WO2010077230A1 - New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers - Google Patents
New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers Download PDFInfo
- Publication number
- WO2010077230A1 WO2010077230A1 PCT/US2008/014144 US2008014144W WO2010077230A1 WO 2010077230 A1 WO2010077230 A1 WO 2010077230A1 US 2008014144 W US2008014144 W US 2008014144W WO 2010077230 A1 WO2010077230 A1 WO 2010077230A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- carbon atoms
- methyl
- zirconium dichloride
- phenyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/6592—Component covered by group C08F4/64 containing a transition metal-carbon bond containing at least one cyclopentadienyl ring, condensed or not, e.g. an indenyl or a fluorenyl ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F17/00—Metallocenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F10/04—Monomers containing three or four carbon atoms
- C08F10/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/642—Component covered by group C08F4/64 with an organo-aluminium compound
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/04—Monomers containing three or four carbon atoms
- C08F210/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/16—Copolymers of ethene with alpha-alkenes, e.g. EP rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65912—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an organoaluminium compound
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65916—Component covered by group C08F4/64 containing a transition metal-carbon bond supported on a carrier, e.g. silica, MgCl2, polymer
Definitions
- the present invention relates to novel metallocene compounds useful as components in polymerization catalysts, to catalysts comprising such metallocene compounds, to a process for the polymerization of olefins and to particularly propylene, and olefin homopolymers, random, and impact copolymers prepared by using the metallocene catalysts.
- Another key requirement of a metallocene catalyst is its capability to produce polypropylene with a high melting point. This is equivalent with a catalyst that has a very high stereospecificity and regioselectivity.
- a catalyst that has a very high stereospecificity and regioselectivity.
- the stereospecificity and regioselectivity has continuously been improved during the last 15 years.
- EP-Al 834 519 relates to metallocenes of the rac-Me 2 Si(2-Me-4-Ar-Ind) 2 ZrCl 2 for the production of high rigid, high Tm polypropylenes with very high stereoregularity and very low amounts of regio errors.
- EP-Al 834 519 Although not tested for their copolymerization performance, the metallocenes disclosed in EP-Al 834 519 anticipated substitution patterns in 2-position that would later be identified as particularly suitable for the production of propylen ⁇ 'ethylene random copolymers when combined with additional substituents in certain positions. However, the highly stereo- and regio regular polypropylenes were not obtained under commercially relevant process conditions and suffered from too low activity/productivity levels.
- US-Al 2001/0053833 discloses metallocenes having substituents in 2-position consisting of an unsubstituted heteroaromatic ring or a heteroaromatic ring having at least one substituent bonded to the ring.
- Such catalysts afford C3/C2 copolymers with reasonably high molar mass, but fail to produce high T m homopolymers under conditions typical for commercial scale production, i. e. on a support and at temperatures from 60 deg C and higher. Also, the productivities of this catalyst family are unsatisfactory.
- WO 01/058970 relates to impact copolymers having a high melting point and a high rubber molar mass, produced by catalysts comprising metallocenes of the rao-Me 2 Si(2-Alk-4-Ar- Ind) 2 ZrCl 2 family. High molar masses in the propylene/ethylene rubber were achieved when both AIk substituents were i-propyl groups.
- WO 02/002576 discloses bridged metallocenes of the (2- R-4-Ph-lnd) 2 ZrCl 2 family having particular combinations of substituents in the 2-positions of the indenyl ligands and substituents in the benzene ring.
- a high polypropylene (PP) melting point is favored if the Ph group exhibits a substitution pattern in the 3 and 5 positions, particularly in case of butyl substituents.
- a combination of high homopolymer melting point and high copolymer molar mass is achieved if both substituents R in 2-position are isopropyl groups.
- the major shortcoming is the very low activity/productivity of the rac-Me 2 Si(2-R-4-Ar-Ind) 2 ZrCl 2 catalysts if both ligands R are branched in the ⁇ -position.
- WO 03/002583 discloses bridged metallocenes of the (2-R-4-Ph-Ind) 2 ZrCl 2 family having particular combinations of substituents in the 2- positions of the indenyl ligands and the 4-Ph substituents.
- a high PP melting point is favored if the Ph group exhibits a substitution pattern in the 2-position, particularly in case of biphenyl substituents.
- a combination of high homopolymer melting point and high copolymer molar mass is achieved if both substituents R in 2-position of the indenyl ligand are isopropyl groups.
- EP-A2 1 250 365, WO 97/40075 and WO 03/045551 relate to metallocenes having substituents in the 2-positions of either of the indenyl ligands with the imperative that at least one of the ligands in 2-position is branched or cyclicized in the ⁇ -position.
- WO 04/106351 relates to metallocenes having substituents in the 2-positions of the indenyl ligands with the proviso that one ligand is unbranched or bound via an sp 2 -hybridized carbon atom and the other ligand is branched in the ⁇ -position.
- Such catalysts afford high Tm homopolymers and high molar mass propylene/ethylene copolymers.
- catalyst activity/productivity and lowest achievable homopolymer melt flow rate there still are limitations with regard to catalyst activity/productivity and lowest achievable homopolymer melt flow rate.
- PCT/US2007/022614 a co-pending applications by the present inventors, demonstrated that metallocenes having ligands being ⁇ -branched in the 2-position created surprising increases in activity of the metallocene catalyst and also created products with unexpectedly superior properties than previously known metallocenes.
- the inventors now have discovered a species of the previously revealed genus that unexpectedly produces even significantly higher increases in catalyst activity and improvements in product properties.
- the main deficiency of supported catalyst systems comprising metallocenes of the above mentioned prior art, is that so far no catalyst has been found that, when used for the homopolymerization of propylene, affords isotactic polypropylene with a high melting point and very high molar mass (or very low melt flow rate) and that, when used for the copolymerization of propylene with ethylene, affords high molar mass propylene/ethylene copolymers, all at very high catalyst productivity.
- An object of the present invention is to address this shortcoming of the state of the art metallocene compounds and to provide metallocenes that increase desirablecharacteristics such as high melting point, high molar mass homopolymers and high molar mass copolymers, and do so at higher productivities when used as components of supported catalysts under industrially relevant polymerization conditions at temperatures of from 50 °C to 100 °C.
- the inventions of the current example provide these advantages by using a metallocene with symmetrically substituted 2 positions on the indenyl group. This is significantly more cost effective, and therefore far more desirable, than the comparative examples that have asymmetric substitution.
- Another objective of the present invention is to provide a process for the polymerization of olefins, particularly propylene, ethylene, and optionally one or more higher 1-olefins.
- olefin polymers particularly propylene homopolymers, random copolymers of propylene with ethylene and/or higher 1-olefins, impact copolymers comprised of propylene, ethylene and/or optionally higher 1-olefins, and random impact copolymers comprised of propylene, ethylene and/or optionally higher 1 -olefins.
- Certain metallocene compounds are provided that, when used as a component in a supported polymerization catalyst under industrially relevant polymerization conditions, afford high molar mass homo polymers or copolymers like polypropylene or propylene/ethylene copolymers without the need for any ⁇ -branched substituent in either of the two available 2- positions of the indenyl ligands.
- the substituent in the 2-position of one indenyl ligand can be any radical comprising hydrogen, methyl, or any other C 2 -C 4O hydrocarbon which is not branched in the ⁇ -position, and the substituent in the 2-position of the other indenyl ligand can be any C 5 -C 4 O hydrocarbon radical with the proviso that this hydrocarbon radical is branched in the ⁇ -position and that the ⁇ -carbon atom is a quarternary carbon atom and part of a mono-cyclic hydrocarbon system.
- the substituents in the 2-position of both indenyl ligands are identical and are any Q-C 4O hydrocarbon radical with the proviso that this hydrocarbon radical is branched in the ⁇ -position and that the ⁇ -carbon atom is a quarternary carbon atom and part of a mono-cyclic hydrocarbon system.
- This metallocene topology affords high melting point, very high molar mass homo polypropylene and very high molar mass propylene-based copolymers.
- the activity/productivity levels of catalysts comprising the metallocenes of the present invention are exceptionally high.
- M 1 is a metal of Group IVb of the Periodic Table of the Elements
- R 1 and R 2 are identical or different and are selected from the group consisting of a hydrogen atom, an alkyl group of from 1 to about 10 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryl group of from 6 to about 20 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an OH group, a halogen atom, or a NR 2 group, where R is an alky 1 group of from 1 to about 10 carbon atoms and an aryl group of from 6 to about 14 carbon atoms, and wherein R 1 and R 2 may form one or more ring system(s),
- R 4 and R 4 are identical or different and are selected from the group consisting of a hydrogen atom and a linear, cyclic or branched hydrocarbon group optionally containing one or more hetero atoms selected from the group consisting of Si, B, Al, O, S, N, P, F, Cl and Br,
- R 10 is a bridging group wherein R 10 is selected from:
- R )40 and I r R>41 can be identical or different and can optionally contain heteroatoms selected from the group consisting of Si, B, Al, O, S, N, P, Cl and Br, and are selected from the group consisting of a hydrogen atom, an alkyl group having from 1 to about 30 carbon atoms, an aryl group of from 6 to about 40 carbon atoms, a fluoroalkyl group of from 1 to about 10 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, a substituted or unsubstituted alkylsilyl, an alkyl(aryl)silyl group, an arylsilyl group, and an ary
- M 12 is silicon, germanium or tin
- R 10 can optionally link two units of the formula 1 to one another,
- R 1 ' and R 1 ' are identical or different and are each a divalent C 2 -C 40 group which together with the cyclopentadienyl ring forms a further saturated or unsaturated ring system having a ring size of from 5 to 7 atoms, where R 11 and R 11 optionally contain the heteroatoms Si, Ge, N, P, O or S within the ring system fused onto the cyclopentadienyl ring, and R 300 has the structure
- R 301 is a linear, cyclic or branched hydrocarbon group selected from the group consisting of an alkyl group of from 1 to about 20 carbon atoms, an alkenyl group of from 2 to about 20 carbon atoms, an aryl group of from 6 to about 20 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, or an arylalkenyl group of from 8 to about 40 carbon atoms, an alkoxy group of from 1 to about 20 carbon atoms, an aryloxy group of from 6 to about 20 carbon atoms, or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group and an arylsilyl group, wherein each of the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br
- R 302 is a hydrocarbon group selected from the group consisting of a substituted or unsubstituted alkyl group of from 2 to about 20 carbon atoms, and an substituted or unsubstituted alkenyl group of from 3 to about 20 carbon atoms, and wherein those groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br, and further wherein R 302 forms a monocyclic ring with the ⁇ carbon atom.
- R 3 has the meaning of R 300 , but R 3 need not be identical to R 300 , or R 3 is a linear, cyclic or branched hydrocarbon group which optionally can contain one or more hetero atoms selected from the group consisting of Si, B, Al, O, S, N, P, F, Cl or Br, or R 3 is selected from the group consisting of an alkyl group of from 1 to about 20 carbon atoms, an alkylalkenyl group of from 3 to about 20 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, and an alkylarylalkenyl group of from 9 to about 40 carbon atoms, with the proviso that, in any case, R 3 is not branched in the ⁇ -position.
- Another embodiment described herein is a process for olefin polymerisation comprising contacting one or more olefins each having from 2 to about 20 carbon atoms under olefin polymerisation reaction conditions with a catalyst system including a bridged metal locene component having Formula 1 shown above.
- M 1 is a metal of Group IVb of the Periodic Table of the Elements
- R 1 and R 2 are identical or different and are selected from the group consisting of a hydrogen atom, an alkyl group of from 1 to about 10 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryl group of from 6 to about 20 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an OH group, a halogen atom, or a NR 2 32 group, where R 32 is an alkyl group of from 1 to about 10 carbon atoms and an aryl group of from 6 to about 14 carbon atoms and R 1 and R 2 may form one or more ring system(s),
- R 4 and R 4 are identical or different and are selected from the group consisting of a hydrogen atom and a linear, cyclic or branched hydrocarbon group optionally containing one or more hetero atoms selected from the group consisting of Si, B, Al, O, S, N, P, F, Cl and Br, R 10 is a bridging group wherein R 10 is selected from:
- R 40 and R 41 can be identical or different and can optionally contain heteroatoms selected from the group consisting of Si, B, Al, O, S, N, P, Cl and Br, and are each selected from the group consisting of a hydrogen atom, an alkyl group having from 1 to about 30 carbon atoms, an aryl group of from 6 to about 40 carbon atoms, a fluoroalkyl group of from 1 to about 10 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, a substituted or unsubstituted alkylsilyl, an alkyl(aryl)silyl group, an arylsilyl group, or an arylalkenyl
- M 12 is silicon, germanium or tin
- R 10 can optionally link two units of the formula 1 to one another, and R 300 has the structure:
- R 301 is a linear, cyclic or branched hydrocarbon groupselected from the group consisting of an alkyl group of from 1 to about 20 carbon atoms, an alkenyl group of from 2 to about 20 carbon atoms, an aryl group of from 6 to about 20 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, or an arylalkenyl group of from 8 to about 40 carbon atoms, an alkoxy group of from 1 to about 20 carbon atoms, an aryloxy group of from 6 to about 20 carbon atoms, or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group and an arylsilyl group, wherein the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and/or may contain halogen atoms like F, Cl or Br,
- R 302 is a hydrocarbon group selected from the group consisting of a substituted or unsubstituted alkyl group of from 2 to about 20 carbon atoms, and an substituted or unsubstituted alkenyl group of from 3 to about 20 carbon atoms, and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and/or may contain halogen atoms like F, Cl or Br, wherein R 302 forms a monocyclic ring with the ⁇ carbon atom.
