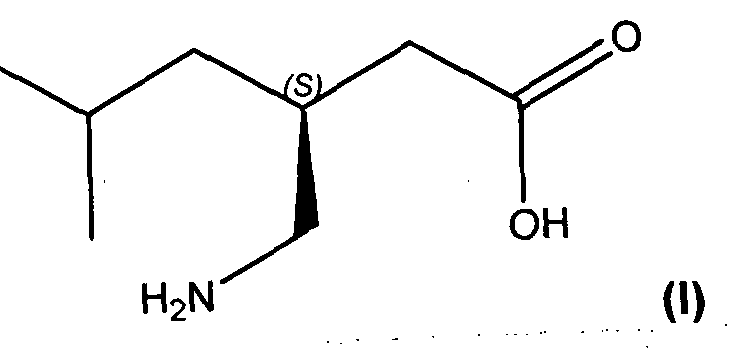
from (f?S)-3-cyano-5-methyl-hexanoic acid (XII); 
(XII)
comprising;
a) treatment of compound (XII) with cinchonidine(XIII) in presence of organic solvent such as methanol, ethanol, 1 ,4-dioxane, ethyl acetate, tetrahydrofuran, 2-methyl tetrahydrofuran, dimethoxy ethane and diglyme at temperature of about 20°C to 80°C to precipitate out (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV), followed by separation of compound (XIV) through known separation techniques such as filtration, centrifugation, sedimentation, followed by optional purification of (S)-3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) in ethyl acetate or through re-salt formation;

(XIII)
(XIV) b) treatment of (S) 3-cyano-5-methylhexanoic acid salt of cinchonidine (XIV) with biphasic mixture of ethyl acetate: dilute hydrochloric acid (1 :1) at room temperature to obtain (S)-3-cyano-5-methylhexanoic acid (II) from ethyl acetate layer optionally

(XIV)

such that at each step the intermediates were optionally isolated and purified by conventional processes.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1235KO2010 | 2010-11-04 | ||
| IN1235/KOL/2010 | 2010-11-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012059797A1 true WO2012059797A1 (en) | 2012-05-10 |
Family
ID=43875371
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2011/000480 WO2012059797A1 (en) | 2010-11-04 | 2011-03-07 | Process for synthesis of (s) - pregabalin |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012059797A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110092733A (en) * | 2018-01-29 | 2019-08-06 | 北京旭阳科技有限公司 | A kind of preparation method of azelaic dinitrile |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4198514A (en) * | 1977-10-11 | 1980-04-15 | Takeda Chemical Industries, Ltd. | Lactam compounds |
| EP0044984A1 (en) * | 1980-07-30 | 1982-02-03 | ALFA FARMACEUTICI S.p.A. | Process for the optical resolution of mixtures of d- and l-2-(6-methoxy-2-naphthyl)-propionic acids, the cinchonidine salt of d-2-(6-methoxy-2-naphthyl)-propionic acid and the preparation thereof |
| US5563175A (en) | 1990-11-27 | 1996-10-08 | Northwestern University | GABA and L-glutamic acid analogs for antiseizure treatment |
| US6001876A (en) | 1996-07-24 | 1999-12-14 | Warner-Lambert Company | Isobutylgaba and its derivatives for the treatment of pain |
| US6127418A (en) | 1997-08-20 | 2000-10-03 | Warner-Lambert Company | GABA analogs to prevent and treat gastrointestinal damage |
| US6194459B1 (en) | 1997-08-19 | 2001-02-27 | Warner-Lambert Company | Methods for treating physiological conditions associated with the use, or sequelae of use, of cocaine or other psychomotors stimulants |
| US6242488B1 (en) | 1997-08-20 | 2001-06-05 | University Of Oklahoma | Method for preventing and treating pain |
| US6306910B1 (en) | 1998-07-09 | 2001-10-23 | Warner-Lambert Company | Use of Gaba-analogues for treating insomnia |
| US6326374B1 (en) | 1998-07-09 | 2001-12-04 | Warner-Lambert Company | Compositions comprising GABA analogs and caffeine |
| US6329429B1 (en) | 1997-06-25 | 2001-12-11 | Warner-Lambert Company | Use of GABA analogs such as Gabapentin in the manufacture of a medicament for treating inflammatory diseases |
| US6359005B1 (en) | 1998-10-16 | 2002-03-19 | Warner-Lambert Company | Method for the treatment of mania and bipolar disorder |
