WO2011110575A1 - Derivatives of 2-[2-(benzo- or pyrido-) thiazolylamino]-6-aminopyridine, useful in the treatment of respiratoric, allergic or inflammatory diseases - Google Patents
Derivatives of 2-[2-(benzo- or pyrido-) thiazolylamino]-6-aminopyridine, useful in the treatment of respiratoric, allergic or inflammatory diseases Download PDFInfo
- Publication number
- WO2011110575A1 WO2011110575A1 PCT/EP2011/053499 EP2011053499W WO2011110575A1 WO 2011110575 A1 WO2011110575 A1 WO 2011110575A1 EP 2011053499 W EP2011053499 W EP 2011053499W WO 2011110575 A1 WO2011110575 A1 WO 2011110575A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- pyridinyl
- benzothiazol
- methyl
- frans
- Prior art date
Links
- 0 Nc1cc(S=C(*2)Nc3cc(Cc4ccccc4)cc(Cl)n3)c2cc1 Chemical compound Nc1cc(S=C(*2)Nc3cc(Cc4ccccc4)cc(Cl)n3)c2cc1 0.000 description 3
- OXQMIXBVXHWDPX-UHFFFAOYSA-N CC(C)(C)N(C)C Chemical compound CC(C)(C)N(C)C OXQMIXBVXHWDPX-UHFFFAOYSA-N 0.000 description 1
- KYVVKZWZFMYZEB-UHFFFAOYSA-N CN(C)Cc1cncc(Br)c1 Chemical compound CN(C)Cc1cncc(Br)c1 KYVVKZWZFMYZEB-UHFFFAOYSA-N 0.000 description 1
- YUGPQNBASYNKRL-UHFFFAOYSA-N Cc1ncc[n]1Cc1cc(NCCCN2CCOCC2)nc(Nc([s]c2c3)nc2ccc3-c2cc(C#N)cnc2)c1 Chemical compound Cc1ncc[n]1Cc1cc(NCCCN2CCOCC2)nc(Nc([s]c2c3)nc2ccc3-c2cc(C#N)cnc2)c1 YUGPQNBASYNKRL-UHFFFAOYSA-N 0.000 description 1
- IBMGDMFSVLCWLJ-UHFFFAOYSA-N Cc1ncc[n]1Cc1cc(NCCCN2CCOCC2)nc(Nc2nc(cccn3)c3[s]2)c1 Chemical compound Cc1ncc[n]1Cc1cc(NCCCN2CCOCC2)nc(Nc2nc(cccn3)c3[s]2)c1 IBMGDMFSVLCWLJ-UHFFFAOYSA-N 0.000 description 1
- LOBJGZYYOPCCPK-MXVIHJGJSA-N Nc(cc1)cc2c1nc(Nc1nc(N[C@H](CC3)CC[C@@H]3O)cc(Cc3ccccc3)c1)[s]2 Chemical compound Nc(cc1)cc2c1nc(Nc1nc(N[C@H](CC3)CC[C@@H]3O)cc(Cc3ccccc3)c1)[s]2 LOBJGZYYOPCCPK-MXVIHJGJSA-N 0.000 description 1
- CMUGDGCSPXSBHM-UHFFFAOYSA-N Nc1nc(Cl)cc(CN2CCOCC2)c1 Chemical compound Nc1nc(Cl)cc(CN2CCOCC2)c1 CMUGDGCSPXSBHM-UHFFFAOYSA-N 0.000 description 1
- YFWPRGNDBHJTMQ-UHFFFAOYSA-N Nc1nc(Cl)cc(C[O-])c1 Chemical compound Nc1nc(Cl)cc(C[O-])c1 YFWPRGNDBHJTMQ-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N OC(C(F)(F)F)=O Chemical compound OC(C(F)(F)F)=O DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- INQPTIMRPPZJBA-HCGLCNNCSA-N O[C@H](CC1)CC[C@@H]1Nc1nc(Nc([s]c2c3)nc2ccc3-c2cnccc2)cc(Cc2ccccn2)c1 Chemical compound O[C@H](CC1)CC[C@@H]1Nc1nc(Nc([s]c2c3)nc2ccc3-c2cnccc2)cc(Cc2ccccn2)c1 INQPTIMRPPZJBA-HCGLCNNCSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention is directed to certain novel compounds which are inhibitors of kinase activity, processes for their preparation, pharmaceutical compositions comprising 5 the compounds, and the use of the compounds or the compositions in the treatment of various disorders. More specifically, the compounds of the invention are inhibitors of the activity or function of Itk (interleukin-2 inducible tyrosine kinase). Compounds which are inhibitors of the activity or function of Itk may be useful in the treatment of disorders such as respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD)
- COPD chronic obstructive pulmonary disease
- allergic diseases including allergic rhinitis and atopic dermatitis
- autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis
- transplant rejection graft versus host disease
- inflammatory disorders including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation
- lnterleukin-2 inducible tyrosine kinase is a non-receptor tyrosine kinase of the Tec family, which is also known as Tsk or Emt.
- Other members of the Tec familiy include: 0 Tec, Btk, Txk and Bmx.
- the Tec family kinases are predominantly expressed in haematopoietic cells, however Bmx and Tec have a wider expression profile.
- the Tec family kinases share a common domain structure: an amino-terminal pleckstrin homology (PH) domain (absent in Txk), a tec homology domain (containing one or two proline rich regions), followed by Src homology SH3 and SH2 domains, and a carboxy-terminal 5 kinase domain.
- the PH domain binds to Ptdln(3,4,5)P3, and is responsible for locating the Tec kinase to the plasma membrane, whilst the PRR, SH3 and SH2 domains are involved in protein-interactions important in formation of the signalling complex.
- Itk expression is restricted to T cells, NK and mast cells. Itk is the predominant Tec family 0 kinase in naive T cells, which also express Txk and Tec. Upon activation via the T cell receptor or interleukin-2 (IL-2), the expression of Itk increases. There is some evidence that Itk is preferentially expressed in Th2 over Th1 cells, in contrast to Txk which is present at higher levels in Th1 cells (1 ). 5 Itk plays a key role in T cell receptor signalling. Itk is recruited to the plasma membrane through interaction with Ptdlns(3,4,5)P3, which is generated by PI3kinase. Itk forms a complex with several signalling and scaffold proteins including SLP76 and LAT.
- Itk is transphorphorylated by Lck. Activated Itk phosphorylates PLCy, leading to the generation of lns(1 ,4,5)P3 (required for calcium flux within the cells) and diacylglycerol (activates members of the protein kinase C family and RAS guanyl-releasing protein. This results in the activation of mitogen-activated protein kinases (including JNK and ERK) and other effectors that regulate gene transcription, leading to the secretion of cytokines (reviewed in ref 2). In addition to the role of Itk in PLCy activation and Ca 2+ mobilisation, Itk may also contribute to TCR-induced actin reorganisation, and formation of the immune synapse.
- Itk may also be activated via the chemokine receptor CXCR4 (4) in T cells, and via the FceRI in mast cells (5).
- CsA Cyclosporin A
- CsA inhibited the late phase but not the early phase response (9), suggesting that effects on mast cells are unlikely to play a key role in the beneficial effect seen of CsA.
- daclizumab an antibody against the anti-IL-2Ra chain (CD25) of activated lymphocytes improved pulmonary function and asthma control in patients with moderate to severe chronic asthma (10), supporting anti-T cell therapy for asthma.
- Inhibition of Itk represents a potential novel therapy for asthma, by inhibiting T cell cytokine release.
- the key role for Itk in T cell receptor signalling has been demonstrated using Itk-/- mice and siRNA.
- In vitro activation of CD4+ cells from Itk knockout mice show reduced levels of Th2 (1 1 ) or both Th1 and Th2 (12) cytokines compared to wild type.
- Naive T cells from Itk knockout mice can differentiate normally into either Th1 or Th2 cells if cultured in vitro under appropriate cytokine conditions, suggesting that Itk is not required for Th2 cell differentiation (12).
- Inhibition of Itk may be beneficial in a variety of T-cell mediated diseases.
- Itk may play a role in other allergic diseases such as allergic rhinitis and atopic dermatitis.
- Single nucleotide polymorphisms in Itk have been associated with atopy (18) and seasonal allergic rhinitis (19).
- Itk mRNA levels in the peripheral blood T cells of atopic dermatitis patient is elevated in T cells from affected patients, compared to healthy controls (20).
- the present inventors have discovered novel compounds which are inhibitors of kinase activity, in particular Itk activity.
- Compounds which are Itk inhibitors may be useful in the treatment of disorders associated with inappropriate kinase activity, in particular inappropriate Itk activity, for example in the treatment and prevention of disorders mediated by Itk mechanisms.
- Such disorders include respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis; allergic diseases including allergic rhinitis and atopic dermatitis; autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis; transplant rejection; graft versus host disease; inflammatory disorders including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation; HIV; aplastic anemia; and pain including inflammatory pain.
- respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis
- allergic diseases including allergic rhinitis and atopic dermatitis
- autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-
- compounds of the invention may show selectivity for Itk over other kinases.
- the invention is directed to certain novel compounds. Specifically, the invention is directed to compounds of formula (I)
- R 1 to R 4 and X are as defined below, and salts thereof.
- the compounds are inhibitors of kinase activity, in particular Itk activity.
- Compounds which are Itk inhibitors may be useful in the treatment of disorders associated with inappropriate Itk activity, such as asthma.
- the invention is further directed to pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the invention is still further directed to methods of inhibiting Itk activity and treatment of disorders associated therewith using a compound of formula (I) or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the invention is yet further directed towards processes for the preparation of the compounds of the invention.
- the invention is directed to compounds of formula (I)
- R 1 is hydrogen, -CH 2 OR 5 , -CH 2 NR 6 R 7 , -CH 2 phenyl or -CH 2 -5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by d -6 alkyl;
- R 2 is hydrogen or methyl
- R 3 is C 3-6 cycloalkyl substituted by -OH or -NR 8 R 9 , or -(CH 2 ) m 6-membered heterocyclyl wherein the 6-membered heterocyclyl contains one or two heteroatoms independently selected from nitrogen and oxygen and is optionally substituted by Ci -6 alkyl, or
- R 2 and R 3 together with the nitrogen atom to which they are attached, are linked to form a piperidinyl substituted by -NR 10 R 11 ;
- R 4 is hydrogen, C -6 alkyl, halo, -NR 12 R 13 , phenyl optionally substituted by -CONR 14 , 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is optionally substituted by one or two substituents independently selected from Ci -6 alkyl, C 2-6 alkynyl, - CN, -(CH 2 ) n OR 15 , -CH 2 phenyl, -(CH 2 ) P NR 16 R 17 and -CONR 18 R 19 , or 9- or 10-membered bicyclic heteroaryl wherein the 9- or 10-membered bicyclic heteroaryl contains one or two nitrogen atoms and is optionally substituted by d -6 alkyl;
- R 5 and R 9 are each independently hydrogen or -COCi -6 alkyl
- R 6 is hydrogen or Ci -6 alkyl
- R 7 is hydrogen, Ci -6 alkyl optionally substituted by -OR 20 or -(CH 2 ) q tetrahydropyran, or R 6 and R 7 , together with the nitrogen atom to which they are attached, are linked to form a 4-, 5- or 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom and the 5- or 6-membered heterocyclyl is optionally substituted by one or two substituents independently selected from C 1-6 alkyl and halo;
- R 8 is hydrogen; R 10 and R 1 1 , together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by Ci -6 alkyl;
- R 12 and R 13 are each hydrogen, or R 12 and R 13 , together with the nitrogen atom to which they are attached, are linked to form a 5- or 6-membered heterocyclyl wherein the 5- or 6- membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by one or two substituents independently selected from oxo, -OH and Ci -6 alkyl optionally substituted by -OH or -NH 2 ; R 14 is C 3 - 6 cycloalkyl;
- R 15 is hydrogen, Ci -6 alkyl optionally substituted by -OH, C 3 - 6 cycloalkyl, -(CH 2 ) r phenyl optionally substituted by halo, or -CH 2 pyridinyl;
- R 16 and R 17 are each independently hydrogen or Ci -6 alkyl optionally substituted by -OR 21 ;
- R 18 is hydrogen
- R 19 is hydrogen or C 1-6 alkyl, or
- R 18 and R 19 together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom;
- R and R are each independently hydrogen or d -6 alkyl;
- X is -N- or -CH-;
- m is 1 , 2 or 3;
- n, p, q and r are each independently 0, 1 or 2; and salts thereof (hereinafter "compounds of the invention").
- R 1 is -CH 2 NR 6 R 7 or -CH 2 -5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by C 1-6 alkyl.
- R 1 is -CH 2 NR 6 R 7 .
- R 1 is -CH 2 -5-membered heteroaryl wherein the 5-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by Ci -6 alkyl.
- R 2 is hydrogen
- R 3 is C 3-6 cycloalkyl substituted by -OH or -NR 8 R 9 . In a further embodiment, R 3 is C 3-6 cycloalkyl substituted by -OH.
- R 4 is hydrogen, -NR 12 R 13 or 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is optionally substituted by Ci -6 alkyl, C 2-6 alkynyl, - CN, -(CH 2 ) n OR 15 , -CH 2 phenyl, -(CH 2 ) P NR 16 R 17 or -CONR 18 R 19 .
- R 4 is hydrogen, -NR 12 R 13 or 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by -CN.
- R 4 is -NR 12 R 13 or 5- or 6-membered heteroaryl wherein the 5- or 6- membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by - CN.
- R 4 is 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is substituted by one or two substituents including -OH
- the R 4 group may be drawn as the corresponding keto tautomer.
- 2,4- dihydro-3H-1 ,2,4-triazo-3-one may be drawn as follows:
- R 5 is hydrogen or -COCi -4 alkyl.
- R 6 is hydrogen or C 1-4 alkyl. In another embodiment, R 6 is hydrogen. In a further embodiment, R 6 is methyl or ethyl. In one embodiment, R 7 is hydrogen or C 1-6 alkyl optionally substituted by -OR 20 . In a further embodiment, R 7 is C 1-6 alkyl.
- R 6 and R 7 together with the nitrogen atom to which they are attached, are linked to form a 4-, 5- or 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom and the 5- or 6-membered heterocyclyl is optionally substituted by one or two substituents independently selected from d -6 alkyl and halo.
- R 6 and R 7 together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom and is optionally substituted by one or two substituents independently selected from Ci -6 alkyl and halo.
- R 9 is hydrogen or -COCi -4 alkyl.
- R 10 and R 11 together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl contains an oxygen atom or a further nitrogen atom and is optionally substituted by methyl.
- R 12 and R 13 together with the nitrogen atom to which they are attached, are linked to form a 5- or 6-membered heterocyclyl wherein the 5- or 6- membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by one or two substituents independently selected from oxo, -OH and C 1-6 alkyl optionally substituted by -OH or -NH 2 .
- R 12 and R 13 together with the nitrogen atom to which they are attached, are linked to form a 5- membered heterocyclyl wherein the 5-membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by one or two oxo substituents.
- R 14 is cyclopentyl.
- R 15 is hydrogen, Ci -4 alkyl optionally substituted by -OH, C 3- 6 cycloalkyl, -(CH 2 ) r phenyl optionally substituted by halo, or -CH 2 pyridinyl. In a further embodiment, R 15 is hydrogen or Ci -4 alkyl optionally substituted by -OH.
- R 16 and R 17 are each independently hydrogen or C -4 alkyl optionally substituted by -OR 21 . In another embodiment, R 16 and R 17 are each independently C-i. 4 alkyl optionally substituted by -OR 21 . In a further embodiment, R 16 is hydrogen and R 17 is d ⁇ alkyl optionally substituted by -OR 21 .
- R 18 is hydrogen
- R 19 is hydrogen or d -6 alkyl. In a further embodiment, R 19 is Ci -4 alkyl. In one embodiment, R 18 and R 19 , together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom. In a further embodiment, R 18 and R 19 , together with the nitrogen atom to which they are attached, are linked to form a morpholinyl.
- R 20 is hydrogen or Ci -4 alkyl. In a further embodiment, R 20 is hydrogen or methyl.
- R 21 is hydrogen or Ci -4 alkyl. In a further embodiment, R 21 is methyl.
- X is -N-. In a further embodiment, X is -CH-. In one embodiment, m is 2 or 3 In one embodiment, n is 0 or 1.
- p is 0 or 1. In one embodiment, q is 0 or 1. In one embodiment, r is 0 or 1.
- Compounds of the invention include the compounds of Examples 1 to 164 and salts thereof.
- the compound of the invention is:
- the compound of the invention is: frans-4-[(4-[(2-methyl-1 H-imidazol-1-yl)methyl]-6- ⁇ [6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2- yl]amino ⁇ -2-pyridinyl)amino]cyclohexanol
- AlkyI refers to a saturated hydrocarbon chain having the specified number of member atoms.
- C 1-6 alkyl refers to an alkyl group having from 1 to 6 member atoms, for example 1 to 4 member atoms.
- Alkyl groups may be optionally substituted with one or more substituents as defined herein.
- Alkyl groups may be straight or branched. Representative branched alkyl groups have one, two, or three branches.
- Alkyl includes methyl, ethyl, propyl (n-propyl and isopropyl), butyl (n-butyl, isobutyl, and t-butyl), pentyl (n-pentyl, isopentyl, and neopentyl), and hexyl.
- Alkynyl refers to a hydrocarbon chain having the specified number of member atoms and at least one triple bond.
- C 2-6 alkynyl refers to an alkynyl group having from 2 to 6 member atoms, for example 2 to 4 member atoms.
- Alkynyl groups may be straight or branched.
- Alkynyl includes ethynyl, 1 -propynyl, 1-butynyl, 2-butynyl, 1 - pentynyl, 2-pentynyl, 3-pentynyl, 1 -hexynyl, 2-hexynyl and 3-hexynyl.
- Cycloalkyl refers to a saturated hydrocarbon ring having the specified number of member atoms. Cycloalkyl groups are monocyclic ring systems. For example, C 3- 6 cycloalkyl refers to a cycloalkyl group having from 3 to 6 member atoms. Cycloalkyl groups may be optionally substituted with one or more substituents as defined herein. Cycloalkyl includes cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- “Enantiomerically enriched” refers to products whose enantiomeric excess is greater than zero. For example, enantiomerically enriched refers to products whose enantiomeric excess is greater than 50% ee, greater than 75% ee, and greater than 90% ee.
- Enantiomeric excess or "ee” is the excess of one enantiomer over the other expressed as a percentage. As a result, since both enantiomers are present in equal amounts in a racemic mixture, the enantiomeric excess is zero (0% ee). However, if one enantiomer was enriched such that it constitutes 95% of the product, then the enantiomeric excess would be 90% ee (the amount of the enriched enantiomer, 95%, minus the amount of the other enantiomer, 5%).
- Enantiomerically pure refers to products whose enantiomeric excess is 99% ee or greater.
- Half-life refers to the time required for half of a quantity of a substance to be converted to another chemically distinct species in vitro or in vivo.
- Halo refers to the halogen radical fluoro, chloro, bromo, or iodo.
- Heteroaryl refers to an aromatic ring containing from 1 to 3 heteroatoms as member atoms in the ring or rings. Heteroaryl groups containing more than one heteroatom may contain different heteroatoms. Heteroaryl groups may be optionally substituted with one or more substituents if so defined herein.
- the heteroaryl groups herein are monocyclic ring systems or are fused bicyclic ring systems. Monocyclic heteroaryl rings have 5 or 6 member atoms. Bicyclic heteroaryl rings have 9 or 10 member atoms.
- Monocyclic heteroaryl includes pyrrolyl, furanyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, oxadiazolyl, triazolyl, pyridinyl, pyrimidinyl, pyridazinyl and pyrazinyl.
- Bicyclic heteroaryl includes indolyl, isoindolyl, indolizinyl, indazolyl, benzimidazolyl, pyrrolopyridinyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl and naphthridinyl.
- Heteroatom refers to a nitrogen, sulphur, or oxygen atom.
- Heterocyclyl refers to a saturated or unsaturated ring containing 1 or 2 heteroatoms as member atoms in the ring. However, heterocyclyl rings are not aromatic. In certain embodiments, heterocyclyl is saturated. In other embodiments, heterocyclyl is unsaturated but not aromatic. Heterocyclyl groups containing more than one heteroatom may contain different heteroatoms.
- the heterocyclyl groups herein are monocyclic ring systems having 4, 5 or 6 member atoms. Heterocyclyl groups may be optionally substituted with one or more substituents as defined herein.
- Heterocyclyl includes azetidinyl, pyrrolidinyl, pyrazolidinyl, imidazolinyl, oxazolidinyl, isoxazolidinyl, piperidinyl, piperazinyl and morpholinyl.
- Member atoms refers to the atom or atoms that form a chain or ring. Where more than one member atom is present in a chain and within a ring, each member atom is covalently bound to an adjacent member atom in the chain or ring. Atoms that make up a substituent group on a chain or ring are not member atoms in the chain or ring.
- Optionally substituted indicates that a group, such as heteroaryl, may be unsubstituted or substituted with one or more substituents as defined herein.
- Substituted in reference to a group indicates that a hydrogen atom attached to a member atom within a group is replaced. It should be understood that the term “substituted” includes the implicit provision that such substitution be in accordance with the permitted valence of the substituted atom and the substituent and that the substitution results in a stable compound (i.e. one that does not spontaneously undergo transformation such as by rearrangement, cyclization, or elimination). In certain embodiments, a single atom may be substituted with more than one substituent as long as such substitution is in accordance with the permitted valence of the atom. Suitable substituents are defined herein for each substituted or optionally substituted group.
- “Pharmaceutically acceptable” refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- solvates including hydrates
- complexes including hydrates
- polymorphs including hydrates
- prodrugs including radiolabeled derivatives, stereoisomers and optical isomers of the compounds of formula (I) and salts thereof.
- the compounds of the invention may exist in solid or liquid form. In the solid state, the compounds of the invention may exist in crystalline or noncrystalline form, or as a mixture thereof.
- pharmaceutically acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization.
- Solvates may involve nonaqueous solvents such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and EtOAc, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent that is incorporated into the crystalline lattice are typically referred to as "hydrates.” Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
- polymorphs may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs".
- the invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification.
- polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymorphs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
- the invention also includes isotopically-labelled compounds, which are identical to the compounds of formula (I) and salts thereof, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number most commonly found in nature.
- isotopes that can be incorporated into the compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen and fluorine, such as 3H, 1 1 C, 14C and 18F.
- the compounds according to formula (I) may contain one or more asymmetric center (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereomers, or other stereoisomeric forms, or as mixtures thereof.
- Chiral centers such as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the stereochemistry of a chiral center present in formula (I), or in any chemical structure illustrated herein, is not specified the structure is intended to encompass any stereoisomer and all mixtures thereof. Thus, compounds according to formula (I) containing one or more chiral center may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers.
- Individual stereoisomers of a compound according to formula (I) which contain one or more asymmetric center may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1 ) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer- specific reagent, for example by enzymatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral enviornment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
- the compounds according to formula (I) may also contain centers of geometric asymmetry. Where the stereochemistry of a center of geometric asymmetry present in formula (I), or in any chemical structure illustrated herein, is not specified, the structure is intended to encompass the trans geometric isomer, the cis geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included in formula (I) whether such tautomers exist in equilibrium or predominately in one form.
- references herein to compounds of formula (I) and salts thereof covers the compounds of formula (I) as free acids or free bases, or as salts thereof, for example as pharmaceutically acceptable salts thereof.
- the invention is directed to compounds of formula (I) as the free acid or free base.
- the invention is directed to compounds of formula (I) and salts thereof.
- the invention is directed to compounds of formula (I) and pharmaceutically acceptable salts thereof.
- pharmaceutically acceptable salts of the compounds according to formula (I) may be prepared. Indeed, in certain embodiments of the invention, pharmaceutically acceptable salts of the compounds according to formula (I) may be preferred over the respective free base or free acid because such salts impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage form. Accordingly, the invention is further directed to compounds of formula (I) and pharmaceutically acceptable salts thereof.
- pharmaceutically acceptable salts refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
- Salts and solvates having non-pharmaceutically acceptable counter-ions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
- one embodiment of the invention embraces compounds of formula (I) and salts thereof.
- compounds according to formula (I) may contain an acidic functional group.
- Suitable pharmaceutically-acceptable salts include salts of such acidic functional groups.
- Representative salts include pharmaceutically acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a pharmaceutically acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, TEA, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
- pharmaceutically acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts
- carbonates and bicarbonates of a pharmaceutically acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc
- pharmaceutically acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic
- compounds according to formula (I) may contain a basic functional group and are therefore capable of forming pharmaceutically acceptable acid addition salts by treatment with a suitable acid.
- suitable acids include pharmaceutically acceptable inorganic acids and pharmaceutically acceptable organic acids.
- Representative pharmaceutically acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphate, acetate, hydroxyacetate, phenylacetate, propionate, butyrate, isobutyrate, valerate, maleate, hydroxymaleate, acrylate, fumarate, malate, tartrate, citrate, salicylate, p- aminosalicyclate, glycollate, lactate, heptanoate, phthalate, oxalate, succinate, benzoate, o-acetoxybenzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, nap
- the compounds of the invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention are prepared in the Examples section.
- R 1 , R 4 and X are as defined above and Y is halo such as fluoro or chloro, with an amine of formula (III)
- the process may be carried out under microwave irradiation, using neat amine or amine in a suitable solvent such as ethylene glycol, and at a suitable temperature such as from about 160 to about 220°C.
- a suitable solvent such as ethylene glycol
- a suitable temperature such as from about 160 to about 220°C.
- Compounds of formula (II) wherein Y is fluoro may be prepared by a process comprising reacting of a compound of formula (IV)
- the process may be carried out in a suitable solvent such as tetrahydrofuran or DMF, in the presence of a suitable base such as sodium hydride, and at a suitable temperature such as about 0°C then allowing the reaction mixture to warm to ambient temperature.
- a suitable solvent such as tetrahydrofuran or DMF
- a suitable base such as sodium hydride
- R 1 is as defined above and Y and Y 3 are both chloro, with concentrated aqueous ammonia.
- the process may be carried out under microwave irradiation, and at a suitable temperature such as from about 160 to about 220°C.
- Suitable conditions for the Suzuki coupling include microwave irradiation, in the presence of a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium (0) or 2'- (dimethylamino)-2-biphenyl-palladium (II) chloride dinorbornylphosphine complex, in a suitable solvent such as aqueous 1 ,4-dioxane, in the presence of a suitable base such as caesium carbonate or potassium phosphate, and at a suitable temperature such as from about 100 to about 150°C.
- a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium (0) or 2'- (dimethylamino)-2-biphenyl-palladium (II) chloride dinorbornylphosphine complex
- a suitable solvent such as aqueous 1 ,4-dioxane
- a suitable base such as
- R 1 to R 3 and X are as defined above and Y 5 is halo such as bromo, with 4,4,4' ,4',5,5,5',5'-octamethyl-2,2'-bi-1 ,3,2-dioxaborolane.
- the process may be carried out under microwave irradiation, in the presence of a suitable palladium catalyst such as 1 ,1 '-bis(diphenylphosphino)ferrocene-palladium(ll)dichloride dichloromethane adduct, in a suitable solvent such as tetrahydrofuran, in the presence of a suitable base such as potassium acetate, and at a suitable temperature such as from about 1 10 to about 170°C, for example about 120°C.
- a suitable palladium catalyst such as 1 ,1 '-bis(diphenylphosphino)ferrocene-palladium(ll)dichloride dichloromethane adduct
- a suitable solvent such as tetrahydrofuran
- a suitable base such as potassium acetate
- Suitable conditions for the Suzuki coupling include microwave irradiation, in the presence of a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium (0) or 2'- (dimethylamino)-2-biphenyl-palladium (II) chloride dinorbornylphosphine complex, in a suitable solvent such as aqueous 1 ,4-dioxane, in the presence of a suitable base such as caesium carbonate or potassium phosphate, and at a suitable temperature such as from about 100 to about 150°C.
- a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium (0) or 2'- (dimethylamino)-2-biphenyl-palladium (II) chloride dinorbornylphosphine complex
- a suitable solvent such as aqueous 1 ,4-dioxane
- a suitable base such as
- Compounds of formula (I) wherein R 1 to R 4 and X are as defined above, and salts thereof, may also be prepared by a process comprising final stage modification of one compound of formula (I), or a salt thereof, into another compound of formula (I), or a salt thereof.
- Suitable functional group transformations for converting one compound of formula (I) into another compound of formula (I) are well known in the art and are described in, for instance, Comprehensive Heterocyclic Chemistry II, eds. A. R. Katritzky, C. W. Rees and E. F. V. Scriven (Pergamon Press, 1996), Comprehensive Organic Functional Group Transformations, eds. A.R. Katritzky, O. Meth-Cohn and C.W.
- Compounds of formula (I) wherein R 1 , R 2 , R 3 and X are as defined above and R 4 is - NR 12 R 13 wherein R 12 and R 13 , together with the nitrogen atom to which they are attached, are linked to form 2,5-pyrrolidinone, may be prepared by a process comprising reacting a compound of formula (I) wherein R 1 , R 2 , R 3 and X are as defined above and R 4 is NH 2 , with succinic anhydride.
- Suitable reaction conditions include microwave irradiation, in a suitable solvent such as acetonitrile, in the presence of HCI, at a suitable temperature such as about 150°C.
- Compounds of formula (I) wherein R 4 is chloro may be reduced to produce compounds of formula (I) wherein R 4 is hydrogen.
- Suitable reduction conditions include treatment with ammonium formate under microwave irradiation, in the presence of a suitable catalyst such as palladium on activated carbon, in a suitable solvent such as methanol and at a suitable temperature such as about 130 °C.
- compounds of formula (I) wherein R 1 , R 2 , R 4 and X are as defined above and R 3 is C 3- 6 cycloalkyl substituted by -NR 8 R 9 wherein R 8 is hydrogen and R 9 is -COC 1-6 alkyl may be prepared by a process comprising reacting a compound of formula (I) wherein R 1 , R 2 , R 4 and X are as defined above and R 3 is C 3-6 cycloalkyl substituted by -NH 2 , with Ci_ 6 alkylCOCI such as acetyl chloride.
- Suitable reaction conditions include reaction in the presence of a suitable base such as triethylamine, in a suitable solvent such as tetrahydrofuran, and at a suitable temperature such as about 0°C.
- Compounds of formula (I) wherein R 2 to R 4 and X are as defined above and R 1 is - CH 2 NR 6 R 7 may be prepared from compounds of formula (I) wherein R 1 is -CH 2 OH by conversion of the alcohol to the corresponding aldehyde by treatment with, for example, manganese dioxide in the presence of a suitable solvent such as tetrahydrofuran and at a suitable temperature such as about 70°C, followed by reaction with an amine of formula R 6 R 7 NH 2 in the presence of sodium triacetoxyborohydride, a suitable solvent such as dichloromethane and at a suitable temperature such as ambient temperature.
- a suitable solvent such as tetrahydrofuran
- a suitable temperature such as about 70°C
- Compounds of formula (I) wherein R 2 to R 4 and X are as defined above and R 1 is - CH 2 NH 2 may be prepared from the abovementioned aldehyde by conversion to the corresponding oxime by treatment with, for example, hydroxylamine hydrochloride and sodium acetate in the presence of a suitable sovent such as aqueous ethanol and at a suitable temperature such as ambient temperature, followed by treatment with zinc powder in glacial acetic acid at a suitable temperature such as ambient temperature.
- a suitable sovent such as aqueous ethanol
- a suitable temperature such as ambient temperature
- the invention provides a process for preparing a compound comprising: a) reacting a compound of formula (II)
- R 4 is as defined above, or d) final stage modification of one compound of formula (I), or a salt thereof, into another compound of formula (I), or a salt thereof.
- the compounds of the invention are inhibitors of kinase activity, in particular Itk activity.
- Compounds which are Itk inhibitors may be useful in the treatment of disorders wherein the underlying pathology is (at least in part) attributable to inappropriate Itk activity, such as asthma.
- "Inappropriate Itk activity” refers to any Itk activity that deviates from the normal Itk activity expected in a particular patient. Inappropriate Itk may take the form of, for instance, an abnormal increase in activity, or an aberration in the timing and or control of Itk activity. Such inappropriate activity may result then, for example, from overexpression or mutation of the protein kinase leading to inappropriate or uncontrolled activation. Accordingly, in another aspect the invention is directed to methods of treating such disorders.
- Such disorders include respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis; allergic diseases including allergic rhinitis and atopic dermatitis; autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis; transplant rejection; graft versus host disease; inflammatory disorders including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation; HIV; aplastic anemia; and pain including inflammatory pain.
- respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis
- allergic diseases including allergic rhinitis and atopic dermatitis
- autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-
- the methods of treatment of the invention comprise administering a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof to a patient in need thereof.
- Individual embodiments of the invention include methods of treating any one of the above-mentioned disorders by administering a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof to a patient in need thereof.
- treat in reference to a disorder means: (1 ) to ameliorate or prevent the disorder or one or more of the biological manifestations of the disorder, (2) to interfere with (a) one or more points in the biological cascade that leads to or is responsible for the disorder or (b) one or more of the biological manifestations of the disorder, (3) to alleviate one or more of the symptoms or effects associated with the disorder, or (4) to slow the progression of the disorder or one or more of the biological manifestations of the disorder.
- prevention of a disorder includes prevention of the disorder.
- prevention is not an absolute term. In medicine, “prevention” is understood to refer to the prophylactic administration of a drug to substantially diminish the likelihood or severity of a disorder or biological manifestation thereof, or to delay the onset of such disorder or biological manifestation thereof.
- safe and effective amount in reference to a compound of formula (I) or a pharmaceutically acceptable salt thereof or other pharmaceutically-active agent means an amount of the compound sufficient to treat the patient's condition but low enough to avoid serious side effects (at a reasonable benefit/risk ratio) within the scope of sound medical judgment.
- a safe and effective amount of a compound will vary with the particular compound chosen (e.g.
- patient refers to a human (including adults and children) or other animal. In one embodiment, “patient” refers to a human.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered by any suitable route of administration, including both systemic administration and topical administration.
- Systemic administration includes oral administration, parenteral administration, transdermal administration and rectal administration.
- Parenteral administration refers to routes of administration other than enteral or transdermal, and is typically by injection or infusion.
- Parenteral administration includes intravenous, intramuscular, and subcutaneous injection or infusion.
- Topical administration includes application to the skin as well as intraocular, otic, intravaginal, inhaled and intranasal administration. Inhalation refers to administration into the patient's lungs whether inhaled through the mouth or through the nasal passages.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered orally.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered topically. In another embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered by inhalation. In a further embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered intranasally.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered once or according to a dosing regimen wherein a number of doses are administered at varying intervals of time for a given period of time. For example, doses may be administered one, two, three, or four times per day. In one embodiment, a dose is administered once per day. In a further embodiment, a dose is administered twice per day. Doses may be administered until the desired therapeutic effect is achieved or indefinitely to maintain the desired therapeutic effect. Suitable dosing regimens for a compound of formula (I) or a pharmaceutically acceptable salt thereof depend on the pharmacokinetic properties of that compound, such as absorption, distribution, and half- life, which can be determined by the skilled artisan.
- suitable dosing regimens including the duration such regimens are administered, for a compound of formula (I) or a pharmaceutically acceptable salt thereof depend on the disorder being treated, the severity of the disorder being treated, the age and physical condition of the patient being treated, the medical history of the patient to be treated, the nature of concurrent therapy, the desired therapeutic effect, and like factors within the knowledge and expertise of the skilled artisan. It will be further understood by such skilled artisans that suitable dosing regimens may require adjustment given an individual patient's response to the dosing regimen or over time as individual patient needs change.
- Typical daily dosages may vary depending upon the particular route of administration chosen. Typical daily dosages for oral administration range from 0.001 mg to 50mg per kg of total body weight, for example from 1 mg to 10mg per kg of total body weight. For example, daily dosages for oral administration may be from 0.5mg to 2g per patient, such as 10mg to 1g per patient.
- the compounds of formula (I) may be administered as prodrugs.
- a "prodrug" of a compound of formula (I) is a functional derivative of the compound which, upon administration to a patient, eventually liberates the compound of formula (I) in vivo.
- a compound of formula (I) as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of the activity of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome a side effect or other difficulty encountered with the compound.
- Typical functional derivatives used to prepare prodrugs include modifications of the compound that are chemically or enzymatically cleavable in vivo. Such modifications, which include the preparation of phosphates, amides, esters, thioesters, carbonates, and carbamates, are well known to those skilled in the art.
- the invention thus provides a method of treating a disorder mediated by inappropriate Itk activity comprising administering a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof to a patient in need thereof.
- the disorder mediated by inappropriate Itk activity is selected from the group consisting of respiratory diseases (including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis); allergic diseases (including allergic rhinitis and atopic dermatitis); autoimmune diseases (including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis); transplant rejection; graft versus host disease; inflammatory disorders (including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation); HIV; aplastic anemia; and pain including inflammatory pain.
- respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis
- allergic diseases including allergic rhinitis and atopic dermatitis
- autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes
- the disorder mediated by inappropriate Itk activity is a respiratory disease. In a further embodiment, the disorder mediated by inappropriate Itk activity is asthma.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in medical therapy.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in the treatment of a disorder mediated by inappropriate Itk activity.
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof in the manufacture of a medicament for use in the treatment of a disorder mediated by inappropriate Itk activity.
- the invention provides a pharmaceutical composition for the treatment or prophylaxis of a disorder mediated by inappropriate Itk activity comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the compounds of formula (I) and pharmaceutically acceptable salts thereof will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable excipients. In a further aspect the invention is directed to pharmaceutical compositions for the treatment or prophylaxis of a disorder mediated by inappropriate Itk activity comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof can be extracted and then given to the patient such as with powders or syrups.
- the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form wherein each physically discrete unit contains a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the pharmaceutical compositions of the invention typically may contain, for example, from 0.5mg to 1 g, or from 1 mg to 700mg, or from 5mg to 100mg of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- compositions of the invention typically contain one compound of formula (I) or a pharmaceutically acceptable salt thereof.
- pharmaceutically acceptable excipient means a pharmaceutically acceptable material, composition or vehicle involved in giving form or consistency to the pharmaceutical composition.
- Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled such that interactions which would substantially reduce the efficacy of the compound of formula (I) or a pharmaceutically acceptable salt thereof when administered to a patient and interactions which would result in pharmaceutical compositions that are not pharmaceutically acceptable are avoided.
- each excipient must of course be pharmaceutically acceptable eg of sufficiently high purity.
- the compound of formula (I) or a pharmaceutically acceptable salt thereof and the pharmaceutically acceptable excipient or excipients will typically be formulated into a dosage form adapted for administration to the patient by the desired route of administration.
- dosage forms include those adapted for (1 ) oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixers, suspensions, solutions, emulsions, sachets, and cachets; (2) parenteral administration such as sterile solutions, suspensions, and powders for reconstitution; (3) transdermal administration such as transdermal patches; (4) rectal administration such as suppositories; (5) inhalation such as aerosols, solutions, and dry powders; and (6) topical administration such as creams, ointments, lotions, solutions, pastes, sprays, foams, and gels.
- oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixers, suspensions, solutions, emulsions, sachets, and cachets
- parenteral administration such as sterile solutions, suspensions, and powders for reconstitution
- transdermal administration such as transdermal patches
- rectal administration such as
- Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen.
- suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition.
- certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of formula (I) or pharmaceutically acceptable salts thereof once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
- Suitable pharmaceutically-acceptable excipients include the following types of excipients: Diluents, fillers, binders, disintegrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweetners, flavoring agents, flavor masking agents, coloring agents, anticaking agents, hemectants, chelating agents, plasticizers, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents.