- R has the meaning of R , but R need not be identical to R , or R is a linear, cyclic or branched hydrocarbon group which optionally can contain one or more hetero atoms selected from the group consisting of Si, B, Al, O, S, N, P, F, Cl or Br or R 3 is selected from the group consisting of an alkyl group of from 1 to about 20 carbon atoms, an alkylalkenyl group of from 3 to about 20 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, and an alkylarylalkenyl group of from 9 to about 40 carbon atoms, with the proviso that R 3 , in any case, is not branched in the ⁇ -position, and where R 5 , R 6 , R 7 and R 8 and also R 5' , R 6' , R 7' and R 8' are identical or different and are each selected from the group consisting of a hydrogen atom, a linear, cyclic or branched hydrocarbon group or
- Fig. 1 illustrates individual steps of the process for producing transition metal compounds of the bridged metallocene compound of the invention.
- Fig. 2 is a photograph of the copolymer obtained in Comparative Example 20.
- Fig. 3 is a photograph of the copolymer obtained in Example 19.
- a supported catalyst system comprising at least one specifically substituted and bridged metallocene, at least one cocatalyst, at least one support and, if desired, at least one metal compound and a further additive component.
- the catalyst system is prepared by mixing at least one specifically substituted and bridged metallocene, at least one cocatalyst, at least one support and if desired at least one metal compound and a further additive component.
- the first embodiment of the invention relates to a substituted, bridged metallocene component of the general Formula 1 below,
- M 1 is a metal of Group IVb of the Periodic Table of the Elements, preferably zirconium or hafnium, and particularly preferably zirconium.
- R 1 and R 2 are identical or ⁇ different and are each a hydrogen atom, an alkyl group of from 1 to about 10 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryl group of from 6 to about 20 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an OH group, a halogen atom, or a NR 2 32 group, where R 32 is an alkyl group of from 1 to about 10 carbon atoms or an aryl group of from 6 to about 14 carbon atoms and R 1 and R 2 may form one or more ring system(s).
- R 1 and R 2 are identical or different and are an alkyl group of from 1 to about 10 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms or a halogen atom, or R 1 and R 2 together may form one or more ring system(s).
- R 1 and R 2 are identical or different and are methyl, chbrine or phenolate.
- R 4 and R 4 are identical or different and are each a hydrogen atom, a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 1 to 20 carbon atoms, an alkenyl group of from 2 to 20 carbon atoms, an aryl group of from 6 to 20 carbon atoms, an arylalkyl group of from 7 to 40 carbon atoms, an alkylaryl group of from 8 to about 40 carbon atoms, or an arylalkenyl group of from 8 to about 40 carbon atoms or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group or an arylsilyl group.
- a linear, cyclic or branched hydrocarbon group for example an alkyl group of from 1 to 20 carbon atoms, an alkenyl group of from 2 to 20 carbon atoms, an aryl group of from 6 to 20 carbon atoms, an arylalkyl group of from 7 to
- the group may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R 4 and R 4 are identical or different and are each a hydrogen atom, a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 1 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an aryl group of from 6 to about 10 carbon atoms, an arylalkyl group of from 7 to about 20 carbon atoms, an alkylaryl group of from 8 to about 20 carbon atoms, or an arylalkenyl group of from 8 to about 20 carbon atoms or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group or an arylsilyl group.
- the groups may be halogenated. Particularly preferably, R 4 and R 4 are both hydrogen.
- R 10 is a bridging group wherein R 10 is selected from:
- R 40 and R 41 can be identical or different and are each a hydrogen atom, a Ci-C 40 group such as an alkyl group having from 1 to about 30 carbon atoms, an aryl group of from 6 to about 40 carbon atoms, a fluoroalkyl group of from 1 to about 10 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, a substituted or unsubstituted alkylsilyl, alkyl(aryl)silyl or arylsilyl group, or an aryl alkenyl group of from 8 to about 40 carbon atoms.
- a Ci-C 40 group such as an alkyl group having from 1 to about 30 carbon atoms, an ary
- R 40 and R 41 together with the atoms connecting them can form one or more cyclic systems or R 40 and/or R 41 can contain additional hetero atoms (i.e., non-carbon atoms) like Si, B, Al, O, S, N or P or halogen atoms like Cl or Br, x is an integer from 1 to 18,
- M 12 is silicon, germanium or tin
- R 10 may also link two units of the formula 1 to one another.
- R 40 and R 41 are identical or different and are each a hydrogen atom, a hydrocarbon group of from 1 to about 30 carbon atoms, in particular an alkyl group of from 1 to about 10 carbon atoms, an aryl group of from 6 to about 40 carbon atom
- R 1 ' and R 1 ' are identical or different and are each a divalent C 2 -C 40 group which together with the cyclopentadienyl ring forms a further saturated or unsaturated ring system having a ring size of from 5 to 7 atoms, where R 1 1 and R 11 may contain the heteroatoms Si, Ge, N, P, O or S within the ring system fused onto the cyclopentadienyl ring.
- the groups R 1 ' and R 1 ' are identical or different and are each a divalent group selected from those given in Formulae 1 .(ft, ⁇ , ⁇ , ⁇ , and v and Formulae 1 q ⁇ ', ⁇ ', ⁇ ', ⁇ ', and v', respectively.
- R 1 1 is represented by Formula l ⁇ and R 11 is represented by Formula l ⁇ ', then the structure given in Formula Ia (see below) is obtained.
- R 1 ' and R 1 ' are identical or different and R 1 ' is a divalent group according to Formula l ⁇ and R 1 ' is selected from the divalent groups in Formulae l ⁇ ', ⁇ ', and ⁇ ' or R 1 ' and R 1 ' are identical or different and are divalent groups according to Formula l ⁇ and l ⁇ ' or Formula l ⁇ and l ⁇ ' or Formula l ⁇ and l ⁇ ' or Formula l ⁇ and l ⁇ ' or Formula l ⁇ and 1 ⁇ ' or Formula Iv and 1 v', respectively,
- R 5 , R 6 , R 7 , R 8 , and R 9 and also R 5' , R 6' , R 7' , R 8' and R 9' as well as R 55 , R 66 , R 77 , R 88 and R 99 and also R 55' , R 66' , R 77' , R 88' and R 99 are identical or different and are each a hydrogen atom, a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 2 to about 20 carbon atoms, an alkenyl group of from 2 to about 20 carbon atoms, an aryl group of from 6 to about 40 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, or an arylalkenyl group of from 8 to about 40 carbon atoms or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl
- the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain
- R 55 , R 66 , R 77 , R 88 and R 99 and also R 55' , R 66' , R 77' , R 88' and R 99' are each a hydrogen atom and R 5 , R 6 , R 7 , R 8 and R 9 and also R 5' , R 6' , R 7' , R 8' and R 9' are identical or different and are each a hydrogen atom, a substituted or unsubstituted alkylsilyl or arylsilyl group, a linear, cyclic or branched alkyl group of from 1 to about 10 carbon atoms, or an aryl group of from 6 to about 40 carbon atoms and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R 5 /R 6 and also R 5 /R 6 may form a hydrocarbon ring system or R 5 and R 5 are identical or different and are each a substituted or unsubstituted aryl group of from 6 to about 40 carbon atoms.
- R 55 , R 66 , R 77 , R 88 and R 99 and also R 55' , R 66' , R 77' , R 88' and R 99' are each a hydrogen atom andR 5 , R 6 , R 7 , R 8 and R 9 and also R 5' , R 6' , R 7' , R 8> and R 9' are identical or different and are each a hydrogen atom or a linear, cyclic or branched alkyl group of from 1 to about 10 carbon atoms, or an aryl group of from 6 to about 40 carbon atoms.
- R 5 , R 6 and also R 5 , R 6 together may form a ring system or R 5 and R 5 are identical or different and are each a substituted or unsubstituted aryl group of from 6 to about 40 carbon atoms.
- R 300 is a
- R 301 is a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 1 to about 20 carbon atoms, an alkenyl group of from 2 to about 20 carbon atoms, an aryl group of from 6 to about 20 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, or an arylalkenyl group of from 8 to about 40 carbon atoms, an alkoxy group of from 1 to about 20 carbon atoms, an aryloxy group of from 6 to about 20 carbon atoms, or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group or an arylsilyl group, and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R 301 is
- R301 is a is a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 1 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an aryl group of from 6 to about 10 carbon atoms, an arylalkyl group of from 7 to about 20 carbon atoms, an alkylaryl group of from 7 to about 20 carbon atoms, or an arylalkenyl group of from 8 to about 20 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group or an arylsilyl group, and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R301 is a is a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 1 to about 4 carbon atoms, an aryl group of from 6 to about 10 carbon atoms, an arylalkyl group of from 7 to about 20 carbon atoms, an alkylaryl group of from 7 to about 20 carbon atoms, an alkoxy group of from 1 to about 10 carbon atoms, an aryloxy group of from 6 to about 10 carbon atoms, and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- an alkyl group of from 1 to about 4 carbon atoms for example an alkyl group of from 1 to about 4 carbon atoms, an aryl group of from 6 to about 10 carbon atoms, an arylalkyl group of from 7 to about 20 carbon atoms, an alkylaryl group of from 7 to about 20 carbon
- R301 is a linear or branched hydrocarbon group, for example an alkyl group of from 1 to about 4 carbon atoms,
- R 302 is a hydrocarbon group which is building a mono-cyclic ring with the ⁇ -carbon atom, for example a substituted or unsubstituted alkyl group of from 2 to about 20 carbon atoms, an substituted or unsubstituted alkenyl group of from 3 to about 20 carbon atoms, and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R302 is a hydrocarbon group which is building a mono-cyclic ring with the ⁇ - carbon atom, for example a substituted or unsubstituted alkyl group of from 2 to about 10 carbon atoms, and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, arid / or may contain halogen atoms like F, Cl or Br.
- R302 is a hydrocarbon group which is building a mono-cyclic ring with the ⁇ -carbon atom, for example a substituted or unsubstituted alkyl group of from 3 to about 7 carbon atoms, and the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br, with the proviso that R 300 contain more than 6 carbon atoms.
- R may be chosen from the same set that described R 300 (but R 3 need not be identical to R ) or R 3 is a hydrogen atom, a linear, cyclic or branched hydrocarbon group which may be halogenated and/or may contain one or more hetero atoms like Si, B, Al, O, S, N or P, for example an alkyl group of from 1 to about 20 carbon atoms, an alkylalkenyl group of from 3 to about 20 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, or an alkylarylalkenyl group of from 9 to about 40 carbon atoms, with the proviso that R 3 is not cyclic or branched in ⁇ -position.
- R 3 is chosen from the same set that described R 300 (but R 3 need not be identical to R 300 ) or R 3 is a linear, cyclic or branched hydrocarbon group of from 1 to about 20 carbon atoms, for example an alkyl group of from 1 to 20 carbon atoms, an alkylaryl group of from 7 to about 20 carbon atoms, an alkylalkenyl group of from 3 to about 20 carbon atoms or an alkylarylalkenyl group of from 9 to about 20 carbon atoms with the proviso that R 3 is not cyclic or branched in ⁇ -position.
- R 3 and R 300 are identical or R 3 is a methyl group or a linear, cyclic or branched hydrocarbon group of from 7 to about 10 carbon atoms which may be halogenated, an alkylaryl group of from 7 to about 10 carbon atoms or an alkylalkenyl group of from 7 to about 10 carbon atoms with the proviso that R 3 is not cyclic or branched in ⁇ -position.
- R 3 and R 300 are identical with the proviso that R 3 and R 300 contain more than 6 carbon atoms.
- either of R 3 and R 300 , or both are not cyclic or, in the alternative, are not branced in the ⁇ -carbon position.
- the specifically substituted, bridged metal locene component of the first embodiment of the invention is as given in Formula Ia below.
- M 1 , R 1 , R 2 , R 3 , R 4 , R 4 , R 10 and R 300 have the meaning set forth above with respect to Formula 1.
- R 5 , R 6 , R 7 and R 8 and also R 5' , R 6' , R 7' and R 8' are identical or different and are each a hydrogen atom, a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 1 to about 20 carbon atoms, an alkenyl group of from 2 to about 20 carbon atoms, an aryl group of from 6 to about 40 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, or an arylalkenyl group of from 8 to about 40 carbon atoms or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group or an arylsilyl group.
- a linear, cyclic or branched hydrocarbon group for example an alkyl group of from 1 to about 20 carbon atoms, an alkenyl group of from 2 to about
- the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br, and / or two adjacent radicals R 5 , R 6 or R 6 , R 7 or R 7 , R 8 and also R 5> , R 6' or R 6> , R 7' or R 7' , R 8' in each case may form a hydrocarbon ring system.
- R 5 , R 6 , R 7 and R 8 and also R 5' , R 6> , R 7' and R 8' are identical or different and are each a hydrogen atom, a substituted or unsubstituted alkylsilyl or arylsilyl group, a linear, cyclic or branched alkyl group of from 1 to about 10 carbon atoms, or an aryl group of from 6 to about 40 carbon atoms, which may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br, and / or the two adjacent radicals R 5 , R 6 and also R 5 , R 6 may form a saturated or unsaturated hydrocarbon ring system.
