WO2012025857A1 - Cycloalkyl methoxybenzyl phenyl pyran derivatives as sodium dependent glucose co transporter (sglt2) inhibitors - Google Patents
Cycloalkyl methoxybenzyl phenyl pyran derivatives as sodium dependent glucose co transporter (sglt2) inhibitors Download PDFInfo
- Publication number
- WO2012025857A1 WO2012025857A1 PCT/IB2011/053641 IB2011053641W WO2012025857A1 WO 2012025857 A1 WO2012025857 A1 WO 2012025857A1 IB 2011053641 W IB2011053641 W IB 2011053641W WO 2012025857 A1 WO2012025857 A1 WO 2012025857A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- benzyl
- methoxy
- substituted
- tetrahydro
- pyran
- Prior art date
Links
- -1 Cycloalkyl methoxybenzyl phenyl pyran derivatives Chemical class 0.000 title claims abstract description 71
- 239000011734 sodium Substances 0.000 title abstract description 117
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 title abstract description 28
- 239000008103 glucose Substances 0.000 title abstract description 28
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 title abstract description 5
- 230000001419 dependent effect Effects 0.000 title abstract description 5
- 239000003112 inhibitor Substances 0.000 title abstract description 5
- 229910052708 sodium Inorganic materials 0.000 title abstract description 5
- 102000003673 Symporters Human genes 0.000 title abstract description 4
- 108090000088 Symporters Proteins 0.000 title abstract description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 49
- 201000010099 disease Diseases 0.000 claims abstract description 29
- 238000000034 method Methods 0.000 claims abstract description 27
- 108091006269 SLC5A2 Proteins 0.000 claims abstract 3
- 102000058081 Sodium-Glucose Transporter 2 Human genes 0.000 claims abstract 3
- 150000001875 compounds Chemical class 0.000 claims description 184
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 59
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 59
- 125000000217 alkyl group Chemical group 0.000 claims description 28
- 206010012601 diabetes mellitus Diseases 0.000 claims description 28
- 208000035475 disorder Diseases 0.000 claims description 19
- 229910052739 hydrogen Inorganic materials 0.000 claims description 16
- 239000001257 hydrogen Substances 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 16
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 15
- 150000003839 salts Chemical class 0.000 claims description 15
- 125000003118 aryl group Chemical group 0.000 claims description 13
- 125000004432 carbon atom Chemical group C* 0.000 claims description 12
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 12
- 125000003545 alkoxy group Chemical group 0.000 claims description 11
- 229910052799 carbon Inorganic materials 0.000 claims description 11
- 229910052736 halogen Inorganic materials 0.000 claims description 11
- 150000002367 halogens Chemical class 0.000 claims description 11
- 125000000623 heterocyclic group Chemical group 0.000 claims description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 10
- 125000001072 heteroaryl group Chemical group 0.000 claims description 9
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 9
- 239000003085 diluting agent Substances 0.000 claims description 8
- 230000001404 mediated effect Effects 0.000 claims description 8
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 8
- 239000012453 solvate Substances 0.000 claims description 8
- 125000003342 alkenyl group Chemical group 0.000 claims description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 7
- 239000000651 prodrug Substances 0.000 claims description 7
- 229940002612 prodrug Drugs 0.000 claims description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 6
- 208000017442 Retinal disease Diseases 0.000 claims description 6
- 206010038923 Retinopathy Diseases 0.000 claims description 6
- 125000002252 acyl group Chemical group 0.000 claims description 6
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 6
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 6
- 239000000194 fatty acid Substances 0.000 claims description 6
- 229930195729 fatty acid Natural products 0.000 claims description 6
- 208000011580 syndromic disease Diseases 0.000 claims description 6
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 5
- 125000002619 bicyclic group Chemical group 0.000 claims description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 5
- 230000003111 delayed effect Effects 0.000 claims description 5
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims description 5
- 201000001421 hyperglycemia Diseases 0.000 claims description 5
- 208000017169 kidney disease Diseases 0.000 claims description 5
- 201000001119 neuropathy Diseases 0.000 claims description 5
- 230000007823 neuropathy Effects 0.000 claims description 5
- 208000033808 peripheral neuropathy Diseases 0.000 claims description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 4
- 208000002249 Diabetes Complications Diseases 0.000 claims description 4
- 208000008589 Obesity Diseases 0.000 claims description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 4
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 4
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 claims description 3
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 claims description 3
- 201000001320 Atherosclerosis Diseases 0.000 claims description 3
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 3
- 206010020772 Hypertension Diseases 0.000 claims description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 3
- 229940125758 compound 15 Drugs 0.000 claims description 3
- 229940126142 compound 16 Drugs 0.000 claims description 3
- 150000004665 fatty acids Chemical class 0.000 claims description 3
- 230000014101 glucose homeostasis Effects 0.000 claims description 3
- 208000006575 hypertriglyceridemia Diseases 0.000 claims description 3
- 235000020824 obesity Nutrition 0.000 claims description 3
- 230000029663 wound healing Effects 0.000 claims description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 claims description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 claims description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 claims description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 claims description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 claims description 2
- 229940126657 Compound 17 Drugs 0.000 claims description 2
- 230000036765 blood level Effects 0.000 claims description 2
- 229940125904 compound 1 Drugs 0.000 claims description 2
- 229940125773 compound 10 Drugs 0.000 claims description 2
- 229940125797 compound 12 Drugs 0.000 claims description 2
- 229940126543 compound 14 Drugs 0.000 claims description 2
- 229940125782 compound 2 Drugs 0.000 claims description 2
- 229940126214 compound 3 Drugs 0.000 claims description 2
- 229940125898 compound 5 Drugs 0.000 claims description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 2
- 235000011187 glycerol Nutrition 0.000 claims description 2
- 230000001771 impaired effect Effects 0.000 claims description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 claims description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 2
- 125000003003 spiro group Chemical group 0.000 claims description 2
- LRGREDWGUWRMPI-HFBCXCLPSA-N (2s,3r,4r,5s,6r)-2-[3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-2-fluorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=C([C@H]3[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)C=CC=2)F)C=CC=1OCC1(C)CCC(F)(F)CC1 LRGREDWGUWRMPI-HFBCXCLPSA-N 0.000 claims 1
- YOVBFQICUHUKJY-RTJMFUJLSA-N (2s,3r,4r,5s,6r)-2-[3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-2-methylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C(C)=C1CC(C=C1)=CC=C1OCC1(C)CCC(F)(F)CC1 YOVBFQICUHUKJY-RTJMFUJLSA-N 0.000 claims 1
- 150000001204 N-oxides Chemical class 0.000 claims 1
- 230000035876 healing Effects 0.000 claims 1
- 230000008569 process Effects 0.000 abstract description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 339
- 239000011541 reaction mixture Substances 0.000 description 255
- 239000000243 solution Substances 0.000 description 233
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 199
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 177
- 238000006243 chemical reaction Methods 0.000 description 134
- 230000015572 biosynthetic process Effects 0.000 description 111
- 238000003786 synthesis reaction Methods 0.000 description 111
- 239000012044 organic layer Substances 0.000 description 105
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 100
- 238000005160 1H NMR spectroscopy Methods 0.000 description 99
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 98
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 93
- 239000000543 intermediate Substances 0.000 description 89
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 89
- 235000019439 ethyl acetate Nutrition 0.000 description 86
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 81
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 72
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 68
- 239000012043 crude product Substances 0.000 description 65
- 239000007787 solid Substances 0.000 description 59
- 239000012267 brine Substances 0.000 description 54
- 238000010898 silica gel chromatography Methods 0.000 description 54
- 229910052938 sodium sulfate Inorganic materials 0.000 description 49
- 235000011152 sodium sulphate Nutrition 0.000 description 49
- 239000012299 nitrogen atmosphere Substances 0.000 description 48
- 229920006395 saturated elastomer Polymers 0.000 description 48
- 238000002360 preparation method Methods 0.000 description 46
- 239000000203 mixture Substances 0.000 description 39
- 239000000047 product Substances 0.000 description 39
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 38
- 238000004587 chromatography analysis Methods 0.000 description 36
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 33
- 238000003756 stirring Methods 0.000 description 32
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 30
- 230000002829 reductive effect Effects 0.000 description 28
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 26
- 239000002904 solvent Substances 0.000 description 26
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 25
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 24
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 24
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 24
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 24
- 238000005481 NMR spectroscopy Methods 0.000 description 23
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 230000003197 catalytic effect Effects 0.000 description 21
- 238000004128 high performance liquid chromatography Methods 0.000 description 21
- 239000003039 volatile agent Substances 0.000 description 21
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 20
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 20
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 19
- 229910000024 caesium carbonate Inorganic materials 0.000 description 16
- 239000007788 liquid Substances 0.000 description 16
- 229940123518 Sodium/glucose cotransporter 2 inhibitor Drugs 0.000 description 15
- 239000000725 suspension Substances 0.000 description 15
- 239000010410 layer Substances 0.000 description 14
- 229910015900 BF3 Inorganic materials 0.000 description 13
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 13
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 11
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 11
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 11
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 11
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 10
- 229940098779 methanesulfonic acid Drugs 0.000 description 9
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 9
- 229940086542 triethylamine Drugs 0.000 description 9
- CSQCYSDEAYXXTN-UHFFFAOYSA-N 4-[(5-bromo-2-chlorophenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1CC1=CC(Br)=CC=C1Cl CSQCYSDEAYXXTN-UHFFFAOYSA-N 0.000 description 8
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- ZQWPRMPSCMSAJU-UHFFFAOYSA-N methyl cyclohexanecarboxylate Chemical compound COC(=O)C1CCCCC1 ZQWPRMPSCMSAJU-UHFFFAOYSA-N 0.000 description 8
- 210000002700 urine Anatomy 0.000 description 8
- VNGTZLYNGGLPIZ-WCXIOVBPSA-N (3r,4s,5r,6r)-3,4,5-tris(trimethylsilyloxy)-6-(trimethylsilyloxymethyl)oxan-2-one Chemical compound C[Si](C)(C)OC[C@H]1OC(=O)[C@H](O[Si](C)(C)C)[C@@H](O[Si](C)(C)C)[C@@H]1O[Si](C)(C)C VNGTZLYNGGLPIZ-WCXIOVBPSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 230000029142 excretion Effects 0.000 description 7
- 239000012280 lithium aluminium hydride Substances 0.000 description 7
- 150000003254 radicals Chemical class 0.000 description 7
- 238000001953 recrystallisation Methods 0.000 description 7
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 6
- 0 CCC(C)(*)C(*(C)CC1*(*(C)C)*1**1N(C*)C1*)C(OC)=O Chemical compound CCC(C)(*)C(*(C)CC1*(*(C)C)*1**1N(C*)C1*)C(OC)=O 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 229940043279 diisopropylamine Drugs 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- ILJSQTXMGCGYMG-UHFFFAOYSA-N triacetic acid Chemical compound CC(=O)CC(=O)CC(O)=O ILJSQTXMGCGYMG-UHFFFAOYSA-N 0.000 description 6
- RWHAVOVETBOKQM-OBKDMQGPSA-N (2r,3s,4r,5r,6s)-2-(hydroxymethyl)-6-[3-[(4-hydroxyphenyl)methyl]-4-methylphenyl]oxane-3,4,5-triol Chemical compound CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC1=CC=C(O)C=C1 RWHAVOVETBOKQM-OBKDMQGPSA-N 0.000 description 5
- ZKDSVPHPBZNOPO-UHFFFAOYSA-N (4,4-difluoro-1-methylcyclohexyl)methyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCC1(C)CCC(F)(F)CC1 ZKDSVPHPBZNOPO-UHFFFAOYSA-N 0.000 description 5
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 5
- 102000004877 Insulin Human genes 0.000 description 5
- 108090001061 Insulin Proteins 0.000 description 5
- DUHHQYAYVZZTEE-UHFFFAOYSA-N [1-(trifluoromethyl)cyclobutyl]methyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCC1(C(F)(F)F)CCC1 DUHHQYAYVZZTEE-UHFFFAOYSA-N 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 150000001721 carbon Chemical group 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- 229940125396 insulin Drugs 0.000 description 5
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 5
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- UUBPRIUBEQJUQL-UHFFFAOYSA-N (1-methylcyclohexyl)methanol Chemical compound OCC1(C)CCCCC1 UUBPRIUBEQJUQL-UHFFFAOYSA-N 0.000 description 4
- NEMMESQJOZVCAX-UHFFFAOYSA-N (4,5-diacetyloxyoxan-3-yl) acetate Chemical compound CC(=O)OC1COCC(OC(C)=O)C1OC(C)=O NEMMESQJOZVCAX-UHFFFAOYSA-N 0.000 description 4
- WVBZJYPLDKJFPH-UHFFFAOYSA-N 2h-pyran-3,4,5-triol Chemical compound OC1=C(O)C(O)=COC1 WVBZJYPLDKJFPH-UHFFFAOYSA-N 0.000 description 4
- SWJMLUHTHXZKEV-UHFFFAOYSA-N 4-[(5-bromo-2-fluorophenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1CC1=CC(Br)=CC=C1F SWJMLUHTHXZKEV-UHFFFAOYSA-N 0.000 description 4
- PUQSUZTXKPLAPR-UJPOAAIJSA-N Gastrodin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC=C(CO)C=C1 PUQSUZTXKPLAPR-UJPOAAIJSA-N 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 229910010084 LiAlH4 Inorganic materials 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- AWARFOLZQROJPH-UHFFFAOYSA-N [1-(trifluoromethyl)cyclopropyl]methyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCC1(C(F)(F)F)CC1 AWARFOLZQROJPH-UHFFFAOYSA-N 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 125000000392 cycloalkenyl group Chemical group 0.000 description 4
- NZNMSOFKMUBTKW-UHFFFAOYSA-N cyclohexanecarboxylic acid Chemical compound OC(=O)C1CCCCC1 NZNMSOFKMUBTKW-UHFFFAOYSA-N 0.000 description 4
- JBDSSBMEKXHSJF-UHFFFAOYSA-N cyclopentanecarboxylic acid Chemical compound OC(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-N 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 4
- 239000005457 ice water Substances 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- VUQUOGPMUUJORT-UHFFFAOYSA-N methyl 4-methylbenzenesulfonate Chemical compound COS(=O)(=O)C1=CC=C(C)C=C1 VUQUOGPMUUJORT-UHFFFAOYSA-N 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- HNPIOWQYKWUYIC-UHFFFAOYSA-N (1-methylcyclopentyl)methyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCC1(C)CCCC1 HNPIOWQYKWUYIC-UHFFFAOYSA-N 0.000 description 3
- DTEYKROZZFNUDX-SEFGFODJSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(1-ethylcyclohexyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(CC)CCCCC1 DTEYKROZZFNUDX-SEFGFODJSA-N 0.000 description 3
- CNMIBWITBAZJCP-RTJMFUJLSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(1-ethylcyclopentyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(CC)CCCC1 CNMIBWITBAZJCP-RTJMFUJLSA-N 0.000 description 3
- LWXIAOWPIZUBMQ-RTJMFUJLSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(1-methylcyclohexyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(C)CCCCC1 LWXIAOWPIZUBMQ-RTJMFUJLSA-N 0.000 description 3
- HWJZWMKMLUIMCY-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(3-ethyloxetan-3-yl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(CC)COC1 HWJZWMKMLUIMCY-SJSRKZJXSA-N 0.000 description 3
- LAGPAPSQEVHXSW-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[[1-(trifluoromethyl)cyclopropyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC3(CC3)C(F)(F)F)=CC=2)=C1 LAGPAPSQEVHXSW-BDHVOXNPSA-N 0.000 description 3
- TWCMVXMQHSVIOJ-UHFFFAOYSA-N Aglycone of yadanzioside D Natural products COC(=O)C12OCC34C(CC5C(=CC(O)C(O)C5(C)C3C(O)C1O)C)OC(=O)C(OC(=O)C)C24 TWCMVXMQHSVIOJ-UHFFFAOYSA-N 0.000 description 3
- PLMKQQMDOMTZGG-UHFFFAOYSA-N Astrantiagenin E-methylester Natural products CC12CCC(O)C(C)(CO)C1CCC1(C)C2CC=C2C3CC(C)(C)CCC3(C(=O)OC)CCC21C PLMKQQMDOMTZGG-UHFFFAOYSA-N 0.000 description 3
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 206010012655 Diabetic complications Diseases 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- 206010022489 Insulin Resistance Diseases 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- OYDJIKGLRQRDIK-UHFFFAOYSA-N [1-(ethoxymethyl)cyclopentyl]methanol Chemical compound CCOCC1(CO)CCCC1 OYDJIKGLRQRDIK-UHFFFAOYSA-N 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 239000008298 dragée Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 229930182478 glucoside Natural products 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- PFOARMALXZGCHY-UHFFFAOYSA-N homoegonol Natural products C1=C(OC)C(OC)=CC=C1C1=CC2=CC(CCCO)=CC(OC)=C2O1 PFOARMALXZGCHY-UHFFFAOYSA-N 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- IIHIJFJSXPDTNO-UHFFFAOYSA-N methyl cyclopentanecarboxylate Chemical compound COC(=O)C1CCCC1 IIHIJFJSXPDTNO-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000002792 vascular Effects 0.000 description 3
- MFAVFTVSWZTMIC-UHFFFAOYSA-N (1-ethylcyclobutyl)methanol Chemical compound CCC1(CO)CCC1 MFAVFTVSWZTMIC-UHFFFAOYSA-N 0.000 description 2
- ZOYKMAIVIKIMHH-UHFFFAOYSA-N (1-ethylcyclobutyl)methyl 4-methylbenzenesulfonate Chemical compound C=1C=C(C)C=CC=1S(=O)(=O)OCC1(CC)CCC1 ZOYKMAIVIKIMHH-UHFFFAOYSA-N 0.000 description 2
- SLXAQJGXTIKPJR-UHFFFAOYSA-N (1-ethylcyclohexyl)methanol Chemical compound CCC1(CO)CCCCC1 SLXAQJGXTIKPJR-UHFFFAOYSA-N 0.000 description 2
- WSMQYNZGMHDGDW-UHFFFAOYSA-N (1-ethylcyclohexyl)methyl 4-methylbenzenesulfonate Chemical compound C=1C=C(C)C=CC=1S(=O)(=O)OCC1(CC)CCCCC1 WSMQYNZGMHDGDW-UHFFFAOYSA-N 0.000 description 2
- GLJSDRXLFVQXQU-UHFFFAOYSA-N (1-ethylcyclopentyl)methanol Chemical compound CCC1(CO)CCCC1 GLJSDRXLFVQXQU-UHFFFAOYSA-N 0.000 description 2
- KHBDVWJLSPFJFI-UHFFFAOYSA-N (1-ethylcyclopentyl)methyl 4-methylbenzenesulfonate Chemical compound C=1C=C(C)C=CC=1S(=O)(=O)OCC1(CC)CCCC1 KHBDVWJLSPFJFI-UHFFFAOYSA-N 0.000 description 2
- LYLSFQGMXHJLMM-UHFFFAOYSA-N (1-methylcyclohexyl)methyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCC1(C)CCCCC1 LYLSFQGMXHJLMM-UHFFFAOYSA-N 0.000 description 2
- MXJAKZQKSYGLTK-UHFFFAOYSA-N (1-methylcyclopentyl)methanol Chemical compound OCC1(C)CCCC1 MXJAKZQKSYGLTK-UHFFFAOYSA-N 0.000 description 2
- CNMIBWITBAZJCP-WSGIOKLISA-N (2r,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(1-ethylcyclopentyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(CC)CCCC1 CNMIBWITBAZJCP-WSGIOKLISA-N 0.000 description 2
- RWHAVOVETBOKQM-QUIYGKKVSA-N (2r,3s,4r,5r)-2-(hydroxymethyl)-6-[3-[(4-hydroxyphenyl)methyl]-4-methylphenyl]oxane-3,4,5-triol Chemical compound CC1=CC=C(C2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC1=CC=C(O)C=C1 RWHAVOVETBOKQM-QUIYGKKVSA-N 0.000 description 2
- UKLIWDGMVAJUCH-ZQGJOIPISA-N (2r,3s,4r,5r,6s)-2-(hydroxymethyl)-6-[4-methyl-3-[[4-[[1-(trifluoromethyl)cyclopropyl]methoxy]phenyl]methyl]phenyl]oxane-3,4,5-triol Chemical compound CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(C=C1)=CC=C1OCC1(C(F)(F)F)CC1 UKLIWDGMVAJUCH-ZQGJOIPISA-N 0.000 description 2
- WYRPDDKRHYSSNU-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(1-ethylcyclobutyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(CC)CCC1 WYRPDDKRHYSSNU-RXFVIIJJSA-N 0.000 description 2
- WLHNAEANZVCSLU-SEFGFODJSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[[1-(ethoxymethyl)cyclopentyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(COCC)CCCC1 WLHNAEANZVCSLU-SEFGFODJSA-N 0.000 description 2
- HCTMTIJAPBZKBK-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC3(CCC3)C(F)(F)F)=CC=2)=C1 HCTMTIJAPBZKBK-ZQGJOIPISA-N 0.000 description 2
- ZYPINIUJIBUTEB-UHFFFAOYSA-N (3-ethyloxetan-3-yl)methyl 4-methylbenzenesulfonate Chemical compound C=1C=C(C)C=CC=1S(=O)(=O)OCC1(CC)COC1 ZYPINIUJIBUTEB-UHFFFAOYSA-N 0.000 description 2
- IQZGXXVRCSRUNI-BIFUOPDKSA-N (3r,4r,5s,6r)-2-[3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-4-fluorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)C2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)F)C=CC=1OCC1(C)CCC(F)(F)CC1 IQZGXXVRCSRUNI-BIFUOPDKSA-N 0.000 description 2
- HBACFHQHMLFDJU-BIFUOPDKSA-N (3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(1-methylcyclopentyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)C2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(C)CCCC1 HBACFHQHMLFDJU-BIFUOPDKSA-N 0.000 description 2
- PRTTYUCECYJBOS-BIFUOPDKSA-N (3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)C2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(C)CCC(F)(F)CC1 PRTTYUCECYJBOS-BIFUOPDKSA-N 0.000 description 2
- MUFAVALLTPBWFD-ITJFLMMVSA-N (3r,4s,5s,6r)-2-[3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-4-fluorophenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(F)C(CC=2C=CC(OCC3(C)CCC(F)(F)CC3)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O MUFAVALLTPBWFD-ITJFLMMVSA-N 0.000 description 2
- NFGFFRWJDUDXHC-MLGNGNQOSA-N (3r,4s,5s,6r)-2-[4-chloro-3-[[4-[(1-methylcyclopentyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=CC(OCC3(C)CCCC3)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O NFGFFRWJDUDXHC-MLGNGNQOSA-N 0.000 description 2
- LHDFJUHKIIJLRS-ITJFLMMVSA-N (3r,4s,5s,6r)-2-[4-chloro-3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=CC(OCC3(C)CCC(F)(F)CC3)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O LHDFJUHKIIJLRS-ITJFLMMVSA-N 0.000 description 2
- ULAWITMNOYYRRG-UHFFFAOYSA-N (4,4-difluoro-1-methylcyclohexyl)methanol Chemical compound OCC1(C)CCC(F)(F)CC1 ULAWITMNOYYRRG-UHFFFAOYSA-N 0.000 description 2
- OEURLNJEQCLGPS-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-(4-ethoxyphenyl)methanone Chemical compound C1=CC(OCC)=CC=C1C(=O)C1=CC(Br)=CC=C1Cl OEURLNJEQCLGPS-UHFFFAOYSA-N 0.000 description 2
- BSKHBYQWMYSDHW-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-(4-hydroxyphenyl)methanone Chemical compound C1=CC(O)=CC=C1C(=O)C1=CC(Br)=CC=C1Cl BSKHBYQWMYSDHW-UHFFFAOYSA-N 0.000 description 2
- HDRHVDWRUGOAGF-UHFFFAOYSA-N (5-bromo-2-fluorophenyl)-(4-ethoxyphenyl)methanol Chemical compound C1=CC(OCC)=CC=C1C(O)C1=CC(Br)=CC=C1F HDRHVDWRUGOAGF-UHFFFAOYSA-N 0.000 description 2
- PIOSVMHSKDFKEY-UHFFFAOYSA-N (5-bromo-2-methylphenyl)-(4-ethoxyphenyl)methanone Chemical compound C1=CC(OCC)=CC=C1C(=O)C1=CC(Br)=CC=C1C PIOSVMHSKDFKEY-UHFFFAOYSA-N 0.000 description 2
- YWLSGZYFWIMLRX-UHFFFAOYSA-N (5-bromo-2-methylphenyl)-(4-hydroxyphenyl)methanone Chemical compound CC1=CC=C(Br)C=C1C(=O)C1=CC=C(O)C=C1 YWLSGZYFWIMLRX-UHFFFAOYSA-N 0.000 description 2
- LEWYCBRUDUVOLG-UHFFFAOYSA-N 1,1,2,2-tetrafluoro-2-(1,1,2,2,3,3,4,4-octafluoro-4-iodobutoxy)ethanesulfonyl fluoride Chemical compound FC(F)(I)C(F)(F)C(F)(F)C(F)(F)OC(F)(F)C(F)(F)S(F)(=O)=O LEWYCBRUDUVOLG-UHFFFAOYSA-N 0.000 description 2
- MPCAJMNYNOGXPB-UHFFFAOYSA-N 1,5-anhydrohexitol Chemical compound OCC1OCC(O)C(O)C1O MPCAJMNYNOGXPB-UHFFFAOYSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- VOYQUKFSLPJVEN-UHFFFAOYSA-N 3-[[4-[(5-bromo-2-chlorophenyl)methyl]phenoxy]methyl]-3-ethyloxetane Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(CC)COC1 VOYQUKFSLPJVEN-UHFFFAOYSA-N 0.000 description 2
- DXUIBWCZAAFAGY-UHFFFAOYSA-N 4-[(5-bromo-2-methylphenyl)methyl]phenol Chemical compound CC1=CC=C(Br)C=C1CC1=CC=C(O)C=C1 DXUIBWCZAAFAGY-UHFFFAOYSA-N 0.000 description 2
- QTGSMWMBTQJGJB-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[(1-ethylcyclohexyl)methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(CC)CCCCC1 QTGSMWMBTQJGJB-UHFFFAOYSA-N 0.000 description 2
- VOLPNVSFVPYBFE-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[(1-methylcyclohexyl)methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(C)CCCCC1 VOLPNVSFVPYBFE-UHFFFAOYSA-N 0.000 description 2
- WAAGSNGJQDAASR-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(C)CCC(F)(F)CC1 WAAGSNGJQDAASR-UHFFFAOYSA-N 0.000 description 2
- SVHDEPSWCPNUMA-UHFFFAOYSA-N 4-bromo-2-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-1-fluorobenzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)F)C=CC=1OCC1(C)CCC(F)(F)CC1 SVHDEPSWCPNUMA-UHFFFAOYSA-N 0.000 description 2
- OZJPLYNZGCXSJM-UHFFFAOYSA-N 5-valerolactone Chemical compound O=C1CCCCO1 OZJPLYNZGCXSJM-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 229910015845 BBr3 Inorganic materials 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 239000002841 Lewis acid Substances 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 229910006124 SOCl2 Inorganic materials 0.000 description 2
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 2
- 108010080361 Sodium-Glucose Transport Proteins Proteins 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- GFZIWKFTVNQYQF-MYOTWTRRSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[3-[(4-acetyloxyphenyl)methyl]-4-methylphenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(C)C(CC=2C=CC(OC(C)=O)=CC=2)=C1 GFZIWKFTVNQYQF-MYOTWTRRSA-N 0.000 description 2
- OFKCGBBHAKJAPE-LNALEXQMSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-4-fluorophenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(F)C(CC=2C=CC(OCC3(C)CCC(F)(F)CC3)=CC=2)=C1 OFKCGBBHAKJAPE-LNALEXQMSA-N 0.000 description 2
- YMWFFEBNEDRNEW-LNALEXQMSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(1-ethylcyclobutyl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)Cl)C=CC=1OCC1(CC)CCC1 YMWFFEBNEDRNEW-LNALEXQMSA-N 0.000 description 2
- LIURDPIVHPYION-BBZHULIZSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(1-ethylcyclopentyl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)Cl)C=CC=1OCC1(CC)CCCC1 LIURDPIVHPYION-BBZHULIZSA-N 0.000 description 2
- UUEFFDUOGQBXQU-IXNRCNHTSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[[1-(ethoxymethyl)cyclopentyl]methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)Cl)C=CC=1OCC1(COCC)CCCC1 UUEFFDUOGQBXQU-IXNRCNHTSA-N 0.000 description 2
- FPIRMWNKRJXYIU-WUWJBHAQSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-fluoro-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(F)C(CC=2C=CC(OCC3(CCC3)C(F)(F)F)=CC=2)=C1 FPIRMWNKRJXYIU-WUWJBHAQSA-N 0.000 description 2
- UQSIEHVSHVIAPB-UHFFFAOYSA-N [1-(trifluoromethyl)cyclobutyl]methanol Chemical compound OCC1(C(F)(F)F)CCC1 UQSIEHVSHVIAPB-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 150000005215 alkyl ethers Chemical class 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 2
- 239000012965 benzophenone Substances 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000007822 coupling agent Substances 0.000 description 2
- TXWOGHSRPAYOML-UHFFFAOYSA-N cyclobutanecarboxylic acid Chemical compound OC(=O)C1CCC1 TXWOGHSRPAYOML-UHFFFAOYSA-N 0.000 description 2
- VZFUCHSFHOYXIS-UHFFFAOYSA-N cycloheptane carboxylic acid Natural products OC(=O)C1CCCCCC1 VZFUCHSFHOYXIS-UHFFFAOYSA-N 0.000 description 2
- JBDSSBMEKXHSJF-UHFFFAOYSA-M cyclopentanecarboxylate Chemical compound [O-]C(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-M 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- CZZYITDELCSZES-UHFFFAOYSA-N diphenylmethane Chemical class C=1C=CC=CC=1CC1=CC=CC=C1 CZZYITDELCSZES-UHFFFAOYSA-N 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 2
- MHPHRRKFENGXGZ-UHFFFAOYSA-N ethyl 1-ethylcyclopentane-1-carboxylate Chemical compound CCOC(=O)C1(CC)CCCC1 MHPHRRKFENGXGZ-UHFFFAOYSA-N 0.000 description 2
- 125000005640 glucopyranosyl group Chemical group 0.000 description 2
- 125000002791 glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 2
- 230000002641 glycemic effect Effects 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 2
- 201000008980 hyperinsulinism Diseases 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000003914 insulin secretion Effects 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 150000007517 lewis acids Chemical class 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 2
- JPAPFFYGFBXJAK-UHFFFAOYSA-N methyl 1-ethylcyclohexane-1-carboxylate Chemical compound COC(=O)C1(CC)CCCCC1 JPAPFFYGFBXJAK-UHFFFAOYSA-N 0.000 description 2
- NKZISBBWNQEPAH-UHFFFAOYSA-N methyl 1-methylcyclohexane-1-carboxylate Chemical compound COC(=O)C1(C)CCCCC1 NKZISBBWNQEPAH-UHFFFAOYSA-N 0.000 description 2
- SUCGJXDLZCDFRV-UHFFFAOYSA-N methyl 1-methylcyclopentane-1-carboxylate Chemical compound COC(=O)C1(C)CCCC1 SUCGJXDLZCDFRV-UHFFFAOYSA-N 0.000 description 2
- AKSHOCABCYYRSX-UHFFFAOYSA-N methyl 4,4-difluoro-1-methylcyclohexane-1-carboxylate Chemical compound COC(=O)C1(C)CCC(F)(F)CC1 AKSHOCABCYYRSX-UHFFFAOYSA-N 0.000 description 2
- XHYGYNWRARNCHL-UHFFFAOYSA-N methyl 4,4-difluorocyclohexane-1-carboxylate Chemical compound COC(=O)C1CCC(F)(F)CC1 XHYGYNWRARNCHL-UHFFFAOYSA-N 0.000 description 2
- CBTGNLZUIZHUHY-UHFFFAOYSA-N methyl cyclobutanecarboxylate Chemical compound COC(=O)C1CCC1 CBTGNLZUIZHUHY-UHFFFAOYSA-N 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- DLRJIFUOBPOJNS-UHFFFAOYSA-N phenetole Chemical compound CCOC1=CC=CC=C1 DLRJIFUOBPOJNS-UHFFFAOYSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Chemical group 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000030558 renal glucose absorption Effects 0.000 description 2
- 229910000077 silane Inorganic materials 0.000 description 2
- 125000003107 substituted aryl group Chemical group 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 230000002485 urinary effect Effects 0.000 description 2
- FRWLZZQCDNDJHF-SJSRKZJXSA-N (2r,3s,4r,5r,6s)-2-(hydroxymethyl)-6-[4-methyl-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]oxane-3,4,5-triol Chemical compound CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(C=C1)=CC=C1OCC1(C(F)(F)F)CCC1 FRWLZZQCDNDJHF-SJSRKZJXSA-N 0.000 description 1
- IQZGXXVRCSRUNI-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-4-fluorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)F)C=CC=1OCC1(C)CCC(F)(F)CC1 IQZGXXVRCSRUNI-RXFVIIJJSA-N 0.000 description 1
- UBZKBVUFOXHPLM-RTJMFUJLSA-N (2s,3r,4r,5s,6r)-2-[3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]-4-methylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(C=C1)=CC=C1OCC1(C)CCC(F)(F)CC1 UBZKBVUFOXHPLM-RTJMFUJLSA-N 0.000 description 1
- HBACFHQHMLFDJU-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(1-methylcyclopentyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(C)CCCC1 HBACFHQHMLFDJU-RXFVIIJJSA-N 0.000 description 1
- PRTTYUCECYJBOS-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(C)CCC(F)(F)CC1 PRTTYUCECYJBOS-RXFVIIJJSA-N 0.000 description 1
- YMXYJPABNJCWDV-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[4-fluoro-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(F)C(CC=2C=CC(OCC3(CCC3)C(F)(F)F)=CC=2)=C1 YMXYJPABNJCWDV-ZQGJOIPISA-N 0.000 description 1
- MSFMMGBOPVABSO-QLZLUGFASA-N (2s,3r,4s,5s,6r)-2-[4-chloro-3-[[4-[(1-ethylcyclohexyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@@]2(OC)[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(CC)CCCCC1 MSFMMGBOPVABSO-QLZLUGFASA-N 0.000 description 1
- ISDCILRFWXPVTJ-CBNWRBMVSA-N (2s,3r,4s,5s,6r)-2-[4-chloro-3-[[4-[(1-methylcyclohexyl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=CC(OCC3(C)CCCCC3)=CC=2)=CC=1[C@]1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O ISDCILRFWXPVTJ-CBNWRBMVSA-N 0.000 description 1
- LYIUWASINBVCBV-XCVFGXLCSA-N (2s,3r,4s,5s,6r)-2-[4-chloro-3-[[4-[(3-ethyloxetan-3-yl)methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@@]2(OC)[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(CC)COC1 LYIUWASINBVCBV-XCVFGXLCSA-N 0.000 description 1
- UNMJLQGKEDTEKJ-UHFFFAOYSA-N (3-ethyloxetan-3-yl)methanol Chemical compound CCC1(CO)COC1 UNMJLQGKEDTEKJ-UHFFFAOYSA-N 0.000 description 1
- WLHNAEANZVCSLU-MZIUKZFCSA-N (3r,4r,5s,6r)-2-[4-chloro-3-[[4-[[1-(ethoxymethyl)cyclopentyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)C2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(COCC)CCCC1 WLHNAEANZVCSLU-MZIUKZFCSA-N 0.000 description 1
- YMXYJPABNJCWDV-HZQGQDIUSA-N (3r,4r,5s,6r)-2-[4-fluoro-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)OC1C1=CC=C(F)C(CC=2C=CC(OCC3(CCC3)C(F)(F)F)=CC=2)=C1 YMXYJPABNJCWDV-HZQGQDIUSA-N 0.000 description 1
- FCILJBUKXYKIQO-YCCWVAGXSA-N (3r,4s,5s,6r)-2-[4-chloro-3-[[4-[[1-(ethoxymethyl)cyclopentyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(CC=2C(=CC=C(C=2)C2(OC)[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)Cl)C=CC=1OCC1(COCC)CCCC1 FCILJBUKXYKIQO-YCCWVAGXSA-N 0.000 description 1
- JVSPNWARKKUSOD-JVGOVNBASA-N (3r,4s,5s,6r)-2-[4-chloro-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=CC(OCC3(CCC3)C(F)(F)F)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O JVSPNWARKKUSOD-JVGOVNBASA-N 0.000 description 1
- ZVIVNYKPZRJZLS-NDUFDKTRSA-N (3r,4s,5s,6r)-2-[4-chloro-3-[[4-[[1-(trifluoromethyl)cyclopropyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=CC(OCC3(CC3)C(F)(F)F)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O ZVIVNYKPZRJZLS-NDUFDKTRSA-N 0.000 description 1
- ZBFSSCSXDHCEQU-JVGOVNBASA-N (3r,4s,5s,6r)-2-[4-fluoro-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(F)C(CC=2C=CC(OCC3(CCC3)C(F)(F)F)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O ZBFSSCSXDHCEQU-JVGOVNBASA-N 0.000 description 1
- OQESVVRPHKBHIK-NDUFDKTRSA-N (3r,4s,5s,6r)-2-[4-fluoro-3-[[4-[[1-(trifluoromethyl)cyclopropyl]methoxy]phenyl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(F)C(CC=2C=CC(OCC3(CC3)C(F)(F)F)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O OQESVVRPHKBHIK-NDUFDKTRSA-N 0.000 description 1
- XKBNMNJSZZMEPS-AWGDKMGJSA-N (3r,4s,5s,6r)-6-(hydroxymethyl)-2-[3-[(4-hydroxyphenyl)methyl]-4-methylphenyl]-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(C)C(CC=2C=CC(O)=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O XKBNMNJSZZMEPS-AWGDKMGJSA-N 0.000 description 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 1
- AEBWATHAIVJLTA-UHFFFAOYSA-N 1,2,3,3a,4,5,6,6a-octahydropentalene Chemical compound C1CCC2CCCC21 AEBWATHAIVJLTA-UHFFFAOYSA-N 0.000 description 1
- KTZQTRPPVKQPFO-UHFFFAOYSA-N 1,2-benzoxazole Chemical compound C1=CC=C2C=NOC2=C1 KTZQTRPPVKQPFO-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- LDVVTQMJQSCDMK-UHFFFAOYSA-N 1,3-dihydroxypropan-2-yl formate Chemical class OCC(CO)OC=O LDVVTQMJQSCDMK-UHFFFAOYSA-N 0.000 description 1
- WVUYYXUATWMVIT-UHFFFAOYSA-N 1-bromo-4-ethoxybenzene Chemical compound CCOC1=CC=C(Br)C=C1 WVUYYXUATWMVIT-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- JWXITBZGTLRTLU-UHFFFAOYSA-N 1h-cyclopenta[c]furan Chemical compound C1=C[C]2[CH]OCC2=C1 JWXITBZGTLRTLU-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000006020 2-methyl-1-propenyl group Chemical group 0.000 description 1
- 125000006088 2-oxoazepinyl group Chemical group 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- 125000004637 2-oxopiperidinyl group Chemical group O=C1N(CCCC1)* 0.000 description 1
- YBJDTSRXKGYRNG-UHFFFAOYSA-N 3-benzyl-4-methoxy-2-phenyl-2H-pyran Chemical compound COC1=C(C(OC=C1)C1=CC=CC=C1)CC1=CC=CC=C1 YBJDTSRXKGYRNG-UHFFFAOYSA-N 0.000 description 1
- CGRJJOYCFCCGPX-UHFFFAOYSA-N 3-ethyloxetane Chemical compound CCC1COC1 CGRJJOYCFCCGPX-UHFFFAOYSA-N 0.000 description 1
- HYIUDFLDFSIXTR-UHFFFAOYSA-N 4,4-difluorocyclohexane-1-carboxylic acid Chemical compound OC(=O)C1CCC(F)(F)CC1 HYIUDFLDFSIXTR-UHFFFAOYSA-N 0.000 description 1
- UJGAKNULKVQLEU-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[(1-ethylcyclobutyl)methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(CC)CCC1 UJGAKNULKVQLEU-UHFFFAOYSA-N 0.000 description 1
- KWOKWAJRIUUGJC-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[(1-methylcyclopentyl)methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(C)CCCC1 KWOKWAJRIUUGJC-UHFFFAOYSA-N 0.000 description 1
- PYGFABSVPRRCDE-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[[1-(ethoxymethyl)cyclopentyl]methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(COCC)CCCC1 PYGFABSVPRRCDE-UHFFFAOYSA-N 0.000 description 1
- KSKJRVVOFPKPMN-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(C(F)(F)F)CCC1 KSKJRVVOFPKPMN-UHFFFAOYSA-N 0.000 description 1
- ONSUPPNIEOZTIG-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-[[1-(trifluoromethyl)cyclopropyl]methoxy]phenyl]methyl]benzene Chemical compound C=1C=C(CC=2C(=CC=C(Br)C=2)Cl)C=CC=1OCC1(C(F)(F)F)CC1 ONSUPPNIEOZTIG-UHFFFAOYSA-N 0.000 description 1
- RQQTZUFJUGCQDL-UHFFFAOYSA-N 4-bromo-1-fluoro-2-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]benzene Chemical compound FC1=CC=C(Br)C=C1CC(C=C1)=CC=C1OCC1(C(F)(F)F)CCC1 RQQTZUFJUGCQDL-UHFFFAOYSA-N 0.000 description 1
- MLHCJGCCWAPCRH-UHFFFAOYSA-N 4-bromo-1-fluoro-2-[[4-[[1-(trifluoromethyl)cyclopropyl]methoxy]phenyl]methyl]benzene Chemical compound FC1=CC=C(Br)C=C1CC(C=C1)=CC=C1OCC1(C(F)(F)F)CC1 MLHCJGCCWAPCRH-UHFFFAOYSA-N 0.000 description 1
- BEYBTNQFCZVLQL-UHFFFAOYSA-N 4-bromo-2-[(4-ethoxyphenyl)methyl]-1-fluorobenzene Chemical compound C1=CC(OCC)=CC=C1CC1=CC(Br)=CC=C1F BEYBTNQFCZVLQL-UHFFFAOYSA-N 0.000 description 1
- BFDIPVNNOBOJSZ-UHFFFAOYSA-N 4-bromo-2-[[4-(methoxymethoxy)phenyl]methyl]-1-methylbenzene Chemical compound C1=CC(OCOC)=CC=C1CC1=CC(Br)=CC=C1C BFDIPVNNOBOJSZ-UHFFFAOYSA-N 0.000 description 1
- GZFGOTFRPZRKDS-UHFFFAOYSA-N 4-bromophenol Chemical compound OC1=CC=C(Br)C=C1 GZFGOTFRPZRKDS-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- FGERXQWKKIVFQG-UHFFFAOYSA-N 5-bromo-2-chlorobenzoic acid Chemical compound OC(=O)C1=CC(Br)=CC=C1Cl FGERXQWKKIVFQG-UHFFFAOYSA-N 0.000 description 1
- MMFGGDVQLQQQRX-UHFFFAOYSA-N 5-bromo-2-fluorobenzaldehyde Chemical compound FC1=CC=C(Br)C=C1C=O MMFGGDVQLQQQRX-UHFFFAOYSA-N 0.000 description 1
- SEENCYZQHCUTSB-UHFFFAOYSA-N 5-bromo-2-methylbenzoic acid Chemical compound CC1=CC=C(Br)C=C1C(O)=O SEENCYZQHCUTSB-UHFFFAOYSA-N 0.000 description 1
- JOGHNBXDSOKTPG-UHFFFAOYSA-N 5-bromo-2-methylbenzoyl chloride Chemical compound CC1=CC=C(Br)C=C1C(Cl)=O JOGHNBXDSOKTPG-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 229910002012 Aerosil® Inorganic materials 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 208000031104 Arterial Occlusive disease Diseases 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 229930182476 C-glycoside Natural products 0.000 description 1
- 150000000700 C-glycosides Chemical class 0.000 description 1
- SXJDKMAAHNXQSF-OHQLCTEISA-N CC1(COc2ccc(CC(CC(C)([C@@H]([C@@H]([C@H]3O)O)O[C@H](CO)[C@H]3O)C=C3)=C3Cl)cc2)CCCCC1 Chemical compound CC1(COc2ccc(CC(CC(C)([C@@H]([C@@H]([C@H]3O)O)O[C@H](CO)[C@H]3O)C=C3)=C3Cl)cc2)CCCCC1 SXJDKMAAHNXQSF-OHQLCTEISA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- PHOQVHQSTUBQQK-SQOUGZDYSA-N D-glucono-1,5-lactone Chemical compound OC[C@H]1OC(=O)[C@H](O)[C@@H](O)[C@@H]1O PHOQVHQSTUBQQK-SQOUGZDYSA-N 0.000 description 1
- JVHXJTBJCFBINQ-ADAARDCZSA-N Dapagliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)=CC=C1Cl JVHXJTBJCFBINQ-ADAARDCZSA-N 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 206010017711 Gangrene Diseases 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 201000001431 Hyperuricemia Diseases 0.000 description 1
- 206010052341 Impaired insulin secretion Diseases 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 206010023379 Ketoacidosis Diseases 0.000 description 1
- 208000007976 Ketosis Diseases 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- ITUXVDCQISIYRU-VEIQOZLZSA-N OC[C@H]([C@H]([C@@H]([C@H]1O)O)O)OC1c(cc1Cc(cc2)ccc2OCC2(CC2)C(F)(F)F)ccc1F Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O)OC1c(cc1Cc(cc2)ccc2OCC2(CC2)C(F)(F)F)ccc1F ITUXVDCQISIYRU-VEIQOZLZSA-N 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 229940126902 Phlorizin Drugs 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 108091006299 SLC2A2 Proteins 0.000 description 1
- 102000000070 Sodium-Glucose Transport Proteins Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- LIURDPIVHPYION-VBQYTDQYSA-N [(2r,3r,4r,5s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(1-ethylcyclopentyl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound C=1C=C(CC=2C(=CC=C(C=2)C2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)Cl)C=CC=1OCC1(CC)CCCC1 LIURDPIVHPYION-VBQYTDQYSA-N 0.000 description 1
- UUCXLSGVBPBMSQ-IXNRCNHTSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(1-ethylcyclohexyl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)Cl)C=CC=1OCC1(CC)CCCCC1 UUCXLSGVBPBMSQ-IXNRCNHTSA-N 0.000 description 1
- YNFSIMLVVMRZQJ-BBZHULIZSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(1-methylcyclohexyl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC3(C)CCCCC3)=CC=2)=C1 YNFSIMLVVMRZQJ-BBZHULIZSA-N 0.000 description 1
- AWVIDDFNCAIYDD-LNALEXQMSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(1-methylcyclopentyl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC3(C)CCCC3)=CC=2)=C1 AWVIDDFNCAIYDD-LNALEXQMSA-N 0.000 description 1
- TXZPMTDICRJGTD-OXFURMMHSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(3-ethyloxetan-3-yl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound C=1C=C(CC=2C(=CC=C(C=2)[C@H]2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)Cl)C=CC=1OCC1(CC)COC1 TXZPMTDICRJGTD-OXFURMMHSA-N 0.000 description 1
- IQPRVUWARGOWLU-LNALEXQMSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[(4,4-difluoro-1-methylcyclohexyl)methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC3(C)CCC(F)(F)CC3)=CC=2)=C1 IQPRVUWARGOWLU-LNALEXQMSA-N 0.000 description 1
- JKQDLEYINSHJGO-WUWJBHAQSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[[1-(trifluoromethyl)cyclobutyl]methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC3(CCC3)C(F)(F)F)=CC=2)=C1 JKQDLEYINSHJGO-WUWJBHAQSA-N 0.000 description 1
- BTUIGADEDXDWFQ-MYOTWTRRSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[[4-[[1-(trifluoromethyl)cyclopropyl]methoxy]phenyl]methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(OCC3(CC3)C(F)(F)F)=CC=2)=C1 BTUIGADEDXDWFQ-MYOTWTRRSA-N 0.000 description 1
- YYWSKSKIJXVNTH-UHFFFAOYSA-N [1-(trifluoromethyl)cyclopropyl]methanol Chemical compound OCC1(C(F)(F)F)CC1 YYWSKSKIJXVNTH-UHFFFAOYSA-N 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- 230000009102 absorption Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 238000002266 amputation Methods 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 208000021328 arterial occlusion Diseases 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 210000000467 autonomic pathway Anatomy 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000005872 benzooxazolyl group Chemical group 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 210000003445 biliary tract Anatomy 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 201000008247 brain infarction Diseases 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 1
- 229960001713 canagliflozin Drugs 0.000 description 1
- VHOFTEAWFCUTOS-TUGBYPPCSA-N canagliflozin hydrate Chemical compound O.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1 VHOFTEAWFCUTOS-TUGBYPPCSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 230000023852 carbohydrate metabolic process Effects 0.000 description 1
- 235000021256 carbohydrate metabolism Nutrition 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000004700 cellular uptake Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 229960003834 dapagliflozin Drugs 0.000 description 1
- 125000005507 decahydroisoquinolyl group Chemical group 0.000 description 1
- 238000005661 deetherification reaction Methods 0.000 description 1
- 230000005595 deprotonation Effects 0.000 description 1
- 238000010537 deprotonation reaction Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- JJOYCHKVKWDMEA-UHFFFAOYSA-N ethyl cyclohexanecarboxylate Chemical compound CCOC(=O)C1CCCCC1 JJOYCHKVKWDMEA-UHFFFAOYSA-N 0.000 description 1
- UWSJCCUODNDXOT-UHFFFAOYSA-N ethyl cyclopentanecarboxylate Chemical compound CCOC(=O)C1CCCC1 UWSJCCUODNDXOT-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- 235000012209 glucono delta-lactone Nutrition 0.000 description 1
- 150000002303 glucose derivatives Chemical class 0.000 description 1
- 230000009229 glucose formation Effects 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 230000004190 glucose uptake Effects 0.000 description 1
- 150000008131 glucosides Chemical class 0.000 description 1
- 230000036252 glycation Effects 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 150000002475 indoles Chemical class 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000007915 intraurethral administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- HVGGCSVCKSHOKU-UHFFFAOYSA-N iodomethoxyethane Chemical compound CCOCI HVGGCSVCKSHOKU-UHFFFAOYSA-N 0.000 description 1
- 125000002510 isobutoxy group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])O* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 239000011344 liquid material Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- AXOIIYMMKYHEBV-UHFFFAOYSA-N methyl 1-ethylcyclobutane-1-carboxylate Chemical compound COC(=O)C1(CC)CCC1 AXOIIYMMKYHEBV-UHFFFAOYSA-N 0.000 description 1
- ADUGONOVZSGHBC-UHFFFAOYSA-N methyl 1-ethylcyclopentane-1-carboxylate Chemical compound COC(=O)C1(CC)CCCC1 ADUGONOVZSGHBC-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 206010062198 microangiopathy Diseases 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 125000006606 n-butoxy group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 150000002829 nitrogen Chemical class 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 125000005061 octahydroisoindolyl group Chemical group C1(NCC2CCCCC12)* 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229940054534 ophthalmic solution Drugs 0.000 description 1
- 239000002997 ophthalmic solution Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000036542 oxidative stress Effects 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- IOUVKUPGCMBWBT-UHFFFAOYSA-N phloridzosid Natural products OC1C(O)C(O)C(CO)OC1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 IOUVKUPGCMBWBT-UHFFFAOYSA-N 0.000 description 1
- IOUVKUPGCMBWBT-GHRYLNIYSA-N phlorizin Chemical compound O[C@@H]1[C@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 IOUVKUPGCMBWBT-GHRYLNIYSA-N 0.000 description 1
- 235000019139 phlorizin Nutrition 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 235000013824 polyphenols Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- ZMJJCODMIXQWCQ-UHFFFAOYSA-N potassium;di(propan-2-yl)azanide Chemical compound [K+].CC(C)[N-]C(C)C ZMJJCODMIXQWCQ-UHFFFAOYSA-N 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 239000012056 semi-solid material Substances 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 239000008279 sol Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 150000003461 sulfonyl halides Chemical class 0.000 description 1
- 229910052717 sulfur Chemical group 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000006090 thiamorpholinyl sulfone group Chemical group 0.000 description 1
- 125000006089 thiamorpholinyl sulfoxide group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000002769 thiazolinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 229940100615 topical ointment Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D201/00—Preparation, separation, purification or stabilisation of unsubstituted lactams
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- the present invention relates to cycloalkyl methoxybenzyl phenyl pyran derivatives, which are SGLT2 inhibitors and are useful as therapeutic agents to treat metabolic disorders more particularly diabetes by stimulating excretion of glucose in the urine.
- Type 2 diabetes is characterized by impaired insulin secretion in response to glucose, increased hepatic glucose production and decreased insulin dependent glucose uptake in the peripheral tissues or insulin resistance.
- the burden of diabetes is driven by vascular complications such as cardiovascular disease, stroke, nephropathy, retinopathy, renal failure, and amputations of the extremities. Although these complications result from multiple metabolic derangements, hyper glycemia is central to both the vascular consequences of diabetes and progressive nature of the disease itself.
- Hyperglycemia can be controlled in Individuals by healthy life style changes that increase activity and reduce weight and or by using current non insulin therapies. These therapies target the liver to reduce glucose output, small intestine to decrease glucose absorption, adipose deposits, or muscle to elevate glucose cellular uptake or to promote glucose metabolism, serum proteases to prolong incretion action, and the pancreas to enhance insulin release.
- Patent applications were published describing C-glucosides for which one of the aryl rings in aglycone part had been replaced with heterocycles, primarily benzothiophene (US 7202350; WO 2004/080990, US 2007/0197450, and WO 2005/085237), azulene moiety (WO 2004/013118), N-benzylated indoles (WO 2006/054629), and thiophene (WO 2008/013321).
- Thiaglucosides (WO 2006/073197), compounds arising from modifications of the glucose moiety, replacing the glucosyl hydroxyl methyl moiety with OH, loweralkoxy, SMe and S0 2 Me or replacement of the pyranosil oxygen with acylated nitrogen (US 2008/0221164) were published.
- SGLT2 inhibitors may be relevant with respect to diabetes, diabetic complications or obesity.
- WO 09/026537 describes benzyl-benzene derivatives and methods of use; US 2005/209166 describes glucopyranosyl substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture; WO 2008/055940 describes combination therapy with SGLT2 inhibitors and their pharmaceutical compositions; WO 2007/00445 describes glucopyranosyl substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture; and US 2003/0114390 describes C-aryl glucoside SGLT2 inhibitors and method.
- Dapagliflozin is in phase III clinical trails in where as Canagliflozin is in phase HI.
- the present invention relates to compounds of the formula (1):
- R can be substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkoxy, or substituted or unsubstituted heterocyclic group and preferably methyl, ethyl, propyl, methoxy, ethoxy, isopropoxy, t-butoxy, and the R can be substituted by R a ;
- each R' is independent and can be either the same or a different group and is selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted acyl group, or substituted or unsubstituted alkoxy carbonyl group;
- X can be CRiR 2 , O, NR l5 or S;
- a, b, c, d, e, and f is independent and can be selected from hydrogen, hydroxyl group, halogen, substituted or unsubstituted alkyl, C(0)-R 3 , -C(0)0-R 3 , or -C(0)NR 3 R 4 ; and a and b; or c and d; or e and f can be together with their attached carbon to form Spiro ring or R and one of the a or b; or c and one of the e or f; or d and one of the e or f can be together with their attached carbon atoms to form bicyclic ring.
- Z and Z' is independent and can be selected from hydrogen, hydroxyl group, halogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy group, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkyloxy, -C(0)-R 3 , -C(0)0-R 3 , or -C(0)NR 3 R4;
- R a can be hydrogen, hydroxyl group, halogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy group, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkyloxy, R 3 R 4 , -C(0)-R 3 , -C(0)0- R 3 , -C(0)NR 3 R4, S(0) p NR 3 R 4 , or S(0) p R 3 ;
- each R ⁇ and R 2 can be independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, or substituted or unsubstituted cycloalkyl;
- each R 3 and R 4 can be independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic group, or substituted or unsubstituted heterocyclylalkyl;
- 'p' can be an integer ranging from 0 to 2.
- prodrugs of the compounds of the formula (1) including ester prodrugs.
- R is substituted or unsubstituted alkyl.
- R is methyl, trifluoro-methyl, ethyl and ethoxy-methyl.
- a compound of formula (1) wherein Z' is halogen or substituted or unsubstituted alkyl.
- Z' is chlorine, fluorine and methyl.
- each a, b, c, d, e and f are independently hydrogen or halogen.
- a pharmaceutical preparation which comprises any one of the above cycloalkyl methoxybenzyl phenyl pyran compounds or a pharmaceutically acceptable salt thereof, a solvate thereof, or a hydrate thereof as an active ingredient.
- such a pharmaceutical preparation which is an inhibitor of sodium-dependent glucose transporter 2 activity.
- such a pharmaceutical preparation which is a prophylactic or therapeutic agent for diabetes, diabetes- related diseases or diabetic complications.
- the present invention also provides a pharmaceutical composition that includes at least one compound as described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent).
- the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein.
- the compound(s) present in the composition may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or may be diluted by a carrier, or enclosed within a carrier which may be in the form of a capsule, sachet, paper or other container.
- the compounds and pharmaceutical compositions described herein are useful in the treatment of diseases, conditions and/or disorders mediated by SGLT2 inhibitors.
- the present invention further provides a method of treating diabetes, disease condition and/or disorder mediated by stimulation of glucose excretion in urea/ inhibition of SGLT2 in a subject in need thereof by administering to the subject one or more compounds described herein in the amount effective to cause inhibition of such receptor.
- the present invention provides cycloalkyl methoxybenzyl phenyl pyran derivatives, which may be used as SGLT2 inhibitors and processes for the synthesis of these compounds.
- Pharmaceutically acceptable salts, pharmaceutically acceptable solvates, enantiomers, diastereomers, polymorphs of these compounds that may have the same type of activity are also provided.
- Pharmaceutical compositions containing the described compounds together with pharmaceutically acceptable carriers, excipients or diluents, which can be used for the treatment of diseases, condition and/or disorders mediated by SGLT2 inhibitors are further provided.
- halogen or halo includes fluorine, chlorine, bromine, or iodine.
- alkyl refers to a straight or branched hydrocarbon chain radical consisting solely of carbon and hydrogen atoms, containing no unsaturation, having from one to eight carbon atoms, and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n-propyl, 1-methylethyl (isopropyl), n-butyl, n- pentyl, and 1,1-dimethylethyl (t-butyl).
- alkenyl refers to an aliphatic hydrocarbon group containing a carbon-carbon double bond and which may be a straight or branched chain having from 2 to about 10 carbon atoms, e.g., ethenyl, 1-propenyl, 2-propenyl (allyl), iso- propenyl, 2 -methyl- 1-propenyl, 1-butenyl, and 2-butenyl.
- haloalkyl is used to denote a group comprised of an alkyl group substituted with halogen atom, where alkyl group is as defined above and halogen is used to denote fluorine, chlorine, bromine or iodine, an example of such group is trifluoromethyl, difluoromethyl.
- acyl group is used to denote a linear or branched aliphatic acyl group (preferably a C 2-6 alkanoyl group) or an aromatic acyl group, which contains 2 to 10 carbon atoms.
- examples include an acetyl group, a propionyl group, a pivaloyl group, a butyryl group, an isobutyryl group, a valeryl group and a benzoyl group, with an acetyl group being preferred.
- alkoxy group is used to denote a linear or branched alkoxy group containing 1 to 6 carbon atoms. Preferred are C 1-4 alkoxy groups including a methoxy group, an ethoxy group, a propoxy group, an isopropoxy group, a n-butoxy group, an isobutoxy group and a tert-butoxy group.
- alkoxycarbonyl group isused to denote a structure composed of a linear or branched C 1-5 alkoxy group and a carbonyl group.
- C 2-5 alkoxycarbonyl groups including a methoxycarbonyl group, an ethoxycarbonyl group, a propoxycarbonyl group, an isopropoxycarbonyl group and a butoxycarbonyl group.
- a methoxycarbonyl group is preferred.
- cycloalkyl denotes a non-aromatic mono or multicyclic ring system of from 3 to about 12 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- multicyclic cycloalkyl groups include, but are not limited to, perhydronapththyl, adamantyl and norbornyl groups, bridged cyclic groups and spirobicyclic groups, e.g., spiro (4,4) non-2-yl.
- bicyclic ring denotes a aromatic or non-aromatic bicyclic ring system of from 3 to about 12 carbon atoms, such as benzofused or simple fused.
- bicyclic ring groups include, but are not limited to, benzoisoxazole, benzoxazole, cyclopenta[c]furan, octahydropentalene, or the like.
- cycloalkylalkyl refers to a cyclic ring-containing radical having from 3 to about 8 carbon atoms directly attached to an alkyl group.
- the cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- Non-limiting examples of such groups include cyclopropylmethyl, cyclobutylethyl, and cyclopentylethyl.
- cycloalkenyl refers to a cyclic ring-containing radical having from 3 to about 8 carbon atoms with at least one carbon-carbon double bond, such as cyclopropenyl, cyclobutenyl, and cyclopentenyl.
- aryl refers to an aromatic radical having from 6 to 14 carbon atoms such as phenyl, naphthyl, tetrahydronapthyl, indanyl, and biphenyl.
- arylalkyl refers to an aryl group as defined above directly bonded to an alkyl group as defined above, e.g., -CH 2 C 6 H 5 and -C2H 5 C 6 H 5 .
- heterocyclyl and “heterocyclic ring” refer to a stable 3- to 15- membered ring radical which consists of carbon atoms and from one to five heteroatoms selected from nitrogen, phosphorus, oxygen and sulfur.
- the heterocyclic ring radical may be a monocyclic, bicyclic or tricyclic ring system, which may include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sulfur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states.
- the nitrogen atom may be optionally quaternized; and the ring radical may be partially or fully saturated (i.e., heterocyclic or heteroaryl).
- heterocyclic ring radicals include, but are not limited to, azetidinyl, acridinyl, benzodioxolyl, benzodioxinyl benzodioxanyl, benzofuranyl, carbazolyl, cinnolinyl, dioxolanyl, indolizinyl, naphthyridinyl, perhydroazepinyl, phenazinyl, phenothiazinyl, phenoxazinyl, phthalazinyl, pyridyl, pteridinyl, purinyl, quinazolinyl, quinoxalinyl, quinolinyl, isoquinolinyl, tetrazoyl, imidazolyl, tetrahydroisouinolyl, piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-o
- heterocyclylalkyl refers to a heterocyclic ring radical directly bonded to an alkyl group.
- the heterocyclylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- heteroaryl refers to an aromatic heterocyclic ring radical.
- the heteroaryl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
- heteroarylalkyl refers to a heteroaryl ring radical directly bonded to an alkyl group.
- the heteroarylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- prodrug means a compound that is transformed in vivo to yield a compound of Formula (1) or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms, such as through hydrolysis in blood.
- a discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the
- treating or “treatment” of a state, disease, disorder or condition includes:
- the benefit to a subject receiving treatment is either statistically significant or at least perceptible to the subject or to the physician.
- subject includes mammals (especially humans) and other animals, such as domestic animals (e.g., household pets including cats and dogs) and non-domestic animals (such as wildlife).
- domestic animals e.g., household pets including cats and dogs
- non-domestic animals such as wildlife.
- a “therapeutically effective amount” means the amount of a compound that, when administered to a subject for treating a state, disease, disorder or condition, is sufficient to effect such treatment.
- the “therapeutically effective amount” will vary depending on the compound, the state, disease, disorder or condition and its severity and the age, weight, physical condition and responsiveness of the subject receiving treatment.
- diabetes encompasses type I diabetes, type II diabetes, and other types of diabetes with specific etiology.
- diabetes-related diseases includes adiposis, hyperinsulinemia, abnormal carbohydrate metabolism, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, abnormal lipid metabolism, hypertension, congestive heart failure, edema, hyperuricemia and gout.
- diabetic complications can be classified into acute complications and chronic complications.
- hyperglycemia e.g., ketoacidosis
- infections e.g., skin, soft tissue, biliary system, respiratory system and urinary tract infections
- chronic complications includes microangiopathy (e.g., nephropathy, retinopathy), arteriosclerosis (e.g., atherosclerosis, heart infarction, brain infarction, or lower extremity arterial occlusion), neuropathy (e.g., sensory nerves, motor nerves, or autonomic nerves), foot gangrene, etc.
- microangiopathy e.g., nephropathy, retinopathy
- arteriosclerosis e.g., atherosclerosis, heart infarction, brain infarction, or lower extremity arterial occlusion
- neuropathy e.g., sensory nerves, motor nerves, or autonomic nerves
- foot gangrene e.g., foot gangrene, etc.
- Major complications are diabetic retinopathy, diabetic nephropathy and diabetic neuropathy.
- the compound of the invention may form salts.
- pharmaceutically acceptable salts forming part of this patent application include salts derived from inorganic bases salts of organic bases salts of chiral bases, salts of natural amino acids and salts of non-natural amino acids.
- Certain compounds of present patent application are capable of existing in stereoisomeric forms (e.g. diastereomers and enantiomers). With respect to the overall compounds described by the Formula (1), the present patent application extends to these stereoisomeric forms and to mixtures thereof.
- solvates includes hydrates and other solvents of crystallization (such as alcohols).
- the compounds of the present invention may form solvates with low molecular weight solvents by methods known in the art.
- compositions provided in the present invention include at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent).
- the contemplated pharmaceutical compositions include a compound(s) described herein in an amount sufficient to excrete glucose in urine or inhibit SGLT2 in a subject.
- the subjects contemplated include, for example, a living cell and a mammal, including human mammal.
- the compound of the present invention may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) and can be formulated in to preparations in solid, semi-solid, liquid or gaseous forms.
- suitable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, polyhydroxyethoxylated castor oil, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, cyclodextrin, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, pentaerythritol fatty acid esters, polyoxyethylene, hydroxymethylcellulose and polyvinylpyrrolidone.
- the carrier or diluent may include a sustained release material, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- the pharmaceutical composition may also include one or more pharmaceutically acceptable auxiliary agents, wetting agents, emulsifying agents, suspending agents, preserving agents, salts for influencing osmotic pressure, buffers, sweetening agents, flavoring agents, colorants, or any combination of the foregoing.
- the pharmaceutical composition of the invention may be formulated so as to provide quick, sustained, or delayed release of the active ingredient after administration to the subject by employing procedures known in the art.
- compositions described herein may be prepared, e.g., as described in Remington: The Science and Practice of Pharmacy, 20 th Ed., 2003 (Lippincott Williams & Wilkins).
- the active compound can be mixed with a carrier, or diluted by a carrier, or enclosed within a carrier, which may be in the form of an ampoule, capsule, or sachet.
- the carrier serves as a diluent, it may be a solid, semi-solid, or liquid material that acts as a vehicle, excipient, or medium for the active compound.
- the active compound can be adsorbed on a granular solid container, for example, in a sachet.
- compositions may be, for example, capsules, tablets, aerosols, solutions, suspensions or products for topical application.
- the route of administration may be any route which effectively transports the active compound to the appropriate or desired site of action.
- Suitable routes of administration include, but are not limited to, oral, nasal, pulmonary, buccal, subdermal, intradermal, transdermal, parenteral, rectal, depot, subcutaneous, intravenous, intraurethral, intramuscular, intranasal, ophthalmic (such as with an ophthalmic solution) or topical (such as with a topical ointment).
- the oral route is preferred.
- Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the like are particularly suitable for oral application. Preferable carriers for tablets, dragees, or capsules include lactose, cornstarch, and/or potato starch. A syrup or elixir can be used in cases where a sweetened vehicle can be employed.
- a typical tablet that may be prepared by conventional tabletting techniques may contain: (1) Core: Active compound (as free compound or salt thereof), 250 mg colloidal silicon dioxide (Aerosil®), 1.5 mg microcrystalline cellulose (Avicel®), 70 mg modified cellulose gum (Ac-Di-Sol®), and 7.5 mg magnesium stearate; (2) Coating: HPMC, approx. 9 mg Mywacett 9-40 T and approx. 0.9 mg acylated monoglyceride
- Liquid formulations include, but are not limited to, syrups, emulsions, soft gelatin and sterile injectable liquids, such as aqueous or non-aqueous liquid suspensions or solutions.
- injectable solutions or suspensions preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil.
- the present invention provides compounds and pharmaceutical formulations thereof that are useful in the treatment of diseases, conditions and/or disorders mediated by SGLT2 inhibitors.
- the connection between therapeutic effect and inhibition of SGLT2 is illustrated.
- PCT publication Nos. WO 01//016147, WO 02/08306, or WO 03/020737 J. Clin. Invest. Vol. 79, pp. 1510- 1515 (1987); J.Clin. Invest., Vol. 93, pp. 397-404 (1994); Diabetes; 57, 1723-1729, 2008 and references cited therein, all of which are incorporated herein by reference in their entirety and for the purpose stated.
- the present patent application further provides a method of treating a disease, condition and/or disorder mediated by SGLT2 inhibitors in a subject in need thereof by administering to the subject a therapeutically effective amount of a compound or a pharmaceutical composition of the present invention.
- Diseases, conditions, and/or disorders that are mediated by SGLT2 inhibitors are believed to include, but are not limited to, diabetes, especially type I and type 11 diabetes, including complications of diabetes such as retinopathy, neuropathy, nephropathy and delayed wound healing, and related diseases such as insulin resistance and impaired glucose homeostasis (IGH), hyperglycemia, hyperinsulinemia, elevated blood levels of fatty acids or glycerol, obesity, hyperlipidemia including hypertriglyceridemia, Syndrome X, hypertension, atherosclerosis and related diseases, and for increasing high density lipid levels.
- IGH insulin resistance and impaired glucose homeostasis
- Syndrome X also known as Metabolic Syndrome
- the compounds of the present invention can obtain more advantageous effects than additive effects in the prevention or treatment of the above diseases when using suitably in combination with the above drugs. Also, the administration dose can be decreased in comparison with administration of either drug alone, or adverse effects of co administrated drugs other than SGLT2 inhibitors can be avoided or declined.
- the compounds described herein may be prepared by techniques known in the art.
- the compounds described herein may be prepared by following the reaction sequence as depicted in Scheme- 1 to 2. Further, in the following schemes, where specific bases, acids, reagents, solvents, coupling agents, etc., are mentioned, it is understood that other bases, acids, reagents, solvents, coupling agents etc., known in the art may also be used and are therefore included within the present invention. Variations in reaction conditions, for example, temperature and/or duration of the reaction, which may be used as known in the art, are also within the scope of the present invention. All the stereo isomers of the compounds in these schemes, unless otherwise specified, are also encompassed within the scope of this invention.
- the deprotonation can be carried out under an inert atmosphere at a temperature from -70° C to room temperature and in suitable aprotic organic solvent, for example, tetrahydrofuran or the like, and then R-X can be reacted with the salts of formula 3 or 2 at an initial temperature from -80 °C to- 10 °C either using one or two equivalent of strong base like potassium diisopropylamide, lithium hexamethyl disilazide and preferably, lithium disopropylamide or the like. After a period of stirring at -15°C, the reaction was quenched then the resulted compounds of formula 4 can be isolated and purified by conventional techniques.
- suitable aprotic organic solvent for example, tetrahydrofuran or the like
- Compounds of formula 4 can be reduced to give hydroxyl compounds of formula 5 by a suitable reducing agent for example, lithium aluminum hydride or the like. Converting the hydroxy compounds of formula 5 to compounds of intermediate of formula 6 by the methods known in the art for example, reacting hydroxyl group with sulfonyl halides in the presence of a base such as, potassium carbonate, triethylamine, dimethylaminopyridine or the like.
- a suitable reducing agent for example, lithium aluminum hydride or the like.
- Converting the hydroxy compounds of formula 5 to compounds of intermediate of formula 6 by the methods known in the art for example, reacting hydroxyl group with sulfonyl halides in the presence of a base such as, potassium carbonate, triethylamine, dimethylaminopyridine or the like.
- Compounds of formula 11 can be prepared according to the reaction sequences as shown in scheme-2.
- Compounds of formula 7 can be prepared by following the method described in patent publication US 7,393,836 (as exemplified in the experimental section).
- benzoyl halides either commercially available or prepared insitu from benzoic acid can be coupled with alkoxyaryl compound using a Lewis acid, typically A1C1 3 or the like to give corresponding benzophenone product.
- Reduction of ketone function of benzophenone can be accomplished by trialkyl silane and an appropriate protic or Lewis acid, like trifluoroacetic acid or borontrifluoride etherate, followed by alkyl ether cleavage, using BBr 3 or A1C1 3 to give compounds of formula 7.
- Alkylation of phenol compounds of formula 7 with intermediate compounds of formula 6 can be obtained in the presence of base such as K 2 C0 3 , CsC0 3 , NaOH or the like to give compounds of formula 8 in the solvents such as, dimethyl formamide, dimethyl sulfoxide or the like.
- Compounds of formula 8 can be treated with a base like t-BuLi, n-BuLi to exchange halogen to lithium followed by addition of nascent lithiated aromatic to silyl sugar compounds of formula 9 (as described in Journal of Medicinal Chemistry, 2008, 51, 1 145-1149) in solvents such as, toluene, methane sulfonic acid or the like to give the diastereomeric mixture of lactol compounds, which were converted insitu to the desilylated O-methyl lactol compounds of formula 10 by treatment with methane sulfonicacid in methanol (J. Org. Chem. 1989, 54, 610-612).
- the desired diastereoisomer of compounds of formula 11 can be separated by either column chromatography or peracetylation by the methods known in the art for example, acetic anhydride with catalytic amount of dimethylamino pyridine or the like and recrystallization in a suitable solvent by the methods known in the art for example, 20% methanolic ammonia or the like.
- 5-bromo-2-chlorobenzoic acid 50 g was suspended in dichloromethane (100 ml) and dimethylformamide then it was cooled to 0 °C and to this mixture SOCl 2 (92.2 ml) was added slowly. The reaction mixture was slowly allowed to attain room temperature and stirred for about 12 hours. The solvent was removed under reduced pressure to give 5-bromo-2-chlorobenzoyl chloride as oil.
- step 1 Ethoxybenzene (step 1, 25.8 g) was suspended in dichloromethane (50 ml) and cooled to -5 °C then A1C1 3 (42.3 g) was added slowly by portion wise and above 5- bromo-2-chlorobenzoyl chloride (53.8 g) in dichloromethane (150 ml ) was added drop wise and stirred for about 1 hour at 0 °C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound.
- A1C1 3 (42.3 g) was added slowly by portion wise and above 5- bromo-2-chlorobenzoyl chloride (53.8 g) in dichloromethane (150 ml ) was added drop wise and stirred for about 1 hour at 0 °
- step 2 To a stirred solution of (5-bromo-2-chlorophenyl)(4- ethoxyphenyl)methanone (step 2, 48 g) in dichloromethane (150 ml) at 0°C, A1C1 3 (188.8 g) was added under N 2 atmosphere by portion wise over 30 minutes. Subsequently, the reaction mixture was stirred at room temperature for about 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound.
- step 3 To a stirred solution of (5-bromo-2-chlorophenyl)(4- hydroxyphenyl)methanone (step 3, 41 g) in dichloromethane (150 ml) and acetylnitrile (1: 1), Et 3 SiH (53 ml) was added at 0 °C under N 2 atmosphere followed by BF 3 : Et 2 0 (25 ml) and the reaction was stirred for about 16 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC0 3 Solution. The reaction mixture was extracted with EtOAc.
- Step 2 Synthesis of Ethyl 1 -ethylcyclopentanecarboxylate: To a solution of diisopropylamine (93.6 ml, 0.688 mol) in tetrahydrafuran (200 ml), n-BuLi (2.5M, 213.7 ml, 0.534 mol) in hexane was added at 0°C and the reaction mixture was allowed to stir at same temperature for about 10-15 minutes. The reaction mixture was stirred at room temperature for about 45 minutes.
- Ethyl cyclohexanecarboxylate (step 1, 38 g, 0.267 mol) in tetrahydrafuran (150 ml) was added drop-wise at -78°C to LDA (reaction mixture).
- the reaction mixture was stirred at same temperature for about 2 hours and then ethyl iodide (32.06 ml, 0.400 mol) was added drop-wise.
- the reaction mixture was maintained at -78°C for an hour and allowed to attain room temperature.
- Reaction mixture was stirred overnight at room temperature and quenched at 0°C with saturated NH 4 C1 solution. The solvent was concentrated over vaccum and the compound was extracted with ethyl acetate.
- Ethyl 1-ethylcyclopentanecarboxylate (step 2, 32 g, 0.187 mol) in tetrahydrafuran (120 ml) was added to a suspension of LiAlH 4 (21.4 g, 0.563 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite.
- step 3 To a solution of (l-ethylcyclopentyl)methanol (step 3, 26 g, 0.202 mol) in dichloromethane (400 ml), triethyl amine (141.3 ml, 1.01 mol) was added followed by tosyl chloride (77.3 g, 0.405 mol) at 0°C. After addition of catalytic amount of DMAP (2.47 g, 0.020 mol), the reaction mixture was allowed to stir overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water, saturated brine solution and the organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 27 gm, 47 %.
- Step 5 Synthesis of4-bromo-l-chloro-2-(4-((l-ethylcyclopentyl) methoxy)benzyl) benzene:
- T-BuLi (1.7 M, 17.4 ml, 29.42 mmol) was added drop-wise at -78°C to a solution of intermediate 3 (6 g, 14.71 mmol) in tetrahydrafuran: toluene (90 ml) (1:2). After 20 minutes, the lithiated derivative was added drop- wise to a pre-cooled solution (at -78°C) of silyl sugar (Intermediate 2, 6.88 g, 14.71 mmol) in toluene (30 ml). The reaction mixture was maintained at the same temperature for about 2 hours and quenched with methane sulfonic acid (6 % in MeOH, 90 ml).
- the reaction mixture was stirred at room temperature for an hour, then the reaction mixture was quenched with saturated NaHC0 3 solution and diluted with ethyl acetate. The organic layer was washed with saturated brine solution. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained diastereomeric products were separated by column chromatographic technique.
- Step 1 Synthesis of Methyl cyclobutanecarboxylate: Concentrated H 2 S0 4 (39.2 ml, 1.0 eq) was added drop-wise to a solution of cyclobutanecarboxylic acid (40 g, 0.438 mol) in MeOH (200 ml) at 0°C and the reaction mixture was refluxed for about 36 hours. After completion of the reaction as monitored by TLC, the solvent was removed and the reaction mixture was dissolved in dichloromethane. The dichloromethane layer was washed with saturated NaHC0 3 solution and the organic layer was dried over anhydrous magnesium sulphate, dichloromethane was removed over rotary evaporator. The residue was used directly for the next step.
- Step 3 Synthesis of (l-ethylcyclobutyl)methanol: Methyl 1-ethylcyclopentanecarboxylate (step 2, 25 g, 0.176 mol) in tetrahydrafuran (120 ml) was added to a suspension of L1AIH 4 (21.4 g, 0.563 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at the same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate.
- step 3 To a solution of (l-ethylcyclobutyl)methanol (step 3, 15 g, 0.131 mol) in dichloromethane (300 ml), triethyl amine (73.2 ml, 0.526 mol) was added followed by tosyl chloride (49.9 g, 0.26 mol) at 0°C. After addition of catalytic amount of DMAP (1.6 g, 0.0131 mol), the reaction mixture was allowed to stir overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water, saturated brine solution and the organic layer was dried over anhydrous Na 2 S0 4 and concentrated then the obtained product was purified by column chromatographic technique. Yield: 27 gm, 47 %.
- Step 5 Synthesis of 4-bromo-l-chloro-2-(4-((l-ethylcyclobutyl)methoxy)benzyl) benzene:
- T-BuLi (1.7 M, 15.6 ml, 0.0265 mol) was added drop-wise at -78°C to a solution of Intermediate 5 (5.2 g, 0.0132mol) in tetrahydrafuran: toluene (90 ml) (1:2). After 20 minutes, the lithiated derivative was added drop- wise to a pre-cooled solution (at -78°C) of silyl sugar (Intermediate 2, 4.94 g, 0.0106 mol) in toluene (30 ml). The reaction mixture was maintained at the same temperature for about 2 hours and quenched with methane sulfonic acid (6 % in MeOH, 78 ml).
- reaction mixture was stirred at room temperature for three hours then the reaction mixture was quenched with saturated NaHC0 3 solution and diluted with ethyl acetate. The organic layer was washed with brine solution, dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was separated by column chromatographic technique.
- Step 1 Synthesis of methyl cyclohexanecarboxylate:
- Step 2 Synthesis of methyl 1-ethylcyclohexanecarboxylate:
- Methyl 1-ethylcyclohexanecarboxylate (step 2, 10 g, 0.0588 mol) in tetrahydrafuran (120 ml) was added to a suspension of L1AIH 4 (6.7 g, 0.1764 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at the same temperature for about 30 min. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na 2 S0 4 and concentrated.
- step 3 To a solution of (l-ethylcyclohexyl)methanol (step 3, 8 g, 0.0563 mol) in dichloromethane (200 ml), triethyl amine (31.3 ml, 0.2253 mol) was added followed by tosyl chloride (21.4 g, 0.1126 mol) at 0°C. After addition of catalytic amount of DMAP (0.567 g, 0.0056 mol), the reaction mixture was allowed to stir for overnight at room temperature. After completion of reaction, the reaction mixture was washed with water, saturated brine solution; the organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 8 gm.
- T-BuLi (1.7 M, 23.8 ml, 0.0404mol) was added drop-wise to a solution of 4- bromo- 1 -chloro-2-(4-(( 1 -ethylcyclohexyl)methoxy)benzyl)benzene (Intermediate 7, 8.5 g, 0.0202 mol) in tetrahydrafuran: toluene (150 ml, 1:2) at -78°C. After 20 minutes, the lithiated derivative was added drop- wise to a pre-cooled solution (at - 78°C) of silyl sugar (Intermediate 2, 7.54 g, 0.0161mol) in toluene (70 ml).
- reaction was maintained at the same temperature for about 2 hours then it was quenched with methane sulfonic acid (6 % in MeOH, 127.5 ml).
- methane sulfonic acid (6 % in MeOH, 127.5 ml).
- the reaction mixture was stirred at room temperature for an hour then the reaction mixture was quenched with saturated NaHC0 3 solution and diluted with ethyl acetate.
- the organic layer was washed with saturated brine solution, dried over anhydrous Na 2 S0 4 and concentrated. Diastereomeric products were separated by column chromatographic technique.
- Step 1 Synthesis of methyl cyclohexanecarboxylate:
- Step 2 Synthesis of methyl 1 -methylcyclohexanecarboxylate:
- Methyl 1-methylcyclohexanecarboxylate 3 (15 g, 0.0961 mol) in tetrahydrafuran (100 ml) was added to a suspension of LiAlH 4 (7.3 g, 0.1923 mol) in tetrahydrafuran (100 ml) at 0°C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na 2 S0 4 and concentrated.
- step 3 To a solution of (l-methylcyclohexyl)methanol (step 3, 9 g, 0.0775 mol) in dichloromethane (100 ml), triethyl amine (64.7 ml, 0.2253 mol) was added followed by tosyl chloride (36.85 g, 0.1939 mol) at 0°C. After addition of catalytic amount of DMAP (0.71 g, 0.00703 mol), the reaction mixture was allowed to stir for overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water, saturated brine solution, and the organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 15 gm.
- Step 5 Synthesis of 4-bromo-l-chloro-2-(4-((l- methylcyclohexyl )methoxy )benzyl )benzene:
- T-BuLi (1.7 M, 20.2 ml, 0.0344mol) was added drop-wise to a solution of 4- bromo- 1 -chloro-2-(4-(( 1 -methylcyclohexyl)methoxy)benzyl)benzene (Intermediate 9, 7 g, 0.0172mol) in tetrahydrafuran: toluene (90 ml) (1:2) at -78°C. After 20 minutes, the lithiated derivative was added drop-wise to a pre-cooled solution (at - 78°C) of silyl sugar (Intermediate 2, 6.4 g, 0.0137mol) in toluene (70 ml).
- reaction mixture was maintained at the same temperature for about 2 hours then quenched with methane sulfonic acid (6 % in MeOH, 105 ml).
- the reaction mixture was stirred at room temperature for an hour then quenched with saturated NaHC0 3 solution and diluted with ethyl acetate.
- the organic layer was washed with brine solution, dried over anhydrous Na 2 S0 4 and concentrated.
- the obtained product was separated by column chromatographic technique.
- Step 1 Synthesis of (3-ethyloxetan-3-yl)methyl 4 -methylbenzene sulfonate:
- Step 2 Synthesis of3-((4-(5-bromo-2-chlorobenzyl)phenoxy)methyl)-3- ethyloxetane:
- T-BuLi (1.7 M, 23.8 ml, 0.0406mol) was added drop-wise to a solution of 3- ((4-(5-bromo-2-chlorobenzyl)phenoxy)methyl)-3-ethyloxetane (Intermediate 11, 8 g, 0.0203mol) in tetrahydrafuran: toluene (90 ml) (1:2) at -78°C. After 20 minutes, the lithiated derivative was added drop-wise to a pre-cooled solution (at -78°C) of silyl sugar (Intermediate 2, 7.6 g, 0.0162 mol) in toluene (60 ml).
- the reaction mixture was maintained at the same temperature for about 2 hours then quenched with methane sulfonic acid (6 % in MeOH, 120 ml). The reaction mixture was stirred at room temperature for three hours then it was quenched with saturated NaHC0 3 solution and diluted with ethyl acetate. The organic layer was washed with saturated brine solution. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was separated by column chromatographic technique.
- Step 1 Synthesis of(5-bromo-2-methylphenyl)(4-ethoxyphenyl)methanone:
- step 1 To a stirred solution of (5-bromo-2-methylphenyl)(4- ethoxyphenyl)methanone (step 1, 15 g) in dichloromethane (200 ml), AICI 3 (62.5 g) was added at 0 °C under N 2 atmosphere by portion wise over 30 minutes. Subsequently, the reaction mixture was stirred at room temperature for about 12 hours. After completion of the reaction (monitored by TLC), reaction mixture was poured into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound.
- dichloromethane 200 ml
- step 2 To a stirred solution of (5-bromo-2-methylphenyl)(4- hydroxyphenyl)methanone (step 2, 14 g) in dichloromethane (100 ml) and acetylnitrile (1: 1), Et 3 SiH (39 ml) was added at 0 °C under N 2 atmosphere followed by BF 3 : Et 2 0 (15 ml) then reaction mixture was continued for about 16 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC0 3 Solution. The reaction mixture was extracted with EtOAc.
- Step 4 Synthesis of4-bromo-2-( -(methoxymethoxy)benzyl)-l-methylbenzene:
- Step 5 Synthesis of (3R,4S,5S,6R) ⁇ 2-(3-(4-hydroxybenzyl)-4-methylphenyl)-6- ( hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3, 4, 5-triol:
- step 4 To a stirred solution of 4-bromo-2-(4-(methoxymethoxy)benzyl)-l- methylbenzene (step 4, 11.6 g) in dry tetrahydrafuran (70 ml) and toluene (60 ml, 1: 1 ), n-BuLi (2.3 M in hexane) was added at -78 °C under N 2 atmosphere and stirred for about 30 minutes.
- Step 6 Synthesis of (3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4-methylphenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol:
- reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC0 3 Solution.
- the reaction mixture was extracted with EtOAc.
- the combined organic layers were washed with water, brine, dried over anhydrous sodium sulphate and concentrated to afford the residue.
- the crude product was purified by silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of ⁇ & ⁇ isomers) as white solid (3 g).
- Step 7 Synthesis of (2S,3S,4R,5R,6R)-2-(3-(4-acetoxybenzyl)-4-methylphenyl)-6- (acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate ( ⁇ - isomer):
- Step 8 Synthesis of (2S,3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4-methylphenyl)-6- ( hydroxymethyl )tetrahydro-2H-pyran-3, 4, 5-triol:
- Step 2 Synthesis of (l-(trifluoromethyl)cyclopropyl)methyl 4- methylbenzenesulfonate:
- step 1 To a stirred solution of (l-(trifluoromethyl)cyclopropyl)methanol (step 1, 4.5 g) in dichloromethane (50 ml), Et 3 N (13.5 ml) was added at 0 °C under N 2 atmosphere followed by TsCl (9.1 g) and catalytic amount of DMAP. The reaction mixture was allowed to room temperature and stirred for about 12 hours. After completion of the reaction (monitored by 1H- NMR), reaction mixture was diluted with dichloromethane and organic layer was washed with saturated aq. NHUC1 solution, brine and dried over anhydrous sodium sulphate and concentrated to give the residue.
- Step 1 Synthesis of ( 1 -(trifluoromethyl)cyclobutyl)methanol:
- step 1 To a stirred solution of (l-(trifluoromethyl)cyclobutyl)methanol (step 1, 5.5 g) in dichloromethane (50 ml), Et 3 N (15 ml) was added at 0 °C under N 2 atmosphere followed by TsCl (10 g) and catalytic amount of DMAP. The reaction mixture was allowed to room temperature and stirred for about 12 hours. After completion of the reaction (monitored by 1H- NMR), reaction mixture was diluted with dichloromethane and organic layer was washed with saturated aq. NH 4 C1 solution, brine and dried over anhydrous sodium sulphate and concentrated to afford the residue.
- step 2 To a stirred solution of (l-(trifluoromethyl)cyclobutyl)methanol (step 1, 5.5 g) in dichloromethane (50 ml), Et 3 N (15 ml) was added at 0 °C under N 2 atmosphere followed by TsCl (10 g) and catalytic amount of
- Step 1 Synthesis of methyl 4,4-difluorocyclohexanecarboxylate:
- Step 2 Synthesis of methyl 4,4-difluoro-l-methylcyclohexanecarboxylate:
- Methyl 4,4-difluoro-l -methylcyclohexanecarboxylate (step 2, 5.7 g) in tetrahydrafuran (20 ml) was added to a suspension of LiAlH 4 (2.25 g) in tetrahydrafuran (50 ml) at 0 °C and the reaction mixture was allowed to stir at the same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction (monitored by TLC), the reaction mixture was quenched by cautious addition of water (3.5 ml), 15 % aq. NaOH solution (3.5 ml) and again water (11 ml) at 0 °C and stirred for about 30 minutes.
- Step 4 Synthesis of (4,4-difluoro-l-methylcyclohexyl)methyl 4- methylbenzenesulfonate:
- step 3 To a solution of (4,4-difluoro-l-methylcyclohexyl)methanol (step 3, 4.8 g) in dichloromethane (60 ml), triethyl amine (12.2 ml) was added followed by tosyl chloride (8.3 g) at 0 °C. After addition of catalytic amount of DMAP (2.47 g, 0.020 mol), the reaction mixture was allowed to stir overnight at room temperature. After completion of reaction, the reaction mixture was washed with water, saturated brine solution, dried over anhydrous Na 2 SC1 ⁇ 4 and concentrated. The product was purified by column chromatographic technique to afford the title compound as solid (5 g).
- step 1 4-bromo-l-chloro-2-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)benzene (step 1, 6.2 g) in dry tetrahydrafuran (40 ml) and toulene (60 ml, 1:1 ), n-BuLi (2.3 M, 16.8 ml) was added at -78 °C under N 2 atmosphere and stirred for about 30 minutes at -78°C. To this solution (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-
- Methyl 1-methylcyclopentanecarboxylate (step 1, 26 g) in tetrahydrafuran (200 ml) was added to a suspension of LiAlH 4 (14 g) in tetrahydrafuran (150 ml) at 0 °C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction (monitored by 1 H-NMR), reaction mixture was quenched by cautious addition of water (14 ml), 15 % aq. NaOH solution (14 ml) and again water (42 ml) at 0 °C and stirred for about 1 hour.
- step 2 To a solution of (l-methylcyclopentyl)methanol (step 2, 20 g) in dichloromethane (250 ml), triethyl amine (74 ml) was added followed by tosyl chloride (66.6 g) at 0°C. After addition of catalytic amount of DMAP (Cat), the reaction mixture was allowed to stir overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water and brine solution, dried over anhydrous Na 2 S0 and concentrated then purified by column chromatographic technique to afford the title compound as liquid (14 g).
- DMAP catalytic amount
- Step 2 Synthesis of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl )methoxy )benzyl )phenyl )-6-( hydroxymethyl )-2-methoxytetrahydro- 2H-pyran-3, 4, 5-triol:
- Step 3 Synthesis of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- step 2 3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 3 g) in dichloromethane (30 ml) and acetylnitrile (30 ml, 1: 1), Et 3 SiH (1.9 ml) was added at 0° C under N 2 atmosphere followed by BF 3 : Et 2 0 (1.1 ml) and stirred for about 6 hours. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure.
- reaction mixture was cooled to 0° C and neutralized by cautious addition of saturated aq. NaHC0 3 Solution.
- the reaction mixture was extracted with EtOAc.
- the combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue.
- the crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of ⁇ & ⁇ isomers) as white solid (1.8 g).
- step 1 4-bromo-l-chloro-2-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)benzene (step 1, 3.8 g) in dry tetrahydrafuran (40 ml) and toulene (40 ml, 1: 1), n-BuLi (2.3 M in hexane, 10.7 ml) was added at -78 °C under N 2 atmosphere and stirred for about 30 minutes at -78°C.
- step 2 To a stirred solution of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 2.5 g) in dichloromethane (20 ml) and acetylnitrile (20 ml, 1: 1), Et 3 SiH (1.5 ml) was added at 0 °C under N 2 atmosphere followed by BF 3 : Et 2 0 (0.8 ml) and stirred for about 6 hours at room temperature.
- Step 1 Synthesis of 4-bromo-l-chloro-2-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )benzene:
- Step 2 Synthesis of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )-2-methoxytetrahydro- 2H-pyran-3, 4, 5-triol:
- step 1 4-bromo-l-chloro-2-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)benzene (step 1)) in dry tetrahydrafuran (40 ml) and toulene (40 ml, 1 : 1 ), n-BuLi (2.3 M in hexane) was added at -78 °C under N 2 atmosphere stirred for about 30 minutes.
- Step 3 Synthesis of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- step 2 1.2 g) in dichloromethane (15 ml) and acetylnitrile (15 ml, 1:1), Et 3 SiH (0.7 ml) was added at 0° C under N 2 atmosphere followed by BF 3 : Et 2 0 (0.4 ml) and reaction was stirred for about 6 hours.
- step 1 To a stirred solution of Mg (9.5 g) in dry tetrahydrafuran (50 ml), 1-bromo- 4-ethoxybenzene (step 1, 53.4 g) in tetrahydrafuran (150 ml) was added under N 2 atmosphere at room temperature slowly by drop wise and stirred for about 1 hour. Then 5-bromo-2-fluorobenzaldehyde (11 ml) was added slowly to the above reaction mixture and the reaction mixture was allowed to stir for over night. After completion of the reaction (monitored by TLC), the reaction mixture was poured into saturated NH 4 C1 solution at 0 °C and extracted with ethyl acetate.
- step 2 To a stirred solution of (5-bromo-2-fluorophenyl)(4-ethoxyphenyl)methanol (step 2, 24 g) in dichloromethane (150 ml) and acetylnitrile (150 ml, 1: 1), Et 3 SiH (24 ml) was added at 0 °C under N 2 atmosphere followed by BF 3 : Et 2 0 (14 ml) and the reaction mixture was stirred for about 12 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC0 3 Solution. The reaction mixture was extracted with EtOAc.
- step 3 To a stirred solution of 4-bromo-2-(4-ethoxybenzyl)-l-fluorobenzene (step 3, 15 g) in dichloromethane (80 ml), BBr 3 (53.3 ml, 1M solution in dichloromethane (50 ml)) was added slowly under N 2 atmosphere at 0 °C and stirred for. over night at room temperature. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC0 3 solution. The reaction mixture was extracted with dichloromethane .
- Step 2 Synthesis of (3R,4S,5S,6R)-2-(4-fluoro-.
- step 1 4-bromo-l-fluoro-2-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)benzene (step 1, 5.7 g) in dry tetrahydrafuran (50 ml) and toulene (50 ml, 1: 1), t-BuLi (16 ml, 1.7 M in toluene (30 ml)) was added at -78 °C under N 2 atmosphere and stirred for about 30 minutes at - 78 °C.
- step 2 3R,4S,5S,6R)-2-(4-fluoro-3-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 3g) in dichloromethane (20 ml) and acetylnitrile (20 ml, 1:1) Et 3 SiH (1.8 ml) was added at 0 °C under N 2 atmosphere followed by BF 3 : Et 2 0 (1 ml) and stirred for about 6 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure.
- reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC0 3 solution.
- the reaction mixture was extracted with EtOAc.
- the combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue.
- the crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of ⁇ & ⁇ isomers) as white solid (2.3 g).
- step 1 To a stirred solution of 4-bromo-l-fluoro-2-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)benzene (step 1, 5.3 g) in dry tetrahydrafuran (40 ml) and toulene (40 ml, 1: 1), t-BuLi (2.3 M in hexane, 15.5 ml) was added at -78 °C under N 2 atmosphere and stirred for about 30 minutes.
- step 2 To a stirred solution of (3R,4S,5S,6R)-2-(4-fluoro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 2.8 g) in dichloromethane (20 ml) and acetylnitrile (20 ml, 1: 1), Et 3 SiH (1.7 ml) was added at 0 °C under N 2 atmosphere followed by BF 3 : Et 2 0 (1 ml) and stirred for about 4 hours at room temperature.
- Step 1 Synthesis of 4-bromo-2-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-l-fluorobenzene:
- Step 2 Synthesis of (3R,4S,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl)-4-fluorophenyl)-6- ( hydroxymethyl )-2- methoxytetrahydro-2H-pyran-3,4,5-triol:
- step 1 4-bromo-2-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-l-fluorobenzene (step 1, 2.6 g) in dry tetrahydrafuran (30 ml) and toluene (30 ml, 1: 1 ), t-BuLi (6 ml, 1.7 M in toluene) was added under N 2 atmosphere at -78 °C and stirred for about 30 minutes.
- Step 3 Synthesis of (3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy )benzyl)-4-fluorophenyl )-6-(hydroxymethyl )tetrahydro- 2H-pyran-3, 4, 5-triol:
- step 2 1.6 g) in dichloromethane (15 ml) and acetylnitrile (15 ml, 1 : 1), Et 3 SiH (0.9 ml) was added at 0°C under N 2 atmosphere followed by BF 3 : Et 2 0 (0.5 ml) and reaction was stirred for about 6 hours.
- Step 1 Synthesis of methyl cyclopentanecarboxylate:
- Step 2 Synthesis of methyl l-(ethoxymethyl)cyclopentanecarboxylate:
- Methyl l-(ethoxymethyl)cyclopentanecarboxylate (step 2, 10 g, 0.0588 mol) in tetrahydrafuran (120 ml) was added to a suspension of L1AIH 4 (6.7 g, 0.1764 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na 2 S0 4 and concentrated.
- Step 4 Synthesis of 1 -(ethoxymethyl)cyclopentyl)methyl 4-methylbenzenesulfonate:
- step 3 To a solution of (l-(ethoxymethyl)cyclopentyl)methanol (step 3, 8 g, 0.0563 mol) in dichloromethane (200 ml), triethyl amine (31.3 ml, 0.2253 mol) was added followed by tosyl chloride (21.4 g, 0.1126 mol) at 0°C. After addition of catalytic amount of DMAP (0.567 g, 0.0056 mol), the reaction mixture was allowed to stir for overnight at room temperature. After completion of reaction, the reaction mixture was washed with water and brine solution, dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 8 gm.
- Step 1 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclopentyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol ( ⁇ -isomer):
- Triethylsilane (1.36 ml, 8.46 mmol) was added to a solution of (2S,3R,4R,5S,6R)-2-(4-cMoro-3-(4-((l-ethylcyclopentyl)methoxy)benzyl)phenyl)- 6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (Intermediate 4-P-isomer, 2.2 g, 4.23 mmol) in dichloromethane:MeCN (40:40 ml) at -10°C. The reaction mixture was allowed to stir at same temperature for 10 minutes then BF 3 .OEt 2 (0.79 ml, 6.34 mmol) was added.
- reaction mixture was stirred for about 30 minutes at -10°C and an hour at room temperature. After completion of reaction as monitored by TLC, the reaction mixture was quenched with 10% NaHC0 3 solution. The reaction mixture was washed with water, saturated brine solution and the organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique.
- Step 2 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclopentyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3,4, 5-triyl triacetate:
- step 1 (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclo-pentyl)methoxy)benzyl)phenyl)-tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 1, 1.3 g) in dichloromethane (50 ml) at 0°C was added pyridine (2.1 ml), acetic anhydride (2.5 ml) and catalytic amount of DMAP. The reaction mixture was stirred at room temperature for about an hour.
- reaction mixture was diluted with dichloromethane, washed with water, 1% HC1 solution followed by saturated brine solution. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. Product was purified by flash column chromatographic technique to remove unreacted Ac 2 0. Recrystallisation of the obtained mixture in ethanol crystallized the desired isomer as white solid.
- Step 3 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclopentyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- Step 1 Synthesis of (2R,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclopentyl)methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol ( ⁇ -isomer):
- Step 2 Synthesis of (2R,3R,4R,5S,6R)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclopentyl)methoxy jbenzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate:
- Step 3 Synthesis of (2R,3R,4R5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclopentyl)methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- Step 1 Synthesis of (2S,3R,4R5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclobutyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )-tetrahydro-2H-pyran- 3,4,5-triol:
- Triethylsilane (3.9ml, 0.0242mol) was added to a solution of (2S,3R,4S,5S,6R)-2-(4-cMoro-3-(4-((l-ethylcyclobutyl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)-2-methoxy-tetrahydro-2H-pyran-3,4,5-triol (Intermediate 6, 3.5 g, 0.00691 mol) in dichloromethane: MeCN (40:40 ml) at -10°C. The reaction mixture was allowed to stir at same temperature for 10 minutes then BF 3 .OEt 2 (3.9 ml, 0.0172 mol) was added.
- reaction mixture was stirred at -10°C for about 30 minutes and at room temperature for an hour. After completion of reaction as monitored by TLC, the reaction mixture was quenched with saturated NaHC0 3 solution. The reaction mixture was washed with saturated brine solution; the organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique.
- Step 2 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclobutyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate:
- step 1 To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran- 3,4,5-triol (step 1, 3 g) in dichloromethane (50 ml) at 0°C was added pyridine (5.08 ml), acetic anhydride (5.95 ml) and 76 mg of DMAP. The reaction mixture was stirred at room temperature for about an hour.
- reaction mixture was diluted with dichloromethane, washed with water, 1% HC1 solution, water followed by saturated brine solution. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. Product was purified by flash column chromatographic technique to remove unreacted Ac 2 0. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid.
- Step 3 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclobutyl jmethoxy )benzyl )phenyl )-6-( hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- Step 1 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl jmethoxy )benzyl )phenyl )-6-(hydroxymethyl )tetr hydro-2H-pyran- 3,4,5-triol:
- Triethylsilane (1.5 ml, 0.00963 mol) was added to a solution of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-((l-ethylcyclohexyl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate 8, 2.5 g, 0.0046 mol) in dichloromethane:MeCN (40:40 ml) at -10°C and the reaction mixture was allowed to stir for 10 minutes at same temperature then BF 3 .OEt 2 (0.882 ml, 0.00702 mol) was added.
- reaction mixture was stirred for about 30 minutes at - 10°C and for an hour at room temperature. After completion of reaction as monitored by TLC, the reaction mixture was quenched with 10% NaHC0 3 solution. The reaction mixture was washed with saturated brine solution and the organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique.
- Step 2 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclohexyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate:
- step 1 To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (step 1, 1.8 g) in dichloromethane (20 ml) at 0°C was added pyridine (2.87 ml), acetic anhydride (3.37 ml) and catalytic amount of DMAP. The reaction mixture was stirred at room temperature for about an hour.
- reaction mixture was diluted with dichloromethane, washed with water, 1% HCl solution followed by saturated brine solution. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by flash column chromatographic technique to remove unreacted Ac 2 0. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid.
- Step 3 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- Step 1 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chbro-3-(4-((l- methylcyclohexyl )methoxy )benzyl)phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- Triethylsilane (0.76 ml, 0.0048 mol) was added at -10°C to a solution of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-((l-methylcyclohexyl)methoxy)benzyl)phenyl)- 6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate 10, 1 g, 0.0019 mol) in dichloromethane:MeCN (10: 10 ml). The reaction mixture was allowed to stir at same temperature for 10 min and then BF 3 .OEt 2 (0.365 ml, 0.0028 mol) was added.
- reaction mixture was stirred for about 30 minutes at -10°C and at room temperature for an hour. After completion of reaction as monitored by TLC, reaction mixture was quenched with 10% NaHC0 3 solution. The reaction mixture was washed with saturated brine solution; the organic layer was dried over anhydrous Na 2 S0 4 and concentrated. The obtained product was purified by column chromatographic technique.
- Step 2 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- methylcyclohexyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate:
- step 1 (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (step 1, 0.6 g) in dichloromethane (10 ml), pyridine (0.98 ml), acetic anhydride (1.15 ml) and a catalytic amount of DMAP were added at 0°C. The reaction mixture was stirred at room temperature for about an hour.
- reaction mixture was diluted with dichloromethane, washed with water, 1% HCl solution followed by saturated brine solution. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. Product was purified by flash column chromatographic technique to remove unreacted Ac 2 0. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid.
- Step 3 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol: A solution of 20% methanolic ammonia (15 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- methylcyclohexyl)methoxy)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 2, 0.35g, 0.0005mol) at 0°C and the reaction mixture was stirred for overnight at room temperature.
- Step 1 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- yl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H ⁇
- Triethylsilane (0.32ml, 0.00196mol) was added to a solution of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3-yl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate 12, 0.5 g, 0.0098 mol) in dichloromethane:MeCN (10: 10 ml) at -10°C. The reaction mixture was allowed to stir at same temperature for 10 minutes then BF 3 :OEt 2 (0.19 ml, 0.00147 mol) was added.
- Step 2 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((3- ethyloxetan-3-yl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate:
- Step 3 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- yl jmethoxy jbenzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran-3, 4, 5-triol: A solution of 20% methanolic ammonia (10 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((3-ethyloxetan-3- yl)methoxy)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 2, 0.4 g, 0.000619 mol) at 0°C and the reaction mixture was stirred overnight at room temperature.
- reaction mixture was cooled to room temperature and partitioned between water and EtOAc. Two layers were separated and aqueous layer was again extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the residue.
- the crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 25) to afford the title compound as white solid (198 mg).
- Example 8 Preparation of ( , 2R,3S.4R.5R,6S)-2-( , hvdroxymethyl)-6-(4-methyl-3-(4- ((1 -(trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyr an-3 ,4,5- triol:
- Step 1 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ( trifluoromethyl jcyclobutyl jmethoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate ( ⁇ isomer):
- Step 2 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl)-6- ( hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol: NH 3 in MeOH (70 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (4-chloro-3-(4-((l-(trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)tetrahydro- 2H-pyran-3,4,5-triyl triacetate ( ⁇ isomer, step 1, 0.8 g) at room temperature and stirred for over night.
- Step 1 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- methylcyclopentyljmethoxy )benzyl jphenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate ( ⁇ - isomer):
- Step 2 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy jbenzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- Step 1 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ( trifluoromethyl )cyclopropyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5- triyl triacetate ( ⁇ - isomer):
- Step 2 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3, 4, 5-triol: NH 3 in MeOH (60 ml) was added to(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (4-chloro-3-(4-((l-(trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)tetrahydro- 2H-pyran-3,4,5-triyl triacetate ( ⁇ - isomer, step 1, 1.5 g) at room temperature and stirred over night.
- Step 1 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((4,4- difluoro-l-methylcyclohexyl)methoxy)benzyl)phenyl)tetraty
- reaction mixture was extracted with dichloromethane. The combined organic layers were washed with 3N HC1 solution and brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound.
- the crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound ( ⁇ - isomer) as white solid (690 mg).
- Step 2 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
- Step 1 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-fluoro-3-(4-((l- ( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate ( ⁇ - isomer):
- Step 1 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-fluoro-3-(4-((l- ( trifluoromethyl jcyclopropyl )methoxy )benzyl jphenyl )tetrahydro-2H-pyran-3, 4, 5- triyl triacetate ( ⁇ - isomer):
- Step 2 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ( trifluoromethyl jcyclopropyl )methoxy jbenzyl )phenyl)-6- ( hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol:
- Step 1 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl)-4-fluorophenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate ( ⁇ - isomer):
- Step 2 Synthesis of (2S,3R4R5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl)-4-fluorophenyl )-6-(hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol: NH 3 in MeOH (4 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (3-(4-((4,4-difluoro-l-methylcyclohexyl)methoxy)benzyl)-4- fluorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate ( ⁇ - isomer, step 1, 0.79 g) at room temperature and stirred for over night.
- Step 2 Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ( ethoxymethyl )cyclopentyl)methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate:
- step 1 To a solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (ethoxymethyl)cyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (step 1, 1.0 g) in dichloromethane (30 ml), pyridine (0.67 ml), acetic anhydride (0.79 ml) and 0.0 lg of DMAP were added at 0°C. The reaction mixture was stirred at room temperature for about an hour.
- reaction mixture was diluted with dichloromethane, washed with water, 1% HC1 solution, water followed by saturated brine solution. The organic layer was dried over anhydrous Na 2 S0 4 and concentrated. Obtained product was purified by flash column chromatographic technique to remove unreacted Ac 2 0. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid (900 mg).
- Step 3 Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ( ethoxymethyl )cyclopentyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro- 2H-pyran-3, 4, 5-triol:
- the compounds described herein can be tested for their activity for SGLT2 inhibition following procedures known to a person of ordinary skill in the art. For example, the following protocols may be employed for testing the compounds. These protocols are illustrative and do not limit to the scope of the invention.
- Stable cells CHO expressing h SGLT gene were seeded in 24 well plates.
- Cells were washed with KRH Na buffer, followed by addition of test compound in KRH Na to the micro plate in defined format.
- the final concentration of vehicle (DMSO) is not more than 1%.
- Incubation was carried out for 10-15 minutes at 37° C in an incubator.
- the radiolabel glucose analogue 14 C AMG was added to each well at 0.5 ⁇ Ci concentration. After lhr incubation plates were washed with ice cold KRH Na buffer containing 0.5 Mm phlorizin. Cells were solubilized with 0.2 N NaOH before transferring them to pico plate for radiolabel count.
- Example 19 Effect of compounds on urinary glucose excretion in normal animal:
- Example 20 Effect of compounds on STZ induced diabetic rats:
- test item was administered at the specified dose mentioned above for 14 consecutive days.
- the control group received vehicle during treatment period.
- GTT was conducted in overnight fasted rats.
- Glucose load of 2 g/kg was administered followed by compound dosing. Blood samples were drawn at time intervals for plasma glucose estimation. Effect of the test compound was assessed by comparing the AUC's plasma glucose of control with treated group.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Endocrinology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention relates to the cycloalkyl methoxybenzyl phenyl pyran derivatives as Sodium dependent glucose co transporter (SGLT) inhibitors, particularly SGLT2 and method of treating diseases, conditions and/or disorders inhibited by SGLT2 with them, and processes for preparing them.
Description
CYCLOALKYL METHOXYBENZYL PHENYL PYRAN
DERIVATIVES AS SODIUM DEPENDENT GLUCOSE CO TRANSPORTER (SGLT2) INHIBITORS
This application claims the benefit of Indian Provisional Patent Application No. 2434/CHE/2010 filed on 23rd August 2010, which is incorporated herein by reference.
Field of the Invention
The present invention relates to cycloalkyl methoxybenzyl phenyl pyran derivatives, which are SGLT2 inhibitors and are useful as therapeutic agents to treat metabolic disorders more particularly diabetes by stimulating excretion of glucose in the urine.
Background of the Invention
Diabetes is growing public health problem in developed and developing nations alike. In 2007, the number of people world wide with diabetes was estimated to be 246 million, and this number is estimated to reach 380 million by 2025. Type 2 diabetes is characterized by impaired insulin secretion in response to glucose, increased hepatic glucose production and decreased insulin dependent glucose uptake in the peripheral tissues or insulin resistance. The burden of diabetes is driven by vascular complications such as cardiovascular disease, stroke, nephropathy, retinopathy, renal failure, and amputations of the extremities. Although these complications result from multiple metabolic derangements, hyper glycemia is central to both the vascular consequences of diabetes and progressive nature of the disease itself. Chronically elevated blood glucose levels have been shown to result in higher protein glycation, reduced insulin secretion, beta cell apoptosis, increased oxidative stress, and heightened insulin resistance (Endocrinology 2002, 143, 339- 342). These effects can be reduced by tight glycemic control. Hyperglycemia can be controlled in Individuals by healthy life style changes that increase activity and reduce weight and or by using current non insulin therapies. These therapies target the liver to reduce glucose output, small intestine to decrease glucose absorption, adipose deposits, or muscle to elevate glucose cellular uptake or to promote glucose metabolism, serum proteases to prolong incretion action, and the pancreas to enhance insulin release. Despite the wide range of anti-hyperglycemic agents, achieving glycemic control is difficult for many patients. Since all of these oral therapies ultimately depend on insulin to regulate blood glucose levels. Patients with
type 2 diabetes will eventually require insulin therapy because of an inevitable decline in beta cell function.
The obvious need for new approaches to treat patients with uncontrolled type 2 diabetes has prompted continued exploration of alternative targets in organs involved in maintenance of glucose homeostasis. In the context of type2 diabetes, renal glucose reabsorption contributes to elevated plasma glucose and the attendant micro vascular complications. Evaluation of molecular targets available in the kidney stimulated interest in the development of a new class of anti hyperglycemic agents that promote urinary glucose excretion. Inhibitors of the sodium glucose co transporter (SGLT2) prevent renal glucose reabsorption from the glomerular filtrate and provide an insulin independent means of controlling hyper glcemia ((7. Clin. Invest. Vol. 79, pp. 1510-1515 (1987), (7. Clin. Invest. Vol. 93, pp. 397-404 (1994)).
A good number of patent applications describing O-benzyl phenolic O- glucosides as SGLT2 inhibitors (WO 01/74834; WO 02/28872; WO 01/16147; and US 6414126) were published during 2000-2003. Subsequent applications from other groups reported the successful utilization of heteroaryl aglycone components or modification of the glucose moiety. Hydrolytically more stable C-glycosides were disclosed in US 641426; WO 2001/027128; WO 2002/083066; WO 2008/002824; WO 2003/099836; WO 2004/063209 and WO 2006/034489.
Patent applications were published describing C-glucosides for which one of the aryl rings in aglycone part had been replaced with heterocycles, primarily benzothiophene (US 7202350; WO 2004/080990, US 2007/0197450, and WO 2005/085237), azulene moiety (WO 2004/013118), N-benzylated indoles (WO 2006/054629), and thiophene (WO 2008/013321). Thiaglucosides (WO 2006/073197), compounds arising from modifications of the glucose moiety, replacing the glucosyl hydroxyl methyl moiety with OH, loweralkoxy, SMe and S02Me or replacement of the pyranosil oxygen with acylated nitrogen (US 2008/0221164) were published.
It is thus believed that SGLT2 inhibitors may be relevant with respect to diabetes, diabetic complications or obesity. PCT Publications Nos. WO 01//016147, WO 01/074835, WO 02/068439, WO 02/083066, WO 03/020737, WO 03/080635, WO 04/063209, WO 04/089967, WO 06//034489, WO 08/072726, WO 08/116195, WO 08/122014, WO 08/101939 and WO 08/144346; US patent application/patent Nos. US 7,476,671, US 7,439,232, US 2006/0194809 and US 2008/0132563; EP
patent application/patent Nos. EP 1506211B1, EP 1224195B1, EP 1268502B1, EP 1268503A1, EP 1581543A4 and EP 1685147A4 disclose SGLT2 inhibitors, for treatment diseases mediated by SGLT2 inhibitors.
WO 09/026537 describes benzyl-benzene derivatives and methods of use; US 2005/209166 describes glucopyranosyl substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture; WO 2008/055940 describes combination therapy with SGLT2 inhibitors and their pharmaceutical compositions; WO 2007/00445 describes glucopyranosyl substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture; and US 2003/0114390 describes C-aryl glucoside SGLT2 inhibitors and method.
Also many of the SGLT2 inhibitors have entered in to the clinical studies. For example, Dapagliflozin is in phase III clinical trails in where as Canagliflozin is in phase HI.
In view of the above, we focused primarily on the identification of new glucosides by exploring chemical space available in this class of compounds, with focus on substituents attached to C4 or C4> of the diaryl methane aglycone in an effort to synthesize novel chemical entities which stimulate significant excretion of glucose in the urine and inhibit SGLT2 for treatment of diabetes or treating diseases and/or disorders related to diabetes.
Summary of the Invention
The present invention relates to compounds of the formula (1):