- excipients include the following types of excipients: Diluents, fillers, binders, disintegrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweetners, flavoring agents, flavor masking agents, coloring agents, anticaking agents, hemectants, chelating agents
- compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company). Accordingly, in another aspect the invention is directed to process for the preparation of a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and one or more pharmaceutically-acceptable excipients which comprises mixing the ingredients.
- a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may be prepared by, for example, admixture at ambient temperature and atmospheric pressure.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof will be formulated for oral administration. In another embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof will be formulated for inhaled administration. In a further embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof will be formulated for intranasal administration.
- the invention is directed to a solid oral dosage form such as a tablet or capsule comprising a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof and a diluent or filler.
- Suitable diluents and fillers include lactose, sucrose, dextrose, mannitol, sorbitol, starch (e.g. corn starch, potato starch, and pre-gelatinized starch), cellulose and its derivatives (e.g. microcrystalline cellulose), calcium sulfate, and dibasic calcium phosphate.
- the oral solid dosage form may further comprise a binder. Suitable binders include starch (e.g.
- the oral solid dosage form may further comprise a disintegrant. Suitable disintegrants include crospovidone, sodium starch glycolate, croscarmelose, alginic acid, and sodium carboxymethyl cellulose.
- the oral solid dosage form may further comprise a lubricant. Suitable lubricants include stearic acid, magnesuim stearate, calcium stearate, and talc.
- dosage unit formulations for oral administration can be microencapsulated.
- the composition can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may also be coupled with soluble polymers as targetable drug carriers.
- soluble polymers can include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamide -phenol, polyhydroxyethylaspartamidephenol, or polyethyleneoxidepolylysine substituted with palmitoyl residues.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may be coupled to a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polylactic acid, polepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- biodegradable polymers useful in achieving controlled release of a drug
- a drug for example, polylactic acid, polepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- the invention is directed to a liquid oral dosage form.
- Oral liquids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- Syrups can be prepared by dissolving the compound of formula (I) or a pharmaceutically acceptable salt thereof in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound of formula (I) or a pharmaceutically acceptable salt thereof in a non-toxic vehicle.
- Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
- the invention is directed to a dosage form adapted for administration to a patient by inhalation.
- a dosage form adapted for administration to a patient by inhalation.
- dry powder compositions for delivery to the lung by inhalation typically comprise a compound of formula (I) or a pharmaceutically acceptable salt thereof as a finely divided powder together with one or more pharmaceutically-acceptable excipients as finely divided powders.
- Pharmaceutically-acceptable excipients particularly suited for use in dry powders are known to those skilled in the art and include lactose, starch, mannitol, and mono-, di-, and polysaccharides.
- the finely divided powder may be prepared by, for example, micronisation and milling.
- the size-reduced (eg micronised) compound can be defined by a D 50 value of about 1 to about 10 microns (for example as measured using laser diffraction).
- the dry powder may be administered to the patient via a reservoir dry powder inhaler (RDPI) having a reservoir suitable for storing multiple (un-metered doses) of medicament in dry powder form.
- RDPIs typically include a means for metering each medicament dose from the reservoir to a delivery position.
- the metering means may comprise a metering cup, which is movable from a first position where the cup may be filled with medicament from the reservoir to a second position where the metered medicament dose is made available to the patient for inhalation.
- the dry powder may be presented in capsules (e.g. gelatin or plastic), cartridges, or blister packs for use in a multi-dose dry powder inhaler (MDPI).
- MDPI multi-dose dry powder inhaler
- MDPIs are inhalers wherein the medicament is comprised within a multi-dose pack containing (or otherwise carrying) multiple defined doses (or parts thereof) of medicament.
- the dry powder When the dry powder is presented as a blister pack, it comprises multiple blisters for containment of the medicament in dry powder form.
- the blisters are typically arranged in regular fashion for ease of release of the medicament therefrom.
- the blisters may be arranged in a generally circular fashion on a disc-form blister pack, or the blisters may be elongate in form, for example comprising a strip or a tape.
- Each capsule, cartridge, or blister may, for example, contain between 20 ⁇ g-10mg of the compound of formula (I) or a pharmaceutically acceptable salt thereof.
- Aerosols may be formed by suspending or dissolving a compound of formula (I) or a pharmaceutically acceptable salt thereof in a liquified propellant.
- Suitable propellants include halocarbons, hydrocarbons, and other liquified gases.
- propellants include: trichlorofluoromethane (propellant 1 1 ), dichlorofluoromethane (propellant 12), dichlorotetrafluoroethane (propellant 1 14), tetrafluoroethane (HFA-134a), 1 ,1- difluoroethane (HFA-152a), difluoromethane (HFA-32), pentafluoroethane (HFA-12), heptafluoropropane (HFA-227a), perfluoropropane, perfluorobutane, perfluoropentane, butane, isobutane, and pentane.
- Aerosols comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof will typically be administered to a patient via a metered dose inhaler (MDI). Such devices are known to those skilled in the art.
- MDI metered dose inhaler
- the aerosol may contain additional pharmaceutically-acceptable excipients typically used with MDIs such as surfactants, lubricants, cosolvents and other excipients to improve the physical stability of the formulation, to improve valve performance, to improve solubility, or to improve taste.
- additional pharmaceutically-acceptable excipients typically used with MDIs such as surfactants, lubricants, cosolvents and other excipients to improve the physical stability of the formulation, to improve valve performance, to improve solubility, or to improve taste.
- a pharmaceutical aerosol formulation comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and a fluorocarbon or hydrogen-containing chlorofluorocarbon as propellant, optionally in combination with a surfactant and/or a cosolvent.
- a pharmaceutical aerosol formulation wherein the propellant is selected from 1 ,1 ,1 ,2-tetrafluoroethane, 1 ,1 ,1 ,2,3,3,3-heptafluoro-n-propane and mixtures thereof.
- compositions of the invention may be buffered by the addition of suitable buffering agents.
- Capsules and cartridges for use in an inhaler or insufflator may be formulated containing a powder mix for inhalation of a compound of formula (I) or a pharmaceutically acceptable salt thereof and a suitable powder base such as lactose or starch.
- a powder mix for inhalation of a compound of formula (I) or a pharmaceutically acceptable salt thereof and a suitable powder base such as lactose or starch.
- Each capsule or cartridge may generally contain from 20 ⁇ g to 10mg of the compound of formula (I) or pharmaceutically acceptable salt thereof.
- the compound of formula (I) or pharmaceutically acceptable salt thereof may be presented without excipients such as lactose.
- the proportion of the active compound of formula (I) or pharmaceutically acceptable salt thereof in the local compositions according to the invention depends on the precise type of formulation to be prepared but will generally be within the range of from 0.001 to 10% by weight. Generally, for most types of preparations, the proportion used will be within the range of from 0.005 to 1 %, for example from 0.01 to 0.5%. However, in powders for inhalation or insufflation the proportion used will normally be within the range of from 0.1 to 5%.
- Aerosol formulations are preferably arranged so that each metered dose or "puff" of aerosol contains from 2C ⁇ g to 10mg, preferably from 2C ⁇ g to 200C ⁇ g, more preferably from about 2C ⁇ g to 50C ⁇ g of a compound of formula (I).
- Administration may be once daily or several times daily, for example 2, 3, 4 or 8 times, giving for example 1 , 2 or 3 doses each time.
- the overall daily dose with an aerosol will be within the range from 10C ⁇ g to 10mg, preferably from 20C ⁇ g to 200C ⁇ g.
- the overall daily dose and the metered dose delivered by capsules and cartridges in an inhaler or insufflator will generally be double that delivered with aerosol formulations.
- the particle size of the particulate (e.g., micronised) drug should be such as to permit inhalation of substantially all the drug into the lungs upon administration of the aerosol formulation and will thus be less than 100 microns, desirably less than 20 microns, and in particular in the range of from 1 to 10 microns, such as from 1 to 5 microns, more preferably from 2 to 3 microns.
- the formulations of the invention may be prepared by dispersal or dissolution of the medicament and a compound of formula (I) or a pharmaceutically acceptable salt thereof in the selected propellant in an appropriate container, for example, with the aid of sonication or a high-shear mixer.
- the process is desirably carried out under controlled humidity conditions.
- the chemical and physical stability and the pharmaceutical acceptability of the aerosol formulations according to the invention may be determined by techniques well known to those skilled in the art.
- the chemical stability of the components may be determined by HPLC assay, for example, after prolonged storage of the product.
- Physical stability data may be gained from other conventional analytical techniques such as, for example, by leak testing, by valve delivery assay (average shot weights per actuation), by dose reproducibility assay (active ingredient per actuation) and spray distribution analysis.
- the stability of the suspension aerosol formulations according to the invention may be measured by conventional techniques, for example, by measuring flocculation size distribution using a back light scattering instrument or by measuring particle size distribution by cascade impaction or by the "twin impinger” analytical process.
- twin impinger assay means "Determination of the deposition of the emitted dose in pressurised inhalations using apparatus A” as defined in British Pharmacopaeia 1988, pages A204-207, Appendix XVII C.
- Such techniques enable the "respirable fraction" of the aerosol formulations to be calculated.
- MDI means a unit comprising a can, a secured cap covering the can and a formulation metering valve situated in the cap.
- MDI system includes a suitable channelling device. Suitable channelling devices comprise for example, a valve actuator and a cylindrical or cone-like passage through which medicament may be delivered from the filled canister via the metering valve to the nose or mouth of a patient such as a mouthpiece actuator.
- MDI canisters generally comprise a container capable of withstanding the vapour pressure of the propellant used such as a plastic or plastic-coated glass bottle or preferably a metal can, for example, aluminium or an alloy thereof which may optionally be anodised, lacquer-coated and/or plastic-coated (for example incorporated herein by reference WO96/32099 wherein part or all of the internal surfaces are coated with one or more fluorocarbon polymers optionally in combination with one or more non-fluorocarbon polymers), which container is closed with a metering valve.
- the cap may be secured onto the can via ultrasonic welding, screw fitting or crimping.
- MDIs taught herein may be prepared by methods of the art (e.g.
- the canister is fitted with a cap assembly, wherein a drug-metering valve is situated in the cap, and said cap is crimped in place.
- the metallic internal surface of the can is coated with a fluoropolymer, more preferably blended with a non-fluoropolymer.
- the metallic internal surface of the can is coated with a polymer blend of polytetrafluoroethylene (PTFE) and polyethersulfone (PES).
- PES polyethersulfone
- the whole of the metallic internal surface of the can is coated with a polymer blend of polytetrafluoroethylene (PTFE) and polyethersulfone (PES).
- the metering valves are designed to deliver a metered amount of the formulation per actuation and incorporate a gasket to prevent leakage of propellant through the valve.
- the gasket may comprise any suitable elastomeric material such as, for example, low density polyethylene, chlorobutyl, bromobutyl, EPDM, black and white butadiene- acrylonitrile rubbers, butyl rubber and neoprene.
- Suitable valves are commercially available from manufacturers well known in the aerosol industry, for example, from Valois, France (e.g. DF10, DF30, DF60), Bespak pic, UK (e.g. BK300, BK357) and 3M-
- the MDIs may also be used in conjunction with other structures such as, without limitation, overwrap packages for storing and containing the MDIs, including those described in U.S. Patent Nos. 6,1 19,853; 6,179,1 18; 6,315,1 12; 6,352,152; 6,390,291 ; and 6,679,374, as well as dose counter units such as, but not limited to, those described in U.S. Patent Nos. 6,360,739 and 6,431 ,168.
- a metering valve is crimped onto an aluminium can to form an empty canister.
- the particulate medicament is added to a charge vessel and liquefied propellant together with the optional excipients is pressure filled through the charge vessel into a manufacturing vessel.
- the drug suspension is mixed before recirculation to a filling machine and an aliquot of the drug suspension is then filled through the metering valve into the canister.
- a metering valve is crimped onto an aluminium can to form an empty canister.
- the liquefied propellant together with the optional excipients and the dissolved medicament is pressure filled through the charge vessel into a manufacturing vessel.
- an aliquot of the liquefied formulation is added to an open canister under conditions which are sufficiently cold to ensure the formulation does not vaporise, and then a metering valve crimped onto the canister.
- each filled canister is check- weighed, coded with a batch number and packed into a tray for storage before release testing.
- Suspensions and solutions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may also be administered to a patient via a nebulizer.
- the solvent or suspension agent utilized for nebulization may be any pharmaceutically-acceptable liquid such as water, aqueous saline, alcohols or glycols, e.g., ethanol, isopropylalcohol, glycerol, propylene glycol, polyethylene glycol, etc. or mixtures thereof.
- Saline solutions utilize salts which display little or no pharmacological activity after administration.
- organic salts such as alkali metal or ammonium halogen salts, e.g., sodium chloride, potassium chloride or organic salts, such as potassium, sodium and ammonium salts or organic acids, e.g., ascorbic acid, citric acid, acetic acid, tartaric acid, etc. may be used for this purpose.
- alkali metal or ammonium halogen salts e.g., sodium chloride, potassium chloride or organic salts, such as potassium, sodium and ammonium salts or organic acids, e.g., ascorbic acid, citric acid, acetic acid, tartaric acid, etc.
- organic acids e.g., ascorbic acid, citric acid, acetic acid, tartaric acid, etc.
- the compound of formula (I) or pharmaceutically acceptable salt thereof may be stabilized by the addition of an inorganic acid, e.g., hydrochloric acid, nitric acid, sulphuric acid and/or phosphoric acid; an organic acid, e.g., ascorbic acid, citric acid, acetic acid, and tartaric acid, etc., a complexing agent such as EDTA or citric acid and salts thereof; or an antioxidant such as antioxidant such as vitamin E or ascorbic acid. These may be used alone or together to stabilize the compound of formula (I) or pharmaceutically acceptable salt thereof.
- an inorganic acid e.g., hydrochloric acid, nitric acid, sulphuric acid and/or phosphoric acid
- an organic acid e.g., ascorbic acid, citric acid, acetic acid, and tartaric acid, etc.
- a complexing agent such as EDTA or citric acid and salts thereof
- an antioxidant such as antioxidant such as vitamin E or as
- Preservatives may be added such as benzalkonium chloride or benzoic acid and salts thereof.
- Surfactant may be added particularly to improve the physical stability of suspensions. These include lecithin, disodium dioctylsulphosuccinate, oleic acid and sorbitan esters.
- the invention is directed to a dosage form adapted for intranasal administration.
- Formulations for administration to the nose may include pressurised aerosol formulations and aqueous formulations administered to the nose by pressurised pump. Formulations which are non-pressurised and adapted to be administered topically to the nasal cavity are of particular interest. Suitable formulations contain water as the diluent or carrier for this purpose. Aqueous formulations for administration to the lung or nose may be provided with conventional excipients such as buffering agents, tonicity modifying agents and the like. Aqueous formulations may also be administered to the nose by nebulisation.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may be formulated as a fluid formulation for delivery from a fluid dispenser, for example a fluid dispenser having a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser.
- a fluid dispenser for example a fluid dispenser having a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser.
- Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations.
- the dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity.
- a fluid dispenser of the aforementioned type is described and illustrated in WO05/044354, the entire content of which is hereby incorporated herein by reference.
- the dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation.
- the housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing.
- the fluid dispenser is of the general type illustrated in Figures 30-40 of WO05/044354.
- compositions adapted for intranasal administration wherein the carrier is a solid include a coarse powder having a particle size for example in the range 20 to 500 microns which is administered by rapid inhalation through the nasal passage from a container of the powder held close up to the nose.
- suitable compositions wherein the carrier is a liquid, for administration as a nasal spray or as nasal drops include aqueous or oil solutions of the compound of formula (I) or a pharmaceutically acceptable salt thereof.
- compositions adapted for transdermal administration may be presented as discrete patches intended to remain in intimate contact with the epidermis of the patient for a prolonged period of time.
- the active ingredient may be delivered from the patch by iontophoresis as generally described in Pharmaceutical Research, 3(6), 318 (1986).
- compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- Ointments, creams and gels may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agent and/or solvents.
- bases may thus, for example, include water and/or an oil such as liquid paraffin or a vegetable oil such as arachis oil or castor oil, or a solvent such as polyethylene glycol.
- Thickening agents and gelling agents which may be used according to the nature of the base include soft paraffin, aluminium stearate, cetostearyl alcohol, polyethylene glycols, woolfat, beeswax, carboxypolymethylene and cellulose derivatives, and/or glyceryl monostearate and/or non-ionic emulsifying agents.
- Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents or thickening agents.
- Powders for external application may be formed with the aid of any suitable powder base, for example, talc, lactose or starch.
- Drops may be formulated with an aqueous or nonaqueous base also comprising one or more dispersing agents, solubilising agents, suspending agents or preservatives.
- Topical preparations may be administered by one or more applications per day to the affected area; over skin areas occlusive dressings may advantageously be used. Continuous or prolonged delivery may be achieved by an adhesive reservoir system.
- compositions may be applied as a topical ointment or cream.
- the compound of formula (I) or a pharmaceutically acceptable salt thereof may be employed with either a paraffinic or a water-miscible ointment base.
- the compound of formula (I) or pharmaceutically acceptable salt thereof may be formulated in a cream with an oil-in-water cream base or a water-in-oil base.
- compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the compositions may be presented in unit- dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- the compound and pharmaceutical formulations according to the invention may be used in combination with or include one or more other therapeutic agents, for example selected from anti-inflammatory agents, anticholinergic agents (particularly an M 1 /M 2 /M 3 receptor antagonist), p 2 -adrenoreceptor agonists, antiinfective agents, such as antibiotics or antivirals, or antihistamines.
- other therapeutic agents for example selected from anti-inflammatory agents, anticholinergic agents (particularly an M 1 /M 2 /M 3 receptor antagonist), p 2 -adrenoreceptor agonists, antiinfective agents, such as antibiotics or antivirals, or antihistamines.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with one or more other therapeutically active agents, for example selected from an anti-inflammatory agent, such as a corticosteroid or an NSAID, an anticholinergic agent, a p 2 -adrenoreceptor agonist, an antiinfective agent, such as an antibiotic or an antiviral, or an antihistamine.
- an anti-inflammatory agent such as a corticosteroid or an NSAID
- an anticholinergic agent such as a corticosteroid or an NSAID
- an anticholinergic agent such as a corticosteroid or an NSAID
- an anticholinergic agent such as an antibiotic or an antiviral
- an antiinfective agent such as an antibiotic or an antiviral
- an antihistamine an antihistamine.
- One embodiment of the invention encompasses combinations comprising one or two other therapeutic agents.
- the other therapeutic ingredient(s) may be used in the form of salts, for example as alkali metal or amine salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates to optimise the activity and/or stability and/or physical characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
- the invention encompasses a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a 2 -adrenoreceptor agonist.
- 2 -adrenoreceptor agonists examples include salmeterol (which may be a racemate or a single enantiomer such as the R-enantiomer), salbutamol (which may be a racemate or a single enantiomer such as the R-enantiomer), formoterol (which may be a racemate or a single duastereomer such as the RJ-?-diastereomer), salmefamol, fenoterol carmoterol, etanterol, naminterol, clenbuterol, pirbuterol, flerbuterol, reproterol, bambuterol, indacaterol, terbutaline and salts thereof, for example the xinafoate (1 -hydroxy-2- naphthalenecarboxylate) salt of salmeterol, the sulphate salt or free base of salbutamol or the fumarate salt of formoterol.
- 2 -adrenoreceptor agonists include those described in WO 02/066422, WO 02/070490, WO 02/076933, WO 03/024439, WO 03/072539, WO 03/091204, WO 04/016578, WO 2004/022547, WO 2004/037807, WO 2004/037773, WO 2004/037768, WO 2004/039762, WO 2004/039766, WO01/42193 and WO03/042160.
- Examples of 2 -adrenoreceptor agonists include:
- the agonist may be in the form of a salt formed with a pharmaceutically acceptable acid selected from sulphuric, hydrochloric, fumaric, hydroxynaphthoic (for example 1 - or 3-hydroxy-2-naphthoic), cinnamic, substituted cinnamic, triphenylacetic, sulphamic, sulphanilic, naphthaleneacrylic, benzoic, 4-methoxybenzoic, 2- or 4-hydroxybenzoic, 4-chlorobenzoic and 4-phenylbenzoic acid.
- a pharmaceutically acceptable acid selected from sulphuric, hydrochloric, fumaric, hydroxynaphthoic (for example 1 - or 3-hydroxy-2-naphthoic), cinnamic, substituted cinnamic, triphenylacetic, sulphamic, sulphanilic, naphthaleneacrylic, benzoic, 4-methoxybenzoic, 2-
- Suitable anti-inflammatory agents include corticosteroids.
- Suitable corticosteroids which may be used in combination with the compounds of formula (I) or pharmaceutically acceptable salts thereof are those oral and inhaled corticosteroids and their pro-drugs which have anti-inflammatory activity. Examples include methyl prednisolone,
- corticosteroids include fluticasone propionate, 6a,9a-difluoro-1 i -hydroxy-16a-methyl- 17a-[(4-methyl-1 ,3-thiazole-5-carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17 -carbothioic acid S-fluoromethyl ester, 6a,9a-difluoro-17a-[(2-furanylcarbonyl)oxy]-1 i -hydroxy-16a- methyl-3-oxo-androsta-1 ,4-diene-17 -carbothioic acid S-fluoromethyl ester, 6a, 9a- difluoro-1 1 ⁇ -hydroxy-l 6a-methyl-3-oxo-17a-(2,2,3,3- tetramethycyclopropylcarbonyl)oxy- androsta-1 ,4-diene-17 -carbothioic acid S-cyanomethyl ester and 6a,9a-d
- the corticosteroid is 6a, 9a- difluoro-17a-[(2-furanylcarbonyl)oxy]-1 1 ⁇ -hydroxy-l 6a-methyl-3-oxo-androsta-1 ,4-diene- 17 -carbothioic acid S-fluoromethyl ester.
- corticosteroids may include those described in WO2002/088167, WO2002/100879, WO2002/12265, WO2002/12266, WO2005/005451 , WO2005/005452, WO2006/072599 and WO2006/072600.
- Non-steroidal compounds having glucocorticoid agonism that may possess selectivity for transrepression over transactivation and that may be useful in combination therapy include those covered in the following patents: WO03/082827, W098/54159, WO04/005229, WO04/009017, WO04/018429, WO03/104195, WO03/082787, WO03/082280, WO03/059899, WO03/101932, WO02/02565, WO01/16128, WO00/66590, WO03/086294, WO04/026248, WO03/061651 and WO03/08277. Further non-steroidal compounds are covered in: WO2006/000401 , WO2006/000398 and WO2006/015870. Examples of anti-inflammatory agents include non-steroidal anti-inflammatory drugs (NSAID's).
- NSAID's non-steroidal anti-inflammatory drugs
- NSAID's examples include sodium cromoglycate, nedocromil sodium, phosphodiesterase (PDE) inhibitors (for example, theophylline, PDE4 inhibitors or mixed PDE3/PDE4 inhibitors), leukotriene antagonists, inhibitors of leukotriene synthesis (for example montelukast), iNOS inhibitors, tryptase and elastase inhibitors, beta-2 integrin antagonists and adenosine receptor agonists or antagonists (e.g.
- adenosine 2a agonists adenosine 2a agonists
- cytokine antagonists for example chemokine antagonists, such as a CCR3 antagonist
- inhibitors of cytokine synthesis or 5-lipoxygenase inhibitors.
- An iNOS (inducible nitric oxide synthase inhibitor) is preferably for oral administration.
- iNOS inhibitors include those disclosed in WO93/13055, WO98/30537, WO02/50021 , W095/34534 and W099/62875.
- CCR3 inhibitors include those disclosed in WO02/26722.
- the invention provides the use of the compounds of formula (I) in combination with a phosphodiesterase 4 (PDE4) inhibitor, especially in the case of a formulation adapted for inhalation.
- PDE4-specific inhibitor useful in this aspect of the invention may be any compound that is known to inhibit the PDE4 enzyme or which is discovered to act as a PDE4 inhibitor, and which are only PDE4 inhibitors, not compounds which inhibit other members of the PDE family, such as PDE3 and PDE5, as well as PDE4.
- Compounds include c/s-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-1 - carboxylic acid, 2-carbomethoxy-4-cyano-4-(3-cyclopropylmethoxy-4- difluoromethoxyphenyl)cyclohexan-1-one and c/s-[4-cyano-4-(3-cyclopropylmethoxy-4- difluoromethoxyphenyl)cyclohexan-1-ol].
- anticholinergic agents are those compounds that act as antagonists at the muscarinic receptors, in particular those compounds which are antagonists of the M-i or M 3 receptors, dual antagonists of the M1/M 3 or M 2 /M 3 , receptors or pan-antagonists of the M1/M2/M 3 receptors.
- exemplary compounds for administration via inhalation include ipratropium (for example, as the bromide, CAS 22254-24-6, sold under the name Atrovent), oxitropium (for example, as the bromide, CAS 30286-75-0) and tiotropium (for example, as the bromide, CAS 136310-93-5, sold under the name Spiriva).
- revatropate for example, as the hydrobromide, CAS 262586-79-8) and LAS- 34273 which is disclosed in WO01/041 18.
- Exemplary compounds for oral administration include pirenzepine (CAS 28797-61-7), darifenacin (CAS 133099-04-4, or CAS 133099- 07-7 for the hydrobromide sold under the name Enablex), oxybutynin (CAS 5633-20-5, sold under the name Ditropan), terodiline (CAS 15793-40-5), tolterodine (CAS 124937-51- 5, or CAS 124937-52-6 for the tartrate, sold under the name Detrol), otilonium (for example, as the bromide, CAS 26095-59-0, sold under the name Spasmomen), trospium chloride (CAS 10405-02-4) and solifenacin (CAS 242478-37-1 , or CAS 242478-38-2 for the succinate also known as
- anticholinergic agents include compounds which are disclosed in US patent application 60/487981 including, for example:
- anticholinergic agents include compounds which are disclosed in US patent application 60/51 1009 including, for example:
- the invention provides a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an H1 antagonist.
- H1 antagonists include, without limitation, amelexanox, astemizole, azatadine, azelastine, acrivastine, brompheniramine, cetirizine, levocetirizine, efletirizine, chlorpheniramine, clemastine, cyclizine, carebastine, cyproheptadine, carbinoxamine, descarboethoxyloratadine, doxylamine, dimethindene, ebastine, epinastine, efletirizine, fexofenadine, hydroxyzine, ketotifen, loratadine, levocabastine, mizolastine, mequitazine, mianserin, noberastine, meclizine, norastemizole, olopatad
- the invention provides a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an H3 antagonist (and/or inverse agonist).
- H3 antagonists include, for example, those compounds disclosed in WO2004/035556 and in WO2006/045416.
- Other histamine receptor antagonists which may be used in combination with the compounds of the present invention include antagonists (and/or inverse agonists) of the H4 receptor, for example, the compounds disclosed in Jablonowski et al., J. Med. Chem. 46:3957-3960 (2003).
- the inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor.
- the inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a
- the inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a corticosteroid.
- the inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a non-steroidal GR agonist.
- the inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with an anticholinergic.
- the inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with an antihistamine.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor and a agonist.
- the inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with an anticholinergic and a PDE-4 inhibitor.
- compositions comprising a combination as defined above together with a pharmaceutically acceptable diluent or carrier represent a further aspect of the invention.
- the individual compounds of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations.
- the individual compounds will be administered simultaneously in a combined pharmaceutical formulation.
- Appropriate doses of known therapeutic agents will readily be appreciated by those skilled in the art.
- the invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with another therapeutically active agent.
- the invention thus provides, in a further aspect, a pharmaceutical composition
- a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor.
- the invention thus provides, in a further aspect, a pharmaceutical composition
- a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a 2 -adrenoreceptor agonist.
- the invention thus provides, in a further aspect, a pharmaceutical composition
- a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a corticosteroid.
- the invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a non-steroidal GR agonist.
- a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an anticholinergic.
- the invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an antihistamine.
- a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor and a 2 -adrenoreceptor agonist.
- a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an anticholinergic and a PDE4 inhibitor.
- A 0.1 % v/v solution of formic acid in water.
- the UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
- A 10 mM ammonium bicarbonate in water adjusted to pH 10 with ammonia solution.
- B acetonitrile.
- the UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
- HPLC analysis was conducted on a Sunfire C18 column (150mm x 30mm internal diameter, 5 ⁇ packing diameter) at ambient temperature.
- the solvents employed were:
- A 0.1 % v/v solution of formic acid in water.
- HPLC analysis was conducted on a Sunfire C18 column (150mm x 30mm internal diameter, 5 ⁇ packing diameter) at ambient temperature.
- the solvents employed were:
- A 0.1 % v/v solution of trifluoroacetic acid in water.
- HPLC analysis was conducted on an XBridge C18 column (150mm x 30mm internal diameter, 5 ⁇ packing diameter) at ambient temperature.
- the solvents employed were:
- A 10 mM ammonium bicarbonate in water adjusted to pH 10 with ammonia solution.
- B acetonitrile.
- the gradient employed was dependent upon the retention time of the particular compound undergoing purification as recorded in the analytical LCMS, and was as follows:
- UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
- the chemical names were generated using ACD Name Pro version 6.02 from Advanced Chemistry Development, Inc.
- aqueous phase was extracted with further ethyl acetate (10ml_) and the combined organic extracts were evaporated to dryness.
- the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane followed by 0 to 20% methanol in dichloromethane to afford the title compound (43mg, 0.20mmol, 20% yield).
- Example 10 and Example 11 Example 10 and Example 11 :
- Example 10 (16.5mg, 0.04mmol, 17% yield) LCMS (Method B): Rt 0.65 minutes; m/z 418 (MH+).
- Example 1 1 (8.7mg, 0.026mmol, 1 1 % yield) LCMS (Method B): Rt 0.74 minutes; m/z 341 (MH+).
- trans-4-[(6- ⁇ [6-(3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino ⁇ -2- pyridinyl)amino]cyclohexanol A mixture of trans-4-( ⁇ 6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl ⁇ amino)cyclohexanol [example 3] (100mg, 0.24mmol), caesium carbonate (207mg, 0.63mmol), tetrakis(triphenylphosphine)palladium(0) (14.05mg, 0.012mmol) and
- reaction mixture was sealed and heated in CEM "Discover" microwave at 1 10°C for 20 minutes.
- the reaction mixture was then loaded onto a C18 solid-phase extraction cartridge (pre-conditioned with acetonitrile/0.1 %TFA), the column was flushed through with a further 3ml_ of acetonitrile/0.1 %TFA.
- the recovered crude product was subjected to purification by mass- directed automated preparative HPLC (trifluoroacetic acid modifier) to afford the title compound (9.2mg, 0.022mmol, 22% yield).
- LCMS Method (Method A): Rt 0.99 minutes; m/z 417 (MH+).
- reaction mixture was sealed and heated in CEM "Discover" microwave at 1 10°C for 20 minutes.
- the reaction mixture was then loaded onto a C18 solid-phase extraction cartridge (pre-conditioned with acetonitrile/0.1 %TFA); the column was then flushed through with a further 3ml_ acetonitrile/0.1 %TFA.
- the product was subjected to purification by mass-directed automated preparative HPLC (trifluoroacetic acid modifier) to afford the title compound (14mg, 0.027mmol, 27% yield).
- LCMS Method (Method A): Rt 0.89 minutes; m/z 518 (MH+).
- reaction mixture was then loaded onto a C18 solid-phase extraction cartridge (pre-conditioned with acetonitrile/0.1 %TFA), the column was flushed through with a further 3ml_ acetonitrile/0.1 %TFA.
- the recovered crude product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (19mg, 0.037mmol, 26% yield).
- LCMS Method A: Rt 1 .03minutes; m/z 510 (MH+).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention is directed to certain novel compounds. Specifically, the invention is directed to compounds of formula (I): and salts thereof. The compounds of the invention are inhibitors of kinase activity, in particular Itk activity.
Description
DERIVATIVES OF 2- [2-(BENZO- OR PYRIDO - ) HIAZOLYLAMINO] - 6 -AMINOPYRIDINE , USEFUL IN THE TREATMENT OF RESPIRATORIC, ALLERGIC OR INFLAMMATORY DISEASES
FIELD OF THE INVENTION
The present invention is directed to certain novel compounds which are inhibitors of kinase activity, processes for their preparation, pharmaceutical compositions comprising 5 the compounds, and the use of the compounds or the compositions in the treatment of various disorders. More specifically, the compounds of the invention are inhibitors of the activity or function of Itk (interleukin-2 inducible tyrosine kinase). Compounds which are inhibitors of the activity or function of Itk may be useful in the treatment of disorders such as respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD)
10 and bronchitis; allergic diseases including allergic rhinitis and atopic dermatitis; autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis; transplant rejection; graft versus host disease; inflammatory disorders including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation;
15 HIV; aplastic anemia; and pain including inflammatory pain.
BACKGROUND OF THE INVENTION
lnterleukin-2 inducible tyrosine kinase (Itk) is a non-receptor tyrosine kinase of the Tec family, which is also known as Tsk or Emt. Other members of the Tec familiy include: 0 Tec, Btk, Txk and Bmx. The Tec family kinases are predominantly expressed in haematopoietic cells, however Bmx and Tec have a wider expression profile. The Tec family kinases share a common domain structure: an amino-terminal pleckstrin homology (PH) domain (absent in Txk), a tec homology domain (containing one or two proline rich regions), followed by Src homology SH3 and SH2 domains, and a carboxy-terminal 5 kinase domain. The PH domain binds to Ptdln(3,4,5)P3, and is responsible for locating the Tec kinase to the plasma membrane, whilst the PRR, SH3 and SH2 domains are involved in protein-interactions important in formation of the signalling complex.
Itk expression is restricted to T cells, NK and mast cells. Itk is the predominant Tec family 0 kinase in naive T cells, which also express Txk and Tec. Upon activation via the T cell receptor or interleukin-2 (IL-2), the expression of Itk increases. There is some evidence that Itk is preferentially expressed in Th2 over Th1 cells, in contrast to Txk which is present at higher levels in Th1 cells (1 ). 5 Itk plays a key role in T cell receptor signalling. Itk is recruited to the plasma membrane through interaction with Ptdlns(3,4,5)P3, which is generated by PI3kinase. Itk forms a
complex with several signalling and scaffold proteins including SLP76 and LAT. Itk is transphorphorylated by Lck. Activated Itk phosphorylates PLCy, leading to the generation of lns(1 ,4,5)P3 (required for calcium flux within the cells) and diacylglycerol (activates members of the protein kinase C family and RAS guanyl-releasing protein. This results in the activation of mitogen-activated protein kinases (including JNK and ERK) and other effectors that regulate gene transcription, leading to the secretion of cytokines (reviewed in ref 2). In addition to the role of Itk in PLCy activation and Ca2+ mobilisation, Itk may also contribute to TCR-induced actin reorganisation, and formation of the immune synapse. However, regulation of the actin cytoskeleton may not require kinase activity (3), suggesting the importance of Itk as a scaffold protein. In addition to the T cell receptor, Itk may also be activated via the chemokine receptor CXCR4 (4) in T cells, and via the FceRI in mast cells (5).
There is considerable evidence suggesting that T cells play a key role in the pathogenesis of asthma. The inhibition of T cell cytokines will dampen down the inflammatory cascade involved in the asthmatic response. Cyclosporin A (CsA), which is thought to exert its major effect via inhibition of T cell cytokine release, has shown significant improvement in lung function in two trials with severe asthmatics (6,7). There is also evidence that CsA is steroid sparing and may lead to fewer exacerbations (7). A further trial reported some benefit of CsA but was non-significant (8). CsA does have actions on other cell types (e.g. mast cells) in addition to T cells. However, following allergen challenge in allergic asthmatics, CsA inhibited the late phase but not the early phase response (9), suggesting that effects on mast cells are unlikely to play a key role in the beneficial effect seen of CsA. Furthermore, daclizumab, an antibody against the anti-IL-2Ra chain (CD25) of activated lymphocytes improved pulmonary function and asthma control in patients with moderate to severe chronic asthma (10), supporting anti-T cell therapy for asthma.
Inhibition of Itk represents a potential novel therapy for asthma, by inhibiting T cell cytokine release. The key role for Itk in T cell receptor signalling has been demonstrated using Itk-/- mice and siRNA. In vitro activation of CD4+ cells from Itk knockout mice show reduced levels of Th2 (1 1 ) or both Th1 and Th2 (12) cytokines compared to wild type. Naive T cells from Itk knockout mice can differentiate normally into either Th1 or Th2 cells if cultured in vitro under appropriate cytokine conditions, suggesting that Itk is not required for Th2 cell differentiation (12). Studies differ in the reported effect of Itk knockout on cytokine release upon re-stimulation, showing either a selective reduction in Th2 or reduction in both Th1 and Th2 cytokines (12, 13). Itk siRNA inhibits cytokine release (Th1
and Th2) from human peripheral blood T cells following activation either with anti- CD3/CD28 or in response to recall antigen in vitro.
Itk-/- mice show reduced lung Th2 cytokine production, cell influx, mast cell degranulation and airway hyperreactivity to methacholine in murine Ova challenge models (14,15,16,). In addition to these knockout studies there is also evidence that an Itk inhibitor is effective at reducing cellular influx in an ova murine model of allergic asthma (17). These studies, together with the in vitro profile of Itk inhibitors in human T cells, suggests that Itk is a potential novel target for asthma therapy.
Inhibition of Itk may be beneficial in a variety of T-cell mediated diseases. In addition to asthma, Itk may play a role in other allergic diseases such as allergic rhinitis and atopic dermatitis. Single nucleotide polymorphisms in Itk have been associated with atopy (18) and seasonal allergic rhinitis (19). Itk mRNA levels in the peripheral blood T cells of atopic dermatitis patient is elevated in T cells from affected patients, compared to healthy controls (20).
References
1. Miller et al. Immunity (2004) 21 , 67-80
2. Schwartzberg et al. Nature Reviews Immunol (2005) 5, 284-295
3. Grasis et al. J Immunol (2003) 170, 3971 -3976
4. Fischer et al. J Biol Chem (2004) 279, (28), 29816-29820
5. Kawakami et al. J Immunol (1995) 155, 3556-3562
6. Alexander et al. The Lancet (1992) 339, 324-328
7. Lock et al. Am J Respir Crit Care Med (1996) 153, 509-514
8. Nizankowska et al. Eur Respir J (1995) 8, 1091-1099
9. Sihra et a/.Thorax (1997) 52, 447-452
10. Busse et al. Am J Respir Crit Care med (2008) 178, 1002-1008
1 1 . Fowell et al. Immunity (1999) 11 , 399-409
12. Schaeffer et al. Nature Immunol (2001 ) 2, (12) 1 183-1 188
13. Au-Yeung et al. J Immunol (2006) 176, 3895-3899
14. Ferrara et al. J Allergy Clin Immunol (2006) 117, 780-786
15. Forssell et al. Am J Respir Cell Mol Biol (2005) 32, 51 1-520
16. Mueller and August J Immunol (2003) 170, 5056-5063
17. Lin et al. Biochemistry (2004) 43, (34) 1 1056-1 1062
18. Graves et al. J Allergy Clin Immunol (2005) 116, 650-656
19. Benson et al. Allergy (2009) DOI :10.1 1 1/j.1398-9995.2009.01991 .x
20. Matsumoto et al. Int Arch Allergy Immunol (2002) 129, 327-340
Attempts have been made to prepare compounds which inhibit Itk activity and a number of such compounds have been disclosed in the art. However, in view of the number of pathological responses which are mediated by Itk, there remains a continuing need for inhibitors of Itk which can be used in the treatment of a variety of conditions.
The present inventors have discovered novel compounds which are inhibitors of kinase activity, in particular Itk activity. Compounds which are Itk inhibitors may be useful in the treatment of disorders associated with inappropriate kinase activity, in particular inappropriate Itk activity, for example in the treatment and prevention of disorders mediated by Itk mechanisms. Such disorders include respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis; allergic diseases including allergic rhinitis and atopic dermatitis; autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis; transplant rejection; graft versus host disease; inflammatory disorders including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation; HIV; aplastic anemia; and pain including inflammatory pain.
In one embodiment, compounds of the invention may show selectivity for Itk over other kinases.
SUMMARY OF THE INVENTION
The invention is directed to certain novel compounds. Specifically, the invention is directed to compounds of formula (I)