- R 5 , R 6 , R 7 and R 8 and also R 5 R , R and R 8 are identical or different and are each a hydrogen atom or a linear, cyclic or branched alkyl group of from 1 to about 10 carbon atoms, or an aryl group of from 6 to about 40 carbon atoms and / or the two adjacent radicals R 5 , R 6 and also R 5 , R 6 together may form a saturated or unsaturated ring system.
- R 6 , R 7 , R 8 and also R 6 , R 7 and R 8 are identical or different and are each a hydrogen atom, a linear, cyclic or branched hydrocarbon group, for example an alkyl group of from 1 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an aryl group of from 6 to about 20 carbon atoms, an arylalkyl group of from 7 to about 40 carbon atoms, an alkylaryl group of from 7 to about 40 carbon atoms, or an arylalkenyl group of from 8 to about 40 carbon atoms or a substituted or unsubstituted alkylsilyl group, an alkyl(aryl)silyl group or an arylsilyl group.
- a linear, cyclic or branched hydrocarbon group for example an alkyl group of from 1 to about 10 carbon atoms, an alkenyl group of from 2 to about 10 carbon atoms, an aryl group
- R 6 , R 7 or R 7 , R 8 as well as R 6 , R 7 or R 7 , R 8 in each case may form a hydrocarbon ring system.
- the groups may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R 5 and R 5 are identical or different and are each a substituted or unsubstituted aryl group of from 6 to about 40 carbon atoms. They may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R 6 , R 7 and R 8 and also R 6 , R 7 and R 8 are identical or different and aie each a hydrogen atom, a substituted or unsubstituted alkylsilyl or arylsilyl group, a linear, cyclic or branched alkyl group of from 1 to about 10 carbon atoms, or an aryl group of from 6 to about 10 carbon atoms, which may contain one or more hetero atoms like Si, B, Al, O, S, N or P, and / or may contain halogen atoms like F, Cl or Br.
- R 5 and R 5 are identical or different and are each a substituted or unsubstituted aryl group of from 6 to about 40 carbon atoms.
- R 6 , R 7 and R 8 and also R 6 , R 7 and R 8 are identical or different and are each a hydrogen atom or a linear, cyclic or branched alkyl group of from 1 to about 10 carbon atoms, or an aryl group of from 6 to about 10 carbon atoms.
- R 5 and R 5 are identical or different and are each naphthyl, 4-(Ci-C ⁇ o-alkyl)phenyl or 4-(C 6 -C 2 o-aryl)phenyl such as 4- methyl-phenyl, 4-biphenyl, 4-ethyl-phenyl, 4-n-propyl-phenyl, 4-isopropyl-phenyl, 4-tert-butyl- phenyl, 4-sec-butyl-phenyl, 4-cyclohexyl-phenyl, 4-trimethylsilyl-phenyl, 4-adamantyl-phenyl, 4-(Ci-C 10 -fluoroalkyl)-phenyl, 3-(C
- mixtures of the metallocenes of formulas 1 and Ia and the corresponding meso or pseudomeso metallocenes may be used in the catalyst preparation.
- the preparation of the isomerically pure racemic form is especially preferred for the use of metallocenes in the polymerization of olefins to isotactic polyolefins, since the corresponding meso form may produce undesired atactic polypropylene ("PP").
- the "isomerically pure” racemic form is understood to mean a rac:meso ratio of greater than 5: 1 preferably of at least 10: 1, more preferred of at least 15: 1 and most preferred of at least 20:1.
- racemic includes “pseudoracemic” (or “pseudorac”), and the term “meso” includes “pseudomeso.”
- the present invention also includes a process for producing the transition-metal compounds of formulas 1 and Ia of the invention.
- An object of the invention is thus a process for producing compounds of formula Ia,
- step (a) the compound of formula 2, for example, 2-(l-methyl-cyclohexylmethyl)-7- (4-tert-Butyl-phenyl)-lH-indene in an inert solvent, which consists of one or more aromatic or aliphatic hydrocarbons and/or one or more polar, aprotic solvents, is deprotonated with a strong base, for example, n-butyllithium.
- the deprotonation is carried out at temperatures of-70°C to 80 0 C, and preferably 0 0 C to 80°C.
- the resulting metal salt is then reacted directly, without further isolation, in step (b) with a silicon compound or germanium compound that contains two leaving groups.
- Step (c) Preferential production of the compound of formula 3 or the compound of formula 4 can be achieved by adjustment of the quantitative proportions.
- Compounds of formula 3 are reacted in step (c) with a metaHndenyl compound of formula 5.
- step (d) the bis(indenyl)silanes of formula 4 or 6 are doubly deprotonated with a strong base, such asn- butyllithium, in an inert solvent, which consists of one or more aromatic or aliphatic hydrocarbons and/or one or more polar, aprotic solvents, and the bislithium salt formed in this way is reacted, without isolation, directly with a source of Ti, Zr, or Hf to obtain the compound of formula Ia.
- a strong base such asn- butyllithium
- the deprotonation is carried out at temperatures of-70°C to 8O 0 C, and preferably 0 0 C to 80 0 C.
- the metallocenes can be isolated either directly from the reaction mixture with rac:meso ratios or pseudo-rac:meso ratios of greater than 5:1 preferably of at least 10: 1, more preferred of at least 15:1 and most preferred of at least 20:1 or further rac:meso separation steps have to be applied to reach rac:meso ratios or pseudo-rac:meso ratios of at least 5:1 preferably of at least 10: 1 , more preferred of at least 15:1 and most preferred of at least 20:1 to obtain a suitable catalyst.
- Fig. 1 the individual steps of the process of the invention for producing transition- metal compounds of formulas Ia are shown once again for the example of a preferred embodiment.
- the present invention relates to a catalyst system comprising at least one compound of formulas 1 or Ia and at least one cocatalyst.
- a suitable cocatalyst component which may be presert according to the present invention in the catalyst system comprises at least one compound of the type of an aluminoxane, a Lewis acid or an ionic compound which reacts with a metallocene to convert the latter into a cationic compound.
- Aluminoxanes are oligomeric or polymeric aluminum oxy compounds, which may exist in the form of linear, cyclic, caged or polymeric structures. Although the exact structure(s) of aluminoxanes is still unknown, it is well accepted that alkylaluminoxanes have the general formula 7.
- the radicals R in the formulas (7), (8), (9) and (10) can be identical or different and are each a Q -C 20 group such as an alkyl group of from 1 to about 6 carbon atoms, an aryl group of from 6 to about 18 carbon atoms, benzyl or hydrogen and p is an integer from 2 to 50, preferably from 10 to 35.
- radicals R are identical and are methyl, isobutyl, n-butyl, phenyl or benzyl, particularly preferably methyl.
- radicals R are different, they are preferably methyl and hydrogen, methyl and isobutyl or methyl and n-butyl, with hydrogen, isobutyl or n-butyl preferably being present in a proportion of from 0.01 to 40% (number of radicals R).
- the aluminoxane can be prepared in various ways by known methods.
- One of the methods comprises the reaction of an aluminum-hydrocarbon compound and/or a hydridoaluminum-hydrocarbon compound with water, which may be gaseous, solid, liquid or bound as water of crystallization, in an inert solvent such as toluene.
- water which may be gaseous, solid, liquid or bound as water of crystallization
- an inert solvent such as toluene.
- an aluminoxane having different alkyl groups R two different trialkylaluminums (AlR 3 +A1R' 3 ) corresponding to the desired composition and reactivity are reacted with water, cf. S. Pasynkiewicz, Polyhedron 9 (1990) 429 and EP-A-O 302 424.
- all aluminoxane solutions have in common a variable content of unreacted aluminum starting compound which is present in free form or as an adduct.
- aluminoxane compounds of the formulas 7, 8, 9 or 10 it is also possible to use modified aluminoxanes in which the hydrocarbon radicals or hydrogen atoms have been partly replaced by alkoxy, aryloxy, siloxy or amide radicals.
- aluminoxane and metallocene used in the preparation of the supported catalyst system can be varied within a wide range.
- metallocene compound of formulas 1 or Ia and the aluminoxane compounds in such amounts that the atomic ratio of aluminum from the aluminoxane compounds to the transition metal from the metallocene compound is in the range from 10:1 to 1000:1, preferably from 20:1 to 500:1 and in particular in the range from 30:1 to 400:1.
- M 2 is an element of Group 13 of the Periodic Table of Elements, in particular B, Al or Ga, preferably B or Al
- X 1 , X 2 and X 3 are the same or different and each are a hydrogen atom, an alkyl group of from 1 to about 20 carbon atoms, an aryl group of from 6 to about 15 carbon atoms, alkylaryl, arylalkyl, haloalkyl or haloaryl each having from 1 to 10 carbon atoms in the alkyl radical and from 6-20 carbon atoms in the aryl radical or fluorine, chlorine, bromine or iodine.
- Preferred examples for X 1 , X 2 and X 3 are methyl, propyl, isopropyl, isobutyl or trifluoromethyl, unsaturated groups such as aryl or haloaryl like phenyl, tolyl, benzyl groups, p- fluorophenyl, 3,5-difluorophenyl, pentachlorophenyl, pentafluorophenyl, 3,4,5-trifluorophenyl and 3,5-di(trifluoromethyl)phenyl.
- Preferred Lewis acids are trimethylaluminum, triethylaluminum, triisobutylaluminum, tributylaluminum, trifluoroborane, triphenylborane, tris(4-fluorophenyl)borane, tris(3,5- difluorophenyl)borane, tris(4-fluoromethylphenyl)borane, tris(2,4,6-trifluorophenyl)borane, tris(penta-fluorophenyl)borane, tris(tolyl)borane, tris(3,5-dimethyl-phenyl)borane, tris(3,5- difluorophenyl)borane and/or tris (3,4,5-trifluorophenyl)borane.
- ionic cocatalysts preference is given to using compounds which contain a non- coordinating anion such as tetrakis(pentafluorophenyl)borate, tetraphenylborate, SbF 6 " , CF 3 SO 3 " or ClO 4 " .
- Suitable counterions are either Lewis acid or Broenstedt acid cation.
- protonated amine or aniline derivatives such as methylammonium, anilinium, dimethylammonium, diethylammonium, N- methylanilinium, diphenylammonium, N,N-dimethylanilinium, trimethylammonium, triethylammonium, tri-n-butylammonium, methyldiphenylammonium, pyridinium, p-bromo- N,N-dimethylanilinium or p-nitro-N,N-dimethylanilinium, N,N-dimethylbenzylammonium, N,N-dimethylcyclohexylammonium,
- protonated amine or aniline derivatives such as methylammonium, anilinium, dimethylammonium, diethylammonium, N- methylanilinium, diphenylammonium, N,N-dimethylanilinium, trimethylammonium, triethylammonium, tri-n-
- Suitable Lewis-acid cations are cations of the formula 12
- Qi to Q z are singly negatively charged groups such as Ci-C 28 -alkyl, C 6 -Ci 5 -aryl, alkylaryl, arylalkyl, haloalkyl, haloaryl each having from 6 to 20 carbon atoms in the aryl radical and from 1 to 28 carbon atoms in the alkyl radical, cycloalkyl groups of from 3 to about 10 carbon atoms, which may in turn bear alkyl groups of from 1 to about 10 carbon atoms as substitutents, halogen, alkoxy groups of from 1 to 28 carbon atoms, aryloxy groups of from 6 to 15 carbon atoms, silyl or mercaptyl groups, a is an integer from 1-6, z is an integer from 0 to 5 and d corresponds to the difference a-z, but d is larger than or equal to 1.
- Particulary suitable cations are carbonium cations such as triphenylcarbenium, oxonium cations, sulfonium cations such as tetrahydrothiophenium, phosphonium cations such as triethyl phosphonium, triphenylphosphonium and diphenylphosphonium, and also cationic transition metal complexes such as the silver cation and the 1,1 -dimethylferrocenium cation.
- carbonium cations such as triphenylcarbenium, oxonium cations, sulfonium cations such as tetrahydrothiophenium, phosphonium cations such as triethyl phosphonium, triphenylphosphonium and diphenylphosphonium, and also cationic transition metal complexes such as the silver cation and the 1,1 -dimethylferrocenium cation.
- Preferred ionic compounds which can be used according to the present invention include: triethylammoniumtetra(phenyl)borate, tributylammoniumtetra(phenyl)borate, trimethylammoniumtetra(tolyl)borate, tributylammoniumtetra(tolyl)borate, tributylammoniumtetra(pentafluorophenyl)borate, tributylammoniumtetra(pentaffluorophenyl) aluminate, tripropylammoniumtetra(dimethylphenyl)borate, tributylammoniumtetra(trifluoromethylphenyl)borate, tributylammoniumtetra(4-fluorophenyl)borate, N,N-dimethylcyclohexylammoniumtetrakis(pentafluorophenyl)borate, N,N-di
- triphenylcarbeniumtetrakis(pentafluorophenyl) borate N 5 N- dimethylcyclohexylammoniumtetrakis(pentafluorophenyl)borate or N,N- dimethylbenzylammoniumtetrakis(pentafluorophenyl)borate.
- mixtures of all of the above and below mentioned cation-forming compounds comprise aluminoxanes and an ionic compound, and/or a Lewis acid.