wherein,
R can be substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkoxy, or substituted or unsubstituted heterocyclic group and preferably
methyl, ethyl, propyl, methoxy, ethoxy, isopropoxy, t-butoxy, and the R can be substituted by Ra;
each R' is independent and can be either the same or a different group and is selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted acyl group, or substituted or unsubstituted alkoxy carbonyl group;
X can be CRiR2, O, NRl5 or S;
a, b, c, d, e, and f is independent and can be selected from hydrogen, hydroxyl group, halogen, substituted or unsubstituted alkyl, C(0)-R3, -C(0)0-R3, or -C(0)NR3R4; and a and b; or c and d; or e and f can be together with their attached carbon to form Spiro ring or R and one of the a or b; or c and one of the e or f; or d and one of the e or f can be together with their attached carbon atoms to form bicyclic ring.
Z and Z' is independent and can be selected from hydrogen, hydroxyl group, halogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy group, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkyloxy, -C(0)-R3, -C(0)0-R3, or -C(0)NR3R4;
Ra can be hydrogen, hydroxyl group, halogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy group, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkyloxy, R3R4, -C(0)-R3, -C(0)0- R3, -C(0)NR3R4, S(0)pNR3R4, or S(0)pR3;
each R\ and R2 can be independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, or substituted or unsubstituted cycloalkyl;
each R3 and R4 can be independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic group, or substituted or unsubstituted heterocyclylalkyl;
'p' can be an integer ranging from 0 to 2.
Pharmaceutically acceptable salts of the compounds of the formula (1) are also contemplated. Likewise, pharmaceutically acceptable solvates, including hydrates, of the compounds of the formula (1) are contemplated.
It should be understood that the formula (1) structurally encompasses all stereoisomers, including enatiomers and diastereomers, which may be contemplated from the chemical structure of the genus described herein.
Also contemplated are prodrugs of the compounds of the formula (1), including ester prodrugs.
According to another embodiment, there is provided a compound of formula (1), wherein R is substituted or unsubstituted alkyl. In this embodiment, preferably, R is methyl, trifluoro-methyl, ethyl and ethoxy-methyl.
According to another embodiment, there is provided a compound of formula (1), wherein X is O and CH2.
According to one embodiment, there is provided a compound of formula (1), wherein Z is hydrogen.
According to one embodiment, there is provided a compound of formula (1), wherein Z' is halogen or substituted or unsubstituted alkyl. In this embodiment, preferably, Z' is chlorine, fluorine and methyl.
According to one embodiment, there is provided a compound of formula (1), wherein R' is hydrogen.
According to one embodiment, there is provided a compound of formula (1), wherein R' is substituted or unsubstituted acetyl.
According to another embodiment, there is provided a compound of formula (1), wherein each a, b, c, d, e and f are independently hydrogen or halogen.
According to another embodiment, there is provided a pharmaceutical preparation, which comprises any one of the above cycloalkyl methoxybenzyl phenyl pyran compounds or a pharmaceutically acceptable salt thereof, a solvate thereof, or a hydrate thereof as an active ingredient.
According to another embodiment, there is provided such a pharmaceutical preparation which is an inhibitor of sodium-dependent glucose transporter 2 activity.
According to another embodiment, there is provided such a pharmaceutical preparation which is a prophylactic or therapeutic agent for diabetes, diabetes- related diseases or diabetic complications.
Below are the representative compounds, which are illustrative in nature only and are not intended to limit to the scope of the invention (Nomenclature has been generated from Chem. Draw Ultra 11.0 version):
(2S,3R,4R,5S ,6R)-2-(4-chloro-3-(4-(( 1 - ethylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (Compound 1),
(2R,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (Compound 2),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(( 1 - ethylcyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (Compound 3),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (Compound 4),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (Compound 5),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- yl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound 6),
(2R,3S,4R,5R,6S)-2-(hydroxymethyl)-6-(4-methyl-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5- triol (Compound 7),
(2R,3S,4R,5R,6S)-2-(hydroxymethyl)-6-(4-methyl-3-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triol (Compound 8),
(2S,3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-4-methylphenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Compound 9),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Compound 10),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol (Compound 11),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound 12),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetraliydro-2H-pyran- 3,4,5-triol (Compound 13),
(2S,3R,4R,5S,6R)-2-(4-fluoro-3-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Compound 14),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound 15),
(2S,3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-4-fluorophenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Compound 16),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (ethoxymethyl)cyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Compound 17), or
pharmaceutically acceptable salts, solvates, including hydrates and prodrugs of compounds are also contemplated.
The present invention also provides a pharmaceutical composition that includes at least one compound as described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent). Preferably, the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein. The compound(s) present in the composition may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or may be diluted by a carrier, or enclosed within a carrier which may be in the form of a capsule, sachet, paper or other container.
The compounds and pharmaceutical compositions described herein are useful in the treatment of diseases, conditions and/or disorders mediated by SGLT2 inhibitors.
The present invention further provides a method of treating diabetes, disease condition and/or disorder mediated by stimulation of glucose excretion in urea/
inhibition of SGLT2 in a subject in need thereof by administering to the subject one or more compounds described herein in the amount effective to cause inhibition of such receptor.
Also provided herein are processes for preparing compounds of the present invention.
Detailed Description of the Invention
The present invention provides cycloalkyl methoxybenzyl phenyl pyran derivatives, which may be used as SGLT2 inhibitors and processes for the synthesis of these compounds. Pharmaceutically acceptable salts, pharmaceutically acceptable solvates, enantiomers, diastereomers, polymorphs of these compounds that may have the same type of activity are also provided. Pharmaceutical compositions containing the described compounds together with pharmaceutically acceptable carriers, excipients or diluents, which can be used for the treatment of diseases, condition and/or disorders mediated by SGLT2 inhibitors are further provided.
The following definitions apply to the terms as used herein:
The terms "halogen" or "halo" includes fluorine, chlorine, bromine, or iodine.
The term "alkyl" refers to a straight or branched hydrocarbon chain radical consisting solely of carbon and hydrogen atoms, containing no unsaturation, having from one to eight carbon atoms, and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n-propyl, 1-methylethyl (isopropyl), n-butyl, n- pentyl, and 1,1-dimethylethyl (t-butyl).
The term "alkenyl" refers to an aliphatic hydrocarbon group containing a carbon-carbon double bond and which may be a straight or branched chain having from 2 to about 10 carbon atoms, e.g., ethenyl, 1-propenyl, 2-propenyl (allyl), iso- propenyl, 2 -methyl- 1-propenyl, 1-butenyl, and 2-butenyl.
The term "haloalkyl" is used to denote a group comprised of an alkyl group substituted with halogen atom, where alkyl group is as defined above and halogen is used to denote fluorine, chlorine, bromine or iodine, an example of such group is trifluoromethyl, difluoromethyl.
The term "acyl group" is used to denote a linear or branched aliphatic acyl group (preferably a C2-6 alkanoyl group) or an aromatic acyl group, which contains 2 to 10 carbon atoms. Examples include an acetyl group, a propionyl group, a pivaloyl
group, a butyryl group, an isobutyryl group, a valeryl group and a benzoyl group, with an acetyl group being preferred.
The term "alkoxy group" is used to denote a linear or branched alkoxy group containing 1 to 6 carbon atoms. Preferred are C1-4 alkoxy groups including a methoxy group, an ethoxy group, a propoxy group, an isopropoxy group, a n-butoxy group, an isobutoxy group and a tert-butoxy group.
The term "alkoxycarbonyl group" isused to denote a structure composed of a linear or branched C1-5 alkoxy group and a carbonyl group. Preferred are C2-5 alkoxycarbonyl groups including a methoxycarbonyl group, an ethoxycarbonyl group, a propoxycarbonyl group, an isopropoxycarbonyl group and a butoxycarbonyl group. Among them, a methoxycarbonyl group is preferred.
The term "cycloalkyl" denotes a non-aromatic mono or multicyclic ring system of from 3 to about 12 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. Examples of multicyclic cycloalkyl groups include, but are not limited to, perhydronapththyl, adamantyl and norbornyl groups, bridged cyclic groups and spirobicyclic groups, e.g., spiro (4,4) non-2-yl.
The term "bicyclic ring" denotes a aromatic or non-aromatic bicyclic ring system of from 3 to about 12 carbon atoms, such as benzofused or simple fused. Examples of bicyclic ring groups include, but are not limited to, benzoisoxazole, benzoxazole, cyclopenta[c]furan, octahydropentalene, or the like.
The term "cycloalkylalkyl" refers to a cyclic ring-containing radical having from 3 to about 8 carbon atoms directly attached to an alkyl group. The cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Non-limiting examples of such groups include cyclopropylmethyl, cyclobutylethyl, and cyclopentylethyl.
The term "cycloalkenyl" refers to a cyclic ring-containing radical having from 3 to about 8 carbon atoms with at least one carbon-carbon double bond, such as cyclopropenyl, cyclobutenyl, and cyclopentenyl.
The term "aryl" refers to an aromatic radical having from 6 to 14 carbon atoms such as phenyl, naphthyl, tetrahydronapthyl, indanyl, and biphenyl.
The term "arylalkyl" refers to an aryl group as defined above directly bonded to an alkyl group as defined above, e.g., -CH2C6H5 and -C2H5C6H5.
The terms "heterocyclyl" and "heterocyclic ring" refer to a stable 3- to 15- membered ring radical which consists of carbon atoms and from one to five heteroatoms selected from nitrogen, phosphorus, oxygen and sulfur. For purposes of this invention, the heterocyclic ring radical may be a monocyclic, bicyclic or tricyclic ring system, which may include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sulfur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states. In addition, the nitrogen atom may be optionally quaternized; and the ring radical may be partially or fully saturated (i.e., heterocyclic or heteroaryl). Examples of such heterocyclic ring radicals include, but are not limited to, azetidinyl, acridinyl, benzodioxolyl, benzodioxinyl benzodioxanyl, benzofuranyl, carbazolyl, cinnolinyl, dioxolanyl, indolizinyl, naphthyridinyl, perhydroazepinyl, phenazinyl, phenothiazinyl, phenoxazinyl, phthalazinyl, pyridyl, pteridinyl, purinyl, quinazolinyl, quinoxalinyl, quinolinyl, isoquinolinyl, tetrazoyl, imidazolyl, tetrahydroisouinolyl, piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxoazepinyl, azepinyl, pyrrolyl, 4-piperidonyl, pyrrolidinyl, pyrazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxazolinyl, oxazolidinyl, triazolyl, indanyl, isoxazolyl, isoxasolidinyl, morpholinyl, thiazolyl, thiazolinyl, thiazolidinyl, isothiazolyl, quinuclidinyl, isothiazolidinyl, indolyl, isoindolyl, indolinyl, isoindolinyl, octahydroindolyl, octahydroisoindolyl, quinolyl, isoquinolyl, decahydroisoquinolyl, benzimidazolyl, thiadiazolyl, benzopyranyl, benzothiazolyl, benzooxazolyl, furyl, tetrahydrofurtyl, tetrahydropyranyl, thienyl, benzothienyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiamorpholinyl sulfone, dioxaphospholanyl, oxadiazolyl, chromanyl, and isochromanyl. The heterocyclic ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
The term "heterocyclylalkyl" refers to a heterocyclic ring radical directly bonded to an alkyl group. The heterocyclylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
The term "heteroaryl" refers to an aromatic heterocyclic ring radical. The heteroaryl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
The term "heteroarylalkyl" refers to a heteroaryl ring radical directly bonded to an alkyl group. The heteroarylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
Unless otherwise specified, the term "substituted" as used herein refers to substitution with any one or any combination of the following substituents: hydroxy, halogen, carboxyl, cyano, nitro, oxo (=0), thio (=S), substituted or unsubstituted alkyl, haloalkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted amino, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclylalkyl ring, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring, substituted or unsubstiuted guanidine, -COORx, -C(0)Rx, -C(S)RX, -C(0)NRxRy, -C(0)ONRxRy, -NRxCONRyRz, -N(Rx)SORy, -N(Rx)S02Ry, -(=N- N(Rx)Ry), -NRxC(0)OR , -NRxRy, -NRxC(0)Ry, -NRxC(S)Ry, -NRxC(S)NRyRz, - SONRxRy, -S02NRxRy, -ORx, -ORxC(0)NRyRz, -ORxC(0)ORy, -OC(0)Rx, - OC(0)NRxRy, -RxNRyC(0)Rz, -RxORy, -RxC(0)ORy, -RxC(0)NRyRz, -RxC(0)Ry, - RxOC(0)Ry, -SRX, -SORx, -S02Rx, and -ON02, wherein Rx, Ry and Rz are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted amino, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted heterocyclylalkyl ring, substituted or unsubstituted heteroarylalkyl, or substituted or unsubstituted heterocyclic ring. The substituents in the aforementioned "substituted" groups cannot be further substituted. For example, when the substituent on "substituted alkyl" is "substituted aryl", the substituent on "substituted aryl" cannot be "substituted alkenyl".
The term "prodrug" means a compound that is transformed in vivo to yield a compound of Formula (1) or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms, such as through hydrolysis in blood. A discussion of the use of prodrugs is provided by T.
Higuchi and W. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the
A. C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward
B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
The term "treating" or "treatment" of a state, disease, disorder or condition includes:
(1) preventing or delaying the appearance of clinical symptoms of the state, disease, disorder or condition developing in a subject that may be afflicted with or predisposed to the state, disease, disorder or condition but does not yet experience or display clinical or subclinical symptoms of the state, disease, disorder or condition;
(2) inhibiting the state, disease, disorder or condition, i.e., arresting or reducing the development of the state, disease, disorder or condition or at least one clinical or subclinical symptom thereof; or
(3) relieving the state, disease, disorder or condition, i.e., causing regression of the state, disease, disorder or condition or at least one of its clinical or subclinical symptoms.
The benefit to a subject receiving treatment is either statistically significant or at least perceptible to the subject or to the physician.
The term "subject" includes mammals (especially humans) and other animals, such as domestic animals (e.g., household pets including cats and dogs) and non-domestic animals (such as wildlife).
A "therapeutically effective amount" means the amount of a compound that, when administered to a subject for treating a state, disease, disorder or condition, is sufficient to effect such treatment. The "therapeutically effective amount" will vary depending on the compound, the state, disease, disorder or condition and its severity and the age, weight, physical condition and responsiveness of the subject receiving treatment.
The term "diabetes" encompasses type I diabetes, type II diabetes, and other types of diabetes with specific etiology.
The term "diabetes-related diseases" includes adiposis, hyperinsulinemia, abnormal carbohydrate metabolism, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, abnormal lipid metabolism, hypertension, congestive heart failure, edema, hyperuricemia and gout.
The term "diabetic complications" can be classified into acute complications and chronic complications.
The term "acute complications" includes hyperglycemia (e.g., ketoacidosis), infections (e.g., skin, soft tissue, biliary system, respiratory system and urinary tract infections), etc.
The term "chronic complications" includes microangiopathy (e.g., nephropathy, retinopathy), arteriosclerosis (e.g., atherosclerosis, heart infarction, brain infarction, or lower extremity arterial occlusion), neuropathy (e.g., sensory nerves, motor nerves, or autonomic nerves), foot gangrene, etc. Major complications are diabetic retinopathy, diabetic nephropathy and diabetic neuropathy.
The compound of the invention may form salts. Non-limiting examples of pharmaceutically acceptable salts forming part of this patent application include salts derived from inorganic bases salts of organic bases salts of chiral bases, salts of natural amino acids and salts of non-natural amino acids. Certain compounds of present patent application are capable of existing in stereoisomeric forms (e.g. diastereomers and enantiomers). With respect to the overall compounds described by the Formula (1), the present patent application extends to these stereoisomeric forms and to mixtures thereof. To the extent prior art teaches synthesis or separation of particular stereoisomers, the different stereoisomeric forms of the present patent application may be separated from one another by the method known in the art, or a given isomer may be obtained by stereospecific or asymmetric synthesis. Tautomeric forms and mixtures of compounds described herein are also contemplated.
Pharmaceutically acceptable solvates includes hydrates and other solvents of crystallization (such as alcohols). The compounds of the present invention may form solvates with low molecular weight solvents by methods known in the art.
Pharmaceutical Compositions
The pharmaceutical compositions provided in the present invention include at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent). Preferably, the contemplated pharmaceutical compositions include a compound(s) described herein in an amount sufficient to excrete glucose in urine or inhibit SGLT2 in a subject.
The subjects contemplated include, for example, a living cell and a mammal, including human mammal.
The compound of the present invention may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) and can be formulated in to preparations in solid, semi-solid, liquid or gaseous forms.
Examples of suitable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, polyhydroxyethoxylated castor oil, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, cyclodextrin, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, pentaerythritol fatty acid esters, polyoxyethylene, hydroxymethylcellulose and polyvinylpyrrolidone.
The carrier or diluent may include a sustained release material, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
The pharmaceutical composition may also include one or more pharmaceutically acceptable auxiliary agents, wetting agents, emulsifying agents, suspending agents, preserving agents, salts for influencing osmotic pressure, buffers, sweetening agents, flavoring agents, colorants, or any combination of the foregoing. The pharmaceutical composition of the invention may be formulated so as to provide quick, sustained, or delayed release of the active ingredient after administration to the subject by employing procedures known in the art.
The pharmaceutical compositions described herein may be prepared, e.g., as described in Remington: The Science and Practice of Pharmacy, 20th Ed., 2003 (Lippincott Williams & Wilkins). For example, the active compound can be mixed with a carrier, or diluted by a carrier, or enclosed within a carrier, which may be in the form of an ampoule, capsule, or sachet. When the carrier serves as a diluent, it may be a solid, semi-solid, or liquid material that acts as a vehicle, excipient, or medium for the active compound. The active compound can be adsorbed on a granular solid container, for example, in a sachet.
The pharmaceutical compositions may be, for example, capsules, tablets, aerosols, solutions, suspensions or products for topical application.
The route of administration may be any route which effectively transports the active compound to the appropriate or desired site of action. Suitable routes of administration include, but are not limited to, oral, nasal, pulmonary, buccal, subdermal, intradermal, transdermal, parenteral, rectal, depot, subcutaneous, intravenous, intraurethral, intramuscular, intranasal, ophthalmic (such as with an
ophthalmic solution) or topical (such as with a topical ointment). The oral route is preferred.
Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the like are particularly suitable for oral application. Preferable carriers for tablets, dragees, or capsules include lactose, cornstarch, and/or potato starch. A syrup or elixir can be used in cases where a sweetened vehicle can be employed.
A typical tablet that may be prepared by conventional tabletting techniques may contain: (1) Core: Active compound (as free compound or salt thereof), 250 mg colloidal silicon dioxide (Aerosil®), 1.5 mg microcrystalline cellulose (Avicel®), 70 mg modified cellulose gum (Ac-Di-Sol®), and 7.5 mg magnesium stearate; (2) Coating: HPMC, approx. 9 mg Mywacett 9-40 T and approx. 0.9 mg acylated monoglyceride
Liquid formulations include, but are not limited to, syrups, emulsions, soft gelatin and sterile injectable liquids, such as aqueous or non-aqueous liquid suspensions or solutions.
For parenteral application, particularly suitable are injectable solutions or suspensions, preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil.
Methods of Treatment
The present invention provides compounds and pharmaceutical formulations thereof that are useful in the treatment of diseases, conditions and/or disorders mediated by SGLT2 inhibitors. The connection between therapeutic effect and inhibition of SGLT2 is illustrated. For example in PCT publication Nos. WO 01//016147, WO 02/08306, or WO 03/020737; J. Clin. Invest. Vol. 79, pp. 1510- 1515 (1987); J.Clin. Invest., Vol. 93, pp. 397-404 (1994); Diabetes; 57, 1723-1729, 2008 and references cited therein, all of which are incorporated herein by reference in their entirety and for the purpose stated.
The present patent application further provides a method of treating a disease, condition and/or disorder mediated by SGLT2 inhibitors in a subject in need thereof by administering to the subject a therapeutically effective amount of a compound or a pharmaceutical composition of the present invention.
Diseases, conditions, and/or disorders that are mediated by SGLT2 inhibitors are believed to include, but are not limited to, diabetes, especially type I and type 11 diabetes, including complications of diabetes such as retinopathy, neuropathy, nephropathy and delayed wound healing, and related diseases such as insulin resistance and impaired glucose homeostasis (IGH), hyperglycemia, hyperinsulinemia, elevated blood levels of fatty acids or glycerol, obesity, hyperlipidemia including hypertriglyceridemia, Syndrome X, hypertension, atherosclerosis and related diseases, and for increasing high density lipid levels. The conditions, diseases, and maladies collectively referred to as" Syndrome X" (also known as Metabolic Syndrome) are detailed in Johannsson, J. Clin. Endrocrinol. Metab. , 82, 727-34 (1997) incorporated herein by reference.
The compounds of the present invention can obtain more advantageous effects than additive effects in the prevention or treatment of the above diseases when using suitably in combination with the above drugs. Also, the administration dose can be decreased in comparison with administration of either drug alone, or adverse effects of co administrated drugs other than SGLT2 inhibitors can be avoided or declined.
General Synthesis Methods
The compounds described herein may be prepared by techniques known in the art. In addition, the compounds described herein may be prepared by following the reaction sequence as depicted in Scheme- 1 to 2. Further, in the following schemes, where specific bases, acids, reagents, solvents, coupling agents, etc., are mentioned, it is understood that other bases, acids, reagents, solvents, coupling agents etc., known in the art may also be used and are therefore included within the present invention. Variations in reaction conditions, for example, temperature and/or duration of the reaction, which may be used as known in the art, are also within the scope of the present invention. All the stereo isomers of the compounds in these schemes, unless otherwise specified, are also encompassed within the scope of this invention.
Scheme 1