(I)
wherein R1 to R4 and X are as defined below, and salts thereof.
The compounds are inhibitors of kinase activity, in particular Itk activity. Compounds which are Itk inhibitors may be useful in the treatment of disorders associated with inappropriate Itk activity, such as asthma. Accordingly, the invention is further directed to
pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof. The invention is still further directed to methods of inhibiting Itk activity and treatment of disorders associated therewith using a compound of formula (I) or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof. The invention is yet further directed towards processes for the preparation of the compounds of the invention.
DETAILED DESCRIPTION OF THE INVENTION
In one embodiment, the invention is directed to compounds of formula (I)

(I)
wherein
R1 is hydrogen, -CH2OR5, -CH2NR6R7, -CH2phenyl or -CH2-5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by d-6alkyl;
R2 is hydrogen or methyl,
R3 is C3-6cycloalkyl substituted by -OH or -NR8R9, or -(CH2)m6-membered heterocyclyl wherein the 6-membered heterocyclyl contains one or two heteroatoms independently selected from nitrogen and oxygen and is optionally substituted by Ci-6alkyl, or
R2 and R3, together with the nitrogen atom to which they are attached, are linked to form a piperidinyl substituted by -NR10R11;
R4 is hydrogen, C -6alkyl, halo, -NR12R13, phenyl optionally substituted by -CONR14, 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is optionally substituted by one or two substituents independently selected from Ci-6alkyl, C2-6alkynyl, - CN, -(CH2)nOR15, -CH2phenyl, -(CH2)PNR16R17 and -CONR18R19, or 9- or 10-membered
bicyclic heteroaryl wherein the 9- or 10-membered bicyclic heteroaryl contains one or two nitrogen atoms and is optionally substituted by d-6alkyl;
R5 and R9 are each independently hydrogen or -COCi-6alkyl;
R6 is hydrogen or Ci-6alkyl,
R7 is hydrogen, Ci-6alkyl optionally substituted by -OR20 or -(CH2)qtetrahydropyran, or R6 and R7, together with the nitrogen atom to which they are attached, are linked to form a 4-, 5- or 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom and the 5- or 6-membered heterocyclyl is optionally substituted by one or two substituents independently selected from C1-6alkyl and halo;
R8 is hydrogen; R10 and R1 1 , together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by Ci-6alkyl;
R12 and R13 are each hydrogen, or R12 and R13, together with the nitrogen atom to which they are attached, are linked to form a 5- or 6-membered heterocyclyl wherein the 5- or 6- membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by one or two substituents independently selected from oxo, -OH and Ci-6alkyl optionally substituted by -OH or -NH2; R14 is C3-6cycloalkyl;
R15 is hydrogen, Ci-6alkyl optionally substituted by -OH, C3-6cycloalkyl, -(CH2)rphenyl optionally substituted by halo, or -CH2pyridinyl; R16 and R17 are each independently hydrogen or Ci-6alkyl optionally substituted by -OR21 ;
R18 is hydrogen,
R19 is hydrogen or C1-6alkyl, or
R18 and R19, together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom;
R and R are each independently hydrogen or d-6alkyl; X is -N- or -CH-; m is 1 , 2 or 3; n, p, q and r are each independently 0, 1 or 2; and salts thereof (hereinafter "compounds of the invention").
In one embodiment, R1 is -CH2NR6R7 or -CH2-5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by C1-6alkyl. In another embodiment, R1 is -CH2NR6R7. In a further embodiment, R1 is -CH2-5-membered heteroaryl wherein the 5-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by Ci-6alkyl.
In one embodiment, R2 is hydrogen.
In one embodiment, R3 is C3-6cycloalkyl substituted by -OH or -NR8R9. In a further embodiment, R3 is C3-6cycloalkyl substituted by -OH.
In one embodiment, R4 is hydrogen, -NR12R13 or 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is optionally substituted by Ci-6alkyl, C2-6alkynyl, - CN, -(CH2)nOR15, -CH2phenyl, -(CH2)PNR16R17 or -CONR18R19. In another embodiment, R4 is hydrogen, -NR12R13 or 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by -CN. In a further embodiment, R4 is -NR12R13 or 5- or 6-membered heteroaryl wherein the 5- or 6- membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by - CN.
The skilled artisan will appreciate that when R4 is 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is substituted by one or two substituents including -OH, the R4 group may be drawn as the corresponding keto tautomer. For example, 2,4- dihydro-3H-1 ,2,4-triazo-3-one may be drawn as follows:
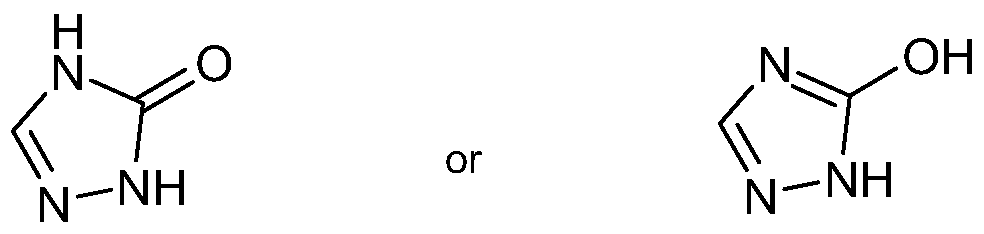
All such tautomeric forms are included whether such tautomers exist in equilibrium or predominantly in one form. In one embodiment, R5 is hydrogen or -COCi-4alkyl.
In one embodiment, R6 is hydrogen or C1-4alkyl. In another embodiment, R6 is hydrogen. In a further embodiment, R6 is methyl or ethyl. In one embodiment, R7 is hydrogen or C1-6alkyl optionally substituted by -OR20 . In a further embodiment, R7 is C1-6alkyl.
In one embodiment, R6 and R7, together with the nitrogen atom to which they are attached, are linked to form a 4-, 5- or 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom and the 5- or 6-membered heterocyclyl is optionally substituted by one or two substituents independently selected from d-6alkyl and halo. In a further embodiment, R6 and R7, together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom and is optionally substituted by one or two substituents independently selected from Ci-6alkyl and halo.
In one embodiment, R9 is hydrogen or -COCi-4alkyl.
In one embodiment, R10 and R11 , together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl contains an oxygen atom or a further nitrogen atom and is optionally substituted by methyl.
In one embodiment, R12 and R13, together with the nitrogen atom to which they are attached, are linked to form a 5- or 6-membered heterocyclyl wherein the 5- or 6- membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by one or two substituents independently selected from oxo, -OH and C1-6alkyl optionally substituted by -OH or -NH2. In a further embodiment, R12 and R13, together with the nitrogen atom to which they are attached, are linked to form a 5-
membered heterocyclyl wherein the 5-membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by one or two oxo substituents. In one embodiment, R14 is cyclopentyl.
In one embodiment, R15 is hydrogen, Ci-4alkyl optionally substituted by -OH, C3- 6cycloalkyl, -(CH2)rphenyl optionally substituted by halo, or -CH2pyridinyl. In a further embodiment, R15 is hydrogen or Ci-4alkyl optionally substituted by -OH.
In one embodiment, R16 and R17 are each independently hydrogen or C -4alkyl optionally substituted by -OR21. In another embodiment, R16 and R17 are each independently C-i. 4alkyl optionally substituted by -OR21. In a further embodiment, R16 is hydrogen and R17 is d^alkyl optionally substituted by -OR21.
In one embodiment, R18 is hydrogen.
In one embodiment, R19 is hydrogen or d-6alkyl. In a further embodiment, R19 is Ci-4alkyl. In one embodiment, R18 and R19, together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom. In a further embodiment, R18 and R19, together with the nitrogen atom to which they are attached, are linked to form a morpholinyl.
In one embodiment, R20 is hydrogen or Ci-4alkyl. In a further embodiment, R20 is hydrogen or methyl.
In one embodiment, R21 is hydrogen or Ci-4alkyl. In a further embodiment, R21 is methyl.
In one embodiment, X is -N-. In a further embodiment, X is -CH-. In one embodiment, m is 2 or 3 In one embodiment, n is 0 or 1.
In one embodiment, p is 0 or 1.
In one embodiment, q is 0 or 1. In one embodiment, r is 0 or 1.
It is to be understood that the present invention covers all combinations of substituent groups described hereinabove.
Compounds of the invention include the compounds of Examples 1 to 164 and salts thereof.
In one embodiment, the compound of the invention is:
frans-4-({6-[(6-methyl-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol;
frans-4-({6-[(6-ethyl-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol;
trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol;
N-(frans-4-aminocyclohexyl)-N'-(6-bromo-1 ,3-benzothiazol-2-yl)-2,6-pyridinediamine;
A/-[frans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexyl]acetamide;
frans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4-b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
frans-4-({6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol;
frans-4-[(6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-[(6-{[5-(1 H-pyrazol-4-yl)[1 ,3]thiazolo[5,4-b]pyridin-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-[(6-{[6-(4-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol; frans-4-{[6-(1 ,3-benzothiazol-2-ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-[(6-{[6-(3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol; trans-4-[(6-{[6-(3-furanyl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol; trans-4-[(6-{[6-(2-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol; frans-4-({6-[(6-phenyl-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol;
frans-4-{[6-({6-[1 -(3-methylbutyl)-1 H-pyrazol-4-yl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(6-{[6-(1 -methyl-1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-{[6-({6-[6-(methyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-({6-[2-(methyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-({6-[4-(methyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(6-{[6-(6-amino-3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-A/- methyl-2-pyridinecarboxamide;
frans-4-[(6-{[6-(4-isoxazolyl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol; /V-cyclopentyl-4-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]benzamide;
frans-4-[(6-{[6-(1 H-indazol-5-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-2- pyridinecarbonitrile;
5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3- pyridinecarbonitrile;
5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3- pyridinecarboxamide;
frans-4-{[6-({6-[5-(methyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-({6-[1 -(phenylmethyl)-l H-pyrazol-4-yl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(6-{[6-(1 H-indazol-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
A/-(1 ,1 -dimethylethyl)-5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)[1 ,3]thiazolo[5,4-b]pyridin-5-yl]-3-pyridinecarboxamide;
frans-4-({6-[(5-phenyl[1 ,3]thiazolo[5,4-b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
frans-4-{[6-({5-[5-(methyloxy)-3-pyridinyl][1 ,3]thiazolo[5,4-b]pyridin-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-({5-[5-(4-morpholinylcarbonyl)-3-pyridinyl][1 ,3]thiazolo[5,4-/b]pyridin-2- yl}amino)-2-pyridinyl]amino}cyclohexanol;
5-[2-({6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridm^
yl]-3-pyridinecarbonitrile;
frans-4-{[6-({6-[5-(phenyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(6-{[6-(1 -ethyl-1 H-indol-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-{[6-({6-[5-(cyclopentyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-({6-[5-(1 -propyn-1 -yl)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-({6-[(6-{5-[(ethylamino)methyl]-3-pyridinyl}-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
frans-4-[(6-{[6-(5-{[(1 -methylethyl)amino]methyl}-3-pyridinyl)-1 ,3-benzothiazol-2-yl]amin 2-pyridinyl)amino]cyclohexanol;
frans-4-[(6-{[6-(1 H-indol-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-({6-[(6-{5-[(methylamino)methyl]-3-pyridinyl}-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
frans-4-[(6-{[6-(5-methyl-3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-{[6-({6-[5-(ethyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-({6-[(6-{5-[(1-methylethyl)oxy]-3-pyridinyl}-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
frans-4-[(6-{[6-(3-quinolinyl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexano frans-4-{[6-({6-[5-(hydroxymethyl)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol;
frans-4-({6-[(6-{5-[(dimethylamino)methyl]-3-pyridinyl}-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
4-[(6-{[6-(1 -ethyl-1 H-pyrrolo[3,2-b]pyridin-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
4-[(6-{[6-(1-methyl-1 H-pyrrolo[3,2-b]pyridin-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-({6-[(6-{5-[(2-pyridinylmethyl)oxy]-3-pyridinyl}-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
frans-4-[(6-{[6-(5-{[(3-fluorophenyl)methyl]oxy}-3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-({6-[(6-{5-[(2-hydroxyethyl)oxy]-3-pyridinyl}-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
frans-4-[(6-{[6-(1 H-pyrrolo[3,2-b]pyridin-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-[(6-{[6-(1 -methyl-1 H-indol-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-{[6-({6-[5-({[2-(methyloxy)ethyl]amino}methyl)-3-pyridinyl]-1 ,3-benzothiazol-2- yl}amino)-2-pyridinyl]amino}cyclohexanol;
4-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3- morpholinone;
3-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3- oxazolidin-2-one;
1-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3- methyl-2-imidazolidinone;
1-(2-aminoethyl)-3-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-2-imidazolidinone;
(4R)-4-hydroxy-1 -[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-2-pyrrolidinone;
(4S)-4-hydroxy-1-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-2-pyrrolidinone;
1-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-(2- hydroxyethyl)-2-imidazolidinone;
frans-4-{[6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-4-(phenylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-([1 ,3]thiazolo[5^-b]pyridin-2-ylamino)-2-pyridinyl]amino}cyclohexanol;
3- [2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyrid^
yl]-1 ,3-oxazolidin-2-one;
4- [2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-5- methyl-2,4-dihydro-3H-1 ,2,4-triazol-3-one;
1-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-2,5- pyrrolidinedione;
4-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-2,4- dihydro-3H-1 ,2,4-triazol-3-one;
frans-4-[(4-(phenylmethyl)-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(2-pyridinylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-4-(2-pyridinylmethyl)-2- pyridinyl]amino}cyclohexanol;
1-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(phenylmethyl)-2-pyridinyl]amino}-1 ,3- benzothiazol-6-yl)-2,5-pyrrolidinedione;
frans-4-{[6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-4-(phenylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-{[6-(3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-4-(2-pyridinylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-
2- pyridinyl}amino)cyclohexanol;
frans-4-{[4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-([1 , 3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-4-(1 H-pyrazol-1 -ylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(4-(1 H-pyrazol-1 -ylmethyl)-6-{[6-(3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
frans-4-[(4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2- yl]amino}-2-pyridinyl)amino]cyclohexanol;
frans-4-[(4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-{[6-(3-pyridinyl)-1 ,3-benzothiazol-2- yl]amino}-2-pyridinyl)amino]cyclohexanol;
5-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-3-pyridinecarbonitrile;
5-(2-{[6-[(frans-4-aminocyclohexyl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-3-pyridinecarbonitrile;
5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({6-[(frans-4-aminocyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
3- (2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-1 ,3-oxazolidin-2-one;
3-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one;
1-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-(2-hydroxyethyl)-2-imidazolidinone;
(4R)-4-hydroxy-1 -[2-({6-[(frans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 - yl)methyl]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-2-pyrrolidinone;
(4S)-4-hydroxy-1-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 - yl)methyl]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-2-pyrrolidinone;
1-(2-aminoethyl)-3-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1- yl)methyl]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-2-imidazolidinone;
4- [(2-methyl-1 H-imidazol-1-yl)methyl]-N-[3-(4-morpholinyl)propyl]-N 1 ,3]thi
b]pyridin-2-yl-2,6-pyridinediamine;
A/-(6-bromo-1 ,3-benzothiazol-2-yl)-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-A/'-[2-(4- morpholinyl)ethyl]-2,6-pyridinediamine;
6-bromo-/V-{4-[(2-methyl-1 H-imidazol-1-yl)methyl]-6-[4-(4-morpholinyl)-1-piperidi pyridinyl}-1 ,3-benzothiazol-2-amine;
6-bromo-/V-{4-[(2-methyl-1 H-imidazol-1-yl)methyl]-6-[4-(4-methyl-1 -piperazinyl)-1 - piperidinyl]-2-pyridinyl}-1 ,3-benzothiazol-2-amine;
N-(6-bromo-1 ,3-benzothiazol-2-yl)-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-N'-[3-(4- morpholinyl)propyl]-2,6-pyridinediamine;
N-(6-bromo-1 ,3-benzothiazol-2-yl)-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-N'-[2-(4-methyl- 1-piperazinyl)ethyl]-2,6-pyridinediamine;
5-{2-[(4-[(2-methyl-1 H-imidazol-1-yl)methyl]-6-{[3-(4-morpholinyl)propyl]amino}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-3-pyridinecarbonitrile;
5- (2-{[6-{[3-(4-morpholinyl)propyl]amino}-4-(1 H-pyrazol-1-ylmethyl)-2-pyridinyl]amino}^ benzothiazol-6-yl)-3-pyridinecarbonitrile;
frans-4-{[4-(hydroxymethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(hydroxymethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(4-(hydroxymethyl)-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol;
{2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-[(frans-4-hydroxycyclohexyl)amino]-4- pyridinyl}methyl acetate;
frans-4-{[4-[(tetrahydro-2H-pyran-4-ylamino)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[(tetrahydro-2H-pyran-4-ylmethyl)am
2-ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[(2,2-dimethylpropyl)amino]methyl}-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylami pyridinyl]amino}cyclohexanol;
frans-4-{[4-({[2-methyl-2-(methyloxy)propyl]amino}methyl)-6-([1 ,3]thiazolo[5^-b]pyridin-2- ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[(2-hydroxy-2-methylpropyl)amino]methyl}-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[(2-methylpropyl)amino]methyl}-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(diethylamino)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[4-(1 -pyrrolidinylmethyl)-6-([1 ,3]thiazolo[5,4-/b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-4-{[(1 ,2,2-trimethylpropyl)amino]methyl}- 2-pyridinyl)amino]cyclohexanol;
frans-4-{[4-(4-morpholinylmethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(2,6-dimethyl-4-morpholinyl)methy^
pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[methyl(1 -methylethyl)amino]methyl}-6-([1 ,3]thiazolo[5,4-/b]pyridin-2-ylamino)- 2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[ethyl(1 -methylethyl)amino]methyl}-6-([1 ,3]thiazolo[5,4-/b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(4,4-dimethyl-1 -piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylami pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3-methyl-1 -pyrrolidinyl)methyl]-6-([1 ,3]thiazolo[5^-b]pyridin-2-ylamin pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3,3-dimethyl-1 -piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylami pyridinyl]amino}cyclohexanol;
frans-4-{[6-([1 ,3]thiazolo[5,4-/b]pyridin-2-ylamino)-4-({[(1 S)-1 ,2,2- trimethylpropyl]amino}methyl)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[6-([1 ,3]thiazolo[5,4-/b]pyridin-2-ylamino)-4-({[(1 R)-1 ,2,2- trimethylpropyl]amino}methyl)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(4,4-difluoro-1-piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamin pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3,3-difluoro-1 -pyrrolidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamin pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[methyl(1 ,2,2-trimethylpropyl)am
ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-(1 -piperidinylmethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[2-(2-methylpropyl)-4-morpholinyl]methyl}-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(2,2-dimethyl-4-morpholin^
pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(2-methyl-1 -pyrrolidinyl)methyl^^
pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-6-([1 ,3]thiazolo[5,4-b]pyridin-2 ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3,3-difluoro-1-piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamin pyridinyl]amino}cyclohexanol;
frans-4-{[6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-4-(4-morpholinylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-({4-(aminomethyl)-6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol;
5-[2-({4-(aminomethyl)-6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1 -pyrrolidinylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-3-pyridinecarbonitrile;
5-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(hydroxymethyl)-2-pyridinyl]amino}-1 ,3- benzothiazol-6-yl)-3-pyridinecarbonitrile;
5-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(4-morpholinylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-3-pyridinecarbonitrile;
5-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1-piperidinylmethyl)-2-pyridinyl]amino}-1 ,3- benzothiazol-6-yl)-3-pyridinecarbonitrile;
5-[2-({4-(1 -azetidinylmethyl)-6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-4-[(methylamino)methyl]-2-pyridinyl}amino)- 1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({4-[(ethylamino)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({4-[(diethylamino)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)^ 1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({4-[(dimethylamino)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}ami 1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-{2-[(6-[(frans-4-hydroxycyclohexyl)amino]-4-{[(1 ,2,2-trimethylpropyl)amino]methyl}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-3-pyridinecarbonitrile;
5-[2-({4-[(3,3-difluoro-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({4-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-6-[(frans-4- hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitri 3-[2-({4-[(3,3-difluoro-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one;
3-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1 -pyrrolidinylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-1 ,3-oxazolidin-2-one;
3-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(4-morpholinylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-1 ,3-oxazolidin-2-one;
3-[2-({4-[(diethylamino)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)^ 1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one;
3-{2-[(6-[(frans-4-hydroxycyclohexyl)amino]-4-{[(1 ,2,2-trimethylpropyl)amino]methyl}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-1 ,3-oxazolidin-2-one;
3-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1-piperidinylmethyl)-2-pyridinyl]amino}-1 ,3- benzothiazol-6-yl)-1 ,3-oxazolidin-2-one;
3-[2-({4-[(4,4-dimethyl-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one;
3-[2-({4-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-6-[(frans-4- hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one;
1-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(4-morpholinylmethyl)-2-pyridinyl]amino}- 1 ,3-benzothiazol-6-yl)-2,5-pyrrolidinedione;
1-{2-[(6-[(frans-4-hydroxycyclohexyl)amino]-4-{[(1 ,2,2-trimethylpropyl)amino]methyl}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-2,5-pyrrolidinedione;
1-[2-({4-[(3,3-difluoro-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-2,5-pyrrolidinedione;
frans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(1-pyrrolidinylmethyl)-2- pyridinyl]amino}cyclohexanol;
frans-4-[(6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-{[(1 ,2,2- trimethylpropyl)amino]methyl}-2-pyridinyl)amino]cyclohexanol;
frans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-[(3,3-difluoro-1 -piperidinyl)methyl]-2- pyridinyl}amino)cyclohexanol;
frans-4-[(6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-{[(2R,6S)-2,6-dimethyl-4- morpholinyl]methyl}-2-pyridinyl)amino]cyclohexanol;
frans-4-{[6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-4-(1-pyrrolidinylmethyl)-2- pyridinyl]amino}cyclohexanol; or
a salt thereof.
In a further embodiment, the compound of the invention is:
frans-4-[(4-[(2-methyl-1 H-imidazol-1-yl)methyl]-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2- yl]amino}-2-pyridinyl)amino]cyclohexanol
frans-4-{[4-(4-morpholinylmethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-^
pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3,3-dimethyl-1 -piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-^
pyridinyl]amino}cyclohexanol;
frans-4-{[6-([1 ,3]thiazolo[5,4-/b]pyridin-2-ylamino)-4-({[(1 S)-1 ,2,2- trimethylpropyl]amino}methyl)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(2,2-dimethyl-4-morpholinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridi
pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-6-([1 ,3]thiazolo[5,4-b]p ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3,3-difluoro-1-piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylam pyridinyl]amino}cyclohexanol;
5-{2-[(6-[(frans-4-hydroxycyclohexyl)amino]-4-{[(1 ,2,2-trimethylpropyl)amino]methyl}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-3-pyridinecarbonitrile;
5-[2-({4-[(3,3-difluoro-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({4-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-6-[(frans-4- hydroxycyclohexyl)amino]-2-pyridinyl}ami
3-[2-({4-[(3,3-difluoro-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one; or
a salt thereof.
Terms and Definitions
"AlkyI" refers to a saturated hydrocarbon chain having the specified number of member atoms. C1-6alkyl refers to an alkyl group having from 1 to 6 member atoms, for example 1 to 4 member atoms. Alkyl groups may be optionally substituted with one or more substituents as defined herein. Alkyl groups may be straight or branched. Representative branched alkyl groups have one, two, or three branches. Alkyl includes methyl, ethyl, propyl (n-propyl and isopropyl), butyl (n-butyl, isobutyl, and t-butyl), pentyl (n-pentyl, isopentyl, and neopentyl), and hexyl.
"Alkynyl" refers to a hydrocarbon chain having the specified number of member atoms and at least one triple bond. For example, C2-6alkynyl refers to an alkynyl group having from 2 to 6 member atoms, for example 2 to 4 member atoms. Alkynyl groups may be
straight or branched. Alkynyl includes ethynyl, 1 -propynyl, 1-butynyl, 2-butynyl, 1 - pentynyl, 2-pentynyl, 3-pentynyl, 1 -hexynyl, 2-hexynyl and 3-hexynyl.
"Cycloalkyl" refers to a saturated hydrocarbon ring having the specified number of member atoms. Cycloalkyl groups are monocyclic ring systems. For example, C3- 6cycloalkyl refers to a cycloalkyl group having from 3 to 6 member atoms. Cycloalkyl groups may be optionally substituted with one or more substituents as defined herein. Cycloalkyl includes cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. "Enantiomerically enriched" refers to products whose enantiomeric excess is greater than zero. For example, enantiomerically enriched refers to products whose enantiomeric excess is greater than 50% ee, greater than 75% ee, and greater than 90% ee.
"Enantiomeric excess" or "ee" is the excess of one enantiomer over the other expressed as a percentage. As a result, since both enantiomers are present in equal amounts in a racemic mixture, the enantiomeric excess is zero (0% ee). However, if one enantiomer was enriched such that it constitutes 95% of the product, then the enantiomeric excess would be 90% ee (the amount of the enriched enantiomer, 95%, minus the amount of the other enantiomer, 5%).
"Enantiomerically pure" refers to products whose enantiomeric excess is 99% ee or greater.
"Half-life" (or "half-lives") refers to the time required for half of a quantity of a substance to be converted to another chemically distinct species in vitro or in vivo.
"Halo" refers to the halogen radical fluoro, chloro, bromo, or iodo.
"Heteroaryl", unless otherwise defined, refers to an aromatic ring containing from 1 to 3 heteroatoms as member atoms in the ring or rings. Heteroaryl groups containing more than one heteroatom may contain different heteroatoms. Heteroaryl groups may be optionally substituted with one or more substituents if so defined herein. The heteroaryl groups herein are monocyclic ring systems or are fused bicyclic ring systems. Monocyclic heteroaryl rings have 5 or 6 member atoms. Bicyclic heteroaryl rings have 9 or 10 member atoms. Monocyclic heteroaryl includes pyrrolyl, furanyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, oxadiazolyl, triazolyl, pyridinyl, pyrimidinyl, pyridazinyl and pyrazinyl.
Bicyclic heteroaryl includes indolyl, isoindolyl, indolizinyl, indazolyl, benzimidazolyl, pyrrolopyridinyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl and naphthridinyl.
"Heteroatom" refers to a nitrogen, sulphur, or oxygen atom.
"Heterocyclyl", unless otherwise defined, refers to a saturated or unsaturated ring containing 1 or 2 heteroatoms as member atoms in the ring. However, heterocyclyl rings are not aromatic. In certain embodiments, heterocyclyl is saturated. In other embodiments, heterocyclyl is unsaturated but not aromatic. Heterocyclyl groups containing more than one heteroatom may contain different heteroatoms. The heterocyclyl groups herein are monocyclic ring systems having 4, 5 or 6 member atoms. Heterocyclyl groups may be optionally substituted with one or more substituents as defined herein. Heterocyclyl includes azetidinyl, pyrrolidinyl, pyrazolidinyl, imidazolinyl, oxazolidinyl, isoxazolidinyl, piperidinyl, piperazinyl and morpholinyl.
"Member atoms" refers to the atom or atoms that form a chain or ring. Where more than one member atom is present in a chain and within a ring, each member atom is covalently bound to an adjacent member atom in the chain or ring. Atoms that make up a substituent group on a chain or ring are not member atoms in the chain or ring.
"Optionally substituted" indicates that a group, such as heteroaryl, may be unsubstituted or substituted with one or more substituents as defined herein.
"Substituted" in reference to a group indicates that a hydrogen atom attached to a member atom within a group is replaced. It should be understood that the term "substituted" includes the implicit provision that such substitution be in accordance with the permitted valence of the substituted atom and the substituent and that the substitution results in a stable compound (i.e. one that does not spontaneously undergo transformation such as by rearrangement, cyclization, or elimination). In certain embodiments, a single atom may be substituted with more than one substituent as long as such substitution is in accordance with the permitted valence of the atom. Suitable substituents are defined herein for each substituted or optionally substituted group.
"Pharmaceutically acceptable" refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity,
irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
As used herein the symbols and conventions used in these processes, schemes and examples are consistent with those used in the contemporary scientific literature, for example, the Journal of the American Chemical Society or the Journal of Biological Chemistry. Unless otherwise noted, all starting materials were obtained from commercial suppliers and used without further purification. Specifically, the following abbreviations may be used in the examples and throughout the specification:
DMF dimethylformamide
DMSO dimethylsulphoxide
Et ethyl
HPLC high performance liquid chromatography
LCMS liquid chromatography-mass spectrometry
MDAP mass-directed autopreparative HPLC
Me methyl
min minutes
mg milligrams
ml_ millilitres
mM millimolar
mmol millimoles
m/z mass/charge ratio
NMR nuclear magnetic resonance
Pr n-propyl
Rt retention time
TFA trifluoroacetic acid
THF tetrahydrofuran
UPLC ultra performance liquid chromatography
UV ultraviolet
All references to brine are to a saturated aqueous solution of NaCI.
Included within the scope of the "compounds of the invention" are all solvates (including hydrates), complexes, polymorphs, prodrugs, radiolabeled derivatives, stereoisomers and optical isomers of the compounds of formula (I) and salts thereof.
The compounds of the invention may exist in solid or liquid form. In the solid state, the compounds of the invention may exist in crystalline or noncrystalline form, or as a mixture thereof. For compounds of the invention that are in crystalline form, the skilled artisan will appreciate that pharmaceutically acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization. Solvates may involve nonaqueous solvents such as ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and EtOAc, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent that is incorporated into the crystalline lattice are typically referred to as "hydrates." Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
The skilled artisan will further appreciate that certain compounds of the invention that exist in crystalline form, including the various solvates thereof, may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs". The invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification. The skilled artisan will appreciate that different polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymorphs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
The invention also includes isotopically-labelled compounds, which are identical to the compounds of formula (I) and salts thereof, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number most commonly found in nature. Examples of isotopes that can be incorporated into the compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen and fluorine, such as 3H, 1 1 C, 14C and 18F. The compounds according to formula (I) may contain one or more asymmetric center (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereomers, or other stereoisomeric forms, or as mixtures thereof. Chiral centers, such
as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the stereochemistry of a chiral center present in formula (I), or in any chemical structure illustrated herein, is not specified the structure is intended to encompass any stereoisomer and all mixtures thereof. Thus, compounds according to formula (I) containing one or more chiral center may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers.
Individual stereoisomers of a compound according to formula (I) which contain one or more asymmetric center may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1 ) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer- specific reagent, for example by enzymatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral enviornment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent. The skilled artisan will appreciate that where the desired stereoisomer is converted into another chemical entity by one of the separation procedures described above, a further step is required to liberate the desired form. Alternatively, specific stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
The compounds according to formula (I) may also contain centers of geometric asymmetry. Where the stereochemistry of a center of geometric asymmetry present in formula (I), or in any chemical structure illustrated herein, is not specified, the structure is intended to encompass the trans geometric isomer, the cis geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included in formula (I) whether such tautomers exist in equilibrium or predominately in one form.
It is to be understood that the references herein to compounds of formula (I) and salts thereof covers the compounds of formula (I) as free acids or free bases, or as salts thereof, for example as pharmaceutically acceptable salts thereof. Thus, in one embodiment, the invention is directed to compounds of formula (I) as the free acid or free base. In another embodiment, the invention is directed to compounds of formula (I) and salts thereof. In a further embodiment, the invention is directed to compounds of formula (I) and pharmaceutically acceptable salts thereof.
The skilled artisan will appreciate that pharmaceutically acceptable salts of the compounds according to formula (I) may be prepared. Indeed, in certain embodiments of
the invention, pharmaceutically acceptable salts of the compounds according to formula (I) may be preferred over the respective free base or free acid because such salts impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage form. Accordingly, the invention is further directed to compounds of formula (I) and pharmaceutically acceptable salts thereof.
As used herein, the term "pharmaceutically acceptable salts" refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
Salts and solvates having non-pharmaceutically acceptable counter-ions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts. Thus one embodiment of the invention embraces compounds of formula (I) and salts thereof. In certain embodiments, compounds according to formula (I) may contain an acidic functional group. Suitable pharmaceutically-acceptable salts include salts of such acidic functional groups. Representative salts include pharmaceutically acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a pharmaceutically acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, TEA, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
In certain embodiments, compounds according to formula (I) may contain a basic functional group and are therefore capable of forming pharmaceutically acceptable acid addition salts by treatment with a suitable acid. Suitable acids include pharmaceutically acceptable inorganic acids and pharmaceutically acceptable organic acids. Representative pharmaceutically acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphate, acetate, hydroxyacetate, phenylacetate, propionate, butyrate, isobutyrate, valerate, maleate,
hydroxymaleate, acrylate, fumarate, malate, tartrate, citrate, salicylate, p- aminosalicyclate, glycollate, lactate, heptanoate, phthalate, oxalate, succinate, benzoate, o-acetoxybenzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, naphthoate, hydroxynaphthoate, mandelate, tannate, formate, stearate, ascorbate, palmitate, oleate, pyruvate, pamoate, malonate, laurate, glutarate, glutamate, estolate, methanesulfonate (mesylate), ethanesulfonate (esylate), 2- hydroxyethanesulfonate, benzenesulfonate (besylate), p-aminobenzenesulfonate, p- toluenesulfonate (tosylate), and napthalene-2-sulfonate. Compound Preparation
The compounds of the invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention are prepared in the Examples section.
Process a
Compounds of formula (I) wherein R1 to R4 and X are as defined above, and salts thereof, may be prepared by a process comprising reacting of a compound of formula (II)

(II)
wherein R1, R4 and X are as defined above and Y is halo such as fluoro or chloro, with an amine of formula (III)
HNR2R3
(HI) wherein R2 and R3 as defined above.
The process may be carried out under microwave irradiation, using neat amine or amine in a suitable solvent such as ethylene glycol, and at a suitable temperature such as from about 160 to about 220°C.
Compounds of formula (II) wherein Y is fluoro, may be prepared by a process comprising reacting of a compound of formula (IV)

(IV)
wherein R1 is as defined above and Y and Y1 are both fluoro, with a compound of formula (V)

(V)
wherein R and X are as defined above.
The process may be carried out in a suitable solvent such as tetrahydrofuran or DMF, in the presence of a suitable base such as sodium hydride, and at a suitable temperature such as about 0°C then allowing the reaction mixture to warm to ambient temperature.
Compounds of formula (II) wherein Y is chloro, may be prepared by a process comprising reacting of a compound of formula

(VI)
wherein R1 is as defined above and Y is chloro, with a compound of formula (VII)

wherein R4 and X are as defined above and Y2 is chloro.
The process may be carried out in a suitable solvent such as tetrahydrofuran or DMF, in the presence of a suitable base such as sodium hydride, and at a suitable temperature such as 0°C then heating the reaction mixture to about 50°C. Compounds of formula (VI) wherein R1 is as defined above and Y is chloro, may be prepared by a process comprising reacting a compound of formula (VIII)

(VIII)
wherein R1 is as defined above and Y and Y3 are both chloro, with concentrated aqueous ammonia.
The process may be carried out under microwave irradiation, and at a suitable temperature such as from about 160 to about 220°C. Process b
Compounds of formula (I) wherein R1 to R4 and X are as defined above, and salts thereof, may also be prepared by a process comprising reacting a compound of formula (IX)

(IX)
wherein R1 to R3 and X are as defined above, with a compound of formula
(X)
wherein R4 is as defined above and Y4 is halo such as bromo.
Suitable conditions for the Suzuki coupling include microwave irradiation, in the presence of a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium (0) or 2'- (dimethylamino)-2-biphenyl-palladium (II) chloride dinorbornylphosphine complex, in a suitable solvent such as aqueous 1 ,4-dioxane, in the presence of a suitable base such as caesium carbonate or potassium phosphate, and at a suitable temperature such as from about 100 to about 150°C. Compounds of formula (IX) wherein wherein R1 and X are as defined above may be prepared by a process comprising reacting a compound of formula (XI)

(XI)
wherein R1 to R3 and X are as defined above and Y5 is halo such as bromo, with 4,4,4' ,4',5,5,5',5'-octamethyl-2,2'-bi-1 ,3,2-dioxaborolane.
The process may be carried out under microwave irradiation, in the presence of a suitable palladium catalyst such as 1 ,1 '-bis(diphenylphosphino)ferrocene-palladium(ll)dichloride dichloromethane adduct, in a suitable solvent such as tetrahydrofuran, in the presence of a suitable base such as potassium acetate, and at a suitable temperature such as from about 1 10 to about 170°C, for example about 120°C.
Process c
Compounds of formula (I) wherein R1 to R4 and X are as defined above, and salts thereof, may also be prepared by a process comprising reacting a compound of formula (XI) as defined above, with a compound of formula XIIA) or (XIIB)

(XIIA)
— B
OH wherein R is as defined above. Suitable conditions for the Suzuki coupling include microwave irradiation, in the presence of a suitable palladium catalyst such as tetrakis(triphenylphosphine)palladium (0) or 2'- (dimethylamino)-2-biphenyl-palladium (II) chloride dinorbornylphosphine complex, in a suitable solvent such as aqueous 1 ,4-dioxane, in the presence of a suitable base such as caesium carbonate or potassium phosphate, and at a suitable temperature such as from about 100 to about 150°C.
Process d
Compounds of formula (I) wherein R1 to R4 and X are as defined above, and salts thereof, may also be prepared by a process comprising final stage modification of one compound of formula (I), or a salt thereof, into another compound of formula (I), or a salt thereof. Suitable functional group transformations for converting one compound of formula (I) into another compound of formula (I) are well known in the art and are described in, for instance, Comprehensive Heterocyclic Chemistry II, eds. A. R. Katritzky, C. W. Rees and E. F. V. Scriven (Pergamon Press, 1996), Comprehensive Organic Functional Group Transformations, eds. A.R. Katritzky, O. Meth-Cohn and C.W. Rees (Elsevier Science Ltd., Oxford, 1995), Comprehensive Organic Chemistry, eds. D. Barton and W.D. Ollis (Pergamon Press, Oxford, 1979), and Comprehensive Organic Transformations, R.C. Larock (VCH Publishers Inc., New York, 1989). For example, compounds of formula (I) wherein R4 is halo such as chloro may be reacted with a suitable amine compound to produce compounds of formula (I) wherein R4 is - NR12R13.
Compounds of formula (I) wherein R1, R2, R3 and X are as defined above and R4 is - NR12R13 wherein R12 and R13, together with the nitrogen atom to which they are attached, are linked to form 2,5-pyrrolidinone, may be prepared by a process comprising reacting a compound of formula (I) wherein R1, R2, R3 and X are as defined above and R4 is NH2, with succinic anhydride. Suitable reaction conditions include microwave irradiation, in a suitable solvent such as acetonitrile, in the presence of HCI, at a suitable temperature such as about 150°C.
Compounds of formula (I) wherein R4 is chloro may be reduced to produce compounds of formula (I) wherein R4 is hydrogen. Suitable reduction conditions include treatment with ammonium formate under microwave irradiation, in the presence of a suitable catalyst such as palladium on activated carbon, in a suitable solvent such as methanol and at a suitable temperature such as about 130 °C.
Compounds of formula (I) wherein R3 is C3-6cycloalkyl substituted by -NH2 may be modified to produce further compounds of formula (I) wherein R3 is C3-6cycloalkyl substituted by -NR8R9 wherein R8 is hydrogen and R9 is -COC1 -6alkyl. For example, compounds of formula (I) wherein R1 , R2, R4 and X are as defined above and R3 is C3- 6cycloalkyl substituted by -NR8R9 wherein R8 is hydrogen and R9 is -COC1-6alkyl may be prepared by a process comprising reacting a compound of formula (I) wherein R1 , R2, R4 and X are as defined above and R3 is C3-6cycloalkyl substituted by -NH2, with Ci_ 6alkylCOCI such as acetyl chloride. Suitable reaction conditions include reaction in the presence of a suitable base such as triethylamine, in a suitable solvent such as tetrahydrofuran, and at a suitable temperature such as about 0°C.
Compounds of formula (I) wherein R2 to R4 and X are as defined above and R1 is - CH2NR6R7 may be prepared from compounds of formula (I) wherein R1 is -CH2OH by conversion of the alcohol to the corresponding aldehyde by treatment with, for example, manganese dioxide in the presence of a suitable solvent such as tetrahydrofuran and at a suitable temperature such as about 70°C, followed by reaction with an amine of formula R6R7NH2 in the presence of sodium triacetoxyborohydride, a suitable solvent such as dichloromethane and at a suitable temperature such as ambient temperature. Compounds of formula (I) wherein R2 to R4 and X are as defined above and R1 is - CH2NH2 may be prepared from the abovementioned aldehyde by conversion to the corresponding oxime by treatment with, for example, hydroxylamine hydrochloride and sodium acetate in the presence of a suitable sovent such as aqueous ethanol and at a suitable temperature such as ambient temperature, followed by treatment with zinc powder in glacial acetic acid at a suitable temperature such as ambient temperature.
Thus, in one embodiment, the invention provides a process for preparing a compound comprising: a) reacting a compound of formula (II)

(II)
wherein R1, R4 and X are as defined above and Y is halo, with an amine of formula (III)

(Hi) wherein R2 and R3 as defined above, b) reacting a compound of formula (IX)

(IX)
wherein R1 to R3 and X are as defined above, with a compound of formula (X)
RY
(X)
wherein R4 is as defined above and Y4 is halo, c) reacting a compound of formula (XI) as defined above, with a compound of formula (XI IA) or (XIIB)