- the amount of Lewis acids or ionic compounds having Lewis-acid or Broensted-acid cations is preferably from 0.1 to 20 equivalents, preferably from 1 to 10 equivalents, based on the metallocene compound of the formulas 1 or Ia.
- Combinations of at least one Lewis base with bimetallic compounds of the type Rj 17 M 3 (- O-M 3 R j 18 ) v or Rj' 8 M 3 C-O-M 3 R j 17 ) v (formula 13), as described in Patent Application WO 99/40129, are likewise important as cocatalyst systems.
- R 17 and R 18 are the same or different and represent a hydrogen atom, a halogen atom, a Ci-C 40 carbon-containing group, especially an alkyl group of from 1 to about 20 carbon atoms, haloalkyl of from 1 to about 20 carbon atoms, alkoxy of from 1 to about 10 carbon atoms, aryl of from 6 to about 20 carbon atoms, haloaryl of from 6 to about 20 carbon atoms, aryloxy of from 6 to about 20 carbon atoms, arylalkyl of from 7 to about 40 carbon atoms, haloarylalkyl of from 7 to about 40 carbon atoms, alkylaryl of from 7 to about 40 carbon atoms, or haloalkylaryl of from 7 to about 40 carbon atoms.
- R 17 may also be an -OSiR 51 3 group, in which the R 51 groups are the same or different and have the same meaning as R 17 , M 3 is the same or different and represents an element of main group III of the periodic table of elements, i, j, and v each stands for a whole number 0, 1, or 2, and i + j + v is not equal to 0.
- Suitable cocatalyst systems according to formula 13 are compounds of formulas (A) and (B)
- R 17 and R 18 have the same meaning as specified above.
- compounds that are generally to be regarded as preferred are those formed by the reaction of at least one compound of formulas (C) and/or (D) and/or (E) with at least one compound of formula (F).
- R 27 may be a hydrogen atom or a boron-free Ci-C 40 carbon-containing group, such as an alkyl of from 1 to about 20 carbon atoms, aryl of from 6 to about 20 carbon atoms, arylalkyl of from 7 to about 40 carbon atoms, and alkylaryl of from 7 to about 40 carbon atoms, and in which R 17 , R 18 have the same meaning as specified above,
- D is an element of main Group VI of the periodic table of elements or an NR 61 group, where R 61 is a hydrogen atom or a Ci-C 2O hydrocarbon group, such as alkyl of from 1 to about 20 carbon atoms or aryl of from 6 to about 20 carbon atoms, f is a whole number from 0 to 3, g is a whole number from 0 to 3 where f + g corresponds to the valency of Boron, and h is a whole number from 1 to 10.
- the bimetallic compounds of formula 13 are possibly combined with an organometallic compound of formula 14, i.e., [M 4 R 19 ,, ⁇ , in which M 4 is an element of main Group 1, II, or III of the periodic table of the elements, R 19 is the same or different and represents a hydrogen atom, a halogen atom, a Ci-C 40 carbon-containing group, an alkyl group of from 1 to about 20 carbon atoms, an aryl group of from about 6 to about 40 carbon atoms, arylalkyl of from 7 to about 40 carbon atoms, and alkylaryl of from 7 to about 40 carbon atoms, q is a whole number from 1 to 3, and k is a whole number from 1 to 4.
- M 4 is an element of main Group 1, II, or III of the periodic table of the elements
- R 19 is the same or different and represents a hydrogen atom, a halogen atom, a Ci-C 40 carbon-containing group, an alkyl group of from 1 to about 20 carbon
- the organometallic compounds of formula 14 are preferably neutral Lewis acids, in which M 4 stands for lithium, magnesium, and/or aluminum, especially aluminum.
- M 4 stands for lithium, magnesium, and/or aluminum, especially aluminum.
- preferred organometallic compounds of formula 14 are trimethylaluminum, triethylaluminum, triisopropylaluminum, trihexylaluminum, trioctylaluminum, tri-w-butylaluminum, tri- «- propylaluminum, triisoprene aluminum, dimethyl aluminum monochloride, aluminum monochloride, diisobutyl aluminum monochloride, methyl aluminum sesquichloride, ethyl aluminum sesquichloride, dimethyl aluminum hydride, aluminum hydride, diisopropyl aluminum hydride, dimethyl aluminum(trimethylsiloxide), dimethyl aluminum(triethylsiloxide), phenylalan, pentafluorophenylalan, and o-
- the catalyst system of the invention contains an organoboroaluminum compound, which contains units of formula 13, as the cocatalytically active chemical compound.
- Compounds of formula 13 in which M 3 stands for boron or aluminum are preferred.
- the compounds that contain units of formula 13 may be present as monomers or as linear, cyclic, or cage-like oligomers. Two or more chemical compounds that contain units of formula 13 may also form dimers, trimers, or higher combinations among themselves by Lewis acid-base interactions.
- Preferred cocatalytically active bimetallic compounds correspond to formulas 15 and 16,
- EP-A-924,223, DE 196 22 207.9, EP-A-601,830, EP-A-824,1 12, EP- A-824,1 13, WO 99/06,414, EP-A-811,627, WO 97/11,775, DE 196 06 167.9 and DE 198 04 970 can be used as additional cocatalysts, which may be present in unsupported or supported form.
- the amount of cocatalysts of formula 13 and/or 15 and/or 16 used in the catalyst of the present invention can vary from 0.1 to 500 equivalents, preferably from 1 to 300 equivalents, most preferably from 5 to 150 equivalents, based on the used amount of metallocene compound of the formulas 1 or Ia.
- the catalyst system of the present invention can further comprise, as additional component, a metal compound of the formula 17, (Formula 17)
- M 5 is an alkali, an alkali earth metal or a metal of Group 13 of the Periodic Table of the Elements
- R 22 is a hydrogen atom, alkyl of from 1 to about 10 carbon atoms, aryl of from
- alkylaryl or arylalkyl each having from 1 to 10 carbon atoms in the alkyl part and from 6 to 20 carbon atoms in the aryl part,
- R 23 and R 24 are each a hydrogen atom, a halogen atom, alkyl of from 1 to about 10 carbon atoms, C 6 -Ci 5 -aryl of from about 6 to about 15 carbon atoms, or alkylaryl, arylalkyl or alkoxy each having from 1 to 10 carbon atoms in the alkyl part and from 6 to 20 carbon atoms in the aryl radical, r is an integer from 1 to 3 and s and t are integers from 0 to 2, where the sum r+s+t corresponds to the valency of M 5 , where this component is not identical with the above mentioned cocatalyst compounds. It is also possible to use mixtures of various metal compounds of the formula 17.
- metal compounds of the formula 17 preference is given to those in which M 5 is lithium, magnesium or aluminum and R 23 and R 24 are each alkyl of from 1 to about 10 carbon atoms.
- Particularly preferred metal compounds of the formula 17 are n-butyllithium, n-butyl-n- octyl-magnesium, n-butyl-n-heptylmagnesium, tri-n-hexylaluminum, triisobutylaluminum, triethylaluminum, trimethylaluminum or mixtures thereof.
- a metal compound of the formula 17 is used, it is preferably present in the catalyst system in such an amount that the molar ratio of M 5 to the transition metal from the metallocene compound of formulas 1 or Ia is from 800:1 to 1 :1, in particular from 200:1 to 2:1.
- the support component of the catalyst system of the present invention can be any organic or inorganic inert solid or a mixture of such solids, in particulate porous solids such as hydrotalcites, talc, inorganic oxides and finely divided polymer powders.
- Suitable inorganic oxides which are preferably employed include from the Periodic Table of Elements Groups 1, 2, 3, 4, 5, 12, 13 and 14, metal oxides such as silicon dioxide, aluminum oxide, aluminosilicates, zeolites, MgO, ZrO 2 , TiO 2 or B 2 O 3 , CaO, ZnO, ThO 2 , Na 2 O, K 2 O, LiO 2 or mixed oxides like Al/Si oxides, Mg/Al oxides or Al/Mg/Si oxides.
- metal oxides such as silicon dioxide, aluminum oxide, aluminosilicates, zeolites, MgO, ZrO 2 , TiO 2 or B 2 O 3 , CaO, ZnO, ThO 2 , Na 2 O, K 2 O, LiO 2 or mixed oxides like Al/Si oxides, Mg/Al oxides or Al/Mg/Si oxides.
- suitable inorganic support materials are Na 2 CO 3 , K 2 CO 3 , CaCO 3 , MgCl 2 , Na 2 SO 4 , A1 2 (SO 4 ) 3 , BaSO 4 , KNO 3 , Mg(NO 3 ) 2 and A1(NO 3 ) 3 .
- Suitable polymer powders are homopolymers, copolymers, crosslinked polymers or polymer blends.
- polymers are polyethylene, polypropylene, polybutene, polystyrene, divinylbenzene-crosslinked polystyrene, polyvinyl chloride, acrylonitrile-butadiene- styrene copolymer, polyamide, polymethacrylate, polycarbonate, polyester, polyacetal or polyvinyl alcohol.
- the preferred support materials have a specific surface area in the range from 10 to 1000 m /g, a pore volume in the range from 0.1 to 5 cm /g and a mean particle size of from 1 to 500 ⁇ m.
- Particular preference is given to supports having a specific surface area in the range from 200 to 400 m 2 /g, a pore volume in the range from 0.8 to 3.0 cmVg and a mean particle size of from 10 to 100 ⁇ m.
- the support materials can be thermally and/or chemically be pretreated in order to adjust certain properties of the carrier such as the water and/or the hydroxyl group content.
- the support material has a low moisture content or residual solvent content, dehydration or drying before use can be omitted. If this is not the case, as when using silica gel as support material, dehydration or drying is advisable.
- Thermal dehydration or drying of the support material can be carried out under reduced pressure with or without simultaneous inert gas blanketing (nitrogen).
- the drying temperature is in the range from 80 0 C to 1000 0 C, preferably from 150 °C to 800 0 C and most preferred from 150 °C to 400 0 C.
- the duration of the drying process can be from 1 to 24 hours. But shorter or longer drying periods are also possible.
- support materials with a weight loss on dryness (LOD) of 0.5 wt.% or less, and even more preferred with a LOD of 0.3 wt% or less are used. Higher amounts of physically adsorbed water up to 1 wt% are possible, but result in reduced catalyst activities.
- the loss on ignition (LOI) of the support material is preferably 1 wt% or greater or even more preferred between 1.5 and 3.5 wt%.
- dehydration or drying of the support material can also be carried out by chemical means, by reacting the adsorbed water and/or the surface hydroxyl groups with suitable passivating agents. Reaction with the passivating reagent can convert the hydroxyl groups completely or partially into a form, which does not show any adverse interaction with the catalytically active centers.
- suitable passivating agents are silicon halicbs, silanes or amines, eg.
- silicon tetrachloride chlorotrimethylsilane, dichlorodialkylsilanes, dimethylaminotrichlorosilane, N,N-dimethyIanilin or N,N-dimethylbenzylamine or organometallic compounds of aluminum, boron and magnesium, eg. aluminoxanes, trimethylaluminum, triethylaluminum, triisobutylaluminum, diisobutylaluminum hydride, triethylborane or dibutylmagnesium.
- aluminoxanes trimethylaluminum, triethylaluminum, triisobutylaluminum, diisobutylaluminum hydride, triethylborane or dibutylmagnesium.
- organic support materials such as finely divided polymer powders, can also be used and should, before use, likewise be freed from any adhering moisture, solvent residues or other impurities by means of appropriate purification and drying operations.
- silica gels having the defined parameters as support materials Preference is given to using silica gels having the defined parameters as support materials.
- Spray dried silica grades, which inherently exhibit meso and macro pores, cavities and channels are preferred over granular silica grades.
- the supported catalyst system according to this invention can be made in various ways.
- At least one of the above-described metallocene components of formulas 1 or Ia is brought into contact in a suitable solvent with at least one cocatalyst component, preferably giving a soluble reaction product, an adduct or a mixture.
- the obtained composition is mixed with the dehydrated or passivated support material, the solvent is removed and the resulting supported metallocene catalyst system is dried to ensure that the solvent is completely or mostly removed from the pores of the support material.
- the supported catalyst is obtained as a free-flowing powder.
- the process for preparing a free-flowing and, if desired, prepolymerized supported catalyst system comprises the following steps: a) preparing a metallocene/cocatalyst mixture in a suitable sdvent or suspension medium, where the metallocene component has one of the above-described structures, b) applying the metallocene/cocatalyst mixture to a porous, preferably inorganic, if necessary thermally or chemically pretreated support, c) removing the major part of solvent from the resulting mixture, d) isolating the supported catalyst system and e) if desired, prepolymerizing the resulting supported catalyst system with one or more olefinic monomer(s), to obtain a prepolymerized supported catalyst system.
- the metallocene / cocatalyst composition is mixed with the dehydrated or passivated support material, the supported catalyst is recovered and optionally washed with an aromatic hydrocarbon and/or paraffinic hydrocarbon solvent.
- the isolated catalyst is then dispersed in a non-reactive suspension media such as a paraffinic hydrocarbon solvent, a mineral oil or a wax or mixtures thereof.
- the catalyst is prepared according to the procedure disclosed in WO 06/60544 (an application by the present inventors), WO 00/05277, WO 98/01481, US 7,355,058, US 7,193,100, US 6,492,292, US 6,107,230 or US 6,355,594 using at least one compound of formulas 1 or Ia as the metallocene component.