6
5
Compounds of intermediate 6 (wherein, L is leaving group, for example, halo, methyl sulfonyl, tolyl sulfonyl, 4-nitrobenzyl sulfonyl or the like, a, b, c, d, e, f, R and X are same as defined above) can be prepared as shown in the scheme 1. Cycloalkyl carboxylic acids of formula 2 can be converted to esters of formula 3 by conventional methods. The deprotonation can be carried out under an inert atmosphere at a temperature from -70° C to room temperature and in suitable aprotic organic solvent, for example, tetrahydrofuran or the like, and then R-X can be reacted with the salts of formula 3 or 2 at an initial temperature from -80 °C to- 10 °C either using one or two equivalent of strong base like potassium diisopropylamide, lithium hexamethyl disilazide and preferably, lithium disopropylamide or the like. After a period of stirring at -15°C, the reaction was quenched then the resulted compounds of formula 4 can be isolated and purified by conventional techniques. Compounds of formula 4 can be reduced to give hydroxyl compounds of formula 5 by a suitable reducing agent for example, lithium aluminum hydride or the like. Converting the hydroxy compounds of formula 5 to compounds of intermediate of formula 6 by the methods known in the art for example, reacting hydroxyl group with sulfonyl halides in the presence of a base such as, potassium carbonate, triethylamine, dimethylaminopyridine or the like.