| WO2006000904A2 (en) * | 2004-06-21 | 2006-01-05 | Warner-Lambert Company Llc | Preparation of pregabalin and related compounds |
| WO2007143152A2 (en) | 2006-05-31 | 2007-12-13 | Teva Pharmaceutical Industries Ltd. | Preparation of (s)-pregabalin-nitrile |
| WO2008009897A1 (en) * | 2006-07-15 | 2008-01-24 | Pliva Hrvatska D.O.O. | Process for preparing pregabalin and its opposite enantiomer |
| WO2008062460A2 (en) * | 2006-10-06 | 2008-05-29 | Cadila Healthcare Limited | Crystalline forms of pregabalin |
| US20100292506A1 (en) * | 2009-05-07 | 2010-11-18 | Dipharma Francis S.R.L. | Process for the synthesis of pregabalin |
-
2011
- 2011-03-07 WO PCT/IB2011/000480 patent/WO2012059797A1/en active Application Filing
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4198514A (en) * | 1977-10-11 | 1980-04-15 | Takeda Chemical Industries, Ltd. | Lactam compounds |
| EP0044984A1 (en) * | 1980-07-30 | 1982-02-03 | ALFA FARMACEUTICI S.p.A. | Process for the optical resolution of mixtures of d- and l-2-(6-methoxy-2-naphthyl)-propionic acids, the cinchonidine salt of d-2-(6-methoxy-2-naphthyl)-propionic acid and the preparation thereof |
| US5563175A (en) | 1990-11-27 | 1996-10-08 | Northwestern University | GABA and L-glutamic acid analogs for antiseizure treatment |
| US6001876A (en) | 1996-07-24 | 1999-12-14 | Warner-Lambert Company | Isobutylgaba and its derivatives for the treatment of pain |
| US6329429B1 (en) | 1997-06-25 | 2001-12-11 | Warner-Lambert Company | Use of GABA analogs such as Gabapentin in the manufacture of a medicament for treating inflammatory diseases |
| US6194459B1 (en) | 1997-08-19 | 2001-02-27 | Warner-Lambert Company | Methods for treating physiological conditions associated with the use, or sequelae of use, of cocaine or other psychomotors stimulants |
| US6127418A (en) | 1997-08-20 | 2000-10-03 | Warner-Lambert Company | GABA analogs to prevent and treat gastrointestinal damage |
| US6242488B1 (en) | 1997-08-20 | 2001-06-05 | University Of Oklahoma | Method for preventing and treating pain |
| US6426368B2 (en) | 1997-08-20 | 2002-07-30 | Warner-Lambert Company | Method for preventing and treating alcoholism |
| US6326374B1 (en) | 1998-07-09 | 2001-12-04 | Warner-Lambert Company | Compositions comprising GABA analogs and caffeine |
| US6306910B1 (en) | 1998-07-09 | 2001-10-23 | Warner-Lambert Company | Use of Gaba-analogues for treating insomnia |
| US6359005B1 (en) | 1998-10-16 | 2002-03-19 | Warner-Lambert Company | Method for the treatment of mania and bipolar disorder |
| WO2006000904A2 (en) * | 2004-06-21 | 2006-01-05 | Warner-Lambert Company Llc | Preparation of pregabalin and related compounds |
| WO2007143152A2 (en) | 2006-05-31 | 2007-12-13 | Teva Pharmaceutical Industries Ltd. | Preparation of (s)-pregabalin-nitrile |
| WO2008009897A1 (en) * | 2006-07-15 | 2008-01-24 | Pliva Hrvatska D.O.O. | Process for preparing pregabalin and its opposite enantiomer |
| WO2008062460A2 (en) * | 2006-10-06 | 2008-05-29 | Cadila Healthcare Limited | Crystalline forms of pregabalin |
| US20100292506A1 (en) * | 2009-05-07 | 2010-11-18 | Dipharma Francis S.R.L. | Process for the synthesis of pregabalin |
Non-Patent Citations (6)
| Title |
|---|
| ALEXANDER, E.R. ET AL.: "A simultaneous condensation-reduction method for the preparation of ethyl monoalkylcyanoacetates", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 66, no. 6, 1944, pages 886 - 888, XP055006286, ISSN: 0002-7863, DOI: 10.1021/ja01234a012 * |