(XIIA)
OH
4 '
R— B
OH
(XIIB)
wherein R4 is as defined above, or d) final stage modification of one compound of formula (I), or a salt thereof, into another compound of formula (I), or a salt thereof.
Methods of Use
The compounds of the invention are inhibitors of kinase activity, in particular Itk activity. Compounds which are Itk inhibitors may be useful in the treatment of disorders wherein the underlying pathology is (at least in part) attributable to inappropriate Itk activity, such as asthma. "Inappropriate Itk activity" refers to any Itk activity that deviates from the normal Itk activity expected in a particular patient. Inappropriate Itk may take the form of, for instance, an abnormal increase in activity, or an aberration in the timing and or control of Itk activity. Such inappropriate activity may result then, for example, from overexpression or mutation of the protein kinase leading to inappropriate or uncontrolled activation. Accordingly, in another aspect the invention is directed to methods of treating such disorders.
Such disorders include respiratory diseases including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis; allergic diseases including allergic rhinitis and atopic dermatitis; autoimmune diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis; transplant rejection; graft versus host disease; inflammatory disorders including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation; HIV; aplastic anemia; and pain including inflammatory pain.
The methods of treatment of the invention comprise administering a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof to a patient in need thereof. Individual embodiments of the invention include methods of treating any one of the above-mentioned disorders by administering a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof to a patient in need thereof.
As used herein, "treat" in reference to a disorder means: (1 ) to ameliorate or prevent the disorder or one or more of the biological manifestations of the disorder, (2) to interfere with (a) one or more points in the biological cascade that leads to or is responsible for the disorder or (b) one or more of the biological manifestations of the disorder, (3) to alleviate one or more of the symptoms or effects associated with the disorder, or (4) to slow the progression of the disorder or one or more of the biological manifestations of the disorder.
As indicated above, "treatment" of a disorder includes prevention of the disorder. The skilled artisan will appreciate that "prevention" is not an absolute term. In medicine, "prevention" is understood to refer to the prophylactic administration of a drug to substantially diminish the likelihood or severity of a disorder or biological manifestation thereof, or to delay the onset of such disorder or biological manifestation thereof.
As used herein, "safe and effective amount" in reference to a compound of formula (I) or a pharmaceutically acceptable salt thereof or other pharmaceutically-active agent means an amount of the compound sufficient to treat the patient's condition but low enough to avoid serious side effects (at a reasonable benefit/risk ratio) within the scope of sound medical judgment. A safe and effective amount of a compound will vary with the particular compound chosen (e.g. consider the potency, efficacy, and half-life of the compound); the route of administration chosen; the disorder being treated; the severity of the disorder being treated; the age, size, weight, and physical condition of the patient being treated; the medical history of the patient to be treated; the duration of the treatment; the nature of concurrent therapy; the desired therapeutic effect; and like factors, but can nevertheless be routinely determined by the skilled artisan.
As used herein, "patient" refers to a human (including adults and children) or other animal. In one embodiment, "patient" refers to a human.
The compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered by any suitable route of administration, including both systemic administration and topical administration. Systemic administration includes oral administration, parenteral administration, transdermal administration and rectal administration. Parenteral administration refers to routes of administration other than enteral or transdermal, and is typically by injection or infusion. Parenteral administration
includes intravenous, intramuscular, and subcutaneous injection or infusion. Topical administration includes application to the skin as well as intraocular, otic, intravaginal, inhaled and intranasal administration. Inhalation refers to administration into the patient's lungs whether inhaled through the mouth or through the nasal passages. In one embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered orally. In another embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered topically. In another embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered by inhalation. In a further embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered intranasally.
The compounds of formula (I) or pharmaceutically acceptable salts thereof may be administered once or according to a dosing regimen wherein a number of doses are administered at varying intervals of time for a given period of time. For example, doses may be administered one, two, three, or four times per day. In one embodiment, a dose is administered once per day. In a further embodiment, a dose is administered twice per day. Doses may be administered until the desired therapeutic effect is achieved or indefinitely to maintain the desired therapeutic effect. Suitable dosing regimens for a compound of formula (I) or a pharmaceutically acceptable salt thereof depend on the pharmacokinetic properties of that compound, such as absorption, distribution, and half- life, which can be determined by the skilled artisan. In addition, suitable dosing regimens, including the duration such regimens are administered, for a compound of formula (I) or a pharmaceutically acceptable salt thereof depend on the disorder being treated, the severity of the disorder being treated, the age and physical condition of the patient being treated, the medical history of the patient to be treated, the nature of concurrent therapy, the desired therapeutic effect, and like factors within the knowledge and expertise of the skilled artisan. It will be further understood by such skilled artisans that suitable dosing regimens may require adjustment given an individual patient's response to the dosing regimen or over time as individual patient needs change.
Typical daily dosages may vary depending upon the particular route of administration chosen. Typical daily dosages for oral administration range from 0.001 mg to 50mg per kg of total body weight, for example from 1 mg to 10mg per kg of total body weight. For example, daily dosages for oral administration may be from 0.5mg to 2g per patient, such as 10mg to 1g per patient.
Additionally, the compounds of formula (I) may be administered as prodrugs. As used herein, a "prodrug" of a compound of formula (I) is a functional derivative of the compound which, upon administration to a patient, eventually liberates the compound of formula (I) in vivo. Administration of a compound of formula (I) as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of the activity of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome a side effect or other difficulty encountered with the compound. Typical functional derivatives used to prepare prodrugs include modifications of the compound that are chemically or enzymatically cleavable in vivo. Such modifications, which include the preparation of phosphates, amides, esters, thioesters, carbonates, and carbamates, are well known to those skilled in the art.
The invention thus provides a method of treating a disorder mediated by inappropriate Itk activity comprising administering a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof to a patient in need thereof.
In one embodiment, the disorder mediated by inappropriate Itk activity is selected from the group consisting of respiratory diseases (including asthma, chronic obstructive pulmonary disease (COPD) and bronchitis); allergic diseases (including allergic rhinitis and atopic dermatitis); autoimmune diseases (including rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivities, Guillain-Barre Syndrome and Hashimoto's thyroiditis); transplant rejection; graft versus host disease; inflammatory disorders (including conjunctivitis, contact dermatitis, inflammatory bowel disease and chronic inflammation); HIV; aplastic anemia; and pain including inflammatory pain.
In one embodiment, the disorder mediated by inappropriate Itk activity is a respiratory disease. In a further embodiment, the disorder mediated by inappropriate Itk activity is asthma.
In one embodiment, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in medical therapy. In another embodiment, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in the treatment of a disorder mediated by inappropriate Itk activity. In another embodiment, the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof in the manufacture of a medicament for use in the treatment of a disorder mediated by inappropriate Itk activity. In a further
embodiment, the invention provides a pharmaceutical composition for the treatment or prophylaxis of a disorder mediated by inappropriate Itk activity comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
Compositions
The compounds of formula (I) and pharmaceutically acceptable salts thereof will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable excipients. In a further aspect the invention is directed to pharmaceutical compositions for the treatment or prophylaxis of a disorder mediated by inappropriate Itk activity comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof.
The pharmaceutical compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof can be extracted and then given to the patient such as with powders or syrups. Alternatively, the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form wherein each physically discrete unit contains a compound of formula (I) or a pharmaceutically acceptable salt thereof. When prepared in unit dosage form, the pharmaceutical compositions of the invention typically may contain, for example, from 0.5mg to 1 g, or from 1 mg to 700mg, or from 5mg to 100mg of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
The pharmaceutical compositions of the invention typically contain one compound of formula (I) or a pharmaceutically acceptable salt thereof.
As used herein, "pharmaceutically acceptable excipient" means a pharmaceutically acceptable material, composition or vehicle involved in giving form or consistency to the pharmaceutical composition. Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled such that interactions which would substantially reduce the efficacy of the compound of formula (I) or a pharmaceutically acceptable salt thereof when administered to a patient and interactions
which would result in pharmaceutical compositions that are not pharmaceutically acceptable are avoided. In addition, each excipient must of course be pharmaceutically acceptable eg of sufficiently high purity. The compound of formula (I) or a pharmaceutically acceptable salt thereof and the pharmaceutically acceptable excipient or excipients will typically be formulated into a dosage form adapted for administration to the patient by the desired route of administration. For example, dosage forms include those adapted for (1 ) oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixers, suspensions, solutions, emulsions, sachets, and cachets; (2) parenteral administration such as sterile solutions, suspensions, and powders for reconstitution; (3) transdermal administration such as transdermal patches; (4) rectal administration such as suppositories; (5) inhalation such as aerosols, solutions, and dry powders; and (6) topical administration such as creams, ointments, lotions, solutions, pastes, sprays, foams, and gels.
Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen. In addition, suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition. For example, certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of formula (I) or pharmaceutically acceptable salts thereof once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body. Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
Suitable pharmaceutically-acceptable excipients include the following types of excipients: Diluents, fillers, binders, disintegrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweetners, flavoring agents, flavor masking agents, coloring agents, anticaking agents, hemectants, chelating agents, plasticizers, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents. The skilled artisan will appreciate that certain pharmaceutically-acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other excipients are present in the formulation.
Skilled artisans possess the knowledge and skill in the art to enable them to select suitable pharmaceutically-acceptable excipients in appropriate amounts for use in the invention. In addition, there are a number of resources that are available to the skilled artisan which describe pharmaceutically-acceptable excipients and may be useful in selecting suitable pharmaceutically-acceptable excipients. Examples include Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
The pharmaceutical compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company). Accordingly, in another aspect the invention is directed to process for the preparation of a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and one or more pharmaceutically-acceptable excipients which comprises mixing the ingredients. A pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may be prepared by, for example, admixture at ambient temperature and atmospheric pressure.
In one embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof will be formulated for oral administration. In another embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof will be formulated for inhaled administration. In a further embodiment, the compounds of formula (I) or pharmaceutically acceptable salts thereof will be formulated for intranasal administration.
In one aspect, the invention is directed to a solid oral dosage form such as a tablet or capsule comprising a safe and effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof and a diluent or filler. Suitable diluents and fillers include lactose, sucrose, dextrose, mannitol, sorbitol, starch (e.g. corn starch, potato starch, and pre-gelatinized starch), cellulose and its derivatives (e.g. microcrystalline cellulose), calcium sulfate, and dibasic calcium phosphate. The oral solid dosage form may further comprise a binder. Suitable binders include starch (e.g. corn starch, potato starch, and pre-gelatinized starch), gelatin, acacia, sodium alginate, alginic acid, tragacanth, guar gum, povidone, and cellulose and its derivatives (e.g. microcrystalline
cellulose). The oral solid dosage form may further comprise a disintegrant. Suitable disintegrants include crospovidone, sodium starch glycolate, croscarmelose, alginic acid, and sodium carboxymethyl cellulose. The oral solid dosage form may further comprise a lubricant. Suitable lubricants include stearic acid, magnesuim stearate, calcium stearate, and talc.
Where appropriate, dosage unit formulations for oral administration can be microencapsulated. The composition can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
The compounds of formula (I) or pharmaceutically acceptable salts thereof may also be coupled with soluble polymers as targetable drug carriers. Such polymers can include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamide -phenol, polyhydroxyethylaspartamidephenol, or polyethyleneoxidepolylysine substituted with palmitoyl residues. Furthermore, the compounds of formula (I) or pharmaceutically acceptable salts thereof may be coupled to a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polylactic acid, polepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
In another aspect, the invention is directed to a liquid oral dosage form. Oral liquids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof. Syrups can be prepared by dissolving the compound of formula (I) or a pharmaceutically acceptable salt thereof in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle. Suspensions can be formulated by dispersing the compound of formula (I) or a pharmaceutically acceptable salt thereof in a non-toxic vehicle. Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
In another aspect, the invention is directed to a dosage form adapted for administration to a patient by inhalation. For example, as a dry powder, an aerosol, a suspension, or a solution composition.
Dry powder compositions for delivery to the lung by inhalation typically comprise a compound of formula (I) or a pharmaceutically acceptable salt thereof as a finely divided powder together with one or more pharmaceutically-acceptable excipients as finely divided powders. Pharmaceutically-acceptable excipients particularly suited for use in dry powders are known to those skilled in the art and include lactose, starch, mannitol, and mono-, di-, and polysaccharides. The finely divided powder may be prepared by, for example, micronisation and milling. Generally, the size-reduced (eg micronised) compound can be defined by a D50 value of about 1 to about 10 microns (for example as measured using laser diffraction).
The dry powder may be administered to the patient via a reservoir dry powder inhaler (RDPI) having a reservoir suitable for storing multiple (un-metered doses) of medicament in dry powder form. RDPIs typically include a means for metering each medicament dose from the reservoir to a delivery position. For example, the metering means may comprise a metering cup, which is movable from a first position where the cup may be filled with medicament from the reservoir to a second position where the metered medicament dose is made available to the patient for inhalation. Alternatively, the dry powder may be presented in capsules (e.g. gelatin or plastic), cartridges, or blister packs for use in a multi-dose dry powder inhaler (MDPI). MDPIs are inhalers wherein the medicament is comprised within a multi-dose pack containing (or otherwise carrying) multiple defined doses (or parts thereof) of medicament. When the dry powder is presented as a blister pack, it comprises multiple blisters for containment of the medicament in dry powder form. The blisters are typically arranged in regular fashion for ease of release of the medicament therefrom. For example, the blisters may be arranged in a generally circular fashion on a disc-form blister pack, or the blisters may be elongate in form, for example comprising a strip or a tape. Each capsule, cartridge, or blister may, for example, contain between 20μg-10mg of the compound of formula (I) or a pharmaceutically acceptable salt thereof.
Aerosols may be formed by suspending or dissolving a compound of formula (I) or a pharmaceutically acceptable salt thereof in a liquified propellant. Suitable propellants include halocarbons, hydrocarbons, and other liquified gases. Representative propellants include: trichlorofluoromethane (propellant 1 1 ), dichlorofluoromethane (propellant 12), dichlorotetrafluoroethane (propellant 1 14), tetrafluoroethane (HFA-134a), 1 ,1-
difluoroethane (HFA-152a), difluoromethane (HFA-32), pentafluoroethane (HFA-12), heptafluoropropane (HFA-227a), perfluoropropane, perfluorobutane, perfluoropentane, butane, isobutane, and pentane. Aerosols comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof will typically be administered to a patient via a metered dose inhaler (MDI). Such devices are known to those skilled in the art.
The aerosol may contain additional pharmaceutically-acceptable excipients typically used with MDIs such as surfactants, lubricants, cosolvents and other excipients to improve the physical stability of the formulation, to improve valve performance, to improve solubility, or to improve taste.
There is thus provided as a further aspect of the invention a pharmaceutical aerosol formulation comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof and a fluorocarbon or hydrogen-containing chlorofluorocarbon as propellant, optionally in combination with a surfactant and/or a cosolvent.
According to another aspect of the invention, there is provided a pharmaceutical aerosol formulation wherein the propellant is selected from 1 ,1 ,1 ,2-tetrafluoroethane, 1 ,1 ,1 ,2,3,3,3-heptafluoro-n-propane and mixtures thereof.
The formulations of the invention may be buffered by the addition of suitable buffering agents.
Capsules and cartridges for use in an inhaler or insufflator, of for example gelatine, may be formulated containing a powder mix for inhalation of a compound of formula (I) or a pharmaceutically acceptable salt thereof and a suitable powder base such as lactose or starch. Each capsule or cartridge may generally contain from 20μg to 10mg of the compound of formula (I) or pharmaceutically acceptable salt thereof. Alternatively, the compound of formula (I) or pharmaceutically acceptable salt thereof may be presented without excipients such as lactose.
The proportion of the active compound of formula (I) or pharmaceutically acceptable salt thereof in the local compositions according to the invention depends on the precise type of formulation to be prepared but will generally be within the range of from 0.001 to 10% by weight. Generally, for most types of preparations, the proportion used will be within the range of from 0.005 to 1 %, for example from 0.01 to 0.5%. However, in powders for
inhalation or insufflation the proportion used will normally be within the range of from 0.1 to 5%.
Aerosol formulations are preferably arranged so that each metered dose or "puff" of aerosol contains from 2C^g to 10mg, preferably from 2C^g to 200C^g, more preferably from about 2C^g to 50C^g of a compound of formula (I). Administration may be once daily or several times daily, for example 2, 3, 4 or 8 times, giving for example 1 , 2 or 3 doses each time. The overall daily dose with an aerosol will be within the range from 10C^g to 10mg, preferably from 20C^g to 200C^g. The overall daily dose and the metered dose delivered by capsules and cartridges in an inhaler or insufflator will generally be double that delivered with aerosol formulations.
In the case of suspension aerosol formulations, the particle size of the particulate (e.g., micronised) drug should be such as to permit inhalation of substantially all the drug into the lungs upon administration of the aerosol formulation and will thus be less than 100 microns, desirably less than 20 microns, and in particular in the range of from 1 to 10 microns, such as from 1 to 5 microns, more preferably from 2 to 3 microns.
The formulations of the invention may be prepared by dispersal or dissolution of the medicament and a compound of formula (I) or a pharmaceutically acceptable salt thereof in the selected propellant in an appropriate container, for example, with the aid of sonication or a high-shear mixer. The process is desirably carried out under controlled humidity conditions. The chemical and physical stability and the pharmaceutical acceptability of the aerosol formulations according to the invention may be determined by techniques well known to those skilled in the art. Thus, for example, the chemical stability of the components may be determined by HPLC assay, for example, after prolonged storage of the product. Physical stability data may be gained from other conventional analytical techniques such as, for example, by leak testing, by valve delivery assay (average shot weights per actuation), by dose reproducibility assay (active ingredient per actuation) and spray distribution analysis.
The stability of the suspension aerosol formulations according to the invention may be measured by conventional techniques, for example, by measuring flocculation size distribution using a back light scattering instrument or by measuring particle size
distribution by cascade impaction or by the "twin impinger" analytical process. As used herein reference to the "twin impinger" assay means "Determination of the deposition of the emitted dose in pressurised inhalations using apparatus A" as defined in British Pharmacopaeia 1988, pages A204-207, Appendix XVII C. Such techniques enable the "respirable fraction" of the aerosol formulations to be calculated. One method used to calculate the "respirable fraction" is by reference to "fine particle fraction" which is the amount of active ingredient collected in the lower impingement chamber per actuation expressed as a percentage of the total amount of active ingredient delivered per actuation using the twin impinger method described above.
The term "metered dose inhaler" or MDI means a unit comprising a can, a secured cap covering the can and a formulation metering valve situated in the cap. MDI system includes a suitable channelling device. Suitable channelling devices comprise for example, a valve actuator and a cylindrical or cone-like passage through which medicament may be delivered from the filled canister via the metering valve to the nose or mouth of a patient such as a mouthpiece actuator.
MDI canisters generally comprise a container capable of withstanding the vapour pressure of the propellant used such as a plastic or plastic-coated glass bottle or preferably a metal can, for example, aluminium or an alloy thereof which may optionally be anodised, lacquer-coated and/or plastic-coated (for example incorporated herein by reference WO96/32099 wherein part or all of the internal surfaces are coated with one or more fluorocarbon polymers optionally in combination with one or more non-fluorocarbon polymers), which container is closed with a metering valve. The cap may be secured onto the can via ultrasonic welding, screw fitting or crimping. MDIs taught herein may be prepared by methods of the art (e.g. see Byron, above and WO96/32099). Preferably the canister is fitted with a cap assembly, wherein a drug-metering valve is situated in the cap, and said cap is crimped in place. In one embodiment of the invention the metallic internal surface of the can is coated with a fluoropolymer, more preferably blended with a non-fluoropolymer. In another embodiment of the invention the metallic internal surface of the can is coated with a polymer blend of polytetrafluoroethylene (PTFE) and polyethersulfone (PES). In a further embodiment of the invention the whole of the metallic internal surface of the can is coated with a polymer blend of polytetrafluoroethylene (PTFE) and polyethersulfone (PES).
The metering valves are designed to deliver a metered amount of the formulation per actuation and incorporate a gasket to prevent leakage of propellant through the valve. The gasket may comprise any suitable elastomeric material such as, for example, low density polyethylene, chlorobutyl, bromobutyl, EPDM, black and white butadiene- acrylonitrile rubbers, butyl rubber and neoprene. Suitable valves are commercially available from manufacturers well known in the aerosol industry, for example, from Valois, France (e.g. DF10, DF30, DF60), Bespak pic, UK (e.g. BK300, BK357) and 3M-
TM
Neotechnic Ltd, UK (e.g. Spraymiser ). In various embodiments, the MDIs may also be used in conjunction with other structures such as, without limitation, overwrap packages for storing and containing the MDIs, including those described in U.S. Patent Nos. 6,1 19,853; 6,179,1 18; 6,315,1 12; 6,352,152; 6,390,291 ; and 6,679,374, as well as dose counter units such as, but not limited to, those described in U.S. Patent Nos. 6,360,739 and 6,431 ,168.
Conventional bulk manufacturing methods and machinery well known to those skilled in the art of pharmaceutical aerosol manufacture may be employed for the preparation of large-scale batches for the commercial production of filled canisters. Thus, for example, in one bulk manufacturing method for preparing suspension aerosol formulations a metering valve is crimped onto an aluminium can to form an empty canister. The particulate medicament is added to a charge vessel and liquefied propellant together with the optional excipients is pressure filled through the charge vessel into a manufacturing vessel. The drug suspension is mixed before recirculation to a filling machine and an aliquot of the drug suspension is then filled through the metering valve into the canister. In one example bulk manufacturing method for preparing solution aerosol formulations a metering valve is crimped onto an aluminium can to form an empty canister. The liquefied propellant together with the optional excipients and the dissolved medicament is pressure filled through the charge vessel into a manufacturing vessel. In an alternative process, an aliquot of the liquefied formulation is added to an open canister under conditions which are sufficiently cold to ensure the formulation does not vaporise, and then a metering valve crimped onto the canister.
Typically, in batches prepared for pharmaceutical use, each filled canister is check- weighed, coded with a batch number and packed into a tray for storage before release testing.
Suspensions and solutions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof may also be administered to a patient via a nebulizer. The solvent or suspension agent utilized for nebulization may be any pharmaceutically-acceptable liquid such as water, aqueous saline, alcohols or glycols, e.g., ethanol, isopropylalcohol, glycerol, propylene glycol, polyethylene glycol, etc. or mixtures thereof. Saline solutions utilize salts which display little or no pharmacological activity after administration. Both organic salts, such as alkali metal or ammonium halogen salts, e.g., sodium chloride, potassium chloride or organic salts, such as potassium, sodium and ammonium salts or organic acids, e.g., ascorbic acid, citric acid, acetic acid, tartaric acid, etc. may be used for this purpose.
Other pharmaceutically-acceptable excipients may be added to the suspension or solution. The compound of formula (I) or pharmaceutically acceptable salt thereof may be stabilized by the addition of an inorganic acid, e.g., hydrochloric acid, nitric acid, sulphuric acid and/or phosphoric acid; an organic acid, e.g., ascorbic acid, citric acid, acetic acid, and tartaric acid, etc., a complexing agent such as EDTA or citric acid and salts thereof; or an antioxidant such as antioxidant such as vitamin E or ascorbic acid. These may be used alone or together to stabilize the compound of formula (I) or pharmaceutically acceptable salt thereof. Preservatives may be added such as benzalkonium chloride or benzoic acid and salts thereof. Surfactant may be added particularly to improve the physical stability of suspensions. These include lecithin, disodium dioctylsulphosuccinate, oleic acid and sorbitan esters.
In a further aspect, the invention is directed to a dosage form adapted for intranasal administration.
Formulations for administration to the nose may include pressurised aerosol formulations and aqueous formulations administered to the nose by pressurised pump. Formulations which are non-pressurised and adapted to be administered topically to the nasal cavity are of particular interest. Suitable formulations contain water as the diluent or carrier for this purpose. Aqueous formulations for administration to the lung or nose may be provided with conventional excipients such as buffering agents, tonicity modifying agents and the like. Aqueous formulations may also be administered to the nose by nebulisation.
The compounds of formula (I) or pharmaceutically acceptable salts thereof may be formulated as a fluid formulation for delivery from a fluid dispenser, for example a fluid dispenser having a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser. Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations. The dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity. A fluid dispenser of the aforementioned type is described and illustrated in WO05/044354, the entire content of which is hereby incorporated herein by reference. The dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation. The housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing. In one embodiment, the fluid dispenser is of the general type illustrated in Figures 30-40 of WO05/044354.
Pharmaceutical compositions adapted for intranasal administration wherein the carrier is a solid include a coarse powder having a particle size for example in the range 20 to 500 microns which is administered by rapid inhalation through the nasal passage from a container of the powder held close up to the nose. Suitable compositions wherein the carrier is a liquid, for administration as a nasal spray or as nasal drops, include aqueous or oil solutions of the compound of formula (I) or a pharmaceutically acceptable salt thereof.
Pharmaceutical compositions adapted for transdermal administration may be presented as discrete patches intended to remain in intimate contact with the epidermis of the patient for a prolonged period of time. For example, the active ingredient may be delivered from the patch by iontophoresis as generally described in Pharmaceutical Research, 3(6), 318 (1986).
Pharmaceutical compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
Ointments, creams and gels, may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agent and/or solvents. Such bases may thus, for example, include water and/or an oil such as liquid paraffin or a vegetable oil such as arachis oil or castor oil, or a solvent such as polyethylene glycol. Thickening agents and gelling agents which may be used according to the nature of the base include soft paraffin, aluminium stearate, cetostearyl alcohol, polyethylene glycols, woolfat, beeswax, carboxypolymethylene and cellulose derivatives, and/or glyceryl monostearate and/or non-ionic emulsifying agents. Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents or thickening agents.
Powders for external application may be formed with the aid of any suitable powder base, for example, talc, lactose or starch. Drops may be formulated with an aqueous or nonaqueous base also comprising one or more dispersing agents, solubilising agents, suspending agents or preservatives.
Topical preparations may be administered by one or more applications per day to the affected area; over skin areas occlusive dressings may advantageously be used. Continuous or prolonged delivery may be achieved by an adhesive reservoir system.
For treatments of the eye or other external tissues, for example mouth and skin, the compositions may be applied as a topical ointment or cream. When formulated in an ointment, the compound of formula (I) or a pharmaceutically acceptable salt thereof may be employed with either a paraffinic or a water-miscible ointment base. Alternatively, the compound of formula (I) or pharmaceutically acceptable salt thereof may be formulated in a cream with an oil-in-water cream base or a water-in-oil base. Pharmaceutical compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. The compositions may be presented in unit- dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use. Extemporaneous
injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
The compound and pharmaceutical formulations according to the invention may be used in combination with or include one or more other therapeutic agents, for example selected from anti-inflammatory agents, anticholinergic agents (particularly an M1/M2/M3 receptor antagonist), p2-adrenoreceptor agonists, antiinfective agents, such as antibiotics or antivirals, or antihistamines. The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with one or more other therapeutically active agents, for example selected from an anti-inflammatory agent, such as a corticosteroid or an NSAID, an anticholinergic agent, a p2-adrenoreceptor agonist, an antiinfective agent, such as an antibiotic or an antiviral, or an antihistamine. One embodiment of the invention encompasses combinations comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a 2-adrenoreceptor agonist, and/or an anticholinergic, and/or a PDE-4 inhibitor, and/or an antihistamine.
One embodiment of the invention encompasses combinations comprising one or two other therapeutic agents.
It will be clear to a person skilled in the art that, where appropriate, the other therapeutic ingredient(s) may be used in the form of salts, for example as alkali metal or amine salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates to optimise the activity and/or stability and/or physical characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
In one embodiment, the invention encompasses a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a 2-adrenoreceptor agonist.
Examples of 2-adrenoreceptor agonists include salmeterol (which may be a racemate or a single enantiomer such as the R-enantiomer), salbutamol (which may be a racemate or a single enantiomer such as the R-enantiomer), formoterol (which may be a racemate or a single duastereomer such as the RJ-?-diastereomer), salmefamol, fenoterol carmoterol, etanterol, naminterol, clenbuterol, pirbuterol, flerbuterol, reproterol, bambuterol, indacaterol, terbutaline and salts thereof, for example the xinafoate (1 -hydroxy-2-
naphthalenecarboxylate) salt of salmeterol, the sulphate salt or free base of salbutamol or the fumarate salt of formoterol. In one embodiment, long-acting 2-adrenoreceptor agonists, for example, compounds which provide effective bronchodilation for about 12 hrs or longer, are preferred.
Other 2-adrenoreceptor agonists include those described in WO 02/066422, WO 02/070490, WO 02/076933, WO 03/024439, WO 03/072539, WO 03/091204, WO 04/016578, WO 2004/022547, WO 2004/037807, WO 2004/037773, WO 2004/037768, WO 2004/039762, WO 2004/039766, WO01/42193 and WO03/042160.
Examples of 2-adrenoreceptor agonists include:
3-(4-{[6-({(2R)-2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)phenyl]ethyl}amino)
hexyl] oxy} butyl) benzenesulfonamide;
3- (3-{[7-({(2R)-2-hydroxy-2-[4-hydroxy-3-hydroxymethyl) phenyl] ethyl}-amino) heptyl] oxy} propyl) benzenesulfonamide;
4- {(1 R)-2-[(6-{2-[(2, 6-dichlorobenzyl) oxy] ethoxy} hexyl) amino]-1-hydroxyethyl}-2- (hydroxymethyl) phenol;
4- {(1 R)-2-[(6-{4-[3-(cyclopentylsulfonyl)phenyl]butoxy}hexyl)amino]-1 -hydroxyethyl}-2- (hydroxymethyl)phenol;
N-[2-hydroxyl-5-[(1 R)-1-hydroxy-2-[[2-4-[[(2R)-2-hydroxy-2- phenylethyl]amino]phenyl]ethyl]amino]ethyl]phenyl]formamide;
N-2{2-[4-(3-phenyl-4-methoxyphenyl)aminophenyl]ethyl}-2-hydroxy-2-(8-hydroxy-2(1 H)- quinolinon-5-yl)ethylamine; and
5- [(R)-2-(2-{4-[4-(2-amino-2-methyl-propoxy)-phenylamino]-phenyl}-ethylamino)-1 - hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one.
The
 agonist may be in the form of a salt formed with a pharmaceutically acceptable acid selected from sulphuric, hydrochloric, fumaric, hydroxynaphthoic (for example 1 - or 3-hydroxy-2-naphthoic), cinnamic, substituted cinnamic, triphenylacetic, sulphamic, sulphanilic, naphthaleneacrylic, benzoic, 4-methoxybenzoic, 2- or 4-hydroxybenzoic, 4-chlorobenzoic and 4-phenylbenzoic acid.
agonist may be in the form of a salt formed with a pharmaceutically acceptable acid selected from sulphuric, hydrochloric, fumaric, hydroxynaphthoic (for example 1 - or 3-hydroxy-2-naphthoic), cinnamic, substituted cinnamic, triphenylacetic, sulphamic, sulphanilic, naphthaleneacrylic, benzoic, 4-methoxybenzoic, 2- or 4-hydroxybenzoic, 4-chlorobenzoic and 4-phenylbenzoic acid.
Suitable anti-inflammatory agents include corticosteroids. Suitable corticosteroids which may be used in combination with the compounds of formula (I) or pharmaceutically acceptable salts thereof are those oral and inhaled corticosteroids and their pro-drugs
which have anti-inflammatory activity. Examples include methyl prednisolone,
prednisolone, dexamethasone, fluticasone propionate, 6a,9a-difluoro-1 i -hydroxy-16a- methyl-17a-[(4-methyl-1 ,3-thiazole-5-carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17β- carbothioic acid S-fluoromethyl ester, 6a,9a-difluoro-17a-[(2-furanylcarbonyl)oxy]-1 1 β- hydroxy-16a-methyl-3-oxo-androsta-1 ,4-diene-17 -carbothioic acid S-fluoromethyl ester (fluticasone furoate), 6a,9a-difluoro-1 i p-hydroxy-16a-methyl-3-oxo-17a-propionyloxy- androsta-1 ,4-diene-17 -carbothioic acid S-(2-oxo-tetrahydro-furan-3S-yl) ester, 6α,9α- difluoro-1 1 β-hydroxy-l 6a-methyl-3-oxo-17a-(2,2,3,3- tetramethycyclopropylcarbonyl)oxy- androsta-1 ,4-diene-17 -carbothioic acid S-cyanomethyl ester and 6a,9a-difluoro-1 i - hydroxy-16a-methyl-17a-(1 -methycyclopropylcarbonyl)oxy-3-oxo-androsta-1 ,4-diene-17β- carbothioic acid S-fluoromethyl ester, beclomethasone esters (for example the 17- propionate ester or the 17,21 -dipropionate ester), budesonide, flunisolide, mometasone esters (for example mometasone furoate), triamcinolone acetonide, rofleponide, ciclesonide (16a,17-[[(R)-cyclohexylmethylene]bis(oxy)]-1 i ,21-dihydroxy-pregna-1 ,4- diene-3,20-dione), butixocort propionate, RPR-106541 , and ST-126. Preferred
corticosteroids include fluticasone propionate, 6a,9a-difluoro-1 i -hydroxy-16a-methyl- 17a-[(4-methyl-1 ,3-thiazole-5-carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17 -carbothioic acid S-fluoromethyl ester, 6a,9a-difluoro-17a-[(2-furanylcarbonyl)oxy]-1 i -hydroxy-16a- methyl-3-oxo-androsta-1 ,4-diene-17 -carbothioic acid S-fluoromethyl ester, 6a, 9a- difluoro-1 1 β-hydroxy-l 6a-methyl-3-oxo-17a-(2,2,3,3- tetramethycyclopropylcarbonyl)oxy- androsta-1 ,4-diene-17 -carbothioic acid S-cyanomethyl ester and 6a,9a-difluoro-1 i - hydroxy-16a-methyl-17a-(1 -methycyclopropylcarbonyl)oxy-3-oxo-androsta-1 ,4-diene-17β- carbothioic acid S-fluoromethyl ester. In one embodiment the corticosteroid is 6a, 9a- difluoro-17a-[(2-furanylcarbonyl)oxy]-1 1 β-hydroxy-l 6a-methyl-3-oxo-androsta-1 ,4-diene- 17 -carbothioic acid S-fluoromethyl ester.
Examples of corticosteroids may include those described in WO2002/088167, WO2002/100879, WO2002/12265, WO2002/12266, WO2005/005451 , WO2005/005452, WO2006/072599 and WO2006/072600.
Non-steroidal compounds having glucocorticoid agonism that may possess selectivity for transrepression over transactivation and that may be useful in combination therapy include those covered in the following patents: WO03/082827, W098/54159, WO04/005229, WO04/009017, WO04/018429, WO03/104195, WO03/082787, WO03/082280, WO03/059899, WO03/101932, WO02/02565, WO01/16128,
WO00/66590, WO03/086294, WO04/026248, WO03/061651 and WO03/08277. Further non-steroidal compounds are covered in: WO2006/000401 , WO2006/000398 and WO2006/015870. Examples of anti-inflammatory agents include non-steroidal anti-inflammatory drugs (NSAID's).
Examples of NSAID's include sodium cromoglycate, nedocromil sodium, phosphodiesterase (PDE) inhibitors (for example, theophylline, PDE4 inhibitors or mixed PDE3/PDE4 inhibitors), leukotriene antagonists, inhibitors of leukotriene synthesis (for example montelukast), iNOS inhibitors, tryptase and elastase inhibitors, beta-2 integrin antagonists and adenosine receptor agonists or antagonists (e.g. adenosine 2a agonists), cytokine antagonists (for example chemokine antagonists, such as a CCR3 antagonist) or inhibitors of cytokine synthesis, or 5-lipoxygenase inhibitors. An iNOS (inducible nitric oxide synthase inhibitor) is preferably for oral administration. Examples of iNOS inhibitors include those disclosed in WO93/13055, WO98/30537, WO02/50021 , W095/34534 and W099/62875. Examples of CCR3 inhibitors include those disclosed in WO02/26722.
In one embodiment, the invention provides the use of the compounds of formula (I) in combination with a phosphodiesterase 4 (PDE4) inhibitor, especially in the case of a formulation adapted for inhalation. The PDE4-specific inhibitor useful in this aspect of the invention may be any compound that is known to inhibit the PDE4 enzyme or which is discovered to act as a PDE4 inhibitor, and which are only PDE4 inhibitors, not compounds which inhibit other members of the PDE family, such as PDE3 and PDE5, as well as PDE4.
Compounds include c/s-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-1 - carboxylic acid, 2-carbomethoxy-4-cyano-4-(3-cyclopropylmethoxy-4- difluoromethoxyphenyl)cyclohexan-1-one and c/s-[4-cyano-4-(3-cyclopropylmethoxy-4- difluoromethoxyphenyl)cyclohexan-1-ol]. Also, c/s-4-cyano-4-[3-(cyclopentyloxy)-4- methoxyphenyl]cyclohexane-1 -carboxylic acid (also known as cilomilast) and its salts, esters, pro-drugs or physical forms, which is described in U.S. patent 5,552,438 issued 03 September, 1996; this patent and the compounds it discloses are incorporated herein in full by reference.
Other compounds include AWD-12-281 from Elbion (Hofgen, N. et al. 15th EFMC Int Symp Med Chem (Sept 6-10, Edinburgh) 1998, Abst P.98; CAS reference No.
247584020-9); a 9-benzyladenine derivative nominated NCS-613 (INSERM); D-4418 from Chiroscience and Schering-Plough; a benzodiazepine PDE4 inhibitor identified as Cl- 1018 (PD-168787) and attributed to Pfizer; a benzodioxole derivative disclosed by Kyowa Hakko in W099/16766; K-34 from Kyowa Hakko; V-1 1294A from Napp (Landells, L.J. et al. Eur Resp J [Annu Cong Eur Resp Soc (Sept 19-23, Geneva) 1998] 1998, 12 (Suppl. 28): Abst P2393); roflumilast (CAS reference No 162401-32-3) and a pthalazinone (WO99/47505, the disclosure of which is hereby incorporated by reference) from Byk- Gulden; Pumafentrine, (-)-p-[(4aR*,10bS*)-9-ethoxy-1 ,2,3,4,4a, 10b-hexahydro-8- methoxy-2-methylbenzo[c][1 ,6]naphthyridin-6-yl]-N,N-diisopropylbenzamide which is a mixed PDE3/PDE4 inhibitor which has been prepared and published on by Byk-Gulden, now Altana; arofylline under development by Almirall-Prodesfarma; VM554/UM565 from Vernalis; or T-440 (Tanabe Seiyaku; Fuji, K. et al. J Pharmacol Exp Ther,1998, 284(1 ): 162), and T2585. Further compounds are disclosed in the published international patent application WO04/024728 (Glaxo Group Ltd), WO04/056823 (Glaxo Group Ltd) and WO04/103998 (Glaxo Group Ltd) (e.g. Example 399 or 544 disclosed therein). Further compounds are also disclosed in WO2005/058892, WO2005/090348, WO2005/090353, and WO2005/090354, all in the name of Glaxo Group Limited.
Examples of anticholinergic agents are those compounds that act as antagonists at the muscarinic receptors, in particular those compounds which are antagonists of the M-i or M3 receptors, dual antagonists of the M1/M3 or M2/M3, receptors or pan-antagonists of the M1/M2/M3 receptors. Exemplary compounds for administration via inhalation include ipratropium (for example, as the bromide, CAS 22254-24-6, sold under the name Atrovent), oxitropium (for example, as the bromide, CAS 30286-75-0) and tiotropium (for example, as the bromide, CAS 136310-93-5, sold under the name Spiriva). Also of interest are revatropate (for example, as the hydrobromide, CAS 262586-79-8) and LAS- 34273 which is disclosed in WO01/041 18. Exemplary compounds for oral administration include pirenzepine (CAS 28797-61-7), darifenacin (CAS 133099-04-4, or CAS 133099- 07-7 for the hydrobromide sold under the name Enablex), oxybutynin (CAS 5633-20-5, sold under the name Ditropan), terodiline (CAS 15793-40-5), tolterodine (CAS 124937-51- 5, or CAS 124937-52-6 for the tartrate, sold under the name Detrol), otilonium (for example, as the bromide, CAS 26095-59-0, sold under the name Spasmomen), trospium chloride (CAS 10405-02-4) and solifenacin (CAS 242478-37-1 , or CAS 242478-38-2 for the succinate also known as YM-905 and sold under the name Vesicare).
Additional compounds are disclosed in WO 2005/037280, WO 2005/046586 and WO 2005/104745, incorporated herein by reference. The present combinations include, but are not limited to:
(3-en- o)-3-(2,2-di-2-thienylethenyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1 ]octane iodide; (3-en- o)-3-(2-cyano-2,2-diphenylethyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1 ]octane bromide;
4- [hydroxy(diphenyl)methyl]-1 -{2-[(phenylmethyl)oxy]ethyl}-1 -azoniabicyclo[2.2.2]octane bromide; and
(1 R,5S)-3-(2-cyano-2,2-diphenylethyl)-8-methyl-8-{2-[(phenylmethyl)oxy]ethyl}-8- azoniabicyclo[3.2.1]octane bromide.
Other anticholinergic agents include compounds which are disclosed in US patent application 60/487981 including, for example:
(3-en- o)-3-(2,2-di-2-thienylethenyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1]octane bromide; (3-en- o)-3-(2,2-diphenylethenyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1 ]octane bromide;
(3-en- o)-3-(2,2-diphenylethenyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1]octane 4- methylbenzenesulfonate;
(3-en- o)-8,8-dimethyl-3-[2-phenyl-2-(2-thienyl)ethenyl]-8-azoniabicyclo[3.2.1]octane bromide; and/or
(3-en- o)-8,8-dimethyl-3-[2-phenyl-2-(2-pyridinyl)ethenyl]-8-azoniabicyclo[3.2.1 ]octane bromide.
Further anticholinergic agents include compounds which are disclosed in US patent application 60/51 1009 including, for example:
(en- o)-3-(2-methoxy-2,2-di-thiophen-2-yl-ethyl)-8,8-dimethyl-8-azonia- bicyclo[3.2.1]octane iodide;
5- iiendoJ-S-methyl-S-aza-bicyclotS^.l loct-S-yl^^-diphenyl-propionitrile;
(en- o)-8-methyl-3-(2,2,2-triphenyl-ethyl)-8-aza-bicyclo[3.2.1]octane;
S-iiendoJ-S-methyl-S-aza-bicyclotS^.l loct-S-yl^^-diphenyl-propionamide;
S-iiendoJ-S-methyl-S-aza-bicyclotS^.l loct-S-yl^^-diphenyl-propionic acid;
(en- o)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide;
(en- o)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane bromide;
S-iiendoJ-S-methyl-S-aza-bicyclotS^.l loct-S-yl^^-diphenyl-propan-l -ol;
/V-benzyl-S-iiendoJ-S-methyl-S-aza-bicyclotS^.lloct-S-yl^^-diphenyl-propionamide; (en- o)-3-(2-carbamoyl-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1 ]octane iodide;
l -benzyl-S-ES-iiendoJ-S-methyl-S-aza-bicyclotS^.l loct-S-yl^^-diphenyl-propy -urea;
1-ethyl-3-[3-((endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propyl]-urea;
/V-[3-((endo)-8-methyl-8-aza-bicyclo[3.2.1 ]oct-3-yl)-2,2-diphenyl-propyl]-acetamide;
/V-[3-((endo)-8-methyl-8-aza-bicyclo[3.2.1 ]oct-3-yl)-2,2-diphenyl-propyl]-benzamide;
3-((endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-di-thiophen-2-yl-propionitrile;
(endo)-3-(2-cyano-2,2-di-thiophen-2-yl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]o iodide;
\/-[3-((endo)-8-methyl-8-aza-bicyclo[3.2.1 ]oct-3-yl)-2,2-diphenyl-propyl]- benzenesulfonamide;
^-((endoJ-S-methyl-S-aza-bicyclotS^.l loct-S-yl^^-diphenyl-propyll-urea;
A/-[3-((endo)-8-methyl-8-aza-bicyclo[3.2.1 ]oct-3-yl)-2,2-cliphenyl-propyl]- methanesulfonamide; and/or
(endo )-3-{2,2-diphenyl-3-[(1-phenyl-methanoyl)-amino]-propyl}-8,8-dimethyl-8-azoni bicyclo[3.2.1]octane bromide. Further compounds include:
(endo )-3-(2-methoxy-2, 2-di-thiophen-2 -yl-ethyl)-8, 8-dimethyl-8-azonia- bicyclo[3.2.1]octane iodide;
(en- o)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide; (en- o)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane bromide; (en- o)-3-(2-carbamoyl-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1 ]octane iodide;
(en- o)-3-(2-cyano-2,2-di-thiophen-2-yl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide; and/or
(endo )-3-{2,2-diphenyl-3-[(1-phenyl-methanoyl)-amino]-propyl}-8,8-dimethyl-8-azonia- bicyclo[3.2.1]octane bromide.
In one embodiment the invention provides a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an H1 antagonist. Examples of H1 antagonists include, without limitation, amelexanox, astemizole, azatadine, azelastine, acrivastine, brompheniramine, cetirizine, levocetirizine, efletirizine, chlorpheniramine, clemastine, cyclizine, carebastine, cyproheptadine, carbinoxamine, descarboethoxyloratadine, doxylamine, dimethindene, ebastine, epinastine, efletirizine, fexofenadine, hydroxyzine, ketotifen, loratadine, levocabastine, mizolastine, mequitazine, mianserin, noberastine, meclizine, norastemizole, olopatadine, picumast, pyrilamine, promethazine, terfenadine, tripelennamine, temelastine, trimeprazine and triprolidine, particularly cetirizine, levocetirizine, efletirizine and fexofenadine. In a further embodiment the invention provides a combination comprising a compound of formula (I) or a
pharmaceutically acceptable salt thereof together with an H3 antagonist (and/or inverse agonist). Examples of H3 antagonists include, for example, those compounds disclosed in WO2004/035556 and in WO2006/045416. Other histamine receptor antagonists which may be used in combination with the compounds of the present invention include antagonists (and/or inverse agonists) of the H4 receptor, for example, the compounds disclosed in Jablonowski et al., J. Med. Chem. 46:3957-3960 (2003).
The inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor.
The inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a

agonist.
The inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a corticosteroid.
The inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with a non-steroidal GR agonist.
The inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with an anticholinergic.
The inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with an antihistamine.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor and a
 agonist.
agonist.
The inven ion thus provides, in a further aspect, a combination comprising a compound of formula (I or a pharmaceutically acceptable salt thereof together with an anticholinergic and a PDE-4 inhibitor.
The combinations referred to above may conveniently be presented for use in the form of a pharmaceutical composition and thus pharmaceutical compositions comprising a
combination as defined above together with a pharmaceutically acceptable diluent or carrier represent a further aspect of the invention.
The individual compounds of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations. In one embodiment, the individual compounds will be administered simultaneously in a combined pharmaceutical formulation. Appropriate doses of known therapeutic agents will readily be appreciated by those skilled in the art. The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with another therapeutically active agent.
The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor.
The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a 2-adrenoreceptor agonist.
The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a corticosteroid.
The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a non-steroidal GR agonist. The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an anticholinergic.
The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an antihistamine.
The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a PDE4 inhibitor and a 2-adrenoreceptor agonist. The invention thus provides, in a further aspect, a pharmaceutical composition comprising a combination of a compound of formula (I) or a pharmaceutically acceptable salt thereof together with an anticholinergic and a PDE4 inhibitor.
The invention will now be illustrated by way of the following non-limiting examples.
EXAMPLES
The following examples illustrate the invention. These examples are not intended to limit the scope of the present invention, but rather to provide guidance to the skilled artisan to prepare and use the compounds, compositions, and methods of the present invention. While particular embodiments of the present invention are described, the skilled artisan will appreciate that various changes and modifications can be made without departing from the spirit and scope of the invention. General Methods
Unless stated otherwise, starting materials were commercially available. All solvents and commercial reagents were of laboratory grade and were used as received. In the examples 1H NMR spectra were recorded on a Bruker DRX 400 (400MHz) instrument. The following abbreviations have been used: s, singlet; d, doublet; t, triplet; Hz, Hertz.
Unless stated otherwise, flash chromatography was carried out using pre-packed Biotage "Isolute" flash silica cartridges on a Biotage "Flashmaster 2" system.
The following methods were used for LCMS (liquid chromatography - mass spectral) analysis:
LCMS Method A:
The analysis was conducted on an Acquity UPLC BEH C18 column (50mm x 2.1 mm internal diameter 1 .7μιη packing diameter) at 40°C.
The solvents employed were:
A = 0.1 % v/v solution of formic acid in water.
B = 0.1 % v/v solution of formic acid in acetonitrile.
The gradient employed was as follows