Compounds of formula 11 (formula 1, when R' is H; wherein, a, b, c, d, e, f, R, Z, Z' and X are same as defined above) can be prepared according to the reaction sequences as shown in scheme-2. Compounds of formula 7 can be prepared by following the method described in patent publication US 7,393,836 (as exemplified in the experimental section). For example, benzoyl halides either commercially available or prepared insitu from benzoic acid can be coupled with alkoxyaryl compound using a Lewis acid, typically A1C13 or the like to give corresponding benzophenone product. Reduction of ketone function of benzophenone can be accomplished by trialkyl silane and an appropriate protic or Lewis acid, like trifluoroacetic acid or borontrifluoride etherate, followed by alkyl ether cleavage, using BBr3 or A1C13 to give compounds of formula 7. Alkylation of phenol compounds of formula 7 with intermediate compounds of formula 6 can be obtained in the presence of base such as K2C03, CsC03, NaOH or the like to give compounds of formula 8 in the solvents such as, dimethyl formamide, dimethyl sulfoxide or the like. Compounds of formula 8 can be treated with a base like t-BuLi, n-BuLi to exchange halogen to lithium followed by addition of nascent lithiated aromatic to silyl sugar compounds of formula 9 (as described in Journal of Medicinal Chemistry, 2008, 51, 1 145-1149) in solvents such as, toluene, methane sulfonic acid or the like to give the diastereomeric mixture of lactol compounds, which were converted insitu to the desilylated O-methyl lactol compounds of formula 10 by treatment with methane sulfonicacid in methanol (J. Org. Chem. 1989, 54, 610-612). Reduction of anomeric methoxy group of the desilylated O-methyl lactols
compounds of formula 10 using a reducing agent such as alky silane, for example, triethyl silane or the like in the presence of acid such as BF3.0Et2, trifluroacetic acid, methane sulfonic acid or the like to give the inventive compounds of formula 11 (Formula 1, when R' is H). Finally, the desired diastereoisomer of compounds of formula 11 can be separated by either column chromatography or peracetylation by the methods known in the art for example, acetic anhydride with catalytic amount of dimethylamino pyridine or the like and recrystallization in a suitable solvent by the methods known in the art for example, 20% methanolic ammonia or the like.
Experimental
The present invention is further illustrated by the following examples, which are not to be construed in any way as imposing limitations upon the scope of this disclosure, but rather are intended to be illustrative only. On the contrary, it is to be clearly understood that resort may be had to various other embodiments, modifications, and equivalents thereof which, after reading the description herein, may suggest themselves to one of ordinary skill in the art without departing from the spirit of the present invention. Thus, the skilled artisan will appreciate how the experiments and Examples may be further implemented as disclosed by variously altering the following examples, substituents, reagents, or conditions.
Intermediates
Intermediate 1: Preparation of 4-(5-bromo-2-chlorobenzyl)phenol:

Step 1: Synthesis ofethoxy benzene

Potassium carbonate (264 g, 0.95 mol) was added to a solution of Phenol (60 g, 0.63 mol) in acetone (350 ml) and allowed to stir at room temperature for about 30 minutes then ethyl iodide (77 ml, 0.95 mol) was added drop wise. The reaction mixture was refluxed for about 3 hours and completion of reaction was monitored by TLC. The reaction mixture was filtered and acetone was removed over rotary evaporator. The residue was dissolved in ethyl acetate and washed with 10% NaOH solution to remove any unreacted phenol. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated and filtered
to yield the title compound. Yield: 38 g, 48 %. 1H NMR (CDC13, 300 MHz): 6 7.30- 7.25 (m, 2H); 6.95-6.88 (m, 3H); 4.02 (q, 2H, J = 6.9 Hz); 1.41 (t, 3H, J = 6.9 Hz).
Step 2: Synthesis of(5-bromo-2-chlorophenyl)(4-ethoxyphenyl)methanone:

5-bromo-2-chlorobenzoic acid (50 g) was suspended in dichloromethane (100 ml) and dimethylformamide then it was cooled to 0 °C and to this mixture SOCl2 (92.2 ml) was added slowly. The reaction mixture was slowly allowed to attain room temperature and stirred for about 12 hours. The solvent was removed under reduced pressure to give 5-bromo-2-chlorobenzoyl chloride as oil. Ethoxybenzene (step 1, 25.8 g) was suspended in dichloromethane (50 ml) and cooled to -5 °C then A1C13 (42.3 g) was added slowly by portion wise and above 5- bromo-2-chlorobenzoyl chloride (53.8 g) in dichloromethane (150 ml ) was added drop wise and stirred for about 1 hour at 0 °C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. Ethanol was added to the crude compound at 0 °C and stirred for about 30 minutes. White solid was formed, then filtered off and washed with cold EtOH to afford the title compound as white solid (48 g). Yield: 67.6 ; 1H NMR (CDC13, 300 MHz): δ 7.76 (dd, J = 1.5, 6.9 Hz, 2H), 7.55- 7.47 (m, 2H), 7.32 (d, J = 8.4 Hz, 1H), 6.93 (d, J = 9 Hz, 2H), 4.11 (q, / = 6.9 Hz, 2H), 1.45 (t, J = 6.9 Hz, 3H).
Step 3: Synthesis of(5-bromo-2-chlorophenyl)(4-hydroxyphenyl)methanone:

To a stirred solution of (5-bromo-2-chlorophenyl)(4- ethoxyphenyl)methanone (step 2, 48 g) in dichloromethane (150 ml) at 0°C, A1C13 (188.8 g) was added under N2 atmosphere by portion wise over 30 minutes. Subsequently, the reaction mixture was stirred at room temperature for about 12
hours. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. Ethanol was added to the crude compound at 0 °C and stirred for about 30 minutes. White solid was formed, then filtered off and washed with cold EtOH to afford the title compound as white solid (41 g). Yield: 93 %; 1H NMR (CDC13, 300 MHz): δ 8.50 (s, 1H), 6.43 (d, J = 8.7 Hz, 2H), 6.28- 6.20 (m, 2H), 6.06 (d, J = 8.4 Hz, 1H), 5.64 (d, J = 8.7 Hz, 2H); ES Mass: (M+H) 310.85.
Step 4: Synthesis of 4-(5-bromo-2-chlorobenzyl)phenol:
To a stirred solution of (5-bromo-2-chlorophenyl)(4- hydroxyphenyl)methanone (step 3, 41 g) in dichloromethane (150 ml) and acetylnitrile (1: 1), Et3SiH (53 ml) was added at 0 °C under N2 atmosphere followed by BF3: Et20 (25 ml) and the reaction was stirred for about 16 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water, brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. Hexane was added to the crude compound at 0 °C and stirred for about 30 minutes. White solid was formed and filtered off to afford the title compound as white solid (37 g). Yield: 94.8 ; 1H NMR (CDC13, 300 MHz): δ 7.27- 7.24 (m, 3H), 7.06 (d, J = 8.4 Hz, 2H), 6.78 (d, / = 8.4 Hz, 2H), 4.69 (s, 1H), 3.98 (s, 2H); ES Mass: (M+H) 296.85
Intermediate 2: Preparation of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-

To a stirred solution of (3R,4S,5S,6R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-one (50 g) in tetrahydrafuran (1.3 L), NMM (N-methyl morpholine, 214 ml) and TMSC1 (trimethyl silyl chloride, 247 ml) were
added at 0 °C under N2 atmosphere and the reaction mixture temperature was slowly raised to about 50 °C and stirred for about 5 to 6 hours. After completion of the reaction (monitored by TLC), toluene and water were added to the reaction mixture and organic layer was separated. Aqueous layer was again extracted with toluene. The combined organic layers were washed with potassium dihydrogen phosphate, water and brine, dried over anhydrous sodium sulphate and concentrated to give the title compound as liquid (120 g). Yield: 91.6 ; 1H NMR (CDC13, 300 MHz): δ 4.19- 4.15 (m, 1H), 3.99 (d, J = 8.1 Hz, 1H), 3.91 (dd, J = 7.5, 7.5 Hz, 1H ), 3.84- 3.72 (m, 3H), 0.20- 0.13 (m, 36H); ES Mass: (M+ Na) 489.
Intermediate 3: Preparation of 4-bromo-l-chloro-2-(4-((l-ethylcvclopentyl) methoxy)benzyl) benzene:

Step-1: Synthesis of Ethyl cyclopentanecarboxylate:

Concentrated H2S04 (43 ml, 1.0 equalent) was added drop-wise to a solution of cyclopentanecarboxylic acid (50 g, 0.438 mol) in EtOH (500 ml) at 0°C and the reaction mixture was refluxed for about 48 hours. After the completion of the reaction, the solvent was removed and the reaction mixture was dissolved in dichloromethane. The dichloromethane layer was washed with saturated NaHC03 solution and the organic layer was dried over anhydrous magnesium sulphate. Dichloromethane was removed over rotary evaporator without vaccum and the resulting residue of the title compound was used directly for the next step. Yield: 38 gm, 61 ; 1H NMR (CDC13, 300 MHz): δ 4.11 (q, 2H, J = 7.2 Hz); 2.75-2.65 (m, 1H); 1.89-1.56 (m, 8 H); 1.25 (t, 3H, J = 7.2 Hz). ESI Mass: 143 (M+l).
Step 2: Synthesis of Ethyl 1 -ethylcyclopentanecarboxylate:
 To a solution of diisopropylamine (93.6 ml, 0.688 mol) in tetrahydrafuran (200 ml), n-BuLi (2.5M, 213.7 ml, 0.534 mol) in hexane was added at 0°C and the reaction mixture was allowed to stir at same temperature for about 10-15 minutes. The reaction mixture was stirred at room temperature for about 45 minutes. Ethyl cyclohexanecarboxylate (step 1, 38 g, 0.267 mol) in tetrahydrafuran (150 ml) was added drop-wise at -78°C to LDA (reaction mixture). The reaction mixture was stirred at same temperature for about 2 hours and then ethyl iodide (32.06 ml, 0.400 mol) was added drop-wise. The reaction mixture was maintained at -78°C for an hour and allowed to attain room temperature. Reaction mixture was stirred overnight at room temperature and quenched at 0°C with saturated NH4C1 solution. The solvent was concentrated over vaccum and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 then concentrated and the resulted crude product of title compound was directly used for the next step. Crude yield: 32 gm, 70 %; 1H NMR (CDC13, 300 MHz): δ 4.11 (q, 2H, J = 7.2 Hz); 2.13-1.41 (m, 13H); 1.21 (t, 3H, J = 7.2 Hz); 0.81 (t, 3H, J = 7.5 Hz). ESI Mass: 171 (M+l).
To a solution of diisopropylamine (93.6 ml, 0.688 mol) in tetrahydrafuran (200 ml), n-BuLi (2.5M, 213.7 ml, 0.534 mol) in hexane was added at 0°C and the reaction mixture was allowed to stir at same temperature for about 10-15 minutes. The reaction mixture was stirred at room temperature for about 45 minutes. Ethyl cyclohexanecarboxylate (step 1, 38 g, 0.267 mol) in tetrahydrafuran (150 ml) was added drop-wise at -78°C to LDA (reaction mixture). The reaction mixture was stirred at same temperature for about 2 hours and then ethyl iodide (32.06 ml, 0.400 mol) was added drop-wise. The reaction mixture was maintained at -78°C for an hour and allowed to attain room temperature. Reaction mixture was stirred overnight at room temperature and quenched at 0°C with saturated NH4C1 solution. The solvent was concentrated over vaccum and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 then concentrated and the resulted crude product of title compound was directly used for the next step. Crude yield: 32 gm, 70 %; 1H NMR (CDC13, 300 MHz): δ 4.11 (q, 2H, J = 7.2 Hz); 2.13-1.41 (m, 13H); 1.21 (t, 3H, J = 7.2 Hz); 0.81 (t, 3H, J = 7.5 Hz). ESI Mass: 171 (M+l).
Step 3: Synthesis of (l-ethylcyclopentyl)methanol:

Ethyl 1-ethylcyclopentanecarboxylate (step 2, 32 g, 0.187 mol) in tetrahydrafuran (120 ml) was added to a suspension of LiAlH4 (21.4 g, 0.563 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate and the organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 then concentrated and the resulted crude product of title compound was directly used for the next step. Crude yield: 24 gm, 100 %. 1H NMR (CDC13, 300 MHz): δ 3.93 (s, 2H); 1.59-1.36 (m, 10H); 0.85 (t, 3H, J = 7.5 Hz).
Step 4: Synthesis of (l-ethylcyclopentyl)methyl 4-methylbenzenesulfonate:

To a solution of (l-ethylcyclopentyl)methanol (step 3, 26 g, 0.202 mol) in dichloromethane (400 ml), triethyl amine (141.3 ml, 1.01 mol) was added followed by tosyl chloride (77.3 g, 0.405 mol) at 0°C. After addition of catalytic amount of DMAP (2.47 g, 0.020 mol), the reaction mixture was allowed to stir overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water, saturated brine solution and the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 27 gm, 47 %. 1H NMR (CDC13, 300 MHz): δ 7.79 (d, 2H, J = 8.1 Hz); 7.35 (d, 2H, J = 8.1 Hz); 3.76 (s, 2H); 2.45 (s, 3H); 1.54- 1.35 (m, 10H); 0.72 (t, 3H, / = 7.5 Hz).
Step 5: Synthesis of4-bromo-l-chloro-2-(4-((l-ethylcyclopentyl) methoxy)benzyl) benzene:
To a suspension of Cs2C03 (89.20 g, 0.273 mol) in dimethylformamide (300 ml) 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate 1, 27 g, 0.091 mol) in dimethylformamide (100 ml) was added followed by (l-ethylcyclopentyl)methyl 4- methylbenzenesulfonate (step 4, 38.5 g, 0.136 mol) in dimethylformamide (100 ml) and the reaction mixture was stirred overnight at 120°C. After completion of the reaction as monitored by TLC, the reaction mixture was acidified with 10% HC1 solution. Compound was extracted with ether and washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated then the obtained product was purified by column chromatographic technique. Yield: 28 gm, 75 %. 1H NMR (CDC13, 300 MHz): δ 7.37-7.24 (m, 3H); 7.02-6.98 (m, 2H); 6.78-6.49 (m, 2H); 4.17-4.15 (m, 1H); 3.90 (m, 2H); 3.76-3.74 (m, 1H); 3.56 (s, 3H); 3.48-3.43 (m, 1H); 2.86 (s, 2H); 2.81 (s, 1H); 1.48-1.31 (m, 10H); 0.71 (t, 3H, J = 7.2 Hz). ESI Mass: 543.20 (M+Na).
Intermediate 4: Preparation of (2S.3Rt4S,5S,6RV2-(4-chloro-3-(4-(f l- ethylcyclopentyl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl)-2-methoxytetrahvdro- 2H-pyran-3.4,5-triol (β-isomer and (2R,3R,4S,5S,6R -2-(4-chloro-3-(4-((l-
emylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydro^
2H-pyran-3,4,5-tri -isomer):

T-BuLi (1.7 M, 17.4 ml, 29.42 mmol) was added drop-wise at -78°C to a solution of intermediate 3 (6 g, 14.71 mmol) in tetrahydrafuran: toluene (90 ml) (1:2). After 20 minutes, the lithiated derivative was added drop- wise to a pre-cooled solution (at -78°C) of silyl sugar (Intermediate 2, 6.88 g, 14.71 mmol) in toluene (30 ml). The reaction mixture was maintained at the same temperature for about 2 hours and quenched with methane sulfonic acid (6 % in MeOH, 90 ml). The reaction mixture was stirred at room temperature for an hour, then the reaction mixture was quenched with saturated NaHC03 solution and diluted with ethyl acetate. The organic layer was washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained diastereomeric products were separated by column chromatographic technique. 1H NMR (CDC13, 300 MHz): β-isomer: δ 7.33-7.28 (m, 3H); 7.05 (d, 2H, J = 8.4 Hz); 6.79 (d, 2H, J = 8.7 Hz); 4.10-3.88 (m, 7H); 3.62 (s, 4H); 3.21-3.18 (m, 2H); 2.99 (s, 2H); 2.53 (bs, 1H); 1.74-1.40 (m, 13H); 0.83 (t, 3H, J = 7.5 Hz). ESI Mass: 543.30 (M+Na).
oc-isomer: 6 7.40-7.29 (m, 3H); 7.08 (d, 2H, J = 8.4 Hz); 6.86-6.82 (m, 2H); 4.49 (t, 1H, / = 5.7 Hz); 4.18-4.14 (m, 1H); 4.04-3.77 (m, 7H); 3.66 (s, 3H); 3.31 (bs, 1H); 3.18 (bs, 1H); 3.08 (s, 2H); 2.94-2.91 (m, 1H); 2.30 (bs, 1H); 2.21 (bs, 1H); 1.64- 1.44 (m, 10H); 0.85 (t, 3H, J = 7.2 Hz).
Intermediate 5j Preparation of 4-bromo- 1 -chloro-2-( 4-(( 1 - ethylcvclobutyl)methoxy)benzyl) benzene:

Step 1: Synthesis of Methyl cyclobutanecarboxylate:
 Concentrated H2S04 (39.2 ml, 1.0 eq) was added drop-wise to a solution of cyclobutanecarboxylic acid (40 g, 0.438 mol) in MeOH (200 ml) at 0°C and the reaction mixture was refluxed for about 36 hours. After completion of the reaction as monitored by TLC, the solvent was removed and the reaction mixture was dissolved in dichloromethane. The dichloromethane layer was washed with saturated NaHC03 solution and the organic layer was dried over anhydrous magnesium sulphate, dichloromethane was removed over rotary evaporator. The residue was used directly for the next step. Yield: 30 gm, 61.2 %. 1H NMR (CDC13, 300 MHz): δ 3.67 (s, 3H),17-3.11 (m, 1H) 2.32-2.16 (m, 4H), 1.92-1.90 (m, 2H).
Concentrated H2S04 (39.2 ml, 1.0 eq) was added drop-wise to a solution of cyclobutanecarboxylic acid (40 g, 0.438 mol) in MeOH (200 ml) at 0°C and the reaction mixture was refluxed for about 36 hours. After completion of the reaction as monitored by TLC, the solvent was removed and the reaction mixture was dissolved in dichloromethane. The dichloromethane layer was washed with saturated NaHC03 solution and the organic layer was dried over anhydrous magnesium sulphate, dichloromethane was removed over rotary evaporator. The residue was used directly for the next step. Yield: 30 gm, 61.2 %. 1H NMR (CDC13, 300 MHz): δ 3.67 (s, 3H),17-3.11 (m, 1H) 2.32-2.16 (m, 4H), 1.92-1.90 (m, 2H).
Step 2: Synthesis of methyl 1-ethylcyclobutanecarboxylate:

To a solution of diisopropylamine (110.6ml) and tetrahydrafuran (200ml), was added n-BuLi (2.5M, 263.7 ml, 0.6578 mol) in hexane at 0°C and the reaction mixture was allowed to stir at same temperature for about 10-15 minutes. The reaction mixture was stirred at room temperature for about 45 minutes. Methyl cyclobutanecarboxylate (step 1, 30 g, 0.263 mol) in tetrahydrafuran (150 ml) was added drop-wise at -78°C to LDA (reaction mixture). The reaction mixture was stirred at same temperature for about 2 hours then neat ethyl iodide (31.56 ml, 0.3947 mol) was added drop-wise. The reaction was maintained at -78°C for an hour and allowed to attain room temperature. Reaction mixture was stirred overnight at room temperature and quenched at 0°C with saturated NH4C1 solution. The solvent was concentrated over vaccum and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution and then dried over anhydrous Na2S04 and concentrated then the obtained crude product was directly used for the next step. Crude yield: 32 gm, 70 %. 1H NMR (CDC13, 300 MHz): 6 3.67 (s, 3H); 2.40-2.36 (q, 2H); 1.89-1.74 (m, 6H); 0.81- 0.76(t, 3H, J = 7.5 Hz).
Step 3: Synthesis of (l-ethylcyclobutyl)methanol:
 Methyl 1-ethylcyclopentanecarboxylate (step 2, 25 g, 0.176 mol) in tetrahydrafuran (120 ml) was added to a suspension of L1AIH4 (21.4 g, 0.563 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at the same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. Compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used for the next step. Crude yield: 24 gm, 100 %. 1H NMR (CDC13, 300 MHz): 6 3.77-3.72 (t, 1H); 3.53(s, 2H) 1.87-1.68 (m, 4H); 1.55-1.48 (q, 2H, J=7.5Hz), 0.84-0.79 (t, 3H, J=7.5 Hz). ESI Mass: (M-l) 113.
Methyl 1-ethylcyclopentanecarboxylate (step 2, 25 g, 0.176 mol) in tetrahydrafuran (120 ml) was added to a suspension of L1AIH4 (21.4 g, 0.563 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at the same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. Compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used for the next step. Crude yield: 24 gm, 100 %. 1H NMR (CDC13, 300 MHz): 6 3.77-3.72 (t, 1H); 3.53(s, 2H) 1.87-1.68 (m, 4H); 1.55-1.48 (q, 2H, J=7.5Hz), 0.84-0.79 (t, 3H, J=7.5 Hz). ESI Mass: (M-l) 113.
Step 4: Synthesis of (l-ethylcyclobutyl)methyl 4-methylbenzenesulfonate:

To a solution of (l-ethylcyclobutyl)methanol (step 3, 15 g, 0.131 mol) in dichloromethane (300 ml), triethyl amine (73.2 ml, 0.526 mol) was added followed by tosyl chloride (49.9 g, 0.26 mol) at 0°C. After addition of catalytic amount of DMAP (1.6 g, 0.0131 mol), the reaction mixture was allowed to stir overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water, saturated brine solution and the organic layer was dried over anhydrous Na2S04 and concentrated then the obtained product was purified by column chromatographic technique. Yield: 27 gm, 47 %. 1H NMR (CDC13, 300 MHz): δ 7.81-7.79 (d, 2H, J = 8.4 Hz); 7.36 (d, 2H, J = 8.4 Hz); 3.9 (s, 2H); 2.45 (s, 3H); 1.80-1.71 (m, 6H); 1.53-1.45 (q, 2H), 0.70-0.65 (t, 3H, J=l .5 Hz).
Step 5: Synthesis of 4-bromo-l-chloro-2-(4-((l-ethylcyclobutyl)methoxy)benzyl) benzene:
To a suspension of intermediate 1 (8g, 0.027 mol) in dimethylformamide (60 ml), Cs2C03 (26.4 g, 0.081 mol) was added followed by (l-ethylcyclobutyl)methyl 4- methylbenzene sulfonate (step 4, 10.8 g, 0.0405 mol) in dimethylformamide (60 ml). The reaction mixture was stirred overnight at 120°C. After completion of the
reaction as monitored by TLC, the reaction mixture was acidified with 10% HC1 solution. Compound was extracted with ether and washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 28 gm, 75 . %. 1H NMR (CDC13, 300 MHz): 6 7.26-7.24 (m, 3H); 7.11-7.08 (d, 2H, J=8.7Hz); 6.89-6.86 (d, 2H, 8.7Hz); 4.00 (s, 2H), 3.82 (s, 2H), 1.95-1.85 (m, 6H); 1.82-1.80 (m, 2H); 1.68-1.61 (q, 2H, J=7.5Hz); 0.86-0.81 (t, 3H, 7 = 7.2 Hz).
Intermediate 6: Preparation of (2S.3R,4S.5S,6R -2-(,4-chloro-3-('4-(('l- ethylcyclobutyl methoxy)benzyl)phenyl)-6-(hvdroxymethyl)-2-methoxy-tetrahydro- 2H-pyran-3,4,5-triol:

T-BuLi (1.7 M, 15.6 ml, 0.0265 mol) was added drop-wise at -78°C to a solution of Intermediate 5 (5.2 g, 0.0132mol) in tetrahydrafuran: toluene (90 ml) (1:2). After 20 minutes, the lithiated derivative was added drop- wise to a pre-cooled solution (at -78°C) of silyl sugar (Intermediate 2, 4.94 g, 0.0106 mol) in toluene (30 ml). The reaction mixture was maintained at the same temperature for about 2 hours and quenched with methane sulfonic acid (6 % in MeOH, 78 ml). The reaction mixture was stirred at room temperature for three hours then the reaction mixture was quenched with saturated NaHC03 solution and diluted with ethyl acetate. The organic layer was washed with brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was separated by column chromatographic technique. 1H NMR (CDCI3, 300 MHz): δ 7.52 (s, 1H), 7.39 (s, 2H); 7.09-7.06 (d, 2H, J=8.4Hz); 6.87 -6.85 (d, 2H, 7=8.4 Hz); 5.00-4.98 (d, 1H, 7 = 5.4 Hz); 4.81-4.75 (m, 2H), 4.57-4.53 (m, 1H), 4.57-4.53 (q, 2H, J=6.3Hz), 4.01 (s, 2H), 3.98 (m, 1H); 3.80-3.73 (m, 3H), 3.23-3.21 (m, 1H), 2.92 (s, 3H), 2.88-2.85 (m, 1H); 1.86-1.73 (m, 6H); 1.60-1.53 (q, 2H, J=7.5Hz); 0.80-0.75 (t, 3H, 7=7.5 Hz). ESI Mass: 529 (M+Na).
Intermediate 7: Preparation of 4-bromo- 1 -chloro-2-(4-(( 1 - ethylcyclohexyl)methoxy)benzyl)benzene:

Step 1: Synthesis of methyl cyclohexanecarboxylate:

Concentrated H2S04 (19.1 ml, 1.0 eq) was added drop- wise to a solution of cyclohexanecarboxylic acid (25 g, 0.1953 mol) in MeOH (125 ml) at 0°C and the reaction mixture was refluxed for about 48 hours. After completion of the reaction, the solvent was removed and the reaction mixture was dissolved in dichloromethane. The organic layer was washed with saturated NaHC03 solution and the organic layer was dried over anhydrous magnesium sulphate. Dichloromethane was removed over rotary evaporator. The obtained residue with least moisture content (mc) was used directly for the next step. Yield: 16 gm.
Step 2: Synthesis of methyl 1-ethylcyclohexanecarboxylate:

To a solution of diisopropylamine (44.4 ml, 0.3169 mol) in tetrahydrafuran (200 ml), was added n-BuLi (2.5M, 105.6 ml, 0.264 mol) in hexane at 0°C and the reaction mixture was allowed to stir at same temperature for about 10-15 min. The reaction mixture was stirred at room temperature for about 45 min. methyl cyclohexanecarboxylate (step 1, 15 g, 0.15067 mol) in tetrahydrafuran (150 ml) was added drop-wise at -78°C to LDA (reaction mixture). The reaction mixture was stirred at same temperature for about 2 h and then neat ethyl iodide (12.67 ml, 0.1584 mol) was added drop-wise. The reaction was maintained at -78°C for an hour and allowed to attain room temperature. The reaction mixture was stirred overnight at room temperature and quenched at 0°C with saturated NH4C1 solution. The solvent was concentrated over vaccum and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used
for the next step. Crude yield: 10 gm. Ή NMR (CDC13, 300 MHz): δ 4.11 (q, 2H, J = 7.2 Hz); 2.13-1.41 (m, 13H); 1.21 (t, 3H, J = 7.2 Hz); 0.81 (t, 3H, 7 = 7.5 Hz). ESI Mass: 171 (M+l).
Step 3: Synthesis of (l-ethylcyclohexyl)methanol:

Methyl 1-ethylcyclohexanecarboxylate (step 2, 10 g, 0.0588 mol) in tetrahydrafuran (120 ml) was added to a suspension of L1AIH4 (6.7 g, 0.1764 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at the same temperature for about 30 min. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used for the next step. Crude yield: 8 gm. 1H NMR (CDC13, 300 MHz): 6 3.41 (s, 2H); 1.42-1.36 (m, 12H); 1.30-1.26 (t, 3H).
Step 4: Synthesis of (l-ethylcyclohexyl)methyl 4-methylbenzenesulfonate:

To a solution of (l-ethylcyclohexyl)methanol (step 3, 8 g, 0.0563 mol) in dichloromethane (200 ml), triethyl amine (31.3 ml, 0.2253 mol) was added followed by tosyl chloride (21.4 g, 0.1126 mol) at 0°C. After addition of catalytic amount of DMAP (0.567 g, 0.0056 mol), the reaction mixture was allowed to stir for overnight at room temperature. After completion of reaction, the reaction mixture was washed with water, saturated brine solution; the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 8 gm. 1H NMR (CDC13, 300 MHz): δ 7.80-7.77 (d, 2H, J = 8.1 Hz); 7.35-7.33 (d, 2H, J = 8.1 Hz); 3.78 (s, 2H); 2.45 (s, 3H); 1.39- 1.27 (m, 12H); 0.68-0.63 (t, 3H, J = 7.5 Hz). ESI Mass: 319.17 (M+Na).
Step 5: Synthesis of4-bromo-l-chloro-2-(4-((l- ethylcyclohexyl )methoxy )benzyl )benzene:

To a suspension of Cs2C03 (39.6 g, 0.1216 mol) in dimethylformamide (150 ml), Intermediate 1 (12 g, 0.0405 mol) in dimethylformamide (100 ml) was added followed by (l-ethylcyclohexyl)methyl 4-methylbenzenesulfonate (step 4, 17.9 g, 0.0608 mol) in dimethylformamide (50 ml) then the reaction mixture was stirred for overnight at 120°C. After completion of the reaction as monitored by TLC, the reaction mixture was extracted with ether and washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 8.5 gm. 1H NMR (CDC13, 300 MHz): δ 7.27-7.23 (m, 3H); 7.09-7.06 (d, 2H, J=8.7Hz); 6.86- 6.83 (d, 2H, J=8.7Hz); 3.98 (s, 3H); 3.67 (s, 2H), 1.45-1.40 (m, 12H); 0.88-0.77 (t, 3H, J = 7.8 Hz).
Intermediate 8: Preparation of (2S.3R.4S.5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl)methoxy benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro- 2H-pyran-3A5-triol (β-isomer) & (2R,3R.4S,5S.6R)-2-(4-chloro-3-(4-(il- ethylcvclohexyl)methoxy benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro- 2H-pyran-3A5-triol oc-isomer):

T-BuLi (1.7 M, 23.8 ml, 0.0404mol) was added drop-wise to a solution of 4- bromo- 1 -chloro-2-(4-(( 1 -ethylcyclohexyl)methoxy)benzyl)benzene (Intermediate 7, 8.5 g, 0.0202 mol) in tetrahydrafuran: toluene (150 ml, 1:2) at -78°C. After 20 minutes, the lithiated derivative was added drop- wise to a pre-cooled solution (at - 78°C) of silyl sugar (Intermediate 2, 7.54 g, 0.0161mol) in toluene (70 ml). The reaction was maintained at the same temperature for about 2 hours then it was quenched with methane sulfonic acid (6 % in MeOH, 127.5 ml). The reaction
mixture was stirred at room temperature for an hour then the reaction mixture was quenched with saturated NaHC03 solution and diluted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. Diastereomeric products were separated by column chromatographic technique. 1H NMR (CDC13, 300 MHz): oc-isomer: 6 7.39-7.29 (m, 3H); 7.08 -7.06 (m, 2H); 6.85-6.81 (m, 2H); 4.49-4.17 (m, 3H); 4.16-3.97 (m, 6H); 3.67 (s, 2H); 3.11-2.93 (d, 2H); 1.51-1.36 (m, 10H), 0.81-0.76 (t, 3H).
β-isomer: 5 7.48 (s, 1H), 7.34 (s, 3H), 7.04-7.01 (d, 2H, J=8.7Hz); 6.82- 6.79 (d, 2H, J = 8.7 Hz); 4.96-4.94 (d, 2H, J=5.4Hz), 4.77-4.72 (m, 1H), 4.54-4.50 (m, 1H), 4.01- 3.88 (q, 2H); 3.71 (m, 1H), 3.67 (s, 2H); 3.56-3.45 (m, 3H); 3.19-3.13 (m, 1H); 2.88 (s, 3H); 2.85-2.82 (m, 1H), 1.40 -1.27 (m, 10H); 0.73-0.68 (t, 3H, J = 7.2 Hz). ESI Mass: 557.30 (M+Na).
Intermediate 9j Preparation of 4-bromo- 1 -chloro-2-(4-(( 1 - methylcyclohexyI)methoxy)benzyl)benzene:

Step 1: Synthesis of methyl cyclohexanecarboxylate:

Concentrated H2S04 (19.1 ml, 1.0 eq) was added drop-wise to a solution of cyclohexanecarboxylic acid (25 g, 0.1953 mol) in MeOH (125 ml) at 0°C and the reaction mixture was refluxed for about 48 hours. After completion of the reaction, the solvent was removed and the reaction mixture was dissolved in dichloromethane. The organic layer was washed with saturated NaHC03 solution and the organic layer was dried over anhydrous magnesium sulphate. The obtained residue with least moisture content (mc) was used directly for the next step. Yield: 16gm.
Step 2: Synthesis of methyl 1 -methylcyclohexanecarboxylate:
O
OMe
To a solution of diisopropylamine (44.3 ml, 0.3169 mol) in tetrahydrafuran (200 ml), n-BuLi (2.5M, 106 ml, 0.264 mol) in hexane was added at 0°C and the reaction mixture was allowed to stir at same temperature for about 10-15 min. The reaction mixture was stirred at room temperature for about 45 minutes; methyl cyclohexanecarboxylate (step 1, 15 g, 0.15067 mol) in tetrahydrafuran (150 ml) was added drop-wise at -78°C to LDA (reaction mixture). The reaction mixture was stirred at same temperature for about 2 h and then neat methyl iodide (13.6 ml, 0.2112 mol) was added drop-wise. The reaction was maintained at -78°C for an hour and allowed to attain room temperature. Reaction mixture was stirred overnight at room temperature and quenched at 0°C with saturated NH4C1 solution. The solvent was concentrated over vaccum and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution and then dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used for the next step. Crude yield: 15 gm. Ή NMR (CDC13, 300 MHz): 3.65 (s, 3H); 2.33-2.26 (m, 1H); 1.91-1.87 (m, 2H, / = 7.2 Hz); 1.76-1.65 (m, 3H), 1.45-1.30 (m, 3H), 1.13 (s, 3H).
Step 3: Synthesis of (l-methylcyclohexyl)methanol:

Methyl 1-methylcyclohexanecarboxylate 3 (15 g, 0.0961 mol) in tetrahydrafuran (100 ml) was added to a suspension of LiAlH4 (7.3 g, 0.1923 mol) in tetrahydrafuran (100 ml) at 0°C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used for the next step. Crude yield: 9 gm. Ή NMR (CDC13, 300 MHz): δ 3.34 (s, 2H); 1.49-1.44 (m, 4H); 1.31-1.25 (m, 6H), 0.91 (s, 3H).
Step 4: Synthesis of (l-methylcyclohexyl)methyl 4-methylbenzenesulfonate :

To a solution of (l-methylcyclohexyl)methanol (step 3, 9 g, 0.0775 mol) in dichloromethane (100 ml), triethyl amine (64.7 ml, 0.2253 mol) was added followed by tosyl chloride (36.85 g, 0.1939 mol) at 0°C. After addition of catalytic amount of DMAP (0.71 g, 0.00703 mol), the reaction mixture was allowed to stir for overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water, saturated brine solution, and the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 15 gm. 1H NMR (CDC13, 300 MHz): δ 7.80-7.77 (d, 2H, J = 7.8 Hz); 7.36-7.33 (d, 2H, J = 8.1 Hz); 3.72 (s, 2H); 2.45 (s, 3H); 1.40- 1.34 (m, 6H); 1.28-1.24 (m, 4H), 0.88 (s, 3H).
Step 5: Synthesis of 4-bromo-l-chloro-2-(4-((l- methylcyclohexyl )methoxy )benzyl )benzene:

To a suspension of Cs2C03 (36.5 g, 0.112 mol) in dimethylformamide (150 ml), Intermediate 1 (11.05 g, 0.037 mol) in dimethylformamide (100 ml) was added followed by (l-methylcyclohexyl)methyl 4-methylbenzenesulfonate (step 4, 15.8 g, 0.056 mol) in dimethylformamide (50 ml) and the reaction mixture was stirred overnight at 120°C. After completion of reaction as monitored by TLC, the reaction mixture was extracted with ether and washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 4 gm. Ή NMR
(CDC13, 300 MHz): 6 7.23-7.22 (m, 3H); 7.09-7.06 (d, 2H, J=8.7Hz); 6.83 (d, 2H, J=8.7Hz); 3.97 (s, 2H); 3.62 (s, 2H), 1.49-1.36 (m, 10H); 1.02 (s, 3H).
Intermediate 10: Preparation of (2S,3R,4S.5S.6R)-2-(4-chloro-3-(4-((l- methylcvclohexyl)methoxy)benzyl phenyl)-6-(hydroxymethyl)-2- methoxytetrahvdro-2H-pyran-3,4,5-triol:

T-BuLi (1.7 M, 20.2 ml, 0.0344mol) was added drop-wise to a solution of 4- bromo- 1 -chloro-2-(4-(( 1 -methylcyclohexyl)methoxy)benzyl)benzene (Intermediate 9, 7 g, 0.0172mol) in tetrahydrafuran: toluene (90 ml) (1:2) at -78°C. After 20 minutes, the lithiated derivative was added drop-wise to a pre-cooled solution (at - 78°C) of silyl sugar (Intermediate 2, 6.4 g, 0.0137mol) in toluene (70 ml). The reaction mixture was maintained at the same temperature for about 2 hours then quenched with methane sulfonic acid (6 % in MeOH, 105 ml). The reaction mixture was stirred at room temperature for an hour then quenched with saturated NaHC03 solution and diluted with ethyl acetate. The organic layer was washed with brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was separated by column chromatographic technique. Ή NMR (CDC13, 300 MHz): δ 7.48 (s, 1H), 7.34 (s, 2H), 7.04-7.01 (d, 2H, J=8.4Hz); 6.81- 6.78 (d, 2H, / = 8.7Hz); 5.06 (bs, 1H), 4.82 (bs, 2H), 4.57 (bs, 1H), 3.96-3.93 (q, 2H); 3.67 (m, 1H), 3.62 (s, 3H); 3.58-3.50 (m, 2H); 3.18-2.87 (m, 3H); 1.40-1.25 (m, 6H); 1.19-1.16 (m, 4H), 0.9 (s, 3H); ESI Mass: 543.30 (M+Na).
Intermediate 11: Preparation of 3-((4-(5-bromo-2-chlorobenzyl)phenoxy)methyl)-3- ethyloxetane:

Step 1: Synthesis of (3-ethyloxetan-3-yl)methyl 4 -methylbenzene sulfonate:

To a solution of (3-ethyloxetan-3-yl)methanol (15 g, 0.129 mol) in dichloromethane (300 ml), triethyl amine (144 ml, 1.0344 mol) followed by tosyl chloride (123 g, 0.646 mol) was added at 0°C. After addition of catalytic amount of DMAP (16 g, 0131 mol), the reaction mixture was allowed to stir overnight at room temperature. After completion of the reaction as monitored by TLC, the reaction
mixture was washed with water, saturated brine solution; the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 15 gm. 1H NMR (CDC13, 300 MHz): δ 7.83-7.80 (d, 2H, J = 8.4 Hz); 7.39- 7.36 (d, 2H, J = 8.4 Hz); 4.38-4.36 (d, 2H, J=6.3Hz), 4.31-4.29 (d, 2H, J=6.3Hz), 4.17 (s, 2H), 2.47 (s, 3H), 1.78-1.71 (q, 2H), 0.83-0.78 (t, 3H).
Step 2: Synthesis of3-((4-(5-bromo-2-chlorobenzyl)phenoxy)methyl)-3- ethyloxetane:

To a suspension of Cs2C03 (32.8 g, 0.1010 mol) in dimethylformamide (60 ml), Intermediate 1 (10 g, 0.033 mol) in dimethylformamide (100 ml) was added followed by (3-ethyloxetan-3-yl)methyl 4-methylbenzenesulfonate (step 1, 9 g, 0.033 mol) in dimethylformamide (50 ml) and the reaction mixture was stirred for overnight at 100°C. After completion of reaction as monitored by TLC, the reaction mixture was extracted with ether and washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 8 gm. !H NMR (CDC13, 300 MHz): δ 7.30-7.21 (m, 3H); 7.12-7.09 (d, 2H, J=8.4Hz); 6.90-6.87 (d, 2H, 8.4Hz); 4.58-4.56 (d, 2H, J=5.7Hz), 4.49-4.47 (d, 2H, J=5.7), 4.06 (s, 2H), 4.00 (s, 2H), 1.91-1.84 (q, 2H); 0.95-0.90 (t, 3H).
Intermediate 12: Preparation of (2S,3R.4S.5S.6R)-2-(4-chloro-3-(4-((3-ethyloxetan-
3-yl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahvdro-2H-pyran-
3A5-triol:

T-BuLi (1.7 M, 23.8 ml, 0.0406mol) was added drop-wise to a solution of 3- ((4-(5-bromo-2-chlorobenzyl)phenoxy)methyl)-3-ethyloxetane (Intermediate 11, 8 g, 0.0203mol) in tetrahydrafuran: toluene (90 ml) (1:2) at -78°C. After 20 minutes,
the lithiated derivative was added drop-wise to a pre-cooled solution (at -78°C) of silyl sugar (Intermediate 2, 7.6 g, 0.0162 mol) in toluene (60 ml). The reaction mixture was maintained at the same temperature for about 2 hours then quenched with methane sulfonic acid (6 % in MeOH, 120 ml). The reaction mixture was stirred at room temperature for three hours then it was quenched with saturated NaHC03 solution and diluted with ethyl acetate. The organic layer was washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was separated by column chromatographic technique. 1H NMR (DMSO-D6, 300 MHz): δ 7.48 (s, 1H), 7.35 (s, 2H), 7.05-7.02 (d, 2H, J=8.7Hz); 6.83-6.80 (d, 2H, J = 8.7Hz); 4.96-4.95 (d, 1H, J = 5.4 Hz); 4.79- 4.52 (m, 3H), 3.97 (m, 1H); 3.94-3.70 (m, 4H), 3.67-3.46 (m, 3H), 3.41 (s, 1H), 3.21-3.16 (m, 1H), 2.87-2.81 (m, 3H); 1.40-1.38 (q, 2H); 0.82-0.77 (t, 3H, J = 7.5 Hz). ESI Mass: 531 (M+Na).
Intermediate 13: Preparation of (2S,3R,4R,5S,6R)-2-(3-(4-hvdroxybenzyl)-4- methylphenyl)-6-(hvdroxymethyl)tetrahydro-2H-pyran-3,4,5-triol:

Step 1: Synthesis of(5-bromo-2-methylphenyl)(4-ethoxyphenyl)methanone:

5-bromo-2-methylbenzoic acid (13 g) was suspended in dichloromethane (100 ml) and dimethylformamide (100 ml) then it was cooled to 0 °C and SOCl2 (26.5 ml) was added slowly. The reaction mixture was slowly allowed to room temperature and stirred for about 12 hours. The solvent was removed under reduced pressure to give 5-bromo-2-methylbenzoyl chloride as oil. Ethoxybenzene (7 ml) was suspended in dichloromethane and cooled to -5 °C then the mixture was added slowly to A1C13 (10.4 g) by portion wise and the above acid chloride in dichloromethane (— ) was added drop wise and stirred for about 1 hour at 0 °C. After completion of the reaction (monitored by TLC), the reaction mixture was poured
into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. The crude product was purified via silica gel column chromatography with EtOAc and n- Hexane (1: 7) to afford the title compound as light yellow solid (15 g). Yield: 77.8 %; 1H NMR (CDCI3, 300 MHz): δ 7.76 (d, J = 8.7 Hz, 2H), 7.48 (dd, J = 2.1, 8.1 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.15 (d, / = 8.1 Hz, 1H), 6.92 (d, J = 8.7 Hz, 2H), 4.11 (q, J = 6.9 Hz, 2H), 2.22 (s, 3H), 1.45 (t, J = 6.9 Hz, 3H).
Step 2: Synthesis of (5-bromo-2-methylphenyl)(4-hydroxyphenyl)methanone:

To a stirred solution of (5-bromo-2-methylphenyl)(4- ethoxyphenyl)methanone (step 1, 15 g) in dichloromethane (200 ml), AICI3 (62.5 g) was added at 0 °C under N2 atmosphere by portion wise over 30 minutes. Subsequently, the reaction mixture was stirred at room temperature for about 12 hours. After completion of the reaction (monitored by TLC), reaction mixture was poured into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1 : 5) to afford title compound as pale yellow solid (14 g). Yield: 81.3 %; 1H NMR (CDC13, 300 MHz): δ 7.74 (d, J = 8.7 Hz, 2H), 7.48 (dd, J = 2.1, 8.4 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.15 ( d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.7 Hz, 2H), 5.53 (s, 1H), 2.23 (s, 3H).
Step 3: Synthesis of4-(5-bromo- -methylbenzyl)phenol:

To a stirred solution of (5-bromo-2-methylphenyl)(4- hydroxyphenyl)methanone (step 2, 14 g) in dichloromethane (100 ml) and
acetylnitrile (1: 1), Et3SiH (39 ml) was added at 0 °C under N2 atmosphere followed by BF3: Et20 (15 ml) then reaction mixture was continued for about 16 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water, brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1: 5) to afford the title compound as liquid (12.2 g). Yield: 92.3 %; 1H NMR (CDC13, 300 MHz): 6 7.25- 7.24 (m, 1H), 7.20 (d, J = 1.8 Hz, 1H), 7.03- 6.96 (m, 3H), 6.75 (dd, J = 1.8, 6.6 Hz, 2H), 4.63 (s, 1H), 3.85 (s, 2H), 2.17 (s, 3H); ES Mass: (M+ H) 276.90 (100 %).
Step 4: Synthesis of4-bromo-2-( -(methoxymethoxy)benzyl)-l-methylbenzene:

To a stirred solution of NaH (3.1 g) in dimethylformamide (110 ml), 4-(5- bromo-2-methylbenzyl)phenol (step 3, 12.2 g) was added under N2 atmosphere at 0 °C then. MOM-C1 (4.3 ml) was added after 30 minutes at 0 °C. The reaction mixture was slowly allowed to attain to room temperature and continued for over night. After completion of the reaction (monitored by TLC), reaction mixture was cooled to 0 °C and quenched with by cautious addition of sat. aq. NH4C1 solution. The reaction mixture was extracted with EtOAc twice and combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford compound as liquid (11.6 g). Yield: 82 %; 1H NMR (CDCI3, 300 MHz): δ 7.27- 7.21 (m, 2H), 7.03- 6.94 (m,5H), 5.15 (s,2H), 3.87 (s, 2H), 3.47 (s, 3H), 2.18 (s, 3H).
Step 5: Synthesis of (3R,4S,5S,6R)~2-(3-(4-hydroxybenzyl)-4-methylphenyl)-6- ( hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3, 4, 5-triol:

To a stirred solution of 4-bromo-2-(4-(methoxymethoxy)benzyl)-l- methylbenzene (step 4, 11.6 g) in dry tetrahydrafuran (70 ml) and toluene (60 ml, 1: 1 ), n-BuLi (2.3 M in hexane) was added at -78 °C under N2 atmosphere and stirred for about 30 minutes. To this solution (3R,4S,5R,6R)-3,4,5- tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (Intermediate2, 13.4 g) and stirred for about 2 hours at -78 °C then 6 % CH3S02H in MeOH (150 ml) was added slowly at same temperature then the reaction was allowed to room temperature and continued for about 12 hours. After completion of the reaction (monitored by TLC), reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water, brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to afford the title compound as White solid and Hygroscopic (5.2 g). Yield: 36.8 %; 1H NMR (DMSO- D6, 300 MHz): δ 9.14 (s, 1H), 7.32 (s, 1H), 7.27 (d, / = 7.8 Hz, 1H), 7.08 (d, J = 7.8 Hz, 1H), 6.89 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 4.93 (d, J = 5.4 Hz, 1H), 4.68 (d, J = 5.1 Hz, 1H), 4.62 (d, J = 7.5 Hz, 1H), 4.50 (dd, J = 6.0, 6.0 Hz, 1H), 3.83- 3.81 (m, 2H), 3.76- 3.71 (m, lH), 3.59- 3.50 (m, 3H), 3.26- 3.16 (m, 1H), 2.93 (s, 3H), 2.90- 2.87 (m, 1H), 2.13 (s, 3H); ES Mass: (M+ Na) 413.20 (100 %).
Step 6: Synthesis of (3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4-methylphenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol:

To a stirred solution of (3R,4S,5S,6R)-2-(3-(4-hydroxybenzyl)-4- methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (step
5, 5.2 g) in dichloromethane (30 ml) and AcCN (30 ml) (1:1), Et3SiH (8.5 ml) was added at 0 °C under N2 atmosphere followed by BF3: Et20 (3.3 ml) then reaction mixture was stirred for about 6 hours. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water, brine, dried over anhydrous sodium sulphate and concentrated to afford the residue. The crude product was purified by silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of α & β isomers) as white solid (3 g). Yield: 62.5 %; HPLC: 83 %; 1H NMR (DMSO- D6, 300 MHz): δ 9.15 (s, 1H), 7.10- 7.07 (m, 3H), 6.91 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 4.91- 4.90 (m, 2H), 4.70 (d, J = 5.7 Hz, 1H), 4.42 (dd, J = 5.7, 5.7 Hz, 1H), 3.93 (d, J = 9.0 Hz, 1H), 3.81 (s, 2H), 3.75- 3.66 (m, 1H), 3.46- 3.38 (m, 1H), 3.23- 3.14 (m, 4H), 2.15 (s, 3H); ES Mass: (M+Na) 383.11
Step 7: Synthesis of (2S,3S,4R,5R,6R)-2-(3-(4-acetoxybenzyl)-4-methylphenyl)-6- (acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (β- isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4- methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (step 6, 3 g) in dichloromethane (70 ml), Et3N was added at 0 °C under N2 atmosphere followed by AC20 (9.4 ml) and DMAP then the reaction temperature was slowly raised to room temperature and stirred for over night. After completion of the reaction (monitored by TLC), reaction mixture was diluted with dichloromethane and organic layer was washed with 3N HC1 solution, brine and dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (6 g). Yield: 96 %; 1H NMR (CDC13, 300 MHz): δ 7.15 (s, 2H), 7.07- 7.05 (m, 3H), 6.97 (d, J = 8.4 Hz, 2H), 5.34- 5.10 (m, 3H), 4.34 (d, J = 9.9 Hz, 1H), 4.30- 4.25 (m, 1H), 4.17- 4.11 (m, 1H), 3.95 (s, 2H), 3.84- 3.78 (m,
1H), 2.27 (s, 3H), 2.20 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H), 1.74 (s, 3H); ES Mass: (M+ Na) 592.98
Step 8: Synthesis of (2S,3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4-methylphenyl)-6- ( hydroxymethyl )tetrahydro-2H-pyran-3, 4, 5-triol:
NH3 in MeOH (120 ml) was added to (2S,3S,4R,5R,6R)-2-(3-(4- acetoxybenzyl)-4-methylphenyl)-6-(acetoxymethyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (β- isomer, step 7, 6g) at room temperature and stirred for over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title product as white solid (2.8 g). Yield: 93 %; 1H NMR (DMSO- D6, 300 MHz): δ 9.15 (s, 1H), 7.10- 7.07 (m, 3H), 6.91 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 4.91- 4.90 (m, 2H), 4.70 (d, J = 5.7 Hz, 1H), 4.42 (dd, J = 5.7, 5.7 Hz, 1H), 3.93 (d, / = 9.0 Hz, 1H), 3.81 (s, 2H), 3.75- 3.66 (m, 1H), 3.46- 3.38 (m, 1H), 3.23- 3.14 (m, 4H), 2.15 (s, 3H); ES Mass: (M+ Na) 383.10; HPLC: 96.3 %
Intermediate 14: Preparation of (l-(trifluoromethyl)cvclopropyl)methyl 4- methylbenzenesulf onate :

Step 1: Synthesis of (l-(trtfluoromethyl)cyclopropyl)methanol:

To a stirred solution of Lithium aluminium hydride (2.4 g) in dry ether (70 ml) l-(trifluoromethyl)cyclopropanecarboxylic acid (5 g) was added at 0 °C under N2 atmosphere and the reaction mixture was stirred for about 1 hour. After completion of the reaction (monitored by Ή- NMR), the reaction mixture was quenched by cautious addition of water (1 ml), 15 % aq. NaOH solution (1 ml) and again water (3 ml) and stirred for 30 minutes. The reaction mixture was filtered and cake was washed with ether twice. Due to volatile nature of the compound, only half of the ether volume was reduced on hot water both using without any vacuum and
next reaction was proceeded as such. 1H NMR (CDC13, 300 MHz): δ 3.73 (s, 2H), 1.83 (br s, 1H), 1.05- 1.01 (m, 2H), 0.80- 0.76 (m, 2H); ES Mass: (M- H) 138.91
Step 2: Synthesis of (l-(trifluoromethyl)cyclopropyl)methyl 4- methylbenzenesulfonate:
To a stirred solution of (l-(trifluoromethyl)cyclopropyl)methanol (step 1, 4.5 g) in dichloromethane (50 ml), Et3N (13.5 ml) was added at 0 °C under N2 atmosphere followed by TsCl (9.1 g) and catalytic amount of DMAP. The reaction mixture was allowed to room temperature and stirred for about 12 hours. After completion of the reaction (monitored by 1H- NMR), reaction mixture was diluted with dichloromethane and organic layer was washed with saturated aq. NHUC1 solution, brine and dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:39) to afford the title compound as liquid (8.2 g). Yield: 86 %; 1H NMR (CDC13, 300 MHz): δ 7.79 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 4.09 (s, 2H), 2.46 (s, 2H), 1.14- 1.10 (m, 2H), 0.85- 0.84 (m, 2H); ES Mass: (M+ Na) 317.08.
Intermediate 15: Preparation of (l-(trifluoromethyl)cyclobutyl)methyl 4- methylbenzenesulfonate:

Step 1: Synthesis of ( 1 -(trifluoromethyl)cyclobutyl)methanol:

To a stirred solution of Lithium aluminium hydride (2.7 g) in dry ether (80 ml), l-(trifluoromethyl)cyclobutanecarboxylic acid (6 g) was added at 0 °C under N2 atmosphere and the reaction mixture was stirred for about 1 h at 0 °C. After completion of the reaction (monitored by !H- NMR), the reaction mixture was quenched by cautious addition of water (1 ml), 15 % aq. NaOH solution (1 ml) and again water (3 ml), with stirring continued for 30 min. The reaction mixture was filtered and cake was washed with ether twice. Due to volatile nature of the
compound, only half of the ether volume was reduced on hot water both using without any vacuum and next reaction was proceeded as such. 1H NMR (CDC13, 300 MHz): δ 3.81 (s, 2H), 2.30- 2.26 (m, 2H), 2.06- 1.91 (m, 4H), 1.71 (br s, 1H); ES Mass: (M- H) 152.95.
Step 2: Synthesis of (l-(trifluoromethyl)cyclobutyl)methyl 4-methylbenzenesulfonate:
To a stirred solution of (l-(trifluoromethyl)cyclobutyl)methanol (step 1, 5.5 g) in dichloromethane (50 ml), Et3N (15 ml) was added at 0 °C under N2 atmosphere followed by TsCl (10 g) and catalytic amount of DMAP. The reaction mixture was allowed to room temperature and stirred for about 12 hours. After completion of the reaction (monitored by 1H- NMR), reaction mixture was diluted with dichloromethane and organic layer was washed with saturated aq. NH4C1 solution, brine and dried over anhydrous sodium sulphate and concentrated to afford the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:39) to afford the title compound as liquid (7 g). Yield: 63 ; 1H NMR (CDC13, 300 MHz): δ 7.81 (d, J = 8.4 Hz, 2H), 7.37 (d, / = 8.4 Hz, 2H), 4.13 (s, 2H), 2.46 (s, 3H), 2.32- 2.27 (m, 2H), 2.03- 1.96 (m, 4H); ES Mass: (M+ Na) 331.
Intermediate 16: Preparation of (4,4-difluoro-l-methylcyclohexyl)methyl 4- methylbenzenesulfonate:

Step 1: Synthesis of methyl 4,4-difluorocyclohexanecarboxylate:
e

Cone. H2S04 was added drop-wise to a solution of 4,4- difluorocyclohexanecarboxylic acid (6 g) in MeOH (50 ml) at 0 °C and the reaction mixture was refluxed for about 16 hours. After the completion of reaction, the
solvent was removed and the reaction mixture was dissolved in dichloromethane. The organic layer was washed with saturated NaHC03 solution and the organic layer was dried over anhydrous sodium sulphate. The solvent was removed over rotary evaporator without vacuum to give the crude compound (7.4 g) which was used directly for the next step. 1H NMR (CDC13, 300 MHz): δ 3.69 (s, 3H), 2.42 (m, 1H), 2.10- 1.97 (m, 4H), 1.91- 1.72 (m, 4H).
Step 2: Synthesis of methyl 4,4-difluoro-l-methylcyclohexanecarboxylate:

To a solution of diisopropylamine (11 ml) in tetrahydrafuran (50 ml), n-BuLi (26 ml) was added at 0 °C and the reaction mixture was allowed to stir at same temperature for about 10-15 minutes. The reaction mixture was stirred at room temperature for about 45 minutes. Methyl 4,4-difluorocyclohexanecarboxylate (step 1, 5.3 g) in tetrahydrafuran (40 ml) was added drop-wise to above mixture at -78 °C. The reaction mixture was stirred at same temperature for about 2 hours and then methyl iodide was added drop-wise. The reaction was maintained at -78 °C for an hour and allowed to attain room temperature. Reaction mixture was stirred overnight at room temperature and quenched at 0 °C with saturated aq. NH4C1 solution and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated then crude product was directly used for the next step. 1H NMR (CDC13, 300 MHz): δ 3.71 (s, 3H), 2.20- 2.16 (m, 2H), 1.96- 1.71 (m, 4H), 1.57- 1.47 (m, 2H), 1.22 (s, 3H).
Step 3: Synthesis of (4,4-difluoro-l -methylcyclohexyl)methanol:

Methyl 4,4-difluoro-l -methylcyclohexanecarboxylate (step 2, 5.7 g) in tetrahydrafuran (20 ml) was added to a suspension of LiAlH4 (2.25 g) in tetrahydrafuran (50 ml) at 0 °C and the reaction mixture was allowed to stir at the
same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction (monitored by TLC), the reaction mixture was quenched by cautious addition of water (3.5 ml), 15 % aq. NaOH solution (3.5 ml) and again water (11 ml) at 0 °C and stirred for about 30 minutes. The reaction mixture was filtered and the residue was washed with diisopropylether. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. Crude product was directly used for the next step. 1H NMR (CDC13, 300 MHz): δ 3.41 (s, 2H), 1.96- 1.86 (m, 4H), 1.62- 1.54 (m, 2H), 1.46- 1.41 (m, 2H), 0.98 (s, 3H).
Step 4: Synthesis of (4,4-difluoro-l-methylcyclohexyl)methyl 4- methylbenzenesulfonate:
To a solution of (4,4-difluoro-l-methylcyclohexyl)methanol (step 3, 4.8 g) in dichloromethane (60 ml), triethyl amine (12.2 ml) was added followed by tosyl chloride (8.3 g) at 0 °C. After addition of catalytic amount of DMAP (2.47 g, 0.020 mol), the reaction mixture was allowed to stir overnight at room temperature. After completion of reaction, the reaction mixture was washed with water, saturated brine solution, dried over anhydrous Na2SC¼ and concentrated. The product was purified by column chromatographic technique to afford the title compound as solid (5 g). Yield: 51.11 %; 1H NMR (CDC13, 300 MHz): δ 7.78 (d, J = 7.8 Hz, 2H), 7.36 (d, J = 7.8 Hz, 2H), 3.75 (s, 2H), 2.46 (s, 3H), 1.88-1.80 (m, 4H), 1.50- 1.45 (m, 4H), 0.97 (s, 3H); ES Mass: (M+ Na) 340.95.
Intermediate 17: Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l-
(trifluoromethyl)cvclobutyl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl)tetrahydro-
2H-pyran-3,4,5-triol:
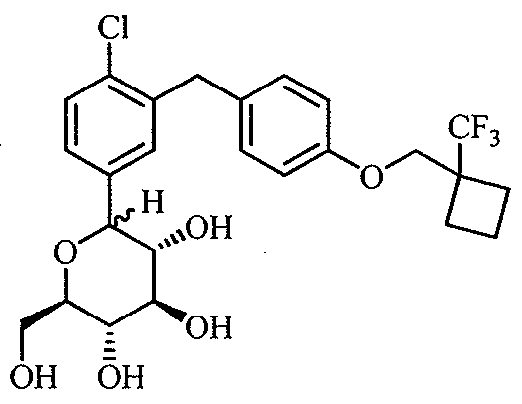
( trifluoromethyl )cyclobutyl )methoxy )benzyl )benzene:

To a stirred solution of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate 1, 5.4 g) in dimethylformamide (100 ml), Cs2C03 (17.8 g) was added under N2 atmosphere followed by (l-(trifluoromethyl)cyclobutyl)methyl 4- methylbenzenesulfonate (Intermediate 15, 6.8 g) at room temperature and the reaction mixture was heated to 110 °C then stirred for about 5 hours. After completion of the reaction (monitored by TLC), reaction mixture was cooled to room temperature and filtered through celite and the cake was washed with EtOAc. The filtrate was washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford the title compound as white solid (6.2 g). Yield: 79.4 %; 1H NMR (CDC13, 300 MHz): δ 7.30- 7.21 (m, 3H), 7.11 (d, 7 = 8.7 Hz, 2H), 6.89 (d, 7 = 8.7 Hz, 2H), 4.05 (s, 2H), 4.00 (s, 2H), 2.38- 2.31 (m, 2H), 2.26- 2.17 (m, 2H), 2.07- 1.99 (m, 2H).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l-
( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )-2- methoxytetrahydro-2H-pyran-3, 4, 5-triol:

To a stirred solution of 4-bromo-l-chloro-2-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)benzene (step 1, 6.2 g) in dry tetrahydrafuran (40 ml) and toulene (60 ml, 1:1 ), n-BuLi (2.3 M, 16.8 ml) was added at -78 °C under N2 atmosphere and stirred for about 30 minutes at -78°C. To this solution (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-
((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (Intermediate 2, 5.3 g) in
toluene was added at -78 °C and stirred for about 2 hours at same temperature then 6 % CH3S02H in MeOH (105 ml) was added slowly. The reaction mixture was allowed to room temperature and stirred for about 12 hours. After completion of the reaction (monitored by TLC), reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to afford the title compound as White solid (3.6 g). Yield: 46 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.52 (s, 1H), 7.39 (s, 2H), 7.10 (d, / = 8.4 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H), 4.99 (d, J = 5.4 Hz, 1H), 4.81- 4.76 (m, 2H), 4.56 (dd, / = 5.7, 5.7 Hz, 1H), 4.15 (s, 2H), 4.00 (ABq, 7 = 14.7 Hz, 2H ), 3.76- 3.71 (m, 1H), 3.60- 3.48 (m, 2H), 3.35- 3.32 (m, 1H), 3.27- 3.17 (1H), 2.91 (s, 3H), 2.88- 2.85 (m, 1H), 2.31- 2.20 (m, 2H), 2.09- 1.89 (m, 4H); ES Mass: (M+Na) 469.
Step 3: Synthesis of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l-
( trifluoromethyl )cyclobutyl)methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol:
To a stirred solution of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 3.6 g) in dichloromethane (25 ml) and acetylnitrile (25 ml, 1: 1), Et3SiH (2.1 ml) was added at 0 °C under N2 atmosphere followed by BF3: Et20 (1.2 ml) and stirred for about 6 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of two α, β isomers) as White solid (3 g). Yield: 88.2 %; HPLC: 79.88 ; 1H NMR (DMSO- D6, 300 MHz): δ 7.38-7.32 (m, 2H), 7.24- 7.14 (m, 1H), 7.12 (d, J = 8.5 Hz, 2H),
6.90 (d, J = 8.5 Hz, 2H), 4.97- 4.95 (m, 2H), 4.84 (d, J = 5.7 Hz, 1H), 4.46 (dd, J = 5.7 Hz, 5.7 Hz, 1H), 4.15 (s, 2H), 4.00-3.97 (m, 3H), 3.71- 3.66 (m, 1H), 3.45-3.40 (m, 1H), 3.26- 3.09 (m, 4H), 2.29- 2.24 (m, 2H), 2.10- 1.93 (m, 4H); ES Mass: (M+Na) 539.
Intermediate 18: Preparation of (l-methylcyclopentyl)methyl 4- methylbenzenesulfonate:

Step 1: Synthesis of methyl 1-methylcyclopentanecarboxylate:

To a solution of di-isopropylamine (69 ml) in tetrahydrafuran (100 ml), n- BuLi (166 ml) in hexane was added at 0 °C and the reaction mixture was allowed to stir at same temperature for about 10-15 minutes. The reaction mixture was stirred at room temperature for about 45 minutes. Methyl cyclopentanecarboxylate (25 g) in tetrahydrafuran (200 ml) was added drop-wise at -78 °C to above reaction mixture. The reaction mixture was stirred at same temperature for about 2 hours and then methyl iodide was added drop-wise. The reaction was maintained at -78°C for an hour and allowed to attain room temperature. The reaction mixture was stirred overnight at room temperature and quenched at 0 °C with saturated aq. NH4C1 solution and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. Crude product was directly used for the next step. 1H NMR (CDC13, 300 MHz):™ 3.66 (s, 3H), 2.10- 2.03 (m, 2H), 1.69- 1.64 (m, 4H), 1.50- 1.42 (m, 2H), 1.23 (s, 3H).
Step 2: Synthesis of (l-methylcyclopentyl)methanol:

Methyl 1-methylcyclopentanecarboxylate (step 1, 26 g) in tetrahydrafuran (200 ml) was added to a suspension of LiAlH4 (14 g) in tetrahydrafuran (150 ml) at
0 °C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of the reaction (monitored by 1 H-NMR), reaction mixture was quenched by cautious addition of water (14 ml), 15 % aq. NaOH solution (14 ml) and again water (42 ml) at 0 °C and stirred for about 1 hour. The reaction mixture was filtered and the residue was washed with diisopropylether. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. Crude product was directly used for the next step. 1H NMR (CDC13, 300 MHz):™ 3.37 (s, 2H), 1.68- 1.46 (m, 7H), 1.35- 1.98 (m, 2H), 1.05 (s; 3H).
Step 3: Synthesis of (l-methylcyclopentyl)methyl 4-methylbenzenesulfonate:
To a solution of (l-methylcyclopentyl)methanol (step 2, 20 g) in dichloromethane (250 ml), triethyl amine (74 ml) was added followed by tosyl chloride (66.6 g) at 0°C. After addition of catalytic amount of DMAP (Cat), the reaction mixture was allowed to stir overnight at room temperature. After completion of the reaction, the reaction mixture was washed with water and brine solution, dried over anhydrous Na2S0 and concentrated then purified by column chromatographic technique to afford the title compound as liquid (14 g). Yield: 27 %; 1H NMR (CDC13, 300 MHz):™ 7.78 (d, J = 8.1 Hz, 2H), 7.34 (d, J = 8.1 Hz, 2H), 3.74 (s, 2H), 2.44 (s, 3H), 1.60-1.41 (m, 6H), 1.31- 1.29 (m, 2H), 0.96 (s, 3H).
Intermediate 19: Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- pyran-3A5-triol:

Step 1: Synthesis
methylcyclopentyl )methoxy )benzyl )benzene:

To a stirred solution of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate 1, 10 g) in dimethylformamide (100 ml), Cs2C03 (33 g) was added under N2 atmosphere followed by (l-methylcyclopentyl)methyl 4-methylbenzenesulfonate (Intermediate 18, 13.6 g) at room temperature. The reaction mixture was heated to 120 °C and stirred for about 5 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and filtered through celite and the cake was washed with EtOAc. The filtrate was washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford the title compound as liquid (7 g). Yield: 53.8 %; 1H NMR (CDCI3, 300 MHz): δ 7.28- 7.22 (m, 3H), 7.08 (d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 3.98 (s, 2H), 3.65 (s, 2H), 1.72- 1.59 (m, 64H), 1.42- 1.37 (m, 2H), 1.11 (s, 3H).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl )methoxy )benzyl )phenyl )-6-( hydroxymethyl )-2-methoxytetrahydro- 2H-pyran-3, 4, 5-triol:

To a stirred solution of 4-bromo-l-chloro-2-(4-((l- methylcyclopentyl)methoxy)benzyl)benzene (step 1, 7 g) in dry tetrahydrafuran (40 ml) and toulene (80 ml, 1 : 1 ), t-BuLi (1.7 M in toluene, 24 ml) was added at -78 °C under N2 atmosphere and stirred for about 30 minutes. To this solution (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-
((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (Intermediate 2, 6.6 g) in toluene (80 ml) was added at -78 °C and stirred for about 2 hours then 6 % CH3S02H in MeOH (105 ml) was added slowly at same temperature. Then the
reaction was allowed to attain room temperature and stirred for about 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 0°C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to afford the title compound as white solid (3 g). Yield: 33.3 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.51 (s, 1H), 7.38 (s, 2H), 7.06 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.4 Hz, 2H), 4.98 (d, J = 5.4 Hz, 1H), 4.80- 4.74 (m, 2H), 4.54 (dd, J = 6.3 Hz, 1H), 3.98 (ABq, / = 15.0 Hz, 2H), 3.76- 3.71 (m, 1H), 3.64 (s, 2H), 3.56- 3.48 (m, 2H), 3.40- 3.34 (m, 1H), 3.25- 3.20 (m, 1H), 2.91 (s, 3H), 2.88- 2.85 (m, 1H), 1.68- 1.55 (m, 6H), 1.34- 1.32 (m, 2H), 1.05 (s, 3H); ES Mass: (M+Na) 529.
Step 3: Synthesis of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
To a stirred solution of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 3 g) in dichloromethane (30 ml) and acetylnitrile (30 ml, 1: 1), Et3SiH (1.9 ml) was added at 0° C under N2 atmosphere followed by BF3: Et20 (1.1 ml) and stirred for about 6 hours. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0° C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of α & β isomers) as white solid (1.8 g). Yield: 64.2 ; HPLC: 88.16 ; 1H NMR (DMSO- D6, 300 MHz): δ 7.36 (d, 7 = 8.1 Hz, 1H), 7.31 (d, 7 = 1.8 Hz, 1H), 7.22 (dd, J = 1.8, 8.1 Hz, 1H), 7.08 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 4.97- 4.95 (m, 2H), 4.83 (d, J
= 5.7 Hz, 1H), 4.45 (dd, J = 6.0 Hz, 1H), 3.99- 3.96 (m, 3H), 3.71- 3.69 (m, 1H), 3.65 (s, 2H), 3.45- 3.41 (m, 1H), 3.29- 3.05 (m, 4H), 1.62- 1.58 (m, 6H), 1.34- 1.32 (m, 2H), 1.05 (s, 3H); ES Mass: (M+Na) 499.
Intermediate 20: Preparation of (3R.4R.5S.6R)-2-(4-chloro-3-(4-((l-
(trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-
(hvdroxymethyl)tetrahvdro-2 -pyran-3A5-triol:

Step 1: Synthesis of 4-bromo-l-chloro-2-(4-((l-
( trifluoromethyl )cyclopropyl j :

To a stirred solution of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate 1, 6 g) in dimethylformamide (50 ml), Cs2C03 (19.82 g) was added under N2 atmosphere followed by (l-(trifluoromethyl)cyclopropyl)methyl 4-methylbenzenesulfonate (Intermediate 14, 7.2 g) at room temperature. The reaction mixture was heated to 120 °C and stirred for about 5 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and filtered through celite then the cake was washed with EtOAc. The filtrate was washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford the title compound as liquid (7.4 g). Yield: 88 %; Ή NMR (CDC13, 300 MHz): δ 7.29- 7.20 (m, 3H), 7.09 (d, J = 8.5 Hz, 2H), 6.83(d, J = 8.5 Hz, 2H), 4.06 (s, 2H), 3.98 (s, 2H), 1.14- 1.10 (m, 2H), 0.91- 0.90 (m, 2H).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l-
( trifluoromethyl )cyclopropyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )-2- methoxytetrahydro-2H-pyran-3,4,5-triol:
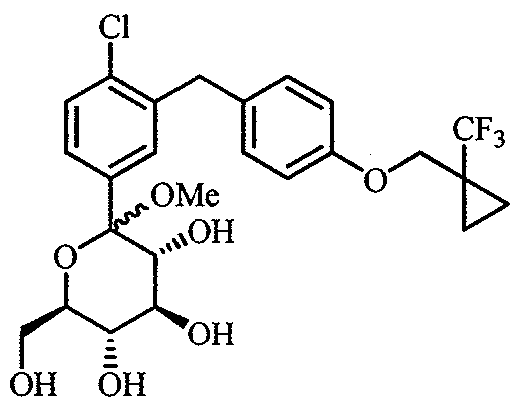
To a stirred solution of 4-bromo-l-chloro-2-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)benzene (step 1, 3.8 g) in dry tetrahydrafuran (40 ml) and toulene (40 ml, 1: 1), n-BuLi (2.3 M in hexane, 10.7 ml) was added at -78 °C under N2 atmosphere and stirred for about 30 minutes at -78°C. To this solution (3R,4S,5R,6R) -3,4,5- tris (trimethylsilyloxy) -6- ((trimethylsilyloxy) methyl)tetrahydro-2H-pyran-2-one (Intermediate 2, 3.4 g) in toluene (20 ml) was added at -78 °C and stirred for about 2 hours at -78 °C then 6 % CH3S02H in MeOH (57 ml) was added slowly at same temperature. Then the reaction was allowed to room temperature and continued for over night. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to afford the title compound as white solid (2.5 g). Yield: 52 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.52 (s, 1H), 7.39 (s, 2H), 7.08 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 4.97 (d, J = 5.4 Hz, 1H), 4.78- 4.73 (m, 2H), 4.53 (dd, J = 5.7 Hz, 1H), 4.05- 3.93 (m, 4H), 3.77- 3.72 (m, 1H), 3.60- 3.52 (m, 2H), 3.39 (m, 1H), 3.25- 3.18 (m, 1H), 2.92 (s, 3H), 2.92- 2.85 (m, 1H), 1.06 (m, 2H), 0.97 (m, 2H); ES Mass: (M+Na) 555.
Step 3: Synthesis of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l-
( trifluoromethyljcyclopropyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro- 2H-pyran-3, 4, 5-triol:
To a stirred solution of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 2.5 g) in dichloromethane (20 ml) and acetylnitrile (20 ml, 1: 1), Et3SiH (1.5 ml) was added at 0 °C under N2
atmosphere followed by BF3: Et20 (0.8 ml) and stirred for about 6 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc twice and the combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of α & β isomers) as white solid (1.8 g). Yield: 76 %; HPLC: 60.38 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.36 (d, J = 8.1 Hz, 1H), 7.32 (d, J = 1.8 Hz, 1H), 7.22 (dd, J - 1.8, 8.1 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 4.97- 4.95 (m, 2H), 4.84 (d, J = 5.7 Hz, 1H), 4.46 (dd, J = 5.7 Hz, 5.7 Hz, 1H), 4.06 (s, 2H), 3.99-3.97 (m, 3H), 3.71- 3.66 (m, 1H), 3.45-3.39 (m, 1H), 3.24- 3.10 (m, 4H), 1.07- 1.05 (m, 2H), 0.97 (m, 2H); ES Mass: (M-H) 501.
Intermediate 21: Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((4.4-difluoro-l- methylcyclohexyl)methoxy)benzyl phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3A5-triol:

Step 1: Synthesis of 4-bromo-l-chloro-2-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )benzene:

To a stirred solution of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate 1 , 4 g) in dimethylformamide (50 ml), Cs2C03 (13.2 g) was added under N2 atmosphere followed by (4,4-difluoro-l-methylcyclohexyl)methyl 4-methylbenzenesulfonate (Intermediate 16, 4.7 g) at room temperature. The reaction mixture was heated to 120 °C and stirred for about 5 hours. After completion of the reaction (monitored by
TLC), the reaction mixture was cooled to room temperature and filtered through celite and the cake was washed with EtOAc. The filtrate was washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford the title compound as solid (4.6 g). Yield: 77.9 %; 1H NMR (CDC13, 300 MHz): δ 7.23- 7.20 (m, 3H), 7.09 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2H), 3.98 (s, 2H), 3.67 (s, 2H), 1.99- 1.89 (m, 4H), 1.79- 1.70 (m, 2H), 1.58 (m, 2H), 1.09 (s, 3H).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )-2-methoxytetrahydro- 2H-pyran-3, 4, 5-triol:

To a stirred solution of 4-bromo-l-chloro-2-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)benzene (step 1, 10.4 g) in dry tetrahydrafuran (40 ml) and toulene (40 ml, 1 : 1 ), n-BuLi (2.3 M in hexane) was added at -78 °C under N2 atmosphere stirred for about 30 minutes. To this solution (3R,4S,5R,6R)- 3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2- one (Intermediate 2, 3.9 g) in toluene (30 ml) was added at -78 °C and stirred for about 2 hours at same temperature then 6 % CH3SO3H in MeOH (69 ml) was added slowly. Then the reaction mixture was allowed to room temperature and stirred for about 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 0°C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to afford the title compound as white solid (1.2 g). Yield: 21 %; 1H NMR (DMSO- D6, 300
MHz): 5 7.52 (s, 1H), 7.38 (s, 2H), 7.07 (d, J = 8.1 Hz, 2H), 6.85 (d, J = 8.1 Hz, 2H), 4.96 (d, J = 5.4 Hz, 1H), 4.78- 4.72 (m, 2H), 4.52 (dd, J = 6.3, 6.3 Hz, 1H),
3.99 (ABq, J = 15.3 Hz, 2H), 3.70 (m, 3H), 3.60- 3.51 (m, 2H), 3.25- 3.20 (m, 2H), 2.91 (s, 3H), 2.88- 2.85 (m, 1H), 1.96- 1.87 (m, 4H), 1.67- 1.63 (m, 2H), 1.48- 1.44 (m, 2H), 1.03 (s, 3H); ES Mass: (M+Na) 579.1.
Step 3: Synthesis of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
To a stirred solution of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 1.2 g) in dichloromethane (15 ml) and acetylnitrile (15 ml, 1:1), Et3SiH (0.7 ml) was added at 0° C under N2 atmosphere followed by BF3: Et20 (0.4 ml) and reaction was stirred for about 6 hours. After completion of the reaction (monitored by 1H-NMR), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of α & β isomers) as white solid (670 mg). Yield: 61 %; HPLC: 88.61 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.37 (d, / = 8.4 Hz, 1H), 7.31 (s, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz; 2H), 4.98- 4.95 (m, 2H), 4.84 (d, J = 5.7 Hz, 1H), 4.44 (dd, J = 5.7 Hz, 5.7 Hz, 1H), 3.99- 3.97 (m, 3H), 3.71- 3.66 (m, 3H), 3.47- 3.06 (m, 5H), 1.97- 1.87 (m, 4H), 1.70- 1.61 (m, 2H), 1.49- 1.44 (m, 2H), 1.03 (s, 3H); ES Mass: (M+Na) 549.
Intermediate 22: Preparation of 4-(5-bromo-2-fluorobenzyl)phenol:

Step 1: Synthesis of l-bromo-4-ethoxybenzene:

Potassium carbonate (118 g) was added to a solution of 4-bromophenol (50 g) in acetone (250 ml) and allowed to stir at room temperature for about 30 minutes then ethyl iodide (67.6 g) was added drop wise. The reaction mixture was refluxed for about 10 hours and completion of reaction was confirmed by TLC. The reaction mixture was filtered and acetone was removed over rotary evaporator. The residue was dissolved in ethyl acetate and washed with water and brine solution, dried over anhydrous Na2S04 and concentrated then filtered and product was purified via silica gel column chromatography with EtOAc and n-Hexane (1: 7) to afford the title compound as liquid (57 g). Yield: 98.27 ; 1H NMR (CDC13, 300 MHz): δ 7.35 (d, / = 8.7 Hz, 2H), 6.76 (d, J = 8.7 Hz, 2H), 3.99 (q, J = 6.9 Hz, 2H), 1.40 (t, J = 6.9 Hz, 3H).
Step 2: Synthesis of (5-bromo-2-fluorophenyl)(4-ethoxyphenyl)methanol:

To a stirred solution of Mg (9.5 g) in dry tetrahydrafuran (50 ml), 1-bromo- 4-ethoxybenzene (step 1, 53.4 g) in tetrahydrafuran (150 ml) was added under N2 atmosphere at room temperature slowly by drop wise and stirred for about 1 hour. Then 5-bromo-2-fluorobenzaldehyde (11 ml) was added slowly to the above reaction mixture and the reaction mixture was allowed to stir for over night. After completion of the reaction (monitored by TLC), the reaction mixture was poured into saturated NH4C1 solution at 0 °C and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane to afford the title compound as liquid (24 g). Yield: 85.7 ; Ή NMR (CDC13, 300 MHz): δ 7.745- 7.71 (m, 1H), 7.37- 7.27 (m, 3H), 6.91- 6.84 (m, 3H), 6.02 (d, J = 3.3 Hz, 1H), 4.01 (q, J = 7.2 Hz, 2H), 2.20 (d, J = 3.3 Hz, 1H), 1.39 (t, J = 7.2 Hz, 3H).
Step 3: Synthesis of4-bromo-2-(4-ethoxybenzyl)-l-fl orobenzene:

To a stirred solution of (5-bromo-2-fluorophenyl)(4-ethoxyphenyl)methanol (step 2, 24 g) in dichloromethane (150 ml) and acetylnitrile (150 ml, 1: 1), Et3SiH (24 ml) was added at 0 °C under N2 atmosphere followed by BF3: Et20 (14 ml) and the reaction mixture was stirred for about 12 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound and purified by silica gel column chromatography with EtOAc and n- Hexane to afford the title compound as liquid (15 g). Yield: 65.7 %; !H NMR (CDC13, 300 MHz): δ 7.29- 7.21 (m, 2H), 7.10 (d, J = 8.4 Hz, 2H), 6.91 (dd, / = 9.0, 9.0 Hz, 1H), 6.83 (d, J = 8.4 Hz, 2H), 4.00 (q, J = 6.9 Hz, 2H), 3.88 (s, 2H), 1.39 (t, J = 6.9 Hz, 3H).
Step 4: Synthesis of 4-(5-bromo-2-fluorobenzyl)phenol:
To a stirred solution of 4-bromo-2-(4-ethoxybenzyl)-l-fluorobenzene (step 3, 15 g) in dichloromethane (80 ml), BBr3 (53.3 ml, 1M solution in dichloromethane (50 ml)) was added slowly under N2 atmosphere at 0 °C and stirred for. over night at room temperature. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with dichloromethane. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound and purified via silica gel column chromatography with EtOAc and n-Hexane to afford the title compound as solid (12 g). Yield: 88.2 %; 1H NMR (CDC13, 300 MHz): 6 7.29- 7.21 (m, 2H), 7.07 (d, J = 8.4 Hz, 2H), 6.91 (dd, J = 9.0, 9.0 Hz, 1H), 6.77 (d, J = 8.4 Hz, 2H), 4.68 (s, 1H), 3.87 (s, 2H); ES Mass: (M-H) 278.90.
Intermediate 23: Preparation of (3R^R.5S.6RV2-(4-fluoro-3-(4-((l-
(trifluoromethyl)cvclobutyl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl tetrahydro-
2H-pyran-3.4.5-triol:
4-bromo-l-fluoro-2-(4-((l-

To a stirred solution of 4-(5-bromo-2-fluorobenzyl)phenol (Intermediate 22, 4 g) in dimethylformamide (60 ml), Cs2C03 (13.9 g) was added under N2 atmosphere followed by (l-(trifluoromethyl)cyclobutyl)methyl 4- methylbenzenesulfonate (Intermediate 15, 15.6 g) at room temperature. The reaction mixture was heated to 120 °C and stirred for about 6 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and filtered through celite then the cake was washed with EtOAc. The filtrate was washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford the title compound as liquid (5.7 g). Yield: 96.6 %; 1H NMR (CDC13, 300 MHz): δ 7.30- 7.22 (m, 2H), 7.13 (d, J = 8.1 Hz, 2H), 6.94- 6.87 (m, 3H), 4.04 (s, 2H), 3.90 (s, 2H), 2.41- 2.31 (m, 2H), 2.25- 2.16 (m, 2H), 2.07- 1.99 (m, 2H).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(4-fluoro-.
( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl )-6-( hydroxymethyl )-2 - methoxytetrahydro-2H-pyran-3,4,5-triol:

To a stirred solution of 4-bromo-l-fluoro-2-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)benzene (step 1, 5.7 g) in dry tetrahydrafuran (50 ml) and toulene (50 ml, 1: 1), t-BuLi (16 ml, 1.7 M in toluene (30 ml)) was added at -78 °C under N2 atmosphere and stirred for about 30 minutes at - 78 °C. To this solution (3R,4S,5R,6R)- 3,4,5-tris (trimethylsilyloxy)- 6- ((trimethylsilyloxy)methyl) tetrahydro-2H-pyran-2-one (Intermediate 2, 5.1 g) in toluene was added at -78 °C and stirred for about 2 hours at same temperature then 6 % CH3SO3H in MeOH (90 ml) was added slowly at -78 °C. Then the reaction mixture was allowed to attain room temperature and stirred for about 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to afford the title compound as white solid (3 g). Yield: 41.6 %; Ή NMR (DMSO- D6, 300 MHz): δ 7.48 (d, J = 7.2 Hz, 1H), 7.39 (m, 1H), 7.14- 7.08 (m, 3H), 6.91 (d, J = 7.8 Hz, 2H), 4.99 (d, J = 5.1 Hz, 1H), 4.79- 4.76 (m, 2H), 4.56 (dd, J = 5.7, 5.7 Hz, 1H), 4.15 (s, 2H), 3.91 (ABq, 7 = 15 Hz, 2H ), 3.75- 3.73 (m, 1H), 3.60- 3.48 (m, 2H), 3.38 (m, 1H), 3.25- 3.20 (m, 1H), 2.91 (s, 3H), 2.91- 2.84 (m, 1H), 2.26 (m, 2H), 2.07- 1.92 (m, 4H); ES Mass: (M+Na) 553.3.
Step 3: Synthesis of (3R,4R,5S,6R)-2-(4-fluoro-3-(4-((l-
( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro- 2H-pyran-3, 4, 5-triol:
To a stirred solution of (3R,4S,5S,6R)-2-(4-fluoro-3-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-
methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 3g) in dichloromethane (20 ml) and acetylnitrile (20 ml, 1:1) Et3SiH (1.8 ml) was added at 0 °C under N2 atmosphere followed by BF3: Et20 (1 ml) and stirred for about 6 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of α & β isomers) as white solid (2.3 g). Yield: 82.1 %; 1H NMR (DMSO- D6, 300 MHz): 6 7.28-7.20 (m, 2H), 7.16- 7.06 (m, 3H), 6.91 (d, J = 8.4 Hz, 2H), 4.96 (d, 7 = 2.1 Hz, 2H), 4.80 (d, 7 = 5.4 Hz, 1H), 4.46 (dd, 7 = 5.7 Hz, 5.7 Hz, 1H), 4.15 (s, 2H), 3.97 (d, 7 = 9.3 Hz, 1H), 3.89- 3.88 (m, 2H), 3.71-3.65 (m, 1H), 3.51- 3.11 (m, 5H), 2.26- 2.24 (m, 2H), 2.10- 1.89 (m, 4H); ES Mass: (M+Na) 523.15.
Intermediate 24: Preparation of (3R,4R,5S,6R -2-(4-fluoro-3-(4-((l-
(trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-
(hydroxymethyl)tetrahvdro-2H-pyran-3A5-triol:
4-bromo-l-fluoro-2-(4-( (1-

To a stirred solution of 4-(5-bromo-2-fiuorobenzyl)phenol (Intermediate 22, 4 g) in dimethylformamide (50 ml), Cs2C03 (13.9 g) was added under N2 atmosphere followed by (l-(trifluoromethyl)cyclopropyl)methyl 4- methylbenzenesulfonate (Intermediate 14, 4.6 g) at room temperature. The reaction
mixture temperature was raised to 120 °C and stirred for about 6 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and filtered through celite then the cake was washed with EtOAc. The filtrate was washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford the title compound as liquid (5.3 g). Yield: 92.7 %; 1H NMR (CDC13, 300 MHz): δ 7.29- 7.21 (m, 2H), 7.11 (d, 7 = 8.1 Hz, 2H), 6.91 (dd, J = 9.0, 9.0 Hz, 1H), 6.82 (d, J = 8.1 Hz, 2H), 4.05 (s, 2H), 3.88 (s, 2H), 1.13- 1.10 (m, 2H), 0.93- 0.88 (m, 2H).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(4-fluoro-3-(4-((l-
( trifluoromethyl)cyclopropyl)methoxy jbenzyl )phenyl )-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3, 4, 5-triol:

To a stirred solution of 4-bromo-l-fluoro-2-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)benzene (step 1, 5.3 g) in dry tetrahydrafuran (40 ml) and toulene (40 ml, 1: 1), t-BuLi (2.3 M in hexane, 15.5 ml) was added at -78 °C under N2 atmosphere and stirred for about 30 minutes. To this solution (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6- ((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (Intermediate 2, 4.9 g) in toluene (30 ml) was added at -78 °C and stirred for about 2 hours then 6 % CH3S03H in MeOH (80 ml) was added slowly at -78 °C. The reaction mixture was allowed to room temperature and stirred for over night. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 0°C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1 :24) to afford the title compound as white solid (2.8
g). Yield: 41.7 %; 1H NMR (DMSO- D6, 300 MHz): 6 7.47 (d, J = 7.5 Hz, 1H), 7.39 (m, 1H), 7.11- 7.09 (m, 3H), 6.85 (d, J = 8.1 Hz, 2H), 4.99 (d, J = 5.1 Hz, 1H), 4.78- 4.76 (m, 2H), 4.55 (dd, J = 5.4, 5.4 Hz, 1H), 4.05 (s, 2H), 3.89 (Abq, J = 15 Hz, 2H), 3.74- 3.72 (m, 1H), 3.60- 3.47 (m, 2H), 3.24- 3.19 (m, 2H), 2.90 (s, 3H), 2.90- 2.84 (m, 1H), 1.06 (m, 2H), 0.97 (m, 2H); ES Mass: (M+Na) 539.15.
Step 3: Synthesis of (3R,4R,5S,6R)-2-(4-fluoro-3-(4-((l-
(trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxy
2H-pyran-3, 4, 5-triol:
To a stirred solution of (3R,4S,5S,6R)-2-(4-fluoro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 2.8 g) in dichloromethane (20 ml) and acetylnitrile (20 ml, 1: 1), Et3SiH (1.7 ml) was added at 0 °C under N2 atmosphere followed by BF3: Et20 (1 ml) and stirred for about 4 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc and the combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of a & β isomers) as white solid (2 g). Yield: 77 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.27- 7.21 (m, 2H), 7.13- 7.05 (m, 3H), 6.85 (d, J = 8.4 Hz, 2H), 4.96 (d, J = 2.1 Hz, 2H), 4.80 (d, J = 5.7 Hz, 1H), 4.46 (dd, J = 5.7 Hz, 5.7 Hz, lH), 4.05 (s, 2H), 3.96 (d, J = 9.3 Hz, 1H), 3.88- 3.87 (m, 2H), 3.71-3.65 (m, 1H), 3.51- 3.13 (m, 5H), 1.06 (m, 2H), 0.97 (m, 2H); ES Mass: (M+Na) 509.25.
Intermediate 25: Preparation of (3R.4R.5S.6R -2-(3-(4-((4.4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-4-fluorophenyl)-6-(hydroxymethyl)tetrahydro-
2H-pyran-3,4.5-triol:

Step 1: Synthesis of 4-bromo-2-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-l-fluorobenzene:

To a stirred solution of 4-(5-bromo-2-fluorobenzyl)phenol (Intermediate 22, 2.6 g) in dimethylformamide (40 ml), Cs2C03 (9 g) was added under N2 atmosphere followed by (4,4-difluoro-l-methylcyclohexyl)methyl 4-methylbenzenesulfonate (Intermediate 16, 3.2 g) at room temperature. The reaction temperature was raised to 120 °C and stirred for about 6 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and filtered through celite then the cake was washed with EtOAc. The filtrate was washed with water and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and n-Hexane (1:49) to afford the title compound as liquid (2.6 g). Yield: 66.6 %; 1H NMR (CDC13, 300 MHz): δ 7.23- 7.21 (m, 2H), 7.10 (d, J = 8.1 Hz, 2H), 6.90 (dd, J = 8.7, 8.7 Hz, 1H), 6.83 (d, J = 8.1 Hz, 2H), 3.88 (s, 2H), 3.66 (s, 2H), 1.99- 1.86 (m, 4H), 1.78- 1.69 (m, 2H), 1.58 (m, 2H), 1.09 (s, 3H).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl)-4-fluorophenyl)-6- ( hydroxymethyl )-2- methoxytetrahydro-2H-pyran-3,4,5-triol:

To a stirred solution of 4-bromo-2-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-l-fluorobenzene (step 1, 2.6 g) in dry
tetrahydrafuran (30 ml) and toluene (30 ml, 1: 1 ), t-BuLi (6 ml, 1.7 M in toluene) was added under N2 atmosphere at -78 °C and stirred for about 30 minutes. To this solution (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6- ((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (Intermediate 2, 2.3 g) in toluene (40 ml) was added at -78 °C and stirred for about 2 hours then 6 % CH3S03H in MeOH (39 ml) was added slowly at -78 °C. Then the reaction mixture was allowed to room temperature and stirred for about 12 hours. After completion of the reaction (monitored by TLC), reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to afford the title compound as white solid (1.6 g). Yield: 50 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.47 (d, J = 6.3 Hz, 1H), 7.39 (m, 1H), 7.13- 7.08 (m, 3H), 6.86 (d, J = 8.4 Hz, 2H), 4.95 (d, J = 5.4 Hz, 1H), 4.73- 4.71 (m, 2H), 4.52 (dd, J = 5.7, 5.7 Hz, 1H), 3.89 (ABq, J = 15 Hz, 2H), 3.70 (m, 3H), 3.60- 3.51 (m, 2H), 3.25- 3.20 (m, 2H), 2.91 (s, 3H), 2.88- 2.85 (m, 1H), 1.96- 1.87 (m, 4H), 1.69- 1.63 (m, 2H), 1.48- 1.44 (m, 2H), 1.03 (s, 3H); ES Mass: (M+Na) 563.15.
Step 3: Synthesis of (3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy )benzyl)-4-fluorophenyl )-6-(hydroxymethyl )tetrahydro- 2H-pyran-3, 4, 5-triol:
To a stirred solution of (3R,4S,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-4-fluorophenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (step 2, 1.6 g) in dichloromethane (15 ml) and acetylnitrile (15 ml, 1 : 1), Et3SiH (0.9 ml) was added at 0°C under N2 atmosphere followed by BF3: Et20 (0.5 ml) and reaction was stirred for about 6 hours. After completion of the reaction (monitored by Ή-NMR), volatiles were concentrated under reduced pressure. The reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 Solution. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with
water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of α & β isomers) as white solid (890 mg). Yield: 59.3 %; HPLC: 87.06 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.27- 7.19 (m, 2H), 7.13- 7.05 (m, 3H), 6.86 (d, J = 8.7 Hz, 2H), 4.96- 4.94 (m, 2H), 4.79 (d, J = 5.7 Hz, 1H), 4.45 (dd, J = 5.7 Hz, 5.7 Hz, 1H), 3.96 (d, J = 9.0 Hz, 1H), 3.93- 3.87 (m, 2H), 3.71- 3.65 (m, 3H), 3.44- 3.11 (m, 5H), 1.96- 1.87 (m, 4H), 1.67- 1.63 (m, 2H), 1.48- 1.46 (m, 2H), 1.03 (s, 3H); ES Mass: (M+Na) 533.10.
Intermediate 26: Preparation of Synthesis of l-(ethoxymethyl)cvclopentyl)methyl 4- methylbenzenesulfonate:

Step 1: Synthesis of methyl cyclopentanecarboxylate:
OMe
Concentrated H2S04 (19.1 ml, 1.0 eq) was added drop-wise to a solution of cyclopentanecarboxylic acid (25 g, 0.1953 mol) in MeOH (125 ml) at 0°C and the reaction mixture was refluxed for about 48 hours. After completion of the reaction, the solvent was removed and the reaction mixture was dissolved in dichloromethane. The dichloromethane layer was washed with saturated NaHC03 solution and the organic layer was dried over anhydrous magnesium sulphate. The solvent was removed over rotary evaporator. The obtained residue with least moisture content was used directly for the next step. Yield: 16 gm.
Step 2: Synthesis of methyl l-(ethoxymethyl)cyclopentanecarboxylate:

To a solution of diisopropylamine (44.4 ml, 0.3169 mol) in tetrahydrafuran (200 ml), n-BuLi (2.5M, 105.6 ml, 0.264 mol) was added in hexane at 0°C and the
reaction mixture was allowed to stir at same temperature for about 10-15 minutes. The reaction mixture was stirred at room temperature for about 45 minutes. Methyl cyclopentanecarboxylate (step 1, 15 g, 0.15067 mol) in tetrahydrafuran (150 ml) was added drop-wise at -78°C to reaction mixture. The reaction mixture was stirred at same temperature for about 2 hours and then (iodomethoxy)ethane (12.67 ml, 0.1584 mol) was added drop- wise. The reaction was maintained at -78°C for an hour and allowed to attain room temperature. Reaction rnixture was stirred overnight at room temperature and quenched at 0°C with saturated NH4C1 solution. Tetrahydrafuran was concentrated over vaccum and the compound was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used for the next step. Crude yield: 10 gm. 1H NMR (CDC13, 300 MHz): δ 4.11 (q, 2H, J = 7.2 Hz); 2.13-1.41 (m, 13H); 1.21 (t, 3H, J = 7.2 Hz); 0.81 (t, 3H, J = 7.5 Hz). ESI Mass: 187 (M+l)
Step 3: Synthesis of ( 1 -(ethoxymethyl)cyclopentyl)methanol:

Methyl l-(ethoxymethyl)cyclopentanecarboxylate (step 2, 10 g, 0.0588 mol) in tetrahydrafuran (120 ml) was added to a suspension of L1AIH4 (6.7 g, 0.1764 mol) in tetrahydrafuran (200 ml) at 0°C and the reaction mixture was allowed to stir at same temperature for about 30 minutes. The reaction mixture was stirred overnight at room temperature. After completion of reaction, the reaction mixture was quenched with ice cold water at 0°C and filtered over celite. The residue was washed with hot ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained crude product was directly used for the next step. Crude yield: 8 gm. 1H NMR (CDC13, 300 MHz): δ 3.41 (s, 2H); 1.42-1.36 (m, 12H); 1.30-1.26 (t, 3H).
Step 4: Synthesis of 1 -(ethoxymethyl)cyclopentyl)methyl 4-methylbenzenesulfonate:
To a solution of (l-(ethoxymethyl)cyclopentyl)methanol (step 3, 8 g, 0.0563 mol) in dichloromethane (200 ml), triethyl amine (31.3 ml, 0.2253 mol) was added
followed by tosyl chloride (21.4 g, 0.1126 mol) at 0°C. After addition of catalytic amount of DMAP (0.567 g, 0.0056 mol), the reaction mixture was allowed to stir for overnight at room temperature. After completion of reaction, the reaction mixture was washed with water and brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 8 gm. 1H NMR (CDC13, 300 MHz): 5 7.80-7.77 (d, 2H, J = 8.1 Hz); 7.35-7.33 (d, 2H, J = 8.1 Hz); 3.78 (s, 2H); 2.45 (s, 3H); 1.39-1.27 (m, 12H); 0.68-0.63 (t, 3H, 7 = 7.5 Hz). ESI Mass: 313 (M+l).
Intermediate 27: Preparation of (3R,4S.5S.6R)-2-(4-chloro-3-(4-((l-
(ethoxymethyl)cvclopentyl methoxy)benzyl)phenyl)-6-(hvdroxymethyl)-2- methoxytetrahydro-2H-pyr -3,4,5-triol:

Step 1: Synthesis of 4-bromo-l -chloro-2-( 4-((l-
( ethoxymethyl )cyclopentyl )methoxy )benzyl jbenzene:

To a suspension of Cs2C03 (39.6 g, 0.1216 mol) in dimethylformamide (150 ml), 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate 1, 12 g, 0.0405 mol) in dimethylformamide (100 ml) was added followed by 1- (ethoxymethyl)cyclopentyl)methyl 4-methylbenzenesulfonate (Intermediate 26, 17.9 g, 0.0608 mol) in dimethylformamide (50 ml) then the reaction mixture was stirred for overnight at 120°C. After completion of the reaction as monitored by TLC, the reaction mixture was extracted with ether and washed with saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. Yield: 8.5 gm. 1H NMR (CDC13, 300 MHz): δ 7.27-7.23 (m, 3H); 7.09-7.06 (d, 2H, J=8.7Hz); 6.86-
6.83 (d, 2H, J=8.7Hz); 3.98 (s, 3H); 3.67 (s, 2H), 1.45-1.40 (m, 12H); 0.88-0.77 (t, 3H, J = 7.8 Hz).
Step 2: Synthesis of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l-
( ethoxymethyl )cyclopentyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )-2- methoxytetrahydro-2H-pyran-3,4,5-triol:
T-BuLi (1.7 M, 20.2 ml, 0.0344mol) was added drop-wise to a solution of 4- bromo- 1 -chloro-2-(4-(( 1 -(ethoxymethyl)cyclopentyl)methoxy)benzyl)benzene (Step 1, 7 g, 0.0172mol) in tetrahydrafuran: toluene (90 ml) (1:2) at -78°C. After 20 minutes, to this solution (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6- ((trimethylsilyloxy)methyl) tetrahydro-2H-pyran-2-one (Intermediate 2, 6.4 g, 0.0137mol) in toluene (70 ml) was added at -78°C. The reaction mixture was maintained at the same temperature for about 2 hours then quenched with methane sulfonic acid (6 % in MeOH, 105 ml). The reaction mixture was stirred at room temperature for an hour then quenched with saturated NaHC03 solution and diluted with ethyl acetate. The organic layer was washed with brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was separated by column chromatographic technique. 1H NMR (CDC13, 300 MHz): δ 7.48 (s, 1H), 7.34 (s, 2H), 7.04-7.01 (d, 2H, J=8.4Hz); 6.81- 6.78 (d, 2H, J = 8.7Hz); 5.06 (bs, 1H), 4.82 (bs, 2H), 4.57 (bs, 1H), 3.96-3.93 (q, 2H); 3.67 (m, 1H), 3.62 (s, 3H); 3.58-3.50 (m, 2H); 3.18-2.87 (m, 3H); 1.40-1.25 (m, 6H); 1.19-1.16 (m, 4H), 0.9 (s, 3H); ESI Mass: 552 (M+l).
Examples
Example 1: Preparation of (2S.3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcvclopentyl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl)tetrahydro-2H-pyran- 3 ,4,5-triol (β-isomer):


Triethylsilane (1.36 ml, 8.46 mmol) was added to a solution of (2S,3R,4R,5S,6R)-2-(4-cMoro-3-(4-((l-ethylcyclopentyl)methoxy)benzyl)phenyl)- 6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (Intermediate 4-P-isomer, 2.2 g, 4.23 mmol) in dichloromethane:MeCN (40:40 ml) at -10°C. The reaction mixture was allowed to stir at same temperature for 10 minutes then BF3.OEt2 (0.79 ml, 6.34 mmol) was added. The reaction mixture was stirred for about 30 minutes at -10°C and an hour at room temperature. After completion of reaction as monitored by TLC, the reaction mixture was quenched with 10% NaHC03 solution. The reaction mixture was washed with water, saturated brine solution and the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. (DMSO-D6+D20, 300 MHz): β-isomer: 6 7.35-7.28 (m, 2H); 7.23-7.16 (m, 1H); 7.08-7.05 (m, 2H); 6.81-6.79 (m, 2H); 4.09- 3.11 (m, 10H); 1.53-1.33 (m, 10H); 0.75 (t, 3H, J = 7.2 Hz). ESI Mass: 513.05 (M+Na).
Step 2: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclopentyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3,4, 5-triyl triacetate:

To a solution of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclo-pentyl)methoxy)benzyl)phenyl)-tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 1, 1.3 g) in dichloromethane (50 ml) at 0°C was added pyridine (2.1 ml), acetic anhydride (2.5 ml) and catalytic amount of DMAP. The reaction mixture was stirred at room temperature for about an hour. After completion of reaction as
indicated by TLC, reaction mixture was diluted with dichloromethane, washed with water, 1% HC1 solution followed by saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. Product was purified by flash column chromatographic technique to remove unreacted Ac20. Recrystallisation of the obtained mixture in ethanol crystallized the desired isomer as white solid. 1H NMR (CDC13, 300 MHz): β-isomer: δ 7.37-7.25 (m, 1H); 7.20-7.17 (m, 1H); 7.07- 7.04 (m, 3H); 6.84-6.81 (m, 2H); 5.32-5.17 (m, 3H); 5.08-5.02 (m, 1H); 4.33-3.77 (m, 6H); 3.64 (m, 2H); 2.07 (s, 3H); 2.05 (s, 3H); 1.99 (s, 3H); 1.71 (s, 3H); 1.63- 1.45 (m, 10H); 0.85 (t, 3H, / = 7.2 Hz); ESI Mass: 681.25 (M+Na).
Step 3: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclopentyl )methoxy )benzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
A solution of 20% methanolic ammonia (10 ml) was added to (2R,3R,4R,5S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclopentyl)methoxy)benzyl)phenyl)-tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 2, 600 mg, 0.91 mmol) at 0°C and the reaction mixture was stirred overnight at room temperature. After completion of the reaction as indicated by TLC, solvent was removed over a rotary evaporator and the residue was dissolved in ethyl acetate. The organic layer was washed with saturated brine solution and dried over anhydrous Na2S04 and concentrated. The obtained title product was purified by column chromatographic technique. 1H NMR (DMSO-D6+D20, 300 MHz): β- isomer: 6 7.38-7.32 (m, 2H); 7.23 (d, 1H, J = 8.1 Hz); 7.08 (d, 2H, J = 8.4 Hz); 6.84 (d, 2H, J = 8.4 Hz); 4.96 (t, 2H, J = 4.2 Hz); 4.83 (d, 1H, J = 5.7 Hz); 4.45 (t, 1H, J = 5.7 Hz); 3.99-3.98 (m, 3H); 3.72-3.64 (m, 3H); 3.21-3.06 (m, 5H); 1.57-1.37 (m, 10H); 0.80 (t, 3H, J = 7.5 Hz); ESI Mass: 513.10 (M+Na);
Example 2: Preparation of (2R,3R.4R,5S.6R)-2-(4-chloro-3-(4-((l- ethylcvclopentyl)methoxy)benzyl)phenyl -6-(hydroxymethyl)tetrahvdro-2H-pyran- 3 A5-triol (oc-isomer):


The experimental procedure similarly as described in Example 1-step 1. H NMR (CDC13, 300 MHz): °c-isomer: δ 7.32-7.29 (m, IH); 7.16-7.10 (m, 2H); 7.05- 7.02 (m, 2H); 6.81-6.76 (m, 2H); 4.52-3.33 (m, 14H); 1.96-1.89 (m, 3H); 1.58-1.38 (m, 10H); 0.83 (t, 3H, J = 7.2 Hz).
Step 2: Synthesis of (2R,3R,4R,5S,6R)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclopentyl)methoxy jbenzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate:

The experimental procedure similarly as described in Example 1-step 2 1H NMR (CDCI3, 300 MHz): ^-isomer: a gummy substance, δ 7.36-7.30 (m, IH); 7.25- 7.11 (m, IH); 7.08-7.05 (m, 3H); 6.85-6.81 (m, 2H); 5.36-5.26 (m, 2H); 4.92-4.88 (m, IH); 4.66-4.61 (m, IH); 4.58-4.50 (m, IH); 4.40-4.36 (m, IH); 4.18-3.97 (m, 3H); 3.64 (s, 2H); 2.14-2.09 (m, 9H); 1.83 (s, 2H); 1.58-1.41 (m, 10H); 0.84 (t, 3H, J = 12 Hz).
Step 3: Synthesis of (2R,3R,4R5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclopentyl)methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
The experimental procedure similarly as described in Example 1-step 3' !H NMR (DMSO-D6+D20, 300 MHz): °c-isomer 5 7.32-7.20 (m, 3H); 7.03 (d, 2H, J = 8.4 Hz); 6.78 (d, 2H, J = 8.7 Hz); 4.45-4.44 (m, IH); 3.93-3.89 (m, 3H); 3.78-3.74 (m, 3H); 3.58 (s, 2H); 3.39-3.34 (m, IH); 3.11 (s, 4H); 1.51-1.33 (m, 10H); 0.74 (t, 3H, J = 7.5 Hz).
Example 3: Preparation of (2S,3R,4R,5S,6R -2-(4-chloro-3-(4-(q- ethylcvclobuty methoxy)benzyl)phenyl -6-(hvdroxwethyl)tetrahydro-2H-pyran-
3.4.5-triol:

Step 1: Synthesis of (2S,3R,4R5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclobutyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )-tetrahydro-2H-pyran- 3,4,5-triol:

Triethylsilane (3.9ml, 0.0242mol) was added to a solution of (2S,3R,4S,5S,6R)-2-(4-cMoro-3-(4-((l-ethylcyclobutyl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)-2-methoxy-tetrahydro-2H-pyran-3,4,5-triol (Intermediate 6, 3.5 g, 0.00691 mol) in dichloromethane: MeCN (40:40 ml) at -10°C. The reaction mixture was allowed to stir at same temperature for 10 minutes then BF3.OEt2 (3.9 ml, 0.0172 mol) was added. The reaction mixture was stirred at -10°C for about 30 minutes and at room temperature for an hour. After completion of reaction as monitored by TLC, the reaction mixture was quenched with saturated NaHC03 solution. The reaction mixture was washed with saturated brine solution; the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (DMSO-D6, 300 MHz): δ 7.38-7.31 (m, 1H); 7.24-7.23 (m, 1H); 7.10-7.08 (d, 2H, J=8.7); 6.87-6.85 (d, 2H, J=8.7Hz); 4.98-4.96 (m, 2H); 4.86-4.84 (d, lH,J=5.7Hz); 4.49-4.45(m, 1H), 3.99- 3.96 (m, 1H), 3.80 (s, 3H), 3.71 (s, 2H), 3.70-3.66 (m, 1H), 3.21-3.08 (m, 5H), 1.86- 1.73 (m, 6H), 1.60-1.53 (q, 2H, J=7.5Hz), 0.80-0.75 (t, 3H); ESI Mass: 499.04 (M+Na).
Step 2: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclobutyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate:

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran- 3,4,5-triol (step 1, 3 g) in dichloromethane (50 ml) at 0°C was added pyridine (5.08 ml), acetic anhydride (5.95 ml) and 76 mg of DMAP. The reaction mixture was stirred at room temperature for about an hour. After completion of the reaction as monitored by TLC, reaction mixture was diluted with dichloromethane, washed with water, 1% HC1 solution, water followed by saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. Product was purified by flash column chromatographic technique to remove unreacted Ac20. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid. 1H NMR (CDC13, 300 MHz): δ 7.37-7.34 (m, 1H); 7.20-7.19 (m, 1H); 7.08-7.04 (m, 3H); 6.86-6.83 (m, 2H); 5.31-5.17 (m, 2H); 5.08-5.02 (m, 1H); 4.33- 4.23 (m, 3H); 4.16-4.15(m, 1H), 4.03-4.00 (q, 2H), 3.81 (s, 2H); 3.79-3.76 (m, 1H), 2.08 (s, 2H), 2.05 (s, 2H), 1.99 (s, 3H), 1.92-1.90 (m, 4H), 1.90-1.86 (m, 2H), 1.84- 1.59 (m, 5H), 0.85-0.80 (t, 3H, J=7.5Hz). ESI Mass: 667.2 (M+Na).
Step 3: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclobutyl jmethoxy )benzyl )phenyl )-6-( hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
A solution of 20% methanolic ammonia (30 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclobutyl)methoxy)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 2, 2.5 g, 0.0038 mol) at 0°C and the reaction mixture was stirred overnight at room temperature. After completion of the reaction as monitored by TLC, solvent was removed over rotary evaporator and the residue was dissolved in ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (DMSO-D6+D20, 300 MHz): δ 7.34-7.28 (m, 2H); 7.20-7.18 (m, 1H); 7.07-7.04 (d, 2H, J=8.7Hz); 6.84-6.81 (d, 2H, J=8.7 Hz);
4.95-4.93 (m, 2H); 4.82-4.80 (d, 1H, 7=5.7 Hz); 4.45-4.41 (t, 1H, 7=5.7 Hz); 3.96- 3.93 (m, 3H); 3.76 (s, 2H); 3.68-3.62 (m, 1H), 3.15-3.13 (m, 5H); 1.83-1.69 (m, 6H); 1.54-1.52 (q, 2H), 0.77-0.72 (t, 3H, J=7.2Hz); 0.80 (t, 3H, J = 7.5 Hz); ESI Mass: 513.10 (M+Na).
Example 4: Preparation of (2S R,4R,5S,6R)-2-(4-chloro-3-(4-(q- ethylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-
3,4.5-triol:

Step 1: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl jmethoxy )benzyl )phenyl )-6-(hydroxymethyl )tetr hydro-2H-pyran- 3,4,5-triol:
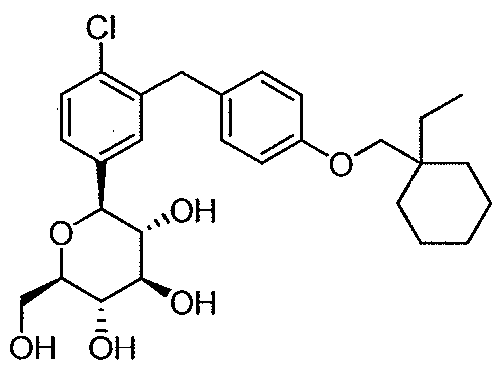
Triethylsilane (1.5 ml, 0.00963 mol) was added to a solution of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-((l-ethylcyclohexyl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate 8, 2.5 g, 0.0046 mol) in dichloromethane:MeCN (40:40 ml) at -10°C and the reaction mixture was allowed to stir for 10 minutes at same temperature then BF3.OEt2 (0.882 ml, 0.00702 mol) was added. The reaction mixture was stirred for about 30 minutes at - 10°C and for an hour at room temperature. After completion of reaction as monitored by TLC, the reaction mixture was quenched with 10% NaHC03 solution. The reaction mixture was washed with saturated brine solution and the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (CDC13, 300 MHz): (DMSO-D6+D20, 300 MHz): 6 7.38-7.32 (m, 3H); 7.24-7.20 (m, 2H); 7.10-7.07 (d, 2H, J=8.4Hz); 6.86-6.83 (d, 2H, J=8.4Hz); 6.18 (bs, 1H), 4.98-4.96 (m, 1H); 4.85-
4.83 (d, 1H, J=5.7Hz); 4.48-4.44 (m, 4H), 4.04-3.97 (m, 3H), 3.66 (s, 2H), 3.21-3.15 (m, 4H), 2.66-2.60 (m, 1H), 1.44-1.31 (m, 12H), 0.77-0.75 (t, 3H). ESI Mass: 527.25 (M+Na).
Step 2: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ethylcyclohexyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate:

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (step 1, 1.8 g) in dichloromethane (20 ml) at 0°C was added pyridine (2.87 ml), acetic anhydride (3.37 ml) and catalytic amount of DMAP. The reaction mixture was stirred at room temperature for about an hour. After completion of the reaction as monitored by TLC, the reaction mixture was diluted with dichloromethane, washed with water, 1% HCl solution followed by saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by flash column chromatographic technique to remove unreacted Ac20. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid. 1H NMR (CDC13, 300 MHz): δ 7.37-7.34 (d, 1H); 7.20-7.08 (m, 1H); 7.07-7.04 (m, 3H); 6.84-6.81 (d, 2H); 5.32-5.17 (m, 2H); 5.09- 5.02 (m, 1H); 4.33-4.24 (m, 2H), 4.16-3.94 (m, 3H), 3.82-3.77 (m, 1H); 3.66 (s, 2H); 2.08-2.05 (d, 6H); 1.99 (s, 3H); 1.70 (s, 3H); 1.51-1.44 (m, 12H); 0.82-0.77 (t, 3H, J = 7.5 Hz). ESI Mass: 695.2 (M+Na).
Step 3: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ethylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
A solution of 20% methanolic ammonia (10 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l-ethylcyclohexyl) methoxy)benzyl) phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 2, 0.8g, 0.0011 mol) at 0°C and the reaction mixture was stirred overnight at room temperature. After completion of reaction as monitored by TLC, solvent was
removed over rotary evaporator and the residue was dissolved in ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (DMSO-D6+D20, 300 MHz): δ 7.38-7.32 (m, 2H); 7.24-7.21 (m, 1H); 7.09 (d, 2H, / = 8.4 Hz); 6.85 (d, 2H, J = 8.7 Hz); 4.98-4.95 (m, 2H, J = 4.2 Hz); 4.84 (d, 1H, J = 6.0 Hz); 4.46 (t, 1H, J = 6.0 Hz); 4.00-3.97 (m, 3H); 3.75-3.67 (m, 3H); 3.45-3.43 (m, 1H); 3.24-3.08 (m, 4H), 1.45-1.31 (m, 12H), 0.75 (t, 3H, J = 7.2 Hz). ESI Mass: 527.25 (M+Na).
Example 5: Preparation of (2S,3R.4R,5S.6R)-2-(4-chloro-3-(,4-(('l- methylcvclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4.5-triol:

Step 1: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chbro-3-(4-((l- methylcyclohexyl )methoxy )benzyl)phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:

Triethylsilane (0.76 ml, 0.0048 mol) was added at -10°C to a solution of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-((l-methylcyclohexyl)methoxy)benzyl)phenyl)- 6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate 10, 1 g, 0.0019 mol) in dichloromethane:MeCN (10: 10 ml). The reaction mixture was allowed to stir at same temperature for 10 min and then BF3.OEt2 (0.365 ml, 0.0028 mol) was added. The reaction mixture was stirred for about 30 minutes at -10°C and at room temperature for an hour. After completion of reaction as monitored by TLC, reaction mixture was quenched with 10% NaHC03 solution. The reaction mixture
was washed with saturated brine solution; the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (CDC13, 300 MHz): δ 7.38-7.35 (m, 2H); 7.31 (m, 1H); 7.24-7.21 (d, 2HJ=8.4Hz); 6.85-6.83 (d, 2H, J=8.4Hz); 6.17 (bs, 1H), 4.97- 4.96 (m, 1H); 4.85-4.83 (d, 2H, J=5.7Hz); 4.48-4.44 (m, 1H), 3.98-3.96 (m, 2H), 3.62 (s, 2H), 3.23-3.19 (m, 4H), 2.62-2.60 (m, 2H), 1.44-1.29 (m, 10H), 1.11-1.06 (m, 3H), 0.97 (s, 3H); ESI Mass: 527.25 (M+Na).
Step 2: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- methylcyclohexyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate:

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (step 1, 0.6 g) in dichloromethane (10 ml), pyridine (0.98 ml), acetic anhydride (1.15 ml) and a catalytic amount of DMAP were added at 0°C. The reaction mixture was stirred at room temperature for about an hour. After completion of the reaction as monitored by TLC, the reaction mixture was diluted with dichloromethane, washed with water, 1% HCl solution followed by saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. Product was purified by flash column chromatographic technique to remove unreacted Ac20. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid. 1H NMR (CDC13, 300 MHz): δ 7.36-7.34 (d, 1H, J=8.1Hz); 7.20-7.16 (m, 1H); 7.06-7.03 (d, 3H); 6.83-6.81 (d, 2H, J=8.7Hz); 5.31- 5.17 (m, 2H); 5.08-5.02 (m, 1H); 4.32-4.23 (m, 3H), 4.16-3.99 (m, 2H), 3.81-3.77 (m, 1H); 3.60 (s, 2H); 2.07 (s, 3H); 2.05(s, 3H), 1.99 (s, 3H); 1.71 (s, 3H); 1.49-1.35 (m, 10H); 1.01 (s, 3H). ESI Mass: 680.92 (M+Na).
Step 3: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
A solution of 20% methanolic ammonia (15 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- methylcyclohexyl)methoxy)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 2, 0.35g, 0.0005mol) at 0°C and the reaction mixture was stirred for overnight at room temperature. After completion of reaction as monitored by TLC, solvent was removed over rotary evaporator and the residue was dissolved in ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (DMSO-D6+D20, 300 MHz): 6 7.34-7.27 (m, 2H); 7.20-7.17 (m, 1H); 7.06-7.03 (d, 2H, J = 8.7 Hz); 6.82-6.79 (d, 2H, J = 8.7 Hz); 4.95-4.92 (m, 2H, ); 4.82-4.80 (d, 1H, J = 5.7 Hz); 4.45-4.41 (t, 1H, J = 6.0 Hz); 3.99-3.93 (m, 3H); 3.66-3.62 (m, 1H); 3.58 (s, 2H), 3.15 (m, 5H); 1.40-1.25 (m, 10H), 0.93 (s, 3H). ESI Mass: 513.25 (M+Na).
Example 6: Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- yl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl)tetrahydro-2H-pyran-3,4,5-triol:

Step 1: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- yl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H^

Triethylsilane (0.32ml, 0.00196mol) was added to a solution of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3-yl)methoxy)benzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate 12, 0.5 g, 0.0098 mol) in dichloromethane:MeCN (10: 10 ml) at -10°C. The reaction mixture was allowed to stir at same temperature for 10 minutes then BF3:OEt2 (0.19 ml, 0.00147 mol) was added. The reaction mixture was stirred for about 30 minutes at -
10°C and at room temperature for an hour. After completion of reaction as monitored by TLC, the reaction mixture was quenched with saturated NaHC03 solution. The reaction mixture was washed with saturated brine solution; the organic layer was dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (DMSO-D6, 300 MHz): δ
7.37-7.31 (m, 1H); 7.23-7.20 (m, 1H); 7.10-7.08 (d, 2H, J=8.4); 6.87-6.84 (d, 2H, J=8.4Hz); 4.98-4.96 (m, 2H); 4.85-4.81 (m, 2H,J=5.7Hz); 4.47 (m, 1H), 3.99-3.96 (m, 3H), 3.74-3.67 (m, 2H), 3.65 (s, 2H), 3.20-3.16 (m, 4H), 1.44-1.39 (q, 2H), 1.13- 1.02 (m, 2H), 0.85-0.80 (t, 3H).
Step 2: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((3- ethyloxetan-3-yl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate:

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- yl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (step 1, 0.4 g) in dichloromethane (50 ml), pyridine (0.67 ml), acetic anhydride (0.79 ml) and O.Olg of DMAP were added at 0°C. The reaction mixture was stirred at room temperature for about an hour. After completion of reaction as monitored by TLC, the reaction mixture was diluted with dichloromethane, washed with water, 1% HC1 solution, water followed by saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. Obtained product was purified by flash column chromatographic technique to remove unreacted Ac20. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid. Ή NMR
(CDC13, 300 MHz): δ 7.37-7.34 (m, 1H); 7.20-7.19 (m, 1H); 7.08-7.04 (m, 3H); 6.86-6.83 (m, 2H); 5.31-5.17 (m, 2H); 5.08-5.02 (m, 1H); 4.33-4.23 (m, 3H); 4.16- 4.15 (m, 1H), 4.03-4.00 (q, 2H), 3.81 (s, 2H); 3.79-3.76 (m, 1H), 2.08 (s, 2H), 2.05 (s, 2H), 1.99 (s, 3H), 1.92-1.90 (m, 4H), 1.90-1.86 (m, 2H), 1.84-1.59 (m, 5H), 0.85- 0.80 (t, 3H, J=7.5Hz); ESI Mass: 667.2 (M+Na).
Step 3: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- yl jmethoxy jbenzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran-3, 4, 5-triol:
A solution of 20% methanolic ammonia (10 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((3-ethyloxetan-3- yl)methoxy)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (step 2, 0.4 g, 0.000619 mol) at 0°C and the reaction mixture was stirred overnight at room temperature. After completion of the reaction as monitored by TLC, the solvent was removed over rotary evaporator and the residue was dissolved in ethyl acetate. The organic layer was washed with saturated brine solution, dried over anhydrous Na2S04 and concentrated. The obtained product was purified by column chromatographic technique. 1H NMR (DMSO-D6+D20, 300 MHz): δ 7.34-7.28 (m, 2H); 7.20-7.18 (m, 1H, ); 7.07-7.04 (d, 2H, J=8.7Hz); 6.84-6.81 (d, 2H, J = 8.7 Hz); 4.95-4.93 (m, 2H); 4.82-4.80 (d, 1H, J = 5.7 Hz); 4.45-4.41 (t, 1H, /=5.7Hz); 3.96- 3.93 (m, 3H); 3.76 (s, 2H); 3.68-3.62 (m, 1H), 3.15-3.13 (m, 5H); 1.83-1.69 (m, 6H); 1.54-1.52 (q, 2H), 0.77-0.72 (t, 3H, J=7.2Hz), 0.80 (t, 3H, J = 7.5 Hz); ESI Mass: 513.10 (M+Na).
Example 7: Preparation of (2R.3S.4R.5R.6S)-2-(hydroxymethyl)-6-(4-methyl-3-(4-
((l-(trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-
3A5-triol:

To a stirred solution of (2S,3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4- methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate 13, 0.3 g) in dimethylformamide (10 ml), Cs2C03 (0.81 g) and (1- (trifluoromethyl)cyclopropyl)methyl 4-methylbenzenesulfonate (intermediate 14, 0.31 g) were added under N2 atmosphere at room temperature. The reaction mixture was heated to 120 °C and stirred for about 8 hours. After completion of the reaction (monitored by TLC), reaction mixture was cooled to room temperature and partitioned between water and EtOAc. Two layers were separated and aqueous layer was again extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the residue.
The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 25) to afford the title compound as white solid (198 mg). Yield: 49.2 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.11- 7.08 (m, 3H), 7.03 (d, J = 8.7 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 4.92 (d, J = 4.5 Hz, 2H), 4.72 (d, J = 5.7 Hz, 1H), 4.43 (dd, J = 5.7, 5.7 Hz, 1H), 4.05 (s, 2H), 3.93 (d, J = 9.3 Hz, 1H), 3.86 (s, 2H), 3.71- 3.66 (m, 1H), 3.46- 3.38 (m, 1H), 3.25- 3.12 (m, 4H), 2.14 (s, 3H), 1.09- 1.05 (m, 2H), 1.00- 0.97 (m, 2H); ES Mass: (M+ Na) 505.11.
Example 8: Preparation of (,2R,3S.4R.5R,6S)-2-(,hvdroxymethyl)-6-(4-methyl-3-(4- ((1 -(trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyr an-3 ,4,5- triol:

To a stirred solution of (2S,3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4- methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3 ,4,5-triol (intermediate 13 , 0.3 g) in dimethylformamide (10 ml), Cs2C03 (0.81 g) and (1- (trifluoromethyl)cyclobutyl)methyl 4-methylbenzenesulfonate (intermediate 15, 0.33 g) were added under N2 atmosphere at room temperature. The reaction mixture was heated to 120 °C and stirred for about 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and partitioned between water and EtOAc. Two layers were separated and aqueous layer was again extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1 : 25) to afford the title compound as white solid (160 mg).
Yield: 41 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.12- 7.04 (m, 5H), 6.89 (d, J = 8.9 Hz, 2H), 4.92 (d, J = 4.8 Hz, 2H), 4.72 (d, J = 5.7 Hz, 1H), 4.43 (dd, J = 5.7, 5.7 Hz, 1H), 4.15 (s, 2H), 3.93 (d, J = 9.3 Hz, 1H), 3.87 (s, 2H), 3.72- 3.66 (m, 1H), 3.44- 3.40 (m, 1H), 3.26- 3.15 (m, 4H), 2.32- 2.21 (m, 4H), 2.15 (s, 3H), 2.10- 2.07 (m, 2H); ES Mass: (M+ Na) 519.10.
Example 9: Preparation of (2S R lR.5S,6R)-2-(3-(4-((4^-difluoro-l- methylcyclohexyl)methoxy)benzyl)-4-methylphenylV6-(hvdroxymethyl)tetrahy
2H-pyran-3A5-triol:

To a stirred solution of (2S,3R,4R,5S,6R)-2-(3-(4-hydroxybenzyl)-4- methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3 ,4,5-triol (intermediate 13 , 0.2 g) in dimethylformamide (8 ml), Cs2C03 (0.54 g) and (4,4-difluoro-l- methylcyclohexyl)methyl 4-methylbenzenesulfonate (intermediate 16, 0.22 g) were added under N2 atmosphere at room temperature. The reaction mixture was heated to 120 °C and stirred for about 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature and partitioned between water and EtOAc. Two layers were separated and aqueous layer was again extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1 : 25) to afford the title compound as white solid (1 19 mg). Yield: 42.5 %; 1H NMR (DMSO- D6, 300 MHz): 5 7.11- 7.08 (m, 3H), 7.03 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 4.92 (d, / = 4.5 Hz, 2H), 4.71 (d, J = 5.7 Hz, 1H), 4.43 (dd, J = 5.7, 5.7 Hz, 1H), 3.93 (d, J = 9.0 Hz, 1H), 3.86 (s, 2H), 3.71 (s, 2H), 3.68- 3.66 (m, 1H), 3.46- 3.38 (m, 1H), 3.26- 3.12 (m, 4H), 2.15 (s, 3H), 1.97- 1.85 (m, 4H), 1.70- 1.61 (m, 2H), 1.49- 1.44 (m, 2H), 1.03 (s, 3H); ES Mass: (M+ Na) 529.20.
Example 10: Preparation of (2S.3R.4R,5S,6R)-2-(4-chloro-3-(4-((l-
(trifluoromethyl)cvclobutyl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl)tetrahydro-
2H-pyran-3,4,5-triol:

Step 1: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ( trifluoromethyl jcyclobutyl jmethoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate (β isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclobutyl)rnethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Intermediate 17, 3 g, mixture of α & β isomers ) in dichloromethane (80 ml), pyridine (4.7 ml) and AC20 (5.5 ml) followed by DMAP (catalytic) were added at 0° C under N2 atmosphere. The reaction temperature was slowly raised to room temperature and stirred for over night. After completion of the reaction (monitored by TLC), the reaction mixture was extracted with dichloromethane. The combined organic layers were washed with saturated 2N HC1 solution and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (3.8 g). HPLC: 97.97 %; H NMR (CDC13, 300 MHz): δ 7.36 (d, / = 8.1 Hz, 1H), 7.19 (dd, / = 1.8, 8.1 Hz, 1H), 7.17- 7.07 (m, 3H), 6.87 (d, / = 8.7 Hz, 2H), 5.32- 5.17 (m, 2H), 5.06 (dd, J = 9.6 Hz, 1H), 4.34-4.24 (m, 2H), 4.17- 4.02 (m, 5H), 3.83- 3.78 (m, 1H), 2.38- 2.31 (m, 2H), 2.23- 2.19 (m, 2H), 2.08 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H), 2.08- 1.99 (m, 2H), 1.72 (s, 3H); ES Mass: (M+Na) 707.
Step 2: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl)-6- ( hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol:
NH3 in MeOH (70 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (4-chloro-3-(4-((l-(trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)tetrahydro- 2H-pyran-3,4,5-triyl triacetate (β isomer, step 1, 0.8 g) at room temperature and stirred for over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title compound as white solid (470 mg). Yield: 78.1 %; 1H NMR (DMSO- D6, 300 MHz): 5 7.38-7.32 (m, 2H), 7.24- 7.14 (m, 1H), 7.12 (d, / = 8.5 Hz, 2H), 6.90 (d, / = 8.5 Hz, 2H), 4.97- 4.95 (m, 2H), 4.84 (d, J = 5.7 Hz, 1H), 4.46(dd, 7 = 5.7 Hz, 5.7 Hz, 1H), 4.15 (s, 2H), 4.00-3.97 (m, 3H), 3.71- 3.66 (m, 1H), 3.45-3.40 (m, 1H), 3.26- 3.09 (m, 4H), 2.29- 2.24 (m, 2H), 2.10- 1.93 (m, 4H); ES Mass: (M+Na) 539; HPLC: 96.63 .
Example 11: Preparation of (2S.3R.4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl)tetrahydro-2H- pyran-3A5-triol:

Step 1: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- methylcyclopentyljmethoxy )benzyl jphenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate (β- isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol (Intermediate 19, mixture of α & β isomers, 1.8 g) in dichloromethane (40 ml), pyridine (3.6 ml) was added at 0°C under N2 atmosphere followed by AC20 (3.6 ml) and DMAP (catalytic). The reaction temperature was
slowly raised to room temperature and stirred for over night. After completion of the reaction (monitored by TLC), the reaction mixture was extracted with dichloromethane. The combined organic layers were washed with 3N HC1 solution and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (1.2 g). Yield: 50 %; HPLC: 97.57 ; 1H NMR (CDC13, 300 MHz) of compound 15: 6 7.35 (d, J = 8.1 Hz, 1H), 7.18 (dd, 7 = 2.1, 8.1 Hz, 1H), 7.07- 7.04 (m, 3H), 6.83 (d, J = 8.4 Hz, 2H), 5.32- 5.17 (m, 2H), 5.05 (dd, J = 9.3 Hz, 1H), 4.33- 4.24 (m, 2H), 4.14 (dd, J = 2.1, 12.34 Hz, 1H), 4.02- 4.0 (m, 2H), 3.82- 3.77 (m, 1H), 3.64 (s, 2H), 2.08 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H), 1.71 (s, 3H), 1.62- 1.56 (m, 6H), 1.41- 1.39 (m, 2H), 1.11 (s, 3H); ES Mass: (M+Na) 667.
Step 2: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- methylcyclopentyl)methoxy jbenzyl )phenyl )-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
NH3 in MeOH (100 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)- 6-(4-chloro-3-(4-((l-methylcyclopentyl)methoxy)benzyl)phenyl)tetrahydro-2H- pyran-3,4,5-triyl triacetate (β- isomer, Step 1, 1.2 g) at room temperature and stirred over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1 : 19) to afford the title compound as white solid (700 mg). Yield: 79 %; HPLC: 93.16 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.36 (d, J = 8.1 Hz, 1H), 7.31 (d, J = 1.8 Hz, 1H), 7.22 (dd, J = 1.8, 8.1 Hz, 1H), 7.08 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 4.97- 4.95 (m, 2H), 4.83 (d, J = 5.7 Hz, 1H), 4.45 (dd, J = 6.0 Hz, 1H), 3.99- 3.96 (m, 3H), 3.71- 3.69 (m, 1H), 3.65 (s, 2H), 3.45- 3.41 (m, 1H), 3.29- 3.05 (m, 4H), 1.62- 1.58 (m, 6H), 1.34- 1.32 (m, 2H), 1.05 (s, 3H); ES Mass: (M+Na) 499.
Example 12: Preparation of (,2S.3R.4R.5S.6R -2-(4-chloro-3-(,4-f('l-
(trifluoromethyl)cvclopropyl)methoxy)benzyl)phenyl)-6-
(hvdroxymethyl)tetrahydro-2H-pyran-3A5-triol:

Step 1: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ( trifluoromethyl )cyclopropyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5- triyl triacetate (β- isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-
(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate 20, mixture of α & β isomers, 1.8 g) in dichloromethane (40 ml), pyridine (2.9 ml) was added at 0 °C under N2 atmosphere followed by AC20 (3.4 ml) and DMAP (catalytic). The reaction temperature was slowly raised to room temperature and stirred over night. After completion of the reaction (monitored by TLC), the reaction mixture was diluted with dichloromethane and the organic layer was washed with 3N HCl solution and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (1.5 g). Yield: 62.5 %; 1H NMR (CDC13, 300 MHz): δ 7.34 (d, J = 8.4 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 7.06- 7.03 (m, 3H), 6.80 (d, J = 8.4 Hz, 2H), 5.30- 5.15 (m, 2H), 5.04 (dd, J = 9.4 Hz, 1H), 4.31-4.22 (m, 2H), 4.15- 3.94 (m, 5H), 3.80- 3.77 (m, 1H), 2.06 (s, 3H), 2.03 (s, 3H), 1.97 (s, 3H), 1.69 (s, 3H), 1.10- 1.08 (m, 2H), 0.90 (m, 2H); ES Mass: (M+Na) 693.
Step 2: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3, 4, 5-triol:
NH3 in MeOH (60 ml) was added to(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (4-chloro-3-(4-((l-(trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)tetrahydro- 2H-pyran-3,4,5-triyl triacetate (β- isomer, step 1, 1.5 g) at room temperature and stirred over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title compound as white solid (0.70 g). Yield: 63.6 %; 1H NMR (DMSO- D6, 300 MHz): 6 7.36 (d, J = 8.1 Hz, 1H), 7.32 (d, J = 1.8 Hz, 1H), 7.22 (dd, J = 1.8, 8.1 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.85 (d, / = 8.4 Hz, 2H), 4.97- 4.95 (m, 2H), 4.84 (d, J = 5.7 Hz, 1H), 4.46(dd, J = 5.7 Hz, 5.7 Hz, 1H), 4.06 (s, 2H), 3.99-3.97 (m, 3H), 3.71- 3.66 (m, 1H), 3.45-3.39 (m, 1H), 3.24- 3.10 (m, 4H), 1.07- 1.05 (m, 2H), 0.97 (m, 2H); ES Mass: (M+Na) 525; HPLC: 97.31 %.
Example 13: Preparation of (2S.3R.4R.5S.6R)-2-(4-chloro-3-(4-((4<4-difluoro-l- methylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3A5-triol:

Step 1: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((4,4- difluoro-l-methylcyclohexyl)methoxy)benzyl)phenyl)tetraty
triacetate (β- isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- 3,4,5-triol (Intermediate 21, mixture of α & β isomers, 0.67 g) in dichloromethane (20 ml), Pyridine (1 ml) was added at 0 °C under N2 atmosphere followed by AC20 (1.2 ml) and DMAP (catalytic). The reaction temperature was slowly raised to room
temperature and stirred for over night. After completion of the reaction (monitored by TLC), reaction mixture was extracted with dichloromethane. The combined organic layers were washed with 3N HC1 solution and brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (690 mg). Yield: 78.1 %; 1H NMR (CDCI3, 300 MHz) of compound: δ 7.35 (d, J = 8.4 Hz, 1H), 7.18 (dd, J = 1.5, 8.4 Hz, 1H), 7.08- 7.04 (m, 3H), 6.81 (d, J = 8.7 Hz, 2H), 5.31- 5.17 (m, 2H), 5.05 (dd, J = 9.6 Hz, 1H), 4.33-4.23 (m, 2H), 4.15- 4.11 (m, 1H), 4.02- 4.00 (m, 2H), 3.82- 3.77 (m, 1H), 3.66 (s, 2H), 2.07 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H), 1.69 (s, 3H), 2.07- 1.70 (m, 6H), 1.60- 1.58 (m, 2H), 1.09 (s, 3H); HPLC: 91.18 %; ES Mass: (M+Na) 717.
Step 2: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro-2H-pyran- 3,4,5-triol:
NH3 in MeOH (20 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (4-chloro-3-(4-((4,4-difluoro- 1 - methylcyclohexyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (β- isomer, step 1, 0.69 g) at room temperature and stirred for over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title compound as white solid (270 mg). Yield: 51.7 %; HPLC: 91.03%; 1H NMR (DMSO- D6, 300 MHz): 6 7.37 (d, J = 8.4 Hz, 1H), 7.31 (s, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.86 (d, / = 8.4 Hz, 2H), 4.98- 4.95 (m, 2H), 4.84 (d, J = 5.7 Hz, 1H), 4.44 (dd, J = 5.7 Hz, 5.7 Hz, 1H), 3.99- 3.97 (m, 3H), 3.71- 3.66 (m, 3H), 3.47- 3.06 (m, 5H), 1.97- 1.87 (m, 4H), 1.70- 1.61 (m, 2H)( 1.49- 1.44 (m, 2H), 1.03 (s, 3H); ES Mass: (M+Na) 549.
Example 14: Preparation of (2S.3R.4R.5S.6R)-2-(4-fluoro-3-r4-('il- (trifluoromethyl)cvclobutyl methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-
2H-pyran-3.4.5-triol:

Step 1: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-fluoro-3-(4-((l- ( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate (β- isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(4-fluoro-3-(4-((l- (trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Intermediate 23, mixture of α & β isomers, 2.3 g) in dichloromethane 960 ml), pyridine (3.9 ml) was added at 0°C under N2 atmosphere followed by AC20 (4.3 ml) and DMAP (catalytic). The reaction temperature was slowly raised to room temperature and stirred for over night. After completion of the reaction (monitored by TLC), the reaction mixture was extracted with dichloromethane. The combined organic layers were washed with saturated 2N HC1 solution and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (1.2 g). Yield: 40 %; HPLC: 99.91 %; lR NMR (CDC13, 300 MHz) of compound 16: δ 7.20 (m, 1H), 7.12- 6.98 (m, 4H), 6.86 (d, / = 8.4 Hz, 2H), 5.31- 5.17 (m, 2H), 5.06 (dd, J = 9.6, 9.6 Hz, 1H), 4.33- 4.23 (m, 2H), 4.15- 4.10 (m, 2H), 4.02 (s, 2H), 3.92- 3.91 (m, 2H), 3.81- 3.78 (1H), 2.36- 2.30 (m, 2H), 2.22- 2.19 (m, 2H), 2.07 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H), 2.07- 1.99 (m, 2H), 1.70 (s, 3H); ES Mass: (M+Na) 691.20.
Step 2: Synthesis of (2S,3R,4R,5S,6R)-2-(4-fluoro-3-(4-((l-
( trifluoromethyl )cyclobutyl )methoxy )benzyl )phenyl )-6-( hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol:
NH3 in MeOH (70 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (4-fluoro-3-(4-((l-(trifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)tetrahydro- 2H-pyran-3,4,5-triyl triacetate (β- isomer, step 1, 1.2 g) at room temperature and stirred for over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title compound as white solid (810 mg). Yield: 90.2 %; HPLC: 99.45 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.28-7.20 (m, 2H), 7.16- 7.06 (m, 3H), 6.91 (d, J = 8.4 Hz, 2H), 4.96 (d, J = 2.1 Hz, 2H), 4.80 (d, J = 5.4 Hz, 1H), 4.46 (dd, J = 5.7 Hz, 5.7 Hz, 1H), 4.15 (s, 2H), 3.97 (d, J = 9.3 Hz, 1H), 3.89- 3.88 (m, 2H), 3.71-3.65 (m, 1H), 3.51- 3.11 (m, 5H), 2.26- 2.24 (m, 2H), 2.10- 1.89 (m, 4H); ES Mass: (M+Na) 523.20.
Example 15: Preparation of (2S,3R,4R.5S,6RV2-(4-chloro-3-(4-((l-
(trifluoromethyl)cvclopropyl)methoxy')benzyl)phenyl)-6-
(hvdroxvmethvl tetrahvdro-2H-pvran-3,4,5-triol:

Step 1: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-fluoro-3-(4-((l- ( trifluoromethyl jcyclopropyl )methoxy )benzyl jphenyl )tetrahydro-2H-pyran-3, 4, 5- triyl triacetate (β- isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(4-fluoro-3-(4-((l- (trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-
(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate 24, mixture of α & β isomers, 2 g) in dichloromethane (20 ml), pyridine (3.3 ml) was added at 0°C under N2 atmosphere followed by AC20 (3.9 ml) and DMAP (catalytic). The reaction
temperature was slowly raised to room temperature and stirred for over night. After completion of the reaction (monitored by TLC), the reaction mixture was diluted with dichloromethane and the organic layer was washed with 3N HCl solution and brine, dried over anhydrous sodium sulphate and concentrated to give the residue. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (1.8 g). Yield: 66.9 %; HPLC: 96.70 ; 1H NMR (CDC13, 300 MHz): δ 7.22- 7.20 (m, IH), 7.10- 6.98 (m, 4H), 6.81 (d, J = 8.1 Hz, 2H), 5.32- 5.17 (m, 2H), 5.05 (dd, J = 9.3 Hz, IH), 4.33- 4.23 (m, 2H), 4.16- 4.11 (m, IH), 4.04 (s, 2H), 3.91- 3.84 (m, 2H), 3.81- 3.78 (m, IH), 2.07 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H), 1.69 (s, 3H), 1.13- 1.10 (m, 2H), 0.93- 0.88 (m, 2H); ES Mass: (M+Na) 677.15.
Step 2: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ( trifluoromethyl jcyclopropyl )methoxy jbenzyl )phenyl)-6- ( hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol:
NH3 in MeOH (100 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)- 6-(4-fluoro-3-(4-((l-
(trifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5- triyl triacetate (β- isomer, step 1, 1.8 g) at room temperature and stirred over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title compound as white solid (0.90 g). Yield: 69.2 %; HPLC: 98.21 %; 1H NMR (DMSO- D6, 300 MHz): δ 7.27- 7.21 (m, 2H), 7.13- 7.05 (m, 3H), 6.85 (d, J = 8.4 Hz, 2H), 4.96 (d, J = 2.1 Hz, 2H), 4.80 (d, J = 5.7 Hz, IH), 4.46 (dd, J = 5.7 Hz, 5.7 Hz, IH), 4.05 (s, 2H), 3.96 (d, J = 9.3 Hz, IH), 3.88- 3.87 (m, 2H), 3.71-3.65 (m, IH), 3.51- 3.13 (m, 5H), 1.06 (m, 2H), 0.97 (m, 2H); ES Mass: (M+Na) 509.05.
Example 16: Preparation of (2S,3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcvclohexyl)methoxy)benzyl)-4-fluorophenyl)-6-(hvdroxymethyl)tetrahydro-
2H-pyran-3,4,5-triol:
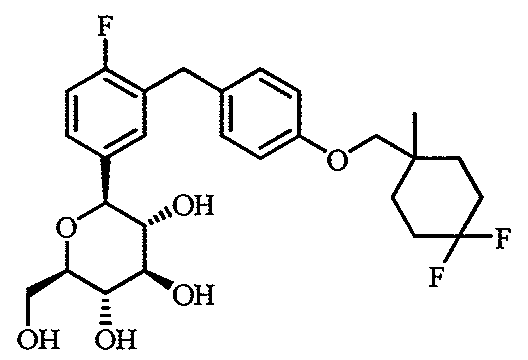
Step 1: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl)-4-fluorophenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate (β- isomer):