| J. ORG. CHEM, vol. 83, 1961, pages 2354 |
| J. ORG. CHEM., vol. 51, 1986, pages 3165 - 31369 |
| J.ORG. CHEM, vol. 83, 1961, pages 2354 |
| KEINAN, E. ET AL.: "An improved method for Sn2-type demethoxycarbonylation of activated esters with 4-aminothiophenol and a cesium catalyst", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 51, no. 16, 1986, pages 3165 - 3169, XP055006249, ISSN: 0022-3263, DOI: 10.1021/jo00366a017 * |
| NAGATA, K. ET AL.: "Catalytic asymmetric alkylation of alpha-cyanocarboxylates and acetoacetates using a phase-transfer catalyst", TETRAHEDRON ASYMMETRY, vol. 20, no. 21, 4 November 2009 (2009-11-04), pages 2530 - 2536, XP026791013, ISSN: 0957-4166, [retrieved on 20091122] * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110092733A (en) * | 2018-01-29 | 2019-08-06 | 北京旭阳科技有限公司 | A kind of preparation method of azelaic dinitrile |
| CN110092733B (en) * | 2018-01-29 | 2022-04-19 | 北京旭阳科技有限公司 | Preparation method of nonane dinitrile |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016191435A1 (en) | Processes to produce brivaracetam | |
| JP2010540614A (en) | Formation of monatin enantiomers | |
| EP3148968B1 (en) | Processes for the preparation of beta-aminosulfone compounds | |
| EP3812371B1 (en) | A process for preparing brivaracetam | |
| WO2013057743A1 (en) | Process for the preparation of an aryl oxime and salts thereof | |
| CA2889650C (en) | Process and intermediates for preparing lacosamide | |
| TW200831478A (en) | Chromane derivatives, synthesis thereof, and intermediates thereto | |
| WO2012059797A1 (en) | Process for synthesis of (s) - pregabalin | |
| CN109422654B (en) | Method for synthesizing fatty aminomethylated compounds | |
| KR20100118747A (en) | Improved preparation method of sarpogrelate hydrochloride | |
| US6414180B1 (en) | Synthesis of chiral β-amino acids | |
| Roy et al. | Eco-friendly, industrial process for synthesis of (S)-3-(aminomethyl)-5-methylhexanoic acid [pregabalin] | |
| TWI333944B (en) | Process for preparing (s)-(+)-2-(substituted phenyl)-2-hydroxy-ethyl carbamates | |
| CN109651437B (en) | Chiral nitrogen-phosphorus ligand, preparation method thereof and method for resolving racemic menthol | |
| CN109265385B (en) | Synthesis process of chiral catalyst | |
| US20210061757A1 (en) | Process for the synthesis of optically active beta-amino alcohols | |
| JP4904945B2 (en) | Process for producing optically active 1- (fluoro, trifluoromethyl or trifluoromethoxy substituted phenyl) alkylamine N-monoalkyl derivatives | |
| JP2005023055A (en) | New optically active amine | |
| CN108191625B (en) | (E) Preparation method of (E) -1- (4-hydroxy-3-methoxyphenyl) -4-en-3-decanone | |
| Singh et al. | Concise synthesis of (S)-(-)-propranolol: using acid catalysed kinetic resolution | |
| JP2003137835A (en) | Method for producing (r)-3-hydroxy-3-(2-phenyl)hexanoic acid | |
| WO2008010578A1 (en) | Process for production of styrene derivative | |
| JP2022110339A (en) | METHOD FOR PRODUCING α-(MERCAPTOMETHYL) ACRYLATE | |
| CN117430527A (en) | Preparation method of 2-alkyl-2-cyano valerate | |
| CN110894184A (en) | Green and environment-friendly ticagrelor intermediate preparation method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11712326 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11712326 Country of ref document: EP Kind code of ref document: A1 |