The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
LCMS Method B:
The analysis was conducted on an XBridge C18 column (50mm x 4.6mm internal diameter 3.5μιη packing diameter) at 30°C.
The solvents employed were:
A = 10 mM ammonium bicarbonate in water adjusted to pH 10 with ammonia solution. B = acetonitrile.
The typical gradient employed was as follows:
Time Flow Rate
% A % B
(minutes) (mL/min)
0 3 99 1
0.1 3 99 1
4.0 3 3 97
5.0 3 3 97
The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization.
The following illustrates the mobile phases and gradients used when compounds underwent purification by mass-directed autopreparative HPLC. Mass-Directed Autopreparative HPLC (Formic Acid Modifier)
The HPLC analysis was conducted on a Sunfire C18 column (150mm x 30mm internal diameter, 5μιη packing diameter) at ambient temperature. The solvents employed were:
A = 0.1 % v/v solution of formic acid in water.
B = 0.1 % v/v solution of formic acid in acetonitrile. Mass-Directed Autopreparative HPLC (Trifluoroacetic Acid Modifier)
The HPLC analysis was conducted on a Sunfire C18 column (150mm x 30mm internal diameter, 5μιη packing diameter) at ambient temperature. The solvents employed were:
A = 0.1 % v/v solution of trifluoroacetic acid in water.
B = 0.1 % v/v solution of trifluoroacetic acid in acetonitrile. Mass-Directed Autopreparative HPLC (Ammonium Bicarbonate Modifier)
The HPLC analysis was conducted on an XBridge C18 column (150mm x 30mm internal diameter, 5μιη packing diameter) at ambient temperature.
The solvents employed were:
A = 10 mM ammonium bicarbonate in water adjusted to pH 10 with ammonia solution. B = acetonitrile.
For each of the mass-directed autopreparative purifications, irrespective of the modifier used, the gradient employed was dependent upon the retention time of the particular compound undergoing purification as recorded in the analytical LCMS, and was as follows:
For compounds with an analytical LCMS retention time below 0.6 minutes (LCMS method A) or below 1.5 minutes (LCMS method B) the following gradient was used:

For compounds with an analytical LCMS retention time between 0.6 and 0.9 minutes (LCMS method A) or between 1.5 and 2.2 minutes (LCMS method B) the following gradient was used:
Time Flow Rate
% A % B
(minutes) (mL/min)
0 40 85 15
1 40 85 15
10 40 45 55
1 1 40 1 99
15 40 1 99
For compounds with an analytical LCMS retention time between 0.9 and 1 .2 minutes (LCMS method A) or between 2.2 and 3.0 minutes (LCMS method B) the following gradient was used:

For compounds with an analytical LCMS retention time between 1.2 and 1 .4 minutes (LCMS method A) or between 3.0 and 3.6 minutes (LCMS method B) the following gradient was used:

For compounds with an analytical LCMS retention time greater than 1 .4 minutes (LCMS method A) or greater than 3.6 minutes (LCMS method B) the following gradient was used
Time Flow Rate
% A % B
(minutes) (mL/min)
0 40 20 80
1 40 20 80
10 40 1 99
1 1 40 1 99
15 40 1 99
The UV detection was an averaged signal from wavelength of 210nm to 350nm and mass spectra were recorded on a mass spectrometer using alternate-scan positive and negative mode electrospray ionization. The chemical names were generated using ACD Name Pro version 6.02 from Advanced Chemistry Development, Inc.
Intermediate 1
W-(6-chloro-2-pyridinyl)-6-methyl-1 ,3-benzothiazol-2-amine

Under an atmosphere of nitrogen, an ice-cooled solution of 6-chloro-2-pyridinamine (300mg, 2.33mmol) in tetrahydrofuran (15ml_) was treated portionwise with sodium hydride (60% w/w in oil) (121 mg, 3.03mmol). After 20 minutes, the mixture was treated with 2-chloro-6-methyl-1 ,3-benzothiazole (471 mg, 2.57mmol) and allowed to stir and warm to ambient temperature overnight. The mixture was then cautiously treated with saturated aqueous ammonium chloride (20ml_) and dichloromethane (50ml_). The organic phase was collected, evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (155mg, 0.56mmol, 24% yield). LCMS (Method A): Rt 1 .33 minutes; m/z 276 (MH+)
Intermediate 2
-bromo-N-(6-chloro-2-pyridinyl)-1 ,3-benzothiazol-2-amine

Under an atmosphere of nitrogen, an ice-cooled solution of 6-chloro-2-pyridinamine (2.6g, 20.2mmol) in anhydrous tetrahydrofuran (50ml_) was treated portionwise with sodium hydride (60% in oil) (2.43g, 60.7mmol). The reaction mixture was stirred for 10 minutes and then 6-bromo-2-chloro-1 ,3-benzothiazole (5.53g, 22.3mmol) was added portionwise. The reaction mixture was stirred for a further 15 minutes then allowed to warm up to ambient temperature, then heated at 65°C overnight. Water (100ml_) was added to the cooled reaction mixture which was then filtered; the filtered solid was washed with water
and thoroughly dried to afford the title compound (7.16g, 21.1 mmol, quantitative yield). LCMS (Method B): Rt 3.45 minutes; m/z 340 (MH+).
Intermediate 3
6-ethyl-W-(6-fluoro-2-pyridinyl)-1 ,3-benzothiazol-2-

Under an atmosphere of nitrogen, an ice-cooled solution of 6-ethyl-1 ,3-benzothiazol-2- amine (387mg, 2.17mmol) in Ν,Ν-dimethylformamide (15ml_) was treated portionwise with sodium hydride (60% in oil) (130mg, 3.26mmol) and the mixture was stirred for 15 minutes. A solution of 2,6-difluorpyridine (250mg, 2.17mmol) in N,N-dimethylformamide (5mL) was added and the mixture was slowly allowed to warm to ambient temperature overnight. The mixture was treated cautiously with saturated aqueous ammonium chloride (20ml_) and ethyl acetate (20ml_). The organic phase was dried over magnesium sulfate, filtered and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 50% ethyl acetate in cyclohexane to afford the title compound (58mg, 0.21 mmol, 10% yield). LCMS (Method A): Rt 1 .33 minutes; m/z 274 (MH+)
The compounds shown in the table were prepared in an analogous manner to that for 6- ethyl-N-(6-fluoro-2-pyridinyl)-1 ,3-benzothiazol-2-amine by reacting 2,6-difluoropyridine with the appropriate 2-aminobenzothiazole or 2-aminothiazolopyridine:
Purification
Intermediate Structure Name Analytical Data
Method
5-chloro-N-(6- fluoro-2- Chromatography
LCMS (Method A):
pyridinyl)[1 ,3]thi on silica: 0-100%
4 Rt 1.10 minutes;
azolo[5,4- ethyl acetate in
F CI m/z 281 (MH+)
b]pyridin-2- cyclohexane amine
6-bromo-N-(6-
Chromatography fluoro-2- LCMS (Method A):
on silica: 0-100%
5 pyridinyl)-1 ,3- Rt 1.24 minutes;
F ethyl acetate in benzothiazol-2- m/z 324,326 (MH+)
cyclohexane amine
Intermediate 6:
trans-4-[(6-{[6-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 ,3-benzothiazol-2- yl]amino}-2-pyridinyl)amino]cyclohexanol
A mixture 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi-1 ,3,2-dioxaborolane (0.727g, 2.86mmol), trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol
[example 3] (1 g, 2.39mmol) , diphenylphosphinoferrocene (0.066g, 0.1 19mmol), 1 ,1 '- bis(diphenylphosphino)ferrocene-palladium(ii)dichloride dichloromethane complex (0.097g, 0.1 19mmol) and potassium acetate (0.702g, 7.15mmol) in anhydrous tetrahydrofuran (10mL) was thoroughly degassed by the alternate application of vacuum and nitrogen pressure via a syringe needle through the septum of the sealed reaction vessel. The reaction mixture was heated in a Biotage "Initiator" microwave at 120°C for 90 minutes. Additional 4,4,4',4', 5,5,5', 5'-octamethyl-2,2'-bi-1 ,3,2-dioxaborolane (0.727g, 2.86mmol) and 1 ,1 '-bis(diphenylphosphino)ferrocene-palladium(ii)dichloride dichloromethane complex (0.097g, 0.1 19mmol) were added, the vessel was resealed and the mixture was heated in the microwave for a further 90 minutes at 120°C. The reaction mixture was partitioned between saturated aqueous ammonium chloride (30ml_) and ethyl acetate (30ml_), separated and the aqueous phase was extracted with further ethyl acetate (2x50ml_). The combined organic extracts were dried over magnesium sulfate and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (1.5g, 3.22mmol, >100% theoretical yield). LCMS (Method B): Rt 2.63 minutes; m/z 467 (MH+).
Intermediate 7:
-bromo-3-pyridinyl)methyl]dimethyl

A solution of 3-bromo-5-(chloromethyl)pyridine hydrochloride (500mg, 2.06mmol) in methanol (5ml_) was cooled to 0°C and dimethylamine (2M in tetrahydrofuran, 10.3ml_, 20.6mmol) was added. The reaction mixture was sealed and stirred at ambient
temperature overnight. The mixture was then evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane followed by 0 to 20% methanol in dichloromethane. A methanolic solution of the recovered product was added to an SCX (sulfonic acid) solid-phase extraction cartridge which was washed with methanol and the product then eluted from the column using ammonia in methanol (2M) to afford the title compound (260mg, 1.21 mmol, 59% yield). LCMS (Method B): Rt 1 .99 minutes; m/z 215,217 (MH+).
Intermediate 8:
6-bromo-1 -methyl-1 H-pyrrolo[3,2-b]pyridine

Under an atmosphere of nitrogen, a solution of 6-bromo-1 H-pyrrolo[3,2-b]pyridine (200mg, 1.02mmol) in anhydrous tetrahydrofuran (5ml_) was cooled to 0°C and then sodium hydride (60% in oil) (29mg, 1 .22mmol) was added portionwise. The reaction mixture was stirred at 0°C for 10 minutes then iodomethane (0.063ml_, 1 .02mmol) in anhydrous THF (1 ml_) was added and the mixture was stirred at ambient temperature overnight. The reaction mixture was then partitioned between water (10ml_) and ethyl acetate (10ml_). The aqueous phase was extracted with further ethyl acetate (10ml_) and the combined organic extracts were evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane followed by 0 to 20% methanol in dichloromethane to afford the title compound (43mg, 0.20mmol, 20% yield). LCMS (Method B): Rt 1 .10 minutes; m/z 21 1 ,213 (MH+). The compound shown in the table was prepared in an analogous manner to that for 6- bromo-1 -methyl-1 H-pyrrolo[3,2-/b]pyridine by reacting 6-bromo-1 H-pyrrolo[3,2-b]pyridine with iodoethane:

-[(5-bromo-3-pyridinyl)oxy]ethanol

A mixture of 1 ,3-dioxolan-2-one (1.01 g, 1 1.5mmol), 5-bromo-3-pyridinol (1g, 5.75mmol) and potassium carbonate (1 .19g, 8.62mmol) in Ν,Ν-dimethylformamide (10mL) was heated at 86°C overnight. The cooled reaction mixture was partitioned between water (20ml_) and ethyl acetate (20ml_). The ethyl acetate extract was evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane followed by 0 to 20% methanol in dichloromethane to afford the title compound (600mg, 2.75mmol, 48% yield). LCMS (Method A): Rt 0.63 minutes; m/z 218,220 (MH+).
Intermediate 11 :
-bromo-5-{[(3-fluorophenyl)methyl]oxy}pyridine

A mixture of 1 -(bromomethyl)-3-fluorobenzene (1 .09g, 5.75mmol), 5-bromo-3-pyridinol (1g, 5.75mmol), and potassium carbonate (1.19g, 8.62mmol) in acetonitrile (10ml_) was stirred at 80°C overnight. The cooled reaction mixture was partitioned between water (20ml_) and ethyl acetate (20ml_). The aqueous phase was extracted with further ethyl acetate (2x20ml_) and the combined organic extracts were evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (329mg, 1.17mmol, 20% yield). LCMS (Method B): Rt 1 .24 minutes; m/z 282,284 (MH+).
Intermediate 12:
-bromo-5-[(2-pyridinylmethyl)oxy]pyridine

A mixture of 2-(bromomethyl)pyridine hydrobromide (1 .45g, 5.75mmol), 5-bromo-3- pyridinol (1 g, 5.75mmol) and potassium carbonate (2.38g, 17.2mmol) in acetonitrile (10ml_) was stirred at 80°C overnight. The cooled reaction mixture was partitioned
between water (20ml_) and ethyl acetate (20ml_). The aqueous phase was extracted with further ethyl acetate (2x20ml_) and the combined organic extracts were evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (250mg, 0.94mmol, 16% yield). LCMS (Method B): Rt 1.78 minutes; m/z 265,267 (MH+).
Intermediate 13:
-[(5-bromo-3-pyridinyl)methyl]-2-(methyloxy)ethanamine

A mixture of 3-bromo-5-(chloromethyl)pyridine hydrochloride (500mg, 2.06mmol), 2- (methyloxy)ethanamine (464mg, 6.17mmol), potassium carbonate (284mg, 2.06mmol) in acetonitrile (10ml_) was heated at 80°C for 4 hours. Water (10ml_) was added to the cooled reaction mixture and the mixture was extracted with ethyl acetate (2x20ml_). The combined ethyl acetate extracts were evaporated to dryness. The product was purified by ion exchange chromatography using an SCX (sulfonic acid) solid-phase extraction cartridge and eluting with methanol and then with ammonia in methanol (2M) to afford the title compound (452mg, 1.84mmol, 90% yield). LCMS (Method B): Rt 1 .80 minutes; m/z 245,247 (MH+).
Intermediate 14:
N-(6-chloro-2-pyridinyl)-6-nitro-1 ,3-benzothiazol-2-

Under an atmosphere of nitrogen, an ice-cooled solution of 6-chloro-2-pyridinamine (2g, 15.56mmol) in tetrahydrofuran (100mL) was treated portionwise with sodium hydride (60% w/w in oil, 0.81g, 20.2mmol) and the resulting mixture was stirred at 0°C for 10 minutes. The mixture was treated dropwise with a solution of 2-chloro-6-nitro-1 ,3- benzothiazole (4.01g, 18.7mmol) in tetrahydrofuran (150mL) and then the reaction mixture was allowed to warm to ambient temperature and stirred overnight. Additional sodium hydride (60% w/w in oil, 0.40g, 10.1 mmol) was added and reaction mixture was heated to 50°C for 2 hours. The cooled reaction mixture was treated cautiously with methanol and evaporated to dryness. The residual solid was stirred in dichloromethane overnight, filtered and dried to afford the title compound (1 .96g, 6.38mmol, 41 % yield).
The filtrate was evaporated to dryness and the residual solid was stirred with dichloromethane to afford a second crop of the title compound (1 .06g, 3.44mmol, 22% yield). LCMS (Method A): Rt 1 .17 minutes; m/z 307 (MH+). Intermediate 15:
A^-(6-chloro-2-pyridinyl)-1 ,3-benzothiazole-2,6-diamine

Under an atmosphere of nitrogen, a stirred solution of N-(6-chloro-2-pyridinyl)-6-nitro-1 ,3- benzothiazol-2-amine [intermediate 14] (2g, 6.52mmol) in acetic acid (10ml_) was treated portionwise with zinc dust (4.26g, 65.2mmol). The reaction mixture was stirred for 1 hour, then taken up in water (100ml_) and chloroform (100ml_) and filtered. The aqueous layer of the filtrate was extracted with chloroform (100ml_) and the combined organics were evaporated to dryness to afford the title compound (1.9g, 6.87mmol, >100% yield). LCMS (Method A): Rt 0.69 minutes; m/z 277 (MH+).
Intermediate 16:
frans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4-b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol

A mixture of 5-chloro-N-(6-fluoro-2-pyridinyl)[1 ,3]thiazolo[5,4-b]pyridin-2-amine [intermediate 17] (3.45g, 12.3mmol) and trans-4-aminocyclohexanol (14.1 g, 123mmol) was heated at 190°C for 90 minutes. To the cooled mixture was added ethyl acetate (20ml_), dichloromethane (100ml_) and water (100ml_). The mixture was separated and the organic phase was dried over magnesium sulfate, filtered and evaporated to dryness. The product was purified by flash chromatography on silica using a gradient elution from 0 to 1 10% ethyl acetate in dichloromethane then 0 to 20% methanol in dichloromethane to afford the title compound (2.1 g, 5.59mmol, 46% yield). LCMS (Method A): Rt 0.89 minutes; m/z 376 (MH+). Intermediate 17:
5-chloro-N-(6-fluoro-2-pyridinyl)[1 ,3]thiazolo[5,4-b]pyridin-2 -amine

Under an atmosphere of nitrogen, a solution of 5-chloro[1 ,3]thiazolo[5,4-b]pyridin-2-amine (4.75g, 25.6mmol) in dry Ν,Ν-dimethylformamide (50ml_) was treated with 2,6- difluoropyridine (3g, 26.1 mmol) and the mixture was cooled with an ice/water bath. Sodium hydride (60% in oil) (2.09g, 52.1 mmol) was added portionwise and the mixture was stirred whilst being allowed to slowly warm to ambient temperature overnight. The mixture was treated cautiously with aqueous ammonium chloride solution (5%, 20ml_) and water (100ml_). The mixture was then extracted with chloroform (2 x 150ml_) and the combined organic fraction was evaporated to dryness. The product was purified by flash chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (4.9g, 17.5mmol, 67% yield). LCMS (Method A): Rt 1 .10 minutes; m/z 281 (MH+). Intermediate 18:
2-[6-chloro-4-(phenylmethyl)-2-pyridinyl]-1 ,3-benzothiazole-2,6-diamine

A suspension of N-[6-chloro-4-(phenylmethyl)-2-pyridinyl]-6-nitro-1 ,3-benzothiazol-2- amine [intermediate 19] (495mg, 1.25mmol) in acetic acid (30ml_) was treated with tetrahydrofuran (15ml_) and zinc dust (816mg, 12.5mmol) and the mixture was stirred at ambient temperature for 1 hour. The mixture was filtered and the filtrate was evaporated to dryness. The residue was partitioned between dichloromethane (30ml_) and water (20ml_). The organic phase was collected, evaporated to dryness and the product was purified by flash chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in isohexane to afford the title compound (212mg, 0.58mmol, 46% yield). LCMS (Method A): Rt 1 .21 minutes; m/z 367 (MH+).
Intermediate 19:
W-[6-chloro-4-(phenylmethyl)-2-pyridinyl]-6-nitro-1 ,3-benzothiazol-2-amine

Under an atmosphere of nitrogen, a solution of a mixture of 6-chloro-4-(phenylmethyl)-2- pyridinamine [intermediate 22] (410mg, 1.88mmol) and 2-chloro-6-nitro-1 ,3-benzothiazole (402mg, 1 .88mmol) in tetrahydrofuran (25mL) was treated with sodium hydride (60% in oil) (187mg, 4.69mmol) and the mixture was stirred at ambient temperature for 1 hour and then at 50°C overnight. The cooled mixture was treated with tetrahydrofuran (20ml_), ethyl acetate (50ml_) and saturated aqueous ammonium chloride (50ml_). The organic fraction was dried over magnesium sulfate, filtered and evaporated to dryness and the product was purified by flash chromatography on silica using a gradient elution from 0 to 50% ethyl acetate in cyclohexane to afford the title compound (502mg, 1.27mmol, 68% yield). LCMS (Method A): Rt 1 .40 minutes; m/z 397 (MH+).
Intermediate 20:
frans-4-{[6-[(6^romo-1 ,3-benzothiazol-2-yl)amino]-4-(phenylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of 6-bromo-N-[6-chloro-4-(phenylmethyl)-2-pyridinyl]-1 ,3-benzothiazol-2-amine [intermediate 21 ] (390mg, 0.905mmol) and frans-4-aminocyclohexanol (521 mg, 4.53mmol) was sealed and heated in a Biotage "Initiator" microwave at 160°C for 5 hours. The mixture was then partitioned between dichloromethane (50ml_) and saturated aqueous ammonium chloride (50ml_). The organic fraction was evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane followed by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (81 mg, 0.159mmol, 18% yield). LCMS (Method A): Rt 1 .16 minutes; m/z 509,51 1 (MH+).
Intermediate 21 :
6-bromo-N-[6-chloro-4-(phenylmethyl)-2-pyridinyl]-1 ,3-benzothiazol-2-amine

Under an atmosphere of nitrogen, an ice-cooled solution of 6-chloro-4-(phenylmethyl)-2- pyridinamine [intermediate 22] (330mg, 1 .51 mmol) in tetrahydrofuran (30ml_) was treated portionwise with sodium hydride (60% in oil) (121 mg, 3.02mmol) and stirred with cooling for 20 minutes. To the mixture was then added 6-bromo-2-chloro-1 ,3-benzothiazole (375mg, 1 .51 mmol) and the reaction mixture was allowed to warm to ambient temperature whereupon it was stirred for 12 hours and then heated at 50°C for a further 18 hours. The cooled mixture was cautiously treated with saturated aqueous ammonium chloride (20ml_) and ethyl acetate (20ml_). The organic phase was dried over magnesium sulfate, filtered and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (390mg, 0.91 mmol, 60% yield). LCMS (Method A): Rt 1 .51 minutes; m/z 430,432 (MH+).
Intermediate 22:
-chloro-4-(phenylmethyl)-2-pyridinamine

A solution of 2,6-dichloro-4-(phenylmethyl)pyridine [intermediate 23] (1.85g, 7.77mmol) in isopropanol (8ml_) was treated with concentrated aqueous ammonia (5ml_, 231 mmol) and the mixture was sealed and heated in a Biotage "Initiator" microwave at 170°C (pressure limited to 19 Bar) for 14 hours. The mixture was then evaporated to dryness and the residue partitioned between dichloromethane (40ml_) and water (30ml_). The organic phase was evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% diethyl ether in petroleum ether (40-60°C boiling fraction) to afford the title compound (1.12q, 5.12mmol, 66% yield). LCMS (Method A): Rt 1 .02 minutes; m/z 219 (MH+).
Intermediate 23:
2,6-dichloro-4-(phenylmethyl)pyrid
Under an atmosphere of nitrogen, a mixture of 2,6-dichloro-4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridine [intermediate 24] (2.5g, 9.13mmol), sodium carbonate (3.87g, 36.5mmol), tetrakis(triphenylphosphine)palladium(0) (1.06g, 0.913mmol) and benzyl bromide (1 .3ml_, 1 1.0mmol) in a mixture of toluene (50ml_) and ethanol (25ml_) was heated to 80°C for 1.5 hours. The cooled mixture was then treated with water (200ml_) and ethyl acetate (150ml_) and separated. The organic phase was dried over magnesium sulfate, filtered and evaporated to dryness and the product was purified by flash chromatography using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (1.87g, 7.85mmol, 86% yield). LCMS (Method A): Rt 1 .28 minutes; m/z 238,240 (MH+).
Intermediate 24:
-dichloro-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridine

Under an atmosphere of nitrogen, a mixture of 2,6-dichloropyridine (3g, 20.3mmol), 4,4,4,,4,,5,5,5,,5'-octamethyl-2,2,-bi-1 ,3,2-dioxaborolane (5.92g, 23.3mmol), 1 ,10- phenanthroline (145mg, 0.81 mmol) and chlorobis(1 ,5-cyclooctadiene)iridium(l) dimer (267mg, 0.30mmol) was treated with 1 ,2-dichloroethane (20ml_). Nitrogen was passed through the mixture for 5 minutes after which time the mixture was heated at 100°C for 1 hour. The cooled reaction mixture was poured into a mixture of diethyl ether (150ml_) and aqueous sodium hydroxide solution (4M, 200ml_) and the phases were separated. The aqueous phase was ice-cooled and acidified with aqueous hydrochloric acid (5M) and the resulting precipitate was filtered, washed with water and dried to afford the title compound (4.9g, 17.9mmol, 88% yield). LCMS (Method A): Rt 0.74 minutes; m/z 192,194 (ionised as the boronic acid) (MH+). NMR (400MHz, dmso-d6) δ 1.28 (12H, s), 7.57 (2H, s).
Intermediate 25:
6-bromo-W-[6-fluoro-4-(2-pyridinylmethyl)-2-pyridinyl]-1 ,3-benzothiazol-2 -amine

Under an atmosphere of nitrogen, an ice-cooled stirred solution of 6-bromo-1 ,3- benzothiazol-2-amine (722mg, 3.15mmol) in tetrahydrofuran (20ml_) was treated portionwise with sodium hydride (60% in oil) (242mg, 6.06mmol). The reaction mixture was allowed to stir for 15 minutes then treated dropwise with a solution of 2,6-difluoro-4- (2-pyridinylmethyl)pyridine [intermediate 26] (500mg, 2.43mmol) in tetrahydrofuran (10ml_). The reaction mixture was allowed to warm to ambient temperature and stirred overnight. Water (100ml_) was added and the reaction mixture was extracted with ethyl acetate (2 x 100ml_). The combined organic fractions were evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane followed by 0 to 20% methanol in dichloromethane to afford the title compound (589mg, 1 .42mmol, 59% yield). LCMS (Method A): Rt 1 .37 minutes; m/z 415,417 (MH+).
Intermediate 26:
-difluoro-4-(2-pyridinylmethyl)pyrid

Under an atmosphere of nitrogen, a mixture of 2-(chloromethyl)pyridine hydrochloride (2.042g, 12.5mmol) and potassium carbonate (6.88g, 49.8mmol) in acetone (150ml_) and water (50ml_) was stirred for 5 minutes whereupon 2,6-difluoro-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)pyridine [intermediate 27] (3g, 12.5mmol) was added. The reaction mixture was stirred for 5 minutes, then cooled to 0°C and palladium(ll)chloride (0.221g, 1.25mmol) was added. The reaction mixture was heated at reflux overnight. Water (200ml_) was added to the cooled mixture which was then extracted with ethyl acetate (2 x 200ml_). The organic fractions were combined, evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (500mg, 2.43mmol, 19% yield). LCMS (Method A): Rt 0.99 minutes; m/z 207 (MH+). Intermediate 27:
2 -difluoro-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridine

Under an atmosphere of nitrogen, a mixture of 2,6-difluoropyridine (7.89ml_, 87mmol), 4,4,4,,4,,5,5,5,,5,-octamethyl-2,2'-bi-1 ,3,2-dioxaborolane (25.4g, 100mmol), 1 ,10- phenanthroline (0.626g, 3.48mmol) and chlorobis(1 ,5-cyclooctadiene)iridium(l) dimer (0.574g, 0.634mmol) was treated with 1 ,2-dichloroethane (80mL). Nitrogen was passed through the mixture for 5 minutes after which time the mixture was heated at 100°C for 1 hour. The cooled reaction mixture was added to a mixture of diethyl ether (600ml_) and aqueous sodium hydroxide solution (4M, 800ml_) and the phases were separated. The aqueous phase was ice-cooled and acidified with aqueous hydrochloric acid (5M) and the resulting precipitate was filtered, washed with water and dried to afford the title compound (15g, 62.2mmol, 72% yield). LCMS (Method A): Rt 0.63 minutes; m/z 160 (ionises as the boronic acid) (MH+). Intermediate 28:
6-bromo-W-[6-chloro-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]-1 ,3-benzothiazol-2- amine

Under an atmosphere of nitrogen, a stirred solution of 6-chloro-4-(1 H-pyrazol-1-ylmethyl)- 2-pyridinamine [intermediate 29] (0.61g, 2.92mmol) in anhydrous tetrahydrofuran (20ml_) was treated with 6-bromo-2-chloro-1 ,3-benzothiazole (0.799g, 3.22mmol) and, portionwise over 5 minutes, with sodium hydride (60% in oil) (0.257g, 6.43mmol). After 30 minutes the mixture was heated to 50°C for 6 hours. The cooled mixture was treated cautiously with saturated aqueous ammonium chloride (30ml_) and tetrahydrofuran (30ml_). The resulting precipitate was filtered off, washed with ethyl acetate and diethyl ether and dried to afford the title compound (878mg, 2.09mmol, 71 % yield). LCMS (Method A): Rt 1 .26 minutes; m/z 420,422 (MH+).
Intermediate 29:
-chloro-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinamine

A mixture of 2,6-dichloro-4-(1 H-pyrazol-1 -ylmethyl)pyridine [intermediate 30] (0.9g, 3.95mmol) and concentrated aqueous ammonia (7ml_, 362mmol) was sealed and heated in a Biotage "Initiator" microwave at 170°C for 9 hours. The cooled suspension was added to water (120ml_) and then filtered. The filtered solid was washed with water and dried to afford the title compound (0.66g, 3.16mmol, 80% yield). LCMS (Method A): Rt 0.64 minutes; m/z 209 (MH+).
Intermediate 30:
-dichloro-4-(1 H-pyrazol-1 -ylmethyl)pyrid

An ice-cooled solution of pyrazole (0.691g, 10.2mmol) in tetrahydrofuran (10ml_) under an atmosphere of nitrogen was treated portionwise with sodium hydride (60% in oil) (0.425g, 10.6mmol). After 15 minutes the mixture was treated with 2,6-dichloro-4- (chloromethyl)pyridine (1 .9g, 9.67mmol) and then allowed to warm slowly to ambient temperature whereupon it was stirred for 16 hours. The mixture was treated with saturated aqueous ammonium chloride (20ml_) and ethyl acetate (30ml_). The organic phase was dried over magnesium sulfate, filtered and the filtrate was evaporated to dryness. The product was purified by flash chromatography on silica using a gradient elution from 0 to 50% ethyl acetate in cyclohexane to afford 2,6-dichloro-4-(1 H-pyrazol-1- ylmethyl)pyridine (1.1 g, 4.82mmol, 50% yield). LCMS (Method A): Rt 0.87 minutes; m/z 228,230 (MH+). Intermediate 31 :
W-(6-bromo-1 ,3-benzothiazol-2-yl)-W-[3-(4-morpholinyl)propyl]-4-(1H-pyrazol-1 - ylmethyl)-2,6-pyridinediamine

Intermediate 32:
6-bromo-W-{6-chloro-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2-pyridinyl}-1 ,3- ine

Under an atmosphere of nitrogen, an ice-cooled, stirred suspension of 6-chloro-4-[(2- methyl-1 H-imidazol-1-yl)methyl]-2-pyridinamine [intermediate 34] (1 .6g, 7.19mmol) in anhydrous tetrahydrofuran (40ml_) was treated with 6-bromo-2-chloro-1 ,3-benzothiazole (1.96g, 7.90mmol) and, portionwise over 5 minutes, with sodium hydride (60% in oil) (0.632g, 15.8mmol). After 30 minutes the mixture was heated to 50°C for 6 hours. The cooled mixture was treated with saturated aqueous ammonium chloride (30ml_) and tetrahydrofuran (30ml_). The resulting precipitate was filtered off, washed with ethyl acetate and diethyl ether and dried to afford the title compound (2.2g, 5.06mmol, 70% yield). LCMS (Method A): Rt 0.94 minutes; m/z 434,436 (MH+).
Intermediate 33:
W-{6-chloro-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2-pyridinyl}[1 ,3]thiazolo[5,4- £>]pyridin-2 -amine

Under an atmosphere of nitrogen, an ice-cooled, stirred suspension of 6-chloro-4-[(2- methyl-1 H-imidazol-1-yl)methyl]-2-pyridinamine [intermediate 34] (600mg, 2.69mmol) in anhydrous tetrahydrofuran (20ml_) was treated with 2-bromo[1 ,3]thiazolo[5,4-b]pyridine (600mg, 2.79mmol) and, portionwise over 5 minutes, with sodium hydride (60% in oil) (216mg, 5.39mmol). After 30 minutes the mixture was heated to 50°C for 4 hours. The
cooled mixture was treated with saturated aqueous ammonium chloride (30ml_) and tetrahydrofuran (30ml_). The mixture was separated and the organic phase dried over magnesium sulfate, filtered and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 30% methanol in dichloromethane to afford the title compound (522mg, 1 .46mmol, 54% yield). LCMS (Method A): Rt 0.64 minutes; m/z 357 (MH+).
Intermediate 34:
-chloro-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2-pyridinamine

A mixture of 2,6-dichloro-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]pyridine [intermediate 35] (3.6g, 14.9mmol) and concentrated aqueous ammonia (10ml_, 517mmol) was sealed and heated in a Biotage "Initiator" microwave at 160°C (pressure limited to 19 Bar) for 9 hours. The cooled suspension was added to water (120ml_) and then filtered. The filtered solid was washed with water and dried to afford the title compound (3.1g, 13.9mmol, 94% yield). LCMS (Method A): Rt 0.33 minutes; m/z 223,225 (MH+).
Intermediate 35:
-dichloro-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]pyridine

Under an atmosphere of nitrogen, an ice-cooled solution of 2-methyl-1 H-imidazole (1 .7g, 20.7mmol) in tetrahydrofuran (30ml_) was treated portionwise with sodium hydride (60% in oil) (1g, 25.0mmol). After 15 minutes the mixture was treated with 2,6-dichloro-4- (chloromethyl)pyridine (4g, 20.4mmol) and then allowed to warm slowly to ambient temperature whereupon it was stirred for 8 hours. The mixture was then treated with saturated aqueous ammonium chloride (40ml_) and extracted with ethyl acetate (2 x 40ml_). The combined organics were dried over magnesium sulfate, filtered and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane followed by a gradient of 0 to
20% methanol in dichloromethane to afford the title compound (3.7g, 15.3mmol, 75% yield). LCMS (Method A): Rt 0.47 minutes; m/z 242,244 (MH+).
Intermediate 36:
-chloro-6-([1 ,3]thiazolo[5,4^]pyridin-2-ylamino)-4-pyridinyl]methanol

A suspension of N-[6-chloro-4-({[(1 ,1 -dimethylethyl)(dimethyl)silyl]oxy}methyl)-2- pyridinyl][1 ,3]thiazolo[5,4-b]pyridin-2-amine [intermediate 37] (2.85g, 7.0mmol) in tetrahydrofuran (40ml_) was treated with tetrabutylammonium fluoride (1 M in THF) (10.5ml_, 10.5mmol) and then stirred at ambient temperature for 1 hour. The mixture was evaporated to dryness and then partitioned between dichloromethane (50ml_) and water (50ml_). The product was only partially soluble and so the mixture was poured into hexane (200ml_) and then filtered. The filtered solid was washed with water, then diethyl ether and dried to afford the title compound (1.98g, 6.76mmol, 97% yield). LCMS (Method A): Rt 0.82 minutes; m/z 293 (MH+).
Intermediate 37:
W-[6-chloro-4-({[(1 ,1 -dimethylethyl)(dimethyl)silyl]oxy}methyl)-2- pyridinyl][1 ,3]thiazolo[5,4-£>]pyridin-2 -amine

Under an atmosphere of nitrogen, an ice-cooled mixture of 6-chloro-4-({[(1 ,1 - dimethylethyl)(dimethyl)silyl]oxy}methyl)-2-pyridinamine [intermediate 40] (2g, 7.33mmol) and 2-bromo[1 ,3]thiazolo[5,4-b]pyridine (1 .58g, 7.33mmol) in dry N,N-dimethylformamide (20mL) was treated portionwise over 5 minutes with sodium hydride (60% in oil) (0.586g, 14.7mmol) and the mixture was stirred for 3 hours whilst being allowed to warm to ambient temperature. The mixture was then treated cautiously with saturated aqueous ammonium chloride (20mL) and water (30mL). The precipitated solid was filtered, washed with water and dried to afford the title compound (2.87g, 7.05mmol, 96% yield). LCMS (Method A): Rt 1 .51 minutes; m/z 407 (MH+).
Intermediate 38:
-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-chloro-4-pyridinyl}methanol

Under an atmosphere of nitrogen, a stirred solution of 6-bromo-N-[6-chloro-4-({[(1 ,1 - dimethylethyl)(dimethyl)silyl]oxy}methyl)-2-pyridinyl]-1 ,3-benzothiazol-2-amine
[intermediate 39] (2.3g, 4.74mmol) in tetrahydrofuran (100mL) was treated dropwise with tetrabutylammonium fluoride (1 M) (2.75ml_, 9.49mmol). The mixture was stirred at ambient temperature for 3 hours whereupon water (200ml_) was added and the resulting precipitate was filtered, washed with water (2 x 10ml_) and dried to afford the title compound (1.18g, 3.19mmol, 67% yield). LCMS (Method A): Rt 1.17 minutes; m/z 370,372 (MH+).
Intermediate 39:
6 )romo-N-[6 :hloro-4-({[(1,1 limethylethyl)(dim
-benzothiazol-2 -amine

Under an atmosphere of nitrogen, an ice-cooled mixture of 6-chloro-4-({[(1 ,1 - dimethylethyl)(dimethyl)silyl]oxy}methyl)-2-pyridinamine [intermediate 40] (1.3g, 4.76mmol) and sodium hydride (60% in oil) (0.572g, 14.3mmol) in tetrahydrofuran (50ml_) was treated dropwise with a solution of 6-bromo-2-chloro-1 ,3-benzothiazole (1 .30g, 5.24mmol) in tetrahydrofuran (50ml_). The mixture was then heated at reflux overnight. Water (100ml_) was added to the cooled mixture which was then extracted with ethyl acetate (2 x 100ml_). The combined organics were evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 25% ethyl acetate in cyclohexane to afford the title compound (2.0g, 4.12mmol, 87% yield). LCMS (Method A): Rt 1 .72 minutes; m/z 484,486 (MH+).
Intermediate 40:
6-chloro-4-({[(1,1 -dimethylethyl)(dimethyl)silyl]oxy}methyl)-2-pyridinamine

Under an atmosphere of nitrogen and at ambient temperature a stirred solution of (2- amino-6-chloro-4-pyridinyl)methanol [intermediate 41] (48g, 0.30mol) and imidazole (24.6g, 0.36mol) in anhydrous Ν,Ν-dimethylformamide (800mL), was treated with chloro(1 ,1-dimethylethyl)dimethylsilane (46.4g, 0.31 mol). The reaction mixture was stirred for 90 minutes and then concentrated in vacuo whilst maintaining the temperature below 40°C. Water was added and the mixture was extracted with dichloromethane (3 x 500ml_). The combined organics were washed with water, dried over magnesium sulfate and filtered through a pad of silica. The filtrate was evaporated to dryness to afford the title compound (49.3g, 0.18mol, 60% yield). LCMS (Method A): Rt 1.58 minutes; m/z 273 (MH+).
Intermediate 41 :
(2-amino-6-chloro-4-pyridinyl)methanol

Under an atmosphere of nitrogen and at ambient temperature, a solution of lithium aluminium hydride (98.2g, 1.25mol) in tetrahydrofuran (2200ml_) was treated portionwise with 2-amino-6-chloro-4-pyridine carboxylic acid [intermediate 42] (98.2g, 569mmol) whilst maintaining the temperature of the reaction below 20°C. The mixture was stirred overnight at ambient temperature and then cooled to 0°C. Water (70ml_) was slowly added dropwise. After a further 5 minutes, aqueous potassium hydroxide solution (15% w/v, 70ml_) was slowly added, followed by additional water (200ml_). The mixture was allowed to warm to ambient temperature before magnesium sulfate was added and then stirred for 30 minutes. The mixture was then filtered, the filtered solid was washed with ethyl acetate (2000ml_) and the filtrate was evaporated to dryness to afford the crude product. The crude product was suspended in dichloromethane (500ml_), filtered and the filtered solid was dried to afford the title compound (80g, 0.506mol, 88% yield). LCMS (Method A): Rt 0.66 minutes; m/z 159 (MH+).
Intermediate 42:
-amino-6-chloro-4-pyridinecarboxylic acid

A mixture of 2,6-dichloro-4-pyridine carboxylic acid (1 10g, 0.58mol) and aqueous ammonia solution (26%, 440ml_) was sealed and heated in a microwave reactor (split into 1 1 separate vessels) at 165°C (pressure limited to 19bar) for 8 hours. The cooled mixtures were combined and evaporated to dryness. The residue was dissolved in water (2.5L) and the pH was adjusted to 3 by the addition of concentrated aqueous hydrochloric acid. The resulting precipitate was filtered, washed with water and thoroughly dried to afford the title compound (100g, 0.58mol, 100% yield). LCMS (Method A): Rt 0.52 minutes; m/z 173,175 (MH+).
Intermediate 43:
2-[(frans-4-hydroxycyclohexyl)amino]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-4- pyridinecarbaldehyde

A mixture of trans-4-{[4-(hydroxymethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol [example 103] (3.36g, 8.14mmol) and activated manganese dioxide (7.08g, 81 mmol) in tetrahydrofuran (200ml_) was heated at 70°C overnight. The cooled mixture was filtered through a layer of silica and celite and the filtrate was evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 25% methanol in dichloromethane to afford the title compound (1.01 g, 2.73mmol, 34% yield). LCMS (Method A): Rt 0.76 minutes; m/z 370 (MH+).
Intermediate 44:
2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-[(frans-4-hydroxycyclohexyl)amino]-4- pyridinecarbaldehyde

5-[2-({4-formyl-6-[(frans-4-hydroxycyclohexyl)amino]-2^yridinyl}amino)-1 ,3- inecarbonitrile

A mixture of 2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-[(trans-4- hydroxycyclohexyl)amino]-4-pyridinecarbaldehyde [intermediate 44] (357mg, 0.798mmol), tetrakis(triphenylphosphine)palladium(0) (46.1 mg, 0.04mmol), potassium phosphate (254mg, 1 .20mmol) and 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinecarbonitrile (367mg, 1 .60mmol) in a mixture of 1 ,4-dioxane (6ml_) and water (2ml_) was sealed and heated in a Biotage "Initiator" microwave at 100°C for 3.5 hours. The reaction mixture was evaporated to dryness and the residue was taken up in tetrahydrofuran and filtered. The filtrate was evaporated to dryness and the product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in dichloromethane followed by 0 to 20% methanol in dichloromethane to afford the title compound (147mg, 0.31 mmol, 39% yield). LCMS (Method A): Rt 0.92 minutes; m/z 471 (MH+).
Intermediate 46:
2-[(frans-4-hydroxycyclohexyl)amino]-6-{[6-(2-oxo-1 ,3-oxazolidin-3-yl)-1 ,3- benzothiazol-2-yl]amino}-4-pyridinecarbaldehyde