To a stirred solution of (3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl)methoxy)benzyl)-4-fluorophenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (Intermediate 25, mixture of α & β isomers, 0.89 g) in dichloromethane (20 ml), pyridine (1.4 ml) was added at 0 °C under N2 atmosphere followed by AC20 (1.5 ml) and DMAP (catalytic). The reaction temperature was slowly raised to room temperature and stirred for over night. After completion of the reaction (monitored by TLC), the reaction mixture was extracted with dichloromethane. The combined organic layers were washed with 3N HC1 solution and brine, dried over anhydrous sodium sulphate and concentrated to give the crude compounds. The crude product was purified via silica gel column chromatography with EtOAc and Hexane (2: 4) to afford the title compound (β- isomer) as white solid (790 mg). Yield: 71.8 %; HPLC: 96.20 %; 1H NMR (CDC13, 300 MHz): δ 7.24- 7.18 (m, 1H), 7.10- 6.98 (m, 4H), 6.81 (d, J = 8.7 Hz, 2H), 5.32- 5.17 (m, 2H), 5.06 (dd, J = 9.6, 9.6 Hz, 1H), 4.33- 4.23 (m, 2H), 4.19- 4.11 (m, 1H), 3.91- 3.90 (m, 2H), 3.82- 3.77 (m, 1H), 3.65 (s, 2H), 2.07 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 2.02- 1.83 (m, 4H), 1.78- 1.70 (m, 2H), 1.70 (s, 3H), 1.59- 1.53 (m, 2H), 1.08 (s, 3H); ES Mass: (M+Na) 701.15.
Step 2: Synthesis of (2S,3R4R5S,6R)-2-(3-(4-((4,4-difluoro-l- methylcyclohexyl )methoxy )benzyl)-4-fluorophenyl )-6-(hydroxymethyl )tetrahydro- 2H-pyran-3,4,5-triol:
NH3 in MeOH (4 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6- (3-(4-((4,4-difluoro-l-methylcyclohexyl)methoxy)benzyl)-4- fluorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (β- isomer, step 1, 0.79 g) at room temperature and stirred for over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title compound as white solid (270 mg). Yield: 51.7 %; HPLC: 93.16%; JH NMR (DMSO- D6, 300 MHz): δ 7.27- 7.19 (m, 2H), 7.13- 7.05 (m, 3H), 6.86 (d, J = 8.7 Hz, 2H), 4.96- 4.94 (m, 2H), 4.79 (d, J = 5.7 Hz, 1H), 4.45 (dd, J = 5.7 Hz, 5.7 Hz, 1H), 3.96 (d, J = 9.0 Hz, 1H), 3.93- 3.87 (m, 2H), 3.71- 3.65 (m, 3H), 3.44- 3.11 (m, 5H), 1.96- 1.87 (m, 4H), 1.67- 1.63 (m, 2H), 1.48- 1.46 (m, 2H), 1.03 (s, 3H); ES Mass: (M+Na) 533.15.
Example 17: Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l-
(ethoxymethyl)cyclopentyl)methoxy)benzyl)phenyl)-6-(hvdroxymethyl)tetrahydro-
2H-pyran-3,4.5-triol:

Step 1: (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l-
( ethoxymethyl )cyclopentyl )methoxy )benzyl )phenyl )-6-( hydroxymethyl )tetrahydro- 2H-pyran-3, 4, 5-triol:

To a stirred solution of (3R,4S,5S,6R)-2-(4-chloro-3-(4-((l- (ethoxymethyl)cyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)-2- methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate 27, 2.8 g) in dichloromethane (20 ml) and acetylnitrile (20 ml, 1: 1), Et3SiH (1.7 ml) was added at 0 °C under N2
atmosphere followed by BF3: Et20 (1 ml) and stirred for about 4 hours at room temperature. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure. Then the reaction mixture was cooled to 0 °C and neutralized by cautious addition of saturated aq. NaHC03 solution. The reaction mixture was extracted with EtOAc and the combined organic layers were washed with water and brine, dried over anhydrous sodium sulphate and concentrated to get the residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1:24) to give mixture compound (mixture of a & P isomers) as white solid (1.3 g). !H NMR (DMSO- D6, 300 MHz): 5 0.70-0.84 (t, 3H); 1.38-1.50 (m, 10H); 1.95-1.98 (m, 3H); 2.91 (s, 1H); 3.27-3.30 (m, 2H); 3.37- 3.54 (m, 5H); 3.75 (s, 2H); 3.95-4.02 (m, 3H); 4.53 (s, 1H); 6.75-6.78 (d, J=9Htz, 2H); 7.01-7.04 (d, J=9Htz, 2H); 7.13 (s, 2H); 7.31 (s, 1H); ESI Mass: 522 (M+l).
Step 2: Synthesis of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-((l- ( ethoxymethyl )cyclopentyl)methoxy )benzyl )phenyl )tetrahydro-2H-pyran-3, 4, 5-triyl triacetate:

To a solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- (ethoxymethyl)cyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro- 2H-pyran-3,4,5-triol (step 1, 1.0 g) in dichloromethane (30 ml), pyridine (0.67 ml), acetic anhydride (0.79 ml) and 0.0 lg of DMAP were added at 0°C. The reaction mixture was stirred at room temperature for about an hour. After completion of reaction as monitored by TLC, the reaction mixture was diluted with dichloromethane, washed with water, 1% HC1 solution, water followed by saturated brine solution. The organic layer was dried over anhydrous Na2S04 and concentrated. Obtained product was purified by flash column chromatographic technique to remove unreacted Ac20. Recrystallisation of the obtained mixture in ethanol gives the desired isomer as white solid (900 mg). ]H NMR (CDC13, 300 MHz): δ 0.70-0.84 (t, 3H) ; 1.38-1.50 (m, 10H); 1.95-1.98 (m, 3H); 2.91 (s, 1H);
3.27-3.30 (m, 2H); 3.37-3.54 (m, 5H); 3.75 (s, 2H); 3.95-4.02 (m, 3H); 4.53 (s, 1H); 6.75-6.78 (d, J=9Htz, 2H); 7.01-7.04 (d, J=9Htz, 2H); 7.13 (s, 2H); 7.31 (s, 1H); 1.90-1.86 (m, 2H), 1.84-1.59 (m, 5H), 0.85-0.80 (t, 3H, J=7.5Hz); ESI Mass: 712 (M+Na).
Step 3: Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ( ethoxymethyl )cyclopentyl )methoxy )benzyl )phenyl)-6-(hydroxymethyl )tetrahydro- 2H-pyran-3, 4, 5-triol:
NH3 in MeOH (100 ml) was added to (2R,3R,4R,5S,6S)-2-(acetoxymethyl)- 6-(4-chloro-3 -(4-(( 1 -(ethoxymethyl)cyclopentyl)methoxy)benzyl)phenyl)tetrahydro- 2H-pyran-3,4,5-triyl triacetate (β- isomer, step 2, 900 mg) at room temperature and stirred for over night. After completion of the reaction (monitored by TLC), volatiles were concentrated under reduced pressure to give residue. The crude product was purified via silica gel column chromatography with MeOH and dichloromethane (1: 19) to afford the title compound as white solid (350 mg). HPLC: 95 %; 1H MR (DMSO- D6, 300 MHz): 8 0.70-0.84 (t, 3H); 1.38-1.50 (m, 10H); 1.95-1.98 (m, 3H); 2.91 (s, 1H); 3.27-3.30 (m, 2H); 3.37-3.54 (m, 5H); 3.75 (s, 2H); 3.95-4.02 (m, 3H); 4.53 (s, 1H); 6.75-6.78 (d, J=9Htz, 2H); 7.01-7.04 (d, J=9Htz, 2H); 7.13 (s, 2H); 7.31 (s, 1H); ESI Mass: 522 (M+l).
Pharmacological activity
The compounds described herein can be tested for their activity for SGLT2 inhibition following procedures known to a person of ordinary skill in the art. For example, the following protocols may be employed for testing the compounds. These protocols are illustrative and do not limit to the scope of the invention.
Example 18: Effect of compounds on SGLT inhibitory activity:
Stable cells (CHO) expressing h SGLT gene were seeded in 24 well plates. Cells were washed with KRH Na buffer, followed by addition of test compound in KRH Na to the micro plate in defined format. The final concentration of vehicle (DMSO) is not more than 1%. Incubation was carried out for 10-15 minutes at 37° C in an incubator. The radiolabel glucose analogue (14C AMG) was added to each well at 0.5μ Ci concentration. After lhr incubation plates were washed with ice cold
KRH Na buffer containing 0.5 Mm phlorizin. Cells were solubilized with 0.2 N NaOH before transferring them to pico plate for radiolabel count.
Compound preparation:
10 Mm stock was made by dissolving test compound in DMSO. Subsequent dilutions were made with DMSO to make necessary working stocks (10X) and the results are in the following table 1.
Table 1

Example 19: Effect of compounds on urinary glucose excretion in normal animal:
Animals: SD rats, male Age: 8-9 weeks, n=4
Route of administration: Oral
Dose volume: lOml/kg
Vehicle: 0.5% Tween-20+0.25%CMC.
For glucoseuria assessment, overnight fasted SD rats (12 weeks of ages) were placed into metabolism cages for baseline urine collection over 24 hours. Rats were weighted, randomized into experimental groups and orally administered with aqueous glucose solution (2g/kg) and drug in 0.25% CMC. Rats were returned to metabolism cages for 24 hours urine collection. Urine samples were tested for glucose concentration at 6, 24 and 48 hour post dosing and urine volume was measured at the end of 6, 24 and 48 hours. The efficacies of the compounds were evaluated based on glucose excretion in urine and the obtained results were shown in the following table 2.
Table 2
Example Total glucose excretion(mg/200gm body weight)
Dose (mg/kg) 24 hrs 48 hrs
3 30 111.65 —
6 30 205 —
7 30 262 265
8 30 234 285
9 30 277 285
11 30 249 —
12 30 358 605
13 30 387 713
Example 20: Effect of compounds on STZ induced diabetic rats:
Male Sprague Dawley rats were made diabetic by single intra peritoneal injection of streptozotocin at 45 mg kg dose, prepared in 0.1M citrate buffer. Induction of diabetes was confirmed by measuring the random blood glucose levels after 48 hours of STZ administration. Animals with more than 300 mg/dl of plasma glucose were considered as diabetic.
One week after induction of diabetes animals were grouped based on fasting plasma glucose levels. The test item was administered at the specified dose mentioned above for 14 consecutive days. The control group received vehicle during treatment period. On day 14 GTT was conducted in overnight fasted rats. Glucose load of 2 g/kg was administered followed by compound dosing. Blood samples were drawn at time intervals for plasma glucose estimation. Effect of the test compound was assessed by comparing the AUC's plasma glucose of control with treated group.
Table3 - Glucose concentration (mg/dl)

Although the invention herein has been described with reference to particular embodiments, it is to be understood that these embodiments are merely illustrative of the principles and applications of the present invention. It is therefore to be understood that numerous modifications may be made to the illustrative embodiments and that other arrangements may be devised without departing from the spirit and scope of the present invention as described above.
All publications and patent applications cited in this application are herein incorporated by reference to the same extent as if each individual publication or patent application was specifically and individually indicated to be incorporated herein by reference.
Claims
1. A compound of the formula (1):

'herein,
R is substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, abstituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkoxy, or abstituted or unsubstituted heterocyclic group and preferably methyl, ethyl, propyl, lethoxy, ethoxy, isopropoxy, t-butoxy, and the R is optionally substituted by Ra;
each R' is independent and either the same or a different group and is selected om hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted acyl group, tid substituted or unsubstituted alkoxy carbonyl group;
X is CRiR2, O, NR], or S;
a, b, c, d, e, and f is independently selected from hydrogen, hydroxyl group, alogen, substituted or unsubstituted alkyl, C(0)-R3, -C(0)0-R3, or -C(0)NR3R4; and a tid b; or c and d; or e and f can be together with their attached carbon to form Spiro ring r R and one of the a or b; or c and one of the e or f; or d and one of the e or f can be )gether with their attached carbon atoms to form bicyclic ring.
Z and Z' is independently selected from hydrogen, hydroxyl group, halogen, abstituted or unsubstituted alkyl, substituted or unsubstituted alkoxy group, substituted r unsubstituted cycloalkyl, substituted or unsubstituted cycloalkyloxy, -C(0)-R3, - !(0)0-R3, or -C(0)NR3R4;
Ra is hydrogen, hydroxyl group, halogen, substituted or unsubstituted alkyl, abstituted or unsubstituted alkoxy group, substituted or unsubstituted cycloalkyl,
.ttorney Docket No. HI 089/20117 lbstituted or unsubstituted cycloalkyloxy, NR3ILt, -C(0)-R3, -C(0)0-R3, -C(0)NR3R4, (0)pNR3R4, or S(0)pR3;
each R] and R2 is independently selected from hydrogen, substituted or tisubstituted alkyl, substituted or unsubstituted alkenyl, or substituted or unsubstituted cloalkyl;
each R3 and ^ is independently selected from hydrogen, substituted or tisubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted cloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted y'cloalkenyl substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, ibstituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, lbstituted or unsubstituted heterocyclic group, or substituted or unsubstituted sterocyclylalkyl;
'p' is an integer ranging from 0 to 2, a pharmaceutically acceptable salt thereof, a tiarmaceutically acceptable solvate thereof, a pharmaceutically acceptable hydrate lereof, an N-oxide thereof, a tautomer thereof, a regioisomer thereof, a stereoisomer lereof, a prodrug thereof or a polymorph thereof.
. A compound selected from the group consisting of:
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(( 1 - :hylqyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5- iol (Compound 1),
(2R,3R,4R,5S,6R)-2-(4-chloro-3-(4-(( 1 - :hylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5- iol (Compound 2),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l-ethylcyclobutyl)methoxy)benzyl)phenyl)- -(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound 3),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l-ethylcyclohexyl)methoxy)benzyl)phenyl)- -(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound 4),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(( 1 - iethylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- ,4,5-triol (Compound 5),
.ttorney Docket No. HI 089/20117
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((3-ethyloxetan-3- [)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
Compound 6),
(2R,3S,4R,5R,6S)-2-(hydroxymethyl)-6-(4-methyl-3-(4-((l- rifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triol Compound 7),
(2R,3S,4R,5R,6S)-2-(hydroxymethyl)-6-(4-methyl-3-(4-((l- rifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triol Compound 8),
(2S,3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l-methylcyclohexyl)methoxy)benzyl)- -methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3 ,4,5-triol (Compound 9),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- rifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- yran-3,4,5-triol (Compound 10),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- iethylcyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- ,4,5-triol (Compound 11),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- rifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- yran-3,4,5-triol (Compound 12),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((4,4-difluoro-l- iethylcyclohexyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran- ,4,5-triol (Compound 13),
(2S,3R,4R,5S,6R)-2-(4-fluoro-3-(4-((l- rifluoromethyl)cyclobutyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- yran-3,4,5-triol (Compound 14),
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- rifluoromethyl)cyclopropyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- yran-3,4,5-triol (Compound 15),
(2S,3R,4R,5S,6R)-2-(3-(4-((4,4-difluoro-l-methylcyclohexyl)methoxy)benzyl)- -fluorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound 16),
attorney Docket No. H1089/20117
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-((l- ;thoxymethyl)cyclopentyl)methoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H- yran-3,4,5-triol (Compound 17),
isomers thereof, and pharmaceutically acceptable salts thereof.
3. A pharmaceutical composition comprising a compound according to any one of laims 1-2, and a pharmaceutically acceptable excipient.
4. The pharmaceutical composition according to claim 3, wherein the harmaceutically acceptable excipient is a carrier or diluent.
5. A method for preventing, ameliorating or treating an SGLT2 mediated disease, isorder or syndrome in a subject in need thereof comprising administering to the subject therapeutically effective amount of a compound according to any one of claims 1-2.
6. The method according to claim 5, wherein the disease, disorder or syndrome is sleeted from the group consisting of type I diabetes, type II diabetes, including omplications of diabetes such as retinopathy, neuropathy, nephropathy, and delayed Ound healing.
7. The method according to claim 5, wherein the disease, disorder or syndrome is sleeted from the group consisting of diabetes, diabetes type I, diabetes type II, omplications of diabetes, retinopathy, neuropathy, nephropathy, delayed wound healing, lsulin resistance, impaired glucose homeostasis (IGH), hyperglycemia, yperinsulinemia, elevated blood levels of fatty acids or glycerol, obesity, hyperlipidemia lcluding hypertriglyceridemia, Syndrome X, hypertension, and atherosclerosis.
8. A method of treating type I diabetes in a subject in need thereof comprising dministering to the subject a therapeutically effective amount of a compound according ) any one of claims 1-2.
9. A method of treating type II diabetes in a subject in need thereof comprising dministering to the subject a therapeutically effective amount of a compound according ) any one of claims 1-2.
10. A method of treating diabetes complications such as retinopathy, neuropathy, ephropathy and delayed wound healing in a subject in need thereof comprising dministering to the subject a therapeutically effective amount of a compound according ) any one of claims 1-2.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2434/CHE/2010 | 2010-08-23 | ||
| IN2434CH2010 | 2010-08-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012025857A1 true WO2012025857A1 (en) | 2012-03-01 |
Family
ID=44653379
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2011/053641 WO2012025857A1 (en) | 2010-08-23 | 2011-08-17 | Cycloalkyl methoxybenzyl phenyl pyran derivatives as sodium dependent glucose co transporter (sglt2) inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012025857A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012109996A1 (en) * | 2011-02-18 | 2012-08-23 | 上海璎黎科技有限公司 | Aryl glycoside compound, preparation method and use thereof |
| WO2013177224A1 (en) | 2012-05-22 | 2013-11-28 | Genentech, Inc. | N-substituted benzamides and their use in the treatment of pain |
| WO2015044849A1 (en) * | 2013-09-27 | 2015-04-02 | Ranbaxy Laboratories Limited | Process for the purification of dapagliflozin |
| JP2015129106A (en) * | 2014-01-03 | 2015-07-16 | 山東軒竹医薬科技有限公司 | Optically pure benzyl-4-chlorophenyl-c-glucoside derivative |
| WO2015158206A1 (en) * | 2014-04-14 | 2015-10-22 | 上海迪诺医药科技有限公司 | C-aryl indican derivative, and pharmaceutical composition thereof, preparation method therefor and uses thereof |
| US9243022B2 (en) | 2012-12-21 | 2016-01-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9315438B2 (en) | 2014-01-03 | 2016-04-19 | Xuanzhu Pharma Co., Ltd | Optically pure benzyl-4-chlorophenyl-C-glucoside derivative |
| US9394329B2 (en) | 2013-09-27 | 2016-07-19 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
| US9422323B2 (en) | 2012-05-25 | 2016-08-23 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| JP2016533357A (en) * | 2013-10-12 | 2016-10-27 | セラコス サブ,リミティド ライアビリティ カンパニー | Preparation of hydroxy-benzylbenzene derivatives |
| US9562029B2 (en) | 2011-06-25 | 2017-02-07 | Xuanzhu Pharma Co., Ltd. | C-glycoside derivatives |
| CN106892948A (en) * | 2015-12-17 | 2017-06-27 | 广东东阳光药业有限公司 | Glucopyranosyl derivatives and its in application pharmaceutically |
| US11198699B2 (en) | 2019-04-02 | 2021-12-14 | Aligos Therapeutics, Inc. | Compounds targeting PRMT5 |
| CN115417836A (en) * | 2022-09-21 | 2022-12-02 | 安庆奇创药业有限公司 | Method for synthesizing lean hypoglycemic drug intermediate by using continuous flow |
Citations (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US641426A (en) | 1897-06-16 | 1900-01-16 | Arthur M Allen | Tricycle. |
| WO2001016147A1 (en) | 1999-08-31 | 2001-03-08 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 (en) | 1999-10-12 | 2001-04-19 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors |
| WO2001074835A1 (en) | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-glucosylated benzamide sglt2 inhibitors and method |
| WO2001074834A1 (en) | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-aryl glucoside sglt2 inhibitors and method |
| WO2002008306A1 (en) | 2000-07-26 | 2002-01-31 | Mitsui Chemicals, Inc. | Polymer and process for producing the same |
| WO2002028872A1 (en) | 2000-09-29 | 2002-04-11 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| WO2002068439A1 (en) | 2001-02-26 | 2002-09-06 | Kissei Pharmaceutical Co., Ltd. | Glycopyranosyloxypyrazole derivatives and medicinal use thereof |
| WO2002083066A2 (en) | 2001-04-11 | 2002-10-24 | Bristol-Myers Squibb Company | Amino acid complexes of c-aryl glucosides for treatment of diabetes and method |
| WO2003020737A1 (en) | 2001-09-05 | 2003-03-13 | Bristol-Myers Squibb Company | O-pyrazole glucoside sglt2 inhibitors and method of use |
| US20030114390A1 (en) | 2001-03-13 | 2003-06-19 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| WO2003080635A1 (en) | 2002-03-22 | 2003-10-02 | Kissei Pharmaceutical Co., Ltd. | Crystals of glucopyranosyloxybenzyl benzene derivative |
| WO2003099836A1 (en) | 2002-05-20 | 2003-12-04 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors and method |
| WO2004013118A1 (en) | 2002-08-05 | 2004-02-12 | Yamanouchi Pharmaceutical Co., Ltd. | Azulene derivatives and salts thereof |
| WO2004063209A2 (en) | 2003-01-03 | 2004-07-29 | Bristol-Myers Squibb Company | Methods of producing c-aryl glucoside sglt2 inhibitors |
| WO2004080990A1 (en) | 2003-03-14 | 2004-09-23 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| WO2004089967A1 (en) | 2003-04-01 | 2004-10-21 | Taisho Pharmaceutical Co., Ltd. | HETEROARYL 5-THIO-β-D-GLUCOPYRANOSIDE DERIVATIVES AND REMEDIES FOR DIABETES CONTAINING THE SAME |
| WO2005085237A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| US20050209166A1 (en) | 2004-03-16 | 2005-09-22 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2006034489A2 (en) | 2004-09-23 | 2006-03-30 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors and method for their production |
| WO2006054629A1 (en) | 2004-11-18 | 2006-05-26 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2006073197A1 (en) | 2005-01-07 | 2006-07-13 | Taisho Pharmaceutical Co., Ltd. | 1-thio-d-glucitol derivatives |
| EP1685147A2 (en) | 2003-11-03 | 2006-08-02 | Isis Pharmaceuticals, Inc. | Modulation of sglt2 expression |
| US20060194809A1 (en) | 2003-04-01 | 2006-08-31 | Hiroyuki Kakinuma | Heteroaryl 5-thio-beta-d-gucopyranoside derivatives and therapeutic agents for diabetes containing the same |
| WO2007000445A1 (en) | 2005-06-29 | 2007-01-04 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008002824A1 (en) | 2006-06-28 | 2008-01-03 | Bristol-Myers Squibb Company | Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| WO2008013321A1 (en) | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
| WO2008042688A2 (en) * | 2006-09-29 | 2008-04-10 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| WO2008055940A2 (en) | 2006-11-09 | 2008-05-15 | Boehringer Ingelheim International Gmbh | Combination therapy with sglt-2 inhibitors and their pharmaceutical compositions |
| WO2008072726A1 (en) | 2006-12-14 | 2008-06-19 | Taisho Pharmaceutical Co., Ltd. | 1-phenyl 1-thio-d-glucitol derivative |
| US7393836B2 (en) | 2004-07-06 | 2008-07-01 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008101939A1 (en) | 2007-02-21 | 2008-08-28 | Boehringer Ingelheim International Gmbh | Tetrasubstituted glucopyranosylated benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20080221164A1 (en) | 2007-03-08 | 2008-09-11 | Goodwin Nicole C | Inhibitors of Sodium Glucose Co-Transporter 2 and Methods of Their Use |
| WO2008116195A2 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Compositions comprising an sglt2 ingibitor for treating obesity |
| WO2008122014A1 (en) | 2007-04-02 | 2008-10-09 | Theracos, Inc. | Benzylic glycoside derivatives and methods of use |
| WO2008144346A2 (en) | 2007-05-18 | 2008-11-27 | Bristol-Myers Squibb Company | Crystal structures of sglt2 inhibitors and processes for their preparation |
| US7476671B2 (en) | 2004-04-10 | 2009-01-13 | Boehringer Ingelheim International Gmbh | 2-amino-imidazo[4,5-d]pyridazin-4-ones, their preparation and their use as pharmaceutical compositions |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2010092125A1 (en) * | 2009-02-13 | 2010-08-19 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition comprising a sglt2 inhibitor, a dpp-iv inhibitor and optionally a further antidiabetic agent and uses thereof |
-
2011
- 2011-08-17 WO PCT/IB2011/053641 patent/WO2012025857A1/en active Application Filing
Patent Citations (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US641426A (en) | 1897-06-16 | 1900-01-16 | Arthur M Allen | Tricycle. |
| WO2001016147A1 (en) | 1999-08-31 | 2001-03-08 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| EP1224195B1 (en) | 1999-10-12 | 2005-05-18 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors |
| WO2001027128A1 (en) | 1999-10-12 | 2001-04-19 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors |
| US6414126B1 (en) | 1999-10-12 | 2002-07-02 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| WO2001074835A1 (en) | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-glucosylated benzamide sglt2 inhibitors and method |
| WO2001074834A1 (en) | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-aryl glucoside sglt2 inhibitors and method |
| EP1268502B1 (en) | 2000-03-30 | 2006-02-01 | Bristol-Myers Squibb Company | O-aryl glucoside sglt2 inhibitors and method |
| EP1268503A1 (en) | 2000-03-30 | 2003-01-02 | Bristol-Myers Squibb Company | O-glucosylated benzamide sglt2 inhibitors and method |
| WO2002008306A1 (en) | 2000-07-26 | 2002-01-31 | Mitsui Chemicals, Inc. | Polymer and process for producing the same |
| WO2002028872A1 (en) | 2000-09-29 | 2002-04-11 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| WO2002068439A1 (en) | 2001-02-26 | 2002-09-06 | Kissei Pharmaceutical Co., Ltd. | Glycopyranosyloxypyrazole derivatives and medicinal use thereof |
| US20030114390A1 (en) | 2001-03-13 | 2003-06-19 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| WO2002083066A2 (en) | 2001-04-11 | 2002-10-24 | Bristol-Myers Squibb Company | Amino acid complexes of c-aryl glucosides for treatment of diabetes and method |
| WO2003020737A1 (en) | 2001-09-05 | 2003-03-13 | Bristol-Myers Squibb Company | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003080635A1 (en) | 2002-03-22 | 2003-10-02 | Kissei Pharmaceutical Co., Ltd. | Crystals of glucopyranosyloxybenzyl benzene derivative |
| WO2003099836A1 (en) | 2002-05-20 | 2003-12-04 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors and method |
| EP1506211B1 (en) | 2002-05-20 | 2007-02-07 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors and method |
| WO2004013118A1 (en) | 2002-08-05 | 2004-02-12 | Yamanouchi Pharmaceutical Co., Ltd. | Azulene derivatives and salts thereof |
| EP1581543A2 (en) | 2003-01-03 | 2005-10-05 | Bristol-Myers Squibb Company | Methods of producing c-aryl glucoside sglt2 inhibitors |
| WO2004063209A2 (en) | 2003-01-03 | 2004-07-29 | Bristol-Myers Squibb Company | Methods of producing c-aryl glucoside sglt2 inhibitors |
| WO2004080990A1 (en) | 2003-03-14 | 2004-09-23 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| US7202350B2 (en) | 2003-03-14 | 2007-04-10 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| US7439232B2 (en) | 2003-04-01 | 2008-10-21 | Taisho Pharmaceutical Co., Ltd. | Heteroaryl 5-thio-β-D-glucopyranoside derivatives and therapeutic agents for diabetes containing the same |
| US20060194809A1 (en) | 2003-04-01 | 2006-08-31 | Hiroyuki Kakinuma | Heteroaryl 5-thio-beta-d-gucopyranoside derivatives and therapeutic agents for diabetes containing the same |
| WO2004089967A1 (en) | 2003-04-01 | 2004-10-21 | Taisho Pharmaceutical Co., Ltd. | HETEROARYL 5-THIO-β-D-GLUCOPYRANOSIDE DERIVATIVES AND REMEDIES FOR DIABETES CONTAINING THE SAME |
| EP1685147A2 (en) | 2003-11-03 | 2006-08-02 | Isis Pharmaceuticals, Inc. | Modulation of sglt2 expression |
| WO2005085237A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| US20070197450A1 (en) | 2004-03-04 | 2007-08-23 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| US20050209166A1 (en) | 2004-03-16 | 2005-09-22 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US7476671B2 (en) | 2004-04-10 | 2009-01-13 | Boehringer Ingelheim International Gmbh | 2-amino-imidazo[4,5-d]pyridazin-4-ones, their preparation and their use as pharmaceutical compositions |
| US7393836B2 (en) | 2004-07-06 | 2008-07-01 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2006034489A2 (en) | 2004-09-23 | 2006-03-30 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors and method for their production |
| WO2006054629A1 (en) | 2004-11-18 | 2006-05-26 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2006073197A1 (en) | 2005-01-07 | 2006-07-13 | Taisho Pharmaceutical Co., Ltd. | 1-thio-d-glucitol derivatives |
| US20080132563A1 (en) | 2005-01-07 | 2008-06-05 | Hiroyuki Kakinuma | 1-Thio-D-Glucitol Derivatives |
| WO2007000445A1 (en) | 2005-06-29 | 2007-01-04 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008002824A1 (en) | 2006-06-28 | 2008-01-03 | Bristol-Myers Squibb Company | Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| WO2008013321A1 (en) | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
| WO2008042688A2 (en) * | 2006-09-29 | 2008-04-10 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| WO2008055940A2 (en) | 2006-11-09 | 2008-05-15 | Boehringer Ingelheim International Gmbh | Combination therapy with sglt-2 inhibitors and their pharmaceutical compositions |
| WO2008072726A1 (en) | 2006-12-14 | 2008-06-19 | Taisho Pharmaceutical Co., Ltd. | 1-phenyl 1-thio-d-glucitol derivative |
| WO2008101939A1 (en) | 2007-02-21 | 2008-08-28 | Boehringer Ingelheim International Gmbh | Tetrasubstituted glucopyranosylated benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20080221164A1 (en) | 2007-03-08 | 2008-09-11 | Goodwin Nicole C | Inhibitors of Sodium Glucose Co-Transporter 2 and Methods of Their Use |
| WO2008116195A2 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Compositions comprising an sglt2 ingibitor for treating obesity |
| WO2008122014A1 (en) | 2007-04-02 | 2008-10-09 | Theracos, Inc. | Benzylic glycoside derivatives and methods of use |
| WO2008144346A2 (en) | 2007-05-18 | 2008-11-27 | Bristol-Myers Squibb Company | Crystal structures of sglt2 inhibitors and processes for their preparation |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2010092125A1 (en) * | 2009-02-13 | 2010-08-19 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition comprising a sglt2 inhibitor, a dpp-iv inhibitor and optionally a further antidiabetic agent and uses thereof |
Non-Patent Citations (11)
| Title |
|---|
| "Remington: The Science and Practice of Pharmacy", 2003, LIPPINCOTT WILLIAMS & WILKINS |
| BAIHUA XU ET. AL.: "Ortho-Substituted C-aryl Glucosides as Highly Potent and Selective Renal Sodium-dependent Glucose Co-transporter (SGLT2) Inhibitors.", BIOORGANIC AND MEDICINAL CHEMISTRY, vol. 18, no. 12, 29 April 2010 (2010-04-29), pages 4422 - 4432, XP027072360, DOI: 10.1016/j.bmc.2010.04.088 * |
| DIABETES, vol. 57, 2008, pages 1723 - 1729 |
| ENDOCRINOLOGY, vol. 143, 2002, pages 339 - 342 |
| J. CLIN. INVEST., vol. 79, 1987, pages 1510 - 1515 |
| J. CLIN. INVEST., vol. 93, 1994, pages 397 - 404 |
| J. ORG. CHEM., vol. 54, 1989, pages 610 - 612 |
| JOHANNSSON, J. CLIN. ENDROCRINOL. METAB., vol. 82, 1997, pages 727 - 734 |
| JOURNAL OF MEDICINAL CHEMISTRY, vol. 51, 2008, pages 1145 - 1149 |
| T. HIGUCHI, W. STELLA: "Bioreversible Carriers in Drug Design", vol. 14, 1987, AMERICAN PHARMACEUTICAL ASSOCIATION AND PERGAMON PRESS, article "Pro-drugs as Novel Delivery Systems" |
| WEI MENG ET. AL.: "Discovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes.", JOURNAL OF MEDICINAL CHEMISTRY, vol. 51, 2 September 2008 (2008-09-02), pages 1145 - 1149, XP002491733, DOI: 10.1021/jm701272q * |
Cited By (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014505709A (en) * | 2011-02-18 | 2014-03-06 | シャンハイ インリ サイエンス アンド テクノロジー カンパニー,リミティド | Aryl glucoside compounds, their preparation and use |
| US8980829B2 (en) | 2011-02-18 | 2015-03-17 | Shanghai Yingli Science And Technology Co., Ltd | Aryl glycoside compound, preparation method and use thereof |
| WO2012109996A1 (en) * | 2011-02-18 | 2012-08-23 | 上海璎黎科技有限公司 | Aryl glycoside compound, preparation method and use thereof |
| US9562029B2 (en) | 2011-06-25 | 2017-02-07 | Xuanzhu Pharma Co., Ltd. | C-glycoside derivatives |
| US10253010B2 (en) | 2011-06-25 | 2019-04-09 | Sihuan Pharmaceutical Holdings Group Ltd. | C-glycoside derivative |
| WO2013177224A1 (en) | 2012-05-22 | 2013-11-28 | Genentech, Inc. | N-substituted benzamides and their use in the treatment of pain |
| US10774106B2 (en) | 2012-05-25 | 2020-09-15 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US10544184B2 (en) | 2012-05-25 | 2020-01-28 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US10301347B2 (en) | 2012-05-25 | 2019-05-28 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US10040814B2 (en) | 2012-05-25 | 2018-08-07 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US9845336B2 (en) | 2012-05-25 | 2017-12-19 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US9422323B2 (en) | 2012-05-25 | 2016-08-23 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US9249174B2 (en) | 2012-12-21 | 2016-02-02 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10487104B2 (en) | 2012-12-21 | 2019-11-26 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US11485753B2 (en) | 2012-12-21 | 2022-11-01 | Janssen Pharmaceutica Nv | Substituted nucleosides, nucleotides and analogs thereof |
| US10793591B2 (en) | 2012-12-21 | 2020-10-06 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10683320B2 (en) | 2012-12-21 | 2020-06-16 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10112966B2 (en) | 2012-12-21 | 2018-10-30 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10144755B2 (en) | 2012-12-21 | 2018-12-04 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9243022B2 (en) | 2012-12-21 | 2016-01-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9394329B2 (en) | 2013-09-27 | 2016-07-19 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
| WO2015044849A1 (en) * | 2013-09-27 | 2015-04-02 | Ranbaxy Laboratories Limited | Process for the purification of dapagliflozin |
| JP2016533357A (en) * | 2013-10-12 | 2016-10-27 | セラコス サブ,リミティド ライアビリティ カンパニー | Preparation of hydroxy-benzylbenzene derivatives |
| US9914688B2 (en) | 2014-01-03 | 2018-03-13 | Sihuan Pharmaceutical Holdings Group Ltd. | Optically pure benzyl-4-chlorophenyl-C-glucoside derivative |
| JP2015129106A (en) * | 2014-01-03 | 2015-07-16 | 山東軒竹医薬科技有限公司 | Optically pure benzyl-4-chlorophenyl-c-glucoside derivative |
| US9315438B2 (en) | 2014-01-03 | 2016-04-19 | Xuanzhu Pharma Co., Ltd | Optically pure benzyl-4-chlorophenyl-C-glucoside derivative |
| WO2015158206A1 (en) * | 2014-04-14 | 2015-10-22 | 上海迪诺医药科技有限公司 | C-aryl indican derivative, and pharmaceutical composition thereof, preparation method therefor and uses thereof |
| US9914724B2 (en) | 2014-04-14 | 2018-03-13 | Shanghai De Novo Pharmatech Co., Ltd. | C-aryl glycosid derivatives, pharmaceutical composition, preparation process and uses thereof |
| JP2017511383A (en) * | 2014-04-14 | 2017-04-20 | 上海▲ディー▼▲ノア▼医▲薬▼科技有限公司 | C-aryl glycoside derivative, drug composition, preparation method and application thereof |
| CN106892948A (en) * | 2015-12-17 | 2017-06-27 | 广东东阳光药业有限公司 | Glucopyranosyl derivatives and its in application pharmaceutically |
| US11198699B2 (en) | 2019-04-02 | 2021-12-14 | Aligos Therapeutics, Inc. | Compounds targeting PRMT5 |
| CN115417836A (en) * | 2022-09-21 | 2022-12-02 | 安庆奇创药业有限公司 | Method for synthesizing lean hypoglycemic drug intermediate by using continuous flow |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012025857A1 (en) | Cycloalkyl methoxybenzyl phenyl pyran derivatives as sodium dependent glucose co transporter (sglt2) inhibitors | |
| AU2005265772B2 (en) | Novel cyclohexane derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes | |
| RU2156247C2 (en) | Propiophenone derivatives and methods of preparation thereof | |
| EP3056507B1 (en) | C-aryl glucoside derivative, preparation method for same, and medical applications thereof | |
| JP5833011B2 (en) | Process for preparing compounds useful as inhibitors of SGLT2 | |
| CA2189603C (en) | Propiophenone derivative and processes for preparing the same | |
| CN102482290A (en) | C-aryl glucoside derivatives, preparation rpocess and pharmaceutical use thereof | |
| BRPI0717156B1 (en) | sodium glucose co-transporter 2 inhibitors and pharmaceutical composition comprising them | |
| WO2006080421A1 (en) | Spiroketal derivative and use thereof as diabetic medicine | |
| EP1783122A1 (en) | Process for production of azulene derivatives and intermediates for the synthesis of the same | |
| FR2929615A1 (en) | C-ARYL GLYCOSIDE COMPOUNDS FOR THE TREATMENT OF DIABETES AND OBESITY | |
| AU616335B2 (en) | Derivatives of alpha, d-glucofuranose or alpha d-allofuranose and intermediates for preparing these derivatives | |
| RU2678327C2 (en) | Mannose derivatives for treating bacterial infections | |
| JP2014517032A (en) | Novel SGLT inhibitor | |
| JP2009528299A (en) | Glucosidase inhibitor and synthesis method thereof | |
| JP6667008B2 (en) | C-Glucoside derivative containing a fused phenyl ring or a pharmaceutically acceptable salt thereof, a method for producing the same, and a pharmaceutical composition containing the same | |
| KR920000646B1 (en) | Fluoro-substituted epipodophyllo-toxin glucosides | |
| Popsavin et al. | Enantiodivergent synthesis of cytotoxic styryl lactones from d-xylose. The first total synthesis of (+)-and (−)-crassalactone C | |
| KR100556335B1 (en) | 6r-3,6-dideoxy-l-arabino-hexopyranosyl heptanoic acid, preparation process for the same and dauer effect thereof | |
| JP5801723B2 (en) | Method for producing neoponcoranols | |
| FR2699535A1 (en) | Etoposide derivatives, process for their preparation, their use as a medicament and their use for the preparation of a medicament for anticancer treatment | |
| CN108774274B (en) | Sialylmethyl ester methyl glycoside derivative and synthesis method and application thereof | |
| FR2903698A1 (en) | NOVEL 5-THIOXYLOPYRANOSE DERIVATIVES. | |
| CN115003661A (en) | Aryl glucoside derivatives and their use in medicine | |
| FR2906247A1 (en) | NEW 5-THIOXYLOPYRANOSE DERIVATIVES |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11757948 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11757948 Country of ref document: EP Kind code of ref document: A1 |