- a free flowing and, if desired, prepolymerized supported catalyst system comprising the following steps: a) contacting at least one support material with a first portion of at least one co-catalyst in a suitable solvent b) impregnating the co-catalyst loaded support with a suspension or solution, which comprises at least one metallocene and a second portion of at least one co-catalyst in a suitable solvent c) isolating the supported catalyst system and f) if desired, prepolymerizing the resulting supported catalyst system with one or more olefinic monomer(s), to obtain a prepolymerized supported catalyst system.
- the process according to WO 06/60544 for preparing a free-flowing and, if desired, prepolymerized supported catalyst system comprises the following steps: a) Contacting a support material with a first composition which includes at least one aluminoxane in a first solvent at a temperature of about 10 to 30°C followed by keeping the mixture at about 20°C for 0 to 12 hours, subsequently heating the resulting mixture to a temperature of 30 to 200°C and keeping the mixture at 30 to 200°C for 30 minutes to 20 hours, optionally followed by removing all or part of the first solvent and/or optionally followed by one or more washing step(s) using a suitable solvent, b) Suspending and/or dissolving, respectively, at least one metallocene of formula 1 and/or Ia and a second portion of an aluminoxane or of a mixture of aluminoxanes or of an ionic compound and/or a Lewis acid in a second solvent or suspension medium at a temperature of
- the process according to WO 06/60544 for preparing a free-flowing and, if desired, prepolymerized supported catalyst system comprises the following steps: a) Contacting a support material with a first composition which includes at least 5 mmol of an aluminoxane or of a mixture of aluminoxanes per g support material in a first solvent at a temperature of about 20°C followed by keeping the mixture at about 2O 0 C for 0.15 to 2 hours, subsequently heating the resulting mixture to a temperature of 50 to 160°C and keeping the mixture at 50 to 160 0 C for 1 to 6 hours, optionally followed by removing all or part of the first solvent and/or optionally followed by one or more washing step(s) using a suitable solvent, b) Suspending and/or dissolving, respectively, at least 0.5 mmole of a second portion of an aluminoxan
- the process according to WO 06/60544 for preparing a free-flowing and, if desired, prepolymerized supported catalyst system comprises the following steps: a) Contacting an optionally thermally pretreated silica support material with at least 10 mmol of an aluminoxane per g support material in toluene at a temperature of about 20 0 C followed by subsequently heating the resulting mixture to a temperature of 50 to 110°C and keeping the mixture at 50 to 110 0 C for 1 to 6 hours, optionally followed by removing all or part of the toluene, and/or optionally followed by one or more washing step(s) using a suitable solvent, k b) Suspending and/or dissolving, respectively, at least 0.5 mmole of a second portion of an aluminoxane per g support material and at least 0.1 mol% of the employed second portion of an aluminoxane or of a mixture of aluminoxanes
- the process according to WO 06/60544 for preparing a free-flowing and, if desired, prepolymerized supported catalyst system comprises the following steps: a) Contacting an optionally thermally pretreated silica support material with a weight loss on dryness (LOD) of 0.5 wt.% or less and a weight loss on ignition (LOI) of 1.0 wt.% or greater with a first composition which includes at least 10 mmol of methylaluminoxane per g support material in toluene at a temperature of about 20 0 C followed by subsequently heating the resulting mixture to a temperature of 1 10 0 C and keeping the mixture at 1 10 0 C for 1 to 6 hours, optionally followed by removing all or part of the toluene, and/or optionally followed by one or more washing step(s) using a suitable solvent, b) Suspending and/or dissolving, respectively, at least 1 mmole of a second portion of methyl
- the process according to WO 06/60544 for preparing a free-flowing and, if desired, prepolymerized supported catalyst system comprises the following steps: a) Contacting an optionally thermally pretreated silica support material with a weight loss on dryness (LOD) of 0.3 wt.% or less and a weight loss on ignition (LOI) between 1.5 and 3.5 wt.%, with at least 10 mmol of methylaluminoxane per g support material in toluene at a temperature of about 20°C followed by subsequently heating the resulting mixture to a temperature of HO 0 C and keeping the mixture at 11O 0 C for 1 to 6 hours, optionally followed by removing all or part of the toluene, and/or optionally followed by one or more washing step(s) using a suitable solvent, b) Suspending and/or dissolving, respectively, at least 1 mmole of a second portion of methylaluminoxane
- step b) of the catalyst preparations instead of an aluminoxane or a mixture of aluminoxanes, at least one alkyl compound of elements of main Groups I to III of the Periodic Table, for example a magnesium alkyl, a lithium alkyl or an aluminum alkyl like trimethylaluminum, triethylaluminum, triisobutyllaluminum, triisopropylaluminum, trihexylaluminum, trioctylaluminum, tri-M-butylaluminum, tri-n- propylaluminum, triisoprene aluminum, dimethyl aluminum monochloride, aluminum monochloride, diisobutyl aluminum monochloride, methyl aluminum sesquichloride, ethyl aluminum sesquichloride, dimethyl aluminum hydride, aluminum hydride,
- Preferred aluminum alkyls are trimethylaluminum, triethylaluminum, triisobutylaluminum.
- a free flowing and, if desired, prepolymerized supported catalyst system is prepared comprising the following steps: a) preparing a trialkylaluminium/borinic acid mixture in a suitable solvent or suspension medium b) applying the trialkylaluminium/borinic acid mixture to a porous, preferably inorganic, if necessary thermally or chemically pretreated support which was prior treated with a base like N,N-diethylbenzylamine, N,N-dimethylbenzylamine, N-benzyldimethylamine, N- benzyldiethylamine, N-benzylbutylamine, N-benzyl tertbutylamine, N-benzylisopropylamine, N- benzylmethylamine, N-benzylethylamine, N-benzyl- 1-pheny
- Preferred solvents for the preparation of the metallocene/cocatalyst mixture are hydrocarbons and hydrocarbon mixtures, which are liquid at the selected reaction temperature and in which the individual components preferably dissolve.
- the solubility of the individual components is, however, not a prerequisite as long as it is ensured that the reaction product of metallocene and cocatalyst components is soluble in the solvent selected.
- Suitable solvents are alkanes such as pentane, isopentane, hexane, isohexane, heptane, octane and nonane, cycloalkanes such as cyclopentane and cyclohexane and aromatics sich as benzene, toluene, ethylbenzene and diethylbenzene. Very particular preference is given to toluene, heptane and ethylbenzene.
- the metallocene in the form of a solid is dissolved in a solution of the cocatalyst in a suitable solvent. It is also possible to dissolve the metallocene separately in a suitable solvent and subsequently to combine this solution with the cocatalyst solution. Preference is given to using toluene.
- the preactivation time is from 1 minute to 200 hours.
- the preactivation can take place at room temperature of 25 0 C. In individual cases, the use of higher temperatures can reduce the required preactivation time and give an additional increase in activity. Elevated temperatures in this case refer to a range from 25 °C to 100 °C.
- the preactivated solution or the metallocene/cocatalyst mixture is subsequently combined with an inert support material, usually silica gel, which is in the form of a dry powder or as a suspension in one of the above mentioned solvents.
- the support material is preferably used as powder.
- the preactivated metallocene/cocatalyst solution or the metallocene/cocatalyst mixture can be either added to the initially charged support material, or else the support material can be introduced into the initially charged solution.
- the volume of the preactivated solution or the metallocene/cocatalyst mixture can exceed 100% of the total pore volume of the support material used or else be up to 100% of the total pore volume.
- the temperature at which the preactivated solution or the metallocene/cocatalyst mixture is brought into contact with the support material can vary within the range from 0 0 C to 100 0 C. However, lower or higher temperatures are also possible.
- the mixture can be stirred and, if desired, also heated.
- both the visible portion of the solvent and the portion in the pores of the support material are removed.
- the removal of the solvent can be carried out in a conventional way using reduced pressure and/or purging with inert gas.
- the mixture can be heated until the free solvent has been removed, which usually takes from 1 to 3 hours at a preferred temperature of from 30 °C to 60 0 C.
- the free solvent is the visible portion of the solvent in the mixture.
- residual solvent is the portion present in the pores.
- the supported catalyst system can also be dried until only a certain residual solvent content is left, with the free solvent having been completely removed. Subsequently, the supported catalyst system can be washed with a low-boiling hydrocarbon such as pentane or hexane and dried again.
- a low-boiling hydrocarbon such as pentane or hexane
- the supported catalyst system prepared according to the present invention can be used either directly for the polymerization of olefins or be prepolymerized with one or more olefinic monomers, with or without the use of hydrogen as molar mass regulating agent, prior to use in a polymerization process.
- the procedure for the prepolymerization of supported catalyst systems is described in WO 94/28034.
- an olefin preferably an alpha-olefin such as styrene or phenyldimethylvinylsilane as activity-increasing component or an antistatic, as described in U.S. Ser. No. 08/365,280.
- the molar ratio of additive to metallocene component of formulas 1 or Ia is preferably from 1:1000 to 1000:1, very particularly preferably from 1 :20 to 20: 1.
- the present invention also provides a process for preparing a polyolefin by polymerization of one or more olefins in the presence of the catafyst system of the present invention comprising at least one transition metal component of the formulas 1 or Ia.
- polymerization refers to both homopolymerization and copolymerization and the term copolymerization includes terpolymerisation or copolymerisation of more than three different monomers.
- R m and R" are identical or different and are each a hydrogen atom or a radical havhg from 1 to 20 carbon atoms, in particular from 1 to 10 carbon atoms, and R m and R" together with the atoms connecting them can form one or more rings.
- Suitable olefins are 1-olefins, eg. ethene, propene, 1-butene, 1-pentene, 1-hexene, 4- methyl-1-pentene or 1-octene, styrene, dienes such as 1 ,3-butadiene, 1,4-hexadiene, vinylnorbornene, norbornadiene, ethylnorbornadiene and cyclic olefins such as norbornene, tetracyclododecene or methylnorbornene.
- 1-olefins eg. ethene, propene, 1-butene, 1-pentene, 1-hexene, 4- methyl-1-pentene or 1-octene, styrene, dienes such as 1 ,3-butadiene, 1,4-hexadiene, vinylnorbornene, norbornadiene, ethyl
- Very suitable copolymers are ethene-propene copolymers, propene- 1-pentene copolymers and ethene-propene- 1-butene, ethene-propene- 1- pentene or ethene-propene- 1,4-hexadiene terpolymers.
- the polymerization is carried out at from -60 °C to 300 °C, preferably from 50 °C to 200 0 C, very particularly preferably from 50 °C to 95 °C.
- the pressure is from 0.5 to 2000 bar, preferably from 5 to 100 bar.
- the polymerization can be carried out in solution, in bulk, in suspension or in the gas phase, continuously or batchwise, in one or more stages.
- impact copolymers are preferably produced in more than one stage.
- the homopolymer or random copolymer content of such a polymer can be produced in (a) first stage(s) and the copolymer rubber content can be produced in (a) consecutive stage(s).
- the supported catalyst system prepared according to the present invention can be used as sole catalyst component for the polymerization of olefins or preferably in combination with at least one alkyl compound of elements of main Groups I to III of the Periodic Table, for example an aluminum alkyl, magnesium alkyl or lithium alkyl or an aluminoxane.
- the alkyl compound is added to the monomer or suspension medium and serves to free the monomer of substances, which can impair the catalytic activity.
- the amount of alkyl compound added depends on the quality of the monomers used.
- a catalyst system comprising two or more different metallocenes and/or two or more different cocatalysts.
- two or more different catalyst systems of the present invention can be used as a mixture.
- hydrogen is added if required.
- the catalyst system may be supplied to the polymerization system as a solid or in the form of a paste or suspension in a hydrocarbon or may be treated with inert components, such as paraffins, oils, or waxes, to achieve better metering. If the catalyst system is to be metered into the reactor together with the monomer to be polymerized or the monomer mixture to be polymerized, the mixing unit and the metering line are preferably cooled.
- an additive such as an antistatic or an alcohol can be used in the process of the present invention, for example to improve the particle morphology of the olefin polymer.
- an additive such as an antistatic or an alcohol can be used in the process of the present invention, for example to improve the particle morphology of the olefin polymer.
- the polymers prepared using the catalyst systems of the present invention display an uniform particle morphology and contain no fines. No agglomerates or deposits are obtained in the polymerization using the catalyst system of the present invention.
- the catalyst systems of the present invention give polymers such as polypropylene having high molecular weight and cover a broad range of stereospecificity and regiospecificity.
- the copolymers which can be prepared using the catalyst system based on metallocenes of formula 1 or Ia of the present invention have a significantly higher molar mass compared to the prior art. At the same time, such copolymers can be prepared using the catalyst system of the present invention at a high productivity and at industrially relevant process parameters without deposit formation.
- the polymers prepared by the process of the present invention are suitable, in particular, for producing products such as fibers, filaments, injection-molded parts, films, sheets, caps, closures, bottles or large hollow bodies such as pipes with excellent properties.
- the preparation and handling of the organometallic compounds were carried out under argon using Schlenk techniques or in a glove box. All solvents were purged with argon and dried over molecular sieves before use.
- the metallocenes produced were characterized by 1 H-NMR spectroscopy using a Bruker DMX 500 spectrometer, operating at 500 MHz using CDCl 3 as the solvent.
- the polymers produced were characterized by 1 H-NMR, 13 C-NMR, DSC, GPC, TREF/ATREF, Melt Flow Rate and IR spectroscopy.