Under an atmosphere of nitrogen, a mixture of 2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]- 6-[(trans-4-hydroxycyclohexyl)amino]-4-pyridinecarbaldehyde [intermediate 44] (100mg, 0.224mmol) , 1 ,3-oxazolidin-2-one (58.4mg, 0.671 mmol), caesium carbonate (146mg, 0.45mmol) and copper(l) iodide (85mg, 0.45mmol) in dry Ν,Ν-dimethylformamide (3ml_) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with Ν,Ν'-dimethylethylenediamine (0.095ml_, 0.89mmol) and heated at 1 10°C for 1 hour. The mixture was cooled to ambient temperature, filtered and evaporated to dryness. The product was purified by chromatography on silica from 0 to 15% methanol in dichloromethane to afford the title compound (98mg, 0.216mmol, 97% yield). LCMS (Method A): Rt 0.78 minutes; m/z 454 (MH+).
Intermediate 47:
-[6-chloro-4-(4-morpholinylmethyl)-2^yridinyl]-1 ,3^enzothiazole-2,6-diami

A solution of N-[6-chloro-4-(4-morpholinylmethyl)-2-pyridinyl]-6-nitro-1 ,3-benzothiazol-2- amine [intermediate 48] (370mg, 0.91 mmol) in acetic acid (10ml_) was treated with zinc dust (298mg, 4.6mmol) and stirred for 30 minutes. The mixture was filtered and the filtrate was evaporated to dryness. The residue was then partitioned between saturated aqueous sodium bicarbonate (30ml_) and dichloromethane (30ml_). The organic phase was dried over magnesium sulfate, filtered and evaporated to dryness to afford the title compound (249mg, 0.66mmol, 73% yield). LCMS (Method A): Rt 0.46 minutes; m/z 376 (MH+).
Intermediate 48:
W-[6-chloro-4-(4-morpholinylmethyl)-2-pyridinyl]-6-nitro-1 ,3-benzothiazol-2-

Under an atmosphere of nitrogen, an ice-cooled solution of 6-chloro-4-(4- morpholinylmethyl)-2-pyridinamine [intermediate 49] (650mg, 2.85mmol) in tetrahydrofuran (15mL) was treated portionwise with sodium hydride (60% in oil) (343mg, 8.56mmol). After 20 minutes 2-chloro-6-nitro-1 ,3-benzothiazole (613mg, 2.85mmol) was added and the mixture was stirred at ambient temperature overnight. The mixture was treated with saturated aqueous ammonium chloride (100ml_) and ethyl acetate (100ml_). The precipitated solid was filtered, washed with ethyl acetate and dried to afford the title compound (389mg, 0.96mmol, 34% yield). LCMS (Method A): Rt 0.85 minutes; m/z 406 (MH+).
Intermediate 49:
-chloro-4-(4-morpholinylmethyl)-2-pyridinamine

A solution of 4-[(2,6-dichloro-4-pyridinyl)methyl]morpholine [intermediate 50] (2g, 8.1 mmol) in ethylene glycol (5ml_) was treated with concentrated aqueous ammonia (5.7ml_, 262mmol) and the mixture was sealed and heated in a Biotage "Initiator" microwave at 200°C for 24 hours. The cooled reaction mixture was filtered and the solid was dried, then suspended in diethyl ether, filtered and dried to afford the title compound (816mg, 3.6mmol, 44% yield). LCMS (Method A): Rt 0.29 minutes; m/z 228 (MH+).
Intermediate 50:
-[(2,6-dichloro-4-pyridinyl)methyl]morpholine

Under an atmosphere of nitrogen, an ice-cooled solution of 4-[(2,6-dichloro-4- pyridinyl)carbonyl]morpholine [intermediate 55] (2.63g, 10.1 mmol) in dichloromethane
(50ml_) was treated with a solution of borane in tetrahydrofuran (1 M, 30.2ml_, 30.2mmol) and stirred with cooling for 1 hour. Additional borane in tetrahydrofuran (30.2ml_, 30.2mmol) was added and the reaction was stirred until complete. The mixture was then carefully acidified with 5M aqueous hydrochloric acid and then heated at reflux. The reaction mixture was then neutralised with 10% aqueous sodium hydroxide. The aqueous phase was extracted twice with ethyl acetate (+10% methanol) and the combined organic fraction was dried over magnesium sulfate, filtered and evaporated to dryness to afford the title compound (2.89g, 1 1 .7mmol, >100% yield). LCMS (Method A): Rt 0.49 minutes; m/z 248,250 (MH+).
Intermediate 51 :
W-(frans-4-aminocyclohexyl)-W-(6-bromo-1 ,3-benzothiazol-2-yl)-4-(1 H-pyrazol-1 - ylmethyl)-2,6-pyridinediamine

A mixture of 6-bromo-N-[6-chloro-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]-1 ,3-benzothiazol- 2-amine [intermediate 28] (250mg, 0.59mmol) and trans-1 ,4-diaminocyclohexane (339mg, 2.97mmol) was sealed and heated in a Biotage "Initiator" microwave at 190°C for 5 hours. Water (50ml_) and ethyl acetate (50ml_) were added and the reaction mixture was stirred for 5 minutes, sonicated for a further 5 minutes and then filtered. The filtered solid was washed with ethyl acetate and water then dried to afford the title compound (290mg, 0.58mmol, 98% yield). LCMS (Method B): Rt 2.42 minutes; m/z 498,500 (MH+).
Intermediate 52:
N-(trans-4-aminocyclohexyl)-N'-(6-bromo-1 ,3-benzothiazol-2-yl)-4-[(2-methyl-1 H- i ]-2,6-pyridinediamine

A mixture of 6-bromo-N-{6-chloro-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-2-pyridinyl}-1 ,3- benzothiazol-2-amine [intermediate 32] (250mg, 0.58mmol) and trans-1 ,4-
diaminocyclohexane (197mg, 1 .73mmol) was sealed and heated in a Biotage "Initiator" microwave at 190°C for 5 hours. Water (50ml_) and ethyl acetate (50ml_) were added and the reaction mixture was stirred for 5 minutes, sonicated for a further 5 minutes and then filtered. The filtered solid was washed with ethyl acetate and water then dried to afford the title compound (290mg, 0.57mmol, 98% yield). LCMS (Method B): Rt 2.41 minutes; m/z 512,514 (MH+).
Intermediate 53:
2-[(6-bromo-1 ,3^enzothiazol-2-yl)amino]-6-[(frans-4-hydroxycyclohexyl)amino]-4- pyridinecarbaldehyde oxime

A mixture of 2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-[(trans-4- hydroxycyclohexyl)amino]-4-pyridinecarbaldehyde [intermediate 43] (350mg, 0.78mmol) and sodium acetate (128mg, 1 .57mmol) in a mixture of ethanol (20ml_) and water (5ml_) was treated with hydroxylamine hydrochloride (65.2mg, 0.94mmol) and the mixture was allowed to stir at ambient temperature for 2 hours. The mixture was then treated with water (40ml_) and extracted with chloroform (2 x 30ml_). The combined organics were evaporated to dryness to afford the title compound (320mg, 0.69mmol, 88% yield). LCMS (Method A): Rt 0.93 minutes; m/z 462,464 (MH+).
Intermediate 54:
[2-(methyloxy)ethyl]{[5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinyl]methyl}amine

A mixture of 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi-1 ,3,2-dioxaborolane (622mg, 2.45mmol), 1 ,1 '-bis(diphenylphosphino)ferrocene-palladium(ll)dichloride dichloromethane complex (66.6mg, 0.08mmol), potassium acetate (601 mg, 6.1 mmol), N-[(5-bromo-3- pyridinyl)methyl]-2-(methyloxy)ethanamine (500mg, 2.0mmol) in anhydrous acetonitrile (10ml_) was sealed and heated in a Biotage "Initiator" microwave at 160°C for 15 minutes. After cooling the reaction was filtered and the filtrate was evaporated to dryness to afford
the title compound (877mg, 3.0mmol, >100% yield). LCMS (Method B): Rt 1.40 minutes; m/z 293 (MH+).
Intermediate 55:
-[(2,6-dichloro-4-pyridinyl)carbonyl]morpholine

An ice-cooled mixture of 2,6-dichloro-4-pyridinecarbonyl chloride (2.85 g, 13.5mmol) and pyridine (5.70ml_, 70.4mmol) in dichloromethane (5ml_) was treated with morpholine (2.36g, 27.1 mmol) and the mixture was stirred with cooling for 2 hours. The mixture was treated with saturated aqueous sodium hydrogen carbonate (20ml_) and then extracted with ethyl acetate (2 x 20ml_). The combined organic fractions were washed with brine (20ml_), dried over magnesium sulfate, filtered and the solvent evaporated to dryness. The residue was then purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane to afford the title compound (2.93g, 1 1 .2mmol, 83% yield). LCMS (Method A): Rt 0.76 minutes; m/z 261 ,263 (MH+).
Example 1 :
trans-4-({6-[(6-methyl-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol

A mixture of N-(6-chloro-2-pyridinyl)-6-methyl-1 ,3-benzothiazol-2-amine [intermediate 1] (50mg, 0.18mmol) and trans-4-aminocyclohexanol (313mg, 2.72mmol) in ethylene glycol (1 ml_) was sealed and heated and stirred in a Biotage "Initiator" microwave at 220°C for 3 hours. The product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (37mg, 0.103mmol, 57% yield). LCMS (Method A): Rt 0.96 minutes; m/z 355 (MH+)
The compound shown in the table was prepared in an analogous manner to that for trans- 4-({6-[(6-methyl-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol by reacting trans-4-aminocyclohexanol with 6-ethyl-N-(6-fluoro-2-pyridinyl)-1 ,3-benzothiazol-2-amine:
Purification
Example Structure Name Analytical Data
Method trans-4-({6-[(6- ethyl-1 ,3-
LCMS (Method A):
benzothiazol-2- MDAP formic
2 Rt 0.83 minutes;
yl)amino]-2- acid modifier m/z 369 (MH+)
pyridinyl}amino)cycl
ohexanol
Example 3:
trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol

Under an atmosphere of nitrogen, a mixture of 6-bromo-N-(6-chloro-2-pyridinyl)-1 ,3- benzothiazol-2-amine [intermediate 2] (3g, 8.81 mmol) and trans-4-aminocyclohexanol (9g, 78mmol) was treated with ethylene glycol (2ml_) and then heated to 195°C for 18 hours. The cooled mixture was partitioned between dichloromethane (+5% methanol) (50ml_) and water (50ml_). Saturated aqueous sodium bicarbonate (10ml_) was added and the mixture then separated. The organic fraction was evaporated to dryness and the product was purified by flash chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in dichloromethane to afford the title compound (2.14g, 5.10mmol, 58% yield). LCMS (Method A): Rt 0.94 minutes; m/z 419,421 (MH+) Example 4:
N-(trans-4-aminocyclohexyl)-N'-(6-bromo-1 ,3-benzothiazol-2-yl)-2,6-pyridinediamine

A solvent-free mixture of 6-bromo-N-(6-fluoro-2-pyridinyl)-1 ,3-benzothiazol-2-amine [intermediate 5] (410mg, 1.27mmol) and trans-1 ,4-cyclohexanediamine (722mg, 6.32mmol) was sealed and heated in a Biotage "Initiator" microwave at 170°C for 3 hours. The reaction mixture was then partitioned between dichloromethane and saturated aqueous sodium carbonate; the organic phase was dried over magnesium sulfate, filtered
and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 30% methanol (+1 % triethylamine) in dichloromethane to afford the title compound (444mg, 1 .06mmol, 84% yield). LCMS (Method A): Rt 0.75 minutes; m/z 418,420 (MH+)
Example 5:
W-[frans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexyl]acetamide

An ice-cooled solution of N-(trans-4-aminocyclohexyl)-N'-(6-bromo-1 ,3-benzothiazol-2-yl)- 2,6-pyridinediamine [example 4] (100mg, 0.24mmol) in tetrahydrofuran (10ml_) was treated with triethylamine (0.167ml_, 1 .20mmol) and stirred for 10 minutes. Acetyl chloride (18.8mg, 0.24mmol) was added and the reaction mixture stirred for a further 1 hour. The mixture was then treated with saturated aqueous sodium bicarbonate and left to stir for 15 minutes. The organic phase was then collected, evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (12mg, 0.03mmol, 1 1 % yield). LCMS (Method A): Rt 0.97 minutes; m/z 460,462 (MH+). Example 6:
frans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4-b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol

A mixture of 5-chloro-N-(6-fluoro-2-pyridinyl)[1 ,3]thiazolo[5,4-b]pyridin-2-amine [intermediate 4] (3.45g, 12.3mmol) and trans-4-aminocyclohexanol (14.1g, 123mmol) was heated at 190°C for 90 minutes. Ethyl acetate (10mL) and dichloromethane (40mL) were added to the cooled mixture which was then washed with water (60mL). The organic phase was dried over magnesium sulfate, filtered and evaporated to dryness. The product was purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in dichloromethane followed by 0 to 20% methanol in dichloromethane to afford
the title compound (2.1g, 5.59mmol, 46% yield). LCMS (Method A): Rt 0.89 minutes; m/z 376 (MH+)
Example 7:
trans-4-({6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol

Under an atmosphere of nitrogen, a mixture of N2-(6-chloro-2-pyridinyl)-1 ,3- benzothiazole-2,6-diamine [intermediate 15] (100mg, 0.36mmol) and trans-4- aminocyclohexanol (499mg, 4.3mmol) in ethylene glycol (0.5ml_) was heated at 200°C overnight. The cooled reaction mixture was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (38mg, 0.1 1 mmol, 30% yield). LCMS (Method A): Rt 0.56 minutes; m/z 356 (MH+).
Example 8:
trans-4-[(6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol

A mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 3] (162mg, 0.39mmol), 1 ,1-dimethylethyl 4- (4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole-1-carboxylate (250mg, 0.85mmol), tetrakis(triphenylphosphine)palladium(0) (22.8mg, 0.02mmol) and caesium carbonate (335mg, 1 .028mmol) in 1 ,4-dioxane (2.4mL) and water (0.6mL) was sealed and heated in a Biotage "Initiator" microwave at 140°C for 1 hour. Additional 1 ,1 -dimethylethyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole-1 -carboxylate (200mg) and tetrakis(triphenylphosphine)palladium(0) (10mg) were added and the mixture was heated at 150°C for a further 75 minutes. The reaction mixture was then added to a mixture of dichloromethane (+10% methanol) (50mL) and water (30mL). The organic phase was collected, evaporated to dryness and the residue was subjected to purification by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (69mg, 0.17mmol, 44% yield). LCMS (Method A): Rt 0.69 minutes; m/z 407 (MH+)
Example 9:
frans-4-[(6-{[5-(1H-pyrazol-4-yl)[1 ,3]thiazolo[5,4-b]pyridin-2-yl]amino}-2- pyridinyl)amino]cyclohexanol

A mixture of trans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4-b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 6] (100mg, 0.27mmol), 1 ,1-dimethylethyl 4- (4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole-1-carboxylate (157mg, 0.53mmol), potassium phosphate (85mg, 0.40mmol) and tetrakis(triphenylphosphine)- palladium(O) (61.5mg, 0.053mmol) in 1 ,4-dioxane (1 .5mL) and water (0.5ml_) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 2 hours. The reaction mixture was then partitioned between ethyl acetate (25ml_) and water (25ml_). The aqueous phase was re-extracted with ethyl acetate (25ml_) and the combined organic fractions were dried over magnesium sulfate, filtered and evaporated to dryness. The residue was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (28mg, 0.07mmol, 26% yield). LCMS (Method A): Rt 0.68 minutes; m/z 408 (MH+)
Example 10 and Example 11 :
trans-4-[(6-{[6-(4-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol and trans-4-{[6-(1 ,3-benzothiazol-2-yl pyridinyl]amino}cyclohexanol

A mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 3] (100mg, 0.24mmol), caesium carbonate (207mg, 0.63mmol), tetrakis(triphenylphosphine)palladium(0) (14.1 mg, 0.012mmol) and 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridine (35.2mg, 0.286mmol) in a mixture of 1 ,4-dioxane (2.4ml_) and water (0.6ml_) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 30 minutes. The cooled reaction mixture was evaporated to dryness and the residue was subjected to purification by mass-directed automated
preparative HPLC (formic acid modifier) to afford the title compounds. Example 10: (16.5mg, 0.04mmol, 17% yield) LCMS (Method B): Rt 0.65 minutes; m/z 418 (MH+). Example 1 1 : (8.7mg, 0.026mmol, 1 1 % yield) LCMS (Method B): Rt 0.74 minutes; m/z 341 (MH+).
Example 12:
trans-4-[(6-{[6-(3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol
 A mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 3] (100mg, 0.24mmol), caesium carbonate (207mg, 0.63mmol), tetrakis(triphenylphosphine)palladium(0) (14.05mg, 0.012mmol) and
A mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 3] (100mg, 0.24mmol), caesium carbonate (207mg, 0.63mmol), tetrakis(triphenylphosphine)palladium(0) (14.05mg, 0.012mmol) and
3- pyridinylboronic acid (35.2mg, 0.29mmol) in a mixture of 1 ,4-dioxane (2.4mL) and water (0.6mL) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 30 minutes. The cooled reaction mixture was evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (49mg, 0.12mmol, 49%) LCMS (Method B): Rt 0.67 minutes; m/z 418 (MH+) The compound shown in the table was prepared in an analogous manner to that for trans-
4- [(6-{[6-(3-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol by reacting trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 3] with 2-(3-furanyl)-4,4,5,5-tetramethyl-1 ,3,2- dioxaborolane:
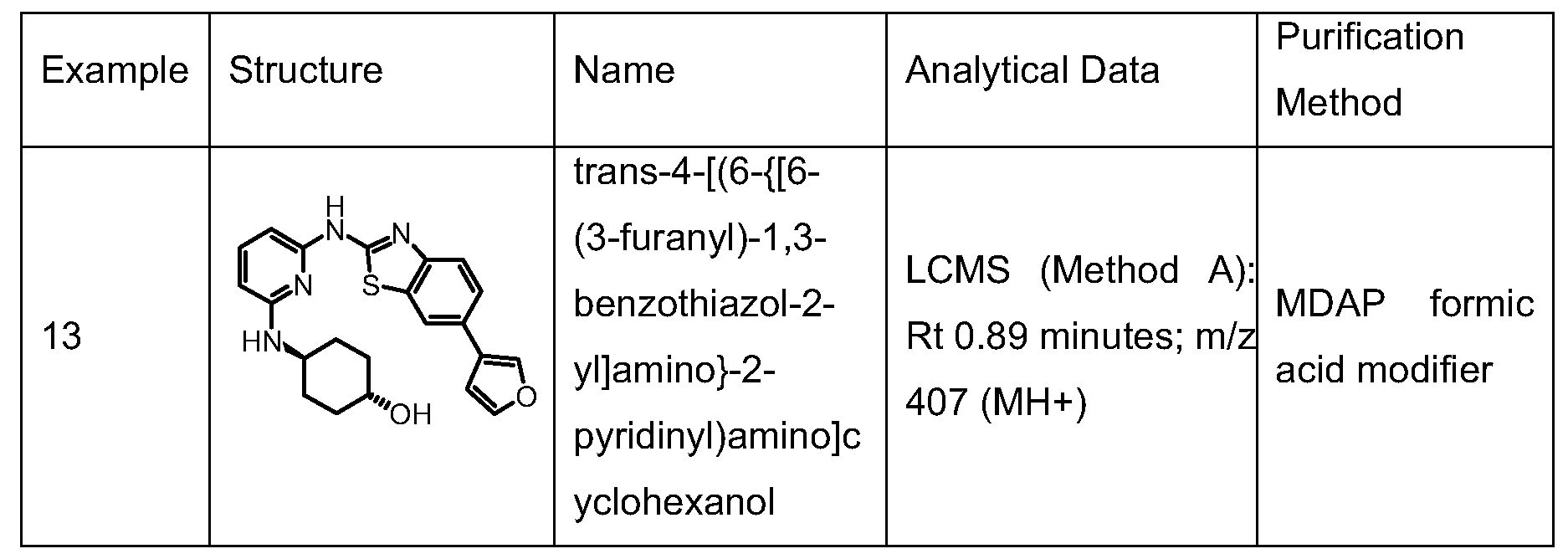
trans-4-[(6-{[6-(2-pyridinyl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol

A mixture of trans-4-[(6-{[6-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 ,3-benzothiazol- 2-yl]amino}-2-pyridinyl)amino]cyclohexanol [intermediate 6] (100mg, 0.21 mmol), 2- bromopyridine (37.3mg, 0.24mmol), tetrakis(triphenylphosphine)palladium(0) (25mg, 0.021 mmol) and caesium carbonate (210mg, 0.64mmol) in a mixture of N,N- dimethylformamide (2mL) and water (0.67ml_) was heated in a sealed tube in a Biotage "Initiator" microwave at 140°C for 20 minutes. The reaction mixture was poured into a mixture of dichloromethane (20ml_) and water (20ml_). The organic phase was evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (45mg, 0.1 1 mmol, 50% yield). LCMS (Method B): Rt 0.76 minutes; m/z 418 (MH+)
Example 15:
frans-4-({6-[(6-phenyl-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol trifluoroacetate (salt)

A mixture of frans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 3] (42mg, 0.1 mmol) and phenylboronic acid (25mg, 0.2mmol) in 1 ,4-dioxane (0.8ml_) was treated with 2'-(dimethylamiono)-2-biphenyl- palladium(ii) chloride dinorbornylphosphine complex (2.2mg, 0.004mmol) and a solution of potassium phosphate (32mg, 0.15mmol) in water (0.2ml_). The reaction mixture was sealed and heated in CEM "Discover" microwave at 1 10°C for 20 minutes. The reaction mixture was then loaded onto a C18 solid-phase extraction cartridge (pre-conditioned with acetonitrile/0.1 %TFA), the column was flushed through with a further 3ml_ of acetonitrile/0.1 %TFA. The recovered crude product was subjected to purification by mass- directed automated preparative HPLC (trifluoroacetic acid modifier) to afford the title
compound (9.2mg, 0.022mmol, 22% yield). LCMS (Method A): Rt 0.99 minutes; m/z 417 (MH+).
The compounds shown in the table was prepared in an analogous manner to that for frans-4-({6-[(6-phenyl-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol trifluoroacetate (salt) by reacting frans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol with the appropriate boronic acid or boronic ester:




Example 32:
yV-(1 ,1 -dimethylethyl)-5-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)[1 ,3]thiazolo[5,4-jb]pyridin-5-yl]-3-pyridinecarboxamide trifluoroacetate (salt)

A mixture of frans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4-/b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 6] (38mg, O.l mmol) and (5-{[(1 ,1- dimethylethyl)amino]carbonyl}-3-pyridinyl)boronic acid (44mg, 0.2mmol) in 1 ,4-dioxane (0.8mL) was treated with 2'-(dimethylamiono)-2-biphenyl-palladium(ii) chloride dinorbornylphosphine complex (2.2mg, 0.004mmol) and a solution of potassium phosphate (32mg, 0.15mmol) in water (0.2mL). The reaction mixture was sealed and heated in CEM "Discover" microwave at 1 10°C for 20 minutes. The reaction mixture was then loaded onto a C18 solid-phase extraction cartridge (pre-conditioned with acetonitrile/0.1 %TFA); the column was then flushed through with a further 3ml_ acetonitrile/0.1 %TFA. The product was subjected to purification by mass-directed automated preparative HPLC (trifluoroacetic acid modifier) to afford the title compound (14mg, 0.027mmol, 27% yield). LCMS (Method A): Rt 0.89 minutes; m/z 518 (MH+). The compounds shown in the table was prepared in an analogous manner to that for N- (1 ,1-dimethylethyl)-5-[2-({6-[(trans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)[1 ,3]thiazolo[5,4-b]pyridin-5-yl]-3-pyridinecarboxamide trifluoroacetate (salt) by reacting frans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4-/b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 6] with the appropriate boronic acid or boronic ester:

hexanol
trifluoroacetate (salt)
formic acid - trans-4- {[6-({5-[5-(4- morpholinylcarbonyl)- MDAP TFA 3- LCMS (Method A): modifier,
35
pyridinyl][1 ,3]thiazolo[ Rt 0.74 minutes; then MDAP 5,4-b]pyridin-2- m/z 532 (MH+) formic acid yl}amino)-2- modifier pyridinyl]amino}cyclo
hexanol (1 :1 )
formic acid - 5-[2-({6- [(trans-4- hydroxycyclohexyl)a MDAP TFA mino]-2- LCMS (Method A): modifier,
36
pyridinyl}amino)[1 ,3]t Rt 0.88 minutes; then MDAP hiazolo[5,4-b]pyridin- m/z 444 (MH+) formic acid 5-yl]-3- modifier pyridinecarbonitrile
(1 :1 )
Example 37:
formic acid - frans-4-{[6-({6-[5-(phenyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl} 2-pyridinyl]amino}cyclohexanol (1 :1)

Under an atmosphere of nitrogen, a mixture of frans-4-[(6-{[6-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol
[intermediate 6] (56mg, 0.12mmol), 3-bromo-5-(phenyloxy)pyridine (25mg, O.l mmol). 2'- (dimethylamiono)-2-biphenyl-palladium(ii) chloride dinorbornylphosphine complex (2.2mg, 0.004mmol) and potassium phosphate (21 mg, O.l mmol) in 1 ,4-dioxane (0.8ml_) and water (0.2ml_) was heated to 100°C for 18 hours. The reaction mixture was then loaded onto a C18 solid-phase extraction cartridge (pre-conditioned with acetonitrile/0.1 %TFA), the
column was flushed through with a further 3ml_ acetonitrile/0.1 %TFA. The recovered crude product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (19mg, 0.037mmol, 26% yield). LCMS (Method A): Rt 1 .03minutes; m/z 510 (MH+).
The compounds shown in the table were prepared in an analogous manner to that for formic acid - frans-4-{[6-({6-[5-(phenyloxy)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol (1 :1 ) by reacting frans-4-[(6-{[6-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol
[intermediate 6] with the appropriate aryl bromide:



Example 49:
frans-4-{[6-({6-[5-(hydroxymethyl)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}
pyridinyl]amino}cyclohexanol

A mixture of (5-bromo-3-pyridinyl)methanol (40.3mg, 0.214mmol), trans-4-[(6-{[6-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol [intermediate 6] (100mg, 0.21 mmol), tetrakis(triphenylphosphine)palladium(0) (74.3mg, 0.06mmol), caesium carbonate
(210mg, 0.64mmol) in 1 ,4-dioxane (2mL) and water (0.5ml_) was heated in a sealed tube in a Biotage "Initiator" microwave at 130°C for 30 minutes. The reaction mixture was partitioned between water (20ml_) and ethyl acetate (20ml_). The aqueous phase was extracted with further ethyl acetate (20ml_) and the combined ethyl acetate extracts were evaporated to dryness. The product was purified by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (23mg, 0.05mmol, 24% yield). LCMS (Method B): Rt 2.18minutes; m/z 448 (MH+).
The compounds shown in the table was prepared in an analogous manner to that for frans-4-{[6-({6-[5-(hydroxymethyl)-3-pyridinyl]-1 ,3-benzothiazol-2-yl}amino)-2- pyridinyl]amino}cyclohexanol by reacting trans-4-[(6-{[6-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 ,3-benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol
[intermediate 6] with the appropriate aryl bromide:

hexanol
trans-4-({6-[(6-{5-[(2- pyridinylmethyl)oxy]-
MDAP
3-pyridinyl}-1 ,3- LCMS (Method A):
ammonium
53 benzothiazol-2- Rt 0.77 minutes;
bicarbonate yl)amino]-2- m/z 525 (MH+)
modifier pyridinyl}amino)cyclo
hexanol
trans-4-[(6-{[6-(5-{[(3- fluorophenyl)methyl]o
MDAP
xy}-3-pyridinyl)-1 ,3- LCMS (Method A):
ammonium
54 benzothiazol-2- Rt 0.79 minutes;
bicarbonate yl]amino}-2- m/z 542 (MH+)
modifier pyridinyl)amino]cyclo
hexanol
trans-4-({6-[(6-{5-[(2- hydroxyethyl)oxy]-3-
MDAP
pyridinyl}-1 ,3- LCMS (Method A):
ammonium
55 benzothiazol-2- Rt 0.66 minutes;
bicarbonate yl)amino]-2- m/z 478 (MH+)
modifier pyridinyl}amino)cyclo
hexanol
Example 56:
frans-4-[(6-{[6-(1H^yrrolo[3,2-b]pyridin-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol

A mixture of 6-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrrolo[3,2-b]pyridine (58.2mg, 0.238mmol), trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 3] (100mg, 0.24mmol), tetrakis(triphenylphosphine)-palladium(0) (83mg, 0.072mmol) and caesium carbonate (233mg, 0.72mmol) in 1 ,4-dioxane (2mL) and water (0.5mL) was sealed and heated in a
Biotage "Initiator" microwave at 130°C for 30 minutes. The reaction mixture was partitioned between water (20ml_) and ethyl acetate (20ml_). The aqueous phase was extracted with further ethyl acetate (20ml_) and the combined ethyl acetate extracts were evaporated to dryness. The residue was subjected to purification by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (16mg, 0.035mmol, 15% yield). LCMS (Method B): Rt 1.47minutes; m/z 457 (MH+).
The compounds shown in the table were prepared in an analogous manner to that for frans-4-[(6-{[6-(1 H-pyrrolo[3,2-b]pyridin-6-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol by reacting trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-2-pyridinyl}amino)cyclohexanol [example 3] with the appropriate boronic acid or boronic ester:

4-[2-({6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6- yl]-3-morpholinone

3-[2-({6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6- -1 ,3-oxazolidin-2-one

Under an atmosphere of nitrogen, a mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-2-pyridinyl}amino)cyclohexanol [example 3] (49mg, 0.12mmol), 1 ,3-oxazolidin- 2-one (30.5mg, 0.35mmol), caesium carbonate (76mg, 0.23mmol) and copper(l) iodide (44.5mg, 0.23mmol) in dry Ν,Ν-dimethylformamide (3mL) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with Ν,Ν'-dimethylethylenediamine (0.050ml_, 0.47mmol) and the mixture was heated at 1 10°C for 4 hours. The mixture was cooled to ambient temperature, filtered and evaporated to dryness. The residue was taken up in DMSO (1 ml_) and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (6.9mg, 0.016mmol, 14% yield) as an off-white solid. LCMS (Method A): Rt 0.72 minutes; m/z 426 (MH+)
Example 61 :
1 -[2-({6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6- yl]-3-methyl-2-imidazolidinone

Under an atmosphere of nitrogen, a mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-2-pyridinyl}amino)cyclohexanol [example 3] (100mg, 0.24mmol), 1-methyl-2- imidazolidinone (71 .6mg, 0.715mmol), caesium carbonate (233mg, 0.72mmol) and copper(l) iodide (136mg, 0.72mmol) in dry Ν,Ν-dimethylformamide (3ml_) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with Ν,Ν'-dimethylethylenediamine (0.102ml_, 0.95mmol) and the mixture was heated at 1 10 °C for 1 hour. The mixture was cooled to ambient temperature, filtered and evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford 1-[2-({6-[(trans-4- hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-methyl-2- imidazolidinone (33mg, 0.075mmol, 32% yield). LCMS (Method A): Rt 0.71 minutes; m/z 439 (MH+) The compounds shown in the table was prepared in an analogous manner to that for 1-[2- ({6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-methyl- 2-imidazolidinone by reacting trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol with the appropriate lactam or cyclic urea:

(4S)-4-hydroxy-1 -[2- ({6-[(trans-4-
LCMS (Method
hydroxycyclohexyl)a
A): Rt 0.61 MDAP formic
64 mino]-2-
HNV\ minutes; m/z 440 acid modifier
OH pyridinyl}amino)-1 ,3- (MH+)
benzothiazol-6-yl]-2- pyrrolidinone
1 -[2-({6-[(trans-4- hydroxycyclohexyl)a
LCMS (Method
mino]-2-
HN — A): Rt 0.64 MDAP formic
65 pyridinyl}amino)-1 ,3- minutes; m/z 469 acid modifier
OH benzothiazol-6-yl]-3- (MH+)
OH (2-hydroxyethyl)-2- imidazolidinone
Example 66:
trans-4-{[6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-4-(phenylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of N2-[6-chloro-4-(phenylmethyl)-2-pyridinyl]-1 ,3-benzothiazole-2,6-diamine [intermediate 18] (340mg, 0.93mmol), trans-4-aminocyclohexanol (1 .3 g, 1 1.3mmol) and ethylene glycol (0.5ml_) was heated at 190°C overnight. The cooled mixture was partitioned between 5% methanol in dichloromethane (40ml_) and water (40ml_). The organic fraction was evaporated to dryness and the product was purified by flash chromatography on silica using a gradient elution from 0% to 100% ethyl acetate in isohexane followed by 5% methanol in dichloromethane to afford the title compound (145mg, 0.33mmol, 35% yield) LCMS (Method A): Rt 0.55 minutes; m/z 446 (MH+) Example 67:
trans-4-{[6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2-pyridinyl]amino}cyclohexanol

A mixture of trans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4-b]pyridin-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [intermediate 16] (50mg, 0.13mmol), ammonium formate (71 .3mg, 1 .13mmol) and palladium (10% by weight on activated carbon) (28.3mg, 0.27mmol) in methanol (5ml_) was sealed and heated in a Biotage "Initiator" microwave at 130°C for a total of 15 hours. The mixture was filtered through celite, the filtrate was evaporated to dryness and the residue was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (3.1 mg, 0.009mmol, 7% yield). LCMS (Method A): Rt 0.71 minutes; m/z 342 (MH+).
Example 68:
3-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)[1,3]thiazolo[5,4- b]pyridin-5-yl]-1,3-oxazolidin-2-one
 Under an atmosphere of nitrogen, a mixture of trans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4- b]pyridin-2-yl)amino]-2-pyridinyl}amino)cyclohexanol [example 6] (150mg, 0.399mmol),1 ,3-oxazolidin-2-one (104mg, 1 .20mmol), caesium carbonate (390mg, 1.20mmol) and copper(l) iodide (228mg, 1 .20mmol) in dry Ν,Ν-dimethylformamide (3ml_) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with Ν,Ν'-dimethyl ethylenediamine (0.170ml_, 1 .60mmol) and the mixture was heated at 1 10°C for 1 hour. The cooled mixture was evaporated to dryness. The residue was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (33mg, 0.077mmol, 19% yield). LCMS (Method A): Rt 0.74 minutes; m/z 427 (MH+).
Under an atmosphere of nitrogen, a mixture of trans-4-({6-[(5-chloro[1 ,3]thiazolo[5,4- b]pyridin-2-yl)amino]-2-pyridinyl}amino)cyclohexanol [example 6] (150mg, 0.399mmol),1 ,3-oxazolidin-2-one (104mg, 1 .20mmol), caesium carbonate (390mg, 1.20mmol) and copper(l) iodide (228mg, 1 .20mmol) in dry Ν,Ν-dimethylformamide (3ml_) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with Ν,Ν'-dimethyl ethylenediamine (0.170ml_, 1 .60mmol) and the mixture was heated at 1 10°C for 1 hour. The cooled mixture was evaporated to dryness. The residue was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (33mg, 0.077mmol, 19% yield). LCMS (Method A): Rt 0.74 minutes; m/z 427 (MH+).
Example 69:
4-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1,3-benzothiazol-6- yl]-5-methyl-2,4-dihydro-3H-1,2,4-triazol-3-one

A mixture of trans-4-({6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 7] (70mg, 0.20mmol) and methyl (2Z)-2-[1- (ethyloxy)ethylidene]hydrazinecarboxylate (31 .5mg, 0.20mmol) in acetonitrile (2ml_) was sealed and heated in a Biotage "Initiator" microwave at 200°C for 6 hours. The reaction mixture was evaporated to dryness and the residue was subjected to purification by mass directed automated preparative HPLC (formic acid modifier) to afford the title compound (5mg, 0.01 1 mmol, 6% yield). LCMS (Method A): Rt 0.66 minutes; m/z 439 (MH+). Example 70:
1 -[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6- yl]-2,5-pyrrolidinedione

A mixture of trans-4-({6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol [example 7] (50mg, 0.14mmol) and succinic anhydride (14.08mg, 0.14mmol) in acetonitrile (2ml_) was treated with aqueous hydrochloric acid (2M, 2 drops) and then sealed and heated in a Biotage "Initiator" microwave at 150°C for 6 hours. The reaction mixture was evaporated to dryness and the residue was subjected to purification by mass directed automated preparative HPLC (formic acid modifier) to afford the title compound (1 1 mg, 0.025mmol, 18% yield). LCMS (Method A): Rt 0.67 minutes; m/z 439 (MH+).
Example 71 :
4-[2-({6-[(frans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6- yl]-2,4-dihydro-3H-1 ,2,4-triazol-3-one

frans-4-[(4-(phenylmethyl)-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol

A mixture of trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(phenylmethyl)-2- pyridinyl]amino}cyclohexanol [intermediate 20] (68mg, 0.13mmol), tetrakis(triphenylphosphine)-palladium(0) (46.3mg, 0.04mmol), caesium carbonate (130mg, 0.40mmol) and 1 ,1-dimethylethyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)- 1 H-pyrazole-1-carboxylate (39.3mg, 0.13mmol) in a mixture of DMF (4mL) and water (1.3mL) was sealed and heated in a Biotage "Initiator" microwave at 130°C for 30 minutes. The reaction mixture was added to water (50mL). After stirring for 15 minutes, the mixture was filtered and the solid washed with DCM and dried. The residue was then subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (18mg, 0.036mmol, 27% yield). LCMS (Method A): Rt 0.91 minutes; m/z 497 (MH+)
Example 73:
trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(2-pyridinylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of 6-bromo-N-[6-fluoro-4-(2-pyridinylmethyl)-2-pyridinyl]-1 ,3-benzothiazol-2- amine [intermediate 25] (589mg, 1.42mmol) and trans-4-aminocyclohexanol (1.96g, 17.0mmol) in ethylene glycol (0.5ml_) was heated at 200°C overnight. The cooled reaction mixture was taken up in methanol, treated with silica (8g) and evaporated to dryness. The product was then purified by chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in cyclohexane, then from 0-20% methanol in dichloromethane to afford the title compound (277mg, 0.54mmol, 38% yield). LCMS (Method A): Rt 0.83 minutes; m/z 510,512 (MH+)
Example 74:
frans-4-{[6-{[6-(1H^yrazol-4-yl)-1 ,3^enzothiazol-2-yl]amino}-4-(2^yridinylmethyl)- -pyridinyl]amino}cyclohexanol

A mixture of 1 ,1-dimethylethyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole-1-carboxylate (101 mg, 0.343mmol), trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-(2-pyridinylmethyl)-2-pyridinyl]amino}cyclohexanol [example 73] (70mg, 0.137mmol) and caesium carbonate (134mg, 0.41 1 mmol), tetrakis(triphenylphosphine)- palladium(O) (47.5mg, 0.04mmol) in 1 ,4-dioxane (10mL) and water (1 ml_) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 30 minutes. The cooled reaction mixture was taken up in water (20ml_) and ethyl acetate (20ml_) and filtered. The filtrate was separated and the aqueous phase was extracted with ethyl acetate (50ml_). The organic phases were combined and evaporated to dryness. The residue was then subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (10mg, 0.020mmol, 15% yield). LCMS (Method A): Rt 0.63 minutes; m/z 498 (MH+)
Example 75:
1-(2-{[6-[(trans-4-hydroxycyclohexyl)amino]-4-(phenylmethyl)-2^yridinyl]amino}- -benzothiazol-6-yl)-2,5-pyrrolidinedione
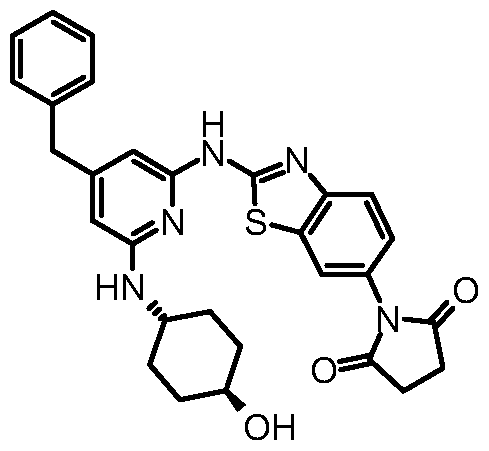
A mixture of trans-4-{[6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-4-(phenylmethyl)-2- pyridinyl]amino}cyclohexanol [example 76] (65mg, 0.15mmol) and succinic anhydride (60mg, 0.60mmol) in acetonitrile (2mL) was treated with aqueous hydrochloric acid (2M, 2 drops), sealed and heated in a Biotage "Initiator" microwave at 150°C for 1 hour. The mixture was evaporated to dryness and the product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (38mg, 0.072mmol, 49% yield). LCMS (Method A): Rt 0.93 minutes; m/z 528 (MH+).
Example 76:
frans-4-{[6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-4-(phenylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of N2-[6-chloro-4-(phenylmethyl)-2-pyridinyl]-1 ,3-benzothiazole-2,6-diamine [intermediate 18] (340mg, 0.93mmol), trans-4-aminocyclohexanol (1 .3g, 1 1.3mmol) and ethylene glycol (0.5ml_) was heated at 190°C overnight. The cooled mixture was partitioned between 5% methanol in dichloromethane (40ml_) and water (40ml_). The organic fraction was evaporated to dryness and the product was purified by flash chromatography on silica using a gradient elution from 0 to 100% ethyl acetate in isohexane followed by 5% methanol in dichloromethane to afford the title compound (145mg, 0.33mmol, 35% yield). LCMS (Method A): Rt 0.80 minutes; m/z 446 (MH+).
Example 77:
trans-4-{[6-{[6-(3^yridinyl)-1,3-benzothiazol-2-yl]amino}-4-(2-pyridinylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of 3-pyridineboronic acid (6.26mg, 0.051 mmol), trans-4-{[6-[(6-bromo-1 ,3- benzothiazol-2-yl)amino]-4-(2-pyridinylmethyl)-2-pyridinyl]amino}cyclohexanol [example 73] (26mg, 0.05mmol), caesium carbonate (49.8mg, 0.15mmol) and tetrakis(triphenylphosphine)-palladium(0) (17.7mg, 0.015mmol) in N,N- dimethylformamide (2mL) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 30 minutes. The cooled reaction mixture was taken up in water (20ml_) and ethyl acetate (20ml_) and filtered. The filtrate was separated and the aqueous phase extracted twice with ethyl acetate (2 x 50ml_). The organic phase was combined and evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (5.1 mg, 0.01 mmol, 20% yield). LCMS (Method A): Rt 0.76 minutes; m/z 510 (MH+).
Example 78:
trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of 6-bromo-N-[6-chloro-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]-1 ,3-benzothiazol- 2-amine [intermediate 28] (0.82g, 1 .95mmol), trans-4-aminocyclohexanol (1 .6g, 13.9mmol) and ethylene glycol (1 ml_) was heated at 195°C under an atmosphere of nitrogen for 18 hours. The cooled mixture was partitioned between dichloromethane (with 5% methanol) (60ml_), water (30ml_) and saturated aqueous sodium bicarbonate (30ml_). The insoluble product was filtered off, washed with water, then with diethyl ether, and dried to afford the title compound (467mg, 0.94mmol, 48% yield). LCMS (Method A): Rt 0.96 minutes; m/z 499,501 (MH+)
Example 79:
trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-[(2-methyl-1 H-imidazol-1 - yl)methyl]-2-pyridinyl}amino)cyclohexanol

A mixture of 6-bromo-N-{6-chloro-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2-pyridinyl}-1 , 3- benzothiazol-2-amine [intermediate 32] (1.3g, 2.99mmol), trans-4-aminocyclohexanol (2.76g, 23.9mmol) and ethylene glycol (1 .5ml_) was heated at 195°C under an atmosphere of nitrogen for 18 hours. The cooled mixture was partitioned between dichloromethane (with 5% methanol) (60ml_), water (30ml_) and saturated aqueous sodium bicarbonate (30ml_). The insoluble product was filtered off, washed with water, then with diethyl ether, and dried to afford the title compound (1.1 g, 2.14mmol, 72% yield). LCMS (Method A): Rt 0.75 minutes; m/z 513,515 (MH+)
Example 80:
trans-4-{[4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-2-pyridinyl]amino}cyclohexanol methanesulfonate (salt)

A mixture of N-{6-chloro-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}[1 ,3]thiazolo[5,4-b]pyridin-2-amine [intermediate 33] (170mg, 0.48mmol) and trans-4-aminocyclohexanol (400mg, 3.47mmol) in ethylene glycol (1 ml_) was sealed and heated and stirred in a Biotage "Initiator" microwave at 200°C for 2 hours. Water (5ml_) was added and the product was extracted with chloroform (+10% methanol) (3 x 20ml_). The combined organics were evaporated to dryness and the product was subjected to purification by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford trans-4-{[4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-([1 ,3]thiazolo[5,4- b]pyridin-2-ylamino)-2-pyridinyl]amino}cyclohexanol. The product was then dissolved in methanol (5ml_) and treated with 1 molar equivalent of methanesulfonic acid. The mixture was then evaporated to dryness, then triturated with diethyl ether, filtered and dried to
afford the title compound (1 10mg, 0.21 mmol, 43% yield). LCMS (Method A): Rt 0.57 minutes; m/z 436 (MH+)
Example 81 :
trans-4-{[6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-4-(1 H-pyrazol-1 - ylmethyl)-2-pyridinyl]amino}cyclohexanol

A mixture of trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(1 H-pyrazol-1 -ylmethyl)- 2-pyridinyl]amino}cyclohexanol [example 78] (101 mg, 0.20mmol), caesium carbonate (197mg, 0.606mmol), tetrakis(triphenylphosphine)palladium(0) (70mg, 0.06mmol) and
1 ,1-dimethylethyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole-1- carboxylate (143mg, 0.49mmol) in a mixture of DMF (4ml_) and water (1 .3ml_) was sealed and heated in a Biotage "Initiator" microwave at 130°C for 30 minutes. The reaction mixture was added to a mixture of dichloromethane (50ml_) and water (50ml_). After stirring for 15 minutes, the mixture was filtered and the solid washed with water and dried. The solid was dissolved in DMSO (2ml_) and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (12.7mg, 0.026mmol, 13% yield). LCMS (Method A): Rt 0.73 minutes; m/z 487 (MH+)
The compound shown in the table was prepared in an analogous manner to that for trans- 4-{[6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-4-(1 H-pyrazol-1 -ylmethyl)-2- pyridinyl]amino}cyclohexanol by reacting trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]amino}cyclohexanol [example 78] with 3- pyridinylboronic acid:

hexanol
Example 83:
trans-4-[(4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-{[6-(1 H-pyrazol-4-yl)-1 ,3- benzothiazol-2-yl]amino}-2-pyridinyl)amino]cyclohexanol

A mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-[(2-methyl-1 H-imidazol-
1-yl)methyl]-2-pyridinyl}amino)cyclohexanol [example 79] (100mg, 0.20mmol), tetrakis(triphenylphosphine)palladium(0) (67.5mg, 0.06mmol), caesium carbonate
(190mg, 0.58mmol) and 1 ,1-dimethylethyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)- 1 H-pyrazole-1-carboxylate (143mg, 0.49mmol) in a mixture of DMF (4ml_) and water (1.3ml_) was sealed and heated in a Biotage "Initiator" microwave at 130°C for 30 minutes. The reaction mixture was added to a mixture of dichloromethane (50ml_) and water (50ml_). After stirring for 15 minutes, the mixture was filtered and the recovered solid was washed with water and dried. The solid was dissolved in DMSO (2ml_) and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (98mg, 0.20mmol, 79% yield). LCMS (Method A): Rt 0.58 minutes; m/z 501 (MH+)
The compound shown in the table was prepared in an analogous manner to that for trans- 4-[(4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2- yl]amino}-2-pyridinyl)amino]cyclohexanol by reacting trans-4-({6-[(6-bromo-1 ,3- benzothiazol-2-yl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)cyclohexanol [example 79] with 3-pyridinylboronic acid:

benzothiazol-2- yl]amino}-2- pyridinyl)amino]cyclo
hexanol
Example 85:
5-(2-{[6-[(trans-4-hydroxycyclohexyl)amino]-4-(1H-pyrazol-1 -ylmethyl)-2- pyridinyl]amino}-1,3-benzothiazol-6-yl)-3-pyridinecarbonitrile

A mixture of trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(1 H-pyrazol-1 -ylmethyl)- 2-pyridinyl]amino}cyclohexanol [example 78] (100mg, 0.20mmol), tetrakis(triphenylphosphine)palladium(0) (34.7mg, 0.03mmol), potassium phosphate (85mg, 0.40mmol) and 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinecarbonitrile (83, 0.36mmol) in a mixture of 1 ,4-dioxane (4mL) and water (1.3mL) was sealed and heated in a Biotage "Initiator" microwave at 130°C for 30 minutes. The reaction mixture was added to a mixture of dichloromethane (50ml_) and water (50ml_). After stirring for 15 minutes, the mixture was filtered and the filtered solid washed with water and dried. The solid was dissolved in DMSO (2ml_) and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (57mg, 0.1 1 mmol, 55% yield). LCMS (Method B): Rt 0.88 minutes; m/z 523 (MH+).
The compound shown in the table was prepared in an analogous manner to that for 5-(2- {[6-[(trans-4-hydroxycyclohexyl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]amino}-1 ,3- benzothiazol-6-yl)-3-pyridinecarbonitrile by reacting A/-(frans-4-aminocyclohexyl)-A/'-(6- bromo-1 ,3-benzothiazol-2-yl)-4-(1 H-pyrazol-1 -ylmethyl)-2,6-pyridinediamine [intermediate 51] with 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3-pyridinecarbonitrile:
Analytical Purification
Example Structure Name
Data Method

Example 87:
5-[2-({6-[(trans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)-1,3-benzothiazol-6-yl]-3-pyridinecarbonitrile

A mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-[(2-methyl-1 H-imidazol- 1-yl)methyl]-2-pyridinyl}amino)cyclohexanol [example 79] (100mg, 0.20mmol), tetrakis(triphenylphosphine)palladium(0) (45.0mg, 0.039mmol), potassium phosphate (62.0mg, 0.29mmol) and 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinecarbonitrile (90mg, 0.39mmol) in a mixture of 1 ,4-dioxane (1.5ml_) and water (0.5ml_) was sealed and heated in a Biotage "Initiator" microwave at 100°C for 45 minutes. The reaction mixture was added to a mixture of chloroform (50ml_), methanol (5ml_) and water (50ml_). After stirring for 15 minutes, the mixture was filtered and the filtrate separated. The organic phase was evaporated to dryness, combined with the filtered solid and purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (58.4mg, 0.1 1 mmol, 56% yield). LCMS (Method B): Rt 0.72 minutes; m/z 537 (MH+).
The compound shown in the table was prepared in an analogous manner to that for 5-[2- ({6-[(trans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile by reacting N-(trans-4- aminocyclohexyl)-A/'-(6-bromo-1 ,3-benzothiazol-2-yl)-4-(1 /-/-pyrazol-1-ylmethyl)-2,6-
pyridinediamine [intermediate 52] with 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinecarbonitrile:

3-(2-{[6-[(trans-4-hydroxycyclohexyl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2- pyridinyl]amino}-1 ,3-benzothiazol-6-yl)-1 ,3-oxazolidin-2-one

Under an atmosphere of nitrogen, a mixture of trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-(1 H-pyrazol-1 -ylmethyl)-2-pyridinyl]amino}cyclohexanol [example 78] (1 10mg, 0.22mmol), 1 ,3-oxazolidin-2-one (57.5mg, 0.66mmol), caesium carbonate (215mg, 0.66mmol) and copper(l) iodide (126mg, 0.66mmol) in dry Ν,Ν-dimethylformamide (3mL) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with Ν,Ν'-dimethylethylenediamine (0.094ml_, 0.88mmol) and the mixture was heated at 1 10°C for 1 hour. The mixture was cooled to ambient temperature, filtered and evaporated to dryness. The residue was taken up in DMSO (2ml_) and the product was subjected to purification by mass-directed automated preparative HPLC
(formic acid modifier) to afford the title compound (58mg, 0.12mmol, 52% yield). LCMS (Method A): Rt 0.74 minutes; m/z 506 (MH+)
Example 90:
3-[2-({6-[(trans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one

Under an atmosphere of nitrogen, a mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2-pyridinyl}amino)cyclohexanol [example 79] (150mg, 0.29mmol), 1 ,3-oxazolidin-2-one (76mg, 0.88mmol), caesium carbonate (286mg, 0.88mmol) and copper(l) iodide (167mg, 0.88mmol) in dry N,N- dimethylformamide (3mL) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with N,N'-dimethylethylenediamine (0.125ml_, 1.17mmol) and the mixture was heated at 1 10°C for 1 hour. The mixture was cooled to ambient temperature, filtered and evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (122mg, 0.24mmol, 80% yield). LCMS (Method A): Rt 0.59 minutes; m/z 520 (MH+) Example 91 :
1 -[2-({6-[(trans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-(2-hydroxyethyl)-2-imidazolidinone

Under an atmosphere of nitrogen, a mixture of trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-2-pyridinyl}amino)cyclohexanol [example 79] (100mg, 0.20mmol), 1-(2-hydroxyethyl)-2-imidazolidinone (76mg, 0.58mmol), caesium carbonate (190mg, 0.58mmol) and copper(l) iodide (1 1 1 mg, 0.58mmol) in dry N,N- dimethylformamide (3mL) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with N,N'-dimethylethylenediamine
(0.083ml_, 0.78mmol) and the mixture was heated at 1 10°C for 1 hour. The mixture was cooled to ambient temperature, filtered and evaporated to dryness. The residue was taken up in DMSO (2ml_) and the product was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (45mg, 0.08mmol, 41 % yield). LCMS (Method A): Rt 0.57 minutes; m/z 563 (MH+)
The compounds shown in the table was prepared in an analogous manner to that for 1-[2- ({6-[(trans-4-hydroxycyclohexyl)amino]-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-(2-hydroxyethyl)-2-imidazolidinone by reacting trans-4-({6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-[(2-methyl-1 H-imidazol-1-yl)methyl]- 2-pyridinyl}amino)cyclohexanol [example 79] with the appropriately-substituted lactam or cyclic urea:

1 H-imidazol-1- yl)methyl]-2- pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-2- imidazolidinone
Example 95:
4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-N-[3-(4-morpholinyl)propyl]-N,- ]pyridin-2-yl-2,6-pyridinediamine

A mixture of N-{6-chloro-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-2- pyridinyl}[1 ,3]thiazolo[5,4-b]pyridin-2-amine [intermediate 33] (170mg, 0.48mmol) and [3- (4-morpholinyl)propyl]amine (350mg, 2.42mmol) were sealed and heated in a Biotage "Initiator" microwave at 190°C for 2 hours. Water (50ml_) and ethyl acetate (50ml_) was added and the reaction mixture was stirred for 5 minutes and then sonicated in an ultrasonic bath for 5 minutes. The mixture was then filtered, washed with water and ethyl acetate to afford the title compound (81 mg, 0.17mmol, 37% yield). LCMS (Method A): Rt 0.45 minutes; m/z 465 (MH+). Example 96:
W-(6-bromo-1 ,3-benzothiazol-2-yl)-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-W-[2-(4- morpholinyl)ethyl]-2,6-pyridinediamine

A mixture of 6-bromo-N-{6-chloro-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-2-pyridinyl}-1 ,3- benzothiazol-2-amine [intermediate 32] (60mg, 0.14mmol) and 4-(2- aminoethyl)morpholine (200mg, 1 .54mmol) was sealed and heated in a Biotage "Initiator"
microwave at 170°C for 3.5 hours. The reaction mixture was evaporated to dryness and the residue was subjected to purification by mass directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (43mg, 0.08mmol, 59% yield) LCMS (Method A): Rt 0.79 minutes; m/z 528,530 (MH+).
The compounds shown in the table were prepared in an analogous manner to that for N- (6-bromo-1 ,3-benzothiazol-2-yl)-4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-N'-[2-(4- morpholinyl)ethyl]-2,6-pyridinediamine by reacting 6-bromo-N-{6-chloro-4-[(2-methyl-1 H- imidazol-1-yl)methyl]-2-pyridinyl}-1 ,3-benzothiazol-2-amine [intermediate 32] with the appropriate amine:


Example 101 :
5-{2-[(4-[(2-methyl-1H-imidazol-1-yl)methyl]-6-{[3-(4-morpholinyl)propyl]amino}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-3-pyridinecarbonitrile

A mixture of A/-(6-bromo-1 ,3-benzothiazol-2-yl)-4-[(2-methyl-1 H-imidazol-1-yl)methyl]-A/'- [3-(4-morpholinyl)propyl]-2,6-pyridinediamine [example 99] (165mg, 0.30mmol), 3- cyanopyridine-5-boronic acid pinacol ester (140mg, 0.61 mmol), tetrakis(triphenylphosphine)palladium(0) (70.3mg, 0.061 mmol) and potassium phosphate (97mg, 0.46mmol) in 1 ,4-dioxane (7ml_) and water (2mL) was sealed and heated in a Biotage "Initiator" microwave at 100°C for 45 minutes. Water (100ml_) was added and the reaction mixture was filtered. The filtered solid was washed with water, dried and subjected to purification by mass-directed automated preparative HPLC (trifluoroacetic acid modifier) followed by passing the recovered salt through an aminopropyl solid-phase
extraction cartridge using methanol as eluant to afford the title compound (60mg, 0.106mmol, 35% yield). LCMS (Method B): Rt 2.42 minutes; m/z 566 (MH+).
The compound shown in the table was prepared in an analogous manner to that for 5-{2- [(4-[(2-methyl-1 H-imidazol-1-yl)methyl]-6-{[3-(4-morpholinyl)propyl]amino}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-3-pyridinecarbonitrile by reacting N-(6-bromo-1 ,3- benzothiazol-2-yl)-N'-[3-(4-morpholinyl)propyl]-4-(1 H-pyrazol-1 -ylmethyl)-2,6- pyridinediamine [intermediate 31 ] with 3-cyanopyridine-5-boronic acid pinacol ester:

Example 103:
trans-4-{[4-(hydroxymethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol

A mixture of [2-chloro-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-4-pyridinyl]methanol [intermediate 36] (1 .98g, 6.76mmol) and trans-4-aminocyclohexanol (4.67g, 40.6mmol) were heated together at 190°C overnight. The mixture was cooled to 70°C and treated with saturated aqueous sodium bicarbonate (100ml_) and chloroform/5% methanol (70ml_). The organic phase was collected and evaporated to dryness. The product was purified by flash chromatography on silica using a gradient elution from 0 to 25%
methanol in dichloromethane to afford the title compound (1.9g, 5.1 1 mmol, 76% yield). LCMS (Method A): Rt 0.59 minutes; m/z 372 (MH+)
Example 104:
trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(hydroxymethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of {2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-chloro-4-pyridinyl}methanol [intermediate 38] (1 .18g, 3.18mmol) and trans-4-aminocyclohexanol (3.67g, 31 .8mmol) was sealed and heated in a Biotage "Initiator" microwave at 200°C for 2 hours. The reaction mixture was taken up in water (100ml_) and dichloromethane (100ml_) and filtered. The filtered solid was washed with water (50ml_) followed by dichloromethane (50ml_) and dried under reduced pressure to afford the title compound (706mg, 1 .57mmol, 49% yield). LCMS (Method A): Rt 0.82 minutes; m/z 449,451 (MH+).
Example 105:
frans-4-[(4-(hydroxymethyl)-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-2- pyridinyl)amino]cyclohexanol

A mixture of 1 ,1-dimethylethyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole-1-carboxylate (380mg, 1.29mmol), trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-(hydroxymethyl)-2-pyridinyl]amino}cyclohexanol [example 104] (580mg, 1.29mmol), caesium carbonate (1 .26g, 3.87mmol) and tetrakis(triphenylphosphine)- palladium(O) (447mg, 0.387mmol) in Ν,Ν-dimethylformamide (6mL) and water (2ml_) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 30 minutes. The cooled reaction mixture was taken up in water (20ml_) and ethyl acetate (20ml_) and filtered. The filtrate was separated and the aqueous phase was extracted with ethyl acetate (50ml_). The organic extracts were combined and evaporated to dryness and the residue was subjected to purification by mass directed automated preparative HPLC (formic acid
modifier) to afford the title compound (273mg, 0.63mmol, 49% yield). LCMS (Method A): Rt 0.60 minutes; m/z 437 (MH+).
Example 106:
{2-[(6^romo-1 ,3-benzothiazol-2-yl)amino]-6-[(frans-4-hydroxycyclohexyl)amino]-4- pyridinyljmethyl acetate

7rans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(hydroxymethyl)-2- pyridinyl]amino}cyclohexanol [example 104] (100mg, 0.22mmol) was refluxed in acetic acid (27.8ml_, 49mmol) for 5 hours. The solvent was removed under vacuum and the product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (7.1 mg, 0.014mmol, 7% yield). LCMS (Method A): Rt 1 .00 minutes; m/z 491 , 493 (MH+). Example 107:
trans-4-{[4-[(tetrahydro-2H^yran-4-ylamino)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-2-pyridinyl]amino}cyclohexanol (2Z)-2-butenedioate (salt)

A mixture of 2-[(trans-4-hydroxycyclohexyl)amino]-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-4-pyridinecarbaldehyde [intermediate 43] (50mg, 0.135mmol) and tetrahydro-2H- pyran-4-amine acetate (27.4mg, 0.271 mmol) in dichloromethane (4ml_) was treated with sodium triacetoxyborohydride (45.3mg, 0.203mmol) and stirred at ambient temperature for 3 hours. The mixture was then treated cautiously with saturated aqueous sodium bicarbonate and stirred rapidly for 30 minutes. The mixture was then treated with methanol (0.5ml_) to effect a solution of the precipitated product and then separated. The aqueous phase was extracted with chloroform (+ 10% methanol) (3 x 10ml_) and the combined organic phase was evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (ammonium bicarbonate
modifier) to afford trans-4-{[4-[(tetrahydro-2H-pyran-4-ylamino)methyl]-6-([1 ,3]thiazolo[5,4- b]pyridin-2-ylamino)-2-pyridinyl]amino}cyclohexanol (47mg, 0.103mmol, 76% yield). The product was then treated with a solution of (2Z)-2-butenedioic acid (1 1.96mg, 0.103mmol) in methanol (8mL) and the resulting solution evaporated to dryness with a stream of nitrogen. The product was triturated in diethyl ether and blown to dryness in a stream of nitrogen to afford the title compound (58mg, 0.102mmol, 75% yield). LCMS (Method A): Rt 0.56 minutes; m/z 455 (MH+)
The compounds shown in the table was prepared in an analogous manner to that for trans-4-{[4-[(tetrahydro-2H-pyran-4-ylamino)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-2-pyridinyl]amino}cyclohexanol (2Z)-2-butenedioate by reacting 2-[(trans-4- hydroxycyclohexyl)amino]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-4- pyridinecarbaldehyde [intermediate 43] with the appropriate amine:


Example 114:
frans-4-{[4-(1^yrrolidinylmethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol

A mixture of 2-[(trans-4-hydroxycyclohexyl)amino]-6-([1 ,3]thiazolo[5,4-b]pyridin-2- ylamino)-4-pyridinecarbaldehyde [intermediate 43] (35mg, 0.095mmol) and pyrrolidine (0.016ml_, 0.19mmol) in dichloromethane (4mL) was treated with sodium triacetoxyborohydride (31 .7mg, 0.14mmol) and stirred at room temperature for 3 hours. The mixture was then treated cautiously with saturated aqueous sodium bicarbonate (5ml_) and stirred rapidly for 30 minutes. The mixture was treated with methanol (0.5ml_) to affect a solution of the precipitated product and then separated. The aqueous phase was extracted with chloroform (+ 10% methanol) (3 x 10ml_) and the combined organic phase was blown to dryness under a stream of nitrogen. The product was purified by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (30.7mg, 0.07mmol, 76% yield) as a white solid. LCMS (Method A): Rt 0.56 minutes; m/z 425 (MH+).
The compounds shown in the table were prepared in an analogous manner to that for trans-4-{[4-(1 -pyrrolidinylmethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol by reacting 2-[(trans-4-hydroxycyclohexyl)amino]-6- ([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-4-pyridinecarbaldehyde [intermediate 43] with the appropriate amine:
Purification
Example Structure Name Analytical Data
Method





Example 134:
frans-4-{[6-[(6-amino-1,3^enzothiazol-2-yl)amino]-4-(4-morpholinylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of N2-[6-chloro-4-(4-morpholinylmethyl)-2-pyridinyl]-1 ,3-benzothiazole-2,6- diamine [intermediate 47] (22.5mg, 0.06mmol) and trans-4-aminocyclohexanol (103mg,
0.90mmol) in ethylene glycol (1 ml_) was sealed and heated in a Biotage "Initiator" microwave at 220°C for 12 hours. The product was subjected to purification by mass- directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (3.5mg, 0.0077mmol, 13% yield). LCMS (Method A): Rt 0.42 minutes; m/z 455 (MH+)
Example 135:
trans-4-({4-(aminomethyl)-6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2- pyridinyl}amino)cyclohexanol

A solution of 2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-[(trans-4- hydroxycyclohexyl)amino]-4-pyridinecarbaldehyde oxime [intermediate 53] (320mg, 0.69mmol) in glacial acetic acid (10mL) was treated with zinc powder (785mg, 12mmol) and the mixture was stirred for 1 hour. The mixture was then filtered and the filtrate was evaporated to dryness. The product was subjected to purification by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (144mg, 0.32mmol, 46% yield). LCMS (Method A): Rt 0.74 minutes; m/z 448,450 (MH+) Example 136:
5-[2-({4-(aminomethyl)-6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-3-pyridinecarbonitrile

A mixture of tetrakis(triphenylphosphine)palladium(0) (41 .2mg, 0.036mmol), trans-4-({4- (aminomethyl)-6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-2-pyridinyl}amino)cyclohexanol [example 135] (80mg, 0.178mmol), potassium phosphate (56.8mg, 0.27mmol) and 5- (4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3-pyridinecarbonitrile (82mg, 0.36mmol) in a mixture of 1 ,4-dioxane (1.5mL) and water (0.5mL) was sealed and heated in a Biotage "Initiator" microwave at 100°C for 45 minutes. The reaction mixture was evaporated to dryness and purified by mass-directed automated preparative HPLC (ammonium
bicarbonate modifier) to afford the title compound (29mg, 0.061 mmol, 35% yield). LCMS (Method A): Rt 0.68 minutes; m/z 472 (MH+)
Examples 137 and 138:
5-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1 -pyrrolidinylmethyl)-2- pyridinyl]amino}-1 ,3-benzothiazol-6-yl)-3-pyridinecarbonitrile and 5-(2-{[6-[(frans-4- hydroxycyclohexyl)amino]-4-(hydroxymethyl)-2-pyridinyl]amino}-1 ,3-benzothiazol- -yl)-3-pyridinecarbonitrile
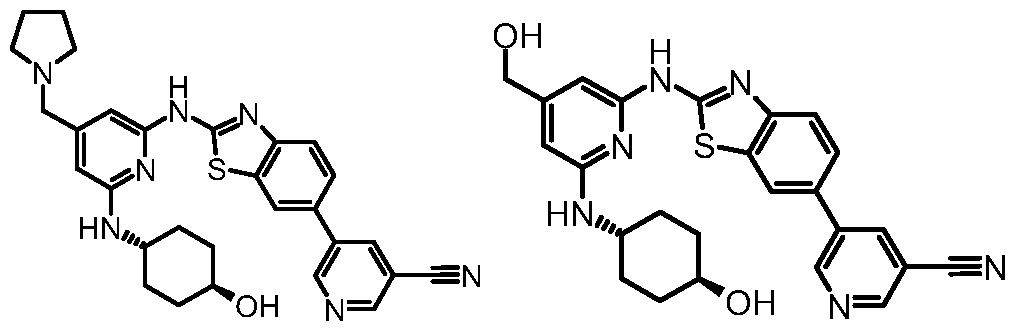
5-[2-({4-formyl-6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol- 6-yl]-3-pyridinecarbonitrile [intermediate 45] (51.8mg, 0.1 10mmol) and pyrrolidine (0.018ml_, 0.220mmol) were dissolved in methanol (10ml_) under nitrogen and sodium cyanoborohydride (10.2mg, 0.154mmol) was added as a solid. The reaction mixture was stirred overnight. The mixture was then blown down under a stream of nitrogen to remove the solvent and purified by mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compounds. Example 137: (8.2mg, 0.017mmol, 16% yield). LCMS (Method B): Rt 2.62 minutes; m/z 526 (MH+). Example 138: (4.9mg, 0.0093mmol, 8% yield). LCMS (Method B): Rt 2.19 minutes; m/z 473 (MH+). Example 139:
5-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(4-morpholinylmethyl)-2- pyridinyl]amino}-1 ,3-benzothiazol-6-yl)-3-pyridinecarbonitrile

To a mixture of 5-[2-({4-formyl-6-[(trans-4-hydroxycyclohexyl)amino]-2-pyridinyl}amino)- 1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile [intermediate 45] (62.8mg, 0.13mmol) and morpholine (0.023mL, 0.27mmol) in dichloromethane (10mL) was added acetic acid (0.5mL), the mixture was stirred for 10 minutes. Sodium triacetoxyborohydride (44.7mg, 0.20mmol) was then added and the mixture was stirred at room temperature overnight. The mixture was evaporated to dryness and the residue was subjected to purification by
mass-directed automated preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (39.6mg, 0.07mmol, 55% yield). LCMS (Method B): Rt 2.39 minutes; m/z 542 (MH+). The compounds shown in the table were prepared in an analogous manner to that for 5- (2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(4-morpholinylmethyl)-2-pyridinyl]amino}-1 ,3- benzothiazol-6-yl)-3-pyridinecarbonitrile by reacting 5-[2-({4-formyl-6-[(trans-4- hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile [intermediate 45] with the appropriate amine:


5- [2-({4-{[cis-2,6- dimethyl-4-
X morpholinyl]methyl}-
LCMS (Method MDAP
VX H 6- [(trans-4- A): Rt 0.73 ammonium
148 hydroxycyclohexyl)a
minutes; m/z bicarbonate mino]-2- 570 (MH+) modifier pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-3- pyridinecarbonitrile
Example 149:
3-[2-({4-[(3,3-difluoro-1 ^iperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one

Under an atmosphere of nitrogen, a mixture of frans-4-({6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-[(3,3-difluoro-1-piperidinyl)methyl]-2-pyridinyl}amino)cyclohexanol [example 162] (50mg, 0.09mmol), 1 ,3-oxazolidin-2-one (15.8mg, 0.18mmol), potassium phosphate (38.4mg, 0.181 mmol) and copper(l) iodide (34.5mg, 0.181 mmol) in dry N,N- dimethylformamide (3mL) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with N, N'-dimethylethylenediamine (0.039ml_, 0.36mmol) and the mixture was sealed and heated in a Biotage "Initiator" microwave at 100°C for 1.25 hours. Water (10ml_) and ethyl acetate (10ml_) was added to the mixture and the organic phase was separated and washed with water until no blue colour was observed in the aqueous phase. The organic phase was dried over magnesium sulfate, filtered and evaporated to dryness and the residue was subjected to purification by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (16mg, 0.03mmol, 32% yield). LCMS (Method A): Rt 0.66 minutes; m/z 559 (MH+).
Example 150:
3-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1-pyrrolidinylmethyl)-2- pyridinyl]amino}-1 ,3-benzothiazol-6-yl)-1 ,3-oxazolidin-2-one (2Z)-2-butenedioate (salt)

Under an atmosphere of nitrogen, a mixture of 2-[(trans-4-hydroxycyclohexyl)amino]-6- {[6-(2-oxo-1 ,3-oxazolidin-3-yl)-1 ,3-benzothiazol-2-yl]amino}-4-pyridinecarbaldehyde
[intermediate 46] (52mg, 0.12mmol) and pyrrolidine (0.019ml_, 0.23mmol) in dichloromethane (10mL) was treated with acetic acid (0.007ml_, 0.12mmol) and the mixture was stirred for 10 minutes. Sodium triacetoxyborohydride (38.4mg, 0.172mmol) was added and the mixture was stirred at ambient temperature overnight. The mixture was then treated with chloroform (+ 10% methanol) (20ml_) and saturated aqueous sodium bicarbonate (20ml_). The aqueous phase was extracted a further three times with chloroform (+10% methanol) and the combined organic phases were evaporated to dryness. The product was purified by mass-directed automated preparative HPLC (trifluoroacetic acid modifier). Product-containing fractions were added to an SCX (sulfonic acid) solid-phase extraction cartridge which was eluted with methanol. The product was then eluted from the column using a solution of ammonia in ethanol (2M) to afford the title compound as its free base. An equimolar amount of (2Z)-2-butenedioate (2.2mg, 0.019mmol) in methanol (2ml_) was added and the resulting solution was evaporated to dryness to afford the title compound (1 1 .2mg, 0.022mmol, 16% yield). LCMS (Method B): Rt 2.1 1 minutes; m/z 509 (MH+).
The compounds shown in the table were prepared in an analogous manner to that for 3- (2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(1 -pyrrolidinylmethyl)-2-pyridinyl]amino}-1 ,3- benzothiazol-6-yl)-1 ,3-oxazolidin-2-one (2Z)-2-butenedioate (salt) by reacting 2-[(trans-4- hydroxycyclohexyl)amino]-6-{[6-(2-oxo-1 ,3-oxazolidin-3-yl)-1 ,3-benzothiazol-2-yl]amino}- 4-pyridinecarbaldehyde [intermediate 46] with the appropriate amine and then optionally preparing the (2Z)-2-butenedioate salt:
Purification
Example Structure Name Analytical Data
Method
3-(2-{[6-[(frans-4- hydroxycyclohexyl)
0 amino]-4-(4- MDAP
LCMS (Method A):
morpholinylmethyl)- ammonium
151 Rt 0.59 minutes;
2-pyridinyl]amino}- bicarbonate m/z 525 (MH+)
1 ,3-benzothiazol-6- modifier yl)-1 ,3-oxazolidin-2- one
3-[2-({4-
[(diethylamino)meth
yl]-6-[(trans-4- hydroxycyclohexyl) MDAP
V S,-0H LCMS (Method A):
amino]-2- ammonium
152 Rt 0.59 minutes;
pyridinyl}amino)- bicarbonate m/z 51 1 (MH+)
1 ,3-benzothiazol-6- modifier yl]-1 ,3-oxazolidin-2- one (2Z)-2- butenedioate (salt)
3-{2-[(6-[(frans-4- hydroxycyclohexyl)
amino]-4-{[(1 ,2,2-
MDAP
trimethylpropyl)ami LCMS (Method A):
ammonium
153 no]methyl}-2- Rt 0.69 minutes;
bicarbonate pyridinyl)amino]- m/z 539 (MH+)
modifier
1 ,3-benzothiazol-6- yl}-1 ,3-oxazolidin-2- one
3-(2-{[6-[(frans-4- hydroxycyclohexyl)
amino]-4-(1- MDAP
LCMS (Method B):
piperidinylmethyl)- ammonium
154 Rt 2.54 minutes;
2-pyridinyl]amino}- bicarbonate m/z 523 (MH+)
1 ,3-benzothiazol-6- modifier yl)-1 ,3-oxazolidin-2- one
3-[2-({4-[(4,4- dimethyl-1- piperidinyl)methyl]-
6-[(trans-4- MDAP
LCMS (Method B):
hydroxycyclohexyl) ammonium
155 Rt 2.84 minutes;
amino]-2- bicarbonate m/z 551 (MH+)
pyridinyl}amino)- modifier
1 ,3-benzothiazol-6- yl]-1 ,3-oxazolidin-2- one
3-[2-({4-{[(2 6S)-
2,6-dimethyl-4- morpholinyl]methyl}
-6-[(trans-4- MDAP
LCMS (Method A):
hydroxycyclohexyl) ammonium
156 Rt 0.63 minutes;
amino]-2- bicarbonate m/z 553 (MH+)
pyridinyl}amino)- modifier
1 ,3-benzothiazol-6- yl]-1 ,3-oxazolidin-2- one
Example 157:
1-(2-{[6-[(frans-4-hydroxycyclohexyl)amino]-4-(4-morpholinylmethyl)-2- pyridinyl]amino}-1,3-benzothiazol-6-yl)-2,5-pyrrolidinedione
A mixture of trans-4-{[6-[(6-amino-1 ,3-benzothiazol-2-yl)amino]-4-(4-morpholinylmethyl)-2- pyridinyl]amino}cyclohexanol [example 134] (80mg, 0.18mmol) and succinic anhydride (17.6mg, 0.18mmol) in acetonitrile (2ml_) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 15mins. A few drops of aqueous hydrochloric acid (2M) were added to the cooled reaction mixture which was then resealed and heated in the microwave at 150°C for 2 hours. The product was purified by mass-directed automated
preparative HPLC (ammonium bicarbonate modifier) to afford the title compound (21 mg, 0.04mmol, 22% yield). LCMS (Method A): Rt 0.54 minutes; m/z 537 (MH+).
Example 158:
1 -{2-[(6-[(frans-4-hydroxycyclohexyl)amino]-4-{[(1 ,2,2- trimethylpropyl)amino]methyl}-2-pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-2,5- pyrrolidinedione

Under an atmosphere of nitrogen, a mixture of frans-4-[(6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-{[(1 ,2,2-trimethylpropyl)amino]methyl}-2-pyridinyl)amino]cyclohexanol
[example 161] (30mg, 0.056mmol), succinimide (1 1.16mg, 0.1 13mmol), potassium phosphate (23.9mg, 0.1 13mmol) and copper(l) iodide (21 .5mg, 0.1 13mmol) in dry N,N- dimethylformamide (3ml_) was thoroughly degassed by the repeated alternate application of vacuum and nitrogen pressure, then treated with Ν,Ν'-dimethylethylenediamine (24μΙ_, 0.225mmol) and the mixture was sealed and heated in a Biotage "Initiator" microwave at 100°C for 1 .25 hours. Water (10ml_) and ethyl acetate (10ml_) were added to the mixture and the organic phase was separated and washed with water until no blue colour was observed in the aqueous phase. The organic phase was dried over magnesium sulfate, filtered and the solvent was evaporated to dryness. The product was purified by mass- directed automated preparative HPLC (formic acid modifier) to afford the title compound (1 1 .1 mg, 0.02mmol, 36% yield). LCMS (Method A): Rt 0.64 minutes; m/z 551 (MH+).
The compound shown in the table was prepared in an analogous manner to that for 1-{2- [(6-[(frans-4-hydroxycyclohexyl)amino]-4-{[(1 ,2,2-trimethylpropyl)amino]methyl}-2- pyridinyl)amino]-1 ,3-benzothiazol-6-yl}-2,5-pyrrolidinedione by reacting frans-4-({6-[(6- bromo-1 ,3-benzothiazol-2-yl)amino]-4-[(3,3-difluoro-1 -piperidinyl)methyl]-2- pyridinyl}amino)cyclohexanol [example 162] with succinimide:
Purificatio
Example Structure Name Analytical Data
n Method
1 -[2-({4-[(3,3- difluoro-1- piperidinyl)methyl]-
MDAP
6-[(trans-4- LCMS (Method B):
formic
159 hydroxycyclohexyl)a Rt 2.48 minutes;
acid
H xc>° mino]-2- m/z 571 (MH+)
modifier pyridinyl}amino)-1 ,3- benzothiazol-6-yl]-
2,5-pyrrolidinedione
Example 160:
frans-4-{[6-[(6-bromo-1,3-benzothiazol-2-yl)amino]-4-(1 -pyrrolidinylmethyl)-2- pyridinyl]amino}cyclohexanol

A mixture of 2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6-[(trans-4- hydroxycyclohexyl)amino]-4-pyridinecarbaldehyde [intermediate 44] (90mg, 0.20mmol) and pyrrolidine (0.166ml_, 0.20mmol) in dichloromethane (1 ml_) was treated with sodium triacetoxyborohydride (59.7mg, 0.28mmol) and stirred at room temperature for 1 hour. The mixture was then treated cautiously with saturated aqueous sodium bicarbonate (5ml_) and dichloromethane (10ml_) added. The aqueous layer was extracted a further three times and the combined organic layers passed through a hydrophobic frit and the solvent removed under vacuum. The product was purified by mass-directed automated preparative HPLC (formic acid modifier) to afford the title compound (47mg, 0.086mmol, 43% yield) as a brown solid. LCMS (Method A): Rt 0.79 minutes; m/z 502,504 (MH+).
The compounds shown in the table were prepared in an analogous manner to that for trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-4-(1 -pyrrolidinylmethyl)-2- pyridinyl]amino}cyclohexanol by reacting 2-[(6-bromo-1 ,3-benzothiazol-2-yl)amino]-6- [(trans-4-hydroxycyclohexyl)amino]-4-pyridinecarbaldehyde [intermediate 44] with the appropriate amine:
Example Structure Name Analytical Data Purification
Method
trans-4-[{6-[{6- bromo-1 ,3- benzothiazol-2-
MDAP
yl)amino]-4- LCMS (Method B):
ammonium
161 {[(1.2,2- Rt 3.24 minutes;
bicarbonate trimethylpropyl)a m/z 532,534 (MH+)
modifier mino]methyl}-2- pyridinyl)amino]cy
clohexanol
trans-4-{{6-[{6- bromo-1 ,3- Silica
benzothiazol-2- chromatography yl)amino]-4-[(3,3- LCMS (Method A): gradient
162 difluoro-1- Rt 0.89 minutes; elution from 0 to piperidinyl)methyl m/z 552,554 (MH+) 15%
]-2- MeOH(+1 %Et3N pyridinyl}amino)c ) in DCM yclohexanol
trans-4-[{6-[{6- bromo-1 ,3- Silica
benzothiazol-2- chromatography yl)amino]-4-{[cis- LCMS (Method A): gradient
163 2,6-dimethyl-4- Rt 0.79 minutes; elution from 0 to morpholinyl]meth m/z 546,548 (MH+) 15%
yl}-2- MeOH(+1 %Et3N pyridinyl)amino]cy ) in DCM clohexanol
Example 164:
frans-4-{[6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2-yl]amino}-4-(1 - pyrrolidinylmethyl)-2-pyridinyl]amino}cyclohexanol