- 0.05 wt% solutions of the samples in 1 ,2,4-trichlorobenzene were analyzed at a temperature of 145 0 C using a Mixed B light scattering quality column (Polymer Labs 1 1 10-6100LS) and a Mixed B guard column (Polymer Labs 1 1 10-1 120).
- Weight average molar mass (Mw) and the ratio of weight average molar mass to number average molar mass (Mw/Mn) were calculated using the Cumulative Matching % Broad Standard procedure that is available in the Waters Millenium 3.2 GPC software module. 2. NMR Spectroscopy of Polymers
- Samples were prepared by weighing 0.32 g of polymer into 2.5 ml of a 1,2,4- trichlorobenzene/deuterobenzene-d6 (4:1 volume) mixture. Samples were heated to 125°C and mixed until a homogeneous solution was formed (typically 1-4 hours). Spectra were obtained at 12O 0 C on a Varian Inova 500 instrument (Varian Inc., 3120 Hansen Way, Palo Alto, CA, 94304, USA) operating at a 13 C-spectro meter frequency of 125.7 MHz and using a 10 mm probe.
- Spectra were obtained using 5000 scans employing a ⁇ /2 pulse of 10.0 ⁇ s, a recycle delay of 10.0 s and an acquisition time of 2.5 s. Waltz- 16 decoupling remained on throughout the pulse sequence to gain the signal to noise enhancement due to the effects of nOe (Nuclear Overhauser enhancement). Spectra were processed with 1 Hz of line broadening. The mmmm peak in the methyl region of the spectrum was used as an internal chemical shift reference and was set to 21.85 ppm.
- DSC measurements were carried out using a Mettler Toledo DSC 822e (Mettler-Toledo Inc., 1900 Polaris Parkway, Columbus, OH, 43240, USA). 4 mg of sample were weighed into a standard aluminum pan and subjected to the following temperature schedule:
- the samples were heated from room temperature to 220 0 C at a heating rate of 20 °C/min, maintained at this temperature for 5 min, then cooled down to -55 0 C at a cooling rate of 20 °C/min, maintained at the same temperature for 5 min, then heated to 220 °C at a heating rate of 20 °C/min.
- the melting point was determined from the second heating run as the temperature where the main peak was observed in the curve.
- the MFR of the samples were determined according to ISO 1 133 at 230°C. Two diffent loads were used: 2.16 kg and 5 kg. Values are reported as MFR(230 / 2.16) and MFR(230/5), respectively. 5.
- the productivity of a catalyst is determined by dividing the produced mass of polypropylene by the mass of catalyst used and the reaction time.
- the yield of a sample is determined by dividing the isolated amount of the desired product divided by the theoretical achievable amount of the product.
- T g glass transition temperature in ⁇ C, determined by differential scanning calorimcfxy (DSC, conditions see above)
- the Loss on Dryness and the Loss on Ignition arc deteraiinec by thcrmogravimetric measurement.
- the Loss on Dryness is the weight loss lhiL the support material experiences by heating it from room temperature to 300 s C and keeping this temperature until a constant weight is obtained. Loss on Dryness is that weight loss as expressed as percentage of the weight of the original support! material. Subsequently the support material is heated from 300 D C to K)OO 0 C and is kept i. this temperature until a constant weight is obtained.
- the Loss of Ignition is defined es the weight loss between the weight of the sample at 1000 ⁇ C and at room temperature expressed as a percentage of Lhe weight of the original support material.
- a 10 gram sample (at room terripetature) is heated to 300 C where it maintains a constant weight at that temperaltur ⁇
- the Loss on Dryness is 10.0% , which is:
- the solvent amount was reduced in vacuo to a total of 150 ml volume and the solution was added dropwise over a period of 2.5 h to 660 g of hot (70 - 75 0 C) concentrated sulphuric acid. After stirring for an additional 30 min at 75°C the mixture was allowed to stand overnight at room temperature. The mixture was poured onto approx. 500 g of ice and extracted three times with 150 ml dichloromethane exh. The organic layer was washed twice with 150 ml of a saturated sodium bicarbonate solution and once with 100 ml of a saturated sodium chloride solution. After drying over magnesium sulphate the solvent was removed in vacuo.
- a premixed catalyst solution consisting of 103 mg (0.2 mole %) palladium acetate, 3 ml NaTPPTS (2.6 M in water, 0.8 mole %) and 2 ml of water was added and the mixture was refluxed at 125°C until complete conversion (approx. 4 h). 300 ml of water were added and the mixture was extracted three times with 150 ml of toluene each. The combined organic layers were washed twice with 100 ml water and once with 100 ml of a saturated sodium chloride solution. Drying over magnesium sulphate and evaporation of the solvert in vacuo yielded 87.2 g (quant.) of the desired product as a yellow sticky oil.
- the solvent amount was reduced in vacuo to a total of 250 ml volume and the solution was added dropwise over a period of 2.5 h to 1000 g of hot (70 - 75°C) concentrated sulphuric acid. After stirring for an additional 30 min at 75°C the mixture was allowed to stand overnight at room temperature. The mixture was poured onto approx. 800 g of ice and extracted three times with 250 ml dichloromethane each. The organic layer was washed twice with 250 ml of a saturated sodium bicarbonate solution and once with 200 ml of a saturated sodium chloride solution. After drying over magnesium sulphate the solvent was removed in vacuo.
- a premixed catalyst solution consisting of 67 mg (0.2 mole %) palladium acetate, 1.94 ml NaTPPTS (2.6 M in water, 0.8 mole %) and 2 ml of water was added and the mixture was refluxed at 125°C until complete conversion(approx. 4 h). 100 ml of water were added and the mixture was extracted three times with 100 ml of toluene each. The combined organic layers were washed twice with 100 ml water and once with 100 ml of a saturated sodium chloride solution. Drying over magnesium sulphate and evaporation of the solvent in vacuo yielded 58.82 g (quant.) of the desired product as a yellow sticky oil.
- the solvent amount was reduced in vacuo to a total of 150 ml volume and the solution was added dropwise over a period of 2.5 h to 660 g of hot (70 - 75°C) concentrated sulphuric acid. After stirring for an additional 30 min at 75°C the mixture was allowed to stand overnight at room temperature. The mixture was poured onto approx. 50O g of ice and extracted three times with 150 ml dichloromethane each. The organb layer was washed twice with 150 ml of a saturated sodium bicarbonate solution and once with 100 ml of a saturated sodium chloride solution. After drying over magnesium sulphate the solvent was removed in vacuo.
- a premixed catalyst solution consisting of 103 mg (0.2 mole %) palladium acetate, 3 ml NaTPPTS (2.6 M in water, 0.8 mole %) and 2 ml of water was added and the mixture was refluxed at 125°C until complete conversion (approx. 6 h). 300 ml of water were added and the mixture was extracted three times with 150 ml of toluene each. The combined organic layers were washed twice with 100 ml water and once with 100 ml of a saturated sodium chloride solution. Drying over magnesium sulphate and evaporation of the solvent in vacuo yielded 94.1 g (quant.) of the desired product as a yellow sticky oil.
- the residue was dried in an oil-pump vacuum, and the product was obtained in a yield of 155 g (80%) and with a rac:meso ratio of 1 : 1.
- the isomers must be separated in an additional step to obtain selective catalysts for propylene pol>merization.
- the mixture was allowed to warm to room temperature and was stirred for another two hours at this temperature.
- the precipitate that forms was separated by filtration through a G3 fritted glass filter and was washed with two 50 mL portions of tetrahydrofuran and with one 70 mL portion of pentane.
- the residue was dried in an oil-pump vacuum, and the product was obtained in a yield of 23.5 g (50%) and with a rac:meso ratio of about 1 :1.
- the isomers must be separated in a subsequent step to obtain selective catalysts for propylene polymerization.
- EtOOC COOEt adding 25.5 g (1.1 1 mol) of sodium cut in small pieces to the reaction mixture. At a reaction temperature of 40-45 °C the sodium addition and reaction is finished after ⁇ 1.5 h. The solution is cooled to room temperature and 192.5 g (1.20 mol) of malonic acid diethyl ester are added. The mixture is stirred at this temperature for 15 minutes.
- the water phase is extracted with 300 and 2 times 150 ml of diethyl ether.
- the combined organic phases are washed with 200 ml of water.
- the ether is removed under reduced pressure and the crude product is distilled in a vacuum at a pressure of 1 mbar.
- the reaction temperature is carefully monitored and the addition of the potassium hydroxide solution is interrupted as soon as a significantly exothermic reaction starts. The temperature may rise up to 80°C.
- the remaining potassium hydroxide solution is added as soon as the initial reaction starts to subside. It may happen that no change in temperature is observed while adding the potassium hydroxide solution. If this happens, the reaction mixture is carefully and slowly heated to 8O 0 C. The exothermic reaction may suddenly start. As soon as the initial reaction starts to subside, the reaction mixture is heated to 100 °C and stirred for 6 h at this temperature.
- the organic layer is separated.
- the aqueous layer is acidified with concentrated hydrochloric acid and extracted twice with methylene chloride.
- the combined organic layers are washed once with 2 M HCl, once with water, dried over anhydrous magnesium sulphate and the solvent is removed under reduced pressure (110.0 g, 87 %).
- the GC-determined purity of the crude product is 92 %.
- Pure product can be obtained by treating the oily residue with 35 ml of ethanol at room temperature.
- Solid 4-(4-tert-Butyl-phenyl)-indan-l-one precipitates and is filtered of.
- the filtrate still contains significant amounts of product which can be isolated by evaporating the filtrate to dryness mixing it with 50 ml of heptane and storing the mixture at -30 0 C over night.
- the filtrate is filtered off, washed with two portions of cold (-30 0 C) heptane and dried in a vacuum (Combined yield: 23.71 g, 82 %;).
- the aqueous layer was extracted once with 60 ml of toluene and the combined organic layers were washed once with 100 ml of saturated NaCl- solution, dried over magnesium sulphate and the solvent was evaporated in a vacuum.
- the crude product was purified via a column chromatography to yield 9.4 g (67 %) of the desired product.
- the filter cake was extracted once with 30 ml, once with 20, once with 15 and once with 10 ml of hot toluene, from the filtrate 2.95 g of the metallocene with a rac/meso-ratio of 4: 1 were obtained. For further r/m enrichment the product was crystallized from toluene.
- Pd / C one (GC: -83 % purity) are dissolved in 250 ml of ethyl acetate in a flask with gas inlet and stirrer. 1.75 g of palladium on activated carbon (10 wt-% palladium) are added. The system is evacuated and refilled with argon three times to remove oxygen and then evacuated and refilled with hydrogen three times. The stirrer is started and the reaction mixture is vigorously stirred to help the diffusion of hydrogen gas into the liquid reaction mixture. The hydrogen uptake is monitored and stirring is continued until the hydrogen uptake ceases.
- Butyl-phenyl)-2-(( 1 -ethyl cyclohexyl)methyl)- 1 H-indene were dissolved in 245 ml of 1.
- the crude reaction mixture was filtered over a G4 frit and the residue was washed twice with 10 ml of diethyl ether.
- the filter cake was extracted once with 30 ml, once with 20, once with 15 and once with 10 ml of hot toluene, from the filtrate 2.80 g of the metallocene with a rac/meso- ratio of 5:1 were obtained.
- the product was crystallized from toluene.
- Example 10 there is no Example 10 in this application. In order to make it easier for the reader to correlate the metallocenes made in Examples 1 thru 8, to the catalysts made in Examples 1 1 through 18, example 10 has been omitted. In this manner, the metallocene of Example 1 is used to prepare the catalyst in Example 1 1, similarly, the metallocene of Comaprative Example 4 is now used to produce the catalyst of Comparative Example 14, etc.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pocketsin the column. The excess toluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing reddish powder in a yield of 12.0 g.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing reddish powder in a yield of 1 1.4 g.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing reddish powder in a yield of 11.8 g.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing orange powder in a yield of 11.9 g.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing orange powder in a yield of 11.9 g.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing orange powder in a yield of 11.3 g.
- Example 9 10.0 g of the methylaluminoxane treated silica prepared in Example 9 are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- Example 19 and Comparative Example 20 are separated from the comparisons made between all other inventive Examples and Comparative Examples.
- the reason for this is that the catalysts for both inventive Example 18 and Comparative Example 19 were prepared by a special process previously revealed by the inventors in U.S. Patent 7,169,864 that has been demonstrated to increase the activity of metallocene catalysts over standard preparation methods. It would be inaccurate to compare catalysts made by the '864 method to those made by standard metallocene preparation methods.
- the residue is washed with two 500 mL portions of toluene and three 500 mL portions of isohexane and dried in vacuum to constant weight.
- the methylaluminoxane treated silica is obtained as a free- flowing powder in a yield of 180 g.
- methylaluminoxane treated silica 10.0 g are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excess toluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing powder in a yield of 12 g.
- the residue is washed with two 500 mL portions of toluene and three 500 mL portions of isohexane and dried in vacuum to constant weight.
- the methylaluminoxane treated silica is obtained as a free-flowing powder in a yield of 180 g.
- methylaluminoxane treated silica 10.0 g are placed in a fritted glass filter as a column with a smooth surface. A minimal amount of toluene is added and the treated silica is carefully stirred with a spatula to remove any air pockets in the column. The excesstoluene is removed by filtration leaving a smooth surface.