A mixture of 1 ,1-dimethylethyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole-1-carboxylate (17.6mg, 0.06mmol), trans-4-{[6-[(6-bromo-1 ,3-benzothiazol-2- yl)amino]-4-(1-pyrrolidinylmethyl)-2-pyridinyl]amino}cyclohexanol [example 160] (30mg, 0.060mmol), caesium carbonate (58.4mg, 0.179mmol), and tetrakis(triphenylphosphine)- palladium(O) (20.7mg, 0.018mmol) in Ν,Ν-dimethylformamide (6mL) and water (2mL) was sealed and heated in a Biotage "Initiator" microwave at 150°C for 30 minutes. The cooled reaction mixture was taken up in water (20ml_) and dichloromethane (20ml_) and filtered. The filtrate was separated and the aqueous phase was extracted with dichloromethane (50ml_). The organic fractions were combined, evaporated to dryness and the residue was suspended in minimal ethyl acetate. The solid was filtered, washed with ethyl acetate (10ml_) and dried to afford the title compound (8.5mg, 0.017mmol, 29% yield). LCMS (Method A): Rt 0.61 minutes; m/z 490 (MH+).
BIOLOGICAL DATA
Itk Homogeneous Time Resolved Fluorescence (HTRF)
The activity of recombinant human Itk (full length) is assessed using an HTRF assay with truncated human SAM-68 (R331-Y443) as the substrate.
Recombinant human Itk (full length) is expressed in insect cells (in pFastBac-1 vector Invitrogen) fused to a Flag tag at its N terminus. The sequence of the Itk part is identical to Genbank entry L10717. The FLAG-ltk fusion protein is extracted from insect cells and purified by immunoaffinity chromatography on anti-FLAG (M2) agarose affinity resin. Further purification is by size exclusion chromatography. Purified protein is stored at - 80°C in Tris-HCI (50mM), NaCI (200mM), sorbitol (500mM), DTT (2mM), pH 8.0.
Truncated human SAM-68 (R331-Y443) is expressed in E. coli (using a pGex-4T vector Pharmacia) as a GST-thrombin cleavage site-Avi-tag-Sam68:331 -443 fusion. The Sam68 part of the fusion (R331-Y443) is identical to the sequence of Genbank database entry NM_006559. GST-SAM68 is purified by affinity chromatography on glutathione-
sepharose. Specific biotinylation of the Avitag sequence of GST-SAM68 is performed at room temperature in the presence ofmg:ATP, (5mM), D-biotin, (1 mM), DTT, (1 mM) and biotin ligase, (1 uM), and is complete in 2 hours. The biotinylated protein is further purified by size exclusion chromatography and stored at -80°C in Tris-HCI (50mM), NaCI (250mM), glycerol (10%), DTT (2mM), pH 8.0.
Itk (typically 5-50μΜ) is pre-activated by incubation with 100μΜ ATP and 10mMmgCI2 for 30 minutes at room temperature before dilution in assay buffer (50mM HEPES, 1 mM dithiothreitol, 0.0025% Tween-20, pH7.4) to give a concentration which ensures linearity proportional to time and enzyme concentration (typically a 5nM final concentration in the assay).
Compounds at various concentrations (typical range 25pM - 25μΜ) or DMSO vehicle (at less than 5% final assay concentration) are incubated with 3μΙ substrate (final assay concentrations 50nM biotinylated GST SAM68, 10mMmgCI2, 20μΜ ATP in 50mM HEPES, 1 mM DTT, 0.0025% Tween 20, pH7.4). The activated Itk enzyme (3μΙ volume,) is added to initate the phosphorylation reaction. Following an incubation at 20°C, (for a time determined to ensure the assay remains in linear initital rate phase, typically 30 minutes), the reaction is halted by adding stop/read reagent (3μΙ). The stop/read reagent comprises streptavidin APC (50nM final assay concentration; Perkin Elmer), europium- anti-phosphotyrosine antibody (0.5nM final assay concentration; Wallac) diluted in 40mM HEPES, 150mM NaCI, 0.03%w/v BSA, 60mM EDTA. The assay plates are left to equilibrate for at least 45 minutes at 20°C, before reading on a suitable HTRF reader. The compounds of Examples 1 to 164 were tested in the above or a similar assay and were found to have a mean pKi of 5 or greater.
Claims
1. A compound of formula

(I)
wherein R1 is hydrogen, -CH2OR5, -CH2NR6R7, -CH2phenyl or -CH2-5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by C1-6alkyl;
R2 is hydrogen or methyl,
R3 is C3-6cycloalkyl substituted by -OH or -NR8R9, or -(CH2)m6-membered heterocyclyl wherein the 6-membered heterocyclyl contains one or two heteroatoms independently selected from nitrogen and oxygen and is optionally substituted by C1-6alkyl, or
R2 and R3, together with the nitrogen atom to which they are attached, are linked to form a piperidinyl substituted by -NR10R11;
R4 is hydrogen, C -6alkyl, halo, -NR12R13, phenyl optionally substituted by -CONR14, 5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is optionally substituted by one or two substituents independently selected from Ci-6alkyl, C2-6alkynyl, - CN, -(CH2)nOR15, -CH2phenyl, -(CH2)PNR16R17 and -CONR18R19, or 9- or 10-membered bicyclic heteroaryl wherein the 9- or 10-membered bicyclic heteroaryl contains one or two nitrogen atoms and is optionally substituted by d-6alkyl;
R5 and R9 are each independently hydrogen or -COCi-6alkyl;
R6 is hydrogen or Ci-6alkyl,
R7 is hydrogen, Ci-6alkyl optionally substituted by -OR20 or -(CH2)qtetrahydropyran, or R6 and R7, together with the nitrogen atom to which they are attached, are linked to form a 4-, 5- or 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom and the 5- or 6-membered heterocyclyl is optionally substituted by one or two substituents independently selected from d-6alkyl and halo;
R8 is hydrogen;
R10 and R1 1 , together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by C1 -6alkyl;
R12 and R13 are each hydrogen, or R12 and R13, together with the nitrogen atom to which they are attached, are linked to form a 5- or 6-membered heterocyclyl wherein the 5- or 6- membered heterocyclyl optionally contains an oxygen atom or a further nitrogen atom and is optionally substituted by one or two substituents independently selected from oxo, -OH and Ci-6alkyl optionally substituted by -OH or -NH2;
R14 is C3-6cycloalkyl; R15 is hydrogen, Ci-6alkyl optionally substituted by -OH, C3-6cycloalkyl, -(CH2)rphenyl optionally substituted by halo, or -CH2pyridinyl;
R16 and R17 are each independently hydrogen or Ci-6alkyl optionally substituted by -OR21 ; R18 is hydrogen,
R19 is hydrogen or Ci-6alkyl, or
R18 and R19, together with the nitrogen atom to which they are attached, are linked to form a 6-membered heterocyclyl wherein the 6-membered heterocyclyl optionally contains an oxygen atom;
R20 and R21 are each independently hydrogen or Ci-6alkyl; X is -N- or -CH-; m is 1 , 2 or 3; n, p, q and r are each independently 0, 1 or 2; or a salt thereof.
2. A compound according to claim 1 , or a salt thereof, wherein R1 is -CH2NR6R7 or - CH2-5- or 6-membered heteroaryl wherein the 5- or 6-membered heteroaryl contains one or two nitrogen atoms and is optionally substituted by d-6alkyl.
3. A compound according to claim 1 or claim 2, or a salt thereof, wherein R2 is hydrogen.
4. A compound according to any one of the preceding claims, or a salt thereof, wherein R3 is C3-6cycloalkyl substituted by -OH or -NR8R9.
5. A compound according to any one of the preceding claims, or a salt thereof, wherein R4 is hydrogen, -NR12R13 or 5- or 6-membered heteroaryl wherein the 5- or 6- membered heteroaryl contains from one to three heteroatoms independently selected from oxygen and nitrogen and is optionally substituted by Ci-6alkyl, C2-6alkynyl, -CN, - (CH2)nOR15, -CH2phenyl, -(CH2)PNR16R17 or -CONR18R19.
6. A compound according to any one of the preceding claims, or a salt thereof, wherein X is -N-.
7. A compound according to any one of claims 1 to 5, or a salt thereof, wherein X is - CH-.
8. A compound substantially as described in any one of Examples 1 to 164, or a salt thereof.
9. A compound which is:
frans-4-[(4-[(2-methyl-1 H-imidazol-1 -yl)methyl]-6-{[6-(1 H-pyrazol-4-yl)-1 ,3-benzothiazol-2- yl]amino}-2-pyridinyl)amino]cyclohexanol
frans-4-{[4-(4-morpholinylmethyl)-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3,3-dimethyl-1 -piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylamino)-2- pyridinyl]amino}cyclohexanol;
frans-4-{[6-([1 ,3]thiazolo[5,4-/b]pyridin-2-ylamino)-4-({[(1 S)-1 ,2,2- trimethylpropyl]amino}methyl)-2-pyridinyl]amino}cyclohexanol; frans-4-{[4-[(2,2-dimethyl-4-morpholinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyri pyridinyl]amino}cyclohexanol;
frans-4-{[4-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-6-([1 ,3]thiazolo[5,4-b]p ylamino)-2-pyridinyl]amino}cyclohexanol;
frans-4-{[4-[(3,3-difluoro-1-piperidinyl)methyl]-6-([1 ,3]thiazolo[5,4-b]pyridin-2-ylam pyridinyl]amino}cyclohexanol;
5-{2-[(6-[(frans-4-hydroxycyclohexyl)amino]-4-{[(1 ,2,2-trimethylpropyl)amino]meth pyridinyl)arnino]-1 ,3-benzothiazol-6-yl}-3-pyridinecarbonitrile;
5-[2-({4-[(3,3-difluoro-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinecarbonitrile;
5-[2-({4-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-6-[(frans-4- hydroxycyclohexyl)amino]-2-pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-3-pyridinec^
3-[2-({4-[(3,3-difluoro-1-piperidinyl)methyl]-6-[(frans-4-hydroxycyclohexyl)amino]-2- pyridinyl}amino)-1 ,3-benzothiazol-6-yl]-1 ,3-oxazolidin-2-one; or
a salt thereof.
10. A compound according to any one of claims 1 to 9 in the form of a pharmaceutically acceptable salt thereof.
1 1. A pharmaceutical composition comprising a compound as defined in any one of claims 1 to 9, or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable excipients.
12. A compound as defined in any one of claims 1 to 9, or a pharmaceutically acceptable salt thereof, for use in medical therapy
13. A compound as defined in any one of claims 1 to 9, or a pharmaceutically acceptable salt thereof, for use in the treatment of a disorder mediated by inappropriate Itk activity.
14. Use of a compound as defined in any one of claims 1 to 9, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for use in the treatment of a disorder mediated by inappropriate Itk activity.
15. A method of treating a disorder mediated by inappropriate Itk activity comprising administering a safe and effective amount of a compound as defined in any one of claims 1 to 9, or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
16. A method according to claim 15 wherein the disorder mediated by inappropriate Itk activity is a respiratory disease; an allergic disease; an autoimmune disease; transplant rejection; graft versus host disease; an inflammatory disorder; HIV; aplastic anemia; or pain.
17. A method according to claim 15 wherein the disorder mediated by inappropriate Itk activity is asthma, chronic obstructive pulmonary disease (COPD), bronchitis, allergic rhinitis, atopic dermatitis, rheumatoid arthritis, multiple sclerosis, psoriasis, type I diabetes, T cell mediated hypersensitivity, Guillain-Barre Syndrome, Hashimoto's thyroiditis, transplant rejection, graft versus host disease, conjunctivitis, contact dermatitis, inflammatory bowel disease, chronic inflammation, HIV, aplastic anemia, or inflammatory pain.
18. A method according to claim 15 wherein the disorder mediated by inappropriate Itk activity is asthma.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US31293310P | 2010-03-11 | 2010-03-11 | |
| US61/312,933 | 2010-03-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011110575A1 true WO2011110575A1 (en) | 2011-09-15 |
Family
ID=44170219
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/053499 WO2011110575A1 (en) | 2010-03-11 | 2011-03-09 | Derivatives of 2-[2-(benzo- or pyrido-) thiazolylamino]-6-aminopyridine, useful in the treatment of respiratoric, allergic or inflammatory diseases |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011110575A1 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013544256A (en) * | 2010-11-19 | 2013-12-12 | リガンド ファーマシューティカルズ インコーポレイテッド | Heterocyclic amines and uses thereof |
| CN103800337A (en) * | 2012-11-07 | 2014-05-21 | 韩冰 | Compound for treating neurodegenerative diseases and application thereof |
| WO2014202505A1 (en) | 2013-06-20 | 2014-12-24 | Bayer Cropscience Ag | Aryl sulfide derivatives and aryl sulfoxide derivatives as acaricides and insecticides |
| US20160207928A1 (en) * | 2013-06-10 | 2016-07-21 | Bayer Pharma Aktiengesellschaft | Novel compounds for the treatment of cancer |
| WO2017157885A1 (en) | 2016-03-16 | 2017-09-21 | Bayer Cropscience Aktiengesellschaft | N-(cyanobenzyl)-6-(cyclopropyl-carbonylamino)-4-(phenyl)-pyridine-2-carboxamide derivatives and related compounds as pesticides and plant protection agents |
| CN107987097A (en) * | 2017-12-17 | 2018-05-04 | 沧州普瑞东方科技有限公司 | The synthesis technique of 2,6- dichloropyridine -4- boric acid pinacol esters |
| US10807983B2 (en) | 2015-03-16 | 2020-10-20 | Ligand Pharmaceuticals, Inc. | Imidazo-fused heterocycles and uses thereof |
Citations (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993013055A1 (en) | 1991-12-24 | 1993-07-08 | The Wellcome Foundation Limited | Amidino derivatives and their use as nitric oxide synthase inhibitors |
| WO1995034534A1 (en) | 1994-06-15 | 1995-12-21 | The Wellcome Foundation Limited | Enzyme inhibitors |
| US5552438A (en) | 1992-04-02 | 1996-09-03 | Smithkline Beecham Corporation | Compounds useful for treating allergic and inflammatory diseases |
| WO1996032099A1 (en) | 1995-04-14 | 1996-10-17 | Glaxo Wellcome Inc. | Metered dose inhaler for albuterol |
| WO1998030537A1 (en) | 1997-01-13 | 1998-07-16 | Glaxo Group Limited | Nitric oxide synthase inhibitors |
| WO1998054159A1 (en) | 1997-05-30 | 1998-12-03 | Schering Aktiengesellschaft | Non-steroidal (hetero) cyclically substituted acylanilides with mixed gestagen and androgen activity |
| WO1999016766A1 (en) | 1997-10-01 | 1999-04-08 | Kyowa Hakko Kogyo Co., Ltd. | Benzodioxole derivatives |
| WO1999047505A1 (en) | 1998-03-14 | 1999-09-23 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Phthalazinone pde iii/iv inhibitors |
| WO1999062875A1 (en) | 1998-05-30 | 1999-12-09 | Glaxo Group Limited | Nitric oxide synthase inhibitors |
| US6119853A (en) | 1998-12-18 | 2000-09-19 | Glaxo Wellcome Inc. | Method and package for storing a pressurized container containing a drug |
| WO2000066590A2 (en) | 1999-05-04 | 2000-11-09 | Ligand Pharmaceuticals, Inc. | Tetracyclic progesterone receptor modulator compounds and methods |
| WO2001004118A2 (en) | 1999-07-14 | 2001-01-18 | Almirall Prodesfarma S.A. | Quinuclidine derivatives and their use as muscarinic m3 receptor ligands |
| WO2001016128A1 (en) | 1999-09-01 | 2001-03-08 | Abbott Laboratories | Dibenzopyrans as glucocorticoid receptor antagonists for treatment of diabetes |
| WO2001042193A1 (en) | 1999-12-08 | 2001-06-14 | Theravance, Inc. | β2-ADRENERGIC RECEPTOR AGONISTS |
| US6315112B1 (en) | 1998-12-18 | 2001-11-13 | Smithkline Beecham Corporation | Method and package for storing a pressurized container containing a drug |
| WO2002002565A2 (en) | 2000-07-05 | 2002-01-10 | Abbott Laboratories | Glucocortiocoid-selective antiinflammatory agents |
| WO2002012266A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 17.beta.-carbothioate 17.alpha.-arylcarbonyloxyloxy androstane derivative as anti-inflammatory agents |
| US6352152B1 (en) | 1998-12-18 | 2002-03-05 | Smithkline Beecham Corporation | Method and package for storing a pressurized container containing a drug |
| US6360739B1 (en) | 1997-06-10 | 2002-03-26 | Smithkline Beecham Corporation | Dispenser with doses counter |
| WO2002026722A1 (en) | 2000-09-29 | 2002-04-04 | Glaxo Group Limited | Morpholin-acetamide derivatives for the treatment of inflammatory diseases |
| US6390291B1 (en) | 1998-12-18 | 2002-05-21 | Smithkline Beecham Corporation | Method and package for storing a pressurized container containing a drug |
| WO2002050021A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Nitric oxide synthase inhibitor phosphate salt |
| WO2002066422A1 (en) | 2001-02-14 | 2002-08-29 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2002070490A1 (en) | 2001-03-08 | 2002-09-12 | Glaxo Group Limited | Agonists of beta-adrenoceptors |
| WO2002076933A1 (en) | 2001-03-22 | 2002-10-03 | Glaxo Group Limited | Formailide derivatives as beta2-adrenoreceptor agonists |
| WO2002088167A1 (en) | 2001-04-30 | 2002-11-07 | Glaxo Group Limited | Anti-inflammatory 17.beta.-carbothioate ester derivatives of androstane with a cyclic ester group in position 17.alpha |
| WO2002100879A1 (en) | 2001-06-12 | 2002-12-19 | Glaxo Group Limited | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| WO2003008277A2 (en) | 2001-07-17 | 2003-01-30 | Riverwood International Corporation | Carton with improved dispenser and handle |
| WO2003024439A1 (en) | 2001-09-14 | 2003-03-27 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003042160A1 (en) | 2001-11-13 | 2003-05-22 | Theravance, Inc. | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003059899A1 (en) | 2002-01-14 | 2003-07-24 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical formulations containing them and uses thereof |
| WO2003061651A1 (en) | 2002-01-22 | 2003-07-31 | The Regents Of The University Of California | Non-steroidal ligands for the glucocorticoid receptor, compositions and uses thereof |
| WO2003072539A1 (en) | 2002-02-28 | 2003-09-04 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003082827A1 (en) | 2002-04-02 | 2003-10-09 | Schering Aktiengesellschaft | Quinoline and isoquinoline derivatives, method for the production thereof and use thereof as anti-inflammatory agents |
| WO2003082280A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003082787A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003086294A2 (en) | 2002-04-11 | 2003-10-23 | Merck & Co., Inc. | 1h-benzo[f]indazol-5-yl derivatives as selective glucocorticoid receptor modulators |
| WO2003091204A1 (en) | 2002-04-25 | 2003-11-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2003101932A2 (en) | 2002-05-29 | 2003-12-11 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003104195A1 (en) | 2002-06-06 | 2003-12-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | 4-(aryl or heteroaryl) -2-butylamine derivatives and their use as glucocorticoid ligans |
| WO2004005229A1 (en) | 2002-07-08 | 2004-01-15 | Pfizer Products Inc. | Modulators of the glucocorticoid receptor |
| WO2004009017A2 (en) | 2002-07-18 | 2004-01-29 | Bristol-Myers Squibb Company | Modulators of the glucocorticoid receptor and method |
| WO2004016578A2 (en) | 2002-07-25 | 2004-02-26 | Glaxo Group Limited | Arylethanolamine beta2-adrenoreceptor agonist compounds |
| WO2004018429A2 (en) | 2002-08-21 | 2004-03-04 | Boehringer Ingelheim Pharmaceuticals, Inc. | Substituted hihydroquinolines as glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2004022547A1 (en) | 2002-09-06 | 2004-03-18 | Glaxo Group Limited | Phenethanolamine derivatives and their use in the treatment of respiratory diseases |
| WO2004024728A2 (en) | 2002-09-16 | 2004-03-25 | Glaxo Group Limited | Pyrazolo[3,4-b]pyridine compounds, and their use as phosphodiesterase inhibitors |
| WO2004026248A2 (en) | 2002-09-20 | 2004-04-01 | Merck & Co., Inc. | Octahydro-2-h-naphtho[1,2-f] indole-4-carboxamide derivatives as selective glucocorticoid receptor modulators |
| WO2004035556A1 (en) | 2002-10-16 | 2004-04-29 | Glaxo Group Limited | Substituted piperazines, (1,4) diaszepines, and 2,5-diazabicyclo (2.2.1) heptanes as histamine h1 and/or h3 antagonists or histamine h3 reverse antagonists |
| WO2004037768A2 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2004037807A2 (en) | 2002-10-22 | 2004-05-06 | Glaxo Group Limited | Medicinal arylethanolamine compounds |
| WO2004037773A1 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivative for the treatment of respiratory diseases |
| WO2004039762A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenethanolamine derivatives for the treatment of respiratory diseases |
| WO2004039766A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenylethanolamine derivatives for the treatment of respiratory diseases |
| WO2004056823A1 (en) | 2002-12-23 | 2004-07-08 | Glaxo Group Limited | PYRAZOLO[3,4-b]PYRIDINE COMPOUNDS, AND THEIR USE AS PHOSPHODIESTERASE INHIBITORS |
| WO2004103998A1 (en) | 2003-05-21 | 2004-12-02 | Glaxo Group Limited | Quinoline derivatives as phosphodiesterase inhibitors |
| WO2005005451A1 (en) | 2003-07-11 | 2005-01-20 | Glaxo Group Limited | Specific glucocorticosteroid compound having anti- inflammatory activity |
| WO2005037280A1 (en) | 2003-10-14 | 2005-04-28 | Glaxo Group Limited | Muscarinic acetycholine receptor antagonists |
| WO2005044354A1 (en) | 2003-11-03 | 2005-05-19 | Glaxo Group Limited | A fluid dispensing device |
| WO2005046586A2 (en) | 2003-11-04 | 2005-05-26 | Glaxo Group Limited | M3 muscarinic acetylcholine receptor antagonists |
| WO2005058892A1 (en) | 2003-12-19 | 2005-06-30 | Glaxo Group Limited | Pyrazolo [3,4-b] pyridine compounds, and their use as phosphodiesterase inhibitors |
| WO2005090353A1 (en) | 2004-03-16 | 2005-09-29 | Glaxo Group Limited | PYRAZOLO[3,4-b]PYRIDINE COMPOUNDS, AND THEIR USE AS PDE4 INHIBITORS |
| WO2005090348A1 (en) | 2004-03-16 | 2005-09-29 | Glaxo Group Limited | Pyrazolo ’3,4-b! pyridine compounds, and their use as phosphodiesterase type 4 (pde4) inhibitors |
| WO2005090354A1 (en) | 2004-03-16 | 2005-09-29 | Glaxo Group Limited | PYRAZOLO[3,4-b] PYRIDINE COMPOUNDS, AND THEIR USE AS PDE4 INHIBITORS |
| WO2005104745A2 (en) | 2004-04-27 | 2005-11-10 | Glaxo Group Limited | Muscarinic acetylcholine receptor antagonists |
| WO2006000398A1 (en) | 2004-06-28 | 2006-01-05 | Glaxo Group Limited | 2,3-benzoxazin derivatives as non-steroidal glucocorticoid receptor modulators |
| WO2006000401A1 (en) | 2004-06-28 | 2006-01-05 | Glaxo Group Limited | Substituted oxazines as glucocorticoid receptor modulators |
| WO2006015870A1 (en) | 2004-08-12 | 2006-02-16 | Glaxo Group Limited | Tetrahydro-naphthalene derivatives as glucocorticoid receptor modulators |
| WO2006045416A1 (en) | 2004-10-19 | 2006-05-04 | F. Hoffmann-La Roche Ag | Quinoline derivatives |
| WO2006072599A2 (en) | 2005-01-10 | 2006-07-13 | Glaxo Group Limited | Androstane 17-alpha carbonate derivatives for use in the treatment of allergic and inflammatory conditions |
| WO2006072600A1 (en) | 2005-01-10 | 2006-07-13 | Glaxo Group Limited | Androstane 17-alpha-carbonate for use in the treatment of inflammatory and allergic conditions |
| WO2008025821A1 (en) * | 2006-08-30 | 2008-03-06 | Cellzome Limited | Triazole derivatives as kinase inhibitors |
-
2011
- 2011-03-09 WO PCT/EP2011/053499 patent/WO2011110575A1/en active Application Filing
Patent Citations (76)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993013055A1 (en) | 1991-12-24 | 1993-07-08 | The Wellcome Foundation Limited | Amidino derivatives and their use as nitric oxide synthase inhibitors |
| US5552438A (en) | 1992-04-02 | 1996-09-03 | Smithkline Beecham Corporation | Compounds useful for treating allergic and inflammatory diseases |
| WO1995034534A1 (en) | 1994-06-15 | 1995-12-21 | The Wellcome Foundation Limited | Enzyme inhibitors |
| WO1996032099A1 (en) | 1995-04-14 | 1996-10-17 | Glaxo Wellcome Inc. | Metered dose inhaler for albuterol |
| WO1998030537A1 (en) | 1997-01-13 | 1998-07-16 | Glaxo Group Limited | Nitric oxide synthase inhibitors |
| WO1998054159A1 (en) | 1997-05-30 | 1998-12-03 | Schering Aktiengesellschaft | Non-steroidal (hetero) cyclically substituted acylanilides with mixed gestagen and androgen activity |
| US6431168B1 (en) | 1997-06-10 | 2002-08-13 | Smithkline Beecham Corporation | Dispenser with doses′ counter |
| US6360739B1 (en) | 1997-06-10 | 2002-03-26 | Smithkline Beecham Corporation | Dispenser with doses counter |
| WO1999016766A1 (en) | 1997-10-01 | 1999-04-08 | Kyowa Hakko Kogyo Co., Ltd. | Benzodioxole derivatives |
| WO1999047505A1 (en) | 1998-03-14 | 1999-09-23 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Phthalazinone pde iii/iv inhibitors |
| WO1999062875A1 (en) | 1998-05-30 | 1999-12-09 | Glaxo Group Limited | Nitric oxide synthase inhibitors |
| US6315112B1 (en) | 1998-12-18 | 2001-11-13 | Smithkline Beecham Corporation | Method and package for storing a pressurized container containing a drug |
| US6179118B1 (en) | 1998-12-18 | 2001-01-30 | Glaxo Wellcome Inc. | Method and package for storing a pressurized container containing a drug |
| US6390291B1 (en) | 1998-12-18 | 2002-05-21 | Smithkline Beecham Corporation | Method and package for storing a pressurized container containing a drug |
| US6679374B2 (en) | 1998-12-18 | 2004-01-20 | Smith Kline Beecham Corporation | Package for storing a pressurized container containing a drug |
| US6352152B1 (en) | 1998-12-18 | 2002-03-05 | Smithkline Beecham Corporation | Method and package for storing a pressurized container containing a drug |
| US6119853A (en) | 1998-12-18 | 2000-09-19 | Glaxo Wellcome Inc. | Method and package for storing a pressurized container containing a drug |
| WO2000066590A2 (en) | 1999-05-04 | 2000-11-09 | Ligand Pharmaceuticals, Inc. | Tetracyclic progesterone receptor modulator compounds and methods |
| WO2001004118A2 (en) | 1999-07-14 | 2001-01-18 | Almirall Prodesfarma S.A. | Quinuclidine derivatives and their use as muscarinic m3 receptor ligands |
| WO2001016128A1 (en) | 1999-09-01 | 2001-03-08 | Abbott Laboratories | Dibenzopyrans as glucocorticoid receptor antagonists for treatment of diabetes |
| WO2001042193A1 (en) | 1999-12-08 | 2001-06-14 | Theravance, Inc. | β2-ADRENERGIC RECEPTOR AGONISTS |
| WO2002002565A2 (en) | 2000-07-05 | 2002-01-10 | Abbott Laboratories | Glucocortiocoid-selective antiinflammatory agents |
| WO2002012265A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 6.ALPHA., 9.ALPHA.-DIFLUORO-17.ALPHA.-`(2-FURANYLCARBOXYL) OXY!-11.BETA.-HYDROXY-16.ALPHA.-METHYL-3-OXO-ANDROST-1,4,-DIENE-17-CARBOTHIOIC ACID S-FLUOROMETHYL ESTER AS AN ANTI-INFLAMMATORY AGENT |
| WO2002012266A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 17.beta.-carbothioate 17.alpha.-arylcarbonyloxyloxy androstane derivative as anti-inflammatory agents |
| WO2002026722A1 (en) | 2000-09-29 | 2002-04-04 | Glaxo Group Limited | Morpholin-acetamide derivatives for the treatment of inflammatory diseases |
| WO2002050021A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Nitric oxide synthase inhibitor phosphate salt |
| WO2002066422A1 (en) | 2001-02-14 | 2002-08-29 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2002070490A1 (en) | 2001-03-08 | 2002-09-12 | Glaxo Group Limited | Agonists of beta-adrenoceptors |
| WO2002076933A1 (en) | 2001-03-22 | 2002-10-03 | Glaxo Group Limited | Formailide derivatives as beta2-adrenoreceptor agonists |
| WO2002088167A1 (en) | 2001-04-30 | 2002-11-07 | Glaxo Group Limited | Anti-inflammatory 17.beta.-carbothioate ester derivatives of androstane with a cyclic ester group in position 17.alpha |
| WO2002100879A1 (en) | 2001-06-12 | 2002-12-19 | Glaxo Group Limited | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| WO2003008277A2 (en) | 2001-07-17 | 2003-01-30 | Riverwood International Corporation | Carton with improved dispenser and handle |
| WO2003024439A1 (en) | 2001-09-14 | 2003-03-27 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003042160A1 (en) | 2001-11-13 | 2003-05-22 | Theravance, Inc. | Aryl aniline beta-2 adrenergic receptor agonists |
| WO2003059899A1 (en) | 2002-01-14 | 2003-07-24 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical formulations containing them and uses thereof |
| WO2003061651A1 (en) | 2002-01-22 | 2003-07-31 | The Regents Of The University Of California | Non-steroidal ligands for the glucocorticoid receptor, compositions and uses thereof |
| WO2003072539A1 (en) | 2002-02-28 | 2003-09-04 | Glaxo Group Limited | Phenethanolamine derivatives for treatment of respiratory diseases |
| WO2003082787A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003082280A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003082827A1 (en) | 2002-04-02 | 2003-10-09 | Schering Aktiengesellschaft | Quinoline and isoquinoline derivatives, method for the production thereof and use thereof as anti-inflammatory agents |
| WO2003086294A2 (en) | 2002-04-11 | 2003-10-23 | Merck & Co., Inc. | 1h-benzo[f]indazol-5-yl derivatives as selective glucocorticoid receptor modulators |
| WO2003091204A1 (en) | 2002-04-25 | 2003-11-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2003101932A2 (en) | 2002-05-29 | 2003-12-11 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003104195A1 (en) | 2002-06-06 | 2003-12-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | 4-(aryl or heteroaryl) -2-butylamine derivatives and their use as glucocorticoid ligans |
| WO2004005229A1 (en) | 2002-07-08 | 2004-01-15 | Pfizer Products Inc. | Modulators of the glucocorticoid receptor |
| WO2004009017A2 (en) | 2002-07-18 | 2004-01-29 | Bristol-Myers Squibb Company | Modulators of the glucocorticoid receptor and method |
| WO2004016578A2 (en) | 2002-07-25 | 2004-02-26 | Glaxo Group Limited | Arylethanolamine beta2-adrenoreceptor agonist compounds |
| WO2004018429A2 (en) | 2002-08-21 | 2004-03-04 | Boehringer Ingelheim Pharmaceuticals, Inc. | Substituted hihydroquinolines as glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2004022547A1 (en) | 2002-09-06 | 2004-03-18 | Glaxo Group Limited | Phenethanolamine derivatives and their use in the treatment of respiratory diseases |
| WO2004024728A2 (en) | 2002-09-16 | 2004-03-25 | Glaxo Group Limited | Pyrazolo[3,4-b]pyridine compounds, and their use as phosphodiesterase inhibitors |
| WO2004026248A2 (en) | 2002-09-20 | 2004-04-01 | Merck & Co., Inc. | Octahydro-2-h-naphtho[1,2-f] indole-4-carboxamide derivatives as selective glucocorticoid receptor modulators |
| WO2004035556A1 (en) | 2002-10-16 | 2004-04-29 | Glaxo Group Limited | Substituted piperazines, (1,4) diaszepines, and 2,5-diazabicyclo (2.2.1) heptanes as histamine h1 and/or h3 antagonists or histamine h3 reverse antagonists |
| WO2004037807A2 (en) | 2002-10-22 | 2004-05-06 | Glaxo Group Limited | Medicinal arylethanolamine compounds |
| WO2004037768A2 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivatives |
| WO2004037773A1 (en) | 2002-10-28 | 2004-05-06 | Glaxo Group Limited | Phenethanolamine derivative for the treatment of respiratory diseases |
| WO2004039766A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenylethanolamine derivatives for the treatment of respiratory diseases |
| WO2004039762A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenethanolamine derivatives for the treatment of respiratory diseases |
| WO2004056823A1 (en) | 2002-12-23 | 2004-07-08 | Glaxo Group Limited | PYRAZOLO[3,4-b]PYRIDINE COMPOUNDS, AND THEIR USE AS PHOSPHODIESTERASE INHIBITORS |
| WO2004103998A1 (en) | 2003-05-21 | 2004-12-02 | Glaxo Group Limited | Quinoline derivatives as phosphodiesterase inhibitors |
| WO2005005451A1 (en) | 2003-07-11 | 2005-01-20 | Glaxo Group Limited | Specific glucocorticosteroid compound having anti- inflammatory activity |
| WO2005005452A1 (en) | 2003-07-11 | 2005-01-20 | Glaxo Group Limited | Specific glucocorticosteroid compound having anti- inflammatory activity |
| WO2005037280A1 (en) | 2003-10-14 | 2005-04-28 | Glaxo Group Limited | Muscarinic acetycholine receptor antagonists |
| WO2005044354A1 (en) | 2003-11-03 | 2005-05-19 | Glaxo Group Limited | A fluid dispensing device |
| WO2005046586A2 (en) | 2003-11-04 | 2005-05-26 | Glaxo Group Limited | M3 muscarinic acetylcholine receptor antagonists |
| WO2005058892A1 (en) | 2003-12-19 | 2005-06-30 | Glaxo Group Limited | Pyrazolo [3,4-b] pyridine compounds, and their use as phosphodiesterase inhibitors |
| WO2005090348A1 (en) | 2004-03-16 | 2005-09-29 | Glaxo Group Limited | Pyrazolo ’3,4-b! pyridine compounds, and their use as phosphodiesterase type 4 (pde4) inhibitors |
| WO2005090353A1 (en) | 2004-03-16 | 2005-09-29 | Glaxo Group Limited | PYRAZOLO[3,4-b]PYRIDINE COMPOUNDS, AND THEIR USE AS PDE4 INHIBITORS |
| WO2005090354A1 (en) | 2004-03-16 | 2005-09-29 | Glaxo Group Limited | PYRAZOLO[3,4-b] PYRIDINE COMPOUNDS, AND THEIR USE AS PDE4 INHIBITORS |
| WO2005104745A2 (en) | 2004-04-27 | 2005-11-10 | Glaxo Group Limited | Muscarinic acetylcholine receptor antagonists |
| WO2006000398A1 (en) | 2004-06-28 | 2006-01-05 | Glaxo Group Limited | 2,3-benzoxazin derivatives as non-steroidal glucocorticoid receptor modulators |
| WO2006000401A1 (en) | 2004-06-28 | 2006-01-05 | Glaxo Group Limited | Substituted oxazines as glucocorticoid receptor modulators |
| WO2006015870A1 (en) | 2004-08-12 | 2006-02-16 | Glaxo Group Limited | Tetrahydro-naphthalene derivatives as glucocorticoid receptor modulators |
| WO2006045416A1 (en) | 2004-10-19 | 2006-05-04 | F. Hoffmann-La Roche Ag | Quinoline derivatives |
| WO2006072599A2 (en) | 2005-01-10 | 2006-07-13 | Glaxo Group Limited | Androstane 17-alpha carbonate derivatives for use in the treatment of allergic and inflammatory conditions |
| WO2006072600A1 (en) | 2005-01-10 | 2006-07-13 | Glaxo Group Limited | Androstane 17-alpha-carbonate for use in the treatment of inflammatory and allergic conditions |
| WO2008025821A1 (en) * | 2006-08-30 | 2008-03-06 | Cellzome Limited | Triazole derivatives as kinase inhibitors |
Non-Patent Citations (35)
| Title |
|---|
| "Comprehensive Heterocyclic Chemistry 11", 1996, PERGAMON PRESS |
| "Comprehensive Organic Chemistry", 1979, PERGAMON PRESS |
| "Comprehensive Organic Functional Group Transformations", 1995, ELSEVIER SCIENCE LTD. |
| "Remington's Pharmaceutical Sciences", MACK PUBLISHING COMPANY |
| "Reminqton's Pharmaceutical Sciences", MACK PUBLISHING COMPANY |
| "The Handbook of Pharmaceutical Additives", GOWER PUBLISHING LIMITED |
| "The Handbook of Pharmaceutical Excipients", THE AMERICAN PHARMACEUTICAL ASSOCIATION AND THE PHARMACEUTICAL PRESS |
| ALEXANDER ET AL., THE LANCET, vol. 339, 1992, pages 324 - 328 |
| AU-YEUNG ET AL., J IMMUNOL, vol. 176, 2006, pages 3895 - 3899 |
| BENSON ET AL., ALLERGY, 2009 |
| BRITISH PHARMACOPAEIA, 1988, pages A204 - 207 |
| BUSSE ET AL., AM J RESPIR CRIT CARE MED, vol. 178, 2008, pages 1002 - 1008 |
| DAS, J. ET AL.: "Discovery of 2-amino-heteroaryl-benzothiazole-6-anilides as potent p56lck inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 13, no. 15, 2003, Pergamon, Elsevier Science B.B.; NL, pages 2587 - 2590, XP002646268, ISSN: 0960-894X * |
| FERRARA ET AL., J ALLERGY CLIN IMMUNOL, vol. 117, 2006, pages 780 - 786 |
| FISCHER ET AL., J BIOL CHEM, vol. 279, no. 28, 2004, pages 29816 - 29820 |
| FORSSELL ET AL., AM J RESPIR CELL MOL BIOL, vol. 32, 2005, pages 511 - 520 |
| FOWELL ET AL., IMMUNITY, vol. 11, 1999, pages 399 - 409 |
| GRASIS ET AL., J IMMUNOL, vol. 170, 2003, pages 3971 - 3976 |
| GRAVES ET AL., J ALLERGY CLIN IMMUNOL, vol. 116, 2005, pages 650 - 656 |
| HOFGEN, N. ET AL., 15TH EFMC INT SYMP MED CHEM, 1998 |
| JABLONOWSKI ET AL., J. MED. CHEM., vol. 46, 2003, pages 3957 - 3960 |
| KAWAKAMI ET AL., J IMMUNOL, vol. 155, 1995, pages 3556 - 3562 |
| LANDELLS, L.J. ET AL., EUR RESP J [ANNU CONG EUR RESP SOC, vol. 12, 19 September 1998 (1998-09-19) |
| LIN ET AL., BIOCHEMISTRY, vol. 43, no. 34, 2004, pages 11056 - 11062 |
| LOCK ET AL., AM J RESPIR CRIT CARE MED, vol. 153, 1996, pages 509 - 514 |
| MATSUMOTO ET AL., INT ARCH ALLERGY IMMUNOL, vol. 129, 2002, pages 327 - 340 |
| MILLER ET AL., IMMUNITY, vol. 21, 2004, pages 67 - 80 |
| MUELLER; AUGUST, J IMMUNOL, vol. 170, 2003, pages 5056 - 5063 |
| NIZANKOWSKA ET AL., EUR RESPIR J, vol. 8, 1995, pages 1091 - 1099 |
| PHARMACEUTICAL RESEARCH, vol. 3, no. 6, 1986, pages 318 |
| R.C. LAROCK: "Comprehensive Organic Transformations", 1989, VCH PUBLISHERS INC. |
| SCHAEFFER ET AL., NATURE IMMUNOL, vol. 2, no. 12, 2001, pages 1183 - 1188 |
| SCHWARTZBERG ET AL., NATURE REVIEWS IMMUNOL, vol. 5, 2005, pages 284 - 295 |
| SIHRA ET AL., THORAX, vol. 52, 1997, pages 447 - 452 |
| TANABE SEIYAKU; FUJI, K. ET AL., J PHARMACOL EXP THER, vol. 284, no. 1, 1998, pages 162 |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2632900C2 (en) * | 2010-11-19 | 2017-10-11 | Лиганд Фармасьютикалс Инкорпорейтед | Heterocyclic amines and their application |
| CN108864151A (en) * | 2010-11-19 | 2018-11-23 | 利亘制药公司 | heterocyclic amine and application thereof |
| JP2013544256A (en) * | 2010-11-19 | 2013-12-12 | リガンド ファーマシューティカルズ インコーポレイテッド | Heterocyclic amines and uses thereof |
| US11773110B2 (en) | 2010-11-19 | 2023-10-03 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US11186593B2 (en) | 2010-11-19 | 2021-11-30 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US10604533B2 (en) | 2010-11-19 | 2020-03-31 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US10030034B2 (en) | 2010-11-19 | 2018-07-24 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US20140155379A1 (en) * | 2010-11-19 | 2014-06-05 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| US9266874B2 (en) * | 2010-11-19 | 2016-02-23 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| CN103800337A (en) * | 2012-11-07 | 2014-05-21 | 韩冰 | Compound for treating neurodegenerative diseases and application thereof |
| US20160207928A1 (en) * | 2013-06-10 | 2016-07-21 | Bayer Pharma Aktiengesellschaft | Novel compounds for the treatment of cancer |
| JP2016521737A (en) * | 2013-06-10 | 2016-07-25 | バイエル・ファルマ・アクティエンゲゼルシャフト | Novel compounds for treating cancer |
| WO2014202505A1 (en) | 2013-06-20 | 2014-12-24 | Bayer Cropscience Ag | Aryl sulfide derivatives and aryl sulfoxide derivatives as acaricides and insecticides |
| US10807983B2 (en) | 2015-03-16 | 2020-10-20 | Ligand Pharmaceuticals, Inc. | Imidazo-fused heterocycles and uses thereof |
| US11858938B2 (en) | 2015-03-16 | 2024-01-02 | Ligand Pharmaceuticals, Inc. | Imidazo-fused heterocycles and uses thereof |
| WO2017157885A1 (en) | 2016-03-16 | 2017-09-21 | Bayer Cropscience Aktiengesellschaft | N-(cyanobenzyl)-6-(cyclopropyl-carbonylamino)-4-(phenyl)-pyridine-2-carboxamide derivatives and related compounds as pesticides and plant protection agents |
| CN107987097A (en) * | 2017-12-17 | 2018-05-04 | 沧州普瑞东方科技有限公司 | The synthesis technique of 2,6- dichloropyridine -4- boric acid pinacol esters |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10946025B2 (en) | Compounds | |
| EP2280705B1 (en) | Novel compounds | |
| EP2300437B1 (en) | Benzpyrazol derivatives as inhibitors of pi3 kinases | |
| EP2280959B1 (en) | 4-amino-indazoles | |
| US20120058984A1 (en) | Pyrimidine derivatives used as itk inhibitors | |
| US20120238571A1 (en) | Indazole derivatives as pi 3-kinase | |
| US20120245171A1 (en) | Benzpyrazole derivatives as inhibitors of pi3 kinases | |
| US20110112070A1 (en) | 4-carboxamide indazole derivatives useful as inhibitors of p13-kinases | |
| WO2012035055A1 (en) | Novel compounds | |
| WO2011067364A1 (en) | Novel compounds | |
| WO2011110575A1 (en) | Derivatives of 2-[2-(benzo- or pyrido-) thiazolylamino]-6-aminopyridine, useful in the treatment of respiratoric, allergic or inflammatory diseases | |
| US10793559B2 (en) | Chemical compounds | |
| EP3612536B1 (en) | Oxepinopyrazole derivatives as inhibitors of pi3-kinase activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11706847 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11706847 Country of ref document: EP Kind code of ref document: A1 |