- the filtration process is stopped and the filter cake is carefully and thoroughly stirred by means of a spatula.
- the catalyst is then allowed to rest for one hour.
- the residual solvent is filtered off and the catalyst is washed twice with isohexane (20 mL) and dried in a nitrogen purge to constant weight.
- the catalyst is obtained as free-flowing powder in a yield of 11.8 g.
- supported metallocene catalyst suspended in 5 cm 3 of white oil, is injected with liquid propylene (one-half of total amount used for the run).
- the reactor is heated to the internally measured run temperature (65, 60 or 30 °C) within 11 minutes.
- the polymerization reaction is allowed to proceed at the run temperature for either 15 or 60 minutes.
- the reactor pressure was maintained by continuous feeding of ethylene and propylene.
- the polymerization is stopped by releasing the monomer and cooling down the reactor.
- the polymer is discharged and dried under reduced pressure.
- Table 1 and Table 2 represent the raw data presented by sixty-one polymerization test runs.
- the remaining tables 3 - 13 break that data out by the ratio of propylene to ethylene (or if it is a propylene homopolymer) and whether hydrogen was used in the polymerization process. Only those results can be directly compared, where essentially the same polymerisation conditions, like temperature, polymerisation time etc. were applied. Further more many of the tables contain two sets of data that are not comparable with each other. The difference between those data sets is the way the catalyst preparation was performed As explained above, the catalysts of Comparative Examples 1 1-15 and Examples 16-18 were prepared by a different catalyst preparation procedure than Comparative Example 20 and Example 19 and hence different polymerisation conditions are reflected by these sets of data. These data sets can only be compared among themselves.
- Table 3 shows the results of four experimental Metallocene catalysts conforming to the requirements of the invention compared to six comparative examples.
- a general comparison of the molecular weight shows that the highest molecular weights of 920 kg/mol and 910 kg/mol are reached by the comparative example 13 and the inventive example 16, respectively. These molecular weights can be considered equal within the experimental error.
- the activity of the inventive example 16 is almost twice as high as the activity of the comparative example 13, while the melting points of these two examples both are 152°C.
- Table 3 shows that, in the case of propylene without a hydrogen moderator, the species catalysts of the current invention, on average, are an astonishing 51.2% more active than the comparative catalysts claimed under the genus application Further, the species catalysts of the present invention also demonstrate the more desirable properties of a higher T m and MFR 2.16 and MFR 5 values over the tested catalysts that would fall under the claims of the co-pending genus application.
- the second set of comparable results consists ofthe catalyst derived from example 19 and comparative example 20.
- the catalyst preparation of these two catalysts differs from the one ofthe already discussed results and hence they need to be compared seperately.
- the inventive example shows a superior performance compared to the comparative example 20.
- the productivity ofthe inventive example 19 is around 22% increased compared to the comparison example 20, while the molecular weights and the MFR values are comparable.
- the melting point of the inventive example of 153°C is two degrees higher than the one of the comparative example 20.
- Table 4 shows the results of four experimental Metallocene catalysts conforming to the requirements of the invention compared to six comparative examples. However, in this case, hydrogen was added during the polymerization process to enhance catalyst productivity and to regulate the Molecular Weight.
- inventive example 16 is reaching a similar molecular weight, but at a much higher productivity level (56 % increase compared to Comp. 13).
- the melting point for this set of comparable results is ranging between 150 and 155°C.
- the inventive catalyst not only show a higher activity than all the comparative catalyst, they also provide with 152°C- 153 °C melting points of in the upper region of this range.
- the only catalyst that is able to achieve a higher melting point is comparative catalyst 15 with 155 0 C, but this is achieved at the lowest activity (9,520 g/ g h, 36% lower than inventive example 16) and the lowest molecular weight (146 kg/g, 37% lower than inventive example 16) of all the catalyst in this set of results.
- the second set of comparable results consists of the catalyst derived from example 19 and comparative example 20.
- the catalyst preparation of these two catalysts differs from the one of the already discussed results and hence they need to be compared seperately.
- the inventive catalyst 19 clearly shows a dramatically improved performance over the comparative catalyst20.
- the productivity of the inventive catalyst 19 is almost doubled compared to the one of the comparative catalyst 20, while obtaining almost identical molecular weights and MFR values.
- the melting point obtained with the inventive catalyst 19 with 153°C by 2°C higher than the melting point of the comparative catalyst 20.
- the inventive catalyst 16 and the comparative catalyst 11 were tested under two different polymerisation conditions.
- the first set of conditions is defined by a polymerization temperature of 6O 0 C and a polymerization time of 60 minutes, while the second set of conditions is determined by a polymerization temperature of 65°C and a polymerization time of only 15 minutes.
- the results are summarized in table 6.
- the inventive catalyst shows a 76% increase in productivity compared to the comparative example 1 1. While the C2-incorporation and the melting point of the polymers are similar, the MFR 2.16 value of the inventive example 16 is only one-third of the comparative example 1 1. This is also reflected in a 25% increase of the molecular weight compared to the comparative example 1 1.
- the second set of conditions again shows the superior performance of the inventive catalyst.
- the productivity of the inventive example 16 shows an 11% increase compared to the comparative example 1 1.
- the slightly lower melting point of 133°C for the inventive catalyst 16 is a result of the higher C2-incorporation of the inventive catalyst under this set of conditions (2.4 compared to 2.1 % for the comparative example 11).
- the MFR 2.16 value of the inventive example 16 is only one-fifth of the comparative example 11. This is also reflected in a 32% increase of the molecular weight compared to the comparative example 11.
- inventive catalyst examples 16
- comparative catalyst comparative example 1 1
- productivity of the inventive example 16 shows a 78% increase compared to comparative example 1 1.
- molecular weight of the inventive example 16 shows an increase for the inventive catalyst, namely 59 % compared to the comparative example 1 1. This is also reflected in the values for the MFR 2.16, which is for the inventive example only one-fourth of the comparative example 1 1. Since the C2-incorporation of the inventive catalyst 16 is slightly higher than for the comparative catalyst 1 1, it is not surprising, that the melting point for the inventive example is slightly lower. Under these conditions the superiority of the inventive catalyst is clearly visible.
- inventive catalyst 16 and the comparative catalyst 11 were tested, the results being presented in Table 8. As with all the analyses before, the inventive catalyst 16 in this example achieves better productivity (27 %). This is achieved at a similar C2-incorporation and the same melting point of 118°C as the comparative example 11.
- the molecular weight of the inventive example is significantly enhanced by 54% compared to the comparative example 11. This fact can also be seen in the value of the MFR 2.16 for the inventive example 16, that is only one-fourth of the value of the comparative catalyst.
- inventive catalyst examples 16
- comparative catalyst comparative example 1 1
- the inventive catalyst shows a large enhancement of the productivity and the molecular weight comparedto the comparative catalyst 1 1, while showing a similar C2-incorporation and a similar melting point.
- productivity is doubled compared to the comparative catalyst 1 1 and the molecular weight is increased by a factor of 1.5. This is also reflected by the MFR 2.16 value, which is for the inventive example 16 only one-sixth of the value obtained for the comparative catalyst 1 1.
- inventive catalyst 16 and the comparative catalyst 1 1 were tested, and the results are presented in Table 10. As with all the analysis before, the inventive catalyst 16 in this example achieves a better productivity by 41%. This is achieved at a similar C2-incorporation and the slightly lower melting point (1°C) of inventive example 1 1 can be rationalised by the higher C2- incorporation of 7.2 % compared to 6.5 % of the comparative example 11. The molecular weight of the inventive catalyst 16 is increased by 35 % compared to the comparative catalyst 1 1. The higher molecular weight is also reflected in the MFR 2.16 value, which is only one-fourth of the value of the comparative catalyst 11.
- Table 1 1 shows the results of the inventive catalyst 16 and the comparative example 1 1 at a propylene/ethylene ratio of approximately 0.55.
- the productivity of these samples could not be determined because in all of these examples and comparative examples amorphous propylene/ethylene rubbers have been produced.
- Such polymers generally stick to the autoclace walls and to the stirrer and a quantitative discharge of the autoclave was not possible which makes the determination of the productivities unreliable.
- Commercially, polymers containing such rubber components are produced in a two step polymerisation where in a first step a homo polymer is produced and in a second step the rubber is produced. This measure reduces the stickiness of the material and allows the commercial production and use of sudi important materials for applications where low temperature toughness is required (applications like bumpers for cars, frigerator and deep freezer food packaging, crates and pails).
- the C2-incorporation of the catalysts is quite similar, although the inventive catalyst 16 shows a slightly higher value of 41.3 % compared to 39.7 % for the comparative catalyst 11.
- the example of the inventive catalyst shows a dramatic improvement over the comparative examples in gains of molecular weight, and reductions in the MFR values.
- the molecular weight of the comparative example 1 1 is more than two times lower than the one obtained with the inventive catalyst 1 1 (280 kg/mol vs. 659 kg/mol). This result is also reflected in the MFR 2.16 values, which is 0.1 g/10' for the inventive catalyst and 6.9 g/10' for the comparative example 1 1.
- the inventive catalyst in this example achieves a much higher molecular weight, while still creating a product with comparable C2-incorporation.
- the molecular weight of the inventive catalyst 1 1 is by a factor 2.3 higher than the molecular weight of the comparative catalyst. This result is also reflected in the MFR 2.16 values, which is 0.2 g/10' for the inventive catalyst and 1 1.5 g/ 10' for the comparative example 1 1.
- the inventive catalyst shows a much superior performance under these conditions, since it delivers a polymer with a much higher molecular weigth at a similar C2 incorporation.
- a generally applicable commercial catalyst has to show a high versatility, meaning a high performance under all the conditions relevant for producing commercially available materials Under the conditions applied here this means that copolmyers with very low MFR 2.16 values below 0.5 g/10' have to be obtained in order to obtain materials where toughness at very low temperatures is required. All the inventive catalysts fulfill this requirement, even more, they are able in all cases to obtain an MFR 2.16 value of 0.2 g/10' or lower (example 16, 17, 18 and also 19).
- the only comparative catalyst that obtains a lower MFR 2.16 ( ⁇ 0.1 g/10') is the one from comparative example 13. Since a MFR 2.16 value of ⁇ 0.1 g/10' does not mean a commercial advantange compared to values of up to 0.5 g/10' and this catalyst has been shown to have a much lower productivity under all other conditions tested, it can be rationalized, that it has not such a high versatility as the inventive catalysts.
- the molecular weights of the inventive catalysts 16, 17, 18 are all in the range of 600 and 700 kg/mol, and only the comparative examples 13 and 15 can reach such high molecular weights. But as explained before at much lower productivities for all other conditions tested.
- the second set of comparable results consists of the catalyst derived from example 19 and comparative example 20.
- the catalyst preparation of these two catalysts differs from the one of the already discussed results and hence they need to be compared separately.
- This second set of conditions showed an unexpected result.
- the copolymer obtained was not sticky and it showed a nice morphology, as shown in the photograph of Fig. 3.
- This photo shows the copolymer rubber obtained in polymerisation example 45 using the catalyst from Example 19.
- the yield could be determined to be 8,700 g / g h.
- the comparative example 20 produced a sticky polymer, shown in Fig. 2.
- This photo shows the copolymer rubber obtained in polymerisation Example 61 using catalyst from Comparative Example 20.
- the inventive example 19 was producing a much higher molecular weight and a much lower MFR value than the comparative example 20 at comparable C2-introduction.
- the molecular weight obtained for the inventive example 19 is by a factor 4.6 higher than that of the comparative example 20. This is also reflected by a much lower MFR value of 0.1 g/10' compared to 18 g/10' for the comparative example 20.
- the different polymerization conditions tested represent the most applied conditions to obtain commercially relevant polypropylene-based materials.
- the inventive catalysts have shown under all conditions higher productivities than the corresponding comparative catalysts and most of the time better or at least similar properties of the resulting materials. Hence the inventive catalysts show a much higher versatility than the state-of-the-art catalysts and a better performance under all commercially relevant polymerization conditions.
Abstract
Description























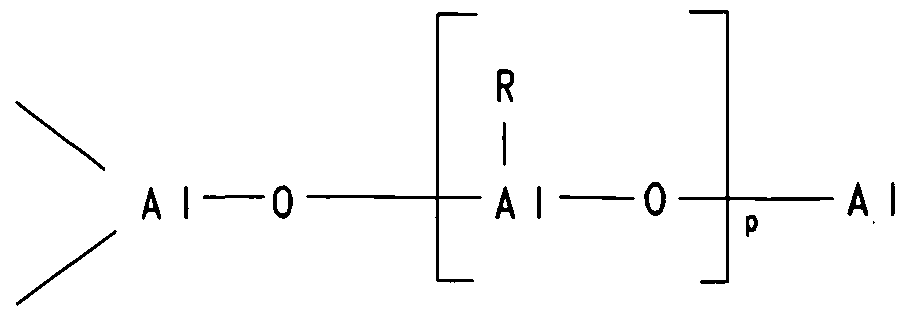
























































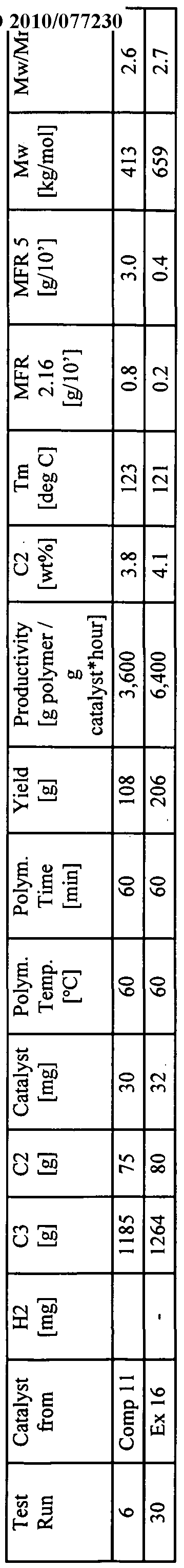






Claims
























Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200880132439.2A CN102257020B (en) | 2008-12-31 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
| EP08876396A EP2384343A1 (en) | 2008-12-31 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
| PCT/US2008/014144 WO2010077230A1 (en) | 2008-12-31 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
| MX2011006667A MX2011006667A (en) | 2008-12-31 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers. |
| JP2011543489A JP2012513463A (en) | 2008-12-31 | 2008-12-31 | New metallocene compound, catalyst containing the same, process for producing olefin polymer using the catalyst, and olefin homopolymer and copolymer |
| EA201170909A EA201170909A1 (en) | 2008-12-31 | 2008-12-31 | NEW METAL COMPOUNDS, CATALYSTS, INCLUDING SUCH CONNECTIONS, METHOD FOR OBTAINING AN OLEFIN POLYMER USING SUCH CATALYSTS AND OLEFIN HOMO AND COPOLYMERS |
| SG2011048345A SG172454A1 (en) | 2008-12-31 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
| KR1020117016447A KR101351103B1 (en) | 2008-12-31 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
| BRPI0823407-8A BRPI0823407A2 (en) | 2008-12-31 | 2008-12-31 | Novel metallocene compounds, catalysts comprising them, Process for producing an olefin polymer by use of olefin catalysts and homo- and copolymers |
| US13/107,594 US8415492B2 (en) | 2007-10-25 | 2011-05-13 | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
| ZA2011/05497A ZA201105497B (en) | 2008-12-31 | 2011-07-26 | New metallocene compounds,catalysts comprising them,process for producing an olefin polymer by use of the catalysts,and olefin homo-and copolymers |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2008/014144 WO2010077230A1 (en) | 2008-12-31 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
Related Parent Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/739,078 Continuation-In-Part US9240888B2 (en) | 2003-03-05 | 2007-04-23 | Authentication system for gaming machines |
| PCT/US2007/022614 Continuation-In-Part WO2009054832A1 (en) | 2007-10-25 | 2007-10-25 | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo-and copolymers |
| US12/739,078 Continuation-In-Part US8299287B2 (en) | 2007-10-25 | 2007-10-25 | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/107,594 Continuation US8415492B2 (en) | 2007-10-25 | 2011-05-13 | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010077230A1 true WO2010077230A1 (en) | 2010-07-08 |
Family
ID=40936621
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/014144 WO2010077230A1 (en) | 2007-10-25 | 2008-12-31 | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP2384343A1 (en) |
| JP (1) | JP2012513463A (en) |
| KR (1) | KR101351103B1 (en) |
| CN (1) | CN102257020B (en) |
| BR (1) | BRPI0823407A2 (en) |
| EA (1) | EA201170909A1 (en) |
| MX (1) | MX2011006667A (en) |
| SG (1) | SG172454A1 (en) |
| WO (1) | WO2010077230A1 (en) |
| ZA (1) | ZA201105497B (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2573091A1 (en) | 2011-09-23 | 2013-03-27 | Lummus Novolen Technology Gmbh | Process for recycling of free ligand from their corresponding metallocene complexes |
| US8865848B2 (en) | 2010-07-01 | 2014-10-21 | Borealis Ag | Process for olefin polymerisation using group 4 metallocene as catalysts |
| US8933256B2 (en) | 2010-07-01 | 2015-01-13 | Borealis Ag | Catalysts |
| US9458260B2 (en) | 2013-07-17 | 2016-10-04 | Exxonmobil Chemical Patents Inc. | Process using substituted metallocene catalysts and products therefrom |
| US9464145B2 (en) | 2013-07-17 | 2016-10-11 | Exxonmobil Chemical Patents Inc. | Metallocenes and catalyst compositions derived therefrom |
| US9469708B2 (en) | 2012-10-18 | 2016-10-18 | Borealis Ag | Polymerisation process |
| US9469700B2 (en) | 2012-10-18 | 2016-10-18 | Borealis Ag | Polymerisation process and catalyst |
| US9475890B2 (en) | 2012-10-18 | 2016-10-25 | Borealis Ag | Catalyst |
| US9644047B2 (en) | 2013-07-17 | 2017-05-09 | Exxonmobil Chemical Patents Inc. | Metallocenes and catalyst compositions derived therefrom |
| US9701772B2 (en) | 2013-07-24 | 2017-07-11 | Borealis Ag | Process for preparation of propylene copolymer |
| US9834628B2 (en) | 2013-07-17 | 2017-12-05 | Exxonmobil Chemical Patents Inc. | Cyclopropyl substituted metallocene catalysts |
| US9938364B2 (en) | 2013-07-17 | 2018-04-10 | Exxonmobil Chemical Patents Inc. | Substituted metallocene catalysts |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6176015B2 (en) | 2012-11-01 | 2017-08-09 | 日本ポリプロ株式会社 | Metallocene complex and olefin polymerization method |
| JP5710035B2 (en) | 2013-02-27 | 2015-04-30 | 日本ポリエチレン株式会社 | Metallocene compound and process for producing olefin polymer |
| JP5695687B2 (en) | 2013-03-05 | 2015-04-08 | 日本ポリエチレン株式会社 | Process for producing ethylene / α-olefin copolymer |
| KR102405286B1 (en) * | 2017-12-06 | 2022-06-02 | 주식회사 엘지화학 | Method for preparing supported metallocene catalyst, the supported metallocene catalyst prepared by the same method, and polypropylene prepared by using the same |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001048034A2 (en) * | 1999-12-23 | 2001-07-05 | Basell Polyolefine Gmbh | Transition metal compound, ligand system, catalyst system and the use of the latter for the polymerisation and copolymerisation of olefins |
| WO2002002576A1 (en) * | 2000-06-30 | 2002-01-10 | Exxonmobil Chemical Patents Inc. | Bridged bis (indenyl) metallocene compounds |
| WO2009054832A1 (en) * | 2007-10-25 | 2009-04-30 | Novolen Technology Holdings, C.V. | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo-and copolymers |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE454098T1 (en) * | 1998-02-10 | 2010-01-15 | Artemis Medical Inc | OCCLUSION, ANCHORING, CHIPING OR POWER CONTROL DEVICE |
| US7232869B2 (en) * | 2005-05-17 | 2007-06-19 | Novolen Technology Holdings, C.V. | Catalyst composition for olefin polymerization |
| RU2476449C2 (en) * | 2007-10-25 | 2013-02-27 | Люммус Новолен Текнолоджи Гмбх | Racemoselective synthesis of ansa-metallocene compounds, ansa-metallocene compounds, catalysts containing said compounds, method of producing olefin polymer using catalysts and olefin homo- and copolymers |
-
2008
- 2008-12-31 EP EP08876396A patent/EP2384343A1/en not_active Withdrawn
- 2008-12-31 SG SG2011048345A patent/SG172454A1/en unknown
- 2008-12-31 CN CN200880132439.2A patent/CN102257020B/en not_active Expired - Fee Related
- 2008-12-31 WO PCT/US2008/014144 patent/WO2010077230A1/en active Application Filing
- 2008-12-31 KR KR1020117016447A patent/KR101351103B1/en not_active IP Right Cessation
- 2008-12-31 BR BRPI0823407-8A patent/BRPI0823407A2/en not_active IP Right Cessation
- 2008-12-31 EA EA201170909A patent/EA201170909A1/en unknown
- 2008-12-31 MX MX2011006667A patent/MX2011006667A/en not_active Application Discontinuation
- 2008-12-31 JP JP2011543489A patent/JP2012513463A/en not_active Ceased
-
2011
- 2011-07-26 ZA ZA2011/05497A patent/ZA201105497B/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001048034A2 (en) * | 1999-12-23 | 2001-07-05 | Basell Polyolefine Gmbh | Transition metal compound, ligand system, catalyst system and the use of the latter for the polymerisation and copolymerisation of olefins |
| WO2002002576A1 (en) * | 2000-06-30 | 2002-01-10 | Exxonmobil Chemical Patents Inc. | Bridged bis (indenyl) metallocene compounds |
| WO2009054832A1 (en) * | 2007-10-25 | 2009-04-30 | Novolen Technology Holdings, C.V. | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo-and copolymers |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8865848B2 (en) | 2010-07-01 | 2014-10-21 | Borealis Ag | Process for olefin polymerisation using group 4 metallocene as catalysts |
| US8933256B2 (en) | 2010-07-01 | 2015-01-13 | Borealis Ag | Catalysts |
| WO2013041619A1 (en) | 2011-09-23 | 2013-03-28 | Lummus Novolen Technology Gmbh | Process for recycling of free ligand from their corresponding metallocene complexes |
| EP2573091A1 (en) | 2011-09-23 | 2013-03-27 | Lummus Novolen Technology Gmbh | Process for recycling of free ligand from their corresponding metallocene complexes |
| US9469700B2 (en) | 2012-10-18 | 2016-10-18 | Borealis Ag | Polymerisation process and catalyst |
| US9475890B2 (en) | 2012-10-18 | 2016-10-25 | Borealis Ag | Catalyst |
| US9469708B2 (en) | 2012-10-18 | 2016-10-18 | Borealis Ag | Polymerisation process |
| US9458260B2 (en) | 2013-07-17 | 2016-10-04 | Exxonmobil Chemical Patents Inc. | Process using substituted metallocene catalysts and products therefrom |
| US9464145B2 (en) | 2013-07-17 | 2016-10-11 | Exxonmobil Chemical Patents Inc. | Metallocenes and catalyst compositions derived therefrom |
| US9644047B2 (en) | 2013-07-17 | 2017-05-09 | Exxonmobil Chemical Patents Inc. | Metallocenes and catalyst compositions derived therefrom |
| US9745390B2 (en) | 2013-07-17 | 2017-08-29 | Exxonmobil Chemical Patents Inc. | Metallocenes and catalyst compositions derived therefrom |
| US9834628B2 (en) | 2013-07-17 | 2017-12-05 | Exxonmobil Chemical Patents Inc. | Cyclopropyl substituted metallocene catalysts |
| US9938364B2 (en) | 2013-07-17 | 2018-04-10 | Exxonmobil Chemical Patents Inc. | Substituted metallocene catalysts |
| US9951155B2 (en) | 2013-07-17 | 2018-04-24 | Exxonmobil Chemical Patents Inc. | Process using substituted metallocene catalysts and products therefrom |
| US10487161B2 (en) | 2013-07-17 | 2019-11-26 | Exxonmobil Chemical Patents Inc. | Cyclopropyl substituted metallocene catalysts |
| US9701772B2 (en) | 2013-07-24 | 2017-07-11 | Borealis Ag | Process for preparation of propylene copolymer |
Also Published As
| Publication number | Publication date |
|---|---|
| SG172454A1 (en) | 2011-08-29 |
| JP2012513463A (en) | 2012-06-14 |
| ZA201105497B (en) | 2012-04-25 |
| BRPI0823407A2 (en) | 2015-06-16 |
| EA201170909A1 (en) | 2012-01-30 |
| EP2384343A1 (en) | 2011-11-09 |
| MX2011006667A (en) | 2011-07-20 |
| CN102257020B (en) | 2014-04-02 |
| KR101351103B1 (en) | 2014-01-14 |
| CN102257020A (en) | 2011-11-23 |
| KR20110094349A (en) | 2011-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8415492B2 (en) | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers | |
| KR101351103B1 (en) | New metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers | |
| US7285608B2 (en) | Metallocene ligands, metallocene compounds and metallocene catalysts, their synthesis and their use for the polymerization of olefins | |
| EP2203486B9 (en) | Racemoselective synthesis of ansa-metallocene compounds, ansa-metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo- and copolymers | |
| CZ434598A3 (en) | Metallocene compounds | |
| EP2235071A2 (en) | Metallocene compounds, catalysts comprising them, process for producing an olefin polymer by use of the catalysts, and olefin homo and copolymers | |
| JP2014196319A (en) | Novel metallocene compound, catalyst containing the compound, process of producing olefin polymer by using the catalyst and olefin homopolymer and copolymer | |
| JP2022528411A (en) | Propylene-ethylene random copolymer | |
| US6365763B1 (en) | Method for producing metallocenes | |
| JP6196581B2 (en) | Metallocene compound, catalyst containing the same, process for producing olefin polymer using the catalyst, and olefin homopolymer and copolymer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880132439.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08876396 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11054021 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2011/006667 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2011543489 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4958/DELNP/2011 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20117016447 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201170909 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008876396 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI0823407 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI0823407 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110630 |